Хиральные γ-кетосульфанильные производные пинановой структуры и способ их получения - RU2783164C1
Код документа: RU2783164C1
Описание
Изобретение относится к получению хиральных γ-кетосульфанильных производных пинановой структуры, в частности 2-кетотиоацетата (RSАс), 3-кетотиобензоата (RSBz), 2- и 3-кетотиолов (RSН) и -дисульфидов (RSSR).
В литературе описан синтез монофунциональных карановых, п-ментановых, пинановых и борнановых тиолов, дисульфидов и тиоацетильных производных [Banach A., Ścianowski Ja., Ozimek P. Phosphorus, Sulfur, Silicon Rel. Elements. 2014, 189, 274-284. DOI: 10.1080/10426507.2013.819867]. Известны полифункциональные серосодержащие соединения карановой, п-ментановой, пинановой и борнановой структур, где наряду с сульфанильной группой присутствует гидрокси-группа [О.А.Банина, Д.В.Судариков, Ю.В.Крымская, Л.Л.Фролова, А.В.Кучин, ХПС, 2015, 2, 231] или двойная связь [Вакуленко И.А., Старцева В.А., Никитина Л.Е., Артемова Н.П., Фролова Л.Л., Кучин А.В. ХПС. 2005, 6, 585; Gadras, J. Dungues, R. Calas, G. Deleris. J. Org. Chem. 1984, 49, 442]. Из серосодержащих терпеноидов с кето-группой известна сульфокамфорная кислота и некоторые сульфиды с кето-группой в пинановом углеродном скелете [D. M. Vialemaringe, Marie-Josephe Bourgeois, M. Campagnole, E. Montaudon, Helvetica Chimica Acta, 2000, 83(3), 616].
В работе [О.А.Банина, Д.В.Судариков, Ю.В.Крымская, Л.Л.Фролова, А.В.Кучин, ХПС, 2015, 2, 231] (наиболее близкий аналог) получена неразделимая смесь диастереомеров 3-кетотиоацетатов пинановой структуры. Способ включает получение пинокарвона из β-пинена, далее ацилирование тиоуксусной кислотой и дезацилирование алюмогидридом лития (LiAlH4). Однако известный способ не позволяет получить диастереомерно чистые 3-кетотиоацетаты пинановой структуры, так как в описанных условиях (-5°C, без растворителя, 3 ч) образуется смесь тиоацетатов (R)- и (S)-конфигурации по атому С(2) (de 33%). При деацилировании полученных тиоацетатов LiAlH4 до тиолов кето-группа восстанавливается до гидрокси-группы, следовательно, данный способ не позволяет получить 3-кетотиол и -дисульфид.
Прямых аналогов получения 2-кетосульфанильных производных пинановой структуры не выявлено. Для их получения необходимо из β-пинена получить 2-норпинанон с последующим ацилированием тиоуксусной кислотой. Известен способ [M. Gianini, A. von Zelewsky. Synthesis. 1996, June, 702. Doi: 10.1055/s-1996-4280], в котором из β-пинена получают нопинон, затем - кетоенол, и далее - 2-норпинанон. Однако стадия получения кетоенола протекает в присутствии NaNH2 при кипячении в течение 15 ч, а выход продукта не превышает 71%. Стадии ацилирования и дезацилирования по способу [О.А.Банина, Д.В.Судариков, Ю.В.Крымская, Л.Л.Фролова, А.В.Кучин, ХПС, 2015, 2, 231] (наиболее близкий аналог стадий) требует охлаждения до -5°С, времени реакции до 3 ч, а при дезацилировании полученного кетотиоацетата LiAlH4 до тиола кето-группа восстанавливается до гидрокси-группы.
Задачей настоящего изобретения является синтез новых хиральных γ-кетосульфанильных производных пинановой структуры и разработка эффективных способов их получения.
Технический результат заключается в расширении арсенала новых хиральных γ-кетосульфанильных производных пинановой структуры, полученных в индивидуальном виде, являющихся перспективными субстратами для получения сульфинил- и сульфонилсодержащих соединений - потенциальных биологически активных веществ.
Технический результат способа получения хиральных 2-кетосульфанильных производных пинановой структуры заключается в оптимизации стадии получения кетоенола с повышением его выхода на 25%, сокращением времени реакции в 3 раза и упрощением процесса за счет исключения кипячения; в упрощении стадии ацилирования 2-норпинанона за счет исключения охлаждения и в сокращении времени реакции в 18 раз; а также в хемоселективности стадии деацилирования 2-кетотиоацетата и сохранении кето-группы у целевых продуктов.
Технический результат способа получения хиральных 3-кетосульфанильных производных пинановой структуры заключается в увеличении стереоселективности образования 3-кетотиоацетата (2S)-конфигурации в 3 раза (de 93%), а также в хемоселективности деацилирования 3-кетотиоацетата и сохранении кето-группы у целевых продуктов.
Технический результат достигается получением хиральных γ-кетосульфанильных производных пинановой структуры (RSR1) формулы (I):

