Производные нитратоалкановых кислот или их фармацевтически приемлемые соли - RU2017748C1
Код документа: RU2017748C1
Чертежи

Описание
Изобретение относится к производным нитратоалканкарбоновых кислот или их фармацевтически приемлемым солям.
Органические нитраты (сложные эфиры азотной кислоты) оказались пригодными в терапии заболеваний сердца.
Их действие проявляется не только в разгрузке сердца через понижение предварительной и последующей нагрузок, но и в повышенном снабжении сердца кислородом в результате расширения коронарных сосудов.
Однако в последние годы установили, что применявшиеся до сих пор в терапии органические нитраты, такие как тринитрат глицерина (ТНГ), изосорбид-5-мононитрат или изосорбид-динитрат, будучи непрерывно введенными в организм в больших количествах, уже в течение короткого периода времени теряют значительную часть своего действия, т.е. наблюдается привыкание организма к этим веществам. Результаты многочисленных опытов свидетельствуют о том, что присутствие сульфгидрильных групп может воспрепятствовать развитию процесса привыкания к нитратам и замедлить уже наступивший процесс привыкания.
В настоящее время под механизмом развития привыкания понимается следующее.
Согласно современному уровню знаний фармакологическое действие органических нитросоединений зависит от присутствия цистеина. С ним органический нитрат образует промежуточный продукт, из которого вследствие распада наряду с другими продуктами высвобождаются радикалы NO, которые в свою очередь активируют целевой энзим - растворимую гуанилциклазу гладкой мышечной клетки. Наконец, последующие реакции, вызываемые образованием GMP, приводят к расслаблению напряжения и расширению сосудов.
Предполагают, что вышеназванный реакционноспособный короткоживущий промежуточный продукт представляет собой тиоэфир азотной кислоты или тионитрат. Далее, считают, что в результате внутримолекулярной перегруппировки и последующих, пока еще не выясненных, реакций образуется нитрозотиол, из которого высвобождается оксид азота или ионы нитрита. С другой стороны, предполагают, что энзиматический распад с помощью GSH-редуктазы, ведущий исключительно к образованию ионов нитрита, не играет никакой роли в проявлении фармакологического действия. Итак, как выше излагалось, для неэнзиматического распада промежуточного продукта необходим цистеин, причем этот распад в зависимости от дозы подлежит истощению (истощение запаса сульфгидрильных групп), так что со временем уменьшается количество высвобождающихся радикалов NO, активирующих гуанилциклазу, что клинически выражается в уменьшении фармакологического действия.
В Европейском патенте N 0362575 описаны соединения со специфической структурой, составленные из нитратожирных кислот (нитратоалканкарбоновых кислот) и серусодержащих аминокислот или пептидов.
Присутствие сульфгидрильных групп предназначено для предотвращения или замедления развития процесса привыкания к действию нитратов.
В процитированном патенте описаны, например, соединения, содержащие такие серусодержащие аминокислоты, как цистеин или метионин, в виде их метиловых, этиловых или пропиловых эфиров. Наконец, сульфгидрильная группа цистеина может быть этерифицирована с низшей алканкарбоновой кислотой с числом атомов С 2-8.
Хотя эти соединения уже имеют ценные фармакологические свойства в том смысле, что они препятствуют развитию процесса привыкания к нитратам и замедляют уже наступивший процесс привыкания, им свойственны определенные недостатки. Они имеют низкую температуру плавления, плохо растворяются в воде, и их трудно получить в чистом виде.
Итак, задача настоящего изобретения заключается в получении и предоставлении специалисту новых органических соединений, не имеющих вышеназванных недостатков.
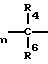
Задача изобретения решена тем, что предлагаемые соединения являются производными нитратоалканкарбоновых кислот общей формулы
O2
N






(I) где R - низкий алкокси или присоединенный пептидной связью пентапептид Tyr-Pro-Phe-Pro-Gly (OMe);
R1 - водород, алкил (С1-С6), ациламино;
R2 - водород, низший алкил;
R3 - водород;
R4 - водород, низший алкил;
R5 - (СН2)kSX, где Х - водород, низший алкил или Х-CORII, в которой RII - водород, низший алкил или остаток N-ациламинокислоты;
m - 0-10, n и о - 0-2, или их фармацевтически приемлемыми солями.
Предлагаемые производные нитратоалканкарбоновых кислот общей формулы I из ряда серусодержащих аминокислот содержат предпочтительно такие аминокислоты, как цистеин, метионин или гомоцистеин.
Согласно другому варианту выполнения изобретения, аминокислоты имеются в стериохимической L-форме.
Серусодержащие аминокислоты на С-конце могут быть этерифицированы.
Согласно предпочтительному варианту выполнения изобретения, аминокислоты - цистеин и/или метионин, могут быть в виде метиловых, этиловых или пропиловых эфиров.
В частности предпочитаются
этиловый эфир N-нитратопивалоил-S-(N-ацетилглицил)-L-цистеина,
этиловый эфир N-нитратопивалоил-S-(N-ацетилаланил)-L-цистеина,
этиловый
эфир N-нитратопивалоил-S-(N-ацетиллейцил)-L-цистеина.
Предлагаемые соединения формулы I получают известным образом тем, что соединение общей формулы O2N


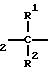

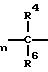

(II) где R1-R4, m, n, o имеют вышеуказанные значения;
R6 - низший алкилтио, подвергают известной реакции образования тиоэфира, т. е. реакции с аминокислотами, N-ациламинокислотами, пептидами или N-ацилпептидами с 2-5 присоединенными пептидной связью аминокислотными остатками. Реакцию получения соединения общей формулы II осуществляют, например, так, как указано в Европейском патенте N 0362575.
Нитратожирные кислоты общей формулы
O2N


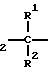

H
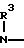

Для перевода соединений общей формулы I в их фармакологически совместимые соли их подвергают реакции предпочтительно в органическом или водном органическом растворителе с эквивалентным количеством неорганической или органической кислоты, например соляной, бромистоводородной, азотной, фосфорной, серной, муравьиной, уксусной, пропионовой, щавелевой, фумаровой, малеиновой, янтарной, адипиновой, бензойной, салициловой, о-ацетоксибензойной, коричной, нафтойной, миндальной, лимонной, яблочной, винной, аспарагиновой, глутаминовой, метансульфоновой или n-толуолсульфоновой.
Предлагаемые новые соединения общей формулы I и их соли можно ввести энтеральным или парентеральным путем в жидком или твердом виде.
В качестве среды для введения препаратов предпочтительно используют воду, содержащую обычно применяемые в инъекционных растворах добавки, такие как стабилизаторы, агенты растворения или буферы. Такими добавками являются, например тертратный и цитратный буферы, этанол, комплексообразователи (например, этилендиаминтетрауксусная кислота и ее нетоксичные соли), высокомолекулярные полимеры (например, жидкий полиэтиленоксид), используемые для регулирования вязкости. Твердыми носителями являются крахмал, лактоза, маннит, метилцеллюлоза, тальк, высокодисперсные силикагели, высокомолекулярные жирные кислоты (например, стеариновая), желатины, агар-агар, фосфат кальция, стеарат магния, животные и растительные жиры и твердые высокомолекулярные полимеры (например, полиэтиленгликоли). Пригодные для орального введения препараты могут содержать еще вкусовые и подслащивающие вещества.
Согласно другому варианту выполнения изобретения, лекарства содержат определенное количество одного из предлагаемых соединений и/или их смесь.
Подобного рода лекарства можно использовать для лечения сердечно-сосудистых заболеваний, например, в качестве коронародилятирующих средств, средств для лечения гипертонии, недостаточности сердца и расширения периферических сосудов, включая сосуды мозга и почек.
Фармацевтические препараты, содержащие предопределенное количество одного или нескольких предлагаемых соединений, можно ввести раз в сутки в виде препаратов замедленного действия или неоднократно (2-3 раза) в сутки через регулярные промежутки времени. Суточные дозы действующего начала составляют 20-300 мг, считая на массу тела 75 кг. Предлагаемые соединения можно также вводить 1-8 раз в сутки в виде инъекций или путем длительного внутривенного вливания, причем обычно достаточно количество 5-200 мг/сутки.
Типичная таблетка может иметь следующий
состав, мг:
1) Этиловый эфир N-нитра-
топивалоил-S-(N-ацетил- глицил)-L-цистеина 25 2) Крахмал, USP* 57 3) Лактоза, USP* 73 4) Тальк, USP* 9 5) Стеариновая кислота 6
*USP - фармацевтической чистоты согласно
фармакопее США
Вещества 1, 2 и 3 просеивают через сито, гранулируют и затем перемешивают с веществами 4 и 5 до достижения однородной смеси.
Смесь затем прессуют в таблетки.
Примеры выполнения иллюстрируют изобретение, не ограничивая его.
П р и м е р 1. Получение этилового эфира N-нитратопивалоил-S-(N-ацетилглицил)-L-цистеина.
48 г (0,41 моль) N-ацетилглицина с перемешиванием при комнатной температуре взвешивают в 300 мл метиленхлорида (CH2Cl2), а затем охлаждают до 10оС. С перемешиванием добавляют раствор 109,8 г (0,373 моль) этилового эфира N-нитратопивалоил-L-цистеина в 300 мл CH2Cl2, причем реакция протекает слабо экзотермически. Реакционную массу с перемешиванием охлаждают до 5оС, а затем, продолжая перемешивать, к ней медленно по каплям добавляют 84,6 г (0,41 моль) дициклогексилкарбодиимида (ДЦК) в 200 мл CH2Cl2 так, чтобы температура лежала в пределах 5-10оС. После нагревания массы до комнатной температуры ее перемешивают 4 сут при комнатной температуре. Отсасывают мочевину - ДЦК и промывают CH2 Cl2 2 раза по 100 мл.
Собранные метиленхлоридные фазы последовательно промывают 200 мл 9%-ного раствора бикарбоната натрия, 300 мл 1н. раствора хлористого водорода и 300 мл дист. воды. Наконец метиленхлоридную фазу высушивают безводным сульфатом натрия и концентрируют на ротационном испарителе типа Rotavapor

