Оксидный катализатор и способ его получения, а также способы получения ненасыщенного альдегида, диолефина и ненасыщенного нитрила - RU2615762C2
Код документа: RU2615762C2
Чертежи







Описание
Область техники
[0001] Настоящее изобретение относится к оксидному катализатору и способу его получения, а также способам получения ненасыщенного альдегида, диолефина и ненасыщенного нитрила с использованием оксидного катализатора.
Уровень техники
[0002] К настоящему времени сообщалось о большом числе оксидных катализаторов для использования в получении ненасыщенного альдегида в качестве основного компонента. Например, старейший оксидный катализатор найден компанией Standard Oil Co. of Ohio и известен как композитный оксидный катализатор, который содержит Мо и Bi в качестве обязательных компонентов. В патентной литературе 1 описан катализатор посредством сосредоточения на Mo, Bi, Се, K, Fe, Со, Mg, Cs и Rb в качестве металлов, образующих катализатор.
[0003] Способ получения ненасыщенного альдегида используют, например, в способе получения (мет)акрилата, такого как метилакрилат или метилметакрилат, через реакцию окислительной этерификации с использованием по меньшей мере одного исходного материала, выбранного из группы, состоящей из пропилена, изобутилена, изобутанола и т-бутилового спирта и промежуточного ненасыщенного альдегида, такого как акролеин или метакролеин. Этот способ получения (мет)акрилата также известен как так называемый процесс прямой метилэтерификации, состоящий из двух реакционных стадий, или как так называемый процесс прямого окисления, состоящий из трех реакционных стадий. Процесс прямого окисления позволяет получать (мет)акрилат посредством трех стадий (см., например, непатентную литературу 1). Первая стадия окисления процесса прямого окисления представляет собой стадию получения ненасыщенного альдегида, такого как акролеин или метакролеин, через реакцию каталитического окисления в газовой фазе по меньшей мере одного исходного материала, выбранного из группы, состоящей из пропилена, изобутилена и т-бутилового спирта, с молекулярным кислородом в присутствии катализатора. Вторая стадия окисления этого процесса представляет собой стадию получения (мет)акриловой кислоты через реакцию каталитического окисления в газовой фазе ненасыщенного альдегида, получаемого на первой стадии окисления, с использованием молекулярного кислорода в присутствии катализатора. Конечная стадия этерификации представляет собой стадию дополнительной этерификации (мет)акриловой кислоты, получаемой на второй стадии окисления, для того, чтобы получать (мет)акрилат. Этерификация с использованием спирта, такого как метанол, может давать метилакрилат или метилметакрилат.
[0004] В отличие от этого, процесс прямой метилэтерификации состоит из двух каталитических реакционных стадий: первая реакционная стадия получения ненасыщенного альдегида, такого как акролеин или метакролеин, через реакцию каталитического окисления в газовой фазе по меньшей мере одного исходного материала, выбранного из группы, состоящей из пропилена, изобутилена, изобутанола и т-бутилового спирта, с использованием содержащего молекулярный кислород газа; и вторая реакционная стадия реакции ненасыщенного альдегида, таким образом полученного, со спиртом, таким как метанол и молекулярный кислород, для того, чтобы получать сразу (мет)акрилат, такой как метилакрилат или метилметакрилат.
[0005] Реакционные системы с использованием таких оксидных катализаторов включают реакционные системы в неподвижном слое, псевдоожиженном слое и подвижном слое. Из них реакционную систему в неподвижном слое часто используют в промышленности за счет следующего преимущества: высокого выхода реакции можно достичь посредством подачи газа, текущего в состоянии, близком к течению выдавливания.
[0006] Однако реакционная система в неподвижном слое имеет низкую теплопроводность и, следовательно, не подходит для экзотермической реакции или эндотермической реакции, которая требует отведения тепла или нагрева. В частности, сильная экзотермическая реакция, такая как реакция окисления, в которой температура возрастает внезапно, нежелательно выходит из-под контроля реакционной системы, что возможно ведет к неуправляемой реакции. Кроме того, такое внезапное повышение температуры повреждает катализатор, что ведет к неблагоприятному раннему разложению катализатора.
[0007] В отличие от этого, реакционная система в псевдоожиженном слое имеет высокую теплопроводность, поскольку частицы катализатора бурно текут в реакторе. Таким образом, температуру в реакторе поддерживают почти постоянной даже во время реакции, в которой происходит значительное выделение или поглощение тепла. Реакционная система в псевдоожиженном слое предпочтительно предотвращать чрезмерное развитие реакции. Эта реакционная система также имеет такое преимущество, что по причине пониженного локального накопления энергии, можно проводить реакцию подаваемого газа в пределах взрыва, чтобы повышать концентрацию исходного материала для того, чтобы повышать производительность. Таким образом, реакционная система в псевдоожиженном слое подходит для реакции каталитического окисления олефина и/или спирта, которая представляет собой сильную экзотермическую реакцию. Несмотря на эти известные преимущества реакционной системы в псевдоожиженном слое, в патентной литературе 2 и 3, например, констатируют, что использование катализаторов в неподвижном слое в целом является предпочтительным для превращения ненасыщенного углеводорода в ненасыщенный альдегид. В этих литературных источниках констатируют, что катализаторы, описанные в них, можно использовать в каком-либо из способов получения ненасыщенного альдегида в неподвижном слое, подвижном слое и псевдоожиженном слое через реакцию каталитического окисления олефина и/или спирта, но не уделяют никакого особого внимания реакционным системам, отличным от систем в неподвижном слое.
[0008] Несмотря на то, что пиролиз лигроина является основным способом получения диолефина, такого как 1,3-бутадиен, существует растущая потребность в получении, основанном на реакции окисления в газовой фазе наряду с современным сдвигом в сторону альтернативных ресурсов для нефти. Примеры способа получения диолефина с использованием реакции окисления в газовой фазе включают способы, которые включают реакцию каталитической окислительной дегидрогенизации моноолефина, который имеет 4 или больше атома углерода, такого как н-бутен или изопентен, и молекулярного кислорода в присутствии катализатора для того, чтобы получать сопряженный диолефин, такой как 1,3-бутадиен или изопрен, соответствующий моноолефину. Что касается катализатора, используемого в такой реакции, например, в патентной литературе 4 описан оксидный катализатор, содержащий Mo, Bi, Fe, Се, Ni, Mg и Rb в качестве катализатора для реакции окислительной дегидрогенизации моноолефина.
[0009] Известный способ получения ненасыщенного нитрила, такого как акрилонитрил или метакрилонитрил, включает реакцию одного или нескольких, выбранных из группы, состоящей из пропилена, изобутилена, изобутанола и т-бутилового спирта, с молекулярным кислородом и аммиаком в присутствии катализатора. Этот способ широко известен как «процесс аммоксидирования» и в настоящее время применяется в промышленном масштабе.
[0010] Катализаторы для использования в процессе аммоксидирования тщательно изучены с целью более эффективного осуществления способа получения ненасыщенного нитрила в промышленном масштабе. Например, Mo-Bi-Fe-Ni или Mo-Bi-Fe-Sb композитные металлооксидные катализаторы известны в качестве таких катализаторов для аммоксидирования. Также с целью повышения эффективности часто проводили исследования композиции, состоящей из этих обязательных металлов с добавлением других компонентов. Например, в патентной литературе 5 раскрыт катализатор, содержащий молибден, висмут, железо, церий и никель с добавлением других компонентов. Также, в патентной литературе 6 раскрыт катализатор, содержащий молибден, висмут, железо, сурьму, никель и хром с добавлением других компонентов.
[0011] Согласно непатентной литературе 2, неупорядоченная фаза, которая представляет собой метастабильную структуру, относится к структуре, содержащей Мо центры, случайно замещенные с помощью Fe, например, в случае Bi-Mo-Fe 3-компонентного композитного оксида, и отличается тем, что Мо и атомы Fe образуют ту же кислородную тетраэдрическую структуру. С другой стороны, упорядоченная фаза, которая представляет собой стабильную структуру, имеет ту же композицию, что и неупорядоченная фаза, но структурно отличается от нее, и получают ее посредством тепловой обработки при более высоких температурах, чем для неупорядоченной фазы. В упорядоченной фазе, атомы Fe и Мо индивидуально образуют тетраэдры. Это обозначает, что атом Fe образует кислородный тетраэдр, тогда как атом Мо образует другой кислородный тетраэдр, независимо от атома Fe. В непатентной литературе 2 констатируют, что формирование неупорядоченной фазы Bi3Fe1Mo2O12 происходит при 450°С, но она подвергается фазовому переходу в упорядоченную фазу при температуре реакции 475°С.
[0012] 1) Bi3Fe1Mo2O12 с неупорядоченной фазой
На фиг. 1 представлена кристаллическая структура Bi3Fe1Mo2O12с неупорядоченной фазой, описанная в непатентной литературе 2. Эта неупорядоченная фаза представляет собой тетрагональную систему по типу кристалла шеелита (тип CaWO4) с двумя равными длинами сторон и тремя осевым углами 90 градусов в постоянной решетке элементарной ячейки (А=В≠С и α=β=γ=90 градусов). Эта фаза имеет два центра: центры X, которые заключены в кислородные тетраэдры, и центры Y, которые не заключены в атомы кислорода. Центры X заняты с помощью Мо и Fe или случайно или с определенным распределением вероятности. Центры Y заняты с помощью Bi и другими элементами или дефектами решетки или случайно или с определенным распределением вероятности. В каждом слое в плоскости АВ центры X и центры Y образуют плоские квадратные решетки с длинами, равными постоянным решетки в направлениях оси А и оси В, соответственно, и занимают положения, смещенные в направлениях оси А и оси В, соответственно, на 1/2 постоянной решетки в плоскости. Эти слои в плоскости АВ уложены стопкой в направлении оси С, и при этом повторно смещены на (А/2,0) и (0.В/2), соответственно. В этой укладке стопкой кислородные тетраэдры расположены вокруг центров X и при этом повернуты вокруг оси С на 90 градусов, причем встроенные в них атомы находятся в центре.
[0013] На фиг. 3 представлена рентгеновская дифракция (XRD) Bi3Fe1Mo2O12 с неупорядоченной фазой. Эта неупорядоченная фаза демонстрирует отдельные пики по меньшей мере в плоскостях 18,30°±0,2° (101), 28,20°±0,2° (112), 33,65°±0,2° (200) и 46,15°±0,2° (204) в диапазоне углов рентгеновской дифракции 2θ = от 10° до 60°, измеряемые посредством рентгеновской дифракции на кристаллах (XRD).
[0014] 2) Bi3Fe1Mo2O12 с упорядоченной фазой
Для сравнения, кристаллическая структура Bi3Fe1Mo2O12 с упорядоченной фазой представлена на фиг. 2. Эта упорядоченная фаза представляет собой моноклинную кристаллическую систему, которая имеет искривленную структуру шеелита с различными длинами сторон в постоянной решетки элементарной ячейки. Два из трех углов, образованных основными векторами, составляют 90 градусов, а другой угол отличается (А≠В≠С, α=γ=90 градусов и β≠90 градусов). Упорядоченная фаза имеет три центра: два неэквивалентных центра X1 и Х2, которые заключены в кислородные тетраэдры, и центры Y, которые не заключены атомами кислорода. Центры X1 заняты с помощью Мо и другими элементами или дефектами решетки. Центры Х2 заняты с помощью Fe и другими элементами или дефектами решетки. Центры Y заняты с помощью Bi и другими элементами или дефектами решетки.
[0015] 3) Структурная разница между Bi3Fe1Mo2O12 с неупорядоченной фазой и Bi3Fe1Mo2O12 с упорядоченной фазой
В Bi3Fe1Mo2O12 с неупорядоченной фазой центры X или центры Y эквивалентны друг другу или элементы различных типов скоординированы случайно. В упорядоченной фазе центры или центры Y заняты регулярно и определенно с помощью элементов различных типов или дефектами с тем, чтобы различались центры этих двух типов. Следовательно, упорядоченная фаза демонстрирует разделение пиков при рентгеновской дифракции, тогда как неупорядоченная фаза отличается тем, что обнаруживают отдельные пики (указанные стрелками на фиг. 3). При измерении в диапазоне углов рентгеновской дифракции 2θ=от 10° до 60°, пик в плоскости 18,30°±0,05° (101) в Bi3Fe1Mo2O12 с неупорядоченной фазой разделяется на плоскости 18,15°±0,05° (310) и 18,50°±0,05° (111); пик в плоскости 28,20°±0,05° (112) в неупорядоченной фазе разделяется на плоскости 28,05°±0,05° (221) и 28,40°±0,05° (42-1); пик в плоскости 33,65°±0,05° (200) в неупорядоченной фазе разделяется на плоскости 33,25°±0,05° (600) и 34,10°±0,05° (202); и пик в плоскости 46,15°±0,05° (204) в неупорядоченной фазе разделяется на плоскости 45,85°±0,05° (640) и 46,50°±0,05° (242).
Список цитируемой литературы
Патентная литература
[0016] Патентная литература 1: международная публикация №WO 95/35273
Патентная литература 2: выложенный японский патент №S49-14392
Патентная литература 3: публикация японского патента №S61-12488
Патентная литература 4: выложенный японский патент №2010-120933
Патентная литература 5: выложенный японский патент №2006-61888
Патентная литература 6: выложенный японский патент №2010-253414
Непатентная литература
[0017] Непатентная литература 1: Chemical Process of Petroleum, ed. by The Japan Petroleum Institute, p. 172-176, Kodansha Scientific Ltd.
Непатентная литература 2: Acta. Cryst (1976). B32, p. 1163-1170
Сущность изобретения
Техническая проблема
[0018] Оксидные катализаторы, раскрытые в патентной литературе 4, 5 и 6, главным образом улучшают начальный выход реакции, но не способны давать достаточно удовлетворительный выход при промышленной эксплуатации в течение длительного времени. Эти катализаторы, когда их используют в течение длительного времени в способе получения ненасыщенного альдегида, диолефина или ненасыщенного нитрила, подвергаются нежелательному разложению из-за восстановления пропиленом, изобутиленом, изобутанолом, н-бутеном, т-бутиловым спиртом и аммиаком и т.д.
[0019] Условия реакции с высокой концентрацией изобутилена и высокой температурой реакции являются желаемыми с точки зрения производительности ненасыщенного альдегида. В этом случае катализаторы более восприимчивы восстановительному разложению. Кроме того, получаемый ненасыщенный альдегид также может разлагаться, что ведет к неблагоприятному снижению выхода ненасыщенного альдегида.
[0020] Авторы настоящего изобретения сделали следующее предположение относительно причины того, почему на практике используют реакционную систему в неподвижном слое, хотя в целом принята реакционная система в псевдоожиженном слое, которая с точки зрения промышленной практики подходит в отношении управления температурой в реакторе: ненасыщенный альдегид, который представляет собой продукт, представляющий интерес, легко подвергается разложению горением в атмосфере с высокой температурой, которая содержит кислород, в реакторе прежде, чем достичь выпуска реактора, из-за его очень высокой реакционной способность, и последовательно разлагается на ненасыщенную карбоновую кислоту или диоксид углерода. Кроме того, для реакционной системы в псевдоожиженном слое, в которой происходит контакт между продуктами и катализаторами, необходим: обогащенный слой, в котором большинство катализаторов присутствуют в состоянии потока; и разбавленный слой, который представляет собой пространство для снижения линейной скорости для разделения катализатора. Время пребывания продуктов в реакторе после выхода из каталитического слоя (обогащенного слоя), следовательно, составляет по меньшей мере в 10 раз больше, чем в реакторе с неподвижным слоем. Как можно представить, это дополнительно способствует разложению ненасыщенного альдегида (продукта, представляющего интерес) в реакторе. Как результат, снижение выхода ненасыщенного альдегида является неизбежной проблемой для реакционной системы в псевдоожиженном слое.
[0021] До настоящего времени реакционную систему в псевдоожиженном слое, которую считают промышленно благоприятной с точки зрения управления температурой, не использовали на практике при получении ненасыщенного альдегида через реакцию каталитического окисления олефина и/или спирта, и вместо нее использовали реакционную систему в неподвижном слое. Вероятно, это обусловлено отсутствием средства для того, чтобы предотвращать разложение ненасыщенного альдегида с высокой реакционной способностью. Предположительно, не было другого выбора, кроме как использовать реакционную систему в неподвижном слое, превосходную в отношении сбора продуктов, даже если поступаться промышленной эффективностью для предотвращения разложения продукта и обеспечивать необходимый выход.
[0022] Настоящее изобретение выполнено в свете описанных выше проблем, и цель настоящего изобретения состоит в том, чтобы предоставить оксидный катализатор, который предотвращает восстановительное разложение катализатора, даже во время промышленной эксплуатации в течение длительного времени и меньше снижает выход ненасыщенного альдегида, выхода диолефина или выход ненасыщенного нитрила, и способ его получения, а также способы получения ненасыщенного альдегида, диолефина и ненасыщенного нитрила с использованием оксидного катализатора.
Решение проблемы
[0023] Как показано в непатентной литературе 1, неупорядоченная фаза термически нестабильна. Ранее принято во внимание, что неупорядоченная фаза, которую несет катализатор для использования в реакции окисления в газовой фазе, которую осуществляют при высокой температуре, не имеет ценности.
[0024] Тем не менее, исследования, проведенные авторами настоящего изобретения, выявили, что некоторые неупорядоченные фазы стабильно присутствуют даже при высокой температуре и могут стабильно присутствовать в течение очень длительного времени даже в восстанавливающей атмосфере. На основе этого открытия авторы настоящего изобретения проводили тщательные исследования и, следовательно, выполнили настоящее изобретение, обнаружив, что катализатор, имеющий неупорядоченную фазу, которая стабильна даже при высокой температуре, и имеет высокую устойчивость к восстановлению, можно получать посредством успешного встраивания элемента, имеющего предварительно определяемый радиус иона, в кристаллическую структуру.
[0025] В частности, настоящее изобретение представляет собой следующее:
[1] Оксидный катализатор для использования в получении ненасыщенного альдегида, диолефина или ненасыщенного нитрила из олефина и/или спирта, оксидный катализатор отвечает следующим с (1) до (3):
(1) оксидный катализатор содержит молибден, висмут, железо, кобальт и элемент А, который имеет радиус иона больше чем 0,96

(2) атомное соотношение а висмута к 12 атомам молибдена составляет 1≤а≤5, атомное соотношение b железа к 12 атомам молибдена составляет 1,5≤b≤6, атомное соотношение с элемента А к 12 атомам молибдена составляет 1≤с≤5 и атомное соотношение d кобальта к 12 атомам молибдена составляет 1≤d≤8; и
(3) оксидный катализатор содержит неупорядоченную фазу, состоящую из кристаллической системы, которая содержит молибден, висмут, железо и элемент А.
[2] Оксидный катализатор согласно приведенному выше пункту [1], где оксидный катализатор имеет отдельный пик в каждом диапазоне дифракционных углов (2θ) 18,30°±0,2°, 28,20°±0,2°, 33,65°±0,2° и 46,15°±0,2° при рентгеновской дифракции, и
соотношение интенсивностей (Ia/Ib) интенсивности (Ia) пика а при 2θ=33,65°±0,2° к интенсивности (Ib) пика b при 2θ=34,10°±0,2° составляет 2,0 или более.
[3] Оксидный катализатор согласно приведенному выше пункту [1] или [2], где оксидный катализатор имеет композицию, представленную с помощью следующей формулы композиции (1):
Mo12BiaFebAcCodBeCfOg (1)
где Мо представляет молибден; Bi представляет висмут; Fe представляет железо; элемент А представляет элемент, имеющий радиус иона больше чем 0,96

[4] Оксидный катализатор согласно какому-либо одному из приведенных выше пунктов [1]-[3], дополнительно содержащий по меньшей мере одно, выбранное из группы, состоящей из диоксида кремния, оксида алюминия, оксида титана и оксида циркония в качестве носителя.
[5] Способ получения оксидного катализатора, включающий: стадию смешивания для смешивания исходного материала, образующего катализатор, который содержит молибден, висмут, железо, кобальт и элемент А, который имеет радиус иона больше чем 0,96