где звездочкой обозначена связь, к которой присоединяется атом серы (-S-), исключая:

Технический результат способа получения хиральных 2-кетосульфанильных производных пинановой структуры достигается тем, что из β-пинена получают нопинон, далее - кетоенол, затем - 2-норпинанон, из которого реакцией присоединения тиоуксусной кислоты получают 2-кетотиоацетат, из которого деацилированием - соответствующие тиол и дисульфид; при этом, по изобретению, стадию получения кетоенола модифицируют, а именно к раствору (-)-нопинона в тетрагидрофуране, охлажденному до 0°С, прибавляют t-BuOK, затем прикапывают раствор изоамилформиата в тетрагидрофуране, перемешивают 6 часов при комнатной температуре, стадию ацилирования 2-норпинанона проводят в тетрагидрофуране при комнатной температуре в течение 5-15 мин, стадию деацилирования 2-кетотиоацетата проводят с использованием гидразингидрата (NH2NH2·H2O).
Технический результат способа получения хиральных 3-кетосульфанильных производных пинановой структуры достигается тем, что окислением (-)-β-пинена получают транс-пинокарвеол, затем - пинокарвон, далее пинокарвон ацилируют тиокислотами для получения тиокарбоксилатов, последние деацилируют до тиолов и дисульфидов, при этом, по изобретению, стадию ацилирования пинокарвона проводят при температуре - 50 ÷ - 65°C в тетрагидрофуране или дихлорметане, стадию деацилирования тиокарбоксилатов проводят с использованием гидразингидрата (NH2NH2·H2O).
Получение соединений осуществляется следующим образом.
Для получения хиральных γ-кетосульфанильных производных пинановой структуры использовали метод, включающий синтез α-,β-ненасыщенных карбонильных соединений на основе (-)-β-пинена, далее синтез тиокарбоксилатов реакцией присоединения тиокислот по двойной связи α-,β-ненасыщенных карбонильных соединений и последующее деацилирование (схема 1, 2). Способ позволяет получить целевые продукты с сохранением структуры терпенового фрагмента и кето-группы в пинановом скелете с хорошими выходами.
Синтез 2-кетосульфанильных производных на основе β-пинена осуществляли по схеме 1. Из (-)-β-пинена 1 многостадийным синтезом получили 2-норпинанон 2 по методике [M. Gianini, A. von Zelewsky. Synthesis. 1996, June, 702. Doi: 10.1055/s-1996-4280], в которой стадия получения кетоенола 3 была модифицирована, а именно к раствору (-)-нопинона 4 в тетрагидрофуране, охлажденному до 0°С, прибавляли t-BuOK; после растворения t-BuOK в смесь прикапывали раствор изоамилформиата в тетрагидрофуране, перемешивали 6 часов при комнатной температуре. В дальнейшем соединение 3 использовали без дополнительной очистки.

Схема 1. Синтез 2-кетосульфанильных производных на основе β-пинена
Присоединение тиоуксусной кислоты к 2-норпинанону 2 проводили по модифицированной методике [F. Martínez-Ramos, M. E. Vargas-Díaz, L. Chacón-García, J. Tamariz, P. Joseph-Nathan, L. G. Zepeda, Tetrahedron: Asymmetry, 2001, 12, 3095] в присутствии каталитических количеств пиридина в тетрагидрофуране при комнатной температуре в течение 5-15 мин. Синтез кетотиоацетата 5 проходит диастереоселективно с образованием единственного (3R)-изомера (схема 1). Деацилирование тиоацетата 5 гидразингидратом (NH2NH2·H2O) в течение 4 ч приводит к 2-кетотиолу (3R)-6 (далее 6) и дисульфиду (3R)-7 (далее 7) в соотношении 3:1 соответственно.
Синтез 3-кетосульфанильных производных на основе β-пинена осуществляли по схеме 2. По методике [M. A. Umbreit, K. B. Sharpless, J.Am.Chem.Soc. 1977, 99, 5526] (-)-β-пинен 1 окисляли t-BuOOH в присутствии каталитических количеств SeO2 при комнатной температуре до транс-пинокарвеола 8, который затем окисляли активным MnO2в СH2Cl2до пинокарвона 9.

