Новый способ синтеза (е)-стильбеновых производных, который позволяет получить ресвератрол и писатаннол - RU2443671C2
Код документа: RU2443671C2
Чертежи

Описание
Объектом данного изобретения является новый способ синтеза (Е)-стильбеновых производных, предназначенных, в частности, для получения ресвератрола и писатаннола.
Более конкретно, изобретение относится к способу синтеза (Е)-стильбеновых производных формулы (VI), описанной в данной патентной заявке, в частности (Е)-триметилресвератрола, (Е)-трибензилресвератрола и (Е)-тетраметилписатаннола, которые позволяют получить ресвератрол и писатаннол.
Полигидроксистильбены представляют собой соединения, которые найдены в различных растениях и которым уделено особенное внимание, так как они проявляют большое разнообразие терапевтических свойств.
Данные производные включают ресвератрол ((Е)-3,5,4'-тригидроксистильбен) и писатаннол ((Е)-3,5,3',4'-тетрагидроксистильбен) формул:

ресвератрол

писатаннол
Ресвератрол и писатаннол представляют собой соединения, принадлежащие классу полифенолов, известных по проявлению антиоксидантных эффектов, способных предотвращать или замедлять вредные воздействия окислительного стресса.
В терапевтической области ресвератрол каталогизирован как средство против агрегации тромбоцитов, противовоспалительное средство или вазодилатор или как ингибитор пролиферации клеток.
Данные соединения приведены в разработке многих синтетических путей, но последние являются неудовлетворительными с производственной точки зрения.
Предусмотренные синтетические пути требуют в большинстве случаев защиты фенольных функциональных групп, либо в виде простых эфирных производных (обычно метильных, изопропильных, бензильных или силильных производных), либо в виде сложноэфирных производных (обычно ацетильных или бензоильных производных), и полигидроксистильбены затем получают высвобождением указанных функциональных групп известными способами.
Самый широко применяемый способ получения ресвератрола или писатаннола описан в ряде публикаций и патентных заявок, включающих следующие: ЕР 1466884; WO 2003/086414. Он заключается в конденсации по Виттигу или Виттигу-Хорнеру защищенного гидроксиароматического (или полигидроксиароматического) альдегида, такого как защищенный 3,5-дигидроксибензальдегид, с солью фосфония или фосфонатом, таким как защищенный бромид 4-гидроксибензилтрифенилфосфония.
Однако реакции Виттига или Виттига-Хорнера приводят к смеси (Е)- и (Z)-стильбеновых изомеров, которые трудно разделить, поэтому требуется дополнительная стадия для преобразования нежелательного Z-изомера в Е-изомер или с йодом в качестве катализатора, как описано в US 2004/00115020, или же взаимодействием с диарилдисульфидом, как описано в Chem. Pharm. Bull., (1992), 40(10), 2842-2844. Эта дополнительная стадия приводит к образованию побочных продуктов, поэтому требуется сложная стадия очистки, которая не очень желательна в производственном плане. Преобразование Z-изомера в E-изомер может быть осуществлено путем взаимодействия Z-изомера с комплексом палладия(II), как описано в J. Org. Chem., (2002), 67, 4627-4629. Однако большое количество указанного комплекса, который будет использоваться [20 мол.% (MeCN)2PdCl2], делает способ очень дорогим.
Другой общепринятый путь получения ресвератрола или писатаннола заключается в получении α-фенилкоричной кислоты по реакции Перкина, как описано в WO 2000/69430 и в Tetrahedron, 59, (2003), 3315-21, путем взаимодействия гидрокси- (или полигидрокси)фенилуксусной кислоты (или простого эфирного/сложноэфирного производного) с (защищенным или незащищенным) гидрокси- (или полигидрокси)ароматическим альдегидом. Декарбоксилирование производного коричной кислоты (Cu/хинолин при 260°С) затем дает производное стильбена.
Однако последняя реакция подразумевает жесткие условия (высокая температура, загрязнение металлического катализатора) для декарбоксилирования и обычно приводит к преобладанию (Z)-изомера, при этом требуется проведение дополнительной стадии изомеризации.
В другом пути синтеза ресвератрола или писатаннола используют реакции типа реакции Хека, такие как конденсация 3,5-диацетоксистирола с 4-ацетоксибромбензолом, как описано в WO 2005/023740, или же конденсация 4-ацетоксистирола с 3,5-диметоксибензоилхлоридом, как описано в WO 2001/60774, или же вновь конденсация с 3,5-диацетоксибензоилхлоридом, раскрытая в WO 2005/069998.
Однако данные реакции требуют использования исходных соединений, которые трудны для получения, таких как 3,5-диацетоксистирол, а также катализаторов на основе солей палладия, которые являются дорогостоящими и не очень устойчивыми при требуемых условиях реакции, что приводит к низким и невоспроизводимым выходам.
Для устранения недостатков вышеприведенных синтетических путей и снижения затрат на получение ресвератрола и писатаннола изобретатели разработали альтернативный путь синтеза полигидроксистильбенов.
Этот новый путь заключается в получении (Е)-стильбеновых производных из производных 1,2-диарилэтанона.
Полученные (Е)-стильбеновые производные представляют собой изомеры Е-типа, с которых затем снимают защиту с получением соединений, представляющих интерес, таких как, например, ресвератрол или писатаннол.
Новый синтетический путь обладает, в дополнение к освобождению от стадии разделения Е- и Z-изомеров, которая прежде представляла проблему в данной области, преимуществом применения, в качестве исходных соединений, производных 1,2-диарилэтанона, которые могут быть получены с низкими затратами из реагентов, таких как гидроксиароматические кислоты, необязательно этерифицированные, и гидроксиароматические сложные эфиры.
Фигура 1 демонстрирует новую схему синтеза полигидроксистильбенов, раскрываемую в данной патентной заявке.
Первым объектом данной заявки, таким образом, является способ синтеза (Е)-стильбеновых производных формулы (VI)

в которой
А представляет собой водород или группу OR2, и
R1, R2, R'1 и R'2 представляют собой, независимо друг от друга, линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(СН2)n- c n=1-3,
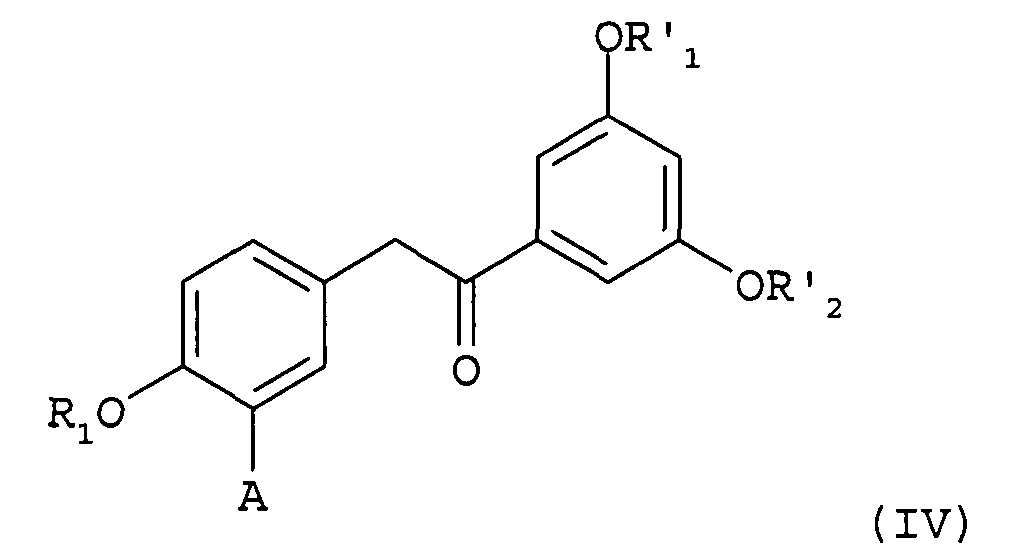
отличающийся тем, что производное 1,2-диарилэтанона формулы (IV)

в которой
А представляет собой водород или группу OR2, и
R1, R2, R'1 и R'2 представляют собой, независимо друг от друга, линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(СН2)n- c n=1-3;
подвергают взаимодействию в качестве промежуточного соединения синтеза.
В рамках настоящего изобретения, когда R1, R2, R'1 и R'2 представляют собой, независимо друг от друга, линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, это означает, например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, пентильную или гексильную группу. Когда R1, R2, R'1 и R'2 представляют собой аралкильную группу, содержащую от 7 до 16 атомов углерода, это означает, например, бензильную, 1-фенилэтильную, нафтилметильную или 1-нафтилэтильную группу.
В рамках настоящего изобретения, что касается заместителей, термин “алкокси” означает, например, радикал метокси, этокси, пропокси или бутокси.
Галогеновый радикал означает Cl, Br, F или I.
Предпочтительный аспект изобретения заключается в синтезе следующих (Е)-стильбеновых производных формулы (VI):
- (Е)-триметилресвератрола, в котором А представляет собой атом водорода, R1, R'1 и R'2 являются метильными группами, или
- (Е)-трибензилресвератрола, в котором А представляет собой атом водорода, R1, R'1 и R'2являются бензильными группами,
для получения ресвератрола, и
- (Е)-тетраметилписатаннола, в котором А представляет собой -ОСН3, R1, R'1 и R'2 являются метильными группами,
для получения писатаннола.
Такие соединения формулы (VI) описаны в литературе.
Промежуточные соединения формулы (IV), в которой А представляет собой водород и каждый из R1, R'1 и R'2 представляет собой бензильную группу, или в которой А представляет собой группу -ОСН3 и каждый из R1, R'1 и R'2 представляет собой метильную группу, или в которой А представляет собой водород, R1 и R'2 представляют собой метильную группу и R1 представляет собой изопропильную группу, или в которой А представляет собой группу -OR2, R'1 и R'2 представляют собой метильную группу и R1 и R2 образуют углеводородную цепь -(СН2)n- c n=1, являются новыми соединениями, которые представляют собой следующий объект изобретения.
Данные новые соединения формулы (IV) включают, в частности, следующие:
- 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанон (или 3,5,4'-трибензилоксидезоксибензоин);
- 1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этанон (или 3,5,3',4'-тетраметоксидезоксибензоин);
- 1-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)этанон, и
- 1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этанон,
которые применяются для получения следующих производных формулы (VI) соответственно:
- (E)-трибензилресвератрола, который позволяет получить ресвератрол в процессе стадии, описанной ниже;
- (Е)-тетраметилписатаннола, который позволяет получить писатаннол в процессе стадии, описанной ниже;
- (Е)-3,5-диметокси-4'-изопропилоксистильбена, и
- (Е)-3,5-диметокси-3',4'-метилендиоксистильбена.
Другой предпочтительный аспект изобретения заключается в синтезе (Е)-триметилресвератрола (соединения формулы (VI)) из соединения формулы (IV), 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанона (или 3,5,4'-триметоксидезоксибензоина).
Соединения формулы (IV) можно использовать в двух различных схемах для получения производного (Е)-стильбена формулы (VI), что и является основой данного изобретения:
- либо восстановлением соединения формулы (IV) с получением спирта формулы (V) и затем дегидратацией образовавшегося спирта (путь А);
- либо синтезом производных арилсульфонилгидразона путем взаимодействия соединения формулы (IV) c арилсульфонилгидразидом, а затем взаимодействием образовавшихся арилсульфонилгидразонов с основанием (путь В).
Синтез 1,2-диарилэтанонов формулы (IV)
1,2-Диарилэтаноны формулы (IV), используемые в способе согласно изобретению, предпочтительно получают реакцией декарбоксилирования исходя из сложных β-кетоэфиров формулы (III)

