Мезопористые материалы с активными металлами - RU2334554C2
Код документа: RU2334554C2
Чертежи














Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящее изобретение является частичным продолжением заявки США с серийным номером №09/955227 от 27 ноября 2001, включенной сюда посредством ссылки, которая является частичным продолжением заявки США с серийным номером №09/390276 от 7 сентября 1999, принятой сейчас как патент США №6358486 В1, которой заявлен приоритет.
Предпосылки изобретения
1. Область техники, к которой относится изобретение
Настоящее изобретение касается мезопористых материалов, особенно каталитических материалов, и использования мезопористых материалов для конверсии органических соединений, особенно углеводородов.
2. Предшествующий уровень техники
Большинство сегодняшних технологий переработки углеводородов базируется на цеолитных катализаторах. Цеолитные катализаторы хорошо известны в данной области техники и представляют собой хорошо упорядоченные пористые системы с однородными размерами пор. Тем не менее, данные материалы имеют тенденцию к тому, чтобы иметь либо только микропоры, либо только мезопоры. Микропоры определены как поры, имеющие диаметр меньше, чем примерно 2 нм. Мезопоры определены как поры, имеющие диаметр в диапазоне от примерно 2 нм до примерно 50 нм.
Поскольку данные реакции превращения углеводородов лимитируются массопереносом, катализатор с идеальным размером пор облегчит перенос реагентов к активным каталитическим центрам и перенос продуктов из катализатора.
Все еще существует необходимость в улучшенных материалах с функционирующими центрами внутри пористой структуры для процессов, направленных на каталитическую конверсию и/или адсорбцию углеводородов и других органических соединений.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В данной заявке предлагается способ обработки органических соединений. Способ включает: (а) обеспечение композиции, которая включает в себя, по существу, мезопористую структуру диоксида кремния, содержащего, по меньшей мере, 97 об.% пор с размером пор в диапазоне от примерно 15 Å до примерно 30 Å и с объемом микропор, по меньшей мере, примерно 0,01 см3/г, где в мезопористую структуру внедрены каталитически и/или химически активные гетероатомы в количестве, по меньшей мере, примерно 0,02 мас.%, выбранные из группы, состоящей из Al, Ti, V, Cr, Zn, Fe, Sn, Mo, Ga, Ni, Co, In, Zr, Mn, Cu, Mg, Pd, Pt и W, и где упомянутый катализатор имеет рентгенограмму с одним пиком от 0,3° до 3,5° для 2θ; и (b) взаимодействие органического сырья в условиях реакции с упомянутым катализатором, где способ обработки выбран из группы, состоящей из алкилирования, ацилирования, олигомеризации, селективного окисления, гидроочистки, изомеризации, деметаллирования, каталитической депарафинизации, гидроксилирования, гидрирования, аммоксимирования, дегидрирования, крекинга и адсорбции.
В одном аспекте данное изобретение связано с улучшенным каталитическим процессом для деметаллирования и десульфуризации минеральных масел, предпочтительно остаточных фракций с нежелательно высоким содержанием тяжелых металлов и/или серы, и/или азота, и/или углеродного остатка по Конрадсону (КУО). Особенно данное изобретение касается способа гидроочистки для снижения содержания тяжелых металлов, серы, азота и КУО минеральных масел, опять-таки предпочтительно содержащих остаточные углеводородные компоненты.
Остаточные фракции минерального масла получают путем атмосферной или вакуумной дистилляции сырой нефти; они обычно имеют высокое содержание металлов, серы, азота и КУО. Это происходит потому, что все металлы и КУО, присутствующие в исходной сырой нефти, остаются в остаточной фракции, и непропорциональное количество серы и азота исходной сырой нефти также остается в этой фракции. Основными загрязняющими металлами являются никель и ванадий с присутствующим иногда железом и небольшими количествами меди.
