Способ приготовления кремнеалюмофосфатных (sapo) молекулярных сит, катализаторы, содержащие упомянутые сита, и способы каталитической дегидратации с использованием упомянутых катализаторов - RU2469792C2
Код документа: RU2469792C2
Чертежи
Описание
Область техники, к которой относится изобретение
Объектом настоящего изобретения является главным образом получение из метанола ценных продуктов дегидратации, таких как олефины (способ МвО) и диметиловый эфир (ДМЭ).
Объектом настоящего изобретения является, в частности, способ получения продуктов дегидратации из метанола с использованием катализатора на основе кремнеалюмофосфатных (SAPO) молекулярных сит.
Далее, объектом настоящего изобретения являются катализатор на основе кремнеалюмофосфатного молекулярного сита, приемлемый для применения при получении продуктов дегидратации из метанола, а также способы приготовления упомянутого катализатора и упомянутого сита и регенерирования упомянутого катализатора.
Описание известного уровня техники
Увеличение стоимости нефти привело к поискам альтернативных способов получения углеводородов из других источников.
Метанол может быть получен из угля или природного газа путем получения синтез-газа. Далее реакциями дегидратации метанол может быть превращен в углеводороды.
Способы превращения метанола в углеводороды известны хорошо. Вначале в качестве катализаторов для этого превращения используют алюмосиликаты или цеолиты.
При этом было установлено, что кремнеалюмофосфаты (SAPO) способны катализировать некоторые процессы дегидратации, включающие метанол. Так, например, в ЕР 1140743 описан способ МвО с использованием катализатора, содержащего в качестве активного компонента SAPO-34. Этот патент касается, кроме того, важности пути синтеза каталитического SAPO-34.
Многие разработчики по опыту знают, что исключительно решающее значение имеет процесс приготовления SAPO. Так, в частности, для свойств получаемых материалов определяющими являются процесс синтеза, включая используемые реагенты (источники Al, Si и Р и темплат), соотношения элементов (Al, Si и Р), темплат и отмечаемые значения рН. Иногда, как отмечают, в зависимости от используемого пути синтеза даже при применении одних и тех же реагентов могут быть получены разные материалы, причем эти материалы, обладающие одинаковым химическим составом и структурой SAPO, как это определяют по дифракции рентгеновских лучей, характеризуются разными каталитическими свойствами.
Важным параметром в синтезе SAPO-34 является также продолжительность реакции в смеси реагентов в автоклаве, как это изложено в работах S.H.Jhung, J.S.Chang, J.S.Hwang, S.E.Park в Micro.Meso.Mater., 64 (2003) 33 и J.W.Yoon, S.P.Jung, Y.P.Kim, S.E.Park, J.S.Chang в Bull. Korean Chem. Soc., 26(4) (2005) 558. Так, в частности, в соответствии с этими ссылками при кратковременных реакциях отмечали образование SAPO-5, и было сделано заключение о том, что SAPO-5 должно быть предшественником образования SAPO-34, вследствие чего увеличение времени реакции в автоклаве всегда должно приводить к чистому SAPO-34.
По-другому считают O.B.Vistad, E.Akporiaye, K.P.Lillerud, которые в J.Phys.Chem. В, 105 (2001) 12437 пишут, что для синтеза SAPO ключевой характеристикой является скорость нагрева в автоклаве, и поэтому при применении высоких скоростей нагрева в автоклаве в качестве кристаллического продукта отмечали наличие только SAPO-5.
Все эти разные и частично противоречащие друг другу утверждения и наблюдения на путях синтеза SAPO-34 вместе со всегда разными химикатами, темплатами и соотношениями используемых элементов указывают на то, что между химикатами, соотношениями элементов, значением рН суспензии и скоростью нагрева в автоклавах существуют, по-видимому, очень сложные взаимосвязи.
Более того, в соответствии с известным уровнем техники осуществление многих известных методов синтеза SAPO требует относительно больших количеств темплата, поскольку, согласно учению, присутствие темплата в избытке относительно потребностей продукта частично служит для регулирования реакционного значения рН в растворе (H.Van Bekkum, E.M.Flanigen, J.C.Jansen, Studies in Surface Science and Catalysis, 58 (1991) 142).
Так, в частности, когда в качестве темплата используют гидроксид тетраэтиламмония (ТЭА-ОН), обычно применяют соотношение ТЭА-ОН/Аl выше 0,5:1 (см. например, US 5126308 и US 5912393).
Однако высокой оказывается стоимость реагентов, которые должны быть использованы для синтеза SAPO. Известно, в частности, что стоимость таких темплатов, как ТЭА-ОН, очень высока. Это сильно ограничивает применимость вышеуказанных методов синтеза SAPO-34 в промышленном масштабе, поскольку их осуществление требует больших количеств реагентов.
Были предприняты попытки уменьшить количество темплата, используемого при синтезе, добавлением менее дорогостоящего органического основания, но это приводило к повышенному давлению (органические основания, такие как дипропиламин, летучи) и вызывало проблемы, связанные с экологией и безопасностью. Кроме того, отмечают образование продукта нежелательной плотности (см. US 4440871).
Было также заявлено об эффективности применения полимерных оснований, таких как полиэтиленимин, взамен некоторого количества ТЭА-ОН (US 2004/0082466 А1) в качестве попытки каким бы то ни было образом уменьшить соотношение ТЭА-ОН/Аl до уровня ниже указанного выше, с получением SAPO-34, загрязненного неидентифицированной фазой.
Другой недостаток ранее известных способов приготовления SAPO заключается в образовании внутри SAPO нежелательных островков или доменов Si-Al (цеолитных зон), которые изменяют более мягкий кислый характер SAPO, если сравнивать с Si/Al цеолитами (см. H.Van Bekkum, E.M.Flanigen, P.A.Jacobs, J.C.Jansen, Studies in Surface Science and Catalysis, 137 (2001) 378).
В связи с этим следует заметить, что SAPO материалы образуются из АlРО (алюмофосфатных) материалов, которые обладают моновариантным составом каркаса Аl/Р=1. АlРО основаны на каркасе тетраэдра АlO-2 и РО2+, связанных друг с другом обобществленными атомами кислорода. АlРО являются нейтральными и, следовательно, характеризуются ничтожной ионообменной способностью. Когда один SiO2 тетраэдр (который не обладает зарядом) замещает РО2+, тогда один АlO2- несет не уравновешенный заряд, который уравновешивается, по-видимому, дополнительным каркасным катионом. Когда этим катионом является Н, внутри молекулярного сита возникает кислотная (по Бренстеду) боковая поверхность. Тот факт, что Si занимает структурную ячейку Р, продемонстрирован для низких концентраций SiO2 (см. работу Ashtekar S., Chilukuri S. V. V., Chakrabarty D.K. в J.Phys.Chem. 98 (1994) 4878).
Краткое изложение сущности изобретения
Техническая проблема, лежащая в основе создания настоящего изобретения, при этом заключается в разработке способа приготовления SAPO молекулярных сит, обладающих высокой каталитической активностью в реакции дегидратации, который свободен от недостатков известных технических решений, упомянутых выше. Так, в частности, техническая проблема заключается в разработке способа приготовления SAPO молекулярных сит, осуществление которого включает компоненты низкой стоимости, благодаря чему он приемлем для промышленности и экономически выверен при одновременном сохранении как чистоты продукта, так и высокого выхода молекулярного сита и благодаря которому образование доменов Si-Al внутри SAPO оказывается затруднительным.
Эту проблему позволяет разрешить осуществление способа приготовления SAPO молекулярного сита, включающего следующие стадии:
смешение источника Р с источником Аl с получением смеси,
добавление в упомянутую смесь источника Si и темплата с получением суспензии или шлама,
гидротермическая обработка упомянутой суспензии или шлама с получением упомянутой суспензии SAPO молекулярного сита,
выделение из упомянутой суспензии упомянутого SAPO молекулярного сита и его сушка,
характеризующегося тем, что перед добавлением упомянутого источника Si и упомянутого темплата упомянутую смесь варят при перемешивании с выдержкой при повышенной температуре.