в которой
А представляет собой водород или же группу OR2,
R1, R2, R'1 и R'2 представляют собой, независимо друг от друга, линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(СН2)n- c n=1-3,
R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода.
В рамках данного изобретения, когда R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, это означает, например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, пентильную или гексильную группу.
Реакцию декарбоксилирования сложных β-кетоэфиров формулы (III) с получением кетонов формулы (IV) можно проводить в кислых условиях в присутствии, например, пар кислота/растворитель, таких как следующие пары: концентрированная хлористоводородная кислота/уксусная кислота, концентрированная хлористоводородная кислота/этанол или серная кислота/уксусная кислота, или же без растворителя, в присутствии борной кислоты или ангидрида, как описано в Advanced Organic Chemistry, Reaction, Mechanisms and Structure, John Wiley & Sons, 4th edition, page 629.
Предпочтительно, реакцию декарбоксилирования проводят без растворителя в присутствии 1-5 эквивалентов борной кислоты или ангидрида борной кислоты при температуре от 100 до 180°С, более предпочтительно в присутствии 1-2 эквивалентов борной кислоты или еще более предпочтительно с 1 эквивалентом борной кислоты.
В данном изобретении предлагаются, с этой целью, новые соединения формулы (III), в которой:
R представляет собой метильную группу, и
или А представляет собой водород и группы R1, R'1 и R'2 представляют собой метильные группы или бензильные группы,
или А представляет собой группу -ОСН3 и каждая из групп R1, R'1 и R'2 представляет собой метильную группу,
или А представляет собой водород, R'1 и R'2 представляют собой метильную группу и R1 представляет собой изопропильную группу,
или А представляет собой группу -OR2, R'1 и R'2 представляют собой метильную группу и R1 и R2 образуют углеводородную цепь -(СН2)n- c n=1;
включающие, в частности, следующие соединения:
- метил 3-(3,5-диметоксифенил)-2-(4-метоксифенил)-3-оксопропионат;
- метил 3-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)-3-оксопропионат;
- метил 3-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)-3-оксопропионат;
- метил 3-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)-3-оксопропионат;
- метил 3-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)-3-оксопропионат;
причем эти соединения позволяют получить соответственно следующие соединения формулы (IV):
- 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанон;
- 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанон;
- 1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этанон;
- 1-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)этанон; и
- 1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этанон;
используемые соответственно для получения (Е)-триметилресвератрола, (Е)-трибензилресвератрола, (Е)-тетраметилписатаннола, (Е)-3,5-диметокси-4'-изопропилоксистильбена и (Е)-3,5-диметокси-3',4'-метилендиоксистильбена.
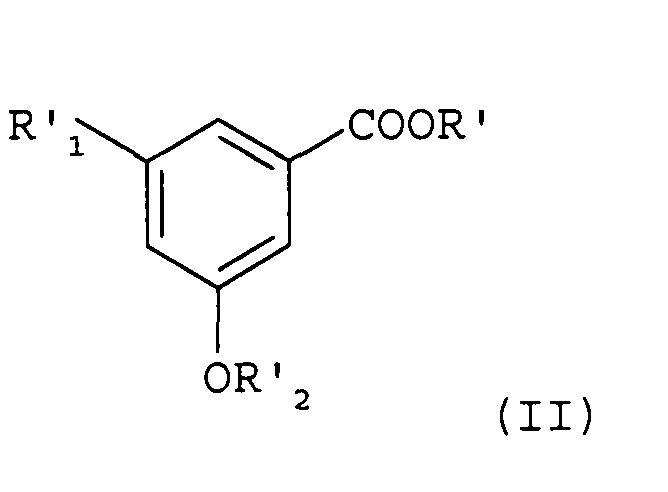
Сложные β-кетоэфиры формулы (III) можно получить, предпочтительно, по реакции конденсации типа реакции Клайзена между простыми эфирными/сложноэфирными производными (I) и простыми эфирными/сложноэфирными производными (II), такими как описано, например, в Advanced Organic Chemistry, Reaction, Mechanisms and Structure, John Wiley & Sons, 4th edition, page 491-493, как представлено ниже:

В формулах (I) и (II)
А представляет собой водород или же группу OR2,
R1, R2, R'1 и R'2 представляют собой, независимо друг от друга, линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(СН2)n- c n=1-3, и
R и R' представляют собой, независимо друг от друга, линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода.
В данном изобретении, когда R' представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, это означает, например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, пентильную или гексильную группу.
Данную реакцию конденсации обычно проводят в присутствии сильного основания при температуре кипения реакционной среды с обратным холодильником со стехиометрическими количествами простых эфирных/сложноэфирных производных (I) и (II).
В качестве примеров сильного основания можно назвать алкоксиды щелочных металлов, такие как этоксид натрия, или гидриды щелочных металлов, такие как гидрид натрия.
В предпочтительных условиях осуществления способа, описанного выше, используют от 2 до 5 эквивалентов сильного основания, в частности от 2 до 2,5 эквивалентов.
Исходные простые эфиры/сложные эфиры (I) и (II) можно синтезировать из соответствующих гидроксиароматических кислот, гидроксиароматических сложных эфиров или этерифицированных гидроксиароматических кислот известными способами, такими как описано в J. Med. Chem., 30(11), (1987), 2121-26; Tetrahedron, 59, (2003), 3315-22; Chem. Lett., 11, (1999), 1193-94; J. Am. Chem. Soc., 126(32), (2004), 9882-83. Данные исходные соединения представляют собой недорогие реагенты, которые являются простыми для использования специалистом в данной области.
4-Гидроксифенилуксусную кислоту, резорциловую кислоту и 3,4-дигидроксифенилуксусную кислоту можно привести в качестве примеров гидроксиароматических кислот.
Путь А для синтеза соединений формулы (VI)
Данный путь заключается в восстановлении кетонов формулы (IV), описанной выше, для получения производных 1,2-диарилэтанола формулы (V), указанной ниже:
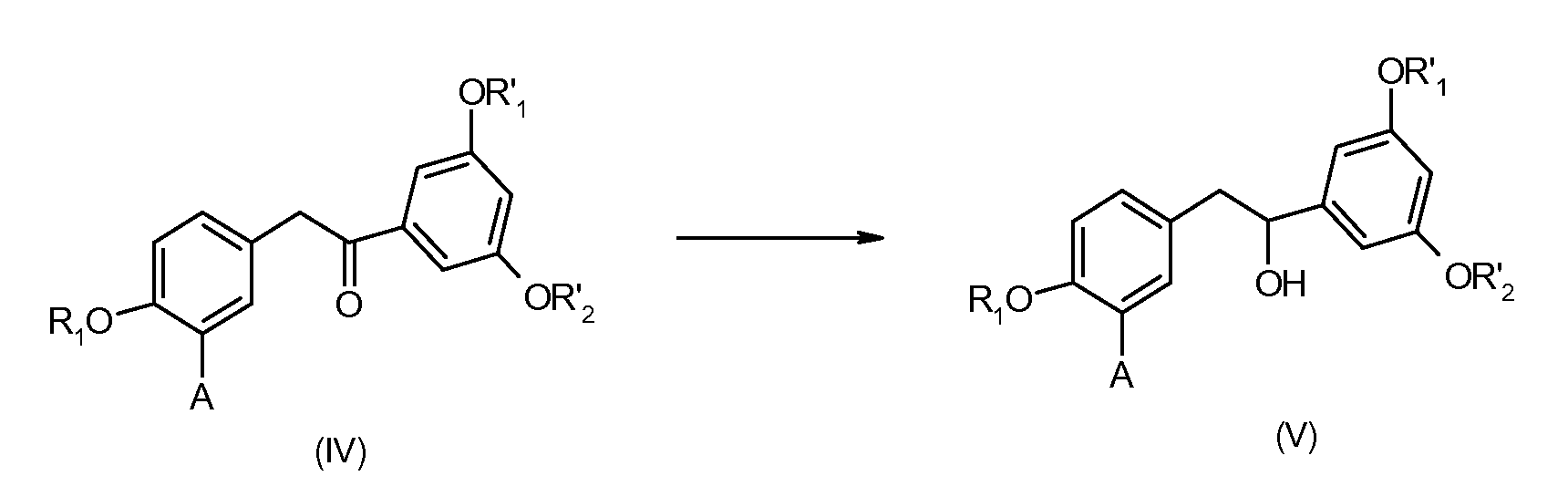
в которой
А представляет собой водород или же группу OR2,
R1, R2, R'1 и R'2 представляют собой, независимо друг от друга, линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(СН2)n- c n=1-3.
Кетоны формулы (IV) могут быть восстановлены путем применения или адаптации способов, описанных, например, в Advanced Organic Chemistry, Reaction, Mechanisms and Structure, John Wiley & Sons, 4th edition, page 910-918.
В предпочтительных условиях способа, описанного выше, кетоны формулы (IV) восстанавливают с образованием спиртов формулы (V) действием гидрида металла, такого как LiAlH4 или NaBH4. Это восстановление обычно осуществляют, используя от 0,25 до 3 эквивалентов гидрида металла. В частности, можно использовать 1 эквивалент NaBH4.
Альтернативно, для кетонов формулы (IV), в которой А представляет собой водород или же группу OR2 и R1, R2, R'1 и R'2 представляют собой, независимо друг от друга, линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, причем для R1 и R2 также возможно образование углеводородной цепи -(СН2)n- c n=1-3, восстановление можно осуществить путем гидрирования. В предпочтительных условиях гидрирование проводят в присутствии катализаторов, таких как Pd/C, в растворителе, таком как метанол или этанол, при давлении водорода порядка от 3×105 Па (3 бар) до 50×105 Па (50 бар), при температуре от температуры окружающей среды до приблизительно 50°С. В частности, указанную реакцию гидрирования проводят при давлении водорода от 5×105 Па до 10×105 Па, при температуре окружающей среды, в присутствии от 5 до 20 мас.% Pd/C по отношению к кетону формулы (IV).
Данная реакция позволяет получить, в частности, следующие предпочтительные соединения формулы (V):
- 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанол,
- 1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этанол
и, более конкретно, новое соединение формулы (V), в которой А представляет собой водород и R1, R'1 и R'2 представляют собой бензильную группу, включающее, в частности, 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанол. Данное новое соединение получено взаимодействием соединения формулы (IV), 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанона, как указано выше.
После того как образуются спирты формулы (V), последние подвергают дегидратации в присутствии каталитических количеств сильной кислоты, такой как, например, серная кислота, п-толуолсульфоновая кислота или фосфорная кислота.
Предпочтительно, реакцию дегидратации проводят в ароматическом растворителе, таком как толуол, при температуре кипения с обратным холодильником, в присутствии каталитических количеств п-толуолсульфоновой кислоты от 1 до 20 мол.% по отношению к спирту формулы (V) и более предпочтительно от 5 до 10 мол.%. Воду, образовавшуюся в ходе реакции, обычно удаляют азеотропной перегонкой. Согласно данной методике получают (Е)-стильбеновые производные формулы (VI) по изобретению.
Путь В для синтеза соединений формулы (VI)
Этот другой путь заключается, на первой стадии, в синтезе арилсульфонилгидразоновых производных формулы (VII) путем взаимодействия соединения формулы (IV) с арилсульфонилгидразидом, как указано на следующей схеме:
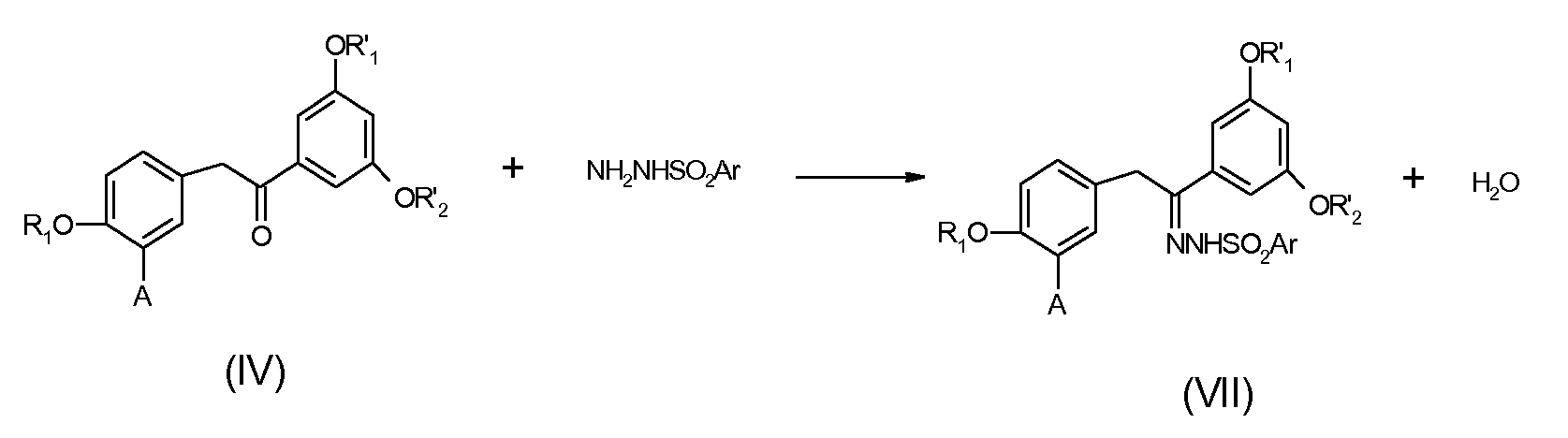
Данную реакцию обычно проводят в спиртовом растворителе, таком как метанол или этанол, или в ароматическом растворителе, таком как толуол, в присутствии каталитических количеств кислоты, такой как серная кислота или хлористоводородная кислота, если необходимо.
Производные арилсульфонилгидразида известны из литературы или являются коммерчески доступными. В качестве примеров можно назвать фенилсульфонилгидразид и п-толуолсульфонилгидразид, продаваемые фирмой Aldrich.
В предпочтительных условиях данную реакцию проводят при температуре кипения с обратным холодильником в этаноле или толуоле, используя избыток арилсульфонилгидразида от 1,1 до 1,5 эквивалентов. Предпочтительным является п-толуолсульфонилгидразид.
Соединения формулы (VII)