В результате получают 162,9 г (вычисленный выход - 146,7 г) целевого продукта в виде светло-желтого масла.
162,9 г целевого продукта при комнатной температуре растворяют в 470 мл этилацетата. После перемешивания в течение 15 мин при комнатной температуре фильтруют образовавшийся нерастворимый белый осадок. Затем к прозрачному светло-желтому фильтрату при комнатной температуре с перемешиванием медленно добавляют 390 мл n-гексана.
К полученному раствору добавляют затравочные кристаллы и перемешивают в течение ночи при комнатной температуре. Выпавшие кристаллы отсасывают и при комнатной температуре 2 раза промывают 100 мл смеси 20 мл этилацетата и 80 мл n-гексана.
Кристаллы высушивают до постоянства массы в вакуумной сушилке при комнатной температуре и вакууме 2 Торр.
Выход целевого продукта составляет 85,4 г (вычисленный выход 146,74 г).
Т.пл. 71,8оС.
П р и м е р 2. Получение этилового эфира N-нитратопивалоил-S-(N-ацетилаланил)-L-цистеина.
53,8 г (0,41 моль) N-ацетилаланина с перемешиванием при комнатной температуре взвешивают в 300 мл метиленхлорида (CH2Cl2), а затем охлаждают до 10оС. С перемешиванием добавляют раствор 109,8 г (0,373 моль) этилового эфира N-нитратопивалоил-L-цистеина в 300 мл СH2 Cl2, причем реакция протекает слабо экзотермически. Реакционную массу с перемешиванием охлаждают до 5оС, а затем, продолжая перемешивать, к ней медленно по каплям добавляют 84,6 г (0,41 моль) дициклогексилкарбодиимида (ДЦК) в 200 мл CH2Cl2 так, чтобы температура лежала в пределах 5-10оС. После нагревания массы до комнатной температуры ее перемешивают 4 сут при комнатной температуре. Отсасывают мочевину - ДЦК и промывают метиленхлоридом 2 раза по 100 мл.
Собранные метиленхлоридные фазы последовательно промывают 200 мл 9%-ного раствора NaHCО3, 300 мл 1н. раствора HCl и 300 мл дист. воды. Наконец метиленхлоридную фазу высушивают безводным сульфатом натрия и концентрируют на ротационном испарителе типа Rotavapor

В результате получают 160,5 г (вычисленный выход 151, 84 г) целевого продукта в виде светло-желтого масла.
160,5 г целевого продукта при комнатной температуре растворяют в 345 мл этилацетата. После перемешивания в течение 15 мин при комнатной температуре фильтруют образовавшийся нерастворимый белый осадок. Затем к прозрачному светло-желтому фильтрату при комнатной температуре с перемешиванием медленно добавляют 345 мл н-гексана.
К полученному раствору добавляют затравочные кристаллы и перемешивают в течение ночи при комнатной температуре. Выпавшие кристаллы отсасывают и при комнатной температуре два раза промывают 100 мл смеси 20 мл этилацетата и 80 мл n-гексана.
Кристаллы высушивают до постоянства массы в вакуумной сушилке при комнатной температуре и вакууме 2 Торр.
Выход целевого продукта составляет 78,2 г (вычисленный выход 151,84 г).
Т.пл. 76,6оС.
П р и м е р 3. Получение этилового эфира N-нитратопивалоил-S-(N-ацетил-лейцил)-L-цистеина.
0,02 моль (6 г) цистеинэтилового эфира нитратопивалиновой кислоты растворяют в 100 мл дихлорметана. При 10оС с подачей азота медленно добавляют 0,03 моля (5,19 г) N-ацетил-лейцина и 0,1 г диметиламинопиридина (ДМАП). Затем по каплям добавляют 0,03 моль (6,15 г) дициклогексилкарбодиимида (ДЦК), растворенного в 80 мл дихлорметана. Полученную смесь перемешивают в течение ночи при комнатной температуре.
Переработка.
Вышеназванную смесь отсасывают. Раствор подряд экстрагируют эквивалентными количествами 0,1н. раствора хлористого водорода, насыщенного раствора бикарбоната натрия и дист. воды. Затем на ротационном испарителе типа Rotavapor

