Полиакрилонитрильные (пан) полимеры c низким индексом полидисперсности (ипд) и получаемые из них углеродные волокна - RU2647861C2
Код документа: RU2647861C2
Чертежи

Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение в целом относится к синтезу полиакрилонитрильных (ПАН) полимеров и способам получения углеродных волокон из ПАН-полимеров.
УРОВЕНЬ ТЕХНИКИ
Благодаря своим свойствам, таким как высокая удельная прочность и жесткость, высокая устойчивость к химическому воздействию и низкий коэффициент термического расширения, углеродное волокно широко применяют в аэрокосмической, спортивной и коммерческой промышленности в автомобильной области, ветроэнергетике и других областях энергосбережения. Как правило, углеродные волокна получают из полиакрилонитрильных (ПАН) полимеров.
Свободнорадикальная полимеризация
Традиционно, ПАН-полимеры получают методом свободнорадикальной полимеризации. При свободнорадикальной полимеризации катализатор или инициатор первым инициирует реакцию с образованием начальных частиц свободных радикалов. Эти радикальные частицы начинают реагировать с мономерами, создавая активные центры с образованием свободных радикалов мономеров. Затем радикалы мономеров реагируют с другими мономерами, в результате чего происходит рост молекулярной цепи с образованием полимерных радикалов.
Иногда в процессе полимеризации один радикал реагирует с другим радикалом, что приводит к их соединению и образованию длинной мертвой цепи, например, как обрыв цепи в результате рекомбинации, в то время как некоторые радикалы на конце одной цепи могут атаковать атом водорода на втором по счету от последнего атома углерода во второй радикальной цепи, что приводит к обрыву цепи в результате диспропорционирования. Полимерный радикал также может реагировать с другим соединением, таким как агент передачи цепи, с прекращением реакции роста полимерного радикала и с образованием нового радикала из агента передачи цепи. Этот вновь образованный радикал агента передачи цепи начинает рост своей новой цепи. Таким образом, агент передачи цепи уменьшает длину растущей цепи полимерного радикала. Если скорость этого обрыва цепи намного выше, чем скорость роста, то образуются очень маленькие полимеры с небольшой длиной цепи. Таким образом, агент передачи цепи применяют для регулирования длины молекулы или молекулярной массы полимера. Вследствие различия механизмов обрыва цепи, полученные молекулярные цепи имеют различные длины или различные молекулярные массы. Таким образом, молекулярная масса полимеров имеет распределение. Данное распределение может быть определено с помощью индекса полидисперсности (ИПД) полимера следующим образом:

Альтернативно, ИПД может быть выражен следующим образом:

Mw, Mn, Mz измеряют посредством ГПХ (гельпроникающей хроматографии). Здесь, Mw представляет собой средневесовую молекулярную массу. Mn представляет собой среднечисленную молекулярную массу, а Mz представляет собой Z-среднюю молекулярную массу или усредненную по размерам молекулярную массу.
Высокий ИПД указывает на то, что полимер имеет широкое молекулярно-массовое распределение, которое означает, что полимер содержит молекулы с очень высокой молекулярной массой или молекулы с очень низкой молекулярной массой или оба вида молекул. Другими словами, полимер состоит из молекулярных цепочек, которые сильно различаются по длине. Присутствие молекул со слишком высокой молекулярной массой или слишком низкой молекулярной массой будет влиять на пригодность полимера для переработки в волокна путем формования и на итоговые свойства волокна, особенно влияют молекулы со слишком низкой молекулярной массой, по причине того, что молекулы с низкой молекулярной массой являются разновидностью молекулярного дефекта для механических свойств полимера.
ПАН-полимер, полученный с помощью обычной радикальной полимеризации, не позволяет контролировать процесс полимеризации. Полученный полимер имеет широкое распределение по молекулярной массе. Таким образом, существуют трудности с точки зрения улучшения механических свойств волокон, сформованных из таких ПАН-полимеров.
КРАТКОЕ ОПИСАНИЕ
Согласно настоящему описанию предложен способ получения полиакрилонитрильного (ПАН) полимера с узким молеклярно-массовым распределением и способ получения предшественников углеродного волокна из такого полимера. Предпочтительный ПАН-полимер имеет ИПД (Mw/Mn), составляющий примерно 2 или менее. Такой ПАН-полимер синтезируют путем контролируемой/живой радикальной полимеризации с применением специального агента передачи цепи с обратимым присоединением-фрагментацией RAFT (Reversible Addition-Fragmentation Chain Transfer).
