1
Изобретение относится к способу получения новых производных хиноксалинов
, обладающих гербицидной активностью , которые могут найти применение в сельском хозяйстве.
Целью изобретения является способ получения новых производных хиноксалинов
, обладающих улучшенной гербицидной активностью.
Пример 1. Получение метил-2- 4- (6-хлор-хиноксалинилокси)-фено - си -пропионата (1).
Смесь 2,6-дихлррхиноксалина (10 ммоль), полученного из 4-хлор-1,
-динитробензола и этилового эфира аминоуксусной кислоты, метил-2-(4-гидриксифенокси )-прописната
(10 ммоль), безводного карбоната натрия (11 ммоль) и диметилформамида
(40 мл), нагревают в режиме кипячения с обратным холодильником в течение
3 ч. Затем реакционную смесь охлаждают и выливают в воду. Водноорганическую
смесь экстрагируют диэтиловым эфиром. Эфирный экстракт сушат над безводным сульфатом натрия и
растворитель удаляют отгонкой при пониженном давлении, при этом получают
твердьв остаток. Твердый остаток
перекристаллизовывают из метанола,
получают метил-2- 4-(6-хлор-2-хиноксалинилокси )-пропионат в виде белых
игл (2,5 г, 70%) с т,пл. 126°С.
Предполагаемая структура соединения
подтверждена спектроскопией протонного магнитного резонанса и массспектрометрией .
Пример 2. Получение метил-2- 4-(6-xлop-1-oкcид-xинoкcaлин-2-илoкcи
)-фeнoкcи -пpoпиoнaтa (4) .
А. Персульфат калия (7,42 г медленно
прибавляют к смеси 2,6-дихлорхинолина (5,0 г) и концентрированной
серной кислоты (25 мл) при перемешивании при 10 С. По окончании прибавления
персульфата калия реакционной смеси дают нагреться до комнатной
температуры и перемешивание продолжают еще в течение 24 ч. После этого
реакционную смесь выливают в ледяную воду (400 мл), нейтрализуют водным
раствором бикарбоната натрия и экстрагируют хлористым метиленом. Органический
экстракт промывают насьш1енным раствором поваренной соли, высушивают над безводным сульфатом натрия
и затем растворитель упаривают. Твердый остаток перекристаллизовывают из этилового спирта, получают 2,6-дихлорхиноксалин-1-оксид в виде коричневых игл с т.пл. 185°С.
Б. Смесь 2,6-дихлорхиноксалин-1-оксида (1,0 г; 4,7 ммоль), мет1ш-2- (4-гидроксифенокси)-пропионата
(0,92 г; 4,7 ммоль)-, безводного карбоната калия (0,65 г- 4,7 ммоль) и метилэтилкетона (70 мл), нагревают
в режиме кипячения с обратным холодильником в течение 30 ч. Затем растворитель
удаляют отгонкой при пониженном давлении и остаток распределяют между хлористым метиленом и водой
. Органический слой отделяют и высушивают, растворитель упаривают, в результате получают темное масло.
Маслообразный остаток хроматографируют на колонке, заполненной силикагелем (40 г), используя в качестве
злюента хлористый метилен. В итоге получают 2- 4-(6-хлор-оксихоноксалин
-2-илокси)-фенокси -пропионат в виде твердого вещества бледно-коричневого
цвета (0,75 г; 43%), имеюпр й т.пл. 110°С. Предполагаемая структура подтверждена
спектроскопией протонного магнитного резонанса и масс-спектрометрией .
