Способ получения высших линейных альфа-олефинов и/или алкилразветвленных альфа-олефинов, композицияна их основе (варианты) - RU2275349C2
Код документа: RU2275349C2
Чертежи

Описание
Настоящее изобретение относится к способу соолигомеризации этилена и альфа-олефинов и к композициям продуктов, получаемых в данном изобретении.
Известны различные способы получения высших линейных альфа-олефинов (например, D. Vogt, Oligomerisation of ethylene to higher α-olefins in Applied Homogeneous Catalysis with Organometallic Compounds Ed. B. Cornils, W. A. Herrmann Vol. 1, Ch. 2.3.1.3, page 245, VCH 1996).
Данные коммерческие способы позволяют получать распределение олигомерных продуктов либо по Пуассону, либо по Шульцу-Флори. Для того чтобы получить распределение Пуассона, во время олигомеризации не должно происходить обрыва цепи. Однако, в противоположность этому, в способе Шульца-Флори обрыв цепи происходит и он не зависит от длины цепи. Катализируемая Ni стадия олигомеризации этилена в способе получения высших олефинов компании Shell (SHOP) представляет собой типичный пример способа Шульца-Флори.
В способе Шульца-Флори обычно получают широкий спектр олигомеров, где содержание фракции каждого олефина можно определить, проведя вычисления на основе так называемого К-фактора. К-фактор, который указывает на относительные доли олефиновых продуктов, представляет собой мольное отношение [Cn+2]/[Cn], рассчитанное из наклона графика для log[Cn мол.%] в зависимости от n, где n представляет собой количество атомов углерода в конкретном олефиновом продукте. К-фактор по определению представляет собой одну и ту же величину для каждого n. В результате изменения лиганда и регулировки параметров реакции можно приводить К-фактор к бульшим или к меньшим значениям. Таким образом, можно управлять процессом для получения определенного набора продуктов с оптимизированной экономической выгодностью.
В WO-A-99/02472 описываются новые катализаторы олигомеризации этилена на основе железа, которые характеризуются высокой активностью и высокой селективностью по отношению к линейным альфа-олефинам. Основой катализаторов являются комплексы железа и выбранного 2, 6-пиридиндикарбоксальдегидбисимина или выбранного 2,6-диацилпиридинбисимина.
В настоящем изобретении термин «бис(арилиминоалкил)пиридин» или, если кратко, «бисарилиминпиридин» используют для описания обоих классов лигандов.
В одновременно находящейся на рассмотрении европейской патентной заявке № 00301036.0 тех же заявителей такие системы улучшены дополнительно, в частности, в отношении распределения олигомерных продуктов.
Было показано, что катализаторы на основе бисарилиминпиридин-FeCl2 обладают высокой реакционной способностью в отношении этилена, но, как было обнаружено, реакционная способность по отношению к другим олефинам, таким как пропилен или высшие альфа-олефины, ниже на несколько порядков.
B.L. Small и M. Brookhart описали в работе J. Am. Chem. Soc. 1998, 120, 7143-7144, что олигомеризация этилена при избыточном давлении 400 фунт/дюйм2 (2,76 МПа) в присутствии смеси с объемным отношением 1-пентена к толуолу 50:50 в качестве растворителя и катализатора на основе бисарилиминпиридин-FeCl2 привела к получению только приблизительно 3 мол.% олигомеров с нечетным числом атомов углерода, что, таким образом, демонстрирует очень высокую селективность такого катализатора в отношении внедрения этилена в сравнении с альфа-олефинами.
Дополнительные эксперименты в данной работе с другим катализатором на основе бисарилиминпиридин-FeCl2 показали еще более высокую селективность в отношении внедрения этилена по сравнению с внедрением альфа-олефинов, при этом были получены только следы (<1%) нечетных олигомеров.
Высокая селективность данных катализаторов в отношении этилена была подтверждена исследованиями V.C. Gibson et al., описанными в работе Chem. Eur. J. 2000, 6, 2221-2231.
Поэтому нет ничего удивительного в том, что применение таких каталитических систем было сфокусировано на продуктах и способах, где в качестве исходного сырья выступал этилен и где, предпочтительно, в продуктах не было или было мало разветвлений, например, на получении линейных альфа-олефинов.
Для процесса олигомеризации основные стадии реакции - стадия роста цепи и стадия обрыва цепи - сбалансированы таким образом, что образуются продукты с ограниченной молекулярной массой, то есть, иначе говоря, количество продуктов с высокими молекулярными массами минимально.
С упрощенной точки зрения можно считать, что рост цепи происходит в результате внедрения этилена в связь металл-водород (для первого мономера, приводящего к получению металлэтильного соединения) и в связи металл-углерод (для второго мономера и далее).
Общим явлением является возможность участия в реакциях со связями металл-водород или металл-углерод других олефинов наряду с этиленом. В частности, реакционной способностью обладают монозамещенные альфа-олефины. На результат реакции оказывают влияние структуры активных промежуточных соединений, тот способ, по которому альфа-олефины вступают в реакцию с ними, и способ, по которому образованные металлалкильные соединения вступают в реакцию дальше.
В реакциях олигомеризации этилена образование побочных продуктов, таких как разветвленные олефины, 2,2-замещенные альфа-олефины (олефины винилиденового типа) и олефины с внутренним расположением двойной связи, можно легко объяснить существованием данных промежуточных соединений. Должно быть очевидно, что с учетом распределения олигомерных альфа-олефинов, образуемых при олигомеризациях этилена, можно получить широкий ассортимент побочных продуктов, что приведет к потере качества продуктов и к напрасному расходованию ценного этиленового сырья. Однако катализаторы, которые сочетают особую реакционную способность в отношении альфа-олефинов с пригодностью для олигомеризации этилена, имели бы большое значение при создании новых технологий получения альфа-олефинов из альтернативных видов исходного сырья или получения (смесей) альфа-олефиновых продуктов со специальными структурами, спроектированными для достижения желательных свойств.
Например, получение 1-гексена, 1-октена или 1-децена в результате гомологизации 1-бутена при помощи этилена можно предположить для систем, которые после обрыва цепи начинают с «1,2»-внедрения 1-бутена в связь металл-водород (образованную после обрыва цепи), но которые после этого перед обрывом цепи не вступают в реакцию в значительной мере ни с каким другим олефином, кроме как с этиленом. Таким образом, дешевый 1-бутен, получаемый на нефтеперерабатывающем заводе, можно превратить в высокоценные альфа-олефины.
Другая интересная возможность представляет собой образование алкилразветвленных альфа-олефинов с хорошо определенной структурой разветвления, являющееся следствием свойств катализатора и условий реакции. Например, метилразветвленные альфа-олефины можно получить, используя системы, которые после обрыва цепи, предпочтительно, начинают с «2,1»-внедрения олефина в связь металл-водород и которые после этого перед обрывом цепи не вступают в реакцию в значительной мере ни с каким другим олефином, кроме как с этиленом.
В настоящем изобретении под «метилразветвленным альфа-олефином» подразумевается олефин, образованный в результате «2,1»-внедрения альфа-олефина формально в связь металл-водород системы, и при том в дальнейшем эта система перед обрывом цепи не вступает в реакцию в значительной мере ни с каким другим олефином, кроме как с этиленом. Альтернативным образом данное «2,1»-внедрение олефина в связь металл-водород можно объяснить обрывом цепи в результате переноса водорода на координированный олефин с получением металл(2-алкильного) соединения в качестве исходного соединения для процесса олигомеризации. В последующем тексте, ради простоты, изложение будет придерживаться механизма, упомянутого первым.
Получение С8-С16 метилразветвленных альфа-олефинов имеет большое экономическое значение, поскольку они могут служить в качестве исходного сырья при алкилировании бензола, представляя собой таким образом исходный материал для получения высокорастворимых поверхностно-активных веществ на основе алкилбензолсульфонатов, и в качестве исходного сырья для процессов гидроформилирования, приводящих к получению высокорастворимых поверхностно-активных веществ на основе спиртов и их производных.
Кроме того, если, например, в качестве «растворителя» при (со)олигомеризации этилена использовать 1-децен, то один единственный процесс приведет к получению линейных 1-алкенов в С4-С10 диапазоне, а также линейных и/или разветвленных 1-алкенов в диапазоне >С12.
Наряду с продуктами с конкретным метильным разветвлением экономический интерес представляют и продукты с конкретным этильным разветвлением. Как можно предвидеть, предпочтительность этильного разветвления будет подтверждаться в каталитических системах, в которых реакция передачи цепи, предпочтительно, происходит на мономерный этилен. В получающихся в результате металлэтильных соединениях рост цепи может иметь место в результате внедрения либо дополнительного этилена, либо другого олефинового сомономера.
В настоящем изобретении под «этилразветвленным альфа- олефином» подразумевается олефин, образованный в результате «1, 2»-внедрения альфа-олефина формально в связь металл-этил системы, и притом после этого данная система перед обрывом цепи не вступает в реакцию в значительной мере ни с каким другим олефином, кроме как с этиленом.
Удостовериться в том, имеют ли место в ходе олигомеризации этилена предложенные реакции и образование желательных молекулярных структур, описанных выше, мешает тот факт, что один и тот же продукт можно получить по более чем одному пути реакции.
Например, линейные альфа-олефины можно получить не только в результате чистой олигомеризации этилена, но также и в результате гомологизации этиленом меньшего альфа-олефина, полученного через «1,2»-внедрение.
Более детальное представление о продуктах и стадиях реакции можно получить из экспериментов по соолигомеризации, в которых сомономером является альфа-олефин с нечетным числом атомов углерода. В результате сопоставления продуктов с нечетным и четным числом атомов углерода и получения их характеристик процессы олигомеризации этилена, которые имеют место в присутствии альфа-олефинов с нечетным числом атомов углерода, позволят получить информацию о внедрении олефинов в продукты. Например, олигомеризация этилена в присутствии 1-гептена может привести к получению обычных С2n альфа-олефинов, а также линейных нечетных альфа-олефинов, начиная с 1-нонена, С9. Соотношение количеств нечетных и четных линейных олефинов представляет собой меру относительной реакционной способности этилена и альфа-олефинов на первой стадии роста цепи в экспериментах.
Важную информацию относительно структур (побочных) продуктов при процессах олигомеризации этилена можно получить, проводя реакцию в присутствии большого избытка конкретного альфа-олефина, например, проводя соолигомеризацию. Это создает эффект упрощения получаемого обычно распределения олигомеров за счет единственного олефина с одной и той же реакционной способностью. Как результат, после этого становятся очевидными образование (побочных) продуктов вследствие внедрения полученных на основе единственного сомономера альфа-олефинов и выход хорошо определенных структур.
Характеристики данных структур получить относительно легко, даже при их присутствии в небольших количествах, проведя сравнение1Н- и13 С-ЯМР-спектров образцов, отличающихся различными уровнями содержания (побочных) продуктов. Из литературы известны и могут быть использованы характеристические резонансы ЯМР для ненасыщенных концевых групп в альфа-олефинах, 2,2-двузамещенных альфа-олефинах (олефинах винилиденового типа), для одиночных метильных и этильных групп вдоль алифатической цепи.
Присутствие 2, 2-двузамещенных альфа-олефинов можно объяснить «1,2»-внедрением альфа-олефина в связь металл-углерод в растущей цепи с последующим обрывом цепи (β-Н элиминирование). Существование распределения метилразветвленных альфа-олефинов соответствует процессу роста цепи, в котором первая стадия реакции включает «2,1»-внедрение сомономера формально в гидрид металла с получением металл(2-алкильного) промежуточного соединения, которое впоследствии претерпевает процессы олигомеризации этилена. Подобным же образом существование распределения для этилразветвленных альфа-олефинов можно объяснить исходя из такого предположения, что обрыв цепи происходит в результате переноса водорода на координированный мономерный этилен с получением металлэтильного соединения в качестве исходного сырья для процесса олигомеризации, в котором первая стадия представляет собой «1,2»-внедрение альфа-олефина в данную связь металл-этил, что приводит к получению металл(3-алкильного) промежуточного соединения, которое впоследствии претерпевает олигомеризации этилена. Само собой разумеется, что тип наблюдаемых побочных продуктов должен характеризоваться подобной структурой для альфа-олефинового сомономера с нечетным и четным числом атомов углерода.
В настоящее время к удивлению было обнаружено, что в результате регулирования условий проведения реакции, в частности, при использовании в реакции соолигомеризации этилена подходящих олефинов при соответствующих концентрациях и специальных каталитических систем на основе бисарилиминпиридинметалла, применяемых в настоящем изобретении, можно в значительной степени увеличить образование линейных альфа-олефинов в результате гомологизации при помощи этилена меньших линейных альфа-олефинов и образование алкилразветвленных, в частности метилразветвленных и/или этилразветвленных, альфа-олефинов.
Под «алкилразветвленным альфа-олефином» в настоящем изобретении, предпочтительно, подразумевается «метилразветвленный альфа-олефин», «этилразветвленный альфа-олефин» или их комбинация.
Должно быть оценено то, что в то время, как алкилразветвленные альфа-олефины настоящего изобретения можно получать по предположительным механизмам, описанным выше, не исключается и то, что упомянутые олефины можно получать и по альтернативному механизму реакции.
Общая структура «алкилразветвленных альфа-олефинов» описывается формулой, приведенной ниже:
С=С[-C-C]n[-C]m(R16)-R,
где R16=метил; n=0, 1, 2 и так далее; m=1; R=необязательно замещенный гидрокарбил, предпочтительно, содержащий от 1 до 30 атомов углерода, или же R16=этил; n=0, 1, 2 и так далее; m=0; R=необязательно замещенный гидрокарбил, предпочтительно, содержащий от 1 до 30 атомов углерода.
Настоящее изобретение предлагает способ получения высших линейных альфа-олефинов и/или алкилразветвленных альфа-олефинов, который включает соолигомеризацию одного или нескольких альфа-олефинов с этиленом в присутствии металлсодержащей каталитической системы, использующей один или несколько комплексов бисарилиминпиридин-МХа и/или один или несколько комплексов [бисарилиминпиридин-MYp·Lb+][NC-]q, причем упомянутые бисарилиминпиридиновые комплексы содержат лиганд, описываемый формулой:

где М представляет собой атом металла, выбираемого из Fe или Со; а равно 2 или 3; Х представляет собой галогенид, необязательно замещенный гидрокарбил, алкоксид, амид или гидрид; Y представляет собой лиганд, который может позволить пройти внедрению олефина; NC- представляет собой некоординирующий анион; р+q равно 2 или 3 в соответствии с формальной степенью окисления упомянутого атома металла; L представляет собой молекулу нейтрального донора Льюиса; b=0, 1 или 2; каждый из R1-R5 независимо от других представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу или же любые два из R1-R3, вицинальные друг по отношению к другу, взятые вместе, могут образовывать кольцо; каждый из Z, которые могут быть идентичными или различными, представляет собой необязательно замещенное ароматическое углеводородное кольцо; необязательно замещенный полиароматический углеводородный фрагмент; необязательно замещенный гетерогидрокарбильный фрагмент или же необязательно замещенное ароматическое углеводородное кольцо в комбинации с металлом, причем упомянутое необязательно замещенное ароматическое углеводородное кольцо π-координировано с металлом; и упомянутый способ реализуют при давлении этилена, меньшем чем 2,5 МПа.
В предпочтительном варианте реализации настоящего изобретения предлагается способ получения высших линейных альфа-олефинов и/или алкилразветвленных альфа-олефинов, который включает соолигомеризацию одного или нескольких альфа-олефинов с этиленом в присутствии металлсодержащей каталитической системы, использующей один или несколько комплексов бисарилиминпиридин-МХа и/или один или несколько комплексов [бисарилиминпиридин-MYp·Lb+][NC-]q, причем упомянутые бисарилиминпиридиновые комплексы содержат лиганд, описываемый формулой:

где М представляет собой атом металла, выбираемого из Fe или Со; а равно 2 или 3; Х представляет собой галогенид, необязательно замещенный гидрокарбил, алкоксид, амид или гидрид; Y представляет собой лиганд, который может позволить пройти внедрению олефина; NC- представляет собой некоординирующий анион; р+q равно 2 или 3, в соответствии с формальной степенью окисления упомянутого атома металла; L представляет собой молекулу нейтрального донора Льюиса; b=0, 1 или 2; каждый из R1-R10 независимо от других представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу, или же любые два из R1-R3, R6-R10, вицинальные друг по отношению к другу, взятые вместе, могут образовать кольцо; R6 может быть взят вместе с R4 с образованием кольца; R10 может быть взят вместе с R4 с образованием кольца; Z представляет собой необязательно замещенное ароматическое углеводородное кольцо; необязательно замещенный полиароматический углеводородный фрагмент; необязательно замещенный гетерогидрокарбильный фрагмент или же необязательно замещенное ароматическое углеводородное кольцо в комбинации с металлом, причем упомянутое необязательно замещенное ароматическое углеводородное кольцо π-координировано с металлом; и упомянутый способ реализуют при давлении этилена, меньшем чем 2,5 МПа.
В предпочтительном варианте реализации настоящего изобретения предлагается способ получения высших линейных альфа-олефинов и/или алкилразветвленных альфа-олефинов, который включает соолигомеризацию одного или нескольких альфа-олефинов с этиленом в присутствии металлсодержащей каталитической системы, использующей один или несколько комплексов бисарилиминпиридин-МХа и/или один или несколько комплексов [бисарилиминпиридин-MYp·Lb+][NC-]q, причем упомянутые бисарилиминпиридиновые комплексы содержат лиганд, описываемый формулой:

где М представляет собой атом металла, выбираемого из Fe или Со; а равно 2 или 3; Х представляет собой галогенид, необязательно замещенный гидрокарбил, алкоксид, амид или гидрид; Y представляет собой лиганд, который может позволить пройти внедрению олефина; NC- представляет собой некоординирующий анион; р+q равно 2 или 3 в соответствии с формальной степенью окисления упомянутого атома металла; L представляет собой молекулу нейтрального донора Льюиса; b=0, 1 или 2; каждый из R1-R5, R7-R9 и R12-R14 независимо от других представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу, или же любые два из R1-R3, R7-R9 и R12-R14, вицинальные друг по отношению к другу, взятые вместе, могут образовать кольцо; R6 представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу или же, взятый вместе с R7 или с R4, образует кольцо; R10 представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу или же, взятый вместе с R9 или с R4, образует кольцо; R11 представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу или же, взятый вместе с R5 или с R12, образует кольцо; и R15 представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу или же, взятый вместе с R5 или с R14, образует кольцо; и упомянутый способ реализуют при давлении этилена, меньшем чем 2,5 МПа.
В одном варианте реализации настоящего изобретения в применяемой металлсодержащей каталитической системе используется один или несколько комплексов бисарилиминпиридин-МХа и второе соединение, которое способно передавать необязательно замещенный гидрокарбил или гидридную группу на атом металла М, выбираемый из Fe или Со, и которое также способно отрывать от упомянутого атома металла группу Х-.
В другом варианте реализации настоящего изобретения в применяемой металлсодержащей каталитической системе используется один или несколько комплексов бисарилиминпиридин-МХа и второе соединение, которое способно передавать необязательно замещенный гидрокарбил или гидридную группу на атом металла М, выбираемый из Fe или Со, и третье соединение, которое способно отрывать от упомянутого атома металла группу Х- .
В настоящем изобретении некоторые термины используются следующим образом:
Под «высшими» в высших линейных альфа-олефинах и высших алкилразветвленных альфа-олефинах подразумеваются молекулы, содержащие от 4 до 30 углеродных атомов.
Примеры необязательно замещенных ароматических углеводородных колец и необязательно замещенных полиароматических углеводородных фрагментов включают фенил, нафтил, антраценил, фенантраценил и тому подобное и их замещенные производные.
Термин «необязательно замещенное ароматическое углеводородное кольцо в комбинации с металлом, причем упомянутое необязательно замещенное ароматическое углеводородное кольцо π-координировано с металлом, включает металлоценовые фрагменты и сэндвичевые и металлареновые комплексы. Таким образом, специалистом в соответствующей области должно быть оценено то, что металл необязательно может быть дополнительно π-координирован с еще одним необязательно замещенным ароматическим углеводородным кольцом, которое может быть отличным от необязательно замещенного ароматического углеводородного кольца в Z, которое непосредственно связано с иминовым атомом азота, и/или координирован с другими лигандами, широко известными на современном уровне техники. Кроме этого, должно быть оценено то, что необязательно замещенное ароматическое углеводородное кольцо в Z, которое непосредственно связано с иминовым атомом азота и которое также π-координировано с металлом, может содержать в кольце один или несколько гетероатомов, то есть так, что упомянутое необязательно замещенное ароматическое углеводородное кольцо будет представлять собой необязательно замещенную ароматическую гетероциклическую группу. Подобным же образом еще одна необязательно замещенная арильная группа, с которой дополнительно может быть π-координирован металл, может содержать в кольце один или несколько гетероатомов. Упомянутый атом металла в удобном случае может представлять собой железо, кобальт, никель, хром, титан и ванадий. Примеры таких звеньев включают радикал, полученный из ферроцена, кобальтоцена, никелоцена, хромоцена, титаноцена, ванадоцена, бис-π-аренванадиевого комплекса, моно-π-аренхромтрикарбонильного комплекса и подобных гетероаренметаллсодержащих комплексов, то есть бис- или моно-π-тиен- или -пирролжелезо- или -хромсодержащих комплексов.
Термин «гетерогидрокарбил» означает гидрокарбильную группу, дополнительно содержащую один или несколько гетероатомов. Упомянутые гетероатомы в гетерогидрокарбильной группе, предпочтительно, связаны, по меньшей мере, с двумя углеродами. Предпочтительными гетероатомами являются азот, кислород и сера.
Упомянутая гетерогидрокарбильная группа может представлять собой необязательно замещенный ароматический гетероциклический фрагмент; необязательно замещенный полиароматический гетероциклический фрагмент; необязательно замещенный алифатический гетероциклический фрагмент или же необязательно замещенный алифатический гетерогидрокарбильный фрагмент.
Примеры гетерогидрокарбильных групп включают 1-пирролил, 2-пирролил, 3-пирролил, фурил, тиенил, инденил, имидазолил, триазолил, оксазолил, изоксазолил, карбазолил, тиазолил, бензотиазолил, тиадиазолил, пиримидинил, пиридил, пиридазинил и тому подобное и их замещенные производные.
Гидрокарбильная группа: группа, содержащая только углерод и водород. Если только не будет утверждаться другого, то количество углеродных атомов, предпочтительно, будет находиться в диапазоне от 1 до 30.
В настоящем изобретении фраза «необязательно замещенный гидрокарбил» используется для описания гидрокарбильных групп, необязательно содержащих одну или несколько «инертных» гетероатомсодержащих функциональных групп. Под термином «инертный» подразумевается то, что функциональные группы не вовлекаются в какой-либо значительной степени в процесс соолигомеризации. Неограничивающими примерами таких инертных групп являются фторид, хлорид, силаны, станнаны, простые эфиры и амины с соответствующей стерической защитой, все хорошо известные специалистам в соответствующей области. Упомянутый необязательно замещенный гидрокарбил может включать содержащие первичный, вторичный и третичный атом углерода группы описанной ниже природы.
Инертная функциональная группа: группа, отличная от необязательно замещенного гидрокарбила, которая является инертной в условиях реализации способа. Под термином «инертная» подразумевается то, что функциональная группа в какой-либо значительной степени не вовлекается в процесс соолигомеризации. Примеры инертных функциональных групп включают галогенид, простые эфиры и амины, в частности третичные амины.
Группа, содержащая первичный атом углерода: группа -СН2-R, где R может быть водородом, необязательно замещенным гидрокарбилом, инертной функциональной группой. Примеры групп, содержащих первичный атом углерода, включают -СН3, -С2Н5, -СН2Cl, -СН2ОСН3, -СН2N(С2Н5)2, -СН2Ph.
Группа, содержащая вторичный атом углерода: группа -СН-R2, где R может быть необязательно замещенным гидрокарбилом, инертной функциональной группой. Примеры групп, содержащих вторичный атом углерода, включают -СН(СН3)2, -CHCl2, -CHPh2, -СН=СН2, циклогексил.
Группа, содержащая третичный атом углерода: группа -С-R3, где R может быть необязательно замещенным гидрокарбилом, инертной функциональной группой. Примеры групп, содержащих третичный атом углерода, включают -С(СН3)3, -CCl3, -С≡CPh, 1-адамантил, -С(СН3)2(ОСН3).
Под «лигандом, который может позволить пройти внедрению олефина» подразумевается лиганд, который координирован с ионом металла с образованием такой связи, в которую для инициирования или продолжения реакции соолигомеризации могут быть внедрены молекула этилена или альфа-олефин. В комплексах [бисарилиминпиридин-MYp·Lb+][NC-]q, соответствующих настоящему изобретению, Y может быть гидридом, алкилом или любым другим анионным лигандом, который может позволить пройти внедрению олефина.
Под «некоординирующим анионом» подразумевается анион, который, по существу, не координируется с атомом металла М. Некоординирующие анионы (NC-), которые можно подходящим образом использовать, включают объемные анионы, такие как тетракис [3, 5-бис(трифторметил)фенил]борат (BAF-), (C6F5)4B-, и анионы алюминийалкильных соединений, в том числе R3 AlX-, R2 AlClX-, RAlCl2X- и «RAlOX-», где R представляет собой водород, необязательно замещенный гидрокарбил или инертную функциональную группу, а Х представляет собой галогенид, алкоксид или кислород.
Специалистами в соответствующей области должно быть оценено то, что в диапазоне граничных условий, описанных здесь и ранее в настоящем документе, заместители R1-R15 можно будет легко выбирать для оптимизации эксплуатационных характеристик каталитической системы и ее экономичного приложения.
Заместители R1-R5, R7-R9, R12-R14 могут независимо друг от друга быть связаны вместе и образовывать циклические структуры.
В одном варианте реализации настоящего изобретения каждый из R1-R5, R7-R9 и R12-R14 независимо от других представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу, или же любые два из R1-R3, R7-R9 и R12-R14, вицинальные друг по отношению к другу, взятые вместе, могут образовывать кольцо; R6 представляет собой группу, содержащую первичный углерод, группу, содержащую вторичный углерод, или же группу, содержащую третичный углерод; и при том условии, что:
если R6 представляет собой группу, содержащую первичный углерод, ни один из R10, R11 и R15 не представляет собой группы, содержащей первичный углерод, или же один или два из R10, R11 и R15 представляют собой группу, содержащую первичный углерод, а остальные группы из R10, R11 и R15 представляют собой водород;
если R6 представляет собой группу, содержащую вторичный углерод, ни один из R10, R11 и R15 не представляет собой группы, содержащей первичный углерод, или группы, содержащей вторичный углерод, или же один из R10, R11 и R15 представляет собой группу, содержащую первичный углерод, или группу, содержащую вторичный углерод, а остальные группы из R10, R11 и R15 представляют собой водород;
если R6 представляет собой группу, содержащую третичный углерод, все группы из R10, R11 и R15 представляют собой водород; и
любые два из R6, R7, R8, R9, R10, R11, R12, R13, R14 и R15, вицинальные по отношению друг к другу, взятые вместе, могут образовывать кольцо.
В еще одном варианте реализации настоящего изобретения каждый из R1 -R5, R7-R9 и R12-R14 независимо от других представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу, или же любые два из R1-R3, R7-R9 и R12-R14, вицинальные друг по отношению к другу, взятые вместе, могут образовывать кольцо; R6 представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу, или же, взятый вместе с R7 или с R4, образует кольцо; R10 представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу, или же, взятый вместе с R9 или с R4, образует кольцо, взятый вместе с R7 или с R4, образует кольцо; R11 и R15 независимо друг от друга представляют собой водород или инертную функциональную группу.
В другом варианте реализации настоящего изобретения каждый из R1-R5, R7-R9 и R12-R14 независимо от других представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу, или же любые два из R1-R3, R7-R9 и R12-R14, вицинальные друг по отношению к другу, взятые вместе, могут образовывать кольцо; R6, R10, R11 и R15 идентичны и каждый их них выбирается из фтора или хлора.
В другом варианте реализации способа настоящего изобретения бисарилиминпиридиновые комплексы, использованные в настоящем изобретении, содержат лиганд, описываемый формулой (IV):