Углеродные волокна, полученные из предшественников волокна, демонстрируют хорошие свойства, такие как постоянное поперечное сечение, небольшие микродефекты и молекулярные дефекты. Такие хорошие свойства обусловлены тем, что полимер с низким ИПД имеет однородную Mw и обеспечивает небольшие молекулярные и микродефекты во время процессов производства углеродного волокна.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
ФИГ. 1 представляет собой график, полученный методом ртутной порометрии, показывающий распределение размеров микропор лиофилизированного коагулированного ПАН-волокна, полученного из ПАН-полимера с низким ИПД.
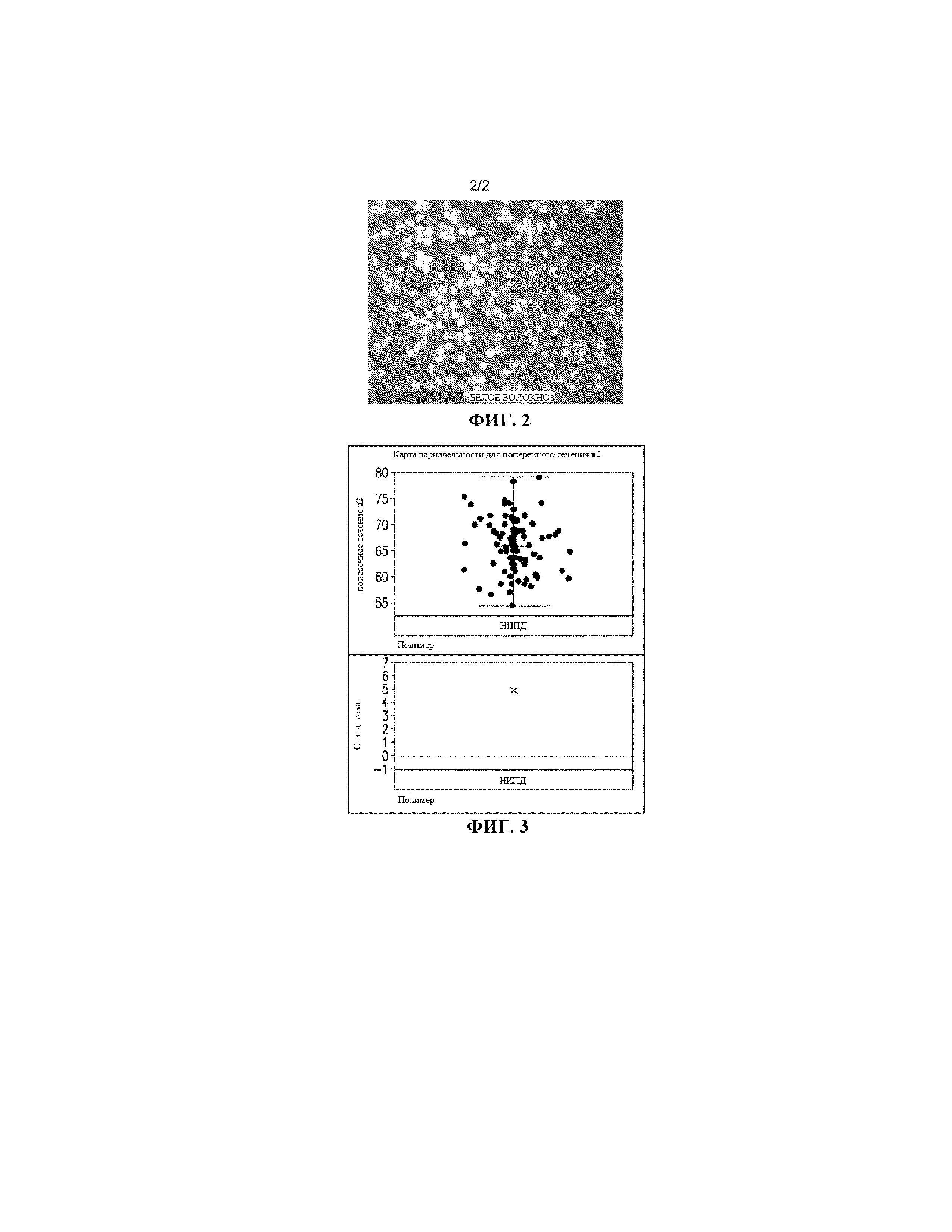
ФИГ. 2 представляет собой микрофотографию поверхности поперечного сечения предшественника ПАН-волокна, полученного из ПАН-полимера с низким ИПД.
