Способ получения катализатора гидроконверсии, содержащего по меньшей мере один цеолит nu-86 - RU2617987C2
Код документа: RU2617987C2
Чертежи
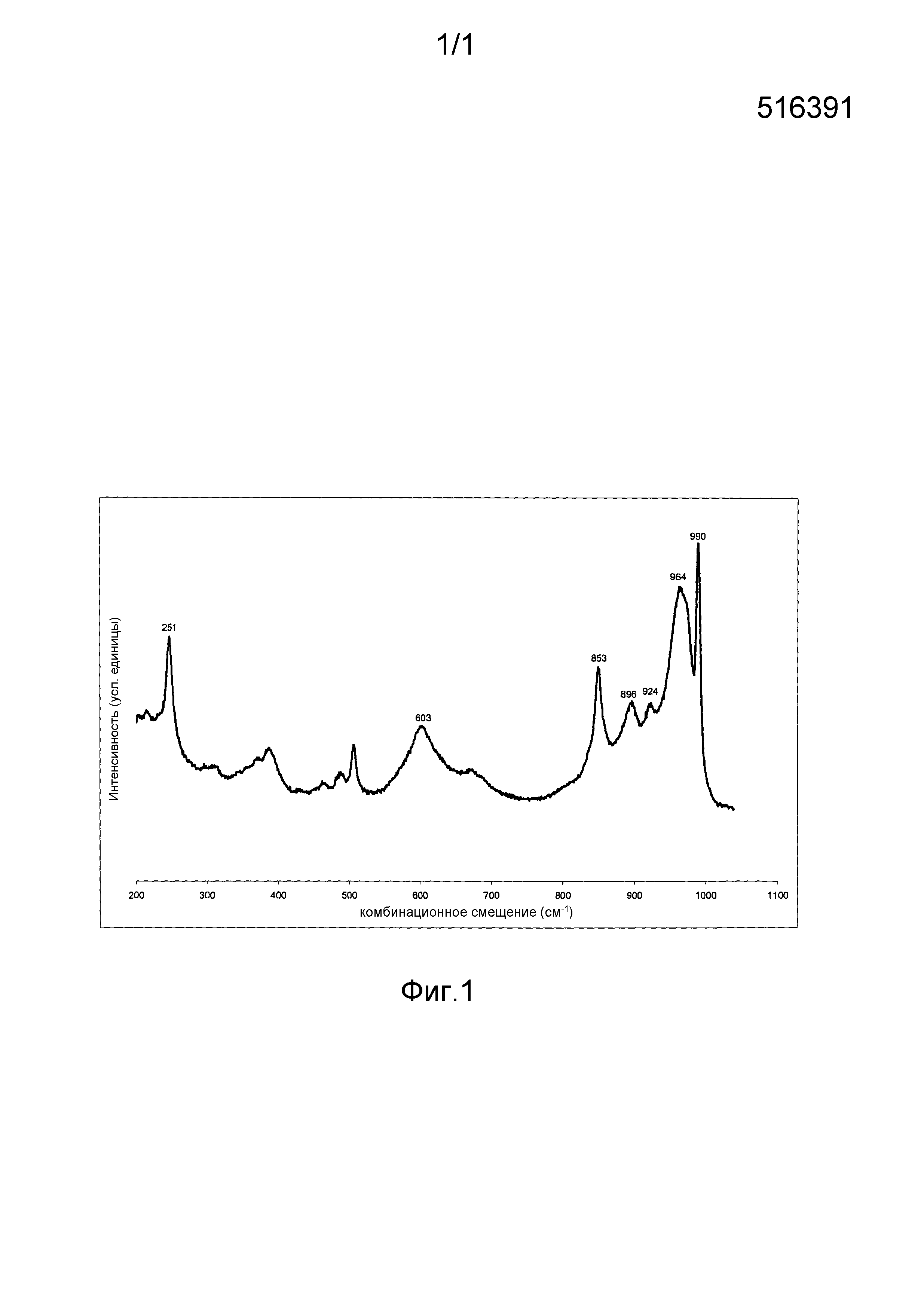
Описание
Изобретение относится к способу получения катализатора, содержащего по меньшей мере один металл, выбранный из группы, состоящей из металлов группы VIII и металлов группы VIB, используемых по отдельности или в смеси, и подложку, содержащую от 0,2 до 30 масс.% цеолита NU-86 и от 70 до 99,8 масс.% неорганической пористой матрицы, причем массовые доли выражены в расчете на общую массу указанной подложки. Настоящее изобретение относится также к способу гидрокрекинга, в котором используется катализатор, приготовленный особым способом получения согласно изобретению.
Уровень техники
Гидрокрекинг тяжелых нефтяных фракций является очень важным способом рафинирования, позволяющим получить из избыточных и малоценных тяжелых фракций более легкие фракции, такие как бензин, реактивное топливо и дизельное топливо, к которым стремятся на нефтеперерабатывающих предприятиях, чтобы адаптировать свою продукцию в соответствии со спросом. Этот способ широко описан в литературе.
Гидрокрекинг является способом, гибкость которого обусловлена тремя основными компонентами, а именно, используемыми рабочими условиями, типами применяемых катализаторов и тем, что гидрокрекинг углеводородного сырья можно реализовать в одну или две стадии.
Все катализаторы, использующиеся в процессах гидрокрекинга, являются бифункциональными, сочетающими кислотную функцию с гидрирующей функцией. Кислотная функция обеспечивается кислотными подложками, поверхность которых составляет обычно 150-800 м2/г, такими, как галогенированные оксиды алюминия (в частности, хлорированные или фторированные), комбинациями оксидов бора и алюминия и, чаще, аморфными алюмосиликатами и цеолитами в комбинации со связующим, обычно алюминиевым. Гидрирующая функция вносится либо одним или несколькими металлами группы VIB Периодической системы элементов, либо комбинацией по меньшей мере одного металла группы VIB Периодической системы и по меньшей мере одного металла группы VIII, осажденных на подложку.
Бифункциональность катализатора, то есть соотношение, сила и расстояние между кислотными и гидрирующими функциями, является ключевым параметром, известным специалисту, для воздействия на активность и селективность катализатора. Слабая кислотная функция и сильная гидрирующая функция дают малоактивные катализаторы, работающие обычно при высокой температуре (больше или равной 390-400°C) и при низкой объемной скорости подачи (VVH, выраженная в объеме сырья, подлежащего обработке, на единицу объема катализатора в час, обычно меньше или равна 2 ч-1), но обладающие очень хорошей селективностью по средним дистиллятам (реактивное топливо и газойли). Напротив, сильная кислотная функция и слабая гидрирующая функция дают активные катализаторы, но имеющие худшую селективность по средним дистиллятам.
Катализаторы, содержащие цеолиты, имеют хорошую каталитическую активность, но часто они имеют недостаточную селективность по средним дистиллятам (реактивное топливо и газойли).
Предшествующий уровень техники представлен многочисленными работами, направленными на улучшение селективности цеолитных катализаторов по средним дистиллятам. Эти катализаторы состоят из гидрирующей фазы с очень разным составом (разные металлы), обычно осажденной на подложку, содержащую цеолит, чаще всего цеолит Y. Гидрирующая фаза является активной в форме сульфида.
Патентная заявка FR 2775293 описывает также применение катализатора, содержащего по меньшей мере одну матрицу, по меньшей мере один цеолит NU-86 или NU-87 и по меньшей мере одну активную фазу, в процессе гидрокрекинга углеводородного сырья. Приготовление указанных катализаторов во всех случаях завершается этапом обжига при температуре от 250 до 600°C.
Целью настоящей заявки является предложить способ получения катализатора, позволяющий улучшить каталитическую активность указанного катализатора, применяющегося в процессе гидроконверсии углеводородного сырья, при сохранении или улучшении селективности цеолитных катализаторов по средним дистиллятам.
Сущность изобретения
Настоящее изобретение относится к способу получения катализатора, включающему по меньшей мере следующие последовательные этапы:
a) по меньшей мере приготовление подложки, содержащей от 0,2 до 30 масс.% цеолита NU-86 и от 70 до 99,8 масс.% неорганической пористой матрицы, причем массовые содержания выражены в расчете на общую массу указанной подложки,
b) по меньшей мере один этап пропитки указанной подложки, полученной на этапе a), по меньшей мере одним раствором, содержащим по меньшей мере один предшественник по меньшей мере одного металла, выбранного из группы, состоящей из металлов группы VIII и металлов группы VIB, используемых по отдельности или в смеси,
c) по меньшей мере один этап созревания,
d) по меньшей мере один этап сушки, проводимый при температуре менее 150°C, без последующего этапа обжига.
