Новое соединение, специфически связывающееся с рецептором ampa - RU2705595C2
Код документа: RU2705595C2
Чертежи



Описание
ОБЛАСТЬ ТЕХНИКИ
[0001]
Настоящее изобретение относится к новому соединению, которое специфически связывается с рецептором α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (AMPA), к его фармацевтически приемлемой соли и его сольватам, а также к композиции, содержащей эти соединения, способу получения этих соединений, и интермедиату, используемому для получения этих соединений.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
[0002]
Известно, что рецепторы AMPA широко распространены в центральной нервной системе и участвуют в процессах обучения, памяти, нервной дегенерации, клеточной смерти и т.п. В недавние годы были проведены исследования, связанные с лечением психических и неврологических заболеваний с использованием рецепторов AMPA в качестве мишеней (Патентные Документы 1-3). Для изучения связи между рецепторами AMPA и этими заболеваниями необходимо оценить уровень экспрессии и распространение рецепторов AMPA в мозге. Существуют, однако, различные проблемы, связанные с тем, что в настоящее время для изучения уровня экспрессии и тому подобного этих рецепторов AMPA мозг неизбежно приходится использовать post mortem, и невозможно проводить сравнение со здоровым человеком.
[0003]
Способ молекулярной визуализации, например, позитронно-эмиссионная томография (ПЭТ) - это способ, способный визуализировать поведение молекул в живых организмах in vivo. Для визуализации поведения рецепторов AMPA в живых организмах in vivo до настоящего времени были синтезированы определённые молекулы-зонды (непатентные документы 1-3). Однако, поскольку обычные молекулы-зонды характеризуются недостаточно специфическим связыванием с рецепторами AMPA и плохо проникают в мозг, эти молекулы-зонды сложно использовать для визуализации рецепторов AMPA in vivo. Соответственно, необходима разработка нового соединения, способного специфически связываться с рецептором AMPA и накапливаться в мозге в высоких концентрациях.
[0004]
Патентный документ 1: Японская нерассмотренная патентная заявка, публикация № 2012-207021
Патентный документ 2: Японская нерассмотренная патентная заявка, публикация № 2010-202525
Патентный документ 3: Японская нерассмотренная патентная заявка (перевод заявки согласно РСТ), публикация № 2006-525292
Непатентный документ 1: Gao M. et al., Synthesis of carbon-11 and fluorine-18 labeled N-acetyl-1-aryl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline derivatives as new potential PET AMPA receptor ligands., Bioorg. Med. Chem. Lett. 2006 Apr 15;16(8):2229-2233.
Непатентный документ 2: Langstrom B. et al., Endogenous compounds labeled with radionuclides of short half-life-some perspectives., J. Labelled Comp. Radiopharm. 2013 Mar-Apr; 56(3-4): 251-262.
Непатентный документ 3: Arstad E. et al., Closing in on the AMPA receptor: synthesis and evaluation of 2-acetyl-1-(4'-chlorophenyl)-6-methoxy-7-[11C]methoxy-1,2,3,4-tetrahydro- isoquinoline as a potential PET tracer., Bioorg. Med. Chem. 2006 Jul 15;14(14):4712-4717.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Проблемы, решаемые с помощью настоящего изобретения
[0005]
Задачей настоящего изобретения является получение нового соединения, специфически связывающегося с рецептором AMPA и хорошо проникающего в мозг. В особенности, задачей настоящего изобретения является получение нового соединения, используемого для визуализации рецепторов AMPA in vivo.
Средства для решения проблем
[0006]
Заявители настоящего изобретения провели интенсивные исследования, и в результате им удалось успешно синтезировать новое соединение, способное специфически связываться с рецептором AMPA. Более того, заявители настоящего изобретения установили на основе полученных результатов, связанных с участком взаимодействия между 2-[2,6-дифтор-4-({2-[(фенилсульфонил)-амино]этил}тио)фенокси]ацетамидом и рецептором AMPA с помощью анализа кристаллической структуры (Biochemistry, 2010, Vol. 49, pp. 2843-2850), что соединение содержит сульфонамидную (-SO2N-) и амидную группу (-CON-) на обоих его концах, и что к атому азота сульфонамидной группы можно присоединить заместитель, не нарушая при этом способности соединения связываться с рецептором AMPA, что позволяет улучшить способность этого соединения к накоплению в мозге. Таким образом, в соответствии с настоящим изобретением, предлагается соединение, представленное следующей Формулой (I), или его фармацевтически приемлемая соль или сольват.

где:
каждый из A и Z независимо представляет собой CO, SO или SO2;
каждый из X и Y независимо представляет собой S или O;
каждый из R1-R4 независимо представляет собой водород, алкил, алкенил, алкинил или гало;
каждый R5 независимо представляет собой алкил, алкенил, алкинил или гало; и
n представляет собой целое число от 0 до 4.
В варианте осуществления в соединении, представленном Формулой (I), один или более атомов являются радиоактивным изотопом атома или атомов.
Технические результаты изобретения
[0007]
Соединение по настоящему изобретению способно специфически связываться с рецептором AMPA и характеризуется чрезвычайно высокой степенью накопления в мозге. В частности, соединение по настоящему изобретению может использоваться в качестве молекулы-зонда, например, в качестве зонда для проведения ПЭТ, и позволяет визуализировать рецептор AMPA в живых организмах in vivo. Более того, соединение по настоящему изобретению легко синтезируется и может быть получено с высоким выходом.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0008]
Фиг. 1 - диаграмма, демонстрирующая степень накопления различных соединений в тканях гиппокампа.
Фиг. 2 - диаграмма, демонстрирующая отношение тока через рецепторы AMPA после введения соединений PEPA или K-2 к его контрольному значению.
Фиг. 3 - диаграмма, демонстрирующая изменение количества рецепторов AMPA после введения K-2 или носителя в живой организм. Левая диаграмма: Количество рецепторов AMPA, присутствующих на поверхности клеточной мембраны; Правая диаграмма: Общее количество рецепторов AMPA.
Фиг. 4 - полученное in vivo ПЭТ-изображение крысы с использованием радиоактивно меченого K-2. Левая диаграмма: Крыса, которой был введён носитель, Правая диаграмма: Крыса, которая подверглась блокированию введением нерадиактивно меченого K-2 в дозировке 0,5 мг/кг.
Фиг. 5 - графики зависимости активности от времени (TAC) для K-2 в гиппокампе и в стволе мозга крысы.
(a) Гиппокамп после введения носителя, (b) Гиппокамп после блокирования, (c) Ствол мозга после введения носителя, и (d) Ствол мозга после блокирования. На этой диаграмме линия графика (c) и линия графика (d) перекрывают друг друга.
Фиг. 6 - полученное in vivo ПЭТ-изображение крысы, которая подверглась блокированию низкой концентрацией (0,05 мг/кг) нерадиактивно меченого K-2.
Фиг. 7 - график зависимости активности от времени (TAC) специфического связывания с использованием ствола мозга в качестве контроля.
Фиг. 8 - диаграмма, на которой количественно определяется специфичность связывания радиоактивно меченого K-2 in vivo. Слева: Полосатое тело, Справа: Гиппокамп. Чёрный столбец: Крыса, которой введён носитель, Серый столбец: Крыса, которая подверглась блокированию.
Фиг. 9 - диаграмма, демонстрирующая сравнение общего уровня экспрессии рецептора AMPA в каждом участке мозга.
Фиг. 10 - график, демонстрирующий корреляцию между биохимическим уровнем экспрессии рецептора AMPA в каждом участке мозга и значением показателя %SUV на ПЭТ-изображении.
Фиг. 11 - полученное in vivo ПЭТ-изображение крысы, которой вводилась shРНК в обе стороны полосатого тела. shРНК для GluA1 - GluA3 (РНК, подавляющая экспрессию белка рецептора AMPA) экспрессируется в левой части полосатого тела той же особи, а скремблированная РНК (РНК, не оказывающая какого-либо заметного воздействия) экспрессируется в правой части полосатого тела той же особи.
Фиг. 12 - диаграмма, демонстрирующая сравнение значений ПЭТ-изображения на стороне shРНК и скремблированной РНК крысы, которой вводилась shРНК в обе стороны полосатого тела.
ПРЕДПОЧТИТЕЛЬНЫЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
[0009]
1. Определения
Термин «алкил» означает моновалентную группу, получаемую, когда у насыщенного алифатического углеводорода отсутствует один атом водорода. Алкил содержит, например, от 1 до 15 (C1-C15) атомов углерода, и обычно содержит от 1 до 10 (C1-C10), от 1 до 8 (C1-C8), от 1 до 6 (C1-C6), от 1 до 5 (C1-C5), от 1 до 4 (C1-C4), от 1 до 3 (C1-C3), от 1 до 2 (C1-C2) или от 2 до 6 (C2-C6) атомов углерода. Алкил может быть прямоцепочечным или разветвлённым. Примеры алкилов включают в себя, без ограничений, метил, этил, пропил, изопропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-1-бутил, 3-метил-1-бутил, 2-метил-3-бутил, 2,2-диметил-1-пропил, 2-метил-1-пентил, 3-метил-1-пентил, 4-метил-1-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-1-бутил, 2-этил-1-бутил, н-бутил, изобутил, трет-бутил, пентил, изопентил, неопентил и гексил. Алкил может дополнительно замещаться подходящим заместителем. Термин «алкил» может включать в себя алкил, содержащий радиоизотоп, например, [11C]алкил.
[0010]
Термин «алкенил» означает ненасыщенную алифатическую углеводородную группу, содержащую по меньшей мере одну двойную связь. Алкенил содержит, например, от 2 до 15 (C2-C15) атомов углерода, и обычно содержит от 2 до 10 (C2-C10), от 2 до 8 (C2-C8), от 2 до 6 (C2-C6), от 2 до 5 (C2-C5), от 2 до 4 (C2-C4), от 2 до 3 (C2-C3), от 3 до 6 (C3-C6), от 3 до 8 (C3-C8), от 4 до 6 (C4-C6), от 4 до 7 (C4-C7) или от 4 до 8 (C4-C8) атомов углерода. Алкенил может быть прямоцепочечным или разветвленным. Примеры алкенилов включают в себя, без ограничения, в частности, винил (-CH=CH2), аллил (-CH2CH=CH2), -CH=CH(CH3), -CH=C(CH3)2, -C(CH3)=CH2, -C(CH3)=CH(CH3), -C(CH2CH3)=CH2, 1,3-бутадиенил (-CH=CH-CH=CH2) и гепта-1,6-диен-4-ил (-CH2-(CH2CH=CH2)2). Алкенил может дополнительно замещаться подходящим заместителем. Термин «алкенил» может включать в себя алкенил, содержащий радиоизотоп, например, [11C]алкенил.
[0011]
Термин «алкинил» означает ненасыщенную алифатическую углеводородную группу, содержащую по меньшей мере одну тройную связь. Алкинил содержит, например, от 2 до 15 (C2-C15) атомов углерода, и типично содержит от 2 до 10 (C2-C10), от 2 до 8 (C2-C8), от 2 до 6 (C2-C6), от 2 до 5 (C2-C5), от 2 до 4 (C2-C4), от 2 до 3 (C2-C3), от 3 до 6 (C3-C6), от 3 до 8 (C3-C8), от 4 до 6 (C4-C6), от 4 до 7 (C4-C7) или от 4 до 8 (C4-C8) атомов углерода. Алкинил может быть прямоцепочечным или разветвленным. Примеры алкинилов включают в себя, без ограничения, этинил (-C≡CH), -C≡CH(CH3), -C≡C(CH2CH3), -CH2C≡CH, -CH2C≡C(CH3) и -CH2C≡C(CH2CH3). Алкинил может дополнительно замещаться подходящим заместителем. Термин «алкинил» может включать в себя алкинил, содержащий радиоизотоп, например, [11C]алкинил.
[0012]
Термин «[11C]алкил» означает алкил, в котором один или более атомов углерода в углеродных атомах, составляющих алкил, являются11C. Аналогичным образом, термин «[11C]алкенил» и термин «[11C]алкинил» означают, соответственно, алкенил, в котором один или более атомов углерода в углеродных атомах, составляющих алкенил, являются11C, и алкинил, в котором один или более атомов углерода в углеродных атомах, составляющих алкинил, являются11C.
[0013]
Термин «галоген» или «гало» означает фтор (-F), хлор (-Cl), бром (-Br) или йод (-I).
[0014]
Термин «фармацевтически приемлемая соль» означает соль, которая не является вредной для млекопитающих, в особенности для людей. Фармацевтически приемлемые соли могут быть получены с использованием нетоксичных кислот или оснований, включая неорганические кислоты и неорганические основания или органические кислоты и органические основания. Примеры фармацевтически приемлемых солей включают в себя соли металлов, образованные алюминием, кальцием, литием, магнием, калием, натрием, цинком и т.п., и органические соли, образованные лизином, N,N'-дибензилэтилендиамином, хлорпрокаином, холином, диэтаноламином, этилендиамином, меглюмином (N-метилглюкамином), прокаином и т.п. Фармацевтически приемлемые соли могут дополнительно включать в себя аддитивные соли кислот и аддитивные соли оснований.
[0015]
Термин «сольват» означает содержащее растворитель соединение, образованное связыванием одной или множества молекул растворителя с соединениями по настоящему изобретению. Сольваты включают в себя, например, моносольваты, дисольваты, трисольваты и тетрасольваты. Далее, сольваты включают в себя гидраты.
[0016]
2. Соединение и радиоактивно меченое соединение
Настоящее изобретение предоставляет собой соединение, представленное следующей Формулой (I), или его фармацевтически приемлемую соль или сольват.
[0017]
В этой формуле каждый из A и Z независимо представляет собой CO, SO или SO2, и для этих групп ожидается, что существует взаимодействие между этими группами и рецептором AMPA. При этом предпочтительно каждый из A и Z независимо представляет собой CO или SO2; более предпочтительно, A представляет собой SO2, а Z представляет собой CO. Каждый из X и Y независимо представляет собой S или O; предпочтительно, X представляет собой S, а Y представляет собой O. Каждый из R1-R4 независимо представляет собой водород, алкил, алкенил, алкинил или гало. В варианте осуществления, все из R1-R4 не являются водородом, т.е. по меньшей мере один из R1-R4 представляет собой элемент, отличный от водорода. В варианте осуществления, R2 представляет собой алкил. В другом варианте осуществления, R1 представляет собой алкил или гало. R1 может находиться в любом из положений - в орто-положении, в мета-положении или в пара-положении. Предпочтительно, R1 находится в пара-положении. В ещё одном варианте осуществления, один из R3 и R4 представляет собой водород, а другой является алкилом. Каждый из R5 независимо представляет собой алкил, алкенил, алкинил или гало. Предпочтительно, R5 представляет собой гало, в частности, предпочтительно фтор. Кроме того, предпочтительно R5 находится в обоих орто-положениях относительно группы Y (т.е. обоих мета-положениях относительно группы X).
n представляет собой целое число от 0 до 4. Предпочтительно, n равен 2.
[0018]
В ещё одном варианте осуществления, в качестве комбинации соответствующих заместителей в соединении, представленном Формулой (I), предпочтительна комбинация, в которой A представляет собой SO2, Z представляет собой CO, X представляет собой S, Y представляет собой O, R2 представляет собой алкил, R1 представляет собой водород, алкил или гало, и в случае, если R1 представляет собой алкил или гало, R1 находилась в пара-положении, один из R3 и R4 представляет собой водород, а другой - алкил, каждый из R5 независимо представляет собой алкил, алкенил, алкинил или гало, а n представляет собой целое число от 0 до 4.
[0019]
В ещё одном из воплощений настоящего изобретения для комбинации соответствующих замещающих групп в соединении, представленном Формулой (I), предпочтительна комбинация, в которой A представляет собой SO2, Z представляет собой CO, X представляет собой S, Y представляет собой O, R2 представляет собой алкил, R1 представляет собой водород, алкил или гало, и в случае, если R1 представляет собой алкил или гало, R1 находится в пара-положении, один из R3 и R4 представляет собой водород, а другой является алкилом, R5 представляет собой гало, в частности фтор, R5 находится в обоих орто-положениях относительно группы Y (т.е. обоих мета-положениях относительно группы X), а n равно 2.
[0020]
В ещё одном варианте осуществления, в качестве комбинации соответствующих заместителей в соединении, представленном Формулой (I), предпочтительна комбинация, в которой A представляет собой SO2, Z представляет собой CO, X представляет собой S, Y представляет собой O, R2 представляет собой алкил, R1 представляет собой водород, алкил или гало, и в случае, если R1 представляет собой алкил или гало, R1 находится в пара-положении, оба из R3 и R4 представляют собой водород, каждый из R5 независимо представляет собой алкил, алкенил, алкинил или гало, а n представляет собой целое число от 0 до 4.
[0021]
В варианте осуществления из соединения, представленного Формулой (I), исключаются 2-[2,6-дифтор-4-({2-[(фенилсульфонил)амино]этил}тио)фенокси]ацетамид (PEPA), 4-[2-(4-хлорфенилсульфониламино)этилтио]-2,6-дифтор-феноксиацетамид, N,N-диметил-4-[2-(4-хлорфенилсульфониламино) этилтио]-2,6-дифторфеноксиацетамид, 4-[2-(4-хлорфенилсульфониламино)этилтио]-2-фторфеноксиацетамид, N,N-диметил-4-[2-(4-хлорфенилсульфониламино)этилтио]-2-фторфеноксиацетамид, N,N-диметил-4-[2-(фенилсульфониламино)этилтио]-2,6-дифторфеноксиацетамид, 4-[2-(фенилсульфониламино)этилтио]-2-фторфенокси-ацетамид и N,N-диметил-4-[2-(фенилсульфониламино)этилтио]-2-фторфеноксиацетамид, не содержащие радиоактивного изотопа.
[0022]
Конкретные примеры соединения, представленного Формулой (I), включают в себя следующие соединения:
[Таблица 1]