синтезированные таким образом, отличаются тем, что:
Ar представляет собой фенильную или о-, м- или п-толильную группу,
А представляет собой водород или же группу OR2,
R1, R2, R'1 и R'2 представляют собой, независимо друг от друга, линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(СН2)n- c n=1-3.
В частности, в данном изобретении предлагаются новые соединения формулы (VII), отличающиеся тем, что:
Ar представляет собой п-толильную группу, и
или А представляет собой водород и все три группы R1, R'1 и R'2 представляют собой метильные группы или бензильные группы,
или А представляет собой группу -ОСН3 и каждая из групп R1, R'1 и R'2 представляет собой метильную группу,
или А представляет собой водород, R'1 и R'2 представляют собой метильную группу и R1 представляет собой изопропильную группу,
или А представляет собой группу -OR2, R'1 и R'2 представляют собой метильную группу и R1 и R2 образуют углеводородную цепь -(СН2)n- c n=1.
Данные новые соединения формулы (IV) включают, в частности, следующие:
- N-[1-(3,5-диметоксифенил)-2-(4-метоксифенил)этилиден]-N'-тозилгидразин,
- N-[1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этилиден]-N'-тозилгидразин и
- N-[1-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)этилиден]-N'-тозилгидразин;
причем эти соединения особенно применимы для получения ресвератрола согласно данному изобретению, и
- N-[1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этилиден]-N'-тозилгидразин и
- N-[1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этилиден]-N'-тозилгидразин;
которые особенно применимы для получения писатаннола согласно данному изобретению.
Согласно способу по изобретению, на второй стадии, арилсульфонилгидразоны формулы (VII) подвергают взаимодействию в условиях Шапиро или Бамфорда-Стевенса путем применения или адаптации способов, описанных, например, в Organic Chemistry, Reactions, Mechanisms and Structure, John Wiley & Sons, 4th edition, pages 1019-1021.
Данную реакцию обычно проводят в растворителе в присутствии основания и в присутствии каталитических количеств катализатора фазового переноса или поверхностно-активного вещества, если они необходимы.
В предпочтительных условиях осуществления способа, описанного выше, используют избыток сильного основания от 2 до 3 эквивалентов, такого как производные лития, например метиллитий, этиллитий, бутиллитий или диизопропиламид лития, в негидроксилированных растворителях, таких как диэтиловый эфир, изопропиловый эфир, метил-трет-бутиловый эфир, ТГФ или диоксан, при температуре от 0°С до 5°С.
В других предпочтительных условиях используют, по меньшей мере, один эквивалент сильного основания, более предпочтительно избыток сильного основания от 2 до 5 эквивалентов, такого как алкоксиды щелочных металлов, например метоксид натрия, трет-бутоксид калия или амид натрия, или гидриды щелочных металлов, например гидрид натрия или гидрид калия, или щелочные основания, например гидроксид натрия или калия или карбонат натрия или калия, при температуре кипения реакционной среды с обратным холодильником, в гидроксилированных или негидроксилированных растворителях, предпочтительно негидроксилированных растворителях, имеющих температуру кипения, по меньшей мере, 90°С, предпочтительно, по меньшей мере, 100°С, таких как ароматические растворители, например толуол, ксилолы, мезитилен, этилбензол или хлорбензол, диоксан или этиленгликоль, или в простых гликолевых эфирах с температурой кипения, по меньшей мере, 100°С.
Для ускорения растворения оснований в реакционной среде можно использовать катализаторы фазового переноса, такие как четвертичные аммониевые соли, например триэтилбензиламмонийхлорид, или простые полигликолевые эфиры, например тритон Х100®.
В других предпочтительных условиях реакцию проводят в негидроксилированном растворителе, имеющем температуру кипения, по меньшей мере, 100°С, при температуре кипения реакционной среды с обратным холодильником, в присутствии от 2,1 до 2,2 эквивалентов трет-бутоксида калия и в присутствии от 1 до 10 мол.% тритона Х100® по отношению к арилсульфонилгидразону формулы (VII).
(Е)-Стильбеновые производные формулы (VI), определенной выше, получены таким образом и могут быть преобразованы в полигидроксистильбены, как описано ниже.
В частности, в данном изобретении предлагается новое соединение формулы (VI), отличающейся тем, что:
А представляет собой водород, R'1 и R'2 представляют собой метильную группу и R1 представляет собой изопропильную группу.
Данное новое соединение представляет собой следующее:
- (Е)-3,5-диметокси-4'-изопропилоксистильбен.
Особенно предпочтительный аспект данного изобретения заключается в осуществлении синтеза (Е)-триметилресвератрола, (Е)-трибензилресвератрола и (Е)-тетраметилписатаннола из соединений формулы (VII), раскрытой выше.
Получение полигидроксистильбенов (ресвератрола и писатаннола) из (Е)-стильбеновых производных формулы (VI)
C (Е)-стильбеновых производных формулы (VI) может быть снята защита способами, известными в литературе. Удаление защиты может быть осуществлено путем применения или адаптации способов, описанных, например, в WO 2003/086414, WO 2001/060774, EP 1466884 или Tetrahedron, 59(18), (2003), 3315-3321.
В предпочтительных технологических условиях используют от 3 до 10 молярных эквивалентов трибромида бора при температуре от -30°С до температуры окружающей среды.
(Е)-Гидроксистильбеновые производные формулы (VIII), например, в виде ресвератрола (В означает Н) получали, таким образом, согласно схеме, показанной ниже:

Согласно данному методу возможно согласно способу по изобретению получить ресвератрол и писатаннол из соединений формулы (IV) и, более конкретно, из соединений формулы (VII), определенной выше.
Данное изобретение также относится к способу синтеза соединения формулы (VII), описанной выше, с использованием, по меньшей мере, одного соединения, выбранного из соединений формул (I), (II), (III) и (IV), определенных в данной патентной заявке.
Настоящее изобретение также включает любое применение соединения формулы (I), (II), (III), (IV) или (VII), как определено выше, в качестве промежуточного соединения в синтезе (Е)-стильбенового производного формулы (VI), в частности (Е)-триметилресвератрола, (Е)-трибензилресвератрола или (Е)-тетраметилписатаннола, или в качестве промежуточного соединения в синтезе (Е)-полигидроксистильбенового производного, такого как ресвератрол или писатаннол.
Цель последующих примеров состоит в том, чтобы завершить данное описание без ограничения объема изобретения.
Пример 1
Синтез метил 3-(3,5-диметоксифенил)-2-(4-метоксифенил)-3-оксопропионата
24,1 г 60% гидрида натрия в минеральном масле (0,601 моль), которое промывали 2 раза 60 мл циклогексана и 60 мл ТГФ, помещают в трехгорлую круглодонную колбу на 1000 мл. Затем при температуре окружающей среды вводят раствор 48,2 г метил 3,5-диметоксибензоата (0,243 моль) в 100 мл ТГФ. Смесь кипятят с обратным холодильником и добавляют раствор 43,8 г метил п-метоксифенилацетата (0,243 моль), растворенного в 60 мл ТГФ, на протяжении 10 ч. Смесь поддерживают при кипении с обратным холодильником в течение 5 ч. Смесь охлаждают до температуры 0-5°С и добавляют раствор уксусной кислоты (38,0 г, т.е. 0,633 моль) в 100 мл ТГФ на протяжении 1/2 часа при этой температуре. Затем при температуре окружающей среды добавляют 150 мл воды и ТГФ отгоняют. Реакционную среду экстрагируют 500 мл метил-трет-бутилового эфира (МТВЕ) и органическую фазу промывают 100 мл насыщенного водного раствора бикарбоната натрия, промывают 50 мл воды и концентрируют на роторном испарителе с выделением 74,4 г сырого сложного β-кетоэфира в виде желтого масла, т.е. выход сырого продукта 89%.
200 мл метанола добавляют к 60 г данного сырого сложного β-кетоэфира и эту смесь поддерживают при температуре окружающей среды при перемешивании в течение 1 ч. Затем полученный осадок отфильтровывают и операцию повторяют со 150 мл метанола. Выделяют 24,7 г твердого вещества белого цвета.
5 г данного осадка поглощают 50 мл МТВЕ, нагретого до температуры кипения с обратным холодильником, температуру доводят до температуры окружающей среды и нерастворимый продукт (0,5 г) отфильтровывают. Фильтрат концентрируют досуха и полученный осадок повторно суспендируют в 20 мл метанола, нагретого до температуры кипения с обратным холодильником. После охлаждения до температуры окружающей среды осадок, который образовался, отфильтровывают и промывают на фильтре 5 мл метанола. Таким образом, выделяют 3,6 г твердого вещества белого цвета, и это твердое вещество имеет температуру плавления 76°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,75 с (3H); δ 3,8 с (9H); δ 5,5 с (1H); δ 6,6 т 1H); δ 6,9 д (2H); δ 7,1 д (2H); δ 7,35 д (2H);
C13 (Dept 135): δ 52,5 (COOCH3); δ 55,07 и 55,37 (OCH3); δ 59,49 (CH); δ 105,65 (аром. CH); δ 106,60; 114,20; 130,45 (аром. CH).
Пример 2
Синтез метил 3-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)-3-оксопропионата
18,9 г 60% гидрида натрия в минеральном масле (0,47 моль), которое промывали 2 раза 50 мл циклогексана и затем 100 мл ТГФ, помещают в трехгорлую круглодонную колбу на 1000 мл и затем вводят 65,7 г метил 3,5-дибензилоксибензоата (0,189 моль) в 100 мл ТГФ. Смесь кипятят с обратным холодильником и добавляют раствор 48,3 г метил 4-бензилоксифенилацетата (0,189 моль) в 120 мл ТГФ на протяжении 10 ч. Смесь поддерживают при кипении с обратным холодильником в течение 4 часов, затем охлаждают до 0-5°С и при этой температуре добавляют раствор 29,4 г уксусной кислоты (0,49 моль) в 240 мл ТГФ. Затем добавляют 360 мл воды и отгоняют ТГФ при атмосферном давлении. Добавляют 360 мл МТВЕ, проводят разделение отстаиванием и органическую фазу выделяют и промывают 100 мл насыщенного водного раствора бикарбоната натрия. Органическую фазу концентрируют на роторном испарителе с получением 106,5 г сложного β-кетоэфира в виде вязкого желтого масла, т.е. выход сырого продукта 98%.
1 г данного продукта, элюированного на колонке с силикагелем (этилацетат/гептан 20/80), приводит к выделению 0,5 г метил 3-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)-3-оксопропионата в виде вязкого светло-желтого масла.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,75 с (3H); δ 5,05 с (6H); δ 5,48 с (1H); δ 6,78 т (1H); δ 6,95 д (2H); δ 7,15 д (2H); δ 7,28 д (2H);
C13: δ 52,8 (COOCH3); δ 59,6 (CH); 70,1 и 70,4 (CH2OPh); δ 107-160 (аром. CH); δ 169,6 (C=O).
Пример 3
Синтез метил 3-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)-3-оксопропионата
23,8 г 60% NaH в минеральном масле (0,59 моль) помещают в трехгорлую круглодонную колбу на 1 л и промывают в круглодонной колбе 2 раза 60 мл циклогексана, а затем добавляют 46,7 г метил 3,5-диметоксибензоата (0,238 моль), растворенного в 200 мл ТГФ. Смесь кипятят с обратным холодильником и добавляют 50 г метил 3,4-диметоксифенилацетата (0,238 моль), растворенного в 120 мл ТГФ, на протяжении 10 ч. Смесь поддерживают при кипении с обратным холодильником в течение 2 часов, охлаждают до 0-5°С и при этой температуре добавляют по каплям 37,1 г (0,61 моль) уксусной кислоты, разведенной в 120 мл ТГФ. Затем добавляют 300 мл воды и отгоняют ТГФ. Смесь охлаждают до температуры окружающей среды и экстрагируют 400 мл МТВЕ, органическую фазу затем промывают 100 мл воды и реакционную среду концентрируют с выделением 92,9 г сырого метил 3-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)-3-оксопропионата в виде вязкого желтого масла.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,75 с (3H); δ 3,80 с (6H); δ 3,95 с (3H); δ 3,98 с (3H); δ 5,5 (1H); δ 6,5-7,3 м (6H);
C13: δ 52,5 (COOCH3); δ 56,46 и 56,53 (OCH3); δ 59,88 (CH); δ 99,67; 106,24; 111,50; 112,02; 121,02; 125,98; 146,48; 148,72; 149,29; 159,27 (аром. CH); δ 167,89 (C=O); δ 198,97 (COOCH3).
Пример 4
Синтез 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанона
26,7 г борной кислоты (0,43 моль) и 74,4 г сырого метил 3-(3,5-диметоксифенил)-2-(4-метоксифенил)-3-оксопропионата (0,216 моль), полученного по методике примера 1, помещают в круглодонную колбу, снабженную дистилляционной насадкой. Проводят нагревание, при этом температуру доводят постепенно до 100°С в течение 1 часа, 120°С в течение 1 часа, 140°С в течение 1 часа и затем 160°С в течение 4 часов при отгонке легких продуктов. Смесь охлаждают до 80°С, добавляют 250 мл воды и затем 200 мл толуола, смесь перемешивают при 60°С в течение 1 часа, затем проводят разделение отстаиванием и фазу толуола выделяют, промывают 100 мл насыщенного водного раствора бикарбоната натрия и концентрируют на роторном испарителе. Полученный сырой маслянистый продукт поглощают 200 мл МТВЕ для осаждения продукта, который отфильтровывают и сушат. Таким образом, после сушки получают 33,6 г 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанона в виде твердого вещества кремовато-белого цвета, т.е. выход 54,4% по отношению к сырому исходному сложному β-кетоэфиру. Т.пл.: 93-4°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,5 с (3H); δ 3,6 м.д. (ppm) с (6H); δ 4,2 с (2H); δ 6,62 т (1H); δ 6,85 д (2H); δ 7,15 д (2H); δ 7,18 д (2H);
C13 (Dept 135): δ 44,6 (CH2); δ 55,1 и 55,4 (OCH3); δ 105,2; 106,3; 114; 130,2 (аром. CH).
Пример 5
Синтез 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанона
34,8 г сырого метил 3-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)-3-оксопропионата (0,0608 моль), полученного по методике примера 2, и 7,51 г борной кислоты (0,121 моль) помещают в круглодонную колбу на 250 мл. Реакционную среду нагревают до 100°С в течение 1 часа, 120°С в течение 1 часа, 140°С в течение 1 часа и затем 150-155°С в течение 5 часов при отгонке легких продуктов. Смесь охлаждают до 60°С и добавляют водный раствор 8,5 г гранул гидроксида натрия, растворенных в 175 мл воды. Смесь затем кипятят с обратным холодильником в течение 3 часов и вновь охлаждают до 60°С, добавляют 250 мл толуола, проводят разделение отстаиванием, органическую фазу выделяют и промывают 75 мл воды, а затем фазу толуола концентрируют на роторном испарителе. Выделяют 19,4 г сырого 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанона.
15 г данного сырого продукта кипятят с обратным холодильником в 140 мл метанола и смесь охлаждают и выдерживают при 20-25°С в течение 1 ч. Полученный осадок отфильтровывают, повторно суспендируют в 75 мл метанола, отфильтровывают и сушат при 40°С с получением 7 г 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанона в виде твердого вещества белого цвета, температура плавления которого равна 87°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 4,15 с (1H); δ 5,05 с (2H); δ 5,1 с (4H); δ 6,8 т (1H); δ 6,95 д (2H); δ 7,2 д (2H); δ 7,75 д (2H); δ 7,45 широкий пик (15H);
C13: δ 44,5 (CH2); δ 69,9 (O-CH2-Ph); δ 70,2 (O-CH2-Ph); δ 106,9-159,9 (аром. CH); δ 197,3 (C=O).
Пример 6
Синтез 1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этанона
90,9 г сырого метил 3-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)-3-оксопропионата, полученного по методике примера 3, и 30 г борной кислоты помещают в трехгорлую круглодонную колбу на 250 мл. Смесь нагревают при перемешивании при 100°С в течение 1 часа, 120°С в течение 1 часа, 140°С в течение 1 часа и затем 160°С в течение 4 часов при отгонке легких продуктов. Смесь охлаждают приблизительно до 60°С, добавляют по каплям 226 г 15% раствора гидроксида натрия и смесь поддерживают при кипении с обратным холодильником в течение 2 часов при перемешивании. Реакционную среду экстрагируют при температуре окружающей среды 350 мл толуола, промывая 100 мл воды. Органическую фазу концентрируют, выделяя 51,70 г 1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этанона в виде масла темно-коричневого цвета.