Перекристаллизация.
10 г полученного выше маслянистого вещества, слегка нагревая, растворяют в 45 мл этанола и 40 мл воды. Потом полученное вещество в течение ночи выкристаллизовывают в холодильнике. Полученные кристаллы отсасывают и высушивают в вакуумной сушилке.
Масс-спектр вещества подтверждает его структуру. Температура плавления: 91,
4оС. Анализ БЖХ: 98,7%. Выход 5 г = 0,012 моль =
= 57,4%
от теорети-
ческого.
П р и м е р 4. Получение L-метионинэтилового эфира 2-нитроксиизомасляной кислоты.
10,6 г основания этилового эфира L-метионина 60,0
ммоль
8,9 г 2-нитроксиизомасляная кислота 60,0 ммоль
13,0 г 98%-ного (ДЦК) (дициклогексилкарбодиимида) 61,5
ммоль
20 мг DMAП (диметиламинопиридина)
185 мл CH2Cl2
Проведение испытания и переработку осуществляют как при этиловом эфире
N-(2-нитратоизобутирил)-L-цистеина в каждом случае в атмосфере азота.
Выход 20,53 г L-метионинэтилового эфира 2-нитроксиизомасляной кислоты в виде светло-желтого масла = 111,0% от теорет.
Из сырого продукта L-метионинэтилового эфира 2-нитроксиизомасляной кислоты получают раствор из 50 мг/мл метанола для анализа тонкослойной хроматографии. Нанесенный объем - 1 мкл = 50 мкг вещества. Проведение тонкослойной хроматографии и условия соответствуют тем, использованным для этилового эфира N-(2-нитратоизобутирил)-L-цистеина в атмосфере азота.
Результат тонкослойной хроматографии.
Сырой продукт содержит примерно 10% примеси, содержащей -O-NO2. Очистку сырого продукта осуществляют хроматографией на колонке согласно системе MPLC (СЖХ) фирмы Buechi: 20,5 г 2-метионинэтилового эфира 2-нитроксиизомасляной кислоты растворяют в 51,5 мл метанола и 20 мл дистиллированной воды. Отфильтрованный раствор затем подают на колонку.
Концентрированная 3. Фракция:
13,8 г этилового эфира
2-метионина 2-нитроксиизомасляной кислоты (в виде бесцветного масла) = 74,6% от теорет.
Концентрированная 4. Фракция:
2,95 г L-метионинэтилового эфира 2-нитроксиизомасляной
кислоты (в виде бесцветного масла) = 15,9% от теорет.
П р и м е р 5. Получение тиопропионата этилового эфира нитроксипивалиновой кислоты-L-цистеина.
10,3 г этилового
эфира
L-цистеина нитроксипи- валиновой кислоты 35,0 ммоль
5,5 г ангидрида
пропи- оновой кислоты 42,0 ммоль
5,0 г триэтиламина D = =0,73 (6,80 мл) 49,0 ммоль
60 мл
CH2Cl2
Проведение испытания и переработку осуществляют как при
S-карбонате этилового эфира N-(3-нитратопивалоил)-цистеина (всегда в атмосфере N2).
Выход 12,46 г N-Piv-Cy-Et-S-Prop в виде светло-коричневого масла = 101,6% от теорет.
Результат масс-спектрометрии: подтверждение идентичности.
П р и м е р 6. Получение тиоизобутирата этилового эфира L-цистеина нитроксипивалиновой кислоты.
10,3 г этилового эфира
L-цистеина нитроксипи- валиновой кислоты 35,0 ммоль
6,7 г
ангидрида изомас- ляной кислоты 42,0 ммоль
5,0 г триэтиламина D = =0,73 (6,80 мл) 49,0
ммоль
60 мл CH2Cl2
Проведение испытания и переработку
осуществляют как при этиловом эфире N-(2-нитратоизобутирил)-L-цистеина (всегда в атмосфере N2
).
Выход 13,39 г N-Piv-Cy-Et-S-i-But в виде светло-коричневого масла = 105,0% от теорет.
Результат масс-спектра: подтверждение идентичности.
П р и м е р 7. Получение этилового эфира цистеина 2-нитроксигексановой кислоты.
Исходная
смесь:
0,015 мол = 2,74 г 2-нитроксигексановой кислоты
0,015 мол = 2,78 г этилового эфира
цистеина х HCl
1 объем шпателя DMAП растворяют в 50 мл диоксана, перемешивают и
нагревают (температура ванны ≈ 80оС)
По каплям добавляют 0,015 мль = 3,09 г ДЦК,
растворенный в 20 мл диоксана. Подача N2.
Получают прозрачный раствор, из которого после охлаждения выпадает осадок.
Переработка.
Раствор отсасывают и подвергают ротационной обработке, его смешивают с 100 мл воды и экстрагируют 100 мл этилацетата с подачей N2, раствор полностью подвергают ротационной обработке.
Выход 5,58 г. (Вычисленный выход 4,63 г).
Перекристаллизацию
осуществляют 50 мл ЕТОН
Выход примерно 60 мг.
П р и м е р 8. Получение этилового эфира цистеина 2-нитроксимасляной кислоты.
Исходная смесь:
0,033
моль = 5 г нитроксимасляной кислоты
0,046 моль = 8,59 этилового эфира цистеина х HCl
1 объем шпателя DMAП
(диметиламинопиридина) при нагревании перемешивают в 100 мл диоксана и
растворяют с подачей N2.
Растворяют 0,046 моль = 9,49 г ДЦК (дициклогексилкарбдиимида) в 80 мл диоксана и по каплям медленно добавляют. Перемешивают в течение ночи.
Раствор отсасывают и подвергают ротационной обработке, к остатку добавляют воду и при высаливании экстрагируют этилацетатом. Раствор азеотропически подвергают ротационной обработке.
Выход 8,03 г (Вычисленный выход 9,24 г).
П р и м е р 9. Получение S-ацетата этилового эфира цистеина 3-нитратомасляной кислоты.
Исходная смесь:
0,
025 моль = 7 г 3-N-But-Cy-Et медленно растворяют в 150 мл CH2Cl2, добавляют 0,3 моль = 27,9 мл
Ас2О. При подаче N2 и при температуре 5-10оС по
каплям медленно добавляют 0,3 моль = 30,4 г триэтиламина, растворенного в 100 мл CH2Cl2.
Перемешивают в течение ночи.
Промывают раствор эквивалентным количеством NaHCО3, а затем водой. Раствор осторожно подвергают ротационной обработке.
Выход 9,8 г.
Перекристаллизуют из 50 мл EtOH.
Отсасывают кристаллы.
Выход в сухом состоянии 3,13 г = 38,84% от теорет.
П р и м е р 10. Получение нитрато-Piv-CyEt-S-бензоата.
Получение:
Растворяют 0,
034 моль = 10 г N-Piv-CyEt в 100 мл CH2Cl2 при температуре 5-10оС и при подаче N2, затем медленно добавляют 0,0408 моль = 4,8 мл бензоилхлорида, а затем по
каплям медленно добавляют 0,034 моль = 4,7 мл (Et)3N, растворенного в 50 мл CH2Cl2.
Перемешивают в течение 3 дней.
Переработка:
Исходную
смесь подают на ледяную воду, экстрагируют ее и подвергают ее ротационной обработке. Осуществляют очистку хроматографией
на колонке.
Растворитель 75 МеОН, 25 Н2О.
Фракцию подвергают ротационной обработке.
Тонкослойная хроматография:
Нет примесей
Выход ≈ 10,7 г = 79,0% от теорет.
П р и м е р 11. Получение S-этилкарбоната 3-N-Piv-CyEt.
Растворяют 0,023 моль

Переработка: раствор подают на воду и тщательно перемешивают, его экстрагируют и подвергают его ротационной обработке. Ставят в холодильник.
Выход 9,3 г; Тпл. 36,2оС.
П р и м е р 12. Получение N-(3-нитратопивалоил)-гомоцистеинтиолактона.
O-
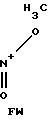




Количества исходной смеси: 5,0 г

линовой кислоты 4,7 г
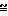
олакто х HCl 2,4 г
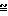

5 г нитратопивалиновой кислоты и 4,7 г гомоцистеинтиолактона х HCl суспендируют в 50 мл ТГФ. После добавления 2,4 г пиридина при температуре 5-10оС по каплям добавляют раствор, состоящий из 6,2 г ДЦК и 20 мл ТГФ. Исходную смесь дополнительно смешивают при комнатной температуре в течение ночи и затем фильтруют ее. Фильтрат подвергают ротационной обработке, а остаток перемешивают с 150 мл 1н. NaOH и дважды экстрагируют его по 100 мл CH2Cl2. Органическую фазу промывают 100 мл 1н. HCl и полностью подвергают ее ротационной обработке. Остаток 2,6 г масла.
Сырой продукт перекристаллизуют из 20 мл n-гексана и 10 мл EtOH. Выход 510 мг (примерно 6,5% от теорет.).
П р и м е р 13. Получение этилового эфира N-(2-нитратопропионил)-L-цистеина.


Количество исходной смеси:
26,9 г

28,6 г

41,3 г

375 мл CH2Cl2
26,9 г нитратомолочной кислоты и 28,6 г этилового эфира цистеина растворяют в 300 мл CH2Cl2. При температуре 15-25оС по каплям добавляют раствор, состоящий из 41,3 г ДЦК и 75 мл CH2Cl2. Исходную смесь дополнительно перемешивают в течение ночи и затем ее фильтруют. Фильтрат полностью подвергают ротационной обработке. Остаток 50,5 г. Из 100 мл EtOH перекристаллизуют 18,5 г сырого продукта. Выход 9,2 г (примерно 49,64% от теорет.).
П р и м е р 14. Получение этилового эфира N-(3-нитратогексаноил)-L-цистеина. H3C



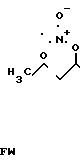

Исходная смесь: 5,1 г

сановой кислоты 5,94 г

этилового эфи-
ра цистеина 6,58 г ≈ 0,032 моль ДЦК Кат. DMAП 70 мл диоксана
5,1 г нитратогексановой кислоты, 5,94 г гидрохлорида этилового эфира цистеина и каталитические количества DMAП cуспендируют в 50 мл диоксана. При комнатной температуре по каплям добавляют раствор, состоящий из 6,58 г ДЦК в 20 мл диоксана. Исходную смесь в течение двух дней перемешивают при комнатной температуре и затем ее фильтруют. Удаляют диоксан, добавляют остаток к 100 мл AcOEt и дважды экстрагируют его по 100 мл 1н. HCl. Органическую фазу подвергают ротационной обработке. Остаток 4,6 г (51,44% от теорет.).
П р и м е р 15. Получение этилового эфира L-цистеина N-(2-нитратоуксусной кислоты).