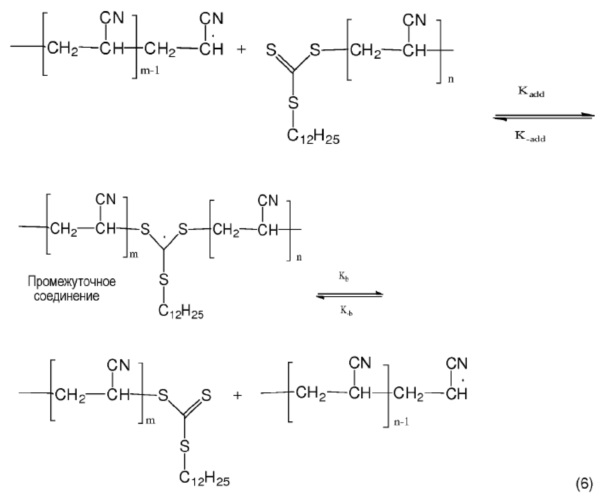
ФИГ. 3 представляет собой карту вариабельности для поверхности поперечного сечения того же предшественника ПАН-волокна, упомянутого на ФИГ. 2.
ПОДРОБНОЕ ОПИСАНИЕ
Один аспект настоящего изобретения относится к механизму регулирования молекулярно-массового распределения или ИПД ПАН-полимера путем контролируемой/живой радикальной полимеризации с применением специальных агентов RAFT. Требуемое значение ИПД (Mw/Mn) составляет примерно 2 или менее, предпочтительно ИПД (Mw/Mn) составляет от I,2 до 1,9 (или альтернативный ИПД (Mz/Mw) составляет от 1,2 до 1,7).
Контролируемая/живая радикальная полимеризация
Если обрыв цепи происходит только после того, как все мономеры потреблены в процессе радикальной полимеризации, эта полимеризация называется живой полимеризацией. В этой реакции полимеризации рост цепи может продолжаться, если в реакцию добавляют большее количество мономера. В идеальном процессе живой полимеризации все цепи инициированы в начале реакции и растут с одинаковой скоростью. При этом не происходит необратимой передачи или обрыва цепи. Если инициирование происходит быстро относительно роста, распределение по молекулярной массе является очень узким, и цепи можно удлинять путем дальнейшего добавления мономеров в реакцию. Однако при радикальной полимеризации все цепи не могут быть одновременно активными. Таким образом, некоторые реагенты применяются для регулирования роста и его скорости за счет формирования стадии покоя. С помощью обратимой деактивации или активации роста может быть достигнуто быстрое равновесие между активными и покоящимися цепями для поддержания роста цепи с одинаковой скоростью таким образом, что может быть получено узкое молекулярно-массовое распределение. Это процесс называется "контролируемой/живой радикальной полимеризацией". Химический агент, применяемый в данном процессе, называется агентом RAFT (агентом передачи цепи с обратимым присоединением/фрагментацией).
Синтез ПАН-полимера
Способ получения ПАН-полимеров, имеющих узкое молекулярно-массовое распределение, представляет собой способ полимеризации в растворе, который включает:
a. объединение акрилонитрильного (АН) мономера с растворителем, одним или более сомономерами и агентом RAFT (определенном в настоящем описании) с образованием раствора;
b. нагревание раствора до температуры выше комнатной температуры, т.е. >25°C, например, 40°C-85°C; и
c. добавление инициатора к раствору для инициирования реакции полимеризации.
После завершения полимеризации непрореагировавшие АН-мономеры удаляют, например, путем деаэрации под высоким вакуумом, и полученный раствор ПАН-полимера охлаждают. На этой стадии ПАН-полимер находится в форме раствора или формовочного раствора, готового для формования.
Полимеризация АН-мономеров зависит от контролируемой/живой радикальной полимеризации с применением агента RAFT, который представляет собой тиокарбонилтиосоединение, имеющее следующую структуру:

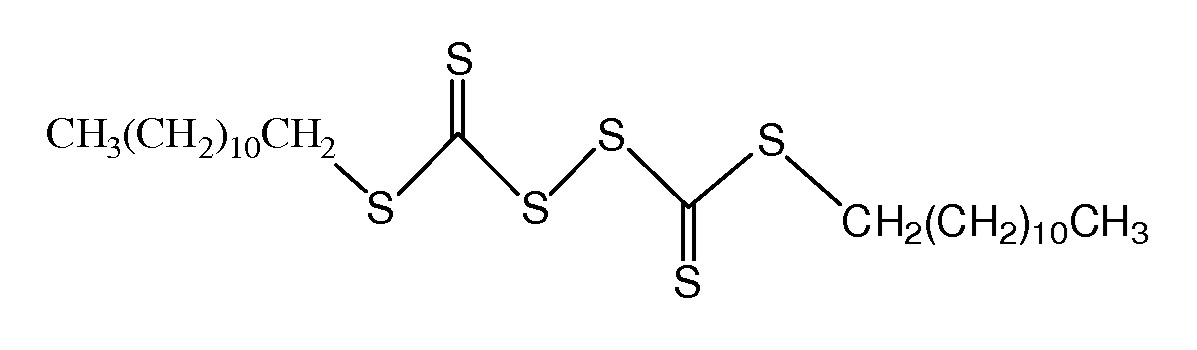
Эффективность агента RAFT зависит от заместителей R и Z. Заместители влияют на кинетику реакции полимеризации и степень контроля структуры. Группа R представляет собой свободнорадикальную уходящую группу. Она регулирует повторное инициирование полимеризации в процессе RAFT-полимеризации. А группа Z регулирует стабильность реакционной способности связи C=S и влияет на скорость присоединения и фрагментации радикала.
