Производные лантионина - RU2518890C2
Код документа: RU2518890C2
Чертежи











Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому соединению, проявляющему активность агониста CaSR, и пищевой композиции, содержащей новое соединение, а также придающее кокуми средство.
УРОВЕНЬ ТЕХНИКИ
В последние годы возрастают потребности, связанные с вкусовыми ощущениями, вследствие, например, диверсификации пищевых привычек человека, и это соответственно ведет к росту потребности в разработке превосходного придающего кокуми средства, которое может придавать «кокуми» различным продуктам питания. При этом кокуми нельзя выразить просто в понятиях пяти основных вкусов, т.е. сладкого вкуса, соленого вкуса, кислого вкуса, горького вкуса и вкуса, называемого «умами», и, таким образом, он обозначает вкусовые ощущения, которые усиливаются даже в пограничных вкусах, относящихся к указанным выше пяти основным вкусам, таких как густая консистенция, рост (заполненность рта), непрерывность и гармония, в дополнение к предшествующим пяти основным вкусам.
С другой стороны, «кальцийчувствительный рецептор» (CaSR), также называемый «кальциевым рецептором», сигналы, испускаемые кальцийчувствительным рецептором, могут управлять различными видами функций в живом организме, и вещества, обладающие активностью агониста CaSR, таким образом, можно использовать в качестве придающего кокуми средства в продуктах питания или т.п. (см. патентный документ 1 и непатентный документ 1, указанные ниже).
Кроме того, в течение длительного времени глутатион был известен в качестве соединения, обладающего придающей кокуми активностью. Однакой глутатион содержит цистеин в своей молекуле, который представляет собой содержащую атом серы аминокислоту, и, следовательно, он будет вызывать множество проблем, подлежащих преодолению, связанных, например, со стабильностью и испусканием плохого запаха.
Соответственно, желательно проводить поиск разнообразных соединений, которые обладают активностью агониста CaSR, чтобы найти вещество, которое обладает более превосходной придающей кокуми функцией, в частности, придающей начальный вкус кокуми функцией, которое обладает превосходной стабильностью, которое можно легко получать при низкой стоимости и которое может придавать кокуми различным продуктам питания; и обеспечить придающее кокуми средство, которое содержит такое полезное вещество, и композицию придающего кокуми средства, которая аналогичным образом содержит это вещество и другое вещество, обладающее активностью агониста CaSR, в комбинации.
ЛИТЕРАТУРА ИЗВЕСТНОГО УРОВНЯ ТЕХНИКИ
Патентный документ
Патентный документ 1: брошюра опубликованного международного патента № 2007/055393.
Непатентный документ
Непатентный документ 1: The Journal of Biological Chemistry, 2010, 285 (2), pp. 1016-22.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Главная цель настоящего изобретения состоит в поиске разнообразных соединений, обладающих активностью агониста CaSR, для того, чтобы получить вещество, обладающее более превосходной придающей кокуми функцией, и более конкретно, в том, чтобы предоставить придающее кокуми средство, которое содержит указанное выше вещество, и композицию придающего кокуми средства, которая аналогичным образом содержит указанное выше вещество и другое вещество, также обладающее активностью агониста CaSR, в комбинации. Дополнительная цель настоящего изобретения состоит в том, чтобы предоставить пищевую композицию, содержащую вещество в предварительно определяемой концентрации.
Авторы данного изобретения провели тщательное исследование различных соединений и в результате обнаружили, что производное лантионина, имеющее структуру, представленную следующей общей формулой (I), которое является новым соединением (далее это соединение будет обозначаться как соединение по настоящему изобретению или производное лантионина по (или в соответствии с) настоящим изобретением), обладает высокой активностью агониста CaSR и чрезвычайно превосходной придающей кокуми функцией. Авторы данного изобретения также обнаружили, что добавление соединения по настоящему изобретению, обнаруженного таким образом, сделает возможным получение подходящей пищевой композиции, у которой кокуми (богатый вкус) усилен или улучшен. Таким образом, авторы настоящего изобретения выполнили настоящее изобретение.
Более конкретно, настоящее изобретение в настоящем документе предусматривает соединение, которое имеет структуру, представленную следующей общей формулой (I), или его съедобную соль:

(I)
где каждый R1 и R2 независимо представляет собой атом водорода или низшую алкильную группу, содержащую от 1 до 3 углеродных атомов;
A представляет собой метиленовую группу или оксигруппу (-О-); и
X представляет собой алкиленовую группу, которая содержит от 1 до 5 углеродных атомов, при условии, что одна из метиленовых групп, в составе алкиленовой группы, может быть заменена на тиогруппу (-S-), дисульфидную группу (-S-S-), оксигруппу (-O-), иминогруппу (-NH-) или алкилиминогруппу, содержащую от 1 до 3 углеродных атомов (-NRa-, где Ra представляет собой алкильную группу, содержащую от 1 до 3 углеродных атомов), и что алкиленовая группа может быть дополнительно замещена 1-6 алкильными группами, каждая из которых содержит от 1 до 3 углеродных атомов.
Кроме того, настоящее изобретение относится к пищевой композиции, содержащей соединение, представленное указанной выше формулой (I), или его съедобную соль в количестве в диапазоне от 10 частей на миллиард до 99,9% по массе (далее ее также обозначают как «пищевая композиция по настоящему изобретению»).
Настоящее изобретение дополнительно относится к придающему кокуми средству, содержащему, в качестве эффективного компонента, соединение, представленное указанной выше формулой (I), или его съедобную соль (далее его также обозначают как «придающее кокуми средство по настоящему изобретению»).
Кроме того, настоящее изобретение аналогичным образом предусматривает композицию придающего кокуми средства, которая содержит (a) соединение, представленное указанной выше общей формулой (I), или его съедобную соль; и (b) одну или по меньшей мере две аминокислоты или пептида, выбранные из группы, состоящей из γ-Glu-X-Gly (где X представляет собой аминокислоту или производное аминокислоты), γ-Glu-Val-Y (где Y представляет собой аминокислоту или производное аминокислоты), γ-Glu-Abu, γ-Glu-Ala, γ-Glu-Gly, γ-Glu-Cys, γ-Glu-Met, γ-Glu-Thr, γ-Glu-Val, γ-Glu-Orn, Asp-Gly, Cys-Gly, Cys-Met, Glu-Cys, Gly-Cys, Leu-Asp, D-Cys, γ-Glu-Met (O), γ-Glu-γ-Glu-Val, γ-Glu-Val-NH2, γ-Glu-Val-ol, γ-Glu-Ser, γ-Glu-Tau, γ-Glu-Cys (S-Me) (O), γ-Glu-Leu, γ-Glu-Ile, γ-Glu-t-Leu и γ-Glu-Cys (S-Me).
Кроме того, настоящее изобретение также относится к соединению, имеющему структуру, представленную следующей общей формулой (IA), или его химически приемлемой соли, которые можно использовать в качестве промежуточных соединений для получения соединения, представленного указанной выше общей формулой (I), или его съедобной соли:

(IA)
где каждый R1' и R2' независимо представляет собой атом водорода или алкильную группу, содержащую от 1 до 3 углеродных атомов;
R3' представляет собой атом водорода, алкильную группу, содержащую от 1 до 4 углеродных атомов, бензильную группу или 9-флуоренилметильную группу;
R4' представляет собой трет-бутоксикарбонильную группу, бензилоксикарбонильную группу или 9-флуоренилметилоксикарбонильную группу;
R5' представляет собой гидроксильную группу, алкоксигруппу, содержащую от 1 до 4 углеродных атомов, бензилоксигруппу, аминогруппу (-NH2) или алкиламиногруппу, содержащую от 1 до 3 углеродных атомов;
A представляет собой метиленовую группу или оксигруппу; и
X представляет собой алкиленовую группу, содержащую от 1 до 5 углеродных атомов, при условии, что одна из метиленовых групп, включенных в алкиленовую группу, может быть заменена на тиогруппу, дисульфидную группу, оксигруппу, иминогруппу или алкилиминогруппу, содержащую от 1 до 3 углеродных атомов, и что алкиленовая группа может быть дополнительно замещена 1-6 алкильными группами, каждая из которых содержит от 1 до 3 углеродных атомов.
Настоящее изобретение также может предусматривать придающее кокуми средство и композицию придающего кокуми средства, которые обладают чрезвычайно превосходной придающей кокуми функцией и превосходной стабильностью и которые легко получать при низкой стоимости. Кроме того, настоящее изобретение аналогичным образом может предусматривать превосходную пищевую композицию, которая содержит вещество, обладающее превосходной придающей кокуми функцией, в концентрации, равной или превышающей предварительно определяемый уровень.
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение описано более подробно.
В настоящем изобретении, используемый в настоящем документе термин «алкильная группа, содержащая от 1 до 3 углеродных атомов» обозначает линейную или разветвленную алкильную группу, и более конкретно, алкильная группа, содержащая от 1 до 3 углеродных атомов, может представлять собой, например, метильную группу, этильную группу, н-пропильную группу и изопропильную группу, и предпочтительно используемое в настоящем документе включает, например, метильную группу или этильную группу.
Кроме того, используемый в настоящем документе термин «алкилиминогруппа, содержащая от 1 до 3 углеродных атомов» обозначает иминогруппу, замещенную алкильной группой, содержащей от 1 до 3 углеродных атомов, такой как те, что перечислены выше.
Предпочтительно используемое в настоящем изобретении в качестве соединений, представленных указанной выше общей формулой (I), включают те, что точно определены ниже:
Соединения, представленные общей формулой (I), где каждый R1 и R2 предпочтительно представляет собой атом водорода;
A предпочтительно представляет собой метиленовую группу;
X предпочтительно представляет собой триметиленовую группу, в которой одна из метиленовых групп, включенных в нее, заменена на тиогруппу и, в частности, группу: -CH2-S-CH2-; или
X предпочтительно представляет собой тетраметиленовую группу, в которой одна из ее метиленовых групп заменена на тиогруппу, или триметиленовую группу, которая замещена алкильной группой, содержащей от 1 до 3 углеродных атомов, и одна из метиленовых групп которой заменена на тиогруппу, и X, в частности, предпочтительно представляет собой группу, выбранную из -СН2-S-СН2-CH2-, -CH(CH3)-S-CH2- или -CH2-S-CH(CH3)-; или
X аналогичным образом предпочтительно представляет собой триметиленовую группу.
В отношении углеродных атомов a и b, присутствующих в структуре кольца в соединении, представленном общей формулой (I), соединения, имеющие любую возможную стерическую конфигурацию, можно использовать в настоящем изобретении, но их предпочтительная конфигурация представляет собой ту, что представлена следующими общими формулами (I-1) и (I-2), при этом конфигурация, представленная формулой (I-1), является особенно предпочтительной. Кроме того, в отношении углерода с, присутствующего в соединении, предпочтительными являются соединения, каждое из которых имеет S-конфигурацию:


Более конкретно, соединения, точно определенные ниже, или их съедобные соли предпочтительно используют в настоящем изобретении в качестве соединений, представленных общей формулой (I), или их съедобных солей:
Соединения, представленные общей формулой (I), в которой каждый R1 и R2 представляет собой атом водорода; A представляет собой метиленовую группу; и X представляет собой триметиленовую группу, замещенную тиогруппой;
Соединения, представленные следующей общей формулой (I-1а):

(I-1a)
Соединения, представленные следующей общей формулой (IIa):

(IIa)
Соединения, имеющие следующую стерическую конфигурацию и представленные указанной выше общей формулой (IIa); среди этих соединений любое из соединений, представленных следующими структурными формулами 8a-8d, можно использовать в настоящем изобретении, при этом, в частности, предпочтительно в настоящем документе используют соединение структурной формулы 8b:

Соединения, представленные указанной выше общей формулой (I), в которых каждый R1 и R2 представляет собой атом водорода; A представляет собой метиленовую группу; X представляет собой тетраметиленовую группу, замещенную тиогруппой, или триметиленовую группу, замещенную алкильной группой, содержащей от 1 до 3 углеродных атомов, и в которой одну из ее метиленовых групп заменяют на тиогруппу;
Соединения, представленные следующими общими формулами (IIb) и (IIc):

(IIb)