где каждый из А1-А6 независимо от других представляет собой углерод, азот, кислород или серу; группа атомов
необязательно может отсутствовать, так что А1 будет непосредственно связан с А5; а каждый из R1-R12, R14-R15 и, в случае наличия, R13 независимо от других представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу или же любые два из R1-R15, вицинальные друг по отношению к другу, взятые вместе, могут образовывать кольцо; при том условии, что, если А1-А5 и, в случае наличия, А6 все будут углеродами, то упомянутые атомы будут составлять циклопентадиенильную или арильную часть π-координированного металла.
В предпочтительном варианте реализации настоящего изобретения в формуле (IV) каждый из R1-R3, R7 -R9, R12, R14 и, в случае наличия, R13 независимо от других представляет собой водород, необязательно замещенный гидрокарбил, инертную функциональную группу, или же любые два из R1-R3, R7-R9, R12-R14, вицинальные друг по отношению к другу, взятые вместе, могут образовывать кольцо; и
а) R6 представляет собой инертную функциональную группу или необязательно замещенный гидрокарбил, а R10, R11 и R15 независимо друг от друга представляют собой водород или галогенид; или
b) R11 представляет собой инертную функциональную группу или необязательно замещенный гидрокарбил, а R6, R10 и R15 независимо друг от друга представляют собой водород или галогенид; или
с) каждый из R6 и R10 независимо друг от друга представляет собой инертную функциональную группу или группу, содержащую первичный или вторичный атом углерода, при том условии, что R6 и R10 не представляют собой оба сразу группу, содержащую вторичный атом углерода, а R11 и R15 независимо друг от друга представляют собой водород или галогенид; или
d) каждый из R11 и R15 независимо от другого представляет собой инертную функциональную группу или группу, содержащую первичный или вторичный атом углерода, при том условии, что R11 и R15 не представляют собой оба сразу группу, содержащую вторичный атом углерода, а R6 и R10 независимо друг от друга представляют собой водород или галогенид; или
е) R6, взятый вместе с R7, образует кольцо, R10 представляет собой группу, содержащую первичный атом углерода, инертную функциональную группу или водород, а R11 и R15 независимо друг от друга представляют собой водород или галогенид; или
f) R11, взятый вместе с R12, образует кольцо, R15 представляет собой группу, содержащую первичный атом углерода, инертную функциональную группу или водород, а R6 и R10 независимо друг от друга представляют собой водород или галогенид; или
g) R6 и R10, взятые вместе с R7 и R9, соответственно, образуют кольца, а R11 и R15 независимо друг от друга представляют собой водород или галогенид; или
h) R11 и R15, взятые вместе с R12 и R14, соответственно, образуют кольца, а R6 и R10 независимо друг от друга представляют собой водород или галогенид.
В формуле (IV) заместители R1-15, в случае наличия, могут независимо друг от друга быть связаны вместе с образованием циклических структур. Примеры таких структур включают связь, например, R6 c R7 с образованием базовой нафтильной структуры или тетрагидронафтильного фрагмента.
Кроме этого, любым специалистом, хорошо разбирающимся в основных принципах гомогенного катализа, легко должно быть оценено то, что во всех из упомянутых выше лигандов, предназначенных для применения в бисарилиминпиридиновых комплексах, используемых в способе настоящего изобретения, вариации заместителей R1-5, R7-9 и R12-14, в случае их наличия, можно выбирать таким образом, чтобы улучшить другие желательные свойства предшественников катализаторов и каталитических систем, такие как растворимость в неполярных растворителях или расширение диапазона подходящих исходных веществ при их синтезе.
Предпочтительные варианты реализации настоящего изобретения используют лиганды, соответствующие формуле (I), и их производные, в которых присутствуют следующие группы R:
R1-R3 представляют собой водород; и/или R4 и R5 представляют собой метил, водород, бензил или фенил, предпочтительно, метил, фенил или водород.
Предпочтительные варианты реализации настоящего изобретения используют лиганды, соответствующие формулам (I), (II), (III) и (IV) и их производные, в которых присутствуют следующие группы R:
R1-R3 представляют собой водород; и/или R4 и R5 представляют собой метил, водород, бензил или фенил, предпочтительно метил, фенил или водород.
Предпочтительные варианты реализации представляют собой лиганды, соответствующие (IV), и их производные, в которых присутствуют следующие группы R:
R1-R3 представляют собой водород; и/или
R4 и R5 представляют собой метил, водород или фенил, предпочтительно метил; и/или группа

