Производные l-аргинина, способ их получения, фармацевтическая композиция, обладающая ингибирующей no-синтазу активностью - RU2168493C2
Код документа: RU2168493C2
Чертежи




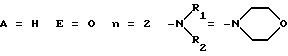
Описание
Изобретение относится к производным L-аргинина, способу их получения и фармацевтическим составам на их основе.
Соединения по настоящему изобретению действуют как биологические блокаторы NO-синтазы. С учетом потенциальной роли NO-синтазы в физиопатологии (С.Монкала, Р.М.Дж.: Пальмер с сотр. Окись азота: физиология, патофизиология и фармакология. Фармацевтические обзоры, т. 43, N 2, стр. 109-142)(S.Monca1a, R.M.J; Palmer et a1, Nitric Oxid: Physiology, Pathophysiology and Pharmacology. Pharmaceutical Reviews, vol. 43, N 2, pp. 109-142) такие соединения могут представлять интерес в качестве гипотензивных, антибактериальных, иммуносупрессивных, антиатеросклеротических, вазотропных, анальгезирующих, антимигреневых, офтальмологических или антидиабетических препаратов. Таким образом, применение этих соединений можно рассматривать как активный подход в медикаментозном лечении патологических состояний центральной и периферической нервной системы, таких как церебральные инфаркты, мигрени и головные боли, эпилепсия, церебральные или спинальные травмы, нейродегенеративные и/или аутоиммунные заболевания, такие как болезнь Альцгеймера, заболевание Паркинсона, пляска святого Вита, амиотрофический боковой склероз, инфекционные церебральные нейропатии (БИЧ), острые и хронические боли, совместимость и привыкание к производным морфина, психотропным лекарствам и алкоголю, глазная нейропатия и депрессия. Более того, м.б. рассмотрено применение этих соединений при лечении других заболеваний, которые относятся к дисфункциям гастроинтестинальной мочевой системы воспалительного и невоспалительного типа, таких как язвенный колит, гастрит, болезнь Крона, нарушение мочеиспускания, пищеводно-желудочный рефлюкс, понос, но также патологий, связанных с сердечно-сосудистой или бронхиальной системой, таких как гипертензия, атеросклероз, фиброз легкого, склеродермия, астма, легочная гипертензия, цирроз, диабеты, воспалительных или инфекционных заболеваний, таких как артрозы, полиартриты, сепсис, васкулиты или даже при нарушении эрекции, приапизма или контрацепции.
Изобретение относится к производным
L-аргинина общей формулы 1:
где А представляет собой атом водорода, низший алкил или нитрогруппу; E представляет собой атом кислорода или ковалентную связь; n равно нулю или целому числу от 1 до 12 и либо R1 и R2 независимо представляют собой разветвленный или линейный низший алкил, либо R1 и R2 образуют вместе с атомом азота, к которому они присоединены, пяти- или шестичленное кольцо, насыщенное или ненасыщенное, формулы

в котором X представляет собой атом кислорода, серы или азота, имино, алкилимино или метиленовую группу, за исключением соединений общей формулы 1, в которых n равно нулю, E представляет собой ковалентную связь и либо А представляет собой атом водорода и R1 и R2 независимо представляют собой низшую алкильную группу или образуют вместе пиперадиновое, морфолиновое или имидазольное ядро, или А представляет собой нитрогруппу и R1 и R2 образуют пиперадиновое ядро.
Выражение "низший алкил" используется для обозначения алкильных групп с числом атомов углерода от 1 до 6. Предпочтение отдается алкильным группам, линейным или разветвленным, которые содержат от одного до четырех атомов углерода, выбираемым из таких, как метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутилгруппы.
R1 и R2 вместе с атомом азота, к которому они присоединены, могут образовывать насыщенное или ненасыщенное пяти- или шестичленное кольцо. Таким образом, получаемые кольца могут выбираться из числа таких, как пиррол, пиперидин, пирролидин, имидазол, имидиазолин, пиразол, пиразолидин, пиридин, пиперазон, пиразин, пиримидин, пиридазин, изотиазол или морфолин.
Более предпочтительно настоящее изобретение относится к соединениям общей формулы 1, в которой n представляет собой целое число, равное от 1 до 12, и предпочтительно, когда E представляет атом кислорода, A представляет нитрогруппу или атом водорода и R1 и R2 образуют морфолиновое, пиперидиновое или имидазольное кольцо. Аналогично, изобретение относится более предпочтительно к соединению общей формулы 1, в которой n равно нулю, E представляет ковалентную связь, А - нитрогруппу и R1 и R2 образуют морфолиновое ядро.
Изобретение также касается фармацевтически приемлемых солей производных по настоящему изобретению. Соли образуются из органических или неорганических кислот, таких как соляная кислота, бромистоводородная кислота, уксусная, фумаровая, сульфоновая, толуолсульфоновая, малеиновая, карбоновая или фосфорная кислота. В качестве карбоновых кислот могут применяться, например, ацетилсалициловая, салициловая, мефенаминовая, ибупрофеновая, сулиндак [(Z)-5-фтор-2-метил-1-[пара- (метилсульфинил)-бензилиден] инден-3-уксусная кислота; препарат фирмы Мерк энд Ko] или индометациновая кислота.
В литературе описаны соединения общей формулы (1), в которой n равно нулю, E представляет собой ковалентную связь, либо А представляет собой атом водорода, R1 и R2 вместе образуют пиперидиновое или морфолиновое кольцо, или А представляет собой нитрогруппу и R1 и R2 образуют пиперидиновое кольцо. В европейском журнале "Медицинская химия", т. 23, N 6 (1988), "Биохимия", т. 23, N 1 (1984 г.) (European Journal of Medicinal Chemistry, vol. 23, N 6 (1988), Biocemistry, vol. 23, N 1 (1984), патентные заявки FR 2320088 и FR 2240720) эти производные использовались для получения производных L-аргинина в качестве антитромботических препаратов. Соединение формулы 1, в котором n равно нулю, E представляет собой ковалентную связь, А представляет собой атом водорода и R1 и R2 вместе образуют имидазольное кольцо, описано в качестве промежуточного вещества при синтезе (Origin of Life, vol. 14 (1984), pp. 351-357). Однако ни о какой фармакологической активности этих соединений в этих источниках не говорится. Активность других производных формулы 1, в которых n равно нулю, E представляет собой ковалентную связь, А представляет собой атом водорода и R1 и R2 представляют собой независимо низшую алкильную группу, сравнивали с активностью инсулина (Z. Naturforsch. Teil. С., vol. 28, N 5-6, 1973; Hope-Seyler's Z.Physiol. Chem., vol. 353, # 11, 1972, pp. 1661-1670).
Но область действия тромбозов и диабетов существенно отличается от области действия NO-синтазы, раскрываемой настоящим изобретением.
Настоящее
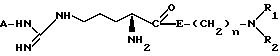
изобретение касается способа получения соединений формулы 1, и этот способ включает реакцию соединения общей формулы (2):
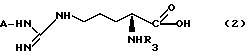
в которой А представляет низшую алкильную группу или нитрогруппу и R3 - защитную группу, с соединением общей формулы (3):
H-E-(CH2 )n-NR1R2 (3)
в которой n, E, R1 и R2 определены выше, в присутствии сочетающего агента, при температуре в диапазоне от 0 до 30oC, с последующим удалением защитной группы R3 из полученного соединения общей формулы (4):

