Соединения для лечения метаболических заболеваний - RU2341513C2
Код документа: RU2341513C2
Чертежи


Описание
Сахарный диабет является основной причиной заболеваемости и смертности. Хронически повышенный уровень глюкозы в крови приводит к подрывающим силы осложнениям: нефропатии, при которой часто требуется диализ или пересадка почки; периферической нейропатии; ретинопатии, приводящей к слепоте; изъязвлению ног и ступней, что приводит к ампутации; ожирению печени, иногда прогрессирующему до цирроза, и подверженности заболеванию коронарной артерии и инфаркту миокарда.
Существует два основных типа диабета. Тип I, или инсулинозависимый сахарный диабет (IDDM), связан с аутоиммунным уничтожением продуцирующих инсулин бета-клеток в панкреатических островках (островках Лангерганса). Данное заболевание обычно возникает в детстве или пубертатном периоде. Лечение в первую очередь заключается в многократных ежедневных инъекциях инсулина в сочетании с частым определением уровня глюкозы в крови для регулирования дозировок инсулина, поскольку избыток инсулина может вызвать гипогликемию и последующее ухудшение функции мозга и других функций.
Тип II, или инсулинонезависимый сахарный диабет (NIDDM), обычно развивается во взрослом возрасте. NIDDM связан с устойчивостью (невосприимчивостью) использующих глюкозу тканей, таких как жировая ткань, мышцы и печень, к действию инсулина. Первоначально бета-клетки панкреатических островков компенсируют это с помощью секреции избытка инсулина. В конечном итоге несостоятельность островка приводит к декомпенсации и хронической гипергликемии. Наоборот, средняя недостаточность островка может предшествовать или совпадать по времени с периферической резистентностью к инсулину. Существует несколько классов лекарственных средств, которые можно использовать для лечения NIDDM: 1) высвобождающие инсулин средства, которые непосредственно стимулируют высвобождение инсулина, вызывая риск гипогликемии; 2) «обеденные» высвобождающие инсулин средства, которые потенцируют индуцированную глюкозой секрецию инсулина и которые следует принимать перед каждым приемом пищи; 3) бигуаниды, включая метформин, которые смягчают глюконеогенез печени (которая парадоксально увеличивается при диабете); 4) сенсибилизаторы инсулина, например производные тиазолидиндиона розиглитазон и пиоглитазон, которые улучшают периферическую ответную реакцию на инсулин, но которые обладают таким побочным действием, как увеличение веса, отек и преходящая токсикация печени; 5) инъекции инсулина, которые часто необходимы на более поздних стадиях NIDDM, когда островки выходят из строя в условиях хронической гиперстимуляции.
Резистентность к инсулину также может происходить без выраженной гипергликемии и обычно связана с атеросклерозом, ожирением, гипергликемией и существенным повышением кровяного давления. Такая совокупность аномалий составляет «метаболический синдром» или «синдром резистентности к инсулину». Резистентность к инсулину также связана с ожирением печени, которое может прогрессировать до хронического воспаления (NASH; «неалкогольный стеатогепатит''), фиброза и цирроза. Совокупно синдромы резистентности к инсулину, включая, но не ограничиваясь диабетом, лежат в основе многих основных случаев заболеваемости и смерти людей, возраст которых превышает 40 лет.
Несмотря на существование таких лекарственных средств, диабет остается основной и все возрастающей проблемой здоровья человека. Осложнения диабета на поздних стадиях потребляют большую долю ресурсов здравоохранения. Существует необходимость в новых перорально активных терапевтических агентах, которые эффективно адресованы первичным дефектам резистентности к инсулину и повреждения островков, обладая меньшим или более мягким побочным действием, чем существующие лекарственные средства.
В настоящее время не имеется безопасных и эффективных способов лечения ожирения печени. Следовательно, такое лечение могло бы оказаться ценным для лечения такого заболевания.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Данное изобретение относится к биологически активному агенту, где агент представляет собой соединение формулы:

где n равно 1 или 2; m равно 0 или 1; q равно 0 или 1; t равно 0 или 1; R5представляет собой алкил, содержащий от 1 до 3 атомов углерода; R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода; A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых углерода независимо являются монозамещенными метилом или этилом; или 5 или 6 членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы I через кольцевой атома углерода; и X представляет собой -CH2-, Q представляет собой -OR1, и R1 представляет собой этил; или X представляет собой -CH2CR12R13- или CH2CH(NHAc)-, где каждый из R12 и R13 независимо представляет собой водород или метил, Q представляет собой OR1, и R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; или X представляет собой -CH2CH2-, и Q представляет собой NR10R11, где один из R10 и R11 представляет собой водород, алкил, содержащий от 1 до 3 атомов углерода, или гидрокси, а другой представляет собой водород или алкил, содержащий от 1 до 3 атомов углерода; или когда R1 представляет собой водород, фармацевтически приемлемую соль соединения.
Данное изобретение относится к биологически активному агенту, где агент представляет собой соединение формулы:

где n равно 1 или 2; t равно 0 или 1; m равно 0, и r равно 1, или m равно 1, и r равно 0; A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых атома углерода независимо являются монозамещенными метилом или этилом; или 5 или 6-членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы II с помощью кольцевого атома углерода; Z представляет собой

R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; R4 представляет собой водород; -NHCOOC(CH3)3; -NHCH3 или -NHCH2CH3; или когда R1 представляет собой водород, фармацевтически приемлемую соль соединения.
Данное изобретение относится к биологически активному агенту, где агент представляет собой соединение формулы:

где n равно 1 или 2; A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где один или два кольцевых углерода независимо являются монозамещенными метилом или этилом; или 5 или 6 членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы III через кольцевой атом углерода.
Данное изобретение относится к биологически активному агенту, где агент представляет собой соединение формулы:

где R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; или когда R1 представляет собой водород, фармацевтически приемлемую соль соединения.
Данное изобретение относится к биологически активному агенту, где агент представляет собой соединение формулы:

где n равен 1 или 2; R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; R14 представляет собой гидрокси или водород; и A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых атома углерода независимо являются монозамещенными метилом или этилом; или 5 или 6-членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы V' с помощью кольцевого атома углерода;
или фармацевтически приемлемую соль соединения.
Данное изобретение относится к биологически активному агенту, где агент представляет собой соединение формулы:

где n равен 1 или 2; R1 представляет собой водород или алкил, содержащий от 1 до 3 атомов углерода; и A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых атома углерода независимо являются монозамещенными метилом или этилом; или 5 или 6-членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы XCI с помощью кольцевого атома углерода; или фармацевтически приемлемую соль соединения.
Данное изобретение относится к биологически активному агенту, где агент представляет собой соединение формулы:

где n равен 1 или 2; R1 представляет собой водород или алкил, содержащий от 1 до 3 атомов углерода; и A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых атома углерода независимо являются монозамещенными метилом или этилом; или 5 или 6-членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы CXVI с помощью кольцевого атома углерода; или фармацевтически приемлемую соль соединения.
Данное изобретение относится к биологически активному агенту, где агент представляет собой соединение формулы:

где n равен 1 или 2; R1 представляет собой водород или алкил, содержащий от 1 до 3 атомов углерода; R15 представляет собой водород или алкил, содержащий от 1 до 3 атомов углерода; R9 представляет собой водород, галоген, гидрокси или алкокси, содержащий от 1 до 3 атомов углерода; и A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых атома углерода независимо являются монозамещенными метилом или этилом; или 5 или 6-членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы CXVII с помощью кольцевого атома углерода; или фармацевтически приемлемую соль соединения.
Описанные выше биологически активные агенты обладают активностью в одном или нескольких из описанных далее анализов на биологическую активность, в которых созданы животные модели диабета человека и синдрома резистентности к инсулину. Таким образом, такие агенты могут оказаться полезными для лечения диабета и синдрома резистентности к инсулину. Все протестированные иллюстративные соединения продемонстрировали активность при исследовании биологической активности или проведенных анализах.
Данное изобретение относится к применению описанных выше биологически активных агентов для получения лекарственного средства для лечения синдрома резистентности к инсулину, диабета, кахексии, гиперлипидемии, ожирения печени, ожирения, атеросклероза или артериосклероза. Данное изобретение также относится к способам лечения субъекта-млекопитающего, подверженного синдрому резистентности к инсулину, диабету, кахексии, гиперлипидемии, ожирению печени, ожирению, атеросклерозу или артериосклерозу, включающим введение субъекту эффективного количества биологически активного агента согласно изобретению. Данное изобретение также относится к фармацевтической композиции, включающей биологически активный агент по изобретению и фармацевтически приемлемый носитель.
Данное изобретение также относится к некоторым новым промежуточным соединениям, которые могут использоваться при получении биологически активных агентов по данному изобретению. Изобретение также относится к способам получения биологически активных соединений и промежуточных продуктов.
Краткое описание чертежей
Фиг.1: Уровни инсулина в сыворотке крови мышей C57B1/6J, получающих высококалорийную диету, которые получали носитель (отрицательный контроль), Соединение BI, Соединение BL,Wyl4643 или розиглитазон.Фиг.2: Уровни лептина в сыворотке крови мышей C57B1/6J, получающих высококалорийную диету, которые получали носитель (отрицательный контроль), Соединение BI, Соединение BL,Wyl4643 или розиглитазон.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
В данном описании термин «алкил» означает линейную или разветвленную алкильную группу. Алкильная группа, указанная как содержащая определенное число атомов углерода, означает любую алкильную группу, имеющую указанное число атомов углерода. Например, алкил, имеющий три атома углерода, может представлять собой пропил или изопропил; и алкил, имеющий четыре атома углерода, может представлять собой н-бутил, 1-метилпропил, 2-метилпропил или трет-бутил.
В данном описании термин «галоген» относится к одному или нескольким из фтора, хлора, брома и иода.
В данном описании термин «перфтор», как, например, в перфторметиле или перфторметокси, означает что рассматриваемая группа имеет атомы фтора вместо всех атомов водорода.
Как использовано в данном описании, «Ас» относится к группе CH3C(O)-.
Ниже перечислены примеры биологически активных соединений настоящего изобретения. Данные соединения упоминаются в настоящем описании по их химическим названиям или с помощью приведенного ниже двухбуквенного кода.
АА 4-(4-(2-Фторбензилокси)фенил)-4-оксобутановая кислота;
АВ 4-(4-(2-Метоксибензилокси)фенил)-4-оксобутановая кислота;
АС 3-[(4-(2-Фторбензилокси)фенил)-метилтио]пропионовая кислота;
AD 4-(4-(3-Фторбензилокси)фенил)-4-оксобутановая кислота;
АЕ 4-(4-(4-Фторбензилокси)фенил)-4-оксобутановая кислота;
AF 4-(4-((2-Пиридил)метокси)фенил)-4-оксобутановая кислота;
AG 4-(4-(Бензилокси)фенил)-4-оксобутановая кислота;
AH 4-(4-(2,6-Дифторбензилокси)фенил)-4-оксобутановая кислота;
AI 4-(4-(2-Хлорбензилокси)фенил)-4-оксобутановая кислота;
AJ 4-(4-(2-(2-Фторфенил)этокси)фенил)-4-оксобутановая кислота;
АК Этил 4-(4-(2-фторбензилокси)фенил)-4-оксобутират;
AL 4-(4-(2-Метилбензилокси)фенил)-4-оксобутановая кислота;
AM 4-[4-(2-(N-(2-фторбензил)-N-метиламино)этокси)фенил]-4-оксобутановая кислота;
AN 4-(3-(2-Метилбензилокси)фенил)-4-оксобутановая кислота;
АО Этил 4-(3-(2-фторбензилокси)фенил)-4-оксобутират;
AP Этил 4-(4-(2-метилбензилокси)фенил)-4-оксобутират;
AQ Этил 4-(4-(2,6-дифторбензилокси)фенил)-4-оксобутират;
AR 4-(4-(2-(2-Тиенил)этокси)фенил)-4-оксобутановая кислота;
AS 4-(2,6-Дифторфенил)-4-оксобутановая кислота;
АТ 4-(4-(2,5-Диметилбензилокси)фенил)-4-оксобутановая кислота;
AU 4-(4-(2,5-Дифторбензилокси)фенил)-4-оксобутановая кислота;
AV 4-(4-(2,4-Дифторбензилокси)фенил)-4-оксобутановая кислота;
AW 4-(3-(2,6-Дифторбензилокси)фенил)-4-оксобутановая кислота;
AX 4-(4-((Циклопропил)метокси)фенил)-4-оксобутановая кислота;
AY 4-(4-(2-Трифторметилбензилокси)фенил)-4-оксобутановая кислота;
AZ 3-[(4-(2,6-Дифторбензилокси)фенил)метилтио]пропионовая кислота;
BA 4-(2-(2,6-Дифторбензилокси)фенил)-4-оксобутановая кислота;
BB Этил 4-(4-(2,6-дифторбензилокси)фенил)метил-3-оксобутират;
ВС 3-(2-(4-(2,6-Дифторбензилокси)фенил)-2-оксоэтил)тио-1H-1,2,4-триазол;
BD 5-[(4-(2,6-Дифторбензилокси)фенил)-метил]-1H-тетразол;
BE (2RS) 2-(N-Boc)-3-[2-(4-(2,6-дифторбензилокси)фенил)-2-оксоэтил]тиопропионовая кислота;
BF Этил 2-гидрокси-4-оксо-4-(4-(2,6-дифторбензилокси)фенил)бут-2-еноат;
BG (2RS) 2-(N-Ацетил)-4-(4-(2,6-дифторбензилокси)фенил)-4-оксобутановая кислота;
BH 4-(3-((Циклопропил)метокси)фенил)-4-оксобутановая кислота;
BI 4-(3-(2,6-Диметилбензилокси)фенил)-4-оксобутановая кислота;
BJ 4-(3-(2-Фтор-6-метилбензилокси)фенил)-4-оксобутановая кислота;
BK Этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутират;
BL Натриевая соль 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты;
BM 4-(4-(2,6-Диметилбензилокси)фенил)-4-оксобутановая кислота;
BN Калиевая соль 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты;
BO 4-(3-(2,6-Диметоксибензилокси)фенил)-4-оксобутановая кислота;
BP 4-(3-(2,6-Диметилбензилокси)фенил)-4-оксо-2,2-диметилбутановая кислота;
BQ 4-(3-(4-Трифторметилбензилокси)фенил)-4-оксобутановая кислота;
BR 4-(3-((Циклобутил)метокси)фенил)-4-оксобутановая кислота;
BS 4-(3-(2,6-Диметилбензилокси)фенил)бутановая кислота;
BT 4-[[4-(2,6-Диметилбензилокси)-3-метокси]фенил]-4-оксобутановая кислота;
BU 4-{3-[((4-Трифторметилбензиламино)карбонил)-4-метокси]фенил}-4-оксобутановая кислота;
BV 4-{3-[((2,6-Диметилбензиламино)карбонил)-4-метокси]фенил}-4-оксобутановая кислота;
BW 4-(3-(2,6-Диметилбензилокси)фенил)-4-оксобутанкарбогидроксамовая кислота;
BX 4-(3-(2,6-Диметилбензилокси)фенил)-4-оксобутирамид;
BY 4-(3-(2,6-Диметилбензилокси)фенил)-4-оксо-2-бутановая кислота; и
BZ 4-(3-(2,6-Диметилбензилокси)фенил)-3-бутеновая кислота.
В данном описании термин «включающий» обозначает открытое множество. Пункт формулы изобретения, в котором такой термин используется, может содержать элементы в дополнение к указанным в данном пункте.
ПОДРОБНОЕ ОПИСАНИЕ АКТИВНЫХ СОЕДИНЕНИЙ
В одном варианте осуществления агента формулы I агент представляет собой соединение формулы:

где n равен 1 или 2; m равно 0 или 1; q равно 0 или 1; t равно 0 или 1; R5 представляет собой алкил, содержащий от 1 до 3 атомов углерода; A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых атома углерода независимо являются монозамещенными метилом или этилом; или 5 или 6-членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы I с помощью кольцевого атома углерода; и X представляет собой -CH2-, и R1 представляет этил; или X представляет -CH2CH2 -или-CH2CH(NHAc)-, и R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; или когда R1 представляет собой водород, фармацевтически приемлемую соль соединения.
В других вариантах осуществления агента формулы I Rl представляет собой водород или этил; q равно 0; или X представляет собой -CH2CH2-.
В другом варианте осуществления агента формулы I A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси, каждый галоген независимо представляет собой фтор или хлор. В конкретном варианте осуществления каждый галогеновый заместитель в фенильном кольце А представляет собой фтор. В более конкретном варианте осуществления фенильное кольцо А замещено 2 группами фтора. В конкретном варианте осуществления алкил, перфторалкил, алкокси или перфторалкокси имеет один атом углерода.
В другом варианте осуществления агента формулы I A представляет собой циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых атома углерода независимо являются монозамещенными метилом или этилом. В конкретном варианте осуществления циклоалкил является незамещенным, или один или оба кольцевых атома углерода, смежных с кольцевым углеродом, ковалентно связанным с остатком соединения формулы I, независимо являются монозамещенными метилом или этилом. В более конкретном варианте осуществления А представляет собой незамещенный циклопропил.
В другом варианте осуществления агента формулы I q равно 1 и R5 представляет собой метил.
В другом варианте осуществления агент представляет собой соединение формулы:

где n равно 1 или 2; m равно 0 или 1; q равно 0 или 1; t равно 0 или 1; каждый из R2и R3 независимо выбран из водорода, галогена, алкила, имеющего 1 или 2 атома углерода, перфторметила, алкокси, имеющего 1 или 2 атома углерода, и перфторметокси; R5 представляет собой алкил, содержащий от 1 до 3 атомов углерода; и X представляет собой -CH2-, и R1 представляет собой этил; или X представляет собой -CH2CH2- или -CH2CH(NHAc)-, и R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; или когда R1 представляет собой водород, фармацевтически приемлемую соль соединения. В более конкретном варианте осуществления R1 представляет собой водород или этил. Примеры соединений формулы IA включают соединение AM и соединение BG.
В конкретном варианте осуществления агент представляет собой соединение формулы:

где n равно 1 или 2; m равно 0 или 1; р равно 1, и R1 представляет собой этил; или р равно 2, и R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; каждый из R2 и R3 независимо выбран из водорода, галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или когда R1 представляет собой водород, фармацевтически приемлемую соль соединения. В более конкретном варианте осуществления R1 представляет собой водород или этил. В еще более конкретном варианте осуществления один из R2 и R3 представляет собой водород или галоген, а другой представляет галоген. Примеры таких соединений включают соединение AD, соединение AE и соединение AI. В другом еще более конкретном варианте осуществления R2 представляет собой фтор и R3 представляет собой водород. Примеры таких соединений включают соединение AA, соединение AJ, соединение AK и соединение AO. В следующем еще более конкретном варианте осуществления R2 представляет собой фтор и R3 представляет собой фтор. Примеры таких соединений включают соединение AU, соединение AV и соединение BB.
В более конкретном варианте осуществления агент представляет собой соединение формулы:

где n равно 1 или 2; m равно 0; R1 представляет собой Н или алкил, содержащий от 1 до 7 атомов углерода; или когда R1 представляет собой водород, фармацевтически приемлемую соль соединения. Примеры таких соединений включают соединение AH, соединение AQ, соединение AW и соединение BA. В еще более конкретном варианте осуществления один из R2 и R3 представляет собой метил, метокси или перфторметил, а другой представляет собой водород или метил. В одном варианте осуществления R2 представляет собой метил, метокси или перфторметил и R3 представляет собой водород. Примеры таких соединений включают соединение AB, соединение AL, соединение AN, соединение AP и соединение AY. В другом варианте осуществления R2 представляет собой метил и R3 представляет собой метил. Примеры таких соединений включают соединение AT и соединение BI. В следующем варианте осуществления R2 представляет собой водород и R3 представляет собой водород. Примеры таких соединений включают соединение AG.
В другом варианте осуществления агент представляет собой соединение формулы:
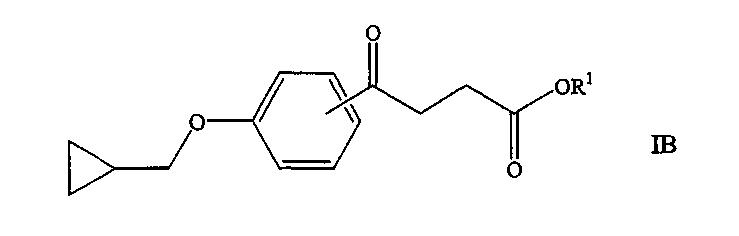
где R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; или когда R1 представляет собой водород, фармацевтически приемлемую соль соединения. В конкретном варианте осуществления R1 представляет собой водород или этил. Примеры таких соединений включают соединение AX и соединение BH. В другом варианте осуществления агент представляет собой соединение формулы:

где n равен 1 или 2; R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; и Het представляет собой 5 или 6-членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы IC с помощью кольцевого атома углерода. В конкретном варианте осуществления R1 представляет собой водород или этил. Примеры таких соединений включают соединение AF и соединение AR.
В варианте осуществления агента формулы II A представляет собой циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых атома углерода, смежных с остатком соединения формулы II являются монозамещенными метилом или этилом. В другом варианте осуществления агента формулы II A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: фтора, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси.
В другом варианте осуществления агент представляет собой соединение формулы:

где m равно 0 или 1; r равно 0 или 1; Z представляет собой

R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; R4 представляет собой водород; -NHCOOC(CH3)3; -NHCH3 или -NHCH2CH3; R3 представляет собой водород или галоген; или когда R1 представляет собой водород, фармацевтически приемлемую соль соединения. В конкретном варианте осуществления R1 представляет собой водород или этил. Примеры таких соединений включают соединение AC, соединение AZ, соединение BC и соединение BE.
В варианте осуществления агента формулы III A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из: галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси. Примеры таких соединений включают соединение BD.
В варианте осуществления агента формулы IV R1 представляет собой водород или этил. Примеры таких соединений включают соединение AS.
В варианте осуществления агента формулы V' агент представляет собой соединение формулы:

где n равен 1 или 2; R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; A представляет собой фенил, незамещенный или замещенный 1 или 2 группами, выбранными из галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, имеющий от 3 до 6 кольцевых атомов углерода, где циклоалкил является незамещенным, или один или два кольцевых атома углерода независимо являются монозамещенными метилом или этилом; или 5 или 6-членное гетероароматическое кольцо, содержащее 1 или 2 гетероатома в кольце, выбранных из N, S и O, и гетероароматическое кольцо ковалентно связано с остатком соединения формулы I с помощью кольцевого атома углерода; или фармацевтически приемлемую соль соединения.
В варианте осуществления агента формулы V агент представляет собой соединение формулы:

где n равен 1 или 2; R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода; каждый из R2 и R3 независимо выбран из водорода, галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси, или фармацевтически приемлемую соль соединения. В конкретном варианте осуществления R1представляет собой водород или этил. Примеры таких соединений включают соединение BF.
ПРИМЕНЕНИЕ В СПОСОБАХ ЛЕЧЕНИЯ
Данное изобретение относится к способу лечения субъекта млекопитающего, страдающего заболеванием, выбранным из группы, состоящей из синдрома резистентности к инсулину и диабета (оба основных важных диабета, такие как диабет типа I или диабет типа II, и вторичные неидиопатические диабеты), включающему введение субъекту эффективного для лечения заболевания количества описанного выше биологически активного агента. В соответствии со способом данного изобретения могут уменьшаться симптомы диабета или возможность развития симптомов диабета, таких как атеросклероз, ожирение, повышенное кровяное давление, гиперлипидемия, ожирение печени, нефропатия, нейропатия, ретинопатия, изъязвление ступней и катаракта, при этом каждый такой симптом связан с диабетом. Данное изобретение также относится к способу лечения гиперлипидемии, включающему введение субъекту биологически активного агента, описанного здесь, в количестве, эффективном для лечения заболевания. Как показано в примерах, соединения понижают содержание триглицеридов и свободных жирных кислот в сыворотке крови у гиперлипидемических животных. Данное изобретение также относится к способу лечения кахексии, включающему введение субъекту биологически активного агента, описанного здесь, в количестве, эффективном для лечения кахексии. Данное изобретение также относится к способу лечения ожирения, включающему введение субъекту биологически активного агента, описанного здесь, в количестве, эффективном для лечения заболевания. Данное изобретение также относится к способу лечения заболевания, выбранного из атеросклероза или артериосклероза, включающему введение субъекту биологически активного агента, описанного здесь, в количестве, эффективном для лечения заболевания. Активные агенты данного изобретения эффективны для лечения гиперлипидемии, ожирения печени, кахексии, ожирения, атеросклероза или артериосклероза вне зависимости от того, есть ли у субъекта диабет или синдром резистентности к инсулину. Агент можно вводить любым обычным путем системного введения. Предпочтительно агент вводят перорально. Другие пути введения, которые можно использовать в соответствии с изобретением, включают ректальный, парентеральный, инъекционный (например, внутривенная, подкожная, внутримышечная или внутрибрюшинная инъекция) или назальный пути.
Дальнейшие варианты осуществления каждого из применений и способов лечения по данному изобретению включают введение любого одного из описанных выше биологически активных агентов. Для того чтобы избежать излишней перегруженности, каждый такой агент и группа агентов не повторяются, но они включены в данное описание применений и способов лечения, как если бы они повторялись.
Многие из заболеваний или нарушений, которым адресованы соединения по изобретению, попадают в две широкие категории: синдромы резистентности к инсулину и последствия хронической гипергликемии. Нарушение регулирования энергетического метаболизма, особенно резистентность к инсулину, которое может возникать в отсутствие диабета (постоянная гипергликемия), само по себе связано со множеством симптомов, включая гиперлипидемию, атеросклероз, ожирение, идиопатическую гипертензию, ожирение печени (NASH, неалкогольный стеатогепатит) и, особенно в контексте ракового или системного воспалительного заболевания, кахексию. Кахексия также может возникать в связи с диабетом типа I или поздней стадией диабета типа II. За счет улучшения энергетического метаболизма в тканях активные агенты по изобретению полезны для профилактики или облегчения протекания заболеваний и симптомов, связанных с резистентностью к инсулину, как это показано в примерах на животных. Хотя у индивидуального пациента совокупность признаков и симптомов, связанных с невосприимчивостью к инсулину, может сосуществовать, во многих случаях может доминировать только один симптом вследствие индивидуальных различий в уязвимости многих физиологических систем, на которые влияет резистентность к инсулину. Тем не менее, поскольку резистентность к инсулину вносит основной вклад во многие болезненные состояния, лекарственные средства, которые направлены на этот клеточный и молекулярный дефект, полезны для профилактики или облегчения практически любого симптома в любой системе органов, который является следствием или обостряется за счет невосприимчивости к инсулину.
Когда резистентность к инсулину или сопутствующее неадекватное продуцирование инсулина островками поджелудочной железы являются достаточно серьезными, возникает хроническая гипергликемия, определяющая начало сахарного диабета типа II (NIDDM). В дополнение к метаболическим нарушениям, связанным с указанной выше резистентностью к инсулину, у пациентов с NIDDM также возникают вторичные относительно гипергликемии симптомы заболевания. Они включают нефропатию, периферическую нейропатию, ретинопатию, заболевание капилляров, изъязвление конечностей и последствия неферментативного гликозилирования белков, то есть повреждение коллагена и других соединительных тканей. Ослабление гипергликемии снижает скорость начала и серьезность таких последствий диабета. Поскольку, как это продемонстрировано в примерах, активные агенты и композиции изобретения помогают снизить гипергликемию при диабете, они полезны для профилактики и облегчения осложнений при хронической гипергликемии.
Субъектов-млекопитающих, как людей, так и животных, можно лечить в соответствии со способом лечения по данному изобретению. Оптимальная доза конкретного активного агента по изобретению для конкретного субъекта может быть определена в клинической практике квалифицированным лечащим врачом. В случае перорального введения человеку для лечения заболеваний, связанных с резистентностью к инсулину, диабетом, гиперлипидемией, ожирением печени, кахексией или ожирением, агент обычно вводят в суточной дозе от 1 мг до 400 мг, которую вводят один или два раза в день. Для перорального введения человеку предполагаемая предпочтительная суточная доза соединения АН составляет от 100 до 400 мг; соединения AW - от 30 до 300 мг и соединения BI - от 10 до 200 мг. В случае перорального введения мышам агент обычно вводят в суточной дозе от 1 до 300 мг агента на килограмм веса тела. Активные агенты по изобретению используют в качестве монотерапии диабета или синдрома резистентности к инсулину или в сочетании с одним или несколькими другими лекарственными средствами, находящими применение при таких типах заболеваний, например, с высвобождающими инсулин агентами, «обеденными» высвобождающими инсулин средствами, бигуанидами или самим инсулином. Такие дополнительные лекарственные средства вводят в соответствии со стандартной клинической практикой. В некоторых случаях агенты по изобретению будут улучшать эффективность некоторых классов лекарственных средств, давая возможность введения пациентам пониженных (и как следствие менее токсичных) дозировок таких агентов при удовлетворительных терапевтических результатах. Установленные безопасные и эффективные диапазоны дозировки для людей иллюстративных соединений составляют: метформин - от 500 до 2550 мг/день; глубурид - от 1,25 до 20 мг/день; Глюкованс (GLUCOVANCE) (комбинированный препарат метформина и глубурида) - от 1,25 до 20 мг/день глубурида и от 250 до 2000 мг/день метформина; аторвастатин - от 10 до 80 мг/день; ловастатин - от 10 до 80 мг/день; правастатин - от 10 до 40 мг/день; и симвастатин - 5-80 мг/день; клофибрат - 2000 мг/день; гемфиброзил - 1200-2400 мг/день; розилитазон - от 4 до 8 мг/день; пиоглитазон - от 15 до 45 мг/день; акарбоза - 75-300 мг/день; репаглинид - от 0,5 до 16 мг/день.
Сахарный диабет типа I: Пациент с диабетом типа I контролирует свое заболевание в первую очередь путем самостоятельного введения от одной до нескольких доз инсулина в сутки, при частом контроле за уровнем глюкозы в крови, чтобы подходящим образом регулировать дозировку и время введения инсулина. Хроническая гипергликемия приводит к осложнениям, таким как нефропатия, нейропатия, ретинопатия, изъязвление ступней и преждевременная смерть; гипогликемия вследствие избыточной дозы инсулина может вызвать познавательную дисфункцию или бессознательное состояние. Пациент, страдающий диабетом типа I, получает от 1 до 400 мг/день активного агента по данному изобретению, например от 50 до 400 мг/день соединения АН, в виде таблетки или капсулы в виде однократной дозы или разделенных доз. Ожидаемым эффектом будет снижение дозы или частоты введения инсулина, требующегося для поддержания в удовлетворительном диапазоне уровня глюкозы в крови, и снижение доли и тяжести гипогликемических случаев. Клинический результат контролируется измерением содержания глюкозы и гликозилированного гемоглобина в крови (индекс адекватности гликемического контроля, интегрированного за период в несколько месяцев), а также по снижению доли и тяжести типичных осложнений диабета. Биологически активный агент по данному изобретению можно вводить в сочетании с трансплантацией островков поджелудочной железы для поддержания антидиабетической эффективности трансплантации островков.
Сахарный диабет типа II: Пациент с диабетом типа II контролирует свое заболевание программами питания и физических упражнений, а также приемом лекарственных средств, таких как метформин, глубурид, репаглинид, розиглитазон или акарбоза, все из которых приводят к некоторому улучшению гликемического контроля для некоторых пациентов, но ни одно из которых не лишено побочных действий или возможности неудачи лечения из-за развития заболевания. Нарушение функции островков происходит с течением времени у пациентов с NIDDM, приводя к необходимости инъекций инсулина у большой части пациентов. Ожидается, что ежедневное лечение с использованием активного агента по изобретению (с использованием или без использования дополнительных классов противодиабетических лекарственных средств) улучшит гликемический контроль, снизит скорость нарушения функции островков и снизит долю и серьезность типичных симптомов диабета. Кроме того, активные агенты по изобретению будут уменьшать повышенные уровни триглицеридов и жирных кислот в сыворотке крови, снижая таким образом риск сердечно-сосудистых заболеваний, являющихся основной причиной смерти пациентов, страдающих диабетом. Подходящие суточные диапазоны дозировки для выбранных соединений изобретения для лечения NIDDM (либо в виде монотерапии, либо в сочетании с другими антидиабетическими лекарственными средствами) составляют от 50 до 400 мг/день соединения АН, от 15 мг до 300 мг соединения AW и от 5 мг до 200 мг соединения BI. Как и в случае всех других терапевтических агентов для диабета, оптимизацию дозы проводят для индивидуального пациента в соответствии с необходимостью, клиническим эффектом и восприимчивостью к побочным действиям.
Гиперлипидемия: Повышенные уровни триглицеридов и свободных жирных кислот в крови поражают значительную долю населения и представляют собой важный фактор риска для атеросклероза и инфаркта миокарда. Активные агенты по изобретению можно использовать для снижения циркулирующих триглицеридов и свободных жирных кислот у гиперлипидемических пациентов. Подходящие суточные диапазоны дозировки для выбранных соединений изобретения для лечения гипертриглицеридемии составляют от 50 до 400 мг/день соединения АН, от 15 мг до 300 мг соединения AW и от 5 мг до 200 мг соединения BI. У пациентов, страдающих гиперлипидемией, часто также наблюдается повышенный уровень холестерина в крови, что также увеличивает риск сердечно-сосудистого заболевания. Снижающие уровень холестерина лекарственные средства, такие как ингибиторы HMG-CoA редуктазы, можно вводить страдающим гиперлипидемией пациентам в дополнение к агентам по изобретению, необязательно включенным в состав одной и той же фармацевтической композиции.
Ожирение печени: Значительная часть населения подвержена такому заболеванию, как ожирение печени, также известному как неалкогольный стеатогепатит (NASH); NASH часто связан с ожирением и диабетом. Гепатический стеатоз, присутствие капелек триглицеридов вместе с клетками печени (гепатоцитами), приводит к предрасположенности печени к хроническому воспалению (обнаруживаемому в образцах для биопсии в виде инфильтрации воспалительных лейкоцитов), что может привести к фиброзу и циррозу. Ожирение печени обычно обнаруживается путем наблюдения повышенных уровней в сыворотке крови печень-специфических ферментов, таких как трансаминазы ALT и AST, которые служат индикаторами повреждения клеток печени, а также по присутствию симптомов, которые включают усталость и боль в области печени, хотя для установления точного диагноза часто требуется биопсия. Как показано в примерах, соединения по изобретению, например соединение AW, снижают содержание печеночных трансаминаз в сыворотке крови и жира в печени, как было установлено на животной модели NASH (склонные к ожирению мыши ob/ob), и, следовательно, полезны для лечения ожирения печени. Подходящий диапазон дозировки для соединения AW для лечения ожирения печени составляет от 15 до 300 мг/день. Ожидаемым преимуществом является снижение воспаления печени и содержания жира, что приведет к ослаблению, прекращению или обратимости прогрессирования NASH к фиброзу и циррозу.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Данное изобретение относится к фармацевтической композиции, включающей биологически активный агент, как указано в данном описании, и фармацевтически приемлемый носитель. Следующие варианты осуществления фармацевтической композиции по данному изобретению включают любой из вариантов осуществления описанных выше биологически активных агентов. Для того чтобы исключить ненужную чрезмерную информацию, каждый такой агент и группа агентов не повторяются, но они включены в данное описание фармацевтических композиций, как если бы они повторялись.
Предпочтительно композиция приспособлена для перорального введения, например, в форме таблетки, покрытой оболочкой таблетки, драже, твердой или мягкой желатиновой капсулы, раствора, эмульсии или суспензии. Обычно пероральная композиция будет включать от 1 мг до 400 мг такого агента. Субъекту будет удобно проглотить одну или две таблетки, покрытых оболочкой таблетки, драже или желатиновых капсул в день. Соответственно, предпочтительные пероральные композиции для лечения людей включают от 50 мг до 400 мг соединения AH, от 15 мг до 300 мг соединения AW или от 5 мг до 200 мг соединения BI. Однако композиция также может быть приспособлена для введения любым другим обычным способом системного введения, включая ректальный, например в виде суппозиториев, парентеральный, например в виде растворов для инъекций или назальный.
Биологически активные соединения могут быть переработаны вместе с фармацевтически инертными неорганическими или органическими носителями для получения фармацевтических композиций.
В качестве таких носителей для таблеток, покрытых оболочкой таблеток, драже и твердых желатиновых капсул можно использовать, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и тому подобное. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие многоатомные спирты и тому подобное. Однако в зависимости от природы активного ингредиента в случае мягких желатиновых капсул носители, отличающиеся от самого мягкого желатина, обычно не требуются. Подходящими носителями для получения растворов и сиропов являются, например, вода, многоатомные спирты, глицерин, растительные масла и тому подобные. Подходящими носителями для суппозиториев являются, например, природные или отвержденные масла, воски, жиры, полужидкие или жидкие многоатомные спирты и тому подобные.
Кроме того, фармацевтические композиции могут содержать консерванты, солюбилизаторы, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, отдушки, соли для изменения осмотического давления, буферы, покрывающие агенты и антиоксиданты. Они также могут содержать другие терапевтически значимые вещества, особенно антидиабетические или гиполипидемические агенты, механизм действия которых отличается от механизма, по которому действуют соединения изобретения. Агенты, которые преимущественно могут быть объединены с соединениями по изобретению в единой композиции, включают, но не ограничиваются этим: бигуанидины, такие как асметформин, инсулин-высвобождающие агенты, такие как высвобождающая инсулин сульфонилмочевина глубурид и другие инсулин-высвобождающие сульфонилмочевины, лекарственные средства, снижающие уровень холестерина, такие как «статиновые» ингибиторы HMG-CoA редуктазы, такие как атровастатин, ловастатин, правастатин и симвастатин, PPAR-альфа агонисты, такие как клофибрат и гемфиброзил, PPAR-гамма агонисты, такие как тиазолидиндионы, например розиглитазон и пиоглитазон, ингибиторы альфа-глюкозидазы, такие как акарбоз (который ингибирует переваривание крахмала), и «обеденные» инсулин-высвобождающие агенты, такие как репаглинид.
Количества дополнительных агентов, объединяемых с соединениями по изобретению в единой композиции, соответствуют дозировкам, используемым в стандартной клинической практике. Разработанные безопасные и эффективные диапазоны дозировки для некоторых иллюстративных соединений указаны выше.
СХЕМЫ РЕАКЦИЙ
Биологически активные соединения по настоящему изобретению могут быть получены в соответствии со следующими схемами реакций.
Соединение формулы I', где X представляет собой -CH2CR12R13-, q и m равны 0, t равно 0 или 1, и n равно 1 или 2, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, Q представляет собой OR1, где R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, т.e. соединения формулы:

где A является таким, как описано выше, и R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, R12 и R13 независимо представляют собой водород или метил, может быть получено из соединения формулы VI по реакционной схеме, показанной на Схеме 1.
На реакционной схеме, представленной на Схеме 1, A, t, n и R9 являются такими, как указано выше. R6 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, R12 и R13 независимо представляют собой водород или метил, и Y представляет собой уходящую группу.
Соединение формулы VI преобразовывают в соединение формулы VIII с помощью реакции стадии (a) с использованием конденсации Мицунобу VI с VII с применением трифенилфосфина и диэтилазодикарбоксилата. Для проведения данной реакции стадии (а) можно использовать любые условия, обычно используемые в реакциях Мицунобу.
Соединение формулы VIII также может быть получено этерификацией или алкилированием соединения формулы VI соединением формулы IX, как в реакции стадии (b). В соединении формулы IX Y может представлять собой любую обыкновенную уходящую группу, такую как мезилокси, тозилокси или галогенид. Для проведения реакции стадии (b) можно использовать любой обычный способ этерификации гидроксильной группы с помощью реакции с галогенидом или уходящей группой. Если соединение формулы IX легко доступно, реакция стадии (b) более предпочтительна, чем реакция стадии (а).
Соединение формулы VIII преобразовывают в соединение формулы XI реакцией стадии (c) с помощью алкилирования соединения формулы VIII соединением формулы X. Данную реакцию проводят с использованием обычного основания, которое превращает ацетофенон в 3-кето сложный эфир (т.е. гамма-кето сложный эфир). Для этих целей в реакции стадии (с) можно использовать любое обычное основание. При проведении данной реакции обычно является предпочтительным использование в качестве основания солей щелочных металлов гексаметилдисилазана, таких как бис(триметилсилил)амид лития. Обычно данную реакцию проводят в инертном растворителе, например смеси тетрагидрофуран:1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинон (5:1). Для проведения реакции стадии (с) можно использовать любые условия, обычные для таких реакций алкилирования.
Соединение формулы XI представляет собой соединение формулы I', где Rl представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XI может быть преобразовано в свободную кислоту, то есть соединение формулы I', где R1 представляет собой H, гидролизом сложноэфирной группы. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы I', где R1 представляет собой H.
Соединение общей формулы VII можно получать восстановлением соответствующей кислоты формулы A-(CH2)t+n-CO2H. Реакцию осуществляют, первоначально проводя этерификацию соединения формулы A-(CH2)t+n-CO2H иодистым метилом с последующим восстановлением с использованием обычного основания, например литийалюминий гидрида или тому подобного, в инертном органическом растворителе, например тетрагидрофуране или тому подобном. Для проведения данной реакции можно использовать любые условия, традиционные для таких реакций восстановления.
Схема 1

Соединение формулы VII, где A представляет собой 2,6-диметилфенил, может быть получено из соединения формулы XCI по реакционной схеме, показанной на схеме 2.
На схеме 2 соединение формулы XCI может быть преобразовано в соединение формулы VII этерификацией с использованием иодистого метила с последующим восстановлением литийалюминий гидридом по реакции стадии (r''). Реакцию стадии (r'') можно проводить с использованием обычного восстановителя. При проведении данной реакции обычно является предпочтительным использовать в качестве восстановителя литийалюминий гидрид. Любые условия, общепринятые для реакций восстановления, можно использовать для проведения данной реакции.
Схема 2

Соединение формулы I, где X представляет собой -CH2-, q равно 0, m равно 1, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где A является таким, как описано выше, R1представляет собой этил, и R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, может быть получено из соединения формулы XII, где m является таким же, как и ранее, по реакционной схеме, показанной на схеме 3.
На схеме 3 A является таким, как указано выше, Y представляет собой уходящую группу, такую как галогенид, мезилокси или тозилокси. Y1 представляет собой хлор.
На схеме 3 соединение формулы XII превращают в этиловый сложный эфир формулы XIII с использованием этанола реакцией стадии (d). Для проведения данной реакции можно использовать любой общепринятый способ превращения кислоты в ее этиловый сложный эфир.
Соединение формулы XIII может быть преобразовано в соединение формулы XIV таким же способом, как описано ранее в связи с реакцией стадии (a) или (b).
На стадии (f) соединение формулы XIV гидролизуют, получая соединение формулы XV. Для проведения данной реакции можно использовать любой общепринятый способ щелочного гидролиза для гидролиза сложных эфиров.
Соединение формулы XV преобразовывают в хлорангидрид кислоты формулы XVI по реакции стадии (g) с помощью взаимодействия с хлористым тионилом. Для проведения данной реакции стадии (g) можно использовать любой общепринятый способ превращения кислоты в хлорангидрид кислоты.
Соединение формулы XVII взаимодействует с хлорангидридом кислоты формулы XVI с образованием соединения формулы XVIII по реакции стадии (h). Для проведения данной реакции можно использовать любое обычное основание, при этом предпочтительным основанием является пиридин. Полученные ацилированные кислоты Мелдрума не выделяли и вместо обработки их нагревали при кипении в абсолютном этаноле с получениеием 2-кетоэфиров. Для проведения реакции стадии (h) можно использовать любые общепринятые условия.Соединение формулы XVIII представляет собой соединение формулы I, где R1 представляет собой этил.
Схема 3

Соединение формулы I', где q равно 1, R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, где X представляет собой -CH2CR12R13-, m равно 0, t равно 0 или 1, и n равно 1 или 2, т.e. соединения формулы:

где A является таким, как указано выше, R1представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, и R12 и R13 независимо представляют собой водород или метил, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, Q представляет собой OR1, где R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, может быть получено из соединения формулы XIX, где t и A являются такими, как указано выше, по реакционной схеме, представленной на схеме 4.
На схеме 4, t, n, A, R1, R9, R12, R13 и R5 являются такими, как указано выше. R6 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Y1 представляет собой хлор.
На схеме 4 соединение формулы XIX мезилируют для получения соединения формулы XX по реакции стадии (i). Для проведения мезилирования можно использовать любые обычные условия. Соединение формулы XX затем нагревают с соединением формулы XXI для получения соединения формулы XXII. Для реакции стадии (j) можно использовать любые условия, обычные для получения аминоспирта.
В соединении формулы XXII спиртовую группу затем заменяют на хлор с помощью обработки соединения формулы XXII хлористым тионилом, получая соединение формулы XXIII по реакции стадии (k). Для проведения данной реакции можно использовать любой способ для замены спиртовой группы на галоген.
Затем проводят взаимодействие соединения формулы XXIII с соединением формулы VI в присутствии основания с использованием диметилформамида в качестве растворителя по реакции стадии (l), получая соответствующее соединение формулы XXIV. Положение заместителей в соединении формулы VI определяется положением заместителей в соединении формулы XXIV. Для проведения реакции стадии (l) можно использовать любой обычный способ этерификации гидроксильной группы галогенидом в присутствии основания (предпочтительным основанием является карбонат калия). Соединение формулы XXIV превращают в соединение формулы XXV реакцией стадии (m) с использованием алкилирования соединения формулы XXIV соединением формулы X в присутствии силиламида металла (например, гексаметилдисилан лития или гексаметилдисилан натрия). Данную реакцию проводят таким же образом, как описано для реакции стадии (c) для схемы 1.
Соединение формулы XXV представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XXV может быть превращено в свободную кислоту, т.е. соединение формулы I, в котором R1 представляет собой H, путем гидролиза сложного эфира. Любые обычные способы гидролиза сложноэфирной группы будут приводить к соединению формулы I', где R1 представляет собой H.
Схема 4

Соединение формулы I', где X представляет собой -CH2CH(NHAc)-, m равно 0, q равно 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где A является таким, как указано выше, R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, и R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, Q представляет собой OR1, где R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, может быть получено из соединения формулы VIII по реакционной схеме, представленной на схеме 5.
На схеме 5, t, n, A, R9 и R1 являются такими, как указано выше. R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Соединение формулы VIII получают таким же способом, как описано ранее в связи с реакцией стадии (a) или (b) на схеме 1.
Соединение формулы VIII превращают в соединение формулы XXVI селективным бромированием метилкетонного фрагмента реакцией стадии (n) обработкой соединения формулы VIII CuBr2. Для проведения реакции стадии (n) можно использовать любые условия селективного бромирования для превращения метилкетона в 1-бромкетон.
Соединение формулы XXVI может быть преобразовано в соединение формулы XXVIII реакцией стадия (o) обработкой соединения формулы XXVI натриевой солью соединения формулы XXVII в этаноле. Для проведения данной реакции можно использовать любые обычные условия реакции алкилирования.
Соединение формулы XXVIII превращают в соединение формулы XXIX реакцией стадии (p), представляющей деэтерификацию с использованием 4 эквивалентов гидроксида натрия. Наблюдали первоначальную монодеэтерификацию с последующим медленным гидролизом оставшегося этилового сложного эфира. Удаление растворителя и выдерживание остатка в уксусной кислоте приводит к соединению формулы XXIX.
Соединение формулы XXIX представляет собой соединение формулы I', в котором R1 представляет собой H.
Соединение формулы XXIX может быть преобразовано в соединение формулы XXXI, где R7 представляет собой алкильную цепь, содержащую от 1 до 7 атомов углерода, этерификацией карбоновой кислоты соединением формулы XXX с использованием N,N-дициклогексилкарбодиимида в качестве дегидратирующего конденсирующего агента. Для проведения реакции стадии (q) можно использовать любые условия, подходящие для данной реакции.
Соединение формулы XXXI представляет собой соединение формулы I', где Rlпредставляет собой алкильную цепь, содержащую от 1 до 7 атомов углерода.
Схема 5


где t, n и A являются такими, как описано выше, R9представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, и R1 представляет собой этил, может быть получено из соединения формулы LX по реакционной схеме, представленной на схеме 6.
На реакционной схеме, представленной на схеме 6, A, t, R9и n являются такими, как указано выше, Y представляет собой уходящую группу и Y1 представляет собой хлор.
На схеме 6 соединение формулы LX превращают в соединение формулы LXI таким же образом, как описано ранее в связи с реакциями стадий (a) или (b) на схеме 1.
На стадии (q') соединение формулы LXI гидролизуют для получения соединения формулы LXII таким же образом, как описано в связи с реакцией стадии (f) на схеме 3.
Соединение формулы LXII превращают в соединение формулы LXIII по реакции стадии (r') таким же образом, как описано в связи с реакцией стадии (g) на схеме 3.
Соединение формулы LXIV первоначально обрабатывают 2 эквивалентами н-бутиллития при низкой температуре и затем добавляют соединение формулы LXIII для получения соединения формулы LXV (Weirenga, W.; Skulnick, H.I, J. O. C. 1979, 44, 310-311). Соединение формулы LXV представляет собой соединение формулы 1, где R1 представляет собой этил.
Схема 6

Соединение формулы I, где q равно 1, R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, где X представляет собой -CH2-, m равно 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где A является таким же, как описано выше, R9 представляет собой водород, галоген, или алкокси, содержащий от 1 до 3 атомов углерода, и R1 представляет собой этил, может быть получено из соединения формулы LX по реакционной схеме, представленной на схеме 7.
На реакционной схеме, представленной на схеме 7, A, t, R9 и n являются такими, как указано выше, Y1 представляет собой хлор. R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода.
На схеме 7 соединение формулы LX взаимодействует с соединением формулы XXIII (получают таким же образом, как описано на схеме 4) для получения соединения формулы LXVI по реакции стадии (t'). Данную реакцию проводят таким же образом, как описано ранее в связи с реакцией стадии (l) на схеме 4.
На стадии (u') соединение формулы LXVI гидролизуют для получения соединения формулы LXVII таким же образом, как описано для реакции стадии (f) на схеме 3.
Соединение формулы LXVII превращают в соединение формулы LXVIII по реакции стадии (v') таким же образом, как описано в связи с реакцией стадии (g) на схеме 3.
Соединение формулы LXIV первоначально обрабатывают 2 эквивалентами н-бутиллития при низкой температуре и затем добавляют соединение формулы LXIII для получения соединения формулы LXV (Weirenga, W.; Skulnick, H.I, J.O. C. 1979, 44, 310-311). Соединение формулы LXIX представляет собой соединение формулы I, где R1 представляет собой алкильную группу, содержащую 2 атома углерода.
Схема 7