(IIc)
где R представляет собой алкильную группу, содержащую от 1 до 3 углеродных атомов.
Конкретные примеры указанных выше съедобных солей включают, например, соли аммония, соли щелочных металлов (их примерами являются, например, соли натрия и соли калия, которые предпочтительно используют в настоящем изобретении) и соли щелочноземельных металлов (их примерами являются, например, соли кальция и соли магния, которые предпочтительно используют в настоящем изобретении); и соли органических оснований, такие как соли лизина и альгинаты для достаточно кислых соединений в соответствии с настоящим изобретением. Кроме того, съедобные соли могут аналогичным образом включать, например, неорганические соли, например, соляной кислоты; или соли органических кислот, таких как уксусная кислота, лимонная кислота, молочная кислота, янтарная кислота, фумаровая кислота и яблочная кислота для достаточно основных соединений в соответствии с настоящим изобретением.
Кроме того, примеры указанных выше химически приемлемых солей включают те, что перечислены выше в связи со съедобными солями.
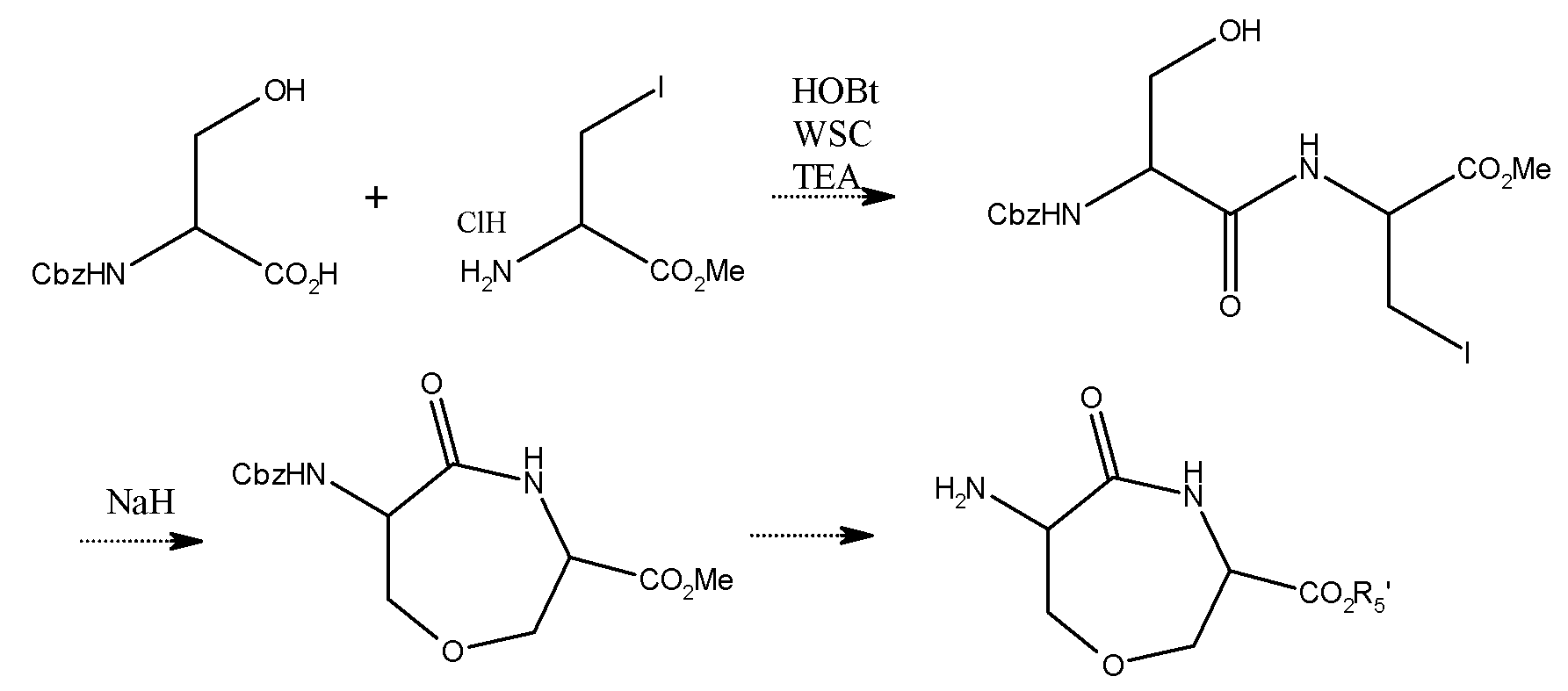
Способы получения
Типичные способы получения соединений по настоящему изобретению подробно описаны ниже:
В этой связи, иногда будет эффективным, с точки зрения методики получения, в следующих способах получения, замещать некоторые функциональные группы, входящие в исходные вещества или промежуточные соединения, подходящими защитными группами, т.е. группами, каждую из которых можно легко превратить в исходные функциональные группы, в зависимости от типов функциональных групп. Впоследствии, в случае необходимости, защитные группы можно удалить для того, чтобы таким образом получить каждое желаемое соединение. В качестве функциональных групп могут быть перечислены, например, аминогруппа, гидроксильная группа и карбоксильнаЯ группа, и примеры защитных групп для них включают, например, трет-бутоксикарбонильную группу (Boc), бензилоксикарбонильную группу (Cbz) и 9-флуоренилметоксикарбонильную группу (Fmoc) в качестве защитных групп для аминогрупп; и трет-бутильную группу (t-Bu) и бензильную группу (Bn или Bzl) в качестве защитных групп для карбоксильной группы. Эти защитные группы, которые могут быть использованы в настоящем документе, более подробно описаны в статье, названной: «Protective Groups in Organic Synthesis», третье издание, авторы T.W. Green & P.G.M. Wuts, публикация JOHN WILLY & SONS, INC. Эти защитные группы можно соответствующим образом выбирать и использовать, при этом принимая во внимание конкретные условия реакции, подлежащие использованию. Способ, раскрытый в указанной выше ссылочной статье, можно соответствующим образом применять для введения защитной группы и для ее удаления (разблокирование). Например, это показывает, что функциональные группы Prot 1 и Prot 2, описанные в следующем способе получения, используют в качестве таких функциональных групп, но настоящее изобретение не ограничено этими конкретными примерами.
Соединение, представленное общей формулой (I) в соответствии с настоящим изобретением, например, можно получать в соответствии со схемой синтеза I, подробно изложенной ниже:
Схема синтеза I

Где определения заместителей, встречающихся в этих формулах, аналогичны тем, что точно определены выше в связи с указанной выше общей формулой (I) или (IA).
Соединение (X) конденсируют с производным глутаминовой кислоты (XI) в присутствии основания, при этом используя конденсирующий агент, чтобы таким образом получить соединение γ-глутамила (XII). Впоследствии, все защитные группы для карбоксильных и аминогрупп соединения (XII) удаляют, чтобы таким образом получить желаемое соединение (I).
Соединение, представленное общей формулой (I), полученное в соответствии с указанным выше способом, можно выделять и очищать любым известным способом, таким как концентрация при пониженном давлении, экстракция растворителем, кристаллизация и/или хроматографический способ.
Кроме того, соединение, представленное указанной выше общей формулой, в которой каждый R1' и R2' представляет собой атом водорода, и X представляет собой группу: -CH2-S-СН2-, в качестве примера исходного вещества (X), например, можно получать в соответствии со следующей схемой синтеза II, приведенной ниже:
Схема синтеза II

где Prot 1-Prot 4 независимо представляют собой подходящие защитные группы, соответственно.
Если объяснять подробно, соединение (III) сначала восстанавливают трифенилфосфином или т.п., с образованием тиола (IV). Затем соединение простого тиоэфира (VI) получают посредством реакции между полученным соединением (IV) и алкилгалогенидом в присутствии основания. После частичного удаления защитных групп с получаемого соединения (VI), последнее превращают в циклическое соединение (VIII) в присутствии основания, при этом используя конденсирующий агент. После удаления защитной группы аминогруппы, присутствующей на соединении (VIII), полученное соединение конденсируют с производным глутаминовой кислоты (X) с использованием подходящего конденсирующего агента.
Соединение, представленное общей формулой (X), полученное в соответствии с указанными выше процедурами, может быть выделено и очищено любым известным способом, таким как концентрация при пониженном давлении, экстракция растворителем, кристаллизация и/или хроматографический способ.
Указанное выше исходное вещество (X), в котором X представляет собой тетраметиленовую группу, замещенную тиогруппой, можно синтезировать в соответствии, например, со следующей схемой синтеза III, и в соответствии с аналогичным способом, используемым для получения указанного выше соединения:
Схема синтеза III

Где определения заместителей, встречающихся в этих соединениях, аналогичны тем, что уже точно определены выше, и n равно 2.
Указанное выше исходное вещество (X), в котором X представляет собой триметиленовую группу, замещенную тиогруппой, которая замещена алкильной группой, содержащей от 1 до 3 углеродных атомов, можно синтезировать в соответствии со следующей схемой синтеза IV, и в соответствии с аналогичными процедурами, использованными выше, в связи с получением указанного выше соединения:
Схема синтеза IV

Где определения заместителей, встречающихся в этих соединениях, аналогичны тем, что уже точно определены выше.
Указанное выше исходное вещество (X), в котором X представляет собой триметиленовую группу, замещенную оксигруппой, можно синтезировать в соответствии, например, со следующей схемой синтеза V:
Схема синтеза V
1) Способ синтеза циклических соединений с использованием производных азиридина:

2) Способ синтеза циклических соединений посредством межмолекулярной этерификации:

3) Способ синтеза циклических соединений посредством внутримолекулярной этерификации:
Производное лантионина в соответствии с настоящим изобретением обладает превосходным придающим кокуми эффектом по отношению к другому веществу, и, следовательно, производное можно использовать в качестве придающего кокуми средства. Производное лантионина в соответствии с настоящим изобретением можно использовать таким образом, чтобы встраивать его в пищевую композицию, которой следует придать желаемый кокуми, в количестве в диапазоне от 10 частей на миллиард до 99,9% по массе, предпочтительно от 0,05 м.д. до 99,9% по массе и более предпочтительно от 0,1 м.д. до 99,9% по массе, основываясь на общей массе пищевой композиции. Более конкретно, согласно другому аспекту, настоящее изобретение относится к пищевой композиции, содержащей производное лантионина, и предпочтительно к пищевой композиции, содержащей производное лантионина в количестве в диапазоне от 0,05 м.д. до 99,9%.
Кроме того, если используют производное лантионина в соответствии с настоящим изобретением в комбинации с по меньшей мере одним другим исходным веществом для приправы, выбранным из группы, состоящей из аминокислот, таких как глутамат натрия (MSG), нуклеиновых кислот, таких как инозинмонофосфат (IMP), неорганических солей, таких как хлорид натрия, органических кислот, таких как лимонная кислота, и различных типов дрожжевых экстрактов, упомянутое выше может предоставлять предпочтительную приправу, которая обладает усиленным кокуми по сравнению с таковым, наблюдаемым при использовании самого по себе такого другого исходного вещества для приправы. Специалист в данной области может соответствующим образом задать концентрацию производного лантионина в соответствии с настоящим изобретением, когда используют то же самое в комбинации с указанным выше другим исходным веществом для приправы, при этом принимая во внимание результаты сенсорной или органолептической оценки. Однако в одном из примеров будет достаточно использовать производное лантионина в соответствии с настоящим изобретением в количестве в диапазоне приблизительно от 0,1 м.д. приблизительно до 500 м.д., как выражают в единицах конечной концентрации.
В настоящем изобретении термин «кокуми» обозначает вкус, который нельзя выразить посредством пяти основных вкусов, т.е. сладкого вкуса, соленого вкуса, кислого вкуса, горького вкуса и умами (восхитительность), и более конкретно, термин обозначает вкус, который усиливается даже в пограничных вкусах по отношению к указанным выше пяти основным вкусам, таких как густая консистенция, рост (заполненность рта), непрерывность и гармония, в дополнение к усилению указанных выше пяти основных вкусов. В этом отношении, термин «придающий кокуми» обозначает, что усиливаются не только пять основных вкусов, представленных сладким вкусом, соленым вкусом, кислым вкусом, горьким вкусом и вкусом умами, но также пограничные вкусы по отношению к указанным выше пяти основным вкусам, такие как густая консистенция, рост (заполненность рта), непрерывность и гармония, придаются любой желаемой пище. Альтернативно, это аналогичным образом можно называть как «эффект, усиливающий вкус». Соответствующим образом, соединение по настоящему изобретению можно аналогичным образом обозначать как «усилитель вкуса». Соединение по настоящему изобретению можно использовать в качестве усилителя сладкого вкуса, усилителя соленого вкуса, усилителя кислого вкуса, усилителя горького вкуса или усилителя умами.
Кроме того, вкусовые ощущения должны варьировать с течением времени после еды, и это можно обозначить как начальный вкус, средний вкус и остаточный вкус по порядку с течением времени после еды. Это просто относительная концепция. Однако, говоря в общем, начальный вкус, средний вкус и остаточный вкус определяют как воспринимаемые посредством вкусов в течение времени в диапазоне от 0 до 2 секунд, в течение времени в диапазоне от 2 до 5 секунд или после 5 секунд и далее, после еды, соответственно. Кроме того, комбинированный начальный и средний вкусы совместно обозначают как «начально-средний вкус», и комбинированные средний и остаточный вкусы совместно обозначают как «средне-остаточный вкус». Кроме того, «начально-средний вкус» определяют как вкус, воспринимаемый в течение времени в диапазоне от 0 до 5 секунд после еды, а «средне-остаточный вкус» определяют как вкус, воспринимаемый в течение времени в диапазоне от 2 секунд приблизительно до 30 секунд после еды. Что касается оценки на основе указанных выше трех делений, дегустаторам (людям, которые съедают образец и оценивают его вкус) будет сложно сконцентрировать свое внимание на оценке каждого конкретного образца, и, следовательно, обыкновенным является то, что, как правило, используют оценку на основе двух делений.
Эффект вещества, обладающего CaSR активностью, оказываемый на ощущение кокуми и аромата, можно подтвердить с помощью способа, такого как органолептический тест для оценки вкуса образца дегустаторами. В качестве органолептического теста для оценки вкуса образца можно отметить, например, тест, раскрытый в примерах в настоящем описании патента, но настоящее изобретение не ограничено этими конкретными способами.
В этом описании термин «CaSR» обозначает кальцийчувствительный рецептор, который относится к классу С семидоменного трансмембранного рецептора, и его также обозначают как кальциевый рецептор. В этом описании термин «агонист CaSR» обозначает вещество, которое связывается с CaSR, чтобы тем самым активировать его. Кроме того, термин «активировать CaSR», используемый в этом описании, обозначает, что лиганд связан с CaSR, чтобы активировать связанный нуклеотидом гуанином белок и тем самым передавать сигналы. Кроме того, термин «активность агониста CaSR» обозначает такие свойства вещества, что он может быть связан с CaSR, чтобы таким образом активировать его.
Способ скрининга соединения, обладающего такой активностью агониста CaSR, который содержит следующие стадии, будет конкретно описан ниже, но настоящее изобретение не ограничено этими стадиями.
1) Стадия добавления тестируемого вещества в систему для определения активности CaSR для определения активности CaSR и определения активности CaSR тестируемого вещества;
2) Стадия сравнения активности CaSR, наблюдаемой при добавлении тестируемого вещества, с таковой, наблюдаемой без добавления тестируемого вещества;
3) Стадия выбора конкретного тестируемого вещества, которое проявляет активность агониста CaSR при добавлении тестируемого вещества.
Активность CaSR можно определить, например, посредством использования системы для определения, в которой используют клетку, способную экспрессировать CaSR. Клетка может представлять собой клетку, способную эндогенно экспрессировать CaSR, или рекомбинантную клетку, несущую ген CaSR, экзогенно введенный в нее. Указанная выше система для определения активности CaSR не ограничена какой-либо одной конкретной, поскольку она может определять связь (реакцию) между активацией вещества и CaSR при добавлении внеклеточного лиганда (активирующего вещества), специфичного к CaSR; или она может передавать поддающиеся обнаружению сигналы в клетке в ответ на формирование связи (реакции) между активирующим веществом и CaSR. Когда CaSR активность обнаруживают посредством реакции с тестируемым веществом, о тестируемом веществе можно таким образом судить, что оно обладает CaSR-стимулирующей активностью.
В качестве указанного выше CaSR предпочтительным является CaSR человека, кодируемый геном CaSR человека, зарегистрированным под номером доступа GenBank NM_000388. В этой связи, CaSR не ограничен белком, кодируемым геном, имеющим указанную выше последовательность гена, и может представлять собой белки, каждый из которых кодируется любым геном, обладающим гомологией с указанной выше последовательностью не менее чем 60%, предпочтительно не менее чем 80% и более предпочтительно не менее чем 90%, поскольку ген может кодировать белок, обладающий функцией CaSR. В то же время функцию CaSR можно проверять посредством экспрессии этих генов внутри клетки и определения какого-либо изменения электрического тока, наблюдаемого при добавлении кальция, или какого-либо изменения концентрации ионов кальция внутри клеток.
Источники указанного выше CaSR не ограничены конкретными источниками, и их конкретные примеры включают не только CaSR, полученные от человека, но также те, что получены от животных других видов, включая, например, мышь, крысу и собаку.
Как описано выше, активность CaSR можно подтверждать посредством, например, живых клеток, которые могут экспрессировать CaSR или его фрагмент, клеточных мембран, которые могут экспрессировать CaSR или его фрагмент, или системы in vitro, содержащей CaSR или белок в качестве его фрагмента.
Далее приведен пример такого способа для подтверждения активности CaSR, в котором используют живую клетку, но настоящее изобретение не ограничено этим способом.
Экспрессию CaSR осуществляют посредством культивирования клеток, таких как овоциты, взятые у ксенопуса, клетки яичника, полученные от хомяка, или эмбриональные клетки почек человека. Более конкретно, экспрессию CaSR можно реализовать посредством введения в клетки-хозяева продукта, полученного посредством трансформации плазмиды, содержащей экзогенные гены с клонированным геном CaSR в форме рекомбинантной плазмиды per se, или кРНК, полученной посредством рекомбинантной плазмиды, в качестве матрицы. Можно использовать электрофизиологический способ или флуоресцентный индикатор для обнаружения какого-либо повышения содержания кальция в клетках для обнаружения возникновения какой-либо желаемой реакции.
Сначала экспрессию CaSR подтверждают посредством обнаружения ответа на кальций или специфическое средство активации. В настоящем документе используют овоциты, которые показывают образование внутриклеточного электрического тока в ответ на концентрацию кальция порядка приблизительно 5 мМ; или культивируемые клетки, для которых наблюдают испускание флуоресценции вследствие флуоресцентного индикатора. Затем повторяют аналогичные процедуры, использованные выше, в то время как изменяется концентрация кальция, чтобы таким образом определить зависимость от концентрации кальция. Впоследствии получают раствор тестируемого вещества, обладающего концентрацией в диапазоне приблизительно от 1 мкМ приблизительно до 1 мКМ, получаемый раствор добавляют к овоцитам или культивируемым клеткам и измеряют активность CaSR в присутствии указанного выше тестируемого вещества, чтобы таким образом определить активность агониста CaSR для тестируемого вещества.
Кроме того, в качестве тестов для определения активности CaSR, можно перечислить, например, те, которые описаны в дальнейших примерах тестов в этом описании, но настоящее изобретение вовсе не ограничено этими конкретными тестами.
В композиции придающего кокуми средства в соответствии с настоящим изобретением аминокислоты или пептиды, используемые в комбинации с производным лантионина по настоящему изобретению, представляют собой одну или по меньшей мере две аминокислоты или пептида, выбранные из группы, состоящей из γ-Glu-X-Gly, где X представляет собой аминокислоту или производное аминокислоты, γ-Glu-Val-Y, где Y представляет собой аминокислоту или производное аминокислоты, γ-Glu-Abu, γ-Glu-Ala, γ-Glu-Gly, γ-Glu-Cys, γ-Glu-Met, γ-Glu-Thr, γ-Glu-Val, γ-Glu-Orn, Asp-Gly, Cys-Gly, Cys-Met, Glu-Cys, Gly-Cys, Leu-Asp, D-Cys, γ-Glu-Met (O), γ-Glu-γ-Glu-Val, γ-Glu-Val-NH2, γ-Glu-Val-ol, γ-Glu-Ser, γ-Glu-Tau, γ-Glu-Cys (S-Me) (O), γ-Glu-Leu, γ-Glu-Ile, γ-Glu-t-Leu и γ-Glu-Cys (S-Me). В этом отношении термин «аминокислота» включает нейтральные аминокислоты, такие как Gly, Ala, Val, Leu, Ile, Ser, Thr, Cys, Met, Asn, Gln, Pro, Hyp и t-Leu; кислые аминокислоты, такие как Asp и Glu; основные аминокислоты, такие как Lys, Arg и His; ароматические аминокислоты, такие как Phe, Tyr и Trp; и гомосерин, цитруллин, орнитин, α-аминомасляную кислоту, норвалин, норлейцин и таурин. Кроме того, аминокислоты или пептиды, используемые в комбинации с производным лантионина по настоящему изобретению, могут аналогичным образом представлять собой, например, искусственно синтезированные аминокислоты (каждая обладает небелковой конфигурацией), такие как трет-лейцин, циклолейцин, α-аминоизомасляная кислота, L-пеницилламин, аллотреонин и аллоизолейцин. В этой связи символ X, встречающийся в пептиде: γ-Glu-X-Gly, может представлять собой одну из указанных выше аминокислот или их производных, но предпочтительно он представляет собой аминокислоту или ее производное, отличное от цистеина (Cys).
В этом описании аминокислотные остатки обозначают в терминах следующих сокращений, соответственно:
(1) Gly: Глицин; (2) Ala: Аланин; (3) Val: Валин; (4) Leu: Лейцин; (5) Ile: Изолейцин; (6) Met: Метионин; (7) Phe: Фенилаланин; (8) Tyr: Тирозин; (9) Trp: Триптофан; (10) His: Гистидин; (11) Lys: Лизин; (12) Arg: Аргинин; (13) Ser: Серин; (14) Thr: Треонин; (15) Asp: Аспарагиновая кислота; (16) Glu: Глутаминовая кислота; (17) Asn: Аспарагин; (18) Gin: Глутамин; (19) Cys: Цистеин; (20) Pro: Пролин; (21) Orn: Орнитин; (22) Sar: Саркозин; (23) Cit: Цитруллин; (24) N-Val (или Nva): Норвалин (2-аминовалериановая кислота); (25) N-Leu (или Nle): Норлейцин; (26) Abu: α-Аминомасляная кислота; (27) Tau: Таурин; (28) Hyp: Гидроксипролин; (29) t-Leu: Трет-лейцин; (30) Cle: Циклолейцин; (31) Aib: α-Аминоизомасляная кислота (2-метилаланин); (32) Pen: L-Пеницилламин; (33) allo-Thr: Аллотреонин; (34) allo-Ile: Аллоизолейцин.
Кроме того, термин «производное аминокислоты» обозначает различные типы производных указанных выше аминокислот, и такие производные включают, например, особые аминокислоты, искусственно синтезированные аминокислоты, аминоспирты или указанные выше аминокислоты, в которых концевые карбонильные группы и/или аминогруппы или их боковые цепи, такие как тиоловая группа цистеина, замещены различными заместителями. Конкретные примеры таких заместителей включают алкильные группы, ацильные группы, гидроксильную группу, аминогруппы, алкиламиногруппы, нитрогруппы, сульфонильные группы и различные типы защитных групп. Конкретные примеры указанных выше производных аминокислот включают N-γ-нитроаргинин: Arg (NO2); S-нитроцистеин: Cys (SNO); S-метилцистеин: Cys (S-Me); S-аллилцистеин: Cys (S-аллил); валинамид: Val-NH2; и валинол (2-амино-3-метил-1-бутанол): Val-ol. В этой связи пептид: γ-Glu-Cys (SNO)-Gly, используемый в этом описании, представляет собой пептид, представленный следующей структурной формулой, и символ (O), встречающийся в указанных выше формулах: γ-Glu-Met (O) и γ-Glu-Cys (S-Me) (O) обозначает, что каждый из этих пептидов имеет сульфоксидную структуру. Символ (γ), встречающийся в γ-Glu, обозначает, что другой аминокислотный остаток связан с глутаминовой кислотой посредством карбоксильной группы, присутствующей в γ-положении последней.

S-Нитрозоглутатион (GNSO)
Производные лантионина и указанные выше аминокислоты или пептиды, используемые в комбинации с производными лантионина, при наличии, могут являться коммерчески доступными. Кроме того, их можно аналогичным образом получать, в случае необходимости, в соответствии с любым известным способом, таким как (1) химический способ получения или (2) способ получения с использованием фермента, причем способ химического синтеза является более удобным. Когда химически синтезируют производное лантионина и аминокислоту или пептид, используемый в комбинации с ним, пептид может быть полусинтетическим или синтетическим с использованием устройства пептидного синтеза. Можно перечислить, например, способ твердофазного синтеза пептидов в качестве указанного выше способа химического синтеза. Пептид, синтезированный в соответствии с указанным выше способом, можно очистить посредством обычного способа, такого как способ ионообменной хроматографии, способ высокоэффективной жидкостной хроматографии с обращенными фазами или способ аффинной хроматографии. Такой способ твердофазного синтеза пептидов и последующий способ очистки пептидов хорошо известен в данной области.
Кроме того, при получении производного лантионина и аминокислоты или пептида, используемого в комбинации с ним, посредством реакции с использованием фермента, производное лантионина и аминокислоту или пептид можно получать в соответствии, например, со способом, раскрытым в брошюре опубликованной международной патентной заявки № WO 2004/011653. Другими словами, аминокислота или дипептид, концевую карбоксильную группу которого превращают в сложноэфирную или амидную форму, вступает в реакцию с другой аминокислотой, которая находится в своем свободном состоянии (таком как аминокислота, карбоксильная группа которой защищена) в присутствии пептид-продуцирующего фермента, и затем получаемый дипептид или трипептид очищают, чтобы таким образом получить желаемый продукт. Пептид-продуцирующие ферменты, которые можно использовать в настоящем документе, включают, например, культуру микроорганизма, обладающего способностью продуцировать предполагаемый пептид; клеточные тела микроорганизма, выделенного из культуры, или продукт, полученный посредством обработки клеточных тел микроорганизма; или пептид-продуцирующий фермент, полученный от микроорганизма.
Кроме того, пептиды, которые можно использовать в настоящем изобретении, иногда присутствуют в растениях, таких как овощи и фрукты, микроорганизмы, такие как дрожжи, и другие встречающиеся в природе вещества, в дополнение к тем, которые синтезируют в соответствии с указанными выше способами ферментативного синтеза и химического синтеза. Если они встречаются в природе, их также можно извлечь из встречающегося в природе вещества и использовать в настоящем изобретении.
Придающее кокуми средство или композицию придающего кокуми средства в соответствии с настоящим изобретением можно использовать в качестве приправы, не подвергая их какой-либо дополнительной обработке, или после его смешивания с носителем, приемлемым в качестве ингредиента для продуктов питания и напитков, и/или другими ингредиентами приправы. Примеры таких других ингредиентов приправы включают вкусо-ароматическое вещество, сахариды, подсластители, съедобные волокна, витамины, аминокислоты, такие как глутамат натрия (MSG), нуклеиновые кислоты, такие как инозинмонофосфат (IMP), неорганические соли, такие как хлорид натрия, и органические кислоты, такие как лимонная кислота, а также различные дрожжевые экстракты.
Производное лантионина и аминокислота или пептид, используемые в комбинации с ним, могут быть в форме солей. Когда производное лантионина и аминокислота или пептид, используемые в комбинации с ним, могут образовывать соли, достаточно, чтобы соли являлись фармацевтически приемлемыми и съедобными, и конкретные примеры таких солей включают соли аммония, соли щелочных металлов, таких как натрий и калий, соли щелочноземельных металлов, таких как кальций и магний, соли алюминия, соли цинка, соли органических аминов, таких как триэтиламин, этаноламин, морфолин, пирролидин, пиперидин, пиперазин и дициклогексиламин, и соли основных аминокислот, таких как аргинин и лизин, для кислых групп указанного выше производного и аминокислоты или пептида, таких как карбоксильная группа. Кроме того, конкретные примеры таких солей включают соли неорганических кислот, таких как соляная кислота, серная кислота, фосфорная кислота, азотная кислота и бромистоводородная кислота, соли органических карбоновых кислот, таких как уксусная кислота, лимонная кислота, бензойная кислота, малеиновая кислота, фумаровая кислота, винная кислота, янтарная кислота, дубильная кислота, масляная кислота, хибензойная кислота, памовая кислота, энантовая кислота, декановая кислота, тиоктовая кислота, салициловая кислота, молочная кислота, щавелевая кислота, миндальная кислота и яблочная кислота, и соли органических сульфоновых кислот, такие как метансульфоновая кислота, бензолсульфоновая кислота и п-толуолсульфоновая кислота, для основных групп указанного выше производного и аминокислоты или пептида.
Производное лантионина, придающее кокуми средство, пищевую композицию или композицию придающего кокуми средства в соответствии с настоящим изобретением можно использовать в любой форме, такой как сухая порошковая форма, паста и раствор без какого-либо ограничения их физических свойств.
Производное лантионина, придающее кокуми средство, пищевую композицию или композицию придающего кокуми средства в соответствии с настоящим изобретением можно использовать при введении в различные продукты питания и напитки, такие как продукт питания, напиток и приправа.
При использовании производного лантионина, придающего кокуми средства, пищевой композиции или композиции придающего кокуми средства в соответствии с настоящим изобретением при введении в различные продукты питания и напитки, такие как продукт питания, напиток и приправа, конечное количество производного лантионина и тех аминокислот или пептидов, используемых в комбинации с упомянутым выше, не ограничено конкретными количествами, поскольку они могут показывать желаемый эффект по настоящему изобретению, но количество каждого из производного лантионина и/или аминокислоты или пептида варьирует приблизительно от 10 частей на миллиард приблизительно до 99,9% по массе, предпочтительно приблизительно от 0,05 м.д. приблизительно до 99,9% по массе и более предпочтительно приблизительно от 0,1 м.д. приблизительно до 99,9% по массе, соответственно, основываясь на общей массе продукта питания, напитка или приправы или т.п.
В настоящем изобретении также можно вводить другие добавки, приемлемые для продуктов питания и напитков, такие как любые твердые или жидкие носители и подходящие ингредиенты приправ, в различные продукты питания и напитки, такие как продукт питания, напиток и приправа, которые содержат производное лантионина, придающее кокуми средство, пищевую композицию или композицию придающего кокуми средства в соответствии с настоящим изобретением, включенного в нее.
Примеры указанных выше носителей включают глюкозу, лактозу, сахарозу, крахмал, маннит, декстрин, глицериды жирных кислот, полиэтиленгликоль, гидроксиэтил крахмал, этиленгликоль, сложные эфиры жирных кислот и полиоксиэтиленсорбитана, желатин, альбумин, аминокислоты, воду и физиологический раствор.
Указанные выше ингредиенты приправ не ограничены конкретными представителями и могут представлять собой любые известные в данной области представители, но их конкретные примеры могут представлять собой те, что уже описаны выше.
Содержание каждого из указанных выше носителей и других ингредиентов приправ не ограничено каким-либо конкретным диапазоном.
Среди указанных выше ингредиентов приправ дрожжевой экстракт может являться любым, и он не ограничен клеточными телами, из которых его получают, условиями их культивирования и способами их экстракции и способами их обработки. Кроме того, дрожжевой экстракт, который можно использовать в настоящем документе, может представлять собой тот, который подвергают какой-либо обработке, например, тепловой обработке, обработке ферментом, концентрационной обработки и/или пульверизационной обработки.
Далее настоящее изобретение описано более подробно со ссылкой на следующие примеры, но следующие примеры ни при каких условиях не ограничивают объем настоящего изобретения.
ПРИМЕРЫ
Пример 1: Синтез соединения 1
(Fmoc-L-Cys-Ot-Bu)2 (ди-трет-бутиловый эфир N,N'-дифлуоренилметоксикарбонил-L-цистина, 4,81 ммоль) растворяли в смешанном растворителе из тетрагидрофурана (58,5 мл) и воды (1,5 мл). Затем трибутилфосфин (5,28 ммоль) добавляли в полученный раствор с охлаждением льдом, и температуру полученной смеси (реакционная жидкость) приводили обратно к комнатной температуре, с последующим перемешиванием в течение 4 часов. Реакционную жидкость охлаждали, и затем 10% водный раствор лимонной кислоты (60 мл) добавляли в реакционную жидкость. Температуру полученной мутной жидкости приводили обратно к комнатной температуре и жидкость экстрагировали с использованием этилацетата (60 мл). Полученную таким образом органическую фазу промывали с использованием 60 мл водного раствора хлорида натрия и затем концентрировали, чтобы таким образом получить маслянистый остаток. Маслянистый остаток очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы таким образом получить соединение 1 в виде маслянистого продукта.
Выход: 97%.
ESI MS m/z 422,4 (M+Na)+.
1H ЯМР (400 МГц, CDCl3) δ: 1,50 (9H, с), 2,99 (2H, м), 4,23 (1H, т, J=6,8 Гц), 4,41 (2H, м), 4,54 (1H, м), 5,68 (1H, д, J=7,2 Гц), 7,32 (2H, м), 7,41 (2H, т, J=7,2 Гц), 7,61 (2H, д, J=7,6 Гц), 7,77 (2H, д, J=7,2 Гц).

