Селенорганические соединения и их применение - RU2371437C2
Код документа: RU2371437C2
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к новому селенорганическому соединению, способу его получения и активатору рецептора, активируемого пероксисомальным пролифератором, δ (PPARδ), который включает это соединение.
Уровень техники
Из известных в настоящее время 48 ядерных рецепторов сообщают о трех подтипах рецептора, активируемого пероксисомальным пролифератором (PPARs). Ими являются PPARα, PPARγ и PPARδ (Nature, 1990, 347, p. 645-650; Proc. Natl. Acad. Sci. USA, 1994, 91, p. 7335-7359). PPARα, PPARγ и PPARδ проявляют различные биологические функции и экспрессируются в различных местах. PPARα в основном экспрессируется в сердце, почках, скелетной мышце и толстой кишке (Mol. Pharmacol. 1998, 53, p. 14-22; Toxicol. Lett. 1999, 110, p. 119-127; J. Biol. Chem. 1998, 273, p. 16710-16714) и связан с β-окислением пероксисомы и митохондрии (Biol. Cell 1993, 77, p. 67-76; J. Biol. Chem. 1997, 272, p. 27307-27312). PPARγ слабо экспрессируется в скелетной мышце, но он сильно экспрессируется в жировой ткани. Известно, что он принимает участие в дифференциации жировых клеток, аккумуляции энергии в виде жира и регулировании гомеостаза инсулин-глюкоза (Moll. Cell 1999, 4, p. 585-594, p. 597-609, p. 611-617). В то время как PPARα и PPARγ гены отличаются тканеспецифичной экспрессией, PPARδ экспрессируется почти на всех тканях, хотя и в различном количестве (J. Bio. Chem. 1995, 270, p. 2367-2371; Endocrinology 1996, 131, p. 354-366). Согласно проведенным к настоящему времени исследованиям, известно, что PPARδ играет ключевую роль в экспрессии гамет (Genes Dev. 1999, 13, p. 1561-1574) и участвует в дифференциации нервных клеток в центральной нервной системе (CNS) (J. Chem. Neuroanat 2000, 19, p. 225-232) и заживлении ран в результате противовоспалительного действия (Genes Dev. 2001, 15, p. 3263-3277; Proc. Natl. Acad. Sci. USA 2003, 100, p. 6295-6296). Согласно последнему исследованию, было доказано, что PPARδ участвует в дифференциации жировых клеток и жировом обмене (Proc. Natl. Acad. Sci. USA 2002, 99, p. 303-308; Mol. Cell. Biol. 2000, 20, p. 5119-5128). Было доказано, что PPARδ активирует экспрессию разобщающих белков (UCPs), гены которых являются основными генами, вовлеченными в β-окисление жирных кислот и энергетический метаболизм (Nature 2000, 406, p. 415-418; Cell 2003, 113, p. 159-170; PLoS Biology 2004, 2, p. 1532-1539). Соответственно, регулирование UCP с помощью PPARδ может быть эффективным путем лечения ожирения.
Действие PPARδ было открыто не только в результате генетических исследований, но также в результате разработки специфических лигандов PPARδ. GW2433, первый синтезированный фактор активации PPARδ, был предложен в качестве средства для лечения атеросклероза фирмой Glaxo Smith Kline (WO 92/10468). И L-165041 фирмы Merck проявил действие по снижению уровня глюкозы и триглицеридов в крови (TG) (J. Biol. Chem. 1999, 274, p. 6718-6725). Модельное испытание на мышах (db/db) показало, что он может быть использован для лечения ожирения и диабета, так как при этом может увеличиваться в некоторой степени холестерин липопротеинов высокой плотности (FEBS Lett. 2000, 473, p. 333-336; WO 97/28115). YM-16638, который был разработан фирмой Yamanouchi Pharma of Japan, проявлял действие по снижению уровня холестерина и холестерина липопротеинов низкой плотности в крови (WO 99/04815). GW501516, ([2-метил-4-[[[4-метил-2-[4-(трифторметил)фенил]-1,3-тиазол-5-ил]метил]сульфанил]фенокси] уксусная кислота), селективный лиганд PPARδ, недавно разработанный фирмой Glaxo Smith Kline, показал значительно более сильное физиологическое действие, чем предшествующий лиганд. GW501516 проявил отличную эффективность при лечении ожирения у мышей (Cell 2003, 113, p. 159-170), и было доказано, что он является эффективным при лечении сердечно-сосудистых заболеваний у приматов за счет эффективного повышения содержания липопротеинов высокой плотности (HDLs) и снижения содержания липопротеинов низкой плотности (LDLs) (Proc. Natl. Acad. USA 2001, 98, p. 5306-5311, 2003, 100, p. 1268-1273). Соединение и способ его получения раскрыты в публикации международной заявки и литературных источниках (WO 01/00603; Bioorg. Med. Chem. Lett. 2003, 13, p. 1517- 1521; J. Org. Chem. 2003, 68, p. 9116-9118). Родственные GW501516 соединения, раскрытые в заявках WO 01/00603, WO 02/50048 и WO 02/062774, ограничены соединениями, имеющими эфирные, тиоэфирные или алкильные
((CR10R11)n, n=1 или 2) группы. Недавно было сообщено, что хотя активация PPARδ не вызывает рак толстой кишки, она может, по меньшей мере, способствовать пролиферации существующих раковых клеток толстой кишки (Nat. Med. 2004, 10, p. 245-247, p. 481-483). В этой публикации предлагается проводить больше исследований по установлению взаимосвязи между раком и PPARδ и разработке безопасного соединения.
Martin L. Smith с соавторами из Университета штата Индиана (США) сообщили, что селенометионин, аминокислота, содержащая неметаллический элемент селен (Se), вызывает активацию р53, гена супрессора опухоли, и таким образом снижает риск возникновения рака (Proc. Natl. Acad. USA 2002, 99, p. 14548-14553). Противоопухолевая активность селена была подтверждена с помощью экспериментов, которые показали, что потребление этого вещества является очень важным. Селен является важным неметаллическим рассеянным элементом, принадлежащим к группе VIA наряду с кислородом (O) и серой (S) (Eur. J. Clin. Nutr. 2004, 58, p. 391-402), и известно, что он защищает клетки от свободных радикалов, образующихся в результате нормального метаболизма кислорода, в качестве антиоксиданта (Regul. Toxicol. Pharm. 2003, 38, p. 232-242) и способствует нормальному функционированию иммунной системы и щитовидной железы (Pharmacol. Ther. 1998, 19, p. 179-192; Immunol. Today 1998, 19, p. 342-245).
Таким образом, если селен использовать для разработки лиганда, проявляющего активность к PPARδ, весьма вероятно, что могут быть разработаны методы лечения сердечно-сосудистых заболеваний, лекарства, снижающие уровень холестерина, методы лечения диабета и методы лечения ожирения без побочных эффектов, связанных с раком.
Раскрытие изобретения
Целью настоящего изобретения является разработка нового селенорганического соединения, нового лиганда, имеющего активность к PPARδ, и способа его получения.
Другой целью настоящего изобретения является разработка удобного способа получения селенорганического соединения, включающего стадии защиты фенольной группы фенольного соединения с помощью реактива Гриньяра без введения специальной защитной группы; осуществление реакции с металлоорганическим реагентом без специального процесса разделения и последующее осуществление реакции с селеном (Se).
Предпочтительный вариант осуществления изобретения
Настоящее изобретение относится к новому селенорганическому соединению, представленному ниже формулой I, и способу его получения:

R1 является независимо C1-C4 алкилом, C1-C4 алкилокси, C1-C4 алкилтиокси, C1-C4 алкиламином, фтором или хлором;
R2 является независимо C1-C4 алкилом, C1-C4 алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном;
R3 является водородом, C1-C4 алкилом или

R4 является водородом, ионом щелочного металла или C1-C7 алкилом или арилом;
R5 является независимо C1-C4 алкилом, C1-C4 алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном;
m является целым числом от 0 до 4;
n является целым числом от 0 до 5; и
p является целым числом от 0 до 5.
Селенорганическое соединение настоящего изобретения включает соединение, представленное ниже формулой II или IIa:

где
R1 и m являются такими, как определено в формуле I;
и
X2 является хлором, бромом или йодом.
Селенорганическое соединение, представленное формулой II или IIa, используют в качестве промежуточного соединения для получения различных селенорганических соединений.
Настоящее изобретение также включает селенорганическое соединение, представленное формулой III, которое является рацемическим соединением или оптическим изомером, полученным из соединения, представленного формулой II или IIa, и соединения, представленного формулой IV, которое может быть получено из соединения, представленного формулой:

где
R1-R3, R5, m, n и p являются такими, как определено в формуле I.

где
R1-R5, m, n и p являются такими, как определено в формуле I.
Селенорганическое соединение, представленное формулой IV, отличается тем, что оно имеет активность по отношению к рецептору, активируемому пероксисомальным пролифератором, δ (PPARδ).
Селенорганические соединения, представленные формулой IV, в которых R4 является метильной, этильной, н-пропильной, изопропильной, н-бутильной, втор-бутильной, трет-бутильной, фенильной или бензильной группой для защиты карбоксильной группы и замещена C1-C7 алкилом или арилом, являются особенно предпочтительными соединениями, имеющими активность по отношению к рецептору, активируемому пероксисомальным пролифератором, δ (PPARδ). Особенно предпочтительными являются метил, этил или трет-бутил. Ионом щелочного или щелочноземельного металла является Li+, Na+, K+, Ca2+ и так далее, предпочтительно водород или Na+.
Новые соединения настоящего изобретения могут быть получены по ниже приведенной схеме 1 и схеме 2.
Как видно на схеме 1, 4-галогенфенольное соединение, представленное формулой V, превращают в соединение, представленное формулой Va, после защиты спиртовой группы с помощью реактива Гриньяра. Галогеновую группу соединения, представленного формулой Va, замещают на литий, и соединение взаимодействует с селеном с получением промежуточного соединения, металл-селенового спирта, представленного формулой IIa. Затем данное соединение взаимодействует с соединением, представленным формулой VI (где X3 является хлором, бромом, йодом или другой уходящей группой, имеющей хорошую реакционную способность при нуклеофильном замещении), без выделения или очистки, с получением селенового эфирного соединения, представленного формулой IIIa. Это соединение взаимодействует с алкилгалогенацетатом с получением соединения, представленного формулой IVb, которое является гидролизованным эфиром, с получением соединения, представленного формулой IVc, с высокой чистотой и хорошим выходом.

В качестве варианта бензил может быть введен в α-положение по отношению к селену. В этом случае гидроксильная группа соединения, представленного формулой IIIa, является защищенной, и соединение взаимодействует с соединением, представленным формулой VII (где X4 является хлором, бромом, йодом или другой уходящей группой, имеющей хорошую реакционную способность при нуклеофильном замещении). Затем защитную группу удаляют с получением соединения, представленного формулой IIIb, которое реагирует с алкилгалогенацетатом, так же как в схеме 1, с получением соединения, представленного формулой IVd, которое является гидролизованным эфиром, с получением соединения, представленного формулой IVe.