1 г сырого кетона очищают из 25 мл гептана, причем нерастворимое тяжелое масло удаляют и осадок, который появляется в гептановом растворе после стояния в течение ночи, отфильтровывают. Получают 0,17 г очищенного кетона, т.пл. 66°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,8 с (6H); δ 3,85 с (6H); δ 4,2 с (2H); δ 6,6-7,7 мультиплет (6H);
C13: δ 45,25 (CH2); δ 55,59; 55,91 (OCH3); δ 105,35; 106,64; 111,56; 112,57; 121,62; 127,04; 138,56; 148,08; 149,12; 161,03 (аром. CH); δ 197,56 (C=O).
Пример 7
Синтез 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанола
160 мл метанола, 32,6 г перекристаллизованного 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанона, полученного по методике примера 4 (114 ммоль), и 3,25 г 5% Pd/C JM тип 87L помещают в реактор для гидрирования и вводят водород при давлении от 5 до 6 бар при температуре окружающей среды в течение 10 ч. Катализатор отфильтровывают при 40°С и температуру снова доводят до температуры окружающей среды, чтобы выделить 29 г 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанола, т.е. выход 88,4% по отношению к исходному кетону, который показывает температуру плавления 101-102°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 2,9 м (2H); δ 3,8 с (9H); δ кв. (1H); δ между 6,3 и 7,2 м (7H);
C13 (Dept 135): δ 44,9 (CH2); δ 55,2 (OCH3); δ 75,2 (CHOH); δ 99,9; 103,7; 113,8; 130,3 (аром. CH).
Пример 8
Синтез 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанола
5 г осажденного 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанона (1,74 ммоль), полученного по методике примера 4, вносят в 75 мл метанола и 62,5 мл ТГФ в трехгорлой круглодонной колбе на 250 мл. 0,78 г боргидрида натрия (1,1 экв.) добавляют при температуре окружающей среды на протяжении приблизительно 1 ч. Реакционную среду поддерживают при перемешивании в течение 1 часа, концентрируют и остаток поглощают 50 мл смеси вода/метанол (50/50 по объему). Полученный осадок отфильтровывают и промывают на фильтре 25 мл смеси вода/метанол (50/50 по объему). Выделяют 5 г осадка белого цвета, и этот осадок, по данным ЯМР, соответствует ожидаемому 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанолу, т.е. фактически количественный выход.
Пример 9
Синтез 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанола
2 г (3,9 ммоль) перекристаллизованного 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанона, полученного по методике примера 5, помещают в трехгорлую круглодонную колбу на 100 мл и растворяют в 30 мл метанола и 25 мл ТГФ. Приблизительно 0,147 г боргидрида натрия добавляют небольшими порциями при температуре окружающей среды на протяжении 1 ч. Реакционную среду поддерживают при перемешивании в течение 1 часа, концентрируют, добавляют 30 мл воды и реакционную среду экстрагируют 60 мл МТВЕ. МТВЕ фазу концентрируют с получением 2 г светло-желтого масла, которое со временем кристаллизуется. К данному продукту добавляют 5 мл метанола, смесь поддерживают при перемешивании в течение 1 часа и затем полученный осадок белого цвета отфильтровывают. После сушки в вакууме при 35°С выделяют 1,3 г 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанола в виде твердого вещества белого цвета с температурой плавления 80-81°С, т.е. выход 65% по отношению к исходному кетону.
ЯМР (CDCl3) 200 МГц
Протон: δ 2 д (1H); δ 2,95 мультиплет (2H); δ 4,8 мультиплет (1H); δ 5,05 с (4H); δ 5,1 с (2H); δ 6,05 т (1H); δ 6,15 д (2H); δ 6,95 д (2H); δ 7,15 д (2H); δ 7,2-7,6 широкий пик (15H);
C13: δ 44,88 (CH2); δ 69,92 (O-CH2); δ 75,2 (CHOH); δ 101,17; 104,98; 114,78; 127,31; 127,80; 128,44; 130,11; 130,40; 136,97; 146,36; 157,54; 159,86 (аром. CH).
Пример 10
Синтез 1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этанола
0,16 г (0,5 ммоль) 1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этанона, синтезированного и очищенного из гептана по методике примера 6, растворяют в 5 мл метанола и добавляют при перемешивании 0,02 г боргидрида натрия при температуре окружающей среды. Реакционную среду поддерживают при перемешивании в течение 1 часа, концентрируют досуха, добавляют 5 мл воды и реакционную среду экстрагируют 10 мл МТВЕ. Органическую фазу промывают 5 мл воды и концентрируют с получением 0,16 г 1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этанола в виде бесцветного масла, т.е. количественный выход.
ЯМР (CDCl3) 200 МГц
Протон: δ 2,95 мультиплет (2H); δ 3,75 с (6H); δ 3,85 с (3H); δ 3,97 с (3H); δ 4,8 мультиплет (1H); δ 6,35-7,28 широкий неразрешенный пик (6H);
C13: δ 44,51 (CH2); δ 54,34; 54,78; 54,88 (OCH3); δ 74,29 (CH-OH); δ 98,51; 102,80; 110,22; 111,69; 120,49; 129,35; 145,42; 146,81; 147,83; 159,81 (аром. CH).
Пример 11
Синтез (Е)-триметилресвератрола
2 г 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанола, полученного по методике примера 7 или примера 8 (6,9 ммоль), и 0,019 г моногидрата п-толуолсульфоновой кислоты (PTSA) (6 мол.%) вносят в 200 мл толуола, находящегося в трехгорлой круглодонной колбе на 250 мл. Смесь кипятят с обратным холодильником в течение 2 ч 30 минут при удалении воды азеотропной отгонкой. Смесь охлаждают до температуры окружающей среды, добавляют 30 мл насыщенного раствора бикарбоната натрия, промывают 30 мл воды и фазу толуола концентрируют с получением 1,95 г желтого масла. Данное масло поглощают 3,8 мл метанола и смесь кипятят с обратным холодильником и дают охладиться до температуры окружающей среды. Полученный осадок отфильтровывают и промывают на фильтре 1 мл метанола. Выделяют 1,31 г осадка светло-коричневого цвета, который имеет температуру плавления 55-56°С и соответствует, по данным ЯМР, (Е)-триметилресвератролу, т.е. выход 70%.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,83 с (9H); δ 6,40 т (1H); δ 6,68 д (2H); δ 6,9 д (1H); δ 6,92 д (2H); δ 7,10 д (1H); δ 7,48 д (2H);
C13: δ 55,32; 55,37 (OCH3); δ 99,73; 104,49; 114,27; (аром. CH); 126,68; 128,84 (этилен. CH); δ 127,97; 130,04; 139,84; 159,54; 161,13 (аром. CH).
Пример 12
Синтез (Е)-трибензилресвератрола
2 г 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанола (3,9 ммоль), полученного по методике примера 9, и 0,04 г моногидрата PTSA в 200 мл толуола (6 мол.%) помещают в трехгорлую круглодонную колбу на 250 мл. Смесь кипятят с обратным холодильником в течение 4 часов при отгонке воды азеотропной перегонкой. Смесь охлаждают до температуры окружающей среды, промывают 20 мл насыщенного раствора бикарбоната натрия и затем 10 мл воды и фазу толуола концентрируют с получением 1,95 г твердого вещества кремовато-белого цвета. Данное твердое вещество поглощают 8 мл МТВЕ и смесь поддерживают при перемешивании при температуре окружающей среды в течение 2 ч. Осадок отфильтровывают и промывают на фильтре 2 мл МТВЕ. Получают 0,99 г коричневого масла, которое со временем кристаллизуется. Продукт поглощают при температуре окружающей среды 4 мл МТВЕ. Полученный продукт отфильтровывают и промывают на фильтре небольшим количеством МТВЕ с получением 0,42 г белого, слегка кремового осадка 3,5,4'-трибензилресвератрола с температурой плавления 117-118°С, ЯМР-спектр которого соответствует спектру (Е)-трибензилресвератрола.
ЯМР (CDCl3) 200 МГц
Протон: δ 5,12 (6H); δ 6,63 т (1H); δ 6,83 д (2H); δ 6,95 д (1H); δ 7,02 д (1H); δ 7,1 д (1H); δ 7,3-7,6 широкий неразрешенный пик (16H);
C13: δ 70,16; 70,24 (O-CH2); δ 101,40; 105,78; 115,24 (аром. CH); δ 126,77 (этилен. CH); δ 127,63; 128,00; 128,96 (аром. CH); δ 130,29 (этилен. CH); δ 137,03; 139,90; 158,76; 160,31 (аром. CH).
Пример 13
Синтез N-[1-(3,5-диметоксифенил)-2-(4-метоксифенил)этилиден-N'-тозилгидразина
105,7 г (0,37 моль) осажденного 1-(3,5-диметоксифенил)-2-(4-метоксифенил)этанона, полученного по методике примера 4, и 75,6 г (0,407 моль) п-толуолсульфонилгидразида вносят в 950 мл этанола, находящегося в трехгорлой круглодонной колбе на 2 л. Смесь кипятят с обратным холодильником в течение 8 часов, охлаждают до температуры окружающей среды и затем полученный осадок отфильтровывают и промывают на фильтре небольшим количеством этанола. Получают 133,6 г п-тозилгидразона (выход 80%) в виде слегка кремового осадка. Продукт повторно суспендируют в 400 мл и затем в 860 мл МТВЕ с получением 123,6 г белого осадка N-[1-(3,5-диметоксифенил)-2-(4-метоксифенил)этилиден-N'-тозилгидразина, т.е. выход 74% по отношению к исходному кетону, который имеет температуру плавления 120-121°С.
ЯМР (CDCl3) 100 МГц
Протон: δ 2,4 с (3H); δ 3,8 с (9H); δ 2,4 с (3H); δ 3,9 с (2H); δ 2,4 с (3H); δ 6,4-7,8 м (11H);
C13: δ 21,4 (CH3); δ 32,5 (CH2); δ 55,04 и 55,23 (OCH3); δ 143,9 (C=N); δ 100,3 и 161,4 (CH).
Пример 14
Синтез N-[1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этилиден-N'-тозилгидразина
5 г 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанона, полученного по методике примера 5 (9,7 ммоль), 2,35 г п-толуолсульфонилгидразида (12,6 ммоль) в 25 мл этанола и 20 капель 35% хлористоводородной кислоты помещают в трехгорлую круглодонную колбу на 100 мл. Реакционную среду кипятят с обратным холодильником в течение 3 часов и концентрируют, остаток поглощают 50 мл МТВЕ при температуре окружающей среды в течение 1 часа при перемешивании, а затем осадок отфильтровывают и промывают на фильтре небольшим количеством МТВЕ. Получают 5,1 г N-[1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этилиден-N'-тозилгидразина в виде осадка белого цвета, т.е. выход 77% по отношению к исходному кетону, который имеет температуру плавления 147°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 2,4 с (3H); δ 3,85 с (2H); δ 2,4 с (3H); δ 5,0 с (4H); δ 5,05 с (2H); δ 2,4 с (3H); δ 6,5-7,7 м (26H);
C13: δ 21,4 (CH3); δ 32,5 (CH2); δ 55,04 и 69,9 (OCH2Ph); δ 143,8 (C=N); δ 100,3-159,8 (аром. CH).
Пример 15
Синтез N-[1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этилиден-N'-тозилгидразина
5 г (15,8 ммоль) сырого 1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этанона, полученного по методике примера 6, и 3,24 г п-толуолсульфонилгидразида (17,4 ммоль) вносят в 33 мл абсолютного этанола, находящегося в трехгорлой круглодонной колбе на 100 мл. Смесь кипятят с обратным холодильником в течение 3 часов, охлаждают до температуры окружающей среды и смесь оставляют перемешиваться в течение 2 ч. Полученный осадок отфильтровывают и затем промывают на фильтре 5 мл этанола. Получают 5,25 г осадка, который поглощают 50 мл МТВЕ, кипятят с обратным холодильником в течение 2 часов, отфильтровывают после охлаждения до температуры окружающей среды и затем промывают на фильтре 10 мл МТВЕ. Выделяют 5 г N-[1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этилиден-N'-тозилгидразина в виде белого, слегка коричневого твердого вещества, т.е. выход 65,4% по отношению к сырому исходному кетону, который имеет температуру плавления 132-133°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 2,5 с (3H); δ 3,6 с (3H); δ 3,75 с (6H); δ 3,8 с (3H); δ 3,85 с (2H); δ 6,4-7,8 м (10H);
