Способ получения замещенных инденов, промежуточный продукт для получения металлоценов и металлоцены - RU2103250C1
Код документа: RU2103250C1
Описание
Изобретение относится к способу получения производных индена, замещенных в пятом и шестом кольце.
Соединения такого типа представляют собой важные промежуточные продукты при получении комплексов металлоценов. Большое значение, в частности, играют соответствующие хиральные производные циркония с наличием мостичных связей между молекулами в качестве высокоактивных катализаторов при полимеризации олефинов (ЕР 129368). Изменением состава лигандной системы, например замещением, можно целенаправленно влиять на свойства катализаторов. Таким образом можно по желанию изменять выход полимера, молекулярную массу, тактичность или температуру плавления полимеров (ЕР 316155; ЕР 351392).
Кроме этого, индены можно использовать в качестве мономеров при гомополимеризации или сополимеризации с другими олефинами (Bull. Soc. Chim.Fr. 6 (1969) 2039).
Описанные в литературе немногочисленные замещенные индены можно получать только проведением многоступенчатого синтеза при небольшом выходе продукта. В большинстве случаев их получают восстановлением замещенных 1-инданонов и последующей дегидратацией. Соответствующие инданоны получают многоступенчатым синтезом замещенных ароматических веществ (Bull Soc. Chem. Fr. 6 (1969) 2039).
Описанные в литературе немногочисленные замещенные индены можно получать только проведением многоступенчатого синтеза при небольшом выходе продукта. В большинстве случаев их получают восстановлением замещенных 1-инданонов и последующей дегидратацией. Соответствующие инданоны получают многоступенчатым синтезом замещенных ароматических веществ ( Bull. Soc. Chim. Fr 6 (1969) 1981; Acta Chem. Scand B 30 (1976) 527; Austr. F.Chem. 29 (1970) 2572; Chem. Lett (1981) 729; Ber. 97(12) (1964) 3461).
Задачей изобретения являлась разработка способа получения вышеуказанных инденов, который был бы лишен недостатков, известных из современного уровня техники. Такие индены дают подход к новым комплексам металлоценов.
Было обнаружено, что ароматические соединения нижеприведенной формулы 1 реагируют при высоком выходе продукта с производными пропионовой кислоты, имеющими в α-положении отщепляемую группу, в присутствии катализатора Фриделя-Крафтса с образованием замещенных 1-инданонов. Такой результат был полностью неожиданным, так как получение этих продуктов можно было бы ожидать только при реагировании с производными пропионовой кислоты, имеющими отщепляемую группу в β-положении (J. Amer. Chem. Soc. 72 (1950) 3286).
Кроме этого, такой синтез представляет собой технически просто осуществимый одностадийный процесс. Инданоны могут затем переводиться в соответствующие индены известными методами. Одновременно с этим предложенный в изобретении способ позволяет получать новые соединения указанного структурного типа.
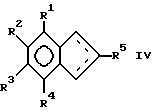
Следовательно, изобретение касается способа поручения соединений формулы IV или его изомера формулы IVa.

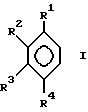
где R1, R2 , R3, R4 и R5 одинаковы или различны и означают водород, низший алкил, низший алкоксил, C6-C14-арил, или соседние остатки R1 - R4 образуют вместе со связанными с ними атомами одно или несколько колец; способ по изобретению отличается тем, что проводят реакцию между соединением формулы I

и соединением формулы II

или его ангидридом в присутствии катализатора Фриделя-Крафтса с образованием соединения формулы III или формулы IIIa,

причем радикалы R1 - R5 имеют указанные значения и X1 и X2 одинаковы или различны и представляют собой нуклеофильную отщепляемую группу, с последующим восстановлением и дегидратацией в соединение формулы IV или IVa.
Индоны в зависимости от положения заместителя в ароматических соединениях могут образовываться в форме двух структурных изомеров формулы III или формулы IIIa. Они могут восстанавливаться до соответствующих инданолов в чистой форме или в виде смеси известными по литературе методами с использованием восстановителей, таких как NaBH4 или LiAlH4 и затем дегидратироваться обработкой кислотами, такими как серной кислотой, щавелевой кислотой, пара-толуолсульфоновой кислотой или обработкой водопоглощающими веществами, как, например, сульфатом магния, сульфатом натрия, окисью алюминия, силикагелем или молекулярными ситами до образования инденов формулы IV или формулы IVa (Bull. Soc. Chim. Fr 11 (1973) 3092; Organomet 9 (1990) 3098.
X1 и X2 предпочтительнее означают атом галогена, гидроксильную группу, группу тозила или (C1-C10) алкоксильную группу; в частности атом галогена, особое предпочтение отдается брому или хлору.
Подходящими катализаторами Фриделя-Крафтса являются, например, AlCl3, AlBr3, FeCl3, SbCl5, SnCl4, BF3, TiCl4, ZnCl2 HF, H2 SO4, полифосфорная кислота, H3PO4 или расплав AlCl3/NaCl; в частности AlCl3.
Предпочтительны соединения формулы VI или IVa; если R1, R2, R3, R4 одинаковы или различны и означают водород или низший алкил, или радикалы R1 и R2 или R3 и R4 образуют со связанными с ними атомами кольцо, и R5 означает метил.
Исходные вещества формул I и II известны и существуют в продаже, их можно синтезировать известными из литературы способами.
Реакция проводится в инертном растворителе. Предпочтение отдается метиленхлориду или CS2. Если исходные компоненты жидкие, то от растворителя можно отказаться.
Молярные соотношения исходных соединений, включая и катализатор Фриделя-Крафтса, могут изменяться в широких пределах. Предпочтительное молярное соотношение соединения I : II : катализатор = 1 : 0,5-1,5 : 1-5; в частности 1 : 1 : 2,5-3.
Реакционная температура предпочтительно составляет О - 130oC, в частности 25 - 80oC.
Время реакции, как правило, находится в пределах от 30 мин до 100 ч, предпочтительнее от 2 до 30 ч.
Предпочтительным вариантом является использование смеси соединений I и II и дозирование катализатора Фриделя-Крафтса. Также возможна и обратная последовательность.
Инданоны формулы III или IIIa могут очищаться дистилляцией, хроматографией или кристаллизацией.
Замещенные индены можно получать в виде изомеров по двойным связям (IV/IVa). Их можно очищать от побочных продуктов методами дистилляции, хроматографии или кристаллографии.
Особое отличие предложенного в изобретении способа заключается в том, что различно замещенные индены можно получить с высоким выходом путем проведения простого и непродолжительного по времени синтеза. При этом существуют широкие возможности для введения в пятое и шестое кольцо заместителей в самые различные положения. Благодаря этому получают новые производные индена.
Объектом изобретения являются также производные индена IV/IVa в качестве промежуточного продукта при получении комплексов металлоценов, в частности комплексов металлоценов нижеприведенной формулы VI, которые также являются предметом предлагаемого изобретения.

