Добавки к катализаторам аммоксидирования на основе смесей оксидов металлов, вводимые перед кальцинированием - RU2619943C2
Код документа: RU2619943C2
Описание
Область техники
Изобретение относится к усовершенствованному катализатору для применения при аммоксидировании ненасыщенного углеводорода с получением соответствующего ненасыщенного нитрила. В частности, настоящее изобретение направлено на создание способа получения усовершенствованного катализатора для аммоксидирования пропилена и/или изобутилена с получением акрилонитрила и/или метакрилонитрила, соответственно и окисления пропилена и/или изобутилена с получением акролеина/акриловой кислоты и/или метакролеина/метакриловой кислоты, соответственно. Более конкретно, данное изобретение относится к способу получения усовершенствованного многокомпонентного катализатора аммоксидирования, который включает комплекс оксидов металлов, при этом во время осуществления способа получения катализатора перед кальцинированием катализатора добавляют разлагающееся при нагревании азотсодержащее соединение.
Уровень техники
Катализаторы, содержащие оксиды железа, висмута и молибдена, промотированные подходящими элементами, уже давно применяются для конверсии пропилена и/или изобутилена при повышенных температурах в присутствии аммиака и кислорода (обычно в виде воздуха) для получения акрилонитрила и/или метакрилонитрила. В частности, патент Великобритании №1436475; патенты США №№4766232; 4377534; 4040978; 4168246; 5223469 и 4863891 каждый направлен на получение катализаторов на основе висмута-молибдена, которые могут быть промотированы элементами II группы для получения акрилонитрила. Кроме того, патенты США №№5093299, 5212137, 5658842 и 5834394 относятся к промотированным катализаторам на основе висмута-молибдена, обеспечивающим высокие выходы акрилонитрила.
Настоящее изобретение относится, в частности, к способу получения катализаторов на основе висмута-молибдена-железа и применению добавки при осуществлении способа. Обычно такие катализаторы получают периодическим способом путем простого смешения и взаимодействия исходных компонентов для различных соединений металлов. Однако использовали более сложные и многостадийные способы. Например, в патенте США №4040978 описан способ получения катализаторов, когда молибдаты каждого металла получали в отдельности и затем их соединяли и осуществляли реакцию, в патенте США №4148757 описан способ получения катализатора, когда висмут и молибден вначале реагируют с образованием молибдата висмута и затем молибдат висмута соединяется со смесью исходных соединений для получения различных других соединений металлов.
Добавление добавок, промотирующих катализаторы аммоксидирования на основе железа-висмута-молибдена, помимо добавления элементов промотора и применения различных материалов носителя, таких как диоксид кремния, вообще является нетипичным. Это менее верно при применении других каталитических систем. Например, в патенте США №5128114 описано добавление цитрата аммония или мочевины к водному золю оксида кремния, сушка этой смеси и затем кальцинирование высушенного порошка для удаления летучих компонентов. Применение такой добавки обеспечивает получение износостойких микросферических частиц. В патенте США №5128114 описано применение этого способа для получения катализаторов на основе палладия и платины-палладия на носителе, представляющем собой оксид кремния, для получения перекиси водорода путем реакции водорода и кислорода. В патентах США №7288669 и 6943135 предложено добавление источника NOx к другим ингредиентам, применяемым при получении катализатора для окисления пропана до акриловой кислоты или для аммоксидирования пропана до акрилонитрила (в качестве примеров описаны каталитические системы типа Mo-V-Te-Nb-O).
Сущность изобретения
Данное изобретение направлено на создание усовершенствованного катализатора на основе смеси оксидов металлов для аммоксидирования пропилена и/или изобутилена. Этот усовершенствованный катализатор, полученный, как описано в данной заявке, обеспечивает более высокую степень общей конверсии пропилена и/или изобутилена в нитрилы (то есть соединения, содержащие функциональную группу "-CN", такие как акрилонитрил, метакрилонитрил, ацетонитрил и цианистый водород), более высокий выход цианистого водорода и большую эффективность использования аммиака. Настоящее изобретение относится также к усовершенствованному катализатору на основе смеси оксидов металлов для окисления пропилена и/или изобутилена.
Согласно одному из вариантов настоящее изобретение относится к способу получения катализатора, включающего комплекс оксидов металлов, причем отношение элементов в указанном катализаторе определяется по следующей формуле:

где А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия, и
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящий из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка и ванадия;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящий из лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, холмия, эрбия, тулия, иттербия, лютеция, скандия, иттрия, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящий из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
а равен от 0.05 до 7,
b равен от 0.1 до 7,
с равен от 0.01 до 5,
d равен от 0.1 до 12,
е равен от 0 до 5,
f равен от 0 до 5,
g равен от 0 до 0.2,
h равен от 0 до 5, и
x обозначает количество атомов кислорода, требующееся для насыщения валентности других имеющихся элементов;
при этом азотсодержащее соединение, разлагающееся при нагревании, добавляют в процессе получения катализатора.
Способ получения катализатора обычно включает:
(a) соединение исходных соединений металлов, которые включают катализатор на основе оксида металла, с образованием предшественника катализатора,
(b) сушку предшественника катализатора с образованием частиц катализатора и
(c) кальцинирование частиц катализатора с получением катализатора.
Азотсодержащее соединение, разлагающееся при нагревании, добавляют в процессе получения катализатора перед стадией кальцинирования. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, добавляют в процессе получения предшественника катализатора, в соответствии с одним из вариантов азотсодержащее соединение, разлагающееся при нагревании, добавляют к предшественнику катализатора перед сушкой при распылении. Согласно другому варианту азотсодержащее соединение, разлагающееся при нагревании, добавляют к частицам катализатора перед кальцинированием.
Настоящее изобретение относится также к способам конверсии олефина, выбранного из группы, состоящей из пропилена и изобутилена или из их смесей, с получением акрилонитрила и/или метакрилонитрила и других побочных нитрилов (то есть соединений, содержащих функциональную группу "-CN", таких как ацетонитрил и цианистый водород) и их смесей путем взаимодействия в паровой фазе при повышенной температуре и давлении с газом, содержащим молекулярный кислород, и аммиаком в присутствии катализатора на основе смеси оксидов металлов, при этом катализатор получают, как описано в данной заявке.
Подробное описание изобретения
Настоящее изобретение относится к способу получения катализатора на основе оксидов металлов для каталитического аммоксидирования пропилена, изобутилена или их смесей с получением акрилонитрила, метакрилонитрила и их смесей, соответственно, и для каталитического окисления пропилена, изобутилена или их смесей с получением акролена/акриловой кислоты, метакролеина/метакриловой кислоты и их смесей, соответственно. В процессе получения катализатора добавляют по меньшей мере одно азотсодержащее соединение, разлагающееся при нагревании.
Согласно одному из вариантов способ получения катализатора включает:
(a) соединение исходных соединений металлов, которые включают катализатор на основе оксида металла, с образованием предшественника катализатора,
(b) сушку предшественника катализатора с образованием частиц катализатора и
(c) кальцинирование частиц катализатора с получением катализатора,
при этом азотсодержащее соединение, разлагающееся при нагревании, добавляют в процессе получения катализатора.
Азотсодержащее соединение, разлагающееся при нагревании, добавляют в процессе получения катализатора перед стадией кальцинирования. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, добавляют в процессе получения предшественника катализатора. В соответствии с одним из вариантов азотсодержащее соединение, разлагающееся при нагревании, добавляют к предшественнику катализатора перед сушкой при распылении. Согласно другому варианту азотсодержащее соединение, разлагающееся при нагревании, добавляют к частицам катализатора перед кальцинированием.
Согласно одному из вариантов способ получения катализатора включает:
(a) соединение исходных соединений металлов, которые включают катализатор на основе оксида металла, с образованием предшественника катализатора,
(b) сушку при распылении предшественника катализатора с образованием микросферических частиц катализатора и
(c) кальцинирование микросферических частиц частиц катализатора с получением катализатора,
при этом азотсодержащее соединение, разлагающееся при нагревании, добавляют в процессе получения катализатора.