[0023]
В варианте осуществления в соединении, представленном Формулой (I), или в его фармацевтически приемлемой соли или сольвате один или более атомов, составляющих это соединение, являются радиоактивным изотопом атома или атомов, т.е. соединение, представленное Формулой (I), или его фармацевтически приемлемая соль или сольват являются соединением, представленным приведённой ниже Формулой (I), или его фармацевтически приемлемой солью или сольватом:
где: каждый из A и Z независимо представляет собой CO, SO или SO2;
каждый из X и Y независимо представляет собой S или O;
каждый из R1-R4 независимо представляет собой водород, алкил, алкенил, алкинил или гало;
каждый из R5 независимо представляет собой алкил, алкенил, алкинил или гало;
n представляет собой целое число от 0 до 4; и
один или более атомов являются радиоактивным изотопом атома или атомов.
[0024]
В соединении, представленном Формулой (I), радиоактивный изотоп выбирается из группы, состоящей из15O,13N,11C,18F и т.п., но без конкретного ограничения. С точки зрения величины периода полураспада предпочтительно, чтобы радиоактивный изотоп являлся11C или18F.
[0025]
Предпочтительно, один, два, три или четыре, но предпочтительнее, один из R1 - R4 является группой, содержащей радиоактивный изотоп (например, [11C]алкил (предпочтительно,11CH3), [11C]алкенил или [11C]алкинил или18F).
[0026]
Что касается соединения, представленного Формулой (I), предпочтительно, A представляет собой SO2, Z представляет собой CO, X представляет собой S, Y представляет собой O, R2 представляет собой алкил, R1 представляет собой водород, алкил или гало, и в случае, если R1 представляет собой алкил или гало, R1 находится в пара-положении, один из R3 и R4 представляет собой водород, а другой является алкилом, R5 представляет собой гало, в частности фтор, R5 находится в обоих орто-положениях относительно группы Y (т.е. обоих мета-положениях относительно группы X), n равно 2, а один из R1 - R4 является группой, содержащей радиоактивный изотоп (например, [11C]алкил (предпочтительно,11CH3), [11C]алкенил, [11C]алкинил или18F). В ещё одном варианте осуществления, в отношении соединения, представленного Формулой (I), более предпочтительно, A представляет собой SO2, Z представляет собой CO, X представляет собой S, Y представляет собой O, R2 представляет собой алкил, R1 представляет собой водород, алкил или гало, и в случае, если R1 представляет собой алкил или гало, R1 находится в пара-положении, один из R3 и R4 представляет собой водород, а другой является алкилом, R5 представляет собой гало, в частности, фтор, R5 находится в обоих орто-положениях относительно группы Y (т.е. обоих мета-положениях относительно группы X), n равно 2, а один из R1 - R4 является группой, содержащей радиоактивный изотоп (например, [11C]алкил (предпочтительно,11CH3), [11C]алкенил, [11C]алкинил или18F).
[0027]
Конкретные примеры соединения, содержащего радиоактивный изотоп, включают в себя следующие соединения:
[Таблица 2]




[0028]
3. Способ получения и интермедиат
Пример 1 синтеза
Соединение, представленное Формулой (I), или его фармацевтически приемлемая соль или сольват, в котором R2 представляет собой алкил, алкенил или алкинил, может быть получено, например, с помощью реакции соединения, представленного приведённой ниже Формулой (II), или его фармацевтически приемлемой соли или сольвата, где A, X, Y, Z, R1, R3, R4, R5 и n такие же, как были определены для соединения, представленного Формулой (I) с X1-R2, где R2 представляет собой алкил, алкенил или алкинил, а X1 представляет собой галоген):

В варианте осуществления, оба R3 и R4 в Формуле (I) и в Формуле (II) представляют собой водород. В варианте осуществления R2 представляет собой [11C]алкил, [11C]алкенил или [11C]алкинил, а R2 предпочтительно представляет собой [11C]алкил, в особенности11CH3. В варианте осуществления X1 представляет собой йод. В качестве конкретного примера соединения, представленного Формулой (II), можно привести 2-[2,6-дифтор-4-({2-[(фенилсульфонил)амино]этил}тио)фенокси]ацетамид (PEPA).
[0029]
Реакция может проводиться в полярном апротонном растворителе, например в диметилформамиде (ДМФ), тетрагидрофуране, ацетонитриле, ацетоне или диметилсульфоксиде (ДМСО). Кроме того, эту реакцию предпочтительно проводить в щелочных условиях с использованием основания, например NaOH. Температура для проведения реакции может быть от комнатной до температуры нагрева обратного холодильника, и в частности предпочтительно, чтобы она была в диапазоне от 60°C до 100°C, и ещё более предпочтительно, 80°C. Время проведения реакции составляет от 1 до 10 минут; в частности, 5 минут.
[0030]
Зонд для ПЭТ необходимо приготовить за короткое время и с высоким выходом, поскольку радиоактивный изотоп обычно имеет короткий период полураспада. Вышеупомянутая реакция пригодна для производства зонда для ПЭТ, поскольку эта реакция проходит количественно за короткое время.
[0031]
Заявители настоящего изобретения установили, что реакция соединения, представленного Формулой (II), с X1-R2 протекает количественно по группе NH, расположенной рядом с группой A в соединении, представленном Формулой (II). Таким образом, даже если R3 и R4 представляют собой водород, замещение на группу N-R2 может проходить только по группе NH без использования какой-либо защитной группы.
[0032]
Соединение, представленное Формулой (II), или его фармацевтически приемлемая соль или сольват могут использоваться в качестве интермедиата для получения соединения, представленного Формулой (I), или его фармацевтически приемлемой соли или сольвата, в котором R2 представляет собой алкил, алкенил или алкинил. Кроме того, соединение, представленное Формулой (II), или его фармацевтически приемлемая соль или сольват, могут использоваться в качестве интермедиата для получения радиоактивно меченого соединения, представленного Формулой (I), или его фармацевтически приемлемой соли или сольвата, в которых R2 представляет собой [11C]алкил, [11C]алкенил или [11C]алкинил.
[0033]
Пример 2 синтеза
Соединение, представленное Формулой (I), или его фармацевтически приемлемая соль или сольват, в которых R1 представляет собой алкил, алкенил или алкинил, может быть получено, например, с помощью реакции соединения, представленного приведённой ниже Формулой (III), или его фармацевтически приемлемой соли или сольвата