Настоящее изобретение относится также к способу гидрокрекинга углеводородного сырья с использованием катализатора, полученного указанным способом по изобретению, осуществляемым в присутствии водорода при температуре выше 200°C, давлении выше 1 МПа, объемной скорости от 0,1 до 20 ч-1 и при таком количестве вводимого водорода, чтобы объемное отношение "литры водорода / литры углеводорода" составляло от 80 до 5000 л/л.
Подробное описание изобретения
Этап a):
В соответствии с этапом a) способа получения согласно изобретению, готовят подложку, содержащую от 0,2 до 30 масс.% цеолита NU-86 и от 70 до 99,8 масс.% неорганической пористой матрицы, причем массовые доли выражены в расчете на общую массу указанной подложки.
Указанная подложка, приготовленная согласно этапу a) способа по изобретению, предпочтительно содержит от 0,5 до 25 масс.%, предпочтительно, от 1 до 20 масс.% цеолита NU-86 и от 75 до 99,5 масс.%, предпочтительно, от 80 до 99 масс.% неорганической пористой матрицы, причем массовые доли выражены в расчете на общую массу указанной подложки.
Цеолит NU-86 в его водородной форме, обозначенной H-NU-86, полученный обжигом и/или ионным обменом из цеолита NU-86 в состоянии непосредственно после синтеза, а также способ его получения описаны в патенте EP-0463768 A2. Указанный цеолит NU-86 характеризуется рентгеноструктурными параметрами, раскрытыми Casci et al. в патентной заявке EP 0463768.
Цеолит NU-86 обычно синтезируют в присутствии катионов натрия и органического структурообразующего агента, которым является либо октаметоний-дибромид, либо нонаметоний-бромид.
Цеолит NU-86 содержит кремний и по меньшей мере один элемент T, выбранный из группы, состоящей из алюминия, железа, галлия, бора, германия, предпочтительно T является алюминием.
Цеолит NU-86 не имеет определенного структурного типа, согласно правилам IZA (Международная ассоциация по цеолитам).
Структурный тип не был еще официально присвоен этому цеолиту комиссией IZA по синтезу. Однако, согласно работам, опубликованным J.L. Casci, P.A. Box, M.D. Shannon на 9-ом Международном конгрессе по цеолитам ("Proceedings of the 9th International Zeolite Conference, Montreal 1992, Eds R. Von Ballmoos et al., 1993 by Butterworth), оказывается, что по своим свойствам:
- цеолит NU-86 обладает трехмерной системой микропор;
- эта трехмерная система микропор состоит из прямолинейных каналов, ширина раскрытия пор которых ограничивается 11 атомами T (тетраэдрическими атомами: Si, Al, Ga, Fe и др.), прямых каналов, ограниченных по очереди шириной раскрытия из 10 и 12 атомов T, и синусоидальных каналов, также ограниченных по очереди шириной раскрытия из 10 и 12 атомов T.
Под термином "ширина раскрытия пор из 10, 11 или 12 тетраэдрических атомов (T)" понимаются поры, вмещающие 10, 11 или 12 атомов кислорода.
Цеолит NU-86, содержащийся в катализаторе согласно изобретению, по меньшей мере частично, а предпочтительно практически полностью, находится в форме кислоты, то есть в водородной форме (H+), при этом содержание натрия предпочтительно таково, что атомное отношение Na/T менее 10%, предпочтительно менее 5%, еще более предпочтительно менее 1%.
Цеолит NU-86, использующийся согласно изобретению, имеет мольное отношение Si/T меньше 150, предпочтительно меньше 100, предпочтительно меньше 50, очень предпочтительно меньше 35, более предпочтительно меньше 20 и еще более предпочтительно меньше 15.
Отношение Si/Al может быть получено при синтезе, без последующей модифицирующей обработки. Оно может быть также получено известными специалисту методами деалюминирования, такими, например, как паровая обработка, то есть термообработка под водяным паром, и/или кислотная обработка. Патентная заявка EP 0939673 описывает варианты осуществления деалюминирования цеолита NU-86.
Предпочтительно, цеолит NU-86, использующийся в изобретении, не подвергают этапу деалюминирования перед его формованием в структуре подложки катализатора согласно настоящему изобретению.
Неорганическая пористая матрица, входящая в состав подложки катализатора, приготовленного согласно изобретению, предпочтительно содержит по меньшей мере алюминий и/или по меньшей мере кремний.
Предпочтительно, указанная матрица содержит по меньшей мере один оксид алюминия или по меньшей мере один оксид кремния. Указанная матрица предпочтительно может быть кислотной или нет. Указанная матрица предпочтительно может быть мезоструктурированной или нет.
Указанная неорганическая пористая матрица предпочтительно может быть выбрана из переходных оксидов алюминия, легированных оксидов алюминия, предпочтительно фосфором, бором и/или фтором, из силикалита и оксидов кремния, алюмосиликатов, предпочтительно аморфных или слабо кристаллизованных, кристаллических нецеолитных молекулярных сит, таких, как силикоалюмофосфаты, алюмофосфаты, ферросиликаты, силикоалюминаты титана, боросиликаты, хромосиликаты и алюмофосфаты переходных металлов, использующихся по отдельности или в смеси.
В случае, когда указанная неорганическая пористая матрица выбрана из переходных оксидов алюминия, силикалита и оксидов кремния, таких, например, как мезопористые оксиды кремния, указанная матрица не является кислотной. Под переходным оксидом алюминия понимается, например, оксид алюминия фазы альфа, оксид алюминия фазы дельта, оксид алюминия фазы гамма или смесь оксида алюминия этих разных фаз.
В случае, когда указанная неорганическая пористая матрица выбрана из алюмосиликатов, предпочтительно аморфных или слабокристаллизованных, не цеолитных кристаллических молекулярных сит, таких, как силикоалюмюфосфаты, алюмофосфаты, ферросиликаты, силикоалюминаты титана, боросиликаты, хромосиликаты и алюмофосфаты переходных металлов, легированных оксидов алюминия, предпочтительно легированных фосфором, бором и/или фтором, то указанная матрица является кислотной. Для изобретения годится любой силикоалюминат или алюмосиликат, известный специалисту.
Предпочтительно кислотная пористая неорганическая матрица может также содержать, в дополнение к по меньшей мере одному из названных выше оксидных соединений, по меньшей мере одну простую синтетическую или натуральную глину типа диоктаэдрического филлосиликата 2:1 или триоктаэдрического филлосиликата 3:1, такую, как каолинит, антигорит, хризотил, монтмориллонит, бейделлит, вермикулит, тальк, гекторит, сапонит, лапонит. Эти глины при необходимости могут быть деламинированы.
Указанная неорганическая пористая матрица предпочтительно имеет количество катионных примесей менее 0,1 масс.%, предпочтительно менее 0,05 масс.% и еще более предпочтительно менее 0,025 масс.%. Под содержанием катионных примесей понимается общее содержание щелочей. Матрица предпочтительно имеет количество анионных примесей менее 1 масс.%, предпочтительно менее 0,5 масс.% и еще более предпочтительно менее 0,1 масс.%.
В случае, когда указанная неорганическая пористая матрица содержит по меньшей мере кремний, массовое содержание SiO2 в указанной неорганической пористой матрице предпочтительно составляет от 1 до 99 масс.%, предпочтительно от 5 до 95 масс.%, предпочтительно от 10 до 90 масс.%, более предпочтительно от 10 до 50 масс.% и еще более предпочтительно от 20 до 50 масс.%.
Предпочтительно, указанная неорганическая пористая матрица выбрана из оксида алюминия и алюмосиликата, используемых по отдельности или в смеси.
Этап a) приготовления подложки катализатора в способе получения по изобретению предпочтительно проводится любым методом, известным специалисту.
Предпочтительно цеолит NU-86, используемый согласно изобретению, можно обрабатывать для его стабилизации или создания в нем микропоры. Цеолит NU-86 предпочтительно модифицируют по меньшей мере одним из методов деалюминирования, известных специалисту, например, гидротермической обработкой или кислотным травлением. Предпочтительно, цеолит NU-86 модифицируют комбинацией следующих трех типов операций: гидротермическая обработка, ионный обмен и кислотное травление. Указанные операции специалисту известны. Указанный цеолит NU-86 можно также подвергнуть обработкам, называемым десиликацией, с помощью растворов оснований. Можно назвать, в частности, но без ограничений, обработку посредством NaOH или Na2CO3, сочетаемую или нет с деалюминирующей обработкой.
