Производные 4,4-дифторбут-3-ена и способ их получения, содержащие их сельскохозяйственные композиции и использование их в качестве инсектицидов, акарицидов или нематоцидов - RU2151147C1
Код документа: RU2151147C1
Чертежи





































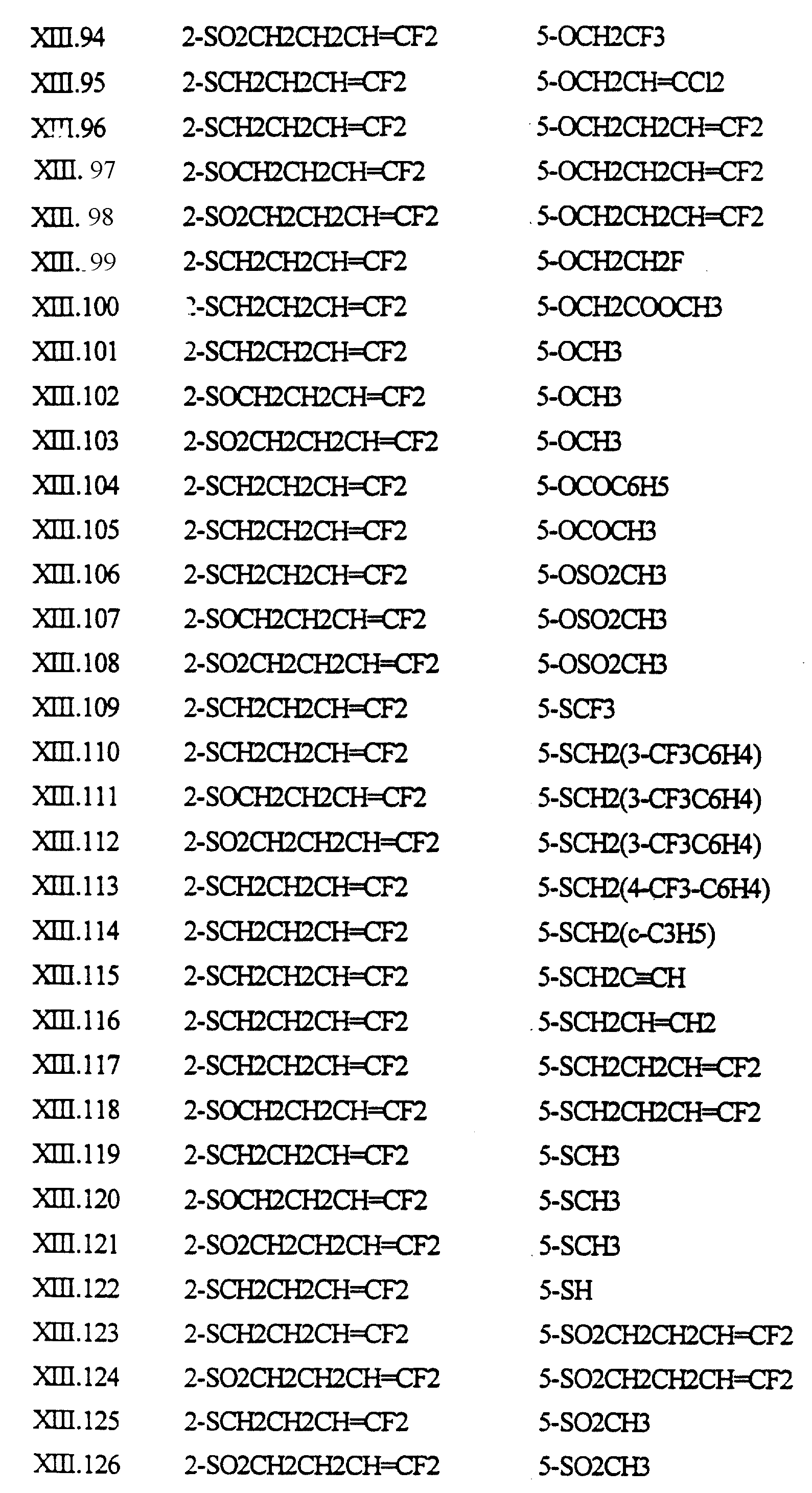



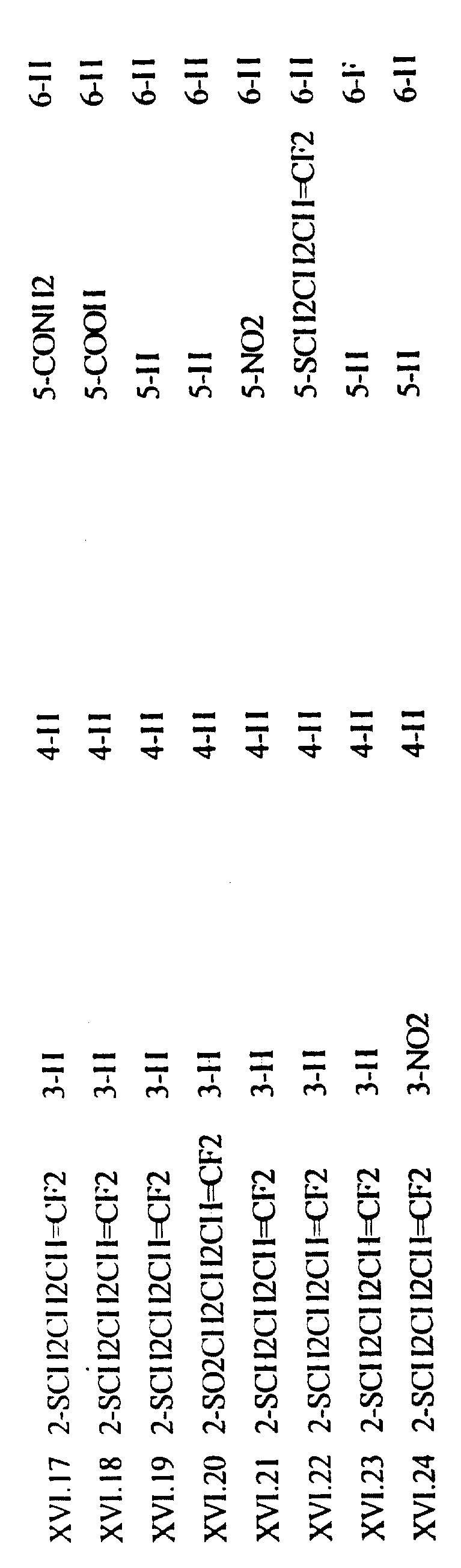



























Описание
Изобретение относится к новым гетероциклическим или карбоциклическим производным, обладающим нематоцидной, инсектицидной и акарицидной активностью, к способу их получения, к содержащим их фармацевтическим композициям и к использующим их способам уничтожения или подавления нематод, вредных насекомых или акарицид.
В соответствии с настоящим изобретением предложено соединение формулы (I) или его соль, где
n = 0, 1 или 2; и R является группой формул II - XXI, где S(O)nCH2CH2CH= CF2 группа является, по крайней мере, одной из R1 (когда он присоединен к атому углерода), R2, R3, R4, R5 или R6;
R1
(когда он присоединен к атому углерода), R2, R3, R4, R5 или R6, каждый, независимо, обозначают водород, необязательно замещенный алкил, необязательно замещенный алкенил, алкинил, циклоалкил,
алкилциклоалкил, алкокси, алкенилокси, алкинилокси, гидроксиалкил, алкоксиалкил, необязательно замещенный арил, необязательно замещенный арилалкил, необязательно замещенный гетероарил, необязательно
замещенный арилокси, необязательно замещенный арилалкокси, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилокси, необязательно замещенный гетероарилалкокси, необязательно
замещенный гетероарилоксиалкил, галогеналкил, галогеналкенил, галогеналкинил, галогеналкокси, галогеналкенилокси, галогеналкинилокси, галоген, гидрокси, циано, нитро, -NR7R8, -NR7COR8, -NR7CSR8,
-NR7SCO2R8, -N(SO2R7)(SO2R8), -COR7, -CONR7R8, -алкилСОNR7R8, -CR7NR8, -COOR7, -OCOR7, -SR7, -SOR7, -SO2R7, -алкилSR7, -алкилSOR7, -алкилSO2R7, -OSO2R7, -SO2NR7R8, -CSNR7R8, -SiR7R8R9, -OCH2CO2R7,
-OCH2CH2CO2R7, -CONR7SO2R8, -алкилCONR7SO2R8, -NHCOR7R8, -NHCSNR7R8 или пара соседних R1, R2, R3, R4, R5 и R6, взятых вместе, образует 5- или 6-членное карбоциклическое или гетероциклическое кольцо;
R1 (когда он присоединен к атому азота) обозначает водород, необязательно замещенный алкил, циклоалкил, алкилциклоалкил, гидроксиалкил, алкоксиалкил, необязательно замещенный арил,
необязательно замещенный арилалкил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарил, необязательно замещенный гетероарилалкил, гидрокси, циано, нитро, -NR7R8, -NR7COR8,
-NR7CSR8, -NR7SCOOR8, -NR7SO2R8, -N(SO2R7)(SO2R8), -COR7, -CONR7R8, -алкилCONR7R8, -CR7NR8, -COOR7, -OCOR7, -SOR7, -SO2R7, -алкилSR7, -алкилSOR7, -алкилSO2R7, -OSO2R7, -SO2NR7R8, -SR7, -SOR7, -SO2R7,
-CSNR7R8, -SiR7R8R7, -OCH2CO2R7, -OCH2CH2CO2R7, -CONR7SO2R8, -алкилCONR7SO2R8, -NHCOR7R8 или -NHCSNR7R8; и
R7, R8 и R9, каждый, независимо, обозначают водород, необязательно замещенный алкил,
необязательно замещенный алкенил, алкинил, необязательно замещенный арил, необязательно замещенный арилалкил, галогеналкил, галогеналкенил, галогеналкинил, галоген или гидрокси.
Следует учесть, что только для удобства отсылок, заместитель в группу R наименован в соответствии с положением в данной группе R. Например, когда R имеет формулу (II), заместители R2, R3, R4 и R5 находятся в положениях 2, 3, 4 и 5 кольца, соответственно. Для того, чтобы избежать дублирования, группа - S(O)nCH2CH2CH=CF2 группа может находиться при любом положении заместителя, обозначаемом R1 (когда он присоединен к атому углерода) - R6.
Когда какой-нибудь из R1-R9 является алкильной группой или содержит алкильный радикал, он может быть прямой или разветвленной цепью и предпочтительно является C1-6-алкилом, даже, более предпочтительно, C1-4-алкилом, например, метилом, этилом, пропилом, изо-пропилом, н-бутилом, изо-бутилом, втор-бутилом или трет-бутилом. Когда алкильная группа действует как "связующая" группа, то есть, R-алкил, например, в R-алкилSR7, особенно предпочтительны C1-4-алкил или C1-2-алкил.
Когда какой-нибудь из R1-R8 является замещенной алкильной группой или содержит замещенный алкильный радикал, он может содержать один или несколько заместителей, выбранных из галогена, циано, -COOR7 или его соли, гидрокси, алкокси, алкоксимино, алкоксикарбонила, карбамоила, моно- или ди-алкилкарбамоила, амино, моно- или ди-алкиламино, ациламидо (предпочтительно, C1-6-ациламидо), алкансульфонила и арилсульфонила, который, в свою очередь, может быть замещен галогеном, алкокси или нитро.
Когда какой-нибудь из R1-R8 является алкенильной или алкинильной группой или содержит алкенильный или алкинильный радикал, он может быть прямой или разветвленной цепью и предпочтительно является C2-6-алкенилом или C2-6-алкинилом, даже, более предпочтительно, C2-4-алкенилом или C2-4-алкинилом, например, винилом, аллилом, бут-3-енилом, 3-метилбут-3-енилом, этинилом или пропаргилом.
Когда какой-нибудь из R1-R8 является замещенной алкенильной группой или содержит замещенный алкенильный радикал, он может содержать один или несколько заместителей, выбранных из галогена, -COOR7 или его соли, гидрокси, нитро или циано.
Когда какой-нибудь из R1-R6 является циклоалкильной или алкилциклоалкильной группой или содержит такой радикал, он предпочтительно является Cl-6-алкокси, например, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, втор- бутокси и трет-бутокси; C2-6-алкинилoкcи, например, винилокси, аллилокси, бут-3-енилокси и 3-метилбут-3-енилокси, C2-6- моноалкоксиалкил, например, метоксиметил, метоксиэтил и этоксиметил; или C3-6 диалкоксиалкил, например, диметоксиметил и диэтоксиметил.
Когда какой-нибудь из R1-R9 является арилом или содержит арильный радикал, он, предпочтительно, является C6-10-арилом, более предпочтительно, он является фенилом. Когда какой-нибудь из R1-R9 является арилалкилом, он, предпочтительно, является C6-10-арил-метилом, даже более предпочтительно, он является бензилом или фенетилом.
Когда какой-нибудь из R1-R6 является гетероарилом или содержит гетероарильный радикал, он, предпочтительно, является 5- или 6-членным циклом, содержащим, по крайней мере, один О, N или S атом в качестве гетероатома, например, пиридином, пирролом, пиразином, фураном или тиофеном. Когда какой-нибудь из R1-R6 является гетероарилом, более предпочтительно, он является гетероарил-C1-2-алкилом.
Когда какой-нибудь из R1-R9 является замещенной арильной, арилалкильной, гетероарильной или гетероарилалкильной группой, он может содержать один или несколько заместителей, выбранных из алкила, алкокси, галогеналкила, галогена, гидрокси, -COOR7 (или его соли), аминосульфонила, циано или нитро. Примерами этих групп являются 4-метилфенил, 4-хлорфенил, 4-фторфенил, 4- нитрофенил, 3-трифторметилфенил, 4-трифторметилфенил, 4- аминосульфонилфенил, 4-хлорбензил, 4-фторбензил, 3-трифторметилбензил, 4-трифторметилбензил, 4-нитробенэил и 4-метилбензил.
Когда какой-нибудь из R1-R6 является арилокси или арилалкокси группой, он, предпочтительно, является фенокси, бензилокси или фенетокси.
Когда какой-нибудь из R1-R6 является замещенной алкокси, арилалкокси, гетероарилокси или гетероарилокси группой, он может содержать один или несколько заместителей, выбранных из алкила, алкокси, галогеналкила, галогена, гидрокси, циано или нитро. Примерами этих групп являются 4-метилфенокси, 4-хлорфенокси, 4-фторфенокси, 4- нитрофенокси, 3-трифторметилфенокси, 4-трифтор-метилфенокси, 4- хлорбензилокси, 4-фторбензилокси, 3-трифторметил-бензилокси, 4- трифторметилбензилокси, 4-нитробензилокси и 4-метилбензилокси.
Когда какой-нибудь из R1-R6 является арилокси или арилалкокси группой, он, предпочтительно, является фенокси, бензилокси или фенетокси.
Когда какой-нибудь из R1-R6 является галогеном или содержит радикал галоген, он, предпочтительно, является фтором, хлором, бромом или иодом. Даже более предпочтительно, он является фтором, хлором или бромом.
Когда какой-нибудь из R1-R6 является галогеналкильной, галогеналкенильной или галогеналкинильной группой, он может содержать один или несколько атомов галогена, предпочтительно, хлор, фтор или бром. Примерами этих групп являются фторметил, дифторметил, трифторметил, хлорметил, дихлорметил, трихлорметил, 2-фторэтил, 2,2,2-трифторэтил, пентафторэтил, 2, 2-дифторэтил, 3,3-дихлорпроп-2-енил, 2-хлор-2-енил, 3,4,4-трифторбут-3-енил, 4-фторбут-3-енил, 4,4-дифторбут-3-енил и 3-метил-4,4- дифторбут-3-енил.
Когда какой-нибудь из R1-R6 является галогеналкенилоксигруппой или галогеналкинилоксигруппой, он может содержать один или несколько атомов галогена, предпочтительно, хлор, фтор или бром. Примерами предпочтительных C1-6-алкокси, C2-6-алкенилокси и C2-6-алкинилокси групп являются трихлорметокси, фторметокси, дифторметокси, трифторметокси, 2- фторэтокси, 2,2,2- трифторэтокси, пентафторэтокси, 1,1,2,2- тетрафторэтокси, 2-дифторэтенилокси, 3,4,4-трифторбут-3- енилокси, 4-фторбут-3-енилокси, 4,4-дифторбут-3-енилокси, 3- метил-4,4-дифторбут-3-енилокси, 2-хлорпроп-2-енилокси и 3,3- дихлорпроп-2-енилокси.
Когда какой-нибудь из R1-R6 является группой -NR7R8, он, предпочтительно, является -NH2; моноалкиламиногруппой, например, метиламино и этиламино; или диалкиламиногруппой, например, диметиламино и диэтиламино.
Когда какой-нибудь из R1- R6 является группой -NR7COR8, он, предпочтительно, является - NHCHO; С2-6-ациламиногруппой, например, -NHCOCH3, -NHCOC2H5; или бензамидо, который может быть замещен одним или более заместителями, выбранными из галогена, например, хлора, фтора и брома; алкилом, например, метилом и этилом; алкокси, например, метокси и этокси; галогеналкилом, например, хлорметилом, фторметилом, трифторметилом и 2,2,2-трифторэтилом; галогеналкокси, например, трифторметокси и 2,2,2-трифторэтокси; гидрокси; циано и нитро.
Когда какой-нибудь из R1-R6 является группой -NR7CSR8, R7 и R8, предпочтительно, являются метилом и этилом.
Когда какой-нибудь из R1-R6 является группой -NR7SO2R8, он, предпочтительно, является алкансульфонамидогруппой, например, -NR7SO2CH3 и -NR7SO2C2H5.
Когда какой-нибудь из R1-R6 является группой -N(SO2R7)(SO2R8), он, предпочтительно, является ди(алкансульфонил) аминогруппой, например, -N(SO2CH3)2 и -N(SO2C2H5)2.
Когда какой-нибудь из R1-R6 является группой -COR7, он, предпочтительно, является C1-6-ацильной группой; или необязательно замещенной бензоильной группой. Бензоил может быть замещен одним или более заместителями, выбранными из галогена, например, хлора, фтора и брома; алкила, например, метила и этила; алкокси, например, метокси и этокси; галогеналкила, например, хлорметила, фторметила, трифторметила и 2,2,2-трифторэтила; галогеналкокси, например, трифторметокси и 2,2,2- трифторэтокси; гидрокси; циано и нитро. Примерами предпочтительных -COR7 групп являются ацетил, пропионил, н-бутаноил, 4-хлорбензоил, 4-фторбензоил, 4-бромбензоил, 4- метилбензоил и 4-трифторметилбензоил.
Когда какой-нибудь из R1-R6 является группой -CONR7R8, он, предпочтительно, является - CONH2; -N-алкилкарбоксамидогруппой, например, -CONHCH3, - CONHC2H5 и -CONHCH2CH2CH3; или группой N,N-диaлкилкapбoкcaмидo, например, -CON(CH3)2, -CON(CH3)(C2H5) и -CON(C2H5)2.
Когда какой-нибудь из R1-R6 является группой -алкилCONR8, он, предпочтительно, обозначает -C1-4 aлкилCONR7R8.
Когда какой-нибудь из R1-R6 является группой CR7NR8, он, предпочтительно, обозначает -CH=NOH.
Когда какой-нибудь из R1-R6 является группой -COOR7, он, предпочтительно, обозначает -COOH; алкоксикарбонильную группу, например, метоксикарбонил и этоксикарбонил; или галогеналкенилоксикарбонильную группу, например, 3, 4,4-трифторбут-3-енилоксикарбонил, 4-фторбут-3-енилоксикарбонил, 4, 4-дифторбут-3-енилкарбонил и 3-метил-4,4- дифторбут-3-енилоксикарбонил.
Когда какой-нибудь из R1-R6 является группой -OCOR7, он, предпочтительно, обозначает C2-6 ацилоксигруппу, например, -OCOCH3 и -OCOC2H5; или необязательно замещенную бензоилоксигруппу. Бензоилоксигруппа может содержать один или несколько заместителей, выбранных из галогена, например, хлора, фтора и брома; алкила, например, метила и этила; алкокси, например, метокси и этокси; галогеналкила, например, хлорметила, фторметила, трифторметила и 2,2,2-трифторэтила; галогеналкокси, например, трифторметокси и 2, 2,2-трифторэтокси; гидрокси; циано и нитро.
Когда какой-нибудь из R1-R6 является группой -SR7, R7, предпочтительно, обозначает водород, необязательно замещенный алкил, необязательно замещенный алкенил, алкинил, галогеналкил, галогеналкенил, галогеналкинил, необязательно замещенный арил или необязательно замещенный арилалкил. Примерами предпочтительных C1-6 алкилтио (C1-4 алкил является особенно предпочтительным), C2-6 алкенилтио или C2-6 алкинилтио групп являются метилтио, этилтио, н-пропилтио, изо-пропилтио, н- бутилтио, изо-бутилтио, втор-бутилтио, аллилтио, бут-3-енилтио, 3-метилбут-3-енилтио и пропаргилтио. Примерами предпочтительных C1-6 галогеналкилтио (C1-4 алкил является особенно предпочтительным), C2-6 галогеналкенилтио или C2-6 галогеналкинилтио групп являются фторметилтио, дифторметилтио, трифторметилтио, трихлорметилтио, 2-фторэтилтио, 2,2,2- трифторэтилтио, 3-фтор-н-пропилтио, пентафторэтилтио, 2- хлорпроп-2-енилтио, 3,3-дихлорпроп-2-енилтио, 3,4, 4-трифторбут- 3-енилтио, 4-фторбут-3-енилтио, 4,4-дифторбут-3-енилтио и 3- метил-4,4-дифторбут-3-енилтио. Примером предпочтительных C6-10 арилтио и C6-10 арил-C1-2 алкилтио групп является 3- трифторметилбензилтио.
Когда какой-нибудь из R1-R6 является группой -SOR7, он, предпочтительно, обозначает алкансульфинил, алкенсульфинил или алкинилсульфинилгруппу, например, метансульфинил или этансульфинил; или галогеналкансульфинил, галогеналкенилсульфинил или галогеналкинилсульфинил группу, например, трифторметан сульфинил. При другом предпочтительном осуществлении -SOR7, предпочтительно, является -SOF, -SOBr или -SOCl.
Когда какой-нибудь из R1-R6 является группой -SO2R7, он, предпочтительно, обозначает алкансульфонильную, алкенсульфонильную, алкинилсульфонильную, галогеналкансульфонильную, галогеналкенилсульфонильную, галогеналкинилсульфонильную группу; или необязательно замещенную бензолсульфонильную группу. Бензолсульфонильная группа может содержать один или несколько заместителей, выбранных из галогена, например, хлора, фтора и брома; алкила, например, метила и этила; алкокси, например, метокси и этокси; галогеналкила, например, хлорметила, фторметила, трифторметила и 2,2,2-трифторэтила; галогеналкокси, такого как трифторметокси и 2,2,2-трифторэтокси; гидрокси; циано и нитро. Примерами таких групп являются метансульфонил, этансульфонил, трифторметансульфонил и 4-метилбензолсульфонил. При другом предпочтительном осуществлении -SO2R7 предпочтительно обозначает -SO2F, -SO2Br или -SO2Cl.
Таким образом, можно ожидать, что R группа в формулах (II) - (XXI) может содержать более чем одну группу S(O)nCH2CH2CH=CF2. Предпочтительно, R группа содержит один или два таких заместителя.
Когда какой-нибудь из R1-R6 является группой -OSO2R7, он, предпочтительно, обозначает алкансульфонилокси группу или необязательно замещенную бензолсульфонилокси группу. Бензолсульфонильная группа может быть замещена одним или несколькими заместителями, выбранными из галогена, например, хлора, фтора и брома; алкила, например, метила и этила; алкокси, например, метокси и этокси; галогеналкила, например, хлорметила, фторметила, трифторметила и 2,2,2-трифторэтила; галогеналкокси, например, трифторметокси и 2,2,2-трифторэтокси; гидрокси; циано и нитро.
Когда какой-нибудь из R1-R6 является группой -SO2NR7NR8, он, предпочтительно, обозначает -SO2NH2; алкиламиносульфонилокси группу, например, -SO2NHCH3 и - SO2NHC2H5; или диалкиламиносульфонильную группу, например, -SO2N(CH3)2 и -SO2N(C2H5)2.
Когда какой-нибудь из R1-R6 является группой -CSNR7NR8, он, предпочтительно, обозначает -CS2NH2, -CSNHCH3 или -CSN(CH3)2.
Когда какой-нибудь из R1-R6 является группой -SiR7R8R9, он, предпочтительно, является триалкилсилильной группой, например, триметилсилилом и триэтилсилилом.
Когда какой-нибудь из R1-R6 является группой -OCH2CO2R7, он, предпочтительно, является алкоксикарбонилметокси группой, например, метоксикарбонилметокси и этоксикарбонилметокси.
Когда какой-нибудь из R1-R6 является группой -OCH2CH2CO2R7, он, предпочтительно, является алкоксикарбонилэтокси группой, например, метоксикарбонилэтокси и этоксикарбонилэтокси.
Когда какой-нибудь из R1-R6 является группой -CONR7SO2R8, он, предпочтительно, является N-алкансульфонилкарбоксамидо группой или N-алкил-N-алкансульфонилкарбоксамидо группой, например, N-(метансульфонил) карбоксамидо и N-метил-N-(метансульфонил) карбоксамидо.
Когда какой-нибудь из R1-R6 является группой -алкилCONR7SO2R8, R7 и R8, предпочтительно, являются алкильными группами, например, этилом и метилом.
Когда какой-нибудь из R1-R6 является -NHCONR7R8, R7 и R8, предпочтительно, являются алкильными группами, например, этилом и метилом.
Когда какой-нибудь из R1-R6 является -NHCSNR7R8, R7 и R8, предпочтительно, являются алкильными группами, например, этилом и метилом.
Когда пара соседних R1, R2, R3, R4, R5 и R6, взятые вместе, образуют конденсированное 5- или 6-членное карбоциклическое или гетероциклическое кольцо, предпочтительно содержащее два атома кислорода, пара заместителей, взятая вместе, обозначает, предпочтительно, -(CH2)3-, -(CH2)4-, -CH=CH- CH=CH-, -O-CH2-O-, необязательно замещенные одним или двумя атомами галогена или метильными группами, например, -O-CHF-O- или -O-CF2-O-, -O-CH(CH3)-O- или -O-C(CH2)2-O-.
В соответствии с предпочтительным
осуществлением настоящего изобретения радикал из R1 (когда он присоединен к атому углерода) - R6, каждый, независимо, обозначает водород; нитро; галоген; циано; -CH=NOH; C1-4 алкил; C1-4 галогеналкил;
C1-4 алкенил; C1-4 галогеналкинил; циклопропил; гидрокси; C1-4 алкокси; C2-4 алкоксиалкил; -COOH; C2-4 алкоксикарбонил; C2-4 галогеналкенилоксикарбонил; -CONH2; моно или ди-C1-2 алкиламинокарбонил;
C2-4 алканкарбонил; -CONHSO2 C1-4 алкил, предпочтительно, -CONHSO2CH3; фенил, необязательно моно- или ди- замещенный группами, независимо выбранными из галогена, нитро, C1-4 алкила, C1-4 алкокси или
аминосульфонила; бензил, необязательно моно- или ди- замещенный группами, независимо выбранными из галогена, нитро, C1-4 алкила или C1-4 алкокси; фенокси, необязательно моно- или ди- замещенный
группами, независимо выбранными из галогена, нитро, C1-4 алкила или C1-4 алкокси; амино, необязательно моно- или ди- замещенный C1-4 алкильными группами; -SH; C1-4 алкилтио; бензилтио, необязательно
моно- или ди- замещенный группами, независимо выбранными из галогена или C1-4 галогеналкила; C1-4 алкенилтио; C2-4 галогеналкенилтио; вторая S(О)nCH2CH2CH=CF2 группа; C1-4 алкансульфонил; C1-4
галогеналкансульфонил; фторсульфонил; моно- или ди- C1-4 алкилсульфамоил; 5- или 6-членная гетероарильная группа, например, фурил, пиридинил или тиенил, необязательно замещенные галогеном; либо любая
соседняя пара образует конденсированное 5- или 6-членное карбоциклическое или гетероциклическое кольцо; и
R1 (когда он присоединен к атому азота) обозначает водород; нитро; циано; -CH= NOH;
C1-4 алкил; C1-4 галогеналкил; циклопропил; гидрокси; -COOH; C2-4 алкоксикарбонил; C2-4 галогеналкенилоксикарбонил; -CONH2; моно или ди-C1-2 алкиламинокарбонил; C2-4 алканкарбонил; -CONHSO2 C1-4 алкил,
предпочтительно, -CONHSO2CH3; фенил, необязательно моно- или ди- замещенный группами, независимо выбранными из галогена, нитро, C1-4 алкила, C1-4 алкокси или аминосульфонила; бензил, необязательно
моно- или ди- замещенный группами, независимо выбранными из галогена, нитро, C1-4 алкила или C1-4 алкокси; фенокси, необязательно моно- или ди- замещенный группами, независимо выбранными из галогена,
циано, C1-4 алкила или C1-4 алкокси; амино, необязательно моно- или ди- замещенный C1-4 алкильными группами; -SH; C1-4 алкилтио; бензилтио, необязательно моно- или ди- замещенный группами, независимо
выбранными из галогена или C1-4 галогеналкила; C1-4 алкенилтио; C2-4 галогеналкенилтио; вторая S(O)nCH2CH2CH= CF2 группа; C1-4 алкансульфонил; C1-4 галогеналкансульфонил; фторсульфонил; моно- или
ди- C1-4 алкилсульфамоил; 5- или 6-членная гетероарильная группа, например, фурил, пиридинил или тиенил, необязательно замещенные галогеном.
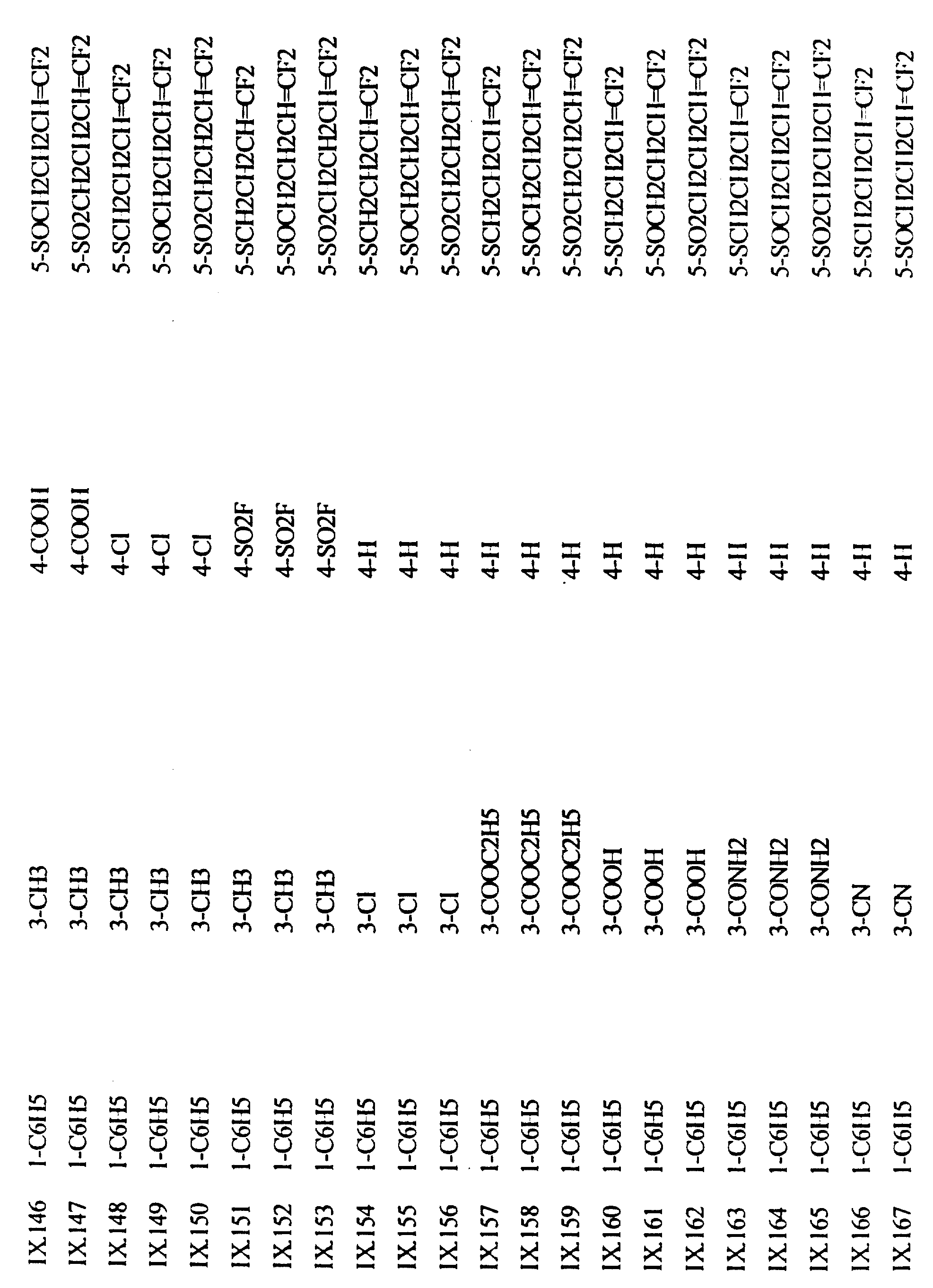
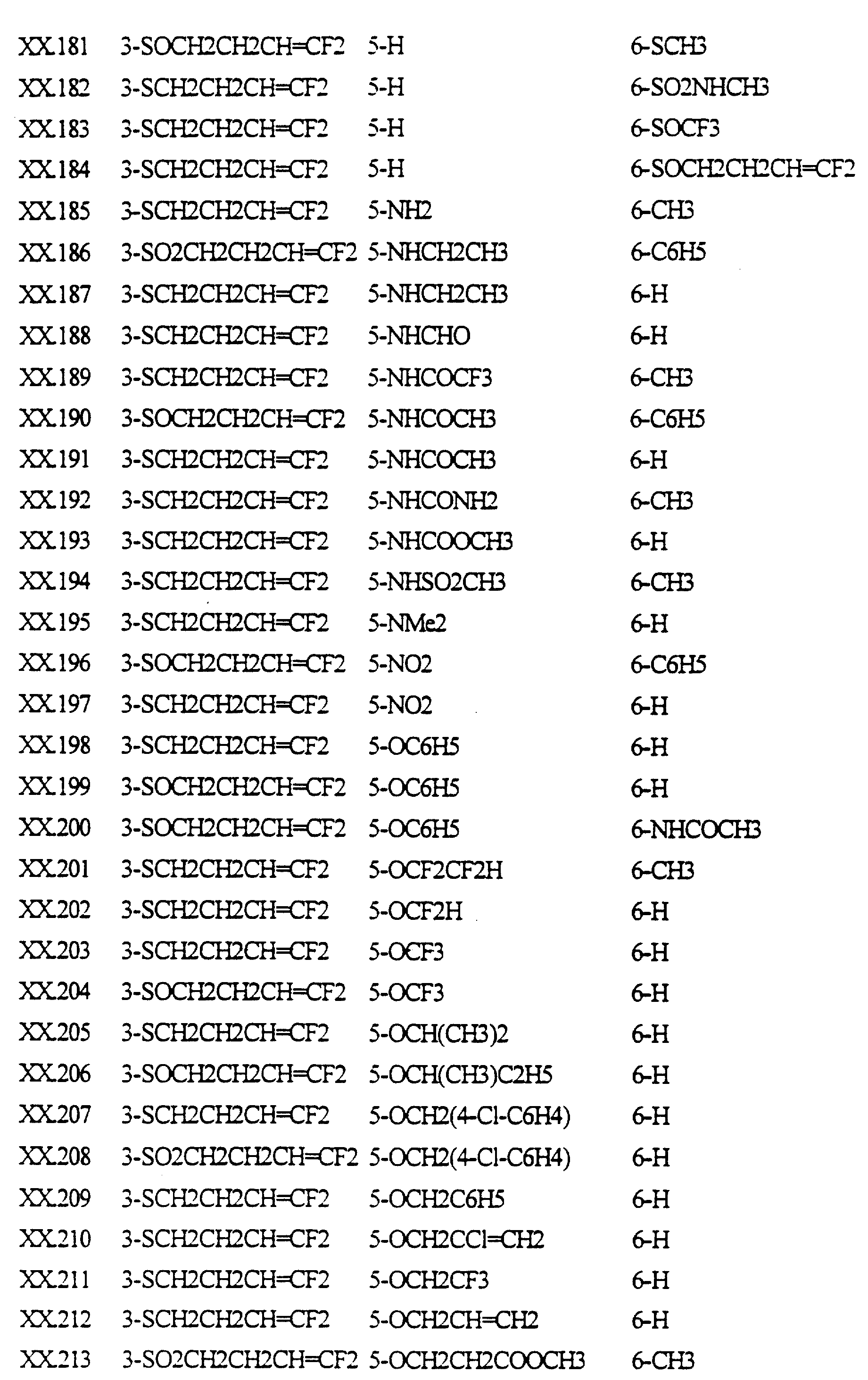
В следующих таблицах даны примеры соединений по изобретению. Примеры соединений формулы (II) в соответствии с изобретением представлены в таблице I-II.
Примеры соединений формулы (III) в соответствии с изобретением представлены в таблице III.
Примеры соединений формулы (IV) в соответствии с изобретением представлены в таблице IV.
Примеры соединений формулы (V) в соответствии c изобретением представлены в таблице V.
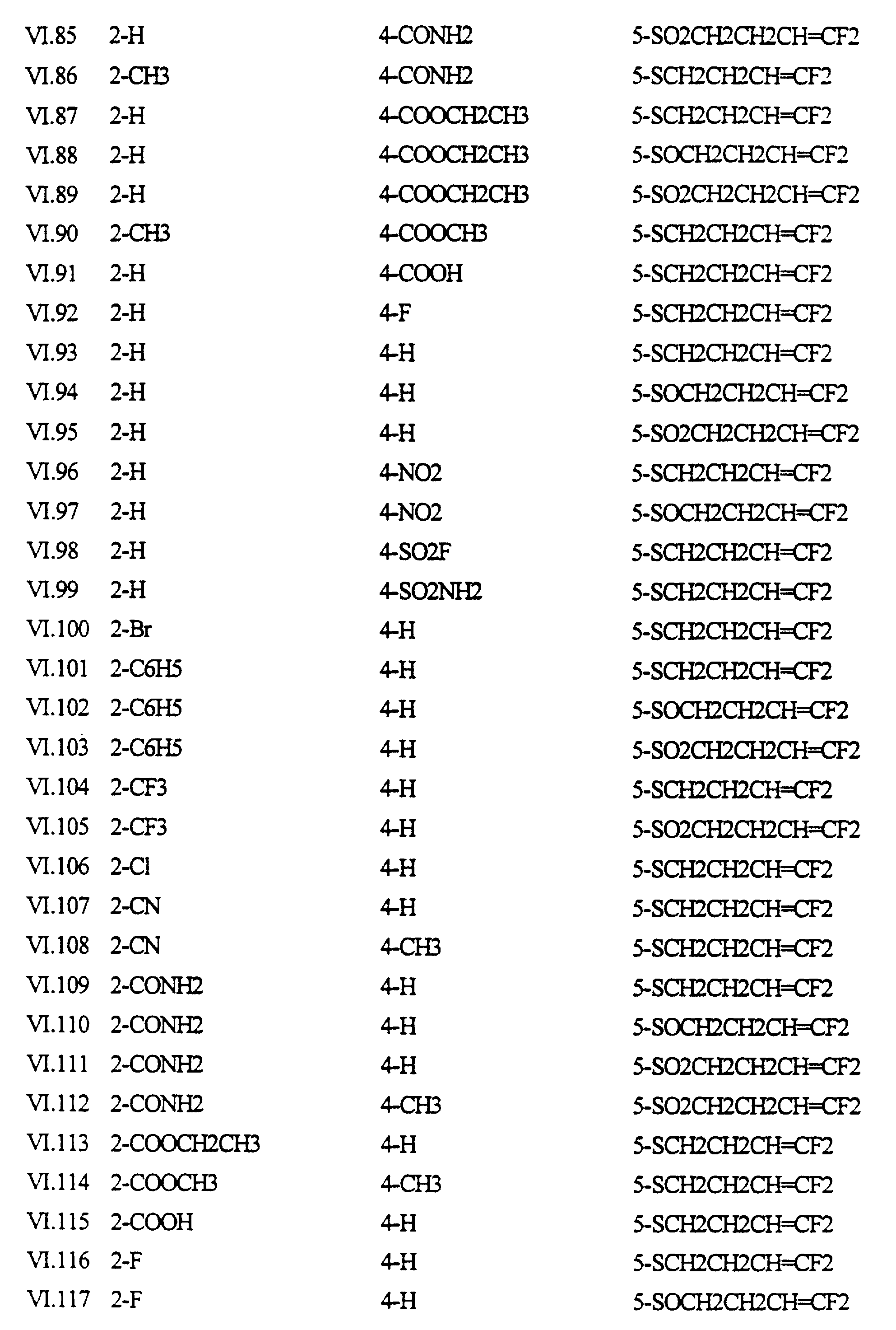
Примеры соединений формулы (VI) в соответствии с изобретением представлены в таблице VI.
