Нейротензин активные производные 2,3-диарилпиразолидина - RU2304578C2
Код документа: RU2304578C2
Описание
Изобретение относится к группе новых производных 2,3-диарилпиразолидина, обладающих ингибирующей активностью по отношению к ферментам, которые разлагают нейропептид нейротензин.
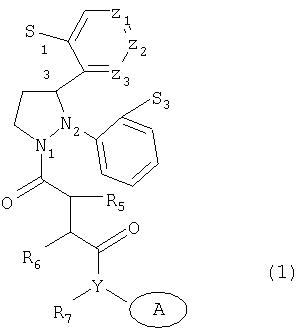
Было обнаружено, что соединения, имеющие формулу (1):

где
S1 представляет водород, галоген, гидрокси или С1-С3 алкокси;
S2 представляет водород или галоген;
S3 представляет водород, галоген, гидрокси или С1-С3 алкокси;
S4 представляет водород, галоген или С1-С6 алкил, необязательно замещенный гидрокси, С1-С3 алкокси, амино, моно- или диалкиламино, имеющим от 1 до 3 атомов углерода в алкильной группе (группах), SH или S-(С1-С3)алкил;
X представляет азот или углерод;
Y представляет азот или кислород, если Х представляет азот, или Y представляет азот, если Х представляет углерод;
R3 и R4 независимо друг от друга представляют водород или С1-С3 алкил;
R5 представляет водород или С1-С6 алкил, который может быть замещен галогеном, CN, CF3, гидрокси, С1-С3 алкокси, С1-С3 сульфонилалкилом, амино, моно- или диалкиламино, имеющим от 1 до 3 атомов углерода в алкильной группе (группах), когда Х представляет углерод или азот, или R5 представляет С1-С6 алкокси, SH или S-(С1-С3)алкил, когда Х представляет углерод;
R5' представляет водород или С1-С3 алкил;
R6 представляет водород или С1-С3 алкил;
R7 представляет водород или С1-С3 алкил;
R5 и R6 вместе или R5' и R6 вместе могут образовывать (3-7)-членную циклическую группу, которая может быть замещена низшим алкилом, галогеном, CN или CF3, и R5+R5' вместе могут образовывать (3-7)-членное кольцо;
Z1, Z2 и Z3 представляют углерод, или Z1 представляет азот, а Z2 и Z3 представляют углерод, или Z1 и Z3 представляют углерод, а Z2 представляет азот, или Z1 и Z2 представляют углерод, а Z3 представляет азот;