В предпочтительном варианте упомянутый источник Аl выбирают среди гидратированного оксида алюминия, предпочтительно псевдобемита, и нитрата алюминия.
В предпочтительном варианте упомянутый источник Р представляет собой ортофосфорную кислоту, предпочтительно ее водный раствор, а упомянутый источник Si представляет собой стабилизированный аммиаком коллоидный раствор кремнекислоты.
В предпочтительном варианте упомянутый темплат выбирают среди алифатических аминов и солей четвертичного аммония, в частности ТЭА-ОН (гидроксид тетраэтиламмония).
Понятие "темплаты" относится, как правило, к структуронаправляющим средствам, которые используют для формирования каналов или туннелеподобных структур (также называемых микропористыми структурами) в композиции молекулярного сита. Однако для открытия каналов или туннелеподобных структур в случаях SAPO материалов, которые должны быть использованы в качестве каталитических композиций, темплат должен быть удален. Это осуществляют во время приготовления содержащего SAPO катализатора кальцинированием содержащего SAPO порошка так, как это лучше разъяснено ниже.
В предпочтительном варианте соответствующие молярные соотношения вышеприведенных реагентов (источников) в конечной суспензии или шламе, выраженные в пересчете на оксиды Р, Аl и Si, при использовании ТЭА-ОН (гидроксид тетраэтиламмония) в качестве темплата, являются нижеследующими: Р2O5:Аl2O3:SiO2:ТЭА-ОН:Н2O=1:(0,8-1,5):(0,1-0,4):(0,6-1,4):(80-130).
В более предпочтительном варианте упомянутые молярные соотношения являются нижеследующими:
Р2O5:Аl2O3:SiO2:ТЭА-ОН:Н2O=1:(0,8-1,4):(0,1-0,4):(0,6-1,15):(80-130).
В предпочтительном варианте молярное соотношение вышеприведенных реагентов выбирают таким образом, чтобы соотношение Si:Al было ниже 0,11 (эквивалент SiO2/Аl2O3=0,22), а соотношение ТЭА-ОН:Аl было ниже 0,5. В предпочтительном варианте все упомянутые реагенты характеризуются очень низким содержанием Na (<0,01%). В предпочтительном варианте коллоидный раствор кремнекислоты стабилизируют аммиаком. В соответствии с настоящим изобретением стадию варки осуществляют перемешиванием смеси источников Аl и Р при температуре в интервале от 50 до 100°С, предпочтительно при 75°С, в течение периода времени от 10 до 30 ч, предпочтительно 24 ч.
В соответствии с одним вариантом выполнения настоящего изобретения способ может также включать стадию регулирования рН суспензии или шлама, приготовленного подмешиванием источника Si и темплата в подвергнутую варке смесь. В этом случае в предпочтительном варианте рН доводят до слегка кислотного уровня, например в пределах от 5 до 6, 8.
Что касается стадии гидротермической обработки, то конечную суспензию или шлам заключают в герметичный автоклав и выдерживают с перемешиванием при температуре от 170 до 220°С, предпочтительно от 180 до 190°С.Продолжительность варьируют, как правило, в интервале от примерно 12 до примерно 80 ч, предпочтительно от примерно 50 до примерно 72 ч.
Выделение SAPO молекулярного сита из суспензии после гидротермической обработки может быть осуществлено обычным путем, например фильтрованием или центрифугированием, и выделенный SAPO, как правило, сушат.
В противоположность известным методам процесс, как было установлено при создании настоящего изобретения, проводят с использованием технических исходных материалов и при очень низком содержании темплата, предусматривая стадию варки смеси источника Аl (такого как оксид алюминия) и источника Р при данной температуре в течение данного времени с последующими добавлением диоксида кремния и темплата, регулированием конечного значения рН, обычной гидротермической обработкой полученной суспензии или шлама при более высокой температуре в автоклаве и получением с очень высоким выходом очень чистого высококристаллического SAPO молекулярного сита, в частности SAPO-34.
Не основываясь на какой-либо теории, полагают, что стадия варки источника Аl (например, оксида алюминия) в источнике Р (например, в водном растворе ортофосфорной кислоты) для приготовления чистого SAPO при самом низком содержании темплата и диоксида кремния в суспензии и самом высоком выходе молекулярного сита имеет существенное значение. Поскольку темплат является самым дорогостоящим исходным материалом, расходы на синтез можно свести к минимуму.
Более того, осуществление стадии варки позволяет также получать SAPO с очень низким содержанием диоксида кремния. Таким образом можно регулировать кислотность, после того как Si замещает Р при синтезе SAPO при низких мольных фракциях диоксида кремния в каркасе, генерирующих поэтому регулируемое число более мягких кислотных боковых поверхностей (см. теоретические аспекты в работе Barthomeuf D. в Zeolites 14 (1994) 394-401).
Решающим фактором для образования чистого SAPO-34 фактически является, по-видимому, образование фазы стабильного и структуронаправляющего предшественника, которую получают в соответствии с изобретением осуществлением стадии варки при данной температуре, предпочтительно в интервале от 50 до 100°С, наиболее предпочтительно при 75°С.
Напротив, в противоположность изложенному в известной литературе скорость нагрева на стадии варки не является, по-видимому, решающей для образования чистого SAPO-34.
Более того, в способе в соответствии с изобретением точное регулирование общего соотношения Al:P:Si:TЭA-OH:H2O может быть выполнено в основных режимах.
В предпочтительном варианте в способе в соответствии с изобретением кристаллы SAPO материала характеризуются размерами в интервале от 0,2 до 5 мкм, предпочтительно от 1 до 3 мкм.
Объектом настоящего изобретения далее является SAPO молекулярное сито, которое может быть получено по способу, описанному выше.
Это SAPO молекулярное сито в соответствии с изобретением характеризуется следующим химическим составом: (SixAlyPz)O2 где х, у и z обозначают мольные доли соответственно Si, Al и Р, причем х обозначает мольную долю Si и обладает значением в интервале от 0,001 до 0,1, у обозначает мольную долю Al и обладает значением в интервале от 0,25 до 0,5, z обозначает мольную долю Р в интервале от 0,4 до 0,8.
В предпочтительном варианте SAPO молекулярное сито представляет собой SAPO-34.
В сравнении с ранее известными материалами SAPO молекулярное сито в соответствии с изобретением обладает низким молярным соотношением Si/Al, a также характеризуется исключительной каталитической активностью в реакциях дегидратации метанола.
Тогда другой объект настоящего изобретения представляет собой катализатор, включающий SAPO молекулярное сито по изложенному выше, который может быть эффективно использован в реакциях дегидратации метанола.
Этот катализатор может быть получен по способу, который включает следующие стадии:
приготовление SAPO молекулярного сита так, как изложено выше,
подмешивание или внедрение упомянутого SAPO молекулярного сита в матричный/связующий материал с получением пасты или суспензии,
обработка упомянутой пасты или суспензии с получением твердых частиц,
кальцинирование упомянутых твердых частиц с получением упомянутого катализатора.
Вышеуказанный способ может дополнительно включать стадию кальцинирования упомянутого SAPO молекулярного сита перед его подмешиванием или внедрением в матричный/связующий материал.
В этом способе матричный материал действует как разбавитель, но также и как непрерывная связующая фаза между мелкими кристаллами молекулярного сита с образованием пасты или суспензии. Затем эту пасту обрабатывают с получением твердых частиц экструзией по методам прессования или атомизацией с получением экструдатов, гранул или микросфер и эти твердые частицы подвергают обработке по методу кальцинирования.
