Клеточные и генные способы улучшения сердечной функции - RU2608957C2
Код документа: RU2608957C2
Чертежи





Описание
[0001] Настоящее изобретение было создано при поддержке правительства в рамках грантов №HL61683, R21 HL091368, HL07828, HL65497, R01 HL64387, R01 HL084642 и Р01 HL004374, присуждаемых Национальными институтами здравоохранения. Правительство имеет определенные права на настоящее изобретение.
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0002] Настоящая заявка испрашивает приоритет в соответствии со Сводом законов США, разделом 35 §119(e), согласно предварительной заявке на патент США №61/490450, поданной 26 мая 2011 года, которая полностью включена в настоящую заявку посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
[0003] Заболевания сердца являются ведущей причиной заболеваемости и смертности в Соединенных Штатах Америки, их распространенность в мире быстро растет
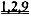
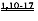
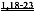
[0004] Многие сердечные патологии, а также ишемически-реперфузионное повреждение и инфаркт миокарда, приводят к снижению систолической функции из-за повреждения и/или отмирания части миокарда, которые значительно нарушают сердечную функцию. Пораженное инфарктом сердце часто не справляется с потребностями организма в работе сердечно-сосудистой системы, и организм пытается компенсировать этот недостаток повышением β-адренергической стимуляции. Хроническая β-адренергическая стимуляция вызывает истощение сократительных резервов, может приводить к повышению диастолического уровня Ca2+ и в конечном итоге к подавлению адренергической чувствительности, приводящей к конечной стадии сердечной недостаточности8-10*. Важно, что в ряде исследований, проведенных на животных моделях и на пациентах, после инфаркта отмечали изменения в содержании и фосфорилировании белков как миофиламентов11-16*, так и саркоплазматического ретикулума (SR) и сарколеммы17,18*, которые могут изменять чувствительность к Ca2+ силы сокращения миофиламентов и транзиторное высвобождение/обратный захват Ca2+. Подобные изменения наблюдали в сердцах, несущих мутации, связанные с дилатационной кардиомиопатией (ДКМП) и гипертрофической кардиомиопатией (ГКМП)
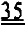
[0005] Сердечная функция нарушена при ряде сердечно-сосудистых заболеваний, включая инфаркт миокарда, ишемически-реперфузионное повреждение, диабет, повышенное кровяное давление и гипертрофическую и дилатационную кардиомиопатию. Указанные патологические состояния часто приводят к изменению цикла Са2+1, β-адренергической чувствительности2 и/или сократительного аппарата кардиомиоцитов3,4. Имеющиеся на сегодняшний день терапевтические подходы в основном направлены на повышение уровня [Ca2+]i, которое, как правило, вызывает про-аритмогенный эффект, нарушение наполнения желудочков путем замедления диастолического расслабления5 и перегрузку Ca2+ саркоплазматического ретикулума, вызывающий триггерную активность19*. Другие подходы, включающие адренергические агенты, могут иметь нежелательные долговременные побочные эффекты, например, значительное действие лекарственного средства на области, не являющиеся мишенями, про-аритмогенную триггерную активность и вероятность ускоренного прогрессирования в сердечную недостаточность2. Таким образом, требуются новые подходы для борьбы с сердечной дисфункцией.
[0006] Альтернативный подход включает применение Ca2+-сенсибилизирующих соединений, которые усиливают связывание Ca2+ с сердечным тропонином С (cTnC) и повышают силу сокращения4. Прилагались значительные усилия для разработки фармацевтических агентов, таких как кальмидазолиум, бепридил и левосимендан, которые повышают связывание Ca2+ с N-концом (II сайтом связывания, «триггерным сайтом») cTnC и усиливают активацию сокращения. Некоторые недостатки, связанные с такими соединениями, включают отсутствие у них специфичности к cTnC и отрицательное воздействие на белки, вовлеченные во взаимодействия с Ca2+ 21,22, и на другие аспекты электромеханического сопряжения5.
[0007] Учитывая недостатки, связанные с различными стандартными фармакологическими и хирургическими подходами, которые в целом направлены на замедление прогрессирования сердечной недостаточности в отличие от восстановления функции, существует необходимость в новых способах улучшения сердечной функции. Согласно настоящему изобретению, предложены более направленно действующие подходы для усиления сердечного сокращения без воздействия на механизм электромеханического сопряжения6, т.е. создание новых рекомбинантных вариантов cTnC с измененными свойствами связывания Ca2+ или введение или продукция in situ дезокси-АТФ с помощью рибонуклеотидредуктазы для ее использования в качестве заместительного субстрата сердечного сокращения.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0008] Один аспект изобретения основан на неожиданно обнаруженном факте, что даже незначительные повышения общего уровня внутриклеточной дезокси-АТФ (даже 1,0%-2% от общего пула адениннуклеотидов) приводит к существенному улучшению сократимости сердца. Повышение уровня дезокси-АТФ достигается с помощью способов согласно настоящему изобретению с использованием векторов, экспрессирующих запускаемую промотором Rrm1 (субъединицу 1 рибонуклеотидредуктазы) или Rrm2 (субъединицу 2 рибонуклеотидредуктазы), которые вводят субъекту, или доставляют путем трансплантации донорских клеток, трансдуцированных указанными векторами, в миокард хозяина. Также была неожиданно обнаружена способность способов согласно настоящему изобретению улучшать сократимость, и, по-видимому, повышать чувствительность к кальцию при отсутствии соответствующих неблагоприятных эффектов на сердечное расслабление или Ca2+-токи. Сердечное расслабление, напротив, существенно ускорялось в результате повышения уровня дезокси-АТФ. Улучшение сердечной функции подтверждалось повышением относительного укорочения левого желудочка и повышением степени и скорости укорочения и расслабления изолированных кардиомиоцитов, что наблюдали с помощью эхокардиографии и видеомикроскопии с использованием системы IonOptix. Преимущества настоящего изобретения подтверждали с помощью исследований на мышах и крысах с нормальными или пораженными инфарктом сердцами. Повышенный уровень дезокси-АТФ приводил к повышению чувствительности к предварительной нагрузке в исследованиях на работающем сердце по Лангердорффу и избавлению от сердечной недостаточности. Механизм саморегуляции рибонуклеотидредуктазы, т.е. аллостерическая активация (с помощью АТФ) и ингибирование (с помощью дезокси-АТФ), также обеспечивает другое существенное преимущество благодаря предотвращению достижения токсического уровня дезокси-АТФ при гиперэкспрессии RR. Таким образом, согласно настоящему изобретению предложен новый терапевтический подход, не имеющий таких недостатков, как аритмогенез, нарушение наполнения желудочков из-за замедленного диастолического расслабления, побочное влияние на области, не являющиеся мишенями, и возможное ускорение прогрессирования сердечной недостаточности, связанных со стандартными терапевтическими подходами, воздействующими на уровень Ca2+ или адренергическую передачу сигнала.
[0009] Другой аспект изобретения основан на обнаружении эффектов, которые изменили связывания Ca2+ миофиламентами (и связанные с этим изменения сократительных свойств) оказывало на 1) функцию сердечного саркоплазматического ретикулума (СПР) и 2) работоспособность сердца в нормальной и больной сердечной мышце. Для проведения указанных исследований мы получали несколько вариантов cTnC, обладающих повышенной или пониженной аффинностью связывания Ca2+. При замещении нативного cTnC в сердечной мышце с химически разрушенной мембраной или в сердечных миофибриллах, указанные варианты cTnC приводили к повышению и снижению чувствительности к Ca2+ силы сокращения, соответственно

[0010] Согласно одному аспекту изобретения предложены способы и композиции для улучшения сердечной функции, сократимости миокарда и расслабления миокарда у млекопитающего, например, человека. Кардиомиоциты, содержащие первый вектор экспрессии, содержащий первую последовательность нуклеиновой кислоты, кодирующую субъединицу R1 рибонуклеотидредуктазы, и второй вектор экспрессии, содержащий вторую последовательность нуклеиновой кислоты, кодирующую субъединицу R2 рибонуклеотидредуктазы, трансплантируют в миокард млекопитающего, нуждающегося в лечении, например, млекопитающего, страдающего инфарктным повреждением миокарда. Первая последовательность нуклеиновой кислоты и вторая последовательность нуклеиновой кислоты функционально связаны с промотором, который индуцирует гиперэкспрессию R1 и R2. Согласно некоторым вариантам реализации изобретения кодирующие R1 и R2 конструкции являются частью одного вектора экспрессии или находятся на разных векторах экспрессии. В некоторых случаях промотор может быть выбран из кардиоспецифичного промотора, например, cTnT455, для обеспечения селективной экспрессии в сердце. Специалистам в данной области техники очевидно, что можно использовать другие промоторы, подходящие для индукции экспрессии в эукариотических клетках, например, клетках млекопитающего. Согласно некоторым вариантам реализации изобретения, гиперэкспрессия запускается промотором СК7 или промотором цитомегаловируса (CMV).
[0011] Клеточная продукция дезокси-АТФ в норме осуществляется в клетках млекопитающего под действием фермента рибонуклеотидредуктазы (R1R2), который удаляет гидроксильный фрагмент в положении 2 на рибозном кольце АДФ с образованием дезокси-АДФ. Дезокси-АДФ затем быстро превращается в дезокси-АТФ. Преимущества согласно настоящему изобретению обеспечиваются гиперэкспрессией R1 и R2, которые образуют рибонуклеотидредуктазный комплекс, приводящий в конце концов к продукции дезокси-АТФ in situ.
[0012] Согласно некоторым вариантам реализации изобретения вектор экспрессии представляет собой вирусный вектор, такой как вектор на основе аденоассоциированного вируса, например, AAV6, AAV2, rAAV2/1, rAAV2/2, rAAV2/3, rAAV2/4, rAAV2/5, rAAV2/6, rAAV2/7 rAAV2/8, rAAV2/9, rAAV2/10, rAAVM41, dsAAV и т.д. Согласно различным вариантам реализации изобретения вектор экспрессии также включает репортер трансдукции. Кардиомиоциты могут быть получены из эмбриональных стволовых клеток (ЭСК), индуцированных плюрипотентных стволовых клеток (иПСК), или мезенхимальных стволовых клеток, полученных из организма млекопитающего. Согласно некоторым вариантам реализации способа согласно изобретению используют кардиомиоциты, происходящие из (являющиеся производными) стволовых клеток человека. иПСК могут происходить из клеток, например, фибробластов, полученных из организма млекопитающего, которого предполагается лечить.
[0013] Согласно некоторым вариантам реализации изобретения способ включает трансдукцию кардиомиоцитов R1- и R2-кодирующими первым и вторым вектором экспрессии ex vivo перед трансплантацией указанных кардиомиоцитов. Трансплантацию кардиомиоцитов можно осуществлять посредством доставки через катетер. Согласно некоторым вариантам реализации изобретения, включающим лечение пораженного инфарктом миокарда, кардиомиоциты можно доставлять в область миокарда, содержащую живые клетки, например, в не затронутую инфарктом зону миокарда.
[0014] В альтернативном варианте улучшение сердечной функции и сократимости и расслабления миокарда у млекопитающего, например, человека, может быть достигнуто путем введения первого вирусного вектора, содержащего первую последовательность нуклеиновой кислоты, кодирующую субъединицу R1 рибонуклеотидредуктазы, и второго вирусного вектора, содержащего вторую последовательность нуклеиновой кислоты, кодирующую субъединицу R2 рибонуклеотидредуктазы, причем указанные первая последовательность нуклеиновой кислоты и вторая последовательность нуклеиновой кислоты функционально связаны с кардиоспецифичным промотором, например, cTnT455. R1- и R2-кодирующие конструкции могут являться частью одного вирусного вектора или находиться в разных вирусных векторах. Согласно некоторым вариантам реализации изобретения, вирусный вектор (векторы) представляет собой вектор на основе аденоассоциированного вируса. Вирусный вектор (векторы) можно вводить системно, например, внутривенно, или местно, например, посредством инъекции в миокард. Вирусный вектор (векторы) также может кодировать нацеливающий агент, который, в частности, связывается с кардиоспецифичным маркером. Согласно некоторым вариантам реализации изобретения, вирусный вектор (векторы) дополнительно содержит репортер трансдукции. Согласно другим вариантам реализации изобретения, способ также включает введение вирусного вектора, содержащего последовательность нуклеиновой кислоты, кодирующую вариант L48Q cTnC, где указанный вариант cTnC обладает повышенной аффинностью связывания Ca2+, и указанная последовательность нуклеиновой кислоты функционально связана с кардиоспецифичным промотором.
[0015] Согласно другому аспекту изобретения, предложены способы доставки дезокси-АТФ в миокард млекопитающего-хозяина, например, человека. Донорские клетки, в которых происходит повышенная экспрессия (гиперэкспрессия) субъединицы рибонуклеотидредуктазы R1 и субъединицы рибонуклеотидредуктазы R2, трансплантируют в миокард. Согласно некоторым вариантам реализации изобретения, донорские клетки содержат первый вектор экспрессии, содержащий первую последовательность нуклеиновой кислоты, кодирующую субъединицу R1 рибонуклеотидредуктазы, и второй вектор экспрессии, содержащий вторую последовательность нуклеиновой кислоты, кодирующую субъединицу R2 рибонуклеотидредуктазы, причем указанные первая последовательность нуклеиновой кислоты и вторая последовательность нуклеиновой кислоты функционально связаны с промотором, который индуцирует гиперэкспрессию R1 и R2. Специалистам в данной области техники очевидно, что R1- и R2-кодирующие конструкции могут являться частью одного вектора экспрессии.
[0016] Дезокси-АТФ образуется под действием рибонуклеотидредуктазного комплекса in situ и поступает в кардиомиоциты млекопитающего-хозяина через щелевые контакты, образовавшиеся между донорскими клетками и кардиомиоцитами хозяина. Согласно некоторым вариантам реализации изобретения, дезокси-АТФ доставляется в пораженный инфарктом миокард. Согласно предпочтительным вариантам реализации изобретения дезокси-АТФ доставляется в область миокарда, содержащую живые клетки, например, в не затронутую инфарктом зону. Трансплантация донорских клеток, например, кардиомиоцитов, фибробластов, может быть осуществлена посредством доставки через катетер. Специалистам в данной области техники очевидно, что кардиомиоциты могут происходить из стволовых клеток, например, плюрипотентных ЭСК, иПСК, мезенхимальных стволовых клеток, выбранных из источника, совместимого с организмом-хозяином. Например, иПСК происходят из клетки, например, фибробласта, полученного из организма млекопитающего-хозяина.
[0017] Согласно другому аспекту изобретения, предложены способы улучшения сердечной функции у субъекта путем введения вирусного вектора, кодирующего вариант L48Q cTnC, где последовательность нуклеиновой кислоты, кодирующая указанный вариант cTnC, функционально связана с кардиоспецифичным промотором. Вариант L48Q cTnC обладает повышенной аффинностью связывания Сайта II связывания Ca+. Согласно некоторым вариантам реализации изобретения, субъект страдает состоянием сердца, приводящим к пониженной сократимости. Указанные способы могут применяться для лечения субъектов, у которых диагностирована кардиомиопатия, например, ишемическая болезнь сердца, кардиомиопатия, инфаркт миокарда, или кардиомиопатия, которая определяется по пониженной систолической функции. Примеры кардиопатологии включают, но не ограничиваются указанными, первичную кардиомиопатию, генетически обусловленную кардиомиопатию и дилатационную кардиомиопатию. Согласно некоторым вариантам реализации изобретения, субъект, нуждающийся в лечении, имеет одну или более генетических мутаций, связанных с пониженной Ca2+-чувствительности сокращения миофибрилл и/или фенотипами дилатационной кардиомиопатии. Примеры таких генетических мутаций включают, но не ограничиваются указанными, миссенс-мутации Ser532Pro и Phe764Leu, делецию в cTnT (deltaLys210) или мутации в генах MYH7, MYBPC3, TNNT2, TNNI3, ТРМ1, АСТС, MYL2, MYL3, или их комбинации, как описано в литературе.
[0018] В альтернативном варианте сердечная функция может быть улучшена у субъектов, имеющих одну или более генетических мутаций в саркомерном белке, выбранном из бета-сердечной тяжелой цепи миозина, сердечного актина, сердечного тропонина T, альфа-тропомиозина, сердечного тропонина I, сердечного миозин-связывающего белка С и легкой цепи миозина, которые связаны с повышенной Ca2+-чувствительностью сокращения миофибрилл и/или фенотипами гипертрофической кардиомиопатии. Примеры указанных генетических мутаций включают, но не ограничиваются указанными, мутации остатка 92 в CTnT, например, R92W, R92Q и R92L cTnT, или мутации по MYH7, MYBPC3, TNNT2, TNNI3, ТРМ1, АСТС, MYL2, MYL3, и их комбинации, как описано в литературе. Способ улучшения сердечной функции включает введение указанным субъектам, имеющим Ca2+-сенсибилизирующую генетическую мутацию (мутации), вирусного вектора, содержащего последовательность нуклеиновой кислоты, кодирующую вариант L57Q или I61Q cTnC, где указанная последовательность нуклеиновой кислоты функционально связана с кардиоспецифичным промотором. Варианты L57Q и I61Q cTnC обладают пониженной аффинностью связывания Сайта II связывания Ca2+ и могут применяться для лечения субъектов с диагностированной гипертрофической кардиомиопатией. Согласно некоторым вариантам реализации изобретения вирусные векторы, кодирующие вариант L48Q, L57Q или I61Q cTnC, вводят, например, путем липофекции, в виде покрытия на стенте или путем прямой инъекции, например, через катетер. Вирусные векторы могут быть выбраны из векторов на основе аденоассоциированного вируса.
[0019] Согласно другому аспекту изобретения предложены фармацевтические композиции для улучшения сердечной функции и/или сократимости миокарда и/или расслабления миокарда. Фармацевтические композиции могут содержать первый вирусный вектор, содержащий первую последовательность нуклеиновой кислоты, кодирующую субъединицу R1 рибонуклеотидредуктазы, и второй вирусный вектор, содержащий вторую последовательность нуклеиновой кислоты, кодирующую субъединицу R2 рибонуклеотидредуктазы, причем указанные первая последовательность нуклеиновой кислоты и вторая последовательность нуклеиновой кислоты функционально связаны с кардиоспецифичным промотором. Согласно некоторым вариантам реализации изобретения кодирующие R1 и R2 конструкции находятся в одном вирусном векторе или в разных вирусных векторах. В альтернативном варианте фармацевтические композиции содержат кардиомиоциты, которые были трансдуцированы кодирующим R1 и R2 вирусным вектором (векторами). Другие фармацевтические композиции, описанные в настоящей заявке включают вирусный вектор, содержащий последовательность нуклеиновой кислоты, кодирующую вариант L48Q cTnC и/или I61Q вариант cTnC и/или вариант L57Q cTnC. Вирусные векторы могут быть выбраны из векторов на основе аденоассоциированного вируса, например, AAV6, AAV2, rAAV2/1, rAAV2/2, rAAV2/3, rAAV2/4, rAAV2/5, rAAV2/6, rAAV2/7 rAAV2/8, rAAV2/9, rAAV2/10, rAAVM41, dsAAV и т.д. Согласно некоторым вариантам реализации изобретения вирусный вектор (векторы) дополнительно содержит промотор CMV, функционально связанный с последовательностью нуклеиновых кислот, кодирующей вариант cTnC. Предполагается, что вектор также может содержать последовательность нуклеиновой кислоты, кодирующую нацеливающий агент, на усмотрение специалиста в данной области техники.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
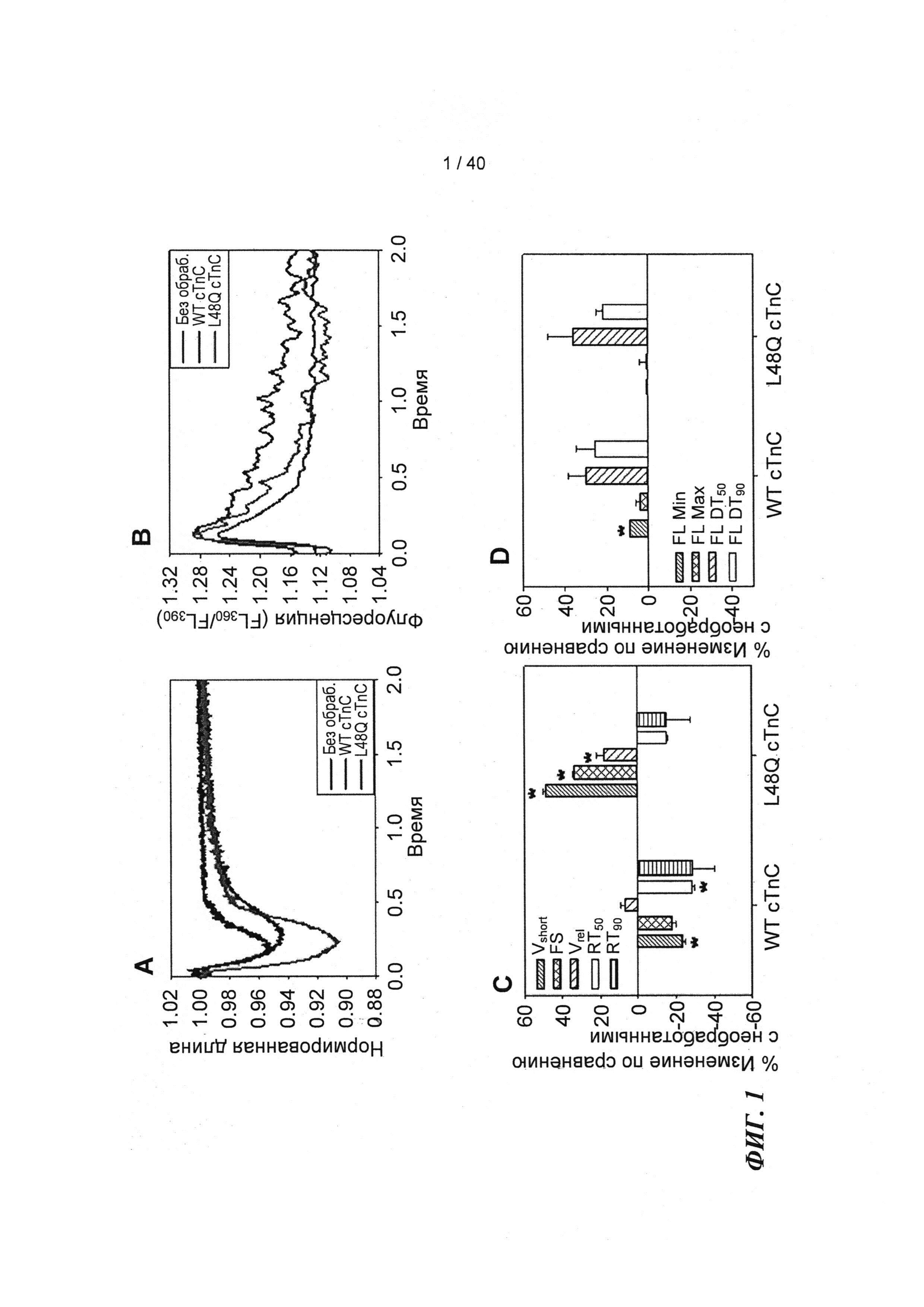
[0020] ФИГ.1. Типичные кривые длины клеток (А) и Ca2+-тока (В, Fura-2 - флуоресценция) для необработанных (черный), WT (дикий тип) cTnC+GFP (синий) и L48Q cTnC+GFP (красный) Процентное изменение сократительных свойств (С) и Ca2+-тока (D) cTnC ДТ+GFP и L48Q cTnC+GFP миоцитов, стимулированных при частоте 0,5 Гц, по сравнению с необработанными миоцитами. Vshort=скорость укорочения; FS=относительное укорочение; Vrel=максимальная скорость расслабления; RT50,90=время до достижения 50% и 90% расслабления, соответственно; FL=флуоресценция; DT50,90=время до достижения 50% и 90% снижения Ca2+, соответственно. *р<0,05 по сравнению с необработанными, †=р<0,05 по сравнению с cTnC ДТ.
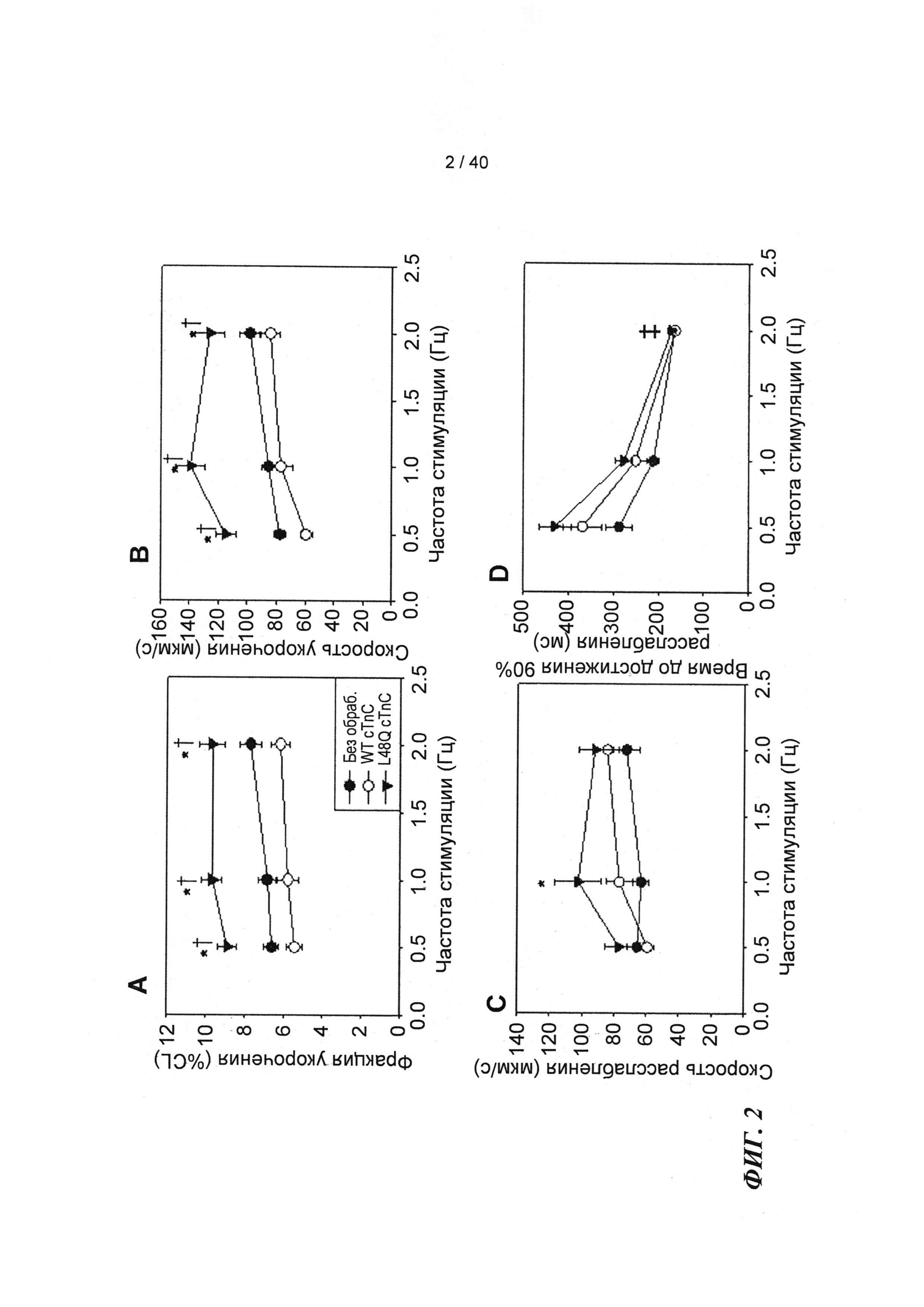
[0021] ФИГ.2. Эффект частоты стимуляции на сократительные свойства. L48Q cTnC-трансдуцированные миоциты (закрашенные треугольники) отвечали так же на частоту стимуляции, как и cTnC ДТ (незакрашенные кружки) и необработанные миоциты (закрашенные кружки), но имели повышенную фракцию укорочения (А) и скорость укорочения (В) при всех частотах. Скорость расслабления (С) и время до достижения 90% расслабления (D) также были одинаковыми во всех группах, при этом время до достижения расслабления укорачивалось при повышении частоты стимуляции. *=р<0,05 по сравнению с необработанными, †=р<0,05 по сравнению с cTnC ДТ, ‡=р<0,05 по сравнению со стимуляцией 0,5 Гц для всех групп.
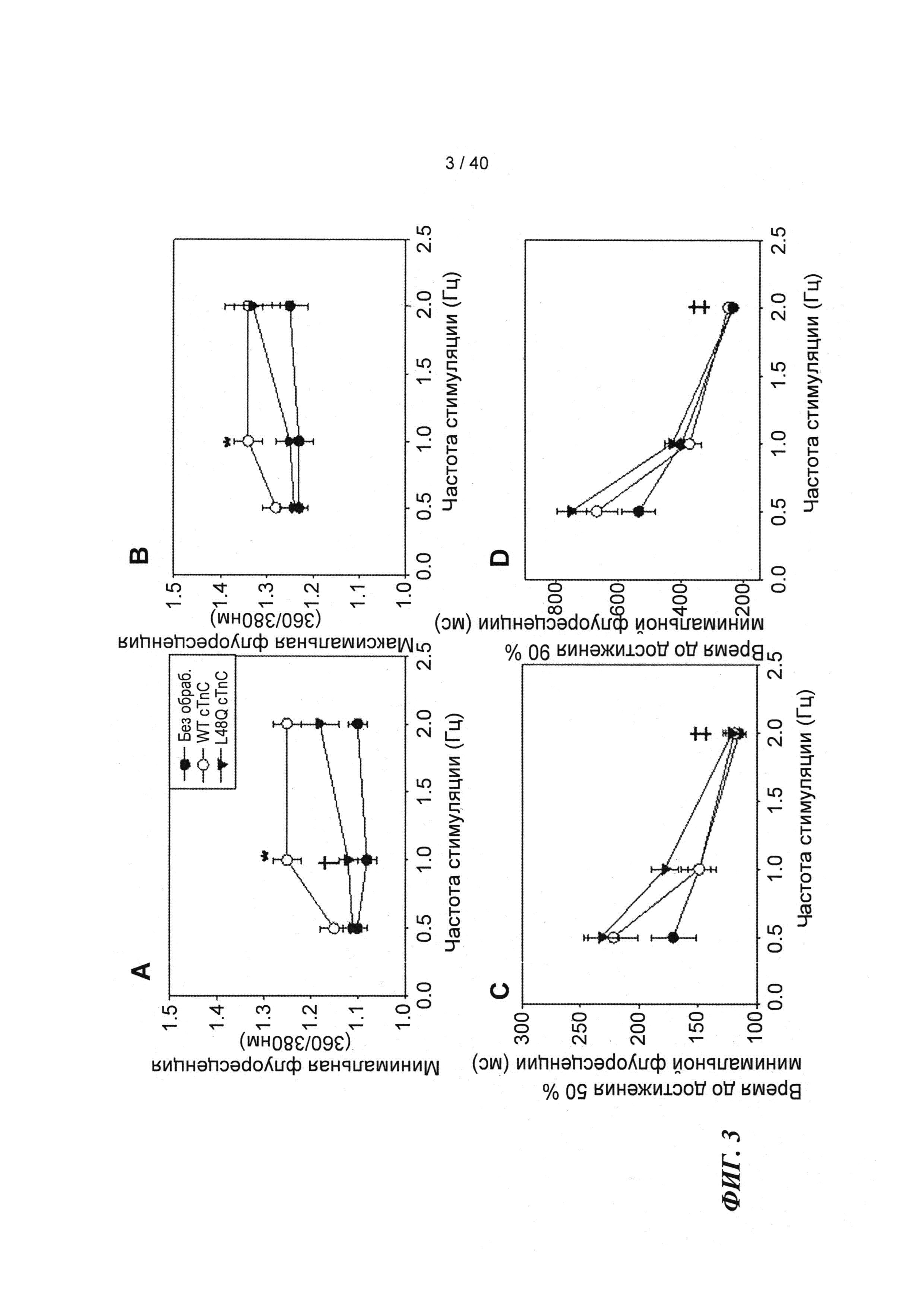
[0022] ФИГ.3. Эффект частоты стимуляции на характер регуляции Ca2+. L48Q cTnC трансдуцированные миоциты (закрашенные треугольники) отвечали на частоту стимуляции так же, как и cTnC ДТ-трансдуцированные (незакрашенные кружки) и необработанные миоциты (закрашенные кружки) при минимальной (А) и максимальной (В) флуоресценции. Как и в случае расслабления кардиомиоцитов, время до снижения Ca2+ тока на 50% (С) и 90% (D) уменьшалось при повышении частоты стимуляции. *=р<0,05 по сравнению с необработанными, †=р<0,05 по сравнению с cTnC ДТ, ‡=р<0,05 по сравнению со стимуляцией 0,5 Гц для всех групп.
