Способ получения сополимера этилен-α-олефиндиена - RU2599626C2
Код документа: RU2599626C2
Описание
Область техники
Настоящее изобретение относится к способу получения сополимера этилен-α-олефиндиена и полученному таким способом сополимеру этилен-α-олефиндиена, и, более конкретно, к способу получения сополимера этилен-α-олефиндиена и полученному таким способом сополимеру этилен-α-олефиндиена с использованием соединения переходного металла на основе циклопента[b]флуоренильной группы в качестве катализатора.
Предшествующий уровень техники
В известном уровне техники так называемый катализатор Зиглера-Натта, состоящий из соединения титана или ванадия в качестве компонента первичного катализатора, и соединение алкилалюминия в качестве компонента сокатализатора обычно используются для получения гомополимеров этилена или сополимеров этилена и α-олефина. Несмотря на то, что каталитическая система Зиглера-Натта показывает высокую активность в полимеризации этилена, недостатком данной каталитической системы является широта молекулярно-массового распределения получаемого полимера ввиду неравномерной активации катализатора, и, в особенности, распределение его композиции является неравномерным в сополимерах этилена и α-олефина.
В недавнем времени были разработаны так называемые металлоценовые каталитические системы, состоящие из металлоценового соединения переходного металла 4 группы периодической таблицы элементов, такого как титан, цирконий и гафний, и метилалюминоксана в качестве сокатализатора. Металлоценовая каталитическая система представляет собой гомогенный катализатор с мономодальной активацией катализатора, который, таким образом, может обеспечить получение полиэтилена с более узким молекулярно-массовым распределением и более гомогенным распределением композиции по сравнению с существующей каталитической системой Зиглера-Натта. Например, согласно данным, приведенным в открытой публикации европейского патента 320762 и 372632; открытой публикации Японского Патента Sho 63-092621, Hei 02-84405 и Hei 03-2347, этилен может быть полимеризован с высокой активностью посредством активации метеллоценовых соединений, таких как Cp2TiCl2, Cp2ZrCl2, Cp2ZrMeCl, Cp2ZrMe2, этилен(IndH4)2ZrCl2, с использованием метилалюминоксана в качестве сокатализатора для получения полиэтилена с молекулярно-массовым распределением (Mw/Mn) в диапазоне от 1,5 до 2,0. Однако трудность представляет получение полимеров с высокой молекулярной массой с использованием указанной выше каталитической системы, а также при полимеризации в растворе, проводимой при высокой температуре, составляющей 100°C или выше, полимеризующая активность резко уменьшается, и преобладает β-дегидрирование. Таким образом, стало известно, что данная система является неподходящей для получения полимеров с высокой молекулярной массой, средневзвешенная молекулярная масса (Mw) которых составляет 100000 или более.
В то же время, были опубликованы данные о так называемых гео-ограничительных неметаллоценовых катализаторах (также обозначаемых как катализаторы одиночной активации), где переходные металлы соединены по кольцевому типу, в качестве катализаторов для получения полимеров с высокой молекулярной массой, имеющих высокую каталитическую активность в гомополимеризации этилена или сополимеризации этилена и α-олефина в условиях полимеризации в растворе. Согласно примерам Европейских патентов 0416815 и 0420436, амидная группа связана с одним циклопентадиеновым лигандом по кольцевом типу, и в Европейском патенте 0842939 приведен пример катализатора, где лиганд на основе фенола в качестве электродонорного соединения связан с циклопентадиеновым лигандом по кольцевом типу. Данный гео-ограничительный катализатор может значительно улучшить реакционную способность с более высокими α-олефинами благодаря сниженному эффекту стерической изоляции самого катализатора, но представляет трудности его коммерческого использования. Таким образом, важным является обеспечение более конкурентными каталитическими системами при необходимости коммерциализированных катализаторов на основе экономической целесообразности, то есть, с отличной высокотемпературной активностью, отличной реакционной способностью в отношении более высоких α-олефинов и способностью получать полимеры с высокой молекулярной массой.
Описание изобретения
Техническая проблема
С целью преодоления проблем известного уровня техники, авторы настоящего изобретения провели обширные исследования и обнаружили, что соединение переходного металла, имеющее структуру, где переходный металл 4 группы периодической таблицы элементов в качестве основного металла связан с циклопента[b]флуоренильной группой, имеющей жесткую плоскую структуру, не смотря на ее отсутствие в гетерокольце; имеет многочисленные широко нелокализованные электроны; и допускает заместитель, вносящий вклад в улучшение растворимости и результативность для легкого индуцирования в положении 9, посредством амидогруппы, замещенной силильной группой, было преимущественно при получении высокоэффективных полимеров и с высокой молекулярной массой в полимеризации сополимера этилен-α-олефиндиена, и таким образом осуществили настоящее изобретение.
Задачей настоящего изобретения является обеспечение способа получения сополимера этилен-α-олефиндиена, с использованием соединения переходного металла на основе циклопента[b]флуоренильной группы в качестве катализатора.
Другой задачей настоящего изобретения является обеспечение сополимера этилен-α-олефиндиена, полученного данным способом.
Решение проблемы
Аспект настоящего изобретения для решения указанных выше задач обеспечивает способ получения сополимера этилен-α-олефиндиена с использованием соединения переходного металла на основе циклопента[b]флуоренильной группы, представленного ниже химической формулой 1. Более конкретно, соединение переходного металла, имеющее структуру, где переходный металл 4 группы периодической таблицы элементов в качестве основного металла, связан с циклопента[b]флуоренильной группой, имеющей жесткую плоскую структуру, не смотря на ее отсутствие в гетерокольце; имеет многочисленные широко нелокализованные электроны; и допускает заместитель, вносящего вклад в улучшение растворимости и результативность для его индуцирования в положении 9, посредством амидогруппы, замещенной силильной группой, чтобы, таким образом, иметь преимущественную структуру для получения сополимеров этилен-α-олефиндиена с высокой эффективностью и высокой молекулярной массой, используется в качестве первичного катализатора.
[Химическая формула 1]

где в химической формуле 1 M представляет собой переходный металл 4 группы периодической таблицы элементов;
n равно целому числу 1 или 2, каждый R1 может быть одинаковым или различным, когда n равно 2;
R1 представляет собой водород, (C1-C50)алкил, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, -NRaRb, -SiRcRdRe или 5-7-членный N-гетероциклоалкил, содержащий, по меньшей мере, один атом азота;
R2 и R3, каждый независимо, представляет собой водород, (C1-C50)алкил, (C1-C50)алкокси, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арилокси, (C1-C50)алкил(C6-C30)арилокси, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арли)(C1-C50)алкил, -NRaRb или -SiRcRdRe;
R4, R5, R10, R11 и R12, каждый независимо, представляет собой (C1-C50)алкил, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, -NRaRb или -SiRcRdRe, и R11 и R12 могут быть связаны через (C4-C7)алкилен с образованием кольца;
R6, R7, R8 и R9, каждый независимо, представляет собой водород, (C1-C50)алкил, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C1-C50)алкокси, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, (C6-C30)арилокси, (C1-C50)алкил(C6-C30)арилокси, N-карбазолил, -NRaRb или -SiRcRdRe, или могут быть связаны с соседним заместителем через (C1-C5)алкилен с образованием кольца, и по меньшей мере, один -CH2- алкилена может быть замещен гетероатомом, выбранным из -O-, -S- и -NR'-, и алкилен может быть дополнительно замещен (C1-C50)алкилом;
арил R1-R12 может быть дополнительно замещен, по меньшей мере, одним заместителем, выбранным из группы, состоящей из (C1-C50)алкила, галоген(C1-C50)алкила, (C1-C50)алкокси, (C6-C30)арилокси, (C6-C30)арила, (C1-C50)алкил(C6-C30)арила и (C6-C30)арил(C1-C50)алкила;
R' и Ra-Re, каждый независимо, представляет собой (C1-C50)алкил или (C6-C30)арил; и
X1 и X2, каждый независимо, представляет собой галоген, (C1-C50)алкил, (C2-C50)алкенил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, (C1-C50)алкокси, (C6-C30)арилокси, (C1-C50)алкил(C6-C30)арилокси, (C1-C50)алкокси(C6-C30)арилокси, (C1-C50)алкилиден или анионный или дианионный лиганд, состоящий из 60 или менее атомов, содержащих N, P, O, S, Si и галоген, за исключением водорода, при условии, что один из X1 и X2 представляет собой дианионный лиганд, другим пренебрегают.
Пример соединения переходного металла на основе циклопента[b]флуоренильной группы, представленного выше химической формулой 1, может включать соединение переходного металла, представленное ниже химическими формулами 2 или 3:
[Химическая формула 2]

[Химическая формула 3]

где в химических формулах 2 и 3 M, R2-R12, X1 и X2 являются такими, как определено для химической формулы 1; R21 и R22, каждый независимо, представляет собой водород, (C1-C50)алкил, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, -NRaRb, -SiRcRdRe или 5-7-членный N-гетероциклоалкил, содержащий, по меньшей мере, один атом азота; арил R1 может быть дополнительно замещен, по меньшей мере, одним заместителем, выбранным из группы, состоящей из галогена, (C1-C50)алкила, галоген(C1-C50)алкила, (C1-C50)алкокси, (C6-C30)арилокси, (C6-C30)арила, (C1-C50)алкил(C6-C30)арила и (C6-C30)арил(C1-C50)алкила; и Ra-Re, каждый независимо, представляет собой (C1-C50)алкил или (C6-C30)арил.
Другой аспект настоящего изобретения для решения приведенных выше задач обеспечивает сополимер этилен-α-олефиндиена, полученный способом получения сополимера этилен-α-олефиндиена с использованием каталитической композиции переходного металла, содержащей соединение переходного металла.
Здесь и далее настоящее изобретение будет описано более подробно.
Переходный металл 4 группы периодической таблицы элементов M представляет собой предпочтительно титан (Ti), цирконий (Zr) или гафний (Hf).
Термин «алкил», используемый в настоящем описании, включает неразветвленный или разветвленный тип цепи.
Термин «арил», используемый в настоящем описании, означает органический радикал, являющийся производным ароматического углеводорода посредством удаления одного атома водорода, и может включать одиночное кольцо или конденсированное кольцо, содержащее 4-7 атомов кольца, и предпочтительно 5 или 6 атомов кольца. Их конкретные примеры включают, но не ограничиваются ими, фенил, нафтил, бифенил, антрил, флуоренил, фенантрил, трифениленил, пиренил, периленил, хризенил, нафтаценил, флуорантенил или т.п.
Например, (C1-C50)алкилом может быть метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, неопентил, амил, н-гексил, н-октил, н-децил, н-додецил, н-тетрадецил, н-гексадецил, н-пентадецил, н-октадецил, н-икозил или н-докозил; (C3-C50)циклоалкилом может быть, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил или циклододецил; (C6-C30)арилом или (C1-C50)алкил(C6-C30)арилом может быть, например, фенил, 2-толил, 3-толил, 4-толил, 2,3-ксилил, 2,4-ксилил, 2,5-ксилил, 2,6-ксилил, 3,4-ксилил, 3,5-ксилил, 2,3,4-триметилфенил, 2,3,5-триметилфенил, 2,3,6-триметилфенил, 2,4,6-триметилфенил, 3,4,5-триметилфенил, 2,3,4,5-тетраметилфенил, 2,3,4,6-тетраметилфенил, 2,3,5,6-тетраметилфенил, пентаметилфенил, этилфенил, н-пропилфенил, изопропилфенил, н-бутилфенил, втор-бутилфенил, трет-бутилфенил, н-пентилфенил, неопентилфенил, н-гексилфенил, н-октилфенил, н-децилфенил, н-додецилфенил, н-тетрадецилфенил, бифенил, флуоренил, трифенил, нафтил или антраценил; (C6-C30)арил(C1-C50)алкилом или ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкилом может быть, например, бензил, (2-метилфенил)метил, (3-метилфенил)метил, (4-метилфенил)метил, (2,3-диметилфенил)метил, (2,4-диметилфенил)метил, (2,5-диметилфенил)метил, (2,6-диметилфенил)метил, (3,4-диметилфенил)метил, (4,6-диметилфенил)метил, (2,3,4-триметилфенил)метил, (2,3,5-триметилфенил)метил, (2,3,6-триметилфенил)метил, (3,4,5-триметилфенил)метил, (2,4,6-триметилфенил)метил, (2,3,4,5-тетраметилфенил)метил, (2,3,4,6-тетраметилфенил)метил, (2,3,5,6-тетраметилфенил)метил, (пентаметилфенил)метил, (этилфенил)метил, (н-пропилфенил)метил, (изопропилфенил)метил, (н-бутилфенил)метил, (втор-бутилфенил)метил, (трет-бутилфенил)метил, (н-пентилфенил)метил, (неопентилфенил)метил, (н-гексилфенил)метил, (н-октилфенил)метил, (н-децилфенил)метил, (н-додецилфенил)метил, (н-тетрадецилфенил)метил, нафтилметил или антраценилметил; и (C1-C50)алкокси может быть, например, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси, неопентилокси, н-гексилокси, н-октилокси, н-додецилокси, н-пентадецилокси или н-эйкозилокси.
Предпочтительно, каждый R1 представляет собой независимо водород, метил, этил, н-пропил, изопропил, н-бутил, фенил, нафтил, бифенил, 2-изопропилфенил, 3,5-ксилил, 2,4,6-триметилфенил, бензил, диметиламино или пирролидино;
предпочтительно, R2 и R3 представляют собой независимо водород, метил, этил, н-пропил, изопропил, н-бутил, фенил, нафтил, бифенил, 2-изопропилфенил, 3,5-ксилил, 2,4,6-триметилфенил, бензил, метокси, этокси, изопропокси, фенокси, 4-трет-бутилфенокси или нафтокси;
предпочтительно, R4 и R5, каждый независимо, представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил, 2-метилбутил, втор-бутил, трет-бутил, н-пентил, неопентил, амил, н-гексил, н-октил, н-децил, н-додецил, н-тетрадецил, н-гексадецил, н-пентадецил, н-октадецил, н-икозил, н-докозил, фенил, 2-толил, 3-толил, 4-толил, 2,3-ксилил, 2,4-ксилил, 2,5-ксилил, 2,6-ксилил, 3,4-ксилил, 3,5-ксилил, 2,3,4-триметилфенил, 2,3,5-триметилфенил, 2,3,6-триметилфенил, 2,4,6-триметилфенил, 3,4,5-триметилфенил, 2,3,4,5-тетраметилфенил, 2,3,4,6-тетраметилфенил, 2,3,5,6-тетраметилфенил, пентаметилфенил, этилфенил, н-пропилфенил, изопропилфенил, н-бутилфенил, втор-бутилфенил, трет-бутилфенил, н-пентилфенил, неопентилфенил, н-гексилфенил, н-октилфенил, н-децилфенил, н-додецилфенил, н-тетрадецилфенил, бифенил, флуоренил, трифенил, нафтил, антраценил, бензил, (2-метилфенил)метил, (3-метилфенил)метил, (4-метилфенил)метил, (2,3-диметилфенил)метил, (2,4-диметилфенил)метил, (2,5-диметилфенил)метил, (2,6-диметилфенил)метил, (3,4-диметилфенил)метил, (4,6-диметилфенил)метил, (2,3,4-триметилфенил)метил, (2,3,5-триметилфенил)метил, (2,3,6-триметилфенил)метил, (3,4,5-триметилфенил)метил, (2,4,6-триметилфенил)метил, (2,3,4,5-тетраметилфенил)метил, (2,3,4,6-тетраметилфенил)метил, (2,3,5,6-тетраметилфенил)метил, (пентаметилфенил)метил, (этилфенил)метил, (н-пропилфенил)метил, (изопропилфенил)метил, (н-бутилфенил)метил, (втор-бутилфенил)метил, (трет-бутилфенил)метил, (н-пентилфенил)метил, (неопентилфенил)метил, (н-гексилфенил)метил, (н-октилфенил)метил, (н-децилфенил)метил, (н-додецилфенил)метил, (н-тетрадецилфенил)метил, нафтилметил, антраценилметил, 4-метоксифенил, 3,4-диметоксифенил или 4-(гексилокси)-3,5-диметилфенилом;
предпочтительно, R6-R9, каждый независимо, представляет собой водород, метил, этил, н-пропил, изопропил, н-бутил, изобутил, 2-метилбутил, втор-бутил, трет-бутил, н-пентил, неопентил, амил, н-гексил, н-октил, н-децил, н-додецил, н-пентадецил, фенил, 2-толил, 3-толил, 4-толил, 2,3-ксилил, 2,4-ксилил, 2,5-ксилил, 2,6-ксилил, 3,4-ксилил, 3,5-ксилил, 2,3,4-триметилфенил, 2,3,5-триметилфенил, 2,3,6-триметилфенил, 2,4,6-триметилфенил, 3,4,5-триметилфенил, 2,3,4,5-тетраметилфенил, 2,3,4,6-тетраметилфенил, 2,3,5,6-тетраметилфенил, пентаметилфенил, этилфенил, н-пропилфенил, изопропилфенил, н-бутилфенил, втор-бутилфенил, трет-бутилфенил, н-пентилфенил, неопентилфенил, н-гексилфенил, н-октилфенил, н-децилфенил, н-додецилфенил, н-тетрадецилфенил, бифенил, флуоренил, 2,7-ди-трет-бутил-9-п-толил-9H-флуорен-9-ил, трифенил, нафтил, антраценил, бензил, (2-метилфенил)метил, (3-метилфенил)метил, (4-метилфенил)метил, (2,3-диметилфенил)метил, (2,4-диметилфенил)метил, (2,5-диметилфенил)метил, (2,6-диметилфенил)метил, (3,4-диметилфенил)метил, (4,6-диметилфенил)метил, (2,3,4-триметилфенил)метил, (2,3,5-триметилфенил)метил, (2,3,6-триметилфенил)метил, (3,4,5-триметилфенил)метил, (2,4,6-триметилфенил)метил, (2,3,4,5-тетраметилфенил)метил, (2,3,4,6-тетраметилфенил)метил, (2,3,5,6-тетраметилфенил)метил, (пентаметилфенил)метил, (этилфенил)метил, (н-пропилфенил)метил, (изопропилфенил)метил, (н-бутилфенил)метил, (втор-бутилфенил)метил, (трет-бутилфенил)метил, (н-пентилфенил)метил, (неопентилфенил)метил, (н-гексилфенил)метил, (н-октилфенил)метил, (н-децилфенил)метил, (н-додецилфенил)метил, (н-тетрадецилфенил)метил, нафтилметил, антраценилметил, 4-метоксифенил, 3,4-диметоксифенил, метокси, этокси, изопропокси, н-бутокси, н-гексилокси, 2-метилбутил, фенокси, 4-трет-бутилфенокси, нафтокси, триметилсилил, трифенилсилил, диметиламино, дифениламино или 9H-карбазол-9-ил или может быть связан с соседним заместителем через

