Новые лекарственные вещества - RU2237657C2
Код документа: RU2237657C2
Описание
Настоящее изобретение относится к новым лекарственным веществам, предназначенным как для систематического, так и для несистематического применения, а также композициям на их основе для использования в случаях окислительного стресса и/или эндотелиальных дисфункций.
Под окислительным стрессом подразумевается возникновение свободных радикалов или радикальных соединений, которые наносят повреждения как клеткам, так и окружающим их тканям (Pathophysiology: the biological basis for disease in adults and children, McCance&Huether 1998, pp.48-54).
Под эндотелиальными дисфункциями подразумевают дисфункции, относящиеся к сосудистому эндотелию. Известно, что повреждение сосудистого эндотелия является одной из важнейших причин, которые могут вызвать серию патологических процессов, затрагивающих различные органы и системы организма, как описано далее (Pathophysiology: the biological basis for disease in adults and children, McCance&Huether 1998, p.1025).
Известно, что окислительный стресс и/или эндотелиальные дисфункции связаны с различными патологиями, как описано далее. Окислительный стресс также может быть обусловлен токсичностью большого числа различных лекарственных веществ, что значительно сказывается на эффективности их действия.
Указанные патологические явления имеют хронический характер, подрывающий силы организма, и очень часто являются типичными для людей пожилого возраста. Как уже было сказано, при указанных патологических состояниях применяемые лекарственные вещества обладают заметным негативным действием.
Можно привести следующие примеры патологических состояний, обусловленных окислительным стрессом и/или эндотелиальными дисфункциями или характерных для людей пожилого возраста.
- Для сердечно-сосудистой системы: миокардиальная и сосудистая ишемия в целом, гипертония, инсульт, атеросклероз и т.д.
- Для соединительной ткани: ревматоидный артрит и связанные с ним воспалительные заболевания и т.д.
- Для легочной системы: астма и связанные с ней воспалительные заболевания и т.д.
- Для желудочно-кишечной системы: язвенные и неязвенные диспепсии, кишечные воспалительные заболевания и т.д.
- Для центральной нервной системы: болезнь Альцгеймера и т.д.
- Для мочеполовой системы: импотенция, недержание.
- Для кожных покровов: экзема, нейродерматит, угри.
Инфекционные заболевания в целом (см. Schwarz, Brady, "Oxidative stress during viral infection: a review" Free Radical Biol. Med. 21/5, 641-649, 1996).
Кроме того, сам процесс старения может рассматриваться как действительное патологическое состояние (см. Pathophysiology: the biological basis for disease in adults and children, pp.71-77).
Известные лекарственные вещества, при введении их пациентам, имеющим патологии, связанные с окислительным стрессом и/или эндотелиальными дисфункциями, проявляют сниженную эффективность и/или повышенную токсичность.
Это происходит, например, в случае таких лекарственных веществ, как противовоспалительные, сердечно-сосудистые лекарственные вещества, лекарственные вещества для дыхательной системы, лекарственные вещества для центральной нервной системы, лекарственные вещества для костной системы, антибиотики, лекарственные вещества для мочеполовой, эндокринной системы и т.д.
Исследования лекарственных веществ направлены на поиск новых молекул, имеющих улучшенный терапевтический индекс (соотношение эффективность/токсичность) или пониженное соотношение риск/полезное действие, в том числе и для указанных выше патологических состояний, при которых терапевтический индекс значительного числа лекарственных веществ оказывается низким. Фактически, при указанных выше состояниях окислительного стресса и/или эндотелиальных дисфункций многие лекарственные вещества проявляют низкую активность и/или высокую токсичность.
В частности, противовоспалительные лекарственные вещества, такие как NSAIDs и лекарственные вещества против колик, например 5-аминосалициловая кислота и ее производные, имеют следующие недостатки. NSAIDs проявляют токсические свойства, особенно в тех случаях, когда организм ослаблен или находится под влиянием патологических состояний, связанных с окислительным стрессом. К указанным состояниям можно отнести, например, следующие: преклонный возраст, ранее перенесенная язва, ранее перенесенное желудочное кровотечение, хронические заболевания, подрывающие силы организма, в частности, воздействующие на сердечно-сосудистую систему, почечный аппарат, состояние крови и т.д. ("Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving non-steroidal anti-inflammatory drugs. A randomized, double blind, placebo-controlled trial." F.E.Silverstein et al. Ann. Intern. Med. 123/4, 241-9, 1995; Martindale 31 a ed. 1996, page 73, Current Medical Diagnosis and Treatment 1998, pages 431 and 794).
Введение противовоспалительных лекарственных веществ пациентам, находящимся в указанных выше патологических состоящих, можно осуществить, только используя более низкие дозы лекарственного вещества, по сравнению с теми, которые обычно используются для терапии, чтобы избежать заметных токсических проявлений. Поэтому противовоспалительная активность проявляется слабо.
Бета-блокаторы, используемые для лечения стенокардии, гипертонии и сердечной аритмии, оказывают побочное воздействие на дыхательную систему (одышка, бронхостеноз), поэтому из-за них могут возникнуть проблемы у пациентов с патологиями указанных органов (астмой, бронхитом). Таким образом бета-блокаторы еще больше ухудшают состояние при таких заболеваниях дыхательной системы, как астма. Поэтому пациентам, страдающим от астмы, следует назначать уменьшенные дозы указанных лекарственных веществ, чтобы не подвергать еще большей опасности их дыхательную систему. В результате эффективность бета-блокаторов значительно снижается.
Антитромботические средства, такие как, например, дипиридамол, аспирин, и т.д., используемые для профилактики явлений тромбоза, имеют те же недостатки. У пациентов, имеющих патологии, связанные с окислительным стрессом и/или эндотелиальными дисфункциями, терапевтическое действие или переносимость этих лекарственных средств значительно снижены, в частности, в случае аспирина.
Для лечения астмы и бронхита используют бронхолитические средства, например сальбутамол и т.д., а при патологиях типа холинэргического недержания мочи используют лекарственные вещества, действующие на холинэргическую систему. При их введении могут возникнуть аналогичные побочные эффекты, влияющие на сердечнососудистую систему и порождающие проблемы у пациентов, страдающих от сердечной недостаточности и гипертонии. Сердечная недостаточность и гипертония являются патологиями, связанными, как упоминалось выше, с окислительным стрессом и/или эндотелиальными дисфункциями. И эти лекарственные вещества также проявляют те же недостатки, как и перечисленные ранее.
Отхаркивающие и муколитические лекарственные вещества, которые применяются для лечения воспалительных заболеваний органов дыхания, проявляют те же недостатки у пациентов в описанных выше состояниях. Их введение может вызвать изжогу и раздражение желудка, особенно у людей пожилого возраста.
Ингибиторы костной ресорбции, такие как дифосфонаты (алендронат и т.д.), являются лекарственными веществами, проявляющими повышенную желудочно-кишечную токсичность. Следовательно, эти лекарственные вещества также могут иметь такие же недостатки, которые описаны выше.
В случае ингибиторов фосфодиэстераз, таких как, например, силденафил, запринаст, используемых для лечения заболеваний сердечно-сосудистой и дыхательной систем, возникают аналогичные проблемы с переносимостью и/или эффективностью при упомянутых выше патологических состояниях окислительного стресса и/или эндотелиальных дисфункций.
Антиаллергические лекарственные вещества, например цетиризин, монтелукаст и т.д., вызывают аналогичные проблемы при упомянутых патологических состояниях, в особенности, в отношении эффективности.
Антиангиотензиновые лекарственные вещества, т.е. ингибиторы АСЕ, например эналаприл, каптоприл и т.д., или ингибиторы рецепторов, например лосартан и т.д., используют для лечения сердечно-сосудистых заболеваний. Их недостатком является побочное воздействие на органы дыхания (например, возникновение кашля и т.д.) при указанных выше патологических состояниях.
Противодиабетические лекарственные вещества как повышающие чувствительность к инсулину, так и снижающие уровень глюкозы, такие как, например, сульфонилмочевины, толбутамид, глипирид, гликлазид, глибурид, никотинамид и т.д., являются неэффективными для профилактики диабетических осложнений. При их введении могут возникать побочные эффекты, например поражение желудка. Эти явления усиливаются при указанных выше патологических состояниях.
В случае антибиотиков, например ампициллина, кларитромицина и т.д., и противовирусных лекарственных веществ, например ацикловира и др., возникают проблемы, связанные с их переносимостью, в частности они вызывают раздражение желудочно-кишечного тракта.
Противоопухолевые лекарственные вещества, например доксорубицин, даунорубипин, цисплатин и др., обладают высокой токсичностью по отношению к различным органам, в том числе к желудку и кишечнику. Это токсическое воздействие еще более усиливается при указанных выше патологических состояниях окислительного стресса и/или эндотелиальных дисфункций.
Лекарственные вещества против слабоумия, например никотин и колиномиметики, характеризуются слабой переносимостью, особенно в случае указанных выше патологий.
Таким образом, существует необходимость разработки доступных лекарственных веществ, обладающих улучшенным терапевтическим воздействием, т.е. обеспечивающих и пониженную токсичность и/или повышенную эффективность, чтобы их можно было вводить пациентам при патологических состояниях окислительного стресса и/или эндотелиальных дисфункций, без проявления недостатков, характерных для лекарственных веществ, известных из уровня техники.
Неожиданно было обнаружено, что вышеупомянутые проблемы, возникающие при введении лекарственных веществ пациентам, страдающим от окислительного стресса и/или эндотелиальных дисфункций, или пожилым людям в общем случае, можно решить при помощи нового класса лекарственных веществ, описанных далее.
Объектом изобретения являются соединения или их соли, имеющие следующую общую формулу (I):

где A=R-T1-, где R представляет собой радикал лекарственного вещества, как определено ниже, имеющий формулу A=R-T1 -Z или A=R-T1-OZ, где Z представляет собой Н или C1-C5 алкил, выбранный из следующих групп:
противовоспалительные лекарственные вещества: ацетилсалициловая кислота, 5-аминоацетилсалициловая кислота, карпрофен, диклофенака натриевая соль, дифлунизал, этодолак, флуфенаминовая кислота, флуниксин, флурбипрофен, ибупрофен, индометацин, индопрофен, кетопрофен, кеторолак, лорноксикам, локсопрофен, меклофенаминовая кислота, мефенаминовая кислота, мелоксикам, мезаламин, напроксен, нифлуминовая кислота, олсалазин, пироксикам, салсалат, сулиндак, супрофен, теноксикам, тиапрофеновая кислота, толфенаминовая кислота, толметин и зомепирак;
болеутоляющие лекарственные вещества: ацетаминофен, ацетилсалицилсалициловая кислота, беноксапрофен и трамадол;
бронходилататоры: албутерол, карбутерол, кленбутерол, дифиллин, этофиллин, фенотерол, метапротенерол, пирбутерол, сальметерол и тербуталин;
отхаркивающие лекарственные вещества: амброксол, бромгексин и гваякол;
антигистаминные лекарственные вещества: цетиризин, левокабастин и терфенадин;
АСЕ-ингибиторы: каптоприл, эналаприл, лизиноприл и рамиприл;
бета-блокаторы: алпренолол, атенолол, бупранолол, лабеталол, метипранолол, метопролол, пиндолол, пропранолол и тимолол;
антитромботические и вазоактивные лекарственные вещества: аргатробан, клопидогрель, далтепарин, дипиридамол, эноксапарин, илопрост, озагрель, трифлузал и бенфуродил гемисукцинат;
противодиабетические лекарственные вещества: никотинамид;
противоопухолевые лекарственные вещества: антрамицин, даунорубицин, доксорубицин и эпирубицин;
противоязвенные лекарственные вещества: циметидин, омепразол и пантопразол;
антигиперлипидемические лекарственные вещества: ловастатин, правастатина натриевая соль, симвастатин, аторвастатин и флувастатин;
антибиотики: амоксициллин, ампициллин, азтреонам, биапенем, карбенициллин, цефаклор, цефадроксил, цефамандол, цефатризин, цефокситин, диклоксациллин, имипенем, меклоциклин, метациклин, моксалактам, панипенем, бакампициллин, апициклин, кломоциклин и окситетрациклин;
противовирусные лекарственные вещества: ацикловир, фамцикловир, ганцикловир, пенцикловир, видарабин и зидовудин;
ингибиторы костной ресорбции: алендроновая кислота, этидроновая кислота и памидроновая кислота.
Лекарственные вещества против слабоумия: такрин.
T1=(CO), O, S, N или NR1c, где R1c представляет собой Н или C1-C5 алкил,
В=-ТB-Х2-ТBI,
где ТB и ТBI одинаковы или различны и выбраны из (СО), О, S, N или NR1c, где R1c определен выше;
X2 представляет собой бивалентную мостиковую группу, такую как соответствующий предшественник В, имеющий формулу
Z’-TB-X2Z’’,
где Z', Z’’ представляют собой независимо Н или ОН, выбраны из следующих соединений:
аминокислоты: L-карнозин (CI), пеницилламин (CV), N-ацетилпеницилламин (CVI), цистеин (CVII), N-ацетилцистеин (CVIII):