Содержание тяжелых металлов, серы, азота и КУО в остаточных фракциях обычно ограничивает их эффективное использование в качестве исходного сырья для последующей каталитической переработки, такой как каталитический крекинг и гидрокрекинг. Металлические загрязнения откладываются на специальных катализаторах для данных способов крекинга и вызывают преждевременное старение катализатора и/или нежелательные побочные реакции, такие как крекинг до кокса, нефтяного газа и водорода. Во время процесса FCC (флюид-каталитический крекинг) способа большая часть серы попадает в кокс FCC-катализатора, который сгорает при регенерации, приводя к существенному выделению SOx. Другая значительная часть остаточной серы попадает в продукты крекинга, такие как газолин и легкий рецикловый газойль (дополнительный компонент для дизельного топлива и топлива для домашнего отопления). Часть азота приводит к выделению NOx, а часть азота (основные азотные соединения) связывается с активными центрами FCC-катализатора и делает их неэффективными. КУО, являющийся мерой тенденции молекул к коксообразованию в большей степени, чем к крекингу и/или дистилляции, также является нежелательным свойством для загружаемых потоков в ходе каталитического крекинга. При высокой температуре, применяемой в каталитическом крекинге, молекулы, склонные к КУО, термически и/или каталитически разрушаются до кокса, легких газов и водорода. Каталитический крекинг обычно превращает углеводородное сырье легче, чем остаточные фракции, которые обычно имеют плотность согласно API меньше 20. Чаще всего сырье для крекинга представляет собой газойль установки для коксования и/или неочищенный газойль, верхний погон вакуумной колонны и т.д., причем это сырье имеет API-плотность от примерно 15 до примерно 45. Так как данные виды сырья для крекинга являются дистиллятами, они не содержат значительной доли больших молекул, в которых сконцентрированы металлы. Такой крекинг обычно осуществляют в реакторе, работающем при температуре примерно от 425 до 800°С, давлении примерно от 1 до 5 атмосфер и скорости потока примерно от 1 до 1000 WHSV (среднечасовая массовая скорость потока).
Загрязняющие металлы и сера создают аналогичные проблемы в операциях гидрокрекинга, которые обычно осуществляются на сырье, даже более легком, чем сырье для крекинга. Типичные условия реактора гидрокрекинга составляют температуру от 200 до 550°С и давление от 700 до 20000 кПа.
Очевидно, что существует необходимость в эффективном способе снижения содержания металлов и/или серы, и/или азота, и/или КУО в углеводородах и, особенно, в остаточных нефтяных фракциях. Хотя технология для достижения этого в случае перегнанных фракций в значительной степени разработана, попытки применить данную технологию к остаточным фракциям терпят неудачу из-за очень быстрой дезактивации катализатора, главным образом, из-за загрязнений металлами и отложения кокса.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Различные варианты осуществления описаны ниже со ссылками на чертежи, в которых
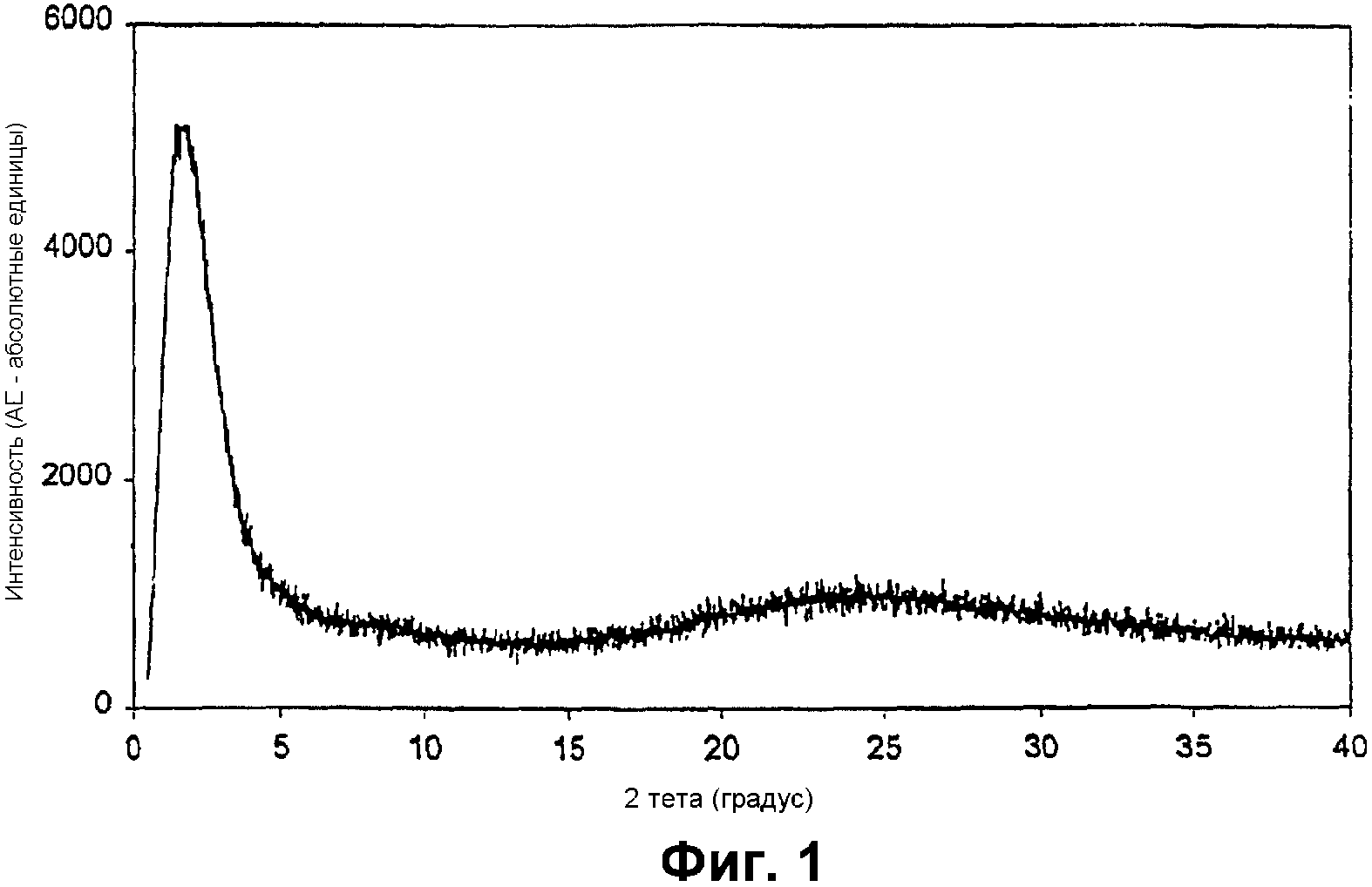
Фиг.1 представляет собой рентгенограмму ("РДГ") мезопористого материала примера 1;
Фиг.2 представляет собой снимок просвечивающего электронного микроскопа ("ПЭМ") мезопористого материала примера 1;
Фиг.3 представляет собой график, показывающий распределение размеров пор мезопористого материала примера 2;
Фиг.4 представляет собой РДГ мезопористого материала примера 2;
Фиг.5 представляет собой РДГ мезопористых материалов примеров 3А, 3В и 3С;
Фиг.6 представляет собой график, показывающий распределение размеров пор мезопористого материала примера 3А;
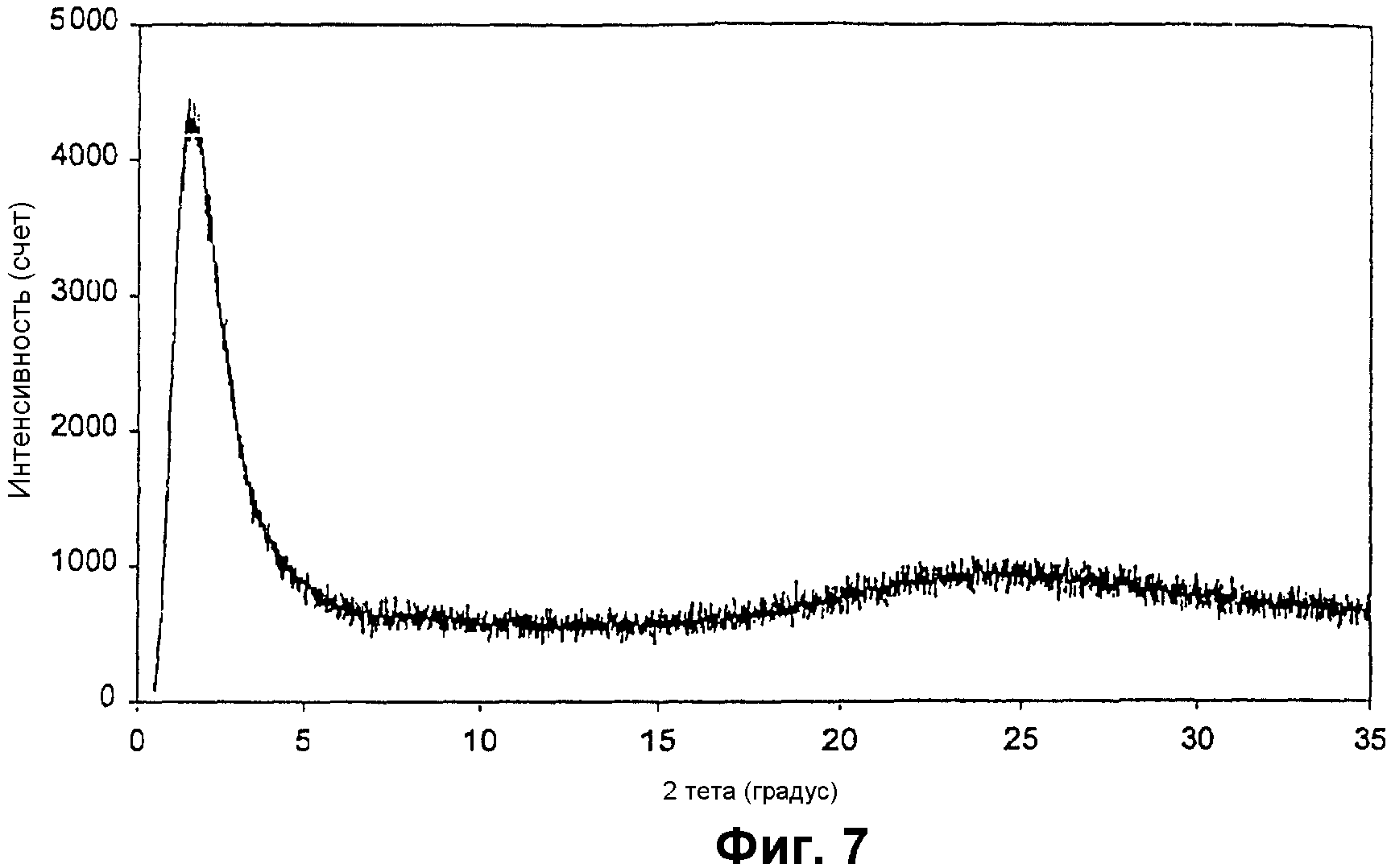
Фиг.7 представляет собой РДГ ванадийсодержащего мезопористого материала примера 5;
Фиг.8 представляет собой РДГ титансодержащего мезопористого материала примера 6;
Фиг.9 представляет собой график, показывающий изотермы сорбции азота титан-содержащего мезопористого материала примера 6;
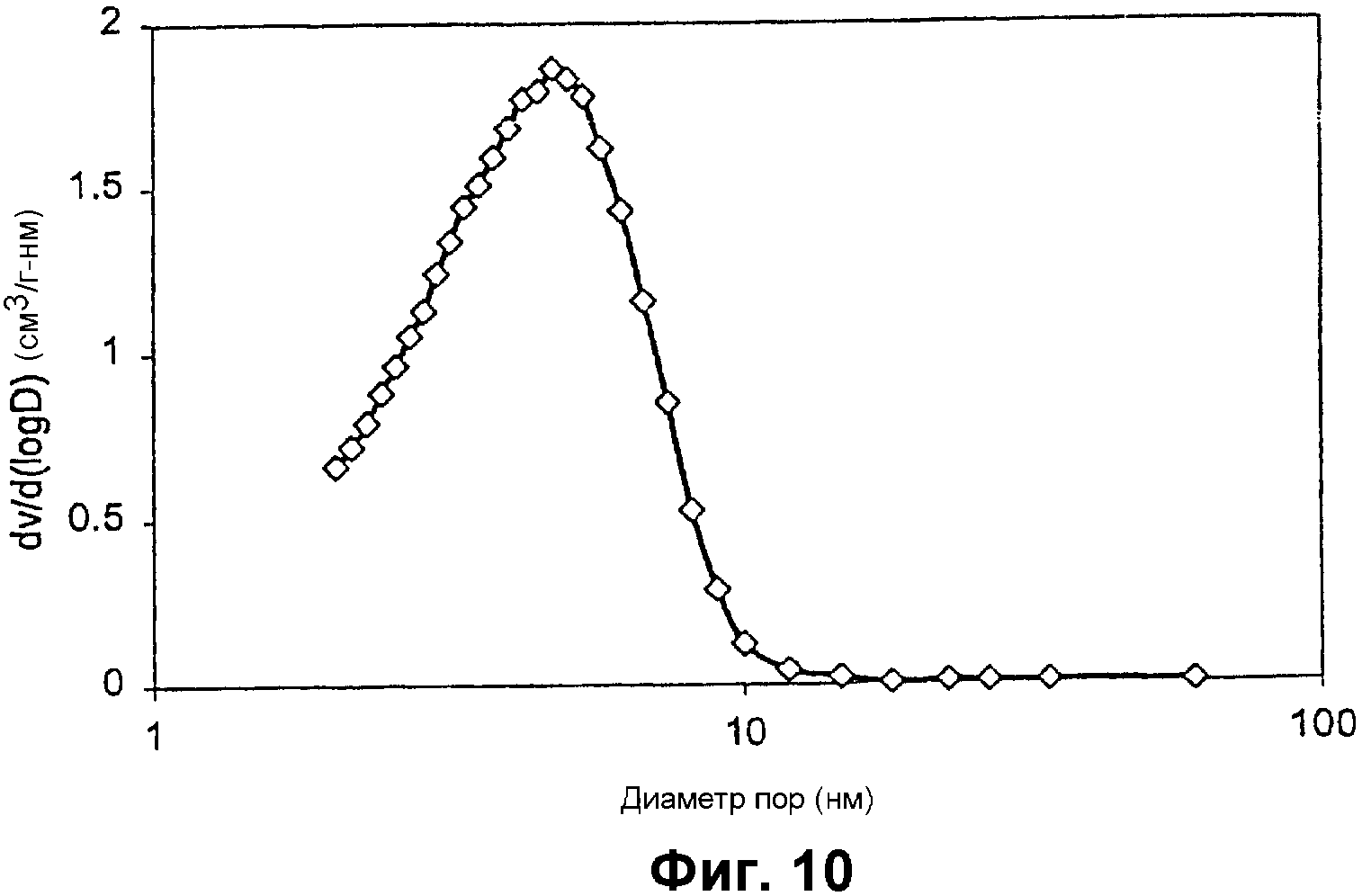
Фиг.10 представляет собой график, показывающий распределение размеров пор титан-содержащего мезопористого материала примера 6;
Фиг.11 представляет собой РДГ мезопористых материалов примеров 7, 8 и 9;
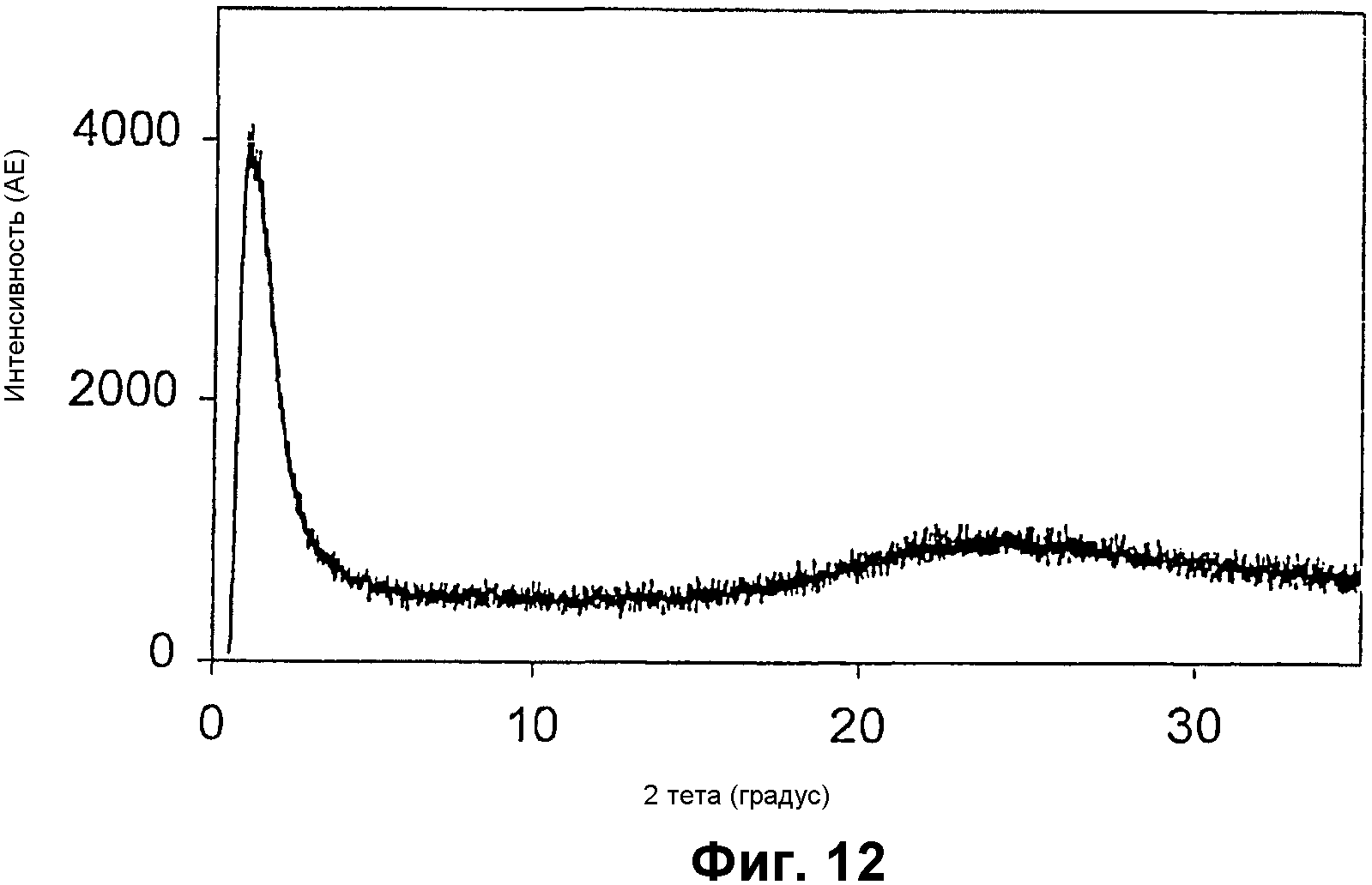
Фиг.12 представляет собой РДГ алюминий- и ванадийсодержащего мезопористого материала примера 10;
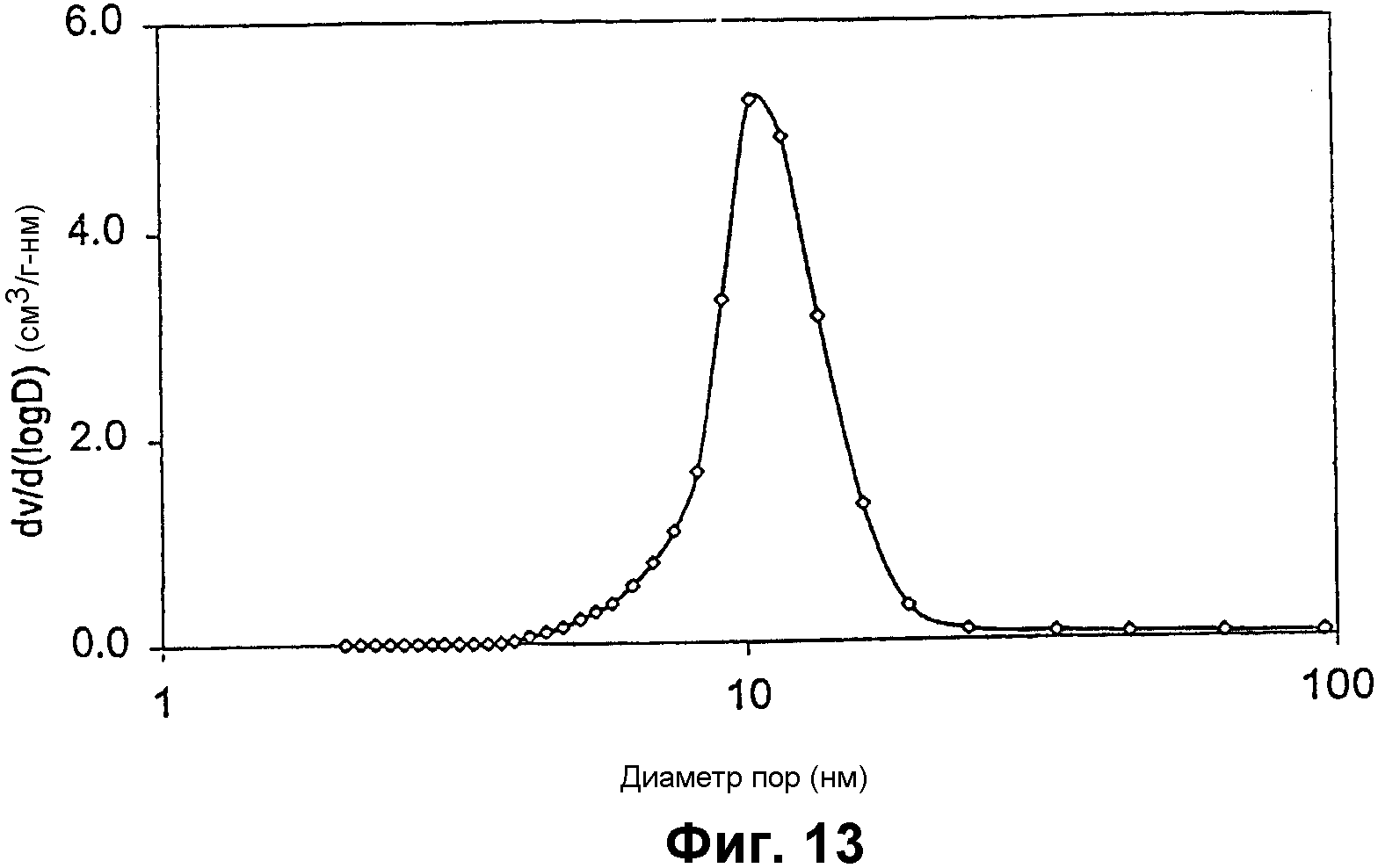
Фиг.13 представляет собой график, показывающий распределение размеров пор алюминий- и ванадийсодержащего мезопористого материала примера 10;
Фиг.14 представляет собой РДГ железосодержащего мезопористого материала примера 11;