предпочтительно, R11 и R12, каждый независимо, представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил, 2-метилбутил, втор-бутил, трет-бутил, н-пентил, неопентил, амил, н-гексил, н-октил, н-децил, н-додецил, н-пентадецил, фенил, 2-толил, 3-толил, 4-толил, 2,3-ксилил, 2,4-ксилил, 2,5-ксилил, 2,6-ксилил, 3,4-ксилил, 3,5-ксилил, 2,3,4-триметилфенил, 2,3,5-триметилфенил, 2,3,6-триметилфенил, 2,4,6-триметилфенил, 3,4,5-триметилфенил, 2,3,4,5-тетраметилфенил, 2,3,4,6-тетраметилфенил, 2,3,5,6-тетраметилфенил, пентаметилфенил, этилфенил, н-пропилфенил, изопропилфенил, н-бутилфенил, втор-бутилфенил, трет-бутилфенил, н-пентилфенил, неопентилфенил, н-гексилфенил, н-октилфенил, н-децилфенил, н-додецилфенил, н-тетрадецилфенил, бифенил, флуоренил, трифенил, нафтил, антраценил, бензил, (2-метилфенил)метил, (3-метилфенил)метил, (4-метилфенил)метил, (2,3-диметилфенил)метил, (2,4-диметилфенил)метил, (2,5-диметилфенил)метил, (2,6-диметилфенил)метил, (3,4-диметилфенил)метил, (4,6-диметилфенил)метил, (2,3,4-триметилфенил)метил, (2,3,5-триметилфенил)метил, (2,3,6-триметилфенил)метил, (3,4,5-триметилфенил)метил, (2,4,6-триметилфенил)метил, (2,3,4,5-тетраметилфенил)метил, (2,3,4,6-тетраметилфенил)метил, (2,3,5,6-тетраметилфенил)метил, (пентаметилфенил)метил, (этилфенил)метил, (н-пропилфенил)метил, (изопропилфенил)метил, (н-бутилфенил)метил, (втор-бутилфенил)метил, (трет-бутилфенил)метил, (н-пентилфенил)метил, (неопентилфенил)метил, (н-гексилфенил)метил, (н-октилфенил)метил, (н-децилфенил)метил, (н-тетрадецилфенил)метил, нафтилметил, антраценилметил, 4-метоксифенил или 3,4-диметоксифенил, или R11 и R12 могут быть связаны друг с другом через бутилен или пентилен с образованием кольца; и
R10 представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил, 2-метилбутил, втор-бутил, трет-бутил, н-пентил, неопентил, амил, н-гексил, н-октил, н-децил, н-додецил, н-пентадецил, циклогексил, фенил, 2-толил, 3-толил, 4-толил, 2,3-ксилил, 2,4-ксилил, 2,5-ксилил, 2,6-ксилил, 3,4-ксилил, 3,5-ксилил, 2,3,4-триметилфенил, 2,3,5-триметилфенил, 2,3,6-триметилфенил, 2,4,6-триметилфенил, 3,4,5-триметилфенил, 2,3,4,5-тетраметилфенил, 2,3,4,6-тетраметилфенил, 2,3,5,6-тетраметилфенил, пентаметилфенил, этилфенил, н-пропилфенил, изопропилфенил, н-бутилфенил, втор-бутилфенил, трет-бутилфенил, н-пентилфенил, неопентилфенил, н-гексилфенил, н-октилфенил, н-децилфенил, н-додецилфенил, н-тетрадецилфенил, бифенил, флуоренил, трифенил, нафтил, антраценил, бензил, (2-метилфенил)метил, (3-метилфенил)метил, (4-метилфенил)метил, (2,3-диметилфенил)метил, (2,4-диметилфенил)метил, (2,5-диметилфенил)метил, (2,6-диметилфенил)метил, (3,4-диметилфенил)метил, (4,6-диметилфенил)метил, (2,3,4-триметилфенил)метил, (2,3,5-триметилфенил)метил, (2,3,6-триметилфенил)метил, (3,4,5-триметилфенил)метил, (2,4,6-триметилфенил)метил, (2,3,4,5-тетраметилфенил)метил, (2,3,4,6-тетраметилфенил)метил, (2,3,5,6-тетраметилфенил)метил, (пентаметилфенил)метил, (этилфенил)метил, (н-пропилфенил)метил, (изопропилфенил)метил, (н-бутилфенил)метил, (втор-бутилфенил)метил, (трет-бутилфенил)метил, (н-пентилфенил)метил, (неопентилфенил)метил, (н-гексилфенил)метил, (н-октилфенил)метил, (н-децилфенил)метил, (н-додецилфенил)метил, (н-тетрадецилфенил)метил, нафтилметил, антраценилметил, 2-метоксифенил или 3,4-диметоксифенил.
В определениях заместителей X1 и X2 примеры атома галогена могут включать атом фтора, хлора, брома и йода; примеры (C1-C50)алкила могут включать метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, неопентил, амил, н-гексил, н-октил, н-децил, н-додецил, н-пентадецил и н-эйкозил; примеры (C3-C50)циклоалкила могут включать циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и адамантил; примеры (C6-C30)арила могут включать фенил и нафтил; примеры (C6-C30)арил(C1-C50)алкила или ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкила могут включать бензил, (2-метилфенил)метил, (3-метилфенил)метил, (4-метилфенил)метил, (2,3-диметилфенил)метил, (2,4-диметилфенил)метил, (2,5-диметилфенил)метил, (2,6-диметилфенил)метил, (3,4-диметилфенил)метил, (4,6-диметилфенил)метил, (2,3,4-триметилфенил)метил, (2,3,5-триметилфенил)метил, (2,3,6-триметилфенил)метил, (3,4,5-триметилфенил)метил, (2,4,6-триметилфенил)метил, (2,3,4,5-тетраметилфенил)метил, (2,3,4,6-тетраметилфенил)метил, (2,3,5,6-тетраметилфенил)метил, (пентаметилфенил)метил, (этилфенил)метил, (н-пропилфенил)метил, (изопропилфенил)метил, (н-бутилфенил)метил, (втор-бутилфенил)метил, (трет-бутилфенил)метил, (н-пентилфенил)метил, (неопентилфенил)метил, (н-гексилфенил)метил, (н-октилфенил)метил, (н-децилфенил)метил, (н-додецилфенил)метил, (н-тетрадецилфенил)метил, нафтилметил и антраценилметил; примеры (C1-C50)алкокси могут включать метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси, неопентилокси, н-гексилокси, н-октилокси, н-додецилокси, н-пентадецилокси и н-эйкозилокси; примеры (C6-C30)арилокси могут включать фенокси, 4-трет-бутилфенокси или 4-метоксифенокси, анионным или дианионным лигандом, состоящим из 60 или менее атомов, содержащих N, P, O, S, Si и галоген, за исключением водорода, может быть -OSiRfRgRh, -SRi [Rf-Ri, каждый независимо, представляет собой (C1-C50)алкил, (C6-C30)арил, (C3-C50)циклоалкил], -NRjRk или -PRlRm [Rj-Rm, каждый независимо, представляет собой (C1-C50)алкил, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, (C3-C20)циклоалкил, три(C1-C50)алкилсилил или три(C6-C30)арилсилил]. Примеры -OSiRfRgRh могут включать триметилсилокси, триэтилсилокси, три-н-пропилсилокси, триизопропилсилокси, три-н-бутилсилокси, три-втор-бутилсилокси, три-трет-бутилсилокси, триизобутилсилокси, трет-бутилдиметилсилокси, три-н-пентилсилокси, три-н-гексилсилокси или трициклогексилсилокси; примеры -NRjRk могут включать диметиламино, диэтиламино, ди-н-пропиламино, диизопропиламино, ди-н-бутиламино, ди-втор-бутиламино, ди-трет-бутиламино, диизобутиламино, трет-бутилизопропиламино, ди-н-гексиламино, ди-н-октиламино, ди-н-дециламино, дифениламино, дибензиламино, метилэтиламино, метилфениламино, бензилгексиламино, бистриметилсилиламино или бис-трет-бутилдиметилсилиламино; примеры -PRlRm могут включать диметилфосфин, диэтилфосфин, ди-н-пропилфосфин, диизопропилфосфин, ди-н-бутилфосфин, ди-втор-бутилфосфин, ди-трет-бутилфосфин, диизобутилфосфин, трет-бутилизопропилфосфин, ди-н-гексилфосфин, ди-н-октилфосфин, ди-н-децилфосфин, дифенилфосфин, дибензилфосфин, метилэтилфосфин, метилфенилфосфин, бензилгексилфосфин, бистриметилсилилфосфин и бис-трет-бутилдиметилсилилфосфин; примеры -SRiмогут включать метилтио, этилтио, пропилтио, изопропилтио, 1-бутилтио или изопентилтио.
X1 и X2, каждый независимо, представляет собой фтор, хлор, бром, метил, этил, изопропил, амил, бензил, метокси, этокси, изопропокси, трет-бутокси, фенокси, 4-трет-бутилфенокси, триметилсилокси, трет-бутилдиметилсилокси, диметиламино, дифениламино, диметилфосфино, диэтилфосфин, дифенилфосфин, этилтио или изопропилтио.
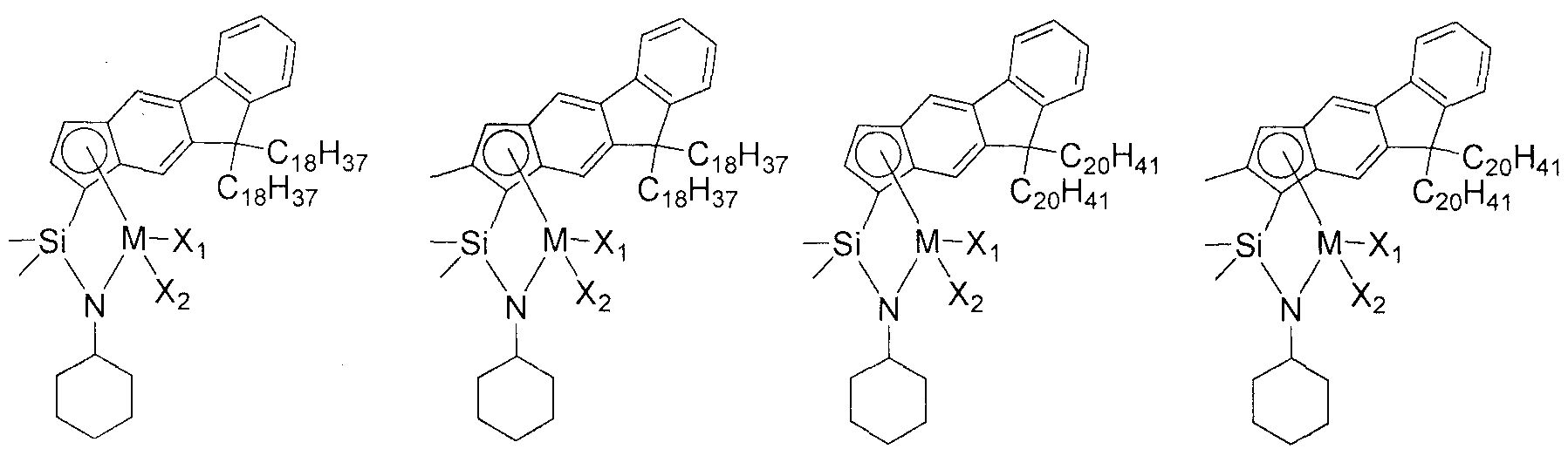
Приведенное выше соединение переходного металла может быть выбрано из соединений, имеющих, но, не ограничиваясь ими, структуры, представленные ниже:













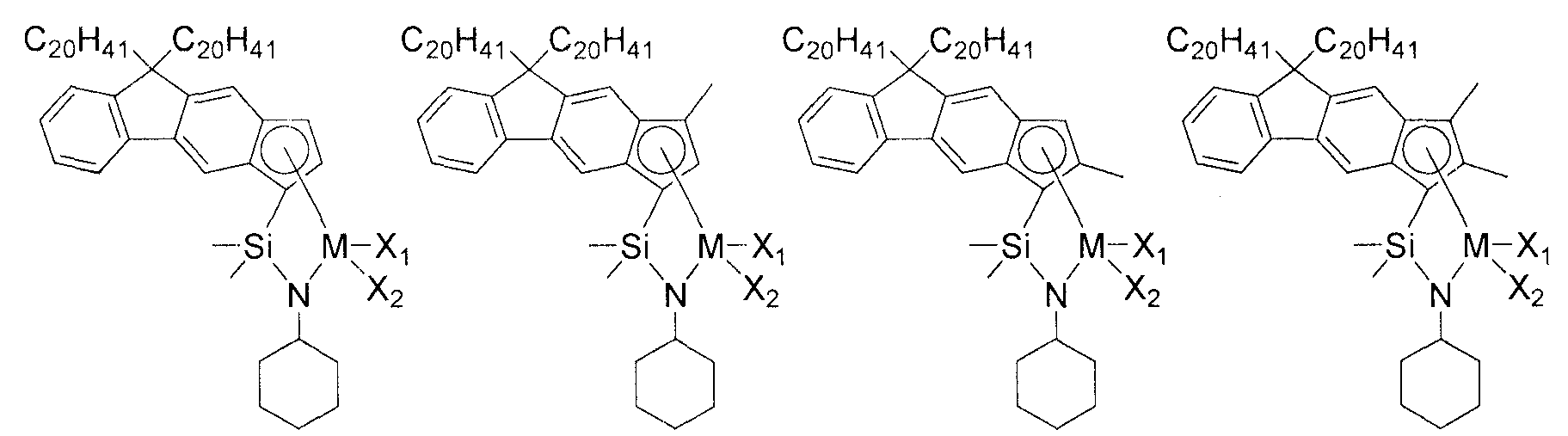











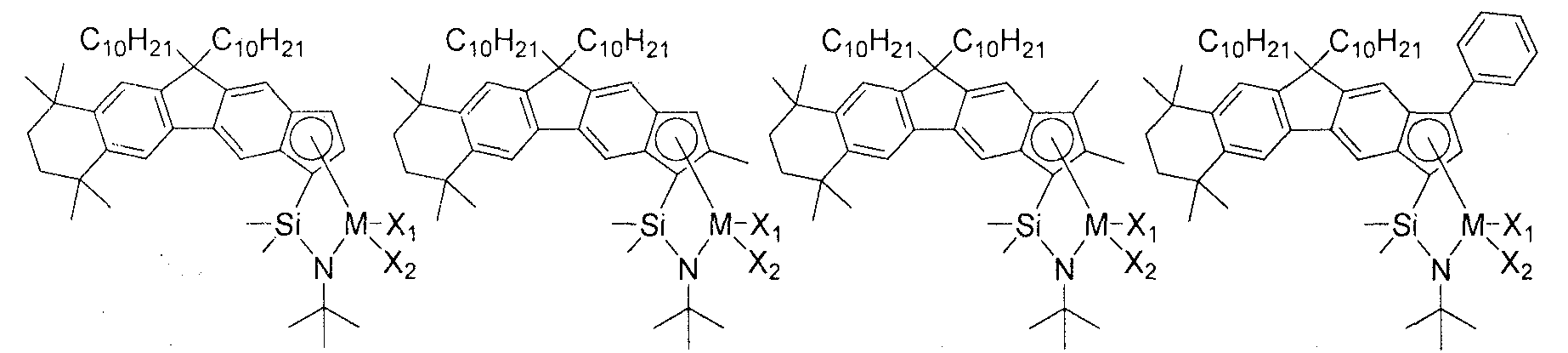










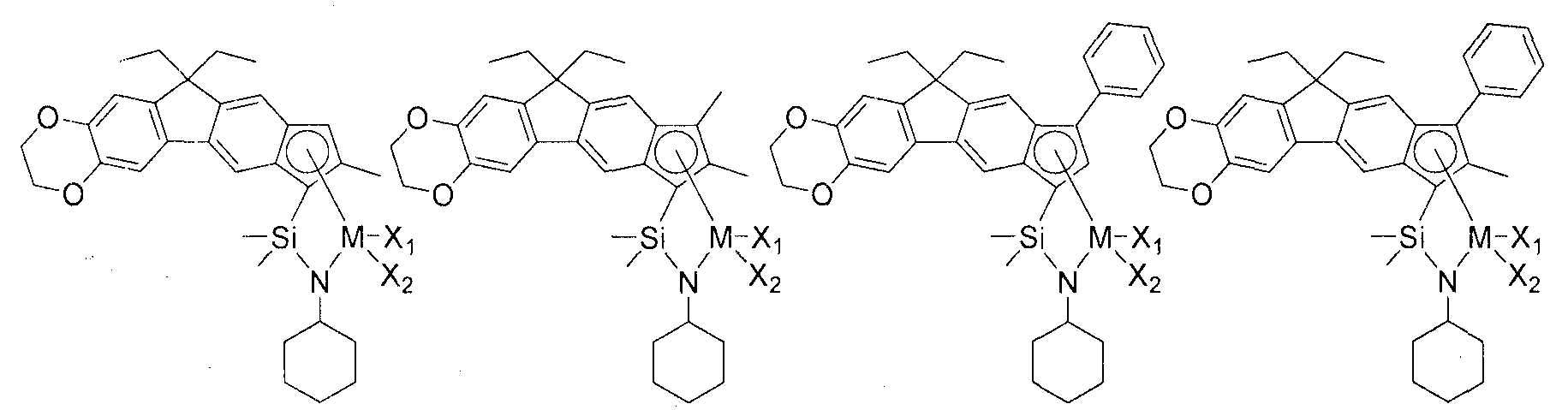







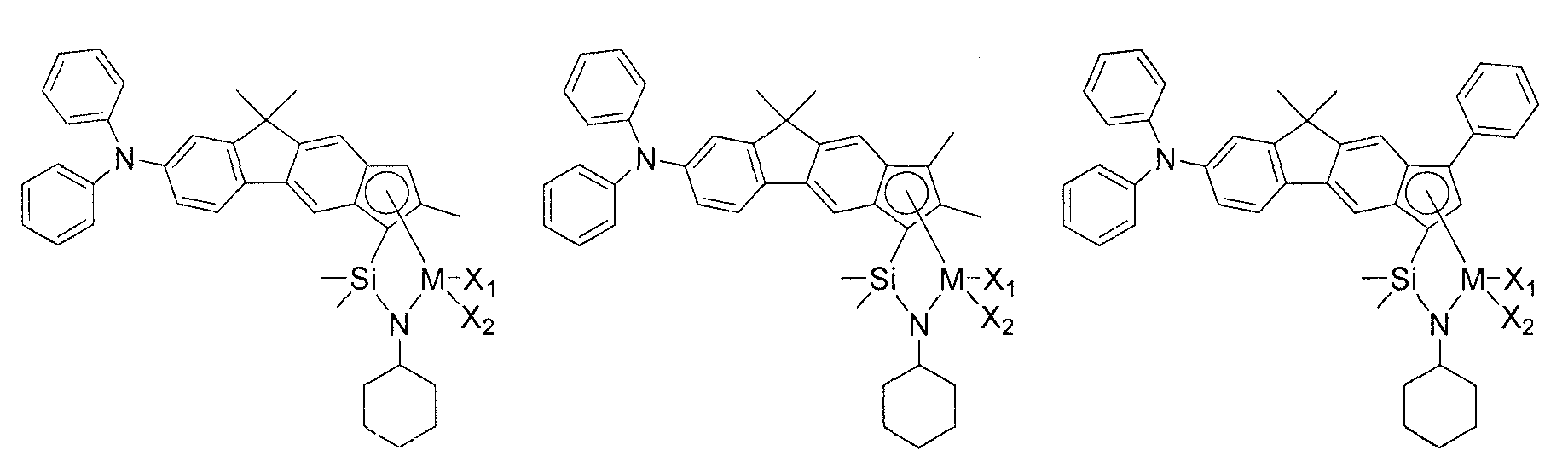




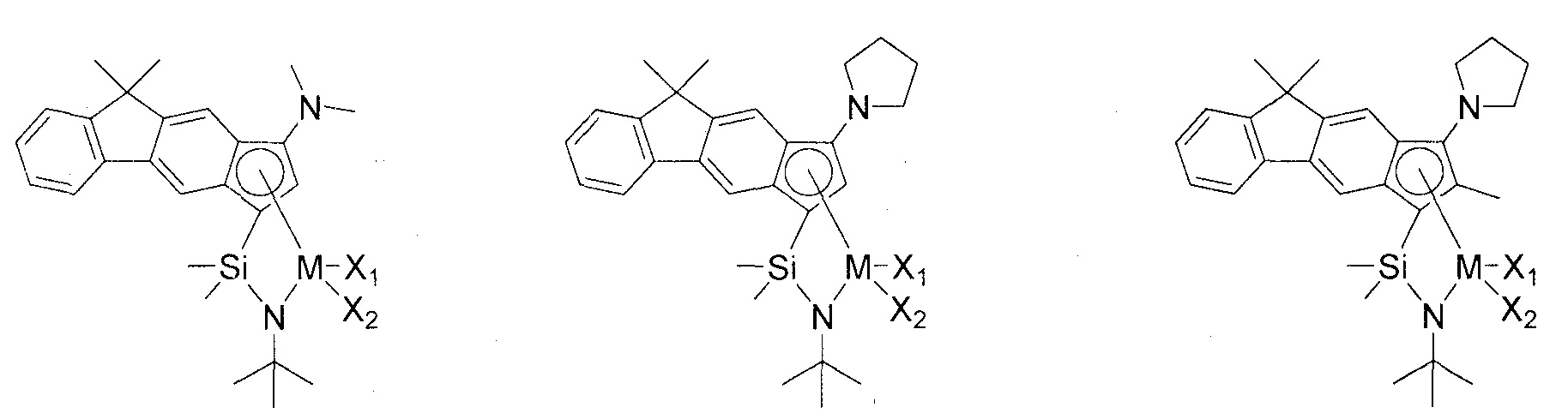
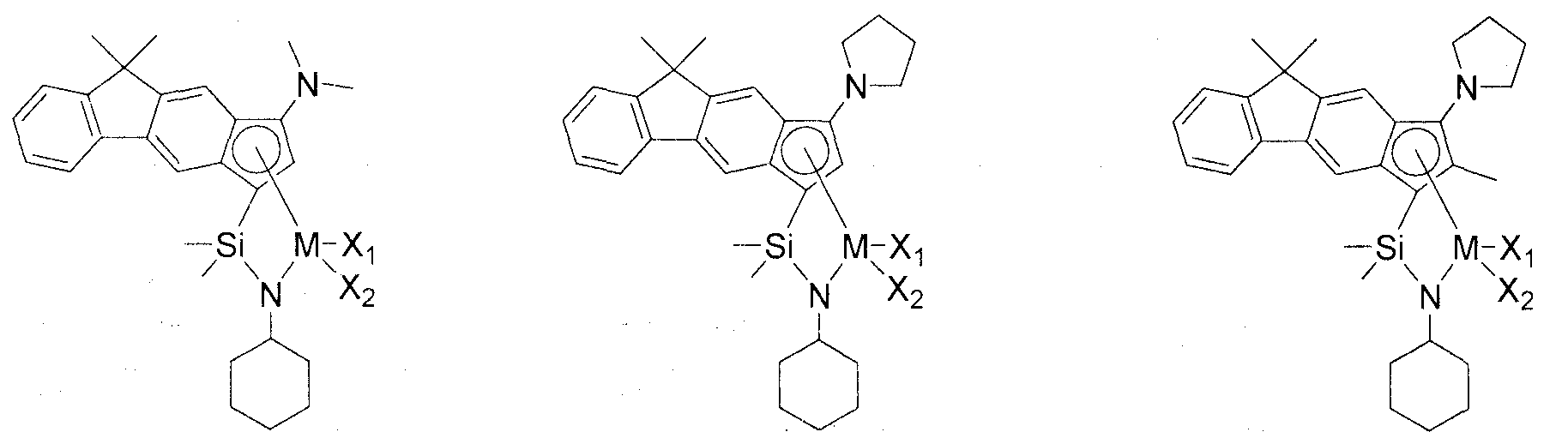




