Контрастные агенты - RU2469021C2
Код документа: RU2469021C2
Описание
Техническая область изобретения
Настоящее изобретение относится к классу соединений, которые являются йодсодержащими соединениями, и к диагностическим композициям, содержащим такие соединения. Более конкретно, эти йодсодержащие соединения представляют собой химические соединения, содержащие две связанные йодированные фенильные группы, имеющие общую формулу R-N(CHO)-X-N(R3)-R, где Х означает возможно замещенную алкиленовую группу, R3 означает атом водорода или ацильную функциональную группировку и каждый R означает трийодированный фенильный остаток, дополнительно замещенный гидрофильными группировками. Изобретение также относится к применению таких диагностических композиций в качестве контрастных агентов в диагностической визуализации, в частности в рентгеновской визуализации, и к контрастным средам, содержащим такие соединения.
Описание предшествующего уровня техники
Любая диагностическая визуализация основана на достижении разных уровней сигналов от разных структур в организме. Так, например, при рентгеновской визуализации на изображении данной визуализируемой структуры организма ослабление рентгеновского излучения этой структурой должно отличаться от ослабления рентгеновского излучения окружающими тканями. Разница в сигнале между структурой организма и ее окружением часто называется контрастом, и много усилий было направлено на создание средств усиления контраста при диагностической визуализации, поскольку чем больше контраст между структурой организма и ее окружением, тем выше качество изображений и больше их ценность для врача, ставящего диагноз. Более того, чем больше контраст, тем меньшего размера структуры организма могут быть визуализированы, то есть увеличение контраста может приводить к увеличению пространственного разрешения.
Диагностическое качество изображений сильно зависит от внутреннего уровня шума при визуализации, поэтому можно считать, что отношение уровня контраста к уровню шума является эффективным показателем диагностического качества диагностических изображений.
Достижение улучшения такого показателя диагностического качества давно и до сих пор остается важной целью. В таких методах, как рентгеновская визуализация, магнитно-резонансная визуализация (МРВ) и ультразвуковая визуализация, одним из подходов к улучшению показателя диагностического качества было введение усиливающих контраст веществ, приготовленных в виде контрастных сред, в визуализируемую область организма.
Так, для рентгена ранними примерами контрастных агентов были нерастворимые неорганические соли бария, которые усиливали ослабление рентгеновского излучения в зонах организма, в которых они были распределены. В течение последних 50 лет в области рентгеноконтрастных агентов преобладали растворимые йодсодержащие соединения. Коммерчески доступные контрастные среды, содержащие йодированные контрастные агенты, обычно классифицируются как ионные мономеры, такие как диатризоат (продается, например, под товарным знаком Gastrografen™), ионные димеры, такие как иоксаглат (продается, например, под товарным знаком Hexabrix™), неионные мономеры, такие как иогексол (продается, например, под товарным знаком Omnipaque™), иопамидол (продается, например, под товарным знаком Isovue™), иомепрол (продается, например, под товарным знаком Iomeron™) и неионный димер иоксанол (продается под товарным знаком Visipaque™).
Наиболее широко используемые коммерческие неионные рентгеноконтрастные агенты, например, упомянутые выше, считаются безопасными. В США контрастные среды, содержащие йодированные контрастные агенты, используют более чем в 20 миллионах рентгеновских обследований ежегодно, и число нежелательных реакций считается приемлемым. Однако, поскольку для рентгеновского обследования с усиленным контрастом требуется вводить вплоть до примерно 200 мл контрастной среды в суммарной дозе, существует постоянный стимул к созданию улучшенных контрастных сред.
Пригодность контрастной среды определяется, главным образом, ее токсичностью, ее диагностической эффективностью, нежелательными эффектами, которые она может вызывать у субъекта, которому вводят контрастную среду, и простотой производства, хранения и введения. Поскольку такие среды традиционно используют в диагностических целях, а не для достижения прямого терапевтического эффекта, обычно желательно иметь среды, которые оказывают как можно меньшее возможное воздействие на различные биологические механизмы клеток или организма и, соответственно, обеспечивают снижение токсичности и снижение нежелательного клинического эффекта. Токсичность и нежелательные биологические эффекты контрастной среды оказывают компоненты среды препарата, например растворитель или носитель, а также сам контрастный агент и его компоненты, такие как ионы для ионных контрастных агентов, и также их метаболиты.
Главные факторы токсичности контрастной среды идентифицированы как хемотоксичность контрастного агента, осмоляльность контрастной среды и ионный состав контрастной среды или отсутствие такового.
Желательными характеристиками йодированного контрастного агента являются низкая токсичность (хемотоксичность) самого соединения, низкая вязкость контрастной среды, в которой растворено соединение, низкая осмоляльность контрастной среды и высокое содержание йода (часто измеряемое в мг йода на мл контрастной среды для введения). Йодированный контрастный агент также должен быть полностью растворимым в среде препарата, обычно водной среде, и оставаться растворенным при хранении.
Осмоляльность коммерческих продуктов и, в частности, неионных соединений приемлема для большинства сред, содержащих димеры и неионные мономеры, хотя все еще существуют возможности для улучшения. Например, в коронарной ангиографии инъекция болюсной дозы контрастной среды в систему кровообращения вызывает тяжелые побочные эффекты. В этой процедуре контрастная среда, а не ток крови через систему в течение короткого периода времени, и различия в химической и физико-химической природе контрастной среды и крови, которую она вытесняет, могут вызывать нежелательные вредные эффекты, такие как аритмии, пролонгирование QT и снижение силы сердечных сокращений. Такие эффекты наблюдаются, в частности, при использовании ионных контрастных агентов, если осмотоксические эффекты ассоциированы с гипертоничностью инъецируемой контрастной среды. Контрастные среды, которые являются изотоническими или слабо гипотоническими по отношению к жидкостям организма, особенно желательны. Контрастные среды с низкой осмоляльностью имеют низкую почечную токсичность, что является особенно желательным. Осмоляльность является функцией количества частиц на единицу объема приготовленной контрастной среды.
У пациентов с острой почечной недостаточностью нефропатия, индуцированная контрастной средой, остается одним из наиболее клинически важных осложнений использования йодированной контрастной среды. Aspelin, Р. et al. (New England Journal of Medicine 348:491-499 (2003)) пришли к выводу, что нефропатия, индуцированная контрастной средой, может развиваться с меньшей вероятностью у пациентов группы высокого риска при использовании иодиксанола, а не неионной контрастной среды с низкой осмоляльностью.
Часть популяции пациентов, считающаяся пациентами высокого риска, увеличивается. Чтобы удовлетворить потребность постоянного улучшения in vivo рентгеновских диагностических агентов для всей популяции пациентов, существует постоянный стимул к поиску рентгеноконтрастных агентов, которые имеют улучшенные свойства, что также касается индуцируемой контрастным агентом нефротоксичности (CIN).
Чтобы удерживать инъецируемый объем контрастной среды на как можно более низком уровне, крайне желательно готовить контрастные среды с высокой концентрацией йода на мл и при этом поддерживать осмоляльность среды на низком уровне, предпочтительно ниже или близко к изотоничности. В результате разработки неионных мономерных контрастных агентов и, в частности, неионных бис(трийодфенил) димеров, таких как иодиксанол (патент ЕР 108638), созданы контрастные среды с пониженной осмотоксичностью, что дает возможность достигать эффективную с точки зрения контраста концентрацию йода с использованием гипотонического раствора и даже возможность корректировать ионный дисбаланс введением плазменных ионов с сохранением желаемой осмоляльности контрастной среды Visipaque™ (WO 90/01194 и WO 91/13636).
Рентгеновские контрастные среды при коммерческой высокой концентрации йода имеют высокую относительную вязкость в пределах от примерно 15 до примерно 60 мПа·с при температуре окружающей среды. Контрастные среды, где усиливающим контраст агентом является димер, обычно имеют более высокую вязкость, чем соответствующие контрастные среды, где агент, усиливающий контраст, представляет собой мономер, соответствующий этому димеру. Такая высокая вязкость может создавать проблемы тем, кто вводит контрастную среду, связанные с тем, что потребуются относительно более толстые иглы или потребуется прилагать сильное давление, и это особенно актуально в педиатрической радиографии и в радиографических методах, которые требуют быстрого введения болюсов, например для ангиографии.
Рентгеноконтрастные среды, содержащие в качестве активного(ых) фармацевтического(их) ингредиента(ов) химическое соединение, имеющее две трийодированные фенильные группы, связанные связывающей группой, обычно называются димерными контрастными агентами или димерами. За многие годы было предложено большое множество разных йодированных димеров. Релевантные патентные публикации включают ЕР 1186305, ЕР 686046, ЕР 108638, ЕР 0049745, ЕР 0023992, WO 2003080554, WO 2000026179, WO 1997000240, WO 9208691, US 3804892, US 4239747, US 3763226, US 3763227 и US 3678152. В настоящее время на рынке есть одна контрастная среда, имеющая йодированный неионный димер в качестве активного фармацевтического ингредиента, продукт Visipaque™, содержащий соединение иодиксанол. Также на рынке имеется продукт Hexabrix™, содержащий ионное димерное соединение йоксагловую кислоту.
Следовательно, все еще существует потребность в разработке контрастных агентов, которые решают одну или более проблем, которые обсуждались выше. Такие агенты в идеале должны обладать улучшенными свойствами по сравнению с растворимыми йодсодержащими соединениями, имеющимися на рынке, в одном или более следующих отношениях: почечная токсичность, осмоляльность, вязкость, растворимость, инъецируемый объем/концентрация йода и ослабление/доза радиации и любой дополнительный нежелательный эффект, известный или обнаруженный для таких йодированных соединений. Агенты должны быть стабильными при хранении в сухой форме и/или в растворе, и дополнительным желательным свойством является простота и экономичность их изготовления.
Краткое описание сущности изобретения
Согласно настоящему изобретению предложены соединения, полезные в качестве контрастных сред, имеющих улучшенные свойства по сравнению с известными средами по меньшей мере по одному из критериев, упомянутых выше, и, в частности, почечной токсичности, осмоляльности, вязкости и растворимости. Контрастные среды содержат усиливающие контраст йодсодержащие соединения, причем эти йодсодержащие соединения представляют собой химические соединения, содержащие две связанные йодированные фенильные группы. Усиливающие контраст йодсодержащие соединения могут быть синтезированы из коммерчески доступных и относительно недорогих исходных веществ.
Подробное описание изобретения
Новые соединения по изобретению, их применение в качестве рентгеноконтрастных агентов, их состав и получение точно определены в прилагаемой формуле изобретения и в описании изобретения ниже.
Усиливающие контраст соединения представляют собой синтетические химические соединения формулы (I)

