Трийод-5-аминоизофталдиамиды, способы их получения и радиологическая композиция - RU2060246C1
Код документа: RU2060246C1
Чертежи


Описание
Изобретение относится к неионным контрастным средам и способу их получения.
Контрастная среда, используемая в рентгенографии для визуализации сердечно-сосудистой системы и внутренних полостей тела, должна иметь низкую вязкость, высокую растворимость в воде, быть нетоксичной и иметь высокое содержание йода. Низкая вязкость является обязательным требованием для быстроты доставки, для мгновенной замены быстропротекающей крови, например, в ангиокардиографии или урографии с высокими дозами, или контрастного усиления компьютерной томографии. Для того, чтобы соединение было нетоксичным, оно должно обладать высокой гидрофильностью, быть неионным и обладать осмотичностью, близкой к среде организма. Несмотря на то, что неионные мономерные среды, известные в этой области техники, имеют приемлемую биологическую толерантность и приемлемую вязкость, они имеют повышенную осмотичность по отношению к среде организма. Обычно приемлемые для диагностики концентрации порядка 300 мг/мл существенно превышают физиологическое значение 310 мОсм. Повышенная осмотичность этих растворов неизбежно вытеснит воду из клеток, разрушит клеточные мембраны, нарушит общий электролитический балланс и повредит контуры сосудов или полости органов. Кроме того, известно, что повышенная осмотичность является одной из причин, вызывающих боль в сосудах, постоянно вызываемую контрастными средами с повышенной осмотичностью.
Предлагается неионная контрастная среда на основе асимметричного трийодизофталевого диамида, в котором остальные позиции кольца занимают замещенный азот, одна из карбоксильных групп, являющаяся незамещенным амидом, и карбоксильные группы, являющиеся, по меньшей мере, моно-гидроксиалкилзамещенным амидом. Молекула имеет, по меньшей мере, две гидроксильные группы.
Предлагается неионная контрастная среда на основе акриламидозамещенного трийодизофталевого диамида, в котором только один из амидных азотов является, по меньшей мере, монозамещенным. Эти соединения могут быть синтезированы при проведении недорогостоящего синтеза с высоким выходом и высокой частотой. Было обнаружено, что эти соединения обеспечивают низкую осмотичность, в то же время имеют низкую вязкость.
Большая часть заявленных соединений имеет формулу
I

где R1 водород;
R2 представляет гидроксиалкил, содержащий от 2 до 4 атомов углерода, и имеющий, по меньшей мере, одну гидроксильную группу и не больше n-1 гидроксильных групп, где n число атомов углерода;
R3 низший алкил или низший алкокси (низший алкил), где алкил содержит от 1 до 2 атомов углерода;
R3 имеет формулу

R4 водород или алкил с 1-3 атомами углерода, имеющий от 0 до n-1 гидроксильных групп, где n число атомов углерода.
Карбамиды могут быть получены с использованием 3-амино-1,2-пропандиола, серинола или аминотетритолов, т. е. тритол и эритрат, или в D,L-смеси или оптически чистых формах этаноламина, или диэтаноламина, или триметамина, или их производных, где гидроксильные группы являются обратимо защищенными. R1 представляет водород или низший алкил, предпочтительно водород или метил. R2 низший алкил или оксиалкил с 1-6, обычно 1-4 атомами водорода, предпочтительно метил, гидроксиметил или гидроксиэтил. Также можно привести в примере алкоксиалкилы, содержащие алкоксильные группы с 1-3 атомами углерода, предпочтительно от 1 до 2 атомов, и алкилы, содержащие 1-3 атома углерода, более частный вариант метоксиметил. Альтернативно две группы R3 могут быть взяты вместе для образования связи, метилен или этилен, более предпочтительно метилен. R4 водород, алкил или моно- или полигидроксиалкил, содержащий от 1 до 6, обычно от 1 до 4 атомов углерода, включая метил, этил, пропил, гидроксиэтил и дигидроксипропил.
Мономерные соединения, также представляющие интерес, включают: 5-[N-(2-гидроксиэтил)ацетамидо] -2,4,6-трийод-3-[N-(1, 3,
4-тригидроксибут-2-ил)] карбамоилбензамид; 5-[N-(2,3-дигидроксипропил)ацетамидо]-2,4,6-трийод-3-[N-(2,3-дигидроксипропи л)] карбамоилбензамид; 5-[N-(2,3-дигидроксипропил)ацетамидо] -2,4,
6-трийод-3-[N-(2-гидроксиэтил)] кар бамоилбензамид; 5-[N-(метил)-2-гидроксиацетамидо] -2,4,6-трийод-3-[N-(1,3,4-тригидроксиэритро бут-2-ил]карбамоилбензамид;
5-[N-(2-гидроксиэтил)ацетамидо]
-2,4,6- трийод-3-[N-(1,3,4-тригидрокситреобут-2- ил)]карбамоилбензамид и
бис-[3-N-(1,3,4-тригидрокси-бут-2-ил- карбамоил)-5-карбамоил] -2,4,6- трийод-N-метиланилид малоновую кислоту.
Предлагаемые соединения содержат от около 50 до 52% йода, обычно около 51% имеют вязкость раствора 300 мг I/мл при 37оС в пределах от около 4 до 5 сП и осмотичность для водного раствора 300 мг I/мл при температуре 37оС в пределах от около 275 до 400 мОсм, чаще около 285 до 375, при этом в фармацевтической композиции осмотичность находится в пределах от около 300 до 400, чаще от около 325 до около 390.
Заявленные соединения получают в соответствии с традиционным способом. Обычно композиции содержат водную среду, которая включает физиологически приемлемую хелатную кальциевую соль, например ЭДТА, буфер для поддержания рН среды в пределах от около 6,5 до 7,5, предпочтительно около 7, где буфер может включать трис, карбонат, цитрат или их комбинации. Другими добавками, которые могут быть включены в композицию, являются бикарбонат, фосфат и т.д. Хелатный кальций может присутствовать в количестве от около 5 до 15, обычно около 10 мг/100 мл, при этом буфер может находиться в количестве от около 2 до 10 ммоль.
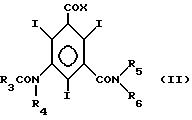
Изобретение касается также способа получения соединений формулы I

имеющей, по меньшей мере, две оксигруппы, и где
R1 водород, R2 гидроксиалкил с 2-4 атомами углерода и имеющий от 1 до n-1 гидроксильных групп, где n число атомов углерода,
R3 низший алкил, гидроксизамещенный низший алкил или низший алкоксизамещенный низший алкил с 1-2 атомами углерода, или R3 имеет формулу

R4 водород или алкил с 1 до 3 атомов углерода, имеющий от 0 до n-1 гидроксильных групп, где n число атомов углерода, который состоит в том, что соединения общей формулы II

в которой в R1 и R2 гидроксильные группы могут быть защищены, в результате взаимодействия с R3CO-X, причем X означает галоген или сложноэфирный остаток, в катализирующем растворителе, например пиридин, диметиламид или диметилформамид, и при необходимости в результате последующего отщепления защитных групп переводят в конечный продукт общей формулы I c R4 водород или в промежуточный продукт общей формулы I с R4, означающим R3CO, и последний, при желании с защищенными гидроксильными группами, в результате алкилирования в щелочных условиях превращают с R4 содержащими реагентами, при необходимости после отщепления защитных групп, в конечный продукт общей формулы I, в которой R4 не означает водород.
Способ получения трийод-5-аминоизофтал-диамидов общей формулы I