В предпочтительном варианте паста содержит от 40 до 98 мас.% (в пересчете на сухое вещество) матричного материала и от 2 до 60 мас.% (в пересчете на сухое вещество) молекулярного сита. В наиболее предпочтительном варианте, когда катализатор используют для ДМЭ, - от 70 до 98 мас.% матричного материала и от 2 до 30 мас.% молекулярного сита, а когда катализатор используют для МвО, - от 40 до 70 мас.% матричного материала и от 60 до 30 мас.% молекулярного сита.
Приемлемые матричные материалы, которые не влияют на каталитические свойства молекулярного сита, представляют собой активированный оксид алюминия, диоксид кремния, силикат алюминия, диоксид кремния/оксид алюминия и природный каолин. В предпочтительном варианте упомянутые матричные материалы характеризуются большой удельной площадью поверхности и хорошими связующими свойствами, обусловленными реакционноспособными поверхностными группами.
Предпочтительные матричные материалы для ДМЭ состоят из активированных глиноземов, содержащих по меньшей мере 50 мас.% псевдобемита, диоксида кремния и диоксида кремния/оксида алюминия для МвО.
В предпочтительном варианте упомянутые твердые частицы находятся в форме гранул, таблеток, экструдатов или микросфер.
В соответствии с одним объектом настоящего изобретения кальцинирование смеси молекулярного сита и матричного материала и необязательно одного молекулярного сита проводят при температурах, не превышающих 600°С. В предпочтительном варианте упомянутое кальцинирование начинают при комнатной температуре, температуру постепенно повышают в течение от 4 до 10 ч, в один или несколько этапов доводят до целевого уровня и поддерживают эту температуру в течение последующих от 4 до 8 ч.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения упомянутое кальцинирование проводят в присутствии инертного газа, такого как аргон или азот, до 500°С.
Этим путем было установлено, что процесс кальцинирования является более кратковременным, оно может быть достигнуто при более низких температурах и одновременно с этим инертный газ действует как разновидность десорбирующего агента, наличие которого позволяет по существу полностью удалять темплат. Более того, приготовленный таким образом катализатор демонстрирует увеличенные стойкость и срок службы во время его применения в реакциях дегидратации метанола, таких как превращение метанола в ДМЭ или олефины.
В соответствии с другим вариантом выполнения настоящего изобретения катализатор находится в форме гранул или экструдатов, обладающих диаметром от 1 до 6 мм и длиной от 1 до 5 мм.
В соответствии с еще одним вариантом выполнения настоящего изобретения катализатор находится в форме микросфер, обладающих средним диаметром от 0,05 до 0,08 мм.
Объектом настоящего изобретения является также способ дегидратации метанола до ценных химикатов, таких как олефины и ДМЭ, который включает стадию контактирования метанола с катализатором, таким как описанный выше, в условиях, эффективных для превращения метанола в один из упомянутых химикатов.
Способ превращения метанола в продукты дегидратации, такие как олефины и ДМЭ, может быть осуществлен в реакторе, обладающем неподвижным слоем или псевдоожиженным слоем упомянутого катализатора. Этот реактор может быть изотермическим, с охлаждающими поверхностями в прямом контакте с формованным катализатором, или адиабатическим, с несколькими каталитическими слоями и промежуточным охлаждением между слоями.
Рабочие условия должны быть оптимизированы для выбранной дегидратации. Более мягкие условия, т.е. температуры в интервале от 150 до 250°С, приводят к селективному образованию ДМЭ, более высокие температуры (превышающие 250°С) приводят к олефинам.
В предпочтительном варианте рабочее давление в реакторе находится в интервале от атмосферного до 20 бар, предпочтительно от 5 до 15 бар, а объемная скорость потока метанола, выраженная в литрах подаваемого жидкого метанола в час на объем катализатора в литрах, находится в интервале от 1 до 50 ч-1, предпочтительно от 1 до 5 ч-1.
В одном варианте выполнения настоящего изобретения реакционные продукты в виде парообразных потоков направляют в ректификационные колонны.
Объектом настоящего изобретения далее являются способы регенерирования каталитической активности катализатора, который описан выше. После каждого цикла его применения в способе в соответствии с изобретением при превращении метанола в ДМЭ или олефины способ регенерирования включает стадию пропускания через катализатор по меньшей мере одного потока регенерирующего агента, выбранного среди азота, аргона, водорода, воздуха, водяного пара и их смесей, при температуре максимум 550°С в течение периода от 1 до 12 ч.
В способе превращения метанола в олефины регенерирование осуществляют в регенераторе с непрерывной циркуляцией закоксованного катализатора из реактора в регенератор. В плотный слой катализатора в регенераторе инжектируют воздух. Продувкой водяным паром или азотом вертикальных трубных реакторов катализатор очищают метанолом и углеводородами, адсорбированными в реакторе, и кислородом, адсорбированным в регенераторе.
Другие характеристики и преимущества настоящего изобретения должны стать более очевидными из примеров, представленных в настоящем описании ниже, которые приведены с иллюстрирующей, а не ограничивающей целью, со ссылкой на прилагаемые чертежи.
Краткое описание чертежей
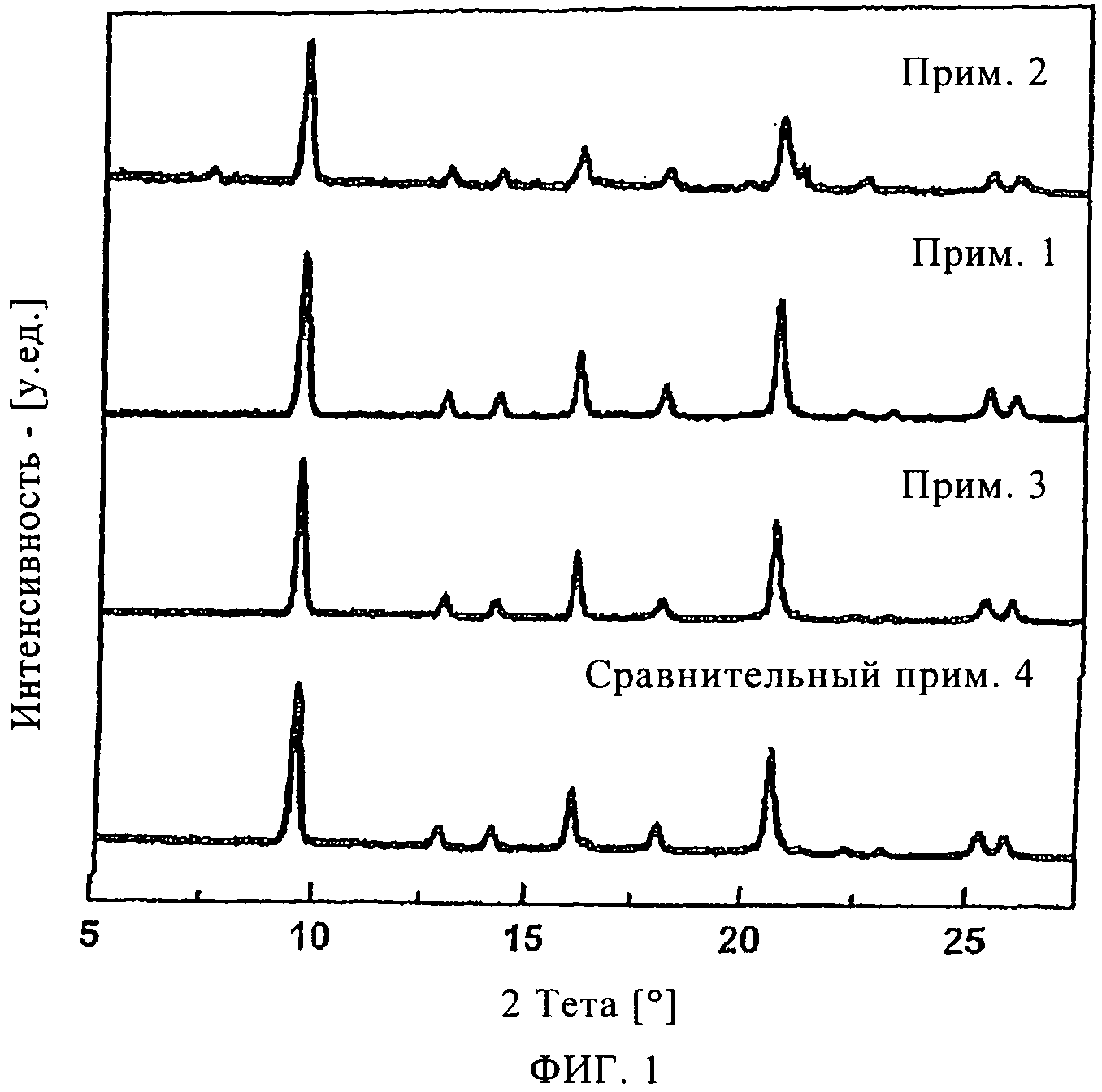
На фиг.1 продемонстрирована рентгенограмма SAPO-34 материалов в соответствии с изобретением, приготовленных согласно методам, изложенным в примерах с 1 по 3, приведенных в настоящем описании ниже, и SAPO-34 материала, приготовленного согласно методу, изложенному в сравнительном примере 4, приведенном в настоящем описании ниже.