A представляет (поли)циклоалкильную систему, состоящую из (4-10)-членных колец, которые могут быть замещены галогеном, CF3, алкилом или С1-С3 алкокси, CN, OH или SH;
и их соли обладают способностью ингибировать ферменты, разлагающие нейротензин.
Более конкретно, соединения ингибируют ферменты олигопептидазу Тимета ЕС 3.4.24.15 и нейролизин ЕС 3.4.24.16, которые разрушают нейропептид нейротензин.
Благодаря ингибированию разрушающей нейротензин активности этих ферментов концентрации эндогенного нейротензина должны возрастать, оказывая благоприятное воздействие в лечении заболеваний, при которых нарушены уровни нейротензина.
Соединения согласно изобретению являются активными при ингибировании вышеупомянутых ферментов в интервале 5, 0-8,0 (значения pIC50) при испытании согласно методам, описанным в Biochem.J. 280, 421-426 и Eur.J.Biochem. 202, 269-276.
Соединения согласно изобретению могут быть использованы для лечения поражений и заболеваний, вызываемых нарушениями медиируемой нейротензином передачи, таких как периферические расстройства, подобные расстройствам регуляции кровяного давления и опорожнения желудка, неврологические расстройства, подобные болезни Паркинсона, и расстройства центральной нервной системы (ЦНС), такие как тревожность, депрессии, психоз и другие психические расстройства.
Соединения, имеющие формулу (1), могут быть получены согласно по меньшей мере одному из следующих четырех способов A, B, C и D. Исходными соединениями для этих четырех способов являются замещенные 2,3-диарилпиразолидины, имеющие одну из структур, показанных на фигуре 1:
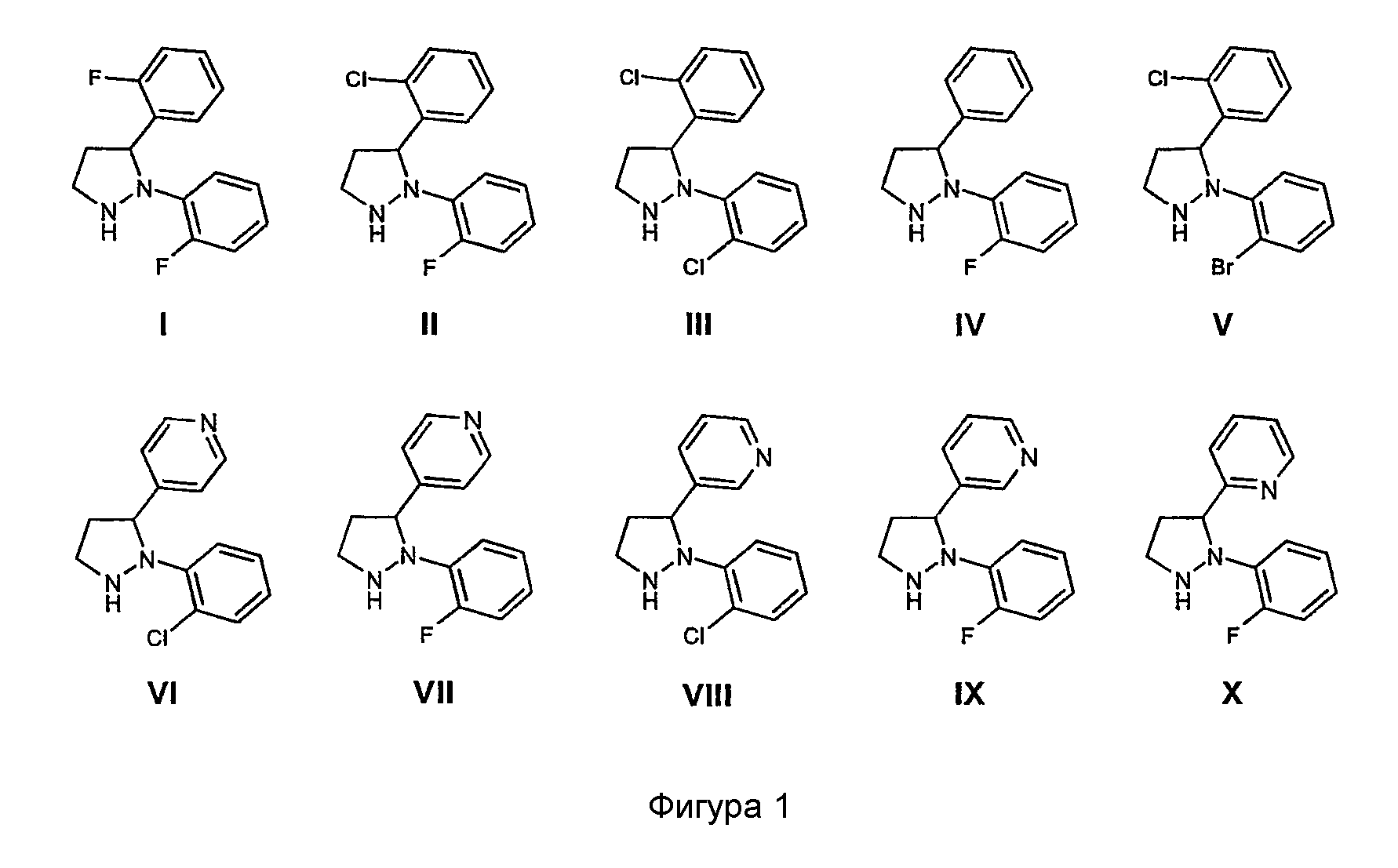
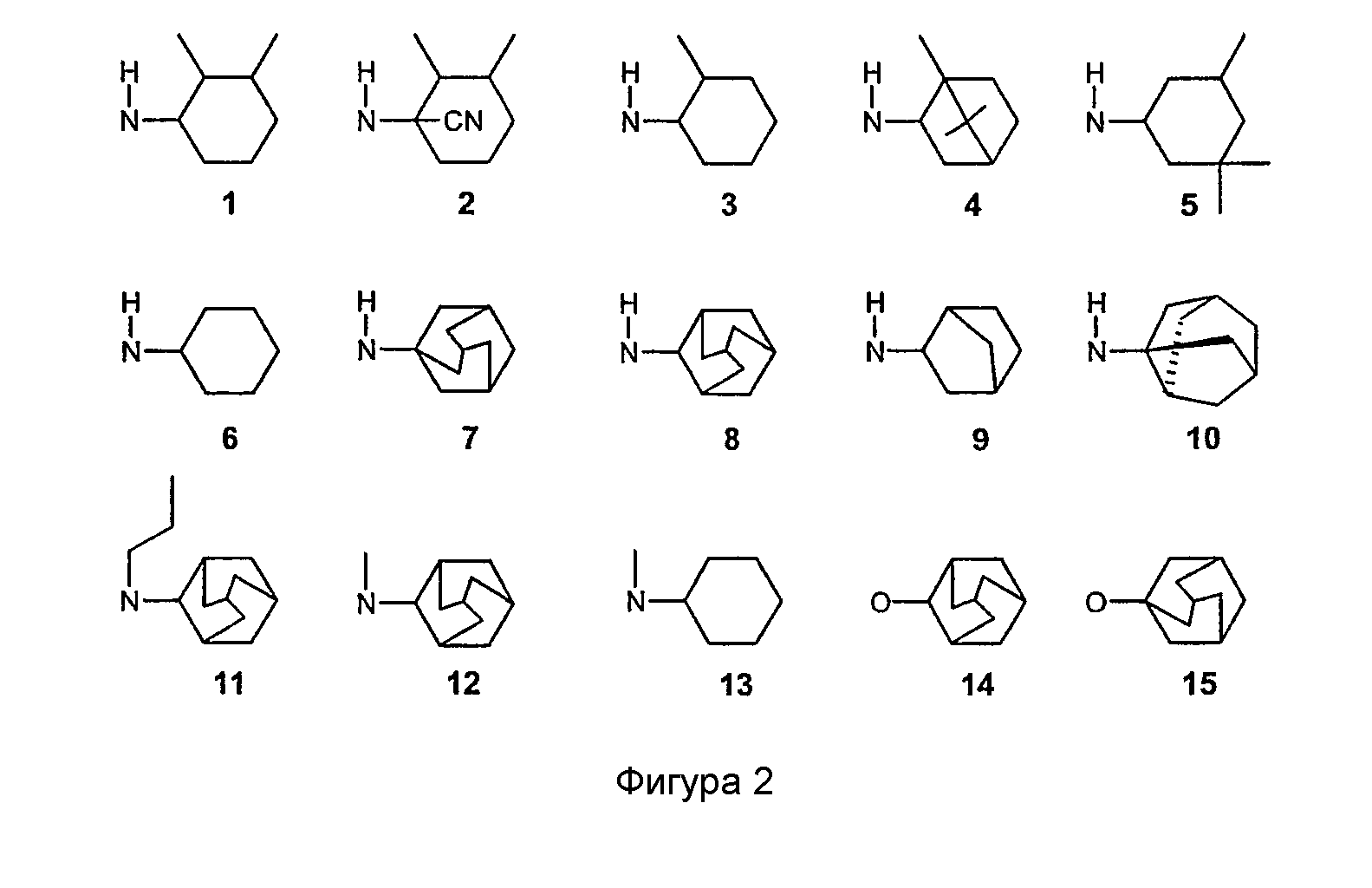
Часть R7-Y-A соединений, имеющих формулу (1), может иметь структуры групп, показанных на фигуре 2.
