Способ получения усовершенствованных катализаторов аммоксидирования на основе смешанных оксидов металлов - RU2560878C2
Код документа: RU2560878C2
Чертежи


Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка является родственной (i) заявке на патент США регистрационный №12/661705, озаглавленной "Improved Mixed Metal Oxide Ammoxidati Ox Catalysts", поданной 23 марта 2010 года Brazdil et al. под регистрационным номером патентного поверенного 50042, и ii) заявке на патент США регистрационный №12/661720, озаглавленной "AttritiOx Resistance Mixed Metal Oxide Catalysts", поданной 23 марта 2010 года Brazdil et al. под регистрационным номером патентного поверенного 50047, причем обе заявки поданы в один день с данной заявкой и включены в настоящее изобретение посредством отсылки для всех целей.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к усовершенствованному катализатору для применения в реакции аммоксидирования ненасыщенного углеводорода в соответствующий ненасыщенный нитрил. В частности, настоящее изобретение относится к способу получения усовершенствованного катализатора аммоксидирования пропилена и/или изобутилена в акрилонитрил и/или метакрилонитрил, соответственно. Более конкретно, изобретение относится к способу получения усовершенствованного многокомпонентного катализатора аммоксидирования, в соответствии с которым предварительно готовят часть катализатора, содержащую комплекс висмута, молибдена, церия и по меньшей мере одного из металлов: натрия, калия, цезия, рубидия, кальция, лантана или иттрия, а затем объединяют с остальными элементами катализатора.
УРОВЕНЬ ТЕХНИКИ
Катализаторы, содержащие оксиды железа, висмута и молибдена, промотируемые соответствующими элементами, уже долгое время применяются для превращения пропилена и/или изобутилена в акрилонитрил и/или метакрилонитрил в присутствии аммиака и кислорода (обычно в виде воздуха). В частности, патент Великобритании 1436475; патенты США 4766232; 4377534; 4040978; 4168246; 5223469 и 4863891, каждый, относятся к катализаторам на основе висмута-молибдена-железа, которые могут промотироваться элементами II для получения ацетонитрила. Помимо этого, патенты США 5093299, 5212137, 5658842 и 5834394 относятся к промотированным висмут-молибденовым катализаторам, обеспечивающим высокие выходы акрилонитрила.
В частности, настоящее изобретение относится к катализаторам на основе висмута-молибдена-железа. Обычно эти катализаторы получают периодическим способом путем простого соединения и взаимодействия исходных (источников) для получения различных металлических компонентов. Однако используют более сложные и многостадийные способы. Например, в патенте США 4040978 раскрыт способ получения катализатора, в котором молибдаты каждого металла получали отдельно и затем соединяли для осуществления их взаимодействия; патент США 4148757 описывает способ получения катализатора, согласно которому висмут и молибден сначала вводят в реакцию с получением молибдата висмута, а затем молибдат висмута соединяют со смесью соединений, являющихся исходными для получения различных других металлических компонентов.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к усовершенствованному катализатору на основе смешанных оксидов металлов. Этот усовершенствованный катализатор, полученный, как представлено в данном описании, обеспечивает более полную конверсию пропилена и/или изобутилена в нитрилы (т.е. соединения, содержащие функциональную группу "-CN", такие как акрилонитрил, метакрилонитрил, ацетонитрил и цианистый водород).
В одном варианте изобретение относится к способу получения указанного катализатора, причем соотношение элементов в указанном катализаторе представлено следующей формулой:
Mo12BiaFebAcDdЕеFfGgCehOx,
где:
А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
a, b, с, d, е, f, g, h и x, соответственно, обозначают число атомов висмута (Bi), железа (Fe), A, D, Е, F, церия (Се) и кислорода, приходящееся на 12 атомов молибдена (Мо), где:
а имеет значение от 0,05 до 7,
b имеет значение от 0,1 до 7,
с имеет значение от 0,01 до 5,
d имеет значение от 0,1 до 12,
е имеет значение от 0 до 5,
f имеет значение от 0 до 5,
g имеет значение от 0 до 0,2,
h имеет значение от 0,01 до 5 и
х обозначает число атомов кислорода, необходимое для насыщения валентности других имеющихся элементов;
и где элементы в указанном катализаторе объединяются вместе в водной суспензии предшественника катализатора, полученную таким образом суспензию предшественника катализатора сушат с образованием предшественника катализатора, а предшественник катализатора прокаливают с образованием указанного катализатора, причем процесс включает:
(i) смешивание, в водном растворе, исходных соединений Bi и Се и, необязательно, одного элемента из группы: Na, К, Rb, Cs, Са, редкоземельного элемента, Pb, W и Y с образованием смеси,
(ii) добавление к смеси исходного соединения молибдена для взаимодействия со смесью и образования суспензии осадка и
(iii) смешивание суспензии осадка с исходными соединениями остальных элементов катализатора и остального молибдена с образованием водной суспензии предшественника катализатора.
Настоящее изобретение также относится к процессам конверсии олефина, выбранного из группы, состоящей из пропилена и изобутилена или их смесей, в акрилонитрил и/или метакрилонитрил и являющиеся побочными продуктами другие нитрилы (т.е. соединения с функциональной группой "-CN", такие как ацетонитрил и цианистый водород) и их смеси, при взаимодействии указанного олефина в паровой фазе при повышенной температуре и повышенном давлении с газом, содержащим молекулярный кислород и аммиак, в присутствии катализатора на основе смешанных оксидов металлов, который получают по данному описанию.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
На Фигуре 1 представлен график исторических тенденций разработки катализаторов для получения акрилонитрила, показывающий выход акрилонитрила по оси х и выход цианистого водорода по оси у. Этот график показывает, что со временем выход акрилонитрила повышался, а соответствующий выход цианистого водорода снижался. Катализаторы по настоящему изобретению не подтверждают этой тенденции. Катализаторы по настоящему изобретению обеспечивают повышение выхода акрилонитрила без значительного снижения выхода цианистого водорода.