Используемый в подложке цеолит NU-86, возможно модифицированный, может находиться, без ограничений, например, в виде порошка, дробленого порошка, суспензии, суспензии, подвергшейся дезагломерирующей обработке. Так, например, цеолит можно вводить в суспензию, подкисленную или нет, в концентрации, соответствующей желаемому конечному содержанию цеолита на подложке. Эту суспензию, обычно называемую тестом, затем предпочтительно смешивают с предшественниками матрицы.
Цеолит NU-86, возможно модифицированный, предпочтительно смешивают с гелем, пастой или суспензией оксида. Смешение проводят так, чтобы получить указанные выше пропорции цеолита NU-86 и неорганической пористой матрицы в указанной подложке. Полученную так смесь затем формуют. Формование указанной подложки можно осуществить, например, экструзией, таблетированием, способом капельной коагуляции ("oil-drop"), грануляцией на вращающемся барабане или любым другим способом, хорошо известным специалисту.
Один предпочтительный вариант осуществления этапа a) способ получения по настоящему изобретению состоит в размешивании порошка цеолитов NU-86 во влажном геле в течение нескольких десятков минут, затем в проведении полученной таким способом пасты через фильеру, чтобы образовать экструдаты диаметром от 0,4 до 4 мм.
На этапе a) приготовления подложки, предпочтительно во время указанного смешения можно необязательно ввести по меньшей мере один гидрирующий-дегидрирующий металл, выбранный из группы, состоящей из металлов группы VIB и группы VIII Периодической системы, используемых по отдельности или в смеси.
Кроме того, для облегчения формования и/или для улучшения конечных механических свойств подложек, в указанную смесь можно добавлять добавки. В качестве примера добавок можно назвать, в частности, целлюлозу, карбоксиметилцеллюлозу, карбоксиэтилцеллюлозу, талловое масло, ксантановую камедь, ПАВы, флокулянты, как полиакриламиды, сажу, крахмалы, стеариновую кислоту, полиакриловый спирт, поливиниловый спирт, биополимеры, глюкозу, полиэтиленгликоли и т.д.
Регулирование характеристик пористости подложек по изобретению осуществляется частично на этом этапе формования частиц подложки.
Формованную подложку предпочтительно подвергают затем одной или нескольким термообработкам.
Указанную формованную подложку предпочтительно подвергают этапу сушки. Указанный этап сушки осуществляют любым методом, известным специалисту. Предпочтительно, сушку проводят в потоке воздуха. Указанную сушку можно также провести в потоке любого окислительного, восстановительного или инертного газа. Сушку выгодно проводить при пониженном давлении. Предпочтительно, сушку осуществляют при температуре от 50 до 180°C, предпочтительно от 60 до 150°C, более предпочтительно от 80 до 130°C.
Затем указанную подложку, возможно высушенную, предпочтительно подвергают этапу обжига.
Указанный этап обжига предпочтительно реализуют в присутствии молекулярного кислорода, например, проводя продувку воздухом, при температуре предпочтительно выше 220°C и меньше или равной 1100°C. Указанный этап обжига можно провести в проницаемом слое, в орошаемом слое или в статической среде. Например, в качестве печи можно использовать вращающуюся печь или вертикальную печь с радиальными проницаемыми слоями. Предпочтительно, указанный этап обжига проводится в течение более одного часа при 200°C до менее часа при 1100°C. Обжиг можно провести в присутствии водяного пара и/или в присутствии паров кислоты или основания. Например, обжиг можно осуществить при парциальном давлении аммиака.
Необязательно после обжига можно проводить дополнительные обработки для улучшения свойств подложки.
Так, указанную подложку можно необязательно подвергнуть гидротермической обработке в неподвижной среде или в потоке водяного пара. Под «гидротермической обработкой в неподвижной среде» понимается обработка в автоклаве в присутствии воды при температуре выше температуры окружающей среды.
В случае, когда указанная гидротермическая обработка проводится в неподвижной среде, указанную формованную подложку, содержащую неорганическую пористую матрицу и цеолит NU-86, можно обрабатывать разными способами. Так, указанную подложку можно пропитать кислотой перед ее подачей в автоклав, причем автоклавирование проводится в паровой фазе или в жидкой фазе, и эта паровая фаза или жидкая фаза в автоклаве может быть кислотной или нет. Эта пропитка до автоклавирования может быть кислотной или нет. Эта пропитка перед автоклавированием может осуществляться сухим способом или путем погружения указанной подложки в водный раствор кислоты. Предпочтительно, проводят сухую пропитку.
Автоклав предпочтительно представляет собой автоклав с вращающейся корзиной, какой определен в патентной заявке EP-A-0387109.
Температура во время автоклавирования предпочтительно составляет от 100 до 250°C в продолжении периода времени от 30 минут до 3 часов.
Этап b):
В соответствии с этапом b) способа получения согласно изобретению, осуществляют по меньшей мере один этап пропитки указанной подложки, приготовленной согласно этапу a), по меньшей мере одним раствором, содержащим по меньшей мере один предшественник по меньшей мере одного металла, выбранного из группы, состоящей из металлов группы VIII и металлов группы VIB, используемых по отдельности или в смеси.
Предпочтительно, указанный этап b) реализуют путем одной пропитки или последовательных пропиток по меньшей мере одним раствором, содержащим все или часть предшественников по меньшей мере одного металла, выбранного из группы, состоящей из металлов группы VIII и металлов группы VIB, используемых по отдельности или в смеси.
Указанный этап b) необязательно можно проводить при пониженном давлении.
Предпочтительно в случае, когда этап b) пропитки в способе получения согласно изобретению осуществляется путем последовательных пропиток, между этими этапами пропиток можно провести по меньшей мере один этап d) сушки.
Растворы, используемые на разных этапах пропитки или последовательных пропиток, необязательно могут содержать по меньшей мере один предшественник легирующего элемента, выбранного из бора, фосфора и кремния, и/или по меньшей мере одно органическое соединение.
Предшественники легирующего элемента, выбранного из бора, фосфора и кремния, и органические соединения также можно добавлять в пропиточные растворы, не содержащие предшественников по меньшей мере одного металла, выбранного из группы, состоящей из металлов группы VIII и металлов группы VIB, используемых по отдельности или в смеси.
Указанную органическую добавку можно вводить путем пропитки перед пропиткой предшественниками металлов, путем совместной пропитки с предшественниками металлов или путем дополнительной пропитки после пропитки предшественниками металлов.
Органические соединения, использующиеся в качестве промотирующих элементов гидрирующей функции, предпочтительно выбраны из хелатообразующих агентов, агентов, не являющихся хелатообразующими, из восстановителей и добавок, известных специалисту. Указанные органические соединения предпочтительно выбраны из одно-, двух- или многоатомных спиртов, возможно этерифицированных, карбоновых кислот, сахаров, нециклических моно-, ди- или полисахаридов, таких, как глюкоза, фруктоза, мальтоза, лактоза или сахароза, сложных эфиров, простых эфиров, краун-эфиров, циклодекстринов и соединений, содержащих серу или азот, как нитрилоуксусная кислота, этилендиаминтетрауксусная кислота, или диэтилентриамин, по отдельности или в смеси.
Указанные предшественники металлов группы VIII и металлов группы VIB, предшественники легирующих элементов и органические соединения предпочтительно вводят в пропиточный раствор или растворы в таком количестве, чтобы содержания в них элементов группы VIII, VIB, легирующего элемента и органических добавок на конечном катализаторе были такими, как определено менее.
Предпочтительно, указанные предшественники металлов группы VIII и металлов группы VIB, предшественники легирующих элементов и органические соединения предпочтительно вводят в пропиточный раствор или растворы в количествах, соответствующих:
- мольному отношению элемента группы VIII к элементу группы VIB от 0,1 до 0,8, предпочтительно от 0,15 до 0,5,
- мольному отношению легирующего элемента к элементу группы VIB от 0 до 1, предпочтительно от 0,08 до 0,5,
- мольному отношению органического соединения к элементу группы VIB от 0 до 5, предпочтительно от 0,2 до 3.
Предпочтительно, указанный или указанные этапы пропитки осуществляют способом пропитки, называемым сухим, который хорошо известен специалисту.
Подходящие для применения источники элементов группы VIII специалисту хорошо известны. Предшественники неблагородного металла или металлов группы VIII предпочтительно выбраны из оксидов, гидроксидов, гидроксикарбонатов, карбонатов и нитратов. Предпочтительно использовать гидроксикарбонат никеля, нитрат никеля, нитрат кобальта, карбонат никеля или гидроксид никеля, карбонат кобальта или гидроксид кобальта.
Предшественники благородных металлов группы VIII предпочтительно выбраны из галогенидов, например, хлоридов, нитратов, кислот, таких как платинохлористоводородная кислота, оксихлоридов, таких как аммиачный оксихлорид рутения.