гидроксикислоты: галловая кислота (DI), феруловая кислота (DII), гентизиновая кислота (DIII), кофеиновая кислота (DV), гидрокофеиновая кислота (DVI), п-кумариновая кислота (DVII), ванилиновая кислота (DVIII), сиреневая кислота (DXI):






ароматические многоатомные спирты: гидрохинон (EVIII), метоксигидрохинон (EXI), гидроксигидрохинон (EXII), конифериловый спирт (EXXXII), 4-гидроксифенетиловый спирт (EXXXIII), п-кумариновый спирт (EXXXIV):





С представляет собой бивалентный радикал -Tc-Y-,
где Тс=(СО), О, S, N или NR1c, R1c имеет значения, как определено выше;
Y имеет следующие значения:
- линейную или разветвленную C1-C20 алкиленоксигруппу или циклоалкилен, содержащий от 5 до 7 атомов углерода, причем в циклоалкиленовом кольце один или более атомов углерода могут быть замещены гетероатомами, а кольцо может содержать боковые цепи типа R’, причем R’ такой, как определено выше, или

где nIX представляет собой целое число от 0 до 3;
nIIX представляет собой целое число от 1 до 3;
RTIX, RTIX’, RTIIX, RTIIX’, одинаковые или отличающиеся друг от друга, представляют собой Н или линейный, или разветвленный C1-C4 алкил;
Y3 представляет собой насыщенное, ненасыщенное или ароматическое гетероциклическое кольцо, содержащее по меньшей мере один атом азота, причем указанное кольцо состоит из 5 или 6 атомов;

где n3 представляет собой целое число от 0 до 3 и n3’ представляет собой целое число от 1 до 3;

где n3 и n3’ имеют указанное выше значение;

где nf’ представляет собой целое число от 1 до 6;

где R1f=H, СН3 и nf представляет собой целое число от 1 до 6.
В формуле (III) Y3 предпочтительно следует выбирать из следующих радикалов:


Наиболее предпочтительным из значений Y3 является Y12 (пиридил), замещенный в положениях 2 и 6. Связи могут также находиться в асимметричных положениях, например Y12 (пиридил) может быть также замещен в положениях 2 и 3; Yl (пиразол) может быть 3, 5-дизамещенным.
Соединения настоящего изобретения, имеющие формулу (I), могут быть трансформированы в соответствующие соли. Например, один из способов образования солей таков: если в молекуле один из атомов азота является достаточно основным, чтобы образовывать соль, то в органическом растворителе, например, таком как ацетонитрил или тетрагидрофуран, он взаимодействует с эквимолярным количеством соответствующей органической или неорганической кислоты. Для образования соли в формулах соединений по изобретению предпочтительно присутствует радикал Y или Y' формулы (III).
Примеры органических кислот: щавелевая, винная, малеиновая, янтарная, лимонная кислоты.
Примеры неорганических кислот: азотная, соляная, серная, фосфорная кислоты.
Производные соединений настоящего изобретения могут быть использованы согласно терапевтическим показаниям, при которых применяются лекарственные вещества-предшественники, что позволяет добиться преимуществ, которые приводятся далее в качестве примера для некоторых групп этих соединений.
- Противовоспалительные лекарственные вещества NSAIDs:
соединения настоящего изобретения отличаются очень хорошей переносимостью и эффективностью, даже в случаях, когда организм ослаблен и находится в состоянии окислительного стресса. Указанные лекарственные вещества могут быть также использованы при патологиях, при которых воспаление играет важную патогенетическую роль, например, таких как рак, астма, инфаркт миокарда, но не ограничиваясь ими.
- Адренэргические блокаторы β-типа: спектр действия соединений, имеющих формулу (I), становится более широким, чем у исходных лекарственных веществ; к непосредственному воздействию на гладкую мускулатуру добавляется ингибирование нервных бета-адренергических сигналов, управляющих сокращением кровеносных сосудов. Уменьшается количество побочных эффектов (затрудненное дыхание, сокращение бронхов), воздействующих на дыхательный аппарат.
- Антитромботические лекарственные вещества: повышается антитромбоцитарная активность и в случае производных аспирина также улучшается желудочная переносимость.
- Бронходилататоры и лекарственные вещества, действующие на холинергическую систему: снижаются побочные эффекты, воздействующие на сердечно-сосудистый аппарат (тахикардия, гипертония).
- Отхаркивающие лекарственные вещества: улучшается желудочно-кишечная переносимость.
- Дифосфонаты: токсичность по отношению к желудочно-кишечном тракту существенно снижается.
Ингибиторы фосфодиэстеразы (PDE) (бронходилататоры): улучшается терапевтическая эффективность при тех же дозах, следовательно, становится возможным, используя соединения настоящего изобретения, введение пониженных доз лекарственного вещества и снижение побочных эффектов.
- Антилейкотриеновые лекарственные вещества: улучшенная эффективность.
- АСЕ-ингибиторы: улучшенная терапевтическая эффективность и пониженные побочные эффекты (затрудненное дыхание, кашель).
- Противодиабетические лекарственные вещества (повышающие чувствительность к инсулину и гипогликемические), антибиотики, противовирусные, противоопухолевые, противоколитные лекарственные вещества, лекарственные вещества против слабоумия: улучшенная эффективность и/или переносимость.
Из примеров лекарственных веществ-предшественников, которые могут быть использованы, могут быть упомянуты следующие.
Из противовоспалительных/обезболивающих лекарственных веществ могут быть упомянуты следующие:
противовоспалительные лекарственные вещества: ацетилсалициловая кислота, 5-аминоацетилсалициловая кислота, карпрофен, диклофенака натриевая соль, дифлунизал, этодолак, флуфенаминовая кислота, флуниксин, флурбипрофен, ибупрофен, индометацин, индопрофен, кетопрофен, кеторолак, лорноксикам, локсопрофен, меклофенаминовая кислота, мефенаминовая кислота, мелоксикам, мезаламин, напроксен, нифлуминовая кислота, олсалазин, пироксикам, салсалат, сулиндак, супрофен, теноксикам, тиапрофеновая кислота, толфенаминовая кислота, толметин, зомепирак;
болеутоляющие лекарственные вещества: ацетаминофен, ацетилсалицилсалициловая кислота, беноксапрофен, трамадол.
Из лекарственных веществ для дыхательной и мочеполовой системы (бронходилататоры и лекарственные вещества, действующие на холинэргическую систему, отхаркивающие лекарственные вещества, антиастматические/антиаллергические антигистаминные лекарственные вещества) могут быть упомянуты следующие:
бронходилататоры и лекарственные вещества, действующие на холинергическую систему: албутерол, карбутерол, кленбутерол, дифиллин, этофиллин, фенотерол, метапротеренол, пирбутерол, сальметерол, тербуталин;
отхаркивающие лекарственные вещества: амброксол, бромгексин, гваякол;
антигистаминные лекарственные вещества: цетиризин, левокабастин, терфенадин;
из сердечно-сосудистых лекарственных веществ (АСЕ-ингибиторы, бета-блокаторы, антитромботические лекарственные вещества и вазодилататоры, противодиабетические и гипогликемические лекарственные вещества) могут быть упомянуты следующие:
АСЕ-ингибиторы: каптоприл, эналаприл, лизиноприл, рамиприл;
бета-блокаторы: алпренолол, атенолол, бупранолол, лабеталол, метипранолол, метопролол, пиндолол, пропранолол, тимолол;
антитромботические и вазоактивные лекарственные вещества: аргатробан, бенфуродил гемисукцинат, клопидогрель, далтепарин,
дипиридамол, эноксапарин, илопрост, озагрель, трифлузал;
противодиабетические лекарственные вещества: никотинамид;
из противоопухолевых лекарственных веществ: антрамицин, даунорубицин, доксорубицин, эпирубицин, эпитиостанол;
из противоязвенных лекарственных веществ могут быть упомянуты следующие: циметидин, омепразол, пантопразол;
среди антигиперлипидемических лекарственных веществ (статинов) могут быть упомянуты следующие: аторвастатин, флувастатин, ловастатин, привастатина натриевая соль, симвастатин;
среди антибиотиков/противовирусных лекарственных веществ могут быть упомянуты следующие:
антибиотики: амоксициллин, ампициллин, азтреонам, биапенем, карбенициллин, цефаклор, цефадроксил, цефамандол, цефатризин, цефокситин, диклоксациллин, имипенем, меклоциклин, метациклин, моксалактам, панипенем, бакампициллин, апициклин, кломоциклин, окситетрациклин;
противовирусные лекарственные вещества: ацикловир, фамцикловир, ганцикловир, пенцикловир, видарабин, зидовудин;
среди ингибиторов костной резорбции (дифосфонаты) могут быть упомянуты следующие:
алендроновая кислота, этидроновая кислота, памидроновая кислота;
среди лекарственных веществ против слабоумия могут быть упомянуты следующие: такрин.
Предпочтительными являются следующие соединения:
среди противовоспалительных веществ: ацетилсалициловая кислота, 5-аминоацетилсалициловая кислота, карпрофен, диклофенака натриевая соль, дифлунизал, этодолак, флуфенамовая кислота, флуниксин, флурбипрофен, ибупрофен, индометацин, индопрофен, кетопрофен, кеторолак, лорноксикам, локсопрофен, меклофенаминовая кислота, мефенаминовая кислота, мелоксикам, мезаламин, напроксен, нифлуминовая кислота, олзалазин, пироксикам, салалат, сулиндак, супрофен, теноксикам, тиапрофеновая кислота, толфенамовая кислота, толметин, зомепирак;
болеутоляющие лекарственные вещества: ацетаминофен, ацетилсалицилсалициловая кислота, беноксапрофен, трамадол;
среди лекарственных веществ для дыхательной и мочеполовой систем (бронходилататоры, отхаркивающие лекарственные вещества, антигистаминные лекарственные вещества):
бронходилататоры: албутерол, карбутерол, кленбутерол, дифиллин, этофиллин, фенотерол, метапротеренол, пирбутерол, сальметерол, тербуталин;
отхаркивающие лекарственные вещества: амброксол, бромгексин, гваякол;
антигистаминные лекарственные вещества: цетиризин, левокабастин, терфенадин;
среди сердечно-сосудистых лекарственных веществ:
АСЕ-ингибиторы: каптоприл, эналаприл, лизиноприл, рамиприл;
бета-блокаторы: алпренолол, атенолол, бупранолол, лабеталол, метипранолол, метопролол, пиндолол, пропранолол, тимолол;
антитромботические и вазоактивные лекарственные вещества: аргатробан, клопидогрель, далтепарин, дипиридамол, эноксапарин, илопрост, озагрель, трифузал;
противодиабетические лекарственные вещества: никотинамид;
среди противоопухолевых лекарственных веществ: антрамицин, даунорубицин, доксорубицин, эпирубицин;
среди противоязвенных лекарственных веществ; циметидин, омепразол, пантопразол;
среди антигиперлипидемических лекарственных веществ: ловастатин, правастатина натриевая соль, симвастатин;
среди антибиотиков/противовирусных лекарственных веществ:
антибиотики: амоксициллин, ампициллин, азтреонам, биапенем, карбенециллин, цефаклор, цефадроксил, цефамандол, цефатризин, цефокситин, диклоксациллин, имипенем, меклоциклин, метациклин, моксалактам, панипенем, бакампициллин, апициклин, кломоциклин, окситетрациклин;
противовирусные лекарственные вещества: ацикловир, фамцикловир, ганцикловир, пенцикловир, видарабин, зидовудин;
среди ингибиторов костной резорбции: алендроновая кислота, этидроновая кислота, памидроновая кислота;
среди лекарственных веществ против слабоумия: такрин.
Вышеупомянутые лекарственные вещества-предшественники получают при помощи методов, известных из уровня техники. Примеры можно найти в "The Merck Index, 12a Ed. (1996)", включенном сюда в качестве ссылки. В случае доступности могут быть использованы соответствующие изомеры, включая оптические изомеры.
Соединения, имеющие формулу (I), получают при помощи синтетических методов, описанных ниже.
Выбор реакции для каждого метода зависит от реакционноспособных групп, присутствующих в молекуле лекарственного вещества-предшественника, в предшественнике соединения В, который может быть, как указано выше, бивалентным или моновалентным, и в предшественнике соединения С.
Реакции проводят, используя методы, широко известные из уровня техники, которые позволяют образовывать связи между лекарственным веществом-предшественником, предшественником соединения В и предшественником соединения С.
В случае, когда реакционноспособная функциональная группа (например, -СООН, -ОН) лекарственного вещества-предшественника образует ковалентную связь, например сложноэфирную, амидную, простую эфирную, указанная функциональная группа может быть регенерирована при помощи методов, хорошо известных из уровня техники.
Далее приведены некоторые схемы синтеза для получения соединений настоящего изобретения.
А. Синтез соединений, имеющих формулу (I).
1. Синтез соединения, полученного путем реакции между лекарственным веществом-предшественником и предшественником соединения В.
1а. Если лекарственное вещество имеет общую формулу R-COOH, а функциональная группа предшественника соединения В, которая образует связь с карбоксильной функцией лекарственного вещества, имеет формулу XZ, причем Х является таким, как определено выше, a Z=Н, то протекающая реакция зависит от природы второй реакционноспособной группы, присутствующей в предшественнике соединения В.
1a.1. Если вторая реакционноспособная группа, присутствующая в предшественнике соединения В, является карбоксильной группой, то общая схема синтеза предполагает первоначальное образование галогенангидрида кислоты R-COHal (Hal=Cl, Br) и последующую реакцию с НХ группой предшественника соединения В