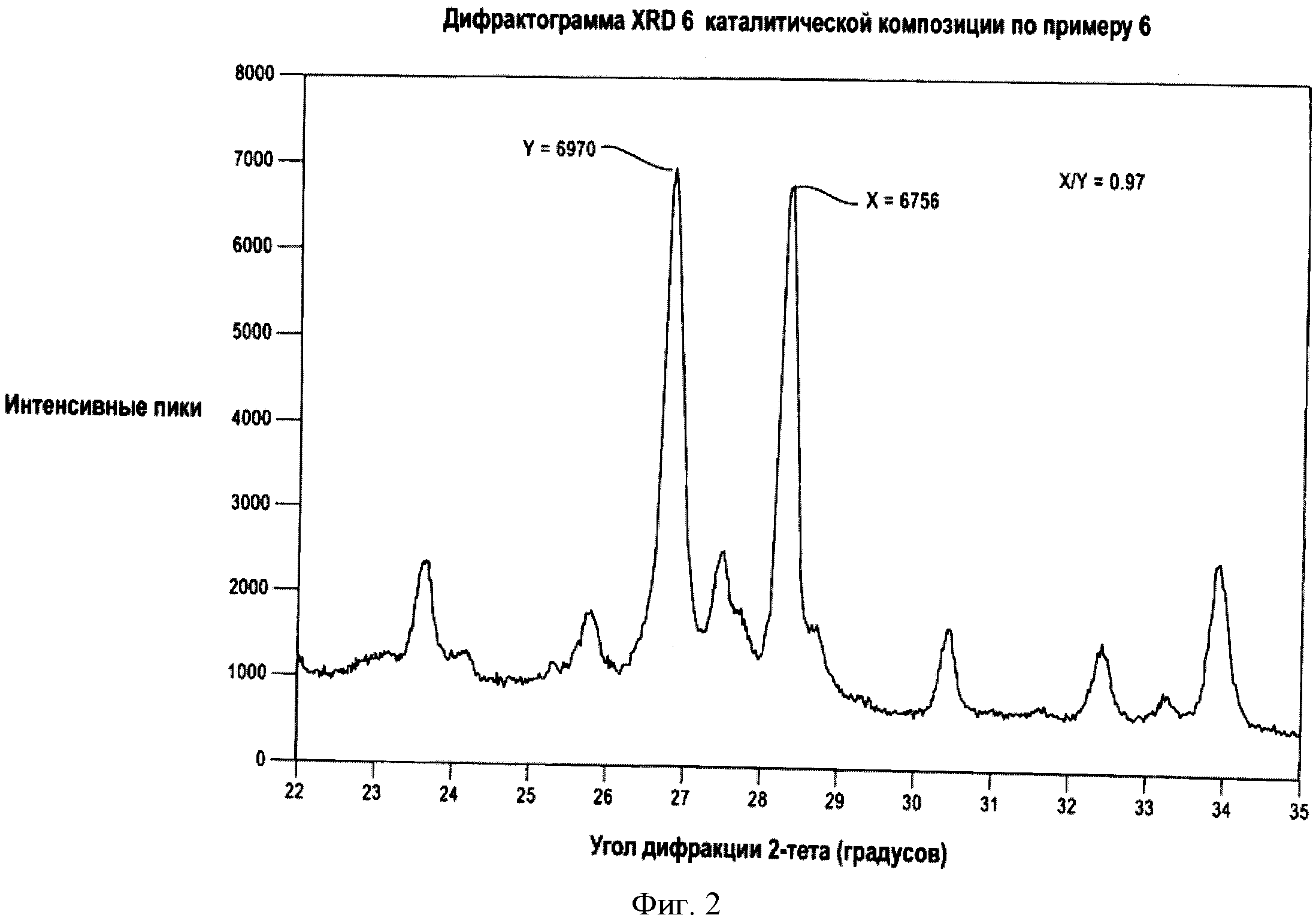
На Фигуре 2 представлена картина рентгеновской (XRD) дифракции или рентгеновская (XRD) дифрактограмма катализатора, входящего в объем настоящего изобретения. На этой дифрактограмме интенсивный пик с величиной угла 2θ 28±0.3 градусов (с интенсивностью, определяемой как "X") и интенсивный дифракционный пик с углом 2θ, равным 26.5±0.3 градусов (с интенсивностью, определяемой как "Y"). Отношение X/Y равно 0.97.
На Фигуре 3 представлена кривая сопротивления истиранию в зависимости от соотношения Ce/Fe в катализаторе. Найдено, что катализаторы, имеющие состав по данному описанию и отношение Ce/Fe, большее или равное 0.8 и меньшее или равное 5, проявляют тенденцию к тому, чтобы становиться более стойкими (прочными, выносливыми) в том смысле, что у них наблюдается меньшая (более низкая) потеря массы на истирание, определяемая в тесте на истирание с помощью затопленной струи.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Предметом настоящего изобретения является новый катализатор, содержащий уникальную комбинацию и уникальное соотношение промоторов, обеспечивающих повышенные рабочие характеристики катализатора в процессе каталитического аммоксидирования пропилена, изобутилена или их смесей в акрилонитрил, метакрилонитрил и их смеси, соответственно.
Катализатор
Настоящее изобретение относится к многокомпонентной (каталитической) композиции катализатора аммоксидирования на основе смешанных оксидов металлов, содержащей каталитические оксиды, причем соотношение элементов в указанной каталитической композиции представлено следующей формулой:
Mo12BiaFebAcDdЕеFfGgCehOx,
где:
А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
а, b, с, d, е, f, g, h и х, соответственно, обозначают число атомов висмута (Bi), железа (Fe), A, D, Е, F, церия (Се) и кислорода, приходящееся на 12 атомов молибдена (Мо),
где:
а имеет значение от 0,05 до 7,
b имеет значение от 0,1 до 7,
с имеет значение от 0,01 до 5,
d имеет значение от 0,1 до 12,
е имеет значение от 0 до 5,
f имеет значение от 0 до 5,
g имеет значение от 0 до 0,2,
h имеет значение от 0,01 до 5 и
х обозначает число атомов кислорода, необходимое для насыщения валентности других имеющихся элементов.
В одном варианте описанной выше каталитической композиции 0.15≤(a+h)/d≤1. В другом варианте описанной выше каталитической композиции 0.8≤h/b≤5. Еще в одном варианте описанной выше каталитической композиции картина дифракции рентгеновских лучей вышеуказанной каталитической композиции включает дифракционные пики при угле 2θ 28±0.3 градусов и угле 2θ 26.5±0.3 градусов, и если отношение интенсивности наиболее интенсивного дифракционного пика при 2θ угле 28±0.3 градусов к интенсивности наиболее интенсивного дифракционного пика при угле 2θ 26.5±0.3 градусов определяется как X/Y, тогда отношение X/Y больше или равно 0.7. В других независимых вариантах вышеуказанной каталитической композиции 0.2≤(a+h)/d≤0.6; 0.3≤(a+h)/d≤0.5; 1≤h/b≤3; 1.5≤h/b≤2; отношение X/Y больше или равно 0.8 и/или отношение X/Y больше или равно 0.90.
В одном варианте изобретения (в котором 0.8≤h/b≤5) "h/b" представляет собой отношение церия к железу в катализаторе, и для любого каталитического состава это отношение представляет собой просто число молей церия (представленное нижним индексом при атоме церия в формуле), деленное на число молей железа (представленное нижним индексом при атоме железа в формуле). Было найдено, что катализаторы, описываемые вышеприведенной формулой, в которой 0.8≤h/b≤5, проявляют тенденцию к тому, чтобы становиться более стойкими (прочными, выносливыми) в том смысле, что у них наблюдается меньшая (более низкая) потеря на истирание, определяемая в тесте на истирание с помощью затопленной струи.
В одном варианте изобретения, характеризуемом картиной рентгеновской дифракции, вышеуказанная каталитическая композиция имеет дифракционные пики при угле 2θ 28±0.3 градусов и угле 2θ 26.5±0.3 градусов, и, если отношение интенсивности наиболее интенсивного дифракционного пика при угле 2θ 28±0.3 градусов к интенсивности наиболее интенсивного дифракционного пика при угле 2θ 26.5±0.3 градусов определяется как X/Y, тогда отношение X/Y больше или равно 0.7, и было найдено, что такие катализаторы обеспечивают более высокую общую конверсию при аммоксидировании пропилена и/или изобутилена в нитрилы (т.е. соединения, содержащие функциональную группу "-CN", такие как акрилонитрил, метакрилонитрил, ацетонитрил и цианистый водород).
Применяемые в данном описании термины "каталитическая композиция" и "катализатор" являются синонимами и употребляются на равных основаниях. Употребляемый в данном описании термин "редкоземельный элемент" означает по меньшей мере один из элементов: лантан, церий, празеодим, неодим, прометий, самарий, европий, гадолиний, тербий, диспрозий, гольмий, эрбий, туллий, иттербий, скандий и иттрий. Применяемый в данном описании угол "2θ" является синонимом угла "2 тета".
Катализатор по настоящему изобретению может применяться в виде катализатора на носителе (подложке) или в виде катализатора без носителя (т.е. катализатор может содержать носитель (подложку)). Подходящими носителями являются диоксид кремния, оксид алюминия, цирконий и диоксид титана или их смеси. Обычно носитель служит в качестве связующего для катализатора и позволяет получать более стойкий катализатор (т.е. с большим сопротивлением истиранию). Однако для промышленного применения соответствующая смесь активной фазы (т.е. описанного выше комплекса каталитических оксидов) и носителя (подложки) является принципиально важной для достижения приемлемой активности и прочности (твердости, сопротивления истиранию) катализатора. Обычно носитель составляет от 40 до 60% вес. от веса катализатора на носителе. В одном варианте данного изобретения носитель может составлять примерно до 30% вес. от веса катализатора на носителе. В другом варианте настоящего изобретения носитель может составлять примерно до 70% вес. от веса катализатора на носителе.