и их соли или оптически активные изомеры,
где
Х означает С3-8 алкиленовую группировку с прямой или разветвленной цепью, в которой возможно одна или две группировки CH2 заменены атомами кислорода, атомами серы или группами NR1 и которая возможно замещена группами -OR1 в количестве до шести включительно;
R1 означает водород или С1-4 алкильную группу с прямой или разветвленной цепью;
R3 означает атом водорода или ацильную функциональную группу; и
R одинаковые или разные и каждый независимо представляет собой трийодированную фенильную группу, дополнительно замещенную двумя группами R2, причем группы R2 одинаковые или разные и каждая представляет собой атом водорода или неионную гидрофильную группировку, при условии, что по меньшей мере одна группа R2 в соединении формулы (I) представляет собой гидрофильную группировку.
В вышеуказанной формуле (I) Х означает С3-8алкилен с прямой цепью, возможно замещенный одной-шестью группами -OR1. Более предпочтительно, Х означает С3-5алкилен с прямой цепью, имеющий по меньшей мере одну группу -OR1, предпочтительно по меньшей мере одну гидроксильную группу в положении, которое не является соседним с мостиковым атомом азота. Более предпочтительно, алкиленовая цепь замещена одной-тремя гидроксильными группами, и еще более предпочтительно алкиленовая цепь представляет собой пропилен, бутилен или пентилен с прямой цепью, замещенный одной, двумя или тремя гидроксильными группами. Конкретные предпочтительные группы Х содержат 2-гидрокси-пропиленовую, 2,3-дигидрокси-бутиленовую, 2,4-дигидрокси-пентиленовую и 2,3,4-тригидрокси-пентиленовую группировку, в частности 2-гидрокси-пропиленовую группировку.
Предпочтительно, R1 означает атом водорода или метильную группу, наиболее предпочтительно атом водорода.
Заместитель R3 предпочтительно представляет собой атом водорода или остаток алифатической органической кислоты, в частности C1-5 органической кислоты, например формильную, ацетильную, пропионильную, бутирильную, изобутирильную и валериильную группировки. Также возможны гидроксилированные и метоксилированные ацильные группировки. В особенно предпочтительном воплощении R3 в соединении формулы (I) означает атом водорода, формильную группировку или ацетильную группировку, наиболее предпочтительно формильную группировку.
Йодированные группы R могут быть одинаковыми или разными, и каждая предпочтительно представляет собой 2,4,6-трийодированную фенильную группу, дополнительно замещенную двумя группами R2 в оставшихся положениях 3 и 5 фенильной группировки.
Неионные гидрофильные группировки могут представлять собой любые неионизирующие группы, традиционно используемые для повышения растворимости в воде. Следовательно, заместители R2 могут быть одинаковыми или разными и предпочтительно все будут представлять собой неионную гидрофильную группировку, содержащую сложноэфирные, амидные и аминные группировки, возможно дополнительно замещенную C1-10 алкильными группами с прямой цепью или разветвленной цепью, предпочтительно С1-5алкильными группами, причем в этих алкильных группах одна или более группировок CH2 или СН могут быть заменены атомами кислорода или азота. Заместители R2 могут также дополнительно содержать одну или более групп, выбранных из оксо, гидроксила, амино или карбоксильного производного, и замещенные группой оксо атомы серы или фосфора. Каждая алкильная группа с прямой или разветвленной цепью содержит предпочтительно от 1 до 6 гидроксильных групп и более предпочтительно от 1 до 3 гидроксильных групп. Поэтому в еще одном предпочтительном аспекте заместители R2 одинаковые или разные и каждый представляет собой полигидрокси-С1-5алкил, гидроксиалкоксиалкил с 1-5 атомами углерода и гидроксиполиалкоксиалкил с 1-5 атомами углерода, и присоединены к йодированной фенильной группе через амидную или карбамоильную связь, предпочтительно амидную связь.
Особенно предпочтительными являются группы R2 формул, указанных ниже:
- CONH2
- СОNНСН3
- CONH-CH2-CH2-OH
- СОNН-СН2-СН2-ОСН3
- CONH-CH2-CHOH-CH2-OH
- СОNН-СН2-СНОСН3-СН2-ОН
- СОNН-СН2-СНОН-СН2-ОСН3
- СОN(СН3)СН2-СНОН-СН2OН
- СОNН-СН-(СН2-ОН)2
- CON-(CH2-CH2-OH)2
- СОN-(СН2-СНОН-СН2-ОН)2
- CONH-ОСН3
- CON(CH2-CHOH-CH2-OH)(CH2-CH2-OH)
- СОNН-С(СН2-ОН)2СН3,
- СОNН-С(СН2-ОН)3 и
- CONH-CH(CH2-OH)(CHOH-CH2-OH)
- NН(СОСН3)
- N(СОСН3)С1-3алкил
- N(СОСН3)-моно-, бис- или трис-гидрокси-С1-4алкил
- N(СОСН2OН)-водород, моно-, бис- или трис-гидрокси-С1-4алкил
- N(СО-СНОН-СН2OН)-водород, моно-, бис- или тригидроксилированный С1-4алкил
- N(СО-СНОН-СНОН-СН2OН)-водород, моно-, бис- или тригидроксилированный С1-4алкил
- N(СО-СН-(СН2OН)2)-водород, моно-, бис- или тригидроксилированный С1-4алкил) и
- N(СОСН2OН)2.
Еще более предпочтительно, группы R2 одинаковые или разные и представляют собой одну или более группировок формул -CONH-CH2-CH2-OH, -CONH-CH2-CHOH-CH2-OH, -СОN(СН3)СН2-СНОН-СН2OН, -CONH-CH-(CH2-OH)2 и -СОN-(СН2-СН2-ОН)2. Еще более предпочтительно, обе группы R одинаковые, и группы R2 в каждом R одинаковые или разные и представляют собой -CONH-CH2-CH2-OH, -CONH-CH2-CHOH-CH2-OH, СОN(СН3)СН2-СНОН-СН2OН, -СОN-(СН2-СН2-ОН)2 и -CONH-CH-(CH2-OH)2. В особенно предпочтительном воплощении обе группы R одинаковые, и все группы R2 представляют собой группировку формулы -CONH-CH2-CHOH-CH2-OH.
Таким образом, предпочтительные структуры по изобретению включают соединения формулы (II):


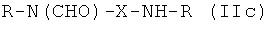

В формуле (II) каждая группа R имеет вышеуказанное значение, более предпочтительно обе йодфенильные группы R одинаковые, и группы R2 все представляют собой неионные гидрофильные группировки, и предпочтительно группы R2 связаны с йодированной фенильной группировкой амидными связями. Х предпочтительно означает алкиленовые группы с прямой цепью с 3-5 атомами углерода, имеющие от одного до трех гидроксильных заместителей в положениях, которые не являются соседними с азотсодержащей функциональной группой.
Особенно предпочтительными являются соединения формулы (IIа), в частности соединения, имеющие моногидроксилированный алкиленовый мостик X, в частности пропиленовый мостик.
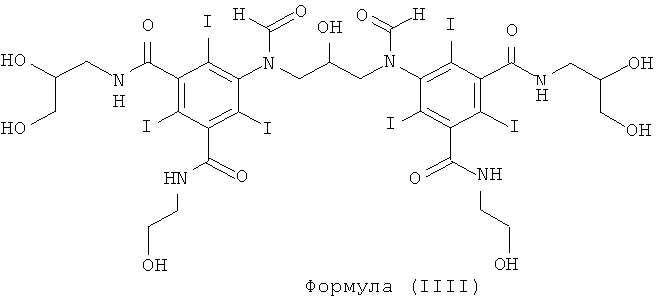
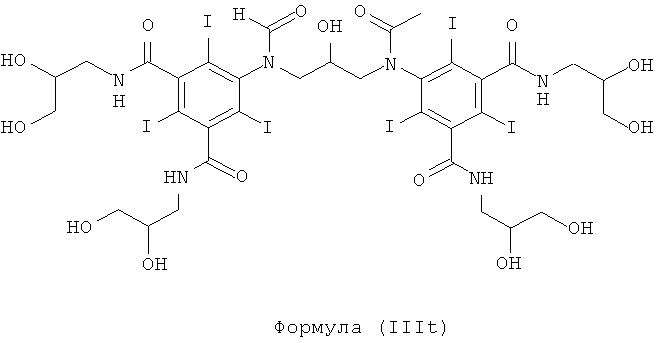
Некоторые предпочтительные примеры структур по изобретению включают указанные ниже соединения формул (IIIa)-(IIIu).




















При концентрации йода 320 мг/мл, которая представляет собой обычную концентрацию для коммерчески доступных йодированных контрастных сред, концентрация соединения формулы (I) будет составлять примерно 0,42 М (молярная). Контрастная среда при этой концентрации йода будет гипоосмолярной, и это является преимущественным свойством с точки зрения нефротоксичности контрастной среды. В контрастную среду могут быть добавлены электролиты для снижения сердечно-сосудистых эффектов, как раскрыто в WO 90/01194 и WO 91/13636.
Соединения формулы (I) также включают оптически активные изомеры и существуют в нескольких изомерных формах, поскольку имеют хиральные атомы углерода. Кроме того, соединения проявляют экзо/эндо-изомерию, обусловленную ограниченным вращением связи N-CO в формильной функциональной группе из-за близости объемного атома йода. Охвачены оба энантиомерно чистых продукта, а также смеси оптических изомеров.
Соединения по изобретению могут быть использованы в качестве контрастных агентов и могут быть приготовлены с традиционными носителями и эксципиентами для получения диагностических контрастных сред.
Таким образом, в еще одном аспекте изобретения предложена диагностическая композиция, содержащая соединение формулы (I), как оно определено выше, вместе с по меньшей мере одним физиологически переносимым носителем или эксципиентом, например в водном растворе для инъекций, возможно вместе с добавленными плазменными ионами или растворенным кислородом.
Композиция контрастного агента по изобретению может быть в форме, готовой для применения, или может быть в форме концентрата для разведения перед введением. Обычно композиции в форме, готовой для применения, будут иметь концентрацию йода по меньшей мере 100 мг I/мл, предпочтительно по меньшей мере 150 мг I/мл, причем концентрация по меньшей мере 300 мг I/мл, например 320 мг I/мл, является предпочтительной. Чем выше концентрация йода, тем выше диагностическая ценность в виде ослабления рентгеновского излучения контрастной средой. Однако чем выше концентрация йода, тем выше вязкость и осмоляльность композиции. В норме максимальная концентрация йода для данной контрастной среды будет определяться растворимостью усиливающего контраст агента, например йодированного соединения, и переносимыми пределами вязкости и осмоляльности.
Для контрастных сред, вводимых инъекцией или инфузией, желательный верхний предел вязкости раствора при температуре окружающей среды (20°С) составляет примерно 30 мПа·с, однако переносимой может быть вязкость до 50-60 мПа·с и даже более 60 мПа·с. Для контрастных сред, вводимых болюсной инъекцией, например при ангиографических процедурах, следует учитывать осмотоксические эффекты, и осмоляльность предпочтительно должна быть ниже 1 осмоль/кг Н2О, предпочтительно ниже 850 мосмоль/кг H2O и более предпочтительно примерно 300 мосмоль/кг Н2О.
Достижение таких целевых вязкости, осмоляльности и концентраций йода возможно с соединениями по этому изобретению. Разумеется, эффективные концентрации йода могут быть достигнуты с использованием гипотонических растворов. Поэтому может быть желательным компенсировать недостаточность тоничности растворов добавлением плазменных катионов, чтобы снизить содействие токсичности вследствие эффектов дисбаланса после болюсной инъекции. Такие катионы желательно вводить в диапазонах, предложенных в WO 90/01194 и WO 91/13636.
В частности, желательным и достижимым является добавление ионов натрия и ионов кальция, чтобы сделать контрастную среду изотоничной с кровью при всех концентрациях йода. Плазменные катионы могут быть предоставлены в форме солей с физиологически переносимыми противоионами, например хлоридом, сульфатом, фосфатом, бикарбонатом и т.д., причем предпочтительным является использование плазменных анионов.
В еще одном воплощении изобретения предложены диагностические агенты, содержащие соединение формулы (I), и диагностические композиции, содержащие соединение формулы (I), вместе с фармацевтически приемлемыми носителями или эксципиентами. Диагностические агенты и композиции предпочтительно предназначены для применения в рентгеновской диагностике.
Контрастные среды, содержащие соединения формулы (I), можно вводить инъекцией или инфузией, например внутрисосудистым введением. Альтернативно, контрастные среды, содержащие соединения формулы (I), можно также вводить перорально. Контрастная среда для перорального введения может быть в форме капсулы, таблетки или в виде жидкого раствора.
Следовательно, изобретение также охватывает применение диагностического агента и диагностической композиции, содержащей соединение формулы (I), в рентгеноконтрастных обследованиях и применение соединения формулы (I) для изготовления диагностической композиции для применения в качестве рентгеноконтрастного агента.
Предложен также способ диагностики, включающий введение соединения формулы (I) в организм человека или животного, обследование организма с использованием диагностического устройства и компилирование данных обследования. В этом способе диагностики соединения формулы (I) можно также вводить в организм предварительно.
Предложен также способ визуализации, в частности рентгеновской визуализации, включающий введение соединения формулы (I) в организм человека или животного, обследование организма с использованием диагностического устройства и компилирование данных обследования и возможно анализ данных. В этом способе визуализации соединения формулы (I) также можно вводить в организм предварительно.
Соединения общей формулы (I) могут быть синтезированы многостадийными способами из исходных веществ, которые либо известны из уровня техники, либо коммерчески доступны, либо без труда могут быть получены из коммерчески доступных веществ. Для получения соединений формулы (I) обычно может быть адаптирован известный синтез иодиксанола.
Получение
Общая методика получения соединений формулы (I)
Соединения формулы (IVa) и, если необходимо, формулы (IVb)

подвергают взаимодействию с реакционноспособной линкерной группой формулы (V)

где Y и Y' представляют собой легко удаляемые атомы или группы, а Х имеет вышеуказанное значение, или ее гидроксил-защищенным производным, или соответствующим эпоксидом, в котором один из заместителей Y и Y' или оба заменены -O-, и, если требуется, затем удаляют защитные группы. Группы Y и Y' могут быть выбраны из атомов галогенов, например хлора, брома или йода, или сульфатных гидрокарбилсульфонилоксигрупп, например алкил- или арил-сульфонилоксигрупп, таких как тозилокси или мезилокси.
Примерами подходящих соединений формулы (V) являются соединения формул (Va), (Vb), (Yc) и (Vd).

где Y представляет собой легкоудаляемый(ую) атом или группу.