имеющей, по меньшей мере, две оксигруппы, и где R1 водород, R2 гидроксиалкил с 2-4 атомами углерода и имеющий от 1 до n-1 гидроксильных групп, где n число атомов углерода;
R3 низший алкил, гидроксизамещенный низший алкил или низший алкоксизамещенный низший алкил с 1-2 атомами углерода, или R3 имеет формулу
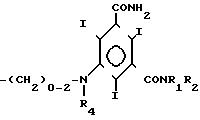
R4 водород или алкил с 1 до 3 атомов углерода, имеющий от 0 до n-1 гидроксильных групп, где n число атомов углерода, состоит в том, что соединения общей формулы III

(III) где R5 означает R1, R6 означает R2 или водород и
Х галоген или сложноэфирный остаток, подвергают взаимодействию с аммиаком и, в случае необходимости с галоидалканолом,
гидроксильные группы которого могут быть защищены, и непосредственно после этого в соответствующем случае защитные группы отщепляют.
Соединения согласно изобретению общей формулы I являются стабильными в водных растворах, они легко образуют супернасыщенные растворы, которые также сохраняют стабильность. При концентрациях йода, требуемых для диагностики, соединения имеют осмотичность, которая очень близка к физиологическим значениям. В то же время вязкость раствора очень низкая. Таким образом, достигнута цель изобретения, указанная выше, т.е. одновременность двух факторов низкой осмотичности и низкой вязкости. В результате новые соединения имеют превосходную совместимость с организмом. Соединения имеют хорошую биологическую совместимость и высокое содержание йода, в частности, если сравнить с известными неионными радиографическими контрастными средами.
В табл. 1 приведены данные по свойствам полученных соединений.
Благодаря своим физико-химическим и фармакологическим свойствам новые соединения пригодны в качестве водорастворимых контрастных сред для визуализации мочевыводящих путей и сердечно-сосудистой системы, а также для контрастного усиления в компьютерной томографии. Вводимые растворы новых соединений могут быть приготовлены путем разбавления их водой и добавления стандартного физиологически приемлемого буфера и стабилизаторов, например хелатных агентов. Эти соединения пригодны также для внутреннего введения при соединении с носителями, используемыми обычно в фармацевтике.
Для внутривенного использования соединения, согласно изобретению, содержат 20-80% от объема при концентрации йода 150-400 мг/мл предпочтительно.
П р и м е р 1. Амидирование монометилового эфира 5-нитроизофталевой кислоты (1) (трео)-2-амино-1,3,4-бутантриолом до
5-нитро-3-[N-(1,3,
4-тригидрокси-трео- бут-2-ил)] карбамоилбензойной кислоты (2).
Исходный продукт (1) (22,5 г, 0,1 моль) смешивают с (трео)-2-амино-1,3,4-бутантриолом (30,25 г, 0,25 моль) и затем
суспензию нагревают до 110-120оС в течение 30 мин. Полное превращение в указанный продукт определяется методом тонкослойной жидкостной хроматографии, а затем раствор выливают в 1
н.хлористоводородную кислоту (200 мл) для осаждения продукта. После охлаждения, продолжающегося ночь, продукт отфильтровывают и промывают ледяной водой (2 раза по 20 мл). При сушке в вакууме
получается белый твердый продукт (2) 21,0 г, выход 67%
П р и м е р 2. Этерификация 5-нитро-3-[N-(1,3,4-тригидрокси-трио-бут-2-ил)] карбамоил бензойной кислоты (2) диметилсульфатом в
метил-5-нитро-3-[N-(1,3,4-тригидрокси-трео-бут-2-ил)]карбамоилбензоат (3).
Соединение (2) (15,7 г, 0,05 моль) растворяют в 1 н.растворе (55 мл) гидроокиси натрия и раствор охлаждают до
температуры ниже 20оС. Диметилсульфат (9,45 г, 0,075 моль) добавляют к раствору через 5 мин, при этом рН среды поддерживают в пределах 8-10 путем добавления 5 н.раствора гидроксида натрия.
Раствор перемешивают в течение 12 ч при комнатной температуре, после чего нерастворимый твердый продукт отфильтровывают. Пастообразный твердый продукт промывают холодной водой (дважды по 50 мл) и
высушивают в вакууме с образованием порошка (3) 11,8 г, выход 72%
П р и м е р 3. Амидирование метил-5-нитро-3-[N-(1,3,4-тригидрокси-трео-бут-2- ил)]карбамоил бензоата (3) аммиаком до
5-нитро-3-[N-(1,3,4-тригидрокси-трео- бут-2-ил)]карбамоилбензамид (4).
Эфир (3) (10,0 г, 0,0305 моль) растворяют в метаноле (50 мл) и к раствору добавляют концентрированный гидроксид
аммония (20 мл, приблизительно 0,3 моль). Суспензию нагревают в герметичной емкости до 50-60оС в течение 30 мин, завершение реакции определяют методом тонкослойной жидкостной хроматографии.
Метанол и гидроокись аммония удаляют путем дистилляции и разбавляют водой (50 мл). Смесь охлаждают всю ночь, после чего нерастворимый продукт отфильтровывают и промывают холодной водой (дважды по 5
мл). Сушкой в вакууме получают белый смешанный амид (4) 7,15 г, выход 75%
П р и м е р 4. Хлорирование сложного монометилового эфира 5-нитроизофталевой кислоты (1) тионилхлоридом в
хлорангидрид сложного монометилового эфира 5-нитроизофталевой кислоты.
Соединение (1) (225 г, 1 моль) растворяют в этилацетате (0,5 л), в качестве катализатора добавляют N,
N-диметилформамид. Раствор нагревают до 70оС и через 1,25 ч добавляют тионилхлорид (219 мл, 3 моль). Температуру поддерживают 70оС в течение 2 ч. Тионилхлорид отгоняют с
этилацетатом (трижды по 200 мл) и продукт растворяют в горячем этилацетате (250 мл), а затем осаждают циклогексаном (1 л), отфильтровывают и промывают циклогексаном (дважды по 200 мл). Затем продукт
высушивают при 50оС под вакуумом и получают твердый продукт (5) 216 г, выход 89%
П р и м е р 5. Амидирование хлорида сложного монометилового эфира 5-нитроизофталевой кислоты (5) с
аминодиоксепаном в метил-5-ниро-3-[N-(транс-2,2-диметил-6-гидрокси-1,3-диоксепан-5-ил)] карбамоилбензоат (6).
Хлорид моноэфира (5) (100 г, 0,411 моль) растворяют в сухом
тетрагидрофуране (1 л), к раствору порциями через 15 мин добавляют твердый аминодиоксепан (132,8 г, 0,825 моль) при поддержании температуры ниже 25оС при помощи ледяной бани. После этого
гетерогенную смесь перемешивают в течение 30 мин при комнатной температуре, завершение определяют тонкослойной жидкостной хроматографией. Нерастворимый амингидрохлорид отфильтровывают, а
тетрагидрофуран удаляют из фильтрата дистилляцией. Остаток растворяют в этилацетате (400 мл) при температуре, близкой к точке кипения, а затем раствор выдерживают в течение нескольких дней до
завершения кристаллизации продукта. Твердый продукт отфильтровывают, промывают холодным этилацетатом (дважды по 50 мл) и высушивают в вакуумной сушилке, при этом получают не совсем белый продукт (6)