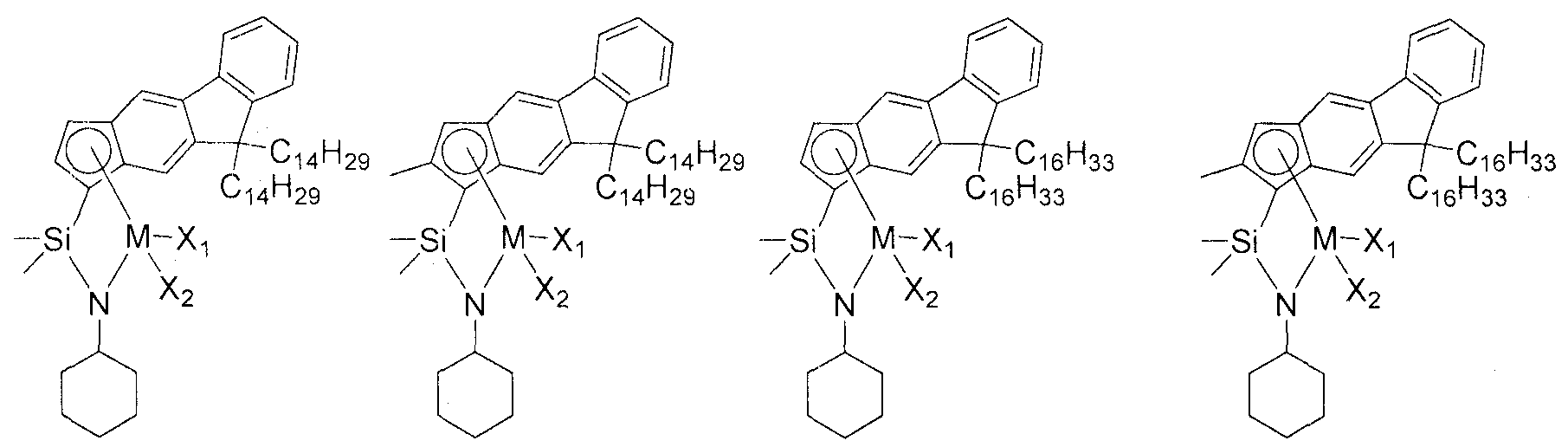




















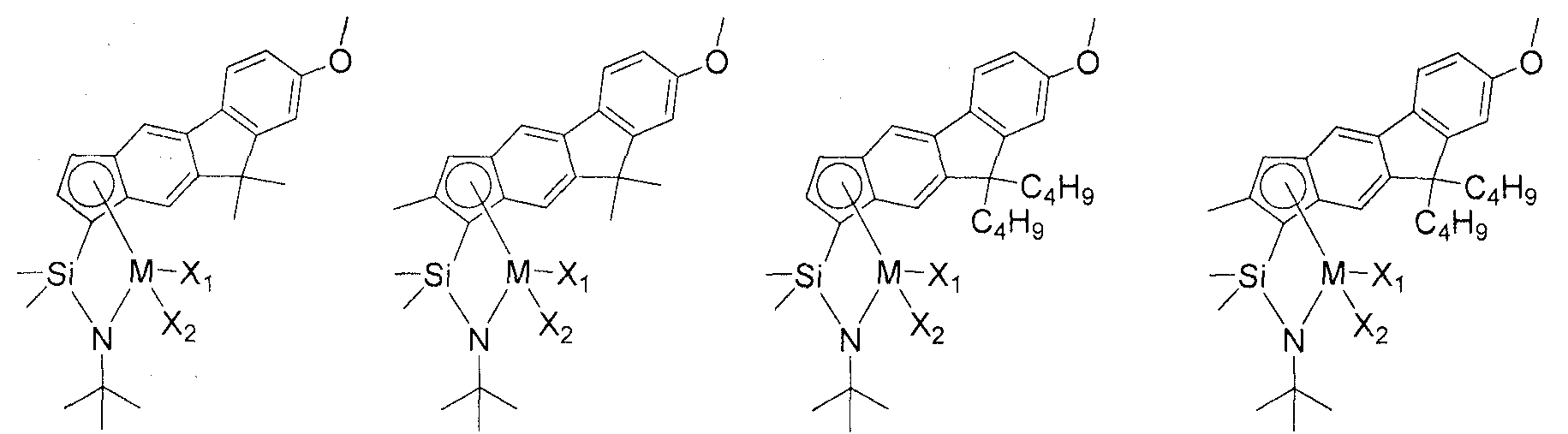



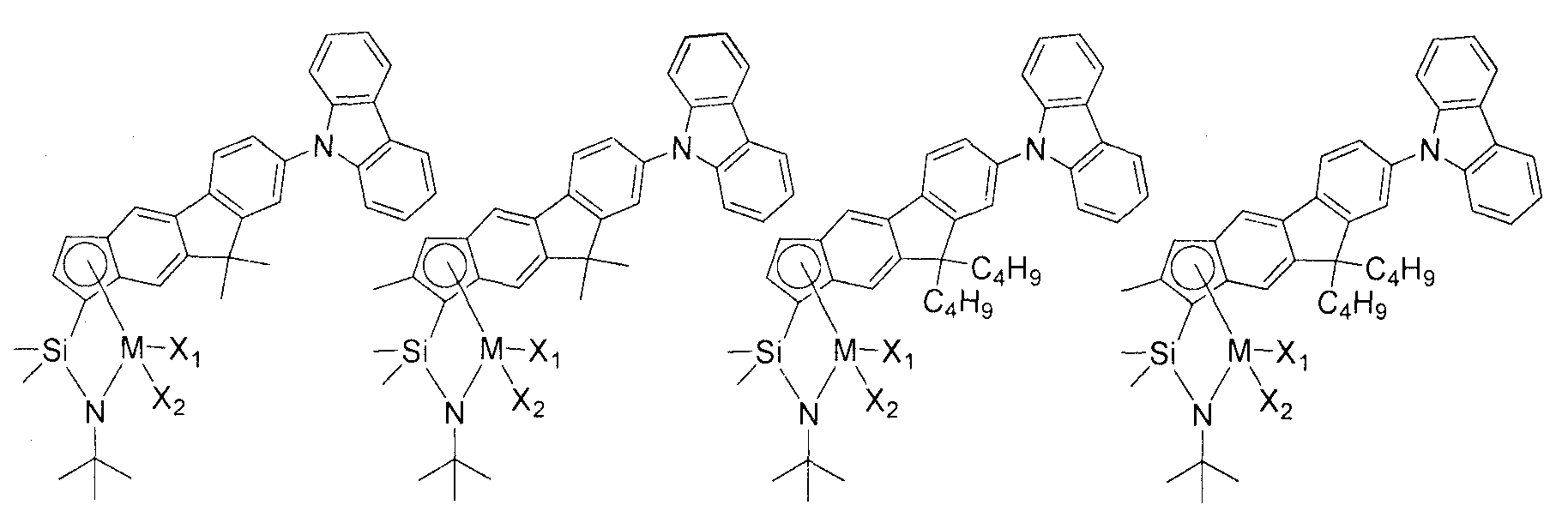







M представляет собой Ti, Zr или Hf; и X1 и X2, каждый является таким, как определено для химической формулы 1.
При этом для того, чтобы быть активным компонентом катализатора для применения в получении сополимера этилен-α-олефиндиена, соединение переходного металла указанной выше химической формулы 1 может предпочтительно действовать в качестве сокатализатора вместе с соединением алюминия, соединением бора или их смесью, которое может извлекать лиганд X1 или X2 из соединения переходного металла для того, чтобы катионировать основной металл и выступать в качестве противоиона, имеющего слабую прочность связи, то есть, аниона.
Иными словами, каталитическая композиция переходного металла может, предпочтительно, дополнительно содержать сокатализатор, выбранный из соединения алюминия, соединения бора или их смеси, а также соединения переходного металла химической формулы 1.
Соединение бора, используемое в качестве сокатализатора в настоящем изобретении, известно из патента США 5198401 и может быть выбрано из соединения бора, представленного ниже химическими формулами 4-6:
[Химическая формула 4]
B(R41)3
[Химическая формула 5]
[R42]+[B(R41)4]-
[Химическая формула 6]
[(R43)pZH]+[B(R41)4]-
В химических формулах 4-6 B представляет собой атомом бора;
R41 представляет собой фенил, и фенил может быть дополнительно замещен 3-5 заместителями, выбранными из атома фтора, (C1-C50)алкила, замещенного или незамещенного атомом фтора, или (C1-C50)алкокси, замещенного или незамещенного атомом фтора;
R42 представляет собой (C5-C7)ароматический радикал или (C1-C50)алкил(C6-C20)арильный радикал, (C6-C30)арил(C1-C50)алкильный радикал, например, трифенилметильный радикал;
Z представляет собой атомом азота или фосфора;
R43 представляет собой (C1-C50)алкильный радикал или радикал анилиния, замещенный атомом азота и двумя (C1-C50)алкильными группами; и p равно целому числу 2 или 3.
Предпочтительные примеры сокатализатора на основе бора могут включать трис(пентафторфенил)боран, трис(2,3,5,6-тетрафторфенил)боран, трис(2,3,4,5-тетрафторфенил)боран, трис(3,4,5-трифторфенил)боран, трис(2,3,4-трифторфенил)боран, фенилбис(пентафторфенил)боран, тетракис(пентафторфенил)борат, тетракис(2,3,5,6-тетрафторфенил)борат, тетракис(2,3,4,5-тетрафторфенил)борат, тетракис(3,4,5-тетрафторфенил)борат, тетракис(2,2,4-трифторфенил)борат, фенилбис(пентафторфенил)борат и тетракис(3,5-бистрифторметилфенил)борат. Кроме того, их некоторые комбинированные примеры могут включать
тетракис(пентафторфенил)борат ферроцения, тетракис(пентафторфенил)борат 1,1′-диметилферроцения, тетракис(пентафторфенил)борат, трифенилметилтетракис(пентафторфенил)борат, трифенилметилтетракис(3,5-бистрифторметилфенил)борат, тетракис(пентафторфенил)борат триэтиламмония, тетракис(пентафторфенил)борат трипропиламмония, тетракис(пентафторфенил)борат три(н-бутил)аммония, тетракис(3,5-бистрифторметилфенил)борат три(н-бутил)аммония, тетракис(пентафторфенил)борат N,N-диметиланилиния, тетракис(пентафторфенил)борат N,N-диэтиланилиния, тетракис(пентафторфенил)борат N,N-2,4,6-пентаметиланилиния, тетракис(3,5-бистрифторметилфенил)борат N,N-диметиланилиния, тетракис(пентафторфенил)борат диизопропиламмония, тетракис(пентафторфенил)борат дициклогексиламмония, тетракис(пентафторфенил)борат трифенилфосфония, тетракис(пентафторфенил)борат три(метилфенил)фосфония и тетракис(пентафторфенил)борат три(диметилфенил)фосфония. В том числе, предпочтительными являются тетракис(пентафторфенил)борат N,N-диметиланилиния, трифенилметилтетракис(пентафторфенил)борат и трис(пентафторфенил)боран.
В настоящем изобретении соединения алюминия, используемые в качестве сокатализатора, могут быть выбраны из соединений алюминоксана химических формул 7 или 8, органических соединений алюминия химической формулы 9 или органических соединений гидрокарбилоксида алюминия химических формул 10 или 11:
[Химическая формула 7]
(-Al(R51)-O-)m
[Химическая формула 8]
(R51)2Al-(-O(R51)-)q-(R51)2
[Химическая формула 9]
(R52)rAl(E)3-r
[Химическая формула 10]
(R53)2AlOR54
[Химическая формула 11]
R53Al(OR54)2
где в химических формулах 7-11 R51 представляет собой (C1-C50)алкил, предпочтительно метил или изобутил; m и q, каждый независимо, равен целому числу от 5 до 20; R52 и R53, каждый независимо, представляет собой (C1-C50)алкил; E представляет собой атом водорода или галогена; r равно целому числу от 1 до 3; и R54 представляет собой (C1-C50)алкил или (C6-C30)арил.
Конкретные примеры соединения алюминия могут включать соединения алюминоксана, такие как метилалюминоксан, модифицированный метилалюминоксан, тетраизобутилалюминоксан; органические соединения алюминия, такие как триалкилалюминий, включая триметилалюминий, триэтилалюминий, трипропилалюминий, триизобутилалюминий и тригексилалюминий; хлорид диалкилалюминия, включая хлорид диметилалюминия, хлорид диэтилалюминия, хлорид дипропилалюминия, хлорид диизобутилалюминия и хлорид дигексилалюминия; дихлорид алкилалюминия, включая дихлорид метилалюминия, дихлорид этилалюминия, дихлорид пропилалюминия, дихлорид изобутилалюминия и дихлорид гексилалюминия; и гидрид диалкилалюминия, включая гидрид диметилалюминия, гидрид диэтилалюминия, гидрид дипропилалюминия, гидрид диизобутилалюминия и гидрид дигексилалюминия. В том числе, предпочтительным является триалкилалюминий, и более предпочтительными являются триэтилалюминий и триизобутилалюминий.
В каталитической композиции переходного металла, содержащей как первичный катализатор, так и сокатализатор, соединение переходного металла в качестве первичного катализатора и сокатализатор имеют предпочтительно молярное отношение переходного металла (M):атом бора (B):атом алюминия (Al) в диапазоне 1:0~100:1~2,000, и более предпочтительно 1:0,5~5:10~500. Указанное выше соотношение способствует получению сополимера этилен-α-олефиндиена, и диапазон соотношения может варьировать в зависимости от чистоты реакции.
Способ получения сополимера этилен-α-олефиндиена с использованием каталитической композиции переходного металла можно осуществить посредством приведения в контакт катализатора переходного металла, сокатализатора и сополимеров этилена, α-олефина и мономера диена, в присутствии подходящего органического растворителя. В данном случае, соединение переходного металла (первичный катализатор) и компоненты сокатализатора могут быть раздельно поданы в реактор, или эти компоненты могут быть предварительно смешаны и затем поданы в реактор. Условия смешивания, такие как порядок подачи, температура или концентрация, не являются особенно ограниченными.
Предпочтительные примеры органических растворителей, используемых в способе получения, могут включать (C3-C20) углеводород, и их особые примеры могут включать бутан, изобутан, пентан, гексан, гептан, октан, изооктан, нонан, декан, додекан, циклогексан, метилциклогексан, бензол, толуол, ксилол и т.п.
Сополимер этилен-α-олефиндиена, полученный по настоящему изобретению, характеризуется вязкостью по Муни (ASTM D1646-94, ML1+4 при 125°C) всего сополимера в диапазоне от 1 до 250, и содержит от 30 до 85% масс. этилена, от 1 до 15% масс. диена, и восстановлением α-олефина.