чтобы получить соединение общей формулы (1), в которой А представляет собой нитрогруппу или низший алкил. При необходимости соединение общей формулы (1), в котором А представляет собой атом водорода, можно получить из соответствующего соединения общей формулы 1, в котором А представляет собой нитрогруппу, отщеплением нитрогруппы; отщепление может быть осуществлено гидрогенолизом.
Исходные соединения (3) и (2), в которых А представляет собой нитрогруппу, известны и выпускаются промышленностью: соединение (2), в котором А представляет собой низшую алкильную группу, может быть получено известным способом (Synth. Commun. 21 (1), 99 (1991)). Первую стадию осуществляют при температуре в диапазоне от 0 до 30oC и предпочтительно в диапазоне от 0o C до температуры окружающей среды. Используемый растворитель должен растворять производное нитроаргинина формулы 2: предпочтительно используется ДМФ. Реакцию осуществляют в присутствии сочетающего агента. В качестве сочетающего агента могут применяться таковые, обычно используемые в пептидном синтезе; так, могут использоваться N,N-дициклогексилкарбодиимид(DСС), бензотриазол-1-ил-окситрис(диметиламино)фосфоний гексафторфосфат (ВОР) или бензотриазол-1-ил-окси-трис-пирролидинофосфоний гексафторфосфат (РуВОР). Также возможно применение вместе с сочетающим агентом добавок, таких как, например, 1-гидроксибензотриазол (HOBt) или 4-диметил-аминопиридина (DМАР), которые в пептидном синтезе используются для ускорения реакции сочетания, в которой принимает участие карбодиимид.
В качестве защитной группы R3 может использоваться любая защитная группа, которая обычно применяется при пептидном синтезе, например бензилоксикарбонил (Z) и трет-бутоксикарбонил (ВОС). При получении соединения (1), в котором А представляет атом водорода, R3 предпочтительно представляет собой группу, которая может отщепляться в условиях гидрогенолиза, используемого для отщепления нитрогруппы. В этом случае отщепление группы R3 и нитрогруппы может осуществляться одновременно.
Изобретение также относится к
фармацевтическим смесям, включающим эффективное количество по крайней мере одного соединения общей формулы 1:

в которой А - атом водорода, низшая алкильная группа или нитрогруппа; E - атом кислорода или ковалентная связь; n равно нулю или целому числу от 1 до 12 и либо R1 и R2 независимо представляют разветвленные или линейные алкильные цепи, либо R1 и R2 вместе с атомом азота, с которым они связаны, образуют пяти- или шестичленное кольцо, насыщенное или ненасыщенное, формулы