Исходная смесь: 11,35 г

ной кислоты
(растворенной
в 200 мл AcOEt) 12,60 г

ра цистеина 19,40 г
Растворяют 12,6 г этилового эфира цистеина в растворе, состоящем из 11,35 г 2-нитратоуксусной кислоты и 200 мл AcOEt. При температуре 20-25оС по каплям добавляют раствор, состоящий из 19,4 г ДЦК в 20 мл CH2 Cl2. Исходную смесь дополнительно перемешивают в течение ночи при комнатной температуре, затем отсасывают ее и фильтрат подвергают ротационной обработке. Остаток 17,9 г. Сырой продукт дважды перекристаллизуют из изопропанола. Выход 2,5 г (11,66% от теорет.). Тпл.71,6о С.
П р и м е р 16. Получение этилового эфира N-(3-нитратобутирил)-цистеина.
I стадия.
Омыление этилового эфира 3-гидрокимасляной кислоты.
13,2 г (0,1 моль) этилового эфира 3-гидрокимасляной кислоты (Aldrich) перемешивают 4,0 г (0,1 моль) растворенного в 100 мл воды NaOH. Реакция закончилась, пока раствор не стал гомогенным. При переработке полученный раствор подкисляют 10 мл конц. HCl и дважды экстрагируют его по 100 мл этилацетата. Затем раствор подвергают ротационной обработке, причем получают жидкотекучее масло.
Выход 8,81 г (теорет. 10,4 г) 3-гидроксимасляной кислоты.
II стадия.
Нитрование 3-гидроксимасляной кислоты.
8,81 г (0,08 моль) 3-гидроксимасляной кислоты и 50 мг мочевины растворяют в 50 мл уксусной кислоты при температуре 5оС. Во-первых по каплям добавляют 6,27 мл (0, 15 моль) HNO3, а затем при охлаждении по каплям добавляют 14,17 мл (0,15 моль) Ac2O. Реакционную смесь перемешивают в течение ночи. При переработке к полученному раствору добавляют 200 мл ледяной воды и экстрагируют его этилацетатом. Органическую фазу экстрагируют NaHCO3. Фазу NaHCO3 подкисляют конц. HCl и ее экстрагируют этилацетатом. Затем раствор подвергают ротационной обработке, причем получают жидкотекучее масло. Выход 9,4 г (теорет. 11,9 г) 3-нитратомасляной кислоты.
III стадия. Получение этилового эфира N-(3-нитратобутирил)-цистеина.
16 г (0,11 моль) 3-нитратомасляной кислоты растворяют в 100 мл дихлорметана. При подаче N2 и температуре 15оС медленно добавляют 17,9 г (0,12 моль) этилового эфира цистеина. Затем медленно по каплям добавляют 24,7 г (0,12 моль) растворенного в 80 мл дихлорметана дициклогексилкарбодиимид (ДЦК) при температуре 15оС и подаче N2. После завершения реакции отсасывают полученную дициклогексилмочевину и промывают раствор 150 мл 0,1 н.HCl. Затем раствор подвергают ротационной обработке.
Осуществляют очистку вещества путем препаративной хроматографии на колонке и путем перекристаллизации из этанола/n-гексана.
Выход 6,88 г (теорет. 30,83 г).
Т.пл. 77,8оС.
П р и м е р 17. Получение этилового эфира N-(3-нитратобутирил)-метионина.
6,35 г (0,043 моль) 3-нитратомасляной кислоты, 7,47 г (0,043 моль) этилового эфира метионина и объем шпателя диметиламинопиридина (DMAП) при перемешивании и охлаждении до 10оС растворяют в 100 мл дихлорметана. 10,31 г (0,05 моль) ДЦК растворяют в 80 мл CH2Cl2 и при одновременной подаче азота по каплям медленно добавляют его. После завершения реакции отсасывают раствор, и промывают его NaHCO3 и затем HCl. Раствор подвергают ротационной обработке, причем получают масло.
Осуществляют очистку вещества путем препаративной хроматографии на колонке и путем перекристаллизации в низкотемпературных условиях.
Выход 1,95 г (теорет. 12,05 г) этилового эфира N-(3-нитратобутирил)-метионина в виде бесцветного масла.
П р и м е р 18. Получение этилового эфира N-(3-нитратопивалоил)-цистеина.
I стадия. Получение метилового эфира нитратопивалиновой кислоты.
25,0 г (0,19 моль) метилового эфира гидроксипивалиновой кислоты и 0, 12 г мочевины растворяют при комнатной температуре в 250 мл CH2Cl2 и охлаждают с перемешиванием до 5оС. К этой смеси с перемешиванием добавляют по каплям 23,8 г (0,38 моль) 100% -ного HNO3 таким образом, чтобы температура не превысила 10оС. Затем охлаждают до 5оС и перемешивая добавляют по каплям 38,6 г (0,38 моль) ангидрида уксусной кислоты таким образом, чтобы температура не превысила 10оС. В течение 15 мин при охлаждении перемешивают смесь в ледяной ванне. Затем медленно нагревают ее до комнатной температуры и в течение ночи продолжают перемешивание при комнатной температуре. Исходную смесь медленно подают при одновременном перемешивании на 500 мл ледяной воды. Отделяют СH2Сl2-фазу и один раз промывают ее 100 мл дист. H2O, один раз 100 мл насыщенного, водяного раствора NaHCO3 и еще раз 100 мл дист. H2O. Экстракт CH2Cl2 затем концентрируют в ротавапоре при температуре ванны в размере макс. 40оС в вакууме, получаемом с помощью водоструйного насоса, до сухого состояния. Светло-желтый, маслянистый остаток перегоняют в вакууме насоса масла при температуре ванны 60оС с получением прозрачного, жидкотекучего масла.
Выход 31,5 г = 94,0% от теорет.
II стадия. Получение нитратопивалиновой кислоты.
14,0 г (0,350 моль) NaOH растворяют в дист. H2O и охлаждают до примерно 10оС. К этой смеси с перемешиванием добавляют раствор, состоящий из 31,0 г (0,175 моль) метилового эфира нитратопивалиновой кислоты в 250 мл метаноле, причем реакционная смесь окрашивается в желтый цвет и температура повышается до 25оС. После перемешивания в течение 90 мин исходную смесь нейтрализуют 29,5 мл (0, 35 моль) 37% -ной HCl. Метанол на ротационном испарителе (ротавапор) полностью отгоняют. Водяную фазу дважды экстрагируют по 200 мл метиленхлорида. Объединенные экстракты метиленхлорида один раз промывают 50 мл дист. H2O и метиленхлоридную фазу концентрируют на ротационном испарителе (ротавапор) до сухого состояния. Бесцветный, маслянистый остаток растворяют в 100 мл этилацетата и опять концентрируют его на ротационном испарителе (ротавапор) до сухого состояния, причем получают твердый, белый остаток, из которого удаляют остатки растворителя в вакууме насоса масла (0,4 Торр) при температуре ванны 40оС и в течение 15 мин на ротационном испарителе (ротавапор). Твердый и белый остаток - 25,44 г (89,1% от теорет.), растворяют в 100 мл кипящего n-гексана и к нему добавляют 2 мл диизопропилового эфира. После охлаждения до комнатной температуры и после добавления затравочных кристаллов, продукт выкристаллизовывается. Продукт поддерживают в течение 72 ч при температуре 0оС, кристаллы отсасывают и после двойной промывки по 10 мл n-гексана охлаждают его при примерно 2 Торр и при комнатной температуре в вакуумной сушилке до постоянства массы.
Т.пл. 54,2оС.
Выход 23,66 = 82,9% от теорет.
III стадия. Получение этилового эфира N-(3-нитратпивалоил)-цистеина.
10,7 г (71,7 ммоль) основания этилового эфира L-цистеина с перемешиванием растворяют в атмосфере азота в 200 мл метиленхлорида при комнатной температуре. К смеси добавляют 11,4 г (70,0 ммоль) кристаллической нитроксипивалиновой кислоты, которую растворяют с перемешиванием при комнатной температуре. При комнатной температуре и в течение 15 мин к этой смеси по каплям добавляют с перемешиванием и в атмосфере азота раствор, состоящий из 14,8 г (71,7 ммоль) N,N-дициклогексилмочевину (ДЦК) в 50 мл метиленхлорида, причем температура повышается до 35оС. После дальнейшего перемешивания осаждается белая дициклогексилмочевина. Исходную смесь охлаждают до комнатной температуры и перемешивают в атмосфере азота в течение ночи. Затем дициклогексилмочевину отсасывают через пористый стеклянный фильтр и один раз его промывают 50 мл CН2Cl2. Объединенные метиленхлоридные растворы один раз промывают 100 мл 1н. HCl и дважды по 100 мл дист. H2O (в атмосфере азота). Затем их концентрируют в ротавапоре при температуре ванны примерно 40оС и в вакууме, получаемом с помощью водоструйного насоса, от сначала 550 мБар до примерно 20 мБар. Получают светло-коричневое масло.