стадию сушки для сушки суспензии, таким образом полученной, для того, чтобы получать высушенный продукт; и
стадию прокаливания высушенного продукта, таким образом полученного, где
стадия прокаливания включает стадию нагревания для постепенного нагревания высушенного продукта от 100°С до 200°С в течение 1 часа или дольше.
[6] Способ получения оксидного катализатора согласно приведенному выше пункту [5], где суспензия имеет рН 8 или ниже.
[7] Способ получения оксидного катализатора согласно приведенному выше пункту [5] или [6], где стадия прокаливания включает:
предварительную стадию прокаливания высушенного продукта при температуре от 200 до 300°С для того, чтобы получать предварительно прокаленный продукт; и
стадию конечного прокаливания предварительно прокаленного продукта, таким образом полученного, при температуре 300°С или выше для того, чтобы получать катализатор.
[8] Способ получения ненасыщенного альдегида, который включает стадию получения ненасыщенного альдегида для окисления олефина и/или спирта с использованием оксидного катализатора согласно какому-либо одному из приведенных выше пунктов [1]-[4] для того, чтобы получать ненасыщенный альдегид.
[9] Способ получения ненасыщенного альдегида согласно приведенному выше пункту [8], где олефин и/или спирт представляет собой по меньшей мере одно, выбранное из группы, состоящей из пропилена, изобутилена, пропанола, изопропанола, изобутанола и т-бутилового спирта.
[10] Способ получения ненасыщенного альдегида согласно приведенному выше пункту [8] или [9], где стадия получения ненасыщенного альдегида включает стадию выгрузки для выгрузки газообразного продукта, который содержит ненасыщенный альдегид, из реактора с псевдоожиженным слоем через реакцию каталитического окисления в газовой фазе олефина и/или спирта с использованием источника кислорода в реакторе с псевдоожиженным слоем.
[11] Способ получения ненасыщенного альдегида согласно какому-либо одному из приведенных выше пунктов [8]-[10], где
реакцию каталитического окисления в газовой фазе осуществляют при температуре реакции от 400 до 500°С, и
газообразный продукт, выгружаемый из реактора с псевдоожиженным слоем, имеет концентрацию кислорода от 0,03 до 0,5% по объему.
[12] Способ получения диолефина, включающий стадию получения диолефина для окисления моноолефина, который имеет 4 или больше углеродных атомов, с использованием оксидного катализатора согласно какому-либо одному из приведенных выше пунктов [1]-[4] для того, чтобы получать диолефин.
[13] Способ получения ненасыщенного нитрила, включающий стадию получения ненасыщенного нитрила для проведения реакции по меньшей мере одного, выбранного из группы, состоящей из пропилена, изобутилена, пропанола, изопропанола, изобутанола и т-бутилового спирта, с молекулярным кислородом и аммиаком в реакторе с псевдоожиженным слоем с использованием оксидного катализатора согласно какому-либо одному из приведенных выше пунктов [1]-[4] для того, чтобы получать ненасыщенный нитрил.
Полезные эффекты изобретения
[0026] Настоящее изобретение может предоставлять оксидный катализатор, который препятствует восстановительному разложению катализатора, даже во время промышленной эксплуатации в течение длительного времени, и меньше снижает выход ненасыщенного альдегида, выход диолефина или выход ненасыщенного нитрила, и способ его получения, а также способы получения ненасыщенного альдегида, диолефина и ненасыщенного нитрила с использованием оксидного катализатора.
Описание вариантов осуществления
[0027] На фиг. 1 представлен схематический вид, который иллюстрирует кристаллическую структуру неупорядоченной фазы в Bi3Fe1Mo2O12;
на фиг. 2 представлен схематический вид, который иллюстрирует кристаллическую структуру упорядоченной фазы в Bi3Fe1Mo2O12;
на фиг. 3a представлена диаграмма, которая иллюстрирует рентгеновскую дифракцию неупорядоченной фазы в Bi3Fe1Mo2O12, а на фиг. 3b представлена диаграмма, которая иллюстрирует рентгеновскую дифракцию упорядоченной фазы в Bi3Fe1Mo2O12;
на фиг. 4 представлена диаграмма, которая иллюстрирует зависимость между уровнем содержания неупорядоченной фазы и пиками а и b;
на фиг. 5 представлена диаграмма, которая иллюстрирует рентгеновскую дифракцию катализаторов, получаемых в примере А1 и сравнительном примере A3;
на фиг. 6 представлена увеличенная диаграмма, которая иллюстрирует диапазон 2θ = от 15 до 30° при рентгеновской дифракции на фиг. 5;
на фиг. 7 представлена увеличенная диаграмма, которая иллюстрирует диапазон 2θ = от 30 до 50° при рентгеновской дифракции на фиг. 5;
на фиг. 8 представлена диаграмма, которая иллюстрирует рентгеновскую дифракцию (2θ = от 25 до 27°) оксидного катализатора до и после реакции каталитического окисления в газовой фазе олефина в сравнительном примере А6;
на фиг. 9 представлена диаграмма, которая иллюстрирует пики рентгеновской дифракции катализаторов, получаемых в примерах В1 и сравнительном примере В1;
на фиг. 10 представлена увеличенная диаграмма, которая иллюстрирует диапазон 2θ = от 15 до 30° в пиках рентгеновской дифракции на фиг. 9;
на фиг. 11 представлена увеличенная диаграмма, которая иллюстрирует диапазон 2θ = от 30 до 50° в пиках рентгеновской дифракции на фиг. 9;
на фиг. 12 представлена диаграмма, которая иллюстрирует XRD (2θ = от 10 до 60°) оксидного катализатора до и после реакции каталитического окисления в газовой фазе олефина в примере С1;
на фиг. 13 представлена увеличенная диаграмма, которая иллюстрирует диапазон 2θ = от 25 до 27° на фиг. 12;
на фиг. 14 представлена диаграмма, которая иллюстрирует XRD (2θ = от 10 до 60°) оксидного катализатора до и после реакции каталитического окисления в газовой фазе олефина в сравнительном примере С1;
на фиг. 15 представлена увеличенная диаграмма, которая иллюстрирует диапазон 2θ = от 25 до 27° на фиг. 14; и
на фиг. 16 представлена диаграмма, которая иллюстрирует пики рентгеновской дифракции катализаторов, получаемых в примерах С1 и сравнительном примере С2.
Вариант осуществления изобретения
[0028] Далее в настоящем документе подробно описаны с первого до третьего вариантов осуществления для выполнения настоящего изобретения. Не предполагают, что настоящее изобретение ограничено вариантами осуществления, описанными ниже, и в нем можно выполнять различные изменения или модификации, не отступая от сущности настоящего изобретения.
[0029] [Первый вариант осуществления]
Оксидный катализатор
Описан оксидный катализатор согласно первому варианту осуществления.
Оксидный катализатор согласно первому варианту осуществления представляет собой оксидный катализатор для использования в получении ненасыщенного альдегида или диолефина из олефина и/или спирта, оксидный катализатор отвечает следующим с (1) до (3):
(1) оксидный катализатор содержит молибден (далее в настоящем документе также обозначаемый как «Мо»), висмут (далее в настоящем документе также обозначаемый как «Bi»), железо (далее в настоящем документе также обозначаемое как «Fe»), кобальт (далее в настоящем документе также обозначаемый как «Со») и элемент А, который имеет радиус иона больше чем 0,96

(2) атомное соотношение а висмута к 12 атомам молибдена составляет 1≤а≤5, атомное соотношение b железа к 12 атомам молибдена составляет 1,5≤b≤6, атомное соотношение с элемента А к 12 атомам молибдена составляет 1≤с≤5 и атомное соотношение d кобальта к 12 атомам молибдена составляет 1≤d≤8; и
(3) оксидный катализатор содержит неупорядоченную фазу, состоящую из кристаллической системы, которая содержит молибден, висмут, железо и элемент А.
[0030] Исходный материал
Примеры олефина, выполняющего функцию исходного материала для использования в получении ненасыщенного альдегида или диолефина, включают, но не ограничиваясь этим, пропилен, н-бутен, изобутилен, н-пентен, н-гексен и циклогексен. Среди них, пропилен и изобутилен являются предпочтительными.
[0031] Примеры спирта, выполняющего функцию исходного материала для использования в получении ненасыщенного альдегида или диолефина, включают, но не ограничиваясь этим, пропанол, изопропанол, бутанол, изобутанол и т-бутиловый спирт. Среди них, изобутанол и т-бутиловый спирт являются предпочтительными.
[0032] В первом варианте осуществления, например, акролеин или акриловую кислоту можно получать с использованием пропилена, пропанола или изопропанола в качестве исходного материала, тогда как метакролеин или метакриловую кислоту можно получать с использованием изобутилена, изобутанола или т-бутилового спирта в качестве исходного материала.
[0033] Альтернативно, бутадиен можно получать с использованием н-бутена в качестве исходного материала. Исходные материалы олефин и спирт могут содержать воду, азот и алканы, такие как пропан, бутан и изобутан.
[0034] Эти олефины и/или спирты можно использовать отдельно или в комбинации.
[0035] (I) Композиция
Оксидный катализатор согласно первому варианту осуществления содержит молибден, висмут, железо, кобальт и элемент А, который имеет радиус иона больше чем 0,96

[0036] Атомное соотношение а для Bi к 12 атомам Мо составляет 1≤а≤5. Атомное соотношение а предпочтительно составляет 1≤а≤4, более предпочтительно 1≤а≤3, с точки зрения дополнительного повышения избирательности к ненасыщенному альдегиду и/или диолефину.
[0037] С точки зрения повышения каталитической активности без снижения избирательности к ненасыщенному альдегиду и/или диолефину, Fe является обязательным элементом, как и в случае Мо и Bi, для промышленного синтеза ненасыщенного альдегида и/или диолефина. Однако при высоком содержании Fe образуется Fe2O3 и возникает склонность к увеличению побочных продуктов, таких как СО или СО2, что ведет к сниженной избирательности к ненасыщенному альдегиду и/или диолефину. Альтернативно, при высоком содержании Fe может не образовываться Fe2O3, но вместо этого образуется Fe-Мо-O 2-компонентый композитный оксид, который представляет собой неактивный компонент, который не проявляет каталитическую активность. С этих точек зрения атомное соотношение b для Fe к 12 атомам Мо в оксидном катализаторе согласно первому варианту осуществления составляет 1,5≤b≤6, предпочтительно 1,5≤b≤5, более предпочтительно 1,5≤b≤4.
[0038] Bi и Мо склонны к образованию композитного оксида, такого как Bi2Mo3O12 или Bi2MoO6, который по имеющимся сообщениям представляет собой активные частицы при каталитическом окислении в газовой фазе, аммоксидировании, реакции окислительной дегидрогенизации и т.п. Катализатор, который состоит из такого композитного оксида, дает высокую избирательность к ненасыщенному альдегиду или диолефину, но обладает низкой активностью. С другой стороны, Fe и Мо образуют композитный оксид, такой как Fe2Mo3O12. Катализатор, который состоит из такого композитного оксида, демонстрирует как низкую активность, так и низкую избирательность. Тем не менее, подходящая композиция из Мо, Bi и Fe образует 3-компонентный композитный оксид, содержащий Bi3Fe1Mo2O12 с неупорядоченной фазой с высокой активностью и высокой избирательностью к ненасыщенному альдегиду и диолефину.
[0039] Авторы настоящего изобретения проводили тщательные исследования для того, чтобы получить оксидный катализатор, превосходный в отношении термостойкости, который сохраняет описанную выше характерную структуру даже при высокой температуре, и, следовательно, обнаружили, что улучшение термостойкости оксидного катализатора происходит посредством встраивания элемента А, который имеет радиус иона больше чем 0,96
[0040] Элемент А без ограничений может представлять собой какой-либо элемент, который имеет радиус иона больше чем 0,96

[0041] Атомное соотношение с элемента А к 12 атомам Мо составляет 1≤с≤5, предпочтительно 1≤с≤4, более предпочтительно 1≤с≤3. При условии, что атомное соотношение с попадает в описанный выше диапазон, формируют 4-компонентный композитный оксид, содержащий неупорядоченную фазу Bi3-xAxFe1Mo2O12. Например, в случае использования La в качестве элемента А, формируют 4-компонентный композитный оксид, который содержит неупорядоченную фазу Bi3-xLaxFe1Mo2O12. La можно надлежащим образом вводить в композицию 3-компонентного композитного оксида, который содержит Bi3Fe1Mo2O12 с неупорядоченной фазой, с тем, чтобы Bi3+ (радиус иона: 0,96

[0042] Со является необходимым для того, чтобы придавать устойчивость к восстановлению оксидному катализатору согласно первому варианту осуществления. В присутствии Со, двухвалентное железо встраивают в CoMoO4 для того, чтобы формировать Co2+-Fe2+-Мо-O 3-компонентную кристаллическую структуру. Это встроенное железо легко окисляется до трехвалентного железа в реакционной атмосфере по причине его метастабильной структуры. Следовательно, во время реакции образуется окислительно-восстановительный цикл, предположительно препятствующий восстановительному разложению.
[0043] Fe в Fe2Mo3O12 или 4-компонентном композитном оксиде, который содержит неупорядоченную фазу Bi3-xAxFe1Mo2O12, является трехвалентным сразу после начала реакции, но восстанавливается до двухвалентного железа через окислительно-восстановительные циклы во время реакции. В отсутствие Со происходит образование композитного оксида двухвалентного железа и молибдена (FeMoO4) и MoO2. Этот FeMoO4 представляет собой редко встречающийся тривалент в реакционной атмосфере, по причине его стабильной структуры. Образование этих стабильных соединений стабилизирует железо с тем, чтобы во время реакции не протекал окислительно-восстановительный цикл, что предположительно вызывает восстановительное разложение.
[0044] Атомное соотношение d для Со к 12 атомам Мо составляет 1≤d≤8, предпочтительно 2≤d≤8, более предпочтительно 2≤d≤6, более предпочтительно 3≤d≤5. При атомном соотношении d, превышающем 8, 4-компонентный композитный оксид, который содержит неупорядоченную фазу Bi3-xAxFe1Mo2O12, образуется с трудом.
[0045] (2) Кристаллическая структура
Оксидный катализатор согласно первому варианту осуществления предпочтительно содержит 4-компонентный композитный оксид, который содержит неупорядоченную фазу Bi3-xAxFe1Mo2O12. Рентгеновскую дифракцию (XRD) можно использовать в качестве показателя для образования 4-компонентного композитного оксида, который содержит неупорядоченную фазу Bi3-xAxFe1Mo2O12. Оксидный катализатор согласно первому варианту осуществления предпочтительно имеет отдельные пики по меньшей мере в плоскостях 18,30°±0,2° (101), 28,20°±0,2° (112), 33,65°±0,2° (200) и 46,15°±0,2° (204), измеренные в диапазоне дифракционных углов 2θ = от 10° до 60° при рентгеновской дифракции на кристаллах, как в 3-компонентном композитном оксиде, который содержит Bi3Fe1Mo2O12 с неупорядоченной фазой. Особенно предпочтительно, оксидный катализатор согласно первому варианту осуществления имеет отдельный пик в положении в пределах каждого эталонного значения ±0,05°.
[0046] Соотношение интенсивностей (Ia/Ib) для интенсивности (Ia) пика а при 2θ=33,65°±0,2° к интенсивности (Ib) пика b при 2θ=34,10°±0,2° предпочтительно составляет 2,0 или более, более предпочтительно 2,5 или более, более предпочтительно 3,0 или более, с точки зрения дополнительного предотвращения разложения катализатора и получения ненасыщенного альдегида и/или диолефина с более высоким выходом. Соотношение интенсивностей (Ia/Ib) составляет 1,1 при 100% упорядоченной фазе и составляет 3,3 при 100% неупорядоченной фазе. Интенсивность (Ib) пика b при 2θ=34,10°±0,2° также содержит интенсивность пика, полученного из Fe2Mo3O12, присутствующего в следовых количествах. При соотношении интенсивностей (Ia/Ib) 2,0 или более, оксидный катализатор содержит предварительно определяемую долю неупорядоченной фазы и, следовательно, дает сниженный выход ненасыщенной карбоновой кислоты и улучшенный выход ненасыщенного альдегида и/или диолефина. На фиг. 4 представлена зависимость содержания неупорядоченной фазы и пиков а и b.
[0047] Механизм образования неупорядоченной фазы Bi3-xAxFe1Mo2O12 не определен. Композитный оксид Bi и Мо формируют в качестве промежуточного продукта на стадии тепловой обработки. Дополнительная тепловая обработка, вероятно, является причиной тепловой диффузии и замещающего твердого раствора Fe и элемента А в композитном оксиде Bi и Мо для того, чтобы формировать композитную неупорядоченную фазу Bi3-xAxFe1Mo2O12.
[0048] Отдельный пик
Термин «отдельный пик», описанный в настоящем документе, не следует понимать в строгом смысле, и основной пик, обнаруживаемый при каждом дифракционном угле, можно понимать как отдельный пик, если он не расщеплен. Это обозначает, что даже пик, имеющий точку перегиба, можно рассматривать в качестве отдельного пика. Исключают явно меньший пик, чем основной пик, если он присутствует, и затем по желанию определяют, является основной пик отдельным или расщепленным пиком. «Меньший пик» относится к пику, который имеет интенсивность меньше чем 50% от интенсивности основного пика в предварительно определяемом диапазоне дифракционных углов.
[0049] «Основной пик» относится к большему пику среди пиков, присутствующих в предварительно определяемом диапазоне дифракционных углов. Например, что касается дифракционного угла в диапазоне 18,30°±0,2°, больший пик можно обнаруживать в положении 18,35°. В таком случае, этот пик определяют как основной пик при 18,30°±0,2°.
[0050] Оксидный катализатор согласно первому варианту осуществления предпочтительно имеет композицию, представленную с помощью формулы композиции (1), представленной ниже. Оксидный катализатор, который имеет композицию, представленную с помощью формулы композиции (1), представленной ниже, склонен предотвращать образование ненасыщенной карбоновой кислоты или диоксида углерода и дополнительно повышать избирательность к ненасыщенному альдегиду и/или диолефину. Формула композиции (1)

где Мо представляет молибден; Bi представляет висмут; Fe представляет железо; элемент А представляет элемент, который имеет радиус иона больше чем 0,96

Мо, где атомное соотношение а для Bi составляет 1≤а≤5, атомное соотношение b для Fe составляет 1,5≤b≤6, атомное соотношение с для элемента А составляет 1≤с≤5, атомное соотношение d для Со составляет 1≤d≤8, атомное соотношение е для элемента В составляет 0≤е<3, атомное соотношение f для элемента С составляет 0≤f≤2 и соотношение Fe/Co составляет 0,8≤b/d; и g представляет атомность кислорода, которую определяют посредством валентностей составляющих элементов, отличных от кислорода.
[0051] В формуле композиции (1) элемент В представляет по меньшей мере один элемент, выбранный из группы, состоящей из магния, цинка, меди, никеля, марганца, хрома и олова, и предположительно замещает некоторые атомы кобальта в оксидном катализаторе. Атомное соотношение е элемента В предпочтительно составляет 0≤е≤3, более предпочтительно 0≤е<2, с точки зрения сохранения баланса с образованием кристаллов неупорядоченной фазы. Элемент В не является обязательным, но вносит вклад в повышение активности катализатора или стабилизацию кристаллической структуры CoMoO4 в катализаторе. Например, медь имеет эффект повышения активности катализатора. Никель, магний, цинк и марганец оказывают эффекты на стабилизацию кристаллической структуры CoMoO4 и ингибирование, например, фазового перехода, объясняемого давлением или температурой.
[0052] В оксидном катализаторе согласно первому варианту осуществления, Со представляет собой обязательный элемент, как и в случае Мо, Bi и Fe, с точки зрения промышленного синтеза ненасыщенного альдегида и/или диолефина. Со играет роль в качестве носителя для формирования композитного оксида CoMoO4 и высокодисперсных активных частиц, таких как Bi-Mo-О, а также играет роль в захвате кислорода из газовой фазы и доставки этого кислорода к Bi-Mo-О или тому подобное. Со и Мо предпочтительно вводят в композицию для того, чтобы формировать композитный оксид CoMoO4, с точки зрения получения ненасыщенного альдегида и/или диолефина с высоким выходом. Атомное соотношение d для Со предпочтительно составляет 1≤d≤8, более предпочтительно 2≤d≤8, более предпочтительно 2≤d≤7, еще более предпочтительно 2≤d≤5, с точки зрения снижения образования простых оксидов, таких как CO3O4 или СоО.
[0053] Соотношение Fe/Co предпочтительно составляет 0,8≤b/d, более предпочтительно 0,8≤b/d≤1,5, более предпочтительно 0,9≤b/d≤1,2. При условии, что соотношение Fe/Co попадает в диапазон, описанный выше, простые оксиды, такие как Co3O4 или СоО, склонны к редкому образованию.
[0054] Элемент А представляет элемент, который имеет радиус иона больше чем 0,96

