Производные амидопиразолов или их соли с органическими или минеральными кислотами или с органическими или неорганическими основаниями и фармацевтическая композиция, обладающая ингибирующей активностью в отношении нейротензина - RU2066317C1
Код документа: RU2066317C1
Чертежи
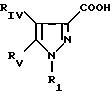
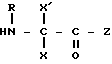
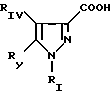
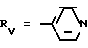
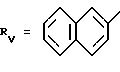
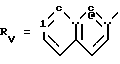

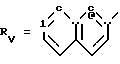
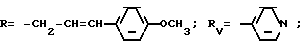
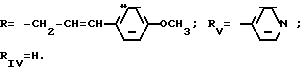
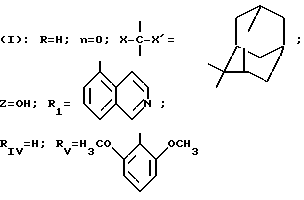
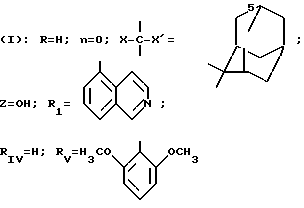
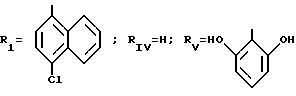
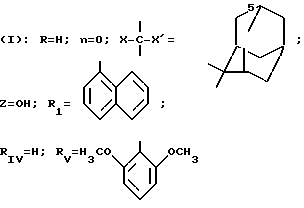
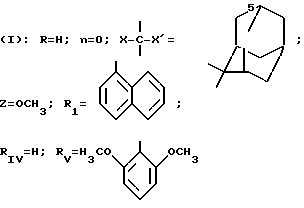
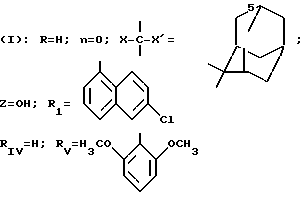
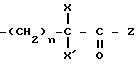
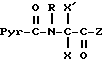
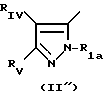
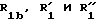
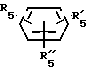
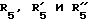
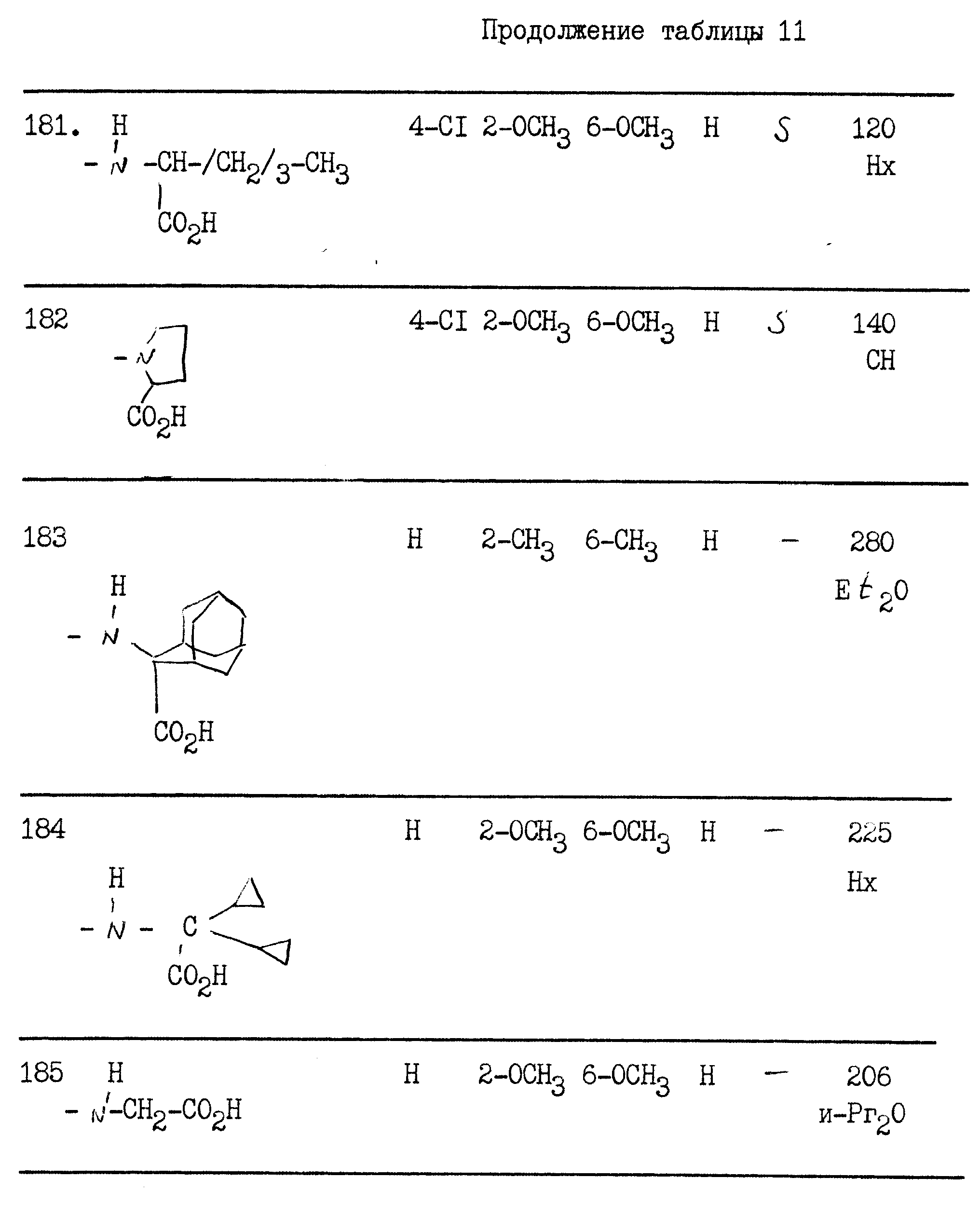
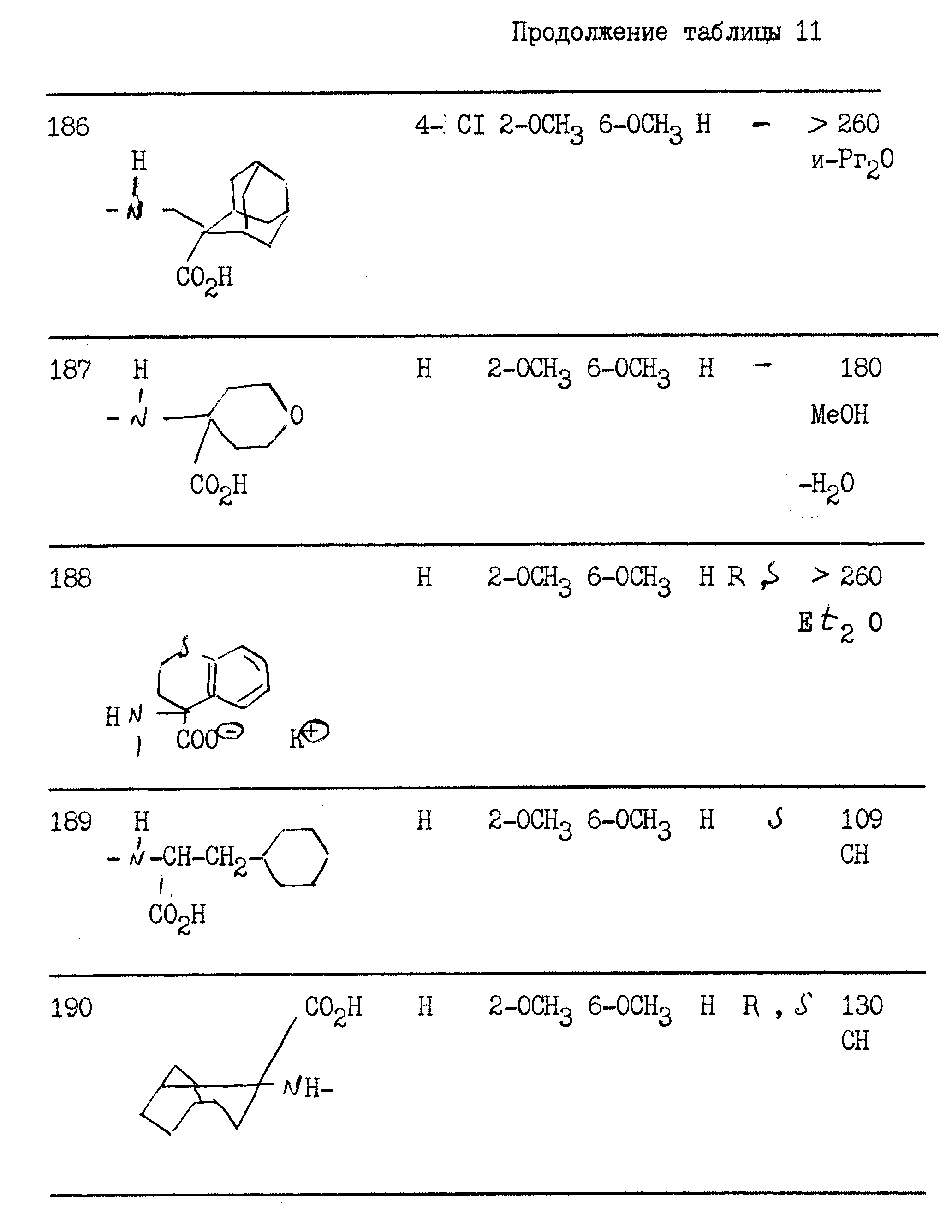
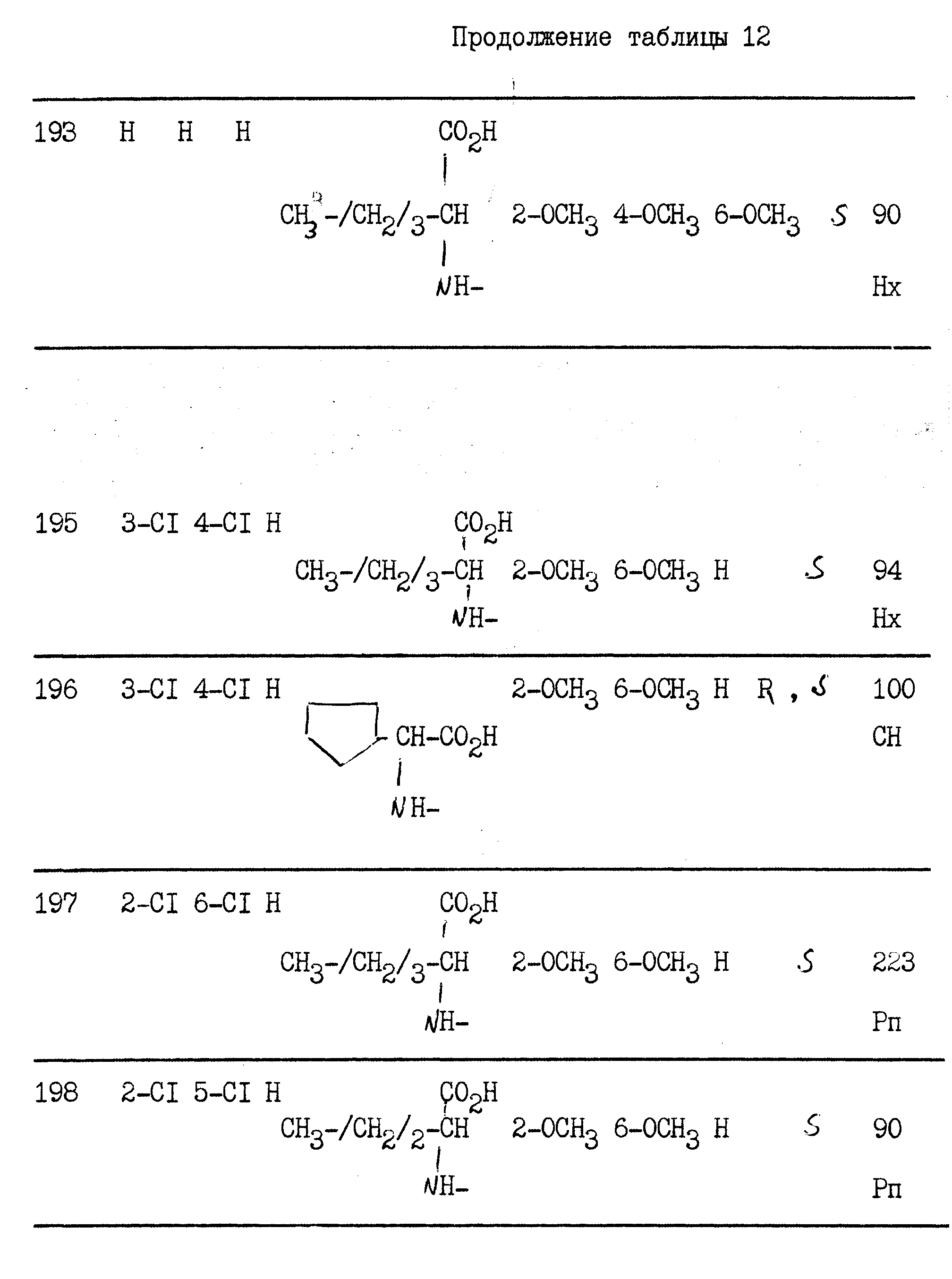
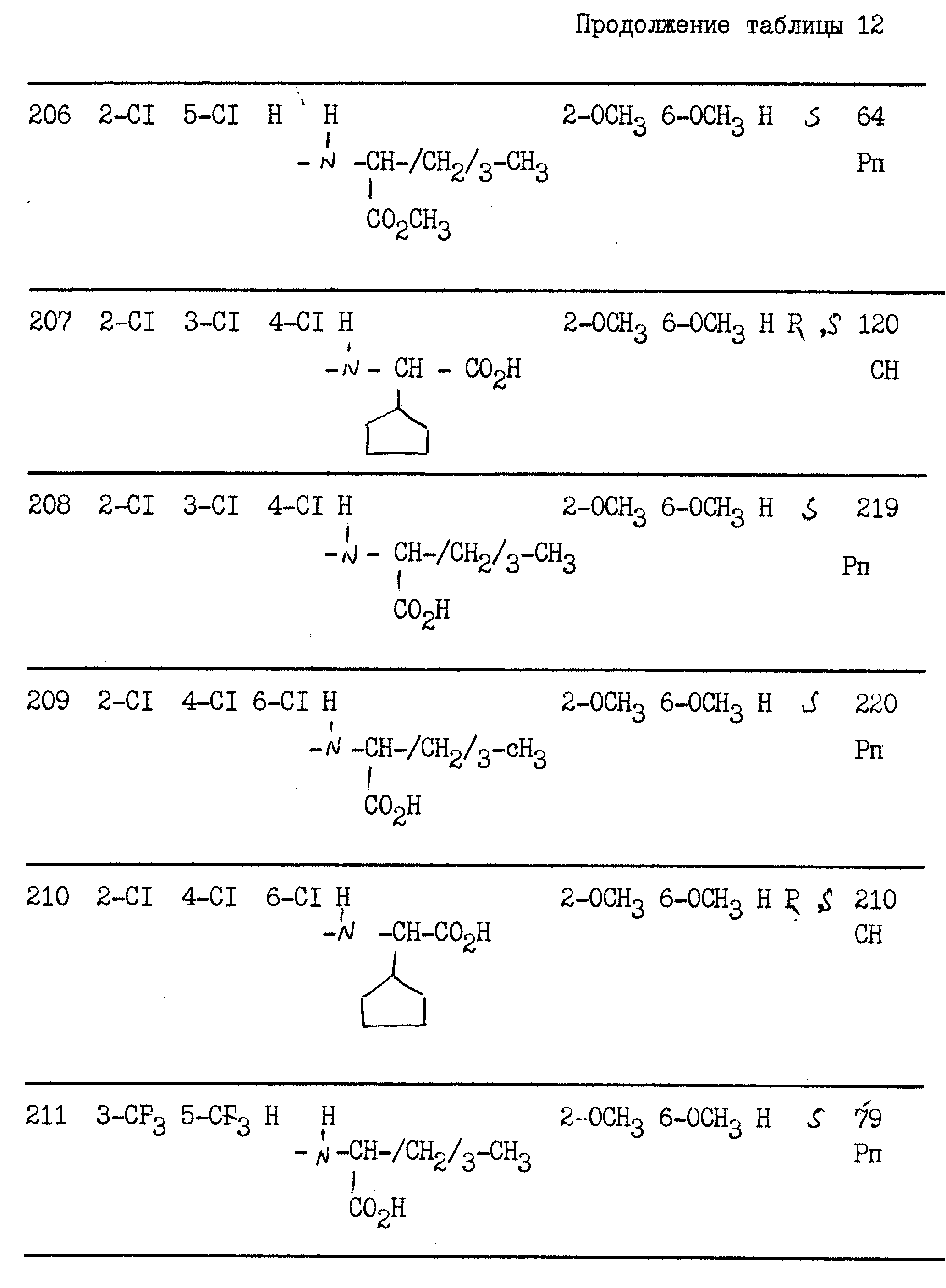
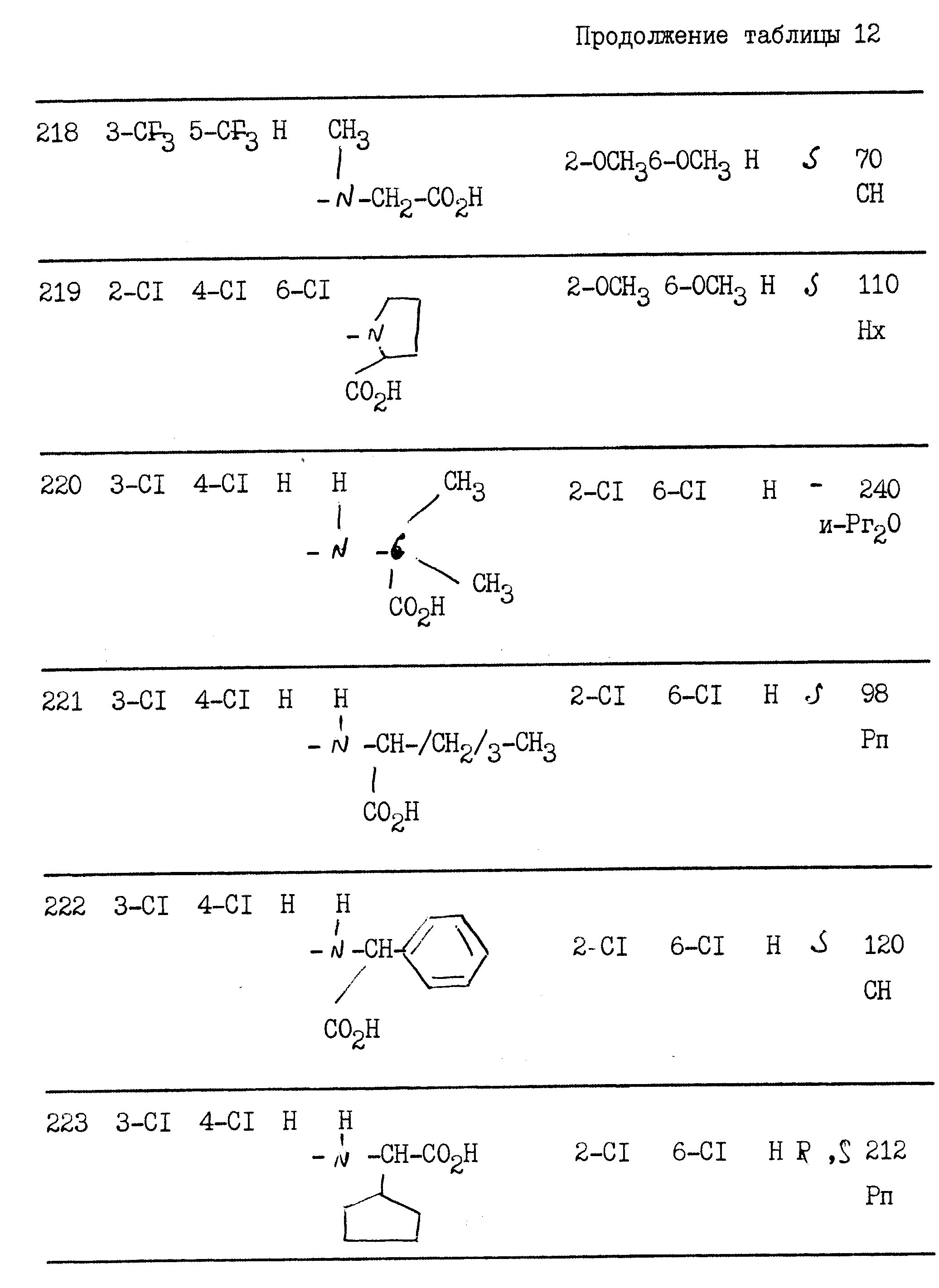

Описание
Изобретение относится к новым производным 3-амидопиразола, обладающим биологической активностью, и к их использованию в фармкомпозициях.
Известны из литературы (ЕП 0248594) 1,5-диарилпиразолы, замещенные в положении 3 алкил (C2-CF16) амидной группой, которые проявляют противовоспалительную активность и активность в отношении сердечно-сосудистой системы.
Заявитель разработал новый класс производных пиразола, которые обладают более широким спектром биологической активности.
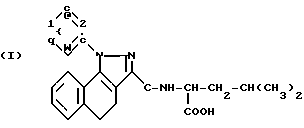
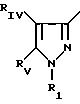
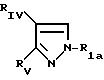
В частности, изобретение относится к новым производным амидопиразола общей формулы I:
Производные амидопиразолов общей формулы I
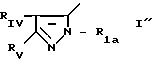
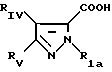
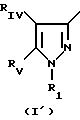
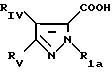
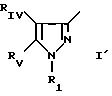
в которой Pyr означает группу формулы II или III:
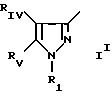
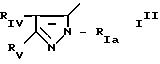
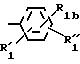
где RI представляет собой группу
где RIв, R и R является каждый независимо друг от друга атомом водорода, атомом галогена, (C1-C4) алкилом с прямой или разветвленной цепью, (С1-C4) алкоксигруппой, трифторметильной группой, трифторметоксигруппой, нитрогруппой или аминогруппой, или RI представляет собой группу, выбранную из ряда содержащего (C3-C6) циклоалкил, тетрагидронафтил, пиридил, нафтил, при необходимости замещенный на галоген, хинолил или изохинолил, при необходимости замещенные на галоген, 2-бензотиазолил, бензотиадиазоил и фталазинилдионовую группу
RIa является бензильной группой, при необходимости замещенной галогеном,
RIV является водородом или атомом галогена, алкилом (С1-C6),
Rv означает группу
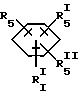
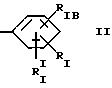
где R5, R и R является каждый независимо друг от друга атомом водорода, атомом галогена, (C1-C4), алкилом с прямой или разветвленной цепью, гидроксилом, (C1-C4) алкокси, фенилом или Ry означает нафтильную или пиридильную или замещенную (C1-C4 ) алкилом стирильную группу, или RIV и RV, взятые вместе означают группу
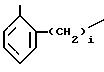
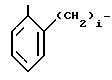