В одном варианте изобретения катализатор наносят, используя золь кремниевой кислоты. Обычно силикатные золи кремниевой кислоты содержат некоторое количество натрия. В одном варианте изобретения золь кремниевой кислоты содержит менее 600 частей на миллион (м.д.) натрия. В другом варианте изобретения золь кремниевой кислоты содержит менее 200 частей на миллион (м.д.) натрия. Обычно средний диаметр коллоидных частиц золя кремниевой кислоты составляет около 15-50 нм. В одном варианте настоящего изобретения средний диаметр коллоидных частиц силикатного золя составляет около 10 нм и может быть до 4 нм. В другом варианте настоящего изобретения средний диаметр коллоидных частиц золя кремниевой кислоты составляет около 100 нм. В другом варианте настоящего изобретения средний диаметр коллоидных частиц золя кремниевой кислоты составляет около 20 нм.
Приготовление катализатора
В одном варианте изобретения элементы композиции вышеуказанного катализатора смешивают в виде водной суспензии предшественника катализатора, полученную таким образом суспензию предшественника катализатора сушат с образованием предшественника катализатора, а предшественник катализатора прокаливают с образованием указанного катализатора. Однако уникальностью процесса по настоящему изобретению является следующее:
(i) смешивание, в водном растворе, исходных соединений (источников) Bi и Се и, необязательно, одного или более элементов из группы: Na, K, Rb, Cs, Ca, редкоземельного элемента, Pb, W и Y с образованием смеси (т.е. первой смеси),
(ii) добавление к смеси (т.е. первой смеси) исходного соединения (источника) молибдена для взаимодействия со смесью и образования "суспензии осадка" и
(iii) смешивание "суспензии осадка" с исходными соединениями остальных элементов катализатора и остального молибдена с образованием водной суспензии предшественника катализатора.
Термины по данному описанию "исходные соединения", "соединения-источники", "исходные" означают соединения, которые содержат и/или предоставляют один или более металлов для каталитической композиции на основе оксидов металлов. Выражение "остальные элементы" или "остальные элементы в катализаторе" относятся к элементам и к количеству этих элементов, представленных символами "A", "D", "Е", "F" и "G" в вышеприведенной формуле, которые не входят в состав первой смеси. В одном варианте изобретения некоторые элементы могут входить в состав как первой, так и второй смеси. Помимо этого, выражения "остальной молибден" или "остальной молибден в катализаторе" относятся к необходимому количеству молибдена в конечном катализаторе, которое не входит в состав суспензии осадка (т.е. не включено при получении этой суспензии). И наконец, суммарное количество молибдена, предоставляемого исходными молибдена, добавляемыми на стадиях (ii) и (iii), равно общему количеству молибдена в катализаторе.
В композиции катализатора по данному описанию исходные соединения остальных элементов и остального молибдена, которые смешиваются с суспензией осадка, можно объединять в любом порядке или в любой комбинации таких остальных элементов и оставшегося молибдена. В одном варианте изобретения смесь исходных соединений остальных элементов и остального молибдена смешивают с суспензией осадка с образованием водной суспензии предшественника катализатора. В другом варианте изобретения (i) смесь исходных соединений остальных элементов смешивают с суспензией осадка и (ii) исходные соединения остального молибдена отдельно добавляют к суспензии осадка с образованием водной суспензии предшественника катализатора. Порядок добавления не важен. В другом варианте изобретения исходные соединения остальных элементов и остального молибдена добавляют по отдельности (т.е. одновременно добавляют одно соединение) к суспензии осадка. В другом варианте изобретения сложные, многокомпонентные (т.е. содержащие более одного компонента) смеси исходных соединений остальных элементов и остального молибдена, где каждая смесь содержит одно или более исходных соединений остальных элементов или остального молибдена, добавляют по отдельности (т.е. одновременно добавляют одну смесь или многокомпонентные смеси) к суспензии осадка с образованием водной суспензии предшественника катализатора. Еще в одном варианте изобретения смесь исходных соединений остальных элементов смешивают с исходным соединением молибдена, а затем полученную смесь добавляют к суспензии осадка с образованием суспензии предшественника катализатора. Еще в одном варианте изобретения носителем является диоксид кремния (SiO2), и диоксид кремния смешивают с исходным соединением остального молибдена, а затем смешивают остальной молибден с суспензией осадка (т.е. диоксид кремния и исходное соединение остального молибдена смешивают с образованием смеси, а затем эту смесь добавляют к "суспензии осадка", индивидуально или в комбинации с одним или более исходных соединений остальных элементов).
При получении катализатора по данному описанию молибден добавляют как при приготовлении суспензии осадка, так и при приготовлении водной суспензии предшественника осадка. На атомном уровне минимальное количество молибдена, добавляемого для образования суспензии осадка, определяется нижеприведенным соотношением
Мо=1.5(Bi+Се)+0.5(Rb+Na+K+Cs)+(Са)+1.5 (сумма числа атомов редкоземельных элементов)+(Pb)+3(W)+1.5(Y).
Причем в вышеприведенном соотношении "Мо" обозначает число атомов молибдена, которое следует добавить к первой смеси, a "Bi", "Се", "Rb", "Na", "K", "Cs", "Са", "Pb", "W" и "Y" обозначают число атомов висмута, церия, рубидия, натрия, калия, цезия, кальция, свинца, вольфрама и иттрия, соответственно, в первой смеси.
Обычно количество молибдена, добавляемого к первой смеси для образования суспензии осадка, составляет около 20-35% от всего молибдена в конечном катализаторе. В одном варианте изобретения исходное соединение остального молибдена в катализаторе добавляют к смеси исходных соединений остальных элементов (т.е. второй смеси) перед объединением смеси остальных элементов с суспензией осадка с образованием суспензии предшественника катализатора. В других вариантах изобретения исходное соединение молибдена, содержащее остальной молибден в катализаторе, добавляют к суспензии осадка либо до, либо после добавления, либо одновременно с добавлением смеси исходных соединений остальных элементов (т.е. второй смеси) для получения суспензии предшественника катализатора.
В процессе приготовления по настоящему изобретению исходные соединения Bi и Се и, необязательно, одного или более элементов из группы: Na, K, Rb, Cs, Са, редкоземельный элемент, Pb, W и Y смешивают в водном растворе с образованием смеси. В одном варианте изобретения нитрат висмута и, необязательно, нитраты других металлов (т.е. нитраты Na, K, Rb, Cs, Са, редкоземельного элемента, Pb, W и/или Y) растворяют в водном растворе нитрата церия-аммония.
К смеси, содержащей висмут и церий (и, необязательно, один или более элементов, выбранных из Na, K, Rb, Cs, Са, редкоземельного элемента, Pb, W и Y), добавляют источник молибдена. После добавления исходного соединения молибдена к смеси, содержащей висмут и церий, идет реакция, в результате которой образуется осадок, а полученная смесь представляет собой суспензию осадка.
Затем суспензию осадка объединяют со смесью исходных соединений остальных элементов катализатора и исходного соединения молибдена, при этом образуется суспензия предшественника катализатора. Смесь исходных соединений остальных элементов и исходного соединения молибдена можно приготовить, смешивая объединенные исходные соединения остальных элементов в водном растворе (например, исходные соединения смешивают в воде), а затем добавляя исходное соединение молибдена. В одном варианте изобретения этим исходным соединением молибдена является гептамолибдат аммония, растворенный в воде. При смешении суспензии осадка со смесью остальных элементов с молибденом порядок добавления не важен, т.е. можно добавлять суспензию осадка к смеси остальных элементов с молибденом или смесь остальных элементов с молибденом можно добавлять к суспензии осадка. Водную суспензию предшественника катализатора поддерживают при повышенной температуре.