Пример 2: Синтез соединения 2
Соединение 1 (6,04 ммоль), полученное в примере 1, растворяли в безводном диметилформамиде (60 мл) с последующим добавлением Boc-йод-D-Ala-OMe (метиловый эфир N-трет-бутоксикарбонил-3-йод-D-аланина) (6,20 ммоль) в полученный раствор, а затем карбоната цезия (6,02 ммоль) и с последующим перемешиванием полученной смеси (реакционная жидкость) при комнатной температуре в течение ночи. Затем реакционную жидкость охлаждали, с последующим добавлением 10% водного раствора лимонной кислоты (50 мл) и воды (30 мл), экстрагированием смеси с использованием этилацетата (60 мл) и экстрагированием во второй раз водной фазы с использованием этилацетата (60 мл). Полученные таким образом органические фазы объединяли вместе, объединенную органическую фазу промывали, по порядку, с использованием 10% водного раствора лимонной кислоты (50 мл) и водного раствора хлорида натрия (50 мл), а затем концентрировали органическую фазу. Полученный маслянистый остаток очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы таким образом получить соединение 2 в виде маслянистого продукта.
Выход: 66%.
ESI MS m/z: 601,2 (M+H)+.
1H ЯМР (400 МГц, CDCl3) δ: 1,45 (9H, с), 1,49 (9H, с), 3,01 (4H, м), 3,73 (3H, с), 4,24 (1H, т, J=7,2 Гц), 4,39 (2H, д, J=7,2 Гц), 4,48-4,55 (2H, м), 5,37 (1H, ушир.д, J=6,8 Гц), 5,80 (1H, ушир.д, J=6,8 Гц), 7,32 (2H, м), 7,40 (2H, т, J=7,2 Гц), 7,63 (2H, м), 7,77 (2H, д, J=7,6 Гц).

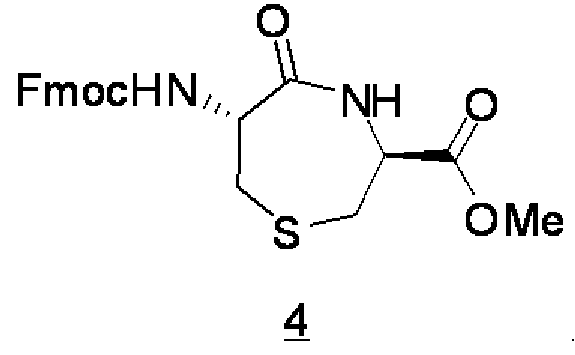
Пример 3: Синтез соединения 4
(Стадия 1): Соединение 2 (4,01 ммоль), полученное в примере 2, растворяли в 70 мл дихлорметана, затем в полученный раствор добавляли трифторуксусную кислоту (70 мл), полученную смесь (реакционная жидкость) перемешивали при комнатной температуре в течение одного часа, и реакционную жидкость концентрировали, чтобы таким образом получить остаток, содержащий соединение 3. Остаток, содержащий соединение 3, использовали на последующей стадии без какой-либо предварительной обработки, исходя из того, что выход соединения полагали равным 100%.

(Стадия 2): Безводный диметилформамид (60 мл) добавляли к соединению 3 (эквивалент 4,01 ммоль), полученному на указанной выше стадии 1, с охлаждением льдом, чтобы получить гомогенный раствор, и затем по каплям добавляли диизопропилэтиламин (8,04 ммоль). Температуру полученной смеси (реакционная жидкость) приводили обратно к комнатной температуре, в смесь добавляли карбонил бис-имидазол (8,10 ммоль), и затем реакционную жидкость перемешивали в течение ночи без какой-либо обработки. 10% Водный раствор лимонной кислоты (50 мл) добавляли в реакционную жидкость в условиях охлаждения и перемешивания, температуру реакционной жидкости приводили обратно к комнатной температуре, затем реакционную жидкость экстрагировали с использованием этилацетата (100 мл), водную фазу дополнительно экстрагировали с использованием этилацетата, и полученные органические фазы объединяли вместе. Эту органическую фазу промывали два раза с использованием 10% водного раствора лимонной кислоты (50 мл × 2) и затем один раз с использованием водного раствора хлорида натрия, с последующей концентрацией органической фазы, чтобы получить маслянистый остаток. Полученный остаток очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы таким образом получить соединение 4 в виде маслянистого продукта.
Выход: 39% (общий выход для указанных выше двух стадий).
ESI MS m/z 448,5 (M+Na)+.
1H ЯМР (400 МГц, CDCl3) δ: 2,71 (1H, дд, J=9,2, 14,4 Гц), 2,78-2,90 (2H, м), 3,02 (1H, д, J=14,4 Гц), 3,86 (3H, с), 4,20 (1H, т, J=6,8 Гц), 4,40 (2H, д, J=6,8 Гц), 4,56 (1H, дд, J=5,6, 9,2 Гц), 4,68 (1H, м), 6,30 (1H, д, J=5,6 Гц), 7,32 (2H, т, J=7,6 Гц), 7,40 (2H, т, J=7,6 Гц), 7,60 (2H, д, J=7,6 Гц), 7,77 (2H, д, J=7,6 Гц).
Пример 4: Синтез соединения 6
(Стадия 1): К соединению 4 (1,54 ммоль), полученному в примере 3, добавляли 10% раствор морфолин-диметилформамида (14 мл), и полученную смесь (реакционная жидкость) перемешивали при комнатной температуре в течение 30 минут. Затем реакционную жидкость концентрировали, чтобы получить остаток, содержащий соединение 5. Остаток использовали в последующей реакции без какой-либо предварительной обработки, исходя из того, что выход соединения 5 полагали равным 100%.

(Стадия 2): Boc-L-Glu-OtBu (α-трет-бутиловый эфир N-трет-бутоксикарбонил-L-глутаминовой кислоты) (1,70 ммоль) растворяли в безводном диметилформамиде (9 мл), и затем в полученный раствор добавляли HOBt·H2O (1-гидроксибензотриазолгидрат) (1,85 ммоль) и WSC·HCl (1-(3-диметиламинопропил)-3-этоксикарбодиимидгидрохлорид) (1,90 ммоль), а затем полученную смесь перемешивали при комнатной температуре в течение 15 минут. В смесь добавляли соединение 5 (эквивалент 1,54 ммоль), суспендированное в диметилформамиде (20 мл), и реакцию продолжали при комнатной температуре в течение ночи. После того как реакционную жидкость концентрировали, затем в полученный остаток добавляли этилацетат (50 мл) и воду (50 мл), чтобы таким образом разделить смесь на фазы и удалить органическую фазу, и водную фазу дополнительно экстрагировали с использованием этилацетата (50 мл). Органические фазы объединяли, промывали с использованием водного раствора бикарбоната натрия (50 мл) и водного раствора хлорида натрия (50 мл), с последующей концентрацией органической фазы, чтобы получить пастообразный остаток. Полученный пастообразный остаток очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы таким образом получить соединение 6 в виде маслянистого продукта.
Выход: 81% (общий выход для указанных выше двух стадий).
ESI MS m/z 490,0 (M+H)+.
1H ЯМР (300 МГц, CDCl3) δ: 1,44 (9H, с), 1,46 (9H, с), 1,90 (1H, м), 2,17 (1H, м), 2,32 (2H, м), 2,60 (1H, дд, J=10,5, 14,1 Гц), 2,92-2,98 (2H, м), 3,18 (1H, дд, J=6,0, 14,7 Гц), 3,84 (3H, с), 4,46 (1H, м), 4,80 (1H, м), 5,19 (1H, д, J=8,1 Гц), 6,32 (1H, д, J=8,1 Гц), 7,09 (1H, ушир.с).

Пример 5: Синтез соединений 8a и 8b
(Стадия 1): Соединение 6 (1,25 ммоль), полученное в примере 4, растворяли в тетрагидрофуране (30 мл), и в полученный раствор добавляли 0,2 M водного раствора гидроксида лития (2,50 ммоль) в условиях охлаждения льдом и перемешивания. Через 30 минут полученную смесь нейтрализовали до значения pH приблизительно 6, используя 10% водный раствор лимонной кислоты. Температуру смеси (реакционная жидкость) приводили обратно к комнатной температуре, затем реакционную жидкость концентрировали, и концентрат экстрагировали три раза с использованием этилацетата (20 мл × 3), чтобы получить экстракт или органическую фазу. Водную фазу дополнительно экстрагировали три раза с использованием этилацетата (20 мл × 3), полученные органические фазы объединяли, промывали с использованием водного раствора хлорида натрия (10 мл) и затем концентрировали, чтобы получить соединение 7. Полученное соединение 7 использовали в последующей реакции без какой-либо предварительной обработки, исходя из того, что его выход полагали равным 100%.

(Стадия 2): К соединению 7 (соответствует 1,25 ммоль), полученному в указанной выше стадии 1, добавляли раствор 4 н. соляной кислоты/диоксана (25 мл), с последующей реакцией этих компонентов при комнатной температуре в течение ночи, и затем реакционную жидкость концентрировали. Полученный пастообразный остаток очищали с использованием сильнокислотной ионообменной смолы (Amberlite IRA 400 OH AG), чтобы таким образом получить две фракции. Часть фракции, элюированной раньше, дополнительно очищали посредством препаративной ВЭЖХ с обращенной фазой (колонка: Develosil RPAQUEOUS-AR-5; подвижная фаза: линейный градиент воды/ацетонитрила, содержащего 0,1% муравьиной кислоты), чтобы таким образом получить соединение 8a в виде клейкого продукта. С другой стороны, фракцию, элюированную позже, концентрировали, чтобы получить белое твердое вещество. Белое твердое вещество растворяли в воде, и полученный водный раствор лиофилизировали, чтобы получить остаток. Полученный остаток промывали водой в соответствии со способом промывания суспензии, чтобы таким образом получить соединение 8b в виде белого твердого вещества.
Соединение 8a
ESI MS m/z 318,3 (M-H)-.
1H ЯМР (600 МГц, D2O) δ: 2,13 (2H, м), 2,50 (2H, т, J=7,8 Гц), 2,64 (1H, д, J=15,0 Гц), 2,83 (1H, дд, J=10,8, 15,0 Гц), 3,02 (1H, дд, J=2,4, 15,0 Гц), 3,15 (1H, дд, J=5,4, 15,0 Гц), 3,83 (1H, т, J=6,0 Гц), 4,55 (1H, дд, J=2,4, 5,4 Гц), 4,91 (1H, дд, J=2,4, 10,8 Гц).
Соединение 8b
Выход: 26% (общий выход для указанных выше двух стадий)
ESI MS m/z 318,0 (M-H)-.
1H ЯМР (600 МГц, D2O) δ 2,12 (2H, м), 2,48 (2H, т, J=7,2 Гц), 2,72 (1H, д, J=14,4 Гц), 2,76 (1H, дд, J=10,2, 14,4 Гц), 2,89 (1H, дд, J=9,6, 14,4 Гц), 3,05 (1H, д, J=14,4 Гц), 3,78 (1H, т, J=6,0 Гц), 4,42 (1H, д, J=9,6 Гц), 4,91 (1H, д, J=10,2 Гц).