Фиг.15 представляет спектр в УФ и видимой областях железосодержащего мезопористого материала примера 11;
Фиг.16 представляет собой график, показывающий распределение размеров пор железосодержащего мезопористого материала примера 11;
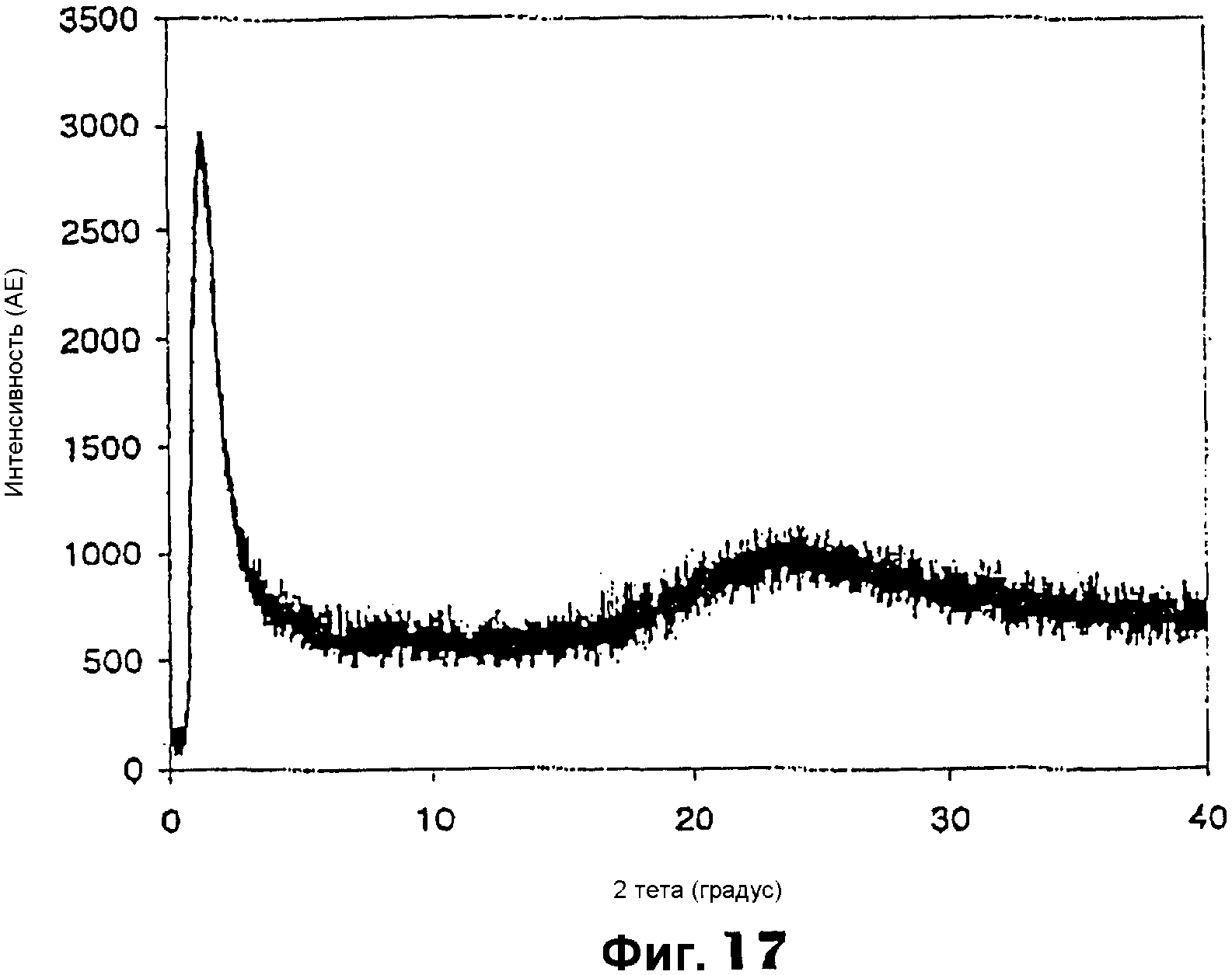
Фиг.17 представляет собой РДГ хромсодержащего мезопористого материала примера 13;
Фиг.18 представляет собой спектр в УФ и видимой областях хромсодержащего мезопористого материала примера 13;
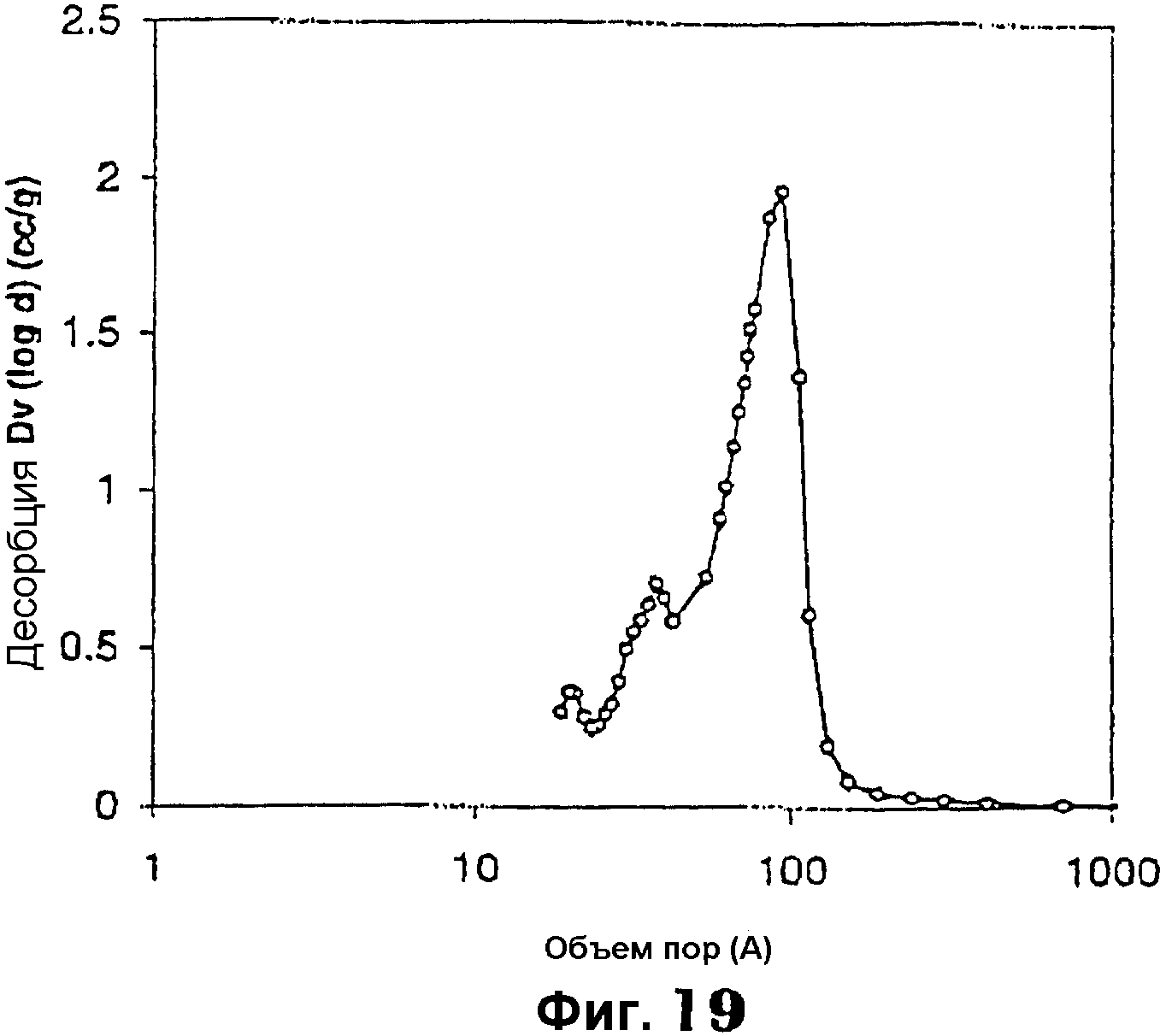
Фиг.19 представляет собой график, показывающий распределение размеров мезопор мезопористого материала примера 13;
Фиг.20 представляет собой РДГ молибденсодержащего мезопористого материала примера 15;
Фиг.21 представляет собой спектр в УФ и видимой областях мезопористого материала примера 15; и
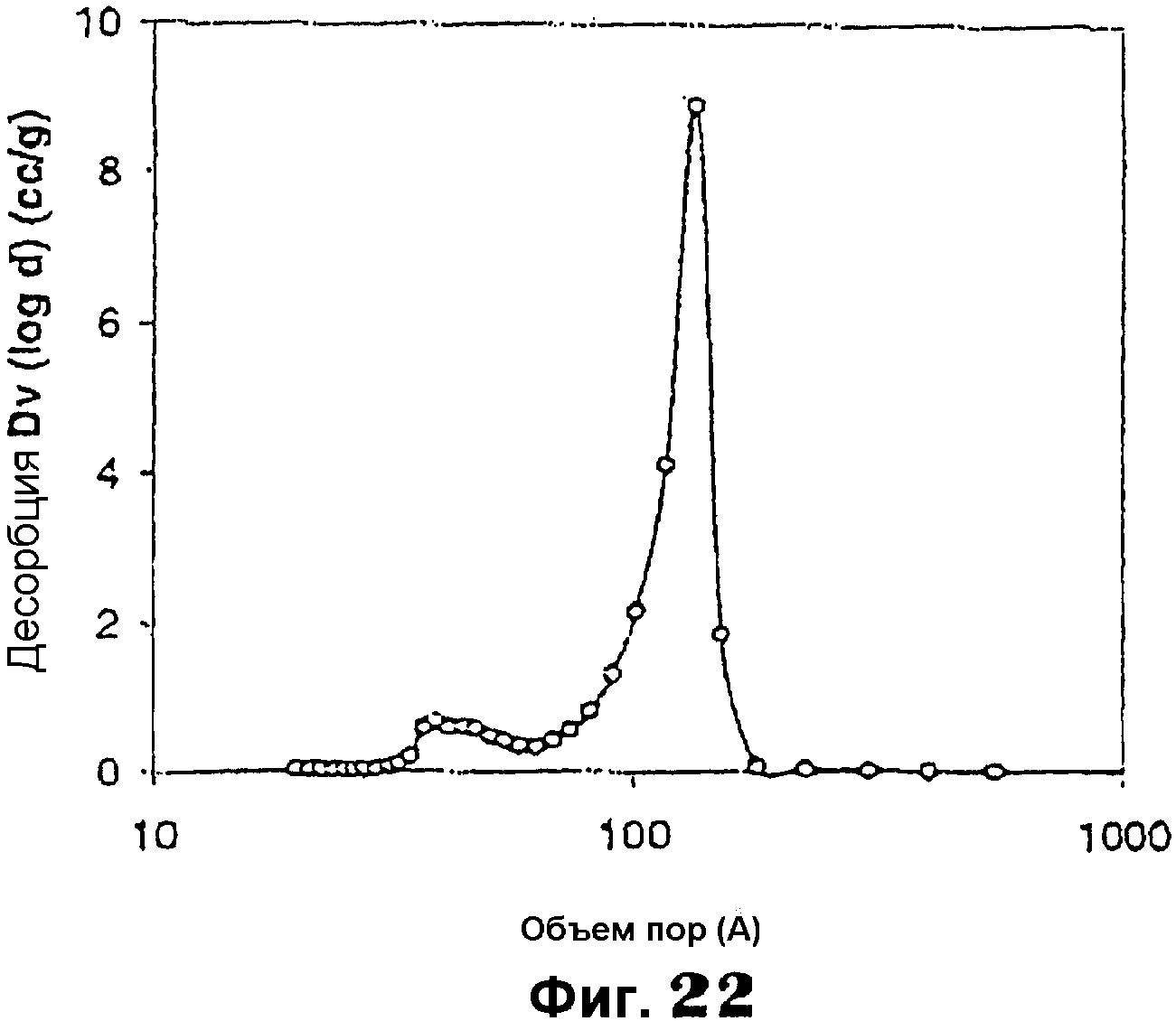
Фиг.22 представляет собой график, показывающий распределение размеров мезопор мезопористого материала примера 15.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА(ОВ) ОСУЩЕСТВЛЕНИЯ
Катализатор по настоящему изобретению включает в себя трехмерный стабильный пористый диоксид кремния с, по существу, мезопористой структурой. Данный диоксид кремния имеет некристаллическую, но регулярную (псевдокристаллическую) структуру. Мезопористые материалы описаны в патенте США №6358486 В1, который включен сюда посредством ссылки во всей своей полноте.
Аморфный материал на основе диоксида кремния по настоящему изобретению обычно содержит как мезопоры, так и микропоры. Микропоры определены как поры с диаметром менее примерно 2 нм. Мезопоры определены как поры с диаметром от примерно 2 нм до примерно 50 нм. Неорганический оксидный материал настоящего изобретения имеет объемный процент мезопор, по меньшей мере, примерно 97% и предпочтительно, по меньшей мере, примерно 98%.
Способ приготовления предпочтительного пористого каталитического носителя, содержащего диоксид кремния, описан в патенте США №6358486 В1. Средний размер мезопор предпочтительного катализатора, определенный с помощью N2-порометрии, находится в диапазоне от примерно 2 нм до примерно 25 нм.
Катализатор включает в себя, и является функционализированным, или один, или несколько гетероатомов каталитически активных металлов, внедренных в пористую силикатную структуру. Гетероатомы каталитически активных металлов (то есть не кремниевые атомы) могут быть выбраны из групп IB, IIB, IIIB, IVB, VB, VIB, VIIB, VIII, IVA и IIIA Периодической системы элементов. Подходящие гетероатомы металлов включают в себя, например, алюминий (Al), титан (Ti), ванадий (V), хром (Cr), цинк (Zn), железо (Fe), олово (Sn), молибден (Mo), галлий (Ga), никель (Ni), кобальт (Co), индий (In), цирконий (Zr), марганец (Mn), медь (Cu), магний (Mg), палладий (Pd), платину (Pt) и вольфрам (W). Внедренные гетероатомы могут быть изолированными и/или распределенными в виде кластеров в пористой матрице. Они могут быть в атомарной форме или молекулярной форме (например, в виде оксида). Содержание гетероатомов в катализаторе составляет, по меньшей мере, примерно 0,02 мас.%. Атомное отношение гетероатомов к атомам кремния в катализаторе может меняться вплоть до примерно 0,9, предпочтительно от примерно 0,0001 до примерно 0,5.
Композиция по настоящему изобретению имеет характерную рентгенограмму ("РДГ"), на которой присутствует, по меньшей мере, один пик 2θ от 0,3° до 3,5°, что соответствует межплоскостным расстояниям от 25 Å до 440 Å. Тесты с адсорбцией азота показывают, что регулируемый размер пор находится в диапазоне от примерно 15 Å (1,5 нм) до примерно 300 Å (30 нм), а площадь поверхности - в диапазоне от примерно 300 м2/г до примерно 1,250 м2/г, и объем пор - от примерно 0,3 см3/г до примерно 2,5 см3/г.
Композиция по настоящему изобретению имеет трехмерную случайно-связанную систему мезопор, которая способствует массопереносу реагентов и продуктов, и предотвращает блокировку пор.
Как правило, мезопористый материал на основе диоксида кремния по настоящему изобретению готовят из синтетической смеси, содержащей, по меньшей мере, один источник диоксида кремния, по меньшей мере, один источник гетероатомов и, по меньшей мере, один пороформирующий органический стандартный агент.
На первом этапе способа приготовления катализатора по изобретению источник диоксида кремния, источник гетероатомов и органический стандартный агент(ты) объединяют в водном растворе с образованием синтетической смеси (обычно геля).