отсутствует, а А1-А5 представляют собой атомы углерода, таким образом образующие циклопентадиенилидную часть ферроценильного фрагмента; или
А3 представляет собой атом азота, группа
отсутствует, а А1, А2, А4, А5 представляют собой атомы углерода, таким образом образующие 1-пирролильное кольцо; и/или
комбинации ортозаместителей, в которых R6 представляет собой метил, этил, изопропил, фенил, третичный бутил или же связан с R7 с образованием нафтильной структуры; R10 представляет собой водород, фторид или хлорид; R11 и R15 независимо друг от друга представляют собой водород, фторид или хлорид, и/или
комбинации ортозаместителей, в которых R6 и R10 независимо друг от друга представляют собой метил, этил или же связаны с R7 и R9 соответственно, с образованием антраценовой структуры, и предпочтительно, R6 и R10 представляют собой метил; R11 и R15 независимо друг от друга представляют собой водород, фторид или хлорид.
В особенности предпочтительно, если в формуле (IV) R11 и R15 независимо друг от друга представляют собой водород или фторид.
Предпочтительные лиганды включают:
лиганд, описываемый формулой (II), где R1-R3 представляют собой водород; R4 и R5 представляют собой метил; R6, R8, R10 представляют собой метил; R7, R9 представляют собой водород, а Z представляет собой 1-пирролил;
лиганд, описываемый формулой (II), где R1-R3 представляют собой водород; R4 и R5 представляют собой метил; R6, R8, R10 представляют собой метил; R7 и R9 представляют собой водород, а Z представляет собой ферроценил;
лиганд, описываемый формулой (III), где R1 -R3 представляют собой водород; R4 и R5 представляют собой метил; R6, R8 и R10 представляют собой метил; R7 и R9 представляют собой водород; R11 и R15 представляют собой водород; R12 и R14 представляют собой водород; а R13 представляет собой трет-бутил;
лиганд, описываемый формулой (III), где R1-R3 представляют собой водород; R4 и R5 представляют собой метил; R6 и R7, взятые вместе, образуют шестичленное ароматическое кольцо; R8 и R10 представляют собой водород; R9 представляет собой водород; R11 и R15 представляют собой водород; R12 и R14 представляют собой водород; а R13 представляет собой трет-бутил;
лиганд, описываемый формулой (III), где R1-R3 представляют собой водород; R4 и R5 представляют собой метил; R6 представляет собой трет-бутил; R7-R10 представляют собой водород; R11 и R15 представляют собой водород; R12 и R14 представляют собой водород; а R13 представляет собой трет-бутил;
лиганд, описываемый формулой (III), где R1-R3 представляют собой водород; R4 и R5 представляют собой метил; R6, R8 и R10 представляют собой метил; R7 и R9 представляют собой водород; R11 представляет собой фтор; а R12-R15 представляют собой водород;
лиганд, описываемый формулой (III), где R1-R3 представляют собой водород; R4 и R5 представляют собой метил; R6 представляет собой трет-бутил; R7-R10 представляют собой водород; R11, R13 и R15 представляют собой водород; а R12 и R14 представляют собой метил;
лиганд, описываемый формулой (III), где R1-R3 представляют собой водород; R4 и R5 представляют собой метил; R6 и R10 представляют собой фтор; R7-R9 представляют собой водород; R12 и R15 представляют собой метил; а R11, R13 и R14 представляют собой водород;
лиганд, описываемый формулой (III), где R1-R3 представляют собой водород; R4 и R5 представляют собой метил; R7-R9 и R12-R14 представляют собой водород; а R6, R10, R11 и R15 представляют собой фтор; и
лиганд, описываемый формулой (III), где R1-R3 представляют собой водород; R4 и R5 представляют собой метил; R7-R10 представляют собой водород; R6 представляет собой метил; R11-R14 представляют собой водород; а R15 представляет собой метил.
В комплексе бисарилиминпиридин-МХа Х в удобном случае может представлять собой галогенид, предпочтительно, хлорид.
В предпочтительном варианте реализации комплекса бисарилиминпиридин-МХа атом металла представляет собой Fe, а «а» равно 2. В еще одном предпочтительном варианте реализации атом М представляет собой Fe, а "a" равно 3.
Соединения, которые способны передавать к атому металла М необязательно замещенный гидрокарбил или гидридную группу и которые также способны отрывать от атома металла М группу Х-, включают алюминийалкильные соединения, такие как алкилалюмоксан и алкилалюминийгалогениды. Предпочтительным соединением является метилалюмоксан.
Соединения, которые способны передавать к атому металла М необязательно замещенный гидрокарбил или гидридную группу, включают алюминийалкильные соединения, в том числе алкилалюмоксаны, литийалкильные соединения, реактивы Гриньяра, оловоалкильные и цинкалкильные соединения.
Соединения, которые способны отрывать от атома металла М группу Х-, включают сильные нейтральные кислоты Льюиса, такие как SbF5, BF3 и Ar3B, где Ar представляет собой арильную группу, сильно оттягивающую на себя электронную плотность, такую как C6F5 или 3,5-(CF3)2C6H3.
Молекула - нейтральный донор Льюиса - представляет собой соединение, которое в подходящем случае может выступать в роли основания Льюиса, такое как простые эфиры, амины, сульфиды и органические нитрилы.
Использование молекул доноров (оснований Льюиса), таких как триэтиламин или 2,6-ди-трет-бутилпиридин, и/или молекул акцепторов (кислоты Льюиса), таких как диэтилцинк, может оказать положительное воздействие на селективность способа соолигомеризации этилена.
Кроме этого, кислоты Льюиса, такие как триизобутилалюминий (TIBA), могут улучшить непрерывный вариант соолигомеризации этилена с использованием катализаторов на основе Fe или Со, делая возможным получение стабильных и прозрачных растворов предшественников катализаторов, в противоположность активированным с использованием МАО и солюбилизированным растворам предшественников катализаторов, которые могут становиться мутными при стоянии.
В комплексе [бисарилиминпиридин-MYp·Ln+][NC-]q, соответствующем настоящему изобретению, L может представлять собой молекулу нейтрального донора Льюиса, способную быть замещенной этиленом, или же вакантное координационное положение.
В комплексе [бисарилиминпиридин-MYp·Ln+][NC-]q, соответствующем настоящему изобретению, атомом металла М, предпочтительно, является Fe, а формальная степень окисления упомянутого атома металла может быть равна 2 или 3.
Каталитическую систему можно образовать в результате смешивания друг с другом комплекса и необязательных дополнительных соединений, предпочтительно, в растворителе, таком как толуол или изооктан.
Мольное соотношение комплекса МХn, второго соединения и необязательно третьего соединения в настоящем изобретении не ограничивается.
Существует возможность увеличения гибкости реакции соолигомеризации в результате использования смеси одной или нескольких каталитических систем, соответствующих настоящему изобретению.
В реакционной смеси для соолигомеризации обычно используют такое количество каталитической системы, чтобы на один моль вступающих в реакцию этилена и/или альфа-олефина иметь от 10-4 до 10-9 грамм атома металла М, в частности металла Fe[II] или [III].
В удобном случае реакцию соолигомеризации можно проводить в диапазоне температур от -100 до 300°С, предпочтительно, в диапазоне от 0 до 200°С, а более предпочтительно в диапазоне от 50 до 150°С.
Реакцию соолигомеризации предпочтительно проводят при давлении этилена, меньшем чем 2,0 МПа (20 бар абсолютного давления), а более предпочтительно при давлении этилена в диапазоне от 0,1 МПа (1 бар (абсолютного давления)) до 1,6 МПа (16 бар (абсолютного давления)).
Альфа-олефиновый сомономер в общем случае присутствует с концентрацией, большей чем 1 моль·л-1 , предпочтительно с концентрацией, большей чем 2,5 моль·л-1, а более предпочтительно с концентрацией, большей чем 5 моль·л-1.
Условия по температуре и давлению, предпочтительно, выбирают так, чтобы получить набор продуктов с К-фактором, находящимся в диапазоне от 0,40 до 0,90, предпочтительно в диапазоне от 0,45 до 0,90. Как считается в настоящем изобретении, полимеризация произошла тогда, когда набор продуктов характеризуется К-фактором, большим чем 0,9.
Реакцию соолигомеризации можно проводить в газовой фазе либо в жидкой фазе, либо в смешанной газожидкостной фазе в зависимости от летучести олефиновых исходного сырья и продуктов.
Реакцию соолигомеризации можно проводить в присутствии инертного растворителя, который также может выступать и в роли носителя для катализатора и/или олефинов исходного сырья. Подходящие растворители включают алканы, алкены, циклоалканы и ароматические углеводороды.
Например, растворители, которые можно использовать в подходящем случае, включают гексан, изооктан, бензол, толуол и ксилол.
Было обнаружено, что подходящими являются времена реакции в диапазоне от 0,1 до 10 часов в зависимости от активности катализатора. Реакцию, предпочтительно, проводят в отсутствие воздуха или воды.
Реакцию соолигомеризации можно проводить обычным образом. Ее можно проводить в реакторе смешения, где в реактор смешения непрерывно добавляют олефины и катализаторы или предшественники катализаторов, а реагенты, продукты, катализаторы и неиспользованные реагенты из реактора смешения отводят, отделяя при этом продукты и отправляя катализаторы и неиспользованные реагенты обратно на рецикл в реактор смешения.
В альтернативном варианте реакцию можно проводить в реакторе периодического действия, где предшественники катализаторов и олефиновые реагенты загружают в автоклав, а после прохождения реакции в течение соответствующего промежутка времени продукты выделяют из реакционной смеси обычным способом, таким как перегонка.
По истечении подходящего времени реакции реакцию соолигомеризации можно прекратить в результате быстрого удаления этилена продувкой, чтобы деактивировать каталитическую систему.
Получаемая в результате композиция продуктов может содержать линейные альфа-олефины и/или алкилразветвленные альфа-олефины.
В предпочтительном варианте реализации композиция продуктов может содержать линейные альфа-олефины и/или метил разветвленные альфа-олефины и/или этилразветвленные альфа-олефины, то есть, например, где R16 представляет собой метил или этил.
Композиция продуктов настоящего изобретения в общем случае будет содержать более 5 масс.%, предпочтительно более 10 масс.%, более предпочтительно более 15 масс.%, а наиболее предпочтительно более 25 масс.% алкилразветвленных альфа-олефинов в расчете на полную массу линейных альфа-олефинов и алкилразветвленных альфа-олефинов в композиции продуктов.
Упомянутые линейные альфа-олефины и/или алкилразветвленные альфа-олефины могут иметь длину цепи в диапазоне от 4 до 100 атомов углерода, предпочтительно от 4 до 30 атомов углерода, а наиболее предпочтительно от 4 до 20 атомов углерода.
Олефиновые продукты можно удобным образом извлекать при помощи перегонки, а далее разделять, как это желательно, используя методики перегонки в зависимости от предполагаемого конечного использования олефинов.
Далее настоящее изобретение будет проиллюстрировано следующими примерами, которые никоим образом не должны рассматриваться в качестве ограничения объема настоящего изобретения, со ссылкой на сопутствующие чертежи, где:
фиг.1 представляет собой графическое представление для регрессионного анализа для примера 4;
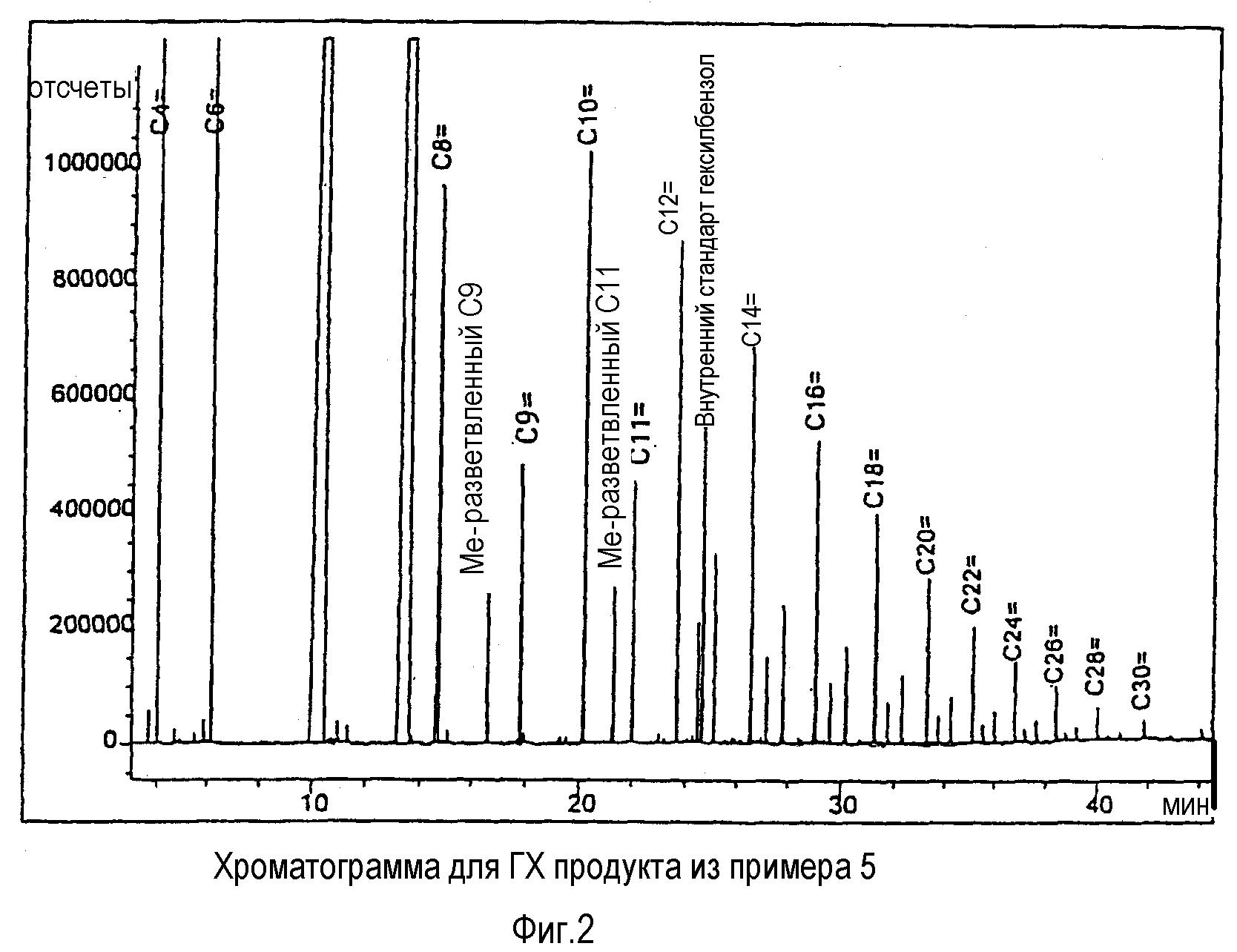
фиг.2 представляет собой ГХ-хроматограмму для продукта из примера 5; и
фиг.3 представляет собой часть хроматограммы для газовой хроматографии (ГХ) продукта из примера 9.
Общие методики и получение характеристик
Все операции с каталитическими системами проводили в атмосфере азота. Все использованные растворители высушивали при помощи стандартных методик.
Безводный толуол (степень чистоты 99,8%) (от компании Aldrich) высушивали над молекулярными ситами 4Е (конечное содержание воды приблизительно равно 3 млн-1).
Этилен (степень чистоты 99,5%) очищали, пропуская через колонку, содержащую молекулярные сита 4Е и катализатор BTS (от компании BASF), для того чтобы уменьшить содержание воды и кислорода до <1 млн-1.
1-октен (содержание 1-октена 99,8%; оставшуюся часть образуют 0,1% 1-гексена и 0,1% 1-децена) и 1-гексадецен (содержание 1-гексадецена 94, 1%; оставшуюся часть образуют 3,6% 1-тетрадецена и 2,3% 1-октадецена) представляли собой альфа-олефины SHOP, полученные от компании Shell Chemicals, и их очищали, проводя обработку основным оксидом алюминия и впоследствии высушивая над молекулярными ситами 4Е в атмосфере азота. 1-гептен (содержание 1-гептена 99,3%; оставшуюся часть образуют изомеры гептена) получали от компании Aldrich и использовали после высушивания над молекулярными ситами 4Е в атмосфере азота.
1-аминонафталин, 2,6-диацетилпиридин, 3,5-диметиланилин, 2,5-диметиланилин, 2,4,6-триметиланилин, 2-трет-бутиланилин, 4-трет-бутиланилин, 2,6-дифторанилин, 2-фторанилин и безводный хлорид железа(II) можно приобрести у компании Aldrich. 1- аминопиррол приобретали у компании TCI, Япония.
Ферроцениламин получали в соответствии со способом, описанным в литературе (D. van Leusen and B. Hessen, Organometallics, 2001, 20, 224-226).
Для того чтобы оценить распределение олигомеров при помощи газовой хроматографии (ГХ), устанавливали характеристики полученных олигомеров, используя аппарат HP 5890 series II и следующие условия проведения хроматографии:
Колонка: НР-1 (сшитый метилсилоксан), толщина пленки=0,25 мкм, внутренний диаметр=0,25 мм, длина 60 м (согласно данным компании Hewlett Packard); температура инжекции: 325°С; температура детектирования: 325°С; начальная температура: 40°С в течение 10 минут; скорость программирования температуры: 10,0°С/минута; конечная температура: 325°С в течение 41,5 минуты; внутренний стандарт: н-гексилбензол. Факторы отклика для четных линейных альфа-олефинов по отношению к н-гексилбензолу (внутреннему стандарту) определяли, используя стандартную калибровочную смесь. Факторы отклика для разветвленных альфа-олефинов с четным числом атомов углерода, линейных и разветвленных альфа-олефинов с нечетным числом атомов углерода предполагались равными по величине токовым для четных линейных альфа-олефинов с тем же самым или же подобным числом атомов углерода. Выходы С4-С30 олефинов получали из анализа методом ГХ, по результатам которого определяли К-факторы (по линейным соединениям), используя регрессионный анализ, в общем случае используя С10-С28 данные для линейных альфа-олефинов. Для соолигомеризации этена/1-октена содержание 1-октена рассчитывали по данным регрессионного анализа для линейных альфа-олефинов в диапазоне С10-С28 . Для соолигомеризации этена/1-гексадецена содержание 1-гексадецена рассчитывали по данным регрессионного анализа для линейных альфа-олефинов в диапазоне С18-С28.
Относительные количества линейного (лин.) 1-гексена среди всех изомеров гексена и относительное количество линейного (лин.) 1-додецена среди всех изомеров додецена, найденные из анализа методом ГХ, использовали в качестве меры селективности катализатора в отношении образования линейных альфа-олефинов.
Выходы разветвленных С10-С30 альфа-олефинов в случае соолигомеризации этена/1-октена или разветвленных С18-С30 альфа-олефинов в случае соолигомеризации этена/1-гексадецена получали из анализа методом ГХ, по результатам которого определяли К-факторы (по разветвленным соединениям), используя регрессионный анализ. В случае соолигомеризации этена и 1-гептена выходы нечетных линейных и разветвленных С9-С29 альфа-олефинов получали из анализа методом ГХ, по результатам которого определяли их К-фактор для линейных соединений и их К-фактор для разветвленных соединений, используя регрессионный анализ.
Массовое соотношение алкилразветвленного 1-ундецена (ундеценов) и алкилразветвленных и линейных 1-ундеценов, массовое соотношение алкилразветвленного 1-додецена (додеценов) и алкилразветвленных и линейных 1-додеценов и массовое соотношение алкилразветвленного 1-эйкозена (эйкозенов) и алкилразветвленных и линейных 1-эйкозенов, определенные из анализа методом ГХ, использовали в качестве меры селективности катализатора в отношении образования алкилразветвленных альфа-олефинов.
Данные спектроскопии ЯМР получали при комнатной температуре, используя аппарат Varian с частотой 300 или 400 МГц. Отнесения структур линейных альфа-олефинов и побочных продуктов получали, проводя сравнение спектров1Н- и13С-ЯМР образцов реакционной смеси, содержащих различные количества различных компонентов. Характеристические резонансы для олефиновых и линейных и разветвленных алифатических групп брали из литературы. Для получения дополнительного доказательства структуры там, где это считалось необходимым, использовали методики, дающие возможность идентифицировать связности углерод-углерод в структуре.
Компоненты катализатора
1. Получение комплекса хлорида 2,6-бис[1-(2-метилфенилимино)этил]пиридинжелеза[II] (X).
Комплекс Х получали в соответствии со способом, описанным в WO-A-99/02472.
2. Получение 2-[1-(2,4,6-триметилфенилимино)этил]-6-ацетилпиридина (1).