Конкретно, давление в реакторе для получения сополимера этилен-α-олефиндиена составляет 1~1000 атм, и более предпочтительно 6~150 атм. Также, преимущественно, температура реакции полимеризации может составлять от 25°C до ~200°C и предпочтительно от 50°C до ~180°C.
В качестве сомономера α-олефина, используемого в настоящем изобретении, можно использовать α-олефин с прямой или разветвленной цепью (C3-C18), (C5-C20) циклоолефин, стирол или производные стирола. Предпочтительные примеры α-олефина с прямой или разветвленной цепью (C3-C18) могут включать пропилен, 1-бутен, 1-пентен, 4-метил-1-пентен, 1-гексен, 1-октен, 1-децен, 1-ундецен, 1-додецен, 1-тетрадецен, 1-гексадецен, 1-октадецен и т.п.; предпочтительные примеры (C5-C20) циклоолефина могут включать циклопентен, циклогексен, норборнен, фенилнорбонен и т.п.; и предпочтительные примеры стирола и его производных могут включать стирол, α-метилстирол, п-метилстирол, 3-хлорметилстирол и т.п.
Мономер диена, используемый в настоящем изобретении, имеет две двойные связи, и для этого можно использовать C4~C20 диолефин с прямой и разветвленной цепью или C5~C20 циклодиолефин. Предпочтительные примеры C4~C20 диолефина с прямой или разветвленной цепью могут включать 1,3-бутадиен, 1,4-пентадиен, 2-метил-1,3-бутадиен, 1,4-гексадиен, 1,5-гексадиен, 1,5-гептадиен, 1,6-гептадиен, 1,6-октадиен, 1,7-октадиен, 1,7-нонадиен, 1,8-нонадиен, 1,8-декадиен, 1,9-декадиен, 1,12-тетрадекадиен, 1,13-тетрадекадиен, 3-метил-1,4-гексадиен, 3-метил-1,5-гексадиен, 3-этил-1,4-гексадиен, 3-этил-1,5-гексадиен, 3,3-диметил-1,4-гексадиен, 3,3-диметил-1,5-гексадиен и т.п.; и предпочтительные примеры C5~C20 циклодиолефина могут включать циклопентадиен, циклогексадиен, 5-винил-2-норборнен, 2,5-норборнадиен, 7-метил-2,5-норборнадиен, 7-этил-2,5-норборнадиен, 7-пропил-2,5-норборнадиен, 7-бутил-2,5-норборнадиен, 7-фенил-2,5-норборнадиен, 7-гексил-2,5-норборнадиен, 7,7-диметил-2,5-норборнадиен, 7-метил-7-этил-2,5-норборнадиен, 7-хлор-2,5-норборнадиен, 7-бром-2,5-норборнадиен, 7-фтор-2,5-норборнадиен, 7,7-дихлор-2,5-норборнадиен, 1-метил-2,5-норборнадиен, 1-этил-2,5-норборнадиен, 1-пропил-2,5-норборнадиен, 1-бутил-2,5-норборнадиен, 1-хлор-2,5-норборнадиен, 1-бром-2,5-норборнадиен, 5-изопропил-2-норборнен, 5-винилиден-2-норборнен (VNB), 5-метилен-2-норборнен (MNB), 5-этилиден-2-норборнен (ENB) и т.п.
Как описано выше, эластомер этилен/пропилен/диена (EPDM) может быть отлично получен с использованием катализатора по настоящему изобретению. В особенности, ввиду легкости инъецирования дорогостоящего диена, продукты EPDM с вязкостью по Муни (ASTM D1646-94, ML1+4 при 125°C), скорректированной в диапазоне от 1 до 250, и предпочтительно от 10 до 200, можно легко получить экономически выгодным образом.
Кроме того, при получении сополимера этилен-α-олефиндиена по настоящему изобретению водород можно использовать в качестве регулятора молекулярной массы с целью регулирования молекулярной массы.
Как правило, при осуществлении полимеризации в растворе при высокой температуре, как описано выше, катализатор может трансформироваться или разрушаться по причине повышения температуры, которая может вызвать понижение активности катализатора, и, таким образом, невозможно получить полимеры с желаемыми физическими свойствами. Однако, поскольку каталитическая композиция, предлагаемая настоящим изобретением, присутствует в гомогенном состоянии в реакторе полимеризации, каталитическую композицию можно предпочтительно использовать в процессе полимеризации в растворе, проводимом при температуре выше температуры плавления соответствующего полимера. Однако, как описано в патенте США 4752597, соединение переходного металла и сокатализатор могут быть закреплены на пористом металлооксидном носителе, чтобы быть использованными, таким образом, для суспензионной полимеризации или процесса газофазной полимеризации в качестве гетерогенной каталитической композиции.
Полезный эффект изобретения
Как указано выше, в способе получения сополимера этилен-α-олефиндиена по настоящему изобретению соединение переходного металла на основе циклопента[b]флуоренильной группы используют в качестве катализатора полимеризации, и, таким образом, сополимеры этилен-α-олефиндиена с высоким содержанием диена, высоким коэффициентом конверсии и высокой степенью вязкости по Муни можно получать в высокотемпературных (120°C или выше) условиях полимеризации с высоким выходом. Более того, каталитическую композицию, содержащую соединение переходного металла, можно легко получить с высоким выходом синтеза экономически выгодным образом. Кроме того, соединение переходного металла или каталитическая композиция по настоящему изобретению может иметь отличную сополимеризующую реакционную способность в отношении других олефинов, сохраняя при этом высокую каталитическую активность даже при высокой температуре благодаря их отличной термостабильности, и позволяет получить полимеры с высокой молекулярной массой с высоким выходом, приводя к более высокой коммерческой целесообразности, по сравнению с уже известными металлоценовыми и неметаллоценовыми катализаторами с единственной точкой активации.
Осуществление изобретения
Далее в настоящем описании будут подробно описаны варианты осуществления настоящего изобретения со ссылкой на сопутствующие примеры, которые не предназначены для ограничения объема изобретения.
Если не указано иное, все эксперименты для синтеза лигандов и катализаторов проводили в атмосфере азота с использованием стандартных методов Schlenk или перчаточной камеры. Органические растворители, использованные в реакции, подвергали кипячению с обратным холодильником над металлическим натрием и бензофеноном для удаления влаги, и затем незамедлительно перегоняли перед использованием. Анализы1H-ЯМР синтезированных лигандов и катализаторов проводили с использованием Bruker 500 МГц при комнатной температуре.
Перед использованием н-гептан в качестве растворителя для полимеризации пропускали через трубку, наполненную молекулярным ситом 5Å и активированным оксидом алюминия, и барботировали азотом высокой степени очистки, чтобы, таким образом, удалить в достаточной мере влагу, кислород и другие вещества, отравляющие катализатор. Полимеризованные полимеры анализировали методами измерения, описанными ниже.
1. Показатель текучести расплава(MI)
Измерение проводили в соответствии с ASTM D 2839.
2. Плотность
Измерение проводили с использованием градиентных трубок, в соответствии с ASTM D 1505.
3. Температура плавления (Tm)
Измерение проводили в условиях 2го нагревания при скорости 10°C/мин в атмосфере азота с использованием Dupont DSC 2910.
4. Молекулярная масса и молекулярно-массовое распределение
Измерение проводили при 135°C при скорости 1,0 мл/мин в присутствии 1,2,3-трихлорбензольного растворителя с использованием PL210 GPC, оборудованного PL Mixed-BX2+preCol, и молекулярную массу калибровали с использованием PL полистирольных стандартов.
5. Содержание α-Олефин (% масс.) в сополимере
Измерение проводили с использованием смешанного растворителя 1,2,4-трихлорбензол/C6D6 (7/3 по массе) при 120°C в13C-ЯМР режиме посредством Bruker DRX500 ЯМР спектрометра при 125 МГц. (Ссылка: Randal, J. C. JMS-Rev. Macromol. Chem. Phys. 1980, C29, 201.)
Соотношение содержания этилена и α-олефина и диена в EPDM полимерах количественно определяли с использованием инфракрасного спектрометра.
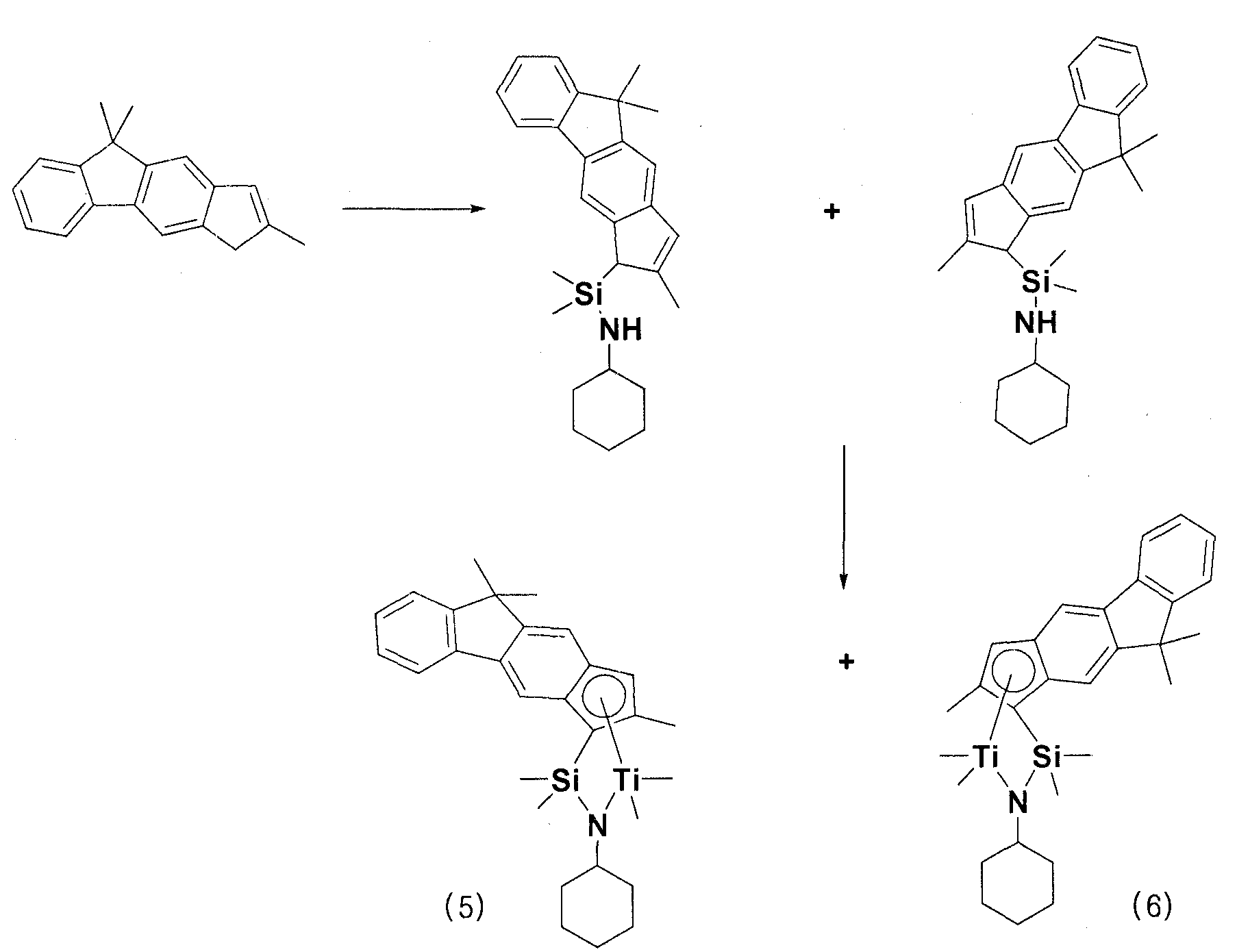
[Пример получения 1] Получение смеси комплекса 1 и комплекса 2