(в этой формуле A, X, Y, Z, R2, R3, R4 и R5 и n такие же, как было определено выше, и каждый Ra независимо представляет собой алкил, алкенил или алкинил) с X1-R1 (в этой формуле, R1 такой же, как определено выше, а X1 представляет собой галоген). В варианте осуществления все Ra являются n-бутилом. В варианте осуществления R1 представляет собой [11C]алкил, [11C]алкенил или11C]алкинил, и R1 предпочтительно представляет собой [11C]алкил, в особенности11CH3. В варианте осуществления X1 представляет собой йод.
[0034]
Конкретные примеры соединения, представленного Формулой (III), включают в себя следующие:
[Таблица 3]

[0035]
Эта реакция может проводиться в присутствии палладиевого катализатора, фосфинового лиганда, карбоната и галида меди. Палладиевый катализатор может являться, например, трис(дибензилиденацетон)дипалладием и т.п. Кроме того, фосфиновый лиганд может являться, например, три(орто-толил)фосфином, (ди-трет-бутил)метилфосфином и т.п. Карбонат может являться K2CO3 и т.п. Галид меди может являться CuCl и т.п. Реакция может проводиться в полярном апротонном растворителе, например в диметилформамиде (ДМФ), тетрагидрофуране, ацетонитриле, ацетоне или диметилсульфоксиде (ДМСО). Температура для проведения реакции может быть от комнатной до температуры кипения при нагреве с обратным холодильником, и особенно предпочтительно, от 60°C до 100°C, и ещё более предпочтительно, 80°C. Время проведения реакции составляет от 1 до 10 минут; в особенности, 5 минут.
[0036]
Зонд для ПЭТ необходимо приготовить за короткое время и с высоким выходом, поскольку радиоактивный изотоп обычно имеет короткий период полураспада. Вышеупомянутая реакция пригодна для производства зонда для ПЭТ, поскольку реакция проходит количественно за короткое время.
[0037]
Соединение, представленное Формулой (III), или его фармацевтически приемлемая соль или сольват могут использоваться в качестве интермедиата для получения соединения, представленного Формулой (I), или его фармацевтически приемлемой соли или сольвата, в котором R1 представляет собой алкил, алкенил или алкинил. Далее соединение, представленное Формулой (III), иди его фармацевтически приемлемая соль или сольват, могут использоваться в качестве интермедиата для получения радиоактивно меченого соединения, представленного Формулой (I), или его фармацевтически приемлемой соли или сольвата, в которых R1 представляет собой [11C]алкил, [11C]алкенил или [11C]алкинил.
[0038]
Соединение, представленное Формулой (I), или его фармацевтически приемлемая соль или сольват, могут быть получены способом, описанным в приведённых ниже Примерах.
[0039]
4. Применение
Соединение, представленное Формулой (I), или его фармацевтически приемлемая соль или сольват, могут специфически связываться с рецептором AMPA. Таким образом, соединение, представленное Формулой (I), или его фармацевтически приемлемая соль или сольват, могут использоваться для визуализации рецептора AMPA. В частности, это соединение может использоваться в качестве молекулы-зонда, например, в качестве молекулы-зонда для ПЭТ.
[0040]
Визуализация включает в себя молекулярную визуализацию, например, позитронно-эмиссионную томографию (ПЭТ), способ мультифотонной визуализации, способ двухфотонной визуализации, способ визуализации с помощью флуоресценции в ближней инфракрасной области, авторадиографию, однофотонную эмиссионную компьютерную томографию (SPECT) и т.п. Эта визуализация предпочтительно является ПЭТ-визуализацией.
[0041]
Настоящее изобретение предоставляет собой композицию для визуализации рецептора AMPA, содержащую соединение, представленное Формулой (I), или его фармацевтически приемлемую соль или сольват. Эта композиция может содержать фармацевтически приемлемый носитель. Фармацевтически приемлемый носитель не имеет конкретных ограничений, и его примеры включают в себя стерилизованную воду, водный солевой раствор, водный физиологический раствор или водный солевой раствор с добавлением фосфатного буфера (PBS), раствор хлорида натрия для инъекций, раствор Рингера для инъекций, изотонический раствор декстрозы для инъекций, стерильный водный раствор для инъекций и раствор Рингера для инъекций с добавлением лактата.
[0042]
Содержание соединения, представленного Формулой (I), или его фармацевтически приемлемой соли или сольвата и фармацевтически приемлемого носителя в Композиции не имеют конкретных ограничений, они определяются на основе различных факторов, например: типа используемого соединения; возраста, пола, массы тела, состояния здоровья и диеты млекопитающих, которым вводится эта Композиция; количества введений и пути введения; продолжительности лечения и других медикаментов, которые применяются параллельно. Содержание соединения, представленного Формулой (I), или его фармацевтически приемлемой соли или сольвата, не имеет конкретных ограничений, пока оно остаётся в тех пределах, которые пригодны для визуализации рецептора AMPA. Композиция предпочтительно производится таким образом, чтобы соединение, представленное Формулой (I), или его фармацевтически приемлемая соль или сольват, могли быть введены. Содержание фармацевтически приемлемого носителя может составлять, например, от 1% до 99% по весу от всего состава.
[0043]
Далее настоящее изобретение является соединением, представленным Формулой (I), или его фармацевтически приемлемой солью или сольватом, применяемым для визуализации рецептора AMPA. Более того, настоящее изобретение предоставляет применение соединения, представленного Формулой (I), или его фармацевтически приемлемой соли или сольвата, для получения фармакологического средства для визуализации рецептора AMPA.
[0044]
Далее настоящее изобретение предоставляет способ визуализации рецептора AMPA, способ, включающий в себя введение эффективной дозы соединения, представленного Формулой (I), или его фармацевтически приемлемой соли или сольвата, млекопитающему. Млекопитающее включает в себя, например, крысу, мышь, морскую свинку, хомяка и т.п. Способ введения не имеет конкретных ограничений, и можно использовать, например, парентеральное введение, внутривенное введение или внутрибрюшинное введение. Предпочтительно можно использовать внутривенное введение. Вводимое количество не имеет конкретных ограничений при условии, что оно достаточно для обеспечения визуализации рецептора AMPA.
[0045]
Кроме того, настоящее изобретение предоставляет набор, используемый для визуализации рецептора AMPA, содержащий соединение, представленное Формулой (I), или его фармацевтически приемлемую соль или сольват. Более того, настоящее изобретение предоставляет интермедиат, используемый для получения соединения, представленного Формулой (I), или его фармацевтически приемлемой соли или сольвата, например, набор, используемый для визуализации рецептора AMPA, набор, содержащий соединение, представленное Формулой (II), или его фармацевтически приемлемую соль или сольват; и/или соединение, представленное Формулой (III), или его фармацевтически приемлемую соль или сольват. Набор может дополнительно содержать инструкцию с указанием вводимого количества, способа введения, способа применения и способа хранения этого соединения, и/или способа визуализации рецептора AMPA. Далее набор может содержать реагент для мечения радиоактивным изотопом, например, галогенированный [11C]алкил, галогенированный [11C]алкенил, галогенированный [11C]алкинил и т.п. Более того, настоящее изобретение предоставляет способ визуализации рецептора AMPA, включающий в себя этап детектирования излучения, испускаемого головным мозгом субъекта, которому было введено соединение, представленное Формулой (I), или его фармацевтически приемлемая соль или сольват.
ПРИМЕРЫ
[0046]
5 Примеров
Примеры будут описаны ниже. Приведённые ниже Примеры будут описаны только для углубления понимания формулы настоящего изобретения, и ни в коем случае не ограничивают формулу настоящего изобретения.
[0047]
Пример 1
(Синтез соединений K-1 и K-2)
2-[2,6-дифтор-4-({2-[(фенилсульфонил)амино]этил}тио)фенокси]ацетамид (K-1, PEPA) и {4-[2-(бензолсульфонилметиламино)этилсульфанил]-2,6-дифтор-фенокси}ацетамид (K-2) были синтезированы по приведённой ниже схеме.1H-ЯМР-спектр каждого из этих соединений был записан с помощью аппарата Bruker Avance III 400 МГц или аппарата Varian Mercury plus-300 МГц с использованием тетраметилсилана в качестве внутреннего эталона.

[0048]
Этап (i): Синтез метилового эфира (2,6-дифторфенокси)-уксусной кислоты (2).

В (75 мл) раствора 2,6-дифторфенола (1) (5,00 г, 38,5 ммоль) в ацетоне был добавлен K2CO3 (8,40 г, 60,7 ммоль), и через 10 мин в реакционный раствор был добавлен метилбромацетат (5,80 г, 38,5 ммоль). Реакционный раствор перемешивают при комнатной температуре в течение ночи. После завершения реакции раствор реакционной смеси был влит в смесь концентрированной соляной кислоты (20 мл) и ледяной воды (200 мл). Полученная смесь была экстрагирована этилацетатом (100 мл × 3), после чего органический слой был промыт водой (50 мл × 3) и рассолом (100 мл × 2), осушен с помощью Na2SO4 и профильтрован. После этого полученный продукт был конденсирован под вакуумом, что позволило получить соединение (2) в виде жёлтой маслянистой жидкости (7,50 г, выход 97%).
1H-ЯМР (300 МГц, в CDCl3): δ 3,78 (s, 3H), 4,74 (s, 2H), 6,86-6,99 (m, 3H).
[0049]
Этап (ii): Синтез метилового эфира (4-хлорсульфонил-2,6-дифтрофенокси)уксусной кислоты (3).

В раствор метилового эфира (2,6-дифторфенокси)уксусной кислоты (2) (5,00 г, 24,7 ммоль) в дихлорметане на ледяной бане была по каплям добавлена хлорсульфоновая кислота (17,2 г, 24,7 ммоль), после чего реакционный раствор был нагрет до 45°C и перемешивался в течение 1,5 ч. После завершения реакции раствор реакционной смеси был разбавлен 50 мл ледяной воды для остановки реакции, и органический слой был отделён и промыт водой (300 мл × 3). Полученный продукт был осушен с помощью Na2SO4 и профильтрован, а затем его конденсируют под вакуумом, что позволило получить соединение (3) в виде жёлтой маслянистой жидкости (5,50 г, выход 74%).
1H-ЯМР (300 МГц, в CDCl3): δ 3,81 (s, 3H), 4,96 (s, 2H), 7,61 (s, 1H), 7,64 (s, 1H).
[0050]
Этап (iii): Синтез метилового эфира (2,6-дифтор-4-меркаптофенокси)уксусной кислоты (4).