Исходные производные пиразолидина фигуры 1 могут быть получены способом в соответствии со схемой 1:

как объяснено в примере 5.
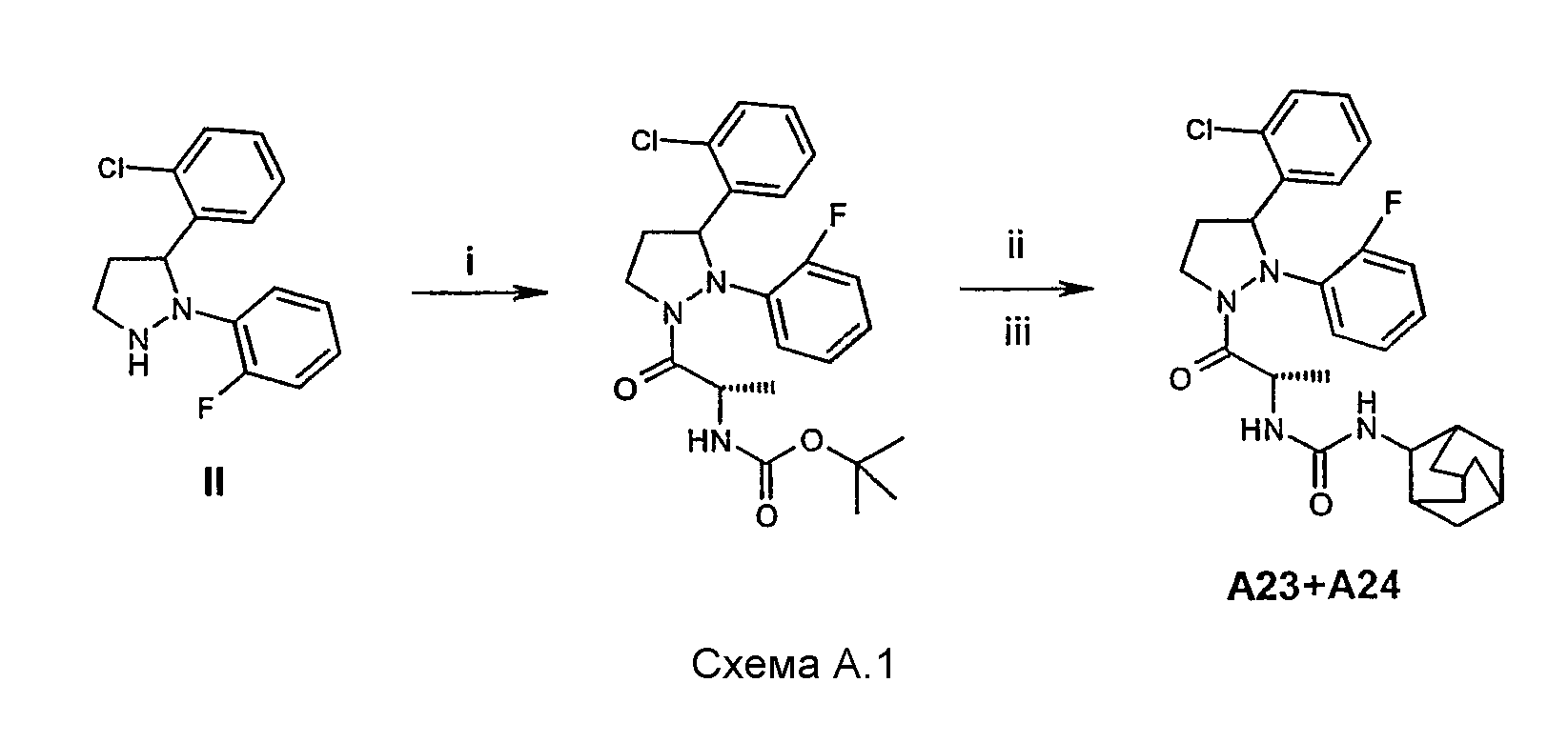
Способ А
Соединения, упомянутые в таблице А, могут быть синтезированы в соответствии с синтезом соединения А23/А24. После стадии i возникают два диастереомера, которые после проведения стадии iii могут быть разделены колоночной хроматографией на энантиомерно чистые диастереомеры А23 и А24 (см. схему А.1).
Способ В
Соединения, упомянутые в таблице В могут быть получены согласно синтезу, показанному на схеме В.1.

Реакционные стадии i и ii схемы В.1 идентичны процедурам, описанным на схеме А.1, стадии i и ii соответственно.