Пример 3. Соединения 18-25, 35-38, 40-43, 48 и 49, подро но описанные в табл. 1, бьши получе
ны из соответствующего хиноксалина и соответствующего алкил-(гидроксифенокси
)-алканкарбоксш1ата, следуя . по существу методике, описанной в примере 1 или в примере 2, Соединение
47 (см. табл. 1) получено взаимодействием 2,6-дихлорхиназолина и диэтил-2-(4-гидроксифенокси)-2-метил
мапоната, следуя той же методике, что описана в примере 1. Предполагаемая
структура полученных соединений подтверждена спектроскопией . протонного магнитного резонанса и
масс-спектроскопией. Пример 4. Получение (6-хлор-2-хиноксалинилокси)-фенокси
-пропионовой кислоты (29). Смесь 2,6-дихлорхиноксалина (10 ммоль), 2-(4-гидроксифенокси)-пропионовой
кислоты (10 ммоль), без водного карбоната калия (20 ммоль) и сухого диметилформамида (50 мп)
нагревают в режиме кипячения с обратным холодильником в течение 3 ч. Затем реакционную смесь охлаждают
и выпивают в воду. В результате под- 644
кисления водно-органической смеси 15%-ным водным раствором HCR образуется
твердый осадок, который отделяют фильтрованием. Высушенньй продукт перекристаллизовывают из толуола
, в итоге получают (6-хлор-2-хиноксалинилокси )-феокси -пропионовую
кислоту в виде белого кристаллического вещества, имеющую т.пл. 130 С (
с разложением). Предполагаемая структура подтверждена спектроскопией протонного
магнитного резонанса и массспектрометрией . Пример 5. (6-хлор-2-хиноксалинокси
)-фенокси -пррпионат натрия (30) получают путем нейтрализации сортветствующей кислоты (пример
4) водным раствором гидроокиси натрия и удаления растворителя под пониженным давлением.
Пример 6. Получение н-пропил-2- 4- (6-хлор-хиноксалинилокси)-фенокси -пропионата (26).
А. Смесь (6-хлор-2-хинокса-линoкcи )-фeнoкcиJпpoпиoнoвoй кислоты
(2,0 г, пример 4) и хлористого тионила (15 мл) нагревают в режиме кипячения
с обратным холодильником в течение 1 ч. После удаления избытка . хлористого тионила отгонкой получают
.(6-хлор-хиноксалинилокси)-фенокси -пропионилхлорид . Б. Смесь хлористого пропионила.
полученного как описано в пункте А, н-пропанола (10 мл) и триэтиламина
(2 мл) перемешивают при комнатной температуре в течение 30 мин. После
этого смесь выливают в воду (100 мп) и полученную водно-органическую смесь
экстрагируют диэтиловым эфиром. Эфирный экстракт сушат над безводным
сульфатом натрия и затем растворитель удаляют отгонкой при пониженном
давлении, в итоге получают н-проПШ1-2- 4-(6-хлор-хиноксалинилокси) -
-феноксиТ-пропионат в виде белого кристаллического вещества с т.пл.
80 С. Предполагаемая структура продукта подтверждена спектроскопией протонного магнитного резонанса и
масс-спектрометрией. Пример 7. Соединения 17, 2628 , 33 и 46, подробно описанные в
табл. 1, были получены из (6-хлор-хиноксалинилокси )-фенокси -пропионолхлорида (см.пример 6,А) и
соответствующего спирта или тиоспирта , следуя той же методике, которая
описана в примере 6 Б, Соединение 32 (см.табл.1) было аналогичным способом
приготовлено из (6-хлор-2-хиноксалинилокси )-фенокси -пропионилхлорида и ацетоноксима. Предполагаемые
структуры для каждого из упомянутых соединений подтвер ждены спектроскопией протонного магнитного резонанса
и масс-спектрометрией и соответствующие физические данные приведены в табл. 2.
Пример 8. 2-(Н,Ы,М-триметш1аммоний )-этил-2- A-(6-xлop-2-xинoкcaлинилoкcн ) -фенокси -пропионат иодид
(34) получают взаимодействием 2-(N,N-диметиламино )этШ1-2L4-(6-xлop-2-xинoкcaлинилoкcи
)-фeнoкcи -пpoпиoнaтa (33, см. пример 7) с йодистым метилом. Полученная соль имеет т.пл.
68°С.
Пример 9. Получение этил-2 4- (6-хлор-2-хиноксалинилтио)-фенокси
-пропионата (31).
А. Раствор 4-меркаптофенола
(10 ммоль) в этиловом спирте (IjO мл) прибавляют одной порцией к раствору
этилата натрия (10 ммоль) в этиловом спирте (30 мл). После перемешивания
полученной реакционной смеси в. течение 15 минут было добавлено 10 ммоль
2,6-дихлорхиноксалина и реакционную смесь перемешивают еще в течение 30 мин. По прошествии этого времени
реакционную смесь разбавляют водой (500 мл), выпавший при этом кристаллический
осадок отделяют фильтрованием и высушивают, в итоге получают 4-(6-хлор-2-хиноксалинтио)-фенол,
имеющий т.пл, 204 С.