в которой X представляет собой атом кислорода, серы или азота, имино, алкилимино или метиленовую группу, за исключением соединений общей формулы 1, в которой n равно нулю, E представляет собой ковалентную связь и либо А - атом водорода и R1 и R2 независимо представляют собой низшую алкильную группу или вместе образуют имидазольное, морфолиновое или пиперидиновое кольцо, либо А представляет собой нитрогруппу и R1 и R2 образуют пиперидиновое кольцо, или соль этих соединений в сочетании по крайней мере с одним разбавителем или фармацевтически приемлемым носителем.
Настоящее изобретение
относится к фармацевтическим составам, содержащим в качестве существенного признака эффективное количество по крайней мере одного соединения формулы 1A:

в которой А представляет собой атом водорода, низшую алкильную или нитрогруппу; E представляет собой атом кислорода или ковалентную связь; n равно нулю или целому числу от одного до 12 и либо R1 и R2 независимо представляют собой разветвленные или линейные низшие алкилы, или R1 и R2 вместе с атомом азота, с которым они связаны, образуют насыщенное или ненасыщенное пяти- или шестичленное кольцо формулы
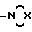
в которой X представляет собой атом кислорода, серы или азота, метиленовую, алкилимино или иминогруппу, за исключением соединений общей формулы 1A, в которой n равно нулю, E представляет собой ковалентную связь, А представляет собой атом водорода и R1 и R2 независимо представляют собой низшие алкильные группы, или соль этих соединений в сочетании по крайней мере с одним разбавителем или фармацевтически приемлемым носителем.
Фармацевтические композиции по настоящему изобретению могут подбираться в соответствии с принятой схемой лечения, особенно в формах, пригодных для орального или парентерального приема. Они могут формоваться в виде капсул, пилюль, гелей, растворов, принимаемых внутрь, растворов для инъекций или в формах с пролонгированным выделением. Соединение по настоящему изобретению может применяться в суточных дозах от 0,1 до 1000 мг, предпочтительно от 1 до 100 мг.
И наконец, предлагаемое изобретение касается использования производных L-аргинина общей формулы 1B:

в которой А представляет собой атом водорода, низший алкил или нитрогруппу; E представляет собой атом кислорода или ковалентную связь; n равно нулю или целому числу от 1 до 12 и либо R1 и R2 независимо представляют разветвленные или линейные низшие алкильные группы, либо R1 и R2 вместе с атомом азота, с которым они связаны, образуют насыщенное или ненасыщенное пяти- или шестичленное ядро формулы

в которой X представляет собой атом кислорода, серы или азота, метиленовую, алкилимино- или иминогруппу, в качестве медицинских препаратов для лечения патологий центральной и периферической нервной системы, заболеваний желудочно-кишечной и мочевой систем воспалительного и невоспалительного типа, патологий сердечно-сосудистой или бронхиальной системы.
Далее изобретение иллюстрируется следующими примерами.
ПРИМЕР 1. Сложный морфолиноэтиловый эфир L-NG-нитроаргинина (L-NNA)

1-я стадия
В 30 мл безводного диметилформамида (ДМФ) растворяли эквимолярную смесь (10-2 М) L-N-Вос-NG-нитроаргинина и N-β -гидроксиэтилморфолина, затем добавляли 3 x 10-3 М диметиламинопиридина (DMAP). Раствор охлаждали до 0oC и добавляли 10-2 М дициклогексилкарбодиимида (DDC). Смесь перемешивали 10 минут при 0oC и затем продолжали перемешивание при комнатной температуре 24 часа. Отфильтровывали образующийся осадок дициклогексилмочевины и удаляли растворитель. Остаток извлекали этилацетатом, однократно промывали водой и насыщенным раствором поваренной соли. При удалении растворителя получали вязкий продукт, который хроматографировали через кремнеземную колонку (элюирующий растворитель CHCl3/метанол 95:95, затем 90:10). Получали соединение (4) в виде масла, которое кристаллизовалось.
Точка плавления: 128oC.
CCM:rf: 0,38 (CHCl3/этанол 85:15)
МК-спектр (см-1), нуйол, γ - NH: 3400-3200, γ - сложный эфир: 1740; γ - BOC: 1710
Масс-спектр:
MH+ = 433
1H-ЯМР, 100 МГц, CDCI3, TMC, δ млн-1: 7,8 (2H, NH2); 5,5 (дуплет, 1H, NHBoc); 4,3 (мультиплет, 3H,


2-я стадия. Удаление защиты NH2 функции
Соединение (4), полученное на предшествующей стадии, растворяли в безводном диоксане и в диоксан при комнатной
температуре по каплям добавляли избыток 4 н. HCl. Раствор становился мутным, затем его энергично перемешивали в течение 15 часов, пока медленно не образовывался белый осадок. Диоксан декантировали;
дважды промывали безводным простым эфиром и сушили в роторном испарителе.
Твердое вещество растворяли в воде и лиофилизовали.
Соединение 1 получали в виде дихлоргидрата.
Точка плавления: 230oC, гигроскопичный белый порошок.
CCML:rf: 0,39 (2- пропанол/H2O/NH4OH 7:3:0,1)
MH+ = 333 (основ.)
[α0] = + 3,81 (H2O)
1H-ЯМР, 100 МГц, D2O, δ млн-1: 4,5 (мультиплет, 2H, CO2CH2); 4,1 (триплет, 1H,

