Способ получения пара-ксилола (варианты) - RU2352550C2
Код документа: RU2352550C2
Чертежи




Описание
Область, к которой относится изобретение
Данное изобретение относится к способу получения пара-ксияола из исходной ароматической фракции C8, содержащей смесь изомеров ксилола.
Уровень техники
Смеси углеводородов, содержащие ароматические соединения C8+ являются побочными продуктами некоторых процессов переработки нефти, включая, но без ограничения, процессы каталитического реформинга. Эти углеводородные смеси, как правило, содержат до 30 весовых процентов (вес.%) С9+ ароматических углеводородов, до 10 вес.% неароматических углеводородов, примерно, до 50 вес.% этилбензола, остальное (баланс) (например, приблизительно, до 100 вес.%) представляет собой смесь изомеров ксилола. Чаще всего среди этих C8 ароматических углеводородов присутствуют этилбензол ("ЕВ") и изомеры ксилола, включая мета-ксилол ("mX"), орто-ксилол ("оХ") и пара-ксилол ("рХ"). Как правило, если в смеси C8 ароматических углеводородов присутствует этилбензол, его концентрация составляет, примерно, до 20 вес.% от общего веса C8 ароматических углеводородов. Обычно три изомера ксилола представляют собой остальные C8 ароматические углеводороды и находятся в равновесной смеси в весовом соотношении 1:2:1 (oX:mX:pX). Таким образом применяемое в данном описании выражение "равновесная смесь изомеров ксилола" относится к смеси, содержащей изомеры в примерном весовом соотношении 1:2:1 (oX:mX:pX).
Этилбензол применяют для получения стирола. Мета-ксилол используют для получения изофталевой кислоты, которая, в свою очередь, применяется для получения специальных полиэфирных волокон, красок и смол. Орто-ксилол используют в производстве фталевого ангидрида, который, в свою очередь, применяется в производстве пластификаторов на основе фталатов. Хотя мета-ксилол и орто-ксилол являются полезным сырьем, требования к этим изомерам и материалам на их основе не так высоки, как к пара-ксилолу и материалам на основе пара-ксилола. Пара-ксилол является сырьем для производства терефталевых кислот и эфиров, используемых для получения полимеров, таких как полибутилентерефталат, полиэтилентерефталат и полипропилентерефталат.
Вследствие их полезности эффективное разделение этилбензола и индивидуальных различных изомеров ксилола вызывает особый и не снижающийся интерес. В зависимости от концентраций, в которых они находятся в смеси С8 ароматических углеводородов, и от потребности в конкретном изомере по сравнению с другими изомерами или с этилбензолом, или в материале на его основе, одного разделения может быть недостаточно для получения адекватных количеств каждого конкретного изомера. Например, так как, как правило, существует более высокая потребность в пара-ксилоле по сравнению с другими изомерами или этилбензолом, желательно повысить или даже максимально увеличить производство пара-ксилола из конкретной смеси C8 ароматических соединений. Поэтому к отделению (выделению) пара-ксилола добавляется изомеризация мета- и орто-ксилола в требуемый пара-ксилол и, необязательно, превращение этилбензола.
Стадия выделения способов получения пара-ксилола, как правило, состоит из двух процессов, один из которых представляет собой кристаллизацию, а другой является жидкостно-адсорбционной хроматографией. Процесс кристаллизации первоначально разработан в Amoco Corporation, усовершенствован и модифицирован позднее в Institut Francais du Petrole ("IFF"), Mobil Corporation ("Mobil"), UOP Inc. ("UOP") и др. Как более подробно описано далее, кристаллизация имеет свои ограничения и может быть очень дорогостоящей, так как различные изомеры ксилола кристаллизуются при очень низких температура (например, около -70°С÷0°С), для этого, как правило, требуются многостадийные охладительные системы, снабженные большими газовыми компрессорами. Жидкостно-адсорбционная хроматография, также называемая адсорбционной хроматографией в системе имитированных подвижных слоев ("SiMBAC"), в промышленных масштабах реализована в IFP и UOP. Процесс S1MBAC также имеет свои ограничения и является дорогим, так как требует собственной рециркуляции больших объемов различных углеводородных десорбентов. Кроме того, потоки эффлюента после стадии (операции) адсорбции следует отделять от нужного продукта на последующих стадиях дистилляции. Следовательно, вышеуказанные обычные процессы кристаллизации и жидкостно-адсорбционной хроматографии являются невыгодными вследствие значительной стоимости и больших энергетических затрат.
Один способ производства пара-ксилола из смеси C8+ ароматических углеводородов включает пропускание смеси через разделительную колонну для удаления тяжелых веществ, таких как С9+ углеводороды. Более легкий верхний погон (дистиллят), преимущественно содержащий смесь C8+ углеводородов, состоящую из изомеров ксилола и этилбензола, можно разделить на разделительной установке. Так как этилбензол, мета-, орто- и пара-ксилолы имеют одинаковые молекулярные массы и сходные температуры кипения (около 136°С, около 139°С, около 138°С и около 144°С соответственно), разделение с помощью фракционной перегонки непрактично. Альтернатива фракционной перегонке включает низкотемпературную кристаллизацию, которая использует разницу температур замерзания или кристаллизации различных компонентов - пара-ксилол кристаллизуется (примерно при 13.3°С) раньше других изомеров ксилола (орто- и мета-ксилол кристаллизуются примерно при -25.2°С и -47.9°С соответственно). В физической системе трех изомеров ксилола имеется две имеющие значение бинарные эвтектики: бинарная эвтектика пара-ксилол/мета-ксилол и бинарная эвтектика пара-ксилол/орто-ксилол. Когда пара-ксилол кристаллизуется из смеси, оставшаяся смесь приближается к одной из этих бинарных эвтектик, в зависимости от исходного состава смеси. Поэтому в промышленных процессах пара-ксилол кристаллизуется таким образом, чтобы приблизить, но не достичь бинарных эвтектик, чтобы избежать сокристаллизации других изомеров ксилола, что понизило бы чистоту полученного пара-ксилола. Из-за этих бинарных эвтектик количество пара-ксилола, извлеченного (полученного) за кристаллизационный цикл, как правило, не выше 65°С от количества пара-ксилола в смеси, подаваемой в кристаллизатор.
Или же некоторые компоненты C8 углеводородной смеси можно выделять из смеси перед любой кристаллизацией, например, жидкофазной адсорбцией (например, процесс PAREXTM фирмы UOP и процесс ELUXILTM института IFP) с применением фожазита (цеолита) для хроматографического выделения пара-ксилола из C8 смесей, содержащих пара-ксилол. В обедненном пара-ксилолом потоке, выходящем из сепаратора, как правило, создают давление, и он реагирует в присутствии катализатора с образованием равновесной смеси изомеров ксилола, которая затем возвращается в абсорбер. Выделяя пара-ксилол из смеси С8 углеводородов перед кристаллизацией, можно повысить выход пара-ксилола в кристаллизаторе от 65% и ниже примерно до 85%, что позволяет преодолеть трудности, вызванные бинарными эвтетиками. См. в целом Swift (UOP) Патент США 5329060.
Другой способ получения пара-ксилола из смеси C8 углеводородов включает пропускание смеси в газовой фазе через адсорбционный слой, содержащий адсорбент, селективный к пара-ксилолу и этилбензолу, получают после соответствующей десорбции раздельные потоки, один из которых содержит, преимущественно, мета- и орто-ксилолы, а другой - пара-ксилол, этилбензол, и десорбированный материал находится в пустом пространстве адсорбента. Адсорбцию проводят при температуре 140-370°С, давление поддерживают в пределах от атмосферного до 300 килопаскалей (абсолютное) (кПа) (около 65 фунтов на квадратный дюйм (пси) абсолютное), применяют цеолитные молекулярные сита (типа Mobil-5 (MFI), включая цеолитные молекулярные сита ZSM-5 (цеолитные молекулярные сита Zeolite Socony Mobil, выпускаемые фирмой ExxonMobil Chemicals), молекулярные сита из феррьерита и силикалита-1. Десорбцию пара-ксилола и этилбензола можно проводить с помощью газообразного десорбента, содержащего воду, при температуре в том же интервале, в каком проводят адсорбцию и при давлении от атмосферного до 1000 кПа (около 145 пси абс.). Или же десорбцию можно проводить без десорбента, просто снижая давление до 1кПа-4кПа (около 0.15-0.58 пси абс.). При таком способе в пустотах адсорбента остается значительное количество исходного продукта, который, в конечном счете, удаляется на стадии десорбции, но, к сожалению, загрязняет десорбированный продукт (поток), содержащий пара-ксилол и этилбензол. В целом см. Long et al. (China Petrochemical Company and Fudan University) китайская опубликованная патентная заявка 1136549 А. Другие исследователи использовали ZSM-8 и ZSM-5 (каждые, необязательно, реагировали с силанами) для разделения ароматических углеводородов, таких как изомеры ксилола и этилбензол, как, например, описано в Патентах США 3699182, 3729523 и 4705909 и в английском патенте 1420796.
Вышеупомянутые стадии кристаллизации и SIMBAC можно сделать более привлекательными, если исходное сырье для этих стадий переработать таким образом, чтобы концентрация в нем пара-ксилола была выше равновесной. Такую переработку можно осуществить, например, с помощью селективного диспропорционирования в толуоле, как описано, например, в Международных заявках WO 00/69796 и WO 93/17987. Как стадию кристаллизации, так и стадию SIMBAC специалисты в данной области техники могут также разработать и провести таким образом, чтобы сделать потоки пара-ксилола на последующих стадиях концентрированными. Однако даже такие стадии страдают многими (или всеми) недостатками, которые обсуждались выше. Другой способ получения исходного с концентрацией пара-ксилола выше равновесной -это процессы адсорбции с переменным давлением (PSA). Такие процессы широко применялись на практике для разделения газов, таких как воздух, на азот и кислород, удаления воды из воздуха и очистки водорода, и в общем описаны в Ralf Т. Yang, Gas "Separation by Adsorption Processes", pp. 237-274 (Butterworth Publishers, Boston, 1987) (TP242.Y36).
Однако меньшее количество описанных процессов по PSA применено на практике для очистки углеводородов, в частности очистки углеводородов, жидких при комнатной температуре. Одним заметньм исключением является способ (процесс ISOVIVTM), разработанный в корпорации Union Carbide, который применим для разделения линейных или нормальных парафиновых углеводородов и разветвленных или изопарафиновых углеводородов. По способу ISOVTVTM давление при адсорбции сохраняется практически постоянным (т.е. постоянное общее давление в адсорбере на стадии адсорбции) и для продувки, очистки (удаления) или осуществления десорбции адсорбированных углеводородов из адсорбента каким-либо другим способом используется инертный газ (например, водород). См. Патент США 3700589. Со временем способ ISOVIVTM был усовершенствован с помощью дополнительного адсорбера на стадии адсорбции, что позволило повторно использовать исходное с целью повышения чистоты в целом и получить нужный изопарафин. См. Патент США 4176053. Однако применение на стадии адсорбции нескольких адсорберов имеет свои ограничения, так как уменьшается возврат (return), когда используют слишком много аппаратов. См. Патенты США 4476345 и 4595490 (в которых показаны преимущества меньшего количества аппаратов и ступенчатых циклов адсорбции/десорбции). Способ ISOVIVTM можно сделать более привлекательным, если концентрация н-парафинов в смеси парафинов, поступающей в адсорбер, выше равновесной. Так, Патент США 4210771 включает аппарат для изомеризации, предшествующий адсорбции, для превращения изопарафинов в н-парафины. В процессах ISOVIVTM с применением расположенного "downstream" реактора для изомеризации (за адсорбером, "downstream" относительно адсорбера) именно адсорбированный материал десорбируется и в конечном счете изомеризуется - рафинат, поступающий со слоя адсорбента, не является изомеризованным. В целом в способе ISOVIVTM были сделаны другие усовершенствования. См., например. Патенты США 4372022 и 4709117. Однако стоит отметить, что в способе ISOVIVTM ничего не предпринималось для того, чтобы обеспечить разделение иных углеводородов, нежели парафиновые.
Ясно, что для потоков эффлюента в процессе ISOVIVTM в дальнейшем ("downstream") требуются дорогостоящие и энергоемкие оборудование и переработка. Кроме того, применение способа ISOVIVTM и относящихся к нему указаний для получения пара-ксилола имеет свои трудности, на которые в уровне техники не обращалось достаточного внимания. Например, для обедненного пара-ксилолом эффлюента требуется дорогостоящий процесс создания давления (или повторного создания давления), когда он поступает в расположенный далее ("downstream") аппарат для изомеризации. Как отмечалось выше, разделение изомеров ксилола часто сочетается с изомеризацией мета- и орто-ксилолов в требующийся пара-ксилол. Такую изомеризацию проводят в реакторе высокого давления. Даже если известен способ ISOVIVTM и попытки применить этот способ для получения пара-ксилола, отсутствует руководство, как ввести обедненный пара-ксилолом рафинат в реактор для изомеризации без дорогостоящей стадии создания давления (или повторного создания давления) между адсорбером и реактором для изомеризации.
Например, Deckman et al. (Exxon Chemical Company) в опубликованной патентной заявке 2002/0065444 A1 описывает метод получения пара-ксилола из смеси ксилолов с применением аппарата для адсорбции с переменным давлением (PSA) или переменной температурой (TSA) и, по меньшей мере, одного реактора для изомеризации. В публикации Deckman раскрывается реактор для изомеризации непосредственно перед PSA адсорбером и отсутствует стадия сжатия (компрессии) между реактором и PSA адсорбером. См. Фиг. 1 и 2 публикации Deckman. Такое раскрытие для специалиста в данной области техники означает, что реактор работает и эффлюенты реактора выходят из реактора при давлении, превышающем давление аппарата PSA. Рафинат на выходе из аппарата PSA имеет более высокую температуру, но пониженное давление (вследствие снятия давления для завершения десорбции), для которого нужен компрессор (или эжектор, вентилятор, воздуходувная машина) для создания давления в потоке перед тем, как пустить его в реактор для изомеризации. Хотя в публикации Deckman отсутствует точное описание, рафинат выходит из PSA при температурах, слишком высоких для того, чтобы сделать сжатие в компрессоре практичным. Поэтому рафинат нужно охладить до подходящей температуры перед сжатием. При охлаждении рафината некоторые его составляющие (например, изомеры ксилола) будут конденсироваться. Весь конденсируемый материал следует отделить от неконденсируемого газа. Затем конденсируемый материал нагревают и подают насосом в реактор для изомеризации, тогда как вновь охлажденный неконденсируемый газ подвергают сжатию в компрессоре, а затем подают в реактор для изомеризации. Следовательно, раскрытие в публикации Deckman является несколько неполной, так как в ней отсутствует конкретное описание необходимого теплообменника (конденсатора), газо-жидкостного сепаратора и жидкостного насоса для введения PSA рафината под соответствующим давлением в реактор для изомеризации. Тем не менее, в раскрываемом способе требуется эжектор (воздуходувная машина, вентилятор) или компрессор на каждом из потоков процесса с переменным давлением. Как легко понять специалисту в данной области техники, покупка и работа такие эжекторов (воздуходувных машин) или компрессоров очень дороги, и там, где это возможно, их следует избегать.