На промежуточном этапе способа летучие компоненты синтетической смеси (например, вода, спирт) удаляются обычными способами, такими как сушка в присутствии или отсутствии принудительного потока воздуха. Сушку можно проводить, например, в диапазоне от 40°С до примерно 130°С течение до примерно 72 часов, более предпочтительно от примерно 60°С до примерно 120°С в течение от 6 до 36 часов.
На конечном этапе органический пороформирующий стандартный агент(ты) удаляют обычными способами, такими как прокаливание или экстракция. Обычно прокаливание осуществляют при температуре от примерно 450°С до примерно 900°С в кислород-содержащем газе (например, воздухе) в течение от 2 до 20 часов, предпочтительно от 540°С до примерно 700°С в течение примерно от 4 до 15 часов. Экстракцию можно проводить, используя органические растворители при температуре от примерно 30°С до примерно 100°С в зависимости от использованного растворителя. Некоторые нетоксичные или слаботоксичные спирты являются предпочтительными в качестве растворителей.
Дополнительно способ может включать в себя старение синтетической смеси при 10°С в течение до 24 часов перед удалением летучих компонентов синтетической смеси.
Дополнительно синтетическую смесь можно нагревать в автоклаве при температуре от примерно 100°С до примерно 220°С в течение примерно до 10 дней, предпочтительно при температуре от примерно 120°С до примерно 200°С в течение до 96 часов, перед удалением пороформирующего агента. Этап нагревания в автоклаве может отрегулировать мезопористость, так чтобы удовлетворять конкретным требованиям. Во время нагревания неорганические частицы, такие как кремний или алюминий, коалесцируют с образованием неорганического каркаса, в то время как пороформирующий агент образует агрегаты, придавая форму неорганическому каркасу. Распределение размеров агрегатов определяет распределение размеров мезопор. Однако размер агрегатов, в основном, зависит от природы пороформирующего агента температуры нагрева и длительности времени нагрева. Поэтому при определенном пороформирующем агенте мезопористость конечного материала может быть отрегулирована путем манипулирования температурой и временем нагрева.
В частности, на первом этапе источник диоксида кремния, или предшественник диоксида кремния, может быть кремниевым соединением, содержащим некоторые органические группы. Такие соединения могут быть алкоксидами, например тетраэтилортосиликат ("ТЭОС"), или силатранами, например триэтаноламин-замещенными силатранами. Альтернативно, источник диоксида кремния может быть неорганическим, таким как безводные или водные силикагели или гидрогели диоксида кремния. Источник диоксида кремния также может быть геотермальным кремнеземом, но для обеспечения реакционной способности предпочтителен не кристаллический источник.
Органический пороформирующий стандартный агент предпочтительно содержит гидроксильные (-ОН) группы, которые образуют водородные связи с неорганическими частицами (то есть диоксидом кремния и гетероатомом). Они могут иметь атомы с парой электронов, которая связывается с кремнием или гетероатомами. Такие органические стандартные агенты включают в себя гликоли (например, пропиленгликоль, глицерин, диэтиленгликоль, триэтиленгликоль, тетраэтиленгликоль), алканоламины (например, триэтаноламин ("ТЭА"), триизопропаноламин), дибензоат диэтилгликоля, триэтиленпентамин, крахмал и сульфолан. Органический стандартный агент должен иметь точку кипения выше 150°С, предпочтительно выше примерно 180°С.
Источник гетероатомов может содержать или не содержать органические группы и обычно добавляется в виде раствора. Например, в случае алюминия источник может быть алкоксидом алюминия (например, изопропоксид алюминия), оксидом алюминия, гидроксидом алюминия, нитратом алюминия, сульфатом алюминия или хлоридом алюминия.
Синтетическая смесь может также включать в себя щелочь или кислоту для подстройки рН смеси. Щелочи обычно включают в себя органические щелочи, такие как гидроксид тетраэтиламмония ("ТЭАОН") и другие гидроксиды тетраалкиламмония, мочевину и тому подобное, или неорганические щелочи, такие как гидроксид аммония, гидроксид натрия, карбонат натрия и тому подобное.