Б. Смесь тиоэфира, полученного
как описано в части А, (1,0 г), этил-2-бромпропионата (0,6 г) и метилэтилкетона
(30 мл) нагревают в режиме кипячения с-обратным холодильником в течение 8ч. После охлаждения
реакционную смесь профильтровывают и растворитель удаляют отгонкой при пониженном давлении, в итоге
получают 2- 4-(6-хлор-2-хиноксалинилтио )-феноксиЗ-пропионат, имеющий
т.пл. 110-115 С. Предполагаемая струтура полученного соединения подтверждена
спектроскопией протонного магнитного резонанса и масс-спектрометрией .
Пример 10. Получение этил-2- 4- (6-хлор-4-оксихиноксапин-2-илокси )--фенокси -пропионата (39 .
А. мета-хлорбензойную кислоту (10,32 г 90%-ного активного ингредиента
) добавляют порциями в течение 40 мин в смесь 2,6-дихлорхиноксапина (9,85 г) и хлористого метилена
(100 мл) при перемешивании и охлаждении ледяной баней. По завершении прибавления хлорбензойной кислоты
реакционной смеси дают нагреться до комнатной температуры и перемешивание продолжают еще в течение
4 дней, К образовавшейся суспензии прибавляют хлористый метилен и затем
реакционную смесь промывают 5%ным водным раствором бикарбоната натрия (3x500 мл). Раствор хлористого
метилена высушивают безводным сульфатом натрия и растворитель упаривают
. Полученный остаток перекристаллизовывают из метилового спирта,
в итоге получают 2,6-дихлорхиноксаЛИН-4-ОКСИД (6,1 г; 57%) в виде кристаллич ского
вещества бледно-оранжевого цвета, имеющего т.пл. }76°С.
Б) 2,6-Дихлорхиноксалин-4-оксид вводят во взаимодействие с этил-2-
(4-гидроксифенокси)-пропионатом,следуя той же методике, которая описана
в примере 2 Б), в результате получают ЭТШ1-2- 2-(6-хлор-4-оксид-хиноксалин-2-илокси
)-фенокси -пропионат, имеющий т.пл. 105 С. Предполагаемая структура соединения подтверждена
спектроскопией протонного магнитного резонанса и масс-спектрометрией.
Пример 11. Получение этш1-2- 4-(6-амино-2-хиноксаЛинилокСи)-фенокси -пропионата (44).
Этил-2- 4-(6-нитро-2-хиноксалинилокси )-фенокси -у1ропионат (20 г, соединение
21, полученное как описано в примере 3), растворили в этилацетате (600 мл) и гидрируют при комнатной
температуре и атмосферном давлений, используя в качестве катализатора
палладий на активированном угле. По завершении поглощения водорода раствор
профильтровывают через слой Целитов (диатомитовая земля) и растворитель из фильтрата удаляют отгонкой
при пониженном давлении. Остаток хроматографируют на колонке, заполненной окисью алю№1ния, используя
в качестве элюента хлористьй кетилен После упаривания растворителя полу- ,
чают этил-2- 4-(6-амино-2-хиноксалинилокси )-фенокси}-пропионат в виде
желтого кристаллического вещества.
71
имеющий т.пл. 134°С. Предполагаемая структура соединения подтверждена
спектроскопией протонного магнитного резонанса и масс-спектрометрией.
Пример 12. Получение этил-2- 4-Гб- (диметиламино)-2-хиноксалинилокси -фенокси -пропионата (45).
Смесь этил-2- 4-(fi-амино-2-хиноксалинилокси )-фенокси -пропионата
(10 ммоль), йодистого метила (25 ммоль ацетона (50 мл) и карбоната калия
(25 ммоль) нагревают в режиме кипячения с обратным холодильником в течение 24 ч. После этого реакционную
смесь профильтровывают и раствориталь упаривают. Остаток хроматографируют на колонке, заполненной окисью
алюминия, используя в качестве; элюента хлористый метилен, в итоге получают
2-{4- б-(диметиламино)-2-хинскса линилокси)-фенокси -пропионат в виде
желтого масла. Предлагаемая структура соединения подтверждена спектроскопией
протонного магнитного резонанса и масс-спектрометрией.