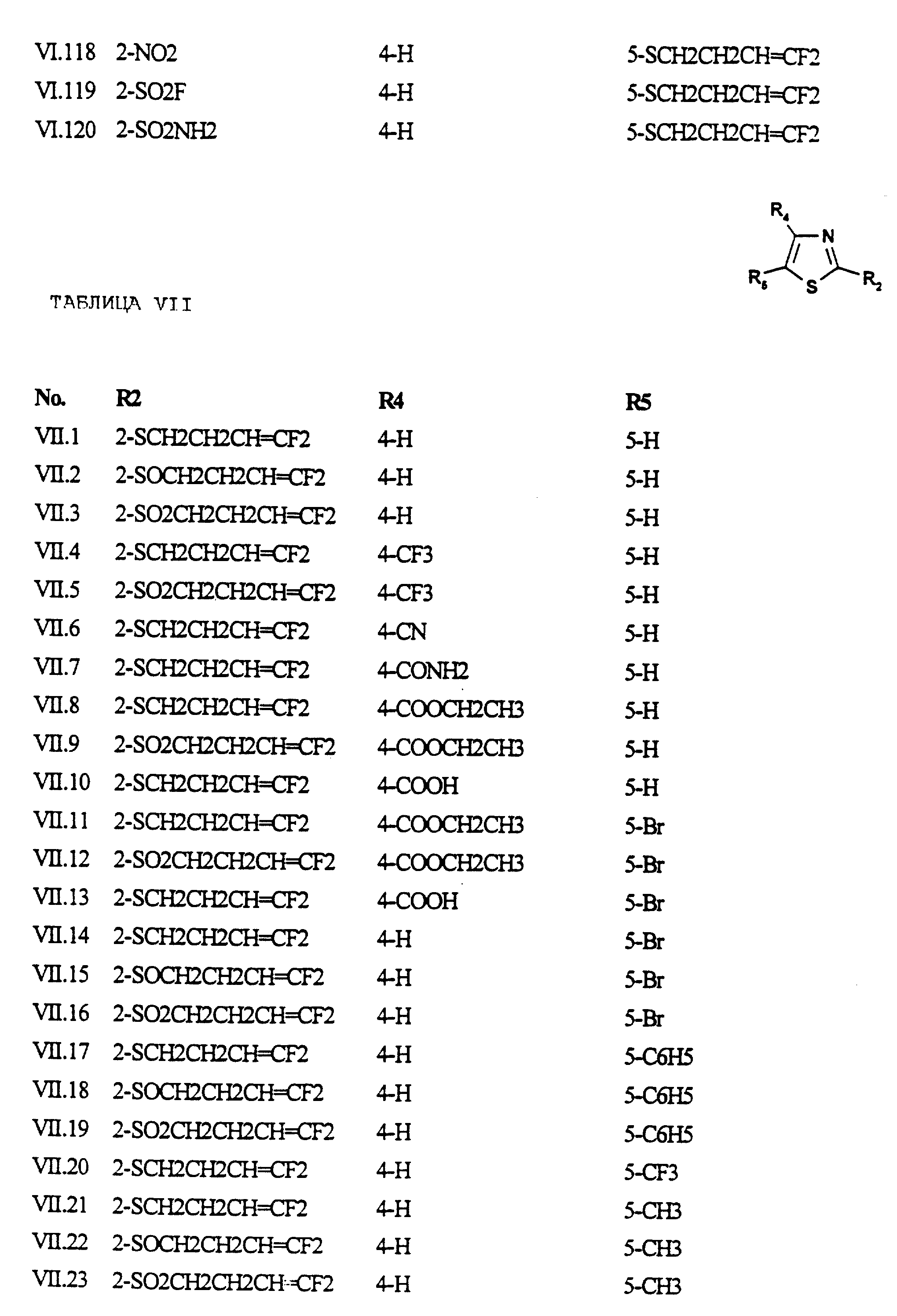
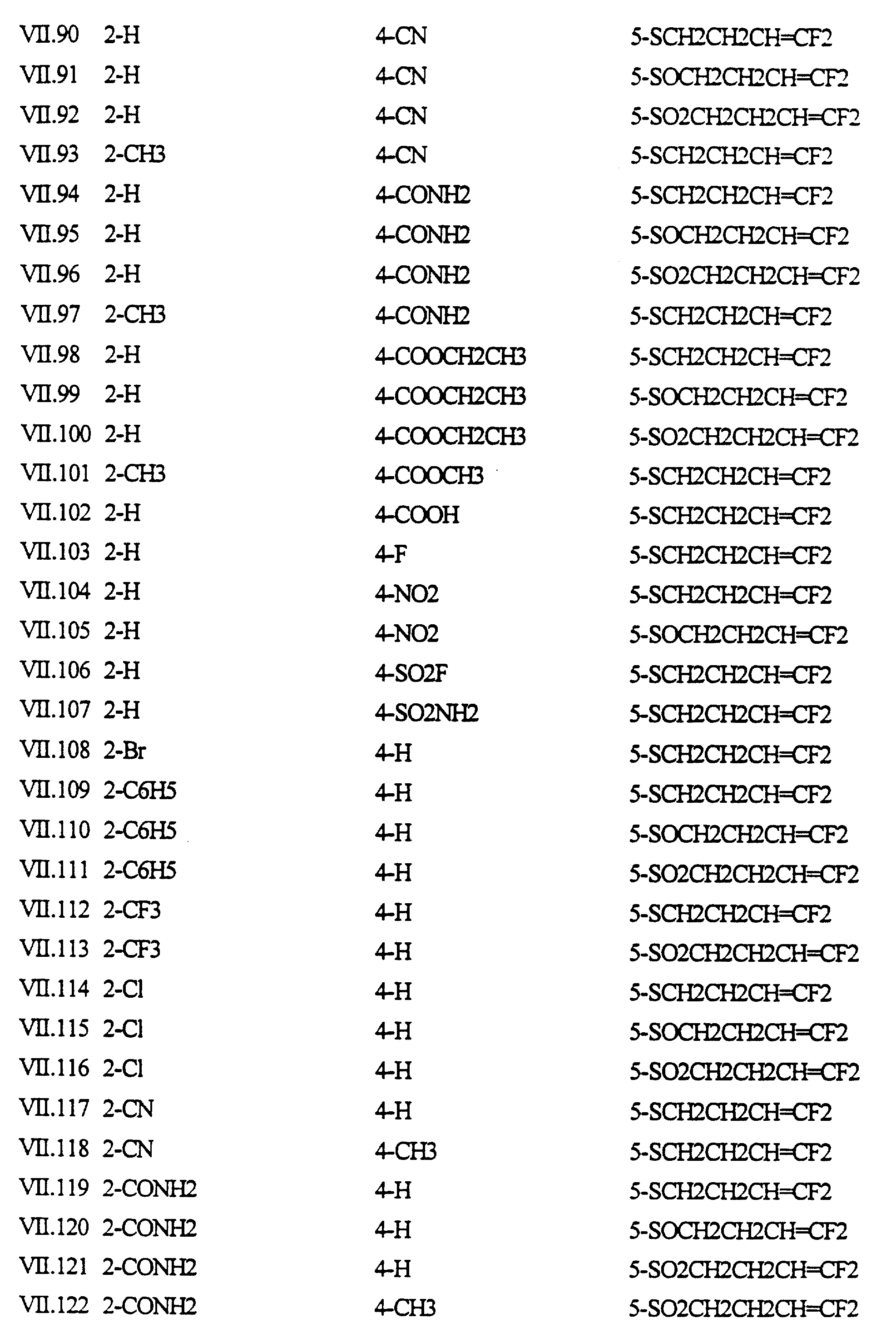
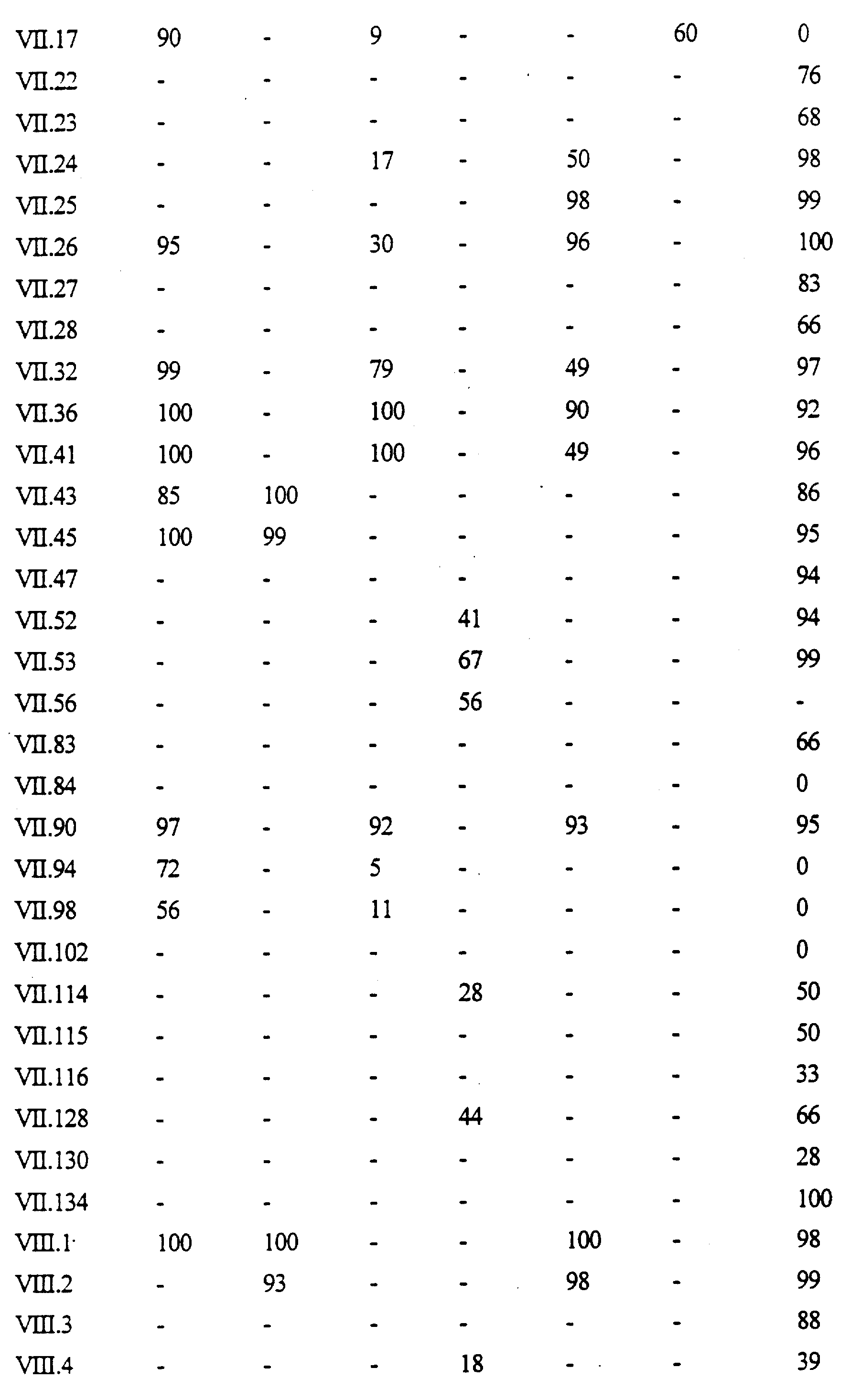
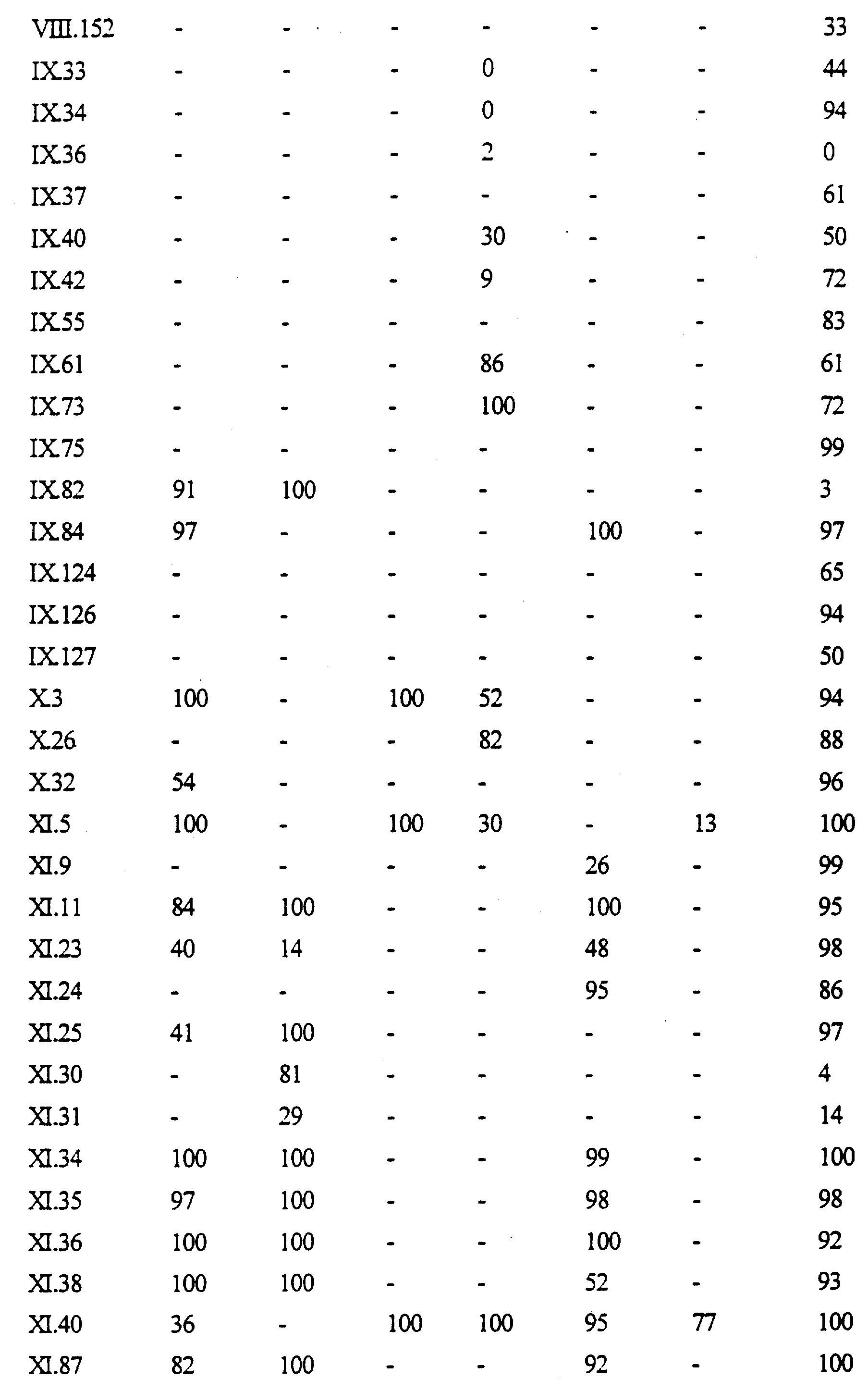
Примеры соединений формулы (VII) в соответствии с изобретением представлены в таблице VII.
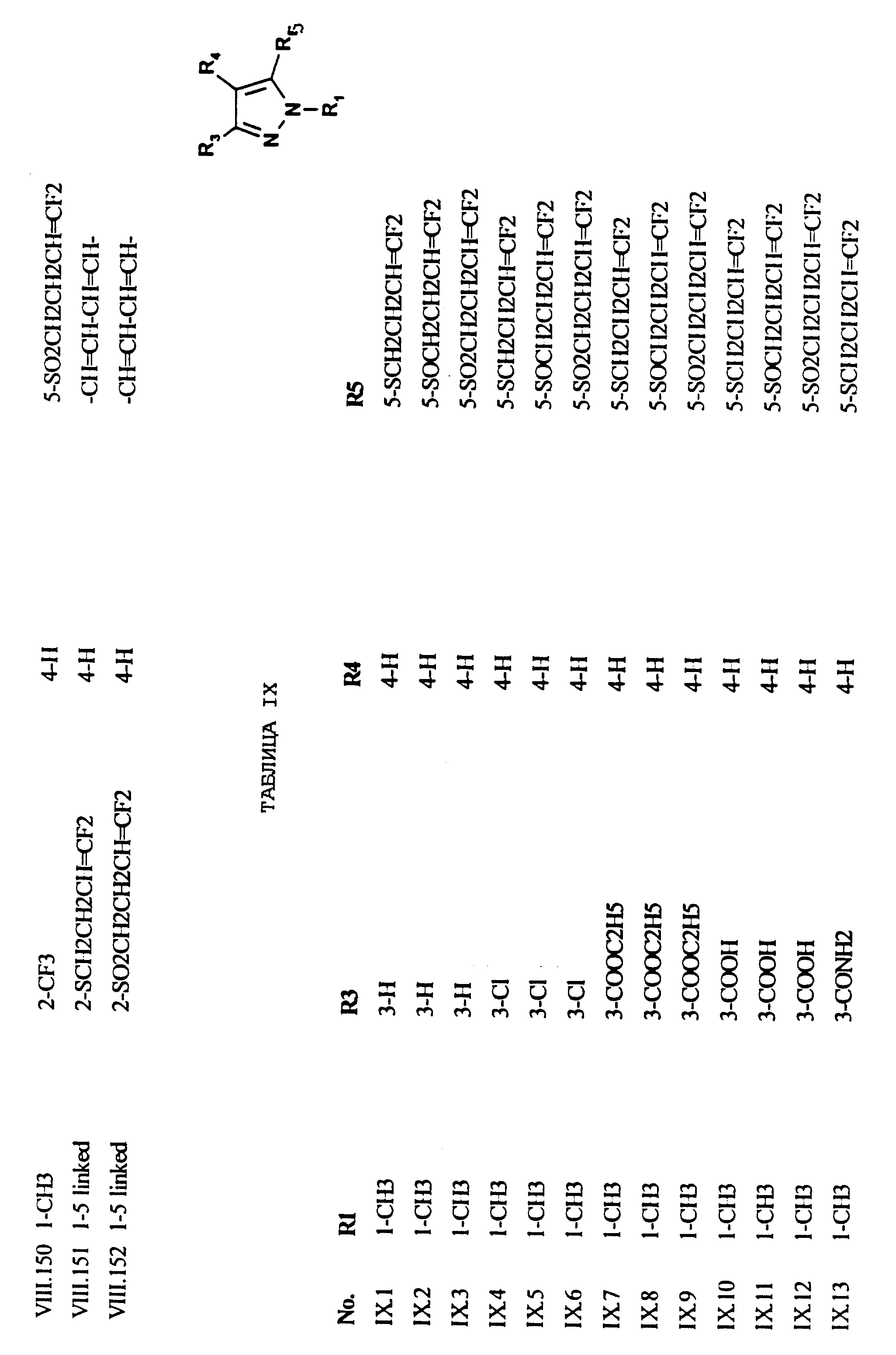
Примеры соединений формулы (VIII) в соответствии с изобретением представлены в таблице VIII.
Примеры соединений формулы (IX) в соответствии с изобретением представлены в таблице IX.
Примеры соединений формулы (X) в соответствии с изобретением представлены в таблице X.
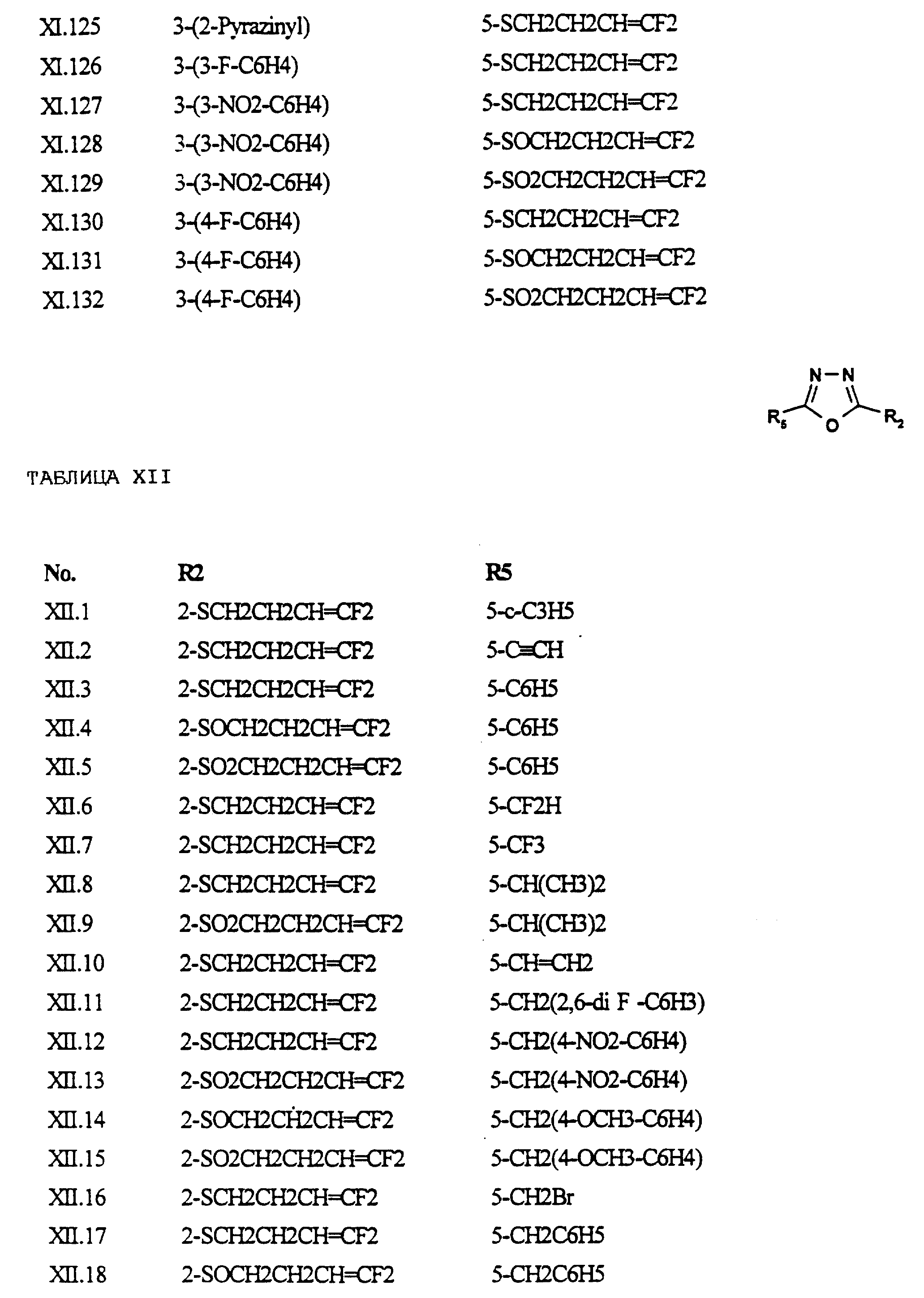
Примеры соединений формулы (XI) в соответствии с изобретением представлены в таблице XI.
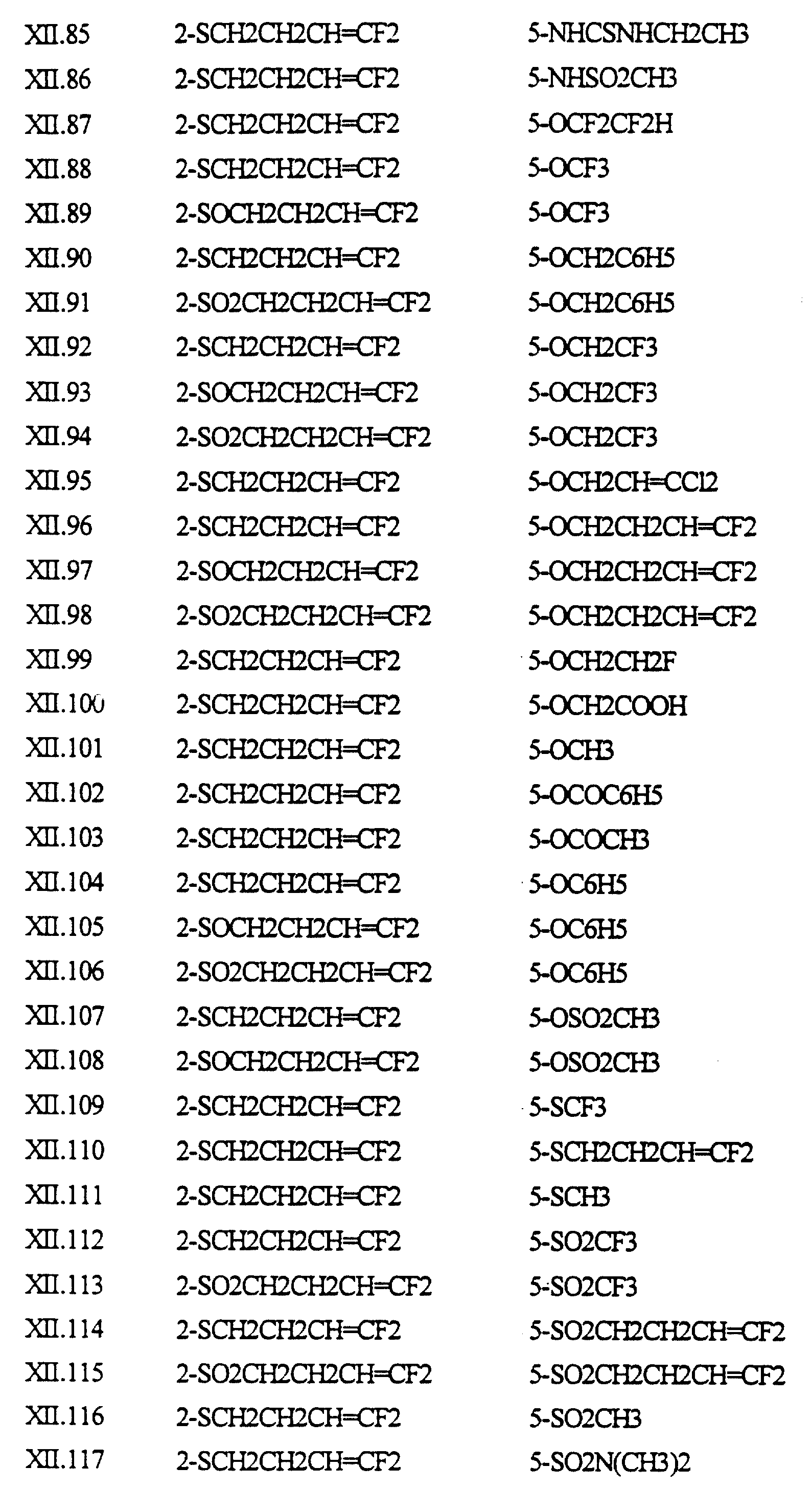
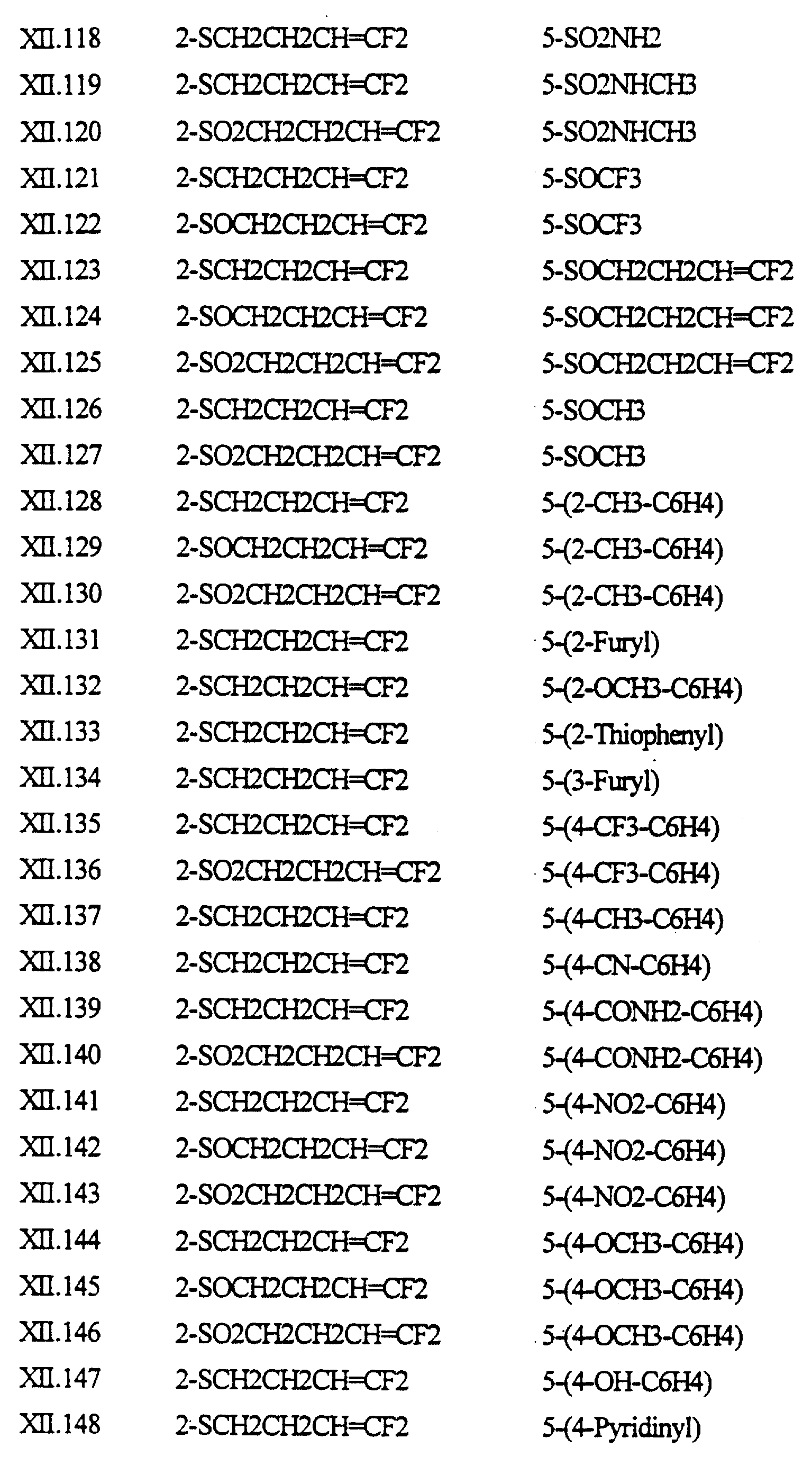
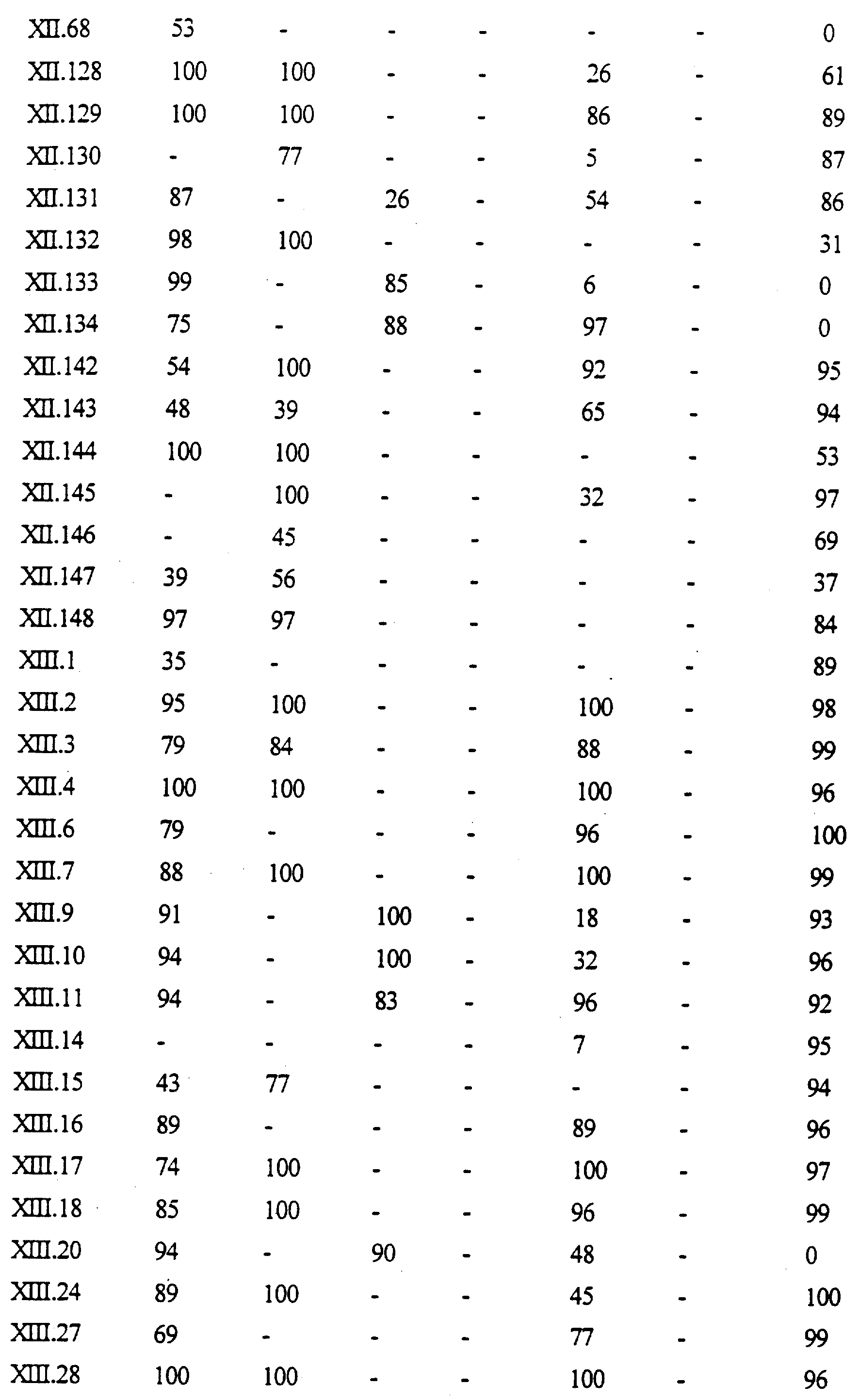
Примеры соединений формулы (XII) в соответствии с изобретением представлены в таблице XII.
Примеры соединений формулы (XIII) в соответствии с изобретением представлены в таблице XIII.
Примеры соединений формулы (XIV) в соответствии с изобретением представлены в таблице XIV.
Примеры соединений формулы (XV) в соответствии с изобретением представлены в таблице XV.
Примеры соединений формулы (XVI) в соответствии с изобретением представлены в таблице XVI.
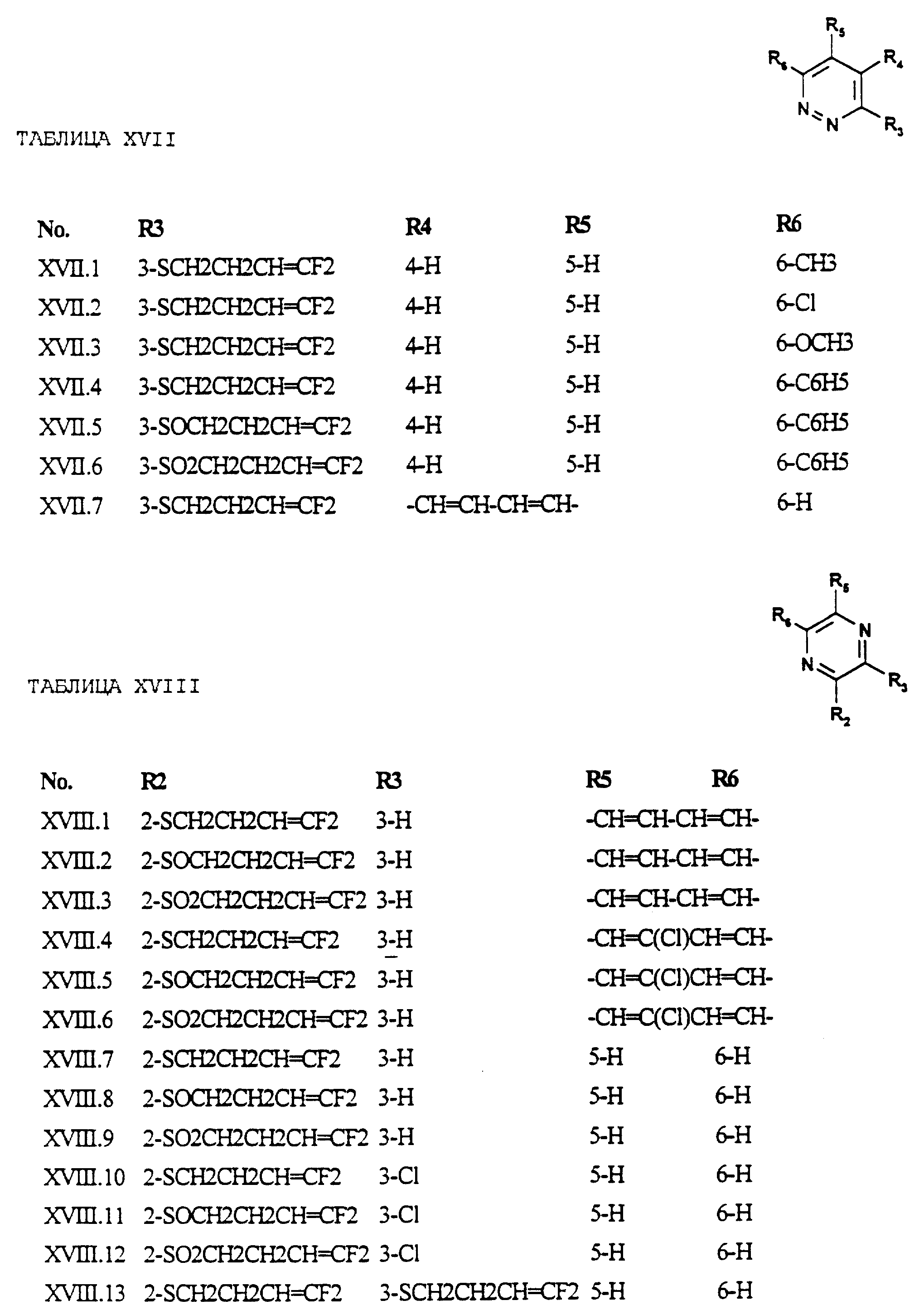
Примеры соединений формулы (XVII) в соответствии с изобретением представлены в таблице XVII.
Примеры соединений формулы (XVIII) в соответствии с изобретением представлены в таблице XVIII.
Примеры соединений формулы (XIX) в соответствии с изобретением представлены в таблице XIX.
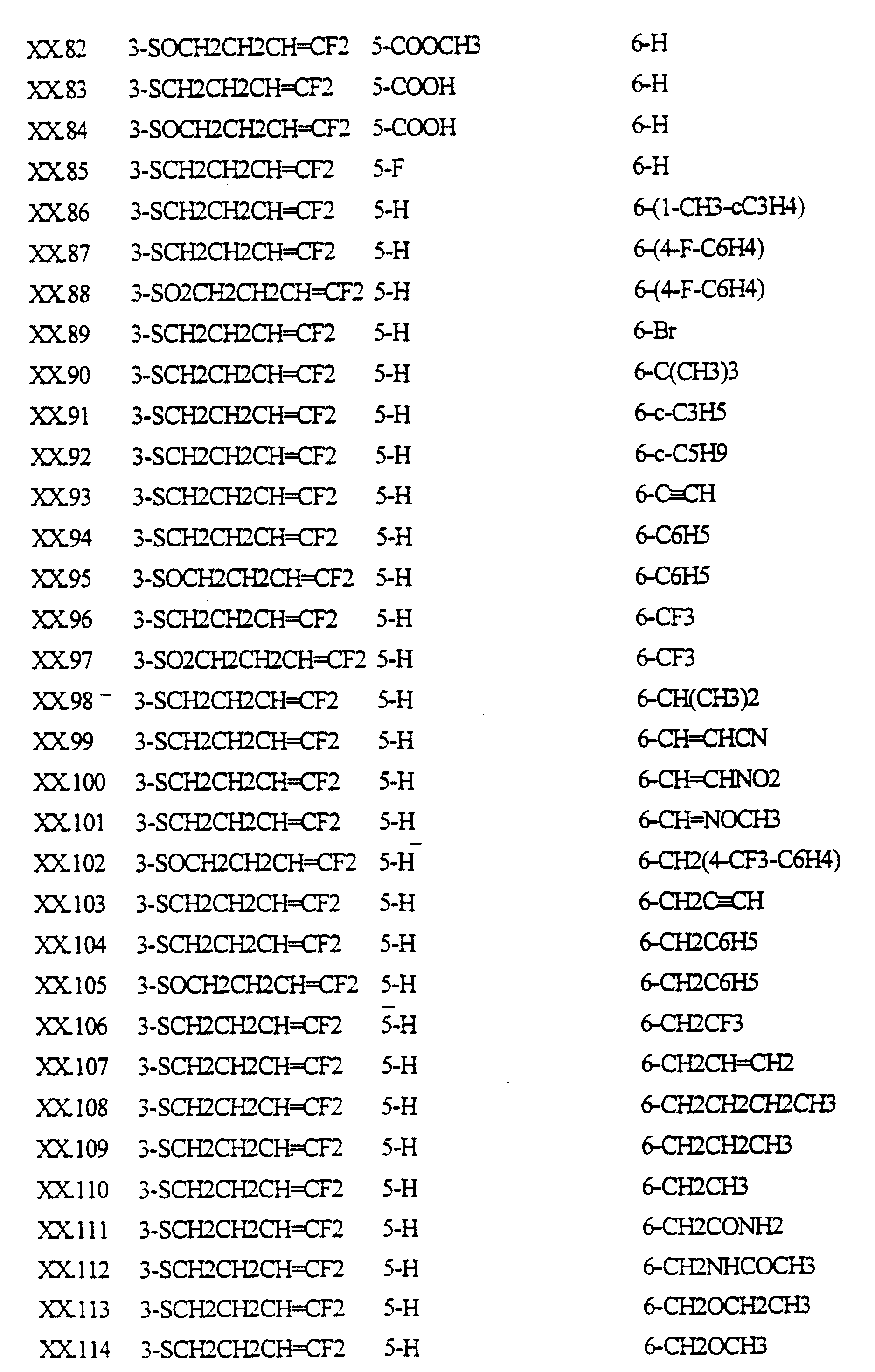
Примеры соединений формулы (XX) в соответствии с изобретением представлены в таблице XX.
Примеры соединений формулы (XXI) в соответствии с изобретением представлены в таблице XXI.
Соединения формулы (I), когда n = 0, может быть получено различными способами.
Они могут быть получены, например, путем взаимодействия соответствующего производного тиола формулы (XIII) с подходящим дифторбут-1-еновым алкилирующим агентом формулы (XXIV), где L является уходящей группой. Это взаимодействие предпочтительно осуществляют в присутствии мягкого основания, такого как карбонат щелочного металла, например, карбонат натрия или калия, в инертном растворителе при температуре от 0 до 200oC. Обычно взаимодействие можно проводить при температуре кипения с обратным холодильником в подходящем инертном растворителе, например, в ацетоне, который имеет температуру кипения в этой области.
В формуле (XXIV) уходящая группа L представляет, предпочтительно, галоген или сложноэфирную группу сульфоновой кислоты, имеющей формулу OSО2Rb, как показано формулой (XXV), где Rb обозначает C1-4 алкильную группу, необязательно замещенную C1-4 алкильную группу. Более предпочтительно, L обозначает бром, как показано в формуле (XXVI).
Сульфоновый эфир формулы (XXV) может быть получен путем взаимодействия 1,4-дибром-1,1, 2-трифторбутана с серебряной солью сульфоновой кислоты и дебромфторирования полученного промежуточного продукта, 4-толуолсульфонового эфира.
В родственной международной патентной заявке PCT/GB94/01570 описан способ получения соединения формулы (XXVI), а именно: 4-бром-1,1-дифторбут-1-ена, по которому бромистый водород подвергают взаимодействию с коммерчески доступным 4-бром-1, 1,2-трифторбут- 1-еном в инертном растворителе с получением 1,4-дибром-1,1,2- трифторбутаном. Этот промежуточный продукт может быть обработан с дебромфторирующим агентом в соответствующем растворителе, например, ацетоне или воде, с получением соединения формулы (XXVI).
Специалисту в данной области понятно, что соединения формулы (XXIII) могут быть в таутомерном равновесии между эквивалентными меркапто и тионовой формами. Для удобства эти соединения указаны здесь в виде их меркапто формы, если не указано иного.
Соединения формулы (XXIII) являются коммерчески доступными или могут быть получены из коммерчески доступных предшественников обычными способами, хорошо известными специалистам.
Альтернативно, соединения формулы (I) могут быть получены взаимодействием соответствующего соединения формулы (XXVII), где L вновь обозначает хорошо уходящую группу, с меркаптопроизводным формулы (XXVIII), в условиях, хорошо известных в данной области для подобных реакций замещения. Предпочтительно, L обозначает галоген или нитрогруппу. Удобно, когда взаимодействие проводят с системе двухфазного растворителя, такого как вода/дихлорметан, в присутствии катализатора межфазного переноса, например, тетра-н- бутиламмонийбромида, при комнатной температуре в атмосфере азота.
Меркаптопроизводное формулы (XXVIII) удобно подвергать взаимодействию в виде его S-ацетил или его изотиоурониевой бромистоводородной соли, которая легко гидролизуется до меркаптопроизводного формулы (XXVIII).
Производное формулы (I) также может быть получено из соответствующего аминопроизводного формулы (XXIX), которое может быть диазотировано, например, алкилнитритом, таким как трет-бутилнитрит, в присутствии дисульфида формулы (XXX) в подходящем растворителе, таком как дихлорметан или ацетонитрил.
Производное формулы (I), где n = 1 или 2, может быть получено путем окисления соответствующим образом замещенного соединения формулы (I), где n = 0, используя обычные методы, например, подходящим окислительным агентом в инертном органическом растворителе. Обычно, окисление соединения формулы (I) одним эквивалентом подходящего окислительного агента приводит к соответствующему производному, где n = 1, а окисление с использованием двух окислительных агентов приводит к соответствующему производному, где n = 2. Подходящие окислительные агенты включают органические и неорганические пероксиды, такие как надоксикарбоновые кислоты или их соли, например, метахлорнадбензойная кислота, надбензойная кислота, магниевая соль монопероксифталевой кислоты или калий пероксимоносульфат.
Таким образом, в соответствии со следующим аспектом настоящего изобретения предложен способ получения соединений формулы (I), где n = 1 или 2, который включает окисление соответствующим образом замещенного соединения формулы (I), где n = 0.
Следует учесть, что как и для соединений формулы (I), получаемых из соответствующих замещенных соединений формулы (XXIII), (XXVII) или (XXIX), последовательные преобразования функциональных групп можно проводить, используя известные способы для осуществления требуемого замещения в кольце. Примеры таких преобразований функциональных групп включают восстановление нитрогрупп до аминогрупп, галогенирование, например хлорирование, гидролиз эфира до кислоты, окисление спирта до кислоты, образование соли.
Различные дальнейшие предпочтительные признаки и варианты осуществления настоящего изобретения описаны далее подробно в следующих иллюстративных примерах, в которых проценты приведены по массе и используются следующие сокращения: т. пл. = температура плавления; т.кип. = температура кипения; г = граммы; ГХ = газовая хроматография; ЯМР = ядерно-магнитный резонанс; с = синглет; д = дублет; дд = дублет дублетов; т = триплет; кв = квартет; м = мультиплет; ушир. = уширенный; М = моль; мМ = ммоль; CDCl3 = дейтерохлороформ. Химические сдвиги (δ) измерены в частях на миллион относительно тетраметилсилана. CDCl3 был использован в качестве растворителя для ЯМР спектра, если не указано иного. М+ = молекулярный ион, как определено масс-спектроскопией; FAB = бомбардировка быстрыми атомами; ТСХ = тонкослойная хроматография.
Синтез ряда промежуточных соединений, используемых при получении соединений по изобретению, приведен далее. Некоторые из этих соединений известны в данной области.
ПРИГОТОВЛЕНИЕ 1
В данном
Приготовлении описан 3-стадийный способ получения 4-бром-4,4-дифторбутилметансульфоната.
Стадия 1: 4- бром-4,4-дифторбутановая кислота.
К перемешиваемому раствору акриловой кислоты (1,44 г) и ацетонитрила (80 см3) прибавляют дитионит натрия (4,18 г), бикарбонат натрия (2,01 г), воду (20 см3) и, наконец, дибромдифторметан (5 см3 ). Двухфазную смесь перемешивают при комнатной температуре при постепенном растворении неорганических солей. ГХ анализ через 4 часа показывает полный расход акриловой кислоты. Водную фазу насыщают раствором хлорида натрия. Органическую фазу отделяют, сушат над сульфатом магния, фильтруют и упаривают при пониженном давлении с получением бледно-желтого масла с небольшим содержанием белого твердого продукта. Эту смесь обрабатывают этилацетатом, фильтруют и растворитель упаривают при пониженном давлении с получением бледно-желтого масла (2,54 г).1H ЯМР (DMSO-d6): δ 2,45 (2H, т); 2,65 (2H, м).
Стадия 2: 4-бром-4,4-дифторбутанол.
Раствор литийалюминийгидрида в диэтиловом эфире (5 см3, 5 мМ) охлаждают до 0oC. Поддерживая эту температуру, по каплям при перемешивании прибавляют 4-бром-4,4-дифторбутановую кислоту (1 г), растворенную в сухом диэтиловом эфире (5 см3). Через час при 0oC реакционную смесь осторожно гасят прибавлением 2М соляной кислоты. Органическую фазу отделяют, промывают насыщенным раствором бикарбоната натрия, сушат над сульфатом магния, фильтруют и упаривают при пониженном давлении с получением бесцветного масла (0,57 г).1H ЯМР: δ 1,82-1,96 (2H, м); 2,40- 2,60 (2H, м); 3,74 (2H, т).
Стадия 3: 4-бром-4, 4-дифторбутилметансульфонат.
Перемешиваемый раствор 4-бром-4,4- дифторбутанола (0,57 г) в сухом диэтиловом эфире (5 см3) охлаждают до 0oC. Поддерживая эту температуру, прибавляют триэтиламин (1,7 см3). Через десять минут прибавляют метансульфонилхлорид (0,3 см3) и смесь перемешивают в течение еще часа при 0oC. Реакционную смесь выливают в 2М соляную кислоту (2 см3) и диэтиловый эфир (20 см3). Органическую фазу отделяют, промывают насыщенным раствором соли, затем пропускают через плаг из силикагеля, элюируя дополнительным количеством диэтилового эфира. Диэтиловые фракции упаривают при пониженном давлении с получением бледно-желтого масла (0,705 г).1H ЯМР: δ 2, 04-2,18 (2H, м); 2,46-2,64 (2H, м); 3,04 (3H, с), 4,32 (2H, т).
