Катализатор для синтеза ненасыщенной карбоновой кислоты путем газофазного окисления ненасыщенного альдегида - RU2678847C2
Код документа: RU2678847C2
Чертежи


Описание
Катализаторы из формованного изделия-носителя и нанесенной на наружную поверхность изделия-носителя и содержащей по меньшей мере элементы Mo, V и Cu оболочки из каталитически активной оксидной массы известны (ср., например, европейские, германские и международные заявки ЕР-А 714700, DE-A 19927624, DE-A 10360057 и WO 2011/134932 А1). Их используют в основном как катализаторы для частичного газофазного окисления акролеина до акриловой кислоты с гетерогенным катализом.
Эти катализаторы, однако, имеют недостатки. При использовании в качестве катализаторов для частичного газофазного окисления акролеина до акриловой кислоты с гетерогенным катализом селективность образования акриловой кислоты удовлетворительна не в полном объеме. В качестве побочной реакции проходит, в частности, переокисление до СО и СО2 (которое в дальнейшем совместно обозначаются как COx).
Достаточно высокой конверсии акролеина при использовании катализаторов нынешнего уровня техники часто достигают только при тех условиях, при которых селективность образования акриловой кислоты неудовлетворительна. Так, при температурах, при которых достигается достаточно высокая конверсия акролеина, часто происходит избыточное окисление и в результате уменьшается селективность формирования акриловой кислоты.
В международной заявке WO 2011/134932 раскрыт оболочечный катализатор, состоящий из изделия-носителя в форме полого цилиндра, а также нанесенной на наружную поверхность изделия-носителя оболочки из обладающей каталитической активностью оксидной массы, равно как и способ синтеза акриловой кислоты путем катализируемого газофазного окисления акролеина на твердом слое катализатора, включающем оболочечный катализатор. В примерах исполнения по прошествии 100 часов эксплуатации достигают значений селективности образования акриловой кислоты вплоть до 97,5%.
Задача состояла в том, чтобы представить катализатор, с применением которого при по-прежнему высокой конверсии акролеина можно снизить переокисление до COx и повысить селективность образования акриловой кислоты.
Эту задачу решают посредством катализатора для получения α,β-ненасыщенной карбоновой кислоты путем газофазного окисления α,β-ненасыщенного альдегида, причем катализатор включает формованное изделие-носитель с нанесенной на него активной массой, отличающейся тем, что степень покрытия активной массой q
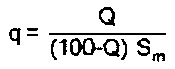
составляет самое большее 0,3 мг/мм2, причем Q - это доля катализатора в активной массе в % масс., a Sm - удельная геометрическая поверхность формованного изделия-носителя в мм2/мг.
Предпочтительно, чтобы степень покрытия активной массой q составляла самое большее 0,26 мг/мм2, предпочтительно самое большее 0,22 мг/мм2. В общем случае степень покрытия активной массой q составляет по меньшей мере 0,10 мг/мм2, предпочтительно по меньшей мере 0,15 мг/мм2.
Предпочтительно, чтобы формованное изделие-носитель обладало заданной геометрической формой.
Предпочтительные формованные изделия-носители - это кольца, шары, таблетки, перфорированные таблетки, трилистники, звездчатые тяжи, звездчатые таблетки, колеса, экструдаты, пилюли, цилиндры и полые цилиндры. Целесообразно, чтобы продольная протяженность (то есть самое длинное прямолинейное соединение двух точек, находящихся на поверхности формованного изделия) формованного изделия-носителя составляла от 1 до 10 мм.
Особо предпочтительные формованные изделия-носители - это полые цилиндры. Предпочтительно, чтобы формованные изделия-носители в форме полых цилиндров обладали высотой от 2 до 5 мм и наружным диаметром от 4 до 8 мм, в то время как половинное значение разности между наружным диаметром и внутренним диаметром составляет от 1 до 2 мм. Половинное значение разности между наружным диаметром и внутренним диаметром соответствует толщине стенки. Особо предпочтительны геометрические параметры с наружным диаметром в 7 мм, высотой в 3 мм и внутренним диаметром в 4 мм.
Предпочтительно, чтобы формованное изделие-носитель состояло из инертного материала. "Инертный" означает, что в условиях газофазного окисления материал формованного изделия-носителя существенно не изменяется и не обладает или в крайнем случае обладает пренебрежимо малой каталитической активностью относительно газофазного окисления по сравнению с нанесенной активной массой. В качестве инертного материала можно использовать, в частности, оксид алюминия, оксид кремния, карбид кремния, диоксид циркония, диоксид тория, силикаты, как то: глину, каолин, стеатит, силикат алюминия и силикат магния и их смеси. Предпочтителен стеатит. Стеатит типа С 220 особо предпочтителен. Крайне предпочтителен стеатит типа С 220 производства фирмы CeramTec.
Предпочтительно, чтобы формованное изделие-носитель обладало отчетливо проявленной шероховатостью поверхности (например, полые цилиндры с накладкой мелкокускового материала). Целесообразно, чтобы поверхность формованного изделия-носителя в форме полого цилиндра была шероховатой, поскольку повышенная шероховатость поверхности обусловливает, как правило, повышенную прочность сцепления оболочки из активной массы и/или массы-предшественника, нанесенной на поверхность обладающего формой полого цилиндра формованного изделия-носителя. Предпочтительно, чтобы шероховатость поверхности RZ изделия-носителя составляла 30-60 мкм, особо предпочтительно 40-50 мкм (определяется согласно DIN 4768, лист 1, посредством „Hommel Tester" поверхностных параметров DIN-ISO производства фирмы Hommelwerke).
Инертный материал может быть пористым или не обладать пористостью. Предпочтительно, чтобы инертный материал по существу не имел пор (общий объем пор составляет менее 1% об. относительно объема изделия-носителя). Геометрическая плотность инертного материала в общем случае находится в пределах от 0,5 до 8,0 г/см3, предпочтительно от 1,0 до 7,0 г/см3, более предпочтительно от 1,5 до 6,0 г/см3, особо предпочтительно от 2,0 до 5,0 г/см3. Геометрическую плотность химически инертного материала рассчитывают путем деления массы формованного изделия-носителя на его геометрический объем.
Геометрический объем можно рассчитать на основании соответствующих данных измерений идеальных геометрических форм, лежащих в основе. Например, геометрический объем полого цилиндра можно рассчитать, положив в основу высоту Н цилиндра, наружный диаметр AD и диаметр внутреннего отверстия ID.
Доля активной массы Q (в % масс.) катализатора - это масса активной массы, в пересчете на сумму масс активной массы и формованного изделия-носителя. Для определеиния массы активной массы можно вычесть из определеной взвешиванием массы катализатора (после тепловой обработки для удаления связывающего агента, см. ниже) известную массу формованного изделия-носителя. Для повышения точности измерений можно определить массу многих катализаторов либо же, соответственно, формованных изделий-носителей и взять среднее значение. Так массу активной массы заданного числа катализаторов можно определить посредством определения общей массы катализаторов за вычетом массы формованных изделий-носителей, которая получается по результатам умножения массы формованного изделия-носителя на количество формованных изделий-носителей. Кроме того, определение доли активной массы Q возможно путем смыва активной массы с формованного изделия-носителя. Для этого катализатор с покрытием, например, несколько раз вываривают в водном растворе аммиака, а полученную жидкость декантируют. Затем можно высушить остающийся носитель. Долю активной массы получают как разность массы катализатора (определенной до смыва активной массы) и массы носителя (определенную после смыва активной массы и сушки), в пересчете на массу катализатора.
Соответственно, доля массы носителя катализатора в % масс. равна (100-Q).
Удельная геометрическая площадь поверхности формованного изделия-носителя Sm - это отнесенная на массу формованного изделия-носителя геометрическая площадь поверхности формованного изделия-носителя.
Геометрическую площадь можно рассчитать на основании соответствующих данных измерений идеальных геометрических форм, лежащих в основе. Геометрическая площадь поверхности - это идеализированная величина, она не учитывает увеличение площади поверхности, обусловленное пористостью или шероховатостью поверхности формованных изделий.
В случае шаровидного формованного изделия-носителя геометрическая площадь поверхности составляет
4πr2
причем r означает радиус шарообразного формованного изделия-носителя. В случае формованного изделия-носителя в форме полого цилиндра геометрическая площадь поверхности составляет
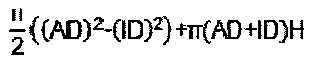
причем Н означает высоту, AD наружный диаметр, a ID внутренний диаметр формованного изделия-носителя в форме полого цилиндра.
Предпочтительно, чтобы средняя толщина активной массы, нанесенной на формованное изделие-носитель составляла от 50 до 400 мкм, предпочтительно от 75 до 350 мкм, особо предпочтительно от 100 до 300 мкм, а крайне предпочтительно от 100 до 200 мкм.
Предпочтительно, чтобы толщина нанесенной на формованное изделие-носитель активной массы была по возможности однородной. Толщина нанесенной активной массы также по возможности однорода при сравнении различных формованных изделий-носителей.
Активные массы для синтеза α,β-ненасыщенной карбоновой кислоты путем газофазного окисления α,β-ненасыщенного альдегида сами по себе известны. Можно, например, использовать обладающие каталитической активностью мультиэлементные оксидные массы, которые содержат элементы Mo и V, причем молярная доля элемента Мо в общем количестве всех отличных от кислорода элементов каталитически активной мультиэлементной оксидной массы составляет от 20% мол. до 80% мол., молярное соотношение между содержащимся в каталитически активной мультиэлементной оксидной массе Mo и содержащимся в каталитически активной мультиэлементной оксидной массе V, Mo/V, составляет от 15:1 до 1:1. Кроме того, предпочтительно, чтобы мультиэлементный оксид содержал по меньшей мере один из элементов Nb и W; соответствующее молярное соотношение Mo/(общее количество W и Nb) предпочтительно составляет от 80:1 до 1:4. Также такие мультиэлементные оксидные массы часто содержат Cu в соответствующем молярном соотношении Mo/Cu от 30:1 до 1:3.
Вышеуказанные мультиэлементные оксидные массы, помимо элементов Mo, V и при необходимости Nb и/или W или Cu, могут дополнительно содержать, например, элементы Та, Cr, Се, Ni, Со, Fe, Mn, Zn, Sb, Bi, щелочные металлы (Li, Na, K, Rb, Cs), H, щелочно-земельные металлы (Mg, Са, Sr, Ва), Si, Al, Ti и Zr. Разумеется, мультиэлементная оксидная активная масса может, однако, состоять и только из элдементов Mo, V, О, а также Cu и при необходимости W и/или Nb. Их можно использовать, в частности, в качестве активных масс для катализаторов для частичного газофазного окисления акролеина до акриловой кислоты с гетерогенным катализом.
Массы, в чрезвычайной степени пригодные для применения в качестве активных масс для катализаторов для частичного газофазного окисления акролеина до акриловой кислоты с гетерогенным катализом, включают мультиэлементные оксидные массы следующей общей формулы (I)
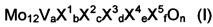
где
X1 означает W, Nb, Та, Cr и/или Ce,
X2 означает Cu, Ni, Co, Fe, Mn и/или Zn,
X3 означает Sb и/или Bi,
X4 означает один или несколько щелочных и/или щелочно-земельных металлов и/или N,
X5 означает Si, Al, Ti и/или Zr,
a означает число в пределах от 1 до 6,
b означает число в пределах от 0,2 до 4,
c означает число в пределах от 0 до 18, предпочтительно от 0,5 до 18,
d означает число в пределах от 0 до 40,
e означает число в пределах от 0 до 4,
f означает число в пределах от 0 до 40, и
n означает стехиометрический коэффициент элемента кислорода, который определяется стехиометрическими коэффициентами отличных от кислорода элементов, а также их валентностью в (I).
Переменные предпочтительно выбирать в заданных диапазонах с таким расчетом, чтобы молярная доля элемента Mo в общей массе всех отличных от кислорода элементов мультиэлементной оксидной массы (I) составляла от 20% мол. до 80% мол.
Предпочтительно, чтобы мультиэлементная оксидная масса соответствовала общей формуле (II)
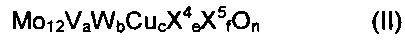
где
X4 означает один или несколько щелочных и/или щелочно-земельных металлов,
X5 означает один или несколько элементов из группы Si, Al, Ti и Zr,
а означает число в пределах от 2 до 4, целесообразно - число в пределах от 2,5 до 3,5,
b означает число в пределах от 0 до 3, целесообразно - число в пределах от 0,2 до 3, предпочтительно число в пределах от 0,5 до 2, особо предпочтительно - число в пределах от 0,75 до 1,5,
с означает число в пределах от 0,5 до 3, целесообразно - число в пределах от 0,7 до 2,7, предпочтительно число в пределах от 0,9 до 2,4, особо предпочтительно - число в пределах от 1 до 1,5,
e означает число в пределах от 0 до 4, целесообразно - число в пределах от 0 до 2, предпочтительно означает число в пределах от 0 до 1, особо предпочтительно - число в пределах от 0 до 0,2,
f означает число в пределах от 0 до 40, целесообразно - число в пределах от 0 до 15, предпочтительно означает число в пределах от 0 до 8, особо предпочтительно 0, и
n означает стехиометрический коэффициент элемента кислорода, который определяется стехиометрическими коэффициентами отличных от кислорода элементов, а также их валентностью в (II).
Элементы X4 и X5 не обязательно являются составной частью активной массы общей формулы (II). В пределах активной массы они в общем случае действуют как инертные разбавители. Благодаря введению их в активную массу можно довести до желательного уровня удельную объемную активность катализатора.
В одной из форм исполнения активная масса может присутствовать в форме тонкодисперсной смеси мультиэлементной оксидной массы, содержащей элементы Mo и V, например, формулы I или II, с источником оксида молибдена, как это изложено в германском патенте DE 102007010422. Источник оксидов молибдена надлежащим образом выбирают из оксидов молибдена и соединений молибдена, из которых под воздействием повышенной температуры и молекулярного кислорода образуется оксид молибдена. К ним относятся оксиды молибдена, например, MoO3, Mo18O52, Мо8О23 и Mo4O11, или такие соединения как молибдат аммония [(NH4)2MoO4], а также полимолибдаты аммония, как, например, аммония гептамолибдат тетрагидрат [(NH4)6Mo7O24 4Н2О]. Альтернативный пример - это гидрат оксида молибдена (MoO3 xH2O). MoO3 - это предпочтительный источник оксида молибдена.
Согласно изобретению целесообразно, чтобы зернистость (диаметр частиц или соответсвенно распределение частиц по размеру) тонкодисперсного источника оксида молибдена была идентична таковой тонкодисперсного мультиэлементного оксида, содержащего элементы Мо и V (это дает возможность особо гомогенного смешивания субстанции с тонкодисперсным мультиэлементным оксидом). В особенности это справедливо тогда, когда тонкодисперсный источник оксида молибдена представляет собой оксид молибдена (в особенности MoO3).
Параллельное применение источника оксида молибдена может профилактически противодействовать деактивации катализатора в процесса частичного газофазного окисления акролеина до акриловой кислоты с гетерогенным катализом или соответственно отсрочить начало деактивации.
В общем случае катализатор пористый. Предпочтительно, чтобы катализатор характеризовался некоторым конкретным распределением пор различного среднего диаметра. Объемная доля pvol макропор катализатора предпочтительно составляет по меньшей мере 0,35, причем pvol определяется как
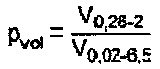
где
V0,26-2 означает объем пор со средними диаметрами в пределах от 0,26 до 2 мкм, а
V0,02-6,5 означает объем пор со средними диаметрами в пределах от 0,02 до 6,5 мкм.
Объем пор со средним диаметром в нанометровом и микрометровом диапазоне можно определить методом ртутной порозиметрии (например, согласно DIN №66133). По отношению к большинству твердых тел ртуть ведет себя как не смачивающая жидкость. Поэтому ртуть не абсорбируется пористым материалом спонтанно, а проникает в поры образца твердого тела только под внешним давлением. Значение (высота) этого давления зависит от размера пор. Это поведение используют в методе ртутной порозиметрии, чтобы по измеряемой волюметрическим методом интрузии при воздействующем извне давлении зарегистрировать диаметр пор.
При этом пористую систему (подлежащий исследованию образец), предварительно дегазированную (чтобы дегазировать присутствующую при необходимости в пористой структуре жидкость), погружают в ртутную ванну, давление которой можно изменять.
Поскольку ртуть не смачивает материал образца, ртуть необходимо вдавливать в поры образца (при каждом конкретном давлении ожидают наступления равновесия). Проникновение ртути в поры с большей площадью сечения происходит при сравнительно низких значениях давления, в то время как для проникновения ртути в более мелкие поры требуется сравнительно более высокое давление. Исходя из предположения о наличии пор в форме круглого цилиндра, можно с помощью уравнения Уошберна установить соотношение между внешним давлением, которое необходимо, чтобы вдавить жидкую ртуть (интрудировать; интрузия ртути) в поры соответствующего диаметра, противодействуя поверхностному натяжению ртути, и указанным диаметром. Диапазон давлений, используемый при ртутной порозиметрии, коррелирует с шириной (полосы) измеренного диаметра пор.
Затем на основании определенных экспериментальным путем кривых интрузии ртути при 25°С можно через измеренную ширину (полосы) диаметра пор с помощью расчетов определить распределение пор по диаметру, общую площадь внутренней поверхности пор и общий внутренний объем пор (суммарный объем интрузии, суммарный объем пор) (ср. диссертацию на соискание ученой степени „Eigenschaften und Einsatzmöglichkeiten von Aerogelfenstern im Vergleich mit konventionellen sowie evakuierten Fenstern" Georges Reber (1991), на философско-естественнонаучном факультете Университета Базеля). Описанный ниже измерительный прибор Auto Pore IV 9520 производства фирмы Micromeritics включает подходящие для этих целей стандартные расчетные программы.