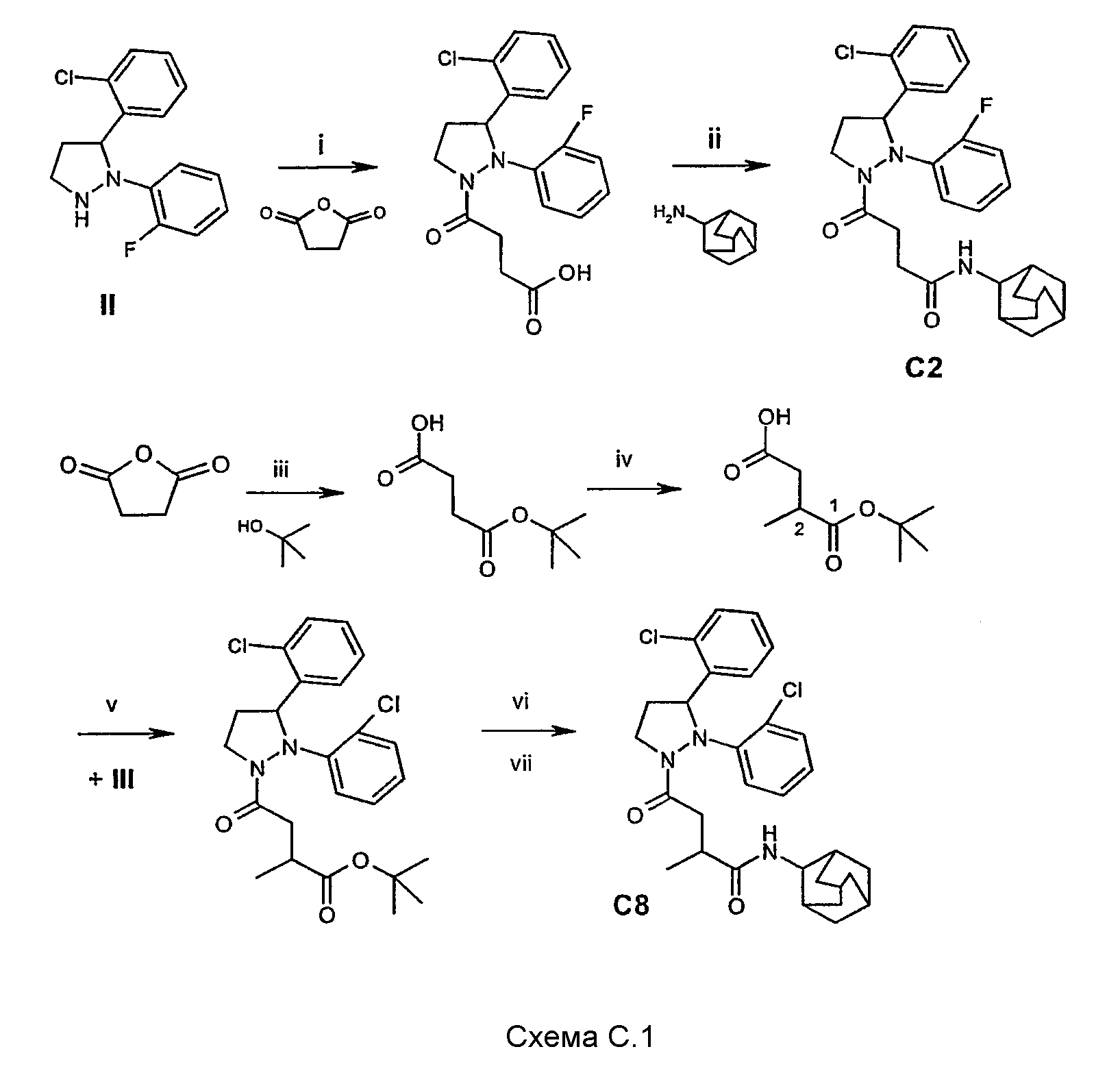
Способ C
Соединения, упомянутые в таблице С, могут быть получены в соответствии с синтезом соединений С2 и С8, как изображено на схеме С.1:
Способ D
Соединения, упомянутые в таблице D, могут быть получены в соответствии с синтезом соединения D1, как показано на схеме D.1:
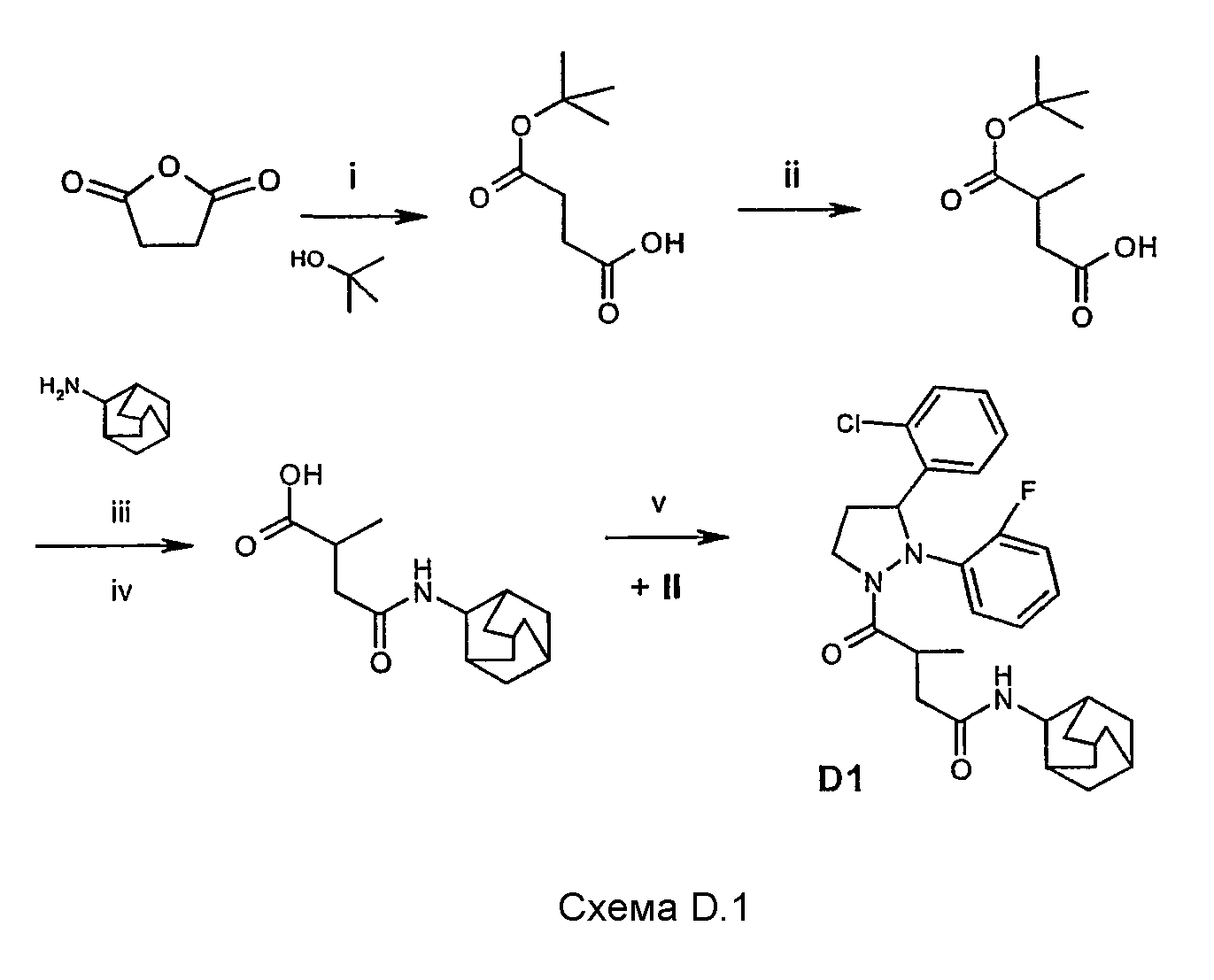
Реакционные стадии i и ii схемы D.1 идентичны процедурам, описанным на схеме C.1, стадии iii и iv соответственно.