где X2, T1, Тв такие, как указано выше.
Если в двух соединениях, вступающих в реакцию, присутствуют другие функциональные группы СООН и/или НХ, они должны быть защищены перед проведением реакции в соответствии с методами, хорошо известными из уровня техники, например, как описано в публикации Th. W. Greene: "Protective groups in organic synthesis", Harward University Press, 1980.
Галогенангидрид RCOHal получают в соответствии с методами, хорошо известными из уровня техники, например при помощи тионил- или оксалилхлорида, PIII или PV галогенидов, проводя реакцию в инертных растворителях, например, таких как толуол, хлороформ, DMF и т.д.
В особых случаях, если НХ группой в предшественнике соединения В, является NH2, ОН или SH, то лекарственное вещество-предшественник формулы R-COOH сначала конвертируют в соответствующий галогенангидрид RCOHal, как описано выше, и затем проводят реакцию с НХ группой предшественника соединения В в присутствии органического основания, такого как триэтиламин, пиридин и т.д., в инертном растворителе, например, таком как толуол, тетрагидрофуран и т.д. при температуре в интервале от 0 до 25°С.
В качестве альтернативы предыдущему синтезу лекарственное вещество-предшественник, имеющее формулу R-COOH, может быть обработано агентом, активирующим карбоксильную группу, выбранным из N,N’-карбонилдиимидазола (CDI), N-гидроксибензотриазола и дициклогексилкарбодиимида, в таком растворителе, как, например, DMF, THF, хлороформ и т.д., при температуре в диапазоне от -5 до 50°С, после чего полученное соединение вводят во взаимодействие in situ с реакционноспособной функциональной группой предшественника соединения В для получения соединения, имеющего формулу (IA.1).
1a.2. Если предшественник соединения В содержит две функциональные группы XZ, одинаковые или отличные друг от друга, причем Х такой, как указано выше, и Z=Н, то лекарственное вещество-предшественник, имеющее формулу R-COOH, вначале обрабатывают агентом, активирующим карбоксильную группу, как описано выше в пункте 1a.1, а затем проводят реакцию с предшественником соединения В, у которого одна из двух реакционноспособных НХ групп блокирована, например, ацетильной или трет-бутилоксикарбонильной защитой; удаление защит в конце синтеза позволяет регенерировать исходные функциональные группы. Схема синтеза следующая:

где X, T1, ТB, X2 такие, как указано выше, a G является защитной группой функциональной группы НХ.
2. Синтез нитроксипроизводных.
2а.1. Если соединение, полученное в конце предыдущей стадии 1а, имеет формулу (IА.1), то кислота может быть превращена в соответствующую натриевую соль, которую используют для получения конечного соединения, следуя известным методам, например в соответствии с одной из следующих схем синтеза:

где T1, ТB, X2, ТB1, Тс такие, как указано выше, R4 выбирают из Cl, Вr, Y такой, как указано выше, X1 является радикалом Y1, не содержащим атома кислорода, R3 является Сl, Вr, I, ОН. Если R3=ОН, то соединение формулы (IA.1b) подвергают галогенированию, например, такими агентами как РВr3, PCl5, SOCl2, РРh3+I2, и затем оно взаимодействует с АgNО3 в органическом растворителе, таком как ацетонитрил, тетрагидрофуран. Если R3 является Сl, Вr, I, то соединение формулы (1A.1b) напрямую взаимодействует с АgNО3, как указано выше.


где R5=ОН или NHR1C; R1C, R3 и другие обозначения определены выше.
Вышеуказанные реакции хорошо известны из уровня техники. Примерами могут служить патентные заявки заявителя: WO 94/12463, WO 95/09831 и WO 95/30641.
Если X1 является линейным С4алкилом, то соответствующая кислота R-T1-TB-X2-COOH взаимодействует с трифенилфосфином в присутствии галогенирующего агента, такого как СВr4 или N-бромсукцинимид в тетрагидрофуране, что приводит к соединению (IA.1c), где R3=Вr.
2а.2. Если соединение, полученное в конце предыдущей стадии 1а, имеет формулу (IA.2), то соответствующее нитроксипроизводное получают обработкой галогенкарбоновой кислоты, имеющей формулу Hal-X1-COOH (X1 такой, как указано выше), сначала агентом, активирующим карбоксильную группу, как описано выше в параграфе 1а.1, и затем соединением, имеющим формулу (IA.2), что приводит к галогенпроизводному, которое выделяют, растворяют в органическом растворителе (см. параграф 2а.1) и затем обрабатывают нитратом серебра. Общая реакционная схема такова:

где T1, ТB, X2, ТB1, Тc, Y такие, как указано выше.
Альтернативным методом является использование галогенида Hal-Xl-COCl, где Hal является предпочтительно Вr, который может взаимодействовать с соединением формулы (IA.2).
1b. Если лекарственное вещество-предшественник имеет реакционноспособную функциональную группу НХ, где Х такой, как указано выше, вместо карбоксильной группы, то две функциональные группы, присутствующие в предшественнике соединения В, могут быть следующими.
1b.1. Карбоксильная группа, которая взаимодействует с функциональной группой НХ лекарственного вещества-предшественника, и НХ группа, причем последняя реакционноспособная группа предшественника соединения В является одинаковой или отличной от функциональной группы лекарственного вещества-предшественника. Формула предшественника соединения В, является формулой следующего типа: Н-Х-Х2-СООН, где Х и Х2 такие, как указано выше. Функциональную группу Н-Х-предшественника соединения В защищают в соответствии с методами, известными из уровня техники, и карбоксильная группа вступает в реакцию, как указано выше, по следующей схеме:

По окончании реакции функциональную группу НХ предшественника соединения В регенерируют.
1b.2. Если предшественник соединения В содержит две карбоксильные группы, то его обрабатывают эквимолярным количеством агента, активирующего карбоксильную группу, в условиях, ранее описанных в параграфе 1a.1, и затем проводят реакцию с реакционноспособной группой НХ молекулы лекарственного вещества-предшественника. Другие возможные реакционноспособные функциональные группы НХ-типа, присутствующие в этих двух соединениях, должны быть защищены, как указывалось ранее. В итоге получают соединение, имеющее формулу К-Т1-Тв-Х2-СООН (IB.2).
2b. Синтез нитроксипроизводных.
2b.1. Для получения конечного нитроксипроизводного из исходного соединения, имеющего формулу R-T1-Tв-X2-X-H (IB.1), полученного в конце синтеза, описанного в параграфе 1b.1, соединение (IВ.1) вводят в реакцию с галогенкислотой, имеющей формулу Hal-X1-COOH, которую обрабатывают так, как описано ранее в параграфе 1a.1, или с соответствующим хлорангидридом галогенкислоты. Полученное соединение растворяют в органическом растворителе, например ацетонитриле или тетрагидрофуране, и затем проводят реакцию с нитратом серебра.
2b.2. Для получения конечного нитроксипроизводного из исходного соединения, имеющего формулу R-T1-Tв-X2-COOH (IB.2), полученного в конце синтеза, описанного в параграфе 1b.2, кислоту превращают в соответствующую натриевую соль, которая взаимодействует с соединением формулы R4-X1-R3, ранее определенном в схеме реакции А параграфа 2а.1, получая в соответствии с тем же упомянутым процессом конечное нитроксипроизводное. Альтернативным образом, если X1 является линейным С4 алкилом, то кислота (IВ.2) реагирует с трифенилфосфином в присутствии галогенирующего агента, такого как СВr4 или N-бромсукцинимид в тетрагидрофуране, и полученное в результате соединение, растворенное в органическом растворителе, например ацетонитриле или тетрагидрофуране, взаимодействует с нитратом серебра.
2b.3. В качестве альтернативы синтетическому процессу, описанному в параграфах 1b.1 и 2b.1, на первой стадии возможна реакция НХ-функции предшественника соединения В НХ-Х2-СООН с хлорангидридом галогенкислоты, имеющим формулу Hal-X1-CO-Cl, где Hal предпочтительно является Вr, затем карбоксильная функция полученного таким образом соединения вступает во взаимодействие с лекарственным веществом-предшественником R-X. На третьей и последней стадии Hal-группу замещают группой -ONO2 в соответствии с процессом, описанным в параграфе 2b.1.
Реакционная схема является следующей:

где ТС, ТB1, ТB, T1, X2, X1, Y такие, как указано выше.
В предыдущей схеме, в качестве альтернативы, может быть проведено нитрование кислоты формулы (2B.3).
Следующие примеры приведены для иллюстрации настоящего изобретения и не должны рассматриваться как ограничивающие его объем.
ПРИМЕР 1
Синтез 4-нитроксибутилового эфира (S,S)-N-ацетил-S-(6-метокси-α-метил-2-нафтилацетил)цистеина (NCX 2101), имеющего формулу

Предшественником является напроксен (формула VI), предшественником В является N-ацетилцистеин (формула СVIII)

а) Синтез (S, S)-N-ацетил-S-(6-метокси-α-метил-2-нафтилацетил)цистеина
К раствору 6-метокси-α-метил-2-нафтилуксусной кислоты (10 г, 34,4 ммоль) в хлороформе (100 мл) и N, N-диметилформамиде (6 мл) добавляют 1,1’-карбонилдиимидазол (CDI) (7,04 г, 43,4 ммоль). Через 15 мин к полученному раствору добавляют (S)-N-ацетилцистеин (7,08 г, 43,4 ммоль) и выдерживают реакционную смесь при комнатной температуре в течение 12 часов. Промывают реакционную смесь 5% НСl, водой, а затем насыщенным водным раствором NaCl. Органическую фазу высушивают сульфатом натрия и затем упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя этилацетатом. Получают 11,66 г целевого продукта в виде белого твердого вещества с т.пл.122-126°С.
1H-ЯМР (CDCl3): 7,71-7,65 (3Н, м), 7,34 (1Н, дд), 7,16-7,09 (2Н, м), 6,36 (1Н, д), 4,67 (1Н, м), 4,00 (1Н, к), 3,90 (3Н, с), 3,32 (2Н, т), 1,84 (3Н, с), 1,59 (3Н, д).
b) Синтез 4-бромбутилового эфира (S,S)-N-ацетил-S-(6-метокси-α-метил-2-нафтилацетил)цистеина
К раствору (S, S)-N-ацетил-S-(6-метокси-α-метил-2-нафтилацетил)цистеина (11,3 г, 30,1 ммоль) в тетрагидрофуране (200 мл) добавляют трифенилфосфин (23,7 г, 90,3 ммоль) и тетрабромметан (28,85 г, 90,3 ммоль). Перемешивают реакционную смесь при комнатной температуре в течение 24 часов. Растворитель упаривают при пониженном давлении. Полученный неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 7/3. Получают 4 г целевого эфира в виде белого твердого вещества с т.пл.67-71°С.
c) Синтез 4-нитроксибутилового эфира (S, S)-N-ацетил-S-(6-метокси-α-метил-2-нафтилацетил)цистеина
К раствору эфира, полученного в результате предыдущей стадии (1 г, 1,96 ммоль), в ацетонитриле (20 мл) добавляют нитрат серебра (0,66 г, 3,92 ммоль). Кипятят реакционную смесь в течение 7 часов без доступа света. Образующуюся соль удаляют фильтрацией и раствор упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 7/3. Получают 0,47 г 4-нитроксибутилового эфира (S,S)-N-ацетил-S-(6-метокси-α-метил-2-нафтилацетил)цистеина в виде белого твердого вещества с т.пл.56-59°С.
1H-ЯМР (CDCl3): 7,80-7,68 (3Н, м), 7,37 (1Н, д), 7,20-7,13 (2Н, м), 6,12 (1Н, д), 4,40 (2Н, дд), 4,26 (1Н, м), 4,15-3,87 (3Н, м), 3,92 (3Н, с), 3,33 (2Н, д), 1,86 (3Н, д), 1,74-1,67 (4Н, м), 1,61 (3Н, д).
Элементный анализ
Рассчитано: С 56,08%; Н 5,73%; N 5,71%; S 6, 51%.
Найдено: С 55,99%; Н 5,68%; N 5,60%; S 6,35%.
ПРИМЕР 2
Синтез 4-нитроксибутилового эфира (S)-N-ацетил-S-{α -метил-[4-(2-метилпропил)фенил]ацетил}цистеина (NCX 2111), имеющего формулу