Далее приводится подробное описание способа получения в соответствии с настоящим изобретением.
[Стадия A] Получение соединения, представленного формулой IIIa
Для получения соединения, представленного формулой IIIa, фенольную группу соединения, представленного формулой V, защищают с помощью реактива Гриньяра без специального процесса выделения, и соединение взаимодействует с металлоорганическим реагентом и селеном. Более подробно, эта стадия включает четыре подстадии, которые происходят одновременно.
Эти подстадии являются следующими.
(Подстадия A-1): По меньшей мере, один растворитель, выбранный из диэтилового эфира, тетрагидрофурана, гексана, гептана и так далее, используют в качестве безводного растворителя. Среди них диэтиловый эфир, тетрагидрофуран или смешанный растворитель из диэтилового эфира и тетрагидрофурана является предпочтительным.
В качестве реактива Гриньяра используют метил-, этил-, н-пропил-, изопропил-, н-бутил-, втор-бутилмагнийхлорид (R2MgCl) или алкилмагнийбромид (R2MgBr). Среди них изопропилмагнийхлорид ((CH3)2CHMgCl) является наиболее предпочтительным.
Реакционная температура может быть различной в зависимости от конкретно используемого растворителя. Обычно реакцию проводят в интервале температур от -20°C до 40°C, предпочтительно от 0°C до комнатной температуры (25°C). Время реакции может быть различным в зависимости от температуры реакции и конкретного используемого растворителя. Обычно реакцию проводят в течение 10-60 минут, предпочтительно в течение 10-30 минут.
(Подстадии A-2 и A-3): В качестве металлоорганического реагента для замещения галоген-металл может быть использован н-бутиллитий, втор-бутиллитий, трет-бутиллитий и так далее. Среди них трет-бутиллитий является предпочтительным.
В качестве селена используют одну из его форм тонкоизмельченного порошка. Селен непосредственно добавляют в растворитель.
Температура реакции может быть различной в зависимости от конкретного используемого растворителя. Обычно реакцию проводят при температуре от -78 до 25°C.
Предпочтительно, чтобы замещение галоген-металл осуществлялось при -75°C и введение селена начиналось при -75°C и осуществлялось при нагревании до комнатной температуры (25°C). Замещение галоген-металл проводят в течение 10-30 минут и введение селена проводят в течение 30-90 минут.
(Подстадия A-4): Добавляют кислоту к соединению, представленному формулой IIa, для идентификации присутствия -SeH. В качестве кислотного раствора является предпочтительным 1-3 N раствор хлористоводородной кислоты. Реакцию проводят при 0-25°C в течение 5-20 минут.
(Подстадия A-5): Синтезируют соединение, представленное формулой VI, 5-галогенметил-4-метил-2-[4-(трифторметил)фенил]- тиазол согласно известному способу (WO 03/106442). Галогеном (X) в соединении, представленном формулой VI, может быть хлор, бром или йод. Среди них хлор является предпочтительным.
Температура реакции может быть различной в зависимости от конкретного используемого растворителя. Обычно реакцию проводят от -78 до 25°C, предпочтительно от 0 до 10°C. Реакцию проводят в течение 10-120 минут, предпочтительно в течение 10-60 минут.
[Стадия B] Получение соединения, представленного формулой IVb
Для получения соединения, представленного формулой IVb, соединение, представленное формулой IIIa, взаимодействует с алкиловым эфиром галогенуксусной кислоты в присутствии основания.
Алкиловый эфир галогенуксусной кислоты является легко доступным известным соединением, и галогеном может быть хлор, бром, йод и так далее. Метилбромацетат и/или этилбромацетат являются наиболее предпочтительными алкиловыми эфирами галогенуксусной кислоты (алкилгалогенацетатами).
В качестве растворителя используют водорастворимый индивидуальный растворитель, выбранный из N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида, ацетонитрила, ацетона, этанола, метанола и так далее, или смесь растворителя с 1-10% воды. Среди них смешанный растворитель из ацетона или диметилсульфоксида с 1-5% воды является наиболее предпочтительным.
В качестве основания может быть использовано или слабое основание, или сильное основание, если оно не отрицательно влияет на реакцию. Например, может быть использован гидрид щелочного металла, такой как гидрид натрия или гидрид лития, гидрид щелочноземельного металла, такой как гидрид кальция, в качестве сильного основания может быть использован гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия, или карбонат щелочного металла, такой как карбонат лития, карбонат калия, гидрокарбонат калия и карбонат цезия. Предпочтительным является карбонат щелочного металла, и более предпочтительным - карбонат калия.