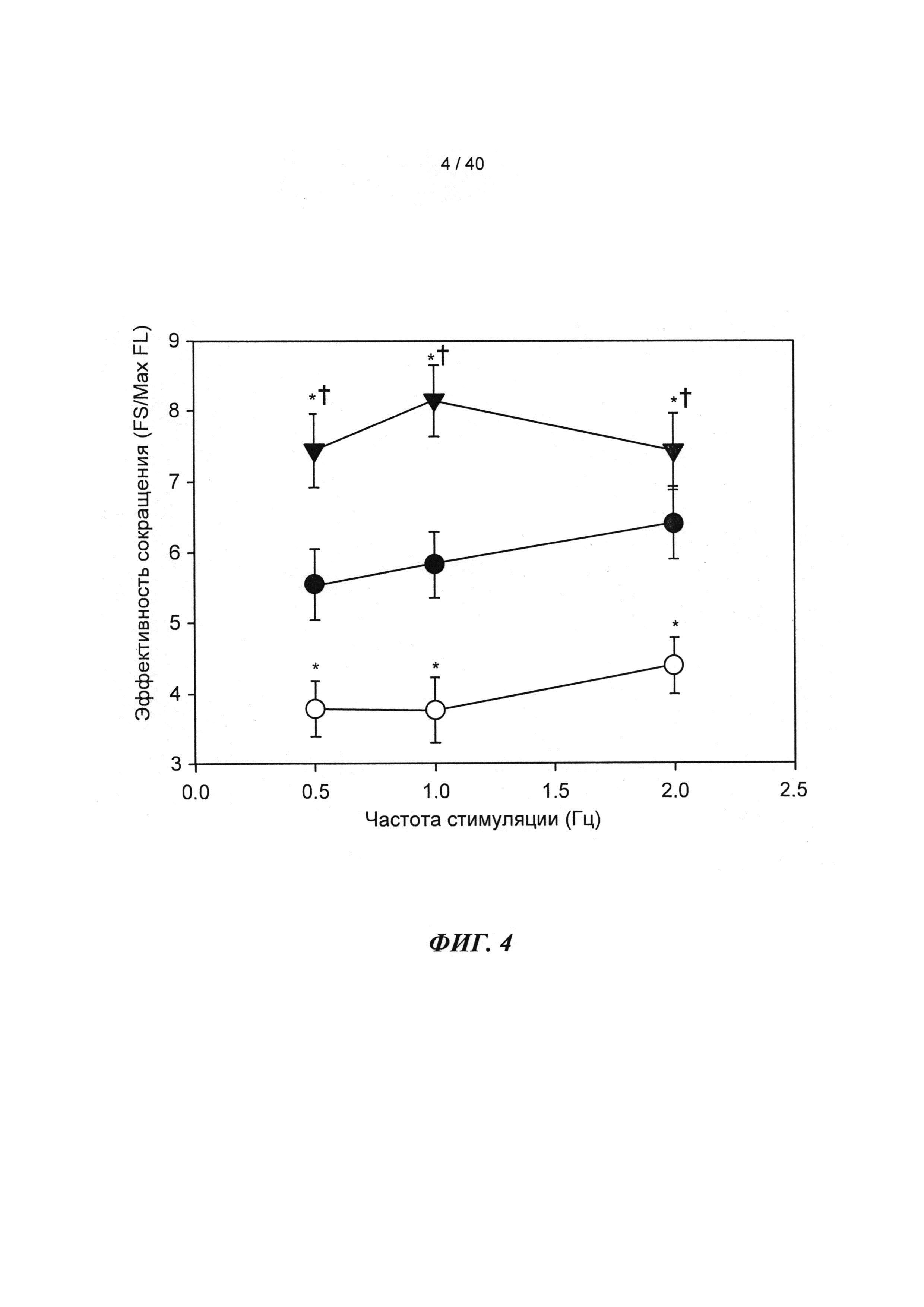
[0023] ФИГ.4. Эффективность сокращения, определенная как относительное укорочение, деленная на максимальную Fura-флуоресценцию (пик Ca2+), указывает на то, что L48Q cTnC-трансдуцированные кардиомиоциты (закрашенные треугольники) значительно более чувствительны к Ca2+ при всех частотах стимуляции, тогда как cTnC ДТ-трансдуцированные кардиомиоциты (незакрашенные кружки) менее чувствительны к Ca2+ по сравнению с необработанными кардиомиоцитами (закрашенные кружки). *=р<0,05 по сравнению с с необработанными, †=р<0.05 по сравнению с cTnC ДТ.
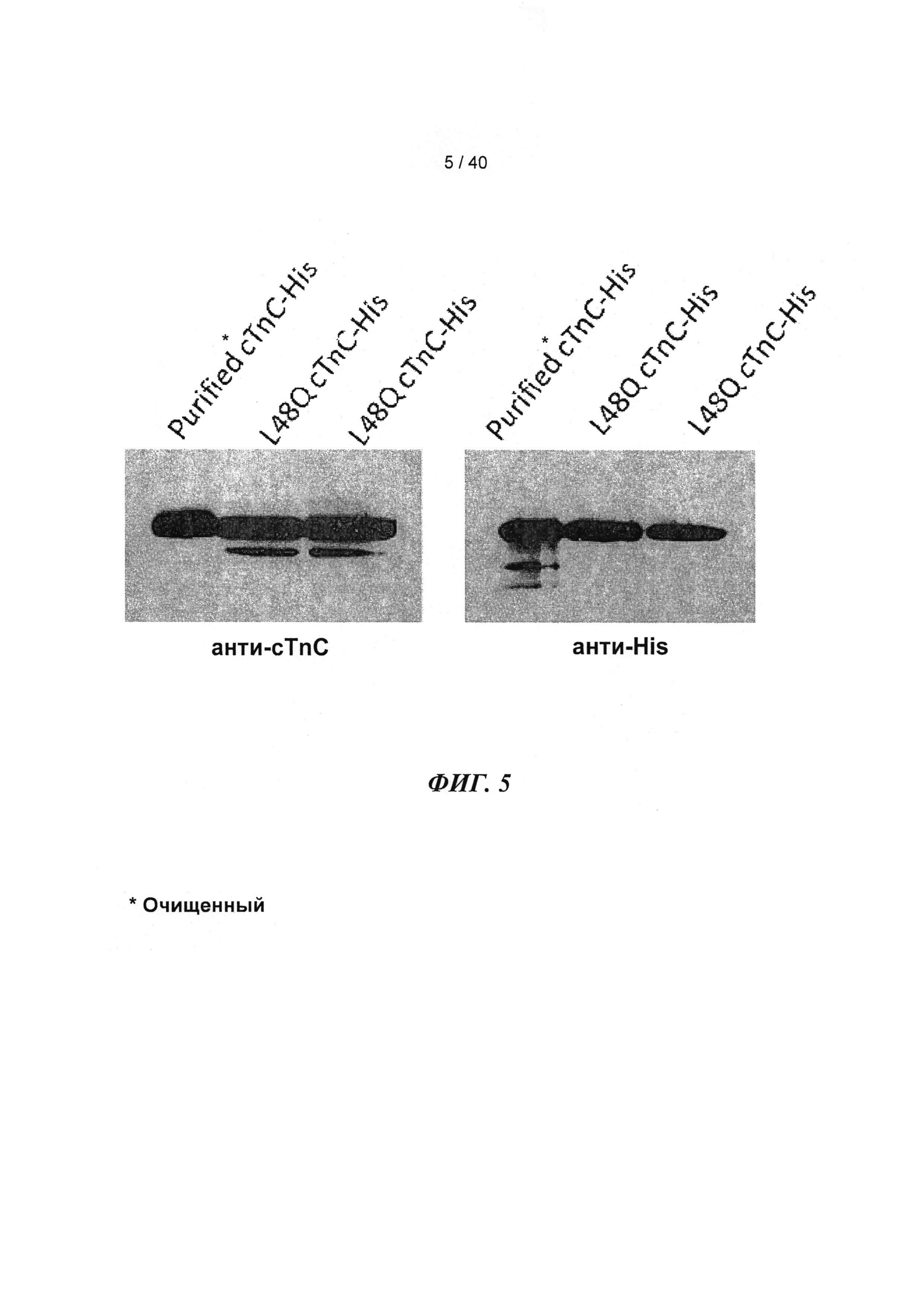
[0024] ФИГ.5. Вестерн-блоты WT и L48Q cTnC-His трансдуцированных кардиомиоцитов навороженных животных, меченные анти-cTnC (А), показывали общее содержание cTnC в миофиламентах, тогда как Вестерн-блоты кардиомиоцитов, меченные анти-His (В), показывали включение L48Q cTnC в тонкие филаменты. Денситометрический анализ показал, что L48Q cTnC замещал 58±7% нативного cTnC.
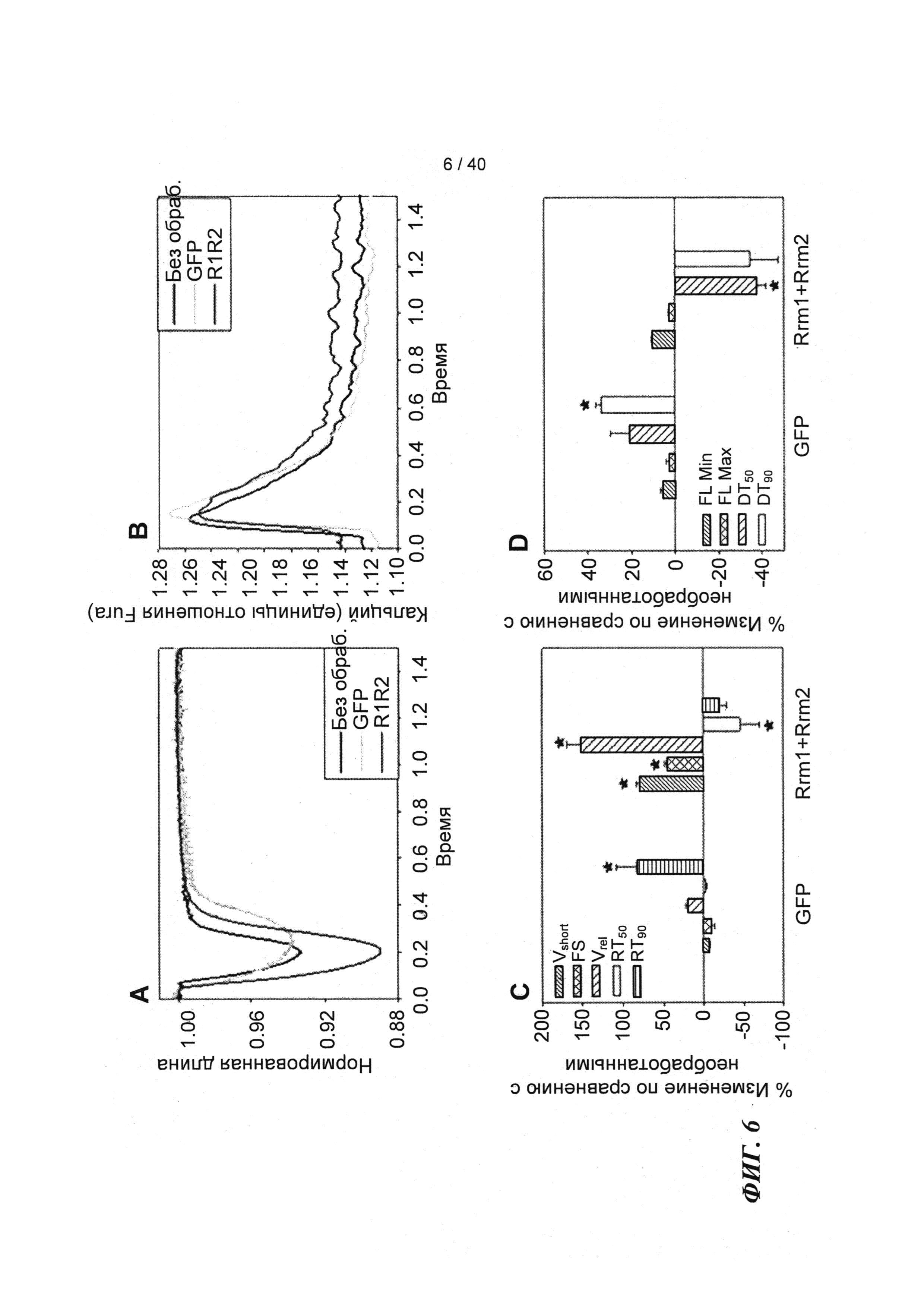
[0025] ФИГ.6. Типичные кривые длины клеток (а) и Ca2+-токов (b, Fura-2 - флуоресценция) необработанных (черный), только GFP (зеленый) и Rrm1+Rrm2+GFP (красный) трансдуцированных кардиомиоцитов. Процентное изменение сократительных свойств (с) и Ca2+-тока (d) у только GFP-трансдуцированных и Rrm1+Rrm2+GFP-трансдуцированных миоцитов, стимулированных при частоте 0,5 Гц, по сравнению с необработанными миоцитами. Vshort=скорость укорочения; FS = фракции укорочения; Vrel=максимальная скорость расслабления; RT50,90=время до достижения 50% и 90% расслабления, соответственно; FL=флуоресценция; DT50,90=время до достижения 50% и 90% снижения Ca2+, соответственно. *р<0,05 по сравнению с необработанными.
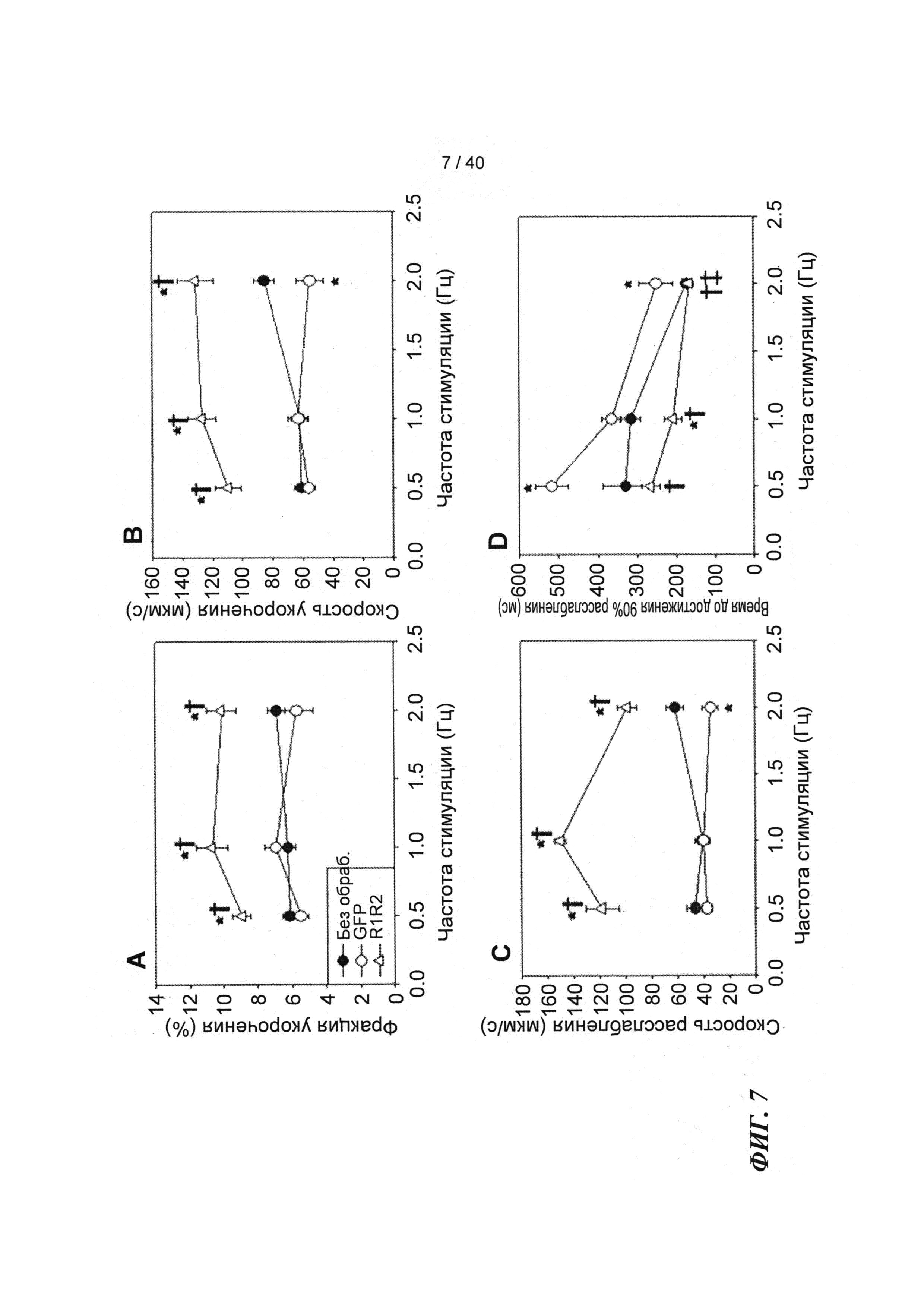
[0026] ФИГ.7. Эффект частоты стимуляции на сократительные свойства. Rrm1+Rrm2-трансдуцированные миоциты (незакрашенные треугольники) отвечали на частоту стимуляции так же, как и только GFP-трансдуцированные (незакрашенные кружки) и необработанные миоциты (закрашенные кружки), но имели повышенную фракцию укорочения (А) и скорость укорочения (В) при всех частотах. Скорость расслабления (С) и время до достижения 90% расслабления (D) также были одинаковыми во всех группах, при этом время до достижения расслабления укорачивалось при повышении частоты стимуляции. *=р<0,05 по сравнению с необработанными, †=р<0,05 по сравнению с GFP-трансдуцированными, ‡=р<0,05 по сравнению со стимуляцией 0,5 Гц для всех групп.
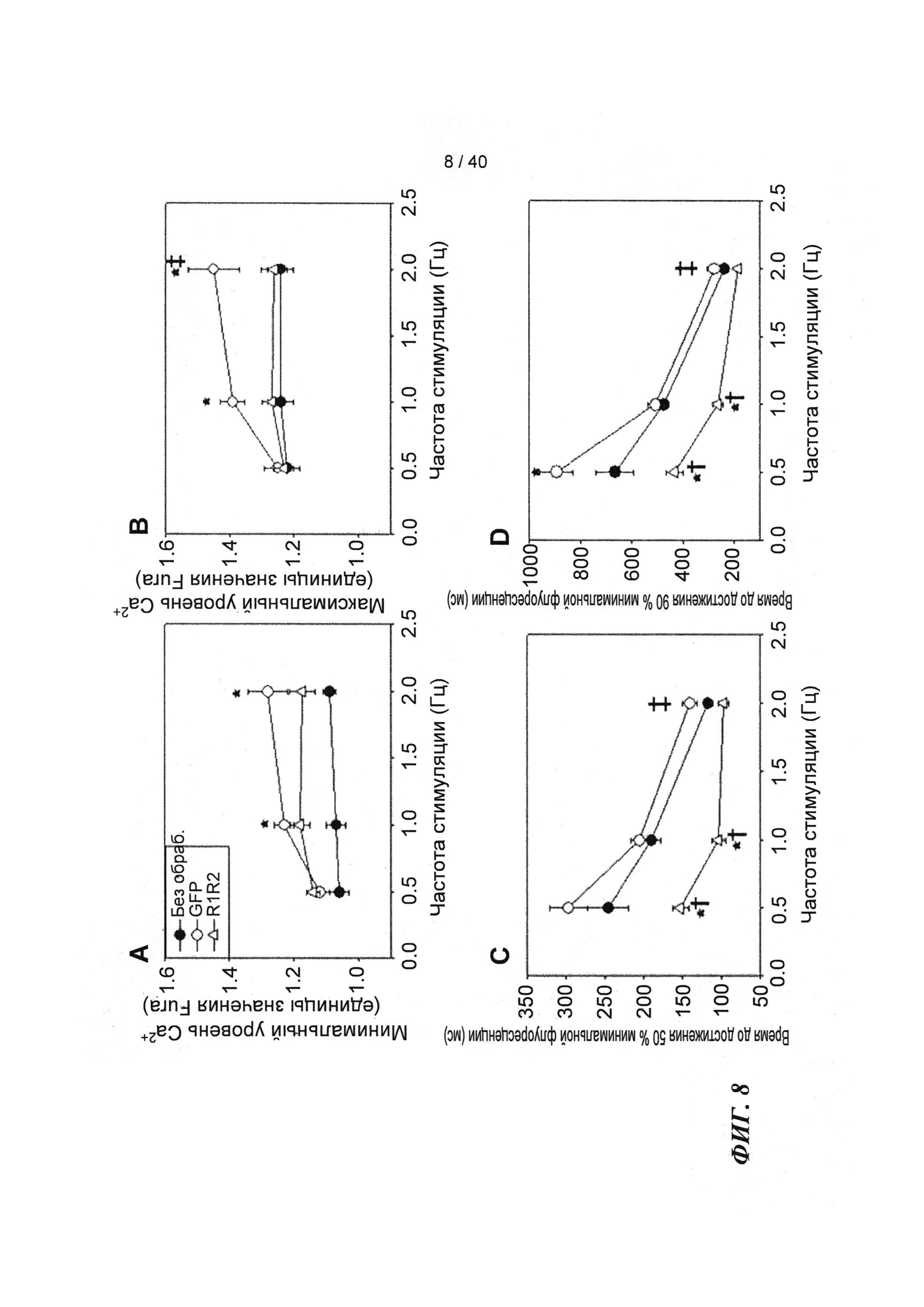
[0027] ФИГ.8. Эффект частоты стимуляции на характер регуляции Ca2+. Rrm1+Rrm2-трансдуцированные миоциты (незакрашенные треугольники) отвечали на частоту стимуляции так же, как необработанные миоциты (закрашенные кружки) при минимальной (А) и максимальной (В) флуоресценции, тогда как только GFP-трансдуцированные миоциты (закрашенные кружки) имели большее повышение по обоим показателям при повышении частоты. Как и в случае расслабления кардиомиоцитов, время снижения тока Ca2+(DT) до 50% (С) и 90% (D) уменьшалось при повышении частоты стимуляции, но оба показателя были значительно снижены у R1R2-трансдуцированных кардиомиоцитов. *=р<0,05 по сравнению с необработанными, †=р<0,05 по сравнению с GFP-трансдуцированными, ‡=р<0,05 по сравнению со стимуляцией 0,5 Гц для всех групп.
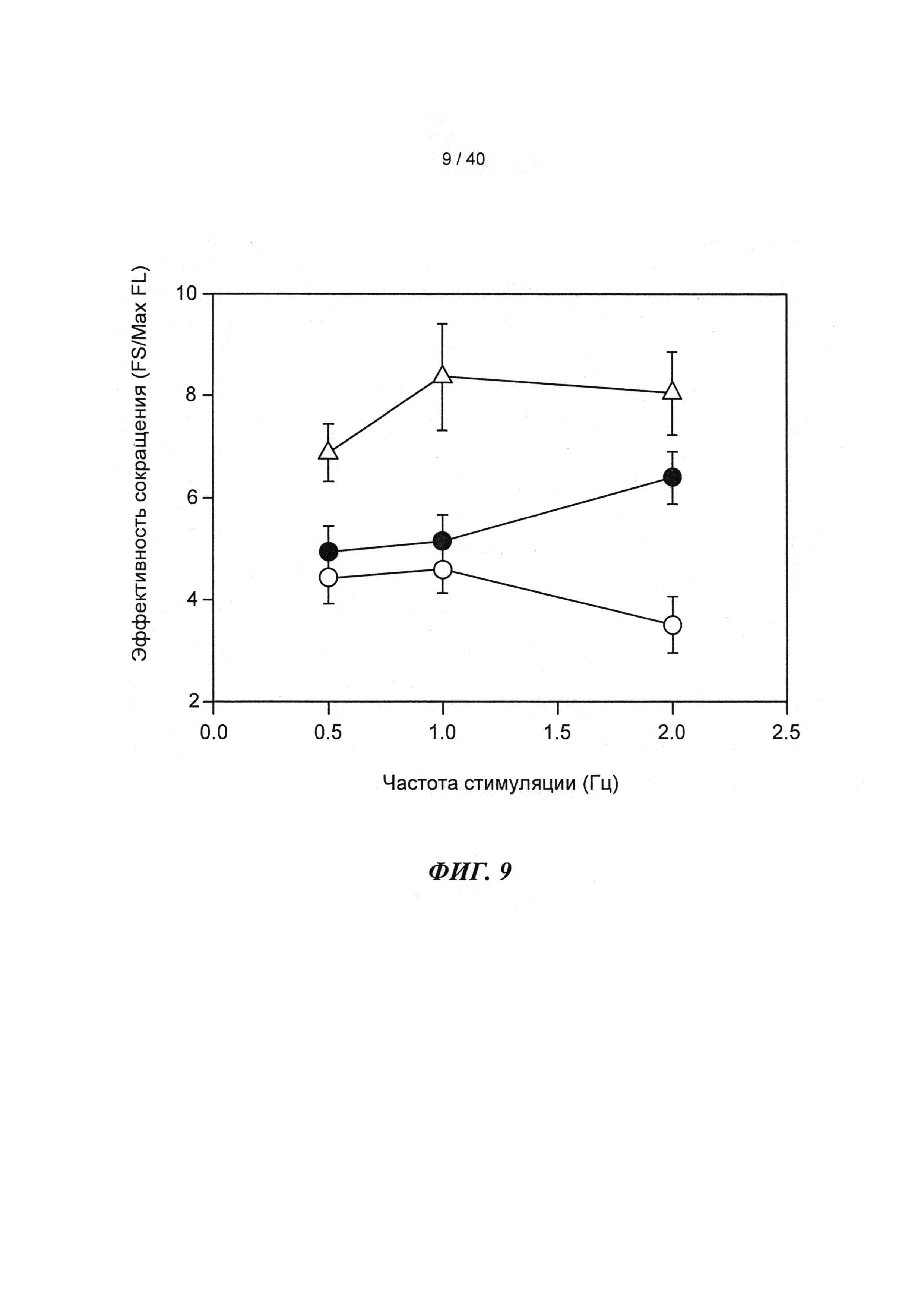
[0028] ФИГ.9. Эффективность сокращения, определенная как относительное укорочение, деленная на максимальную Fura-флуоресценцию (пик Ca2+), указывает на то, что Rrm1+Rrm2-трансдуцированные кардиомиоциты (незакрашенные треугольники) значительно более чувствительны к Ca2+ при всех частотах стимуляции, тогда как только GFP-трансдуцированные кардиомиоциты (незакрашенные кружки) менее чувствительны к Ca2+ по сравнению с необработанными кардиомиоцитами (закрашенные кружки) только при частоте стимуляции 2 Гц. *=р<0,05 по сравнению с необработанными, †=р<0,05 по сравнению с GFP-трансдуцированными.
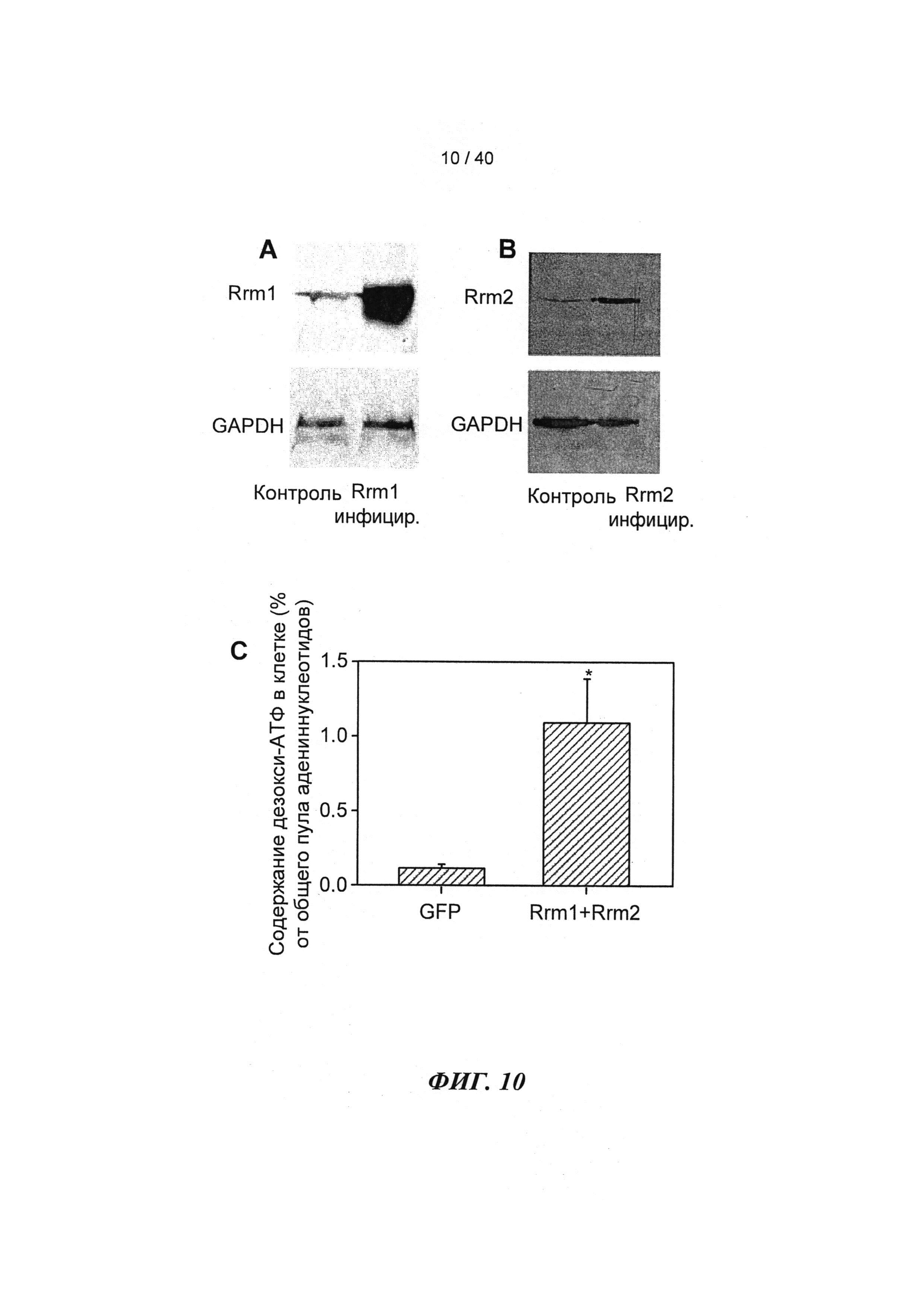
[0029] ФИГ.10. (А) Вестерн-блоттинг Rrm1-трансдуцированных кардиомиоцитов новорожденных крыс, меченных антителами против Rrm1, указывает на повышение уровня Rrm1 в >24 раз. (В) Вестерн-блоттинг Rrm2-трансдуцированных кардиомиоцитов новорожденных крыс, меченных антителами против Rrm2, указывает на повышение уровня Rrm2 в >46 раз. Гиперэкспрессия Rrm1+Rrm2 значительно повышает внутриклеточную [дезокси-АТФ] в >10 раз в кардиомиоцитах новорожденных крыс, как было показано с помощью ВЭЖХ-анализа. *=р<0,05 по сравнению с GFP-трансдуцированными кардиомиоцитами.
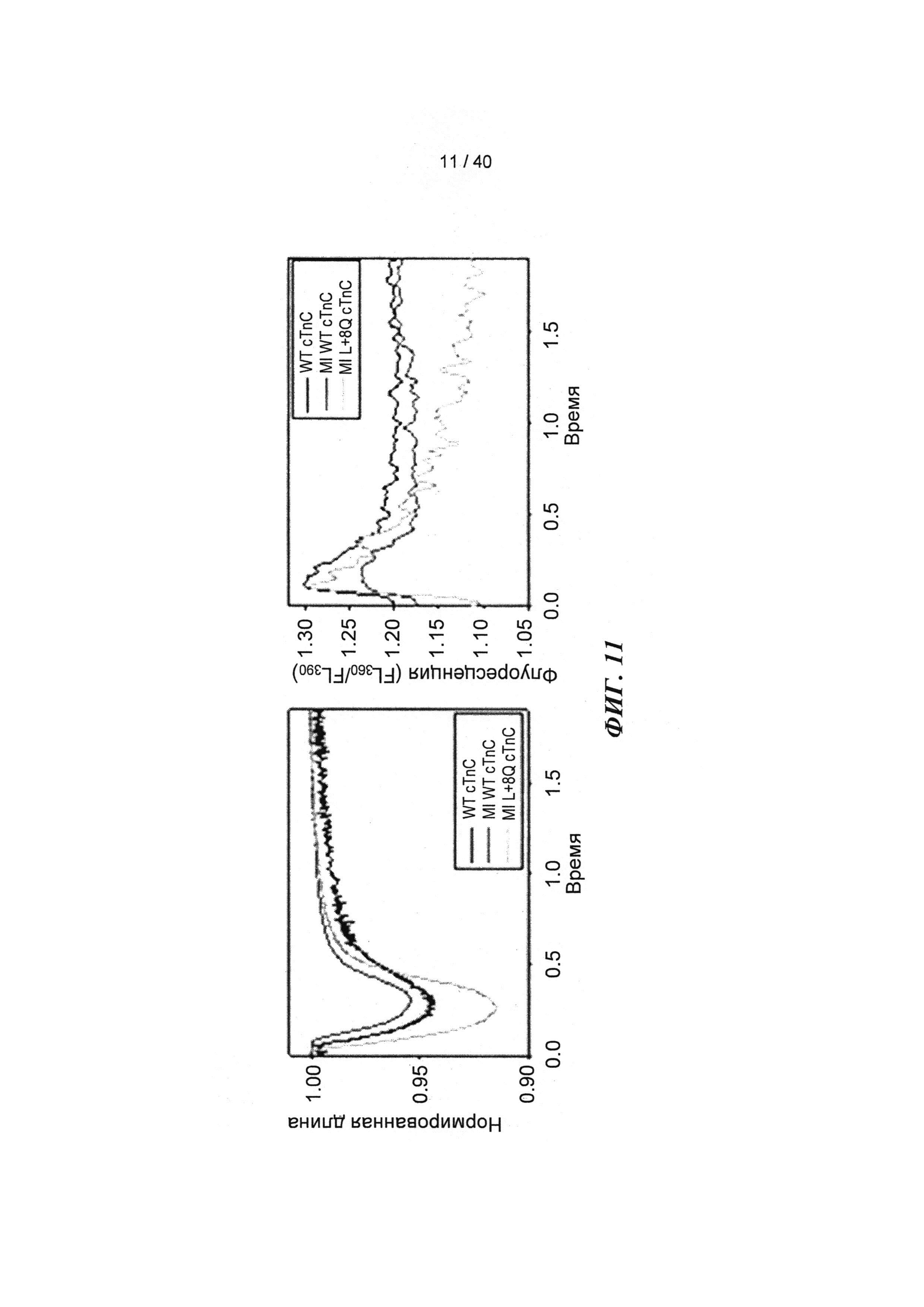
[0030] ФИГ.11. Кривые укорочения (А) и тока Ca2+ (В) для AV-cTnC-трансдуцированных миоцитов: не пораженные инфарктом клетки (cTnC ДТ, черные кривые), пораженные инфарктом контрольные клетки (cTnC ДТ, красн.) и пораженные инфарктом клетки (L48Q cTnC, заленый).