C13: δ 21,95 (CH3); δ 33,67 (CH2); δ 55,80; 56,31; 56,41 (OCH3); δ 102,04; 105,21; 110,97; 112,08; 120,12; 126,16; 128,33; 135,63; 139,62; 129,95 (аром. CH); δ 144,43 (C=N); δ 148,74; 150,06; 154,03; 161,16 (аром. CH).
Пример 16
Синтез (Е)-триметилресвератрола
50 г N-[1-(3,5-диметоксифенил)-2-(4-метоксифенил)этилиден]-N'-тозилгидразина, полученного по методике примера 13 (0,110 моль), и 27,16 г трет-бутоксида калия (0,242 моль) в 1 л толуола, содержащем 2,5 г тритона Х100®, помещают в трехгорлую круглодонную колбу на 2 л. Смесь кипятят с обратным холодильником в течение 3 часов, охлаждают до температуры окружающей среды, добавляют 1 л воды, проводят разделение отстаиванием и отделяют органическую фазу. Водную фазу повторно экстрагируют 0,4 л толуола. Объединенные органические фазы концентрируют на роторном испарителе с получением 29,6 г сырого продукта в виде твердого вещества желтого цвета. Данный продукт поглощают 90 мл этанола при температуре окружающей среды при перемешивании в течение ночи, получая 17,4 г 3,5,4'-триметилресвератрола в виде твердого вещества слегка оранжевого цвета. Продукт перекристаллизовывают из 34 мл метанола и фильтруют при 0-5°С с получением 16,6 г (Е)-триметилресвератрола в виде твердого вещества белого цвета, т.е. выход 58% по отношению к исходному гидразону, который имеет температуру плавления 56-57°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,83 с (9H); δ 6,40 т (1H); δ 6,68 д (2H); δ 6,9 д (1H); δ 6,92 д (2H); δ 7,10 д (1H); δ 7,48 д (2H);
C13: δ 55,32; 55,37 (OCH3); δ 99,73; 104,49; 114,27 (аром. CH); 126,68; 128,84 (этилен. CH); δ 127,97; 130,04; 139,84; 159,54; 161,13 (аром. CH).
Пример 17
Синтез (Е)-триметилресвератрола
Осуществляют такую же реакцию, как в примере 16, с 2,5 г N-[1-(3,5-диметоксифенил)-2-(4-метоксифенил)этилиден]-N'-тозилгидразина в 12,5 мл толуола в присутствии 1,36 г трет-бутоксида калия и 0,125 г тритона Х100®, получая 1,47 г сырого 3,5,4'-триметилресвератрола, который перекристаллизовывают из 5 мл метанола с получением 0,91 г (Е)-триметилресвератрола в виде твердого вещества белого цвета, т.е. выход 63,6% по отношению к исходному гидразону.
Пример 18
Синтез (Е)-трибензилресвератрола
28 г N-[1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этилиден]-N'-тозилгидразина (41 ммоль), полученного по методике примера 14, и 10,1 г трет-бутоксида калия (100 ммоль) в 560 мл толуола, содержащего 1,4 г тритона Х100®, помещают в трехгорлую круглодонную колбу на 2 л. Смесь кипятят с обратным холодильником в течение 3 часов, охлаждают до температуры окружающей среды, добавляют 500 мл воды и органическую фазу отделяют отстаиванием. Водную фазу повторно экстрагируют 400 мл толуола. Объединенные толуольные фазы промывают и концентрируют с получением 21,8 г сырого продукта в виде желтого осадка, который поглощают 70 мл МТВЕ, отфильтровывают и промывают на фильтре МТВЕ, получая 11,9 г (Е)-трибензилресвератрола, который имеет температуру плавления 118°С, т.е. выход 58% по отношению к исходному гидразону.
ЯМР (CDCl3) 200 МГц
Протон: δ 5,12 (6H); δ 6,63 т (1H); δ 6,83 д (2H); δ 6,95 д (1H); δ 7,02 д (1H); δ 7,1 д (1H); δ 7,3-7,6 широкий неразрешенный пик (16H);
C13: δ 70,16 и 70,24 (O-CH2); δ 101,40; 105,78; 115,24 (аром. CH); δ 126,77 (этилен. CH); δ 127,63; 128,00; 128,96 (аром. CH); δ 130,29 (этилен. CH); δ 137,03; 139,90; 158,76; 160,31 (аром. CH).
Пример 19
Синтез (Е)-трибензилресвератрола
1,46 г N-[1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этилиден]-N'-тозилгидразина (2,1 ммоль), полученного по методике примера 14, и 0,28 г гранул трет-бутоксида калия (85%) в 30 мл толуола, содержащего 0,07 г тритона Х100®, помещают в трехгорлую круглодонную колбу на 100 мл. Смесь кипятят с обратным холодильником в течение 2 ч. Добавляют при температуре окружающей среды 30 мл воды, осуществляют разделение отстаиванием и фазу толуола отделяют и промывают 15 мл воды. Толуольную фазу концентрируют с получением 1 г твердого вещества в виде желтого осадка, который поглощают 4 мл МТВЕ, получая после фильтрования 0,8 г (Е)-трибензилресвератрола, т.е. выход 76% по отношению к исходному гидразону.
Пример 20
Синтез (Е)-тетраметилписатаннола
2,6 г (5,0 ммоль) N-[1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этилиден]-N'-тозилгидразина, полученного по методике примера 15, 1,24 г (11,4 ммоль) трет-бутоксида калия и 0,13 г тритона Х100® вносят в 25 мл толуола, находящегося в трехгорлой круглодонной колбе на 100 мл. Смесь кипятят с обратным холодильником в течение 3 ч. Температуру доводят приблизительно до 90°С, добавляют по каплям 15 мл воды, среду разделяют отстаиванием приблизительно при 60°С, органическую фазу промывают 15 мл воды и концентрируют ее на роторном испарителе с выделением 1,55 г слегка коричневого масла. Данное масло поглощают 10 мл метанола. Температуру доводят до температуры окружающей среды и смесь выдерживают при температуре окружающей среды в течение 2 ч. Осадок отфильтровывают и промывают на фильтре 3 мл метанола. Выделяют 1,03 г (Е)-тетраметилписатаннола в виде белого, слегка кремового осадка, т.е. выход 68,6% по отношению к исходному гидразону, который имеет температуру плавления 68°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,8 с (6H); δ 3,88 с (3H); δ 3,92 с (3H); δ 6,40 т (1H); δ 6,68 д (2H); 6,85 д (1H); δ 6,95 д (1H); δ 6,98-7,12 м (3H);
C13 (Dept 135): δ 55,38 и 55,97 (OCH3); δ 99,75; 104,40; 108,84; 120,08 (аром. CH); δ 126,81; 129,03 (этилен. CH).
Пример 21
Синтез (Е)-тетраметилписатаннола
2 г (3,86 ммоль) N-[1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этилиден]-N'-тозилгидразина, полученного по методике примера 15, 0,97 г трет-бутоксида калия (2,1 экв.) и 0,10 г тритона Х100® вносят в 20 мл мезитилена, находящегося в трехгорлой круглодонной колбе на 50 мл. Реакционную среду кипятят с обратным холодильником в течение 2 часов и затем охлаждают до температуры 60°С, добавляют 10 мл воды, выполняют разделение отстаиванием и органическую фазу отделяют и промывают 5 мл воды. Органическую фазу концентрируют при 80°С при 5 мм Hg. Выделяют 1,06 г желтого масла. Данное масло кипятят с обратным холодильником в 6 мл метанола, охлаждают до температуры окружающей среды, смесь выдерживают при перемешивании в течение 2 часов и осадок отфильтровывают и промывают на фильтре 2 мл метанола. Выделяют 0,55 г (Е)-тетраметилписатаннола в виде белого, слегка желтого осадка, т.е. выход 47,8% по отношению к исходному гидразону.
Пример 22
Синтез (Е)-тетраметилписатаннола
2 г (3,86 ммоль) N-[1-(3,5-диметоксифенил)-2-(3,4-диметоксифенил)этилиден]-N'-тозилгидразина, полученного по методике примера 15, 0,431 г метоксида натрия и 0,10 г тритона Х100® вносят в 25 мл толуола, находящегося в трехгорлой круглодонной колбе на 50 мл. Реакционную среду кипятят с обратным холодильником в течение 5 часов и охлаждают приблизительно до 60°С, медленно добавляют 10 мл воды, выполняют разделение отстаиванием при данной температуре. Фазу толуола промывают 5 мл воды и органическую фазу концентрируют на роторном испарителе с получением 1,18 г оранжевого масла. Данное масло кипятят с обратным холодильником в 6 мл метанола, охлаждают до температуры окружающей среды, смесь выдерживают при перемешивании в течение 2 часов и осадок отфильтровывают и промывают на фильтре 2 мл метанола. Выделяют 0,68 г (Е)-тетраметилписатаннола в виде белого, слегка желтого осадка, т.е. выход 59,1% по отношению к исходному гидразону.
Пример 23
Синтез (Е)-тетраметилписатаннола
Осуществляют такую же реакцию, как в примере 22, но с 0,34 г 60% NaH в масле (8,5 ммоль), которое промывали заранее в круглодонной колбе 2 раза 5 мл циклогексана. Смесь поддерживают при кипении с обратным холодильником в течение 3 часов и обрабатывают по методике примера 22 с получением 1,32 г оранжево-желтого масла. После осаждения из 7 мл метанола по методике примера 22 выделяют 0,66 г белого, слегка оранжевого, образовавшегося твердого (Е)-тетраметилписатаннола, т.е. выход 57,4% по отношению к исходному гидразону.
Пример 24
Синтез (Е)-ресвератрола
37,9 мл трибромида бора (приблизительно 100 г, 400 ммоль) вводят в 100 мл метиленхлорида в атмосфере азота в трехгорлой круглодонной колбе. Реакционную среду охлаждают приблизительно до -20°С и при данной температуре вносят 10,8 г (приблизительно 40 ммоль) (Е)-триметилресвератрола, растворенного в 20 мл метиленхлорида, на протяжении 1 ч 30 минут. Реакционной среде дают охладиться до температуры окружающей среды при перемешивании и оставляют перемешиваться при данной температуре в течение 4 часов. Реакционную среду затем медленно выливают в 800 г смеси лед/вода. Среду экстрагируют 325 мл, а затем 200 мл МТВЕ и органические фазы промывают 2 раза 75 мл насыщенного раствора бикарбоната натрия и затем 75 мл воды. Объединенные органические фазы концентрируют на роторном испарителе. Твердый остаток поглощают 100 мл метиленхлорида, отфильтровывают и сушат с получением 8,1 г сырого ресвератрола.
Осадок растворяют в этаноле при 60°С и осаждают добавлением воды, получая ресвератрол с температурой плавления 262-264°С. Протон и С13 ЯМР спектр соответствуют ожидаемому продукту.
Пример 25
Синтез метил 3-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)-3-оксопропионата
14,4 г 60% гидрида натрия в минеральном масле (0,36 моль), которое промывали 2 раза 50 мл толуола, помещают в трехгорлую круглодонную колбу. Затем вносят 90 мл толуола и 28,3 г метил 3,5-диметоксибензоата (0,144 моль). Смесь кипятят с обратным холодильником и добавляют раствор 30 г метил п-изопропилоксифенилацетата (0,144 моль), растворенного в 95 мл толуола, на протяжении 10 ч. Смесь поддерживают при кипении с обратным холодильником в течение 2 ч. Смесь охлаждают до температуры 0-5°С и медленно добавляют раствор ледяной уксусной кислоты (22,5 г, т.е. 0,374 моль) в 25 мл толуола. Затем при температуре окружающей среды медленно добавляют 135 мл воды.