ПРИМЕР 2. Сложный пиперидиноэтиловый эфир L-NNA
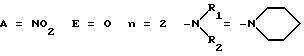
1-я стадия
Соответствующее соединение общей формулы (4) получали способом, описанным в Примере 1 1-ой стадии, используя вместо N-(β-гидроксиэтил)-морфолина N-(β-гидроксиэтил)пиперидин. Получали белый порошок с точкой плавления 98oC.
CCM: rf: 0,22 (CHCl3-этанол 8:2)
1H-ЯМР, 100 МГц, CDCl3, TMC, δ млн-1: 2 7,
6 (2H, NH2); 5,5 (триплет, 1H,
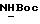

2-я стадия
Соединение 1 получали в виде дигидрохлорида по методике, описанной в Примере 1, 2-й стадии.
Точка плавления: 250oC, гигроскопичное белое вещество.
CCM:rf: 0,36 (2-пропанол/H2O/NH4OH 7:3:0,1)
MH+ = 331 (основ.)
[α0] = +5,78 (H2O)
1H-ЯМР, 100 МГц, D2O, δ млн-1: 4,5
(мультиплет, 2H, CO2CH2); 4,1 (триплет, 1H,
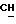
ПРИМЕР 3. 3-(диметиламино)пропиловый сложный эфир L-NNA
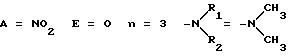
1-я стадия
Соответствующее соединение общей формулы (4) получали по методике, описанной в Примере 1 стадии 1, используя вместо N-(β-гидроксиэтил)-морфолина 3-(диметиламино)-пропанол. Соединение (4) очищали методом флеш-хроматографии (CHCl3/этанол 8:2).
Получали вязкий продукт.
CCM:rf: 0,17 (CHCl3/метанол 7:3)
1H-ЯМР, 100 МГц, CDCl3, TMC, δ млн-1: 5,4 (дуплет, 1H NHBoc); 4,2 (мультиплет, 3H,
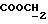



2-я стадия
Соединение 1 получали в виде дигидрохлорида по методике, описанной в Примере 1,
2-й стадии.
Точка плавления: 178oC, гидроскопический белый порошок.
CCM:rf: 0,28 (2-пропанол/H2O/NH4OH 7:3:0,1)
MH+
(основ.) = 378
1H-ЯМР, 100 МГц, D2O, δ млн-1: 1 4,15 (мультиплет, 3H, CH(NH2)CO,
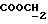

ПРИМЕР 4. Морфолинопропиловый сложный эфир L-NNA
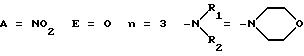
1-ая стадия
Следуя методике Примера 1 стадии 1 и используя вместо N-(β-гидроксиэтил)-морфолина 3-морфолинопропанол, получали соответствующее соединение (4). Последнее очищали методом флеш-хроматографии.
Точка плавления: 106oC.
CCM:rf: 0,34 (CHCl3/этанол 85:15)
1H-ЯМР,
100 МГц, CDCl3, TMC, δ млн-1: 7,8 (2H, NH2); 5,4 (дуплет, 1H,




2-я стадия
Следуя методике по Примеру 1 стадии 2, получали соединение 1 в виде дигидрохлорида.
Точка плавления: 232oC, гидроскопичный белый порошок.
CCM:rf: 0,39 (2-пропанол/H2O/NH4OH 7:3:0,1)
[α0] = +8,2 (H2O)
MH+ = 347 (основ.)
1H - ЯМР, 100 МГц, D2O, δ млн-1: 4,15 (мультиплет, 3H,


ПРИМЕР 5. Морфолиногексиловый сложный эфир L-NNA
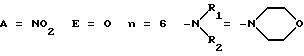
1-я стадия
Следуя методике по Примеру 1 стадии 1 и используя вместо N-(β-гидроксиэтил)-морфолина 6-морфолиногексанол, получали соответствующее соединение (4). Последнее очищали методом флеш-хроматографии (CHCl3/этанол 90:10).
Получали вязкий продукт.
Точка плавления: 99oC.
CCM:rf: 0,32 (CHCl3/этанол 85:14)
1H-ЯМР, 100 МГц, CDCl3, TMC, δ млн-1: 7,7 (2H, NH2); 5,4 (дуплет, 1H, NHBoc); 4,25 (мультиплет, 3H,

2-я стадия
Следуя методике Примера 1 стадии 2, получали дигидрохлорид соединения 1.
Точка плавления: 214oC, гигроскопичное соединение.
CCM:rf: 0,52 (2-пропанол/H2O/NH4OH 7:3:0,1)
MH+ = 389 (основ.)
[α0] = +6,67 (H2O)
1H-ЯМР, 100 МГц, D2O, δ млн-1: 4,1 (мультиплет, 3CH,

ПРИМЕР 6. Имидазолоэтиловый сложный эфир L-NNA

1-я стадия
Следуя методике Примера 1 стадии 1 и используя вместо N-(β -гидроксиэтил)-морфолина 2-гидроксиэтил-3-имидазол, получали соответствующее соединение (4). Последнее очищали на колонке с кремнеземом (CHCl3/этанол 9:1, затем 88:12).
Точка плавления: 102oC.
CCM:rf: 0,19 (CHCl3/этанол/NH4OH 85:15:0,1)
1H-ЯМР, 100 МГц, CDCl3, TMC, δ: 8 (2H, NH2); 7,6 (1H, NH); 7 (дуплет, 2H, имидазол); 5,7 (дуплет, 1H, NHBoc); 4,3-4,1 (мультиплет, 5H,