Пример 6: Синтез соединения 9
D-цистин (5,20 ммоль) растворяли в 60% водном растворе перхлорной кислоты (2,1 мл), в полученный раствор по каплям добавляли трет-бутилацетат (12,6 мл), смесь (реакционная жидкость) перемешивали при комнатной температуре в течение двух суток, реакционную жидкость охлаждали льдом, и значение pH жидкости корректировали до уровня приблизительно 11, используя 4 н. водный раствор гидроксида натрия. Температуру реакционной жидкости приводили обратно к комнатной температуре, реакционную жидкость экстрагировали 6 раз с использованием этилацетата (50 мл), и полученные органические фазы объединяли с последующей сушкой объединенной органической фазы над сульфатом натрия и последующей концентрацией органической фазы, чтобы таким образом получить соединение 9 в виде маслянистого продукта.
Выход: 72%.
ESI MS m/z 353,2 (M+H)+.
1H ЯМР (400 МГц, CDCl3) δ: 1,48 (18H, с), 2,88 (2H, дд, J=8,0, 13,2 Гц), 3,14 (2H, дд, J=4,4, 13,2 Гц), 3,69 (2H, дд, J=4,4, 8,0 Гц).

Пример 7: Синтез соединения 10
Соединение 9 (3,70 ммоль), полученное в примере 6, растворяли в тетрагидрофуране (40 мл), и затем в полученный раствор добавляли Fmoc-OSc (N-(9-флуоренил-метокси-карбонилокси)сукцинимид) (7,40 ммоль). Эту реакционную жидкость охлаждали льдом, в реакционную жидкость по каплям добавляли N-метилморфолин (7,46 ммоль), и смесь перемешивали в течение ночи без какой-либо обработки. В эту реакционную жидкость добавляли этилацетат (50 мл) и 10% водный раствор лимонной кислоты (25 мл), чтобы таким образом разделить смесь на различные фазы, чтобы получить органическую фазу. Полученную органическую фазу дополнительно промывали два раза 10% водным раствором лимонной кислоты (25 мл) и водным раствором хлорида натрия (25 мл), и затем концентрировали, чтобы получить остаток, похожий на суспензию. Похожий на суспензию остаток очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы таким образом получить соединение 10 в виде белого твердого вещества.
Выход: 54%.
ESI MS m/z 819,1 (M+Na)+.
1H ЯМР (400 МГц, CDCl3) δ: 1,48 (18H, с), 3,21 (4H, м), 4,20 (2H, м), 4,35 (4H, м), 4,56 (2H, м), 5,72 (2H, д, J=7,2 Гц), 7,28 (4H, м), 7,38 (4H, м), 7,28 (4H, д, J=7,6 Гц), 7,28 (4H, д, J=7,6 Гц).

Пример 8: Синтез соединений 8c и 8d
Повторяли те же процедуры, используемые в примерах 1-5, за исключением использования соединения 10, описанного в примере 7, в качестве исходного вещества, чтобы таким образом синтезировать соединения 8c и 8d.
Соединение 8c
ESI MS m/z 317,9 (M-H)-.
1H ЯМР (600 МГц, D2O) δ: 2,10 (2H, м), 2,44 (2H, м), 2,59 (1H, ушир.д, J=14,4 Гц), 2,79 (1H, дд, J=10,4, 14,4 Гц), 2,98 (1H, дд, J=2,8, 14,8 Гц), 3,11 (1H, дд, J=6,0, 14,8 Гц), 3,80 (1H, т, J=6,0 Гц), 4,50 (1H, дд, J=2,8, 6,0 Гц), 4,81 (1H, дд, J=2,0, 10,4 Гц).
Соединение 8d
ESI MS m/z 317,8 (M-H)-.
1H ЯМР (600 МГц, D2O) δ: 2,09 (2H, м), 2,43 (2H, м), 2,67 (1H, дд, J=1,6, 14,4 Гц), 2,72 (1H, дд, J=9,6, 14,4 Гц), 2,84 (1H, дд, J=9,6, 14,4 Гц), 3,02 (1H, д, J=14,4 Гц), 3,77 (1H, т, J=6,0 Гц), 4,42 (1H, дд, J=1,6, 9,6 Гц), 4,87 (1H, дд, J=2,4, 9,6 Гц).

Пример 9: Синтез соединения 11
К H-D-Asp-OMe (α-метиловый эфир D-аспарагиновой кислоты) (6,81 ммоль) добавляли тетрагидрофуран (14 мл) и воду (14 мл), чтобы растворить первое в последних. В полученный раствор добавляли раствор, полученный посредством растворения Boc2O (ди-трет-бутилдикарбонат) (8,38 ммоль) в тетрагидрофуране (5 мл), триэтиламине (13,63 ммоль) и DMAP (N,N-диметил-4-аминопиридин) (1,36 ммоль) в условиях охлаждения льдом. Температуру смеси (реакционной жидкости) приводили обратно к комнатной температуре, смесь перемешивали в течение 6 часов, и реакционную жидкость концентрировали, чтобы таким образом удалить тетрагидрофуран. К оставшейся реакционной жидкости добавляли 1-2 н. раствор соляной кислоты, чтобы корректировать ее значение pH до уровня приблизительно 2, и затем реакционную жидкость экстрагировали с использованием этилацетата (50 мл). Полученную органическую фазу промывали водным раствором хлорида натрия (25 мл) и затем концентрировали, чтобы таким образом получить предполагаемое соединение 11 в виде клейкого продукта.
Выход: 76%.
1H ЯМР (400 МГц, CDCl3) δ: 1,48 (9H, с), 2,91 (1H, дд, J=4,4, 8,0 Гц), 3,10 (1H, дд, J=3,6, 8,0 Гц), 3,79 (3H, с), 4,61 (1H, м), 5,52 (1H, ушир.д, J=8,8 Гц).

Пример 10: Синтез соединения 13
(Стадия 1): Соединение 11 (5,19 ммоль), полученное в примере 9, растворяли в этилацетате (21 мл), и затем в полученный раствор добавляли HOSu (N-гидроксисукцинимид) (5,73 ммоль). После добавления DCC (дициклогексилкарбодиимид) (5,71 ммоль) в смесь при охлаждении льдом, температуру полученной реакционной жидкости приводили обратно к комнатной температуре, и жидкость перемешивали в течение 4 часов. Нерастворимое вещество, отделенное от реакционной жидкости, отфильтровывали, и фильтрат концентрировали, чтобы таким образом получить гелеобразный остаток, содержащий соединение 12. Полученный остаток использовали в последующей реакции без какой-либо предварительной обработки, исходя из того, что выход соединения 12 полагали равным 100%.

(Стадия 2): Смешанную жидкость, содержащую тетрагидрофуран (20 мл) и воду (5 мл), охлаждали льдом, и затем в смешанную жидкость добавляли борогидрид натрия (9,47 ммоль) с последующим перемешиванием полученной смеси в течение 10 минут и последующим постепенным добавлением по каплям раствора соединения 12 (эквивалент 5,19 ммоль), полученного выше, в тетрагидрофуране (20 мл). Через 10 минут насыщенный водный раствор хлорида аммония (12 мл) добавляли в указанную выше реакционную систему, и затем температуру реакционной системы приводили обратно к комнатной температуре. Реакционную систему экстрагировали три раза с использованием этилацетата (30 мл × 3), чтобы получить органическую фазу, последнюю затем концентрировали, и полученный остаток очищали с использованием колонки с силикагелем (дихлорметан-метанол), чтобы таким образом получить соединение 13 в виде гелеобразного продукта.
Выход: 67% (общий выход для указанных выше 2 стадий).
1H ЯМР (400 МГц, CDCl3) δ: 1,47 (9H, с), 1,63 (1H, м), 2,17 (1H, м), 3,73 (2H, м), 3,80 (3H, с), 4,50 (1H, м), 5,39 (1H, м).

Пример 11: Синтез соединения 14
Соединение 13 (3,47 ммоль), полученное в примере 10, растворяли в безводном дихлорметане (10 мл), и полученный раствор затем по каплям добавляли в раствор трифенилфосфина (4,18 ммоль), имидазола (4,17 ммоль) и йода (4,16 ммоль) в безводном метиленхлориде (10 мл). После перемешивания смеси (реакционная жидкость) при комнатной температуре в течение 2 часов, реакционную жидкость концентрировали, чтобы получить остаток, и в полученный остаток добавляли этилацетат (35 мл). После перемешивания смеси в форме суспензии в течение одного часа, нерастворимое вещество удаляли посредством фильтрования, и затем фильтрат концентрировали, чтобы таким образом получить масло коричневого цвета. Затем масло очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы получить соединение 14 в виде маслянистого продукта.
Выход: 56%.
1H ЯМР (400 МГц, CDCl3) δ: 1,47 (9H, с), 2,20 (1H, м), 2,45 (1H, м), 3,20 (2H, т, J=7,6 Гц), 3,79 (3H, с), 4,38 (1H, м), 5,13 (1H, ушир.с).

Пример 12: Синтез соединения 15
Fmoc-Cys-Ot-Bu (трет-бутиловый эфир N-флуоренил-метоксикарбонил-L-цистеина) (1,90 ммоль) растворяли в безводном диметилформамиде (10 мл) с последующим добавлением в полученный раствор раствора соединения 14 (1,94 ммоль), полученного в примере 11, в безводном диметилформамиде (10 мл). Затем карбонат цезия (1,92 ммоль) добавляли в полученную смесь, эту смесь (реакционная жидкость) перемешивали при комнатной температуре в течение 5 часов, и реакционную жидкость разделяли на фазы посредством добавления этилацетата (20 мл) и 10% водного раствора лимонной кислоты (10 мл), чтобы таким образом получить органическую фазу. Оставшуюся водную фазу снова экстрагировали с использованием этилацетата (20 мл), органические фазы объединяли, и затем промывали 10% водным раствором лимонной кислоты (10 мл) и насыщенным водным раствором хлорида натрия (10 мл). Полученную органическую фазу концентрировали, чтобы получить маслянистый остаток, и последний очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы получить соединение 15 в виде маслянистого продукта.
Выход: 62%.
1H ЯМР (400 МГц, CDCl3) δ 1,45 (9H, с), 1,51 (9H, с), 1,92 (1H, м), 2,12 (1H, м), 2,63 (2H, м), 2,99 (2H, м), 3,74 (3H, с), 4,26 (1H, т, J=7,2 Гц), 4,41 (2H, м), 4,50 (1H, м), 5,17 (1H, ушир.), 7,34 (2H, м), 7,43 (2H, т, J=7,2 Гц), 7,65 (2H, д, J=7,2 Гц), 7,79 (2H, д, J=7,6 Гц).

Пример 13: Синтез соединения 16
(Стадия 1): Соединение 15 (0,55 ммоль), полученное в примере 12, растворяли в безводном дихлорметане (8 мл), затем в раствор добавляли трифторуксусную кислоту (4 мл), и смесь (реакционная жидкость) перемешивали при комнатной температуре в течение 2 часов. Эту реакционную жидкость концентрировали, и в концентрат добавляли безводный диметилформамид (4 мл), и смесь отгоняли в виде азеотропной смеси, чтобы таким образом получить раствор в диметилформамиде, содержащий соединение 16. Полученную азеотропную смесь использовали в последующей реакции без какой-либо предварительной обработки, исходя из того, что выход соединения 16 полагали равным 100%.

(Стадия 2): Дополнительный безводный диметилформамид (24 мл) добавляли в раствор, содержащий соединение 16 (эквивалент 0,55 ммоль), в диметилформамиде, и затем в смесь добавляли PyBOP (бензотриазол-1-ил-окси-трис-пирролидинофосфонийгексафторфосфат) (0,82 ммоль) и WSC·HCl (1-(3-диметиламинопропил)-3-этоксикарбодиимидгидрохлорид) (0,83 ммоль), при перемешивании. Затем в полученную реакционную систему добавляли триэтиламин (0,65 ммоль) с последующим перемешиванием реакционной системы при комнатной температуре в течение 24 часов и концентрированием реакционной жидкости. В другой контейнер добавляли воду (10 мл) и этилацетат (10 мл), полученный смешанный растворитель перемешивали, и указанный выше концентрат реакционной жидкости добавляли в смешанный растворитель. Затем полученную смесь промывали и экстрагировали с использованием этилацетата (10 мл), чтобы таким образом получить органическую фазу. Оставшуюся водную фазу дополнительно экстрагировали два раза с использованием этилацетата (10 мл × 2), полученные таким образом органические фазы объединяли, и объединенную органическую фазу промывали насыщенным водным раствором бикарбоната натрия (10 мл) и насыщенным водным раствором хлорида натрия (10 мл). К белому твердому веществу, полученному после концентрирования органической фазы, добавляли этилацетат, чтобы получить суспензию, с последующим перемешиванием суспензии и удалением нерастворимого вещества посредством фильтрования. Маслянистый остаток, полученный посредством концентрирования фильтрата, очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы получить соединение 17 в виде белого твердого вещества.
Выход: 15% (общий выход для указанных выше двух стадий).
ESI MS m/z 257,5 (M+H)+.

Пример 14: Синтез соединения 19
(Стадия 1): К соединению 17 (0,08 ммоль), полученному в примере 13, добавляли 5% раствор морфолин/диметилформамид (0,70 мл), смесь (реакционная жидкость) перемешивали при комнатной температуре в течение одного часа, и затем реакционную жидкость концентрировали, чтобы получить раствор, содержащий соединение 18. Полученный раствор использовали в последующей реакции без какой-либо предварительной обработки, исходя из того, что выход соединения 18 полагали равным 100%.