Синтез 9,9-дигексил-9H-флуорена
В 2000 мл круглодонную колбу загружали 9H-флуорен (50 г, 300,1 ммоль) и трет-бутоксид калия (77,0 г, 721,9 ммоль), и затем туда медленно вводили 700 мл ДМСО. К полученному медленно через капельную воронку добавляли 1-бромогексан (119 г, 721,9 ммоль) в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 24 часов, и реакцию завершали добавлением 500 мл дистиллированной воды. Органический слой, собранный экстракцией н-гексаном, сушили над сульфатом магния, с последующим удалением летучих веществ и затем очищали н-гексаном с использованием колоночной хроматографии на силикагеле, с последующей сушкой и длительным выдерживанием при комнатной температуре, с получением, таким образом, 90,0 г 9,9-дигексил-9H-флуорена (выход: 72,40%) в виде твердого вещества.
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 0,625-0,628 (м, 4H), 0,759-0,785 (м, 6H), 1,050-1,125 (м, 12H), 1,953-1,983 (т, 4H), 7,293-7,340 (м, 6H), 7,706-7,720 (д, 2H).
Синтез 9,9-дигексил-2-метил-2,3-дигидроциклопента[b]флуорен-1(9H)-она
В 2000 мл круглодонную колбу загружали 9,9-дигексил-9H-флуорен (79 г, 236,2 ммоль) и 2-бром-2-метилпропионилбромид (54,3 г, 236,2 ммоль) и затем растворяли введенными туда 600 мл дисульфида углерода. Затем реактор охлаждали ледяной водой. В атмосфере азота к полученному медленно десятью порциями добавляли трихлорид алюминия (78,7 г, 590,4 ммоль) в течение 2 часов. Смесь перемешивали при комнатной температуре в течение 8 часов, и затем реакцию завершали добавлением 500 мл дистиллированной воды, с последующей трех разовой промывкой 500 мл дистиллированной воды. Органический слой сушили над сульфатом магния, с последующим удалением летучих веществ и сушкой, с получением, таким образом, 89,0 г 9,9-дигексил-2-метил-2,3-дигидроциклопента[b]флуорен-1(9H)-она (выход: 93,6%) в виде масла высокой вязкости.
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 0,601-0,627 (м, 4H), 0,741-0,774 (м, 6H), 1,000-1,126 (м, 12H), 1,366-1,380 (д, 3H), 1,961-2,202 (м, 4H), 2,789-2,801 (д, 2H), 3,445-3,498 (м, 1H), 7,375-7,383 (м, 3H), 7,731 (с, 2H), 7,764-7,779 (д, 1H).
Синтез 9,9-дигексил-2-метил-1,2,3,9-тетрагидроциклопента[b]флуорен-1-ола
В 1000 мл круглодонной колбе 9,9-дигексил-2-метил-2,3-дигидроциклопента[b]флуорен-1(9H)-он (85 г, 211,1 ммоль) растворяли в 400 мл ТГФ и 400 мл этанола и затем перемешивали. Боргидрид натрия (NaBH4) (10 г, 265,0 ммоль) пятью порциями добавляли к реакционному продукту и затем перемешивали в течение 12 часов. Полученную смесь после удаления растворителя растворяли в этилацетате и затем промывали водой три раза. Органический слой сушили над сульфатом магния, с последующим удалением летучих веществ и сушкой, с получением, таким образом, 82,0 г 9,9-дигексил-2-метил-1,2,3,9-тетрагидроциклопента[b]флуорен-1-ола (выход: 96,0%) (два изомера) в виде масла высокой вязкости.
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 0,628-0,631 (м, 8H), 0,762-0,788 (м, 12H), 1,109-1,136 (м, 24H), 1,198-1,212 (д, 3H), 1,314-1,327 (д, 3H), 1,522-1,535 (д, 1H), 1,830-1,846 (д, 1H), 1,956-1,963 (м, 8H), 2,323-2,352 (м, 1H), 2,525-2,572 (м, 1H), 2,628-2,655 (м, 1H), 2,733-2,779 (м, 1H), 3,011-3,057 (м, 1H), 3,164-3,210 (м, 1H), 4,783-4,812 (т, 1H), 5,052-5,077 (т, 1H), 7,289-7,380 (м, 8H), 7,525 (с, 1H), 7,558 (с, 1H), 7,672-7,685 (д, 2H).
Синтез 9,9-дигексил-2-метил-3,9-дигидроциклопента[b]флуорена
В 500 мл круглодонной колбе 9,9-дигексил-2-метил-1,2,3,9-тетрагидроциклопента[b]флуорен-1-ол (80 г, 197,7 ммоль) и п-толуолсульфоновую кислоту (0,2 г) растворяли в 320 мл толуола, и затем воду полностью удаляли кипячением с обратным холодильником с использованием аппарата Dean-Stark. Полученное в результате вещество охлаждали до комнатной температуры, и затем туда инъецировали водный раствор хлорида аммония (150 мл) и 200 мл простого диэтилового эфира, с последующим разделением органического слоя. Органический слой, собранный экстракцией остатка простым диэтиловым эфиром, сушили над сульфатом магния, с последующим удалением летучих веществ и затем очищали с использованием колоночной хроматографии на силикагеле, с получением, таким образом, 74,0 г 9,9-дигексил-2-метил-3,9-дигидроциклопента[b]флуорена (выход: 96,8%).
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 0,611-0,671 (м, 4H), 0,755-0,784 (м, 6H), 1,041-1,140 (м, 12H), 1,943-1,976 (м, 4H), 2,200 (с, 3H), 3,373 (с, 2H), 6,556 (с, 1H), 7,208-7,381 (м, 4H), 7,653-7,668 (д, 1H), 7,700 (с, 1H).
Синтез N-трет-бутил-1-(9,9-дигексил-2-метил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-трет-бутил-1-(9,9-дигексил-2-метил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина
В 500 мл круглодонной колбе 9,9-дигексил-2-метил-3,9-дигидроциклопента[b]флуорен (40,0 г, 103,5 ммоль) растворяли в 320 мл простого диэтилового эфира, и затем температуру снижали до -78°C. Затем туда медленно инъецировали н-бутиллитий (2,5M раствор гексана, 42 мл), с последующим перемешиванием при комнатной температуре в течение 12 часов. После удаления летучих веществ в вакууме, 350 мл н-гексана добавляли к смеси для снижения температуры реактора до -78°C, с последующим добавлением дихлордиметилсилана (40 г). Температуру снова повышали до комнатной температуры, с последующим перемешиванием в течение 24 часов, и затем соли удаляли фильтрованием. Затем летучие вещества удаляли в вакууме. Продукт снова вводили в 500 мл круглодонную колбу и растворяли в 320 мл простого диэтилового эфира. Температуру снижали до -78°C, и к полученному добавляли трет-бутиламин (22,7 г, 310,4 ммоль). Температуру повышали до комнатной температуры, с последующим перемешиванием в течение 12 часов, и затем летучие вещества полностью удаляли в вакууме. Затем 200 мл н-гексана добавляли для растворения полученного в результате вещества, и соли удаляли фильтрованием. Растворитель удаляли, с получением, таким образом, 48 г смеси N-трет-бутил-1-(9,9-дигексил-2-метил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-трет-бутил-1-(9,9-дигексил-2-метил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина (соотношение=~1:1), (выход: 88,9%) в виде вязкого вещества.
1H-ЯМР (500 МГц, C6D6, м.д.): δ 0,132 (с, 3H), 0,177-0,198 (д, 6H), 0,270 (с, 1H), 0,804-0,879 (м, 12H), 0,973-1,295 (м, 50H), 2,170-2,348 (м, 14H), 3,398-3,428 (д, 2H), 6,745 (с, 2H), 7,337-7,434 (м, 6H), 7,518-7,908 (м, 6H)
Синтез (трет-бутиламидо)диметил(9,9-дигексил-2-метил-3,9-дигидроциклопента[b]флуорен-3-ил)силантитан(IV)диметила (комплекс 1) и (трет-бутиламидо)диметил(9,9-дигексил-2-метил-1,9-дигидроциклопента[b]флуорен-1-ил)силантитан(IV)диметила (комплекс 2)
В 250 мл круглодонной колбе смесь N-трет-бутил-1-(9,9-дигексил-2-метил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-трет-бутил-1-(9,9-дигексил-2-метил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина (в соотношении=~1:1) (8,64 г, 16,75 ммоль) растворяли в 130 мл простого диэтилового эфира, и затем температуру снижали до -78°C. Затем к полученному медленно инъецировали метиллитий (1,5M раствор простого диэтилового эфира, 49,4 мл). Температуру повышали до комнатной температуры, с последующим перемешиванием в течение 12 часов, с получением литиевой соли. В дополнение, в сухой камере TiCl4 (16,75 ммоль) и 150 мл безводного н-гексана вводили в 500 мл круглю колбу, и затем температуру снижали до -78°C. Затем туда медленно добавляли полученную литиевую соль. Температуру снова повышали до комнатной температуры, с последующим перемешиванием в течение 4 часов, и растворитель удаляли в вакууме. Полученное в результате вещество растворяли в н-гексане, и затем фильтрат извлекали фильтрованием. Снова растворитель удаляли в вакууме, с получением, таким образом, 8,1 г смеси комплекса 1 и комплекса 2 (соотношение приблизительно 1:1), в виде твердого вещества.
1H-ЯМР (500 МГц, C6D6, м.д.): δ 0,079-0,091 (д, 6H), 0,623-0,645 (д, 6H), 0,813-1,336 (м, 56H), 1,601-1,619 (д, 18H), 2,071-2,514 (м, 14H), 7,025-7,035 (д, 2H), 7,330-8,099 (м, 12H).
[Пример получения 2] Получение смеси комплекса 3 и комплекса 4