82,4 г, выход 55%
П р и м е р 6. Амидирование метил-5-нитро-3-[N-(транс-2,2-диметил-6-гидрокси-1,3-диоксепан-5- ил)] карбамоилбензоата (6) гидроокисью аммония в 5-нитро-3-[N-(транс-2,
2-ди-метил-6-гидрокси-1,3-диоксипан-5-ил)] карбамоилбензамид (7).
Реактор Парра, работающий под давлением (800 мл), заполняют соединением (6), (80 г, 0,22 моль) метанолом (110 мл) и 15
г. гидроокисью аммония (225 мл, 3,38 моль). Реакционную емкость герметизируют и погружают в баню с водой при температуре 50оС на 2 ч, хроматография фиксирует завершение реакции.
Гетерогенную реакционную смесь затем смешивают с водой (100 мл) и омыляют. Пену суспендируют водой (100 мл), отфильтровывают и промывают два раза по 50 мл воды для получения белого твердого продукта
(7) 60,4 г, выход 79%
П р и м е р 7. Восстановление 5-нитро-3-[N-(транс-2,2-диметил-6-гидрокси-1,3-ди-оксепан-5-ил)] карбамоилбензамида (7) водородом и палладием на угле и хлористоводородной
кислотой в 5-амино-гидрохлорид-3-[N-(3,4-тригидрокси-трео-бут-2-ил)]карбамоилбензамид (8).
Реактор Парра, работающий под давлением (2,0 л), заполняют соединением (7) (58 г, 0,16 моль), 1 н. хлористоводородной кислоты (410 мл) и катализатором палладий-на-угле (10% Pd/C, 5,8 г, 1% Pd). Реакционную емкость соединяют с гидрогенератором и встряхивают при давлении 50 пси газообразного водорода в течение 2 ч, т.е. до тех пор, пока жидкостная хроматография не покажет 90% превращения в продукт (8). Затем палладиевый катализатор отфильтровывают, а ацетон, образовавшийся при восстановлении, удалялся вакуумом при температуре 50оС. Полученный чистый раствор (8) (450 мл, выход 90%) направляют непосредственно на стадию иодирования.
П р и м е р 8. Иодирование 5-амино-(гидрохлорид)-3-[N-(1,3,4- тригидрокси-трео-бут-2-ил)] карбамоилбензамида (8) монохлоридом йода в 5-амино-2,4,6-трийод-3-[N-(1,3,4-тригидрокси-трео-бут-2- ил)]карбамоилбензамид (9).
Соединение (8) (0,144 моль) в 1 н. хлористоводородной кислоте (450 мл) нагревают до 85оС и добавляют монохлорид йода (135 мл, 0,49 моль). Реакционную смесь нагревают до 85оС в течение 2 ч, окончание реакции контролируют. Реакционную смесь охлаждают до 25оС и экстрагируют дважды циклогексаном (200 мл), трижды дихлорметаном (300 мл) и 5 раз хлороформом (200 мл) до тех пор, пока не исчезает пурпурный цвет водного слоя. Полученный светло-желтый раствор направляют на рециркуляцию в колонну, содержащую Дуолит-А340 (800 г) и Доуэкс 50W-Х8 (266 г). Смолу промывают водой (6 л) и раствор концентрируют до объема 300 мл до тех пор, пока не начинают образовываться белые твердые кристаллы. Затем продукт отфильтровывают и получают белый твердый продукт (9) (40 г, 0,06 моль, выход 43%).
П р и м е р 9. Ацетилирование 5-амино-2,4,6-трийод-3-[N-(1,3,4-тригидрокси-трео-бут-2-ил)] карбамоилбензамида (9) ацетангидридом в 5-диацетиламино-2,4,6-трийодо-3-[N-(1,3,4-триацетокси-трео-бут-2-ил)] карбамоилбензамид (10).
Соединение (9) (90 г, 0,14 моль) смешивают с ацетангидридом (500 мл, 4,95 моль) при 70оС при интенсивном перемешивании. Добавляют катализатор перхлорная кислота (0,36 мл, 0,004 моль, что вызывает подъем температуры до 85оС. Реакционную смесь перемешивают при 85оС в течение 1 ч, до тех пор, пока она не становилась гомогенной, окончание реакции регистрируют хроматографией. Затем добавляют ацетат натрия (0,33 г, 0,004 моль) для нейтрализации перхлорной кислоты, и растворитель удаляют для получения вязкого коричневого масла. Масло разбавляют бутилацетатом (200 мл) при 70оС, что осуществляют сразу после удаления растворителя. Процедуру очистки повторяют два раза для получения коричневой пены (10) (113 г, 0,13 моль, выход 93%).
П р и м е р 10. Деацетилирование и алкилирование 5-диацетиламино-2,4,6-трийод-3-[N-(1,3, 4-триацетокси-трео-бут-2- ил)]карбамоилбензамида (10) метоксидом натрия и 2-хлорэтанолом в 5-[N-(2-гидроксиэтилацетамидо)]-2,4,6-трийод-3-[N-(1,3,4- тригидрокси-трео-бут-2-ил)]карбамоилбензамид (11).
Соединение (10) (113 г, 0,13 моль) растворяют в метаноле (500 мл), к которому добавляют 25% метоксид натрия (55 г, 0,25 моль) при 50оС. Через 5 ч хроматография показала, что деацетилирование завершено и раствор нейтрализуют смолой Доуэкс 50W-Х4 (10 г). Смолу отфильтровывают и фильтрат концентрируют до объема 400 мл. Нейтральный метаноловый раствор нагревают до 45оС и обрабатывают тринатрийфосфатдодекагидратом (129 г, 0,34 моль) и 2-хлорэтанолом (18,2 мл, 0,272 моль). Реакционную смесь перемешивают в течение 48 ч при 45оС, после чего добавляют хлорэтанол (4,7 мл, 0,07 моль) и натрий метоксид (14,7 г, 0,07 моль). После 71 ч хроматография показывает, что реакция завершена. Нерастворимые соли (89 г) удаляют путем фильтрации, а раствор нейтрализуют хлористоводородной кислотой (6 н. 7 мл). Затем раствор концентрируют для получения коричневой пены (11) (94 г, 0,12 моль, выход 92%).
П р и м е р 11. Амидирование монометилового эфира 5-нитроизофталевой кислоты (1) 3-амино-1,2-пропандиолом в 5-нитро-3-[N-(2,3-дигидроксипропил)] карба-моилбензойную кислоту (12).
Исходный продукт (1) (225 г, 1 моль) смешивают в 3-амино-1,2-пропандиолом (227,8 г, 2,5 моль) и гетерогенную смесь нагревают до 110-120оС в течение 1 ч. При этой температуре реакция завершается и гомогенную смесь смешивают с водой (1 л) и концентрированной НCl (170 мл). Смесь охлаждают в течение нескольких дней для полного осаждения продукта, а затем твердые частицы отфильтровывают и промывают холодной водой (дважды по 50 мл). После высушивания в вакууме получают белый твердый продукт (12) (193 г, выход 68% ).
П р и м е р 12. Восстановление 5-нитро-3-[N-(2, 3-дигидроксипропил)]карбамоилбензойной кислоты (12) водородом и палладием-на-угле в 5-амино-(гидрохлорид)-3-[N- (2,3-дигидроксипропил)]карбамоилбензойную кислоту (13).
Нитрокислоту (12) (180 г, 0,634 моль) смешивают с водой (1 л) и концентрированной HCl (60 мл), а также к раствору добавляют 10% палладий на угле (18 г). Суспензию гидрируют при давлении 2-4 атм. до тех пор, пока давление не остается постоянным, после чего хроматография показывает завершение реакции. Палладий-на-угле удаляется фильтрацией и гомогенный раствор используют без выделения продукта для последующих реакций (13) (приблизительный выход 98%).
П р и м е р 13. Йодирование 5-амино-(гидрохлорид)-3-[N-(2,3-ди- гидроксипропил)] карбамоилбензойной кислоты (13) монохлоридом йода в 5-амино-2, 4,6-трийод-3-[N-(2,3-дигидроксипропил)]карбамоил- бензойную кислоту (14).