Многие предлагаемые соединения подробно описанные в табл. 1, являются кристаллическими веществами и
могут быть идентифицированы по температурам плавления (для удобства температуры плавления соединений
сведены в таблицу и представлены в табл. 1).
Некоторые предлагаемые соединения
подробно описанные в таблице 1, являются- маслообразными веществами и могут
быть идентифицированы по их ПКРспёктрам (для удобства данные спектроскопии
протонного магнитного резонанса сведены в табл. 2).
Данные, иллюстрирующие гербицидную активность соединений, полученных по
предлагаемому способу, представлены в таблицах, приведенных ниже. В табл.
3 и 4 - степень повреждения или ущерба , нанесенного растениям: , выражается
по шкале с оценками от О до 3, причем оценка О обозначает степень повреждения растений в пределах от
О до 25%, оценка 3 выражает 75-99% гибель растений, а оценка 3+ служит
для обозначения 100% гибели подопыт564 8
ных растений. Прочерк (-) означает, что в данном случае эксперимент не проводился.
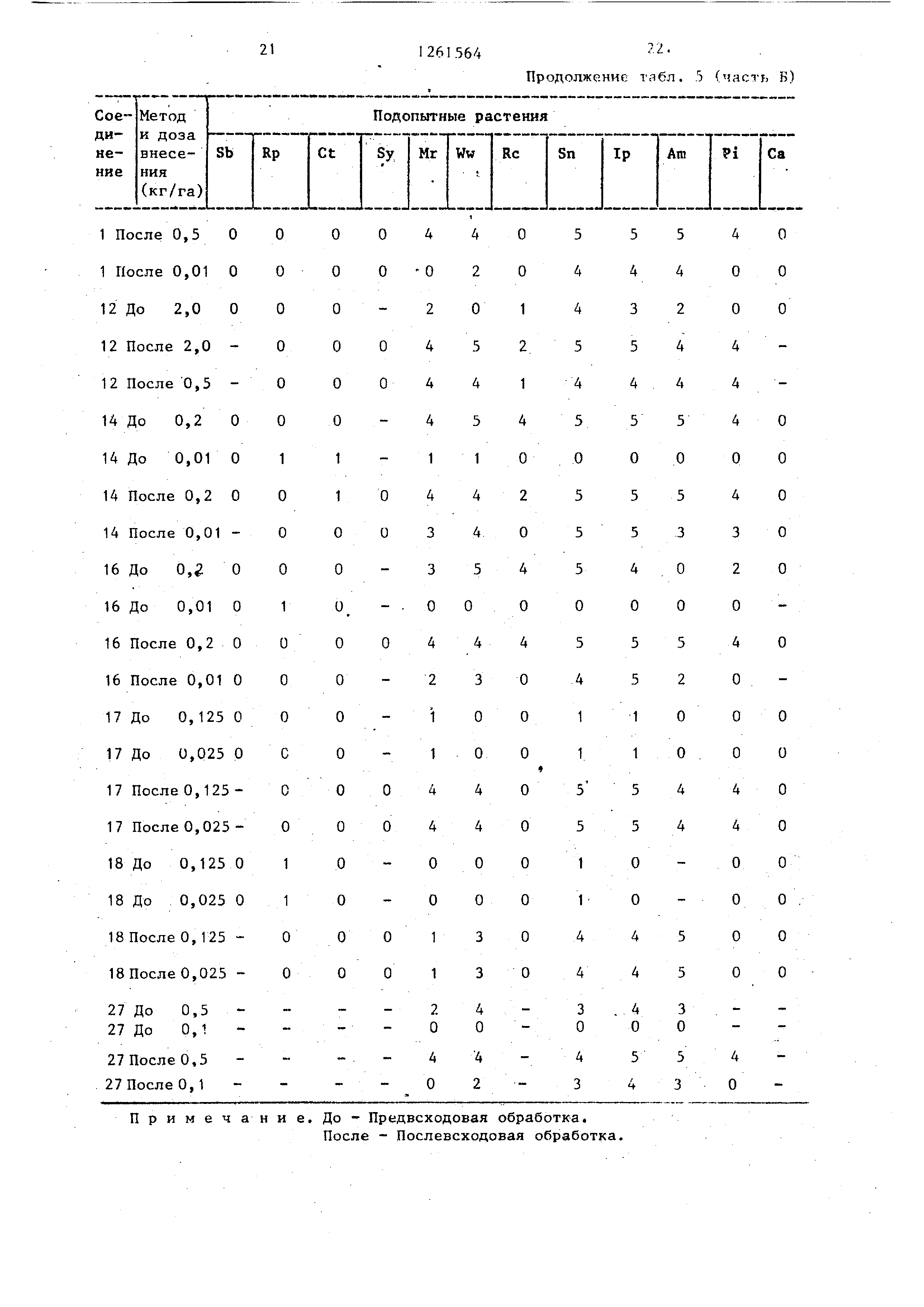
Степень повреждения подопытных ратений в табл, 5 оценена по шкале с
оценками от О до 5, где оценка О соответствует 20% повреждению, тогда
как оценка 5 означает гибель растения .