Получение соединений формулы (1) и ряда промежуточных соединений согласно способам A-D будет далее подробно описано в следующих примерах.
Пример 1
Стадия i (схема А.1):
К 50 мл перемешиваемого сухого ацетонитрила при комнатной температуре и в атмосфере азота добавляли 4 г (14,5 ммоль) соединения II, 2,7 г (14,3 ммоль) N-Boc-L-Alanine и 3,8 г (18,4 ммоль) DCC (дициклогексилкарбодиимида). Сразу образовывался осадок. Перемешивание продолжали в течение одной ночи. Тонкослойная хроматография реакционной смеси показала подобное восьмерке двойное пятно, содержащее два возможных диастереомера. Осадок отделяли фильтрацией. К фильтрату добавляли около 20 г диоксида кремния и концентрировали его в вакууме. Полученный порошок помещали в верх сухой колонки (SiO2), после чего проводили элюирование (элюент: CH2Cl2/MeOH, 98/2). Часть насадки колонки, содержащую два диастереомера, отбирали в МеОН. Суспензию в последнем фильтровали, остаток промывали еще один раз МеОН. Соединенные МеОН-фракции концентрировали в вакууме и полученный остаток поглощали CH2Cl2, после чего его сушили над MgSO4. В результате удаления осушающего агента фильтрацией и растворителя выпариванием в вакууме выделили 5 г (80%) сырого продукта.
Стадия ii (схема А.1)
При перемешивании 5 г (около 10 ммоль) продукта, полученного на стадии i, растворяли в 100 мл раствора, состоящего из трифторуксусной кислоты/CH2Cl2/воды 70/25/5. Перемешивание продолжали в течение 2 часов. После этого реакционную смесь концентрировали в вакууме, полученный остаток поглощали CH2Cl2. Последний раствор обрабатывали насыщенным водным раствором K2CO3, промывали водой и раствором соли и наконец сушили над MgSO4.
После удаления осушающего агента фильтрацией и растворителя выпариванием в вакууме выделяли 4 г (около 100%) сырого амина.
Стадия iii (схема А.1)
При комнатной температуре и в атмосфере азота 0,50 г (1,44 ммоль) сырого амина со стадии ii суспендировали в 10 мл ацетонитрила при перемешивании. После этого добавляли 0,26 г (1,44 ммоль) 2-адамантилизоцианата. Реакцию продолжали в течение 2 часов. К реакционной смеси добавляли около 2 г диоксида кремния и смесь концентрировали в вакууме. Полученный порошок помещали на верх сухой колонки (SiO2), после чего проводили элюирование (элюент: EtOAc/петролейный эфир 1/1). Участки насадки колонки, содержавшие диастереомеры, отбирали по отдельности и помещали в МеОН. Полученные две суспензии по отдельности фильтровали, каждый из двух остатков промывали один раз МеОН. Для каждого диастереомера объединяли соответствующие МеОН-фракции и концентрировали в вакууме, после чего каждый остаток поглощали CH2Cl2 и оба раствора сушили над MgSO4. После удаления осушающего агента и растворителя в вакууме получали два твердых вещества, каждое из которых содержало один диастереомер: 0,16 г А23 (21%), температура плавления 140-143°С, и 0,22 г А24 (29%), температура плавления 145-148°С
Примечание:
Соединение А12 было получено энантиомерно чистым. Промежуточное соединение после стадии ii (схема А.1) было разделено на энантиомеры, после чего была проведена стадия iii (схема А.2). (+)- Энантиомер А12 был эутомером.