Выход 21,2 г = 102,9% от теорет.
Вещество очищают с помощью кристаллизации из этанола/гексана в низкотемпературных условиях.
Выход 13,42 г = 65,1% от торет. этилового эфира N-(3-нитратопивалоил)-цистеина в виде светло-розового масла.
IV стадия. Получение этилового эфира N-(3-нитратопивалоил)-S-ацетилцистеина.
К 10,3 г (35,0 ммоль) этилового эфира N-(3-нитратопивалоил)-цистеина, растворенного в 70 мл дихлорметана, в охлажденных условиях с перемешиваним по каплям добавляют раствор, состоящий из 4,3 г (42,0 ммоль) ангидрида уксусной кислоты в 10 мл дихлорметана. Затем по каплям добавляют с перемешиванием и в охлажденных условиях раствор, состоящий из 5,0 г (49,0 ммоль) триэтиламина в 20 мл дихлорметана. После завершения реакции исходную смесь промывают 1н. HCl 10%-ным водным раствором бикарбоната натрия и водой. Дихлорметановый экстракт концентрируют на ротационном испарителе (ротавапор) до сухого состояния. Получают 11,6 г светло-желтого, маслянистого продукта, из которого получают путем кристаллизации из этанола/воды в охлажденных условиях и путем добавления затравочных кристаллов 7,8 г кристаллического продукта (66,3% от теорет.).
Т.пл. < 5оС.
IV стадия (Вариант 1). Получение этилового эфира N-3(нитратопивалоил)-S-бутирилцистеина.
При использовании 6,7 г (42,0 ммоль) ангидрида масляной кислоты вместо ангидрида уксусной кислоты (см. стадия 4), при тех же самых условиях реакции и переработки получают 13,0 г светло-желтого маслянистого продукта, из которого как описано выше, (IV стадия) путем кристаллизации в низкотемпературных условиях получают 9,7 г кристаллического продукта (76,2% от теорет.).
Т.пл. < 5оС.
IV стадия (Вариант 2). Получение этилового эфира N-(3-нитратопивалоил)-S-пивалоилцистеина.
При использовании 7,8 г (42,0 ммоль) ангидрида пивалиновой кислоты вместо ангидрида уксусной кислоты (см.стадия IV), при тех же самых условиях реакции и переработки получают 14,1 г светло-желтого маслянистого продукта, из которого как описано выше (IV стадия) путем кристаллизации в низкотемпературных условиях получают 10,5 г кристаллического продукта (79,5% от теорет.).
Т.пл. 45оС.
IV стадия (Вариант 3). Получение этилового эфира N-(3-нитратопивалоил)-S-цистеина-S-карбоната.
При использовании 4,3 г (42,0 ммоль) этилового эфира хлормуравьиной кислоты вместо ангидрида уксусной кислоты (см.стадия IV), при тех же самых условиях реакции и переработки получают 11,5 г светло-желтого маслянистого продукта, из которого как описано выше (IV стадия) путем кристаллизации в низкотемпературных условиях получают 9,5 г кристаллического продукта (= 74,1% от теорет.).
Т.пл. 36оС.
П р и м е р 19. Получение этилового эфира N-(3-нитратопивалоил)-метионина.
Растворяют с перемешиванием и при комнатной температуре 12,4 г (70,0 ммоль) основания этилового эфира L-метионина в атмосфере азота в 250 мл метиленхлорида. Добавляют 11,4 г (70,0 ммоль) кристаллической нитропивалиновой кислоты, которую растворяют с перемешиванием при комнатной температуре. К этой смеси с перемешиванием и в атмосфере азота по каплям добавляют 14,8 г (71,7 ммоль) N-N-дициклогексилмочевину (ДЦК) в 50 мл метиленхлорида при комнатной температуре и в течение 15 мин, причем температура повышается до 35оС. После дальнейшего перемешивания осаждается белая мочевина ДЦК. Исходную смесь охлаждают до комнатной температуры и ее перемешивают в течение ночи в атмосфере азота. ДЦК-мочевину затем отсасывают через пористый стеклянный фильтр и ее один раз промывают 50 мл CH2Cl2. Объединенные растворы метиленхлорида один раз промывают 100 мл 1н. HCl и дважды по 100 мл дист. H2O (в атмосфере азота), а затем их концентрируют на ротационном испарителе (ротавапор) при температуре ванны примерно 40оС в вакууме, получаемом с помощью водоструйного насоса, от ≈550 мБар до ≈20 мБар. Получают светло-желтое масло.
Выход 24,9 г = 110,3% от теорет. сырого этилового эфира N-(3-нитратопивалоил)-L-метионина.
Сырой продукт очищают путем препаративной хроматографии на колонке.
Выход 17,6 г = 78,0% от теорет. этилового эфира N-(3-нитратопивалоил)-метионина в виде бесцветного масла.
П р и м е р 20. Получение N-(12-нитратоауроил)-S-ацетилцистеина.
I стадия. Получение 12-нитратолауриновой кислоты.
Растворяют 54,1 г (0,250 моль) 12-гидроксилауриновой кислоты и 0,3 г мочевины при слабом нагревании в 1,3 л CHCl3 и охлаждают смесь с перемешиванием до 20оС 23,6 г (0,375 моль) HNO3 (100%-ный) с перемешиванием медленно по каплям добавляют, причем температура повышается до 27оС. Затем смесь охлаждают до 20оС и, перемешивая, по каплям добавляют 38,3 г (0,375 моль) ангидрида уксусной кислоты при охлаждении, причем поддерживают температуру по крайней мере на 25оС. В течение ночи перемешивают смесь при комнатной температуре. Затем промывают 5 раз по 0,5 л дист. H2O. После высушивания над Na2SO4 и осветления порошкообразным активным углем концентрируют CHCl3 фазу на ротационном испарителе (ротавапор) при температуре ванны 50о С и при вакууме, получаемом с помощью водоструйного насоса, до сухого состояния. Маслянистый остаток 60,8 г растворяют в 500 мл кипящего н-гексана и после охлаждения до комнатной температуры его оставляют в течение ночи в холодильнике при температуре 0оС. Выкристаллизованный продукт отсасывают и промывают дважды по 50 мл n-гексана. Затем высушивают продукт в вакуумной сушилке при комнатной температуре и примерно 2 Торр до постоянства массы.
Т.пл. 29оС.
Выход 39,4 г = 60,3% от теорет.
II стадия. Получение хлорида 12-нитратолауриновой кислоты.
Растворяют 2,61 г (10 ммоль) нитратолауриновой кислоты в 50 мл метиленхлорида и с перемешиванием при комнатной температуре по каплям добавляют 4, 44 г (35 ммоль) оксалилхлорида в 50 мл метиленхлорида. Смесь в течение ночи перемешивают. Затем продукт концентрируют в ротационном испарителе до сухого состояния.
Выход 3 г = 93,2% от теорет.
III стадия. Получение N-(12-нитратолауроил)-цистеина.
В атмосфере азота добавляют с перемешиванием 6,06 г (50 ммоль) L-цистеина в 300 мл DМФ. 5,60 г (20 ммоль) хлорида 12-нитратолауриновой кислоты в 50 мл дихлорметана добавляют по каплям. Ввиду того, что не получают прозрачный раствор, его нагревают до 60оС. Затем еще добавляют 100 мл дист. H2O и смесь перемешивают в течение ночи при комнатной температуре. Затем разбавляют 300 мл дист. H2O и четыре раза экстрагируют по 200 мл этилацетата. Органическую фазу высушивают над Na2SO4 и затем ее концентрируют. Остаток добавляют к 100 мл эфира и ставят в холодильник для выкристаллизации в течение ночи и при температуре 0оС. Получают белые кристаллы.
Т.пл. 74-75оС.
Выход 4,1 г N-(12-нитратолауроил)-цистеина.
IV стадия. Получение N-(12-нитратолауроил)-S-ацетилцистеина.
В атмосфере азота приготовляют 1,82 г (5 ммоль) N-(12-нитратолауроил)-цистеина в 20 мл этилацетата. Затем охлаждают смесь до 0оС и по каплям добавляют 2,5 мл ангидрида уксусной кислоты. При температуре -5оС по каплям медленно добавляют 1,52 г (15 ммоль) триэтиламина, растворенного в 5 мл этилацетата. Реакционный раствор промывают водой и концентрируют до сухого состояния.
Т.пл. при комнатной температуре - масло.
Выход: 2 г = 98,4% от теорет.
П р и м е р 21. Получение этилового эфира N-(12-нитратолауроил)-цистеина.
Растворяют 4 г (26,8 ммоль) основания этилового эфира цистеина в 50 мл метиленхлорида и с перемешиванием по каплям добавляют 2,8 г (10 ммоль) хлорида 12-нитратолауриновой кислоты, растворенного в 50 мл метиленхлорида, и в течение ночи смесь перемешивают. Отсасывают осажденный этиловый эфир цистеина HCl и удаляют растворитель на ротационном испарителе (ротавапор). Маслянистый остаток (6 г) растворяют в 100 мл эфира и оставляют в холодильнике в течение ночи при температуре 0оС. Осажденный продукт отсасывают.