2-я стадия
Следуя методике, ранее описанной в Примере 1 стадии 2, получали целевое
соединение 1 в виде дигидрохлорида.
Точка плавления: 228oC, гигроскопичное желтое твердое вещество.
MH+ = 314 (основ.)
CCM:rf: 0,39 (2
пропанол/H2O/NH4OH 7:3:0,1)
1H-ЯМР, 100 МГц, D2O, δ млн-1: 7,3 (дуплет, 2H, имидазол); 4,4 (мультиплет, 4H,


ПРИМЕР 7. Морфолиноэтиловый сложный эфир L-аргинина

1-ая стадия
Следуя методике Примера 1 стадии 1, получали соответствующее соединение (4) путем реакции L-N(Z)-NG-нитроаргинина, т. е. соединения 2, в котором группа R3 представляет собой Z защитную группу, с N-(β-гидроксиэтил)-морфолином. Соединение очищали методом флеш-хроматографии (CHCl3/этанол 9:1, затем 89:11). Получали вязкий продукт.
Точка плавления: 65oC.
CCM:rf: 0,28 (CHCl3/EtOH 85:15)
1H-ЯМР, 100 МГц, CDCl3, TMC, δ млн-1: 8,5 (1H, NH); 7,3 (2H, NH2); 7,2 (синглет, 5H, φ); 5,7 (дуплет, 1H,


2-ая стадия
Приготовленное на предшествующих стадиях соединение (4) растворяли в этаноле и подвергали гидрогенолизу; гидрогенолиз осуществляли в аппарате
ПАРР в присутствии 10%
палладия на угле при давлении 344,7 КПа и окружающей температуре. Исчезновение предшественника определяли хроматографически, и реакция завершалась через четыре часа.
После фильтрования растворитель испаряли при 40oC. Вязкий остаток извлекали минимальным количеством этанола. Добавляли однонормальную соляную кислоту в серном эфире и смесь перемешивали три
часа. Медленно выделялся осадок. Твердое вещество декантировали и дважды промывали сухим эфиром. Твердый продукт сушили в вакууме и лиофилизовали. Получали слегка желтоватый продукт, содержащий
хлористоводородную кислоту, и этот продукт был очень гигроскопичен.
Точка плавления: выше 240oC (с разложением).
MH+ = 288 (основ.)
1H-ЯМР, 100 МГц, D2
O, δ млн-1: 4,4 (мультиплет, 2H, CO2CH2); 4,1 (триплет, 1H,

ПРИМЕР 8. Пиперидиноэтиловый сложный эфир L-аргинина

1-ая стадия
Соответствующее соединение общей формулы (4) может быть получено по методике, описанной в Примере 7 стадии 1, используя вместо N-(β-гидроксиэтил)-морфолина N-(β- гидроксиэтил)пиперидин. Соединение может быть очищено методом флеш-хроматографии.
2-ая стадия
Гидрохлорид соединения 1 может быть получен в соответствии с ранее
описанной методикой (Пример 7, стадия 2).
ПРИМЕР 9. N-имидазолоэтиловый сложный эфир L-аргинина

1-ая стадия
Соответствующее соединение общей формулы (4) может быть получено по методике, описанной в Примере 7 стадии 1, используя вместо N-(β -гидроксиэтил)-морфолина N-(β- гидроксиэтил)-пиперидин. Соединение может быть очищено методом флеш-хроматографии.
2-ая стадия
Гидрохлорид соединения 1 может быть
получен по методике Примера 7 стадии 2.
ПРИМЕР 10. Морфолиноэтиловый сложный эфир L-NG-метиларгинина (L-NMA)

1-ая стадия
Соответствующее соединение общей формулы (4) может быть получено по методике Примера 1 стадии 1, осуществляя реакцию L-N(Z)-NG-метиларгинина и N-(β-гидроксиэтил)- морфолина. Соединение может быть очищено хроматографически на колонке с силикагелем.
2-ая стадия
Гидрохлорид
соединения 1 можно получить в соответствии с методикой Примера 7 стадией 2.
ПРИМЕР 11. Морфолинамид L-NNA

1-ая стадия. Амид L-N-Boc-NG-нитроаргинина
В безводном диметилформамиде растворяли эквимолярные количества L-NBoc-NG -нитроаргинина, морфолина и 1-гидрокси-бензотриазола (HOBT), охлаждали до 0oC и добавляли эквивалент DDC. Смесь выдерживали при 0oC 10 минут. Реакцию продолжали в течение 20 минут при 0oC, затем шесть часов при температуре окружающей среды. После обычной обработки флеш-хроматографией получали масло, которое кристаллизовалось (элюент CHCl3/метанол 95:5).
CCM:rf: 0,66 (CHCl3/метанол (8:2)
ИК(нуйол)см-1: γNH 3400-3300; γBoc 1700; γамид 1630
1H-ЯМР, 100 МГц, CDCl3, TMC, δ млн-1: 5,8 (1H, дуплет, NHBoc); 3,7 (мультиплет, 1H,