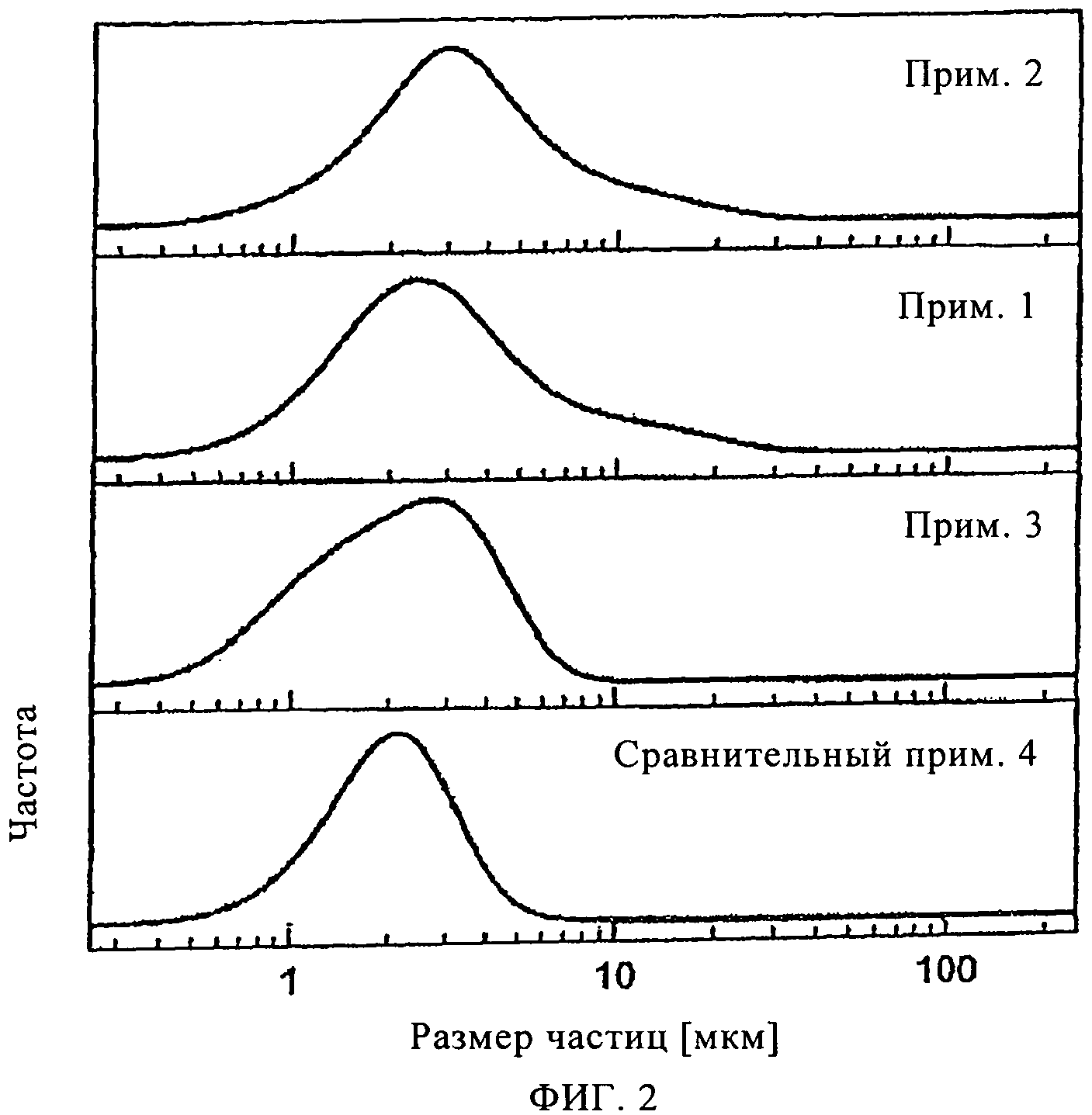
На фиг.2 показано распределение по размерам частиц SAPO-34 материалов, показанных на фиг.1.
На фиг.3 продемонстрированы результаты катализа для катализатора, включающего SAPO-34 по изобретению, приготовленный в соответствии с примером 1, приведенным в настоящем описании ниже.
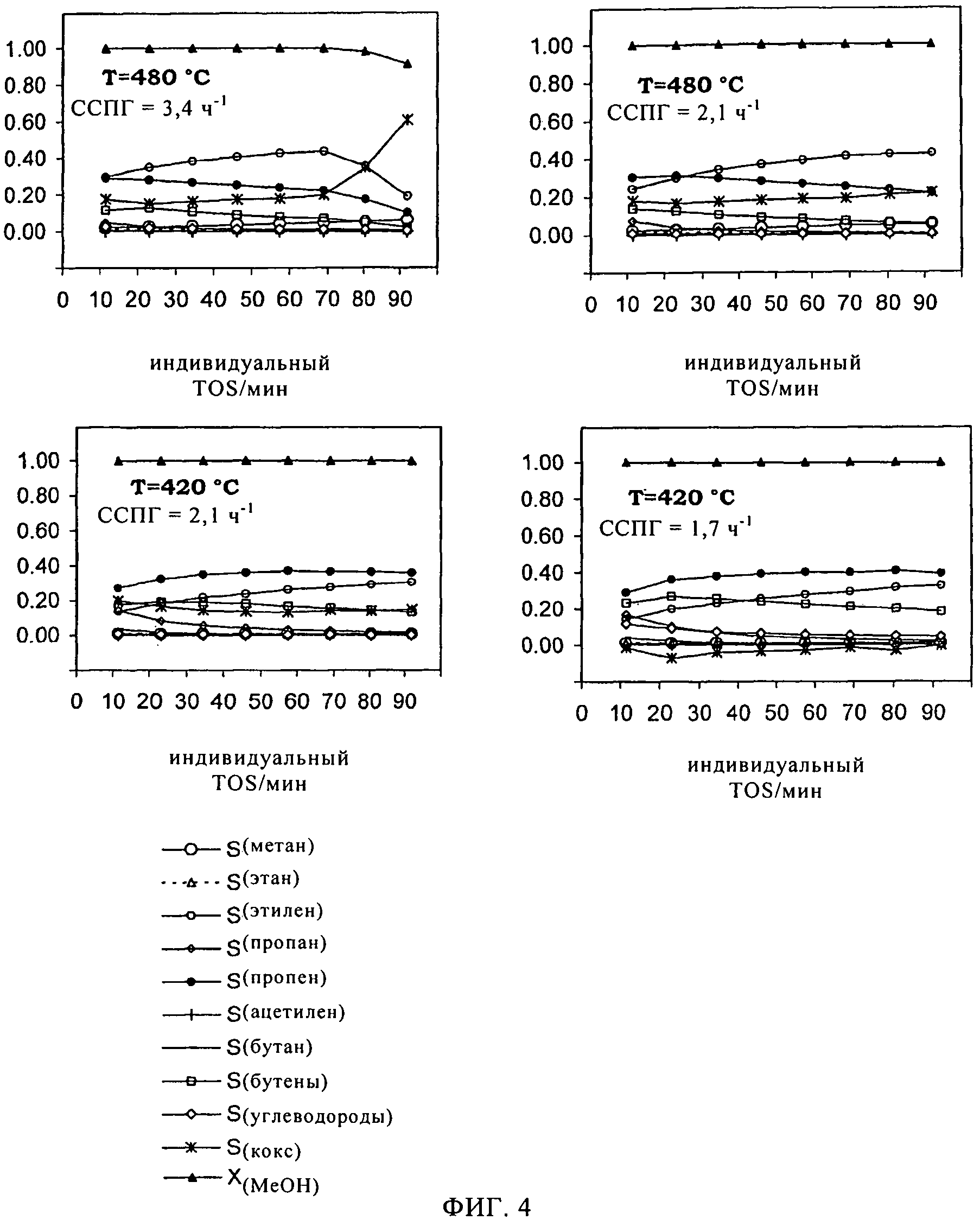
На фиг.4 продемонстрированы результаты катализа для катализатора, включающего SAPO-34 по изобретению, приготовленный в соответствии с примером 2, приведенным в настоящем описании ниже.