Предпочтительные агенты RAFT выбраны из группы, состоящей из тиокарбонилтиосоединений, имеющих следующие структуры:



Конкретные примеры агентов RAFT, имеющих вышеприведенные структуры I, II и III, представляет собой, соответственно:
1) Тритиокарбонат: 2-циано-2-пропилдодецилтритиокарбонат (CPDTC)

2) Дитиобензоат: 2-циано-2-пропил бензодитиоат (CPBZ)

3) Тиокарбонилдисульфид: бис-додецилсульфанилтиокарбонилдисульфид (BDSTD)

Подходящие растворители для полимеризации включают: диметилсульфоксид (ДМСО), диметилформамид (ДМФА), диметилацетамид (DMAc), этиленкарбонат (ЭК), хлорид цинка (ZnCl2)/вода и тиоцианат натрия (NaSCN)/вода.
Сомономеры, подходящие для синтеза ПАН-полимеров, могут представлять собой одну или более виниловых кислот, включая: метакриловую кислоту (МАК), акриловую кислоту (АК), итаконовую кислоту (ИТК), виниловые сложные эфиры, например, метакрилат (МА), метилметакрилат (ММА), винилацетат (ВА), этилакрилат (ЭА), бутилакрилат (БА), этилметакрилат (ЭМА) и другие виниловые производные, например, винилимидазол (ВИМ), акриламид (ААм) и диацетонакриламид (ДААм).
ПАН-полимеризация может быть инициирована с помощью инициатора (или катализатора) на основе азосоединения, например: азобисизобутиронитрила (АИБН), азобисциановалериановой кислоты (АЦВК) и 2,2'-азобис-(2,4-диметил)валеронитрила (АБВН) или других соединений, или органического пероксида, например, дилауроилпероксида (ЛПО), ди-трет-бутилпероксида (ТБПО), диизопропилпероксидикарбоната (ИПП) и др.
В соответствии с предпочтительным вариантом реализации ПАН-полимеризацию осуществляют на основе следующего состава, % по массе (масс. %): >90% АН-мономера; < 5% сомономера; < 1% инициатора; < 1% агента RAFT из расчета на общую массу этих четырех компонентов; и достаточного количества растворителя для получения раствора, содержащего от 5 масс. % до 28 масс. % конечного ПАН-полимера, предпочтительно от 15 масс. % до 25 масс. %.
Метод контролируемой/живой радикальной полимеризации обеспечивает регулирование архитектуры полимера. Это включает молекулярную массу, распределение по молекулярной массе (т.е. полидисперсность), функциональность и состав. Описанные выше агенты RAFT функционируют в качестве агентов передачи цепи в процессе контролируемой/живой радикальной полимеризации АН-мономеров в ПАН.
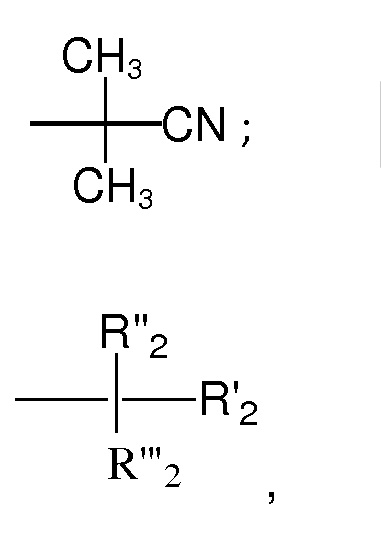
Механизм RAFT-полимеризации включает четыре стадии реакции: инициирование, присоединение-фрагментацию, повторное инициирование и установление равновесия, как показано ниже с применением, в качестве примера, CPDTC в качестве агента RAFT. В процессе ПАН-полимеризации азобисизобутиронитрил (АИБН) применяется в качестве инициатора, а ДМСО - в качестве растворителя.