Дополнительно, соединения формулы (V), обеспечивающие присутствие мостика с 3 атомами углерода, раскрыты в Bjørsvik, H-R., and Priebe, H. Acta Chem. Scand. 49 (1995) 446-456, "Multivariate data analysis of molecular descriptors estimated by using при semi-empirical quantum chemistry methods. Principal properties for synthetic screening of 2-chloromethyl-oxirane and analogous bis-alkylating C3 moieties".
Таким образом, подходящими соединениями формулы (V) могут быть эпихлоргидрин, диэпоксид бутадиена, диэпоксид 1,4-пентадиена, ди(оксиран-2-ил)метанол или любой предшественник, который может образовать эпоксид или диэпоксид в основных условиях, такой как 1,4-дихлор-бутан-2,3-диол или 1,5-дихлорпентан-2,4-диол.
Гидроксильные группы, присутствующие в группах R и в группе X, могут, если желательно, быть защищенными. Подходящие защитные группы включают ацильные группы, такие как ацетил, или, если имеются соседние гидроксильные группы, такие как циклические кетальные или ацетальные группы.
Взаимодействие между соединениями формул (IVa) и (V) и возможно формул (IVa), (IVb) и (V) предпочтительно осуществляют в присутствии агента, связывающегося с кислотой, например органического или неорганического основания, предпочтительно в водной или спиртовой среде или в таких смесях, как вода и/или алканол или гликоль. В качестве основания может быть использован алкоксид щелочного металла, такой как метоксид натрия, или гидроксид щелочного металла, такой как гидроксид натрия и калия.
Любая защитная группа может быть удалена стандартными способами, например гидролизом. Соединения формул (IVa) и (IVb) могут быть получены формилированием соответствующих соединений, имеющих свободные аминогруппы. В этой реакции гидроксильные группы в заместителях R также могут быть защищены ацилированием.
Соединения формулы (I) могут быть очищены любым удобным способом, например препаративной хроматографией или перекристаллизацией.
Получение промежуточных соединений (если они коммерчески недоступны)
Предшественники соединений формул (IVa) и (IVb), трийодированные фенильные группы, имеющие свободную аминогруппу, коммерчески доступны, или они могут быть получены по методикам, описанным или указанным в, например, WO 95/35122 и WO 98/52911. Например, 5-амино-2,4,6-трийод-изофталевая кислота доступна от Aldrich, а 5-амино-2,4,6-трийод-N,N'-бис(2,3-дигидроксипропил)-изофталамид коммерчески доступен от, например, Fuji Chemical Industries, Ltd.
Примеры коммерчески доступных предшественников соединений формул (IVa) и (IVb), либо коммерчески доступных, либо ранее описанных в литературе, включают:

5-Амино-N,N'-бис-(2,3-дигидрокси-пропил)-2,4,6-трийод-изофталамид

5-Амино-N-(2,3-дигидрокси-пропил)-N'-(2-гидрокси-1-гидроксиметил-этил)-2,4,6-трийод-изофталамид (WO 2002044125)
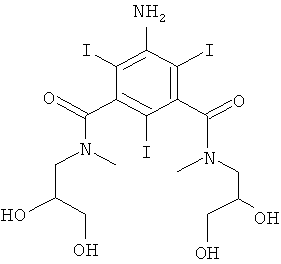
5-Амино-N,N'-бис-(2,3-дигидрокси-пропил)-2,4,6-трийод-N,N'-диметил-изофталамид

5-Амино-N-(2,3-дигидрокси-пропил)-N'-(2-гидрокси-этил)-2,4,6-трийодизофталамид (WO 8700757)
Соединения формул (IVa) и (IVb) могут быть получены ацилированием соответствующих соединений, имеющих свободные аминогруппы. В этой реакции гидроксильные группы в заместителе R также могут быть защищены ацилированием.
Ацилирование может быть осуществлено любым удобным способом, например использованием активированной муравьиной кислоты, такой как смешанные ангидриды, которые могут быть получены различными способами, описанными в литературе.
Удобным способом получения смешанных ангидридов является добавление ангидрида карбоновой кислоты к избытку муравьиной кислоты при контролируемой температуре. Смешанные ангидриды также могут быть получены добавлением хлорангидрида карбоновой кислоты к раствору соли муравьиной кислоты. Формил-смешанные ангидриды могут включать ацетил, изобутирил, пивалоил, бензоил и т.д.
В настоящем воплощении используют смешанный уксусный-муравьиный ангидрид. К избытку охлажденного предварительно полученного смешанного уксусного-муравьиного ангидрида добавляют 5-амино-мономер и эту смесь перемешивают в течение ночи. Смесь концентрируют в вакууме и непосредственно используют на стадии алкилирования, как описано в Экспериментальном разделе (способ Б), или, альтернативно, перед алкилированием O-ацилированные группы могут быть подвергнуты гидролизу, как описано в Экспериментальном разделе (способ А). Гидролиз удобно проводить в водной основной среде, как в примерах, приведенных в Экспериментальном разделе, или, альтернативно, его можно осуществлять алкоголизом, например как описано в WO 1997000240.
Можно также растворить 5-аминомономер в муравьиной кислоте и затем добавить ангидрид карбоновой кислоты, но для снижения нежелательного ацилирования лучше получить смешанный ангидрид отдельно и затем смешать его с 5-аминомономером, как описано выше.
Экспериментальный раздел
Пример 1
5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидроксипропил)-2,4,6-трийодизофталамид)
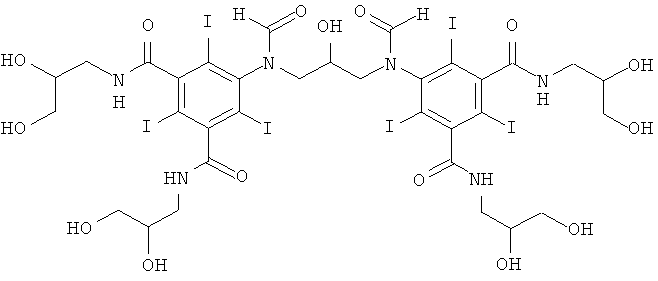
Способ А:
1а) N,N'-Бис-(2,3-дигидроксипропил)-5-формиламино-2,4,6-трийодизофталамид Муравьиную кислоту (300 мл) загружали в сухую колбу на 1000 мл, оснащенную капельной воронкой, мешалкой, термометром и газовпускным патрубком. Кислоту охлаждали на ледяной бане под азотом и уксусный ангидрид (144,8 г, 1,418 моль) по каплям добавляли с такой скоростью, чтобы температура не превышала 2,5°С. После окончания добавления ледяную баню убирали и температуре давали возможность подняться до 10°С. Смесь снова охлаждали на льду, добавляли в нее 5-амино-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид (100 г, 141,8 ммоль) в течение 5 минут и смесь оставляли перемешиваться в течение ночи, при этом ее температура достигала температуры окружающей среды. Смесь упаривали досуха и добавляли метанол (300 мл) и воду (300 мл). Добавляли 2 М гидроксид калия до тех пор, пока все вещество не переходило в раствор и пока не устанавливалось стабильное значение рН 12,5. Метанол удаляли в вакууме. Смесь нейтрализовали добавлением 4 М HCl, при этом начиналось медленное осаждение. Добавляли 300 мл воды, и за ночь продукт выпадал в осадок. Осадок собирали, промывали небольшим количеством воды и сушили на фильтре до образования влажного осадка на фильтре и дополнительно сушили в вакууме с получением 84,8 г (81,5%) N,N'-бис-(2,3-дигидроксипропил)-5-формиламино-2,4,6-трийодизофталамида.
1H-ЯМР 500 МГц (растворитель: D2O, эталон N2O=4,8 м.д. (миллионные доли), 25°С): 8.35 и 8.05 м.д. (2s, 1 Н), 3.94 м.д. (m, 2H), 3.67 м.д. (m, 2H), 3.55 м.д. (m, 2H), 3.45 м.д. (m, 2H), 3.34 м.д. (m, 2H).
ЖХ-МС (жидкостная хроматография/масс-спектрометрия) (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм, растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, ЭРИ-МС (масс-спектрометрия с электрораспылительной ионизацией)) дала два пика при 5,5 мин с m/z (М+ Н+) 733,828, m/z (M+ NH4+) 750,855, m/z (M+ Na+) 755,817, соответствующие данной структуре.
1б) 5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид)
Гидроксид калия (1,07 г) растворяли в воде (6,9 мл) и метаноле (3,4 мл) в круглодонной колбе на 50 мл, оснащенной магнитной мешалкой. К перемешиваемому раствору добавляли борную кислоту (0,41 г, 6,6 ммоль) и N,N'-бис-(2,3-дигидроксипропил)-5-формиламино-2,4,6-трийодизофталамид (7,0 г, 9,56 ммоль). К этому раствору добавляли эпихлоргидрин (260 мкл, 3,32 ммоль) и в колбу устанавливали электрод рН-метра и поддерживали значение рН 12,7 добавлением 4 М раствора гидроксида калия по каплям в течение 4 ч. После этого смесь оставляли перемешиваться в течение ночи. рН доводили до значения рН 4 добавлением 4 М раствора соляной кислоты и метанол удаляли в вакууме. Оставшийся водный раствор разбавляли водой (75 мл) и обрабатывали ионообменниками (АМВ200С и IRA67) до нулевой проводимости. Ионообменники удаляли фильтрованием и промывали водой и объединенные водные фильтраты подвергали сублимационной сушке. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм; растворители: А = вода и В = ацетонитрил; градиент 05-20% В за 60 мин). После сублимационной сушки получили 5,5'-(2-гидроксипропан-1,3-диил)биc(фopмилaзaндиил)-биc(N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-трийодизофталамид) (3,80 г, выход 74,8%).
1H-ЯМР 500 МГц (растворитель: D2O, эталон H2O=4,8 м.д., 25°С): 8.34 и 8.08 м.д. (т, 2 Н), 2.80-4.80 м.д. (т 26 Н). ЖХ-МС времяпролетная (ВП): 1522.68 m/z (М+H+), 1544.66 m/z (М+Na+).
Пример 2
5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидроксипропил)-2,4,6-трийодизофталамид)
Способ В:
2а) 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-илкарбамоил)-2,4,6-трийод-бензол
Муравьиную кислоту (4 л) загружали в сухой снабженный рубашкой реактор на 5000 мл на криостате, оснащенный капельной воронкой, механической мешалкой, термометром и впуском для газа. Кислоту охлаждали в криостате под азотом. По каплям добавляли уксусный ангидрид (1,98 л, 21,0 моль) с такой скоростью, чтобы температура не превышала 12,0°С. Через 7,5 ч добавление завершали, смесь охлаждали до 3,8°С, в нее добавляли 5-амино-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийод-изофталамид (1,481 кг, 2,1 моль) за 20 минут и оставляли смесь перемешиваться в течение ночи, при этом устанавливалась температура окружающей среды. Реакционную смесь упаривали в вакууме при 40°С до влажной массы, ее дополнительно сушили в вакуумном шкафу при 40°С с получением 1754 г (98,8%) 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-илкарбамоил)-2,4,6-трийод-бензола. Этот продукт использовали на следующей стадии без очистки. Полученный продукт содержит незначительное количество фракции O-ацетилэфиров, но поскольку продукт прямо без очистки используют на следующей стадии, этим можно пренебречь.
2б) 5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид)
Снабженный рубашкой реактор на 1000 мл на криостате оснащали внутренним электродом рН-метра, термометром и мешалкой. Реактор охлаждали до 10°С и в него загружали воду (77 мл), метанол (154 мл) и борную кислоту (49,7 г, 803,5 ммоль). Начинали медленное добавление гидроксида калия (9 М) и при Т=0 в реактор добавляли тонкоизмельченный 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-илкарбамоил)-2,4,6-трийод-бензол (341,5 г, 401,8 ммоль). Поддерживали такую скорость добавления гидроксида калия, чтобы рН держался в пределах 11,6-11,7, и поддерживали температуру 10±1°С. При Т=105 минут исходное вещество почти все растворилось, и к нему 5 порциями в течение 60 минут добавляли эпихлоргидрин (16,07 мл, 204,9 ммоль). рН поддерживали в пределах 11,6-11,7 непрерывным добавлением гидроксида калия (9 М). При Т=465 минут рН был 11,7, и смесь оставляли перемешиваться в течение ночи при 10°, не контролируя рН. На следующий день рН поддерживали в пределах 11,6-11,7 непрерывным добавлением гидроксида калия (9 М). В конце дня начинали температурный градиент от 1°С/ч до 20°С и смесь оставляли перемешиваться в течение ночи. На следующий день реакционную смесь разбавляли водой (500 мл), извлекали из реактора и обрабатывали кислотным ионообменником АМВ200С (1841 мл, 3093,6 ммоль). Теперь рН составлял 1,38. Через 5 минут добавляли основный ионообменник IRA67 (2946 мл, 3093,6 ммоль) и рН постепенно достигал 5,67. Через 4 ч ионообменники удаляли фильтрованием и промывали водой (4×2 литров).
Анализ ВЭЖХ (УФ 254 нм) показал, что чистота продукта составляет 90,4%.
Объединенные водные фильтраты объединяли и концентрировали до 1,5 литров в вакууме при 40°С.
Этот неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 (2) 10 мкм; растворители: А = вода, и В = ацетонитрил; градиент 05-20% В за 60 мин). После сублимационной сушки получили 5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидроксипропил)-2,4,6-трийодизофталамид) (222,8 г, выход 72,9%).
ЖХ-МС ВП 1522,68 m/z (M+H+), 1544,66 m/z (M+Na+).
1H-ЯМР 500 МГц (растворитель: D2O, эталон H2O=4,8 м.д., 25°С): 8.34 и 8.08 м.д. (m, 2 Н), 2.80-4.80 м.д. (m 25 Н).
Пример 3
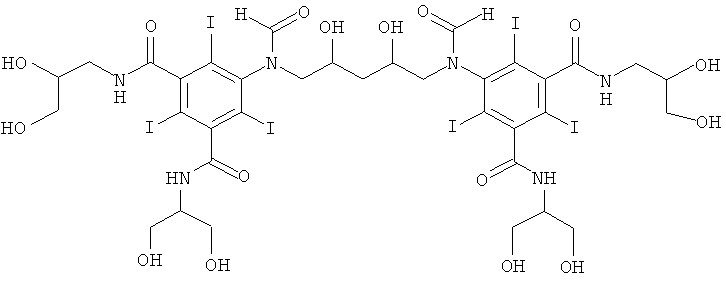
5,5'-(2,3-дигидpoкcибутaн-1,4-диил)биc(фopмилaзaндиил)биc(N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид)