Количество водного растворителя в каждой из вышеописанных смесей и суспензий может меняться в зависимости от растворимости исходных соединений, смешиваемых с образованием конкретного смешанного оксида металла. Количество водного растворителя должно быть по меньшей мере достаточным для получения суспензии или смеси твердых и жидких веществ, которое способно перемешиваться.
Во всяком случае, смешивание и/или реакцию исходных соединений, предпочтительно, проводят в соответствии с протоколом, который включает смешивание исходных соединений на стадии объединения и/или реакции. Конкретный механизм смешения не имеет принципиального значения и может включать, например, смешивание (например, перемешивание или взбалтывание) компонентов в процессе реакции любым эффективным методом. Такие методы включают, например, взбалтывание содержимого сосуда, например, встряхиванием, вращением или вибрацией сосуда, содержащего компоненты. Такие методы включают также, например, перемешивание с помощью мешалки, расположенной, по меньшей мере частично, внутри реакционного сосуда, а движущая сила прилагается к мешалке или к реактору, чтобы обеспечить движение мешалки и реактора относительно друг друга. Мешалка может приводиться в движение приводом или без привода (непосредственно от мотора). Движущая сила может прикладываться к мешалке непосредственно или опосредованно (например, с помощью магнита). Обычно при смешении компонентов перемешивания бывает достаточно для того, чтобы осуществить эффективную реакцию между компонентами реакционной среды с образованием более гомогенной реакционной среды (например, для получения более гомогенного предшественника смешанных оксидов металлов) по сравнению с реакцией без перемешивания. Это приводит к более эффективному расходу исходных и дает более однородный продукт на основе смешанных оксидов металлов. Перемешивание суспензии осадка на стадии реакции также вызывает предпочтительное образование осадка в объеме раствора, а не на стенках реакционного сосуда. Более предпочтительно, когда осадок образуется в растворе, это делает возможным равномерный рост частиц по всем боковых поверхностям, а не ограничивается экспонированными поверхностями, как при росте на стенке реакционного сосуда.
Исходное соединение молибдена может включать оксид молибдена (VI) (МоО3), гептамолибдат аммония или молибденовую кислоту. Исходное соединение молибдена можно вводить с применением любого оксида молибдена, такого как диоксид, триоксид, пентаоксид или гептаоксид. Однако предпочтительно в качестве исходного соединения молибдена использовать гидролизующуюся или разрушающуюся соль молибдена.
Типичными исходными соединениями висмута, церия и остальных элементов катализатора являются нитраты металлов. Такие нитраты легкодоступны и хорошо растворимы.
Исходное соединение висмута может включать оксид или соль, которая при прокаливании дает оксид. Предпочтительными являются водорастворимые соли, которые легко диспергируются, но образуют устойчивые при нагревании оксиды. В одном варианте изобретения исходным соединением висмута является нитрат висмута, Bi(NO3)3·5H2O.
Исходное соединением церия может включать оксид или соль, которая при прокаливании дает оксид. Предпочтительными являются водорастворимые соли, которые легко диспергируются, но образуют устойчивые при нагревании оксиды. В одном варианте изобретения исходным соединением церия является нитрат аммония церия, (NH4)2Се(NO3)6.
Исходным соединением железа может являться любое соединение железа, которое при прокаливании дает оксид. Что касается других элементов, предпочтительными являются водорастворимые соли из-за легкости, с которой они могут равномерно диспергироваться в катализаторе. Наиболее предпочтительным является нитрат трехвалентного железа.
Исходные соединения остальных элементов могут быть из любого подходящего источника. Например, кобальт, никель и магний можно вводить в катализатор, используя нитраты. Кроме того, можно вводить эти элементы в катализатор в виде нерастворимого карбоната или гидроксида, которые при нагревании дают оксид. Фосфор можно вводить в катализатор в виде соли щелочного или щелочноземельного металла или аммониевой соли, но предпочтительно вводить его в виде фосфорной кислоты.
Исходные соединения щелочных компонентов катализатора могут представлять собой оксид или соль, которая при прокаливании дает оксид.
Растворители, помимо воды, которые можно вводить для приготовления смешанных оксидов металлов по изобретению, включают, но без ограничения, спирты, такие как метанол, этанол, пропанол, диолы (например, этиленгликоль, пропиленгликоль и т.д.), органические кислоты, такие как уксусная кислота, а также другие полярные растворители, известные в уровне техники. Исходные соединения металлов по меньшей мере частично растворимы в растворителе.
Как отмечалось ранее, катализатор по настоящему изобретению может быть на носителе и без носителя (т.е. катализатор может содержать носитель). Подходящими носителями являются диоксид кремния, оксид алюминия, диоксиды циркония, титана или их смеси. Время добавления носителя не играет важной роли при приготовлении катализатора. Носитель можно добавлять в любое время до процесса сушки суспензии предшественника катализатора. Носитель можно добавлять в любой момент во время или после приготовления любой смеси элементов, суспензии осадка или суспензии предшественника катализатора. Кроме того, носитель необязательно добавлять в одно время или на одной стадии (т.е. в процессе приготовления катализатора носитель можно добавлять неоднократно в различные моменты). В одном варианте изобретения носитель смешивают с другими ингредиентами во время приготовления водной суспензии предшественника катализатора. В одном варианте изобретения носитель добавляют к суспензии осадка (т.е. после того, как получена суспензия осадка). В одном варианте изобретения носитель смешивают с исходным соединением молибдена до момента смешения исходного соединения молибдена с исходными соединениями остальных элементов в катализаторе с образованием указанной выше "второй смеси".
Суспензию предшественника катализатора сушат и "денитрифицируют" (т.е. удаляют нитраты), с получением предшественника катализатора. Предпочтительно, суспензию предшественника катализатора сушат методом распылительной сушки при температуре на выходе из сушилки между 110°С и 350°С, предпочтительно между 110°С и 250°С, наиболее предпочтительно между 110°С и 180°С. Интервал температуры денитрификации составляет от 100°С до 500°С, предпочтительно от 250°С до 450°С.
Наконец, высушенный предшественник катализатора прокаливают. В одном варианте изобретения прокаливание проводят на воздухе. В другом варианте изобретения прокаливание проводят в атмосфере инертного газа, такого как азот. Предпочтительные условия прокаливания включают интервалы температур примерно от 300°С примерно до 700°С, более предпочтительно примерно от 300°С примерно до 650°С и в некоторых вариантах изобретения прокаливание можно проводить примерно при 600°С.
Катализаторы по настоящему изобретению можно готовить любым из многочисленных методов приготовления катализаторов, которые известны специалистам в данной области техники. В одном варианте изобретения компоненты катализатора могут смешиваться с носителем в виде суспензии с последующей сушкой или диоксид кремния или другие носители могут пропитываться компонентами катализатора.
Процесс аммоксидирования:
Катализаторы по настоящему изобретению применимы в процессах аммоксидирования для конверсии олефинов, выбранных из группы, состоящей из пропилена, изобутилена или их смесей, в акрилонитрил, метакрилонитрил и их смеси, соответственно, реакцией указанного олефина с газом, содержащим молекулярный кислород и аммиак, в паровой фазе при повышенной температуре и повышенном давлении в присутствии катализатора. Катализаторы по настоящему изобретению применимы также для аммоксидирования метанола в цианистый водород и аммоксидирования этанола в ацетонитрил. В одном варианте изобретения, в котором применяются катализаторы по данному описанию, метанол и/или этанол могут подаваться совместно для аммоксидирования пропилена, изобутилена или их смесей в акрилонитрил, метакрилонитрил и/или их смеси для повышения выхода цианистого водорода и/или ацетонитрила, образующихся в этом процессе в качестве побочных продуктов.