(Стадия 2): Boc-Glu-Ot-Bu (α-трет-бутиловый эфир N-трет-бутоксикарбонил-L-глутаминовой кислоты) (0,096 ммоль) растворяли в безводном диметилформамиде (1 мл) и затем в полученный раствор добавляли HOBt·H2O (1-гидроксибензотриазолгидрат) (0,12 ммоль) и WSC·HCl (1-(3-диметиламинопропил)-3-этоксикарбодиимидгидрохлорид) (0,12 ммоль) с последующим перемешиванием полученной смеси (реакционная жидкость) при комнатной температуре в течение 10 минут. В эту реакционную жидкость добавляли раствор соединения 18 (эквивалент 0,08 ммоль) в безводном диметилформамиде (2 мл) с последующим перемешиванием полученной смеси при комнатной температуре в течение ночи. К остатку, полученному посредством концентрирования реакционной жидкости, добавляли этилацетат (20 мл) и воду (10 мл), чтобы разделить жидкость на фазы. Полученную органическую фазу промывали насыщенным водным раствором бикарбоната натрия (10 мл) и насыщенным водным раствором хлорида натрия (10 мл) и затем концентрировали. Полученный остаток очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы таким образом получить соединение 19 в виде клейкого продукта.
Выход: 90%.
1H ЯМР (400 МГц, CDCl3) δ: 1,46 (9H, с), 1,48 (9H, с), 1,88 (1H, м), 2,40-2,15 (5H, м), 2,50 (1H, м), 2,78 (1H, дд, J=10,0, 14,4 Гц), 3,09 (1H, дд, J=4,8, 15,6 Гц), 3,37 (1H, дд, J=4,8, 14,4 Гц), 3,68 (1H, т, J=4,8 Гц), 3,79 (3H, с), 4,18 (1H, м), 4,51 (1H, м), 5,01 (1H, м), 5,20 (1H, ушир.д, J=6,8 Гц).

Пример 15: Синтез соединения 21
(Стадия 1): Соединение 19 (0,092 ммоль), полученное в примере 14, растворяли в тетрагидрофуране (1,84 мл), в полученный раствор добавляли 0,2 н. водный раствор гидроксида лития (0,18 ммоль) при охлаждении льдом, температуру смеси приводили обратно к комнатной температуре и смесь (реакционная жидкость) перемешивали при этой температуре в течение одного часа. После подтверждения исчезновения исходных веществ с помощью способа TLC, 0,2 н. раствор соляной кислоты добавляли в реакционную жидкость, чтобы контролировать ее значение pH на слабокислом уровне, с последующей концентрацией реакционной жидкости, чтобы удалить тетрагидрофуран. Оставшуюся жидкость экстрагировали три раза с использованием этилацетата (10 мл × 3), и полученную органическую фазу промывали насыщенным водным раствором хлорида натрия. Органическую фазу концентрировали, чтобы получить клейкое соединение 20, в виде смеси диастереомеров. Полученную смесь диастереомеров использовали в последующей реакции без какой-либо предварительной обработки, исходя из того, что его выход полагали равным 100%.

(Стадия 2): К соединению 20 (эквивалент 0,092 ммоль), полученному на указанной выше стадии 1, добавляли 4 н. раствор соляной кислоты/диоксана (1,8 мл), и смесь (реакционная жидкость) перемешивали при комнатной температуре в течение ночи. Остаток, полученный посредством концентрирования реакционной жидкости, растворяли в воде, и водный раствор пропускали через анионную ионообменную смолу (Amberlite IRA 400 OH AG). После промывания смолы ионообменной водой, ее элюировали с использованием 1-3 н. раствора уксусной кислоты с последующей лиофилизацией элюата, чтобы таким образом получить соединение 21 в виде смеси диастереомеров.
Выход: 56% (в виде общего выхода для указанных выше двух стадий).
ESI MS m/z 334,0 (M+H)+.
1H ЯМР (400 МГц, D2O) δ 2,07 (2H, м), 2,42 (2H, м), 2,86-3,17 (2H, м), 3,75 (1H, т, J=6,0 Гц), 4,27-4,41 (1H, м), 4,75-5,24 (1H, м).

Пример 16: Синтез соединения 22
(Boc-L-Cys-OH)2 (N,N'-ди-трет-бутоксикарбонил-L-цистин) (2,51 ммоль) растворяли в тетрагидрофуране (29,2 мл) и воде (0,8 мл) и затем в раствор добавляли трибутил-фосфин (2,76 ммоль) в условиях охлаждения льдом. Температуру этой реакционной жидкости приводили обратно к комнатной температуре с последующим перемешиванием реакционной жидкости в течение одного часа при этой температуре и концентрации реакционной жидкости. В полученный остаток добавляли этилацетат (20 мл) и 10% водный раствор лимонной кислоты (10 мл), чтобы фракционировать остаток, и полученную органическую фазу промывали насыщенным водным раствором хлорида натрия (20 мл). Органическую фазу концентрировали, и полученный остаток очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы таким образом получить соединение 22 в виде маслянистого продукта.
Выход: 99%.
ESI MS m/z 220,1 (M-H)-.
1H ЯМР (400 МГц, CDCl3) δ: 5,47 (1H, ушир.с), 4,65 (1Н, ушир.с), 3,09-2,97 (2H, м), 1,48 (9H, с).

Пример 17: Синтез соединения 23
Соединение 18 (4,97 ммоль), полученное в примере 14 (стадия 1), и ангидрид уксусной кислоты (49,90 ммоль) объединяли, полученную смесь охлаждали льдом, и в охлажденную льдом смесь по каплям добавляли раствор гидрокарбоната калия (5,93 ммоль) в воде (2,4 мл). Температуру реакционной системы (указанная выше смесь) приводили обратно к комнатной температуре с последующим перемешиванием реакционной системы в течение 2 часов и добавлением воды (5 мл) и этилацетата (20 мл) для экстрагирования, которое приводило к формированию водной фазы и органической фазы. Полученную водную фазу дополнительно экстрагировали с использованием этилацетата (20 мл), чтобы получить дополнительную органическую фазу. Эти органические фазы объединяли вместе и затем промывали насыщенным водным раствором хлорида натрия (5 мл). Объединенную органическую фазу концентрировали, и полученный остаток очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы таким образом получить соединение 23 в виде клейкого продукта.
Выход: 76%.
ESI MS m/z 261,9 (M-H)-.
1H ЯМР (400 МГц, CDCl3) δ 5,33 (1H, д, J=6,4 Гц), 4,53 (1H, м), 3,47 (1H, дд, J=4,0, 14,0 Гц), 3,34 (1H, дд, J=6,8, 14,0 Гц), 2,40 (3H, с), 1,48 (9H, с).

Пример 18: Синтез соединения 24
Соединение 23 (3,76 ммоль), полученное в примере 17, растворяли в безводном диметилформамиде (30 мл) с последующим добавлением в полученный раствор HOBt·H2O (1-гидроксибензотриазолгидрат) (4,14 ммоль) и CMC (1-циклогексил-3-(2-морфолиноэтил)-карбодиимид мето-п-толуолсульфонат) (4,13 ммоль) и перемешиванием полученной смеси при комнатной температуре в течение 15 минут. Впоследствии, L-Thr-OMe·HCl (метиловый эфир L-треонина·HCl) (3,77 ммоль) и триэтиламин (3,80 ммоль) добавляли в смесь, и полученную смесь (реакционная жидкость) перемешивали при комнатной температуре в течение 2 часов. Реакционную жидкость концентрировали, и в полученный остаток добавляли воду (20 мл) и этилацетат (40 мл), чтобы фракционировать ее и чтобы получить органическую фазу. Органическую фазу промывали водным раствором бикарбоната натрия (20 мл) и насыщенным водным раствором хлорида натрия (20 мл), и затем органическую фазу концентрировали. Полученный остаток очищали с использованием колонки с силикагелем (дихлорметан-метанол), чтобы таким образом получить соединение 24 в виде белого твердого вещества.
Выход: 74%.
ESI MS m/z 401,3 (M+Na)+.
1H ЯМР (400 МГц, CDCl3) δ 7,14 (1H, д, J=7,6 Гц), 5,37 (1H, д, J=7,2 Гц), 4,60 (1H, дд, J=2,8, 8,8 Гц), 4,37 (2H, м), 3,80 (3H, с), 3,39 (1H, дд, J=4,4, 14,0 Гц), 3,24 (1H, дд, J=8,0, 14,0 Гц), 2,40 (3H, с), 1,47 (9H, с), 1,24 (3H, д, J=6,4 Гц).

Пример 19: Синтез соединений 25a и 25b
Соединение 24 (4,20 ммоль), полученное в примере 18, растворяли в безводном дихлорметане (5 мл), и затем в полученный раствор добавляли диизопропилэтиламин (8,38 ммоль) и метансульфонилхлорид (8,00 ммоль). После перемешивания смеси (реакционная жидкость) при комнатной температуре в течение полутора часов, реакционную жидкость концентрировали, чтобы таким образом получить маслянистый остаток. С другой стороны, гидрид литийалюминия (33,6 ммоль) добавляли в безводный тетрагидрофуран (50 мл) при охлаждении льдом. Затем безводный метанол (101,48 ммоль) постепенно по каплям добавляли в смесь. В эту реакционную жидкость дополнительно добавляли безводный тетрагидрофуран (50 мл), и смесь дополнительно перемешивали. Через 10 минут раствор в безводном тетрагидрофуране (15 мл), содержащий остаток, полученный выше, по каплям добавляли в указанную выше отдельно полученную смесь, и эту реакцию продолжали в течение 2 часов. Эту реакционную жидкость добавляли в смешанную жидкость, содержащую этилацетат (150 мл) и 0,5 н. раствор соляной кислоты (120 мл), малыми порциями, чтобы таким образом фракционировать реакционную жидкость. Полученную таким образом органическую фазу промывали 0,5 н. раствором соляной кислоты (120 мл) и насыщенным водным раствором хлорида натрия (120 мл), и затем концентрировали. Полученный остаток очищали с использованием колонки с силикагелем (н-гексан-этилацетат), чтобы таким образом получить два соединения 25a и 25b, которые являлись изомерами по отношению друг к другу.
25a: Выход 13%; ESI MS m/z 341,1 (M+Na)+;1H ЯМР (400 МГц, CDCl3) δ: 6,50 (1H, д, J=5,6 Гц), 6,01 (1H, д, J=5,2 Гц), 4,87 (1H, дд, J=1,2, 6,0 Гц), 4,61 (1H, м), 3,87 (3H, с), 3,30 (1H, м), 2,95 (1H, дд, J=10,0, 14,8 Гц), 2,76 (1H, дд, J=1,2, 14,8 Гц), 1,47 (9H, с), 1,24 (3H, д, J=6,8 Гц).
25b: Выход 20%; ESI MS m/z 341,4 (M+Na)+;1H ЯМР (400 МГц, CDCl3) δ: 6,05 (1H, д, J=8,8 Гц), 5,99 (1H, ушир.с), 4,63 (1H, м), 4,15 (1H, м), 3,87 (3H, с), 3,48 (1H, м), 2,73 (2H, м), 1,49 (3H, д, J=7,2 Гц), 1,47 (9H, с).

Пример 20: Синтез соединения 27
(Стадия 1): 4 н. раствор соляной кислоты/диоксана (2,26 мл) добавляли к соединению 25a (0,45 ммоль), полученному в примере 19, и смесь (реакционная жидкость) перемешивали при комнатной температуре в течение ночи. Затем реакционную жидкость концентрировали, чтобы таким образом получить остаток, содержащий соединение 26. Полученный остаток использовали в последующей реакции без какой-либо предварительной обработки исходя из того, что выход соединения 26 полагали равным 100%.

(Стадия 2): Boc-Glu-Ot-Bu (α-трет-бутиловый эфир N-трет-бутоксикарбонил-L-глутаминовой кислоты) (0,52 ммоль) растворяли в безводном диметилформамиде (4 мл), затем в полученный раствор добавляли HOBt·H2O (1-гидроксибензотриазолгидрат) (0,65 ммоль) и WSC·HCl (1-(3-диметиламинопропил)-3-этоксикарбодиимидгидрохлорид) (0,66 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 10 минут. К этой смеси добавляли раствор соединения 26 (эквивалент 0,45 ммоль), полученного в указанной выше стадии 1, в безводном диметилформамиде (2,5 мл), и затем триэтиламин (0,77 ммоль), и смесь (реакционная жидкость) перемешивали при комнатной температуре в течение 2 часов. Реакционную жидкость концентрировали, затем к полученному остатку добавляли этилацетат (40 мл) и воду (20 мл), чтобы фракционировать его, и полученную органическую фазу промывали насыщенным водным раствором бикарбоната натрия (20 мл) и насыщенным водным раствором хлорида натрия (20 мл). Органическую фазу затем концентрировали, и полученный остаток очищали с использованием колонки с силикагелем (дихлорметан-метанол), чтобы таким образом получить соединение 27.
Выход: 98% (общий выход для этих двух стадий).
ESI MS m/z 526,1 (M+Na)+;1H ЯМР (400 МГц, CDCl3) δ: 7,02 (1H, м), 6,53 (1H, д, J=6,0 Гц), 5,18 (1H, д, J=7,6 Гц), 4,88 (1H, дд, J=1,2, 5,6 Гц), 4,81 (1H, м), 4,19 (1H, м), 3,87 (3H, с), 3,32 (1H, м), 2,90 (1H, дд, J=10,0, 14,8 Гц), 2,78 (1H, дд, J=2,0, 14,8 Гц), 2,33 (2H, м), 2,21 (1H, м), 1,90 (1H, м), 1,49 (9H, с), 1,46 (9H, с), 1,25 (3H, д, J=6,8 Гц).

Пример 21: Синтез соединения 29
(Стадия 1): Соединение 27 (0,38 ммоль), полученное в примере 20 (стадия 2), растворяли в тетрагидрофуране (7,6 мл), затем при охлаждении льдом в раствор добавляли 0,2M водный раствор гидроксида лития (0,76 ммоль), и смесь (реакционная жидкость) перемешивали в течение 2 часов. Затем в реакционную жидкость добавляли 0,5 н. раствор соляной кислоты, чтобы сделать реакционную жидкость слабокислой, температуру реакционной жидкости приводили обратно к комнатной температуре, и затем реакционную жидкость концентрировали, чтобы удалить тетрагидрофуран. Оставшуюся жидкость экстрагировали три раза с использованием этилацетата (20 мл × 3), полученную органическую фазу промывали насыщенным водным раствором хлорида натрия (20 мл) и затем концентрировали, чтобы получить соединение 28. Полученный концентрат или остаток использовали в последующей реакции без какой-либо предварительной обработки, исходя из того, что выход соединения 28 полагали равным 100%.