Предшественником является ибупрофен (формула VII), предшественником В является N-ацетилцистеин (формула CVIII)


а) Синтез (S)-N-ацетил-S-{α -метил-[4-(2-метилпропил)фенил]ацетил}цистеина
К раствору α-метил-[4-(2-метилпропил)фенил]уксусной кислоты (10 г, 48,48 ммоль) в хлороформе (100 мл) и N,N-диметилформамиде (6 мл) добавляют 1,1’-карбонилдиимидазол (7,86 г, 48,48 ммоль). Через 1 час к полученному раствору добавляют (S)-N-ацетилцистеин (7,91 г, 48,47 ммоль) и выдерживают реакционную смесь при комнатной температуре в течение 24 часов. Промывают реакционную смесь 5% НСl, водой, а затем насыщенным водным NaCl. Органическую фазу высушивают сульфатом натрия и затем упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя этилацетатом. Получают 13,3 г целевого продукта в виде масла.
1H-ЯМР (CDCl3): 10,17 (1H, с), 7,13 (2Н, д), 6,54 (1Н, д), 4,76 (1H, м), 3,93 (1Н, к), 3,42-3,30 (2Н, м), 2,49 (2Н, д), 1,85-1,83 (4Н, м), 1,55 (3Н, д), 0,93 (6Н, д).
b) Синтез 4-бромбутилового эфира (S)-N-ацетил-S-{α-метил-[4-(2-метилпропил)фенил] ацетил}цистеина
К раствору (S)-N-ацетил-S-{α-метил-[4-(2-метилпропил)бензил]ацетил}цистеина (12,8 г, 36,4 ммоль) в тетрагидрофуране (100 мл) добавляют трифенилфосфин (28,65 г, 109,23 ммоль) и тетрабромметан (36,23 г, 109,23 ммоль). Перемешивают реакционную смесь при комнатной температуре в течение 48 часов. Растворитель упаривают при пониженном давлении. Полученный неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью циклогексан/этилацетат 1/1. Получают 5,79 г целевого эфира в виде масла.
c) Синтез 4-нитроксибутилового эфира (S)-N-ацетил-S-{α-метил-[4-(2-метилпропил)фенил]ацетил}цистеина
К раствору эфира, полученного в результате предыдущей стадии (5,5 г, 11,3 ммоль), в ацетонитриле (100 мл) добавляют нитрат серебра (2,69 г, 15,8 ммоль). Кипятят реакционную смесь в течение 24 часов без доступа света. Образующуюся соль удаляют фильтрацией и раствор упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью циклогексан/этилацетат 7/3. Получают 1,18 г 4-нитроксибутилового эфира (S)-N-ацетил-S-{α-метил-[4-(2-метилпропил)фенил]ацетил}цистеина в виде масла.
1H-ЯМР (CDCl3): 7,27-7,09 (4Н, м), 6,19 (1H, д), 4,75 (1H, м), 4,47 (2Н, т), 4,15-4,02 (2Н, м), 3,86 (1H, к), 3,31 (2Н, д), 2,44 (2Н, д), 1,89 (3Н, д), 1,86-1,76 (5Н, м), 1,51 (3Н, д), 0,89 (6Н, д).
Элементный анализ
Рассчитано: С 56,39%; Н 6,88%; N 6,00%; S 6,84%.
Найдено: С 56,22%; Н 6,79%; N 5,88%; S 6,92%.
ПРИМЕР 3
Синтез 4-нитроксибутилового эфира (S)-N-ацетил-S-[1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индолил-3-ацетил]цистеина (NCX 2121), имеющего формулу

Предшественником является индометацин (формула VIII), предшественником В является N-ацетилцистеин (формула СVIII)


а) Синтез (S)-N-ацетил-S-[1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индолил-3 -ацетил]цистеина
К раствору 1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индолил-3-уксусной кислоты (10 г, 28,00 ммоль) в хлороформе (100 мл) и N,N-диметилформамиде (2 мл) добавляют 1,1’-карбонилдиимидазол (4,53 г, 28,00 ммоль). Через 1 час к полученному раствору добавляют (S)-N-ацетилцистеин (4,56 г, 28,00 ммоль) и выдерживают реакционную смесь при комнатной температуре в течение 24 часов. Промывают реакционную смесь 5% НСl, водой, а затем насыщенным водным раствором NaCl. Органическую фазу высушивают сульфатом натрия и затем упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя этилацетатом. Получают 7,79 г целевого продукта в виде желтого твердого вещества с т.пл.129°С.
1H-ЯМР (DMSO-d6): 12,90 (1Н, с), 8,21 (1H, д), 7,69-7,64 (4Н, м), 7,06 (1H, д), 6,96 (1H, д), 6,73 (1H, дд), 4,33 (1H, м), 4, 02 (2Н, с), 3,77 (3Н, с), 3,33-2,96 (2Н, м), 2,22 (3Н, с), 1,78 (3Н, с).
b) Синтез 4-бромбутилового эфира (S)-N-ацетил-S-[1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индолил-3-ацетил]цистеина
К раствору (S)-N-ацетил-S-[1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индолил-3-ацетил]цистеина (3,09 г, 6, 14 ммоль) в N,N-диметилформамиде (50 мл) добавляют этилат натрия (0,42 г, 6,14 ммоль), а затем через 30 минут добавляют 1,4-дибромбутан (2,18 мл, 18,00 ммоль), растворенный в 25 мл N, N-диметилформамида. Перемешивают реакционную смесь при комнатной температуре в течение 20 часов, затем разбавляют ее этиловым эфиром и промывают водой. После высушивания органической фазы сульфатом натрия растворитель упаривают при пониженном давлении. Полученный неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью циклогексан/этилацетат 1/1. Получают 1,7 г целевого эфира в виде желтого твердого вещества с т.пл.130-134°С.
c) Синтез 4-нитроксибутилового эфира (S)-N-ацетил-S-[1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индолил-3-ацетил]цистеина
К раствору эфира, полученного в результате предыдущей стадии (1,6 г, 2,5 ммоль), в ацетонитриле (30 мл) добавляют нитрат серебра (0,6 г, 3,51 ммоль). Кипятят реакционную смесь в течение 8 часов без доступа света. Образующуюся соль удаляют фильтрацией и раствор упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью циклогексан/этилацетат 4/6. Получают 1,2 г 4-нитроксибутилового эфира (S)-N-ацетил-S-[1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индолил-3-ацетил]цистеина в виде масла.
1 H-ЯМР (CDCl3): 7,66 (2Н, д), 7,48 (2Н, д), 6,90 (2Н, м), 6,68 (1Н, м), 6,14 (1Н, д), 4,77 (1Н, м), 4,43 (2Н, т), 4,08 (2Н, м), 3,87 (2Н, с), 3,83 (3Н, с), 3,34 (2Н, д), 2,38 (3Н, с), 1,90 (3Н, с), 1,78-1,70 (4Н, м).
Элементный анализ
Рассчитано: С 54,24%; Н 4,88%; N 6,80%; S 5,17%; Cl 5,72%.
Найдено: С 54,32%; Н 4,93%; N 6,91%; S 5,13%; Cl 5,84%.
ПРИМЕР 4
Синтез 4-нитроксибутилового эфира (S)-N-ацетил-[2-фтор-α-метил-(1,1’-бифенил)-4-ацетил]цистеина (NCX 2131), имеющего формулу

Предшественником явлется флурбипрофен (формула IX), предшественником В является N-ацетилцистеин (формула СVIII)


Соединение NCX 2131 синтезируют в соответствии с методикой, описанной в примере 1. Соединение получают в виде масла. Выход 26%.
1H-ЯМР (CDCl3): 7,41-7,38 (6Н, м), 7,10 (2Н, м), 6,22 (1Н, д), 4,78 (1H, м), 4,46 (2Н, т), 4,13 (2Н, т), 3,92 (1Н, к), 3,36 (2Н, д), 1,93 (3Н, д), 1,76 (4Н, д), 1,55 (3Н, д).
Элементный анализ
Рассчитано: С 56,91%; Н 5,37%; N 5,55%; S 6,33%; F 3,75%.
Найдено: С 56,99%; Н 5,41%; N 5,66%; S 6,41%; F 3,83%.
ПРИМЕР 5
Получение 4-нитроксибутилового эфира транс-3-[4-[α-метил-[4-(2-метилпропил)фенил]ацетилокси]-3-метоксифенил]-2-пропеновой кислоты (NCX 2210), имеющего формулу

Предшественником является ибупрофен (формула VII), предшественником В является феруловая кислота (формула DII)

a) Синтез транс-3-[4-[α -метил-[4-(2-метилпропил)фенил]ацетилокси]-3-метоксифенил]-2-пропеновой кислоты
К раствору α-метил-[4-(2-метилпропил)фенил]уксусной кислоты (5,03 г, 24,4 ммоль) в тетрагидрофуране (100 мл) и N,N-диметилформамиде (5 мл) добавляют 1,1’-карбонилдиимидазол (4,25 г, 24,8 ммоль). Через 1 час к полученному раствору добавляют феруловую кислоту (4,90 г, 25 ммоль) и этилат натрия (89 мг) и перемешивают реакционную смесь при комнатной температуре в течение 12 часов. Промывают реакционную смесь 5% НСl, водой, а затем насыщенным водным раствором NaCl. Органическую фазу высушивают сульфатом натрия и затем упаривают при пониженном давлении.
Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью этилацетат/н-гексан 7/3. Получают 5,1 г транс-3-[4-[α-метил-[4-(2-метилпропил)фенил]ацетилокси]-3-метоксифенил]-2-пропеновой кислоты в виде белого твердого вещества с т.пл.131-137°С.
1H-ЯМР (CDCl3): 7,72 (1Н, д), 7,32 (2Н, дд), 7,26 (1Н, м), 7,16-7,07 (4Н, м), 6,98 (1Н, д), 6,37 (1H, д), 3,99 (1Н, к), 3,73 (3Н, с), 2,47 (2Н, д), 1,88 (1Н, м), 1,63 (3Н, д), 0,92 (6Н, д).
b) Синтез 4-бромбутилового эфира транс-3-[4-[α-метил-[4-(2-метилпропил)фенил]ацетилокси]-3-метоксифенил]-2-пропеновой кислоты
К раствору транс-3-[4-[α-метил-[4-(2-метилпропил)бензил]ацетилокси]-3-метоксифенил]-2-пропеновой кислоты (5,33 г, 14 ммоль) в N,N-диметилформамиде (130 мл) добавляют при перемешивании этилат натрия (1,2 г, 16 ммоль). Через 1 час к полученной смеси добавляют 1,4-дибромбутан (10 г, 46 ммоль) и выдерживают смесь при комнатной температуре в течение 12 часов. Промывают реакционную смесь 5% НСl, водой, а затем насыщенным водным раствором NaCl, высушивают органическую фазу сульфатом натрия и затем упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 8/2. Получают 4,46 г 4-бромбутилового эфира транс-3-[4-[α-метил-[4-(2-метилпропил)фенил]ацетилокси]-3-метоксифенил]-2-пропеновой кислоты.
с) Синтез 4-нитроксибутилового эфира транс-3-[4-[α-метил-[4-(2-метилпропил)фенил]ацетилокси]-3-метоксифенил]-2-пропеновой кислоты
К раствору 4-бромбутилового эфира транс-3-[4-[α-метил-[4-(2-метилпропил)фенил]ацетилокси]-3-метоксифенил]-2-пропеновой кислоты (4 г, 7,72 ммоль) в ацетонитриле (70 мл) добавляют нитрат серебра (2,58 г, 15 ммоль). Кипятят реакционную смесь в течение 2 часов без доступа света. Образующуюся соль удаляют фильтрацией и раствор упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 8/2. Получают 2,4 г 4-нитроксибутилового эфира транс-3-[4-[α-метил-[4-(2-метилпропил)бензил]ацетилокси]-3-метоксифенил]-2-пропеновой кислоты в виде масла.
1H-ЯМР (CDCl3): 7,62 (1Н, д), 7,32 (2Н, д), 7,15 (2Н, д), 7,16-7,05 (2Н, м), 6,96 (1Н, д), 6,35 (1Н, д), 4,51 (2Н, т), 4,24 (2Н, т), 3,99 (1Н, к), 3,74 (3Н, с), 2,48 (2Н, д), 1,89-1,83 (5Н, м), 1,62 (3Н, д), 0,92 (6Н, д).
Элементный анализ
Рассчитано: С 64,91%; Н 6,66%; N 2,82%.
Найдено: С 64, 83%; Н 6,52%; N 2,69%.
ПРИМЕР 6
Синтез 4-нитроксибутилового эфира транс-3-[4-[2-фтор-α-метил-(1,1’-бифенил)-4-ацетокси]-3-метоксифенил]-2-пропеновой кислоты (NCX 2216), имеющего формулу

Предшественником является флурбипрофен (формула IX), предшественником В является феруловая кислота (формула DII)