Предпочтительно, реакцию аммоксидирования проводят в реакторе с псевдоожиженным слоем, хотя предусматриваются также реакторы других типов, таких как реакторы с транспортной линией. Реакторы с псевдоожиженным слоем для получения акрилонитрила хорошо известны в уровне техники. Например, применим дизайн реактора, представленный в патенте США №3230246, содержание которого включается в настоящее изобретение посредством отсылки.
Условия проведения реакции аммоксидирования также хорошо известны в уровне техники, о чем свидетельствуют патенты США №5093299; 4863891; 4767878 и 4503001; содержание которых вводится в настоящее изобретение посредством отсылки. Как правило, процесс аммоксидирования осуществляют контактированием пропилена или изобутилена в присутствии аммиака и кислорода с псевдоожиженным слоем катализатора при повышенной температуре с образованием акрилонитрила или метакрилонитрила. Можно использовать любой источник кислорода. Однако по экономическим причинам предпочтительно использовать воздух. Обычно молярное соотношение кислорода и олефина в подаваемой смеси должно составлять от 0.5:1 до 4:1, предпочтительно от 1:1 до 3:1.
Молярное соотношение аммиака к олефину в смеси, подаваемой в реактор, может варьироваться от 0.5:1 до 2:1. На самом деле не существует верхнего предела соотношения аммиак-олефин, но обычно по экономическим причинам нет никакого резона превышать соотношение 2:1. Соответствующие соотношения в подаваемой смеси для применения с катализатором по настоящему изобретению для получения акрилонитрила из пропилена составляют: соотношение аммиака и пропилена в интервале от 0.9:1 до 1.3:1, а соотношение воздуха и пропилена в интервале от 8.0:1 до 12.0:1. Катализатор по настоящему изобретению способен обеспечивать высокие выходы акрилонитрила при сравнительно низком соотношении аммиака к пропилену в подаваемой смеси от около 1:1 до около 1.05:1. Такое "низкое содержание аммиака" позволяет уменьшить содержание непрореагировавшего аммиака в продуктах реакции, результат, известный как "шаг вперед" в применении аммиака, который впоследствии сделает возможным уменьшить промышленные отходы. Конкретно, непрореагировавший аммиак следует удалять из продуктов реакции перед регенерацией (улавливанием) акрилонитрила. Непрореагировавший аммиак обычно удаляют взаимодействием продуктов реакции с серной кислотой с образованием сульфата аммония или взаимодействием продуктов реакции с акриловой кислотой с образованием акрилата аммония, что в обоих случаях приводит в результате к обработке и/или сбросу отходов производства.
Реакцию проводят при в примерном температурном интервале 260-600°С, предпочтительно в интервале 310-500°С, особенно предпочтительно в интервале 350-480°С. Время контакта, хотя это не является особо важным, обычно составляет диапазон от 0.1 до 50 секунд, предпочтительно от 1 до 15 секунд.
Продукты реакции можно выделять и очищать любыми методами, известными специалистам в данной области техники. Один такой метод включает промывку выходящих из реактора газов холодной водой или подходящим растворителем с целью удаления продуктов реакции, а затем очистку продукта реакции перегонкой.
Главным применением катализатора, полученного в процессе по настоящему изобретению, является применение для аммоксидирования пропилена в акрилонитрил. Другие варианты применения включают аммоксидирование пропана в акрилонитрил и аммоксидирование этанола в ацетонитрил. Катализатор, полученный способом по настоящему изобретению, можно также применять для окисления пропилена в акриловую кислоту. Такие процессы являются обычно двухстадийными процессами, в которых на первой стадии пропилен в присутствии катализатора на первой стадии превращается главным образом в акролеин, а на второй стадии акролеин в присутствии катализатора превращается в основном в акриловую кислоту. Катализатор по данному описанию применим для окисления пропилена в акролеин на первой стадии.
КОНКРЕТНЫЕ ВАРИАНТЫ ИЗОБРЕТЕНИЯ
С целью иллюстрации настоящего изобретения катализатор, приготовленный в соответствии с настоящим изобретением, изучали и сравнивали, в аналогичных условиях реакции, с аналогичными катализаторами, полученными методами, известными в уровне техники, не входящими в объем настоящего изобретения. Эти примеры приводятся лишь с целью иллюстрации.
Катализаторы, имеющие состав Cs0.1K0.1Ni5Mg2Na0.05Fe1.8Bi0.45Ce1.1Mo12.55O50.35+45% вес. Na SiO2, получали различными препаративными методами, описанными ниже, и испытывали в лабораторном реакторе в реакции аммоксидирования пропилена в акрилонитрил. Все испытания проводили в реакторе с псевдоожиженным слоем катализатора на 40 мл. Пропилен подавали в реактор со скоростью 0.06 WWH (т.е. вес пропилена/вес катализатора/час). Давление внутри реактора поддерживали при 10 psig (68.95 кПа). Температура реакции 430°С. После периода стабилизации, ~20 часов или более, проводили отбор образцов продуктов реакции. Продукты реакции собирали в скрубберах барботажного типа с холодным раствором HCl. Скорость отходящих газов измеряли с помощью пленочного расходомера, а состав отходящих газов определяли в конце опыта на газовом хроматографе, снабженном газоанализатором с разделенными потоками (с колонки). По окончании извлечения всю жидкость со скруббера разводили дистиллированной водой примерно до 200 г. В качестве внутреннего стандарта использовали 2-бутанон в аликвоте ~50 грамм разбавленного раствора. Образец 2 мкл анализировали на газовом хроматографе (ГХ), снабженном пламенно-ионизационным детектором и колонкой Carbowax. Количество NH3 определяли титрованием избытка свободной HCl раствором NaOH.
Сравнительный пример С1: Традиционный метод
Реакционную смесь А готовили, нагревая 224 млм деионизированной воды до 65°С, а затем добавляли при перемешивании в течение 30 минут гептамолибдат аммония (203.75 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (625 г, 32.5% диоксида кремния).
Реакционную смесь В готовили, нагревая 30 мл деионизированной воды до 55°С, а затем добавляя Fe(NO3)3·9H2O (66.9 г), Ni(NO3)2·6H2O (133.7 г), Mg(NO3)2·6H2O (47.2 г), Bi(NO3)3·5H2O (20.1 г), CsNO3 (1.79 г), KNO3 (0.93 г) и NaNO3 (0.39 г). Затем при перемешивании прибавляли 110.0 г 50% вес. водного раствора (NH4)2Се(NO3)6.
Затем при перемешивании к реакционной смеси А прибавляли реакционную смесь В, при этом получали суспензию предшественника катализатора. Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили распылительной сушкой при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, а затем еще в течение 3 часов при температуре 425°С. Затем порошок прокаливали на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена в микрореакторе на 40 мл (см). Результаты испытания приведены в Таблице 1.
Сравнительный пример С2: Приготовление в соответствии с патентом США 4212766 (т.е. при отсутствии суспензии осадка Bi-Ce-Mo, полученного на отдельной стадии)
Реакционную смесь А готовили, нагревая 233 мл деионизированной воды до 65°С, добавляли при перемешивании в течение 30 минут гептамолибдат аммония (212.1 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (692 г, 32.5% диоксида кремния).
Реакционную смесь В готовили, нагревая 33 мл деионизированной воды до 55°С, а затем последовательно при перемешивании добавляли Fe(NO3)3·9H2O (73.6 г), Ni(NO3)2·6H2O (147.1 г), Mg(NO3)2·6H2O (51.9 г), CsNO3 (1.97 г), KNO3 (1.02 г) и NaNO3 (0.43 г) и 122.0 г 50% вес. водного раствора (NH4)2Се(NO3)6.