Соединение (13) (0,62 моль в 1,5 л воды) дополнительно разбавляют водой до общего объема 4 л и нагревают до 85оС. Через 20 мин к раствору добавляют монохлорид йода (4,1 М, 499 мл, 2,05 моль) и в течение 6-8 ч температуру поддерживают 90оС, хроматография показывает завершение реакции. Гомогенную смесь охлаждают, экстрагируют смесью 1,2-дихлорэтан-циклогексан (9:1) 500 мл один раз, а затем дважды 1,2-дихлорэтаном (250 мл). Йодный слой затем концентрируют дистилляцией до 0,9 л и охлаждают в течение нескольких дней для завершения осаждения твердых частиц. После фильтрации, промывания холодной водой (дважды по 100 мл) и вакуумной сушки получают рыжевато-коричневый продукт (14) (286 г, выход 73%).
П р и м е р 14. Ацетилирование 5-амино-2,4,6-трийод-3-[N-(2,3-дигидроксипро-пил)] карбамоилбензойной кислоты (14) ацетангидридом в 5-диацетиламино-2,4, 6-трийод-3-[N- (2,3-диацетоксипропил)]карбамоилбензойную кислоту (15).
Исходный продукт (14) (100 г, 0,158 моль) смешивают с ацетангидридом (300 мл, 3,16 моль) и 70% перхлорной кислотой (0,2 мл), а затем нагревают до 80-90оС в течение 8 ч. Смесь нейтрализуют безводным ацетатом натрия (0,25 г) и ацетангидрид и уксусную кислоту удаляют путем дистилляции при 70-80оС. Масляный остаток превращают в азеотропную смесь бутилацетатом (дважды по 100 мл), затем растворяют в этилацетате (250 мл), и подвергают непосредственно хлорированию (15) (приблизительный выход 90%).
П р и м е р 15. Хлорирование 5-диацетиламино-2,4,6-трийод-3-[N- (2,3-диацетоксипропил)] карбамоилбензойной кислоты (15) тионилхлоридом в 5-диацетиламино-2, 4,6-трийод-3-[N-(2,3-диацетоксипропил)] карбамоилбензоилхло рид (16).
К исходному продукту (15) (0,142 моль) в этилацетате (225 мл) добавляют тионилхлорид (57 мл, 0,78 моль) при 65-70оС в течение часа. Тионилхлорид и этилацетат подвергают вакуумной дистилляции. Остаток превращают в азеотропную смесь бутилацетатом (дважды по 100 мл) и вакуумной сушкой. Коричневую пену (16) (130 г, выход 95%) направляют прямо на стадию амидирования.
П р и м е р 16. Амидирование 5-диацетиламино-2,4,6-трийод-3- [N-(2,3-диацетоксипропил)] карбамоилбензоилхлорида (16) аммиаком в 5-ацетиламино-2,4,6-трийод-3-[N-(2,3-диацетоксипропил)]карбамоилбен-замид (17).
Хлорид кислоты (16) (0,135 моль) растворяют в сухом N,N-диметилацетамиде (150 мл). Этот раствор охлаждают от 0-5оС, безводный аммиак (приблизительно 20 мл) конденсируют в смеси при использовании смеси сухой лед ацетон, и температуру реакционной смеси поддерживают равной комнатной температуре в течение 24 ч. Аммиак и диметилацетамид удаляют вакуумной дистилляцией. С помощью 1-пентанола (500 мл) осаждают твердые частицы, которые отфильтровывают и промывают дважды 1-пентанолом по 150 мл. Вакуумной сушкой получают твердый порошок (17) (82 г, выход 80,2%).
П р и м е р 17. Деацетилирование 5-ацетиламино-2,4,6-трийод-3- [N-(2,3-диацетоксипропил)] карбамоилбензамида (17) в 5-ацетиламино-2,4,6-трийод-3-[N-(2,3-дигид-роксипропил)]карбамоилбензамид (18).
Соединение (17) (81,2 г, 0,107 моль) суспендируют в воде (203 мл) и затем обрабатывают, добавляя по каплям 50%-ный раствор гироксида натрия в воде (16,9 мл, 0,322 моль). При перемешивании получают раствор. Раствор дегазируют в вакууме в течение 30 мин, в это время добавляют 12 М HCl (15 мл, 0,18 моль). После выдержки при 4оС полученный осажденный твердый продукт отфильтровывают, промывают ледяной водой трижды по 50 мл, этанолом (80 мл) и высушивают в вакууме, получают продукт (18) (54,1 г, 75 выход).
П р и м е р 18. Алкилиpование 5-ацетиламино-2,4,6-трийод-3-[N- (2,3-дигидроксипропил)] карбамоилбензамида (18) в 5-[N-(2, 3- дигидроксипропил)ацетамидо] -2,4,6-трийод-3-[N-(2,3-дигидроксипропил)] карбамо илбензамид (19).
Соединение (18) (39,7 г, 0,059 моль) растворяют в пропиленгликоле (16,7 мл), этаноле (120 мл) и 25 мас. метоксида натрия (17,6 мл, 0,077 моль). Затем добавляют хлорпропандиол (9,78 г, 0,0885 моль) и смесь перемешивают при 25оС в течение часа. Реакционную смесь нагревают до 33оС и перемешивают еще 19 ч, во время этого перемешивания к смеси добавляют 25 мас. метоксида натрия (3,4 мл, 0,015 моль). Реакцию подавляют добавлением 12 М HCl, реакционную смесь перегоняют в вакууме, добавляют воду (200 мл) и перегоняют снова для получения водного раствора, который пропускают через Доуэкс 50 Н+ (62 г) и Дуолит А-340 ОН- (140 г). Смолу промывают при помощи воды и концентрируют и получают 150 г раствора, который обрабатывают древесным углем Норит Ультра SX (1,0 г) при 60оС в течение 14 ч. Древесный уголь отфильтровывают с получением водного раствора, который перемешивают в течение 2 ч с Доуэксом 50 Н+ (1,0 г) и Дуолитом А-340 ОН- (4 г). Смолу отфильтровывают и водный раствор упаривают до 50,3 г маслянистого продукта (19) (32,8 г, выход 74%) в смеси глицерин-пропиленгликоль. Это масло очищают, как указано на последующей стадии.
П р и м е р 19. Переацетилирование, очистка в колонке с двуокисью кремния и последующее деацетилирование 5-[N-(2,3-дигидроксипропил)ацетамидо]-2,4,6- трийод-3-[N-(2,3-дигидрокси- пропил)]карбамоилбензамида (19).
Соединение (19) (16,4 г, 0,022 моль) растворяют в глицерин-пропиленгликоле (общая масса 25,15 г), затем разбавляют пиридином (1,74 г, 0,022 моль) и ацетангидридом (115 г, 1,12 моль) и затем нагревают до 60о С в течение 18 ч. Реакционную смесь перегоняют при пониженном давлении с получением масла, растворяют в CHCl3 (100 мл) и экстрагируют дважды 0,1 н. HCl и дважды по 50 мл 15 мас. рассолом. Слой CHCl3 высушивают над MgSO4, отфильтровывают с получением масла. Это масло очищают на 900 г двуокиси кремния в колонке, где устанавливается градиент растворителя от 5% уксусной кислоты, 95% хлороформа до 5% уксусной кислоты, 4% метанола, 91% хлороформа. Очищенные фракции объединяют, перегоняют до образования пены и затем обрабатывают метанолом (30 мл) и 25 мас. метоксида натрия в метаноле (0,98 г, 0,0054 моль). Через 30 мин раствор перегоняют, восстанавливают метанолом (20 мл) и перемешивают со смолой Доуэкс 50 Н+ (1,3 г). После снижения рН с 12 до 5 смолу отфильтровывают с получением раствора, который упаривают до пены, добавляют воду (25 мл) и выпаривают до получения твердого продукта (19) (8,12 г, выход 49%).
П р и м е р 20. Деацитилирование 5-диацетиламино-2,4,6-трийод- 3-[N-(2,3-диацетоксипропил)] карбамоилбензойной кислоты (15) в 5-ацетиламино-2,4,6-трийод-3-[N-(2,3-дигидроксипропил)]карбамоил- бензойную кислоту (20).