где фенильная группа замещает пиразол в положении 5 и группа (CH2)i, в которой i равно целому числу от 1 до 3, замещает пиразол в положении 4, R является водородом, (С1-C4) алкилом с прямой или разветвленной цепью, Z является гидроксильной группой, (C1-C6) алкокси группой, аминогруппой, Х и ХI оба являются водородом или один из них является водородом, а другой (C1-C6) алкилом с прямой или разветвленной цепью, (С3-C6) циклоалкил (С1-C4) алкилом, амино (С1-C4) алкилом, гидрокси (C1-C4) алкилом, карбокси (С1-C4) алкилом, ацетамидо (С1-C4) алкилтиометилом, гуанидино (C1-C4) алкилтиометилом, гуанидино (С1-C4)
алкилом, нитрогуанидино (C1-C4) алкилом, (С3-C7) циклоалкилом, фенил (C1-C4) алкилом, при необходимости замещенным галогеном, или гидроксилом, гетероарил (C1-C4) алкилом, где гетероарил означает имидазолил или индолил, или Х является водородом и XI и R, взятые вместе, образуют с атомом азота, с которым R связан, цикл формулы
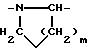
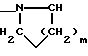
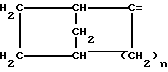
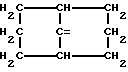
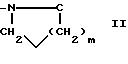
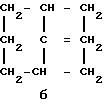
где m равно целому числу 1 или 2, или цикл индолинила или 4,5,6, 7-тетрагидро-(2,3-С)-тиенопиридина, или Х и ХI каждый означает (C1-C4) алкил, (C3-C6 циклоалкил или фенил, или Х и ХI вместе с атомом углерода, с которым они связаны образуют (C3-C12) циклоалкилиденовую группу, при необходимости замещенную (C1-C3) алкилом, адамантилиденовую группу, хинуклидинилиденовую группу, 4-пиперидинилиденовую группу, при необходимости N-замещенную бензильной группой, тетрагидронафтилиденовую группу, 4-тетрагидропиранилиденовую группу, дигидро 2,3 (4Н)-4-бензотиопиранилиденовую группу дигидро 2,3(4Н)-4-бензопиранилиденовую группу или группу формулы а
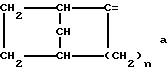
где n 1 или 2, или группу формулы б

или их соли с органическими или минеральными кислотами или с органическими или неорганическими основаниями.
Возможные соли продуктов формулы (I) в соответствии с настоящим изобретением включают как соли с минеральными или органическими кислотами, которые обеспечивают разделение или подходящую кристаллизацию соединений с формулой (I) или (I'), такими как пикриновая кислота или щавелевая кислота, так и соли, которые являются фармацевтически приемлемыми, такие как хлоргидрат, бромгидрат, сульфат, гидросульфат, дигидрофосфат, метансульфонат, метилсульфонат, малеат, фумарат, 2-нафталинсульфонат.
Возможные соли продуктов формулы (I) включают также соли с катионами, например соли щелочных или щелочно-земельных металлов, такие как соли натрия, калия, кальция, причем соль натрия является предпочтительной, когда указанный продукт формулы (I) содержит группу карбоновой кислоты.
Способ получения соединений формулы (I) заключается в том, что
функциональное
производное пиразолкарбоновой кислоты формулы (II) или формулы (II')
в которых RI, RIV, RV и RIa являются такими; как определено выше, обрабатывают аминокислотой, защищенной в случае необходимости защитными группами, обычными для пептидного синтеза формулы:
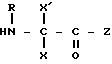
в которой R, X, X' и Z являются такими, как определено выше, или в случае необходимости защищенными.
В качестве функционального производного пиразолкарбоновой кислоты формулы (III) или (II') можно использовать хлорангидрид, ангидрид, смешанный ангидрид, сложный эфир, активированный сложный эфир, например, сложный эфир п-нитрофенила, или свободную кислоту в необходимый момент активированную, например, при помощи N,N-дициклогексилкарбодиимида или при помощи гексафторфосфата бензотриазолил-N-окситрис-(диметиламино)-фосфония (БОФ).
Полученные таким образом соединения (I) могут потом в случае необходимости подвергнуться снятию защиты с целью получения соответствующих свободных кислот.
Сложные эфиры, предшественники карбоновых кислот (II) и (II'),
определенных выше, синтезируются при использовании метода,
описанного в
Chem.
Pharm, Bull, 1984, 32, 4, 1577.
Если аминокислота содержит в качестве заместителя гидроксильную группу, то последняя может быть защищена обычно используемой О-защищающей группой с последующим снятием защиты в соответствии с обычными методами.