где М1 является цирконием, гафнием, R1, R2, R3, R4 и R5 одинаковы или различны и означают водород, низший алкил, низший алкоксил, С6-С14 -арил, или соседние остатки R1-R4 образуют со связанными с ними атомами углерода одно или несколько колец,
R7 означают остаток

причем M2 означает углерод, кремний, германий;
R8 и R9 одинаковы или различны и означают водород, низший алкил, фенил;
R10 и R11 одинаковы или различны и означают галоген;
p = 1 или 2.
Предпочтителен вариант, при котором:
М1 является цирконием
М2 означает углерод или кремний, в частности кремний.
Кроме этого, для соединений формулы VI предпочтительнее, чтобы радикалы R5 и R3; R1, R3 и R5; R1, R2, R3 и R5 или все остатки R1-R5 не означали водород, а преимущественно являлись (C1-C4) алкилом. Особенно предпочтителен вариант, при котором радикалы R1, R3 и R5 не являются водородом, когда они одинаковы или различны и означают (C1-C4) алкил.
Предпочтительными положениями заместителей в остатках инденила являются положения 2,6-, 2,4,6-, 2,4,5-, 2,4,5, 6- и 2,4,5,6,7-, и в частности 2,4,6-и 2,4,5-. При этом положение 2- в остатках инденила предпочтительнее замещается группой метила. Кроме этого, для соединения формулы VI предпочтительны остатки инденила, являющиеся бензоаннелированными.
Особое значение имеют соединения формулы VI, указанные в примерах осуществления.
Исходя из инденов формул IV и IVa, которые
могут использоваться в виде смеси изомеров, получение металлоценов VI протекает в соответствии с известными по
литературе способами (сравним
AU-А-31478/89, J.Organomet. Chem. 342 (1988) 21,
ЕП-А-284707 по следующей реакционной схеме:

(X3 - нуклеофильная отщепляемая группа, например, Cl, Br, O-тозил).
Из галогенидов металлоценов формулы VI можно получать производные, соответствующие моно- или диалкилсоединения, используя известные по литературе методы, например, реакции с алкилирующими средствами, например, литийалкиленом (J. Amer. Chem. Soc. 95 (1973) 6263).
Лигандные системы с мостичными связями между молекулами формулы I могут получаться в зависимости от положения заместителей в индене в виде структурных изомеров. Если они не разделяются, то образуются структурные изомеры металлоценов формулы VI. Металлоцены формулы VI получаются в виде смеси рацемической формы с мезоформой. Процессы разделения изомерных форм, в частности отделение нежелательной для полимеризации олефинов мезоформы, в принципе известны (AU-A-31478/89. J. Organomet. Chem. 342 (1988) 21; ЕП-А 284707). Оно осуществляется как правило экстракцией или перекристаллизацией с различными растворителями.
Металлоцены VI являются высокоактивными катализаторами и используются для получения полимеров олефинов с высокой изотактичностью и большой молекулярной массой.
Полимеризацию или сополимеризацию проводят в растворителе, суспензии или газовой фазе проведением непрерывного или периодического одно- или многоступенчатого процесса при температуре 0 - 150oC, предпочтительнее 30 - 80oC. Полимеризации или сополимеризации подвергают олефины формулы Rа -CH=CH-Rб. В этой формуле Rа и Rб одинаковы или различны и означают атом водорода или алкил с числом атомов С от 1 до 14. Остатки Rа и Rб вместе со связанными с ними атомами С могут образовывать кольцо. Примерами таких олефинов являются этилен, пропилен, 1-бутен, 1-гексен, 4-метил-1-пентен, 1-октен, норборндиметаноктагидронафталин или норборнадиен. В частности, подвергаются полимеризации пропилен и этилен.
В качестве сокатализаторов предпочтительнее применение находят алюминоксаны.
В соответствии с изобретением вместо алюминоксана или вместе с ним в качестве сокатализаторов могут использоваться соединения формулы RxNH4-xBR4,
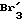
Пример A. 2,5,7-Триметил-1-инданон (1).
К раствору, состоящему из 34,4 г (324 мМ) м-ксилола (99%) и 74 г (324 мМ) бромида 2-бромизомасляной кислоты (98%) в 600 мл метиленхлорида высшей степени очистки медленно добавляли через дозирующую воронку для твердых веществ 107 г (810 мМ) AlCl3, добавление проводилось при комнатной температуре и интенсивном перемешивании, причем начиналось бурное газовыделение. Смесь перемешивалась в течение 15 ч при комнатной температуре, выливалась в ледяную воду, подкисленную 25 мл конц. HCl и производилось многократное экстрагирование. Объединенные органические фазы сначала промывались насыщенным раствором NaHCO3 и затем насыщенным раствором NaCl и высушивались сульфатом магния. Масло, остающееся после извлечения растворителя при пониженном давлении, подвергалось дистилляции через короткий дистилляционный мостик. При 81 - 90oC/0,1-0,2 мбар образовывалось 52,4 г инданона I в форме бесцветного масла, которое кристаллизовалось при комнатной температуре. Выход продукта составлял 93%.
1H-ЯМР-спектр (100 МГц, CDCI3: 7,05 (1, s), 6,87 (1, s) 3,25 (1, q), 2,43-2,80 (2, m), 2,57 (3, s), 2,35 (3, s), 1,25 (3, d).
Масс-спектр: 174 M+, правильный продукт распада.
Пример В. 2,4, 6-Триметилинден (2).
20,4 г (117 мМ) 2,5,7-Триметил-1-инданона (1) были растворены в 300 мл смеси из тетрагидрофурана с метанолом (2 : 1) и к смеси было добавлено при комнатной температуре 6,6 г (175 мМ) NaBH4, Смесь еще перемешивалась в течение 1 ч, к ней добавили 50 мл полуконц. HCl и экстрагировали. Объединенные органические фазы высушивались сульфатом натрия и освобождались от растворителя. Остаток был перенесен в дистилляционный аппарат и к нему было добавлено 13 г сульфата магния. Был создан вакуум величиной примерно 10 мбар и дистиллятор был нагрет до очень высокой температуры, пока не произошла перегонка продукта (130 - 150oC). При перегонке получилось 17,7 г индена 2 в виде бесцветного масла. Выход продукта составил 96%.
1H-ЯМР-спектр (100 МГц, CDCl3): изомеры по двойным связям А : В = 2 : 1
Изомер А: 6,97 (1, s), 6,80 (1, s),
6,50 (1, m), 3,20 (2, m), 2,1-2,3 (9, m).
Изомер В: 6,87 (1, s), 6,70 (1, s), 6,37 (1, m), 3,07 (2, m), 2,1-2,3 (9, m).
Масс-спектр: 158 М+, правильный продукт распада.
Пример C. 2-Метил-5, 7-диизопропил-1-инданон (3) или 2-метил-4, 6-диизопропил-1-инданон (3а).
К раствору, состоящему из 84,8 г (523 мМ) 1, 3-диизопропиленбензола и 120 г (523 мМ) бромида 2-бромизомасляной кислоты (98%) в 600 мл метиленхлорида высшей степени очистки медленно добавляли через дозирующую воронку для твердых веществ 174 г (1300 мМ) AlCl3, добавление производилось при комнатной температуре. Смесь нагревалась последующие 20 ч при обратном потоке и затем перерабатывалась аналогично примеру А. Сырой продукт очищался методом хроматографии через 3 кг силикагеля 60. Инданоны 3 и 3а проявлялись элюированием при помощи смеси растворителей, состоящей из гексана и 15%-ного уксусного эфира. С тем же растворителем в дальней зоне проявлялось в качестве побочного продукта соединение: 2-метил-5-изопропил-1-инданон. Разделять оба изомера для проведения последующих реакций обмена не требуется. Общий выход составил 93 г (78 %).
1H-ЯМР-спектр (360 МГц, CDCl3): смесь изомеров (3:2) 7,49 (d), 7,36 (d) 7,13 (s), 7,10 (s), 4,15 (sept.), 3,25-3,40 (m), 3,10 (sept.), 2,90-3,00 (m), 2,60-2,73 (m), 1,22-1,30 (m).
Масс-спектр: 230 М+, правильный продукт распада.
Пример D. 2-Метил-4,6-диизопропиленден (4) или 2-метил-5, 7-диизопропиленден (4а), вариант I.
К раствору, состоящему из 78,5 г (341 мМ) смеси изомеров 3/3а в 700 мл смеси растворителей тетрагидрофурана с метанолом высшей степени очистки (2 : 1) при комнатной температуре добавляли 19,3 г (511 мМ) NaBH4. После перемешивания в течение 2 ч добавляли при комнатной температуре 120 - 130 мл полуконц. HCl и экстрагировали. Объединенные органические фазы высушивались Na2SO4. Остающийся после извлечения растворителя остаток был растворен в 500 мл метиленхлорида и смесь нагревалась вместе с 6,5 г (34 мМ) пара-толуолсульфоновой кислоты в течение 15 мин до образования обратного потока. Остающийся после извлечения растворителя остаток очищался методом хроматографии через 1,5 кг силикагеля 60. Смесью растворителей гексана с диизопропиловым эфиром 20 : 1 выделялись 56 г смеси изомеров 4/4а в форме бесцветного масла. Общий выход составлял 86%.