Соединение (15) (720 г, 0,9 моль) растворяют в 500 мл метанола и добавляют 25 мас. метоксида натрия в метаноле (345 мл, 1,5 моль). Через 4 ч при температуре 45-50оС реакционную смесь упаривают при пониженном давлении, подкисляют 12 М HCl (124 мл, 1,5 моль), и соль отфильтровывают. Фильтрат упаривают при пониженном давлении с получением масла, которое разбавляют н-пропанолом (680 мл). После кристаллизации при 4оС полученный твердый продукт (20) отфильтровывают, промывают н-пропанолом дважды по 300 мл и высушивают в вакууме. Выход составляет 391 г (64%).
П р и м е р 21. Алкилирование 5-ацетиламино-2,4,6-трийод-3- [N-(2,3-дигидроксипропил)] карбамоилбензойной кислоты (20) в натриевую соль 5-[N-(2,3-дигидроксипропил)ацетамидо] -2, 4,6- трийод-3-[N-(2,3-дигидроксипропил)]карбамоилбензойной кислоты (21).
Соединение (20) (100 г, 0,148 моль) растворяют в 400 мл метанола. К раствору добавляют твердый Na3 PO4·12 Н2О (140,6 г, 0,37 моль), затем хлорпропандиол (32,7 г, 0,296 моль) и 25 мас. метоксида натрия в метаноле (24,1 г, 0,111 моль), по каплям. Реакционную смесь нагревают до 40оС в течение 10 ч, в течение которых к смеси частями добавляют 25% метоксида натрия (8,0 г, 0,368 моль). Соль отфильтровывают, метанольный фильтрат подкисляют 12 М HCl (3,5 мл), превращают в вязкий маслянистый продукт (21) и направляют непосредственно на следующую реакцию.
П р и м е р 22. Ацетилирование натриевой соли 5-[N-(2,3-дигидроксипропил)ацетамидо] -2,4,6-трийод-3-[N-(2,3- дигидроксипропил)]карбамоилбензойной кислоты (21) в 5-[N-(2,3- диацетоксипропил)ацетамидо]-2,4,6-трийод-3-[N-(2,3-диа-цетоксипропил)]карбам оилбензойную кислоту (22).
Соединение (21) (114 г, 0,148 моль) (масло) разбавляют пиридином (11,7 г, 0,148 моль) и ацетангидридом (605 г, 5,92 моль), а затем смесь перемешивают при температуре 65-70оС в течение 2 ч. Реакционную смесь упаривают до получения масла, затем азеотропируют бутилацетатом дважды по 100 мл и обрабатывают водой (300 мл) и смесью толуол-этилацетат (3:1) (200 мл). Водный слой экстрагируют смесью толуол-этилацетат (3:1) (трижды по 100 мл) и подкисляют НCl (22,5 мл) в присутствии этилацетата (300 мл). Подкисленный слой воды отделяют и дважды экстрагируют этилацетатом (100 мл). Три последних экстракта этилацетата объединяют, высушивают над MgSO4, отфильтровывают и упаривают до твердого продукта (22) (118 г, выход 87%).
П р и м е р 23. Хлорирование 5-[N-(2,3-диацетоксипропил)ацетамидо]-2,4,6-трийод- 3-[N-(2,3- диацетоксипропил]карбамоилбензойной кислоты (22) в 5-[N-(2,3- диацетоксипропил)ацетамидо] -2,4,6-трийод-3-[N-(2, 3-диацетоксипропил)]карбамоилбензоил- хлорид (23).
Соединение (22) (113,6 г, 0,124 моль) растворяют в этилацетате (100 мл) при 55оС, к нему по каплям добавляют тионилхлорид
(44 г, 0,37 моль) и смесь нагревают с обратным холодильником в течение 2 ч, подвергают роторному испарению до получения масла, а затем азеотропируют бутилацетатом (2х50 мл) с получением пены, которая
растворяется в хлороформе (200 мл), и экстрагируют 0,2 М фосфатным буфером с рН 6,7 (100 мл). Органический слой высушивают над MgSO4, отфильтровывают и выпаривают с образованием твердого
продукта (23,115 г, выход 98%
П р и м е р 24. Амидирование 5-[N-(2,3-диацетоксипропил)- ацетамидо]-2,4,6-трийод-3-[N-(2,3-диацетоксипропил)] карбамоилбензоилхлорида (23) в 5-[N-(2,
3-диацетоксипропил) ацетамидо]-2,4,6-трийод-3-[N-(2,3-диацетоксипропил)]карбамоил- бензамид (24).
Соединение (23) (105 г, 0,111 моль) растворяют в ацетонитриле (400 мл), к которому добавляют безводный аммиак, с использованием сухого льда при 25оС. Через 3 ч аммиак испаряют, реакция завершилась. Соль отфильтровывают и путем выпаривания получают твердый продукт (24) (98, 9 г, выход 96%).
П р и м е р 25. Деацетилирование 5-[N-(2,3-диацетоксипропил) ацетамидо] -2,4,6-трийод-3-[N-(2,3-диацетоксипропил)] карба- моилбензамида (24) в 5-[N-(2, 3-дигидроксипропил)ацетамидо] -2,4,6-трийод- 3-[N-(2,3-дигидроксипропил)]карбамоилбенза-мид (19).
Соединение (24) (98,7 г, 0,106 моль) растворяют в метаноле (250 мл), к которому при 25оС добавляют 25 мас. метоксида натрия в метаноле (2,30 г, 0,0106 моль). Через 15 мин раствор перегоняют под вакуумом до масла, обрабатывают метанолом (200 мл) и перемешивают со смолой Доуэкс 50 Н+ (6,0 г) до тех пор, пока значение рН не снизится от 12 до 6. Смолу отфильтровывают и получают раствор, который упаривают до образования пены, которую обрабатывают водой (320 мл) и древесным углем Норит SX (3,0 г), кипятят с обратным холодильником в течение 7 ч, фильтруют, деионизируют путем перемешивания со смолой Доуэкс 50 Н+ (3 г) и смолой Доуэкс XUS40123 (12 г), после чего смолу отфильтровывают и остаток упаривают с образованием твердого продукта (19) (79,2 г, выход 96%).
П р и м е р 26. Метоксиацетилирование 5-амино-2,4,6-трийод-3- [N-(1, 3,4-тригидрокси-трео-бут-2-ил)]карбамоилбензамида (9) метоксиацетилхлоридом в 5-метоксиацетиламино-2,4,6-трийод-3- [N-(1,3,4-тригидрокси-трео-бут-2-ил)]карбамоилбензамид (25).
Соединение (9) (100 г, 0,15 моль) суспендируют в N,N-диметилацетамиде (250 мл) при 25оС, к которому добавляют метоксиацетилхлорид (68 мл, 0,75 моль) через 30 мин. Реакционную смесь перемешивают при 35оС в течение 5 ч, хроматографией определяют завершение реакции. К реакционной смеси добавляют метоксид натрия (97 г, 0,45 моль) и смесь перемешивают при 40оС в течение 2 ч. Раствор нейтрализуют смолой Доуэкс 50W-Х4, фильтруют и разбавляют н-бутанолом (700 мл). Сразу образуется белый осадок, который отфильтровывают для получения не совсем белого твердого продукта (25) (80,6 г, 0,11 моль, выход 73%).
П р и м е р 27. Алкилирование иоксифталамовой кислоты (26) в 5-[N-(2,3-дигидроксипропил)ацетамило] -2,4, 6-трийод-3-[N-(2- гидроксиэтил)]карбамоилбензоат натрия (27).
Иоксифталамовую кислоту (26) (966 г, 1,5 моль) растворяют в 1 н. гидроокиси натрия (1,5 л) при комнатной температуре, нагревают до 75оС и к ней добавляют 3-хлор-1,2-пропандиол (223,8 г, 2,03 моль) и 5 н. гидрохлорида натрия (приблизительно 0,4 л) одновременно свыше 1,25 ч. Реакционную смесь нагревают до 80-90оС в течение следующих 2,5 ч, хроматографией определяют завершение реакции (приблизительно 90% превращения продукта). Реакционную смесь нейтрализуют концентрированной хлористоводородной кислотой (приблизительно 3 мл) и упаривают. Приблизительно половину полученного остатка помещают в воду (0,4 л). При охлаждении осаждается белый кристаллический твердый осадок, который отфильтровывают и промывают холодной ледяной водой. При сушке получают кристаллический продукт (27) (249 г).