Когда продукт формулы (I) имеет основную функцию и получается в форме свободного основания, то его переводят в соль обработкой с помощью выбранной кислоты в органическом растворителе. В результате обработки свободного основания, растворенного, например, в спирте, таком как изопропанол, раствором выбранной кислоты в том же самом растворителе получают соответствующую соль, которая выделяется в соответствии с обычными методами. Таким образом, получают например, хлоргидрат, бромгидрат, сульфат, гидросульфат, дигидрофосфат, метансульфонат, метил-сульфат, оксалат, малеат, фумарат, 2-нафталинсульфонат.
Когда соединение формулы (I) имеет основную функцию и выделяется в форме одной из его солей, например хлоргидрата или оксалата, то свободное основание может быть получено в результате нейтрализации указанной соли при помощи минерального или органического основания, такого как гидроксид натрия или триэтиламин, или при помощи щелочного карбоната или биокарбоната, такого как карбонат или бикарбонат натрия или калия.
Когда продукт формулы (I)
содержит
кислотную группу, то полученное таким образом соединение может быть превращено в соль металла, в частности щелочного, такую как натрия, или
щелочно-земельного, такую как соль
кальция, в
соответствии с классическими способами.
Новые производные амидопиразола проявляют активность по отношению к центральной нервной системе и, в частности, по отношению к системам регуляции нейропептидов, вытесняя, например, меченный тритием или иодзамещенный нейротенсин из его рецептора в оболочке головного мозга морской свинки в соответствии с методом, описанным S/oul J.Z. u comp. Biochemical and Biophysical Research communication, 1984, 120, 3, 821-819.
Соединения по настоящему изобретению являются малотоксичными; в частности, их низкая токсичность совместима с их применением в качестве медикаментов. Для такого применения млекопитающих вводят эффективное количество соединения с формулой (I) или (I') или одной из их фармацевтически приемлемых солей.
Таким образом соединения, заявленные в изобретении, могут стать первыми потенциальными медикаментами, способными связываться с рецептором нейтротензина и которые могут быть полезны при патологических состояниях, а также при дисфункции допаминергических систем, например, в качестве антипсихотических средств, а также при расстройствах сердечно-сосудистой или желудочно-кишечной систем.
Таким образом, другим объектом изобретения являются фармацевтические композиции, содержащие в качестве активных компонентов соединения формулы (I) или их возможные фармацевтически приемлемые соли.
В фармацевтических композициях по настоящему изобретению для орального, подъязычного, внутримышечного, внутривенного, трансдермического или ректального введения активные компоненты могут вводиться в виде единичных форм введения, в смеси с классическими фармацевтическими носителями. Соответствующие единичные формы введения включают формы для орального пути, такие как таблетки, желатиновые капсулы, порошки, гранулы и оральные растворы или суспензии, формы подъязычного и ротового введения, формы подкожного, внутримышечного или внутривенного введения и формы для ректального введения.
Чтобы получить желаемый эффект доза активного компонента может варьироваться между 1 и 1000 мг в день, предпочтительно между 2 и 500 мг.
Каждая единичная доза может содержать от 1 до 250 мг активного компонента, предпочтительно от 2 до 125 мг, в комбинации с фармацевтическим носителем. Эта единичная доза может вводиться 1-4 раза в день.
Когда готовят твердую композицию в форме таблеток, то смешивают активный компонент с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик. Можно наносить на таблетки покрытие из сахарозы или других соответствующих веществ или можно их обрабатывать таким образом, чтобы они имели пролонгированную или замедленную активность и чтобы они непрерывно высвобождали предопределенное количество активного компонента.
Препарат в желатиновых капсулах получают, смешивая активный компонент с разбавителем и разливая полученную смесь в мягкие или твердые желатиновые капсулы.
Препарат в форме сиропа или эликсира может содержать активный компонент совместно с подсластителем, предпочтительно некалорийным, и в качестве антисептика с метилпарабеном и пропилпарабеном, а также с агентом, придающим вкус, и с соответствующим красителем.
Порошки или гранулы, диспергируемые в воде, могут содержать активный компонент в смеси с агентами для диспергирования или со смачивающими агентами или с суспендирующими агентами, такими как поливинилпирролидон и ему подобные, а также с подсластителями или корректорами вкуса.
Для ректального введения используют свечи, которые приготавливают при помощи связующих, плавящихся при ректальной температуре, например масла какао или полиэтиленгликолей.
Для парентерального введения применяют водные суспензии, солевые изотонические растворы или стерильные растворы, пригодные для инъекций, которые содержат агенты для диспергирования и/или смачивающие агенты, фармакологически совместимые, например, пропиленгликоль или бутиленгликоль.
Активный компонент может также находиться в композиции в форме микрокапсул, в случае необходимости с одним или несколькими носителями или добавками.
В качестве примера приведен состав таблетки, мг:
соединение 13 250
гидроксипропиленцеллюлоза 6
лактоза 62
микрокристаллическая целлюлоза 60
карбоксиметилкрахмал 12
полиоксиэтиленгликоль 600 10
Покрытие, мг:
bndraget L 100 1
дибутилфталат 1
изопропиловый спирт (испаренный) 28
Последующие примеры иллюстрируют изображение, не ограничивая его.
Температуры плавления
(Tпл) кристаллизованных продуктов были определены на нагревающей установке Кофлера и выражаются в градусах Цельсия. В последующих таблицах были использованы
следующие сокращения:
CH циклогексан
CH2Cl2 дихлорметан
EtOH этанол
Et2O диэтиловый эфир
Hx гексан
Рп пентан
и-Рг2O
диизопропиловый эфир
и-РгОН изопропинол
AcOEt этилацетат
MeOH метанол
Cx означает конфигурацию асимметричного углерода
В спектрах ЯМР
используются следующие сокращения:
M мультиплет
C синглет
PC расширенный синглет
Д дублет
Нар ароматический Н
о орто
м
мета
Получение промежуточных соединений для синтеза
А. Получение производных гидразина (RINH NH2).
Большое число производных гидразина
являются
коммерческими
продуктами. Другие были получены в соответствии с известными методами путем диазотирования соответствующего ароматического амина с последующим восстановлением соли диазония.
Так, в
качестве примера
можно указать получение:
5,6,7,8-тетрагидро-1-нафтилгидразина в соответствии с R,Fusco, Gazz, Chim. Ital, 1974, 104, 813-817;
8-гидразинохинолина в
соответствии с
A.Albert и др.
J.Chem. Soc. 1967, 1533-1541;
5-гидразинохинолина и 5-гидразиноизохинолина в соответствии с M.G.Ferlin, Il Farmaco, 1989, 44 (12), 1141-1155.
В. Получение
пиразолкарбоновых
кислот (II):

Метод получения описан выше.
Приведенная ниже таблица А показывает в качестве примера (неограничивающего) характеристики кислот с общей формулой (II).
С. Получение аминокислот.
Некоммерческие продукты получаются в соответствии с синтезом по Strecker (Ann. 75, 27, 1850) или в соответствии с синтезом по Н.Т.Bucherer, J.Pract, Chem. 1934, 141,5, с последующим гидролизом для получения аминокислот; например, -аминоадамантан-2-карбоновая кислота получается в соответствии с Н.Т.Nasanta. и др. J.Med.Chem. 1973б 16 (7), 823.
Альфа-аминоциклоалканкарбоновые кислоты получаются в соответствии с J.W. Tsaang и др. J.Med.Chem. 1984, 27, 1663.
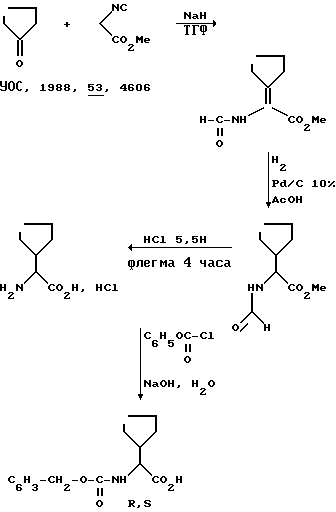
Циклопентилглицины R и S получаются расщеплением бензилоксикарбонилциклопентилглицина.
1) Получение рацемического бензилоксикарбонилциклопентилглицина.
Это соединение получают согласно следующей схеме I:
Схема I
2) Хлоргидрат циклопентилглицина R, S.
Растворяют 80% -ный NaOH (1,8 г) в безводном ТГФ (50 мл). Прибавляют по каплям, перемешивая, смесь циклопентанола (4,2 г) и изоцианометилацетата (5 г) в ТГФ (50 мл). По окончании прибавления оставляют на 2 ч. Охлаждают до 5oС и медленно прибавляют 10% водный раствор уксусной кислоты (50 мл). Выпаривают ТГФ под вакуумом. Водный остаток экстрагируется хлороформом (3 х 120 мл). Сушат над сульфатом натрия и концентрируют под вакуумом. Остаток извлекают пентаном, фильтруют и промывают в пентане.
Твердое вещество (7,6 г) растворяют в уксусной кислоте (100 мл). 10% -ный палладий на угле (3 г) и перемешивают при атмосферном давлении и комнатной температуре под водородом в течение 24 ч (поглощают 1 л водорода). Фильтруют на целите, промывают несколько раз уксусной кислотой. Выпаривают под вакуумом. Остаток извлекается 5,5 н.хлорводородной кислоте (70 мл). Нагревают до образования флегмы в течение 4 ч. Концентрируют досуха, образуют несколько раз азеотропную смесь с толуолом и сушат под вакуумом. Получают целевой продукт.
m 7,2 г
ЯМР
Д2O: 8Н при 1,6/М, СН2 цикла); 1H при 2,20 (М, СН цикла); 1Н при 3,80 (Д, J 7 СНСО2H); 3 H при 8,60 (РС, NH3+.
3) Ацилирование при помощи хлорформиата бензила.
Растворяют хлоргидрат циклопентилглицина R, S (7,2 г) в 2 н. растворе гидроксида натрия (65 мл). Прибавляют по капле хлорформиат бензила (8,5 г) в ТГФ (30 мл), охлаждая до 5oС. Оставляют с перемешиванием на ночь при комнатной температуре. Охлаждают во льду. Подкисляют при помощи концентрированной HCl до рН 2 (T ≅ 5oC). Извлекают в хлороформе, сушат и выпаривают. Остаток извлекается пентаном. Получают бензилоксикарбонилциклопентилглицин R,S.
Тпл 110oС.
4) Расщепление бензилоксикарбонилциклопентилглицина.
Растворяют бензилоксикарбонилциклопентилглицин (5,54 г) в абсолютном этаноле (65 мл). Прибавляют 1,2-дифенил-1-этанол-2-амин (1 R, 2S) (-), полученный в соответствии с J.Weijlasd и др. J.Am.Chem.Soc. 1951, 73, 1216. Нагревают до растворения. Оставляют для осаждения на ночь и фильтруют. Получают 2,8 г соли (Тпл 175o С). Маточные растворы сохраняют.
Полученная соль извлекается водой (20 мл) HCl (30 мл) и эфиром (100 мл). Перемешивают до растворения. Органическая фаза декантируется, сушится, выпаривается. Получают бензилоксикарбонилциклопентилглицин, который тут же обрабатывают концентрированной HCl (15 мл), АсОН (15 мл. Нагревают до образования флегмы в течение 3 ч. Выпаривают досуха. Остаток извлекается сухим эфиром, фильтруется и сушится. Получают хлоргидрат (S)-циклопентилглицина.
[α
] +10,4oС (с 0,5 HCl 1н)
m 0,6 г
Маточные растворы выпаривают
досуха и поглощают водой (50 мл), HCl (60 мл), Et2O (300 мл).
Перемешивают и все растворяют. Декантируют эфирную фазу, сушат и выпаривают. Извлекают бензилоксикарбонилциклопентиглицин (4,
3 г), вводят его в абсолютный метанол (50 мл), с 1,
2-дифенил-1-этанол-2-амином (1S, 2R) (+) (3,30 г). Нагревают до растворения, оставляют в покое на ночь, фильтруют. Получают 4,15 г соли.
Tпл 175oС.
Эту соль поглощают водой (20 мл), 1н HCl (40 мл) и эфиром (200 мл). Перемешивают. Эфирную фазу сушат, выпаривают, затем остаток обрабатывают концентрированной HCl (10 мл) уксусной кислотой (100 мл). Нагревают смесь в течение 3 ч до образования флегмы, концентрируют под вакуумом, извлекают безводным эфиром для получения хлоргидрата (R) циклопентилглицина.
m 1,2 г.
[α] -10,5 (с 0,85 HCl 1н)
Оптическая чистота R-циклопентилглицина.
Растворяют 0,10 г полученного выше хлоргидрата в абсолютном метаноле. Охлаждают до температуры -40oС, прибавляют 0,5 мл тионилхлорида и оставляют смесь в течение 24 ч при комнатной температуре. Концентрируют под вакуумом, извлекают остаток безводным хлороформом (20 мл), прибавляют триэтиламин (0,2 мл) и (S)-фенилметилизоцианат (0,074 мл). Оставляют в течение 24 ч, затем выпаривают хлороформ. Остаток хроматографируют на силикагеле, элюант: этилацетат. После концентрирования чистых фракций получают 0, 1 г метилового сложного эфира.
Спектр ЯМР в
СДСl3 показывает около 3,8 ч/млн. наличие двух сигналов для -CO2CH3. Интегрирование показывает, что
наиболее слабый сигнал составляет 4% а наиболее интенсивный сигнал
96%
Таким образом, энантиомерный избыток равен 92%
Можно также получить альфа-аминоциклоалкилкислоты с
конфигурацией R или S путем стереоспецифического энзиматического гидролиза
соответствующих рацемических N-ацетилированных производных в соответствии с J.Hill и др. J. Org.Chem. 1965, 1321.
Пример 1. Метиловый сложный эфир 2-{[1-фенил-5-(4-пиридил)-3-пиразолил] -карбониламино}-4- метилпентановой кислоты (S).