Прочерк (-) означает, что в отмеченном
случае эксперимент не проводился .
Использованные в табл. 5 сокращенные
, названия подопытных растений расшифровываются следующим образом: Sb Сахарная свекла
RP Редька Ct Хлопчатник ЗУ Соя культурная
Mr Кукуруза (маис) Ww Озимая пшеница RC Рис
Ot Овес и овсюг
Овес использовали в опытах по предвсходовой
обработке, а овсюг использовали в опытах по послевс содовой об работке.
Сокращенные названия подопытных растений, приведенные в табл. 6, расшифровываются следующим образом:
Sy Соя культурная (сорт Pethal) Ct Хлопчатник (сорт Dalta Pine 16
Pn Земляной орех (арахис, сорт
. Испанский красный)
Мг Кукуруза (маис, сорт XL 45) SS Setaria anceps Dg Digitaria Sanguinalis
Sc Echinochloa crus-galli Sg Sorghum Goldrush Sh Sorghum halepense
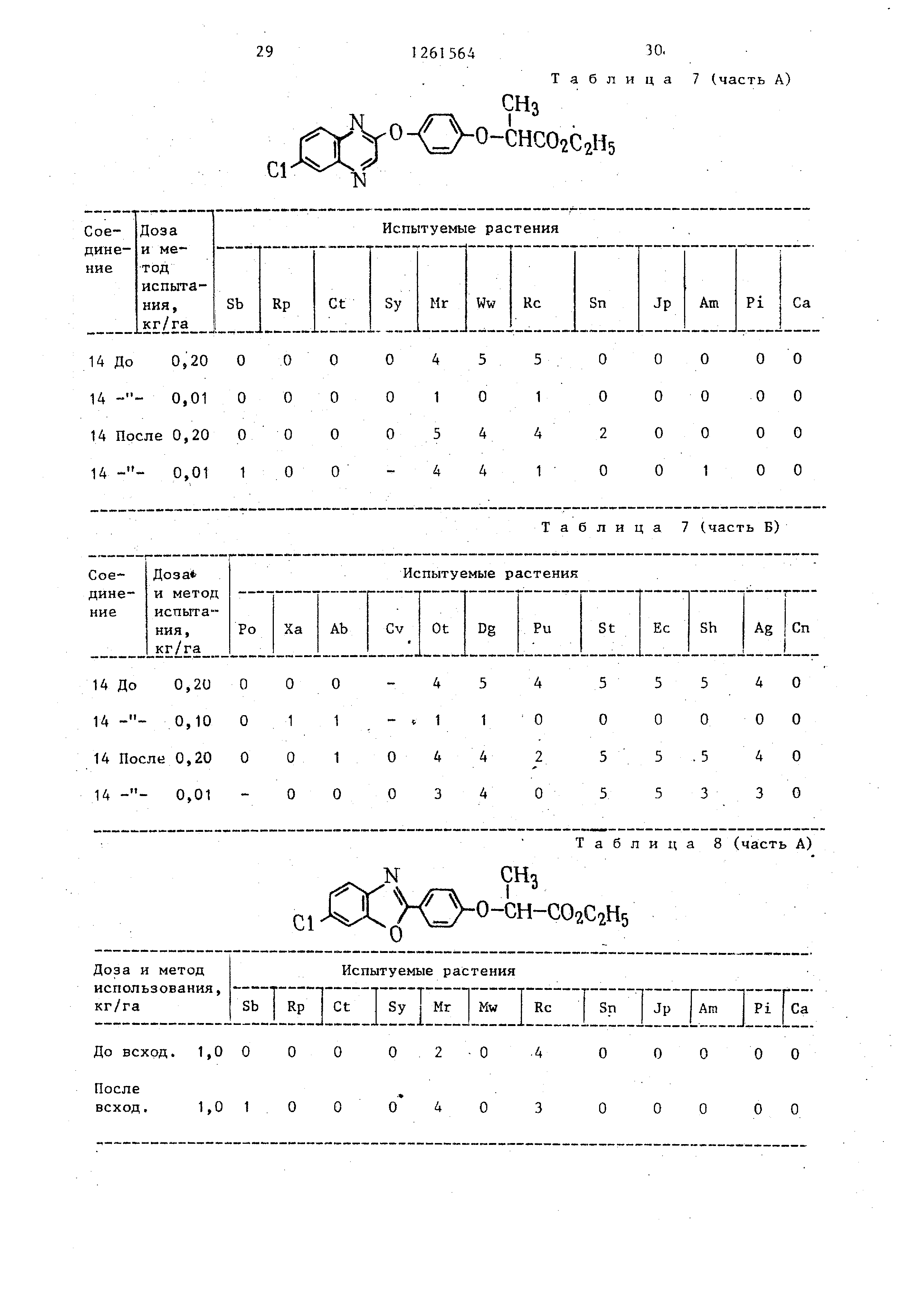
Активность предлагаемого соединения п оказана в табл. 7.
Активность одного из наиболее известных соединений показана в табл. 8
Активность другого известного соединения показана в табл. 9.
Сравнение активности известных соединений с активностью предлагаемых
четко показывает, что последние имеют более высокий уровень послевсходовой
гербицидной активности против однодольных растений (см.табл.7,8 и 9)
1261564It)
Таблица 1
Таблица 2
11 8,7 синглет,1Г:8,1 уширен-1,7
ный синглет,1Н 7,7 уширен-дублет, ный синглет,2Н; 7,1 мульти-ЗН плет, 4Н8