A. Инициирование с помощью АИБН (азобисизобутиронитрила)

B. Присоединение-фрагментация с участием CPDTC

С. Повторное инициирование

D. Установление равновесия
Полимеризация инициируется с помощью АИБН. Он разлагается с образованием двух свободных радикалов (уравнение 1), а затем радикалы начинают вступать в реакцию с АН-мономером, что приводит к инициированию полимеризации (уравнение 2). Большее количество (АН) вступает в реакцию с радикалами и образует живой полимер или полимерный радикал Pn• (уравнение 3). CPDTC, в качестве агента RAFT, реагирует или присоединяется к Pn• с образованием радикального аддукта RAFT. Этот радикальный аддукт RAFT может вести реакцию фрагментации в направлении получения либо исходных частиц, либо нового радикала и полимерного RAFT-Pn (уравнение 4). Это обратимая стадия. В уравнении реакции 5 вновь образованный радикал повторно инициирует рост полимера с получением другого живого полимера или полимерного радикала Pm•. Этот живой полимер Pm• реагирует с полимерным RAFT-Pn с образованием промежуточного радикала - аддукта с RAFT (уравнение 6). Этот промежуточный продукт может фрагментироваться в любом направлении, что обеспечивает регулирование цепей, имеющих равные возможности для роста в направлении Pn• или Pm•, и узкое распределение по ИПД. Полимеризация заканчивается, когда потребляются все мономеры и сомономеры.
Молекулярная масса ПАН-полимеров, полученных описанным выше способом, может находиться в диапазоне от 60 до 500 кг/моль, предпочтительно от 90 до 250 кг/моль и наиболее предпочтительно от 115 до 180 кг/моль, при этом ИПД составляет примерно 2 или менее. Молекулярную массу измеряют с помощью системы для гельпроникающей хроматографии (ГПХ) Viscotek GPCmax. В процессе определения характеристик в качестве подвижной фазы применяют ДМФА (диметилформамид) с 0,02 М LiBr при скорости потока 1 мл/мин. Температуру колонки устанавливают на уровне 45°C.
Производство углеродного волокна
Описанные выше ПАН-полимеры с низким ИПД подходят для мокрого формования и формования с воздушным зазором (или попеременного "сухого-струйного мокрого формования") для получения непрерывных предшественников углеродного волокна (т.е. белых волокон). Было обнаружено, что ПАН-полимеры с низким ИПД обладают хорошей способностью к формованию, что означает легкость получения волокон из таких полимеров с помощью процесса формования. Итоговые предшественники волокна, полученные из таких полимеров, демонстрируют постоянное поперечное сечение, предел прочности на разрыв >5 г/денье и начальный модуль при растяжении >125 г/денье согласно стандарту ASTM 2256 Американского общества по испытанию материалов ASTM (American Society for Testing Materials).
Для получения белых ПАН-волокон раствор ПАН-полимера (т.е. формовочный "раствор") подвергают обычному мокрому формованию и/или формованию с воздушным зазором после удаления пузырьков воздуха с помощью вакуума. Формовочный "раствор" может иметь концентрацию полимера в диапазоне 5%-28% по массе, предпочтительно от 15 масс. % до 25 масс. % из расчета на общую массу раствора. В процессе мокрого формования формовочный раствор фильтруют и подвергают экструзии через отверстия фильеры (из металла) в коагуляционную ванну с жидкостью для образования нитей полимера. Отверстия фильеры определяют желаемое количество нитей ПАН-волокна (например, 3000 отверстий для углеродного волокна 3K). В процессе формования с воздушным зазором обеспечивают вертикальный воздушный зазор размером от 1 до 50 мм, предпочтительно от 2 до 15 мм, между фильерой и коагуляционной ванной. При этом способе формования раствор полимера фильтруют и подвергают экструзии в воздух из фильеры, и затем экструдированные нити коагулируют в коагуляционной ванне. Коагуляционная жидкость, применяемая в этом процессе, представляет собой смесь растворителя и осадителя. В качестве осадителя обычно применяют воду или спирт. Соотношение растворителя и осадителя и температуру ванны используют для регулирования скорости затвердевания образующихся экструдированных нитей при коагуляции.