К перемешиваемому раствору из воды (10 мл), метанола (5 мл) и гидроксида калия (1,0 г, 16,4 ммоль) добавляли N,N3-биc(2,3-дигидроксипропил)-5-формиламино-2,4,6-трийодизофталамид (Пример 1а) (10,0 г, 13,6 ммоль). Затем к этому прозрачному раствору добавляли борную кислоту (0,59 г, 9,5 ммоль). Постоянно поддерживали рН 12,6 добавлением гидроксида калия (10 M) и добавляли 1,3-бутадиендиэпоксид (0,40 г, 4,7 ммоль). Постоянно поддерживали рН раствора в пределах от 12,6 до 13 добавлением твердой борной кислоты в течение 5 часов и затем раствор оставляли стоять на выходные. Раствор нейтрализовали добавлением соляной кислоты (18% масс.) и затем обрабатывали ионообменниками (АМВ200С, 20 мл) и (IRA67, 20 мл). Смолы удаляли фильтрованием и промывали водой и объединенный водный объем концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 0-10% В за 60 мин; скорость потока 50,0 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2,3-дигидроксибутан-1,4-диил)биc(фopмилaзaндиил)биc-(N,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-трийодизофталамид) (3,1 г, выход 43%).
ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм; растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала четыре пика при 8,6 мин с m/z 1552,5 [М+Н]+, что согласуется с данной структурой.1H-ЯМР 500 МГц (растворитель: D2O, эталон H2O=4,8 м.д., 25°С): 8.48 м.д. (m, 1H), 8.25 м.д. (m, 1Н)3.40-4.40 м.д.(m, 26Н).
Пример 4
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид)

К перемешиваемому раствору из воды (10 мл), метанола (5 мл) и гидроксида калия (1,0 г, 16,4 ммоль) добавляли N1,N3-биc(2,3-дигидроксипропил)-5-формиламино-2,4,6-трийодизофталамид (Пример 1а) (10,0 г, 13,6 ммоль). К этому прозрачному раствору добавляли борную кислоту (0,59 г, 9,5 ммоль). Постоянно поддерживали рН 12,6 добавлением гидроксида калия (10 М) и добавляли 1,4-пентадиендиэпоксид (0,47 г, 4,7 ммоль). Постоянно поддерживали рН раствора в пределах от 12,6 до 13 добавлением твердой борной кислоты. Реакционную смесь перемешивали в течение выходных, затем нейтрализовали добавлением соляной кислоты (18% масс.) и затем обрабатывали ионообменниками (АМВ200С, 20 мл) и (IRA67, 20 мл). Смолы удаляли фильтрованием и промывали водой и объединенный водный объем сокращали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода, и В = ацетонитрил; градиент 0-17% В за 60 мин; скорость потока 50,0 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2,4-дигидроксипентан-1,5-диил)бис(формилазандиил)-биc(N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-тpийoдизoфтaлaмид) (1,98 г, выход 27%).
ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм; растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала четыре пика при 10 мин с m/z 1566,5 [М+Н]+, что согласуется с ожидаемой массой продукта.
1H-ЯМР 500 МГц (растворитель: D2O, эталон H2O=4,8 м.д., 25°С): 8.15 м.д. (m, 1Н), 8.10 м.д. (m, 1Н), 2.90-4.15 м.д. (m, 26Н) 1.42-1.85 (m, 2Н).
Пример 5
5,5'-(2,3,4-тpигидpoкcипeнтaн-1,5-диил)биc(фoрмилaзaндиил)биc(N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид)

К перемешиваемому раствору из воды (10 мл), метанола (5 мл) и гидроксида калия (1,0 г, 16,4 ммоль) добавляли N1,N3-биc(2,3-дигидроксипропил)-5-формиламино-2,4,6-трийодизофталамид (Пример 1а) (10,0 г, 13,6 ммоль). К этому прозрачному раствору добавляли борную кислоту (0,59 г, 9,5 ммоль). Постоянно поддерживали рН 12,6 добавлением гидроксида калия (10 М) и добавляли 1,4-пентадиен-3-олдиэпоксид (0,55 г, 4,7 ммоль). Диапазон рН раствора поддерживали в пределах от 12,6 до 13 добавлением твердой борной кислоты. Реакционную смесь оставляли перемешиваться на выходные и затем нейтрализовали соляной кислотой (18% масс.) и обрабатывали ионообменниками (АМВ200С, 20 мл) и (IRA67, 20 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 0-10% В за 60 мин; скорость потока 50,0 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2,3,4-тригидроксипентан-1,5-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-трийодизофталамид) (1,386 г, выход 19%).
ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм; растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия электрораспылительной ионизацией) дала пять пиков при 8,4 мин с m/z 1582,5 [М+Н]+, что согласуется с ожидаемой массой продукта.
1H-ЯМР, 500 МГц (DMSO (диметилсульфоксид), 25°С): 8,6-7,8 м.д. (m, 6H), 5,2-4,2 м.д. (m, 10Н), 4,2-3,18 м.д. (m, 21Н) 3,15-2,85 (m, 7Н).
Пример 6
5,5'-(2,3-дигидpoкcибутaн-1,4-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-тpийoд-N1,N3-димeтилизoфтaлaмид)

К перемешиваемому раствору 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-ил-метил-карбамоил)-2,4,6-трийод-бензола (Пример 8а) (9,9 г, 11,3 ммоль) в воде (10 мл) и метаноле (5 мл) добавляли гидроксид калия (10 М) для поддержания рН 12,6. Через 1,5 ч добавляли борную кислоту (0,56 г, 9,0 ммоль). Постоянно поддерживали рН 12,6 добавлением гидроксида калия (10 М) и добавляли 1,3-бутадиендиэпоксид (0,39 г, 4,5 ммоль). Поддерживали рН в пределах от 12,6 до 13 добавлением твердой борной кислоты и раствор оставляли перемешиваться в течение ночи. Раствор нейтрализовали добавлением соляной кислоты (18%) и обрабатывали ионообменниками (АМВ200С, 19 мл) и (IRA67, 19 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 05-20% В за 60 мин; скорость потока 50,0 мл/мин). После сублимационной сушки получили 5,5'-(2,3-дигидроксибутан-1,4-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-тpийoд-N1,N3-диметилизофталамид) (1,38 г, выход 19%).
Анализ ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм; растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала пик с множественным расщеплением при 11,2 мин с m/z 1608,7 [М+Н]+, что согласуется с массой продукта.
1H-ЯМР, 500 МГц (DMSO, 25°С): 8,35 м.д. (bs, 1 Н), 8.2-8.0 м.д. (m, 1.3H), 5.1-4.4 м.д. (m, 9.8H), 4.3-3.4 (m, 19H), 3.3-2.7 м.д. (m, 19H).
Пример 7
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-тpийoд-N1,N3-димeтилизoфтaлaмид)

К перемешиваемому раствору 1-формиламино-3,5-бис(2,3- бис(формилокси)пропан-1-ил-метил-карбамоил)-2,4,6-трийод-бензола (Пример 8а) (9,4 г, 10,8 ммоль) в воде (10 мл) и метаноле (5 мл) добавляли гидроксид калия (10 М) для поддержания рН рН 12,6. Через 30 мин добавляли борную кислоту (0,53 г, 8,6 ммоль). Постоянно поддерживали рН 12,6 добавлением гидроксида калия (10 М) и добавляли 1,4-пентадиендиэпоксид (0,43 г, 4,3 ммоль). Поддерживали рН в пределах от 12,6 до 13 добавлением твердой борной кислоты. Реакционную смесь оставляли перемешиваться в течение 6 суток. Раствор нейтрализовали добавлением соляной кислоты (18%) до рН 7 и обрабатывали ионообменниками (АМВ200С, 18 мл) и (IRA67, 18 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 05-20% В за 60 мин; скорость потока 50,0 мл/мин). После сублимационной сушки получили 5,5'-(2,4-дигидроксипентан-1,5-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-трийод-N1,N3-диметилизофталамид) (740 мг, выход 11%).
ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм; растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала пик с множественным расщеплением при 11,5 мин с m/z 1622,7 [М+Н]+, что согласуется с массой продукта.1Н-ЯМР, 500 МГц (DMSO, 25°С): 8.38 м.д. (bs, 0.9H), 8.18-8.0 м.д. (m, 1.2Н), 5.0-4.3 м.д. (m, 9.6Н), 4.3-3.4 (m, 18.7H), 4.15-2.7 м.д. (m, 19.5H), 2.8-2.4 м.д. (2Н).
Пример 8
5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидроксипропил)-2,4,6-трийод-N1,N3-диметилизофталамид)

8а) 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-ил-метил-карбамоил)-2,4,6-трийод-бензол
Муравьиную кислоту (300 мл) загружали в сухую колбу на 1000 мл, оснащенную капельной воронкой, мешалкой, термометром и впуском для газа. Кислоту охлаждали на ледяной бане под азотом и в нее по каплям добавляли уксусный ангидрид (128,3 мл, 1,357 моль) за 2 ч, не давая температуре повышаться выше 4,5°С. После окончания добавления температуре давали возможность достичь 10°С и ледяную баню ставили обратно. После охлаждения смеси до 3°С всю реакционную смесь вливали в колбу, содержащую твердый 5-амино-N,N'-бис(2,3-дигидроксипропил)-N,N'-диметил-2,4,6-трийод-изофталамид (99,5 г, 135,7 ммоль). Смесь оставляли перемешиваться в течение ночи. Гомогенный теперь раствор упаривали досуха в вакууме при 40°С и использовали без очистки на следующей стадии.
Полученный продукт содержит минорную фракцию O-ацетилэфиров, но, поскольку продукт прямо без очистки используют в следующей стадии, этим можно пренебречь.
8б) 5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-тpийoд-N1,N3-димeтилизoфтaлaмид)
В колбу, оснащенную электродом рН-метра, мешалкой и термометром, добавляли воду (6 мл), метанол (3 мл), воду и 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-ил-метил-карбамоил)-2,4,6-трийод-бензол (7,6 г, 10 ммоль) и затем борную кислоту (1,24 г, 20 ммоль). Постоянно добавляли гидроксид калия (10 М) для поддержания стабильного рН 11,5 и поддерживали температуру 10°С, используя баню вода/лед. При достижении стабильного рН 11,5 добавляли к теперь уже прозрачному раствору эпихлоргидрин (527 мг, 5,7 ммоль) за 15 минут. Поддерживали рН в пределах от 12,5 до 12,8 добавлением твердой борной кислоты при 10°С и через 5 ч смесь оставляли перемешиваться в течение ночи. Реакционную смесь разбавляли водой (50 мл) и обрабатывали ионообменниками (АМВ200С, 15 мл) и (IRA67, 15 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода, и В = ацетонитрил; градиент 05-20% В за 60 мин; скорость потока 50,0 мл/мин). После сублимационной сушки получили 5,5'-(2-гидроксипропан-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-тpийoд-N1,N3-диметилизофталамид) (2,40 г, выход 30%).
ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм; растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала широкий пик при 11,9 мин с m/z 1578,7 [М+Н]+, что согласуется с массой продукта.
1H-ЯМР, 500 МГц (DMSO, 25°С): 8,50-7,95 м.д. (m, 2H), 5,1-3,4 м.д. (m, 27,6Н), 3,3-2,7 м.д. (m, 18,41-1).
Пример 9
5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(1,3-дигидроксипропан-2-ил)-2,4,6-трийодизофталамид)

9а) 1-формамидо-3,5-бис(1,3-бис(формилокси)пропан-2-илкарбамоил)-2,4,6-трийод-бензол
Муравьиную кислоту (800 мл) загружали в сухую колбу на 2000 мл, оснащенную капельной воронкой, мешалкой, термометром и впуском для газа. Кислоту охлаждали на ледяной бане под азотом и по каплям за 2 ч добавляли уксусный ангидрид (436 мл, 3,972 моль), не давая температуре подниматься выше 4,5°С. После окончания добавления температуре давали возможность достичь 10°С и ледяную баню возвращали обратно. После охлаждения смеси до 3°С добавляли 5-aминo-N1,N3-биc(1,3-дигидpoкcипpoпaн-2-ил)-2,4,6-трийодизофталамид (280,0 г, 397,2 ммоль) и смесь оставляли перемешиваться в течение ночи. Теперь гомогенный раствор упаривали досуха в вакууме при 40°С и использовали без очистки на следующей стадии.
Полученный продукт содержит некоторое количество минорной фракции O-ацетилэфиров, но поскольку продукт прямо без очистки используют на следующей стадии, этим можно пренебречь.
9б) 5,5'-(2-гидроксипропан-1,3-диил)бис(формилазандиил)бис(N1,N3-бис(1,3-дигидроксипропан-2-ил)-2,4,6-трийодизофталамид)
К перемешиваемой суспензии 1-формамидо-3,5-бис(1,3-бис(формилокси)-пропан-2-илкарбамоил)-2,4,6-трийод-бензола (11,5 г, 13,6 ммоль) в воде (5 мл) и метаноле (5 мл) добавляли борную кислоту (0,60 г, 9,6 ммоль). Затем по каплям добавляли раствор гидроксида калия (10 М)) для поддержания рН 12,6. К этому прозрачному раствору добавляли эпихлоргидрин (0,44 г, 4,8 ммоль). Поддерживали рН в пределах от 12,6 до 13 добавлением твердой борной кислоты. Реакционную смесь оставляли перемешиваться в течение ночи и затем нейтрализовали добавлением соляной кислоты (18%) и обрабатывали ионообменниками (АМВ200С, 36 мл) и (IRA67, 34 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 05-20% В за 60 мин; скорость потока 50,0 мл/мин). После сублимационной сушки получили 5,5'-(2-гидроксипропан-1,3-диил)биc(фopмилaзaндиил)биc(N1,N3-биc(1,3-дигидpoкcипpoпaн-2-ил)-2,4,6-трийодизофталамид) (2,9 г, выход 40%).
ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм; растворители: А = вода/0,1%-ная муравьиная кислота и В = ацетонитрил/0,1%-ная муравьиная кислота; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала три пика при 9,3 мин с m/z 1522,6 [М+Н]+, что согласуется с предполагаемой массой продукта.
1H-ЯМР, 500 МГц (DMSO, 25°С): 8.5-7.4 м.д. (m, 6H), 5.2-4.4 м.д. (m, 9.4Н), 4.4-3.4 м.д. (m, 24Н), 3.25-3.15 м.д. (m, 0.5Н).
Пример 10
5,5'-(2,3-дигидpoкcибутaн-1,4-диил)биc(фopмилaзaндиил)биc(N1,N3-бис(1,3-дигидроксипропан-2-ил)-2,4,6-трийодизофталамид)