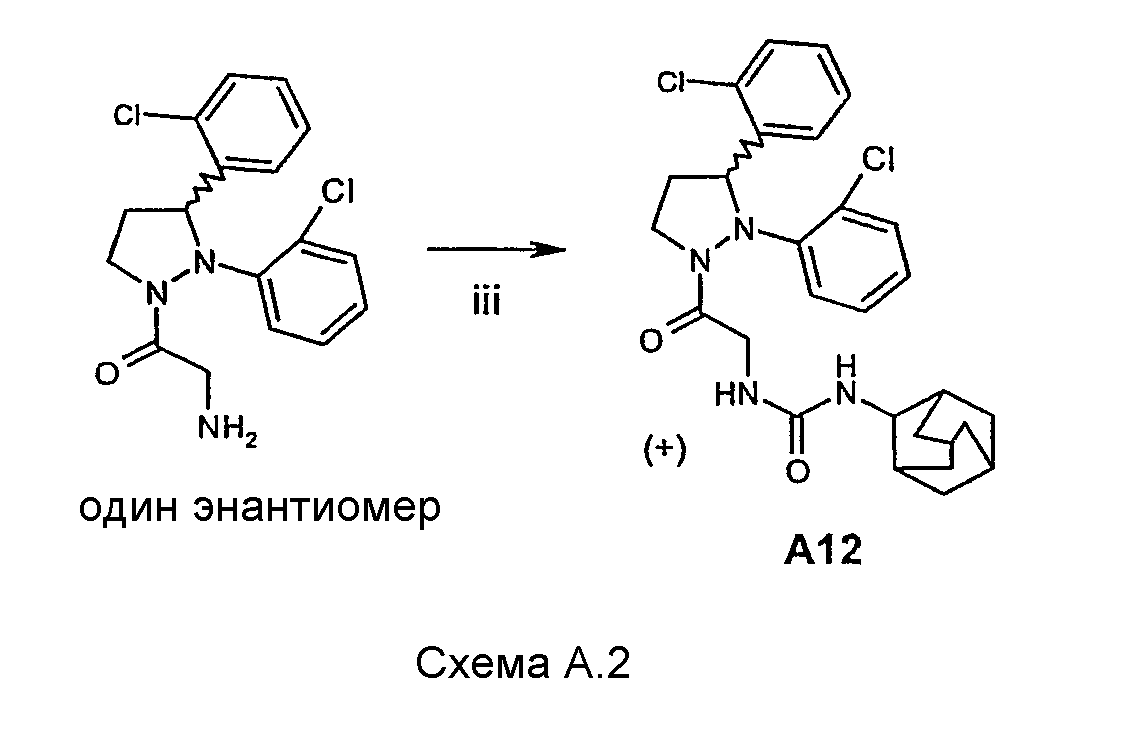
Разделение на энантиомеры промежуточного соединения после стадии ii (схема А.1) проводили с использованием колонки Chiracel CD (25×5 см2, 20 μ, элюент: гексан/этанол 4/1).
Соединения таблицы А были получены аналогичным образом.
Пример 2
Стадия iii (схема В.1)
0,20 г (0,67 ммоль) трифосгена растворяли в 10 мл сухого дихлорметана. К данной смеси за период 45 минут добавляли раствор 0,70 г (2,0 ммоль) производного пиразолидина и 0,42 мл (2,4 ммоль) диизопропилэтиламина. Реакционную смесь непрерывно перемешивали. После этого в реакционную смесь за 5 минут добавляли раствор, содержавший 0,33 г (2,0 ммоль) метил-2-адамантиламина и 0,42 мл (2,4 ммоль) диизопропилэтиламина в 5 мл сухого дихлорметана. Реакционной смеси давали возможность реагировать в течение одной ночи, после чего растворитель выпаривали в вакууме. Остаток поглощали этилацетатом и полученный раствор обрабатывали 5% водным NaHCO3 и раствором соли соответственно. Органический слой отделяли и сушили над MgSO4. Фильтрация осушающего агента и удаление растворителя в вакууме дают масло, которое подвергают колоночной хроматографии (SiO2, элюент: CH2Cl2/MeOH 99/1). Отбор фракций, содержавших продукт, и последующее удаление элюента в вакууме дали масло, которое кристаллизовали при перемешивании из диизопропилового эфира. Фильтрация и сушка на воздухе дают 0,69 г (64%) твердого В2 (т.пл. 184-186°С).
Примечание: Использованный метил-2-адамантиламин может быть легко получен обычными методиками восстановительного аминирования исходя из 2-адамантанона и гидрохлорида метиламина с использованием NaBH(OAc)3 в качестве восстанавливающего агента.
Соединения таблицы В были получены аналогичным образом.
Примечание: Необходимое промежуточное соединение после стадии ii (схема В.1) в случае В3 (R6=Me) может быть получено аналогично стадиям i и ii в соответствии со схемой A.1.
Пример 3
Стадия i (схема С.1)
16 г (160 ммоль) янтарного ангидрида растворяли в сухом диэтиловом эфире. После этого к раствору янтарного ангидрида при перемешивании добавляли по каплям 44 г (160 ммоль) соединения II, растворенного в диэтиловом эфире. После того как добавление завершилось, реакционную смесь доводили до температуры кипения с обратным холодильником, которое продолжали в течение одной ночи. Образовался осадок, который отфильтровывали, остаток дважды промывали диэтиловым эфиром. Сушка на воздухе дала 45,6 г (75%) требуемого промежуточного соединения.
Стадия ii (схема С.1)
В атмосфере азота 4,5 г (12 ммоль) промежуточного соединения стадии i и 7,9 г (61 ммоль, 5,1 экв.) диизопропилэтиламина растворяли в 50 мл сухого CH2Cl2, полученный раствор при перемешивании доводили до 4°С. После этого добавляли 0,90 г (7,0 ммоль) 1-гидрокси-7-азабензтриазола и 4,20 г (15 ммоль) гексафторфосфата 2-хлор-1,3-диметилимидазолиния. Затем 2,19 г (15 ммоль) 2-аминоадамантана добавляли к реакционной смеси, которой давали возможность реагировать в течение одного часа при комнатной температуре.
К реакционной смеси добавляли около 4 г диоксида кремния и концентрировали ее в вакууме. Полученный порошок помещали на верх сухой колонки (SiO2), после чего проводили элюирование (элюент: EtOAc/петролейный эфир 1/1). Участок насадки колонки, содержавший продукт, отбирали и помещали в МеОН. Полученную суспензию фильтровали, остаток промывали один раз МеОН. МеОН-фракции объединяли и концентрировали в вакууме, после чего остаток поглощали CH2Cl2 и полученный раствор сушили над MgSO4. После удаления осушающего агента и растворителя в вакууме получали твердое вещество: 2,0 г С2 (32%), температура плавления 192-195°С.