В 450 мл толуола растворяли 2,6-диацетилпиридин (7,3 г, 44,8 ммоль) и 2,4,6-триметиланилин (5,74 г, 42,55 ммоль). К данному раствору добавляли молекулярные сита 4Е и небольшое количество п-толуолсульфоновой кислоты (0,22 ммоль). Смесь кипятили в колбе с обратным холодильником в течение 16 часов. После фильтрования растворитель удаляли в вакууме. Несколько кристаллизаций из этанола дали 3,42 г (28,7%) моноимина (1).
1Н-ЯМР (CDCl3) δ 8,55 (д, 1H, Py-Нm), 8, 11 (д, 1H, Py-Нm), 7,92 (т, 1H, Py-Hp), 6,89 (с, 2H, ArH), 2,77 (с, 3Н, Me), 2,27 (с, 3H, Me), 2,22 (с, 3Н, Me), 1,99 (с, 6H, Me).
3. Получение 2-[1-(2,4, 6-триметилфенилимино)этил]-6-[1-(4-трет-бутилфенилимино)этил]пиридина (2).

В 100 мл толуола растворяли моноимин (1, 2,8 г, 10 ммоль) и 4-трет-бутиланилин (1,49 г, 10 ммоль). К данному раствору добавляли молекулярные сита 4Е и небольшое количество п-толуолсульфоновой кислоты (0, 1 ммоль). После стояния смеси в течение 5 дней при добавлении дополнительного количества молекулярных сит 4Е смесь кипятили в колбе с обратным холодильником в течение 2 часов. После фильтрования растворитель удаляли в вакууме. Остаток промывали метанолом и перекристаллизовывали из этанола. Выход смешанного диимина (2) 2,4 г (58%).
1Н-ЯМР (CDCl3) δ 8,42 (д, 1H, Py-Hm), 8,34 (д, 1H, Py-Hm), 7,86 (т, 1H, Py-Hp), 7,38 (д, 2H, ArH), 6,89 (с, 2H, ArH), 6,78 (д, 2H, ArH), 2,42 (с, 3H, Me), 2,29 (с, 3H, Me), 2, 22 (с, 3H, Me), 2,00 (с, 6H, Me), 1,34 (с, 9H, But).
4. Получение комплекса хлорид 2-[1-(2,4,6-триметилфенилимино)этил]-6-[1-(4-трет-бутилфенилимино)этил]пиридинжелеза[II] (3).

В инертной атмосфере к 420 мг FeCl2 (3,3 ммоль) в 150 мл дихлорметана добавляли раствор 1,5 г диимина (2, 3,6 ммоль) в 100 мл дихлорметана. Смесь перемешивали в течение одной недели. Образованный голубой осадок выделяли, используя фильтрование, и высушивали в вакууме. Выход комплекса железа (3) 1,5 г (84%).
1Н-ЯМР (Cl2CDCDCl2, широкие сигналы) δ 79,3 (1H, Py-Hm), 77,7 (1H, Py-Hm), 27,0 (1H, Py-Hp), 20,7 (3H, Me), 17,3 (6H, Me), 15,0 (2H, ArH), 14,3 (2H, ArH), 1,2 (9H, But), -2,6 (3H, MeC=N), -17,9 (2H, o-ArH), -32,1 (3H, MeC=N).
5. Получение 2, 6-бис[1-(2,6-дифторфенилимино)этил]пиридина (4).

В 50 мл толуола растворяли 2, 6-диацетилпиридина (1,76 г, 10,8 ммоль) и 2,6-дифторанилина (2,94 г, 22,8 ммоль). К данному раствору добавляли молекулярные сита 4Е. После стояния смеси в течение 3 дней при добавлении дополнительного количества молекулярных сит 4Е смесь отфильтровывали. Растворитель удаляли в вакууме. Остаток кристаллизовали из этанола. Выход 4: 1 г (24%).
1Н-ЯМР (CDCl3) δ 8,44 (д, 2H, Py-Hm), 7,90 (т, 1H, Py-Hp), 7,05 (м, 2H, ArH) 6,96 (м, 4H, ArH), 2,44 (с, 6H, Me).19F-ЯМР (CDCl3 ) δ -123,6.
6. Комплекс хлорид 2,6-бис[1-(2,6-дифторфенилимино)-этил]пиридинжелеза[II] (5).

В инертной атмосфере в 50 мл ТГФ растворяли 493 г диимина (4, 1,27 ммоль). Добавляли FeCl2 (162 мг, 1,28 ммоль) в 10 мл ТГФ. После перемешивания в течение 16 часов при комнатной температуре растворитель удаляли в вакууме. Добавляли толуол (100 мл). Голубой осадок выделяли, используя фильтрование, промывали его пентаном и высушивали в вакууме. Выделяли 0,5 г (76%) комплекса железа 5.
1Н-ЯМР (Cl2CDCDCl2, широкие сигналы) δ 75,5 (2H, Py-Нm), 39,6 1Н, Py-Hp), 15,7 (4Н, ArH), -11,6 (2Н, ArH), -22,4 (6Н, MeC=N).19F-ЯМР (Cl2CDCDCl2) δ -70,3.
7. Альтернативный вариант получения комплекса хлорид 2,6-бис[1-(2, 6-дифторфенилимино)этил]пиридинжелеза[II] (5').

В инертной атмосфере к раствору 260 мг диимина (4, 0,67 ммоль) в смеси растворителей, состоящей из 10 мл толуола и 6 мл пентана, медленно добавляли раствор 60 мг FeCl2 (0,47 ммоль) в 0,5 мл этанола. Полученный в результате голубой осадок выделяли, используя центрифугирование, три раза промывали его толуолом и высушивали в вакууме. Выход комплекса железа 5' 210 мг (87%).
1Н-ЯМР (CD2Cl2, широкие сигналы) δ 76,7 (2H, Py-Hm), 37,6 (1H, Py-Hp), 16,8 (4H, ArH), -10,2 (2H, ArH), -20,3 (6H, MeC=N).19F-ЯМР (CD2 Cl2) δ -75.
8. Получение 2-[1-(1-нафтилимино)этил]-6-ацетилпиридина (6).

В 100 мл толуола растворяли 2,6-диацетилпиридин (5,49 г, 33,6 ммоль) и 1-аминонафталин (4,8 г, 33,5 ммоль). К данному раствору добавляли молекулярные сита 4Е. После стояния смеси в течение 20 часов при комнатной температуре смесь отфильтровывали. Растворитель удаляли в вакууме. Получаемую в результате смесь 2, 6-диацетилпиридина, 2, 6-бис[1-(1-нафтилимино)этил]пиридина и 2-[1-(1-нафтилимино)этил]-6-ацетилпиридина растворяли в 50 мл ТГФ. В результате селективного комплексообразования с галогенидом металла удаляли дииминпиридиновый побочный продукт 2,6-бис[1-(1-нафтилимино) этил]пиридин. В инертной атмосфере добавляли FeCl2 (0,79 г, 6,23 ммоль). После перемешивания в течение 16 часов при комнатной температуре растворитель удаляли в вакууме. К полученной в результате смеси добавляли толуол (100 мл). Осажденный комплекс отфильтровывали через небольшой слой диоксида кремния с получением желтого раствора. Растворитель удаляли в вакууме. Кристаллизация из этанола дала 3,25 г 2-[1- 1-нафтилимино)этил]-6-ацетилпиридина (6) (33,6%).
1Н-ЯМР (CDCl3) δ 8,65 (д, 1H, Py-Hm), 8,15 (д, 1H, Py-Hm), 7,95 (т, 1H, Py-Hp), 7,87 (д, 1H, ArH), 7,76 (д, 1H, ArH), 7,64 (д, 1H, ArH), 7,4-7,6 (м, 3H, ArH), 6,82 (д, 1H, ArH), 2,79 (с, 3H, Me), 2, 38 (с, 3H, Me).
9. Получение 2-[1-(1-нафтилимино)этил]-6-[1-(4-трет-бутилфенилимино)этил]пиридина (7).

В 50 мл толуола растворяли моноимин (6, 1,25 г, 4,34 ммоль) и 4-трет-бутиланилин (0,65 г, 4,34 ммоль). К данному раствору добавляли молекулярные сита (4Е). После стояния смеси в течение 16 часов смесь отфильтровывали. Растворитель удаляли в вакууме. Остаток перекристаллизовывали из этанола. Выход смешанного диимина (7, степень чистоты 96% согласно анализу методом ЯМР) 0,44 г (24%).
1Н-ЯМР (CDCl3) δ 8,51 (д, 1H, Py-Hm), 8,38 (д, 1H, Py-Hm), 7,91 (т, 1H, Py-Hp), 7,86 (д, 1H, ArH), 7,78 (д, 1H, ArH), 7,63 (д, 1H, ArH), 7,4-7,6 (м, 5H, ArH), 6,8-6,9 (м, 3H, ArH), 2,43 (с, 3Н, Me), 2,37 (с, 3H, Me), 1,34 (с, 9H, But).
10. Получение комплекса хлорид 2-[1-(1-нафтилимино)этил]-6- [1-(4-трет-бутилфенилимино)этил]пиридинжелеза[II] (8).

В инертной атмосфере к 130 мг FeCl2 (1,03 ммоль) в 20 мл дихлорметана добавляли раствор 440 мг диимина (7, 1,05 ммоль) в 5 мл дихлорметана. Смесь перемешивали в течение 9 дней. Добавление 10 мл пентана приводило к получению голубого осадка, который выделяли, используя центрифугирование, и высушивали в вакууме. Выход комплекса железа (8) 480 мг (85%). В анализе методом1H-ЯМР (Cl2CDCDCl2) получили широкие сигналы, отнесение которых после этого не провели.
11. Получение 2-[1-(2-трет-бутилфенилимино)этил]-6- ацетилпиридина (9).

В 100 мл толуола растворяли 2,6-диацетилпиридин (4,37 г, 26,78 ммоль) и 2-трет-бутиланилин (4,0 г, 26,8 ммоль). К данному раствору добавляли молекулярные сита (4Е). После стояния смеси в течение 20 часов при комнатной температуре смесь отфильтровывали. Растворитель удаляли в вакууме.
Получаемую в результате смесь 2,6-диацетилпиридина, 2,6-бис[1-(2-трет-бутилфенилимино)этил]пиридина и 2-[1-(2-трет- бутилфенилимино)этил]-6-ацетилпиридина растворяли в 50 мл ТГФ. В результате селективного комплексообразования с галогенидом металла удаляли дииминпиридиновый побочный продукт 2, 6-бис[1-(2-трет-бутилфенилимино)этил]пиридин.
В инертной атмосфере добавляли FeCl2 (0,79 г, 6,23 ммоль). После перемешивания в течение 16 часов при комнатной температуре растворитель удаляли в вакууме.
К полученной в результате смеси добавляли толуол (100 мл). Осажденный комплекс отфильтровывали через небольшой слой диоксида кремния с получением желтого раствора. Растворитель удаляли в вакууме.
Кристаллизация из этанола дала 2,8 г 2-[1-(2-трет-бутилфенилимино)этил]-6-ацетилпиридина (9) (36%).
1Н-ЯМР (CDCl3) δ 8,48 (д, 1H, Py-Hm), 8,10 (д, 1H, Py-Hm),
7,93 (т, 1H, Py-Hp), 7,41 (д, 1H, ArH), 7,17 (т, 1H, ArH), 7,07 (т, 1H, ArH), 6,51 (д, 1H, ArH), 2,77 (с, 3H, Me), 2,38 (с, 3H, Me), 1,33 (с, 9H, But ).
12. Получение 2-[1-(2-трет-бутилфенилимино)этил]-6-[1-(4-трет-бутилфенилимино)этил]пиридина (10).
В 25 мл толуола растворяли моноимин (9, 1,06 г, 3,6 ммоль) и 4-трет-бутиланилин (0,56 г, 3,75 ммоль). К данному раствору добавляли молекулярные сита (4Е). После стояния смеси в течение 60 часов смесь отфильтровывали. Растворитель удаляли в вакууме. Остаток перекристаллизовывали из этанола. Выход смешанного диимина (10) 0,81 г (53%).
1Н-ЯМР (CDCl3) δ 8,36 (д, 1H, Py-Hm), 8,34 (д, 1H, Py-Hm), 7,88 (т, 1H, Py-Hp), 7,4 (м, 3H, ArH), 7,18 (т, 1H, ArH), 7,07 (т, 1H, ArH), 6,78 (д, 2H, ArH), 6,54 (д, 1H, ArH), 2,42 (с, 3H, Me), 2,38 (с, 3H, Me), 1,35 (с, 9H, But ), 1,34 (с, 9H, But).
13. Получение комплекса хлорид 2-[1-(2-трет-бутилфенилимино)этил]-6-[1-(4-трет-бутилфенилимино)этил]пиридинжелеза[II] (11).