Соединение NCX 2216 синтезируют в соответствии с методикой, описанной в примере 5. Суммарный выход 32%. Соединение имеет вид твердого аморфного вещества.
1H-ЯМР (CDCl3): 7,40-7,25 (9Н, м), 7,07-7,01 (2Н, д), 6,98 (1Н, м), 6,38 (1Н, д), 4,44 (2Н, т), 4,21 (2Н, т), 4,04 (1Н, к), 3, 73 (3Н, с), 1,72 (4Н, м), 1,65 (3Н, д).
Элементный анализ
Рассчитано: С 64,79%; Н 5,25%; N 2,62%; F 3,53%.
Найдено: С 64,85%; Н 5,31%; N 2,74%; F 3, 48%.
ПРИМЕР 7
Получение 4-ацетамидофенилового эфира N-(4-нитроксибутирил)-β-аланил-(L)-гистидина (NCX 2160), имеющего формулу

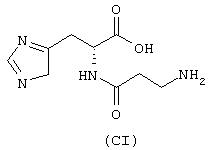
где предшественником является ацетаминофен (парацетамол), имеющий формулу (X), а предшественником В является (L)-карнозин (NCX 2053), имеющий формулу (CI)

а) Синтез N-(4-бромбутирил)-β-аланил-(L)-гистидина
К раствору карнозина (5 г, 22,1 ммоль) в N,N-диметилформамиде (80 мл) добавляют триэтиламин (4,62 мл, 33,1 ммоль) и 4-бромбутирилхлорид (хлорангидрид 4-бромбутановой кислоты, 83,85 мл, 33,1 ммоль). Перемешивают раствор при комнатной температуре в течение 24 часов, затем разбавляют его этилацетатом и промывают органическую фазу водой. Высушивают органическую фазу сульфатом натрия и упаривают при пониженном давлении. Полученный неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя этилацетатом, что дает в результате конечный продукт.
b) Синтез 4-ацетамидофенилового эфира N-(4-бромбутирил)-β-аланил-(L)-гистидина
К раствору N-(4-бромбутирил)-β -аланил-(L)-гистидина (3 г, 8 ммоль) в хлороформе (50 мл) и N,N-диметилформамиде (4 мл) добавляют при перемешивании парацетамол (1,21 г, 8 ммоль), N,N-дициклогексилкарбодиимид (1,65 г, 8 ммоль) и диметиламинопиридин (0,04 г, 0,36 ммоль). Выдерживают реакционную смесь при комнатной температуре в течение 6 часов. Далее смесь фильтруют, разбавляют хлороформом и промывают водой. Высушивают органическую фазу сульфатом натрия и упаривают при пониженном давлении. Полученный неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью этилацетат/н-гексан 7/3. Получают 4-ацетамидофениловый эфир N-(4-бромбутирил)-β-аланил-(L)-гистидина.
c) Синтез 4-ацетамидофенилового эфира N-(4-нитроксибутирил)-β-аланил-(L)-гистидина
К раствору 4-ацетамидофенилового эфира N-(4-бромбутирил)-β-аланил-(L)-гистидина (4 г, 7,87 ммоль) в ацетонитриле (70 мл) добавляют при перемешивании нитрат серебра (1,87 г, 11 ммоль). Кипятят реакционную смесь в течение 5 часов без доступа света. Образующуюся соль удаляют фильтрацией и раствор упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 3/7. Получают целевой продукт с выходом 17%.
Элементный анализ
Рассчитано: С 51,39%; Н 5,34%; N 17,19%.
Найдено: С 51,28%; Н 5,28%; N 17,06%.
ПРИМЕР 8
Получение 4-нитроксибутилового эфира N-ацетил-S-[(S)-α-(2-хлорфенил)-6,7-дигидротиено[3, 2-с]пиридин-5(4Н)ацетил]-(S)-цистеина (NCX 2136)

где предшественником является клопидогрель, имеющий формулу (XI), а предшественником В является N-ацетилцистеин, имеющий формулу (СVIII)


Соединение синтезируют по методу, описанному в примере 1. Выход 23%.
Элементный анализ
Рассчитано: С 50,55%; Н 4,95%; N 7,40%; S 11,24%; Cl 6,22%.
Найдено: С 50,70%; Н 4,99%; N 7,60%; S 11,20%; Cl 6,15%.
ПРИМЕР 9
Получение 4-[(2-амино-3,5-дибромфенил)метиламино]циклогексилового эфира [3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеновой кислоты (NCX 2161)

где предшественником является амброксол, имеющий формулу (ХII), а предшественник В представлен феруловой кислотой, имеющей формулу (DII)


а) Синтез 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]-транс-циклогексанола
К раствору 4-[(2-амино-3, 5-дибромфенил)метиламино]циклогексанола (5 г, 13,22 ммоль) в смеси диоксана (35 мл) и воды (50 мл) добавляют при перемешивании триэтиламин (3,31 мл, 23,7 ммоль) и ди-трет-бутилдикарбонат (3,46 г, 15, 86 ммоль). Через 24 часа раствор концентрируют в вакууме, добавляют 1% раствор НСl до достижения нейтрального рН (рН 7), затем экстрагируют органическую фазу этилацетатом. Высушивают органическую фазу сульфатом натрия и упаривают в вакууме. Получают 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]-транс-циклогексанол, который используют без дальнейшей очистки.
b) Синтез 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]циклогексилового эфира [3-метокси-4-гидроксифенил]-2-транс-пропеновой кислоты
К раствору феруловой кислоты (4 г, 20,5 ммоль) в тетрагидрофуране (40 мл) при 0°С добавляют 1,1’-карбонилдиимидазол (3,34 г, 20,5 ммоль). Через 10 минут к раствору добавляют 4-[(2-трет-бутилоксикарбониламино-3, 5-дибромфенил)метиламино]циклогексанол (9,8 г, 20,5 ммоль) и выдерживают реакционную массу при комнатной температуре в течение 4 часов. Концентрируют реакционную смесь в вакууме, добавляют дихлорметан, после чего промывают ее 1% раствором НСl, а затем водой. Высушивают органическую фазу сульфатом натрия и затем упаривают в вакууме. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 1/1. Получают 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]циклогексиловый эфир [3-метокси-4-гидроксифенил]-2-транс-пропеновой кислоты.
c) Синтез 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]циклогексилового эфира [3-метокси-4-(4-бромбутирилокси)фенил]-2-транс-пропеновой кислоты
К раствору 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]циклогексилового эфира [3-метокси-4-гидроксифенил]-2-транс-пропеноеой кислоты (4 г, 6,11 ммоль) в тетрагидрофуране (80 мл) добавляют при перемешивании триэтиламин (0,85 мл, 6,11 ммоль) и бромбутирилхлорид (0,7 мл, 6,11 ммоль). Реакцию ведут при комнатной температуре в течение 8 часов, после чего упаривают органический растворитель при пониженном давлении. Полученный неочищенный продукт обрабатывают этилацетатом и органическую фазу промывают водой. Затем органическую фазу сушат сульфатом натрия и упаривают в вакууме. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 7/3. Получают 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]циклогексиловый эфир [3-метокси-4-(4-бромбутирилокси)фенил]-2-транс-пропеновой кислоты.
d) Синтез 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]циклогексилового эфира [3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеновой кислоты
К раствору 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]циклогексилового эфира [3-метокси-4-(4-бромбутирилокси)фенил]-2-транс-пропеновой кислоты (4 г, 4,98 ммоль) в ацетонитриле (70 мл) добавляют при перемешивании нитрат серебра (0,87 г, 4,98 ммоль). Кипятят реакционную смесь в течение 7 часов без доступа света, после чего образующуюся соль удаляют фильтрацией. Раствор упаривают при пониженном давлении. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 7/3. Получают 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил)метиламино]циклогексиловый эфир [3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеновой кислоты.
e) Синтез 4-[(2-амино-3,5-дибромфенил)метиламино]циклогексилового эфира [3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеновой кислоты
К раствору 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил) метиламино]циклогексилового эфира [3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеновой кислоты (2 г, 2,54 ммоль) в этилацетате (50 мл), охлажденному до 0°С, при перемешивании добавляют 5 н. раствор НСl в этилацетате (3,17 мл). Перемешивают раствор при 0°С в течение 4 часов, после чего отфильтровывают выпавший осадок. К полученному неочищенному продукту приливают этилацетат, к которому добавлен 5% раствор бикарбоната натрия. Смесь встряхивают, затем заменяют раствор бикарбоната равным объемом воды, после чего встряхивают снова. Отделяют органическую фазу, высушивают ее сульфатом натрия и упаривают при пониженном давлении. Получают 4-[(2-амино-3,5-дибромфенил)метиламино]циклогексиловый эфир [3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеновой кислоты. Выход 36%.
Элементный анализ
Рассчитано: С 47,30%; Н 4,56%; N 6,15%; Br 23,31%.
Найдено: С 47,26%; Н 4,53%; N 6,00%; Вr 23,42%.
ПРИМЕР 10
Получение [4-амино-[[3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеноил]-1-гидроксибутилиден]бисфосфоновой кислоты (NCX 2211)

где предшественником является алендроновая кислота, имеющая формулу (ХIII), а предшественником В является феруловая кислота (формула DII)


a) Синтез [3-метокси-4-(4-бромбутирилокси)фенил]-2-транс-пропеновой кислоты
К раствору феруловой кислоты (1,2 г, 6,11 ммоль) в тетрагидрофуране (80 мл) при перемешивании добавляют триэтиламин (0,85 мл, 6,11 ммоль) и 4-бромбутирилхлорид (0,7 мл, 6,11 ммоль). Ведут реакцию при комнатной температуре в течение 3 часов, а затем упаривают при пониженном давлении. К полученному неочищенному продукту добавляют этилацетат и промывают органическую фазу водой. Затем высушивают органическую фазу сульфатом натрия и упаривают в вакууме. Полученный остаток очищают при помощи хроматографии на силикагеле, элюируя смесью хлороформ/метанол 8/2. В итоге выделяют [3-метокси-4-(4-бромбутирилокси)фенил]-2-транс-пропеновую кислоту.
b) Синтез [3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеновой кислоты
К раствору [3-метокси-4-(4-бромбутирилокси)фенил]-2-транс-пропеновой кислоты (1,5 г, 4,5 ммоль) в ацетонитриле (70 мл) добавляют при перемешивании нитрат серебра (0,87 г, 4,98 ммоль). Кипятят реакционную смесь в течение 3 часов без доступа света и при перемешивании. Образующуюся соль удаляют фильтрацией и упаривают органическую фазу при пониженном давлении. Полученный остаток очищают при помощи хроматографии на колонке с силикагелем, элюируя смесью хлороформ/метанол 8/2. Получают [3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеновую кислоту.
с) Синтез [4-амино-[[3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеноил]-1-гидроксибутилиден]бисфосфоновой кислоты
К раствору [3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеновой кислоты (2 г, 6,4 ммоль) в N,N-диметилформамиде (30 мл), охлажденному до 0°С, добавляют при перемешивании N, N’-дициклогексилкарбодиимид (1,3 г, 6,4 ммоль) и 1-гидроксибензотриазол (1,04 г, 7,68 ммоль). Через 30 минут добавляют алендроновую кислоту (1,6 г, 6,4 ммоль). Перемешивают реакционную смесь при комнатной температуре в течение 7 часов. После этого подкисляют ее 5% раствором НСl и экстрагируют органическую фазу этилацетатом. Промывают органическую фазу насыщенным водным раствором NaCl, высушивают сульфатом натрия и затем упаривают при пониженном давлении. Неочищенный продукт очищают при помощи хроматографии на колонке с силикагелем, элюируя смесью дихлорметан/метанол 8/2. Получают [4-амино-[[3-метокси-4-(4-нитроксибутирилокси)фенил]-2-транс-пропеноил]-1-гидроксибутилиден]бисфосфоновую кислоту. Выход 11%.
Элементный анализ
Рассчитано: С 19,71%; Н 4,36%; N 5,07%; Р 11,17%.
Найдено: С 19,56%; Н 4,28%; N 5,04%; Р 11,25%.
ПРИМЕР 11
Получение 4-нитроксибутилового эфира S-[[2-[4-(4-хлорфенил)фенилметил)-1-пиперазинил]этокси]ацетил]пеницилламина (NCX 2060), имеющего формулу

где предшественником является цетиризин, имеющий формулу XIV, а предшественником В является пеницилламин (формула CV)

a) Синтез 4-нитроксибутилового эфира S-[[2-[4-(4-хлорфенил)фенилметил)-1-пиперазинил]этокси]ацетил]-N-трет-бутилоксикарбонилпеницилламина
Соединение получают в соответствии с методом, изложенным в примере 1, используя N-трет-бутилоксикарбонилпеницилламин вместо N-ацетилцистеина.
b) Синтез 4-нитроксибутилового эфира S-[[2-[4-(4-хлорфенил)фенилметил)-1-пиперазинил]этокси]ацетил]пеницилламина
Соединение получают из предыдущего соединения по методу, описанному в примере 9 (стадия е), удаляя N-трет-бутилоксикарбонильную защиту и деблокируя таким образом аминогруппу. Выход 26%.
Элементный анализ
Рассчитано: С 55,78%; Н 6,49%; N 8,43%; S 4,80%; Cl 5,31%.
Найдено: С 55,61%; Н 6,31%; N 8,29%; S 4,93%; Cl 5,43%.
ПРИМЕР 12
Получение 4-нитроксибутилового эфира N-ацетил-S-[(S)-1-[N-[1-(этоксикарбонил)-3-фенилпропил]-L-аланил]-L-пролин]цистеина (NCX 2134)