(I): R H n 0; X' H; X -CH2-CH-(CH3)2; Z OCH3; R1 C6H5; RIV H;
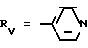
Растворяют 0,35 г 1-фенил-5-(4-пиридил)-пиразол-3-карбоновой кислоты в 5 мл диметилформамида в присутствии 0,45 мл диизопропилэтиламина (ДИПЭА) и 0,59 г гексафторфосфата бензотриазолил-N-окситрисдиметиламинофосфония (БОФ). Затем прибавляют 0,23 г (1 эквивалент) хлоргидрата метилового сложного эфира (S)-лейцина, растворенного в 0,4 мл ДИПЭА, и реакционную смесь оставляют в течение ночи при комнатной температуре. Растворители концентрируются под вакуумом, остаточная маслянистая жидкость экстрагируется дихлорметаном, этот раствор промывается водой, затем раствором бикарбоната натрия и опять водой. Органическая фаза сушится над сульфатом натрия, затем концентрируется под вакуумом. Остаток хроматографируют на силикагеле, элюент-этилацетат.
m 0,18 г.
Спектр ЯМР протона соединения I: 3Н при 8,82 (М, Нар оN и CONH); 5Н при 7,50 (М, Нар Phe); 3Н при 7,27 (Нар мN и Н4 пиразола); 1Н при 4,60 (М, Н альфа Leu); 3Н при 3,77 (С, СО2CH3); 1Н при 2,00 (М, Н гамма Lue); 2H при 1,70 (М, Н бета Leu); 6Н при 1,00 (2Д, СН3 Leu).
Пример 2. 2-{[1-Фенил-5(2-нафтил)3-пиразолил] карбониламино}-3-фенилпропановая кислота (S).
(I): R H; n 0; X' H; X -CH2-C6H5; Z OH; RI C6
H5; RIV H
Получение хлорангидрида 5-(2-нафтил)1-фенилпиразол-3-карбоновой кислоты.
Растворяют 5 г 5-(2-нафтил)1-фенилпиразол-3-карбоновой кислоты в 56 мл толуола и прибавляют по каплям 3,5 мл сульфинилхлорида к этому раствору. Смесь нагревают при 90oС в течение 2,5 ч, затем концентрируют под вакуумом. Остаточную маслянистую жидкость дважды извлекают в толуоле и концентрируют под вакуумом.
m 5 г.
Получение соединения 2.
К 60 мл 2 н.раствора гидроксида натрия прибавляют 4,9 г (S)-фенилаланина, а затем прикапывают раствор 4 г хлорангидрида полученной выше кислоты, растворенного в 65 мл тетрагидрофурана. Реакционную смесь оставляют на ночь при комнатной температуре, затем концентрируют под вакуумом. Остаток извлекают водой и рН доводят до 1 путем прибавления хлорводородной кислоты. Раствор экстрагируют дихлорметаном и органическую фазу промывают водой с насыщенным раствором хлорида натрия, сушат на сульфате натрия, фильтруют и концентрируют под вакуумом. Остаток перекристаллизовывают из пентана.
m 2 г.
Tпл 226oС.
Пример 3. N,
N-Диэтил-2{[1-фенил-5-(2-нафтил)-3-пиразолил]-карбониламино} -3- фенилпропанамид (S) (I)
R H,
n 0; X' H; X
-CH2-C6H5; Z -N-(C2H5)2; RI C6H5; RIV H;
Растворяют 2 г продукта, полученного в соответствии с примером 2, 0,88 г дициклогексилкарбодиимида (ДЦКДИ) и 1,14 г 1-гидроксибензотриазола (ГОБТ) в 68 мл тетрагидрофурана и перемешивают смесь в течение 3/4 ч при комнатной температуре. Затем прибавляют 0,4 г диэтиламина и оставляют реакционную смесь при комнатной температуре в течение 24 ч.
Дициклогексилкарбамид отделяют путем фильтрования, а маточные растворы концентрируют под вакуумом. Остаток хроматографируют на силикагеле, элюент - этилацетат. Фракции чистого продукта концентрируют под вакуумом, а остаток перекристаллизовывают из пентана.
m 1,46 г.
Tпл 70oС.
Пример 4. 2{[1-Фенил-4,
5-дигидробенз-(g)-3-индазолил]-карбониламино}-4- метилпентановая кислота (S)

А) Натриевая соль бета-кетокарбэтокси-альфа-тетралона.
Это промежуточное соединение получается в соответствии с методом, описанным D.Ramech et al. Indian Journal of Chemistry, 1989, 28В, 76-78.
В) Этиловый сложный эфир 1-фенил-4,5-дигидробенз-(g)-индазол-3-карбоновой кислоты.
Растворяют 8,04 г полученной выше натриевой соли в 100 мл уксусной кислоты. Прибавляют 3, 3 мл фенилгидразина и нагревают реакционную смесь с образованием флегмы в течение 8 ч. Охлажденную смесь приливают к ледяной воде, осадок отделяют фильтрованием, промывают водой, затем в пентане.
m 10,5 г.
С) 1-Фенил-4, 5-дигидробенз-(g)-индазол-3-карбоновая кислота.
Растворяют 9,5 г полученного выше продукта в 100 мл метанола и 100 мл воды. Прибавляют 4,2 г гидроксида калия и нагревают реакционную смесь с образованием флегмы в течение 5 ч. Смесь приливают к ледяной воде, затем промывают этилацетатом. Водную фазу подкисляют до рН 2 путем прибавления хлорводородной кислоты, осадок отделяют фильтрованием, промывают в воде, затем в пентане.
m 7,3 г.
Д) Хлорангидрид 1-фенил-4, 5-дигидробенз-(g)-индазол-3-карбоновой кислоты.
Растворяют 2,8 г полученной выше кислоты в 100 мл толуола, затем прибавляют 2,2 мл сульфинилхлорида и нагревают при 100oС в течение 5 ч. Раствор концентрируют под вакуумом, прибавляют 20 мл толуола и концентрируют под вакуумом. Дважды повторяют ту же самую операцию.
Е) Соединение 4
Растворяют 0,88
г S-лейцина в растворе 1,33 г гидроксида натрия в 20 мл воды. Этот раствор
охлаждают, затем к нему прибавляют 0,99 г полученного выше хлорангидрида кислоты, растворенного в 16 мл
тетрагидрофурана, и
оставляют реакционную смесь при перемешивании в течение 18 ч при комнатной
температуре. Раствор концентрируют под вакуумом, остаток извлекают льдом и подкисляют до рН 2 путем
прибавления
хлорводородной кислоты, затем экстрагируют этилацетатом. Органическую фазу сушат на
сульфате натрия, фильтруют и концентрируют под вакуумом. Остаток перекристаллизовывают из
изопропилового эфира.
m 1 г.
Tпл 100oС.
Пример 5. 2{[1-Бензил-3-(2-нафтил)-5-пиразолил]-карбониламино}-3- фенилпропановая кислота
(S)
(I') R H; n
0; X' H; X -CH2H
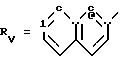
А) Реакция 2-нафтоилметилпуривата с хлоргидратом бензилгидразина приводит к смеси следующих сложных эфиров: меловый сложный эфир 1-бензил-5-(нафтил)-пиразол-3-карбоновой кислоты и метиловый сложный эфир 1-бензил-3-(2-нафтил)-пиразол-5-карбоновой кислоты.
Хроматография на силикагеле позволяет разделить два изомера. Метиловый сложный эфир 1-бензил-5-(2-нафтил)-пиразол-3-карбоновой кислоты элюируется первым смесью этилацетат/гексан 50/50 (об/об). Метиловый сложный эфир 1-бензил-3-(2-нафтил)-пиразол-5-карбоновой кислоты элюируется во второй фракции.
В) 1-Бензил-3-(2-нафтил)-пиразол-5-карбоновая кислота
Кислота
получена в результате омыления
полученного выше сложного эфира.
С) Хлорангидрид 1-бензил-3-(2-нафтил)-пиразол-5-карбоновой кислоты.
Хлорангидрид кислоты получается при действии сульфинилхлорида на полученную выше кислоты и не выделяется.
Д) Соединение 5
Растворяют 0,28 г фенилаланина (S) в охлажденном растворе гидроксида натрия. Затем
прибавляют раствор 0,3 г
полученного выше хлорангидрида кислоты в 5 мл ТГФ, и оставляют реакционную смесь при комнатной температуре в течение 24 ч. ТГФ концентрируют под вакуумом, остаток извлекают
водой, нейтрализуют путем
прибавления концентрированной хлорводородной кислоты. Экстрагируют этилацетатом, сушат на сульфате натрия и концентрируют под вакуумом. Остаток перекристаллизовывают из
циклогексана.
m 1 г.
Tпл 100oС.
Пример 6.
Метиловый сложный эфир
2{[1-(4'-метоксициннамил)-5-(4-пиридил)-3-пиразолил]-карбониламино}- 4-метилпентановой кислоты (S)
(I) R H; n 0; X' H; X -CH2-CH-(CH3)2; Z OCH3
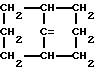
А) Метиловый сложный эфир 1-(4'-метоксициннамоил)-5-(4-пиридил)-пиразол-3-карбоновой кислоты.
Растворяют 4,6 г метилового сложного эфира 5-(4-пиридил)-(1Н)-пиразол-3-карбоновой кислоты в 60 мл диметилформамида, затем прибавляют 0,63 г 80% масляной суспензии гидрида натрия и нагревают реакционную смесь при 40oС в течение 1 ч. Затем к охлажденной смеси прибавляют раствор 5,2 г 4'-метоксибромида 1-циннамила растворенного в 60 мл диметилформамида и оставляют реакционную смесь при комнатной температуре в течение 12 ч. Концентрируют диметилформамид под вакуумом, извлекают остаток водой, экстрагируют этилацетатом, сушат органическую фазу на сульфате натрия, фильтруют и концентрируют под вакуумом. Остаточную маслянистую жидкость хроматографируют на силикагеле, элюент этилацетата/циклогексан 50/50 (об/об). Фракции чистого продукта концентрируют под вакуумом.
m 2,6 г.
Tпл 118oС.
B) Соединение 6
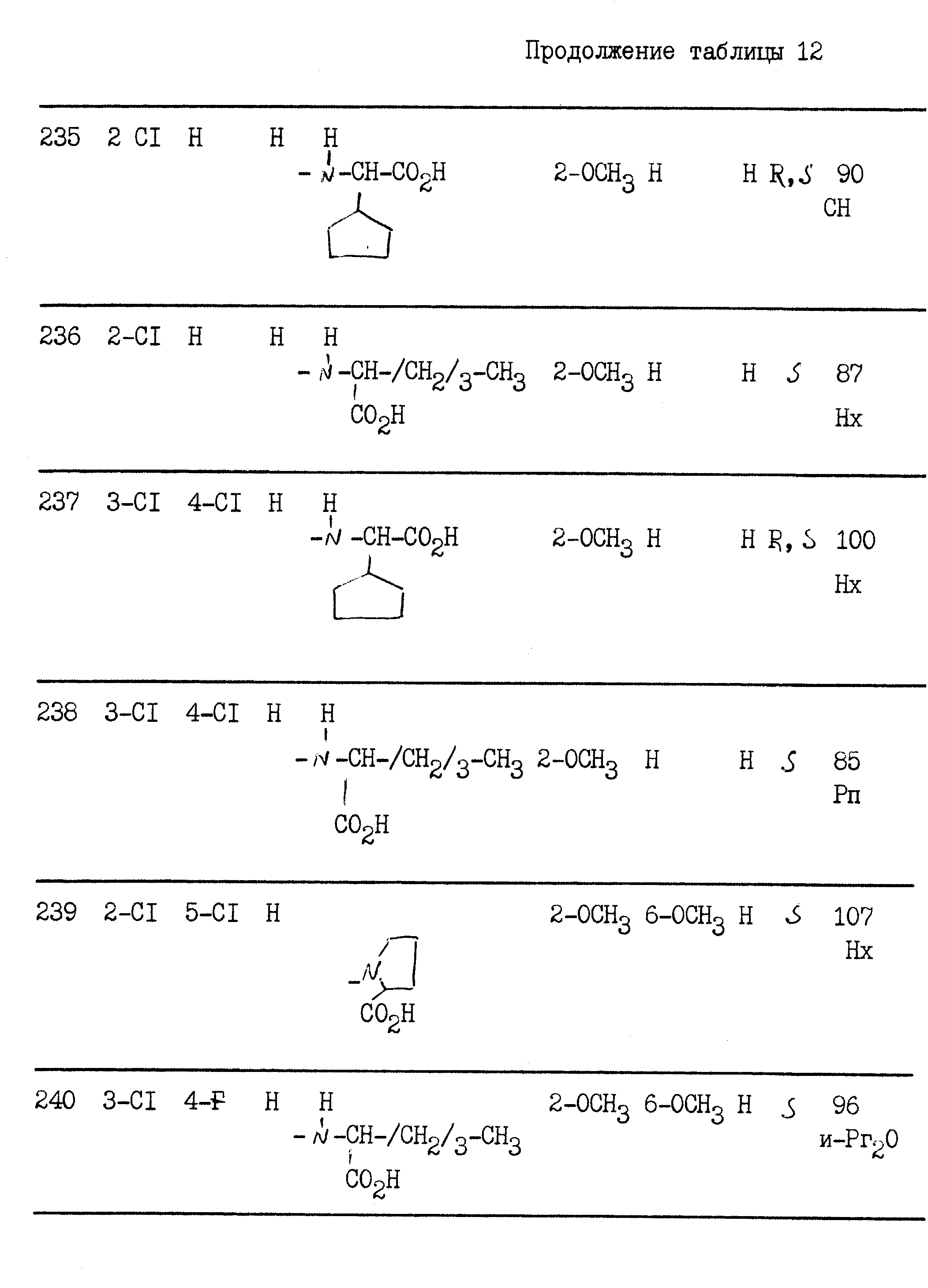
Растворяют 0,4 г полученной выше кислоты в 12 мл
диметилформамида в
присутствии 0,63 мл ДИПЭА и 0,53 г БОФ. Затем прибавляют 0,22 г хлоргидрата метилового сложного эфира (S)-лейцина, растворенного в 0,63 мл ДИПЭА, и реакционную смесь оставляют на
ночь при комнатной
температуре. Диметилформамид концентрируют под вакуумом и остаток извлекают водой. Экстрагируют этилацетатом, сушат органическую фазу на сульфате натрия, фильтруют и концентрируют
под вакуумом.