Сформованные нити затем извлекают из коагуляционной ванны с помощью роликов через промывную ванну для удаления избытка коагулянта и растягивают в ваннах с горячей (например, от 40°С до 100°С) водой для придания нитям молекулярной ориентации в качестве первого этапа регулирования диаметра волокна. Растянутые нити затем сушат, например, на сушильных вальцах. Сушильные вальцы могут состоять из множества вращающихся вальцов, расположенных последовательно и в змеевидной конфигурации, над которыми нити проходят последовательно от вальца к вальцу и под натяжением, достаточным для обеспечения растягивания или релаксации нитей на вальцах. По меньшей мере некоторые из вальцов нагревают с помощью пара под давлением, который циркулирует внутри или через вальцы, или электрических нагревательных элементов внутри вальцов. Перед сушкой на растянутые волокна можно наносить масло для отделки с целью предотвращения прилипания нитей друг к другу в последующих процессах.
В качестве второго этапа регулирования диаметра волокна за первым вытягиванием волокна следует сверхрастягивание. Этот процесс сверхрастягивания проводят при температуре от 100°C до 185°C, выше температуры стеклования волокна, предпочтительно при температуре от 135°C до 175°C. Такое растягивание дополнительно ориентирует молекулы в нити. Сверхрастянутое волокно может иметь диаметр, составляющий от 0,4 до 1,5 денье, предпочтительно 0,5-1,0 денье.
Условия обработки (включая состав формовочного раствора и коагуляционной ванны, общее количество растягиваний, температуры и скорости образования нитей) соотносят для обеспечения нитей желаемой структуры и диаметра. После стадии сверхрастягивания нити волокна могут пропускать над одним или более горячими вальцами и затем наматывать на бобины.
Для превращения белых ПАН-волокон в углеродные волокна, ПАН-волокна подвергают окислению и карбонизации.
На стадии окисления ПАН-волокна подают под напряжением через одну или более специализированных печей, в которые подают нагретый воздух. Температура печи для окисления может находиться в диапазоне от 200°С до 300°С, предпочтительно от 220 до 285°С. Процесс окисления соединяет молекулы кислорода из воздуха с ПАН-волокном и вызывает начало образования поперечных связей между полимерными цепями, что приводит к увеличению плотности волокна до значения от 1,3 г/см3 до 1,4 г/см3. В процессе окисления натяжение, прикладываемое к волокну, как правило, необходимо для регулирования растягивания или сокращения волокна при коэффициенте растяжения, составляющем от 0,8 до 1,35, предпочтительно от 1,0 до 1,2. Когда коэффициент растяжения равен 1, растягивания нет. И когда коэффициент растяжения больше 1, приложенное натяжение вызывает растягивание волокна. Такое окисленное ПАН-волокно имеет неплавкую лестничную ароматическую молекулярную структуру и готово для обработки карбонизацией.
Карбонизация происходит в инертной (бескислородной) атмосфере внутри одной или более специально разработанных печей. В предпочтительном варианте реализации окисленное волокно пропускают через печь для предварительной карбонизации, в которой волокно подвергают нагреву при температуре от примерно 300°С до 900°С, предпочтительно от 350 до 750°С, при воздействии инертного газа, например азота, с последующей карбонизацией путем пропускания волокна через печь, нагретую до более высокой температуры, составляющей от примерно 700°С до 1650°C, предпочтительно от 800 до 1450°С при воздействии инертного газа. При этом указанная вторая температура выше, чем первая температура.
На протяжении всего процесса предварительной карбонизации и карбонизации должно быть добавлено натяжение волокна. В процессе предварительной карбонизации прикладываемое натяжение волокна достаточно для регулирования коэффициента растяжения таким образом, чтобы он находился в диапазоне от 0,9 до 1,2, предпочтительно от 1,0 до 1,15. В процессе карбонизации применяемое натяжение достаточно для обеспечения коэффициента растяжения, составляющего от 0,9 до 1,05. Карбонизация приводит к кристаллизации молекул углерода и, как следствие, обеспечивает готовое углеродное волокно, содержание углерода в котором составляет более 90 процентов.
Адгезия между смолой матрицы и углеродным волокном является важным критерием для композита полимера, армированного углеродным волокном. Таким образом, в процессе производства углеродного волокна после окисления и карбонизации может быть проведена обработка поверхности для увеличения этой адгезии.
Обработка поверхности может включать протягивание карбонизированного волокна через электролитическую ванну, содержащую электролит, такой как бикарбонат аммония или гипохлорит натрия. Химические агенты из электролитической ванны протравливают или придают шероховатость поверхности волокна, что приводит к увеличению площади поверхности, доступной для связывания на границе раздела волокно/матрица, и добавлению реакционноспособных химических групп.