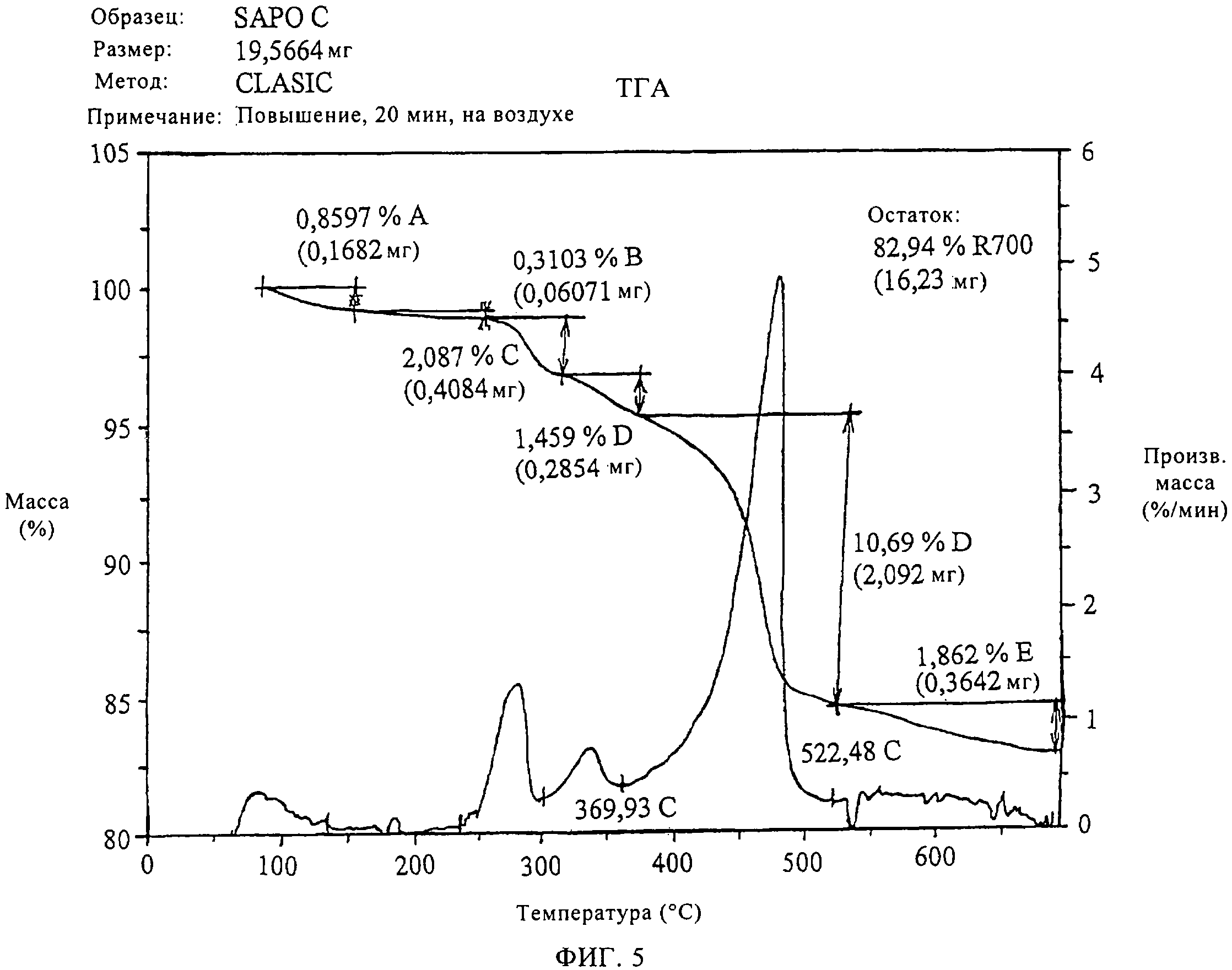
На фиг.5 продемонстрированы данные термогравиметрического анализа только что синтезированного SAPO-34 молекулярного сита в соответствии с изобретением на воздухе.
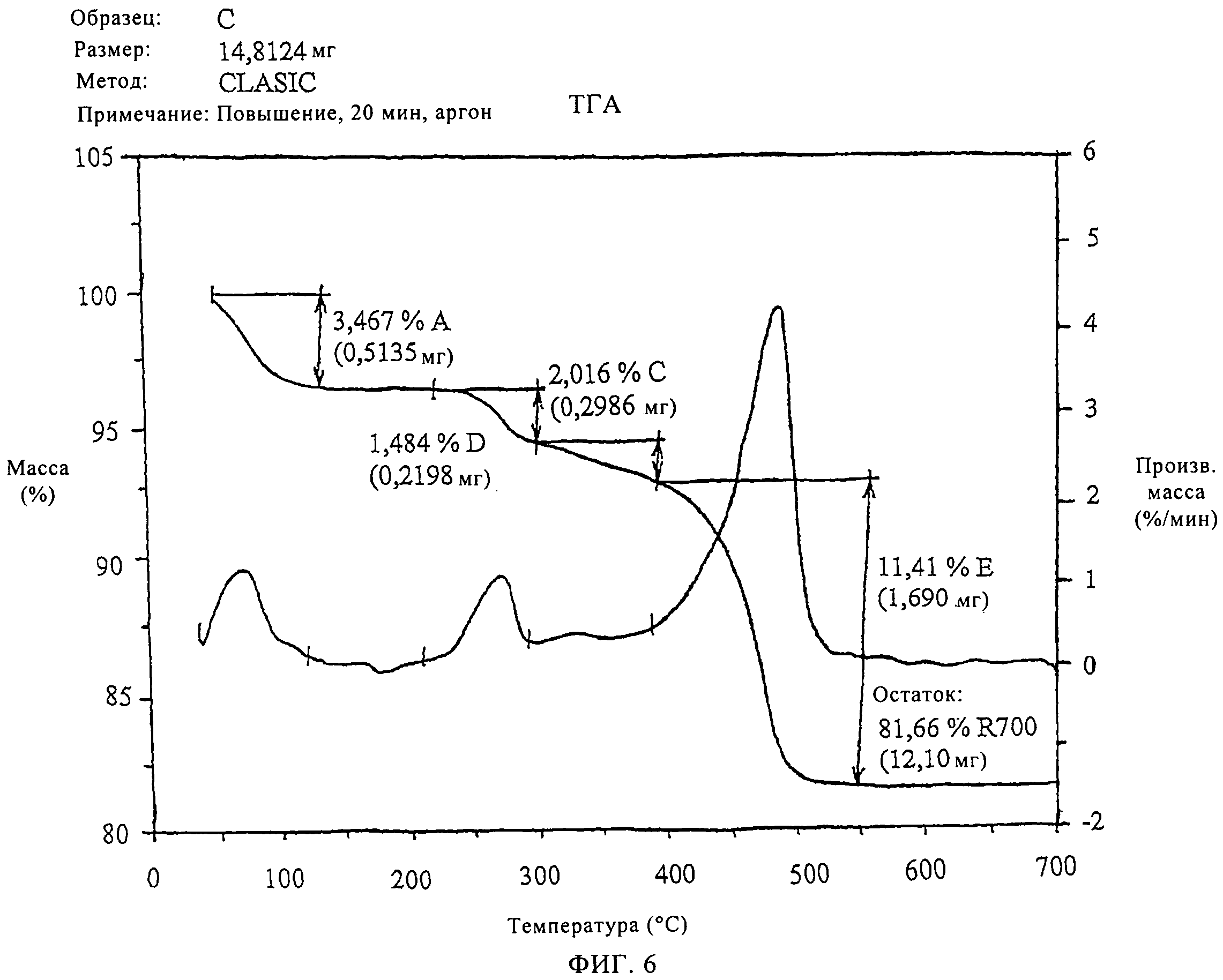
На фиг.6 продемонстрированы данные термогравиметрического анализа только что синтезированного SAPO-34 молекулярного сита в соответствии с изобретением в аргоне.