ПРИГОТОВЛЕНИЕ 2
В данном Приготовлении описан 3-стадийный способ получения 4,4- дифторбут-3-енилового эфира
4-метилбензолсульфоновой кислоты из коммерчески доступного 4-бром-1,1,2-трифторбут-1-ена.
Стадия 1: 1,4-дибром-1,1,2-трифторбутана.
Перед использованием 4-бром- 1,1, 2-трифторбут-1-ен (Fluorochem Ltd.) (240 г) промывают водой (300 см3) и сушат (MgSO4). Одной порцией прибавляют бензилпероксид (приблизительно 0,7 г) и через смесь барботируют газообразный бромистый водород с такой скоростью, чтобы температура поддерживалась при 30-40oC. Через 2 часа ГХ образца реакционной смеси показывает, что немного исходного материала осталось. Реакционную смесь промывают водой (300 см3), затем насыщенным раствором бикарбоната натрия (300 см3), сушат (MgSO4) и фильтруют с получением бледножелтого масла (296,7 г), идентифицированного как 1,4-дибром-1,1,2-трифторбутан. По данным ГХ анализа чистота продукта выше, чем 98%.1H ЯМР: δ 2,38 (2H, м); 3,57 (2H, м); 4,90 (1H, м).
Стадия 2: Получение 4-бром-3,4,4-трифторбут-4- метилбензолсульфонат.
Продукт со стадии 1 (1 г) прибавляют по каплям к перемешиваемому раствору тозилата серебра (1,03 г) в ацетонитриле (10 см3) при комнатной температуре, предохраняя от попадания света. Реакционную смесь нагревают с обратным холодильником в течение 24 часов, после чего ГХ анализ показывает полный расход исходного продукта. Реакционную смесь охлаждают до комнатной температуры и выпавший осадок отфильтровывают и промывают этилацетатом. Фильтрат и этилацетатные промывки объединяют и промывают водой и водный слой экстрагируют этилацетатом. Объединенные этилацетатные слои промывают водой и насыщенным раствором соли, сушат над сульфатом магния и упаривают при пониженном давлении с получением коричневого масла (1,21 г). ГХ анализ показывает, что чистота продукта > 99%.1H ЯМР: δ 2,20 (2H, м); 2,46 (3H, с); 4,19 (2H, м); 4,74 (1H, м); 7,38 (2H, д); 7,80 (2H, д).
Стадия 3: Получение 4,4-дифторбут-3-енил-4- метилбензолсульфоната.
К перемешиваемой суспензии порошка цинка (1,41 г) и иода (один гран, катализатор) в метаноле (3 см3) прибавляют раствор 4-бром-3,4,4-трифторбутил-п-толилсульфоната (0,71 г) в метаноле (2 см3). Реакционную смесь нагревают с обратным холодильником в течение 2 1/2 часов, после чего ГХ анализ показывает полный расход исходного продукта. Органическую фазу отбирают пипеткой от суспензии цинка и цинк промывают 3 порциями этилацетата. Объединенные этилацетатные части промывают 2М соляной кислотой, сушат над сульфатом магния и упаривают при пониженном давлении с получением коричневой жидкости (0,47 г). ГХ анализ показывает, что чистота продукта > 99%.1H ЯМР: δ 2,35 (2H, м); 2,46 (3H, с); 4,01 (2H, м); 4,15 (1H, м); 7,38 (2H, д); 7,79 (2H, д).
ПРИГОТОВЛЕНИЕ 3
В данном Приготовлении описан способ получения
4-бром-1,1-дифторбут-1-ена из 1,4-дибром- 1,1,2-трифторбутана.
Порошок цинка (0,88 г) прибавляют к перемешиваемому раствору 1,4-дибром-1,1,2-трифторбутана (1,38 г) в ацетоне (6 см3), содержащем воду (1 капля), в атмосфере азота. Через 45 минут ГХ анализ показывает, что большая часть исходного продукта израсходована. Затем смесь прибавляют к дополнительному количеству порошка цинка (3 г) в ацетоне, содержащем следы волы, предварительно нагретому до 55oC. Через еще 20 минут при этой температуре ГХ анализ показывает, что весь исходный продукт израсходован, свидетельствуя о том, что реакция дебромфторирования началась. К реакционной смеси в течение более 75 минут при поддержании температуры реакционной смеси при 55oC прибавляют еще исходного продукта (12,34 г). Затем нагревание продолжают еще 95 минут. ГХ анализ образца показывает, что около 3% исходного дибромпроизводного остается неизмененными. Добавляют еще порошок цинка (0,16 г) и нагревание продолжают до тех пор, пока ГХ анализ будет показывать, что израсходован весь исходный продукт. Ацетоновый раствор декантируют от осадка цинка с получением раствора 4-бром-1, 1-дифторбут-1-ена, подходящего для использования в дальнейших химических реакциях.
ПРИГОТОВЛЕНИЕ 4
В данном Приготовлении описан способ получения тиоацетата 4,4
- дифторбут-3-енила.
Тиоацетат калия (1,98 г) 4-бром-1,1- дифторбут-1-ена (3,0 г) и тетра-н-бутиламмоний бромид (0,3 г, катализатор) перемешивают при комнатной температуре в атмосфере азота в течение 5 часов и оставляют на 18 часов. Смесь перегоняют в аппарате Кугельрора (Kugelrohr) с получением тиоацетата 4,4-дифторбут-3-енила в виде бесцветной жидкости (1,12 г).1H ЯМР: δ 2,25 (2H, м); 2,30 (3H, с); 2,90 (2H, т); 4,20 (1H, м); (т.кип. 115oC при 120 мм рт. ст.).
ПРИГОТОВЛЕНИЕ 5
В данном Приготовлении описан способ
получения бензолсульфонатной соли 4,4-дифторбут-3-енила.
Тиомочевину (0,29 г) и 4-метилбензолсульфоната 4,4-дифторбут-3- енила (1,0 г) нагревают вместе при кипячении с обратным холодильником в этаноле (20 см3) в течение 24 часов. Реакционную смесь охлаждают и растворитель упаривают при пониженном давлении с получением масла, которое медленно кристаллизуется. Растирание с гексаном дает (4,4-дифторбут-3- енил) тиомочевину в виде ее 4-метилбензолсульфонатной соли (1,14 г). MH+ (FAB) = 167;1H ЯМР (DMSO-d6): δ 2,48 (3H, с); 2,46-2,58 (2H, м); 3,42 (2H, т); 4,46-4,84 (1H, м); 7,32 (2H, д); 7,68 (2H, д); 9,10-9,40 (3H, ушир.).
ПРИГОТОВЛЕНИЕ 6
В данном Приготовлении описан способ получения
гидробромида 4,4- дифторбут-3-енилизотиомочевины.
Тиомочевину (18,5 г) прибавляют к раствору 4-бром-1,1-дифторбут-3-ена (41,5 г) в этаноле (150 см3) и нагревают с обратным холодильником при перемешивании в течение 18 часов. Реакционную смесь охлаждают и растворитель упаривают при пониженном давлении. Полученный воскообразный продукт промывают диэтиловым эфиром, фильтруют, промывают дополнительным количеством диэтилового эфира и отсасывают досуха с получением требуемого продукта в виде бесцветного твердого продукта (57 г). MH+ (FAB) = 167;1H ЯМР (DMSO-d6): δ 2,20 (2H, м); 2,20 (2H, т); 4,50 (1H, м); 8,95 (4H, широкий сигнал).
N-Метилпроизводное продукта выше получают по вышеописанному способу, но используя N-метилтиомочевину вместо тиомочевины. Оно имеет MH+ = 181;1H ЯМР (DMSO-d6): δ 2,45-2,55 (2H, м); 3,0-3,05 (3H, д); 3,4-3,5 (2H, т); 4,30-4,45 (1H, м); (т.пл. 74-77,2oC).
ПРИГОТОВЛЕНИЕ 7
В данном Приготовлении описан способ получения бис-(4,4-дифторбут-3-енил)дисульфида.
Раствор дисульфида натрия (предварительно полученный из нонагидрата сульфида натрия (53 г) и серы (7,0 г)) в этаноле (250 см3) прибавляют к 1-бром-4,4-дифторбут-3-ену (50 г) в этаноле (100 см3). Смесь ступенчато нагревают и перемешивают при нагревании с обратным холодильником в течение 2 часов, затем охлаждают и упаривают при пониженном давлении. Остаток экстрагируют диэтиловым эфиром, органическую фазу фильтруют для удаления бромида натрия и эфир упаривают при пониженном давлении с получением жидкого продукта, который перегоняют при 16 мм рт.ст., т.кип. 120oC с получением бис-(4,4-дифторбут-3-енил) дисульфида (24 г) в виде бесцветного жидкого продукта.
ПРИМЕР II.1
В данном примере описан способ получения 2-(4,
4-дифторбут-3-енилтио)фурана (соединение II.1)
Бутиллитий (6,5 см3, 2,5М в эфире) прибавляют по каплям к раствору фурана (1 г) в диэтиловом эфире (40 см3). Через 90
минут реакционную смесь нагревают с обратным холодильником в течение 30 минут и затем охлаждают до комнатной температуры. При перемешивании порциями прибавляют порошок серы (0,48 г). Через 2 часа
прибавляют 4-бром-1,1- дифторбут-1-ен (3,0 г) и перемешивание продолжают при комнатной температуре в течение 18 часов. Реакционную смесь гасят водой и продукт экстрагируют диэтиловым эфиром.
Объединенные органические экстракты сушат, фильтруют и упаривают с получением темно-коричневой жидкости. Хроматография на силикагеле (элюент - смеси гексан-диэтиловый эфир) дает соединение II.1 (0,965
г).1H ЯМР: δ 2,2-2,3 (2H, м); 2,7-2,8 (2H, т); 4,15-4,35 (1H, м); 6,4 (1H, дд); 6,53 (1H, д); 7,5 (1H, д); (масло).
ПРИМЕР II.2
В данном примере описан
способ получения 2-(4,4-дифторбут-3-енилтио)-5-метилфурана (соединение II.4)
Бутиллитий (5,4 см3, 2,5М в эфире) прибавляют по каплям к раствору фурана (1 г) в диэтиловом эфире (40
см3). Через 90 минут реакционную смесь нагревают с обратным холодильником в течение 30 минут и затем охлаждают до комнатной температуры. При перемешивании порциями прибавляют порошок серы
(0,38 г). Через 2 часа прибавляют 4-бром-1,1-дифторбут-1-ен (2,05 г) и перемешивание продолжают при комнатной температуре в течение 18 часов. Реакционную смесь гасят водой и продукт экстрагируют
диэтиловым эфиром. Объединенные органические экстракты сушат, фильтруют и упаривают. Хроматография на силикагеле (элюент - смеси гексан-эфир) дает соединение II.4 (1,25 г).1H ЯМР: δ
2,2-2,3 (2H, м); 2,3 (3H, с); 2,7-2,77 (2H, т); 4,16-4,34 (1H, м); 5,97 (1H, м); 6,43 (1H, д); (масло).
ПРИМЕР II.3
В данном примере описан способ получения 3-(4,
4-дифторбут-3- енилтио)-2-метилфурана (соединение II.7)
Раствор, содержащий 2-метил-3-фурантиол (2,0 г), 4-бром-1,1-дифторбут-1-ен (3,24 г) и карбонат калия (2,48 г) в ацетоне (50 см3) нагревают с обратным холодильником в течение 2 часов и затем оставляют стоять 18 часов. Реакционную смесь гасят водой и продукт экстрагируют диэтиловым эфиром. Объединенные органические
экстракты сушат, фильтруют и упаривают. Хроматография на силикагеле (элюент - 10% эфир в гексане) дает соединение II.7 (3,338 г).1H ЯМР: δ 2,1-2,25 (2H, м); 2,35 (3H, с); 2,65 (2H,
т); 4,1-4,3 (1H, м); 6,3 (1H, д); 7,3 (1H, д); (масло).
ПРИМЕР II.4
В данном примере описан способ получения 2-(4,4-дифторбут-3-енилсульфинил)фурана (соединение II.2)
3-Хлорнадбензойную кислоту (0,54 г 50% по весу твердого продукта, 1,58 мМ) порциями прибавляют к раствору соединения II.1 (0,30 г) в дихлорметане (5 см3) при охлаждении на ледяной бане.
После перемешивания в течении 4 часов реакционную смесь распределяют между этилацетатом и 2М раствором NaOH. Отделяют органический слой, промывают дополнительным количеством 2М раствора NaOH, сушат
над сульфатом магния, фильтруют и упаривают с получением соединения II. 2 (0,210 г).1H ЯМР: δ 2,3-2,5 (2H, м); 3,1-3,4 (2H, м); 4,15-4,35 (1H, м); 6,5 (1H, дд); 7,0 (1H, д); 7,7 (1H,
д); (масло).
Вышеописанными методами получают следующие соединения:
(1) 2-(4,4-дифторбут-3-енилсульфонил)фуран (соединение II.3);1H ЯМР: δ 2,4-2,55 (2H,
м); 3,25-3,3 (2H, т); 4,1-4,3 (1H, м); 6,6 (1H, м); 6,6 (1H, дд); 7,23 (1H, д); 7,67 (1H, д); (масло) из соединения II.1, используя 2,1 эквивалента окислителя.
(2) 2-(4, 4-дифторбут-3-енилсульфинил)-5-метилфуран (соединение II.5);1H ЯМР: δ 2,3-2,45 (5H, м); 3,0- 3,15 и 3,3-3,4 (всего, 2H, м); 4,2-4,3 (1H, м); 6,6 (1H, д); 6,85 (1H, д); (масло) из соединения II.4, используя 1 эквивалент окислителя.
(3) 2-(4,4-дифторбут-3-енилсульфонил)-5-метилфуран (соединение II.6);1H ЯМР: δ 2,4-2,55 (5H, м); 3,2-3,28 (2H, т); 4,15-4,3 (1H, м); 6,2 (1H, д); 7,1 (1H, д); (масло) из соединения II.4, используя 2,1 эквивалента окислителя.
(4) 3-(4,4-дифторбут-3-енилсульфинил)-2-метилфуран (соединение II.8);1H ЯМР: δ 2,3-2,45 (5H, м); 2,8-2,9 и 3,1-3,2 (всего, 2H, м); 4,2-4,35 (1H, м); 6,66 (1H, д); 7,4 (1H, д); из соединения II.4, используя 1 эквивалент окислителя.
(5) 3-(4,4-дифторбут-3-енилсульфонил)-2-метилфуран (соединение II.9);1H ЯМР: δ 2,42-2,52 (2H, м); 2,6 (3H, с), 3,2-3,28 (2H, т); 4,1-4,23 (1H, м); 6,6 (1H, д); 7,36 (1H, д) из соединения II.4, используя 2,1 эквивалента окислителя.
ПРИМЕР III.1
В данном примере описан способ получения 2-(4,4-дифторбут- 3-енилтио)тиофена (соединение III.1)
Раствор, содержащий 2-меркаптотиофен (10 г), 4-бром-1,1-дифторбут-1-ен (15,47 г) и карбонат калия (11,87 г) в ацетоне (250 см3), нагревают при кипячении с обратным холодильником в течение 2
часов и затем оставляют стоять в течение 18 часов. Реакционную смесь гасят водой и несколько раз экстрагируют диэтиловым эфиром. Объединенные органические экстракты сушат над сульфатом магния,
фильтруют и упаривают с получением янтарного масла. Хроматография на силикагеле (элюент 5% эфир в гексане) дает Соединение III.1 (9,5 г); М+ = 206;1H ЯМР: δ 2,2-2,4 (2H,
м); 2,8 (2H, т); 4,1-4,3 (1H, м); 6,95-7,0 (1H, дд); 7,15 (1H, д); 7,35 (1H, д); (масло).
Вышеописанными методами в соответствии с изобретением получают следующее соединение:
(1) 2-(4,4-дифторбут-3-енилтио)бензо[b] тиофен (соединение III.10);1H ЯМР: δ 2,2-2,3 (2H, м); 2,7-2,8 (2H, т); 4,15-4,35 (1H, м); 6,4 (1H, дд); 6,53 (1H, д); 7,5 (1H, д) из
соединения бензотиофена.
ПРИМЕР III.2
В данном примере описан способ получения 2-(4,4-дифторбут-3-енилтио)-5-формилтиофена (соединение III.4)
Соединение III. 1 (1,0
г) медленно прибавляют к раствору, содержащему диметилформамид (0,48 см3) и фосфорилхлорид (0,56 см3). Реакционную смесь нагревают при 100oC в течение 2 часов,
охлаждают на ледяной бане и затем нейтрализуют 2М раствором NaOH. Водный раствор дважды экстрагируют диэтиловым эфиром и объединенные органические слои промывают водой и раствором NaHCO3.
Органические экстракты сушат над сульфатом магния, фильтруют и упаривают с получением темной жидкости. Фильтрование через силикагель (элюент 20% диэтиловый эфир в гексане) дает соединение III.4 (0,91
г). М+ = 234;1H ЯМР: δ 2,4 (2H, м); 3,0 (2H, т); 4,2-4,4 (1H, м); 7,1 (1H, д); 7,7 (1H, д); 9,8 (1H, с); (масло).
ПРИМЕР III.3
В данном примере
описан способ получения 2-(4,4-дифторбут-3-енилтио)-5- гидроксиметилтиофена (соединение III.5)
Боргидрид натрия (0,065 г) прибавляют к раствору соединения III.4 (0,75 г) в этаноле (21 см3) и воде (9 см3). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, гасят 2М хлористоводородной кислотой и затем распределяют между водой и диэтиловым
эфиром. Органическую фазу сушат над сульфатом магния, фильтруют и упаривают с получением зеленоватой жидкости. Хроматография на силикагеле (элюент 20% этилацетат в гексане) дает соединение III. 5 (0,
44 г). М+ = 236;1H ЯМР: δ 1,8-2,0 (1H, ушир. с); 2,3 (2H, м); 2,8 (2H, т); 4,1-4,3 (1H, м); 4,8 (2H, с); 6,8 (1H, д); 7,0 (1H, д); (масло).
ПРИМЕР
III.4
В данном примере описан способ получения (E)- и (Z)-(4,4- дифторбут-3-енилтио)-5-гидроксиметилтиофена (соединения III.6 и III.7)
В течение 5 минут в этаноле (15 см3)
и воде (15 см3) перемешивают гидрохлорид гидроксиламина (0,9 г) и гидрокарбонат натрия (1,09 г). Прибавляют соединение III. 4 (3 г) и реакционную смесь перемешивают при комнатной
температуре в течение 2 часов и затем оставляют стоять в течение 18 часов. Реакционную смесь распределяют между водой и диэтиловым эфиром. Органическую фазу сушат над сульфатом магния, фильтруют и
упаривают с получением янтарной жидкости. Хроматография на силикагеле (элюент 20% этилацетат в гексане) дает соединение III. 6 (1,5 г) и III.7 (1,3 г). М+ = 249;1H ЯМР: δ
2,25- 2,35 (2H, м); 2,85 (2H, т); 4,14-4,35 (1H, м); 7,05 (2H, м); 7,52 (1H, ушир. с); 8,18 (1H, с); (масло) и М+ = 249; δ 2,25-2,38 (2H, м); 2,85-2,95 (2H, т); 4,18-4,35 (1H, м); 7,
08 (1H, д); 7,25 (1H, д); 7,64 (1H, с); (масло).
ПРИМЕР III.5
В данном примере описан способ получения 5-циано-2-(4,4-дифторбут-3-енилтио)тиофена (соединение III.8)
1,1'-Карбонилдиимидазол (0,326 г) прибавляют к раствору соединения III.6 (0,5 г) и затем реакционную смесь перемешивают при комнатной температуре в течение 10 минут, затем нагревают с обратным
холодильником в течение 2 часов и оставляют стоять в течение 18 часов. Прибавляют еще эквивалент 1,1'-карбонилдиимидазола и реакционную смесь нагревают с обратным холодильником в течение 1 часа.
Реакционную смесь фильтруют через целит и затем упаривают с получением соединения III.8 (0,28 г).1H ЯМР: δ 2,27-2,38 (2H, м); 2,94 (2H, т); 4,15-4,33 (1H, м); 7,05 (1H, д); 7,5 (1H,
д); (масло).
ПРИМЕР III.6
В данном примере описан способ получения 5-ацетил-2-(4,4- дифторбут-3-енилтио)тиофена (соединение III.9)
К раствору метилмагнийбромида (1,3
см3, 2М раствор в диэтиловом эфире, 3 эквивалента) медленно прибавляют к раствору соединения III.8 (0,3 г) в тетрагидрофуране (10 см3). Реакционную смесь перемешивают при
комнатной температуре в течение 3 часов и растворитель упаривают. Остаток распределяют между раствором гидроксида аммония и хлороформом. Органический слой промывают водой, сушат над Na2
SO4 и упаривают. Остаток хроматографируют на силикагеле (элюент 10% этилацетат в гексане) с получением соединения III.9 (0,16 г).1H ЯМР: δ 2,30-2,40 (2H, м); 2,50 (3H, с);
2,97 (2H, т); 4,15-4,33 (1H, м); 7,04 (1H, д); 7,55 (1H, д); (масло).
ПРИМЕР III.7
В данном примере описан способ получения 2-(4,4-дифторбут-3-енилсульфинил)тиофена
(соединение III.2)
Соединение III. 1 (0,50 г) перемешивают при комнатной температуре в дихлорметане (5 см3) и прибавляют 3-хлорнадбензойную кислоту (0,834 г 50%-ной твердой по весу,
1 эквивалент). После того, как ТСХ показывает полное расходование исходного продукта, реакционную смесь гасят добавлением насыщенного водного раствора бикарбоната натрия и продукт затем экстрагируют
дихлорметаном. Органическую фазу отделяют, промывают насыщенным раствором соли и сушат над сульфатом магния. После фильтрации и концентрирования упариванием при пониженном давлении получают жидкий
продукт (0,584 г), который затем очищают хроматографией на силикагеле, используя 20%-ный этилацетат в гексане в качестве элюента и затем диэтиловый эфир для вымывания соединения III.2 (0,29 г).1H ЯМР: δ 2,3-2,55 (2H, м); 2,9-3,2 (2H, м); 4,2-4,5 (1H, м); 7,15 (1H, м); 7,5 (1H, м); 7,7 (1H, м); (масло).
Вышеописанным способом получают следующие соединения в
соответствии с изобретением:
(1) 2-(4,4-дифторбут-3-енилсульфонил)тиофен (соединение III.3);1H ЯМР: δ 2,4-2,6 (2H, м); 3,2-3,4 (2H, т); 4,1-4,3 (1H, м); 7,15 (1H, дд); 7,
7-7,8 (1H, м); (масло) из соединения III.1, используя два эквивалента окислителя.
(2) 2-(4,4-дифторбут-3-енилсульфинил)бензо[b]тиофен соединение III.11);1H ЯМР: δ 2, 3-2,4 (2H, м); 3,0-3,2 (2H, м); 4,2-4,4 (1H, м); 7,45 (1H, м); 7,9 (2H, м); 7,75 (1H, с) из соединения III.10, используя один эквивалент окислителя.
(3) 2- (4, 4-дифторбут-3-енилсульфонил)бензо[b] тиофен (соединение III. 12);1H ЯМР: δ 2,5 (2H, м); 3,3 (2H, т); 4,2-4,35 (1H, м); 7,5 (2H, м); 7,9 (3H, м) из соединения III.10, используя два эквивалента окислителя.
ПРИМЕР IV.1
В данном примере описан способ получения этил 5-(4,4- дифторбут-3-енилтио) метилизоксазол-4-карбоксилат (соединение IV.8)
Раствор
4,4-дифторбут-3-енилтиоацетата (2 г) в 50%-ном растворе гидроксида натрия (6,7 см3) энергично перемешивают в течение 30 минут. Прибавляют этил 5-хлор-4-метилизоксазол (2,2 г) и затем
тетра-н-бутиламмонийбромид (катализатор) и реакционную смесь перемешивают при комнатной температуре в атмосфере азота. Через 3 часа слои разделяют и органическую фазу промывают солевым раствором,
сушат (MgSO4), фильтруют и упаривают при пониженном давлении. Остаток перемешивают с 880 аммиаком, вызывая кристаллизацию. Кристаллы отделяют фильтрацией с получением соединения IV.8 (2,87
г).1H ЯМР: δ 1,35 (3H, т); 2,45 (3H, с); 2,50 (2H, м); 3,20 (2H, т); 4,25 (1H, м); 4,30 (2H, кв); (т.пл. 41-42oC).
ПРИМЕР IV.2
В данном примере
описан способ получения 5-(4,4-дифторбут- 3-енилтио)метилизоксазол-4-карбоновой кислоты (соединение IV.9)
Раствор соединения IV.8 (0,5 г) в изопропаноле (5 см3) и 2М NaOH (1 см3) перемешивают в течение 3 часов. Смесь затем выливают в воду и промывают этилацетатом. Водный слой затем подкисляют 2М HCl и продукт экстрагируют этилацетатом. Экстракт затем сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением соединения IV.9 (0,16 г). М+ = 249;1H ЯМР: δ 2,45 (3H, с); 2,50 (2H, м); 3,20 (2H, т); 4,20-4,40 (1H,
м); (т.пл. 132-133oC).
ПРИМЕР IV.3
В данном примере описан способ получения 5-(4,4-дифторбут-3-енилтио)-3-метилизоксазол-4- карбоксамида (соединение IV.7)
Триэтиламин (0,33 см3) и этилхлорформиата (0,24 см3) прибавляют к соединению IV.9 (0,56 г) в дихлорметане (15 см3) при 0oC. Реакционной смеси дают нагреться
до комнатной температуры и перемешивают в течение 2 часов. Через раствор барботируют аммиак до насыщения и затем реакционную смесь перемешивают в течение 1 часа. Прибавляют водный аммиак и продукт
экстрагируют дихлорметаном. Органическую фазу промывают водой, сушат (MgSO4), фильтруют и упаривают при пониженном давлении. Очистка дистилляцией в аппарате кугельрора с получением
соединения IV.7 (0,069 г).1H ЯМР: δ 2,45 (2H, м); 2,50 (3H, с); 3,25 (2H, т); 4,25 (1H, м); (т.пл. 87oC).
ПРИМЕР IV.4
В данном примере описан
способ получения 3-(5-хлорфур-2-ил)-5-(4,4-дифторбут-3-енилтио)-3-изоксазол (соединение IV.23)
Через перемешиваемый раствор метоксида калия (1,9 г) в этаноле (10 см3), охлажденном
на бане ацетон со льдом, барботируют сероводород. Прибавляют 5-хлор-3-(5-хлорфур-2-ил)изоксазол (2,2 г) и реакционную смесь затем нагревают с обратным холодильником в течение 1 часа, в течение
которого упаривают растворитель. Прибавляют ацетон (10 см3) и 4-бром-1,1-дифторбут-1-ен (2 г) и смесь нагревают с обратным холодильником еще в течение 2 часов. Полученный раствор охлаждают,
выливают в диэтиловый эфир и солевой раствор и слои разделяют. Водный слой экстрагируют эфиром. Объединенные органические фазы промывают солевым раствором, сушат (MgSO4), фильтруют и
упаривают при пониженном давлении с получением темного твердого продукта. Очистка колоночной хроматографией на силикагеле с использованием 10%- ного эфира в гексане в качестве элюента дает соединение
IV.23 (2 г). М+ = 291;1H ЯМР: δ 2,35-2,50 (2H, м); 3,10 (2H, т); 4,30 (1H, м); 6,30 (1H, д); 6,43 (1H, с); 6,90 (1H, д); (т.пл. 80-82oC).
Следующее соединение в соответствии с изобретением получают вышеописанным способом:
(1) 5-(4,4-дифторбут-3-енилтио)-3- фенилизоксазол) (соединение IV.1); М+ = 267;1H
ЯМР: δ 2,43 (2H, м); 3,10 (2H, т); 4,28 (1H, м); 6,50 (1H, с); 7,45 (3H, м); 7,78 (2H, м); (масло) из 5-хлор-3-фенилизоксазола.
ПРИМЕР IV.5
В данном примере описан
способ получения 2-(4,4-дифторбут-3-енилтио)-3-метилизоксазол (соединение IV.10)
К перемешиваемому раствору оксима ацетона (0,365 г) в сухом тетрагидрофуране (20 см3) при 0oC в атмосфере азота прибавляют н-бутиллитий (4,6 см3 2,5 М раствора в гексане) с получением бледно-желтого осадка. После перемешивания при 0oC в течение 30 минут
прибавляют дисульфид углерода (0,3 см3) с получением ярко-оранжевого раствора. Еще через 10 минут прибавляют 3М HCl (20 см3) и реакционную смесь нагревают с обратным
холодильником в течение 3 часов и затем охлаждают. Слои разделяют и водный слой экстрагируют хлороформом. Объединенные органические фазы сушат (MgSO4), фильтруют и упаривают при пониженном
давлении с получением коричневого масла. Масло затем обрабатывают ацетоном (11 см3) и прибавляют 1-бром-4,4- дифторбут-3-ен (0,77 г) и карбонат калия (0,87 г) и реакционную смесь нагревают
с обратным холодильником в течение 3,5 часов и затем охлаждают. Затем смесь выливают в этилацетат и 1М HCl и слои разделяют. Водный слой экстрагируют этилацетатом и объединенные органические фазы
сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением коричневого масла. Очистка колоночной хроматографией на силикагеле с использованием 1:9 этилацетата:гексана дает
2-(4,4-дифторбут-3- енилтио)-3-метилизоксазола (0,105 г). М+ = 205;1H ЯМР: δ 2,3 (3H, с); 2,40 (3H, с); 2,40 (2H, м); 3,05 (2H, т); 4,25 (1H, м); 6,00 (1H, с);
(масло).
ПРИМЕР IV.6
В данном примере описан способ получения 3-(5-хлорфур-2-ил)-5- (4,4-дифторбут-3-енилсульфинил)изоксазол (соединение IV.24)
К перемешиваемому
раствору соединения IV.23 (2 г) в метаноле (40 см3), охлаждаемому на бане из ацетона со льдом, прибавляют магний монопероксифталат (9,4 г). После перемешивания в течение 30 минут
охлаждающую баню удаляют и реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 1 часа. Смесь выливают в диэтиловый эфир и солевой раствор и слои разделяют. Объединенные
органические фазы промывают 2М NaOH, водой и солевым раствором и слои разделяют. Объединенные органические фазы промывают 2М NaOH, водой и насыщенным раствором соли, сушат (MgSO4),
фильтруют и упаривают при пониженном давлении с получением соединения IV.24 (1,9 г). М+ = 323;1H ЯМР: δ 2,50-2,60 (2H, м); 3,40 (2H, т); 4,25 (1H, м); 6,38 (1H, д); 7,05
(1H, д); 7,20 (1H, с); (т.пл. 86,5-88,5oC).
Вышеописанным способом получают следующие соединения в соответствии с изобретением:
(1) 5-(4,
4-дифторбут-3- енилсульфинил)-3-фенилизоксазол (соединение IV. 2); М+ = 283;1H ЯМР: δ 2,53 (2H, м); 3,25 (2H, т); 4,29 (1H, м); 7,18 (1H, с); 7,50 (3H, м); 7,80 (2H, м);
(масло) из соединения IV.1 и одного эквивалента окислителя.
(2) 5-(4,4-дифторбут-3-енилсульфонил)-3-фенилизоксазол (соединение IV. 3); М+ = 299;1H ЯМР: δ 2,57 (2H, м); 3,43 (2H, т); 4,27 (1H, м); 7,2 (1H, с); 7,51 (3H, м); 7,80 (2H, м); (т.пл. 57-58,5oC) из соединения IV.1 и двух эквивалентов окислителя.
(3) 3-(тиен-2-ил)-5-(4,4-дифторбут-3-енилсульфонил)-изоксазол (соединение IV. 26); М+ = 306;1H ЯМР: δ 2,50-2,63 (2H, м); 3,41 (2H, т); 4,27 (1H, м); 7,20 (2H, с); 7,14-7,23; 7,52 (2H, м); (т.пл. 55-57oC) из 3-(тиен-2-ил)-5-(4,4-дифторбут-3-енилтио)изоксазола, соединения IV.25, в свою очередь полученного из 5-хлор-3- (тиен-2-ил) изоксазола по способу примера IV.4.
ПРИМЕР V.1
В данном примере описан способ получения 3-хлор-4-циано-5-(4,4- дифторбут-3-енилтио)изотиазола (соединение V.2)
Раствор 4-циано-3,5-дихлоризотиазола
(1 г) в метаноле (10 см3) прибавляют в течение более 15 мин к раствору наногидрата сульфида натрия (1,3 г) в воде (2,6 см3) и метаноле (25 см3), нагретом до 50oC. Реакционную смесь перемешивают в течение 1 часа и затем растворитель упаривают при пониженном давлении с получением желтого твердого продукта. Остаток растворяют в ацетоне (20 см3
), прибавляют 4-бром-1,1-дифторбут-1-ен (0,68 г) и реакционную смесь перемешивают в течение 12 часов. Полученную смесь выливают в воду и слои разделяют. Водный слой экстрагируют этилацетатом.
Объединенные органические фазы сушат (MgSO4), фильтруют и упаривают при пониженном давлении. Очистка колоночной хроматографией на силикагеле с использованием в качестве элюента 1: 1
этилацетата:гексана дает соединение V.2 (0,88 г). М+ = 266;1H ЯМР: δ 2,50 (2H, м); 3,20 (2H, т); 4,20-4,40 (1H, м); (масло).
По вышеуказанному способу
получают следующее соединение по изобретению, но используя 2 эквивалента нонагидрата сульфида натрия и 4-бром-1,1-дифторбут-1-ен:
(1)
3,5-бис-(4,4-дифторбут-3-енилтио)-4-цианоизотиазол
(соединение V. 12).1H ЯМР: δ 2,40-2,60 (4H, м); 3,20 (2H, т); 3,30 (2H, т); 4,20-4,40 (2H, м); (масло).
ПРИМЕР V.2
В данном примере описан способ получения
5-(4,4-дифторбут-3- енилсульфонил)-4-цианоизотиазола (соединение V. 6) и 3, 5-(4,4- дифторбут-3-енилсульфонил)-4-цианоизотиазола (соединение V.15)
К раствору соединения V. 12 (0,1 г) в
дихлорметане (5 см3) прибавляют 3-хлорнадбензойную кислоту (0,42 г) и реакционную смесь перемешивают до тех пор, пока исходный продукт не израсходуют. Реакционную смесь выливают в
этилацетат и воду и слои разделяют. Водный слой экстрагируют этилацетатом. Объединенные этилацетатные фазы промывают гидрокарбонатом натрия, сушат (MgSO4), фильтруют и упаривают при
пониженном давлении. Остаток обрабатывают диэтиловым эфиром и прибавляют триэтиламин, вызывающий помутнение раствора. Эфирный раствор затем промывают водой, сушат (MgSO4), фильтруют и
упаривают. Очистка остатка колоночной хроматографией на силикагеле с использованием в качестве элюента 1:1 этилацетата:гексана дает соединение V.15 (0,01 г). М+= 418;1H ЯМР:
δ 2,60-2,70 (4H, м); 3,60 (4H, т); 4,30 (2H, м); (масло) и соединение V. 6 (0,08 г). М+ = 264;1H ЯМР: δ 2,60 (2H, м); 3,60 (2H, т); 4,30 (1H, м); 9,40 (1H, с);
(масло).
ПРИМЕР VI.1
Оксазолы, замещенные группой 4,4-дифторбут-3-енилтио по 2, 4 и 5-му положению, могут быть получены исходя из соответственно замещенного меркаптооксазола
и подходящего дифторбут-1- енового алкилирующего агента. Это иллюстрировано следующим получением 2-(4,4-дифторбут-3-енилтио)-5-фенилоксазола (соединение VI.18).
К раствору 2-меркапто-5-фенилоксазола (0,44 г) в ацетоне (15 см3) прибавляют 4,4-дифтор-3-бутен-4- метилбензолсульфонат (0,7 г) и карбонат калия (0,369 г) и реакционную смесь нагревают с обратным холодильником в течение всего 8 часов, после чего часть исходного тозилата остается. Прибавляют еще 2-меркапто-5-фенилоксазола (0,05 г) и нагревание продолжают в течение 5 часов. Реакционную смесь охлаждают, выливают в диэтиловый эфир и воду и слои разделяют. Водный слой экстрагируют эфиром и объединенные органические фазы сушат (MgSO4) и упаривают при пониженном давлении с получением желтой жидкости. Хроматография на силикагеле с использованием 5%-ного трет-бутилдиметилового эфира в гексане дает соединение VI.18;1H ЯМР: (CDCl3) δ 2,51 (2H, м); 3,23 (2H, т); 4,30 (1H, м); 7,23-7,47 (4H, м); 7,58 (2H, д); (масло).
По вышеуказанному способу получают следующие соединения по изобретению:
(1) 2-(4,
4-дифторбут-3-енилтио)-оксазол (соединение VI. 1).1H ЯМР (CDCl3) δ 2,45 (2H, м); 3,20 (2H, т); 4,25 (1H, м); 7,10 (1H, с); 7,66 (1H, с); (масло) изоксазол-2-тиона.
(2) 2-(4,4-дифторбут-3-енилтио)-4-метилоксазол (соединение VI. 6). M+ 205;1H ЯМР: δ 2,04 (3H, с); 2,45 (2H, м); 3,18 (2H, т); 4,25 (1H, м); 7,38 (1H, кв); (масло) из 2-меркапто-4-метилоксазола.
ПРИМЕР VI.2
В данном примере описан 3-стадийный способ получения метил 2-(4,4-дифторбут-3-енилтио)метилизоксазол-5-карбоксилата
(соединение VI.32)
Стадия 1:
Метил-2-амино-4-метилоксазол-5-карбоксилат.
Метил 3-хлорацетоацетат (75 г) и мочевину (90 г) в метаноле (200 см3) перемешивают и нагревают с обратным холодильником в течение 24 часов. Реакционную смесь охлаждают до комнатной температуры и осадок отфильтровывают от раствора, промывают холодным метанолом и отсасывают досуха. Этот твердый продукт обрабатывают 2М гидроксидом натрия и продукт экстрагируют этилацетатом (несколько порций). Упаривание растворителя при пониженном давлении дает бесцветный твердый продукт (14,5 г), который перекристаллизовывают из ацетонитрила, т. пл. 225oC (разл.).1H ЯМР (DMSO-d6): δ 2,15 (3H, с); 3,75 (3H, с); 7,4 (2H, шир.).
Стадия 2: Метил-2-хлор-4-метилоксазол-5-карбоксилат.
Продукт со стадии 1 (1,56 г) частично растворяют в сухом ацетонитриле (40 см3) и прибавляют по частям при 8oC к перемешиваемой смеси хлорида меди (2) (1,61 г) и третичного бутилнитрита в ацетонитриле (сухой, 20 см3) в атмосфере азота. Полученный коричневый раствор перемешивают при 20oC в течение 2 часов и упаривают при пониженном давлении. Остаток обрабатывают водной 2М соляной кислотой и продукт экстрагируют диэтиловым эфиром. Органическую фазу сушат (MgSO4) и затем промывают на короткой колонке с силикагелем еще эфиром. Фильтрат упаривают при пониженном давлении с получением требуемого промежуточного продукта (0,9 г) в виде желтого твердого продукта. М+ = 175.
Стадия 3: Метил-(4,4-дифторбут-3-енилтио)- 4-метилоксазол-5-карбоксилат.
Продукт со стадии 2 (0,176 г) и тиомочевину (0,084 г) в этаноле (5 см3) перемешивают и нагревают с обратным холодильником в течение 5 часов. Реакционную смесь охлаждают и растворитель удаляют путем упаривания при пониженном давлении с получением желтой смолы, которую растворяют в ацетоне, содержащем 4-бром-1,1-дифторбут-1- ена (0,17 г) и карбонат калия (0,3 г). Эту смесь перемешивают в течение 1 часа в атмосфере азота при комнатной температуре и оставляют стоять в течение 18 часов. Растворитель упаривают при пониженном давлении и остаток обрабатывают водой и диэтиловым эфиром. Органическую фазу отделяют, сушат (MgSO4) и упаривают с получением соединения VI.32 (0,095 г); М+ = 175;1H ЯМР: δ 2,45 (5H, м); 3,22 (2H, т); 3,90 (3H, с); 4,25 (5H, м); (масло).
ПРИМЕР VI.3
В данном примере описан способ получения 2-(4,4-дифторбут-3-енилтио)-4- метилоксазол-5-карбоновой кислоты (соединение VI.37).
Соединение VI.32 (0,4 г) растворяют в пропан-2-оле (10 см3), содержащем водный гидроксид натрия (2 см3 2М раствора) и перемешивают в течение 5 часов при комнатной температуре. Смесь упаривают при пониженном давлении и остаток разбавляют водой, экстрагируют этилацетатом, подкисляют разбавленной хлористоводородной кислотой и вновь экстрагируют этилацетатом (3 х 100 см3). Последние экстракты объединяют, промывают насыщенным солевым раствором, сушат (MgSO4) и упаривают при пониженном давлении с получением требуемого продукта в виде бесцветного твердого продукта (0,3 г). М+ = 249;1H ЯМР: δ 2,5 (5H, м); 3,27 (2H, т); 4,27 (1H, м); 6,5 (1H, ушир.с); (т.пл. 66-68oC). Натриевая соль этого соединения получена обработкой образца (0,7 г) раствором метоксида натрия в сухом метаноле (0,061 г металлического натрия, растворенного в метаноле (10 см3)) при комнатной температуре. Упаривание раствора при пониженном давлении дает натриевую соль соединения VI.37 в виде бесцветного твердого продукта; M+(FAB) = 271; (т.пл. 211-212oC).
По вышеуказанному способу получают следующее
соединение по изобретению:
(1) 2-(4,4-дифторбут-3-енилтио)-4-трифторметилоксазол-5- карбоновая кислота (соединение VI.36).1H ЯМР: δ 2,54 (2H, м); 3,32 (2H, т); 4,28 (1H, м);
7,65 (1H, ушир. с) из соединения VI.31.
ПРИМЕР VI.4
В данном примере описан способ получения 2-(4,4-дифторбут- 3-енилтио)-4-трифторметилоксазола (соединение VI.4).
2-Амино-4-трифторметилоксазол (0,84 г) в дихлорметане (25 см3), содержащем бис (4,4-дифторбут-3-енил)сульфид (2,71 г), перемешивают при 0oC и обрабатывают по каплям трет- бутилнитритом (0,62 г) в атмосфере азота. Реакционный раствор упаривают при пониженном давлении и остаток фракционируют с помощью хроматографии (силикагель, элюент - гексан) с получением соединения VI.4 (0,35 г). М+ = 259;1H ЯМР: δ 2,50 (2H, м); 3,26 (3H, т); 4,28 (1H, м); 7,95 (1H, кв); (масло).
По вышеуказанному способу получают
следующее соединение по изобретению:
(1) Этил 2-(4,4-дифторбут-3-енилтио)- 4-трифторметилоксазол-5- карбоксилат (соединение VI.31). МH+ = 322;1H ЯМР: δ 1,40 (3H,
т); 2,52 (2H, м); 3,30 (2H, м); 4,28 (1H, м); 4,43 (2H, кв); (масло) из этил 2-амино-4-трифторметилоксазол-5-карбоксилата (полученного из 1,1,1-трифторметилацетоацетата и мочевины по способу,
аналогичному примеру VI.2).
ПРИМЕР VI.5
В данном примере описан способ получения 2-(4,4-дифторбут-3-енилтио)-5-хлороксазола (соединение VI.13).
Соединение VI. 1 (2,0 г) растворяют в ацетонитриле (50 см3), содержащем N-хлорсукцинимид (1,50 г), и перемешивают при комнатной температуре в течение 24 часов. Смесь упаривают при пониженном давлении, экстрагируют гексаном (50 см3), фильтруют и фильтрат упаривают при пониженном давлении. Остаток фракционируют с помощью хроматографии (силикагель, элюент - 20%-ный диэтиловый эфир в гексане) с получением соединения VI.13 (0,75 г). М+ = 225;1H ЯМР: δ 2,46 (2H, м); 3,16 (2H, т); 4,28 (1H, м); 6,86 (1H, с); (масло).
По вышеуказанному
способу получают следующее соединение по изобретению из соединения VI.6:
(1) 2-(4,4-дифторбут-3-енилтио)-4-метил-5-хлороксазол (соединение VI. 15). МH+ = 239;1H ЯМР:
δ 2,05 (3H, с); 2,45 (2H, м); 3,15 (2H, т); 4,25 (1H, м); (масло).
ПРИМЕР VI.6
В данном примере описан способ получения 2-(4,
4- дифторбут-3-енилтио)-4-метилоксазол-5-карбоксамида (соединение VI.40).
Соединение VI. 32 (1,5 г) растворяют в метаноле (10 см3) и обрабатывают водным аммиаком (35 см3, плотность 0,88) при комнатной температуре. Смесь перемешивают в течение 5 часов, разбавляют солевым раствором и продукт экстрагируют этилацетатом (2 х 100 см3). Объединенные органические фазы промывают солевым раствором (4 х 50 см3), сушат (MgSO4) и упаривают при пониженном давлении; остаток промывают гексаном с получением соединения VI.40 (1 г). М+ = 248;1H ЯМР: δ 2,50 (5H, м); 3,26 (2H, т); 3,85 (3H, с); 4,28 (1H, м); 5,6, 6,0 (2H, ушир.с); (т.пл. 72-73oC).
ПРИМЕР VI.7
В
данном примере описан способ получения 5-циано-2-(4,4- дифторбут-3-енилтио)-4-метилоксазола (соединение VI.25).
Соединение VI.40 (0,64 г) растворяют в дихлорметане (10 см3), содержащем сухой пиридин (1 см3), при комнатной температуре и обрабатывают метансульфонилхлоридом (0,5 см3). Раствор перемешивают в течение 5 часов, пиридин (1 см3), выдерживают в течение 72 часов, прибавляют еще метансульфонилхлорид (0,25 см3) и пиридин (0,5 см3), перемешивают в течение 48 часов. Смесь обрабатывают разбавленной хлористоводородной кислотой и продукт экстрагируют этилацетатом. Объединенные органические фазы промывают солевым раствором и сушат (MgSO4). После фильтрации растворитель упаривают при пониженном давлении и остаток фракционируют с помощью хроматографии (силикагель, элюент -10%-ный этилацетат в гексане) с получением соединения VI.25 (0,46 г). М+ = 230;1H ЯМР: δ 2,32 (3H, с); 2,50 (2H, м); 2,58 (3H, с); 3,25 (2H, т); 3,85 (3H, с); 4,28 (1H, м); (масло).