Можно предположить, что адсорбер с переменным давлением, раскрываемый в публикации Deckman, работает под давлением, достаточно высоким для того, чтобы давление, пониженное с целью осуществления десорбции, оставалось достаточно высоким, и не требовалось дополнительного сжатия перед подачей материалов в расположенные далее аппараты, такие как реактор для изомеризации. Однако на практике работа адсорбера при таком высоком давлении в процессе десорбции будет резко и невыгодно снижать производительность адсорбента.
Таким образом, существуют различные способы получения пара-ксилола из смесей C8 ароматических углеводородов, эти способы очень сложны и включают необходимые и дорогостоящие начальные и конечные стадии (предыдущие и последующие стадии).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В данном описании раскрывается усовершенствованный способ получения пара-ксилола из смеси изомеров ксилола. В одном варианте изобретения способ включает контактирование при практически не снижающемся общем давлении газообразной смеси, содержащей изомеры ксилола и этилбензола, с адсорбентом, селективным к пара-ксилолу, с целью получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом. Способ также включает изомеризацию, по меньшей мере, части обедненного пара-ксилолом рафината.
В альтернативном варианте изобретения способ включает контактирование при первом давлении смеси, содержащей изомеры ксилола и этилбензол, с адсорбентом, селективным в отношении пара-ксилола, с целью получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом. В этом варианте изобретения способ также включает изомеризацию при втором давлении, по меньшей мере, части обедненного пара-ксилолом рафината, причем первое давление равно второму давлению или выше его.
В другом альтернативном варианте изобретения способ включает контактирование смеси, содержащей изомеры ксилола, этилбензол и неадсорбируемый нереакционноспособный (не реагирующий) газ, с адсорбентом, селективным к пара-ксилолу, с целью получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом. В этом варианте изобретения способ также включает изомеризацию, по меньшей мере, части обедненного пара-ксилолом рафината. Газ присутствует в количестве, достаточном для того, чтобы гарантировать давление рафината, равное или более высокое, чем давление на стадии изомеризации, сохраняя при этом парциальное давление изомеров ксилола и этилбензола равньм соответствующему конденсационному давлению или более низким, чем соответствующее конденсационное давление изомеров ксилола и этилбензола.
В другом альтернативном варианте изобретения способ включает контактирование газообразной смеси, содержащей изомеры ксилола и этилбензол, с адсорбентом, селективньм к пара-ксилолу, для получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом. В этом варианте изобретения способ также включает изомеризацию, по меньшей мере, части обедненного пара-ксилолом рафината. Сумма парциальных давлений изомеров ксилола и этилбензола ниже общего давления смеси.
Еще в одном альтернативном варианте изобретения способ включает контактирование изомеров ксилола и неадсорбируемого газа с адсорбентом, селективньм к пара-ксилолу, для получения обедненного пара-ксилолом рафината, включающего углеводороды, практически не содержащие пара-ксилол, и эффлюент после десорбции, содержащий продукт, обогащенный пара-ксилолом. Способ также включает изомеризацию, по меньшей мере, части обедненного пара-ксилолом рафината. Газ не способен реагировать со смесью на стадии контактирования и присутствует в количестве, достаточном для того, чтобы молярное соотношение газа и углеводорода в рафинате, обедненном пара-ксилолом, составляло, примерно, от 0.1:1 до 10:1.
Дополнительные признаки изобретения, возможно, станут очевидными специалистам в данной области техники из нижеприведенного подробного описания вместе с чертежами, примерами и прилагаемой формулой изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Для более полного понимания изобретения следует сделать ссылку на нижеприведенное подробное описание и сопровождающие его чертежи,
где на фиг.1 дана блок-схема процесса, иллюстрирующая один цикл работы двухступенчатой системы адсорбции с переменным парциальным давлением (PPSA), пригодной для осуществления раскрываемого способа;
на фиг.2 представлена блок-схема процесса, иллюстрирующая один цикл работы трехступенчатой системы PPSA, пригодной для осуществления раскрываемого способа;
на фиг.3 представлена блок-схема процесса, иллюстрирующая один цикл работы четырехступенчатой системы PPSA, пригодной для осуществления раскрываемого способа;
на фиг.4 представлена блок-схема процесса, иллюстрирующая один цикл работы другой трехступенчатой системы PPSA, пригодной для осуществления раскрываемого способа;
на фиг.5 представлена блок-схема процесса, иллюстрирующая один пример процесса, объединяющий системы PPSA, показанные на фиг.1-4 и представленные в данном описании;
на фиг.6 дан график: выход пара-ксилола и этилбензола/выделение в Примере 3; и
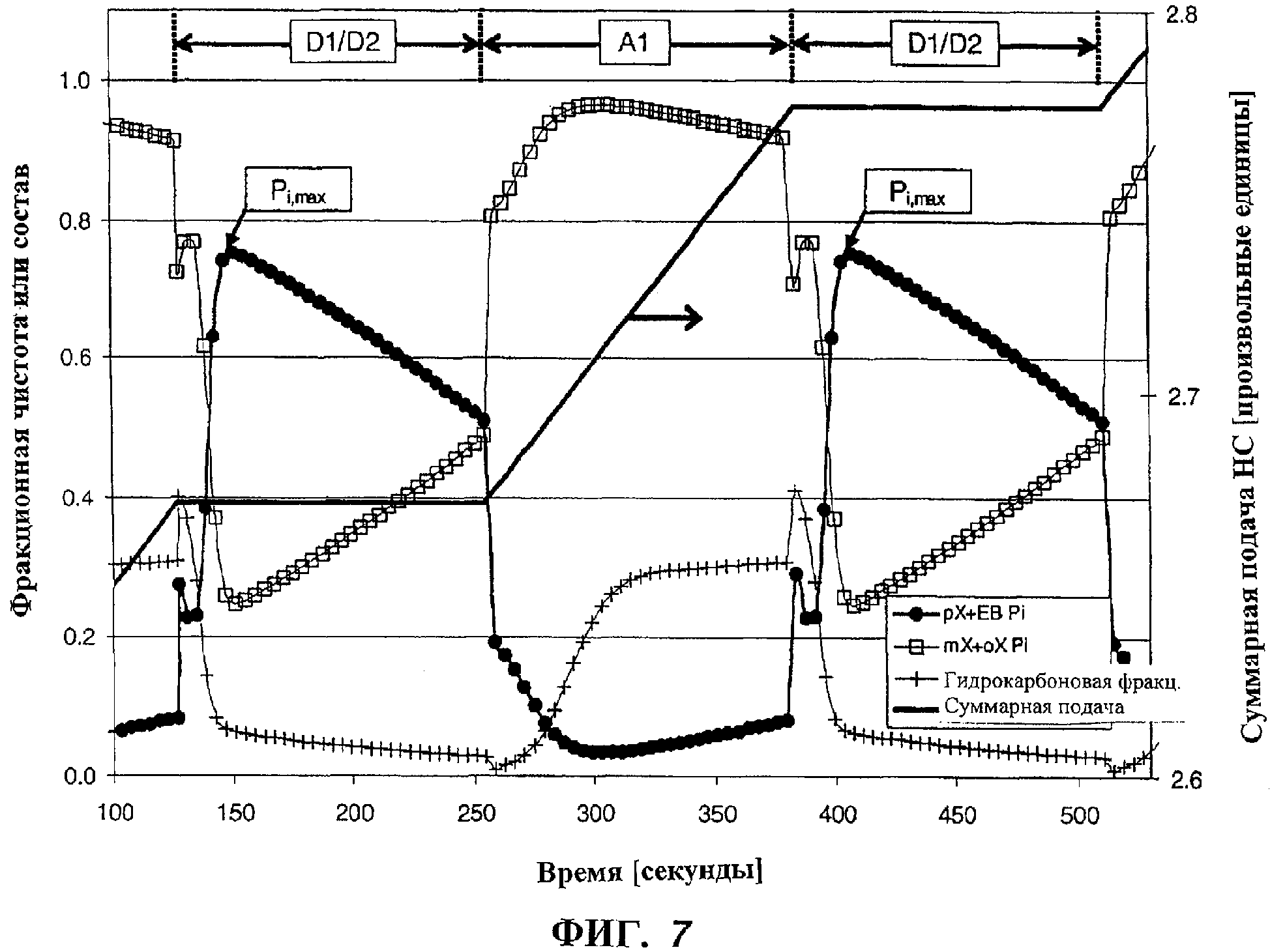
на фиг.7 изображен график: составы различных потоков в системе имитированной PPSA в Примере 4.
Хотя раскрываемый способ допускает различные формы воплощения изобретения, на фиг. проиллюстрированы (а ниже и описаны) конкретные варианты изобретения, при этом следует понимать, что предполагается, что раскрытие лишь иллюстрирует изобретение и не претендует на то, чтобы ограничиваться конкретными вариантами изобретения, описанными и проиллюстрированными в данном описании.
ПОДРОБНОЕ ОПИСАНИЕ ДАННОГО ИЗОБРЕТЕНИЯ
Данное изобретение в целом относится к способу адсорбции пара-ксилола - без спада общего давления/с переменным парциальным давлением - из исходной смеси C8 ароматических углеводородов (сырье), содержащей изомеры ксилола, десорбции и улавливания адсорбированного продукта, обогащенного пара-ксилолом, и изомеризации неадсорбированной (обедненной пара-ксилолом) доли исходной смеси с целью получения смеси изомеров ксилола, которую можно объединить с исходной смесью.
В одном варианте изобретения способ включает контактирование газообразной смеси практически без спада общего давления, содержащей изомеры ксилола и этилбензол, с адсорбентом, селективным к пара-ксилолу, с образованием обедненного пара-ксилолом рафината, и десорбцию эффлюента, содержащего продукт, обогащенный пара-ксилолом. Способ также включает изомеризацию, по меньшей мере, части обедненного пара-ксилолом рафината, предпочтительно, при давлении, равном или меньшем, чем практически не пониженное общее давление и, более предпочтительно, при давлении, меньшем, чем не сниженное (не снижающееся) общее давление. Сумма парциальных давлений изомеров ксилола и этилбензола, предпочтительно, меньше, чем практически не сниженное (не снижающееся) общее давление, и, более предпочтительно, сумма парциальных давлений составляет, примерно, 15%-99.5% от практически постоянного общего давления. Более предпочтительно, сумма парциальных давлений составляет, примерно, 35%-75% от практически постоянного общего давления, и, наиболее предпочтительно, сумма парциальных давлений составляет, примерно, 45%-60% от практически постоянного общего давления. Способ также включает выделение практически чистого пара-ксилола из эффлюента после десорбции, предпочтительно, при давлении, равном или меньшем, чем практически не сниженное (не снижающееся) общее давление и, более предпочтительно, при давлении, меньшем, чем не сниженное (не снижающееся) общее давление.
В этом варианте изобретения способ также может включать объединение со смесью неадсорбируемого газа, который не реагирует со смесью на стадии контактирования. Предпочтительно, газ присутствует в количестве, достаточном для создания молярного соотношения газ: углеводороды в обедненном пара-ксилолом рафинате, примерно, от 0.1:1 до 10:1. Или же, или помимо этого, газ присутствует в количестве, достаточном для того, чтобы гарантировать, что практически не сниженное (не снижающееся)общее давление равно давлению или выше, чем давление на стадии изомеризации, при этом значения парциального давления изомеров ксилола и этилбензола сохраняются равными соответствующим значениям конденсационного давления или меньшими, чем значения соответствующего конденсационного давления изомеров ксилола и этилбензола. Предпочтительно, газ присутствует в количестве, достаточном для того, чтобы гарантировать, что практически не снижающееся общее давление выше, чем давление на стадии изомеризации. Кроме того, газ, предпочтительно, присутствует в количестве, достаточном для того, чтобы избежать конденсации изомеров ксилола.
В альтернативном варианте изобретения способ включает контактирование при первом давлении смеси (предпочтительно, газообразной смеси), содержащей изомеры ксилола и этилбензол, с адсорбентом, селективным к пара-ксилолу, для получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом. Способ также включает изомеризацию при втором давлении, по меньшей мере, части обедненного пара-ксилолом рафината, при этом, предпочтительно, первое давление выше второго давления. Сумма парциальных давлений изомеров ксилола и этилбензола, предпочтительно, меньше, чем первое давление, и, более предпочтительно, составляет около 15-99.5% от первого давления. Предпочтительно, первое давление является не снижающимся давлением. Способ также включает выделение практически чистого пара-ксилола из эффлюента после десорбции, практически при давлении, равном или меньшем, чем первое давление, и, более предпочтительно, при давлении, меньшем, чем первое давление.
В этом варианте изобретения способ также может включать объединение со смесью неадсорбируемого газа, который не реагирует со смесью на стадии контактирования. Предпочтительно, газ присутствует в количестве, достаточном для создания молярного соотношения газ: углеводороды в обедненном пара-ксилолом рафинате около 0.1:1-10:1. Или же, или помимо этого, газ присутствует в количестве, достаточном для того, чтобы значения парциального давления изомеров ксилола и этилбензола сохранялись равными соответствующим значениям конденсационного давления изомеров ксилола и этилбензола или меньшими, чем значения соответствующего конденсационного давления изомеров ксилола и этилбензола. Предпочтительно, газ присутствует в количестве, достаточном для того, чтобы избежать конденсации изомеров ксилола.