Органическую фазу декантируют и водную фазу экстрагируют 25 мл толуола. Объединенные толуольные фазы концентрируют с получением 58 г сырого метил 3-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)-3-оксопропионата в виде вязкого масла темно-коричневого цвета.
ЯМР (CDCl3) 200 МГц
Протон: δ 1,35 д (6H); δ 3,75 с (3H); δ 3,8 с (6H); δ 4,5 гепт. (1H); δ 5,5 с (1H); δ 6,6 т (1H); δ 6,85 д (2H); δ 7,10 д (2H); δ 7,30 д (2H);
C13 (Dept 135): δ 22 (CH3-CH); δ 52,7 (COOCH3); δ 55,5 (2 OCH3); δ 59,7 (CH-COOMe); δ 69,9 (CH-CH3); δ 105,8; 106,8; 116,0; 130,7 (аром. CH).
Пример 26
Синтез метил 3-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)-3-оксопропионата
19,7 г 60% гидрида натрия в минеральном масле (0,49 моль), которое промывали 2 раза 60 мл толуола, помещают в трехгорлую круглодонную колбу. Затем вносят 120 мл толуола, 4 г тритона Х100® и 38,6 г метил 3,5-диметоксибензоата (0,197 моль). Смесь кипятят с обратным холодильником и добавляют раствор 38,2 г метил 3,4-метилендиоксифенилацетата (0,197 моль), растворенного в 130 мл толуола, на протяжении 10 ч. Смесь поддерживают при кипении с обратным холодильником в течение 2 ч. Смесь охлаждают до температуры 0-5°С и добавляют по каплям раствор ледяной уксусной кислоты (30,7 г, т.е. 0,51 моль) в 30 мл толуола. Смесь поддерживают 1 час при перемешивании, при этом охлаждая до температуры окружающей среды, затем медленно добавляют 180 мл воды и органическую фазу отделяют. Водную фазу повторно экстрагируют 40 мл толуола. Объединенные органические фазы концентрируют с получением 76,5 г сырого метил 3-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)-3-оксопропионата в виде вязкого масла темного цвета.
1 мл метанола добавляют к 1 г данного сырого продукта, получая осадок белого цвета, который фильтруют, промывают на фильтре метанолом и сушат, причем осадок имеет температуру плавления 51°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,75 с (3H); δ 3,8 с (6H); δ 5,5 с (1H); δ 5,95 м (2H); δ 6,6 т (1H); δ 6,8-7,3 аром. (5H);
C13 (Dept 135): δ 52,8 (COOCH3); δ 55,6 (2 OCH3); δ 59,9 (CH-COOMe); δ 101,3 (O-CH2-O); δ 105,9; 106,8; 108,6; 109,8; 123,1 (аром. CH).
Пример 27
Синтез 1-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)этанона
9,65 г борной кислоты (0,16 моль) и 58 г сырого метил 3-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)-3-оксопропионата (0,16 моль), полученного по методике примера 25, помещают в круглодонную колбу. Смесь нагревают при перемешивании при 150-155°С в течение 3 часов и затем охлаждают приблизительно до 80°С. Добавляют 200 мл толуола и затем 150 мл воды. Проводят разделение отстаиванием и органическую фазу отделяют. Водную фазу повторно экстрагируют 20 мл толуола. Органические фазы концентрируют, выделяя 48,8 г сырого 1-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)этанона в виде вязкого масла темно-коричневого цвета.
ЯМР (CDCl3) 200 МГц
Протон: δ 1,35 д (6H); δ 3,8 с (6H); δ 4,2 с (2H); δ 4,5 гепт. (1H); δ 6,6 т (1H); δ 6,85 д (2H); δ 7,15 д (2H); δ 7,20 д (2H);
C13 (Dept 135): δ 22,1 (CH3-CH); δ 44,8 (CH2); δ 55,5 (2 OCH3); δ 69,9 (CH-CH3); δ 105,3; 106,5; 116,1; 130,5 (аром. CH).
Пример 28
Синтез 1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этанона
13 г борной кислоты (0,21 моль) и 75 г сырого метил 3-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)-3-оксопропионата (0,21 моль), полученного по методике примера 26, помещают в круглодонную колбу. Реакционную среду доводят до 140°С в течение 1 часа и затем нагревают при 150-155°С в течение 3 ч. Смесь охлаждают до 80°С, добавляют 360 мл толуола и затем 190 мл воды. Проводят разделение отстаиванием и водную фазу повторно экстрагируют 70 мл толуола. Органические фазы концентрируют, выделяя 63,6 г твердого вещества коричневого цвета, которое поглощают 100 мл этанола. Осадок отфильтровывают, промывают на фильтре 25 мл этанола и сушат с получением 44,7 г сырого 1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этанона в виде твердого вещества кремового цвета, т.е. выход 71% по отношению к сырому исходному сложному кетоэфиру.
2 г данного продукта перекристаллизовывают из 20 мл этанола с получением осадка белого цвета, имеющего температуру плавления 123°С.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,85 с (6H); δ 4,2 с (2H); δ 5,95 с (2H); δ 6,65 т (1H); δ 6,7-6,8 м (3H); δ 7,15 д (2H);
C13 (Dept 135): δ 45,3 (CH2-CO); δ 55,6 (2 OCH3); δ 101,0 (O-CH2-O); δ 105,4; 106,5; 108,5; 109,9; 122,6 (аром. CH).
Пример 29
Синтез N-[1-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)этилиден-N'-тозилгидразина
48,5 г (0,14 моль) сырого 1-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)этанона, полученного по методике примера 27, и 31,8 г (0,17 моль) п-толуолсульфонилгидразида вносят в 250 мл этанола, находящегося в трехгорлой круглодонной колбе. Смесь кипятят с обратным холодильником в течение 6 часов, охлаждают до температуры окружающей среды и смесь оставляют перемешиваться в течение 5 ч. Затем полученный осадок отфильтровывают и промывают на фильтре 25 мл этанола. Осадок поглощают 100 мл МТВЕ при температуре окружающей среды в течение 1 часа при перемешивании, затем осадок отфильтровывают и промывают на фильтре 25 мл МТВЕ и сушат. Выделяют 31,4 г N-[1-(3,5-диметоксифенил)-2-(4-изопропилоксифенил)этилиден-N'-тозилгидразина в виде твердого вещества белого цвета, которое имеет температуру плавления 101°С, т.е. выход 42% по отношению к сырому исходному кетону.
ЯМР (CDCl3) 200 МГц
Протон: δ 1,35 д (6H); δ 2,4 с (3H); δ 3,8 с (6H); δ 3,9 с (2H); δ 4,5 гепт. (1H); δ 6,4 т (1H); δ 6,7 д (2H); δ 6,83 д (2H); δ 6,85 д (2H); δ 7,25 д (2H); δ 7,68 д (2H);
C13 (Dept 135): δ 21,7 (CH3); δ 22,0 (CH3-CH); δ 32,7 (CH2); δ 55,4 (2 OCH3); δ 69,9 (CH-CH3); δ 101,7; 104,8; 116,7; 128,0; 128,8; 129,5 (аром. CH).
Пример 30
Синтез N-[1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этилиден-N'-тозилгидразина
42,7 г (0,142 моль) 1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этанона, полученного по методике примера 28, и 29 г (0,155 моль) п-толуолсульфонилгидразида вносят в 270 мл этанола, находящегося в трехгорлой круглодонной колбе. Смесь кипятят с обратным холодильником в течение 5 часов, охлаждают до температуры окружающей среды, оставляют перемешиваться в течение 3 ч и затем 2 ч при 0-5°С. Полученный осадок отфильтровывают и промывают на фильтре 20 мл этанола, охлажденного до 0-5°С. Осадок поглощают 100 мл МТВЕ, отфильтровывают и сушат, выделяя 44,6 г N-[1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этилиден-N'-тозилгидразина в виде кремового твердого вещества, которое имеет температуру плавления 143-144°С, т.е. выход 67% по отношению к исходному кетону.
ЯМР (CDCl3) 200 МГц
Протон: δ 2,4 с (3H); δ 3,75 с (6H); δ 3,85 с (2H); δ 5,95 с (2H); δ 6,35 д (1H); δ 6,45 т (1H); δ 6,6 д (1H); δ 6,8 д (2H); δ 7,25 д (2H); δ 7,25 с (1H); δ 7,7 д (2H);
C13 (Dept 135): δ 21,7 (CH3); δ 33,1 (CH2-CN-); δ 55,5 (2 OCH3); δ 101,3 (O-CH2-O); δ 101,7; 104,8; 108,2; 108,8; 120,6; 128,0; 129,5 (аром. CH).
Пример 31
Синтез (Е)-3,5-диметокси-4'-изопропилоксистильбена
19 г N-[1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этилиден-N'-тозилгидразина (0,39 моль), полученного по методике примера 30, 9,3 г трет-бутоксида калия (0,83 моль) и 1,9 г тритона Х100® вносят в трехгорлую круглодонную колбу, содержащую 100 мл толуола. Смесь кипятят с обратным холодильником в течение 3 часов, охлаждают приблизительно до 90°С и вносят 70 мл воды. Выполняют разделение отстаиванием и водную фазу повторно экстрагируют 50 мл толуола. Фазы толуола промывают 50 мл воды и концентрируют с получением 12,3 г вязкого продукта желтого цвета, который поглощают 10 мл метанола и выдерживают при перемешивании в течение 3 ч. Осадок отфильтровывают, промывают на фильтре метанолом и сушат. Выделяют 6,5 г (Е)-3,5-диметокси-4'-изопропилоксистильбена в виде твердого вещества белого цвета, которое имеет температуру плавления 61°С, т.е. выход 55,6% по отношению к исходному гидразону.
ЯМР (CDCl3) 200 МГц
Протон: δ 1,35 д (6H); δ 3,85 с (6H); δ 4,6 гепт. (1H); δ 6,4 т (1H); δ 6,7 д (2H); δ 6,9 д (1H); δ 6,93 д (2H); δ 7,09 д (1H); δ 7,45 д (2H);
C13 (Dept 135): δ 22,5 (CH3-CH); δ 55,7 (2 OCH3); δ 70,3(CH-CH3); δ 100,0; 104,8; 116,4 (аром. CH); δ 126,9; 128,2 (CH=); δ 129,2 (аром. CH).
Пример 32
Синтез (Е)-3,5-диметокси-3',4'-метилендиоксистильбена
39,4 г N-[1-(3,5-диметоксифенил)-2-(3,4-метилендиоксифенил)этилиден-N'-тозилгидразина (0,084 моль), полученного по методике примера 30, 19,9 г трет-бутоксида калия (0,177 моль) и 3,9 г тритона Х100® вносят в трехгорлую круглодонную колбу, содержащую 190 мл толуола. Смесь кипятят с обратным холодильником в течение 4 часов, охлаждают приблизительно до 80°С и вносят 130 мл воды. Выполняют разделение отстаиванием и водную фазу повторно экстрагируют 90 мл толуола. Фазы толуола промывают 50 мл воды и концентрируют с получением 27,8 г коричневого твердого вещества, которое поглощают 30 мл метанола и выдерживают при перемешивании в течение 1 ч. Осадок отфильтровывают, промывают на фильтре метанолом и сушат. Выделяют 16,6 г (Е)-3,5-диметокси-3',4'-метилендиоксистильбена в виде коричневого твердого вещества, которое имеет температуру плавления 96°С, т.е. выход 69% по отношению к исходному гидразону.
ЯМР (CDCl3) 200 МГц
Протон: δ 3,8 с (6H); δ 6 с (2H); δ 6,4 т (1H); δ 6,65 т (2H); δ 6,8 д (1H); δ 6,9 д (1H); δ 6,95 д (1H); δ 7,05 д (1H); δ 7,25 с (1H);
C13 (Dept 135): δ 55,5 (2 OCH3); δ 99,8 (аром. CH); δ 101,2 (O-CH2-O); δ 104,4; 105,6; 108,5; 121,7; 127,0; 128,9 (аром. CH).
Реферат
Настоящее изобретение относится к вариантам нового способа получения (E)-стильбенового производного формулы (VI), которые используются для получения полигидроксистильбенов, в частности ресвератрола или писатаннола, проявляющих антиоксидантный эффект, к новым промежуточным соединениям формулы (III), (IV), (V) и (VII), используемым в данных способах, а также к применению соединения формулы (I), (II), (III), (IV) или (VII) в качестве промежуточного соединения в синтезе (E)-стильбенового производного формулы (VI) или полигидроксистильбена. ! ! ! ! ! Значения заместителей R1, R1', R2', R, A, Ar, R' такие, как указаны в формуле изобретения. 8 н. и 18 з.п. ф-лы, 1 ил., 32 пр.
Формула

