Покрытие для восстановления оксидов азота - RU2648045C2
Код документа: RU2648045C2
Чертежи







Описание
Данное изобретение представляет собой каталитическое покрытие для применения в качестве гидролитического катализатора (Г-катализатора) для восстановления оксидов азота. Изобретение также представляет собой способ изготовления такого покрытия, а также состав катализатора и его применение.
Сгорание топлива при избытке кислородсодержащей газовой смеси, является эффективным способом производства энергии в стационарных и мобильных практических применениях. Топливные эффективные дизельные двигатели используются в исключительных случаях в грузовиках и в большей степени в автомобилях, особенно в Европе. При сгорании бедной смеси, выбросы обычно имеют довольно низкое содержание окиси углерода (СО) или углеводородов (УВ), но, что касается оксидов азота (NOx) и частиц (твердых частиц, ТЧ), проблемы могут возникнуть в достижении стандартных выбросов, введенных властями. Кроме того, выбросы окиси углерода и углеводородов могут быть эффективно устранены с помощью окислительного катализатора, но восстановление оксидов азота и частиц требует применения других типов способов последующей обработки. Частицы могут быть эффективно удалены с помощью различных фильтров твердых частиц. Восстановление оксидов азота при избытке кислородсодержащего выхлопного газа затруднительно, потому что возможные восстановители, как правило, предпочтительно окисляются, вместо того, чтобы вступить в реакцию с оксидами азота.
Восстановления оксидов азота в мусоросжигательных установках стал актуальным вопросом в начале 1970-х годов в Японии, где были наложены ограничения на выброс NOX для снижения образования смога, вызывающего проблемы, особенно в крупных городах. Селективное каталитическое восстановление (СКВ - от англ. selective catalytic reduction, SCR) оксидов азота с помощью аммиака (NH3) было разработано для подобных целей. В катализаторе, аммиак в первую очередь реагирует с NOx, несмотря на присутствие избытка кислорода. В самом деле, кислород поддерживает реакцию в СКВ-катализаторах, производимые с некоторых пор на основе TiO2 в коммерческих продуктах, включающих ванадий, вольфрам и оксиды молибдена в качестве активных компонентов и стабилизаторов. Есть также многочисленные публикации о других видах СКВ-катализаторов, производимые на основе оксида, цеолита или углерода или их смесей. СКВ-катализаторы в настоящее время почти всегда сетчатого типа, в результате чего падение давления и загрязнение остаются умеренными. Катализаторы могут быть экструдированы из массы СКВ-катализатора или нанесены на поверхность материала-носителя сетчатого типа. Материал-носитель, как правило, керамический или металлический.
Основная реакция СКВ при избытке кислородсодержащей смеси может быть представлена следующим образом:

Аммиак может быть введен путем специально построенных распылительных форсунок в виде газа или водного раствора в выхлопной газ немного впереди катализатора. В случае применения аммиака в качестве восстановителя, он сразу оказывается в правильном составе, а ограничивающим фактором может быть смешивание в потоке газа или испарение водного раствора.
В 1980-х годах было обнаружено, что аммиак можно заменить путем применения других восстановителей, таких как мочевина или циануровая кислота, с содержанием производных аммония или азота. СКВ системы, предназначенные для автомобилей с самого начала были преимущественно основаны на применении мочевины в качестве восстановителя, так как применение, хранение и транспортировка мочевины и раствора мочевины являются безопасными по сравнению с аммиаком. Мочевина (CO(NH2)2) включает в себя две NH2 группы и распад одной молекулы мочевины производит две молекулы аммиака в водосодержащей газовой смеси. Чистая мочевина представляет собой твердое белое вещество, легко растворяющееся в воде в высоких концентрациях. В системах мочевина-СКВ для практического применения в грузовиках и на электростанциях работающим восстановителем действительно является раствор мочевины в воде:


Мочевина содержится в виде 32,5-процентного раствора для практического применения в грузовиках, и данный раствор вводят в выхлопной газ вместе с воздухом или отдельно в виде раствора. Применение воздуха с раствором обеспечивает смесь, которая может быть доставлена под давлением в горячий выхлопной газ. Когда используемый восстановитель представляет собой раствор мочевины, необходимо дать раствору мочевины достаточное количество времени, чтобы смешаться в трубке для испарения, также как и для термолиза (реакция 2) и гидролиза (реакция 3) мочевины. Раствор мочевины должен быть введен в достаточном удалении от переднего края СКВ каталитического нейтрализатора, чтобы мочевина прореагировала до аммиака последовательно в радиальном направлении. В случае практического применения в грузовиках (двигатель объемом 4-20 литров), количества выхлопных газов настолько велики, что, как правило, необходима круговая ячейка 250-400 мм в диаметре для поддержания линейных скоростей и падения давления в пределах нормативного диапазона, и для предоставления возможности СКВ-катализатору функционировать без нарушения работы двигателя. Таким образом, смешивание мочевины в радиальном направлении имеет большое значение. Термически проходящие термолиз и гидролиз требуют достаточного количества времени, поэтому точка введения мочевины может находиться на расстоянии нескольких метров от переднего края СКВ-катализатора.
Грузовики могут также включать применение дизельного окислительного катализатора, чтобы способствовать окислению углеводородов, окиси углерода и NO до NO2. В окислительных катализаторах обычно используют высокоустойчивую к сере платину (Pt) в качестве активного металла. В связи с использованием тепла, желательно установить катализаторы настолько близко к двигателю, насколько представляется возможным. Окислительный катализатор полезен для работы СКВ-катализатора, потому что удаление УВ и получаемый NO2 обеспечивают поразительное поддержание СКВ реакций. Было высказано предположение, что специальный гидролитический катализатор (Г-катализатор) можно использовать в передней части СКВ-катализатора, чтобы способствовать смешиванию мочевины и гидролизу при различных температурах (Döring и Jacob, 21-й симпозиум Vienna Motor Symposium, 2000 год). В то же время было предложено, что гидролитический катализатор и катализатор предварительного окисления могут быть установлены бок о бок, в результате чего мочевину вводят только в боковой поток выхлопных газов. Таким образом, не исключено, что автомобиль, имеющий систему СКВ, может быть снабжен катализатором предварительного окисления, гидролитическим катализатором, СКВ-катализатором, и катализатором повторного окисления, с целью удаления в дальнейшем возможного аммиака, оставшегося в выхлопном газе после реакции СКВ (ЕР 0896831). Устройство смешивающего типа, также упоминаемое как испаритель с каталитическим покрытием на его поверхности. Еще одним аргументом, упоминаемым в пользу применения Г-катализатора, является то, что объем СКВ-катализатора может быть уменьшен, например, на 10-30% (ЕР 0555746).
Применение гидролитического катализатора было предложено либо отдельно, либо в комбинации вниз по течению от отдельного элемента испарителя (ЕР 0487886). Упоминаемые как каталитические покрытия TiO2, оксид Al, SiO2 или их смесь, которая также может сопровождаться SO3 или WO3, например, для кислотных свойств или термальной стабилизации. Было сказано, что удельная площадь поверхности превышает 10 м2/г (ЕР 0487886). Кроме того, было отмечено, что, в дополнение к вышесказанному, гидролитический катализатор включает цеолит (Н-морденит, H-ZSM5) (ЕР 0555746). Г-катализатор должен обладать настолько незначительной результирующей активностью разложения аммиака насколько возможно (ЕР 0487886), в противном случае образуется дополнительная потеря в потреблении мочевины.
Гидролиз мочевины и ее смешивание с потоком газа могут быть ускорены путем улучшения фактического введения мочевины, которое может быть ассистировано путем применением различных форсунок, с регулированием нагнетающего давления и техникой автоматического управления. Наиболее важным аспектом является то, как далеко, и в какой точке в устройстве производится введение мочевины. Аспект, обязательный к рассмотрению в отношении размеров заключается в разработке распылителя мочевины и проточного канала такой формы, чтобы не было распыления мочевины на холодные стенки. Независимо от того, окажется ли мочевина в конечном итоге в виде капли на стенке или на холодной стенке, существует опасность получения нежелательных побочных продуктов, таким образом, увеличивая потери мочевины. В СКВ, эксплуатационная эффективность мочевины при восстановлении оксидов азота должна быть больше 90%, потому что, в стандартном европейском испытательном цикле, расход мочевины составляет, например, 3-6% от расхода дизельного топлива, таким образом, представляя собой основные затраты.
Было сделано предложение о введении твердой мочевины в виде порошка, что позволяет избежать необходимости содержать воду вместе с раствором. Кроме того система может включать гидролитический катализатор так же, как при введении жидкого раствора мочевины (ЕР 0615777). Основной трудностью в данных системах часто является соответствие дозы порошка в выхлопном газе при различных условиях.