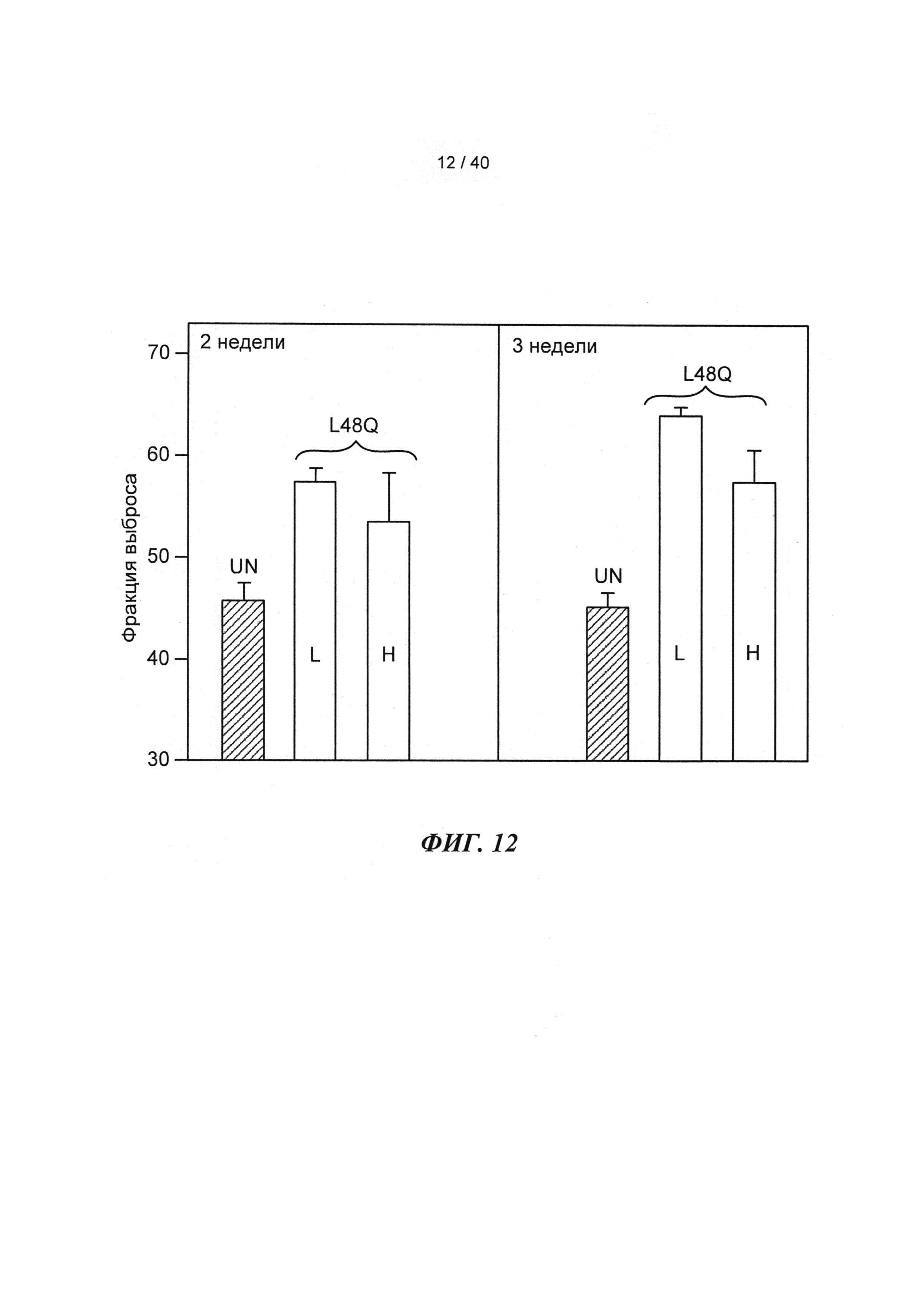
[0031] ФИГ.12. Фракция выброса ЛЖ через 2 недели (слева) и 3 недели (справа) для необработанных животных (UN; n=5) по сравнению с введением низкой (L; n=3) или высокой (Н; n=3) дозы AAV6-L48Q.
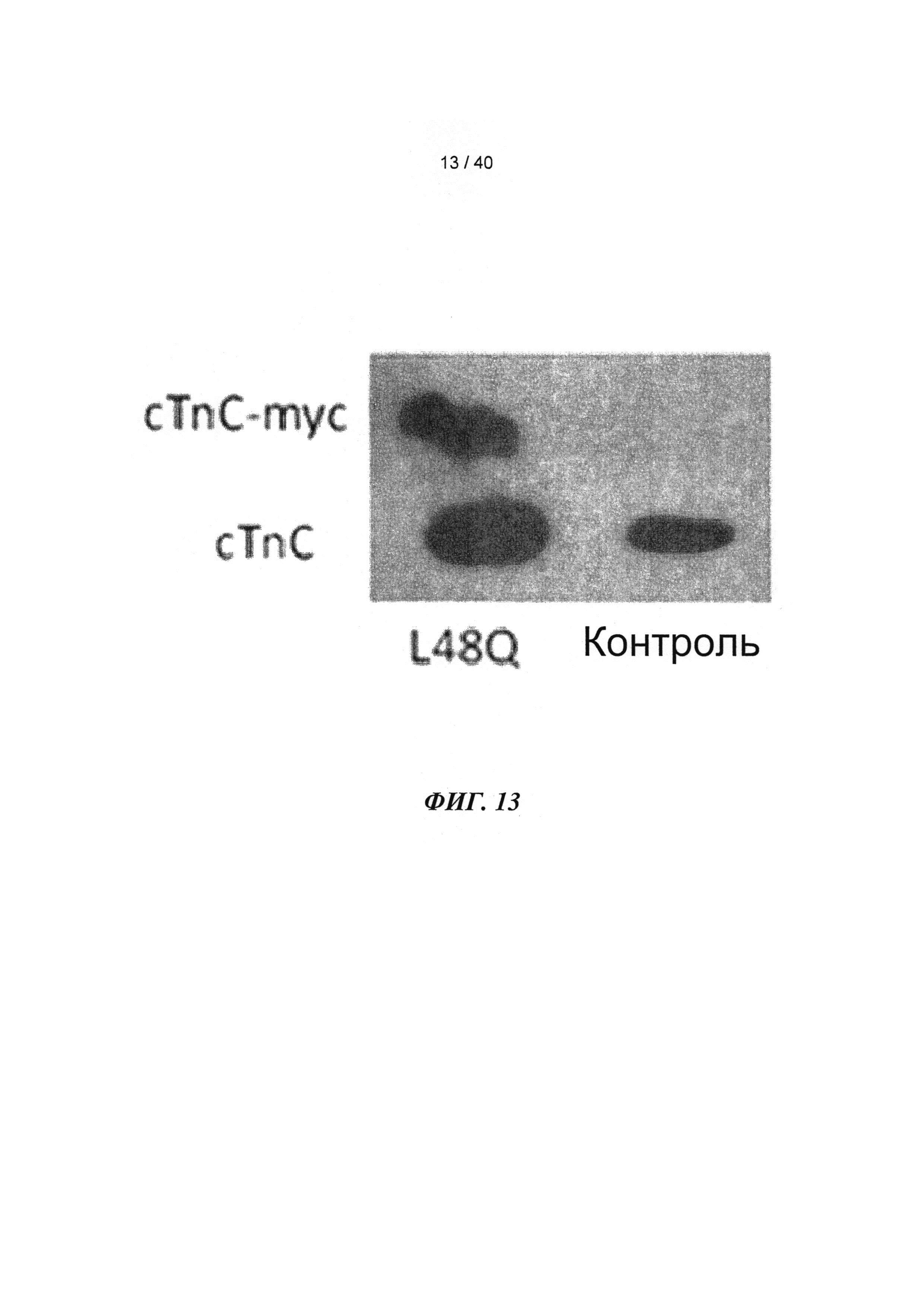
[0032] ФИГ.13. Вестерн-блот, меченный анти-cTnC, для AAV6 L48Q cTnC-транедуцированной мышиной сердечной ткани (слева) и нетранедуцированного контроля (справа).
[0033] ФИГ.14. Изображения, полученные методом светлого поля (слева), и флуоресцентные изображения (справа) AV-трансдуцированной клетки.
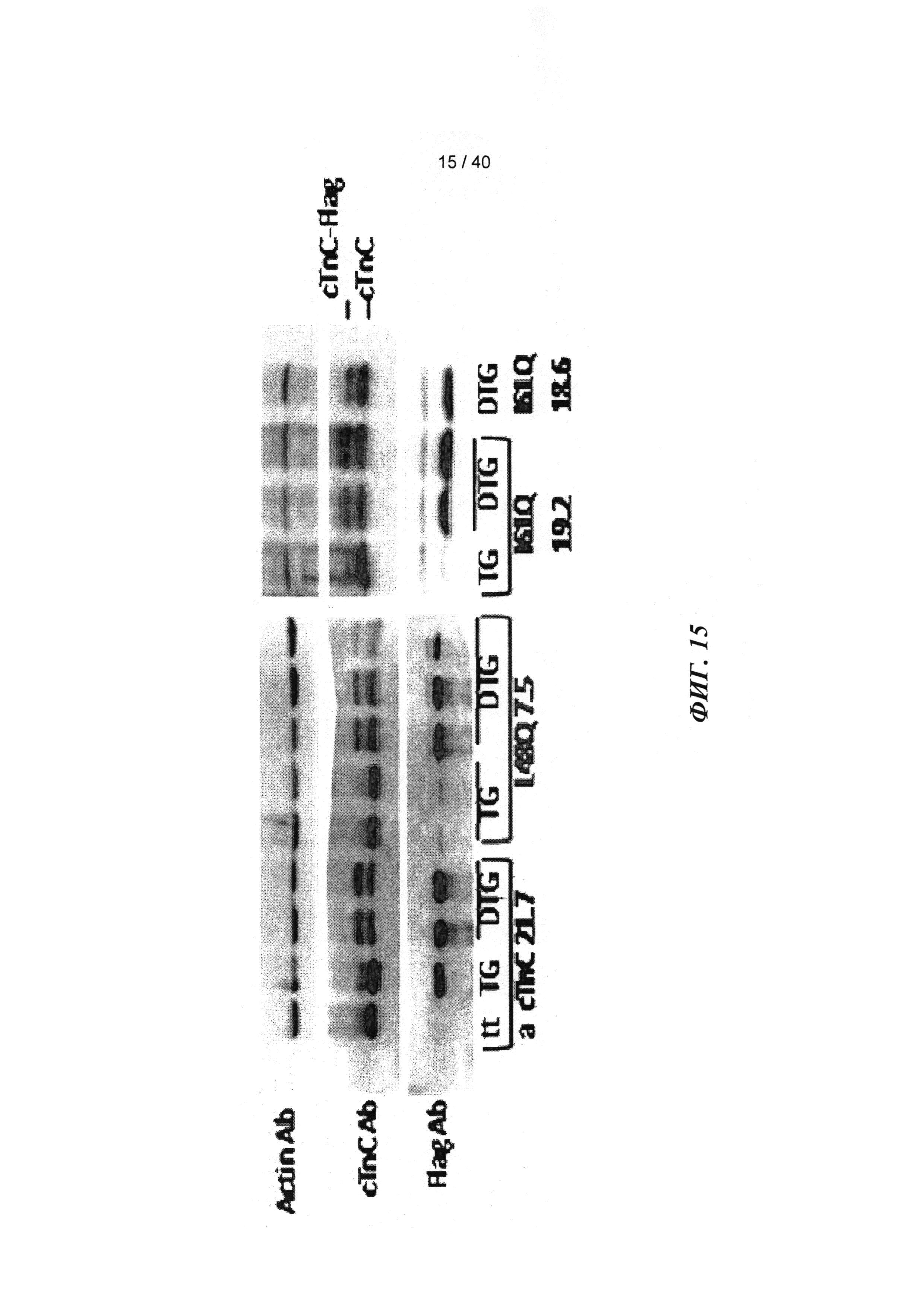
[0034] ФИГ.15. Вестерн-блот ткани миокарда от трансгенных животных (меченный). Flag-меченный cTnC выявляется как белок более высокой молекулярной массой, и денситометрический анализ flag-меченного cTnC по сравнению с немеченым cTnC указывает на 40-50% замещение нативного cTnC у Tg-животных.
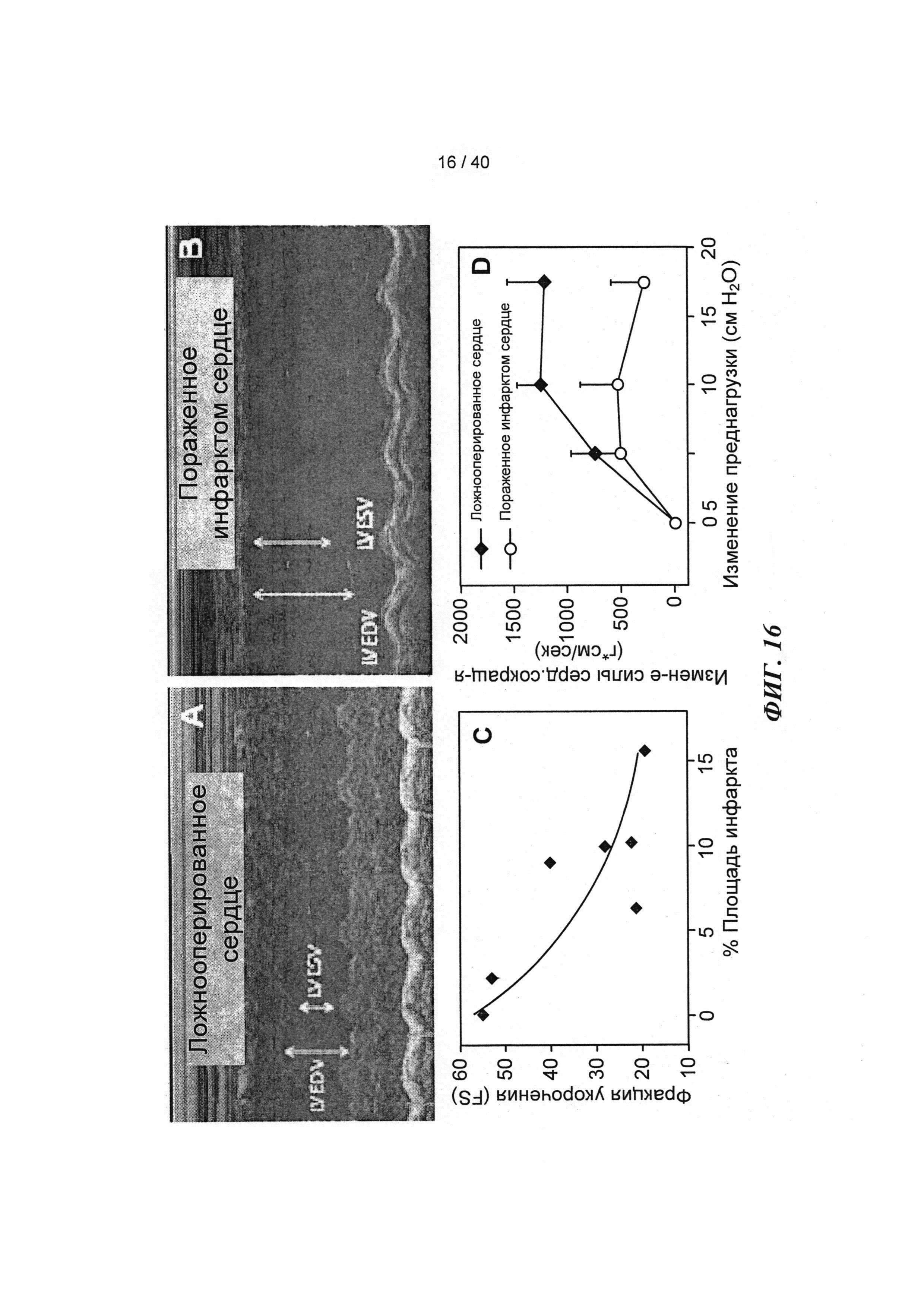
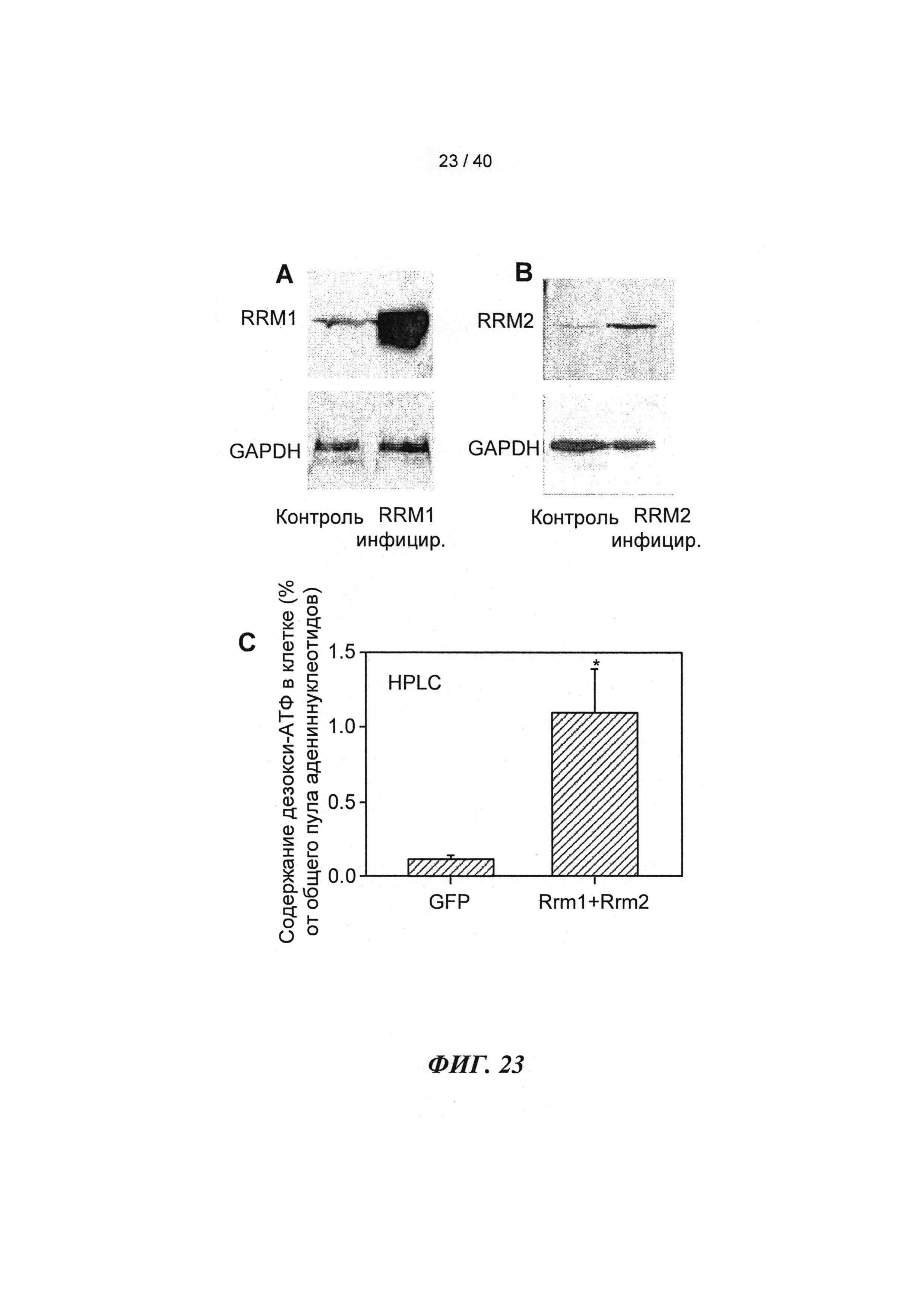
[0035] ФИГ.16. Эхокардиография (А-С) и измерения на работающем сердце (D) функции ЛЖ демонстрируют потерю функции после инфаркта.
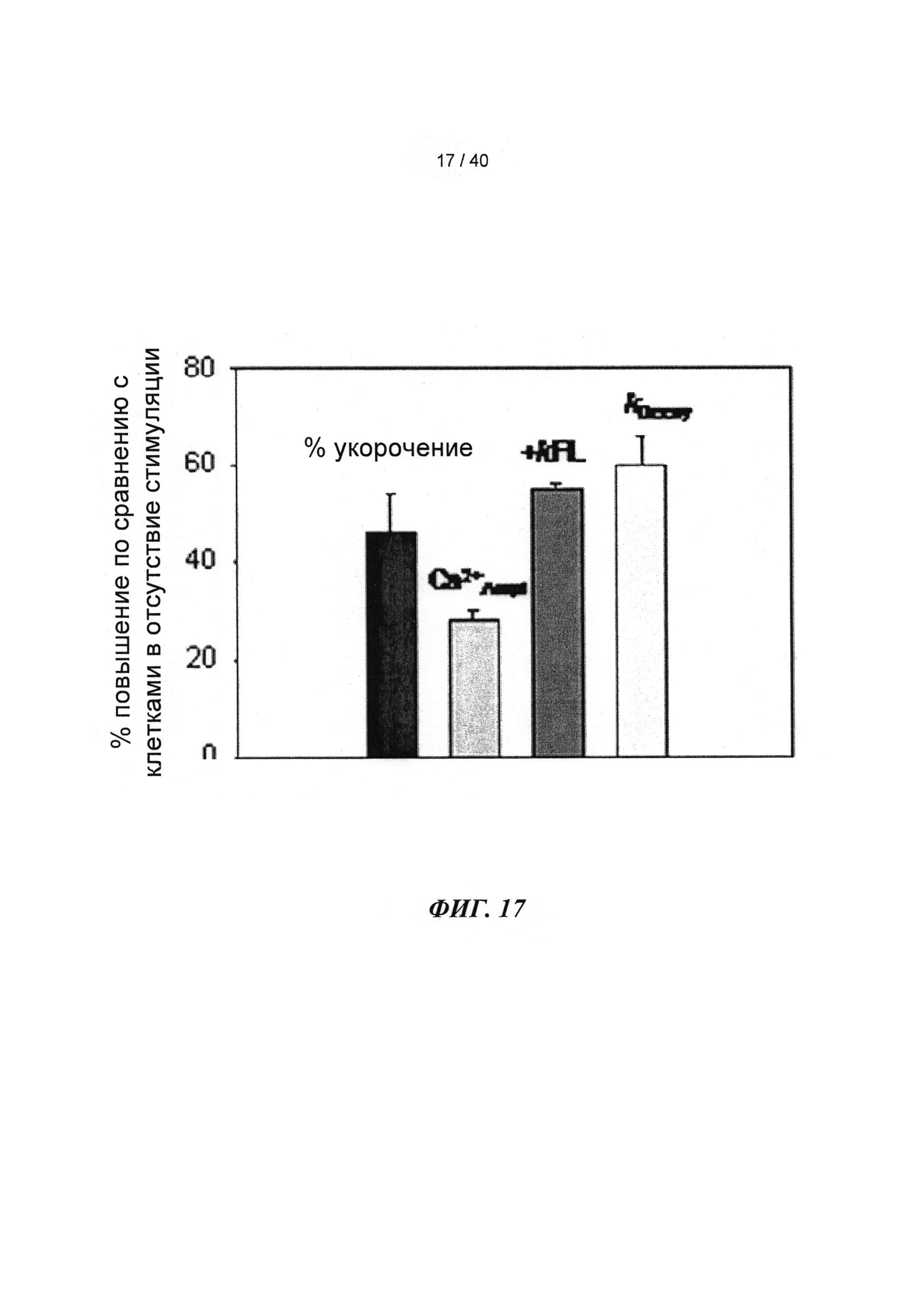
[0036] ФИГ.17. Процентное повышенное сократительных свойств и Ca2+-тока клеток при стимуляции 1 Гц (30 часов) по сравнению с отсутствием стимуляции.
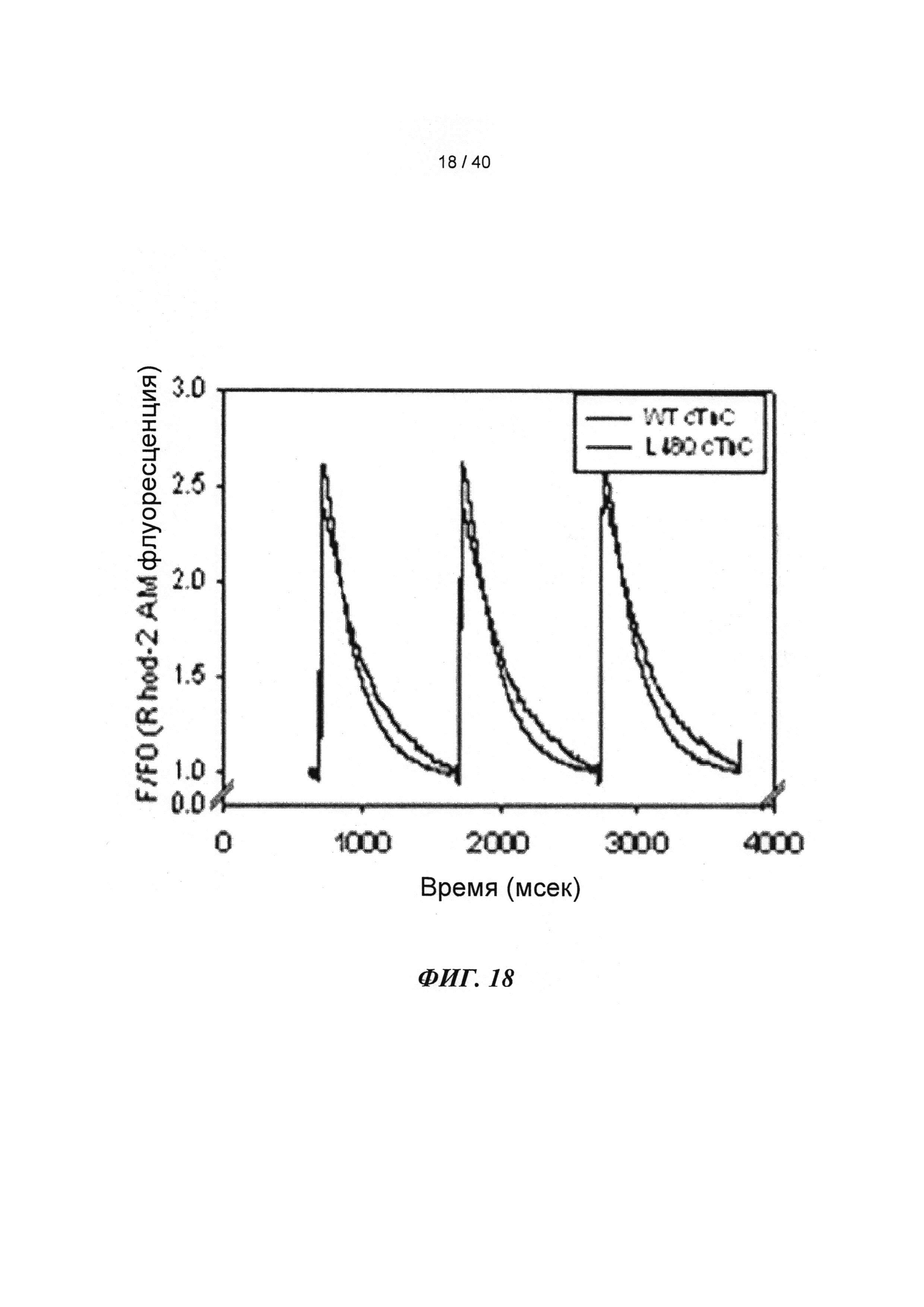
[0037] ФИГ.18. Ca2+-ток (rhod-2) показывает более быстрое угасание в случае L48Q (синий) по сравнению с WT (черный) cTnC.