В раствор смеси метилового эфира (4-хлорсульфонил-2,6-дифторфенокси)уксусной кислоты (3) (5,50 г, 18,3 ммоль) и SnCl2 (14,5 г, 64,2 ммоль) в 50 мл метанола была по каплям добавлена концентрированная соляная кислота (25 мл). Раствор реакционной смеси кипят до кипения с обратным холодильником и перемешивают в течение 2 ч. После охлаждения раствор реакционной смеси был влит в ледяную воду (100 мл), и полученная смесь была экстрагирована дихлорметаном (100 мл × 3). Органический слой был промыт водой (100 мл × 3) и рассолом (100 мл × 2), осушен с помощью Na2SO4 и профильтрован. После этого полученный продукт был конденсируют под вакуумом, что позволяет получить соединение (4) в виде жёлтой маслянистой жидкости (3,30 г, выход 77%).
1H-ЯМР (300 МГц, в CDCl3): δ 3,52 (s, 1H), 3,77 (s, 3H), 4,71 (s, 2H), 6,83 (s, 1H), 6,86 (s, 1H).
[0051]
Этап (iv): Синтез метилового эфира [4-(2-бензолсульфониламино-этилсульфанил)-2,6-дифторфенокси]уксусной кислоты (5).

Раствор смеси метилового эфира (2,6-дифтор-4-меркапто-фенокси)уксусной кислоты (4) (1,10 г, 4,7 ммоль) и карбоната калия (778 мг, 5,6 ммоль) в 15 мл ацетона перемешивают в атмосфере N2 при комнатной температуре в течение 20 мин. Затем в реакционный раствор был добавлен N-(2-бромэтил)-бензолсульфонамид (9) (1,30 г, 4,90 ммоль), после чего реакционный раствор перемешивают при комнатной температуре в течение ночи. После завершения реакции реакционный раствор был вылит в 30 мл 2 Н раствора HCl, и полученный продукт был экстрагирован этилацетатом (50 мл × 3). Органический слой был промыт водой (50 мл × 3) и рассолом (100 мл × 2), осушен с помощью Na2SO4 и профильтрован. После этого полученный продукт конденсируют под вакуумом до получения сухого остатка. Этот остаток был очищен с помощью хроматографии на колонке с силикагелем (объёмное соотношение петролейного эфира и этилацетата от 10/1 до 3/1), что позволило получить соединение (5) в виде жёлтой маслянистой жидкости (1,60 г, выход 84%).
1H-ЯМР (300 МГц, в CDCl3): δ 2,95 (t, J = 6,6 Гц, 2H), 3,12 (q, J = 6,3 Гц, 2H), 3,78 (s, 3H), 4,72 (s, 2H), 5,20 (t, J = 6,0 Гц, 1H), 6,76-6,83 (m, 2H), 7,47-7,60 (m, 3H), 7,82-7,84 (m, 2H).
[0052]
Этап (v): Синтез метилового эфира {4-[2-(бензолсульфо-нилметиламино)этилсульфанил]-2,6-дифторфенокси}уксусной кислоты (6).

К 10 мл раствора смеси метилового эфира [4-(2-бензолсульфонил-аминоэтилсульфанил)-2,6-дифторфенокси]уксусной кислоты (5) (300 мг, 0,72 ммоль) и K2CO3 (397 мг, 2,88 ммоль) в диметилформамиде был добавлен метилйодид (255 мг, 1,80 ммоль) при температуре 0°C. После этого реакционный раствор перемешивают при комнатной температуре в течение 1 ч. После окончания реакции реакционный раствор был разбавлен 20 мл воды и экстрагирован этилацетатом (30 мл × 3). Органический слой промывают водой (30 мл × 3) и рассолом (20 мл × 2), осушен с помощью Na2SO4 и профильтрован. После этого полученный продукт конденсируют под вакуумом, что позволяет получить соединение (6) в виде жёлтой маслянистой жидкости (285 мг, выход 92%).
1H-ЯМР (300 МГц, в CDCl3): δ 2,81 (s, 3H), 3,04-3,09 (m, 2H), 3,19-3,24 (m, 2H), 3,79 (s, 3H), 4,74 (s, 2H), 6,90-6,94 (m, 2H), 7,50-7,60 (m, 3H), 7,74-7,77 (m, 2H).
[0053]
Этап (vi): Синтез {4-[2-(бензолсульфонилметиламино)этил-сульфанил]-2,6-дифторфенокси}ацетамида (K-2).
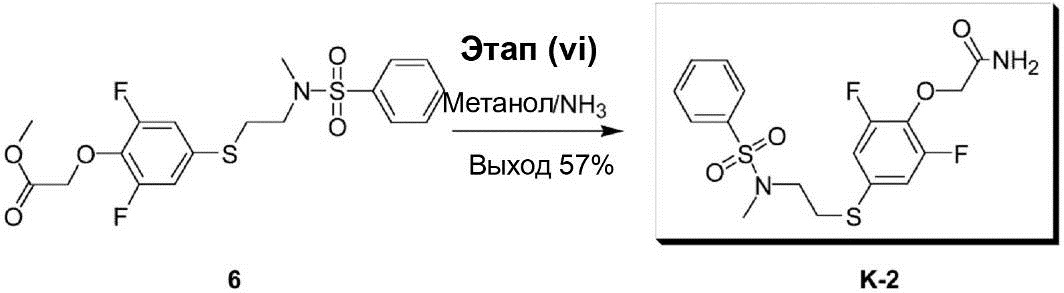
Раствор смеси метилового эфира {4-[2-(бензолсульфонилметил-амино)этилсульфанил]-2,6-дифторфенокси}уксусной кислоты (6) (40,0 мг, 0,09 ммоль) и 13 мл 4 N метанола/NH3 перемешивают при комнатной температуре в течение 18 ч. После завершения реакции раствор реакционной смеси конденсируют под вакуумом до получения сухого остатка. Этот остаток был очищен с помощью препаративной ВЭЖХ, что позволило получить соединение (K-2) в виде белого твёрдого вещества (22,0 мг, выход 57%).
1H-ЯМР (300 МГц, в CDCl3): δ 2,82 (s, 3H), 3,08-3,13 (m, 2H), 3,20-3,26 (m, 2H), 4,58 (s, 2H), 6,93-6,99 (m, 2H), 7,50-7,63 (m, 3H), 7,75-7,78 (m, 2H).
[0054]
Этап (vii): Синтез 2-[2,6-дифтор-4-({2-[(фенилсульфонил)-амино]этил}тио)фенокси]ацетамида (K-1).

Раствор смеси метилового эфира [4-(2-бензолсульфониламино-этилсульфанил)-2,6-дифторфенокси]уксусной кислоты (5) (200 мг, 0,48 ммоль) и 10 мл 4 N метанола/NH3 перемешивался при комнатной температуре в течение 18 ч. После завершения реакции раствор реакционной смеси конденсируют под вакуумом до получения сухого остатка. Этот остаток был очищен с помощью препаративной ВЭЖХ, что позволило получить соединение K-1 в виде белого твёрдого вещества (110 мг, выход 57%).
1H-ЯМР (300 МГц, в CDCl3 + D2O): δ 2,97-3,02 (m, 2H), 3,11-3,16 (m, 2H), 4,56 (s, 2H), 6,82-6,90 (m, 2H), 7,48-7,61 (m, 3H), 7,82-7,87 (m, 2H).
[0055]
Этап (viii): Синтез N-(2-бромэтил)бензолсульфонамида (9).

К 30 мл раствора бензолсульфонилхлорида (7) (3,00 г, 17,0 ммоль) и гидробромида 2-бромэтиламина (8) (3,80 г, 18,7 ммоль) в дихлорметане на ледяной бане был добавлен диизопропилэтиламин (4,80 г, 37,4 ммоль). После этого реакционный раствор перемешивали на ледяной бане в течение 1,5 ч. После завершения реакции реакционный раствор был разбавлен 20 мл воды и экстрагирован этилацетатом (30 мл × 3). Органический слой был промыт водой (30 мл × 3) и рассолом (20 мл × 2), осушен с помощью Na2SO4 и профильтрован. Затем полученный продукт конденсируют под вакуумом, что позволяет получить соединение (9) в виде белого твёрдого вещества (4,40 г, выход 98%).
1H-ЯМР (300 МГц, в CDCl3): δ 3,36-3,39 (m, 4H), 5,09 (s, 1H), 7,50-7,63 (s, 3H), 7,87-7,89 (s, 2H).
[0056]
Пример 2
(Синтез M-1, M-2 и M-3)
2-[4-(2-бензолсульфониламиноэтилсульфанил)-2,6-дифторфе-нокси]-N-метилацетамид (M-1), 2-{2,6-дифтор-4-[2-(4-фторбензолсульфониламино)-этилсульфанил]-фенокси}-ацетамид (M-2) и 2-{2,6-дифтор-4-[2-(4-метилбензолсульфо-ниламино)этилсульфанил]фенокси}ацетамид (M-3) синтезируют согласно приведённой ниже схеме.1H-ЯМР-спектр каждого из этих соединений был записан на аппарате Varian Mercury plus-400 МГц с использованием тетраметилсилана в качестве внутреннего эталона. Для проведения жидкостной хроматографии с масс-спектрометрией использовалось следующее оборудование: установка Agilent 1200A, колонка: C18; размер колонки: 4,6*50 минут; подвижная фаза: B (ацетонитрил), A (0,05% водный раствор NH3); градиент (B%): как описано в Примере.

[0057]
Этап (i): Синтез 3,5-дифтор-4-гидроксибензолсульфонилхлорида (10).

В раствор соединения (1) (5,00 г) в дихлорметане (50 мл) была по каплям добавлена хлорсульфоновая кислота (15 мл). Реакционную смесь перемешивают при температуре 25°C в течение 1 ч. Завершение реакции было определено с помощью тонкослойной хроматографии (петролейный эфир/этилацетат: 20/1). После этого раствор был вылит в наколотый лёд. Органический слой был отделён и профильтрован через целит. Фильтрат был осушен и дистиллирован под вакуумом, что позволило получить соединение (10) в виде жёлтой маслянистой жидкости (5 г, выход 57%).
1H-ЯМР (400 МГц, в CDCl3): δ 6,30 (s, 1H), 7,66-7,68 (m, 2H).
[0058]
Этап (ii): Синтез 2,6-дифтор-4-меркаптофенола (11).

В (3 мл) раствора трифенилфосфина (3,4 г, 13,1 ммоль) и диметилформамида (0,1 мл) в дихлорметане в азотной атмосфере по каплям добавляют (4 мл) раствора соединения (10) (1,0 г, 4,3 ммоль) в дихлорметане при температуре 0°C. Раствор реакционной смеси перемешивают при температуре 25°C в течение 2 ч. После этого в раствор реакционной смеси был добавлен 1 Н раствор HCl для доведения значения pH до 3, после чего раствор реакционной смеси экстрагируют этилацетатом. Органический слой был осушен с помощью Na2SO4 для удаления растворителя, что позволило получить неочищенное соединение (11) в виде жёлтой маслянистой жидкости.
[0059]
Этап (iii): Синтез 2-[2-(3,5-дифтор-4-гидроксифенилсульфанил)этил]изоиндол-1,3-диона (12).