ПРИМЕР VI.8
В данном примере описан способ получения
N-метилсульфонил-2-(4,4-дифторбут-3-енилтио)-4-метилоксазолкарбоксамида (соединение VI.38).
Натриевую соль соединения VI. 37 (0,52 г) перемешивают в гексане (6,5 см3) и обрабатывают оксалилхлоридом (0,275 г) при комнатной температуре. Смесь перемешивают в течение 6 часов, выдерживают в течение 18 часов и упаривают при пониженном давлении. Остаток, содержащий производное оксазолкарбонилхлорид, обрабатывают раствором метансульфонамида (0,20 г) в сухом бутан-2-оне (5 см3), нагревают с обратным холодильником при перемешивании в течение 18 часов, охлаждают до комнатной температуры, выдерживают в течение 18 часов. Смесь упаривают при пониженном давлении, остаток растворяют в воде, подкисляют 2М хлористоводородной кислотой и продукт экстрагируют диэтиловым эфиром (2 х 150 см3). Эфирные экстракты объединяют, промывают водным насыщенным раствором хлорида натрия, сушат (MgSO4), упаривают при пониженном давлении и остаток очищают с помощью хроматографии (силикагель, элюент ацетонитрил) с получением соединения VI.38 (0,20 г). М+ = 326;1H ЯМР: δ 2,50 (5H, м); 3,27 (2H, т); 3,40 (3H, с); 4,28 (1H, м); (т.пл. 60-62oC).
ПРИМЕР VI.9
В данном примере описан способ получения 2-(4,4-дифторбут-3-енилсульфинил)-5-фенилоксазола (соединение VI.19).
К раствору соединения VI. 18 (1 г) в дихлорметане (40 см3) прибавляют 3-хлорнадбензойную кислоту (1,3 г 50%-ной твердой по весу, (1 экв.)) и реакционную смесь перемешивают при комнатной температуре в течение 5 часов. Реакционную смесь выливают в смесь диэтилового эфира и водного бикарбоната натрия и слои разделяют. Органический слой сушат (MgSO4) и упаривают при пониженном давлении с получением белого твердого продукта, который очищают с помощью хроматографии на силикагеле, элюируя 1:4 этилацетатом: гексаном с получением соединения VI. 19 (0,567 г). M+ = 283;1H ЯМР: δ 2,50 (2H, м); 3,41 (2H, т); 4,29 (1H, м); 7,27 (1H, с); 7,38-7,55 (3H, м); 7,71 (2H, д); (масло).
По общему описанному выше способу из
соответствующих тио-эфиров, но используя 2 эквивалента 3-хлорнадбензойной кислоты получают следующие соединения по изобретению:
(1) 2-(4,4-дифторбут-3-енилсульфонил)-4-трифторметилоксазол
(соединение VI. 5).1H ЯМР: δ 2,65 (2H, м); 3,58 (2H, т); 4,28 (1H, м); 8,24 (1H, кв); (масло).
(2) 5-хлор-2-(4,4-дифторбут-3-енилсульфонил)оксазол (соединение VI.14). MNH4+ = 275;1H ЯМР: δ 2,60 (2H, м); 3,47 (2H, т); 4,26 (1H, м); 7,18 (1H, с); (масло).
(3) 5-хлор-2-(4, 4-дифторбут-3-енилсульфонил)-4- метилоксазол (соединение VI. 16).1H ЯМР: δ 2,25 (3H, с); 2,60 (2H, м); 3,45 (2H, т); 4,26 (1H, м); (масло).
(4) 2-(4, 4-дифторбут-3-енилсульфонил)-5-фенилоксазол (соединение VI.20). М+ = 299;1H ЯМР: δ 2,62 (2H, м); 3,51 (2H, т); 4,27 (1H, м); 7,26 (1H, с); 7,42-7,55 (3H, м); 7,69-7,79 (2H, д); (т.пл. 55-59oC).
ПРИМЕР VII.1
Данный пример иллюстрирует общий способ получения тиазолов, замещенных 4,4-дифторбут-3-енилтио группой по 2, 4 или 5-му
положению, исходя из соответствующим образом замещенного меркаптотиазола и подходящего дифторбут-1-енового алкилирующего агента. Это продемонстрировано следующим получением 2-(4,
4- дифторбут-3-енилсульфинил)-5-фенилоксазола (соединение VII.17).
К раствору 2-меркапто-5-фенилтиазола (0,483 г) в ацетоне (15 см3) прибавляют 4, 4-дифтор-3-бутенил-4-метилбензолсульфоната (0,7 г) и карбонат калия (0,369 г) и реакционную смесь нагревают с обратным холодильником в течение всего 8 часов, после чего часть исходного тозилата остается. Прибавляют еще 2-меркапто-5- фенилоксазола (0,05 г) и нагревание продолжают в течение 5 часов. Реакционную смесь охлаждают, выливают в диэтиловый эфир и воду и слои разделяют. Водный слой экстрагируют эфиром и объединенные органические фазы сушат (MgSO4) и упаривают при пониженном давлении с получением желтой жидкости. Хроматография на силикагеле с использованием 5%-ного трет-бутил- диметилового эфира в гексане дает соединение VII.17 (0,582 г). М+ = 283;1H ЯМР: δ 2,51 (2H, м); 3,31 (2H, т); 4,32 (1H, м); 7,26 (1H, с); 7,30-7,48 (3H, м); 7, 89 (2H, д); (масло).
По общему описанному выше способу получают следующие соединения по изобретению:
(1) 2-(4,4-дифторбут-3-енилтио)тиазол (соединение VI.5). М+ =
207;1H ЯМР: δ 2,47 (2H, м); 3,62 (2H, т); 4,27 (1H, м); 7,22 (1H, д); 7,68 (1H, д); (масло) из 2- меркаптотиазола.
(2) 2-(4,4-дифторбут-3-енилтио)тиазолин (соединение VII.134). М+ = 209;1H ЯМР: δ 2,40 (2H, м); 3,15 (2H, т); 3,4 (2H, т); 4,18-4,31 (1H, м); 4,2 (2H, т); (масло) из 2-меркаптотиазолина.
ПРИМЕР
VII.2
Данный пример иллюстрирует двухстадийный способ получения 2-(4,4-дифторбут-3-енилтио)-4-трифторметилтиазола (соединение VII.4).
Стадия 1. Получение
2-меркапто-4-трифторметилтиазола
1-Бром-3,3,3-трифторпропан-2-он (5,0 г) в трет-бутаноле (20 см3) обрабатывают дитиокарбаматом (2,9 г), смесь перемешивают при комнатной температуре
в течение 18 часов, выливают в воду, экстрагируют этилацетатом и органическую фазу сушат (MgSO4). Растворитель упаривают при пониженном давлении и остаток фракционируют с помощью
хроматографии (силикагель; элюент гексан:этилацетат от 17:3 до 7:3 по объему) с получением гидрата (2,16 г) требуемого меркаптотиазола. Часть этого продукта (1,0 г) прибавляют к толуолу (20 см3), содержащему пара-толуолсульфоновую кислоту (0,005 г, каталитическое количество) и нагревают с обратным холодильником в течение 4 часов. Образующуюся в реакции воду удаляют с помощью насадки
Дина-Старка. Раствор охлаждают до комнатной температуры, промывают водой, сушат (MgSO4), упаривают при пониженном давлении с получением требуемого промежуточного продукта (0,37 г).1H ЯМР: δ 7,10 (1H, с), 7,8 (1H, с).
Стадия 2. Получение соединения VII.4.
Продукт со стадии 1 (0,37 г) в ацетоне (14 см3), содержащий безводный карбонат калия (0,3 г) и 4-бром-1,1- дифторбут-1-ен (0,34 г) перемешивают и нагревают с обратным холодильником в течение 4 часов. Смесь охлаждают, выливают в воду, экстрагируют этилацетатом, сушат (MgSO4) и упаривают при пониженном давлении с получением Соединения VII.4 (0,30 г). М+ = 275;1H ЯМР: δ 2,50 (2H, м); 3,32 (2H, т); 4,28 (1H, м); 7,60 (1H, с); (масло).
По описанной выше стадии 2 получают следующие соединения по изобретению:
(1) этил 2-(4,4-дифторбут-3- енилтио)тиазол-4-карбоксилат (соединение VII. 8).1H ЯМР: δ 1,40 (3H, т); 2,48 (2H, м); 4,28 (1H, м); 4,40 (2H, кв); 8,03 (1H, с); (масло) из этил 2-меркаптотиазол-4-карбоксилата.
(2) метил 2-(4, 4-дифторбут-3-енилтио)-4-метилтиазол- 5-карбоксилат (соединение VII. 41).1H ЯМР: δ 2,48 (2H, м); 2,68 (3H, с); 3,26 (2H, т); 3,85 (3H, с); 4,28 (1H, м); (масло) из метил 2-меркапто-4-метилтиазол-5-карбоксилата.
(3) 2-(4,4-дифторбут-3-енилтио) -5-нитротиазол (соединение VII.47). М+ = 252;1H ЯМР: δ 2,52 (2H, м); 3,35 (2H, м); 4,27 (1H, м); 8,35 (1H, с); (масло) из 2-меркапто-5-нитротиазола (полученного из 2-бром-5-нитротиазола и тиомочевины).
ПРИМЕР VII.3
Данный пример иллюстрирует
трехстадийный способ получения 5-хлор-2-(4,4-дифторбут-3-енилтио)тиазола (соединение VII.24).
Стадия 1. 2-(4-бром-3,4,4-трифторбутилтио)тиазол
2-Меркаптотиазол (11,7 г) в
ацетоне (30 см3), содержащем 1,4- дибром-1,1,2-трифторбутан (27,0 г), обрабатывают безводным карбонатом калия (13,8 г) в атмосфере азота. Реакционную смесь перемешивают в течение 1,5 часа,
фильтруют и нерастворенный продукт промывают еще ацетоном (4 х 25 см3). Фильтрат упаривают при пониженном давлении и остаток фракционируют с помощью хроматографии (силикагель, элюент
- 10%-ный этилацетат в гексане) с получением 2-(4-бром-3,4,4-трифторбутилтио)тиазола (29,5 г).1H ЯМР: δ 2,2-2,5 (2H, м); 3,2-3,6 (2H, м); 4,7-5,0 (1H, м); 7,23 (1H, д); 7,68 (1H,
д).
Стадия 2. 2-(4-бром-3,4,4-трифторбутилтио)-5-хлортиазол
Продукт со стадии 1 (30,6 г) в дихлорметане (130 см3) обрабатывают при комнатной температуре
сульфурилхлоридом (9,6 см3) в дихлорметане (30 см3) в течение более 1 часа при перемешивании в атмосфере азота. Реакционную смесь перемешивают еще 1 час, медленно выливают в воду
(250 см3) и перемешивают в течение 0,25 часа. Органическую фазу отделяют, водную фазу экстрагируют дихлорметаном (2 х 75 см3), объединенные органические фазы промывают водным
раствором гидрокарбоната натрия, солевым раствором и сушат (MgSO4). Растворитель упаривают при пониженном давлении и остаток фракционируют с помощью хроматографии (силикагель; элюент 5%-ный
диэтиловый эфир в гексане) с получением 2-(4-бром-3,4,4-трифторбутилтио)-5-хлортиазола (28,0 г).1H ЯМР: δ 2,20-2,45 (2H, м); 3,25-3,50 (2H, м); 4,70-5,0 (1H, м); 7,45 (1H, с);
(масло).
Стадия 3. Соединение VII.24
Порошок цинка (33 г) в воде (600 см3) перемешивают с иодом (0,17 г, каталитическое количество), нагревают до 80oC и
прибавляют концентрированную хлористоводородную кислоту (0,5 см3) и далее соединение со стадии 2 (125 г) в течение более 1,5 часов в атмосфере азота. Еще порциями прибавляют цинк (16,6 г),
иод (0,1 г) и хлористоводородную кислоту (0,6 см3) в течение более 4 часов для завершения реакции. Смесь охлаждают до комнатной температуры, фильтруют через кизельгур с использованием
дихлорметана в качестве растворителя и фильтрат экстрагируют дихлорметаном (5 х 250 см3). Объединенные органические фазы сушат (MgSO4), упаривают при пониженном давлении и
остаток фракционируют с помощью хроматографии на силикагеле, элюируя гексаном с получением соединения VII.24 (140 г). М+ = 241;1H ЯМР: δ 2,42 (2H, м); 3,20 (2H, т); 4,25
(1H, м); 7,45 (1H, с); (масло).
ПРИМЕР VII.4
В данном примере описан способ получения 5-бром-2-(4,4-дифторбут-3-енилтио)тиазола (соединение VII.14).
Гидробромид 2-амино-5-бромтиазола (11 г) обрабатывают водным гидрокарбонатом калия, экстрагируют дихлорметаном (2 х 250 см3) и сушат (MgSO4). Смесь фильтруют и фильтрат прибавляют к бис- (4,4-дифторбут-3-енил)дисульфиду (20 г). По каплям к перемешиваемому раствору при комнатной температуре в атмосфере азота прибавляют трет-бутилнитрит (9,6 см3) в дихлорметане (40 см3). Реакционную смесь перемешивают в течение 18 часов, упаривают на силикагеле, остаток помещают на короткую колонку с силикагелем, которую элюируют (1) гексаном и (2) гексан: диэтиловым эфиром; 20:1 по объему с получением соединения VII.14 (6,4 г). М+ = 285;1H ЯМР: δ 2,46 (2H, м); 3,24 (3H, т); 4,27 (1H, м); 7,54 (1H, с); (масло).
ПРИМЕР VII.5
В данном примере описан способ получения 2-(4,4-дифторбут-3-енилтио)-4-метилтиазол-5-сульфонилфторида (соединение VII.52) с применением альтернативного способа
диазотирования, представленного в примере VII.4.
2-Амино-4-метилтиазолсульфонилфторид (1,5 г) в ацетонитриле (10 см3) прибавляют по каплям к перемешиваемой смеси трет-бутилнитрита (1,65 см3) и бис-(4,4-дифторбут-3- енил)дисульфида (2,25 г) в ацетонитриле (50 см3) при 60oC в атмосфере азота. Смесь нагревают 1 час, упаривают при пониженном давлении и остаток фракционируют с помощью хроматографии (силикагель, элюент гексан:этилацетат 4:1 по объему) с получением соединения VII.52 (1,84 г). М+ = 303;1H ЯМР: δ 2,50 (2H, м); 2,68 (3H, с); 3,32 (2H, т); 4,28 (1H, м); (масло).
По описанному выше способу получают следующие соединения по изобретению:
(1) 2-(4,
4-дифторбут-3-енилтио)-5-метилтиазол (соединение VII.21). М+ = 221;1H ЯМР: δ 2,45 (5H, м); 3,22 (2H, т); 4,26 (1H, м); 7,30 (1H, с); (масло) из
2- амино-5-метилтиазола.
(2) 5-хлор-2-(4,4-дифторбут-3-енилтио)тиазол (соединение VII.24). М+ = 241;1H ЯМР: δ 2,45 (2H, м); 3,22 (2H, т); 4,26 (1H, м); 3, 22 (H, т); 4,26 (1H, м); 7,45 (1H, с); (масло) из 2-амино-5-хлортиазола с применением альтернативного способа диазотирования, представленного в примере VII.3 выше.
(3) 5-хлор-2-(4, 4-дифторбут-3-енилтио)-4-метилтиазол (соединение VII. 27). М+ = 255;1H ЯМР: δ 2,35 (3H, с); 2,42 (2H, м); 3,18 (2H, т); 4,26 (1H, м); (масло) из 2- амино-5-хлор-4-метилтиазола.
ПРИМЕР VII.6
В данном примере описан двухстадийный способ получения этил 5-бром-2-(4,4-дифторбут-3-енилтио) тиазол-4-карбоксилата (соединение
VII.11).
Стадия 1. Получение этил 2-амино-5-бромтиазол-4-карбоксилата
Этил 2-амино-4-карбоксилат (5,0 г) (полученный из этилбромпирувата и тиомочевины по способу, описанному
в J. Med Chem., 1971, 14, 1075, из соответствующего оксазола) в концентрированной бромистоводородной кислоте (9 см3) перемешивают при комнатной температуре и обрабатывают по каплям бромом
(3,2 г), затем нагревают до 60oC в течение 2 часов, нейтрализуют карбонатом натрия и продукт экстрагируют этилацетатом. Органическую фазу сушат (MgSO4) и упаривают при пониженном
давлении с получением этил 2-амино-5-бромтиазол-4-карбоксилата (1,54 г). МH+ = 251;1H ЯМР: δ 1,40 (3H, т); 4,40 (2H, т); 5,6 (2H, ушир. с).
Стадия 2. Получение соединения VII.11.
Продукт со стадии 1 подвергали реакции диазотирования, как описано в примере VII. 5 выше, и получали соединение VII.11; М+ = 357;1H ЯМР: δ 1,40 (3H, т); 2,50 (2H, м); 3,30 (2H, т); 4,30 (1H, м); 4,25 (2H, кв); (масло).
ПРИМЕР VII.7
В данном примере описан трехстадийный способ получения N,N-диэтил
2-(4,4-дифторбут-3-енилтио)-4-метилтиазол-5-сульфонамида (соединение VII.56).
Стадия 1. N,N-диэтил 2-ацетамидо-4-метилтиазол-5-сульфонамид
2-Ацетамидо-4-метилтиазол-5-сульфонилхлорид (5,2 г) в тетрагидрофуране (100 см3) перемешивают при комнатной температуре и обрабатывают по каплям диэтиламином (4,5 см3). Смесь
перемешивают в течение 4 часов, упаривают при пониженном давлении и остаток экстрагируют этилацетатом (200 см3), промывают водой (2х100 см3), сушат (MgSO4) и вновь
упаривают при пониженном давлении с получением N, N-диэтил 2-ацетамидо-4-метилтиазол-5-сульфонамида (4,9 г).1H ЯМР: δ 1,20 (6H, т); 2,28 (3H, с); 2,57 (3H, с); 3,32 (2H, кв); 9,7
(1H, ушир.с); (твердый продукт).
Стадия 2. N,N-Диэтил 2-амино-4-метилтиазол-5-сульфонамид
Продукт со стадии 1 (2,5 г) растворяют в метаноле (25 см3) и охлаждают до
5oC при перемешивании в атмосфере азота. Прибавляют по каплям метоксид натрия в метаноле (2 см3 25%-ного раствора (вес/объем)) и смеси дают нагреться до комнатной температуры в
течение 18 часов. Реакционную смесь нагревают с обратным холодильником в течение 1 часа, охлаждают, разбавляют водой (250 см3), экстрагируют диэтиловым эфиром (2 х 100 см3),
сушат (MgSO4) и упаривают при пониженном давлении с получением N,N-диэтил 2-амино-4-метилтиазол-5-сульфонамида (0,48 г). М+ = 249;1H ЯМР: δ 1,20 (6H, т); 2,48
(3H, с); 3,28 (4H, кв); 5,25 (2H, ушир. с); (твердый продукт).
Стадия 3. Получение соединения VII.56.
Продукт со стадии 2 подвергали реакции диазотирования, как описано в примере VII. 5 выше, и получали соединение VII.56; М+ = 356;1H ЯМР: δ 1,15 (6H, т); 2,45 (2H, м); 2,60 (3H, с); 3,25 (2H, т); 3,26 (H, кв); 4,25 (1H, м); (масло).
ПРИМЕР VII.8
В данном примере описан способ получения 2-амино-5-(4,4- дифторбут-3-енилтио)тиазола (соединение VII.128).
Гидробромид 4, 4-дифторбут-3-енилизотиоурония (16,87 г) прибавляют к раствору гидроксида калия (18,0 г) в этаноле (150 см3) при комнатной температуре и перемешивают в течение 0,2 часа в атмосфере азота. Порциями прибавляют гидробромид 2-амино-5-бромтиазол (17,76 г) в этаноле (150 см3), смесь нагревают до 40oC в течение 2 часов, нейтрализуют хлористоводородной кислотой и упаривают при пониженном давлении. Остаток растворяют в 2М хлористоводородной кислоте, экстрагируют диэтиловым эфиром, подщелачивают 2М гидроксидом натрия и вновь экстрагируют диэтиловым эфиром. Последние эфирные экстракты объединяют, сушат (MgSO4) и упаривают при пониженном давлении с получением соединения VII. 128 (4,9 г). М+ = 222;1H ЯМР: δ 2,28 (2H, м); 2,67 (2H, т); 4,24 (1H, м); 5,3 (2H, два ушир. с); 7,08 (1H, с); (т. пл. 34,6-35,4oC).
ПРИМЕР VII.9
В данном примере описан способ получения 5-(4,
4- дифторбут-3-енилтио)тиазола (соединение VII.82).
Соединение VII.128 (0,30 г) растворяют в сухом тетрагидрофуране (14 см3) и нагревают с обратным холодильником в атмосфере азота. Прибавляют по каплям трет-бутилнитрит (0,52 см3) в тетрагидрофуране (8 см3) в течение 0,25 ч. Смесь нагревают в течение 2 часов. Прибавляют еще трет-бутилнитрит (0,52 см3) и нагревание продолжают еще в течение 2 часов. Раствор охлаждают, упаривают при пониженном давлении и остаток фракционируют с помощью хроматографии (силикагель, элюент гексан:этилацетат 1:1 по объему) с получением соединения VII. 128 (0,10 г).1H ЯМР: δ 2,28 (2H, м); 2,82 (2H, т); 4,24 (1H, м); 2,82 (2H, т); 4,24 (1H, м); 7,86 (1H, с); 8,86 (1H, с); (масло).
ПРИМЕР VII.10
В данном примере описан способ получения 2-хлор-5-(4,4- дифторбут-3-енилтио)тиазола (соединение VII.114).
Соединение VII.128 (4,0 г) в ацетонитриле (50 см3) прибавляют при 0oC к перемешиваемой смеси хлорида меди (2) (5,38 г) и трет-бутилнитрита (3,71 г) в ацетонитриле (50 см3) и дают медленно нагреться до комнатной температуры в течение более 18 часов. Растворитель упаривают при пониженном давлении, продукт растворяют в диэтиловом эфире, фильтруют и фильтрат вновь упаривают с получением желто-коричневой жидкости, который фракционируют с помощью хроматографии (силикагель, элюент гексан:диэтиловый эфир 4:1 по объему) с получением соединения VII.114 (2,43 г). М+ = 241;1H ЯМР: δ 2,28 (2H, м); 2,68 (2H, т); 4,22 (1H, м); 7,52 (1H, с); (масло).
ПРИМЕР VII.10
В данном примере описан способ получения 2-(4-цианфенокси)-5-(4,
4-дифторбут-3-енилтио)тиазола (соединение VII.130).
Соединение VII.114 (0,483 г), 4-цианфенол (0,238 г), безводный карбонат калия (0,276 г) и фторид цезия (0,304 г) в N-метилпирролидин-2-оне (3 см3) перемешивают вместе в атмосфере азота и нагревают при 90oC в течение 36 часов. Реакционную смесь разбавляют водой, продукт экстрагируют диэтиловым эфиром, сушат (MgSO4), упаривают при пониженном давлении и остаток фракционируют с помощью тонкослойной хроматографии (силикагель, элюент гексан:диэтиловый эфир 4:1 по объему) с получением соединения VII.130 (0,125 г). М+ = 324;1H ЯМР: δ 2,28 (2H, м); 2,80 (2H, т); 4,20 (1H, м); 7,40 (2H, м); 7,75 (2H, м); (масло).
ПРИМЕР VII.12
В
данном примере описан способ получения 5-(4,4-дифторбут-3-енилтио)тиазол-4-карбоксилата (соединение VII.98).
Этилизоцианат (2,3 г) в сухом тетрагидрофуране (15 см3) прибавляют к перемешиваемой смеси трет-бутоксида (2,24 г) при -40oC в атмосфере азота. Через 10 минут реакционную смесь охлаждают до -78oC и медленно прибавляют дисульфид углерода (1,52 г) в тетрагидрофуране (20 см3). После окончания прибавления реакционной температуре дают подняться до -10oC и прибавляют 4, 4-дифторбут-3-енил-4-метилбензолсульфамид (5,24 г) в тетрагидрофуране (10 см3). Смеси дают нагреться до комнатной температуры и перемешивают в течение 24 часов, нагревают с обратным холодильником в течение 3 часов и охлаждают до комнатной температуры. Реакционную смесь выливают в водную 2М хлористоводородную кислоту и продукт экстрагируют этилацетатом. Органические фазы сушат (MgSO4) и растворитель удаляют упариванием при пониженном давлении. Колоночная хроматография остатка на силикагеле, элюируя 1:1 гексаном:этилацетатом с получением соединения VII.98 (3,15 г).1H ЯМР: δ 1,45 (3H, т); 2,50 (2H, м); 3,10 (2H, т); 4,3-4,4 (1H, м); 4,50 (2H, кв); 8,65 (1H, с); (масло).
ПРИМЕР VI.13
В данном примере описан способ
получения 5-(4,4- дифторбут-3-енилтио)-тиазол-4-карбоксамида (соединение VII.94).
Соединение VII.98 (0,5 г) в метаноле (8 см3) перемешивают с водным аммиаком (35 см3, плотность 0,88) в течение 4 часов. Соединение VII.94 получают в виде твердого продукта, который отфильтровывают от раствора и отсасывают досуха (0,27 г).1H ЯМР: δ 2,50 (2H, м); 3,00 (2H, т); 4,30-4,41 (1H, м); 5,5 и 7,0 (2H, ушир. с); (т.пл. 141-142oC).
По вышеуказанному способу получают следующее соединение по изобретению:
(1) 2-(4,
4-дифторбут-3-енилтио) тиазол-4-карбоксамид (соединение VII.7). М+ = 250;1H ЯМР: δ 2,50 (2H, м); 3,27 (2H, т); 4,28 (1H, м); 5,9 и 7,1 (2H, ушир. с); 8,03 (1H, с); (т.пл.
57-58oC) из соединения VII.8.
(2) 2-(4,4-дифторбут-3-енилтио)-4-метилтиазол-5- карбоксамид (соединение VII. 36).1H ЯМР: δ 2,48 (2H, м); 2,66 (3H, с); 3, 26 (2H, т); 3,85 (3H, с); 4,28 (1H, м); 5,7 (2H, ушир. с); (т.пл. 99-100oC) из соединения VII.41.
ПРИМЕР VI. 14
В данном примере описан способ получения
4-циано-5-(4,4-дифторбут-3-енилтио)тиазола (соединение VII.90).
Соединение VII. 94 (0,27 г) в сухом дихлорметане (13 см3) обрабатывают пиридином (1 см3) и метансульфурилхлоридом (0,26 см3). Смесь перемешивают в течение 5 дней, затем прибавляют метансульфурилхлорид (0,2 см3) и опять перемешивают в течение 2 часов. Реакционную смесь выливают в водную 2М хлористоводородную кислоту и продукт экстрагируют этилацетатом. Органическую фазу сушат (MgSO4) и упаривают при пониженном давлении. Хроматография остатка на силикагеле дает соединение VII.90 (0,142 г).1H ЯМР: δ 2,40 (2H, м); 3,20 (2H, т); 4,30 (1H, м); 8,80 (1H, с).
По вышеуказанному способу получают следующее соединение по
изобретению:
(1) 4-циано-2-(4,4-дифторбут-3- енилтио)тиазол (соединение VII.6).1H ЯМР: δ 2,50 (2H, м); 3,32 (2H, т); 4,26 (1H, м); 7,86 (1H, с); (масло) из соединения
VII.7.
(2) 5-циано-2-(4,4-дифторбут-3-енилтио)-4-метилтиазол (соединение VII. 32).1H ЯМР: δ 2,48 (2H, м); 2,58 (3H, с); 3,85 (3H, с); 4,28 (1H, м); (масло) из соединения VII.36.
ПРИМЕР VII.15
В данном примере описан способ получения 5-бром- 2-(4,4-дифторбут-3-енилтио)тиазол-4-карбоновой кислоты (соединение VII.13).
Соединение VII.11 (0,30 г) в метаноле (5 см3), содержащем водный раствор гидроксида натрия (1,2 см3 2М раствора), перемешивают при комнатной температуре в течение 18 часов, выливают в водную 2М хлористоводородную кислоту. Продукт экстрагируют этилацетатом, сушат (MgSO4) и упаривают при пониженном давлении с получением соединения VII.13 (0,18 г). М+ = 239;1H ЯМР: δ 2,48 (2H, м); 3,30 (2H, т); 4,28 (1H, м); 7,0 (1H, широкий сигнал); (т.пл. 86,5-87,5oC).
По вышеуказанному способу из соответствующего
эфира получают следующее соединение по изобретению:
(1) 2-(4,4-дифторбут-3-енилтио)тиазол-4-карбоновая кислота (соединение VII. 10). М+ = 251;1H ЯМР: δ 2,50 (2H,
м); 3,35 (2H, т); 4,28 (1H, м); 8,18 (1H, с); (т.пл. 114- 115oC) из соединения VII.8.
(2) 2-(4,4-дифторбут-3-енилтио)-4-метилтиазол-5-карбоновая кислота (соединение VII. 45). М+ = 265;1H ЯМР: δ 2,50 (2H, м); 2,70 (3H, с); 3,27 (2H, т); 4,28 (1H, м); 9,8 (1H, широкий сигнал); (т.пл. 52,0-53,5oC) из соединения VII.41.
(3) 5-(4,4-дифторбут-3-енилтио)-тиазол-4-карбоновая кислота (соединение VII.102). М+ = 251;1H ЯМР: δ 2,50 (2H, м); 3,10 (2H, т); 4,28 (1H, м); 8,70 (1H, с); (т.пл. 128,5oC из соединения VII.98).
ПРИМЕР VII.15
В данном примере описаны способы, подходящие для получения соединений по изобретению, в которых атом серы из 4,
4-дифтор-3-енилтио заместителя соответствующего неокисленного соединения окисляют до сульфоксида (сульфинила) или сульфона (сульфонила).
Метод A: Использование пероксимоносульфата калия в качестве окислителя.
Получение 5-хлор-2-(4,4-дифторбут-3-енилсульфонил) тиазола (соединение VII.26)
Перемешиваемый раствор соединения VII.24 (4,83 г) в метаноле (50
см3) при 8oC обрабатывают по каплям пероксимоносульфатом калия (27,0 г) в воде (100 см3) при охлаждении в течение более 0,25 часов и прибавляют еще метанол (50 см3). Реакционную смесь перемешивают при комнатной температуре в течение 18 часов, нерастворимые продукты отфильтровывают, фильтрат экстрагируют дихлорметаном (4 х 50 см3) и сушат
(MgSO4). Растворитель удаляют при пониженном давлении и остаток фракционируют с помощью хроматографии (силикагель, элюент гексан:этилацетат 4:1 по объему) с получением соединения VII.26 (3,
91 г). M(NH4)+ = 291;1H ЯМР: δ 2,60 (2H, м); 3,50 (2H, т); 4,25 (1H, м); 7,85 (1H, с); (масло).
Метод B: Использование мономагниевой соли пероксифталевой кислоты.
Получение 5-бром-2-(4,4-дифторбут-3-енилсульфинил) тиазола (соединение VII.15)
Соединение VII. 14 (1,50 г) растворяют в дихлорметане (10 см3) и обрабатывают гексагидратом мономагниевой соли пероксифталевой кислоты (1,6 г, 80% надкислота) и водой (15 см3). Смесь перемешивают при комнатной температуре в течение 1 часа,
разбавляют дихлорметаном (90 см3) и органическую фазу промывают водным гидрокарбонатом натрия и водой. Органическую фазу сушат (MgSO4), упаривают при пониженном давлении и
остаток фракционируют с помощью хроматографии (силикагель, элюент гексан : этилацетат 10 : 1 по объему) с получением соединения VII.15 (1,0 г). M(NH4)+ = 321;1H ЯМР:
δ 2,38 (1H, м); 2,60 (2H, м); 4,20 (1H, м); 7,85 (1H, с); (масло).
Из соответствующих тиоэфиров получают следующие соединения по изобретению, применяя метод B, описанный
выше:
(1) 2-(4,4-дифторбут-3-енилсульфинил)тиазол (соединение VII.2). MH+ = 224;1H ЯМР: δ 2,36 (1H, м); 2,50-2,70 (1H, м); 3,20 (2H, м); 4,22 (1H, м); 7,67 (1H,
д); 7,98 (1H, д); (масло) из соединения VII.1 и одного эквивалента окислителя.
(2) 2-(4,4-дифторбут-3-енилсульфонил)тиазол (соединение VII.3). MH+ = 240;1H ЯМР: δ 2,55 (2H, м); 3,45 (2H, т); 4,24 (1H, м); 7,78 (1H, д); 8,08 (1H, д); (масло) из соединения VII.1 и двух эквивалентов окислителя.
(3) 5-бром-2-(4,4-дифторбут-3-енилсульфонил) тиазол (соединение VII.6). M(NH4)+ = 335;1H ЯМР: δ 2,58 (2H, м); 3,46 (2H, т); 4,25 (1H, м); 7,96 (1H, с); (масло) из соединения VII.14 и двух эквивалентов окислителя.
(4) 2-(4,4-дифторбут-3-енилсульфинил)-5-метилтиазол (соединение VII.22).1H ЯМР: δ 2,38 (1H, м); 2,50-2,65 (4H, м); 3,15 (1H, м); 4,23 (1H, м); 7,60 (1H, кв); (масло) из соединения VII.21 и одного эквивалента окислителя.
(5) 2-(4,4-дифторбут-3-енилсульфонил)-5-метилтиазол (соединение VII.23).1H ЯМР: δ 2,55 (2H, м); 3, 45 (2H, т); 4,25 (1H, м); 7,73 (1H, кв); (масло) из соединения VII.21 и двух эквивалентов окислителя.
(6) 5-хлор-2-(4,4-дифторбут-3-енилсульфинил)тиазол (соединение VII.25). M(NH4)+ = 240;1H ЯМР: δ 2,38 (1H, м); 3,18 (2H, м); 4,25 (1H, м); 7,74 (1H, с); (масло) из соединения VII.24 и одного эквивалента окислителя.
(7) 5-(4, 4-дифторбут-3-енилсульфинил)тиазол (соединение VII.83).1H ЯМР: δ 2,50 (2H, м); 3,05 (1H, м); 3,20 (1H, м); 4,28 (1H, м); 8,20 (1H, с); 9,12 (1H, с); (масло) из соединения VII.82 и одного эквивалента окислителя.
(8) 5-(4,4-дифторбут-3-енилсульфонил)тиазол (соединение (VII. 84).1H ЯМР: δ 2,50 (2H, м); 3,30 (2H, м); 4,25 (1H, м); 8,20 (1H, с); 9, 12 (1H, с); (масло) из соединения VII.82 и двух эквивалентов окислителя.
(9) 2-хлор-5-(4,4-дифторбут-3-енилсульфинил)тиазол (соединение VII.115).1H ЯМР: δ 2,50 (2H, м); 3,05 (1H, м); 3,20 (1H, м); 4,28 (1H, м); 7,85 (1H, с); (масло) из соединения VII.114 и одного эквивалента окислителя.
(10) 2-хлор-5-(4,4-дифторбут-3-енилсульфонил)тиазол (соединение VII. 116).1H ЯМР: δ 2,52 (2H, м); 3,30 (2H, м); 4,28 (1H, м); 8,08 (1H, с); (масло) из соединения VII.114 и двух эквивалентов окислителя.
Метод C: Использование 3-хлорнадбензойной кислоты.
Из соответствующих тиоэфиров получают следующие соединения по изобретению, применяя метод В, описанный выше, но с использованием
3-хлорнадбензойной кислоты вместо гексагидратов мономагниевой соли пероксифталевой кислоты:
(11) этил 2-(4,4-дифторбут-3-енилсульфинил)тиазол-4- карбоксилат (соединение VII. 9). М+
= 311;1H ЯМР: δ 1,43 (3H, т); 2,60 (2H, м); 3,56 (2H, т); 4,28 (1H, м); 4,48 (2H, кв); 8,50 (1H, с); (т.пл. 64-64oC) из соединения VII.8 и двух эквивалентов
окислителя.
(12) этил 5-бром-2-(4,4-дифторбут-3-енилсульфонил)тиазол- 4-карбоксилат (соединение VII. 12). М+ = 389;1H ЯМР: δ 1,43 (3H, т); 2,60 (2H, м); 3,56 (2H, т); 4,28 (1H, м); 4,48 (2H, кв); (т.пл. 72-73oC) из соединения VII.11 и двух эквивалентов окислителя.
(13) 5-хлор-2-(4,4-дифторбут-3-енилсульфонил)-4- метилтиазол (соединение VII. 28).1H ЯМР: δ 2,55 (2H, м); 2,45 (3H, с); 3,40 (2H, т); 4,25 (1H, м); (масло) из соединения VII. 27 и двух эквивалентов окислителя.
(14) метил 2- (4, 4-дифторбут-3-енилсульфонил)-4- метилтиазол-5-карбоксилат (соединение VII. 43).1H ЯМР: δ 2,60 (2H, м); 2,85 (3H, c); 3,50 (2H, т); 3,95 (3H, с); 4,25 (1H, м); (масло) из соединения VII.41 и двух эквивалентов окислителя.
(15) 2-(4,4-дифторбут-3-енилсульфонил)-4-метилтиазол-5- сульфонилфторид (соединение VII. 53).1H ЯМР: δ 2,60 (2H, м); 2,85 (3H, с); 3,55 (2H, т); 4,28 (1H, м); (т.пл. 67oC) из соединения VII.52 и двух эквивалентов окислителя.
ПРИМЕР VIII.1
В данном примере описан способ
получения 2-(4,4-дифторбут- 3-енилтио)-1-метилимидазола (соединение VIII, 5).
К раствору 2-меркапто-1-метилимидазола (9,78 г) в ацетоне (300 см3) прибавляют карбонат калия (14,2 г) и 4-бром-1,1- дифторбут-1-ен (16,12 г) в виде раствора в ацетоне (100 см3). Смесь нагревают с обратным холодильником в течение 18 часов и оставляют остывать. Нерастворимые твердые продукты удаляют фильтрацией реакционной смеси через плаг из силикагеля sorbsil-C30, промывая этилацетатом. Фильтрат упаривают при пониженном давлении с получением соединения VIII. 5 (17,8 г), которое подходит для дальнейшей реакции (см. пример VIII.7).
Часть (1 г) очищают с помощью хроматографии на силикагеле sorbsil-C30, элюируя этилацетатом: гексаном 3: 7, и получают чистое соединение VIII.5 (0,776 г).1H ЯМР: δ 2,3-2,4 (2H, м); 3,05-3,15 (2H, т); 3,60 (3H, с); 4,15-4,35 (1H, м); 7,05 (1H, с); (масло).
Используя соответствующие
меркаптоимидазолы, получают следующие соединения по изобретению, применяя способ, описанный выше:
(1) 2-(4,4-дифторбут-3-енилтио)-1-фенилимидазол (соединение VIII.3). M+ = 266;1H ЯМР: δ 2,3-2,4 (2H, м); 3,1-3,15 (2H, т); 4,1-4,25 (1H, м); 7,1-7,2 (2H, м); 7,3-7,55 (5H, м); (масло).
(2) 2-(4,4-дифторбут-3-енилтио)-1-этилимидазол (соединение VIII.10). M+ = 218;1H ЯМР: δ 1,4 (3H, т); 2,3-2,4 (2H, м); 3,15 (2H, т); 4,0 (2H, кв); 4,2-4,35 (1H, м); 6,95 (1H, с); 7,1 (1H, с); (масло).
(3) 2-(4, 4-дифторбут-3-енилтио)-4-этил-5-метилимидазол (соединение VIII. 27). M+ = 232;1H ЯМР: δ 1,15 (3H, т); 2,15 (3H, с); 2,35-2,45 (2H, м); 2,5-2,6 (2H, кв); 2,95 (2H, т); 4, 1-4,3 (1H, м); (т. пл. 54-56oC). 2-(4,4-дифторбут-3-енилтио)-4-метилимидазол (соединение VIII.58). M+ = 204;1H ЯМР: δ 2,25-2,35 (5H, м); 3,0 (2H, т); 4,15-4,3 (1H, м); 6,75 (1H, с); (масло).
(5) 2-(4,4-дифторбут-3-енилтио)-4-этоксикарбонилимидазол (соединение VIII.64). M+ = 262;1H ЯМР: δ 1,3-1,35 (3H, т); 2, 25-2,35 (2H, м); 3,05-3,15 (2H, т); 4,1- 4,25 (1H, м); 4,3-4,4 (2H, кв); 7,8 (1H, ушир. с); (т.пл. 57,8-61oC).
(6) 3-(4,4-дифторбут-3-енилтио)-[1,5-а] -пиридин (соединение VIII.151).1H ЯМР: δ 2,30 (2H, м); 3,00 (2H, т); 4,20 (1H, м); 6,65 (1H, м); 6,80 (1H, м); 7,45 (1H, дт); 7,55 (1H, с); 8,15 (1H, дд); (масло) из 2,3-дигидроимидазо-[1, 5-a]-пиридин-3-тиона.
ПРИМЕР VIII.2
В данном примере описан способ получения 2-(4,4-дифторбут- 3-енилтио)имидазола (соединение VIII. 1) и 1-(4,4-дифтоpбут-3- енилтио)-2-(4,
4-дифторбут-3-енилтио)имидазола (соединение VIII. 8) в виде смеси продуктов, которые можно разделить хроматографически.
К раствору 2-меркаптоимидазола (10,01 г) в ацетоне (400 см3) прибавляют карбонат калия (20,73 г) и 4-бром-1,1- дифторбут-1-ен (16,79 г). Смесь нагревают с обратным холодильником в течение 18 часов и оставляют остывать. Нерастворимые твердые продукты удаляют фильтрацией реакционной смеси через плаг из силикагеля sorbsil-C30, промывая этилацетатом. Фильтрат упаривают при пониженном давлении с получением бледно-коричневого масла (19,2 г), которое хроматографируют на силикагеле, элюируя 15%-ным этилацетатом в гексане, поднимая до 50% этилацетата в гексане. Получают две основные фракции, первая из которых (7,4 г), согласно данным ТСХ, содержит два продукта. Вторая фракция (10,75 г), полученная в виде белого твердого продукта, как показано, является чистым соединением VIII.1. Первую фракцию подвергают дальнейшей хроматографии, как описано выше, с получением соединения VIII.8 (1,61 г). M+ = 280;1H ЯМР: δ 2,3-2,45 (4H, м); 3,1-3,15 (2H, т); 3,9-4,0 (2H, т); 4,05-4,3 (1H, с); 7,05 (1H, с); (масло), и соединения VIII.1 (5,31 г). Образец соединения VIII.1 перекристаллизовывали из этилацетата и гексана с получением 4,2 г, которые имели M+ = 190;1H ЯМР: δ 2,3-2,4 (2H, м); 3,0-3,1 (2H, т); 4,15-4,3 (1H, м); 7,0-7,1 (2H, ушир. с); 9,2 (1H, ушир. с); (т.пл. 58,6- 59,6oC).
ПРИМЕР VIII.3
В данном примере описан способ получения 2-(4,
4-дифторбут- 3-енилтио)-4-фенилимидазола (соединение VIII.19).
Фенилбромид (1,611 г) в хлороформе (7 см3) прибавляют к гидробромиду 4,4-дифторбут-3-енилизотиоурония (2 г) в 84% этаноле/воде (20 см3) и при перемешивании медленно прибавляют бикарбонат натрия (2,72 г). Полученную желтую суспензию нагревают с обратным холодильником в течение 3 часов. Смесь охлаждают и растворитель удаляют упариванием при пониженном давлении. Остаток дважды промывают теплой водой (2 х 20 см3), который декантируют от нерастворимых твердых продуктов. Полученный таким образом сырой продукт очищают хроматографией на силикагеле, элюируя 20%-ным этилацетатом в гексане, и продукт, содержащий фракции, вновь пропускают через колонку с использованием 5%-ного этилацетата в толуоле. Это дает соединение VIII.19 (0,78 г). M+ = 266;1H ЯМР: δ 2,3-2,4 (2H, м); 3,05-3,1 (2H, т); 4,15-4,3 (1H, м); 7,2-7,4 (4H, м); 7,6-7,75 (2H, ушир. с); не содержащее побочного продукта, соответствующее N-фенацилпроизводное.
Применяя способ, описанный выше, получают следующие соединения по изобретению:
(1) 2-(4,
4-дифторбут-3-енилтио)-1-метил-4-фенилимидазол (соединение VIII. 22). M+ = 280;1H ЯМР: δ 2,35-2,45 (2H, м); 3,10-3,18 (2H, т); 3,65 (3H, с); 4,19-4,35 (1H, м); 7,2-7,25
(2H, м); 7,33-7,38 (2H, т); 7,72- 7,78 (2H, д); (масло), используя гидробромид N-метил- 4,4-дифторбут-3-енилизотиоурония.
(2) 2-(4, 4-дифторбут-3-енилтио)-5-этил-4-метоксикарбонил- 1-метилимидазол (соединение VIII.68). М+ = 290;1H ЯМР: δ 1,21-1,25 (3H, т); 2,35-2,45 (2H, м); 2,9-2,9 (2H, кв); 3,2-3,25 (2H, т); 3,78 (3H, с); 3,85 (3H, с); 4,16-4,32 (1H, м); (масло), используя гидробромид N-метил-4,4-дифторбут-3-енилизотиоурония и метил 2 бром-3-оксопентаноат.
ПРИМЕР VIII.4
В данном примере описан способ получения 2-(4,4-дифторбут- 3-енилтио)-4,5-диметилфенилимидазола (соединение VIII.29).