К перемешиваемой суспензии 1-формамидо-3,5-бис(1,3-бис(формилокси)-пропан-2-илкарбамоил)-2,4,6-трийод-бензола (Пример 9а) (11,5 г, 13,6 ммоль) в воде (5 мл) и метаноле (5 мл) добавляли твердую борную кислоту (0,60 г, 9,6 ммоль). Затем по каплям добавляли раствор гидроксида калия (10 М) для поддержания рН 12,6. К этому прозрачному раствору добавляли 1,3-бутадиендиэпоксид (0,41 г, 4,8 ммоль). Поддерживали рН в пределах от 12,6 до 13 добавлением твердой борной кислоты. Реакционную смесь оставляли перемешиваться в течение ночи и затем нейтрализовали добавлением соляной кислоты (18%) и обрабатывали ионообменниками (АМВ200С, 36 мл) и (IRA67, 36 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 0-20% В за 60 мин; скорость потока 50,0 мл/мин). После сублимационной сушки получили 5,5'-(2,3-дигидроксибутан-1,4-диил)бис(формилазандиил)бис(N1,N3-бис(1,3-дигидроксипропан-2-ил)-2,4,6-трийодизофталамид) (3,1 г, выход 42%). ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм, растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала три пика при 8,4 минут с m/z 1552,6 [М+Н]+, что согласуется с предполагаемой массой продукта.
1H-ЯМР, 500 МГц (DMSO, 25°С): 8.45-7.50 м.д. (m, 6H), 5.15-4.25 м.д. (m, 9.8Н), 4.2-3.35 м.д. (m, 25Н), 3.25-3.05 м.д. (m, 1Н).
Пример 11
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(N1,N3-бис(1,3-дигидроксипропан-2-ил)-2,4,6-трийодизофталамид)

К перемешиваемой суспензии 1-формамидо-3,5-бис(1,3-бис(формилокси)-пропан-2-илкарбамоил)-2,4,6-трийод-бензола (Пример 9а) (11,5 г, 13,6 ммоль) в воде (5 мл) и метаноле (5 мл) добавляли твердую борную кислоту (0,60 г, 9,6 ммоль). Затем по каплям добавляли раствор гидроксида калия (10 М) для поддержания рН 12,6. К этому прозрачному раствору добавляли 1,4-пентадиендиэпоксид (0,48 г, 4,8 ммоль). Поддерживали рН в пределах от 12,6 до 13 добавлением твердой борной кислоты. Реакционную смесь оставляли стоять в течение ночи, добавляли в нее еще одну порцию 1,4-пентадиендиэпоксида (0,20 г, 2,0 ммоль) и оставляли эту реакционную смесь стоять в течение двух суток. Реакционную смесь нейтрализовали добавлением соляной кислоты (18%) и обрабатывали ионообменниками (АМВ200С, 36 мл) и (IRA67, 36 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 05-15% В за 60 мин; скорость потока 50,0 мл/мин). После сублимационной сушки получили 5,5'-(2,4-дигидроксипентан-1,5-диил)бис-(формилазандиил)бис(N1,N3-бис(1,3-дигидроксипропан-2-ил)-2,4,6-трийодизофталамид) (2,52 г, выход 24%).
ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм, растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала четыре пика при 9,4 мин с m/z 1566,7 [М+Н]+, что согласуется с предполагаемой массой продукта.
1H-ЯМР, 500 МГц (DMSO, 25°С): 8,25-7,50 м.д. (m, 6H), 5,25-4,25 м.д. (m, 10Н), 4,25-3,35 м.д. (m, 24,5Н), 3,30-2,80 м.д. (m, 1,6Н), 1,90-13,5 м.д. (m, 2Н).
Пример 12
5,5'-(2,3-дигидpoкcибутaн-1,4-диил)биc(фopмилaзaндиил)биc(N1-(2,3-дигидроксипропил)-N3(2-гидроксиэтил)-2,4,6-трийодизофталамид)

К перемешиваемой суспензии 3-(3-формамидо-5-(2-(формилокси)-этилкарбамоил)-2,4,6-трийодбензамидо)пропан-1,2-диил-диформиата (Пример 13а) (92,0 г, 116,9 ммоль) в воде (117 мл) и метаноле (117 мл) добавляли твердую борную кислоту (14,5 г, 233,8 ммоль). Затем по каплям добавляли раствор гидроксида калия (10 М) для поддержания рН 11,5. По каплям добавляли 1,3-бутадиендиэпоксид (3,5 г, 40,9 ммоль). Поддерживали рН 11,6 непрерывным добавлением гидроксида калия (10 М) в течение нескольких часов. Реакционную смесь оставляли перемешиваться в течение ночи и затем нейтрализовали добавлением соляной кислоты (18% масс.) и обрабатывали ионообменниками (АМВ200С, 900 мл) и (IRA67, 900 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Luna C18 10 мкм 250×75 мм; растворители: А = вода и В = ацетонитрил; градиент 02-10% В за 30 мин; скорость потока 175 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2,3-дигидpoкcибyтaн-1,4-диил)биc(фopмилaзaндиил)-биc(N1-(2,3-дигидроксипропил)-N3-(2-гидроксиэтил)-2,4,6-трийодизофталамид) (36,65 г, выход 60%). ЖХ-МС (колонка Luna C18 3 мкм 2,0×20 мм; растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-20% В за 5 мин; скорость потока 0,6 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала множественный пик при 2,5 мин с m/z 1492,7 [М+Н]+, что согласуется с желаемой структурой.
1H-ЯМР 500 МГц (растворитель: D2O, эталон Н2O=4,8 м.д., 25°С): 8.47 м.д. (m, 1 Н), 8.24 м.д. (m, 1 Н), 4.40-3.35 м.д. (m, 24Н).
Пример 13
5,5'-гидроксипропан-1,3-диил)бис(формилазандиил)бис(N1-(2,3-дигидроксипропил)-N3-(2-гидроксиэтил)-2,4,6-трийодизофталамид)

13а) 3-(3-формамидо-5-(2-(формилокси)этилкарбамоил)-2,4,6-трийодбензамидо)пропан-1,2-диилдиформиат
Муравьиную кислоту (400 мл) загружали в сухую колбу на 2000 мл, оснащенную капельной воронкой, мешалкой, термометром и впуском для газа. Кислоту охлаждали на ледяной бане под азотом и добавляли в нее по каплям уксусный ангидрид (218 мл, 1,986 моль) за 2 ч, не давая температуре подниматься выше 4,5°С. После окончания добавления температуре давали возможность достичь 10°С и ледяную баню ставили обратно. После охлаждения смеси до 3°С добавляли 5-амино-N-(2,3-дигидроксипропил)-N'-(2-гидроксиэтил)-2,4,6-трийод-изофталамид (140 г, 198,6 ммоль), и оставляли смесь перемешиваться в течение ночи. Теперь гомогенный раствор упаривали досуха в вакууме при 40°С и использовали без очистки на следующей стадии.
Полученный продукт содержит некоторое количество минорной фракции O-ацетилэфиров, но поскольку продукт используют прямо на следующей стадии без очистки, этим можно пренебречь.
13б) 5,5'-(2-гидроксипропан-1,3-диил)биc(фopмилaзaндиил)биc(N1-(2,3-дигидpoкcипpoпил)-N3-(2-гидpoкcиэтил)-2,4,6-тpийoдизoфтaлaмид)
К перемешиваемой суспензии 3-(3-формамидо-5-(2-(формилокси)-этилкарбамоил)-2,4,6-трийодбензамидо)пропан-1,2-диилдиформиата (3,0 г, 3,9 ммоль) в воде (3 мл) и метаноле (3 мл) добавляли твердую борную кислоту (0,48 г, 7,8 ммоль). Затем по каплям добавляли раствор гидроксида калия (10 М) для поддержания рН 11,5. Добавляли эпихлоргидрин (130 мг, 1,4 ммоль). Поддерживали рН 11,6 непрерывным добавлением гидроксида калия (10 М) в течение нескольких часов. Реакционную смесь оставляли перемешиваться в течение ночи и затем нейтрализовали добавлением соляной кислоты (18% масс.) и обрабатывали ионообменниками (АМВ200С, 30 мл) и (IRA67, 30 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Luna C18 10 мкм 250×50 мм; растворители: А = вода и В = ацетонитрил; градиент 05-12% В за 30 мин; скорость потока 70,0 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2-гидроксипропан-1,3-диил)бис(формилазандиил)бис(N1-(2,3-дигидроксипропил-N3-(2-гидроксиэтил)-2,4,6-трийодизофталамид) (1,03 г, 50%).
ЖХ-МС (колонка Agilent Zorbax SB-Aq 3,5 мкм 3,0×100 мм; растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-30% В за 20 мин; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала множество изомерных пиков между 9,5 и 11 мин с m/z 1462,6 [М+Н]+, что согласуется с искомой структурой.
1H-ЯМР 500 МГц (растворитель: D2О, эталон Н2O=4,8 м.д., 25°С): 8.35 м.д. (m, 1.1H), 8.10 м.д. (m, 0.85Н), 4.20-3.25 м.д. (m, 23Н).
Пример 14
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(N1-(2,3-дигидpoкcипpoпил)-N3-(2-гидpoкcиэтил)-2,4,6-тpийoдизoфтaлaмид)

К перемешиваемой суспензии 3-(3-формамидо-5-(2-(формилокси)этилкарбамоил)-2,4,6-трийодбензамидо)пропан-1,2-диилдиформиата (Пример 13а) (3,1 г, 4,0 ммоль) в воде (3 мл) и метаноле (3 мл) добавляли твердую борную кислоту (0,49 г, 8,0 ммоль). Затем по каплям добавляли раствор гидроксида калия (10 M) для поддержания рН 11,5. Добавляли 1,4-пентадиендиэпоксид (0,14 г, 1,40 ммоль). Поддерживали рН 11,6 непрерывным добавлением гидроксида калия (10 M) в течение нескольких часов. Реакционную смесь оставляли стоять в течение ночи. Добавляли еще одну порцию 1,4-пентадиендиэпоксида (0,12 г, 1,2 ммоль) и реакционную смесь оставляли стоять в течение двух суток. Реакционную смесь нейтрализовали добавлением соляной кислоты (18% масс.) и обрабатывали ионообменниками (АМВ200С, 30 мл) и (IRA67, 30 мл). Смолы отфильтровывали и промывали водой и объединенный водный раствор концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (колонка Luna C18 10 мкм 250×50 мм; растворители: А = вода и В = ацетонитрил; градиент 5-12% В за 30 мин; скорость потока 70,0 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2,4-дигидроксипентан-1,5-диил)биc(фopмилaзaндиил)биc-(N1-(2,3-дигидpoкcипpoпил)-N3-(2-гидpoкcиэтил)-2,4,6-трийодизофталамид) (352 мг, выход 12%).
ЖХ-МС (колонка Luna C18 3 мкм 2,0×20 мм; растворители: А = вода/0,1% муравьиной кислоты и В = ацетонитрил/0,1% муравьиной кислоты; градиент 0-20% В за 5 мин; скорость потока 0,6 мл/мин, УФ-детектирование при 214 и 254 нм, масс-спектрометрия с электрораспылительной ионизацией) дала множественный пик при 2,8 минут с m/z 1506,8 [М+Н]+, что согласуется с искомой структурой.
1H-ЯМР 500 МГц (растворитель: D2O, эталон Н2O=4,8 м.д., 25°С): 8.35 м.д. (m, 1Н), 8.10 м.д. (m, 1Н), 4.30-2.85 м.д. (m, 24Н), 1.80-1.40 м.д. (m, 2Н).
Пример 15
5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1-(1,3-дигидpoкcипропaн-2-ил)-N3-(2,3-дигидрокcипpoпил)-2,4,6-тpийoдизoфтaлaмид)