Реакция может быть проведена в любом интервале температур, в котором температура поддерживается ниже температуры кипения растворителя. Однако относительно высокая температура не является предпочтительной в результате возможности протекания побочной реакции. Обычно реакцию проводят при 0-60°C. Время реакции может быть различным в зависимости от температуры реакции. Обычно реакцию проводят в течение времени от 30 минут до одного дня, предпочтительно в течение 30-90 минут.
[Стадия C] Получение соединения, представленного формулой IVc
Соединение, представленное формулой IVc, получают из соединения, представленного формулой IVb, гидролизом эфира карбоновой кислоты в растворе водорастворимой неорганической соли и спирта.
В качестве растворителя используют спирт, смешивающийся с водой, например метанол и этанол.
Основание, гидроксид щелочного металла, такой как гидроксид лития, гидроксид натрия и гидроксид калия, используют в виде 0,1-3 N водного раствора, в зависимости от конкретного карбоксилата щелочного металла. Уксусную кислоту или 0,1-3 N раствор хлористоводородной кислоты используют для получения карбоксилатной формы соединения, представленного формулой IVc.
Предпочтительно, чтобы реакцию проводили при относительно низкой температуре для предотвращения любой побочной реакции. Обычно реакцию проводят в интервале температур от 0°C до комнатной температуры. Время реакции может быть различным в зависимости от температуры реакции. Обычно реакцию проводят в течение времени от 10 минут до 3 часов, предпочтительно от 30 минут до 1 часа.
Получающееся соединение, содержащее селен, представленное формулой IVc, является важным продуктом в качестве лиганда белков типа PPARδ.
[Стадия D] Получение соединения, представленного формулой IIIb
Соединение, представленное формулой IIIb, в котором α-положение по отношению к селену замещено бензильной группой, может быть получено из соединения, представленного формулой IIIa, полученного на стадии A, путем защиты его фенольной группы с помощью TMSCl или TBDMSCl, удаления активного водорода в α-положении по отношению к селену с помощью основания, такого как LDA, добавления производного бензилгалогенида и удаления защитной группы TMS или TBDMS.
Соединения, представленные формулами IVd и IVe, получают на стадиях E и F, так же как соединения, представленные формулами IVb и IVc соответственно.
Подробно описанные выше селенорганические соединения настоящего изобретения имеют лиганды с активностью по PPARδ и, таким образом, они являются с высокой вероятностью лекарственными средствами для лечения сердечно-сосудистых заболеваний, снижения содержания холестерина, лечения диабета и лечения ожирения. Способ получения в соответствии с настоящим изобретением используют для получения селенорганических соединений.
ПРИМЕРЫ
Далее настоящее изобретение подробно описывается со ссылкой на предпочтительные примеры. Однако следующие примеры служат только для разъяснения настоящего изобретения и настоящее изобретение не ограничивается ими.
Пример 1: Получение 4-гидроселено-2-метилфенола (II)
400 мг (1,7 ммоль) 4-йод-2-метилфенола растворяли в 30 мл безводного тетрагидрофурана в атмосфере азота и поддерживали температуру при 0°C. Медленно добавляли 935 мкл изопропилмагнийхлорида (2 M эфирный раствор, 1,1 эквивалента) и проводили реакцию в течение 10 минут. Реакционный раствор охлаждали до -78°C и медленно по каплям добавляли 2,2 мл трет-бутиллития (1,7 M гептановый раствор, 2,2 эквивалента), реакцию проводили в течение 20 минут. Медленно добавляли 134 мг селена (1,7 ммоль, 1,0 эквивалент) и реакцию проводили до тех пор, пока температура реакционной массы не достигала комнатной температуры. Добавляли 30 мл раствора хлорида аммония и 1 N хлористоводородную кислоту. Органический слой отделяли и удаляли из него влагу с помощью сульфата магния. После фильтрации растворитель удаляли отгонкой при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем в системе гексан/этилацетат (об./об.=2/1) с получением 302 мг требуемого соединения (выход: 95%).
1H ЯМР (300 MГц, CDCl3) δ 7,35 (д, 1H, J=1,3 Гц), 7,29 (дд, 1H, J=8,2, 2,2 Гц), 6,67 (д, 1H, J=8,2 Гц), 4,84 (шир. с, 1H), 2,21 (с, 3H).
13C ЯМР (75,5 MГц, CDCl3) δ 154,8, 137,3, 133,6, 125,2, 122,4, 115,9, 16,0.
Пример 2: Получение 4-[[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]метилселанил]-2-метилфенола (IIIa)