Т.пл. 59-60оС.
Выход 1,6 г = 40,0% от теорет.
П р и м е р 22. Получение этилового эфира N-(2-нитратопропионил)-цистеина.
I стадия. Получение этилового эфира нитратомолочной кислоты.
33 г (0,28 моль) этилового эфира молочной кислоты растворяют в 300 мл дихлорметана. После добавления 100 мг мочевины при температуре 5-10оС по каплям добавляют 22,5 мл (0,56 моль) 100%-ной азотной кислоты. Охлаждают раствор до температуры 0оС. Затем по каплям добавляют 52,8 мл (0,56 моль) ацетангидрида таким образом, чтобы температура не превысила 5оС. Оставляют раствор в течение ночи при комнатной температуре, а затем промывают его 250 мл воды. Органическую фазу отделяют и сушат ее над сульфатом натрия. После фильтрации отгоняют дихлорметан. Полученный, маслянистый остаток обрабатывают дистилляцией.
Выход 30, 34 г = 66,4% от теорет.
Т.кип. 34оС (0,25 торр).
II стадия. Получениe нитратомолочной кислоты.
30 г (0,18 моль) этилового эфира нитратомолочной кислоты растворяют в 80 мл диоксана. К раствору добавляют 30 мл воды и 2 г (0,02 моль) серной кислоты и потом его подвергают дефлегмации в течение 19 ч. Раствор концентрируют до объема в размере примерно 50 мл и затем разбавляют его 300 мл воды. Значение рН доводят до 7-8 добавлением бикарбоната натрия. Непреобразованный эфир отделяют экстракцией с дихлорметаном. Водную фазу доводят до значения рН 1 конц. соляной кислотой и три раза экстрагируют ее по 150 мл этилацетата. Соединяют экстракты и их сушат над сульфатом натрия. После фильтрации этилацетат полностью удаляют на ротационном испарителе.
Выход 14,6 г бесцветного масла = 59,2% от теорет.
III стадия. Получение этилового эфира N-(2-нитратопропионил)-цистеина.
В атмосфере азота растворяют 17 г (0,13 моль) нитратомолочной кислоты и 18,9 г (0,13 моль) этилового эфира цистеина при температуре 10-15оС в 200 мл дихлорметана. При температуре 15-20оС по каплям добавляют раствор, состоящий из 28,6 (0,14 моль) N,N-дициклогексилкарбодиимид и 75 мл дихлорметана. После 1 ч отфильтровывают осажденную N,N-дициклогексилмочевину и дополнительно промывают ее 75 мл дихлорметана. Фильтрат дважды экстрагируют по 50 мл 0,1н. соляной кислоты. Органическую фазу полностью концентрируют на роторном испарителе. Кристаллический сырой продукт (22,4 г) перекристаллизуют из 100 мл этанола/н-гексана (1:1).
Выход 7,6 г = 22,6% от теорет.
Т.пл. 92,8оС.
Примерами для значений
указанных радикалов может служить следующее:
А) заместитель
1) R1 = водород, С1-С4-алкил, амино, ациламино, окси, ацилокси.
В качестве примера для R1 = ациламино заявитель указывает на получение этилового эфира N-(нитрато-N'-ацетил-D,L-серил)-цистеина.
Соединение О-ацил и оксисоединения могут быть получены таким же образом.
R1 = ациламино
Получение этилового эфира N-(нитрато-N'-ацетил-D,L-серил)-цистеина.
I. Стадия. Получение нитрато-N-ацетил-D,L-серина.
11,8 г (0,08 моль) N-ацетил-D,L-серина растворяют в 50 мл уксусной кислоты. При температуре 5-10оС по каплям добавляют 6,27 мл (0,15 моль) HNO3, а затем 14,17 мл (0,15 моль) Ас2О. Реакционную смесь перемешивают в течение ночи. К полученному раствору добавляют 200 мл ледяной воды и экстрагируют его этилацетатом. Органическую фазу экстрагируют раствором NaHCO3. Подкисляют фазу NaHCO3 с конц. HCl и экстрагируют ее этилацетатом. Полученный экстракт подвергают ротационной обработке, причем получают вязкотекучее масло.
Физических параметров нет, потому что продукт является промежуточным продуктом.
Выход 11,98 г = 72% от теорет.
II стадия. Получение этилового эфира N-(нитрато-N'-ацетил-D,L-серин)-цистеина.
10,7 г (71,7 ммоль) основания этилового эфира L-цистеина в атмосфере азота и с перемешиванием растворяют в 200 мл метиленхлорида при комнатной температуре. Добавляют 11,98 г нитрато-N-ацетил-D,L-серина, который растворяют с перемешиванием при комнатной температуре. При комнатной температуре и в течение 15 мин к этой смеси по каплям добавляют с перемешиванием и в атмосфере азота раствор 14,8 г (71,7 ммоль) N,N-дициклогексилмочевины (DСС) в 50 мл метиленхлорида, причем температура повышается до 35оС. После дальнейшего перемешивания осаждается белая дициклогексилмочевина. Исходную смесь охлаждают до комнатной температуры и перемешивают в атмосфере азота в течение ночи. Затем дициклогексилмочевину отсасывают через пористый стеклянный фильтр и один раз его промывают 50 мл CH2Cl2. Объединенные метиленхлоридные растворы один раз промывают 100 мл 1 н.HCl и дважды по 100 мл дист. H2O (в атмосфере азота). Затем их концентрируют в ротавапоре при температуре ванны примерно 40оС и в вакууме, получаемом с помощью водоструйного насоса, от сначала 550 мБар до примерно 20 мБар. Получают светло-коричневое масло. Согласно СЖХ (MPLC) получают 4,26 г масла++ = 21% от теорет. (ВЖХ: 98,3%-ный).
++МС: m+ 323 и (m+1)+ 324 получено.
Фрагментация подтверждает структуру.
2) R2 = водород или С1-С4-алкил;
3)
R3 = водород;
4) R4 = водород, С1-С4-алкил;
5) R5 = меркапто-С1-С4-алкил, С1-С6
-алканоил-тио-С1-С4-алкил, в частности алкилен-S-пивалат, алкилен-S-бутират, алкилен-S-пропионат, алкилен-S-ацетат, алкилен-S-гексаноат, алкилен-S-октаноат, алкилен-S-бензоат,
алкилен-S-сукциноилэтиловый эфир, алкилен-тиокарбонат, алкилен-тиокарбамат, алкилен-тионизший алкил, низший алкил-тионизший алкилкарбоновая кислота и/или их сложные эфиры и/или их амиды.
Следующие примеры подтверждают вывшеуказанные значения R5:
R5 = метилен-S-гексаноил, метилен-S-октаноил, метилен-S-бензоил, метилен-S-сукциноилэтиловый эфир,
метилентиокарбонат (СH2-S-CO2-R).
Получение этилового эфира N-(3-нитратопивалоил)-S-гексаноилцистеина.
Растворяют 20 г (0,07 моль) этилового эфира N-(3-нитратопивалоил)-цистеина в 200 мл дихлорметана и добавляют 11,2 мл (0,08 моль) гексаноилхлорида. Потом при температуре 5-10оС по каплям добавляют 11 мл (0,08 моль) триэтиламина. Исходную смесь дополнительно перемешивают в течение 15 ч и затем промывают ее подряд 5%-ным HCl, конц. раствором NaHCO3 и водой. Дихлорметановую фазу концентрируют. Согласно СЖХ (MPLC) получают 8,28 г масла++ (31,25% от теорет. , БЖХ: 97,3%-ный);++МС: m+392 и (n+1)+ 393 видно. Фрагментация подтверждает структуру.
R5 = СH2-S-CH2-COOET.
Получение этилового эфира N-(3-нитратопивалоил)-S-(карбэтоксиметил)-цисте- ина.
Растворяют 14,4 г (0,061 моль) этилового эфира карбэтоксиметилцистеина и 11,05 г (0,068 моль) 3-нитратопивалиновой кислоты в 150 мл дихлорметана. При температуре 15-25оС по каплям добавляют 14 г (0,068 моль) дициклогексилкарбодиимида (ДЦК), растворенного в 50 мл дихлорметана. Исходную смесь дополнительно перемешивают при комнатной температуре в течение 15 ч. Затем отфильтруют мочевину и фильтрат концентрируют. После переработки путем хроматографии на колонке получают 8,5 г масла++ = 36,6% от теорет.
++МС: m+380 и (m+1)+ 381 видно. Фрагментация подтверждает структуру.
R5 = CH2-S-CONH2.
Получение этилового эфира N-(3-нитратопивалоил)-S-карбамоилцистеина.
По 0,05 моль этилового эфира карбамоилцистеина и 3-нитратопивалиновой кислоты растворяют в 100 мл дихлорметана. При комнатной температуре по каплям добавляют 0,5 моль дициклогексилкарбодиимида, растворенного в 50 мл дихлорметана. После переработки путем хроматографии на колонке получают продукт с выходом в размере 24%; Т.пл. 58оС.
6) R5 и R с тиолактоновым мостиком.
Получение тиолактона N-(3-нитратопивалоил)-гомоцистеина.