Реакционную смесь С готовили, нагревая 152 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (12.1 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, растворяя Bi(NO3)3·5H2O (22.1 г) в 160 г 10% вес. водного раствора HNO3.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь D к реакционной смеси С. При этом выпадал бесцветный осадок. Перемешивание продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, а затем еще в течение 3 часов при температуре 425°С. Затем порошок прокаливали на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена в микрореакторе на 40 мл (см3). Результаты испытания приведены в Таблице 1.
Пример 1: Приготовление катализатора в соответствии с изобретением
Реакционную смесь А готовили, нагревая 198 мл деионизированной воды до 65°С, а затем добавляли при перемешивании в течение 30 минут гептамолибдат аммония (180.4 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (692 г, 32.5% диоксида кремния).
Реакционную смесь В готовили, нагревая 33 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (73.6 г), Ni(NO3)2·6H2O (147.1 г) и Mg(NO3)2·6H2O (51.9 г).
Реакционную смесь С готовили, нагревая 48 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (43.75 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 122.0 г 50% вес. водного раствора (NH4)2Ce(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно добавляли Bi(NO3)3·5H2O (22.1 г), CsNO3 (1.97 г), KNO3 (1.02 г) и NaNO3 (0.43 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь С к реакционной смеси D. При этом выпадал оранжевый осадок. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при скорости 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, а затем еще в течение 3 часов при температуре 425°С. Затем порошок прокаливали на воздухе при 560°С в течение 3 часов. Затем полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена в микрореакторе на 40 мл (см3). Результаты испытания приведены в Таблице 1.
Пример 2: Приготовление катализатора в соответствии с изобретением
Реакционную смесь А готовили, нагревая 198 мл деионизированной воды до 65°С, а затем добавляли при перемешивании в течение 30 минут гептамолибдат аммония (180.4 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь В готовили, нагревая 33 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (73.6 г), Ni(NO3)2·6H2O (147.1 г) и Mg(NO3)2·6H2O (51.9 г).
Реакционную смесь С готовили, нагревая 48 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (43.75 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 122.0 г 50% вес. водного раствора (NH4)2Се(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно добавляли Bi(NO3)3·5H2O (22.1 г), CsNO3 (1.97 г), KNO3 (1.02 г) и NaNO3 (0.43 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, (i) добавляя реакционную смесь С к реакционной смеси D, что сопровождалось выпадением оранжевого осадка (эта полученная смесь представляла собой суспензию осадка), (ii) перемешивание суспензии осадка продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С, и (iii) при перемешивании прибавляли золь кремниевой кислоты (692 г, 32.5% вес. диоксида кремния).
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при скорости 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, а затем еще в течение 3 часов при температуре 425°С. Затем порошок прокаливали на воздухе при 560°С в течение 3 часов. Затем полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена в микрореакторе на 40 мл (см3). Результаты испытания приведены в Таблице 1.
Как можно видеть в Таблице 1, композиции катализатора, приготовленного методом по настоящему изобретению, обуславливали более высокую конверсию в акрилонитрил и HCN, когда пропилен аммоксидировали над такими катализаторами при повышенных температурах в присутствии аммиака в смеси с воздухом, по сравнению с идентичными катализаторами, полученными методами, не входящими в объем настоящего изобретения.
Для дальнейшей иллюстрации настоящего изобретения приготовили катализатор, входящий в объем настоящего изобретения (Примеры 3-7), катализатор проверяли и сравнивали в аналогичных условиях реакции с аналогичными катализаторами, не входящими в объем настоящего изобретения (Сравнительные примеры С3-С7). Эти примеры приводятся только с целью иллюстрации.
Сравнительный пример 3 - Катализатор С-49МС для получения акрилонитрила
Результаты испытания и другие данные для катализатора С-49МС, предназначенного для получения акрилонитрила, представлены в Таблице 2. "Катализатор С-49МС" - это обозначение изделия - продажного катализатора, производимого и продаваемого кампанией INEOS USA LLC. Композиция С-49МС является производственным секретом кампании INEOS USA LLC. "С-49МС" является товарным знаком INEOS USA LLC.
Сравнительный пример С4 - Ni7.5Mg3Fe1.8Rb0.12Cr0.1Bi0.45Ce1.1Mo15.85O63.835+50% вес. 27 м.д. (ppm) Na, SiO2 с размером частиц 39 нм
Реакционную смесь А готовили, нагревая 225.1 мл деионизированной воды до 65°С, а затем в течение 30 минут при перемешивании прибавляли гептамолибдат аммония (204.6 г), получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты(27 м.д. (ppm) Na, средний размер частиц 39 нм, 595.2 г, 42% диоксида кремния).
Реакционную смесь В готовили, нагревая 32.2 мл деионизированной воды до 55°С, а затем при перемешивании прибавляли Fe(NO3)2·9H2O (53.2 г), Ni(NO3)2·6H2O (159.6 г), Mg(NO3)2·6H2O (56.2 г), Bi(NO3)3·5H2O (16 г), Cr(NO3)3·9H2O (2.9 г) и RbNO3 (1.3 г).
К раствору В прибавляли 88.2 г 50% вес. водного раствора (NH4)2Се(NO3)6, а затем при перемешивании при ~55°С прибавляли полученную смесь к раствору А в течение одного часа с последующим охлаждением до 40°С. Затем полученную суспензию предшественника катализатора гомогенизировали в смесителе в течение 3 мин при скорости перемешивания 5000 об/мин. Затем суспензию сушили распылительной сушкой при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, а затем еще в течение 3 часов на воздухе при температуре 425°С, а затем прокаливали на воздухе при 560°С в течение 3 часов. Затем полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена. Результаты испытания приведены в Таблице 2.
Сравнительный пример С5 - Ni7.5Mg3Fe1.8Rb0.12Cr0.1Bi0.45Ce1.1Mo15.85O63.835+50% вес. SiO2
Реакционную смесь А готовили, нагревая 191.1 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (173.8 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (27 м.д. Na, средний размер частиц 39 нм, 595.2 г, 42% вес. диоксида кремния).
Реакционную смесь В готовили, нагревая 32.1 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (53.2 г), Ni(NO3)2·6H2O (159.5 г), Mg(NO3)2·6H2O (56.2 г) и Cr(NO3)3·9H2O (2.9 г).
Реакционную смесь С готовили, нагревая 33.9 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (30.8 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 88.2 г 50% вес. водного раствора (NH4)2Се(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно прибавляли Bi(NO3)3·5Н2О (16 г) и RbNO3 (1.3 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь С к реакционной смеси D. При этом выпадал оранжевый осадок. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, а затем еще в течение 3 часов на воздухе при температуре 425°С. Затем порошок прокаливали на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена. Результаты испытания приведены в Таблице 2.
Сравнительный пример С6 - Ni5Mg2Fe1.8Rb0.12Cr0.1Bi0.45Ce1.1Mo12.35O49.835+50% вес. SiO2
Реакционную смесь А готовили, нагревая 1619.7 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (1472.5 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (27 м.д. Na, средний размер частиц 39 нм, 5357.1 г, 42% вес. диоксида кремния).
Реакционную смесь В готовили, нагревая 276.2 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (608.6 г), Ni(NO3)2·6H2O (1216.8 г), Mg(NO3)2·6H2O (429.2 г) и Cr(NO3)3·9H2O (33.5 г).