Проблемы в описанных гидролитических катализаторах могут включать в себя тот факт, что с одним Г-катализатором очень трудно достичь одновременно эффективного низкотемпературного гидролиза, смешивания и небольшого разложения NH3 на азот или оксиды азота во всем требуемом температурном диапазоне (100-600°C). Состав и пространственный дизайн Г-катализатора, эффективного при гидролизе при температуре 150-200°C, часто слишком активны при высоких температурах и NH3 разлагается до начала реакции СКВ. Гидролитические катализаторы были описаны как высокотемпературные катализаторы с большой площадью поверхности (10 м2/г) и избыточной пористостью, в особенности с небольшими порами. Кроме того, было описано, что Г-катализатор специально включает применение соединений, обеспечивающих поверхностную кислотность для адсорбции NH3. В данном случае, время выдержки для аммиака становится, тем не менее, больше в результате адсорбции, пор и объема, а самым продолжительным при низких температурах, в результате чего кинетически NH3 имеет больше шансов разложиться и остается слишком долго в Г-катализаторе. Г-катализаторы были описаны как смешивающие структуры, отличающиеся тем, что смешивание, прежде всего, происходит в одиночном канале ячейки, и апертурные числа были около 150 cpsi (ячеек на квадратный дюйм, от англ. cells per square inch) и достаточно высокое количество покрытия, то есть около 150-200 г/л (ЕР 0896831). Некоторое смешивание внутри ячейки канала достигается с помощью различных барьеров потока и зубцов, но смешивание в радиальном направлении реактора остается незначительным, в результате чего несоответствие в радиальном направлении потока и, в особенности, температуры может быть даже усилено. Такие структуры напоминают ячейки сетчатого катализатора, которые, по сравнению с пустой выхлопной трубой, имеют большую площадь геометрической поверхности (ПГП) и количество катализатора, низкое число Рейнольдса (→ эффективность обмена массы) в каналах, слабое смешивание в радиальном направлении. Подобные структуры хороши с точки зрения поддержания каталитической реакции, но мочевина и раствор должны испаряться, и смешиваться в потоке газа до достижения какого-либо преимущества путем каталитического поддержания гидролиза. Желательно чтобы одновременно, несмотря на хорошее смешивание, происходило и введение мочевины как можно ближе к СКВ-катализатору или лицевой поверхности Г-катализатора. Если Г-катализатор имеет чрезмерно большое апертурное число, существует опасность попадания брызг смеси мочевина-вода на лицевую поверхность плотной ячейки с негативными последствиями, аналогичными результату попадания брызг на стенки трубы. Следующей опасностью в подобном случае является то, что фронтальная поверхность ячейки Г-катализатора и ее покрытие изнашиваются механически от капель или плотная ячейка забита твердыми побочными продуктами. Общей проблемой в отношении раздельного гидролиза и СКВ реакторов является полное преобразование мочевины в аммиак, передача полученного NH3 в СКВ реакторе для восстановления оксидов азота с аммиаком.
Если гидролитический катализатор установлен таким образом, что только часть потока проходит через него, то будет трудно регулировать скорость потока через гидролитический катализатор с подходящей разреженной или плотной камерой таким образом, чтобы обеспечить в то же время необходимое пространство для распыления мочевины в передней части ячейки и установить линейную скорость в пределах приемлемого диапазона. Обратное давление разреженной ячейки ниже, и слишком большой поток проходит через ячейку. В случае плотной ячейки, обратное давление слишком высокое с низким значением потока, и мочевина не может быть введена слишком близко к переднему краю ячейки. По этим причинам, реактор гидролиза требует дальнейшего совершенствования.
Еще одно предложение было сделано для 3D смешивающих структур, которые были применены в непокрытом состоянии или были покрыты типичным СКВ-катализатором (статические смесители). В данном случае, смешивание является наиболее эффективным с большими размерами каналов, что хорошо для применения на электростанциях с участием большого количества частиц. Таким образом, обмен массы и распределение мочевины/аммиака согласуются, но есть проблемы, в том числе, например, небольшое количество материала катализатора на стенках традиционных, с большими каналами статических смесителей, а гидролиз мочевины основан на реакциях, происходящих термически или на СКВ-катализаторе.
Описание изобретения
Задачей настоящего изобретения является обеспечение практического применения для выхлопных или отходящих газов высокопроизводительного каталитического покрытия для применения в качестве гидролитического катализатора, а в гидролитическом блоке катализатора, имеющего рабочий диапазон настолько широкий, насколько возможно, и также способного обладать некоторой возможностью восстановления оксидов азота. Другой задачей настоящего изобретения является способ изготовления подобного покрытия, а также состав катализатора и его применение.
Что касается композиции, основной концепцией изобретения является то, что гидролитический катализатор (Г-катализатор), установленный ниже по течению от точки введения восстановителя, применяемого в восстановлении мочевины или других оксидов азота, включает щелочные вещества, адсорбирующие HNCO и/или адсорбирующие оксиды азота, например, щелочные и щелочно-земельные металлы, лантан, и/или иттрий, и/или гафний, и/или празеодим, и/или галлий, и/или цирконий для содействия восстановлению, например, для содействия гидролизу мочевины и образованию аммиака и/или селективному восстановлению оксидов азота.
Предпочтительно, устройство содержит элементы, смешивающие поток в трех измерениях, и поверхность которых снабжена указанной композицией для покрытия и системой каналов, и композиции для покрытия таких элементов, также способные изменяться в направлении потока. Гидролитический катализатор по настоящему изобретению позволяет сократить расстояние (уменьшает объем трубы) от точки введения восстановителя в СКВ-катализатор, в силу повышенного смешивания, а также термического и каталитического гидролиза. Кроме того, СКВ-катализатор, возможно, содержащий Г-катализатор, обеспечивает возможность начала восстановления NOx, как только NH3 образуется в реакции. Преимущество в особенности было получено при низких температурах (150-300°C), и применяемые элементы изобретения позволяют использовать меньший объем в расположенном ниже по течению СКВ-катализаторе.
Сферы применения изобретения включают выхлопной, отработавший и отходящий газы в мобильных или стационарных работах при нормальном, положительном или отрицательном давлении. Давления выше нормального существовуют, например, в герметичных топках котлов, а также в двигателях вверх по течению турбокомпрессора. Сжигание, производящее оксиды азота, позволило применение любого газообразного (например, метан, пропан, биогаз), жидкого (тяжелая или легкая топливная нефть, дизельное топливо, бензин, биотопливо) или твердого топлива (уголь, биотопливо) или их смесей. В среднем, условия их применения просты (избыток кислорода), но есть полезные функции, включающие обогащение, например, благодаря регенерации фильтра твердых частиц или адсорбента NOx. Обогащения (недостаточность или стехиометрическое соотношение кислорода), также могут быть использованы для удаления сульфатов и нитратов из Г-катализатора, если те, в свою очередь, не десорбируются.
Как правило, восстановителем оксидов азота является мочевина или некоторое другое случайное азотсодержащий твердый, жидкий или газообразный восстановитель, как, например, в виде раствора или в виде смеси нескольких восстановителей. В дополнение к мочевине, такие восстановители могут включать циануровую кислоту, гидразин, различные аммониевые соединения, различные аминные соединения, различные азотсодержащие органические или неорганические соединения (например, пиридин). Катализатор согласно изобретению может быть использован не только для мочевины, но и для данных других источников аммиака.
В изобретении композиция гидролитического катализатора была усовершенствована по отношению к предшествующему уровню техники с тем, чтобы получить более эффективную реакцию до NH3 и меньше побочных реакций, а также, чтобы обеспечить гидролитический блок с некоторым СКВ оборудованием, интегрированным в него. Ранее известные гидролитические катализаторы были на основе ТiO2, включающей Si, Al, цеолит, и W в качестве добавок. Цель ранее известных способов заключалась в том, чтобы обеспечить кислотную поверхность катализатора, способную генерировать настолько много NH3, насколько представляется возможным без побочной реакции. Кислотная поверхность, тем не менее, адсорбирует аммиак, которые может остаться на ней в течение неоправданно длительного времени, особенно при низких температурах. Изобретение использует свойства противоположные таковым из предшествующего уровня техники. Целью является установление щелочных сайтов на поверхности катализатора путем дополнения катализатора, например, лантаном (La), иттрием (Y), щелочноземельными металлами и/или щелочными металлами. Щелочные сайты обнаруживают мощную адсорбцию кислотных соединений, таких как HNCO, разгрузочное количество которого настолько мало, насколько представляется возможным, особенно при низких температурах, когда адсорбция сильна и формирование HNCO в качестве побочного продукта находится на пике. В то же время, количество адсорбции аммиака на щелочной поверхности значительно уменьшается. Адсорбция HNCO при низких температурах (>180-250°C) отсрочивает реакцию гидролиза HNCO до состояния, когда катализатор чуть более прогретый. Те же самые химические соединения также функционируют в качестве адсорбентов для оксидов азота, чьи возможности могут быть использованы в определенных условиях. Кроме того, соединения Zr, среди прочего, также могут присутствовать. Приведенные в качестве примеров поддерживающие вещества были на основе ТiO2, но также можно использовать другие пористые носители (цеолиты, диоксид кремния, оксиды алюминия) для аналогичной цели совместно со щелочными добавками и адсорбентами.