В инертной атмосфере к 182 мг FeCl2 (1,44 ммоль) в 20 мл дихлорметана добавляли раствор 640 мг диимина (10, 1,5 ммоль) в 10 мл дихлорметана. Смесь перемешивали в течение 16 часов. Добавление 20 мл пентана приводило к получению голубого осадка. Выделение и высушивание в вакууме позволило получить 650 мг (82%) комплекса железа (11).
1Н-ЯМР (CD2Cl2), широкие сигналы) δ 81,9 (1H, Py-Hm), 77,5 (1H, Py-Hm), 30,4 (1Н, Py-Hp), 16,4 (1H, ArH), 13,8 (2Н, ArH), 6,3 (1H, ArH), 1,5 (9Н, But), 1,1 (9Н, But), -1,0 (3Н, MeC=N), -12,7 (1H, ArH), -21,3 (2Н, о-ArH), -33,1 (3Н, MeC=N), -33,7 (1H, о-ArH).
14. Получение 2-[1-(2-трет-бутилфенилимино)этил]-6-[1-(3,5-диметилфенилимино)этил]пиридина (12).

В 25 мл толуола растворяли моноимин (9, 1,13 г, 3,87 ммоль) и 3,5 диметиланилин (0,5 г, 4,13 ммоль). К данному раствору добавляли молекулярные сита (4Е). После стояния смеси в течение 60 часов смесь отфильтровывали. Растворитель удаляли в вакууме. Остаток перекристаллизовывали из этанола. Выход смешанного диимина (12) 0,79 г (52%).
1Н-ЯМР (CDCl3) δ 8,37 (д, 1H, Py-Hm), 8,32 (д, 1H, Py-Hm), 7,87 (т, 1H, Py-Hp), 7,42 (д, 1H, ArH), 7,18 (т, 1H, ArH), 7,07 (т, 1H, ArH), 6,76 (с, 1H, ArH), 6,54 (д, 1H, ArH), 6,46 (с, 2H, ArH), 2,40 (с, 3H, Me), 2,39 (с, 3H, Me), 2,33 (с, 3H, Me), 1,36 (с, 9H, But).
15. Получение комплекса хлорид 2-[1-(2-трет-бутилфенилимино)этил]-6-[1-(3, 5-диметилфенилимино)этил]пиридинжелеза[II] (13).

В инертной атмосфере к 187 мг FeCl2 (1,48 ммоль) в 20 мл дихлорметана добавляли раствор 617 мг диимина (12, 1,55 ммоль) в 10 мл дихлорметана. Смесь перемешивали в течение 16 часов. Добавление 20 мл пентана приводило к получению голубого осадка. Охлаждение до -30°С приводило к получению второй порции голубого осадка. Выделение и высушивание в вакууме позволило получить 660 мг (85%) комплекса железа (13).
1Н-ЯМР (CD2Cl2, широкие сигналы) δ 81,5 (1H, Py-Hm), 76,9 (1H, Py-Hm), 37,6 (1H, Py-Hp), 16,1 (1H, ArH), 1,2 (1H, ArH), 1,0 (9H, But), -2,7 (3H, MeC=N), -5,6 (6H, Me), -11,7 (1H, ArH), -13,5 (1H, ArH), -25,6 (2H, о-ArH), -35,7 (3H, MeC=N), -37,4 (1H, о-ArH).
16. Получение 2-[1-(2,4, 6-триметилфенилимино)этил]-6-[1-(2- фторфенилимино)этил]пиридина (14).

В 50 мл толуола растворяли моноимин (1, 1,0 г, 3,57 ммоль) и 2-фторанилин (398 мг, 3,57 ммоль). К данному раствору добавляли молекулярные сита 4Е. После стояния смеси в течение 20 часов при добавлении дополнительного количества молекулярных сит смесь отфильтровывали. Растворитель удаляли в вакууме, а маслянистый остаток нагревали в этаноле (50°С). Желтую твердую фазу, которую высаживали после охлаждения при -20°С, отфильтровывали и высушивали в вакууме. Выход смешанного диимина (14) 300 мг (23%).
1Н-ЯМР (CDCl3) δ 8,45 (д, 1H, Py-Hm), 8,38 (д, 1H, Py-Hm), 7,88 (т, 1H, Py-Hp), 7,1 (м, 4H, ArH), 6,93 (дд, 2H, ArH), 6, 89 (с, 2H, ArH), 2,41 (с, 3H, Me), 2,29 (с, 3H, Me), 2,22 (с, 3H, Me), 2,00 (с, 6H, Me).19F-ЯМР (CDCl3) δ -126,8.
17. Получение комплекса хлорид 2-[1-(2,4,6-триметилфенилимино)этил]-6-[1-(2-фторфенилимино)этил]пиридинжелеза[II] (15).

В инертной атмосфере к 87 мг FeCl2 (0,67 ммоль) в 20 мл дихлорметана добавляли раствор 270 мг диимина (14, 0,72 ммоль) в 5 мл дихлорметана. Смесь перемешивали в течение 20 часов. Добавление 10 мл пентана приводило к получению голубого осадка, который выделяли при помощи центрифугирования и высушивали в вакууме. Выход комплекса железа (15) 175 мг (51%).
1Н-ЯМР (CD2Cl2, широкие сигналы, избранные данные) δ 84,5 (1H, Py-Hm), 80,4 (1H, Py-Hm), 21,2 (1H, Py-Hp), 4,5 (3H, Mec=N), -24, 5 (1H, o-ArH), -38,1 (3Н, MeC=N).19F-ЯМР (CD2Cl2) δ -95,0.
18. Получение 2-[1-(2,4, 6-триметилфенилимино)этил]-6-[1-(1- пирролилимино)этил]пиридина (16).

В 50 мл толуола растворяли моноимин ((1), 3,0 г, 10,7 ммоль) и 1-аминопиррол (1,0 г, 12,18 ммоль). К данному раствору добавляли молекулярные сита (4Е). После стояния смеси в течение 40 часов смесь отфильтровывали. Растворитель удаляли в вакууме. Осадок перекристаллизовывали из этанола. Выход смешанного диимина (16) 1,85 г (50%).
1Н-ЯМР (CDCl3) δ 8,42 (д, 1H, Py-Hm), 8,29 (д, 1H, Py-Hm), 7,86 (т, 1H, Py-Hp), 6,93 (м, 2H, пиррол-H), 6,88 (с, 2H, ArH), 6,26 (м, 2H, пиррол-H), 2,67 (с, 3H, Me), 2,28 (с, 3H, Me), 2,20 (с, 3H, Me), 2,00 (с, 6H, Me).
19. Получение комплекса хлорид 2-[1-(2,4, 6-триметилфенилимино)этил]-6-[1-(1-пирролилимино)этил]пиридинжелеза[II] (17).

В инертной атмосфере к раствору 400 мг диимина ((16), 1,16 ммоль) в смеси растворителей, образованной 10 мл толуола и 6 мл пентана, медленно добавляли раствор 103 мг FeCl2 (0,81 ммоль) в 0, 7 мл этанола. Зелено-коричневый осадок выделяли при помощи центрифугирования, три раза промывали толуолом и высушивали в вакууме. Выход комплекса железа (17) 375 мг (98%).
1 Н-ЯМР (CD2Cl2, широкие сигналы, не отнесены) δ 88,1 (1H), 72,4 (1H), 29,9 (3Н), 19,5 (3Н), 16,9 (6Н), 13,5 (2Н), 8-8 (2Н), 5,8 (2Н), 2,9 (1H), -45,1 (3Н).
20. Получение 2-[1-(2,6-дифторфенилимино)этил]-6-ацетилпиридина (18).
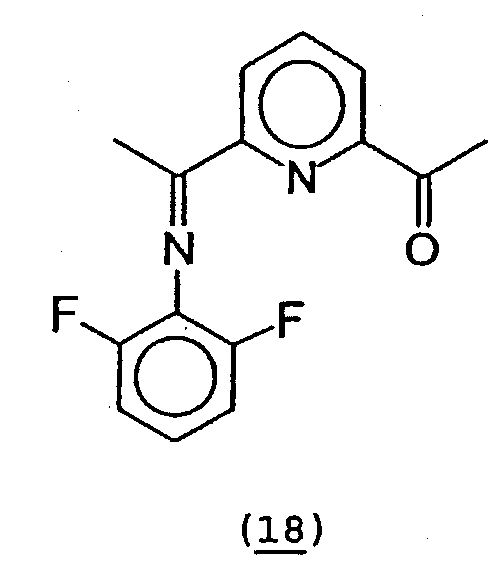
В 50 мл толуола растворяли 2,6-диацетилпиридин (4,04 г, 24,7 ммоль) и 2, 6-дифторанилин (3,2 г, 24,7 ммоль). К данному раствору добавляли молекулярные сита (4Е). После стояния смеси в течение 5 дней при комнатной температуре смесь отфильтровывали. Растворитель удаляли в вакууме. Из получаемой в результате смеси 2,6-диацетилпиридина, моноимина и диимина. Самую значительную часть 2,6-диацетилпиридина удаляли в результате проведения сублимации в вакууме при 80-90°С. Согласно данным, полученным анализом по методу1Н-ЯМР, остаток содержал 0,35 ммоль 2,6-диацетилпиридина, 1,28 ммоль диимина и 5,46 ммоль моноимина. Данную смесь вводили в реакцию с 162 мг (1,28 ммоль) FeCl2 в 10 мл ТГФ для удаления диимина. После перемешивания в течение 16 часов при комнатной температуре растворитель удаляли в вакууме. К полученной в результате смеси добавляли толуол (50 мл). Осажденный комплекс отфильтровывали через небольшой слой диоксида кремния с получением желтого раствора. Растворитель удаляли в вакууме. Кристаллизация из этанола дала 1,35 г 2-[1-(2,6-дифторфенилимино)этил]-6-ацетилпиридина (18) (19,8%).
1Н-ЯМР (CDCl3) δ 8,52 (д, 1H, Py-Hm), 8,12 (д, 1H, Py-Hm),
7,92 (т, 1H, Py-Hp), 7,03 (м, 1H, ArH), 6,97 (м, 2H, ArH), 2,77 (с, 3H, Me), 2,43 (с, 3H, Me).19F-ЯМР (CDCl3) δ -123,6.
21. Получение 2-[1-(2,6-дифторфенилимино)этил]-6-[1-(2,5-диметилфенилимино)этил]пиридина (19).

В 25 мл толуола растворяли моноимин (18, 0,86 г, 3,13 ммоль) и 2,5-диметиланилин (0,40 г, 3,3 ммоль). К данному раствору добавляли молекулярные сита (4Е). После стояния смеси в течение 3 дней смесь отфильтровывали. Растворитель удаляли в вакууме. Остаток кристаллизовали из этанола. Выделяли смесь 2-[1-(2, 6- дифторфенилимино)этил]-6-[1-(2,5-диметилфенилимино)этил]пиридина и 2,6-бис{2-[1-(2,5-диметилфенилимино)этил]}пиридина. В ТГФ из 2,6-бис{2-[1-(2,5-диметилфенилимино)этил]}пиридина и FeCl2 получали координационное соединение. Растворитель удаляли в вакууме. К получаемой в результате смеси добавляли толуол (10 мл). Осажденный комплекс отфильтровывали через небольшой слой диоксида кремния с получением желтого раствора. Растворитель удаляли в вакууме. Кристаллизация из этанола дала 40 мг (3%) 2-[1-(2,6-дифторфенилимино)этил]-6-[1-(2,5-диметилфенилимино)этил]пиридина (19).
1Н-ЯМР (CDCl3) δ 8,41 (д, 2H, Py-Hm), 7,89 (т, 1H, Py-Hp), 6,8-7,2 (м, 5H, ArH), 6,50 (с, 1H, ArH), 2,44 (с, 3H, Me), 2,32
(с, 6H, Me), 2,05 (с, 3H, Me).19F-ЯМР (CDCl3 ) δ -123,4.
22. Получение комплекса хлорид 2-[1-(2,6-дифторфенилимино)этил]-6-[1-(2, 5-диметилфенилимино)этил]пиридинжелеза[II] (20).