Реакционную смесь С готовили, нагревая 387.7 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (352.4 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 1009.4 г 50% вес. водного раствора (NH4)2Ce(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно прибавляли Bi(NO3)3·5H2O (182.7 г) и RbNO3 (14.8 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь С к реакционной смеси D. При этом выпадал оранжевый осадок. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, в течение 3 часов на воздухе при температуре 425°С, а затем порошок прокаливали на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена. Результаты испытания приведены в Таблице 2.
Сравнительный пример C7 - Ni4Mg3Fe1.8Rb0.192Cr0.1Bi0.45Ce1.1Mo12.386O49.979+50% вес. SiO2
Реакционную смесь А готовили, нагревая 181.5 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (165 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (90 м.д. Na, средний размер частиц 39.2 нм, 606.8 г, 41.2% вес. диоксида кремния).
Реакционную смесь В готовили, нагревая 30.7 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (68.2 г), Ni(NO3)2·6H2O (109.1 г), Mg(NO3)2·6H2O (72.1 г) и Cr(NO3)3·9H2O (3.8 г).
Реакционную смесь С готовили, нагревая 44.1 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (40.1 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 113.1 г 50% вес. водного раствора (NH4)2Ce(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно прибавляли Bi(NO3)3·5Н2О (20.5 г) и RbNO3 (2.7 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь С к реакционной смеси D. При этом выпадал оранжевый осадок. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, в течение 3 часов на воздухе при температуре 425°С, а затем прокаливали на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена. Результаты испытания приведены в Таблице 2.
Пример 3 - Ni4Mg3Fe0.9Rb0.192Cr0.05Bi0.72Ce1.76Mo12.806O51.539+50% вес. SiO2
Реакционную смесь А готовили, нагревая 154.5 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (140.4 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (27 м.д. Na, средний размер частиц 39 нм, 595.2 г, 42% вес. диоксида кремния).
Реакционную смесь В готовили, нагревая 26.5 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (32.2 г), Ni(NO3)2·6H2O (102.9 г), Mg(NO3)2·6H2O (68 г) и Cr(NO3)3·9H2O (1.8 г).
Реакционную смесь С готовили, нагревая 65.5 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (59.6 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 170.6 г 50% вес. водного раствора (NH4)2Се(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно прибавляли Bi(NO3)3·5H2O (30.9 г) и RbNO3 (2.5 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь С к реакционной смеси D. При этом выпадал оранжевый осадок. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, в течение 3 часов на воздухе при температуре 425°С, а затем прокаливали на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена. Результаты испытания приведены в Таблице 2.
Пример 4 - Ni4Mg3Fe0.9Rb0.192Cr0.05Bi0.72Ce1.76Mo12.335O50.126+50% вес. SiO2
Реакционную смесь А готовили, нагревая 149.9 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (136.3 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (27 м.д. Na, средний размер частиц 39 нм, 595.2 г, 42% вес. диоксида кремния).
Реакционную смесь В готовили, нагревая 27.1 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (32.9 г), Ni(NO3)2·6H2O (105.4 г), Mg(NO3)2·6H2O (69.7 г) и Cr(NO3)3·9H2O (1.8 г).
Реакционную смесь С готовили, нагревая 67.1 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (61 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 174.8 г 50% вес. водного раствора (NH4)2Ce(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно прибавляли Bi(NO3)3·5H2O (31.6 г) и RbNO3 (2.6 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь С к реакционной смеси D. При этом выпадал оранжевый осадок. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, в течение 3 часов на воздухе при температуре 425°С, а затем прокаливали на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена. Результаты испытания приведены в Таблице 2.
Пример 5 - Ni4.26Mg3.195Fe0.9Rb0.192Cr0.05Bi0.72Ce1.76Mo12.806O51.994+50% вес. SiO2
Реакционную смесь А готовили, нагревая 152.9 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (139 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (27 м.д. Na, средний размер частиц 39 нм, 595.2 г, 42% вес. диоксида кремния).
Реакционную смесь В готовили, нагревая 27.4 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (31.8 г), Ni(NO3)2·6H2O (108.5 г), Mg(NO3)2·6H2O (71.7 г) и Cr(NO3)3·9H2O (1.8 г).
Реакционную смесь С готовили, нагревая 64.9 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (59 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 169 г 50% вес. водного раствора (NH4)2Ce(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно прибавляли Bi(NO3)3·5H2O (30.6 г) и RbNO3 (2.5 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь С к реакционной смеси D. При этом выпадал оранжевый осадок. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, в течение 3 часов на воздухе при температуре 425°С, а затем прокаливали на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена. Результаты испытания приведены в Таблице 2.
Пример 6 - Ni4Mg3Fe0.9Rb0.192Cr0.05Bi0.72Ce1.76Mo12.5026O50.627+50% вес. SiO2
Реакционную смесь А готовили, нагревая 1363.6 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (1239.6 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (90 м.д. Na, средний размер частиц 39 нм, 5461.2 г, 41.2% вес. диоксида кремния).
Реакционную смесь В готовили, нагревая 241.9 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (239.9 г), Ni(NO3)2·6H2O (940.2 г), Mg(NO3)2·6H2O (621.8 г) и Cr(NO3)3·9H2O (16.2 г).
Реакционную смесь С готовили, нагревая 599.1 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (544.6 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 1559.9 г 50% вес. водного раствора (NH4)2Ce(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно прибавляли Bi(NO3)3·5H2O (282.3 г) и RbNO3 (22.9 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь С к реакционной смеси D. При этом выпадал оранжевый осадок. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, в течение 3 часов на воздухе при температуре 425°С, а затем прокаливали на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена. Результаты испытания приведены в Таблице 2.
Пример 7 - Ni4Mg3Fe0.9Rb0.192Cr0.05Bi0.72Ce1.76Mo12.806O51.539+50% вес. SiO2 с размером частиц 22 нм
Реакционную смесь А готовили, нагревая 154.5 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (140.4 г), при этом получали прозрачный бесцветный раствор. Затем при перемешивании прибавляли золь кремниевой кислоты (568 м.д. Na, средний размер частиц 22 нм, 625 г, 40% вес. диоксида кремния).
Реакционную смесь В готовили, нагревая 26.5 мл деионизированной воды до 55°С, а затем при перемешивании добавляли Fe(NO3)3·9H2O (32.2 г), Ni(NO3)2·6H2O (102.9 г), Mg(NO3)2·6H2O (68 г) и Cr(NO3)3·9H2O (1.8 г).
Реакционную смесь С готовили, нагревая 65.5 мл деионизированной воды до 65°С, а затем при перемешивании в течение 30 минут добавляли гептамолибдат аммония (59.6 г), при этом получали прозрачный бесцветный раствор.
Реакционную смесь D готовили, (i) нагревая 170.6 г 50% вес. водного раствора (NH4)2Се(NO3)6 до 55°С, и (ii) при перемешивании и нагревании раствора последовательно прибавляли Bi(NO3)3·5H2O (30.9 г) и RbNO3 (2.5 г), получали прозрачный оранжевый раствор.
Реакционную смесь Е готовили, прибавляя при перемешивании реакционную смесь В к реакционной смеси А.
Реакционную смесь F готовили, добавляя реакционную смесь С к реакционной смеси D. При этом выпадал оранжевый осадок. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 минут, поддерживая при этом температуру в интервале 50-55°С.
Затем реакционную смесь F прибавляли к реакционной смеси Е, получая целевую суспензию предшественника катализатора.
Суспензию предшественника катализатора оставляли охлаждаться примерно до 40°С при перемешивании в течение одного часа. Затем гомогенизировали в смесителе в течение 3 мин при 5000 об/мин. Затем суспензию сушили в распылительной сушилке при температуре на входе/выходе 325/140°С. Полученный порошок денитрифицировали, нагревая в течение 3 часов на воздухе при температуре 290°С, в течение 3 часов на воздухе при температуре 425°С, а затем прокаливали (кальцинировали) на воздухе при 560°С в течение 3 часов. Полученный прокаленный порошок испытывали в качестве катализатора аммоксидирования пропилена. Результаты испытания приведены в Таблице 2.