Синтез 9,9-диметил-9H-флуорена
В 2000 мл круглодонную колбу загружали 9H-флуорен (50 г, 300,1 ммоль) и трет-бутоксид калия (77,0 г, 721,9 ммоль), и затем туда медленно инъецировали 700 мл ДМСО. В атмосфере азота йодометан (113,5 г, 800 ммоль) медленно капали через капельную воронку, при этом температуру реактора поддерживали при 10°C или ниже. Смесь перемешивали при комнатной температуре в течение 24 часов, и реакцию завершали добавлением 500 мл дистиллированной воды. Органический слой, собранный экстракцией н-гексаном, сушили над сульфатом магния, с последующим удалением летучих веществ и очищали н-гексаном с использованием трубки колоночной хроматографии на силикагеле, с последующей сушкой, с получением, таким образом, 47,5 г 9,9-диметил-9H-флуорена (выход: 81,50%) в виде белого твердого вещества.
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 1,547 (с, 6H), 7,368-7,393 (т, 4H), 7,488-7,499 (д, 2H), 7,777-7,791 (д, 2H).
Синтез 2,9,9-триметил-2,3-дигидроциклопента[b]флуорен-1(9H)-она
В 2000 мл круглодонную колбу загружали 9,9-диметил-9H-флуорен (50 г, 257,4 ммоль) и 2-бром-2-метилпропионилбромид (61,0 г, 265,1 ммоль) и затем растворяли введенными 700 мл дисульфида углерода. Затем реактор охлаждали ледяной водой. В атмосфере азота десятью партиями туда медленно добавляли трихлорид алюминия (85,8 г, 643,4 ммоль) в течение 2 часов. Смесь перемешивали при комнатной температуре в течение 8 часов, и затем реакцию завершали добавлением 500 мл дистиллированной воды. Полученную в результате смесь разбавляли добавлением 500 мл метилхлорида и три раза промывали 500 мл дистиллированной воды. Органический слой сушили над сульфатом магния, с последующим удалением летучих веществ и сушкой, и затем перекристаллизовывали с использованием метилхлорида и метанола, с получением, таким образом, 64,0 г 2,9,9-триметил-2,3-дигидроциклопента[b]флуорен-1(9H)-она (выход: 94,8%) в виде белого твердого вещества.
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 1,354-1,369 (д, 3H), 1,517 (с, 6H), 2,784-2,811 (д, 2H), 3,444-3,496 (м, 1H), 7,376-7,429 (м, 2H), 7,471-7,485 (д, 2H), 7,763 (с, 1H), 7,795-7,808 (д, 2H), 7,832 (с, 1H).
Синтез 2,9,9-триметил-3,9-дигидроциклопента[b]флуорена
В 1000 мл круглодонной колбе 2,9,9-триметил-2,3-дигидроциклопента[b]флуорен-1(9H)-он (50 г, 190,6 ммоль) растворяли в 400 мл ТГФ и 400 мл этанола, и затем перемешивали. Боргидрид натрия (NaBH4) (9,4 г, 247,8 ммоль) добавляли к реакционному продукту пятью партиями, и затем перемешивали в течение 12 часов. Полученную в результате смесь после удаления растворителя растворяли в этилацетате и затем три раза промывали водой. Органический слой сушили над сульфатом магния, с последующим удалением летучих веществ. Высушенный реакционный продукт растворяли в 320 мл толуола и затем вводили в 500 мл круглодонную колбу. Затем туда вводили п-толуолсульфоновую кислоту (0,2 г), и воду полностью удаляли кипячением с обратным холодильником с использованием аппарата Dean-Stark. Полученное в результате вещество охлаждали до комнатной температуры, и затем туда инъецировали водный раствор хлорида аммония (150 мл) и 200 мл простого диэтилового эфира, с последующим разделением органического слоя. Органический слой, собранный экстракцией остатка простым диэтиловым эфиром, сушили над сульфатом магния, с последующим удалением летучих веществ и затем очищали с использованием колоночной хроматографии на силикагеле, с получением, таким образом, 42,0 г 2,9,9-триметил-3,9-дигидроциклопента[b]флуорена (выход: 89,42%).
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 1,515 (с, 6H), 2,203 (с, 3H), 3,375 (с, 2H), 6,559 (с, 1H), 7,279-7,332 (м, 3H), 7,425-7,440 (д, 1H), 7,697-7,711 (д, 1H), 7,740 (с, 1H).
Синтез N-трет-бутил-1-(2,9,9-триметил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-трет-бутил-1-(2,9,9-триметил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина
В 500 мл круглодонной колбе 2,9,9-триметил-3,9-дигидроциклопента[b]флуорен (15,0 г, 60,9 ммоль) растворяли в 300 мл простого диэтилового эфира, и затем температуру снижали до -78°C. Затем туда медленно инъецировали н-бутиллитий (2,5M раствор гексана, 24,8 мл), с последующим перемешиванием при комнатной температуре в течение 12 часов. После удаления летучих веществ в вакууме, 350 мл н-гексана добавляли к смеси для понижения температуры реактора до -78°C, с последующим добавлением дихлордиметилсилана (23 г). Температуру снова повышали до комнатной температуры, с последующим перемешиванием в течение 24 часов, и затем соли удаляли фильтрованием. Затем летучие вещества удаляли в вакууме. Продукт снова вводили в 500 мл круглодонную колбу и растворяли в 320 мл простого диэтилового эфира. Температуру снижали до -78°C, и туда добавляли трет-бутиламин (16,1 г, 152,2 ммоль). Температуру повышали до комнатной температуры, с последующим перемешиванием в течение 12 часов, и летучие вещества полностью удаляли в вакууме. Затем 200 мл толуола добавляли для растворения полученного в результате вещества, и соли удаляли фильтрованием. Растворитель удаляли, с получением, таким образом, 21,0 г смеси N-трет-бутил-1-(2,9,9-триметил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-трет-бутил-1-(2,9,9-триметил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина (выход: 91,8%) в виде вязкого вещества.
1H-ЯМР (500 МГц, C6D6, м.д.): δ 0,085-0,098 (д, 6H), 0,229-0,253 (д, 6H), 0,555 (с, 2H), 1,161-1,179 (д, 18H), 1,534-1,559 (д, 12H), 2,304 (с, 6H), 3,385-3,422 (д, 2H), 6,747 (с, 2H), 7,303-8,049 (м, 12H).
Синтез (трет-бутиламидо)диметил(2,9,9-триметил-3,9-дигидроциклопента[b]флуорен-3-ил)силантитан(IV)диметила (комплекс 3) и (трет-бутиламидо)диметил(2,9,9-триметил-1,9-дигидроциклохлорпента[b]флуорен-1-ил)силантитан(IV)диметила (комплекс 4)
В 250 мл круглодонной колбе смесь N-трет-бутил-1-(2,9,9-триметил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-трет-бутил-1-(2,9,9-триметил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина (10,4 г, 27,69 ммоль) растворяли в 200 мл простого диэтилового эфира, и затем температуру снижали до -78°C. Затем туда медленно инъецировали метиллитий (1,5M раствор простого диэтилового эфира, 75,6 мл). Температуру повышали до комнатной температуры, с последующим перемешиванием в течение 12 часов, с получением литиевой соли. В дополнение, в сухой камере TiCl4 (5,25 г, 27,69 ммоль) и 150 мл безводного н-гексан вводили в 500 мл круглодонную колбу, и затем температуру снижали до -78°C. Затем туда медленно добавляли полученную литиевую соль. Снова температуру повышали до комнатной температуры, с последующим перемешиванием в течение 4 часов, и растворитель удаляли в вакууме. Полученное в результате вещество снова растворяли в толуоле, и затем нерастворившуюся часть удаляли фильтрованием. Снова толуол удаляли в вакууме, с получением, таким образом, 10,8 г смеси комплекса 3 и комплекса 4 в виде твердого вещества.
1H-ЯМР (500 МГц, C6D6, м.д.): δ (-0,019)-(-0,010) (д, 6H), 0,641-0,647 (д, 6H), 0,794-2,212 (м, 48H), 7,004-7,025 (д, 2H), 7,106-8,092 (м, 12H).
[Пример получения 3] Получение смеси комплекса 5 и комплекса 6
Синтез N-циклогексил-1-(2,9,9-триметил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-циклогексил-1-(2,9,9-триметил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина
В 250 мл круглодонной колбе 2,9,9-триметил-3,9-дигидроциклопента[b]флуорен (7,5 г, 30,5 ммоль) растворяли в 300 мл простого диэтилового эфира, и затем температуру снижали до -78°C. Затем туда медленно инъецировали н-бутиллитий (2,5M раствор гексана, 12,4 мл), с последующим перемешиванием при комнатной температуре в течение 12 часов. После удаления летучих веществ в вакууме 200 мл н-гексана добавляли к смеси для снижения температуры реактора до -78°C, с последующим добавлением дихлордиметилсилана (11,8 г, 91,4 ммоль). Температуру снова повышали до комнатной температуры, с последующим перемешиванием в течение 24 часов, и затем соли удаляли фильтрованием. Затем летучие вещества удаляли в вакууме. Продукт снова вводили в 200 мл круглодонную колбу и растворяли в 150 мл простого диэтилового эфира. Температуру снижали до -78°C, и туда добавляли циклогексанамин (9,05 г, 91,4 ммоль). Температуру повышали до комнатной температуры, с последующим перемешиванием в течение 12 часов, и летучие вещества полностью удаляли в вакууме. Затем 100 мл толуола добавляли для растворения полученного в результате вещества, и соли удаляли фильтрованием. Растворитель удаляли, с получением, таким образом, 10,6 г смеси N-циклогексил-1-(2,9,9-триметил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-циклогексил-1-(2,9,9-триметил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина в виде вязкого вещества.
Синтез (циклогексиламидо)диметил(2,9,9-триметил-3,9-дигидроциклопента[b]флуорен-3-ил)силантитан(IV)диметила (комплекс 5) и (циклогексиламидо)диметил(2,9,9-триметил-1,9-дигидроциклохлорпента[b]флуорен-1-ил)силантитан(IV)диметила (комплекс 6)
В 250 мл трехгорлой круглодонной колбе хорошо высушенную смесь N-циклогексил-1-(2,9,9-триметил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-циклогексил-1-(2,9,9-триметил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина (10,6 г, 26,39 ммоль) растворяли в 200 мл простого диэтилового эфира, и затем температуру снижали до -78°C. Затем туда медленно инъецировали метиллитий (1,5M раствор простого диэтилового эфира, 72,1 мл). Температуру повышали до комнатной температуры, с последующим перемешиванием в течение 12 часов, с получением литиевой соли. В дополнение, в сухой камере TiCl4 (5,00 г, 26,39 ммоль) и 150 мл безводного н-гексана водили в 500 мл круглодонную колбу, и затем температуру снижали до -78°C. Затем туда медленно добавляли полученную литиевую соль. Снова температуру повышали до комнатной температуры, с последующим перемешиванием в течение 4 часов, и растворитель удаляли в вакууме. Полученное в результате вещество снова растворяли в толуоле, и затем нерастворившуюся часть удаляли фильтрованием. Снова толуол удаляли в вакууме, с получением, таким образом, 11,5 г смеси комплекса 5 и комплекса 6 в виде твердого вещества.
1H-ЯМР (500 МГц, C6D6, м.д.): δ (-0,070)-(-0,049) (д, 6H), 0,628-0,634 (д, 6H), 0,764-2,195 (м, 50H), 4,779 (м, 2H), 6,985-7,002 (д, 2H), 7,100-8,095 (м, 12H).
[Пример получения 4] Получение смеси комплекса 7 и комплекса 8