П р и м е р 28. Ацетилирование 5-[N-(2, 3-дигидроксипропил)ацетамидо]-2,4,6-трийод- 3-[N-(2,3-гидроксиэтил)]карбамоилбензо- ата натрия (27) в 5-[N-(2,3-диацетоксипропил)ацетамидо] -2,4,6- трийод-3-[N-(2-ацетоксиэтил)] карбамоилбензойную кислоту (28).
Соединение (27) (50 г, 0,067 моль) добавляют к перемешиваемому ацетангидриду (102 мл, 1,080 моль, 16,0) при 25оС, к смеси добавляют пиридин (5,4 мл, 0,067 моль, 1,0) и затем температуру поднимают до 85оС в течение часа, по хроматографии наблюдают, что реакция идет к завершению. Гомогенную реакционную смесь выпаривают в вакууме до вязкого масла, растворяют в бутилацетате (50 мл) и снова выпаривают. Масло обрабатывают водой (260 мл) и экстрагируют смесью толуол-этилацетат (2:1) 4 раза по 100 мл. Водный слой подкисляют 12 н. хлористоводородной кислотой (11 мл) и экстрагируют этилацетатом 3 раза по 50 мл. Органический слой высушивают над сульфатом магния, выпаривают до образования пены (28) и направляют непосредственно на следующую стадию (55 г, 0,065 моль, выход 97%).
П р и м е р 29. Хлорирование 5-[N-(2,3-диацетоксипропил)ацетамидо]-2,4,6-трий- од-3-[N-(2-ацетоксиэтил)] карбамоилбензой- ной кислоты (28) в 5-[N-(2, 3-диацетоксипропил)ацетамидо] -2,4,6- трийод-3-[N-(2-ацетоксиэтил)] карбамоилбензоилхлорид (29).
Соединение (28) (55 г, 0,065 моль) растворяют в 1,2-дихлорэтане (170 мл) и нагревают до 85оС. Тионилхлорид (9,8 мл, 0,134 моль, 2,0) добавляют к раствору и через 3 ч наблюдают хроматографией завершение реакции. Реакционную смесь выпаривают под вакуумом с получением масла, повторно растворяют в бутилацетате (50 мл) и повторно выпаривают. Продукт выделяют в виде желтой пены (29) (51,9 г, 0,060 моль, выход 92%).
П р и м е р 30. Амидирование 5-[N-(2, 3-диацетоксипропил)ацетамидо]-2,4,6-трий-од-3-[N-(2- ацетоксиэтил)]карбамоилбензоилхлорида (29) в 5-[N-(2,3- диацетоксипропил)ацетамидо] -2,4,6-трийод-3-[N-(2-ацето- ксиэтил)] карбамоилбензамид (30).
Соединение (29) (51,9 г, 0,060 моль) растворяют в ацетонитриле (200 мл) и безводный аммиак (взятый в избытке) добавляют к смеси при температуре 10оС. Через 4 ч хроматографией наблюдают завершение реакции. Реакционную смесь фильтруют для удаления солей хлорида аммония и растворитель удаляют для получения желтого масла (30) (47 г, 0,056 моль, выход 93%).
П р и м е р 31. Деацетилирование 5-[N-(2,3-диацетоксипропилацетамидо)] -2,4,6- трийод-3-[N-(2-ацетоксиэтил)] карбамоилбен- замида (30) в 5-[N-(2,3-дигидроксипропил- ацетамидо)] -2,4, 6-трийод-3-[N-(2-гидрокси- этил)] карбамоилбензамид (31).
Соединение (30) (47 г, 0,056 моль) растворяют в метаноле (240 мл) и 25% -ный метоксид натрия добавляют к раствору для повышения рН до приблизительно 12. Раствор перемешивают при 25оС в течение 1 ч, после чего хроматография показала, что деацетилирование завершено. Реакционную смесь нейтрализуют 1 н. HCl (10 мл) и из удаленного растворителя получают почти белую пену (31) (39 г, 0,054 моль, выход 97% чистота 98%), перекристаллизовывают из горячего метанола (5 г, в 15 мл, с затравкой).
П р и м е р 32. Амидирование 5-амино-2,4,6-трийод-изофталоилхлорида (32) в 5-амино-2,4,6-трийод-3-хлоркарбонилбенза-мид (33).
Исходное вещество (32) (300 г, 0,503 моль) растворяют в тетрагидрофуране (900 мл) и гомогенный раствор охлаждают льдом до 5-10оС. Концентрированную гидроокись аммония (92,3 мл, 1,38 моль) добавляют к раствору через 10 мин, температура возрастает до 30оС. Реакционную смесь перемешивают при комнатной температуре в общей сложности в течение 90 ч с последующим добавлением гидроокиси аммония (общее количество 25,2 мл, 0,38 моль), затем охлаждают и нерастворимые соли удаляют фильтрацией. Фильтрат промывают насыщенным раствором NaCl (2 раза по 200 мл). Тетрагидрофуран выпаривают до получения вязкого масла. Осажденное этилацетатом (800 мл) золотисто-коричневого цвета вещество отфильтровывают, затем промывают этилацетатом (дважды по 100 мл) и высушивают с получением соединения (35) (193 г, выход 66,5%).
П р и м е р 33. Димеризация 5-амино-2,4,6-трийод-3-хлоркарбонилбензамида (33) в бис-(3-хлоркарбонил-5-карбамоил)-2,4,6-трийоданилид малоновой кислоты (34).
Соединение (33) (20,0 г, 34,7 ммоль) растворяют в сухом тетрагидрофуране (100 мл), нагревают до 45оС и через 3 мин к нему добавляют малонил дихлорид (2,53 мл, 26 ммоль) для получения гетерогенной смеси. Затем добавляют сухой ТГФ (100 мл) и суспензию перемешивают в течение 1 ч, после чего хроматографией определяют завершение реакции. Смесь разбавляют бутилацетатом (150 мл) и твердую часть отфильтровывают, промывают бутилацетатом (50 мл х 2), высушивают под вакуумом с получением продукта (34) (13,18 г, выход 62).
П р и м е р 34. Амидирование бис-[(3-хлоркарбонил-5-карбамоил)-2,4,6-трийода-нилид] малоновой кислоты (34) в бис-{[3-N-(1,3,4-тригидрокси-трео-бут-2-ил)карбамо- ил-5-карбамоил] -2,4,6-трийоданилид} мало-новую кислоту (35).
Соединение (34) (8,0 г, 6,56 ммоль) растворяют в сухом N,N-диметилацетамиде (10 мл), затем добавляют триэтиламин (2,83 мл, 13,12 ммоль) и раствор охлаждают до 20оС. Через 3 мин добавляют транс-5-амино-2,2-диметил-6-гидрокси-1, 3-диоксепан (2,64 г, 16,4 ммоль), и гомогенную смесь перемешивают при комнатной температуре в течение 6 ч, хроматографией определяют завершение реакции. Растворитель выпаривают, добавляют воду (50 мл) и смесь нагревают до 75оС в течение 15 мин для расслоения ацетонидов. Продукт получают путем выпаривания и осаждения изопропанолом (100 мл). Твердый остаток отфильтровывают, промывают изопропанолом (дважды по 20 мл) и высушивают с получением 8,6 г (35) (выход 94%).
П р и м е р 35. Амидирование 5-N-метиламино-2,4,6-трийодизофталоилхлорида (36) в 5-N-метиламино-2,4, 6-трийод-3-хлоркарбонилбензамид (37).
Исходный продукт (36) (305 г, 0,5 моль) растворяют в тетрагидрофуране (1 л) и охлаждают до 10оС. Через 5 мин добавляют концентрированный гидроксид аммония (100 мл, 1,5 моль); температура возрастает до приблизительно 25оС. Реакционную смесь перемешивают при комнатной температуре в течение 65 ч, затем добавляют дополнительные порции концентрированного NH4ОН через 20 ч (3,5 мл) и через 44 ч (3,5 мл). После охлаждения нерастворимые соли и бисамид отфильтровывают и тетрагидрофурановый фильтрат промывают насыщенным раствором хлорида натрия (дважды по 100 мл). Тетрагидрофуран упаривают и продукт осаждают из густого масла этилацетатом (500 мл). После фильтрации, промывки этилацетатом и сушки получают продукт (37) (132,1 г, выход 45%).