К (100 мл) раствора неочищенного соединения (11) (14 г, 86 ммоль) в диметилформамиде были добавлены 2-(2-бромэтил)-изоиндол-1,3-дион (13,2 г, 51,8 ммоль) и K2CO3 (23,8 г, 172,4 ммоль). Раствор смеси перемешивался при температуре 25°C в течение ночи. После этого в раствор смеси был добавлен 1 Н раствор HCl для доведения значения pH до 3, и затем раствор смеси был экстрагирован этилацетатом. Органический слой был осушен с помощью сульфата натрия для удаления растворителя, что позволило получить соединение (12) в виде жёлтого твёрдого вещества (8 г, выход 27%).
1H-ЯМР (400 МГц, в ДМСО_D6): δ 3,20-3,23 (t, 2H), 3,75-3,79 (t, 2H), 7,08-7,10 (d, 2H), 7,84 (s, 4H).
[0060]
Этап (iv): Синтез {4-[2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этилсульфанил]-2,6-дифторфенокси}этилового эфира уксусной кислоты (13).
[Соединение 19]
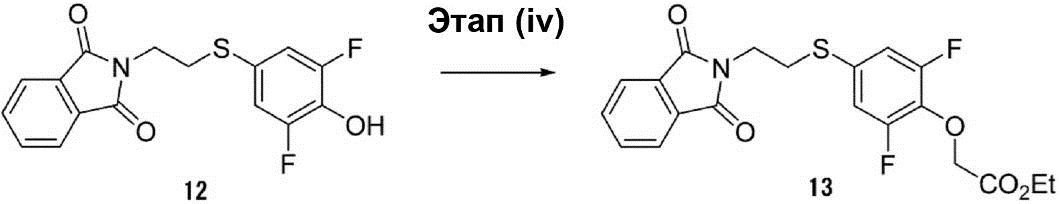
В раствор, полученный растворением соединения (12) (5,0 г, 15 ммоль) в диметилформамиде (30 мл), были добавлены этиловый эфир 3-бромпропионовой кислоты (2,5 г, 15 ммоль) и K2CO3 (3,0 г, 22,5 ммоль). Раствор смеси перемешивался при температуре 25°C в течение ночи. После этого раствор смеси экстрагируют этилацетатом. Органический слой был осушен с помощью сульфата натрия для удаления растворителя, что позволило получить соединение (13) в виде белого твёрдого вещества (6 г, выход 97%).
1H-ЯМР (400 МГц, в CDCl3): δ 1,21-1,24 (t, 3H), 3,11-3,14 (t, 2H), 3,84-3,88 (t, 2H), 4,18-4,20 (d, 2H), 4,61 (s, 2H), 6,91-6,94 (d, 2H), 7,66-7,68 (m, 2H), 7,77-7,79 (m, 2H).
[0061]
Этап (v): Синтез 2-{4-[2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этилсульфанил]-2,6-дифторфенокси}-N-метилацетамида (14).

(10 мл) Раствора соединения (13) (0,5 г, 1,2 ммоль) в метиламиновом спирте перемешивают при температуре 100°C в течение 30 мин. После этого раствор смеси конденсируют, что позволяет получить неочищенное соединение (14) в виде жёлтой маслянистой жидкости (1 г).
[0062]
Этап (vi): Синтез 2-[4-(2-аминоэтилсульфанил)-2,6-дифторфенокси]-N-метилацетамида (15).

К (10 мл) раствора неочищенного соединения (14) (1 г, 2,5 ммоль) в этаноле был добавлен гидрат гидразина (0,25 г, 5 ммоль) при температуре 90°C. Раствор был нагрет до температуры 90°C, перемешивался в течение 30 мин, а затем охлаждён до комнатной температуры. Полученный продукт был профильтрован и промыт этанолом. Органический слой был осушен с помощью сульфата натрия для удаления растворителя, а затем конденсирован для получения таким образом неочищенного соединения (15) в виде жёлтой маслянистой жидкости (0,5 г).
[0063]
Этап (vii): Синтез 2-[4-(2-бензолсульфониламиноэтилсульфанил)-2,6-дифторфенокси]-N-метилацетамида (M-1).

К (10 мл) раствора неочищенного соединения (15) (0,5 г, 1,8 ммоль) в дихлорметане были добавлены бензолсульфонилхлорид (0,4 г, 2,2 ммоль) и триэтиламин (0,2 г, 2,2 ммоль). После этого раствор смеси перемешивают при температуре 25°C в течение 1 ч, а затем экстрагируют этилацетатом. Органический слой был осушен с помощью сульфата натрия и конденсирован. Остаток очищают с помощью флэш-хроматографии, что позволило получить соединение (M-1) в виде белого твёрдого вещества (20 мг).
1H-ЯМР (400 МГц, в ДМСО_D6): δ 2,65-2,66 (d, 3H), 2,91-2,94 (t, 2H), 2,01-3,04 (t, 2H), 4,50 (s, 2H), 7,10-7,12 (d, 2H), 7,57-7,65 (m, 3H), 7,76-7,78 (d, 2H), 7,92-7,95 (t, 1H), 8,05 (s, 1H).
Результаты масс-спектрометрии: m/z 417 (M+1)+
Результаты LCMS (жидкостной хроматографии с масс-спектрометрией) [подвижная фаза: с 90% воды (с 0,1% NH4OH) и 10% CH3CN к 5% воды (с 0,1% NH4OH) и 95% CH3CN, 6,0 мин, в конце 0,5 мин в указанных условиях]: чистота 97,4%, Rt = 3,341 мин; расчётное значение для результатов масс-спектрометрии: 416; экспериментальное значение для результатов масс-спектрометрии: 417 ([M+1]+).
[0064]
Этап (viii): Синтез 2-{4-[2-(1,3-диоксо-1,3-дигидроизоин-дол-2-ил)этилсульфанил]-2,6-дифторфенокси}ацетамида (16).

(100 мл) Этанольно-аммиачного раствора соединения (13) (5,0 г, 11,8 ммоль) перемешивают при температуре 25°C в течение 2 ч. После этого раствор конденсируют для получения таким образом полученного неочищенного соединения (16) в виде жёлтой маслянистой жидкости (6,0 г).
[0065]
Этап (ix): Синтез 2-[4-(2-аминоэтилсульфанил)-2,6-дифторфенокси]ацетамида (17).

К 50 мл раствора неочищенного соединения (16) (6,0 г, 15,3 ммоль) в этаноле добавляют гидрат гидразина (1,5 г, 30 ммоль) при температуре 90°C. Раствор нагревают до температуры 90°C, перемешивают в течение 30 мин, а затем охлаждают до комнатной температуры. Полученный продукт был профильтрован и промыт этанолом. Органический слой был осушен с помощью сульфата натрия для удаления растворителя, после чего его конденсируют, чтобы получить таким образом неочищенное соединение (17) в виде жёлтой маслянистой жидкости (4,0 г).
[0066]
Этап (x): Синтез 2-{2,6-дифтор-4-[2-(4-фторбензолсульфо-ниламино)этилсульфанил]фенокси}ацетамида (M-2).

К 10 мл раствора неочищенного соединения (17) (0,5 г, 1,9 ммоль) в диметилформамиде добавляют 4-фторбензолсульфонилхлорид (0,4 г, 2,3 ммоль) и триэтиламин (0,2 г, 2,2 ммоль). После этого раствор смеси перемешивают при температуре 25°C в течение 1 ч, а затем экстрагируют этилацетатом. Органический слой был осушен с помощью сульфата натрия и конденсирован. Остаток очищают с помощью флэш-хроматографии для получения соединения (M-2) в виде белого твёрдого вещества (20 мг).
1H-ЯМР (400 МГц, в ДМСО_D6): δ 2,92-2,95 (t, 2H), 3,01-3,04 (t, 2H), 4,45 (s, 2H), 7,09-7,11 (d, 2H), 7,40-7,44 (m, 3H), 7,47 (s, 1H), 7,81-7,85 (m, 2H), 7,95-7,98 (t, 1H).
Результаты масс-спектрометрии: m/z 421 (M+1)+
Результаты LCMS [подвижная фаза: с 90% воды (с 0,1% NH4OH) и 10% CH3CN к 5% воды (с 0,1% NH4OH) и 95% CH3CN, 6 мин, в конце 0,5 мин в указанных условиях]: чистота 95,1%, Rt = 3,284 мин; расчётное значение для результатов масс-спектрометрии: 420; экспериментальное значение для результатов масс-спектрометрии: 421 ([M+1]+).
[0067]
Этап (xi): Синтез 2-{2,6-дифтор-4-[2-(4-метилбензолсуль-фониламино)этилсульфанил]фенокси}ацетамида (M-3).

К 10 мл раствора неочищенного соединения (17) (0,5 г, 1,9 ммоль) в диметилформамиде были добавлены 4-метилбензолсульфонилхлорид (0,5 г, 2,3 ммоль) и триэтиламин (0,2 г, 2,2 ммоль). После этого раствор смеси перемешивался при температуре 25°C в течение 1 ч, а затем он был экстрагирован этилацетатом. Органический слой был осушен с помощью сульфата натрия и конденсирован. Остаток очищают с помощью флэш-хроматографии для получения соединения (M-3) в виде белого твёрдого вещества (20 мг).
1H-ЯМР (400 МГц, в ДМСО_D6): δ 2,38 (s, 3H), 2,88-2,91 (t, 2H), 2,99-3,02 (t, 2H), 4,49 (s, 2H), 7,08-7,10 (d, 2H), 7,37-7,48 (m, 4H), 7,64-7,66 (d, 2H), 7,81-7,84 (t, 1H).
Результаты масс-спектрометрии: m/z 417 (M+1)+
Результаты LCMS [подвижная фаза: с 90% воды (с 0,1% NH4OH) и 10% CH3CN к 5% воды (с 0,1% NH4OH) и 95% CH3CN, 6,0 мин, в конце 0,5 мин в указанных условиях]: чистота 96,6%, Rt = 3,365 мин; расчётное значение для результатов масс-спектрометрии: 416; экспериментальное значение для результатов масс-спектрометрии: 417 ([M+1]+).
[0068]
Пример 3
Синтез M-3pre
2-(2,6-Дифтор-4-((2-(4-(трибутилстаннил)фенилсульфонамид) этил)тио)фенокси)ацетамид (M-3pre) был синтезирован в соответствии с приведённой ниже схемой.1H-ЯМР-спектр каждого из соединений был записан с помощью аппаратов Bruker Avance III 400 МГц и Bruker Fourier 300 МГц с использованием тетраметилсилана в качестве внутреннего эталона. Для проведения жидкостной хроматографии с масс-спектрометрией использовались: квадрупольный масс-спектрометр серии Agilent LC/MSD 1200 (колонка: ODS 2000 (50 × 4,6 мм, 5 мкм)), используемый в режиме ионизации ES (+) или (-); T = 30°C; скорость протекания = 1,5 мл/мин; детектирование на длине волны 254 нм.

[0069]
Этап (i): Синтез этилового эфира 2-(2,6-дифторфенокси)-уксусной кислоты (19).

Раствор смеси соединения (1) (39,0 г, 0,30 моль), K2CO3 (62,0 г, 0,45 моль), соединения (18) (50,1 г, 0,30 моль) и ацетона (200 мл) перемешивают при комнатной температуре в течение приблизительно 16 ч. После этого раствор реакционной смеси был влит в 3% HCl и экстрагирован этилацетатом (90 мл × 3). Объединённый органический слой был осушен с помощью безводного сульфата натрия, профильтрован и конденсирован. Остаток был очищен с помощью хроматографии на силикагельной колонке (соотношение петролейного эфира и этилацетата 10 : 1), что позволило получить соединение (19) (57 г, выход 87%).
1H-ЯМР (300 МГц, в CDCl3): δ 1,19 (t, J = 7,2 Гц, 3H), 4,17 (q, J = 7,2 Гц, 2H), 4,82 (s, 2H), 7,06-7,13 (m, 3H).
[0070]
Этап (ii): Синтез этилового эфира 2-(4-(хлорсульфонил)-2,6-дифторфенокси)уксусной кислоты (20).