В другом альтернативном варианте изобретения способ включает контактирование смеси (предпочтительно, газообразной смеси), содержащей изомеры ксилола, этилбензола и неадсорбируемого нереагирующего газа с адсорбентом, селективным к пара-ксилолу, для получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего обогащенный пара-ксилолом продукт, изомеризацию, по меньшей мере, части обедненного пара-ксилолом рафината. Газ присутствует в количестве, достаточном для того, чтобы гарантировать, что давление рафината равно давлению или выше давления на стадии изомеризации, при сохранении парциального давления изомеров ксилола и этилбензола равным соответствующим значениям или ниже соответствующих значений конденсационного давления изомеров стирола и этилбензола. Или же, или кроме того, предпочтительно, газ присутствует в количестве, достаточном для того, чтобы гарантировать, что давление рафината выше, чем давление на стации изомеризации, более предпочтительно, газ присутствует в количестве, достаточном для того, чтобы избежать конденсации изомеров ксилола.
В этом варианте способа изобретения стадию контактирования, предпочтительно, проводят при практически не снижающемся общем давлении. Способ также может включать выделение практически чистого пара-ксилола из эффлюента после десорбции, предпочтительно, при давлении, равном или меньшем, чем практически не снижающееся общее давление, и, более предпочтительно, при давлении, меньшем, чем практически не снижающееся общее давление.
В другом альтернативном варианте изобретения способ включает контактирование газообразной смеси, содержащей изомеры ксилола и этилбензол, с адсорбентом, селективным к пара-ксилолу, для получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего обогащенный пара-ксилолом продукт, и изомеризацию, по меньшей мере, части обедненного пара-ксилолом рафината. В этом варианте изобретения сумма парциальных давлений изомеров ксилола и этилбензола ниже, чем общее давление смеси, и предпочтительно, составляет около 15-99.5% общего давления. Общее давление, предпочтительно, является практически не снижающимся. Обедненный пара-ксилолом рафинат изомеризуется, предпочтительно, при давлении, равном или меньшем, чем общее давление, и, более предпочтительно, при давлении, меньшем, чем общее давление. Способ также может включать выделение практически чистого пара-ксилола из эффлюента после десорбции, предпочтительно, при давлении, равном или более низком, чем общее давление, и, более предпочтительно, при давлении, более низком, чем общее давление.
В этом варианте изобретения способ может также включать объединение (смешение) со смесью неадсорбируемого газа, который не реагирует со смесью на стадии контактирования. Предпочтительно, газ присутствует в количестве, достаточном для того, чтобы гарантировать, что тотальное давление равно давлению или выше, чем давление на стадии изомеризации, при этом значения парциального давления изомеров ксилола и этилбензола сохраняется равным давлению или более низким, чем соответствующие значения конденсационного давления изомеров ксилола или этилбензола. Предпочтительно, газ присутствует в количестве, достаточном для того, чтобы гарантировать, что общее давление выше, чем давление на стадии изомеризации. Или же, или кроме того, предпочтительно, газ присутствует в количестве, достаточном для того, чтобы избежать конденсации изомеров ксилола.
Еще в одном альтернативном варианте изобретения способ включает контактирование смеси изомеров ксилола (предпочтительно, газообразной смеси) с адсорбентом, селективным к пара-ксилолу, для получения обедненного пара-ксилолом рафината, включающего углеводороды, практически не содержащие пара-ксилол, и эффлюент после десорбции, содержащий продукт, обогащенный пара-ксилолом, и изомеризацию, по меньшей мере, части обедненного пара-ксилолом рафината. В этом варианте изобретения газ не реагирует со смесью на стадии контактирования, и газ присутствует в количестве, достаточном для создания молярного соотношения газ: углеводороды в обедненном пара-ксилолом рафинате, примерно, 0.1:1-10:1. Предпочтительно, газ присутствует в количестве, достаточном для того, чтобы гарантировать давление рафината, равное или более высокое, чем давление на стадии изомеризации, при этом сохраняются значения парциального давления изомеров ксилола и этилбензола, равные соответствующим значениям или более низкие, чем соответствующие значения конденсационного давления изомеров ксилола и этилбензола. Или же, или кроме того, предпочтительно, газ присутствует в количестве, достаточном для того, чтобы гарантировать давление рафината более высокое, чем давление при изомеризации, и, более предпочтительно, газ присутствует в количестве, достаточном для того, чтобы избежать конденсации изомеров ксилола.
В этом варианте способа стадию контактирования, предпочтительно, проводят при практически не снижающемся давлении. Кроме того, способ может включать выделение практически чистого пара-ксилола из эффлюента после десорбции, предпочтительно, при давлении, равном или более низком, чем практически не снижающееся давление, и, более предпочтительно, при давлении, более низком, чем практически не снижающееся давление.
Неадсорбируемый газ, о котором говорится выше, может представлять собой газ с достаточно малым размером молекулы, позволяющим входить в пустоты внутри кристаллов молекулярных сит (адсорбента), но он необязательно должен иметь такие размеры. Предпочтительно, неадсорбируемый газ представляет собой газ, который адсорбируется адсорбентом недостаточно активно, чтобы в значительной степени замещать адсорбированные углеводороды. Подходящие неадсорбируемые газы включают, но без ограничения, аргон, диоксид углерода, гелий, водород, азот и легкие парафиновые углеводороды, такие как метан, этан, пропан, бутан и их смеси. Однако предпочтительным неадсорбируемым газом является один или более веществ, выбранных из группы, состоящей из водорода, азота и легких парафиновых углеводородов. Более предпочтительно, неадсорбируемым газом является водород. Тем не менее, неадсорбируемый газ может включать следовые количества примесей, таких как бензол, толуол, С9 парафиновые и нафтеновые и С8+ ароматические углеводороды. Газ можно соединять со смесью (подаваемой на адсорбент) для повышения чистоты и выхода пара-ксилола. Предпочтительно, чтобы газ присутствовал в количестве, достаточном для создания молярного соотношения газ: углеводород в обедненном пара-ксилолом рафинате около 0.1:1-10:1. Так как неадсорбируемый газ может включать углеводороды (например, легкие парафиновые углеводороды), следует понимать, что ссылка на "углеводород" в выражении "молярное отношение газ: углеводород" относится к иным углеводородам, нежели углеводороды, которые могут входить в неадсорбируемый газ. Или же, или, кроме того, газ присутствует в количестве, достаточном для того, чтобы гарантировать, чтобы давление, при котором проводится стадия контактирования, равно давлению или выше давления, при котором проводится стадия изомеризации. В настоящем описании газ также называется " продувочный" газ.
Как отмечалось выше, неадсорбируемый газ, объединяемый со свежей подаваемой смесью С8 ароматических углеводородов, поступающей в адсорбер, должен присутствовать в количествах, достаточных для того, чтобы гарантировать градиент давления между содержимым и потоками, выходящими из слоя (адсорбента), и последующими операциями (например, изомеризацией, конденсацией и т.д.). Кроме того, и в некоторых предпочтительных вариантах изобретения, газ присутствует в количестве, достаточном для того, чтобы сохранить парциальное давление изомеров ксилола и этилбензола равным соответствующим значениям или более низким, чем соответствующие значения конденсационного давления изомеров ксилола и этилбензола. Или же, или кроме того, газ присутствует в количестве, достаточном для того, чтобы избежать конденсации изомеров ксилола. Присутствие этого газа на стадии контактирование позволяет проведение селективной адсорбции пара-ксилола при более высоком давлении, чем то, которое допускает в настоящее время простая адсорбция с переменным давлением. Конкретно, присутствие газа делает возможным более высокое общее давление в адсорбере (в слое адсорбера), в то же время, что является преимуществом, сохраняется постоянным парциальное давление изомеров ксилола (а не заметно выше, чем парциальное давление изомеров ксилола при простой адсорбции с переменным давлении). В отсутствие газа адсорбер не может работать при столь высоком общем давлении и при этом не происходила конденсация изомеров ксилола, что является недостатком. Таким образом, присутствие газа делает возможной успешную работу слоя адсорбента при более высоком общем давлении, при этом не происходит наносящей ущерб конденсации ксилола в слое. В отсутствие газа при работе в слое адсорбента с высоким общем давлением требуется одновременное повышение температуры для того, чтобы сохранить изомеры ксилола в газообразном состоянии. Однако такие более высокие температуры ухудшают селективность адсорбента к пара-ксилолу - также адсорбируются более высокие количества других изомеров, тем самым понижается чистота пара-ксилола. Итак, более высокое общее давление в слое адсорбента в результате присутствия газа не оказывает отрицательного воздействия на селективность адсорбента, так как нет заметного изменения величин парциального давления изомеров ксилола.
Предполагается, что термин "рафинат" по данному описанию относится к части текучей среды (жидкости или газа), которая остается после удаления, например, с помощью адсорбции, других нужных компонентов. Обедненный пара-ксилолом рафинат, как правило, включает одну или более составляющих смеси (сырье, исходная смесь), контактирующей с адсорбентом, но имеет более низкую концентрацию пара-ксилола, чем исходная смесь. Термин "обедненный пара-ксилолом" в выражении "обедненный пара-ксилолом рафинат" означает просто, что концентрация пара-ксилола ниже, предпочтительно, значительно ниже, чем концентрация пара-ксилола в смеси, контактирующей с адсорбентом. Термин не означает, что рафинат не содержит пара-ксилола. Таким образом, обедненный пара-ксилолом рафинат, по-видимому, будет содержать более низкую концентрацию пара-ксилола, более высокие концентрации других изомеров ксилола, этилбензол и отличные от C8 ароматические углеводороды, которые могут присутствовать в исходной (поступающей в адсорбер) смеси.
Как правило, обедненный пара-ксилолом рафинат включает углеводороды, практически не содержащие пара-ксилол (например, мета-ксилол, орто-ксилол, толуол, С9+ ароматические и С9 парафиновые и нафтеновые углеводороды). Так как неадсорбируемый газ, не реагирующий со смесью, может объединяться со смесью (подаваемой на адсорбент), обедненный пара-ксилолом рафинат также может включать неадсорбируемый газ. Следовательно, обедненный пара-ксилолом рафинат может включать одно или более веществ, выбранных из группы, состоящей из аргона, диоксида углерода, гелия, водорода, азота и легких парафиновых углеводородов, таких как метан, этан, пропан, бутан и их смеси. Обедненный пара-ксилолом рафинат, предпочтительно, включает углеводороды, практически не содержащие пара-ксилол, и, более предпочтительно, он включает мета-ксилол, орто-ксилол и водород. Предпочтительно, обедненный пара-ксилолом рафинат содержит орто-ксилол и мета-ксилол и менее 25 мольных процентов пара-ксилола и этилбензола от общего количества С8 ароматических углеводородов в рафинате, и более предпочтительно, менее десяти мольных процентов пара-ксилола и этилбензола от общего количества C8 ароматических углеводородов в рафинате.
Любую неизомеризуемую часть рафината можно пустить в последующие операции для очистки содержащихся в рафинате мета-ксилола и/или орто-ксилола. Как отмечалось выше, способ включает изомеризации, по меньшей мере, части обедненного пара-ксилолом рафината. Предпочтительно, проводят изомеризацию с целью получения смеси углеводородов, содержащей равновесную смесь изомеров ксилола. Для повышения чистоты и выхода пара-ксилола часть (или все количество) изомеров ксилола, полученных изомеризацией, можно объединять со смесью, контактирующей с адсорбентом (т.е. рециркулировать, снова пустить в цикл). Стадия изомеризации более подробно описана далее.
Осуществление раскрываемого способа (и его различных вариантов) находится в компетенции специалистов в области соответствующего оборудования и контроля, необходимого для осуществления способа. Такое оборудование для проведения процесса включает, но без ограничения, систему трубопроводов, насосы, вентили, рабочее оборудование (например, реакторы с соответствующими впускными и выпускными клапанами (вентилями), теплообменники, сепараторы и т.д.), соответствующее оборудование для контроля процесса и контроля качества, если это необходимо. Описание любого другое оборудования для проведения процесса, особенно там, где это предпочтительно, представлено в данном описании.
Адсорбенты, наиболее применимые в способе, как правило, содержат некислые молекулярные сита и связующее. Молекулярные сита должны селективно адсорбировать пара-ксилол в своих пустотах, каналах и порах и в то же время не адсорбировать мета- и орто-ксилолы и неароматические C8 углеводороды (т.е. тотальное исключение или более низкая скорость адсорбции мета- и орто-ксилолов по сравнению с пара-ксилолом). Среди молекулярных сит, подходящих для применения по настоящему описанию, те молекулярные сита, которые селективно адсорбируют не только пара-ксилол, но также и этилбензол, так как размер молекул и структура/конфигурация этилбензола аналогичны размеру молекул и структуре/конфигурации пара-ксилола. Подходящие сита, которые селективно адсорбируют только пара-ксилол, являются наиболее предпочтительными. Адсорбенты не должны адсорбировать С8 ароматические углеводороды и не должны обладать каталитической активностью (например, активностью в реакции изомеризации или активностью в реакциях конверсии) в отношении C8 ароматических соединений. В случае адсорбента, не являющегося каталитически активным, как правило, наблюдается конверсия пара-ксилола в мета-ксилол или орто-ксилол менее десяти процентов, предпочтительна конверсия менее пяти процентов, и более предпочтительна конверсия менее одного процента, при температурах, при которых в данном способе имеет место стадия контактирования. Следовательно, адсорбенты не должны быть кислыми и, предпочтительно, совершенно некислыми.
В целом, молекулярные сита являются упорядоченными, пористыми кристаллическими материалами. Более конкретно, молекулярные сита, как правило, образованы из тетраэдров кремнезема, оксида алюминия (глинозема) и оксида фосфора (РО4), которые образуют кристаллическую структуру с пустотами и/или порами, соединенными между собой проходами, каналами. Пустоты, поры и каналы, как правило, однородны по размеру и допускают селективное разделение углеводородов в зависимости от определенных молекулярных характеристик - наиболее часто, разделение по размеру молекул или по конфигурации молекул. Обычно термин "молекулярные сита" включает различные натуральные и синтетические кристаллические пористые материалы, как правило, на основе тетраэдров кремнезема в сочетании с тетраэдрами других оксидов, таких как оксиды алюминия, бора, галлия, железа, титана и т.п. В таких структурах решетки кремнезема и таких элементов, как алюминий, связаны общими атомами кислорода. Замены в структуре молекулярных сит элементов, таких как алюминий, на кремний сообщает структуре (или каркасу в целом) отрицательный заряд, который должен быть уравновешен положительными ионами, такими как ионы щелочных, щелочно-земельных металлов или ионом аммония для того, чтобы сита гарантированно не были кислыми.