Суспендируют 5 г (0,03 моль) нитратопивалиновой кислоты и 4,7 г (0,03 моль) гомоцистеинтиолактона х HCl в 50 мл ТГФ. После добавления 2,4 г пиридина при температуре 5-10оС по каплям добавляют раствор 6,2 г дициклогексилкарбодиимида (ДЦК) в 20 мл ТГФ. Продукт подвергают переработке путем хроматографии на колонке и затем его перекристаллизуют из гексана/EtOH.
Выход 0,51 г = 6,5% от теорет. Т.пл. 88, 6оС.
Элементарный анализ показал: 41,43% С (41,21%); 5,44% Н (5,38%); 30,1% О (30,5%).
7) R = гидрокси, С1-С4-алкокси, амино, С1 -С4-алкиламино, ди-С1-С4-алкиламино, низший-алкиламинонизший-алкокси, динизший алкиламино-низший алкокси, ацилокси-низший алкокси, арилнизший алкокси, замещенный арилнизший алкокси, где заместитель означает метил, галоген или метокси или аминокислотные остатки, выбранные из группы цистеина, метионина или гомоцистеина, присоединенные пептидной связью.
Соответственные примеры могут служить дополнением к примеру 16, III стадия.
R = динизший-алкил-аминонизший алкокси
ацилокси-низший алкокси
арилнизший
алкокси
Дополнение к примеру 16, III стадия.
При использовании соответственных эфиров цистеина таким же образом получают этиловый эфир N-(3-нитратобутирил)-цистеиндиметиламино (масло, МС: m+ 323 и (m+1)+ 324 видно. Фрагментация подтверждает структуру), бензиловый эфир N-(3-нитратобутирил)-цистеина (Т.пл. 33оС) и этиловый эфир гликолевой кислоты N-(3-нитратобутирил)-цистеинила (Т.пл. 48оС).
8). Пример, иллюстрирующий R = пептид.
Растворяют 4 г (10 ммоль) N-12-нитратолауроил-S-ацетилцистеина и 10 ммоль пептида в 80 мл дихлорметана. При комнатной температуре по каплям добавляют раствор 2,06 г (10 ммоль) дициклогексилкарбодиимида (ДЦК) в 20 мл дихлорметана. Исходную смесь дополнительно перемешивают при комнатной температуре в течение 15 ч. Мочевину ДЦК отфильтруют и дихлорметан полностью удаляют. Остаток перерабатывают с помощью СЖХ.
Выход 3,87 г. Т.пл. 68,4оС (диизопропиловый эфир).
Присоединение пептида N-концом к С-концу N-12-нитратолауроил-S-ацетилцистеина осуществляется аналогичным образом, что и синтез нижеописанного пентапептида - Tyr-Pro-Phe-Pro-Gly-метиловый эфир.
9) Синтез пентапептида, имеющего последовательность: Tyr-Pro-Phe-Pro-Gly-метиловый эфир.
а) План синтеза.
Аминовый компонент взаимодействует при температуре -15оС или ниже в диметилформамиде (ДМФ) с 0,5 молярным избытком смешанного ангидрида изобутилкарбоновой кислоты Z-аминокислоты (причем Z служит защитной группой и означает остаток N-бензилоксикарбонила) в течение 2-4 ч.
Смешанный ангидрид образуют в ДМФ при температуре -15оС или ниже в течение 10-15 мин при использовании 6%-ного избытка у производного Z-аминокислоты и N-метилморфолина через изобутиловый эфир хлормуравьиной кислоты. Избыток смешанного ангидрида затем разрушают. При температуре 0оС водным и насыщенным раствором KHCO3 доводят рН реакционного продукта до значения 8 и перемешивают его в течение 30 мин.
Пептиды экстрагируют этилацетатом. Для удаления соли калия Z-аминокислоты промывают смесь этилацетата и пептида три раза хлоридом натрия/водой и три раза водой и потом выпаривают его. Полученный таким образом пептид, который еще имеет защитную группу Z, гидрируют в метаноле. При этом добавляют 100-500 мг Pd/активный уголь-катализатор на ммоль пептида. Отщепление СО2 контролируют раствором Ва(ОН)2. Катализатор отфильтруют (бумажный фильтр N 595 фирмы Schleicher & Schull), тщательно промывают его водой и фильтрат испаривают на ротационном испарителе (типа Rotavapor, фирмы Buchi). Желаемый деблокированный пептид находится в остатке.
б) Синтез пентапептида
Tyr-Pro-Phe-Pro-Gly-метиловый эфир.
Стадия I.
а) Получение смешанного ангидрида (Z-Phe-Pro-смешанный ангидрид).
640 мг (1,6 ммоль = 6%-ный избыток) дипептида Z-L-Phe-L-Pro (причем Z представляет собой остаток N-бензилоксикарбонила, который имеет функцию защитной группы) подвергают взаимодействию в 20 мл диметилформамида (ДМФ) 200 мкл (1,5 ммоль) изобутилового эфира хлормуравьиной кислоты при температуре -15оС и в течение 15 мин после добавления 170 мкл (1,6 ммоль) N-метилморфолина.
б) Приготовление аминового компонента.
125,6 мг (1,0 ммоль) глицинметилэфиргидрохлорида растворяют в 20 мл ДМФ при добавлении 100 мкл (1 ммоль) N-метилморфолина при температуре -15оС.
Стадия II.
Реакция смешанного ангидрида из стадии 1а с аминовым компонентом 1б. Z-Phe-Pro-смешанный ангидрид подвергают взаимодействию с глицинметиловым эфиром в 40 мл ДМФ при температуре -15оС и в течение 4 ч.
z-Phe-Pro-смешанный ангидрид+Gly-метэфир -15оС - - - - -> z-Phe-Pro-метэфир 4 ч
До переработки разрушают
50%-ный избыток смешанного ангидрида. При температуре 0оС водным, насыщенным раствором КНСО3 доводят рН реакционного продукта до значения 8 и перемешивают его в течение 30 мин
при температуре 0оС. Затем экстрагируют пептид 50-100 мл этилацетата (EtAc). Смесь этилацетата и пептида промывают насыщенным, водным раствором хлорида натрия. После дополнительной
окончательной промывки водой выпаривают фазу этилацетата.
Стадия III. Отщепление защитной группы путем гидрирования.
Растворяют пептид в 30 мл метанола и добавляют 100 мг палладия на активном угле (Merck). После вытеснения воздуха с помощью азота подают водород в реакционный сосуд. Гидрирование осуществляют при температуре 25-30оС. Гидрирование закончено, когда больше не освобождается СО2, т.е. когда после контроля в водном растворе гидроксида бария больше не образуется осадок. Раствор фильтруют, промывают его водой и подвергают его ротационной обработке на ротационном испарителе. Оставшийся промежуточный продукт используют в качестве аминового компонента на IV стадии.
Стадия IV.
а) Получение смешанного ангидрида (Z-Pro-смеш. ангидрид).
Растворяют 374 мг (1,5 ммоль) Z-L-пролина в 15 мл ДМФ при добавлении 170 мкл (1,5 ммоль) N-метилморфолина и реагируют раствор 180 мкл (1,4 ммоль) изобутилового эфира хлормуравьиной кислоты при температуре -15оС в течение 15 мин.
б) Реакция смешанного ангидрида из стадии IVа с аминовым компонентом
стадии III.
Z-L-Pro-ангидрид + Phe-Pro-Gly-метэфир (в
-15оС 15 мл ДМФ) - - - - - - - - -> Z-Pro-Phe--Pro-Gly-метэфир.
4 ч Разрушение избыточного смешанного ангидрида, экстракцию, гидрирование осуществляют в соответствии с вышеописанным.
Конечный продукт IVб служит аминовым компонентом для стадии V. Контроль по составу аминокислот после гидролиза показал, что пептид имеет правильное молярное соотношение аминокислот.
Стадия V.
а) Образование смешанного ангидрида (Z-Tyr-смеш. ангидрид).
Растворяют 629,24 (1,4 ммоль) N,O-ди-Z-L-тирозина в 15 мл ДМФ при добавлении 165 мкл (1,4 ммоль) N-метилморфолина и раствор подвергают взаимодействию с 175 мкл (1,3 ммоль) изобутилового эфира хлормуравьиной кислоты при температуре -15оС и в течение 15 мин.
б) Реакция смешанного ангидрида стадии Vа с конечным продуктом стадии IVб. Конечный продукт стадии IVб растворяют в 15 мл ДМФ и его подвергают взаимодействию с смешанным ангидридом стадии 5а при температуре -15оС и в течение 4 ч.
Разрушение избыточного смешанного ангидрида, экстракцию и гидрирование осуществляют в соответствии с вышеописанным.
Анализ аминокислот, проведенный после гидролиза в кислой среде, показал правильное молярное соотношение аминокислот в соответствии с пентапептидом Tyr-Pro-Phe-Pro-Gly-метиловый эфир.
10) m, n и о имеют значение 0 и/или 1-10.
б) Стереохимические формы использованных аминокислот.
Использованные аминокислоты, например цистеин, были получены от фирмы Fluka. Они получили D-аминокислоты, а также L-аминокислоты.
Принципиально использовали L-аминокислоты. Рацемические D/L-аминокислоты или D-аминокислоты могут быть использованы таким же образом.