1Н-ЯМР-спектр (100 МГц, CDCI3): изомеры по двойным связям (1 : 1) 7,1 (m), 6,95 (m), 6,60 (m), 6,43 (m), 3,25 (br), 2,75 - 3,20 (m), 2,12 (d), 1,28 (d), 1,25 (d).
Масс-спектр: 214 М+, правильный продукт распада.
Пример Е. 2-Метил-4, 6-диизопропилинден (4) или 2-метил-5,7-диизо-пропилинден (4а), вариант II.
К раствору, состоящему из 78,5 г (341 мМ) смеси изомеров 3/3а в 700 мл смеси растворителей тетрагидрофурана с метанолом высшей степени очистки (2 : 1) при комнатной температуре добавляли 19,3 г (511 мМ) NaBH4. После перемешивания в течении 2 ч добавляли при комнатной температуре 120 - 130 мл полуконц. HCl и экстрагировали. Объединенные органические фазы высушивались Na2SO4. Остающийся после извлечения растворителя остаток переносили в дистилляционный аппарат и туда добавляли 50 г сульфата магния. После создания вакуума величиной примерно 1 мбар производилось нагревание, пока продукт не перегонялся (примерно 130oC). Получали 65 г смеси изомеров 4/4а в виде бесцветного масла. Выход составлял 90%.
Пример F. 2-Метил-1-инданон (5).
К раствору, состоящему из 3,91 г (50 мМ) бензола в 30 мл метиленхлорида высшей степени очистки добавляли при охлаждении льдом 17,3 г (125 мМ) AlCl3. Затем добавляли 11,9 г (52 мМ) бромида 2-бромизомасляной кислоты и продолжали перемешивание в течение 1 ч при 0oC и 2 ч при комнатной температуре. Смесь еще нагревали в течение 15 ч при обратном потоке, после чего перерабатывали аналогично примеру А. Сырой продукт очищали методом хроматографии через 100 г силикагеля (гексан/метиленхлорид 1 : 1). Выход продукта составлял 5,1 г (70%).
1H-ЯМР-спектр (100 МГц, CDCl3): 7,5 (m), 3,33 (q), 2,73 (m), 1,30 (d).
Масс-спектр: 146 М+, правильный продукт распада.
Пример G. 2-Метилинден (6).
Аналогично примеру D восстанавливали 5,0 г (34 мМ) 2-метил-1-инданона (5) при помощи 1,94 г (51 мМ) NaBH4. Не очищающийся дальше спирт подвергали затем реакции в присутствии 0,2 г пара-толуолсульфоновой кислоты в 100 мл толуола при 80oC. После очистки методом хроматографии через 100 г силикагеля (гексан-метиленхлорид 9 : 1) получали 3,68 г (82%) 2-метилиндена (6).
1H-ЯМР-спектр (100 МГц, CDCl3): 7,2 (4,m), 6,45 (1,m) 3,25 (2,m), 2,1 (3,m).
Масс-спектр: 130 М+, правильный продукт распада.
Пример Н. 2-Метил-5-изобутил-1-инданон (7).
К раствору, состоящему из 6,71 г (50 мМ) изобутилбензола в 30 мл метиленхлорида высшей степени очистки добавляли при охлаждении льдом 17,3 г (125 мМ) AlCl3. Затем быстро добавляли 11,9 г (52 мМ) бромида 2-бромизомасляной кислоты и продолжали перемешивание в течение 1 ч при 0oC и 2 ч при комнатной температуре. Смесь дальше нагревали еще в течение 15 ч при обратном потоке и затем перерабатывали аналогично примеру А. Сырой продукт очищали методом хроматографии через 100 г силикагеля (гексан/метиленхлорид 1 : 1). Выход составлял 8,42 г (83%).
1H-ЯМР-спектр (100 МГц, CDCl3): 7,7 (m), 7,2 (m), 3,35 (q), 2,70 (m), 2,58 (d), 1,95 (q), 1,25 (d), 0, 93 (d).
Масс-спектр: 202 М+, правильный продукт распада.
Пример J. 2-Метил-6-изобутилинден (8).
Аналогично примеру D восстанавливали 8,3 г (41 мМ) 2-метил-5-изобутил-1-инданона (7) при помощи 2,4 г (62 мМ) NaBH4. Не очищающийся дальше спирт подвергали затем реакции обмена в присутствии 0,4 пара-толуолсульфоновой кислоты в 100 мл толуола при 80oC. После очистки методом хроматографии через 400 г силикагеля (гексан) получали 7,17 (95 %) 2-метил-6-изобутилиндена (8).
1-H-ЯМР-спектр (100 МГц, CDCI3: 7,1 (m), 6,45 (m), 3,25 (m), 2, 45 (d), 2,88 (q), 2,10 (d), 0,95 (d).
Масс-спектр: 184 M+, правильный продукт распада.
Пример К. 2,5,6, 7-Тетраметил-1-инданон (9).
К раствору, состоящему из 6,01 r (50 мМ) 1,2,3-триметилбензола в 30 мл метиленхлорида высшей степени очистки добавляли при охлаждении льдом 17,3 г (125 мМ.) AlCI3. Затем быстро добавляли 11, 9 г (52 мМ) бромида 2-бромизомасляной кислоты и продолжали перемешивание в течение 1 ч при 0oC и 2 ч при комнатной температуре. Смесь выдерживалась еще в течение 15 ч при комнатной температуре и затем перерабатывалась аналогично примеру А. Сырой продукт очищали дистилляцией (0,05 Торр/96 - 107oC). Выход составлял 8,1 г (86%).
1H-ЯМР-спектр (100 МГц, CDCl3): 7,0 (m), 3,20 (q), 2,60 (m), 2,20 (m), 1,25 (d).
Масс-спектр: 188 М+, правильный продукт распада.
Пример L. 2,4,5, 6-Тетраметилинден (10).
Аналогично примеру D восстанавливали 1,50 г (8 мМ) 2,5,6,7-тетраметид-1-инданона (9) при помощи 0,45 г (12 мМ) NaBH4. Не очищающийся дальше спирт подвергали затем реакции обмена в присутствии 0,1 г пара-толуолсульфоновой кислоты в 100 мл толуола. После очистки методом хроматографии через 100 г силикагеля (гексан/метиленхлорид 9 : 1) получали 0,88 г (65%) 2,4,5,6-тетраметилиндена (10).
1H-ЯМР-спектр (100 МГц, CDCl3): 7,0 (s), 6,45 (m), 3,25 (m), 2,60 (m), 2,20 (m), 2,10 (d).
Масс-спектр: 170 М+, правильный продукт распада.
Пример М. Диметилбис-(2-метил-4,6-диизопропилинденил)силан (II).
К раствору, состоящему из 4,9 г (22,8 мМ) смеси изомеров 4/4a в 25 мл ТГФ, добавляли при 0oC в среде аргона (Ar) 9,2 мл (22,8 мМ) 2,5 М раствора бутиллития в гексане и раствор нагревали в течение 1 ч до образования обратного потока. Раствор красного цвета прикапывали затем при комнатной температуре к раствору из 1,5 г (11,4 мл) диметилдихлорсилана в 10 мл ТГФ и нагревали в течение 8 ч при обратном потоке. Смесь выливали в ледяную воду и экстрагировали. Высушенная через сульфат магния эфирная фаза упаривалась при пониженном давлении. Оставшееся желтоватое масло подвергалось затем очистке через 500 г силикагеля 60. Обработкой смесью растворителей из гексана с 5%-ным метиленхлоридом сначала стало возможным проявить элюированием 1,4 г смеси инденов 4/4а. При обработке гексаном с 8%-ным метиленхлоридом следовала лигандная система II. Смешиванием с метанолом в ледяной бане провели кристаллизацию остающегося после извлечения растворителя вязкого масла. Было получено 3,1 г желтоватого твердого вещества. Выход составил 56% или 84% относительно индена, подвергшегося реакции обмена.
1H-ЯMR-спектр (100 МГц, CDCl3): изомеры по двойным связям (3 : 1) 6,28-7,32 (m), 6,70 (m), 6,62 (m), 6,52 (m), 3,75 (s, br), 3,65 (s, br), 3,35 (s), 2,70-3,30 (m), 2,05-2,25 (m), 1,10-1,45 (m), 0,10-0,22 (m), от -0,15 до -0,32 (m).
Масс-спектр: 484 М+, правильный распад.
Пример N. Дихлорид диметилсиландиилбис(2-метил-4, 6-диизопропилинденил)циркония (12).