Связующее, предпочтительно, выбирают из группы, состоящей из оксида алюминия (глинозема), фосфата алюминия, глины, оксида кремния (кремнезема), кремнезема-глинозема, кремнезема - глинозема - магнезии, кремнезема - глинозема-оксида тория, кремнезема - глинозема - оксида циркония, кремнезема-оксида бериллия, кремнезема - магнезии, кремнезема - магнезии-оксида циркония, кремнезема-оксида тория, кремнезема-оксида титана, кремнезема-оксида циркония, оксида титана, оксида циркония и их смесей.
Адсорбент, селективный к пара-ксилолу, должен преимущественно адсорбировать пара-ксилол по сравнению с мета-ксилолом и орто-ксилолом при пропускании через него эквимолярной смеси ксилолов в паровой (или газовой) фазе при 50°С, так что общее количество пара-ксилола в эффлюенте после десорбции по данному способу составляло, по меньшей мере, около 75 мольных процентов (%) от общего количества C8 ароматических углеводородов в эффлюенте после десорбции, предпочтительно, по меньшей мере, около 80%, более предпочтительно, по меньшей мере, около 85%, более предпочтительно, по меньшей мере, около 90%, еще более предпочтительно, по меньшей мере, около 95% и, наиболее предпочтительно, по меньшей мере, около 97% от общего количества C8 ароматических углеводородов в эффлюенте после десорбции. Преимущественная селективность подходящих адсорбентов должна быть аналогичной селективности или превышающей селективность под действием равновесной смеси изомеров ксилола или смеси с более чем эквимолярным соотношением пара-ксилола к мета- и орто-ксилолу. Емкость адсорбента определяется как количество грамм адсорбата (т.е. адсорбированного материала), деленное на количество грамм адсорбента, ее также можно выразить в весовых процентах, умножив на 100. Таким образом, в соответствии с различными вариантами раскрываемого способа, предпочтительно, по меньшей мере, около 0.01 г пара-ксилола адсорбируется на 1 г адсорбента. Более предпочтительно, по меньшей мере, около 0.015 г пара-ксилола адсорбируется на 1 г адсорбента, еще более предпочтительно, по меньшей мере, около 0.02 г пара-ксилола адсорбируется на 1 г адсорбента, и наиболее предпочтительно, по меньшей мере, около 0.03 г пара-ксилола адсорбируется на 1 г адсорбента.
В каждом из вышеприведенных вариантов изобретения эффлюент после десорбции, предпочтительно, содержит, по меньшей мере, около 50 мольных процентов (%) от количества пара-ксилола в (исходной, подаваемой) смеси, более предпочтительно, по меньшей мере, около 75% от количества пара-ксилола в исходной смеси, еще более предпочтительно, по меньшей мере, около 90% от количества лара-ксилола в исходной смеси, и, наиболее предпочтительно, по меньшей мере, около 95% от количества пара-ксилола в исходной смеси. В данном описании выражение "продукт, обогащенный пара-ксилолом" означает продукт, содержащий пара-ксилол и в целом менее 25 мольных процентов мета-ксилола и орто-ксилола от общего количества C8 ароматических углеводородов в продукте. Предпочтительно, продукт, обогащенный пара-ксилолом, содержит менее десяти мольных процентов мета-ксилола и орто-ксилола от общего количества C8 ароматических углеводородов в продукте, и, более предпочтительно, продукт, обогащенный пара-ксилолом, содержит менее пяти мольных процентов мета-ксилола и орто-ксилола от общего количества C8 ароматических углеводородов в продукте.
Адсорбент может содержаться в одном или более контейнерах или сосудах (также называемых в данном описании адсорбционные колонны или слои), в которых выделение практически чистого потока пара-ксилола и этилбензола из смеси изомеров ксилола и этилбензола возможно с применением регулируемого (или программируемого) потока в контейнеры или сосуды и из контейнеров или сосудов. Разделение компонентов в адсорбере является разделением с помощью модифицированной адсорбции с переменным парциальным давлением (PPSA), где продолжительность (время) цикла определяется как интервал времени, начиная с того момента, когда исходная смесь попадает в слой (адсорбер), и, кончая моментом, когда адсорбер готов к приему следующей порции исходной смеси. Следовательно, "продолжительность цикла" можно описать как временной интервал, за который порция исходного вводится в слой (адсорбционный) (адсорбер), например, каждые 30 секунд, каждую минуту, каждые пять минут, каждые десять минут, каждые пятнадцать минут и т.д. Цикл представляет собой полный процесс PPSA (т.е. сумма всех стадий). Как более подробно описано ниже, стадии представляют собой отдельные операции PPSA, такие как операция (стадия) адсорбции (Ах) и различные операции (стадии) десорбции (Dx), где х обозначает целое число, равное одному или больше одного.
Стадию контактирования следует проводить в условиях (например, при температуре и давлении), эффективно гарантирующих, что различные потоки исходной смеси, эффлюента и рафината находятся в газообразном/парообразном, но не в жидком состоянии. Так, например, давление следует сохранять, чтобы потоки газа/пара не конденсировались. Как правило, это условие удовлетворяется при поддержании величины давления, составляющей, примерно, менее 80% от критического давления основного компонента потока с наиболее высокой температурой кипения, или, примерно, менее 60% от давления при точке росы потоков при различных температурах процесса, каким бы ни было это более низкое значение. Как отмечалось выше, присутствие неадсорбируемого газа на стадии контактирования может гарантировать, что изомеры ксилола, как минимум, не будут конденсироваться на стадии контактирования.
Стадию контактирования, предпочтительно, следует проводить в условиях изотермального процесса при рабочей температуре, по меньшей мере, около 175°С, и, более предпочтительно, по меньшей мере, около 200°С. Рабочая температура (изотермальный процесс) на стадии (в операции) контактирования, предпочтительно, должна быть в интервале около 175-400°С, и, более предпочтительно, около 200-300°С. Стадию контактирования (изобарный процесс) следует проводить при рабочем давлении, по меньшей мере, около 345 кПа (около 50 пси абс.), и, более предпочтительно, по меньшей мере, около 448 кПа (около 65 пси абс.). Рабочее давление при изобарном процессе контактирования должно быть в интервале около 345-6895 кПа (около 50-1000 пси абс.), предпочтительно, около 345-3000 кПа (около 50-435 пси абс.), более предпочтительно, около 448-3000 кПа (около 65-435 пси абс.), и наиболее предпочтительно, около 448-2068 кПа (около 65-300 пси абс.).
Термин "изотермальный" означает, что рабочая температура в течение каждой из операций - адсорбции и десорбции - стадии контактирования остается практически постоянной. Выражение "практически постоянная" во фразе "практически постоянная общая температура" означает, что в ходе последовательных операций не наблюдается заметного изменения температуры слоя (адсорбента). Принимая во внимание то, что раскрывается в данном описании, специалисты в данной области поймут, что могут быть некоторые слабые колебания температуры вследствие меняющегося расхода и/или вследствие нагревания при адсорбции и десорбции. Так, температура потока в начале операции (адсорбции или десорбции) может составлять, примерно, от 85% до 115% от температуры в конце той же операции. Кроме того, температура потоков: смеси, подаваемой в адсорбер, рафината и эффлюента, получаемых в адсорбере, и "продувочного" газа (если таковой имеется), предпочтительно, остается одинаковой в качестве рабочей температуры слоя в ходе той операции стадии контактирования, на которой находятся потоки.
Термин "изобарный" ("изобарический") подразумевает, что рабочее давление в ходе каждой операции-адсорбции и десорбции-стадии контактирования следует держать практически постоянным. Выражение "практически постоянный" во фразе "практически постоянное общее давление" означает, что в ходе последовательных операций не наблюдается заметного спада давления в слое. Принимая во внимание то, что раскрывается в данном описании, специалисты в данной области поймут, что могут быть некоторые слабые колебания давления вследствие меняющегося расхода, или что парциальное давление адсорбированного и десорбированного компонентов меняются на стадии контактирования. Так, давление потока в начале операции (адсорбции или десорбции) может составлять, примерно, от 95% до 105% от давления в конце той же операции. Кроме того, давление потоков: смеси, подаваемой в адсорбер, рафината и эффлюента, получаемых в адсорбере, и "продувочного" газа (если таковой имеется), предпочтительно, остается одинаковым в качестве рабочего давления слоя в ходе той операции стадии контактирования, на которой находятся потоки. Однако парциальное давление различных компонентов в каждом потоке меняется в зависимости от временного расположения компонентов на стадии контактирования - отсюда характер этого способа - адсорбция с переменным "парциальным давлением".
В некоторых вариантах способа стадию контактирования проводят при "практически не снижающемся общем давлении", это означает, что последовательные операции десорбции следует проводить при давлениях, которые превышают давление или практически равны давлению на предыдущей стадии адсорбции. Таким образом, хотя каждую операцию стадии контактирования можно проводить при различных давлениях в изобарном режиме, изобарное давление последующей операции десорбции не должно быть меньше, чем изобарное давление предыдущих операций десорбции и адсорбции. Такой рабочий показатель, что является преимуществом, позволяет снова пускать выбранные фракции эффлюентов после десорбции (как более подробно описано ниже) на слой (адсорбента) в операции адсорбции и комбинировать (объединять) со свежей исходной смесью C8 ароматических углеводородов, при этом не требуются дорогостоящие насосы и компрессоры для повторного сжатия эффлюента до давления свежей исходной смеси C8 ароматических углеводородов или давления в операции адсорбции. Более высокое давление в эффлюенте после десорбции (более высокое относительно давления в ходе адсорбции) выгодно освобождает от необходимости использовать насосы и компрессоры для транспортировки эффлюента в другие, расположенные далее аппараты для осуществления процесса. Кроме того, более высокое давление в эффлюенте после десорбции (более высокое относительно давления в ходе адсорбции) облегчает удаление из него более легких углеводородов, так как эти углеводороды, по-видимому, будут более легко конденсироваться при более высоких давлениях, при которых происходит отделение C8 ароматических углеводородов от не адсорбирующегося газа, и сводит к минимуму сжатие, необходимое для возвращения неадсорбирующегося газа в адсорбер. Кроме того, преимущество этого способа состоит в том, что рафинат, полученный на стадии контактирования, не нужно подвергать сжатию перед последующей изомеризацией. Так как, среди прочего, раскрываемый способ делает ненужным дорогостоящее компрессионное оборудование, он этим отличается от способа ISOSIVТМ и способы, раскрываемые в патентной заявке США 2002/0065444 А1 на имя Deckman et al. (Exxon Chemical Company) и в китайской опубликованной патентной заявке 1136549 A Long et al. (China Petrochemical Company and Fudan University).
Как отмечалось выше, способ включает изомеризацию обедненного пара-ксилолом рафината. Предпочтительно, и как ранее отмечалось в данном описании, изомеризацию проводят с целью получения смеси углеводородов, содержащую равновесную смесь изомеров ксилола. Изомеризацию можно проводить в любом подходящем реакторе, например таком, как реакторы, описанные в Патентах США 4899011 и 4236996, которые вводятся в данное описание в качестве ссылки. Реактор должен содержать катализатор, пригодный для проведения заданной изомеризации. В зависимости от типа требуемой изомеризации можно использовать различные катализаторы. Например, некоторые катализаторы более пригодны для того, чтобы уравновесить обедненный пара-ксилолом рафинат с изомерами ксилола (или с четырьмя основными C8 ароматическими углеводородами, включая этилбензол) с концентрациями, полученными за счет термодинамики процесса в условиях изомеризации. Такие катализаторы включают, но без ограничения, катализаторы, описанные в Европейском патенте 138617, Патентах США 4098836, 4899011, 5011296 и Re 31782, содержание которых вводится в данное описание в качестве ссылки. Продажные катализаторы включают, но без ограничения, катализаторы IFPP/Engelhard Octafimng и Octafining II.
Другие катализаторы боле пригодны для конверсии некоторого количества этилбензола в рафинате и получения бензола и этана в качестве удаляемых побочных продуктов, конверсию этилбензола можно проводить в присутствии любой каталитической системы для деалкилирования, гидродеэтилирования (деэтилирование + гидрирование) и гидроизомеризации этилбензола. Такие катализаторы включают, но без ограничения, катализаторы, раскрываемые в Патентах США 4899011 (гидродеэтилирование), 4908342 (деалкилирование), 5028573 (гидроизомеризация), 5367099 (гидродеэтилирование), 5908967 (гидроизомеризация), 6150292 (гидроизомеризация) и Re 31782 (деалкилирование), содержание которых вводится в данное описание в качестве ссылки. Продажные катализаторы включают, но без ограничения, катализаторы Mobil высокотемпературной изомеризации (MHTI), катализаторы Mobil высокоэффективной изомеризации (MHAI), которые выпускаются ExxonMobil Chemicals, и катализаторы ISOMARTM корпорации UOP 1-9, 1-100, 1-210 и 1-300. Обедненный пара-ксилолом рафинат можно пропускать в блок (установку) для изомеризации, разделенный на несколько зон (или реакторов), причем каждая зона содержит другой катализатор для проведения другой реакции изомеризации в соответствии с находящимся в этой зоне катализатором. Или же, изомеризацию можно проводить только в одной зоне (или реакторе) с единственным катализатором.
Эффективную изомеризацию можно проводить в различных условиях, которые включают широкий интервал температур. В общем понятно, что концентрация пара-ксилола в равновесной смеси ксилолов не очень сильно зависит от температурного интервала, примерно, от 200°С до 500°С. Следовательно, выбор конкретной температуры в интервале для конкретной зоны изомеризации (или реактора) не должен оказывать значительного влияния на концентрацию пара-ксилола в смеси. Выбор конкретной температуры изомеризации в конкретной зоне (или реакторе) установки для изомеризации по данному способу зависит от множества факторов, таких как, присутствует ли этилбензол в смеси, подаваемой на установку, желательна ли или необходима конверсия этилбензола в процессе изомеризации, имеются две или более зон изомеризации (или реакторов), работающие последовательно, температура подаваемой смеси и комбинация этих факторов.
Как правило, интервал температур на стадии изомеризации лежит в интервале около 200-550°С, предпочтительно, около 250-550°С, и более предпочтительно, около 340-430°С. Конверсия этилбензола не происходит в значительной степени при температурах ниже 300°С. Таким образом, если желательна конверсия этилбензола, тогда изомеризацию следует проводить при температуре в вышеуказанных интервалах, примерно, выше 300°С. Напротив, если конверсия этилбензола нежелательна, тогда изомеризацию следует проводить при температуре ниже вышеуказанных интервалов, примерно, ниже 300°С. Если используют установку для изомеризации, имеющую несколько зон (или реакторов), предпочтительно поддерживать температуру в первой зоне (или контейнере) в пределах вышеуказанных температурных интервалов, выше 300°С, а температуру во второй зоне (или реакторе) ниже вышеуказанных температурных интервалов, ниже 300°С.