Растворители, условия реакции, порядок добавления и смешения компонентов и рН могут зависеть от гетероатома и должны выбираться таким образом, чтобы избежать преждевременного отделения (например, осаждения) гетероатома. Преждевременное отделение может привести к неудаче при внедрении гетероатома в структуру диоксида кремния.
Композиция по изобретению может применяться в качестве катализатора, со-катализатора (часть катализатора), каталитического носителя, адсорбента и молекулярных сит. В зависимости от функциональности внедренных гетероатомов композиция может иметь слабую, среднюю или сильную кислотность, соответственно, она может катализировать крекинг, изомеризацию, алкилирование, ацилирование, олигомеризацию/полимеризацию, дегидратирование органических соединений и десульфуризацию. Композиция также может иметь окислительно-восстановительные свойства, которые могут катализировать эпоксидирование алкенов (например, циклогексена, октена, этилена или пропилена), селективное окисление алканов (например, циклододекана, циклогексана), спиртов и аминов, гидроксилирование ароматики и аммоксимирование кетонов. Композиция может применяться в качестве со-катализаторов или каталитических носителей. Например, добавление благородного металла, например Pd и/или Pt, к данной композиции придает функциональность в гидрокрекинге, гидрировании, дегидрировании и десульфуризации. Данная композиция может также содержать все типы цеолитов и цеолитоподобных структур вместе со всеми возможными гетероатомами, упомянутыми выше.
Типичным примером композиции по изобретению, которая обладает кислотностью, является композиция, содержащая алюминий и/или галлий. Алкилирование представляет собой группу промышленно важных реакций, в которых обычно используются коррозионные кислоты Льюиса, такие как AlCl3 и HF, и производят большое количество отходов. Композиция по настоящему изобретению является экологически приемлемой и может заменить обычные катализаторы. Она может катализировать алкилирование алканов или ароматики (включая в себя алкилирование по Фриделю-Крафтсу) с применением олефинов, алкилгалогенидов или спиртов в качестве алкилирующих агентов. Ароматические соединения, главным образом, включают в себя бензол, нафталин, фенантрен и их производные, такие как толуол, ксилол, изопропилнафталин, дифенилоксид или 2,4-ди-трет-бутилфенол. Олефиновые алкилирующие агенты в основном включают в себя альфа-олефины, предпочтительно с числом атомов углерода больше двух, предпочтительно больше четырех. Подходящие олефины включают в себя, например, этилен, пропилен и 1-гексадецен. Спиртовые алкилирующие агенты в основном включают в себя метанол, этанол, изопропанол, бензиловый спирт и коричный спирт. Реакцию алкилирования можно проводить при температуре от примерно 80°С до примерно 400°С при давлении от 1 до 50 бар, предпочтительно от примерно 90°С до примерно 300°С и от 1 до 30 бар.
Олигомеризация и полимеризация олефинов может давать фракции для бензина, топлива для реактивных двигателей, дизельного топлива и смазочного базового масла. Каталитические композиции по изобретению, особенно содержащие гетероатомы алюминия, хрома, галлия или железа, могут использоваться для олигомеризации олефинов, таких как альфа-олефины с числом атомов углерода больше трех. Условия реакции, в зависимости от конкретного сырья и желаемых продуктов, включают в себя температуру в диапазоне от примерно 25°С до примерно 300°С и давление в диапазоне от атмосферного давления до примерно 70 бар.
Каталитическая композиция по данному изобретению может быть использована для селективного окисления органических соединений. Особенно предпочтительны композиции, содержащие один или несколько гетероатомов, выбранных среди переходных металлов, включая, например, медь, цинк, железо, титан, хром, ванадий, молибден и олово. Например, композиция, содержащая титан, цинк, хром, железо и марганец, может катализировать эпоксидирование олефинов, включая ароматику, такую как фенантрен, антрацен и транс-стильбен. Окислители, используемые в данном типе реакции, включают в себя органические и неорганические пероксиды, оксиды азота, кислород и любые газовые смеси, содержащие кислород. Композиция, содержащая медь и цинк, особенно предпочтительна для катализа селективного окисления спиртов в соответствующие альдегиды. Гидроксилирование фенола и 1-нафтола можно проводить, используя каталитическую композицию, содержащую олово, железо, медь, кобальт и ванадий.
В предшествующем уровне техники ацилирование ароматики обычно осуществляют, используя кислоты Льюиса, такие как AlCl3, FeCl3, H2SO4 и т.д., которые производят огромное количество отходов. Напротив, композиция по настоящему изобретению, особенно в тех ее вариантах осуществления, которые содержат алюминий, железо, галлий, индий и т.д., заменяет кислоты Льюиса. Ацилирующие агенты, главным образом, включают в себя ацилгалогениды, ангидриды карбоновых кислот. Ароматические соединения в основном включают в себя бензол, нафталин, фенантрен и их производные. Ацилирование можно проводить при температуре от примерно 40°С до примерно 300°С под давлением от примерно 0,5 бар до примерно 20 бар, предпочтительно от примерно 60°С до примерно 250°С и давлении от примерно 1 до 15 бар.
Будучи внедренными в качестве гетероатомов в мезопористый диоксид кремния по изобретению переходные металлы, такие как кобальт, никель, молибден, вольфрам или их комбинации, или благородные металлы, такие как платина, палладий или их комбинации, обеспечивают катализаторы, особенно подходящие для способа гидроочистки, такого как (1) гидрирование ароматики в бензине, топливе для реактивных двигателей, дизельном топливе и смазочном масле; (2) гидрокрекинг тяжелых фракций, таких как вакуумный газойль, остаточные фракции и жидкости, производимые из угля (угольная нефть); (3) снижение КУО, деазотирование, десульфуризация и деметаллирование углеводородов, включая вышеупомянутые фракции. Деметаллирование особенно подходит для удаления железа, никеля, ванадия и мышьяка. Условия реакции гидроочистки обычно включают в себя температуру реакции в диапазоне от примерно 60°С до примерно 350°С и давление в диапазоне от атмосферного давления до примерно 300 бар.
Изомеризация углеводородов (например, н-бутана, н-пентана, 1-бутена и ксилола) может катализироваться путем применения катализатора по данному изобретению. Предпочтительные каталитические композиции для изомеризации содержат цирконий, вольфрам, галлий, железо, титан и алюминий в качестве гетероатомов.
Дегидрирование насыщенных углеводородов до ненасыщенных углеводородов может катализироваться с применением композиции, содержащей, главным образом, ванадий, железо, галлий, кобальт и хром. Насыщенный углеводород может быть, например, пропаном, изобутаном и этилбензолом. Часовая объемная скорость газа (ЧОСГ) обычно находится в диапазоне от 100 до 2000 ч-1, предпочтительно от 500 до 1000 ч-1. Рабочее давление обычно находится в диапазоне от примерно 7 кПа до примерно 600 кПа, предпочтительно от примерно 7 кПа до примерно 400 кПа. Температура реакции обычно составляет от примерно 350°С до примерно 650°С, предпочтительно от примерно 450°С до примерно 600°С.
Крекинг углеводородов может преимущественно осуществляться с использованием изобретенной каталитической композиции, содержащей никель, вольфрам, молибден, алюминий и/или галлий. Кроме того, каталитическая композиция по данному изобретению может быть использована одна или вместе с цеолитами. Углеводород может быть сырьем для крекинга с псевдоожиженным катализатором, гидрокрекинга и т.д. Данная каталитическая композиция может также катализировать крекинг полимерных отходов для возврата полезных фракций желаемых химических веществ.
Композиция может быть использована в качестве катализатора для способа Фишера-Тропша. Данный способ включает в себя взаимодействие исходного потока, содержащего водород и монооксид углерода, с катализатором в реакционной зоне, поддерживающей ускоряющие конверсию условия, эффективные для получения выходящего потока, содержащего углеводороды. Часовая объемная скорость подачи (ЧОСП) газового потока может находиться в диапазоне от примерно 100 объемов/час/объем катализатора (ч-1) до примерно 10000 ч-1, предпочтительно от примерно 300 ч-1 до примерно 2000 ч-1. Температура реакции обычно составляет от примерно 160°С до примерно 300°С, предпочтительно от примерно 190°С до примерно 260°С. Реакционное давление обычно составляет от примерно 5 бар до примерно 60 бар, предпочтительно от 8 бар до примерно 30 бар.
Композиция может быть использована для эффективной и селективной адсорбции отдельных соединений. Благодаря своим регулируемым порам и функционализированным стенкам пор композиция позволяет различным соединениям проникать в поры и взаимодействовать с функциональными гетероатомными группами на или в стенке. Например, внедренные гетероатомы могут иметь высокие, но ненасыщенные координационные числа, которые позволяют гетероатомам образовывать координационные связи с кислородсодержащими, азотсодержащими и серасодержащими соединениями, таким образом эффективно убирая данные соединения из потоков. Взаимодействие также может быть кислотно-основным. Например, композиция, содержащая алюминий, может удалять из потоков токсичные соединения, такие как циануровая кислота и п-хлорфенол. По существу, композиции могут быть использованы в качестве адсорбентов и молекулярных сит.
В описании настоящего изобретения представлен новый тип мезопористого или мезо-микропористого силиката, содержащего гетероатомы, со случайно-связанной трехмерной структурой пор с регулируемым размером пор. Оно предлагает новый экономичный способ синтеза мезопористого силиката без включения какого-либо поверхностно-активного вещества. И оно обеспечивает способ применения данной композиции в катализе и разделении.
Различные признаки изобретения иллюстрируются примерами, приведенными ниже. Рентгеновские дифрактограммы (РДГ) порошков полученных материалов регистрировали с использованием CuKα излучения на дифрактометре Philips PW 1840 с графитовым монохроматором. Образцы сканировали в диапазоне 2θ от 0,5 до 40° с шагом 0,02°. Просвечивающую электронную микроскопию (ПЭМ) осуществляли с использованием электронного микроскопа Philips CM30T с LaB6 нитью в качестве источника электронов, работавшей при 300 кВ. Изотермы адсорбции азота измеряли на Quantachrome Autosorb-6B при 77К. Мезопористость вычисляли с применением метода Barrett, Joyner и Halenda (BHJ). Все части композиций даны по массе, если не оговорено особо.
ПРИМЕР 1
Данный пример показывает, как внедрить алюминий в диоксид кремния без нагрева в автоклаве перед прокаливанием.
Сначала 1 часть изопропоксида алюминия (Al(изо-ОС3Н6)3) добавляли к 26 частям водного раствора гидроксида тетраэтиламмония (ТЭАОН, 35%) при перемешивании. После растворения добавляли в упомянутый раствор при перемешивании 38 частей триэтаноламина (ТЭА) вместе с 8 частями воды. Затем при энергичном перемешивании добавляли 26 частей тетраэтилортосиликата (ТЭОС). Получали прозрачный раствор. Перемешивание продолжали в течение 1 часа и затем синтетическую смесь оставляли при комнатной температуре на ночь и сушили при 98°С на воздухе в течение 24 часов. В конце синтетическую смесь прокаливали при 570°С в течение 10 часов на воздухе со скоростью нагрева 1°С/мин.
Фиг.1 показывает ее РДГ с интенсивным отражением при примерно 1,1° для 2θ, характерным для мезопористого материала. К тому же отсутствие разрешенных пиков от оксида алюминия означает, что фаза объемного оксида алюминия не образуется. Фиг.2 представляет собой снимок просвечивающей электронной микроскопии (ПЭМ), показывающий случайно-связанную мезопористую структуру. Элементный анализ показал отношение Si/Al примерно 24,8, что согласуется с отношением в исходной синтетической смеси, равным 25. Адсорбция азота дает площадь поверхности 983 м2/г, суммарный объем пор 1,27 см3/г и узкое распределение мезопор с центром при 4,2 нм, показанное на фиг.3.