,7 синглет, 1Н; 6,9-7,7 мультиплет, 7Н (6-(CH|)j, N 3,0 синглет.6Н
8,7 синглет, 1Н, 7,0-8,0 мультиплет, 7Н
8.7синглет, 1Н{ 8,0 уширенный
синглет, 1HJ
7,6 мультиплет, 2Н; 7,2 мультиплет, 4Н
8.8синглет, 1Н; 7,0-8,1 мультиплет, 7Н
Предвсходовая гербицидная активность
12
261564
Продолжение табл.2 4,9 1Н 4,8
1Н дублет, -, ЗН 4,81Н
1,8 синглет,
G
4,4 квадруплет,
ЗН 4Н; 1,3 триплет , 6Н
дублет,4,7 квадруплет, 4,1 квадруплет, ЗН 1Н2Н; 1,1 триплет , ЗН
Таблица 3 уплет, 4,3 триплет 2Н, 2,7 триплет , 2Н,2,3
синглет, 6Н уплет, 4,2 квадруплет, 2Н, 1,2 триплет , ЗН
уплет, 2,9 уширенный триплет, 2Н 1,0-1,8 мультиплет
, ЮН
Продолжение табл.3
15
Послевсходовая гербицидная активность
16
1261564
Таблица 4
Продолжение табл. 4
П р и м е ч а н и е. До - Предвсходовая обработка.
После - Прслевсходовая обработка.
1 До 2,0 О 1 До 0,5 О 1 После 2,0 О
Таблица 5 (часть Б)
Примечание, До- Предвсходовая обработка.
После - Цослевсходовая обработка. 231261564
Предвсходовые ; полевые 24 Таблицаб (часть А)
испытания
25126156426
Послевсходовые полевые испытания Число дней, по прошествии которых {после Еербицидного эффекта
- Необработанные контрольные участки
Таблица 6 (часть Б)
обработки) производилась
27126156428 Послевсходовые полевые испытания
Таблица 6 (часть В)
30,
1261564
Таблица 7 (часть А)
СНз
31
32
1261564 Таблица 8 (часть Б)















Комментарии