Карбонат калия (18,9 г) и гидробромид 4, 4-дифторбут-3- енилизотиоурония (16,9 г) прибавляют к раствору 3-бромбутан-2- она (10,32 г) в диметилформамиде (100 см3) и смесь перемешивают при 60oC в течение 90 минут, затем при 80oC в течение 30 минут. Полученную смесь охлаждают, прибавляют воду (100 см3) и продукт экстрагируют диэтиловым эфиром. Объединенные органические фазы промывают водой и солевым раствором и сушат (MgSO4). Концентрирование упариванием при пониженном давлении дает бледно-оранжевую жидкость (12,2 г), которую очищают хроматографией на силикагеле sorbital C30, элюируя 30%-ным этилацетатом в гексане. Получают три компонента. Первый элюированный продукт был идентифицирован как 3-(4,4-дифторбут-3-енилтио)бутан-2-он (0,68 г). M+ = 194;1H ЯМР: δ 1,4 (3H, д); 2,15-2,3 (5H, м); 2,45 (2H, т); 3,3-3,4 (1H, кв); 4,1-4,35 (1H, м). Второй элюированный продукт был требуемым соединением VIII. 29 (4,1 г). M+ = 218;1 H ЯМР: δ 2,15 (6H, с); 2,25-2,35 (2H, м); 2,9-3,0 (2H, т); 4,15-4,3 (1H, м); (т.пл. 81,4-84,4oC). Третье элюированное соединение было идентифицировано как продукт дальнейшего N-алкилирования соединением VIII.29 дополнительным количеством 3-бромбутан-2-она, а именно, как N-(1-метилпропан-2-он)-2-(4,4-дифторбут-3-енилтио)-4,5- диметилимидазолабутан-2-он (1,11 г). M+ = 217;1H ЯМР: δ 1,55 (3H, с); 2,0 (3H, с); 2,05 (3H, с); 2,15 (3H, с); 2,3-2,4 (2H, м); 3,0-3,05 (2H, т); 4,15-4,35 (1H, м); 5,0-5,1 (1H, м); (масло).
ПРИМЕР VIII.5
В данном примере описан способ получения 2-(4,4-дифторбут- 3-енилтио)-1-пропилимидазола (соединение VIII. 12) из соответствующего N-H имидазола, соединение VIII.1, путем
алкилирования с использованием пропилиодида.
Соединение VIII. 1 (2 г) прибавляют по частям (вскипание) к суспензии гидрида натрия (0,73 г 60%-ного твердого продукта в масле) в диметилформамиде (20 см3) в атмосфере азота. После перемешивания в течение 30 минут прибавляют н-пропил иодид (2,68 г) и реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Прибавляют затем воду и диэтиловый эфир и продукт экстрагируют эфиром. Объединенные органические фазы промывают водой и насыщенным солевым раствором и сушат (MgSО4). После фильтрации удаляют растворитель упариванием при пониженном давлении с получением сырого продукта (2,7 г), который очищают хроматографией на силикагеле sorbital C30, элюируя этилацетатом : гексаном 3 : 7 и получая соединение VIII.12 (2,15 г). M+ = 232;1H ЯМР: δ 0,9-0,95 (3H, т); 1,75-1,85 (2H, м); 2,3-2,45 (2H, м); 3,1-3,15 (2H, т); 4,15-4,35 (1H, м); 6,9 (1H, с); 7,1 (1H, с); (масло).
Применяя способ, описанный выше, используя подходящий алкилирующий агент и исходный N-H имидазол, получают следующие соединения по изобретению:
(1) 2-(4,
4-дифторбут-3-енилтио)-1-(1-метилэтил) имидазол (соединение VIII. 14). M+ = 232;1H ЯМР: δ 1,4 (6H, д); 2,3-2,4 (2H, м); 3,1-3,15 (2H, т); 4,15-4,3 (1H, м); 4,5-4,6 (1H,
м); 7,0 (1H, с); 7,1 (1H, с); (масло) из соединения VIII.1.
(2) 2-(4,4-дифторбут-3-енилтио)-1,4,5-триметилимидазол (соединение VIII. 31).1H ЯМР: δ 2,15 (6H, два с); 2,25-2,35 (2H, м); 2,95 (2H, т); 3,5 (3H, с); 4,15-4,3 (1H, м); (масло) из соединения VIII.29.
(3) 1-этил-2-(4,4-дифторбут-3-енилтио)-4,5-диметилимидазол (соединение VIII. 33).1H ЯМР: δ 1,25 (3H, т); 2,15 (6H, ушир. с); 2,3-2,4 (2H, м); 3,0 (2H, т); 3,9-3,95 (2H, кв); 4,15-4,3 (1H, м); (масло) из соединения VIII.29.
(4) смесь 2-(4, 4-дифторбут-3-енилтио)-1,5-диметилимидазол (соединение VIII.35) и 2-(4,4-дифторбут-3-енилтио)-1,4-диметилимидазол (соединение VIII. 60).1H ЯМР: δ 2,15 (3H, с); 2,3-2,4 (2H, м); 3, 0-3,05 (2H, т); 3,5 и 3,55 (3H, два с); 4,15-4,3 (1H, м); 6,65 и 6,80 (1H, два с); из соединения VIII. 58.
(5) смесь 2- (4,4-дифторбут-3-енилтио)-5-метил-1-(1-метилэтил)-имидазол (соединение VIII. 37) и 2-(4,4-дифторбут-3-енилтио)-4-метил-1-(1- метилэтил)-имидазол (соединение VIII.62); как показано, главным является последний изомер,1H ЯМР: δ 1,35 (6H, д); 2, 2 (3H, с); 2,3-2,4 (2H, м); 3,05 (2H, т); 4,15-4,3 (1H, м); 4,5-4,6 (1H, м); 6,70 (1H, с); из соединения VIII.58.
(6) 2-(4,4-дифторбут-3-енилтио)-5-этоксикарбонил-1-метилимидазол (соединение VIII.52); M+ = 276;1H ЯМР: δ 1,32-1,40 (3H, т); 2,73-2,47 (2H, м); 3,2-3,3 (2H, т); 3,82 (3H, с); 4,17-4,32 (1H, м); 4,25-4,35 (2H, кв); 7,70 (1H, с); (масло) из соединения VIII.64. По данной реакции получают хроматографически разделяемую смесь соединения VIII.52 и VIII.65, первое элюируется первым.
(7) 2-(4, 4-дифторбут-3-енилтио)-4-этоксикарбонил-1- метилимидазол (соединение VIII.65). M+ = 276;1H ЯМР: δ 1,35-1,40 (3H, т); 2,35-2,42 (2H, м); 3,20-3,27 (2H, т); 3,63 (3H, с); 4, 15-4,30 (1H, м); 4,3-4,4 (2H, кв); 7,60 (1H, с); (масло) из соединения VIII.64.
ПРИМЕР VIII.6
В данном примере описан способ получения 2-(4,
4-дифторбут- 3-енилтио)-N-(метансульфонил)имидазола (соединение VIII.18)
Раствор соединения VIII.1 (0,49 г) в сухом тетрагидрофуране (3 см3) прибавляют по каплям к суспензии
гидрида натрия (55% в масле, 0,12 г, промытый гексаном перед использованием) в сухом тетрагидрофуране (5 см3), охлажденном на холодной водяной бане. Реакционной смеси дают перемешиваться
при комнатной температуре в течение 2 часов и затем прибавляют метансульфонилхлорид (0,3 г) и реакционную смесь перемешивают еще в течение 16 часов. Реакционную смесь выливают в этилацетат/воду и слои
разделяют. Водный слой экстрагируют этилацетатом и органические фазы сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением желтого масла. Очистка колоночной
хроматографией на силикагеле с использованием 3 : 7 этилацетата : гексана дает (соединение VIII. 18 (0,31 г).1H ЯМР: δ 2,40-2,50 (2H, м); 3,30 (2H, т); 3,30 (3H, с); 4,25 (1H, м); 7,
05 (1H, д); 7,35 (1H, д); (масло).
ПРИМЕР VIII.7
В данном примере описан способ, подходящий для получения соединений по изобретению, в которых атом серы в 4,
4-дифторбут- 3-енилтио заместителе соответствующего неокисленного соединения окислен до сульфоксида (сульфинил) или сульфона (сульфонил).
Получение соединения VIII.7 из соединения VIII.5.
Соединение VIII. 5 (18,1 г) охлаждают до 0oC в дихлорметане (400 см3) и прибавляют 3-хлорнадбензойную кислоту (61,2 г влажного твердого продукта, 2 экв. ). Смесь перемешивают при комнатной температуре в течение 18 часов и затем выливают в насыщенный водный бикарбонат натрия. Продукт экстрагируют дихлорметаном, органическую фазу промывают водой и насыщенным солевым раствором и сушат (MgSO4). Упаривание растворителя при пониженном давлении дает сырое соединение VIII. 7, которое хроматографируют на силикагеле, элюируя 15%-ным этилацетатом в гексане, увеличивая до 50%-ного этилацетата в гексане с получением чистого соединения VIII.7.1H ЯМР: δ 2,55-2,6 (2H, м); 3,5-3,55 (2H, т); 4,0 (3H, с); 4,15-4,3 (1H, м); 7,0 (1H, с); 7,15 (1H, с); (масло).
Применяя способ, описанный выше в примере VIII, используя два эквивалента окислителя, если не указано другого, вместе с подходящим исходным
продуктом, получают следующие соединения по изобретению:
(1) 2-(4,4-дифторбут-3-енилсульфонил)-имидазол (соединение VIII.2). MH+ = 223;1H ЯМР: δ 2,4-2,6 (2H, м);
3,4-3,45 (2H, т); 4,1-4,3 (IH, м); 7,3-7,4 (3H, ушир. с); (белый твердый продукт, т.пл. 113-114oC).
(2) 2-(4,4-дифторбут-3-енилсульфонил)-1-фенилимидазол (соединение VIII. 4). M+ = 298;1H ЯМР: δ 2,45-2,55 (2H, м); 3,45-3,55 (2H, т); 4,15-4,3 (1H, м); 7,2 (1H, д); 7,25 (1H, д); 7,45-7,55 (5H, м); (масло).
(3) 1-(4, 4-дифторбут-3-енил)-2-(4,4-дифторбут-3- енилсульфонил) имидазол (соединение VIII.9). M+ = 312;1H ЯМР: δ 2,5-2,65 (4H, м); 3,5-3,6 (2H, т); 4,1-4,35 (2H, м); 4,4-4,45 (2H, м); 7,05 (1H, с); 7,15 (1H, с); (масло).
(4) 2-(4,4-дифторбут-3-енилсульфонил)-1-этилимидазол (соединение VIII. 11). M+ = 250;1H ЯМР: δ 1,5 (3H, т); 2, 5-2,65 (2H, м); 3,5-3,6 (2H, т); 4,15-4,35 (1H, м); 4,15-4,35 (1H, М); 4,4-4,45 (2H, кв); 7,05 (1H, с); 7,15 (1H, с); (масло).
(5) 2-(4,4-дифторбут-3-енилсульфонил) 1-пропил-имидазол (соединение VIII. 13). MH+ = 265;1H ЯМР: δ 0,9-1,0 (3H, т); 1,8-2,0 (2H, м); 2,5-2,6 (2H, м); 3,5-3,6 (2H, т); 4,15-4,35 (3H, м); 7,05 (1H, с); 7,15 (1H, с); (масло).
(6) 2-(4,4-дифторбут-3-енилсульфинил)-1- (1-метилэтил)имидазол (соединение VIII. 15); M+ = 264;1H ЯМР: δ 1,5 (6H, д); 2,55-2,65 (2H, м); 3,5-3,6 (2H, м); 4, 15-4,35 (1H, м); 5,15-5,35 (1H, м); 7,15 (2H, широкий); (масло).
(7) 2-(4,4-дифторбут-3-енилсульфонил)-4-фенилимидазол (соединение VIII. 20). M+ = 282;1H ЯМР: δ 2,35-2,65 (2H, м); 3,25-3,4 (2H, т); 4,15-4,3 (1H, м); 7,25-7,5 (4H, кв); 7,6-7,8 (2H, м); (масло), используя 1,5 эквивалента окислителя.
(8) 2-(4, 4-дифторбут-3-енилсульфонил)-4-фенилимидазол (соединение VIII. 21). M+ = 298;1H ЯМР: δ 2,45-2,6 (2H, м); 3,4-3,5 (2H, т); 4,1-4,28 (1H, м); 7,3-7,55 (4H, кв); 7,7-7,75 (2H, д); (т.пл. 109,6-110,4oC), используя 1,5 эквивалента окислителя.
(9) 2-(4,4-дифторбут-3-енилсульфинил)-1-метил-4- фенилимидазол (соединение VIII. 23). M+ = 296;1H ЯМР: δ 2,5-2,65 (2H, м); 3,4-3,65 (2H, м); 4,0 (3H, с); 4,25-4,4 (1H, м); 7,25-7,45 (4H, м); 7,7-7,75 (2H, дд); (т. пл. 106-106,6oC), используя 1,5 эквивалента окислителя.
(10) 2-(4,4-дифторбут-3-енилсульфонил)-1-метил-4- фенилимидазол (соединение VIII. 24). M+ = 312;1H ЯМР: δ 2,6-2,7 (2H, м); 3,58-3,65 (2H, м); 4,02 (3H, с); 4,2-4,35 (1H, м); 7,25-7,42 (4H, м); 7,7-7,75 (2H, дд); (т.пл. 78,6-79,6oC), используя 1,5 эквивалента окислителя.
(11) 2-(4, 4-дифторбут-3-енилсульфонил)-4-этил-5- метилимидазол (соединение VIII.28). M+ = 264;1H ЯМР: δ 1,15-1,3 (3H, кв); 2,25 (3H, два с (таутомеры)); 2,45-2,7 (4H, м); 3,35-3,45 (2H, т); 4,1-4,3 (1H, м); 11,3 (1H, ушир. с); (масло).
(12) 2-(4,4-дифторбут-3-енилсульфонил)-4,5-диметилимидазол (соединение VIII.30). M+ = 250;1H ЯМР: δ 2,25 (6H, два с); 2,45-2,55 (2H, м); 3,35 (2H, т); 4,1-4,25 (1H, м); 10,3-10,6 (1H, ушир. с); (т. пл. 113,4-114,6oC).
(13) 2-(4,4-дифторбут-3-енилсульфонил)-1,4, 5- триметилимидазол (соединение VIII. 32). M+ = 264;1H ЯМР: δ 2,2 (6H, ушир. с); 2,5-2,6 (2H, м); 3,4-3,5 (2H, т); 3,85 (3H, с); 4,15-4,3 (1H, м); (масло).
(14) 1-этил-2-(4,4-дифторбут-3-енилсульфонил)-4,5- диметилимидазол (соединение VIII. 34). M+ = 278;1H ЯМР: δ 1,4 (3H, т); 2,15 (6H, ушир. с); 2,5-2,6 (2H, м); 3,5 (2H, т); 4,15-4,3 (1H, м); (масло).
(15) смесь 2-(4,4-дифторбут-3-енилсульфонил)-1,5- диметилимидазола (соединение VIII. 36) и 2-(4,4- дифторбут-3-енилсульфонил)-1, 4-диметилимидазола (соединение VIII.61);1H ЯМР: δ 2,20 (3H, с); 2,25 (3H, с); 2,5-2,6 (4H, м); 3,45-3,55 (4H, м); 3,95 (3H, с); 4,15-4,3 (2H, м); 6,75 (1H, с); 6,95 (1H, с); из смеси соединений VIII.35 и VIII. 60, полученных по примеру VIII.5 (4) выше.
(16) смесь 18:82 2-(4,4-дифторбут-3-енилсульфонил)-5- метил-(1-метилэтил)имидазола (соединение VIII. 38);1H ЯМР: δ 1,55 (6H, д); 2,4 (3H, с); 2,55-2,65 (2H, м); 3,6 (2H, т); 4,15-4,35 (1H, м); 5,15-5,30 (1H, м); 6,85 (1H, с); и 2-(4, 4-дифторбут-3-енилсульфонил)-4- метил-(1-метилэтил)имидазол (соединение VIII. 63);1H ЯМР: δ 1,45 (6H, д); 2,25 (3H, с); 2,55-2,65 (2H, м); 3,55 (2H, т); 4,15-4,35 (1H, м); 5,15-5,30 (1H, м); 6,90 (1H, с); из смеси соединений VIII.37 и VIII.62, полученных по примеру VIII.5(5) выше.
(17) 2-(4,4-дифторбут-3-енилсульфонил)-5-этоксикарбонил-1- метилимидазол (соединение VIII.53). M+ = 308;1H ЯМР: δ 1,37-1,42 (3H, т); 2,55-2,67 (2H, м); 3,6-3,65 (2H, т); 4,2-4,35 (1H, м); 4,25 (3H, с); 4,32-4,4 (2H, кв); 7,7 (1H, с); (масло).
(18) 2-(4,4-дифторбут-3-енилсульфонил)-4-метилимидазол (соединение VIII. 59). M+ = 2368;1H ЯМР: δ 2,3-2,4 (3H, широкий); 2,45-2,55 (2H, м); 3,4 (2H, т); 4,2-4,4 (1H, м); 7,0 (1H, широкий); (масло).
(19) 2-(4,4-дифторбут-3-енилсульфинил)-4- этоксикарбонил-1-метилимидазол (соединение VIII. 66). M+ = 292;1 H ЯМР: δ 1,35-1,4 (3H, т); 2,49-2,7 (2H, м); 3,35-3,6 (2H, м); 4,02 (3H, с); 4,2-4,38 (1H, м); 4,35-4,42 (4H, кв); 7,67 (1H, с); (масло), используя 1,5 эквивалента окислителя.
(20) 2-(4,4-дифторбут-3-енилсульфонил)-4-этоксикарбонил-1-метилимидазол (соединение VIII. 67). M+ = 308;1H ЯМР: δ 1,35-1,4 (3H, т); 2,55-2,65 (2H, м); 3,65-3,70 (2H, т); 4,02 (3H, с); 4,2-4,38 (1H, м); 4,35-4,42 (2H, кв); 7,65 (1H, с); (масло), используя 1,5 эквивалента окислителя.
(21) 3-(4,4-дифторбут-3-енилсульфонил)-[1,5a]-пиридин (соединение VIII. 152). M+ = 272;1H ЯМР: δ 2,50 (2H, м); 3,50 (2H, т); 4,15 (1H, м); 6,90 (1H, м); 7,10 (1H, м); 7,20 (1H, с); 7,65 (1H, м); 8,95 (1H, дд); (масло).
ПРИМЕР IX.1
В данном примере описан способ получения 5-(4,4-дифторбут- 3-енилтио)-1,3-диметил-4-нитропиразол (соединение IX.82)
Гидробромид 4,4-дифторбут-3-енилизотиоурония (2,46 г)
перемешивают при комнатной температуре водным гидроксидом натрия (1,2 г в 12 см3 воды) в течение 0,3 часа. 2-Хлор-1,3-диметил-4- нитропиразол (1,76 г) в дихлорметане (12 см3),
содержащем тетра-н-бутиламмонийхлорид (0,01 г; каталитическое количество)
прибавляют при комнатной температуре и реакционную смесь перемешивают в течение 18 часов в атмосфере азота.
Реакционную смесь разбавляют водой (100 см3) и продукт экстрагируют дихлорметаном (100 см3). Органические фазы объединяют, промывают водой, сушат (MgSO4) и упаривают
при пониженном давлении с получением желтой жидкости (2,6 г). Образец (0,9 г) фракционируют с помощью хроматографии (силикагель, гексан:этилацетат 10:1 по объему) с получением соединения IX. 82 (0,78
г). M+ = 263;1H ЯМР: δ 2,25 (2H, м); 2,55 (3H, с); 3,08 (2H, т); 3,94 (3H, с); 4,20 (1H, м); (масло).
ПРИМЕР IX.2
В данном примере описан способ
получения 5-(4,4-дифторбут- 3-енилсульфонил)-1,3-диметил-4-нитропиразол (соединение IX.84)
3-Хлорнадбензойную кислоту (1,74 г 50% ного по весу твердого продукта) прибавляют к раствору
соединения IX.82 (0,526 г) в дихлорметане (10 см3) и реакционную смесь перемешивают при комнатной температуре в течение 3 дней. Реакционную смесь разбавляют дихлорметаном (100 см3
), промывают водным раствором гидрокарбоната натрия и затем водой, сушат (MgSO4) и упаривают при пониженном давлении с получением маслянистого твердого продукта. Сырой продукт фракционируют
с помощью хроматографии с получением соединения IX.84 (0,15 г).1H ЯМР: δ 2,52 (3H, м); 2,60 (2H, м); 3,74 (2H, т); 4,20 (3H, с); 4,25 (1H, м); (смола).
ПРИМЕР
IX.3
В данном примере описан 3-стадийный способ получения 5-(4,4-дифторбут-3-енилтио)-1,3-диметил-4-иодпиразол (соединение IX.61)
Стадия 1: Получение 4-амино-5-(4,
4-дифторбут-3-енилтио)- 1,3-диметилпиразола
Соединение IX.82 (1,0 г) растворяют в пропан-2-оле (10 см3), содержащем воду (2 см3) и концентрированную хлористоводородную
кислоту (каталитическое количество, 0,1 см3), и обрабатывают восстановителем, порошком железа (1,0 г). Смесь перемешивают и нагревают с обратным холодильником в течение 3 часов, охлаждают,
нейтрализуют твердым гидрокарбонатом натрия и фильтруют через кизельгель. Нерастворимые остатки промывают пропан-2-олом и фильтрат упаривают при комнатной температуре с получением аминопиразола в виде
красно-коричневого масла (1,0 г). М+ = 233.
Стадия 2: Продукт со стадии 1 (4,6 г) растворяют в сухом дихлорметане (25 см3) и прибавляют к перемешиваемому раствору диэтилэфирата трифторида бора (4,26 г) в сухом дихлорметане (25 см3) при -15oC. По каплям к смеси прибавляют трет-бутилнитрит в дихлорметане (10 см3). Реакционной смеси дают нагреться до 5oC в течение 0,3 часа, разбавляют гексаном и требуемую соль тетрафторбората диазония фильтруют из раствора в виде коричневого твердого продукта (6,7 г).
Стадия 3: Соединение IX.61
Продукт со стадии 1 (1,33 г) прибавляют частями к перемешиваемому раствору иодида калия (1,7 г) в воде (5 см3) при 35oC.
Реакционная смесь в процессе проведения реакции выделяет газ и дает красно-коричневое масло. Через 1 час при 35oC смесь охлаждают, разбавляют водой, экстрагируют диэтиловым эфиром (100
см3), органическую фазу промывают последовательно водным метабисульфитом натрия, затем водой и сушат (MgSO4). Растворитель удаляют при пониженном давлении и оставшееся масло
фракционируют с помощью хроматографии (силикагель, гексан : этилацетат 10 : 1 по объему) с получением соединения IX.61 в виде коричневого масла (0,2 г). М+ = 344.
ПРИМЕР
IX.4
В данном примере описан способ получения 5-(4,4 - дифторбут-3-енилсульфонил)-1,3-диметилпиразола (соединение IX.55)
Диазониевую соль по примеру IX.3, стадия 2 (2,0 г)
перемешивают в метаноле (20 см3) при 0oC и порциями прибавляют боргидрид натрия (порошок, 0,25 г). Выделяется газ и раствор изменяется от бесцветного до оранжево-коричневого.
Смеси дают нагреться до 15oC в течение более 0,5 часов, выдерживают при комнатной температуре в течение 18 часов, разбавляют водой, экстрагируют диэтиловым эфиром, сушат (MgSO4)
и упаривают при пониженном давлении с получением красно-коричневой жидкости. Жидкость фракционируют с помощью хроматографии (силикагель, гексан : диэтиловый эфир 1 : 1 по объему) с получением
соединения IX.55 (0,42 г). M+ = 218;1H ЯМР: δ 2,20-2,30 (5H, м); 2,74 (2H, м); 3,75 (3H, с); 4,23 (2H, м); 6,12 (1H, с); (масло).
ПРИМЕР IX.5
В
данном примере описан способ получения этил 5-(4,4-дифторбут-3-енилтио)-1-метилпиразол-4-ил карбоксилата (соединение IX.34)
Бис-(4,4-дифторбут-3-енил) дисульфид (2,90 г) и трет-бутилнитрит (1,
22 г) в ацетонитриле (40 см3) нагревают при 60oC в атмосфере азота. К перемешиваемому раствору по каплям прибавляют этил 5-амино-1-метилпиразол-4-илкарбоксилат (1,00 г) в
ацетонитриле (10 см3). По завершении прибавления реакционный раствор нагревают при 60oC в течение 2 часов, упаривают при пониженном давлении и фракционируют с помощью
хроматографии (силикагель, гексан : диэтиловый эфир 5 : 1 по объему) с получением соединения IX.34 (выход 42%). М+ = 276;1H ЯМР: δ 1,38 (3H, т); 2,20 (2H, м); 3,05 (2H,
т); 3,97 (3H, с); 4,25 (1H, м); 4,34 (2H, кв); 7,98 (1H, с); (масло).
Применяя способ, описанный выше, и используя подходящий аминопиразол, получают следующие соединения по
изобретению:
(1) 4-бром-5-(4,4-дифторбут-3-енилтио)-1-метилимидазол (соединение IX. 31). MH+ = 282;1H ЯМР: δ 2,32 (2H, м); 2,95 (2H, т); 3,88 (3H, с); 4,30 (1H,
м); 7,38 (1H, с); (масло).
(2) 4-циано-5-(4,4-дифторбут-3-енилтио)-1,3-диметилпиразол (соединение IX. 73).1H ЯМР: δ 1,38 (3H, т); 2,20 (2H, м); 2,38 (3H, с); 3,02 (2H, т); 3,97 (3H, с); 4,25 (1H, м); (масло).
(3) этил 5-(4,4-дифторбут-3-енилтио)-1- фенилпиразол (соединение IX. 124). М+ = 338;1H ЯМР: δ 1,40 (3H, т); 2,05 (2H, м); 2,88 (2H, т); 3,97 (1H, м); 4,37 (2H, кв); 7,50 (5H, м); 8,16 (1H, с); (масло).
ПРИМЕР IX.6
В данном примере описан способ получения 4-циано-5-(4,
4- дифторбут-3-енилсульфонил)-1,3-диметилпиразола (соединение IX.75).
Соединение IX.73 (1,32 г) в дихлорметане (120 см3) обрабатывают 3-хлорнадбензойной кислотой (3,94 г, содержащие 50% надкислоты). Смесь перемешивают в течение 18 часов, разбавляют еще дихлорметаном и промывают последовательно водным раствором карбоната натрия, метабисульфита натрия. Осуществляют промывание водой, карбонатом натрия и водой перед тем, как органическую фазу сушат (MgSO4). Растворитель упаривают при пониженном давлении и остаток фракционируют с помощью хроматографии (силикагель, гексан : диэтиловый эфир 1 : 1 по объему) с получением соединения IX.75 (0,47 г). М+ = 275;1H ЯМР: δ 2,42 (3H, с); 2,30 (2H, м); 2,58 (3H, с); 3,38 (2H, т); 4,14 (3H, с); 4,36 (1H, м); (т. пл. 78-81oC).
Применяя способ, описанный выше, и используя подходящий пиразол, получают следующие соединения по изобретению:
(1)
4-бром-5-(4,4-дифторбут-3-енилсульфонил) -1- метилпиразол (соединение IX.33).1H ЯМР: δ 2,54 (2H, м); 3,35 (2H, т); 4,00 (3H, с); 4,27 (1H, м); 7,55 (1H, с); (т. пл. 42,6-43,6oС) из соединения IX.31.
(2) этил 5-(4,4-дифторбут-3-енилсульфонил)-1-метилпиразол-4-карбоксилат (соединение IX. 36).1H ЯМР: δ 1,38 (3H, т); 2,50 (2H, м); 3, 78 (2H, т); 4,15-4,30 (1H, м); 4,36 (2H, кв); 7,95 (1H, с); (масло) из соединения IX.34.
(3) этил 5-(4,4-дифторбут-3-енилсульфонил)-1- фенилпиразол-4-карбоксилат (соединение IX. 126).1H ЯМР: δ 1,42 (3H, т); 2,45 (2H, м); 3,72 (2H, т); 4,18 (1H, м); 4,40 (2H, кв); 7,35-7,55 (5H, м); 8,13 (1H, с); (т. пл. 62,5-63,0oC) из соединения IX.124.
ПРИМЕР VII.7
В данном примере описан способ получения 5-(4,4-дифторбут-3-енилтио)-1-метилпиразол-4-карбоновой кислоты (соединение IX. 40) и пропан-2-ил 5-(4,
4-дифторбут-3- енилтио)-1-метилпиразол-4-карбоксилат (соединение IX.37).
Соединение IX. 34 (1,5 г) растворяют в пропан-2-оле (40 см3) и обрабатывают 2М водным гидроксидом калия (8 см3) и перемешивают при комнатной температуре в течение 18 часов. Смесь разбавляют 2М водной хлористоводородной кислотой и экстрагируют этилацетатом (3x50 см3). Объединенные органические фазы сушат (MgSO4) и упаривают при пониженном давлении с получением желтого масла, которое затвердевает при обработке гексаном/этиловым эфиром. Твердый продукт отфильтровывают от раствора, промывают гексаном и сушат досуха с получением соединения IX. 40 (0,76 г).1H ЯМР: δ 2,24 (2H, м); 3,06 (2H, т); 4,00 (3H, с); 4,20 (1H, м); 8,08 (1H, с); (т. пл. 59,4-60,0oC).
Гексан/диэтиловый эфирный фильтрат упаривают при пониженном давлении и масло (содержащее соединение IX. 37 с около 10% этилового эфира исходного продукта со стадии 1) обрабатывают пропан-2-олом (20 см3), содержащим метоксид натрия (катализатор переэтерификации, 10 мг), и смесь нагревают с обратным холодильником в течение 5 часов. Реакционную смесь охлаждают, разбавляют водой и продукт экстрагируют диэтиловым эфиром. Органическую фазу сушат (MgSO4) и упаривают при пониженном давлении с получением соединения IX.37 (0, 1 г). MH+ = 291;1H ЯМР: δ 1,35 (5H, д); 2,22 (2H, м); 3,05 (2H, т); 3,97 (3H, с); 4,20 (1H, м); 5,20 (1H, септиплет); 7,95 (1H, с); (масло).
ПРИМЕР
IX.8
В данном примере описан способ получения 5-(4,4-дифторбут-3-енилсульфонил)-1-метилпиразол-4- карбоновой кислоты (соединение IX.42)
Соединение IX. 36 (0,83 г) растворяют в
этаноле (35 см3) и обрабатывают моногидратом гидроксида лития (0,34 г) в воде (7 см3) при комнатной температуре. Реакционную смесь перемешивают в течение 18 часов, этанол
упаривают при пониженном давлении, водную фазу подкисляют 2М хлористоводородной кислотой и продукт экстрагируют этилацетатом. Органическую фазу сушат (MgSO4) и растворитель упаривают при
пониженном давлении с получением смолы, которую растирают с диэтиловым эфиром/гексаном с получением соединения IX.42 (0,43 г). M(NH4)+ = 298;1H ЯМР: δ 2,52 (2H,
м); 3,72 (2H, т); 4,10-4,30 (3H, м); 8,05 (1H, с); (т. пл. 118,4-122,0oC).
ПРИМЕР X.1
Данный пример иллюстрирует два родственных способа получения меркапто-1,2,
4-оксадиазолов, необходимых в качестве промежуточных продуктов для получения соединений по изобретению. Общим способом получения 5-меркапто-1,2,4-оксадиазолов является циклизация амидоксима и
активированного тиокарбонильного соединения, такого как тиофосген или 1,1-тиокарбонилдиимидазол. Применение первого из этих реагентов проиллюстрировано получением 5-меркапто-3-фенил-1,2,
4- оксадиазола.
Бензонитрил (15 г), гидрохлорид гидроксиламина (10 г), карбонат калия (10 г), этанол (150 см3) и воду нагревают вместе с обратным холодильником в течение 6 часов и затем оставляют на холоду в течение ночи. Реакционную смесь фильтруют и твердый остаток промывают этанолом. Фильтрат и промывные жидкости объединяют и упаривают и полученный коричневый осадок распределяют между этилацетатом и водой. Органическую фазу отделяют, промывают насыщенным солевым раствором и сушат (MgSO4). Упаривание дает коричневое масло, которое кристаллизуют при добавлении этилацетата и гексана с получением серого твердого продукта (10,3 г). Твердый продукт (4,4 г) перемешивают в эфире (50 см3) и прибавляют тиофосген (0,55 см3), что дает образование густого белого осадка. Реакционную смесь нагревают с обратным холодильником в течение 1 часа и затем оставляют охлаждаться. Затем прибавляют раствор гидроксида натрия в воде (50 см3) и реакционную смесь нагревают еще в течение 4 часов, затем оставляют охлаждаться для образования двухфазной реакционной смеси. Органическую фазу отделяют и водный слой дважды экстрагируют эфиром. Водный слой экстрагируют этилацетатом и объединенные этилацетатные слои сушат (MgSO4) и упаривают с получением 5-меркапто-3-фенил-1,2,4-оксадиазола в виде оранжево-коричневого твердого продукта (0,554 г), которое используют без дополнительной очистки.1H ЯМР: δ 7,45-7,63 (3H, м); 7,70-7,90 (2H, м).
Использование альтернативного реагента, 1, 1-тиокарбонилдиимидазола, проиллюстрировано получением 5-меркапто-3-метоксиметил-1,2,4-оксадиазола.
Метоксиацетонитрил (7,1 г), гидрохлорид гидроксиламина (7 г), карбонат калия (13,8 г), этанол (90 см3) и воду (9 см3) нагревают вместе при 50oC в течение 9 часов и затем оставляют охлаждаться. Реакционную смесь фильтруют и белый твердый остаток промывают этилацетатом. Фильтрат и промывные жидкости объединяют и упаривают и полученный остаток растворяют в дихлорметане. Нерастворимые продукты удаляют фильтрацией и фильтрат упаривают с получением вязкого масла (9,2 г). Масло прибавляют к толуолу (60 см3) и сухому диметилформамиду (4 см3), содержащему 1,1-тиокарбонилимидазол (5,655 г), и смесь перемешивают при комнатной температуре в течение 2 часов. После выдерживания еще в течение 60 часов образуется твердый продукт бежевого цвета, который отделяют фильтрацией (получают 5,2 г), и ЯМР которого показывает, что он представляет собой нециклизованный продукт взаимодействия между гидроксигруппой амидоксима и тиокарбонильной группой. Часть раствора (2 г) прибавляют к суспензии гидрида натрия (0,33 г) в сухом диметилформамиде (30 см3) (вспенивание) и смесь перемешивают при комнатной температуре в течение 5 часов и оставляют стоять на 18 часов. Продукт реакции выливают в воду и продукт экстрагируют этилацетатом. Объединенные органические фазы отделяют, сушат (MgSO4) и упаривают при пониженном давлении, а в конце в высоком вакууме для удаления следов диметилформамида. Сырой 5-меркапто-3-метоксиметил-1,2,4- оксадиазол имеет1H ЯМР: δ 3,2 (3H, с); 4,10 (2H, с); 6,92-6,98 (1H, ушир. с), и используется далее без дополнительной очистки.
ПРИМЕР X.2
Данный пример иллюстрирует общий способ получения 5-(4,4-дифторбут-3-енилтио)-3-замещенных-1,2, 4-оксадиазолов, используя соответствующие 5-меркаптопроизводные, полученные по
примеру выше. Это показано следующим получением 5-(4,4-дифторбут-3-енилтио)-3-метоксиметил-1,2,4-оксадиазола, соединения X.26.
К раствору 5-меркапто-3-метоксиметил-1,2,4-оксадиазола (2,78 г) в ацетоне (150 см3) прибавляют 4-бром-1,1-дифторбут-1-ен (4,87 г) и карбонат калия (3,15 г) и смесь нагревают вместе с обратным холодильником в течение 18 часов. ГХ показывает, что реакция завершена. Неорганические твердые продукты удаляют фильтрацией реакционной смеси через плаг силикагеля sorbital-С30, промывают ацетоном. Фильтрат упаривают при пониженном давлении и желтый осадок хроматографируют на sorbital-С30, элюируя 5%-ным этилацетатом в гексане с получением соединения X.26 (1,6 г).1H ЯМР: δ 2,47-2,58 (2H, м); 3,28-3,35 (2H, т); 3,49 (3H, с); 4, 18-4,36 (1H, м); 4,54 (2H, с); (масло).
По способу, описанному выше, используя соответствующие промежуточные продукты, получают следующие соединения по изобретению:
(1) 5-(4,
4-дифторбут-3-енилтио)-3-метил-1,2,4- оксадиазол (соединение X. 32).1H ЯМР: δ 2,37 (3H, с); 2,45-2,55 (2H, м); 3,22-3,30 (2H, т); 4,16-4,35 (1H, м); (масло) из 5-меркапто-3-метил-1,
2,4-оксадиазола.
(2) 5-(4,4-дифторбут-3-енилтио)-3-фенил-1,2,4-оксадиазол (соединение X. 1). М+ = 268.1H ЯМР: δ 2,58 (2H, м); 3,35 (2H, т); 4,31 (1H, м); 7,45-7,56 (3H, м); 8,07 (2H, д); (масло) из 5-меркапто-3-фенил-1,2,4-оксадиазола.
ПРИМЕР X.3
Данный пример иллюстрирует способ получения 5-(4,
4- дифторбут-3-енилсульфонил)-3-метоксиметил-1,2,4- оксадиазола (соединение X.27) из соединения X.26.
Соединение X.26 (0,6 г) охлаждают до 0oC в дихлорметане (50 см3) и прибавляют 3-хлорнадбензойную кислоту (2,19, 2,5 экв.) в течение более пяти минут. Смесь перемешивают при комнатной температуре 1 час и выдерживают 40 часов. Реакционную смесь выливают в насыщенный водный раствор бикарбоната натрия и продукт экстрагируют дихлорметаном. Органический слой промывают насыщенным водным раствором бикарбоната натрия, водой и насыщенным солевым раствором и сушат (MgSO4). Упаривание растворителя при пониженном давлении дает светло-коричневое твердое вещество, которое хроматографируют на силикагеле, элюируя 10%-ным этилацетатом в гексане, повышая до 20%-ного этилацетата в гексане, с получением соединения X.27, имеющего1H ЯМР: δ 2,6-2,71 (2H, м); 3,51 (3H, с); 3,58-3,65 (2H, т); 4,21-4,37 (1H, м); 4,69 (2H, с); (масло). Этот продукт, как обнаружено, нестабилен и при стоянии в течение 60 часов заметно гидролизуется.
ПРИМЕР ХI.1А
Данный пример иллюстрирует общие способы получения
5-хлор-3-замещенных-1,2,4-тиадиазолов, как показано на примере следующего получения 5-хлор-3-хлорметил-1,2,4-тиадиазола из гидрохлорида хлорацетамидина.
Суспензию гидрохлорида хлорацетамидина (1,29 г) в дихлорметане (100 см3) охлаждают до 0oC и прибавляют перхлорметилмеркаптан. По каплям прибавляют (выделение тепла) гидроксид натрия в воде (20 г в 30 см3), поддерживая температуру реакционной смеси ниже 5oC. После завершения прибавления реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Смесь разбавляют водой и дихлорметаном и все фильтруют через пад из целита для удаления нерастворенного продукта. Органическую фазу отделяют, промывают насыщенным солевым раствором и сушат над сульфатом магния. Затем раствор продукта фильтруют и упаривают при пониженном давлении с получением коричневого масла (9,72 г), которое используют без дополнительной очистки. М+ = 168;1H ЯМР: δ 4,75 (с).
По способу примера XI.1А получают следующие промежуточные соединения:
(1) 5-хлор-3-трифторметил-1,2,4-тиадиазол. М+ = 188 (т. пл.
50oC при 12 мм рт.ст.).
(2) 5-хлор-3-метилмеркапто-1,2,4- тиадиазол. М+ = 166.
(3) 5-хлор-3-метокси-1,2,4-тиадиазол. М+ = 150.
(4) 5-хлор-3-(2-пиразинил)-1,2,4-тиадиазол. М+ = 198.
Подобный способ был использован для получения соединения XI.102 по изобретению, как это описано далее.
Получение 5-хлор-3-(4,4-дифторбут-3-енилтио)-1,2,4-тиадиазола
Суспензию гидробромида 4,4-дифторбут-3-енилизотиоурония (8,68 г) в воде (200 см3), содержащей натрий
лаурилсульфат (0,1 г, каталитическое количество) и перхлорметилмеркаптан (7,17 г), охлаждают до 0oC и прибавляют по каплям, поддерживая температуру реакционной смеси ниже 5oC,
гидроксид натрия в воде (5,6 г в 200 см3). После завершения прибавления реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Смесь два раза экстрагируют
этилацетатом, органическую фазу отделяют, промывают насыщенным солевым раствором и сушат (MgSO4). Затем раствор продукта фильтруют и упаривают при пониженном давлении с получением
коричневого масла (8,2 г), которое хроматографируют на силикагеле (Sorbsil C30), используя 3%-ный этилацетат в гексане в качестве элюента, и получают соединение XI. 102. М+ = 242;1H ЯМР: δ 2,4-2,6 (2H, м); 3,2-3,3 (3H, т); 4,2-4,4 (1H, м); (масло).
ПРИМЕР XI.1В
Общий способ получения 5-[(4-метилфенил)сульфонил] -3- замещенных-1,2,
4-тиадиазолов проиллюстрирован в следующем двухстадийном способе получения 3-метоксиметил-5-[(4- метилфенил)сульфонил]-1,2,4-тиадиазола из метоксиацетамида.
Стадия 1: Получение
5-метоксиметил-1,3,4-оксатиазол-2-она
Хлоркарбонилсульфенилхлорил (7,35 г) прибавляют к суспензии метоксиацетамида (5 г) в толуоле (30 см3). Реакционную смесь перемешивают и
нагревают при 90-100oC в течение 5 часов, затем охлаждают. Растворитель удаляют упариванием при пониженном давлении с получением коричневой смолы (6,6 г). М+ = 147;1H
ЯМР (ДМСО-d6): δ 3,30 (3H, с); 4,30 (2H, с), которую используют далее без дополнительной очистки.
Стадия 2: Получение 3-метоксиметил-5-[(4-метилфенил) сульфонил]-1,2,
4-тиадиазола
4-Метилбензолсульфонилцианид (16,26 г) прибавляют к эмульсии 5-метоксиметил-1,2,3-оксатиазол-2-она (6,6 г) в додекане (60 см3). Реакционную смесь перемешивают и
нагревают при 150oC в течение 18 часов, затем охлаждают. Прибавляют воду и продукт экстрагируют этилацетатом. Объединенные органические фазы сушат (MgSO4), этилацетат удаляют
путем упаривания при пониженном давлении. Остаток разделяют на коричневую жидкость и прозрачный слой, который удаляют и отбрасывают. Хроматография коричневой жидкости на силикагеле (Sorbsil C30),
используя 3:7 этилацетат:гексан в качестве элюента с получением бледно-оранжевого масла, которое затвердевает при стоянии (6,94 г). М+ = 284;1H ЯМР: δ 2,40 (3H, с); 3,45
(3H, с); 4,7 (2H, с); 7,4 (2H, д); 8,0 (2H, д); (Т. пл. 43,4-45,4oC).
По двухстадийному способу примера XI. 1В получают следующие соединения по изобретению. Исходные продукты были известными соединениями.
(1) 3-этил-5-[(4-метилфенил)сульфонил] -1,2,4-тиадиазол. М+ = 268;1H ЯМР: δ 1,3-1,4 (3H, т); 2,45 (3H, с); 2,95-3, 05 (2H, кв); 7,4 (2H, т); 8,0 (2H, м); из пропионамида.
(2) 3-(Е-проп-1-енил)-5-[(4-метилфенил) сульфонил]- 1,2,4-тиадиазол. М+ = 280.1H ЯМР: δ 1,95 (3H, дд); 2,45 (3H, с); 6,5-6,6 (1H, м); 7,0-7,1 (2H, м); 7,4 (2H, м); 8,0 (2H, м) из кротонамида.
ПРИМЕР XI.2
Данный пример иллюстрирует общие способы получения 5-(4,
4- дифторбут-3-енилтио)-3-замещенных-1,2,4-тиадиазолов, используя либо 5-хлор- или 5-[(4-метилфенил)сульфонил] -1,2,4-тиадиазольное промежуточное соединение, полученное, как указано выше. Общий метод
проиллюстрирован следующим получением 5-(4,4-дифторбут-3-енилтио) -3-метоксиметил-1,2,4-тиадиазол (соединение XI.34).
Гидроксид натрия в воде (0,848 г в 10 см3) прибавляют к 4,4-дифторбут-3-енилизотиуроний гидробромиду (1,75 г) и смесь перемешивают при комнатной температуре в течение 20 минут. Раствор 3-метоксиметил-5-[(4-метилфенил) сульфонил] -1,2,4-тиадиазола (2,01 г) в дихлорметане (10 см3) прибавляют тетрабутиламмонийбромид (0,1 г, каталитическое количество) и смесь перемешивают в течение 20 минут. ТСХ показывает, что продукт образован. Смесь разбавляют еще дихлорметаном (10 см3) и органическую фазу отделяют, промывают насыщенным раствором соли, сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением оранжево-желтой жидкости. Хроматография сырого продукта на силикагеле (Sorbsil C30), используя 1:4 этилацетат в гексане в качестве элюента с получением соединения XI.34 (1,67 г). М+ = 252;1H ЯМР: δ 2,45-2,55 (2H, м); 3,25-3,35 (2H, т); 3,5 (3H, с); 4,15-4,35 (1H, м); 4,65 (2H, с); (масло).
По вышеуказанному способу в соответствии с изобретением, но
используя соответствующие промежуточные продукты, получают следующие соединения:
(1) 5-(4,4-дифторбут-3-енилтио)-3-трифторметил-1,2,4- тиадиазол (соединение XI. 9). М+ = 276.1H ЯМР: δ 2,5-2,6 (2H, м); 3,3-3,4 (2H, т); 4,2-4,4 (1H, м); (масло).
(2) 5-(4,4-дифторбут-3-енилтио)-3-(Е-проп-1-енил)-1,2,4-тиадиазол (соединение XI.II). М+ = 248.1H ЯМР: δ 1,95 (3H, дд); 2,5-2,6 (2H, м); 3,3 (2H, т); 4,2-4,4 (1H, м); 6,45-6,55 (1H, м); 6,9-7,1 (1H, м); (масло).
(3) 3-этил-5-(4,4-дифторбут-3-eнилтиo) -1, 2,4-тиадиазол (соединение XI. 23). М+ = 236.1H ЯМР: δ 1,3-1,4 (3H, т); 2,45-2,55 (2H, м); 2,9-3,0 (2H, кв); 3,3 (2H, т); 4,2-4,4 (1H, м); (масло).