В общем случае катализатор согласно изобретению получают путем нанесения порошкообразной активной массы на формованное изделие-носитель, предпочтительно согласно описанному ниже способу получения.
Полученение порошкообразной активной массы можно осуществлять различным образом. В одной форме исполнения активную массу изготавливают, создавая тонкодисперсную гомогенную смесь из источников элементарных составляющих активной массы, кальцинируют эту смесь при температурах от 350 до 600°С, а затем переводят ее в порошкообразное состояние.
Предпочтительные источники элементарных составляющих активной массы - это оксиды содержащихся в активной массе металлов. В качестве источников элементарных составляющих пригодными являются соединения, которые путем нагревания, по меньшей мере в присутствии кислорода, можно преобразовывать в оксиды, в частности, галогениды, нитраты, формиаты, оксалаты, цитраты, ацетаты, карбонаты, аминные комплексы, аммониевые соли и/или гидроксиды содержащихся в активной массе металлов.
Предпочтительно создавать гомогенную сухую смесь путем гомгенного смешивания источников. Гомогенное смешивание можно осуществлять в сухом или в мокром виде. Если его осуществляют в сухом виде, то целесообразно применять источники в виде тонкодисперсных порошков. Особо гомогенные сухие смеси при смешивании получают тогда, когда имеющиеся источники присутствуют исключительно в растворенном виде. Поэтому предпочтительно проводить гомогенное смешивание источников в мокром виде. Предпочтительно смешивать друг с другом источники в форме растворов и/или суспензий, а получающуюся при этом мокрую смесь затем сушить с образованием гомогенной сухой смеси. В качестве растворителя и/или среды суспендирования предпочтительно применять воду или водный раствор. Сушку мокрой смеси предпочтительно осуществлять путем распылительной сушки при температуре на выходе от 100 до 150°С. Сушащий поток газа предпочтительно представляет собой воздух или молекулярный азот.
Перед кальцинацией можно провести подготовку массы сухой смеси (например, полученной распылительной сушкой сухой смеси) путем перемешивания. Под понятием "перемешивание" подразумевают сухое перемешивание, замес и перемешивание мешалкой, при необходимости - с добавлением жидкости. Благодаря перемешиванию получают гомогенизированную массу с более узким распределением частиц по размеру.
Особо целесообразно проводить операцию перемешивания после добавления жидкости, например, воды, уксусной кислоты и т.п., в виде замеса, причем получают пластическую или пластифицированную массу. Благодаря действующим при этом усилиям сдвига происходит измельчение агломератов. Пластическая масса пригодна к экструзии в тяжи, из нее получаются стабильные полоски, которые можно высушить. Высушенные полоски целесообразно в числе прочего подвергать кальцинации во вращающейся трубе.
Кальцинацию можно проводить как в инертном газе, так и в окислительной атмосфере, а также и в восстановительной атмосфере. Предпочтительно проводить кальцинацию в окислительной атмосфере. В качестве инертного газа можно использовать, в частности, азот, водяной пар, благородные газы и их смеси. Окислительная атмосфера предпочтительно содержит кислород, в частности, воздух. Восстановительная атмосфера предпочтительно содержит Н2, NH3, CO, метан и/или акролеин. Каталитическая активность активной массы в общем случае демонстрирует оптимум в зависимости от содержания кислорода в атмосфере кальцинации. Предпочтительно, чтобы содержание кислорода в атмосфере кальцинации составляло от 0,5 до 10% об., особо предпочтительно от 1 до 5% об. Содержание кислорода выше и ниже вышеупомянутых границ обычно уменьшает итоговую каталитическую активность. Продолжительность кальцинирования может составлять от нескольких минут от нескольких часов, и с ростом температуры кальцинации она обычно сокращается. Хорошо пригодный процесс кальцинации описан, например, в международной заявке WO 95/11081.
При кальцинации сухой смеси получают активную массу. Преобразование в порошкообразное состояние предпочтительно осуществлять путем размола.
В альтернативном способе изготовления катализатора на поверхность формованного изделия-носителя сначала наносят тонкодисперсную массу-предшественник и проводят кальцинацию массы-предшественника с образованием активной массы на поверхности формованного изделия-носителя. Тонкодисперсная масса-предшественник предпочтительно содержит источники элементарных составляющих активной массы. Активная масса - это предпочтительно активная масса общей формулы (I) или (II).
При реализации способа согласно изобретению для изготовления катализатора формованное изделие-носитель покрывают активной массой, для чего в емкости перемешивают множество формованных изделий-носителей, порошкообразную активную массу и жидкий связующий агент, не насыщая порошкообразную активную массу жидким связующим агентом, причем длительность процесса нанесения покрытия составляет менее 30 минут. Насыщения порошкообразной активной массы жидким связующим агентом избегают, выбирая для этого отношение количества жидкого связующего агента к количеству порошкообразной активной массы так, чтобы количество связующего агента оставалось ниже способности порошкообразной активной массы поглощать жидкость.
Способность порошков поглощать жидкость можно определить, например, размешивая порошок в мешалке и подавая на перемешиваемый порошок жидкость и измеряя вращающий момент на двигателе мешалки в зависимости от времени. Из количества жидкости, которое нанесено на порошок до максимума вращающего момента, можно рассчитать поглощающую способность порошка по жидкости.
Предпочтительно, чтобы доля частиц с максимальным продольным размером более 50 мкм в порошкообразной активной массе была менее 1%.
Под понятием связующего агента подразумевают вещества, которые постоянно или временно улучшают сцепление частиц порошка активной массы друг с другом и/или с материалом-носителем. Предпочтительно, чтобы при последующей сушке связующий агент в основном испарялся или сублимировался. При реализации способа согласно изобретению в качестве связующих агентов можно применять, например, полиолы, как то: этиленгликоль, пропиленгликоль, бутиленгликоли, глицерин или амиды, например, формамид, N,N-диметилформамид, N,N-диэтилформамид, N,N-дибутилформамид, ацетамид, пирролидон или N-метилпирролидон. Жидкий связующий агент предпочтительно выбирать из воды, глицерина и растворов глицерина в воде. Предпочтительный жидкий связующий агент - это раствор глицерина в воде, который содержит от 20 до 99% масс. воды. Особо предпочтительный жидкий связующий агент - это раствор глицерина в воде, который содержит 75% масс. воды.
Предпочтительно сначала помещать в емкость формованные изделия-носители и отдельно друг от друга добавлять в емкость на протяжении нанесения покрытия порошкообразную активную массу и жидкий связующий агент. Таким образом контакт порошкообразной активной массы и жидкого связующего агента друг с другом создают только в емкости. Предпочтительно впервые соединять порошкообразную активную массу и жидкий связующий агент только на поверхности предварительно помещенных в емкость формованных изделий-носителей. Этого удается достичь, распыляя жидкий связующий агент в емкость и вводя порошкообразную активную массу в область емкости, находящуюся за пределами конуса распыления жидкого связующего агента. Так избегают локальной перегрузки частиц порошка жидкостью. Порошкообразную активную массу и жидкий связующий агент можно вводить в емкость на протяжении обработки, например, путем непрерывного добавления или путем разделенного во времени добавления по частям.
Перемешивание предпочтительно осуществлять путем непрерывного движения емкости. Предпочтительно, чтобы движение представляло собой вращательное движение.
Для реализации описанного выше способа изготовления катализатора можно использовать прежде всего принцип работы, описанный в германской заявке DE-A 2909671 (ср. также европейскую заявку ЕР-А 714700 и германскую заявку DE-A 102005010645), в каждом случае - с применением желательного жидкого связующего агента.
Т.е., подлежащие покрытию формованные изделия-носители, предпочтительно - формованные изделия-носители в форме полых цилиндров, помещают во вращающуюся емкость (например, во вращающийся плоский бак, или в дражировочный котел, или дражировочный барабан), предпочтительно наклонный (угол наклона, как правило, составляет от 30 до 90°). Вращающиеся емкости, удобные для этой цели - это, в частности, Hi-Coater типа HCF-100, производства фирмы Freund Industrial Co., Ltd, (Токио, Япония), а также Hi-Coater типа LH 100, производства фирмы Gebrüder Lödige Maschinenbau GmbH, Германия, Падерборн.
Вращающийся бак проводит формованные изделия-носители, предпочтительно - формованные изделия-носители в форме полых цилиндров, под двумя дозировочными устройствами, расположенными на целесообразном расстоянии друг за другом. Целесообразно, чтобы первое из этих дозировочных устройств представляло собой сопло, с помощью которого катающиеся по вращающемуся баку (Hi-Coater) формованные изделия-носители опрыскивают и контролируемым образом увлажняют - жидким связующим агентом. С точки зрения техники применения целесообразно, чтобы второе дозирующее устройство было расположено за пределами конуса распыления жидкого связующего агента и предназначено для подачи порошкообразной активной массы (например, по вибролотку). Формованные изделия-носители принимают активную массу, поскольку активная масса благодаря движению качения по поверхности формованных изделий-носителей уплотняется с образованием сплошной оболочки.
При необходимости формованное изделие-носитель (предпочтительно в форме полого цилиндра), прошедшее такое первоначальное покрытие, снова проходит при дальнейшем вращении под распылительным соплом, подвергается контролируемому увлажнению (возможно, другим жидким связующим агентом), чтобы при дальнейшем движении иметь возможность получить еще один слой тонкодисперсной оксидной активной массы (возможно, другой) и/или тонкодисперсной массы предшественника, и т.д. (промежуточная сушка, как правило, не требуется). Согласно техническому решению из европейской заявки ЕР-А 714700 или германской заявки DE-A 102005010645, использованный жидкий связущий агент, можно в заключение по меньшей мере частично удалить, например, путем подачи тепла, например, воздействуя горячими газами, как то: N2 или воздухом (их подачу и отвод осуществляют через расположенные на определенном расстоянии друг от друга сетевидные настенные элементы вращающегося плоского бака, или дражировочного котла, или дражировочного барабана (в общем, вращающейся емкости)).
Для описанного выше варианта исполнения способа нанесения покрытия существенно проводить увлажнение подлежащей покрытию поверхности изделия-носителя контролируемым образом. Говоря кратко, это означает, что поверхность носителя целесообразно увлажнять так, чтобы на ней, несмотря на наличие адсорбированного жидкого связующего агента, нельзя было визуально обнаружить его наличие как такового. Если поверхность формованного изделия-носителя слишком увлажнена, тонкодисперсная активная масса и/или масса-предшественник вместо того, чтобы притягиваться к поверхности, формирует отдельные агломераты. Подробные данные приведены в германском патенте DE-A 2909671, в европейской заявке ЕР-А 714700, а также в германском патенте DE-A 102005010645. Преимущество описанного способа работы состоит в том, что возможно удаление использованного жидкого связующего агента сравнительно контролируемым образом, например, путем испарения и/или сублимации. В простейшем случае это можно осуществить, как уже упомянуто, воздействуя горячими газами, имеющими соответствующую температуру (нередко, 50-150°С). Такое воздействие горячих газов в общем случае обеспечивает предварительную сушку.
Удаление связующего агента можно осуществлять в сушильном устройстве произвольного рода (например, в ленточной сушилке) и/или лишь при нахождении в твердом слое катализатора в реакторе в виде пучка труб, как это рекомендовано, например, в германской заявке DE-A 102005010645. Предпочтительно получать катализатор согласно изобретению, удаляя жидкий связующий агент из формованных изделий-носителей, на которые нанесено покрытие, путем сушки при температуре в пределах от 150 до 400°С, предпочтительно от 250 до 350°С. Сушку предпочтительно проводят в потоке воздуха. Предпочтительно, чтобы продолжительность сушки составляла от 0,5 до 8 ч, предпочтительно от 1 до 4 ч.
Также объектом изобретения является способ получения α,β-ненасыщенной карбоновой кислоты посредством газофазного окисления α,β-ненасыщенного альдегида молекулярным кислородом в твердом слое катализатора, который включает засыпку катализатора согласно изобретению. Предпочтительно создавать контакт молекулярного кислорода и α,β-ненасыщенного альдегида с твердым слоем катализатора, для чего молекулярный кислород и α,β-ненасыщенный альдегид проводят через твердый слой катализатора. Предпочтительно проводить реакционный газ, который содержит молекулярный кислород и α,β-ненасыщенный альдегид, через твердый слой катализатора и преобразовывать реакционный газ в газообразный продукт.
Предпочтительно выбирать α,β-ненасыщенный альдегид из содержащих от 3 до 6 (то есть 3, 4, 5 или 6) атомов кислорода α,β-ненасыщенных альдегидов, в частности из акролеина и метакролеина. Особо предпочтительно, чтобы α,β-ненасыщенный альдегид представлял собой акролеин. Способ особо удобно применять для получения α,β-ненасыщенных карбоновых кислот, в частности, для окисления акролеина до акриловой кислоты и метакролеина до метакриловой кислоты. Предпочтительно, чтобы речь шла о способе получения акриловой кислоты путем газофазного окисления акролеина.
Молекулярный кислород предпочтительно подавать в процесс в виде воздуха.
Доля содержащегося в реакционном газе α,β-альдегида в общем случае составляет от 3 до 15% об., целесообразно от 4 до 10% об., предпочтительно от 5 до 8% об., в каждом случае в пересчете на реакционный газ.
Кроме того, предпочтительно, чтобы реакционный газ содержал по меньшей мере один отличный от водяного пара инертный разбавляющий газ. Под этим понятием подразумевают такие газы, которые в процессе газофазного окисления по меньшей мере на 95% мол., предпочтительно по меньшей мере на 98% мол., остаются химически неизменными. Примерами инертных разбавляющих газов являются N2, CO2 и благородные газы, например, Ar. В качестве инертного разбавляющего газа предпочтительно применять молекулярный азот. Инертный разбавляющий газ может содержать по меньшей мере 20% об., предпочтительно по меньшей мере 40% об., более предпочтительно - по меньшей мере 60% об., особо предпочтительно по меньшей мере 80% об., крайне предпочтительно по меньшей мере 95% об., молекулярного азота.
Реакционный газ может кроме того содержать водяной пар.
Реакционный газ может кроме того содержать циркуляционный газ. Под циркуляционным газом подразумевают остаточный газ, который остается, когда от газообразного продукта газофазного окисления по существу селективно отделяют α,β-ненасыщенную карбоновую кислоту.
Предпочтительно, чтобы способ согласно изобретению для получения α,β-ненасыщенной карбоновой кислоты представлял собой вторую ступень двухступенчатого газофазного окисления алкена до α,β-ненасыщенной карбоновой кислоты. В рамках такого двухступенчатого газофазного окисления газообразный продукт первого этапа предпочтительно подают на второй этап. Перед подачей на второй этап газообразный продукт первого этапа можно, например, охладить и/или добавить кислород (вторичное добавление кислорода; предпочтительно добавление воздуха). Циркуляционный газ предпочтительно подавать на первый из двух этапов.
В реакционном газе молярное соотношение "O2 : α,β-ненасыщенный альдегид" предпочтительно составляет от 1 до 3, предпочтительно от 1 до 2, особо предпочтительно от 1 до 1,5.
Предпочтительно, чтобы реакционный газ содержал α,β-ненасыщенный альдегид : кислород : водяной пар : отличный от водяного пара разбавляющий газ в объемном соотношении 1:(1-3):(0-20):(3-30), предпочтительно 1:(1-3):(0,5-10):(7-10).
Предпочтительно, чтобы нагрузка засыпки α,β-ненасыщенным альдегидом составляла самое большее 600 Н⋅л/(лч), предпочтительно - самое большее 300 Н⋅л/(лч), особо предпочтительно самое большее 250 Н⋅л/(лч), наиболее предпочтительно самое большее 200 Н⋅л/(лч). Предпочтительно, чтобы нагрузка засыпки α,β-ненасыщенным альдегидом составляла по меньшей мере 30 Н⋅л/(лч), предпочтительно - по меньшей мере 70 Н⋅л/(лч), особо предпочтительно по меньшей мере 90 Н⋅л/(лч), наиболее предпочтительно по меньшей мере 120 Н⋅л/(лч). Под выраженной в Н⋅л/(лч) нагрузкой засыпки α,β-ненасыщенным альдегидом подразумевают количество α,β-ненасыщенного альдегида в нормолитрах, которое как составную часть реакционного газа проводят через твердый слой катализатора в час в расчете на литр засыпки. Нормолитр (Н⋅л) - это объем в литрах, который занимало бы при нормальных условиях, то есть при 25°С и 1 бар, количество моль идеального газа, соответствующее количеству моль α,β-ненасыщенного альдегида.
В общем случае суммарное давление в реакционном газе составляет от 0,5 до 100 бар, предпочтительно от 1 до 5 бар, в частности от 1 до 3 бар. Все данные о давлении на этом этапе подразумевают абсолютные значения давления.
Предпочтительно реализовывать способ получения α,β-ненасыщенной карбоновой кислоты в кожухотрубном реакторе, реакционные трубы которого заполнены твердым слоем катализатора.
Засыпка может состоять, например, исключительно из катализаторов согласно изобретению. В засыпке могут также присутствовать в основном гомогенные смеси катализаторов согласно изобретению и формованных изделий-разбавителей, которые демонстрируют по существу инертное поведение относительно газофазного окисления. В качестве материалов для формованных изделий-разбавителей можно использовать, например, пористые или не имеющие пор оксиды алюминия, диоксид кремния, диоксид циркония, карбид кремния, силикаты, как, например, силикат магния или алюмосиликат и/или стеатит (например, типа С220 производства фирмы CeramTec, Германия).