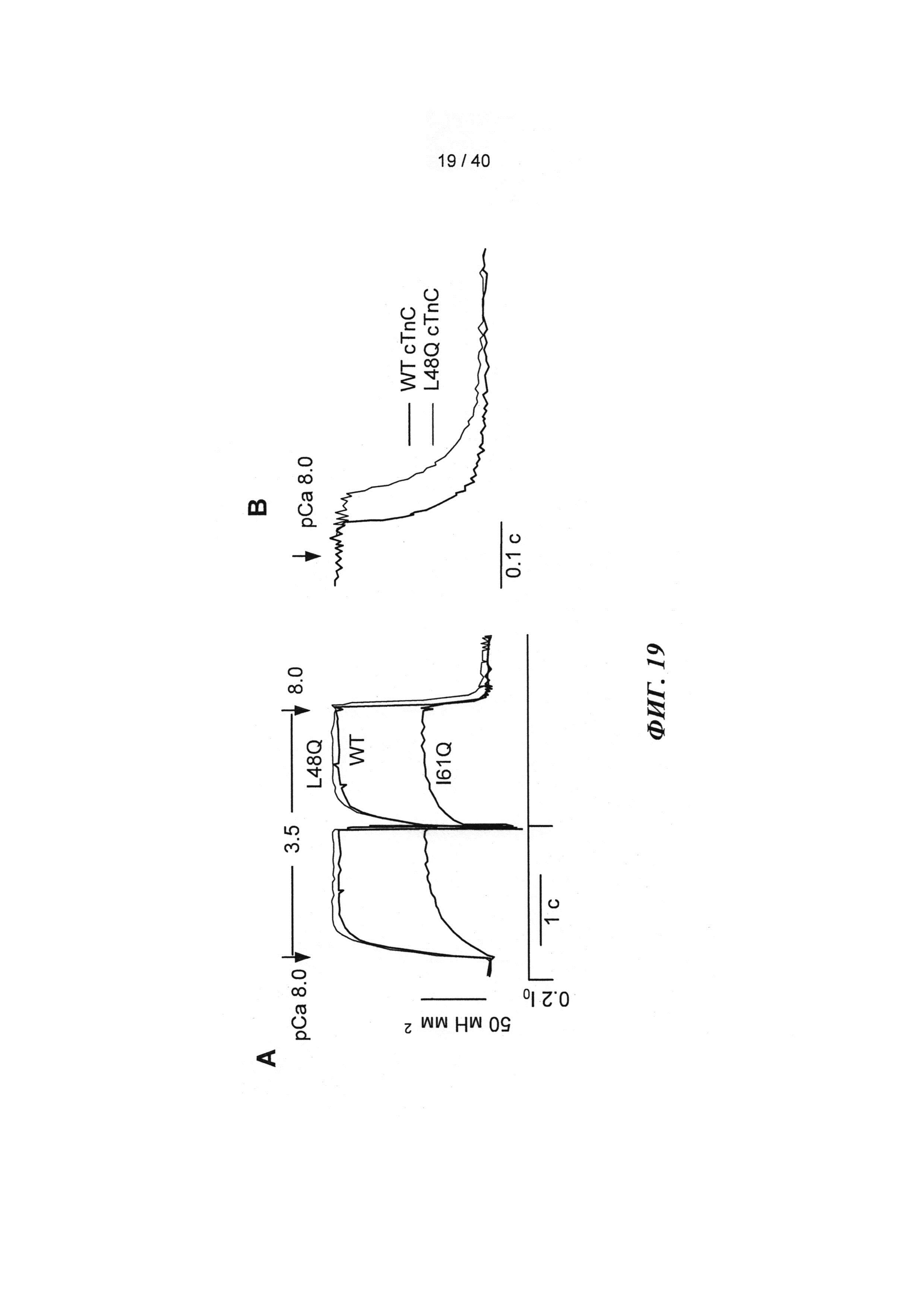
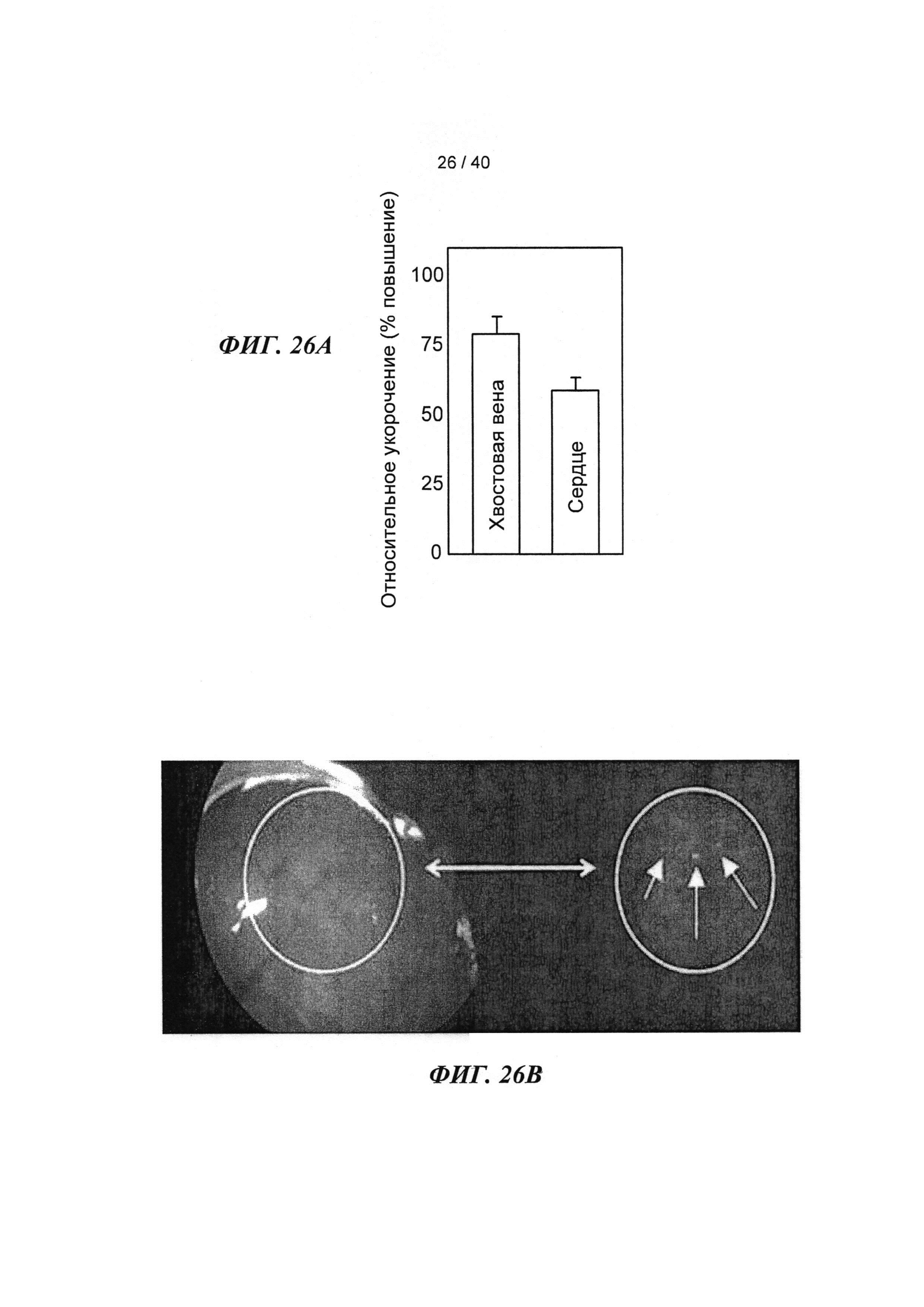
[0038] ФИГ.19. (А) Кинетика активации и расслабления сердечных миофибрилл при быстрой смене растворов от рСа 8 до 3,5 и до 8 после замещения на WT, I61Q (синий) или L48Q (красный) cTnC. (В) Кривые расслабления при более высоком разрешении для L48Q по сравнению с cTnC ДТ.
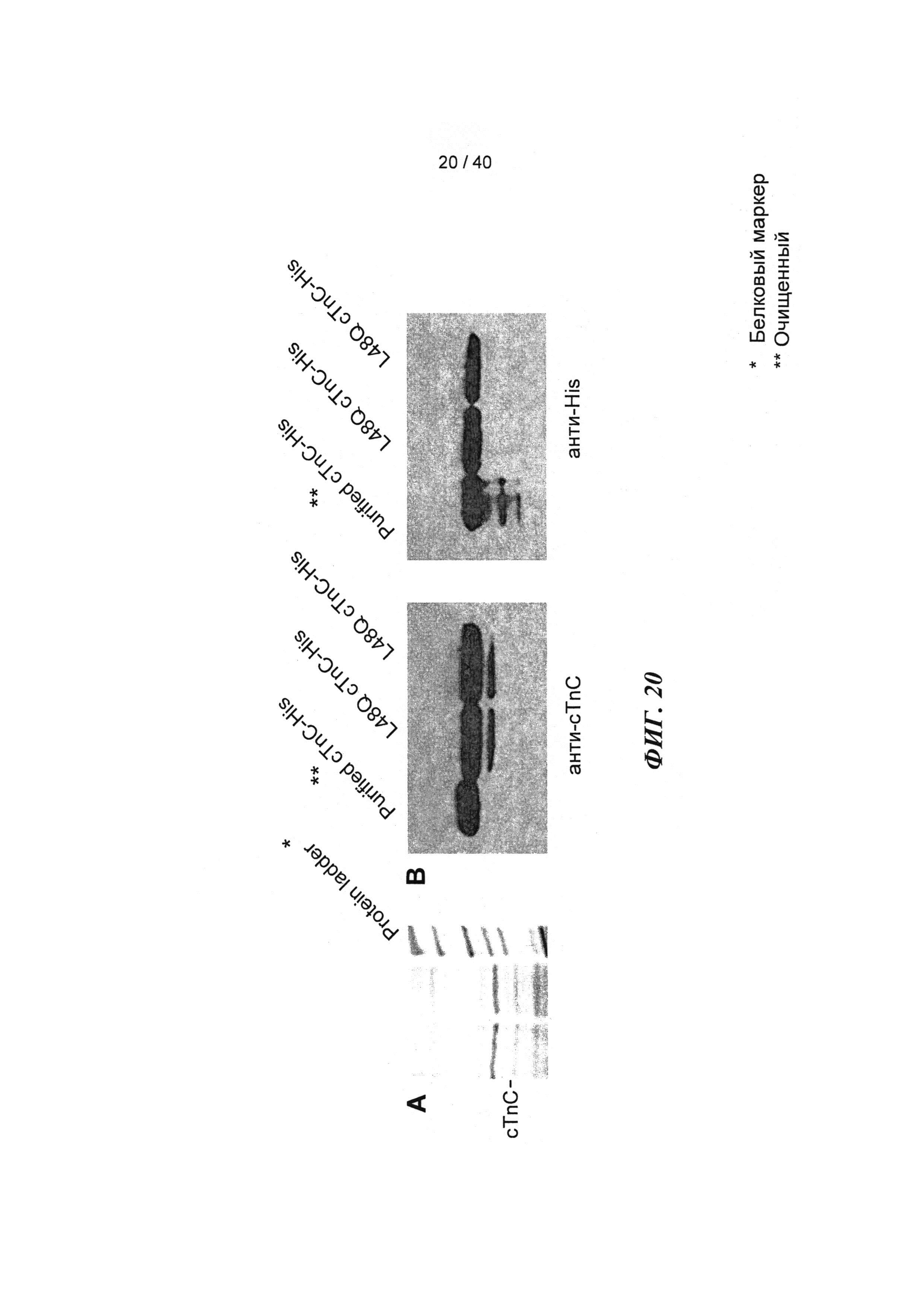
[0039] ФИГ.20. (А) Окраска серебром миофиламентов необработанных и L48Q cTnC-His-трансдуцированных кардиомиоцитов демонстрирует поддержание стехиометрии белков тонких филаментов. (В) Вестерн-блоты с анти-cTnC (левая панель) показывают общее содержание cTnC в миофиламентах, Вестерн-блоты с анти-His (правая панель) показывают 58±7% L48Q cTnC включение в тонкие филаменты (по результатам денситометрии).
[0040] ФИГ.21. Профиль фосфорилирования миофибрилл для WT по сравнению с I61Q cTnC-трансдуцированными кардиомиоцитами. (А) Вестерн-блоттинг с анти-фосфосерином. (В) Окраска Кумасси голубым в присутствии додецилсульфата натрия (ДСН), показывающая общий белок. (С) Окраска Pro-Q diamond, показывающая фосфорилирование. Различий не наблюдалось.
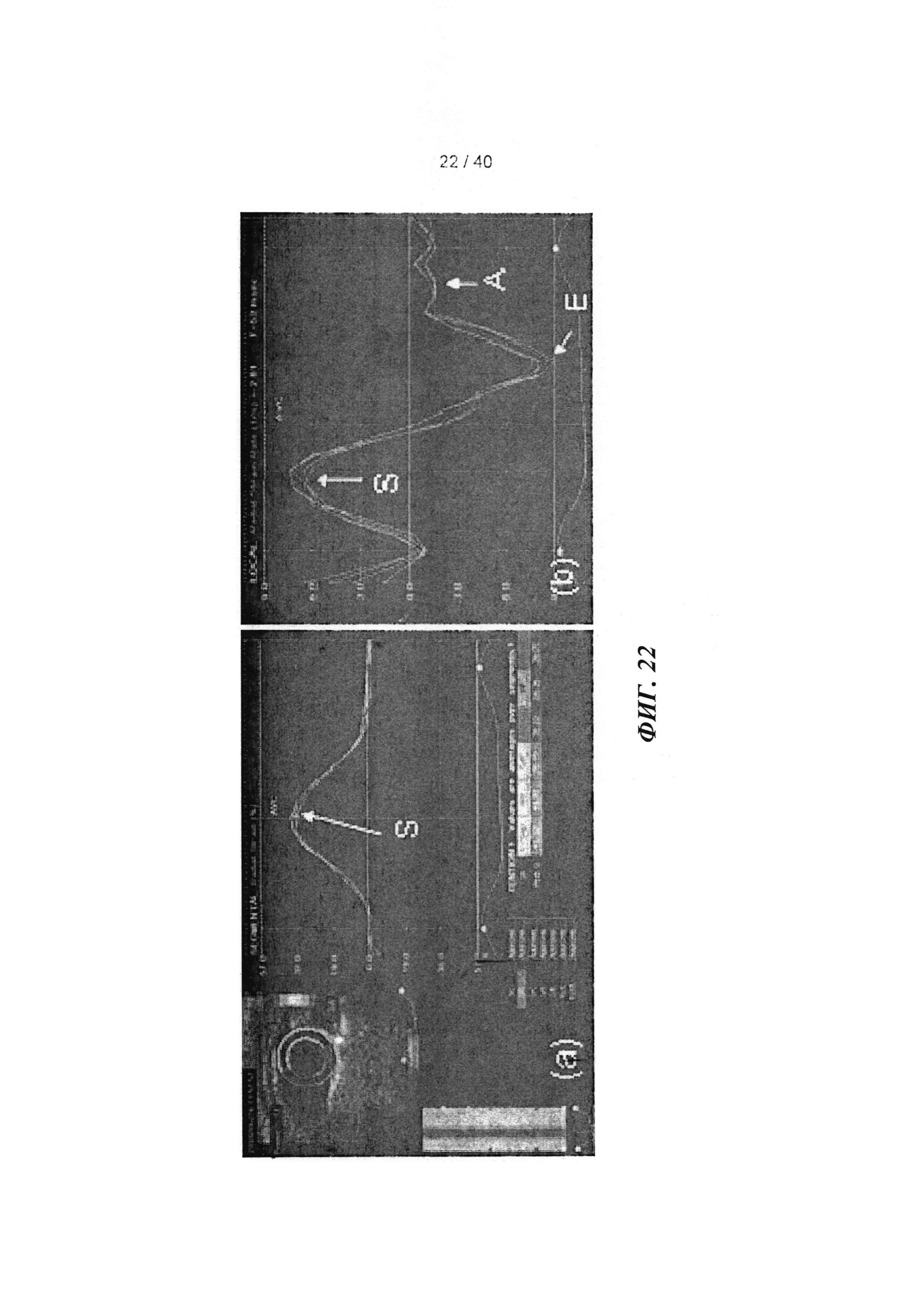
[0041] ФИГ.22. Радиальное растяжение и скорость растяжения с использованием спекл-трекинг эхокардиографии. (а) вид короткой оси средней части желудочка с цветовой маркировкой областей, представляющей передний, боковой, задний, нижний и перегородочный сегменты сердца, для анализа радиального растяжения (сверху левый, стрелка указывает на систолический пик (S); каждая линия соответствует сегментам с цветовой маркировкой. (b) скорость радиального растяжения в систоле и двух диастолических периодов - ранней (Е) и поздней (А) диастоле.
[0042] ФИГ.23. Вестерн-блоттинг для R1 (А) и R2 (В) с использованием GAPDH в качестве контроля нагрузки. ВЭЖХ (С) [дезокси-АТФ] трансдуцированных кардиомиоцитов.
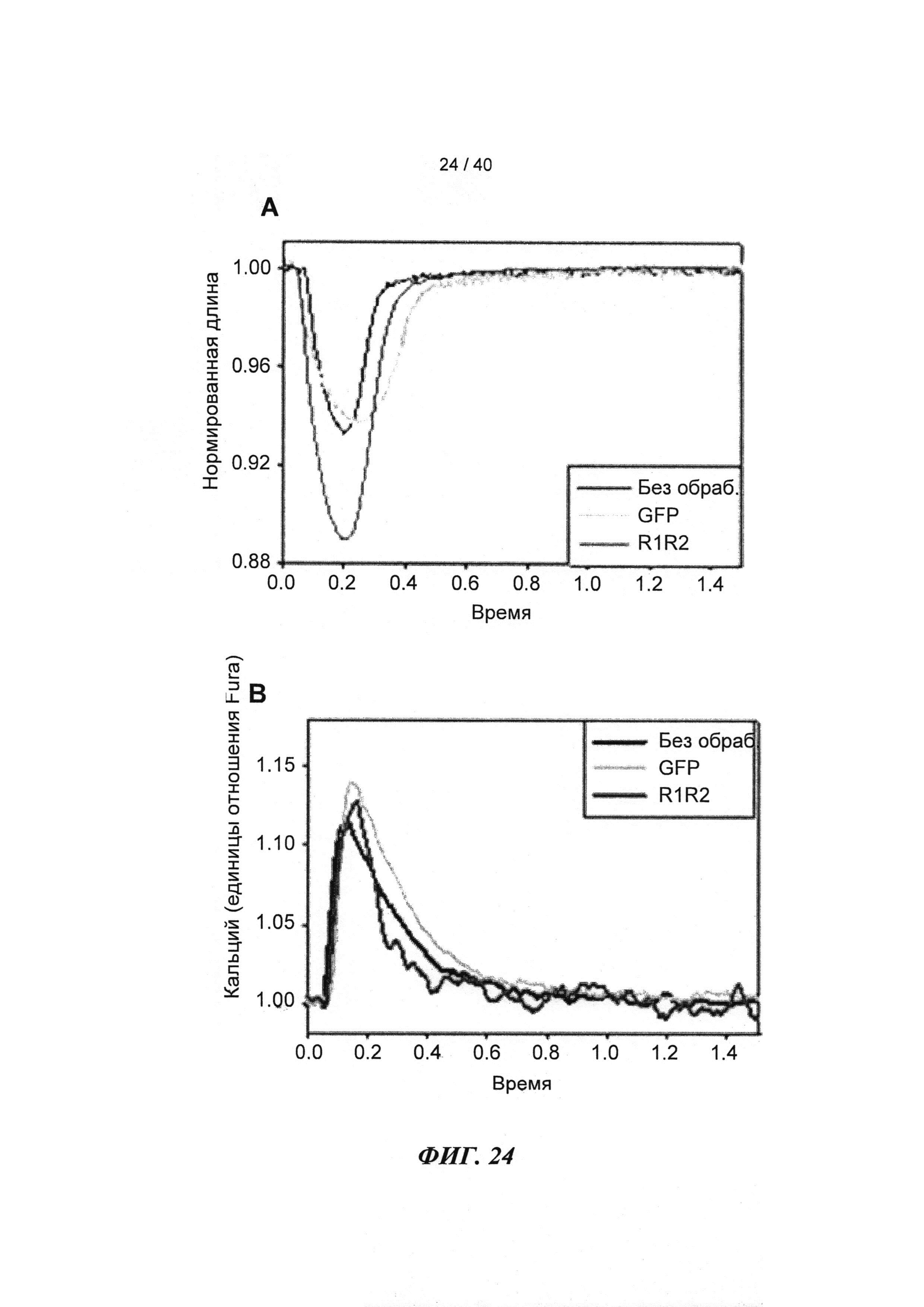
[0043] ФИГ.24. Длина (А) и Ca2+ (В) переходы указывают на повышенное укорочение без изменений в Ca2+ для R1R2-трансдуцированных клеток по сравнению с контролями GFP и неподверженными лечению контролями.
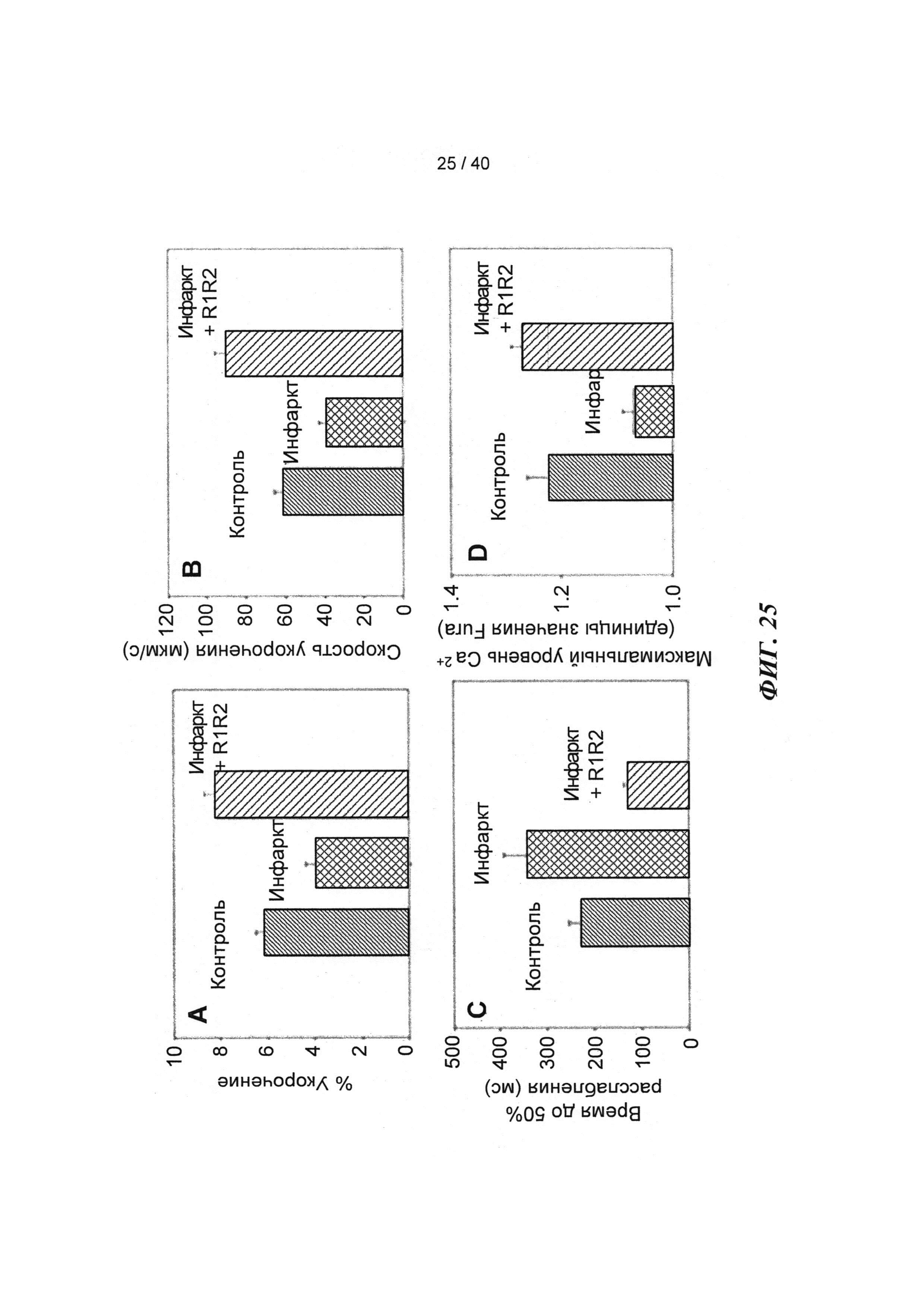
[0044] ФИГ.25. Степень (А) и скорость (В) укорочения, время до достижения 50% расслабления (С) и максимальное высвобождение Ca2+ (D) снижены в кардиомиоцитах из пораженных инфарктом сердец, но восстановлены или улучшены по сравнению с контролями при трансдукции AV-R1R2.
[0045] На ФИГ.26. (А) показано повышение фракции укорочения левого желудочка для мышей, которым инъецировали AV-R1R2 в хвостовую вену и непосредственно в сердце по сравнению с контролем через 4 дня после инъекции; (В) ЛЖ после инъекции, демонстрирующий область локализованной трансфекции, показанной с помощью яркой зеленой флуоресценции (от GFP).
[0046] ФИГ.27. Процентное повышение фракции укорочения (А) и изменение внутреннего диаметра левого желудочка (В) у R1R2-гиперэкспрессирующих мышей по сравнению с контрольными однопометными животными, d-диастола, s-систола.
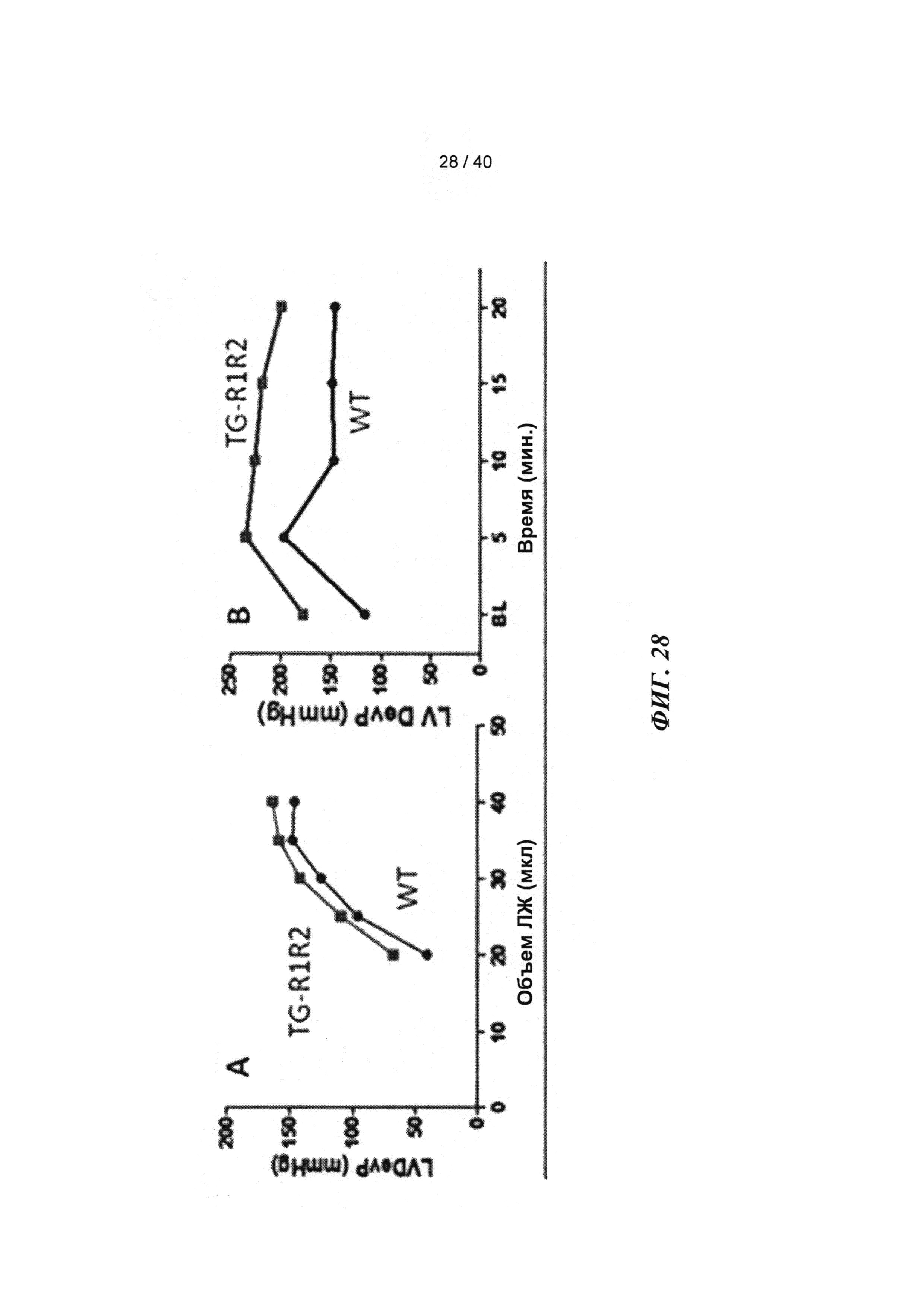
[0047] ФИГ.28. Соотношение между давлением и объемом ЛЖ (А) и ответ давления ЛЖ на стимуляцию [Ca2+] в высокой концентрации (В).
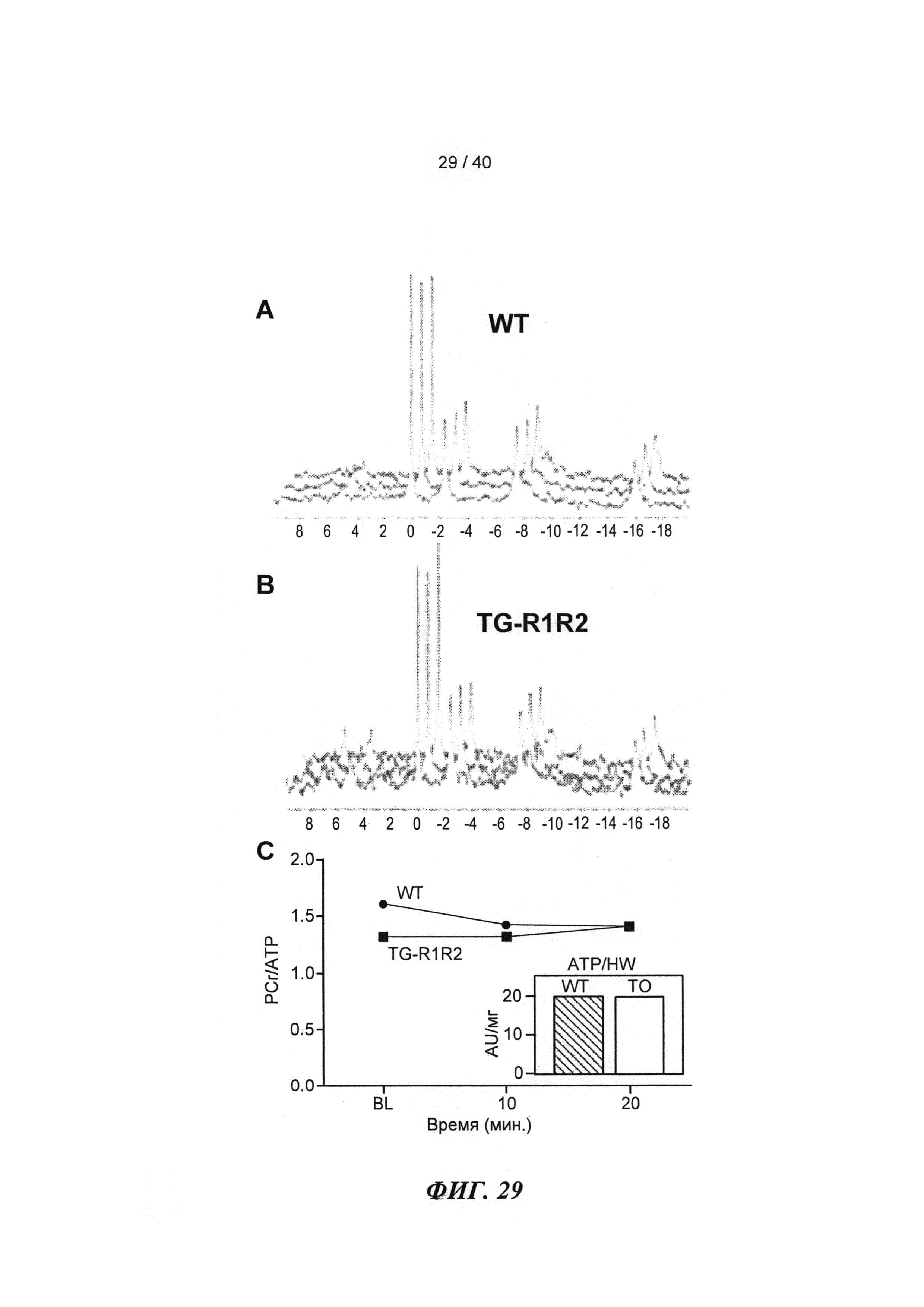
[0048] ФИГ.29. ЯМР-спектры для сердец дикого типа (WT) (А) и TG-R1R2 (В) и отношение PCr/АТР (С) на исходном уровне и после стимуляции [Ca2+] в высокой концентрации. Отношение АТФ/HW на исходном уровне (врезка).