В дополнение к примеру 20, стадия III, следующий вариант может быть введен.
Получение N-(12-нитратолауроил)-D-цистеина.
Если заместить L-цистеин 6,06 г D-цистеина (Fluka), то при тех же самых условиях реакции и переработки получают 3,8 г кристаллического продукта. Т.пл. 59оС.
в) Получение этилового эфира N-(3-нитратопивалоил)-метионина.
12,4 г (70,0 ммоль) основания этилового эфира L-метионина растворяют с перемешиванием в атмосфере азота и при комнатной температуре в 250 мл метиленхлорида. Добавляют 11,4 г (70,0 ммоль) кристаллической нитратопивалиновой кислоты и с перемешиванием растворяют ее при комнатной температуре. Перемешивая, к этой смеси по каплям добавляют в атмосферу азота раствор 14,8 г (71,7 ммоль) N, N-дициклогексилмочевины в 50 мл метиленхлорида в течение примерно 15 мин и при комнатной температуре, причем температура повышается до 35оС. После дополнительного перемешивания осаждается белая дициклогексилмочевина. Исходную смесь охлаждают до комнатной температуры и ее перемешивают в течение ночи в атмосфере азота. N,N-дициклогексилмочевину затем отсасывают через пористый стеклянный фильтр и один раз ее промывают 50 мл СН2СН2. Объединенные растворы метиленхлорида один раз промывают 100 мл 1 н.HCl и дважды по 100 мл дист. Н2О (в атмосфере азота) и затем их концентрируют в ротавапоре при температуре ванны 40оС и при вакууме, получаемом с помощью водоструйного насоса, от ≈550 мБар до ≈20 мБар. Получают светло-желтое масло.
Выход 24,9 г = 110,3% от теорет. сырого этилового эфира N-(3-нитратопивалоил)-L-метионина. Сырой продукт очищают путем препаративной хроматографии на колонке.
Выход 17,6 г = 78,0% от теорет. этилового эфира N-(3-нитратопивалоил)-метионина в виде бесцветного масла.
В данном случае для включения в объем защиты С1-С4 -алкилтио-С1-С4-алкил как значение радикала R5 указывается физический параметр - показатель преломления, который имеет значение 1,4897 при температуре 25о С.
Пример, иллюстрирующий образование солей.
Получение Na-соли N-(12-нитратолауроил)-S-пивалоилцистеина.
446 мг (1 ммоль) N-(12-нитратолауроил)-S-пивалоилцистеина растворяют в 50 мл этилацетата. В атмосфере азота медленно по каплям добавляют раствор, состоящий из 40 мг (1 ммоль) NaOH и 5 мл этанола. Осажденный продукт отсасывают и высушивают в вакууме.
Выход 480 мг. Т.пл. 118-125оС (Z).
Получение триэтаноламиновой соли N-(3-нитратопивалоил)-S-пивалоилцистеина.
1,75 г (5 ммоль) N-(3-нитратопивалоил)-S-пивалоилцистеина растворяют в 20 мл дихлорметана. В атмосфере азота и при комнатной температуре по каплям добавляют 750 мг (5 ммоль) триэтаноламина, растворенного в 10 мл дихлорметана. Дихлорметан полностью удаляют.
Выход 2,5 красного масла++, которое затвердевает при температуре 5-7оС.
++МС: m+ 470 и (m+1)+ 471 видно. Фрагментация подтверждает структуру.
Результаты биологических испытаний соединений по изобретению.
Что касается токсичности, то заявитель отмечает, что соединения согласно изобретению обладают слабой токсичностью. Соединения этиловый эфир N-нитратопивалоил-S-(N-ацетиланил)-L-цистеина и этиловый эфир N-нитратопивалоил-S-(N-ацетилглицил)-L-цистеина были подвергнуты испытаниям по токсичности как представители класса веществ. Согласно результатам испытаний нетоксичная доза составляет 200 мг соединения по изобретению на 1 кг массы тела. В соответствии с строгими правилами по токсичности для лекарственных средств каждый день до 160 мг соединения по изобретению могут быть приняты в течение длинных периодов времени без побочных эффектов.
Фармакологические данные по выбранным соединениям согласно изобретению:
а) Фармакологическая in vivo модель (бодрствующая собака)
Гемодинамический образ действия этилового эфира N-нитратопивалоил-S-(N-ацетилаланил)-L-цистеина и этилового эфира
N-нитратопивалоил-S-(N-ацетилглицил)-L-цистеина после внутривенного и орального введения испытывают путем прямого сравнения с этиловым эфиром N-нитратопивалоил-S-пивалоил-L-цистеина и классическим
органическим нитратом - изосорбид-5-мононитратом (IS-5-N) в бодрствующих натренированных собаках рода Beagle. Параметры по кровообращению регистрируют с помощью атериального катетера-манометра и с
помощью катетера, введенного через V,hugularis. Артериальную систему описывают систолическим, средним и диастолическим кровяным давлением и частотой сердечных сокращений, а венозную систему
- центрально-венозным и легочно-артериальным давлением. Одновременно определяют минутный объем крови путем термодилуции и вычисляют общее периферическое сопротивление и эластичность артериальной
сосудистой системы.
Оба вещества, а именно этиловый эфир N-нитратопивалоил-S-(N-ацетилаланил)-L-цистеина и этиловый эфир N-нитратопивалоил-S-(N-ацетилглицил)-L-цистеина проявляют при
внутривенном, а также при оральном введении, спектр действий, типичный для нитрата, однако, они в общем имеют значительно сильное действие, чем сравнительные вещества, т.е. этиловый эфир
N-нитратопивалоил-S-пивалоил-L-цистеина и IS-5-N. В табл.1 представлено репрезентативное сравнение уровней действия после введения эквимолярных доз соответственного вещества (1,7 мкммоль/кг
внутривенно) в бодрствующую собаку (н = 5):
б) Фармакологическая in vitro модель (изолированные кольца аорты):
Действие вышеописанных нитратов также испытывают in vitro по отношению
их эффекта расслабления сосудов. В качестве модели используют изолированную Aorta thoracalis крысы. После эквилибрации в течение 1 ч сегментов кольца аорты с шириной 4-5 мм, от которых предварительно
удаляют адвентивные ткани, в растворе Krebs-Henseleit температурой 37оС и в атмосфере кислорода, сосудистые кольца при предварительном нагружении в размере 2 г предварительно сокращают
2х10-7 М фенилэфрина. После достижения стабильного уровня сокращения осуществляют кумулятивное добавление соответственных нитратов в ванну, причем изометрически регистрируют развитие сила
для получения кривой по концентрации и действию. В данной модели соединения, а именно этиловый эфир N-нитратопивалоил-S-(N-ацетилаланил)-L-цистеина и этиловый эфир
N-нитратопивалоил-S-(N-ацетилглицил)-L-цистеина также проявляют более сильное действие чем нитраты, т.е. этиловый эфир N-нитратопивалоил-S-пивалоил-L-цистеина и IS-5-N. Ниже следует сравнение
установленных концентраций (ЕС50), которые вызывают полумаксимальное расслабление сокращенных колец аорты:
Вышеописанные фармакологические испытания доказывают, что соединения,
согласно изобретению, благодаря соединительному компоненту, т.е. N-ацетиламинокислоты, в частности N-ацетилглицина или N-ацетилаланина, соответственно обладают значительно высоким действием нитрата,
чем сравнительное вещество - этиловый эфир N-нитратопивалоил-S-пивалоил-L-цистеина. Это обозначает для терапевтического применения пониженное введение вещества при том же самом действии.
В ходе метаболизации этих новых веществ в качестве продуктов гидролиза лишь возникают аминокислоты - глицин и аланин, которые уже присутствуют в микромолярных концентрациях в плазме.
Реферат
Использование: в медицине, при терапии заболеваний сердца. Сущность изобретения: производные нитратоалкановых кислот формулы


Формула
O2N-O-CH2-
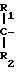




где R -низший алкокси или присоединенный пептидной связью пентапептид Tyr - Pro - Phe - Pro - Gly(OMe);
R1 - водород, С1-С6 - алкил, ациламино;
R2 - водород, низший алкил;
R3 - водород,
R4 - водород, низший алкил;
R5 - (CH2)kSX, где X - водород, низший алкил или X - COR11,
где R11 - водород, низший алкил или остаток N-ациламинокислоты;
m = 0,1 - 10;
n и o = 0 - 2,
или их фармацевтически приемлемые соли.
Комментарии