Геометрические параметры формованных изделий разбавления могут, в принципе, быть любыми. То есть, это могут быть, например, кольца, шары, таблетки, перфорированные таблетки, трилистники, звездчатые тяжи, звездчатые таблетки, колеса, экструдаты, пилюли, цилиндры и полые цилиндры.
Кожухотрубный реактор предпочтительно представляет собой двухзонный кожухотрубный реактор. Предпочтительный двухзонный кожухотрубный реактор раскрыт в германском патенте DE-C 2830765. Кроме того, можно использовать двухзонные кожухотрубные реакторы, представленные в германском патенте DE-C 2513405, заявке США US-A 3,147,084, германской заявке DE-A 2201528, европейской заявке ЕР-А 383224 и германской заявке DE-A 2903218.
Вокруг реакционных труб в двухзонном кожухотрубном реакторе обеспечивают циркуляцию предпочтительно двух по существу отделенных друг от друга в пространстве теплоносителей. Теплоносители предпочтительно представляют собой расплавы солей. Температуру теплоносителя на входе предпочтительно устанавливают на уровне от 230 до 300°С, предпочтительно от 240 до 290°С, особо предпочтительно от 250 до 285°С. Циркуляцию теплоносителей по данной конкретной зоне поддержания температуры можно обеспечивать равнонаправленным потоком или противотоком относительно реакционной газовой смеси. Предпочтительно, чтобы траектория проведения теплоносителя в пределах зоны поддержания температуры имела вид меандров. Скорость течения теплоносителя в пределах данной зоны поддержания температуры предпочтительно выбирают так, чтобы температура агента теплообмена от места входа в температурную зону до места выхода из температурной зоны возрастала на 0-15°С, часто на 1-10°С, или на 2-8°С, или на 3-6°С.
В предпочтительной форме исполнения твердый слой катализатора включает по меньшей мере две следующие друг за другом реакционные зоны, причем по меньшей мере в ближайшей ко входу в реактор реакционной зоне засыпка включает катализатор согласно изобретению.
В другой предпочтительной форме исполнения твердый слой катализатора включает по меньшей мере две следующие друг за другом реакционные зоны, причем по меньшей мере в той реакционной зоне, в которой развивается наивысшая локальная температура, засыпка включает катализатор согласно изобретению.
Отдельные реакционные зоны отличаются друг от друга по меньшей мере по одному свойству, которое можно выбрать из: содержания инертных формованных изделий разбавления, формы катализаторов, степенью плотности упаковки катализаторов, доли активной массы в катализаторах и химического состава активной массы.
Вследствие различных свойств удельная объемная активность катализатора одной реакционной зоны может отличаться от удельной объемной активности катализатора другой реакционной зоны. Предпочтительно, чтобы удельная объемная активность катализатора в направлении от входа в реактор до выхода из реактора возрастала от одной реакционной зоны к следующей.
Удельную (относительную) объемную активность катализатора можно рассчитать как скорость реакции, отнесенную к объему катализаторной засыпки, при постоянных прочих условиях.
Удельную объемную активность катализатора можно варьировать путем разбавления катализатора формованными изделиями-разбавителями. В качестве альтернативы или дополнения можно регулировать удельную объемную активность катализатора, варьируя долю активной массы.
Предпочтительно, чтобы в ближайшей ко входу в реактор реакционной зоне пространственная плотность активной массы, то есть количество активной массы в г на объем свободного пространства в литрах, была ниже, чем в ближайшей к выходу из реактора реакционной зоне.
Начиная с некоторого определенного момента эксплуатации, рост продолжительности эксплуатации сопровождается все большим ухудшением качества катализаторной засыпки. В предпочтительной форме исполнения для восстановления качества засыпки изымают не всю использованную засыпку, а только часть засыпки, в которой отмечается максимальная локальная температура, и заменяют свежей засыпкой. Например, засыпку той реакционной зоны, в которой отмечается наивысшая локальная температура, заменяют свежей засыпкой и сохраняют засыпку в реакционных зонах, расположенных ниже в направлении потока реакционного газа.
Кожухотрубный реактор обычно дополнительно располагает термотрубами для определения температуры газа в слое катализатора. Внутренний диаметр термотруб и диаметр расположенной внутри приемной втулки для термоэлемента целесообразно выбирать так, чтобы отношение объема, в котором вырабатывается теплота реакции, к площади теплоотводящей поверхности у термотруб и реакционных труб было одинаково или лишь незначительно различалось.
Потеря давления у реакционных труб и термотруб должна быть (в расчете на одинаковые GHSV) одинаковой. Компенсировать падение давления в случае термотрубы можно, например, путем добавления к катализатору дробленого катализатора. Целесообразно осуществлять эту компенсацию равномерно по всей длине термотрубы. В остальном заполнение термотруб можно осуществлять так, как это описано в европейской заявке ЕР-А 873783.
Температуры, измеренные в термотрубах, позволяют определить наивысшую локальную температуру твердого слоя катализатора и ее положение в твердом слое катализатора.
Фигуры
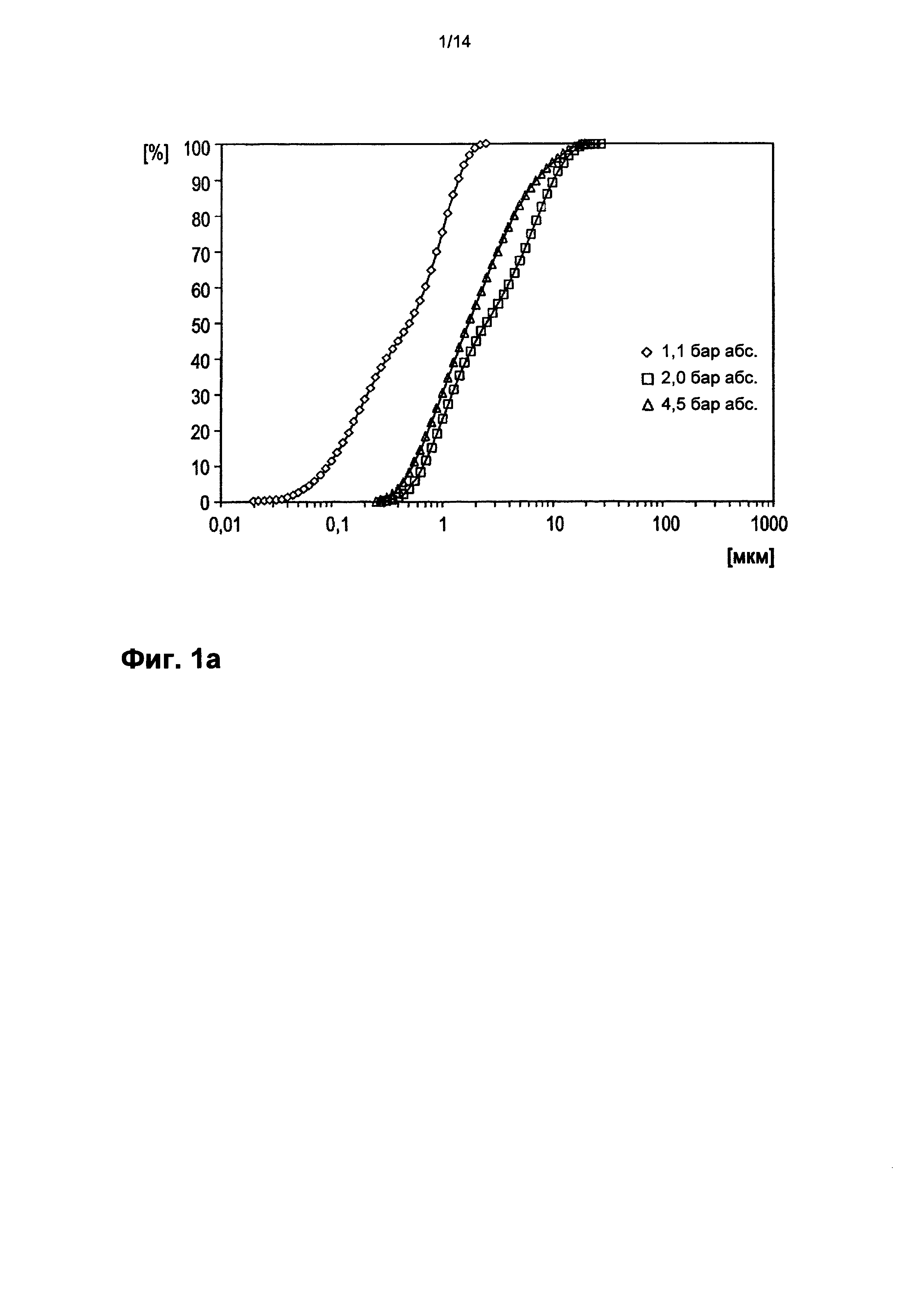
На фигуре 1а показано кумулятивное распределение частиц тонкодисперсного порошка Р по размеру. При этом по оси абсцисс в логарифмическом масштабе приведен диаметр частиц в мкм. Значение ординаты на кривой распределения, соответствующее определенному диаметру частиц, означает процентную долю суммарного объема частиц, состоящую из частиц данного конкретного или меньшего диаметра частиц.
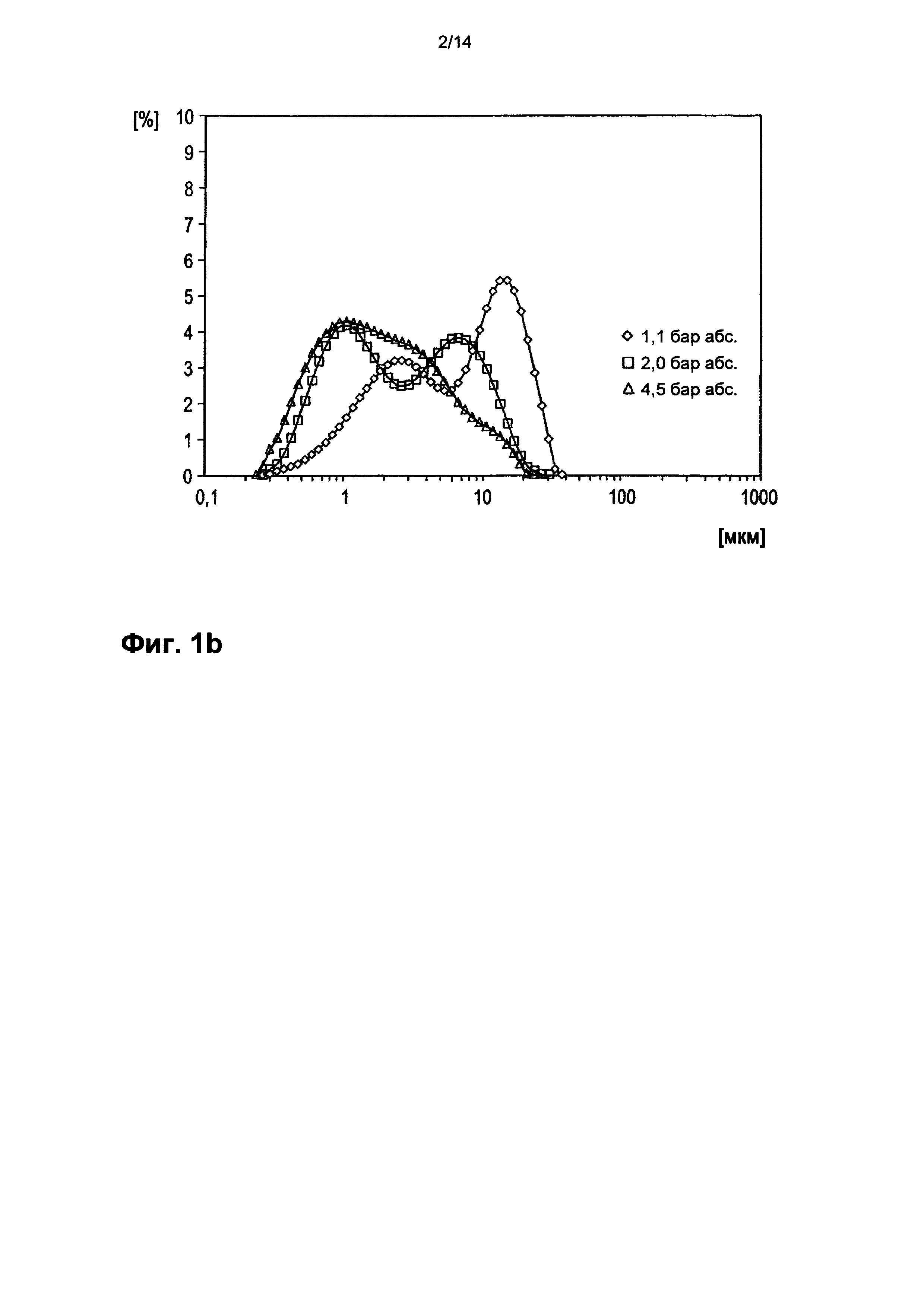
На фигуре 1b показано дифференциальное распределение частиц тонкодисперсного порошка Р по размеру. При этом по оси абсцисс в логарифмическом масштабе приведен диаметр частиц в мкм. Значение ординаты на кривой распределения, соответствующее определенному диаметру частиц, означает процентную долю суммарного объема частиц, состоящую из частиц данного конкретного диаметра частиц.
Фигура 2 демонстрирует дифрактограмму тонкодирперсного порошка Р.
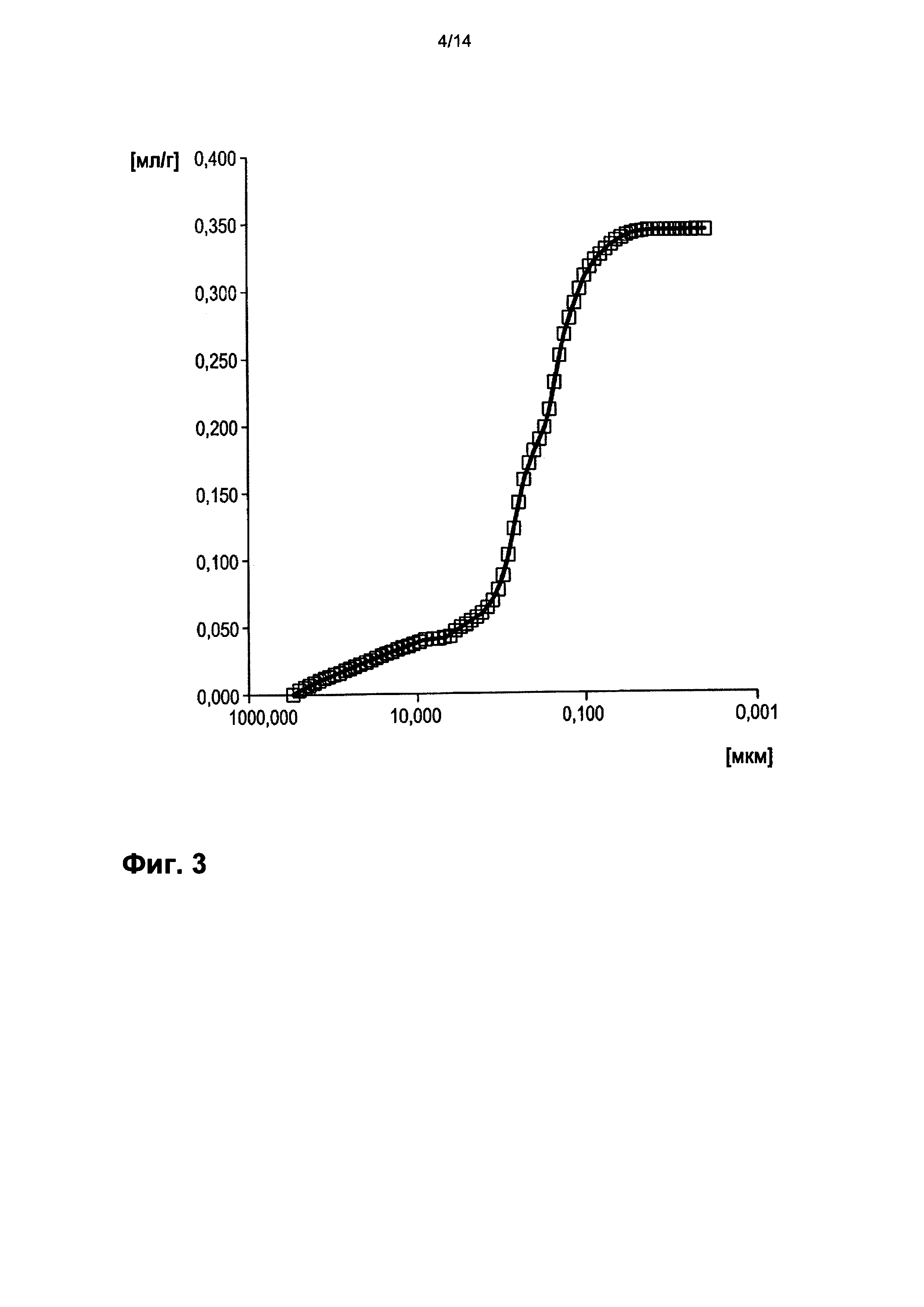
На фигуре 3 показано распределение пор оболочки активной массы С1 по размеру.