Стадия iii (схема С.1)
При перемешивании и в атмосфере азота 6,0 г (60 ммоль) янтарного ангидрида растворяли в 35 мл толуола. После этого добавляли 2,07 г (18 ммоль) N-гидроксисукцинимида, 0,73 г (6 ммоль) 4-диметиламинопиридина, 13,3 г (18 ммоль) сухого трет-бутанола и 1,82 г (18 ммоль) триэтиламина. Реакционную смесь доводили до температуры кипения с обратным холодильником и давали реакции протекать в течение одной ночи. Реакционную смесь охлаждали, после чего добавляли EtOAc. Полученный раствор последовательно обрабатывали 10% водной лимонной кислотой и раствором соли, после чего органические фракции сушили над MgSO4. Удаление осушающего агента и растворителя выпариванием в вакууме дает коричневое масло. Кристаллизация из диэтилового эфира/гексана дала 4,4 г (42%) желаемого моноэфира.
Стадия iv (схема С.1)
Реакцию проводили согласно методике, описанной в Synthesis (2000), p. 1369-71. Моно-трет-бутиловый эфир янтарной кислоты метилировали в положение 2 реакцией с диизопропиламидом лития и метилйодидом в тетрагидрофуране при -78°С. Выход выделенного моно-трет-бутилового эфира 2-метилянтарной кислоты составил 60%.
Стадия v (схема С.1):
При перемешивании 1,8 г (9,8 ммоль) моно-трет-бутилового эфира 2-метилянтарной кислоты (стадия iv) растворяли в 45 мл сухого CH2Cl2, после чего раствор доводили до 4°С. К последнему раствору добавляли 0,9 г (6,4 ммоль) 1-гидрокси-7-азабензтриазола и 4,0 г (15 ммоль) гексафторфосфата 2-хлор-1,3-диметилимидазолиния. Последующее добавление 4,1 г соединения III (14 ммоль) не приводило к повышению температуры, и реакции давали протекать в течение ночи при комнатной температуре. К реакционной смеси добавляли около 3 г силикагеля (SiO2), после чего ее концентрировали в вакууме. Полученный порошок помещали на верх сухой колонки (SiO2), после чего проводили элюирование (элюент: EtOAc/петролейный эфир 1/4). Участок насадки колонки, содержащий продукт, отбирали и помещали в МеОН. Полученную суспензию фильтровали, остаток промывали еще один раз МеОН. Объединенные МеОН-фракции концентрировали в вакууме и полученный остаток поглощали CH2Cl2, после чего его сушили над MgSO4. Удалением осушающего агента фильтрацией и растворителя в вакууме выделили 3 г (66%) нужного промежуточного соединения.
Стадия vi (схема С.1):
Гидролиз трет-бутилового эфира промежуточного соединения стадии v осуществляли следующим образом: 3 г (6,4 ммоль) трет-бутилового эфира растворяли в 30 мл сухого CH2Cl2, после чего по каплям добавляли 10 мл трифторуксусной кислоты. Спустя два часа реакция завершалась, и реакционную смесь концентрировали в вакууме, после чего остаток, растворенный в небольшом количестве диэтилового эфира, помещали на верх короткой колонки (сухой SiO2) и элюировали диэтиловым эфиром. Содержащий продукт элюат концентрировали в вакууме, остаток перемешивали в течение ночи в петролейном эфире. Кристаллы отбирали фильтрацией, после сушки на воздухе получали 2,1 г (80%) нужного промежуточного соединения.
Стадия vii (схема С.1):
В атмосфере азота 2,17 г (5,3 ммоль) промежуточного соединения стадии vi и 4,7 мл (27 ммоль, 5,1 экв.) диизопропилэтиламина растворяли в 25 мл сухого CH2Cl2, полученный раствор при перемешивании доводили до 4°С. После этого добавляли 0,42 г (3,1 ммоль) 1-гидрокси-7-азабензтриазола и 1,85 г (6,6 ммоль) гексафторфосфата 2-хлор-1,3-диметилимидазолиния. Затем 1,0 г (6,6 ммоль) 2-аминоадамантана добавляли в реакционную смесь, которой давали возможность реагировать в течение одного часа при комнатной температуре.
К реакционной смеси добавляли около 4 г диоксида кремния и смесь концентрировали в вакууме. Полученный порошок помещали на верх сухой колонки (SiO2), после чего проводили элюирование (элюент: EtOAc/петролейный эфир 1/2). Участки насадки колонки, содержащие рацематы диастереомеров, отбирали по отдельности и помещали в МеОН. Полученные две суспензии по отдельности фильтровали, каждый из двух остатков промывали один раз МеОН. Для каждого диастереомерного рацемата объединяли соответствующие МеОН-фракции и концентрировали в вакууме, после чего каждый остаток поглощали CH2Cl2, после чего два раствора сушили над MgSO4. После удаления осушающего агента и растворителя в вакууме получали два твердых вещества, каждое из которых содержало один из возможных диастереомерных рацематов; были получены: 1,08 г С8 (37%), активный рацемат, температура плавления 238-240°С, и 1,09 г (37%) другого фармакологически неактивного рацемата, температура плавления 125-130°С (не показан в таблице С).
Соединения таблицы С были получены аналогичным образом.
Пример 4
Стадия iii (схема D.1):
В атмосфере азота 0,92 г (4,9 ммоль) промежуточного соединения стадии ii и 4,4 мл (25 ммоль, 5,1 экв.) диизопропилэтиламина растворяли в 15 мл сухого CH2Cl2, полученный раствор при перемешивании доводили до 4°С. После этого добавляли 0,45 г (3,3 ммоль) 1-гидрокси-7-азабензтриазола и 2,1 г (7,5 ммоль) гексафторфосфата 2-хлор-1, 3-диметилимидазолиния. Затем 1,08 г (7,2 ммоль) 2-аминоадамантана добавляли в реакционную смесь, которой давали возможность реагировать в течение одного часа при комнатной температуре. Данную реакционную смесь использовали на следующей стадии iv.