Схема 2. Синтез 3-кетосульфанильных производных на основе β-пинена
Для получения диастереомерно чистого тиоацетата (2S)-10 оптимизировали методику [F. Martínez-Ramos, M. E. Vargas-Díaz, L. Chacón-García, J. Tamariz, P. Joseph-Nathan, L. G. Zepeda, Tetrahedron: Asymmetry, 2001, 12, 3095]. Нами варьировались такие условия, как растворитель (дихлорметан, ТГФ), реагент (тиоуксусная, тиобензойная кислоты) и температура (-5 ÷ -65°C). Выявлено, что снижение температуры реакции приводит к повышению стереоселективности в отношении соединений (2S)-10, (2S)-11 от de 33% до 93% (схема 2).
Использование тиоуксусной кислоты для ацилирования пинокарвона 9 является предпочтительным, так как деацилирование тиоацетата (2S)-10 до тиола (2S)-12 гидразин гидратом происходит в течение 4-5 ч с выходом до 90%, в то время как тиобензоата (2S)-11 - с выходом 38-50% вследствие неполной конверсии. Увеличение времени деацилирования (2S)-11 до 24 ч приводит к образованию соответствующего дисульфида (2S)-13 (80%) (схема 2).
Таким образом, оптимальными условиями для стадии ацилирования являются использование тиоуксусной кислоты, тетрагидрофуран в качестве растворителя, присутствие каталитических количеств пиридина и температура синтеза - 60 ÷ -65°C.
Структура соединений 5-7 и 10-13 подтверждена методами ЯМР и ИК спектроскопии и данными элементного анализа. ИК спектры соединений регистрировали на ИК-Фурье-спектрометре Shimadzu IR Prestige 21 в тонком слое. Спектры ЯМР1H и13C регистрировали на спектрометре Bruker Avance-300 (300.17 МГц для1Н и 75.48 МГц для13С) в растворах CDCl3и ДМСО-d6. Полное отнесение сигналов1Н и13С выполняли с помощью двумерных гомо- (1H-1H COSY,1H-1H NOESY) и гетероядерных экспериментов (1H-13C HSQC, HMBC). Соотношение (S)- и (R)-производных определяли методом спектроскопии ЯМР1Н по величинам интегральных интенсивностей сигналов соответствующих протонов H10b. Тонкослойную хроматографию выполняли на пластинах Sorbfil, проявитель - раствор ванилина в EtOH. Элементный анализ выполняли с использованием автоматического анализатора марки ЕА 1110 CHNS-O. Все реакции проводились с использованием свежеперегнанных растворителей. Колоночную хроматографию выполняли на силикагеле Alfa Aesar (0.06-0.2 мм). Температуру плавления определяли на приборе Gallenkamp MPD350BM3.5 фирмы Sanyo и не корректировали. Угол оптического вращения измеряли на автоматизированном цифровом поляриметре Optical Activity PolAAr 3001 (Англия).
(-)-β-Пинен (1) - коммерческий продукт производства Sigma Aldrich, чистота 99%,