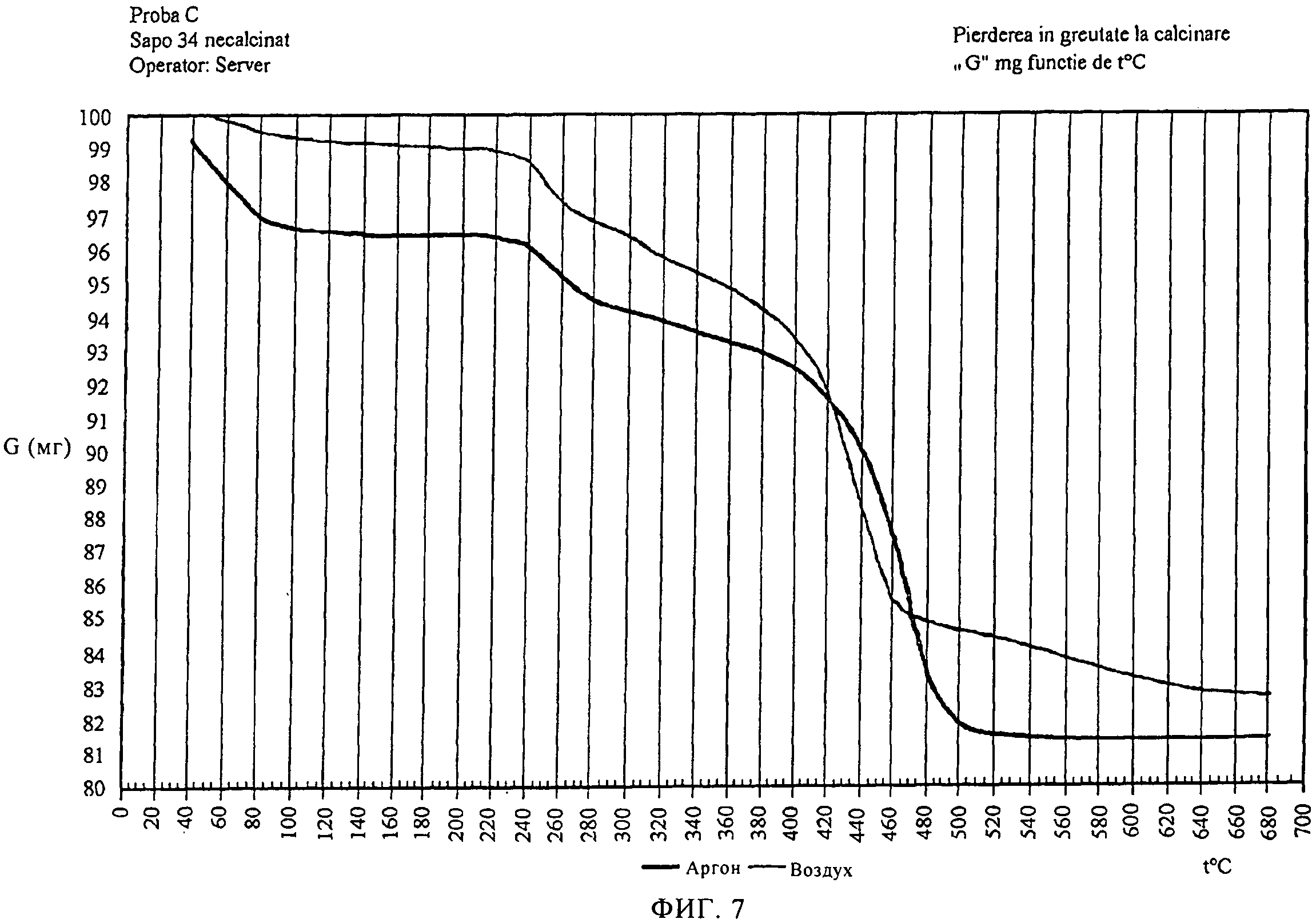
На фиг.7 продемонстрировано совмещение данных термогравиметрического анализа показанного на фиг.5 и 6 только что синтезированного SAPO-34 молекулярного сита соответственно на воздухе и в аргоне.
Пример 1
Синтез чистого SAPO-34 молекулярного сита
100 г Н2О и 15,6 г H3PO4 (85%-ной) смешивали при 25°С. При 25°С медленно с перемешиванием добавляли 11,2 г псевдобемита (АlOOН·Н2О, эквивалентного 65,4% Аl2О3). Приготовленную суспензию нагревали до 75°С и перемешивали в течение дополнительных 24 ч. По прошествии этого периода времени при 75°С с перемешиванием добавляли 3,6 г коллоидного раствора кремнекислоты (25%-ного) и 40,7 г раствора ТЭА-ОН (25%-ного). Сразу же после конечного добавления источников Si- и темплата значение рН конц. HNO3 (~3 мл) при 75°С доводили до 6,8. Приготовленную конечную суспензию выдерживали в течение 3 дней при 185°С в автоклаве, фильтровали, промывали Н2О и сушили при 110°С в течение 12 ч. Выход после сушки составлял 20 г (~80% от теоретического выхода).
Подробности всего элемента, количества темплата и соотношения представлены в таблице 1.
Рентгенограмма, демонстрирующая чистую SAPO-34 фазу, приведена на фиг.1. Распределение частиц по размерам представлено на фиг.2.
Пример 2
Синтез чистого SAPO-34 молекулярного сита
Синтез проводили идентично рецепту синтеза, описанному в примере 1. Значение рН регулировали ~3 мл Н3РO4 (85%-ной). Выход после сушки составлял 20 г (~80% от теоретического выхода). Подробности всего элемента, количества темплата и соотношения представлены в таблице 1.