2-ая стадия. Депротекция NH2 функции
Полученный на стадии 1 продукт (4) растворяли в безводном дихлорметане и
добавляли 20% трифторуксусной кислоты. Смесь перемешивали при температуре окружающей среды, при этом происходило выделение газа. Растворитель удаляли, и остаток извлекали водой. Раствор пропускали
через ионообменную смолу. Получали вязкий продукт, который кристаллизовался при добавлении ацетона.
Точка плавления: 246oC.
MH+ = 289
CCM:rf: 0,18 (CHCl3/метанол 8:2)
[α0] = +17,2 (вода)
ИК-спектр: нуйол, см-1: γNH 3400-3300; γамид 1630
1H-ЯМР, 100 МГц, D2O, δ млн-1: 3,8 (мультиплет, 1H,

ПРИМЕР 12. Соль ибупрофена и морфолиноэтилового сложного эфира L-NG-нитроаргинина
2 x 10-3 М ибупрофена растворяли в 15 мл воды,
содержащей 2 эквивалента гидроксида натрия; затем этот раствор медленно добавляли к водному раствору (7 мл) 2 x 10 М морфолиноэтилового сложного эфира L-NNA. Смесь становилась мутной; ее нагревали при
перемешивании до 60oC в течение 30 минут. Получали прозрачный раствор, который после охлаждения лиофилизовали, получая белый порошок.
Точка плавления: выше 260oC, соль водорастворима.
1H-ЯМР, 100 МГц, D2O, δ млн-1: 7 (4H, φ); 4,6 (мультиплет, CH, CHNCO и