Покрытие согласно изобретению также может быть применено для покрытия одной или нескольких ячеек в сетчатой системе, и некоторые из других ячеек могут быть снабжены кислотным или менее щелочным покрытием. Кислотный слой также может быть СКВ-активным. Способность к высокой адсорбции NH3 способствует реакции СКВ, увеличивая адсорбцию реагента NH3. Кислотность коррелирует, например, с силами адсорбции NH3 в Г-катализаторах. Желательно, чтобы HNCO быстро реагировал до NH3, но NH3 не должен разлагаться. Щелочной NH3 энергично адсорбируется на кислотных поверхностях, которые могут способствовать каталитической реакции или вызывать ингибирование при низких температурах. В наложенных друг на друга слоях, один может быть снабжен кислотным, а другой щелочным или менее кислотным покрытием. Кислотность при определенной температуре может быть оценена, например, с помощью измерений адсорбции NH3или пиридина. Общую щелочность можно оценить, например, с помощью измерений адсорбции СO2. Щелочность коррелирует также с адсорбционной способностью или силой нитратов. Кислотности могут регулироваться, например, количествами сульфата, который может быть стабилизирован с помощью Zr, Sn или других соответствующих производящих сульфат катионов (сульфатированный оксид Zr). Щелочность была увеличена в примерах, например, с помощью La, Са, Ва, Sr и Y. В последовательных ячейках, описанные выше способы могут быть применены к последней ячейке для увеличения, особенно в слое СКВ-катализатора, способности к адсорбции NH3 при высокой температуре, в результате чего полученный из мочевины NH3 реагирует с NOx, а не с кислородом.
Фиг. 1 показывает пример катализаторов в соответствии с изобретением с различными покрытиями. Версия А имеет покрытие с содержанием щелочных соединений на поверхности ячеистой структуры, композиция которого способствует адсорбции и реакции HNCO и NOx. Дополняя ту же структуру с помощью нижнего или верхнего слоя СКВ-катализатора, получают немедленную реакцию СКВ посредством проявления NH3 (фиг. 1, версии B и C). Если СКВ-катализатор представляет собой нижний слой, мочевина или HNCO сначала идет с потоком в направлении гидролитического катализатора, из которого полученный NH3 легко проходит на нижний слой для реакции СКВ. Так как щелочной Г-катализатор связывает меньше NH3, чем ранее известные Г-катализаторы, препятствие эффекту адсорбции NH3 меньше при прохождении NH3 через СКВ слой или блок. Адсорбция оксидов азота также может быть использована когда Г- и СКВ-катализаторы интегрированы. В данном случае адсорбированный NOx способен вступать в реакцию с NH3 в соседнем сайте СКВ-катализатора. Если слой СКВ находится сверху, термически производимый NH3 может сразу реагировать в верхнем слое, поскольку разложение/окисление NH3 является простым процессом, а непрореагировавшие HNCO и NOx могут мигрировать, адсорбироваться и реагировать в нижнем слое. Кроме того, структура может включать в себя покрытие небольшой площади поверхности (4), функцией которого является служить связывающим/экранирующим слоем между ячеистой структурой и другими слоями катализатора (версии D и Е). Данный слой (4) также может включать щелочные соединения. Данное покрытие также может быть использовано само по себе в качестве экранирующего слоя для сетчатых или трубчатых структур, например, вблизи точки введения мочевины, и в практических применениях, где геометрическая площадь поверхности для катализатора является небольшой, а размеры каналов большие (версия F). Щелочная поверхность катализатора связывает кислотный HNCO и его производные на его поверхности, исключая неблагоприятные реакции полимеризации, в том числе и в данной небольшой площади поверхности покрытия. Следовательно, основная задача данной структуры представляет собой поощрение перемещения масс и смешивание. Так как количество, площадь поверхности и время выдержки катализатора являются небольшими, ничто не может усилить недостатки высокой температуры (разложение/окисление NH3). Данная структура особенно подходит для первого разреженной ячеистой структуры в сетках, где основная цель заключается в обеспечении смешивания. Раствор мочевины также может быть в жидком состоянии в момент попадания потока на поверхность катализатора в случае, если испарение еще продолжается, например, из-за большого размера капель раствора, очень низкой рабочей температуры или неправильного введения.
Пористым носителем, используемым для Г-катализатора, может быть, например, TiO2, TiO2-SiO2, цеолит или их смесь. В случае применения цеолитов, отношение Si/Al2, как правило, превышает 10, что делает их прочными, где бы они ни использовались. Таким образом, начальные базовые слои умеренно кислые, что дает такую же хорошую способность к адсорбции NH3. Щелочные добавки могут быть использованы для уменьшения значения данной кислотности и для, соответственно, увеличения щелочности, тем самым способствуя адсорбции HNCO, оксидов азота и оксидов серы. Хотя, накапливаясь на катализаторе, оксиды серы не полностью устраняют щелочность катализатора, и применение сульфатированных соединений позволило осуществить преднамеренную стабилизацию данных катионов. Было обнаружено, что данный процесс оказывает стимулирующий гидролиз эффект, и результаты с сульфатированными Г-катализаторами также были хороши. Слой СКВ-катализатора может быть в виде сегрегированного или интегрированного слоя, в результате чего данные щелочные соединения также могут быть использованы в слое СКВ. Количество щелочных добавок в Г-катализаторе находится в диапазоне от 0,1 до 80% от общей массы, как правило, в пределах от 0,5 до 20% от общей массы. Количество зависит от катиона и желаемых свойств, и от молекулярной массы, а также покрывающей способности элемента. В случае применения легких щелочных металлов, достаточно высокое количество массы получают даже при легковесных фракциях. При больших количествах (>10%), катализатор обладает значительной способностью к адсорбции оксидов азота, которые могут быть использованы в цепи реакций. В дополнение к указанным щелочным соединениям, Г-катализатор может включать применение соединений циркония, находящихся в форме оксидов и сульфатированных гидроксидов, причем соединения Zr могут быть кислыми или щелочными. Чистый ZrO2 дает катализатору кислотность. Zr может быть в форме смеси совместно с указанными компонентами поддерживающей среды, или он может быть добавлен впоследствии, например, путем пропитывания. Оксиды Zr также могут представлять собой основной компонент в поддерживающем веществе, в результате чего его количество может быть выше, чем в упомянутых щелочных добавках. Адсорбированные NOx десорбируются от нескольких щелочных соединений уже в бедных условиях, во время регенерации фильтра твердых частиц, или, самое позднее, в условиях, где А-значение меньше 1,2. Десорбирующиеся NOx могут быть восстановлены в последующем СКВ-катализаторе. Адсорбированный NOx/HNCO также может реагировать непосредственно или путем короткой миграции поверхности, когда Г- и СКВ-катализаторы интегрированы в один и тот же катализатор.
Способствующие гидролизу покрытия катализатора были добавлены в структуры смесителя, предпочтительно способные к 3D смешиванию и состоящие, например, из кусочков гофрированной фольги или сетки, попеременно волнистых в разных направлениях и сложенных друг на друга, и составляющих смешивающую структуру ячеистого типа. Фиг. 2 показывает пример структуры и покрытий в двух последовательных каналах неравных размеров, применяя свойства каждого покрытия.
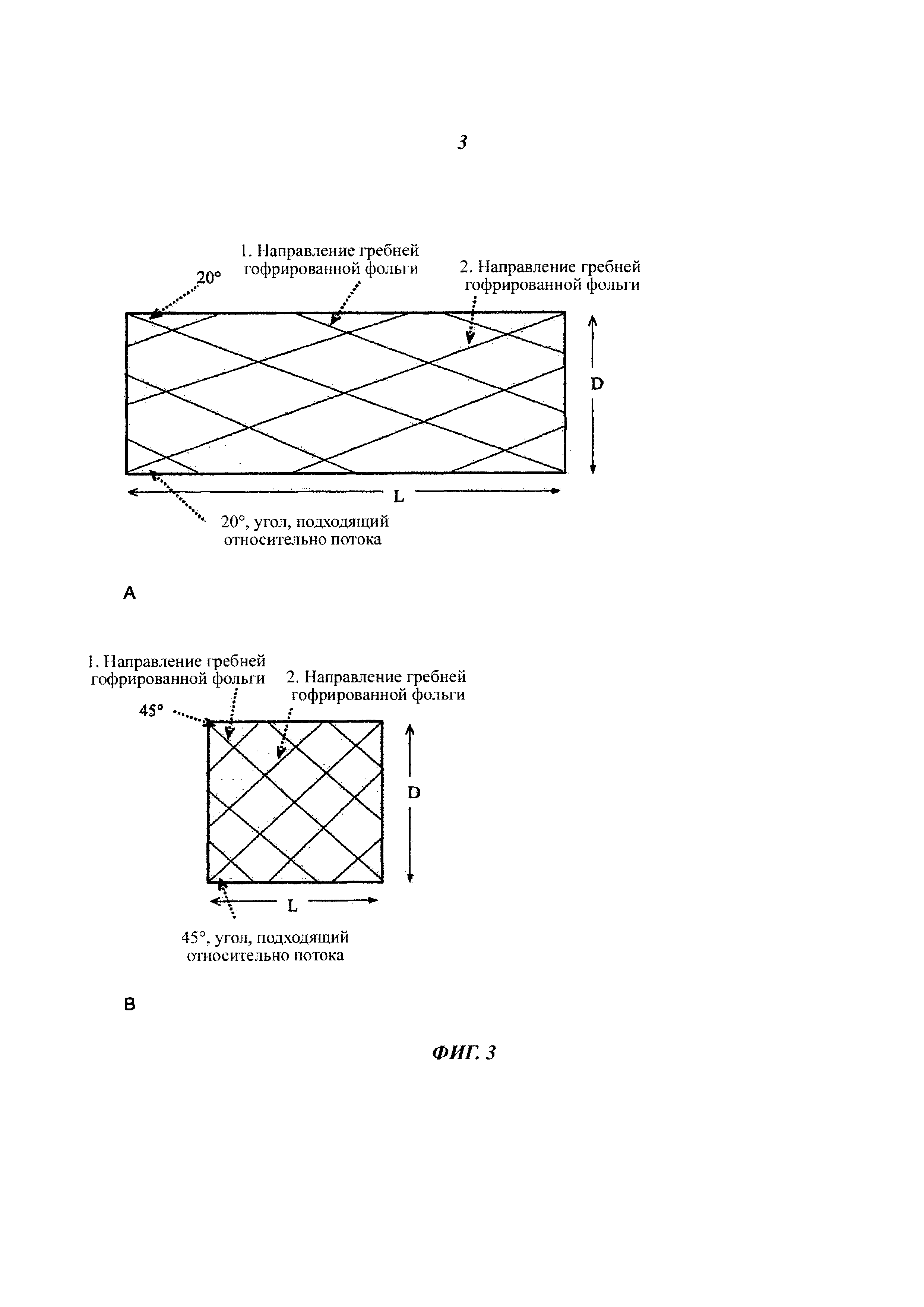
Каталитически покрытый блок статического смесителя может упоминаться как статический смеситель катализатора (ССК). В зависимости от положения фольги, один элемент смеситель осуществляет частично 2D и частично 3D смешивание в различных направлениях. Когда ряд данных смесителей установлен последовательно под углом 90 градусов друг к другу, достигаемым результатом является полное 3D смешивание (фиг. 3). Поток, поступивший в определенное место на внутренней лицевой поверхности ячейки, может появляться в любом месте наружной поверхности трубы. В случае если стенки выполнены из сеток, то также присутствует некоторый поток через стенки, скорость которого зависит от характеристик сетки и потока. Данный пример составляет разительный контраст со смешивающими структурами, обладающими смешиванием только внутри каналов ячеек, в результате чего NH3, мочевина, или HNCO поступает в ячейку неравномерно не в состоянии пройти с канала на канал. СКВ-катализатор должен обладать в каждом канале в точности одинаковым соотношением NH3/NOx, что позволяет применение всего объема катализатора и предотвращает выбросы NH3. По этой причине, Г-катализатор должен выравнивать поток и распределение температуры настолько равномерно, насколько представляется возможным. Если, например, NH3, оказывается направлен на холодные края, может оказаться более чем одно соотношение NH3/NOx, в результате чего произойдут выбросы NH3. Как правило, температура вдоль стенки трубки значительно ниже, чем в середине пустой трубки, такая разница может быть уменьшена с помощью статической смешивающей структуры.