[0055] Элемент С представляет по меньшей мере один элемент, выбранный из группы, состоящей из калия, цезия и рубидия. Элемент С вероятно играет роль в нейтрализации кислотного центра некомпозитного МоО3 или тому подобное в оксидном катализаторе. Присутствие или отсутствие элемента С, содержащегося в нем, непосредственно не оказывает влияния на кристаллическую структуру неупорядоченной фазы, описанной далее. Атомное соотношение f элемента С к 12 атомам Мо предпочтительно составляет 0≤f≤2, более предпочтительно 0,01≤f≤2, более предпочтительно 0,01≤f≤1, с точки зрения каталитической активности. При атомном соотношении f, равном 0 или более, нейтрализующий эффект проявляет тенденцию к дальнейшему улучшению. При атомном соотношении f, равном 2 или меньше, оксидный катализатор склонен становиться основным или нейтральным. Получаемый оксидный катализатор легко адсорбирует исходный материал олефин или спирт и склонен проявлять более высокую каталитическую активность.
[0056] Оксидный катализатор может содержать произвольные компоненты в качестве других металлических компонентов без ингибирования образования неупорядоченной фазы Bi3-xAxFe1Mo2O12.
[0057] Элемент В и элемент С образуют кристаллические структуры отдельно от кристаллической структуры неупорядоченной фазы, описанной далее, и, следовательно, не оказывают непосредственного влияния на кристаллическую структуру неупорядоченной фазы.
[0058] (3) Компонент, отличный от оксида металла
Оксидный катализатор согласно первому варианту осуществления дополнительно может содержать носитель для того, чтобы поддерживать оксид металла. Катализатор, дополнительно содержащий носитель, является предпочтительным по причине высокого диспергирования оксида металла и наделения поддерживаемого оксида металла высокой устойчивостью к износу. В этом контексте, катализатор предпочтительно содержит носитель, когда его формуют способом экструзионного формования. Альтернативно, катализатор, который прессуют в таблетки, для получения метакролеина в реакторе с неподвижным слоем может не обязательно содержать носитель.
[0059] Примеры носителя включают, но не ограничиваясь этим, по меньшей мере одно, выбранное из группы, состоящей из диоксида кремния, оксида алюминия, оксида титана и оксида циркония. Оксидный катализатор на основе такого носителя, склонен иметь дополнительно улучшенные физические свойства, подходящие для реакции в псевдоожиженном слое, такие как геометрическая форма, размер, распределение, текучесть и механическая прочность частицы. Среди них, диоксид кремния предпочтителен в качестве носителя. В целом, носитель из диоксида кремния предпочтителен в отношении его способности придавать физические свойства, подходящие для реакции в псевдоожиженном слое, оксидному катализатору, а также в силу того, что он не активен по сравнению с другими носителями и оказывает благоприятный связывающий эффект на оксид металла без снижения избирательности к ненасыщенному альдегиду и/или диолефину. Кроме того, носитель из диоксида кремния также предпочтителен, поскольку он легко придает высокую устойчивость к износу оксиду металла, который он несет.
[0060] Золь диоксида кремния предпочтителен в качестве источника диоксида кремния. Концентрация золя диоксида кремния в состоянии исходного материала, не смешанного с другими компонентами, предпочтительно составляет от 10 до 50% по массе с точки зрения диспергируемости частиц диоксида кремния. Золь диоксида кремния предпочтительно содержит от 40 до 100% по массе по меньшей мере одного золя диоксида кремния (а), содержащего основные частицы диоксида кремния, которые имеют средний диаметр частицы от 20 нм до меньше чем 55 нм, предпочтительно от 20 до 50 нм, и от 60 до 0% по массе по меньшей мере одного золя диоксида кремния (b), содержащего основные частицы диоксида кремния, которые имеют средний диаметр частицы от 5 нм до меньше чем 20 нм, с точки зрения избирательности к ненасыщенному нитрилу.
[0061] Содержание носителя предпочтительно составляет от 20 до 80% по массе, более предпочтительно от 30 до 70% по массе, более предпочтительно от 40 до 60% по массе, еще более предпочтительно от 5 до 10% по массе, относительно всего 100% по массе носителя и оксидного катализатора. Когда задают содержание носителя, которое попадает в диапазон, описанный выше, выход ненасыщенного альдегида и/или диолефина проявляет тенденцию к дополнительному повышению.
[0062] (4) Формование оксидного катализатора
Оксидный катализатор по первому варианту осуществления можно формовать для использования. В таком случае, формование осуществляют посредством известного в данной области способа, такого как прессование таблеток или экструзионное формование. Примеры геометрических форм, в которых формуют оксидный катализатор, включают таблетки, пеллеты, сферы, геометрические формы, разработанные на компьютере (CDS), из трех долей, из четырех долей, кольца, большие геометрические поверхности (HGS), формы клеверного листа и формы медовых сот. Среди них, CDS и кольца являются предпочтительными с точки зрения прочности.
[0063] Удельная площадь поверхности оксидного катализатора предпочтительно составляет от 2 до 5 м2/г, более предпочтительно от 2 до 4 м2/г. В случае использования носителя, такого как диоксид кремния, удельная площадь поверхности проявляет тенденцию быть больше чем от 2 до 5 м2/г. Удельная площадь поверхности только оксида металла без носителя предпочтительно составляет от 2 до 5 м2/г.
[0064] [2] Способ получения оксидного катализатора
Как указано выше, авторы настоящего изобретения сосредоточились на получении неупорядоченной фазы Bi3-xAxFe1Mo2O12, содержащей элемент A, Bi, Fe и Мо, и всесторонне изучали соотношения в композициях элементов и способ получения неупорядоченной фазы.
[0065] Как видно из названия Bi-Mo катализатор, и Bi и Мо представляют собой обязательные элементы для формирования активных частиц. Катализатор, богатый Bi и Мо, благоприятен с точки зрения активности. Однако показано, что высокое содержание Bi делает катализатор негомогенным. Например, нитрат висмута, который представляет собой стандартный источник Bi, используемый в промышленности, представляет собой плохо водорастворимое вещество и, следовательно, требует большое количество азотной кислоты для его растворения. Как результат, катализатор имеет негомогенную композицию после прокаливания. В этом отношении, содержание Bi ограничено пределами диапазона в общепринятом способе получения катализаторов. Это не позволяет получать гомогенный катализатор из-за образования простых оксидов, таких как Bi2O3, что ведет к неблагоприятному снижению выхода ненасыщенного альдегида и/или диолефина.
[0066] С точки зрения повышения каталитической активности без снижения избирательности к ненасыщенному альдегиду и/или диолефину, ранее сообщалось, что Fe является обязательным элементом, как и в случае Мо и Bi, для промышленного синтеза ненасыщенного альдегида и/или диолефина. Однако, как сообщалось в международной публикации №WO 95/35273, большое количество добавленного Fe склонно увеличивать побочные продукты, такие как СО или СО2, что ведет к сниженной избирательности к ненасыщенному альдегиду и/или диолефину. Следовательно, оптимально добавлять Fe в малом количестве.
[0067] После многих испытаний и ошибок в решении этой проблемы, авторы настоящего изобретения обнаружили, что кристаллы неупорядоченной фазы Bi3-xAxFe1Mo2O12 можно легко формировать при образовании упорядоченной фазы, подавленном с помощью способа получения оксидного катализатора, который описан далее.
[0068] Способ получения оксидного катализатора согласно первому варианту осуществления включает:
стадию смешивания для смешивания исходных материалов, которые образуют катализатор, который содержит молибден, висмут, железо, кобальт и элемент А, который имеет радиус иона больше чем 0,96

стадию сушки для сушки суспензии, таким образом полученной, для того, чтобы получать высушенный продукт; и
стадию прокаливания высушенного продукта, таким образом полученного, где
стадия прокаливания включает стадию нагревания для постепенного нагревания высушенного продукта от 100°С до 200°С в течение 1 часа или дольше.
[0069] (1) Стадия смешивания
Стадия смешивания представляет собой стадию для смешивания исходных материалов металлических элементов, образующих катализатор, которые содержат молибден, висмут, железо, кобальт и элемент А, который имеет радиус иона больше чем 0,96

[0070] В случае использования оксида каждого источника элемента, предпочтительно используют дисперсию, которая содержит оксид, диспергированный в воде или органическом растворителе, и предпочтительно используют оксидную дисперсию, которая содержит оксид, диспергированный в воде. Дисперсия оксида в воде дополнительно может содержать стабилизатор дисперсии, такой как полимер, для диспергирования оксида. Размер частицы оксида предпочтительно составляет от 1 до 500 нм, более предпочтительно от 10 до 80 нм.
[0071] В суспензию можно надлежащим образом добавлять: водорастворимые полимеры, такие как полиэтиленгликоль, метилцеллюлоза, поливиниловый спирт, полиакриловая кислота и полиакриламид; поливалентные карбоновые кислоты, такие как амины, аминокарбоновые кислоты, щавелевая кислота, малоновая кислота и янтарная кислота/ и/или органические кислоты, такие как гликолевая кислота, яблочная кислота, винная кислота и лимонная кислота, с точки зрения единообразного диспергирования исходных материалов катализатора в суспензии. Количество добавляемого водорастворимого полимера и/или органической кислоты составляет, но не ограничиваясь этим, предпочтительно 30% по массе или менее относительно 100% по массе исходных материалов катализатора с точки зрения баланса между однородностью и выходом.
[0072] Суспензию без ограничений можно получать с помощью любого обычно используемого способа и можно получать, например, посредством смешивания раствора, содержащего соль аммония и молибдена, растворенную в горячей воде, с растворами, содержащими нитраты металлических компонентов, отличных от молибдена, таких как висмут, железо, кобальт и элемент А, растворенные в воде или этими металлическими компонентами, растворенными в водном растворе азотной кислоты. В случае оксидного катализатора, содержащего носитель, например, золь диоксида кремния можно добавлять до или после смешивания раствора, содержащего соль аммония и молибдена, растворенную в горячей воде, с растворами, содержащими металлические компоненты, отличные от молибдена, растворенные в воде или водном растворе азотной кислоты. Суспензия, смешанная таким образом, имеет концентрацию металлического элемента обычно от 1 до 50% по массе, предпочтительно от 10 до 40% по массе, более предпочтительно от 20 до 40% по массе, относительно 100% по массе суспензии с точки зрения баланса между однородностью и выходом.
[0073] Стадия получения суспензии, описанной выше, предоставлена в иллюстративных целях и не предназначена для ограничения технического объема настоящего изобретения. Можно изменять порядок, в котором добавляют источники элементов, или можно изменять рН или вязкость суспензии посредством корректировки концентрации азотной кислоты или посредством добавления водного раствора аммиака в суспензию. Предпочтительно, суспензия гомогенна для формирования кристаллической структуры неупорядоченной фазы Bi3-xAxFe1Mo2O12. С этой точки зрения, рН суспензии предпочтительно составляет 8,0 или менее, более предпочтительно 7,0 или менее, более предпочтительно 6,0 или менее. Суспензия, которая имеет рН 8,0 или менее, может предотвращать осаждение соединений висмута и склонна способствовать образованию кристаллической структуры неупорядоченной фазы Bi3-xAxFe1Mo2O12.
[0074] (2) Стадия сушки
Стадия сушки представляют собой стадию сушки суспензии, получаемой на стадии смешивания, для того, чтобы получать высушенный продукт. Сушку можно осуществлять без ограничений с помощью любого обычно используемого способа. Примеры способа сушки включают произвольные способы, такие как испарение досуха, распылительная сушка и сушка при пониженном давлении. Примеры способа распылительной сушки включают, но не ограничиваясь этим, обычные способы, такие как системы центрифугирования, системы сопел с двумя текучими веществами и системы сопел высокого давления, которые осуществляют в промышленности. При этой сушке в качестве источника сухого тепла предпочтительно используют воздух, нагретый паром, электрическим нагревателем или тому подобное. Температура на впуске сушилки аппарата распылительной сушки обычно составляет от 150 до 400°С, предпочтительно от 180 до 400°С, более предпочтительно от 200 до 350°С.
[0075] (3) Стадия прокаливания
Стадия прокаливания представляет собой стадию для прокаливания высушенного продукта, получаемого на стадии сушки. Прокаливание можно осуществлять с использованием печи, такой как карусельная печь, туннельная печь или муфельная печь. Стадия прокаливания включает стадию нагревания для постепенного нагревания высушенного продукта от 100 до 200°С в течение 1 часа или дольше. Через эту стадию нагревания, 4 составляющих элемента Bi, Мо, Fe и элемент А можно единообразно смешивать на атомном уровне для того, чтобы легко формировать кристаллическую структуру неупорядоченной фазы Bi3-xAxFe1Mo2O12. Термин «постепенное нагревание», описанный в настоящем документе, относится к нагреванию до предварительно определяемой температуры, обычно в течение от 1 ч до 10 ч в качестве времени нагрева. Скорость нагрева не обязательно поддерживают постоянной. Время нагрева обычно составляет от 1 ч до 10 ч, предпочтительно от 1 ч до 5 ч, более предпочтительно от 2 ч до 4 ч.
[0076] Способ прокаливания высушенного продукта отличается в зависимости от используемых исходных материалов. Например, высушенный продукт из исходных материалов, содержащих ионы азотной кислоты, предпочтительно прокаливают на двух этапах из предварительного прокаливания и конечного прокаливания. В частности, прокаливание предпочтительно осуществляют посредством: стадии предварительного прокаливания высушенного продукта при температуре от 200 до 300°С для того, чтобы получать предварительно прокаленный продукт; и стадии конечного прокаливания предварительно прокаленного продукта, таким образом полученного, при температуре 300°С или выше для того, чтобы получать катализатор. Стадию нагревания осуществляют до стадии предварительного прокаливания, и затем температуру поднимают до диапазона от 200°С до 300°С, обычно в течение 1 ч или дольше.
[0077] Стадия предварительного прокаливания включает прокаливание высушенного продукта при температуре в диапазоне от 200°С до 300°С. Время предварительного прокаливания обычно составляет от 1 ч до 10 ч, предпочтительно от 2 ч до 8 ч, более предпочтительно от 3 ч до 6 ч. Предварительное прокаливание осуществляют с целью вызывать постепенное выгорание азотной кислоты, остающейся в высушенном продукте. При предварительном прокаливании обычно образуется гомогенная кристаллическая структура неупорядоченной фазы Bi3-xAxFe1Mo2O12. В этом контексте, при предварительном прокаливании в температурном диапазоне от 200°С до 300°С, непосредственно осуществляемой без стадии нагревания от 100 до 200°С, редко образуется кристаллическая структура неупорядоченной фазы Bi3-xAxFe1Mo2O12, и вместо нее может образовываться упорядоченная фаза или 2-компонентные оксиды, такие как Fe2Mo3O12, Bi2Mo3O12 или A2Mo3O12.
[0078] Предварительно прокаленный продукт, таким образом полученный посредством предварительного прокаливания, наконец прокаливают на втором этапе для того, чтобы облегчать образование кристаллической структуры неупорядоченной фазы. В соответствии с находками, сделанными авторами настоящего изобретения, образование кристаллической структуры зависит от произведения температуры прокаливания и времени прокаливания. Предпочтительно, температуру прокаливания и время прокаливания задают надлежащим образом. Температура конечного прокаливания выше, чем температура предварительного прокаливания, и предпочтительно составляет 300°С или выше, более предпочтительно 300°С или выше и 700°С или ниже, более предпочтительно от 300 до 650°С, еще более предпочтительно от 400°С до 600°С, особенно предпочтительно от 450°С до 600°С. Когда задают температуру конечного прокаливания, которая попадает в диапазон, описанный выше, кристаллическая структура неупорядоченной фазы Bi3-xAxFe1Mo2O12 обычно образуется более легко.
[0079] В случае конечного прокаливания при такой температуре, конечное время прокаливания обычно составляет от 0,1 до 72 часов, предпочтительно от 2 до 48 часов, более предпочтительно от 3 до 24 часов, с точки зрения того, что надлежащим образом задают произведение температуры прокаливания и времени прокаливания и способствуют образованию кристаллов.
[0080] Для формирования кристаллической структуры время конечного прокаливания предпочтительно составляет, например, от 24 до 72 часов при температуре конечного прокаливания всего лишь по меньшей мере 400°C с точки зрения того, что надлежащим образом задают температуру прокаливания х время прокаливания, и предпочтительно составляет 3 часа или короче при температуре конечного прокаливания по меньшей мере целых 600°С с точки зрения предотвращения образования упорядоченной фазы.
[0081] Кристаллическую структуру неупорядоченной фазы Bi3-xAxFe1Mo2O12 можно легко формировать посредством осуществления всех этих стадий.
[0082] Успешное образование кристаллической структуры неупорядоченной фазы Bi3-xAxFe1Mo2O12 на стадии конечного прокаливания можно подтвердить посредством рентгеноструктурного анализа оксидного катализатора, получаемого посредством конечного прокаливания. При условии, что оксидный катализатор согласно первому варианту осуществления имеет отдельные пики при дифракционных углах (2θ) по меньшей мере в каждом диапазоне плоскостей 18,30°±0,2° (101), 28,20°±0,2° (112), 33,65°±0,2° (200) и 46,15°±0,2° (204) при рентгеновском дифракционном анализе и что соотношение интенсивностей (Ia/Ib) для интенсивности пика (Ia) при 2θ=33,65°±0,2° к интенсивности пика (Ib) при 2θ=34,10°±0,2° составляет 2,0 или более, можно определять, что сформирована кристаллическая структура неупорядоченной фазы Bi3-xAxFe1Mo2O12.
[0083] [3] Способ получения ненасыщенного альдегида
Ненасыщенный альдегид можно получать через реакцию окисления по меньшей мере одного олефина, выбранного из группы, состоящей из пропилена и изобутилена, и/или по меньшей мере одного спирта, выбранного из изобутанола и т-бутилового спирта, с использованием оксидного катализатора согласно первому варианту осуществления. Далее в настоящем документе, описаны конкретные примеры способа, хотя способ получения ненасыщенного альдегида не ограничен конкретными примерами, приведенными ниже.
[0084] (1) Способ получения метакролеина
Метакролеин можно получать, например, через реакцию каталитического окисления в газовой фазе изобутилена, изобутанола или т-бутилового спирта с использованием оксидного катализатора согласно первому варианту осуществления. Реакцию каталитического окисления в газовой фазе можно осуществлять посредством введения, в каталитический слой в реакторе с неподвижным слоем, подаваемого газа, который состоит из от 1 до 10% по объему изобутилена, изобутанола, т-бутилового спирта или их газовой смеси с добавлением содержащего молекулярный кислород газа и разбавляющего газа при концентрации молекулярного кислорода от 1 до 20% по объему, в присутствии оксидного катализатора. Эту реакцию можно осуществлять при температуре от 250 до 480°С, давлении от обычного давления до 5 атм, и объемной скорости от 400 до 4000/ч [при стандартных условиях (NTP)]. Молярное соотношение кислорода к изобутилену, изобутанолу, т-бутиловому спирту или их газовой смеси (кислород/изобутилен, изобутанол, т-бутиловый спирт или их газовая смесь) обычно составляет от 1,0 до 2,0, предпочтительно от 1,1 до 1,8, более предпочтительно от 1,2 до 1,8, с точки зрения управления концентрацией кислорода на выпуске реактора для того, чтобы повышать выход ненасыщенного альдегида.
[0085] Примеры содержащего молекулярный кислород газа включают, но не ограничиваясь этим, кислородсодержащие газы, такие как чистый газообразный кислород, N2O и воздух. Среди них, воздух предпочтителен с промышленной точки зрения.
[0086] Примеры разбавляющего газа включают, но не ограничиваясь этим, азот, диоксид углерода, водяной пар и их газовые смеси. Соотношение смешения между содержащим молекулярный кислород газом и разбавляющим газом предпочтительно составляет 0,01 < молекулярный кислород/(содержащий молекулярный кислород газ + разбавляющий газ) <0,3 по объему. Содержание молекулярного кислорода предпочтительно составляет от 1 до 20% по объему относительно 100% по объему подаваемого газа.
[0087] Водяной пар в подаваемом газе эффективен для предотвращения спекания катализатора. В отличие от этого, концентрацию водяного пара в разбавляющем газе предпочтительно снижают насколько возможно для того, чтобы ингибировать образование побочных продуктов карбоновых кислот, таких как метакриловая кислота или уксусная кислота. Содержание водяного пара обычно составляет от 0 до 30% по объему относительно 100% по объему подаваемого газа.
[0088] (2) Способ получения акролеина
Акролеин можно получать через каталитическое окисление пропилена в газовой фазе, например, при любых условиях без ограничений, и можно получать с помощью способа, обычно используемого для получения акролеина через каталитическое окисление пропилена в газовой фазе. Например, газовую смесь, которая содержит от 1 до 15% по объему пропилена, от 3 до 30% по объему молекулярного кислорода, от 0 до 60% по объему водяного пара и от 20 до 80% по объему инертного газа, такого как азот и газообразный СО2, можно вводить при от 250 до 450°С и давлении от 0,1 до 1 мПа с объемной скоростью (SV) от 300 до 5000 ч-1 в каталитический слой в реакторе. Используемый реактор может представлять собой обычный реактор с неподвижным слоем, реактор с псевдоожиженным слоем или реактор с подвижным слоем.
[0089] (3) Способ получения диолефина
Способ получения диолефина согласно первому варианту осуществления включает стадию получения диолефина для окисления моноолефина, который содержит 4 или больше атома углерода, с использованием оксидного катализатора согласно первому варианту осуществления для того, чтобы получать диолефин. Более конкретно, стадия получения диолефина представляет собой стадию получения диолефина через реакцию каталитического окисления моноолефина в газовой фазе, который имеет 4 или больше атома углерода, с использованием источника кислорода в присутствии оксидного катализатора согласно первому варианту осуществления.
[0090] Источник кислорода, используемый в реакции каталитического окисления в газовой фазе, может представлять собой, но не ограничиваясь этим, например, газовую смесь содержащего молекулярный кислород газа и разбавляющего газа.
[0091] Примеры моноолефина, который имеет 4 или больше атома углерода, включают, но не ограничиваясь этим, н-бутен.
[0092] Получаемый диолефин отличается в зависимости от используемого моноолефина, который имеет 4 или больше атома углерода. Например, бутадиен получают с использованием н-бутена.
[0093] Примеры содержащего молекулярный кислород газа включают, но не ограничиваясь этим, кислородсодержащие газы, такие как чистый газообразный кислород и воздух. Среди них, воздух предпочтительно используют в качестве содержащего молекулярный кислород газа. Использование воздуха обычно бывает превосходным с промышленной точки зрения, такой как стоимость.
[0094] Примеры разбавляющего газа включают, но не ограничиваясь этим, азот, диоксид углерода, водяной пар и их газовые смеси.
[0095] В подаваемый газ, который подлежит подаче на стадии получения диолефина, предпочтительно добавляют содержащий молекулярный кислород газ и разбавляющий газ при концентрации моноолефина, который имеет 4 или больше атома углерода, от 1 до 10% по объему и концентрации молекулярного кислорода от 1 до 20% по объему.
[0096] Примеры реактора включают, но не ограничиваясь этим, реакторы с неподвижным слоем. Реакцию можно осуществлять при температуре предпочтительно от 250 до 450°С, давлении предпочтительно от обычного давления до 5 атм, и объемной скорости предпочтительно от 400 до 4000/ч [при стандартных условиях (NTP)].
[0097] [Второй вариант осуществления]
Описан оксидный катализатор согласно второму варианту осуществления (далее в настоящем документе также обозначаемому как «катализатор для аммоксидирования»).
Оксидный катализатор согласно второму варианту осуществления представляет собой оксидный катализатор для использования в получении ненасыщенного нитрила из олефина и/или спирта, оксидный катализатор отвечает следующим с (1) до (3):
(1) оксидный катализатор содержит молибден, висмут, железо, кобальт и элемент А, который имеет радиус иона больше чем 0,96