Подходящие для применения предшественники элементов группы VIB хорошо известны специалисту. Например, из источников молибдена можно использовать оксиды и гидроксиды, молибденовые кислоты и их соли, в частности, соли аммония, такие, как молибдат аммония, гептамолибдат аммония, фосфорномолибденовая кислота (H3PMo12O40) и ее соли, и, необязательно, кремнемолибденовая кислота (H4SiMo12O40) и ее соответствующие соли. Источниками молибдена могут быть также любые полиоксиметаллаты типа Кеггина, например, лакунарные гетерополианионы типа Кеггина, замещенные гетерополианионы Кеггина, Доусона, Андерсона, Страндберга. Предпочтительно использовать триокисд молибдена и гетерополианионы типа Страндберга (P2Mo5O236-), Кеггина (PMo12O403-), лакунарного Кеггина или замещенного Кеггина, известные специалисту.
Например, из источников вольфрама можно использовать оксиды и гидроксиды, вольфрамовые кислоты и их соли, в частности, соли аммония, такие, как вольфрамат аммония, метавольфрамат аммония, фосфорновольфрамовая кислота (H3PW12O40) и ее соли, и, необязательно, кремнийвольфрамовая кислота (H4SiW12O40) и ее соли. Источниками вольфрама могут также быть, например, все полиоксометаллаты типа Кеггина, лакунарного Кеггина, замещенного Кеггина, Доусона. Предпочтительно используют оксиды и соли аммония, такие как метавольфрамат аммония, или гетерополианионы типа Кеггина, лакунарного Кеггина или замещенного Кеггина, известные специалисту.
Источником фосфора предпочтительно может быть ортофосфорная кислота H3PO4, соответствующие соли и сложные эфиры, или фосфаты аммония. Фосфор можно также ввести при синтезе указанной матрицы одновременно с элементом или элементами группы VIB в виде гетерополианионов Кеггина, лакунарного Кеггина, замещенного Кеггина или типа Страндберга, например, в виде фосфорномолибденовой кислоты и ее солей, фосфорновольфрамовой кислоты и ее солей. Фосфор, если его вводят не во время синтеза указанной матрицы, а при дополнительной пропитке, можно вводить в виде смеси фосфорной кислоты и основного органического соединения, содержащего азот, такого, как аммиак, первичные и вторичные амины, циклические амины, соединения из семейства пиридинов и хинолеинов и соединений из семейства пиррола.
Можно использовать большое число источников кремния. Так, можно использовать этилортосиликат Si(OEt)4, силоксаны, полисилоксаны, силикатно-галогенидные соединения, как фторсиликат аммония (NH4)2SiF6 или фторсиликат натрия Na2SiF6. Можно также использовать кремниймолибденовую кислоту и ее соли, кремнийвольфрамовую кислоту и ее соли. Кремний можно добавить, например, путем пропитки раствором этилсиликата в смеси вода/спирт. Кремний можно также добавить, например, пропиткой соединением кремния типа силикона, суспендированного в воде.
Предшественником бора может быть борная кислота, предпочтительно ортоборная кислота H3BO3, биборат или пентаборат аммония, оксид бора, сложные борные эфиры. Бор можно также вводить во время синтеза указанной матрицы одновременно с элементом или элементами группы VIB в виде гетерополианионов Кеггина, лакунарного Кеггина, замещенного Кеггина, например, в виде боромолибденовой кислоты и ее солей или боровольфрамовой кислоты и ее солей. Если бор вводится не во время синтеза указанной матрицы, а при дополнительной пропитке, то может быть выгодным вводить его, например, с помощью раствора борной кислоты в смеси вода/спирт или же в смеси вода/этаноламин. Бор можно также ввести в виде смеси борной кислоты, перекиси водорода и основного органического соединения, содержащего азот, такого как аммиак, первичные и вторичные амины, циклические амины, соединения из семейства пиридина и хинолинов, и соединения из семейства пиррола
Этап c):
Согласно этапу c) способа получения по изобретению, осуществляют по меньшей мере один этап созревания пропитанной подложки, полученной на этапе b).
Указанный этап созревания предпочтительно осуществляют, выдерживая пропитанную подложку, полученную на этапе b), во влажной атмосфере при температуре, составляющей предпочтительно от 10 до 80°C.
Указанный этап созревания предпочтительно имеет продолжительность от 15 минут до 48 часов.
Этап d):
Согласно этапу d) способа получения по изобретению, осуществляют по меньшей мере один этап сушки при температуре менее 150°C, без последующего этапа обжига, на пропитанной и дозревшей подложке, полученной на выходе этапа c).
Предпочтительно, указанный этап сушки осуществляют при температуре менее 140°C, предпочтительно, менее 145°C, очень предпочтительно менее 130°C, более предпочтительно составляющей от 100 до 145°C, и еще более предпочтительно от 100 до 130 °C, без последующего этапа обжига.
В случае, когда этап b) пропитки в способе получения согласно изобретению проводится путем последовательных пропиток, между указанными этапами пропитки предпочтительно проводят по меньшей мере один этап d) сушки.
Сушка позволяет удалить растворитель для пропитки без существенной модификации предшественников оксида, осажденных на подложке. Эффективная сушка, позволяющая удалить большую часть растворителя, обычно проводится при температуре от 100 до 150°C, предпочтительно от 100 до 130°C. Температура сушки не должна превышать 150°C, так как при температуре выше этой осажденные на подложке предшественники оксидов будут денатурироваться.
На выходе этапа d) получают сухой катализатор, который не подвергают никакому последующему этапу обжига.
Под этапом обжига понимается этап термообработки, вызывающий частичное или общее разложение органических молекул, возможно присутствующих на подложке, и на котором предшественники оксидов, осажденные на подложке, частично или полностью денатурируются. Этап обжига обычно проводится при температуре выше 150°C, предпочтительно от 250 до 600°C.
Катализаторы, полученные способом по настоящему изобретению, затем предпочтительно формуют в виде зерен разной формы и размеров. Их обычно используют в виде цилиндрических или многодольчатых экструдатов, как двухдольчатые, трехдольчатые, многодольчатые, прямой или скрученной формы, но необязательно их можно изготавливать и использовать в виде дробленого порошка, таблеток, колец, шариков, колес. Они имеют удельную поверхность, измеренную по адсорбции азота согласно методу БЭТ (Brunauer, Emmett, Teller, J. Am. Chem. Soc., vol. 60, 309-316 (1938)), от 50 до 600 м2/г, объем пор, измеренный ртутной порозиметрией, от 0,2 до 1,5 см3/г, причем распределение пор по размерам может быть унимодальным, бимодальным или полимодальным.
Предпочтительно, катализаторы, полученные способом по настоящему изобретению, имеют форму сфер или экструдатов. Однако выгодно, чтобы катализатор находился в виде экструдатов диаметром от 0,5 до 5 мм, в частности, от 0,7 до 2,5 мм. Формы являются цилиндрическими (которые могут быть полыми или нет), скрученными цилиндрическими, многодольчатыми (например, 2, 3, 4 или 5 лепестков), кольцевыми. Предпочтительно применяется трехдольчатая форма, но могут использоваться и любые другие формы.
Полученные, таким образом, катализаторы, находящиеся в оксидной форме, можно необязательно привести, по меньшей мере частично, в форму металла или сульфида.
Предпочтительно, перед применением указанные катализаторы, полученные способом по настоящему изобретению, преобразуют в сульфированные катализаторы, чтобы образовать их активные центры.
Этап e) сульфирования предпочтительно проводится после указанного этапа d) сушки, на указанном сухом катализаторе, полученном на выходе этапа d) способа по изобретению, без промежуточного этапа обжига.
Указанный сухой катализатор предпочтительно сульфируют ex situ или in situ. Сульфирующими агентами являются либо газообразный H2S, либо любое другое соединение, содержащее серу, используемое для активации углеводородного сырья в целях сульфирования катализатора. Указанные серосодержащие соединения предпочтительно выбраны из алкилдисульфидов, таких, например, как диметилдисульфид (DMDS), алкилсульфидов, как, например, диметилсульфид, н-бутилмеркаптан, полисульфидных соединений типа трет-нонилполисульфида, как, например, TPS-37 или TPS-54, выпускаемые в продажу компанией ARKEMA, или из любых других известных специалисту соединений, обеспечивающих хорошее сульфирование катализатора. Предпочтительно, катализатор сульфируют in situ в присутствии сульфирующего агента и углеводородного сырья. Очень предпочтительно, катализатор сульфируют in situ в присутствии углеводородного сырья, в которое добавлен диметилдисульфид.