К раствору из 3,1 г (6,5 мМ) лигандной системы II в 25 мл диэтилового эфира добавляли при комнатной температуре в атмосфере Ar 6,3 мл (16,2 мМ) 2,5 М раствора бутиллитий в гексане и раствор перемешивали в течение ночи. После добавления 10 мл гексана бежевая суспензия фильтровалась и остаток промывался 20 мл гексана. Дилитиевая соль сушилась продолжительное время в вакууме, создаваемом масляным насосом, и затем добавлялась при -78oC к суспензии из 1,6 г (6,8 мМ) в 30 мл метиленхлорида. Смесь в течение 1 ч нагревалась до комнатной температуры и еще 30 мин перемешивалась при этой температуре. После удаления растворителя оранжево-коричневый остаток экстрагировали 50 мл гексана. После удаления растворителя получали 2,6 г (60%) комплекса 12 в форме желтого порошка. Соотношение рацемата к мезоформе было равно 3 : 1. Путем перекристаллизации из гексана смогли получить 1,3 г (30%) комплекса 12 в виде чистого рацемата (желтый кристаллический порошок).
1H-ЯМР-спектр (100 МГц, CDCl3: 7,27 (2, s, аром. -Н), 7,05 (2, s, аром. -Н), 6,80 (2, s, β -Ind-H), 2,6-3,2 (4, m, i-Pr-CH), 2,22 (6, s, Ind -CH3), 1,15-1,40 (30, m, i, -Pr-CH3, Si-CH3).
Масс-спектр: 642 M+ (относ.90Zr), правильный изотопный образец, правильный распад.
Пример O. Диметилбис(2,4,6,-триметилинденил)силан (13).
К раствору из 10,1 г (64 мМ) индена 2 в 50 мл ТГФ добавляли при комнатной температуре и в атмосфере Ar 25,5 мл (63,7 мМ) 2,5 М раствора бутиллития в гексане и смесь нагревали в течение 1 ч до образования обратного потока. Полученный таким образом раствор прикапывали при комнатной температуре к раствору из 4,1 г (32 мМ) диметилдихлорсилана в 20 мл ТГФ и затем производили нагревание в течение 3 ч до образования обратного потока. Реакционная смесь выливалась в ледяную воду и проводилось многократное извлечение эфира. Объединенные органические фазы высушивались сульфатам натрия и упаривались при пониженном давлении. Остаток очищали методом хроматографии через силикагель 60 в количестве 450 г. Обработкой смесью растворителей из гексана с 5%-ным метиленхлоридом сначала стало возможным проявить элюированием 2,5 г индена 2. При обработке гексаном с 8%-ным метиленхлоридом последовало 6,5 г лигандной системы 13 (изомеры). Выход составил 54% или 72% относительно индена 2, подвергшегося реакции обмена.
Пример P. Дихлорид диметилсиландиилбис(2,4, 6-триметилинденил цирконий (14).
К раствору из 2,0 г (5,4 мМ) лигандной системы 13 в 30 мл диэтилового эфира добавляли при комнатной температуре в атмосфере Ar 6,6 мл (16,2 мМ) 2,5 М раствора бутиллития в гексане и при этой же температуре производилось перемешивание в течение 5 - 6 ч. Раствор полностью упаривался. Оставшийся твердый остаток промывался порциями в общем 30 мл гексана и высушивался продолжительное время в вакууме, создаваемом масляным насосом. Подученный таким образом бежевый порошок добавлялся при -78oC к суспензии из 1,23 г 5,5 мМ тетрахлорида циркония в 30 мл метиленхлорида. После нагревания до комнатной температуры реакционная смесь полностью упаривалась и сушилась в вакууме, создаваемом масляным насосом. Твердый остаток состоял из смеси рацемической формы с мезо-формой в соотношении 1 : 1. Сначала он промывался небольшим количеством гексана. Затем экстрагировался толуолом общим количеством 120 мл. Раствор концентрировался и его оставляли при -35oC для кристаллизации. Получалось 800 мг (28%) цирконоцена 14 в виде чистого рацемата в форме оранжевых кристаллов.
1H-ЯМР-спектр рацемата (100 МГц, CDCl3): 7,20 (s, 2, аром. -Н), 6,97 (s, 2, аром. -Н), 6,70 (s, 2, β -Ind -H), 2,32 (s, 6, CH3), 2,27 (s, 6, CH3), 2,20 (s, 6, CH3), 1,27 (s, 6, Si-CH3).
Масс-спектр: 530 М+ (относ.90 Zr), правильный изотопный образец, правильный распад.
Пример R. Метилфенилбис-(2-метил-4, 6-диизопропилинденсилан)силан (15).
К раствору из 10 г (46 мМ) индена 4/4а в 200 мл ТГФ добавляли при комнатной температуре в атмосфере Ar 18,6 мл (46 мМ) 2,5 М раствора бутиллития в гексане и смесь нагревали в течение 1 ч до образования обратного потока. Раствор прикапывали при комнатной температуре к раствору из 4,48 г (23 мМ) метилфенилдихлорсилана в 30 мл ТГФ и производилось нагревание в течение 3 ч до образования обратного потока. Смесь выливалась в ледяную воду и производилось многократное экстрагирование. Объединенные органические фазы высушивались сульфатом натрия и упаривались. Остаток очищался методом хроматографии через 450 г силикагеля 60. С помощью смеси растворителей из гексана и метиленхлорида (10 : 1) сначала удалось извлечь 1,9 г не подвергшегося реакции обмена индена 4/4а. Затем последовало 7,4 лигандной системы 15 (смесь изомеров). Выход составил 57% или 73% относительно подвергшегося реакции обмена индена.
Пример S. Дихлорид метилфенилсилилбис-(2-метил-4,6-диизопропилинденил)циркония (16).
К раствору из 7,4 г (13,5 мМ) лигандной системы 15 в 30 мл диэтилового эфира добавляли при комнатной температуре и в атмосфере Ar 11,2 мл (28 мМ) 2,5 М раствора бутиллития в гексане и производилось перемешивание при комнатной температуре в течение 16 ч. После удаления растворителя оставшийся остаток сушился в течение 3 - 4 ч при 40 - 50oC и затем при -78oC добавлялся к суспензии из 3,2 г (13,5 мМ) тетрахлорида циркония в 40 мл метиленхлорида. После нагревания до комнатной температуры удалялся растворитель при пониженном давлении. Оставшийся твердый остаток высушивался в вакууме, создаваемом масляным насосом, и экстрагировался 100 мл гексана. После удаления растворителя получают 5, 4 (55%) цирконоцена 16 как смесь рацемической формы с мезо-формой в соотношении 2 : 1 (оранжево-коричневый кристаллический порошок). Перекристаллизацией из гексана может получаться чистая рацемическая форма.
1H-ЯМР-спектр смеси изомеров (100 МГц, CDCl3): 6,6-8,2 (m, аром. -Н, β -Ind -H), 2,5-3,2 (m, i -Pr -H), 2,52 (s, CH3), 2,32 (s, CH3), 2,20 (s, CH3), 1,90 (s, CH3), 1, 0-1,5 (m, i -Pr -CH3, Si-CH3).
Масс-спектр: 704 М+ (относ.90Zr), правильный изотопный обюразец, правильный распад.
Пример Т. 1,2-Бис-2-метил-4,6-диизопропилинденил этан 17.
К раствору из 5,0 г (23,3 мМ) индена 4/4а в 50 мл ТГФ добавляли при комнатной температуре и в среде Ar 18,6 мл (46 мМ) 2,5 М раствора бутиллития в гексане и проводилось нагревание в течение 1 ч до образования обратного потока. Этот раствор добавляли при -78o C к раствору из 2,18 г (11,0 мМ) 1, 2-дибромметана. Полученный раствор медленно нагревали до комнатной температуры и при этой температуре перемешивали в течение ночи. Смесь выливали в ледяную воду и производили экстрагирование. Объединенные органические фазы высушивались сульфатом натрия и упаривались. Остаток очищался методом хроматографии через 450 г силикагеля 60. С помощью смеси растворителей из гексана и метиленхлорида (от 20 : 1 до 10 : 1) сначала удалось извлечь 1,2 г не подвергшегося реакции обмена индена 4/4а. Затем последовало 1,7 г липидной системы 17 (бесцветное вещество). Выход составил 35% или 45% относительно подвергшегося реакции обмена индена.