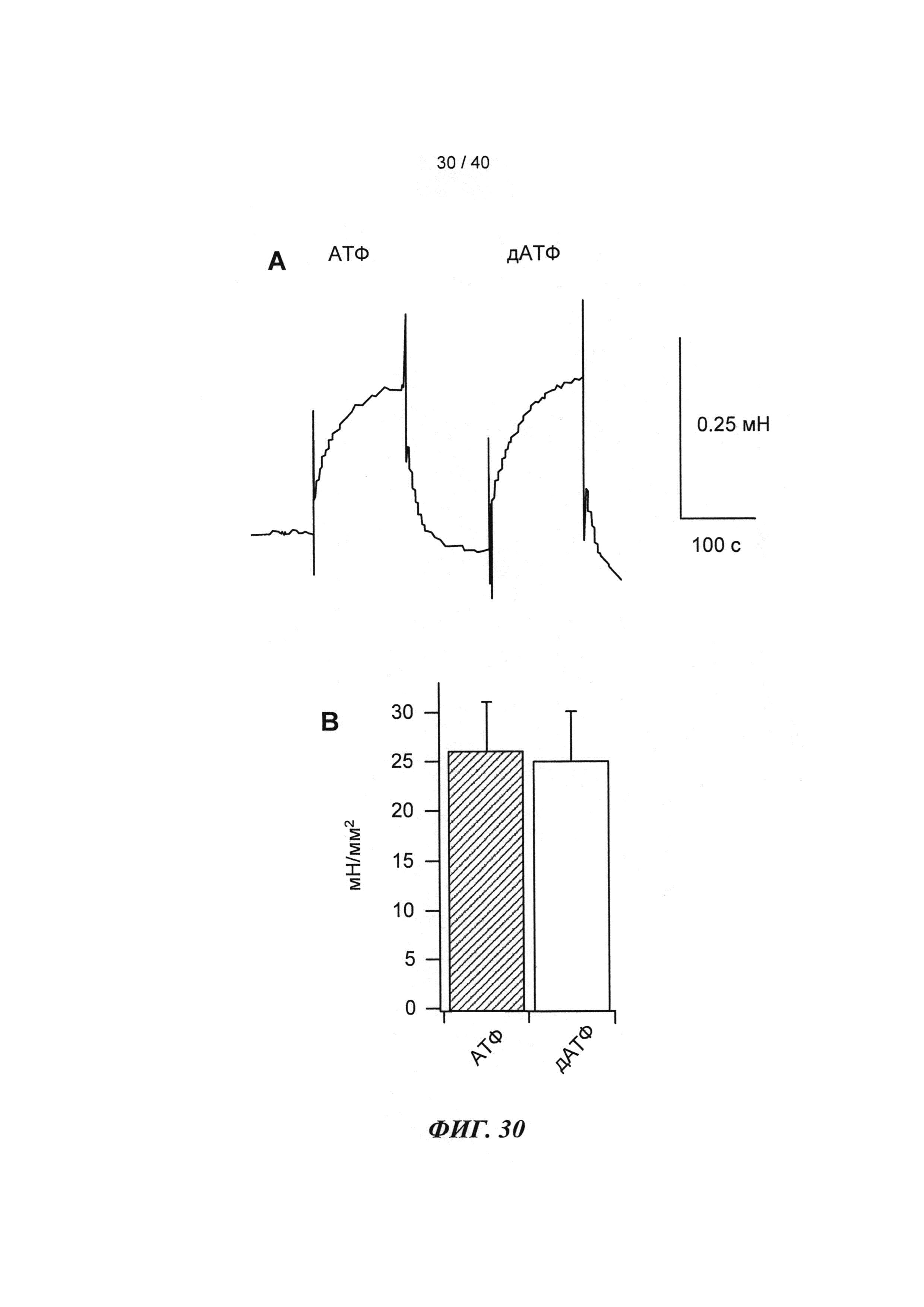
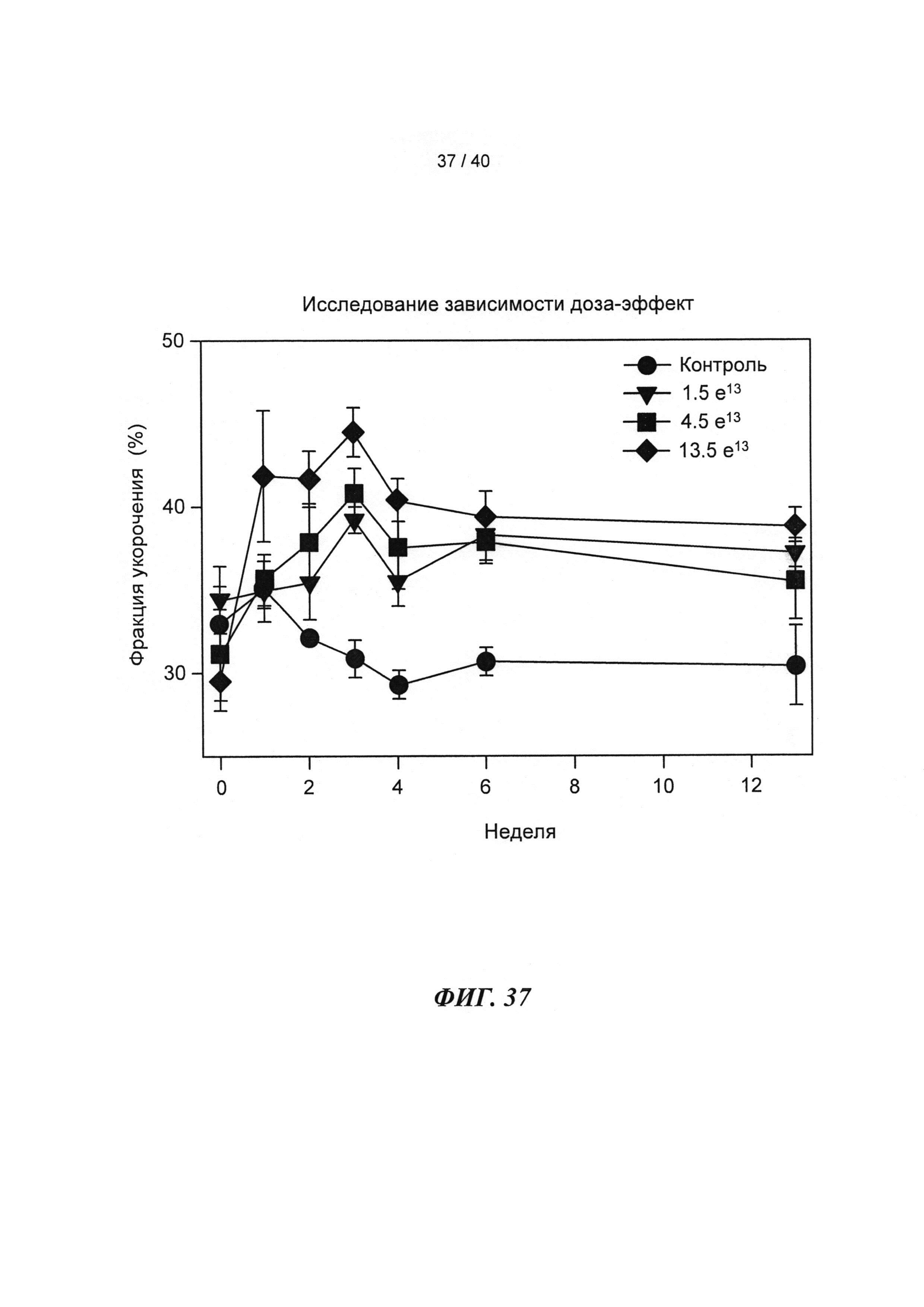
[0049] ФИГ.30. Кривые сокращения гладкой мышцы мышиной аорты при использовании АТФ и дезокси-АТФ (А) и суммированные данные (В).
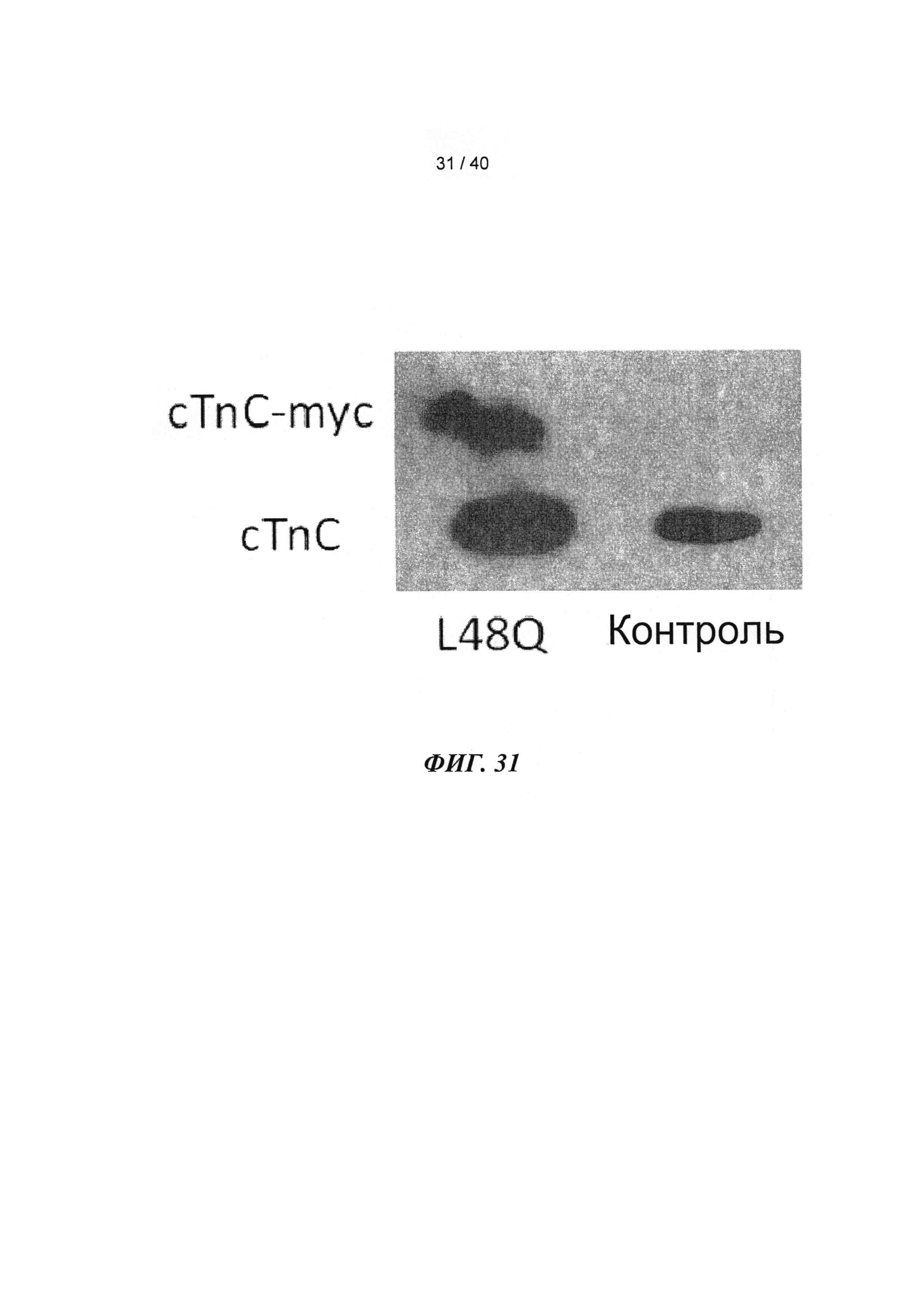
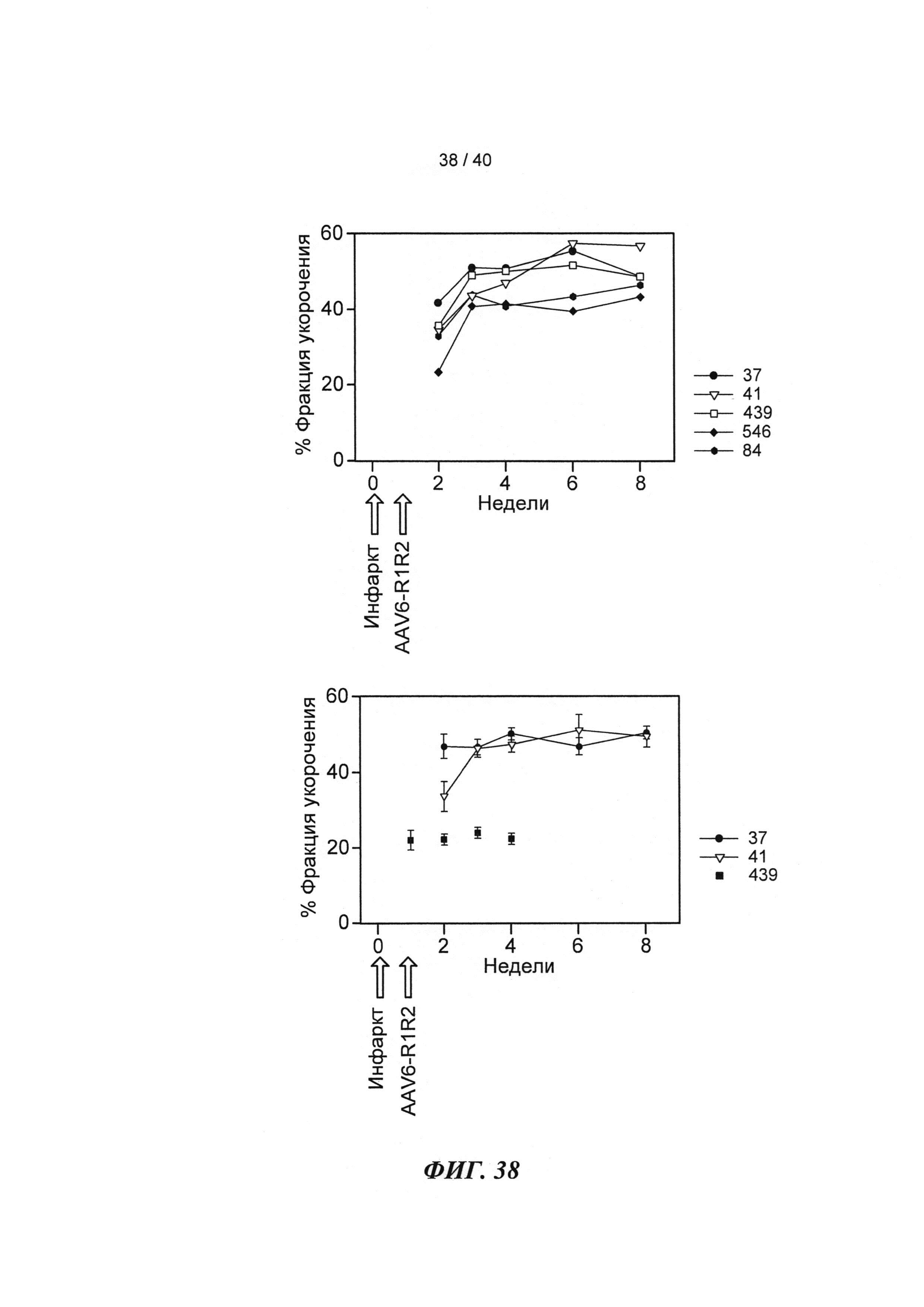
[0050] ФИГ.31. Вестерн-блот с анти-cTnC для AAV6 L48Q cTnC-трансдуцированной ткани мышиного сердца (слева) и нетрансдуцированный контроль (справа).
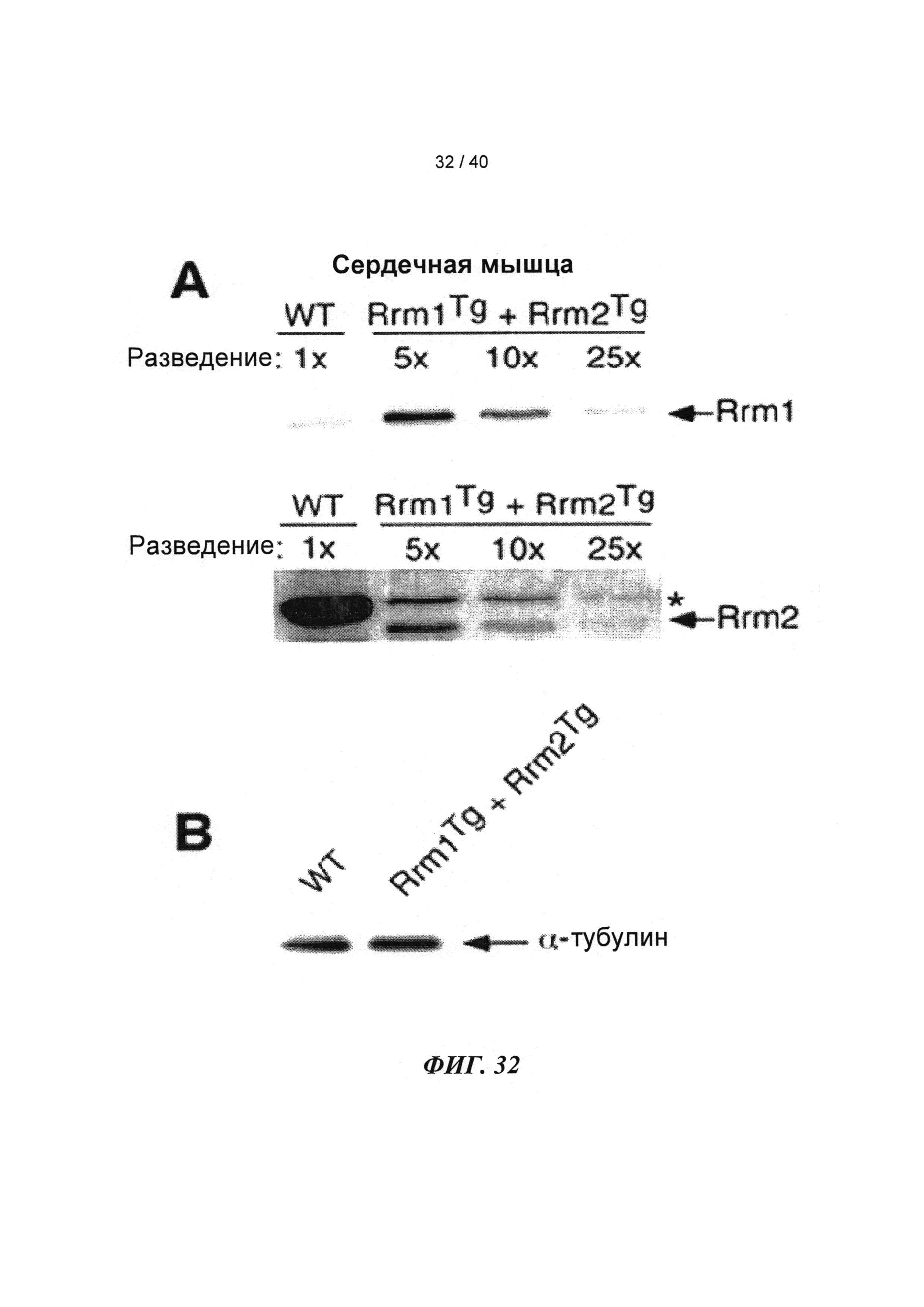
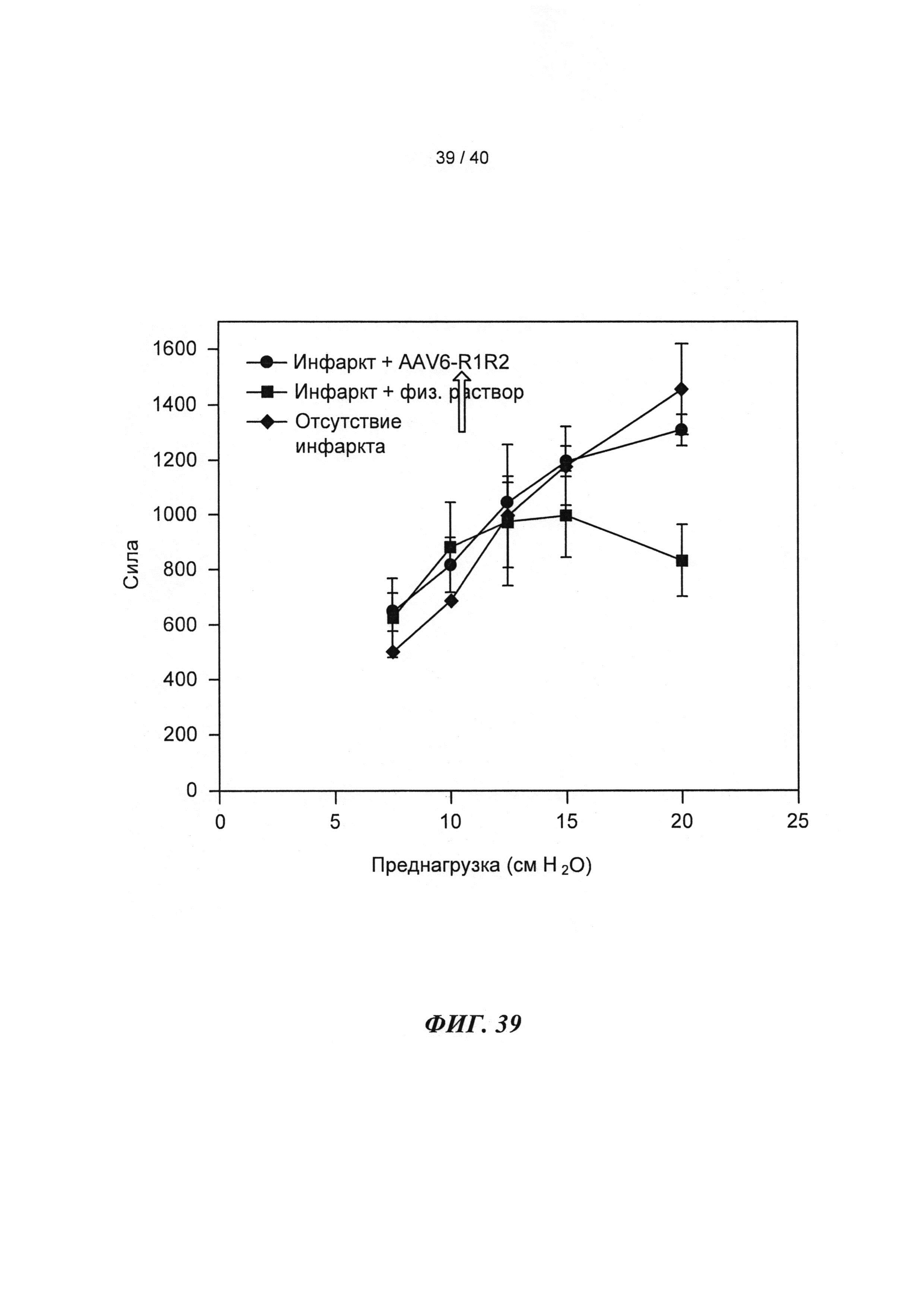
[0051] ФИГ.32. Вестерн-блоттинг для (A) R1 и R2 и (В) α-тубулина в качестве контроля нагрузки.
[0052] ФИГ.33. Кривая активации и расслабления образца сердечной миофибриллы (слева). Увеличение медленной фазы расслабления (справа). Скорость повышения растяжения (kACT). Медленная фаза расслабления (kREL,slow). Быстрая фаза расслабления (kREL,fast). Продолжительность медленной фазы (tREL,slow).
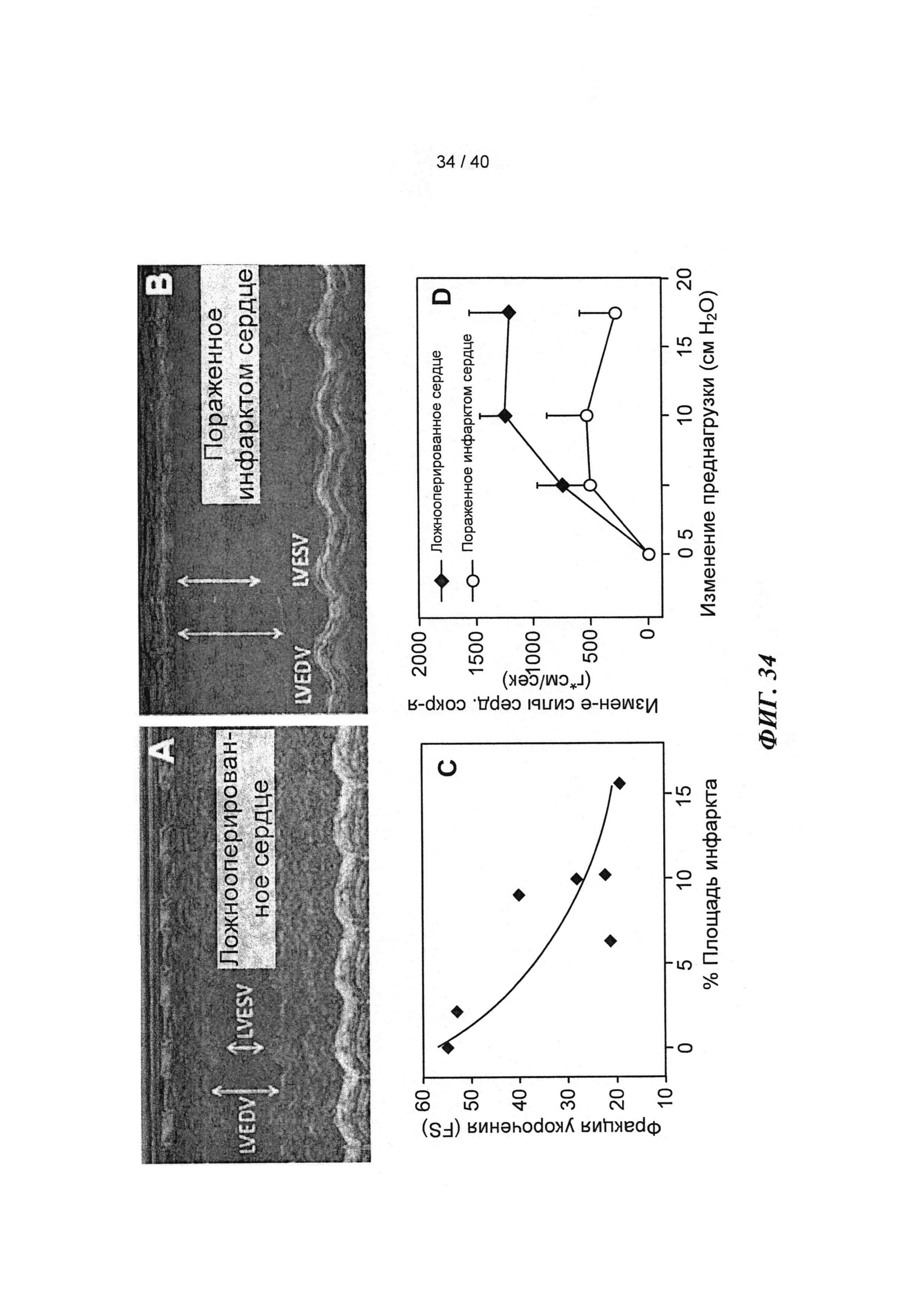
[0053] ФИГ.34. Эхокардиография (А-С) и измерения на работающем сердце (D) функции ЛЖ демонстрируют потерю функции после инфаркта.
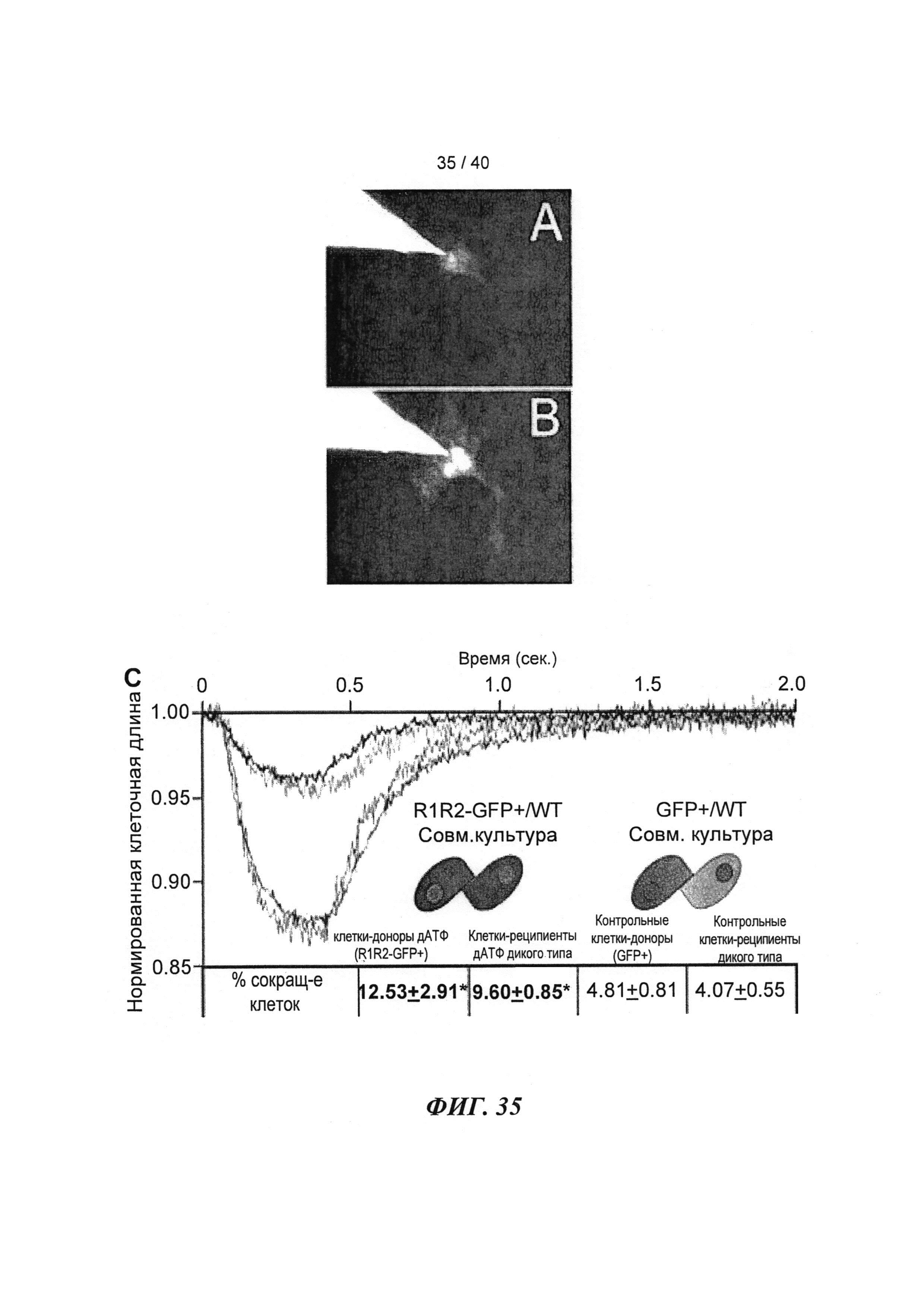
[0054] ФИГ.35. Инъекции меченной дезокси-АТФ в культивируемые чЭСК-кардиомиоциты (чЭСК-КМЦ) через 10 сек (А) и 4 мин (В). Сокультивирование с R1R2-гиперэкспрессирующими кардиомиоцитами усиливает сократительные свойства миоцитов дикого типа (реципиентов) (С). чЭСК-КМЦ трансфицировали либо AV-R1R2, либо AV-GFP, и указанные трансдуцированные миоциты сокультивировали с нетрансдуцированными чЭСК-КМЦ дикого типа (WT). Оба партнера в совместных культурах AV-R1R2+GFP-трансдуцированных миоцитов («дезокси-АТФ-донорских клеток», синяя кривая) и нетрансдуцированных миоцитов (WT дезокси-АТФ-реципиентов, красная кривая) показали повышенную сократимость по сравнению с их соответствующими контролями, т.е. AV-GFP-трансдуцированными («контрольными донорскими клетками», зеленая кривая) или их реципиентами WT (черная кривая). *р<0,05 по сравнению с контролями.
[0055] ФИГ.36. На ФИГ.36А показаны предварительные данные вестерн-блоттинга, подтверждающие уровень экспрессии субъединиц R1 и R2 в скелетной мышце, легком и сердце у AAV6R1R2cTNT455-трансфицирвоанных (4,5 е13) мышей и контрольных мышей. На Фиг.36 также представлены данные для сердечной ткани, полученной от мышей, которым не проводили инъекцию (В) по сравнению с мышами, которым инъецировали AAV6-щелочную фосфатазу (темно-красный; С)36 через 20 месяцев, предполагающие, что AAV6-R1R2cTnT455 должен обеспечивать стабильную длительную гиперэкспрессию R1R2.
[0056] На ФИГ.37 показан эффект геномов 1,5 е13, 4,5 е13, 1,35 е14 вектора AAV6-R1R2cTnT455 или физиологического раствора (контроль), инъецированных системно в приблизительно 10-кратном диапазоне 3-месячным мышам (n=6 на группу), на функцию ЛЖ.
[0057] На ФИГ.38 показано изменение фракции укорочения у крыс, которым инъецировали AAV6-R1R2 непосредственно в сердце на пятый день после инфаркта, измеренное с помощью эхокардиографии, по сравнению с необработанными страдающими инфарктом крысами и неподверженными лечению ложно-оперированными крысами.
[0058] На ФИГ.39 показаны измерения работающего сердца по Нили (Neely) in vitro для крысиных сердец, описанных на ФИГ.38. Сила на оси у приведена в единицах г⋅см/мин. Наблюдали потерю чувствительности к предварительной нагрузке у сердец (с сердечной недостаточностью), которые были поражены инфарктом (необработанных), и восстановление чувствительности к предварительной нагрузке у пораженных инфарктом сердец, получающих векторы, до уровня контрольных не пораженных инфарктом сердец, что демонстрировало, таким образом восстановление сердечной функции.
[0059] На ФИГ.40 Представлена последовательность нуклеиновой кислоты для кардиоспецифичного промотора cTnT455, используемого согласно типичным вариантам реализации настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
I. ВВЕДЕНИЕ
[0060] Многие современные фармацевтические подходы для лечения сердечной недостаточности направлены на изменение метаболизма внутриклеточного [Ca2+] ([Ca2+]i), которое может иметь значительные побочные эффекты, такие как аритмогенез или неблагоприятное влияние на диастолическую функцию. Согласно одному аспекту настоящего изобретения предложен способ, непосредственно направленный на тонкие филаменты сердца для усиления сокращения интактных кардиомиоцитов независимо от [Ca2+]i. В частности, в настоящей заявке показано, что активация тонких филаментов сердца усиливалась в результате опосредованной аденовирусом гиперэкспрессии варианта сердечного тропонина С (cTnC), сконструированного таким образом, что он обладал повышенной аффинностью связывания Ca2+, которая обеспечивалась одной аминокислотной заменой (L48Q). Мы (а также другие авторы) показали, что в очищенных сердечных трабекулах и миофибриллах замена нативного cTnC на L48Q cTnC приводит к повышению чувствительности к Ca2+ силы сокращения и максимальной скорости развития силы сокращения. Таким образом, согласно одному варианту реализации изобретения предложен способ усиления сокращения интактных кардиомиоцитов с помощью прямой направленности на тонкие филаменты сердца.
[0061] Согласно одному аспекту настоящего изобретения предложен новый терапевтический подход, который объединяет клеточную и генную терапию и направлен на улучшение функционирования сердца, например, у пациентов, страдающих сердечной недостаточностью. За исключением трансплантации органа, существующие терапевтические способы лечения сердечной недостаточности нацелены либо на угасание неблагоприятного ремоделирования желудочка, либо на усиление генерации сократительной силы оставшимися кардиомиоцитами, обычно путем фармакологических воздействий на внутриклеточную кальциевую передачу сигнала. Первый из указанных подходов, как правило, помогает пациентам только на ранних стадиях процесса заболевания, тогда как последний подход осложняется побочными эффектами (например, усугублением аритмии). В последние годы сформировался значительный интерес к трансплантации клеток как к альтернативному способу достижения репарации сердца, но попытки применения указанного способа до сих пор были ограничены низким потенциалом к дифференцировке в сердечном направлении используемых клеток и/или относительно маленьким количеством образующихся трансплантатов миокарда. Кардиомиоциты, полученные из плюрипотентных эмбриональных стволовых клеток человека (ЭСК) или связанных с ними индуцированных плюрипотентных стволовых клеток (иПСК), показали многообещающие результаты в доклинических исследованиях. Они образуют трансплантаты миокарда человека, которые по меньшей мере частично встраиваются в мышцу хозяина. Однако и в этом случае образовавшиеся трансплантаты обычно являются достаточно маленькими и, по-видимому, не обеспечивают достаточное количество генерирующих сократительную силу единиц для значительного улучшения сердечного выброса.