При использовании при аммоксидировании пропилена катализаторы, полученные описанным выше способом (а именно, способом получения, который включает добавление азотсодержащего соединения, разлагающегося при нагревании), характеризуются большей степенью полной конверсии пропилена в нитрилы (то есть в соединения, содержащие функциональную группу "-CN"), такие как акрилонитрил, цианистый водород и ацетонитрил, по сравнению с подобными катализаторами, полученными без добавления азотсодержащего соединения, разлагающегося при нагревании. Для любого катализатора, применяемого при аммоксидировании пропилена, этот результат можно оценить количественно путем определения "а" по следующему уравнению:
α=[(%AN+(3×%HCN)+(1.5×%ACN))÷%РС]×100,
где %AN означает выход акрилонитрила,
%HCN означает выход цианистого водорода,
%ACN означает выход ацетонитрила,
%РС означает степень конверсии пропилена.
Параметр "α" обозначает меру "введения азота" или "использования азота" (а именно, азота из аммиака, соединяющегося с пропиленом с образование соединений, содержащих функциональную группу "-CN" в процессе аммоксидирования, например, чем больше "α", тем выше степень общей конверсии пропилена в акрилонитрил, цианистый водород и ацетонитрил).
Используемый в данной заявке термин "выход акрилонитрила" означает выход акрилонитрила в мол. % (выраженный как число без знака %), определенный следующим образом: (количество молей полученного акрилонитрила ÷ количество молей пропилена, подаваемое в реактор)×100. Используемый в данной заявке термин "выход цианистого водорода" означает выход цианистого водорода в мол. % (выраженный как число без знака %), определенный следующим образом: (количество молей цианистого водорода÷3)÷(количество молей пропилена, подаваемое в реактор)×100. Используемый в данной заявке термин "выход ацетонитрила" означает выход ацетонитрила в мол. % (выраженный как число без знака %), определенный следующим образом: (количество молей полученного ацетонитрила 1.5)÷(количество молей пропилена, подаваемое в реактор)×100. Степень конверсии пропилена означает величину степени конверсии пропилена в мол. % в продукты и побочные продукты (выраженную как число без знака %), определенную следующим образом: [(количество молей пропилена, подаваемое в реактор, минус количество молей пропилена, выходящего из реактора) количество молей пропилена, подаваемое в реактор]×100.
Наконец, при использовании при аммоксидировании пропилена такие катализаторы, полученные описанным выше способом (а именно, способом, который включает добавление азотсодержащего соединения, разлагающегося при нагревании), характеризуются меньшим "сгоранием аммиака" по сравнению с подобными катализаторами, полученными без добавления азотсодержащего соединения, разлагающегося при нагревании. Попросту говоря, термин "сгорание аммиака'' представляет собой меру количества аммиака, которое потребляется при осуществлении реакции, которую нельзя объяснить количеством азота, содержащегося в желаемых продуктах реакции. Желательно меньшее сгорание аммиака, потому что тогда большее количество аммиака реагирует с образованием ценных продуктов (например, при введении азота в молекулу нитрила) в противоположность реакции образования побочных продуктов или отходов (например, N2, N2O, NO или NO2). "Сгорание аммиака" обычно выражается как мольное количество аммиака, подаваемого при проведении реакции, которое реагирует, но не приводит к получению акрилонитрила, HCN или ацетонитрила и может быть определено следующим образом: [1-[(количество молей полученных акрилонитрила, HCN и ацетонитрила + количество молей аммиака, выходящего из реактора)÷(количество молей аммиака, подаваемого в реактор)]]×100.
Катализаторы согласно настоящему изобретению характеризуются как большим значением "α" (а именно, более 100), так и низкой величиной "сгорания аммиака" (а именно, менее примерно 15%). В общем, и "α" и "сгорание аммиака" являются мерой эффективности катализатора при использовании аммиака для аммоксидирования пропилена при получении акрилонитрила.
Катализатор:
Настоящее изобретение относится к многокомпонентной каталитической композиции на основе смеси оксидов металлов для аммоксидирования, содержащей комплекс каталитических оксидов, при этом элементы и отношения элементов в указанной каталитической композиции выражены следующей формулой:

где A обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия, и
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящий из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка и ванадия;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящий из лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, холмия, эрбия, тулия, иттербия, лютеция, скандия, иттрия, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящий из серебра, золота, рутения, родия, палладия, осмия, иридия, пдатиныв и ртути; и
a равен от 0.05 до 7,
b равен от 0.1 до 7,
с равен от 0.01 до 5,
d равен от 0.1 до 12,
е равен от 0 до 5,
f равен от 0 до 5,
g равен от 0 до 0.2,
h равен от 0 до 5, и
x обозначает количество атомов кислорода, требующееся для насыщения валентности других имеющихся элементов;
Согласно одному из вариантов катализатор не содержит теллура, сурьмы или селена.
Согласно еще одному варианту компоненты или элементы, обозначенные в приведенной формуле как "Е", могут также включать теллур или сурьму. Согласно одному из вариантов h равен от 0.01 до 5.
Согласно одному из вариантов описанной выше каталитической композиции 0.15≤(a+h)/d≤1. Согласно другому варианту описанной выше каталитической композиции 0.8≤h/b≤5. Согласно еще одному варианту рентгеновская дифракционная картина описанной выше каталитической композиции характеризуется рентгенодифракционными пиками при угле 2 θ (тета), составляющем 28±0.3 градуса и при угле 2 тета, составляющем 26.5±0.3 градуса, и, если отношение интенсивности наиболее интенсивного рентгенодифракционного пика при угле 2 тета 28±0.3 градуса к интенсивности наиболее интенсивного рентгенодифракционного пика при угле 2 тета 26.5±0.3 градуса определено как X/Y, тогда X/Y больше или равно 0.7. В соответствии с другими вариантами описанной выше каталитической композиции 0.2≤(a+h)/d≤0.6; 0.3≤(a+h)/d≤0.5; 1≤h/b≤3; 1.5≤h/b≤2; X/Y больше или равно 0.8.; и/или X/Y больше или равно 0.90.
Согласно одному из вариантов (когда 0.8≤h/b≤5), "h/b" является отношением церия к железу в катализаторе и для любого состава катализатора это отношение равно количеству молей церия (как показывает нижний индекс для церия в формуле), деленному на количество молей железа (как показывает нижний индекс для железа в формуле). Было установлено, что катализаторы, описываемые приведенной выше формулой, где 0.8≤h/b≤5 являются более прочными, они имеют меньшие потери при износе, что показывает тест на определение износа катализатора струей.
Согласно одному из вариантов, когда рентгеновская дифракционная картина описанной выше каталитической композиции характеризуется рентгенодифракционными пиками при угле 2 тета, составляющем 28±0.3 градуса и при угле 2 тета, составляющем 26.5±0.3 градуса, и, если отношение интенсивности наиболее интенсивного рентгенодифракционного пика при угле 2 тета 28±0.3 градуса к интенсивности наиболее интенсивного рентгенодифракционного пика при угле 2 тета 26.5±0.3 градуса определено как X/Y, и X/Y больше или равно 0.7, было установлено, что такие катализаторы обеспечивают большую степень полной конверсии при аммоксидировании пропилена и/или изобутилена в нитрилы (то есть соединения, содержащие функциональную группу "-CN", такие как акрилонитрил, метакрилонитрил, ацетонитрил и цианистый водород).
Используемые в данной заявке термины "каталитическая композиция" и "катализатор" являются синонимами и применяются как взаимозаменяемые. Термин "редкоземельный элемент", применяемый в данной заявке, означает по меньшей мере один элемент из лантана, церия, празеодима, неодима, прометия, самария, европия, гадолиния, тербия, диспрозия, холмия, эрбия, тулия, иттербия, скандия и иттрия (хотя церий представляет собой редкоземельный элемент, он исключен из этого перечня, так как церий указан отдельно как компонент катализатора, описанного в данной заявке). Используемое обозначение "20" является синонимом обозначения "2 тета".