(2) атомное соотношение а висмута к 12 атомам молибдена составляет 1≤а≤5, атомное соотношение b железа к 12 атомам молибдена составляет 1,5≤b≤6, атомное соотношение с элемента А к 12 атомам молибдена составляет 1≤с≤5 и атомное соотношение d кобальта к 12 атомам молибдена составляет 1 (3) оксидный катализатор содержит неупорядоченную фазу, которая состоит из кристаллической системы, которая содержит молибден, висмут, железо и элемент А. [0098] Исходный материал Примеры олефина, выполняющего функцию исходного материала для использования в получении ненасыщенного нитрила, включают, но не ограничиваясь этим, пропилен, н-бутен, изобутилен, н-пентен, н-гексен и циклогексен. Среди них пропилен и изобутилен являются предпочтительными. [0099] Примеры спирта, выполняющего функцию исходного материала для использования в получении ненасыщенного нитрила, включают, но не ограничиваясь этим, пропанол, бутанол, изобутанол и т-бутиловый спирт. Среди них, изобутанол и т-бутиловый спирт являются предпочтительными. [0100] Во втором варианте осуществления, например, акрилонитрил можно получать с использованием пропилена или пропанола в качестве исходного материала, тогда как метакрилонитрил можно получать с использованием изобутилена, изобутанола или т-бутилового спирта в качестве исходного материала. [0101] Эти олефины и/или спирты можно использовать отдельно или в комбинации. [0102] Примеры ненасыщенного нитрила включают, но не ограничиваясь этим, акрилонитрил и метакрилонитрил. [0103] (1) Композиция Оксидный катализатор согласно второму варианту осуществления содержит молибден, висмут, железо, кобальт и элемент А, который имеет радиус иона больше чем 0,96

[0104] Атомное соотношение а для Bi к 12 атомам Мо составляет 1≤а≤5, предпочтительно 1≤а≤4, более предпочтительно 2≤а≤4. При условии, что атомное соотношение а для Bi попадает в диапазон, описанный выше, избирательность к ненасыщенному нитрилу обычно бывает дополнительно повышена.
[0105] С точки зрения повышения каталитической активности без снижения избирательности к ненасыщенному нитрилу, Fe является обязательным элементом, как и в случае Мо и Bi, для промышленного синтеза ненасыщенного нитрила. Однако при высоком содержании Fe образуется Fe2O3 и обычно увеличиваются побочные продукты, такие как СО или СО2, что ведет к сниженной избирательности к ненасыщенному нитрилу. Альтернативно, при высоком содержании Fe может не происходить образование Fe2O3, но вместо этого образуется 2-компонентный композитный оксид Fe2Mo3O12, который представляет собой неактивный компонент, который не проявляет каталитическую активность. С этих точек зрения, атомное соотношение b для Fe к 12 атомам Мо в катализаторе для аммоксидирования согласно второму варианту осуществления предпочтительно составляет 1,5≤b≤6, более предпочтительно 2,0≤b≤5, более предпочтительно 3≤b≤5.
[0106] Bi и Мо обычно образуют композитные оксиды, такие как Bi2Mo3O12 и Bi2MoO6, которые, по имеющимся сообщениям, представляют собой активные частицы в реакции аммоксидирования. Катализатор, который состоит из такого композитного оксида, обычно дает высокую избирательность к ненасыщенному нитрилу, но обладает низкой активностью. С другой стороны, Fe и Мо образуют композитный оксид, такой как Fe2Mo3O12. Катализатор, который состоит из такого композитного оксида, демонстрирует как низкую активность, так и низкую избирательность. При введении Мо, Bi и Fe в композицию образуется Bi3Fe1Mo2O12 с упорядоченной фазой. Показано, что в промышленной реакции аммоксидирования с использованием в течение длительного времени катализатора, который имеет эту структуру, происходит снижение активности катализатора из-за восстановления во время реакции, что ведет к сниженному выходу ненасыщенного нитрила, несмотря на высокий выход в начале. Авторы настоящего изобретения сделали следующее предположение относительно причины этого восстановительного разложения: железо, содержащееся в Bi3Fe1Mo2O12, находится в трехвалентном состоянии сразу после начала реакции, но восстанавливается до двухвалентного железа через окислительно-восстановительные циклы во время реакции для формирования композитного оксида двухвалентного железа и молибдена (FeMoO4). Также Мо или Bi образует MoO2, Bi2O3 или металлический Bi. Образование этих стабильных соединений стабилизирует железо, так что во время реакции не протекают окислительно-восстановительные циклы, что предположительно вызывает восстановительное разложение.
[0107] Авторы настоящего изобретения проводили тщательные исследования оксидного катализатора с превосходной устойчивостью к восстановлению, который сохраняет описанную выше структуру, даже в сильно восстанавливающей атмосфере, и в результате обнаружили, что: неупорядоченную фазу Bi3-xAxFe1Mo2O12 формируют посредством дополнительного встраивания элемента А, который имеет радиус иона больше чем 0,96

[0108] Элемент А может без ограничений представлять собой какой-либо элемент, который имеет радиус иона больше чем 0,96

[0109] Атомное соотношение с элемента А к 12 атомам молибдена составляет 1≤с≤5, предпочтительно 1≤с≤4, более предпочтительно 1,5≤с≤3. При условии, что атомное соотношение с попадает в описанный выше диапазон, 4-компонентный композитный оксид, который содержит композитную неупорядоченную фазу Bi3-xAxFe1Mo2O12, легко образуется. Например, в случае использования La в качестве элемента А, образуется 4-компонентный композитный оксид, содержащий неупорядоченную фазу Bi3-xLaxFe1Mo2O12. La можно надлежащим образом вводить в композицию в 3-компонентный композитный оксид, который содержит Bi3Fe1Mo2O12 с неупорядоченной фазой, с тем, чтобы Bi3+ (радиус иона: 0,96

В этом контексте, радиус иона описан, например, в «Chemistry of Ceramics», ed., Hiroaki Yanagida, стр. 14-17, Maruzene Co., Ltd.
[0110] (2) Кристаллическая структура
Катализатор для аммоксидирования согласно второму варианту осуществления предпочтительно содержит 4-компонентный композитный оксид, который содержит неупорядоченную фазу Bi3-xAxFe1Mo2O12. Рентгеновскую дифракцию (XRD) можно использовать в качестве показателя для образования 4-компонентного композитного оксида, который содержит неупорядоченную фазу Bi3-xAxFe1Mo2O12. При условии, что 4-компонентный композитный оксид, который содержит неупорядоченную фазу Bi3-xAxFe1Mo2O12, образован, можно подтвердить, что катализатор для аммоксидирования согласно второму варианту осуществления имеет отдельные пики по меньшей мере в плоскостях 18,30°±0,2° (101), 28,20°±0,2° (112), 33,65°±0,2° (200) и 46,15°±0,2° (204), измеряемые в диапазоне дифракционных углов 2θ = от 10° до 60° при рентгеновской дифракции на кристаллах, как в 3-компонентном композитном оксиде, который содержит Bi3Fe1Mo2O12 с неупорядоченной фазой. Особенно предпочтительно, катализатор для аммоксидирования согласно второму варианту осуществления имеет отдельный пик в положении в пределах каждого эталонного значения +0,05°.
[0111] Соотношение интенсивностей (Ia/Ib) для интенсивности (Ia) пика а при 2θ=33,65°±0,2° к интенсивности (Ib) пика b при 2θ=34,10°±0,2° предпочтительно составляет 2,0 или более, более предпочтительно 2,5 или более, более предпочтительно 3,0 или более, с точки зрения предотвращения восстановительного разложения катализатора. Соотношение интенсивностей (Ia/Ib) составляет 1,1 при 100% упорядоченной фазе и составляет 3,3 при 100% неупорядоченной фазе. Интенсивность (Ib) пика b при 2θ=34,10°±0,2° также содержит интенсивность пика Ib, получаемого из Fe2Mo3O12, присутствующего в следовом количестве. При соотношении интенсивностей (Ia/Ib) 2,0 или более, оксидный катализатор содержит предварительно определяемую долю неупорядоченной фазы и, следовательно, менее восприимчив к восстановительному разложению при промышленной эксплуатации в течение длительного времени. Таким образом, выход ненасыщенного нитрила обычно бывает дополнительно увеличен. На фиг. 4 представлена зависимость содержания неупорядоченной фазы и пиков а и b.
[0112] Механизм образования неупорядоченной фазы Bi3-xAxFe1Mo2O12 не определен. Композитный оксид Bi и Мо формируют в качестве промежуточного продукта на стадии тепловой обработки.
Дополнительная тепловая обработка, вероятно, служит причиной тепловой диффузии и замещающего твердого раствора Fe и элемента А в композитный оксид Bi и Мо для того, чтобы формировать композитную неупорядоченную фазу Bi3-xAxFe1Mo2O12.
[0113] «Отдельный пик» и «основной пик» определены в первом варианте осуществления.
[0114] Катализатор для аммоксидирования согласно второму варианту осуществления предпочтительно содержит оксид металла, который имеет композицию, представленную с помощью формулы композиции (2), приведенной ниже. Катализатор для аммоксидирования, содержащий оксид металла, имеющий композицию, представленную с помощью формулы композиции (2), приведенной ниже, обычно повышает избирательность к ненасыщенному нитрилу.
Mo12BiaFebAcCodBeCfOg (2)
где Мо представляет молибден; Bi представляет висмут; Fe представляет железо; элемент А представляет элемент, который имеет радиус иона больше чем 0,96

[0115] В катализаторе для аммоксидирования согласно второму варианту осуществления, Со является обязательным элементом, как и в случае Мо, Bi и Fe, с точки зрения промышленного синтеза ненасыщенного нитрила. Со играет роль в качестве носителя для формирования композитного оксида CoMoO4 и высокодисперсных активных частиц, таких как Bi-Mo-O, а также играет роль в захвате кислорода из газовой фазы и переносе этого кислорода на Bi-Mo-O, или тому подобное. Со и Мо предпочтительно вводят в композицию для того, чтобы формировать композитный оксид CoMoO4, с точки зрения получения ненасыщенного нитрила с высоким выходом. Атомное соотношение d для Со предпочтительно составляет 1≤d≤8, более предпочтительно 2≤d≤8, более предпочтительно 2≤d≤6, еще более предпочтительно 2≤d≤4, с точки зрения снижения образования простых оксидов, таких как Co3O4 или СоО.
[0116] В формуле композиции (2), элемент В представляет по меньшей мере один элемент, выбранный из группы, состоящей из магния, цинка, меди, никеля, хрома, марганца и олова и предположительно замещает некоторые атомы кобальта в оксидном катализаторе. Атомное соотношение е элемента В предпочтительно составляет 0≤е<3, более предпочтительно 0≤е≤2, с точки зрения сохранения баланса с образованием кристаллов неупорядоченной фазы. Элемент В не является обязательным, но обычно вносит вклад в стабилизацию кристаллической структуры CoMoO4 в катализаторе.
[0117] Элемент А представляет элемент, который имеет радиус иона больше чем 0,96

[0118] Элемент С представляет по меньшей мере один элемент, выбранный из группы, состоящей из калия, цезия и рубидия. Элемент С вероятно играет роль в нейтрализации кислотного центра некомпозитного MoO3 или тому подобное в катализаторе для аммоксидирования. Присутствие или отсутствие калия, цезия и/или рубидия, содержащегося в нем, не оказывает непосредственного влияния на кристаллическую структуру неупорядоченной фазы, описанную далее. Атомное соотношение f элемента С к 12 атомам Мо предпочтительно составляет 0≤f≤2, более предпочтительно 0,01≤f≤2, более предпочтительно 0,01≤f≤1, с точки зрения каталитической активности. При атомном соотношении f, равном 0 или более, нейтрализующий эффект обычно бывает дополнительно улучшен. При атомном соотношении f, равном 2 или меньше, оксидный катализатор обычно бывает основным или нейтральным. Получаемый оксидный катализатор обычно легко абсорбирует исходный материал пропилен, изобутилен, изобутанол или т-бутиловый спирт, а также обычно проявляет дополнительно повышенную каталитическую активность.
[0119] Элемент В и элемент С образуют кристаллические структуры отдельно от кристаллической структуры неупорядоченной фазы, описанной далее, и, следовательно, не оказывают непосредственного влияния на кристаллическую структуру неупорядоченной фазы.
[0120] Катализатор для аммоксидирования может содержать произвольные компоненты в качестве других металлических компонентов без ингибирования образования неупорядоченной фазы Bi3-xAxFe1Mo2O12.
[0121] (3) Компонент, отличный от оксида металла
Катализатор для аммоксидирования согласно второму варианту осуществления дополнительно может содержать носитель для того, чтобы поддерживать оксид металла. Катализатор, дополнительно содержащий носитель, предпочтителен по причине высокого диспергирования оксида металла и придания высокой устойчивости к износу поддерживаемому оксиду металла.
[0122] Примеры носителя включают, но не ограничиваясь этим, по меньшей мере одно, выбранное из группы, состоящей из диоксида кремния, оксида алюминия, оксида титана и оксида циркония. Оксидный катализатор, поддерживаемый таким носителем, обычно имеет дополнительно улучшенные физические свойства, подходящие для реакции в псевдоожиженном слое, такие как геометрическая форма, размер, распределение, текучесть и механическая прочность частицы. Среди них, в качестве носителя предпочтителен диоксид кремния. В целом, носитель из диоксида кремния предпочтителен в отношении его свойства придавать оксидному катализатору физические свойства, подходящие для реакции в псевдоожиженном слое, а также потому что он сам не активен по сравнению с другими носителями и оказывает благоприятный связывающий эффект на оксид металла без снижения избирательности к ненасыщенному нитрилу. Кроме того, носитель из диоксида кремния также предпочтителен по той причине, что он легко придает высокую устойчивость к износу оксиду металла, который он поддерживает.
[0123] Золь диоксида кремния предпочтителен в качестве источника диоксида кремния. Концентрация золя диоксида кремния в состоянии исходного материала, не смешанного с другими компонентами, предпочтительно составляет от 10 до 50% по массе с точки зрения диспергируемости частиц диоксида кремния. Золь диоксида кремния предпочтительно содержит от 40 до 100% по массе по меньшей мере одного золя диоксида кремния (а), содержащего основные частицы диоксида кремния, имеющие средний диаметр частицы от 20 нм до меньше чем 55 нм, предпочтительно от 20 до 50 нм, и от 60 до 0% по массе по меньшей мере одного золя диоксида кремния (b), содержащего основные частицы диоксида кремния, имеющие средний диаметр частицы от 5 нм до меньше чем 20 нм, с точки зрения избирательности к ненасыщенному нитрилу.
[0124] Содержание носителя предпочтительно составляет от 20 до 80% по массе, более предпочтительно от 30 до 70% по массе, более предпочтительно от 40 до 60% по массе, относительно всего 100% по массе носителя и оксидного катализатора. Задавая содержание носителя, которое попадает в диапазон, описанный выше, выход ненасыщенного нитрила обычно бывает дополнительно повышен.
[0125] [2] Способ получения катализатора для аммоксидирования
Способ получения катализатора для аммоксидирования согласно второму варианту осуществления включает:
стадию смешивания для смешивания исходных материалов, которые образуют катализатор, который содержит молибден, висмут, железо, кобальт и элемент А, который имеет радиус иона больше чем 0,96