П р и м е р 36. Димеризация 5-N-метиламино-2,4,6-трийод-3-хлоркарбонилбенза-мида (37) в бис-[(3-хлоркарбонил-5-карбамоил)-2,4, 6-трийод-N-метиланилид] малоновой кислоты (38).
Соединение (37) (25 г, 42,3 ммоль) растворяют в сухом тетрагидрофуране (100 мл) и гомогенный раствор нагревают до 50оС. Через 2 мин добавляют малонилдихлорид (3,05 мл, 31,3 ммоль), затем тетрагидрофуран (50 мл) и суспензию нагревают в течение 1 ч, хроматографией определяют завершение реакции. После разбавления бутилацетатом (50 мл) продукт отфильтровывают, промывают бутилацетатом (25 мл х 2) и высушивают, при этом получается не совсем белый твердый продукт (38) (15,24 г, выход 58).
П р и м е р 37. Превращение бис-[(3-хлоркарбонил-5-карбамоил)- 2,4,6-трийод-N-метиланилид]малоновой кислоты (38) в бис-{[3-N- (1,3,4-тригидрокси-трео-бут-2-ил-карбамоил)-5-карбамоил]-2,4, 6- трийод-N-метиланилид}малоновой кислоты (39).
Исходный продукт (38) (10 г, 8 ммоль) растворяют в сухом диметилацетамиде (15 мл) и триэтиламине (2,23 мл, 16 ммоль). Через 5 мин добавляют транс-5-амино-2,2-диметил-6-гидрокси-1,3- диоксипан (аминодиоксипан) (3,22 г, 20 ммоль) и гомогенную смесь перемешивают в течение 6 ч, после чего хроматографией определяют завершение реакции. ДМА удаляют вакуумной дистилляцией и изопропилиден расщепляют хлористоводородной кислотой при 50оС. Воду удаляют на роторном испарителе и для осаждения продукта добавляют изопропанол. После фильтрации, промывки изопропанолом (10 мл х 3) и сушки получали димер (39) (9,86 г, выход 87%).
П р и м е р 38. Превращение 5-N-(метил) амино-2,4, 6-трийод-3- хлоркарбонилбензамида (36) в 5-[N-(метил)-2-ацетоксиацетамидо] 2,4,6-трийод-3-хлоркарбонилбензамид (40).
Исходный продукт (36) (25 г, 42,3 ммоль) растворяют в сухом N, N- диметилацетамиде (50 мл) при комнатной температуре, затем добавляют 2-ацетоксиацетилхлорид (6,83 мл, 63,5 ммоль) и после перемешивания в течение ночи хроматографией определяют конец реакции. Продукт осаждают добавлением ледяной воды (200 мл) и отфильтровывают. После промывки водой твердый продукт растворяют в тетрагидрофуране (200 мл) и раствор экстрагируют смесью насыщенный NaCl насыщенный NaHCO3 (3:1 (250 мл), затем насыщенным NaCl (100 мл). Органический слой высушивают (MgSO4) и растворитель удаляют с получением пены (40) (22,1 г, выход 77,2%).
П р и м е р 39. Амидирование и снятие защиты 5-[N-(метил)-2- ацетоксиацетамидо] -2,4,6-трийод-3-хлоркарбонилбензамида (40) в 5-[N-(метил-2-гидроксиацетамидо)] -2,4,6-трийод-3-[N-(1,3, 4-тригидрокси-трео- бут-2-ил)]карбамоилбензамид (41).
Соединение (40) (7,0 г, 10,35 ммоль) растворяют в смеси тетрагидрофурана (40 мл) и триэтиламина (1,44 г, 10,35 ммоль) и охлаждают до 10оС. Добавляют твердый аминодиоксилан (2,0 г, 12,41 ммоль). Охлаждение прекращают и реакционную смесь перемешивают при 25оС. Через 18 ч хроматографией определяют завершение реакции. Реакционную смесь разбавляют тетрагидрофураном (40 мл) и смесью насыщенный NaCl насыщенный NaHCO3 (3:1) 50 мл, и слои разделяют. Органический слой промывают насыщенным NaCl (40 мл х 2), высушивают MgSO4 и растворитель выпаривают, в результате получают пену (6,9 г, выход 82%). Пену растворяют в метаноле (50 мл) и добавляют 4,6 г формальдегидного раствора NaOMe (0,5 мл). Раствор упаривают при температуре 50оС с получением масла, которое затем смешивалось с водой (50 мл) и смолой Доуэкс 50Н+ (10 г). При нагревании до 60оС в течение 30 мин получают гомогенный раствор, хроматографией определяют, что завершилось разложение эфира и изопропилидена. Смолу отфильтровывают и раствор пропускают через последовательные колонки с Дуолитом А 340 ОН- (Доуэкс 50 Н+) до тех пор, пока не завершится деионизация. Соединение вымывают из колонок водой и затем обрабатывают углем (0,4 г) Норит Ультра S-X. Через 1 ч при 70оС уголь отфильтровывают, а воду выпаривают, получают белую пену (41) (3,8 г, выход 50% из 40).
П р и м е р 40. Инъекционный растворы, содержащие 5-[N-(2, 3-дигидроксипропил)-ацетамидо]-2,4,6-трийод-3-[N-(2,3- дигидроксипропил)]карбамоилбензамида (19), приведены в табл.2.
Процедура: натрийкальцевую соль этилендиаминтетрауксусной кислоты, трис-гидроксиметиламинометан и контрастную среду растворяют в воде и рН среды доводят до 7,0 путем добавления 1 н. хлористоводородной кислоты. Раствор разбавляют до 100 мл водой для инъекций, профильтрованной через мембрану 0,22 мкм в стеклянные бутылочки, закрывают крышками и автоклавируют в течение 20 мин при 121оС.
П р и м е р 41. Растворы для инъекций, содержащие 5-[N-(2- гидроксиэтил)ацетамидо] -2,4,6-трийод-3-[N-(1,3,4-тригидрокси- трео-бут-2-ил)]карбамоилбензамида (11), приведены в табл.3.
Процедура: кальцийдинатриевая соль этилендиаминтетрауксусной кислоты, тринатрий цитрат и контрастную среду растворяют в воде для инъекций и рН среды доводят до 5,0-6,0 путем добавления карбоната натрия и двуокиси углерода. Растворы доводять до 100 мл водой для инъекций, фильтруют через мембрану 0,22 мкм в стеклянные бутылочки, закрывают крышками и подвергают автоклавированию в течение 20 мин при 121оС.
Из вышеприведенных данных очевидно, что новая неионная контрастная среда обладает превосходными свойствами по сравнению с известными. Благодаря улучшению физических характеристик, в частности осмотичности и вязкости, появилась возможность исследовать различные части тела. Несмотря на большое число соединений, которые были синтезированы и проверены, заявленные соединения превосходят соединения, раскрытые ранее. Благодаря наличию в молекуле трех различных азотов, только два из которых являются замещенными, достигаются новые свойства. Кроме того, предлагаются синтетические пути, которые являются эффективными и обеспечивают высокий выход при использовании легко доступных соединений.
Реферат
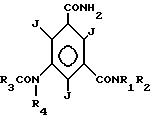
Использование: в качестве неионогенных контрастных сред. Сущность изобретения: продукт-трийод-5-аминоизофталдиамиды ф-лы I. Реагент 1: соединение ф-лы II. Реагент 2: NH3.
Радиологическая композиция содержит физиологически приемлемый носитель и в качестве контрастной среды соединение ф-лы I в количестве 150 - 400 мг/мл. Структура соединений ф-лы I и II