ПРИМЕР 2
Данный пример демонстрирует внедрение гетероатомов с помощью нагрева в автоклаве перед прокаливанием. 3,3 части изопропоксида алюминия добавляли в колбу с 42 частями ТЭОС и перемешивали в течение часа. Смесь 7,6 частей ТЭА и 25,8 частей воды добавляли в смесь ТЭОС и Al(изо-OC3Н6)3 при перемешивании. После 2-часового перемешивания 21 часть ТЭАОН по каплям добавляли в вышеупомянутую смесь, и образовывался плотный гель. Гель высушивали в сушильном шкафу при 98°С в течение 22 часов и затем выдерживали в автоклаве при 190°С в течение 16 часов. В конце гель прокаливали при 600°С в течение 10 часов на воздухе.
Фиг.4 показывает его РДГ с интенсивным отражением при малом угле в 2θ, характерном для мезопористого материала. Элементный анализ показал отношение Si/Al примерно 24,5, что согласуется с отношением в исходной синтетической смеси, равным 25. Адсорбция азота дает площадь поверхности 799 м2/г, суммарный объем пор 1,24 см3/г и узкое распределение мезопор с центром при 4,5 нм.
ПРИМЕР 3А
Пример демонстрирует внедрение алюминия и его стабильность в композиции. 3 части изопропоксида алюминия добавляли в колбу с 38,8 частями ТЭОС и перемешивали в течение 1,5 часов. Смесь 23 частей ТЭА и 21 часть воды добавляли в вышеуказанную смесь при перемешивании. После 2 часового перемешивания 23 части ТЭАОН по каплям добавляли в вышеупомянутую смесь, и после 0,5 часового перемешивания она превращалась в прозрачный раствор. Раствор высушивали в сушильном шкафу при 100°С в течение 4 дней и затем выдерживали в автоклаве при 190°С в течение 7,5 дней. В конце его прокаливали при 600°С в течение 10 часов на воздухе при скорости нагрева 1°С/мин.
Элементный анализ показал отношение Si/Al, равное 99,2. Фиг.5 показывает его РДГ с интенсивным пиком. Адсорбция азота показывает узкое распределение мезопор с центром при 17 нм, как показано на фиг.6, которое демонстрирует площадь поверхности примерно 385 м2/г и объем пор примерно 1,32 см3/г.
ПРИМЕР 3В
Материал, полученный в примере 3А, кипятили в воде в течение 17 часов, но его РДГ, изображенная на фиг.5, все еще показывает интенсивный пик, подобный пику в исходном материале. Это означает, что данная композиция имеет высокую гидротермальную стабильность по сравнению с другими мезопористыми материалами.
ПРИМЕР 3С
Материал, полученный в примере 3А, прокаливали при 900°С на воздухе, но его РДГ (фиг.5) все еще показывает интенсивный пик, демонстрируя, что мезопористая структура сохранилась. Данный результат означает, что данная композиция имеет высокую термическую стабильность вплоть до 900°С.
ПРИМЕР 4
Это пример использования неорганических источников гетероатомов для внедрения алюминия в диоксид кремния. 7,2 частей нонагидрата нитрата алюминия растворяли в 20 частях воды. Затем добавляли 61,4 частей ТЭОС и перемешивали в течение 0,5 часа. Другую смесь из 56,3 частей тетраэтиленгликоля и 24 частей воды добавляли в вышеуказанную смесь при перемешивании. После 1-часового перемешивания добавляли 49 частей водного раствора гидроксида тетраэтиламмония (ТЭАОН, 35% мас.), и после 0,5-часового перемешивания конечная смесь превращалась в плотный гель. Гель сушили в сушильном шкафу при 100°С в течение ночи и затем выдерживали в автоклаве при 180°С в течение 3 часов. В конце его прокаливали при 600°С в течение 10 часов на воздухе при скорости нагрева 1°С/мин.
Элементный анализ показал отношение Si/Al, равное 15,3. Его РДГ показала интенсивный пик около 1 градуса в 2θ. Адсорбция азота показала узкое распределение мезопор с центром при 4,5 нм, удельную площадь поверхности примерно 786 м2/г и полный объем пор примерно 1,02 см3/г.
ПРИМЕР 5
Пример иллюстрирует внедрение ванадия в диоксид кремния. 1 часть ацетилацетоната ванадия (IV) добавляли в колбу с 41 частью ТЭОС и перемешивали в течение 2 часов. Смесь 30 частей ТЭА и 25 частей воды добавляли в вышеупомянутую смесь при перемешивании. После 2-часового перемешивания 20 частей ТЭАОН по каплям добавляли в вышеупомянутую смесь, и после 0,5 часового перемешивания она превращалась в твердый гель. Гель выдерживали при комнатной температуре в течение 24 часов и сушили в сушильном шкафу при 100°С в течение ночи, и затем прокаливали при 700°С в течение 10 часов на воздухе, и в конце он превращался в оранжевый порошок.
Элементный анализ показал отношение Si/V, равное 50,5. Фиг.7 показывает его РДГ с интенсивный пиком для мезоструктуры и без каких-либо пиков от фаз оксида ванадия. Адсорбция азота показала узкое распределение мезопор с центром при 4,1 нм, удельную площадь поверхности примерно 835 м2/г и объем пор примерно 0,91 см3/г.
ПРИМЕР 6
В данном примере демонстрируется внедрение титана. 1 часть бутоксида титана (IV) добавляли в колбу с 31 частью ТЭОС и перемешивали в течение 2 часов. Смесь 22,5 частей ТЭА и 17 частей воды добавляли в вышеупомянутую смесь при перемешивании. После 1-часового перемешивания 18,5 частей ТЭАОН по каплям добавляли в вышеупомянутую смесь, и после 0,5 часового перемешивания она превращалась в плотный гель. Гель выдерживали при комнатной температуре в течение 22 часов и сушили в сушильном шкафу при 98°С в течение ночи, и затем прокаливали при 700°С в течение 10 часов на воздухе, и в конце она превращалась в белый порошок.
Элементный анализ показал отношение Si/Ti, равное 49,6. Фиг.8 показывает его РДГ с интенсивный пиком для мезоструктуры и без разрешенных пиков от оксида титана. Изотермы адсорбции азота показаны на фиг.9, которая демонстрирует распределение мезопор с центром при 4,7 нм, показанное на фиг.10, удельную площадь поверхности примерно 917 м2/г и суммарный объем пор примерно 0,84 см3/г.
ПРИМЕРЫ 7-9
В данных примерах демонстрируется внедрение трех различных гетероатомов. 42 части тетраэтилортосиликата (ТЭОС) смешивали с 30 частями триэтаноламина (ТЭА) в течение 1 часа, получая смесь I. Смесь II готовили растворением источников гетероатомов в 22 частях воды. 1 часть нитрата галлия, 0,54 части хлорида цинка и 0,9 частей хлорида олова использовали в примерах 7,8 и 9 соответственно. Смесь II по каплям добавляли к смеси I при перемешивании. После этого объединенные смеси I и II перемешивали в течение 0,5 часа и во время перемешивания по каплям добавляли 24,5 частей гидроксида тетраэтиламмония. После перемешивания в течения 2 часов каждая из трех смесей становилась прозрачным раствором, и в конце добавляли 0,5 г гидроксида аммония (27-30 мас.%). После перемешивания в течение еще 2 часов смеси статически выдерживали в течение ночи. Смеси высушивали при 98°С в течение 24 часов, и каждая превращалась в высушенный гель. Высушенные гели выдерживали в автоклаве при 180°С в течение 2,5 часов и в конце прокаливали при 600°С на воздухе в течение 10 часов.
Фиг.11 показывает РДГ галлий-, цинк- и оловосодержащих силикатов, полученных в примерах 7, 8 и 9 соответственно. Таблица 1 представляет мезопористость и химический состав трех материалов.
ПРИМЕР 10
Данный пример демонстрирует одновременное внедрение двух типов гетероатомов в диоксид кремния. Сначала 2,7 частей изопропоксида алюминия смешивали с 0,86 частями ацетилацетоната ванадия (IV) и 34 частями тетраэтилортосиликата (ТЭОС) для получения первой смеси. Вторая смесь содержала 34 части ТЭА и 21 часть воды. Затем вторую смесь по каплям добавляли в первую смесь при перемешивании. После перемешивания в течение 1,5 часов по каплям добавляли 16,8 частей гидроксида тетраэтиламмония при перемешивании. Синтетическая смесь превращалась в плотный гель. Данный гель статически выдерживали в течение ночи при комнатной температуре, сушили при 100°С в течение 42 часов и затем нагревали в автоклаве при 180°С в течение 3 дней. В конце его прокаливали при 650°С на воздухе в течение 10 часов.
Фиг.12 показывает РДГ алюминий- и ванадийсодержащего силиката. Адсорбция азота показала, что композиция имеет узкое распределение пор около 11 нм (показанное на фиг.13), площадь поверхности примерно 433 м2/г и суммарный объем пор примерно 1,25 см3/г. Элементный анализ показал, что Si/Al=13,5 и Si/V=49,1.
ПРИМЕР 11
Данный пример демонстрирует получение Fe-содержащего мезопористого силиката. Одну часть нитрата железа (III) растворяли в 5 частях деионизированной воды и затем добавляли к 27,4 частям тетраэтилортосиликата (ТЭОС), и перемешивали в течение 1 часа. Другой раствор, состоящий из 19,8 частей триэтаноламина (ТЭА) и 30,4 частей деионизированной воды, вводили по каплям в первую смесь. После еще 1 часа перемешивания к данной смеси по каплям добавляли 16,2 частей гидроксида тетраэтиламмония (ТЭАОН). Конечный гомогенный бледно-желтый раствор выдерживали при комнатной температуре в течение 24 часов, сушили при 100°С в течение 24 часов и в конце прокаливали при 650°С в течение 10 часов, получая бледно-желтый порошок.
Фиг.14 показывает РДГ с одним интенсивным пиком при малом угле примерно от 0,5 до 2,2°, указывающем на мезоструктурные характеристики. Элементный анализ показал атомное отношение Si/Fe 48,8. Спектроскопия в УФ-видимой области (фиг.15) показала пик около 220 нм, характерный для четырехкоординированного железа, а также плечо в диапазоне от 250 до 350 нм, характерное для октаэдрической координации железа в матрице диоксида кремния. Измерения адсорбции N2 дали площадь поверхности по БЭТ примерно 630 м2/г, средний диаметр мезопор примерно 4,8 нм (сравни фиг.16) и суммарный объем пор примерно 1,24 см3/г.
ПРИМЕР 12
Методика приготовления Fe-содержащего силиката подобна методике в примере 11; однако использовали только 0,52 части нитрата железа (III). После прокаливания элементный анализ показал, что порошок имеет атомное отношение Si/Fe 98,6. Адсорбция азота показала удельную площадь поверхности 580 м2/г, средний диаметр пор 5,96 нм и объем пор 1,82 см3/г.
ПРИМЕР 13
Данный пример демонстрирует получение Cr-содержащего силиката. 1,2 части нонагидрата нитрата хрома растворяли в 5 частях деионизированной воды и затем добавляли к 26,3 частям тетраэтилортосиликата (ТЭОС) и перемешивали в течение 1 часа. Другой раствор, состоящий из 19 частей триэтаноламина (ТЭА) и 22,2 частей деионизированной воды, по каплям вводили в вышеупомянутый раствор. После перемешивания в течение еще 1 часа к данной смеси добавляли по каплям 26,2 части гидроксида тетраэтиламмония. Конечный гомогенный бледно-зеленый раствор выдерживали при комнатной температуре в течение 24 часов, сушили при 100°С в течение 24 часов и в конце прокаливали при 650°С в течение 10 часов, получая желтовато-оранжевый порошок, содержащий хром.