Катализатор согласно данному изобретению может быть на носителе или без носителя (то есть катализатор может включать носитель). Подходящие носители включают оксид кремния, оксид алюминия, оксид циркония, оксид титана или их смеси. Носитель обычно служит как связующее для катализатора и обеспечивает получение более прочного (то есть более износостойкого) катализатора. Однако при промышленном применении соответствующая смесь и активной фазы (то есть комплекса оксидов металлов, описанного в данной заявке), и носителя является решающей для получения приемлемой активности и твердости (износостойкости) катализатора. Обычно носитель включает между 40 и 60 вес. % катализатора на носителе. Согласно одному из вариантов настоящего изобретения носитель может включать небольшое количество катализатора на носителе, например, примерно 30 вес. %. Согласно другому варианту данного изобретения носитель может включать до примерно 70 вес. % катализатора на носителе.
Согласно одному варианту носителем катализатора является золь оксида кремния. Обычно золи оксида кремния содержат некоторое количество натрия. Согласно одному из вариантов золи оксида кремния содержит менее 600 ч. м. натрия. Согласно другому варианту золи оксида кремния содержит менее 200 ч. м. натрия. Обычно величина среднего диаметра коллоидной частицы составляет от примерно 15 нм до примерно 50 нм. Согласно одному варианту данного изобретения величина среднего диаметра коллоидной частицы золя оксида кремния составляет примерно 10 нм и может быть равна примерно 4 нм. Согласно другому варианту настоящего изобретения величина среднего диаметра коллоидной частицы золя оксида кремния составляет примерно 100 нм. Согласно другому варианту настоящего изобретения величина среднего диаметра коллоидной частицы золя оксида кремния составляет примерно 20 нм.
Получение катализатора:
За исключением добавления азотсодержащего соединения, которое разлагается при нагревании, как описано ниже, катализатор по изобретению может быть получен любым из многочисленных способов получения катализаторов, которые известны специалистам в данной области. Типичный способ получения катализатора начинается с приготовления смеси воды, источника молибдена и материала носителя (например, золя оксида кремния). Источники оставшихся элементов катализатора отдельно соединяют в воде с образованием второй смеси. Эти две смеси затем соединяют при перемешивании при слегка повышенной температуре (приблизительно 40°C) с образованием суспензии предшественника катализатора. Затем суспензию предшественника катализатора сушат и удаляют азот и затем кальцинируют, как будет описано ниже.
Согласно одному из вариантов элементы описанного выше катализатора соединяют вместе в водной суспензии предшественника катализатора, полученную таким образом суспензию предшественника катализатора подвергают сушке с получением предшественника катализатора, и предшественник катализатора кальцинируют с получением катализатора. Однако для этого способа уникальным является следующее:
(i) соединение в водном растворе источника Bi и Се и необязательно одного или более элемента, выбранного из Na, K, Rb, Cs, Са, лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, холмия, эрбия, тулия, иттербия, лютеция, скандия, иттрия, Pb и W, с образование смеси (а именно, первой смеси),
(ii) добавление к смеси (а именно, к первой смеси) источника молибдена для осуществления взаимодействия со смесью и образования суспензии осадка и
(iii) соединение суспензии осадка с источниками остальных элементов и остальным количеством молибдена с образованием водной суспензии предшественника катализатора.
Используемый в данной заявке термин "источник соединений" означает соединения, которые содержат и/или обеспечивают наличие одного или более металлов для каталитической композиции на основе смеси оксидов металлов. Применяемый в данной заявке термин "остальные элементы" или "остальные элементы катализатора" относится к таким элементам и к количеству этих элементов, которые обозначены как "A", "D", "Е", "F" и "G" в приведенной выше формуле и которые не были включены в первую смесь. Согласно одному из вариантов некоторые элементы могут быть частью и первой, и второй смесей. Далее, используемый в данной заявке термин "остальное количество молибдена" или "остальное количество молибдена в катализаторе" относится к такому количеству молибдена, требующегося в конечном катализаторе, которое не находилось (то есть не было включено) при приготовлении суспензии осадка. Наконец, сумма количеств молибдена, содержащегося в исходных соединениях молибдена, добавленных на стадиях (ii) и (iii), будет равно общему количеству молибдена, которое содержится в катализаторе.
При осуществлении описанного выше способа источники остальных элементов и остального количества молибдена, которые соединяют с суспензией осадка, могут добавляться в любом порядке или в любой комбинации этих остальных элементов и остального молибдена. Согласно одному из вариантов смесь источников остальных элементов и остальной молибден соединяются в суспензии осадка с образованием водной суспензии предшественника катализатора. Согласно другому варианту (i) смесь источников остальных элементов соединяется с суспензией осадка, и (ii) источники остального молибдена отдельно добавляются к суспензии осадка с образованием водной суспензии предшественника катализатора. Согласно другому варианту источники остальных элементов и остальной молибден добавляются в отдельности (то есть по одному) в суспензию осадка. Согласно еще одному варианту многокомпонентные (то есть содержащие более одного элемента) смеси источников остальных элементов и остальной молибден, причем каждая смесь содержит один или более источников остальных элементов или остальной молибден, добавляются в отдельности (то есть одна смесь в какой-то момент времени или многокомпонентные смеси добавляются одновременно) к суспензии осадка с получением водной суспензии предшественника катализатора. Согласно еще одному варианту смесь источников остальных элементов соединяется с источником остального молибдена, и полученная смесь затем добавляется с образованием суспензии предшественника катализатора. Согласно еще одному варианту носителем является диоксид кремния (SiCl2), и диоксид кремния соединяется с источником остального молибдена до соединения остального молибдена с суспензией осадка (то есть диоксид кремния и источник остального молибдена соединяются с образованием смеси и затем эта смесь добавляется к суспензии осадка, отдельно или в комбинации с одним или более соединениями остальных элементов).
При осуществлении описанного выше способа молибден добавляется и при получении суспензии осадка, и при получении водной суспензии предшественника катализатора.
На уровне атомов минимальное количество молибдена, добавляемое для получения суспензии осадка, определяется по следующему уравнению:
Mo=1.5(Bi+Ce)+0.5(Rb+Na+K+Cs)+(Са)+1.5 (сумму количества атомов лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, холмия, эрбия, тулия, иттербия, лютеция, скандия и иттрия +(Pb)-(W),
где в приведенном выше уравнении "Мо" означает количество атомов молибдена, которое должно быть добавлено к первой смеси, и "Bi", "Се", "Rb", "Na", "К", "Cs", "Са", "Pb" и "W" обозначают количества атомов висмута, церия, рубидия, натрия, калия, цезия, кальция, свинца и вольфрама, соответственно, содержащихся в первой смеси.
При осуществлении описанного выше способа получения катализатора количество молибдена, добавляемого к первой смеси для получения суспензии осадка, обычно составляет примерно от 20 до 35% от общего количества молибдена в конечном катализаторе. Согласно одному из вариантов, источник остального молибдена, находящегося в катализаторе, добавляется к смеси источников остальных элементов (то есть ко второй смеси) до соединения смеси остальных элементов с суспензией осадка с образованием суспензии предшественника катализатора. Согласно другим вариантам источник молибдена, содержащий остальной молибден катализатора, добавляется к суспензии осадка или до, или после, или одновременно со смесью источников остальных элементов (то есть со второй смесью) для получения суспензии предшественника катализатора.
При осуществлении описанного выше способа получения катализатора источники Bi и Се, и необязательно, один или более элементов из Na, К, Rb, Cs, Са, редкоземельного элемента, Pb и W, соединяются в водной суспензии с образованием смеси. Согласно одному из вариантов нитрат висмута и необязательно нитраты других металлов (например, нитраты Na, К, Rb, Cs, Са, редкоземельного элемента и/или Pb) растворяют в водном растворе нитрата церия-аммония. Если добавляется вольфрам, источником его соединения является пара-вольфрамат аммония, (NH4)10H2(W2O7)6.