3 с. и 9 з. п. ф-лы, 3 табл.
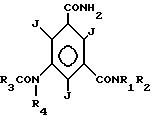
Формула

где R1 водород;
R2 C1-C4-гидроксиалкил, имеющий от 1 до n-1 гидроксильных групп, где n число атомов углерода,
R3 низший алкил, или низший алкоксизамещенный низший C1 -C2-алкил, или R3 имеет формулу

R4 водород или C1 -C3-алкил, имеющий от 0 до n-1 гидроксильных групп, где n число атомов углерода, используемые в качестве неионных контрастных сред.
имеющей по меньшей мере две оксигруппы,
где R1 водород;
R2 C2-C4 -гидроксиалкил, имеющий от 1 до n-1 гидроксильных групп, где n число атомов углерода;
R3 низший алкил, гидроксизамещенный низший алкил или низший алкоксизамещенный низший C1-C3-алкил
или R3 имеет формулу

где R4 водород или C1-C3-алкил, имеющий от 0 до n-1 гидроксильных групп, где n число атомов углерода,
отличающийся тем, что соединения общей формулы II

где в R1 и R2 гидроксильные группы могут быть защищены,
в результате взаимодействия с соединением общей формулы
R3CO-X,
где Х галоген или сложноэфирный остаток,
в катализирующем растворителе, например пиридин, диметиламид или диметилформамид, и при необходимости в результате последующего отщепления защитных групп переводят в конечный продукт общей формулы I, где R4 - водород, или в промежуточный продукт общей формулы I, где R4 - R3CO, и последний, при желании, с защищенными гидроксильными группами, в результате алкилирования в щелочных условиях превращают при необходимости после отщепления защитных групп в конечный продукт общей формулы I, где R4 не означает водород.
имеющей по меньшей мере две оксигруппы,
где R1 водород,
R2 C2-C4-гидроксиалкил, имеющий от 1 до n-1 гидроксильных групп, где n число атомов углерода;
R3 низший алкил, гидроксизамещенный низший алкил или низший алкоксизамещенный низший C1-C2-алкил или R3 имеет общую формулу

R4 водород или C1-C3-алкил, имеющий от 0 до n-1 гидроксильных групп, где n число атомов углерода,
отличающийся тем, что соединения общей формулы III
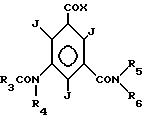
где R5 R1,
R6 R2 или водород,
X галоген или сложноэфирный остаток,
подвергают взаимодействию с аммиаком и, в случае необходимости, с галоидалканолом, гидроксильные группы которого могут буть защищены, и непосредственно после этого в соответствующем случае защитные группы отщепляют.

имеющей по меньшей мере две оксигруппы,
где R1 водород;
R2 C2-C4-гидроксиалкил, имеющий от 1 до n-1 гидроксильных групп, где n число атомов углерода;
R3 низший алкил, гидроксизамещенный низший алкил или низший алкоксизамещенный низший C1-C2-алкил или R3 имеет формулу
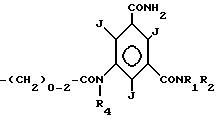
R4 водород или C1-C3-алкил, имеющий от 0 до n-1 гидроксильных групп, где n число атомов углерода,
в количестве 150 400 мг/мл.
03.11.89 при R3 группа формулы

05.07.89 остальное.
Комментарии