7. Катализаторы этого процесса описываются по отношению к "Мо12" (т.е. нижний индекс Мо=12), т.е. чтобы перевести каждую композицию в соответствие с формулой, содержащей "Мо12", просто делят каждый нижний индекс в композиции на нижний индекс при Мо, а затем умножают на 12. Например, композиция в Примере 3 Ni4Mg3Fe0.9Rb0.192Cr0.05Bi0.72Ce1.76Mo12.806O51.539 в расчете на Mo12 эквивалентна композиции Ni3.748Mg2.811Fe0.843Rb0.180Cr0.047Bi0.675Ce1.65Mo12O48.296.
Как можно видеть из Таблиц 2 и 3, каталитические композиции согласно данному изобретению (следует иметь в виду отношение X/Y) приводят (i) к высоким общим выходам акрилонитрила, ацетонитрила и HCN в процессе аммоксидирования пропилена при повышенных температурах в присутствии этой каталитической композиции, аммиака и воздуха и (ii) к меньшим потерям массы при истирании (большей прочности частиц) по сравнению с похожими катализаторами, которые не входят в объем данного изобретения.
Сравнительные пример С 8 и примеры 8-11
С применением способов, описанных в данной заявке, были получены различные составы катализаторов. В Таблице 4 показано, что эти катализаторы с отношениями Ce/Fe менее 0,7 имеют большую степень износа, чем катализаторы с более высокими отношениями Ce/Fe.
В то время как приведенные выше описание и варианты данного изобретения являются типичными для осуществлении данного изобретения, в свете данной заявки, для специалиста в данной области очевидны его многие альтернативы, модификации и изменения. Соответственно, подразумевается, что такие альтернативы, модификации и изменения охватываются данным изобретением и входят в его объем, определяемый прилагаемой формулой изобретения.
Реферат
Изобретение относится к способ получения катализатора, к способу аммоксидирования олефина и к катализатору. Катализатор представлен следующей формулой: MoBiFeADЕFGCeO, где А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия; D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария; Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура; F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца; G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и а, b, с, d, e, f, g, h и n соответственно обозначают число атомов висмута (Bi), железа (Fe), А, D, Е, F, церия (Се) и кислорода (О), приходящееся на 12 атомов молибдена (Мо), где а имеет значение от 0,05 до 7, b имеет значение от 0,1 до 7, с имеет значение от 0,01 до 5, d имеет значение от 0,1 до 12, е имеет значение от 0 до 5, f имеет значение от 0 до 5, g имеет значение от 0 до 0,2, h имеет значение от 0,01 до 5 и х обозначает число атомов кислорода, необходимое для насыщения валентности других имеющихся элементов. Элементы в указанном катализаторе объединяются вместе в водной суспензии предшественника катализатора, полученную таким образом водную суспензию предшественника катализатора сушат с образованием предшественника катал�
Формула
M12BiaFebAcDdEeFfGgCehOx,
где
А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
а, b, с, d, е, f, g, h и х соответственно обозначают число атомов висмута (Bi), железа (Fe), A, D, Е, F, церия (Се) и кислорода (О), приходящееся на 12 атомов молибдена (Мо),
где
а имеет значение от 0,05 до 7,
b имеет значение от 0,1 до 7,
с имеет значение от 0,01 до 5,
d имеет значение от 0,1 до 12,
е имеет значение от 0 до 5,
f имеет значение от 0 до 5,
g имеет значение от 0 до 0,2,
h имеет значение от 0,01 до 5 и
х обозначает число атомов кислорода, необходимое для насыщения валентности других имеющихся элементов;
и где элементы в указанном катализаторе объединяются вместе в водной суспензии предшественника катализатора, причем полученную таким образом водную суспензию предшественника катализатора сушат с образованием предшественника катализатора, а предшественник катализатора прокаливают с образованием указанного катализатора, причем способ включает:
(i) смешивание, в водном растворе, исходных соединений Bi и Се и, необязательно, одного или более элементов из Na, К, Rb, Cs, Са, редкоземельного элемента, Pb, W и Y с образованием смеси,
(ii) добавление исходного соединения молибдена к смеси (i) для взаимодействия со смесью и образования суспензии осадка и
(iii) смешивание суспензии осадка с исходными соединениями остальных элементов катализатора и остального молибдена катализатора с образованием водной суспензии предшественника катализатора.
M12BiaFebAcDdEeFfGgCehOx,
где
А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
а, b, с, d, е, f, g, h и х соответственно обозначают число атомов висмута (Bi), железа (Fe), A, D, Е, F, церия (Се) и кислорода (О), приходящееся на 12 атомов молибдена (Мо),
где
а имеет значение от 0,05 до 7,
b имеет значение от 0,1 до 7,
с имеет значение от 0,01 до 5,
d имеет значение от 0,1 до 12,
е имеет значение от 0 до 5,
f имеет значение от 0 до 5,
g имеет значение от 0 до 0,2,
h имеет значение от 0,01 до 5 и
х обозначает число атомов кислорода, необходимое для насыщения валентности других имеющихся элементов; и
где указанный катализатор получают способом получения катализатора, который включает смешивание элементов катализатора в водной суспензии предшественника катализатора, полученную таким образом водную суспензию предшественника катализатора сушат с образованием предшественника катализатора, а предшественник катализатора прокаливают с образованием указанного катализатора, причем указанный способ дополнительно включает:
(i) смешивание, в водном растворе, исходных соединений Bi и Се и, необязательно, одного или более элементов из Na, К, Rb, Cs, Са, редкоземельного элемента, Pb, W и Y с образованием смеси,
(ii) добавление исходного соединения молибдена к смеси (i) для взаимодействия со смесью и образования суспензии осадка и
(iii) смешивание суспензии осадка с исходными соединениями остальных элементов катализатора и остального молибдена катализатора с образованием водной суспензии предшественника катализатора.
M12BiaFebAcDdEeFfGgCehOx,
где
А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
а, b, с, d, е, f, g, h и х соответственно обозначают число атомов висмута (Bi), железа (Fe), A, D, Е, F, церия (Се) и кислорода (О), приходящееся на 12 атомов молибдена (Мо),
где
а имеет значение от 0,05 до 7,
b имеет значение от 0,1 до 7,
с имеет значение от 0,01 до 5,
d имеет значение от 0,1 до 12,
е имеет значение от 0 до 5,
f имеет значение от 0 до 5,
g имеет значение от 0 до 0,2,
h имеет значение от 0,01 до 5 и
х обозначает число атомов кислорода, необходимое для насыщения валентности других имеющихся элементов;
и где указанный катализатор получают способом получения катализатора, который включает смешивание элементов катализатора в водной суспензии предшественника катализатора, полученную таким образом водную суспензию предшественника катализатора сушат с образованием предшественника катализатора, а предшественник катализатора прокаливают с образованием указанного катализатора, причем указанный способ дополнительно включает:
(i) смешивание, в водном растворе, исходных соединений Bi и Се и, необязательно, одного или более элементов из Na, К, Rb, Cs, Са, редкоземельного элемента, Pb, W и Y с образованием смеси,
(ii) добавление исходного соединения молибдена к смеси (i) для взаимодействия со смесью и образования суспензии осадка и
(iii) смешивание суспензии осадка с исходными соединениями остальных элементов катализатора и остального молибдена катализатора с образованием водной суспензии предшественника катализатора.
Документы, цитированные в отчёте о поиске
Катализатор производства акрилонитрила
Катализатор для производства акрилонитрила
Способ приготовления катализатора для окисления и аммоксидации олефинов
Комментарии