Смешивающая структура может быть сделана в основном из металлической фольги, но также из волокна, сетки или керамической структуры, увенчанной покрытием по настоящему изобретению. Металлическая структура позволяет легче, чем керамические материалы, построить систему смешивающих каналов. Первый и второй кусочки фольги могут представлять собой единую структуру или могут быть уложены поочередно поверх друг друга. Металлическая структура может быть собрана механически посредством сварки или пайки в точках контакта. Опорные элементы могут быть установлены сквозь ячейку, перед ней или позади нее.
Размеры каналов смешивающих элементов в разреженной ячейке, сделанной с применением профиля гофрированной фольги, отличающейся тем, что гофр имеет высоту более 2 мм и ширину более 3 мм (≤100 cpsi). В плотной ячейке, соответственно, профиль смесителя был получен с высотой гофра менее 2 мм и шириной менее 3 мм (>100 cpsi). Подходящий угол гофрированной фольги по отношению к направлению потока должен быть таковым, чтобы достигалось достаточное смешивание, но не приводило к чрезмерному увеличению противодавления (сопротивление потоку). Таким образом, обычно используемый угол для диагонально гофрированной ячеистой структуры составляет около 20-30°, но угол также может быть меньше или больше. Больший угол позволяет применять, соответственно, более короткую ячейку, в результате чего противодавление посредством эффективности смешивания остается достаточно похожим на таковое в более длинной ячейке с меньшим углом. В разреженной ячейке, число Ре (Ре = Рейнольдса) и, следовательно, турбулентность выше, что способствует смешивания в радиальном направлении по всей лицевой поверхности трубки. Корреляция, желательно предумышленная, может быть предусмотрена между подходящим углом (β), а также длиной (L) и диаметром (D) ячейки. Как правило, диаметр определяется на основании других аспектов, так что длина ячейки зависит от желаемой эффективности смешивания и падения давления.
Ячейка может обладать таким пространственным дизайном, что гофрированный гребень, начинающийся у одного края, достигает противоположного края канала, то есть перемещение (s) гофрированного гребня в точности равно диаметру трубки в радиальном направлении (s=D). Данный факт указывает на то, что, с углом, например, 20° в 100 мм трубе, смешивающее устройство будет около 274 мм в длину. Соответственно, с углом 45°, длина смесителя, удовлетворяющая данному критерию, будет 100 мм (фиг. 3 показывает смешивающие структуры при 20 и 45°, нарисованные в соответствии с данным критерием). На данный смеситель можно ссылаться как на смеситель с идеальным соотношением Д/Д (длина/диаметр от англ. L/D, length/diameter) при определенной структуре. Соответственно, Д/Д может также быть меньше, чем данное пороговое значение, а несколько смесителей подряд позволяют осуществить адекватное полное смешивание. 3D сборка смесителя может быть установлена, например, путем прилаживания трех последовательно идущих ячеек в гидролитическом реакторе. Конструкция может присутствовать в прямой трубе или в воронке. Первая ячейка представляет собой разреженную смешивающую структуру (7, гофрирование в соответствии с ≤100 cpsi), вторая представляет собой плотную смешивающую структуру (8, гофрирование в соответствии с>100 cpsi, горизонтальное смешивание), а третья также является плотной смешивающей структурой (8, гофрирование в соответствии с >100 cpsi, вертикальное смешивание под углом 90° по отношению ко второй ячейке).
Таким образом, гидролитический катализатор из системы мочевина-СКВ позволяет использовать структуру, в которой передний сегмент должен быть разреженным, чтобы предотвратить капли от слишком сильного удара о лицевую поверхность и стенки чрезвычайно плотного сетчатого экрана, тем самым вызывая нежелательные реакции. Каплям мочевины сначала должно быть разрешено смешиваться, испаряться и становиться термически гидролизованными, чему больше всего способствует разреженная ячейка. В том случае, когда первая ячейка плотная, было бы необходимо предусмотреть большее расстояние от точки введения до переднего края ячейки, чем в случае с разреженной ячейкой. Разреженная передняя ячейка также обеспечивает возможности для продолжения термического гидролиза без особых помех со стороны поверхностных контактов. В разреженной ячейке, число Ре, представляющее силу турбулентности в канале, может быть достаточно высоким. Данный факт позволяет уменьшить общий объем, требуемый внутри системы трубок от точки введения до СКВ-катализатора. В разреженной ячейке (от 10 до 100 cpsi), поток является турбулентным или менее ламинарным, позволяя происходить реальному смешиванию в канале, что является целью с точки зрения термического гидролиза. В плотной ячейке (>100-200 cpsi), смешивание слабее, но больше каталитическая контактная поверхность и, с точки зрения обмена массы, расстояние до стенки короче, что частично компенсирует эффект (смешивания) более низкого коэффициента обмена массы. Для смешивания адекватно, чтобы присутствовала разреженная ячейка и короткая передняя часть ячейки. В случае стремления способствовать гидролизу смешанной и испаренной мочевины каталитически, выгодно обеспечить большую каталитическую поверхность в задней части. Именно поэтому задняя секция имеет более высокое апертурное число. Обычно, однако, гидролитический катализатор имеет тонкий слой поддерживающего вещества, так как реакции могут протекать в нежелательном направлении в случае слишком продолжительного времени выдержки в порах. Аммиак может, например, распадаться до азота, и данная реакция является проблемой, когда аммиак находится в пористых катализаторах, имеющих плохую селективность в аммиак. Толщина покрытия в Г-катализаторе, например, составляет всего лишь 1-20 мкм, в то время как обычная толщина в покрытых катализаторах составляет около 20-100 мкм. Передняя и задняя секции могут быть равными или неравными по длине. Они обе могут присутствовать в трубке или во впускной/выпускной воронке, или один участок может быть в воронке, а другой в прямой трубке.
Распределение потока через различные ячейки определяется в соответствии с профилем падения давления. Падение давления генерируется в таких каталитических и ячеистых системах совместно с трубкой и локальными сопротивлениями самой ячейки и проточных каналов. Локальное давление различается в разных частях системы канала. Если система каналов является симметричной относительно параллельных ячеек, то о распределении потока можно судить только на основе падения давления в ячейках. Если проточные каналы не являются симметричными и Г-катализатор снабжен шунтом, расчеты будут более сложными, так как сопротивление ячейки и сопротивление системы проточного канала должны быть рассчитаны одновременно. Сопротивлениями ячеек легко оцениваются в случае с 1D моделями, но оптимизация конфигурации системы каналов требует, чтобы профили потока рассматривались на микроуровне с моделью 2D/3D (ВГД, вычислительная гидродинамика, от англ. CFD, computational fluid dynamics).