К этой смеси, содержащей висмут и церий (и необязательно один или более элементов из Na, К, Rb, Cs, Са, редкоземельного элемента, Pb и/или W) добавляется источник молибдена. Согласно одному из вариантов этот источник молибдена представляет собой гептамолибдат аммония, растворенный в воде. После добавления источника молибдена к смеси, содержащей висмут и церий, происходит реакция, которая приводит к образованию осадка, и полученная смесь является суспензией осадка.
Суспензия этого осадка затем соединяется со смесью источников остальных элементов катализатора и источником молибдена с образованием водной суспензии предшественника катализатора. Смесь источников остальных элементов катализатора и источника молибдена может быть получена путем соединения источников остальных элементов в водном растворе (например, источники элементов соединяются в воде) и затем добавления источника молибдена. Согласно одному из вариантов изобретения этот источник молибдена представляет собой гептамолибдат аммония, растворенный в воде. При соединении суспензии осадка со смесью остальные элементы/молибден порядок добавления не имеет значения, то есть суспензия осадка может быть добавлена к смеси остальные элементы/молибден или смесь остальные элементы/молибден может быть добавлена к суспензии осадка. Водная суспензия предшественника катализатора выдерживается при повышенной температуре.
Количество водного растворителя в каждой из описанных выше водных смесей и суспензий может меняться в зависимости от растворимости источников элементов, соединяемых с образованием конкретной смеси оксидов металлов. Количество водного растворителя должно быть по меньшей мере достаточным для получения суспензии или смеси твердых веществ и жидкостей, которые способны смешиваться.
В любом случае источники соединений предпочтительно соединять и/или подвергать взаимодействию по протоколу, который включает смешение источников элементов во время их соединения и/или взаимодействия. Конкретный механизм смешения не является критическим и может включать, например, смешение (например, перемешивание или возбуждение) компонентов во время реакции любым эффективным методом. Такие методы включают, например, перемешивание содержимого сосуда, в том числе, путем встряхивания, опрокидывания или раскачивания реакционного сосуда, содержащего такие компоненты. Такие методы включают также, например, перемешивание при использовании перемешивающего элемента, расположенного по меньшей мере частично внутри реакционного сосуда и движущей силы, соединенной с перемешивающим элементом, или с реакционным сосудом для обеспечения относительного движения между перемешивающим элементом и реакционным сосудом. Перемешивающий элемент может представлять собой приводной или прикрепленный к валу перемешивающий элемент. Движущая сила может непосредственно или косвенно сочетаться с перемешивающим элементом (например, при помощи магнитной муфты). Смешение предпочтительно является достаточным для смешения компонентов и обеспечения эффективного взаимодействия между компонентами реакционной среды с образованием более гомогенной реакционной среды (например, приводя к получению более гомогенного предшественника смеси оксидов металлов) по сравнению с не перемешанной смесью. Это приводит к более эффективному потреблению исходных материалов и к получению более однородного продукта, смеси оксидов металлов. Перемешивание суспензии осадка во время реакции также вызывает образование осадка с получением раствора, а не на стенках реакционного сосуда. Более предпочтительно, образование осадка в растворе приводит к росту частиц со всех сторон этих частиц, а не на ограниченно доступных поверхностях, когда этот рост происходит вне стенки реакционного сосуда.
Источник молибдена может включать оксид молибдена (VI) (MoO3), гептамолибдат аммония или молибденовую кислоту. Источник молибдена может быть введен из любого оксида молибдена, такого как диоксид, триоксид, пентоксид или гептоксид. Однако предпочтительно, чтобы в качестве источника молибдена применялась гидролизующаяся или разлагающаяся соль молибдена.
Типичными источниками висмута, церия и остальных элементов катализатора являются нитраты этих металлов. Такие нитраты являются легко доступными и легко растворимыми.
Источник висмута может включать оксид или соль, которая после кальцинирования образует оксид. Предпочтительными являются растворимые в воде соли, которые легко диспергируются, но образуют стабильные оксиды после нагревания. Согласно одному из вариантов источник висмута представляет собой нитрат висмута, Bi(NO3)3.⋅5H2O Источник церия может включать оксид или соль, которая после кальцинирования образует оксид. Предпочтительными являются растворимые в воде соли, которые легко диспергируются, но образуют стабильные оксиды после нагревания. Согласно одному из вариантов источник церия представляет собой нитрат церия, (NH4)2Се(NO3)6.
Источник железа может быть получен из любого соединения железа, которое после кальцинирования образует оксид. Как и в случае других элементов, предпочтительными являются растворимые в воде соли, которые легко диспергируются в катализаторе. Наиболее предпочтительным является нитрат железа.
Источники остальных элементов могут быть получены из любого подходящего источника. Например, кобальт, никель и магний могут быть введены в катализатор при помощи нитратов. Дополнительно в катализатор может быть введен магний в виде нерастворимого карбоната или гидроксида, которые при нагревании образуют оксид магния. Фосфор может быть введен в состав катализатора в виде соли щелочного металла или соли щелочноземельного металла или в виде соли аммония, но предпочтительно вводится в виде фосфорной кислоты.
Источники щелочных компонентов катализатора могут быть введены в состав катализатора в виде оксидов или соли, которая после кальцинирования образует оксид.
Растворители, в дополнение к воде, которые могут быть использованы для приготовления смеси оксидов металлов согласно изобретению, включают, но без ограничения, спирты, такие как метанол, этанол, пропанол, диолы (например, этиленгликоль, пропиленгликоль и т.п.), органические кислоты, такие как уксусная кислота, а также другие полярные растворители, известные из уровня техники. Источники металлов являются по меньшей мере частично растворимыми в растворителе.
Как указывалось ранее, катализатор согласно данному изобретению может использоваться в виде катализатора на носителе или без носителя (то есть катализатор может включать носитель). Подходящими носителями являются оксид кремния, оксид алюминия, оксид циркония, оксид титана или их смеси. Носитель может быть добавлен в любое время до сушки суспензии предшественника катализатора. Носитель может быть добавлен в любое время во время приготовления или после приготовления любой смеси элементов, суспензии осадка или суспензии предшественника катализатора. Более того, носитель не обязательно добавляют в одной точке или на одной стадии (то есть носитель можно вводить во многих точках в процессе получения катализатора). Согласно одному из вариантов носитель соединяется с другими ингредиентами в процессе приготовления водной суспензии предшественника катализатора. Согласно другому варианту носитель добавляется в суспензию осадка (то есть после получения суспензии осадка). Согласно еще одному варианту носитель соединяют с источником молибдена до смешения источника молибдена с источниками остальных элементов в катализаторе с образованием "второй смеси", описанной выше.
Суспензию предшественника катализатора подвергают сушке и удалению азота (то есть удалению нитратов) с получением предшественника катализатора. Согласно одному из вариантов суспензию предшественника катализатора подвергают сушке с образованием частиц катализатора. Согласно одному из вариантов суспензию предшественника катализатора подвергают сушке распылением до получения микросферических частиц катализатора. Согласно одному из вариантов температура на выходе из распылительной сушилки составляет между 110°C и 350°C, предпочтительно между 110°C и 250°C, наиболее предпочтительно, между 110°C и 180°C. Согласно одному из вариантов распылительная сушилка представляет собой распылительную сушилку с параллельным потоком (то есть частицы катализатора распыляются параллельно потоку газа). Согласно другому варианту распылительная сушилка представляет собой распылительную сушилку с противотоком (то есть частицы катализатора распыляются навстречу потоку газа). Согласно еще одному варианту распылительная сушилка является сушилкой с соплом высокого давления. При осуществлении таких процессов с распылением содержащие воду твердофазные частицы распыляются в горячий газ (обычно воздух), чтобы происходило испарение воды. Сушка регулируется температурой газа и расстоянием, которое проходят частицы до контакта с газом. Вообще нежелательно регулировать эти параметры для осуществления слишком быстрой сушки, так как это приводит к образованию подсушенной корки на частицах твердой фазы, которая затем разрушается по мере того, как вода внутри частиц испаряется и стремится к удалению. К тому же желательно получать катализатор в форме, которая включает как можно меньше воды. Следовательно, когда применяют реактор с псевдоожиженным слоем и желательно получить микросферические частицы, следует выбирать условия сушки при распылении с точки достижения полной сушки без разрушения частиц катализатора. Высушенный катализатор затем нагревают для удаления оставшихся нитратов. Температура удаления нитратов составляет от 100°C до 500°C, предпочтительно, от 250°C до 450°C.