Стадия iv (схема D.1)
К реакционной смеси стадии iii при перемешивании добавляли 45 мл сухого CH2Cl2, а также 11 мл (143 ммоль) трифторуксусной кислоты. Перемешивание продолжали в течение 24 часов. Реакционную смесь концентрировали в вакууме, после чего остаток, растворенный в небольшом количестве диэтилового эфира, помещали на верх короткой колонки (сухой SiO2) и элюировали диэтиловым эфиром. Содержащий продукт элюат концентрировали в вакууме, получая 0,87 г (67%, две стадии) нужного кислого промежуточного соединения.
Стадия v (схема D.1):
При перемешивании 0,87 г (3,28 ммоль) моноамида метилянтарной кислоты (стадия iv) растворяли в 15 мл сухого CH2Cl2, после чего раствор доводили до 4°С. К полученному раствору добавляли 0,3 г (2,2 ммоль) 1-гидрокси-7-азабензтриазола и 1,40 г (5,0 ммоль) гексафторфосфата 2-хлор-1,3-диметилимидазолиния. Последующее добавление 1,33 г соединения II (4,80 ммоль) не приводило к повышению температуры, и реакции давали протекать в течение ночи при комнатной температуре. К реакционной смеси добавляли около 3 г силикагеля (SiO2), после чего ее концентрировали в вакууме. Полученный порошок помещали на верх сухой колонки (SiO2), после чего проводили элюирование (элюент: EtOAc/петролейный эфир 1/1). Участки насадки колонки, содержащие диастереомерные рацематы, отбирали по отдельности и помещали в МеОН. Полученные две суспензии по отдельности фильтровали, каждый из двух остатков промывали один раз МеОН. Для каждого диастереомерного рацемата объединяли соответствующие МеОН-фракции и концентрировали в вакууме, после чего каждый остаток поглощали CH2Cl2, после чего два раствора сушили над MgSO4. После удаления осушающего агента и растворителя в вакууме получали два твердых вещества, каждое из которых содержало один из возможных диастереомерных рацематов; были получены: 0,31 г (18%) неактивного рацемата (не показан в таблице D), особенности (характер) плавления: температура плавления 90-95°С, отверждается при 130°С, повторно плавится при 160-165°С, и 0,40 г (23%) активного рацемата D1, характер плавления: плавление 80-82°С, отверждается при 100°С, повторно плавится при 125-128°С.
Соединения таблицы D были получены аналогичным образом.
Пример 5
2,3-Диарилпиразолидины I-X, использованные в качестве исходных веществ в вышеприведенных примерах 1-4, получали следующим образом:
Стадия i (схема 1):
Смесь 16,9 мл уксусной кислоты и 2,3 мл воды охлаждали (смесью лед/вода), после чего осторожно добавляли 6,8 мл концентрированной серной кислоты. К охлажденному раствору при интенсивном перемешивании и в атмосфере азота порциями добавляли 13,3 г (82 ммоль) 2-фторфенилгидразина. К последнему раствору порциями добавляли смесь, состоящую из 10,0 г (82 ммоль) 2-фторстирола и 2,46 г (82 ммоль) параформальдегида, поддерживая температуру ниже 25°С. Реакция может продолжаться в течение некоторого времени. Интенсивное перемешивание продолжали в течение одной ночи при комнатной температуре. При охлаждении добавляли 50 мл воды, после чего проводили экстракцию диэтиловым эфиром (2×). Оставшуюся водную фракцию подщелачивали 50% водным NaOH и затем экстрагировали диэтиловым эфиром (2×). Последнюю эфирную фракцию промывали водой (3×) и раствором соли (1×) и, наконец, сушили над MgSO4. Фильтрация осушающего агента и удаление растворителя в вакууме дали 16 г (75%) сырого сиропообразного масла. Масло не очищали и хранили в атмосфере азота при -20°С, чтобы предотвратить окисление пирролидинового ядра.


Реферат
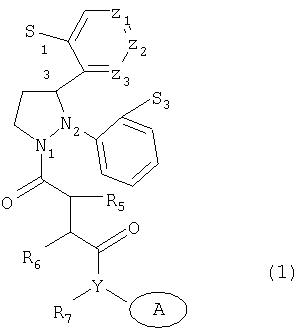
Изобретение относится к группе новых соединений формулы (1).
где S1 представляет водород, галоген; S3 представляет галоген; Х представляет азот или углерод; Y представляет азот или кислород, когда Х представляет азот, или Y представляет азот, когда Х представляет углерод; R5 представляет водород или C1-С6 алкил; R6 представляет водород или C1-С3 алкил; R7 представляет водород или C1-С3 алкил; Z1, Z2 и Z3 представляют углерод, или Z1 представляет азот, а Z2 и Z3 представляют углерод, или Z1 и Z3 представляют углерод, а Z2 представляет азот, или Z1и Z2 представляют углерод, а Z3 представляет азот; А представляет (поли)циклоалкильную систему, состоящую из (4-10)-членных колец, которые могут быть замещены алкилом или CN; и его фармацевтически приемлемые соли. Изобретение также относится к фармацевтической композиции и к применению соединений на основе формулы (1), обладающих ингибирующей активностью по отношению к ферментам, которые разрушают нейропептид нейротензин. Технический результат - получение новых биологически активных соединений и фармацевтических композиций на их основе, обладающих ингибирующей активностью по отношению к ферментам, которые разрушают нейропептид нейротензин. 4 н.п. ф-лы, 4 табл.
Формула
Документы, цитированные в отчёте о поиске
Производные замещенных 1-(7-хлорхинолин-4-ил)пиразол-3-карбоксамид-n-оксидов, способ их получения, промежуточные соединения и фармацевтическая композиция
Производные амидопиразолов или их соли с органическими или минеральными кислотами или с органическими или неорганическими основаниями и фармацевтическая композиция, обладающая ингибирующей активностью в отношении нейротензина
Комментарии