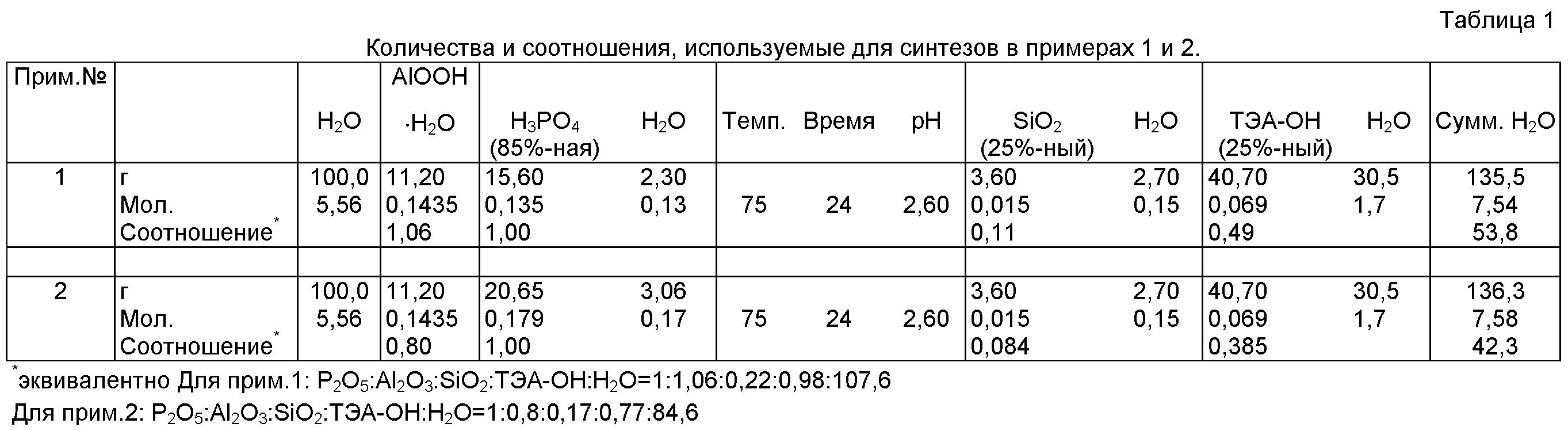
Примечание: общие количества Н2РO4, коллоидного раствора кремнекислоты и раствора ТЭА-ОН в граммах включают отдельно указанное содержание Н2O в этих химикатах (которое является единственно указанным для расчета общего количества Н2O, использованной в рецептуре синтеза).
Рентгенограмма, демонстрирующая чистую SAPO-34 фазу, приведена на фиг.1. Распределение частиц по размерам представлено на фиг.2.
Пример 3
Синтез чистого SAPO-34 молекулярного сита
При 25°С смешивали 100 г Н2O и 11,2 г Н3РO4 (85%-ной). При 25°С медленно с перемешиванием добавляли 11,2 г псевдобемита. Приготовленную суспензию нагревали до 75°С и перемешивали в течение дополнительных 24 ч. По прошествии этого периода времени при 75°С с перемешиванием добавляли 3,6 г коллоидного раствора кремнекислоты (25%-ного) и 40,7 г раствора ТЭА-ОН (25%-ного). Сразу же после конечного добавления источников Si- и темплата значение рН конц. НNO3 (~4 мл) при 75°С доводили до 6,8. Приготовленную конечную суспензию выдерживали в течение 3 дней при 185°С в автоклаве, фильтровали, промывали Н2О и сушили при 110°С в течение 12 ч. Выход после сушки составлял 14 г (~56% от теоретического выхода).
Подробности всего элемента, количества темплата и соотношения представлены в таблице 2.
Примечание: общие количества Н3РO4, коллоидного раствора кремнекислоты и раствора ТЭА-ОН в граммах включают отдельно указанное содержание Н2О в этих химикатах (которое является единственно указанным для расчета общего количества Н2O, использованной в рецептуре синтеза).
Рентгенограмма, демонстрирующая чистую SAPO-34 фазу, приведена на фиг.1. Распределение частиц по размерам представлено на фиг.2.
Пример 4 (сравнительный пример)
Синтез SAPO-34 молекулярного сита
При 25°С смешивали 50 г Н2О и 15,6 г 85%-ной Н3РO4. При 25°С медленно с перемешиванием добавляли 11,2 г псевдобемита и при 25°С перемешивали в течение дополнительных 24 ч. По прошествии этого периода времени с перемешиванием добавляли 3,6 г 25%-ного коллоидного раствора кремнекислоты. Значение рН суспензии с использованием 72 г раствора ТЭА-ОН и 25 г Н2О при 25°С и перемешивании доводили до 6,8. Приготовленную конечную суспензию выдерживали в течение 3 дней при 185°С в автоклаве, фильтровали, промывали Н2О и сушили при 110°С в течение 12 ч. Выход после сушки составлял 11 г (~44% от теоретического выхода).
При применении большего количества ТЭА-ОН предварительное перемешивание всей химической массы при 25°С приводило к чистому SAPO-34 материалу, однако с пониженными значениями выхода.
Подробности всего элемента, количества темплата и соотношения представлены в таблице 3.
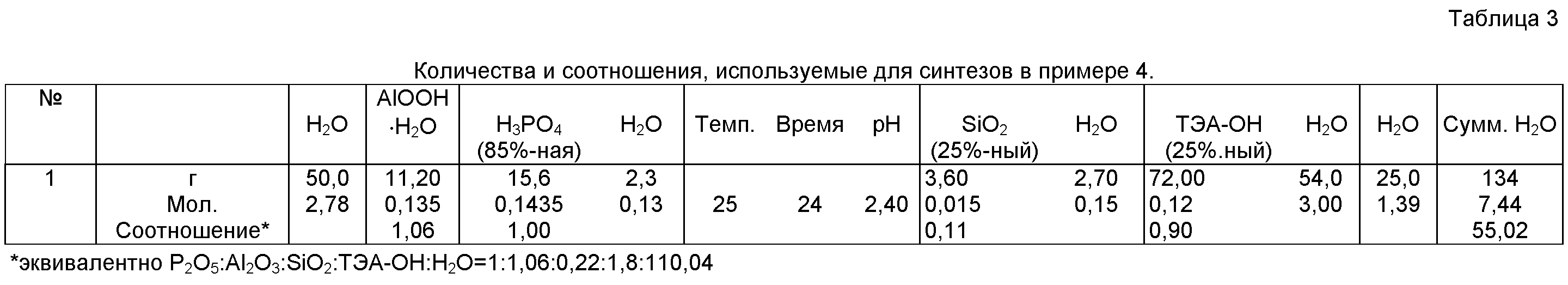
Примечание: общие количества Н2РO4, коллоидного раствора кремнекислоты и раствора ТЭА-ОН в граммах включают отдельно указанное содержание Н2О в этих химикатах (которое является единственно указанным для расчета общего количества H2O, использованной в рецептуре синтеза).
Рентгенограмма, демонстрирующая чистую SAPO-34 фазу, приведена на фиг.1. Распределение частиц по размерам представлено на фиг.2.
Пример 5 (сравнительный пример)
Синтез SAPO-34 молекулярного сита
С использованием соотношений элементов и темплата, идентичных указанным для рецептуры синтеза в примере 1, без стадии перемешивания и нагревания после смешения источников фосфата и оксида алюминия проводили несколько синтезов. Испытывали любую возможную последовательность добавления. Различные продолжительности синтеза в автоклаве в пределах от 2 до 7 дней и скорости нагрева приводили к безуспешному синтезу SAPO-34. В связи с этим понятие "безуспешный" означает, что полученные материалы всегда состояли из чистого SAPO-5 или смеси с различными соотношениями SAPO-5 и SAPO-34.
Пример 6
Каталитическая композиция
140 г только что синтезированного порошкообразного SAPO, полученного в соответствии с методом, приведенным в примере 1 или 2 (содержащего 100 г сухого материала, причем остальное приходится на влагу), смешивают с 800 г стабилизированного аммиаком коллоидного кремнезема с 8,5% SiO2. Приготовленную суспензию перемешивают до гомогенного состояния. Перемешивание суспензии продолжают и ее атомизируют распылительной сушкой в следующих условиях:
температура воздуха на входе: 420°С
температура воздуха на выходе: 150°С
инжекционное давление: 4 бара
Далее продукт подвергают кальцинированию на воздухе при 550°С в течение 10 ч. В первый период в течение 6 ч температуру повышают до 350°С и поддерживают на этом уровне в течение 2 ч, во второй период температуру повышают до 550°С со скоростью нагрева 2°С/мин и ее поддерживают в течение 10 ч. Потеря массы во время кальцинирования составляет примерно 30% от исходной массы. Конечный продукт состоит из бисера со средним размером частиц от 50 до 80 мкм.