(4) 3-хлорметил-5-(4,4-дифторбут-3-енилтио)-1,2,4-тиадиазол (соединение XI. 25). M+ = 256.1H ЯМР: δ 2,45-2,55 (2H, м); 3,35 (2H, т); 4,2-4,35 (1H, м); 4,7 (2H, с); (масло). В этой реакции получены еще два продукта, которые разделяют в процессе хроматографии и охарактеризованы. Это 3-(4,4-дифторбут-3- енилтиометил)-5-(4,4-дифторбут-3-енилтио)-1,2, 4- тиадиазол (соединение XI. 38). М+ = 344.1H ЯМР: δ 2,25-2,35 (2H, м); 2,45-2,55 (2H, м); 2,65 (2H, т); 3,3 (2H, т); 4,15-4,35 (2H, м); (масло) и 5-хлор-3-(4,4-дифторбут-3- енилтиометил)-1,2, 4-тиадиазол. М+ = 256.1H ЯМР: δ 2,2-2,3 (2H, м); 2,65 (2H, т); 2,65 (2H, т); 3,9 (2H, с); 4,1-4,3 (1H, м); (масло).
(5) 5-(4, 4-дифторбут-3-енилтио)-3-метокси-1,2,4- тиадиазол (соединение XI. 87). М+ = 238.1H ЯМР: δ 2,45-2,55 (2H, м); 3,3 (2H, т); 4,1 (3H, с); 4,2-4,35 (1H, м); (масло).
(6) 3,5-бис-(4,4-дифторбут-3-енилтио)-1,2,4- тиадиазол (соединение XI. 109). М+= 330.1H ЯМР: δ 2,45-2,6 (4H, м); 3,2-3,35 (4H, м); 4,2-4,4 (2H, м); (масло) из соединения XI.102.
(7) 5-(4,4-дифторбут-3-енилтио)-3-(2-пиразинил)-1,2,4- тиадиазол (соединение XI. 125). М+ = 286.1H ЯМР: δ 2,55-2,65 (2H, м); 3,35-3,45 (2H, т); 4,25-4,40 (1H, м); 8,65 (1H, д); 8,75 (1H, дд); 9,55 (1H, д); (масло).
ПРИМЕР XI.3
Данный пример иллюстрирует способ получения 3-бутоксиметил- 5-(4,
4-дифтopбут-3-eнилтиo)-1,2,4-тиадиазола (соединение XI.30).
Карбонат калия (0,444 г) и н-бутанол (0,397 г) прибавляют к раствору соединения XI. 25 (0,275 г) в диметилфорамиде (2 см3) и смесь перемешивают при комнатной температуре в течение 18 часов. ТСХ показывает, что соединение XI.25 все еще присутствует в смеси, поэтому прибавляют гидрид натрия (0,1 г) и н-бутанол (0,4 г) и смесь перемешивают при комнатной температуре в течение еще 24 часов. Прибавляют воду и продукт экстрагируют диэтиловым эфиром. Органическую фазу сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением масла (0,442 г), которое очищают хроматографией на силикагеле (Sorbsil C30), используя 10%-ный этилацетат в гексане в качестве элюента с получением соединения XI.30 (0,131 г). М+ = 294;1H ЯМР: δ 0,95- 1,0 (3H, т); 1,4-1,55 (2H, м); 1,75-1,9 (2H, м); 2,25-2,45 (2H, м); 2,6-2,7 (2H, т); 3,75 (2H, с); 4,15-4,3 (1H, м); 4,4-4,5 (1H, т); (масло).
По вышеописанному способу с н-пропанолом вместо н-бутанола получают следующее соединение:
(1) 5-(4,4-дифторбут-3-енилтио)-3-пропоксиметил-1,2,
4- тиадиазола (соединение XI. 31). M+ = 280;1H ЯМР: δ 1,0-1,1 (3H, т); 1,8-1,95 (2H, м); 2,25-2,35 (2H, м); 2,6-2,7 (2H, т); 3,75 (2H, с); 4,15-4,4 (1H, м); 4,4-4,5 (1H,
т); (масло).
ПРИМЕР XI.4
Данный пример иллюстрирует двухстадийный способ получения 5-(4,4-дифторбут-3-енилтио)-3-метил-1,2,4-оксадиазола (соединение XI.40).
К раствору ацетамида (5 г) в метаноле (100 см3) прибавляют дисульфид углерода (4 г), серу (1,7 г) и метоксид натрия (5,7 г) и смесь нагревают с обратным холодильником в течение 6 часов. Смесь охлаждают, фильтруют через hi-flo фильтр для удаления избытка серы и фильтрат распределяют между водой и этилацетатом. Этилацетат упаривают с получением коричневого твердого продукта и при подкислении водного слоя образуется красный твердый продукт и его отфильтровывают. Оба твердых продукта, полученных из фильтрата, являются смесью 3-метил-1,2,4-тиадиазол-5(4H)-тиона и серы. Эти два твердых продукта объединяют и используют на следующей стадии.
Стадия 2: Получение соединения XI.40
К раствору 3-метил-1,2,4-тиадиазол-5(4H)-тиона (1,2 г) в ацетоне (100
см3) 4,4-дифторбут-3-енил-4-метилбензолсульфонат (2,4 г) и карбонат калия (1,2 г) и смесь нагревают с обратным холодильником в течение 4 часов, после чего ТСХ показывает полное расходование
исходного продукта. Реакционную смесь выливают в этилацетат и разделяют воду и слои. Водный слой экстрагируют этилацетатом и объединенные органические слои сушат (MgSO4). Раствор продукта
затем фильтруют и упаривают при пониженном давлении с получением коричневого масла, которое очищают флэш хроматографией (силикагель, 7% этилацетат в гексане) с получением 5-(4,
4-дифторбут-3-енилтио)-3-метил-1,2,4-тиадиазола в виде коричневого масла (0,645 г). М+ = 222;1H ЯМР: δ 2,56 (2H, ушир. кв); 2,63 (3H, с); 3,30 (2H, т); 4,30 (1H, м).
По вышеуказанному способу, но с 3-фенил-1,2,4-тиадиазол-5(4H)-тионом в качестве исходного продукта, получают следующее соединение:
(1) 5- (4,4-дифторбут-3-енилтио)-3-фенил-1,2,
4-тиадиазол (соединение XI. 5).1H ЯМР: δ 2,53 (2H, кв); 3,30 (2H, т); 4,24 (1H, м); 7,38 (3H, м); 8,2 (2H, м).
ПРИМЕР XI.5
Данный пример иллюстрирует
способ получения 3-(4,4-дифторбут-3- енилтио)-5-метокси-1,2,4-тиадиазола (соединение XI.108).
Гидроксид натрия (0,182 г) прибавляют к раствору соединения XI.102 (1 г) в метаноле (5 см3) и смесь перемешивают при комнатной температуре в течение 45 минут, после чего определяется полное расходование исходного продукта. К смеси прибавляют воду и диэтиловый эфир и продукт экстрагируют диэтиловым эфиром. Органическую фазу сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением желтого масла (0,9 г). Очистка флэш хроматографией (силикагель, 5% этилацетат в гексане) дает соединение XI.108 (0,78 г). М+ = 238;1H ЯМР: δ 2,4-2,5 (2H, м); 3,2 (2H, т); 4,15 (3H, с); 4,2-4,35 (1H, м); (масло).
ПРИМЕР XI.6
Данный пример иллюстрирует способ получения 5-(4,4-дифторбут-3-енилтио)-3-метилтио-1,2,4-тиадиазола (соединение XI.110).
Hаногидрат сульфида натрия (0,555 г) прибавляют к 5-хлор-3-метилтио-1,2,4-тиадиазолу (1,5 г) в этаноле (10 см3) и смесь перемешивают и нагревают с обратным холодильником в течение 18 часов. Реакционную смесь охлаждают и растворитель удаляют упариванием при пониженном давлении с получением желтого твердого продукта (1,76 г), которое растворяют в ацетоне (30 см3). Прибавляют карбонат калия (2,22 г) и 4-бром-1,1-дифторбут-1-ен (1,83 г) и смесь перемешивают и нагревают с обратным холодильником в течение 18 часов. Реакционную смесь охлаждают, фильтруют через hi-flo фильтр для удаления неорганических продуктов, промывают этилацетатом и растворитель удаляют упариванием при пониженном давлении с получением коричневой смолы, которую очищают хроматографией на силикагеле (Sorbsil C30), используя 5% этилацетат в гексане в качестве элюента с получением соединения XI.110 (0,392 г). М+ = 254;1H ЯМР: δ 2,45-2,55 (2H, м); 2,65 (2H, м); 3,25-3,35 (2H, т); 4,2-4,35 (1H, м); (масло).
ПРИМЕР XI. 7
Данный пример иллюстрирует способ получения 5- (4,4-дифторбут-3-енилтио)-3-(нитрофенил)-1,2,4-тиадиазола (соединение XI.127).
Газообразный сульфид водорода барботируют в течение 40 минут через смесь метоксида калия (2,2 г) и абсолютного этанола (2,5 см3), охлаждают до ~ -10oC. Колбу снимают с охлаждающей бани, прибавляют 5-хлор-3-(нитрофенил)-1,2,4- тиадиазол (3 г) и смесь нагревают с обратным холодильником в течение 1 часа. Реакционную смесь охлаждают и выливают в эфир и полученный осадок отфильтровывают. Фильтрат, содержащий 5-меркапто-3-(нитрофенил)-1,2,4-тиадиазол, помещают в колбу и прибавляют 4,4-дифторбут-3-енилтио-4-метилбензолсульфонат (1,5 г) и добавляют ~ 1 г карбоната калия и смесь нагревают с обратным холодильником в течение 3 часов, после чего ГХ показывает практически полное расходование тозилата. Реакционную смесь выливают в этилацетат и воду и слои разделяют. Водный слой экстрагируют этилацетатом и объединенные органические слои сушат (MgS04). Упаривание при пониженном давлении дает коричневую жидкость, которую очищают флэш хроматографией (силикагель; элюент 5% этилацетат в гексане) с получением соединения XI.127 (0,901 г). М+ = 329;1H ЯМР: δ 2,60 (2H, широкий кв); 3,44 (2H, т); 4,34 (1H, м); 7,65 (1H, т); 8,32 (1H, дд); 8,6 (1H, д); (масло).
ПРИМЕР XI.8
Данный пример иллюстрирует способ, подходящий для получения соединений по изобретению, в которых атом серы в заместителе 4,
4-дифторбут-3-енилтио соответствующего неокисленного соединения (полученный по способам предшествующих примеров) окисляют до сульфоксида (сульфинил) или сульфона (сульфонил).
Соединение XI. 34 (0,85 г) перемешивают при комнатной температуре в дихлорметане (10 см3) и прибавляют 3-хлорнадбензойную кислоту (0,814 г, 1,4 экв). Через три с половиной часа ГЖХ показывает полное расходование исходных продуктов и образование двух продуктов, реакцию гасят прибавлением насыщенного водного раствора бикарбоната натрия и продукты экстрагируют дихлорметаном. Органические фазы разделяют, промывают насыщенным раствором соли и сушат над сульфатом магния. После фильтрации и концентрирования при пониженном давлении получают белый твердый продукт (1,2 г), который очищают хроматографией на силикагеле, используя 3:7 этилацетат:гексан в качестве элюента с получением сначала 5-[(4,4-дифторбут-3-енил)сульфонил]-3- метоксиметил-1,2,4-тиадиазола (соединение XI.36). М+ = 284;1H ЯМР: δ 2,55-2,65 (2H, м); 3,55 (3H, с); 3,5-3,6 (2H, т); 4,2-4,35 (1H, м); 4,80 (2H, с); (масло). Дальнейшее элюирование дает 5-[(4, 4-дифторбут-3-енил)сульфинил]-3-метоксиметил-1,2,4- тиaдиaзoл (соединение XI.35). М+ = 268;1H ЯМР: δ 2,3-2,75 (2H, м); 3,15-3,4 (2H, м); 3,55 (3H, с); 4,15-4,35 (1H, м); 4,75 (2H, с); (масло).
По вышеуказанному способу в соответствии с изобретением получают следующие соединения:
(1) 5-[(4,4-дифторбут-3-енил)сульфонил] -3-этил-1,2,4-тиадиазола
(соединение XI.24). М+ = 268;1H ЯМР: δ 1,4-1,5 (3H, т); 2,55- 2,65 (2H, м); 3,05-3,15 (2H, кв); 3,5-3,55 (2H, т); 4,2-4,4 (1H, м); (масло).
ПРИМЕР XII.
1
Методы синтеза меркапто-1,3,4-оксадиазолов, используемых в качестве промежуточных продуктов, известны. Далее проиллюстрированы два таких представительных метода.
Метод A
Получение 2-меркапто-5-метил-1,3,4-оксадиазола
К раствору гидразида уксусной кислоты (5 г) в этаноле (10 см3) прибавляют дисульфид углерода (7,7 г) и затем гидроксид калия в
этаноле (5,7 г в 20 см3), что вызывает образование осадка. Реакционную смесь перемешивают при комнатной температуре в течение 2 часов и оставляют стоять в течение ночи. Реакционную смесь
фильтруют с получением белого твердого продукта (11 г). Эту соль (5 г) обрабатывают пиридином (10 см3) и реакционную смесь нагревают с обратным холодильником всего в течение 14 часов. После
охлаждения реакционную смесь подкисляют и дважды экстрагируют диэтиловым эфиром. Эфирный слой затем сушат над сульфатом магния, фильтруют и упаривают при пониженном давлении с получением оранжевого
твердого продукта. Его перекристаллизовывают из этилацетата с получением 2-меркапто-5- метил-1,3,4-оксадиазола (0,655 г). M+ = 116;1H ЯМР: δ 2,43 (3H, с); 10,9 (1H, ушир.
с).
Метод B
Получение 5-(4-метоксибензил)-1,3,4-оксадиазол-2 (3H)тиона
Стадия 1: Получение гидразида (4-метоксифенил) уксусной кислоты
Гидразидгидрат (4,7
см3) прибавляют по каплям к этил (4-метоксифенил) ацетату (3,73 г) и затем добавляют метанол (20 см3) с образованием гомогенной реакционной смеси. Эту смесь перемешивают при
комнатной температуре в течение 18 часов, в течение которых образуется белый осадок. Осадок отделяют фильтрацией и промывают метанолом и водой, затем сушат на воздухе с получением гидразида
(4-метоксифенил)уксусной кислоты (2 г). М+ = 180;1H ЯМР: δ 3,50 (2H, с); 3,80 (3H, с); 3,85 (2H, ушир. с); 6,70 (1H, ушир. с); 6,90 (2H, д); 7,20 (2H, д); (твердый
продукт).
Следующие соединения получены по вышеуказанным методам:
(1) гидразид 2-метилпропионовой кислоты. M+ = 102; (твердый продукт).
(2) гидразид циклопропилуксусной кислоты. М+ = 100; (твердый продукт).
(3) гидразид бутановой кислоты. М+ = 102;1H ЯМР: δ 0,95 (3H, т); 1,60-1,75 (2H, м); 2,15 (2H, т); 3,90 (2H, ушир. с); 6,95 (1H, ушир. с); (твердый продукт).
(4) гидразид пропионовой кислоты. М+ = 188; (твердый продукт).
(5) гидразид пентановой кислоты.1H ЯМР: δ 0,90 (3H, т); 1,30-1,40 (2H, м); 1,60-1,70 (2H, м); 2,20 (2H, т); 3,90 (2H, ушир. с); 6,80 (1H, ушир. с); (твердый продукт).
(6) гидразид гексановой кислоты.1H ЯМР: δ 0,90 (3H, т); 1,60-1,70 (2H, м); 2,15 (2H, т); 3,90 (2H, ушир. с); 6,70 (1H, ушир. с); (твердый продукт).
(7) гидразид (4-нитрофенил)уксусной кислоты.1H ЯМР: δ (ДМСО-d6) 3,50 (2H, с); 7,50 (2H, д); 8,10 (2H, д); (твердый продукт).
(8) гидразид (2,6-дифторфенил) уксусной кислоты. М+ = 186;1H ЯМР: δ 3,60 (2H, с); 6,90-7,00 (2H, м); 7,20-7,30 (1H, м); (твердый продукт).
(9) гидразид 2-метилбензойной кислоты. М+ = 150;1H ЯМР: δ 2,45 (3H, с); 4,10 (2H, ушир. с); 7,00 (1H, ушир. с); 7,20-7,40 (4H, м); (твердый продукт).
Стадия 2: Получение 5-(4-метоксибензил)-1,3,
4-оксадиазол-2(3H) тиона
Раствор гидроксида калия (0,7 г) в воле (2 см3) прибавляют к перемешиваемому раствору гидразида (4-метоксифенил) уксусной кислоты (1,99 г) в этаноле (30
см3). Прибавляют дисульфид углерода (0,7 см3) и реакционную смесь нагревают с обратным холодильником в течение 6 часов и затем оставляют остывать Реакционную смесь упаривают
досуха при пониженном давлении и твердый остаток растворяют в воде. Устанавливают pH 1 с помощью концентрированной хлористоводородной кислоты, при этом образуется белый осадок. Осадок отделяют
фильтрацией, промывают водой и эфиром и сушат на воздухе с получением 5-(4-метоксибензил)- 1,3,4-оксадиазол-2(3H)тиона (1,96 г). М+ = 222;1H ЯМР: δ 3,80 (3H, с); 3,95 (2H,
с); 6,90 (2H, д); 7,20 (2H, д); (твердый продукт).
Следующие соединения получены по вышеуказанным методам, с использованием подходящих промежуточных продуктов (либо известных
соединений, либо полученных по стадии 1):
(1) 1,3,4-тиадиазол-2(3H)тион-5-карбоксамид. М+ = 145; (твердый продукт).
(2) 5-тиенил-1,3, 4-тиадиазол-2(3H)тион-5-карбоксамид. М+ = 184; (твердый продукт).
(3) 5-изопропил-1,3,4-тиадиазол-2(3H)тион. М+ = 144; (смола).
(4) 5-циклопропил-1,3,4-тиадиазол-2(3H)тион. М+ = 142; (смола).
(5) 5-пропил-1,3,4-тиадиазол-2(3H)тион. М+ = 144; (смола).
(6) 5-этил-1,3, 4-тиадиазол-2(3H)тион. М+ = 130; (смола).
(7) 5-(4-пиридил)-1,3,4-тиадиазол-2(3H)тион. М+ = 179;1H ЯМР: δ 7,75 (2H, д); 8,75 (2H, д); (твердый продукт).
(8) 5-бутил-1,3,4-тиадиазол-2(3H)тион.1H ЯМР: δ 0,95 (3H, т); 1,35-1,50 (2H, м); 1,65-1,75 (2H, м); 2,70 (2H, т); 2,90 (1H, ушир. с); (масло).
(9) 5-пентил-1,3,4-тиадиазол-2(3H)тион.1H ЯМР: δ 0,90 (3H, т); 1,25-1,45 (4H, м); 1,75 (2H, м); 2,70 (2H, 2 м); (масло).
(10) 5-(4-нитробензил)-1,3, 4-тиадиазол-2(3H)тион. М+= 237;1H ЯМР: δ 4,20 (2H, с); 7,50 (2H, д); 8,25 (2H, д); (твердый продукт).
(11) 5-(2,6-дифторбензил)-1,3, 4-тиадиазол-2(3H)тион.1H ЯМР: δ 4,10 (2H, с); 7,00 (2H, м); 7,25-7,40 (1H, м); (твердый продукт).
(12) 5-(4-метоксифенил)-1,3,4-тиадиазол-2(3H)тион.1H ЯМР: δ 3,80 (3H, с); 7,10 (2H, д); 7,75 (2H, д); (твердый продукт).
(13) 5-(2-метилфенил)-1,3,4-тиадиазол-2(3H)тион.1H ЯМР (ДМСО-d6): δ 2,50 (3H, с); 7,30-7,40 (2H, м); 7,40-7,50 (1H, м); 7,55 (1H, ушир. д); (твердый продукт).
(14) 5-(2-метоксифенил)-1,3,4-тиадиазол-2(3H)тион. М+= 208; (твердый продукт).
(15) 5-(нитрофенил)-1,3,4-тиадиазол-2(3H)тион. М+ = 233;1H ЯМР: δ 8,05 (2H, д); 8,30 (2H, д); (твердый продукт).
(16) 5-бензил-1,3, 4-тиадиазол-2(3H)тион.1H ЯМР: δ 4,00 (2H, с); 7,25-7,40 (5H, м); (твердый продукт).
ПРИМЕР XII.3
Данный пример иллюстрирует способ получения 2-(4,
4- дифторбут-3-енилтио)-5-фенил-1,3,4-оксадиазола (соединение XII.3).
К раствору 2-меркапто-5-фенил-1,3,4-оксадиазола (0,499 г) в ацетоне (15 см3) прибавляют карбонат калия (0,387 г) и 4,4- дифторбут-3-енил-4-метилбензолсульфонат (0,7 г) и смесь нагревают с обратным холодильником в течение 1,5 часа, за это время происходит полное расходование исходных продуктов. Реакционную смесь выливают в диэтиловый эфир и воду и слои разделяют. Водный слой экстрагируют этиловым эфиром и объединенные органические слои промывают водой и сушат (MgSO4). Упаривание растворителя при пониженном давлении дает бледно-желтую жидкость, которую очищают флэш хроматографией с получением соединения XII.3 в виде бесцветного масла, которое затвердевает при стоянии (0,293 г). М+ = 268;1H ЯМР: δ 2,58 (2H, м); 3,32 (2H, т); 4,31 (1H, м); 7,45-7,57 (3H, м); 8,01 (2H, д); (т. пл. 38-40oC).
Следующие соединения
получены с использованием подходящих промежуточных продуктов (либо известных соединений, либо полученных по примеру XII.1) по вышеописанному методу, но с 4-бром-1,1-дифторбут-2-ен алкилирующим агентом
вместо 4,4-дифторбут-3-ен-4-метилбензолсульфоната:
(1) 5-циклопропил-2-(4,4-дифторбут-3-енилтио)-1,3,4-оксадиазол (соединение XII. 1);1H ЯМР: δ 1,10-1,15 (4H, м); 2,10-2,20
(1H, м); 2,45-2,55 (2H, м); 3,20 (2H, т); 4,30 (1H, м); (масло).
(2) 2-(4,4-дифторбут-3-енилтио)-5-изопропил-1,3,4- оксадиазол (соединение XII. 8); М+ = 268;1H ЯМР: δ 1,19 (6H, д); 2,50-2,60 (2H, м); 3,15 (1H, септет); 3,30 (2H, т); 4,30 (1H, м); (масло).
(3) 5-(2,6-дифторбензил)-2-(4,4-дифторбут-3-енилтио) - 1,3,4-оксадиазол (соединение XII. 11);1H ЯМР: δ 2,45-2,55 (2H, м); 3,25 (2H, т); 4,20 (2H, с); 4,25 (1H, м); 6,90-7,00 (2H, м); 7,25-7,35 (1H, м); (масло).
(4) 2-(4, 4-дифторбут-3-енилтио)-5-изобутил-1,3,4- оксадиазол (соединение XII. 23);1H ЯМР: δ 1,00 (6H, д); 2,10-2,20 (1H, м); 2,50-2,60 (2H, м); 2,70 (2H, д); 3,25 (2H, т); 4,30 (1H, м); (масло).
(5) 2-(4,4-дифторбут-3-енилтио)-5-пентил-1,3,4-оксадиазол
(соединение XII. 25);1H ЯМР: δ 0,90 (3H, т); 1,30-1,40 (4H, м); 1,70-1,80 (2H, м); 2,
50-2,55 (2H, м); 2,80 (2H, т); 3,25 (2H, т); 4,30 (1H, м); (масло).
(6) 5-бутил-2-(4,4-дифторбут-3-енилтио)-1,3,4-оксадиазол (соединение XII. 1);1H ЯМР: δ 1,10-1,15 (4H, м); 2,10-2,20 (1H, м); 2,45-2,55 (2H, м); 3,20 (2H, т); 4,30 (1H, м); (масло).
(7) 5-бутил-2-(4,4-дифторбут-3-енилтио)-1,3,4-оксадиазол (соединение XII. 28);1H ЯМР: δ 0,95 (3H, т); 1,35-1,50 (2H, м); 1,70-1,80 (2H, м); 2,50-2,60 (2H, м); 2,80 (2H, т); 3,25 (2H, т); 4,30 (1H, м); (масло).
(8) 2-(4,4-дифторбут-3-енилтио)-5-пропил-1,3, 4-оксадиазол (соединение XII. 31);1H ЯМР: δ 1,00 (3H, т); 1,75-1,90 (2H, м); 2,50-2,60 (2H, м); 2,80 (2H, т); 3,25 (2H, т); 4,30 (1H, м); (масло).
(9) 2-(4, 4-дифторбут-3-енилтио)-5-этил-1,3,4-оксадиазол (соединение XII. 35);1H ЯМР: δ 1,40 (3H, т); 2,50-2,60 (2H, м); 2,85 (2H, т); 3,25 (2H, т); 4,30 (1H, м); (масло).
(10) 2-(4,4-дифтopбут-3-eнилтиo)-5-метил-1,3,4-оксадиазол (соединение XII. 49);1H ЯМР: δ 2,51 (2H, м); 2,73 (3H, с); 3,27 (2H, т); 4,25 (1H, м); (масло).
(11) 2-(4, 4-дифторбут-3-енилтио)-1,3,4-оксадиазол-5-карбоксамид (соединение XII. 55); М+ = 235;1H ЯМР: δ 2,50-2,60 (2H, м); 3,35 (2H, т); 4,30 (1H, м); (т. пл. 113o C).
(12) 2-(4,4-дифторбут-3-енилтио)-5-(2-метилфенил) - 1,3,4-оксадиазол (соединение XII. 128);1H ЯМР: δ 2,55-2,65 (2H, м); 2,70 (3H, с); 3,35 (2H, т); 4,30 (1H, м); 7,30-7,45 (3H, м); 7,90 (1H, д); (масло).
(13) 2-(4,4-дифторбут-3-енилтио)-5-(2-фурил)-1,3,4- оксадиазол (соединение XII. 131); М+ = 258;1H ЯМР: δ 2, 50-2,60 (2H, м); 3,35 (2H, т); 4,30 (1H, м); 6,60 (1H, м); 7,10 (1H, д); 7,60 (1H, д); (масло).
(14) 2-(4,4-дифторбут-3-енилтио)-5-(метоксифенил)-1,3,4- оксадиазол (соединение XII. 132);1H ЯМР: δ 2,50-2,60 (2H, м); 3,30 (2H, т); 3,95 (3H, с); 4,30 (1H, м); 7,10 (2H, м); 7,50 (1H, дт); 7,90 (2H, дд); (т. пл. 35-37oC).
(15) 2-(4, 4-дифторбут-3-енилтио)-5-(2-тиенил)-1,3,4- оксадиазол (соединение XII. 133); М+ = 274;1H ЯМР: δ 2,50-2,60 (2H, м); 3,30 (2H, т); 4,30 (1H, м); 3,20 (2H, т); 4,30 (1H, м); 7,10-7,20 (3H, м); 7,55 (1H, д); 7,70 (1H, д); (масло).
(16) 2-(4,4-дифторбут-3-енилтио)-5-(3-фурил)-1,3,4- оксадиазол (соединение XII. 134);1H ЯМР: δ 2,50-2,60 (2H, м); 3,30 (2H, т); 4,30 (1H, м); 6,90 (1H, м); 7,50-7,55 (1H, м); 8,05 (1H, ушир. с); (масло).
(17) 2-(4,4-дифторбут-3-енилтио)-5-(4-метоксифенил) - 1,3,4-оксадиазол (соединение XII. 144);1H ЯМР: δ 2,50-2,60 (2H, м); 3,30 (2H, т); 3,90 (3H, т); 3,90 (3H, с); 4,30 (1H, м); 7,00 (2H, д); 8,00 (2H, д); (масло).
(18) 2-(4, 4-дифторбут-3-енилтио)-5-(4-пиридил)-1,3,4-оксадиазол (соединение XII. 148); M+ = 269;1H ЯМР: δ 2,50-2,65 (2H, м); 3,40 (2H, т); 4,30 (1H, м); 7,90 (1H, д); 8,80 (2H, д); (масло).
ПРИМЕР XII.3
Данный пример иллюстрирует способ получения 2-(4,4-дифторбут-3-енилсульфинил)-5-фенил-1,3,4-оксадиазола (соединение XII.4).
К раствору соединения XII.3 (1 г) в сухом дихлорметане, перемешиваемому при 0oC, прибавляют 3-хлорнадбензойную кислоту (1,3 г 50%-ной по весу твердой, 1 эквивалент). Реакционной смеси дают достичь комнатной температуры, перемешивают в течение 3 часов и оставляют на ночь. ТСХ показывает полное расходование исходного продукта. Реакционную смесь фильтруют и фильтрат распределяют между дихлорметаном и раствором бикарбоната натрия. Водный слой экстрагируют дихлорметаном и объединенные органические фазы сушат над сульфатом магния. Упаривание растворителя при пониженном давлении дает желтое масло, которое очищают флэш хроматографией на силикагеле, элюируя 25%-ным этилацетатом в гексане с получением соединения XII.4 (0,474 г). М+ = 284;1H ЯМР: δ 2,67 (2H, м); 3,52 (2H, м); 4,32 (1H, м); 7,50 (3H, м); 8,12 (2H, м); (масло).
Следующие соединения по изобретению получены из соответствующих тиоэфиров с использованием подходящих
промежуточных продуктов по вышеописанному методу:
(1) 2-(4,4-дифторбут-3-енилсульфинил)-5- (4-метоксибензил)-1,3,4-оксадиазол (соединение XII.14);1H ЯМР: δ 2,45-2,70 (2H,
м); 3,30-3,50 (2H, м); 3,80 (3H, с); 4,25 (1H, м); 4,25 (2H, с); 6,90 (2H, д); 7,25 (2H, д); (масло).
(2) 2-(4,4-дифторбут-3-енилсульфинил)-5-пентил-1,3,4- оксадиазол (соединение XII. 26);1H ЯМР: δ 0,95 (3H, т); 1,30-1,40 (2H, м); 1,80-1,90 (2H, м); 2,50-2,75 (2H, м); 2,95 (2H, т); 3,35-3,55 (2H, м); 4,30 (1H, м); (масло).
(3) 5-бутил-2-(4, 4-дифторбут-3-енилсульфинил)-1,3,4- оксадиазол (соединение XII. 29);1H ЯМР: δ 1,00 (3H, т); 1,40-1,50 (2H, м); 1,80-1,90 (2H, м); 2,50-2,75 (2H, м); 3,00 (2H, т); 3,35-3,55 (2H, м); 4,30 (1H, м); (масло).
(4) 2-(4,4-дифторбут-3-енилсульфинил)-5-пропил-1,3,4-оксадиазол (соединение XII. 32);1H ЯМР: δ 1,05 (3H, т); 1,80-1,95 (2H, м); 2,50-2,75 (2H, м); 2,95 (2H, т); 3,35-3,55 (2H, м); 4,30 (1H, м); (масло).
(5) 2-(4,4-дифторбут-3-енилсульфинил)-5-(2-метилфенил) -1,3,4-оксадиазол (соединение XII. 129);1H ЯМР: δ 2,55-2,80 (2H, м); 2,70 (3H, с); 3,40-3,60 (2H, м); 4,30 (1H, м); 7,30-7,40 (1H, м); 7,45-7,50 (1H, м); 8,00 (1H, д); (масло).
(6) 2-(4,4-дифтоpбут-3-eнилcульфинил)-5-(4-нитрофенил) 1,3,4-оксадиазол (соединение XII. 142);1H ЯМР: δ 2,55-2,80 (2H, м); 3,45-3,70 (2H, м); 4,35 (1H, м); 8,35 (2H, д); 8,45 (2H, д); (масло).
(7) 2-(4, 4-дифторбут-3-енилсульфинил)-5-(4-метоксифенил)-1,3,4-оксадиазол (соединение XII.145);1H ЯМР: δ 2,50-2,80 (2H, м); 3,40-3,60 (2H, м); 3,90 (3H, с); 4,30 (1H, м); 7,05 (2H, д); 8,05 (2H, д); (масло).
Следующие соединения по изобретению получены из соответствующих тиоэфиров по общему методу, описанному выше, но с 2 эквивалентами 3-хлорнадбензойной кислоты в
качестве окислителя:
(8) 2-(4,4-дифторбут-3-енилсульфонил)-5-фенил-1,3,4- оксадиазол (соединение XII. 5); М+ = 284;1H ЯМР: δ 2,71 (2H, м); 3,67 (2H, м); 4,33 (1H,
м); 7,52-7,03 (3H, м); 8,25 (2H, д); (т. пл. 104-106oC).
(9) 2-(4,4-дифторбут-3-енилсульфонил)-5-изопропил-1,3,4- оксадиазол (соединение XII. 9);1H ЯМР: δ 1,50 (6H, д); 2,60-3,70 (2H, м); 3,20-3,40 (1H, м); 4,30 (1H, м); (смола).
(10) 2-(4,4-дифторбут-3-енилсульфонил)-5- (4-нитробензил)-1,3,4-оксадиазол (соединение XII.13);1H ЯМР: δ 2,60-2,70 (2H, м); 3,60 (2H, т); 4,30 (1H, м); 4,40 (1H, м); 7,55 (2H, д); 8,25 (2H, д); (масло).
(11) 2-(4, 4-дифтopбут-3-eнилcульфoнил)-5- (4-метоксибензил)-1,3,4-оксадиазол (соединение XII. 15);1H ЯМР: δ 2,60-2,70 (2H, м); 3,65 (2H, т); 3,80 (3H, с); 4,20 (1H, м); 4,25 (2H, с); 6,90 (2H, л); (т. пл. 60-63oC).
(12) 5-бензил-2-(4,4-дифторбут-3-енилсульфонил) -1,3,4-оксадиазол (соединение XII. 19);1H ЯМР: δ 2,60 (2H, м); 3,60 (2H, т); 4,25 (1H, м); 7,25-7,40 (5H, м); (масло).
(13) 2-(4,4-дифторбут-3-енилсульфонил)-5-пентил-1,3,4- оксадиазол (соединение XII. 27);1H ЯМР: δ 0,95 (3H, т); 1,30-1,45 (4H, м); 1,80-1,90 (2H, м); 2,60-2,70 (2H, м); 3,00 (2H, т); 3,60 (2H, т); 4,30 (1H, м); (масло).
(14) 5-бутил-2-(4,4-дифторбут-3-енилсульфонил)-1,3,4- оксадиазол (соединение XII. 30);1H ЯМР: δ 1,00 (3H, т); 1,40-1,50 (2H, м); 1,80-1,90 (2H, м); 2,60-2,70 (2H, м); 3,00 (2H, т); 3,60 (2H, т); 4,30 (1H, м); (масло).
(15) 2-(4, 4-дифтopбут-3-eнилcульфoнил)-5-пропил-1,3,4- оксадиазол (соединение XII. 33);1H ЯМР: δ 1,10 (3H, т); 1,85-2,00 (2H, м); 2,60-2,70 (2H, м); 2,95 (2H, т); 3,60 (2H, т); 4,30 (1H, м); (масло).
(16) 2-(4,4-дифторбут-3-eнилcульфoнил)-метил-1,3,4- оксадиазол (соединение XII. 51);1H ЯМР: δ 2,20-2,30 (2H, м); 2,30 (3H, с); 3,60 (2H, т); 4,30 (1H, м); (масло).
(17) 2-(4,4-дифторбут-3-енилсульфонил)-5-(2-метилфенил)- 1,3,4-оксадиазол (соединение XII. 130);1H ЯМР: δ 2,70-2,80 (2H, м); 2,75 (3H, с); 3,70 (2H, т); 4, 30 (1H, м); 7,40 (1H, д); 7,50 (1H, д); 8,00 (1H, д); (т. пл. 90-93oC).
(18) 2-(4,4-дифторбут-3-енилсульфонил)-5-(4-нитрофенил)- 1,3,4-оксадиазол (соединение XII.143); М+ = 284;1H ЯМР: δ 2,70-2,80 (2H, м); 3,70 (2H, т); 4,35 (1H, м); 8,35 (2H, д); 8,45 (2H, д); (масло).
(19) 2-(4, 4-дифторбут-3-енилсульфонил)-5-(4-метоксифенил) - 1,3,4-оксадиазол (соединение XII.146);1H ЯМР: δ 2,70-2,80 (2H, м); 3,65 (2H, т); 3,90 (3H, с); 4,30 (1H, м); 7,05 (2H, д); 8,10 (2H, д); (т. пл. 60oC).
ПРИМЕР XIII.1
Данный пример иллюстрирует общий способ получения 2-(4,4-дифторбут-3-eнилтио)-5-зaмeщeнныx-1,3,4-тиaдиaзoлoв из соответствующего
тиадиазол-2(3H)-тиона, соединения, которое хорошо известно. Этот способ показан на получении 2-(4,4-дифторбут-3- енилтио)-5-метиламино-1,3,4-оксадиазола (соединения XIII. 70) из соответствующего тиона
и 4,4-дифторбут-3-енил-4 -метилбензолсульфоната. Может быть также использован другой алкилирующий агент, например, 4-бром-1,1-дифторбут-1-ен.
Получение соединения XIII.70.
К раствору 5-метиламино-1,3,4-тиадиазол-2(3H)-тиона (0,393 г) в ацетоне (10 см3) прибавляют карбонат калия (0,396 г) и 4,4-дифторбут-3-енил-4-метилбензолсульфонат (0,7 г) и смесь нагревают с обратным холодильником в течение 3 часов, после чего ГХ показывает полное расходование исходных продуктов. Реакционную смесь фильтруют через hi-flo фильтр и тщательно промывают диэтиловым эфиром. Фильтрат выливают в эфир и воду и слои разделяют. Водный слой дважды экстрагируют эфиром и объединенные органические фазы сушат (MgSO4). Упаривание растворителя при пониженном давлении дает коричневое масло, которое очищают флэш хроматографией на силикагеле, элюируя 1:1 этилацетатом:гексаном с получением соединения XII.70 (0,273 г). М+ = 273;1H ЯМР: δ 2,43 (2H, м); 3,04 (3H, с); 3,15 (2H, т); 4,29 (1H, м); 5,36 (1H, ушир. с); (т. пл. 52,5-53,5oC).
Следующие соединения по изобретению были получены по
вышеуказанному способу с использованием подходящих меркаптотиадиазолов и в некоторых случаях 4-бром-1,1-дифторбут-1- ена в качестве алкилирующего агента
(1) 5-циклопропил-2-(4,
4-дифтopбут-3-eнилтиo)-1,3,4- тиадиазол (соединение XIII. 6); M+ = 248;1H ЯМР: δ 1,10-1,30 (4H, м); 2,30-2,40 (1H, м); 2,45-2,55 (2H, м); 3,30 (2H, т); 4,30 (1H, м);
(масло).
(2) 2-(4,4-дифторбут-3-енилтио)-5-фенил-1,3,4-тиадиазол (соединение XIII. 9); М+ = 284;1H ЯМР: δ 2,50-2,65 (2H, м); 3,40 (2H, т); 4,50 (1H, м); 7,40-7,50 (3H, м); 7,85-7,95 (2H, м); (т. пл. 39oC).
(3) 2-(4,4-дифторбут-3-енилтио)-5-изопропил-1,3,4-тиадиазол (соединение XIII.16); М+ = 250;1H ЯМР: δ 1,40 (3H, с); 1,45 (3H, с); 2,45-2,60 (2H, м); 3,35-3,50 (1H, м); 4,30 (1H, м); (масло).
(4) 5-бензил-2-(4,4-дифторбут-3-енилтио)-1,3,4-тиадиазол (соединение XIII.20); М+ = 298;1H ЯМР: δ 2,45-2,55 (2H, м); 3,30 (2H, т); 4,30 (1H, м); 4,40 (2H, с); 7,25-7,40 (2H, м); (масло).
(5) 2-(4,4-дифтopбут-3-eнилтиo)-5-метил-1,3, 4-тиадиазол (соединение XIII.40); М+ = 222;1H ЯМР: δ 0,50 (2H, м); 2,73 (3H, с); 3,35 (2H, т); 4,29 (1H, м); (масло).
(6) 2-(4,4-дифторбут-3-енилтио)-1,3, 4-тиадиазол-5- карбоксамид (соединение XIII. 45); М+ = 151;1H ЯМР: δ 2,50-2,60 (2H, м); 3,45 (2H, т); 4,30 (1H, м); 6,80 (1H, ушир. с); 7,15 (1H, ушир. с); (т. пл. 168oC).
(7) 2-(4,4-дифторбут-3-енилтио)-1,3,4-тиадиазол (соединение XIII.63); M+ = 208;1H ЯМР: δ 2,54 (2H, м); 3,43 (2H, т); 4,30 (1H, м); 9, 03 (1H, с); (масло).
(8) 5-амино-2-(4,4-дифторбут-3-енилтио) -1,3,4-тиадиазол (соединение XIII.69); М+ = 223;1H ЯМР: δ 2,45 (2H, м); 3,20 (2H, т); 4,30 (1H, м); 5,20 (2H, ушир. с); (т. пл. 138oC).
(9) 2-(4,4-дифторбут-3-енилтио)-5- (3-трифторметилбензилтио)-1,3,4-тиадиазол (соединение XIII.110); М+ = 304;1H ЯМР: δ 2,51 (2H, м); 3,31 (2H, т); 4,27 (1H, м); 4,56 (2H, с); 7,42-7,70 (4H, м); (масло).
(10) 5-циклoпpoпилмeтилтиo-2-(4,4-дифторбут-3-eнилтиo)- 1,3,4-тиадиазол (соединение XIII.114); М+ = 294;1H ЯМР: δ 0,35 (2H, м); 0,65 (2H, м); 1,25 (1H, м); 2,50 (2H, м); 3,25 (2H, т); 4,25 (1H, м); (масло).
(11) 2-(4, 4-дифторбут-3-енилтио)-5-метилтио-1,3,4- тиадиазол (соединение XIII. 119); М+ = 254;1H ЯМР: δ 2,51 (2H, м); 2,77 (3H, с); 3,31 (2H, т); 4,20 (2H, т); (масло).
(12) 2-(4,4-дифторбут-3-eнилтиo)-5-(сульфонамидофенил)- 1,3,4-тиадиазол (соединение XIII.143); М+ = 363;1H ЯМР: δ 2,55-2,65 (2H, м); 3,50 (2H, т); 4,30 (1H, м); 4,95 (2H, ушир. с); 8,05 (4H, м); (т. пл. 154oC).
ПРИМЕР XIII.2
Данный пример иллюстрирует общий способ получения 2-(4,4-дифторбут-3-енилтио)-5-замещенных-1,3,
4- тиадиазолов из 2-амино-5-замещенных-1,3,4-тиадиазолов. Этот способ показан на получении 2- (4, 4-дифторбут-3-енилтио) -5- этил-1,3,4-тиадиазола (соединения XIII.27) из 2-амино-5-этил- 1,3,
4-тиадиазола.
Получение соединения XIII.27.
Раствор 2-амино-5-этил-1,3,4-тиадиазола (0,786 г) и 4,4- дифторбут-3-енилдисульфида (1,5 г) в дихлорметане (25 см3) перемешивают на ледяной бане. Прибавляют трет-бутилнитрит (1,2 г), удаляют охлаждающую баню и реакционную смесь нагревают с обратным холодильником в течение 1,3 часа. Затем реакционную смесь выливают в диэтиловый эфир/воду и слои разделяют. Водный слой экстрагируют эфиром и объединенные органические фазы сушат (MgSO4), фильтруют, упаривают при пониженном давлении с получением коричневого масла. Очистка с помощью колоночной хроматографии на силикагеле с использованием 1: 9 и 2: 8 этилацетата: гексана в качестве элюента дает соединение XIII.27 (0,959 г). М+ = 236;1H ЯМР: δ 1,40 (3H, т); 2,45-2,60 (2H, м); 3,10 (2H, кв); 3,35 (2H, т); 4,30 (1H, м); (масло).
Следующие соединения по изобретению были получены по вышеуказанному
способу с использованием подходящих промежуточных соединений:
(1) 5-бром-2-(4,4-дифторбут-3-енилтио)-1,3,4- тиадиазол (соединение XIII. 1); М+ = 286;1H ЯМР: δ 2,
45-2,60 (2H, м); 3,40 (2H, т); 4,30 (1H, м); (масло).
(2) 2-(4,4-дифторбут-3-енилтио)-5-трет-бутил-1,3,4- тиадиазол (соединение XIII. 3); М+ = 264;1H ЯМР: δ 1,48 (9H, с); 2,45- 2,60 (2H, м); 3,35 (2H, т); 4,30 (1H, м); (масло).
(3) 2-(4,4-дифторбут-3-енилтио)-5-трифторметил-1,3,4- тиадиазол (соединение XIII. 14); М+ = 276;1H ЯМР: δ 2,50-2,60 (2H, м); 3,45 (2H, т); 4,30 (1H, м); (масло).
ПРИМЕР XIII.3
Данный пример иллюстрирует способ получения 2,5-бис-(4,
4-дифторбут-3-енилсульфинил)-1,3,4-оксадиазола (соединение XIII.133) с использованием 3-хлорнадбензойной кислоты в качестве окислителя.
Раствор соединения XIII.17 (0,49 г) в дихлорметане (30 см3) охлаждают на бане из метанола со льдом до ~ -10oC, прибавляют 3-хлорнадбензойную кислоту (1 г 50%-ной по весу твердой, 1 эквивалент) и реакционную смесь перемешивают, давая постепенно достичь комнатной температуры, затем перемешивают в течение 7 часов и оставляют на 18 часов. Затем смесь выливают в раствор бикарбоната натрия и продукт экстрагируют диэтиловым эфиром. Объединенные органические фазы промывают раствором бикарбоната натрия и сушат (MgSO4). Упаривание растворителя при пониженном давлении дает желтую жидкость, которую очищают флэш хроматографией на силикагеле, элюируя 30%-ным этилацетатом в гексане с получением соединения XIII.133 (0,168 г). М+ = 362;1H ЯМР: δ 2,58 (4H, м); 3,36 (4H, м); 4,26 (2H, м); (т. пл. 46-48oC).
Следующие соединения по изобретению получены с использованием подходящего числа эквивалентов 3-хлорпербензойной кислоты в качестве окислителя.