В инертной атмосфере к 11 мг FeCl2 (0,086 ммоль) в 10 мл дихлорметана добавляли раствор 35 мг диимина (19, 0,093 ммоль) в 5 мл дихлорметана. Смесь перемешивали в течение 16 часов. После добавления 5 мл пентана получающийся голубой осадок выделяли при помощи центрифугирования, промывали пентаном и высушивали в вакууме. Выход комплекса железа 20 40 мг (90%).
1Н-ЯМР (Cl2 CDCDCl2, широкие сигналы) δ 78,6 (1H, Py-Hm), 75,0 (1H, Py-Hm), 37,9 (1H, Py-Hp), 19,8 (1H, ArH), 16,6 (3H, Me) 15,8 (1H, ArH), 15,6 (1H, ArH), -8,2 (3H, Me) -9,7 (1H, ArH), -10,8 (3H, MeC=N), -15,7 (1H, ArH), -22,4 (1H, ArH), -29,8 (3H, MeC=N).19F-ЯМР (Cl2CDCDCl2) δ -62,7 и -67,4.
23. Получение 2-[1-(2,4, 6-триметилфенилимино)этил]-6-[1-(ферроценилимино)этил]пиридина (21).
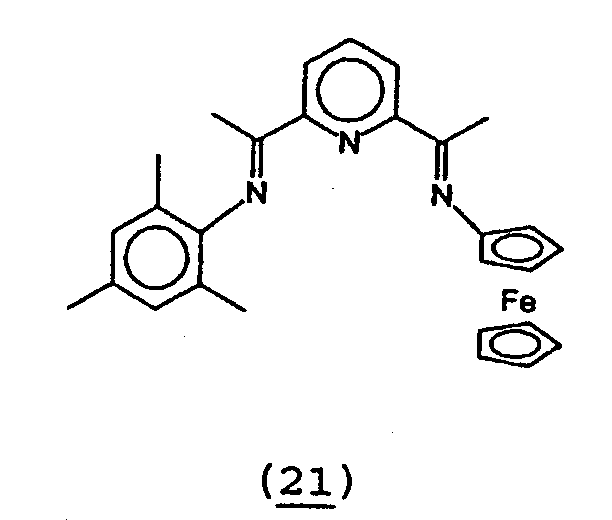
В 40 мл толуола растворяли моноимин 2-[1-(2,4,6-триметилфенилимино)этил]-6-ацетилпиридин (1,263 мг, 0,94 ммоль) и ферроцениламин (280 мг, 1,03 ммоль). К данному раствору добавляли молекулярные сита (4Е). После стояния смеси в течение 16 часов смесь отфильтровывали. Растворитель удаляли в вакууме. Остаток перекристаллизовывали из этанола. Выход смешанного диамина 21 180 мг (41%).
1Н-ЯМР (CD2Cl2) δ 8,36 (дд, 2H, Py-Hm), 7,85 (т, 1H, Py-Hp), 6,88 (с, 2H, ArH), 4,46 (т, 2H, CpH), 4,25 (т, 2H, CpH), 4,20 (с, 5H, CpH), 2, 55 (с, 3H, Me), 2,27 (с, 3H, Me), 2,20 (с, 3H, Me), 1,98 (с, 6H, Me).
24. Получение комплекса хлорид 2-[1-(2,4, 6-триметилфенилимино)этил]-6-[1-(ферроценилимино)этил]пиридинжелеза[II] (22).