Пример U. Дихлорид 1,2-этандиилбис-(2-метил-4,6-диизопропилинденил)циркония (18).
К раствору из 1,6 г (3, 52 мМ) лигандной системы 17 в 20 мл диэтилового эфира добавляли при комнатной температуре и в среде Ar 3,5 мл (8,8 мМ) 2,5 М раствора бутиллития в гексане и производилось перемешивание в течение ночи. Оставшийся после извлечения растворителя остаток промывали гексаном и сушили длительное время в вакууме, создаваемом масляным насосом. Полученный таким образом порошок добавляли при -78oC к суспензии, состоящей из 815 мг (3,5 мМ) тетрахлорида циркония в 15 мл метиленхлорида. После нагревания до комнатной температуры перемешивание продолжали еще 1 ч и удаляли растворитель при пониженном давлении. Остаток, высушенный в вакууме от масляного насоса, экстрагировали толуолом. После удаления растворителя и промывки гексаном получали 1,5 г (70%) цирконоцена 18 в виде смеси рацемический формы с мезо-формой в соотношении 2 : 1 (оранжевый порошок). Путем перекристаллизации из смеси толуола с гексаном возможно было получение 700 мг (32%) чистого рацемата.
1 H-ЯМР-спектр рацемата (100 МГц, CDCl3): 7,3 (s, аром. -Н), 7,0 (s, аром. -Н), 6,55 (s, β/ -Ind -H), 3,6 (s, C2H4), 2,6-3,2 (m, i -Pr -H), 2,2 (s, CH3).
Масс-спектр: 612 М+ (относ.90Zr), правильный изотопный образец, правильный распад.
Пример V. 2-Метил-6,
7-бензоиндан-1-он
(19а) и
2-метил-4,5-бензоиндан-1-он (19б)
К раствору, состоящему из 10 г (83 мМ) нафталина и 19 г (83 мМ) бромида 2-бромизомасляной кислоты в 200 мл CH2Cl2 добавляли
через
дозирующую воронку для твердых веществ 27,5 г (207 мМ) AlCl3; добавление производилось при комнатной температуре в течение 30 мин. Через 4 ч смесь перерабатывали
аналогично примеру
А.
Сырой продукт фильтровали с уксусным эфиром через небольшую, заполненную силикагелем колонку. После удаления растворителя получали 15,5 г (95%) смеси изомеров 19а/19б в виде
желтоватого масла.
Соотношение изомеров 19а : 19б составляло 1 : 2.
1H-ЯМР-спектр (100 МГц, CDCl3): 19a: 9,15 (m, 1H), 7,40-8,10 (m, 5H), 3,47 (dd, 1H), 2,62-2,95 (m, 2H), 1,37 (d, 3H); 19б : 7,4-8,0 (m, 6H), 3,7 (dd, 1H), 2,75-3,10 (m, 2H), 1,40 (d, 3H).
Масс-спектр: 196M+, правильный продукт распада.
Пример W. 2-Метил-6, 7-бензоиндан-1-он (19а).
Были использованы те же количества, что и в примере V. Нафталин вносили вместе с AlCl3 в CH2Cl2 и медленно прикапывали при комнатной температуре бромид бромизомасляной кислоты. Через 1,5 ч смесь перерабатывали как и в примере V. После очистки методом хроматографии через силикагель 60 с использованием смеси гексана и уксусного эфира получали 11 г (67%) инданона 19а.
Пример X. 2-Метил-4,5-бензоинден (20а) и 2-метил-6,7-бензоинден (20б).
К раствору, состоящему из 7,8 г (39, 7 мМ) смеси изомеров инданонов 19а/19б (пример V) в 200 мл смеси ТГФ/метанол (2 : 1) добавляли порциями при комнатной температуре 2,2 г (59,5 мМ) боргидрида натрия и осуществляли перемешивание в течение 14 ч. Раствор выливали в подкисленную HCl ледяную воду и извлекали эфир. Объединенные органические фазы многократно промывали водой и сушили сульфатом натрия. Остающееся после удаления растворителя масло оранжевого цвета растворяли в 240 мл толуола и нагревали вместе с 570 мг (3,15 мМ) паратолуолсульфоновой кислоты в течение 15 мин до температуры 80oC. Раствор промывали многократно водой при комнатной температуре, сушили сульфатом натрия и упаривали. Остаток очищался методом хроматографии через 300 г силикагеля 60. При помощи смеси растворителей из гексана с диизопропиловым эфиром (20 : 1) стало возможным проявить элюированием 4,7 г (65%) смеси изомеров инденов 20а/20б в соотношении 1 : 2 (бесцветное масло).
1H-ЯМР-спектр (360 МГц, CDCl3: смесь изомеров 7,2-8,1 (m), 7,05 (m), 6,57 (m), 3,57 (s), 3,42 (m), 2,25 (d), 2,20 (d).
Молярная масса: 180 М+, правильный образец распада.
Пример Y. Диметилбис-(2-метил-4,5-бензоинденил)силан (21).
К раствору, состоящему из 4,6 г (25,5 мМ) смеси изомеров инденов 20а/20б (пример Х), в 50 мл ТГФ добавляли при комнатной температуре 10,2 мл (25,5 мМ) 2,5 М раствора бутиллития в гексане и раствор нагревали в течение 1 ч до обратного потока. Раствор красного цвета прикапывали затем при комнатной температуре к раствору, состоящему из 1,55 г (12 мМ) диметилдихлорсилана в 10 мл ТГФ и производили нагревание в течение 5 - 6 ч до обратного потока. Реакционный раствор выливали в ледяную воду и осуществляли многократное экстрагирование. Высушенные сульфатам натрия объединенные органические фазы упаривали и сушили в вакууме, создаваемом масляным насосом. Остаток очищали методом хроматографии через 300 г силикагеля 60. При помощи смеси растворителей из гексана и 3%-ного уксусного эфира сначала удалось проявить элюированием 500 г не подвергшегося реакции обмена, исходного продукта 20а/20б. С тем же растворителем далее следовала лигандная система 21. Ее кристаллизацию удалось провести после удаления растворителя путем смешивания с гексаном. Выход составил 1,7 г (34% относительно Si или 44% относительно подвергшейся реакции обмена смеси веществ 20а/20б).
1H-ЯМР-спектр (100 МГц, CDCl3): диастереомеры (1 :1) 7,2-8,2 (m), 4,05 (s), 2,45 (d), 2,35 (d), -0,22 (s), -0, 32 (s), -0,35 (s).
Масс-спектр: 416 М+, правильный продукт распада и изотопный образец.
Пример Z. Рац.-диметилсиландиилбис(2-метил-4, 5-бензоинденил)цирконийдихлорид (22).
К раствору, состоящему из 1,7 г (4,1 мМ) лигандной системы 21, в 20 мл ТГФ добавляли при комнатной температуре и в среде Ar 4,0 мл (10,2 мМ) 2, 5 М раствора бутиллития в гексане и производили перемешивание в течение 14 ч при комнатной температуре. Оставшийся после извлечения растворителя остаток сушили в вакууме, создаваемом масляным насосом, и промывали гексаном. Полученный таким образом светло-коричневый порошок подвергали многочасовой сушке в вакууме масляного насоса при 40 - 50oC, после чего добавляли при -78oC к суспензии из 1,0 г (4,0 мМ) тетрахлорида циркония в 25 мл метиленхлорида. После нагревания до комнатной температуры удаляли растворитель и остаток экстрагировали 20 мл толуола для того, чтобы удалить мезо-форму цирконоцена 22. Остаток толуольной вытяжки затем экстрагировали 40 мл метиленхлорида. Раствор концентрировали до небольшого объема и оставляли кристаллизоваться при -35oC. Несколькими фракциями удалось выделить всего 970 мг (42%) цирконоцена 22 в виде чистого рацемата.