Фиг.17 показывает РДГ с одним интенсивным пиком при малом угле примерно от 0,5 до 2,2°, указывающем на мезоструктурные характеристики. Спектроскопия в УФ-видимой области (фиг.18) показала два различимых пика около 220 и 390 нм, характерных для четырех координированного хрома, а также плечо около 480 нм, характерное для октаэдрической координации полихромата (-Cr-O-Cr-)n в матрице диоксида кремния. Измерения адсорбции N2 дали площадь поверхности по БЭТ примерно 565 м2/г, средний диаметр мезопор 1,96 нм (сравни фиг.19) и суммарный объем пор примерно 1,54 см3/г.
ПРИМЕР 14
Методика получения Cr-мезопористого силиката подобна методике в примере 13; однако использовали 1,31 часть нитрата хрома. После прокаливания элементный анализ показал, что порошок имеет атомное отношение Si/Cr 40,3. Адсорбция азота показала удельную площадь поверхности 572 м2/г, диаметр пор 2,35 нм и объем пор 1,7 см3/г.
ПРИМЕР 15
В данном примере демонстрируется получение композиции, содержащей Мо. 1,6 частей тетрагидрата гептамолибдата аммония [(NH4)6Mo7O24·4H2O] растворяли в 5 частях деионизированной воды и затем добавляли к 27,1 части тетраэтилортосиликата (ТЭОС), и перемешивали в течение 1 часа. В вышеупомянутый раствор добавляли по каплям другой раствор, состоящий из 19,6 частей триэтаноламина (ТЭА) и 30,4 частей деионизированной воды. После другого перемешивания в течение 1 часа 16,1 часть гидроксида тетраэтиламмония (ТЭАОН) добавляли по каплям к данной смеси. Конечный гомогенный бледно-желтый раствор выдерживали при комнатной температуре в течение 24 часов, сушили при 100°С в течение 24 часов и в конце прокаливали при 650°С в течение 10 часов, получая белый порошок.
Фиг.20 показывает РДГ с одним интенсивным пиком при малом угле примерно от 0,5 до 2,2°, указывающем на мезоструктурные характеристики. Спектроскопия в УФ-видимой области (фиг.21) показывает пик около 220 нм, характерный для четырехкоординированного молибдена в матрице диоксида кремния. Измерения адсорбции N2 дали площадь поверхности по БЭТ примерно 500 м2/г, средний диаметр мезопор примерно 8,91 нм (сравни фиг.22) и суммарный объем пор примерно 1,31 см3/г.
ПРИМЕР 16
Методика получения Мо-мезопористого силиката подобна методике в примере 15; однако использовали 3,9 части тетрагидрата гептамолибдата аммония [(NH4)6Mo7O24·4H2O]. После прокаливания элементный анализ показал, что порошок имеет атомное отношение Si/Мо 39,8. Адсорбция азота показала удельную площадь поверхности 565 м2/г, средний диаметр пор 3,93 нм и объем пор 0,98 см3/г.
ПРИМЕР 17
Демонстрируется одновременное внедрение как Ni, так и Мо в мезопористый материал. Сначала 7,7 частей гексагидрата нитрата никеля (II) и 32 части тетрагидрата гептамолибдата аммония растворяли в 54 частях воды при перемешивании. Затем добавляли к вышеупомянутому раствору при интенсивном перемешивании 67 частей тетраэтилортосиликата (ТЭОС). После перемешивания в течение 1,5 часов добавляли по каплям при перемешивании 40 частей гидроксида тетраэтиламмония (ТЭАОН). Синтетическая смесь превращалась в плотный гель. Данный гель неподвижно выдерживали при комнатной температуре в течение ночи, сушили при 100°С в течение 24 часов и затем нагревали в автоклаве при 180°С в течение 3 часов. В конце синтетическую смесь прокаливали при 600°С на воздухе в течение 10 часов.
РДГ конечного порошка показывает интенсивный пик около 1,1° для 2θ, указывающий на характеристики мезопористого материала. Адсорбция азота показывает, что материал имеет узкое распределение пор около 2,3 нм, площадь поверхности, равную примерно 633 м2/г, и суммарный объем пор примерно 0,86 см3/г. Элементный анализ показывает, что конечный порошок содержит 6,1 мас.% Ni и 10,5% Mo.
ПРИМЕР 18
Данный пример демонстрирует одновременное введение как Ni, так и W в мезопористый материал. Сначала 5,8 частей гексагидрата нитрата Ni (II) и 35 частей гидрата метавольфрамата аммония растворяли при перемешивании в 42,3 частях воды. Затем в полученный раствор добавляли 50,5 частей тетраэтилортосиликата (ТЭОС) при энергичном перемешивании. После перемешивания в течение 1,5 часов добавляли по каплям гидроксид тетраэтиламмония при перемешивании. Синтетическая смесь превращалась в плотный гель. Гель выдерживали неподвижно при комнатной температуре в течение ночи, сушили при 100°С в течение 24 часов и затем выдерживали в автоклаве при 180°С в течение 3 часов. Окончательно его прокаливали на воздухе при 600°С в течение 10 часов.
РДГ конечного порошка показывает интенсивный пик около 1,0° для 2θ, указывающий на характеристики мезопористого материала. Адсорбция азота показывает, что материал имеет узкое распределение пор около 2,4 нм, площадь поверхности примерно 649 м2/г и суммарный объем пор примерно 0,81 см3/г. Элементный анализ показывает, что конечный порошок содержит 6,4 мас.% Ni и 12,0% W.
ПРИМЕР 19
Было продемонстрировано получение палладийсодержащих катализаторов. 65 частей материала по примеру 1 смешивали с 35 частями оксида алюминия и к данной смеси добавляли воду, так, чтобы полученный катализатор можно было экструдировать. Катализатор прокаливали при 480°С в токе азота 5 об./об./мин в течение 6 часов с последующей заменой тока азота на воздух 5 об./об./мин. Прокаливание заканчивали при подъеме температуры до 540°С и выдерживании при этой температуре в течение 12 часов. Палладий вводили пропиткой водным раствором тетрааминовой соли палладия, Pd(NH3)4Cl2. Экструдат сушили при 120°С в течение ночи и прокаливали при 300°С на воздухе в течение 3 часов. Конечный катализатор имел 0,81 мас.% палладия, площадь поверхности 630 м2/г, плотность частиц 0,83 г/мл и объем пор 1,21 см3/г.
ПРИМЕР 20
Алкилирование нафталина 1-гексадеканом проводили в колбе с механической мешалкой. Использовали катализаторы примеров 1, 2 и 3А. 1 часть катализатора загружали в колбу и нагревали вплоть до 200°С в вакууме в течение 2 часов. Затем катализатор охлаждали до 90-100°С в атмосфере азота и смесь, состоящую из 6,5 частей мас. нафталина и 26 частей 1-гексадекана, вводили в колбу при перемешивании. Температуру повышали до 200-205°С и держали постоянной. Реакционную смесь анализировали с помощью газовой хроматографии с использованием колонки WAX 52 CB. Результаты реакции с использованием различных катализаторов суммированы в таблице 2.
ПРИМЕР 21
Алкилирование по Фриделю-Крафтсу бензола с хлорбензолом проводили в колбе с магнитной мешалкой. Использовали катализаторы примеров 7, 9, 11 и 12. 1 часть катализатора загружали в колбу и нагревали до 180°С в вакууме в течение 4 часов. Затем катализатор охлаждали до 80°С в атмосфере азота и в колбу вводили смесь, состоящую из 102 частей бензола и 8,2 частей бензилхлорида. Температуру держали постоянной, равной 60°С или 80°C. Реакционную смесь анализировали с помощью газовой хроматографии с использованием колонки WAX 52 СВ. Результаты реакции с использованием различных катализаторов суммированы в таблице 3.
ПРИМЕР 22
Селективное окисление этилбензола до ацетофенона проводили в колбе в атмосфере азота при перемешивании. Использовали катализаторы примеров 13, 14 и 16. Одну часть катализатора активировали при 180°С в течение 4 часов в вакууме и затем охлаждали до 80°С. Затем смесь, состоящую из 100 частей ацетона, 82 частей этилбензола и 9,5 частей трет-бутилгидропероксида (ТБГП), вводили в колбу при перемешивании. Реакционную смесь анализировали с помощью газовой хроматографии с использованием колонки WAX 52 CB. Результаты реакции с использованием различных катализаторов суммированы в таблице 4.
ПРИМЕР 23
Олигомеризацию 1-децена проводили в реакторе периодического действия с перемешиванием. 1 часть катализатора активировали в реакторе при нагревании в атмосфере азота при 200°С в течение 2 часов. 25 частей 1-децена добавляли шприцем в потоке азота. Реакцию проводили при 150°С в течение 24 часов. После охлаждения реактора продукт анализировали с помощью газовой хроматографии (ГХ) с использованием колонки WAX 52 CB. Для каждого теста % мол. конверсии децена и селективность димера представлены в таблице 5.
ПРИМЕР 24
Ацилирование 2-метоксинафталина до 2-ацетил-6-метоксинафталина проводили в реакторе периодического действия с перемешиванием. Реактор с 16 частями катализатора, полученного в примере 2, выдерживали при 240°С в течение 2 часов в вакууме и затем заполняли сухим азотом. После охлаждения реактора до 120°С в реактор вводили 250 частей декалина (как растворитель), 31 часть 2-метоксинафталина, 42 части уксусного ангидрида и 10 частей н-тетрадекана (как внутренний стандарт). После протекания реакции в течение 6 часов реакционную смесь анализировали с помощью (ГХ) с использованием колонки WAX 52 CB, и было найдено, что конверсия 2-метоксинафталина достигает 36,5% с 100% селективностью по 2-ацетил-6-метоксинафталину.
ПРИМЕР 25
Окисление циклогексанола до циклогексанона проводили в реакторе периодического действия с перемешиванием. Реактор с 1 частью катализатора выдерживали при 180°С в вакууме в течение 4 часов и затем заполняли сухим азотом. После охлаждения до 55°С в реактор вводили 100 частей ацетона, 10 частей трет-бутилгидропероксида (ТБГП) и 7,5 частей циклогексанола; температуру протекания реакции поддерживали равной 55°С. После 5 часов протекания реакции реакционную смесь анализировали с помощью (ГХ) с использованием колонки WAX 52 CB, рабочие характеристики различных катализаторов суммированы в таблице 6.
ПРИМЕР 26
Материал примера 17 оценивали в отношении модернизации горючего сланца Paraho при 68 бар H2 и ЧОСП, равном 2. Температуру реакции варьировали от 260 до 400°С. Свойства горючего сланца представлены в таблице 7. Свойства продукта после модернизации представлены в таблице 8.
Оценка показала, что каталитический материал примера 17 является очень активным для насыщения олефинов, удаления железа и никеля, деазотирования и десульфирования. Материал также очень активен для удаления мышьяка.
ПРИМЕР 27
Селективный гидрокрекинг смазочного материала осуществляли на Ni- и W-содержащем мезопористом материале вышеприведенного примера 18. Исходное сырье состояло из тяжелого нейтрального дистиллята со свойствами, представленными ниже в таблице 9 вместе со свойствами масла после депарафинизации растворителем до температуры застывания -18°С (ASTM D-97 или эквивалентный, такой как Autopour). После депарафинизации растворителем содержание азота составляло 1500 м.д. и индекс вязкости дистиллята («ИВ») составил 53. Целью гидрокрекинга смазочного материала является увеличение уровня ИВ не превращенного материала до значений в интервале от 95 до 100 при максимизации выхода смазочного материала.