Синтез 9,9-дитетрадецил-9H-флуорена
В 2000 мл круглодонную колбу загружали 9H-флуорен (15 г, 90,24 ммоль) и трет-бутоксид калия (21,2 г, 198,5 ммоль), и затем туда медленно инъецировали 300 мл ДМСО. В атмосфере азота 1-бромотетрадекан (54 г, 198,5 ммоль) медленно капали через капельную воронку, при этом температуру реактора поддерживали при 10°C или ниже. Смесь перемешивали при комнатной температуре в течение 24 часов, и реакцию завершали добавлением 500 мл дистиллированной воды. Органический слой, собранный экстракцией н-гексаном, сушили над сульфатом магния, с последующим удалением летучих веществ и очищали н-гексаном с использованием трубки колоночной хроматографии на силикагеле, с последующей сушкой, с получением, таким образом, 42,0 г 9,9-дитетрадецил-9H-флуорена (выход: 83,26%) в виде белого твердого вещества.
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 0,616-0,634 (м, 4H), 0,881-0,909 (м, 6H), 1,051-1,323 (м, 44H), 1,951-1,984 (т, 4H), 7,292-7,355 (м, 6H), 7,708-7,722 (д, 2H).
Синтез 2-метил-9,9-дитетрадецил-2,3-дигидроциклопента[b]флуорен-1(9H)-она
В 5000 мл круглодонную колбу загружали 9,9-дитетрадецил-9H-флуорен (30 г, 53,7 ммоль) и 2-бром-2-метилпропионилбромид (12,7 г, 55,3 ммоль), и затем растворяли введенным туда 300 мл дисульфида углерода. Затем реактор охлаждали ледяной водой. В атмосфере азота туда медленно десятью партиями добавляли трихлорид алюминия (15,7 г, 118,1 ммоль) в течение 2 часов. Смесь перемешивали при комнатной температуре в течение 8 часов, и затем реакцию завершали добавлением 100 мл дистиллированной воды, с последующей трех разовой промывкой 500 мл дистиллированной воды. Органический слой сушили над сульфатом магния, с последующим удалением летучих веществ и сушили, с получением, таким образом, 30,0 г 2-метил-9,9-дитетрадецил-2,3-дигидроциклопента[b]флуорен-1(9H)-она (выход: 89,1%) в виде масла высокой вязкости.
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 0,590 (м, 4H), 0,867-0,895 (м, 6H), 1,024-1,295 (м, 44H), 1,367-1,382 (д, 3H), 1,963-2,204 (т, 4H), 2,792-2,826 (д, 2H), 3,448-3,500 (м, 1H), 7,372-7,400 (м, 3H), 7,726-7,780 (м, 3H).
Синтез 2-метил-9.9-дитетрадецил-3,9-дигидроциклопента[b]флуорена
В 500 мл круглодонной колбе 2-метил-9,9-дитетрадецил-2,3-дигидроциклопента[b]флуорен-1(9H)-он (20 г, 31,9 ммоль) растворяли в 150 мл ТГФ и 150 мл этанола, и затем перемешивали. Боргидрид натрия (NaBH4) (1,8 г, 47,8 ммоль) добавляли к реакционному продукту пятью партиями, и затем перемешивали в течение 12 часов. Полученную в результате смесь после удаления растворителя растворяли в этилацетате, и затем три раза промывали водой. Органический слой сушили над сульфатом магния, с последующим удалением летучих веществ. Высушенный продукт растворяли в 150 мл толуола, и затем вводили в круглодонную колбу. После этого туда вводили п-толуолсульфоновую кислоту (0,08 г), и затем воду полностью удаляли кипячением с обратным холодильником с использованием аппарата Dean-Stark. Полученное в результате вещество охлаждали до комнатной температуры, и затем туда инъецировали водный раствор хлорида аммония (100 мл) и 200 мл простого диэтилового эфира, с последующим разделением органического слоя. Органический слой, собранный экстракцией остатка простым диэтиловым эфиром, сушили над сульфатом магния, с последующим удалением летучих веществ, и затем очищали с использованием колоночной хроматографии на силикагеле, с получением, таким образом, 15,3 г 2-метил-9,9-дитетрадецил-3,9-дигидроциклопента[b]флуорена (выход: 78,5%).
1H-ЯМР (500 МГц, CDCl3, м.д.): δ 0,649-0,665 (м, 4H), 0,891-0,918 (м, 6H), 1,059-1,319 (м, 44H), 1,953-1,986 (т, 4H), 2,206 (с, 3H), 3,378 (с, 2H), 6,562 (с, 1H), 7,237-7,332 (м, 4H), 7,663-7,678 (д, 1H), 7,710 (с, 1H).
Синтез N-трет-бутил-1-(9,9-дитетрадецил-2-метил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-трет-бутил-1-(9,9-дитетрадецил-2-метил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина
В 250 мл круглодонной колбе 2-метил-9,9-дитетрадецил-3,9-дигидроциклопента[b]флуорен (4,9 г, 8,0 ммоль) растворяли в 100 мл безводного простого диэтилового эфира, и затем температуру снижали до -78°C. Затем туда медленно инъецировали н-бутиллитий (1,6M раствор гексана, 5,5 мл), с последующим перемешиванием при комнатной температуре в течение 12 часов. После удаления летучих веществ в вакууме 100 мл н-гексана добавляли к смеси для понижения температуры реактора до -78°C, с последующим добавлением дихлордиметилсилана (2,9 г). Температуру снова повышали до комнатной температуры, с последующим перемешиванием в течение 24 часов, и затем соли удаляли фильтрованием. Затем летучие вещества удаляли в вакууме. Продукт снова вводили в 250 мл круглодонную колбу и растворяли в 100 мл простого диэтилового эфира. Температуру снижали до -78°C, и туда добавляли трет-бутиламин (1,8 г, 24,1 ммоль). Температуру повышали до комнатной температуры, с последующим перемешиванием в течение 12 часов, и летучие вещества полностью удаляли в вакууме. Затем 200 мл н-гексана добавляли для растворения полученного в результате вещества, и соли удаляли фильтрованием. Растворитель удаляли, с получением, таким образом, 5,5 г смеси N-трет-бутил-1-(9,9-дитетрадецил-2-метил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-трет-бутил-1-(9,9-дитетрадецил-2-метил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина (соотношение=~1:1), (выход: 92,7%) в виде вещества высокой вязкости.
1H-ЯМР (500 МГц, C6D6, м.д.): δ 0,145 (с, 3H), 0,183-0,204 (д, 6H), 0,290 (с, 3H), 0,552 (с, 1H), 0,603 (с, 1H), 0,998-1,370 (м, 126H), 2,228-2,301 (м, 14H), 3,408-3,435 (д, 2H), 6,749-6,760 (д, 2H), 7,353-7,461 (м, 6H), 7,546-8,073 (м, 6H).
Синтез (трет-бутиламидо)диметил(9,9-дитетрадецил-2-метил-3,9-дигидроциклопента[b]флуорен-3-ил)силантитан(IV)диметила (комплекс 7) и (трет-бутиламидо)диметил(9,9-дитетрадецил-2-метил-1,9-дигидроциклопента[b]флуорен-1-ил)силантитан(IV)диметила (комплекс 8)
В 250 мл круглодонной колбе смесь N-трет-бутил-1-(9,9-детрадецил-2-метил-3,9-дигидроциклопента[b]флуорен-3-ил)-1,1-диметилсиланамина и N-трет-бутил-1-(9,9-дитетрадецил-2-метил-1,9-дигидроциклопента[b]флуорен-1-ил)-1,1-диметилсиланамина (в соотношении=~1:1) (5,0 г, 6,8 мм) растворяли в 100 мл простого диэтилового эфира, и затем температуру снижали до -78°C. Затем туда медленно инъецировали метиллитий (1,5M раствор простого диэтилового эфира, 18,5 мл). Температуру повышали до комнатной температуры, с последующим перемешиванием в течение 12 часов, с получением литиевой соли. В дополнение, в сухой камере вводили TiCl4 (16,75 ммоль) и 50 мл безводного н-гексана в 250 мл круглодонную колбу, и затем температуру снижали до -78°C. Затем туда медленно добавляли полученную литиевую соль. Температуру снова повышали до комнатной температуры, с последующим перемешиванием в течение 4 часов, и растворитель удаляли в вакууме. Полученное в результате вещество растворяли в н-гексане, и затем фильтрат выделяли фильтрованием. Снова н-гексан удаляли в вакууме, с получением, таким образом, 5,2 г смеси комплекса 7 и комплекса 8 (в соотношении приблизительно 1:1) в виде твердого вещества.
1H-ЯМР (500 МГц, C6D6, м.д.): δ 0,093-0,104 (д, 6H), 0,630-0,647 (д, 6H), 0,856-1,392 (м, 120H), 1,609-1,643 (д, 18H), 2,095-2,214 (м, 14H), 7,023-7,041 (д, 2H), 7,305-8,097 (м, 12H).
[Сравнительный пример получения 1] Получение (трет-бутиламидо)диметил(2-метилинденил)силантитан(IV)диметила
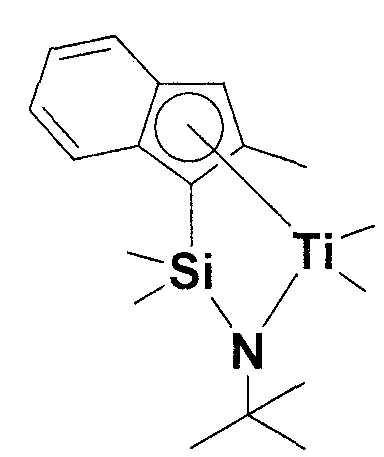
Его получали, начиная с 2-метилиндена способом синтеза, описанного в ссылке "Journal of Organometallic Chemistry 666(2002) 5-26".
Получение EPDM
[Пример 1-Пример 8] Получение EPDM непрерывным процессом полимеризации в растворе
Этилен, пропилен и 5-этилиден-2-норборнен (ENB) полимеризовали над катализатором полученным в настоящем изобретении, с использованием полимеризатора непрерывного действия, с получением, таким образом, EPDM. Катализаторы, синтезированные в примерах получения 1-4 и сравнительном примере получения 1, использовали в качестве катализаторов с единственной точкой активации, и циклогексан использовали в качестве растворителя. Используемое количество катализаторов описано в таблице 1, представленной ниже. Ti, Al и B показывают катализатор с единственной точкой активации, триизобутилалюминий в качестве сокатализатора, и трифенилметилтетракис(пентафторфенил)борат, соответственно. Соответствующие катализаторы инъецировали, при этом каждый из них растворяли в толуоле в концентрации 0,2 г/л, и синтез проводили с использованием пропилена в качестве сомономера α-олефина и 5-этилиден-2-норборнен (ENB) в качестве мономера диена. Оценку коэффициента конверсии реактора можно провести в условиях реакции и градиента температуры в реакторе, когда один вид полимера получали посредством полимеризации в соответствующих условиях реакции. Молекулярную массу, в случае катализатора с единственной точкой активации, контролировали в качестве функции температуры реактора, коэффициента конверсии и содержания водорода, и подробное описание условий полимеризации и результатов полимеризации показано в таблице 1, представленной ниже.
Как видно из приведенной выше таблицы 1, в примерах 1-8 получения с использованием катализатора, разработанного в настоящем изобретении, продукты EPDM, делающие возможным легкое введение сомономеров (C3 и ENB) и высокий коэффициент конверсии, и высокую степень вязкости по Муни, можно получать в высокотемпературных условиях полимеризации. Таким образом, можно утверждать, что EPDM система полимеризации с использованием катализатора единичной точкой активации, разработанного в настоящем изобретении, является более экономичной каталитической системой, чем EPDM система полимеризации с использованием существующего катализатора с единичной активации благодаря легкому контролю физических свойств (молекулярная масса и композиция сомономера) продуктов и высокой активности.
Настоящее изобретение подробно описано со ссылкой на примеры, как указано выше, но специалисты в данной области, к которой относится настоящее изобретение, могут проводить различные модификации без отступления от существа и объема настоящего изобретения, определенного в прилагаемой формуле изобретения. Таким образом, изменения и модификации примеров по настоящему изобретению не будут отступлением от способа по настоящему изобретению.
Промышленная применимость
Как указано выше, в способе получения сополимера этилен-α-олефиндиена по настоящему изобретению соединение переходного металла на основе циклопента[b]флуоренильной группы используют в качестве катализатора полимеризации, и, таким образом, сополимеры этилен-α-олефиндиена с высоким содержанием диена, высоким коэффициентом конверсии и высокой степенью вязкости по Муни можно получать в высокотемпературных (120°C или выше) условиях полимеризации с высоким выходом. Более того, каталитическую композицию, содержащую соединение переходного металла, можно легко получить с высоким выходом экономически выгодным синтезом. Также соединение переходного металла или каталитическая композиция по настоящему изобретению может иметь отличную сополимеризующую реакционную способность в отношении других олефинов, при этом сохраняя высокую каталитическую активность даже при высокой температуре благодаря их отличной термостабильности, и позволяет получить полимеры с высокой молекулярной массой с высоким выходом, приводя к более высокой коммерческой целесообразности, по сравнению с уже известными металлоценовыми и неметаллоценовыми катализаторами с единственной точкой активации.
Реферат
Настоящее изобретение относится к способу получения сополимера этилен-α-олефиндиена и полученному таким способом сополимеру этилен-α-олефиндиена с использованием соединения переходного металла на основе циклопента[b]флуоренильной группы в качестве катализатора. Соединение переходного металла представлено формулой (1), в которой значения радикалов указаны в формуле изобретения. Изобретение позволяет получить сополимер с высоким содержанием диена, высоким коэффициентом конверсии и высокой степенью вязкости по Муни. 2 н. и 11 з.п. ф-лы, 1 табл., 4 пр.Формула (1):
Формула
[Химическая формула 1]
где в химической формуле 1 M представляет собой переходный металл 4 группы периодической таблицы элементов;
n равно целому числу 1 или 2, каждый R1 может быть одинаковым или различным, когда n равно 2;
R1 представляет собой водород, (C1-C50)алкил, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, -NRaRb, -SiRcRdRe или 5-7-членный N-гетероциклоалкил, содержащий по меньшей мере один атом азота;
R2 и R3, каждый независимо, представляет собой водород, (C1-C50)алкил, (C1-C50)алкокси, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арилокси, (C1-C50)алкил(C6-C30)арилокси, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, -NRaRb или -SiRcRdRe;
R4, R5, R10, R11 и R12, каждый независимо, представляет собой (C1-C50)алкил, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, -NRaRb или -SiRcRdRe, и R11 и R12 могут быть связаны через (C4-C7)алкилен с образованием кольца;
R6, R7, R8 и R9, каждый независимо, представляет собой водород, (C1-C50)алкил, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C1-C50)алкокси, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, (C6-C30)арилокси, (C1-C50)алкил(C6-C30)арилокси, N-карбазолил, -NRaRb или -SiRcRdRe или могут быть связаны с соседним заместителем через (C1-C5)алкилен с образованием кольца, и по меньшей мере один -CH2- алкилена может быть замещен гетероатомом, выбранным из -O-, -S- и -NR'-, и алкилен может быть дополнительно замещен (C1-C50)алкилом;
арил R1-R12 может быть дополнительно замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из (C1-C50)алкила, галоген(C1-C50)алкила, (C1-C50)алкокси, (C6-C30)арилокси, (C6-C30)арила, (C1-C50)алкил(C6-C30)арила и (C6-C30)арил(C1-C50)алкила;
R' и Ra-Re, каждый независимо, представляет собой (C1-C50)алкил или (C6-C30)арил; и
X1 и X2, каждый независимо, представляет собой галоген, (C1-C50)алкил, (C2-C50)алкенил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, (C1-C50)алкокси, (C6-C30)арилокси, (C1-C50)алкил(C6-C30)арилокси, (C1-C50)алкокси(C6-C30)арилокси, (C1-C50)алкилиден, или лиганд аниона или дианиона, состоящий из 60 или менее атомов, содержащих N, P, O, S, Si и галоген, за исключением водорода, при условии, что один X1 и X2 представляет собой лиганд дианиона, другим пренебрегают.
[Химическая формула 2]
[Химическая формула 3]
где в химических формулах 2 и 3 M, R2-R12, X1 и X2 являются такими, как определено в химической формуле 1 по п.1; R21 и R22, каждый независимо, представляет собой водород, (C1-C50)алкил, галоген(C1-C50)алкил, (C3-C50)циклоалкил, (C6-C30)арил, (C6-C30)арил(C1-C50)алкил, ((C1-C50)алкил(C6-C30)арил)(C1-C50)алкил, -NRaRb, -SiRcRdRe или 5-7-членный N-гетероциклоалкил, содержащий по меньшей мере один атом азота; арил R1 может быть дополнительно замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из галогена, (C1-C50)алкила, галоген(C1-C50)алкила, (C1-C50)алкокси, (C6-C30)арилокси, (C6-C30)арила, (C1-C50)алкил(C6-C30)арила и (C6-C30)арил(C1-C50)алкила; и Ra-Re, каждый независимо, представляет собой (C1-C50)алкил или (C6-C30)арил.
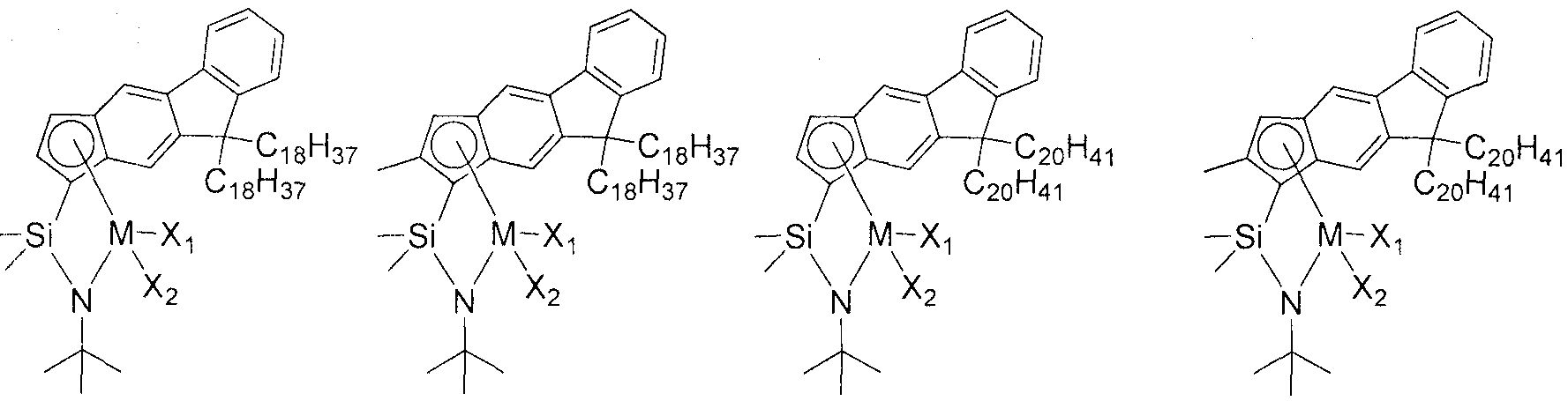


M представляет собой Ti, Zr или Hf; и X1 и X2, каждый имеет значение, как определено в химической формуле 1 по п.1.
мономер диена представляет собой по меньшей мере один выбранный из 1,3-бутадиена, 1,4-пентадиена, 2-метил-1,3-бутадиена, 1,4-гексадиена, 1,5-гексадиена, 1,5-гептадиена, 1,6-гептдиена, 1,6-октадиена, 1,7-октадиена, 1,7-нонадиена, 1,8-нонадиена, 1,8-декадиена, 1,9-декадиена, 1,12-тетрадекадиена, 1,13-тетрадекадиена, 3-метил-1,4-гексадиена, 3-метил-1,5-гексадиена, 3-этил-1,4-гексадиена, 3-этил-1,5-гексадиена, 3,3-диметил-1,4-гексадиена, 3,3-диметил-1,5-гексадиена, циклопентадиена, циклогексадиена, 5-винил-2-норборнена, 2,5-норборнадиена, 7-метил-2,5-норборнадиена, 7-этил-2,5-норборнадиена, 7-пропил-2,5-норборнадиена, 7-бутил-2,5-норборнадиена, 7-фенил-2,5-норборнадиена, 7-гексил-2,5-норборнадиена, 7,7-диметил-2,5-норборнадиена, 7-метил-7-этил-2,5-норборнадиена, 7-хлор-2,5-норборнадиена, 7-бром-2,5-норборнадиена, 7-фтор-2,5-норборнадиена, 7,7-дихлор-2,5-норборнадиена, 1-метил-2,5-норборнадиена, 1-этил-2,5-норборнадиена, 1-пропил-2,5-норборнадиена, 1-бутил-2,5-норборнадиена, 1-хлор-2,5-норборнадиена, 1-бром-2,5-норборнадиена, 5-изопропил-2-норборнена, 5-винилиден-2-норборнена (VNB), 5-метилен-2-норборнена (MNB) и 5-этилиден-2-норборнена (ENB).
Документы, цитированные в отчёте о поиске
Способ получения замещенных инденов, промежуточный продукт для получения металлоценов и металлоцены
Комментарии