[0062] Согласно одному варианту реализации настоящего изобретения предложен новый подход к лечению сердечной недостаточности, позволяющий преодолеть многие из указанных недостатков. Как показано в настоящей заявке, повышенный систолический уровень 2-дезокси-АТР (дезокси-АТФ) повышает связывание поперечных мостиков и кинетику их циклической активности, что приводит к значительному повышению сократительных свойств без воздействия на внутриклеточный кальций. Концентрация дезокси-АТФ может быть повышена в поврежденных кардиомиоцитах путем принужденной гиперэкспрессии рибонуклеотидредуктазы (RR)-фермента, лимитирующего скорость ее продукции, - с использованием геннотерапевтических подходов. Как показано в настоящей заявке, перенос, ограниченный малым участком стенки левого желудочка (ЛЖ) (посредством прямой инъекции вирусного вектора), приводит к существенному повышению функции ЛЖ.
[0063] Согласно другому варианту реализации настоящего изобретения поскольку дезокси-АТФ легко проходит через щелевые контакты, и для усиления силы сокращения требуются ее низкие концентрации (≤1% клеточного пула адениннуклеотида), предложен способ доставки дезокси-АТФ в поврежденный миокард путем трансплантации второго типа клеток, который в результате генной модификации гиперэкспрессирует RR и который способен формировать щелевые контакты с миокардом хозяина, являющимся мишенью. Благодаря чувствительности к такой генетической модификации, чрезвычайно высокой способности к размножению и способности образовывать стабильные внутрисердечные имплантаты, в которых экспрессируются соответствующие изоформы коннексина, кардиомиоциты, произошедшие из человеческих ЭСК или иПСК, являются идеальными донорами дезокси-АТФ. Например, согласно одному варианту реализации изобретения кожные фибробласты получают от пациента, страдающего сердечной недостаточностью, перепрограммируют в иПСК и затем модифицируют путем встраивания конструкции, в которой кардиоспецифичный промотор запускает экспрессию RR. Согласно одному варианту реализации изобретения в указанном способе применяют трансгенез, опосредуемый цинк-пальцевой нуклеазой (которая дает возможность нацеливания на хорошо изученный, «безопасный» локус в геноме) для встраивания конструкции. После скрининга и размножения подходящим образом направленных клонов иПСК, указанные клетки дифференцируют в кардиомиоциты и затем имплантируют (например, посредством использования катетера) в сердце, страдающее сердечной недостаточностью. Преимуществом является то, что указанная стратегия не требует формирования большого сердечного трансплантата и не требует пересадки трансплантата в неблагоприятную область инфарктного рубца. Поскольку задачей трансплантата является доставка дезокси-АТФ, а не генерация силы сокращения, то вместо этого может быть достаточной имплантация маленького трансплантата в хорошо васкуляризированный удаленный участок миокарда. Указанный способ представляет собой один из многих возможных подходов и никоим образом не ограничивает объем настоящего изобретения. Например, согласно некоторым вариантам реализации изобретения в способах, предложенных в настоящей заявке, можно использовать другие типы донорских клеток (например, мезенхимальные стволовые клетки). Подобным образом, согласно некоторым вариантам реализации изобретения, можно использовать другие способы трансгенеза (например, доставку транспозона, плазмиды или вируса).
[0064] Согласно другому аспекту настоящего изобретения предложены аденовирусные векторы, экспрессирующие промотор цитомегаловируса (CMV), запускающий Rrm1 или Rrm2. Согласно одному варианту реализации изобретения векторы также кодируют зеленый флуоресцентный белок (GFP), используемый в качестве репортера трансдукции. Культивируемые кардиомиоциты взрослых крыс трансдуцировали указанными векторами, и за повышением и снижением скорости и степени сокращения и расслабления миоцитов и Ca2+ током (Fura2-флуоресценция) наблюдали с помощью видеомикроскопии после инкубации в присутствии вируса в течение 48 часов. В настоящей заявке показано, что указанная обработка приводит к значительному повышению внутриклеточного уровня дезокси-АТФ, скорости и степени укорочения и скорости расслабления с минимальными эффектами на Ca2+-токи при стимуляции 0,5 Гц, 1 Гц и 2 Гц. Таким образом, согласно одному варианту реализации настоящего изобретения предложен способ усиления сократимости сердца без нарушения диастолической функции путем изменения сердечного уровня внутриклеточного RR и/или пула дезокси-АТФ, который может значительно изменять цикл активности актомиозиновых поперечных мостиков.
[0065] Согласно другому аспекту настоящего изобретения впервые предложено применение манипуляций с нуклеотидами для улучшения сократимости in vivo. Применение дезокси-АТФ как субстрата для сокращения было впервые изучено д-ром Ренье (Dr. Regnier) более 15 лет назад как способ для исследования хемомеханической трансдукции миофиламентов. Однако применение дезокси-АТФ для улучшения функции кардиомиоцитов обеспечивает потенциальную возможность разработка большого количества новых путей исследования, включая, но не ограничиваясь указанными, влияние на взаимодействия с Ca2+, метаболические пути и облегчение заболевания сердца. Также предложен способ направленной доставки, который является минимально инвазивным и может быть масштабирован для крупных животных моделей и человека.
[0066] Первоначальные исследования влияния дезокси-АТФ проводили на скелетной мышце с разрушенной мембраной, и в них наблюдали значительное повышение чувствительности к Ca2+, но максимальная Ca2+-активируемая сила сокращения не изменялась. Последующие исследования на сердечной мышце показали более значительный результат: дезокси-АТФ повышала не только Ca2+-чувствительность силы сокращения, но также повышала величину максимальной Ca2+-активируемой силы на ~40%. Указанные результаты указывали на то, что регуляция сокращения сердечной мышцы более чувствительна к изменениям в актомиозиновой активности, что обеспечивает преимущество кардиоспецифичного направленного на миофиламенты действия. Соответственно, мы искали возможность стимулировать продукцию дезокси-АТФ в кардиомиоцитах. Задача исследований на интактных клетках и исследований in situ заключалась в повышении внутриклеточного уровня [дезокси-АТФ], достаточном для обеспечения повышенной сократимости. Указанную задачу решили с помощью гиперэкспрессии природного фермента (рибонуклеотидредуктазы; R1R2), который превращает АДФ в дезокси-АДФ, которая затем быстро фосфорилируется фосфокреатинкиназой до дезокси-АТФ. Шоффсталль и др. (Schoffstall et al., 2006) показали, что всего 10% [дезокси-АТФ] (90% [АТФ]) обеспечивало повышение сократимости сердечной ткани с разрушенной мембраной и изолированных сократительных белков23*, что дает основания полагать, что незначительные уровни дезокси-АТФ могли бы приводить к повышению сократимости в интактных кардиомиоцитах. В настоящей заявке показано, что неожиданно было обнаружено, что для стимуляции функционирования кардиомиоцитов и сердца требуется значительно меньший уровень дезокси-АТФ.
II. ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ
А. Экспрессия L48Q cTnC увеличивает сократимость сердца
[0067] С использованием видеомикроскопии для наблюдения за длиной клеток (CL) и Ca2+-токами (Fura2) было показано, что экспрессия L48Q cTnC (идентифицированная по ко-экспрессии с GFP) приводит к значительному повышению скорости и степени укорочения (на 33% и 48%, соответственно) без изменения Са - тока по сравнению с нетрансдуцированными кардиомиоцитами взрослых крыс. Указанные различия были еще более существенными по сравнению с трансдуцированными cTnC дикого типа кардиомиоциты. Важно отметить, что присутствие L48Q cTnC не влияло на скорость расслабления по сравнению со всеми другими группами. Экспрессию и встраивание L48Q cTnC в тонкие филаменты подтверждали с помощью Вестерн-блоттинга миофибрилл из трансдуцированных кардиомиоцитов, который показал стехиометрическое замещение ~58% нативного cTnC на L48Q cTnC.
[0068] Указанные эксперименты демонстрируют возможность непосредственного нацеливания на белки тонких филаментов сердца для усиления его сократимости без нарушения расслабления.
[0069] Одной из задач настоящего изобретения было определить, можно ли обеспечить экспрессию L48Q cTnC в интактных кардиомиоцитах и встраивание в миофиламенты для повышения сократимости без отрицательного влияния на расслабление кариомиоцитов или свойства тока. Как показано в Примерах 1-6, гиперэкспрессия L48Q cTnC приводила к замещению 58±7% нативного cTnC на L48Q cTnC, и это сильно повышало степень и скорость укорочения и скорость расслабления миоцитов, при этом не оказывая вдимого влияния на свойства Ca2+ тока. Самый низкий наблюдаемый уровень замещения составил 42%, а самый высокий уровень замещения составил 70%, значительных изменений в функции между указанными уровнями встраивания не выявлено.
[0070] Предварительные эксперименты с использованием очищенных сердечных трабекул показали, что пассивная замена нативного cTnC на L48Q cTnC повышает величину и скорость развития силы и укорочения при всех уровнях активации Ca2+, исключая насыщающую концентрацию [Ca2+] (рСа 4,0), но эти исследования проводили при 100% замещении нативного cTnC в препаратах мышц. В настоящем исследовании с использованием интактных кардиомиоцитов мы не ожидали, что экспрессия L48Q cTnC приведет к 100% замещению нативного cTnC. Однако мы действительно ожидали, что повышение уровня L48Q cTnC будет необходимо для достижения замещения ~15-25% нативного cTnC для повышения функции, поскольку ранее мы определили, что указанное отношение TnC (M80Q sTnCF27W) необходимо для значительного наблюдаемого повышения Ca2+-чувствительности силы в мышечных волокнах поясничной мышцы кролика. Тот факт, что указанный минимум был существенно превышен, был неожиданным и интересным, поскольку он указывает на то, что количество вируса, необходимое для достижения значительно повышения функции, может быть меньше количества, которое применялось в настоящем исследовании. По-видимому, менее устойчивый перенос генов с использованием различных вирусных конструкций, таких как лентивирус или AAV6, или более низкая множественность инфицирования также могли бы обеспечить значительное функциональное улучшение. Это могло бы помочь избежать вероятности возникновения отрицательных побочных эффектов, таких как воспаление ткани. Дальнейшие исследования следует сосредоточить на поиске минимального количества L48Q cTnC, необходимого для обеспечения значительного функционального улучшения, и нагрузки вирусных частиц, необходимой для удовлетворения этому минимальному количествту. Кроме того, было показано, что другие мутанты cTnC изменяют аффинность связывания Ca2+ и повышают Ca2+-чувствительность силы сокращения и скорость развития силы сокращения при субмаксимальной концентрации [Ca2+]17 и, таким образом, также могут иметь терапевтический потенциал для лечения различных патологических состояний.
[0071] Потенциальная возможность использования L48Q cTnC как терапевтического инструмента вдохновляет, поскольку не наблюдалось значительного влияния на характер Ca2+-тока (или авторитмику), в частности, принимая во внимание то, что другие исследования показали, что Ca2+-сенсибилизация миофиламентов путем активации белков18 или фармакологическими19,20 способами приводила к изменению характера потенциала действия и повышенному риску аритмогенеза. Указанные данные были неожиданными, учитывая малые различия (достоверные при 1 Гц) в минимальной и максимальной концентрации Ca2+ у cTnC ДТ (дикого типа)+GFP трансдуцированных кардиомиоцитах. В настоящем исследовании не было проведено специальной оценки того, обусловлен ли указанный эффект cTnC ДТ или GFP, но результаты других исследований показали, что трансфекция кардиомиоцитов взрослых крыс только белком GFP-значительно повышает минимальный и максимальный уровни Ca2+, особенно при более высоких частотах стимуляции21. Таким образом, указанный эффект, который частично устраняется гиперэкспрессией cTnC, может быть, в первую очередь, обусловлен GFP. В любом случае экспрессия L48Q cTnC давала минимальный и максимальный уровни [Ca2+], которые не отличались от нетрансдуцированных кардиомиоцитов. Таким образом, эффект экспрессии L48Q cTnC вероятно недооценивают. В таком случае экспрессия L48Q cTnC в отсутствие GFP имела бы тенденцию 1) снижать минимальную концентрацию [Ca2+], приводя к усилению расслабления, и 2) снижать максимальную концентрацию [Ca2+]. Это повышало бы показатель эффективности сокращения путем большего укорочения для данного количества высвобожденного Са. Примечательно, что Лим и др. (Lim et al.) показали, что кардиомиоциты взрослых крыс, гиперэкспрессирующие мутант TnC (E59D, D75Y), связанный с идиопатической дилатационной кардиомиопатией, заметно снижали сократимость, при этом не оказывая влияния на гомеостаз внутриклеточного Ca2+ 22, демонстрируя возможность управлять аффинностью связывания Ca2+ тонких филаментов без влияния на характер Ca2+-тока.
[0072] Очевидное преимущество применения варианта L48Q cTnC заключается в том, что при улучшении времени сокращение не наблюдалось влияния на расслабление миоцитов, которое даже усиливалось при более высоких частотах стимуляции. Было показано, что повышенная аффинность связывания Ca2+ приводила к продолжительной активации тонких филаментов и, соответственно, более длительному связыванию поперечными мостиками, которое мешало расслаблению23. Пролонгирование систолы и замедление диастолического расслабления могло иметь серьезные функциональные последствия in vivo, приводящие к снижению диастолического наполнения и впоследствии сердечного выброса. Например, мы описали, что скорость расслабления сердечных миофибрилл слабо повышалась после ~100% замены на L48Q cTnC (по сравнению с заменой на cTnC ДТ). Это было связано со незначительным продлением медленной фазы, предположительно зависящим от разъединения поперечных мостиков9. Однако описанные исследования были проведены в изометрических условиях, тогда как в настоящем исследовании интактным миоцитам позволяли свободно укорачиваться. В указанных условиях на скорость расслабления также влияет восстанавливающая сила, обеспечиваемая титаном, которая повышается линейно по мере укорочения саркомера24. Скорости расслабления, наблюдаемые в настоящем исследовании, примерно коррелируют с относительным укорочением клетки. По-видимому, это также указывает на то, что L48Q cTnC не пролонгирует связывание поперечными мостиками, поскольку скорости расслабления в случае L48Q cTnC были больше, чего следовало бы ожидать при повышенном укорочении. Также возможно, что 42-70% замещение эндогенного cTnC на L48Q cTnC было достаточным для улучшения укорочения без влияния на расслабление, тогда как большее замещение может начинать оказывать неблагоприятное влияние на расслабление. Кроме того, любое влияние L48Q cTnC на расслабление может быть снижено или устранено при субмаксимальном уровне [Ca2+], который встречается в инфарктном миокарде, и эффекты могут также отличаться в условиях нагрузки (растяжение), таких как условия, встречаемые in vivo.
[0073] В заключение, указанные эксперименты демонстрируют возможность непосредственного нацеливания на сердечные белки тонких филаментов для усиления сократимости сердца без изменения диастолической функции или характера Ca2+-тока. Согласно одному аспекту описанный выше подход можно использовать для лечения кардиомиоцитов из миокарда с функциональной недостаточностью или заболеванием.
В. Экспрессия экзогенного комплекса RR усиливает сократимость сердца
[0074] В настоящей заявке показано, что инъекции в хвостовую вену или непосредственно в сердце мыши аденовирусного вектора (AV-R1R2), приводящего к повышению [дезокси-АТФ] путем гиперэкспрессии R1 и R2, значительно повышают (на 40-50%) относительное укорочение и фракцию выброса левого желудочка (ЛЖ) (Фигура 27) в пределах 3-4 дней. Примечательно, что эхокардиография, проведенная у 8-месячных трансгенных мышей, гиперэкспрессирующих R1R2, показала аналогичное улучшение функции ЛЖ по сравнению с однопометными животными дикого типа (WT) (Фигура 27), не страдающими видимой сердечной патологией, что дает основания говорить о переносимости настоящего подхода. Указанное улучшение, кроме того, подтверждалась результатами предварительных гемодинамических экспериментов (Millar) и экспериментов на перфузируемыых сердцах по Лангердорффу. Важно отметить, что предварительные ЯМР-измерения указывают на то, что улучшение функции происходит не за счет энергетических резервов, даже при стимуляции [Ca2+] в высокой концентрации или стимуляции добутамином. Наконец, дезокси-АТФ не изменяла сокращение мышиных гладких мышц аорты (по сравнению с АТФ; (Фигура 30)), что дает основания полагать, что изменения сосудистого тонуса, вероятно, не являются причиной каких-либо возможных изменений в гемодинамических параметрах.
[0075] За длиной клеток и логометрической Ca2+-флуоресценцией (Fura2) наблюдали методом видеомикроскопии. При стимуляции на 0,5 Гц в кардиомиоцитах, гиперэкспрессирующих Rrm1+Rrm2, степень укорочения повышалась на ~40%, и максимальная скорость укорочения повышалась на ~80% по сравнению с необработанными кардиомиоцитами. Максимальная скорость расслабления также повышалась на -150% у клеток с гиперэкспрессией Rrm1+Rrm2 (+GFP), что приводило к снижению времени до достижения 50% расслабления по сравнению с необработанными кардиомиоцитами. Указанные различия были даже более значительными по сравнению с трансдуцированными только GFP кардиомиоцитами. Примечательно, что гиперэкспрессия Rrm1+Rrm2 не оказывала влияния на минимальную или максимальную внутриклеточную концентрацию [Ca2+] (Fura2-флуоресценция), указывая на то, что повышенная сократимость, в первую очередь, вызвана повышенной активностью миофиламентов без изменения высвобождения Ca2+ из саркоплазматического ретикулума. Кроме того, улучшение функции поддерживалось при гиперэкспрессии Rrm1+Rrm2 при повышении частоты стимуляции (1 Гц и 2 Гц). ВЭЖХ-анализ показал, что внутриклеточный уровень [дезокси-АТФ] был повышен примерно в 10 раз после трансфекции, и был несколько выше 1% пула адениннуклеотидов.
[0076] Указанные эксперименты демонстрируют возможность непосредственного нацеливания на актомиозиновые поперечные мостики для усиления сократимости и расслабления сердца без влияния на минимальный или максимальный уровень Ca2+.
[0077] Одной из задач настоящего исследования было определить, вызывает ли гиперэкспрессия рибонуклеотидредуктазы (Rrm1+Rrm2) повышение внутриклеточного уровня [дезокси-АТФ], в свою очередь, повышение сократимости в интактных кардиомиоцитах без негативного влияния на расслабление указанных кариомиоцитов. Гиперэкспрессия Rrm1+Rrm2 приводила к повышению внутриклеточного уровня [дезокси-АТФ] до ~1,0-1,5% от общего пула адениннуклеотидов, что значительно повышало степень и скорость укорочения и скорость расслабления миоцитов, тогда как не оказывало очевидного эффекта на характер Ca2+-тока.
[0078] Более ранние эксперименты с использованием очищенных сердечных трабекул показали, что дезокси-АТФ повышала изометрическую силу и скорость развития силы и укорочения при всех уровнях активации Ca2+, включая концентрацию насыщения [Ca2+] (рСа 4,0), но указанные исследования проводили при 100% замещении 5 мМ АТФ на 5 мМ дезокси-АТФ в промывочных расторах7,8,20. В настоящем исследовании на интактных кардиомиоцитах мы не ожидали, что гиперэкспрессия Rrm1+Rrm2 приведет к высоким (мМ) уровням дезокси-АТФ. Однако мы предполагали необходимость повышения уровня дезокси-АТФ для достижения ~10% [АТФ] для повышения функции, поскольку было известно отношение дезокси-АТФ к АТФ, необходимое для наблюдения функционального изменения в миокарде с разрушенной мембраной21. Фактически предыдущие эксперименты на куриных эмбриональных кардиомиоцитах указывали на то, что для достижения значительного усиления сокращения может требоваться замещение ~1/3 АТФ на дезокси-АТФ22. Неожиданно, наблюдаемые большие повышения сократимости происходили при относительно малом повышении [дезокси-АТФ] в кардиомиоцитах. Такие даные указывают на то, что количество вируса, необходимое для достижения значительного повышения функции, может быть меньше количества, которое использовали в настоящем исследовании. Преимуществом укзанных данных является то, что меньшая гиперэкспрессиия Rrm1+Rrm2 может снижать вероятность отрицательных побочных эффектов22,23. Возможно, что присутсвовала небольшая популяция контаминирующих клеткок (например, фибробластов), которые не так хорошо трансдуцировались, или в которых гиперэкспрессия Rrm1+Rrm2 была ниже, что могло бы привести к заниженной оценке уровня [дезокси-АТФ] в кардиомиоцитах по результатам ВЭЖХ-анализа. Однако, принимая во внимание незначительное относительное количество не кардиомиоцитарных клеток в культуре, описанный искажающий оценку эффект должен был быть минимальным.
[0079] Интересно рассуждать, без какого-либо теоретического ограничения, как относительно малое количество общей дезокси-АТФ может оказывать такое выраженное действие на функцию кардиомиоцитов. Оценки эффективности сокращения (Фигура 9) указывают на то, что повышенная сократимость в трансдуцированных Rrm1+Rrm2 миоцитах, в первую очередь, связана с миофиламентами, и, соответственно, вероятно, что эффект дезокси-АТФ, в первую очередь, обусловлен улучшением циклической активности поперечных мостиков. Такие данные согласуются с экспериментом, в котором более быстрый (альфа) миозин экспрессировался в кардиомиоцитах, в которых в норме экспрессируется более медленный (бета) миозин, что приводило к повышению функции при отсутствии влияния на амплитуду Ca2+-тока. Механизм, лежащий в основе повышения активности миофиламентов при повышенном уровне [дезокси-АТФ], которое наблюдалось в настоящем исследовании, неясен, поскольку концентрация [дезокси-АТФ] составляли всего только ~1% по отношению с АТФ.
[0080] В исследованиях на скелетном миозине авторы показали, что равновесие расщепления γ-фосфата по миозину близко для АТФ и дезокси-АТФ, но после гидролиза связывание поперечными мостиками и скорость разъединения поперечных мостиков выше для дезокси-АТФ24. Это может объяснить повышение Ca2+-чувствительности развития растяжения и более высокую скорость развития растяжения и скорость укорочения в очищенной скелетной мышце24-26. Несмотря на то, что мы не проводили подробный химико-механический анализ с использованием дезокси-АТФ в сердечной мышце, мы показали, что она повышает максимальное связывание поперечными мостиками (как следует из измерений жесткости) и изометрическую силу на >40% в дополнение к повышению ktr и скорости укорочения в отсутствии нагрузки20. Мы также показали, что дезокси-АТФ значительно повышает изометрическую силу и ktr в сердечной мышце при всех уровнях Ca2+, независимо от того, экспрессирует сердечная мышца с разрушенной мембраной, в первую очередь, α- или β-тяжелую цепь миозина8. Указанные данные существенны, так как в отличие от скелетной мышцы, в мышце сердца во время сокращения достигается уровень внутриклеточного [Ca2+], который находится примерно в пределах диапазона полумакимальной активации. В случае настоящих экспериментов с использованием культивируемых кардиомиоцитов возможно, что незначительного повышения связывания головок миозина S1, содержащих дезокси-АДФ. Pi, было достаточно для кооперативного повышения активации тонких филаментов, приводящей к повышению величины и скорости укорочения.
[0081] Не наблюдалось неблагоприятного эффекта на расслабление при гиперэкспрессии Rrm1+Rrm2 (и последующим повышении [дезокси-АТФ]); фактически расслабление миоцитов повышалось. Возможно, что это являлось результатом по меньшей мере отчасти, более быстрого угасания Ca2+-тока. Дезокси-АТФ могла бы использоваться другими АТФазами (кроме миозина), такими как саркоплазматическая Ca2+-АТРаза (SERCA), Ca2+-АТФаза плазматической мембраны (РМСА) и натрий/кальциевый обменник (NCX). Повышение активности SERCA могло бы объяснить повышение угасания скорости Ca2+-тока, в частности при стимуляции 0,5 и 1,0 Гц. Однако повышенная активность SERCA, как известно, повышает Ca2+-запасы СПР27, обеспечивая большее количество доступного для высвобождения Ca2+ во время активации, чего не наблюдалось в Rrm1+Rrm2-трансдуцированных кардиомиоцитах (Фигура 6d). Кроме того, повышенная активность РМСА и NCX должна была бы привести к ослаблению Ca2+-тока с течением времени в результате выкачивания Ca2+ из клетки. Так как ~95% активированного Ca2+ высвобождается из СПР в кардиомиоцитах крыс28,29, выкачивание Ca2+ из клетки привело бы прогрессирующему снижению амплитуды Ca2+-тока и сокращения, чего не наблюдалось в указанных экспериментах. Однако конкретный механизм, лежащий в основе повышения скорости угасания Ca2+-тока, требует дальнейших исследований.
[0082] Поскольку дезокси-АТФ повышает скорость разъединения поперечных мостиков20,25, это может также объяснить более высокую скорость расслабления в настоящих экспериментах на культивируемых кардиомиоцитах. Несмотря на то, что специфические механизмы, регулирующие расслабление в интактной сердечной мышце, не известны, было показано, что ранняя фаза расслабления в сердечных и скелетных миофибриллах регулируется скоростью диссоциации поперечных мостиков30-33. Также с обнаруженными фактами согласуется то, что Rrm1+Rrm2 повышали сократимость кардиомиоцитов, так как скорость укорочения в клетках в отсутствие нагрузки (как в случае культуры), в первую очередь, определяется скоростями разъединения поперечных мостиков34-35. Также возможно, что повышенная скорость разъединения поперечных мостиков в случае дезокси-АТФ ускоряет кооперативную инактивацию тонких филаментов в результате более быстрого уменьшения популяции связанных поперечных мостиков по мере снижения Ca2+-связывания тонкими филаментами во время расслабления.
[0083] Таким образом, согласно одному аспекту настоящего изобретения предложены способы, согласно которым дезокси-АТФ может обеспечивать двойное преимущество - положительную инотропии и лузитропии - с незначительным изменением характера Ca2+-тока, таким образом, представляющие собой потенциальную альтернативу доступным на сегодняшний день фармацевтическим терапевтическим подходам.
[0084] Перед началом описания настоящего изобретения также необходимо учесть, что изобретение не ограничивается конкретными вариантами реализации, описанными ниже, так как могут быть реализованы модификации конкретных вариантов реализации изобретения в пределах прилагаемой формулы изобретения. Также необходимо понимать, что используемая терминология применяется для описания конкретных вариантов реализации изобретения и не ограничивает настоящее изобретение. Объем настоящего изобретения определяется прилагаемой формулой изобретения. В настоящей заявке и прилагаемой формуле изобретения упоминание предмета в единственном числе также включает указанный предмет во множественном числе, если иное не следует очевидно из контекста.
[0085] Необходимо понимать, что если приведен диапазон значений, то каждое промежуточное значение, до десятых долей значения нижнего предела, если иное очевидно не следует из контекста, между верхним и нижним пределом указанного диапазона и любое другое указанное или промежуточное значение в указанном установленном диапазоне включено в пределы изобретения. Верхний и нижний пределы указанных более маленьких диапазонов могут независимо быть включены в указанные более маленькие диапазоны и также включены в пределы изобретения, за исключением любого специально исключенного предела в установленном диапазоне. Если установленный диапазон включает один или оба предела, то указанные диапазоны, исключающие любой или оба из указанных включенных пределов, также входят в объем изобретения.
[0086] Если не указано иное, все технические и научные термины, используемые в настоящей заявке, имеют такое же значение, как общепринятое значение для специалистов в области техники, к которой относится настоящее изобретение. Несмотря на то что при практическом осуществлении изобретения или в ходе проверки изобретения можно использовать любые способы, устройства и материалы, подобные или эквивалентные способам, устройствам и материалам, описанным в настоящей заявке, предпочтительными являются способы, устройства и материалы, описаны далее.
III. АББРЕВИАТУРЫ
[0087] cTnC - сердечный тропонин С
[0088] cTnT - сердечный тропонин Т
[0089] koff - скорость диссоциации кальция
[0090] NRC - кардиомиоцит новорожденных крыс
[0091] ARC - кардиомиоцит взрослых крыс
[0092] GFP - зеленый флуоресцентный белок
[0093] RT50, RT90 - время до достижения 50% и 90%) расслабления
[0094] DT50, DT90 - время до достижения 50%) и 90%) снижения Ca2+
[0095] WT - дикого типа
[0096] Rrm1 или R1 - мышечная рибонуклеотидредуктаза 1
[0097] Rrm2 или R1 - мышечная рибонуклеотидредуктаза 2
IV. ОПРЕДЕЛЕНИЯ
[0098] «Сердечная функция» в контексте настоящего изобретения относится к функции сердца, которая отражается в одном или более измеряемых параметрах, например, сокращении миокарда, изменении относительного укорочения, максимальной скорости укорочения, расслабления миокарда, максимальной скорость расслабления миокарда, времени расслабления, эффектах на Ca2+-токи, частоте сердечных сокращений, конечном систолическом давлении, конечном диастолическом давлении, конечном систолическом объеме, конечном диастолическом объеме, сердечном выбросе, систолической работе, ударном объеме сердца, сердечном индексе и т.д. Указанные параметры можно определить с помощью гемодинамических и/или эхокардиографических измерений и/или любых других методов, известных специалистам в данной области техники. Наличие улучшения сердечной функции определяется для каждого конкретного субъекта. Например, для субъекта, нуждающегося в положительном инотропном эффекте, улучшение сократимости миокарда означает повышение в сокращении миокарда. В альтернативном варианте для субъекта, нуждающегося в положительном лузитропном эффекте, улучшение расслабления миокарда означает повышение расслабления миокарда, которое отражается, например, в повышении скорости расслабления.
[0099] Термин «сократимость миокарда» в настоящей заявке используется взаимозаменяемо с термином «инотропия» и относится к силе сокращения желудочка, во время которого кровь выталкивается из сердца. Улучшение сократимости миокарда определяют для каждого конкретного субъекта с использованием одного или более измеримых параметров инотропии. Для субъекта или пациента, нуждающегося в положительном инотропном эффекте, улучшение сократимости миокарда подразумевает повышение сократимости миокарда по измерениям с использованием эхокардиографии. Для субъекта или пациента, нуждающегося в отрицательном инотропном эффекте, улучшение сократимости миокарда подразумевает снижение сократимости миокарда. Примеры измеримых параметров инотропии включают OP/Ot, процентное утолщение, процентное укорочение, относительное укорочение и фракцию выброса.
[0100] Термин «расслабление миокарда», используемый в настоящей заявке взаимозаменяемо с термином «лузитропия», относится к способности сердца расслабляться после электромеханического сопряжения. Улучшение расслабления миокарда определяется для каждого конкретного субъекта с использованием одного или более измеримых параметров лузитропии. Для субъекта или пациента, нуждающегося в положительном лузитропном эффекте, улучшение расслабления миокарда подразумевает повышение скорости расслабления. Для субъекта или пациента, нуждающегося в отрицательном лузитропном эффекте, улучшение сокращения миокарда подразумевает снижение скорости расслабления. Примеры измеримых параметров лузитропии включают скорость снижения давления (-dP/dtmin) во время диастолы, определяемую на основе измерений сенсора давления, скорость снижения силы/растяжения (-dF/dt), определяемая на основе измерений сенсора силы, и время изоволюметрического расслабления (IVRT), определяемое на основе измерений сопротивления сердца или на основе выявляемых звуков в сердце. Например, измерительное устройство может быть запрограммировано на сравнение параметра лузитропии с конкретным порогом для определения нарушения диастолического расслабления и управлять схемой нейронной стимуляции для доставки симпатической стимуляции к сердцу в ответ на нее.
[0101] «Трансплантация» в настоящей заявке относится к помещению клеток в организм субъекта. Клетки могут быть аутологическими (т.е., полученными от субъекта, которого предполагается лечить), изогенными (т.е., полученными от генетически идентичного, но другого субъекта, например, однояйцевого близнеца), аллогенными (т.е., полученными от генетически не идентичного представителя того же вида) и/или ксеногенными (т.е., полученными от представителя другого вида). Клетки могут быть получены от донора (живого донора или трупного материала) или происходить из постоянной клеточной линии. Для получения клеток от донора (например, для потенциального реципиента трансплантата клеточного биокаркаса), могут использоваться стандартные методы биопсии, известные в данной области техники. Типичные методы описаны, например, в патенте США №6536567.
[0102] Термины «терапия», «лечение» и «облегчение» относятся к любому снижению тяжести симптомов или количества амилоидных агрегатов или улучшению когнитивной функции. В настоящей заявке, термины «лечить» и «предотвращать» не являются абсолютными терминами. Лечение может относиться к любому из следующих эффектов: задержка возникновения симптомов, облегчение симптомов, улучшение выживаемости пациента, повышение времени или скорости выживаемости и т.д. Эффект лечения может сравниваться с субъектом или группой субъектов, не получающих лечение.
[0103] Под термином «кардиомиоцит» подразумевается сократимая клетка сердца, которая представляет собой клетку сердечной мышцы. Кардиомиоцит может быть выделен и помещен в культуру in vitro или может являться частью миокарда хозяина.