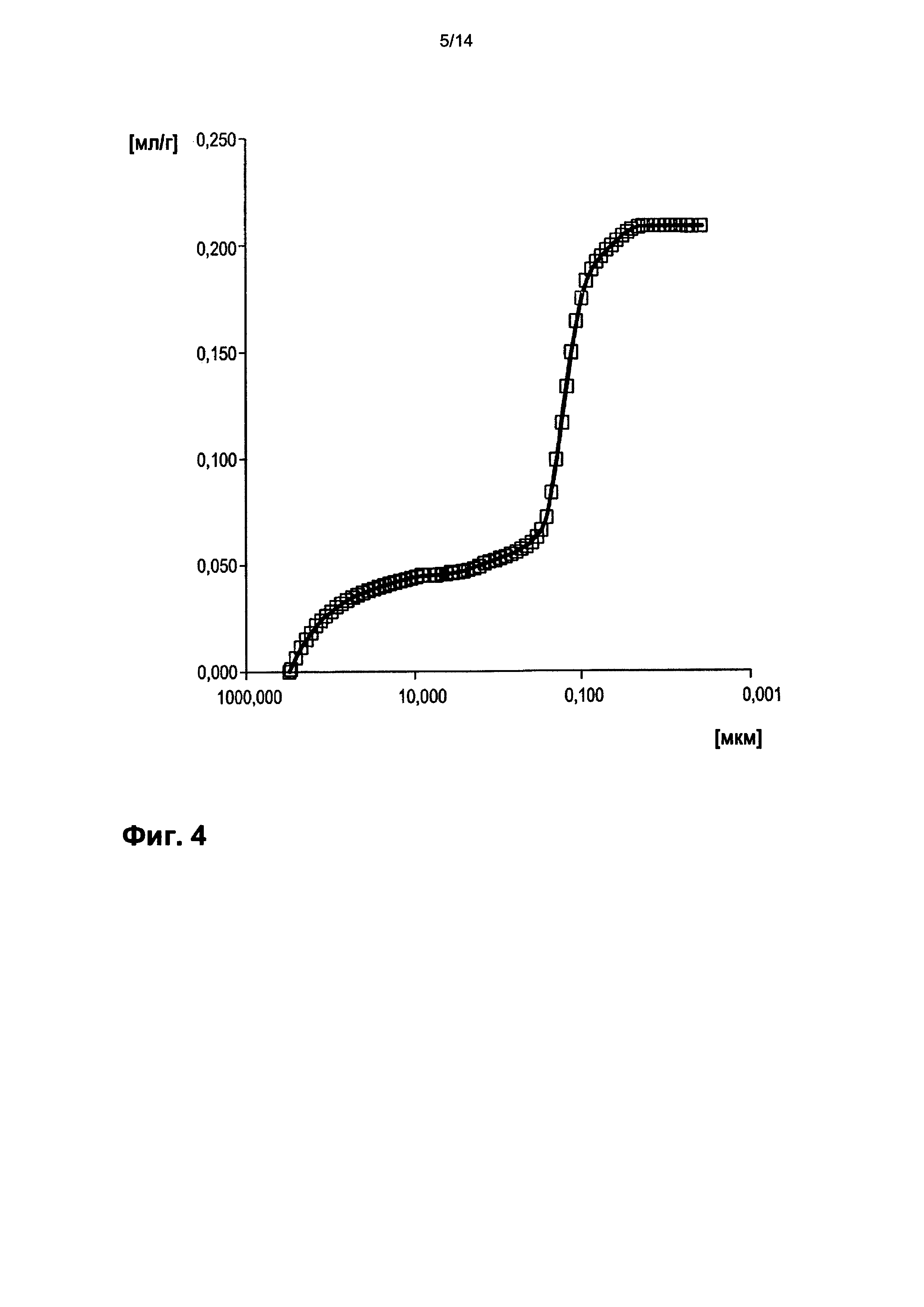
На фигуре 4 показано распределение пор оболочки активной массы С2 по размеру.
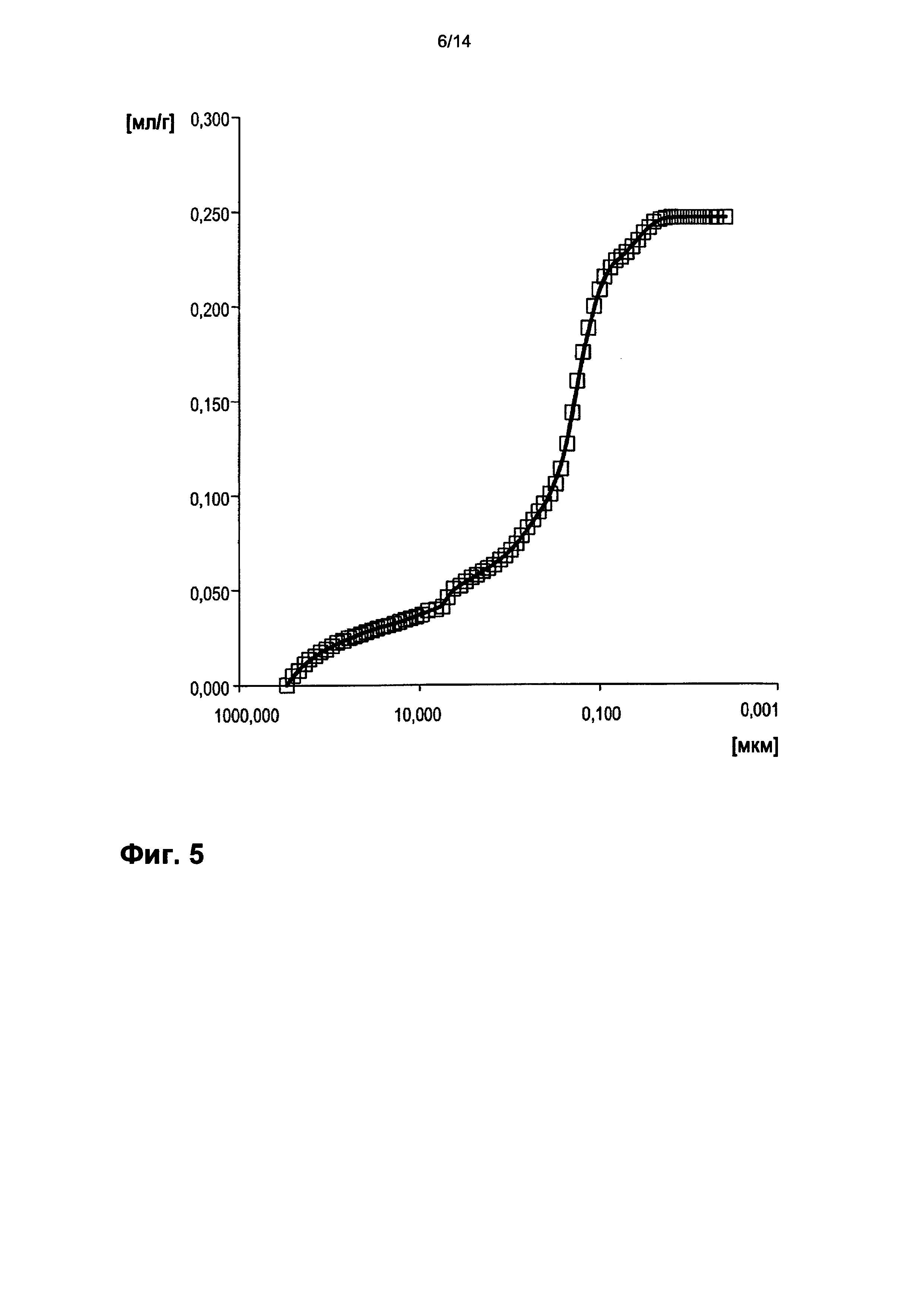
На фигуре 5 показано распределение пор оболочки активной массы С3 по размеру.
На фигуре 6 показано распределение пор оболочки активной массы С4 по размеру.
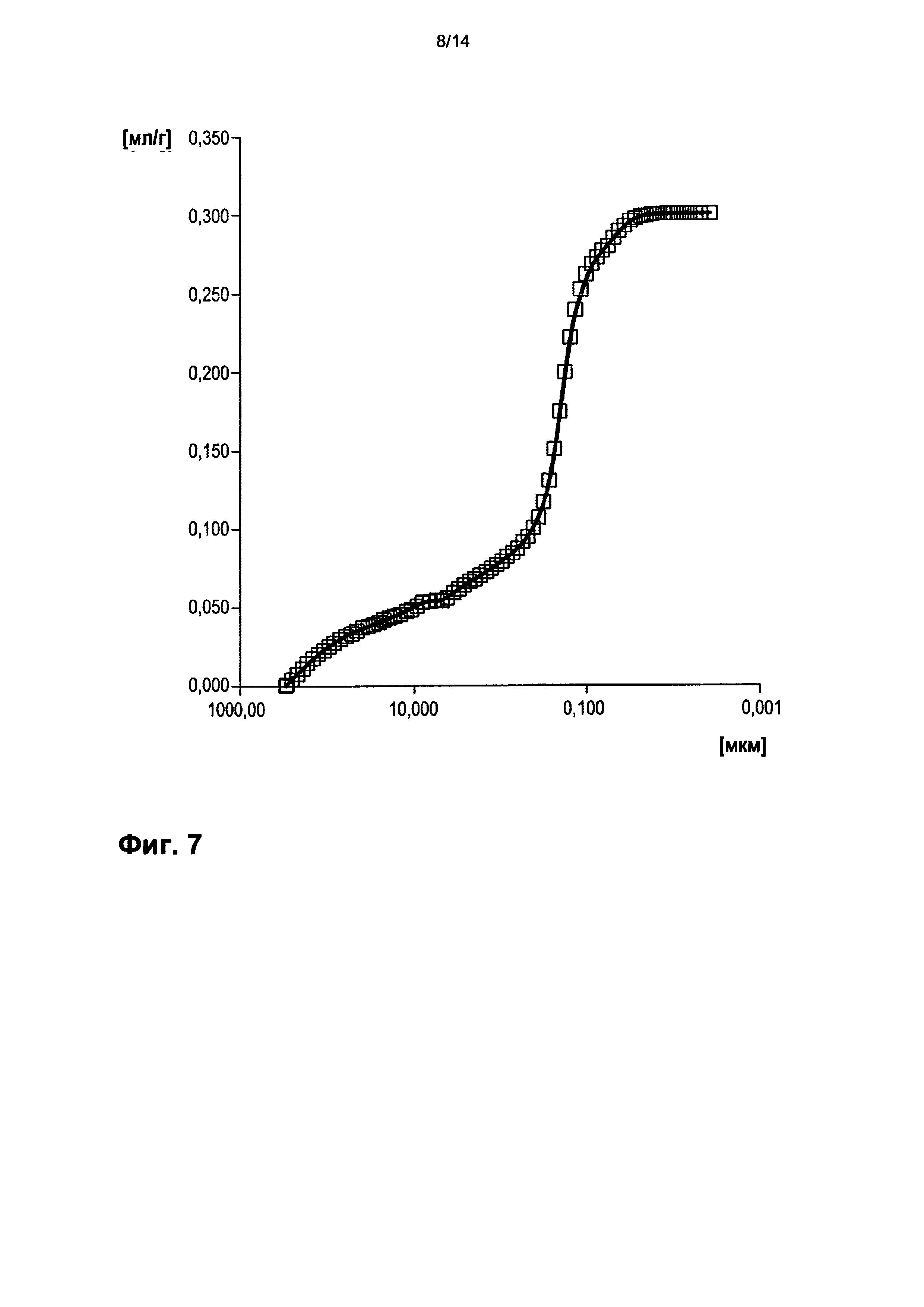
На фигуре 7 показано распределение пор оболочки активной массы С5 по размеру.
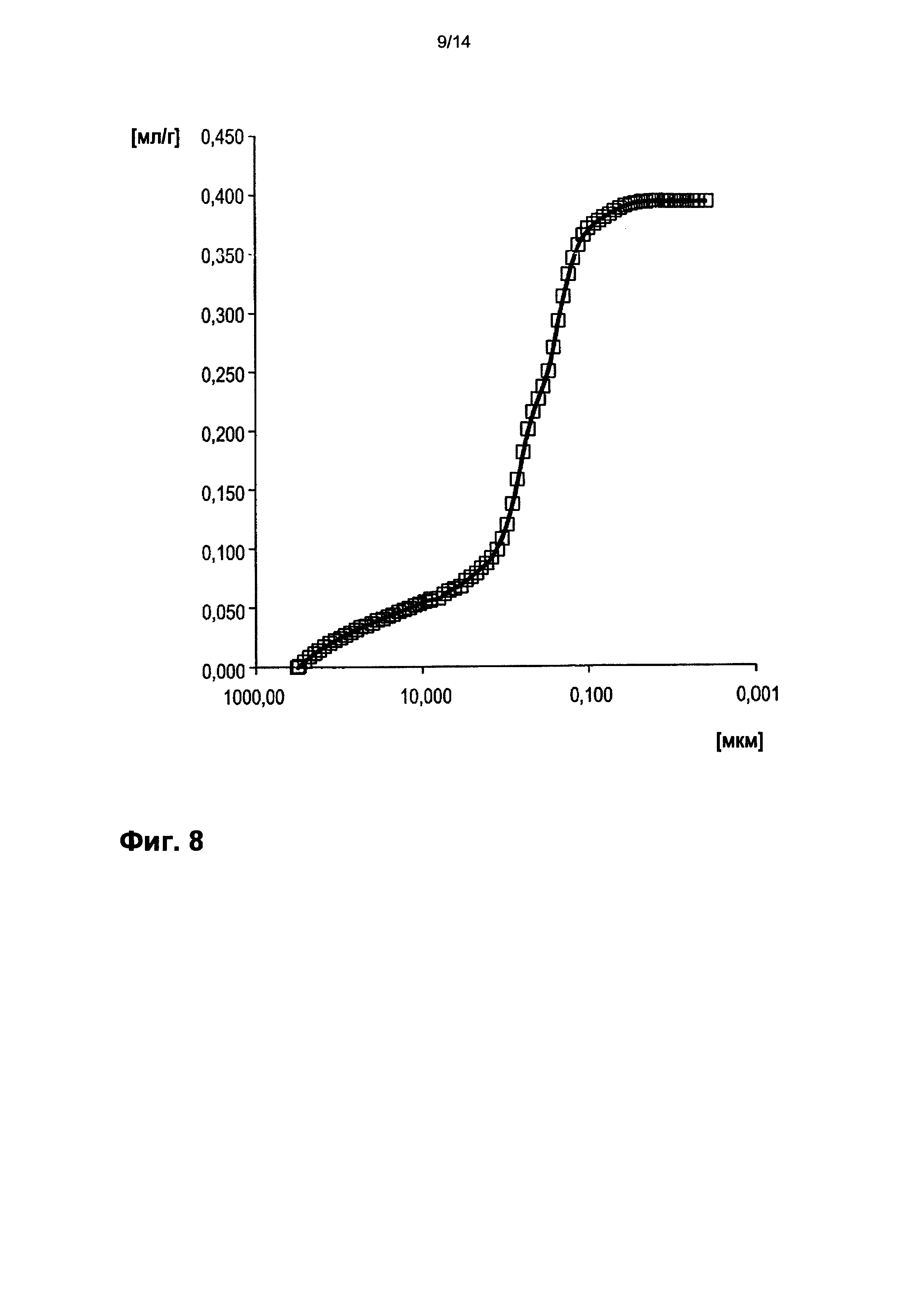
На фигуре 8 показано распределение пор оболочки активной массы С6 по размеру.
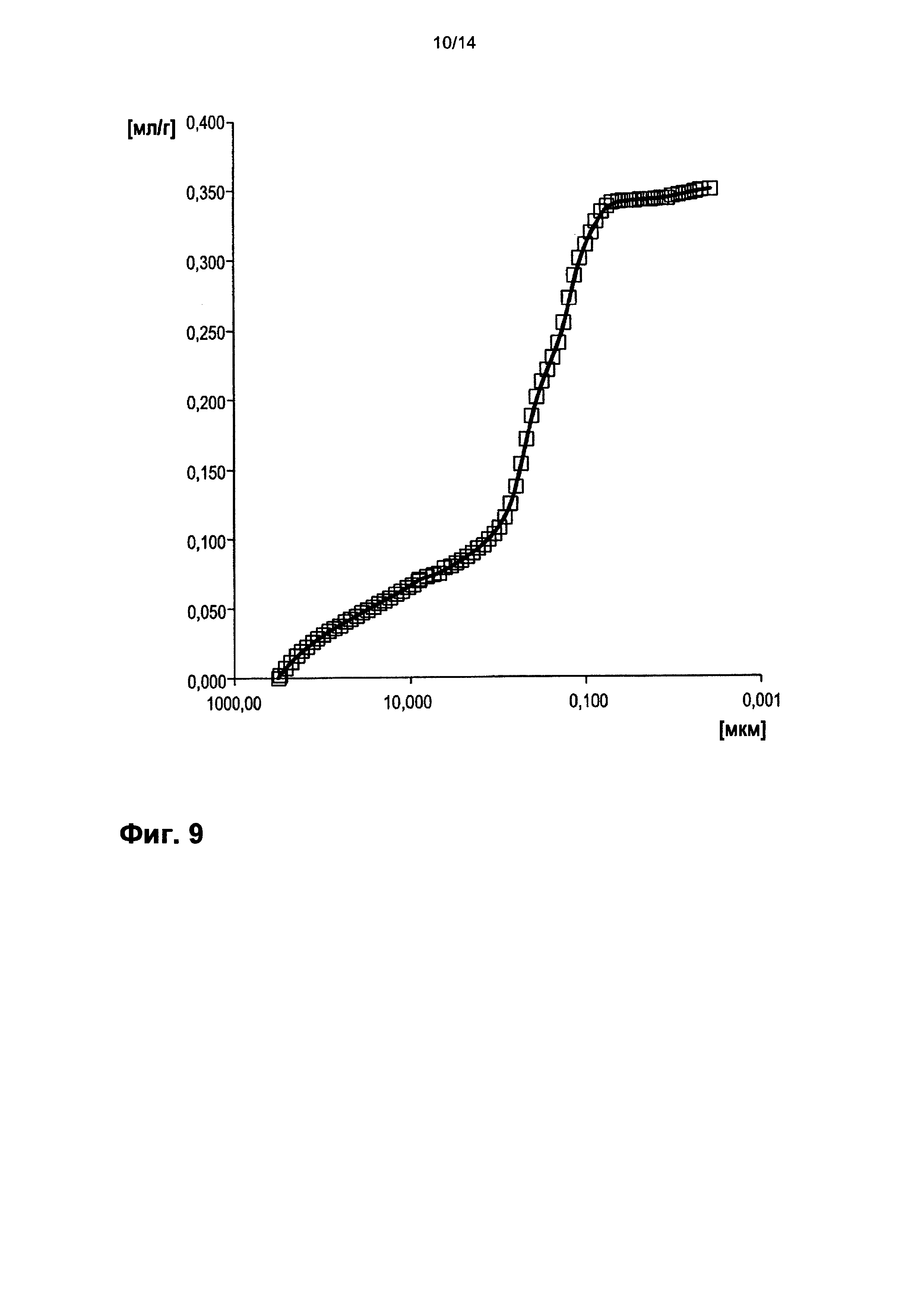
На фигуре 9 показано распределение пор оболочки активной массы С7 по размеру.