Аналогичным способом получали соль сложного эфира по Примеру 1 с салициловой кислотой (Пример 13) и соль того же самого сложного эфира с сулиндаком (Пример 14). Все эти соли гигроскопичны.
ПРИМЕР 15. Соль ацетилсалициловой кислоты и морфолиноамида L-NNA
В 30 мл воды растворяли морфолинамид L-NNA. При перемешивании добавляли ацетилсалициловую кислоту и медленно
растворяли в смеси. Затем прозрачный раствор лиофилизовали и получали белый порошок.
Точка плавления: выше 240oC, соль водорастворима.
Аналогичным способом могут быть получены соли по Примеру 11 с другими карбоновыми кислотами, такими как салициловая кислота, ибупрофен или сулиндак. Все эти соли гигроскопичны.
Биологическая активность соединений по настоящему изобретению иллюстрируется следующими опытами.
1. Измерение биологической активности in vitro
1.1. Эндотелиальная системная NO-синтаза
Блокирование системной эндотелиальной NO-синтазы продуктов устанавливали по их способности действовать как антогонисты эндотелиальной релаксации под действием карбахола на выделенную аорту крысы,
предварительное сокращение которой вызывали фенилэфрином (Auguet et al., 1992).
Материалы и методы
Осуществляли экстракцию грудной аорты у мужских особей крыс Sprague Dawley
(290-350 г), умерщвленных путем разреза цервикальной артерии. Аорту помещали в глюкозусодержащую среду Кребса Хенселейта следующего состава:
компонент - мМ
хлорид натрия - 118
хлорид калия - 4,7
сульфат магния - 1,17
дигидрофосфат калия - 1,18
хлорид кальция - 2,5
бикарбонат натрия - 25
глюкоза - 11
Среду
выдерживали при 37oC и непрерывно пропускали поток карбогена (95% кислорода и 5% углекислого газа).
После удаления избытка соединительной ткани и жира сосуды осторожно разрезали на кольца (шириной 2 мм) и суспендировали при напряжении 2 г в 20 мл емкостях. Регистрация сокращений осуществлялась с помощью изометрических зондов, связанных с системой сбора информации (10S Дей-Лере).
Продукты и обработка
После часового отдыха повышали тонус артерии фенилэфрином (РЕ 10-6 М). Когда сокращения достигали максимума (10 минут), в
емкости вводили карбахол (10-5 М) для оценки целостности эндотелиального слоя. Препараты затем промывали и после отдыха в течение 45 минут повторно вводили в ванну РЕ (10-6 М)
при максимальной концентрации; при достижении максимального расслабления вводили 10-5 М карбахола. Испытуемые продукты вводили в ванну коммулятивными дозами.
Результаты представлены в Таблице 1.
1.2. Нейросистемная NO-синтаза
Введение
Измерение активности NO-синтазы выполнено в соответствии с методикой, описанной Бредом и Снайдером
(Bredt and Snyder) в 1990 г.
Материалы и методы
Быстро извлекали мозжечок у крысы Sprague Dawley (280 г Charles River), сушили при 4oC и гомогенизировали в буферном
объеме, соответствующем 5 мл буфера НЕРЕS на грамм ткани, используя тигель Томаса (Thomas' potter (10 round trips). Гомогенаты затем центрифугировали (21000• g в течение 15 минут при 4oC).
Плавающее вещество удаляли и пропускали через колонку с 1 мл Доуекс AG 50 WX-8 Na+(БиоРед форма), предварительно ополоснутую дистиллированной водой и экстракционным буфером. После прохождения через колонку образцы хранили при 4oC и быстро готовили дозы.
Дозы готовили в стеклянных трубках, в которые помещали 100 мкл выдержанного буфера, содержащего 100 мМ HEPES; pH 7,4 EDTA; 2,5 мМ хлорида кальция; 2 мМ дитиотреитола; 2 мМ восстановленного NADPH, 10 мкG/мл кальмодулина и 10 мкМ тетрагидробиоптерина. Добавляли 25 мкл раствора, содержащего 100 нМ меченного тритием аргинина (удельная активность: 209 1010 расп./ с ммоль (-56,4 Ci/ ммоль, Амершам)), и 40 мкМ нерадиоактивного аргинина. Реакцию запускали добавлением 50 мкл гомогената, причем конечный объем доводили до 200 мкл (недостающие 25 мкл - либо вода, либо испытуемый продукт).
После инкубации при комнатной температуре в течение 30 минут ферментативную реакцию обрывали добавлением 2 мл останавливающего буфера (20 мМ HEPES, pH 5,5; 2 мМ EDTA). Образцы пропускали через колонки объемом 1 мл со смолой Dowex AG 50 WX-8 Na + формы и элюировали 2 мл дистиллированной воды. После добавления 10 мл сцинтиллирующей жидкости определяли радиоактивность с помощью сцинтилляционного спектромера. Пустышки получали по той же методике, используя вместо 50 мкл образца 50 мкл экстрагирующего буфера. Для каждого испытания определяли общую радиоактивность.
Результаты выражали в пикомолях цитруллина, образующегося в минуту, в расчете на мг. Концентрацию протеина определяли в соответствии с методикой Бретфорда (1976) (Таблица 2).
1.3 Индуцируемая NO-синтаза
Макрофаги продуцируют окись
азота из индуцируемой NO-синтазы под действием противовоспалительного стимула. Микрофагом индуцируемая NO-синтаза получается из гомогената J774A1 миеломоноцитов, предварительно
стимулируемых в течение 48 часов LPS (E.coli) и IFNγ.
Материалы и метод
1. Клетки: культуры и индукции NO-синтазы
Клетки J774A1 (АТСС см.
TIB 67) культивировали в DMEM при 10% SVF, 37oC в атмосфере 5% углекислого газа. Их высевали из расчета 5 x 10 клеток/см2 в колбы 150 см2. Выращивание осуществляли в
присутствии LP3 (1 мкг/мл) и IFN-τ мурина (50 U/мл) в DMEM при 10% SVF в течение 24 часов.
2. Получение частично очищенной NO-синтазы
Клетки промывали PBS, затем
помещали на клеточный скрейпер в 4 мл холодного буфера А (4oC) (буфер А: HEPES 50 мМ, дитиотреитол 0,5 мМ при pH, доведен до 4 однонормальным едким натром). Добавляли без подготовки:
пепстатин А 1 мг/мл, леупептин 1 мг/мл, ингибитор трипсина из сои 1 мг/мл, антипаин 1 мг/мл, PMSF (Пефоблок) 10 мг/мл.
После центрифугирования (при 1000 g, 4oC в течение 5 минут) собирали центрифугированные отложения и помещали их в 1 мл буфера А, обрабатывали ультразвуком при 4oC, и гомогенат подвергали ультрацентрифугированию (100000 g, 4oC, 60 минут). К плавающему сверху материалу добавляли 10% глицерина перед замораживанием при -80oC, предварительно фракционированному. Препарат испытывали в тот же день, но активность его сохранялась после хранения в течение 10 дней при -80oC. Фракцию сохраняли для дозировки протеинов микрометодом по Бретфорду.
3. Ферментативное исследование
Исследование включало превращение под действием NO-синтазы L-аргинина в L-цитруллин.
а. Превращение L-аргинина в L-цитруллин
- Реакционный буфер В: HEPES 100 мМ, дитиотреитол
1 мМ, доведенный до pH 7,4 однонормальным едким натром. Этот буфер выдерживали в течение нескольких дней при 4oC. Сразу же добавляли софакторы NO-синтазы, а именно: 10 мкМ
тетрагидробиоптерина, 10 мкМ FAD, 2 мМ NADPH, 2,5 мМ хлорида кальция, 1 мг/мл BSA (чтобы сделать фермент растворимым).
- Раствор L-аргинина
Конечная концентрация L-аргинина 40
мкМ
Для продолжения реакции к раствору добавляли (3Н)-L-аргинин до конечной концентрации 100 нМ, получая изотопный раствор.
- Приготовление фермента
В
соответствии с испытанием, ранее проведенным для определения активности фермента, использовали чистый раствор или при 1/10 градуса.
- Ингибитор
Его готовили в В буфере из
концентрированного раствора в водной среде или диметилсфульфоксиде (ДМСО) и испытывали, сравнивая с контрольным без ингибитора. Контрольный ДМСО добавляли там, где необходимо.
Объем (мкл), используемый при проведении испытаний, приведен в табл. 3.
Культивирование осуществляли в двухкамерном испарителе при 37oC в течение 15 минут.
- Прекращение реакции
Реакцию обрывали добавлением 2 мл буфера С: 20 мМ HEPES, 2 нМ EDTA, pH 5,5 с помощью однонормальной соляной кислоты.
- Разделение L-аргинина и
L-цитруллина
Осуществляли на 0,5 мл катионообменной смолы Dowex 50Х-8, предварительно приведенной в равновесное состояние в буфере С в 2 мл шприцах, снабженных стеклянным шариком, который
пропускал только жидкую фазу.
Катионообменная смола