Конечно, если нет необходимости в конверсии этилбензола, более низкие температуры позволяют создать выгодный способ с меньшим потреблением энергии и меньшими потерями ксилола, связанными с превращением его в побочные продукты. Хотя этилбензол полезен как исходное для получения стирола, его выделение может быть неэкономичным, если он присутствует в минорных количествах. Поэтому более выгодным с экономической точки зрения проводить стадию изомеризации при высоких температурах, а не регенерировать этилбензол, вне зависимости от того, присутствует ли этилбензол. Побочные продукты изомеризации могут, например, включать С9+ углеводороды.
Стадию изомеризации, предпочтительно, проводить при давлении, равном давлению или более низком, чем давление, при котором рафинат получают на предыдущей стадии контактирования, и, предпочтительно, при давлении, традиционно применяемом в обычных реакторах. См. в целом Патент США 5516956 о стандартных условиях изомеризации ксилола. Конкретно, общее давление в установке для изомеризации, содержащей все углеводороды и любые другие газы, которые могут там присутствовать, находится в примерном интервале от атмосферного давления до 6895 кПа (около 1000 пси абс.), предпочтительно, около 345-2757 кПа (около 50-400 пси абс.), и, более предпочтительно, около 413-1516 кПа (около 60-220 пси абс.).
Если обратиться к чертежам, на которых номера (цифры) относятся к идентичным или сходным элементам, встречающимся на разных чертежах, на фиг.1 показан цикл работы двухступенчатой системы адсорбции с переменным парциальным давлением (PPSA) 10, пригодной для осуществления раскрываемого способа и, конкретно, для проведения стадии контактирования по этому способу. Как видно, система 10 включает два идентичных аппарата (слоя) для разделения - идентичных в том смысле, что каждый из них содержит адсорбент (не показан) и идентичные впускные и выпускные отверстия, хотя только некоторые впускные и выпускные отверстия показаны и четко представлены в данном описании для более точной иллюстрации одиночного цикла. Аппараты (слои), как показано, расположены последовательно, причем в первом аппарате (слое) 12 происходит адсорбция ("A1"), a во втором аппарате (слое) 14 происходит одна из двух операций десорбции ("D1" и "D2"). Когда в первом аппарате (слое) 12 происходит адсорбция (А1), во втором аппарате (слое) 14 осуществляют одну из двух операций десорбции (D1 или D2), и соответственно, когда во втором аппарате 14 происходит адсорбция, в первом аппарате (слое) 12 осуществляется одна из двух операций десорбции. Подробнее см. табл. I:
Продолжаем описание фиг.1. Исходная смесь, содержащая изомеры ксилола и этилбензол (также в данном описании называемые "C8 ароматические углеводороды"), подается через впускной трубопровод 16 в нижнюю часть 18 первого аппарата 12 и контактирует в нем с адсорбентом, селективным к пара-ксилолу в указанных выше условиях (например, температура, давление и т.д.). При таком контакте пара-ксилол и этилбензол, содержащиеся в смеси, адсорбируются адсорбентом, тогда как другие компоненты смеси не адсорбируются и в конечном счете вытесняются (поступающей исходной смесью) из верхней части 20 первого аппарата 12 через выпускную линию 22 в виде обедненного пара-ксилолом рафината. Рафинат, при необходимости, можно нагревать и пропускать непосредственно в расположенный далее аппарат для изомеризации для получения равновесной смеси изомеров ксилола, которую объединяют с подаваемой исходной смесью C8 ароматических углеводородов.
Как видно, в то время как в аппарате 12 происходит адсорбция (А1), во втором аппарате 14 осуществляется десорбция через две последовательные стадии D1 и D2.
Конкретно, в процессе стадии десорбции D1 "продувочный" газ подают через впускную линию 24 в верхнюю часть 26 второго аппарата 14, энергично выпуская из нижней части 28 второго аппарата 14 эффлюент, содержащий "продувочный" газ, неадсорбированные углеводороды и другой газ, присутствующий в пустотах адсорбента и второго адсорбера 14. Эффлюент с операции D1 десорбции выходит из второго аппарата 14 через выпускную линию 30, причем эффлюент содержит, главным образом, "продувочный" газ и углеводороды состава, сходного по составу подаваемой исходной смеси (или немного обогащенные пара-ксилолом и этилбензолом). Часть эффлюента на выпускной линии 30 можно объединять со свежей исходной смесью, подаваемой в аппарат (например, через впускную линию 16 первого аппарата 12), с образованием объединенной смеси, подаваемой на операцию адсорбции (А1).
Как и на стадии D1, операции десорбции в ходе последующей стадии D2 операции десорбции "продувочный" газ непрерывно подают через впускную линию 24 в верхнюю часть 26 второго аппарата 14, энергично выдувая через нижнюю часть 28 второго аппарата 14 какие-либо углеводороды и другой газ, находящийся в пустотах адсорбента и второго аппарата 14 и прочно связанный (адгезия) с адсорбентом. Эффлюент после десорбции на стадии D2 операции десорбции выходит из второго аппарата 14 через выпускную линию 32. Однако, в отличие от углеводородов в эффлюенте, полученном на стадии D1 операции десорбции, углеводороды в эффлюенте, полученном на стадии D2 операции десорбции, богаты пара-ксилолом и этилбензолом - практически более высокой чистоты, чем в исходной смеси. Эффлюент после D2 десорбции содержит, главным образом, продукт, который можно вводить в соответствующий последующий процесс для последующей операции на составляющие, а именно пара-ксилол и этилбензол. Продолжительность стадий десорбции в целом равна продолжительности операции адсорбции. Продолжительность различных стадий десорбции можно корректировать в зависимости от порции исходного, подаваемой в аппарат (на слой), и выхода и чистоты нужного продукта.
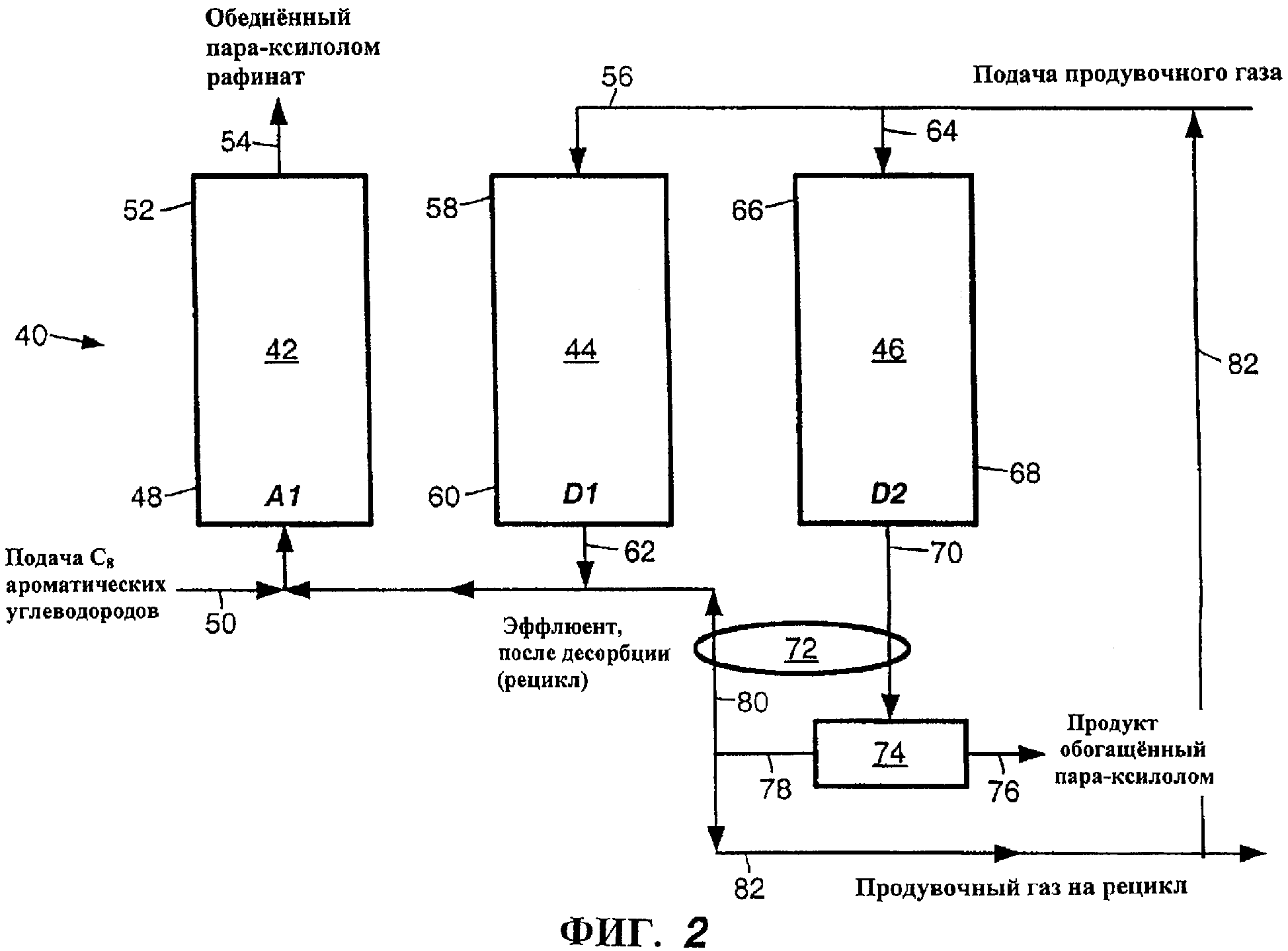
На фиг.2 представлена трехступенчатая система PPSA 40, пригодная для осуществления раскрываемого способа и, конкретно, стадии контактирования по этому способу. Как видно, система 40 включает три идентичных аппарата 42, 44 и 46 - идентичных в том смысле, что каждый из них содержит адсорбент (не показан) и идентичные впускные и выпускные отверстия, хотя только некоторые впускные и выпускные отверстия каждого аппарата показаны и четко представлены в данном описании для более точной иллюстрации одиночного цикла. Подробнее см. табл. II:
Как показано в табл. II и на фиг.2, когда в первом аппарате 42 происходит адсорбция (А1), во втором и третьем аппаратах 44 и 46 соответственно осуществляются стадии D1 и D2 соответственно операции десорбции. Аналогично, когда в третьем аппарате 46 происходит адсорбция (А1), в первом и втором аппаратах 42 и 44 соответственно осуществляются стадии D1 и D2, соответственно операции десорбции. Когда во втором аппарате 44 происходит адсорбция (А1), в третьем и первом аппаратах 46 и 42 соответственно осуществляются стадии D1 и D2, соответственно операции десорбции.
Принимая во внимание, что первый аппарат 42 служит точкой отсчета для начала проведения трехчастного цикла, в первой части исходная смесь, содержащая C8ароматические углеводороды, вводится в нижнюю часть 48 первого аппарата 42 через впускную линию 50 за период времени (А1), в течение которого во втором аппарате 44 происходит десорбция D1, а в третьем аппарате 46 происходит десорбция D2. В первом аппарате 42 исходные C8 ароматические углеводороды контактируют с селективным к пара-ксилолу адсорбентом (не показан) в вышеуказанных условиях, и пара-ксилол и этилбензол в исходной смеси адсорбируются адсорбентом, тогда как другие компоненты исходной смеси не адсорбируются и в конечном счете вытесняются (поступающей исходной смесью) из верхней части 52 первого аппарата 42 через выпускную линию 54 в виде обедненного пара-ксилолом рафината. Рафинат, при необходимости, можно нагревать и пропускать непосредственно в расположенный далее аппарат для изомеризации для получения равновесной смеси изомеров ксилола, которую объединяют с подаваемой исходной смесью C8 ароматических углеводородов.
В то время, как в первом аппарате 42 происходит адсорбция (А1), во втором аппарате 44 осуществляется стадии D1 десорбции. "Продувочный" газ подают через впускную линию 56 в верхнюю часть 58 второго аппарата 44, энергично выпуская из нижней части 60 второго аппарата 44 эффлюент, содержащий "продувочный" газ, неадсорбированные углеводороды и другой газ, присутствующий в пустотах адсорбента и второго адсорбера 42. Эффлюент с операции D1 десорбции выходит из второго аппарата 44 через выпускную линию 62, причем эффлюент содержит, главным образом, "продувочный" газ и углеводороды состава, сходного по составу подаваемой исходной смеси.
В то время, как в первом аппарате 42 происходит адсорбция (А1), а во втором аппарате 44 происходит десорбция (D1), в третьем аппарате 46 осуществляется стадия D2 десорбции. Как и на стадии D1 десорбции, во время последующей стадии D2 десорбции "продувочный" газ непрерывно подают через впускную линию 64 в верхнюю часть 66 третьего аппарата 46, энергично выдувая через нижнюю часть 68 третьего аппарата 46 любые углеводороды и другой газ, находящийся в пустотах адсорбента и третьего аппарата 46 и прочно связанный (адгезия) с адсорбентом. Эффлюент после десорбции на стадии D2 операции десорбции выходит из третьего аппарата 46 через выпускную линию 70. Однако, в отличие от углеводородов в эффлюенте, полученном на стадии D1 операции десорбции, углеводороды в эффлюенте, полученном на стадии D2 операции десорбции, богаты пара-ксилолом и этилбензолом - практически более высокой чистоты, чем в исходной смеси.
Во второй части того же цикла сразу же после первой части смесь C8 ароматических углеводородов описанным выше способом подают в нижнюю часть 68 третьего аппарата 46 в течение времени (А1) адсорбции по данному способу, идентичного времени, в течение которого в первом аппарате 42 происходит стадия D1 десорбции и во втором аппарате 44 протекает стадия D2 десорбции. В третьей и последней части цикла, которую осуществляют непосредственно сразу же после второй части, смесь C8 ароматических углеводородов описанным выше способом вводят в нижнюю часть 60 второго аппарата 44 в течение времени (А1) адсорбции по данному способу, идентичного времени, в течение которого в третьем аппарате 46 происходит стадия D1 десорбции и в первом аппарате 42 протекает стадия D2 десорбции, тем самым завершается цикл относительно первого аппарата 42. Естественно, что при непрерывном процессе описанный цикл повторяется.