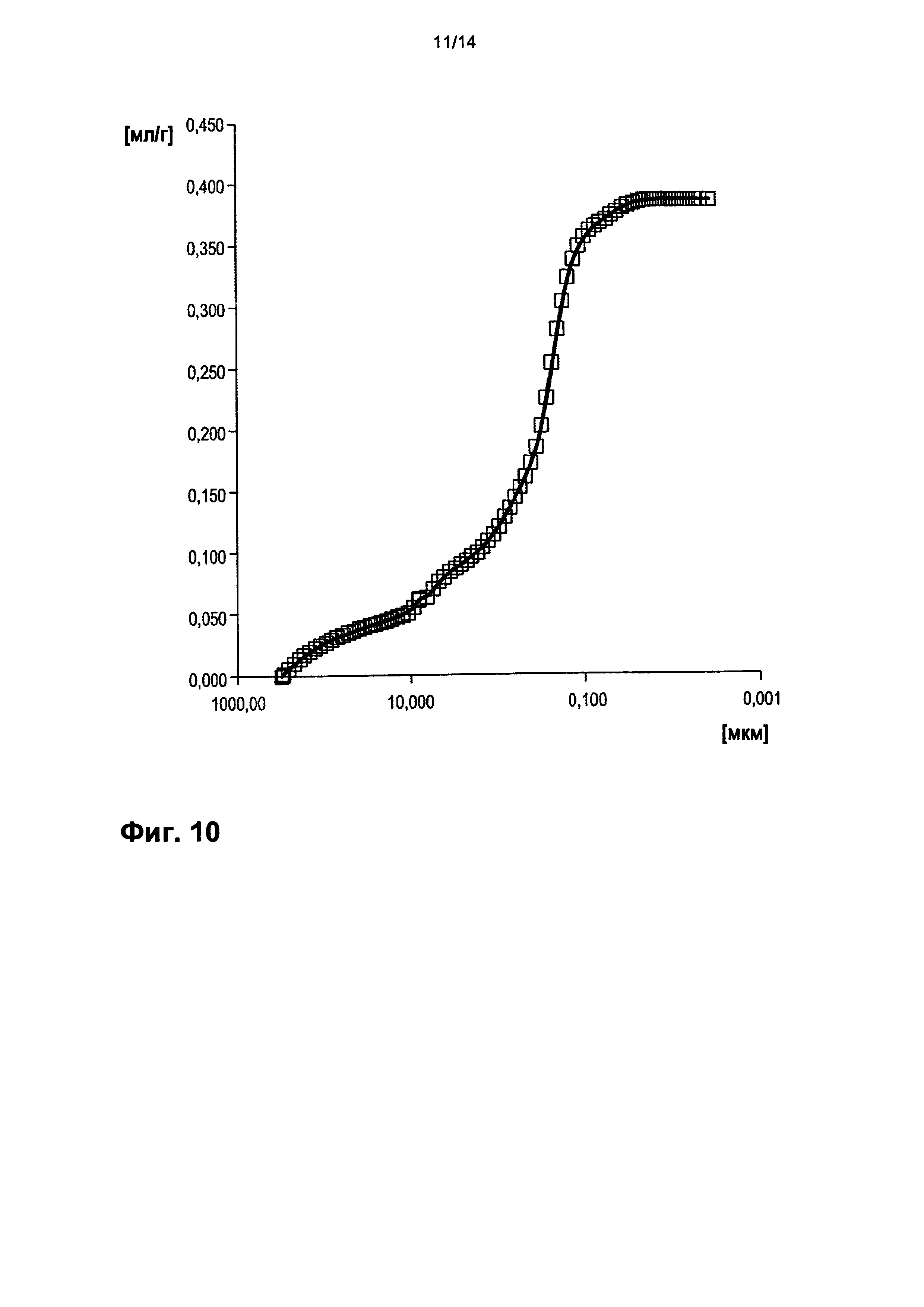
На фигуре 10 показано распределение пор оболочки активной массы С8 по размеру.
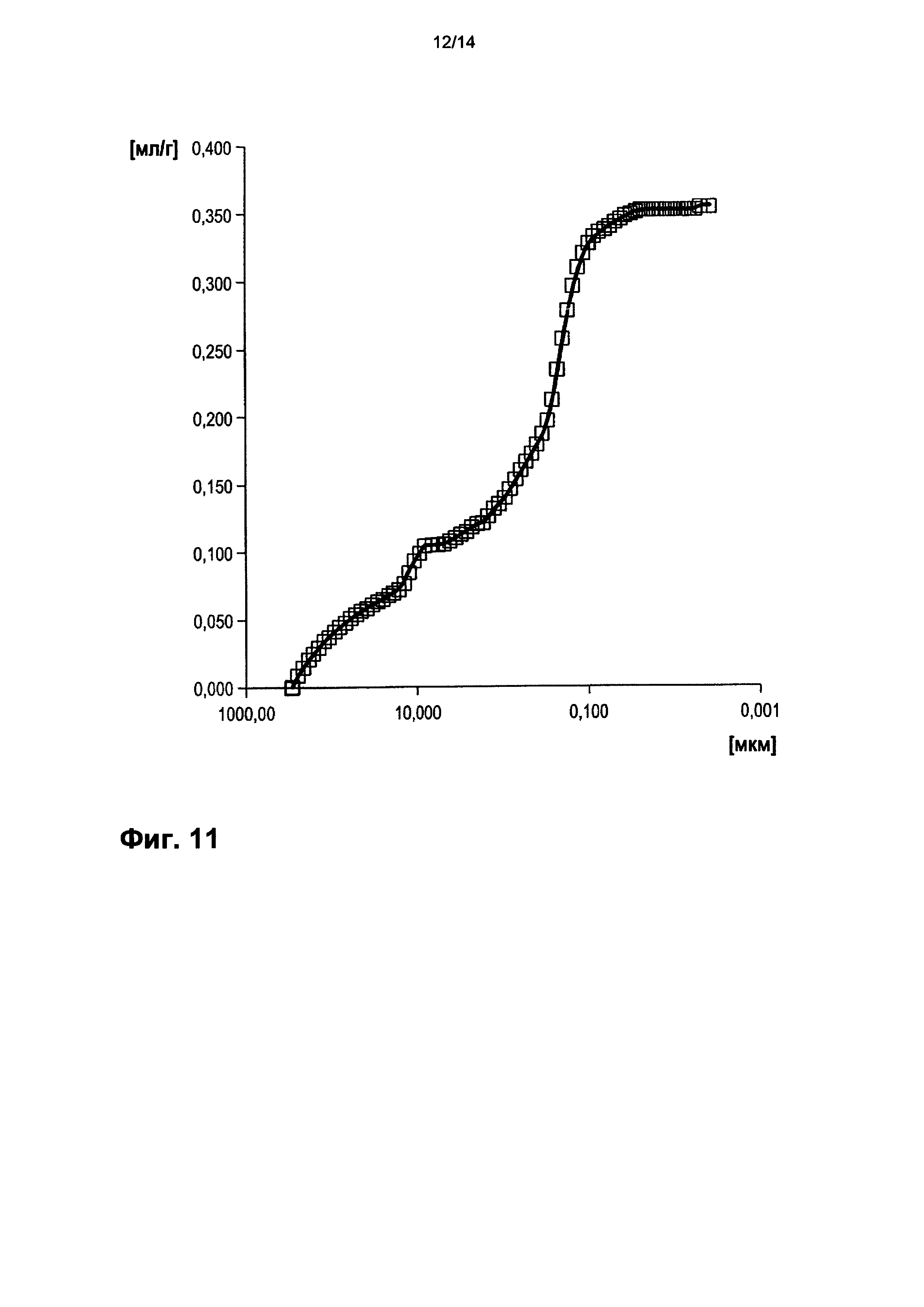
На фигуре 11 показано распределение пор оболочки активной массы С9 по размеру.
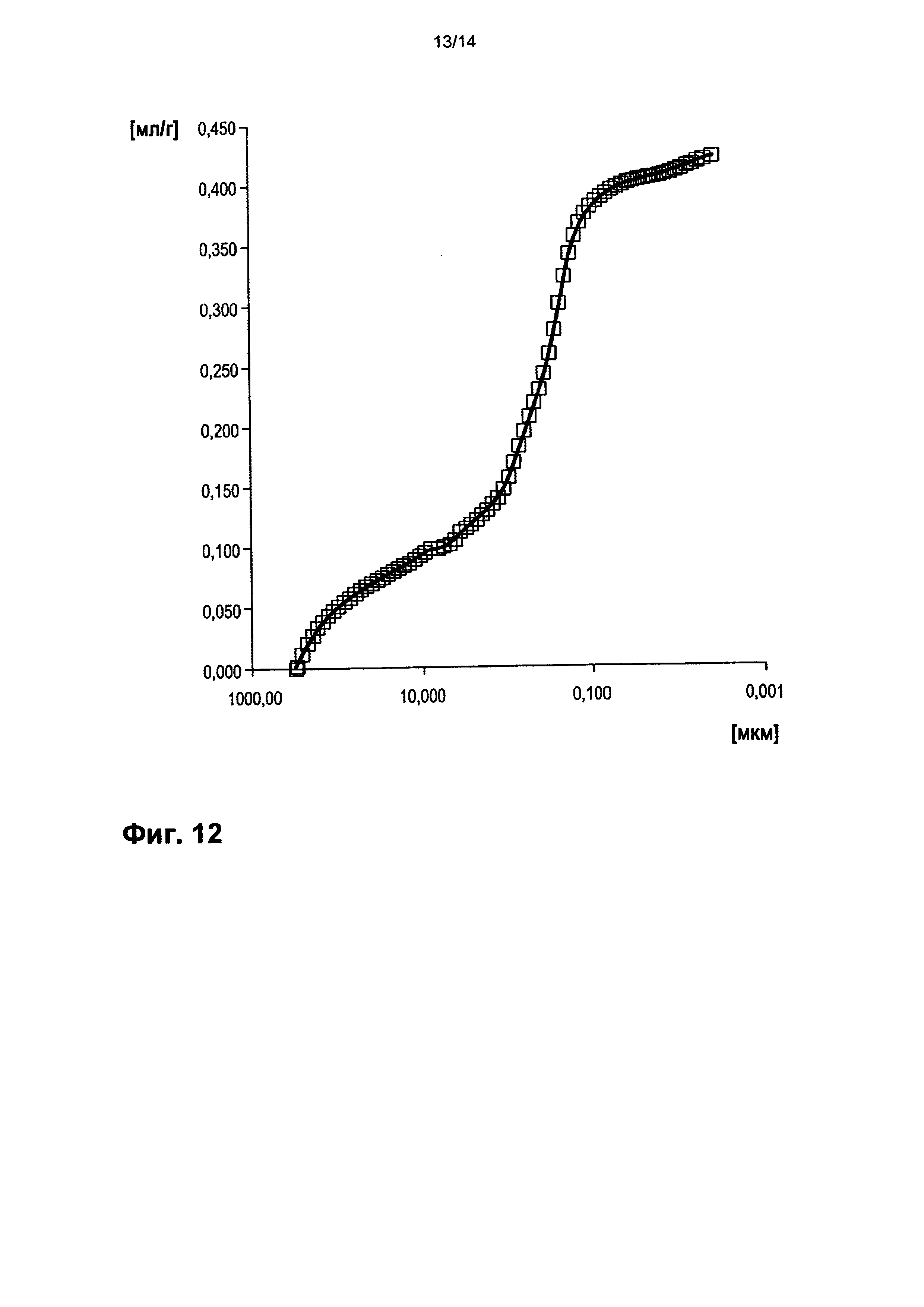
На фигуре 12 показано распределение пор оболочки активной массы С10 по размеру.
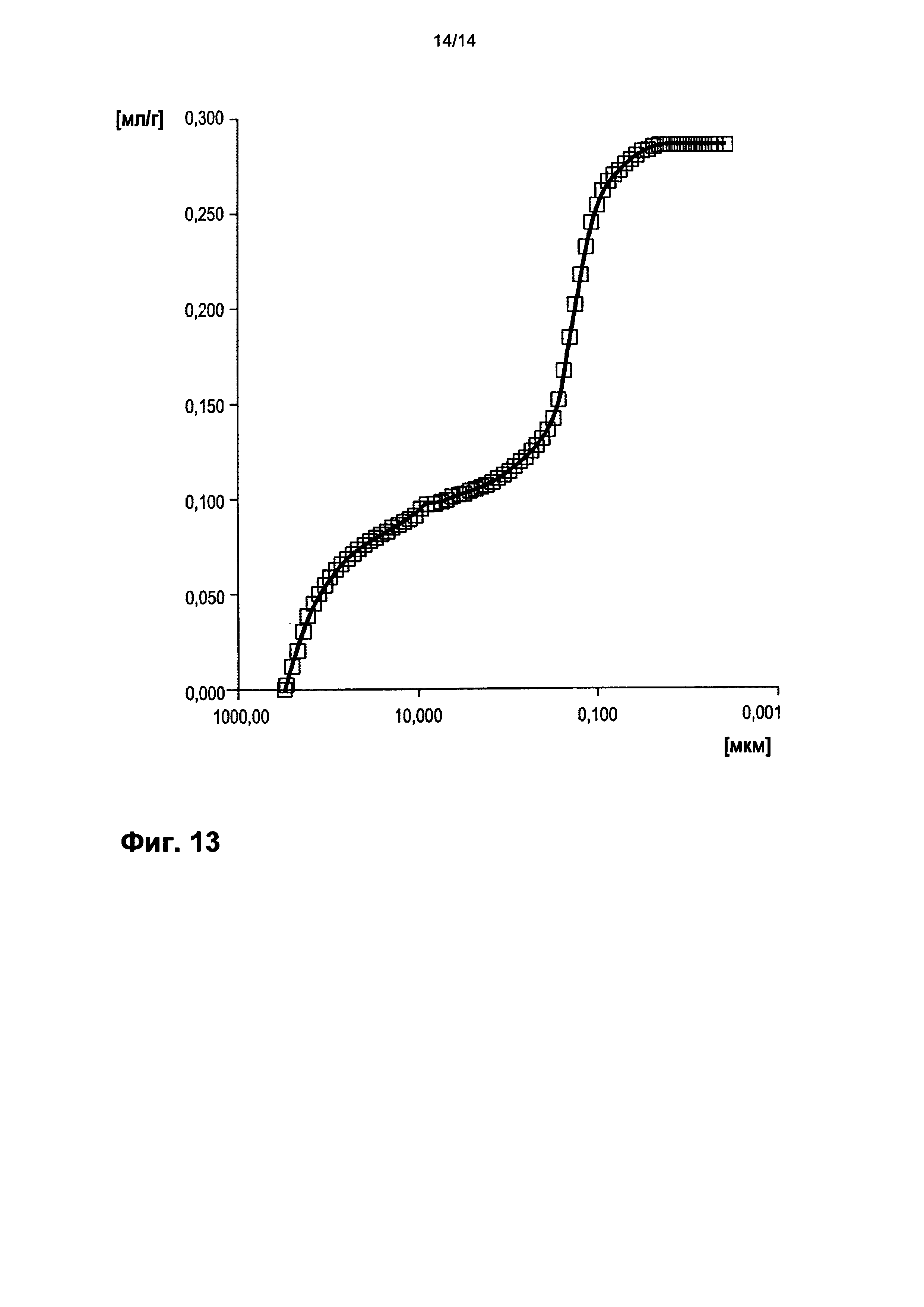
На фигуре 13 показано распределение пор оболочки активной массы С11 по размеру.
На фигурах 3-13 по оси абсцисс отложен соответствующий диаметр пор в мкм (логарифмическая шкала с основанием 10). По оси ординат в единицах (мг/л активной массы) отложен интеграл долей отдельных значений диаметра пор в удельном общем объеме пор (совокупный вклад в удельный общий объем пор) (кривая □). Конечная точка - это отнесенный на активную массу (удельный) общий объем пор (общий объем интрузии).
Примеры
Получение катализаторов
А) Получение массы-предшественника
В баке с двойными стенками из нержавеющей стали емкостью 1,75 м3 с однолопастной мешалкой и поддержанием температуры с помощью воды, при перемешивании (скорость вращения 70 оборотов/мин) растворяют 8,2 кг гидрата ацетата меди (содержание: 32,0% масс. Cu, скорость добавления 50 кг/ч, фирма Goldschmidt) в 274 л воды при комнатной температуре (~25°С). Получили раствор 1. Его перемешивали в течение дальнейших 30 мин.