Собранную пробу сохраняли, и остаток элюировали из цитруллина 2 мл дистиллированной воды.
К 4 мл водной среды добавляли 16 мл Инстагель Плюс и сцинтилляционные колбы были измерены по Пакарду.
- Расчеты
Для каждой величины были выполнены следующие расчеты: [cpm (образец) - cpm (пустышка)] : [cpm (контрольный) - cpm (пустышка)].
Величина IC50 есть среднее значение из двух экспериментов, проведенных для разных ферментативных препаратов. Результаты представлены в табл. 4.
Измерение биологической активности in
vivo
Карагинановый отек
Принцип
Инъекция суспензии карагинана, мукополисахаридов водорослей, в подошвенную часть вызывала местную воспалительную реакцию.
Противовоспалительные соединения могут подавлять этот отек разным способом в соответствии с принципом их действия (Winter et al., 1962). Ингибиторы NO-синтазы показали определенный эффект в этом
эксперименте (Antunes et al., 1991).
Материалы и методы
Группу из восьми мужских особей крыс Sprague Dawley VAF (Charles River, St.Aubin les Elbeuf) массой 140-215 г морили
голодом в течение 18 часов.
Отек вызывали путем инъекций в плантар пульвинус одной из задних лап суспензии карагинана (1%) в физиологической сыворотке в количестве 0,1 мл на каждое животное.
Объем лап измеряли плетисмографически до и после полутора и трех часов после инъекции карагинана. Для каждого животного определяли процент воспаления лапы, используя водный плетисмометр Уго Базиле (Ugo-Basile).
Результаты
Результаты представлены в Таблице 5 (незначительное - Нз;* - значительное;** - очень значительное;*** - сильное).
Реферат
В изобретении описываются соединения формулы 1, а также их фармацевтически приемлемые соли, где заместители имеют указанные в описании значения. Также в изобретении приводится способ получения соединений формулы 1 и фармацевтическая композиция, содержащая в качестве активного ингредиента эффективное количество соединения формулы 1 и фармацевтически приемлемый носитель или разбавитель. Целевые соединения действуют как биологические блокаторы NO-синтазы и пригодны в качестве гипотензивных, антибактериальных, иммуносупрессивных, антиатеросклеротических, вазотропных, анальгезирующих, антимигреневых, офтальмологических или антидиабетических препаратов. 3 с. и 8 з.п. ф-лы, 5 табл.
Формула

где A - атом водорода, низший алкил или нитрогруппа;
Е - атом кислорода или ковалентная связь;
n равно нулю или целому числу от 1 до 12; либо R1 и R2 независимо представляют разветвленную или линейную алкильную цепь, либо R1 и R2 вместе с атомом азота, с которым они связаны, образуют пяти- или шестичленное кольцо, насыщенное или ненасыщенное, формулы

где Х представляет атом кислорода, азота или метиленовую группу, за исключением соединений общей формулы 1, в которой n равно нулю, Е представляет собой ковалентную связь, и либо А представляет собой атом водорода и R1 и R2 независимо представляют алкильные группы, или вместе образуют пиперидиновое, морфолиновое или имидазольное кольцо, либо А представляет нитрогруппу и R1 и R2 образуют пиперидиновое кольцо,
и их фармацевтически приемлемые соли.

где А представляет низший алкил или нитрогруппу;
R3 представляет защитную группу;
с соединением общей формулы 3
H-E-(CH2)n-NR1R2,
где n, Е, R1 и R2 определены выше,
в присутствии сочетающего агента при температуре 0 - 30°С, с последующим отщеплением защитной группы R3 из полученного соединения общей формулы 4

чтобы получить соединение общей формулы 1, где А означает нитрогруппу или низший алкил и, если необходимо получить соединение общей формулы 1, в котором А представляет атом водорода, удаляют защитную группу из соответствующего соединения формулы 1, в которой А представляет нитрогруппу, путем отщепления нитрорадикала.

где А означает атом водорода, низший алкил или нитрогруппу;
Е представляет атом кислорода или ковалентную связь;
n равно нулю или целому числу от 1 до 12; и либо R1 и R2 независимо друг от друга представляют разветвленную или линейную алкильную цепь, либо R1 и R2 вместе с атомом азота, с которым они связаны, образуют пяти- или шестичленное кольцо, насыщенное или ненасыщенное формулы
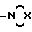
где Х представляет атом кислорода, азота или метиленовую группу, за исключением соединений общей формулы 1, в которой n равно нулю, Е представляет собой ковалентную связь, и либо А представляет собой атом водорода и R1 и R2 независимо представляют собой низший алкил, или соль этого соединения в сочетании с по крайней мере одним разбавителем или фармацевтически приемлемым носителем.
Документы, цитированные в отчёте о поиске
Производные нитратоалкановых кислот или их фармацевтически приемлемые соли
Комментарии