Наконец, высушенный и очищенный от нитратов катализатор подвергают кальцинированию с получением готового катализатора. Согласно одному из вариантов кальцинирование проводят в среде воздуха. Согласно другому варианту кальцинирование осуществляют в инертной атмосфере. Согласно одному из вариантов кальцинирование проводят в среде азота. Условия кальцинирования включают температуры в пределах от примерно 300°C до примерно 700°C, более предпочтительно, от примерно 350°C до примерно 650°C, и согласно некоторым вариантам кальцинирование проводят при температуре примерно 600°C. Согласно одному из вариантов кальцинирование является многостадийным с повышением температуры. Согласно одному из вариантов первую стадию кальцинирования осуществляют при температуре в пределах от примерно 300°C до примерно 450°C, с последующей второй стадией кальцинирования, проводимой при температуре в пределах от примерно 500°C до примерно 650°C.
Азотсодержащие соединение, разлагающиеся при нагревании Используемый в данной заявке термин "азотсодержащее соединение, разлагающееся при нагревании" означает любое азотсодержащее соединение, которое разлагается при температуре кальцинирования катализатора или ниже. Азотсодержащее соединение, разлагающееся при нагревании, не является нитратом металла или нитритом металла, добавляемыми в процессе получения катализатора в качестве источника для металла-промотора в составе катализатора. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, не содержит никаких металлов. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, будет высвобождать аммиак (NH3.)
Подходящие азотсодержащее соединение, разлагающееся при нагревании, выбирают из группы, состоящей из алкиламмония, аминов, амидов, нитробензойных соединений, а также циклических азоторганических соединений. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, представляет собой гидроксид аммония или гидроксид алкиламмония. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, представляет собой соль аммония, выбранную из группы, состоящей из нитрата аммония, нитрита аммония, нитратов моно-, ди-, три- и тетраалкиламмония и нитритов моно-, ди-, три- и тетраалкиламмония. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, является амином, выбранным из группы, состоящей т моно-, ди- и триалкиламинов и моно-, ди- и триариламинов. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, является амидом. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, является мочевиной. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, является нитробензойным соединением. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, является нитробензолом. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, является циклическим азоторганическим соединением, выбранным из группы, состоящей из пиридинов, пирролов, пирролидинов и пиперидинов. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, является гидразином.
Добавление азотсодержащего соединения, разлагающегося при нагревании, в процессе получения катализатора
Ключевым признаком настоящего изобретения является добавление азотсодержащего соединения, разлагающегося при нагревании, в процессе получения катализатора перед стадией кальцинирования. Такое добавление можно осуществлять в любой точке процесса, где азотсодержащее соединение не будет мешать реакции и/или влиять на комбинацию металлов, которая образует активную фазу или активные фазы процесса. Азотсодержащее соединение, разлагающееся при нагревании, добавляют в количестве, которое меньше или равно 0.4 от веса добавки на вес катализатора (то есть 0 < веса добавки на вес катализатора ≤0.4). Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, добавляют в количестве, которое меньше или равно 0.3 от веса добавки на вес катализатора (то есть 0 < веса добавки на вес катализатора ≤0.3). Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, добавляют в количестве, которое больше или равно 0.01 от веса добавки на вес катализатора. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, добавляют в количестве, которое больше или равно 0.03 от веса добавки на вес катализатора. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, добавляют в количестве, которое находится в следующих пределах: 0.025 ≤ вес добавки на вес катализатора ≤0.3. Используемый в данной заявке термин "вес добавки" означает вес добавляемого соединения, кроме любого раствора или растворителей. Используемый в данной заявке термин "вес катализатора" означает вес готового катализатора, включая вес любого носителя катализатора и вес добавки.
Азотсодержащее соединение, разлагающееся при нагревании, можно добавлять к смеси источников металлов, которые образуют суспензию катализатора. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, можно добавлять к предварительно полученному раствору или суспензии, содержащим оксид молибдена-висмута-церия, в процессе получения катализатора. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, можно добавлять к предшественнику катализатора перед его сушкой. Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, можно добавлять к частицам катализатора, образовавшимся при сушке предшественника катализатора перед его кальцинированием.
Согласно одному из вариантов азотсодержащее соединение, разлагающееся при нагревании, можно добавлять к частицам катализатора, образовавшимся при сушке при распылении предшественника катализатора, путем контактирования частиц катализатора с раствором, содержащим азотсодержащее соединение, разлагающееся при нагревании, с образованием частиц катализатора, пропитанных азотсодержащим соединением, разлагающимся при нагревании, и последующей сушки частиц катализатора, пропитанных азотсодержащим соединением, разлагающимся при нагревании. Раствор, содержащий азотсодержащее соединение, разлагающееся при нагревании, может включать водный растворитель или неводный растворитель, или их смесь. Контактирование можно осуществлять пропиткой по влагоемкости или другим известным методом, включая погружение катализатора в раствор, содержащий азотсодержащее соединение, разлагающееся при нагревании, или распыление раствора, содержащего азотсодержащее соединение, разлагающееся при нагревании, на частицы катализатора. Когда частицы катализатора контактируют с раствором, содержащим азотсодержащее соединение, разлагающееся при нагревании, этот раствор абсорбируется в порах частиц катализатора. После такого контактирования частицы являются "сырыми" или увлажненными" раствором, содержащим азотсодержащее соединение, разлагающееся при нагревании. После контактирования или пропитки раствором, содержащим азотсодержащее соединение, разлагающееся при нагревании, влажные частицы подвергают сушке для удаления органического или водного растворителя, использованного в растворе, который содержит азотсодержащее соединение, разлагающееся при нагревании. Влажный катализатор сушат путем нагревания при повышенной температуре в течение времени, достаточного для удаления растворителя. Согласно одному из вариантов пропитанный катализатор сушат в атмосфере азота. Обычно влажные частицы сушат при температуре между 100°C и 300°C в течение промежутка времени между 2 ч и 5 ч. Влажные частицы можно также сушить температуре равной примерно 200°C в течение примерно 3 ч.
Согласно одному из вариантов данного изобретения азотсодержащее соединение, разлагающееся при нагревании, добавляют к микросферическим частицам катализатора, образовавшимся при сушке распылением предшественника катализатора, путем контактирования микросферических частиц катализатора с раствором, содержащим азотсодержащее соединение, разлагающееся при нагревании, с образованием микросферических частиц катализатора, пропитанных азотсодержащим соединением, разлагающимся при нагревании, с последующей сушкой микросферических частиц катализатора, пропитанных азотсодержащим соединением, разлагающимся при нагревании.
Процесс аммоксидирования
Катализаторы согласно настоящему изобретению используют в процессах аммоксидирования для конверсии олефина, выбранного из группы, состоящей из пропилена, изобутилена или их смесей, в акрилонитрил, метакрилонитрил и их смеси, соответственно, путем взаимодействия в паровой фазе при повышенной температуре указанного олефина с газом, содержащим молекулярный кислород и аммиак, в присутствии катализатора. Катализаторы согласно настоящему изобретению используют также при аммоксидировании метанола с получением цианистого водорода и при аммоксидировании этанола с получением ацетонитрила. Согласно одному из вариантов применения катализаторов, описанных в данной заявке, метанол и/или этанол могут подаваться совместно при аммоксидировании пропилена, изобутилена или их смесей с образованием акрилонитрила, метакрилонитрила или их смесей для повышения выхода побочных цианистого водорода и/или ацетонитрила, получающихся при осуществлении указанного процесса. Предпочтительно осуществлять реакцию аммоксидирования в реакторе с псевдоожиженном слое, хотя предусмотрены другие типы реакторов, такие как реакторы с линией транспортировки. Реакторы с псевдоожиженным слоем для получения акрилонитрила известны из уровня техники. Например, подходящим является реактор, описанный в патенте США №3230246, который включен в данную заявку посредством отсылки.