стадию сушки для сушки суспензии, таким образом полученной, для того, чтобы получать высушенный продукт/ и
стадию прокаливания высушенного продукта, таким образом полученного, где
стадия прокаливания включает стадию нагревания для постепенного нагревания высушенного продукта от 100°С до 200°С в течение 1 часа или дольше. Подробности способа получения катализатор для аммоксидирования согласно второму варианту осуществления представляют собой то, что описано в первом варианте осуществления.
[0126] [3] Способ получения ненасыщенного нитрила
Способ получения ненасыщенного нитрила согласно второму варианту осуществления включает стадию получения ненасыщенного нитрила реакцией олефина и/или спирта с молекулярным кислородом и аммиаком в реакторе с псевдоожиженным слоем с использованием оксидного катализатора согласно второму варианту осуществления для того, чтобы получать ненасыщенный нитрил.
[0127] Примеры олефина и/или спирта включают, но не ограничиваясь этим, одно или несколько, выбранных из группы, состоящей из пропилена, изобутилена, пропанола, изопропанола, изобутанола и т-бутилового спирта.
[0128] Одно или несколько, выбранных из группы, состоящей из пропилена, изобутилена, изобутанола и т-бутилового спирта, могут вступать в реакцию с молекулярным кислородом и аммиаком с использованием катализатора для аммоксидирования согласно второму варианту осуществления для того, чтобы получать акрилонитрил или метакрилонитрил. Реакцию предпочтительно осуществляют с использованием реактора с псевдоожиженным слоем. Исходные материалы пропилен, изобутилен, изобутанол, т-бутиловый спирт и аммиак не обязательно должны иметь высокую степень очистки, и можно использовать исходные материалы промышленных сортов.
[0129] Обычно воздух предпочтительно используют в качестве источника молекулярного кислорода. Также можно использовать газ, который имеет повышенную концентрацию кислорода, например, посредством смешивания кислорода с воздухом. В отношении композиции подаваемого газа, молярное соотношение пропилена, изобутилена, изобутанола и/или т-бутилового спирта с аммиаком и молекулярным кислородом [(пропилен, изобутилен, изобутанол и/или т-бутиловый спирт)/аммиак/молекулярный кислород] предпочтительно составляет 1/от 0,8 до 1,4/от 1,4 до 2,4, более предпочтительно 1/от 0,9 до 1,3/от 1,6 до 2,2.
[0130] Температура реакции предпочтительно составляет от 350 до 550°С, более предпочтительно от 400 до 500°С.
[0131] Давление в реакции предпочтительно составляет от обычного давления до 0,3 мПа.
[0132] Длительность контакта между подаваемым газом и катализатором предпочтительно составляет от 0,5 до 20 с×г/см3, более предпочтительно от 1 до 10 с×г/см3.
[0133] [Третий вариант осуществления]
Способ получения ненасыщенного альдегида
Способ получения ненасыщенного альдегида согласно третьему варианту осуществления включает
стадию получения ненасыщенного альдегида для окисления олефина и/или спирта с использованием оксидного катализатора согласно первому варианту осуществления для того, чтобы получать ненасыщенный альдегид.
[0134] Стадия получения ненасыщенного альдегида предпочтительно включает стадию выгрузки для выгрузки газообразного продукта, который содержит ненасыщенный альдегид, из реактора с псевдоожиженным слоем, через реакцию каталитического окисления в газовой фазе олефина и/или спирта с использованием источника кислорода в реакторе с псевдоожиженным слоем.
[0135] Олефин и/или спирт предпочтительно представляет собой по меньшей мере одно, выбранное из группы, состоящей из пропилена, изобутилена, пропанола, изопропанола, изобутанола и т-бутилового спирта.
[0136] Предпочтительно, реакцию каталитического окисления в газовой фазе осуществляют при температуре реакции от 400 до 500°С и газообразный продукт, выгружаемый из реактора с псевдоожиженным слоем, имеет концентрацию кислорода от 0,03 до 0,5% по объему.
Далее в настоящем документе более подробно описан третий вариант осуществления.
[0137] Олефин
Примеры исходного материала олефина включают, но не ограничиваясь этим, пропилен, н-бутен, изобутилен, н-пентен, н-гексен и циклогексен. Среди них, одно или несколько соединений, выбранных из группы, состоящей из пропилена и изобутилена, являются предпочтительными. Использование такого олефина обычно дополнительно повышает выход ненасыщенного альдегида.
[0138] Спирт
Примеры исходного материала спирта включают, но не ограничиваясь этим, пропанол, бутанол, изобутанол и т-бутиловый спирт. Среди них, одно или несколько соединений, выбранных из группы, состоящей из пропанола, изобутанола и т-бутилового спирта, являются предпочтительными. Использование такого спирта обычно дополнительно повышает выход ненасыщенного альдегида.
[0139] Нижний предел концентрации олефина и/или спирта, вводимого в реактор с псевдоожиженным слоем, предпочтительно составляет 5,0% по объему или выше, более предпочтительно 6,0% по объему или выше, более предпочтительно 7,0% по объему или выше от всего газа, вводимого в реактор с псевдоожиженным слоем. Задавая нижний предел концентрации, который попадает в диапазон, описанный выше, производительность ненасыщенного альдегида обычно бывает дополнительно повышена. Верхний предел концентрации предпочтительно составляет 10% по объему или менее, более предпочтительно 9,5% по объему или менее, более предпочтительно 9,0% по объему или менее. Задавая верхний предел концентрации, который попадает в диапазон, описанный выше, выход ненасыщенного альдегида обычно бывает дополнительно повышен.
[0140] Способ получения ненасыщенного альдегида согласно третьему варианту осуществления позволяет получать, например, акролеин, с использованием пропилена или пропанола в качестве исходного материала. Альтернативно, способ получения ненасыщенного альдегида согласно третьему варианту осуществления позволяет получать метакролеин с использованием изобутилена, изобутанола или т-бутилового спирта в качестве исходного материала. Олефин и спирт могут содержать воду, азот и алканы, такие как пропан, бутан и изобутан.
[0141] Источник кислорода
В способе получения ненасыщенного альдегида согласно третьему варианту осуществления ненасыщенный альдегид получают через реакцию каталитического окисления олефина и/или спирта в газовой фазе с использованием источника кислорода и оксидного катализатора. Эта реакция каталитического окисления в газовой фазе не ограничена источником кислорода и, например, можно использовать газовую смесь содержащего молекулярный кислород газа и разбавляющего газа.
[0142] Примеры содержащего молекулярный кислород газа включают, но не ограничиваясь этим, кислородсодержащие газы, такие как чистый газообразный кислород и воздух. Среди них, воздух предпочтительно используют в качестве содержащего молекулярный кислород газа. Использование воздуха обычно бывает превосходным с промышленной точки зрения, такой как стоимость.
[0143] Примеры разбавляющего газа включают, но не ограничиваясь этим, азот, диоксид углерода, водяной пар и их газовые смеси.
[0144] Соотношение смешения между содержащим молекулярный кислород газом и разбавляющим газом в газовой смеси предпочтительно отвечает условиям (по объему), представленным с помощью следующего неравенства:
0,01 < содержащий молекулярный кислород газ / (содержащий молекулярный кислород газ + разбавляющий газ) < 0,3.
[0145] При высокой температуре реакции, реакция каталитического окисления в газовой фазе протекает легко и обычно в ней имеет место недостаток кислорода. По этой причине, источник кислорода предпочтительно подают в оксидный катализатор в большем количестве, чем олефин и/или спирт. Для реакции каталитического окисления в газовой фазе с высокой концентрацией олефина и/или спирта, предпочтительно в оксидный катализатор подают достаточно кислорода, при этом достаточное количество источника кислорода также подают в него для того, чтобы предотвращать чрезмерное восстановление оксидного катализатора. Чрезмерная подача источника кислорода, однако, вызывает реакцию разложения и реакцию горения ненасыщенного альдегида и обычно скорее снижает выход. Таким образом, источник кислорода предпочтительно подают в подходящей пропорции. С этих точек зрения, источник кислорода в способе получения согласно третьему варианту осуществления подают в оксидный катализатор так, что молярное соотношение воздуха, подаваемого в каталитический слой, к олефину и/или спирту предпочтительно составляет от 7,0 до 10,5, более предпочтительно от 8,0 до 9,5, более предпочтительно от 8,0 до 9,0. Когда задают молярное соотношение воздуха, подаваемого в каталитический слой, к олефину и/или спирту, которое составляет 7,0 или более, концентрация олефина и/или спирта обычно бывает низкой, что ведет к дополнительному предотвращению восстановительного разложения оксидного катализатора. Когда задают молярное соотношение воздуха, подаваемого в каталитический слой, которое составляет 10,5 или меньше, концентрация кислорода, подаваемого в каталитический слой, обычно бывает низкой, что ведет к дополнительному предотвращению окислительного разложения оксидного катализатора.
[0146] Олефин, спирт и содержащий молекулярный кислород газ могут содержать водяной пар. Водяной пар, содержащийся в них, обычно дополнительно ингибирует спекание оксидного катализатора.
[0147] Разбавляющий газ может содержать водяной пар. Низкое содержание водяного пара в разбавляющем газе является более предпочтительным. Водяной пар, содержащийся в них, обычно дополнительно ингибирует образование побочных продуктов, таких как уксусная кислота.
[0148] Концентрация водяного пара, содержащегося во всем газе, подаваемом в реактор, предпочтительно составляет от 0,01 до 30% по объему.
[0149] Реактор с псевдоожиженным слоем
В способе получения ненасыщенного альдегида согласно третьему варианту осуществления предпочтительно используют реактор с псевдоожиженным слоем (далее в настоящем документе также просто обозначают как «реактор»). Реактор с псевдоожиженным слоем относится к аппарату, который имеет распределитель газа, интерполятор и циклон в качестве основных составляющих реактора, и имеет структуру, в которой оксидный катализатор контактирует с подаваемым газом, которому при этом позволяют течь. Более конкретно, можно использовать реакторы с псевдоожиженным слоем или тому подобное, описанные, например, в Handbook of Fluidized Bed (опубликовано Baifukan Co., Ltd., 1999). Среди них, реактор с псевдоожиженным слоем на основе системы с псевдоожиженным слоем и барботированием, в частности, подходит для способа. Образуемое тепло реакции можно удалять с использованием трубки охлаждения, установленной в реакторе с псевдоожиженным слоем.
[0150] Температура реакции в реакции каталитического окисления в газовой фазе
В способе получения ненасыщенного альдегида согласно третьему варианту осуществления, температура реакции в реакции каталитического окисления в газовой фазе предпочтительно составляет от 400 до 500°С, более предпочтительно от 420 до 470°С, более предпочтительно от 430 до 450°С. При температуре реакции 400°С или выше, степень превращения и скорость реакции обычно дополнительно повышены, что ведет к дополнительно повышенному выходу ненасыщенного альдегида. При температуре реакции 500°С или менее, разложению образованного ненасыщенного альдегида горением обычно дополнительно препятствуют. Температуру реакции в реакции каталитического окисления в газовой фазе можно измерять термометром, установленным в реакторе с псевдоожиженным слоем.
[0151] Поскольку реакция каталитического окисления в газовой фазе обычно представляет собой экзотермическую реакцию, в реакторе с псевдоожиженным слоем предпочтительно предусмотрен охлаждающий аппарат для удаления тепла для того, чтобы задавать подходящую температуру реакции. Температуру реакции можно корректировать в диапазоне, описанном выше, посредством удаления тепла реакции с использованием трубки охлаждения или посредством подачи тепла с использованием нагревательного аппарата.
[0152] Способы введения олефина и/или спирта и источника кислорода Олефин и/или спирт и источник кислорода можно вводить с помощью любого способа без ограничений. Например, газ, содержащий олефин и/или спирт и воздух, или газ, который имеет повышенную концентрацию кислорода, можно предварительно смешивать и вводить в реактор с псевдоожиженным слоем, заполненный оксидным катализатором. Альтернативно, каждый газ можно вводить в него индивидуально. Каждый газ, который подвергают реакции, можно вводить в реактор и затем нагревать до предварительно определяемой температуры реакции или можно предварительно нагревать и затем вводить в реактор. Среди этих подходов, предпочтительным для непрерывной и эффективной реакции является предварительный нагрева, за которым следует введение в реактор.
[0153] Концентрация кислорода в газообразном продукте, выгружаемом из реактора с псевдоожиженным слоем
Концентрация кислорода в газообразном продукте, выгружаемом из реактора с псевдоожиженным слоем, предпочтительно составляет от 0,03 до 0,5% по объему, более предпочтительно от 0,03 до 0,2% по объему, более предпочтительно от 0,05 до 0,1% по объему. При концентрации кислорода на выпуске реактора 0,5% по объему или менее, обычно дополнительно препятствуют чрезмерному восстановлению катализатора. При концентрации кислорода 0,03% по объему или выше препятствуют чрезмерному окислению катализатора. В любом случае, выход ненасыщенного альдегида обычно бывает дополнительно повышен, Концентрацию кислорода на выпуске реактора можно корректировать в описанном выше диапазоне для того, чтобы предотвращать разложение ненасыщенного альдегида горением, не нарушая баланс степени окисления-восстановления.
[0154] Газообразный продукт, содержащий ненасыщенный альдегид, представляющий интерес, выгружают из выпуска реактора. Далее в настоящем документе концентрацию кислорода в газообразном продукте, выгружаемом из реактора с псевдоожиженным слоем, также обозначают как «концентрация кислорода на выпуске из реактора». В этом контексте, «концентрация кислорода на выпуске из реактора» относится к концентрации кислорода в ненасыщенном содержащем альдегид газообразном продукте, выгружаемом из выпуска реактора. Концентрацию кислорода на выпуске из реактора можно измерять в участке, где содержание кислорода в газообразном продукте является постоянным рядом с выпуском реактора с псевдоожиженным слоем. Участок не должен находиться в центре, где выгружают газообразный продукт из реактора с псевдоожиженным слоем, или вблизи от него в строгом смысле. Таким образом, концентрацию кислорода на выпуске из реактора можно измерять в газе в любой момент времени от времени присутствия ниже по потоку от реактора или непосредственно перед выгрузкой из реактора до непосредственно перед операцией очистки. Например, газообразный продукт можно быстро охлаждать, затем абсорбировать в воде и очищать посредством экстракционной дистилляции. В таком случае, можно брать образцы газообразного продукта для измерения концентрации кислорода на выпуске из реактора в трубке между реактором и колонной быстрого охлаждения, расположенной ниже по потоку от реактора. Концентрацию кислорода на выпуске из реактора можно измерять посредством газовой хроматографии, оборудованной детектором теплопроводности (TCD).
[0155] Важно управлять концентрацией кислорода на выпуске из реактора в описанном выше диапазоне, поскольку концентрация кислорода на выпуске из реактора влияет на разложение ненасыщенного альдегида или вторичную реакцию в реакторе. Концентрацию кислорода на выпуске из реактора можно корректировать посредством изменения молярного соотношения источника кислорода, подаваемого в реактор с псевдоожиженным слоем, к олефину и/или спирту, количества кислородсодержащего газа, подаваемого в реактор с псевдоожиженным слоем, температуры реакции, внутреннего давления в реакторе, длительности контакта между газовой смесью исходных материалов и оксидным катализатором, количества катализатора и количества всего газа, подаваемого в реактор. Среди этих подходов предпочтительным для корректировки является управление количеством содержащего молекулярный кислород газа, например, воздуха, подаваемого в реактор с псевдоожиженным слоем.
[0156] Например, подаваемый газ можно подавать в условиях, включающих температуру реакции 440°С, давление в реакции 0,05 мПа и скорость потока 595 см2/мин (исходя из NTP) с использованием 40 г оксида, представленного с помощью Mo12Bi2,0Ce2,0Fe3,4Co3,0Cs0,16Ox, в качестве катализатора. В таком случае, молярное соотношение в композиции подаваемого газа можно менять от изобутилен/воздух/гелий = 1/9,2/остальное (концентрация изобутилена = 8% по объему) до изобутилен/воздух/гелий = 1/8,1/остальное (концентрация изобутилена = 8% по объему) для того, чтобы изменять концентрацию кислорода в газе на выпуске реактора с 0,4% по объему до 0,05% по объему. В этом контексте, «изобутилен/воздух/гелий = 1/8,1/остальное (концентрация изобутилена = 8% по объему)» обозначает, что количество гелия определяют так, что соотношение изобутилена/воздуха составляет 1/8,1 и концентрация изобутилена составляет 8% по объему.
[0157] Примеры условий для корректировки концентрации кислорода на выпуске из реактора включают степень превращения, а также количество катализатора, длительность контакта, давление в реакции и объемную скорость, как описано выше. Эти условия можно как единое целое корректировать для того, чтобы корректировать концентрацию кислорода на выпуске из реактора до произвольного значения. Например, реакцию можно осуществлять при температуре реакции, корректируемой в диапазоне от 430°С до 500°С, и концентрации олефина и/или спирта, корректируемой в диапазоне от 6 до 10% по объему. В таком случае, длительность контакта, определяемая с помощью следующего выражения для корректировки концентрации кислорода на выпуске из реактора, предпочтительно составляет 5,0 (г×с/см2) или короче, более предпочтительно 4,0 (г×с/см2) или короче, более предпочтительно 3,0 (г×с/см2) или короче:
Длительность контакта (г×с/см2)=W/F*60*273,15/(273,15+Т)*(Р*1000+101,325)/101,325
где W представляет количество (г) катализатора, загруженного в реактор с псевдоожиженным слоем; F представляет скорость потока (см2/мин, исходя из NTP) газовой смеси исходных материалов; Т представляет температуру реакции (°С); и Р представляет давление в реакции (мПа).
[0158] Для корректировки концентрации кислорода на выпуске из реактора в описанном выше диапазоне, давление в реакции предпочтительно составляет от обычного давления до 5 атм. Объемная скорость предпочтительно составляет от 400 до 4000/ч [при стандартных условиях (NTP)].
[0159] Оксидный катализатор
Оксидный катализатор согласно первому варианту осуществления используют в качестве оксидного катализатора в третьем варианте осуществления. Среди оксидных катализаторов, примеры которых приведены выше, предпочтительно используют оксидный катализатор, который содержит молибден, висмут, железо, кобальт и лантаноидный элемент, где атомное соотношение железа к кобальту (Fe/Co) составляет Fe/Co≥1, и который дополнительно содержит носитель.
[0160] Присутствие или отсутствие каждого элемента в оксидном катализаторе и атомное соотношение каждого элемента можно идентифицировать посредством рентгеновского флуоресцентного анализа (XRF).
[0161] Мо, Bi, Fe и Со являются обязательными компонентами для формирования оксидного катализатора. Олефин и/или спирт подвергается реакции окисления кислородом кристаллической решетки в этом оксидном катализаторе для того, чтобы давать ненасыщенный альдегид. Когда кислород кристаллической решетки в оксидном катализаторе расходуется в реакции окисления, обычно возникает пустое место кислорода в оксидном катализаторе. Как результат, восстановление оксидного катализатора протекает по мере протекания реакции окисления. Восстановленный оксидный катализатор является инактивированным. Следовательно, необходимо незамедлительно повторно окислять такой восстановленный оксидный катализатор. Оксид, содержащий Мо, Bi, Fe и Со, обладает способностью реагировать с олефином и/или спиртом и с источником кислорода и также, по-видимому, обладает превосходным эффектом повторного окисления для встраивания молекулярного кислорода в газовой фазе в оксид посредством диссоциативной адсорбции и регенерации израсходованного кислорода кристаллической решетки. Таким образом, вероятно, что этот эффект повторного окисления можно сохранять даже во время реакции окисления в течение длительного времени с тем, чтобы стабильно получать ненасыщенный альдегид из олефина и/или спирта без инактивации оксидного катализатора.
[0162] В Mo-Bi оксиде металла, содержащем Мо, Bi, Fe и Со, эти металлические элементы можно вводить в композицию. В оксидном катализаторе по третьему варианту осуществления, атомное соотношение а для Bi к 12 атомам Мо составляет 1≤а≤5, предпочтительно 1,5≤а≤4, более предпочтительно 1,8≤а≤4. При условии, что атомное соотношение а попадает в диапазон, описанный выше, избирательность к продукту, представляющему интерес, дополнительно повышают.Bi и Мо предпочтительно образуют композитные оксиды Bi-Mo-O, такие как Bi2Mo3O12 и Bi2MoO6, которые, по имеющимся сообщениям, представляют собой активные частицы при каталитическом окислении в газовой фазе, реакции аммоксидирования и т.п.
[0163] Fe является обязательным элементом, как и в случае Мо и Bi, для промышленного синтеза ненасыщенного альдегида. Атомное соотношение b для Fe к 12 атомам Мо в оксидном катализаторе составляет 1,5≤b≤6, предпочтительно 2≤b≤5,5, более предпочтительно 3≤b≤5. При условии, что атомное соотношение b попадает в диапазон, описанный выше, твердый раствор железа в CoMoO4 может возникать за счет использования окисления-восстановления между трехвалентным железом и двухвалентным железом для того, чтобы формировать катализатор, который имеет устойчивость к восстановлению, даже в реакторе, содержащем малое количество кислорода.
[0164] Со представляет собой обязательный элемент, как и в случае элементов, описанных выше, для промышленного синтеза ненасыщенного альдегида. Со образует композитный оксид (например, CoMoO4) с Мо и вероятно играет роль в захвате кислорода из газовой фазы и переносе этого кислорода на Bi-Mo-O или тому подобное. С такой точки зрения, атомное соотношение d для Со предпочтительно составляет 1≤d≤8, более предпочтительно 2≤d≤7, более предпочтительно 2,5≤d≤6.
[0165] В случае твердого раствора железа в CoMoO4, улучшена устойчивость к восстановлению оксидного катализатора. В этом отношении, атомное соотношение Fe/Co железа к кобальту в оксидном катализаторе для использования в третьем варианте осуществления предпочтительно составляет Fe/Co≥0,8, более предпочтительно Fe/Co≥1, более предпочтительно Fe/Co≥1,05, еще более предпочтительно Fe/Co≥1,1. Верхний предел Fe/Co предпочтительно составляет 3≥Fe/Co, более предпочтительно 2≥Fe/Co, более предпочтительно 1,5≥Fe/Co. Обычно происходит окисление железа до трехвалентной формы. Трехвалентное железо менее подвержено образованию твердого раствора в CoMoO4. Оксидный катализатор, который имеет высокую устойчивость к восстановлению, сложно получать, даже если железо просто увеличено. Тем не менее, показано, что реакция каталитического окисления олефина и/или спирта в газовой фазе при температуре 400°С или выше и 500°С или менее с использованием оксидного катализатора, который имеет композицию Fe/Co≥0,8, снижает трехвалентное железо и, тем самым, повышает устойчивость к восстановлению оксидного катализатора посредством твердого раствора получаемого железа в CoMoO4. Успешный твердый раствор железа в CoMoO4 можно определять на основе сдвига пика в XRD для CoMoO4. Более конкретно, твердый раствор железа в CoMoO4 можно подтвердить с помощью способа, описанного в примерах.
[0166] Оксидный катализатор для использования в третьем варианте осуществления предпочтительно имеет композицию, представленную следующей формулой (1):