Фиг. 4 иллюстрирует параллельную систему с меньшим объемом гидролитического катализатора и большим объемом окислительного катализатора. Цель состоит в том, чтобы создать подходящее распределение потока через ячейки по причине того, что даже меньшее количество газа достаточно для гидролиза и дисперсии мочевины. В отличие от этого, важно, чтобы окислительный катализатор обладал достаточным объемом для генерации NO2 в достаточном количестве. Выгодно чтобы гидролитический и окислительный катализаторы располагались параллельно друг другу, так как NO2, производимый в окислительном катализаторе может чрезмерно способствовать реакции окисления в гидролитическом катализаторе, в результате чего мочевина более восприимчива к реакции, идущей до NOx или азота. Падение давления также может увеличиться в значительной степени в системах, установленных последовательно, так что весь выхлопной газ проходит через обе секции. В системе последовательного включения соотношение объем/диаметр Г-катализатора должно быть достаточно большим для обеспечения возможности всех выхлопных газов проходить через него без чрезмерного увеличения Δр (падение давления). Дополнительное падение давления в результате Г-катализатора может быть сведено к минимуму в параллельной системе с комбинацией Г-катализатора и окислительного катализатора. Падение давления в параллельной системе можно поддерживать приблизительно равным таковому только от окислительного катализатора, в результате чего включение Г-катализатора в систему имеет смысл с точки зрения общего сопротивления потоку.
Давление на совместном впускном (pin) и выпускном (pout) пространстве приблизительно одинаково по сравнению с ячейкой и шунтом. Если судить о падении давления и распределении потока в системе по фиг. 4, то необходимо учитывать сопротивление потоку, вызванное воронкой введения мочевины, также как и сопротивление в обводном канале. Преимущества, предлагаемые данной системой на фиг. 4 практически идентичны таковым у системы, имеющей разреженную 3D-ССК ячейку вверх по течению и плотную 3D-CCK ячейку вниз по течению. Тот же размер ячейки позволяет установить максимизированную общую площадь поверхности (ОПП, от англ. total surface area TSA) и оптимальное распределение потока, что было бы невозможно при постоянной плотности ячеек.
Поскольку передняя секция Г-катализатора должна быть в первую очередь разреженная (от 50 до 100 cpsi), то будет трудно регулировать достаточно небольшое количество газа через стандартный размер Г-катализатора. С точки зрения нормального введения и диффузии распылителя, тем не менее, диаметр Г-катализатора должен быть, по меньшей мере, около 100 мм. С другой стороны, апертурное число окислительного катализатора должно быть достаточно высоким (плотная ячейка) для обеспечения его достаточным количеством поддерживающей среды для кинетически ограниченной реакции произведения NO2, что требует достаточно высокой нагрузки на Pt, особенно, когда объем катализатора мал. В структуре согласно изобретению (особенно в параллельной структуре), апертурное число Г-катализатора, как правило, меньше, чем 200 cpsi, а у окислительного катализатора составляет 200 cpsi или больше. Когда используемый окислительный катализатор является плотной ячейкой, падение давления в любом случае выше, чем таковое в смешивающей структуре. Таким образом, диаметр окислительного катализатора должен увеличиться до неприемлемо высокого значение, чтобы получить желаемое распределение потока окислительный катализатор/Г-катализатор: ~80%/20%. Как правило, максимальные диаметры катализаторов продиктованы пространственными ограничениями.
В переднем разреженном ССК было применено очень тонкое (от 0,1 до 10 мкм), с низкой площадью поверхности (<10 м2/г) покрытие, в результате чего адгезия и когезия могут стать хорошими и плотность покрытия может стать низкой. Ранее, до известных систем, было сделан особый акцент на том, что удельная площадь поверхности покрытия составляет более 10 м2/г. Когда площадь поверхности не является чрезмерной, мочевина не допускается в поры, чтобы прореагировать в первой камере, в результате чего образуются задержки или вредные побочные продукты (например, полимеризация). Первая ячейка также может быть полностью без покрытия или поверхность снабжена только собственным материалом смешивающей структуры в качестве необработанного слоя, например в виде оксида. Несмотря на возможность быть настолько тонким, покрытие обеспечивает существенное дополнение к площади поверхности по сравнению с голой ячейкой. Если ячейка 100 cpsi имеет 1,7 м2 поверхности/л ячейки, то площадь поверхности может быть существенно увеличена с применением тонкого покрытия (количество покрытия 5 г/м2 (<10 мкм), БЭТ (удельная поверхность по методу Брунауэра - Эммета - Теллера): 5 м2/г покрытия), то есть до уровня 42 м2 поверхности/л ячейки или, приблизительно, в 25 раз. Данное действие представляет собой способ обеспечения прочности слоя в случае удара капель мочевины о первый слой и значительного увеличения общей площади контакта. Тонкий слой также применяется для обеспечения того, чтобы время выдержки за счет адсорбции не стало слишком продолжительным в ячейке с большой площадью поверхности, что усложнило бы управление системой в переходных режимах, особенно при низких температурах с высокими адсорбционными мощностями, например, в Г-катализаторах на основе ТiO2. Первая ячейка, как правило, обеспечена материалами низкой площади поверхности, такой как TiO2 в виде рутила. Кроме того, существуют соединения, производящие при прокаливании другие соединения низкой площади поверхности, такие как соединения La, Zr или пористых металлов, или керамики.
В последующих ССК-смесителях, с более толстым покрытием (>5 мкм), и имеющим более высокую площадь поверхности (>10 м2/г), в результате чего его каталитические свойства содействуют протеканию кинетически ограниченных реакций. Во втором СКС, ячеистая поверхность может быть снабжена тонким, с низкой площадью поверхности покрытием, аналогичным таковому, используемому в первой ячейке, и способным быть увенчанным другим более толстым покрытием Г-катализатора, имеющим более высокую площадь поверхности и каталитическую активность при гидролизе. То, что было использовано в качестве активного покрытия Г-катализатора представляет собой, например, TiO2 в форме анатаза или цеолит совместно со связующими, активными и щелочными компонентами, имеющими площадь поверхности в покрытии более 10 м2/г, как правило, более, чем 50 м2/г. В слое ниже по течению может также присутствовать некий СКВ-катализатор, в результате чего выделяющийся NH3 способен немедленно вступать в реакцию, также как и выше по течению с фактическим СКВ-реактором. Данный СКВ-катализатор может быть на основе ТiO2, цеолита или оксида алюминия. Покрытие может состоять из, например, V, W, Mo, Zr, La, Sn, В, Mn, Cu, Со, Fe, Се и/или Si, в поддерживающей среде ТiO2 или Fe, Се и/или Cu в цеолитвой поддерживающей среде. Используемый цеолит может быть ZSM-5, бета-цеолитом, феррьеритом, ТС-1 или морденитом, или их смесью, или соответствующими металлическими и алюминиевыми силикатами. Другие вспомогательные среды с большой площадью поверхности, включают, например, оксиды алюминия и фосфаты алюминия (ФАС, фосфат алюмосиликата от англ. SAPO, silico-alumino-phosphate). СКВ покрытие может быть на поверхности или на нижнем слое. Щелочные компоненты также могут быть дополнением к Г-катализатору или находиться исключительно в слое СКВ-катализатора. Вполне возможно, что свойства и композиции Г- и СКВ-катализаторов могут быть интегрированы в один и тот же слой покрытия. Наличие СКВ покрытия в нижнем слое позволяет избежать контакта частиц мочевины/капель с СКВ-слоем, тем самым уменьшая возможные побочные реакции. Таким образом, капли мочевины способны вступать в контакт с покрытием Г-катализатора, но миграция капель в нижний слой перед испарением исключена посредством пор диффузионных барьеров. Данная функция может понадобиться при работе при очень низких температурах, где системе необходимо наибольшее содействие Г-катализатора, а испарение мочевины или другого восстановителя является наиболее медленным с некоторыми каплями, возможно проходящими весь путь до второй ячейки.
Наличие в камере второго катализатора, способствующего высокой температуре СКВ, таким образом, делает возможным увеличение общей СКВ активности и позволяет Г-катализатору применить СКВ-катализатор, предназначенный специально для высокой температуры (например, Fe-цеолитовый катализатор), состав которого отличается от такового у фактического СКВ-катализатора (например, V-W/TiO2-(SiO2) или Cu-цеолитовый катализатор). Г-катализатор является выгодным при низких температурах, но наличие Г-катализатора с добавлением композиции СКВ-катализатора позволяет избежать негативных последствий при высокой температуре, таких как разложение или окисление NH3.
В системе согласно изобретению, первый и второй блоки могут быть использованы таким образом, что один снабжен кислым, а другой щелочным или менее кислым покрытием. Кислотность коррелирует, помимо прочего, с адсорбционной силой NH3 и HNCO в Г-катализаторах. Желательно, чтобы HNCO быстро реагировал до NH3, но NH3 не должен разлагаться, чтобы данная композиция могла быть установлена. Щелочной NH3 энергично адсорбируется на кислых поверхностях, которые могут способствовать каталитической реакции или вызывать ингибирование при низких температурах. Что касается наложенных друг на друга слоев, один из них может быть снабжен кислотным, а другой - щелочным или менее кислотным покрытием. Кислотность при определенной температуре может быть оценена, например, с помощью измерений адсорбции NH3 или пиридина. Кислотности могут регулироваться, например, количествами сульфата, причем указанный сульфат, возможно, может быть стабилизирован с помощью Zr, Sn или других соответствующих производящих сульфат катионов (например, сульфатированный оксид/гидроксид Zr). Щелочность была увеличена, например, с помощью La, Ca, Ва, Sr и/или Y. Во второй ячейке можно увеличить, как описано выше, особенно СКВ-слой катализатора, адсорбционную емкость NH3 при высокой температуре, в результате чего произведенный из мочевины NH3 реагирует с NOx, а не с кислородом. Таким образом, целью является повышение кислотность при высокой температуре в СКВ слое данной структуры, в результате чего фактический СКВ-катализатор (отдельный реактор) может быть оптимизирован на регулярной основе в отношении его состава и пространственной структуры для температурного диапазон от 200 до 500°C, что является наиболее типичным рабочим диапазоном, например для СКВ-катализаторов на основе V-W/TiO2.