К (180 мл) раствора соединения (19) (50 г, 0,23 моль) в дихлорметане был добавлен ClSO3H (106 мл, 1,38 моль) при температуре 35°C. Раствор реакционной смеси кипятят до кипения и перемешивают в течение приблизительно 1,5 ч. После этого раствор реакционной смеси выливают на лёд. Органический слой был отделён, осушен и конденсирован для получения соединения (20) (37 г, выход 50%).
1H-ЯМР (300 МГц, в CDCl3): δ 1,18 (t, J = 6,9 Гц, 3H), 4,16 (q, J = 6,9 Гц, 2H), 4,83 (s, 2H), 7,18-7,21 (m, 2H).
[0071]
Этап (iii): Синтез метилового эфира 2-(2,6-дифтор-4-меркаптофенокси)уксусной кислоты (21).

Раствор смеси соединения (20) (25,0 г, 0,08 моль), SnCl2 (63,3 г, 0,28 моль), концентрированной HCl (46,6 мл, 0,56 моль) и метанола (333 мл) нагревали до рефлюкса и перемешивали в течение приблизительно 1,5 ч. После этого раствор реакционной смеси выливали на лёд и экстрагировали толуолом. Органический слой был трижды промыт 12% HCl, осушен с помощью безводного сульфата натрия и конденсирован. Остаток был очищен с помощью хроматографии на силикагельной колонке (соотношение петролейного эфира и этилацетата 2 : 1), что позволило получить соединение (21) (14 г, выход 75%).
1H-ЯМР (300 МГц, в CDCl3): δ 3,52 (s, 1H), 3,79 (s, 3H), 4,72 (s, 2H), 6,88 (d, J = 6,3 Гц, 2H).
[0072]
Этап (iv): Синтез 4-бром-N-(2-бромэтил)бензолсульфонамида (24).

Соединение (23) (1,35 г, 11,0 ммоль) было добавлено в 40 мл раствора соединения (22) (2,54 г, 10,0 ммоль) в дихлорметане, и затем в смесь был добавлен тетраэтиламмоний (1,52 г, 15,0 ммоль). После этого раствор реакционной смеси перемешивался при комнатной температуре в течение приблизительно 3 ч, а затем был разбавлен водой. Полученный раствор был экстрагирован дихлорметаном (80 мл × 3). Органический слой был промыт рассолом, осушен безводным сульфатом натрия и конденсирован. Полученный неочищенный продукт был очищен с помощью хроматографии на силикагельной колонке (соотношение петролейного эфира и этилацетата 5 : 1), что позволило получить соединение (24) (2,45 г, выход 72%).
1H-ЯМР (300 МГц, в ДМСО-D6): δ 3,12-3,16 (m, 2H), 3,43 (t, J = 3,6 Гц, 2H), 7,69-7,73 (m, 2H), 7,79-7,82 (m, 2H), 8,13 (t, J = 3,9 Гц, 1H).
[0073]
Этап (v): Синтез метилового эфира 2-(4-((2-(4-бромфенил-сульфонамид)этил)тио)-2,6-дифторфенокси)уксусной кислоты (25).
Раствор смеси соединения (21) (1,25 г, 5,36 ммоль), K2CO3 (905 мг, 6,55 ммоль), соединения (24) (1,88 г, 5,50 ммоль) и ацетона (50 мл) перемешивают при комнатной температуре в течение приблизительно 16 ч. Затем раствор реакционной смеси был вылит в 3% HCl и экстрагирован этилацетатом (90 мл × 3). Органический слой был осушен безводным сульфатом натрия и конденсирован. Полученный неочищенный продукт был очищен с помощью хроматографии на силикагельной колонке (соотношение петролейного эфира и этилацетата 5 : 1), что позволило получить соединение (25) (2 г, выход 76%).
1H-ЯМР (300 МГц, в CDCl3): δ 2,94-2,98 (m, 2H), 3,08-3,14 (m, 2H), 3,77 (s, 3H), 4,73 (s, 2H), 5,33 (t, J = 6,0 Гц, 1H), 6,78-6,84 (m, 2H), 7,61-7,70 (m, 4H).
[0074]
Этап (vi): Синтез 2-(4-((2-(4-бромфенилсульфонамид)этил)-тио)-2,6-дифторфенокси)ацетамида (26).

Раствор смеси соединения (25) (3,00 г, 6,06 ммоль) и 2 М NH3/метанола (150 мл, 300 ммоль) перемешивают при комнатной температуре в течение приблизительно 16 ч. Полученный осадок был отделён с помощью фильтрации, что позволило получить соединение (26) (2,3 г, выход 80%).
1H-ЯМР (400 МГц, в ДМСО-D6): δ 2,93-2,96 (m, 2H), 3,00-3,03 (m, 2H), 4,48 (s, 2H), 7,10 (d, J = 9,2 Гц, 2H), 7,40-7,45 (m, 2H), 7,70 (d, J = 8,4 Гц, 2H), 7,80 (d, J = 8,4 Гц, 2H), 8,01 (br s, 1H).
[0075]
Этап (vii): Синтез 2-(2,6-дифтор-4-((2-(4-(трибутилстаннил)фенилсульфонамид)этил)тио)фенокси)ацетамида (M-3pre).

В 50 мл раствора соединения (26) (670 мг, 1,39 ммоль) в ксилоле были добавлены бис(трибутилолово) (0,87 мл, 1,81 ммоль) и Pd(PPh3)4 (40 мг). Раствор реакционной смеси перемешивался в атмосфере N2 при температуре 120°C в течение приблизительно 1 ч. После этого раствор реакционной смеси конденсируют под вакуумом, и полученный остаток очищают с помощью хроматографии на силикагельной колонке (соотношение петролейного эфира и этилацетата 3 : 1), что позволило получить соединение (M-3pre) в виде жёлтой маслянистой жидкости (180 мг, выход 18%).
1H-ЯМР (300 МГц, в CD3OD): δ 0,94 (t, J = 7,2 Гц, 9H), 1,12-1,17 (m, 5H), 1,29-1,39 (m, 8H), 1,52-1,60 (m, 5H), 2,98-3,06 (m, 4H), 4,55 (s, 2H), 7,01 (d, J = 9,0 Гц, 2H), 7,68 (d, J = 8,1 Гц, 2H), 7,77 (d, J = 8,1 Гц, 2H).
Результаты жидкостной хроматографии с масс-спектрометрией [подвижная фаза: с 30% воды (с 0,02% ацетата аммония) и 70% CH3CN к 5% воды (с 0,02% ацетата аммония) и 95% CH3CN, 6 мин, в конце 0,5 мин в указанных условиях]: чистота >95%, Rt = 4,259 мин; расчётное значение для результатов масс-спектрометрии: 692; экспериментальное значение для результатов масс-спектрометрии: 693 ([M+H]+).
[0076]
Пример 4
Синтез радиоактивно меченого K-2
Радиоактивно меченое соединение K-2 синтезируют следующим образом.