Скорость потока "продувочного" газа на стадии D2 десорбции может быть такой же, как скорость потока "продувочного" газа на стадии D1 десорбции или может быть отличной от этой скорости. В зависимости от скорости потока "продувочного" газа эффлюент после стадии D2 десорбции может содержать значительное количество "продувочного" газа. Поэтому для отделения "продувочного" газа от пара-ксилола и этилбензола для конденсации пара-ксилола и этилбензола в жидкость и отделения от "продувочного" газа используют конденсационную систему теплообменников (конденсаторов) 72 и барабан(ы) для улавливания 74. Соответствующие конденсационные системы и оптимизация таких систем известны специалистам в данной области техники. Эффлюенты после конденсационной системы представляют собой жидкий продукт 76- пара-ксилол и этилбензол и отделенный от этого потока неконденсируемый газ со значительно уменьшенным содержанием углеводородов 78. Эффлюент после десорбции D2 можно пускать в соответствующие последующие процессы или же делить продукт на составляющие, а именно пара-ксилол и этилбензол. Хотя этот выделенный газ содержит мало углеводородов, чистота остающихся в нем пара ксилола и этилбензола высока. Поэтому часть выделенного газа 80 можно вернуть в смесь 50, подаваемую на стадию адсорбции (А1). Рециркуляция отделенного газа позволяет дополнительно регенерировать пара-ксилол и этилбензол из подаваемой смеси, а также обеспечивает способ регуляции парциального давления углеводородов на стадии адсорбции (А1). Или же, или кроме того, часть отделившегося газа 82 можно рециркулировать и объединить со свежим "продувочным" газом, подаваемым в аппараты (на слои). Количество отделившегося газа, возвращаемого в цикл и объединяемого со свежей исходной смесью и свежим продувочным газом, может варьироваться от 0% до 100%.
На фиг.3 изображена четырехступенчатая система PPSA 90, пригодная для осуществления раскрываемого способа и, конкретно, стадии контактирования по этому способу. Четырехступенчатая система PPSA 90 включает многие признаки трехступенчатой системы PPSA 40, описанной выше и изображенной на фиг.2. Однако существенным отличием от трехступенчатой системы PPSA 40 является наличие в четырехступенчатой системе PPSA 90 и работа четвертого аппарата 92, и третья стадия десорбции (D3). В процессах, включающих только две стадии десорбции, эффлюент после второй стадии десорбции, как правило, содержит нужный продукт, и в процессе стадии существует момент, когда эффлюент содержит максимальное количество продукта. Преимуществом D3 стадии десорбции является то, что она дает возможность завершить стадию D2 десорбции как раз после момента, когда достигается максимальное количество продукта, и собрать остальной, менее чистый продукт на последующей стадии D3.
Как показано на фиг.3, система 90 включает четыре идентичных аппарата для разделения 42, 44, 46 и 92 - идентичных в том смысле, что каждый из них содержит адсорбент (не показан) и идентичные впускные и выпускные отверстия, хотя только некоторые впускные и выпускные отверстия каждого аппарата показаны и четко представлены в данном описании для более точной иллюстрации одиночного цикла. Подробнее см. табл. III:
Как показано в табл. III и на фиг.3, когда в первом аппарате 42 происходит адсорбция (А1), во втором, третьем и четвертом аппаратах 44, 46 и 92 соответственно осуществляются стадии D1-D3, соответственно операции десорбции. Аналогично, когда в четвертом аппарате 92 происходит адсорбция (А1), в первом, втором и третьем аппаратах 42, 44 и 46 соответственно осуществляются стадии D1-D3, соответственно операции десорбции. Аналогично, когда в четвертом аппарате 92 происходит адсорбция (А1), в первом, втором и третьем аппаратах 42, 44 и 46, соответственно осуществляются стадии D1-D3, соответственно операции десорбции.
Когда в третьем аппарате 46 происходит адсорбция (А1), в четвертом, первом и втором аппаратах 92, 42 и 44 соответственно осуществляются стадии D1-D3, соответственно операции десорбции. Наконец, когда во втором аппарате 44 происходит адсорбция (А1), в третьем, четвертом и первом аппаратах 46, 92 и 42 соответственно осуществляются стадии D1-D3, соответственно операции десорбции.
Принимая во внимание, что первый аппарат 42 служит точкой отсчета для начала проведения четырехчастного (четырехступенчатого) цикла, первые три аппарата работают практически таким же образом, как и в трехчастном цикле, описанном выше и изображенном на фиг.2. В то время, как в первом аппарате 42 происходит адсорбция (А1), а во втором и третьем аппаратах 44 и 46 соответственно происходит десорбция, стадии D1 и D2 соответственно в четвертом аппарате 92 происходит третья стадия, (D3), десорбции. Как и на стадиях D1 и D2 десорбции, на последующей стадии D3 десорбции продувочный газ непрерывно поступает через впускную линию 94 в верхнюю часть 96 четвертого аппарата 92, энергично выдувая через нижнюю часть 98 четвертого аппарата 92 все углеводороды и другой газ, находящийся в пустотах адсорбента и четвертого аппарата 92. Эффлюент после стадии D3 десорбции выходит из четвертого аппарата 92 через выходную линию 100. Углеводороды, содержащиеся в эффлюенте, полученном на стадии D3 десорбции, несмотря на низкое парциальное давление, более богаты пара-ксилолом, чем исходная смесь, но не настолько богаты, как в эффлюенте после стадии D2 адсорбции. Эффлюент после стадии D3 десорбции аналогичен газу, выходящему из сборника(ов) (барабана(ов)) для улавливания 74, в котором(ых) эффлюент содержит небольшую фракцию ксилолов с содержанием пара-ксилола выше равновесного. Вследствие такой чистоты эффлюент после D3 десорбции можно преимущественно репиркулировать и объединить со смесью, подаваемой для дальнейшего повышения чистоты и выхода пара-ксилола в эффлюенте после D2 десорбции. Дополнительным и неявным преимуществом использования четырехступенчатой системы PPSA и D3 стадии десорбции является то, что скорость тока продувочного газа можно корректировать таким образом, чтобы установить заданное соотношение водорода к углеводороду в смеси, подаваемой на операцию адсорбции (А1), и, наконец, в рафинате, подаваемом в расположенный далее аппарат для изомеризации.
На фиг.4 изображена другая трехступенчатая система PPSA 110, пригодная для осуществления раскрываемого способа, и, конкретно, стадии контактирования по этому способу. Как видно, система 110 включает три идентичных аппарата 112, 114 и 116, "идентичных "в том смысле, что каждый из них содержит адсорбент (не показан) и идентичные впускные и выпускные отверстия, хотя только некоторые впускные и выпускные отверстия каждого аппарата показаны и четко представлены в данном описании для более точной иллюстрации одиночного цикла. Подробнее см. табл. IV:
Как показано в табл. IV и на фиг.4, когда в первом аппарате 112 происходит адсорбция (А1), в третьем аппарате 116 осуществляется стадия D3 операции десорбции, а во втором аппарате 114 происходит одна из стадий десорбции D1 или D2. Аналогично, когда в третьем аппарате 116 происходит адсорбция (А1), во втором аппарате 114 осуществляется стадия D3 операции десорбции, а в первом аппарате 112 протекает одна из стадий D1 или D2 операции десорбции. Наконец, когда во втором аппарате 114 происходит адсорбция(А1), в первом аппарате 112 осуществляется стадия D3 операции десорбции, а в третьем аппарате 116 протекает одна из стадий десорбции D1 или D2.
Принимая во внимание, что первый аппарат 112 служит точкой отсчета для начала проведения трехчастного цикла, в первой части исходная смесь, содержащая C8 ароматические углеводороды, вводится в нижнюю часть 118 первого аппарата 112 через впускную линию 120 за период времени (А1), в течение которого во втором аппарате 114 последовательно осуществляются стадии D1 и D2 десорбции, а в третьем аппарате 116 происходит стадия D3 десорбции. В первом аппарате 112 исходные C8 ароматические углеводороды контактируют с селективным к пара-ксилолу адсорбентом в вышеуказанных условиях, и пара-ксилол и этилбензол в исходной смеси адсорбируются адсорбентом, тогда как другие компоненты подаваемой смеси не адсорбируются и в конечном счете вытесняются (поступающей исходной смесью) из верхней части 122 первого аппарата 112 через выпускную линию 124 в виде обедненного пара-ксилолом рафината. Рафинат, при необходимости, можно нагревать и пропускать непосредственно в расположенный далее аппарат для изомеризации для получения равновесной смеси изомеров ксилола, которую объединяют с подаваемой исходной смесью C8 ароматических углеводородов.
Как показано, в то время, как в первом аппарате 112 происходит адсорбция (А1), во втором аппарате 114 осуществляются последовательные стадия D1 и D2 десорбции. Конкретно, во время стадии D1 десорбции продувочный газ подают через впускную линию 126 в верхнюю часть 128 второго аппарата 114, энергично выдувая из нижней части 130 второго аппарата 114 эффлюент, содержащий продувочный газ, неадсорбированные углеводороды и другой газ, присутствующий в пустотах адсорбента и второго аппарата 114. Эффлюент со стадии D1 операции десорбции выходит из второго аппарата 114 через выпускную линию 132, причем эффлюент содержит продувочный газ и углеводороды состава, сходного по составу подаваемой исходной смеси (или немного обогащенный пара-ксилолом и этилбензолом). Эффлюент после D1 десорбции на выпускной линии 132 объединяется с помощью трубопровода (линии) 134 со свежей исходной смесью, подаваемой в аппараты, и с любым эффлюентом, выходящим после стадии D3 (описанной ниже) с образованием объединенной смеси, подаваемой на операцию адсорбции (А1).
Как и на стадии D1 десорбции, во время последующей стадии D2 десорбции продувочный газ непрерывно поступает через впускную линию (трубопровод) 126 в верхнюю часть 128 второго аппарата 114, энергично выдувая через нижнюю часть 130 второго аппарата 114 любые углеводороды и другой газ, находящийся в пустотах адсорбента и второго аппарата 114 и связанный (адгезия) с адсорбентом. Эффлюент после десорбции на стадии D2 выходит из второго аппарата 114 через выпускную линию 132. Однако в отличие от углеводородов в эффлюенте, полученном на стадии D1 десорбции, углеводороды в эффлюенте, полученном на стадии D2 десорбции, богаты пара-ксилолом и этилбензолом - практически, их чистота выше, чем чистота исходной смеси.
Скорость тока продувочного газа на стадии D2 десорбции может быть такой же, как скорость продувочного газа на стадии D1 десорбции, или может отличаться от нее. В зависимости от скорости тока продувочного газа эффлюент из стадии D2 десорбции может содержать значительное количество продувочного газа. Поэтому для отделения продувочного газа от пара-ксилола и этилбензола в эффлюенте после D2 десорбции эффлюент далее транспортируют в конденсационную систему теплообменников (конденсаторов) 72 через линию 136, а затем в барабан(ы) для улавливания 74, которые можно использовать для конденсации пара-ксилола и этилбензола в жидкость и отделения жидкости от продувочного газа. Как ранее указывалось в данном описании, соответствующие конденсационные системы и оптимизация таких систем известны специалистам в данной области техники. Эффлюенты из конденсационной системы представляют собой жидкий продукт пара-ксилол и этилбензол 76 и отделившийся неконденсируемый газ со значительно пониженным содержанием углеводородов 78. Эффлюент после D2 десорбции можно пустить в надлежащие последующие процессы для дальнейшей реакции или для разделения продукта на составляющие, а именно пара-ксилол и этилбензол. Хотя этот выделенный газ содержит мало углеводородов, чистота остающихся в нем пара ксилола и этилбензола высока. Поэтому часть выделенного газа 80 можно вернуть в смесь 120, подаваемую на стадию адсорбции (А1). Рециркуляция отделенного газа позволяет дополнительно регенерировать пара-ксилол и этилбензол из подаваемой смеси, а также обеспечить способ регуляции парциального давления углеводородов на стадии адсорбции (А1). Или же, или кроме того, часть отделившегося газа 82 можно рециркулировать и объединить со свежим "продувочным" газом, подаваемым в аппараты (на слои). Количество отделившегося газа, возвращаемого в цикл и объединяемого со свежей исходной смесью и свежим продувочным газом, может варьироваться от 0% до 100%.
Когда в первом аппарате 112 происходит адсорбция (А1), а во втором аппарате 114 осуществляются последовательные стадия D1 и D2 десорбции, в третьем аппарате протекает стадия D3 десорбции. Как и на стадиях D1 и D2 десорбции, во время последующей стадии D3 десорбции продувочный газ непрерывно поступает через впускную линию (трубопровод) 138 в верхнюю часть 140 третьего аппарата 116, энергично выдувая через нижнюю часть 142 третьего аппарата 116 любые углеводороды и другой газ, находящийся в пустотах адсорбента и второго аппарата 116.
Эффлюент после десорбции на стадии D3 выходит из третьего аппарата 116 через выпускную линию 144. Углеводороды, содержащиеся в эффлюенте, полученном на стадии D3 десорбции, несмотря на низкое парциальное давление, более богаты пара-ксилолом, чем исходная смесь, но не настолько богаты, как в эффлюенте после стадии D2 адсорбции. Эффлюент после стадии D3 десорбции аналогичен газу, выходящему из сборника(ов) (барабана(ов)) для улавливания 74, тем, что эффлюент содержит небольшую фракцию ксилолов с содержанием пара-ксилола выше равновесного. Вследствие такой чистоты эффлюент после D3 десорбции можно преимущественно рециркулировать и объединить со смесью, подаваемой для дальнейшего повышения чистоты и выхода пара-ксилола в эффлюенте после D2 десорбции. Дополнительным и неявным преимуществом использования четырехступенчатой системы PPSA и D3 стадии десорбции является то, что скорость тока продувочного газа (например, водорода) можно корректировать таким образом, чтобы установить заданное соотношение водорода к углеводороду в смеси, подаваемой на операцию адсорбции (А1), и, наконец, в рафинате, подаваемом в расположенный далее аппарат для изомеризации.