Соединение формулы I', где q равно 1, R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, Q представляет собой OR1, где R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, X представляет собой -CH2CH(NHAc)-, m равно 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где t, n, A и R1 являются такими, как указано выше, может быть получено из соединения формулы VI по реакционной схеме, представленной на схеме 8.
На схеме 8 t, n, A, R9 и R1 являются такими, как указано выше. R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода. Y1 представляет собой хлор.
Соединение формулы XXIV получают таким же образом, как описано ранее в связи с реакцией стадии (l) на схеме 4.
Соединение формулы XXIV превращают в соединение формулы LXX селективным бромированием метилкетонного фрагмента по реакции стадии (x') обработкой соединения формулы XXIV CuBr2. Для проведения реакции стадии (x') можно использовать любые условия селективного бромирования для превращения метилкетона в 1-бромкетон.
Соединение формулы LXX может быть преобразовано в соединение формулы LXXI по реакции стадии (y') обработкой соединения формулы LXX натриевой солью соединения формулы XXVII в этаноле. Для проведения реакции алкилирования можно использовать любые общепринятые условия.
Соединение формулы LXXI превращают в соединение формулы LXXII по реакции стадии (z') путем деэтерификации с использованием 4 эквивалентов гидроксида натрия. Это указывает на первоначальную монодеэтерификацию с последующим медленным гидролизом оставшейся этильной сложноэфирной группы. Удаление растворителя и выдерживание остатка в уксусной кислоте приводит к соединению формулы LXXII.
Соединение формулы LXXII представляет собой соединение формулы I', где R1 представляет собой Н.
Соединение формулы LXXII может быть преобразовано в соединение формулы LXXIII, где R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, этерификацией карбоновой кислоты соединением формулы XXX с использованием N,N-дициклогексилкарбодиимида в качестве дегидратирующего конденсирующего агента. Для проведения реакции стадии (a'') можно использовать любые условия, подходящие для данной реакции.
Соединение формулы LXXIII представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Схема 8

Соединение формулы I', где X представляет собой -CH2CH(NHAc)-, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, Q представляет собой OR1, где R1представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, m равно 1, q равно 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединение формулы:

где A является таким, как указано выше, и R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, может быть получено из соединения формулы LXXIV по реакционной схеме, представленной на схеме 9.
На схеме 9 t, n, A, R9 и R1 являются такими, как указано выше. R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода.
Соединение формулы LXXIV может быть получено в соответствии со способом, описанным Murphy et al J.C.С. Perkin 1, 1980, 1555-1566.
Соединение формулы LXXIV можно проалкилировать для получения соединения формулы LXXV по реакции стадии (b'') с использованием либо соединения формулы VII, применяя тот же способ, как описано в связи с реакцией стадии (a) на схеме 1, либо соединения формулы IX с использованием карбоната калия в качестве основания для алкилирования. Реакцию проводят таким же образом, как описано ранее в связи с реакцией стадии (l) на схеме 4.
Соединение формулы LXXV затем селективно бромируют при 0°C с использованием 30 мас.% HBr в уксусной кислоте, добавляемой по каплям, для получения соединения формулы LXXVI по реакции стадии (c''). Для проведения данной реакции стадии (c'') можно использовать любой обычный способ для селективного превращения замещенного ацетона в 1-бромацетон.
Соединение формулы LXXVI превращают в соединение формулы LXXVII по реакции стадии (d'') таким же образом, как описано ранее в связи с реакцией стадии (o) на схеме 5.
Соединение формулы LXXVII превращают в соединение формулы LXXVIII по реакции стадии (e'') путем деэтерификации с использованием 4 эквивалентов гидроксида натрия. Наблюдали первоначальную монодеэтерификацию с последующим медленным гидролизом оставшейся этильной сложноэфирной группы. Удаление растворителя и выдерживание остатка в уксусной кислоте приводит к соединению формулы LXXVIII.
Соединение формулы LXXVIII представляет собой соединение формулы I', где R1 представляет собой Н.
Соединение формулы LXXVIII может быть преобразовано в соединение формулы LXXIX, где R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, этерификацией карбоновой кислоты соединением формулы XXX с использованием N,N-дициклогексилкарбодиимида в качестве дегидратирующего конденсирующего агента. Для проведения реакции стадии (f'') можно использовать любые условия, общепринятые для данной реакции.
Соединение формулы LXXIX представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Схема 9

Соединение формулы I', где q равно 1, R5представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, X представляет собой -CH2CH(NHAc)-, m равно 1, t равно 0 или 1, и n равно 1 или 2, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, Q представляет собой OR1, где R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, т.е. соединения формулы:

где A является таким, как указано выше, и R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, может быть получено из соединения формулы LXXIV по реакционной схеме, представленной на схеме 10.
На схеме 10 t, n, A, R9 и R1 являются такими, как указано выше. R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода. Y1 представляет собой хлор.
Соединение формулы LXXIV может быть получено способом, описанным Murphy et. al. J. C. С. Perkin 1, 1980, 1555-1566.
На схеме 10 соединение формулы LXXIV взаимодействует с соединением формулы XXIII (получают таким же образом, как описано на схеме 4) для получения соединения формулы LXXX по реакции стадии (g''). Данную реакцию проводят таким же образом, как описано ранее в связи с реакцией стадии (l) на схеме 4.
Соединение формулы LXXX затем селективно бромируют при 0° C с использованием 30 мас.% HBr в уксусной кислоте, добавляемой по каплям, для получения соединения формулы LXXXI по реакции стадии (h''). Для проведения реакции стадии (h'') можно использовать любой обычный способ для превращения замещенного ацетона в 1-бромацетон.
Соединение формулы LXXXI преобразовывают в соединение формулы LXXXII по реакции стадии (i'') таким же образом, как описано ранее в связи с реакцией стадии (o) на схеме 5.
Соединение формулы LXXXII превращают в соединение формулы LXXXIII по реакции стадии (j'') таким же образом, как описано для реакции стадии (p) на схеме 5.
Соединение формулы LXXXIII представляет собой соединение формулы I', где R1 представляет собой Н.
Соединение формулы LXXXIII может быть преобразовано в соединение формулы LXXXIV, где R7 представляет собой алкильную цепь, содержащую от 1 до 7 атомов углерода, этерификацией карбоновой кислоты соединением формулы XXX с использованием N,N-дициклогексилкарбодиимида в качестве дегидратирующего конденсирующего агента. Для проведения реакции стадии (k'') можно использовать любые условия, подходящие для данного взаимодействия.
Соединение формулы LXXXIV представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Схема 10

Соединение формулы I', где X представляет собой -CH2CR12R13-, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, Q представляет собой OR1, где R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, q равно 0, m равно 1, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где A является таким же, как описано выше, R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, и R12 и R13независимо представляют собой водород или метил, может быть получено из соединения формулы LXXIV по реакционной схеме, представленной на схеме 11.
На реакционной схеме, представленной на схеме 11, A, t, R9, R12, R13 и n являются такими, как указано выше. R6 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, и Y представляет собой уходящую группу.
Соединение формулы LXXV получают из соединения формулы LXXIV таким же образом, как описано ранее в связи с реакцией стадии (b'') на схеме 9.
Соединение формулы LXXV превращают в соединение формулы LXXXV по реакции стадии (1'') селективным алкилированием соединения формулы LXXV соединением формулы X. Данную реакцию проводят с использованием обычного основания, которое превращает замещенный кетон в гамма-кето сложный эфир. При проведении данной реакции обычно является предпочтительным использование диизопропиламида лития в качестве основания. Алкилирование будет проходить при менее стерически затрудненной метильной группе. Обычно данную реакцию проводят в инертном растворителе, таком как тетрагидрофуран или 1,2-диметоксиэтан, при -78°С.
Соединение формулы LXXXV представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы LXXXV может быть преобразовано в свободную кислоту, т.е. соединение формулы I', в котором R1 представляет собой Н, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы I', где R1представляет собой Н.
Схема 11

Соединение формулы I', где q равно 1, R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, где X представляет собой -CH2-, m равно 1, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где A является таким, как указано выше, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, и R1 представляет собой этил, может быть получено из соединения формулы XIII, где m является таким, как указано выше по реакционной схеме, представленной на схеме 12.
На схеме 12 A является таким, как указано выше. Y1 представляет собой хлор.
Соединение формулы XIII (получают таким же образом, как описано ранее в связи с реакцией стадии (d) на схеме 3) может быть преобразовано в соединение формулы LXXXVI по реакции стадии (m'') таким же образом, как описано выше для реакции стадии (1) на схеме 4.
На стадии (n'') соединение формулы LXXXVI гидролизуют для получения соединения формулы LXXXVII. Для проведения данной реакции можно использовать любой обычный способ основного гидролиза для гидролиза сложноэфирной группы.
Соединение формулы LXXXVII превращают в хлорангидрид кислоты формулы LXXXVIII по реакции стадии (o'') с использованием взаимодействия с хлористым тионилом. Для проведения реакции можно использовать любой обычный способ превращения кислоты в галогенангидрид кислоты.
Соединение формулы XVII подвергают взаимодействию с соединением формулы LXXXVIII для получения соединения формулы LXXXIX по реакции стадии (p''). Для проведения данной реакции можно использовать любое обычное основание, при этом предпочтительным основанием является пиридин. Для проведения реакции стадии (p'') можно использовать любые подходящие условия.Соединение формулы LXXXIX представляет собой соединение формулы I, где R1 представляет собой этил.
Схема 12

Соединение формулы I', где q равно 1, R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, где X представляет собой -CH2CR12R13-, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, Q представляет собой OR1, где R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, m равно 1, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где A является таким же, как описано выше, R1 представляет собой водород или алкил, содержащий от 1 до 7 атомов углерода, и R12 и R13 независимо представляют собой водород или метил, может быть получено из соединения формулы LXXIV по реакционной схеме, представленной на схеме 13.
На реакционной схеме, представленной на схеме 13, R9, R12, R13, A, t и n являются такими, как указано выше. R6представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Соединение формулы LXXX получают из соединения формулы LXXIV таким же образом, как описано ранее в связи с реакцией стадии (g'') на схеме 10.
Соединение формулы LXXX превращают в соединение формулы XC по реакции стадии (q'') с использованием алкилирования соединения формулы LXXX соединением формулы X. Данную реакцию проводят с использованием обычного основания, которое превращает кетон в 3-кето сложный эфир. При проведении данной реакции обычно является предпочтительным использование диизопропиламиида лития в качестве основания. Алкилирование будет протекать при менее стерически затрудненной метильной группе. Обычно данную реакцию проводят в инертном растворителе, таком как тетрагидрофуран или 1,2-диметоксиэтан, при -78°С.
Соединение формулы XC представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XC может быть преобразовано в свободную кислоту, т.е. соединение формулы I', где R1 представляет собой Н, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы I', где R1представляет собой Н.
Схема 13

Соединение формулы II, где Z представляет собой

m равно 0, r равно 1, q равно 0, t равно 0 или 1, и n равно 1 или 2, R4 представляет собой -NHCO2C(CH3)3, -NHCH3 или- NHCH2CH3, т.е. соединения формулы:

где A и R1 являются такими, как указано выше, может быть получено из соединения формулы XXVI по реакционной схеме, представленной на схеме 14.
На схеме 14 t, n, A и R1 являются такими, как указано выше. R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. R8 представляет собой алкильную группу, содержащую 1-2 атома углерода. Y1 представляет собой галоген, предпочтительно бром.
На схеме 14 соединение формулы XXVI (получают таким же образом, как описано ранее в связи с реакцией стадии (n) на схеме 5) подвергают взаимодействию с соединением формулы XXXII в присутствии основания для получения соединение формулы XXXIII по реакции стадии (r). При проведении данной реакции обычно является предпочтительным использовать триэтиламин в качестве основания. Для проведения данной реакции можно использовать любой подходящий способ взаимодействия Boc-cys-OEt с галогенидом.
Соединение формулы XXXIII представляет собой соединение формулы II, где R4 представляет собой -NHCO2C(CH3)3, и R1 представляет собой этил.
Соединение формулы XXXIII может быть преобразовано в свободную кислоту, т.е. соединение формулы II, где R1 представляет собой Н, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы II где R1 представляет собой Н, и R4 представляет собой -NHCO2C(CH3)3.
Соединение формулы XXXIII превращают в соединение формулы XXXV первоначально по реакции стадии (s) снятием защитной трет-бутоксильной группы с использованием трифторуксусной кислоты и заменой на низший алкил, имеющий от 1 до 2 атомов углерода, по реакции стадии (t). Для проведения данной реакции можно использовать любой обычный способ конденсации амина с алкилгалогенидом.
Соединение формулы XXXV представляет собой соединение формулы II, где R4 представляет собой амин, имеющий от 1 до 2 атомов углерода, и R1 представляет собой алкильную группу, имеющую 2 атома углерода. Соединение формулы XXXV может быть преобразовано в свободную кислоту, т.е. соединение формулы XXXVI, где R1 представляет собой Н, основным гидролизом по реакции стадии (u). Соединение формулы XXXVI представляет собой соединение формулы II, где R4 представляет собой -NHCH3 или -NHCH2CH3, и R1 представляет собой Н.
Соединение формулы XXXVI может быть преобразовано в соединение формулы XXXVII, где R7представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, этерификацией карбоновой кислоты соединением формулы XXX с использованием N,N-дициклогексилкарбодиимида в качестве дегидратирующего конденсирующего агента. Для проведения реакции стадии (v) можно использовать любые условия, обычные для данной реакции.
Соединение формулы XXXVII представляет собой соединение формулы II, где R1 представляет собой алкил, содержащий от 1 до 7 атомов углерода, и R4 представляет собой -NHCH3 или -NHCH2CH3.
Схема 14

Соединение формулы II, где Z представляет собой

m и q равны 0, r равно 1, t равно 0 или 1, n равно 1 или 2, т.е. соединения формулы:

A является таким, как указано выше, может быть получено из соединения формулы VIII, где t, n и A являются такими, как указано выше по реакционной схеме, представленной на схеме 15.
На схеме 15 соединение формулы VIII (получают таким же образом, как описано ранее в связи с реакцией стадии (a) или (b) на схеме 1) превращают в соединение формулы XXVI таким же образом, как описано для реакции стадии (n) на схеме 5.
Соединение формулы XXVI подвергают взаимодействию с соединением формулы XXXVIII в присутствии основания, при этом предпочтительным основанием является триэтиламин, для получения соединение формулы XXXIX. Для проведения реакции стадии (w) можно использовать любой общепринятый способ для взаимодействия тиола с галогенидом.
Соединение формулы II, где Z представляет собой

m равно 0, r равно 1, t равно 0 или 1, и n равно 1 или 2, R4представляет собой Н, т.е. соединения формулы:

где t, n, A и R1 являются такими, как указано выше, может быть получено из соединения формулы VIII по реакционной схеме, представленной на схеме 15.
На реакционной схеме, представленной на схеме 15, t, n, A и R1 являются такими, как указано выше. R6 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Соединение формулы VIII получают таким же образом, как описано ранее в связи с реакцией стадии (a) или (b) на схеме 1.
Соединение формулы XXVI получают из соединения формулы VIII таким же образом, как описано ранее в связи с реакцией стадии (n) на схеме 5.
Соединение формулы XXVI подвергают взаимодействию с соединением формулы XL в присутствии основания, при этом предпочтительным основанием является триэтиламин для получения соединение формулы XLI. Для проведения реакции стадии (x) можно использовать любой обычный способ для взаимодействия тиола с 1-бромкетоном.
Соединение формулы XLI представляет собой соединение формулы II, где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XLI может быть преобразовано в свободную кислоту, т.е. соединение формулы II, где R1 представляет собой Н, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы II, где R1представляет собой Н.
Схема 15

Соединение формулы II, где Z представляет собой

r равно 0, m равно 1, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где n, t и A являются такими, как указано выше, может быть получено из соединения формулы XLII по реакционной схеме, представленной на схеме 16.
На схеме 16 t, n, и A являются такими же, как указано выше. Y представляет собой уходящую группу, такую как галогенид, мезилокси или тозилокси. Y1 представляет собой галоген, предпочтительно бром.
На схеме 16 соединение формулы XLII превращают в соединение формулы XLIII реакцией стадии (y) путем селективного замещения гидроксильной группы первичного спирта на галоген. Для проведения данной реакции можно использовать любой обычный галогенирующий агент, при этом предпочтительным галогенирующим агентом является трибромид фосфора. Данную реакцию проводят при низкой температуре. Любые условия, подходящие для данного способа, можно использовать для проведения реакции стадии (y). Соединение формулы XLIII используют сразу же без дополнительной очистки.
Соединение формулы XLIII подвергают взаимодействию с соединением формулы XXXVIII в присутствии основания для получения соединение формулы XLIV. Для проведения реакции стадии (z) можно использовать любой обычный способ конденсации тиола с галогенидом. Для проведения данной реакции можно использовать любое обычное основание, при этом предпочтительным основанием является триэтиламин.
Соединение формулы XLIV превращают в соединение формулы XLV реакцией с соединением формулы VII по реакционной стадии (a'). Данную реакцию проводят таким же образом, как описано ранее в связи с реакцией стадии (a) на схеме 1.Соединение формулы II, где Z представляет собой

r равно 0, m равно 1, t равно 0 или 1, и n равно 1 или 2, R4 представляет собой Н, т.е. соединения формулы:

где A и R1 являются такими, как указано выше, может быть получено из соединения формулы XLII по реакционной схеме, представленной на схеме 16.
На схеме 16, t, n, и A являются такими, как указано выше. Y представляет собой уходящую группу, такую как галогенид, мезилокси или тозилокси. Y1 представляет собой галоген, предпочтительно бром. R6представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Соединение формулы XLII превращают в соединение формулы XLIII таким же образом, как описано ранее в связи с реакцией стадии (y).
Соединение формулы XLIII взаимодействует с соединением формулы XL по реакции стадии (b'), как описано в связи с реакцией стадии (x) на схеме 15. Соединение формулы XLVI превращают в соединение формулы XLVII по реакции стадии (c'). Данную реакцию проводят таким же образом, как описано для реакционных стадий (a) или (b) на схеме 1.
Соединение формулы XLVII представляет собой соединение формулы II, где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XLVII может быть преобразовано в свободную кислоту, т.е. соединение формулы II, где R1 представляет собой Н, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы II, где R1представляет собой Н.
Схема 16

Соединение формулы II, где Z представляет собой

r равно 0, m равно1, t равно 0 или 1, и n равно 1 или 2, т.e. соединения формулы:

где t, n, A и R1являются такими, как указано выше, R4 представляет собой -NHCO2C(CH3)3, -NHCH3, -NHCH2CH3, может быть получено из соединения формулы XLIII по реакционной схеме, представленной на схеме 17.
На схеме 17 t, n, A и R1 являются такими, как указано выше. Y представляет собой уходящую группу, такую как галогенид, мезилокси или тозилокси. R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, и R8 представляет собой алкильную группу, содержащую от 1 до 2 атомов углерода. Y1 представляет собой галоген, предпочтительно бром.
На схеме 17 соединение формулы XLIII (получают таким же образом, как описано ранее для реакции стадии (y) на схеме 16) подвергают взаимодействию с соединением формулы XXXII в присутствии основания для получения соединения формулы XLVIII по реакции стадии (d'). При проведении данной реакции обычно является предпочтительным использовать триэтиламин в качестве основания. Для проведения данной реакции можно использовать любой обычный способ взаимодействия Boc-cyst-OEt с галогенидом.
Соединение формулы XLIX получают взаимодействием соединения формулы XLVIII с соединением формулы VII или IX. Данную реакцию проводят таким же образом, как описано для реакционных стадий (a) или (b) на схеме 1.
Соединение формулы XLIX представляет собой соединение формулы II, где R4 представляет собой -NHCO2C(CH3)3 и R1 представляет собой алкильную группу, содержащую 2 атома углерода.
Соединение формулы XLIX может быть преобразовано в свободную кислоту, т.е. соединение формулы II, где R1 представляет собой Н, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы II, где R1представляет собой Н и R4 представляет собой -NHCO2C(CH3)3.
Соединение формулы XLIX превращают в соединение формулы L первоначально по реакции стадии (f), снимая защитную трет-бутоксильную группу с использованием трифторуксусной кислоты и затем заменяя ее на низший алкил, содержащий от 1 до 2 атомов углерода по реакции стадии (g'). Для проведения данной реакции можно использовать любой обычный способ конденсации амина с алкилгалогенидом.
Соединение формулы L представляет собой соединение формулы II, где R4 представляет собой амин, имеющий 1-2 атома углерода, и R1 представляет собой алкильную группу, содержащую 2 атома углерода.
Соединение формулы L может быть преобразовано в свободную кислоту, т.е. соединение формулы LI, где R1 представляет собой Н, основным гидролизом по реакционной стадии (h').
Соединение формулы LI представляет собой соединение формулы II, где R4 представляет собой -NHCH3 или -NHCH2CH3, и R1 представляет собой Н. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы II, где R1представляет собой Н.
Соединение формулы LI может быть преобразовано в соединение формулы LII, где R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, этерификацией карбоновой кислоты соединением формулы XXX с использованием N,N-дициклогексилкарбодиимида в качестве дегидратирующего конденсирующего агента. Для проведения реакции стадии (i') можно использовать любые обычные для данной реакции условия.
Соединение формулы LII представляет собой соединение формулы II, где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, и R4 представляет собой -NHCH3 или -NHCH2CH3.
Схема 17

Соединение формулы III

где n равно 1 или 2 и A является таким, как указано выше, может быть получено из соединения формулы LIII, по реакционной схеме, представленной на схеме 18, где n, A и Y являются такими, как указано выше.
На схеме 18 соединение формулы LIII превращают в соединение формулы LIV таким же образом, как описано в связи с реакциями стадий (a) или (b) на Схеме 1.
Соединение формулы LIV превращают в соединение формулы III по реакции стадии (k') нагреванием соединения формулы LIV с азидом натрия в присутствии хлорида аммония в диметилформамиде. Для проведения данной реакции можно использовать любой обычный способ для преобразования нитрила в тетразол.
Схема 18

Соединение формулы IV

где R1 является таким, как указано выше, может быть получено из 2',6'-дифторацетофенона по реакционной схеме, представленной на схеме 19.
На схеме 19 R6 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Соединение формулы LV превращают в соединение формулы LVI по реакции стадии (l') таким же образом, как описано ранее в связи с реакцией стадии (c) на схеме 1.
Соединение формулы LVI представляет собой соединение формулы IV, где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы LVI может быть преобразовано в свободную кислоту, т.е. соединение формулы IV, где R1 представляет собой Н, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы IV, где R1представляет собой Н.
Схема 19

Соединение формулы V

где n, A и R1 являются такими, как указано выше, R14 представляет собой гидрокси, может быть получено из соединения формулы VI по реакционной схеме, представленной на схеме 20.
На схеме 20 n, A являются такими, как указано выше. Y представляет собой уходящую группу, такую как галогенид, мезилокси или тозилокси. R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, и R8 представляет собой алкильную группу, содержащую от 1 до 2 атомов углерода.
Соединение формулы VI превращают в соединение формулы VIII таким же образом, как описано ранее в связи с реакциями стадий (a) или (b) на схеме 1.
Соединение формулы VIII подвергают взаимодействию с соединением формулы LVII по реакции стадии (m') в присутствии свежеприготовленного алкоксида натрия при комнатной температуре для получения соединение формулы LVIII. Для проведения реакции можно использовать любые обычные условия для такого алкилирования.
Соединение формулы LVIII представляет собой соединение формулы V, где R1 представляет собой алкильную группу, содержащую от 1 до 2 атомов углерода. Соединение формулы LVIII может быть преобразовано в свободную кислоту, т.е. соединение формулы V, где R1 представляет собой Н, гидролизом сложного эфира по реакции стадии (n'). Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы V, где R1представляет собой Н.
Соединение формулы LVIII может быть преобразовано в соединение формулы LIX, где R7 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, этерификацией карбоновой кислоты соединением формулы XXX с использованием N,N-дициклогексилкарбодимида в качестве дегидратирующего конденсирующего агента. Любые обычные условия для такой реакции можно использовать для проведения реакции стадии (o').
Соединение формулы LIX представляет собой соединение формулы V, где RI представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Схема 20

Соединение формулы I', где X представляет собой -CH2CH2-, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, q и m равны 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где Q представляет собой NR10R11, где R10 представляет собой водород и R11 представляет собой гидроксильную группу; t, n, A и R9 являются такими, как описано выше, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 21.
На реакционной схеме 21 A, t, R9, R6 и n являются такими, как указано выше.
Соединение формулы XI получают таким же образом, как описано для реакционной схемы, представленной на схеме 1.
Соединение формулы XI может быть преобразовано в соединение формулы XCII по реакционной стадии (s'') обработкой соединения формулы XI гидрохлоридом гидроксиламина в органическом растворителе, например этаноле, тетрагидрофуране или тому подобном. Реакцию проводят с использованием органического основания, например гидроксида калия или тому подобного. Для проведения данной реакции можно использовать любые условия, общепринятые для получения гидроксамовых кислот.
Соединение формулы I', где X представляет собой -CH2-CH2-, q и m равны 0, t равно 0 или 1, и n равно 1 или 2, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, т.е. соединения формулы:

где t, n, A и R9 являются такими, как описано выше.
Q представляет собой NR10R11, где R10 и R11представляют собой водород, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 21.
На реакционной схеме, представленной на схеме 21, A, t, R9 и n являются такими, как указано выше. R1 представляет собой Н, R6представляет собой алкил, содержащий от 1 до 7 атомов углерода.
Соединение формулы XI получают таким же образом, как описано для реакционной схемы, представленной на схеме 1. Соединение формулы XI представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XI может быть преобразовано в свободную кислоту, т.е. соединение формулы I', где R1 представляет собой H, гидролизом сложного эфира.
Соединение формулы XI может быть преобразовано в соединение формулы XCIII по реакционной стадии (t'') путем первоначальной активации с использованием, например, гексафторфосфата бензотриазол-1-илокситриспирролидинофосфония или тому подобного в органическом растворителе, например хлористом метилене, N,N-диметилформамиде или тому подобном с последующим добавлением водного гидроксида аммония или аммиака. Реакцию проводят с использованием органического основания, например триэтиламина, диизопропилэтиламина или тому подобного. Для проведения реакции стадии (t'') можно использовать любые обычные для синтеза амида условия.
Соединение формулы I', где X представляет собой -CH2-CH2-, q и m равно 0, t равно 0 или 1, и n равно 1 или 2, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, т.е. соединения формулы:

где t, n, A и R9 являются такими, как описано выше. Q представляет собой NR10R11, где R10 и R11независимо представляют собой водород или алкил, содержащий от 1 до 3 атомов углерода, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 21.
На реакционной схеме, представленной на схеме 21, A, t, R9 и n являются такими, как указано выше. R1 представляет собой H, и R6 представляет собой алкил, содержащий от 1 до 7 атомов углерода.
Соединение формулы XI получают таким же образом, как описано для реакционной схемы, представленной на схеме 1. Соединение формулы XI представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XI может быть преобразовано в свободную кислоту, т.e. соединение формулы I', где R1представляет собой H, гидролизом сложного эфира.
Соединение формулы XI может быть преобразовано в соединение формулы XCIV или первоначальным взаимодействием с хлорирующим реагентом, например хлористым тионилом или тому подобным, а затем взаимодействием галогенангидрида кислоты с соответствующим амином. Для проведения реакции стадии (u'') можно использовать любой подходящий способ конденсации амина с галогенангидридом кислоты. Или можно использовать конденсацию соответствующего амина с соединением формулы XI с использованием 1,3-дициклогексилкарбодиимида в качестве конденсирующего агента.
Для проведения реакции стадии (u'') можно использовать любой обычный способ конденсации амина с кислотой.
Схема 21
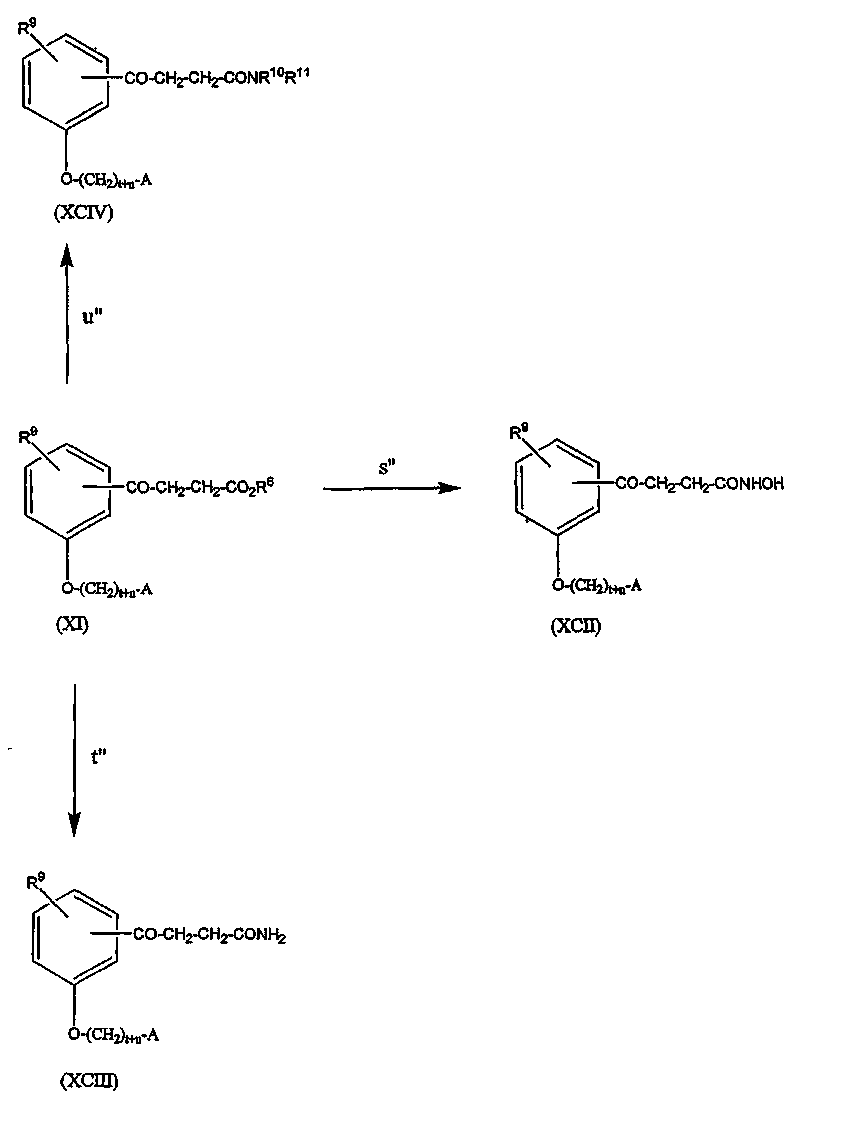
Соединение формулы I', где X представляет собой -CH2-CH2-, q равно 1, R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, m равно 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где Q представляет собой NR10R11, где R10 представляет собой водород, и R11 представляет собой гидроксильную группу; A, t, n и R9 являются такими, как описано выше, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 22.
На реакционной схеме 22 q, A, t, R5, R9 и n являются такими, как описано выше. R6представляет собой алкил, содержащий от 1 до 7 атомов углерода.
Соединение формулы XXV получают таким же образом, как описано для реакционной схемы, представленной на схеме 4.
Соединение формулы XXV может быть преобразовано в соединение формулы XCV по реакционной стадии (v'') таким же образом, как описано для реакционной стадии (s'') схемы 21.
Соединение формулы I', где X представляет собой -CH2-CH2-, q равно 1, R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, m равно 0, t равно 0 или 1, и n равно 1 или 2, R1 представляет собой H, т.е. соединения формулы:

где q, t, n, A, R5 и R9 являются такими, как описано выше; Q представляет собой NR10R11, где R10 и R11 представляют собой водород, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 22.
На реакционной схеме, представленной на схеме 22, q, A, t, R5, R9 и n являются такими, как указано выше. R1 представляет собой H, и R6 представляет собой алкил, содержащий от 1 до 7 атомов углерода.
Соединение формулы XXV получают таким же образом, как описано для реакционной схемы, представленной на схеме 4. Соединение формулы XXV представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XXV может быть преобразовано в свободную кислоту, т.е. соединение формулы I', где R1 представляет собой H, гидролизом сложного эфира.
Соединение формулы XXV может быть преобразовано в соединение формулы XCVI по реакционной стадии (w'') таким же образом, как описано для стадии (t'') реакционной схемы 21.
Соединение формулы I', где X представляет собой -CH2-CH2-, q равно 1, R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, R9 представляет собой водород, галоген, или алкокси, содержащий от 1 до 3 атомов углерода, m равно 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где q, t, n, A, R5 и R9 являются такими, как описано выше; Q представляет собой NR10R11, где R10 и R11 независимо представляют собой водород или алкил, содержащий от 1 до 3 атомов углерода, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 22.
На реакционной схеме 22 q, A, t, R5, R9 и n являются такими, как указано выше. R6 представляет собой алкил, содержащий от 1 до 7 атомов углерода. R1 представляет собой H.
Соединение формулы XXV получают таким же образом, как описано для реакционной схемы, представленной на схеме 4. Соединение формулы XXV представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XXV может быть преобразовано в свободную кислоту, т.е. соединение формулы I', где R1 представляет собой H, гидролизом сложного эфира. Соединение формулы XXV может быть преобразовано в соединение формулы XCVII по реакционной стадии (x'') таким же образом, как описано для стадии (u'') реакционной схемы 21.
Схема 22

Соединение формулы I', где X представляет собой -CH2-CH2-, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, m равно 1, q равно 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где Q представляет собой NR10R11, где R10 представляет собой водород, R11 представляет собой гидроксильную группу. t, n, A и R9 являются такими, как описано выше, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 23.
На реакционной схеме 23 A, t, R9 и n являются такими, как описано выше. R6 представляет собой алкил, содержащий от 1 до 7 атомов углерода.
Соединение формулы LXXXV получают таким же образом, как описано на реакционной схеме, представленной на схеме 11.
Соединение формулы LXXXV может быть преобразовано в соединение формулы XCVIII по реакционной стадии (y'') таким же образом, как описано для реакционной стадии (s'') схемы 21.
Соединение формулы I', где X представляет собой -CH2-CH2-, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, m равно 1, q равно 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где t, n, A и R9являются такими, как описано выше; Q представляет собой NR10R11, где R10 и R11представляют собой водород, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 23.
На реакционной схеме, представленной на схеме 23, A, t, R5, R9 и n являются такими, как указано выше. R1 представляет собой H. R6 представляет собой алкил, содержащий от 1 до 7 атомов углерода.
Соединение формулы LXXXV получают таким же образом, как описано для реакционной схемы, представленной на схеме 11. Соединение формулы LXXXV представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы LXXXV может быть преобразовано в свободную кислоту, т.е. соединение формулы I', где R1 представляет собой H, гидролизом сложного эфира.
Соединение формулы LXXXV может быть преобразовано в соединение формулы XCIX по реакционной стадии (z'') таким же образом, как описано для реакционной стадии (t'') реакционной схемы 21.Соединение формулы I', где X представляет собой -CH2-CH2-, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, m равно 1, q равно 0, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где t, n, A и R9 являются такими, как описано выше, Q представляет собой NR10R11, где R10 и R11 независимо представляют собой водород или алкил, содержащий от 1 до 3 атомов углерода, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 23.
На реакционной схеме 23 A, t, R9 и n являются такими, как указано выше. R1представляет собой H. R6 представляет собой алкил, содержащий от 1 до 7 атомов углерода.
Соединение формулы LXXXV получают таким же образом, как описано для реакционной схемы, представленной на схеме 11. Соединение формулы LXXXV представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы LXXXV может быть преобразовано в свободную кислоту, т.е. соединение формулы I', где R1 представляет собой H, гидролизом сложного эфира.
Соединение формулы LXXXV может быть преобразовано в соединение формулы C по реакционной стадии (a''') таким же образом, как описано для стадии (u'') реакционной схемы 21.
Схема 23

Соединение формулы I', где X представляет собой -CH2-CH2-, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, q равно 1, R5представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, m равно 1, t равно 0 или 1, и n равно 1 или 2, т.e. соединения формулы:

где Q представляет собой NR10R11, где R10 представляет собой водород и R11 представляет собой гидроксильную группу; t, n, A, R5 и R9являются такими, как описано выше, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 24.
На реакционной схеме 24 q, A, t, n, R5, R9 и R6 являются такими, как описано выше.
Соединение формулы XC получают таким же образом, как описано для реакционной схемы, представленной на схеме 13.
Соединение формулы XC может быть преобразовано в соединение формулы CII по реакционной стадии (b''') таким же образом, как описано для реакционной стадии (s'') схемы 21. Соединение формулы I', где X представляет собой -CH2-CH2-, R9 представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, q равно 1, R5представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, m равно 1, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где q, t, n, A, R5 и R9 являются такими, как описано выше. Q представляет собой NR10R11, где R10 и R11представляют собой водород, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 24.
На реакционной схеме, представленной на схеме 24, q, A, t, R5, R9 и n являются такими, как указано выше. R1 представляет собой H. R6 представляет собой алкил, содержащий от 1 до 7 атомов углерода.
Соединение формулы XC получают таким же образом, как описано для реакционной схемы, представленной на схеме 13. Соединение формулы XC представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XC может быть преобразовано в свободную кислоту, т.е. соединение формулы I', где R1 представляет собой H, гидролизом сложного эфира.
Соединение формулы XC может быть преобразовано в соединение формулы CIII по реакционной стадии (c''') таким же образом, как описано для стадии (t'') реакционной схемы 21.
Соединение формулы I', где X представляет собой -CH2-CH2-, R9представляет собой водород, галоген или алкокси, содержащий от 1 до 3 атомов углерода, q равно 1, R5 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, m равно 1, t равно 0 или 1, и n равно 1 или 2, т.е. соединения формулы:

где q, t, n, A, R5 и R9 являются такими, как описано выше, Q представляет собой NR10R11, где R10 и R11 независимо представляют собой водород или алкил, содержащий от 1 до 3 атомов углерода, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 24.
На реакционной схеме 24 A, t, R5, R9 и n являются такими, как указано выше. R1 представляет собой H. R6 представляет собой алкил, содержащий от 1 до 7 атомов углерода.
Соединение формулы XC получают таким же образом, как описано для реакционной схемы, представленной на схеме 13. Соединение формулы XC представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XC может быть преобразовано в свободную кислоту, т.е. соединение формулы I', где R1представляет собой H, гидролизом сложного эфира.
Соединение формулы XC может быть преобразовано в соединение формулы CIV по реакционной стадии (d''') таким же образом, как описано для стадии (u'') реакционной схемы 21.
Схема 24

Соединение формулы V', где n равно 1 или 2, t равно 0, R1, R9 и R14 представляют собой H, т.е. соединения формулы:

где t, n, A, R9, R14 и R1 являются такими, как описано выше, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 25.
На реакционной схеме, представленной на схеме 25, A и n являются такими, как указано выше. R6 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Соединение формулы XI получают таким же образом, как описано для реакционной схемы, представленной на схеме 1.
Соединение формулы XI может быть преобразовано в соединение формулы CV по реакционной стадии (e'''), обработкой соединения формулы XI бромом или тому подобным в органическом растворителе, например простом эфире, четыреххлористом углероде, где предпочтительным органическим растворителем является простой эфир.
Что касается температуры реакции, то реакцию можно проводить при температуре от охлаждения льдом до комнатной температуры, при этом предпочтительным является охлаждение льдом.
Соединение формулы CV может быть преобразовано в соединение формулы CVI по реакционной стадии (f'') дегидробромированием. Реакцию проводят с использованием обычного основания, при этом предпочтительным является триэтиламин и тому подобное, в органическом растворителе, например четыреххлористом углероде или тому подобном. Для проведения реакции стадии (f''') можно использовать любые условия, общепринятые для дегидробромирования.
Соединение формулы CVI представляет собой соединение формулы V', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы CVI может быть преобразовано в свободную кислоту, т.е. соединение формулы V', где R1представляет собой Н, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к получению соединения формулы V', где R1 представляет собой H.
Схема 25

Соединение формулы CXVI, где X представляет собой -CH2-CH2-, t равно 0 или 1, и n равно 1 или 2, R1 и R9 представляют собой H, т.е. соединения формулы:

где t, n, A и R9 являются такими, как описано выше, R1 представляет собой H, может быть получено из соединения формулы

по реакционной схеме, представленной на схеме 26.
На реакционной схеме 26 A, t, n и R9 являются такими, как указано выше. R1 представляет собой H. R6 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода.
Соединение формулы XI получают таким же образом, как описано для реакционной схемы, представленной на схеме 1. Соединение формулы XI представляет собой соединение формулы I', где R1 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода. Соединение формулы XI может быть преобразовано в свободную кислоту, т.е. соединение формулы I', где R1 представляет собой H, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы I', где R1 представляет собой H.
Соединение формулы XI превращают в соединение формулы CVII по реакционной стадии (g''') восстановлением по Вольфу-Кижнеру обработкой соединения формулы XI гидразин гидратом и гидроксидом калия в органическом растворителе, например этиленгликоле или тому подобном. Любые условия, обычные для реакций восстановления по Вольфу-Кижнеру, можно использовать для проведения реакционной стадии (g''').
Схема 26

Соединение формулы XCI, где n равно 1 или 2, R9 представляет собой H, и R1представляет собой водород или алкил, содержащий от 1 до 3 атомов углерода, т.е. соединения формулы:

где n, A, R9 и R1 являются такими, как описано выше, может быть получено из соединения формулы
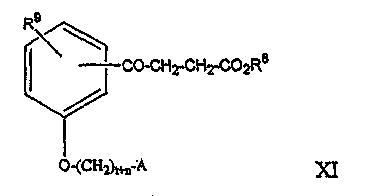
по реакционной схеме, представленной на схеме 27.
На реакционной схеме, представленной на схеме 27, R9 представляет собой атом водорода, t равно 0, R6 представляет собой алкильную группу, содержащую от 1 до 7 атомов углерода, A и n являются такими, как описано выше.
Соединение формулы XI получают таким же образом, как описано для реакционной схемы, представленной на схеме 1.
Соединение формулы XI может быть преобразовано в соединение формулы CVIII по реакционной стадии (h''') селективным восстановлением кетонной группы до спирта. Данную реакцию проводят с использованием обычных восстановителей, например боргидрида натрия в этаноле, бис-3-метил-2-бутил-борана в тетрагидрофуране и тому подобных. Для проведения реакции стадии (h''') можно использовать любые условия, обычные для таких реакций селективного восстановления.
Соединение формулы CVIII может быть преобразовано в соединение формулы CIX по реакционной стадии (i''') бромированием соединения формулы CVIII с помощью бромирующих агентов, например трехбромистого фосфора в тетрагидрофуране или диоксане, бромистого водорода в уксусной кислоте или диоксане, четырехбромистого углерода и бис(1,2-дифенилфосфино)этана или тому подобных. Любые условия, обычные для таких реакций бромирования, можно использовать для проведения реакции стадии (i''').
Соединение формулы CIX может быть преобразовано в соединение формулы CX по реакционной стадии (j''') дегидробромированием. Реакцию проводят с использованием обычного основания, предпочтительным основанием является триэтиламин или тому подобное, в органическом растворителе, например четыреххлористом углерода или тому подобном. Любые условия, обычные для таких реакций дегидробромирования, можно использовать для проведения реакционной стадии (j''').
Соединение формулы CX представляет собой соединение формулы XCI, где R1 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода. Соединение формулы CX может быть преобразовано в свободную кислоту, т.е. соединение формулы XCI, где R1 представляет собой H, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы XCI, где R1 представляет собой H.
Схема 27

Соединение формулы CXVII, где X представляет собой -CH2-CH2-, и n равно 0 или 2, R15 представляет собой водород или низшую алкильную группу, содержащую от 1 до 3 атомов углерода, R9 представляет собой гидрокси, водород, алкоксильную группу, содержащую от 1 до 3 атомов углерода, атом галогена, R1 представляет собой водород или алкил, содержащий от 1 до 3 атомов углерода, т.е. соединения формулы:

где n, A, R9 и R15являются такими, как описано выше, может быть получено взаимодействием соединения формулы

с соединением формулы

по реакционной схеме, представленной на схеме 28.
На реакционной схеме, представленной на схеме 28, A, n, R9, R15 являются такими, как описано выше, R6 представляет собой алкил, содержащий от 1 до 3 атомов углерода.
Соединение формулы CXI может быть преобразовано в соединение формулы CXIII по реакционной стадии (k''') обработкой соединения формулы CXI конденсирующим агентом, например диэтилцианофосфатом, 1-этил-3-(3'-диметиламинопропил)карбодимидом или тому подобным, в органическом растворителе, например хлористом метилене, N,N-диметилформамиде, с последующим добавлением соединения формулы CXII.
Температура реакции может составлять от 0°C до комнатной температуры.
Соединение формулы CXIII может быть преобразовано в соединение формулы CXIV по реакционной стадии (l''') алкилированием соединения формулы CXIII соединением формулы X. Данную реакцию проводят таким же образом, как описано для реакционной стадии (c) реакционной схемы 1.
Соединение формулы CXIV представляет собой соединение формулы CXVII, где R9 представляет собой алкоксильную группу, содержащую от 1 до 3 атомов углерода, атом галогена. Заместитель R9 можно преобразовать в гидроксильную группу деметилированием с использованием, например, трехбромистого бора в хлористом метилене или тому подобного. Для проведения реакции можно использовать любые условия, обычные для таких реакций деметилирования.
Соединение формулы CXIV представляет собой соединение формулы CXVII, где R1 представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода, Соединение формулы CXIV может быть преобразовано в свободную кислоту, т.e. соединение формулы CXVII, где R1 представляет собой Н, гидролизом сложного эфира. Любой обычный способ гидролиза сложного эфира будет приводить к соединению формулы CXVII, где R1 представляет собой Н.
Соединения общей формулы CXI могут быть получены этерификацией соединения формулы CI с использованием галоидного алкила с последующим гидролизом сложного эфира.

где R16 представляет собой низшую алкильную группу, содержащую от 1 до 3 атомов углерода, R9представляет собой гидроксильную группу.
Взаимодействие между соединением формулы CI и алкил галогенидом можно проводить в органическом растворителе, например N,N-диметилформамиде или тому подобном, с использованием основания, например карбоната калия, карбоната цезия или тому подобного. Для проведения данной реакции можно использовать любые условия, обычные для таких реакций алкилирования. Гидролиз сложного эфира можно проводить в кислотных условиях, например с использованием кислоты или хлористоводородной кислоты, смешанной с органическим растворителем, например с этанолом, или с использованием уксусной кислоты или тому подобного. Реакцию можно проводить при температуре от комнатной до температуры кипения растворителя. Для проведения данной реакции можно использовать любые обычные условия для кислотного гидролиза сложного эфира. Кроме того, при необходимости гидролиз сложно эфира может быть проведен с использованием основных условий, например в водном растворе гидроксида натрия или в смешанном растворе гидроксида натрия в органическом растворителе, например этаноле или тому подобном. Для проведения данной реакции можно использовать любые обычные для основного гидролиза условия.
Соединения общей формулы CXII могут быть получены взаимодействием соединения формулы VII с хлорирующим агентом, например триметилсилилхлоридом, хлористым тионилом или тому подобным, в органическом растворителе, например диметилсульфоксиде, N,N-диметилформамиде или тому подобном. Температура реакции может быть от комнатной температуры до температуры кипения органического растворителя. Любые обычные для реакций хлорирования условия можно использовать для проведения реакции.
Хлорметильное промежуточное соединение преобразуют в соединение формулы CXII с использованием синтеза Габриэля при обработке хлорметильного промежуточного соединения фталимидом калия в органическом растворителе, например N,N-диметилформамиде, диоксане или тому подобном. Фталимид затем подвергают взаимодействию с гидразином по обменной реакции в органическом растворителе, например этаноле, диоксане или тому подобном, для получения соединение формулы CXII. Для проведения реакции можно использовать любые условия, обычно используемые для синтеза Габриэля.
Схема 28

Изобретение будет более понятно при ссылке на следующие примеры, которые иллюстрируют, но не ограничивают описанное здесь изобретение.
ПРИМЕРЫ ХИМИЧЕСКОГО СИНТЕЗА
Пример 1: Синтез 4-(4-(2-фторбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2-фторбензилокси)ацетофенона:
Раствор 4-гидроксиацетофенона (2,80 г, 20,6 ммоль) в безводном ДМФ (15 мл) добавляли при комнатной температуре к суспензии NaH (60% в масле, 794 г) в безводном ДМФ (20 мл). После прекращения выделения водорода добавляли по каплям 2-фторбензил бромид (3 г, 15,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 6 часов, гасили насыщенным водным раствором NH4Cl и концентрировали в вакууме. Неочищенный остаток помещали в EtOAc и промывали водой и насыщенным раствором соли. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение в виде не совсем белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,1 (м, 4H); 7,2-7,3 (м, 1H); 7,4 (т, 1H); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-(2-фторбензилокси)фенил)-4-оксобутирата:
К перемешиваемому раствору 4-(2-фторбензилокси)ацетофенона (Стадия A, 1,5 г, 6,1 ммоль) в безводном ТГФ (20 мл) и DMPU (5 мл) добавляли раствор бис(триметилсилил)амида лития (1,0 M, 7 мл) при -60°C в атмосфере аргона. Через 10 минут перемешивания при -60°C, быстро добавляли трет-бутил бромацетат (4,75 г, 24,4 ммоль). Реакционную смесь перемешивали дополнительно в течение 10 минут и затем нагревали до комнатной температуры в течение 4 часов. Неочищенную смесь помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой экстрагировали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,7 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 6,9-7,1 (м, 4H); 7,2-7,3 (м, 1H); 7,4 (т, 1H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(2-фторбензилокси)фенил)-4-оксобутановой кислоты:
Раствор трет-бутил 4-(4-(2-фторбензилокси)фенил)-4-оксобутирата (Стадия B, 1,27 г, 4,2 ммоль) в дихлорметане (25 мл) обрабатывали трифторуксусной кислотой (5 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 3 часов и концентрировали в вакууме. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5, обогащенный уксусной кислотой), получая указанное в заголовке соединение в виде белого порошка.
1H ЯМР (270 МГц, CDCl3:CD3OD): 2,6 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 6,9-7,1 (м, 4H); 7,2-7,3 (м, 1H); 7,4 (т, 2H); 7,9 (д, 2H).
Пример 2: Синтез 4-(4-(2-метоксибензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2-метоксибензилокси)ацетофенона:
Раствор 2-метоксибензилового спирта (2,99 г, 21,7 ммоль) в безводном ТГФ (5 мл) и безводном ДМФ (5 мл) добавляли к перемешиваемому раствору 4-гидроксиацетофенона (3,25 г, 23,8 ммоль), трифенилфосфина (7,36 г, 28,0 ммоль) и диэтилазодикарбоксилата (4,51 г, 25,9 ммоль) в безводном ТГФ (20 мл) при 5-10° C. Реакционную смесь перемешивали при 0°C в течение 2 часов, нагревали до комнатной температуры и концентрировали в вакууме. Остаток помещали в EtOAc и дважды промывали насыщенным раствором NaHCO3. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 99:1), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 3,9 (с, 3H); 5,2 (с, 2H); 6,9-7,1 (м, 4H); 7,3 (м,1H); 7,4 (д,1H); 7,9 (д, 2H).
Стадия B: Получение этил 4-(4-(2-метоксибензилокси)фенил)-4-оксобутирата:
К перемешиваемому раствору 4-(2-метоксибензилокси)ацетофенона (Стадия A, 1,22 г, 4,7 ммоль) в безводном ТГФ (20 мл) и DMPU (5 мл) добавляли раствор бис(триметилсилил)амида лития (1,0 M, 5 мл) в атмосфере аргона при -60°C. Через 10 минут перемешивания при -60°C быстро добавляли этилбромацетат (2,59 г, 15,6 ммоль). Реакционную смесь перемешивали дополнительно в течение 10 минут и затем нагревали до комнатной температуры в течение 2 часов. Неочищенную смесь помещали в EtOAc и промывали водой. Водный слой экстрагировали еще один раз, EtOAc и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 2,6 (т, 2H); 3,2 (т, 2H); 3,8 (с, 3H); 4,1 (кв, 2H); 5,1 (с, 2H); 6,9-7,0 (м, 4H); 7,1-7,3 (м, 2H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(2-метоксибензилокси)фенил)-4-оксобутановой кислоты:
Раствор этил 4-(4-(2-метоксибензилокси)фенил)-4-оксобутирата (Стадия B, 1,49 г, 4,3 ммоль) в абсолютном этаноле (20 мл) обрабатывали 1н NaOH (6 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем подкисляли 1М HCl. Полученное белое твердое вещество отфильтровывали, промывали холодной водой и сушили в вакууме, получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3:CD3OD): 2,6 (т, 2H); 3 2 (т, 2H); 3,8 (с, 3H); 5,1 (с, 2H); 6,9-7,0 (м, 4H); 7,2-7,3 (м, 2H); 7,8 (д, 2H).
Пример 3: Синтез 3-[(4-(2-фторбензилокси)фенил)метилтио]пропионовой кислоты