Дистиллят обрабатывали при температурах от 385 до 401°С, давлении водорода 138 бар, циркуляции водорода 7500 SCF/B feed и от 0,55 до 0,61 ЧОСП. Данные этих экспериментов суммированы в таблице 10 ниже:
Каталитический материал является селективным для модернизации тяжелого нейтрального дистиллята из сырого дистиллята с ИВ от 53 до продукта с ИВ, равным 105, с выходом смазочного материала (не депарафинизированного), равным 65% мас.
ПРИМЕР 28
Данный пример демонстрирует получение катализатора FCC с использованием композиции по данному изобретению и сравнение результатов его крекинга с результатами, полученными при использовании катализатора MCM-41. Получение катализатора было следующим.
Получали примерно 35% мас. композиции примера 4 в матрице диоксид кремния - оксид алюминия - глина. 130 Частей композиции примера 4 обрабатывали в шаровой мельнице в течение 14 часов в 230 мл H2O. Продукт вымывали из мельницы 52,5 мл H2O. Получали суспензию, содержащую 827 г H2O, 33,5 частей каолиновой глины (Georgia Kaolin Kaopaque) и 175,4 частей водного диоксида кремния (Philadelphia Quartz N-brand). Суспензию перемешивали с добавлением 16,4 частей H2SO4 в течение 30 минут. Добавляли по каплям 22,9 частей Al2(SO4)3·16H2O, растворенных в 92,2 частях воды. 396 частей суспензии MCM-41, измельченной в шаровой мельнице (11,36% твердого вещества), добавляли к суспензии диоксид кремния - оксид алюминия - глина и смесь энергично перемешивали при 800 об/мин в течение 30 минут и затем отфильтровывали.
Твердое вещество снова переводили в суспензию в H2O и аэрозоль сушили. Высушенный аэрозольный продукт переводили в суспензию с водой и пылеобразную часть, плавающую над суспензией, отбрасывали. Оставшееся твердое вещество обменивали с 1н NH4NO3 (5 см3 NH4NO3/г твердого вещества). Твердое вещество промывали с H2O, отфильтровывали и сушили в сушильном шкафу при 120°С.
Образец массой 50 г данного материала прокаливали при 540°С в течение одного часа в N2 и в течение 6 часов на воздухе. Остаток твердого вещества после сушки в сушильном шкафу обрабатывали паром в 45% H2O при 650°С в течение 4 часов. До напуска пара в реактор образец нагревали до 650°С в N2. Воздух постепенно увеличивался в течение периода в 1/2 часа, в то время как скорость потока азота увеличивали. После 1/2-часового периода напускали пар на 4-часовой период.
Для сравнения катализатор FCC, содержащий 35 мас.% MCM-41, получали так же, как описано выше. Исходный MCM-41 имел площадь поверхности 980 м2/г, распределение по размеру пор с максимумом около 2,5 нм и объем пор 0,72 см3/г. Он содержал 5,4 мас.%. Al2O3,аналогично материалу примера 4 с 5,3 мас.%. Свойства обработанных паром катализаторов представлены в таблице 11.
Испытания по каталитическому крекингу
Два катализатора, представленные в таблице 11, оценивали по крекингу Joliet Sour Heavy Gas Oil (JSHGO) в установке с неподвижным кипящим слоем псевдоожиженного катализатора при 516°С и одной минуте на поток. Использованный JSHGO имел свойства, представленные в таблице 12. Соотношение катализатор-нефть меняли от 2,0 до 6,0 для рассмотрения широкого интервала конверсий. Выходы, суммированные в таблице 13, даны на основании постоянного коксового числа (4,0 мас.%)
ПРИМЕР 29
Кубовую фракцию, полученную гидрокрекингом при среднем давлении, подвергали депарафинизации и гидрообработке. Исходное сырье обрабатывали с применением каскадной операции в реакторах с неподвижным слоем. Восемьдесят граммов HZSM-5 катализатора депарафинизации загружали в первый реактор и 240 г катализатора по изобретению, описанного в примере 19, загружали во второй реактор. Исходное сырье проходило над обоими катализаторами при 175 бар, 1,0 ЧОСП над катализатором депарафинизации, 0,33 ЧОСП над катализатором гидрообработки. Температуру в первом реакторе поддерживали 307-321°C, чтобы получить целевую точку застывания, равную -6,6°C. Свойства кубовой фракции описаны ниже в таблице 14.
Для определения ароматики в базовом компоненте смазочного вещества использовали УФ-поглощение продукта. Поглощение при 226 нм является мерой суммарной ароматики, в то время как поглощение при 400 нм (×103) является мерой полиядерной ароматики. Для сравнительных целей проводили испытания над катализаторами Pd/MCM-41, полученными по методике, описанной в примере 19. Результаты опытов суммированы в последующей таблице 15.
Сравнивая характеристики Pd/MCM-41 катализатора с катализатором, содержащим Pd в композиции по настоящему изобретению, видно, что композиция по настоящему изобретению является гораздо более эффективной для насыщенной ароматики.
ПРИМЕР 30
Данный пример демонстрирует использование композиции по изобретению как катализатора для гидроочистки угольной жидкости. Хотя конкретная, полученная из угля жидкость, взятая здесь в качестве примера, является жидкой фракцией продукта, полученного способом производства синтетического жидкого топлива на основе гидрогенизации угля (используя Illinois No 6 уголь как исходный материал), другие угольные жидкости (например, экстракты угольной смолы, растворитель после очистки угля и т.д.) могут быть обработаны аналогично. Образец катализатора получали так же, как описано в примере 3А. Однако способ включал в себя гидротермальную обработку в автоклаве при 109°С в течение относительно короткого периода времени, равного 4 дням. Адсорбция азота показала мезопоры размером около 11 нм с площадью поверхности примерно 630 м2/г. Элементный анализ показал атомное отношение Si/Al, равное примерно 99,6.
Материал дополнительно пропитывали раствором гептамолибдата аммония. В частности, 45,12 частей водного раствора, содержащего 6,38 частей гептамолибдата аммония, добавляли к 40 частям описанного выше материала. Полученный влажный материал сушили при 120°С и прокаливали на воздухе при 538°С при условиях, достаточных для того, чтобы разрушить гептамолибдат аммония с образованием MoO3, получая таким образом импрегнированный молибденом материал.
Импрегнированный молибденом материал затем пропитывали раствором нитрата никеля. В частности, 48,2 части водного раствора, содержащего 9,3 части Ni(NO3)2·6H2O, добавляли к импрегнированному молибденом материалу. Полученный влажный материал сушили при 121°С и прокаливали на воздухе при 538°С, чтобы разрушить нитрат никеля с образованием NiO, получая таким образом импрегнированный никелем и молибденом материал. Элементный анализ показал, что конечный материал содержит 15 мас.% MoO3 и 6,4 мас.% NiO.
Для сравнения использовали материал MCM-41, который имел площадь поверхности 992 м2/г, распределение размера пор с максимумом при 3,5 нм и объемом пор, равным 0,72 см3/г. Данный материал пропитывали так же, как описано выше, и окончательно он содержал 15,2 мас.% MoO3 и 6,75 мас. NiO.
Их активность для гидроочистки оценивали с использованием Illinois H-coal в качестве исходного сырья. Таблица 16 показывает свойства исходного сырья.
Данные два катализатора предварительно сульфировали в течение 1 часа при скорости потока, содержащего 10% H2S в H2 при 230°С, равной 500 см3/мин, и суммарном давлении 680 кПа. Гидроочистку проводили при температуре 350°С, давлении 6890 кПа, скорости потока 500 см3/мин, объемной часовой скорости жидкости, равной 0,33. Таблица 17 показывает сравнение активности по деазотированию, восстановлению углеродного остатка по Конрадсону (КУО) и десульфированию.
Катализатор по данному изобретению показывает гораздо более высокую активность, которая частично может быть связана с его уникальной пористой структурой. Он имеет относительно большие поры с трехразмерными сочленениями, которые могут подходить для транспорта больших молекул, таких, которые присутствуют в угольных жидкостях.
ПРИМЕР 31
Данный пример демонстрирует получение катализатора Фишера-Тропша и его каталитические характеристики. Двадцать (20) частей Al-содержащего материала, полученного в примере 1, сушили при 200°С в течение получаса в потоке азота. Материал затем тщательно перемешивали с 2 частями Co2(CO)8 в перчаточной камере. Данную смесь помещали в лодочку для прокаливания в герметично закрытой трубке и вынимали из перчаточной камеры. Затем смесь выдерживали в потоке гелия при 100°С в течение 15 минут, повышали температуру до 200°С в течение 10 минут и выдерживали при 200°С в потоке гелия в течение получаса. Конечный катализатор содержал 16 мас.% Со.
Полученный выше катализатор обрабатывали водородом перед испытанием. Катализатор помещали в маленький кварцевый тигель в камере и продували азотом со скоростью потока 8,5×10-6 нм3/с при комнатной температуре в течение 15 минут. Катализатор затем нагревали со скоростью 1°С/мин до 100°С в потоке водорода 1,8×10-6 нм3/с и выдерживали при 100°С в течение часа. Катализатор затем нагревали со скоростью 1°С/мин до 400°С и выдерживали при 400°С в течение четырех часов в потоке водорода 1,8×10-6 нм3/с. Катализатор охлаждали в водороде и продували азотом перед использованием.
Реактор для работы под давлением, содержащий катализатор и н-октан, нагревали до 225°С под давлением 69 бар в смеси H2:CO (2:1) и выдерживали при данной температуре и давлении в течение 1 часа. Реактор охлаждали льдом, продували и добавляли внутренний стандарт ди-н-бутиловый эфир. Углеводороды в интервале С11-С40 анализировали относительно внутреннего стандарта на ГХ.
Производительность по С11+ (г С11+/час/кг катализатора), рассчитанная на основании суммарной производительности С11-С40-углеводородов на кг катализатора в час, равнялась 234. Логарифм массы фракции для каждого количества углеродных атомов ln(Mn/n) нанесен на ординату против количества углеродных атомов в (Mn/n) на абсциссе. Из наклона получено значение альфа, равное 0,87.
Хотя описание выше содержит много подробностей, они не должны рассматриваться как ограничение рамок изобретения, а только как иллюстрация предпочтительного варианта осуществления. Специалисты в данной области увидят много других возможностей в пределах объема и сути изобретения, как определено прилагаемой формулой изобретения.
Реферат
Изобретение относится к способам обработки органических соединений в присутствии каталитических композиций, включающих диоксид кремния, который имеет мезопористую структуру. Способ включает взаимодействие исходного сырья, содержащего органическое соединение, в условиях реакции с композицией катализатора, при этом способ обработки выбирают из группы, состоящей из алкилирования, ацилирования, гидроочистки, деметаллирования, каталитической депарафинизации, процесса Фишера-Тропша и крекинга. Композиция катализатора включает в себя, по существу, мезопористую структуру диоксида кремния, содержащую, по меньшей мере, 97 об.% пор, имеющих размер пор в интервале от примерно 15 Å до примерно 300 Å, и имеет объем микропор, по меньшей мере, примерно 0,01 см3/г. Мезопористая структура имеет введенный в нее в количестве, по меньшей мере, примерно 0,02 мас.%, по меньшей мере, один каталитически и/или химически активный гетероатом, выбранный из группы, состоящей из Al, Ti, V, Cr, Zn, Fe, Sn, Mo, Ga, Ni, Co, hi, Zr, Mn, Cu, Mg, Pd, Ru, Pt, W и их комбинаций, причем упомянутая композиция катализатора имеет рентгенограмму с одним пиком от 0,3° до примерно 3,5° при 2θ. Технический результат - обеспечение высокоэффективного способа обработки органических соединений в присутствии композиции катализатора, не содержащей цеолит. 19 з.п. ф-лы, 22 ил., 17 табл.
Формула
Документы, цитированные в отчёте о поиске
Способ получения олефиноксидов прямым окислением олефинов, каталитический состав для этого процесса и способ его регенерации
Комментарии