где Мо представляет молибден; Bi представляет висмут; Fe представляет железо; Со представляет кобальт;
А представляет по меньшей мере один лантаноидный элемент, выбранный из группы, состоящей из лантана, церия, празеодимия и неодимия;
В представляет по меньшей мере один элемент, выбранный из группы, состоящей из магния, цинка, меди, никеля, марганца, кальция, стронция, бария, олова и свинца;
С представляет по меньшей мере один элемент, выбранный из группы, состоящей из калия, цезия и рубидия;
каждый с а до g представляет атомное соотношение каждого элемента к 12 атомам молибдена и соответствует 1≤а≤5, 1,5≤b≤6, 1≤d≤6, Fe/Co≥1, 1≤с≤5, 0≤е≤3 и 0,01≤f≤2; и
g представляет атомность кислорода, которую определяют посредством валентностей составляющих элементов, отличных от кислорода.
[0167] В формуле (1) лантаноидный элемент А представляет по меньшей мере один элемент, выбранный из группы, состоящей из лантана, церия, празеодимия и неодимия. Как указано выше, Bi и Мо образуют композитный оксид Bi-Mo-O, который обладает высокой каталитической активностью, но низкой температурой плавления и низкой термостойкостью. С другой стороны, лантаноидный элемент и Мо редко образуют композитный оксид, такой как А-Мо-O, который однако имеет высокую температуру плавления и очень высокую термостойкость. При введении этих оксидов в подходящий состав образуется теплостойкий Bi-A-Mo-О через введение Bi, элемента А и Мо в состав. Образованный композитный оксид обладает как высокой активностью, так и высокой термостойкостью, и подходит в качестве катализатора для псевдоожиженного слоя.
[0168] В формуле (1) элемент В представляет по меньшей мере один элемент, выбранный из группы, состоящей из магния, цинка, меди, никеля, марганца, кальция, стронция, бария, олова и свинца. Элемент В предположительно замещает некоторые атомы кобальта в оксидном катализаторе. Элемент В не является обязательным, но вносит вклад в повышение активности катализатора или стабилизацию кристаллической структуры CoMoO4 в катализаторе. Например, медь оказывает эффект повышения активности катализатора. Никель, магний, цинк и марганец оказывают эффекты стабилизации кристаллической структуры CoMoO4 и ингибирования, например, фазового перехода, объясняемого давлением или температурой. Атомное соотношение е такого элемента В предпочтительно составляет 0≤е<3, более предпочтительно 0≤е<2, более предпочтительно 0≤е<1,5. Когда задают атомное соотношение е, которое попадает в диапазон, описанный выше, эффекты могут проявляться без разрушения структуры с твердым раствором железа в CoMoO4.
[0169] В формуле (1) С представляет по меньшей мере один элемент, выбранный из группы, состоящей из цезия, рубидия и калия. Элемент С вероятно играет роль в нейтрализации кислотного центра некомпозитного МоО3 или тому подобное в оксидном катализаторе. Атомное соотношение f элемента С к 12 атомам Мо предпочтительно составляет 0≤f≤2, более предпочтительно 0,01≤f≤2, более предпочтительно 0,05≤f≤0,5. Когда задают атомное соотношение f, которое попадает в диапазон, описанный выше, каталитическая активность обычно бывает дополнительно повышена. В частности, при атомном соотношении f, равном 2 или меньше, оксидный катализатор редко становится основным. Кроме того, оксидный катализатор легко адсорбирует исходный материал олефин и/или спирт в реакции окисления олефина и/или спирта и обычно дополнительно повышает каталитическую активность.
[0170] Носитель
Оксидный катализатор для использования в третьем варианте осуществления поддерживает носитель. Содержание носителя предпочтительно составляет от 20 до 80% по массе, более предпочтительно от 30 до 70% по массе, более предпочтительно от 40 до 60% по массе, относительно общей массы носителя и оксидного катализатора. Когда задают содержание носителя, которое попадает в диапазон, описанный выше, выход ненасыщенного альдегида обычно бывает дополнительно повышен. Поддерживаемый катализатор, который содержит оксид, содержащий Мо, Bi, Fe, Со и атомы лантаноида, можно получать известным в данной области способом, например, способом, который включает: стадию смешивания для смешивания исходных материалов для того, чтобы получать суспензию; стадию сушки для распылительной сушки суспензии; и стадию прокаливания высушенного продукта, получаемого на стадии сушки.
[0171] Носитель предпочтительно представляет собой, но не ограничиваясь этим, по меньшей мере одно, выбранное из группы, состоящей, например, из диоксида кремния, оксида алюминия, оксида титана и оксида циркония. Поддержка оксидного катализатора с помощью такого носителя обычно улучшает его физические свойства, подходящие для реакции в псевдоожиженном слое, такие как геометрическая форма, размер, распределение, текучесть и механическая прочность частицы. Среди них, в качестве носителя предпочтителен диоксид кремния. Носитель из диоксида кремния обладает свойством придавать оксидному катализатору физические свойства, подходящие для реакции в псевдоожиженном слое. Кроме того, носитель из диоксида кремния неактивен по сравнению с другими носителями и оказывает благоприятный связывающий эффект на катализатор без снижения каталитической активности в отношении продукта, представляющего интерес, или избирательности к нему.
[0172] Способ получения оксидного катализатора
Оксидный катализатор для использования в третьем варианте осуществления можно получать посредством какого-либо известного в данной области способа без ограничений. Оксидный катализатор для использования в третьем варианте осуществления можно получать, например, посредством способа получения, который включает: стадию смешивания для смешивания исходных материалов для того, чтобы получать суспензию; стадию сушки для распылительной сушки суспензии для того, чтобы получать высушенный продукт; и стадию прокаливания высушенного продукта. Далее в настоящем документе описан предпочтительный аспект способа получения оксидного катализатора, который включает эти стадии.
[0173] Стадия смешивания включает получение суспензии с использованием исходных материалов катализатора. Примеры исходных материалов катализатора включают молибден, висмут, железо, кобальт и лантаноидные элементы, такие как лантан, церий, празеодимий и неодимий. Другие примеры исходных материалов катализатора включают, но не ограничиваясь этим, марганец, никель, медь, цинк, свинец, щелочные металлические элементы, магний, кальций, стронций, барий и редкоземельные элементы, отличные от тех, которые описаны выше. Эти исходные материалы можно использовать в форме соли аммония, нитрата, гидрохлорида, сульфата и солей органических кислот, которые растворимы в воде или азотной кислоте. В частности, соль аммония предпочтительна в качестве источника элемента молибден. Источники элементов, отличные от молибдена, предпочтительно представляют собой нитраты, содержащие каждый элемент.
[0174] Как указано выше, диоксид кремния, оксид алюминия, оксид титана, оксид циркония или тому подобное можно использовать в качестве носителя для оксида. Предпочтительно в качестве носителя используют диоксид кремния. Золь диоксида кремния предпочтителен в качестве источника диоксида кремния.
[0175] Концентрация золя диоксида кремния в состоянии, не смешанном с другими компонентами в суспензии или тому подобное, предпочтительно составляет от 10 до 50% по массе, более предпочтительно от 15 до 45% по массе, более предпочтительно от 20 до 40% по массе. Когда задают концентрацию, которая попадает в диапазон, описанный выше, диспергируемость частиц диоксида кремния обычно бывает дополнительно повышена.
[0176] Золь диоксида кремния предпочтительно содержит от 40 до 100% по массе по меньшей мере одного золя диоксида кремния (а), который содержит основные частицы диоксида кремния, которые имеют средний диаметр частицы от 20 до 55 нм, предпочтительно от 20 до 50 нм, и от 60 до 0% по массе по меньшей мере одного золя диоксида кремния (b), который содержит основные частицы диоксида кремния, которые имеют средний диаметр частицы от 5 нм до 20 нм, с точки зрения избирательности к продукту, представляющему интерес.
[0177] Количество носителя из диоксида кремния для того, чтобы поддерживать получаемый оксидный катализатор, предпочтительно составляет от 20 до 80% по массе, более предпочтительно от 30 до 70% по массе, более предпочтительно от 40 до 60% по массе, относительно общей массы оксидного катализатора и носителя из диоксида кремния.
[0178] Суспензию можно получать посредством добавления соли аммония и молибдена, растворенной в воде, в золь диоксида кремния и последующего добавления раствора, содержащего нитрат каждого источника элемента, отличного от молибдена, растворенный в воде или водном растворе азотной кислоты. Порядок добавления этих материалов можно менять надлежащим образом.
[0179] Стадия сушки включает распылительную сушку суспензии, таким образом полученной на стадии смешивания, для того, чтобы получать высушенный продукт (сухие частицы). Суспензию можно распылять посредством какого-либо из обычных способов, таких как системы центрифугирования, системы сопел с двумя текучими веществами и системы сопел высокого давления, которые осуществляют в промышленности. Среди них, система центрифугирования является предпочтительной. Затем частицы, получаемые распылением, сушат. Воздух, нагретый паром, электрическим нагревателем или тому подобное, предпочтительно используют в качестве источника сухого тепла. Температура на впуске сушилки предпочтительно составляет от 100 до 400°С, более предпочтительно от 150 до 300°С.
[0180] Стадия прокаливания включает прокаливание сухих частиц, таким образом полученных на стадии сушки, для того, чтобы получать желаемый катализатор. Для прокаливания предпочтительно сухие частицы, в случае необходимости, предварительно прокаливают при от 150 до 400°С и затем наконец прокаливают при температуре в диапазоне от 400 до 700°С, предпочтительно от 500 до 700°С, в течение от 1 до 20 часов. Прокаливание можно осуществлять с использованием печи, такой как карусельная печь, туннельная печь или муфельная печь. Размеры частиц катализатора предпочтительно распределены в диапазоне от 10 до 150 мкм.
ПРИМЕРЫ
[0181] Пример А
Пример А предоставлен далее для того, чтобы дополнительно иллюстрировать первый аспект по настоящему изобретению, но не предназначен для того, чтобы ограничивать этим объем первого аспекта по настоящему изобретению. Поскольку атомное соотношение кислорода в оксидном катализаторе определяют с помощью состояния атомных валентностей других элементов, атомное соотношение кислорода пропущено в формуле, представляющей композицию катализатора в примерах и сравнительных примерах. Композиционное соотношение каждого элемента в оксидном катализаторе вычисляли по композиционному соотношению для получения.
[0182] Далее в примере А и сравнительном примере А водные дисперсии различных металлов использовали в качестве исходных материалов катализатора. Каждую из водных дисперсий оксида висмута, оксида железа и оксида кобальта для использования получили в CIK Nanotek Corporation, и каждую из водных дисперсий оксида лантана и оксида церия для использования получили в Taki Chemical Co., Ltd.
[0183] <Измерение среднего размера частицы>
Средний размер частицы получали посредством вычисления на основе следующего уравнения:
Средний размер частицы [нм] = 6000/(площадь поверхности [м2/г] × истинная плотность (8,99 г/см3).
[0184] <Измерение рН>
Измерение осуществили с использованием измерителя рН KR5E производства AS ONE.
[0185] <Измерение угла рентгеновской дифракции>
При XRD измерении, измеряли плоскость (111) и плоскость (200) соединения LaB6 в качестве стандартного эталонного материала 660 согласно National Institute of Standards & Technology так, что значения нормализовали по 37,441° и 43,506°, соответственно.
[0186] Использовали XRD аппарат Bruker D8 Advance. Использовали следующие условия XRD измерения: выход рентгеновского излучения 40 кВ-40 мА, щель расходимости (DS) 0,3°, ширина шага 0,02°/шаг, время счета 2,0 с и диапазон измерения 2θ = от 5° до 60°.
[0187] <Степень превращения, избирательность и выход>
В примере А и сравнительном примере А эффективность реакции представлена степенью превращения, избирательностью и выходом, которые определяют с помощью следующих выражений.
Степень превращения = (число молей исходного материала, вступившего в реакцию/число молей поданного исходного материала) × 100
Избирательность = (число молей полученного соединения/число молей соединения, вступившего в реакцию) × 100
Выход = (число молей полученного соединения/число молей поданного исходного материала) × 100
[0188] <Оценка восстанавливаемости>
Восстанавливаемость оценивали для ускоренной оценки устойчивости катализатора к восстановлению. Катализатор восстановили посредством восстанавливающей обработки в атмосфере газа, не содержащего кислород, и повторно окислили в возвращенных условиях оценки реакции. Процесс повторили для того, чтобы достичь ускоренной оценки устойчивость катализатора к восстановлению.
[0189] Газовую смесь 2% по объему олефина и/или спирта и 98% по объему гелия пропустили при скорости потока 3,0 см3/с (в пересчете на NTP) в течение 5 минут для восстанавливающей обработки, и затем вернули условия оценки реакции и сохраняли поток в течение 5 минут. Это составляло одно повторение, а реакцию оценивали после выполнения 100 повторений. После выполнения дополнительных 100 повторений, оценили реакцию и устойчивость к восстановлению. Оценку восстанавливаемости для реакции метакролеина осуществили при 430°С, оценку восстанавливаемости для реакции акролеина осуществили при 320°С и оценку восстанавливаемости для реакции диолефина осуществили при 360°С.
[0190] Когда сохраняется степень превращения и избирательность по сравнению с начальной способностью, определяют отсутствие восстановительного разложения. Когда происходит снижение степени превращения и избирательности, определяют присутствие восстановительного разложения.
[0191] Каждый из элементов А для использования в примере А и сравнительном примере А имеет следующий радиус иона:
La: 1,14
Се: 1,07

Са: 1,03

Pb: 1,24

V: 0,56

[0192] Пример А1
В перемешанную жидкость из 90,5 г ионообменной воды и 127,5 г 30% по массе водного раствора пероксида водорода добавили 54,5 г триоксида молибдена, который взбалтывали и перемешивали приблизительно при 70°С для растворения. Таким образом получили раствор (жидкость А). Кроме того, 204,75 г водной дисперсионной жидкости 10% по массе оксида висмута, который имеет средний размер частицы 51 нм, 54,3 г водной дисперсионной жидкости 15% по массе оксида кобальта, который имеет средний размер частицы 22 нм, 57,1 г водной дисперсионной жидкости 15% по массе оксида железа, который имеет средний размер частицы 39 нм, 71,9 г водной дисперсионной жидкости 10% по массе оксида лантана, который имеет средний размер частицы 40 нм, и 4,3 г жидкости 10% по массе гидроксида цезия смешали для того, чтобы получить раствор (жидкость В).
[0193] Жидкость А и жидкость В смешали для получения перемешанной жидкости, в которую водный раствор аммиака добавили для того, чтобы скорректировать рН до 3,2. Затем жидкость взбалтывали и перемешивали в течение приблизительно 3 часов для того, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 3 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 520°С в течение 6 часов для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0194] Для того чтобы оценить реакцию катализатора, SUS реакционную пробирку в рубашке, имеющую диаметр 14 мм, заполнили 4,0 г катализатора. Газовую смесь из 8% по объему изобутилена, 12,8% по объему кислорода, 3,0% по объему водяного пара и 76,2% по объему азота пропустили через пробирку при 430°С и скорости потока 120 мл/мин (NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Результаты оценки реакции представлены в таблице 3. Результаты оценки реакции после исполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 3.
[0195] Пример А2
В 202,6 г горячей воды при температуре приблизительно 90°С растворили 67,5 г гептамолибдата аммония (жидкость А). Кроме того, 37,0 г нитрата висмута, 22,0 г нитрата церия, 51,3 г нитрата железа, 0,55 г нитрата цезия и 37,2 г нитрата кобальта растворили в 41,9 г 18% по массе водного раствора азотной кислоты, в которую добавили 206,2 г горячей воды приблизительно при 90°С (жидкость В).
[0196] Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,1. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 540°С в течение 6 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0197] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 4,5 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0198] Пример A3
В 199,2 г горячей воды при температуре приблизительно 90°С, растворили 66,4 г гептамолибдата аммония (жидкость А). Кроме того, 47,0 г нитрата висмута, 13,5 г нитрата церия, 7,4 г нитрата кальция, 42,8 г нитрата железа, 1,56 г нитрата рубидия и 32,0 г нитрата кобальта растворили в 41,5 г 18% по массе водного раствора азотной кислоты, в которую добавили 209,0 г горячей воды приблизительно при 90°С (жидкость В). Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,0. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 3 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 3 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 530°С в течение 5 часов на воздухе для получения катализатора. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0199] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 4,8 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0200] Пример А4
В перемешанную жидкость из 90,6 г ионообменной воды и 127,6 г 30% по массе водного раствора пероксида водорода добавили 54,5 г триоксида молибдена, который взбалтывали и перемешивали приблизительно при 70°С для растворения. Таким образом получили раствор (жидкость А). Кроме того, 155,48 г водной дисперсионной жидкости 10% по массе оксида висмута, который имеет средний размер частицы 51 нм, 4,2 г нитрата свинца, 62,4 г водной дисперсионной жидкости 15% по массе оксида кобальта, который имеет средний размер частицы 22 нм, 50,4 г водной дисперсионной жидкости 15% по массе оксида железа, который имеет средний размер частицы 39 нм, 92,6 г водной дисперсионной жидкости 10% по массе оксида лантана, который имеет средний размер частицы 40 нм, и 4,3 г жидкости 10% по массе гидроксида цезия смешали для того, чтобы получить раствор (жидкость В).
[0201] Жидкость А и жидкость В смешали для того, чтобы получить перемешанную жидкость, в которую добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,6. Затем жидкость взбалтывали и перемешивали в течение приблизительно 3 часов для того, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора.
Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 3 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 520°С в течение 6 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0202] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 4,5 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0203] Пример А5
В 207,2 г горячей воды при температуре приблизительно 90°С растворили 69,1 г гептамолибдата аммония (жидкость А). Кроме того, 45,7 г нитрата висмута, 14,0 г нитрата церия, 2,3 г нитрата марганца, 48,5 г нитрата железа, 0,57 г нитрата цезия и 27,6 г нитрата кобальта растворили в 40,9 г 18% по массе водного раствора азотной кислоты, в которую добавили 195,4 г горячей воды приблизительно при 90°С (жидкость В). Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,0. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 3 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 5 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 540°С в течение 5 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0204] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 4,2 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0205] Пример А6
В 211,2 г горячей воды при температуре приблизительно 90°С растворили 70,4 г гептамолибдата аммония (жидкость А). Кроме того, 34,0 г нитрата висмута, 21,6 г нитрата церия, 35,0 г нитрата железа, 0,58 г нитрата цезия и 44,8 г нитрата кобальта растворили в 35,3 г 18% по массе водного раствора азотной кислоты, в которую добавили 140,8 г горячей воды приблизительно при 90°С (жидкость В).
[0206] Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,1. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 540°С в течение 6 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0207] Пример А7
В 193,1 г горячей воды при температуре приблизительно 90°С растворили 64,4 г гептамолибдата аммония (жидкость А). Кроме того, 37,0 г нитрата висмута, 23,7 г нитрата церия, 36,9 г нитрата железа, 0,41 г нитрата цезия и 54,3 г нитрата кобальта растворили в 34,1 г 18% по массе водного раствора азотной кислоты, в которую добавили 128,7 г горячей воды приблизительно при 90°С (жидкость В).
[0208] Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,0. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 3 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 540°С в течение 5 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0209] Сравнительный пример А1
В 208,8 г горячей воды при температуре приблизительно 90°С растворили 69,6 г гептамолибдата аммония (жидкость А). Кроме того, 28,6 г нитрата висмута, 28,3 г нитрата церия, 10,9 г нитрата магния, 38,3 г нитрата железа, 1,43 г нитрата рубидия и 38,5 г нитрата никеля растворили в 41,9 г 18% по массе водного раствора азотной кислоты, в которую добавили 200,3 г горячей воды приблизительно при 90°С (жидкость В). Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,2. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 3 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 5 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 530°С в течение 5 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0210] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 5,0 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0211] Сравнительный пример А2
В 216,9 г горячей воды при температуре приблизительно 90°С растворили 72,3 г гептамолибдата аммония (жидкость А). Кроме того, 28,1 г нитрата висмута, 14,7 г нитрата церия, 0,35 г нитрата калия, 19,2 г нитрата железа, 2,0 г нитрата цезия и 69,8 г нитрата кобальта растворили в 40,6 г 18% по массе водного раствора азотной кислоты, в которую добавили 181,7 г горячей воды приблизительно при 90°С (жидкость В). Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,3. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 3 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 3 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 520°С в течение 5 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0212] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 5,2 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0213] Сравнительный пример A3
В 197,2 г горячей воды при температуре приблизительно 90°С растворили 65,7 г гептамолибдата аммония (жидкость А). Кроме того, 64,5 г нитрата висмута, 42,4 г нитрата железа, 0,54 г нитрата цезия и 30,8 г нитрата кобальта растворили в 40,6 г 18% по массе водного раствора азотной кислоты, в которую добавили 203,6 г горячей воды приблизительно при 90°С (жидкость В). Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,0. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 3 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 3 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 540°С в течение 5 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0214] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 6,1 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0215] Сравнительный пример А4
В 309,9 г горячей воды при температуре приблизительно 90°С растворили 70,0 г гептамолибдата аммония и 5,4 г метаванадата аммония (жидкость А). Кроме того, 46,3 г нитрата висмута, 45,2 г нитрата железа, 0,57 г нитрата цезия и 32,8 г нитрата кобальта растворили в 38,6 г 18% по массе водного раствора азотной кислоты, в которую добавили 170,3 г горячей воды приблизительно при 90°С (жидкость В). Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,5. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 3 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 3 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 460°С в течение 5 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0216] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 6,0 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0217] Сравнительный пример А5
В перемешанную жидкость из 90,2 г ионообменной воды и 127,0 г 30% по массе водного раствора пероксида водорода добавили 54,3 г триоксида молибдена, который взбалтывали и перемешивали приблизительно при 70°С для растворения. Таким образом, получили раствор (жидкость А). Кроме того, 168,9 г водной дисперсионной жидкости 10% по массе оксида висмута, который имеет средний размер частицы 51 нм, 63,7 г водной дисперсионной жидкости 15% по массе оксида кобальта, который имеет средний размер частицы 22 нм, 66,9 г водной дисперсионной жидкости 15% по массе оксида железа, который имеет средний размер частицы 39 нм, 84,9 г водной дисперсионной жидкости 10% по массе оксида церия, который имеет средний размер частицы 20 нм, и 4,2 г жидкости 10% по массе гидроксида цезия смешали для того, чтобы получить раствор (жидкость В).
[0218] Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,0. Затем жидкость взбалтывали и перемешивали в течение приблизительно 3 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 250°С в течение 20 минут на воздухе, и поддерживали при 250°С в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 530°С в течение 5 часов для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0219] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 4,2 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0220] Сравнительный пример А6
Каталитический состав Mo12Bi1,6Ce0,4Fe1,0Co8,0Cs0,4K0,2, который представляет атомное соотношение относительно 12 атомов Мо, получали следующим образом. В 1820 г горячей воды при температуре приблизительно 50°С растворили 364 г гептамолибдата аммония (жидкость А). Кроме того, 133 г нитрата висмута, 29,8 г нитрата церия, 69,4 г нитрата железа, 13,4 г нитрата цезия, 3,46 г нитрата калия и 400 г нитрата кобальта растворили в 290 г водного раствора азотной кислоты 15% масс. (жидкость В).
Жидкость А и жидкость В взбалтывали и перемешивали в течение приблизительно 2 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала подвергали распылительной сушке для того, чтобы получить высушенный распылением предшественник каталитического состава, который затем предварительно прокаливали при 200°С в течение 3 часов. Полученному предварительно прокаленному предшественнику каталитического состава в форме псевдосферических частиц придавали цилиндрическую геометрическую форму, которая имеет диаметр 5 мм и высоту 4 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 460°С в течение 3 часов для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0221] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 4,2 г катализатора и осуществили реакцию синтеза метакролеина при тех же условиях, что и в примере А1. Результаты оценки реакции представлены в таблице 3.
[0222] На фиг. 8 проиллюстрирована XRD (2θ = от 25 до 27°) оксидного катализатора до и после реакции каталитического окисления в газовой фазе олефина в сравнительном примере А6. Оксидный катализатор до реакции каталитического окисления в газовой фазе олефина в сравнительном примере А6 имел XRD пик от CoMoO4 (002) при 2θ=26,46°, и оксидный катализатор после реакции имел XRD пик от CoMoO4 (002) при 2θ=26,46°. Таким образом, обнаружено, что двухвалентное Fe отсутствовало в твердом растворе в CoMoO4. Это объясняли атомным соотношением железа к кобальту (Fe/Co), не отвечающим следующему выражению: Fe/Co≥1.
[0223] <Реакция синтеза акролеина>
Пример А8
В перемешанную жидкость из 90,7 г ионообменной воды и 127,8 г 30% по массе водного раствора пероксида водорода добавили 54,6 г триоксида молибдена, который взбалтывали и перемешивали приблизительно при 70°С для растворения. Таким образом получили раствор (жидкость А). Кроме того, 205,3 г водной дисперсионной жидкости 10% по массе оксида висмута, который имеет средний размер частицы 51 нм, 54,5 г водной дисперсионной жидкости 15% по массе оксида кобальта, который имеет средний размер частицы 22 нм, 57,2 г водной дисперсионной жидкости 15% по массе оксида железа, который имеет средний размер частицы 39 нм, 72,1 г водной дисперсионной жидкости 10% по массе оксида лантана, который имеет средний размер частицы 40 нм, и 1,4 г жидкости 10% по массе гидроксида цезия смешали для того, чтобы получить раствор (жидкость В).
[0224] Жидкость А и жидкость В смешали для того, чтобы получить перемешанную жидкость, в которую добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,8. Затем жидкость взбалтывали и перемешивали в течение приблизительно 3 часов для того, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 5 часов на воздухе, затем до 260°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 510°С в течение 5 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0225] Используя полученный катализатор, синтезировали акролеин из пропилена. SUS реакционную пробирку в рубашке, которая имеет внутренний диаметр 15 мм, заполнили 20 мл катализатора. Газообразный исходный материал из 10% по объему пропилена, 17% по объему водяного пара и 73% по объему воздуха пропустили через пробирку при температуре реакции 320°С с тем, чтобы выполнить реакцию синтеза акролеина. Результаты оценки реакции представлены в таблице 4.
[0226] Сравнительный пример А7
В 203,1 г горячей воды при температуре приблизительно 90°С растворили 67,7 г гептамолибдата аммония (жидкость А). Кроме того, 37,1 г нитрата висмута, 22,0 г нитрата церия, 51,4 г нитрата железа, 0,19 г нитрата цезия и 37,4 г нитрата кобальта растворили в 41,9 г 18% по массе водного раствора азотной кислоты, в которую добавили 205,7 г горячей воды приблизительно при 90°С (жидкость В). Жидкость А и жидкость В смешали, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,0. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 3 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 250°С в течение 20 минут на воздухе, и поддерживали при 250°С в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученному предварительно прокаленному продукту придали геометрическую форму кольца, которое имеет диаметр 5 мм, высоту 4 мм и внутренний диаметр 2 мм посредством прессования таблеток, которые затем подвергли конечному прокаливанию при 460°С в течение 5 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 1 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 2.
[0227] Для того чтобы оценить реакцию катализатора, реакционную пробирку заполнили 20 мл катализатора и осуществили реакцию синтеза акролеина при тех же условиях, что и в примере А8. Результаты оценки реакции представлены в таблице 4.
[0228] <Реакция синтеза бутадиена>
Пример А9
Используя тот же катализатор, что и в примере А2, бутадиен синтезировали из 1-бутена следующим образом. SUS реакционную пробирку в рубашке, которая имеет диаметр 14 мм, заполнили 6,0 г катализатора. Газовую смесь 8% по объему 1-бутена, 12,8% по объему кислорода и 79,2% по объему азота пропустили через пробирку при температуре реакции 360°С при скорости потока 120 мл/мин (NTP) с тем, чтобы осуществить реакцию синтеза бутадиена. Результаты оценки реакции представлены в таблице 5.
[0229] Сравнительный пример А8
Используя тот же катализатор, что и в сравнительном примере А5, реакционную пробирку заполнили 6,0 г катализатора. Бутадиен синтезировали из 1-бутена при тех же условиях реакции, что и в примере А9, следующим образом. Результаты оценки реакции представлены в таблице 5.
[0230]