где предшественником является эналаприл, имеющий формулу XV, а предшественником В является N-ацетилцистеин (формула СVIII)


Соединение синтезируют по методу, изложенному в примере 1, Выход 27%.
Элементный анализ
Рассчитано: С 55,18%; Н 6,79%; N 8,62%; S 4,91%.
Найдено: С 55,30%; Н 6,85%; N 8,71%; S 4,85%.
ПРИМЕР 13
Получение 4-нитроксибутилового эфира 3-[4-[D-α -аминобензилпеницилламиноилокси]-3-метоксифенил]-2-транс-пропеновой кислоты (NCX 2080), имеющего формулу

где предшественник представлен ампициллином (формула XVI), а предшественником В является феруловая кислота (формула DII)


Соединение синтезируют по методу, изложенному в примере 5. Выход 11%.
Элементный анализ
Рассчитано: С 56,04%; Н 5,33%; N 8,75%; S 4,99%.
Найдено: С 56,15%; Н 5,48%; N 8,65%; S 4,83%.
ПРИМЕР 14
Получение 9-[[2-[N-ацетил-S-(4-нитроксибутироил)цистеинил]этокси]метил]гуанина (NCX 2135)

где предшественником является ацикловир (формула XVII), а предшественником В является N-ацетилцистеин (формула СVIII)


а) Синтез N-ацетил-S-(4-бромбутироил)цистеина
Готовят раствор, содержащий 4-бромбутановую кислоту (5,1 г, 30,6 ммоль) и 1,1’-карбонилдиимидазол (5,61 г, 34,6 ммоль) в хлороформе (50 мл), и перемешивают его при комнатной температуре в течение 1 часа. Добавляют к реакционной смеси раствор N-ацетилцистеина (5 г, 30,6 ммоль) в N,N-диметилформамиде (5 мл), содержащий этилат натрия (50 мг). Перемешивают раствор в течение 24 часов, после чего промывают его 1% НСl, а затем насыщенным водным раствором NaCl. Органическую фазу высушивают сульфатом натрия и упаривают при пониженном давлении. Полученный неочищенный продукт очищают при помощи хроматографии на колонке с силикагелем, элюируя смесью этилацетат/хлороформ 7/3. В итоге получают N-ацетил-S-(4-бромбутироил)цистеин.
b) Синтез N-ацетил-S-(4-нитроксибутироил)цистеина
К раствору N-ацетил-S-(4-бромбутироил)цистеина (3 г, 9,6 ммоль) в ацетонитриле (70 мл) добавляют нитрат серебра (1,7 г, 10 ммоль). Кипятят реакционную смесь в течение 2 часов без доступа света. Образующуюся соль удаляют фильтрацией и упаривают раствор при пониженном давлении. Полученный остаток очищают при помощи хроматографии на колонке с силикагелем, элюируя смесью этилацетат/хлороформ 7/3. Получают в итоге N-ацетил-S-(4-нитроксибутироил)цистеин.
c) Синтез 9-[[2-[N-ацетил-S-(4-нитроксибутироил)цистеинил]этокси]метил]гуанина
Готовят раствор N-ацетил-S-(4-нитроксибутироил)цистеина (2,8 г, 9,6 ммоль) и 1,1’-карбонилдиимидазола (1,55 г, 9,6 ммоль) в тетрагидрофуране (50 мл) и перемешивают его при комнатной температуре в течение 1 часа. Добавляют к реакционной смеси ацикловир (2,16 г, 9,6 ммоль). Ведут реакцию при комнатной температуре в течение 6 часов, растворитель упаривают при пониженном давлении, полученный остаток растворяют в этилацетате и промывают насыщенным водным раствором NaCl. Высушивают органическую фазу сульфатом натрия и затем упаривают в вакууме. Полученный остаток очищают при помощи хроматографии на колонке с силикагелем, элюируя этилацетатом. Получают 9-[[2-[N-ацетил-S-(4-нитроксибутироил)цистеинил]этокси]метил]гуанин. Выход 9%.
Элементный анализ
Рассчитано: С 35,25%; Н 3,95%; N 13,76%; S 47,05%.
Найдено: С 35,38%; Н 3,99%; N 13,84%; S 47,20%.
ПРИМЕР 15
Получение 4-нитроксибутилового эфира транс-3-[4-(5-амино-2-гидроксибензоил)-3-метоксифенил]-2-пропеновой кислоты (NCX 2212)

где предшественником является мезаламин (XVIII), а предшественником В является феруловая кислота (формула DII)


а) Синтез 4-нитроксибутилового эфира транс-3-[4-(5-трет-бутилоксикарбониламино-2-гидроксибензоил)-3-метоксифенил]-2-пропеновой кислоты
Соединение синтезируют в соответствии с методикой, изложенной в примере 5, предварительно защитив первичную аминогруппу мезаламина, как описано в примере 9, стадия а).
b) Получение 4-нитроксибутилового эфира транс-3-[4-(5-амино-2-гидроксибензоил)-3-метоксифенил]-2-пропеновой кислоты
Конечное соединение получают путем гидролитического расщепления связи между аминогруппой и N-трет-бутилоксикарбонильной защитной группой, как описано в примере 9, стадия е). Выход 28%.
Элементный анализ
Рассчитано: С 56, 49%; Н 4,96%; N 6,30%.
Найдено: С 56,55%; Н 4,82%; N 6,45%.
ПРИМЕР 16
Получение 6-метилен-5-гидрокси-10-[2-гидрокси-5-(4-нитроксибутирилокси)бензоил]тетрациклина (NCX 2163)

где предшественником является метациклин (XIX), а предшественником В является гентизиновая кислота (DIII)


a) Синтез 5-(4-бромбутирилокси)-2-гидроксибензойной кислоты
К раствору 4-бромбутирилхлорида (3 г, 16,17 ммоль) в тетрагидрофуране (50 мл), охлажденному до 0°С, по каплям добавляют триэтиламин (4,5 мл, 32,34 ммоль), а затем гентизиновую кислоту (2,4 г, 16,16 ммоль). Ведут реакцию при 0°С и при перемешивании в течение 4 часов, затем упаривают реакционную смесь при пониженном давлении. К полученному неочищенному продукту добавляют этилацетат, промывают органическую фазу 1% НСl, затем насыщенным водным раствором NaCl. Высушивают органическую фазу сульфатом натрия и упаривают. Полученный остаток очищают при помощи хроматографии на колонке с силикагелем, элюируя смесью дихлорметан/метанол 95/5. Получают 5-(4-бромбутирилокси)-2-гидроксибензойную кислоту.
b) Синтез 5-(4-нитроксибутирилокси)-2-гидроксибензойной кислоты
К раствору 5-(4-бромбутирилокси)-2-гидроксибензойной кислоты (3 г, 9,6 ммоль) в ацетонитриле (150 мл) добавляют нитрат серебра (1, 7 г, 10 ммоль). Кипятят реакционную смесь в течение 7 часов без доступа света. Образующуюся соль удаляют фильтрацией и упаривают раствор при пониженном давлении. Полученный остаток очищают при помощи хроматографии на колонке с силикагелем, элюируя смесью дихлорметан/метанол 95/5. В результате описанных операций выделяют чистую 5-(4-нитроксибутирилокси)-2-гидроксибензойную кислоту.
с) Синтез 6-метилен-5-гидрокси-10-[2-гидрокси-5-(4-нитроксибутирилокси)бензоил]тетрациклина
Готовят раствор 5-(4-нитроксибутирилокси)-2-гидроксибензойной кислоты (5 г, 16,4 ммоль) и 1, 1’-карбонилдиимидазола (2,67 г, 16,4 ммоль) в тетрагидрофуране (70 мл) и перемешивают его при комнатной температуре в течение 1 часа. Добавляют адриамицин (7,2 г, 16,4 ммоль). Ведут реакцию при комнатной температуре и при перемешивании в течение 12 часов. Затем органический раствор упаривают при пониженном давлении, к полученному остатку приливают этилацетат и промывают смесь насыщенным водным раствором NaCl. Органическую фазу высушивают сульфатом натрия и упаривают в вакууме. Полученный остаток очищают при помощи хроматографии на колонке с силикагелем, элюируя этилацетатом. Получают 6-метилен-5-гидрокси-10-[2-гидрокси-5-(4-нитроксибутирилокси)бензоил]тетрациклин. Выход 19%.
Элементный анализ
Рассчитано: С 55,84%; Н 4,40%; N 5,95%.
Найдено: С 55,95%; Н 4,55%; N 5,98%.
ПРИМЕР 17
Получение 5-[[3-[3-метокси-4-(4-нитрокси)бутирилокси]фенил-2-транс-пропеноил]амино]-1,2,3,4-тетрагидроакридина (NCX 2214)

где предшественником является такрин (XX), а предшественником В является феруловая кислота (формула DII)


Соединение синтезируют в соответствии с методикой, изложенной в примере 10. Выход 7%.
Элементный анализ
Рассчитано: С 64,13%; Н 5,38%; N 8,34%.
Найдено: С 64,28%; Н 5,46%; N 8,47%.
ПРИМЕР 18
Получение 1,2,3,7,8,8-гексагидро-3, 7-диметил-8-[тетрагидро-4-[2-гидрокси-5-(4-нитроксибутирилокси)]бензоилокси[6-оксо-2Н-пиран-2-ил]этил]-1-нафтилового эфира [1S-[1α,3α,7β,8β,(2S*,4S*)]]-2,2-диметилбутановой кислоты (NCX 2164)

где предшественником является симвастатин (формула XXI), а предшественником В является гентизиновая кислота (формула DIII)

Соединение синтезируют по методу, описанному в примере 16. Выход 13%.
Элементный анализ
Рассчитано: С 63,15%; Н 7,06%; N 2,01%.
Найдено: С 63,68%; Н 7,21%; N 2, 19%.
ПРИМЕР 19
Получение 5-метокси-2-[[[4-[N-[4-нитроксибутил-β-аланил]-(L)-гистидинилокси]-3,5-диметил-2-пиридинил]метил]сульфинил]-1Н-бензимидазола (NCX 2062)

где предшественником является 4-гидроксиомепразол (формула ХХП), полученный из омепразола, как описано в Acta Chem. Scand. 43, 6, 1989, стр.549-568, а предшественником В является карнозин (формула СI)

Соединение синтезировано в соответствии с методикой, описанной в примере 7. Выход 25%.
Элементный анализ
Рассчитано: С 51,97%; Н 4,96%; N 16,79%; S 4,78%.
Найдено: С 51,81%; Н 4,80%; N 16,68%; S 4,92%.
ПРИМЕР 20
Получение 4-нитроксибутилового эфира N-никотиноил-β-аланил-(L)-гистидина (NCX 2073)

где предшественником является никотинамид (формула ХХIII), а предшественником В является карнозин (формула CI)


а) Синтез N-никотиноил-β-аланил-(L)-гистидина
К раствору никотиновой кислоты (2,5 г, 20,5 ммоль) в тетрагидрофуране (40 мл), охлажденному до 0°С, добавляют при перемешивании 1,1’-карбонилдиимидазол (3,34 г, 20,5 ммоль). Через 10 мин к этому раствору добавляют (L)-карнозин (4,6 г, 20,5 ммоль) и перемешивают смесь при комнатной температуре в течение 4 часов. Концентрируют реакционную смесь в вакууме, разбавляют дихлорметаном, промывают 1% НСl, а затем водой. Высушивают органическую фазу сульфатом натрия и упаривают в вакууме. Полученный остаток хроматографируют на колонке с силикагелем, элюируя этилацетатом. Выделяют N-никотиноил-β-аланил-(L)-гистидин.
b) Синтез 4-бромбутилового эфира N-никотиноил-β-аланил-(L)-гистидина
К раствору N-никотиноил-β -аланил-(L)-гистидина (9,9 г, 30,1 ммоль) в тетрагидрофуране (200 мл) добавляют при перемешивании трифенилфосфин (23,7 г, 90,3 ммоль) и тетрабромметан (28,85 г, 90,3 ммоль). Перемешивают реакционную смесь при комнатной температуре в течение 24 часов. После этого растворитель упаривают при пониженном давлении. Полученный неочищенный продукт очищают при помощи хроматографии на колонке с силикагелем, элюируя смесью н-гексан/этилацетат 1/1. Получают 4-бромбутиловый эфир N-никотиноил-β-аланил-(L)-гистидина.
c) Синтез 4-нитроксибутилового эфира N-никотиноил-β -аланил-(L)-гистидина
К раствору 4-бромбутилового эфира N-никотиноил-β-аланил-(L)-гистидина (0,91 г, 1,96 ммоль) в ацетонитриле (20 мл) добавляют при перемешивании нитрат серебра (0,66 г, 3,92 ммоль). Кипятят реакционную смесь в течение 4 часов при перемешивании и без доступа света. Образующуюся соль удаляют фильтрацией и упаривают раствор при пониженном давлении. Полученный остаток очищают при помощи хроматографии на колонке с силикагелем, элюируя смесью н-гексан/этилацетат 1/1. Получают 4-нитроксибутиловый эфир N-никотиноил-β-аланил-(L)-гистидина. Выход 32%.
Элементный анализ
Рассчитано: С 49,50%; Н 5,54%; N 19,32%.
Найдено: С 49,35%; Н 5,28%; N 19,17%.
ПРИМЕР 21
Получение 1-[(1-метилэтил)амино]-3-(1-нафталинилокси)-2-пропилового эфира N-ацетил-S-(4-нитроксибутироил)цистеина (NCX 2132)