1 мг Соединения PEPA (около 2,5 мкмоль) растворяют в (0,3 мл) диметилформамида. В полученный раствор добавляют (7 мкл) 0,5 Н водного раствора NaOH. После перемешивания полученная реакционная смесь была помещена в реакционный контейнер в горячей ячейке. После того, как [11C]метилйодид собран нормальным способом реакцию проводят при температуре 80°C в течение 5 мин. Полученный продукт охлаждают приблизительно до комнатной температуры, разбавляют 500 мкл растворителя для жидкостной хроматографии (CH3CN : H2O = 1:1), и затем подвергают разделению с помощью жидкостной хроматографии с использованием колонки Capcell Pak UG-80 (10 x 250 мм) (Shiseido Co., Ltd., Япония). Скорость протекания во время разделения составляет 5,0 мл/мин, а детектирование осуществлялось в ультрафиолете на длине волны 254 нм и с помощью контроля интенсивности радиоактивного излучения. Часть, соответствующая пику с максимальной радиоактивностью (со временем удерживания около 8 мин), была отделена и ее конденсируют в испарителе после добавления Tween 80 (конечная концентрация: 0,8%) и 2,5 мг аскорбиновой кислоты. Остаток был растворён путём добавления к нему 2,5 мл физиологического солевого раствора.
[0077]
Радиоактивно меченое K-2 и немеченое K-2 были подвергнуты сравнению с помощью ВЭЖХ. ВЭЖХ-анализ проводят с использованием колонки Capcell Pak UG-80 (4,6 x 250 мм) (Shiseido Co., Ltd., Япония) и скорости протекания 1,0 мл/мин; детектирование осуществляют в ультрафиолете на длине волны 254 нм и с помощью рефрактометрии. Немеченое K-2 (УФ-детектирование) и радиоактивно меченое K-2 (рефрактометрическое детектирование) давали одинаковый пик со временем удерживания 8 мин. Это свидетельствует, что оба вещества являются одинаковыми, и что можно получить радиоактивно меченое K-2.
[0078]
Было установлено, что прореагировавший метилйодид связывается на 100% с сульфонамидной группой соединения PEPA, и было установлено, что синтез радиоактивно меченого K-2 является очень простым и проходит с высоким выходом.
[0079]
Пример 5
Синтез радиоактивно меченого M-3
Pd2(dba)3(1,74 мг), хлорид меди (I) (1,7 мг) и карбонат калия (2,25 мг) были взвешены и помещены в 1-мл стеклянный флакон. После этого к смеси в атмосфере азота добавляют 300 мкл раствора P(o-tol)3 (1,7 мг) в диметилформамиде. Полученная смесь перемешивалась при комнатной температуре в течение приблизительно 5 мин, а затем раствор был перенесён в ёмкость для проведения реакции мечения. [11C]CH3I собирают в условиях охлаждения, и после того, как радиоактивность насыщается, в раствор добавляют 300 мкл раствора неочищенного соединения трибутилолова соединения preM-3 (1,6 мг) в диметилформамиде. После этого реакция проводилась при температуре 80°C в течение приблизительно 5 мин. Затем реакционная смесь была пропущена через тефлоновый фильтр для удаления твёрдых примесей и подвергнута разделению с помощью ВЭЖХ. Порция, соответствующая пику с максимальной радиоактивностью (со временем удерживания около 7 мин), была отделена, конденсирована и компаундирована.
Pd2(dba)3: трис(дибензилиденацетон)дипалладий
P(o-tolyl)3: три(орто-толил)фосфин
[0080]
Пример 6
(Биологический пример)
Получение и введение соединения, связывающегося с рецептором AMPA
В эксперименте с использованием жидкостной хроматографии с масс-спектрометрией все синтезированные соединения растворялись в 100% ДМСО до концентрации в 2,5 мМ, разбавлялись физиологическим солевым раствором непосредственно перед введением (PEPA - 1,2 пиколмоль/г, M-1 - 12 пиколмоль/г, K-2 - 12 пикомоль/г, M-3 - 60 пикомоль/г, и M-2 - 240 пикомоль/г) и вводились парентеральным путём. В электрофизиологическом эксперименте соединения PEPA и K-2 растворялись в 100% ДМСО до концентрации в 150 мМ, разбавлялись искусственной ЦСЖ, используемой в качестве жидкости для орошения, непосредственно перед началом эксперимента до концентрации в 150 мкм, и затем использовались. В биохимическом эксперименте и в эксперименте с использованием блокирования и позитронно-эмиссионной томографии K-2 растворялось в 50% ДМСО до концентраций в 2,5 мМ и 25 мМ, и затем вводилось крысе в дозировке 1 мкл на грамм веса парентеральным путём для получения дозировки 0,5 мг/кг и 5 мг/кг.
[0081]
Экспериментальное животное
Все эксперименты на животных были рассмотрены и одобрены Комитетами по этике экспериментов на животных Университета Йокогамы и Национального института радиологических наук. Что касается крыс, для экспериментов использовались взрослые самцы линии Sprague-Dawley в возрасте от 6 до 10 недель (крысы линии SD) (Charles River Laboratories International, Inc., Япония).
[0082]
Эксперимент с использованием жидкостной хроматографии с масс-спектрометрией и биохимический эксперимент проводились после того, как крыса засыпала в результате вдыхания изофлурана; в дальнейшем анестезия поддерживалась с использованием специального воздушного смесителя, настроенного на концентрацию в 1,5%. Используемое соединение вводилось в дозировке 1 мкл на грамм массы тела. На шее крысы делался надрез для обнажения яремной вены, и затем соединение вводилось через яремную вену под прямым визуальным контролем с помощью инсулинового шприца (TERUMO CORPORATION, Япония). После введения соединения крыса выдерживалась под анестезией в течение 15 мин, а затем производилась экстракция мозга. В эксперименте с использованием жидкостной хроматографии с масс-спектрометрией участок, в котором расположен гиппокамп, получали из извлечённого цельного мозга в виде фрагмента толщиной около 2 мм с использованием аппарата Brain Matrix (ASI Instruments, США), затем производилось определение массы ткани в этом участке, после чего он помещался 1,5-мл коническую пробирку. В биохимическом эксперименте из извлечённого цельного мозга изготавливался острый срез мозга, включающий в себя гиппокамп и имеющий толщину около 400 мкм, с использованием вибротома (модели VT1000 от компании Leica, Германия).
[0083]
Эксперимент с использованием жидкостной хроматографии с масс-спектрометрией
Для предварительного изучения условий проведения измерений, оптимального увеличения степени разбавителя и оптимальной степени разбавления для каждого из соединений было проведено исследование с использованием ткани гиппокампа (см. Таблицу 4).
[Таблица 4]
Приготовление вводимого соединения и условия приготовления полученной ткани
* acn = Ацетонитрил; FAfree acn = Ацетонитрил, не содержащий муравьиной кислоты; FA = Муравьиная кислота; MeOH = Метанол
[0084]
После получения образца в коническую пробирку добавлялся прошедший предварительную проверку растворитель в заранее определённом количестве. Полученный продукт суспендировался с помощью гомогенизирующего пестика, а затем разрушался до нужной степени с помощью подходящего соникатора (sonicator) (модель UR-20P от компании TOMY SEIKO CO., LTD., Япония). После этого полученный продукт обрабатывался на вортексной мешалке и центрифугировался в заранее определённых условиях, после чего производился отбор супернатанта. Супернатант разбавлялся в заранее определённой степени непосредственно перед проведением измерений с использованием жидкостной хроматографии с масс-спектрометрией. Концентрация соединения, содержащегося в ткани гиппокампа, измерялась с использованием жидкостной хроматографии и квадрупольного масс-спектрометра (UPLC-MS/MS, Aquity UPLC I-Class System, Xevo TQ-S, компании Nihon Waters K.K., Япония). Для проведения СВЭЖХ использовалась колонка с внутренним диаметром 2,1 мм, длиной 100 мм и размером частиц 1,8 мкм (мод. HSS T3 от Nihon Waters K.K., Япония), условие для подвижной фазы, используемое для каждого соединения, прошло предварительную проверку (Таблица 5), и концентрации соединений измерялись при этих условиях.
[0085]
[Таблица 5]
Условие проведения измерений для образцов при проведении жидкостной хроматографии с масс-спектрометрией и Композиция подвижной фазы
* Acn = Ацетонитрил; FA = Муравьиная кислота; AA = Ацетат аммония
[0086]
При проведении масс-спектрометрии используемый МС-способ был заранее подобран для каждого соединения с использованием высококонцентрированного соединения (Таблица 6), разрушение родительских ионов до дочерних ионов осуществлялось в соответствии с протоколом, и концентрации соединений измерялись с использованием способа мониторинга множественных реакций. Далее, что касается калибровочной кривой, использовавшейся для измерения концентраций, использовались те, которые были получены с помощью декапитации подвергнутых анестезии изофлураном крыс в возрасте от 6 до 10 недель, которым не вводились никакие фармакологические средства, извлечения ткани гиппокампа и последующего добавления к собранной ткани каждого соединения в известной концентрации.
[0087]
[Таблица 6]
Условие проведения измерений для каждого соединения при масс-спектрометрии
[0088]
Расчёт коэффициента накопления соединения в ткани
В результате оптимизации условия проведения измерений для каждого соединения было установлено, что доза, вводимая живым организмам in vivo, и степень увеличения разведения во время изменений различны. Соответственно, для представления процентной доли введённого соединения в гиппокампе, рассчитывалось значение %ID/г (процента введённой дозы соединения на грамм ткани) по следующей формуле:
%ID/г = измененное значение (в пикомолях) × степень увеличения разведения × 10/концентрация введённого соединения (в пикомоль/г) × масса тела животного (в г)/масса ткани (в мг)
Крысам вводили через хвостовую вену пять типов соединений, для которых известно, что они связываются с рецептором AMPA. Через 15 минут после введения собирали ткань гиппокампа, и измеряли концентрации соединений, накопившихся в этой ткани, и в результате было установлено, что самым высоким было накопление PEPA. На основе этого результата было предложено, что PEPA характеризуется самым высоким значением коэффициента проникновения из крови в мозг (Фиг. 1). Способность K-2 связываться с различными рецепторами благодаря тому, что метильная группа, присоединённая к являющемуся его основой соединению PEPA, изменяет способности его различных участков к связыванию с рецепторами, была подвергнуты интенсивному исследованию с учетом 60 различных рецепторов-мишеней. В результате было выдвинуто предположение, что специфичность связывания PEPA является высокой по сравнению со специфичностью связывания с другими рецепторами.
[Таблица 7]
[0089]
Электрофизиологический эксперимент
Использовали самца крысы линии SD в возрасте от 7 до 8 недель. Крыса подвергалась декапитации под изофлурановой анестезией. Из извлечённого цельного мозга изготавливался острый срез мозга, включающий в себя гиппокамп и имеющий толщину около 400 мкм, с использованием вибротома (мод. VT1000 от Leica, Германия). Этот срез выдерживали неподвижным при комнатной температуре в искусственной ЦСЖ в течение 60 мин, а затем измеряли ток через рецепторы AMPA с помощью способики регистрации с использованием цельных клеток. Производилось введение пикротоксина в дозировке 100 мкм и DL-APV в дозировке 100 мкм в условиях, при которых проводилось орошение искусственной ЦСЖ со скоростью 3 мл/мин, и затем изолировался и измерялся только ток через рецепторы AMPA.
[0090]
Регистрирующий электрод помещался на пирамидальную клетку в участке CA1, а возбуждающий электрод помещался на шафферово волокно, отстоящее от клетки, на которой проводилась регистрация, на 100-200 мкм. Регистрация с использованием цельных клеток осуществлялась путём фиксации напряжения на клеточной мембране на уровне -80 мВ и подачи стимулирующих 100-мкс импульсов с частотой раз в 30 секунд. Ток через рецепторы AMPA в базовом состоянии регистрировался на протяжении 5 минут, после чего проводилось орошение с использованием искусственной ЦСЖ с добавлением PEPA или K-2 на протяжении 15 минут, и затем проводилась регистрация тока через рецепторы AMPA в жидкости, используемой для орошения, не содержащей PEPA или K-2, на протяжении 30 минут.
[0091]
Во время выдерживания в неподвижном состоянии и орошения среза мозга искусственная ЦСЖ обычно насыщалась 95% O2/5% CO2, и затем использовалась. Искусственная ЦСЖ имела следующий состав: 119 мМ NaCl, 2,5 мМ KCl, 2,5 мМ CaCl2, 1,5 мМ MgSO4, 26 мМ NaHCO3 и 1 мМ NaH2PO4. Регистрирующий электрод изготавливался из стеклянного капилляра (марки GD-1.5 от NARISHIGE Group, Япония) и перед использованием сопротивление его кончика настраивалось так, чтобы оно находилось в диапазоне от 3 до 5 МОм. Жидкость, которой заполнялся регистрирующий электрод, имела следующий состав: 115 мМ CsMeSO4, 20 мМ CsCl, 10 мМ ГЭПЭС, 2,5 мМ MgCl2, 4 мМ Na2АТФ, 0,4 мМ Na3ГТФ, 10 мМ натриевой соли фосфокреатинина и 0,6 мМ ЭГТА. Результат представлялся в виде среднего значения тока через рецепторы AMPA за последние 10 мин по результатам его регистрации на протяжении 30 минут после введения фармакологического средства, когда за единичное значение принималось среднее значение тока через рецепторы AMPA в базовом состоянии по результатам его регистрации на протяжении 5 минут.
[0092]
Биологический эксперимент