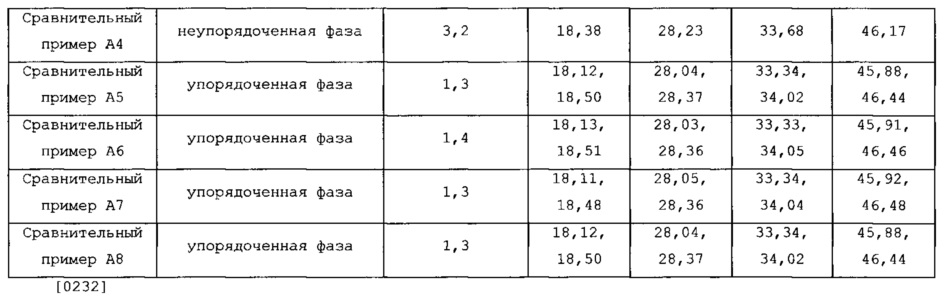
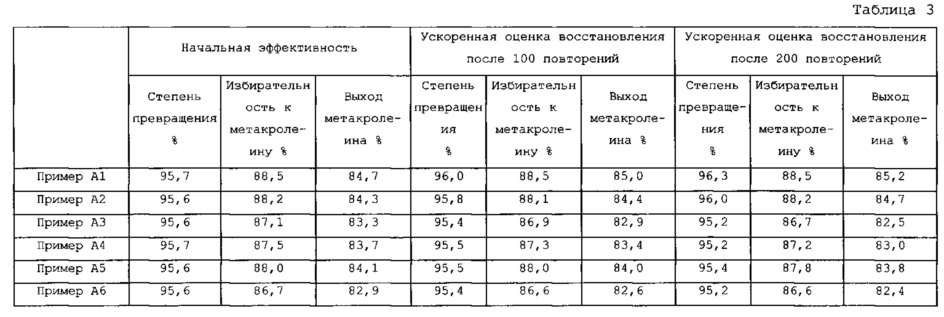



[0235] <Пики рентгеновской дифракции катализаторов, полученных в примере А1 и сравнительном примере А3>
На фиг. 5 проиллюстрированы пики рентгеновской дифракции катализаторов, полученных в примере А1 и сравнительном примере A3. На фиг. 6 проиллюстрирована увеличенная диаграмма пиков рентгеновской дифракции для диапазона 2θ = от 15 до 30° на фиг. 5. На фиг. 7 проиллюстрирована увеличенная диаграмма для диапазона 2θ = от 30 до 50°. На фиг. 6 и фиг. 7 проиллюстрировано, что катализатор, полученный в примере А1, имел пики рентгеновской дифракции по меньшей мере при 2θ=18,34° для плоскости (101), 28,16° для плоскости (112), 33,66° для плоскости (200) и 46,10°, при соотношении интенсивностей пиков (Ia/Ib)=3,3, где Ia представляет интенсивность пика при 2θ=33,66° и Ib представляет интенсивность пика при 2θ=34,06°. Можно делать предположение о том, что кристаллическую структуру неупорядоченной фазы в Bi3-xAxFe1Mo2O12 формировали в катализаторе, полученном в примере А1.
[0236] С другой стороны, при рентгеновской дифракции катализатора, полученного в сравнительном примере A3, не наблюдали пик при 18,30°±0,05° для плоскости (101), 28,20°±0,05° для плоскости (112), 33,65°±0,05° для плоскости (200) и 46,15°±0,05° для плоскости (204). Следовательно, можно предположить, что кристаллическую структуру неупорядоченной фазы в Bi3-xAxFe1Mo2O12 не сформировали в катализаторе, полученном в сравнительном примере A3.
[0237] Сравнили рентгеновскую дифракцию между катализаторами, полученными в примере А1 и сравнительном примере A3 следующим образом. В катализаторе, полученном в сравнительном примере A3, наблюдали расщепленные пики при 18,10° для плоскости (310) и при 18,48° для плоскости (111), соответствующие пику при 18,34° для плоскости (101) катализатора, полученного в примере А1. В катализаторе, полученном в сравнительном примере A3, наблюдали расщепленные пики при 28,02° для плоскости (221) и при 28,35° для плоскости (42-1), соответствующие пику при 28,20°±0,05° для плоскости (112) катализатора, полученного в примере А1. В катализаторе, полученном в сравнительном примере A3, наблюдали расщепленные пики при 33,30° для плоскости (600) и при 34,06° для плоскости (202), соответствующие пику при 33, 65°±0,05° для плоскости (200) катализатора, полученного в примере А1. В катализаторе, полученном в сравнительном примере A3, наблюдали расщепленные пики при 45,90° для плоскости (640) и при 46,46° для плоскости (242), соответствующие пику при 46,15°±0,05° для плоскости (204) катализатора, полученного в примере А1. Катализатор, полученный в сравнительном примере A3, имел соотношение интенсивностей пиков (Ia/Ib)=1,1, где Ia представляет интенсивность пика при 2θ=33,66° и Ib представляет интенсивность пика при 2θ=34,06°. Можно делать предположение о том, что в катализаторе, полученном в сравнительном примере A3, вместо неупорядоченной фазы в Bi3-xAxFe1Mo2O12 формировали упорядоченную фазу.
[0238] Пример В
Пример В предоставлен далее для того, чтобы дополнительно иллюстрировать второй аспект по настоящему изобретению, но не предназначен для того, чтобы ограничивать этим объем второго аспекта по настоящему изобретению. Поскольку атомное соотношение кислорода в катализаторе для аммоксидирования определяют с помощью состояния атомной валентности других элементов, атомное соотношение кислорода пропущено в формуле, представляющей композицию катализатора в примерах и сравнительных примерах. Композиционное соотношение каждого элемента в катализаторе для аммоксидирования вычисляли по композиционному соотношению для получения.
Дальше в примере В и сравнительном примере В водные дисперсии различных металлов использовали в качестве исходных материалов катализатора. Каждую из водных дисперсий оксида висмута, оксида железа и оксида кобальта для использования получили в CIK Nanotek Corporation, и каждую из водных дисперсий оксида лантана и оксида церия для использования получили в Taki Chemical Co., Ltd.
[0239] <Измерение среднего размера частицы>
Средний размер частицы получали посредством вычисления на основе следующего уравнения:
Средний размер частицы [нм] = 6000/(площадь поверхности [м2/г] × истинная плотность (8,99 г/см3).
[0240] <Измерение рН>
Измерение осуществили с использованием измерителя рН KR5E производства AS ONE.
[0241] <Измерение угла рентгеновской дифракции>
При XRD измерении измеряли плоскость (111) и плоскость (200) соединения LaB6 в качестве стандартного эталонного материала 660 согласно National Institute of Standards & Technology, так, что значения нормализовали по 37,441° и 43,506°, соответственно.
Использовали XRD аппарат Bruker D8 Advance. Условия XRD измерения представляли собой следующее: выход рентгеновского излучения 40 кВ-40 мА, щель расходимости (DS) 0,3°, ширина шага 0,02°/шаг, время счета 2,0 с и диапазон измерения 2θ = от 5° до 60°.
[0242] <Условия оценки реакции аммоксидирования>
От 40 до 60 г катализатора помещали в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, в которой размещали 12 листов проволочного сита 10 меш с интервалами в 1 см. Газовую смесь (с объемным соотношением пропилена или изобутилена : аммиака : кислорода : гелия 1:1, 2:1, 85:7,06) пропустили через пробирку при температуре реакции 430°С при нормальном давлении в реакции при скорости потока 3,64 см3/с (в пересчете на NTP).
[0243] <Степень превращения, избирательность и выход>
Эффективность реакции представлена посредством степени превращения, избирательности и выхода, которые определяют с помощью следующих выражений.
Степень превращения = (число молей исходного материала, вступившего в реакцию/число молей поданного исходного материала) × 100
Избирательность = (число молей полученного соединения/число молей исходного материала, вступившего в реакцию) × 100
Выход = (число молей полученного соединения/число молей поданного исходного материала) × 100
[0244] <Время контакта>
Время контакта определяют посредством следующего выражения. Время контакта (с×г/см3)=(W/F)×273/(273+Т)×Р/0,10 В выражении W представляет количество катализатора (г), F представляет скорость потока газовой смеси исходных материалов (Н.см3/с) в нормальном состоянии (0°С, 1 атм), Т представляет температуру реакции (°С) и Р представляет давление в реакции (мПа).
[0245] <Оценка восстанавливаемости>
Восстанавливаемость оценивали для ускоренной оценки устойчивости катализатора к восстановлению. Катализатор восстанавливали в атмосфере газа, не содержащего кислород, и повторно окисляли в возвращенных условиях оценки реакции. Процесс повторяли для того, чтобы достигать ускоренной оценки устойчивости катализатора к восстановлению.
Газовую смесь из 2% по объему пропилена, изобутилена, изобутанола и/или т-бутилового спирта и 98% по объему гелия пропустили при температуре 430°С при скорости потока 3,64 см3/с (в пересчете на NTP) в течение 5 минут для восстанавливающей обработки. Впоследствии условия оценки реакции восстанавливали и поток сохраняли в течение 5 минут. Это составляло одно повторение, и реакцию оценивали после выполнения 100 повторений. После выполнения дополнительных 100 повторений, оценивали реакцию и устойчивость к восстановлению.
[0246] Каждый из элементов А для использования в примере В и сравнительном примере В имеет следующий радиус иона:
La: 1,14

Се: 1,07

Pr: 1,06

Са: 1,03
Pb: 1,24

V: 0,56

[0247] Пример B1
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 211,0 г горячей воды при температуре приблизительно 90°С растворили 70,3 г гептамолибдата аммония (жидкость А). Кроме того, 38,6 г нитрата висмута, 22,9 г нитрата лантана, 42,7 г нитрата железа, 0,51 г нитрата цезия и 31,4 г нитрата кобальта растворили в 36,4 г 18% по массе водного раствора азотной кислоты, в которую добавили 148,1 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,1. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 260°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 55 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 4,3 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0248] Пример В2
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 207,8 г горячей воды при температуре приблизительно 90°С растворили 69,3 г гептамолибдата аммония (жидкость А). Кроме того, 41,1 г нитрата висмута, 19,7 г нитрата церия, 44,7 г нитрата железа, 0,50 г нитрата цезия и 32,5 г нитрата кобальта растворили в 40,9 г 18% по массе водного раствора азотной кислоты, в которую добавили 194,7 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,3. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 55 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 4,2 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0249] Пример В3
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 209,1 г горячей воды при температуре приблизительно 90°С растворили 69,7 г гептамолибдата аммония (жидкость А). Кроме того, 44,6 г нитрата висмута, 17,0 г нитрата празеодимия, 31,8 г нитрата железа, 0,57 г нитрата цезия и 38,4 г нитрата кобальта растворили в 40,2 г 18% по массе водного раствора азотной кислоты, в которую добавили 188,8 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,6. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 270°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 57 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 4,4 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0250] Пример В4
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 223,4 г горячей воды при температуре приблизительно 90°С растворили 74,5 г гептамолибдата аммония (жидкость А). Кроме того, 20,4 г нитрата висмута, 30,3 г нитрата лантана, 45,2 г нитрата железа, 0,54 г нитрата цезия, 6,6 г нитрата кальция и 32,9 г нитрата кобальта растворили в 34,3 г 18% по массе водного раствора азотной кислоты, в которую добавили 114,4 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,6. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 240°С в течение 1 часа и поддерживали температуру в течение 4 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 580°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 57 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 4,5 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0251] Пример В5
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды смешали для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 202,0 г горячей воды при температуре приблизительно 90°С растворили 67,3 г гептамолибдата аммония (жидкость А). Кроме того, 21,5 г нитрата висмута, 24,6 г нитрата церия, 42,2 г нитрата железа, 0,74 г нитрата цезия, 8,4 г нитрата свинца и 47,3 г нитрата кобальта растворили в 39,4 г 18% по массе водного раствора азотной кислоты, в которую добавили 182,0 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,6. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 57 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 4,6 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0252] Пример В6
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 211,8 г горячей воды при температуре приблизительно 90°С растворили 7 0,6 г гептамолибдата аммония (жидкость А). Кроме того, 37,1 г нитрата висмута, 21,5 г нитрата церия, 41,5 г нитрата железа, 0,63 г нитрата рубидия, 10,2 г нитрата магния, 19,5 г нитрата кобальта и 9,8 г нитрата никеля растворили в 41,3 г 18% по массе водного раствора азотной кислоты, в которую добавили 193,1 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,6. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 57 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 4,7 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0253] Пример В7
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 210,8 г горячей воды при температуре приблизительно 90°С растворили 70,3 г гептамолибдата аммония (жидкость А). Кроме того, 27,3 г нитрата висмута, 14,3 г нитрата церия, 20,0 г нитрата железа, 1,94 г нитрата рубидия, 0,67 г нитрата калия, 4,9 г нитрата цинка и 72,7 г нитрата кобальта растворили в 40,5 г 18% по массе водного раствора азотной кислоты, в которую добавили 186,0 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,6. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 57 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 4,6 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0254] Сравнительный пример В1
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 203,4 г горячей воды при температуре приблизительно 90°С растворили 67,8 г гептамолибдата аммония (жидкость А). Кроме того, 61,9 г нитрата висмута, 46,3 г нитрата железа, 0,49 г нитрата цезия и 26,2 г нитрата кобальта растворили в 40,4 г 18% по массе водного раствора азотной кислоты, в которую добавили 195,7 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,1. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 260°С в течение 1 часа и поддерживали температуру в течение 4 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 55 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 5,6 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0255] Сравнительный пример В2
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 252,6 г горячей воды при температуре приблизительно 90°С растворили 84,2 г гептамолибдата аммония (жидкость А). Кроме того, 5,8 г нитрата висмута, 3,4 г нитрата церия, 24,0 г нитрата железа, 0,70 г нитрата рубидия, 26,4 г нитрата магния и 75,6 г нитрата никеля растворили в 41,8 г 18% по массе водного раствора азотной кислоты, в которую добавили 150,8 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,6. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 57 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 5,4 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0256] Сравнительный пример В3
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 249,9 г горячей воды при температуре приблизительно 90°С растворили 83,3 г гептамолибдата аммония (жидкость А). Кроме того, 8,6 г нитрата висмута, 15,2 г нитрата церия, 26,9 г нитрата железа, 0,23 г нитрата рубидия, 20,1 г нитрата магния, 34,5 г нитрата кобальта, 0,36 г нитрата калия и 23,0 г нитрата никеля растворили в 40,9 г 18% по массе водного раствора азотной кислоты, в которую добавили 148,1 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 3,6. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 260°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 57 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 5,3 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0257] Сравнительный пример В4
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
В 216,3 г горячей воды при температуре приблизительно 90°С растворили 72,1 г гептамолибдата аммония и 5,2 г метаванадата аммония (жидкость А). Кроме того, 44,4 г нитрата висмута, 43,8 г нитрата железа, 0,46 г нитрата цезия и 31,8 г нитрата кобальта растворили в 38,3 г 18% по массе водного раствора азотной кислоты, в которую добавили 161,9 г горячей воды приблизительно при 90°С (жидкость В).
Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В, добавили водный раствор аммиака для того, чтобы скорректировать рН до 4,1. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 2 часов на воздухе, затем до 250°С в течение 1 часа и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 580°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 55 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 5,9 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0258] Сравнительный пример В5
Тот же предшественник оксидного катализатора, что и в примере В1, нагревали до 250°С в течение 1 часа на воздухе и поддерживали температуру в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 3 часов на воздухе для того, чтобы получить катализатор. Композиция катализатора представлена в таблице 6 и результаты измерения порошковой рентгеновской дифракции представлены в таблице 7.
Для оценки реакции катализатора, 55 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование пропилена при времени контакта 5,8 (с×г/см3). Результаты оценки реакции представлены в таблице 8. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 8.
[0259] Пример В8
Используя тот же катализатор, что и в примере В1, для оценки реакции, 57 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование изобутилена при времени контакта 5,4 (с×г/см3). Результаты оценки реакции представлены в таблице 9. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 9.
[0260] Сравнительный пример В6
Используя тот же катализатор, что и в сравнительном примере В5, для оценки реакции, 55 г катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм, и осуществили аммоксидирование изобутилена при времени контакта 6,0 (с×г/см3). Результаты оценки реакции представлены в таблице 9. Результаты оценки реакции после выполнения 100 повторений и 200 повторений для ускоренной оценки восстановления также представлены в таблице 9.
[0261]