1,17 г (5,0 ммоль) 4-йод-2-метилфенола растворяли в 80 мл безводного тетрагидрофурана в атмосфере азота и поддерживали температуру при 0°C. Медленно добавляли 2,75 мл изопропилмагнийхлорида (2 M эфирный раствор, 1,1 эквивалента) и проводили реакцию в течение 10 минут. Реакционный раствор охлаждали до -78°C и медленно по каплям добавляли 6,47 мл трет-бутиллития (1,7 M гептановый раствор, 2,2 эквивалента), реакцию проводили в течение 20 минут. Медленно добавляли 395 мг селена (5,0 ммоль, 1,0 эквивалент) и реакцию проводили до тех пор, пока температура реакционной массы не достигала 15°C. Через 40 минут медленно добавляли при этой же температуре 1,46 г соединения, представленного формулой VI, 5-хлорметил-4-метил-2-[(4-трифторметил)фенил]тиазола, (5,0 ммоль, 1,0 эквивалент), растворенного в 5 мл безводного ТГФ. После примерно 30 минут проведения реакции добавляли 100 мл раствора хлорида аммония для прерывания реакции. Органический слой отделяли и удаляли из него влагу с помощью сульфата магния. После фильтрации растворитель удаляли отгонкой при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем в системе гексан/этилацетат (об./об.=3/1) с получением 2,05 г требуемого соединения (выход: 93%).
1H ЯМР (300 MГц, CDCl3) δ 7,95 (д, 2H, J=8,1 Гц), 7,64 (д, 2H, J=8,2 Гц), 7,28 (д, 1H, J=1,4 Гц), 7,09 (дд, 1H, J=8,2, 1,9 Гц), 6,59 (д, 1H, J=8,2 Гц), 4,09 (c, 2H), 2,19 (c, 3H), 2,05 (c, 3H).
13C ЯМР (75,5 MГц, CDCl3) δ 163,7, 155,4, 151,5, 139,3, 135,5, 135,3 (кв, J=33 Гц) 132,5, 126,8, 126,3 (м), 125,7, 118,5, 115,9, 23,3, 16,1, 14,9.
Пример 3: Получение этил-2-[4-[[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]метилселанил]-2-метилфенокси]ацетата (IVb)
1,0 г 4-[[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]метилселанил]-2-метилфенола (2,26 ммоль), полученного в примере 2, тщательно смешивали с 50 мл ацетона, содержащего 5% воды, и 719 мг карбоната калия (5,2 ммоль, 2,3 эквивалента) при комнатной температуре. Медленно добавляли 376 мкл этилового эфира бромуксусной кислоты (3,4 ммоль, 1,5 эквивалента) и смесь сильно перемешивали в течение 4 часов. После окончания реакции проводили экстракцию с использованием рассола и этилацетата и влагу удаляли с помощью сульфата магния. После фильтрации растворитель удаляли отгонкой при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем в системе гексан/этилацетат (об./об.=5:1) с получением 1,19 г требуемого соединения (выход: 99%).
1H ЯМР (300 MГц, CDCl3) δ 7,96 (д, 2H, J=8,1 Гц), 7,65 (д, 2H, J=8,3 Гц), 7,29 (д, 1H, J=1,4 Гц), 7,23 (дд, 1H, J=8,4, 2,1 Гц), 6,56 (д, 1H, J=8,4 Гц), 4,62 (c, 2H), 4,25 (кв, 2H, J=14,3, 7,1 Гц), 4,12 (c, 2H), 2,23 (c, 3H), 2,14 (c, 3H), 1,28 (т, 3H, J=7,1 Гц).
13C ЯМР (75,5 MГц, CDCl3) δ 169,1, 162,9, 157,1, 151,6, 138,8, 137,3, 134,8, 131,9, 131,6 (кв, J=33 Гц), 128,9, 126,2 (м), 120,0, 112,1, 65,9, 61,8, 23,3, 16,4, 15,2, 14,6.
Пример 4: Получение 2-[4-[[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]метилселанил]-2-метилфенокси]уксусной кислоты (IVb, HK101225)
500 мг этил-2-[4-[[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]метилселанил]-2-метилфенокси]ацетата (1,0 ммоль), полученного в примере 3, тщательно смешивали с 50 мл этанола и добавляли 3,5 мл 3 N раствора гидроксида натрия. Перемешивание осуществляли в течение 30 минут при комнатной температуре. Когда реакция заканчивалась, доводили pH до значения 2,0 с помощью 2 N HCl. Удаляли около 80% этанола отгонкой при пониженном давлении. После экстракции с помощью рассола и этилацетата и фильтрации растворитель удаляли отгонкой при пониженном давлении, и остаток очищали хроматографией на колонке LH-20 с получением 495 мг требуемого соединения (выход: 99%).
1H ЯМР (300 MГц, CDCl3) δ 9,92 (шир.с, 1H), 7,93 (д, 2H, J=8,2 Гц), 7,65 (д, 2H, J=8,4 Гц), 7,29 (д, 1H, J=1,4 Гц), 7,19 (дд, 1H, J=8,4, 2,1 Гц), 6,58 (д, 1H, J=8,4 Гц), 4,66 (c, 2H), 4,10 (c, 2H), 2,21 (c, 3H), 2,07 (c, 3H).
13C ЯМР (105,9 MГц, CDCl3) δ 173,2, 163,5, 156,8, 151,5, 138,9, 136,8, 134,9, 132,3, 131,8 (кв, J=33 Гц), 128,9, 126,9, 126,3 (м), 120,1, 112,0, 65,4, 23,1, 16,4, 14,8.
Пример 5: Получение 4-[1-[4-метил-2-(4-трифторметилфенил)- тиазол-5-ил]-2-фенилэтилселанил]-2-метилфенола (IIIb)
A: Получение 5-[4-(трет-бутилдиметилсиланилокси)-3-метил-фенилселанилметил]-4-метил-2-[(4-трифторметил)фенил]тиазола
200 мг 4-[[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]- метилселанил]-2-метилфенола (0,45 ммоль), полученного в примере 2, и 77 мг имидазола (1,13 ммоль, 2,5 эквивалентов) полностью растворяли в безводном диметилформамиде (5 мл). Медленно добавляли 102 мг трет-бутилметилсилилхлорида (0,67 ммоль, 1,5 эквивалента) и смесь перемешивали в течение 4 часов при комнатной температуре. После окончания реакции органический слой экстрагировали водой (20 мл) и этиловым эфиром (15 мл) и промывали водой (20 мл). Органический слой сушили с помощью сульфата магния и получали с помощью хроматографии на силикагеле 238 мг (выход: 95%) 5-[4-(трет-бутилдиметилсиланил- окси)-3-метилфенилселанилметил]-4-метил-2-[(4-трифторметил)- фенил]тиазола с защищенной фенольной группой.