В инертной атмосфере к 41 мг FeCl2 (0,32 ммоль) в 5 мл дихлорметана добавляли раствор 153 мг диимина (21, 0,33 ммоль) в 5 мл дихлорметана. Смесь перемешивали в течение 16 часов. Серо-голубой осадок выделяли при помощи центрифугирования, промывали гексаном и высушивали в вакууме. Выход комплекса железа 22 170 мг (89%).
1Н-ЯМР (CD2Cl2, широкие сигналы, избранные данные) δ 88,6 (1Н, Py-Hm), 76,7 (1H, Py-Hm), 21,3 (3H, Me), 16,3 (6H, Me), 2,8
(5H, CpH), -11,5 (3H, Mec=N).
25. Метилалюмоксан (МАО).
Использовавшийся раствор МАО в толуоле (Eurecen AL 5100/10T, партия: В7683; [Al]=4,88 масс.%, ТМА=35,7 масс.% (по расчету), молекулярная масса=900 г/моль) получали от компании Witco GmbH, Бергкамен, Германия.
Получение каталитической системы
Получение катализатора проводили в атмосфере азота в скафандре Braun MB 200-G.
Комплекс железа (обычно приблизительно 10 мг) помещали в стеклянную колбу, загерметизированную мембраной; вводили раствор МАО (4,0 г) упомянутой выше марки и проводили перемешивание в течение 2 минут. В общем случае это приводило к получению раствора с темной окраской, который иногда содержал некоторое количество осадка. После этого добавляли толуол (9,0 г) и раствор перемешивали еще в течение 10 мин. Сразу же после этого часть данного раствора использовали в реакции олигомеризации (см. в таблице 1 информацию об использованных количествах).
Эксперименты по олигомеризации
Эксперименты по олигомеризации проводили в стальном автоклаве объемом 1 литр, оснащенном рубашкой, охлаждающей при помощи нагревательной/охлаждающей ванны, (от компании Julabo, модель № ATS-2), турбинной мешалкой/пневматическим перемешиванием и перегородками. Для того чтобы удалить из реактора следы воды, его вакуумировали в течение ночи при <10 Па при 70°С. Проводили захватывание примесей в реакторе, вводя в него 250 мл толуола и МАО (раствор с содержанием 0,3-1,2 г) с последующим перемешиванием в течение 30 минут при 70°С в атмосфере азота при давлении 0,4-0,5 МПа. Содержимое реактора сливали через спускное отверстие в основании автоклава. Реактор вакуумировали до 0,4 кПа, осуществляли загрузку приблизительно 250 мл толуола, 1-гептена, 1-октена или 1-гексадецена (точные количества приводятся в таблице 1), проводили нагревание до 40°С и подавали под давлением этилен при величине давления, указанной в таблице 1 или в описании эксперимента. После этого в реактор при помощи толуола вводили раствор МАО (обычно 0,5 г) (полный инжектируемый объем составлял 30 мл, использованная методика была подобна методике инжектирования катализатора; см. ниже) и перемешивание при 800 оборотах в минуту продолжали в течение 30 минут. Каталитическую систему, полученную так, как описывается выше, и в таком количестве, как описывается в таблице 1, вводили в реактор смешения при помощи системы инжектирования с использованием толуола (полный инжектируемый объем был равен 30 мл: инжектировали раствор катализатора, разбавленный толуолом до 10 мл, а систему инжектора два раза промывали, используя 10 мл толуола). Добавление раствора катализатора приводило к выделению тепла (в общем случае 5-20°С), которое достигало своего максимума в течение 1 минуты, а после этого быстро устанавливались температура и давление, указанные в таблице 1. Температуру и давление отслеживали в течение всей реакции, а кроме этого, отслеживали и расходование этилена, сохраняя при этом давление этилена постоянным. После расходования определенного объема этилена олигомеризацию прекращали, быстро удаляя этилен продувкой, декантируя смесь продуктов в сборную колбу, пользуясь спускным отверстием в основании автоклава. Попадание смеси на воздух приводило к быстрому деактивированию катализатора.
После добавления к сырому продукту н-гексилбензола (0,5-3,5 г) в качестве внутреннего стандарта при помощи газовой хроматографии определяли количество С4-С30 олефинов, по которому, используя регрессионный анализ, определяли (наблюдаемый) К-фактор Шульца-Флори для линейного соединения, в общем случае пользуясь С10 -С28 данными для линейных альфа-олефинов. Под термином «наблюдаемый» в данном случае подразумевается то, что имеет место небольшое отклонение от распределения Шульца-Флори. Для соолигомеризации этена/1-октена содержание 1-октена рассчитывали из регрессионного анализа для линейных альфа- олефинов в С10-С28 диапазоне. Для соолигомеризации этена/1- гексадецена содержание 1-гексадецена рассчитывали из регрессионного анализа для линейных альфа-олефинов в С18-С28 диапазоне. Данные приведены в таблице 1.
Количество твердой фазы в продукте определяли следующим образом. Сырой продукт реакции центрифугировали при 4000 оборотах в минуту в течение 30 минут, после чего прозрачный верхний слой декантировали. Нижний слой, состоящий из твердых олефинов, толуола и незначительного количества жидких олефинов, смешивали с 500 мл ацетона, пользуясь мешалкой с большими сдвиговыми усилиями (Ultra-Turrax, тип ТР 18-10). Смесь центрифугировали при упомянутых выше условиях. Нижний слой смешивали с 200 мл ацетона и отфильтровывали через стеклянный фильтр (пористость Р3). Твердый продукт высушивали в течение 24 часов при 70°С при <1 кПа, взвешивали и содержание в нем фракций <С32 определяли при помощи газовой хроматографии раствора твердой фазы в 1, 2-дихлорбензоле или в 1,2,4-трихлорбензоле. Количества твердой фазы, приведенные в таблице 1, представляют собой выделенную твердую фазу с числом атомов углеродов >С30.
Относительное количество линейного (лин.) 1-гексена среди всех изомеров гексена и относительное количество линейного (лин.) 1-додецена среди всех изомеров додецена оценивали, используя анализ методом ГХ, и эти величины приведены в таблице 1.
Выходы разветвленных С10-С30 альфа-олефинов в случае соолигомеризации этена/1-октена или разветвленных С18-С30 альфа-олефинов в случае соолигомеризации этена/1-гексадецена и выходы нечетных линейных и разветвленных С9-С29 альфа-олефинов в случае соолигомеризации этена и 1-гептена получали, используя анализ методом ГХ. К-факторы для линейных соединений и/или К-факторы для разветвленных соединений соответственно определяли, используя регрессионный анализ. Эти данные приведены в таблице 1 и/или в подробном описании экспериментов.
Массовое соотношение алкилразветвленного 1-ундецена (ундеценов) и алкилразветвленных и линейных 1-ундеценов, массовое соотношение алкилразветвленных 1-додецена (додеценов) и алкилразветвленных и линейных 1-додеценов и массовое соотношение алкилразветвленного 1-эйкозена (эйкозенов) и алкилразветвленных и линейных 1-эйкозенов, определенные из анализа методом ГХ, приведены в таблице 1.
Пример 1
Комплекс железа 3, предварительно активированный по способу, описанному в «Получении каталитической системы», использовали в стальном автоклаве объемом 1 литр, загруженном 0,5 г МАО и толуолом (полный объем 310 мл), в эксперименте по олигомеризации этилена при давлении этилена 1,6 МПа. После расходования этилена в количестве 118,2 г реакцию прекращали и получали 110,6 г линейных С4-С30 альфа-олефинов и 2,5 г твердой фазы фракции >С30. Полное количество продукта олигомеризации этилена 113,1 г несколько меньше количества поданного этилена, что приписали потере части летучего 1-бутена и образованию небольших количеств побочных продуктов.
Линейные альфа-олефины характеризовались почти безупречным распределением Шульца-Флори (S-F) с К-фактором, равным 0,72, в соответствии с результатами, полученными из регрессионного анализа при использовании содержаний С10-С28 фракций, определенных при помощи анализа методом ГХ (регрессионная статистика: R2=1,00; среднеквадратическая погрешность=0,01 по 10 наблюдениям).
Число (частота) оборотов реакции (T.O.F.) составляло 4,65Е+07 моль этилена/моль Fe·час.
Степень чистоты (линейных) 1-гексена и 1-додецена была равна 99,5 и 97,7 масс.% соответственно. Количества разветвленного С12 альфа-олефина и разветвленного С20 альфа-олефина были на уровне <2 и <3 масс.% соответственно.
Подробности примера 1 приведены в таблице 1.
Пример 2
Пример 2 представляет собой повторение примера 1 за исключением того, что часть толуола заменили на 1-гептен. Расходование этилена, равное 118,3 г, приводило к получению 110,3 г линейных С4-С30 альфа-олефинов с четным числом атомов углерода, а кроме того, выделили 2,0 г твердой фазы фракции >С30. Помимо данных продуктов, анализы методом ГХ выявили распределения образования линейных и разветвленных альфа-олефинов с нечетным числом атомов углерода. Количество нечетных (С9-С29) линейных альфа-олефинов было равно 1,7 г, в то время как количество нечетных разветвленных альфа-олефинов было равным 1,1 г.
Для линейных С10-С28 альфа-олефинов имело место распределение Шульца-Флори в соответствии с результатами, полученными из регрессионного анализа, с К-фактором (четное линейное соединение), равным 0,69 (R2=1,00; среднеквадратическая погрешность <0,01 по 10 наблюдениям). Регрессионный анализ для линейных С9-С21 альфа-олефинов с нечетным числом атомов углерода и для разветвленных С9-С21 альфа-олефинов с нечетным числом атомов углерода дал в результате распределения Шульца-Флори, у которых К (нечетное линейное соединение) был равен 0,70 (R2=1,00; среднеквадратическая погрешность=0,02 по 7 наблюдениям), а К (нечетное разветвленное соединение) был равен 0,68 (R2=1,00; среднеквадратическая погрешность=0,02 по 7 наблюдениям) соответственно.
T.O.F. было равно 2,13Е+07 моль этилена/моль Fe·час.
Степень чистоты линейных (лин.) 1-гексена и 1-додецена была равна 99,0 и 96,1 масс.% соответственно.
Подробности примера 2 приведены в таблице 1.
Пример 3
Пример 3 представляет собой повторение примера 2, но 1-гептен заменили на 1-октен. После того как израсходовали 118,0 г этилена, реакцию прекращали, получая 125,4 г линейных С4-С30 альфа-олефинов и 9,7 г твердой фазы фракции >С30. Избыточное получение линейных альфа-олефинов приписали включению в конечные продукты исходного 1-октена, как это продемонстрировано в примере 2.
Для линейных альфа-олефинов имело место распределение Шульца-Флори с К-фактором, равным 0,73, в соответствии с результатами, полученными из регрессионного анализа при использовании содержаний С10-С28 фракций, определенных при помощи анализа методом ГХ (регрессионная статистика: R2=1,00; среднеквадратическая погрешность=0,02 по 10 наблюдениям).
T.O.F. было равно 3,43Е+07 моль этилена/моль Fe·час.
Степень чистоты линейного 1-гексена и линейного 1-додецена была равна 99,5 и 91,9 масс.% соответственно.
Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метилразветвленные (Ме-разветвленные) альфа-олефины, для которых К-фактор равен 0,71 (R2=0,98; среднеквадратическая погрешность=0,06 по 10 наблюдениям).
Подробности реакции приведены в таблице 1.
Пример 4
Пример 4 представляет собой повторение примера 3, но в данном случае с использованием 1-гексадецена вместо 1-октена. Количество линейных альфа-олефинов превышало количество израсходованного этилена: 116,3 против 111,5 г соответственно. Для линейных альфа-олефинов имело место распределение Шульца-Флори с К-фактором, равным 0,72, в соответствии с результатами, полученными из регрессионного анализа при использовании содержаний С18-С28 фракций, определенных при помощи анализа методом ГХ (регрессионная статистика: R2=1,00; среднеквадратическая погрешность=0,01 по 6 наблюдениям), как показано на фиг.1. Из результатов, представленных на данной фигуре, ясно, что имеет место 1,2-внедрение 1-гексадецена, как это подтверждает пример 2.
T.O.F. было равно 1,42Е+06 моль этилена/моль Fe·час.
Степень чистоты линейных 1-гексена и 1-додецена была равна 99,6 и 97,9 масс.% соответственно. Количество алкилразветвленного С20 альфа-олефина было равно 11 масс.%, тогда как в отсутствие мономера 1-гексадецена наблюдается <3 масс.%, см. пример 1.
Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метилразветвленные альфа-олефины, для которых К-фактор равен 0,70 (R2=0,99; среднеквадратическая погрешность=0,04 по 6 наблюдениям).
Подробности реакции приведены в таблице 1.
Пример 5
Пример 5 представляет собой повторение примера 2, но в данном случае при более высокой концентрации 1-гептена и при меньшем давлении этилена, равном 0,7 МПа, что иллюстрирует влияние изменения концентраций олефинов. Помимо линейных альфа-олефинов с четным числом атомов углерода, анализы методом ГХ показали распределение при образовании линейных и разветвленных альфа-олефинов с нечетным числом атомов углерода (см. фиг.2, где приведена хроматограмма для ГХ). Количество нечетных (С9-С29) линейных альфа-олефинов было равно 11,9 г, в то время как количество нечетных метилразветвленных альфа-олефинов было равным 6,6 г. Для линейных С10-С28 альфа-олефинов имело место распределение Шульца-Флори в соответствии с результатами, полученными из регрессионного анализа, с К-фактором для четного линейного соединения, равным 0,64 (R2=1, 00; среднеквадратическая погрешность <0,01 по 10 наблюдениям). Регрессионный анализ для линейных С9-С29 альфа-олефинов с нечетным числом атомов углерода и для метилразветвленных С9-С29 альфа-олефинов с нечетным числом атомов углерода дал в результате распределения Шульца-Флори, у которых К (нечетное линейное соединение) был равен 0,64, (R2=1,00; среднеквадратическая погрешность=0,01 по 11 наблюдениям), а К (нечетное разветвленное соединение) был равен 0,63 (R2=1,00; среднеквадратическая погрешность=0,03 по 11 наблюдениям) соответственно.
Дополнительные подробности приведены в таблице 1.
Пример 6
Пример 6 представляет собой повторение примера 3, но при другом давлении этилена, равном 0,7 МПа, что иллюстрирует влияние изменения концентрации олефинов. После того как израсходовали 68,8 г этилена, реакцию прекращали, получая 85,8 г линейных С4-С30 альфа-олефинов и 3,6 г твердой фазы фракции >С30. Избыточное получение линейных альфа-олефинов приписали включению в конечные продукты исходного 1-октена, как это продемонстрировано в примерах 2, 4 и 5.
Для линейных альфа-олефинов имело место распределение Шульца-Флори с К-фактором, равным 0,70, в соответствии с результатами, полученными из регрессионного анализа при использовании содержаний С10-С28 фракций, определенных при помощи анализа методом ГХ (регрессионная статистика: R2=1,00; среднеквадратическая погрешность=0,02 по 10 наблюдениям).
T.O.F. было равно 1,10Е+07 моль этилена/моль Fe·час.
Степень чистоты линейного 1-гексена и линейного 1-додецена была равна 99,2 и 84,7 масс.% соответственно. Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метилразветвленные альфа-олефины, для которых К-фактор равен 0,70 (R2=1,00; среднеквадратическая погрешность=0,04 по 10 наблюдениям).
Подробности в отношении реакций и продуктов приведены в таблице 1.
Пример 7
Пример 7 представляет собой повторение примера 6, но при другой концентрации 1-октена, что иллюстрирует влияние изменения концентрации олефинов. Результаты подобны результатам для примера 6. Подробности в отношении реакций и продуктов приведены в таблице 1.
Следующая серия экспериментов демонстрирует влияние каталитических систем с различными бисиминпиридиновыми лигандами.
Пример 8
Комплекс железа Х (полученный в соответствии с WO-A-99/02472) использовали в реакции, почти идентичной реакции примера 6. Выход линейных альфа-олефинов в С4-С30 диапазоне составил 96,9 г, что превышало расходование этилена, соответствующее 68,7 г, что указывало на включение в продукты 1-октена.
Степень чистоты линейного 1-гексена составляла 98,0 масс.%, а содержание алкилразветвленного 1-додецена соответствовало 14 масс.%. Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метил- и этилразветвленные (Ме- и Et-разветвленные) альфа-олефины с соотношением, приблизительно равным 1:1. Подробности реакции приведены в таблице 1.
Пример 9
Комплекс железа 5 использовали в эксперименте по соолигомеризации 1-октена при давлении этилена, равном 0,7 МПа, и при условиях, подобных условиям для примера 7. Выход линейных альфа-олефинов в С4-С30 диапазоне, равный 60,2 г, превысил расход этилена, соответствующий 53,5 г.
Степень чистоты линейного 1-гексена составляла 94,7 масс.%, а содержание алкилразветвленного 1-додецена соответствовало 28 масс.%. Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метил- и этилразветвленные альфа-олефины с соотношением, приблизительно равным 1:1 (см. фиг.3 с приведенной хроматограммой для ГХ, где А представляет собой винилиденовый олефин, В представляет собой олефины с внутренним расположением двойной связи, С и D представляют собой этилразветвленные олефины). Подробности реакции приведены в таблице 1.
Пример 10
Пример 10 представляет собой повторение примера 9, но в данном случае с использованием комплекса железа 5'. Результаты подобны результатам для примера 9. Подробности приведены в таблице 1.
Пример 11
Комплекс железа 8 использовали в эксперименте по соолигомеризации 1-октена, почти идентичном эксперименту примера 7. Выход линейных альфа-олефинов в С4-С30 диапазоне составил 73,6 г, что превышало расход этилена, соответствующий 68,6 г.
Степень линейности фракции 1-гексена составляла 96,8 масс.%, а содержание алкилразветвленного 1-додецена соответствовало 20 масс.%. Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метил- и этилразветвленные альфа-олефины с соотношением, приблизительно равным 1:1. Подробности реакции приведены в таблице 1.
Пример 12
Комплекс железа 11 использовали в эксперименте по соолигомеризации 1-октена, почти идентичном эксперименту примера 7. Полный выход продуктов был >75,6 г, что превышало расход этилена, соответствующий 68,8 г. Степень чистоты линейного 1-гексена составляла 99,2 масс.%, а содержание алкилразветвленного 1-додецена соответствовало 5 масс.%. Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метилразветвленные альфа-олефины. Подробности реакции приведены в таблице 1.
Пример 13
Комплекс железа 13 использовали в эксперименте по соолигомеризации 1-октена при условиях, подобных условиям для примера 7. Степень чистоты линейного 1-гексена составляла 98,8 масс.%, а содержание алкилразветвленного 1-додецена соответствовало 4 масс.%. Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метилразветвленные альфа-олефины. Подробности реакции приведены в таблице 1.
Пример 14
Комплекс железа 15 использовали в эксперименте по соолигомеризации 1-октена при условиях, почти идентичных условиям примера 6. Степень чистоты линейного 1-гексена составляла 99,1 масс.%, а содержание алкилразветвленного 1-додецена соответствовало 16 масс.%. Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метилразветвленные альфа-олефины. Подробности реакции приведены в таблице 1.
Пример 15
Комплекс железа 17 использовали в эксперименте по соолигомеризации 1-октена в условиях, почти идентичных условиям примера 7. Выход линейных альфа-олефинов в С4-С30 диапазоне составил 69,5 г, что превышало расход этилена, соответствующий 49,8 г. Степень чистоты линейного 1-гексена составляла 98,4 масс.%, а содержание алкилразветвленного 1-додецена соответствовало 17 масс.%. Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном метил- и этилразветвленные альфа-олефины с соотношением, приблизительно равным 1:1. Подробности реакции приведены в таблице 1.
Пример 16
Комплекс железа 20 использовали в эксперименте по соолигомеризации 1-октена при условиях, почти идентичных условиям примера 6. Степень чистоты линейного 1-гексена составляла 97,8 масс.%, а содержание алкилразветвленного 1-додецена соответствовало 21 масс.%. Данные ГХ и ЯМР показали, что побочные продукты представляют собой в основном Ме- и Et-разветвленные альфа-олефины с соотношением, приблизительно равным 1:1. Подробности реакции приведены в таблице 1.
Пример 17
Комплекс железа 22 использовали в эксперименте по олигомеризации 1-октена при условиях, почти идентичных условиям примера 6. К-фактор был очень мал, что говорит о том, что значительная часть этилена превратилась в 1-бутен, который принимает участие в соолигомеризации. Это отразила степень чистоты 1-гексена, равная 54,4 масс.%. Остальное в основном представляло собой разветвленные гексены. Содержание разветвленного 1-додецена соответствовало 33 масс.%. Анализ методом ГХ свидетельствовал о том, что побочные продукты представляют собой в основном Ме- и Et-разветвленные альфа-олефины с соотношением, приблизительно равным 1:1. Подробности реакции приведены в таблице 1.
Эксперименты проводили при 70°С в 1-октене/толуоле с использованием стального автоклава объемом 1 литр, если только не указано другого.
н. о.=не определено.
1 Вместо 1-октена использовали 1-гептен.
2 Вместо 1-октена использовали 1-гексадецен.
3 Относится к альфа-олефинам с четным числом атомов углерода.
4 Относится к альфа-олефинам с нечетным числом атомов углерода.
5 Массовое отношение алкилразветвленных 1-С20= к алкилразветвленным и линейным 1-С20=, в масс.%.
6 Массовое отношение алкилразветвленных 1-С11= к алкилразветвленным и линейным 1-С11=, в масс.%.
7 Катализатор получали в соответствии с WO-A-99/02472.
Реферат
Использование: нефтехимия. Сущность: проводят олигомеризацию одного или нескольких альфа-олефинов с этиленом в присутствии металлсодержащей каталитической системы, использующей один или несколько комплексов бисарилиминпиридин - МХа и/или один или несколько комплексов [бисарилиминпиридин - MYp·Lb+]q. Способ проводят при давлении этилена, меньшем 2,5 МПа. Технический результат - повышение выхода целевых продуктов. 3 н. и 7 з.п. ф-лы, 1 табл., 3 ил.

Формула



Документы, цитированные в отчёте о поиске
Каталитическая система для олигомеризации этилена в линейные альфа-олефины.
Комментарии