1-H-ЯМР-спектр рацемата (300 Мгц, CDCl3): 7,92 (2, m), 7,78 (2, m), 7,60 (2, d), 7,48-7,56 (4, m), 7,36 (2, d), 7,27 (2, s, α -Ind -H), 2,37 (6, s, Ind -CH3), 1,36 (6, s, Si-CH3).
Масс-спектр: 574 М+, правильный распад, правильный изотопный образец.
Пример AA. 2-Метил-α-аценафтиндан-1-он (23).
К раствору из 20 г (129 мМ) α-аценафтена в 320 мл метиленхлорида добавляли при комнатной температуре 29,7 г (129 мМ) бромида 2-бромизомасляной кислоты. Затем в течение 15 мин добавляли через дозирующую воронку для твердых веществ 43,5 г (324 мМ) AlCl3. После перемешивания в течение 30 мин раствор выливали в ледяную воду и экстрагировали метиленхлоридом. Органическая фаза промывалась водой, раствором NaHCO3 и сушилась NaSO4. Остающийся после удаления растворителя остаток фильтровали через небольшую колонку с силикагелем. При помощи смеси гексана с уксусным эфиром (9 : 2) получали 25 г (87%) инданона 23.
1H-ЯМР (CDCl3, 100 МГц): 8,57 (d, 1), 7,60 (t, 1), 7,35 (d, 1), 7,25 (s, 1), 3,45 (q, 1), 3,40 (s, 4), 2,60-2,95 (m, 2), 1,35 (d, 3).
Пример BB. 2-Метил-α-аценафтинден (24).
Раствор из 20 г (90 мМ) соединения 23 в 250 мл смеси ТГФ/метанола (2 : 1) прикапывали к суспензии, состоящей из 3,8 г 100 мМ NaBH4 в 80 мл ТГФ. Смесь перемешивали в течение 2 ч при комнатной температуре и добавляли к ней 100 мл уксусного эфира и 100 мл полуконц. HCl. Затем производили нагревание в течение 10 мин до образования обратного потока и экстракцию уксусным эфиром. Органическую фазу промывали водой и сушили Na2SO4. При концентрировании и охлаждении до -35oC произошла кристаллизация за несколько фракций всего 16,3 г (88 %) соединения 24.
1H-ЯМР (CDCl3, 100 МГц): 7,1-7,8 (m, 4, аромат. -Н), 6,97 (m, 1, олефин-Н), 3,37 (s, 6,
CH2),
2,
25 (d, 3, CH3)
Пример CC. Диметилбис-(2-метил-α-аценафтинденил)силан (25).
Аналогично примеру 24 10,8 г 52,4 мМ соединения 24 подвергали реакции депротонирования и реакции обмена с диметилдихлорсиланом. Органическую фазу упаривали и остаток очищали методом хроматографии через силикагель. С помощью смеси гексана с 4%-ным уксусным эфиром удалось выделить 6,2 г (51%) лигандной системы 25.
1H-ЯМР (CDCl3, 100 МГц): два диастереомера 7,1-7,8 (m, аромат. -Н), 4,0 (s, CH), 3,45 (s, CH2), 2, 47 (d, CH3), 2,40 (d, CH3), -0,25 (s, SiCH3), -0,37 (s, SiCH3).
Пример DD. Рац.-диметилсиландиилбис-(2-метил- α -аценафтинденил)цирконий дихлорид (26).
Аналогично примеру Р подвергали реакции обмена 4,9 г (10,5 мМ) лигандной системы 25. Сырой продукт, состоящий из рацемический формы с мезо-формой в соотношении 1 : 1, перекристаллизовывали из хлороформа. Получали 1,3 г (20%) рацемата 26 в форме оранжевого-желтого порошка.
1H-ЯМР (CDCl3, 100 МГц): 7,0-7,8 (m, аромат. -Н), 3,1-3, 4 (m, CH2), 2,35 (s, CH3), 1,35 (s, SiCH3).
Примеры процесса полимеризации:
Пример 1. Сухой реактор
емкостью 24 дм3 промывали
пропиленом и заполняли 12 дм3 объема реактора жидким пропиленом. Затем добавляли раствор метилалюмоксана в толуоле объемом 35 см3
(соответствует 52 мМ Al, средней степени
олигомеризации р = 20) и смесь перемешивали при 30oC в течение 15 мин.
Параллельно этому растворяли 3,5 мг (0,005 мМ) дихлорида рац.-диметилсилил(2-метил-4, 6-диизопропил-1-инденил)2 циркония в растворе метилалюминоксана в толуоле (20 мМ Al) объемом 13,5 см3 и смесь предварительно активировалась, оставаясь в покое в течение 15 мин.
Бордовый раствор добавляли затем в реактор, подводом тепла нагревали его до 75oC (10oC/мин) и выдерживали полимеризационную систему в течение 1 ч при 70oC, охлаждая реактор. Процесс полимеризации прекращался за счет газовыделения избыточных мономеров. Было получено 2,11 кг полипропилена.
Активность металлоцена составляла тем самым 603 кг ПП/г металлоцена в ч.
Коэф. вязк. = 259 см3/г, Mw= 305000 г/моль; Мw/Мn = 2,0; инд. изотакт. = 96,0 каж. плотн. = 400 г/дм3, инд.плавл. (230/5) = 8,5o/мин.
Пример 2. Сухой реактор емкостью 24 дм3 промывали пропиленом и заполняли 12 дм3 объема реактора жидким пропиленом. Затем добавляли раствор метилалюминоксана в толуоле 35 см3 (соответствует 52 мМ Al, средней степени олигомеризации р = 20) и смесь перемешивали при 30oC в течение 15 мин.
Параллельно растворяли 3,5 мг (0,005 мМ) дихлорида - диметилгермилдиилбис-(2-метил-6-фенил-1-инденил)циркония в растворе метилалюминоксана в толуоле (20 мМ Al) объемом 13,5 см3 и смесь предварительно активировали, оставляя в покое в течение 15 мин.
Раствор добавляли затем в реактор, нагревали его до 75oC (10oC/мин) и выдерживали полимеризационую смесь в течение 1 ч при 70oC. Процесс полимеризации прекращался за счет газовыделения избыточных мономеров.
Активность металлоцена составляла 168 кг ПП/г металлоцена в ч.
Сравнительный пример 1. Пример 1 был повторен с металлоценом, каким является дихлорид рац. -диметилсилил(2-метил-1-инденил)2цирконий. Активность металлоцена была равна 395 кг ПП/г металоцена в ч.
Коэф. вязк. = 159 см3/г, Мw = 158000 г/моль, Мw/Мn = 2,1 и инд. пл. (230/5) была равна 48o/мин. Показатель изоактивности (11) составлял 96%.
Сравнительный пример 2. Пример 1 был повторен с металлоценом, каким является дихлорид рац.-диметилсилил (2-метил-4-изопропил-1-инденил)2 цирконий.
Активность металлоцена была равной 460 кг ПП/г металлоцена в ч, коэф. вязк. = 152 см3/г, Мw = 147500 г/моль, Мw/Mn = 2,3 и инд. пл. (230/5) = 51o/мин.
Сравнительный пример 3. Пример 1 был повторен с дихлоридом рац.-диметилсилил (1-инденил)2 циркония. Активность металлоцена была равной 695 кг ПП/г металлоцена в 1 ч.
Коэф. вязк. = 31 см3/г, Мw = 18500 г/моль, Мw/Мn = 2,2, инд.пл. (230/5) измерить уже не удалось.
Сравнительный пример 4. Пример 1 был повторен с металлоценом, каким является дихлорид рац.-диметилсилил-(4,7-диметил-1-инденил)2циркония. Активность металлоцена была равной 195 кг ПП/г металлоцена х ч., коэф. вязк. = 16 см3/г, Мw = 9500 г/моль, Mw/Mn = 2,0, 11 = 87%, инд. пл. (230/5) измерить уже не удалось.
Четыре сравнительных опыта показывают, что полипропилены, полученные с использованием по-разному замещенных в лигандах инденила металлоценов или с использованием незамещенных металлоценов, значительно отличаются друг от друга по молекулярной массе. Включая предложенный в изобретении металлоцен из примера 1, ее разброс составляет от области молекулярных масс восков (сравнительный пример 4) до очень высокомолекулярного предложенного полимера (пример 1).
Данные опыты подтверждают преимущество замещенных во 2,4,6-положении металлоценов.
Реферат
Изобретение касается способа
получения соединения формулы IV или IVa