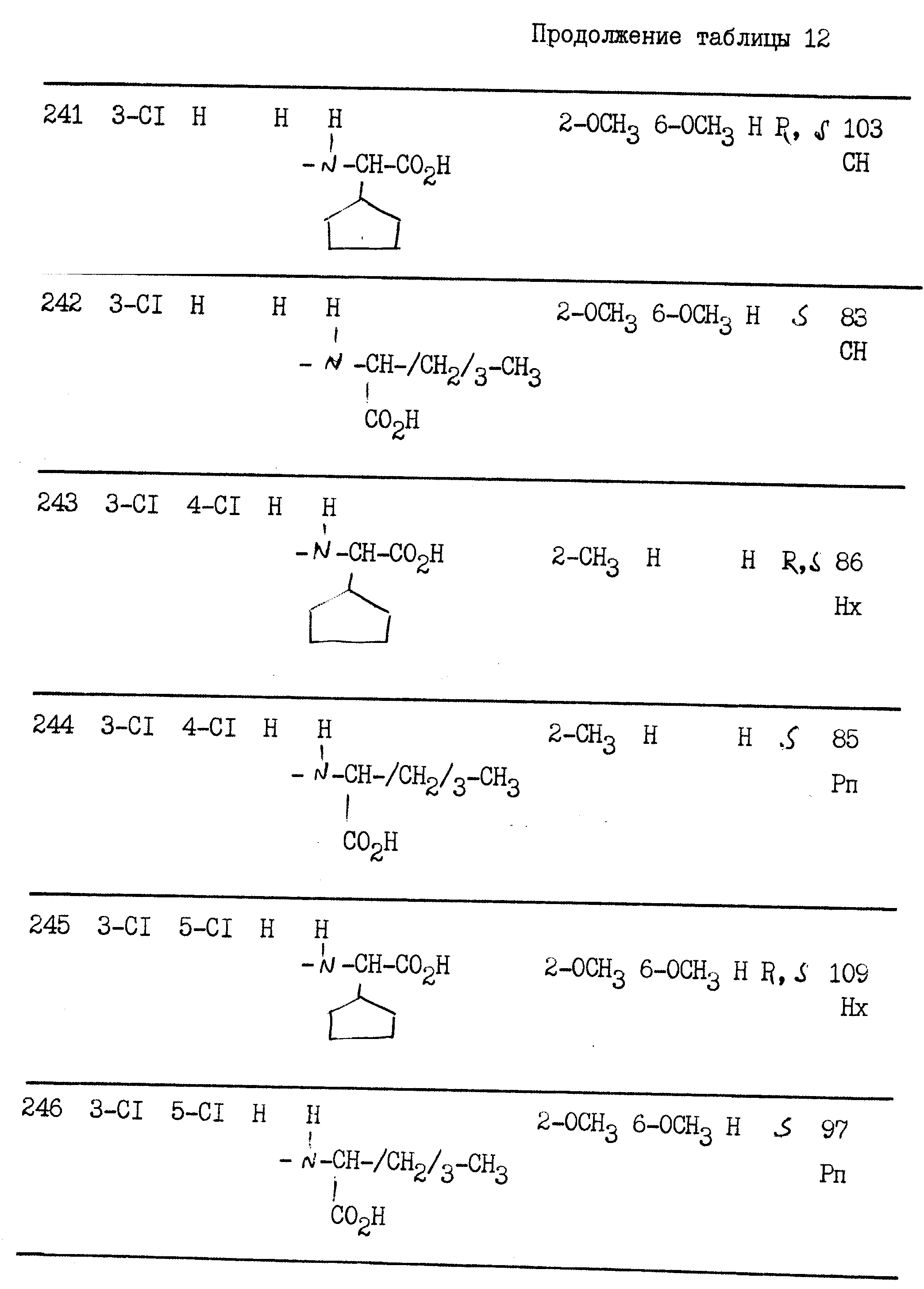
Остаток уплотняется в диизопропиловом эфире.
m 0,15 г.
Tпл 172oС.
Пример 7. Натриевая соль
2{[1-(4'-метоксициннамоил)-5-(4-пиридил)-3-пиразолил]-карбониламино} -3-фенилпропановой кислоты (S)
(I): R H; n O; X' H; X -CH2-C6H5; Z O-Na+;
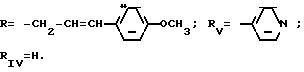
Поступая как в примере 6, но заменяя хлоргидрат метилового сложного эфира (S)-лейцина на хлоргидрат метилового сложного эфира (S)фенилаланина, получают метиловый сложный эфир, который гидролизуют до натриевой соли при помощи 0,9 эквивалента гидроксида натрия в 10 мл 96o этанола. Смесь оставляют на ночь при комнатной температуре, концентрируют под вакуумом и остаток промывают в эфире. После фильтрования получают соединение 7.
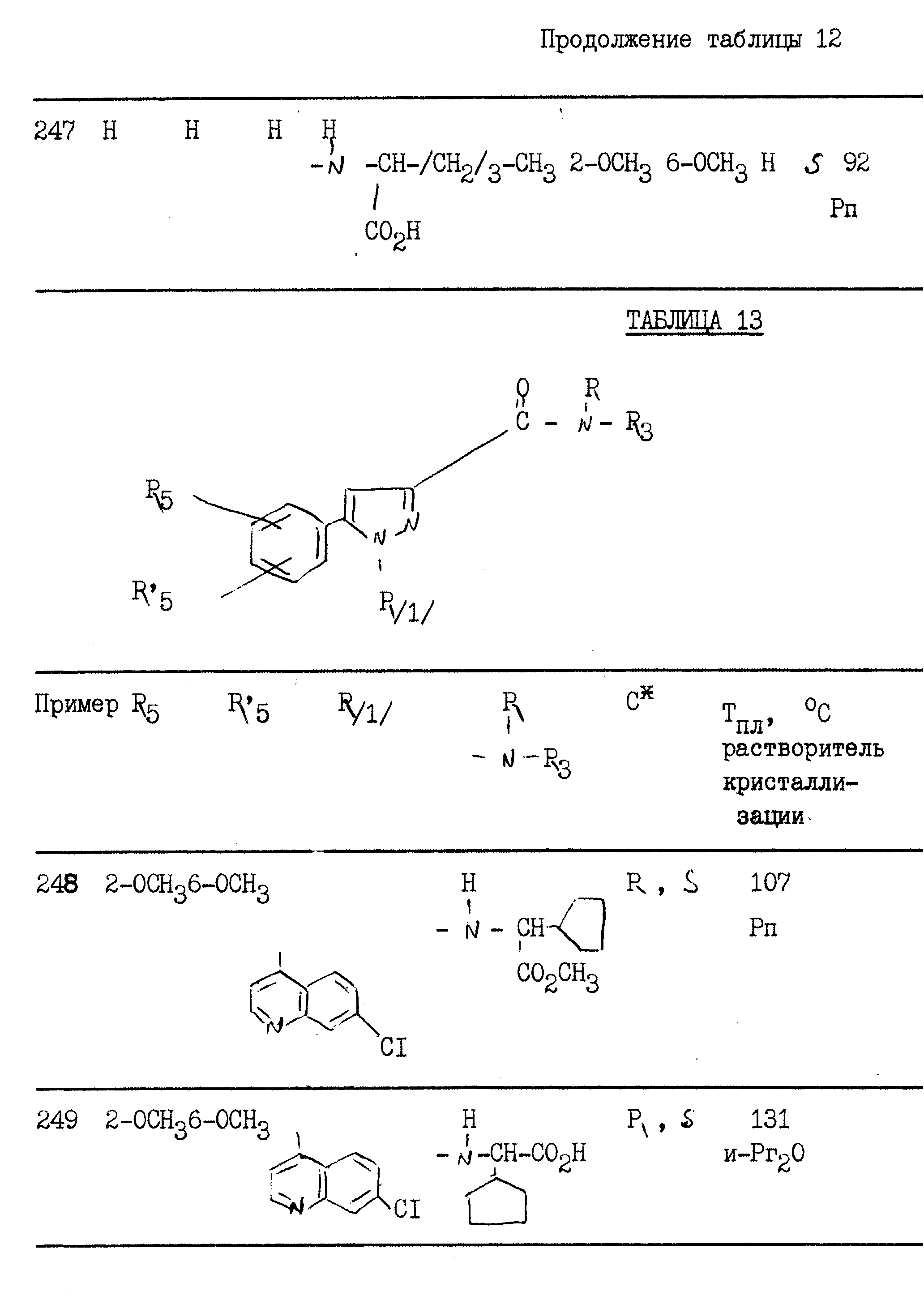
Тпл 137oС.
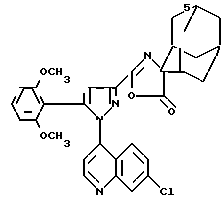
Пример 8. 2{[1-(5-Изохинолил)-5-(2,6-диметоксифенил)-3-пиразолил]-карбониламино} 2-адамантанкарбоновая кислота
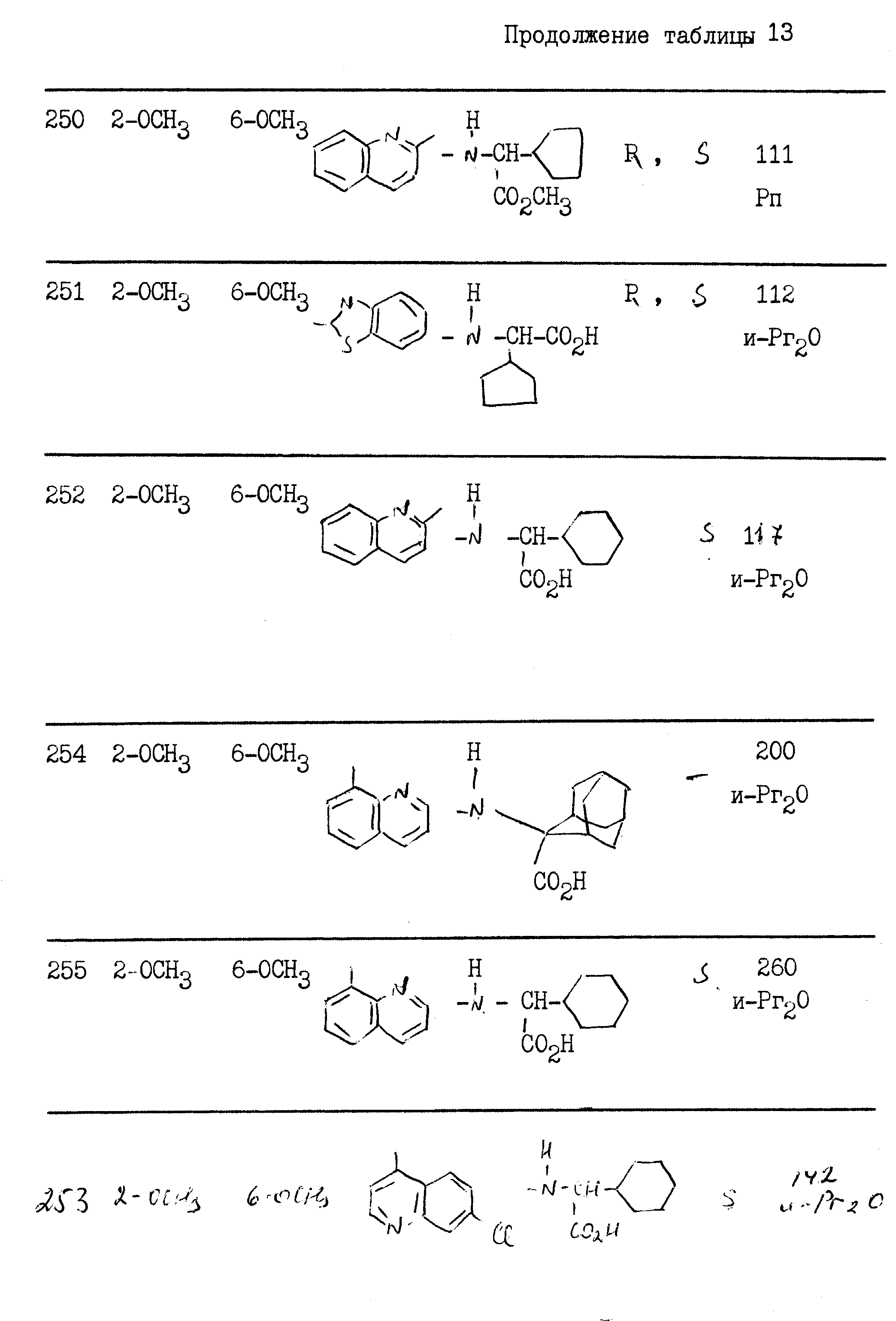
Растворяют 0,75 г 2-амино-2-адамантанкарбоновой кислоты в 20 мл пиридина. Прибавляют 1,4 г хлорангидрида 1-(5-изохинолил)-5-(2, 5-диметоксифенил)-3-пиразолкарбоновой кислоты, растворенного в 20 мл дихлорметана, и оставляют реакционную смесь на ночь при комнатной температуре. Концентрируют под вакуумом, извлекают остаток буферным раствором с рН 2, перемешивают, фильтруют осадок и споласкивают диизопропиловым эфиром.
m 0,4 г.
Tпл выше 260oС.
Пример 9. 2{[1-(5-Хинолил)-5-(2,6-диметоксифенил)-3-пиразолил]- карбониламино}2-адамантанкарбоновая кислота
Растворяют 0,23 г 2-амино-2-адамантанкарбоновой кислоты, 0,5 г хлорангидрида 1-(5-хинолил)-5-(2,6-диметоксифенил)-3-пиразолкарбоновой кислоты и 0,7 г гидроксида калия в 25 мл дихлорметана в присутствии 0,1 г Aliquat 336®
Реакционную смесь перемешивают в течение ночи при комнатной температуре, прибавляют 0,7 г гидроксида калия и перемешивают в течение 4 ч. Смесь фильтруют и получают 0,2 г целевого продукта.