Получение соединений показано на следующих примерах.
(1R,5R,E)-3-(Гидроксиметилен)-6,6-диметилбицикло[3.1.1]гептан-2-он (3). К раствору (-)-нопинона (11.4 ммоль, 1.575 г) в 20 мл ТГФ, охлажденному до 0°С, прибавляли t-BuOK (15.38 ммоль, 1.726 г). После растворения t-BuOK в смесь прикапывали раствор изоамилформиата (15.38 ммоль, 1.726 г) в 10 мл ТГФ. Полученную смесь перемешивали 6 часов при комнатной температуре. ТГФ отгоняли, к сухому остатку прибавляли 30 мл воды и доводили до pH = 6-7 10%-м раствором HCl, экстрагировали Et2O (3x20 мл). Объединенные органические слои промывали насыщенным раствором NaCl, сушили над Na2SO4, растворитель отгоняли. Получили 1.818 г кетоенола 3. Выход 96%. В дальнейшем соединение 3 использовали без дополнительной очистки. Физико-химические характеристики соответствуют литературным данным [L. E. Nikitina, N. P. Artemova, V. A. Startseva, I. V. Fedyunina, V. V. Klochkov, Chem. Nat. Comp. 2017, 53(5), 811].

S-(((1R,3R,5R)-6,6-Диметил-2-оксобицикло[3.1.1]гептан-3-ил)метил) этантиоат (5). К 3.0 г (0.02 моль) 2-норпинанона 2 при комнатной температуре в 18 мл ТГФ при перемешивании добавляли 2 капли пиридина (1 мол. %) и 3.0 г (0.04 моль) тиоуксусной кислоты. Время синтеза 0.08-0.25 ч. Контроль за ходом реакции проводили по ТСХ (элюент - CH2Cl2, проявитель - раствор ванилина в EtOH). Добавляли для экстракции CH2Cl2и насыщенный раствор NaHCO3. Объединенные органические слои сушили над Na2SO4, удаляли растворитель при пониженном давлении. Продукт очищали методом колоночной хроматографии (элюент - CH2Cl2). Выход 3.6 г (79%, de 98%). Светло-желтая жидкость.


(1R,3R,5R)-3-(Тиометил)-6,6-диметилбицикло[3.1.1]гептан-2-он (6). К раствору 0.23 г (1 ммоль) тиоацетата 5 в 2.3 мл ТГФ, охлажденного до 0°С, добавляли по каплям 0.1 г (2 ммоль) гидразин гидрат, перемешивали 4 ч. К реакционной смеси добавляли 1 Н раствор HCl (2 ммоль) для отмывания от гидразин гидрата, экстрагировали Et2O (3×20), промывали насыщенным раствором NaCl. Экстракт сушили над Na2SO4, удаляли растворитель при пониженном давлении. Продукт очищали методом колоночной хроматографии (элюент - CНCl3). Выход 0.14 г (75%). Прозрачная жидкость.


(1R,1'R,3R,3'R,5R,5'R)-3,3'-(Дисульфандиил-бис(метилен))бис(6,6-диметилбицикло[3.1.1]гептан-2-он) (7). Получен аналогично тиолу 6. Выход 0.05 г (25%). Белый порошок. Т. пл. 97-99°С.

S-(((1S,2S,5R)-6,6-Диметил-3-оксобицикло[3.1.1]гептан-2-ил)метил) этантиоат ((2S)-10). Раствор 3.0 г (0.02 моль) пинокарвона 9 в 18 мл ТГФ при перемешивании охлаждали до -65°С, добавляли 2 капли пиридина (~1 мол. %), а затем по каплям раствор 3.0 г (0.04 моль) тиоуксусной кислоты в 18 мл ТГФ. Контроль по ТСХ (элюент - петролейный эфир : этилацетат = 5:1,проявитель - раствор ванилина в EtOH). Смесь перемешивали 6 ч, затем добавляли для экстракции Et2O и насыщенный раствор NaHCO3. Объединенные органические слои сушили над Na2SO4, удаляли растворитель при пониженном давлении. Продукт очищали методом колоночной хроматографии (элюент - CH2Cl2:Et2O = 30:1). Выход 3.8 г (84%, de 92%). Светло-желтая жидкость.


S-(((1S,2S,5R)-6,6-Диметил-3-оксобицикло[3.1.1]гептан-2-ил)метил) бензотиоат ((2S)-11). Получали реакцией пинокарвона 9 с тиобензойной кислотой аналогично тиоацетату (2S)-10. Выход 68% (de 93%). Прозрачная жидкость.
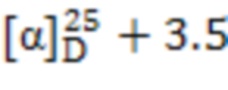

(1S,2S,5R)-2-(Тиометил)-6,6-диметилбицикло[3.1.1]гептан-3-он ((2S)-12). Получали аналогично тиолу 6 из тиоацетата (2S)-10. Продукт очищали методом колоночной хроматографии (элюент - CH2Cl2: Et2O = 30:1). Выход 90%. Прозрачная жидкость.