в которой А представляет собой водород или группу OR2, и
R1, R2, R'1 и R'2 представляют собой независимо друг от друга линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(CH2)n- с n=1-3, отличающийся тем, что соединение формулы (III)

в которой A, R1, R2, R'1 и R'2 имеют значения, определенные выше, R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода,
подвергают взаимодействию в качестве промежуточного соединения, из которого декарбоксилированием получают соединение формулы (IV)

в которой A, R1, R2, R'1 и R'2 имеют значения, определенные выше, затем соединение формулы (IV)
- либо восстанавливают в спирт, тогда образовавшийся спирт подвергают дегидратации,
- или подвергают взаимодействию с арилсульфонилгидразидом, тогда происходит взаимодействие полученного в результате арилсульфонилгидразона с основанием,
с получением вышеназванного (Е)-стильбенового производного формулы (VI).

в которой А представляет собой водород или же группу OR2,
R1, R2, R'1 и R'2 представляют собой независимо друг от друга линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(CH2)n - с n=1-3,
причем указанная реакция представляет собой восстановление соединений формулы (IV), при этом производные 1,2-диарилэтанола формулы (V) затем подвергают дегидратации с образованием соединений формулы (VI), определенной по п.1.
А представляет собой водород или группу OR2,
R1, R2, R'1 и R'2 представляют собой независимо друг от друга линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, причем для R1 и R2 также возможно образование углеводородной цепи -(CH3)n - с n=1-3.

в которой Ar представляет собой фенильную или о-, м- или п-толильную группу,
А представляет собой водород или же группу OR2,
R1, R2, R'1 и R'2 представляют собой независимо друг от друга линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(CH2)n - с n=1-3,
причем указанное соединение формулы (VII) затем подвергают взаимодействию с сильным основанием.

в которых А представляет собой водород или же группу OR2,
R1, R2, R'1 и R'2 представляют собой независимо друг от друга линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи - (CH2)n - с n=1-3,
R и R' представляют собой независимо друг от друга линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, в присутствии сильного основания.

в которой А представляет собой водород или группу OR2, и
R1, R2, R'1, и R'2 представляют собой независимо друг от друга линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 16 атомов углерода, которая необязательно замещена одной или несколькими алкокси- или галогеновыми группами, причем для R1 и R2 также возможно образование углеводородной цепи -(CH2)n - с n=1-3,
отличающийся тем, что соединение формулы (IV)

в которой A, R1, R2, R'1 и R'2 имеют значения, определенные выше, как промежуточное соединение подвергают взаимодействию с арилсульфонилгидразидом с получением арилсульфонилгидразона формулы (VII)

в которой A, R1, R2, R'1 и R'2 имеют значения, определенные выше, и Ar представляет собой фенильную или о-, м- или п-толильную группу,
затем образовавшийся таким образом арилсульфонилгидразон подвергают взаимодействию с основанием
с получением вышеназванного (Е)-стильбенового производного формулы (VI).

в которой R представляет собой метальную группу, и
- или А представляет собой водород, и группы R1, R'1 и R'2 представляют собой метальные группы или бензильные группы,
- или А представляет собой группу -OCH3, и каждая из групп R1, R'1 и R'2 представляет собой метальную группу,
- или А представляет собой водород, R'1 и R'2 представляют собой метальную группу, и R1 представляет собой изопропильную группу,
- или А представляет собой группу -OR2, R'1 и R'2 представляют собой метальную группу, и R1 и R2 образуют углеводородную цепь -(CH2)n- с n=1.
в которой - или А представляет собой водород, и каждая из групп R1, R'1 и R'2 представляет собой бензильную группу,
- или А представляет собой группу -OCH3, и каждая из групп R1, R'1 и R'2 представляет собой метальную группу,
- или А представляет собой водород, R'1 и R'2 представляют собой метильную группу, и R1 представляет собой изопропильную группу,
- или А представляет собой группу -OR2, R'1 и R'2 представляют собой метильную группу, и R1 и R2 образуют углеводородную цепь -(CH2)n - с n=1.
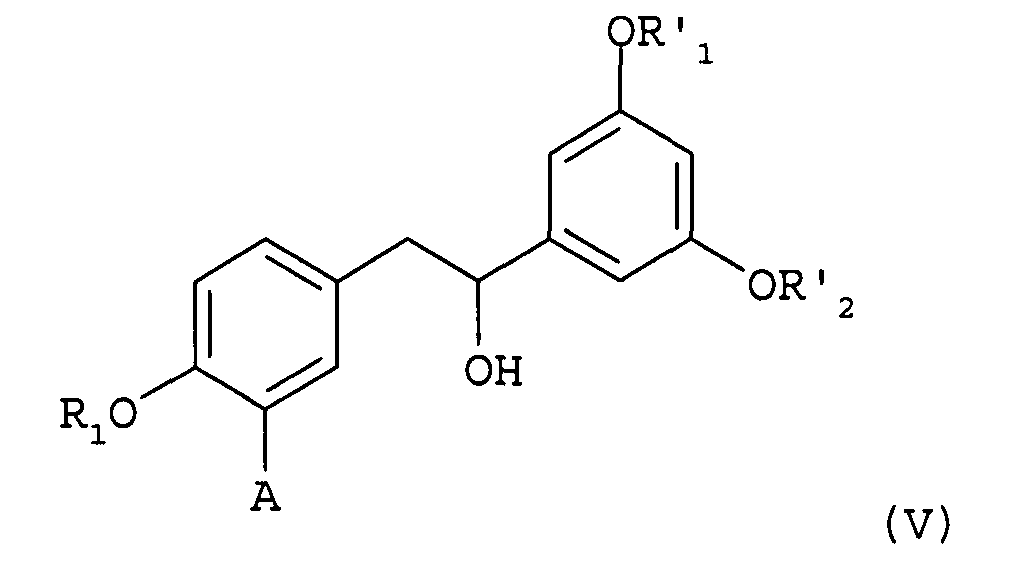
в которой А представляет собой водород, и каждая из групп R1, R'1 и R'2 представляет собой бензильную группу, причем указанное соединение представляет собой 1-(3,5-дибензилоксифенил)-2-(4-бензилоксифенил)этанол.

в которой Ar представляет собой п-толильную группу, и
- или А представляет собой водород, и группы R1, R'1 и R'2 представляют собой метальные группы или бензильные группы,
- или А представляет собой группу -OCH3, и группы R1, R'1 и R'2 представляют собой метальную группу,
- или А представляет собой водород, R'1 и R'2 представляют собой метальную группу, и R1 представляет собой изопропильную группу,
- или А представляет собой группу -OR2, R'1 и R'2 представляют собой метальную группу, и R1 и R2 образуют углеводородную цепь -(CH2)n - с n=1.
Документы, цитированные в отчёте о поиске
Производные бензола и их фармацевтическое применение
Комментарии