В другом месте в бак с двойными стенками из нержавеющей стали емкостью 1,75 м3 с поддержанием температуры с помощью воды и с однолопастной мешалкой (скорость вращения 70 оборотов/мин) поместили 614 л воды, нагрели до 40°С и при перемешивании добавили 73 кг аммония гептамолибдата гидрата (81,5% масс. МоО3, скорость добавления 300 кг/ч, фирма Н.С. Starck GmbH), поддерживая температуру 40°С. Затем содержимое бака при перемешивании за 30 мин нагрели до 90°С и, поддерживая эту температуру, в указанной последовательности друг за другом добавили 12,1 кг метаванадата аммония (77,6% V2O5, скорость добавления 150 кг/ч, время перемешивания после добавления 40 мин), а также 10,7 кг паравольфрамата аммония гептагидрата (89,6% масс. WO3, скорость добавления 150 кг/ч, время перемешивания после добавления 30 мин). Получили раствор 2.
Раствор 2 охладили до 80°С, а затем при перемешивании (при скорости вращения однолопастной мешалки 70 об/мин) быстро ввели раствор 1 в раствор 2. К полученной смеси добавили 133 л 25-%-ного по массе водного раствора NH3, имевшего температуру 25°С. При перемешивании образовался прозрачный раствор, который кратковременно обладал температурой 65°С и значением pH 8,5. Его слили еще в один бак с двойными стенками из нержавеющей стали емкостью 1,75 м3 с поддержанием температуры с помощью воды и однолопастной мешалкой. Содержимое бака нагрели до 80°С при перемешивании со скоростью 40 оборотов/мин и запустили циркуляцию. Значение pH содержимого емкости путем автоматического добавления 25-%-ного по массе водного раствора NH3 удерживали на уровне 8,5. Содержимое бака закачали в ротационную распылительную башню типа FS 15 производства фирмы Niro (Дания) и сушили в равнонаправленном потоке горячего воздуха при входной температуре газа в 350±10°С, скорости вращения диска в 15000 об/мин и объемном потоке воздуха на сгорание в 2300 Нм3/ч, причем в распылительной башне поддерживали пониженное давление в 1 мбар. При этом подаваемый в распылительную башню объемный поток жидкости отрегулировали так, чтобы достичь температуры газа на выходе в 110±5°С. Итоговый распылительный порошок обладал диаметром частиц в 2-50 мкм и характеризовался потерями при прокаливании в 21±2% масс. Потери при прокаливании определили нагреванием в фарфоровом тигеле на воздухе (3 ч при 600°С). Предварительно фарфоровый тигель прокалили при 900°С до постоянства массы. Распылительный порошок пересыпали на хранение в специальные контейнеры или специальные бочки (200 л) с пластмассовой вставкой. При этом для отделения обломков использовали ситовую насадку.
75 кг полученного таким образом распылительного порошка добавили в месильное устройство производства фирмы AMK (Aachener Misch- und Knetmaschinen Fabrik) типа VM 160 (с сигмовидными лопатками) при скорости вращения шнеков в 15 оборотов/мин. В завершение в месильное устройство при скорости вращения шнеков в 15 оборотов/мин добавили 6,5 л уксусной кислоты (100%-ной по массе, ледяная уксусная кислота) и 5,2 л воды. По прошествии 4-5 минут смешивания (скорость вращения шнека 20 оборотов/мин) добавили еще 6,5 л воды и процесс смешивания продолжили до истечения 30 минут (температура замеса ок 40-50°С). Во время процесса смешивания следили за потреблением электрического тока. По превышении потребления тока в 25% при необходимости к перемешиваемому материалу добавляли еще приблизительно 1 л воды. Затем перемешиваемый материал помещали в экструдер и с помощью экструдера (производства фирмы Bonnot Company (штат Огайо) типа G 103-10/D7A-572K (6-дюймовый экструдер, пакер W) формовали из него тяжи (длина 1-10 см, диаметр 6 мм). Тяжи сушили на трехзонной ленточной сушилке при скорости движения ленты 10 см/мин и итоговом времени пребывания в 64 мин, а также входной температуре газа в 155°С. Ожидаемые значения температуры газа составили в зоне 1 90-95°С, в зоне 2 ок. 115°С, а в зоне 3 ок. 125°С.
В) Получение активной массы формулы Mo12V3W1,2Cu1,2On
Выполнение термической обработки проводили в печи в виде вращающейся трубы, описанной в германском патенте DE 10360057 А1, при следующих условиях:
Термическую обработку осуществляли с количеством материала в 306 кг, изготовленного так, как описано в разделе А);
Угол наклона вращающейся трубы относительно горизонтали составлял ≈0°;
Вращающаяся труба вращалась со скоростью 1,5 оборотов/мин вправо;
На протяжении всей термической обработки через вращающуюся трубу проводили поток газа в 205 Нм3/ч, который (после вытеснения исходно содержавшегося воздуха) имел следующий состав, а на выходе из вращающейся трубы был дополнен еще 25 Нм3/ч изолирующего газа - азота: 80 Нм3/ч, состоящие из азота основной нагрузки и высвободившихся во вращающейся трубе газов, 25 Нм3/ч изолирующего газа - азота, 30 Нм3/ч воздуха и 70 Нм3/ч рециркулированного циркуляционного газа. Изолирующий газ - азот подавали при температуре 25°С. Смесь остальных газовых потоков при выходе из нагревателя в каждом случае вводили во вращающуюся трубу с той температурой, которую в данном конкретном случае имел материал во вращающейся трубе.
в течение 10 ч температуру материала с 25°С по существу линейно повысили до 300°С; затем температуру материала в течение 2 ч по существу линейно повысили до 360°С; после этого температуру материала в течение 7 ч по существу линейно снизили до 350°С; потом температуру материала в течение 2 ч по существу линейно повысили до 420°С и поддерживали эту температуру материала в течение 30 мин; затем в проводимом через вращающуюся трубу газовом потоке 30 Нм3/ч воздуха заменили путем соответствующего повышения количества азота основной нагрузки (благодаря чему процесс собственно термической обработки был закончен), нагрев вращающейся трубы отключили, а материал путем включения быстрого охлаждения вращающейся трубы посредством всасывания окружающего воздуха в течение 2 ч охладили до температуры ниже 100°С и наконец до температуры окружающей среды; при этом поток газа подавали во вращающуюся трубу при температуре 25°С; во время всей термической обработки давление после выхода газового потока из вращающейся трубы (непосредственно) находилось на 0,2 ниже давления окружающей среды.
На всех фазах термической обработки содержание кислорода в газовой атмосфере в печи в виде вращающейся трубы составляло 2,9% об. При арифметическом усреднении по общей длительности восстановительной термической обработки концентрация аммиака в газовой атмосфере в печи в виде вращающейся трубы находилась около 4% об.
Полученный каталитически активный материал с помощью биплексной мельницы-сепаратора с поперечными потоками (BQ 500 производства фирмы Hosokawa-Alpine, Аугсбург) размололи в тонкодисперсный порошок Р. При этом в размалывающих лентах установили 24 продольных ножа. Скорость вращения мельницы составила 2500 оборотов в минуту. Дроссельная заслонка вентилятора была открыта полностью. Подачу установили на 2,5 оборота/мин. Объем отводимого воздуха составлял 1300 м3/ч, дифференциальное давление 10-20 мбар. 50% частиц тонкодисперсного порошка, получавшегося при размоле, проходили через сито с размером ячейки от 1 до 10 мкм. Доля частиц с максимальным продольным размером более 50 мкм составляла в тонкодисперсном порошке менее 1%. Распределение частиц вышеуказанного размолотого порошка мультиэлементной оксидной массы, обладающего каталитической активностью, показано на фигурах 1а и 1b в зависимости от диспергирующего давления сжатого воздуха, использованного для сухого диспергирования (◊=1,1 бар (абсолютное значение).; □=2,0 бар (абсолютное значение).; Δ=4,5 бар (абсолютное значение).
Методом измерения, лежащим в основе распределения частиц по размеру на фигурах 1а и 1b, является лазерная дифракция. При этом порошок мультиэлементной оксидной массы провели по желобу для диспергирования в сухой диспергатор RODOS (Sympatec GmbH, System-Partikel-Technik, Am Pulverhaus 1, Клаусталь-Целлерфельд, 38678, Германия), там подвергали сухому диспергированию сжатым воздухом (который характеризовался давлением 1,1, или соответственно 2, или соответственно 4,5 бар (абсолютное значение)) и в виде свободной струи нагнетали в измерительную ячейку. Затем в этой ячейке с помощью лазерно-дифракционного спектрометра Malvern Mastersizer S (Malvern Instruments, Worcestershire WR14 1AT, Великобритания) определяли отнесенное к объему распределение частиц по диаметру (затемнение луча 3-7%).
На фигуре 2 показана рентгеновская дифрактограмма тонкодисперсного порошка Р. На оси абсцисс представлен угол дифракции по шкале 20 [градусов]. По оси ординат указана абсолютная интенсивность.
С) Формовка активной массы
С1 (сравнительный пример)
На 1600 г изделий-носителей в форме полого цилиндра (7 мм наружного диаметра, 3 мм длины, 4 мм внутреннего диаметра, стеатит С220 производства фирмы CeramTec с шероховатостью поверхности Rz 45 мкм (накладка мелкокускового материала)) нанесли покрытие из размолотого тонкодисперсного порошка Р. Покрытие наносили в устройстве HiCoater LHC 25/36 (фирма Lödige, Падерборн, 33102, Германия). Этот HiCoater модифицировали, чтобы дать возможность непрерывной дозировки порошка. Модификация состояла в воронкообразном шаблоне для порошка, который с помощью шланга Tygon (внутренний диаметр 8 мм, наружный диаметр 11,1 мм; фирма Saint-Gobain Performance, 89120 Шарни, Франция) был соединен с барабаном устройства HiCoater. Радиус барабана при этом составлял 18 см. Глубина барабана 20 см. Ось, вокруг которой вращался барабан, была направлена горизонтально. Для нанесения покрытия в шаблон для порошка поместили 750 г размолотого каталитически активного порошка оксидной массы. Дозирование порошка осуществляли путем непрерывной дозировки давления. Клапан с временной пульсацией настроили на 50 мс, а установленное давление составляло 0,7 бар выше давления окружающей среды (~1 атм). Порошок в воронкообразном шаблоне во время нанесения покрытия непрерывно перемешивали, чтобы обеспечить равномерную подачу (время работы мешалки 2 с, продолжительность паузы мешалки 1 с, модифицированная V-образная якорная мешалка собственной конструкции фирмы BASF SE). Связующий агент представлял собой водный раствор из 75% масс. воды и 25% масс. глицерина. Его подавали в барабан отдельно, через устройство дозирования жидкости. Жидкость с помощью насоса для ВЭЖХ производства фирмы Watson-Marlow (тип 323) нагнетали в дозировочную трубку (фан), находящуюся в барабане (давление распыления 3 бар, давление формовки 2 бар, количественный поток 3 г раствора глицерина в воде (1:3)/мин). Подача порошка и подача жидкости расположены параллельно друг другу. Установленное на дозировочной трубке сопло производства фирмы Schlick (Германия) типа 570/0 S75 и закрепленное также на дозировочной трубке ниже выходное отверстие подачи твердого вещества направлены параллельно друг другу, размещены на расстоянии 6 см друг от друга и с помощью угломера направлены под углом 40° к горизонтали. Добавление порошка осуществляют вне пределов распылительного конуса сопла. При этом отверстие сопла и выходное отверстие устройства подачи твердого материала указывают направление вращения барабана. Барабан во время нанесения покрытия вращался со скоростью 15 об/мин по часовой стрелке. Нанесение покрытия проводили при 25°С на протяжении 50 мин. После этого материалы-носители с нанесенным на них покрытием в течение 27 мин сушили при температуре подводимого воздуха 130°С и температуре отходящего воздуха 81°С. Затем на протяжении периода в 30 минут их охладили в неподвижном барабане до 25°С. Во время нанесения покрытия добавленный порошок большей частью закрепился на поверхности носителя. Не зафиксированные части улавливали фильтром после барабана. Формирование дуплетов места не имело, и агломерацию тонкодисперсной оксидной массы не наблюдали.
Формованные изделия-носители с нанесенным на них покрытием обрабатывали в циркуляционном сушильном шкафу производства фирмы Memmert GmbH + Co. KG (тип UM 400; внутренний объем 53 л; поток воздуха 800 л/ч), чтобы удалить еще присутствующий в образце глицерин. Для этого циркуляционный сушильный шкаф за 2 ч разогрели до 300°С (включая температуру воздуха), а затем в течение 2 ч выдерживали при 300°С. Во время сушки высушиваемый материал размещался на расположенном по центру сушильного шкафа перфорированном металлическом листе (диаметр сквозных отверстий, равномерно распределенных по перфорированному металлическому листу, 0,5 см; соотношение отверстий перфорированного металлического листа равнялось 60%; общая площадь сечения перфорированного металлического листа равнялась 35 см × 26 см = 910 см2), послойно (высота слоя 2 см). После этого циркуляционный сушильный шкаф в течение 2-3 ч охладили до 40-50°С и извлекли образец. Извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С1 в форме полых цилиндров имели долю оксидной оболочки в 29,1% масс., в пересчете на их общую массу.
С2 (сравнительный пример)
335 кг размолотого тонкодисперсного порошка Р в течение 10 мин интенсивно перемешивали с 50 кг триоксида молибдена производства фирмы Climax (66,6 вес. % Mo, МоО3 удовлетворяет всем требованиям, приведенным в германском патенте DE 102007010422 А1) в смесителе типа R645 производства фирмы AMK (Ахен, Германия). Он представляет собой наклонный смеситель с режущими лопастями (смеситель высокой интенсивности). Рычаг смесителя вращается со скоростью 39 оборотов/мин. Полученный порошок в дальнейшем называют порошком РМо.
Формовку осуществляли следующим образом: 61 кг изделий-носителей в форме полых цилиндров (7 мм наружного диаметра, 3 мм длины, 4 мм внутреннего диаметра; стеатит типа С220 производства фирмы Ceram Тес с шероховатостью поверхности Rz 45 мкм и суммарным объемом пор относительно объема изделия-носителя не более 1% об.; ср. германский патент DE-A 2135620) засыпали в дражировочный барабан (угол наклона 90°; Hicoater производства фирмы Lödige, Германия) внутренним объемом 200 л. Затем дражировочный барабан привели во вращение со скоростью 16 об/мин. Через сопло типа Schlick 0,5 мм, 90° в течение 40 мин на изделия-носители распыляли от 3,8 до 4,2 литра водного раствора, состоявшего из 75% масс. воды и 25% масс. глицерина, при давлении жидкости до распыления ок. 1,8 бар. Одновременно за тот же период непрерывным образом добавили 18,3 кг порошка РМо (по вибролотку за пределами конуса распыления форсунки). Во время нанесения покрытия подаваемый порошок полностью закреплялся на поверхности изделия-носителя; агломерации тонкодисперсной оксидной активной массы или формирования дуплетов не наблюдали.
По завершении добавления порошка активной массы и и водного раствора в дражировочный котел при скорости вращения 2 об/мин в течение 40 мин (в качестве альтернативы 15-60 мин (нагнетали горячий воздух температурой 110°С (в качестве альтернативы 80-120°С) (ок. 400 м3/ч).
Отобрали пробу ок. 2 кг порошка активной массы с покрытием. Присутствовавший еще в пробе глицерин удалили в циркуляционном сушильном шкафу производства фирмы Memmert GmbH + Co. KG (тип UM 400; внутренний объем 53 л, поток воздуха 800 л/Ч). Условия тепловой обработки были идентичны таковым примера С1. Извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С2 в форме полых цилиндров имели долю оксидной оболочки в 22% масс., в пересчете на их общую массу.
С3 (сравнительный пример)
Формовку осуществляли следующим образом: 70 кг изделий-носителей в форме полых цилиндров (7 мм наружного диаметра, 3 мм длины, 4 мм внутреннего диаметра; стеатит типа С220 производства фирмы Ceram Тес с шероховатостью поверхности Rz 45 мкм и суммарным объемом пор относительно объема изделия-носителя не более 1% об.; ср. германский патент DE-A 2135620) засыпали в дражировочный барабан (угол наклона 90°; Hicoater производства фирмы Lödige, Германия) внутренним объемом 200 л. Затем дражировочный барабан привели во вращение со скоростью 16 об/мин. Через сопло типа Schlick 0,5 мм, 90° в течение 40 мин на изделия-носители распыляли от 3,8 до 4,2 литра водного раствора, состоявшего из 75% масс. воды и 25% масс. глицерина, при давлении жидкости до распыления ок. 1,8 бар. Одновременно за тот же период непрерывным образом добавили 18,2 кг размолотого тонкодисперсного порошка Р, удельная площадь поверхности которого составляла 16 м2/г, (по вибролотку за пределами конуса распыления форсунки). Во время нанесения покрытия подаваемый порошок полностью закреплялся на поверхности изделия-носителя; агломерации тонкодисперсной оксидной активной массы или формирования дуплетов не наблюдали.
По завершении добавления порошка активной массы и воды в дражировочный котел при скорости вращения 2 об/мин в течение 40 мин (в качестве альтернативы 15-60 мин (нагнетали горячий воздух температурой 110°С (в качестве альтернативы 80-120°С) (ок. 400 м3/ч). Получили оболочечные катализаторы в форме полых цилиндров, у которых доля оксидной активной массы в пресчете на общую массу составляла 20% масс.