Один хорошо известный специалисту способ классического сульфирования состоит в нагреве катализатора в присутствии сероводорода (чистого или, например, в потоке смеси водород/сероводород) до температуры в интервале от 150 до 800°C, предпочтительно от 250 до 600°C, обычно в реакционной зоне с проницаемым слоем.
Настоящее изобретение относится также к катализатору, который может быть получен способом получения согласно изобретению.
Катализатор, полученный способом согласно изобретению содержит по меньшей мере один гидрирующий-дегидрирующий металл, выбранный из группы, состоящей из металлов группы VIB и группы VIII Периодической системы, использующихся по отдельности или в смеси, и подложку, содержащую от 0,2 до 30 масс.%, предпочтительно от 0,5 до 25 масс.% и предпочтительно от 1 до 20 масс.% цеолита NU-86 и от 70 до 99,8 масс.%, предпочтительно от 75 до 99,5 масс.% и предпочтительно от 80 до 99 масс.% неорганической пористой матрицы, где массовые процентные доли выражены в расчете на общую массу указанной подложки.
Металлы группы VIB и группы VIII могут присутствовать, по меньшей мере частично, в форме, выбранной из металлической, и/или оксидной, и/или сульфидной форм.
Металлы группы VIII предпочтительно выбраны из благородных или неблагородных металлов, предпочтительно из железа, кобальта, никеля, рутения, родия, палладия, осмия, иридия и платины по одиночке или в смеси, предпочтительно, указанные металлы группы VIII выбраны из никеля, кобальта и железа, платины и палладия, использующихся по отдельности или в смеси.
Неблагородные металлы группы VIII предпочтительно выбраны из никеля, кобальта и железа, использующихся по отдельности или в смеси.
Благородные металлы группы VIII предпочтительно выбраны из платины и палладия, использующихся по отдельности или в смеси.
Металлы группы VIB предпочтительно выбраны из вольфрама и молибдена, использующихся по отдельности или в смеси.
Предпочтительно используют комбинации следующих металлов: никель-молибден, кобальт-молибден, никель-вольфрам, кобальт-вольфрам, предпочтительными комбинациями являются: никель-молибден, кобальт-молибден, кобальт-вольфрам, никель-вольфрам, еще более предпочтительными никель-молибден и никель-вольфрам.
Предпочтительно, указанный катализатор содержит по меньшей мере один гидрирующий-дегидрирующий металл группы VIB в комбинации с по меньшей мере одним неблагородным металлом группы VIII.
В случае, когда катализатор содержит по меньшей мере один металл группы VIB в комбинации с по меньшей мере одним неблагородным металлом группы VIII, содержание металла группы VIB предпочтительно составляет, в эквиваленте по оксиду, от 5 до 40 масс.% от полной массы указанного катализатора, предпочтительно от 10 до 35 масс.% и очень предпочтительно от 15 до 30 масс.%, а содержание неблагородного металла группы VIII предпочтительно составляет, в эквиваленте по оксиду, от 0,5 до 10 масс.% от полной массы указанного катализатора, предпочтительно от 1 до 8 масс.% и очень предпочтительно от 1,5 до 6 масс.%.
В случае, когда катализатор содержит по меньшей мере один благородный металл группы VIII, содержание благородного металла группы VIII предпочтительно составляет, в эквиваленте по оксиду, от 0,05 до 5 масс.% от полной массы указанного катализатора, предпочтительно от 0,1 до 2 масс.% и очень предпочтительно от 0,1 до 1 масс.%.
Катализатор согласно настоящему изобретению необязательно может также содержать промоторы активной фазы, предпочтительно выбранные из легирующих элементов и органических соединений. Указанные компоненты можно добавлять на различных этапах приготовления катализатора согласно изобретению.
Катализатор согласно настоящему изобретению необязательно может также содержать по меньшей мере один легирующий элемент, выбранный из бора, кремния и фосфора, по отдельности или в смеси. Под легирующим элементом понимается добавленный элемент, который сам не обладает каталитическими свойствами, но повышает каталитическую активность катализатора.
Указанный катализатор необязательно имеет содержание легирующего элемента от 0 до 10%, предпочтительно от 0,5 до 8%, более предпочтительно от 0,5 до 6 масс.%, выраженных в оксиде от полной массы указанного катализатора. Содержание кремния как легирующего элемента не учитывается в полном содержании кремния в цеолите или в матрице.
Бор, кремний и/или фосфор могут находиться в неорганической пористой матрице или в цеолите NU-86, или, предпочтительно, их осаждают на катализатор, и тогда они в основном локализованы на указанной неорганической пористой матрице.
Катализатор согласно настоящему изобретению необязательно может также содержать по меньшей мере одну органическую добавку. Под органической добавкой понимается органическая молекула, которая сама не обладает каталитическими свойствами, но повышает каталитическую активность катализатора.
Органические соединения, использующиеся в качестве промотирующих элементов гидрирующей функции, предпочтительно выбраны из хелатообразующих агентов, агентов, не являющихся хелатообразующими, из восстановителей и добавок, известных специалисту. Указанные органические соединения предпочтительно выбраны из одно-, двух- или многоатомных спиртов, возможно этерифицированных, карбоновых кислот, сахаров, нециклических моно-, ди- или полисахаридов, таких, как глюкоза, фруктоза, мальтоза, лактоза или сахароза, сложных эфиров, простых эфиров, краун-эфиров, циклодекстринов и соединений, содержащих серу или азот, таких как нитрилоуксусная кислота, этилендиаминтетрауксусная кислота, или диэтилентриамин, по отдельности или в смеси.
Указанный катализатор необязательно включает органическую добавку в содержании от 0 до 30%, предпочтительно от 5 до 30%, более предпочтительно от 10 до 30 масс.% от полной массы указанного катализатора.
Способ гидроконверсии
Настоящее изобретение относится также к способу гидроконверсии, предпочтительно гидрокрекинга, углеводородного сырья, в котором используется катализатор, приготовленный способом по настоящему изобретению, причем указанный способ гидроконверсии осуществляют в присутствии водорода при температуре выше 200°C, давлении выше 1 МПа, объемной скорости от 0,1 до 20 ч-1 и при таком количестве вводимого водорода, чтобы объемное отношение литры водорода/литры углеводорода составляло от 80 до 5000 л/л.
Предпочтительно, способ гидроконверсии согласно изобретению осуществляют при температуре в интервале от 250 до 480°C, предпочтительно от 320 до 450°C, очень предпочтительно от 330 до 435°C, при давлении в интервале от 2 до 25 МПа, предпочтительно от 3 до 20 МПа, объемной скорости в интервале от 0,1 до 6 ч-1, предпочтительно от 0,2 до 3 ч-1 и при таком количестве вводимого водорода, чтобы объемное отношение "литры водорода/литры углеводорода" составляло от 100 до 2000 л/л.
Использование этих рабочих условий в способе согласно изобретению обычно позволяют достичь за один проход конверсий в продукты с температурами кипения менее 340°C, предпочтительнее менее 370°C, более 15 масс.%, еще более предпочтительно от 20 до 95 масс.%.
Способом согласно изобретению можно обрабатывать самое разное сырье. Оно предпочтительно содержит по меньшей мере 20 об.%, предпочтительно по меньшей мере 80 об.% соединений, кипящих выше 340°C.
Углеводородное сырье, применяемое в способе согласно настоящему изобретению, предпочтительно выбрано из LCO (Light Cycle Oil = легкие газойли, получаемые на установке каталитического крекинга), атмосферных дистиллятов, вакуумных дистиллятов, таких, например, как газойли, полученные в результате прямой перегонки нефти или полученные на установках конверсии, таких, как FCC, установки коксования или висбрекинга, а также из сырья, поступающего с установок экстракции ароматики из базовых смазочных масел, или полученного при депарафинизации базовых смазочных масел растворителем, или же из дистиллятов, полученных в процессах десульфирования или гидроконверсии, в неподвижном слое или в кипящем слое, RAT (остатков атмосферной перегонки), и/или RSV (остатков вакуумной перегонки), и/или деасфальтированных масел, из парафинов, образованных в процессе Фишера-Тропша, из деасфальтированных масел и из сырья, полученного в процессах гидрообработки и гидроконверсии биомассы, где эти типы сырья могут использоваться по отдельности или в смеси. Данный список не является ограничительным. Указанное сырье предпочтительно имеет точку кипения T5 выше 340°C, предпочтительно выше 370°C, то есть 95% соединений, находящихся в сырье, имеют точку кипения выше 340°C, предпочтительно выше 370°C.