B: Получение 5-[1-[4-трет-бутилдиметилсилилокси)-3-метил-фенилселанил]-2-фенилэтил]-4-метил-2-[4-(трифторметил)фенил]-тиазола
150 мг 5-[4-(трет-бутилдиметилсиланилокси)-3-метилфенил-селанилметил]-4-метил-2-[(4-трифторметил)фенил]тиазола (0,27 ммоль), полученного в A, растворяли в безводном тетрагидрофуране (5 мл) в атмосфере азота. Реакционный раствор охлаждали до -78°C и медленно добавляли 240 мкл диизопропиламида лития (2 M раствор диизопропиламида лития в смеси гептан/тетрагидрофуран/этилбензол, 2,0 эквивалента). Реакцию проводили в течение 30 минут при такой же температуре при добавлении 32 мкл бензилбромида (0,27 ммоль, 1,0 эквивалент), сохраняя цвет реакционного раствора темно-синим. Добавляли к реакционному раствору насыщенный раствор хлорида аммония (10 мл). После экстракции этилацетатом (10 мл) органический слой промывали рассолом. Органический слой сушили с помощью сульфата магния и концентрированный остаток очищали хроматографией на силикагеле с получением 141 мг требуемого соединения (выход: 81%).
1H ЯМР (600 МГц, CDCl3) δ 7,99 (д, 2H, J=8,3 Гц), 7,12-7,26 (м, 7H), 6,64 (д, 2H, J=8,2 Гц), 4,73 (дд, 1H, J=9,8, 5,6 Гц), 3,44 (дд, 1H, J=14,0, 5,6 Гц), 3,25 (дд, 1H, J=14,0, 9,8 Гц), 2,12 (c, 3H), 1,91 (c, 3H), 1,03 (c, 9H), 0,21 (c, 6H).
C: Получение 4-[1-[4-метил-2-(4-трифторметилфенил)тиазол-5-ил]-2-фенилэтилселанил]-2-метилфенола
100 мг 5-[1-[4-трет-бутилдиметилсилилокси)-3-метилфенил-селанил]-2-фенилэтил]-4-метил-2-[4-(трифторметил)фенил]тиазола (0,15 ммоль), полученного в B, полностью растворяли в тетрагидрофуране (10 мл) при комнатной температуре. Медленно добавляли при такой же температуре 180 мкл тетрабутиламмонийфторида (TBAF) (0,18 ммоль, 1 M раствор в тетрагидрофуране, 1,2 эквивалента). После 1 часа реакции органический слой экстрагировали насыщенным раствором хлорида аммония (10 мл) и этилацетатом (10 мл) и сушили с помощью сульфата магния. После фильтрации растворитель удаляли отгонкой при пониженном давлении и сконцентрированный остаток очищали хроматографией на силикагеле с получением 79 мг требуемого соединения (выход: 99%).
1H ЯМР (300 МГц, CDCl3) δ 7,94 (д, 2H, J=8,2 Гц), 7,63 (д, 2H, J=8,3), 7,00-7,26 (м, 7H), 6,50 (д, 1H, J=8,1 Гц), 6,32 (шир., 1H), 4,67 (дд, 1H, J=9,8, 5,7 Гц), 3,42 (дд, 1H, J=13,9, 5,7 Гц), 3,22 (дд, 1H, J=13,9, 9,8 Гц), 2,13 (с, 3H), 1,78 (с, 3H).
Пример 6: Получение этил-[4-[1-[4-метил-2-(4-трифторметил-фенил)тиазол-5-ил]-2-фенилэтилселанил]-2-метил-фенокси]ацетата (IVd)
100 мг 4-[1-[4-метил-2-(4-трифторметилфенил)тиазол-5-ил]-2-фенилэтилселанил]-2-метилфенола (0,19 ммоль), полученного в примере 5, полностью растворяли в ацетоне (10 мл), содержащем 5% воды, и добавляли при комнатной температуре 66 мг карбоната калия (0,475 ммоль, 2,5 эквивалента). Добавляли при этой же температуре 28 мкл этилового эфира бромуксусной кислоты (0,25 ммоль, 3 эквивалента) и смесь перемешивали в течение 4 часов. После окончания реакции органический слой экстрагировали рассолом (10 мл) и этилацетатом (10 мл). Затем органический слой сушили с помощью сульфата магния. Растворитель удаляли отгонкой при пониженном давлении и остаток очищали хроматографией на силикагеле с получением 103 мг (выход: 88%) требуемого соединения.
1H ЯМР (300 МГц, CDCl3) δ 7,97 (д, 2H, J=8,2 Гц), 7,65 (д, 2H, J=8,4 Гц), 7,07-7,25 (м, 7H), 6,53 (д, 2H, J=8,1 Гц), 4,69 (дд, 1H, J=9,8, 6,0 Гц), 4,59 (с, 2H), 4,23 (кв, 1H, J=14,2, 7,1 Гц), 3,40 (дд, 1H, J= 13,8, 6,0 Гц), 3,20 (дд, 1H, J=13,8, 9,8 Гц), 2,26 (с, 3H), 1,85 (с, 3H), 1,28 (т, 3H, J=7,0 Гц).
Пример 7: Получение [4-[1-[4-метил-2-(4-трифторметилфенил)-тиазол-5-ил]-2-фенилэтилселанил]-2-метилфенокси]уксусной кислоты (IVe)
100 мг [4-[1-[4-метил-2-(4-трифторметилфенил)тиазол-5-ил]-2-фенилэтилселанил]-2-метилфенокси]ацетата (0,16 ммоль), полученного в примере 6, полностью растворяли в этаноле (10 мл) и добавляли 200 мкл 1 N раствора гидроксида натрия. После перемешивания в течение 30 минут при комнатной температуре, когда реакция заканчивалась, доводили pH до значения 3,0 2 N раствором HCl. Реакционный растворитель удаляли при пониженном давлении и органический слой экстрагировали рассолом и этилацетатом. Остаток очищали хроматографией на колонке со смолой LH-20 с получением 71 мг требуемого соединения (выход: 75%).
1H ЯМР (300 МГц, CDCl3) δ 7,97 (д, 2H, J=8,2 Гц), 7,65 (д, 2H, J=8,4 Гц), 7,07-7,25 (м, 7H), 6,53 (д, 2H, J=8,1 Гц), 4,69 (дд, 1H, J=9,8, 6,0 Гц), 4,59 (с, 2H), 3,40 (дд, 1H, J=13,8, 6,0 Гц), 3,20 (дд, 1H, J=13,8, 9,8 Гц), 2,26 (с, 3H), 1,85 (с, 3H).
Примеры 7-22
Представленные ниже в таблице 1 и таблице 2 селенорганические соединения были получены так же, как в примерах 1-6.