Пример 7
Метод кальцинирования
Молекулярное сито для катализатора в соответствии с изобретением готовили согласно методу примера 1. Кальцинирование проводили в присутствии воздуха или инертного газа (аргон) при температуре до 500°С.Было установлено, что для полного удаления темплата в более мягких условиях кальцинирование в инертном газе более эффективно.
Как можно видеть на фиг.5, термический анализ образца только что синтезированного молекулярного сита, приготовленного в соответствии с примером 1, на воздухе демонстрирует постоянную потерю массы после 550°С до 700°С.Термогравиметрические кривые, полученные в инертной среде, в аргоне, молекулярного сита в соответствии с данным примером (на фиг.6) демонстрируют полное удаление органического материала при 550°С. Совмещение кривых потери массы на фиг.7 демонстрирует эти отличия более четко.
Пример 8
Каталитические данные SAPO-34 молекулярных сит в реакции МвО
По 300 мг каждого катализатора, синтезированного в соответствии с примером 6, содержавшего SAPO-34 в соответствии с примером 1 или 2, испытывали в реакции МвО. Образцы катализатора испытывали в четырех реакционных условиях:
Т: 480°С, ССПГ: 3,4 ч-1
Т: 480°С, ССПГ: 2,1 ч-1
Т: 420°С, ССПГ: 2,1 ч-1
Т: 420°С, ССПГ: 1,7 ч-1
Исходный материал состоял из метанола (80 об.%) и азота (20 об.%). Во всех вышеприведенных реакционных условиях все катализаторы испытывали в течение 90 мин каждый. После этого катализаторы регенерировали в токе воздуха при 550°С в течение 60 мин. Затем проводили второе испытание в тех же реакционных условиях для проверки, достигали ли катализаторы своей полной активности после регенерирования. После второго регенерирования испытание проводили при последующем реакционном условии и т.д. В общей сложности каждый из образцов катализатора испытывали и регенерировали по 8 раз без потери активности.
Результаты представлены на фиг.3 для катализатора, включавшего SAPO-34 в соответствии с примером 1, и на фиг.4 - для катализатора, включавшего SAPO-34 в соответствии с примером 2.
При создаваемых реакционных условиях превращение метанола составляет 100%. По прошествии времени реакции для обоих образцов катализатора, вначале демонстрировавшего дезактивацию после 70 мин при более высокой температуре, практически никакой дезактивации не обнаруживали.
Обычно основными реакционными продуктами являются пропей и этилен, за которыми следуют бутены, пропан и бутан. Метан, этан, ацетилен и углеводороды С5 обнаруживают только в виде следов.
Значения селективности в отношении продуктов, а также соотношения между разными углеводородами испытывают влияние, главным образом, реакционной температуры.
При 480°С этилен является основным реакционным продуктом, тогда как при пониженной реакционной температуре, 420°С, более высокой оказывается селективность в отношении пропена. При 420°С, в сравнении с более высокой температурой, образуется также больше бутанов и пропана. При обеих температурах селективность в отношении пропана и бутена с течением времени уменьшается, тогда как образование пропена улучшается. Селективность в отношении метана, этана, ацетилена и углеводородов С5 обычно низка.
Как показывает расчет по углеродному балансу, вместе с селективностью проявляется коксообразование.
Пример 9
Формование катализатора и результаты катализа с помощью SAPO-34 молекулярных сит в реакции до ДМЭ
10 г высушенного некальцинированного SAPO-34, синтезированного в соответствии с методом примера 1, смешивали с 51,5 г гидратированного оксида алюминия. В эту смесь добавляли 30 г разбавленной азотной кислоты (10%-ной). Приготовленную гомогенную пасту экструдировали с использованием фильеры диаметром 2 мм. Экструдаты сушили при 100°С и в дальнейшем кальцинировали на воздухе. Температуру повышали в течение 6 ч от комнатной до 600°С и эту температуру поддерживали в течение 8 ч.
30 мл катализатора испытывали в реакторе с электрическим нагревом в условиях разных температур и давлений. Создаваемую объемную скорость СЧСЖ выражали в л ч-1 (литров жидкого метанола/литров катализатора в час). После охлаждения и конденсации жидкие продукты анализировали, не сконденсировавшийся газообразный продукт направляли на газовую хроматографию. Полученные результаты представлены в таблице 4.
Реферат
Настоящее изобретение относится к способу приготовления кремнеалюмофосфатных (SAPO) молекулярных сит, к катализаторам, содержащим их, и к способам каталитической дегидратации с использованием упомянутых катализаторов. Описан способ приготовления SAPO молекулярного сита, предпочтительно обладающего кристаллической структурой SAPO-34, включающий следующие стадии: смешение источника Р с источником Аl с получением смеси, добавление в упомянутую первую смесь источника Si и темплата с получением суспензии или шлама, необязательно регулирование рН упомянутой суспензии или шлама до значения в интервале от 5 до 6,8, гидротермическая обработка упомянутой суспензии или шлама с получением суспензии SAPO молекулярного сита, выделение из суспензии упомянутого SAPO молекулярного сита и его сушка, причем перед добавлением упомянутого источника Si и упомянутого темплата упомянутую смесь источников Р и Аl варят при перемешивании, упомянутую стадию варки осуществляют при температуре в интервале от 50 до 100°С, предпочтительно при 75°С, в течение периода времени от 10 до 30 ч, предпочтительно 24 ч. Описано SAPO молекулярное сито, предпочтительно обладающее кристаллической структурой SAPO-34, оно может быть приготовлено согласно описанному способу и характеризуется следующим химическим составом: (SiАlР)O, где x, y и z обозначают мольные доли соответственно Si, Al и Р, причем x обозначает мольную долю Si и обладает значением в интервале от 0,001 до 0,1, у обозначает мольную долю Al и обладает значением в интервале от 0,25 до 0,5, z обозначает мольную долю Р в интервале от 0,4 до 0,8. Описан катализатор, включающий SAPO молекулярное сито и матричный материал, выбранный из гр
Формула
смешение источника Р с источником Аl с получением смеси, добавление в упомянутую первую смесь источника Si и темплата с получением суспензии или шлама,
необязательно регулирование рН упомянутой суспензии или шлама до значения в интервале от 5 до 6, 8,
гидротермическая обработка упомянутой суспензии или шлама с получением суспензии упомянутого SAPO молекулярного сита,
выделение из упомянутой суспензии упомянутого SAPO молекулярного сита и его сушка,
отличающийся тем, что перед добавлением упомянутого источника Si и упомянутого темплата упомянутую смесь источников Р и Аl варят при перемешивании, упомянутую стадию варки осуществляют при температуре в интервале от 50 до 100°С, предпочтительно при 75°С, в
течение периода времени от 10 до 30 ч, предпочтительно 24 ч.
Р2O5:Аl2O3:SiO2:ТЭА-ОН:Н2O=1:(0,8-1,5):(0,1-0,4):(0,6-1,4):(80-130), более предпочтительно Р2O5:Аl2О3: SiO2:ТЭА-ОН:Н2O=1:(0,8-1,4):(0,1-0,4):(0,6-1,15):(80-130).
(SixAlyPz)O2,
где x, y и z обозначают мольные доли соответственно Si, Аl и Р, причем x обозначает мольную долю Si и обладает значением в интервале от 0,001 до 0,1, у обозначает мольную долю Аl и обладает значением в интервале от 0,25 до 0,5, z обозначает мольную долю Р в интервале от 0,4 до 0,8.
приготовление SAPO молекулярного сита по п.6,
смешивание или внедрение упомянутого SAPO молекулярного сита с/в матричный материал с получением пасты или суспензии,
обработка упомянутой пасты или суспензии с получением твердых частиц, кальцинирование упомянутых твердых частиц с получением упомянутого катализатора.
Комментарии