Содержание азота в сырье, обработанном способами согласно изобретению, предпочтительно выше 500 масс.ч./млн., предпочтительно составляет от 500 до 10000 масс.ч./млн., более предпочтительно от 700 до 4000 масс.ч./млн. и еще более предпочтительно от 1000 до 4000 масс.ч./млн. Содержание серы в сырье, обработанном способами согласно изобретению, обычно составляет от 0,01 до 5 масс.%, предпочтительно от 0,2 до 4%, еще более предпочтительно от 0,5 до 3%.
Сырье в известных случаях может содержать металлы. Суммарное содержание никеля и ванадия в сырье, обработанном способами согласно изобретению, предпочтительно менее 1 масс.ч./млн.
Сырье в известных случаях может содержать асфальтены. Содержание асфальтенов обычно менее 3000 масс.ч./млн., предпочтительно менее 1000 масс.ч./млн., еще более предпочтительно менее 200 масс.ч./млн.
Указанное углеводородное сырье может в известных случаях предпочтительно содержать металлы, в частности, никель и ванадий. Суммарное содержание никеля и ванадия в указанном углеводородном сырье, обработанном способом гидрокрекинга согласно изобретению, предпочтительно менее 1 масс.ч./млн. Содержание асфальтенов в указанном углеводородное сырье обычно менее 3000 масс.ч./млн., предпочтительно менее 1000 масс.ч./млн., еще более предпочтительно менее 200 масс.ч./млн.
Перед введением сырья и в случае, когда указанные катализаторы содержат неблагородные металлы, катализаторы, используемые в способе по настоящему изобретению, подвергают сульфирующей обработке, позволяющей превратить по меньшей мере часть металлических соединений в сульфид перед тем, как привести их в контакт с обрабатываемым сырьем. Эта обработка сульфированием хорошо известна специалисту и может быть осуществлена любым способом, уже описанным в литературе, in-situ, то есть в реакторе, или ex-situ.
Защитные слои
В случае, когда сырье содержит соединения типа смол и/или асфальтенов, выгодно сначала пропустить сырье через слой катализатора или адсорбента, отличного от катализатора гидрокрекинга или гидрообработки. Защитные катализаторы или слои, применяющиеся согласно изобретению, имеют форму сфер или экструдатов. Однако, выгодно, чтобы катализатор находился в виде экструдатов диаметром от 0,5 до 5 мм, в частности, от 0,7 до 2,5 мм. Формы являются цилиндрическими (и могут быть полыми или нет), скрученными цилиндрическими, многодольчатыми (например, 2, 3, 4 или 5 лепестков), кольцевыми. Предпочтительно применяется цилиндрическая форма, но могут использоваться и любые другие формы.
Чтобы предотвратить присутствие загрязняющих примесей или ядов в сырье, защитные катализаторы могут, в другом предпочтительном варианте осуществления, иметь особые геометрические формы, чтобы повысить в них долю пустот. Доля пустот в этих катализаторах составляет от 0,2 до 0,75. Их наружный диаметр может варьироваться от 1 до 35 мм. Из конкретных возможных форм можно назвать, без ограничений: полые цилиндры, полые кольца, кольца Рашига, полые зазубренные цилиндры, заершенные полые цилиндры, колесики с пятью спицами, цилиндры с несколькими отверстиями и т.д.
Эти защитные катализаторы или слои могут быть пропитаны или нет активной фазой. Предпочтительно, катализаторы пропитаны гидрирующей-дегидрирующей фазой. Очень предпочтительно, используется фаза CoMo или NiMo.
Эти защитные катализаторы или слои могут иметь макропоры. Защитные слои можно приобрести на рынке, например, у Norton-Saint-Gobain, например, защитные слои MacroTrap®. У компании Axens можно приобрести защитные слои из семейства ACT: ACT077, ACT645, ACT961 или HMC841, HMC845, HMC868 или HMC945. Может быть особенно предпочтительным разместить эти катализаторы друг над другом в по меньшей мере два разных слоя разной высоты. Катализаторы, имеющие наибольшую степень пустот, предпочтительно использовать в первом или первых каталитических слоях на входе каталитического реактора. Можно также использовать для этих катализаторов по меньшей мере два разных реактора.
Варианты осуществления
Способ гидроконверсии, предпочтительно гидрокрекинга согласно изобретению, в котором используется катализатор, полученный способом по изобретению, охватывает диапазоны давлений и конверсий от мягкого гидрокрекинга до гидрокрекинга высокого давления. Под мягким гидрокрекингом понимается гидрокрекинг, ведущий к умеренным конверсиям, обычно менее 40%, и проводимый при низком давлении, обычно от 2 МПа до 6 МПа.
Способ гидрокрекинга по изобретению осуществляют в присутствии по меньшей мере одного катализатора гидрокрекинга согласно изобретению. Способ гидрокрекинга по изобретению может быть реализован в одну или две стадии, независимо от давления, при котором осуществляют указанный способ. Способ осуществляют в присутствии одного или нескольких катализаторов гидрокрекинга, полученных согласно изобретению, на одной или нескольких реакционных установках, оборудованных одним или несколькими реакторами.
В способе гидрокрекинга согласно изобретению может использоваться единственный описанный выше катализатор, в одном или нескольких каталитических слоях, в неподвижном слое, в одном или нескольких реакторах, в схеме гидрокрекинга, называемой одностадийной, с или рециркуляции жидкости из непрореагировавшей фракции, возможно в сочетании с классическим катализатором гидроочистки, находящимся по потоку выше катализатора, используемого в способе по настоящему изобретению.
В способе гидрокрекинга согласно изобретению может использоваться указанный, описанный выше катализатор самостоятельно, в одном или нескольких реакторах с кипящем слоем, в схеме гидрокрекинга, называемой одностадийной, с или без рециркуляции жидкости из непрореагировавшей фракции, возможно в сочетании с классическим катализатором гидроочистки, находящимся в реакторе с неподвижным слоем или с кипящим слоем по потоку выше катализатора, используемого в способе по настоящему изобретению.
Кипящий слой функционирует с удалением отработанного катализатора и ежедневным добавлением нового катализатора, чтобы сохранить стабильной активность катализатора.
Катализатор, описанный согласно изобретению, можно также использовать в первой реакционной зоне гидрообработки, при предварительной конверсионной обработке, один или в сочетании с другим классическим катализатором гидроочистки, находящимся по потоку выше катализатора по изобретению, в одном или нескольких каталитических слоях, в одном или нескольких реакторах с неподвижным слоем или кипящим слоем.
Одностадийный способ
Способ гидрокрекинга согласно изобретению может осуществляться в процессе, называемом одностадийным.
Одностадийный гидрокрекинг обычно включает в первую очередь глубокую гидроочистку, цель которой заключается в осуществлении гидродеазотирования и глубокого десульфирования сырья перед его проведением на катализатор собственно гидрокрекинга, в частности, в случае, когда он содержит цеолит. Эта глубокая гидроочистка сырья приводит лишь к ограниченной конверсии сырья в более легкие фракции, которая остается недостаточной и, следовательно, должна быть завершена на более активном катализаторе гидрокрекинга, описанном выше. Однако следует отметить, что между этими двумя типами катализаторов не проводится никакого разделения. Весь поток на выходе из реактора вводится на указанный катализатор собственно гидрокрекинга, и только после этого осуществляется разделение образованных продуктов. Этот вариант гидрокрекинга, называемый также "Once Through" (одноразовым), имеет модификацию, которая включает возврат непрореагировавшей фракции в реактор в целях получения более высокой конверсии сырья.
Таким образом, катализатор, полученный способом по настоящему изобретению, применяется в одностадийном способе гидрокрекинга в зоне гидрокрекинга, находящейся по потоку за зоной гидроочистки, причем между этими двумя зонами не проводится никакого промежуточного разделения.
Предпочтительно, катализатор гидроочистки, используемый в первой реакционной зоне гидроочистки, один или в комбинации с другим классическим катализатором гидроочистки, находящимся по потоку выше катализатора, полученного способом по настоящему изобретению, является катализатором, возможно содержащим легирующий элемент, выбранный из фосфора, бора и кремния, причем указанный катализатор имеет в основе неблагородные элементы группы VIII, возможно в комбинации с элементами группы VIB, на подложке из оксида алюминия или алюмосиликата, еще более предпочтительно указанный катализатор содержит никель и вольфрам.
Катализатор, полученный способом по настоящему изобретению, может также применяться в первой реакционной зоне гидроочистки, в предварительной конверсионной обработке, один или в комбинации с другим классическим катализатором гидроочистки, находящимся по потоку выше катализатора, описанного в настоящем изобретении, в одном или нескольких каталитических слоях, в одном или нескольких реакторах.