где предшественником является пропранолол (формула XXIV), а предшественником В является N-ацетилцистеин (формула СVIII)


Соединение синтезируют по методу, описанному в примере 14. Выход 7%.
Элементный анализ
Рассчитано: С 56,04%; Н 6,21%; N 7,88%; S 5,98%.
Найдено: С 56,13%; Н 6,35%; N 7,91%; S 6,04%.
ПРИМЕР 22
Получение 2-(трет-бутиламино)-1-[4-гидрокси-3-[N-ацетил-S-(4-нитрокси-бутирил)пеницилламиноил]оксифенил]этанола (NCX 2133)

где предшественником является сальбутамол (албутерол) (формула XXV), а предшественником В является N-ацетилпеницилламин (формула CV)


Соединение синтезируют по методу, изложенному в примере 14, используя N-ацетилпеницилламин вместо N-ацетилцистеина. Выход 43%.
Элементный анализ
Рассчитано: С 53,01%; Н 6,86%; N 7,76%; S 5,89%.
Найдено: С 53,19%; Н 6,80%; N 7,66%; S 5,72%.
ПРИМЕР 23
Получение 7-[2-гидрокси-3-[3-метокси-5-(4-нитроксибутирилокси)бензоил]-транс-2-пропеноил]теофиллина (NCX 2213)

где предшественником является дифиллин (формула XXVI), а предшественником В является феруловая кислота (формула DII)


Соединение синтезируют в соответствии с методикой, описанной в примере 9. Выход 22%.
Элементный анализ
Рассчитано: С 51,31%; Н 4,84%; N 12,52%.
Найдено: С 51,50%; Н 4,91%; N 12,68%.
ПРИМЕР 24
Получение 4-нитроксибутилового эфира N-ацетил-S-(2-ацетилбензоил)цистеина (NCX 2138), имеющего формулу

где предшественником является ацетилсалициловая кислота (формула XXVII), а предшественником В является N-ацетилцистеин (формула СVIII)


Соединение синтезируют в соответствии с методикой, описанной в примере 1. Выход 36%.
Элементный анализ
Рассчитано: С 48,85%; Н 5,01%; N 6,36%; S 7,24%.
Найдено: С 48,75%; Н 5,02%; N 6,28%; S 7,12%.
ПРИМЕР 25
Получение 4-[3-[3-метокси-5-(4-нитроксибутирилокси)фенил]-2-пропеноилокси]-2-метил-N-2-пиридинил-2Н-1,2-бензотиазин-3-карбоксамид-1,1-диоксид (NCX 2215)

где предшественником является пироксикам (формула ХХVIII), а предшественником В является феруловая кислота (формула DII)


Соединение синтезируют в соответствии с методикой, изложенной в примере 9. Выход 18%.
Элементный анализ
Рассчитано: С 55,11%; Н 4,47%; N 8,60%; S 4,90%.
Найдено: С 55,18%; Н 4,52%; N 8,71%; S 4,98%.
ПРИМЕР 26
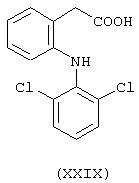
Получение 4-нитроксибутилового эфира S-[2-[(2,6-дихлорфенил)амино]фенилацетилокси]пенипилламина (NCX 2061), имеющего формулу

где предшественником является диклофенак (формула XXIX), а предшественником В является пеницилламин (формула CV)