Далее углеродное волокно может быть подвергнуто проклеиванию, при этом проклеивающее покрытие, например, покрытие на основе эпоксида, наносят на волокно. Проклеивание можно осуществлять путем пропускания волокна через проклеивающую ванну, содержащую жидкий покрывающий материал. Проклеивание защищает углеродное волокно в процессе эксплуатации и переработки в промежуточные формы, такие как сухая ткань и препрег. Проклеивание также удерживает нити вместе в отдельных жгутах для уменьшения распушивания, улучшения пригодности для обработки и увеличения сдвигового напряжения на поверхности раздела между волокном и смолой матрицы.
После проклеивания покрытое углеродное волокно высушивают и затем наматывают на бобины.
Было обнаружено, что углеродные волокна, полученные из описанных выше ПАН-полимеров с низким ИПД, имеют следующие механические свойства: предел прочности на разрыв более чем 700 тыс. фунтов/_В. дюйм (4826 Мпа) и начальный модуль при растяжении более чем 35 млн фунтов/_В. дюйм (241 Гпа), согласно методу испытания ASTM D4018.
Преимущества и свойства описанного выше ПАН-полимера и полученных из них углеродных волокон будут дополнительно проиллюстрированы с помощью следующих примеров.
ПРИМЕРЫ
Пример 1
Синтез ПАН-полимеров
ПАН-полимеры были получены в соответствии с составами для ПАН-полимеризации, представленными в таблицах 1А-1С.
В приведенных выше таблицах CPDTC, CPBZ, BDSTD представляет собой агенты RAFT, где:
CPDTC=2-циано-2-пропилдодецилтритиокарбонат
CPBZ=2-циано-2-пропилбензодитиоат
BDSTD=бис-додецилсульфанилтиокарбонилдисульфид
Примечание:* агент RAFT применяется в моль % из расчета на общее количество мономеров.
Контролируемую/живую радикальную ПАН-полимеризацию проводили следующим образом:
Азобисизобутиронитрил (АИБН) применяли в качестве инициатора/катализатора, а ДМСО - в качестве растворителя. Агенты RAFT применяли в качестве агентов передачи цепи. В процессе полимеризации проводили следующую последовательность стадий:
a) Отмеривание ДМСО из резервуара для хранения ДМСО в реактор, затем АН из резервуара для хранения АН в реактор;
b) Продувка реактора азотом;
c) Предварительное нагревание реактора и добавление сомономеров и агента RAFT в реактор при температуре выше комнатной (25°C);
d) Нагревание реактора и затем добавление инициатора/катализатора в требуемой температурной точке 40-85°С;
e) Запуск полимеризации на период времени 15-23 часа при температуре 60-80°С;
f) Охлаждение до низкой температуры (40-50°С) и сливание раствора полимера.
После полимеризации измеряли молекулярные массы и ИПД полученных ПАН-полимеров, и результаты приведены в таблицах 2A-2C.
Гель-проникающую хроматографию (ГПХ) применяли для анализа молекулярных масс и индекса полидисперсности (ИПД) полученных ПАН-полимеров. Использовали хроматографическую систему Viscotek GPCmax/SEC с детекторами рассеянного света под малым углом и прямым углом и рефрактометрическим детектором (RI). Данные собирали и анализировали с применением программного обеспечения Viscotek OMNISEC, версия 4.06 для определения абсолютной средневесовой молекулярной массы (Mw) и ее распределения.
Все ПАН-полимеры, полученные из составов, содержащих агенты RAFT, обеспечивали ПАН-полимеры с ИПД (Mw/Mn), составляющим примерно 2 или менее. ПАН-полимер, полученный из Состава 6, имеет более высокую молекулярную массу (Mw) 217778 г/моль с ИПД 1,69 после регулирования дозы агента RAFT и концентрации раствора относительно Состава 5.
Пример 2
Производство белых волокон
ПАН-полимер, полученный из Состава 5, как описано в примере 1, использовали для формирования предшественников углеродных волокон (или белых волокон) путем мокрого формования. ПАН-полимеры, полученные из Состава 12, как описано в примере 1, использовали для формирования белых волокон с помощью метода формования с воздушным зазором с применением фильеры с диаметром отверстий 150 мкм.
Свойства белых волокон определяли следующим образом.
Анализ поперечного сечения
Образец пучка белых волокон погружали в акриловую смолу и затем подвергали отверждению. Затвердевший стержень волокна полировали на шлифовальном устройстве со шлифовальной бумагой различных марок для получения гладкого сечения. После этого поперечное сечение волокна измеряли с помощью оптического микроскопа с системой анализа изображений для определения постоянства поперечного сечения.
Порометрия
Для формования с воздушным зазором образец волокна, покидающий коагуляционную ванну, подвергали сублимационной сушке при -60°C, и лиофилизированный образец исследовали с применением ртутного порозиметра для анализа пористости и пористой структуры.