Если есть окислительный катализатор выше по течению от Г-катализатора, гидролиз и СКВ реакции могут простимулированы, в частности, при низких температурах (эффект NO2). Окислительный катализатор также может образовывать мало NO2, в результате чего жидкость, поступающая в Г-катализатор, была в основном очищена от углеводородов, окиси углерода и некоторых из частиц. В данном случае, активным металлом окислительного катализатора является, например, PtPd или Pd, причем формирование NO2 с Pd является очень небольшим по сравнению с Pt. Данное действие способствует содержанию Г-катализатора в чистоте, за исключением остатков, собранных от углеводородов в Г-катализаторе.
Система в соответствии с изобретением обеспечивает снижение количества нежелательных побочных продуктов (HNCO), выходящих из выхлопной трубы. Данное обстоятельство особенно критично при низких температурах за счет замедления скорости реакции, и, возможно, при высоких температурах за счет избирательности.
Некоторые каталитические композиции в соответствии с изобретением были покрыты путем погружения или окунания завершенной, как правило, ячеистого типа, металлической или керамической структуры катализатора в каталитическую суспензию. В качестве альтернативы, гофрированную, открытую ячеистую структуру покрывают с помощью распыления и с помощью упаковки или складывания после процесса покрытия для завершения ячейки. Способствующие щелочности добавки также могут быть применены после пропитывания катализатора, например, из водных растворов, высушены и прокалены.
Каталитическое покрытие в соответствии с изобретением может быть пре- или пост-покрытием на обычных керамических или металлических ячейках или структурах, у которых форма диафрагмы (например, квадрат, треугольник), плотность диафрагмы (по диагонали гофрированные структуры, соответствующее от 10 до 2000 cpsi, диафрагма/квадратный дюйм или эквивалентно высоте гофра) или толщина стенки (от 10 до 500 мкм) может колебаться в широком диапазоне, в зависимости от предполагаемого применения, материала и требований.
Покрывающие ячейки составляют своего рода статическую смешивающую структуру, которая либо имеет отдельные каналы, снабженные зонами смешивания (например, изгибы, барьеры потока или сужения), либо структура установлена путем укладки гофрированных, волнистых листов фольги и/или сетчатых экранов сверху друг на друга таким образом, что направление гребня волны отклоняется от входящего направления газа и гребни волн, наложенных друг на друга листов, расходятся (фиг. 3). В обычной металлической ячейке, гребни волн из гофрированной фольги сонаправлены друг с другом и с основным направлением потока. Эффективность смешения можно регулировать путем изменения угла между гребнем волны и направлением основного потока. Пара диагонально гофрированной фольги/сетки имеет свои гребни волн, обеспеченные точечными контактами, которые могут быть использованы в механическом укреплении (сварка, пайка). Смешивающая структура обеспечивает турбулентность потока в радиальном направлении трубы. Смешивающая структура также предусматривает степени разделения для частиц выше, чем в обычной смешивающей структуре. Покрывающая структура также может состоять полностью или частично из металлического сетчатого экрана, синтерированного пористого металла или ловушки для частиц.
Описанные покрытия гидролитического катализатора также могут быть применены к фильтру твердых частиц, находящемуся в передней части СКВ-катализатора, а введение мочевины происходит выше по течению от фильтра твердых частиц. Покрытие Г-катализатора является термически более долговременным, чем большинство СКВ-катализаторов в условиях фильтра твердых частиц, и количество катализатора не может быть низким в любом случае с точки зрения гидролиза, в результате чего покрытие значительно увеличивает противодавление. Фильтр твердых частиц обеспечивает также основной объем и время выдержки для повышения гидролиза. Особенно подходящим для данного решения является также присутствие покрытия СКВ, большее время выдержки не дает в результате слишком большого разложения или окисления NH3. Возможные осадки и мочевина накапливаются в фильтре вместо выпускания из выхлопной трубы, и разлагаются на аммиак при повышении температуры. Система мониторинга фильтра твердых частиц (ВД (встроенная диагностика, от англ. OBD, on-board diagnostics), основанная на датчиках давления) также чувствует накопление мочевины, в результате чего мониторинг гидролиза достигается без вспомогательного оборудования посредством ВД-способа, интегрированного с мониторингом фильтра твердых частиц. Фильтр может быть фильтром ячеистого типа полного или частичного потока, который также нелегко заблокировать. Поскольку система в любом случае включает в себя блок фильтра и блок СКВ в таком порядке, температура на этапах прогревания в любом случае будет выше перед фильтром, чем перед СКВ-катализатором, что выгодно для реакции СКВ.
Параллельное соединение окислительный катализатор + Г-катализатор также функционирует в качестве системы, предназначенной для восстановления частиц и позволяющей количеству частиц уменьшиться, например, на 20-60%. В данном случае, вполне возможно, что система не требует отдельного фильтра твердых частиц для достижения будущих стандартных выбросов.
Температура блока Г-катализатора может быть разогрета в рабочих условиях, например, электрически или посредством подачи топлива в выхлопной коллектор. В данном случае, Г-катализатору предшествует окислительный катализатор, что способствует сгоранию топлива при низких температурах. Повышение температуры также может быть произведено с помощью В/Т-регулировки (воздух/топливо от англ. A/F = air/fuel) двигателя в сочетании с последующим введением в двигатель или выхлопной коллектор. Дополнительное отопление позволяет Г-катализатору функционировать при критически низких температурах (150-250°C). Обогрев также может применяться к СКВ-катализатору. Стратегия дополнительной системы отопления сочетается с параметрами двигателя или картой введения мочевины.
Гидролитический катализатор также может быть применен для удаления NOx, основанного на термальном, некаталитическом СКВ. В данном случае производное аммиака (например, мочевина) смешивают и гидролизуют в катализаторе согласно изобретению, перед тем как передать его на некаталитическое восстановление. Данная процедура служит для ускорения гидролиза мочевины. Гидролитический блок также может поставляться со способствующими термальному СКВ добавками, такими как водород, углеводороды, СО или их производные. Гидролитический катализатор также может быть использован для газификации газов, включающих не только NH3, но и HNCO. Обработанный газ передается в удаляющий NH3 катализатор для сжигания или в какой-либо другой производящий мощность/топливо блок. Температуры в данных операциях часто находятся в диапазоне от 600 до 900°C, то есть температуры при которых композиции по настоящему изобретению являются устойчивыми. Давление при данных операциях также часто на повышенном уровне (1-30 бар, 105-3×106 Па), что является выгодным с точки зрения пространственного дизайна (большее время выдержки в условиях повышенного давления, таким образом, позволяя применение соответственно меньших объемов катализатора, например 20 бар (2×106 Па) → объем реактора составляет 1/20 часть от такового при работе под нормальным давлением).
Гидролитический катализатор также может быть применен в реакциях жидкость-твердое вещество или жидкость-газ-твердое вещество, включающих восстановление, например, нитратов в жидкостях (твердое вещество = катализатор, жидкость = вода, газ = например аммиак или водород).
Изобретение также может быть применено на предприятиях, где восстановителем является непосредственно аммиак. Описанный катализатор адсорбирует аммиак очень плохо, но эффективен при адсорбции NOx, что может быть использовано в блоке (блок смешивания) выше по течению от реактора СКВ или интегрировано с блоком СКВ.
Катализаторы в соответствии с изобретением были изготовлены для лабораторных испытаний сначала путем получения суспензии из сырья в виде порошка и раствора, также как и из воды. Полученную суспензию смешивали и измельчали в шаровой мельнице. Свойства (качество, чистота, размер частиц, возможные стабилизаторы) сырья были изменены в испытаниях для обеспечения требуемых свойств покрытия. Катализаторы были применены к поверхности металлической фольги 50 мкм в толщину, образцы высушивали при температуре около 110°C и прокаливали в течение 4 ч при 550°C. При кальцинировании в неподвижном воздухе, большинство соединений сформировали соответствующие оксиды.
Пример 1
Гидролитические катализаторы (Г-катализаторы) были испытаны в лабораторных условиях с применением газовой смеси, содержащей 1000 частиц на миллион (от англ. particles per million, ppm) NO, 10% кислорода, 8% воды и оставшиеся проценты - азот, а также 500 ppm мочевины (дающей теоретически 1000 ppm NH3), в виде водного раствора, введенного в выхлопной газ, причем измерение проводилось в диапазоне от 150 до 450°C. Целью было получить вниз по течению гидролитического катализатора настолько много NH3, насколько возможно, а также минимальное количество HNCO без окисления NH3 до азота или оксидов азота. Продукты реакции (NH3, HNCO, NO, NO2) были измерены с FTIR (Fourier transform infrared spectroscopy - инфракрасная спектроскопия с преобразованием Фурье), и линия отбора проб была нагрета с помощью кабеля. Обмен на Г-катализатор занял 100,000 ч-1 и на СКВ-катализатор - 50,000 ч-1.