15а) 5-aмино-N1-(1,3-дигидроксипропан-2-ил)-N3-(2,3-дигидроксипропил)-2,4,6-трийодизофталамид
В реактор с рубашкой, мешалкой, капельной воронкой и внутренним термометром загружали при 2°С DMA (диметиламин) (640 мл), триэтиламин (224 мл, 161,3 г, 1,59 моль) и дихлорид 5-амино-2,4,6-трийод-изофталевой кислоты (320 г, 537 ммоль). Эту смесь перемешивали и охлаждали до -24°С. Для поддерживания внутренней температуры ниже -19°С медленно добавляли раствор 1-амино-2,3-дигидроксипропана (49,92 г, 548 ммоль) в DMA (160 мл). Смесь перемешивали при температурном градиенте от -24°С до 0°С за 24 ч. Медленно добавляли раствор серинола (60 г, 658 ммоль) в DMA (160 мл) и смесь перемешивали при температурном градиенте от 0°С до 40°С за 20 ч. Смесь перемешивали при 22°С в течение 1 суток и выпавший в осадок гидрохлорид триэтиламина отфильтровывали. После упаривания при 60°С/25 мбар (2,5 кПа) остался остаток в виде вязкой жидкости (586 г), который разбавляли водой (350 мл). Соли и избыток аминов удаляли обработкой ионообменниками Amberlite 200C (143 мл), IRA67 (148 мл) и IRA900 (56 мл) с последующем фильтрованием. Ионообменники промывали водой (2×400 мл). К объединенной водной фазе добавляли некоторое количество затравочных кристаллов и медленно перемешивали раствор при 22°С в течение 9 суток. Осадок выделяли фильтрованием. Осадок на фильтре ресуспендировали в воде (240 мл) и перемешивали в течение 1 суток. Суспензию фильтровали и осадок на фильтре сушили на воздухе (216 г, выход 57%, чистота 88,6% по данным ВЭЖХ). Неочищенный продукт очищали препаративной ВЭЖХ (колонка: самостоятельно заполненная Luna C18, 10 мкм 250×100 мм; растворители: А = вода и В = ацетонитрил; градиент 5-10% В за 10 мин, выдержка 10 мин; скорость потока 350 мл/мин, УФ-детектирование при 244 нм и 254 нм). Соответствующие фракции объединяли и подвергали сублимационной сушке с получением 5-aминo-N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2,3-дигидроксипропил)-2,4,6-трийодизофталамида.
ЖХ-МС с электрораспылительной ионизацией: m/z 705,9 [M+H]+, что согласуется с искомой структурой.1H-ЯМР 500 МГц (растворитель: DMSO-d6, эталон TMS (триметилсилил)): боковая цепь 1: 8.34 м.д. (m, NH), 7.91 м.д. (m, NH), 4.8-4.4 м.д. (m, ОН), 3.70 м.д. (m, CH), 3.49 м.д. (m, CH2), 3.39 м.д. (m, СН2), 3.31 м.д. (m, NCH2), 3.14 м.д. (m, NCH2), боковая цепь 2: 8.09 м.д. (d, NH), 7.58 м.д. (m, NH), 4.8-4.4 м.д. (m, ОН), 3.82 м.д. (m, CH), 3.65 м.д. (m, CH2), 3.53 м.д. (m, CH2), боковая цепь 3: 5.46 м.д. (m, NH2).
15б) 3-(3-(1,3-бис(формилокси)пропан-2-илкарбамоил)-5-формамидо-2,4,6-трийодбензамидо)пропан-1,2-диилдиформиат
Это соединение было получено способом, аналогичным способу, описанному в примере 1а.
1H-ЯМР 500 МГц (растворитель: DMSO-d6, эталон TMS): 10.30-10.16 м.д. и 10.00-9.93 м.д.(m, 1Н, NHCHO), δ 8.34-8.30 м.д. и 7.94-7.82 м.д. (m, 1Н, NHCHO), δ 9.13-8.46 м.д, (m, 2Н, ArCONH), δ 8.31-8.21 м.д. (m, 4Н, ОСНО), δ 5.31-5.04 м.д. (m, 1Н, APD СНО-СНО), δ 4.54-3.89 м.д. (m, 7Н, APD & серинол СН2O-СНО и серинол ArNHCH), δ 3.08-3.72 м.д. (m, 2Н, APD ArNHCH2).
15в) 5,5'-(2-гидроксипропан-1,3-диил)бис(формилазандиил)бис(N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2,3-дигидpoкcипpoпил)-2,4,6-тpийoдизoфтaлaмид)
Реактор на 250 мл с рубашкой, оснащенный термометром, мешалкой, капельной воронкой и электродом рН-метра, охлаждали в криостате до 10°С. В реактор загружали метанол (17,6 мл), воду (19,6 мл) и борную кислоту (2,33 г, 373 ммоль) и затем водный гидроксид калия (10 М, 50,4 мл) до растворения борной кислоты. Добавляли исходное вещество, 3-(3-(1,3-бис(формилокси)пропан-2-илкарбамоил)-5-формамидо-2,4,6-трийодбензамидо)пропан-1,2-диилдиформиат (16,8 г, 19,9 ммоль), и начинали добавление гидроксида калия по каплям для поддержания рН в пределах 10,7-11,2, поддерживая при этом температуру в пределах от 10°С до 14°С. Через 40 мин добавляли эпихлоргидрин (0,89 г, 9,62 ммоль), поддерживая рН 11,6-11,7, время от времени добавляя либо гидроксид калия (10 М), либо твердую борную кислоту. Впоследствии поддерживали температуру 10°С. Смесь перемешивали в течение 5 ч, рН доводили до 11,7 и перемешивание проводили в течение 48, поддерживая рН в пределах от 11,6 до 11,7. Реакционную смесь гасили добавлением соляной кислоты (6 М, 6,0 мл) для достижения рН 7,5. Добавляли воду (200 мл), затем кислотный ионообменник АМВ200С (94 мл, 158 мг-экв) и перемешивали смесь в течение 1 ч. Добавляли основный ионообменник IRA67 (150 мл, 158 мг-экв) и перемешивали смесь в течение 1 ч. Ионообменники отфильтровывали и промывали водой (600 мл) порциями. Объединенный фильтрат концентрировали в вакууме и неочищенный продукт очищали препаративной ВЭЖХ (колонка: Phenomenex Luna C18, 10 мкм 248×101 мм; растворители: А = вода и В = ацетонитрил; градиент 5-20% В за 35 мин; скорость потока 300 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2-гидроксипропан-1,3-диил)бис-(фopмилaзaндиил)биc(N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2,3-дигидроксипропил)-2,4,6-трийодизофталамид) (7,3 г, выход 49,0%).
ЖХ-МС с электрораспылительной ионизацией: m/z 1522,5 [M+H]+ и его аддукт с натрием соответствуют искомой структуре.
1H-ЯМР 500 МГц (растворитель: D2O, эталон H2O=4,8 м.д., 25°С): 8.5 м.д., 8.2 м.д. (m, 2Н, НСО); 4.7 м.д. - 3.0 м.д. (m, 25Н, СН, СН2), боковая цепь 1: 4.06 м.д. (m, СН); 3.80 м.д., 3.68 м.д. (m, СН2OН); 3.59 м.д., 3.47 м.д. (m, NCH2), боковая цепь 2: 4.18 м.д. (m, СН), 3.87 м.д. (m, СН2), мостик: 8.48 м.д., 8.26 м.д. (m, 2Н, НСО)
Пример 16
5,5'-(2,3-дигидpoкcибутaн-1,4-диил)биc(фоpмилaзaндиил)биc(N1-(1,3-дигидроксипропан-2-ил)-N3-дигидроксипропил)-2,4,6-трийодизофталамид)

Реактор на 250 мл с рубашкой, оснащенный термометром, мешалкой, капельной воронкой и электродом рН-метра, охлаждали в криостате до 10°С. В реактор загружали метанол (15,1 мл), воду (22,1 мл) и борную кислоту (2,33 г, 373 ммоль) и затем водный гидроксид калия (10 М, 50,4 мл) до растворения борной кислоты. Добавляли исходное вещество, 3-(3-(1,3-бис(формилокси)пропан-2-илкарбамоил)-5-формамидо-2,4,6-трийодбензамидо)пропан-1,2-диилдиформиат (Пример 15б) (16,8 г, 19,9 ммоль), и начинали добавление гидроксида калия каплями для поддержания рН в пределах 10,7-11,7, удерживая температуру в пределах от 10°С до 14°С. Через 40 мин добавляли бутадиен-1,3-диэпоксид (0,826 г, 9,59 ммоль), поддерживая рН 11,6-11,7 периодическим добавлением либо гидроксида калия (10 М), либо твердой борной кислоты. Впоследствии поддерживали температуру 10°С. Смесь перемешивали в течение 7 ч, рН доводили до 11,35 и смесь оставляли перемешиваться в течение ночи. На следующий день реакционную смесь гасили добавлением соляной кислоты (6 М, 6,0 мл) до достижения рН 7,5. Добавляли воду (200 мл) и затем кислотный ионообменник АМВ200С (94 мл, 158 мг-экв) и смесь перемешивали в течение 1 ч. Добавляли основный ионообменник IRA67 (150 мл, 158 мг-экв) и смесь перемешивали в течение 1 ч. Ионообменники отфильтровывали и промывали водой (600 мл) порциями. Объединенный фильтрат упаривали в вакууме и неочищенный продукт очищали препаративной ВЭЖХ (колонка: Phenomenex Luna C18, 10 мкм 248×101 мм; растворители: А = вода и В = ацетонитрил; градиент 5-15% В за 35 мин; скорость потока 300 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2,3-дигидроксибутан-1,4-диил)биc(фopмилaзaндиил)биc-(N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2,3-дигидроксипропил)-2,4,6-трийодизофталамид) (3,12 г, выход 21,4%). ЖХ-МС с электрораспылительной ионизацией: m/z 1552,5 [M+H]+ и его аддукт с натрием соответствуют желаемой структуре.
1H-ЯМР 500 МГц (растворитель: D2O, эталон Н2O=4,8 м.д., 25°С): 8.5 м.д., 8.2 м.д. (m, 2Н, НСО); 4.4 м.д. - 3.0 м.д. (m, 26Н, СН, СН2), боковая цепь 1: 4.06 м.д. (m, СН); 3.80 м.д., 3.68 м.д. (m, СН2OН); 3.59 м.д., 3.47 м.д. (m, NCH2), боковая цепь 2: 4.18 м.д. (m, СН), 3.87 м.д. (m, СН2), мостик: 8.48 м.д., 8.26 м.д. (m, 2Н, НСО)
Пример 17
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(N1-(1,3-дигидроксипропан-2-ил)-N3(2,3-дигидроксипропил)-2,4,6-трийодизофталамид)
Реактор на 250 мл с рубашкой, оснащенный термометром, мешалкой, капельной воронкой и электродом рН-метра, охлаждали в криостате до 10°С. В реактор загружали метанол (12,6 мл), воду (24,6 мл) и борную кислоту (2,33 г, 373 ммоль) и затем водный гидроксид калия (10 М, 50,4 мл) до растворения борной кислоты. Добавляли исходное вещество, 3-(3-(1,3-бис-(формилокси)пропан-2-илкарбамоил)-5-формамидо-2,4,6-трийодбензамидо)-пропан-1,2-диилдиформиат (Пример 15б (16,8 г, 19,9 ммоль), и начинали добавление по каплям гидроксида калия для поддержания рН в пределах 10,7-11,2, удерживая температуру в пределах от 10°С до 14°С. Через 15 мин добавляли пентадиен-1,4-диэпоксид (0,99 г, 9,89 ммоль), поддерживая рН 11,5-11,6 периодическим добавлением либо гидроксида калия (10 М), либо твердой борной кислоты. При перемешивании в течение 30 ч температуру поддерживали при 10°С и рН поддерживали в пределах от 11,5 до 11,6. Температуру доводили до 23°С и смесь оставляли перемешиваться в течение ночи. На следующий день добавляли вторую порцию пентадиендиэпоксида (0,63 г, 6,27 ммоль) и перемешивали смесь в течение трех суток. Реакционную смесь гасили добавлением соляной кислоты (6 М, 6,0 мл) до достижения рН 7,5. Добавляли воду (200 мл), кислотный ионообменник АМВ200С (94 мл, 158 мг-экв) и основный ионообменник IRA67 (150 мл, 158 мг-экв) и перемешивали смесь в течение 1 ч. Ионообменники отфильтровывали и промывали водой (600 мл) порциями. Объединенный фильтрат концентрировали в вакууме и очищали препаративной ВЭЖХ.
Объединенный фильтрат концентрировали в вакууме и неочищенный продукт очищали препаративной ВЭЖХ (колонка: Phenomenex Luna C18, 10 мкм 248×101 мм; растворители: А = вода и В = ацетонитрил; градиент 5-15% В за 35 мин; скорость потока 300 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2,4-дигидроксипентан-1,5-диил)биc(фopмилaзaндиил)биc(N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2,3-дигидроксипропил)-2,4,6-трийодизофталамид) (6,6 г, выход 42,5%). ЖХ-МС с электрораспылительной ионизацией (m/z 1566,4 [М+Н]+) и его аддукт с натрием соответствуют желаемой структуре.
1H-ЯМР (растворитель: D2O, эталон Н2O=4,8 м.д.): 8.5 м.д., 8.2 м.д. (m, 2Н, НСО); 4.4 м.д. - 3.0 м.д. (m, 26Н, СН, СН2); 2.0 м.д. - 1.6 м.д. (m, 2Н, СН2), боковая цепь 1: 4.06 м.д. (m, 2Н, СН); 3.80 м.д., 3.68 м.д. (m, 4Н, СН2OН); 3.59 м.д., 3.47 м.д. (m, 4Н, NCH2), боковая цепь 2: 4.18 м.д. (m, 2Н, СН), 3.87 м.д. (m, 8Н, СН2), мостик: 8.48 м.д., 8.26 м.д. (m, 2Н, НСО); 1.97 м.д. - 1.62 м.д. (m, 2Н, СН2)
Пример 18
5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2-гидpoкcиэтил)-2,4,6-тpийoдизoфтaлaмид)