Одностадийный способ в неподвижном слое с промежуточным разделением
Способ гидрокрекинга согласно изобретению можно осуществлять в одностадийном процессе с неподвижным слоем, включающем промежуточное разделение.
Указанный способ предпочтительно включает зону гидроочистки, т.е. зону, позволяющую частично удалить аммиак, например, путем резкого повышения температуры, и зону, содержащую указанный катализатор гидрокрекинга согласно изобретению. Этот одностадийный способ гидрокрекинга углеводородного сырья для получения средних дистиллятов и, возможно, базовых масел, предпочтительно содержит по меньшей мере одну первую реакционную зону гидроочистки и по меньшей мере одну вторую реакционную зону, в которой проводится гидрокрекинг по меньшей мере части потока, выходящего из первой реакционной зоны. Этот способ предпочтительно включает также неполное удаление аммиака из потока, выходящего из первой зоны. Это разделение осуществляется предпочтительно посредством промежуточного резкого повышения температуры.
Гидрокрекинг, осуществляемый во второй реакционной зоне, предпочтительно проводится в присутствии аммиака в количестве меньше, чем присутствует в сырье, предпочтительно меньше 1500 масс.ч./млн., более предпочтительно меньше 1000 масс.ч./млн. и еще более предпочтительно меньше 800 масс.ч./млн. азота.
Таким образом, катализатор согласно настоящему изобретению применяется в процессе одностадийного гидрокрекинга в неподвижном слое с промежуточным разделением, в зоне гидрокрекинга, находящейся по потоку менее зоны гидроочистки, причем между этими двумя зонами проводится промежуточное разделение для частичного удаления аммиака.
Предпочтительно, катализатор гидроочистки, используемый в первой реакционной зоне гидроочистки, один или в комбинации с другим классическим катализатором гидроочистки, находящийся по потоку выше катализатора, описанного согласно изобретению, является катализатором, необязательно содержащим легирующий элемент, выбранный из фосфора, бора и кремния, причем указанный катализатор имеет в основе неблагородные элементы группы VIII, возможно в комбинации с элементами группы VIB, на подложке из оксида алюминия или алюмосиликата, еще более предпочтительно указанный катализатор содержит никель и вольфрам.
Описанный катализатор по изобретению может также применяться в первой реакционной зоне гидроочистки, при предварительной конверсионной обработке, один или в сочетании с другим классическим катализатором гидроочистки, находящимся по потоку выше катализатора по изобретению, в одном один или нескольких каталитических слоях, в одном или нескольких реакторах.
Двухстадийный способ
Способ гидроконверсии, предпочтительно гидрокрекинга, согласно изобретению может осуществляться в процессе, называемом двухстадийным.
Двухстадийный гидрокрекинг включает первую стадию, цель которой, как и в одностадийном способе, состоит в том, чтобы осуществить гидроочистку сырья, а также достичь конверсии сырья обычно порядка 40-60%. Поток, выходящий с первой стадии, подвергают затем разделению (дистилляции), чаще всего называемому промежуточным разделением, целью которого является отделить продукты конверсии от непрореагировавшей фракции. На второй стадии двухстадийного способа гидрокрекинга обрабатывается только фракция сырья, не прореагировавшая на первой стадии. Это разделение делает двухстадийный способ гидрокрекинга более селективным по средним дистиллятам (керосин + дизель), чем одностадийный способ. Действительно, промежуточное разделение продуктов конверсии предотвращает их чрезмерное крекирование в нафту и газы на второй стадии на катализаторе гидрокрекинга. Кроме того, следует отметить, что непрореагировавшая фракция сырья, обработанного на второй стадии, имеет обычно очень низкое содержание NH3, а также органических азотсодержащих соединений, обычно менее 20 масс.ч./млн. даже менее 10 масс.ч./млн.
Конфигурации неподвижного или кипящего слоя катализатора, описанные для одностадийного способа, могут использоваться на первой стадии двухстадийной схемы, независимо от того, используется ли катализатор согласно изобретению самостоятельно или в комбинации с классическим катализатором гидроочистки.
Таким образом, катализатор, полученный способом по изобретению, может использоваться в процессе гидрокрекинга, называемом двухстадийным, на второй стадии гидрокрекинга, находясь по потоку менее первой стадии гидроочистки, причем между этими двумя зонами осуществляют промежуточное разделение.
Для одностадийных способов и для первой стадии гидроочистки двухстадийных способов гидрокрекинга классическими катализаторами гидроочистки, которые предпочтительно могут использоваться, являются катализаторы, необязательно содержащие легирующий элемент, выбранный из фосфора, бора и кремния, причем указанный катализатор имеет в основе неблагородные элементы группы VIII, возможно в комбинации с элементами группы VIB на подложке оксида алюминия или алюмосиликата, еще более предпочтительно, указанный катализатор содержит никель и вольфрам.
В соответствии с первым вариантом осуществления способа гидрокрекинга согласно изобретению, применяемые в этом способе катализатор или катализаторы предпочтительно используются, одни или в последовательной цепочке, в одном или нескольких каталитических слоях, в неподвижном или в кипящем слое, в одном или нескольких реакторах, в одностадийной схеме гидрокрекинга, с или без рециркуляции жидкости из непрореагировавшей фракции, возможно в комбинации с катализатором гидроочистки, находящимся по потоку выше катализатора или катализаторов гидрокрекинга. Кипящий слой функционирует с удалением отработанного катализатора и ежедневной добавкой нового катализатора, чтобы сохранить стабильной активность катализатора.
Согласно второму варианту осуществления способа гидрокрекинга по изобретению, катализатор или катализаторы гидрокрекинга предпочтительно используются в этом способе, одни или в последовательной цепочке, в одном или нескольких каталитических слоях, на той и/или иной стадии двухстадийной схемы гидрокрекинга. Двухстадийная схема представляет собой схему, в которой имеется промежуточное разделение потоков между двумя реакционными зонами. Эта схема может функционировать с или без рециркуляции жидкости из непрореагировавшей фракции, выходящей из первой реакционной зоны или второй реакционной зоны. Первая реакционная зона функционирует в неподвижном слое или в кипящем слое. В частном случае, когда катализатор или катализаторы гидрокрекинга, полученные согласно изобретению, находятся в первой реакционной зоне, их предпочтительно размещают в комбинации с катализатором гидроочистки, находящимся по потоку выше указанных катализаторов.
Следующие примеры иллюстрируют настоящее изобретение, однако не ограничивают его объем.
Пример 1: Приготовление подложки S1, содержащей цеолит NU-86 и пористую матрицу типа оксида алюминия
Одним из используемых исходных материалов является цеолит NU-86, который получен согласно примеру 2 патента EP 0463768 A2 и имеет отношение полных атомных содержаний Si/Al, равное 11, и атомное отношение Na/Al, равное 0,25.
Цеолит NU-86 в состоянии сразу после синтеза сначала подвергали обжигу, называемому сухим, при 550°C в потоке сухого воздуха в течение 9 часов. Затем полученное твердое вещество подвергали четырем этапам ионного обмена в растворе NH4NO3 (10н), при температуре около 100°C в течение 4 часов для каждого этапа обмена. Полученный таким образом раствор, обозначен NH4-NU-86/1, он имеет отношение Si/Al=11 и отношение Na/Al=0,0012. Другие физико-химические характеристики приведены в Таблице 1.
Кристаллиты цеолита NU-86 имели вид кристаллов с размерами в интервале от 0,4 мкм до 2 мкм.
Затем 23 масс.% цеолита NU-86 смешивали с 77 масс.% матрицы, состоящей из ультратонкого плоского бемита или алюмогеля. К этой смеси добавляли водный раствор, содержащий 66%-ную азотную кислоту (7 масс.% кислоты на грамм сухого геля). Эту смесь размешивали 15 минут. Перемешанную пасту экструдировали затем через фильеру, имеющую трехдольчатые отверстия диаметром 2 мм. Затем экструдаты обжигали 2 часа при 500°C на воздухе. Эти экструдаты образуют подложку S1.
Пример 2: Приготовление подложки S2, содержащей цеолит NU-86 и пористую матрицу алюмосиликатного типа
Порошок алюмосиликата готовили путем совместного осаждения композиции, состоящей из 30% SiO2 и 70% Al2O3. Затем готовили подложку катализатора гидрокрекинга, содержащую этот алюмосиликат и цеолит Nu-86 из примера 1. Для этого использовали 7,6 масс.% цеолита NU-86 из примера 1, который перемешивали с 92,4 масс.% приготовленной выше матрицы, состоящей из алюмосиликата. Затем эту порошковую смесь перемешивали с водным раствором, содержащим 66%-ную азотную кислоту (7 масс.% кислоты на грамм сухого геля), затем перемешивали 15 минут. По окончании этого перемешивания полученную пасту пропускали через фильеру, имеющую трехдольчатые отверстия диаметром 2 мм. Затем экструдаты сушили в течение ночи при 120°C, затем обжигали при 550°C в течение 2 часов на воздухе. Наконец, экструдаты обрабатывали водяным паром при 750°C в течение 2 ч. Эти экструдаты образуют подложку S2.