Испытуемые катализаторы были помещены на поверхности металлической ячеистой структуры (серия испытаний 1 - таблица 1). Существенным аспектом в лабораторном масштабе было изучение влияния композиции, поскольку значение массы и теплообмена становится все более очевидным в полномасштабных испытаниях. Апертурное число составляло около 500-600 cpsi (ячеек на квадратный дюйм), а толщина фольги 50 мкм. Используемое сырье включает анатаз ТiO2, имеющий большую площадь поверхности и содержащий менее 0,6% по массе сульфата, или катализатор №6, содержащий 2% по массе сульфата в своем поверхностном слое. Сульфатированный гидроксид циркония также был использован в качестве исходного материала. La был добавлен в виде нитрата La, либо в виде катализатора №7 где его пропитывали и прокаливали заранее в ТiO2. Si-золь был использован в качестве связующего вещества в образцах, и содержание SiO2 возрастало до 10-16% в конечных катализаторах. Сушка была осуществлена путем просушивания теплым воздухом и в заключение образцы прокаливали при 550°C в течение 4 часов. Катализаторы имели площадь поверхности около 60-100 м2/г.

Испытания включали контроль за образованием HNCO (свести к минимуму), за образованием NH3 (довести до максимума после Г-катализатора, чтобы свести к минимуму после СКВ-катализатора) и за преобразованием NOx (без отрицательного преобразования в Г-катализаторе и доведение до максимума после СКВ-катализатора). В испытаниях расстояние Г- и СКВ-катализаторов друг от друга было коротким для моделирования сложных условий.
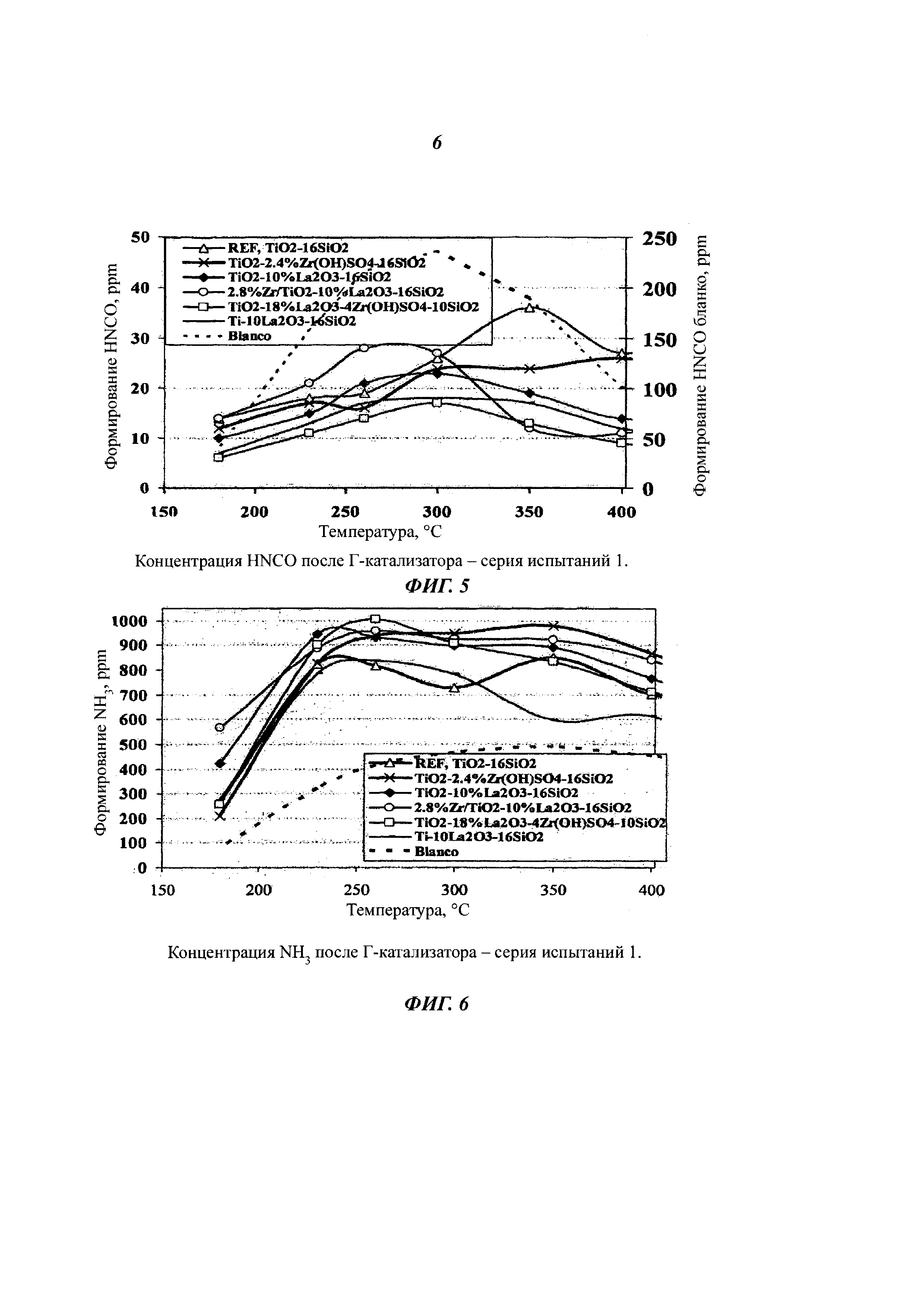
Концентрации HNCO были очень высокими, когда мочевина была введена в пустой реактор (бланко), а время выдержки в горячем реакторе было в данных условиях достаточно коротким. В результате с помощью Г-катализатора в реакторе, концентрации HNCO значительно уменьшились. С известным ранее TiO2-SiO2-катализатором, концентрация HNCO была низкой при низких температурах, но с катализаторами согласно изобретению, концентрация HNCO была заметно ниже при более чем 300°C (фиг. 5). При менее чем 250°C, катализаторы согласно изобретению производят больше NH3 чем эталонный катализатор (фиг. 6). Катализаторы согласно изобретению, имели низкую активность окисления NH3 до NOx (фиг. 7). С двухслойным катализатором (№6) образование HNCO практически отсутствует (фиг. 8). Данный катализатор обладал СКВ-активным нижним слоем и верхним слоем согласно изобретению, что привело к данному результату. Благодаря подобной структуре достигается возможность неразложившейся мочевины выйти на поверхности первой композиции Г-катализатора согласно изобретению, и после этого NH3, находящийся в данный момент в газовой фазе и NOx мигрируют в порах катализатора к нижнему слою СКВ-катализатора, где также восстанавливаются некоторые из оксидов азота.
Сочетание Г-катализатор + СКВ-катализатор используют в попытке смоделировать конечную систему СКВ. Применение Г-катализатора во фронтальной части СКВ-катализатора делает возможным полное устранение HNCO-образования (фиг. 9 и 10) с нулевым уровнем выбросов HNCO производимых выхлопными газами. Преобразование NOx не показало существенных различий, но на активность при низкой температуре положительно влияет Г-катализаторы согласно изобретению. Данные результаты послужили основой для проектирования катализаторов для серии испытаний 2 с учетом оптимизации композиции.
Пример 2
Вторая серия (металлическая фольга 50 мкм, апертурное число 600 cpsi, количество покрытие 20 г/м2) использовала разнообразные добавки в Г-катализаторе в целях содействия гидролизу (таблица 2). Используемые добавки включали, например, Ва, Sr, Y, La, Pr, Zr, Ca, К, и различные их комбинации (концентрации представлены в % по массе в Таблице 2). Добавки были пропитаны раствором нитрата исходного материала в поддерживающей среде, позднее включающей ТiO2 и SiO2 (в весовом соотношении 5:1). После пропитки катализаторы прокаливали при 550°C в течение 2 часов. Используемый эталон представлял собой катализатор, содержащий только ТiO2 и SiO2 без добавок. Испытания проводили, как в примере 1. Окислившиеся образцы подвергали гидротермальной обработке (10% воды в воздухе при 600°C/20 ч) и сульфатировали в течение 20 часов при 400°C в смеси, включающей 25 ppm SO2, 10% кислорода и 8% воды. Применяемая в комбинация Г- и СКВ-катализаторов была керамическим вольфрам-оксидом титана на основе ванадий-СКВ-катализатора (V-W/TiO2), у которого апертурное число 300 cpsi (кордиерит, стенки около 125 мкм), а количество покрытия около 40 г/м2.