Условия реакции аммоксидирования также хорошо известны из уровня техники, см., например, патенты США №№5093299; 4863891; 4767878 и 4503001; которые включены в данную заявку посредством отсылки. Обычно процесс аммоксидирования осуществляют путем контактирования пропилена или изобутилена в присутствии аммиака, кислорода и катализатора в псевдоожиженном слое при повышенной температуре с получением акрилонитрила или метакрилонитрила. Можно применять любой источник кислорода. Однако, по экономическим причинам, предпочтительно использовать воздух. Типичное мольное отношение кислорода к олефину в подаваемом сырье должно быть в пределах от 0.5:1 до 4:1, предпочтительно, от 1:1 до 3:1.
Мольное отношение аммиака к олефину в исходном сырье может колебаться от 0.5:1 до 2:1. В действительности нет верхнего предела для отношения аммиак-олефин, но в общем, по экономическим соображениям, нет оснований превышать отношение 2:1. Подходящими отношениями в сырье при использовании вместе с катализатором согласно изобретению при получении акрилонитрила из пропилена являются отношение аммиака к пропилену в пределах от 0.9:1 до 1.3:1, и отношение воздуха к пропилену в пределах от 8.0:1 до 12.0:1. Катализатор по изобретению способен обеспечивать высокие выходы акрилонитрила при сравнительно низком отношении аммиака к пропилену, составляющем от примерно 1:1 до примерно 1.05:1. Эти "условия с низким содержанием аммиака" помогают уменьшить количество непрореагировавшего аммиака в потоке, выходящем из реактора, это условие известно как "переломное открытие этого способа", которое впоследствии помогает снизить количество отходов. Конкретно, непрореагировавший аммиак должен быть удален из потока, выходящего из реактора, до выделения акрилонитрила. Непрореагировавший аммиак обычно удаляется путем контактирования потока, выходящего из реактора, с серной кислотой с получением сульфата аммония или путем контактирования потока, выходящего из реактора, с акриловой кислотой с получением акрилата аммония, что в обоих случаях приводит к обработке и/или удалению отходящего потока.
Реакция проводится при температуре в пределах между примерно 260°C до 600°C, предпочтительно между 310° и 500°C, особенно предпочтительно между 350° и 480°C. Время контактирования, хотя и не является критическим, обычно составляет от 0.1 до 50 с, предпочтительно, от 1 до 15 с.
Продукты реакции могут быть выделены и очищены способами, известными специалистам в данной области. Один такой метод включает промывание отходящих газов холодной водой или подходящим растворителем для удаления продуктов реакции и затем очистки реакционного продукта путем перегонки.
Основное применение катализатора, полученного описанным способом, состоит в использовании его при аммоксидировании пропилена с получением акрилонитрила. Другие области применения включают аммоксидирование пропана с получением акрилонитрила и аммоксидирование глицерина с получением акрилонитрила. Катализатор, полученный способом согласно данному изобретению, можно также применять при окислении пропилена до акролеина и/или акриловой кислоты. Такие способы обычно являются двухстадийными, при этом пропилен в присутствии катализатора превращается в основном в акролеин на первой стадии, и акролеин в присутствии катализатора превращается в основном в акриловую кислоту на второй стадии. Катализатор, описанный в данной заявке, пригоден для применения на первой стадии для окисления пропилена в акролеин.
Конкретные варианты осуществления изобретения
Для иллюстрации настоящего изобретения катализаторы, полученные в соответствии с данным изобретением, оценивали и сравнивали при похожих условиях реакции с подобными катализаторами, полученными известными способами, не входящими в объем данного изобретения. Эти примеры приведены исключительно в целях иллюстрации данного изобретения.
Катализаторы, имеющие состав Ni4Mg3Fe0.9Rb0.192Cr0.05Bi0.72Ce1.76Mo12.502O50.627+50 вес. % SiO2 (39 нм), были получены следующим способом:
Сравнительный пример 1 - без добавки
Реакционная смесь А была приготовлена путем нагревания 10,309 мл деионизированной воды до температуры 65°C и последующего добавления при перемешивании в течение 30 мин гептамолибдата аммония (9371.5 г) с получением прозрачного бесцветного раствора. Затем при перемешивании добавляли золь оксида кремния (41,486 г, 41 вес. % оксида кремния)
Реакционную смесь В готовили путем нагревания 1,829 мл деионизированной воды до температуры 55°C последующего добавления при перемешивании Fe(NO3)3⋅9H2O (2,221.9 г), Ni(NO3)2⋅6H20 (7,107.9 г), Mg(NO3)2⋅6H2O (4,700.5 г) и Cr(NO3)3⋅9H2O (122.3 г).
Реакционную смесь С приготавливали путем нагревания 2,264.4 мл деионизированной воды до температуры 65°C и последующего добавления при перемешивании в течение 30 мин гептамолибдата аммония (2,058.6 г) с получением прозрачного бесцветного раствора.
Реакционную смесь С2 приготавливали путем нагревания 2,264.4 мл деионизированной воды до температуры 65°C и последующего добавления при перемешивании в течение 30 мин гептамолибдата аммония (2,058.6 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D1 приготавливали путем нагревания 5,896.4 г 50 вес. % водного раствора (NH4)2Ce(NO3)6 до 55°C. При нагревании и перемешивании этого раствора последовательно добавляли Bi(NO3)3⋅5H2O (1,067.1 г) и RbNO3 (86.5 г) с образованием прозрачного раствора оранжевого цвета.
Реакционную смесь D2 приготавливали путем нагревания 5,896.4 г 50 вес. % водного раствора (NH4)2Се(NO3)6 до 55°C. При нагревании и перемешивании этого раствора последовательно добавляли Bi(NO3)3⋅5H2O (1,067.1 г) и RbNO3 (86.5 г) с образованием прозрачного раствора оранжевого цвета.
Реакционную смесь Е готовили путем добавления при перемешивании реакционной смеси В к реакционной смеси А.
Реакционную смесь F1 готовили путем добавления реакционной смеси С1 к реакционной смеси D1. Это привело к осаждению твердого продукта оранжевого цвета.
Перемешивание продолжали в течение 15 мин, в то время как температуру поддерживали равной 50-55°C.
Реакционную смесь F2 готовили путем добавления реакционной смеси С2 к реакционной смеси D2. Это привело к осаждению твердого продукта оранжевого цвета. Перемешивание продолжали в течение 15 мин, в то время как температуру поддерживали равной 50-55°C.
Затем последовательно добавляли реакционную смесь F1 и реакционную смесь F2, к реакционной смеси Е при перемешивании.
Полученную суспензию оставляли при перемешивании в течение 1 ч. в то время как она охлаждалась до примерно 40°C. Затем эту смесь гомогенизировали в блендере в течение 3 мин при перемешивании со скоростью 5000 об/мин. Затем суспензию сушили при распылении при температурах на входе/выходе 325/140°C. Из полученного порошка удаляли азот путем тепловой обработки в течение 1 ч при 350°C и затем кальцинировали в течение 1 ч в атмосфере смеси азот/воздух (50%/50% об/об) при температуре 560°C. Полученный порошок затем испытывали в качестве катализатора аммоксидирования пропилена.
Пример 1 - с добавкой
Реакционная смесь А была приготовлена путем нагревания 153 мл деионизированной воды до 65°C и последующего добавления в течение 30 мин гептамолибдата аммония (138.8 г) с получением прозрачного бесцветного раствора. Затем при перемешивании добавляли золь оксида кремния (609.8 г, 41 вес. % оксида кремния)
Реакционную смесь В готовили путем нагревания 28 мл деионизированной воды до температуры to 55°C последующего добавления при перемешивании Fe(NO3)3⋅9H2O (32.9 г), Ni(NO3)2⋅6H20 (105.3 г), Mg(NO3)2⋅6H2O (69.6 г) и Cr(NO3)3⋅9H2O (1.81 г).