[0104] Термин «эмбриональные стволовые клетки» (ЭСК) относится к клеткам, произошедшим из внутренней клеточной массы бластоцист или морул, которые прошли несколько пассажей как клеточные линии. ЭСК могут быть получены путем оплодотворения яйцеклетки сперматозиодом или ДНК, переноса ядра, партеногенеза или с помощью способов, приводящих к получению чЭСК, гомозиготностным по участку ГКГС. Термин «эмбриональные стволовые клетки человека (чЭСК)» относится к клеткам, произошедшим из внутренней клеточной массы бластоцисты или морулы человека, которые прошли несколько пассажей как клеточные линии. чЭСК могут быть получены путем оплодотворения яйцеклетки спрематозиодом или ДНК, переноса ядра, партеногенеза или с помощью способов, приводящих к получению чЭСК, гомозготным в участке HLA.
[0105] Термин «индуцированные плюрипотентные стволовые клетки» или «иПСК» в настоящей заявке относится к плюрипотентной стволовой клетке, произошедшей из постнатальной соматической клетки в результате форсированной экспрессии любой комбинации перепрограммирующих факторов отдельно или в комбинации с одним или более перепрограммирующими агентами.
[0106] Термин «мезенхимальная стволовая клетка» в настоящей заявке относится к клетке, способной давать начало клеткам, дифференцированным во многих мезенхимальных направлениях, в частности, остеобластам, адипоцитам, миобластам и хондроцитам. В целом, мезенхимальные стволовые клетки также обладают одним или более из следующих свойств: способностью претерпевать асинхронную или симметричную репликацию, когда две дочерние клетки после деления могут иметь различные фенотипы; неограниченной способностью к самообновлению и клональной регенерации ткани, в которой они существуют, например, не-гематопоэтических клеток костного мозга.
[0107] Термины «пациент», «хозяин» и «субъект» используются в настоящей заявке взаимозаменяемо и относятся к любому субъекту, представляющему собой млекопитающее, которому необходимы диагностика или терапия, например, к виду приматов, такому как человек и шимпанзе; крупному рогатому скоту, собакам, кошкам, морским свинкам, кроликам, крысам, мышам, лошадям и т.д. Согласно предпочтительным вариантам реализации изобретения указанный вид представляет собой человека. Особый интерес представляют субъекты, страдающие связанным с миокардом нарушением, которое поддается лечению (например, облегчению симптомов, ассоциированных с нарушением) путем трансплантации указанному субъекту клеток, например, кардиомиоцитов, фибробластов и т.д., которые экспрессируют обе субъединицы рибонуклеотидредуктазы (т.е., R1, R2). Согласно многим вариантам реализации изобретения, хозяин представляет собой человека.
[0108] Термин «Донорские клетки» в контексте настоящего изобретения относится к клеткам, произошедшим из организма млекопитающего, например, вида приматов, такого как человек и шимпанзе; крупного рогатого скота, собак, кошек, морских свинок, кроликов, крыс, мышей, лошадей и т.д., которые обладают способностью образовывать или обеспечивать щелевые контакты с кардиомиоцитами хозяина при трансплантации в миокард указанного хозяина. Примеры донорских клеток включают, но не ограничиваются указанными, фибробласты и кардиомиоциты. Донорские клетки могут являться аутологическими (т.е., полученными из органимзма хозяина, которого предполагается лечить), изогенными (т.е., полученными от генетически идентичного, но другого субъекта, например, от однояйцевого близнеца), аллогенными (т.е., полученными от генетически не идентичного представителя того же вида) и/или ксеногенными (т.е., полученными от представителя другого вида). Клетки могут быть получены от донора (живого донора или трупного материала) или происходить из постоянной клеточной линии. Для получения клеток от донора можно использовать стандартные методы биопсии, известные в данной области техники.
[0109] Термины «вектор экспрессии» или «вектор» относятся к соединению и/или композиции, которая трансдуцирует, трансформирует или инфицирует микроорганизм-хозяина, заставляв результате чего указанная клетка экспрессирует нуклеиновые кислоты и/или белки, отличные от естественных для указанной клетки, или не естественным для указанной клетки образом. «Вектор экспрессии» содержит последовательности нуклеиновой кислоты (обычно РНК или ДНК), которые должны экспрессироваться микроорганизмом-хозяином. Вектор экспрессии также может также включать материалы, способствующие поступлению нуклеиновой кислоты в микроорганизм-хозяин, такие как вирус, липосома, белковые покрытия и т.д. Векторы экспрессии, предполагаемые для использования согласно настоящему изобретению, включают векторы, в которые может быть встроена последовательность нуклеиновой кислоты вместе с любыми предпочтительными или необходимыми функциональными элементами. Кроме того, вектор экспрессии должен представлять собой вектор, который может быть перенесен в микроорганизм-хозяина и способен реплицироваться в нем. Предпочтительные векторы экспрессии представляют собой плазмиды, в частности, плазмиды с точно документированными сайтами рестрикции, которые содержат операционные элементы, предпочтительные или необходимые для транскрипции последовательности нуклеиновых кислот. Такие плазмиды, а также другие векторы экспрессии, хорошо известны специалистам в данной области техники. Согласно некоторым вариантам реализации изобретения вектор экспрессии представляет собой вирусный вектор, например, вектор на основе аденоассоциированного вируса.
[0110] Термин «вектор на основе аденоассоциированного вируса» в настоящей заявке включает как векторы на основе аденоассоциированного вируса дикого типа, так и рекомбинантные векторы на основе аденоассоциированного вируса, которые представляют собой непатогенные безоболочечные ДНК-вирусы, геном которых представлен линейной одноцепочечной молекулой размером примерно 4,6-4,8 т.п.н., которые требуют совместной инфекции вируса-помощника для вирусной репликации. Примеры векторов на основе аденоассоциированного вируса, которые можно применять согласно настоящему изобретению, включают, но не ограничиваются указанными: AAV6, AAV2, rAAV2/1, rAAV2/2, rAAV2/3, rAAV2/4, rAAV2/5, rAAV2/6, rAAV2/7 rAAV2/8, rAAV2/9, rAAV2/10, rAAVM41, dsAAV и т.д. Векторы на основе аденоассоциированного вируса и их применение в способах переноса генов рассмотрены в следующих источниках: Pacak et al. (2011) Molecular Therapy 19(9): 1582-1590; Hansruedi Biieler, Biol. Chem. (June 1999) 380: 612 622; Robbins et al., TIBTECH (January 1998) 16: 35 40; Patjin & Kay, Semin. Liver. Dis. (1999) 19: 61 69. Другие соответствующие ссылки включают следующие источники: Burton, et al., Proc Natl Acad Sci USA (1999) 96: 12725 12730; Fan, et al., Hum. Gene Ther. (1998) 9: 2527 2535; Miao et al., Nat. Genet. (May 1998) 19: 13 15; Nakai et al., J. Virol. (July 1999) 73: 5438 5447; Rendahl, et al., Nat Biotechnol (1998) 16, 757 761; Gregorevic et al. (2004) Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nature Med 10: 828-834; Blankinship et al. (2004) Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther 10: 671-678; Blankinship et al (2006) Gene therapy strategies for Duchenne muscular dystrophy utilizing recombinant adeno-associated virus vectors. Mol Ther 13: 241-249., Salva et al. (2007) Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther 15: 320-329.
[0111] В качестве иллюстративного примера, векторы для генной терапии на основе AAV6 могут быть созданы путем клонирования ДНК-кассеты экспрессии (например, промотор/энхансер, регулирующий экспресиию гена, связанного с последовательностью комплементарной ДНК (кДНК), кодирующей терапевтический белок или РНК, за которой следует сигнал остановки транскрипции, такой как последовательность полиаденилирования; в некоторых случаях, кДНК и промотор разделены интроном) между двумя копиями инвертированного концевого повтора (ITR) аденоассоциированного вируса (AAV). Указанный геном ITR-кассета экспрессии-ITR называется рекомбинантным геномом AAV (rAAV). Последовательности ITR AAV обеспечивают сигнал упаковки для инкапсулирования AAV в рекомбинантную частицу. ITR также обеспечивает ориджин репликации для продукции множества копий рекомбинантного генома AAV. ДНК, содержащие указанный рекомбинантный геном, затем может ко-трансфицироваться в пакующую клеточную линию, экспрессирующую различные аденовирусные вспомогательные белки (как правило, в клетки линии HEK293), вместе с плазмидой (плазмидами), содержащей гены Rep/Cap из генома AAV дикого типа, а также выполняющую дополнительные аденовирусные хелперные функции. AAV не может реплицироваться автономно, а требует ко-трансфекции второго вируса, такого как аденовирус, которые обеспечивает важные хелперные функции для обеспечения возможности транскрипции. ITR AAV могут происходить из различных серотипов AAV дикого типа. Например, обычно используемые ITR происходят из 2 серотипа AAV. При создании рекомбинантных векторов AAV6 (rAAV6) на этапе ко-трансфекции используется ген Сар из 6 серотипа AAV. Таким образом, «AAV6» представляет собой рекомбинантный вектор на основе аденоассоциированного вируса, содержащий ITR AAV2, фланкирующие кассету экспрессии, и инкапсулированный в белки капсида AAV6.
[0112] Под термином «промотор» подразумевается минимальная последовательность, достаточная для стимуляции транскрипции в рекомбинантной клетке. «Промотор» дополнительно включает элементы, достаточные для промотор-зависимой экспрессии гена, контролируемой клеточной специфичностью, тканеспецифичностью или индуцируемой внешними сигналами или агентами; такие элементы могут быть расположены в 5' или 3' областях нативного гена (например, энхансерные элементы). Примеры промоторов включают, но не ограничиваются указанными, промотор СК7 и промотор цитомегаловирусы (CMV).
[0113] Термин «кардиоспецифичный промотор», в контексте настоящего изобретения, относится к промотору дикого типа или рекомбинантному промотору, который селективно запускает экспрессию гена, находящегося под его контролем, в клетках сердца. Примеры кардиоспецифичных промоторов включают промотор α-MHC5.5, промотор α-МНС86, промотор сердечного актина человека и промотор cTnT455, описанные в настоящей заявке.
[0114] Под термином «функционально связанные» или «связанные с возможностью реализации функции» подразумевается, что последовательность ДНК и регуляторная последовательность (последовательности) связаны таким образом, что экспрессия возможна, когда соответствующие молекулы (например, белки-активаторы транскрипции) связаны с указанной регуляторной последовательностью (последовательностями).
[0115] «Процентная идентичность по последовательности», «процентная идентичность по аминокислотной последовательности», «процентная идентичность по последовательности гена» и/или «процентная идентичность по последовательности нуклеиновой кислоты/полинуклеотида» в отношении двух последовательностей аминокислот, полинуклеотидов и/или генов (в зависимости от конкретного случая), относится к проценту остатков, которые идентичны в указанных двух последовательностях при их оптимальном выравнивании. Таким образом, 80% идентичность по аминокислотной последовательности означает, что 80% аминокислот в двух последовательностях полипептидов, которые оптимально выровнены, идентичны.
[0116] Под термином «трансформация», «трансдукция» или «трансфекция» подразумевается постоянное или временное генетическое изменение, предпочтительно, постоянное генетическое изменение, вызванное в клетке введением новой нуклеиновой кислоты (например, ДНК или РНК, экзогенной для указанной клетки). Генетическое изменение может быть осуществлено посредством встраивания новой нуклеиновой кислоты в геном клетки-хозяина или посредством временного или постоянного поддержания новой ДНК в виде эписомного элемента.
[0117] Под терминами «трансформированная клетка», «трансдуцированная клетка» или «трансдуцированная клетка» подразумевается клетка, в которую (или в клетку-предшественник которой) методами рекомбинации ДНК была введена молекула ДНК, кодирующая интересующий белок.
[0118] Под термином «гиперэкспрессируемый» или «гиперэкспрессия» генного продукта (такого как R1 или R2) подразумевается повышенный уровень экспрессии белка по сравнению с нормальным уровнем экспрессии указанного белка для конкретной клетки или типа клеток, например, на конкретной стадии развития или стадии дифференцировки. В некоторых примерах гиперэкспрессия может являться совокупным результатом экспрессии белка с эндогенных и рекомбинантных генов или представлять собой экспрессию белка по существу с рекомбинантного гена. Подразумевается, что гиперэкспрессия R1 или R2 относится к экспрессии соответствующей субъединицы белка рибонуклеотидредуктазы в конкретной клетке, повышенной по сравнению с нормальным уровнем экспрессии, связанным с нормальной клеткой или клеткой дикого типа на определенной стадии дифференцировки. Согласно некоторым вариантам реализации изобретения, предполагается, что гиперэкспрессия генного продукта представляет собой повышение экспрессии по меньшей мере примерно в 2 раза, согласно другим вариантам реализации изобретения по меньшей мере примерно в 5 раз и, согласно другим вариантам реализации изобретения по меньшей мере примерно в 10 раз.
[0119] Под термином «конструкция» подразумевается рекомбинантная нуклеиновая кислота, обычно рекомбинантная ДНК, которая была создана для обеспечения экспрессии конкретной нуклеотидной последовательности (последовательностей), или которую предполагается использовать в конструировании других рекомбинантных нуклеотидных последовательностей.
[0120] В настоящей заявке, термин «полипептид» относится к аминокислотной последовательности рекомбинантного или нерекомбинантного полипептида, содержащего аминокислотную последовательность i) нативного полипептида, ii) биологически активного фрагмента полипептида, iii) биологически активных полипептидных аналогов полипептида или iv) биологически активного варианта полипептида. Полипептиды, которые можно применять в настоящем изобретении, могут быть получены из организма любого вида, например, от млекопитающего или не млекопитающего (например, рептилий, амфибий, птиц (например, курицы)), в частности, млекопитающего, включающего человека, грызунов (например, мышей или крыс), коров, овец, свиней, мышей или лошадей, предпочтительно, крыс или человека, из любого источника, синтетического, полусинтетического или рекомбинантного. Например, подразумевается, что «полипептид R1» относится к аминокислотным последовательностям выделенного человеческого полипептида R1, полученного от человека, и включает все природные аллельные варианты и не ограничивает аминокислотные последовательности полностью нативной аминокислотной последовательностью, характерной для описанной молекулы белка.
[0121] «Вариант» полипептида определяется как аминокислотная последовательность, измененная по одной или более аминокислотам (например, путем делеции, добавления, вставки и/или замены). В целом, «добавление» относится к нуклеотидным или аминокислотным остаткам, присоединяемым к концу молекулы, тогда как «вставка» относится к добавлению нуклеотидных или аминокислотных остатков между остатками природной молекулы. Вариант может включать «консервативные» изменения, где замененная аминокислота имеет такие же структурные или химические свойства, например, в случае замены лейцина на изолейцин. Реже вариант может включать «неконсервативные» замены, например, замену глицина на триптофан. Подобные минорные изменения также могут включать делеции или вставки аминокислот или как делеции, так и вставки. Руководство, помогающее определить, какие и сколько аминокислотных остатков можно заместить, добавить, встроить или удалить, не затрагивая биологическую и иммунологическую активность, можно найти с помощью компьютерных программ, хорошо известных специалистам в данной области техники, например, с помощью программы DNAStar.
[0122] Термин «нацеливающий агент» относится к соединению, которое проявляет селективность в отношении конкретного органа-, ткани- или клеточного типа-мишени. Нацеливающий агент способен нацеливать композицию, с которой он функционально связан, к конкретному органу- или ткани-мишени. Нацеливающий агент может быть функционально связан по меньшей мере с одним катионным полимерным носителем и/или другим агентом.
[0123] Термин «инфаркт миокарда» в настоящей заявке означает процесс, в ходе которого ишемическая болезнь сердца приводит к замещению участка миокарда рубцовой тканью.
[0124] Термин «ишемическая болезнь сердца» в настоящей заявке означает любое расстройство, являющееся результатом нарушенного баланса между потребностями миокарда в кислороде и его адекватным снабжением кислородом. Наибольшее число случаев ишемической болезни сердца являются результатом сужения коронарных артерий, происходящим при атеросклерозе или других сосудистых расстройствах.
[0125] Термин «сердечная недостаточность» в настоящей заявке означает нарушение сердечной функции, которое приводит к неспособности сердца сохранять нормальный выброс крови в состоянии покоя или при нагрузке, или поддерживать нормальный сердечный выброс на фоне нормального давления наполнения сердца. Фракция выброса левого желудочка, составляющая примерно 40% или менее, указывает на сердечную недостаточность (для сравнения, фракция выброса от примерно 55% до 60% считается нормальной). Пациенты, страдающие сердечной недостаточностью, проявляют хорошо известные клинические симптомы и признаки, такие как тахипное, плевральный выпот, утомляемость в состоянии покоя или при нагрузке, нарушение сократительной функции и эдема. Относительную тяжесть и прогрессирование заболевания оценивают с использованием хорошо известных методов, таких как медицинский осмотр, эхокардиография, радионуклеотидная визуализация, инвазивный мониторинг гемодинамических параметров, магнитно-резонансная ангиография и тестирование с нагрузкой на беговой дорожке, совмещенное с исследованием потребления кислорода.
[0126] Термин «кардиомиопатия» в настоящей заявке относится к сердечнососудистому нарушению. Согласно некоторым вариантам реализации изобретения кардиомиопатия выбрана из первичной кардиопатологии или вторичной кардиопатологии. Кардиомиопатия может быть выбрана из генетически обусловленной кардиопатологии, гипертрофической кардиомиопатии, ишемической кардиомиопатии, рестриктивной кардиомиопатии и дилатационной кардиомиопатии. Согласно вариантам реализации изобретения, включающим улучшение сердечной функции у субъекта, страдающего гипертрофической кардиомиопатией, кардиомиопатия может являться результатом: (а) постинфарктного ремоделирования миокарда, (b) заболевания сердечного клапана; (с) длительной сердечной постнагрузки; (d) миокардита или (е) семейной гипертрофической кардиомиопатии.
[0127] В настоящей заявке, термин «сердечный тропонин С» или «cTnC» относится к полипептиду тропонинового комплекса, имеющему множество сайтов связывания кальция. Согласно предпочтительному варианту реализации изобретения cTnC относится к человеческому cTnC (ссылка в базе данных Entrez: NP_003271), закодированному геном TNNC1 (ссылка в базе данных Entrez: NM_003280,2), или его консервативным вариантам, вариантам сплайсинга или меченным вариантам.
[0128] В настоящей заявке, термин «рибонуклеотидредуктазный комплекс», «RR» или «RNR» относится к тетрамерному комплексу гетеродимерных полипептиднов, содержащему субъединицы RNR1 (или «R1») и RNR2 (или «R2»). Согласно предпочтительному варианту реализации изобретения субъединица R1 относится к человеческой субъединице рибонуклеотидредуктазы M1 (ссылка в базе данных Entrez: AAD37491,1), закодированной геном RRM1 (ссылка в базе данных Entrez: AF107045,1), или ее консервативным вариантам, вариантам сплайсинга или меченным вариантам. Субъединица R2 может относиться либо к субъединице рибонуклеотидредуктазы М2, либо к субъединице рибонуклеотидредуктазы М2 В. Согласно одному предпочтительному варианту реализации изобретения, R2 относится к субъединице человеческой рибонуклеотидредуктазы М2 (ссылка в базе данных Entrez: ААК51163), закодированной геном RRM2 (ссылка в базе данных Entrez: AY032750,1). Согласно другим предпочтительным вариантам реализации изобретения, R2 относится к 1 изоформе субъединицы человеческой рибонуклеотидредуктазы М2 В (ссылка в базе данных Entrez: NP_056528,2), закодированной геном RRM2B (ссылка в базе данных Entrez: NM_015713,4); 2 изоформе субъединицы человеческой рибонуклеотидредуктазы М2 В (ссылка в базе данных Entrez: NP_001165948,1), закодированной геном RRM2B (ссылка в базе данных Entrez: NM_001172477); или 3 изоформе субъединицы человеческой рибонуклеотидредуктазы М2 В (ссылка в базе данных Entrez: NP001165949), закодированной геном RRM2B (ссылка в базе данных Entrez: NM_001172478,1); или их консервативным вариантам, вариантам сплайсинга или меченным вариантам.
V. ПРИМЕРЫ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
[0129] Согласно одному аспекту настоящего изобретения предложены способы лечения сердечной функции, включающие введение искусственных (модифицированных) вариантов cTnC, которые не были клинически идентифицированы как ГКМП/ДКМП-мутации, но которые обеспечивают такое же повышение или понижение Ca2+-чувствительности сокращения.
[0130] Согласно одному аспекту настоящего изобретения предложен способ улучшения сердечной функции у субъекта, нуждающегося в этом, включающий введение вектора, кодирующего вариант cTnC, обладающий повышенной аффинностью связывания сайта связывания Ca2+ II, в ткань сердца указанного субъекта. Согласно конкретному варианту реализации изобретения вариант cTnC содержит аминокислотную замену L48Q.
[0131] Согласно конкретному варианту реализации изобретения субъект страдает состоянием сердца, приводящим к снижению сократимости. Согласно конкретному варианту реализации изобретения субъект страдает инфарктным поражением сердца.
[0132] Согласно одному аспекту настоящего изобретения предложен способ улучшения сердечной функции у субъекта, нуждающегося в этом, включающий введение вектора, кодирующего вариант cTnC, обладающий пониженной аффинностью связывания Сайта II связывания Ca2+, в ткань сердца указанного субъекта. Согласно одному варианту реализации изобретения вариант cTnC содержит аминокислотную замену L57Q. Согласно другому варианту реализации изобретения вариант cTnC содержит аминокислотную замену I61Q.
[0133] Согласно некоторым вариантам реализации изобретения, предложен способ улучшения сердечной функции у субъекта, имеющего генетическую предрасположенность к болезням сердца. Более конкретно, генетическая предрасположенность обусловлена одной или более генетическими мутациями в саркомерном белке, выбранным из тяжелой цепи бета сердечного миозина, сердечного актина, сердечного тропонина Т, альфа-тропомиозина, сердечного тропонина I, сердечного миозин-связывающего белка С и легкой цепи миозина. Согласно некоторым вариантам реализации изобретения, генетическая мутация (мутации) связана с повышенной Ca2+-чувствительностью сокращения миофибрилл и/или фенотипами гипертрофической кардиомиопатии. Примеры таких генетических мутаций включают, но не ограничиваются указанными, мутации по остатку 92 cTnT, например, R92W, R92Q и R92L cTnT, или мутации в гене MYH7, MYBPC3, TNNT2, TNNI3, ТРМ1, АСТС, MYL2, MYL3, или их комбинации. Способ улучшения сердечной функции у таких субъектов включает введение указанным субъектам вирусного вектора, содержащего последовательность нуклеиновой кислоты, кодирующую вариант L57Q или I61Q cTnC, где указанная последовательность нуклеиновой кислоты функционально связана с кардиоспецифичным промотором. Согласно другим вариантам реализации изобретения, генетическая мутация (мутации) связана со пониженной Ca2+-чувствительностью сокращения миофибрилл и/или фенотипами дилатационной кардиомиопатии. Примеры таких генетических мутаций включают, но не ограничиваются указанными, миссенс-мутации Ser532Pro и Phe764Leu, делецию в cTnT (deltaLys210) или мутации в генах MYH7, MYBPC3, TNNT2, TNNI3, ТРМ1, АСТС, MYL2, MYL3, или их комбинации. Способ улучшения сердечной функции у таких субъектов включает введение указанным субъектам, несущим Ca2+-десенсибилизирующую генетическую мутацию (мутации), вирусного вектора, содержащего последовательность нуклеиновой кислоты, кодирующую вариант L48Q cTnC, где указанная последовательность нуклеиновой кислоты функционально связана с кардиоспецифичным промотором.
[0134] Согласно конкретным способам, предложенным в настоящей заявке, вектор вводят путем липофекции, в виде покрытия на стенте или путем прямой инъекции (т.е., через катетер). Согласно одному варианту реализации изобретения вектор включает транспозон, плазмиду или вирусный вектор. Согласно одному варианту реализации изобретения вектор включает аденовирусный вектор, содержащий нуклеиновую кислоту, кодирующую вариант cTnC. Согласно конкретному варианту реализации изобретения нуклеиновая кислота, кодирующая вариант cTnC, дополнительно содержит промотор CMV, функционально связанный с нуклеотидной последовательностью, кодирующей вариант cTnC.
[0135] Согласно одному варианту реализации способа согласно изобретению, перенос гена cTnC в клетки обеспечивается не с помощью вирусного вектора, например, путем слияния липосом, с помощью фосфата кальция, микроинъекции, электропорации, поликатионов, бомбардировки частицами и рецептор-опосредованных способов. Согласно конкретному варианту реализации изобретения вектор включает наночастицу, связанную с нуклеиновой кислотой, кодирующей вариант cTnC. Согласно конкретному варианту реализации изобретения наночастица включает липосому, инкапсулирующую нуклеиновую кислоту, кодирующую вариант cTnC. Согласно некоторым вариантам реализации изобретения, вектор также кодирует нацеливающий агент, обладающий аффинностью к сердечному тканеспецифичному маркеру. Согласно одному варианту реализации изобретения нацеливающий агент выбран из группы, состоящей из антитела, фрагмента антитела и аптамера.
[0136] Согласно другому аспекту настоящего изобретения предложен способ улучшения сердечной функции у субъекта, нуждающегося в этом, включающий введение вектора, кодирующего рибонуклеотидредуктазный (RR) комплекс, в ткань сердца указанного субъекта. Согласно одному варианту реализации изобретения субъект страдает состоянием сердца, приводящим к снижению сократимости. Согласно конкретному варианту реализации изобретения субъект страдает инфарктным поражением сердца.
[0137] Согласно конкретным способам, предложенным в настоящей заявке, вектор вводят путем липофекции, в виде покрытия на стенте или путем прямой инъекции (т.е., с помощью катетера). Согласно одному варианту реализации изобретения вектор включает транспозон, плазмиду или вирусный вектор.
[0138] Согласно конкретному варианту реализации изобретения вектор включает клетку, содержащую нуклеиновую кислоту, кодирующую субъединицы R1 и R2 комплекса RR. Согласно одному варианту реализации изобретения клетка представляет собой кардиомиоцит. Согласно конкретному варианту реализации изобретения кардиомиоцит происходит из плюрипотентной эмбриональной стволовой клетки (ЭСК), индуцированной плюрипотентной стволовой клетки (иПСК) или мезенхимальной стволовой клетки. Согласно одному варианту реализации изобретения иПСК происходит из клетки, полученной от субъекта. Согласно конкретному варианту реализации изобретения клетка, полученная от из организма субъекта, представляет собой кожный фибробласт. Согласно некоторым вариантам реализации способов, предложенных в настоящей заявке, нуклеиновая кислота, кодирующая субъединицы R1 и R2 комплекса RR, также включает кардиоспецифичный промотор, функционально связанный с нуклеотидной последовательностью, кодирующей субъединицы R1 и/или R2.
[0139] Согласно конкретному варианту реализации изобретения вектор включает наночастицу, связанную с нуклеиновой кислотой, кодирующей субъединицы R1 и R2 комплекса RR. Согласно одному варианту реализации изобретения вектор также включает последовательность, кодирующую нацеливающий агент, обладающий аффинностью к сердечному тканеспецифичному маркеру. Согласно конкретному варианту реализации изобретения нацеливающий агент выбран из группы, состоящей из антитела, фрагмента антитела и аптамера. Согласно более конкретному варианту реализации изобретения наночастица включает липосому, инкапсулирующую нуклеиновую кислоту, кодирующую вариант cTnC.
[0140] Согласно другому аспекту настоящего изобретения предложен способ улучшения сердечной функции у субъекта, нуждающегося в этом, включающий введение: (i) вектора, кодирующего вариант cTnC, обладающего повышенной аффинностью связывания Сайта II связывания Ca2+, в ткань сердца указанного субъекта; и (ii) вектора, кодирующего рибонуклеотидредуктазный (RR) комплекс, в ткань сердца указанного субъекта.
[0141] Согласно другому аспекту настоящего изобретения предложена фармацевтическая композиция для улучшения сердечной функции у субъекта, нуждающегося в этом, содержащая вектор, кодирующий вариант cTnC, обладающий повышенной аффинностью связывания сайта II связывания Ca2+. Согласно одному варианту реализации изобретения вариант cTnC содержит аминокислотную замену L48Q.
[0142] Согласно другому аспекту настоящего изобретения предложена фармацевтическая композиция для улучшения сердечной функции у субъекта, нуждающегося в этом, содержащая вектор, кодирующий вариант cTnC, обладающий пониженной аффинностью связывания. Согласно одному варианту реализации изобретения вариант cTnC содержит аминокислотную замену L57Q и/или I61Q.
[0143] Согласно некоторым вариантам реализации фармацевтической композиции, предложенной в настоящей заявке, вектор включает аденовирусный вектор, содержащий нуклеиновую кислоту, кодирующую вариант cTnC. Согласно конкретному варианту реализации изобретения нуклеиновая кислота, кодирующая вариант cTnC, также включает промотор цитомегаловируса (CMV), функционально связанный с нуклеотидной последовательностью, кодирующей вариант cTnC.
[0144] Согласно одному аспекту настоящего изобретения предложена фармацевтическая композиция для улучшения сердечной функции у субъекта, нуждающегося в этом, содержащая вектор, кодирующий рибонуклеотидредуктазный (RR) комплекс. Согласно одному варианту реализации изобретения вектор включает клетку, содержащую нуклеиновую кислоту, кодирующую субъединицы R1 и R2 комплекса RR.
[0145] Согласно некоторым вариантам реализации фармацевтических композиций, предложенных в настоящей заявке, клетка представляет собой кардиомиоцит. Согласно конкретному варианту реализации изобретения кардиомиоцит происходит из плюрипотентной эмбриональной стволовой клетки (ЭСК), индуцированной плюрипотентной стволовой клетки (иПСК) или мезенхимальной стволовой клетки. Согласно более одному варианту реализации изобретении, иПСК происходит из клетки, полученной от субъекта, например, из кожного фибробласта.
[0146] Согласно некоторым вариантам реализации фармацевтических композиций, предложенных в настоящей заявке, нуклеиновая кислота, кодирующая субъединицы R1 и R2 комплекса RR, дополнительно содержит кардиоспецифичный промотор, функционально связанный с нуклеотидной последовательностью, кодирующей субъединицы R1 и/или R2.
[0147] Согласно одному аспекту настоящего изобретения предложен способ улучшения сердечной функции у субъекта, нуждающегося в этом, включающий введение вектора, кодирующего вариант cTnC, обладающий повышенной аффинностью связывания Сайта II связывания Ca+, в ткань сердца указанного субъекта. Согласно конкретному варианту реализации изобретения вариант cTnC содержит аминокислотную замену L48Q.
[0148] Согласно некоторым вариантам реализации способов, предложенных выше, субъект страдает состоянием сердца, приводящим к пониженной сократимости. Согласно одному варианту реализации изобретения у субъекта диагностирована ишемическая болезнь сердца, кардиомиопатия или инфаркт миокарда. Согласно одному варианту реализации изобретения кардиомиопатия представляет собой первичную кардиомиопатию, генетически обусловленную кардиомиопатию, дилатационную кардиомиопатию или гипертрофическую кардиомиопатию. Согласно одному варианту реализации изобретения у субъекта диагностировано снижение систолической функции.
[0149] Согласно одному аспекту настоящего изобретения предложен способ улучшения сердечной функции у субъекта, нуждающегося в этом. Более конкретно, субъект характеризуется Ca2+-сенсибилизирующим фенотипом или фенотипом гипертрофической кардиомиопатии, обусловленным одной или более генетическими мутациями в саркомерном белке, выбранном из тяжелой цепи бета сердечного миозина, сердечного актина, сердечного тропонина Т, альфа-тропомиозина, сердечного тропонина I, сердечного миозин-связывающего белка С и легкой цепи миозина. Примеры указанных генетических мутаций включают, но не ограничиваются указанными, мутации по остатку 92 cTnT, например, R92W, R92Q и R92L cTnT, или мутации в MYH7, MYBPC3, TNNT2, TNNI3, ТРМ1, АСТС, MYL2, MYL3, или их комбинации. Способ улучшения сердечной функции у субъектов включает введение указанным субъектам, несущим Ca2+-сенсибилизирующую генетическую мутацию (мутации), вирусного вектора, содержащего последовательность нуклеиновой кислоты, кодирующую вариант L57Q или I61Q cTnC, где указанная последовательность нуклеиновой кислоты функционально связана с кардиоспецифичным промотором.
[0150] Согласно другому аспекту настоящего изобретения предложен способ улучшения сердечной функции у субъекта, имеющего Ca2+-десенсибилизирующий фенотип или фенотип дилатационной кардиомиопатии, связанный с одной или более генетическими мутациями в саркомерном белке, выбранном из тяжелой цепи бета сердечного миозина, сердечного актина, сердечного тропонина Т, альфа-тропомиозина, сердечного тропонина I, сердечного миозин-связывающего белка С и легкой цепи миозина. Примеры таких генетических мутаций включают, но не ограничиваются указанными, миссенс-мутации Ser532Pro и Phe764Leu, делецию в cTnT (дельта Lys210), или мутации в генах MYH7, MYBPC3, TNNT2, TNNI3, ТРМ1, АСТС, MYL2, MYL3, или их комбинации. Способ улучшения сердечной функции включает введение указанным субъектам, имеющим Ca2+-десенсибилизирующую генетическую мутацию (мутации), вирусного вектора, содержащего последовательность нуклеиновой кислоты, кодирующую вариант L48Q cTnC, где указанная последовательность нуклеиновой кислоты функционально связана с кардиоспецифичным промотором.