Во второй части того же цикла сразу же после первой части смесь C8 ароматических углеводородов описанным выше способом подают в нижнюю часть 142 третьего аппарата 116 в течение времени протекающей в нем (А1) адсорбции, идентичного времени, в течение которого в первом аппарате 112 последовательно протекают стадии D1 и D2 десорбции, а во втором аппарате 114 протекает стадия D3 десорбции. В третьей и последней части цикла, которую осуществляют непосредственно сразу же после второй части, смесь C8 ароматических углеводородов описанным выше способом вводят в нижнюю часть 130 второго аппарата 114 в течение времени протекающей в нем (А1) адсорбции, идентичного времени, в течение которого в третьем аппарате 116 последовательно протекают стадии D1 и D2 десорбции, а в первом аппарате 112 протекает стадия D3 десорбции, тем самым завершается цикл относительно первого аппарата 112. Естественно, что при непрерывном процессе описанный цикл повторяется.
Ссылка в приведенном выше обсуждении на "верхнюю" и "нижнюю" части адсорберов не предполагает ограничения. Применяемые в данном описании термины "верхняя" и "нижняя" следует рассматривать как противоположные стороны одного и того же аппарата. С помощью соответствующей системы трубопроводов, вентилей и другого оборудования для регулирования способа (процесса) некоторые впускные и выпускные отверстия можно комбинировать, так как, в зависимости от того, протекает в аппарате (в слое) адсорбция или десорбция, через впускные и выпускные отверстия можно транспортировать (подавать) разный материал. Например, и снова возвращаясь к фиг.1, выходные отверстия 30 и 32 можно объединить в одно выпускное отверстие с помощью вентиля в нем, что позволяет регулировать, куда в последующем процессе следует направить эффлюент (например, рециркулировать или отбросить в случае эффлюента после D1 десорбции, или очищать продукт, содержащий пара-ксилол, в случае эффлюента после D2 десорбции). Аналогично, отверстие 22 (первый аппарат 12) для выпуска обедненного пара-ксилолом рафината может служить в качестве отверстия для впуска продувочного газа на стадиях десорбции, а впускное отверстие 24 (во втором аппарате 14) для ввода продувочного газа может служит в качестве выпускного отверстия для выхода обедненного пара-ксилолом рафината на стадии адсорбции.
Изображенная на фиг.5 блок-схема процесса иллюстрирует один пример способа 150, объединяющего вышеописанные системы PPSA. Способ включает одну из вышеописанных PPSA систем (показанных на фиг.1-4), их или соответствующим образом модифицированных систем, на фиг.5 обозначенных 152. Из PPSA системы 152 через линию 154 выходит обедненный пара-ксилолом рафинат, который проходит в нагреватель/печь 156 для повышения температуры рафината (при необходимости) до температуры, пригодной для последующей изомеризации. По выходе из нагревателя/печи 156 через линию 158 нагретый рафинат проходит в аппарат для изомеризации 160 для получения равновесной смеси изомеров ксилола. Выходящая из аппарата для изомеризации через линию (трубопровод) 162 смесь изомеризованных продуктов содержит вышеуказанную смесь изомеров ксилола, а также побочные продукты. Смесь изомеризованных продуктов на линии 162 проходит через сепаратор 164, в котором отделяются побочные продукты и выводятся из процесса 150 по линии 166, C8 ароматические углеводороды отделяют и их выводят из сепаратора 164 по линии 168, инертный (продувочный) газ, присутствующий в смеси изомеризованных продуктов, отделяют и по линии 170 пропускают в расположенный далее компрессор 172. Свежий продувной газ вводят в процесс (способ) с помощью линии 174. Кроме того, продувочный газ из системы 152 можно собирать (улавливать) и через линию (трубопровод) 176 снова отправить в цикл, объединив с газовыми потоками 170 и 174, которые затем могут поступать в компрессор 172. После сжатия продувочный газ выходит из компрессора 172 и поступает в систему PPSA 152 через линию 178. Естественно, по выходе из PPSA системы поток с нужным продуктом на линии 180 содержит пара-ксилол и этилбензол. Этот нужный продукт можно пустить в последующие процессы (не показаны) для дальнейшего процессирования или разделения продукта на составляющие, а именно пара-ксилол и этилбензол. Кроме того, часть обедненного пара-ксилолом рафината можно вводить в другие подходящие последующие процессы (не показаны) для дальнейшего процессирования или разделения рафината на его основные составляющие, а именно мета-ксилол, пара-ксилол и продувочный газ.
Как отмечалось выше, осуществление раскрываемого способа (и его различных вариантов) находится в компетенции специалистов в области соответствующего оборудования и контроля, необходимого для осуществления способа. Такое оборудование для проведения процесса включает, но без ограничения, соответствующую систему трубопроводов, насосы, вентили, рабочее оборудование (например, реакторы с соответствующими впускными и выпускными вентилями, теплообменники, сепараторы и т.д.), соответствующее оборудование для контроля процесса и контроля качества, если это необходимо. Например, можно использовать несколько стадий разделения и сложное оборудование, необходимое для разделения смеси изомеризованной смеси в сепараторе 164. Важно, однако, что раскрываемый способ имеет преимущество перед другими способами, заключающееся в том, что нет необходимости в компрессионном оборудовании между системой PPSA 152 и расположенным далее аппаратом для изомеризации 160, и поэтому оно не показано на фиг.5.
Свежая порция изомеров ксилола, подаваемая в систему PPSA 152, и любые последующие рециркуляционные потоки (например, из расположенного далее аппарата для очистки пара-ксилола/этилбензола) не показаны на фиг.5. Такие свежие порции исходного и возвращенные в цикл (рециркуляция) потоки можно объединять по способу, изображенному на фиг.5, в различных точках (местах) для достижения различных целей. Например, эти потоки можно вводить непосредственно в систему PPSA 152. Или же, или помимо этого, части этих потоков можно также вводить в сепаратор 164, чтобы в дальнейшем свести к минимуму образование побочных продуктов. Или же, или помимо этого, части этих потоков можно также вводить в печь/нагреватель 156 (если он используется) или в аппарат для изомеризации 160, чтобы несколько разбавить этилбензол, и можно снизить содержание любых не C8 ароматических углеводородов в аппарате для изомеризации 160 и отделить их в сепараторе 164. Любую из этих выбираемых возможностей можно рассматривать как часть настоящего изобретения.
С учетом вышеприведенного описания эти и другие модификации оборудования должны быть в компетенции специалистов в данной области техники.
Примеры
Нижеприведенные примеры даны для иллюстрации способа, но не претендуют на ограничение его объема.
Пример 1 относится к получению адсорбента, селективного к пара-ксилолу, который затем применяют для осуществления раскрываемого способа тем методом, который представлен в последующих примерах. Пример 2 относится к эксперименту по адсорбции с переменньм парциальным давлением ("PPSA") с продувочным газом, который проводят при различных условиях в четырех различных опытах в двухступенчатой PPSA системе. Пример 3 относится к другому эксперименту PPSA с продувочным газом, который проводят при различных условиях в одиннадцати различных опытах в двухступенчатой PPSA системе. Пример 4 относится к моделированию способа и сопутствующим соображениям.
Пример 1
Как отмечалось выше, этот пример относится к получению адсорбента, селективного к пара-ксилолу, который затем применяют для осуществления раскрываемого способа тем методом, который представлен в последующих примерах.
Адсорбент, селективный к пара-ксилолу, получают, готовя 80 вес.% силикалита и 20 вес.% связующего (85 вес.% кальциевой глины и 15 вес.% оксида кремния от общего веса связующего) от общего веса адсорбента. Силикалит имеет структуру типа MFI и его можно получать различными стандартными способами. Типичная методика такова: объединяют 18.4 г гидроксида натрия (NaOH) и 12.8 г тетрапропиламмонийбромида в 227.6 г воды. После растворения прибавляют 122.6 г раствора оксида кремния Nalco 2327 (40 вес.% оксида кремния) и перемешивают в течение двух часов. Медленно прибавляют концентрированную серную кислоту (H2SO4) до рН 13. Полученный раствор нагревают при аутогенном давлении в автоклаве с соединениями из TEFLONTM линии от одного до семи дней при 300°F (149°C).
Пример 2
Как отмечалось выше, данный пример относится к адсорбции с переменным парциальным давлением ("PPSA") с продувочным газом, который проводят в двухступенчатой PPSA системе в условиях, представленных в Таблице V (см. ниже). Адсорбент, полученный в Примере 1, используют в аппаратах для разделения в различных режимах (в различных опытах), описанных ниже. Результаты эксперимента (работы) представлены в нижней части Таблицы V. Выход и выделение компонента Х и выход при рециркуляции определяют по следующим уравнениям:
Вес X, собранного в потоке
Выход Х (%) = Вес Х в исходной смеси ×100
Вес X, собранного в потоке
Улавливание Х (%) = (Вес Х в исх. смеси)-(Вес Х рецикла) ×100
Вес собранного для рецикла
Выход при рециркул. (%) = (Вес Х в исх. смеси)-(Вес Х для рецикла) ×100
Два аппарата для разделения (сепараторы) размещают последовательно, они содержат адсорбент, описанный в Примере 1. Четыре отдельных опыта проводят в условиях, описанных в табл. V (т.е. температура, давление, скорость продувочного газа-азота, массовый расход исходных С8 углеводородов ("НС"), молярный расход инертного газа (азота) на моль НС и состав подаваемой смеси НС), где четыре опыта (режима) определяются как опыты (режимы) A-D. Небольшие вариации указанных условий легко представить из табл. V и принимая во внимание воздействие(я) на процесс, описанный(ые) ниже.
Используемая в данном Примере двухступенчатая PPSA система показана на фиг.1 и подробнее описана в табл. I и сопровождающих описаниях к этой таблице. Более конкретно, ссылаясь на опыт "А", исходную смесь Cg вводят в первый аппарат за 64 секунды (А1), в течение этого периода во втором аппарате протекают две различные стадии адсорбции, первая из которых длится 18 с (D1), а вторая продолжается 46 с (D2). Сразу же после этого исходную смесь C8 вводят во второй аппарат еще в течение 64 с (А1), за это время в первом аппарате протекают две различные стадии десорбции, первая из которых длится 18 с (D1), а вторая продолжается 46 с (D2).
Десорбцию проводят с помощью инертного продувочного газа (азота). Весь эффлюент после десорбции, полученный на первой стадии десорбции D1, собирают в виде продукта для рецикла (рециркуляции) в каждом из опытов А-С, но не пускают в рецикл; однако его пускают в рецикл в опыте D. Обычно в непрерывном процессе части эффлюента после первой стадии десорбции D1 объединяют с исходной смесью с целью дальнейшего повышения чистоты и улавливания в процессе в целом. Рафинат содержит преимущественно мета-ксилол и орто-ксилол. Эффлюент после десорбции D1 содержит преимущественно избыточную исходную смесь, неадсорбированные углеводороды и азот. Эффлюент после D2 десорбции содержит преимущественно пара-ксилол, этилбензол и азот. На практике часть эффлюента после десорбции D2 можно очистить, удаляя азот, а затем пустить в рецикл (повторный цикл) (хотя этого не делают ни в одном из опытов А-D данного примера).
Проводят множество опытов в различных условиях с целью определить достижимые уровни улавливания и чистоты, при этом имеются в виду последующие отдельные операции, необходимые для изомеризации, например, рафината, содержащего преимущественно мета- и орто-ксилол. Как указывается выше в данном описании, продажные реакторы для изомеризации ксилола, как правило, работают при давлении в примерном интервале от атмосферного давления до 6895 кПа (около 1000 пси абс.), и, более обычно, при давлении в примерном интервале около 345-2757 кПа (около 50-400 пси абс.) и при температуре в примерном интервале около 200-550°С.
Хотя результаты опыта А кажутся обещающими, совмещение обедненного пара-ксилолом рафината с реактором для изомеризации потребовало бы многочисленных теплообменников и насосов, а также значительно повышенной производительности печи для того, чтобы адекватно повысить температуру и давление рафината перед подачей его в реактор для изомеризации. С учетом условий для (состояния) рафината и необходимых условий для изомеризации ксилола такое оборудование было бы очень дорогостоящим как с точки зрения капитальных вложений, так и с точки зрения осуществления непрерывной работы.
В опыте В более высокого давления обедненного пара-ксилолом рафината достигают с помощью подачи в аппараты на стадии (в операции) адсорбции инертного газа, конкретно азота, одновременно (совместно) со смесью C8 ароматических углеводородов. По сравнению с опытом А поддерживают примерно постоянный уровень парциального давления, но при более высоком общем давлении в аппарате. Результаты опыта В в основном идентичны результатам, полученным в опыте А, лишь с незначительными изменениями полученных показателей чистоты и улавливания. Преимуществом является, что поток обедненного пара-ксилолом рафината в опыте В выходит из адсорбера при давлении около 807 кПа (около 117 пси абс.) и, следовательно, непосредственно вводится в реакторы для изомеризации ксилола в необходимом интервале рабочего давления.
В опыте С общее давление повышают, повышая только давление углеводорода (например, в подаваемой в аппарат с адсорбентом смеси С8 ароматических углеводородов отсутствует инертный газ). Результаты опыта С показывают, что наблюдаемая чистота пара-ксилола и этилбензола (рХ + ЕВ) понижается с 82.3 вес.% (в опыте А) до 73.1 вес.%. Это уменьшение чистоты показывает, что добавление инертного газа к смеси C8 ароматических углеводородов, подаваемой в адсорбер, имеет заметные, но неочевидные преимущества.
В опыте D сделана попытка имитировать работу в PPSA процессе с продувочным газом с рециклом. Благодаря смеси для рецикла и сопутствующей более высокой концентрации в ней пара-ксилола и этилбензола смесь C8 ароматических углеводородов, подаваемая на адсорбцию, содержит высокую концентрацию пара-ксилола и этилбензола (32.4 вес.%) по сравнению с предыдущими опытами А, В и С (27.4 вес.%). Также количество подаваемого совместно азота увеличивается, что отражает, что смесь для рецикла также содержит газ, который рециркулирует вместе с углеводородами. Для прогнозирования этих изменений используют компьютерную модель (имитацию) материального баланса совместного (объединенного) способа (т.е. способа с рециклом). Результаты опыта D показывают, что при таком варианте (типе) способа достижимы даже более высокие значения температуры и давления, высокая чистота и улавливание. Такое давление является идеальным для интеграции с расположенным далее (последующим) реактором для изомеризации, необходимо только корректировать температуру рафината. Таким образом, не требуются дорогостоящие компрессоры между адсорбером и аппаратом для изомеризации.