Реакционную смесь С приготавливали путем нагревания 68 мл деионизированной воды до температуры 65°C и последующего добавления при перемешивании в течение 30 мин гептамолибдата аммония (60.98 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D готовили путем нагревания 174.6 г 50 вес. % водного раствора (NH4)2Ce(NO3)6 до температуры 55°C. При нагревании и перемешивании этого раствора последовательно добавляли Bi(NO3)3⋅5H2O (31.6 г) и RbNO3 (2.56 г), что привело к образованию прозрачного раствора оранжевого цвета.
Реакционную смесь Е готовили путем добавления при перемешивании реакционной смеси В к реакционной смеси А.
Реакционную смесь F готовили путем добавления реакционной смеси С к реакционной смеси D при перемешивании. Это привело к осаждению твердого продукта оранжевого цвета. Перемешивание продолжали в течение 15 мин, в то время как температуру поддерживали равной 50-55°C.
Реакционную смесь F затем добавляли к реакционной смеси Е при перемешивании. Затем при непрерывном перемешивании добавляли NH4NO3 (24.4 г) с получением суспензии предшественника катализатора.
Полученную суспензию оставляли при перемешивании в течение 1 ч. в то время как она охлаждалась до примерно 40°C. Затем эту смесь гомогенизировали в бленд ере в течение 3 мин при перемешивании со скоростью 5000 об/мин. Затем суспензию сушили при распылении при температурах на входе/выходе 325/140°C. Из полученного порошка удаляли азот путем тепловой обработки в течение 1 ч при 425°C и затем кальцинировали в течение 1 ч при температуре 580°C. Полученный порошок затем испытывали в качестве катализатора аммоксидирования пропилена.
Пример 2 - с добавкой
Реакционная смесь А была приготовлена путем нагревания 153 мл деионизированной воды до 65°C и последующего добавления в течение 30 мин гептамолибдата аммония (138.8 г) с получением прозрачного бесцветного раствора. Затем при перемешивании добавляли золь оксида кремния (609.8 г, 41 вес. % оксида кремния)
Реакционную смесь В готовили путем нагревания 27 мл деионизированной воды до температуры 55°C и последующего добавления при перемешивании Fe(NO3)3⋅9H2O (32.9 г), Ni(NO3)2⋅6H20 (105.3 г), Mg(NO3)2⋅6H2O (69.6 г) и Cr(NO3)3⋅9H2O (1.81 г).
Реакционную смесь С приготавливали путем нагревания 67 мл деионизированной воды до температуры 65°C и последующего добавления при перемешивании в течение 30 мин гептамолибдата аммония (60.98 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D приготавливали путем нагревания 174.7 г 50 вес. % водного раствора (NH4)2Се(NO3)6 до температуры 55°C. При нагревании и перемешивании этого раствора последовательно добавляли Bi(NO3)3⋅5H2O (31.6 г) и RbNO3 (2.56 г), что привело к образованию прозрачного раствора оранжевого цвета.
Реакционную смесь Е готовили путем добавления при перемешивании реакционной смеси В к реакционной смеси А.
Реакционную смесь F готовили путем добавления реакционной смеси С к реакционной смеси D. Это привело к осаждению твердого продукта оранжевого цвета. Перемешивание продолжали в течение 15 мин, в то время как температуру поддерживали равной 50-55°C.
Затем реакционную смесь F добавляли при перемешивании к реакционной смеси Е. Затем при непрерывном перемешивании добавляли NH4NO3 (57.4 г) с получением суспензии предшественника катализатора.
Полученную суспензию оставляли при перемешивании в течение 1 ч, в то время как она охлаждалась до примерно 40°C. Затем эту смесь гомогенизировали в блендере в течение 3 мин при перемешивании со скоростью 5000 об/мин. Затем суспензию сушили при распылении при температурах на входе/выходе 325/140°C. Из полученного порошка удаляли азот путем тепловой обработки в течение 1 ч при 325°C и затем кальцинировали в течение 1 ч при температуре 560°C. Полученный порошок затем испытывали в качестве катализатора аммоксидирования пропилена.
Пример 3 - с добавкой
Реакционная смесь А была приготовлена путем нагревания 153 мл деионизированной воды до 65°C и последующего добавления в течение 30 мин гептамолибдата аммония (138.8 г) с получением прозрачного бесцветного раствора. Затем при перемешивании добавляли золь оксида кремния (609.8 г, 41 вес. % оксида кремния) Реакционную смесь В готовили путем нагревания 28 мл деионизированной воды до температуры 55°C и последующего добавления при перемешивании Fe(NO3)3⋅9H2O (32.9 г), Ni(NO3)2⋅6H20 (105.3 г), Mg(NO3)2⋅6H2O (69.6 г) и Cr(N03)3 9H20 (1.81 г). Реакционную смесь С приготавливали путем нагревания 67 мл деионизированной воды до температуры 65°C и последующего добавления при перемешивании в течение 30 мин гептамолибдата аммония (60.99 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D приготавливали путем нагревания 174.7 г 50 вес. % водного раствора (NH4)2Ce(NO3)6 до температуры 55°C. При нагревании и перемешивании этого раствора последовательно добавляли Bi(NO3)3⋅5H2O (31.6 г) и RbNO3 (2.56 г), что привело к образованию прозрачного раствора оранжевого цвета.
Реакционную смесь Е готовили путем добавления при перемешивании реакционной смеси В к реакционной смеси А.
Реакционную смесь F готовили путем добавления реакционной смеси С к реакционной смеси D. Это привело к осаждению. твердого продукта оранжевого цвета. Перемешивание продолжали в течение 15 мин, в то время как температуру поддерживали равной 50-55°C.
Затем реакционную смесь при перемешивании добавляли к реакционной смеси Е. Затем при непрерывном перемешивании добавляли NH4NO3 (88.1 г) с получением суспензии предшественника катализатора.
Полученную суспензию оставляли при перемешивании в течение 1 ч. в то время как она охлаждалась до примерно 40°C. Затем эту смесь гомогенизировали в блендере в течение 3 мин при перемешивании со скоростью 5000 об/мин. Затем суспензию сушили при распылении при температурах на входе/выходе 325/140°C. Из полученного порошка удаляли азот путем тепловой обработки в течение 1 ч при 325°C и затем кальцинировали в течение 1 ч при температуре 560°C. Полученный порошок затем испытывали в качестве катализатора аммоксидирования пропилена.
Пример 4 - с добавкой, вводимой путем пропитки
Реакционная смесь А была приготовлена путем нагревания 1364 мл деионизированной воды до 65°C и последующего добавления в течение 30 мин гептамолибдата аммония (1239.6 г) с получением прозрачного бесцветного раствора. Затем при перемешивании добавляли золь оксида кремния (5488 г, 41 вес. % оксида кремния)
Реакционную смесь В готовили путем нагревания 242 мл деионизированной воды до температуры 55°C и последующего добавления при перемешивании Fe(NO3)3⋅9H2O (293.9 г), Ni(NO3)2⋅6H20 (940.2 г), Mg(NO3)2⋅6H2O (621.8 г) и Cr(NO3)3⋅9H20 (16.2 г).
Реакционную смесь С приготавливали путем нагревания 599 мл деионизированной воды до температуры 65°C и последующего добавления при перемешивании в течение 30 мин гептамолибдата аммония (544.6 г) с получением прозрачного бесцветного раствора.
Реакционную смесь D приготавливали путем нагревания 1560 г 50 вес. % водного раствора (NH4)2Ce(NO3)6 до температуры 55°C. При нагревании и перемешивании этого раствора последовательно добавляли Bi(NO3)3⋅5H2O (282.3 г) и RbNO3 (22.89 г), что привело к образованию прозрачного раствора оранжевого цвета.
Реакционную смесь Е готовили путем добавления при перемешивании реакционной смеси В к реакционной смеси А.
Реакционную смесь F готовили путем добавления реакционной смеси С к реакционной смеси D. Это привело к осаждению твердого продукта оранжевого цвета. Перемешивание продолжали в течение 15 мин, в то время как температуру поддерживали равной 50-55°C.
Затем реакционную смесь F добавляли при перемешивании к реакционной смеси Е.