(1S,1'S,2S,2'S,5R,5'R)-2,2'-(Дисульфандиил-бис(метилен))бис(6,6-диметилбицикло[3.1.1]гептан-3-он) ((2S)-13). Получен аналогично тиолу (2S)-6 из тиобензоата ((2S)-11. Время синтеза 24 ч. Выход 80%. Белый порошок. Т. пл. 93-95°С.

Обоснование применения.
Синтез ацилсульфанильных и сульфанильных производных открывает пути к получению разнообразных классов соединений: дисульфидов, сульфоксидов, тиолсульфонатов и -сульфинатов, сульфоновых кислот, сульфин- и сульфонхлоридов, сульфин- и сульфонамидов, сульфен-, сульфин- и сульфониминов и др., которые являются потенциальными биологически активными соединениями или полупродуктами в тонком органическом синтезе (схема 3).

Серосодержащие монотерпеноиды проявляют противогрибковую, противовоспалительную, антихеликобактерную, противоопухолевую и другие виды биологической активности [Nikitina L.E., Artemova N.P., Startseva V.A., Fedyunina I.V., Klochkov V.V. Chem. Nat. Comp. 2017, 53 (5), 811-819. DOI: 10.1007/s10600-017-2131-z].
Сульфокамфорная кислота используется в производстве лекарственных препаратов, обладающих коронарорасширяющей способностью, антибактериальной активностью, болеутоляющим свойством (сульфокамфокаин, полусинтетические пенициллины и цефалоспорины). Тиолсульфонаты (RSO2SR) обладают бактерицидной и фунгицидной активностями [D.R. Hogg. In Comprehensive Organic Chemistry, Vol. 3, Sulphur Compounds. (Eds. D. Barton, W.D. Ollis). Pergamon Press: Oxford, 1979]. Сульфокислоты RSO3H и сульфохлориды RSO2Cl используются в качестве полупродуктов в органическом синтезе лекарственных препаратов [Котегов Н. А., Белогурова Н. В., Зырянов В. А., Тупикина В. Г., Петров А. Ю., Пат. 2119332 (1995). РФ. Б.И. 1998, № 27]. Пинановые гидрокситиолсульфонаты (RSO2SR) проявляют антимикробную активность в отношении Candida albicans, Staphylococcus aureus и Cryptococcus neoformans [Гребенкина О.Н., Лезина О.М., Изместьев Е.С., Судариков Д.В., Пестова С.В., Рубцова С.А., Кучин А.В. ЖОрХ. 2017, 53 (6), 844.]. Монотерпеновые сульфонамиды (RSO2NR1) на основе камфоры обладают ингибирующей активностью в отношении вирусов Эбола и Марбург [А.С.Соколова, Д.В.Баранова, О.И.Яровая, Д.С.Баев, О.А.Полежаева и др. Изв. АН, Сер. Хим., 2019, 5, 1041]. Энантиомеры транс-миртанилсульфонамида, содержащие группы NH2, пиперазиновый и хлорфенильный фрагменты, являются антагонистами белок-белкового взаимодействия Bcl-2 [N. Yusuff, M. Dore, C. Joud, M. Visser, C. Springer, X. Xie, K. Herlihy, D. Porter, B. B. Toure. ACS Med. Chem. Lett., 2012, 3(7), 579. doi: 10.1021/ml300095a].
Реферат
Настоящее изобретение относится к хиральному γ-кетосульфанильному производному пинановой структуры (RSR1) формулы (I):

где звездочкой обозначена связь, к которой присоединяется атом серы (-S-), исключая:

Также предлагаемое изобретение относится к способу получение заявленного хирального γ-кетосульфанильного производного пинановой структуры (RSR1) формулы (I). Технический результат - расширение арсенала новых хиральных γ-кетосульфанильных производных пинановой структуры, являющихся перспективными субстратами для получения сульфинил- и сульфонилсодержащих соединений - потенциальных биологически активных веществ. 2 н.п. ф-лы.
Формула


Документы, цитированные в отчёте о поиске
Хиральные трифторметилированные монотерпеновые тиоацетаты и тиолы пинанового ряда
Комментарии