Отобрали пробу ок. 2 кг порошка активной массы с покрытием. Присутствовавший еще в пробе глицерин удалили в циркуляционном сушильном шкафу производства фирмы Memmert GmbH + Co. KG (тип UM 400; внутренний объем 53 л, поток воздуха 800 л/Ч). Условия тепловой обработки были идентичны таковым примера С1. Извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С3 в форме полых цилиндров имели долю оксидной оболочки в 20% масс., в пересчете на их общую массу.
С4 (сравнительный пример)
Формовку катализатора С4 осуществляли так же, как и С1, причем, однако, в отличие от С1 в шаблон для порошка поместили только 600 г порошка, а нанесение покрытия осуществляли за период 40 мин. После тепловой обработки в циркуляционном сушильном шкафу, проведенной, как в С1, извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С4 в форме полых цилиндров имели долю оксидной оболочки в 20,0% масс., в пересчете на их общую массу.
С5 (сравнительный пример)
Нанесение покрытия выполняли во вращающемся дражировочном барабане (внутренний диаметр 25,5 см; 36 об/мин) с гранулятором производства фирмы ERWEKA (Германия). При этом ось вращения барабана была установлена под углом 51,6° относительно горизонтали. Дражировочный барабан заполнили 800 г полых цилиндрических изделий-носителей (7 мм наружного диаметра, 3 мм длины, 4 мм внутреннего диаметра, Steatit С220 производства фирмы CeramTec с шероховатостью поверхности Rz 45 мкм (накладка мелкокускового материала)). В качестве связующего агента использовали водный раствор из 75% масс. воды и 25% масс. глицерина. Приблизительно 76 г жидкого связующего агента распыляли через сопло (диаметр сопла 1 мм) на изделия-носители за время нанесения покрытия 45 мин. Одновременно за тот же период непрерывным образом добавляли 200 г размолотого тонкодисперсного порошка Р (через подающий шнек, за пределами конуса распыления форсунки). Во время нанесения покрытия добавленный порошок полностью закрепился на поверхности изделий-носителей. Агломерации тонкодисперсной оксидной активной массы не наблюдали. Нанесение покрытия повторили. Общее количество из обоих экспериментов по нанесению покрытия объединили в одну пробу. Пробу обрабатывали в циркуляционном сушильном шкафу производства фирмы Memmert GmbH + Co. KG (тип UM 400; внутренний объем 53 л; поток воздуха 800 л/ч), чтобы удалить еще присутствующий в образце глицерин. Условия тепловой обработки были идентичны таковым из примера С1. Извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С5 в форме полых цилиндров имели долю оксидной оболочки в 19,6% масс., в пересчете на их общую массу.
С6 (пример)
Формовку катализатора С6 осуществляли также, как и С1, причем, однако, в отличие от С1 в шаблон для порошка поместили только 451 г порошка, а нанесение покрытия осуществляли за период 30 мин. После тепловой обработки в циркуляционном сушильном шкафу, проведенной, как в С1, извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С6 в форме полых цилиндров имели долю оксидной оболочки в 18,0% масс., в пересчете на их общую массу.
С7 (пример)
Формовку катализатора С7 осуществляли также, как и С1, причем, однако, в отличие от С1 в шаблон для порошка поместили только 377,5 г порошка, а нанесение покрытия осуществляли за период 25 мин. После тепловой обработки в циркуляционном сушильном шкафу, проведенной, как в С1, извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С7 в форме полых цилиндров имели долю оксидной оболочки в 15,8% масс., в пересчете на их общую массу.
С8 (пример)
Формовку катализатора С8 осуществляли так же, как и С5, причем, однако, в отличие от С5 через сопло (диаметр 1 мм) на изделия-носители распылили только приблизительно 44 г жидкого связующего агента, а нанесение покрытия осуществили в течение 27,5 мин. После тепловой обработки в циркуляционном сушильном шкафу, проведенной, как в С1, извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С8 в форме полых цилиндров имели долю оксидной оболочки в 15,5% масс., в пересчете на их общую массу.
С9 (пример)
Формовку катализатора С9 осуществляли так же, как и С5, причем, однако, в отличие от С5 через сопло (диаметр 1 мм) на изделия-носители распылили только приблизительно 29 г жидкого связующего агента, а нанесение покрытия осуществили в течение 16 мин. После тепловой обработки в циркуляционном сушильном шкафу, проведенной, как в С1, извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С9 в форме полых цилиндров имели долю оксидной оболочки в 10,8% масс., в пересчете на их общую массу.
С10 (пример)
Формовку катализатора С10 осуществляли так же, как и С1, причем, однако, в отличие от С1 в шаблон для порошка поместили только 300 г порошка, а нанесение покрытия осуществляли за период 20 мин. После тепловой обработки в циркуляционном сушильном шкафу, проведенной, как в С1, извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С10 в форме полых цилиндров имели долю оксидной оболочки в 10,4% масс., в пересчете на их общую массу.
С11 (пример)
Как в примере С2, тонкодисперсный порошок Р перемешали с MoO3. В отличие от примера С2, однако, на изделия-носители в течение приблизительно 20 минут распыляли только около 1,9-2,1 литра водного раствора (глицерин/вода = 1/3). Одновременно за тот же период непрерывным образом добавляли приблизительно лишь около 8 кг порошка РМо. После нанесения покрытия, как и в примере С2, провели тепловую обработку в оборудовании для нанесения покрытия.
Отобрали пробу ок. 2 кг порошка активной массы с покрытием. Присутствовавший еще в пробе глицерин удалили в циркуляционном сушильном шкафу производства фирмы Memmert GmbH + Co. KG (тип UM 400; внутренний объем 53 л, поток воздуха 800 л/Ч). Условия тепловой обработки были идентичны таковым примера С1. Извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С11 в форме полых цилиндров имели долю оксидной оболочки в 10% масс., в пересчете на их общую массу.
С12 (пример)
Формовку катализатора С12 осуществляли так же, как и С1, причем, однако, в отличие от С1 в шаблон для порошка поместили только 345,5 г порошка, а нанесение покрытия осуществляли за период 23 мин. После тепловой обработки в циркуляционном сушильном шкафу, проведенной, как в С1, извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С12 в форме полых цилиндров имели долю оксидной оболочки в 14% масс., в пересчете на их общую массу. Покрытие оболочечного катализатора С12 согласно изобретению активной массой составляет 0,23 мг/мм2.
С13 (пример)
Формовку катализатора С13 осуществляли так же, как и С1, причем, однако, в отличие от С1 в шаблон для порошка поместили только 360,7 г порошка, а нанесение покрытия осуществляли за период 24 мин. После тепловой обработки в циркуляционном сушильном шкафу, проведенной, как в С1, извлеченные из циркуляционного сушильного шкафа оболочечные катализаторы С13 в форме полых цилиндров имели долю оксидной оболочки в 13,4% масс., в пересчете на их общую массу.
Свойства катализаторов, соответствующих примерам С1-С11, представлены в таблице 1. Во всех примерах удельная геометрическая площадь поверхности Sm формованного изделия-носителя составляла 0,725 мм2/мг. Ее рассчитывали путем деления геометрической площади поверхности формованного изделия-носителя (155,5 мм2) на его массу (214,4 мг).
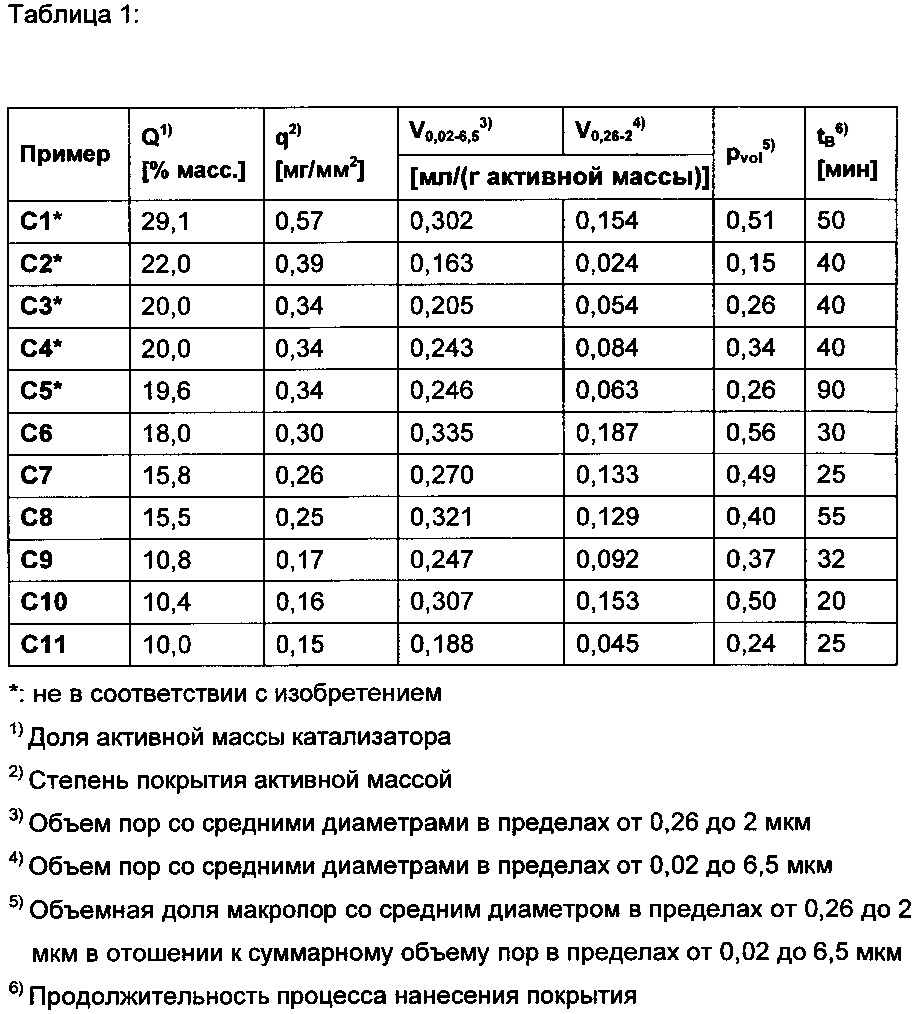
Все приведенные в настоящем тексте данные, касающиеся структуры и качеств пор в твердых веществах, если явно не указано иное, основаны на результатах определения методом ртутной порозиметрии с применением прибора Auto Pore IV 9520 производства фирмы Micromeritics в Норкроссе, 30093-1877, США, штат Джорджия. В случае исследованных порошков количество пробы, вводимое в отделение для образцов, в каждом случае составляло 2,5 г. В случае исследованных оболочечных катализаторов в отделение для образцов в каждом случае помещали по 5 штук данного конкретного оболочечного катализатора (при этом вклад пор геометрического формованного изделия-носителя оболочечного катализатора в исследованных случаях был пренебрежимо мал по сравнению со вкладом пор оболочки из активной массы).
Отделение для образцов продолжалось вытянутыми в длину капиллярами, так что небольшие изменения давления соответствовали существенным изменениям длины выступающей в капилляры ртутной нити. Во всех случаях использованный объем капилляра составлял от 25 до 91% об., совокупного объема капилляра.
До начала исследования каждой конкретной пробы из отделения для образцов (при 25°С) откачивали воздух, в каждом случае до внутреннего давления в 9,3×10-4 бар, и в течение 20 минут дегазировали пробу при этом давлении и при этой температуре. Затем в отделение для образцов нагнетали ртуть с нарастанием давления со временем до конечного значения давления в 4137 бар. Начальное давление составило 0,04 бар. Этому соответствует ширина (полосы) зарегистрированных значений диаметра пор от 0,003 мкм - 360 мкм.
На фигуре 3 настоящей публикации показано распределение пор оболочки активной массы С1 по размеру. По оси абсцисс отложен соответствующий диаметр пор в мкм (логарифмическая шкала с основанием 10). По левой оси ординат в единицах ([мг]/[л активной массы]) отложен интеграл долей отдельных значений диаметра пор в удельном общем объеме пор (совокупный вклад в вышеуказанный удельный общий объем пор) (кривая □). Конечная точка - это отнесенный на активную массу (удельный) общий объем пор (общий объем интрузии).
Газофазного окисление акролеина до акриловой кислоты
Реакционная труба (сталь V2A; наружный диаметр 30 мм, толщина стенок 2 мм, внутренний диаметр 26 мм, длина 464 см) содержала сверху вниз:
Участок 1: 79 см длины, пустая труба;
Участок 2: 62 см длины, предварительная засыпка из колец из стеатита с геометрическими характеристиками 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр, стеатит С220 производства фирмы CeramTec);
Участок 3: 100 см длины, твердый слой катализатора из гомогенной смеси, состоящей из 20% масс. колец из стеатита с геометрическими характеристиками 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр, стеатит С220 производства фирмы CeramTec) и 80% масс. в каждом случае соответствующего катализатора;
Участок 4: 200 см длины, твердый слой катализатора, состоящий исключительно из катализатора, применяемого в каждом случае на участке 3.
Участок 5: 10 см длины, завершающая засыпка из тех же самых колец из стеатита, что и на участке 2;
Участок 6: 14 см длины, сетка для катализатора из стали V2A, для размещения твердого слоя катализатора.
Через реакционную трубу в каждом случае проводили в направлении сверху реакционной трубы вниз поток реакционной газовой смеси, состав которой на входе в реакционную трубу был следующим:
4.3% об. акролеина,
0,2% об. пропена,
0,2% об. пропана,
0,3% об. акриловой кислоты,
5.4%об. O2,
7,0% об. H2O,
0,4% об. СО и CO2,
остальное N2.
Нагрузка твердого слоя катализатора акролеином составляла в каждом случае 75 Нл/л⋅ч.
Вокруг реакционной трубы по ее длине (за исключением последних 10 см пустой трубы на участке 1 и последних 3 см трубы на участке 6) в каждом случае циркулировала перемешиваемая и обогреваемая снаружи электрическим путем соляная баня (смесь 53% масс. нитрата калия, 40% масс. нитрита натрия и 7% масс. нитрата натрия, 50 кг соляного расплава); скорость течения у трубы составляла 3 м/с (в плоскости, перпендикулярной продольной оси трубы).
Температура соляной бани TB соответствует температуре, при которой в соляной бане циркулировал соляной расплав. Во всех случаях ее устанавливали так, чтобы конверсия акролеина в расчете на простое прохождение реакционной смеси через твердый слой катализатора UA в итоге составляла 99,3% мол. Благодаря подогреву температура соляной бани вдоль реакционной трубы была неизменна (соляная баня излучала больше теплоты, чем реакционная труба отдавала соляной бане). На входе в реакционную трубу температура реакционного газа соответствовала данной конкретной температуре соляной ванны TB. Максимальную локальную температуру TH определяли точечным измерением в реакционной трубе. Результаты, полученные с применением различных катализаторов, обобщены в таблице 2.
Под селективностью образования акриловой кислоты (SAS (% мол.)) в настоящей публикации подразумевают:

(значения конверсии в каждом случае в расчете на однократное прохождение реакционной газовой смеси через твердый слой катализатора).
В нижеследующей таблице 2 приведены результаты, полученные в каждом случае по прошествии 100 часов эксплуатации, в зависимости от использованного оболочечного катализатора:
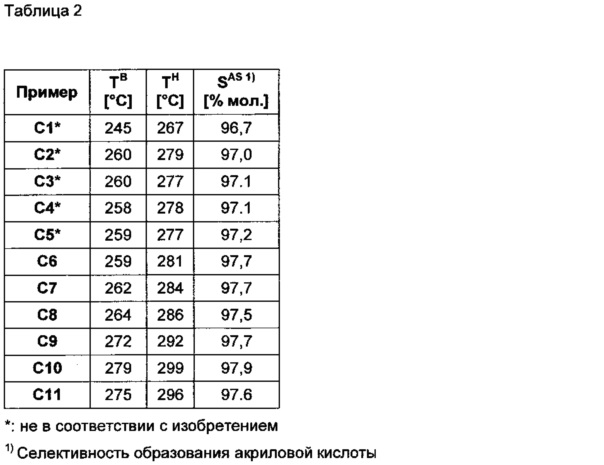
При покрытии катализатора активной массой, равном самое большее 0,3 мг/мм2, SAS составляет 97,6% мол. или более. При более высоком покрытии катализатора активной массой SAS меньше, принимая значения от 96,7 до 97,2% мол. Преимущество покрытия активной массой согласно изобретению проявляется несмотря на более высокие значения температуры горячей точки TH в 281-299°С.
Преимущество предпочтительной согласно изобретению высокой объемной доли макропор (pvol) проявляется при сравнении результатов, которые получили с использованием катализаторов согласно изобретению с по существу одинаковым покрытием активной массой. В группе катализаторов с покрытием активной массой от 0,15 до 0,17 мг/мм2 при pvol=0,5 SAS особо высок (97,9% мол., С10), в то время как при pvol=0,24 SAS снижен (97,6% мол., С11). Из обоих катализаторов с покрытием активной массой в пределах от 0,25 до 0,26 мг/мм2 при pvol=0,49 SASвысок (97,7% мол., С7), в то время как при pvol=0,40 SAS снижен (97,5% мол., С8).