Стадия A: Получение 4-гидроксибензил бромида:
К перемешиваемому раствору PBr3 (1,38 г, 5,0 ммоль) в безводном ТГФ (2 мл) при -5°C добавляли раствор безводного пиридина (0,201 мл) в безводном ТГФ (0,4 мл). К реакционной смеси добавляли по каплям раствор 4-гидроксибензилового спирта (1,89 г, 15,2 ммоль) в безводном ТГФ (23 мл). Реакционную смесь оставляли при комнатной температуре на 18 часов, затем разбавляли ТГФ и фильтровали через слой целита. Фильтрат упаривали, полученное полутвердое вещество повторно растворяли в безводном толуоле (16 мл). Раствор выдерживали при -20°C в течение 2 часов и затем фильтровали через слой целита, получая указанное в заголовке соединение в виде светло-желтого раствора, который использовали без дополнительной очистки.
Стадия B: Получение этил 3-((4-гидроксифенил)метилтио)пропионата:
К раствору NaH (60% диспергированный в масле, 0,731 г, 21,7 ммоль) в безводном ДМФ (15 мл) добавляли этил 3-меркаптопропионат (2,66 г, 19,8 ммоль). После прекращения выделения водорода добавляли 4-гидроксибензил бромид со стадии A. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре, гасили насыщенным раствором NH4Cl и концентрировали в вакууме. Неочищенный остаток помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой промывали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (дихлорметан:этилацетат 95:5), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 2,4-2,6 (м, 4H); 3,6 (с, 2H); 4,1 (кв, 2H); 6,7 (д, 2H); 7,2 (д, 2H).
Стадия C: Получение этил 3-((4-(2-фторбензилокси)фенил)метилтио)пропионата:
К раствору NaH (60% диспергированный в масле, 0,054 г, 1,3 ммоль) в безводном ДМФ (10 мл) добавляли этил 3-((4-гидроксифенил)метилтио)пропионат (Стадия B, 2,5 г, 1,0 ммоль). После прекращения выделения водорода добавляли 2-фторбензил бромид (0,263 г, 1,3 ммоль). Реакционную смесь перемешивали в течение 4 часов при комнатной температуре, гасили насыщенным раствором NH4Cl и концентрировали в вакууме. Неочищенный остаток помещали в EtOAc и промывали дважды водой и насыщенным раствором соли. Водный слой промывали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 2,4-2,6 (м, 4H); 3,6 (с, 2H); 4,2 (кв, 2H); 5,15 (с, 2H); 6,9 (д, 2H); 7,2-7,4 (м, 5H); 7,5 (т, 1H).
Стадия D: Получение 3-((4-(2-фторбензилокси)фенил)метилтио)пропионовой кислоты:
К раствору этил 3-((4-(2-фторбензилокси)фенил)метилтио)пропионата (Стадия C, 0,122 г, 0,35 ммоль) в этаноле (5 мл) добавляли 1н NaOH (0,5 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 3 часов, подкисляли 1M HCl и концентрировали в вакууме, получая белое вещество, которое очищали флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 92,5:7,5, обогащенный уксусной кислотой), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,4-2,6 (м, 4H); 3,7 (с, 2H); 5,1 (с, 2H); 6,9 (д, 2H); 7,2-7,4 (м, 5H); 7,5 (т,1H).
Пример 4: Синтез 4-(4-(3-фторбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(3-фторбензилокси)ацетофенона:
С использованием метода, описанного в примере 1, Стадия A, используя 3-фторбензил бромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,1 (с, 2H); 7,0 (м, 3H); 7,2-7,3 (т, 2H); 7,4 (м, 1Н); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-(3-фторбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 1, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,7 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 7,0 (м, 3H); 7,2 (т, 2H); 7,4 (м, 1H); 8,0 (д, 2H).
Стадия C: Получение 4-(4-(3-фторбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 1, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 6,9-7,1 (м, 3H); 7,2-7,3 (м, 2H); 7,4 (кв, 1H); 7,9 (д, 2H).
Пример 5: Синтез 4-(4-(4-фторбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(4-фторбензилокси)ацетофенона:
С использованием метода, описанного в примере 1, Стадия A, используя 4-фторбензил бромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,1 (с, 2H); 7,0 (д, 2H); 7,1 (т, 2H); 7,4 (м, 2H); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-(4-фторбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 1, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,8 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 7,0 (м, 2H); 7,2 (т, 2H); 7,4 (м, 2H); 8,0 (д, 2H).
Стадия C: Получение 4-(4-(4-фторбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 1, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 6,9-7,1 (м, 2H); 7,2-7,3 (д, 2H); 7,4 (м, 2H); 7,9 (д, 2H).
Пример 6: Синтез 4-(4-((2-пиридинил)метокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-((2-пиридинил)метокси)ацетофенона:
Раствор 4-гидроксиацетофенона (1,99 г, 14,6 ммоль) в безводном ДМФ (5 мл) добавляли при комнатной температуре к суспензии NaH (60% в масле, 604 г) в безводном ДМФ (20 мл). После прекращения выделения водорода добавляли гидрохлорид 2-пиколилхлорида (2 г, 12,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили насыщенным водным раствором NH4Cl и концентрировали в вакууме. Неочищенный остаток помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой дважды промывали EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 1:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 7,0 (д, 2H); 7,2 (м, 1H); 7,5 (д, 1H); 7,7 (т, 1H); 7,9 (д, 2H); 8,6 (с, 1H).
Стадия B: Получение трет-бутил 4-(4-((2-пиридинил)метокси)фенил)-4-оксобутирата:
К перемешиваемому раствору 4-((2-пиридинил)метокси)ацетофенона (Стадия A, 0,968 г, 3,6 ммоль) в безводном ТГФ (16 мл) и DMPU (4 мл) добавляли раствор бис(триметилсилил)амида лития (1,0M, 5 мл) при -60°C в атмосфере аргона. После перемешивания в течение 10 минут при -60° C быстро добавляли трет-бутилбромацетат (2,64 г, 13,5 ммоль). Реакционную смесь перемешивали дополнительно в течение 10 минут и затем нагревали до комнатной температуры в течение 4 часов. Неочищенную смесь помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой экстрагировали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,7 (т, 2H); 3,2 (т, 2H); 5,3 (с, 2H); 7,0 (д, 2H); 7,2 (м, 1H); 7,5 (д, 1H); 7,7 (т, 1H); 7,9 (д, 2H); 8,6 (с, 1H).
Стадия C: Получение 4-(4-((2-пиридинил)метокси)фенил)-4-оксобутановой кислоты:
Раствор трет-бутил 4-(4-((2-пиридинил)метокси)фенил)-4-оксобутирата (Стадия C, 1,27 г, 4,2 ммоль) в дихлорметане (25 мл) обрабатывали трифторуксусной кислотой (5 мл). Смесь перемешивали при температуре окружающей среды в течение 3 часов и концентрировали в вакууме. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5, обогащенный уксусной кислотой), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3:CD3OD): 2,7 (т, 2H); 3,2 (т, 2H); 5,3 (с, 2H); 7,0 (д, 2H); 7,3 (м, 1H); 7,5 (д, 1H); 7,9 (м, 3H); 8,6 (с, 1H).
Пример 7: Синтез 4-(4-(бензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(бензилокси)ацетофенона:
С использованием метода, описанного в примере 1, Стадия A, используя бензилбромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,1 (с, 2H); 7,0 (д, 2H); 7,3-7,5 (м, 5H); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-(бензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 1, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,6 (т, 2H); 3,2 (т, 2H); 5,2 (с, 2H); 7,0 (д, 2H); 7,3-7,5 (м, 5H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(бензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 1, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 7,0 (д, 2H); 7,3-7,5 (м, 5H); 7,9 (д, 2H).
Пример 8: Синтез 4-(4-(2,6-дифторбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2,6-дифторбензилокси)ацетофенона:
Раствор 4-гидроксиацетофенона (3,61 г, 26,5 ммоль) в безводном ДМФ (5 мл) добавляли при комнатной температуре к суспензии NaH (60% в масле, 1,21 г) в безводном ДМФ (40 мл). После прекращения выделения водорода добавляли по каплям 2,6-дифторбензил бромид (5 г, 24,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 6 часов, гасили насыщенным водным раствором NH4Cl и концентрировали в вакууме. Неочищенный остаток помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой экстрагировали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,0 (м, 4H); 7,3-7,4 (м, 1H); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-(2,6-дифторбензилокси)фенил)-4-оксобутирата:
К перемешиваемому раствору 4-(2,6-дифторбензилокси)ацетофенона (Стадия A, 0,6 г, 22,8 ммоль) в безводном ТГФ (60 мл) и DMPU (12 мл) добавляли раствор бис(триметилсилил)амида лития (1,0 M, 30 мл) при -60°C в атмосфере аргона. После перемешивания в течение 10 минут при -60°C быстро добавляли трет-бутил бромацетат (8,97 г, 46 ммоль). Реакционную смесь перемешивали дополнительно в течение 10 миинут и затем нагревали до комнатной температуры в течение 4 часов. Неочищенную смесь помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой экстрагировали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,6 (т, 2H); 3,2 (т, 2H); 5,2 (с, 2H); 6,9-7,0 (м, 4H); 7,3-7,4 (м, 1H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(2,6-дифторбензилокси)фенил)-4-оксобутановой кислоты:
Раствор трет-бутил 4-(4-(2,6-дифторбензилокси)фенил)-4-оксобутирата (Стадия B, 4,76 г, 12,6 ммоль) в дихлорметане (40 мл) обрабатывали трифторуксусной кислотой (20 мл). Смесь перемешивали при температуре окружающей среды в течение 3 часов и концентрировали в вакууме. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5, обогащенный уксусной кислотой), получая указанное в заголовке соединение в виде белого порошка.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,2 (т, 2H); 5,2 (с, 2H); 6,9-7,0 (м, 4H); 7,4 (м, 1H); 7,9 (д, 2H).
Пример 9: Синтез 4-(4-(2-хлорбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2-хлорбензилокси)ацетофенона:
С использованием метода, описанного в примере 1, Стадия A, используя 2-хлорбензил бромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 7,0 (д, 2H); 7,2-7,5 (м, 4H); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-(2-хлорбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 1, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,6 (т, 2H); 3,2 (т, 2H); 5,2 (с, 2H); 7,0 (д, 2H); 7,2-7,5 (м, 4H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(2-хлорбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 1, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3:CD3OD): 2,6 (т, 2H); 3,2 (т, 2H); 5,2 (с, 2H); 7,0 (д, 2H); 7,2 (м,2H); 7,3 (м, 1H); 7,4 (м, 1H); 7,9 (д, 2H).
Пример 10: Синтез 4-(4-(2-(2-фторфенил)этокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2-(2-фторфенил)этокси)ацетофенона:
С использованием метода, описанного в примере 2, Стадия A, используя 2-фторфенэтиловый спирт в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,3 (с, 3H); 2,9 (т, 2H); 4,2 (т, 2H); 6,9 (д, 2H); 7,1 (м, 2H); 7,3 (м, 2H); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-(2-(2-фторфенил)этокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 2, Стадия B, используя трет-бутил бромацетат в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,6 (т, 2H); 3,2 (м, 4H); 4,2 (т, 2H); 6,9 (д, 2H); 7,1 (м, 2H); 7,3 (т, 2H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(2-(2-фторфенил)этокси)фенил)-4-оксобутановой кислоты:
Раствор трет-бутил 4-(4-(2-(2-фторфенил)этокси)фенил)-4-оксобутирата (Стадия 2, 1,2 г, 3,2 ммоль) в дихлорметане (25 мл) обрабатывали трифторуксусной кислотой (10 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 4 часов и концентрировали в вакууме. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5, обогащенный уксусной кислотой), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,8 (т,2H); 3,3 (т, 2H); 4,2 (т, 2H); 6,9 (д, 2H); 7,1 (м, 2H); 7,3 (т, 2H); 7,9 (д, 2H).
Пример 11: Синтез этил 4-(4-(2-фторбензилокси)фенил)-4-оксобутирата

Стадия A: Получение 4-(4-(2-фторбензилокси)ацетофенона:
С использованием метода, описанного в примере 1, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,1 (м, 4H); 7,2-7,3 (м, 1H); 7,4 (т, 1H); 7,9 (д, 2H).
Стадия B: Получение этил 4-(4-(2-фторбензилокси)фенил)-4-оксобутирата:
К перемешиваемому раствору 4-(2-фторбензилокси)ацетофенона (7,26 г, 29,7 ммоль) в безводном ТГФ (80 мл) и DMPU (16 мл) добавляли раствор бис(триметилсилил)амида лития (1,0 M, 35 мл) при -60°C в атмосфере аргона. После перемешивания в течение 10 минут при -60°C быстро добавляли этилбромацетат (10,12 г, 60,5 ммоль). Реакционную смесь перемешивали дополнительно в течение 10 минут и затем нагревали до комнатной температуры в течение 4 часов. Неочищенную смесь помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой экстрагировали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1), получая указанное в заголовке соединение в виде белого порошка.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 2,7 (т, 2H); 3,2 (т, 2H); 4,2 (кв, 2H); 5,1 (с, 2H); 6,9 (д, 2H); 7,2 (м, 2H); 7,4 (м, 1H); 7,5 (м, 1H); 7,9 (д, 2H).
Пример 12: Синтез 4-(4-(2-метилбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2-метилбензилокси)ацетофенона:
С использованием метода, описанного в примере 1, Стадия A, используя 2-метилбензил бромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 3H); 2,5 (с, 3H); 5,2 (с, 2H); 6,9 (д, 2H); 7,2-7,3 (м, 3H); 7,4 (м, 1H); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-(2-метилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 1, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,5 (с, 9H); 2,4 (с, 3H); 2,6 (т, 2H); 3,2 (т, 2H); 5,2 (с, 2H); 6,9 (д, 2H); 7,2-7,3 (м, 3H); 7,4 (м, 1H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(2-метилбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 1, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 3H); 2,8 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 6,9 (д, 2H); 7,2-7,3 (м, 3H); 7,4 (м, 1H); 7,9 (д, 2H).
Пример 13: Синтез 4-[4-(2-(N-(2-фторбензил)-N-метиламино)этокси)фенил]-4-оксобутановой кислоты

Стадия A: Получение 2-фторбензил метансульфоната:
К раствору 2-фторбензилового спирта (10 г, 79,28 ммоль) в безводном дихлорметане (200 мл) добавляли триэтиламин (12,03 г, 118,9 ммоль) в атмосфере аргона при комнатной температуре. К вышеуказанной реакционной смеси при 0°С добавляли метансульфонилхлорид (10,71 г, 93,5 ммоль) и перемешивание продолжали еще в течение 3 часов. К реакционной смеси добавляли воду (100 мл) и смесь дважды экстрагировали дихлорметаном. Объединенные органические слои промывали водой и насыщенным раствором соли. Реакционную смесь сушили над Na2SO4, фильтровали и концентрировали, получая указанное в заголовке соединение в виде желтого масла, которое использовали без дополнительной очистки.
1H ЯМР (270 МГц, CDCl3): 1,3 (т, 3H); 2,4-2,6 (м, 4H); 5,25 (с, 2H); 6,9-7,5 (м, 4H).
Стадия B: Получение 2-(N-(2-фторбензил)-N-метиламино)этанола:
Смесь 2-фторбензилметансульфоната (Стадия A, 5 г, 24,5 ммоль) и 2-(метиламино)этанола (18,4 г, 244,9 ммоль) нагревали в атмосфере аргона при 120°C при перемешивании в течение 7 часов. Смесь охлаждали до комнатной температуры и концентрировали. Неочищенный остаток очищали флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 90:10, обогащенный триэтиламином), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,3 (с, 3H); 2,6 (м, 2H); 3,6 (м, 4H); 6,9-7,5 (м, 4H).
Стадия C: Получение 2-(N-(2-фторбензил)-N-метиламино)этилхлорида:
К раствору 2-(N-(2-фторбензил)-N-метиламино)этанола (Стадия B, 7,51 г, 41 ммоль) в безводном толуоле (50 мл) добавляли хлористый тионил (16 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов и концентрировали. Неочищенную смесь разбавляли хлороформом и промывали водным раствором NaHCO3, водой и насыщенным раствором соли. Органический слой сушили над Na2SO4, фильтровали и концентрировали, получая указанное в заголовке соединение, которое использовали без дополнительной очистки.
1H ЯМР (270 МГц, CDCl3): 2,3 (с, 3H); 2,8 (т, 2H); 3,6 (т, 2H); 3,7 (с, 2H); 7,0-7,15 (м, 2H); 7,25 (м, 1H), 7,4 (т, 1H).
Стадия D: Получение 4-(2-(N-(2-фторбензил)-N-метиламино)этокси)ацетофенона:
К раствору 2-(N-(2-фторбензил)-N-метиламино)этилхлорида (Стадия C, 7,48 г, 37 ммоль) и 4-гидроксиацетофенона (10,07 г, 74 ммоль) в безводном ДМФ (10 мл) добавляли K2CO3 (7,77 г, 56,2 ммоль). Смесь нагревали при 80°C в течение 6 часов, охлаждали, гасили водой и дважды экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный остаток очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение в виде светло-желтого масла.
1H ЯМР (270 МГц, CDCl3): 2,35 (с, 3H); 2,4 (с, 3H); 2,8 (т, 2H); 3,7 (с, 2H); 4,2 (т, 2H); 6,9 (д, 2H); 7,0-7,15 (м, 2H); 7,25(м, 1H), 7,4 (т,1H); 7,9 (д, 2H).
Стадия E: Получение трет-бутил 4-[4-(2-(N-(2-фторбензил)метиламино)этокси)фенил]-4-оксобутирата
Бис(триметилсилил)амид лития (1,0M, 20 мл) медленно добавляли в течение 10 минут к перемешиваемому раствору 4-(2-(N-(2-фторбензил)-N-метиламино)этокси)ацетофенона (Стадия D, 4,91 г, 16,3 ммоль) в безводном ТГФ (60 мл) и DMPU (15 мл) при -65°C в атмосфере аргона. После перемешивания в течение 15 минут быстро добавляли трет-бутил бромацетат (6,35 г, 32,6 ммоль). Перемешивание продолжали дополнительно в течение 10 минут при -65°C и затем реакционную смесь нагревали до комнатной температуры в течение 2 часов, гасили водой и дважды экстрагировали EtOAc. Объединенные органические слои очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 1:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,5 (с, 9H); 2,4 (с, 3H); 2,6 (т, 2H); 2,8 (т, 2H); 3,2 (т, 2H) 3,7 (ушир., 2H); 4,2 (ушир., 2H); 6,9 (д, 2H); 7,0-7,15 (м, 2H); 7,25 (м, 1H), 7,4 (т, 1H); 7,9 (д, 2H).
Стадия F: Получение 4-[4-(2-(N-(2-фторбензил)-N-метиламино)этокси)фенил]-4-оксобутановой кислоты:
Раствор трет-бутил 4-[4-(2-(N-(2-фторбензил)-N-метиламин)этокси)фенил]-4-оксобутирата (Стадия E, 2,23 г, 5,3 моль) в дихлорметане (20 мл) обрабатывали трифторуксусной кислотой (10 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов и концентрировали в вакууме. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 92,5:7,5-90:10, обогащенный уксусной кислотой), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3:CD3OD): 2,5 (т, 2H); 2,6 (с, 3H); 3,0 (т, 2H); 3,4 (т, 2H); 4,2-4,5 (м, 4H); 6,9 (д, 2H); 7,0-7,15 (м, 2H); 7,3 (м, 1H), 7,5 (т, 1H); 7,9 (д, 2H).
Пример 14: Синтез 4-(3-(2-Метилбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 3-(2-метилбензилокси)ацетофенона:
С использованием метода, описанного в примере 12, Стадия A, используя 3-гидроксиацетофенон в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,3 (с, 3H); 2,5 (с, 3H); 5,1 (с, 2H); 7,2-7,3 (м, 4H); 7,4 (м, 2H); 7,6 (м, 2H).
Стадия B: Получение трет-бутил 4-(3-(2-метилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 1, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,5 (с, 9H); 2,4 (с, 3H); 2,6 (т, 2H); 3,2 (т, 2H); 5,2 (с, 2H); 7,2-7,3 (м, 4H); 7,4 (м, 2H); 7,6 (м, 2H).
Стадия C: Получение 4-(3-(2-метилбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 1, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 3H); 2,8 (т, 2H); 3,3 (т, 2Н); 5,1 (с, 2H); 7,2-7,3 (м, 4H); 7,4 (м, 2H); 7,6 (м, 2H).
Пример 15: Синтез этил 4-(3-(2-фторбензилокси)фенил)-4-оксобутирата

Стадия A: Получение 3-(2-фторбензилокси)ацетофенона
С использованием метода, описанного в примере 1, Стадия A, используя 3-гидроксиацетофенон в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 7,1 (м, 4H); 7,3 (м, 2H); 7,6 (м, 2H).
Стадия B: Получение этил 4-(3-(2-фторбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 11, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,3 (с, 9H); 2,8 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 7,1 (т, 2H); 7,2 (д, 2H); 7,4 (м, 1H); 7,5 (т, 1H); 7,6 (д, 2H).
Пример 16: Синтез этил 4-(4-(2-метилбензилокси)фенил)-4-оксобутирата

Стадия A: Получение 4-(2-метилбензилокси)ацетофенона:
С использованием метода, описанного в примере 12, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 3H); 2,5 (с, 3H); 5,2 (с, 2H); 6,9 (д, 2H); 7,2-7,3 (м, 3H); 7,4 (м, 1H); 8,0 (д, 2H).
Стадия B: Получение этил 4-(4-(2-метилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 11, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 2,4 (с, 3H); 2,7 (т, 2H); 3,2 (т, 2H); 4,2 (кв, 2H); 5,1 (с, 2H); 7,0 (д, 2H); 7,2-7,3 (м, 3H); 7,4 (м, 1H); 8,0 (д, 2H).
Пример 17: Синтез этил 4-(4-(2,6-дифторбензилокси)фенил)-4-оксобутирата

Стадия A: Получение 4-(2,6-дифторбензилокси)ацетофенона:
С использованием метода, описанного в примере 8, Стадия A, получали указанное в заголовке соединение
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,0 (м, 4H); 7,3-7,4 (м, 1H); 7,9 (д, 2H).
Стадия B: Получение этил 4-(4-(2,6-дифторбензилокси)фенил)-4-оксобутирата:
К перемешиваемому раствору 4-(2,6-дифторбензилокси)ацетофенона (Стадия A, 6 г, 22,8 ммоль) в безводном ТГФ (60 мл) и DMPU (12 мл) добавляли раствор бис(триметилсилил)амида лития (1,0 M, 30 мл) при -60°C в атмосфере аргона. После перемешивания в течение 10 минут при -60°C быстро добавляли этилбромацетат (7,61 г, 45,6 ммоль). Реакционную смесь дополнительно перемешивали в течение 10 минут и затем нагревали до комнатной температуры в течение 4 часов. Неочищенную смесь помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой экстрагировали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат, 4:1), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 1,3 (т, 3H); 2,8 (т, 3H); 3,2 (т, 2H); 4,1 (кв, 2H); 5,2 (с, 2H); 6,9-7,0 (м, 4H); 7,3-7,4 (м, 1H); 7,9 (д, 2H).
Пример 18: Синтез 4-(4-(2-(2-тиенил)этокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2-(2-тиенил)этокси)ацетофенона:
С использованием метода, описанного в примере 2, Стадия A, используя 2-(2-тиенил)этанол в качестве исходного вещества, и после очистки флэш-хроматографией на колонке с силикагелем (гексан:этилацетат, 3:1) получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 3,3 (т, 2H); 4,2 (т, 2H); 6,9-7,1 (м, 4H); 7,2 (д, 1H); 7,9 (д, 2H).
Стадия B: Получение этил 4-(4-(2-(2-тиенил)этокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 2, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,3 (т, 3H); 2,8 (т, 2H); 3,3 (м, 4H); 4,1 (кв, 2H); 4,2 (т, 2H); 6,9-7,1 (м, 4H); 7,2 (д, 1H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(2-(2-тиенил)этокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 2, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,3 (м, 4H); 4,2 (т, 2H); 6,9-7,1 (м, 4H); 7,2 (д, 1H); 7,9 (д, 2H).
Пример 19: Синтез 4-(2,6-дифторфенил)-4-оксобутановой кислоты