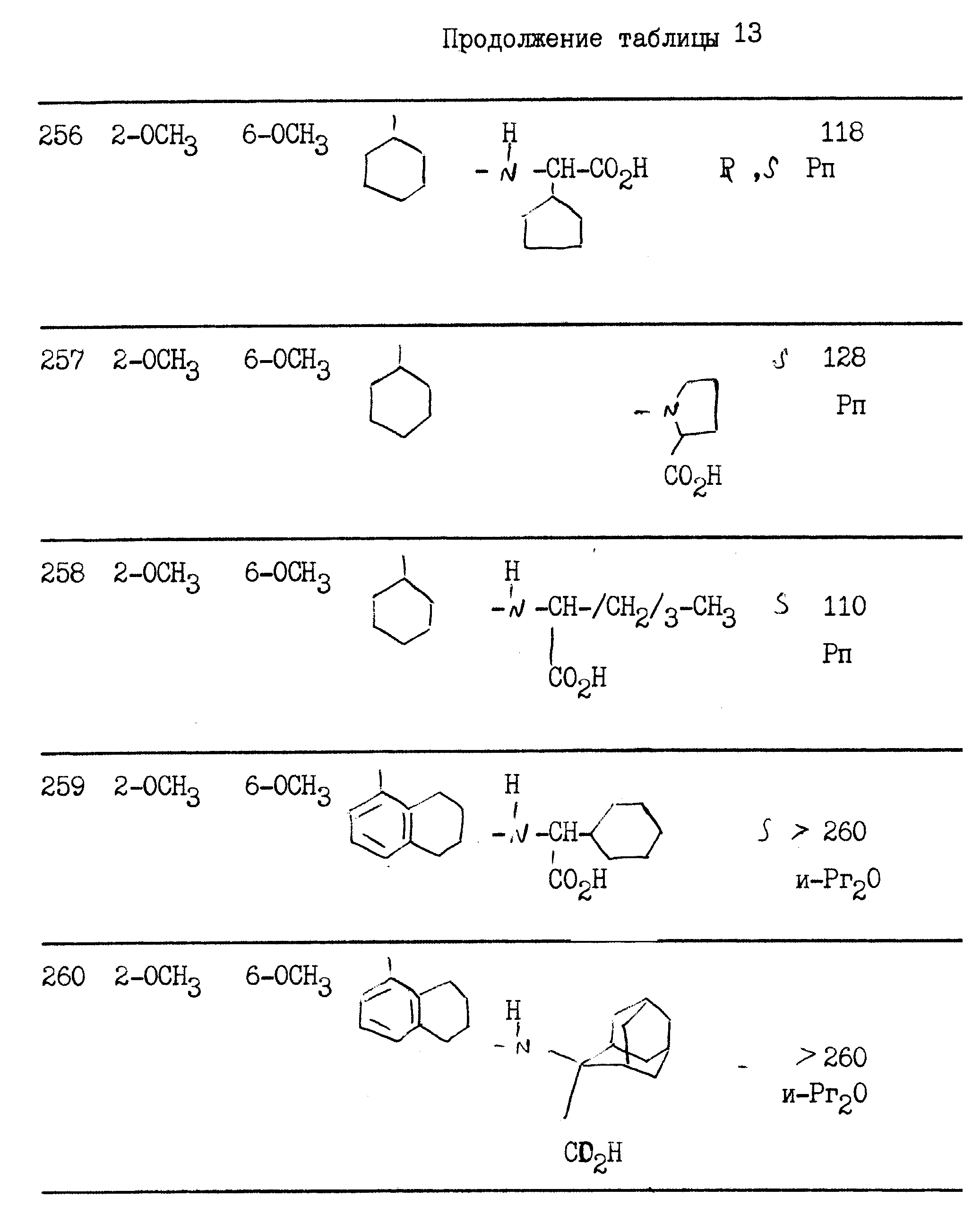
Tпл выше 260oС.
Пример 10. 2{[1-(4-Хлор-1-нафтил)-5-(2,
6-дигидроксифенил)-3-пиразолил]- карбониламино}гексановая кислота (S)
(I): R H; n O; X' H; X (CH2)3-CH3; X OH;
Растворяют 0,3 г 3-[1-(4-хлор-1-нафтил)-5-(2,6-диметоксифенил)-пиразолил]-2- карбониламиногексановой кислоты в 6,7 мл дихлорметана и охлаждают до температуры -70oС. Прибавляют по капле 5,7 мл трибромида бора, растворенного в 20 мл дихлорметана, и оставляют реакционную смесь в течение 2 ч при -70oС. Дают вернуться к комнатной температуре, затем прибавляют, охлаждая, 12 мл воды. Прибавляют концентрированную NaOH до рН 14. Промывают водную фазу в эфире, доводят ее до рН 2, экстрагируют этилацетатом, сушат на сульфате натрия, фильтруют и выпаривают. Кристаллизуют остаток из диизопропилового эфира.
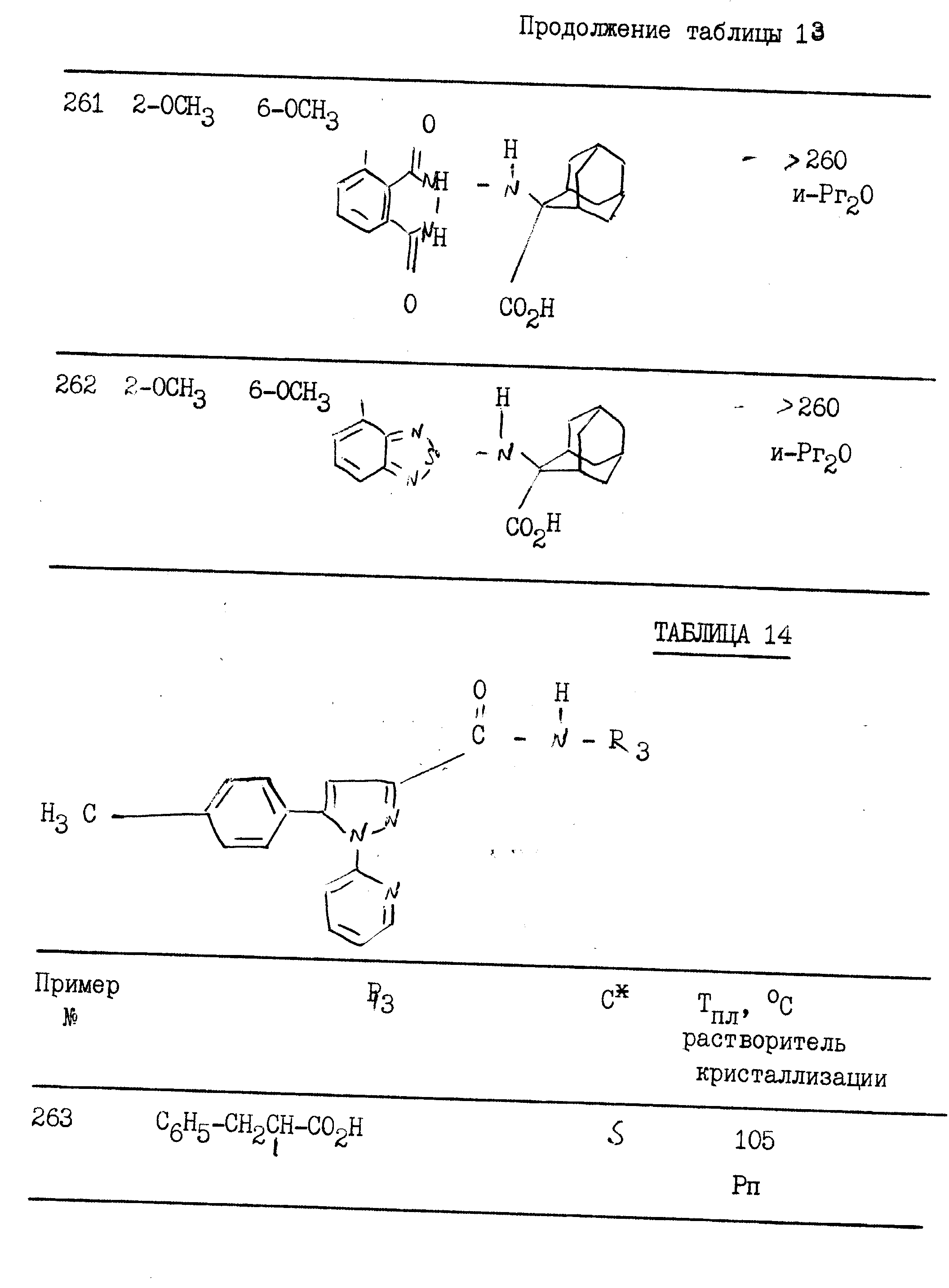
m 0,13 г.
Tпл выше 260oС.
Пример 11. 2{[1-(1-Нафтил)-5-(2,6-диметоксифенил)-3-пиразолил]карбониламино} 2-адамантанкарбоновая кислота
Охлаждают до 0oС 0,107 г гидроксида натрия в 1,36 мл воды и 0,51 мл тетрагидрофурана. Прибавляют одной порцией 0,52 г 2-амино-2-адамантанкарбоновой кислоты, затем по каплям 0,53 г хлорангидрида 1-(1-нафтил)-5-(2,6-диметоксифенил)-3-пиразолкарбоновой кислоты, растворенного в 3 мл тетрагидрофурана. Смесь оставляют на 10 мин, затем снова прибавляют то же самое количество того же хлорангидрида кислоты в 3 мл тетрагидрофурана; одновременно прибавляют 1,32 мл 2 н.гидроксида натрия. Реакционную смесь оставляют на 4 дня при комнатной температуре; прибавляют последовательно ледяную воду, концентрируют хлорводородную кислоту до рН 1 и отфильтровывают осадок. Кристаллы промывают в диизопропиловом эфире.
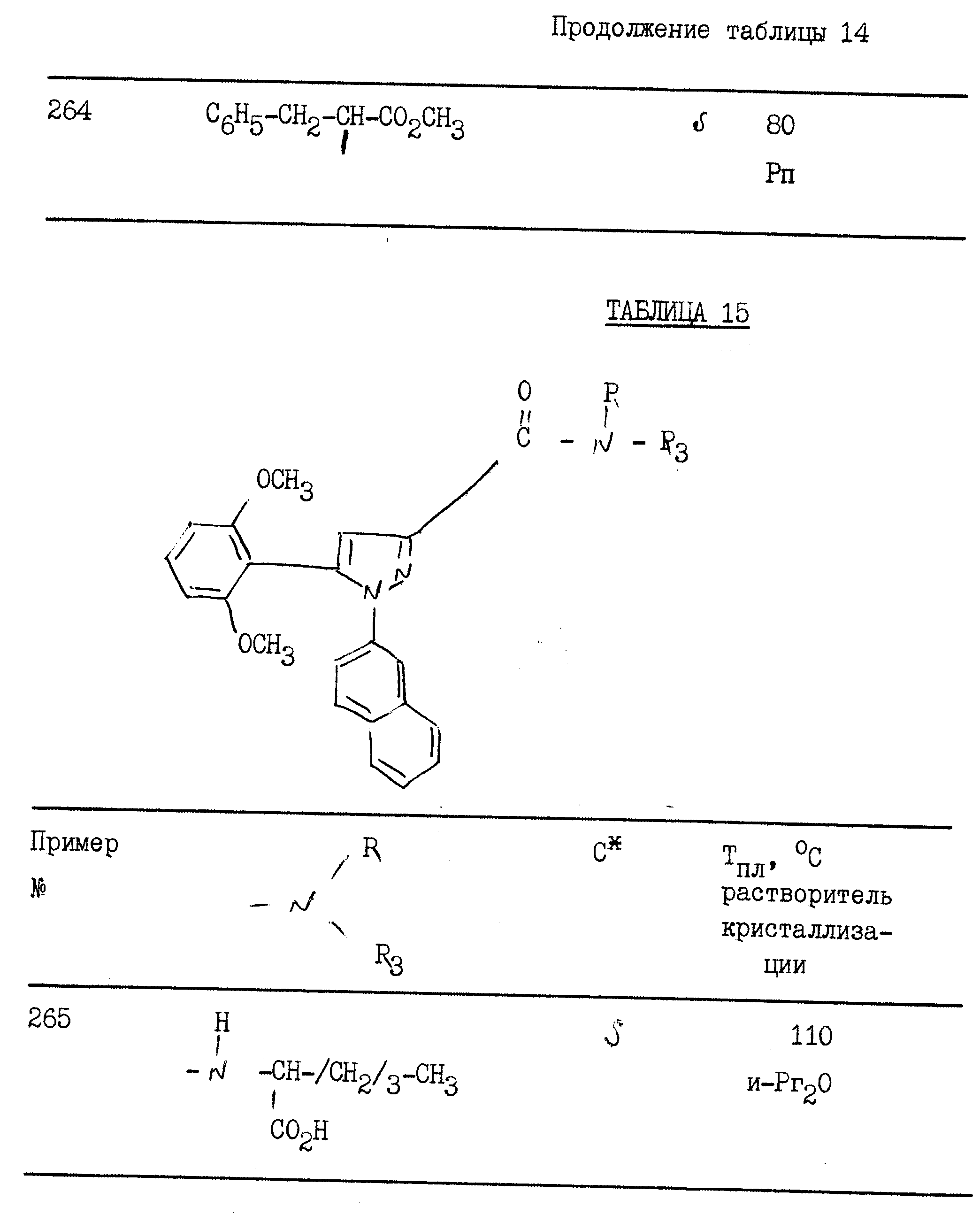
m 0,48 г.