(1) 2-(4,4-дифторбут-3-енилсульфинил)-5-фенил-1,3,4- тиадиазол (соединение XIII.10); М+ = 300;1H ЯМР: δ 2,40-2,80 (2H, м); 3,30-3,40 (2H, м); 4,30 (1H, м); 7,45-7,60 (3H, м); 7,95-8,00 (2H, м); (т. пл. 67oC).
(2) 2-(4,4-дифторбут-3-енилсульфонил)-5-фенил-1,3,4- тиадиазол (соединение XIII. 11); М+ = 316;1H ЯМР: δ 2,60-2,75 (2H, м); 3,65 (2H, т); 4,30 (1H, м); 7,45-7,65 (3H, м); 7,95-8,05 (2H, м); (т. пл. 80oC).
(3) 2-(4, 4-дифторбут-3-енилсульфинил)-5-метил-1,3,4- тиадиазол (соединение XIII.41); М+ = 238;1H ЯМР: δ 2,35-2,55 (1H, м); 2,55-2,75 (1H, м); 2,90 (3H, с); 3,20-3,40 (2H, м); 4,30 (1H, м); (масло).
(4) 2-(4,4-дифторбут-3-енилсульфонил)-5-метил-1,3,4- тиадиазол (соединение XIII. 42); М+ = 255;1H ЯМР: δ 2,55-2,70 (2H, м); 2,95 (3H, с); 3,60 (2H, т); 4,30 (1H, м); (масло).
(5) 2-(4,4-дифторбут-3-енилсульфинил)-1,3,4-тиадиазол (соединение XIII. 64); М+ = 225;1H ЯМР: δ 2,35-2,50 (1H, м); 2,60-2,75 (1H, м); 3,25-3,45 (2H, м); 4,30 (1H, м); 4,30 (1H, м); 9,35 (1H, с); (масло).
(6) 2-(4,4-дифторбут-3-eнилcульфoнил)-1,3,4- тиадиазол (соединение XIII. 65); MH+ = 241;1H ЯМР: δ 2,60-2,70 (2H, м); 3,70 (2H, т); 4,30 (1H, м); 9,40 (1H, с); (масло).
(7) 2,5-бис-(4,4-дифторбут-3-енилсульфонил)-1,3,4- тиадиазол (соединение XIII.124); М+ = 394;1H ЯМР: δ 2,69 (4H, м); 3,70 (4H, т); 4,30 (2H, м); (т. пл. 88-91oC).
(8) 2-(4, 4-дифторбут-3-eнилcульфoнил)-5- (4,4-дифторбут-3-енилсульфинил)-1,3,4-тиадиазол (соединение III. 134); M+ = 378;1H ЯМР: δ 2,38-2,55 (1H, M); 2,60-2,79 (3H, м); 3,38 (2H, м); 3,62-3,74 (2H, м); 4,19- 3,74 (2H, м); (т. пл. 45-47oC).
Следующие соединения по изобретению получены с использованием подходящего числа эквивалентов магний монопероксифталата в качестве окислителя.
(9) 5-бром-2-(4,4-дифторбут-3-енилсульфонил)-1,3,4- тиадиазол (соединение XIII. 2); MH+ = 319;1H ЯМР: δ 2,60-2, 70 (2H, м); 3,65 (2H, т); 4,30 (1H, м); (масло).
(10) 2-(4,4-дифторбут-3-енилсульфонил)-5-трет-бутил-1,3,4- тиадиазол (соединение XIII. 4); MH+ = 297;1H ЯМР: δ 1,55 (9H, с); 2,60-2,70 (2H, м); 3,65 (2H, т); 4,30 (1H, м); (т. пл. 37oC).
(11) 5-циклопропил-2-(4,4-дифторбут-3-енилсульфонил)- 1,3,4-тиадиазол (соединение XIII.7); MH+ = 281;1H ЯМР: δ 1,25-1,45 (4H, м); 2,40-2,55 (1H, м); 2,55-2,65 (2H, м); 3,60 (2H, т); 4,30 (1H, м); (масло).
(12) 2-(4, 4-дифторбут-3-енилсульфонил)-5-трифторметил-1,3,4-тиадиазол (соединение XIII.15); M-SO2H+ = 243;1H ЯМР: δ 2,60-2,75 (2H, м); 3,75 (2H, т); 4,20-4,40 (1H, м); (смола).
(13) 2-(4,4-дифтopбут-3-eнилcульфoнил)-5-изопропил-1,3,4-тиадиазол (соединение XIII. 17); М+ = 283;1H ЯМР: δ 1,50 (3H, с); 1,55 (3H, с); 2,60-2, 70 (2H, м); 3,50- 3,70 (3H, м); 4,30 (1H, м); (т. пл. 43oC).
(14) 2-(4,4-дифторбут-3-енилсульфинил)-5-этил-1,3,4-оксадиазол (соединение XIII. 28);1H ЯМР: δ 1,50 (3H, т); 2,40-2,50 (1H, м); 2,60-2,70 (1H, м); 3,20 (2H, кв); 3,25-3,35 (2H, м); 4,25 (1H, м); (смола).
(15) 2-(4,4-дифторбут-3-енилсульфонил)-5-этил-1,3,4-оксадиазол (соединение XIII. 29); М+ = 269;1H ЯМР: δ 1,50 (3H, т); 2,60-2,70 (2H, м); 3,20 (2H, кв); 3,65 (2H, т); 4,30 (1H, м); (смола).
ПРИМЕР XIII.4
Данный пример иллюстрирует способ получения 2-(4,4-дифторбут-3-енилтио)-5-метокси-1,3,4-тиадиазола (соединение XIII.101) из соединения XIII.1.
К перемешиваемой суспензии гидрида натрия (0,030 г) в толуоле (3 см3) прибавляют метанол (0,022 г), что приводит к выделению газа. После перемешивания в течение 10 минут прибавляют соединение XII.1 (0,20 г) и реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Реакционную смесь анализируют ГХ и затем порциями прибавляют гидрид натрия и метанол до определения полного расходования исходных продуктов. Реакционную смесь выливают в воду и слои разделяют. Продукт экстрагируют диэтиловым эфиром и объединенные органические фазы сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением желтого масла. Очистка с помощью колоночной хроматографии на силикагеле с использованием 3:17 эфира: гексана в качестве элюента дает соединение XIII.101 (0,069 г). М+ = 238;1H ЯМР: δ 2,40-2,55 (2H, м); 3,25 (2H, т); 4,20 (3H, с); 4,30 (1H, м); (масло).
ПРИМЕР XIV.1
Данный пример иллюстрирует способ получения 5-(4,
4-дифторбут-3-енилтио)-1-метилтетразола (соединение XIV.1).
Натриевую соль 5-меркапто-1-метилтетразола алкилируют 4-бром-1,1-дифторбут-1-еном по способу примера XIII.1 с получением соединения XIV.1. М+ = 206;1H ЯМР: δ 2,53 (2H, м); 3,38 (2H, т); 3,92 (3H, с); 4,28 (1H, м); (масло).
ПРИМЕР XV.1
Данный пример иллюстрирует
способ получения 1-(4,4-дифторбут-3-енилтио)-4-нитробензола (соединение XV.1).
4-Hитротиофенол (0,448 г), карбонат калия (0,448 г), 4,4-дифторбут-3-енил-4-метилбензолсульфонат (0,846 г) и иодид калия (0,388 г) перемешивают и нагревают с обратным холодильником в ацетоне (15 см3) всего в течение б часов, после чего никаких исходных продуктов не определяется с помощью ТСХ. Реакционную смесь выливают в воду и экстрагируют 3 порциями этилацетата. Объединенные органические фазы промывают 3 раза 2М NaOH и насыщенным солевым раствором и затем сушат (MgSO4). Удаление растворителя путем упаривания при пониженном давлении дает темно-желтое масло, которое очищают флэш хроматографией на силикагеле, используя 5% этилацетат в гексане в качестве элюента с получением соединения XV.1 (0,474 г). M+ = 245;1H ЯМР: δ 2,42 (2H, м); 3,09 (2H, т); 4,30 (1H, м); 7,35 (2H, д); 8,14 (2H, д); (масло).
ПРИМЕР XVI.1
Данный пример иллюстрирует общий способ получения 2-хлор-4-(4,4-дифторбут-3-енилтио)пиридина (соединение XVI.1).
Трет-бутилнитрит (0,442 г) в дихлорметане (20 см3) прибавляют по каплям к раствору 4-амино-2-хлорпиридина и бис-(4,4-дифторбут-3-енил)дисульфида (1,9 г) в дихлорметане (20 см3), перемешивают смесь при 0oС . Реакционную смесь перемешивают в течение 4 часов и затем оставляют стоять при комнатной температуре в течение 18 часов. Затем прибавляют воду и продукт экстрагируют этилацетатом. Объединенные органические фазы промывают насыщенным раствором соли, сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением оранжево-коричневой смолы. Хроматографии на sorbsil-C30 с использованием 4% этилацетата в гексане в качестве элюента дает соединение XVI.1 (0,134 г). М+ = 235;1H ЯМР: δ 2,35-2,46 (2H, м); 2,98-3,7 (2H, т); 4,18 (1H, м); 7,02 (1H, д); 7,11 (1H, д); 8,14-8,21 (1H, д); (масло).
ПРИМЕР XVI.2
Данный пример иллюстрирует двухстадийный способ получения 4-(4,4-дифторбут-3-енилтио)-2,3,5,6-тетрафторпиридина (соединение
XVI.2).
Стадия 1: Получение натриевой соли 2,3,5,6- тетрафторпиридин-4-тиола
4-Хлор-2,3,5,6-тетрафторпиридина (2 г) и дигидрат гидросульфида натрия перемешивают и нагревают с
обратным холодильником в изопропаноле (40 см3) в течение 3 часов. Смесь затем перемешивают при комнатной температуре в течение 18 часов. Твердый осадок удаляют фильтрацией, промывают
диэтиловым эфиром и отбрасывают. Объединенные органические растворы упаривают при пониженном давлении с получением 2,3,5,6- тетрафторпиридин-4- тиола в виде его натриевой соли (2,21 г), которую
используют на второй стадии без дополнительной очистки.
Стадия 2: Получение соединения XVI.2.
Промежуточный продукт со стадии 2 (1,7 г), 4-бром-1,1-дифторбут-1-ен (1, 99 г) и карбонат калия (1,53 г) перемешивают и нагревают с обратным холодильником в ацетоне (30 см3) в течение 18 часов. Неорганические твердые продукты удаляют фильтрацией и фильтрат упаривают при пониженном давлении с получением темно-коричневого масла. Хроматография на sorbital-C30, с использованием гексана в качестве элюента получают соединение XVI.2 (1,82 г). М+ = 273;1H ЯМР: δ 2,3-2,42 (2H, м); 3,15-3,25 (2H, т); 4,15-4,34 (1H, м); (масло).
Следующие соединения по изобретению получены с использованием способа со стадии 2
выше. Алкилирующим агентом является 4-бром-1,1-дифторбут-2-ен или 4,4-дифторбут-3-ен-4-метилбензолсульфонат:
(1) 4-(4,4-дифторбут-3-енилтио)пиридин (соединение XVI.5); М+ = 201;1H ЯМР: δ 2,40 (2H, м); 3,04 (2H, т); 4,30 (1H, м); 7,11 (2H, д); 8,41 (2H, д); (масло) из 4-меркаптопиридина.
(2) 4, 4-дифторбут-3-енил-2-(4, 4-дифторбут-3- енилтио)пиридин-3-карбоксилат (соединение XVI. 10); М+ = 335;1H ЯМР: δ 2,43 (1H, м); 3,22 (2H, т); 4,29 (2H, м); 4,36 (2H, т); 7,09 (1H, дд); 8,20 (1H, дд); 8,57 (1H, дд); (масло) из 2-меркаптопиридин- 3-карбоновой кислоты. Для преобразования двух эквивалентов 4,4- дифторбут-3-енил-4-метилбензолсульфоната в более реакционноспособный 4-иод-1, 1-дифторбут-1-ен in situ в данной реакции используют иодид калия.
(3) 2-(4,4-дифторбут-3-енилтио)-5-трифторметилпиридин (соединение XVI. 11);1H ЯМР: δ 2,40 (2H, м); 3,25 (2H, т); 4,25 (1H, м); 7,25 (1H, дд); 7,45 (1H, дд); 8,49 (1H, д); (масло) из 2-меркаптопиридин-5-трифторметилпиридина.
(4) 2-(4,4-дифторбут-3-eнилтиo)пиридин (соединение XVI.19); М+ = 201;1H ЯМР: δ 2,40 (2H, м); 3,20 (2H, т); 4,30 (1H, м); 6,90 (1H, дд); 7,20 (1H, д); 7,45 (1H, тд); 8,40 (1H, дд); (масло).
(5) 2-(4, 4-дифторбут-3-енилтио)-5-нитропиридин (соединение XVI.21); М+ = 246;1H ЯМР: δ 2,45 (2H, м); 3,30 (2H, т); 4,28 (1H, м); 7,30 (1H, д); 8,23 (1H, тд); 9,25 (1H, д); (масло) из 2-меркапто-5-нитропиридина.
ПРИМЕР XVI.3
Данный пример представляет общий способ получения 2-(4,4- дифторбут-3-енилтио)-5-замещенных пиридинов из 2-хлор-5-замещенных
пиридинов. Способ проиллюстрирован получением 5-хлор-2-(4,4-дифторбут-3-енилтио)пиридина (соединение XVI.13).
Дигидрат гидросульфида натрия (0,672 г) прибавляют к раствору 2,
5-дихлорпиридина (1,43 г) в диметилформамиде (20 см3),
от чего смесь становится голубой, а затем зеленой при нагревании до 100oC. Реакционную смесь затем нагревают в
течение 2 часов и дают ей остыть. Реакционную смесь выливают в диэтиловый эфир и 2М HCl и слои разделяют. Водный слой экстрагируют эфиром. Объединенные органические фазы промывают 2М HCl, водой и
солевым раствором (3 раза каждым), сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением коричневого масла. Колоночная хроматография на силикагеле с использованием
2%-ного диэтилового эфира в гексане дает соединение XVI.13 (0,805 г). M+ = 235;1H ЯМР: δ 2,40 (2H, м); 3,20 (2H, т); 4,25 (1H, м); 7,15 (1H, дд); 7,45 (1H, дд); 8,40;
(масло).
По вышеописанному способу были получено следующее соединение:
(1) 5-циано-2-(4,4-дифторбут-3-енилтио)пиридин (соединение XVI.15);1H ЯМР: δ 2,40
(2H, м); 3,25 (2H, т); 4,25 (1H, м); 7,25 (1H, дд); 7,70 (1H, дд); 8,65 (1H, м); (т. пл. 34oC).
ПРИМЕР XVI.4
Данный пример представляет общий способ получения 2-(4,
4-дифторбут-3-енилтио)-3-замещенных пиридинов из 2-хлор-3-замещенных пиридинов и гидробромида 4,4-дифтор-3-енилизотиоурония. Способ проиллюстрирован получением 2-(4,
4-дифторбут-3-енилтио)-3-нитропиридина (соединение XVI.24) из 2-хлор-3-нитропиридина.
Гидробромид 4,4-дифтopбут-3-eнилизoтиoуpoния (1,24 г) прибавляют к раствору гидроксида натрия (0, 6 г) в воде (10 см3) и реакционную смесь энергично перемешивают при комнатной температуре в течение 20 минут. Прибавляют раствор 2-хлор-3- нитропиридина (0,795 г) в дихлорметане (10 см3) и затем тетра-н-бутиламмоний бромид (каталитическое количество). Реакционную смесь энергично перемешивают в течение 3 часов. Смесь разбавляют дихлорметаном и слои разделяют. Органический слой промывают насыщенным солевым раствором, сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением желтого масла. Колоночная хроматография на силикагеле с использованием 15%-ного диэтилового эфира в гексане в качестве элюента дает соединение XVI. 24 (0,847 г). М+ = 246;1H ЯМР: δ 2,40 (2H, м); 3,25 (2H, т); 4,30 (1H, м); 7,20 (1H, дд); 8, 50 (1H, дд); 8,70 (1H, дд); (масло).
По вышеописанному способу было получено следующее соединение:
(1) 3-циано-2-(4, 4-дифторбут-3-енилтио)пиридин (соединение XVI.8); М+ = 226;1H ЯМР: δ 2,40 (2H, м); 3,30 (2H, т); 4,25 (1H, м); 7,10 (1H, дд); 7,80 (1H, дд); 8,55 (1H, дд); (масло).
ПРИМЕР XVI.5
Данный пример
представляет общий способ получения соединений по изобретению, в котором атом серы в 4,4-дифторбут-3-енилтио заместителе соответствующего неокисленного соединения (полученного в соответствии со
способом предшествующих примеров) окисляют до сульфоксида (сульфинил) или сульфона (сульфонил).
Получение соединения XVI.3 из соединения XVI.2, используя один эквивалент окислителя.
Соединение XVI. 2 (0,818 г) охлаждают до 0oC в дихлорметане (30 см3) и прибавляют 3-хлорнадбензойную кислоту (0,99 г) в течение более пяти минут. Смесь перемешивают при комнатной температуре в течение 6 часов и оставляют на 40 часов. Реакционную смесь выливают в насыщенный водный раствор бикарбоната натрия и продукт экстрагируют дихлорметаном. Органический слой промывают водой и сушат (MgSO4). Упаривание растворителя при пониженном давлении дает желтое масло, которое хроматографируют на sorbsil-C30, элюируя 15%-ным этилацетатом в гексане с получением 4-(4,4-дифторбут-3- енилсульфинил) -2,3,5,6-тетрагидропиридина (0,711 г).1H ЯМР: δ 2,45-2,8 (2H, м); 3,15- 3,3 (1H, м); 3,5-3,65 (1H, м); 4,2-4,4 (1H, м); (масло).
Получение 5-циано-2-(4,4-дифторбут-3-енилсульфонил)-пиридина (соединение XVI.16) из соединения XVI.15, используя два эквивалента окислителя.
3-Хлорнадбензойную кислоту (3,14 г 50%-ной твердой) порциями прибавляют к перемешиваемому раствору соединения XVI.15 (1,03 г) в дихлорметане (30 см3) при 0oC. Смесь выливают в 2М водный раствор гидроксида натрия и слои разделяют. Водный слой экстрагируют дихлорметаном и объединенные органические слои сушат над сульфатом магния, фильтруют и упаривают при пониженном давлении с получением желтого масла, которое кристаллизуется при стоянии. Колоночная хроматография на силикагеле с использованием 3:7 этилацетата:гексана в качестве элюента дает соединение XVI.16 (0,785 г).1H ЯМР: δ 2,40 (2H, м); 3,25 (2H, т); 4,25 (1H, м); 7,25 (1H, дд); 7,70 (1H, дд); 8,65 (1H, д); (т. пл. 34oC).
По вышеописанному способу были получены
следующие соединения:
(1) 4-(4,4-дифторбут-3-енилсульфонил) пиридин (соединение XVI.6).1H ЯМР: δ 2,50 (2H, м); 3,20 (2H, т); 4,25 (1H, м); 7,80 (2H, д); 8,95 (2H, д);
(масло).
(2) 2-(4,4-дифторбут-3-енилсульфонил)-5-трифторметилпиридин (соединение XVI. 12).1H ЯМР: δ 2,50 (2H, м); 3,50 (2H, т); 4,25 (1H, м); 8,25 (2H, д); 9,00 (1H, ушир. с); (т. пл. 60oC).
(3) 2-(4,4-дифторбут-3-енилсульфонил)пиридин (соединение XVI. 20).1H ЯМР: δ 2,50 (2H, м); 3,50 (2H, т); 4,25 (1H, м); 7,45-7,50 (1H, м); 8,10 (1H, д); 8,75 (1H, д); (масло).
ПРИМЕР XVII.1
Данный пример представляет 2-стадийный способ получения 3-(4,4-дифторбут-3-енилтио)-6-метилпиридазина (соединение
XVII.1).
Стадия 1: Получение 3-меркапто-6-метилпиридазина
3-Хлор-6-метилпиридазин (2,96 г) и тиомочевину перемешивают вместе и нагревают с обратным холодильником в этаноле
(50 см3) в течение 7,5 часов. Реакционную смесь охлаждают и оставляют на 18 часов. Образовавшийся твердый осадок отфильтровывают и промывают диэтиловым эфиром с получением
3-меркапто-6-метилпиридазина (2,3 г), который используют далее без дополнительной очистки.1H ЯМР: δ 2,40 (3H, с); 7,30 (1H, д); 14,5-14,7 (1H, ушир. с).
Стадия 2:
Получение соединения XVII.1
Смесь продукта со стадии 1 (0,337 г), 4,4-дифторбут-3-енил-4- метилбензолсульфоната (0,70 г) и карбонат калия (0,369 г) перемешивают вместе и нагревают с обратным
холодильником в ацетоне (20 см3) в течение 11 часов. Неорганические твердые продукты удаляют фильтрацией и фильтрат упаривают при пониженном давлении с получением коричневого масла.
Хроматография на силикагеле с использованием 1:4 этилацетата:гексана в качестве элюента дает соединение XVII. 1 (0,15 г). М+ = 216;1H ЯМР: δ 2,48 (2H, м); 2,62 (3H, с); 3,
36 (2H, т); 4,20-4,40 (1H, м); 7,10 (1H, д); 7,10 (1H, д); (масло).
По вышеописанным стадиям 1 и 2 получают следующие соединения по изобретению и соответствующие промежуточные
продукты:
(1) 3-(4,4-дифторбут-3-енилтио) -6-хлорпиридазин (соединение XVII.2). М+ = 236;1H ЯМР: δ 2,50 (2H, м); 3,39 (2H, т); 4,20-4,40 (1H, м); 7,27 (2H, с); 8,
95 (2H, с); (масло) из 3,6-дихлорпиридазина.
(2) 3-(4,4-дифторбут-3-енилтио)-6-метоксипиридазин (соединение XVII.3). М+ = 232;1H ЯМР: δ 2,47 (2H, м); 3, 31 (2H, т); 4,09 (3H, с); 4,20-4,40 (1H, м); 6,83 (1H, д); 7,20 (1H, д); (твердый продукт, т. пл. 39,3-40,1oC) из 3-хлор-6-метоксипиридазина.
(3) 3-(4, 4- дифторбут-3-енилтио)-6-фенилпиридазин (соединение XVII.4). М+ = 278;1H ЯМР: δ 2,54 (2H, м); 3,46 (2H, т); 4,25-4,42 (1H, м); 7,39 (1H, д); 7,51 (3H, м); 8,05 (2H, м); (твердый продукт, т. пл. 91,7-92,1oC) из 3-хлор-6- фенилпиридазина.
(4) 1-(4,4-дифторбут-3-енилтио)фталазин (соединение XVII.7). М+= 252;1H ЯМР (CDCl3): δ 2,59 (2H, м); 3,55 (2H, т); 4,28-4,45 (1H, м); 7,89 (3H, м); 8,12 (1H, м); 9,25 (2H, м); (масло) из 1(2H)-фталазинтиона по способу стадии 2, описанной выше.
ПРИМЕР XVII.2
Данный пример представляет способ получения соединения XVII.5 из соединения XVII.4.
Соединение XVII.4 перемешивают при комнатной температуре в изопропаноле (20 см3) и прибавляют гексагидрат магниевой соли монопероксифталевой кислоты (0,89 г в 10 см3 воды). Смесь перемешивают при комнатной температуре в течение 20 часов. Образовавшийся твердый продукт отфильтровывают и промывают водой. Фильтрат выливают в насыщенный водный раствор бикарбоната натрия и затем продукт экстрагируют этилацетатом. Объединенные органические слои промывают насыщенным солевым раствором и сушат (MgSO4). Упаривание растворителя при пониженном давлении дает не совсем белое твердое вещество, которое объединяют с продуктом первого осаждения и хроматографируют на силикагеле, элюируя 30%-ным этилацетатом в гексане. Вновь выделяемым первым продуктом является 3-(4,4-дифторбут-3-енилсульфонил)-6-фенилпиридазин (соединение XVII. 6) (0,2 г). М+ = 310;1H ЯМР: δ 2,60 (2H, м); 3,75 (2H, т); 4,20-4,40 (1H, м); 7,60 (3H, м); 8,11 (1H, д); 8,15 (1H, м); 8,22 (1H, д); (твердый продукт, т. пл. 141,7-144, 3oC). Дальнейшее элюирование дает 3-(4,4-дифторбут-3- енилсульфинил)-6-фенилпиридазин (соединение XVII.5) (0,25 г). M+ = 294;1H ЯМР: δ 2,40 и 2,65 (2H, м); 3, 20-3,40 (2H, м); 4,18-4,32 (1H, м); 7,59 (3H, м); 8,11 (1H, д); 8,15 (1H, м); 8,21 (1H, д); (т. пл. 133-134oC).
ПРИМЕР XVIII.1
Данный пример представляет
3-стадийный способ получения 2-(4,4-дифторбут-3- енилтио)хиноксалина (соединение XVIII.1).
Стадия 1: Получение хиноксалин-2-тиона
2-Хиноксалинол (10 г), пентасульфид фосфора
(16,72 г) и пиридин (20 см3) перемешивают вместе и нагревают с обратным холодильником в течение 7 часов. Реакционной смеси дают охлаждаться и большую часть пиридина удаляют упариванием при
пониженном давлении. Остаток распределяют между этилацетатом и водой и органический слой отделяют. Водный слой экстрагируют тремя порциями этилацетата и объединенные органические фазы промывают
насыщенным солевым раствором, сушат (MgSO4) и упаривают при пониженном давлении с получением коричневого маслянистого твердого продукта, который растирают с горячим этилацетатом:гексаном
(1:1) для того, чтобы растворить продукт и удалить нерастворимые остатки. Растворитель удаляют при пониженном давлении и получают оранжевый твердый продукт, часть которого используют далее на стадии 2
без дополнительной очистки.
Стадия 2: Получение 2-(4-бром-4,4-дифторбутилтио)-хиноксалина.
Смесь продукта со стадии 1 (1 г), 4-бром-4, 4-дифторбутил-4- метилбензолсульфоната (1,65 г) и карбонат калия (0,852 г) перемешивают вместе в ацетоне (20 см3) при комнатной температуре в течение 7 часов. Неорганические твердые продукты удаляют фильтрацией и фильтрат упаривают при пониженном давлении с получением коричневого масла. Хроматография на силикагеле с использованием 1: 4 этилацетата:гексана в качестве элюента дает 2- (4-бром-4,4-дифторбутилтио)хиноксалин (1,375 г). М+ = 332;1H ЯМР: δ 2,19 (2H, м); 2,50-2,70 (2H, м); 3,43 (2H, т); 7,60-7,73 (2H, м); 7,93 (1H, дд); 8,03 (1H, дд); 8, 60 (1H, с); (масло).
Стадия 3: Получение соединения XVIII.1.
1,8-Диазабицикло[5,4,0]ундец-7-ен (DBU) (1,14 см3) и продукт со стадии 2 (1,275 г) перемешивают в толуоле (30 см3) и нагревают с обратным холодильником в течение 5 часов. Смесь охлаждают и прибавляют избыток этилацетата и 2М водную хлористоводородную кислоту и органическую фазу отделяют. Водную фазу экстрагируют этилацетатом и объединенные органические фазы промывают насыщенным солевым раствором, сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением коричневого масла. Хроматография на силикагеле с использованием 1:4 этилацетата:гексана в качестве элюента дает соединение XVIII.1 (0,65 г). М+ = 252;1H ЯМР: δ 2,51 (2H, м); 3,39 (2H, т); 4,30- 4,40 (1H, м); 7,60-7,65 (2H, м); 8,03 (1H, дд); 8,60 (1H, с); (масло).
По вышеописанным стадиям 2 и 3 получают следующие
соединения по изобретению и соответствующие промежуточные продукты:
(1) 6-хлор-2-(4,4-дифторбут-3-енилтио) хиноксалин (соединение XVIII.4). М+ = 286;1H ЯМР: δ 2,
50 (2H, м); 3,35 (2H, т); 4,20-4,40 (1H, м); 7,65 (1H, дд); 7,86 (1H, д); 8,01 (1H, д); 8,59 (1H, с); (масло) из 6-хлорхиноксалин-2-тиона через 2-(4-бром-4,4-дифторбутилтио)-6-хлорхиноксалина.1H ЯМР: δ 2,08-2,21 (2H, м); 2,48-2,68 (2H, м); 3,40 (2H, т); 7,65 (1H, дд); 7,88 (1H, д); 8,00 (1H, д); 8,60 (1H, с); (масло).
ПРИМЕР XVIII.2
Данный пример
представляет 2-стадийный способ получения 2-(4,4-дифторбут-3-енилтио)-пиразина (соединение XVIII.7).
Стадия 1: Получение 3-меркаптопиразина
2-Хлорпиразин (5 г) и тиомочевину
(3,32 г) нагревают с обратным холодильником в этаноле (50 см3) в течение 8 часов. Реакционную смесь охлаждают и этанол удаляют упариванием при пониженном давлении с получением коричневой
смолы (8,31 г), которую перемешивают с 2М водным гидроксидом натрия (50 см3) в течение 16 часов. Твердый продукт отфильтровывают и промывают водой и ацетоном и подвергают вакуумной сушке.
Это дает желтый твердый продукт (0,72 г);1H ЯМР (ДМСО-d6): δ 7,69 (1H, 1); 7,89 (1H, д); 8,6 (1H, с), который используют на второй стадии без дополнительной очистки.
Стадия 2: Получение соединения XVIII.7
Продукт со стадии 1 (0,213 г), 4,4-дифторбут-3-енил-4- метилбензолсульфонат (0,5 г), карбонат калия (0,263 г) и иодид калия (0,317 г)
перемешивают в ацетоне (10 см3) и нагревают с обратным холодильником в течение 9 часов и затем оставляют на холоду на пару дней. Образовавшийся осадок удаляют фильтрацией и фильтрат
упаривают при пониженном давлении с получением коричневого масла. Хроматография на силикагеле с использованием 1:4 этилацетата:гексана в качестве элюента дает соединение XVIII.7 (0,3 г). М+
= 202;1H ЯМР: δ 2,40 (2H, м); 3,21 (2H, т); 4,20-4,40 (1H, м); 8,20 (1H, д); 8,38 (1H, т); 8,48 (1H, с); (масло).
ПРИМЕР XVIII.3
Данный пример представляет
2-стадийный способ получения 3-хлор-2-(4,4-дифторбут-3-енилтио)-пиразина (соединение XVIII.10).
Стадия 1: Получение 2-хлор-3-меркаптопиразина и 2,3-димеркаптопиразина дигидрат гидросульфида натрия (2,5 г) смешивают в изопропаноле (20 см3) и смесь нагревают с обратным холодильником в течение 1 часа. Реакционную смесь охлаждают и оставляют стоять в течение 36 часов. Образовавшийся желтый твердый осадок вновь выделяют фильтрацией, промывают диэтиловым эфиром и сушат в вакууме. Фильтрат упаривают. Твердый продукт растворяют в горячем этаноле и при охлаждении выделяют небольшое количество гидросульфида натрия и удаляют фильтрацией. Оставшийся раствор этанола разбавляют диэтиловым эфиром, при этом выпадает в осадок желтый твердый продукт (0,3 г). Его идентифицируют как бис-натриевую соль 2,3-димеркаптопиразина, MH+ (FAB) = 188;1H ЯМР (ДМСО-d6): δ 7,35 (1H, д); 7,50 (1H, д). Упаривание маточного раствора дает желтый твердый продукт (0,8 г), идентифицируемый как натриевая соль 2-хлор-3-меркаптопиразина, M+ (FAB) = 145;1H ЯМР (ДМСО-d6): δ 7,35 (1H, д); 7,50 (1H, д).
Стадия 2: Получение 3-хлор-2-(4,4-дифторбут-3-енилтио)-пиразина
Моно-тиолатный продукт со стадии 1 (0,8 г), 4,4-дифторбут-3-енил-4-метилбензолсульфонат (1,24 г) и
карбонат калия (0,655 г) перемешивают в ацетоне (25 см3), содержащем диметилформамид (5 см3), и нагревают с обратным холодильником в течение 15 часов, затем дают остыть. Осадок
удаляют фильтрацией и фильтрат упаривают при пониженном давлении с получением коричневого масла. Хроматография на силикагеле с использованием 95:5 смеси гексана:этилацетата в качестве элюента дает
соединение XVIII.10 (0,45 г). M+ = 236;1H ЯМР: δ 2,41 (2H, м); 3,20 (2H, т); 4,20-4,40 (1H, м); 8,05 (1H, д); 8,30 (1H, д); (масло). Продукт содержит (ГХ) в качестве
примеси 5% соединения XVIII.13.
Само соединение XVIII. 13 получают чистым путем обработки бис-тиолированного продукта со стадии 1 по примеру выше с двумя эквивалентами 4, 4-дифторбут-3-енил-4-метилбензолсульфоната в тех же условиях, что и на стадии 2 данного примера. Хроматография на силикагеле с использованием 4:1 смеси гексана: этилацетата в качестве элюента дает 2, 3-бис-(4,4-дифторбут-3-енилтио)- пиразин. М+ = 324;1H ЯМР: δ 2,40 (4H, м); 3,22 (4H, т); 4,20-4,40 (1H, м); 8,07 (2H, с); (масло).
По вышеописанному
способу получают следующее соединение:
(1) 6-хлор-2-(4,4-дифторбут-3-енилтио)-пиразин (соединение XVIII.14). M+ = 236;1H ЯМР: δ 2,42 (2H, м); 3,21 (2H, т); 4,
20-4,40 (1H, м); 8,20 (1H, с); 8,35 (1H, с); (масло) из 2,6-дихлорпиразина.
ПРИМЕР XVIII.4
Данный пример представляет способ получения соединений XVIII.2 и XVIII.3 из
соединения XVIII.1.
Соединение XVIII. 1 (0,25 г) перемешивают при комнатной температуре в этаноле (10 см3) и прибавляют гексагидрат магниевой соли монопероксифталевой кислоты (0,589 г в 5 см3 волы) в течение более пяти минут. Через 30 минут смесь нагревают до 70oC в течение 1 часа. Реакционную смесь охлаждают, выливают в насыщенный водный раствор бикарбоната натрия и продукты экстрагируют этилацетатом. Органический слой промывают водой и сушат (MgSO4). Упаривание растворителя при пониженном давлении дает не совсем белое твердое вещество (0,15 г), которое хроматографируют на силикагеле, элюируя 10%-ным этилацетатом в гексане. Вновь выделяемым основным продуктом является 2-(4,4- дифторбут-3-енилсульфонил)-хиноксалин (соединение XVIII. 3) (0,1 г), т. пл. 86,5-87,5oC. М+ = 284;1H ЯМР: δ 2,61 (2H, м); 3,62 (2H, т); 4,20-4,40 (1H, м); 8,00 (2H, м); 8,25 (2H, м); 9,51 (1H, с); ТСХ показывает наличие в неочищенном реакционном продукте вещества с низким Rf, 2-(4,4-дифторбут-3-енилсульфинил)-хиноксалина (соединение XVIII.2), но его не выделяют в чистом виде.
По вышеописанному способу получают следующее соединение:
(1) 6-хлор-2-(4,4-дифторбут-3-енилсульфинил)-пиразин (соединение XV111.15). М+ = 253;1H ЯМР: δ
2,30-2,70 (2H, м); 3,00-3,30 (2H, м); 4,19-4,35 (1H, м); 8,70 (1H, с); 9,10 (1H, с); (масло) из соединения XVIII.14.
(2) 6-хлор-2-(4,4-дифторбут-3-енилсульфонил)-пиразин (соединение XVIII. 16). М+ = 268;1H ЯМР: δ 2,55 (2H, м); 3,50 (2H, т); 4,20-4,35 (1H, м); 8,90 (1H, с); 9,19 (1H, с); (масло) из соединения XVIII.14.
ПРИМЕР
VIII.1
Данный пример представляет 3-стадийный способ получения 4-(4,4-дифторбут-3-енилтио)-1,2,3-бензотриазина (соединение XIX.1).
Стадия 1: Получение 4-меркапто-1,2, 3-бензотриазина Перемешивают в пиридине (30 см3) 2-аминобензонитрил (5 г), прибавляют триэтиламин (6 см3) и в реакционную смесь барботируют газообразный сульфид водорода в течение более 4 часов. Смесь затем выливают в воду (20 см3) и масло, которое отделяется при встряхивании, отделяют и высушивают с помощью азеотропной отгонки с этанолом и толуолом. Это приводит к получению желтого твердого продукта (2-аминотиобензамид, 3 г), который перемешивают при 0oC с 2М хлористоводородной кислотой (30 см3). По каплям прибавляют нитрит натрия (1,65 г) в воде (10 см3) и реакционную смесь перемешивают на холоду в течение 1 часа, затем дают нагреться до комнатной температуры и перемешивают еще 1 час. Образовавшийся твердый продут отфильтровывают, промывают водой и сушат промывкой диэтиловым эфиром с получением коричневого масла (2,15 г).
Стадия 2: Получение 4-(4-бром-4,4-дифторбутил-3-енилтио)-1,2, 3- бензотриазина.
Продукт со стадии 1 (1 г), 4-бром-4,4- дифторбутил-4-метилбензолсульфонат (1,65 г) и карбонат калия (0,852 г) перемешивают вместе в ацетоне (30 см3) при комнатной температуре в течение 36 часов. Неорганические твердые продукты удаляют фильтрацией и фильтрат упаривают при пониженном давлении с получением коричневого масла. Хроматография на силикагеле с использованием 1: 4 этилацетата:гексана в качестве элюента дает два желтых масла (каждое по 0,5 г). Элюированное первым масло идентифицируют как N-алкилированный продукт, а второй является желаемым S-алкилированным промежуточным 4-(4,4-дифторбутилтио)- 1,2,3-бензотриазином.1H ЯМР: δ 2,18-2,31 (2H, м); 2,58-2,71 (2H, м); 3,63 (2H, т); 7,95 (1H, т); 8,09 (2H, м); 8,40 (1H, д).
Стадия 3: Получение соединения XIX.1.
1,8-Диазабицикло[5,4,0]ундец-7-ен (DBU) (0,45 см3) и продукт со стадии 2 (0,5 г) перемешивают в толуоле (15 см3) и нагревают с обратным холодильником в течение 6 часов. Смесь охлаждают, затем прибавляют избыток этилацетата и 2М водную хлористоводородную кислоту и органическую фазу отделяют. Водную фазу экстрагируют этилацетатом и объединенные органические фазы промывают насыщенным солевым раствором, сушат над сульфатом магния, фильтруют и упаривают при пониженном давлении с получением желтого масла. Хроматография на силикагеле с использованием 1:4 этилацетата:гексана в качестве элюента дает соединение XIX.1 (0,25 г). MH+ = 254;1H ЯМР: δ 2,60 (2H, м); 3, 60 (2H, т); 4,30-4,40 (1H, м); 7,93 (1H, т); 8,09 (2H, м); 8,39 (1H, д); (масло).
ПРИМЕР XX.1
Далее описаны два способа получения (A и B) меркапто-1,2,4-триазинов,
необходимых в качестве промежуточных продуктов для получения соединений по изобретению.
МЕТОД A
Общий синтез меркапто-1,2,4-триазинов представлен реакцией взаимодействия
тиосемикарбазида и 1,2-дикарбонильного производного. Он проиллюстрирован получением 3-меркапто-5-метил- 1,2,4-триазина.
Раствор бикарбоната натрия (8 г) в воде (100 см3) прибавляют к суспензии тиосемикарбазида (8 г) в воде (100 см3). Полученный раствор охлаждают ниже 5oC и прибавляют метилглиоксаль (пировиноградный альдегид) (40 мас.% раствора в воде (20 см3)).
Раствор выдерживают при 5oC в течение 18 часов, затем промывают хлороформом (10 x 50 см3). Устанавливают pH водных слоев 2 с помощью концентрированной хлористоводородной кислоты. Полученный осадок отфильтровывают, промывают обильным количеством воды и сушат с получением оранжевого твердого продукта (4,036 г). 3-Меркапто-5-метил-1,2, 4-триазина используют на следующей стадии без дополнительной очистки.
По вышеописанному способу получают следующие промежуточные меркаптотриазины. Исходные продукты являются коммерчески доступными.
(1) 3-меркапто-1,2,4-триазин из глиоксаля.
(2) 3-меркапто-5-пропил-6-метил-1,2,4-триазин из гексан- 2,3-диона.1H ЯМР: δ 0, 98 (3H, т); 1,54-1,68 (2H, м); 2,08 (3H, с); 3,04 (2H, т); 8,2 (1H, ушир. с).
(3) 3-меркапто-5-фенил-6-метил-1,2,4-триазин.
МЕТОД B
Альтернативный способ
получения меркапто-1,2,4-триазинов включает обработку соответствующего гидрокситриазина (который может существовать в виде различных таутомерных форм) с тиолатирующим реагентом, таким как пентасульфид
фосфора. Далее проиллюстрирован способ получения 1,2,4-триазин-3,5(2H, 4H)-дитиона из 1,2,4-триазин- 3,5(2H,4H)-диона (6-азаурацил).
6-Азаурацил (2 г), пентасульфид фосфора (15,72 г) и пиридин (5 см3) перемешивают вместе и нагревают с обратным холодильником в течение 56 часов. Реакционной смеси дают охлаждаться и большую часть пиридина удаляют упариванием при пониженном давлении. Остаток перемешивают с этиловым эфиром и водой и органический слой отделяют. Водный слой экстрагируют тремя порциями этилового эфира и объединенные органические фазы промывают насыщенным водным солевым раствором, сушат (MgSO4) и упаривают при пониженном давлении с получением коричневого масла, которое хроматографируют на силикагеле, используя 1:5 этилацетата:гексана в качестве элюента. Это приводит к получению оранжевого твердого продукта (1 г), который используют без дополнительной очистки.1H ЯМР (ДМСО-d6): δ 7,95 (1H, с); 13,8-14,1 (1H, ушир. с).
3-Меркапто-1,2,4-бензотриазин получают из 3-гидрокси-1,2,4- бензотриазина и 6-мeтил-1,2,3-тpиaзин-5(4H)-тиoн из 6-метил- 1,2,3-триазин-5(4H)-она, следуя, по-существу, вышеописанному способу.
ПРИМЕР XX.2
Данный пример представляет способ получения соединений по изобретению, которые содержат 1,2,4-триазиновый заместитель в 4,
4-дифторбут-3-енилтио группе в 3, 5 или 6 положении, исходя из соответствующим образом замещенного меркаптотриазина и подходящего дифторбут-1-енового алкилирующего агента. Это показано следующим
получением 3-(4,4-дифторбутил-3- енилтио)-5-гидрокси-1,2,4-триазина (соединение XX.227) из 6-аза-2-тиоурацила и 4,4-дифторбут-3-енил-4-метилбензолсульфоната.
4, 4-Дифторбут-3-енил-4-метилбензолсульфонат (1,5 г) и иодид калия (0,95 г) перемешивают вместе в этаноле (5 см3) и нагревают с обратным холодильником в течение трех часов, затем дают остыть. Согласно этой части способа исходный продукт преобразуется в более химически реакционноспособный и тио-избирательный алкилирующий агент, 4-иод-1,1-дифторбут-1-ен. Прибавляют 6-аза-2-тиоурацил (0,744 г) в виде раствора в 1М водном гидроксиде натрия (5,73 см3) и реакционную смесь перемешивают при комнатной температуре в течение 70 часов. Реакционную смесь обрабатывают путем добавления 1М водного гидроксида натрия и этилацетата. Этилацетатный слой отделяют и водный слой промывают дополнительным количеством этилацетата. Эти экстракты отбрасывают. Затем водный слой подкисляют до pH 4 с помощью 2М водной хлористоводородной кислоты. Продукт экстрагируют этилацетатом (3 порциями) и эти объединенные органические слои промывают водой и насыщенным солевым раствором и сушат (MgSO4). Продукт выделяют из раствора упариванием при пониженном давлении с получением твердого продукта кремового цвета (0,659 г). Часть вновь растворяют в горячем этилацетате и небольшое количество нерастворившегося продукта удаляют фильтрацией. Соединение XX.227, вновь выделенное из раствора упариванием, имеет Т. пл. 102-103oC. М+ = 219;1H ЯМР (ДМСО-d6): δ 2,45 (2H, м); 3,25 (2H, т); 4,55-4,75 (1H, м); 7,69 и 7,79 (всего 1H, каждый с, таутомерные протоны).
Соединение XX.247 получают, следуя следующему способу.
6-Метил-1,2,3-триазин-5(4H)-тион (0,5 г), 4-бром-1,1-дифторбут-1-ен (0,673 г) и карбонат калия (0,543 г) перемешивают вместе в ацетоне (5 см3) в течение 60 часов при комнатной температуре. Неорганические продукты отфильтровывают и промывают ацетоном. Растворитель удаляют из объединенных ацетоновых растворов путем упаривания при пониженном давлении и оставшуюся коричневую смолу хроматографируют на силикагеле, элюируя 20%-ным этилацетатом в гексане с получением 5-(4,4- дифторбут-3-енилсульфинил)-6-метил-1,2,4-триазина (соединение XX. 247) (0,075 г). М+ = 217;1H ЯМР: δ 2,45 (2H, м); 2,60 (3H, с); 3,29 (2H, т); 4,20-4,40 (1H, м); 9,11 (1H, с); (масло).
ПРИМЕР XX.3
Когда меркаптотриазин полностью растворим в
ацетоне и не обладает значительно затрудняющими нуклеофильными группами, может быть использован способ, альтернативный способу по примеру XX.2.
Это показано следующим получением 3, 5-бис-(4,4- дифторбутил-3-енилтио)-1,2,4-триазина (соединение XX.231) из 1,2,4-триазин-3,5(2H,4H)дитиона и 4,4-дифтор-3-бутенил-4- метилбензол-сульфоната.
1,2,4-Триазин-3,5(2H, 4H)дитион (1 г), 4,4-дифторбут-3- енил-4-метилбензолсульфонат (3,6 г) и карбонат калия (0,952 г) перемешивают и нагревают с обратным холодильником в ацетоне (20 см3) в течение 7 часов. Раствор охлаждают и фильтруют для удаления твердых продуктов. Фильтрат и ацетоновые промывки объединяют и упаривают при пониженном давлении с получением коричневого масла. Хроматография на силикагеле, с использованием 15:85 этилацетата:гексана в качестве элюента дает соединение XX.231 (1 г) в виде оранжевого масла. М+ = 325;1H ЯМР: δ 2,40-2,55 (4H, м); 3,25 (4H, м); 4, 15-4,40 (2H, м); 8,70 (1H, с).