Стадия A: Получение трет-бутил 4-(2,6-дифторфенил)-4-оксобутирата:
К перемешиваемому раствору 2,6-дифторацетофенона (5 г, 32 ммоль) в безводном ТГФ (40 мл) и DMPU (8 мл) добавляли раствор бис(триметилсилил)амида лития (1,0 M, 45 мл) при -60°C в атмосфере аргона. После перемешивания в течение 10 минут при -60°C быстро добавляли трет-бутилбромацетат (6,99 г, 35,8 ммоль). Реакционную смесь перемешивали дополнительно в течение 10 минут и затем нагревали до комнатной температуры в течение 4 часов. Неочищенную смесь помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой экстрагировали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,8 (т, 2H); 3,2 (т, 2H); 6,9-7,0 (м, 2H); 7,4 (м, 1H).
Стадия B: Получение соединения AS:
Раствор трет-бутил 4-(2,6-дифторфенил)-4-оксобутирата (Стадия A, 9,52 г, 35,2 ммоль) в дихлорметане (30 мл) обрабатывали трифторуксусной кислотой (20 мл). Смесь перемешивали при температуре окружающей среды в течение 3 часов и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5, обогащенный уксусной кислотой), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,2 (т, 2H); 6,9-7,0 (м, 2H); 7,4 (м, 1H).
Пример 20: Синтез 4-(4-(2,5-диметилбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2,5-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 8, Стадия A, используя 2,5-диметилбензил хлорид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,3 (с, 3H); 2,5 (с, 3H); 5,1 (с,2H); 6,9-7,2 (м, 5H); 7,9 (д, 2H).
Стадия B: Получение этил 4-(4-(2,5-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,3 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,0 (д, 2H); 7,2-7,3 (м, 3H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(2,5-диметилбензилокси)фенил)-4-оксобутановой кислоты:
К раствору этил 4-(4-(2,5-диметилбензилокси)фенил)-4-оксобутирата (Стадия B, 2,62 г, 7,7 ммоль) в абсолютном этаноле (30 мл) добавляли 1н NaOH (10 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 3 часов и затем подкисляли 1M HCl. Образовавшийся белый осадок отфильтровывали, промывали водой и сушили в вакууме, получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,3 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 7,0 (д, 2H); 7,2-7,3 (м, 3H); 8,0 (д, 2H).
Пример 21: Синтез 4-(4-(2,5-дифторбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2,5-дифторбензилокси)ацетофенона:
С использованием метода, описанного в примере 8, Стадия A, используя 2,5-дифторбензил бромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,1 (с, 2H); 6,9-7,0 (м, 3H); 7,2 (м, 2H); 8,0 (д, 2H).
Стадия B: Получение этил 4-(4-(2,5-дифторбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 6,9-7,0 (м, 3H); 7,2 (м, 2H); 8,0 (д, 2H).
Стадия C: Получение 4-(4-(2,5-дифторбензилокси)фенил)-4-оксобутановой кислоты:
К раствору этил 4-(4-(2,5-дифторбензилокси)фенил)-4-оксобутирата (Стадия B, 16,51 г, 47,4 ммоль) в абсолютном этаноле (100 мл) добавляли 1н NaOH (40 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 3 часов, подкисляли 1M HCl и концентрировали в вакууме. Неочищенную смесь помещали в хлороформ и промывали водой. Водный слой промывали еще один раз хлороформом. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5, обогащенный уксусной кислотой), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 6,9-7,0 (м, 3H); 7,2 (м, 2H); 8,0 (д, 2H).
Пример 22: Синтез 4-(4-(2,4-дифторбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-(2,4-дифторбензилокси)ацетофенона:
С использованием метода, описанного в примере 8, Стадия A, используя 2,4-дифторбензил бромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,1 (с,2H); 6,9-7,0 (м, 2H); 7,1 (д, 2H); 7,4 (м, 1H); 8,0 (д, 2H).
Стадия B: Получение этил 4-(4-(2,4-дифторбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,8 (т,2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 6,9-7,0 (м, 2H); 7,1 (д, 2H); 7,4 (м, 1H); 8,0 (д, 2H).
Стадия C: Получение 4-(4-(2,4-дифторбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 21, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 6,9-7,0 (м, 2H); 7,1 (д, 2H); 7,4 (м, 1H); 8,0 (д, 2H).
Пример 23: Синтез 4-(3-(2,6-дифторбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 3-(2,6-дифторбензилокси)ацетофенона:
С использованием метода, описанного в примере 8, Стадия A, используя 3-гидроксиацетофенон в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,0 (м, 2H); 7,2 (м, 1H); 7,4 (м, 2H); 7,9 (д, 2H).
Стадия B: Получение этил 4-(3-(2,6-дифторбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 6,9-7,0 (м, 2H); 7,2 (м, 1H); 7,4 (м, 2H); 7,9 (д, 2H).
Стадия C: Получение 4-(3-(2,6-дифторбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 21, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 6,9-7,0 (м, 2H); 7,2 (м, 1H); 7,4 (м, 2H); 7,9 (д, 2H).
Пример 24: Синтез 4-(4-((циклопропил)метокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 4-((циклопропил)метокси)ацетофенона:
С использованием метода, описанного в примере 8, Стадия A, используя циклопропилметил бромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 0,4 (м, 2H); 0,6 (м, 2H); 1,2 (м, 1H); 2,5 (с, 3H); 3,8 (д, 2H); 6,9 (д, 2H); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-((циклопропил)метокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 8, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 0,4 (м, 2H); 0,6 (м, 2H); 1,2 (м, 1H); 1,4 (с, 9H); 2,6 (т, 2H); 3,2 (т, 2H); 3,8 (д, 2H); 6,9 (д, 2H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-((циклопропил)метокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 8, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 0,4 (м, 2H); 0,6 (м, 2H); 1,2 (м, 1H); 2,8 (т, 2H); 3,2 (т, 2H); 3,8 (д, 2H); 6,9 (д, 2H); 7,9 (д, 2H).
Пример 25: Синтез 4-(4-(2-трифторметилбензилокси)фенил)-4-оксобутановой кислоты
Стадия A: Получение 4-(2-трифторметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 8, Стадия A, используя 2-(трифторметил)бензил бромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,3 (с, 2H); 6,9 (д, 2H); 7,4 (т, 1H); 7,6 (т, 1H); 7,7 (д, 2H); 7,9 (д, 2H).
Стадия B: Получение трет-бутил 4-(4-(2-трифторметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 8, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 2,7 (т, 2H); 3,2 (т, 2H); 5,3 (с, 2H); 6,9 (д, 2H); 7,4 (т, 1H); 7,6 (т, 1H); 7,7 (д, 2H); 7,9 (д, 2H).
Стадия C: Получение 4-(4-(2-трифторметилбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 8, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,2 (т, 2H); 5,3 (с, 2H); 6,9 (д, 2H); 7,4 (т, 1H); 7,6 (т, 1H); 7,7 (т, 2H); 7,9 (д, 2H).
Пример 26: Синтез 3-[(4-(2,6-дифторбензилокси)фенил)метилтио]пропионовой кислоты
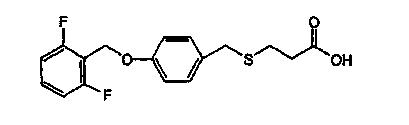
Стадия A: Получение 4-гидроксибензил бромида:
С использованием метода, описанного в примере 3, Стадия A, получали указанное в заголовке соединение, которое использовали без дополнительной очистки.
Стадия B: Получение этил 3-[(4-гидроксифенил)метилтио]пропионата:
С использованием метода, описанного в примере 3, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 2,4-2,6 (м, 4H); 3,6 (с, 2H); 4,1 (кв, 2H); 6,7 (д, 2H); 7,2 (д, 2H).
Стадия C: Получение этил 3-[(4-(2,6-дифторбензилокси)фенил)метилтио]пропионата:
С использованием метода, описанного в примере 3, Стадия C, используя 2,6-дифторбензил бромид в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 2,4-2,6 (м, 4H); 3,6 (с, 2H); 4,2 (кв, 2H); 5,15 (с, 2H); 6,9 (д, 4H); 7,2-7,4 (м, 3H).
Стадия D: Получение 3-[(4-(2,6-дифторбензилокси)фенил)метилтио]пропионовой кислоты:
С использованием метода, описанного в примере 3, Стадия D, получали указанное в заголовке соединение.1H ЯМР (270 МГц, CDCl3): 2,5-2,6 (м, 4H); 3,7 (с,2H); 5,1 (с, 2H); 6,9-7,0 (м, 4H); 7,2-7,4 (м, 3H).
Пример 27: Синтез 4-(2-(2,6-Дифторбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 2-(2,6-дифторбензилокси)ацетофенона:
С использованием метода, описанного в примере 8, Стадия A, используя 2-гидроксиацетофенон в качестве исходного вещества, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,0 (м, 3H); 7,1 (д, 1H); 7,4 (м, 1H); 7,5 (т, 1H); 7,8 (д, 1H).
Стадия B: Получение этил 4-(2-(2,6-дифторбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,6 (т, 2H); 3,2 (т, 2H); 4,1 (кв, 2H); 5,2 (с, 2H); 6,9-7,0 (м, 3H); 7,1 (д, 1H); 7,4 (м, 1H); 7,5 (т, 1H); 7,8 (д, 1H).
Стадия C: Получение 4-(2-(2,6-дифторбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 21, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,6 (т, 2H); 3,2 (т, 2H); 5,2 (с, 2H); 6,9-7,0 (м, 3H); 7,1 (д, 1H); 7,4 (м, 1H); 7,5 (т, 1H); 7,8 (д, 1H).
Пример 28: Синтез этил 4-(4-(2,6-дифторбензилокси)фенил)метил-3-оксобутирата

Стадия A: Получение этил 4-гидроксибензилата:
К перемешиваемому раствору 4-гидроксибензилового спирта (4 г, 26,28 ммоль) в безводном ДМФ (15 мл), пиридине (1 мл) и N,N-дициклогексилкарбодимиде (6,50 г, 31,5 ммоль) добавляли абсолютный EtOH (3,26 г, 78,84 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре и затем фильтровали. Фильтрат концентрировали при пониженном давлении и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат, 2:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 3,5 (с, 2H); 4,1 (кв, 2H); 6,7 (д, 2H); 7,1 (д, 2H).
Стадия B: Получение этил 4-(2,6-дифторбензилокси)бензилата:
К раствору NaH (60% диспергированный в масле, 0,393 г, 9,8 ммоль) в безводном ДМФ (20 мл) добавляли этил 4-гидроксибензилат (Стадия A, 1,59 г, 8,8 ммоль). После прекращения выделения водорода добавляли 2,6-дифторбензил бромид (1,64 г, 7,9 ммоль). Реакционную смесь перемешивали в течение 4 часов при комнатной температуре, гасили насыщенным раствором NH4Cl и концентрировали в вакууме. Остаток помещали в EtOAc и дважды промывали водой и насыщенным раствором соли. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат, 2:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 3,5 (с, 2H); 4,1 (кв, 2H); 5,1 (с, 2H); 6,9 (м, 4H); 7,2-7,4 (м, 3H).
Стадия C: Получение 4-(2,6-дифторбензилокси)бензиловой кислоты:
К перемешиваемому раствору этил 4-(2,6-дифторбензилокси)бензилата (Стадия B, 2,14 г, 6,9 ммоль) в абсолютном EtOH (30 мл) добавляли 1н NaOH (10 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 3 часов, подкисляли 1M HCl и фильтровали. Белый осадок промывали водой и сушили в высоком вакууме, получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 3,6 (с, 2H); 5,1 (с, 2H); 6,9 (м, 4H); 7,2-7,4 (м, 3H).
Стадия D: Получение 4-(2,6-дифторбензилокси)бензилкарбонил хлорида:
Тионилхлорид (10 мл) К 4-(2,6-дифторбензилокси)бензиловой кислоте (Стадия C, 1,61 г, 5,79 ммоль) добавляли хлористый тионил (10 мл). Реакционную смесь нагревали при кипении с обратным холодильником в течение 3 часов и концентрировали в вакууме, получая светло-желтое масло, которое использовали без дополнительной очистки.
Стадия Е: Этил 4-(4-(2,6-дифторбензилокси)фенил)метил-3-оксобутират:
К раствору кислоты Мелдрума (0,846 г, 5,8 ммоль) в дихлорметане (5 мл) добавляли пиридин (2 мл) в течение 10 минут при 0°C. К этому раствору добавляли 4-(2,6-дифторбензилокси)бензилкарбонил хлорид (Стадия D, 1,71 г, 5,7 ммоль) в дихлорметане (5 мл), что приводило к образованию оранжевого раствора. Темно-оранжевый раствор перемешивали в течение 1 часа при 0°C, оставляли нагреваться до комнатной температуры и дополнительно перемешивали в течение часа. Реакционную смесь разбавляли дихлорметаном и выливали на 2M HCl и лед. фазы разделяли и водную фазу дважды экстрагировали дихлорметаном. Объединенные органические слои дважды промывали 2M HCl и насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали, получая твердое вещество. Твердое вещество суспендировали в абсолютном EtOH (15 мл) и нагревали при кипении с обратным холодильником в течение 2,5 часов. Растворитель удаляли в вакууме, получая темное масло. Остаток очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 3,4 (с, 2H); 3,7 (с, 2H); 4,2 (кв, 2H); 5,1 (с, 2H); 6,9 (м, 4H); 7,1 (д, 2H); 7,3 (м, 1H).
Пример 29: Синтез 3-(2-(4-(2,6-дифторбензилокси)фенил)-2-оксоэтил)тио-1H-1,2,4-триазола

Стадия A: Получение 4-(2,6-дифторбензилокси)ацетофенона:
С использованием метода, описанного в примере 8, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,0 (м, 4H); 7,3-7,4 (м, 1H); 7,9 (д,2H).
Стадия B: Получение 2-бром-1-(4-(2,6-дифторбензилокси)фенил)-1-этанона:
К перемешиваемому раствору бромида меди (2) (3,70 г, 16,6 ммоль) в этилацетате (20 мл) добавляли раствор 4-(2,6-дифторбензилокси)ацетофенона (Стадия A, 2,74 г, 10,4 ммоль) в хлороформе (20 мл) при комнатной температуре. Реакционную смесь нагревали при кипении с обратным холодильником в течение 16 часов и затем добавляли воду. Неочищенную смесь дважды экстрагировали EtOAc. Органические слои объединяли и промывали водой, насыщенным раствором соли, сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат, 4:1), получая указанное в заголовке соединение в виде белых хлопьев.
1H ЯМР (270 МГц, CDCl3): 4,4 (с, 2H); 5,2 (с, 2H); 6,9-7,1 (м, 4H), 7,3 (м, 1H); 8,0 (д, 2H).
Стадия C: Получение 3-(2-(4-(2,6-дифторбензилокси)фенил)-2-оксоэтил)тио-1H-1,2,4-триазола:
К раствору 1H-1,2,4-триазол-3-тиола (0,250 г, 2,4 ммоль) и триэтиламина (2,50 г, 2,4 ммоль) в безводном дихлорметане (20 мл) добавляли 2-бром-1-(4-(2,6-дифторбензилокси)фенил)-1-этанон (Стадия B, 0,851 г, 2,4 ммоль) в безводном дихлорметане (5 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 50 минут и затем концентрировали в вакууме. Неочищенный остаток помещали в EtOAc и промывали 1M HCl и насыщенным раствором соли. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 9:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 4,5 (с, 2H); 5,1 (с, 2H); 6,8-7,0 (м, 4H), 7,2 (м, 1H); 7,9 (д, 2H); 8,0 (с, 1H).
Пример 30: Синтез 5-((4-(2,6-дифторбензилокси)фенил)-метил)-1H-тетразола

Стадия A: Получение (4-(2,6-дифторбензилокси)фенил)ацетонитрила:
К раствору 4-гидроксибензилцианида (5 г, 37,5 ммоль) и K2CO3 (6,74 г, 48,8 ммоль) в безводном ДМФ (20 мл) добавляли 2,6-дифторбензил бромид (7,77 г, 37,5 ммоль). Реакционную смесь перемешивали в течение 4 часов при комнатной температуре и концентрировали в вакууме. Неочищенный остаток помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой промывали еще один раз EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали, получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 3,65 (с, 2H); 5,1 (с, 2H); 6,9-7,0 (м, 4H); 7,2-7,4 (м, 3H).
Стадия B: Получение 5-((4-(2,6-дифторбензилокси)фенил)метил)-1H-тетразола:
Смесь (4-(2,6-дифторбензилокси)фенил)ацетонитрила (Стадия A, 5 г, 19,3 ммоль), NaN3 (1,3 г, 20 ммоль) и NH4Cl (1,06 г, 20 ммоль) в безводном ДМФ (60 мл) нагревали при 90°C в течение 16 часов. Растворитель удаляли в вакууме и маслянистый остаток распределяли между EtOAc и водой (подкисленной до pH 1 концентрированной HCl). Органический слой промывали водой, сушили над Na2SO4, фильтровали и концентрировали, получая коричневое полутвердое вещество. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол, 9:1), получая указанное в заголовке соединение в виде светло-кремового твердого вещества.
1H ЯМР (270 МГц, CDCl3): 4,0 (с, 2H); 5,1 (с, 2H); 6,7-6,9 (м, 4H); 7,0 (д, 2H); 7,2 (м, 1H).
Пример 31: Синтез (2RS) 2-(N-Boc)-3-[2-(4-(2,6-дифторбензилокси)фенил)-2-оксоэтил]тиопропионовой кислоты
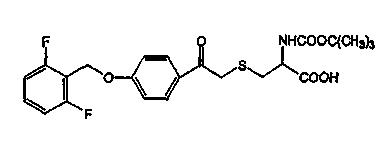
Стадия A: Получение 4-(2,6-дифторбензилокси)ацетофенона:
К раствору 4-гидроксиацетофенона (3,28 г, 24 ммоль) и K2CO3 (4,33 г, 31,3 ммоль) в безводном ДМФ (15 мл) добавляли 2,6-дифторбензил бромид (5 г, 24,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 часов, гасили водой и концентрировали в вакууме. Неочищенный остаток помещали в EtOAc и промывали водой и насыщенным раствором соли. Водный слой дважды экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,0 (м, 4H); 7,3-7,4 (м, 1H); 7,9 (д, 2H).
Стадия B: Получение 2-бром-1-(4-(2,6-дифторбензилокси)фенил)-1-этанона:
С использованием метода, описанного в примере 29, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 4,4 (с, 2H); 5,2 (с, 2H); 6,9-7,1 (м, 4H), 7,3 (м, 1H); 8,0 (д, 2H).
Стадия C: Получение этил (2RS) 2-(N-Boc)-3-[2-(4-(2,6-дифторбензилокси)фенил)2-оксоэтил]тиопропионата:
К перемешиваемому раствору 2-бром-1-(4-(2,6-дифторбензилокси)фенил)-1-этанона (Стадия B, 2,07 г, 8,3 ммоль) в безводном дихлорметане (20 мл) и триэтиламине (8,39 г, 83 ммоль) добавляли Boc-Cys-OEt (2,94 г, 8,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение часа и концентрировали в вакууме. Неочищенный остаток помещали в EtOAc, промывали 1M HCl и насыщенным раствором соли. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 97,5:2,5), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 1,4 (с, 9H); 3,0 (м, 2H); 3,8 (с, 2H); 4,2 (кв, 2H); 4,5 (ушир., 1H); 5,2 (с, 2H); 5,4 (д, 1H); 6,9-7,1 (м, 4H); 7,3 (м, 1H); 7,9 (д, 2H).
Стадия D: Получение (2RS) 2-(N-Boc)-3-[2-(4-(2,6-дифторбензилокси)фенил)-2-оксоэтил]тиопропионовой кислоты:
К раствору этил (2RS) 2-(N-Boc)-3-[2-(4-(2,6-дифторбензилокси)фенил)-2-оксоэтил]тиопропионата (Стадия C, 0,761 г, 1,5 ммоль) в абсолютном EtOH (10 мл) добавляли 1н NaOH (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, подкисляли 1M HCl и концентрировали в вакууме. Неочищенный остаток помещали в хлороформ и промывали водой и насыщенным раствором соли. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 92,7:7,5), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,4 (с, 9H); 3,0 (т, 2H); 4,0 (кв, 2H); 4,5 (ушир., 1H); 5,2 (с, 2H); 5,4 (д, 1H); 6,9-7,1 (м, 4H); 7,3(м, 1H); 7,9 (д, 2H).
Пример 32: Синтез этил 2-гидрокси-4-оксо-4-(4-(2,6-дифторбензилокси)фенил)бут-2-еноата

Стадия A: Получение 4-(2,6-дифторбензилокси)ацетофенона:
С использованием метода, описанного в примере 31, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,0 (м, 4H); 7,3-7,4 (м, 1H); 7,9 (д, 2H).
Стадия B: Получение этил 2-гидрокси-4-оксо-4-(4-(2,6-дифторбензилокси)фенил)бут-2-еноата:
Смесь 4-(2,6-дифторбензилокси)ацетофенона (Стадия A, 5,64 г, 21,5 ммоль) и диэтилоксалата (3,14 г, 21,5 ммоль) добавляли к охлаждаемому льдом раствору NaOEt (0,490 г, 22,4 ммоль металлического Na) в абсолютном EtOH (25 мл). После выдерживания смеси в течение ночи при комнатной температуре, смесь разбавляли водой (50 мл), подкисляли 10%-ной HCl и трижды экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3):1,4 (т, 3H); 4,4 (кв, 2H); 5,2 (с, 2H); 6,9-7,1 (м, 5H); 7,3-7,4 (м, 1H); 8,0 (д, 2H).
Пример 33: Синтез (2RS) 2-(N-ацетил)-4-(4-(2,6-дифторбензилокси)фенил)4-оксобутановой кислоты

Стадия A: Получение 4-(2,6-дифторбензилокси)ацетофенона:
С использованием метода, описанного в примере 31, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,2 (с, 2H); 6,9-7,0 (м, 4H); 7,3-7,4 (м, 1H); 7,9 (д, 2H).
Стадия B: Получение 2-бром-1-(4-(2,6-дифторбензилокси)фенил)-1-этанона:
С использованием метода, описанного в примере 29, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 4,4 (с, 2H); 5,2 (с, 2H); 6,9-7,1 (м, 4H); 7,3 (м, 1H); 8,0 (д, 2H).
Стадия C: Получение диэтил (N-Ацетил)-(2-(4-(2,6-дифторбензилокси)фенил)-2- оксоэтил)пропандиоата:
К раствору диэтилацетамидомалоната (0,949 г, 4,3 ммоль) и NaOEt (0,301 г, 4,4 ммоль) в абсолютном EtOH (25 мл) добавляли 2-бром-1-(4-(2,6-дифторбензилокси)фенил)-1-этанон (Стадия B, 1,42 г, 4,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали в вакууме. Неочищенный остаток распределяли между EtOAc и 0,01н NaOH. Органический слой промывали водой и 0,01М HCl, сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 6H); 2,0 (с, 3H); 4,2-4,3 (м, 6H); 5,2 (с, 2H); 6,9-7,1 (м, 4H), 7,3-7,4 (м, 1H); 7,9 (д, 2H).
Стадия D: Получение (2RS) 2-(N-ацетил)-4-(4-(2,6-дифторбензилокси)фенил)-4-оксобутановой кислоты:
К раствору диэтил (N-ацетил)-(2-(4-(2,6-дифторбензилокси)фенил)-2-оксоэтил)пропандиоата (Стадия C, 1,28 г, 2,6 ммоль) в воде (20 мл) добавляли NaOH (0,529 г, 13,2 ммоль). Реакционную смесь нагревали при кипении с обратным холодильником в течение 16 часов, затем добавляли ледяную уксусную кислоту (18 мл) и нагревание при кипении продолжали дополнительно 3 часа. Смесь концентрировали в вакууме и очищали флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 9:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3:CD3OD): 2,0 (с, 3H); 3,5(м, 2H); 4,8 (т, 1H); 5,1 (с, 2H); 6,9-7,1 (м, 4H), 7,3-7,4 (м, 1H); 7,9 (д, 2H).
Пример 34: Синтез 4-(3-((циклопропил)метокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 3-((циклопропил)метокси)ацетофенона:
С использованием метода, описанного в примере 31, Стадия A, используя циклопропилметил бромид и 3-гидроксиацетофенон в качестве исходных веществ, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 0,4 (м, 2H); 0,6 (м, 2H); 1,2 (м, 1H); 2,5 (с, 3H); 3,8 (д, 2H); 7,1 (м, 1H); 7,4 (м, 1H); 7,5-7,6 (м, 2H).
Стадия B: Получение трет-бутил 4-(3-((циклопропил)метокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 8, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 0,4 (м, 2H); 0,6 (м, 2H); 1,2 (м, 1H); 1,4 (с, 9H); 2,6 (т, 2H); 3,2 (т, 2H); 3,8 (д, 2H); 7,1 (м, 1H); 7,4 (м, 1H); 7,5-7,6 (м, 2H).
Стадия C: Получение 4-(3-((циклопропил)метокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 8, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 0,4 (м, 2H); 0,6 (м, 2H); 1,2 (м, 1H); 2,8 (т, 2H); 3,2 (т, 2H); 3,8 (д, 2H); 7,1 (м, 1H); 7,4 (м, 1Н); 7,5-7,6 (м, 2H).
Пример 35: Синтез 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 2,6-диметилбензилового спирта:
К раствору 2,6-диметилбензойной кислоты (10 г, 66,5 ммоль) и карбоната калия (9,18 г, 66,5 ммоль) в диметилформамиде (67 мл) добавляли иодистый метил (8,28 мл, 133,16 мл) при охлаждении на ледяной бане и смесь перемешивали в течение 16 часов. К реакционной смеси добавляли толуол и воду и органический слой промывали 3%-ным раствором K2CO3, 1н HCl и насыщенным раствором соли. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Маслянистый остаток повторно растворяли в безводном ТГФ (135 мл), добавляли к (3,79 г, 99,8 ммоль) и перемешивали в течение 4 часов на ледяной бане. К реакционной смеси медленно добавляли 1н HCl с последующим прибавлением этилацетата и органический слой промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Маслянистый остаток использовали без дополнительной очистки.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
К перемешиваемому раствору 3'-гидроксиацетофенона (8,07 г, 59,24 ммоль) и трифенилфосфина (16,93 г, 64,5 ммоль) в безводном ТГФ (180 мл) добавляли по каплям 2,6-диметилбензиловый спирт (8,05 г, 59,24 ммоль) и диэтилазодикарбоксилат (11,24 г, 64,57 ммоль) в безводном ТГФ (45 мл) и безводном ДМФ (18 мл) при температуре окружающей среды. После перемешивания в течение 1,5 часов при температуре окружающей среды реакционную смесь разбавляли эфиром и дважды промывали водой, 1н NaOH и насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (дд, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H);2, 8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия D: Получение 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты:
К раствору этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата (Стадия C, 12,31 г, 36,2 ммоль) в абсолютном этаноле (160 мл) добавляли 1н NaOH (50 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 3 часов и затем подкисляли 1M HCl. Образовавшийся белый осадок отфильтровывали, промывали водой и сушили в вакууме, получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,8 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2-7,3 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Пример 36: Синтез 4-(3-(2-фтор-6-метилбензилокси)фенил)-4-оксобутановой кислоты:

Стадия A: Получение 2-фтор-6-метилбензойной кислоты:
Синтезировали, как описано в примере 89 (d) Международной патентной публикации No.WO 97/34893, стр. 43.
Стадия B: Получение 2-фтор-6-метилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 3H); 4,7 (с, 2H); 6,85 (т, 1H); 6,95 (д, 1H); 7,15 (м, 1H).
Стадия C: Получение 3-(2-фтор-6-метилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 3H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (м, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия D: Получение этил 4-(3-(2-фтор-6-метилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 3H); 2,8 (т, 2H); 3,3 (т, 2H); 4,4 (кв, 2H); 5,2 (с, 2H); 6,9-7,1 (м, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия E: Получение 4-(3-(2-фтор-6-метилбензилокси)фенил)-4-оксобутановой кислоты:
К раствору этил 4-(3-(2-фтор-6-метилбензилокси)фенил)-4-оксобутирата (Стадия D, 8,56 г, 24,9 ммоль) в абсолютном этаноле (100 мл) добавляли 1н NaOH (40 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 3 часов, подкисляли 1М HCl и концентрировали. Остаток помещали в хлороформ и промывали 1М HCl, насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5 с добавкой уксусной кислоты), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 3H); 2,8 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 6,9-7,1 (м, 2H); 7,2(м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Пример 37: Синтез этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата:

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (дд, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Пример 38: Синтез натриевой соли 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (дд, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия D: Получение 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,8 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2-7,3 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия E: Получение натриевой соли 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты:
4-(3-(2,6-Диметилбензилокси)фенил)-4-оксобутановую кислоту (Стадия D, 5,5 г, 17,6 ммоль) растворяли в абсолютном этаноле (20 мл) при осторожном нагревании с последующим добавлением NaOH (0,705 г) при температуре 0°C. Реакционную смесь перемешивали в течение одного часа, концентрировали в вакууме и лиофилизовали, получая продукт в виде белого твердого вещества.
1H ЯМР (270 МГц, D2О): 2,0 (с, 6H); 2,5 (т, 2H); 3,0 (т, 2H); 4,8 (с, 2H); 6,8 (д, 2H); 6,9 (м, 2H); 7,2 (т, 1H); 7,5 (д, 2H).
Пример 39: Синтез 4-(4-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 4-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,0-7,2 (м, 5H); 8,0 (д, 2H).
Стадия C: Получение этил 4-(4-(2,6-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,3 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,0-7,2 (м, 5H); 8,0 (д, 2H).
Стадия D: Получение 4-(4-(2,6-Диметилбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,8 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 7,0-7,2 (м,5H); 8,0 (д, 2H).
Пример 40: Синтез калиевой соли 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,3 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,45 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,45 (т, 1H); 7,6 (м,2H).
Стадия D: Получение 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,5 (т, 2H); 3,2 (т, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2-7,3 (м, 2H); 7,45 (т, 1H); 7,6 (м,2H).
Стадия E: Получение калиевой соли 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты:
4-(3-(2,6-Диметилбензилокси)фенил)-4-оксобутановую кислоту (Стадия D, 6,0 г, 19,4 ммоль) растворяли в абсолютном этаноле (20 мл) при осторожном нагревании с последующим добавлением КОН (1,21 г) при температуре 0°C. Реакционную смесь перемешивали в течение одного часа, концентрировали в вакууме и лиофилизовали, получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, D2О): 2,3 (с, 6H); 2,5 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2-7,3 (м, 2H); 7,45 (т, 1H); 7,6 (м, 2H).
Пример 41: Синтез 4-(3-(2,6-диметоксибензилокси)фенил)-4-оксобутановой кислоты:

Стадия A: Получение 2,6-диметоксибензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 3,9 (с, 6H); 4,8 (с, 2H); 6,5 (д, 2H); 7,25 (м, 1H).
Стадия B: Получение 3-(2,6-диметоксибензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,6 (с, 3H); 3,9 (с, 6H); 5,2 (с, 2H); 6,6 (д, 2H); 7,3 (м, 3H); 7,5 (д, 1H); 7,7 (д, 1H).
Стадия C: Получение этил 4-(3-(2,6-диметоксибензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 2,8 (т, 2H); 3,3 (т, 2H); 3,8 (с, 6H); 4,1 (кв, 2H); 5,2 (с, 2H); 6,5 (д, 2H); 7,3-7,4 (м, 3H); 7,6 (д, 1H); 7,7 (д, 1H).
Стадия D: Получение 4-(3-(2,6-диметоксибензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,8 (т, 2H); 3,3 (т, 2H); 3,8 (с, 6H); 5,2 (с, 2H); 6,5 (д, 2H); 7,3-7,4 (м, 3H); 7,6 (д, 1H); 7,7 (д, 1H).
Пример 42: Синтез 4-(3-(2,6-диметилбензилокси)фенил)-4-оксо-2,2-диметилбутановой кислоты

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (дд, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксо-2,2-диметилбутирата:
К перемешиваемому раствору 3-(2,6-диметилбензилокси)ацетофенона (Стадия B, 4,11 г, 16,1 ммоль) в безводном ТГФ (60 мл) и DMPU (12 мл) добавляли раствор бис(триметилсилил)амида лития (1,0M, 17,74 мл) при -60 °C в атмосфере аргона. После перемешивания в течение 10 минут при -60 °C быстро добавляли этил 2-бромизобутират (4,73 г, 24,2 ммоль). Реакционную смесь перемешивали дополнительно в течение 10 минут, а затем нагревали до комнатной температуры в течение 4 часов. Неочищенную реакционную смесь помещали в EtOAc и промывали водой. Водный слой экстрагировали еще один раз EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали, концентрировали и очищали флэш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 1,3 (с, 6H); 2,3 (с, 6H); 3,3 (с, 2H); 4,1 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия D: Получение 4-(3-(2,6-диметилбензилокси)фенил)-4-оксо-2,2-диметилбутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,3 (с, 6H); 2,3 (с, 6H); 3,3 (с, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Пример 43: Синтез 4-(3-(4-(трифторметил)бензилокси)фенил)-4-оксобутановой кислоты:

Стадия A: Получение 3-(4-(трифторметил)бензилокси)ацетофенона:
С использованием метода, описанного в примере 31, Стадия A, с использованием 4-(трифторметил)бензил бромида и 3-гидроксиацетофенона в качестве исходных веществ, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 3H); 5,1 (с, 2H); 7,1 (д, 2H); 7,4-7,6 (м, 6H).
Стадия B: Получение этил 4-(3-(4-(трифторметил)бензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,3 (т, 3H); 2,7 (т, 2H); 3,3 (т, 2H); 4,1 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,4-7,6 (м, 6H).
Стадия C: Получение 4-(3-(4-(трифторметил)бензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,7 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,4-7,6 (м, 6H).
Пример 44: Синтез 4-(3-((циклобутил)метокси)фенил)-4-оксобутановой кислоты:

Стадия A: Получение 3-((циклобутил)метокси)ацетофенона:
С использованием метода, описанного в примере 31, Стадия A, с использованием циклобутилметилбромида и 3-гидроксиацетофенона в качестве исходных веществ, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,9 (м, 4H); 2,1 (м, 2H); 2,5 (с, 3H); 2,7 (м, 1H); 4,0 (д, 2H); 7,1 (дд, 1H); 7,4 (т, 1H); 7,5-7,6 (м, 2H).
Стадия B: Получение этил 4-(3-((циклобутил)метокси)фенил)-4-бутирата:
С использованием метода, описанного в примере 35, Стадия C, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 1,9 (м, 4H); 2,1 (м, 2H); 2,7 (м, 1H); 2,8 (т, 2H); 3,3 (т, 2H); 4,0 (д, 2H); 4,1 (кв, 2H); 7,1 (дд, 1H); 7,4 (т, 1H); 7,5-7,6 (м, 2H).
Стадия C: Получение 4-(3-((циклобутил)метокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,9 (м, 4H); 2,1 (м, 2H); 2,7 (м, 1H); 2,8 (т, 2H); 3,3 (т, 2H); 4,0 (д, 2H); 7,1 (дд, 1H); 7,4 (т, 1H); 7,5-7,6 (м, 2H).
Пример 45: Синтез 4-(3-(2,6-диметилбензилокси)фенил)бутановой кислоты:

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (дд, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия D: Получение 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,8 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2-7,3 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия E: Получение 4-(3-(2,6-диметилбензилокси)фенил)бутановой кислоты:
Раствор 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты (Стадия D, 3 г, 9,6 ммоль), гидразина (1,41 мл, 28,8 ммоль) и гидроксида калия (1,61 г, 28,8 ммоль) в этиленгликоле (12 мл) нагревали при кипении с обратным холодильником в течение 4 часов, к реакционной смеси добавляли воду (18 мл) и 6н HCl (10 мл). Неочищенную реакционную смесь концентрировали и остаток растворяли в EtOAc, промывали водой и насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5 с добавлением уксусной кислоты), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,4 (м, 8H); 2,8 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2-7,3 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Пример 46: Синтез 4-[[4-(2,6-диметилбензилокси)-3-метокси]фенил]-4-оксобутановой кислоты:

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 4-(2,6-диметилбензилокси)-3-метоксиацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 3,9 (с, 3H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,6 (м, 2H).
Стадия C: Получение этил 4-[[4-(2,6-диметилбензилокси)-3-метокси]фенил]-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,3 (т, 2H); 3,9 (с, 3H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,0-7,2 (м, 4H); 7,6 (м,2H).
Стадия D: Получение 4-[[4-(2,6-диметилбензилокси)-3-метокси]фенил]-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,8 (т, 2H); 3,3 (т, 2H); 3,9 (с, 3H); 5,1 (с, 2H); 7,0-7,2 (м, 4H); 7,6 (м, 2H).
Пример 47: Синтез 4-[5-[[N-(4-трифторметилбензил)аминокарбонил]-2-метокси]фенил]-4-оксобутановой кислоты:

Стадия A: Получение метил 2-метокси-5-ацетилбензоата:
К перемешиваемому раствору метил 2-гидрокси-5-ацетилбензоата (12 г, 61,7 ммоль) в ДМФ (200 мл) добавляли карбонат цезия (24,15 г, 74,1 ммоль) и MeI (9,64 г, 68 ммоль). Реакционную смесь перемешивали в течение 16 часов при 0°C и затем разбавляли этилацетатом, промывали водным раствором Na2S2О5, насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (этилацетат:гексан 1:2), получая указанное в заголовке соединение в виде не совсем белого твердого вещества.
1H ЯМР (270 МГц, ДМСО): 2,6 (с, 3H); 3,8 (с, 3H); 3,9 (с, 3H); 7,3 (д, 1H); 8,1 (дд, 1H); 8,2 (с, 1H).
Стадия B: Получение 2-метокси-5-ацетилбензойной кислоты:
Метил 2-метокси-5-ацетилбензоат (Стадия A, 3 г, 14,4 ммоль) растворяли в уксусной кислоте (80 мл) и затем обрабатывали концентрированной HCl (28 мл). Реакционную смесь нагревали при кипении с обратным холодильником в течение 4 часов, концентрировали при пониженном давлении и лиофилизовали, получая указанное в заголовке соединение в виде твердого вещества кремового цвета, которое использовали без дополнительной очистки.
1H ЯМР (270 МГц, ДМСО): 2,6 (с, 3H); 3,9 (с, 3H); 7,3 (д, 1H); 8,1 (дд, 1H); 8,2 (с, 1H).
Стадия C: Получение 5-ацетил-2-метокси-N-[[4-(трифторметил)фенил]метил]бензамида:
К перемешиваемому раствору 2-метокси-5-ацетилбензойной кислоты (Стадия B, 2,5 г, 12,8 ммоль), HOBt•H2О (2,08 г, 15,4 ммоль) и EDC (3,70 г, 19,3 ммоль) в CH2Cl2 (20 мл) и ДМФ (5 мл) добавляли 4-(трифторметил)бензиламин (2,48 г, 14,1 ммоль) и смесь перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и затем опять растворяли в этилацетате. Органический слой промывали 3%-ным раствором K2CO3, 1н HCl и насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,6 (с, 3H); 4,0 (с, 3H); 4,8 (д, 2H); 7,0 (д, 1H); 7,5 (д, 2H); 7,6 (д, 2H); 8,1 (дд, 1H); 8,8 (с, 1Н).
Стадия D: Получение этил 4-[5-[[N-(4-трифторметилбензил)аминокарбонил]-2-метокси]фенил]-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,6 (т, 2H); 3,3 (т, 2H); 4,0 (с, 3H); 4,4 (кв, 2H); 4,8 (с, 2H); 7,0 (д, 1H); 7,4 (д, 2H); 7,6 (д, 2H); 8,1 (дд, 1H); 8,8 (с, 1H).
Стадия E: Получение 4-[5-[[N-(4-трифторметилбензил)аминокарбонил]-2-метокси]фенил]-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3:CD3OD): 2,6 (т, 2H); 3,3 (т, 2H); 4,0 (с, 3H); 4,7 (с, 2H); 7,0 (д, 1H); 7,4 (д, 2H); 7,6 (д, 2H); 8,1 (дд, 1H); 8,8 (с, 1H).
Пример 48: Синтез 4-[5-[[N-(2,6-диметилбензил)аминокарбонил]-метокси]фенил]-4-оксобутановой кислоты:

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение N-(2,6-диметилбензил)фталимида:
К перемешиваемому раствору 2,6-диметилбензилового спирта (Стадия A, 6,59 г, 48,4 ммоль) в ДМСО (20 мл) добавляли хлортриметилсилан (15,75 мл, 145 ммоль) при комнатной температуре и смесь перемешивали в течение одного часа. К данной реакционной смеси добавляли этилацетат и воду, органический слой промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали, получая масло. Маслянистый остаток повторно растворяли в ДМФ (100 мл) и добавляли фталимид калия (10,76 г, 58,1 ммоль). Реакционную смесь перемешивали в течение 16 часов при комнатной температуре, добавляли этилацетат и органический слой промывали 3%-ным раствором Na2CO3, 1н HCl, сушили над Na2SO4, фильтровали и концентрировали, получая белое твердое вещество. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, ДМСО): 2,3 (с, 6H); 4,8 (с, 2H); 7,0 (м, 3H); 7,8 (с, 4H).
Стадия C: Получение 2,6-диметилбензиламина:
К перемешиваемому раствору N-(2,6-диметилбензил)фталимида (Стадия B, 7,77 г, 29,3 ммоль) в этаноле (80 мл) добавляли моногидрат гидразина (2,16 мл, 44,52 ммоль) и реакционную смесь нагревали при кипении с обратным холодильником в течение 3,5 часов. К данной реакционной смеси добавляли концентрированную HCl для доведения pH до 1 и кипячение продолжали еще в течение 3,5 часов, добавляли воду и реакционную смесь фильтровали, фильтрат концентрировали и pH доводили до 10 с помощью 2н NaOH. Остаток помещали в хлористый метилен и промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали, получая масло, которое использовали без дополнительной очистки.
1H ЯМР (270 МГц, ДМСО): 2,3 (с, 6H); 3,8 (с, 2H); 7,0 (м, 3H).
Стадия D: Получение 5-ацетил-2-метокси-N-[[2,6-диметил)фенил]метил]бензамида:
К перемешиваемому раствору 2-метокси-5-ацетилбензойной кислоты (пример 47, Стадия B, 2,5 г, 12,8 ммоль), HOBt (2,08 г, 15,4 ммоль) и EDC (3,70 г, 19,3 ммоль) в CH2Cl2 (20 мл) и ДМФ (5 мл) добавляли 2,6-диметилбензиламин (Стадия C, 1,72 г, 12,8 ммоль) и смесь перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и затем опять растворяли в этилацетате. Органический слой промывали 3%-ным раствором K2CO3, 1н HCl и насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Очистку проводили флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5), получая указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 2,5 (с, 6H); 2,6 (с, 3H); 3,9 (с, 3H); 4,7 (с, 2H); 7,0 (д, 1H); 7,2 (м, 3H); 7,6 (ушир., 1H); 8,1 (дд, 1H); 8,8 (с, 1H).
Стадия E: Получение этил 4-[5-[[N-(2,6-диметилбензил)аминокарбонил]метокси]фенил]-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (т, 3H); 2,4 (с, 6H); 2,7 (т, 2H); 3,3 (т, 2H); 3,9 (с, 3H); 4,4 (кв, 2H); 4,7 (с, 2H); 7,0 (м, 3H); 7,2 (м, 1H); 8,1 (дд, 1H); 8,7 (с, 1H).
Стадия F: Получение 4-[5-[[N-(2,6-диметилбензил)аминокарбонил]-2-метокси]фенил]-4-оксобутановой кислоты:
С использованием метода, описанного в примере 36, Стадия E, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3:CD3OD): 2,4 (с, 6H); 2,7 (т, 2H); 3,3 (т, 2H); 3,9 (с, 3H); 4,7 (с, 2H); 7,0 (м, 3H); 7,2 (м, 1H); 8,1 (дд, 1H); 8,7 (с, 1H).
Пример 49: Синтез 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутанкарбогидроксамовой кислоты

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (дд, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия D: Получение 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутанкарбогидроксамовой кислоты
К раствору гидрохлорида гидроксиламина в безводном этаноле добавляли раствор гидроксида калия в безводном этаноле при 35°C. Смесь охлаждали и добавляли этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутират (Стадия C) и порошкообразный гидроксид калия.
Через несколько часов реакционную смесь можно разбавить водой и нейтрализовать соляной кислотой, отфильтровать и перекристаллизовать, получая указанное в заголовке соединение.
Пример 50: Синтез 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирамида:

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (дд, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия D: Получение 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты:
С использованием метода, описанного в примере 35, Стадия D, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,8 (т, 2H); 3,3 (т, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2-7,3 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия E: Получение 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирамида:
К раствору 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутановой кислоты (Стадия D) в ДМФ добавляли триэтиламин и BOP, через приблизительно 2 часа перемешивания к реакционной смеси можно добавлять жидкий аммиак при -40°C и полученную смесь можно нагревать в течение 16 часов, давая указанное в заголовке соединение.
Пример 51: Синтез 4-(3-(2,6-диметилбензилокси)фенил)-4-оксо-2-бутеновой кислоты

Стадия A: Получение 2,6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (дд, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия D: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксо-3-бром-бутирата:
К охлажденному льдом раствору этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата (Стадия C, 3 г, 9 ммоль) в безводном диэтиловом эфире (70 мл) добавляли по каплям бром (0,7971 г, 9,9 ммоль), разбавленный эфиром (30 мл). Через 4 часа перемешивания реакционную смесь концентрировали и очищали флэш-хроматографией на колонке с силикагелем (EtOAc:гексан 1:4), получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 3,1 (м, 1H); 3,5 (м, 1H); 4,2 (кв, 2H); 5,1 (с, 2H); 5,5 (м, 1H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия E: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксо-2-бутеноата:
Триэтиламин (5,95 г, 58,9 ммоль) добавляли к раствору этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксо-3-бром-бутирата (Стадия D, 2,47 г, 5,8 ммоль) в четыреххлористом углероде (50 мл). После перемешивания в течение 4 часов при комнатной температуре реакционную смесь несколько раз фильтровали через слой силикагеля и концентрировали, получая указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 4,2 (кв, 2H); 5,1 (с, 2H); 6,9 (дд, 1H); 7,1 (д, 2H); 7,2(м, 2H); 7,4 (т, 1H); 7,6 (м, 2H); 7,9 (дд, 1H).
Стадия F: Получение 4-(3-(2,6-диметилбензилокси)фенил)-4-оксо-2-бутеновой кислоты:
К раствору этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксо-2-бутеноата (Стадия E) в абсолютном этаноле при низкой температуре добавляли водный раствор гидроксида натрия, через один час смесь концентрировали и очищали флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5, обогащенный уксусной кислотой).
Пример 52: Синтез 4-(3-(2,6-диметилбензилокси)фенил)-3-бутеновой кислоты:

Стадия A: Получение 2, 6-диметилбензилового спирта:
С использованием метода, описанного в примере 35, Стадия A, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 4,7 (с, 2H); 7,0-7,15 (м, 3H).
Стадия B: Получение 3-(2,6-диметилбензилокси)ацетофенона:
С использованием метода, описанного в примере 35, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 2,4 (с, 6H); 2,6 (с, 3H); 5,1 (с, 2H); 7,1 (дд, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия C: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-бутирата:
С использованием метода, описанного в примере 17, Стадия B, получали указанное в заголовке соединение.
1H ЯМР (270 МГц, CDCl3): 1,2 (с, 3H); 2,4 (с, 6H); 2,8 (т, 2H); 3,2 (т, 2H); 4,4 (кв, 2H); 5,1 (с, 2H); 7,1 (д, 2H); 7,2 (м, 2H); 7,4 (т, 1H); 7,6 (м, 2H).
Стадия D: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-гидрокси-бутирата:
К раствору этил 4-(3-(2,6-диметилбензилокси)фенил)-4-оксобутирата (Стадия C) в тетрагидрофуране добавляли боргидрид натрия, растворенный в воде. После 3-4 часов перемешивания при комнатной температуре смесь гасили кислотой. Органический слой можно экстрагировать дихлорметаном, промыть водой, водным раствором бикарбоната натрия и насыщенным раствором соли, высушить над Na2SO4, профильтровать и сконцентрировать. При необходимости соединение может быть очищено флэш-хроматографией на колонке с силикагелем (EtOAc:гексан).
Стадия E: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-4-бром-бутирата:
К раствору этил 4-(3-(2,6-диметилбензилокси)фенил)-4-гидрокси-бутирата (Стадия D) в диоксане добавляли по каплям трибромид фосфора в диоксане. После перемешивания при комнатной температуре в течение 16 часов смесь гасили водой и хлороформом. Через несколько минут реакционная смесь может быть нейтрализована мягким водным основанием, органический слой высушен на Na2SO4, отфильтрован, сконцентрирован и очищен флэш-хроматографией на колонке с силикагелем (EtOAc:гексан).
Стадия F: Получение этил 4-(3-(2,6-диметилбензилокси)фенил)-3-бутеноата:
К раствору этил 4-(3-(2,6-диметилбензилокси)фенил)-4-бромбутирата (Стадия E) в четыреххлористом углероде добавляют триэтиламин. После перемешивания в течение приблизительно 4 часов смесь может быть профильтрована несколько раз через слой силикагеля и сконцентрирована, давая указанное в заголовке соединение.
Стадия G: Получение 4-(3-(2,6-диметилбензилокси)фенил)-3-бутеновой кислоты:
К раствору этил 4-(3-(2,6-диметилбензилокси)фенил)-3-бутеноата (Стадия F) в абсолютном этаноле при низкой температуре добавляют водный раствор гидроксида натрия, через час смесь концентрируют и очищают флэш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5 с добавкой уксусной кислоты).
ПРИМЕРЫ БИОЛОГИЧЕСКОЙ АКТИВНОСТИ
Пример A. Соединение AH улучшает метаболические аномалии при инсулин-зависимом диабете.
Стрептозотоцин (STZ) представляет собой токсин, который селективно разрушает продуцирующие инсулин панкреатические бета-клетки и широко используется для индуцирования инсулинозависимого диабета у экспериментальных животных.
Самок мышей Balb/C (возраст 8 недель; масса тела 18-20 грамм) обрабатывали стрептозотоцином (STZ) (50 мг/кг i.p. (внутрибрюшинно) в каждый из пяти последовательных дней). Через четырнадцать дней после введения последней дозы STZ изменяли уровень глюкозы в крови для подтверждения того, что у животных развился диабет, и мышей распределяли на две группы, по 5 животных в каждой; животные одной группы получали Соединение AH (250 мг/кг) ежедневно перорально с помощью желудочного зонда, а в другой группе животные получали носитель (0,75% гидроксипропилметилцеллюлоза, суспендирующий агент в воде). Также проводили контроль для группы недиабетических мышей из того же набора мышей, которые не получали STZ. Для определения концентраций глюкозы в крови периодически отбирали образцы крови и также регистрировали массу тела.
После нескольких недель обработки концентрации глюкозы в крови мышей, которые получали перорально соединение AH, начинали снижаться относительно базовой линии, тогда как уровень глюкозы в крови контрольных животных, которые получали носитель, продолжал повышаться. В таблице 1 приведены масса тела, концентрации глюкозы, триглицеридов и холестерина в крови через 14 недель после начала обработки лекарственным средством.
Пероральное введение соединения AH приводило к существенному улучшению метаболических аномалий, связанных с инсулинозависимым диабетом.
Пример B. При пероральном введении Соединение AH улучшает выживаемость у мышей с летальным с инсулинозависимым диабетом
Самок мышей Balb/C (возраст 14 недель) обрабатывали однократной дозой стрептозотоцина (175 мг/кг i.p.) для индуцирования тяжелого инсулинозависимого диабета. Через 7 дней мышей распределяли на три группы обработки: Соединение AH, пиоглитазон и носитель.
Мышей обрабатывали ежедневно путем перорального введения и контролировали выживаемость во времени.
Все диабетические животные, получавшие перорально носитель, умерли от тяжелого неконтролируемого диабета. Двое из пяти животных, которых обрабатывали пиоглитазоном, антидиабетическим инсулиновым сенсибилизатором, используемым для лечения людей с инсулинонезависимым диабетом, остались живы через 12 недель, но потеряли 15-20% массы тела. Четыре из пяти животных, получавших перорально Соединение AH, остались живы через 12 недель, и их вес тела восстановился и поддерживался в нормальном диапазоне.
Пример C: При пероральном введении Соединение AA снижает смертность при тяжелом инсулинозависимом диабете
Самкам мышей balb/C (возраст 19 недель к началу эксперимента) многократно вводили высокие дозы STZ (75 мг/кг i.p. в течение 5 последовательных дней). Затем животных распределяли на две группы (20 мышей/группа), подходящих по тяжести протекания диабета. Через четыре дня после введения последней дозы STZ начинали обработку. В одной группе животные получали носитель (0,4 мл 0,75% HPMC, p.o. (перорально)), а в другой группе животные получали перорально соединение AA (30 мг/кг/день). Через три недели ежедневной обработки совокупная смертность в контрольной группе носителя составила 19/20 мышей. Напротив, только 5/20 мышей в группе, получавшей соединение АА, умерло в течение данного промежутка времени.
Пример D: Соединение AH снижает процент спонтанного диабета и смертность у NOD мышей.
У значительной части NOD ("нетучного диабета") мышей инсулинозависимый диабет развивается как следствие спонтанного аутоимунного разрушения панкреатических островковых клеток. Две группы из 20 NOD мышей (возраст 6 недель) обрабатывали ежедневно либо перорально носителем (0,4 мл 0,75% гидроксипропилметилцеллюлоза в воде; HPMC), либо соединением АН (200 мг/кг/день), суспендированным в HPMC. В течение семи месяцев контролировали случаи смертности из-за спонтанного развития инсулинозависимого диабета. По окончании данного промежутка времени 13/20 мышей, которых обрабатывали носителем, умерло от неконтролируемого диабета, тогда как только 5/20 мышей, которых обрабатывали соединением АН, умерло.
Пример E. Соединение AW снижает гипергликемию и гиперлипидемию и облегчает протекание ожирения печени у ob/ob тучных диабетических мышей.
Мыши ob/ob имеют дефект гена для лептина, белка, включенного в регулирование аппетита и энергетический метаболизм, и являются гиперфаговыми, страдающими ожирением и резистентными к инсулину. У них развивается гипергликемия и ожирение печени.
Самцов тощих (ob/+ гетерозиготные) и толстых (ob/ob гомозиготные) C57BL/6 мышей возрастом приблизительно 8 недель получали от Jackson Labs (Bar Harbor, ME) и случайным образом распределяли на группы по 5 животных, так чтобы вес тела и концентрации глюкозы в крови были подобными между группами. Всех животных содержали при контролируемой температуре (23°C), относительной влажности (50 + 5%) и световом периоде (7:00-19:00), и животные имели свободный доступ к воде и лабораторному питанию (Formulab Diet 5008, качество Lab Products, Elkridge, MD).
Глюкозу в крови определяли в плановом порядке с использованием полосок для определения глюкозы и прибора-глюкометра Elite XL (Bayer Corporation). В выбранные моменты времени отбирали образцы крови (˜100 микролитров) с использованием гепаринизированного капилляра из ретроглазничной пазухи для химического анализа сыворотки крови. Анализы химического состава сыворотки крови (глюкоза, триглицериды, холестерин, BUN, креатинин, AST, ALT, SDH, CPK и свободные жирные кислоты) проводили с использованием анализатора Hitachi 717 и панкреатический инсулин измеряли электрохемилюминисцентным иммуноанализом (Origen Analyzer, Igen, Inc., Gaithersburg, MD).
Группу мышей ob/ob распределяли на группы обработки, как указано ниже, и ежедневно вводили перорально дозы соединения AW (10, 30, 100, 150 или 300 мг), розиглитазона (1, 3, 10 или 30 мг) или пиоглитазона (30 или 100 мг). Два последних соединения представляют собой инсулино-сенсибилизирующие лекарственные средства, используемые для лечения людей с инсулинонезависимым сахарным диабетом, и их использовали для сравнения по эффективности и безопасности с соединениями по изобретению. Диапазон дозировки для соединений в этом эксперименте выбирали таким образом, чтобы включить как условно оптимальные, так и потенциально сверхоптимальные дозы.
Как показано в таблице 3, соединение AW приводило к снижению глюкозы в крови, сравнимому с достигаемым при использовании пиоглитазона и розиглитазона. В дозах от 100 до 300 мг/кг/день соединение AW снижало уровень триглицеридов и жирных кислот в сыворотке крови лучше, чем розиглитазон либо пиоглитазон, в их оптимальных антигипергликемических дозах.
У мышей ob/ob развивается хроническое воспалительное ожирение печени, и их рассматривают как животную модель неалкогольного стеатогепатита (NASH), болезненного состояния, которое может приводить к прогрессирующему циррозу и дисфункции печени. При NASH накопление жира увеличивает подверженность печени воспалительному повреждению. Одним характерным признаком NASH у пациентов являются, в отсутствие вирусной инфекции или алкоголизма, повышенные уровни в сыворотке крови ферментов, которые высвобождаются из поврежденных клеток печени (гепатоцитов), например аланинаминотрансферазы (ALT), аспартат аминотрансферазы (AST) и сорбит дегидрогеназы (SDH). Содержание данных ферментов повышено у мышей ob/ob вследствие ожирения печени и вторичного воспаления. В таблице 4 представлены ALT, AST и SDH в образцах сыворотки крови, полученных у мышей, которых обрабатывали соединением AW, пиоглитазоном и розиглитазоном, а также уровни ферментов в сыворотке крови, полученной у нормальных худых мышей и у диабетических контрольных мышей, которых обрабатывали только носителем. ALT, AST и SDH существенно повышены у страдающих ожирением диабетических ob/ob мышей по сравнению с худыми мышами. Обработка соединением AW в дозе, колеблющейся от 30 мг/кг/день до 300 мг/кг/день, приводила к дозозависимому снижению ферментов печени в сыворотке крови. Напротив, пиоглитазон (30 и 100 мг/кг/день) и розиглитазон (1 до 30 мг/кг/день) индуцировали повышение ALT и AST и не изменяли содержание SDH. Профили ферментов печени в сыворотке коррелировали с гистологией печени. У страдавших ожирением диабетических мышей ob/ob, которых обрабатывали носителем, отмечалось накопление жира в печени в отдельных внутриклеточных капельках. Ежедневное введение соединения AW в течение 4 недель вызывало заметное снижение капелек жира в печени, тогда как ни пиоглитазон, ни розиглитазон не снижали ни размера, ни плотности капелек жира в гепатоцитах.
Мыши ob/ob прибавили в весе во время четырехнедельного периода обработки. Как показано в таблице 5, пиоглитазон и розиглитазон усиливали прибавление в весе относительно мышей, обрабатываемых носителем, тогда как соединение AW индуцировало дозозависимое ослабление набора веса.
Пример F: Сильное гипогликемическое действие соединений по изобретению у диабетических мышей: эксперимент 1
Соединения по изобретению проявляют сильную антигипергликемическую активность у животных с инсулинонезависимым диабетом.
Самцов диабетических мышей ob/ob случайным образом распределяли на группы по пять животных в каждой. Масса тела составляла 50-55 г, и уровень глюкозы в крови составлял приблизительно 300 мг/декалитр в накормленном состоянии. С помощью желудочного зонда вводили однократную пероральную дозу исследуемого вещества, суспендированного в 0,5% карбоксиметилцеллюлозном носителе. Уровень глюкозы в крови измеряли в капельках крови, полученных засечкой хвостовой вены бритвой, с использованием глюкометрических тест-полосок и прибора Glucometer Elite XL (Bayer) через 0, 0,5, 2, 4, 6 и 18 часов после введения первоначальной дозы. 10%-ное снижение уровня глюкозы в крови относительно введенного перорально носителя рассматривалось как положительный результат скрининга. Понижение глюкозы в крови было максимальным через 6 часов после введения лекарственного средства.
Пример G: Сильное гипогликемическое действие соединений по изобретению у диабетических мышей: эксперимент 2
Соединения по изобретению проявляют сильную антигипергликемическую активность у животных, страдающих инсулинонезависимым диабетом.
Самцов мышей ob/ob (50-55 граммов; уровень глюкозы-300 мг/декалитр) распределяли на группы по пять животных в каждой и давали однократную дозу тестируемого лекарственного средства (250 мг/кг), суспендированного в носителе, представлявшем собой 0,5% карбоксиметилцеллюлозу; контрольная группа получала только носитель.
Через шесть часов после перорального введения тестируемых лекарственных средств или носителя (контроль) из хвостовой вены отбирали образцы крови и определяли содержание глюкозы с использованием глюкометра.
Пероральное введение соединений по изобретению выявило сильное антигипергликемическое действие у диабетических мышей, страдающих ожирением.
Пример H: Антидиабетическое действие соединений по изобретению у мышей db/db
Мыши db/db имеют дефект в лептиновой сигнальной передаче, что приводит к гиперфагии, ожирению и диабету. Более того, в отличие от мышей ob/ob, которые имеют относительно здоровые островковые клетки, их продуцирующие инсулин островковые клетки поджелудочной железы подвержены повреждению во время хронической гипергликемии, что приводит к переходу от гиперинсулинемии (связанной с периферической невосприимчивостью инсулина) к гипоинсулинемическому диабету.
Самцам мышей db/db ежедневно перорально вводили носитель (0,75% гидроксипропилметилцеллюлоза) или антидиабетические соединения, как указано ниже. Образцы крови получали из ретроглазничной пазухи для химического анализа сыворотки крови или из хвостовой вены для измерения глюкозы с помощью тест-полосок и глюкометра.
Через четыре недели ежедневного перорального дозирования соединение AW и соединение BH продемонстрировали значительное снижение глюкозы в крови. Хотя пиоглитазон действительно первоначально снижал уровень глюкозы в крови в течение первых 3 недель, его активность сильно падала к 4-й неделе и после этого. О дозе пиоглитазона, использованной в данном эксперименте, в литературе сообщалось как о максимально эффективной дозе для обработки мышей db/db (Shimaya et al. (2000), Metabolism 49: 411-7).
Во втором эксперименте на мышах db/db сравнивали антидиабетическую активность соединения BI и розиглитазона. Через 8 недель обработки содержание глюкозы и триглицеридов в крови заметно снижались у животных, получавших или соединение BI или розиглитазон по сравнению с обрабатываемыми носителем животными. Доза розиглитазона, использованная в данном исследовании, в опубликованной литературе известна как оптимальная доза для поздней стадии для db/db мышей (Lenhard et al., (1999) Diabetologia 42: 545-54). Группы состояли из 6-8 мышей каждая.
Пример I: Антидиабетическое действие соединений по изобретению на мышах db/db
Мыши db/db имеют дефект в лептиновой сигнальной передаче, что приводит к гиперфагии, ожирению и диабету. Более того, в отличие от мышей ob/ob на основе линии C57BL/6J мыши db/db на основе линии C57BL/KS подвержены повреждению продуцирующих инсулин островковых клеток поджелудочной железы, что приводит к развитию от гиперинсулинемии (связанной с периферической невосприимчивостью инсулина) к гипоинсулинемическому диабету.
Самцов C57BL/Ksola мышей, подверженных ожирению (db/db гомозиготные), возрастом примерно 8 недель получали от Jackson Labs (Bar Harbor, ME) и случайным образом подразделяли на группы по 5-7 животных, так что масса тела (50-55 г) и уровень глюкозы в сыворотке крови (>300 мг/декалитр в накормленном состоянии) были схожими между группами; самцы тощих мышей (db/+ гетерозиготные) выступали в качестве контрольных групп. Для адаптации после прибытия оставляли минимум 7 дней. Все животные содержались при контролируемой температуре (23° C), относительной влажности (50 + 5%) и световом периоде (7:00-19:00), и животные имели свободный доступ к лабораторному питанию (Formulab Diet 5008, качество Lab Products, Elkridge, MD) и воде.
Группы обработки получали ежедневно перорально дозы (1% гидроксипропилметилцеллюлозы) соединений BI, BO, BP, BQ или BR в течение 2 недель. В конце периода обработки отбирали по 100 мкл венозной крови в гепаринизированную капиллярную пробирку из ретроглазничной пазухи мышей db/db для химического анализа сыворотки крови.
Влияние соединений по изобретению на уровень глюкозы в крови, отобранной не натощак, представлено в таблице 10, влияние на содержание триглицеридов и свободных жирных кислот в сыворотке крови показано в таблице 11.
Уровни глюкозы у тощих недиабетических db/+ гетерозиготных мышей составлял 225 ± 15 мг/декалитр
Пример J: Антидиабетическое влияние соединений по изобретению на db/db мышей.
Мыши db/db имеют дефект в лептиновой сигнальной передаче, что приводит к гиперфагии, ожирению и диабету. Более того, в отличие от мышей ob/ob на основе линии C57BL/6J мыши db/db на основе линии aC57BL/KS подвержены повреждению продуцирующих инсулин островковых клеток поджелудочной железы, что приводит к развитию от гиперинсулинемии (связанной с периферической невосприимчивостью инсулина) к гипоинсулинемическому диабету. Самцов C57BL/Ksola мышей, подверженных ожирению (db/db гомозиготные), возрастом примерно 8 недель получали от Jackson Labs (Bar Harbor, ME) и случайным образом подразделяли на группы по 5-7 животных, так что масса тела (50-55 г) и уровень глюкозы в сыворотке крови (>300 мг/декалитр в накормленном состоянии) были схожими между группами; самцы тощих мышей (db/+ гетерозиготные) выступали в качестве контрольных групп. Для адаптации после прибытия оставляли минимум 7 дней. Все животные содержались при контролируемой температуре (23°C), относительной влажности (50 + 5%) и световом периоде (7:00-19:00), и животные имели свободный доступ к лабораторному питанию (Formulab Diet 5008, качество Lab Products, Elkridge, MD) и воде.
Группы обработки получали ежедневно перорально дозы носителя (1% гидроксипропилметилцеллюлозы), соединений BI, BS, BT, BU, BV или фенофибрата в течение 2 недель. В конце периода обработки отбирали по 100 мкл венозной крови в гепаринизированную капиллярную пробирку из ретроглазничной пазухи мышей db/db для химического анализа сыворотки крови.
Влияние соединений по изобретению на уровень глюкозы в крови, отобранной не натощак, представлено в таблице 12, влияние на содержание триглицеридов и свободных жирных кислот в сыворотке крови показано в таблице 13.
Уровни глюкозы у тощих недиабетических db/+ гетерозиготных мышей составлял 208,5 ± 6,6 мг/декалитр
Пример K: Ослабление катарактогенеза при использовании соединений по изобретению у диабетических жирных крыс Zucker (ZDF)
Катаракты представляют собой одну из ведущих причин прогрессирования упадка зрения и слепоты, связанных с возрастом и диабетом, и модель диабетических жирных крыс Zucker (ZDF) имеет много общего с катарактогенезом у человека, включая биохимические изменения и окислительный стресс в хрусталиках глаза. Эти крысы, однако, подвержены катарактогенезу обычно в возрасте 14-16 недель.
Самцов крыс ZDF и их подобранных по возрасту тощих двойников - крыс Zucker (ZL) (fa/+ или+/+) получали от Genetic Models, Inc. (Indianapolis,IN) в возрасте 12 недель и акклиматизировали в течение 1 недели перед исследованием. Все животные содержались при контролируемой температуре (23°C), относительной влажности (50 + 5%) и световом периоде (7:00-19:00), и животные имели свободный доступ к стандартному питанию (Formulab Diet 5008, качество Lab Products, Elkridge, MD) и водопроводной воде по необходимости. Обрабатываемые группы получали ежедневно перорально дозу носителя и 100 мг/кг соединений BI или BH в течение 10 недель. Вес тела и содержание глюкозы в крови определяли обычным образом (один раз в неделю, обычно около 10.00 утра), пуская кровь из хвоста, с использованием тест-полосок и устройства Glucometer Elite XL (Bayer Corporation). В конце периода обработки собирали 100 мкл венозной крови (обычно около 10.00 утра) в гепаринизированную пробирку из хвостовой вены для химического анализа сыворотки крови (Anilytics, Inc., Gaithersburg, MD). Химический состав сыворотки крови (глюкоза (GL), триглицериды (TG), аспартат аминотрансфераза (AST), аланин аминотрансфераза (ALT), сорбит дегидрогеназа (SDH) и свободные жирные кислоты (FFA)) анализировали с использованием Hitachi 717 Analyzer (Anilytics, Inc., Gaithersburg, MD). Содержание инсулина в плазме измеряли электрохемолюминисцентным иммуноанализом, ECL (Origen Analyzer, Igen, Inc., Gaithersburg, MD). Животных убивали и ткани и/или органы (хрусталики и печень) удаляли, взвешивали (сырой вес) и обрабатывали для биохимических анализов.
Малонодиальдегид (MDA), основной продукт перокисления жиров, анализировали в хрусталиках по методике Ohkawa et al (1979), Analytical Biochem 95,351-358).
В таблице 14 показана доля видимой катаракты в глазах крыс ZDF. В таблице 15 приведены дополнительные количественные показатели катарактогенеза у тех же животных.
Данные представляют собой средние значения±стандартное отклонение. *p < 0,05 при сравнении с контрольной группой носителя (диабетическая) и обрабатываемыми соединением ВН группами соответственно; **p < 0,05 при сравнении с контрольными группами носителя; ‡p < 0,05 при сравнении с контрольной группой носителя и правыми хрусталиками группы соединения ВН соответственно (One Way ANOVA, Tukey Test). Все парные множественные сравнения.
Пример L: При пероральном введении BI и BL понижают циркулирующие триглицериды, свободные жирные кислоты, инсулин и лептин у мышей C57B1/6J, получающих питание с высоким содержанием жиров
Мыши, получающие питание с высоким содержанием жира, представляют собой модель гипертриглицеридемии и высоких циркулирующих уровней жирных кислот и резистентности к инсулину и лептину, что обнаруживается у людей с предрасположенностью к ожирению или страдающих ожирением, диабетом, сердечно-сосудистыми и другими заболеваниями.
Самцов мышей C57B1/6J, возрастом приблизительно 8 недель, случайным образом распределяли на группы по 6 животных. Их содержали при контролируемой температуре (23°C), относительной влажности (50 + 5%) и световом периоде (7:00-19:00), и животные имели свободный доступ к пище и воде по необходимости. Мыши получали питание с высоким содержанием жиров (номер диеты D12451, содержащая 45% калорий в виде жира (Научно-исследовательская диета (Research Diets), New Brunswick, NJ)) в течение 6 недель. Через 6 недель группы мышей получали либо носитель (гидроксиметилцеллюлоза), либо BI, BL, Wyl4,643 или розиглитазон с помощью желудочного зонда в указанных дозах в течение дополнительных 4 недель, при этом они продолжали получать питание с высоким содержанием жиров. Химический состав плазмы (Anilytics, Inc., Gaithersburg, MD) анализировали через 2 недели обработки лекарственным средством. Содержание в плазме сыворотки крови инсулина (фиг.1) и лептина (фиг.2) измеряли с использованием электрохемолюминисцентного иммуноанализа (Origen Analyzer, Igen, Inc., Gaithersburg, MD) по окончании 4-недельного периода обработки лекарственными средствами.
BI и BL оказались эффективными для понижения содержания триглицеридов и свободных жирных кислот в сыворотке крови, а также уровней инсулина и лептина в сыворотке. Значения в сыворотке крови, полученные для мышей из той же группы ("тощий контроль"), которых содержали на регулярном лабораторном питании (Formulab Diet 5008, Quality Lab Products, Elkridge, MD), приведены для сравнения.
Пример М: При пероральном введении BI понижает циркулирующие триглицериды, свободные жирные кислоты, инсулин и лептин у крыс Sprague Dawley, получающих питание с высоким содержанием жиров.
Крыса, получающая питание с высоким содержанием жиров, представляет собой модель резистентности к инсулину и лептину. Крысы Sprague-Dawley обладают неповрежденной лептиновой системой и дают ответную реакцию на питание с высоким содержанием жиров в виде гиперинсулинемии из-за нарушения регуляции нормального инсулинового ответа в периферических тканях, таких как печень, жировая ткань и мышцы.
Самцов крыс Sprague-Dawley возрастом приблизительно 17 недель получали от Jackson Labs (Bar Harbor, ME) и случайным образом распределяли на группы по 5-7 животных, где вес тела между группами был схожим. Всех животных содержали в температурно-контролируемых (25°C) благоприятных условиях с точным 12-часовым циклом свет/темнота, и они имели свободный доступ к воде и еде. Крыс кормили питанием с высоким содержанием жира (диета номер D12451 (содержащая 45% калорий в виде жира), Research Diets, New Brunswick, NJ) в течение одного месяца перед обработкой лекарственным средством.
Группы из 6 крыс Sprague-Dawley обрабатывали однократной ежедневной дозой носителя (гидроксиметилцеллюлоза), BI (10, 30 и 100 мг/кг) или розиглитазона (3 мг/кг) в течение 6 недель при поддержания диеты с высоким содержанием жира. В указанные моменты времени отбирали образцы крови (˜100 мкл) из хвостовой вены для химического анализа сыворотки крови. BI (30 мг/кг) снижал содержание инсулина и триглицеридов в сыворотке крови, BI во всех дозах снижал содержание свободных жирных кислот.
Реферат
Изобретение относится к новым соединениям формул I', I, II, III, IV, V', XCI, CXVI, CXVII (обозначения всех групп приведены в формуле изобретения), которые используют для лечения различных метаболических заболеваний, таких как синдром резистентности к инсулину, диабет, гиперлипидемия, ожирение печени, кахексия, ожирение, атеросклероз и артериосклероз





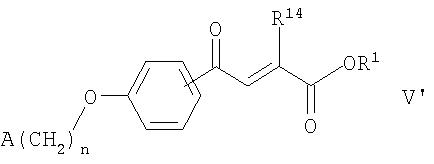



Изобретение также относится к способу получения указанных соединений, применению этих соединений в качестве биологически активного агента, фармацевтическим композициям на основе указанных соединений и к способу лечения с их использованием. 31 н. и 93 з.п. ф-лы, 17 табл., 2 ил.
Формула

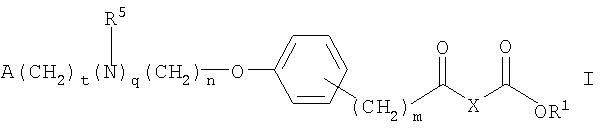






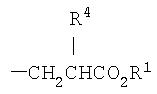

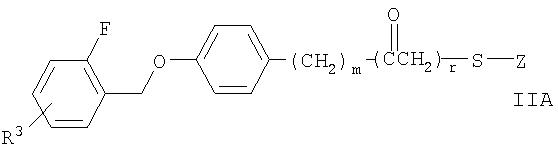


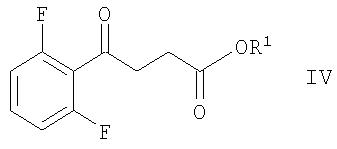






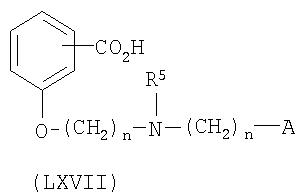





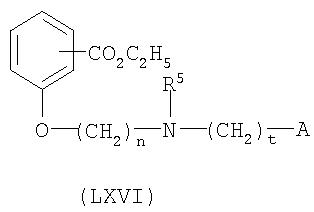



























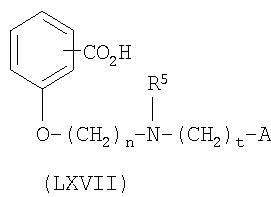




























































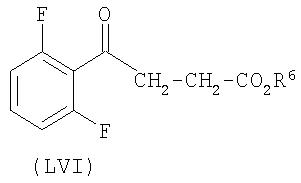





Комментарии