Газофазное окисление акролеина до акриловой кислоты с применением твердого слоя катализатора с двумя следующими друг за другом реакционными зонами
Реакционную трубу (нержавеющая сталь типа 1.4541 (нормативный номер ЕС EN 10088-3; 33,7 мм наружного диаметра; 2 мм толщины стенки; 29,7 мм внутреннего диаметра; 400 см длины, 4 мм термогильза) загрузили (в перечислении снизу вверх) следующим образом:
Участок 1: 70 см длины
Предварительная засыпка из колец из стеатита с геометрическими характеристиками 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр, стеатит С220 производства фирмы CeramTec);
Участок 2: 100 см длины
Засыпка твердым слоем катализатора из данного конкретного оболочечного катализатора
Участок 3: 200 см длины
Засыпка твердым слоем катализатора из данного конкретного оболочечного катализатора
Участок 4: 8 см длины
завершающая засыпка из тех же самых колец из стеатита, что и на участке 1;
Участок 5: 23 см длины
пустая труба
Через реакционную трубу, загруженную так, как описано выше, в каждом случае проводили в направлении сверху реакционной трубы вниз поток реакционной газовой смеси, состав которой был следующим:
4,5% об. акролеина,
0,1% об. пропена,
0,07% об. пропана,
0,5% об. акриловой кислоты,
5,4% об. O2,
7% об. H2O,
0,6% об. СО и CO2, и
остальное N2.
Нагрузка твердого слоя катализатора акролеином (согласно определению в германском патенте DE-A 19927624) составила в каждом случае 75 Нл/л⋅ч.
Вокруг реакционной трубы по ее длине в каждом случае циркулировала перемешиваемая и обогреваемая снаружи электрическим путем соляная баня (смесь 53% масс. нитрата калия, 40% масс. нитрита натрия и 7% масс. нитрата натрия, 50 кг соляного расплава); скорость течения у трубы составляла 3 м/с (в плоскости, перпендикулярной продольной оси трубы).
Температуру соляной бани TB (°С) (при которой подавали соляную баню) устанавливали так, чтобы конверсия акролеина в расчете на простое прохождение реакционной смеси через твердый слой катализатора в итоге составляла 98,3% мол. Благодаря подогреву температура соляной бани вдоль реакционной трубы была неизменна (соляная баня излучала больше теплоты, нежели реакционная труба отдавала соляной бане). Температуру подачи реакционной газовой смеси (на входе в реакционную трубу) в каждом случае устанавали в соответсвии с конкретной температурой соляной бани.
Температуру в слое катализатора непрерывно измеряли с помощью термоэлемента, который был размещен в термогильзе, находившейся внутри реакторной трубы, и с помощью тягового устройства передвигался в слое реактора снизу вверх. Максимальная температура этого измерения соответствовала температуре горячей точки TH.
В нижеследующей таблице 3 показаны полученные через 100 часов эксплуатации результаты, которые устанавливаются в зависимости от загрузки участков реактора 2 и 3 с использованием различных оболочечных катализаторов согласно изобретению и не в соответствии с изобретением.
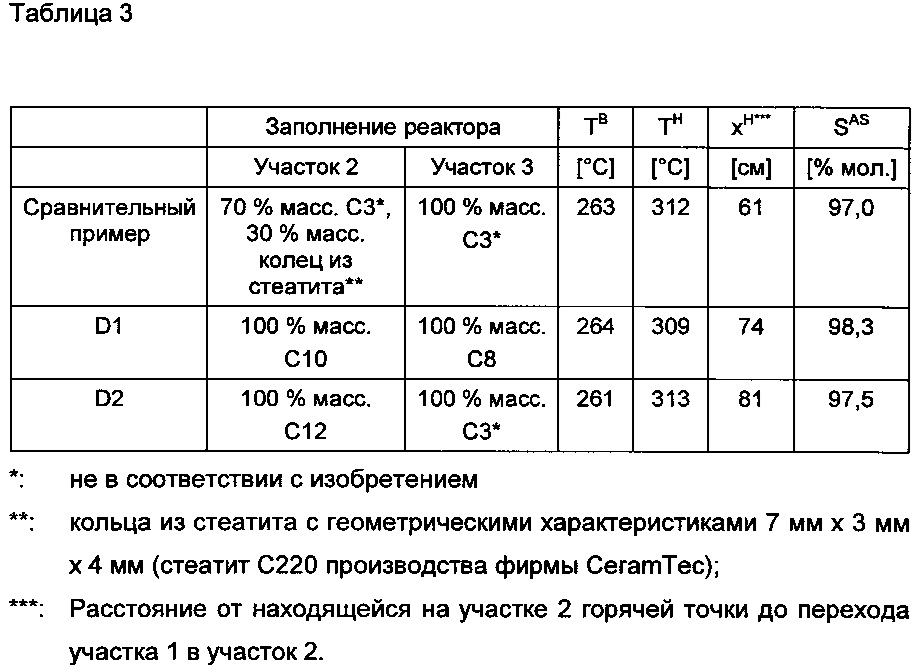
Сравнение примера D2 со сравнительным примером в таблице (97,5% мол.) в силу лучшей селективности по акриловой кислоте (97,5% мол.) у D2 в сравнении с эталоном (97% мол.) доказывает, что заполнение реакционной зоны с наивысшей температурой (горячая точка в D2 и в эталоне находится на участке 2, см. таблицу 3) оболочечными катализаторами согласно изобретению выгодно с точки зрения селективности образования акриловой кислоты.
Частичная замена расположенной выше по потоку реакционной зоны твердого слоя катализатора
В германском патенте DE-A 10232748 описана замена части твердого слоя катализатора свежей катализаторной засыпкой. Ниже приведено исследование, выгодны ли катализаторы согласно изобретению также и для описанной в германском патенте DE-A10232748 частичной замены твердого слоя катализатора.
Реакционную трубу (нержавеющая сталь типа 1.4541 (нормативный номер ЕС EN 10088-3; 33,7 мм наружного диаметра; 2 мм толщины стенки; 29,7 мм внутреннего диаметра; 400 см длины, 4 мм термогильза) загрузили (в перечислении снизу вверх) следующим образом:
Участок 1: 75 см длины
Предварительная засыпка из колец из стеатита с геометрическими характеристиками 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр, стеатит С220 производства фирмы CeramTec);
Участок 2: 110 см длины
Засыпка твердым слоем катализатора из данного конкретного оболочечного катализатора
Участок 3: 190 см длины
Засыпка твердым слоем катализатора, состоящим из оболочечного катализатора (71% масс.), изготовленного в соответствии с примером исполнения 1 германского патента DE 10360057 А1, и 29% масс. колец из стеатита с геометрическими характеристиками 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр, стеатит С220 производства фирмы CeramTec);
Участок 4: 3 см длины
завершающая засыпка из тех же самых колец из стеатита, что и на участке 1;
Участок 5: 23 см длины
пустая труба
Через реакционную трубу, загруженную так, как описано выше, в каждом случае проводили в направлении сверху реакционной трубы вниз поток реакционной газовой смеси, состав которой был следующим:
Нагрузка твердого слоя катализатора акролеином (согласно определению в германском патенте DE-A 19927624) составила в каждом случае 75 Нл/л⋅ч.
Вокруг реакционной трубы по ее длине в каждом случае циркулировала перемешиваемая и обогреваемая снаружи электрическим путем соляная баня (смесь 53% масс. нитрата калия, 40% масс. нитрита натрия и 7% масс. нитрата натрия, 50 кг соляного расплава); скорость течения у трубы составляла 3 м/с (в плоскости, перпендикулярной продольной оси трубы).
Температуру соляной бани TB (°С) (при которой подавали соляную баню) устанавливали так, чтобы конверсия акролеина в расчете на простое прохождение реакционной смеси через твердый слой катализатора в итоге составляла 99,3% мол. Благодаря подогреву температура соляной бани вдоль реакционной трубы была неизменна (соляная баня излучала больше теплоты, нежели реакционная труба отдавала соляной бане). Температуру подачи реакционной газовой смеси (на входе в реакционную трубу) в каждом случае устанавали в соответсвии с конкретной температурой соляной бани.
Температуру в слое катализатора непрерывно измеряли с помощью термоэлемента, который был размещен в термогильзе, находившейся внутри реакторной трубы, и с помощью тягового устройства передвигался в слое реактора снизу вверх. Максимальная температура этого измерения соответствовала температуре горячей точки TH.
В нижеследующей таблице 4 показаны полученные через 100 часов эксплуатации результаты, которые устанавливаются в зависимости от загрузки участка реактора 2 с использованием различных оболочечных катализаторов согласно изобретению и не в соответствии с изобретением.
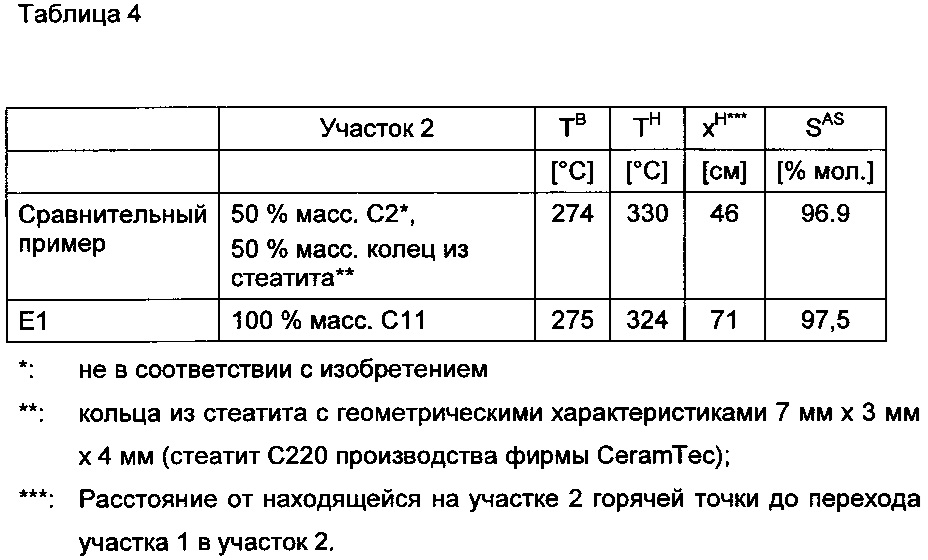
Сравнение примера Е1 со сравнительным примером в таблице (97,5% мол.) в силу лучшей селективности по акриловой кислоте (97,5% мол.) у примера Е1 в сравнении с эталоном (96,9% мол.) доказывает, что при частичной замене катализатора загрузка передней реакционной зоны оболочечными катализаторами согласно изобретению выгодна с точки зрения селективности образования акриловой кислоты.
Газофазное окисление акролеина до акриловой кислоты с применением твердого слоя катализатора с двумя следующими друг за другом зонами нагрева
Реакционную трубу (нержавеющая сталь типа 1.4541 (нормативный номер ЕС EN 10088-3; 33,7 мм наружного диаметра; 2 мм толщины стенки; 29,7 мм внутреннего диаметра; 400 см длины, 4 мм термогильза) загрузили (в перечислении сверху вниз) следующим образом:
Участок 1: 70 см длины
Предварительная засыпка из колец из стеатита с геометрическими характеристиками 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр, стеатит С220 производства фирмы CeramTec)
Участок 2: 100 см длины
Засыпка твердым слоем катализатора из оболочечного катализатора С13;
Участок 3: 200 см длины
Засыпка твердым слоем катализатора из оболочечного катализатора С3;
Участок 4: 8 см длины
Завершающая засыпка из тех же самых колец из стеатита, что и на участке 1;
Участок 5: 23 см длины
Пустая труба
Через реакционную трубу, загруженную так, как описано выше, в каждом случае проводили в направлении сверху реакционной трубы вниз поток реакционной газовой смеси, состав которой следующий:
Значения нагрузки твердого слоя катализатора акролеином LACR (согласно определению в германском патенте DE-A 19927624) составляли 75, 100 и 145 Н⋅лакролеина/(лч).
Реакционную трубу обогревали с помощью двух различных соляных бань, как это описано в германском патенте DE 2010-10048405. На первых 190 см обеспечивали термостатичность посредством соляной бани, перекачиваемой противотоком и подаваемой при температуре ТА. На вторых 210 см обеспечивали термостатичность посредством соляной бани В, перекачиваемой противотоком и подаваемой при температуре TB. Обе соляные бани состояли из смеси 53% масс. нитрата калия, 40% масс. нитрита натрия и 7% масс. нитрата натрия; 50 кг соляного расплава. Скорость течения у трубы составляла 3 м/с (в плоскости, перпендикулярной продольной оси трубы).
Температуры соляной бани ТА и TB (°С) (при которой подавали соляную баню) устанавливали так, чтобы конверсия акролеина в расчете на простое прохождение реакционной смеси через твердый слой катализатора в итоге составляла 99,4% мол. при нагрузках акролеином в 75 и 100 Н⋅лакролеина/(лч), а также 98,5% мол. при нагрузке 145 Н⋅лакролеина/(лч). Благодаря подогреву температура соляной бани вдоль реакционной трубы была неизменна (соляная баня излучала больше теплоты, нежели реакционная труба отдавала соляной бане). Температура подачи реакционной газовой смеси (на входе в реакционную трубу) в каждом случае настраивали на данную конкретную температуру соляной бани.
Температуру в слое катализатора непрерывно измеряли с помощью термоэлемента, который был размещен в термогильзе, находившейся внутри реакторной трубы, и с помощью тягового устройства передвигался в слое реактора снизу вверх. Определенные при этом измерении максимальные температуры соответствовали температурам горячей точки ТН1 и ТН2.
В нижеследующей таблице 5 показаны полученные через 100 часов эксплуатации результаты, которые устанавливаются при различных значениях нагрузки с использованием оболочечного катализатора согласно изобретению.
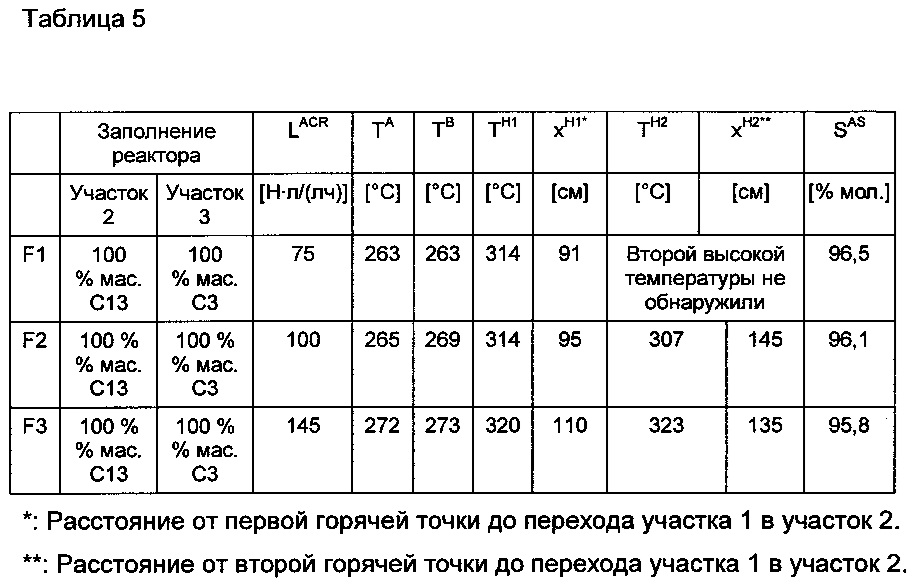
Реферат
Описан катализатор для получения α,β-ненасыщенной карбоновой кислоты путем газофазного окисления α,β-ненасыщенного альдегида, причем катализатор включает формованное изделие-носитель с нанесенной на него активной массой, отличающейся тем, что степень покрытия активной массой q, где, составляет самое большее 0,26 мг/мм, причем Q - это доля активной массы катализатора в мас.%, a S- удельная геометрическая поверхность формованного изделия-носителя в мм/мг, а активная масса включает мультиэлементный оксид общей формулы (II)где Xозначает один или несколько щелочных и/или щелочно-земельных металлов, Xозначает один или несколько элементов из группы Si, Al, Ti и Zr, а означает число в пределах от 2 до 4, b означает число в пределах от 0 до 3, с означает число в пределах от 0,5 до 3, е означает число в пределах от 0 до 2, f означает число в пределах от 0 до 40 и n означает стехиометрический коэффициент элемента кислорода, который определяется стехиометрическими коэффициентами отличных от кислорода элементов, а также их валентностью в (II). Также описан способ получения катализатора, а также способ получения α,β-ненасыщенной карбоновой кислоты путем газофазного окисления α,β-ненасыщенного альдегида молекулярным кислородом на твердом слое катализатора, который включает по меньшей мере две следующие друг за другом реакционные зоны, а засыпка по меньшей мере ближайшей ко входу реактора реакционной зоны включает катализатор. Благодаря катализатору при по-прежнему высокой конверсии акролеина уменьшается переокисление до COи повышается селективность образования акриловой кислоты. 3 н. и 11 з.п. ф-лы, 14 ил., 5 табл., 13 пр.
Формула
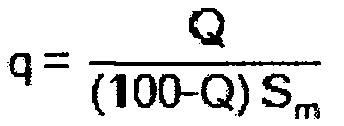
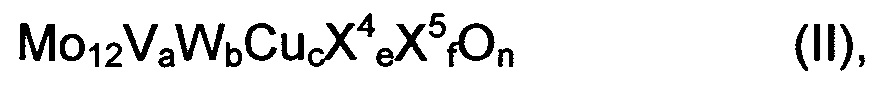
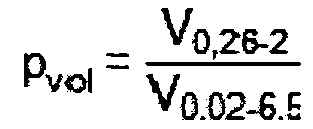
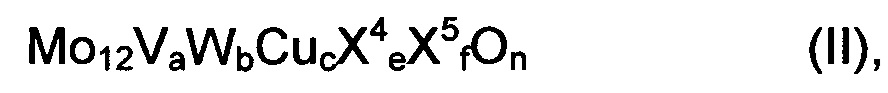
Документы, цитированные в отчёте о поиске
Способ получения акролеина, или акриловой кислоты, или их смесей из пропана
Комментарии