Полученную суспензию оставляли при перемешивании в течение 1 ч. в то время как она охлаждалась до примерно 40°C. Затем эту смесь гомогенизировали в блендере в течение 3 мин при перемешивании со скоростью 5000 об/мин. Затем суспензию сушили при распылении при температурах на входе/выходе 325/140°C. Из полученного порошка удаляли азот путем тепловой обработки в течение 3 ч при 290°C.
Водный раствор нитрата аммония получали путем растворения 39.96 г NH4NO3 в деионизированной воде. Полученный раствор оставляли нагреться до комнатной температуры и разбавляли до конечного объема равного 50.00 мл.
К 150.0 г обработанного путем нагрева катализатора по каплям при перемешивании добавляли 33.0 мл указанного выше раствора нитрата аммония с приращением по 3 мл и энергично перемешивали после добавления каждой порции. Полученный порошок подвергали дальнейшей тепловой обработке в течение 1 ч на воздухе при температуре 350°C и затем кальцинировали на воздухе при 560°C в течение 1 ч. Полученный порошок затем испытывали в качестве катализатора аммоксидирования пропилена.
Испытание катализаторов
Все катализаторы были испытаны в стендовом реакторе при аммоксидировании пропилена с получением акрилонитрила. Испытания проводили в реакторе с псевдоожиженным слоем объемом 48 см3. Пропилен подавали в реактор со скоростью 0.07 WWH (вес пропилена/вес катализатора/час), за исключением Примера 2, где скорость составляла 0.10 WWH. Давление в реакторе поддерживали равным 10 ф/дюйм2 (68.94 кПа). Температура реакции составляла 430°C. Через несколько дней испытаний (через примерно 140-190 ч реакции) отбирали образцы продуктов реакции. Отходящий поток поступал в скрубберы пенного типа, содержащие холодный раствор HCl. Долю отходящего газа определяли при помощи расходомера с мыльной пленкой, и состав отходящего газа определяли в конце пробега с помощью газового хроматографа, снабженного двухканальным газовым анализатором. В конце обработки всю жидкость в скруббере разбавляли до примерно 200 г дистиллированной водой. В качестве внутреннего стандарта применяли взвешенное количество 2-бутанона в аликвоте разбавленного раствора ~50 г. Образец объемом 2 мкл анализировали на газовом хроматографе, снабженном плазменно-ионизационным детектором и колонкой Carbowax™. Количество NH3 определяли путем титрования избытка свободной HCl раствором NaOH.
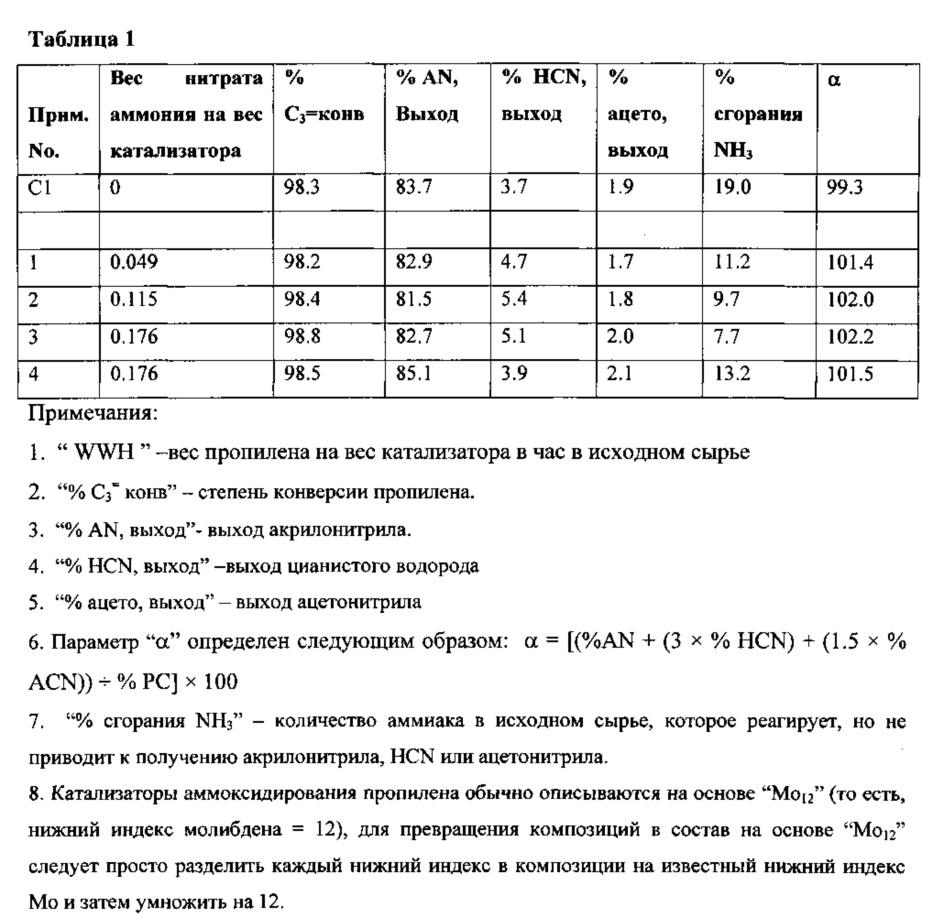
Данные в Таблице 1 ясно показывают преимущества настоящего изобретения. Конкретно, катализаторы (Примеры 1, 2, 3 и 4), полученные с добавкой азотсодержащего соединения, разлагающегося при нагревании, характеризуются повышенной эффективностью использования аммиака, о чем свидетельствуют повышенные величины а и пониженное количество сгоревшего аммиака по сравнению с похожим катализатором (Пример СТ), полученным без добавления азотсодержащего соединения, разлагающегося при нагревании.
Хотя приведенные выше описание и варианты изобретения являются типичными в практике осуществления данного изобретения, специалистам в данной области очевидно, что в свете этого описания могут быть сделаны многочисленные альтернативы, модификации и изменения. Соответственно, подразумевается, что все такие альтернативы, модификации и изменения охвачены и соответствуют сущности и широкому объему изобретения, определяемому формулой изобретения.
Реферат
Описаны способ получения катализаторов для получения акрилонитрила, ацетонитрила и цианистого водорода и способ конверсии олефина, включающий контактирование при повышенной температуре пропилена и/или изобутилена, аммиака и кислорода в паровой фазе в присутствии катализатора. Катализатор содержит комплекс оксидов металлов, причем отношения элементов в указанном катализаторе выражены следующей формулой: MoBiFeADEFGCeO, где А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия, D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария; Е обозначает по меньшей мере один элемент, выбранный из группы, состоящий из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка и ванадия; F обозначает по меньшей мере один элемент, выбранный из группы, состоящий из лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия, тулия, иттербия, лютеция, скандия, иттрия, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца; G обозначает по меньшей мере один элемент, выбранный из группы, состоящий из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; а равен от 0.05 до 7, b равен от 0.1 до 7, с равен от 0.01 до 5, d равен от 0.1 до 12, е равен от 0 до 5, f равен от 0 до 5, g равен от 0 до 0.2, h равен от 0 до 5 и х обозначает количество атомов кислорода, требующееся для насыщения валентности других имеющихся элементов. В процессе получения катализатора добавляют по меньшей мере одно азотсодержащее соединение, разлагающееся при нагревании, причем азотсодержащее соединение, разлагающееся при нагревании, выбрано из группы, состоящей из соединений аммония, нитратных соединений, нитритных соединений, амидных соединений, нитробензойных соединений и соединений гидроксида аммония, и причем азотсодержащее соединение, разлагающееся при нагревании, не является нитратом металла или нитритом металла, добавляемыми в процессе получения катализатора в качестве источника для металла-промотора в составе катализатора. Технический результат – высокая общая степень превращения в нитрильные продукты, меньшее сгорание аммиака и улучшенная эффективность использования аммиака. 2 н. и 14 з.п. ф-лы, 1 табл., 4 пр.
Формула
Документы, цитированные в отчёте о поиске
Способ приготовления оксидного катализатора для окисления или аммоксидирования пропилена

Комментарии