где R1 - R5 предпочтительнее являются водородом или алкилом, отличающегося тем, что проводят реакцию между соединением формулы I

и соединением формулы II
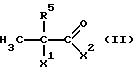
где X1 и X2 предпочтительнее являются галогеном, до образования соответствующих инданонов, которые путем восстановления и дегидратации переводятся в соединения IV и IVa. Соединения IV и IVa являются важными промежуточными продуктами при получении хиральных комплексов из металлоценов, которые могут использоваться в качестве компонентов катализаторов при полимеризации олефинов. 4 с. п. ф-лы.
Формула
где

R1, R2, R3, R4, R5 одинаковые или различные, водород, низший алкил, низший алкоксил, С6 - С14-арил или расположенные рядом радикалы R1 R4 вместе в атомами углерода ядра образуют одно или несколько колец,
отличающийся тем, что осуществляют реакцию между соединением формулы I
где R1, R2, R3, R4 имеют указанные значения,
с соединением формулы II

где Х1 и Х2 одинаковые или различные, отщепляемая нуклеофильная группа;
R5 водород, низший алкил или низший алкоксил,
или его ангидридом в присутствии катализатора Фриделя-Крафтса до образования соединения формулы III или IIIа


где R1 R5 имеют указанные значения,
с последующим его восстановлением и дегидратацией в соединение IV.

где

R1, R2, R3, R4, R5 одинаковые или различные, водород, низший алкил, низший алкоксил, С6 - С14 -арил или расположенные рядом радикалы R1 R4 вместе с атомами углерода ядра образуют одно или несколько колец,
в качестве промежуточных соединений для получения металлоценкомплексов.

где R1, R2, R3, R4, R5 одинаковые или различные, водород, низший алкил, низший алкоксил, С6 - С14-арил или расположенные рядом радикалы R1 R4 вместе с атомами углерода ядра образуют одно или несколько колец;
R7 остаток

где R8 и R9 одинаковые или различные, водород, низший алкил, фенил;
М1 цирконий, гафний;
М2 углерод, кремний, германий;
R10 и R11 одинаковые, галоген;
р 1 или 2.
Комментарии