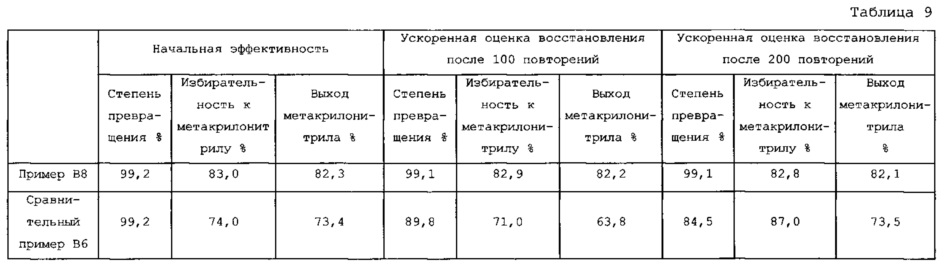
[0265] <Пики рентгеновской дифракции катализаторов, полученных в примере В1 и сравнительном примере В1>
На фиг. 9 проиллюстрированы пики рентгеновской дифракции катализаторов, полученных в примере В1 и сравнительном примере В1. На фиг. 10 проиллюстрирована увеличенная диаграмма пиков рентгеновской дифракции для диапазона 2θ = от 15 до 30° на фиг. 9. На фиг. 11 проиллюстрирован увеличенная диаграмма для диапазона 2θ = от 30 до 50°. На фиг. 9 и фиг. 10 проиллюстрировано, что катализатор, полученный в примере В1, имел пики рентгеновской дифракции по меньшей мере при 2θ=18,28° для плоскости (101), 28,16° для плоскости (112), 33,60° для плоскости (200) и 46,00° при соотношении интенсивностей пиков (Ia/Ib)=3,3, где Ia представляет интенсивность пика при 2θ=33,60° и Ib представляет интенсивность пика при 2θ=34,06°. Можно делать предположение о том, что в катализаторе, полученном в примере В1, формировали кристаллическую структуру неупорядоченной фазы в Bi3-xAxFe1Mo2O12.
[0266] С другой стороны, при рентгеновской дифракции катализатора, полученного в сравнительном примере В1, не наблюдали пик при 18,30°±0,05° для плоскости (101), 28,20°±0,05° для плоскости (112), 33,65°±0,05° для плоскости (200) и 46,15°±0,05° для плоскости (204). Следовательно, можно предположить, что в катализаторе, полученном в сравнительном примере В1, не сформировали кристаллическую структуру неупорядоченной фазы в Bi3-xAxFe1Mo2O12.
Сравнивали рентгеновскую дифракцию между катализаторами, полученными в примере В1 и сравнительном примере В1, следующим образом. В катализаторе, полученном в сравнительном примере В1, наблюдали расщепленные пики при 18,10° для плоскости (310) и при 18,45° для плоскости (111), соответствующие пику при 18,34° для плоскости (101) катализатора, полученного в примере В1. В катализаторе, полученном в сравнительном примере В1, наблюдали расщепленные пики при 28,00° для плоскости (221) и при 28,35° для плоскости (42-1), соответствующие пику при 28,20°±0,2° для плоскости (112) катализатора, полученного в примере В1. В катализаторе, полученном в сравнительном примере В1, наблюдали расщепленные пики при 33,30° для плоскости (600) и при 34,10° для плоскости (202), соответствующие пику при 33, 65°±0,2° для плоскости (200) катализатора, полученного в примере В1. В катализаторе, полученном в сравнительном примере В1, наблюдали расщепленные пики при 45,90° для плоскости (640) и при 46,45° для плоскости (242), соответствующие пику при 46,15°±0,2° для плоскости (204) катализатора, полученного в примере В1. Катализатор, полученный в сравнительном примере В1, имел соотношение интенсивностей пиков (Ia/Ib)=1,0, где Ia представляет интенсивность пика при 2θ=33,60° и Ib представляет интенсивность пика при 2θ=34,06°. Можно делать предположение о том, что в катализаторе, полученном в сравнительном примере В1, упорядоченную фазу формировали вместо неупорядоченной фазы в Bi3-xAxFe1Mo2O12.
[0267] Далее пример С предоставлен для того, чтобы дополнительно иллюстрировать третий аспект по настоящему изобретению, но он не предназначен для того, чтобы ограничивать этим объем третьего аспекта по настоящему изобретению. Поскольку атомное соотношение кислорода в оксидном катализаторе определяют с помощью состояния атомной валентности других элементов, атомное соотношение кислорода пропущено в формуле, представляющей композицию катализаторов в примере С и сравнительном примере С. Композиционное соотношение каждого элемента в оксидном катализаторе вычисляли по композиционному соотношению для получения.
[0268] В примере С и сравнительном примере С эффективность реакции представлена с помощью степени превращения, избирательности и выхода, которые определяют с помощью следующих выражений. Термин «число молей исходного материала» относится к числу молей олефина и/или спирта.
Степень превращения (%) = (число молей олефина и/или спирта, вступивших в реакцию/число поданных молей олефина и/или спирта) × 100
Избирательность (%) = (число молей получаемого ненасыщенного альдегида/число молей олефина и/или спирта, вступивших в реакцию) × 100
Выход (%) = (число молей получаемого ненасыщенного альдегида/число поданных молей олефина и/или спирта) × 100
[0269] Пример С1
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 2 6% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
[0270] В 211,9 г горячей воды при температуре приблизительно 90°С растворили 70,6 г гептамолибдата аммония (жидкость А). Кроме того, 32,3 г нитрата висмута, 28,7 г нитрата церия, 45,6 г нитрата железа, 1,03 г нитрата цезия и 29,2 г нитрата кобальта растворили в 40,9 г 18% по массе водного раствора азотной кислоты, в которую добавили 190,3 г горячей воды приблизительно при 90°С (жидкость В).
[0271] Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 1 часа или больше на воздухе, затем до 250°С в течение всего 2 часов и поддерживали при 250°С в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 600°С в течение 3 часов на воздухе для того, чтобы получить оксидный катализатор. Композиция оксидного катализатора представлена в таблице 10.
[0272] Используя полученный таким образом оксидный катализатор, получили ненасыщенный альдегид. В частности, 40 г оксидного катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/8,1/остальное подали через реакционную пробирку при температуре реакции 440°С при давлении в реакции 0,05 мПа при скорости потока 595 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между оксидным катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0273] Пример С2
Использовали 40 г того же оксидного катализатора, что и в примере С1. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/8,8/остальное подали при температуре реакции 430°С при давлении в реакции 0,05 мПа при скорости потока 725 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0274] Пример С3
Использовали 30 г того же оксидного катализатора, что и в примере С1. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/9,4/остальное подали при температуре реакции 440°С при давлении в реакции 0,05 мПа при скорости потока 794 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между оксидным катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0275] Пример С4
Использовали 40 г того же оксидного катализатора, что и в примере С1. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/7,9/остальное подали при температуре реакции 440°С при давлении в реакции 0,05 мПа при скорости потока 595 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между оксидным катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0276] Пример С5
Использовали 40 г того же оксидного катализатора, что и в примере С1. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/9,5/остальное подали при температуре реакции 440°С при давлении в реакции 0,05 мПа при скорости потока 625 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между оксидным катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0277] Пример С6
Использовали 35 г того же оксидного катализатора, что и в примере С1. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/8,8/остальное подали при температуре реакции 460°С при давлении в реакции 0,05 мПа при скорости потока 595 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между оксидным катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0278] Пример С7
Использовали 40 г того же оксидного катализатора, что и в примере С1. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/8,1/остальное подали при температуре реакции 400°С при давлении в реакции 0,05 мПа при скорости потока 595 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между оксидным катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0279] Пример С8
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
[0280] В 211,8 г горячей воды при температуре приблизительно 90°С растворили 70,6 г гептамолибдата аммония (жидкость А). Кроме того, 25,8 г нитрата висмута, 18,6 г нитрата церия, 18,7 г нитрата лантана, 42,9 г нитрата железа, 1,03 г нитрата цезия и 31,1 г нитрата кобальта растворили в 37,5 г 18% по массе водного раствора азотной кислоты, в которую добавили 157,5 г горячей воды приблизительно при 90°С (жидкость В).
[0281] Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 1 часа или больше на воздухе, затем до 250°С в течение всего 2 часов, и поддерживали при 250°С в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 590°С в течение 3 часов на воздухе для того, чтобы получить оксидный катализатор. Композиция оксидного катализатора представлена в таблице 10.
[0282] Используя полученный таким образом оксидный катализатор, получили ненасыщенный альдегид. В частности, 40 г оксидного катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/8,1/остальное подали в реакционную пробирку при температуре реакции 440°С при давлении в реакции 0,05 мПа при скорости потока 595 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между оксидным катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0283] Пример С9
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
[0284] В 214,5 г горячей воды при температуре приблизительно 90°С растворили 71,5 г гептамолибдата аммония (жидкость А). Кроме того, 18,0 г нитрата висмута, 39,2 г нитрата церия, 4,9 г нитрата никеля, 3,4 г нитрата магния, 43,4 г нитрата железа, 1,48 г нитрата рубидия и 31,6 г нитрата кобальта растворили в 41,6 г 18% по массе водного раствора азотной кислоты, в которую добавили 192,3 г горячей воды приблизительно при 90°С (жидкость В).
[0285] Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали от 100°С до 200°С в течение 1 часа или больше на воздухе, затем до 250°С в течение всего 2 часов, и поддерживали при 250°С в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 580°С в течение 3 часов на воздухе для того, чтобы получить оксидный катализатор. Композиция оксидного катализатора представлена в таблице 10.
[0286] Используя полученный таким образом оксидный катализатор, получили ненасыщенный альдегид. В частности, 40 г оксидного катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/8,1/остальное подали через реакционную пробирку при температуре реакции 440°С при давлении в реакции 0,05 мПа при скорости потока 595 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе, время контакта между катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0287] Пример C10
Использовали 40 г того же оксидного катализатора, что и в примере С1. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/10,2/остальное подали при температуре реакции 440°С при давлении в реакции 0,05 мПа при скорости потока 595 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между оксидным катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0288] Сравнительный пример С1
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
[0289] В 221,4 г горячей воды при температуре приблизительно 90°С растворили 73,8 г гептамолибдата аммония (жидкость А). Кроме того, 27,0 г нитрата висмута, 6,0 г нитрата церия, 14,0 г нитрата железа, 2,68 г нитрата цезия, 0,70 г нитрата калия и 81,4 г нитрата кобальта растворили в 40,4 г 18% по массе водного раствора азотной кислоты, в которую добавили 175,3 г горячей воды приблизительно при 90°С (жидкость В).
[0290] Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали до 250°С в течение 2 часов на воздухе, и поддерживали при 250°С в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 570°С в течение 2 часов на воздухе для того, чтобы получить оксидный катализатор. Композиция оксидного катализатора представлена в таблице 10.
[0291] Используя полученный таким образом оксидный катализатор, получили ненасыщенный альдегид. В частности, 40 г оксидного катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/8,1/остальное подали через реакционную пробирку при температуре реакции 440°С при давлении в реакции 0,05 мПа при скорости потока 595 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе, время контакта между катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0292] Сравнительный пример С2
Сначала смешали 125,0 г 40% по массе золя диоксида кремния, который содержал 26% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 44 нм, и 147,1 г 34% по массе водного золя диоксида кремния, который содержал 30% по массе SiO2 основных частиц диоксида кремния, которые имеют средний диаметр 12 нм, и 61,3 г воды для того, чтобы получить исходный материал 30% по массе диоксид кремния.
[0293] В 202,8 г горячей воды при температуре приблизительно 90°С растворили 67,6 г гептамолибдата аммония (жидкость А). Кроме того, 61,7 г нитрата висмута, 43,6 г нитрата железа, 0,98 г нитрата цезия и 28,0 г нитрата кобальта растворили в 40,3 г 18% по массе водного раствора азотной кислоты, в которую добавили 195,7 г горячей воды приблизительно при 90°С (жидкость В).
[0294] Смешали исходный материал диоксид кремния и как жидкость А, так и жидкость В. Затем жидкость взбалтывали и перемешивали приблизительно при 55°С в течение приблизительно 4 часов с тем, чтобы получить суспензию исходного материала. Суспензию исходного материала транспортировали в распылительную сушилку с тем, чтобы подвергнуть распылительной сушке при входной температуре 250°С и при выходной температуре 140°С. Таким образом, получили предшественник оксидного катализатора. Полученный предшественник оксидного катализатора нагревали до 250°С в течение 2 часов на воздухе, и поддерживали при 250°С в течение 3 часов с тем, чтобы получить предварительно прокаленный продукт. Полученный предварительно прокаленный продукт подвергли конечному прокаливанию при 600°С в течение 3 часов на воздухе для того, чтобы получить оксидный катализатор. Композиция оксидного катализатора представлена в таблице 10.
[0295] Используя полученный таким образом оксидный катализатор, получили ненасыщенный альдегид. В частности, 40 г оксидного катализатора поместили в реакционную пробирку с псевдоожиженным слоем из стекла Vycor, которая имеет внутренний диаметр 25 мм. Перемешанный газообразный исходный материал (концентрация изобутилена: 8% по объему) с молярным композиционным соотношением изобутилена/воздуха/гелия = 1/8,1/остальное подали через реакционную пробирку при температуре реакции 440°С при давлении в реакции 0,05 мПа при скорости потока 595 см2/мин (в пересчете на NTP) с тем, чтобы выполнить реакцию синтеза метакролеина. Концентрация кислорода на выходе реактора, время контакта между оксидным катализатором и газовой смесью и результаты оценки реакции для этого случая представлены в таблице 11.
[0296]



[0298] <Измерение порошковой рентгеновской дифракции (XRD)>
Осуществляли измерение рентгеновской дифракции (XRD) оксидного катализатора для диапазона углов рентгеновской дифракции 2θ = от 5° до 60°. Пик, получаемый для угла рентгеновской дифракции (20) для плоскости (002) композитного оксида из Со и Мо, показан при 26,46°±0,02°. Двухвалентное Fe в твердом растворе в композитном оксиде Со и Мо образует композит, что делает возможным сдвиг пика из-за разности в радиусах ионов между Со2+ и Fe2+. Поскольку двухвалентное Fe в твердом растворе образует композитную структуру, пик, получаемый из композитного оксида, который содержит Со, Мо и двухвалентное Fe, показан не при 26,46°, а при 26,46°-α° (0<α), где α° представляет значение сдвига. Присутствие пика в диапазоне от 26,30 до 26,40 связано с образованием кристалла трехкомпонентной системы Со2+-Fe2+-Mo-O.
[0299] XRD измерение осуществили, исходя из указанного выше. При XRD измерении измеряли плоскость (111) и плоскость (200) соединения LaB6 в качестве стандартного эталонного материала 660 согласно National Institute of Standards & Technology так, что значения нормализовали по 37,441° и 43,506°, соответственно.
[0300] Использовали XRD аппарат Bruker D8 Advance. Условия XRD измерения представляли собой следующее: выход рентгеновского излучения 40 кВ-40 мА, щель расходимости (DS) 0,3°, ширина шага 0,01°/шаг, время счета 2,0 с и диапазон измерения 2θ = от 5° до 45°.
[0301] На фиг. 12 проиллюстрирована XRD (2θ = от 10 до 60°) оксидного катализатора до и после реакции каталитического окисления олефина в газовой фазе в примере С1. На фиг. 13 проиллюстрирована увеличенная диаграмма для диапазона 2θ = от 25 до 27° на фиг. 12. Оксидный катализатор до реакции каталитического окисления олефина в газовой фазе в примере С1 имел XRD пик от CoMoO4 (002) при 2θ=26,46°, и оксидный катализатор после реакции имел XRD пик от CoMoO4 (002) при 2θ=26,34°. Таким образом, обнаружено, что двухвалентное Fe находилось в твердом растворе в CoMoO4.
[0302] На фиг. 14 проиллюстрирована XRD (2θ = от 10 до 60°) оксидного катализатора до и после реакции каталитического окисления олефина в газовой фазе в сравнительном примере С1. На фиг. 15 проиллюстрирована увеличенная диаграмма для диапазона 2θ = от 25 до 27° на фиг. 14. Оксидный катализатор до реакции каталитического окисления олефина в газовой фазе в сравнительном примере С1 имел XRD пик от CoMoO4 (002) при 2θ=26,46° и оксидный катализатор после реакции имел XRD пик от CoMoO4 (002) при 2θ=26,46°. Таким образом, обнаружено, что двухвалентное Fe не находилось в твердом растворе в CoMoO4. Это объясняли атомным соотношением железа к кобальту (b/с), не соответствующим следующему выражению: b/с≥1.
[0303] <Пики рентгеновской дифракции катализаторов, полученных в примере С1 и сравнительном примере С2>
На фиг. 16 проиллюстрированы пики рентгеновской дифракции катализаторов, полученных в примере С1 и сравнительном примере С2. На фиг. 16 проиллюстрировано, что катализатор, полученный в примере С1, имел пики рентгеновской дифракции по меньшей мере при 2θ=18,32° для плоскости (101), 28,18° для плоскости (112), 33,66° для плоскости (200) и 46,12° при соотношении интенсивностей пиков (Ia/Ib)=3,2, где Ia представляет интенсивность пика при 2θ=33,66° и Ib представляет интенсивность пика при 2θ=34,06°. Можно делать предположение о том, что в катализаторе, полученном в примере С1, формировали кристаллическую структуру неупорядоченной фазы в Bi3-xAxFe1Mo2O12.
[0304] При рентгеновской дифракции катализатора, полученного в сравнительном примере С2, не наблюдали пик при 18,30°±0,05° для плоскости (101), 28,20°±0,05° для плоскости (112), 33,65°±0,05° для плоскости (200) и 46,15°±0,05° для плоскости (204). Следовательно, можно предположить, что в катализаторе, полученном в сравнительном примере С2, не сформировали кристаллическую структуру неупорядоченной фазы в Bi3-xAxFe1Mo2O12.
[0305] Проводили сравнение рентгеновской дифракции между катализаторами, полученными в примере С1 и сравнительном примере С2, следующим образом. В катализаторе, полученном в сравнительном примере С2, наблюдали расщепленные пики при 18,06° для плоскости (310) и при 18,44° для плоскости (111), соответствующие пику при 18,30°±0,05° для плоскости (101) катализатора, полученного в примере С1. В катализаторе, полученном в сравнительном примере С2, наблюдали расщепленные пики при 28,00° для плоскости (221) и при 28,32° для плоскости (42-1), соответствующие пику при 28,20°±0,05° для плоскости (112) катализатора, полученного в примере С1. В катализаторе, полученном в сравнительном примере С2, наблюдали расщепленные пики при 33,52° для плоскости (600) и при 33,98° для плоскости (202), соответствующие пику при 33,65°±0,05° для плоскости (200) катализатора, полученного в примере С1. В катализаторе, полученном в сравнительном примере С2, наблюдали расщепленные пики при 45,82° для плоскости (640) и при 46,40° для плоскости (242), соответствующие пику при 46,15°±0,05° для плоскости (204) катализатора, полученного в примере С1. При соотношении интенсивностей пиков (Ia/Ib)=0,94, где Ia представляет интенсивность пика при 2θ=33,52° и Ib представляет интенсивность пика при 2θ=33,98°, можно делать предположение о том, что в катализаторе, полученном в сравнительном примере С2, вместо неупорядоченной фазы в Bi3-xAxFe1Mo2O12 формировали упорядоченную фазу.
[0306] Настоящая заявка основана на японской патентной заявке №2012-216071, поданной в японское патентное бюро 28 сентября 2012 года, японской патентной заявке №2012-253243, поданной в японское патентное бюро 19 ноября 2012 года, и японской патентной заявке №2013-033663, поданной в японское патентное бюро 22 февраля 2013 года, которые полностью включены в настоящий документ посредством ссылки.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0307] Настоящее изобретение представляет собой промышленно применимый оксидный катализатор для использования в получении ненасыщенного альдегида или диолефина из олефина и/или спирта.
Реферат
Изобретение относится к оксидному катализатору для использования в получении ненасыщенного альдегида, диолефина или ненасыщенного нитрила из олефина и/или спирта. Оксидный катализатор имеет следующие характеристики: (1) оксидный катализатор содержит молибден, висмут, железо, кобальт и элемент А, который имеет радиус иона больше чем 0,96(за исключением калия, цезия и рубидия); (2) атомное соотношение а висмута к 12 атомам молибдена составляет 1≤а≤5, атомное соотношение b железа к 12 атомам молибдена составляет 1,5≤b≤6, атомное соотношение с элемента А к 12 атомам молибдена составляет 1≤с≤5, и атомное соотношение d кобальта к 12 атомам молибдена составляет 1≤d≤8; и (3) оксидный катализатор содержит неупорядоченную фазу, которая состоит из кристаллической системы, которая содержит молибден, висмут, железо и элемент А. Также предложены способ получения оксидного катализатора, способ получения ненасыщенного альдегида, способ получения диолефина и способ получения ненасыщенного нитрила. Изобретение позволяет предоставить оксидный катализатор, который препятствует восстановительному разложению катализатора даже во время промышленной эксплуатации в течение длительного времени. 5 н. и 9 з.п. ф-лы, 16 ил., 11 табл., 43 пр.
Формула



Документы, цитированные в отчёте о поиске
Катализатор для окисления и окислительного аммонолиза олефинов @ - @
Комментарии