Пример 3: Приготовление катализатора C1 формулы NiMoP (согласно изобретению) на подложке S2
Экструдаты подложки S2, содержащие цеолит NU-86 и алюмосиликатную матрицу, пропитывали сухим способом водным раствором, в котором заранее были растворены, путем кипячения с обратным холодильником, Ni(OH)2, MoO3 и H3PO4. Экструдаты сушили в течение ночи при 120°C на воздухе и не повергали последующему обжигу. Формула NiMoP катализатора C1 следующая: 2,2-20-4,2 масс.% в расчете на сухую массу катализатора, соответственно для Ni (выражено на NiO), для Mo (выражено на MoO3) и для P (выражено на P2O5). Отношения следующие: Ni/Mo=,21 и P/Mo=0,42.
Пример 4: Приготовление катализатора C2 формулы NiMoP (не по изобретению) на подложке S2
Катализатор C2 соответствует катализатору C1, обожженному при 450°C.
Пример 5: Приготовление катализатора C3 формулы NiMoP (согласно изобретению) на подложке S1
Экструдаты подложки, содержащие цеолит NU-86 и оксид алюминия, пропитывали сухим способом водным раствором, в котором были предварительно растворены, путем кипячения с обратным холодильником, Ni(OH)2, MoO3 и H3PO4. Их сушили в течение ночи при 120°C на воздухе, обжиг позднее не проводили. Формула NiMoP катализатора C3 следующая: 2,6-23,2-4,7 масс.%, в расчете на сухую массу катализатора, соответственно для Ni (выражено на NiO), для Mo (выражено на MoO3) и для P (выражено на P2O5). Отношения следующие: Ni/Mo=0,22 и P/Mo=0,41.
Пример 6: Приготовление катализатора C4 формулы NiMoP (не по изобретению) на подложке S1
Катализатор C4 соответствует катализатору C3, обожженному при 450°C.
Пример 7: Оценка гидрокрекинга высокого давления вакуумного дистиллята на катализаторах C1 и C2
Катализаторы C1 и C2, получение которых описано в примерах 3 и 4, использовались для осуществления гидрокрекинга частично гидрообработанного вакуумного дистиллята, основные характеристики которого указаны в Таблице 2.
Катализаторы C1 и C2 использовали в соответствии со способом по изобретению, применяя пилотную установку, содержащую один реактор с проницаемым слоем, причем среды текли сверху вниз (нисходящий поток).
Перед испытанием на гидрокрекинг катализаторы сульфировали при давлении 14 МПа и температуре 350°C, посредством газойля прямой перегонки, в который было добавлено 2 масс.% диметилдисульфида (DMDS).
После сульфирования осуществляли каталитические испытания в следующих условиях:
- общее давление: 14 МПа,
- расход водорода: 1000 литров газообразного водорода на литр вводимого сырья,
- объемная скорость (VVH) 0,66 ч-1.
Использовалась температура, при которой получают брутто-конверсию 80%.
В сырье добавляли DMDS и анилин, чтобы сохранить в ходе испытания парциальные давления H2S и NH3 были такими, какие образовались бы в результате предварительной гидрообработки исходного сырья, не подвергавшегося гидрообработке.
Каталитические характеристики выражены в терминах брутто-конверсии фракции 370+ (молекулы, точка кипения которых выше 370°C) во фракцию 370- (молекулы, точка кипения которых менее 370°C), и выхода по средним дистиллятам (DM, фракция 150-370°C). Конверсия и выход по DM оценивались из результатов имитированной дистилляции и по анализу газов методом газовой хроматографии.
Брутто-конверсия в продукты с точкой кипения менее 370°C, обозначенная CB 370°C, определена как массовая процентная доля молекул, точка кипения которых в выходящих потоках менее 370°C, т.е. CB 370°C = % 370°C-эффлюенты
Брутто-селективность по средним дистиллятам (фракция, точки кипения которой лежат в интервале от 150 до 370°C) обозначена SB DM и равна:
SB DM = [(фракция 150-370эффлюенты)]/[(% 370°Cэффлюенты)]
Полученные каталитические параметры указаны в Таблице 3 менее.
Результаты показывают, что сушка при 120°C оксидной фазы, являющейся предшественником гидрирующей функции, позволяет создать катализатор C1 (приготовленный согласно изобретению), более активный, чем катализатор C2 (не по изобретению), у которого оксидную фазу обжигали при 450°C, при сохранении той же селективности по средним дистиллятам.
Пример 8: Оценка гидрокрекинга вакуумного дистиллята при высоком давлении на катализаторах C3 и C4
Катализаторы C3 и C4, приготовление которых описано в примерах 5 и 6, использовались для осуществления гидрокрекинга гидрообработанного вакуумного дистиллята, основные характеристики которого приведены в Таблице 4.
Катализаторы C3 и C4 применялись в соответствии со способом по изобретению, используя пилотную установку, содержащую один реактор с проницаемым неподвижным слоем, причем среды двигались сверху вниз (нисходящий поток).
Перед испытанием на гидрокрекинг катализаторы сульфировали при давлении 14 МПа и температуре 350°C посредством газойля прямой перегонки, в который было добавлено 2 масс.% диметилсульфида (DMDS).
После сульфирования были проведены каталитические испытания в следующих условиях:
- общее давление: 14 МПа,
- расход водорода: 1000 литров газообразного водорода на литр введенного сырья,
- объемная скорость (VVH) равна 1 ч-1.
Использовалась температура, при которой получают брутто-конверсию 70%.
В сырье добавляли DMDS и анилин, чтобы сохранить в ходе испытания парциальные давления H2S и NH3 такими, какие образовались бы в результате предварительной гидрообработки исходного сырья, не подвергавшегося гидрообработке.
Каталитические характеристики выражены в терминах брутто-конверсии фракции 370+ (молекулы, точка кипения которых выше 370°C) во фракцию 370- (молекулы, точка кипения которых менее 370°C), и выхода по средним дистиллятам (DM, фракция 150-370°C). Конверсия и выход по DM оценивались из результатов имитированной дистилляции и по анализу газов методом газовой хроматографии.
Брутто-конверсия в продукты с точкой кипения менее 370°C, обозначенная CB 370°C, определена как массовая процентная доля молекул, точка кипения которых в выходящих потоках менее 370°C, т.е. CB 370°C = % 370°C-эффлюенты
Выход по средним дистиллятам (фракция, точки кипения которой составляют от 150 до 370°C) определяется как:
Выход по DM = % молекул, точки кипения которых в выходящих потоках составляют от 150°C до 370°C.
Полученные характеристики катализатора приведены менее в Таблице 5.
Результаты показывают, что сушка при 120°C оксидной фазы, являющейся предшественником гидрирующей функции, позволяет получить катализатор C3 (приготовленный способом согласно изобретению), являющийся более активным, чем катализатор C4 (не по изобретению, у которого оксидную фазу обжигали при 450°C, при сохранении такого же выхода по средним дистиллятам.
Реферат
Изобретение относится к способу получения катализатора, включающему по меньшей мере следующие последовательные этапы: a) по меньшей мере приготовление подложки, содержащей от 0,2 до 30 мас.% цеолита NU-86 и от 70 до 99,8 мас.% неорганической пористой матрицы, причем массовые содержания выражены в расчете на общую массу указанной подложки, b) по меньшей мере один этап пропитки указанной подложки, полученной на этапе а), по меньшей мере одним раствором, содержащим по меньшей мере один предшественник по меньшей мере одного металла, выбранного из группы, состоящей из металлов группы VIII и металлов группы VIB, используемых по отдельности или в смеси, c) по меньшей мере один этап созревания, d) по меньшей мере один этап сушки, проводимый при температуре менее 150°С, без последующего этапа обжига. Изобретение относится также к способу гидрокрекинга углеводородного сырья с применением катализатора, полученного, согласно изобретению, указанным способом получения. Технический результат заключается в увеличении каталитической активности катализатора. 2 н. и 12 з.п. ф-лы, 5 табл., 1 ил., 8 пр.
Формула
Документы, цитированные в отчёте о поиске
Каталитическая композиция для гидрокрекинга и способ мягкого гидрокрекинга и раскрытия колец
Комментарии