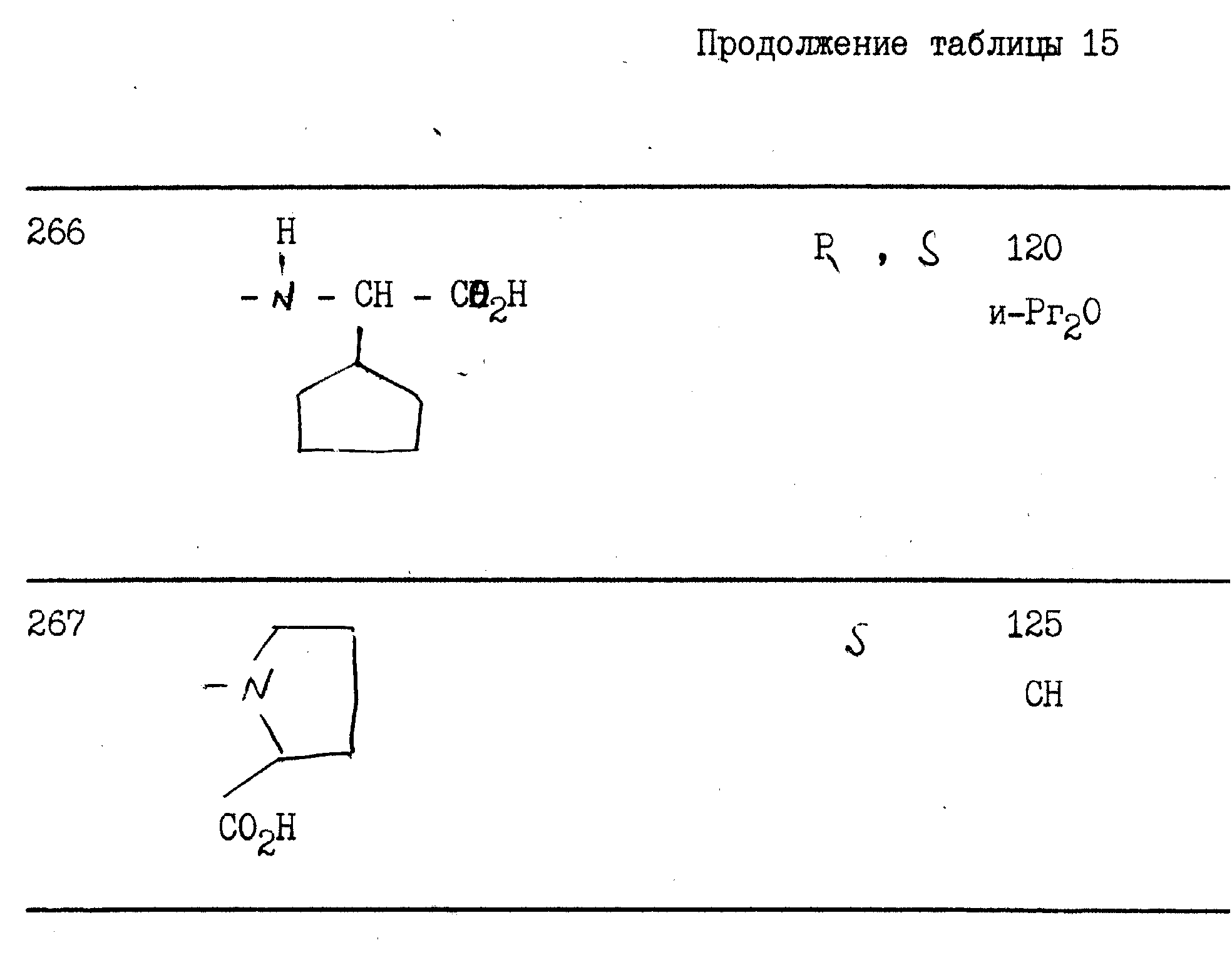
Tпл выше 260oС.
Пример 12. 2{[1-(1-Нафтил)-5-(2,6-диметоксифенил)-3-пиразолил]-карбониламино} -2-адамантанметилкарбоксилат
Растворяют 0,5 г соединения, полученного в примере 11, в 34,6 мл безводного тетрагидрофурана и 4 мл диметилформамида. Прибавляют 3,5 мл воды и 0,208 г карбоната цезия и оставляют реакционную смесь при комнатной температуре в течение 1 ч. Концентрируют под вакуумом и азеотропируют с толуолом. Остаток извлекают с 5 мл тетрагидрофурана. Прибавляют 0,6 мл метилиодида и оставляют реакционную смесь в течение 1 ч при комнатной температуре. Концентрируют под вакуумом, извлекают остаток водой, перемешивают и отделяют осадок путем фильтрования. Осадок промывается водой и пентаном.
m 0,39 г.
Tпл 242-244oС.
Пример 13.
2{[1-(7-Хлор-4-хинолил)-5-(2,
6-диметоксифенил)-3-пиразолил] -карбониламино}-2-адамантанкарбоновая кислота
Поступая в соответствии с примером 8, но заменяя хлорангидридом кислоты на хлорангидрид 1-(7-хлор-4-хинолил)-5-(2,6-диметоксифенил)-3-пиразолкарбоновой кислоты, получают промежуточное соединение с формулой
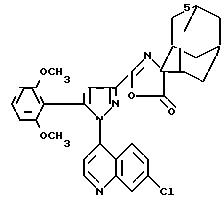
температура плавления которого равна 249oС.
Растворяют 0,1 г этого промежуточного соединения в 5 мл дихлорметана, прибавляют 5 мл трифторуксусной кислоты и оставляют смесь на полчаса при комнатной температуре. Концентрируют под вакуумом с целью получения ожидаемого соединения.
m 0,080 г.
Tпл выше 260oС.
Повторяя какой-либо из вариантов осуществления, описанных в
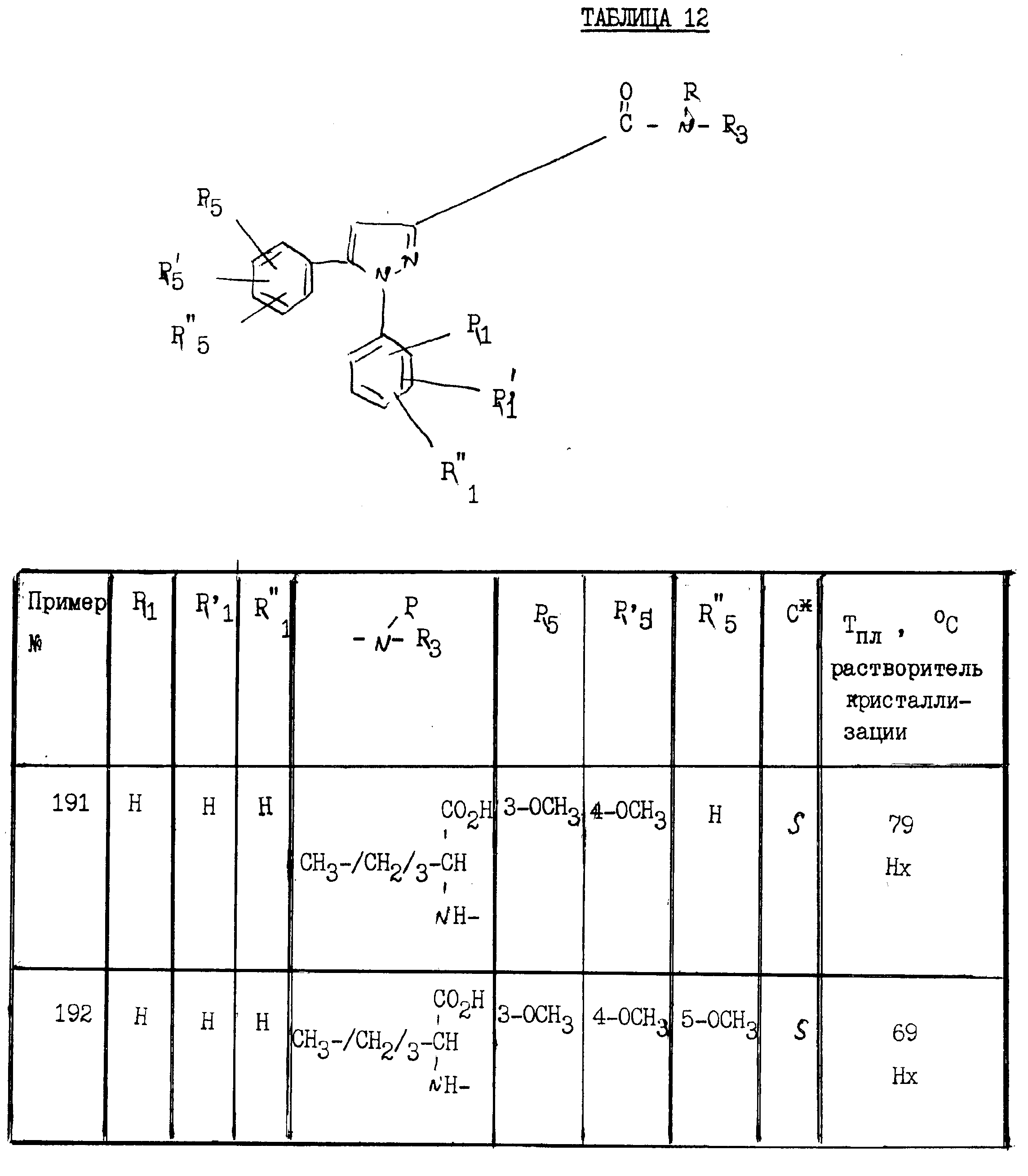
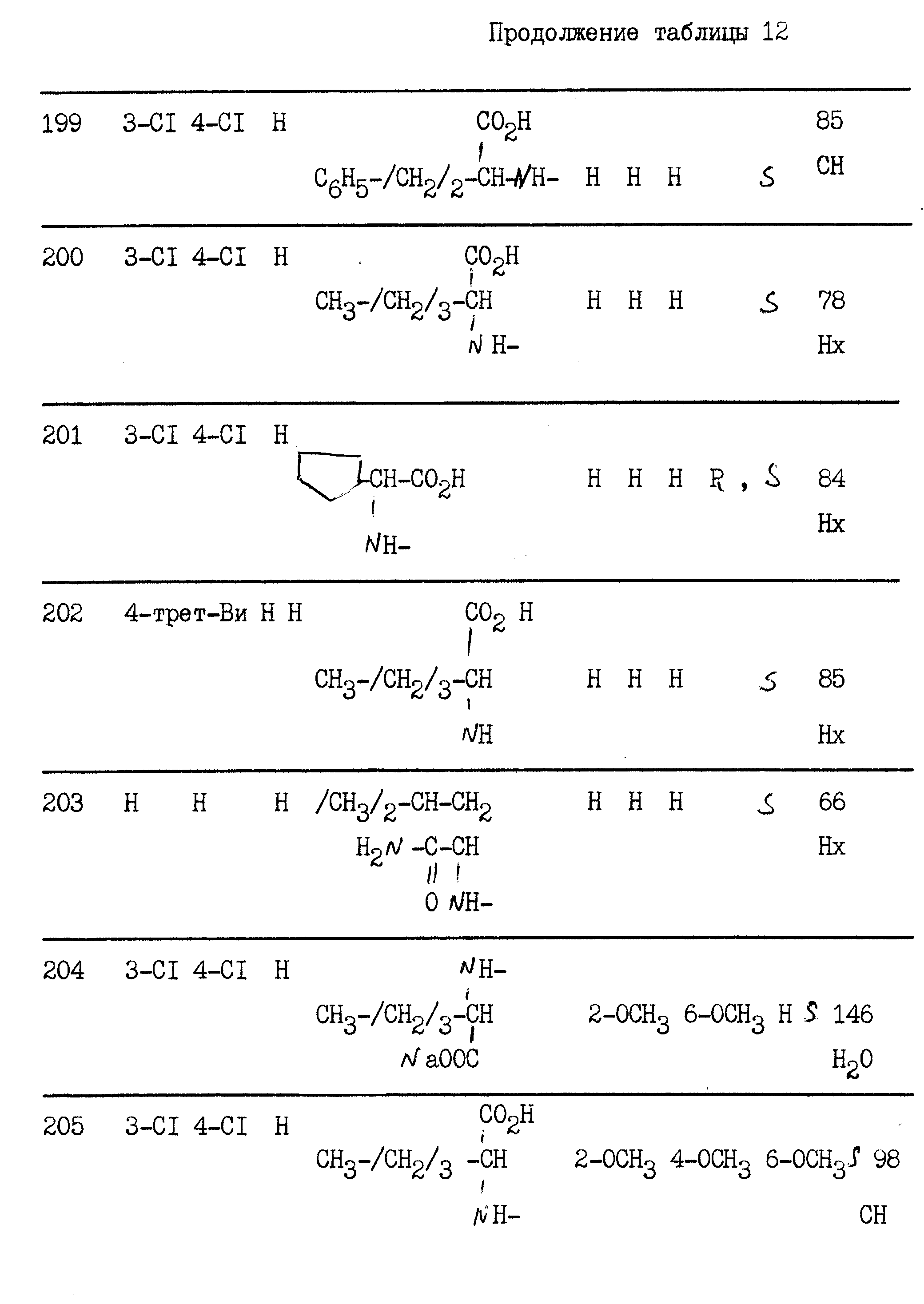
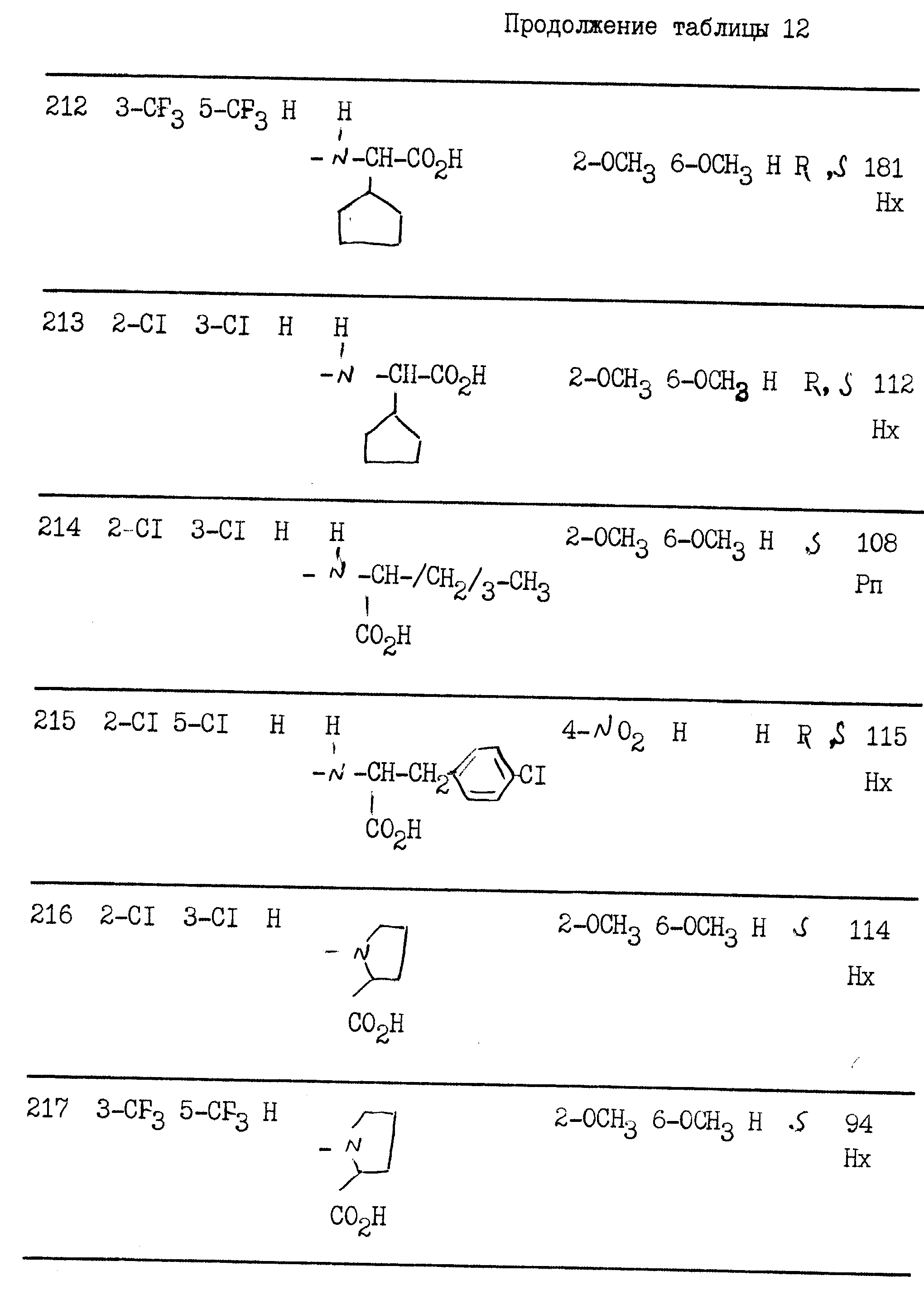
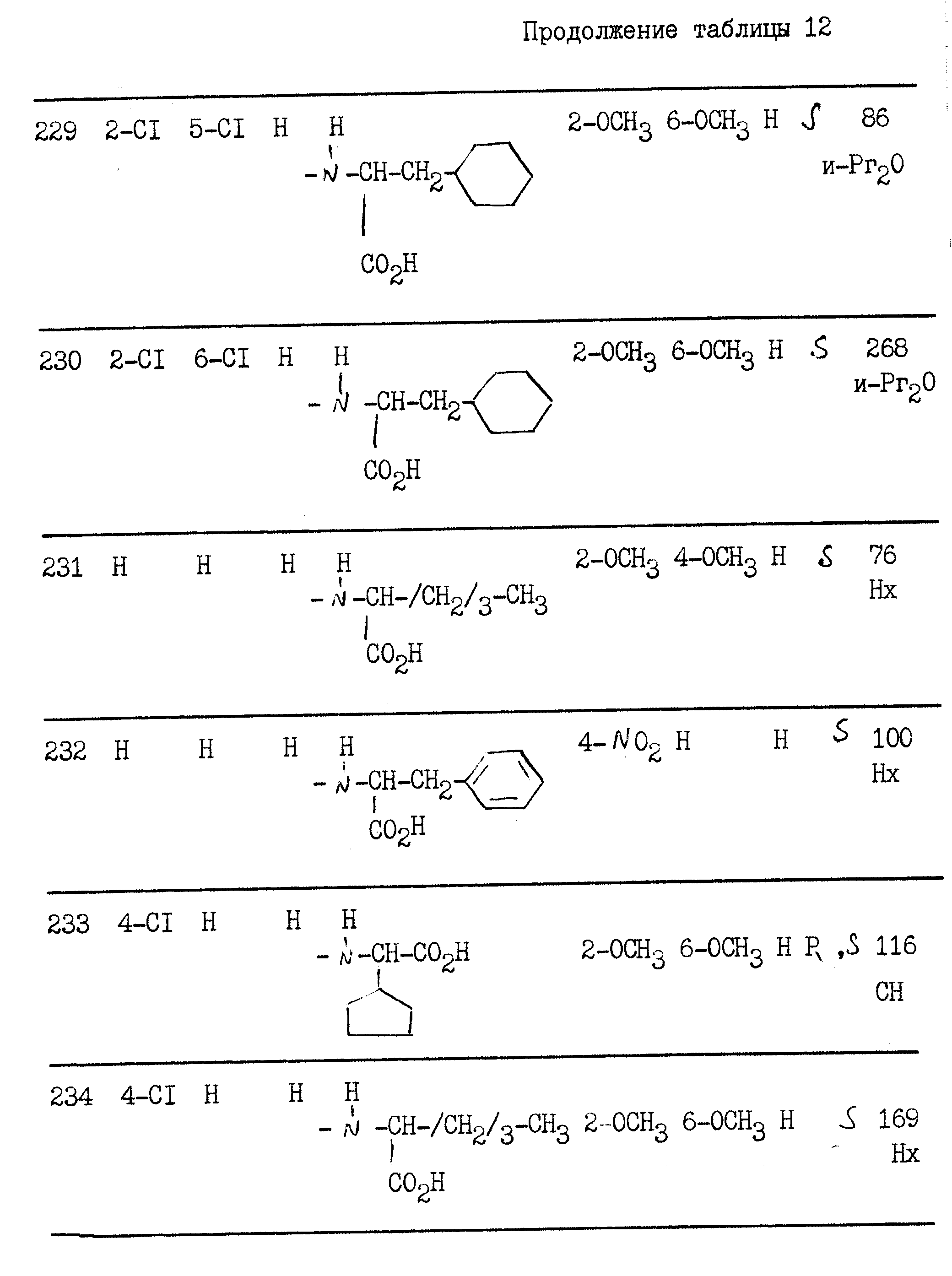
примерах 1 - 13, получили
соединения, указанные в приведенных ниже табл. 1-15. В этих таблицах R3, когда он используется представляет собой заместитель
Описанные ниже биологические и фармакологические испытания иллюстрируют активность новых соединений.
1.
Результаты
биохимических соединений
Соединения согласно изобретению были испытаны на препаратах из мембран головного мозга взрослых морских свинок. Связывание осуществлялось при 20oС в
присутствии 50 рМ (125 1)-Тир-нейротензина. Полное связывание измеряли по методу J.Z.Sasoul и др. (упомянутому выше). В таблицу 1 (приложение) сведены результаты биохимических испытаний.
Из нее
следует, что несколько соединений имеют наномолярное сродство к рецептору в гомогенате мозга морской свинки, идентичное природной лиганде.
Практически все соединения, полученные в примерах, являются ингибиторами нейтротензина при более или менее высоких концентрациях.
2. Воздействие на сердечно-сосудистую систему
Согласно R.Quirion и др.
Can. F.
Physical. Pharmacol, 1978, 56, (3), 671 нейротензин имеет позитивное инотропное и хронотропное действие на спонтанное сокращение изолированного правого предсердия морской свинки. Это
действие
вызывается прямой стимуляцией на уровне рецепторов нейротензина, находящихся в этой ткани (R.Quirion и др. Br.Journ, Pharmacol, 1980, 68, 83-91). Одно из соединений изобретения (соединение
примера
13)
было изучено в соответствии с этим тестом. Вызывают ингибирование положительного инотропного и хронотропного эффекта, вызванного нейротензином с концентрацией 1•10-8 М
на
спонтанные
сокращения изолированного правого предсердия морской свинки при Cl50 4,2 н М и 1,6 н М соответственно.
3. Воздействие на центральную нервную систему
Как это было
доказано для холецистокинина, P.Wouus и др. Life Sci, 1986, 39, 2199-2208 односторонняя и внутрь полосатого тела инъекция нейротензина (10 пг на 1 мл) самкам мышей СД1) Charles River
France),
находящихся в создании, вызывает также вращательное движение в противоположную сторону от места инъекции.
Соединение примера 13, введенное внутрибрюшинно или перорально за 30 и 60 мин соответственно до инъекции нейротензина, вызывает уменьшение вращательных движений, спровоцированных нейротензином, снижение на 80% вращательных движений достигается при дозе 80 мкг/кг (перорально), что доказывает, что это производное является антагонистом нейротензина на уровне центральной нервной системы.
4. Воздействие на желудочно-кишечную систему
Нейротензин при
концентрации 3•10-8 М вызывает эффект возбуждения спонтанных сокращений изолированного фундуса крысы. В этих условиях соединение примера 137 при концентрации
3•10-6
М ингибирует на 46% сокращения, вызванные нейротензином.