Тестовый пример 1: Тест на активность и токсичность
Активность HK101225 по отношению к PPARδ определяли с помощью трансфекционного анализа. Кроме того, определяли селективность к подтипам PPARα и PPARγ. Тест на токсичность проводили с помощью MTT анализа (анализ на основе 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий бромида) и активность in vivo подтверждали тестом на животном.
Трансфекционный анализ
Трансфекционный анализ проводили с использованием CV-1 клеток. Клетки культивировали в среде DMEM, содержащей 10% FBS, DBS (делипидизированный) и 1% пенициллина/стрептомицина при 37°C в культуральной системе для трансфекционного анализа, содержащей 5% диоксида углерода, на 96-луночном планшете. Тест проводили в четыре стадии - посев клеток, трансфекция, обработка соединением настоящего изобретения и подтверждение. CV-1 клетки высевали на 96-луночном планшете, 5000 клеток/лунка. Трансфекцию проводили через 24 часа. Использовались полноразмерная плазмидная ДНК PPARs, репортерная ДНК, которая имеет активность по люциферазе и таким образом позволяет подтверждать активность по отношению к PPARs, и ДНК β-галактозидазы, которая предлагает эффективную информацию по трансфекции. HK101225, полученный в примере 4, растворяли в диметилсульфоксиде (DMSO) и обрабатывали им клетки при различных концентрациях. После 24 часов культивирования в инкубаторе клетки лизировали с помощью лизирующего буфера и измеряли активность по люциферазе и активность по β-галактозидазе с использованием люминометра и микропланшетного ридера. Измеренную активность по люциферазе сопоставляли с активностью по β-галактозидазе. Результат изображали на графике и определяли EC50.
Как видно в таблице 3, селенорганическое соединение настоящего изобретения имело высокую селективность для EC50 PPARδ.
Соединения, представленные в таблице 1 и 2, и их этиловые эфиры проявляли, по меньшей мере, в 10000 раз меньшую селективность по отношению к PPARα и PPARγ, и EC50для PPARδ находилась в интервале от 500 pM до 10 nM.
MTT анализ
Токсичность определяли с помощью MTT анализа для HK101225, полученного в примере 4. MTT является водорастворимым желтым веществом. Однако при введении в живую клетку оно превращается под действием митохондриальной дегидрогеназы в водонерастворимые пурпурные кристаллы. Клеточная токсичность может быть оценена путем растворения этого вещества в диметилсульфоксиде и измерения поглощения света при 550 нм. Тест проводили следующим образом.
CV-1 клетки высевали на 96-луночном планшете, 5000 клеток/лунка. После культивирования в течение 24 часов в увлажненной системе культивирования при 37°C, содержащей 5% диоксида углерода, клетки обрабатывали HK101225 при различных концентрациях. MTT реагент добавляли после 24 часов культивирования. После культивирования в течение около 15 минут пурпурный кристалл растворяли в диметилсульфоксиде и измеряли поглощение с помощью микропланшетного ридера для подтверждения клеточной токсичности.
HK101225 не показал токсичности даже при концентрации в 50000 раз больше величины EC50 (1,87 нM). Другие соединения, представленные в таблице 1 и 2, и их этиловые эфиры не показали токсичности при концентрации в 10000 раз больше значения EC50.
Тест на модели животного
Предотвращение ожирения
Тест на модели животного проводили на мышах, для того чтобы подтвердить in vivo действие HK101225. Использовали мышей C57BL/6 (SLC Co.) 14-недельного возраста и давали им корм, содержащий 35% жира, для вызывания ожирения. Наполнитель, GW501516 (10 мг/кг/день) и HK101225 (10 мг/кг/день) вводили перорально в течение 78 дней при рационе с высоким содержанием жиров. Мыши, которым вводили наполнитель, показали 102% увеличения массы тела, а масса тела мышей, которым вводили GW501516 и HK101225, увеличилась только на 21%, только 1/5 от увеличения массы тела группы, в которой вводили наполнитель. Таким образом, было подтверждено, что HK101225 обладает сильным действием, предотвращающим ожирение.
Улучшение состояния при диабете
GTT (пробу на толерантность к глюкозе) проводили для того, чтобы подтвердить действие соединения настоящего изобретения по улучшению состояния при диабете. Наполнитель, GW501516 и HK101225 перорально вводили мышам в течение 78 дней. Затем абдоминально вводили глюкозу (1,5 г/кг) и контролировали изменение во времени уровня сахара в крови. В группе, которой вводили HK101225, наблюдался более низкий уровень сахара в крови при голодании, чем в группе, в которой вводили наполнитель и GW501516. В группе, в которой вводили HK101225, отмечалось быстрое понижение уровня сахара в крови между 20 и 40 минутами и полное исчезновение глюкозы после 100 минут. Для сравнения, в группе, в которой вводили наполнитель, уровень сахара в крови не приходил в норму даже после 120 минут. В группе, в которой вводили GW501516, уровень сахара в крови был ниже, чем в группе с введением наполнителя, но нормальный уровень не восстанавливался. Таким образом, было подтверждено, что HK101225 является более эффективным для улучшения состояния при диабете, чем GW501516.
Селенсодержащее соединение, представленное формулой IV, или фармацевтически доступную соль соединения используют в качестве композиции активатора рецептора, активируемого пероксисомальным пролифератором, δ (PPARδ). Кроме того, селенсодержащее соединение, представленное формулой IV, или фармацевтически доступную соль соединения используют в качестве фармацевтической композиции для лечения сердечно-сосудистых заболеваний, снижения уровня холестерина, лечения диабета или лечения ожирения, добавки лечебного питания, лечебного напитка, пищевого ингредиента и функциональной косметики для лечения.
Фармацевтически доступной солью соединения, представленного формулой IV, может быть карбоксилат или любая другая фармацевтически доступная соль, причем ионом щелочного и щелочноземельного металла являются Li+, Na+, K+ и Ca2+.
Терапевтическая доза соединения, представленного формулой IV, или фармацевтически доступной его соли может изменяться в зависимости от конкретного соединения, способа введения, конкретного пациента и конкретного заболевания, подвергаемого лечению. Соединение может быть введено перорально или местно в зависимости от типа препарата. В случае перорального введения соединение может быть приготовлено в любой форме, включая таблетку, порошок, сухой сироп, жевательную таблетку, гранулу, жевательную резинку, капсулу, мягкую капсулу, пилюлю, напиток и сублингвальную таблетку. Таблетка, включающая соединение настоящего изобретения, может быть введена пациенту любым методом или способом, который является биологически приемлемым, то есть перорально. В качестве варианта она может быть введена другим методом или способом, в зависимости от особенностей предотвращаемого или подвергаемого лечению заболевания, развития заболевания или других обстоятельств. В случае, когда композицией, включающей соединение настоящего изобретения является таблетка, она может включать, по меньшей мере, один фармацевтически приемлемый наполнитель. Пропорция и свойство наполнителя могут быть определены с учетом растворимости или других химических свойств таблетки, пути введения и других фармацевтических стандартов. Настоящее изобретение предлагает новое селенорганическое соединение, которое используют в качестве лиганда с активностью по отношению к PPARδ, и способ его получения.
Настоящее изобретение предлагает удобный способ получения селенорганического соединения путем защиты фенольной группы фенольного соединения с помощью реактива Гриньяра без введения специальной защитной группы; взаимодействия его с металлоорганическим реагентом без выделения; и последующего взаимодействия его с селеном (Se).
На основе подробного описания настоящего изобретения со ссылками на предпочтительные варианты осуществления для специалистов в этой области очевидно, что различные модификации и замены могут быть сделаны в нем без отступления от сути и объема настоящего изобретения, которые устанавливаются прилагаемой формулой изобретения.
Реферат
Настоящее изобретение относится к новым селеноорганическим соединениям формулы IV. Соединения изобретения обладают активностью по отношению к рецептору, активируемому пероксисомальным пролифератором δ (PPARδ) и могут быть использованы для лечения сердечно-сосудистых заболеваний, снижения уровня холестерина, лечения диабета или ожирения. В формуле IV: ! , ! ! А является ; ! В является ; ! R1 является независимо С1-С4 алкилом; R2 является независимо С1-C4 алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном; R3 является водородом или ; ! R4 является водородом, ионом щелочного металла или ! С1-С7 алкилом; R5 является независимо ! С1-С4 алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном; m является целым числом, равным 1; n является целым числом от 1 до 2; и p является целым числом от 0 до 3. Изобретение также относится к способам получения соединения формулы IV, способу получения промежуточного соединения, фармацевтическим композициям, включающим селеноорганическое соединение в качестве активного компонента, а также к промежуточным соединениям II, IIa и III. 8 н. и 2 з.п. ф-лы, 3 табл.
Формула