(Стадия 2): 4 н. раствор соляной кислоты/диоксана (7,2 мл) добавляли в соединение 28 (эквивалент 0,38 ммоль), полученное на указанной выше стадии 1, и смесь (реакционная жидкость) перемешивали при комнатной температуре в течение ночи. Реакционную жидкость концентрировали, полученный остаток растворяли в воде, и затем полученный водный раствор пропускали через анионную ионообменную смолу (Amberlite IRA 400 OH AG). Смолу промывали ионообменной водой с последующим элюированием в соответствии со способом элюирования в градиенте с использованием 1-3 н. раствора уксусной кислоты и лиофилизацией полученного элюата, чтобы получить соединение 29 в виде белого твердого вещества.
Выход: 65% (общий выход для этих двух стадий).
ESI MS m/z 333,6 (M+H)+;1H ЯМР (400 МГц, D2O) δ: 4,86 (1H, дд, J=2,8, 9,6 Гц), 4,81 (1H, д, J=1,6 Гц), 3,82 (1H, т, J=6,4 Гц), 3,40 (1H, м), 2,95 (1H, дд, J=9,6, 15,2 Гц), 2,60 (1H, дд, J=2,8, 15,2 Гц), 2,46 (2H, т, J=7,2 Гц), 2,08 (2H, м), 1,14 (3H, д, J=7,2 Гц).

Пример 22: Синтез соединения 35

(1) Получение соединения 31
2,6-Диаминопимелиновую кислоту (5,0 г, 26,3 ммоль) растворяли в 42 мл метанола, и затем в полученный раствор медленно по каплям добавляли тионилхлорид (4,2 мл, 57,9 ммоль) при охлаждении льдом. После завершения добавления по каплям, температуре смеси позволяли спонтанно расти до комнатной температуры, и затем смесь перемешивали в течение ночи. После завершения реакции, растворитель отгоняли из реакционной системы, чтобы получить соединение 31 в количественном выходе.
(2) Получение соединения 32
Соединение 31 (0,582 г, 2,0 ммоль), полученное выше, растворяли в смешанном растворителе, содержащем 10 мл воды и 10 мл метанола, затем в раствор добавляли оксид серебра (0,730 г, 3,2 ммоль), и его перемешивали при комнатной температуре в течение ночи. После завершения реакции оксид серебра удаляли посредством фильтрования через целлит, а растворитель отгоняли из фильтрата, чтобы таким образом получить соединение 32 в качестве сырья.
(3) Получение соединения 33
В 5 мл N,N-диметилформамида растворяли Boc-Glu-Ot-Bu (0,303 г, 1,0 ммоль), 1-гидроксибензотриазолмоногидрат (0,169 г, 1,1 ммоль), 1-этил-3-(3-диметиламинопропил)-карбодиимидгидрохлорид (0,211 г, 1,1 ммоль) и соединение 32 (0,205 г, 1,1 ммоль), полученное выше, затем в полученный раствор добавляли триэтиламин (0,198 мл, 1,4 ммоль), и полученную смесь (реакционная жидкость) перемешивали при комнатной температуре в течение ночи. После завершения реакции, растворитель отгоняли из реакционной жидкости, а оставшуюся жидкость разводили посредством добавления этилацетата. Разведенную жидкость промывали два раза 5% водным раствором лимонной кислоты и затем один раз насыщенным водным раствором хлорида натрия, затем промывали два раза 10% насыщенным водным раствором бикарбоната натрия и наконец один раз насыщенным водным раствором хлорида натрия. Полученную органическую фазу сушили над сульфатом магния с последующим ее фильтрованием и удалением растворителя посредством дистилляции, чтобы таким образом получить соединение 33 в качестве неочищенного продукта.
(4) Получение соединения 34
Соединение 33 (0,277 г, 0,59 ммоль), полученное выше, растворяли в 5 мл тетрагидрофурана, 4 мл 1M водного раствора гидроксида лития затем добавляли в полученный раствор, и смесь (реакционная жидкость) перемешивали при комнатной температуре в течение 2 часов. После завершения реакции, pH реакционной жидкости корректировали приблизительно до 2 посредством добавления 1M водного раствора соляной кислоты, и затем добавляли в реакционную жидкость этилацетат, чтобы экстрагировать ее. Полученную органическую фазу промывали насыщенным водным раствором хлорида натрия, затем сушили над сульфатом магния, последний удаляли фильтрованием, и из фильтрата отгоняли растворитель, чтобы таким образом получить соединение 34.
(5) Получение соединения 35
4 н. раствор соляной кислоты-диоксана добавляли к соединению 34 (0,181 г, 0,38 ммоль), полученному выше, и реакцию между ними продолжали при комнатной температуре в течение ночи. После завершения реакции, растворитель отгоняли, чтобы получить остаток, и часть остатка, содержащего соединение 35, очищали посредством препаративной ВЭЖХ с обращенной фазой (колонка: Develosil RPAQUEOUS-AR-5, доступна от NOMURA Chemical Co., Ltd.; подвижная фаза: линейный градиент воды, содержащей систему 0,1% гептафтормасляная кислота/ацетонитрил), чтобы таким образом получить соединение 35 в виде смеси диастереомеров.
1H ЯМР (D2O) δ: 1,41-1,78 (м, 3H), 1,80-1,91 (м, 2H), 1,99-2,03 (м, 1H), 2,08-2,22 (м, 2H), 2,42-2,51 (м, 2H), 3,97-4,02 (м, 1H), 4,31-4,34 (м, 1H), 4,45-4,49 (м, 1H).
MS(ESI) m/z: 302,0 (M+1).
Пример 23: Получение экспрессирующей CaSR плазмиды:
Получение экспрессирующей CaSR плазмиды осуществляли в соответствии со следующими процедурами:
Синтезировали синтетические олигомерные ДНК [прямой праймер (последовательность № 3: ACTAATACGACTCACTATAGGGACCATGGCATTTTATAGCTGCTGCTGG)] и обратный праймер (последовательность № 4: TTATGAATTCACTACGTTTTCTGTAACAG), для использования в процедурах ПЦР, основываясь на последовательности ДНК, зарегистрированной в NCBI [CaSR (кальциевый рецептор): NM_000388, последовательности №№ 1 и 2], в качестве матрицы.
Процедуры ПЦР осуществляли с использованием указанных выше праймеров и ДНК полимеразы Pfu Ultra (доступна от Stratagene Company) при следующих условиях с использованием кДНК, полученной из почки человека (доступна от Clontech Company) в качестве материала или матрицы. В этой связи последовательность цикла репликации содержала стадии обработки системы при 94°C в течение 3 минут, затем при 94°C в течение 30 секунд, при 55°C в течение 30 секунд и при 72°C в течение 2 минут, где последние три стадии повторяли более 35 раз, а последнюю стадию продолжения реакции при 72°C в течение 7 минут. Процедуры электрофореза осуществляли с помощью электролита, удерживаемого в агарозном геле, с последующим окрашиванием продуктов ПЦР, подвергавшихся электрофорезу, окрашивающим ДНК средством и последующим обнаружением, посредством облучения лучами УФ света, произошла ли предполагаемая амплификация или нет. Кроме того, длины цепей продуктов ПЦР также подтверждали посредством их сравнения с таковыми ДНК маркеров, каждый из которых имел известный размер и подвергался процедурам электрофореза одновременно.
Плазмидный вектор pBR322 разрезали рестрикционным ферментом EcoRV (доступен от Takara Company), и фрагменты генов, амплифицированные посредством указанных выше процедур ПЦР, соединяли с плазмидным вектором в его участке разрезания с использованием набора для лигирования (доступен от Promega Company). Штамм Escherichia coli DH5α трансформировали этим реакционным раствором, за чем следовал отбор трансформанта, который сохранял плазмиду, которая способна подвергаться клонированию с продуктом, амплифицированным в ПЦР, и затем продукт, амплифицированный в ПЦР, подтверждали в соответствии анализом последовательности оснований ДНК.
Эту рекомбинантную плазмиду использовали для конструирования или создания экспрессирующей CaSR человека плазмиды: hCaSR/pcDNA3.1.
Пример 24: Оценка (1) активности агониста CaSR
Клетки 293E (экспрессирующие EBNA1 клетки HEK293, ATCC № CRL-10852) культивировали в DMEM/Хэма-F12 (3,15/мл содержащая глюкозу модифицированная Дульбекко среда Игла; доступна от NAKARAI TESK Company), содержащей 10% эмбриональной телячьей сыворотки в присутствие 200 мкг/мл G418 (доступно от Genetisine Company). Культивируемые клетки инокулировали в колбу F25 при плотности 3×106клеток/10 мл, затем содержимое колбы оставляли стоять в течение 24 часов в CO2 инкубаторе (5% CO2, 37°C), и затем экспрессирующую CaSR человека плазмиду: hCaSR/pcDNA3.1 трансфицировали с использованием средства для трансфекции Fugene 6 (доступно от Roche Company). После того как колбы оставляли стоять в CO2 инкубаторе в течение 6-7 часов, клетки извлекали с использованием содержащей 10% эмбриональную телячью сыворотку DMEM/Хэма-F12 и затем инокулировали в каждую лунку покрытого поли-D-лизином 96-луночного планшета (BD-Biocoat) при плотности 70000 клеток/лунка.
После того, как планшет с лунками оставляли стоять в CO2 инкубаторе в течение 24 часов, среду для культивирования удаляли из каждой лунки 96-луночного планшета, в которые инокулировали клетки, в каждую лунку добавляли раствор Ca2+ флуоресцентного индикатора Calcium 4 Assay Kit (доступен от Molecular Devices Company) в Assay Buffer (содержит 146 мМ NaCl, 5 мМ KCl, 1 мМ MgSO4, 1 мг/мл глюкозы, 20 мМ HEPES (pH 7,2) и от 0,75 до 1,25 мМ CaCl2) в количестве 200 мкл/лунка, и каждую лунку оставляли стоять при 37°C в течение одного часа и затем при комнатной температуре в течение 10 минут, чтобы таким образом заставить клетки захватить индикатор.
В каждую лунку 96-луночного планшета добавляли раствор тестируемого соединения в содержащем 0,1% БСА Assay Buffer в количестве 50 мкл/лунка, и лунки проверяли на какое-либо изменение в интенсивности флуоресценции, испускаемой из них в течение 3 минут с использованием FLEX Station (доступна от Molecular Devices Company).
Способ вычисления EC50
Вычисляли разность (ОЕФ (макс-мин) между максимальной и минимальной интенсивностями флуоресценции, наблюдаемой до и после добавления тестируемого соединения, в соответствии с автоматическим вычислением с помощью FLEX Station. В этом отношении вычисляли степень активности тестируемого соединения, причем значение ОЕФ (макс-мин), наблюдаемое при добавлении тестируемого соединения в максимальной концентрации, определяли равным 100%, а значение ОЕФ (макс-мин), наблюдаемое при использовании содержащего 0,1% БСА Assay Buffer, не содержащего какое-либо добавленное тестируемое соединение, определяли равным 0%, затем аппроксимацию кривой осуществляли с использованием программного обеспечения Xfit для электронных таблиц или GraphPad Prism, чтобы таким образом определить каждое соответствующее значение EC50 в виде концентрации каждого тестируемого соединения, при которой наблюдают степень активности 50%.
Пример 25: Оценка (2) активности агониста CaSR
Клетки 293E (экспрессирующие EBNA1 клетки HEK293, ATCC № CRL-10852) культивировали в DMEM/Хэма-F12 (3,15/мл содержащая глюкозу модифицированная Дульбекко среда Игла; доступна от NAKARAI TESK Company), содержащей 10% эмбриональную телячью сыворотку в присутствие 200 мкг/мл G418 (доступно от Genetisine Company). Культивируемые клетки инокулировали в колбу F25 при плотности 3 × 106 клеток/10 мл, затем содержимое колбы оставляли стоять в течение 24 часов в CO2 инкубаторе (5% CO2, 37°C), и затем экспрессирующую CaSR человека плазмиду: hCaSR/pcDNA3.1 трансфицировали с использованием средства для трансфекции Fugene 6 (доступно от Roche Company). После того как колбу оставляли стоять в CO2 инкубаторе в течение 6-7 часов, клетки извлекали с использованием содержащей 10% эмбриональную телячью сыворотку DMEM/Хэма-F12 и затем инокулировали в каждую лунку покрытого поли-D-лизином 384-луночного планшета (BD-Biocoat) при плотности 2×106 клеток/лунка.
После того как планшет с лунками оставляли стоять в CO2 инкубаторе в течение 24 часов, среду для культивирования удаляли из каждой лунки 384-луночного планшета, в которые инокулировали клетки, в каждую лунку добавляли раствор Ca2+ флуоресцентного индикатора Calcium 5 Assay Kit (доступен от Molecular Devices Company) в Assay Buffer (содержит 146 мМ NaCl, 5 мМ KCl, 1 мМ MgSO4, 1 мг/мл глюкозы, 20 мМ HEPES (pH 7,2) и от 0,75 до 1,25 мМ CaCl2 и 2,5 мМ пробенецида (доступен от SIGMA Company)) в количестве 40 мкл/лунка, и каждую лунку оставляли стоять при 37°C в течение 45 минут и затем при комнатной температуре в течение 15 минут, чтобы таким образом заставить клетки захватить индикатор.
В каждую лунку 384-луночного планшета добавляли раствор тестируемого соединения в содержащем 0,1% БСА Assay Buffer в количестве 10 мкл/лунка, и лунки проверяли на какое-либо изменение в интенсивности флуоресценции, испускаемой из них в течение 3 минут с использованием FLIPR (доступно от Molecular Devices Company).
Способ вычисления EC50
Вычисляли разность (ОЕФ (макс-мин) между максимальной и минимальной интенсивностями флуоресценции, наблюдаемой до и после добавления тестируемого соединения в соответствии с автоматическим способом вычисления FLIPR. В этом отношении вычисляли степень активности тестируемого соединения, причем значение ОЕФ (макс-мин), наблюдаемое при добавлении тестируемого соединения в максимальной концентрации, определяли равным 100%, а значение ОЕФ (макс-мин), наблюдаемое при использовании содержащего 0,1% БСА Assay Buffer, не содержащего какое-либо добавленное тестируемое соединение, определяли равным 0%, затем аппроксимацию кривой осуществляли с использованием программного обеспечения Xfit для электронных таблиц или GraphPad Prism, чтобы таким образом определить каждое соответствующее значение EC50 в виде концентрации каждого тестируемого соединения, при которой наблюдают степень активности 50%.
В следующей таблице 1 представлены результаты, определенные в соответствии с указанными выше способами (1) и (2):
Все соединения в соответствии с настоящим изобретением проявляли желаемую активность агониста CaSR. Среди них соединение 8b в качестве предпочтительного производного лантионина в соответствии с настоящим изобретением проявляло чрезвычайно высокую активность агониста CaSR, которая превышала приблизительно в 9 раз таковую γ-Glu-Cys, о котором известно, что он обладает активностью агониста CaSR.
Пример 26: Оценка придающей кокуми активности
Соединение 8b проверяли на интенсивность придающей кокуми активности в соответствии с тестом на количественную органолептическую оценку.
Тест на количественную органолептическую оценку в настоящем документе осуществляли в соответствии со следующими процедурами: интенсивность придающей кокуми активности определяли в случае, когда тестируемое соединение (в количестве в диапазоне от 0,0001 до 0,0005 г/дл) смешивали с дистиллированной водой, содержащей глутамат натрия (0,05 г/дл), инозинмонофосфат (инозиновая кислота) (0,05 г/дл) и хлорид натрия (0,5 г/дл). Значение pH используемых образцов корректировали до такового в контроле, не содержащем какое-либо тестируемое соединение (т.е., значение pH последнего ±0,2). В этом отношении критерии оценки задавали следующим образом: 0: оценка тестируемого соединения, действие которого эквивалентно контролю; 3: оценка тестируемого соединения, действие которого сильнее контроля; и 5: оценка тестируемого соединения, действие которого значительно сильнее контроля. Кроме того, чтобы сделать критерий оценки более ясным, устанавливали следующие стандарты: 2,5: начально-средний вкус, наблюдаемый для γ-Glu-Val-Gly; и 3,0: его остаточный вкус, и оценку осуществляли с использованием 5 дегустаторов для каждого оценочного теста (n=5). В этой связи интенсивность кокуми, которую наблюдали для γ-Glu-Val-Gly в концентрации 0,001 г/дл, соответствует интенсивности кокуми (3,0: начально-средний вкус; 3,0: остаточный вкус), которую наблюдали для γ-Glu-Cys-Gly (глутатион) в концентрации 0,01 г/дл. Оценку осуществляли с использованием способа с линейной шкалой, в котором каждую соответствующую оценку наносили на прямую линию, на которой ясно и точно указаны оценки, равные -5, 0 и +5. Кроме того, в качестве дегустаторов в этих оценочных тестах выбирали людей, которые связаны с разработкой приправ в течение суммарного периода по меньшей мере полгода и которые, таким образом, могли судить, что разница между γ-Glu-Cys-Gly и γ-Glu-Val-Gly, каждый из которых добавляли в раствор, обладающий вкусом умами и соленым вкусом, составляла приблизительно 10 раз (эту способность подтверждали через регулярные интервалы). В этом отношении, термин «начально-средний вкус» обозначает вкус, обнаруживаемый в течение времени в диапазоне от 0 до 5 секунд после держания каждого образца во рту каждого дегустатора, и термин «остаточный вкус» обозначает то, что обнаруживали впоследствии. Соединение 8b, используемое в этих тестах, показало свою придающую кокуми активность в широком диапазоне указанных выше добавляемых концентраций, но результаты, наблюдаемые при типичных концентрациях, приведены в следующей таблице 2.
Кроме того, в таблице 2 также приведены результаты, полученные, когда γ-Glu-Val-Gly оценивали в соответствии с аналогичными процедурами.
Указанные выше результаты ясно указывают на то, что соединение по настоящему изобретению проявляет превосходную придающую кокуми активность, даже в очень низкой концентрации. Это должно быть достаточно полезно с производственной точки зрения.
Пример 27: Оценка соединений в соответствии с настоящим изобретением по придающей кокуми активности в случае высушенного экстракта скумбриевых
Соединение 8b проверяли на интенсивность его придающей кокуми активности в соответствии с тестом на количественную органолептическую оценку.
Тест на количественную органолептическую оценку осуществляли в соответствии со следующими процедурами: интенсивность придающей кокуми активности тестируемого соединения определяли с использованием смеси, полученной посредством разведения коммерчески доступного высушенного экстракта скумбриевых (эквивалент 50% бульона высушенных скумбриевых) горячей водой до концентрации 20,0 г/дл, с добавления хлорида натрия (0,5 г/дл) в разведенный экстракт, чтобы получить раствор высушенного экстракта скумбриевых и затем введения тестируемого соединения в раствор в количестве в диапазоне от 0,0001 до 0,0005 г/дл. Значение pH используемых образцов корректировали до такового контроля, не содержащего какое-либо тестируемое соединение (т.е., значение pH последнего ±0,2). В этом отношении критерии оценки задавали следующим образом: 0: оценка тестируемого соединения, действие которого эквивалентно контролю; 3: оценка тестируемого соединения, действие которого сильнее контроля; и 5: оценка тестируемого соединения, действие которого значительно сильнее контроля. Кроме того, чтобы сделать критерий оценки более ясным, устанавливали следующие стандарты: 2,5: начально-средний вкус, наблюдаемый для γ-Glu-Val-Gly; и 3,0: его остаточный вкус, и оценку осуществляли с использованием 5 дегустаторов для каждого оценочного теста (n=5). В этой связи интенсивность кокуми, которую наблюдали для γ-Glu-Val-Gly в концентрации 0,001 г/дл, соответствует интенсивности кокуми (3,0: начально-средний вкус; 3,0: остаточный вкус), которую наблюдали для γ-Glu-Cys-Gly (глутатион) в концентрации 0,01 г/дл. Оценку осуществляли с использованием способа с линейной шкалой, в котором каждую соответствующую оценку наносили на прямую линию, на которой ясно и точно указаны оценки, равные -5, 0 и +5. Кроме того, в качестве дегустаторов в этих оценочных тестах выбирали людей, которые связаны с разработкой приправ в течение суммарного периода по меньшей мере полгода и которые, таким образом, могли судить, что разница между γ-Glu-Cys-Gly и γ-Glu-Val-Gly, каждый из которых добавляли в раствор, обладающий вкусом умами и соленым вкусом, составляла приблизительно 10 раз (эту способность подтверждали через регулярные интервалы). В этом отношении, термин «начально-средний вкус» обозначает вкус, обнаруживаемый в течение времени в диапазоне от 0 до 5 секунд после держания каждого образца во рту каждого дегустатора, и термин «остаточный вкус» обозначает то, что обнаруживали впоследствии. Соединение 8b, используемое в этих тестах, показало свою придающую кокуми активность в широком диапазоне указанных выше добавляемых концентраций, но результаты, наблюдаемые при типичных концентрациях, приведены в следующей таблице 3.
Кроме того, в таблице 3 также приведены результаты, полученные, когда γ-Glu-Val-Gly оценивали в соответствии с аналогичными процедурами.
По отношению даже к высушенному экстракту скумбриевых обнаружено, что производное лантионина в соответствии с настоящим изобретением может служить в качестве превосходного придающего кокуми средства, которое проявляет превосходную придающую кокуми активность, даже в очень низкой концентрации. Высушенные скумбриевые широко используются, например, в мисо супе, супе для тонкой лапши, пшеничной вермишели и концентрированном бульоне для вермишели. Более конкретно, соединение по настоящему изобретению делает возможным улучшение вкусового ощущения различных продуктов питания, в которых используют высушенные скумбриевые, при низкой стоимости и в очень малом количестве. Соответствующим образом, соединение по настоящему изобретению должно быть достаточно полезным с производственной точки зрения.
Пример 28: Оценка соединений в соответствии с настоящим изобретением по придающей кокуми активности в случае коровьего молока
Соединение 8b проверяли на интенсивность его придающей кокуми активности в соответствии с тестом на количественную органолептическую оценку.
Тест на количественную органолептическую оценку осуществляли в соответствии со следующими процедурами: интенсивность придающей кокуми активности тестируемого соединения определяли с использованием смеси, полученной посредством добавления в коммерчески доступное коровье молоко (содержание жира: 3,6%) тестируемого соединения в количестве в диапазоне от 0,0001 до 0,0005 г/дл. Значение pH используемых образцов корректировали до такового контроля, не содержащего какое-либо тестируемое соединение (т.е., значение pH последнего ±0,2). В этом отношении критерии оценки задавали следующим образом: 0: оценка тестируемого соединения, действие которого эквивалентно контролю; 3: оценка тестируемого соединения, действие которого сильнее контроля; и 5: оценка тестируемого соединения, действие которого значительно сильнее контроля. Кроме того, чтобы сделать критерий оценки более ясным, устанавливали следующие стандарты: 2,5: начально-средний вкус, наблюдаемый для γ-Glu-Val-Gly; и 3,0: его остаточный вкус, и оценку осуществляли с использованием 5 дегустаторов для каждого оценочного теста (n=5). В этой связи интенсивность кокуми, которую наблюдали для γ-Glu-Val-Gly в концентрации 0,001 г/дл, соответствует интенсивности кокуми (3,0: начально-средний вкус; 3,0: остаточный вкус), которую наблюдали для γ-Glu-Cys-Gly (глутатион) в концентрации 0,01 г/дл. Оценку осуществляли с использованием способа с линейной шкалой, в котором каждую соответствующую оценку наносили на прямую линию, на которой ясно и точно указаны оценки, равные -5, 0 и +5. Кроме того, в качестве дегустаторов в этих оценочных тестах выбирали людей, которые связаны с разработкой приправ в течение суммарного периода по меньшей мере полгода и которые, таким образом, могли судить, что разница между γ-Glu-Cys-Gly и γ-Glu-Val-Gly, каждый из которых добавляли в раствор, обладающий вкусом умами и соленым вкусом, составляла приблизительно 10 раз (эту способность подтверждали через регулярные интервалы). В этом отношении, термин «начально-средний вкус» обозначает вкус, обнаруживаемый в течение времени в диапазоне от 0 до 5 секунд после держания каждого образца во рту каждого дегустатора, а термин «остаточный вкус» обозначает то, что обнаруживали впоследствии. Соединение 8b, используемое в этих тестах, показало свою придающую кокуми активность в широком диапазоне указанных выше добавляемых концентраций, но результаты, наблюдаемые при типичных концентрациях, приведены в следующей таблице 4.
Кроме того, в таблице 4 также приведены результаты, полученные, когда γ-Glu-Val-Gly оценивали в соответствии с аналогичными процедурами.
По отношению даже к коровьему молоку обнаружено, что производное лантионина в соответствии с настоящим изобретением может служить в качестве превосходного придающего кокуми средства, которое может проявлять превосходную придающую кокуми активность, даже в очень низкой концентрации. Коровье молоко широко используется, например, в ингредиентах различных продуктов питания, напитков, кондитерских изделий и ферментированных продуктов питания. Более конкретно, соединение по настоящему изобретению делает возможным улучшение вкуса и ощущения на небе различных продуктов питания, в которых используют коровье молоко, при низкой стоимости и в очень малом количестве.
Соответствующим образом, соединение по настоящему изобретению должно быть достаточно полезным с производственной точки зрения.
Реферат
Изобретение относится к соединению общей формулы (I) или к его пищевой соли:, где каждый R1 и R2 независимо представляет собой атом водорода или низшую алкильную группу, содержащую от 1 до 3 углеродных атомов; А представляет собой метиленовую группу или оксигруппу; и X представляет собой алкиленовую группу, содержащую от 1 до 5 углеродных атомов, при условии, что одна из метиленовых групп, присутствующих в алкиленовой группе, может быть заменена на тиогруппу, оксигруппу, и что алкиленовая группа может быть дополнительно замещена 1-6 алкильными группами, каждая из которых содержит от 1 до 3 углеродных атомов. Изобретение также относится к пищевой композиции, обеспечивающей эффект кокуми, на основе указанного соединения и к промежуточному соединению для получения соединения формулы (I). Технический результат: получено новое соединение и композиция на его основе, которые могут найти применение в пищевой промышленности в качестве придающего кокуми средства. 4 н. и 7 з.п. ф-лы, 4 табл., 28 пр.
Формула