Кроме того, крайне важно подчеркнуть, что производные пиразола, заявленные в изобретении, являются первыми непептидными молекулами лиганд рецепторов нейротензина. ТТТ44 ТТТ45 ТТТ46 ТТТ47 ТТТ48 ТТТ49 ТТТ50
Реферат
Использование: в химикофармацевтической промышленности для
получения композиции, обладающей ингибирующей активностью в
отношении нейротензина. Сущность изобретения: производные
амидопиразолов общей формулы I, в которой Pyr означает группу формул II
или III, где RI - фенил, замещенный
определенными заместителями, RIa - бензильная
группа, возможно замещенная Hal, RIY-H, Hal, (C1-C6
)-алкил, Rv-фенильная группа, замещенная
определенными заместителями, или Rv-нафтил, пиридин,
или замещенный (С1-C4) алкилом стирил, или RIV и
Rv взятые вместе, означают группу
- 2Ph-(CH2)i, где фенильная группа замещает
пиразол в 5 положении, и группа (СН2)i, где i = 1 - 3, замещает
пиразол в положении 4, R - H1 (C1 -C4) алкил, Z - OH1 (C1
- C6) алкокси, амино, Х и ХI -H, или один из них Н, а другой
(С1-C6) алкил, (C3
- C6) циклоалкил (С1-C4) алкил,
фенил, амино(C1-C4)алкил, гидрокси(C1-C4
)алкил, карбокси(C1-C4)алкил,
ацетамидо (С1-C4) алкилтиометил,
гуанидино (С1-C4) алкил, карбокси (C1-C4) алкил,
нитрогуанидино (С1-C4) алкил,
(С3-C7) циклоалкил, фенил
(С1-C4), алкил, возможно замещенный Hal, ОН, гетероарил (C1-C4) алкил, где гетероарил - имидазолил или
индолил, или Х является Н и ХI и R,
взятые вместе, образуют с атомами азота цикл формулы II, где m = 1 - 2, или цикл индолинила, или 4,5,6,
7-тетрагидро-[2,3-c] тиенопиридина, или Х и XI каждый означает (С1-C4) алкил (С3-C6) циклоалкил, или фенил, или Х и XI вместе с
атомом углерода, с которым они связаны, образуют (C3 - C12)
циклоалкилиденовую группу, при необходимости замещенную (С1 - C3), алкилом, адамантилиденовую,
хинуклидинилиденовую, 4-пиперидинилиденовую группу,
возможно N-замещенную
бензилом, тетрагидронафтилиденовую, 4-тетрагидропиранилиденовую, дигидро 2,3 (4Н)-4-бензотиопиранилиденовую, или дигидро 2,
3(4Н)-4-бензопиранилиденовую группу, или группу формул
а и б, или их соли с
кислотами или основаниями, а также фармацевтическая композиция на основе указанных соединений. 2 с. и 1 з.п. ф-лы, 16 табл.
Структура соединений и групп:
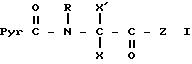
Формула
где Pyr группа общих формул I' или I''
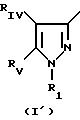
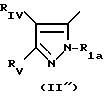
где R1 группа общей формулы
где
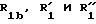
R1a бензильная группа, при необходимости замещенная галогеном;
RIV водород или галоген, C1 С6-алкил;
RV группа общей формулы
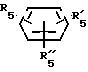
где
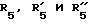
или RV нафтильная, или пиридильная, или замещенная С1 -С4-алкилом стирильная группа,
или RIV и RV, взятые вместе, группа
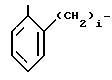
где фенильная группа замещает пиразол в положении 5 и группа (СH2)i, где i 1 3 целое число, от 1 до 3, замещает пиразол в положении 4;
R водород, C1 C4-алкил с прямой или разветвленной цепью;
Z гидроксильная группа, C1 C6-алкоксигруппа, аминогруппа;
Х и Х' оба водород или один из них водород, а другой С1 - С6-алкил с прямой или разветвленной цепью, C3 C6- циклоалкил-С1 C4-алкил, амино-С1 C4-алкил, гидрокси-С1 C4-алкил, карбокси-C1 C4-алкил, ацетамидо-С1 C4 - алкилтиометил, гуанидино-С1 - C4-алкил, нитрогуанидино-С1 C4-алкил, С3 - C7-циклоалкил, фенил-С1 C4-алкил, при необходимости замещенный галогеном, или гидроксил, гетероарил-С1 C4-алкил, где гетероарил означает имидазолил или индолил, или Х водород, а Х' и R, взятые вместе, образуют с атомом азота, с которым R связан, цикл формулы
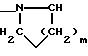
где m 1 или 2, или цикл индолинила или 4,5,6, 7-тетрагидро-(2,3-с)-тиенопиридина, или Х и Х' каждый С1 - C4-алкил, С3 C6-циклоалкил или фенил, или Х и Х' вместе с атомом углерода, с которым они связаны, образуют С3 - C12 -циклоалкилиденовую группу, при необходимости замещенную С1 - C3-алкилом, адамантилиденовую группу, хинуклидинилиденовую группу, 4-пиперидинилиденовую группу, при необходимости N-замещенную бензильной группой, тетрагидронафтилиденовую группу, 4-тетрагидропиранилиденовую группу, дигидро-2,3(4Н)-4-бензотиопиранилиденовую группу, дигидро-2,3(4Н)-4-бензопиранилиденовую группу или группу формулы
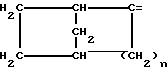
где n 1 или 2, или группу формулы
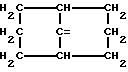
или их соли с органическими или минеральными кислотами или с органическими или неорганическими основаниями.
Комментарии