Пример 3
Как отмечается выше, данный пример относится к эксперименту PPSA, который проводят в двухступенчатой PPSA системе с использованием адсорбента, полученного в Примере 1. Более конкретно, в данном примере проводят одиннадцать опытов (помеченные опыты Е-N, см. ниже), в которых продолжительность стадий десорбции D1 и D2 изменяют в каждом опыте с целью определения оптимальной продолжительности каждой стадии, при которой чистота пара-ксилола и этилбензола максимальна. Продолжительность стадии адсорбции А1 а каждом опыте составляет 128 с. Время стадии десорбции D1 варьируется от 50 с до 123 с, а время адсорбции D2 варьируется от 5 с до 78 с, таким образом, в любом приведенном опыте общее время двух стадий десорбции остается постоянным, равным 128 с. Более подробное описание стадий адсорбции и десорбции дано на фиг.1, в табл. I и в сопровождающих ее описаниях, а также в примере 2.
Проводят одиннадцать различных опытов в условиях, представленных в таблице VI (т.е. температура, давление, скорость продувочного газа-азота, массовый расход исходных C8 углеводородов ("НС"), молярный расход инертного газа (азота) на моль НС и состав подаваемой смеси НС), см. ниже.
С учетом приведенных выше условий каждого из опытов Е-N получены следующие данные улавливания и чистоты при переменной продолжительности стадий D1 и D2 десорбции. Данные представлены в приведенной ниже табл. VII.
Полученные значения чистоты пара-ксилола и этилбензола (Ро) определяют непосредственно из анализа потока продуктов, аккумулированного на стадии D2. Чистоту пара-ксилола и этилбензола в данный момент (Рi) определяют как чистоту пара-ксилола и этилбензола в углеводородном потоке, выходящем из аппарата в данный момент времени. Непосредственное определение чистоты в данный момент (Pi) представляет собой сложную аналитическую проблему, но изменяя продолжительность стадий D1 и D2 десорбции при сохранении постоянного времени десорбции 128 с, можно определять чистоту различных фракций всего материала, собранного в потоке на рецикл и в потоке, содержащем продукты. Выше, в табл. VII, и на фиг.6 суммированы полученные данные.
Как видно из полученных данных, в опытах L-N продолжительность стадий D1 и D2 десорбции составляет 123 с и 5 с соответственно, и получен самый низкий выход продукта. В этих опытах собранный продукт соответствует последней порции углеводорода, выдуваемого из аппарата со слоем адсорбента на стадиях десорбции D1 и D2. Чистота потока продукта в этих опытах составляет около 47 вес.%. Выход продукта и наблюдаемая чистота (Ро) пара-ксилола и этилбензола повышается по мере повышения продолжительности стадии D2 десорбции. Например, при продолжительности стадии десорбции 10 с выход продукта с наблюдаемой чистотой (Рo) составляет около 62%. Так как наблюдаемая чистота (Ро) повышается подобным образом, чистота в данный момент (моментальная) (Рi) ранее полученного материала (например, материала, собранного за предыдущие 123 с) должна быть выше, чем чистота в данный момент (моментальная) (Pi) материала, собранного в период между первыми 123 с и 128 с. Это наблюдение продолжает быть верным для более коротких D1 стадий десорбции (и более продолжительных стадий D2 десорбции).
Не связывая себя какой-либо конкретной теорией, полагаем, что это явление происходит потому, что разделение ксилолов на цеолитах с порами среднего размера является кинетическим и зависит от размера молекул. У цеолитов с порами среднего размера, таких как MFI, как правило, отверстия пор имеют размер примерно от 5.4 Ангстрем (Å)-5.8 Å. Молекулы пара-ксилола и этилбензола имеют кинетические диаметры около 5.9 Å и около 6.0 Å соответственно и, следовательно, будут адсорбироваться быстрее, чем молекулы мета- и орто-ксилола (имеющие кинетические диаметры около 6.8 Å). Хотя молекулы мета-и орто-ксилола адсорбируются до некоторой степени, это происходит значительно медленнее, чем адсорбция пара-ксилола и этилбензола. Обычные конфигурационно-селективные процессы адсорбции, такие как процессы ISOSIVТМ (которые относятся к разделению парафинов), раскрывают, что другие компоненты не адсорбируют до некоторой заметной степени. Это верно в случае изопарафинов на цеолите А с порами малого размера. Или же равновесное разделение наводит на мысль, что чистота продукта скорее продолжает повышаться на стадии десорбции, нежели понижаться, как показывают вышеприведенные данные (Ralph Т. Yang, "Gas Separation by Adsorption Processes", pp. 237-274 (Butterworth Publishers, Boston, 1987) (TP242. Y36)).
Данные показывают, что отбор последней фракции продукта в процессе PSA с продувным газом при кинетическом разделении понижает наблюдаемую чистоту продукта, так как на самом деле эта фракция продукта имеет более низкую чистоту. Этот результат не является очевидным, если исходить из предыдущего уровня техники или данных наблюдаемой чистоты (Ро), собранных в ходе обычного эксперимента по адсорбции с переменным давлением. Предпочтительно, чтобы по применяемому на практике способу можно было собирать продукт в момент, в который чистота (Pi) максимальна, причем определение этой величины специалисты в данной области техники могут проводить соответствующими методами оптимизации.
Пример 4
Процесс PPSA можно моделировать с помощью компьютерной имитации. Модель, применяемая в этом примере, состоит из одного аппарата с адсорбентом (одного слоя адсорбента) в соответствии с условиями, описанными в примере 2. Как часть этой имитации адсорбент в аппарате (слой адсорбента) подразделен по длине на десять равных зон, предположительно хорошо перемешанных. Каждая зона содержит твердый адсорбент и пустое пространство, которое моделируется независимо. Модельный адсорбент может адсорбировать как этилбензол, так и все изомеры ксилола, но скорость адсорбции и десорбции зависит от структуры ароматической молекулы. Оба, пара-ксилол и этилбензол адсорбируются быстро и поэтому рассматриваются как один компонент. Мета-ксилол и орто-ксилол адсорбируются значительно медленнее и поэтому рассматриваются как отдельный единичный компонент. Для каждого из этих отдельных компонентов скорость массообмена между пустым пространством и адсорбентом устанавливают по экспериментальным данным с помощью изотерм адсорбции и движущей силы в линейном приближении. По ходу имитации потоки в верхнюю и нижнюю части аппарата чередуются, имитируя стадии PPSA цикла.
На фиг.7 показан кумулятивный исходный продукт (подача), чистота в данный момент (Pi) эффлюента и состав эффлюента для одной операции абсорбера при постоянном общем давлении, при циклической работе с изменением парциального давления с продувочным газом. Примерно, через 128 с в аппарате начинаются стадии десорбции (например, D1, D2 и т.д.). В течение этого времени исходную смесь не впускают (нет подачи). Примерно через 150 с достигается максимальная чистота (пик) пара-ксилола и этилбензола в эффлюенте. После этого момента чистота пара-ксилола и этилбензола падает. Содержание углеводородов в потоке по мере протекания стадий десорбции быстро падает. Примерно через 254 с снова начинают подавать исходную смесь углеводородов на стадию адсорбции (А1). В этот момент (в этой точке) поток эффлюента становится обогащенным мета-ксилолом и орто-ксилолом, так как пара-ксилол и этилбензол адсорбируются. Примерно через 382 с по мере продолжения циклической работы в аппарате (слое) начинается десорбция.
В процессе стадии адсорбции (А1) композиция, выходящая из верхней части аппарата (слоя), содержит заметно пониженные количества пара-ксилола и этилбензола по сравнению с подаваемой исходной смесью, которая в данном примере содержит фракцию пара-ксилола и этилбензола, равную 0.278. Однако, когда продувочный газ-водород подают через верхнюю часть аппарата (слоя), содержание пара-ксилола и этилбензола, выходящих их верхней части аппарата, повышается, и композиция достигает максимальной чистоты (как показано на фиг.7) перед началом стадии адсорбции (А1).
Предпочтительно, продукт имеет наивысшую возможную степень чистоты, и, как правило, наиболее предпочтительна чистота выше, примерно, 65 вес.% пара-ксилола и этилбензола. Отмечается, что эффлюент с продувочным газом, собираемый в промежутке от 140 с до 220 с (фиг.7), является продуктом, который дает наибольшее количество материала самой высокой чистоты. Это соответствует стадиям D2 десорбции в примерах 2 и 3 и в двух-, трех-и четырехступенчатых PPSA системах, описанных выше. Эффлюент, полученный ранее, чем примерно через 140 с, имеет значительно более низкую чистоту и может направляться непосредственно для объединения с исходной смесью с целью повышения общего уровня улавливания пара-ксилола. Это соответствует стадии D1 десорбции, описанной в примерах 2 и 3 и в двух-, трех- и четырехступенчатых PPSA системах, описанных выше. Эффлюент, полученный позднее, чем примерно через 220 с, по-прежнему имеет более высокую чистоту, чем исходная смесь, но более низкое парциальное давление ксилолов (низкое содержание углеводородной фракции). Эту часть эффлюента также можно пустить на рецикл вместе с исходной смесью как для повышения общего улавливания пара-ксилола, так и для лучшей совместимости (интеграции) с реактором для изомеризации. Это соответствует стадии D3 десорбции, описанной выше, относящейся к трех- и четырехступенчатым PPSA системам.
Рафинат, содержащий преимущественно мета-ксилол и орто-ксилол, выходит из PPSA системы с содержанием углеводородной фракции около 0.3, что соответствует соотношению водород: углеводород (Н2:НС) около 2.3. Соотношение, рассматриваемое как приемлемое для исходной смеси, поступающей в реактор для изомеризации, составляет около 0.1-10. Следовательно, полученное соотношение 2.3 попадает в этот приемлемый интервал. Соотношение Н2:НС мета-ксилола и орто-ксилола можно регулировать, корректируя расход продувочного газа в промежутке между временем 220 с и началом следующей стадии адсорбции (например, в процессе стадии D3 десорбции). Следовательно, PPSA рафинат, содержащий мета-ксилол и орто-ксилол, можно нагревать и непосредственно направлять в реактор для изомеризации, минуя последующие стадии процесса (например, сжатие, компрессию) и не добавляя и не убавляя какой-либо материал. В этих обстоятельствах отсутствует необходимость в дополнительном компрессионном оборудовании и так как улавливание пара-ксилола и этилбензола является высоким (около 87 вес.%), в реакторе для изомеризации достигается высокий выход.
Реферат
В данном описании раскрывается усовершенствованный способ получения пара-ксилола из смеси изомеров ксилола и представлены различные варианты способа. Один из вариантов способа включает стадии: (а) контактирования при практически не снижающемся общем давлении газообразной смеси, содержащей изомеры ксилола, этилбензол и неадсорбируемый газ, с адсорбентом, селективным к пара-ксилолу, содержащим цеолит с порами среднего размера, с целью получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом, причем неадсорбируемый газ содержит водород и не реагирует с изомерами ксилола и этилбензолом; и (б) изомеризации, по меньшей мере, части обедненного пара-ксилолом рафината. Стадию контактирования проводят таким образом, чтобы не надо было подвергать сжатию рафинат перед изомеризацией, тем самым выгодно избегая дорогостоящие стадии сжатия (компрессии). 5 н. и 16 з.п. ф-лы, 7 табл., 7 ил.
Формула
(а) контактирования при практически не снижающемся общем давлении газообразной смеси, содержащей изомеры ксилола, этилбензол и неадсорбируемый газ, с адсорбентом, селективным к пара-ксилолу, содержащим цеолит с порами среднего размера с целью получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом, причем неадсорбируемый газ содержит водород и не реагирует с изомерами ксилола и этилбензолом; и
(б) изомеризации, по меньшей мере, части обедненного пара-ксилолом рафината.
(а) контактирования при первом давлении смеси, содержащей изомеры ксилола и этилбензол и неадсорбируемый газ, с адсорбентом, селективным к пара-ксилолу, содержащим цеолит с порами среднего размера с целью получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом, причем неадсорбируемый газ содержит водород и не реагирует с изомерами ксилола и этилбензолом; и
(б) изомеризации при втором давлении, по меньшей мере, части обедненного пара-ксилолом рафината, притом что газ присутствует в таком количестве, что первое давление равно второму давлению или выше второго давления.
(а) контактирования смеси, содержащей изомеры ксилола, этилбензол и неадсорбируемый, нереакционноспособный газ, с адсорбентом, селективным к пара-ксилолу, с целью получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом; и
(б) изомеризации, по меньшей мере, части обедненного пара-ксилолом рафината, притом что газ присутствует в количестве, достаточном для того, чтобы обеспечить условие, что давление рафината равно давлению на стадии изомеризации или выше давления на стадии изомеризации, при этом парциальное давление изомеров ксилола и этилбензола сохраняется равным соответствующим конденсационным давлениям изомеров ксилола и этилбензола или ниже соответствующих конденсационных давлений изомеров ксилола и этилбензола.
(а) контактирования газообразной смеси, содержащей изомеры ксилола, этилбензол и неадсорбируемый газ с адсорбентом, селективным к пара-ксилолу, содержащим цеолит с порами среднего размера с целью получения обедненного пара-ксилолом рафината и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом, причем неадсорбируемый газ содержит водород и не реагирует с изомерами ксилола и этилбензолом; и
(б) изомеризации, по меньшей мере, части обедненного пара-ксилолом рафината, притом что сумма парциальных давлений изомеров ксилола и этилбензола меньше, чем общее давление смеси, и газ присутствует в таком количестве, что общее давление равно или выше, чем давление на стадии изомеризации.
(а) контактирования смеси изомеров ксилола и неадсорбируемого газа с адсорбентом, селективным к пара-ксилолу, с целью получения обедненного пара-ксилолом рафината, содержащего углеводороды, практически не включающие пара-ксилол, и эффлюента после десорбции, содержащего продукт, обогащенный пара-ксилолом; и
(б) изомеризации, по меньшей мере, части обедненного пара-ксилолом рафината, притом что газ содержит водород и не реагирует со смесью на стадии контактирования (а) и что газ присутствует в количестве, достаточном для того, чтобы обеспечить мольное соотношение газа к углеводородам в обедненном пара-ксилолом рафинате, примерно, 0.1:1-10:1 и давление равное или большее, чем давление на стадии изомеризации.
Документы, цитированные в отчёте о поиске
Способ каталитической изомеризации неравновесной смеси c8- ароматических углеводородов
Комментарии