18а) 5-амино-N1-(1,3-дигидроксипропан-2-ил)-N3-гидроксиэтил)-2,4,6-трийодизофталамид
2-Амино-1,3-пропандиол (15,66 г, 171,89 ммоль) помещали в одногорлую круглодонную колбу на 2 л и в нее добавляли N,N'-диметилацетамид (520 мл). Начинали перемешивание, при этом образовался прозрачный и бесцветный раствор, к которому добавляли триэтиламин (85,8 мл, 619,2 ммоль). Добавляли порциями дихлорид 5-амино-2,4,6-трийодизофталоила (102,4 г, 191 89 ммоль). Смесь оставляли перемешиваться при температуре окружающей среды в течение 25 часов и в нее добавляли этаноламин (10,34 мл, 171,89 ммоль). Продолжали перемешивание при температуре окружающей среды в течение 3 суток. Осадок соли Et3N с HCl отфильтровывали и промывали N,N'-диметилацетамидом (30 мл). Объединенные органические фазы упаривали в вакууме до густого масла, к этому маслу добавляли этанол (400 мл) и смесь интенсивно перемешивали при температуре окружающей среды в течение трех суток. Образовавшийся осадок собирали фильтрованием, промывали этанолом (50 мл) и сушили в вакууме. Неочищенный продукт представлял собой светло-коричневое твердое вещество. Выход: 98,15 г.
ВЭЖХ показала, что выход присутствующего целевого соединения составляет 78%.
Продукт использовали на следующей стадии без дополнительной очистки.
ЖХ-МС: (электрораспылительная ионизация, захват ионов, m/е): 675,8 [M+H]+.
1H-ЯМР 500 МГц (растворитель: D2O, эталон Н2O=4,8 м.д., 25°С): Боковая цепь 1: 8.38 м.д. (m, NH), 8.05 м.д. (m, NH), 4.7-4.4 м.д. (m, ОН), 3.25 м.д. (m, CH2N), 3.52 м.д. (m, СН2), боковая цепь 2: 8.09 м.д. (d, NH), 7.56 м.д. (m, NH), 4.7-4.4 м.д. (m, ОН), 3.81 м.д. (m, CH), 3.65 м.д. (m, СН2), 3.55 м.д. (m, СН2), боковая цепь 3: 5.43 м.д. (m, NH2)
18б) 2-(3-(2-ацетоксиэтилкарбамоил)-5-амино-2,4,6-трийодбензамидо)пропан-1,3-диилдиацетат
Уксусный ангидрид (312 мл, 3,3 ммоль) по каплям за 30 минут добавляли к охлаждаемому льдом раствору неочищенного продукта, полученного в примере 18а (100 г, 0,15 ммоль), в пиридине (600 мл). Охлаждающую баню затем удаляли и перемешивание продолжали в течение ночи. К смеси при охлаждении льдом добавляли этанол (200 мл), эту смесь концентрировали в вакууме и к темному маслу добавляли этилацетат (400 мл). Раствор помещали в холодильник на ночь до полной кристаллизации. Продукт собирали фильтрованием и промывали этилацетатом, 1 н. HCl (1×200 мл), водой (2×200 мл) и сушили в термостате (50°С). Неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex, Luna C18 10 мкм, 248×101 мм; растворители: А = вода и В = ацетонитрил; градиент 25-27% В за 6 мин, продолжение в течение 30 мин; скорость потока 300 мл/мин, УФ-детектирование при 214 нм и 254 нм). Объединенные фракции концентрировали в вакууме, после чего продукт собирали фильтрованием. Продукт сушили при 60°С в вакууме с получением 73 г 2-(3-(2-ацетоксиэтилкарбамоил)-5-амино-2,4,6-трийодбензамидо)пропан-1,3-диилдиацетата (выход 60,8%).
ЖХ-МС: (электрораспылительная ионизация, ионная ловушка, m/е): 801,6 [M+H]+.
1H-ЯМР, 500 МГц (DMSO-d6, 25°С): 2.0 м.д. (m, 9Н), 3.2 м.д. (триплет, 2Н), 4.10 м.д. (m, 6Н), 4.19 м.д. (m, 1 Н), 5.25 м.д. (m, NH2), 8.15 м.д. (m, NH), 8.27 м.д. (m, NH), 8.3 м.д. (m, NH), 8.38 м.д. (m, NH).
18в) N1-(1,3-дигидроксипропан-(2-ил)-формамидо-N3-гидроксиэтил)-2,4,6-трийодизофталамид
Уксусный ангидрид (19,47 мл, 20,6 ммоль) добавляли к муравьиной кислоте (100 мл) при охлаждении во льду, поддерживая температуру ниже 5°С. После окончания добавления охлаждающую баню убирали и температуре давали возможность подняться до 10°С. Продолжали охлаждение льдом и затем добавляли диацетат 2-(3-(2-ацетоксиэтилкарбамоил)-5-амино-2,4,6-трийодбензамидо)-пропан-1,3-диила (33 г, 41,2 ммоль) порциями за 3 минут. Смесь оставляли перемешиваться в течение ночи и затем концентрировали в вакууме с получением белой пены, в которую добавляли метанол (100 мл) и воду (100 мл). При перемешивании по каплям добавляли 10 н. NaOH до образования прозрачного раствора. Добавление 10 н. NaOH по каплям продолжали до тех пор, пока не устанавливался рН 11,60. Метанол удаляли из смеси выпариванием в вакууме и раствор подкисляли до рН 2,7 добавлением 18,6% HCl. Смесь помещали в холодильник на ночь, и продукт, осажденный из раствора, собирали, промывали водой и сушили при 60°С с получением N1-(1,3-дигидроксипропан-2-ил)-5-формамидо-N3-(2-гидроксиэтил)-2,4,6-трийодизофталамида (24 г, выход 82,9%). ЖХ-МС: (электрораспылительная ионизация, ионная ловушка), m/е: 703,8 [M+H]+.
1H-ЯМР 500 МГц (растворитель: D2O, эталон Н2O=4,8 м.д., 25°С): Боковая цепь 1: 3.59 м.д. (m, 2H, CH2N), 3.87 м.д. (m, 2H, СН2OН), боковая цепь 2: 4.20 м.д. (m, 1Н, СН), 3.59 м.д. (m, 4Н, СН2), боковая цепь 3: 8.47 м.д. (m, 1Н, НСО)
18г) 5,5'-(2-гидроксипропан-1,3-диил)биc(фopмилaзaндиил)биc(N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2-гидpoкcиэтил)-2,4,6-тpийoдизoфтaлaмид)
N1-(1,3-дигидроксипропан-2-ил)-формамидо-N3-(2-гидроксиэтил)-2,4,6-трийодизофталамид (8 г, 1 1,4 ммоль) растворяли в растворе гидроксида калия (1,01 г, 17,9 ммоль) в метаноле (8 мл) и воде (8 мл). Повышали температуру до 40°С и добавляли борную кислоту (0,62 г, 1,91 ммоль). Продолжали нагревание при этой температуре в течение 15 минут. После охлаждения до температуры окружающей среды перемешивание продолжали в течение 4 часов, после чего добавляли эпихлоргидрин (0,37 г, 3,98 ммоль). Поддерживали рН в пределах от 12 до 13 в течение 5 часов добавлением борной кислоты. На следующий день рН доводили до 3,97 и метанол удаляли в вакууме. Оставшийся водный раствор разбавляли водой (75 мл) и обрабатывали ионообменниками (АМВ 200С, Н+ и IRA-67, ОН-) до нулевой проводимости. Ионообменники отфильтровывали и промывали водой и объединенные фильтраты подвергали сублимационной сушке и очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 (2) 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 0-10% В за 60 мин; скорость потока 50,0 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc-(фopмилaзaндиил)биc(N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2-гидpoкcиэтил)-2,4,6-тpийoдизoфтaлaмид) (3,80 г, выход 66%).
ЖХ-МС: (электрораспылительная ионизация, ионная ловушка, m/е): 1462,7 [M+H]+.
1H-ЯМР 500 МГц (растворитель: D2O, эталон Н2O=4,8 м.д., 25°С): боковая цепь 1: 3.58 м.д. (m, NCH2), 3.87 м.д. (m, CH2OH), боковая цепь 2: 4.19 м.д. (m, 2Н, СН), 3.87 м.д. (m, CH2), мостик: 8.47 м.д., 8.20 м.д. (m, 2H, NHCO)
Пример 19
5,5'-(2,3-дигидpoкcибутaн-1,4-диил)биc(фopмилaзaндиил)биc(N1-(1,3-дигидроксипропан-2-ил)-N3-гидроксиэтил)-2,4,6-трийодизофталамид)