Соединение синтезируют в соответствии с методикой, описанной в примере 11. Выход 21%.
Элементный анализ
Рассчитано: С 50,72%; Н 5,00%; N 7,75%; S 5,89%; Cl 13,02%.
Найдено: С 50,61%; Н 4,89%; N 7,81%; S 6,01%; Cl 13,21%.
ФАРМАКОЛОГИЧЕСКИЕ ТЕСТЫ
Острая токсичность
Острую токсичность оценивали, вводя группе из 10 крыс весом 20 г однократную дозу каждого из тестируемых соединений в ротовое отверстие, через трубочку, в составе водной суспензии 2 (мас./об.) карбоксиметилцеллюлозы.
За животными вели наблюдение в течение 14 дней. Ни у одного из животных в тестируемой группе не появлялись токсические симптомы, даже после введения дозы 100 мг/кг.
ПРИМЕР F1
Тест 1 - экспериментальная модель in vivo с N-этилмалеимидом (NEM): исследование желудочной переносимости некоторых лекарственных веществ, анализированных в качестве предшественников соединений настоящего изобретения.
Животные (крысы, вес примерно 200 г) были распределены по следующием группам (по 10 животных в каждой группе).
A. Контрольные группы.
Группа 1°. Обработка: только носитель (водная суспензия 1% (мас./об.) карбоксиметилцеллюлозы, доза 5 мл/кг, если лекарственное вещество вводили через рот, физиологический раствор в случае парентерального способа введения).
Группа 2°. Обработка: носитель + NEM.
B. Группы, которым вводили каждое лекарственное вещество.
Группа I. Обработка: носитель + лекарственное вещество.
Группа II. Обработка: носитель + лекарственное вещество + NEM.
Следующие соединения были исследованы в этом эксперименте (таблица I): индометацин, амброксол, мезаламин, алендронат натрия, такрин, омепразол, мизопростол.
Индометапин, амброксол и алендронат вводили через рот, мезаламин вводили в прямую кишку (ректально), а такрин, омепразол и мизопростол -подкожно.
Максимальная переносимая доза, определяемая путем введения каждого вещества указанными способами животным, которым не вводили NEM, указана в таблице I. При превышении указанных доз у животных появлялись энтеропатия, понос, депрессия, дрожь и заторможенные реакции.
В этой экспериментальной модели животным сначала вводили дозу NEM (25 мг/кг) в физиологическом растворе путем подкожной инъекции. Лекарственное вещество вводили спустя 1 час в суспензии носителя. Спустя 24 часа животных умерщвляли и оценивали повреждения, нанесенные слизистым оболочкам желудочно-кишечного тракта, путем подсчета числа крыс в каждой группе, имеющих видимые патологические изменения желудка. Суммарное число таких крыс затем разделяли на суммарное число крыс в группе и умножали на 100%. Полученные таким образом величины в % представлены в таблице I. Эта таблица показывает, что в группах крыс, которым вводили указанные лекарственные вещества без NEM, не было обнаружено желудочных повреждений.
У всех крыс из группы II (которым вводили NEM) были обнаружены повреждения желудка после введения следующих лекарственных веществ: индометацин, амброксол, мезаламин, алендронат натрия, такрин. Следовательно, указанные лекарственные вещества могут быть использованы в синтезе продуктов настоящего изобретения.
По результатам теста 1 омепразол и мизопростол, напротив, не могут быть использованы для получения продуктов настоящего изобретения.
ПРИМЕР F2
Тест 2 (in vitro) - ингибирование апоптоза (фрагментация ДНК), вызываемое в эндотелиальных клетках соединением CIP, в присутствии некоторых лекарственных веществ, анализированных в качестве предшественников соединений настоящего изобретения.
Были протестированы следующие лекарственные вещества-предшественники: индометацин, парацетамол, клопидогрель, сальбутамол, амброксол, алендронат натрия, дифиллин, цетиризин, эналаприл, никотинамид, ампициллин, ацикловир, мезаламин, такрин, симвастатин, омепразол.
Эндотелиальные клетки человека из пупочной вены получают в соответствии со стандартной процедурой. Свежие пупочные вены заполняют 0,1 мас.% раствором коллагеназы и инкубируют при 37°С в течение 5 минут.
После этого вены заливают средой М 199 (GIBCO, Grand Island, NY), pH 7,4, содержащей 0,1% (мас./об.) коллагеназы с добавлением 10% феталыюй сыворотки крупного рогатого скота (10 мкг/мл), натриевой соли гепарина (50 мкг/мл), тимидина (2,4 мкг/мл), глутамина (230 мкг/мл), пенициллина (100 м.е./мл), стрептомицина (100 мкг/мл) и стрептомицина В (0,125 мкг/мл). Собирают клетки из перфузата при помощи центрифугирования (800 об/мин) и выращивают в культуральных колбах Т-75, предварительно обработанных фибронектином человека. Затем выращивают клетки в той же самой среде, к которой добавлен фактор роста гипоталамуса крупного рогатого скота (100 нг/мл). Когда клетки первичной клеточной культуры (то есть полученные напрямую ex vivo из пупочной вены) образуют слитный клеточный монослой (примерно 8000000 клеток на одну колбу), рост культуры останавливают, слои промывают и обрабатывают трипсином. Переносят клеточные суспензии в лунки 24-луночного планшета для клеточных культур, к половине из которых добавляют ту же самую культуральную среду, содержащую лекарственное вещество в концентрации 10-4М, после чего выращивают клетки в термостате при 37°С, постоянной влажности (90%) и 5% концентрации СO2. Если соединение нерастворимо в культуральной среде, его сначала растворяют в небольшом количестве диметилсульфоксида. Максимальное количество диметилсульфоксида, которое может быть добавлено к культуральной среде, составляет 0,5%. Только клетки, произошедшие из указанных первых субкультур, используют для экспериментов с гидропероксидом кумола (СIР). Клетки идентифицируют как эндотелиальные клетки путем морфологического анализа, а также по их специфической иммунологической реакции на фактор VIII; в указанных культурах не обнаруживаются каких-либо загрязнения, происходящие из миоцитов или фибробластов.
Перед началом теста клеточную культуральную среду удаляют и клеточные слои осторожно промывают стандартным физиологическим раствором, содержащим 0,1 М фосфатный буфер (рН 7,0), при температуре 37°С. Содержимое каждой лунки затем инкубируют в течение 1 часа с суспенизией CIP в культуральной среде в концентрации 5 мМ. Оценку клеточных повреждений (апоптоз) проводят путем определения процентного изменения фрагментации ДНК в культурах, содержащих лекарственное вещество и CIP, относительно контрольных групп, которые обрабатывали только CIP. Указанное процентное изменение фрагментации ДНК определяют путем оценки изменения флуоресценции при длинах волн 405-450 нм, при помощи микроскопа ВХ60 Olympus (Olympus Co, Рим), сравнивая оптические плотности опытных и контрольных образцов. Для каждого образца проводят 5 повторных измерений флуоресценции. Статистическую оценку результатов проводят при помощи t теста Стьюдента (р<0,01).
Результаты, приведенные в таблице II, показывают, что индометацин, парацетамол, клопидогрель, сальбутамол, алендронат натрия, дифиллин, цетиризин, эналаприл, никотинамид, ампициллин, ацикловир, такрин и омепразол практически не ингибируют апоптоз. Следовательно, эти лекарственные вещества могут быть использованы для получения продуктов настоящего изобретения.
В противоположность этому амброксол, мезаламин и симвастатин ингибируют апоптоз. Следовательно, по результатам теста 2, эти соединения не могут быть использованы для получения продуктов настоящего изобретения.
ПРИМЕР F3
Тест 3 - экспериментальная in vivo модель, использующая метиловый эфир Nw-нитро-L-аргинина (L-NAME): анализ желудочной переносимости (возникновение повреждений желудочно-кишечного тракта), переносимости для печени (доза GPT, глутамат-пируват трансаминазы) и сердечно-сосудистой системы (кровяное давление) некоторых лекарственных веществ, использованных в качестве предшественников соединений настоящего изобретения.
Экспериментальная модель является адаптированной моделью, описанной в J.Clin. Investigation 90, 278-281,1992.
Эндотелиальные дисфункции оценивают путем определения повреждений слизистых оболочек желудочно-кишечного тракта, повреждений печени (увеличение уровня GPT), а также повреждений сосудистого эндотелия и сердечно-сосудистой системы (увеличение кровяного давления), вызванных введением L-NAME.
Животных (крысы, средний вес 200 г) делят на группы в соответствии с приведенной ниже схемой. Группе, получающей L-NAME, вводят в течение 4 недель указанное соединение, растворенное в питьевой воде, в концентрации 400 мг/л. Выделены следующие группы (по 10 животных в каждой):
A. Контрольные группы.
Группа 1°. Обработка: только носитель (водная суспензия 1% (мас./об.) карбоксиметилцеллюлозы, доза 5 мл/кг, если лекарственное вещество вводится через рот, физиологический раствор, если вводится парентерально).
Группа 2°. Обработка: носитель + L-NAME.
B. Группы, которым вводили лекарственное вещество.
Группа 3°. Обработка: носитель + лекарственное вещество.
Группа 4°. Обработка: носитель + лекарственное вещество + L-NAME.
Были протестированы следующие лекарственные вещества: парацетамол, доксорубицин, симвастатин, омепразол и мизопростол. Каждое соединение вводили один раз в день в течение 4 недель.
Максимальную переносимую дозу лекарственного вещества, вводимого животным, определяли в ходе экспериментов с масштабированием отдельной дозы путем оценки появления у животных симптомов энтеропатии, поноса, депрессии, дрожи и заторможенных реакций.
В конце четвертой недели животным перестают давать воду и по истечении 24 часов их умерщвляют.
За один час до умерщвления определяют кровяное давление и его повышение принимают в качестве показателя повреждения сосудистого эндотелия.
Повреждения слизистой оболочки желудка оценивают так, как проиллюстрировано в тесте 1 (см. пример F1). Повреждения печени определяют путем оценки содержания глутамат-пируват трансаминазы (увеличение уровня GPT) после умерщвления.
Лекарственное вещество отвечает условиям теста 3, то есть может быть использовано для получения соединений настоящего изобретения, если в группах крыс, которых обрабатывали системой L-NAME + лекарственное вещество + носитель, обнаружены более существенные повреждения печени (более высокий уровень GPT) и/или более существенные повреждения желудка и/или сердечно-сосудистые повреждения (повышенное кровяное давление) по сравнению с группой, которую обрабатывали только носителем, или группой, которую обрабатывали носителем и лекарственным веществом, или группой, которую обрабатывали носителем и L-NAME.
Результаты теста представлены в таблице IV. Частоту появления (в %) желудочных повреждений определяли так же, как в тесте 1. Величины (в %) уровня GPT и кровяного давления отнесены к соответствующим величинам, найденным у животных 1-й из контрольных групп. Среднее значение кровяного давления в этой группе составило 105±8 мм рт.ст.
Полученные результаты свидетельствуют о том, что действие парацетамола, доксорубицина и симвастатина приводит к повреждению печени и гастроэнтеропатии (величины уровня GPT и частоты появления повреждений желудка (в %) превышают аналогичные величины у соответствующих групп, которым вводили лекарственное вещество без L-NAME, и контрольных групп, которым вводили L-NAME).
Следовательно, эти лекарственные вещества могут быть использованы для получения продуктов настоящего изобретения.
По результатам этого теста омепразол и мизопростол, напротив, не могут быть использованы для получения продуктов настоящего изобретения.
ПРИМЕР F4
Тест 4 - ингибирование образования радикалов из DPPH некоторыми веществами, предназначенными для использования в качестве предшественников В или B1 (см. формулы I и II настоящего изобретения).
Метод основан на колориметрическом тесте, в котором DPPH (2,2-дифенил-1-пикрилгидразил) используется в качестве соединения, образующего радикалы (M.S.Nenseter et al., Atheroscler. Thromb. 15, 1338-1344, 1995).
Сначала готовят метанольные растворы тестируемых соединений в конечных концентрациях 100 мкМ. 0,1 мл каждого их этих растворов добавляют к аликвотам метанольного раствора 0,1 М DPPH объемом 1 мл, а затем объем доводят до 1,5 мл. После выдерживания раствора при комнатной температуре без доступа света в течение 30 мин измеряют поглощение при длине волны 517 нм. Определяют понижение поглощения по отношению к поглощению раствора, содержащего такую же концентрацию DPPH.
Эффективность тестируемого соединения в отношении ингибирования образования радикалов, или антирадикальная активность, выражается следующей формулой:
(1-As/Ac)х·100, где As и Ас являются соответственно величинами поглощений раствора, содержащего тестируемое вещество и DPPH, и раствора, содержащего только DPPH.
Соединение отвечает условиям теста 4, если подавляет образование радикалов с эффективностью, равной или превышающей 50%.
В таблице V представлены результаты, полученные для следующих соединений: N-ацетилцистеин, цистеин, феруловая кислота, (L)-карнозин, гентизиновая кислота.
Таблица V демонстрирует, что N-ацетилцистеин, цистеин, феруловая кислота, (L)-карнозин, гентизиновая кислота отвечают условиям теста 4, так как они ингибируют образование радикалов из DPPH с эффективностью, превышающей 50%.
ПРИМЕР F5
Противовоспалительная активность и желудочная переносимость соединений настоящего изобретения по сравнению с соответствующими лекарственными веществами-предшественниками в условиях эндотелиальных дисфункций, вызванных действием L-NAME (метиловый эфир Nw-нитро-L-аргинина)
Была использована экспериментальная модель, описанная в Edwards et al, J.Patfaol. 134, 147-156, 1981.
Были образованы группы, содержащие по 10 крыс со средним весом 200 г. Животным в этих группах, за исключением одной контрольной группы, в течение двух недель вводили L-NAME, растворенный в питьевой воде (400 мг/л).
Лекарственные вещества вводили через ротовое отверстие, в дозе 10 мг/кг, используя в качестве носителя 1% карбоксиметилцеллюлозу в воде, 5 мл/кг.
Таким образом, животным из всех групп, за исключением описанных ниже контрольных групп, вводили лекарственное вещество, L-NAME и носитель.
Были образованы следующие контрольные группы.
Контрольная группа 1°. Обработка: носитель.
Контрольная группа 2°. Обработка: носитель + L-NAME.
В эксперименте использовались следующие лекарственные вещества: диклофенак и его соответствующий тиоэфир с остатком 4-нитроксибутирилпеницилламина (Пр.26), пироксикам и соответствующий эфир n-(4-нитрокси)бутирилоксиферуловой кислоты (Пр.25), ацетилсалициловая кислота и соответствующий тиоэфир с остатком N-ацетил-4-нитроксибутирилцистеина (Пр.24).
Спустя две недели после начала эксперимента животным последовательно вводили три инъекции воздуха (подкожно) в область спины в соответствии со следующей процедурой:
первая инъекция 20 мл,
спустя 3 дня после первой инъекции 10 мл,
спустя 6 дней после первой инъекции - ту же самую дозу, 10 мл.
Затем животным перестали давать пищу до утра следующего дня. За один час до подкожной инъекции каррагенина (2 мл 1% водного раствора каррагенина) в воспалительный эксудат животным вводили через ротовое отверстие носитель или одно из тестируемых соединений, растворенных или суспендированных в носителе. Спустя 6 часов после инъекции раствора каррагенина животные были умерщвлены. Воспалительный эксудат был собран и измерен для оценки инфильтрации лейкоцитов.
В таблице VI противовоспалительная активность выражена как ингибирование (в %) инфильтрации лейкоцитов по сравнению с величиной инфильтрации лейкоцитов у животных, которым вводили носитель и предварительно L-NAME, ингибирование (в %) повреждений желудочно-кишечного тракта оценивали так же, как ранее описано для теста 1 (Пр.1), и кровяное давление (в %) оценивали за один час до умерщвления и сравнивали с кровяным давлением в первой контрольной группе (обработка: носитель). В этой группе животных средняя величина давления крови составила 108±10 мм рт.ст.
Таблица VI демонстрирует, что соединения настоящего изобретения обладают такой же активностью, что и соответствующие предшественники в тесте противовоспалительной активности, но, в отличие от последних, они снижают повреждение сердечно-сосудистого эндотелия (менее значительное повышение кровяного давления (в %) по сравнению с таковым для соответствующего предшественника), а кроме того, лишь незначительно повреждают желудок, или совсем не повреждают.
ПРИМЕР F6
Во втором эксперименте по исследованию апоптоза проводили сравнение индометацина и тиоэфира индометацина, содержащего остаток N-ацетил-(4-нитрокси)бутирилцистеина (Пр.3), приготовленного в соответствии с настоящим изобретением. Результаты, представленные в таблице 3, свидетельствуют о том, что соединение настоящего изобретения ингибирует, в отличие от предшественника, апоптоз, вызванный гидропероксидом кумола (СIР).
ПРИМЕР F7
Желудочная переносимость некоторых лекарственных веществ, использованных в качестве предшественников, и соответствующих соединений настоящего изобретения.
Был повторен тест на повреждение желудочно-кишечного тракта (пример F5), за исключением того, что исключено предварительное введение животным L-NAME. Протестированные лекарства, введенные дозы, а также результаты теста представлены в таблице VII. Из таблицы видно, что уровень гастропатии значительно ниже в группах, которым вводили соединения настоящего изобретения, по сравнению с группами, которым вводили соответствующие предшественники.
ПРИМЕР 27
Синтез 4-нитроксибутилового эфира (S)-N-ацетил-S-[[1-[5-(2, 5-дигидро-5-оксо-3-фуранил)-3-метил-2-бензофуранил]этилокси]-4-оксобутаноил]цистеина, имеющего формулу

где предшественником является бенфуродил гемисукцинат (формула XXXI), а предшественником В является N-ацетилцистеин (формула СVIII)


Соединение синтезируют в соответствии с методом, описанным в примере 4. Выход 25%.
Элементный анализ
Рассчитано: С 54,19%; Н 5,20%; N 4,51%; S 5,17%.
Найдено: С 54, 25%; Н 5,22%; N 4,47%; S 5,15%.
ПРИМЕР 28
Синтез (8S-цис)-10-[(3-амино-2,3,6-тридезокси-α-L-ликсо-экзопиранозил)окси]-7,8,9,10-тетрагидро-6,8, 11-тригидрокси-8-[[[3-метокси-4-(4-нитроксибутаноил)фенил]-2-транс-пропеноилокси]метилоксо]-1-метокси-5,12-нафтацендиона, имеющего формулу

где предшественником является доксорубицин (формула ХХХII), а предшественником В является феруловая кислота (формула DII)


Соединение синтезируют в соответствии с методикой, описанной в примере 9. Выход 11%.
Элементный анализ
Рассчитано: С 57,88%; Н 4,98%; N 3,29%.
Найдено С: 57,91%; Н 5,02%; N 3,27%.
ПРИМЕР F8
Пример F1 был повторен с четырьмя группами крыс (в каждой группе по 10 животных), каждая из которых получала NEM, а также следующие вещества, вводимые орально:
а) контрольная группа: носитель, образованный водной суспензией 1% (мас./об.) карбоксиметилцеллюлозы;
b) одна группа (группа сравнения b), животным из которой в то же самое время вводили 5 мг/кг (0,014 ммоль/кг) индометацина, а также 2,3 мг/кг (0,014 ммоль/кг) N-ацетилцистеина в вышеупомянутом носителе;
с) одна группа (группа сравнения с), животным из которой в то же самое время вводили 6,6 мг/кг (0,014 ммоль/кг) 4-нитроксибутилового эфира индометацина, синтезированного в соответствии с методикой, опубликованной в WO 95/09831, а также 2,3 мг/кг (0,014 ммоль/кг) N-ацетилцистеина в вышеупомянутом носителе;
d) одна группа (группа d), животным из которой вводили 8,7 мг/кг (0,014 ммоль/кг) тиоэфира индометацина, содержащего остаток N-ацетил-(4-нитрокси)бутилцистеина (см. Пр.3), в вышеупомянутом носителе.
Результаты, представленные в таблице VIII, показывают, что смеси, вводимые соответственно животным групп b и с (группы сравнения), в отличие от соединений настоящего изобретения, вводимых животным группы d, были практически неэффективны (группа b) или значительно менее эффективны (группа с) в отношении уменьшения повреждений желудка.








Реферат
Изобретение относится к органической химии и может найти применение в медицине. Описываются соединения или их соли, имеющие следующую общую формулу (I):

где A=R-T1-, где R представляет собой радикал лекарственного вещества, такой как определено в формуле изобретения, T1=(CO), О, S, N или NR1c, где R1c представляет собой Н или C1-C5 алкил; В=-ТB -Х2-tBI, где ТB и tBI одинаковы или различны и выбраны из (СО), О, S, N или NR1c, где R1c такой, как определено выше, Х2 представляет собой бивалентную мостиковую группу, такую как соответствующий предшественник В, такой, как определено в формуле изобретения; С представляет собой бивалентный радикал -Tc-Y-, где Тс=(СО), О, S, N или NR1c, где R1c такой, как определено выше, Y имеет значения такие, как представлено в формуле изобретения. Также описывается фармацевтическая композиция на основе соединений формулы (I) для использования в случаях окислительного стресса и/или эндотелиальных дисфункций. Технический результат - получены новые соединения и их соли, обладающие полезными биологическими свойствами. 2 н. и 4 з.п.ф-лы, 8 табл.
Формула
































Документы, цитированные в отчёте о поиске
Производные 2-(2,6-дигалофениламино)фенилуксусной кислоты и способ их получения
Комментарии