ПРИМЕР XX.4
Данный пример представляет общий способ двухстадийного получения соединений по изобретению путем взаимодействия меркаптозамещенного
1,2,4-триазина с 4-бром-4,4- дифторбутилметансульфонатом с последующим дегидробромированием полученного промежуточного продукта, как показано следующим получением 3-(4,
4- дифторбутил-3-енилтио)-5-фенил-6-метил-1,2,4-триазина (соединение XX.4).
Стадия 1: 3-(4-бром-4,4-дифторбутилтио)-5- фенил-6-метил-1,2,4-триазин
Смесь 4-бром-4,
4-дифторбутилметансульфоната (0,7 г), 3-мepкaптo-5-фeнил-6-мeтил-1,2,4-тpиaзинa (0,5 г) и карбонат калия (0,7 г) перемешивают вместе и нагревают с обратным холодильником в ацетоне (40 см3)
в течение 14 часов. Неорганические твердые продукты удаляют фильтрацией и фильтрат упаривают при пониженном давлении с получением коричневого масла (1,058 г). Хроматография на силикагеле с
использованием 1:4 этилацетата:гексана в качестве элюента дает оранжевое масло (0,576 г). Его перемешивают и нагревают с обратным холодильником в трифторуксусной кислоте (3 см3) в течение 6
часов для возвращения в цикл небольшого количества продукта, который подвергается гидролизу и открытию цикла. Продукт вновь выделяют упариванием растворителя при пониженном давлении, и хроматография
на силикагеле дает 3-(4- бром-4,4-дифторбутилтио)-5-фенил-6-метил- 1,2,4-триазин (0,509 г).1H ЯМР: δ 2,12-2,24 (2H, м); 2,48-2,68 (2H, м); 2,78 (3H, с); 3,36 (2H, т); 7,50-7,58 (3H,
м); 7,68-7,74 (2H, м); (масло).
Стадия 2: 3-(4,4-дифторбут-3-енилтио)-5-фенил-6-метил- 1,2,4-триазин
1,8-Диазабицикло[5,4,0] ундец-7-ен (DBU) (1 см3) прибавляют по
каплям к перемешиваемому раствору продукта со стадии 1 (0,5 г) в толуоле (10 см3). Смесь выдерживают при комнатной температуре в течение 20 часов, затем прибавляют избыток этилацетата и
насыщенный водный хлорид аммония и органическую фазу отделяют. Водную фазу экстрагируют этилацетатом и объединенные органические фазы промывают насыщенным водным хлоридом аммония, сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением оранжевого масла. Хроматография на силикагеле с использованием 1:4 этилацетата:гексана в качестве элюента дает соединение XX.4 (0,
281 г). М+ = 293;1H ЯМР: δ 2,48-2,58 (2H, м); 2,76 (3H, с); 3,32 (2H, т); 4,22-4,40 (1H, м); 7,48-7,58 (3H, м); 7,68-7,76 (2H, м); (масло).
ПРИМЕР
XX.5
Данный пример представляет способ, подходящий для получения соединений по изобретению, которые содержат алкокси или замещенную алкокси группу как в 3, так и в 5 положении, как показано
получением 3-(4,4-дифторбутил-3-енилтио)-5-метокси-1,2,4-триазина (соединение XX.221) из соединения XX.231.
Соединение XX. 231 (0,5 г) перемешивают в течение одного часа вместе с метоксидом натрия (0,083 г) в метаноле (15 см3) при комнатной температуре. Растворитель удаляют упариванием при пониженном давлении и остаток хроматографируют на силикагеле, элюируя 15:85 этилацетата:гексана с получением 0,12 г соединения XX. 221. М+ = 233;1H ЯМР: δ 2,50 (2H, м); 3,29 (2H, т); 4,02 (3H, с); 4,22-4,42 (1H, м); 8,55 (1H, с); в виде одного изомера, который идентифицирован как 5-метоксипроизводное, как подтверждено данными ЯМР.
По вышеописанному способу были получены следующие соединения по изобретению с использованием
соответствующего алкоксила в соответствующем спирте в качестве растворителя:
(1) 3-(4,4-дифторбутил-3-енилтио)-5-(1- метилэтокси)-1,2,4-триазин (соединение XX.205). М+ = 261;1H ЯМР: δ 1,4 (6H, д); 2,5 (2H, м); 3,25 (2H, т); 4,2-4,4 (1H, м); 8,45 (1H, с).
(2) 3-(4,4-дифторбутил-3-енилтио)-5-этокси-1,2,4-триазин (соединение XX. 218). М+ = 247;1H ЯМР: δ 1,43 (3H, т); 2,50 (2H, м); 3,26 (2H, т); 4,2-4,4 (1H, м); 4,46 (2H, кв); 8,5 (1H, с).
ПРИМЕР XX.6
Данный пример представляет
2-стадийный способ получения соединений XX.52 и XX.116 в виде смеси 1:1.
Стадия 1: 3-(4-бром-4,4-дифторбутилтио)-5-метил-1,2,4-триазин и 3-(4-бром-4,4-дифторбутилтио)-6-метил-1,2,
4-триазин
4-Бром-4,4-дифторбутилметансульфоната (1 г) и тиосемикарбазид (0,475 г) перемешивают вместе и нагревают с обратным холодильником в этаноле (20 см3) в течение 5 часов. ГХ
свидетельствует о полном расходовании метансульфоната. Смеси дают остыть и прибавляют воду (3 см3) и бикарбонат натрия (1 г) (вспенивание). Прибавляют 40 мас.% раствор пировиноградного
альдегида и смесь перемешивают при комнатной температуре в течение 4 часов. ТСХ показывает, что продукт образован. Его экстрагируют этилацетатом и органический слой сушат (MgSO4), фильтруют
и упаривают с получением масла (1,209 г). Этот промежуточный продукт очищают хроматографией на силикагеле, элюируя 1:4 этилацетата:гексана с получением желтого масла (0,464 г), которое имеет1H ЯМР: δ 2,08-2,20 (2H, м); 2,48-2,62 (2H, м); 2,52 и 2,66 (всего 3H, каждый с); 3,28-3,38 (2H, м); 8,28 и 8,84 (всего 1H, каждый с), который идентифицирован как смесь приблизительно 1:1
5- и 6-метил изомеры.
Стадия 2: 3-(4,4-дифторбут-3-енилтио)-5-метил-1,2,4-триазин и 3-(4,4-дифторбут-3-енилтио)-6-метил-1,2,4-триазин.
Смесь продуктов со стадии 1 (0, 46 г) дегидробромируют с помощью DBU по способу, аналогичному в примере XX.4, и продукт (0,253 г) очищают хроматографией на силикагеле, элюируя дихлор-метаном. В этих условиях 5- и 6-метил изомеры неразделимы и получают 0,232 г желтого масла, которое имеет М+ = 217;1H ЯМР: δ 2,44-2,54 (2H, м); 2,50 и 2,66 (всего 3H, каждый с); 3,26-3,34 (2H, м); 4,22-4,40 (1H, м); 8,28 и 8,82 (всего 1H, каждый с); который идентифицирован как смесь 1:1 5- и 6-метил соединения XX.52 и соединения XX.116.
Соединение XX. 251 в соответствии с изобретением получают по вышеописанному способу, но используя циклогексан-1,2-дион на стадии 1 вместо пировиноградного альдегида.1H ЯМР: δ 1,86- 1,98 (4H, м); 2,42-2,54 (2H, м); 2,84-2,92 (2H, м); 3,04-3,12 (2H, м); 3,28 (2H, т); 4,22-4,38 (1H, м); (масло).
ПРИМЕР XX.7
Данный пример представляет альтернативный способ, используемый для получения соединения XX.52 и соединения
XX.116 в виде хроматографически разделяемой смеси.
4,4-Дифтоpбут-3-eнил-4-мeтилбeнзoлcульфoнaт (1 г) и тиосемикарбазид (0,4 г) перемешивают вместе и нагревают с обратным холодильником в этаноле (20 см3) в течение 6 часов. Смеси дают остывать в течение одной ночи, в течение которой образуется желтый кристаллический осадок S-алкилированного соединения. Его не выделяют, но растворитель удаляют упариванием при пониженном давлении и остаток обрабатывают бикарбонатом натрия (0,928 г), водой (3 см3) и этанолом (20 см3). Затем прибавляют 40 мас.% раствор пировиноградного альдегида (0,9 см3) (легкое вспенивание) и смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь выливают в воду и продукт экстрагируют этилацетатом (3 порции). Объединенные органические фазы сушат (MgSO4), фильтруют и упаривают с получением коричневой смолы (0,85 г). Хроматография на силикагеле, элюируя 10% этилацетатом в гексане, дает фракцию, которую идентифицируют как соединение XX.116 (0,05 г) (содержащее 10% соединение XX. 52 по данным ЯМР). Дальнейшее элюирование дает смешанную фракцию, содержащую смесь приблизительно 1: 1 соединения XX.52 и соединения XX.116 (0,16 г) и далее фракцию идентифицируют как соединение XX.52 (0,1 г) (содержащее 15% соединения XX.116 по данным ЯМР).
ПРИМЕР
XX.8
Данный пример представляет общий способ двухстадийного получения соединений по изобретению путем взаимодействия тиосемикарбазида с 4-бром-1,1-дифторбут-1-еном и затем циклизацией
промежуточного соединения с 1,2-дикарбонильным производным, как показано следующим получением 3-(4,4-дифторбутил- 3-енилтио)-1,2,4-триазина (соединение XX.137).
Тиосемикарбазид (2,6 г) перемешивают с 4-бром-1,1-дифторбут-1-еном в этаноле (75 см3) и смесь нагревают с обратным холодильником в течение 7 часов и дают ей остыть до комнатной температуры. Прибавляют глиоксаль (4,2 г 40% водного раствора) и гидрокарбонат натрия (7,37 г) и смесь перемешивают при комнатной температуре в течение 18 часов. Прибавляют воду и продукт экстрагируют тремя порциями этилацетата. Объединенные органические фазы промывают насыщенным раствором соли, сушат (MgSO4), фильтруют и упаривают при пониженном давлении с получением коричневой смолы. Хроматография на силикагеле, элюируя 20% этилацетатом в гексане, дает соединение XX.137. М+ = 203;1H ЯМР: δ 2,50 (2H, м); 3,30 (2H, т); 4,20-4,49 (1H, м); 8,39 (1H, д); 8,95 (1H, д) (масло).
По вышеописанному способу были получены следующие соединения по изобретению с помощью соответствующего 1,2-дикарбонильного производного вместо глиоксаля:
(1) 3-(4,
4-дифторбут-3-енилтио)-5-пропил-1,2,4-триазин (соединение XX. 27) из 2-оксопентаналя. М+ = 245;1H ЯМР: δ 1,0 (3H, т); 1,7-1,85 (2H, м); 2,45-2,55 (2H, м); 2,45-2,55 (2H,
м); 2,6 (2H, т); 4,2-4,4 (1H, м); 8,8 (1H, с); (масло). Это получение также дает изомерное соединение XX.109; М+ = 245;1H ЯМР: δ 1,0 (3H, т); 1,75-1,92 (2H, м); 2,45-2,55
(2H, м); 2,9 (2H, т); 3,3 (2H, т); 4,2-4,4 (1H, м); 8,25 (1H, с); (масло), последний более быстро проходящий изомер при хроматографии составляет приблизительно 8% полученной смеси.
(2) 3-(4, 4-дифторбут-3-енилтио) -5-этил-1, 2, 4-триазин (соединение XX. 30) из 2-оксобутаналя. M+ = 231;1H ЯМР: δ 1,3-1,4 (3H, т); 2,45-2,55 (2H, м); 2,75-2,85 (2H, кв); 3,25-3,35 (2H, т); 4,2-4,4 (1H, м); 8,85 (1H, с); (масло). Изомерное соединение XX.110 было определено в реакционной смеси, но не было выделено. Однако, он не был главным продуктом циклизации, когда используют 1,1-диэтоксибутан-2-он вместо 2-оксобутаналя. Первоначальное образование имина в воде с последующей циклизацией в водном ацетоне, катализируемое пиридиний тозилатом, дает соединение XX.110. М+ = 231;1H ЯМР: δ 1,4 (3H, т); 2,50 (2H, м); 2,95 (2H, кв); 3,3 (2H, т); 4,2-4,4 (1H, м); 8,29 (1H, с); (масло).
(3) 3-(4, 4-дифторбут-3-енилтио)-6-метил-1,2,4-триазин (соединение XX. 226) из пировиноградной кислоты. М+ = 233;1H ЯМР (ДМСО-d6): δ 2,05 (3H, с); 2,30 (2H, м); 3,12 (2H, т); 4,4-4,6 (1H, м); (смола) и более быстро проходящий изомер при хроматографии 3-(4,4-дифторбут-3-енилтио) - 6-(1-метилэтил)-1,2,4-триазин (соединение XX. 98). М+ = 245;1H ЯМР (ДМСО-d6): δ 1,4 (6H, д); 2,50 (2H, м); 3,18-3,35 (3H, м); 4,20-4,40 (1H, м); 8,30 (1H, с); (масло) из 2,2-дихлор-3-метилбутаналя.
ПРИМЕР XX.9
Данный пример
иллюстрирует способ получения 5-дихлорметил-3-(4,4-дифторбут-3-енилтио)-1,2,4- триазина (соединение XX.65) из соединения XX.52.
Соединение XX. 52 (3,1 г) и N-сукцинимид (2 г) перемешивают и нагревают вместе с обратным холодильником в тетрахлориде углерода (40 см3) в течение 1 часа. Смеси дают остыть и перемешивают при комнатной температуре в течение 4 часов. Растворитель удаляют упариванием при пониженном давлении и темный смолистый остаток суспендируют в диэтиловом эфире (40 см3) и пропускают через прослойку фильтра hi-flo. Нерастворимые продукты промывают еще диэтиловым эфиром и объединенные растворы упаривают при пониженном давлении. Остаток очищают хроматографией на силикагеле, элюируя диэтиловым эфиром:гексаном 1:1 и фракции, содержащие продукт, упаривают при пониженном давлении и еще очищают с помощью препаративной ТСХ, элюируя 30%-ным диэтиловым эфиром в гексане с получением соединения XX.65 (0,2 г).1H ЯМР: δ 2,50 (2H, м); 3,3 (2H, т); 4,20-4,40 (1H, м); 6,50 (1H, с); 9,37 (1H, с).
ПРИМЕР XX.10
Данный пример иллюстрирует способ получения 3-(4,
4-дифторбут-3-енилсульфонил)-5-пропил-1,2,4- триазина (соединение XX.28) из соединения XX.27.
Соединение XX. 27 (0,2 г) перемешивают при 5oC в дихлорметане (5 см3 ) и прибавляют 3-хлорнадбензойную кислоту (0,282 г, 2 эквивалента). Перемешивание продолжают при комнатной температуре в течение 18 часов. Реакционную смесь гасят прибавлением насыщенного водного раствора бикарбоната натрия и продукт экстрагируют дихлорметаном. Органическую фазу отделяют, промывают насыщенным солевым раствором и сушат (MgSO4). После фильтрации и концентрирования путем упаривания при пониженном давлении получают масло, которое очищают хроматографией на силикагеле, используя 3:7 этилацетата:гексана в качестве элюента с получением соединения XX.28 (0,187 г). М+ = 277;1H ЯМР: δ 1,0-1,1 (3H, т); 1,8-1,95 (2H, м); 2,6-2,7 (2H, м); 2,9-3,0 (2H, т); 3,7-3,8 (2H, т); 4,2-4,4 (1H, м); 9,3 (1H, с); (масло).
По
вышеописанному способу были получены следующие соединения по изобретению, используя 1,75 эквивалента окислителя:
(1) 3-(4,4-дифторбут-3-енилсульфинил)-5-метил-1,2,4-триазин (соединение
XX.53). М+ = 233;1H ЯМР: δ 2,35-2,7 (2H, м); 2,75 (3H, с); 3,2-3,4 (2H, м); 4,2-4,35 (1H, м); 9,25 (1H, с); (масло) и 3-(4,4-дифторбут-3- енилсульфонил)-5-метил-1,2,
4-триазин (соединение XX.54). М+ = 250;1H ЯМР: δ 2,6-2,7 (2H, м); 2,8 (3H, с); 3,7-3,8 (2H, т); 4,25-4,4 (1H, м); 9,35 (1H, с); (масло) из соединения XX.52.
(2) 3-(4,4-дифторбут-3-енилсульфинил)-6-метил-1,2,4-триазин (соединение XX. 117). MH+ = 234;1H ЯМР: δ 2,3-2,75 (2H, м); 2,85 (3H, с); 3,2-3,4 (2H, м); 4,15-4,3 (1H, м); 8,7 (1H, с); (масло) и 3-(4,4-дифторбут-3-енилсульфонил)-6-метил-1, 2, 4-триазин (соединение XX.118). MH+ = 250;1H ЯМР: δ 2,6-2,7 (2H, м); 2,9 (3H, с); 3,65-3,75 (2H, т); 4,2-4,4 (1H, м); 8,75 (1H, с); (масло) из соединения XX.116.
ПРИМЕР XXI.1
Данный пример иллюстрирует способ получения 2-(4,4-дифторбут-3-енилтио-1,3,5-триазина
(соединение XXI.1).
(4,4-дифторбут-3-енилтио)тиомочевину (в виде ее 4-метил-бензолсульфонатной соли) (0,9 г) и 1,3,5- триазин (0,216 г) вместе нагревают с обратным холодильником в этаноле (20 см3) в течение 4 часов. Реакционную смесь охлаждают и растворитель удаляют упариванием с получением твердого осадка, который растирают с гексаном. Растворимый в гексане продукт вновь выделяют упариванием при пониженном давлении. Это дает соединение XX.1 (0,4 г). М+ = 203;1H ЯМР: δ 2,45 (2H, м); 3,20 (2H, т); 4,20- 4,40 (1H, м); 8,82 (2H, с); (масло).
Соединения формулы (I) являются нематоцидными и могут использоваться для подавления нематод на растениях. Таким образом, следующим объектом настоящего изобретения является способ уничтожения или подавления нематод, который включает нанесение соединения формулы (I) на нематоду.
Термин "подавление" подразумевает нелетальное действие, результатом которого является предохранение растения-хозяина от поражения и прекращение роста популяции нематод. Эффекты могут быть результатом химически спровоцированного косвенного вредного воздействия организмов друг на друга, обездвиживания или защиты от появления из яиц или индукции. Химическая обработка может также иметь отрицательное воздействие на развитие нематод или их размножение.
Соединения по настоящему изобретению могут использоваться как против паразитирующих на растениях, так и против живущих в почве нематод.
Примерами паразитирующих на растениях нематод являются: эктопаразиты, например, Xiphinema spp., Longidorus spp. и Trichodorus spp.; полупаразиты, например, Tylenchulus spp.; мигрирующие эндопаразиты, например, Pretylenchus spp. , Radopholus spp. и Scutellonema spp.; закрепляющиеся (оседлые) паразиты, например, Heterodera spp., Globodera spp. и Meloidogyne spp.; и стволовые и лиственные эндопаразиты, например, Dikylenchus spp., Aphelenchoides spp. и Hirshmaniella spp.
Соединения формулы (I) также проявляют активность против различного вида нематод, включая цист нематоды.
Соединения по настоящему
изобретению также проявляют активность против других паразитов, вредящих росту и хранению сельскохозяйственных продуктов, лесоводству, урожаю в теплицах, декоративным растениям, урожаю в питомниках,
хранению пищевых и волокнистых продуктов. Эти пестициды включают:
Heteroptera/Homoptera, включая Myzus persicae, Aphis gossypii, Aphis fabae, Rhopalosiphum padi, Aonidiella spp., Trialeurodes
spp., Bemisia tabaci, Nilaparyata lugens, Nephotettix cincticeps, Nezara viridula, Dysdercus suturellus, Dysdercus fasciatus и Lygus lineoralis.
Diptera, включая Ceratitis capitata, Tipula spp., Oscinella frit, Lirimyza spp., Delia spp. и Peromya spp.
Lepidoptera, включая Pieris brassicae, Plutella xylostella, Spodoptera littoralis и другие Spodoptera spp., Heliothis virescens и другие Heliothis и Helicoverpa spp., и Chilo parkellus.
Coleoptera, включая Phaedon cochleariae, Diabrotica spp., Agrotis spp. и Leptinotarsa decemlineata.
Blattodea, включая Blattella germanica. Periplaneta americana и Biatta orientalis.
Orthoptera, включая Chortiocetes terminifera, Schistocerca spp., Locusta spp. и Scapteriscus spp.
Acari, включая Panonychus ulmi, Panoychus urticae, Tetranychus cinnabarinus, Phyllocoptruta oleivora и Brevipalpus spp.
Соединения могут также
использоваться против паразитов, вредных для домашнего скота, домашних животных, для здоровья общества и животных, таких как:
Siphonaptera, включая Ctenocephalides felis, Ctenocephalides
canis, Xenopsylla cheopis и Pulex irritans.
Mallophaga, включая Menopon gallinae и Cuclotogaster heterographus.
Anoplura, включая Pediculus humanus capitis, Pediculus humanus humanus и Phthirus pubis.
Diptera, включая Musca domestica, Aedes aegypti, Anopheles gambiae, Culex quinquefasciatus, Chrysops discalis и Tabanus nigrovittatus.
Sarcophagidae, включая Sarcophaga haemorrhoidalis и Wohlfahrtia magnifica.
Calliphoridae, включая Lucilia cuprina и Cordylobia anthropophaga.
Oestridae, включая Oestrus ovis.
Главным образом соединения могут использоваться для уничтожения и подавления паразитов, поражающих и/или связанных с перенесением заболеваний человека и животных. Паразиты, которые могут быть уничтожены и подавлены с помощью соединений по изобретению, представляют собой паразиты-нематоды животных, включая млекопитающих, которые можно обнаружить в желудочно-кишечном тракте, воздушных протоках или сосудах крови дыхательного тракта и сердца, вместе со связанными с ними кровеносными сосудами.
Соединения формулы (I) могут использоваться для лечения позвоночных, таких как млекопитающие (например, человек, свиньи, овцы, рогатый скот, лошади, кошки и собаки), птицы (например, куры, утки, индейки, гуси, канарейки и волнистые попугайчики) и рыбы (например, семга, форель и декоративные рыбы).
Нематоды и другие вредители могут быть уничтожены/подавлены нанесением эффективного количество одного или более соединений по настоящему изобретению на среду обитания паразитов, на поверхность, которую необходимо защитить, а также непосредственно на паразиты.
При нанесении на локус нематодных, инсектицидных или акарицидных паразитов или на растение, которое чувствительно к воздействию нематоцидных, инсектицидных или акарицидных паразитов, соединение обычно вводят в состав композиции, которая содержит в дополнение к соединению формулы (I) подходящий инертный растворитель или носитель и/или поверхностно-активные агенты. Таким образом, двумя следующими объектами изобретения являются нематоцидная, инсектицидная или акарицидная композиция, содержащая эффективное количество соединения формулы (I), как указано здесь, и инертный разбавитель или носитель и, необязательно, поверхностно-активный агент.
Количество соединения, обычно наносимое для подавления нематодных паразитов, дается для обеспечения уровня нанесения от 0,01 до 10 кг на гектар, предпочтительно от 0,1 до 6 кг на гектар.
Композиции могут наноситься на почву, растение, семена или другую поверхность, которую необходимо защищать, на локус паразитов или на место обитания паразитов, в виде тонких порошков, смачивающихся порошков, гранул (медленного или быстрого высвобождения), концентратов эмульсий или суспензий, жидких растворов, эмульсий, покрытий для семян, составов для задымления/тумана или в виде подавляющих высвобождение композиций, таких как микроинкапсулированные гранулы или суспензии.
Тонкие порошки готовят путем смешивания активного ингредиента с одним или более тонкоизмельчаемым носителями и/или разбавителями, например, природные глины, каолин, пирофиллит, бентонит, окись алюминия, монтмориллонит, кизельгур, шеллак, диатомовые земли, фосфаты кальция и карбонаты магния, сера, известь, мука, тальк и другие органические или неорганические твердые носители.
Гранулы получают либо абсорбированием активного ингредиента в пористый гранулированный материал, например, пемзу, аттапульгитовые глины, фуллерову землю, кизельгур, диатомовые земли, кочерыжка кукурузного початка и тому подобное, или на твердое вещество ядра, такого как песок, силикаты, минеральные карбонаты, сульфаты, фосфаты или тому подобное. Агенты, которые обычно используют для облегчения пропитки, связывания или покрытия твердых носителей активным ингредиентом, включают алифатические или ароматические петролейные растворители, спирты, поливинилацетаты, поливиниловые спирты, простые эфиры, кетоны, сложные эфиры, декстрины, сахара и растительные масла. Другие добавки также можно включить, такие как эмульгирующие агенты, смачивающие агенты или диспергирующие агенты.
Микроинкапсулированные составы (микрокапсулированные суспензии CS) или другие подавляющие высвобождение составы также могут использоваться, в частности, для медленного высвобождения в течение некоторого периода времени и для обработки семян.
Альтернативно, композиции могут быть в виде жидких препаратов для использования путем погружения, орошения или обрызгивания, которые являются обычно дисперсиями или эмульсиями активного ингредиента в присутствии одного или более смачивающих агентов, диспергирующих агентов или эмульгирующих агентов (поверхностно-активных агентов). Композиции, которые предполагается использовать в виде водных дисперсий или эмульсий, обычно хранятся в виде способных к образованию эмульсий концентрата (ЭК) или в виде способных к образованию суспензий концентрата (СК), содержащие большое количество активного ингредиента или ингредиентов. ЭК является гомогенной жидкой композицией, обычно содержащей активный ингредиент, растворенный в, по-существу, органический растворитель. СК является тонкой дисперсией твердого активного ингредиента в воде. Для нанесения концентратов их разбавляют в воде и обычно наносят с помощью разбрызгивания на обрабатываемую поверхность. Для агрономических и садоводческих целей, в частности, используют водный состав, содержащий от 0,0001 до 0,1 мас.% активного ингредиента (приблизительно эквивалент на 5-2000 г/га).
Подходящие жидкие растворители для ЭК включают кетон, метилизобутилкетон, циклогексанон, ксилолы, толуол, хлорбензол, парафины, керосин, белое масло, спирты (например, бутанол), метилнафталин, триметилбензол, трихлорэтилен, N-метил-2-пирролидон и тетрагидрофурфуриловый спирт (THFA-ТГФС).
Смачивающие агенты, диспергирующие агенты и эмульгирующие агенты могут быть катионного, анионного или неионного типа. Подходящие агенты катионного типа включают, например, четвертичные аммониевые соединения, например, цетилтриметиламмонийбромид. Подходящие агенты анионного типа включают, например, мыла, соли алифатических моноэфиров серной кислоты, например, натрийлаурилсульфат, соли сульфонированных ароматических соединений, например, натрий додецилбензолсульфонат, натрий, кальций или аммоний лигносульфонат, или бутилнафталинсульфонат, и смесь натриевых солей диизопропил- и триизопропилнафталинсульфонаты. Подходящие агенты неионного типа включают, например, продукты конденсации этиленоксида с жирными спиртами, такими как олеиловый спирт или цетиловый спирт, или с алкилфенолами, такими как октилфенол, нонилфенол и октилкрезол. Другими неионными агентами являются частичные сложные эфиры, производные длинноцепочечных жирных кислот и ангидридов гекситола, продукты конденсации указанных частичных эфиров с этиленоксидом и лецитины.
Эти концентраты должны быть устойчивы при хранении в течение длительного времени и после такого хранения должны быть способны к разбавлению водой с образованием водных препаратов, которые остаются гомогенными в течение времени, достаточного для нанесения с помощью обычного разбрызгивающего аппарата. Концентраты могут содержать 1-85 мас.% активного ингредиента или ингредиентов. Когда концентраты разбавляют для получения водных препаратов, такие препараты могут содержать различные количества активного ингредиента в зависимости от цели, для которой они предназначены. Для обработки семян соединения формулы (I) могут также быть представлены в виде порошков (сухая обработка семян СС или водорастворимый порошок ВС) или жидкостей (текучий концентрат ТС, жидкая обработка семян ЖС) или микрокапсульных суспензий КС. Составы могут наноситься на семена обычными методами и с помощью обычных устройств для обработки. При использовании композиций для нанесения на нематоды, на локус нематод, на место обитания нематод или на растущие растения, пораженные нематодами, с помощью известных средств для нанесения пестицидных композиций, например, путем распыления, разбрызгивания или нанесения гранул.
Соединения по изобретению могут быть единственным активным ингредиентом композиции или они могут быть смешаны с одним или более дополнительными активными ингредиентами, такими как нематоцидные агенты или агенты, которые изменяют поведение нематод, такие как факторы, препятствующие появлению из яиц, инсектициды, синергисты, гербициды, фунгициды или регуляторы роста растений, что подходит.
Подходящие дополнительные активные ингредиенты для включения в смесь с соединениями по изобретению могут быть соединениями, которые будут иметь более широкий спектр активности соединений по изобретению или повышают их устойчивость в месте нахождения вредителя. Они могут усиливать активность соединения по изобретению или дополнять активность, например, повышением скорости действия или преодолением невосприимчивости. Кроме того, мультикомпонентные смеси такого вида могут помогать преодолевать или предохранять развитие устойчивости к индивидуальным компонентам.
Конкретный дополнительно содержащийся активный ингредиент будет зависеть от предполагаемого использования смеси и типа требуемого
дополнительного действия. Примеры подходящих инсектицидов включают следующее:
а) пиретроиды, такие как перметрин, эсфенвалерат, дельтаметрин, цигалотрин, в частности, лямбда-цигалотрин,
бифентрин, фенпропатрин, цифлутрин, тефлутрин, безопасные для рыб пиретроиды, например, этофенпрокс, природный пиретрин, тетраметрин, s-биоаллетрин, фенфлутрин, праллетрин и 5-бензил- 3-фурилметил-(Е)
-(1R, 3S)-2,2-диметил-3-(2-оксотиолан-3-илилденметил) циклопропанкарбоксилат;
б) органофосфаты, такие как профенофос, сульпрофос, метилпаратион, азинфос-метил, деметон-s-метил, гептенофос,
тиометон, фенамифос, монокротофос, триазофос, метамидофос, диметоат, фосфамидон, малатион, хлоропирифос, фосалон, тебуфос, фенсульфотион, фонофос, форат, фоксим, пиримифосметил, пиримифос-этил,
фенитротион или диазинон;
в) карбаматы (включая арилкарбаматы), такие как пиримикарб, клоэтокарб, карбофуран, фуратиокарб, этиофенкарб, альдикарб, тиофурокс, карбосульфан, бендиокарб,
фенобукарб, пропоксур или оксамил;
г) бензоилмочевины, такие как трифлумурон или хлорофлуазурон;
д) производные органоолова, такие как цигексатин, фенбутатин оксид, азоциклотин;
е) макролиды, такие как авермектины или мильбемицины, например, такие как абамектин, ивермектин и мильбемицин;
ж) гормоны и феромоны;
з) органохлористые соединения, такие как
бензолгексахлорид, ДДТ, эндосульфан, хлордан или диельдрин;
и) амидины, такие как хлордимеформ или амитрац;
к) нитрометилены, такие как имидаклоприд.
Кроме основных химических классов инсектицидов, перечисленных выше, если желательно использование смеси, в ней могут быть и другие инсектициды, имеющие специфичное назначение. Например, могут быть использованы селективные инсектициды для конкретных культур, например, специфичные против стволовых паразитов инсектициды для применения на рисе, такие как каптар или бупрофезин. В композиции могут быть также включены альтернативные инсектициды для конкретных видов насекомых/стадий, например, яйце-личинок, такие как клофентезин, флубензимин, гекситиазокс и тетрадифон, мотилициды, такие как дикофол или пропаргит, основные акарициды, такие как бромопропропилат, хлоробензилат или регуляторы роста, такие как гидраметилнон, циромазин, метопрен, хлорфлуазурон и дифлубензурон.
Примеры подходящих синергистов для применения в композициях включают пиперонилбутоксид, сезамакс, сафроксан и додецилимидазол.
Подходящие гербициды, фунгициды и регуляторы роста растений для включения в композиции будут зависеть от предполагаемого назначения и требуемого эффекта.
Примером гербицидов, селективных в отношении риса, которые могут быть включены, является пропанил, примером регулятора роста растений для использования на хлопке является "Pix", а примеры фунгицидов для использования на рисе включают бластициды, такие как бластицин-S. Отношение соединения по изобретению к другому активному ингредиенту в композиции будет зависеть от множества факторов, включая вид мишени, требуемый от смеси эффект и т.д. Однако, обычно дополнительный активный ингредиент композиции будет наноситься при таком уровне нанесения, как он обычно используется, или при слегка меньшем уровне, если имеет место синергизм.
ПРИМЕР 1
Активность соединений
формулы (I) в соответствии с изобретением определяли на различных паразитах. Паразитов обрабатывали жидкой композицией, содержащей 500 мас. ч. соединения на миллион (миллионные доли - м.д.), если не
указано иного. Композиции были получены растворением соединения в смеси ацетон : этанол (50:50) и разбавлением растворов водой, содержащей 0,05 мас. % смачивающего агента, продаваемого под торговой
маркой "Synperonic" NP8, до такой степени, чтобы жидкая композиция содержала требуемую концентрацию соединения. "Synperonic" является зарегистрированной торговой маркой.
Метод исследования, выбранный по отношению к каждому пестициду, был, в основном, одним и тем же, проводился на ряде паразитов и в среде, которой обычно является растение-хозяин или продукт питания, на котором питаются паразиты, и включал обработку композицией среды и паразитов либо отдельно, либо того и другого одновременно. Гибель паразитов оценивают затем за период, обычно варьирующийся от одного до трех дней после обработки.
Результаты исследований представлены в таблице A для каждого из соединений. Данные выражены в степени гибели, обозначаемые как A, B или C, где A обозначает менее чем 40%-ную гибель, B обозначает 40-70%-ную гибель и C обозначает 80-100%-ную гибель, "-" указывает либо на то, что соединение не испытывалось, либо на то, что никаких значимых результатов не получено.
Сведения о видах паразитов, среде или продуктах, на которых были паразиты, и о виде и продолжительности испытания представлены в таблице B. Образцы паразитов обозначены буквенным кодом.
ПРИМЕР 2
Этот пример иллюстрирует пестицидную активность соединений формулы (I) в соответствии с изобретением.
В таблице C
показаны данные активности исследуемых соединений в отношении четырех видов при различных уровнях нанесения. Методы испытания и подробности опытов TU (Tetranychus urticae, контакт), Mpa (Myzus
persicae, контакт) и DB (Diabrotica balteata, контакт) описаны в примере 1 и таблице B. Уровни нанесения показаны в оглавлении таблицы для каждого вида испытаний. Метод испытаний для исследования Mpb
(Myzus persicae, системный) следующий:
Системно исследуемые соединения оценивают в отношении персиковой картофельной тли, Myzus persicae, путем пропитки почвы 2-3-недельных растений редьки
(cv Cherrybelle) при 10 м.д. или 2,5 м.д. Использовали растения с 1-ми действительными листьями размером приблизительно 2 x 1 см. Семядолю, точку роста и 1 действительный лист удаляют. Почву покрывают
прозрачным колпаком. За день до обработки к каждому растению прибавляют 12-18 зрелых тлей. В день обработки каждый сосуд помещают в 250 см3 пластиковый сосуд с изоляционной лентой для
предотвращения выхода тлей. Каждый сосуд обрабатывают 10 см3 препарата в водном растворе (приготовлен в 1%-ном этанол : ацетон (1:1) и 0,01% Synperonic NP8 - ICI Chemicals and Polymers).
Каждую обработку повторяют три раза. Обработанные растения перемещают в стабильные комнатные условия с 20oC, 60% относительной влажности и 16-часовым светопериодом. Гибель оценивают на 3-й
и 5-й дни обработки.
Активность в отношении нематод корневых наростов Meloidogyne incognita (MI) оценивают путем нанесения возможного нематоцида в виде смачивающего раствора на 2-недельную рассаду огурцов (культура Telegraph) и инфицируют почву нематодами. К каждому растению добавляют 10 см3 водного раствора испытуемого соединения (приготовлен в 1%-ном этанол : ацетон (1:1) и 0,01% Synperonic NP8 - ICI Chemicals and Polymers), так, чтобы конечная концентрация на почве была 2 м.д. Каждую обработку повторяют дважды. Рассаду образцов инокулируют 48 часов после обработки 2 см3 суспензии только что вылупившихся молодых особей в концентрации 350 нематод на см3. Испытание проводят при 25oC при 16-часовом световом периоде в течение 9 дней. Корень каждого растения оценивают в процентах уменьшения корневого нароста относительно необработанного инфицированного контроля, и результаты представлены в таблице C в виде % уменьшения нароста по сравнению с контролем.
Данные таблицы С для четырех образцов, отличающиеся от MI, выражают в виде наблюдаемого % контроля. Черточка указывает либо на то, что соединение не испытывалось, либо на то, что никаких значимых результатов не получено.
ПРИМЕР 3
Спектр нематоцидной активности соединений в соответствии с изобретением
исследуют в контактных опытах в присутствии почвы и растения-хозяина. Наибольшая активность показана в отношении Meloidogyne incognita, Globodera rostochiensis, Pratylenchus brachyurus,
Tylenchorhynchus claytoni, Hoplolaimus columbus и Radopholus similis. Адекватная активность показана в отношении Rotylenchus reniformis и Belonolaimus longicaudatus.
В следующих примерах показаны составы, подходящие для нанесения, соединений по настоящему изобретению. Количество ингредиента выражено в масс. частях или граммах на литр, как указано. A* обозначает торговую марку.
ПРИМЕР 4
В данном примере представлены гранулы, подходящие для нанесения на почву. Гранулы могут быть получены обычными способами, такими как пропитка,
покрытие, экструзия или аггломерация.
Пропитанные гранулы, мас.%/мас.%:
Активный ингредиент - 5
Древесная смола - 2,5
Гипсовые гранулы (20-40 меш) - 92,5
Покрытые гранулы:
Активный ингредиент - 0,5
"Solvesso"* 200 - 0,4
Кальцийкарбонатные гранулы (30-60 меш) - 99,1
Гранулы замедленного высвобождения:
Активный ингредиент - 10
Сополимерный латекс поливинилацетат/винилхлорид - 5
Аттапульгитовые гранулы - 85
ПРИМЕР 5
В данном примере представлены составы для
использования разбрызгиванием. Соединения могут входить в состав в виде смачиваемых порошков, водно-диспергируемых гранул, концентратов суспензий, способных к эмульгированию концентратов, эмульсий или
микрокапсулированных суспензий для нанесения разбавленных в воде.
Способный к эмульгированию концентрат, г/л:
Активный ингредиент - 250
Кальций
додецилбензолсульфонат - 50
Нонилфенолэтоксилат - 50
Алкилбензольный растворитель - До 1 л
Смачиваемый порошок, мас.%/мас.%:
Жидкий активный ингредиент - 40
Лигносульфонатный диспергатор - 5
Силикагель - 25
Натрий лаурилсульфат - 3
Китайский клей (каолин) - 27
Микрокапсулированная суспензия:
Жидкий активный
ингредиент - 250
Диизоцианат толуола - 10
Полиметиленполифенилизоцианат - 20
Нонилфенолэтоксилат - 6
Лигносульфонатный диспергатор - 15
Ксантановая смола
- 1
Бентонит - 10
Биоцид "Proxel"* - 0,1
Карбонат натрия - 5
Вода - До 1 л
Микрокапсулированные суспензии могут использоваться путем
разбрызгивания, пропитки почвы или как промежуточные продукты для получения гранул с замедленным высвобождением для нанесения на почву.
Концентрат суспензии, г/л:
Твердый
активный ингредиент - 400
Лигносульфонатный диспергатор - 50
Натрий лаурилсульфат - 30
Ксантановая смола - 1
Биоцид "Proxel"* - 0,1
Бентонит
- 10
Вода - До 1 л
ПРИМЕР 6
В данном примере представлен состав для использования при обработке семян обычным механическим методом.
Сухая обработка семян,
мас.%/мас.%:
Активный ингредиент - 20
Додецилбензол - 3
Rubine Toner (краситель) - 2,7
Тальк - 53,3
Силикагель - До 100%
При обработке семян в
качестве текучих концентратов могут использоваться концентрат суспензии и микрокапсулированная суспензия по примеру 5.
ПРИМЕР 7
В данном примере представлен состав соединений
для электростатического опрыскивания, г/л:
Активный ингредиент - 200
N-Метилпирролидон - 50
Соевое масло - 120
"Solvesso"* 200 - До 1 л
ПРИМЕР 8
В
данном примере представлен состав, подходящий для применения в качестве приманки, мас.%/мас.%:
Активный ингредиент - 0,25
Сахарная глазурь - 99,65
Бутилированный
гидрокситолуол - 0,10
ПРИМЕР 9
В данном примере представлен состав для пищевых добавок, мг:
Активный ингредиент - 1300
Натрий крахмал гликолят - 300
Микрокристаллическая целлюлоза - 1200
Лактоза - 2920
Повидон - 250
Стеарат магния - 30
ПРИМЕР 10
В данном примере представлен состав, подходящий для
использования в виде суспензии для инъекции, мг:
Активный ингредиент - 40
Натрий метабисульфит - 1
Полисорбат - 1
Натрий метилгидроксибензоат - 2
Вода - До
1 л
Стеарат магния - 30
ПРИМЕР 11
В данном примере представлен состав, подходящий для использования в виде инъекционного раствора, мг:
Активный ингредиент - 20
Натрий цитрат - 6
Лимонная кислота - 1
Натрий хлорид - 7
Хлоркрезол - 1
Вода, мл - До 1
ПРИМЕР 12
В данном примере представлен состав,
подходящий для использования в виде пероральной суспензии, г:
Активный ингредиент - 100
Полисорбат 80 - 2
Ксантановая смола - 5
Коллоидный диоксид кремния - 10
Метилгидроксибензоат - 1,5
Моногидрат лимонной кислоты - 10
Цитрат натрия - 10
Очищенная вода, мл - До 1000
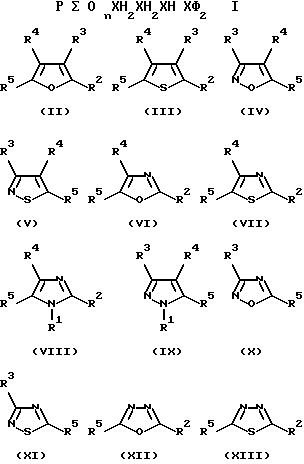

Химические формулы
(в описании)
R - SH (XXIII)
CF2=CHCH2CH2L (XXIV)
CF2=CHCH2CH2OSO2Rb (XXV)
CF2 =CHCH2CH2Br (XXVI)
R - L (XXVII)
CF2=CHCH2CH2SH (XXVIII)
R - NH2 (XXIX)
(CF2=CHCH2CH2S)2 (XXX)с
Реферат
Описываются новые соединения общей формулы (I) R-S(O)nCH2CH2CH=CF2 или его соль, где значения n и R указаны в п.1 формулы изобретения, проявляющие инсектицидную, акарицидную или нематоцидную активность. Описывается также способ их получения и сельскохозяйственные композиции на основе соединений формулы (I). 9 с. и 5 з.п. ф-лы, 24 табл.
Формула
R-S(O)n -CH2-CH2-CH=CF2
или их соли,
где n = 0, 1 или 2;
R является группой формул II - XXI


причем группа S(O)nCH2 CH2CH=CF2 может быть по крайней мере одной из R1 (когда он присоединен к атому углерода), R2, R3, R4, R5 или R6;
R1 (когда он присоединен к атому углерода), R2, R3, R4, R5 или R6 каждый независимо - водород, C1 - C6 -алкил, C1 - C6-галогеналкил, C2 - C6-алкенил, C3 - C6-цикоалкил, гидрокси, C1 - C6-алкокси, C1 - C6-гидроксиалкил, C2 - C6-моноалкоксиалкил, фенил, необязательно моно- или дизамещенный группами, независимо выбранными из галогена, нитро, C1 - C4-алкила, C1 - C4-алкокси или аминосульфонила, группа -CONHSO2- C1 - C4-алкил, CH=NOH, бензил, необязательно моно- или дизамещенный группами, независимо выбранными из галогена, нитро, C1 - C4-алкила или C1 - C4-алкокси, фенокси, необязательно моно- или дизамещенный группами, независимо выбранными из галогена, циано, C1 - C4-алкила или C1 - C4-алкокси, тиофенил, галоген, циано, нитро, NR7R8, COR7, CONR7R8, COOR7, SR7, SO2NR7R8, SO2R7, 5- или 6-членный гетероцикл, содержащий в качестве гетероатома O, S или N, необязательно замещенный галогеном, или пара соседних R1, R2, R3, R4, R5 или R6, взятых вместе, образуют 5- или 6-членное карбоциклическое кольцо;
R7 и R8 каждый независимо - водород, галоген, C2 - C6-алкенил, C2 - C6-алкинил, C1 - C6-алкил, C2 - C6-галогеналкенил;
R1 (когда он присоединен к атому азота) - водород, C1 - C6-алкил, фенил, метилсульфонил.
R-X
подвергают взаимодействию с соединением формулы CF2=CHCH2CH2Y,
где R имеет значения по п.1 или 2,
X и Y - группы SH или L, при условии, что X и Y не могут быть одинаковыми, и является легко удаляемой группой,
с получением соединения формулы I по пп.1 и 2, где n = 0, и, если желательно, с последующим окислением указанного соединения с получением соединения формулы I, где n = 1 или 2.
R-SH
с соединением формулы XXIV
CF2=CHCH2CH2L,
где R имеет значения по п.1 или 2;
L - легко удаляемая группа.
RL
с соединением формулы (XXVIII)
CF2=CHCH2CH2SH
где R имеет значения по п.1 или 2;
L - легко удаляемая группа.

где n = 0, 1 или 2,
или его соль.
27.02.95 (PCT) соединения формулы I, в которой R - группа формул II и XIX.
Комментарии