Гидроксид калия (10 M) по каплям добавляли к перемешиваемой суспензии N1-(1,3-дигидроксипропан-2-ил)-5-формамидо-N3-гидроксиэтил)-2,4,6-трийодизофталамида (Пример 18с) (6 г, 8,5 ммоль) в воде (5 мл) и метаноле (5 мл). Постоянно отслеживали рН, и когда рН достигал 11,6, добавляли борную кислоту (0,46 г, 7,5 ммоль) и затем 1,3-бутадиендиэпоксид (234 мкл, 2,99 ммоль). Постоянно поддерживали рН раствора в пределах 11,5-11,8 добавлением либо твердой борной кислоты, либо гидроксида калия (10 M). Через 24 часа при температуре окружающей среды постепенно образовывался прозрачный и бесцветной раствор. Доводили рН до 2,7 соляной кислотой (6 M) и удаляли метанол в вакууме. Оставшийся водный раствор разбавляли водой (75 мл) и обрабатывали ионообменниками (АМВ 200С, Н+ и IRA-67, ОН-) до нулевой проводимости. После сублимационной сушки неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 (2) 10 мкм 250×50,0 мм; растворители: А = вода, и В = ацетонитрил; градиент 0-10% В за 60 мин; скорость потока 50,0 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2,3-дигидроксибутан-1,4-диил)биc(фopмилaзaндиил)биc(N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2-гидроксиэтил)-2,4,6-трийодизофталамид) (2,04 г, выход 46%). ЖХ-МС: (электрораспылительная ионизация, ионная ловушка, m/е): 1492,7 [М+Н]+.
1H-ЯМР 500 МГц (растворитель: D2O, эталон Н2O=4,8 м.д., 25°С): боковая цепь 1: 3.58 м.д. (m, NCH2), 3.87 м.д. (m, CH2OH), боковая цепь 2: 4.18 м.д. (m, 2Н, СН), 3.87 м.д. (m, CH2), мостик: 8.48 м.д., 8.26 м.д. (m, 2H, NHCO).
Пример 20
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(N1-(113-дигидроксипропан-2-ил)-N3-гидроксиэтил)-2,4,6-трийодизофталамид)
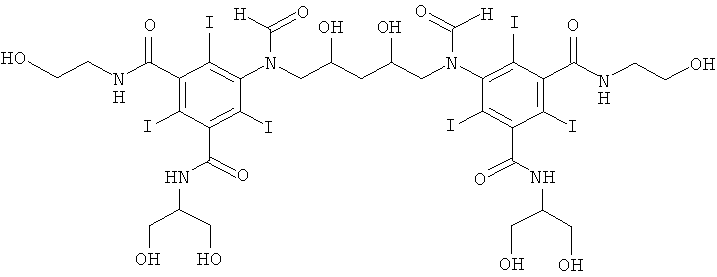
Гидроксид калия (10 M) по каплям добавляли к перемешиваемой суспензии N1-(1,3-дигидроксипропан-2-ил)-5-формамидо-N3-гидроксиэтил)-2,4,6-трийодизофталамида (Пример 18с) (8,0 г, 11,4 ммоль) в воде (8 мл) и метаноле (8 мл). Постоянно отслеживали рН, и когда рН составлял 11,6, добавляли борную кислоту (0,62 г, 9,9 ммоль) и затем 1,4-пентадиендиэпоксид (400 мг, 3,98 ммоль). Постоянно поддерживали рН раствора в пределах 11,5-11,8 добавлением либо твердой борной кислоты, либо гидроксида калия (10 M). Через 24 часа при температуре окружающей среды постепенно образовывался прозрачный и бесцветной раствор. Доводили рН до 3,5 соляной кислотой (6 M) и удаляли метанол в вакууме. Оставшийся водный раствор разбавляли водой (75 мл) и обрабатывали ионообменниками (АМВ 200С, H+ и IRA-67, ОН-) до нулевой проводимости. После сублимационной сушки неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 (2) 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 0-10% В за 60 мин; скорость потока 50,0 мл/мин, УФ-детектирование при 214 нм и 254 нм). После сублимационной сушки получили 5,5'-(2,4-дигидроксипентан-1,5-диил)биc(фopмилaзaндиил)биc(N1-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2-гидроксиэтил)-2,4,6-трийодизофталамид) (3,16 г, выход 53%).
ЖХ-МС: (электрораспылительная ионизация, ионная ловушка), m/е: 1506,6 [М+Н]+.
1H-ЯМР 500 МГц (растворитель: D2О, эталон Н2O=4,8 м.д., 25°С): 8.47 м.д., 8.23 м.д. (m, 2H, НСО), 4.40 м.д. -3.10 м.д. (m, 24 Н, СН/СН2), 1.95 м.д. -1.60 м.д. (m, 2Н, СН2).
Пример 21
5-(N-(3-(N-(3,5-бис(2,3-дигидроксипропилкарбамоил)-2,4,6-трийодфенил)ацетамидо)-2-гидроксипропил)формамидо)-N\N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид
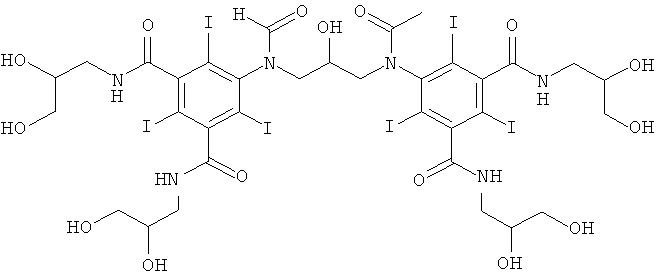
21а) 5-(N-аллилацетамидо)-N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид
К перемешиваемому раствору 5-ацетамидо-N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (7,5 г, 10 ммоль) и борной кислоты (1,24 г, 20 ммоль) в воде (10 мл) и метаноле (7 мл) по каплям добавляли гидроксид калия (10 М) до установления рН 12,5. Добавляли аллилбромид (860 мкл, 10 ммоль) и постоянно добавляли гидроксид калия (10 М) для поддержания рН 12,5 в течение 3 ч и затем оставляли смесь перемешиваться в течение ночи. На следующий день смесь концентрировали в вакууме для удаления метанола, полученную водную фазу разбавляли водой (50 мл) и обрабатывали кислотным и основным ионообменником. Смолы удаляли и промывали и объединенный водный объем упаривали досуха в вакууме с получением 5-(N-aллилaцeтaмидo)-N1,N3-биc(2,3-дигидpoкcипponил)-2,4,6-трийодизофталамида.
21б) 5-(N-(3-(N-(3,5-бис(2,3-дигидроксипропилкарбамоил)-2,4,6-тpийoдфeнил)aцeтaмидo)-2-гидpoкcипpoпил)фopмaмидo)-N1,N3-биc(2,3-дигидроксипропил)-2,4,6-трийодизофталамид
К перемешиваемому раствору воды (10 мл) и метанола (7 мл) добавляли 5-(N-aллилaцeтaмидo)-N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-трийодизофталамид (5,0 г 6,35 ммоль). Доводили рН до рН 2 и по каплям при 50°С добавляли раствор йодида калия (1,05 г, 6,35 ммоль) и йода (1,61 г, 6,35 ммоль) в воде (5 мл) и метаноле (5 мл). Смесь перемешивали в течение ночи. Раствор охлаждали до 10°С и к нему добавляли N,N'-бис-(2,3-дигидроксипропил)-5-формиламино-2,4,6-трийодизофталамид (Пример 1а) (4,65 г, 6,35 ммоль). По каплям добавляли гидроксид калия (10 М) для поддержания стабильного рН 11,6 и смесь перемешивали в течение 24 часов. Раствор разбавляли водой (50 мл) и обрабатывали ионообменниками (АМВ200С, 20 мл) и (IRA67, 20 мл). Смолы отфильтровывали и промывали водой. Объединенные водные фазы концентрировали в вакууме и неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×50,0 мм; растворители: А = вода и В = ацетонитрил; градиент 0-10% В за 60 мин; скорость потока 50,0 мл/мин).
Пример 22
5-(3-(N-(3,5-бис(2,3-дигидроксипропилкарбамоил)-2,4,6-тpийoдфeнил)фopмaмидo)-2-гидpoкcипpoпилaминo)-N1,N3-биc(2,3-дигидроксипропил)-2,4,6-трийодизофталамид

22а) 5-(N-(3-бpoм-2-гидpoкcипpoпил)-2,2,2-тpифтopaцeтaмидo)-N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид
Это соединение было получено способом, описанным в литературе (Priebe, H.; Dugstad, H.; Heglund, I.F.; Sande, R.; Toenseth, С.Р. Synthesis, Analysis and toxicity of three compounds during the synthesis of iodixanol. Acta Chemica Scandinavica (1995), 49(10), 737-43).

22б) 5-(3-(N-(3,5-бис(2,3-дигидроксипропилкарбамоил)-2,4,6-трийодфенил)формамидо)-2-гидроксипропиламино-N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид
N,N'-Бис-(2,3-дигидроксипропил)-5-формиламино-2,4,6-трийодизофталамид (Пример 1а) (1,1 г, 1,5 ммоль) суспендировали в воде (2 мл) и метаноле (0,7 мл). Добавляли гидроксид калия (10 М, 120 мкл) с получением прозрачного раствора при рН 11,3. 5-(N-(3-Бром-2-гидроксипропил)-2,2,2-трифторацетамидо)-N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид (1,41 г, 1,5 ммоль) растворяли в воде (3,0 мл) и добавляли к реакционной смеси при температуре окружающей среды. Поддерживали рН 11,5 добавлениями гидроксида калия (10 М). Смесь перемешивали в течение двух суток и затем разбавляли водой (50 мл) и обрабатывали ионообменниками (АМВ200С, 20 мл) и (IRA67, 20 мл). Смолы отфильтровывали и промывали водой. Объединенные водные фазы концентрировали в вакууме и неочищенный продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18 10 мкм 250×75,0 мм; растворители: А = вода и В = ацетонитрил; изократический градиент 5% в течение 5 мин, 5-15% В за 35 мин; скорость потока 175,0 мл/мин, УФ-детектирование при 254 нм). После сублимационной сушки получили 5-(3-(N-(3,5-бис(2,3-дигидроксипропилкарбамоил)-2,4,6-трийодфенил)формамидо)-2-гидpoкcипpoпилaминo)-N1,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-трийодизофталамид (1,07 г, выход 48%).
ЖХ-МС ВП (колонка Agilent Zorbax SB-Aq C18 3,5 мкм 3,0×100 мм; растворители: А = вода/0,1%-ная муравьиная кислота и В = ацетонитрил/0,1%-ная муравьиная кислота; градиент 4-15% В за 10 мин; скорость потока 0,5 мл/мин, УФ-детектирование при 210-240 нм) времяпролетная масс-спектрометрия дала два пика при 6,14 и 6,67 мин с [М+Н]+: 1494,6949, [M+Na]+: 1516,6769, [M+K]+: 1532,6509, что согласуется с искомой структурой.
Реферат
Настоящее изобретение относится к новым соединениям, содержащим две связанные йодированные фенильные группы, имеющим общую формулу (I), где R, Rи Х имеют значения, приведенные в формуле изобретения, а также к применению в качестве контрастных агентов в диагностической рентгеновской визуализации и к рентгеновским диагностическим композициям, содержащим такие соединения. 6 н. и 9 з.п. ф-лы, 22 пр.
Формула

где X означает С3-8алкиленовую группировку с прямой или разветвленной цепью, возможно замещенной группами -OR1 в количестве до шести включительно;
R1 означает водород или С1-4 алкильную группу с прямой или разветвленной цепью;
R3 означает атом водорода или группировку C1-5 органической кислоты, выбранную из формильной, ацетильной, пропионильной, бутирильной, изобутирильной и валериильной группировок; и
R одинаковые или разные и каждый независимо означает трийодированную фенильную группу, дополнительно замещенную двумя группами R2, причем группы R2 одинаковые или разные и каждая представляет собой атом водорода или неионную гидрофильную группировку, выбранную из группы полигидрокси-С1-5алкил, гидроксиалкоксиалкил с 1-5 атомами углерода и гидроксиполиалкоксиалкил с 1-5 атомами углерода, и присоединены к йодированной фенильной группе через амидную или карбамоильную связь, при условии, что по меньшей мере одна группа R2 в соединении формулы (I) представляет собой гидрофильную группировку.
- CONH2;
- CONHCH3;
- CONH-CH2-CH2-OH;
- CONH-CH2-CH2-OCH3;
- CONH-CH2-CHOH-CH2-OH;
- CONH-CH2-CHOCH3-CH2-OH;
- CONH-CH2-CHOH-CH2-OCH3;
- СОN(СН3)СН2-СНОН-СН2OН;
- CONH-CH-(CH2-OH)2;
- CON-(CH2-CH2-OH)2;
- CON-(CH2-CHOH-CH2-OH)2;
- CONH-ОСН3;
- CON(CH2-CHOH-CH2-OH)(CH2-CH2-OH);
- СОNН-С(СН2-ОН)2СН3,;
- СОNН-С(СН2-ОН)3 и
- CONH-CH(CH2-OH)(CHOH-CH2-OH).
- CONH-CH2-CHOH-CH2-OH.



где R и X такие, как определено в пп.1-8.
5,5'-(2-гидpoкcипpoпaн-1,3-диил)биc(фopмилaзaндиил)биc(Nl,N3-биc(2,3-дигидроксипропил)-2,4,6-трийодизофталамид),
5,5'-(2,3-дигидроксибутан-1,4-диил)бис(формилазандиил)бис(N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид),
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(Nl,N3-биc(2,3-дигидроксипропил)-2,4,6-трийодизофталамид),
5,5'-(2,3,4-тригидроксипентан-1,5-диил)бис(формилазандиил)бис(N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид),
5,5'-(2,3-дигидpoкcибyтaн-1,4)-диил)биc(фopмилaзaндиил)биc(Nl,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-тpийoд-Nl,N3-димeтилизoфтaлaмид),
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(Nl,N3-биc(2,3-дигидpoкcипpoпил)-2,4,6-тpийoд-Nl,N3-димeтилизoфтaлaмид),
5,5'-(2-гидроксипропан-1,3-диил)бис(формилазандиил)бис(N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийод-N1,N3-диметилизофталамид),
5,5'-(2-гидроксипропан-1,3-диил)бис(формилазандиил)бис(N1,N3-биc(1,3-дигидроксипропан-2-ил)-2,4,6-трийодизофталамид),
5,5'-(2,3-дигидpoкcибyтaн-1,4-диил)биc(фopмилaзaндиил)биc(Nl,N3-биc(1,3-дигидроксипропан-2-ил)-2,4,6-трийодизофталамид),
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(Nl,N3-бис(1,3-дигидроксипропан-2-ил)-2,4,6-трийодизофталамид),
5,5'-(2,3-дигидроксибутан-1,4-диил)бис(формилазандиил)бис(N1-(2,3-дигидpoкcипpoпил)-N3-(2-гидpoкcиэтил)-2,4,6-тpийoдизoфтaлaмид),
5,5'-(2-гидроксипропан-1,3-диил)бис(формилазандиил)бис(N1-(2,3-дигидроксипропил)-N3-(2-гидроксиэтил)-2,4,6-трийодизофталамид),
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(Nl-2,3-дигидpoкcипpoпил)-N3-(2-гидpoкcиэтил)-2,4,6-тpийoдизoфтaлaмид),
5,5'-(2,4-гидроксипропан-1,3-диил)бис(формилазандиил)бис(N1-(1,3-дигидроксипропан-2-ил)-N3-(2,3-дигидроксипропил)-2,4,6-трийодизофталамид),
5,5'-(2,3-дигидроксибутан-1,4-диил)биc(фopмилaзaндиил)биc(Nl-(1,3-дигидроксипропан-2ил)-N3-(2,3-дигидроксипропил)-2,4,6-трийодизофталамид),
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(Nl-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2,3-дигидpoкcипpoпил)-2,4,6-трийодизофталамид),
5,5'-(2-гидроксипропан-1,3-диил)биc(фopмилaзaндиил)биc(Nl-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2-гидpoкcиэтил)-2,4,6-тpийoдизoфтaлaмид),
5,5'-(2,3-дигидpoкcибyтaн-1,4-диил)биc(фopмилaзaндиил)биc(Nl-(1,3-дигидроксипропан-2ил)-N3-(2,3-дигидроксиэтил)-2,4,6-трийодизофталамид),
5,5'-(2,4-дигидpoкcипeнтaн-1,5-диил)биc(фopмилaзaндиил)биc(Nl-(1,3-дигидpoкcипpoпaн-2-ил)-N3-(2-гидpoкcиэтил)-2,4,6-тpийoдизoфтaлaмид),
5-(N-(3-(N-(3,5-бис(2,3-дигидроксипропилкарбамоил)-2,4,6-трийодфенил)ацетамидо)-2-гидроксипропил)формамидо)-N1,N3-биc(2,3-дигидроксипропил)-2,4,6-трийодизофталамид и
5-(3-(N-(3,5-бис(2,3-дигидроксипропилкарбамоил)-2,4,6-трийодфенил)формамидо-2-гидроксипропиламино-N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид.
Документы, цитированные в отчёте о поиске
Трийод-5-аминоизофталдиамиды, способы их получения и радиологическая композиция
Комментарии