Поверхностная фракция белков мембран гиппокампа - для её получения использовали самца крысы линии SD в возрасте от 7 до 8 недель. K-2 или 50% ДМСО вводились парентерально под изофлурановой анестезией, и через 15 минут проводилась декапитация крыс. Из извлечённого цельного мозга изготавливался острый срез мозга, включающий в себя гиппокамп и имеющий толщину около 400 мкм, с использованием вибротома, и этот срез выдерживали в неподвижном состоянии при комнатной температуре в искусственной ЦСЖ в течение 60 мин. После этого для биотинилирования поверхностных мембранных белков из этого среза выделялся только участок, содержащий гиппокамп, который подвергался медленному перемешиванию при температуре 4°C в течение 45 мин в искусственной ЦСЖ, содержащей биотин в концентрации 2,0 мг/мл (марки EZ Link Sulfo-NHS-Biotin от Thermo Scientific, США). После проведения биотинилирования выделенный участок среза пять раз промывался 1 мл охлаждённого на льду буфера TBS с pH 7,5, и суспендировался с помощью пестика 150 мкл буфера для гомогенизации (150 мМ NaCl, 0,5 мМ ЭДТА, 0,1 мМ ЭГТА, 1 мМ ГЭПЭС, 20% Triton X100). Далее, к образцу добавлялись 150 мкл буфера для гомогенизации, и затем полученный продукт подвергался фрагментации ультразвуком в подходящем соникаторе. После этого проводилось разделение с помощью центрифугирования при температуре 4°C в течение 15 минут при относительном ускорении 14000 х g, после чего проводилось отделение супернатанта (до 300 мкл). Проводилось количественное определение содержания белка и гомогенизация его концентрации в супернатанте, затем 50 мкл супернатанта смешивались с 10 мкл буфера для образцов в 6 повторах, и полученная смесь нагревалась до 100°C в течение 5 мин для получения общей белковой фракции (суммарной фракции). Далее, для проведения иммунопреципитации биотинилированного поверхностного белка 150 мкл оставшегося супернатанта смешивались со 150 мкл агарозной смолы NeutrAvidin (Thermo Scientific, США), и затем полученная смесь перемешивалась при температуре 4°C в течение 16 ч. После этого проводилось центрифугирование при температуре 4°C в течение 1 минуты при 2000 х g для удаления супернатанта, а оставшиеся шарики смолы пять раз промывались 1000 мкл буфера IP. После этого к ним добавлялись 150 мкл буфера для образцов в двух повторах, и полученный продукт нагревался в течение 5 минут, после чего проводился отбор супернатанта, что позволяло получить фракцию мембранных белков (поверхностную фракцию).
[0093]
Количественный вестерн-блоттинг - общая белковая фракция и фракция мембранных белков подвергались электрофорезу в полиакриламидном геле (готовые гели марки Mini-PROTEAN TGX от компании BIO-RAD, США), а затем переносились на мембрану из ПВДФ. Эта мембрана обрабатывалась в течение 1 ч с использованием блокирующего раствора, изготовленного с использованием блокирующего реагента (Perfectblock от компании Mobitec, США) и буфера TBS-T (137 мМ NaCl, 2,68 мМ KCl, 25 мМ Tris, 0,1% Triton-X, pH 7,6). В качестве первичных антител использовались кроличьи антитела Pan AMPA/GluA2/3/4 (1 : 1000, от Cell Signaling Technology, США) и антитела GAPDH для подтверждения того, что фракция мембранных белков не содержала примесей фракций внутриклеточных белков (1 : 1000, Cell Signaling Technology), разбавленные блокирующим раствором в соотношении 1 : 1000, и реакция проводилась при комнатной температуре в течение 1 ч и в течение 3 ч, соответственно. После этого первичные антитела три раза отмывались буфером TBS-T на протяжении 10 минут, и затем проводилась реакция с антикроличьими вторичными антителами (1 : 1000; от Jackson ImmunoResearch, США) в течение 1 ч при комнатной температуре. После этого полученный продукты трижды промывали буфером TBS-T на протяжении 10 минут, и проводили детектирование полосы соответствующего белка с использованием реагента Amersham ECL (от GE Healthcare Japan, Япония) с использованием фотоаппарата для регистрации хемилюминесценции (LAS4000 mini от GE). Интенсивность сигнала для зарегистрированной полосы количественно анализировалась с использованием программы Multi Gauge V3.0 (FUJIFILM Corporation, Япония).
[0094]
Визуализация in vivo с использованием ПЭТ на крысах
ПЭТ-визуализация выполнялась с использованием способики микро-ПЭТ (Focus 220 от Siemens Medical Solution). Эксперимент по ПЭТ-визуализации на крысе: после усыпления крысы изофлураном (DS Pharma Animal Health Co., Ltd., Япония) анестезия поддерживалась с помощью изофлурана в концентрации 1,5% (расход воздуха 2 л/мин), после чего к хвостовой вене подключалась трубка для внутривенного вливания с помощью имплантируемой иглы 24G Surflo (от TERUMO CORPORATION, Япония). Крыса закреплялась на основании для проведения ПЭТ-визуализации, и затем перед построением основной визуализации проводилась предварительная радиационная визуализация для проверки положения животного. После этого крысе парентерально вводили 50% ДМСО или K-2, растворённые в 50% ДМСО, и через 3 минуты после этого вводили радиоактивно меченое K-2 (около 4 МБк). Во время осуществления ПЭТ-визуализации температура тела крысы поддерживалась равной 37±0,5°C с помощью нагревательной пластины с обратной связью (мод. BWT-100A от Bio Research Center, Япония). После визуализации трубку для внутривенных вливаний удаляли, подача изофлурана останавливалась, и затем крыса отсоединялась от основания для проведения ПЭТ-визуализации и возвращалась в свою клетку. Крыса содержалась в помещении, в котором проводилась ПЭТ-визуализация, в течение одной недели после неё, и затем крыса возвращалась в основное помещение для содержания крыс.
[0095]
Строилось суммарное ПЭТ-изображение, и затем из него вычитался базовый сигнал с использованием 0,5-мм фильтра Ханнинга для реконструкции динамического изображения. Реконструированное изображение анализировалось с помощью программы PMOD для анализа изображений (от PMOD Technologies) путём комбинирования исследуемых объёмных областей, включая множество участков полосатого тела, гиппокампа, мозжечка, ствола мозга и т.п. с участком, образованным на эталонном МРТ-изображении. Расчётным значением, использовавшимся для количественного определения, было %SUV (% от стандартизованной величины накопления), которое рассчитывалось по следующей формуле:
%SUV = количество радиации от каждой ткани, окружённой исследуемой объемной областью (в МБк)/введённое количество радиации (в МБк) × массу (в г)
[0096]
Результат эксперимента
(Оценка характеристик соединения, распознающего рецепторы AMPA)
Для оценки характеристик связывания синтезированных соединений с рецептором AMPA был проведён анализ с использованием электрофизиологических и биохимических способик. При использовании острого среза гиппокампа, полученного от взрослой крысы, было подтверждено, что ток через рецепторы AMPA значительно повышается после введения PEPA в течение 15 минут (2,4±0,13, n = 4, использовали четверых животных; p = 0,01 по сравнению с контролем). Далее, тот же эксперимент был проведён с использованием K-2, и было подтверждено, что ток через рецепторы AMPA значительно увеличивается и при использовании K-2 (1,7±0,22, n = 5, использовали четверых животных; p = 0,01 по сравнению с контролем) (Фиг. 2).
[0097]
Затем был биохимически проанализирован механизм увеличения тока через рецепторы AMPA. Был получен срез мозга, включающий в себя гиппокамп от крысы, которой in vivo вводилось K-2, и было проведено количественное определение числа рецепторов AMPA, присутствующих на поверхности клеточных мембран, с помощью способики биотинилирования. В результате было установлено, что введение K-2 в дозировке 5 мг/кг способствует переносу рецепторов AMPA на поверхностные мембраны клеток (136% ± 14, n = 5, использовались пять животных; p = 0,05 по сравнению с 50% ДМСО). С другой стороны, изменения общего количества рецепторов AMPA в том же самом животном выявлено не было (Фиг. 3). На основании вышеописанных результатов было установлено, что K-2 приводит к резкому увеличению презентации рецепторов AMPA на поверхности клеток.
[0098]
ПЭТ-визуализация на крысах с использованием соединения K-2, способного распознавать рецепторы AMPA
Чётко установлено, что K-2 способно связываться с рецепторами AMPA, и поэтому радиоактивно меченое K-2 вводилось крысам, и затем осуществлялась ПЭТ-визуализация in vivo. В результате было установлено, что у крыс радиоактивно меченое K-2 очень хорошо проникало в мозг и специфически накапливалось в области, включающей в себя гиппокамп, полосатое тело и мозжечок, и известно, что в этой области гистологически подтверждено присутствие большого числа рецепторов AMPA (см. левую панель на Фиг. 4 и панель (a) на Фиг. 5).
[0099]
Затем, для изучения специфичности накопления K-2 был проведён эксперимент по блокированию его связывания, в котором осуществлялось введение радиоактивно немеченого K-2 за 3 минуты перед введением радиоактивно меченого K-2. Предварительное введение нерадиоактивно меченого K-2 в дозировке 0,5 мг/кг позволило чётко установить, что оно приводит к исчезновению специфического накопления радиоактивно меченого соединения K-2, и что K-2 специфически связывается с рецептором AMPA in vivo (см. правую панель на Фиг. 4 и панель (b) на Фиг. 5).
[0100]
Далее, степень потери специфического связывания была незначительной в случае предварительного введения нерадиоактивно меченого K-2 в дозировке 0,05 мг/кг по сравнению с его предварительным введением в дозировке 0,5 мг/кг, и в результате была подтверждена зависимость блокирования связывания от концентрации и выдвинуто предположение о насыщении связывания (Фиг. 6). Накопление в стволе мозга не изменялось при блокировании, и таким образом было установлено, что экспрессия рецептора AMPA в этой области является более низкой (см. панели (c) и (d) на Фиг. 5). Таким образом, когда была рассчитана степень накопления радиоактивно меченого K-2 в гиппокампе с использованием ствола мозга в качестве контрольной области, было установлено, что высоко специфическое связывание наблюдалось в интервале времени от 20 до 60 минут после введения радиоактивно меченого K-2 (Фиг. 7). Количественные характеристики этого специфического связывания были определены с использованием способа Logan Plot (Фиг. 8). Значение BPnd (оценочного значения потенциала связывания) значительно снижалось при блокировании с помощью нерадиоактивно меченого K-2 (серый столбец на Фиг. 8). Кроме того, было чётко установлено, что значение, представляющее специфическое связывание (чёрный столбец на Фиг. 8), характеризуется высокой степенью корреляции со значением, полученным при биохимическом измерении общего уровня экспрессии рецептора AMPA в тканях (Фиг. 9), и что ПЭТ-визуализация in vivo отражает распределение рецепторов AMPA. Далее, биохимически определенный уровень экспрессии (грубое фракционирование) рецептора AMPA в каждой области мозга проявляет высокую степень корреляции со значениями на ПЭТ-изображении в той же области (Фиг. 10).
[0101]
shРНК способна специфически подавлять экспрессию конкретного белка. shРНК, подавляющая экспрессию рецепторов AMPA (GluA1 - GluA3) была эксперссирована в полосатом теле в левой части мозга с помощью лентивируса, а скремблированная РНК, т.е. нефункциональная shРНК, была экспрессирована в полосатом теле в правой части мозга. В результате этого на ПЭТ-изображении, полученном in vivo, было зафиксировано снижение накопления радиоактивно меченого K-2 в той стороне мозга, где экспрессировалась функциональная shРНК (Фиг. 11). Далее, реальное снижение значений на ПЭТ-изображениях для семи крыс составило около 30% (Фиг. 12). На основании этих результатов можно сделать заключение, что радиоактивно меченое K-2 проявляло высокую степень специфичности в отношении рецептора в живых организмах in vivo.
[0102]
Описание сокращений
ACSF: искусственная цереброспинальная жидкость (ЦСЖ)
AMPA: α-амино-3-гидрокси-5-метил-4-изоксазолпропионовая кислота
DIPEA: диизопропилэтиламин
DCM: дихлорметан
EA: этилацетат
PE: петролейный эфир
PEPA: 2-[2,6-дифтор-4-({2-[(фенилсульфонил)амино]этил}тио)фе-нокси]ацетамид
ПЭТ: позитронно-эмиссионная томография
TEA: тетраэтиламмоний
TMS: тетраметилсилан
1-BCP: 1-(1,3-бензодиоксол-5-илкарбонил)пиперидин
SYM 2206: (±)-4-(4-фминофенил)-1,2-дигидро-1-метил-2-пропил-карбамоил-6,7-метилендиоксифталазин
GYKI: 4-(8-метил-9H-[1,3]диоксоло[4,5-h][2,3]бензодиазепин-5-ил)анилин
CX546: 2,3-дигидро-1,4-бензодиоксин-7-ил-(1-пиперидил)метанон
GABA - γ-аминомасляная кислота
ДМСО - диметилсульфоксид
ГЭПЭС - N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота
ЭДТА - этилендиаминтетрауксусная кислота
ЭГТА - этиленгликольтетрауксусная кислота
Реферат
Изобретение относится к соединению формулы (I), которое способно специфически связываться с рецептором α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (АМРА) и характеризуется чрезвычайно высокой степенью накопления в мозге. В формуле (I) А представляет собой СО, SO или SO; Z представляет собой СО; X представляет собой S; Y представляет собой О; каждый R, Rи Rнезависимо представляют собой водород, С-Салкил, С-Салкенил, С-Салкинил или галоген; Rпредставляет собой С-Салкил, С-Салкенил или С-Салкинил; Rпредставляет собой галоген и n представляет собой целое число от 0 до 4. Изобретение относится также к конкретным соединениям формулы (I), соединениям формулы (I), где один или более атомов являются радиоактивным изотопом атома или атомов, способам их получения, содержащим их композициям, применению указанных соединений для визуализации рецептора АМРА в мозге живых организмов и способам визуализации рецептора АМРА с их использованием. 14 н. и 17 з.п. ф-лы, 12 ил., 7 табл., 6 пр.
Формула











Комментарии