Натяжение и модуль
Прочность волокна на разрыв и начальный модуль измеряли по методу ASTM D2256.
Было обнаружено, что ПАН-полимеры на основе Составов 5 и 12 обладают хорошей способностью к формованию. Предшественники белого волокна, полученные как путем влажного формования, так и путем формования с воздушным зазором, также имели хорошую прочность на разрыв и модуль, как можно видеть из таблицы 3.
ФИГ. 1 представляет собой график, полученный методом ртутной порометрии для распределения диаметров пор в лиофилизированном коагулированном волокне. Ось Y представляет собой значения логарифма дифференциальной интрузии в мл/г, или dV/dLog D. V представляет собой объем ртути, проникшей в поры образца. Ось Х представляет собой значения логарифма диаметра пор. Таким образом, на фигуре показана производная от проникшего объема относительно логарифма диаметра пор. Общий объем или пустоты представляет собой площадь под кривой. ФИГ. 1 показывает, что лиофилизированное коагулированное ПАН-волокно, полученное путем формования с воздушным зазором из ПАН-полимера с низким ИПД согласно Составу 12, имеет небольшие дефекты микропор. Микрофотография на ФИГ. 2 и карта вариабельности на ФИГ. 3 показывают, что белое волокно с низким ИПД, сформованное путем формования с воздушным зазором, имеет постоянное поперечное сечение. ФИГ. 3 представляет собой карту вариабельности площади поперечного сечения, показывающую дисперсию или распределение.
Превращение белых волокон в углеродные волокна
Предшественники из белых волокон окисляли на воздухе в диапазоне температур 220°C-285°C, и подвергали карбонизации в атмосфере азота в диапазоне температур 350°C-650°C (предварительной карбонизации), а затем 800°C-1300°C.
Предел прочности на разрыв и модуль упругости при растяжении полученных углеродных волокон были определены и приведены в таблице 4.
Предел прочности углеродного волокна на разрыв и начальный модуль при растяжении определяли согласно ASTM D4018. Углеродное волокно сначала помещали в ванну с эпоксидной смолой и затем подвергали отверждению. Нити отвержденного углеродного волокна исследовали на MTS при скорости ползуна 0,5 дюйм/мин для определения предела его прочности на разрыв и модуля упругости при растяжении.
Плотность волокна определяли методом погружения в жидкость согласно ASTM D3800.
Реферат
Изобретение относится способу получения полиакрилонитрильного (ПАН) полимера с узким молекулярно-массовым распределением и к способу получения углеродного волокна из ПАН-полимера. Способ получения полиакрилонитрильного полимера заключается в том, что объединяют акрилонитрильный мономер с растворителем по меньшей мере одним сомономером и тиокарбонилтиосоединением. Полученный раствор нагревают до температуры от 40°С до 85°С. Затем к раствору добавляют инициатор для регулирования реакции полимеризации. Полимеризация регулируется путем контролируемой/живой радикальной полимеризации, в которой тиокарбонилтиосоединение действует в качестве агента передачи цепи с обратимым присоединением-фрагментацией (RAFT). Сомономер выбирают из группы, состоящей из виниловых кислот, виниловых сложных эфиров и виниловых производных. Инициатор представляет собой азосоединение или органический пероксид. Способ получения углеродного волокна заключается в том, что вначале получают раствор вышеуказанного полимера. Далее осуществляют формование путем мокрого формования или формования с воздушным зазором с образованием предшественника полиакрилонитрильного волокна, который коагулируют в коагуляционной ванне. Далее вытягивают предшественник волокна из коагуляционной ванны роликами через промывную ванну для удаления излишка коалулянта. Затем растягивают предшественник волокна в ваннах с горячей водой для придания молекулярной ориентации в волокнах. После этого осуществляют сушку вытянутых предшественников волокон. Затем проводят окисление предшественника волокна под напряжением в одной или более печах, в которые подают нагретый воздух. Далее проводят карбонизацию окисленного предшественника волокна в одной или нескольких печах, имеющих инертную, свободную от кислорода, атмосферу. Изобретение позволяет получить полиакрилонитрильный полимер, который имеет низкий индекс полидисперсности 2 или менее и молекулярную массу в диапазоне от 60 кг/моль до 500 кг/моль, и получить углеродные волокна с улучшенными механическими свойствами, постоянным поперечным сечением и небольшими микродефектами. 2 н. и 11 з.п. ф-лы, 3 ил., 8 табл., 2 пр.
Формула









Комментарии