где А является

и В является

R1 является независимо С1-С4алкилом;
R2 является независимо C1-C4алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном;
R3 является водородом или

R4 является водородом, ионом щелочного металла или С1-С7алкилом;
R5 является независимо С1-С4алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном;
m является целым числом, равным 1;
n является целым числом от 1 до 2; и
p является целым числом от 0 до 3.
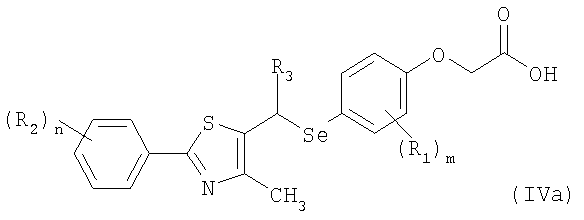
где R1 является независимо С1-С4алкилом;
R2 является независимо С1-С4алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном;
R3 является водородом или
R5 является независимо С1-С4алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном;
m является целым числом, равным 1;
n является целым числом от 1 до 2; и
p является целым числом от 0 до 3.
R1 является независимо метилом или этилом;
R2 является независимо CF3, F или Cl;
R3 является водородом или
R5 является независимо CF3, F или Cl;
m является целым числом, равным 1;
n является целым числом от 1 до 2; и
p является целым числом от 0 до 3.
a) взаимодействия соединения, представленного ниже формулой V, с реактивом Гриньяра, и взаимодействия его с металлоорганическим реагентом с получением металлоорганического соединения, представленного ниже формулой Vb;
b) последующего взаимодействия металлоорганического соединения, представленного формулой Vb, с селеном (Se) с получением металл-селенового соединения, представленного ниже формулой IIa; и
c) последующего взаимодействия селенового соединения, представленного формулой IIa, с тиазольным соединением, представленным ниже формулой VI, с получением соединения, представленного ниже формулой IIIa





где X1 является бромом или йодом, Х2 является хлором, бромом или йодом, Х3 является хлором, бромом, йодом или другой уходящей группой с хорошей реакционной способностью к нуклеофильному замещению, и другие заместители являются такими, как определено в формуле IV в п.1.
a) взаимодействия соединения, представленного ниже формулой V, с реактивом Гриньяра, и взаимодействия его с металлоорганическим реагентом с получением металлоорганического соединения, представленного ниже формулой Vb;
b) последующего взаимодействия металлоорганического соединения, представленного формулой Vb, с селеном (Se) с получением металл-селенового соединения, представленного ниже формулой IIa;
c) последующего взаимодействия селенового соединения, представленного формулой IIa, с тиазольным соединением, представленным ниже формулой VI, с получением соединения, представленного ниже формулой IIIa;
e) взаимодействия соединения, представленного ниже формулой IIIa, с алкилгалогенацетатом с получением эфирного соединения, представленного ниже формулой IVb; и
f) гидролиза эфирного соединения, представленного формулой IVb, с получением соединения, представленного ниже формулой IVc


где X1 является бромом или йодом, Х2 является хлором, бромом или йодом, Х3 является хлором, бромом, йодом или другой уходящей группой с хорошей реакционной способностью к нуклеофильному замещению, R1, R2, m, n и p являются такими, как определено в формуле IV в п.1, и R4 является C1-C7алкилом.
a) взаимодействия соединения, представленного ниже формулой V, с реактивом Гриньяра, и взаимодействия его с металлоорганическим реагентом с получением металлоорганического соединения, представленного ниже формулой Vb;
b) последующего взаимодействия металлоорганического соединения, представленного формулой Vb, с селеном (Se) с получением металл-селенового соединения, представленного ниже формулой IIa;
c) последующего взаимодействия селенового соединения, представленного формулой IIa, с тиазольным соединением, представленным ниже формулой VI, с получением соединения, представленного ниже формулой IIIa;
g) защиты гидроксильной группы соединения, представленного ниже формулой IIIa, взаимодействия указанного соединения с соединением, представленным ниже формулой VII, в основной среде и удаления группы, защищающей гидроксил, с получением соединения, представленного ниже формулой IIIb;
h) взаимодействия соединения, представленного формулой IIIb, с алкилгалогенацетатом с получением эфирного соединения, представленного ниже формулой IVd; и
i) гидролиза эфирного соединения, представленного формулой IVd, с получением соединения, представленного ниже формулой IVe




где X1 является бромом или йодом, Х2 является хлором, бромом или йодом, Х3 является хлором, бромом, йодом или другой уходящей группой с хорошей реакционной способностью к нуклеофильному замещению, R1, R2, m, n и p являются такими, как определено в формуле IV в п.1, R4 является С1-С7алкилом, и Х4 является хлором, бромом, йодом или другой уходящей группой с хорошей реакционной способностью к нуклеофильному замещению.


где R1 независимо является C1-C4алкилом, m равно 1, и Х2 является хлором, бромом или йодом.

где R1 является независимо C1-C4алкилом; R2 является независимо С1-С4алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном; R3 является водородом или
R5 является независимо С1-С4алкилом, замещенным, по меньшей мере, одним галогеном, или галогеном; m является целым числом, равным 1; n является целым числом от 1 до 2; и p является целым числом от 0 до 3.
Документы, цитированные в отчёте о поиске
Производные пропионовой кислоты и фармацевтическая композиция на их основе
Комментарии