где каждый R1 и R2 независимо представляет собой атом водорода или низшую алкильную группу, содержащую от 1 до 3 углеродных атомов;
А представляет собой метиленовую группу или оксигруппу; и
X представляет собой алкиленовую группу, содержащую от 1 до 5 углеродных атомов, при условии, что одна из метиленовых групп, присутствующих в алкиленовой группе, может быть заменена на тиогруппу, оксигруппу, и что алкиленовая группа может быть дополнительно замещена 1-6 алкильными группами, каждая из которых содержит от 1 до 3 углеродных атомов.



где каждый R1' и R2' независимо представляет собой атом водорода или алкильную группу, содержащую от 1 до 3 углеродных атомов;
R3' представляет собой атом водорода, алкильную группу, содержащую от 1 до 4 углеродных атомов, бензильную группу или 9-флуоренилметильную группу;
R4' представляет собой трет-бутоксикарбонильную группу, бензилоксикарбонильную группу или 9-флуоренилметилоксикарбонильную группу;
R5' представляет собой гидроксильную группу, алкоксигруппу, содержащую от 1 до 4 углеродных атомов, бензилоксигруппу, аминогруппу (-NH2) или алкиламиногруппу, содержащую от 1 до 3 углеродных атомов;
А представляет собой метиленовую группу или оксигруппу; и
Х представляет собой алкиленовую группу, содержащую от 1 до 5 углеродных атомов, при условии, что одна из метиленовых групп, присутствующих в алкиленовой группе, может быть заменена на тиогруппу, оксигруппу, и что алкиленовая группа может быть дополнительно замещена 1-6 алкильными группами, каждая из которых содержит от 1 до 3 углеродных атомов.
Документы, цитированные в отчёте о поиске
Лиганды для рецепторов, связанных с g-белком
Комментарии