Экспертиза была впервые проведена на NH3 и HNCO развивается в Г-катализатора без нижнего СКВ-катализатора. То, что было открыто в данном измерении, представляло собой влияние Г-катализатора относительно селективности превращения мочевины в аммиак, HNCO, и окисления до NOx. Анализ результатов был сосредоточен на температурных диапазонах, где присутствие Г-катализатора оказалось наиболее значительным: при низких температурах в диапазоне 205-350°C, чтобы добиться максимального количества NH3 и минимального количества HNCO, также как и при высокой температуре (450°С) настолько малого, насколько возможно окисления мочевины и аммиака в NOx. Температура была измерена выше по течению от Г-катализатора. В присутствии всех катализаторов согласно изобретению, выход NH3 был выше, чем в пустом реакторе, и, по сравнению с эталонным катализатором, и в присутствии всех катализаторов, отличных от тех, что включают Sr, Рг и Ga, выход NH3 был выше. Формирование HNCO было самым высоким в пустом реакторе, и по сравнению с TiO2-SiO2 эталоном, был достигнут приблизительно тот же уровень, за исключением катализаторов с добавками Са, Pr, La-Ba и La-Sr. Окисление NH3 при 450°C в условиях испытаний было приблизительно на том же уровне, что и при использовании эталона, то есть ни один из данных композиций не увеличила активность окисления NH3. Особенно низкая окислительная активность была достигнута при использовании La-Ba добавок. Основным эталоном был бланко, то есть пустой реактор. Целью являлось доказательство того, что хорошие свойства достижимы при низких температурах, но при этом те же добавки не должны обладать чрезмерно высокой окислительной активностью при более высоких температурах.
Те же Г-катализаторы были также использованы в проведении испытаний адсорбционной мощности NH3 и NOx при 200°C (таблица 2). Температура была выбрана в пределах самой критической для гидролиза. Следует отметить, что щелочные добавки отчетливо повышают адсорбцию NOx и уменьшают адсорбцию NH3, за исключением Zr и Ga, использование которых привело к увеличению адсорбции NH3. Zr служил щелочью совместно с La. В двухкомпонентной системе, ролью Zr может быть стабилизация катализатора термически и химически. Особенно высокая адсорбция NOx была достигнута с La и Y. Наименьшая адсорбция NH3 была достигнута в двухкомпонентных системах La+Pr (5% от эталонной мощности), с последующим La+Ва и La+Sr (16% от эталонной емкости). Принцип работы Pr немного отличается от такового у соединений, выбранных из других групп таблицы Менделеева, например, в соответствии с измерениями адсорбции NOx и NH3. Pr функционировал хорошо и с La. Кроме Pr, можно использовать и другие редкоземельные металлы, такие как Се, Nd, Sm, Eu и/или Gd. Вместо или в дополнение к La, Y и Zr, также можно использовать гафний (Hf), влияние которого, предположительно, аналогично таковому у La, Y и Zr. Данные добавки обладают стимулирующим эффектом, даже если СКВ было включено в качестве составной части покрытия. Данные измерения адсорбции предоставляют результат прямого измерения описанных свойств изобретения, приводящих к преимуществам, открытым в испытаниях.


Адсорбция NH3: адсорбция NH3 в эксперименте переходной характеристики N2 → 500 ppm NH3 в азоте и в присутствии 10% кислорода, при температуре 200°C. Рассчитано из эксперимента переходной характеристики, является количеством адсорбированного NH3 на единицу покрытия.
Адсорбция NOx: адсорбция NOx в эксперименте переходной характеристики N2 → 300 ppm NO + 200 ppm NO2 в азоте и в присутствии 10% кислорода, при температуре 200°C. Рассчитано из эксперимента переходной характеристики, является количеством адсорбированного NOx на единицу покрытия.
Преобразование NOx оценивали с помощью смеси, имеющей следующее содержание окислившихся соединений: 1000 ppm NO и 600 ppm NO + 400 ppm NO2 (500 ppm мочевины, теоретически приводящие к 1000 ppm NH3/NH3/NOx=1). NO2-содержащая смесь имитирует систему окислительного катализатора (DOC, от diesel oxidation catalyst - дизельный окислительный катализатор, ДОК) + Г-катализатор + СКВ-катализатор. Реакция СКВ в СКВ-катализаторе поддерживается с помощью NO2, особенно при низких температурах, но в данном эксперименте также есть промежуточный Г-катализатор, в результате чего система отличается от ДОК+СКВ. NO2 может влиять на реакции мочевины, аммиака и HNCO в Г-катализаторе. Таким образом, экспертиза результатов, в основном, сосредоточена на низких температурах.
Что касается всех катализаторов согласно изобретению, преобразование NOx с помощью комбинации СКВ + Г-катализатора была выше при 205-260°C, чем при применении смеси NO без Г-катализатора (таблица 3). Большинство образцов также способствовало преобразованию NOx больше, чем катализатор ТiO2 - 16% SiO2, используемый в качестве эталона. В присутствии СКВ-катализатора, концентрации HNCO со всеми катализаторами были меньше чем 10 ppm, за исключением испытания без Г-катализатора, в котором концентрация HNCO была 15 ppm. Таким образом, выброс HNCO эффективно устраняется с помощью СКВ-катализатора, но возможно наличие между точкой введения мочевины и СКВ-катализатором более высокой концентрации HNCO в газовой фазе и на поверхностях, что оказывает вред. Следует отметить, что результаты, приведенные в таблице 3, были получены с гидротермально окислившегося и сульфатированного (на протяжении 20 ч, в присутствии 10% воды и при 25 ppm SO2 в воздухе) образца, посредством чего было выяснено, что сульфатирование щелочных добавок не препятствует хорошим качествам. Данные сайты адсорбции, таким образом, могут быть также заполнены сульфатами (SOx) или нитратами (NOx) в рабочих условиях или даже после приготовления (проведенного с помощью сульфата сырого материала).

Пример 3
Данный пример включает имитацию соответствующего распределения потока через параллельные и последовательные каталитические ячейки. Распределение потока через параллельные ячейки зависит от падения давления в камере и в проточных каналах. В том случае, когда проточные каналы (впускные и выпускные воронки и трубы) идентичны, падение давления может быть оценено посредством измерения или математически исключительно на основе падения давления в ячейках. Таким образом, может быть рассчитано, сколько газа проходит через параллельные и последовательные ячейки. На основе уравнений падения давления, смоделированных из экспериментальных измерений, преимущества и свойства системы в соответствии с изобретением представлены в таблице. ПГП с точки зрения профиля представляет собой 1,67 м2/л для “100 cpsi смесителя”, и, соответственно, 3,1 м2/л для “400 cpsi смесителя”. Данная ситуация приводит к значительному расхождению в отношении ОПП (общей площади поверхности), которая в системе таблицы 5 на 43% выше, чем в системе таблицы 4, причем в последней Г-катализатор, состоящий из двух идентичных разреженных смесителей. Использование комплекта 100+400 cpsi в Г-катализаторе сделало возможным структуру, в которой 23% потока прошло через Г-катализатор. Тем не менее, общее противодавление выросло только на 1 мбар (102 Па) во всех ячейках системы (8 → 9,4 мбар). Применяемые переменные в оптимизации системы могут включать в себя апертурное число, диаметр (увеличение диаметра окислительного катализатора), тип ячеек (удельное сопротивление потока), а также Г- и окислительный катализаторы неравной длины (окислительный катализатор короче). Фиг. 11 показывает разнородные структуры для применения параллельных окислительного и гидролитического катализаторов согласно изобретению. Данные структуры частично такие же, как на фиг. 4. Можно также обеспечить обводную трубку со структурой как на фиг. 11C, где введению также предшествует сборка ячеек, которые могут быть использованы для регулирования распределения потока. Передняя ячейка (12) также может быть покрыта окислительным катализатором, катализатором НС-СКВ или не иметь покрытия. Платиносодержащий окислительный катализатор может быть использован для произведения соответствующего количества NO2 и для удаления углеводородов перед введением мочевины, что способствует, например, гидролизу мочевины при низких температурах. Данный катализатор также может быть палладийсодержащим или Pd-богатым, в результате чего большое количество NO2 не производится и гидролиз может происходить без NO2. Подобное поведение может оказаться полезным при высокой температуре эксплуатации (уменьшение окисления NН3). Драгоценный металл, включающий катализатор, также полезен, если помещен перед фильтром твердых частиц, который активно регенерируется. Следовательно, данный передний катализатор предотвращает присутствие выбросов углеводородов и частиц в зону гидролиза.
Другой возможностью является применение структуры, в которой вместе с окислительным катализатором присутствует пустая трубка или шунт, тем самым обеспечивая преимущество противодавления и, возможно, редуктор вводящей трубы подогревается в окружающем газе, при этом в блоке гидролиза нет холодных стенок во время работы. Это эффективный способ учета температурных градиентов, производимых на холодных стенках и являющихся вредными для гидролиза (мочевины). Конечно, то же достоинство обнаружено в блоках, содержащих внутри Г-катализатор. Все описанные действия позволяют идти гидролизу так эффективно и избирательно, как это возможно.


Реферат
Изобретение относится к катализатору гидролиза для восстановления оксидов азота, выполненному в форме каталитического покрытия. В качестве соединения, адсорбирующего HNCO и оксиды азота, указанный катализатор гидролиза содержит лантан и дополнительно содержит одно из следующих: щелочноземельный металл, иттрий, празеодим, галлий, цирконий, причем каталитическое покрытие из указанного катализатора гидролиза представляет собой покрытие на основе диоксида титана, на основе SiO, на основе цеолита, и/или на основе двуокиси циркония. Изобретение также относится к каталитической структуре, включающей в себя катализатор гидролиза, а также к ее применению. Технический результат заключается в увеличении эффективности катализатора гидролиза. 3 н. и 14 з.п. ф-лы, 11 ил., 5 табл., 3 пр.
Формула
Документы, цитированные в отчёте о поиске
Устройство и способ для обработки отработавших газов, образующихся при работе двигателя на бедных смесях, селективным каталитическим восстановлением окислов азота
Комментарии