[0151] Согласно некоторым вариантам реализации способов, предложенных выше, вектор вводят путем липофекции, в виде покрытия на стенте или путем прямой инъекции (т.е., с помощью катетера). Согласно одному варианту реализации изобретения вектор включает транспозон, плазмиду или вирусный вектор. Согласно конкретному варианту реализации изобретения вектор включает аденовирусный вектор, содержащий нуклеиновую кислоту, кодирующую вариант cTnC. Согласно одному варианту реализации изобретения нуклеиновая кислота, кодирующая вариант cTnC, дополнительно содержит промотор CMV, промотор СК7, промотор α-MHC5,5, промотор α-МНС86, промотор сердечного актина, промотор cTnT455 или другой кардиоспецифичный промотор, функционально связанный с нуклеотидной последовательностью, кодирующей вариант cTnC. Согласно одному варианту реализации изобретения вектор включает наночастицу, ассоциированную с нуклеиновой кислотой, кодирующей вариант cTnC. Согласно одному варианту реализации изобретения вектор также кодирует нацеливающий агент, обладающий аффинностью к сердечному тканеспецифичному маркеру. Согласно одному варианту реализации изобретения нацеливающий агент выбран из группы, состоящей из антитела, фрагмента антитела и аптамера. Согласно одному варианту реализации изобретения наночастица включает липосому, инкапсулирующую нуклеиновую кислоту, кодирующую вариант cTnC.
[0152] Согласно одному аспекту настоящего изобретения предложен способ улучшения сердечной функции у субъекта, нуждающегося в этом, включающий введение вектора, кодирующего рибонуклеотидредуктазный (RR) комплекс, в ткань сердца субъекта.
[0153] Согласно некоторым вариантам реализации способов, предложенных выше, субъект страдает состоянием сердца, приводящим к снижению сократимости. Согласно одному варианту реализации изобретения субъект страдает инфарктным поражением сердца.
[0154] Согласно некоторым вариантам реализации способов, предложенных выше, вектор вводят путем липофекции, в виде покрытия на стенте или путем прямой инъекции (т.е. с помощью катетера). Согласно одному варианту реализации изобретения вектор включает транспозон, плазмиду или вирусный вектор. Согласно одному варианту реализации изобретения вектор включает клетку, содержащую нуклеиновую кислоту, кодирующую субъединицы R1 и R2 комплекса RR. Согласно одному варианту реализации изобретения клетка представляет собой кардиомиоцит. Согласно одному варианту реализации изобретения кардиомиоцит происходит из плюрипотентной эмбриональной стволовой клетки (ЭСК), индуцированной плюрипотентной стволовой клетки (иПСК) или мезенхимальной стволовой клетки. Согласно одному варианту реализации изобретения иПСК происходит из клетки, полученной от субъекта. Согласно одному варианту реализации изобретения клетка, полученная от из организма субъекта, представляет собой кожный фибробласт.
[0155] Согласно некоторым вариантам реализации способов, предложенных выше, нуклеиновая кислота, кодирующая субъединицы R1 и R2 комплекса RR, дополнительно содержит кардиоспецифичный промотор, функционально связанный с нуклеотидной последовательностью, кодирующей субъединицу R1 и/или R2.
[0156] Согласно некоторым вариантам реализации способов, предложенных выше, вектор включает наночастицу, связанную с нуклеиновой кислотой, кодирующей субъединицы R1 и R2 комплекса RR. Согласно одному варианту реализации изобретения вектор также кодирует нацеливающий агент, обладающий аффинностью к сердечному тканеспецифичному маркеру. Согласно одному варианту реализации изобретения нацеливающий агент выбран из группы, состоящей из антитела, фрагмента антитела и аптамера. Согласно одному варианту реализации изобретения наночастица включает липосому, инкапсулирующую нуклеиновую кислоту, кодирующую вариант cTnC.
[0157] Согласно одному аспекту настоящего изобретения предложен способ улучшения сердечной функции у субъекта, нуждающегося в этом, включающий введение: (i) вектора, кодирующего вариант cTnC, обладающий повышенной аффинностью связывания Сайта II связывания Ca+, в ткань сердца указанного субъекта; и (ii) вектора, кодирующего рибонуклеотидредуктазный (RR) комплекс, в ткань сердца указанного субъекта.
[0158] Согласно одному аспекту настоящего изобретения предложена фармацевтическая композиция для улучшения сердечной функции у субъекта, нуждающегося в этом, содержащая вектор, кодирующий вариант cTnC, обладающий повышенной аффинностью связывания сайта II связывания Ca2+. Согласно одному варианту реализации изобретения вариант cTnC содержит аминокислотную замену L48Q.
[0159] Согласно одному аспекту настоящего изобретения предложена фармацевтическая композиция для улучшения сердечной функции у субъекта, нуждающегося в этом, содержащая вектор, кодирующий вариант cTnC, обладающий пониженной аффинностью связывания сайта II связывания Ca2+. Согласно одному варианту реализации изобретения вариант cTnC содержит аминокислотную замену L57Q и/или I61Q.
[0160] Согласно некоторым вариантам реализации предложенных выше композиций, вектор включает аденовирусный вектор, содержащий нуклеиновую кислоту, кодирующую вариант cTnC. Согласно одному варианту реализации изобретения нуклеиновая кислота, кодирующая вариант cTnC, дополнительно содержит промотор CMV, функционально связанный с нуклеотидной последовательностью, кодирующей вариант cTnC.
[0161] Согласно одному аспекту настоящего изобретения предложена фармацевтическая композиция для улучшения сердечной функции у субъекта, нуждающегося в этом, содержащая вектор, кодирующий рибонуклеотидредуктазный (RR) комплекс. Согласно одному варианту реализации изобретения вектор включает клетку, содержащую нуклеиновую кислоту, кодирующую субъединицы R1 и R2 комплекса RR. Согласно одному варианту реализации изобретения клетка представляет собой кардиомиоцит. Согласно одному варианту реализации изобретения кардиомиоцит происходит из плюрипотентной эмбриональной стволовой клетки (ЭСК), индуцированной плюрипотентной стволовой клетки (иПСК) или мезенхимальной стволовой клетки. Согласно одному варианту реализации изобретения иПСК происходит из клетки, полученной от субъекта. Согласно одному варианту реализации изобретения клетка, полученная от из организма субъекта, представляет собой кожный фибробласт. Согласно одному варианту реализации изобретения нуклеиновая кислота, кодирующая субъединицы R1 и R2 комплекса RR, дополнительно содержит кардиоспецифичный промотор, функционально связанный с нуклеотидной последовательностью, кодирующей субъединицу R1 и/или R2.
VI. ПРИМЕРЫ
ПРИМЕР 1
[0162] Выделение и культивирование клеток взрослых и новорожденных животных. Указанные исследования были одобрены комитетом по содержанию лабораторных животных Вашингтонского университета (UW) и проводились в соответствии с федеральным руководством. С животными обращались в соответствии с политикой Национального института здравоохранения США по уходу за лабораторными животными и их использованию человеком Отдела сравнительной медицины Вашингтонского университета. Кардиомиоциты взрослых крыс линии Fischer 344 (adult rat cardiomyocytes, ARC), выделяли из сердца с помощью ретроградной перфузии через аорту для ферментативной диссоциации клеток (коллагеназа/протеаза)10. Кардиомиоциты новорожденных крыс (NRC) выделяли путем ферментативной диссоциации из 1-3-дневных новорожденных крыс линии Fischer 344, как описано ранее11.
ПРИМЕР 2
[0163] Конструирование плазмид и продукция вируса. Получали продуцированные клетками HEK293 аденовирусные векторы, экспрессирующие меченный гистидином (N-концевой 6-His) L48Q cTnC или cTnC ДТ с промотора CMV. Оба вектора содержали вторую кассету экспрессии для зеленого флуоресцентного белка (green fluorescent protein, GFP) в качестве репортерного белка трансдукции. Мы также экспрессировали вектор для GFP отдельно. Вирус вводили в кардиомиоциты в концентрации ~250 частиц на клетку.
[0164] Рекомбинантный аденовирус, содержащий соответствующие кДНК-конструкции под контролем промотора CMV, использовали для индукции гиперэкспрессии L48Q и WT-cTnC в трансдуцированных культивируемых кардиомиоцитах взрослых и новорожденных крыс. Вторая кассета экспрессии для зеленого флуоресцентного белка (GFP) содержалась в каждом аденовирусе в качестве репортерного белка успешной трансдукции. Кардиомиоциты инфицировали аденовирусом, содержащим гены для [L48Q cTnC+GFP] или [cTnC ДТ+GFP], в течение 2 дней. Мы достигли почти 100% эффективности трансфекции и переноса генов, на что явно указывала наблюдаемая с помощью микроскопа зеленая флуоресценция. Эти данные согласуются с предшествующими исследованиями с использованием кардиомиоцитов13. Выживаемость клеток в течение указанного периода была одинаковой для всех групп, включая нетрансдуцированные контрольные клетки, что дает основания полагать, что указанные вирусные векторы не оказывали отрицательного влияния на жизнеспособность кардиомиоцитов. Количество кардиомиоцитов и длины саркомеров суммированы в Таблица 1. Различия в длине саркомеров в состоянии покоя между группами отсутствовали, что указывает на то, что гиперэкспрессия L48Q или экспрессия cTnC ДТ (+GFP) не повышала кальций-независимую активацию.
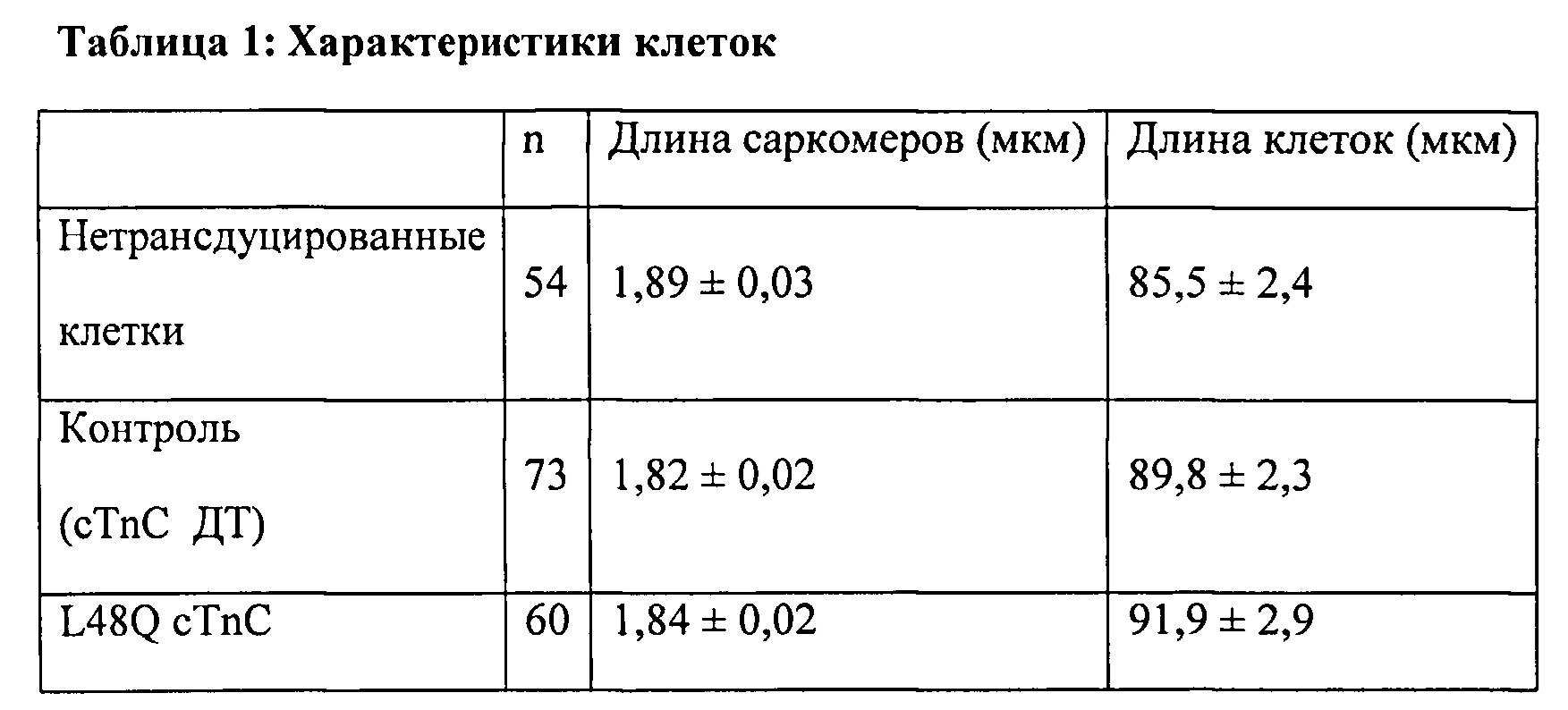
ПРИМЕР 3
[0165] Оценка сократимости. Укорочение и расслабление клеток регистрировали с модифицированном буфере Тирод при комнатной температуре на произвольно выбранных стимулированных кардиомиоцитах с использованием системы видеомикроскопии IonOptix (IonOptix, Милтон, Массачусетс, США). Кальциевые токи, вызванные электрической стимуляцией, измеряли в нагруженных Fura2 клетках с использованием оборудования IonOptix, как описано12. Флуоресценцию Fura2 измеряли с использованием спектрофотометра IonOptix (Stepper Switch), соединенного с флуоресцентным микроскопом. Испускаемую Флуоресценцию Fura2 собирали с помощью объектива 40Х, пропускали через 510 нм фильтр и регистрировали с помощью трубки фотоумножителя.
[0166] Влияние экспрессии L48Q cTnC на степень и скорость стимулированного укорочения-повторного удлинения и высвобождения-обратного захвата Ca2+ кардиомиоцитов взрослых крыс определяли путем видеодетекции длины и флуоресцентной фотометрии, соответственно (IonOptix). На Фигуре 1А показаны типичные кривые укорочения, и на Фигуре 1В показаны типичные Ca2+-токи (Fura2 флуоресценция) для необработанных (черный), cTnC ДТ+GFP (синий), и L48Q cTnC+GFP (красный) трансдуцированных кардиомиоцитов. Данные для всех измерений при стимуляции 0,5 Гц суммированы в Таблица 2. Гиперэкспрессия cTnC ДТ+GFP значительно снижала скорость сокращения, но не оказывала значительного влияния на относительное укорочение (FS) по сравнению с необработанными кардиомиоцитами. Не была проведена оценка того, обусловлено ли указанное снижение GFP или гиперэкспрессией cTnC, тогда как было описано, что GFP оказывает негативное влияния14 или не влияет12,15 на сократимость. В любом случае экспрессия L48Q была больше, чем экспрессия, способная преодолевать потенциальное ингибирование при значительно повышенной скорости и степени укорочения по сравнению с cTnC ДТ трансдуцированными и нетрансдуцированными кардиомиоцитами. Не наблюдалось различий в скорости максимального расслабления или времени до 50% и 90% расслабления между группами при частоте стимуляции 0,5 Гц. На Ca2+-ток также незначительно влияла гиперэкспрессия WT или cTnC, за исключением того, что у cTnC ДТ-трансдуцированных кардиомиоцитов наблюдали слабое незначимое (р=0,55) повышение минимального Ca2+. Это не оказывало влияния на SL в состоянии покоя или максимальное высвобождение Ca2+. На Фигуре 1С показаны % различия в скорости и степени укорочения, скорости расслабления и времени до достижения 50% и 90% расслабления, и на Фигуре 1D показано % различие в характере Ca2+-тока, включая минимальную и максимальную концентрацию Ca2+, и время до достижения 50% и 90% снижения Ca2+. Не наблюдалось значительного эффекта cTnC ДТ+GFP или L48Q cTnC+GFP на максимальную концентрацию Ca2+, что указывает на то, что усиленная сократимость в случае L48Q cTnC была, в первую очередь, опосредована повышенной чувствительностью миофиламентов к активации Ca2+.
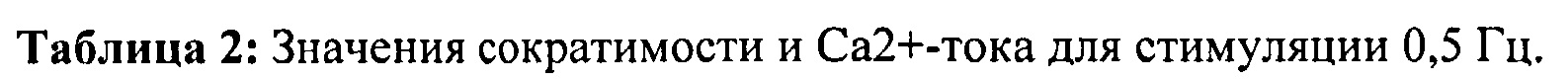
ПРИМЕР 4
[0167] Поскольку изменения в частоте сердечных сокращений являются нормальной физиологической адаптацией к системным требованиям, важно определить, влияет ли гиперэкспрессия L48Q cTnC на гормальный клеточный ответ на повышенную частоту стимуляции. На Фигуре 2 приведены результаты изучения влияния повышенной частоты стимуляции (0,5 до 1 до 2 Гц) на относительное укорочение (2А), скорость укорочения (2В), скорость расслабления (2С) и время до достижения 90% расслабления (2D). Сократительный ответ на частоты стимуляции был одинаковым между группами, и L48Q cTnC-трансдуцированные кардиомиоциты поддерживали усиление функции при всех частотах. Важно отметить, что повышенная частота стимуляции связана с положительным лузитропным эффектом, укорочением времени до достижения 90% расслабления во всех группах. Наблюдалось небольшое различие между нетрансдуцированными миоцитами и cTnC ДТ-трансдуцированными миоцитами, но скорость расслабления была фактически выше в L48Q cTnC-миоцитах при стимуляции 1 Гц и 2 Гц. Также оценивали влияние частоты стимуляции на Ca2+-токи, который суммирован на Фигуре 3 для минимального уровня Ca2+ (3А), максимального уровня Ca2+ (3В) и времени до достижения 50% (3С) и 90% (3D) снижения Ca2+. Как и в случае сокращения, не наблюдалось различия в характере Ca2+-тока при повышенной частоте стимуляции между нетрансдуцированными и L48Q cTnC-трансдуцированными миоцитами. У cTnC ДТ-трансдуцированных кардиомиоцитов наблюдали слабое повышение минимального и максимального уровня Ca2+ при стимуляции 1 Гц по сравнению с нетрансдуцированными и L48Q cTnC-трансдуцированными миоцитами, но время до достижения 50% и 90% расслабления было одинаковым между группами при всех частотах стимуляции.
[0168] Так как наблюдалось малое различие между группами в минимальном и максимальном уровне Ca2+, изменения сократимости могут наилучшим образом объясняться изменением чувствительности миофиламентов к активации Ca2+. На Фигуре 4 это показано как эффективность сокращения, определенная в настоящей заявке как относительное укорочение кардиомиоцита, деленная на максимальную флуоресценцию Fura2 (пик Ca2+). У кардиомиоцитов, экспрессирующих L48Q cTnC, эффективность сокращения была значительно выше, чем у нетрансдуцированных или GFP-трансдуцированных кардиомиоцитов при всех частотах стимуляции. Примечательно, что наблюдалось значительное различие в эффективности сокращения при всех частотах стимуляции между cTnC ДТ и нетрансдуцированными миоцитами, что может указывать на то, что вызванное L48Q cTnC усиление сокращения недооценивают. Результаты стимуляции при 1 Гц и 2 Гц суммированы в Таблица 3 и Таблица 4, соответственно.
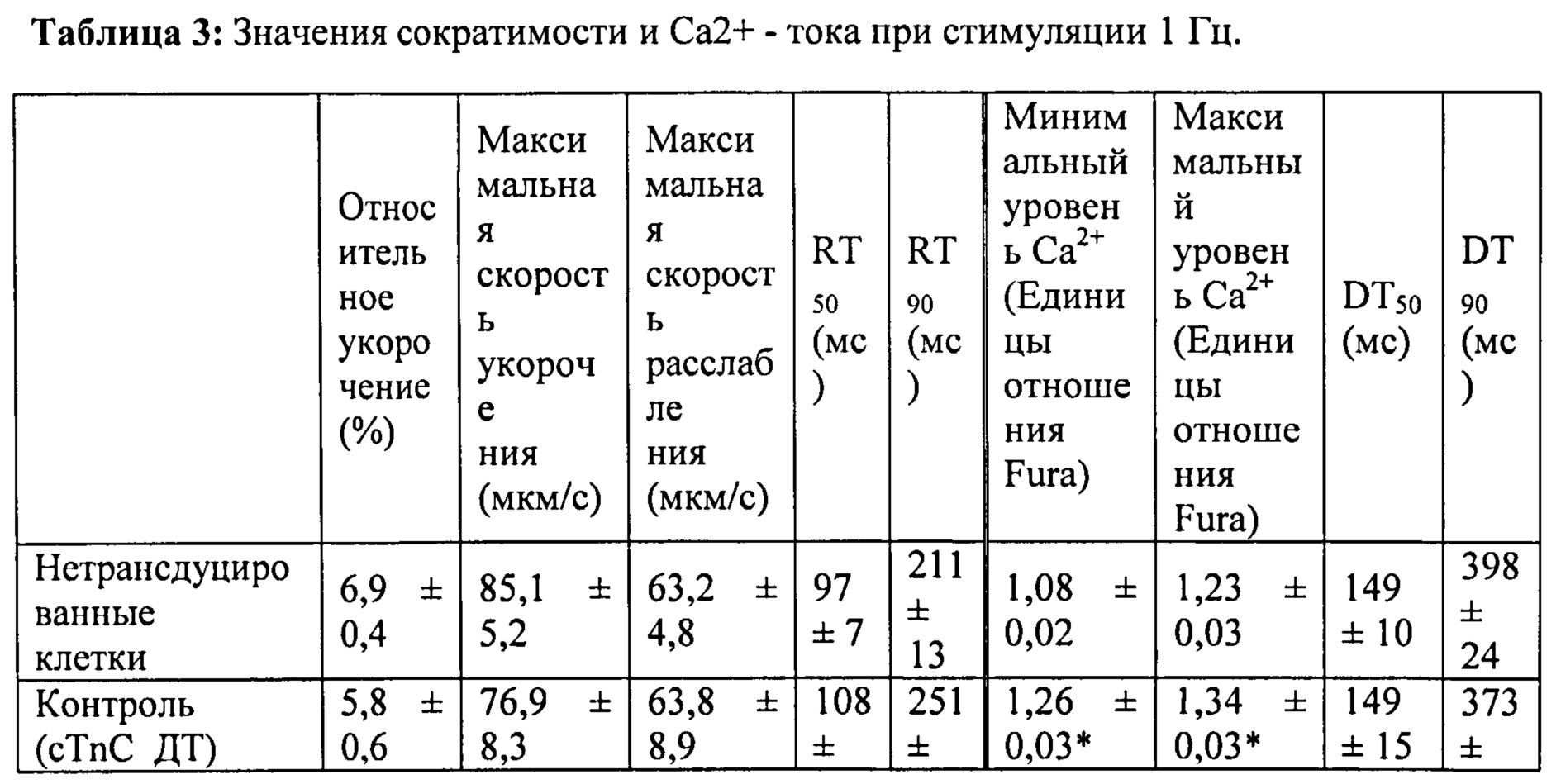
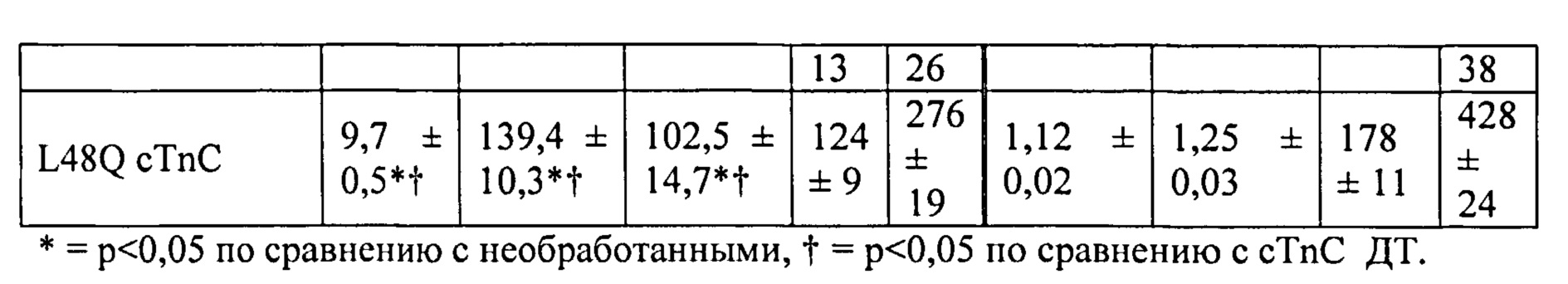
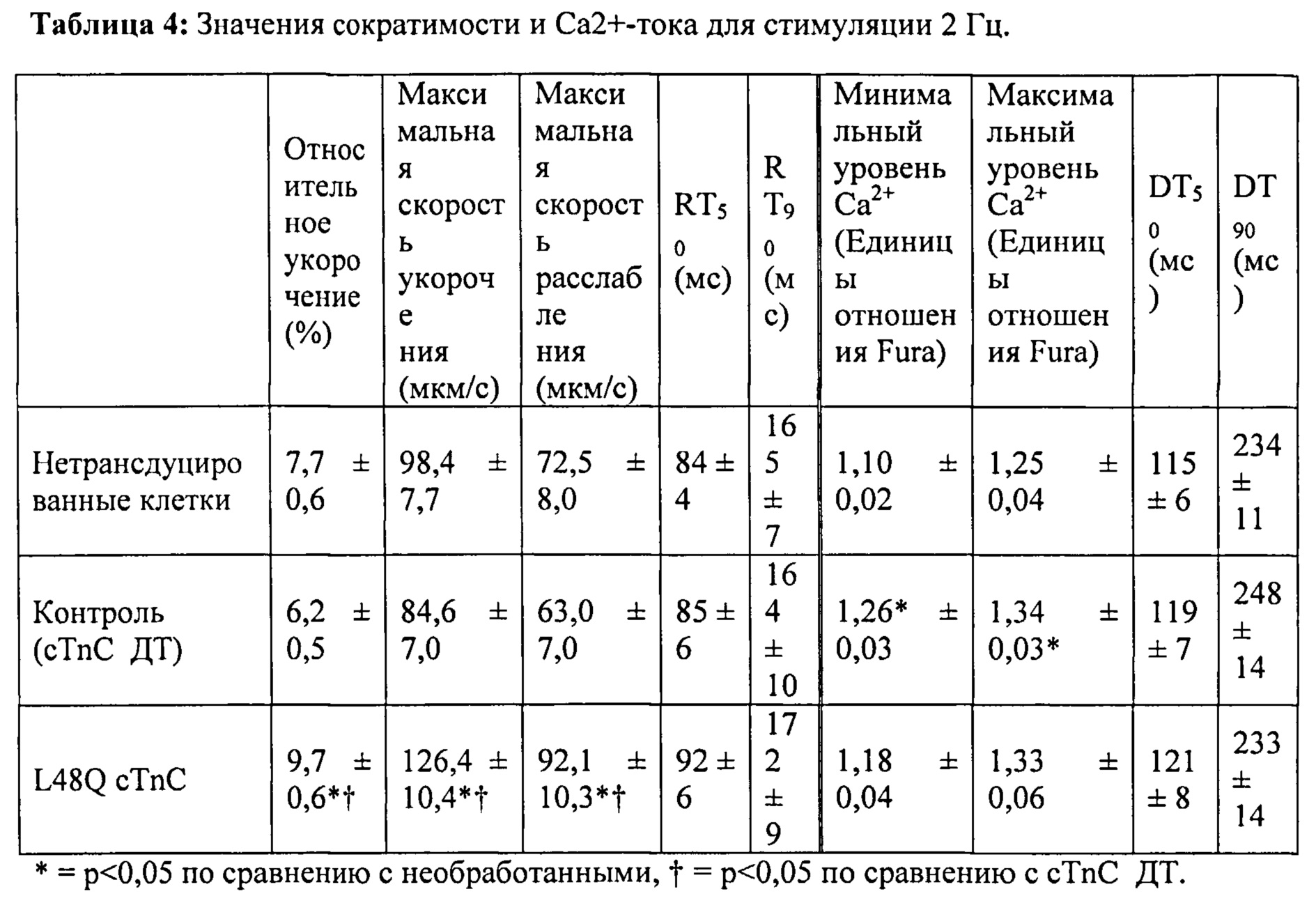
ПРИМЕР 5
[0169] Для определения уровня встраивания L48Q cTnC в миофиламенты культивируемые кардиомиоциты новорожденных крыс инфицировали аденовирусными конструкциями, содержащими гены [L48Q cTnC+GFP] или [cTnC ДТ+GFP], в течение 48 часов, или оставляли без обработки. Как и в случае кардиомиоцитов взрослых, выживаемость клеток в течение этого периода была качественно очень близкой для всех групп, указывая на то, что вирусные векторы не снижали жизнеспособность кардиомиоцитов in vitro. На успешный перенос гена указывала наблюдаемая зеленая флуоресценция (GFP) в инфицированных кардиомиоцитах при наблюдении помощью микроскопа. Гиперэкспрессия ДТ или L48Q cTnC и включение ДТ или L48Q cTnC в тонкие филаменты определяли с помощью Вестерн-блотов очищенных миофибрилл из трансдуцированных миоцитов, меченных анти-cTnC (Santa Cruz Biotechnology, Санта Круз, Калифорния) для определения общего содержания cTnC, или анти-His (Novagen, Gibbstown, NJ) для определения содержания трансдуцированного белка. Для оценки приблизительного уровня включения в миофиламенты трансдуцированных cTnC полосы, полученные с помощью Вестерн-блоттинга для миофибрилл трансдуцированных миоцитов, количественно оценивали с помощью дензитометрического метода для сравнения общего содержания cTnC по сравнению с содержанием His-меченного cTnC (трансдуцированный белок). Денситометрические калибровочные кривые для Вестерн-блоттинга были построены на основе известных количеств cTnC и cTnC-His. На Фигуре 5 показан типичный пример указанного эксперимента для двух популяций клеток с замещением ~70% (средние дорожки) и 56% (правые дорожки) нативного cTnC (Фиг.5А) указанным трансдуцированным cTnC-His (Фиг.5 В) и сравнением с очищенным cTnC-His с известной концентрацией (левые дорожки). Три популяции миоцитов анализировали в двух повторностях, и усредненные результаты указывали на замещение 58±7% нативного cTnC на L48Q cTnC.
ПРИМЕР 6
[0170] Обработка данных и статистический анализ. Скорость укорочения оценивали с использованием нелинейного регрессионного анализа, приведенного к одной трехпараметрической затухающей экспоненциальной функции с использованием уравнения: y=y0+ae-bx. Расслабление оценивали путем измерения времени до достижения 50%, 90% и 100% расслабления (RT50, RT90, и RT100, соответственно) и путем приведения к одной трехпараметрической экспоненциальной функции с использованием уравнения: y=y0+a(1-e-bx). Статистические различия определяли с помощью программы ANOVA. Различия при p-значении <0,05 считали статистически значимыми. Данные представляли как среднее ± стандартная ошибка среднего.
ПРИМЕР 7
[0171] Выделение и культивирование клеток взрослых и новорожденных животных. Указанные исследования были одобрены комитетом по содержанию лабораторных животных Вашингтонского университета (UW) и проводились в соответствии с федеральным руководством. За животными ухаживали в соответствии с политикой Национального института здравоохранения США по уходу за лабораторными животными и их использованию человеком Отдела сравнительной медицины Вашингтонского университета. Кардиомиоциты взрослых крыс (Fischer 344), ARC, выделяли из сердца с использованием аортальной ретроградной перфузии для ферментативной (коллагеназа/протеаза) диссоциации клеток13. Кардиомиоциты новорожденных крыс (NRC) выделяли путем ферментативной диссоциации из 1-3-дневных новорожденных крыс Fischer 344, как описано ранее14.
ПРИМЕР 8
[0172] Конструирование плазмид и вирусная продукция. Клетки HEK293 использовали для создания аденовирусных векторов15, экспрессирующих Rrm1 или Rrm2 с промотора CMV. Оба вектора содержали вторую кассету экспрессии для зеленого флуоресцентного белка (GFP) в качестве белка-репортера трансдукции. Мы также экспрессировали вектор только для GFP. Вирусы вводили в кардиомиоциты в концентрации ~250 частиц на клетку.
[0173] Для индукции гиперэкспрессии мышечной рибонуклеотидредуктазы 1 (Rrm1) и рибонуклеотидредуктазы 2 (Rrm2) в культивируемых кардиомиоцитов взрослых и новорожденных крыс использовали трансдукцию рекомбинантного аденовируса, содержащего соответствующие конструкции кДНК, запускаемые промотором CMV. Каждый аденовирус также содержал вторую кассету экспрессии для зеленого флуоресцентного белка (GFP), который использовали в качестве репортерного белка, идентифицирующего успешную трансдукцию. Кардиомиоциты инфицировали аденовирусом, содержащим гены для [Rrm1+GFP и Rrm2+GFP] или [GFP], в течение 2 дней. Успешный перенос гена, наблюдаемый по зеленой флуоресценции, наблюдаемая с помощью микроскопа, указывал приблизительно на 100% эффективность трансфекции. Это согласуется с предыдущими исследованиями с использованием кардиомиоцитов17. Выживаемость клеток во время этого периода была одинаковой для всех групп, включая нетрансдуцированные контрольные клетки, что указывало на то, что указанные вирусные векторы не снижали жизнеспособность кардиомиоцитов. Число кардиомиоцитов и длины саркомеров суммированы в Таблице 5. Не наблюдалось различий в длине саркомеров в состоянии покоя между группами, что указывает на то, что гиперэкспрессия Rrm1+Rrm2 (или GFP) не приводила к повышению кальций-независимой активации.
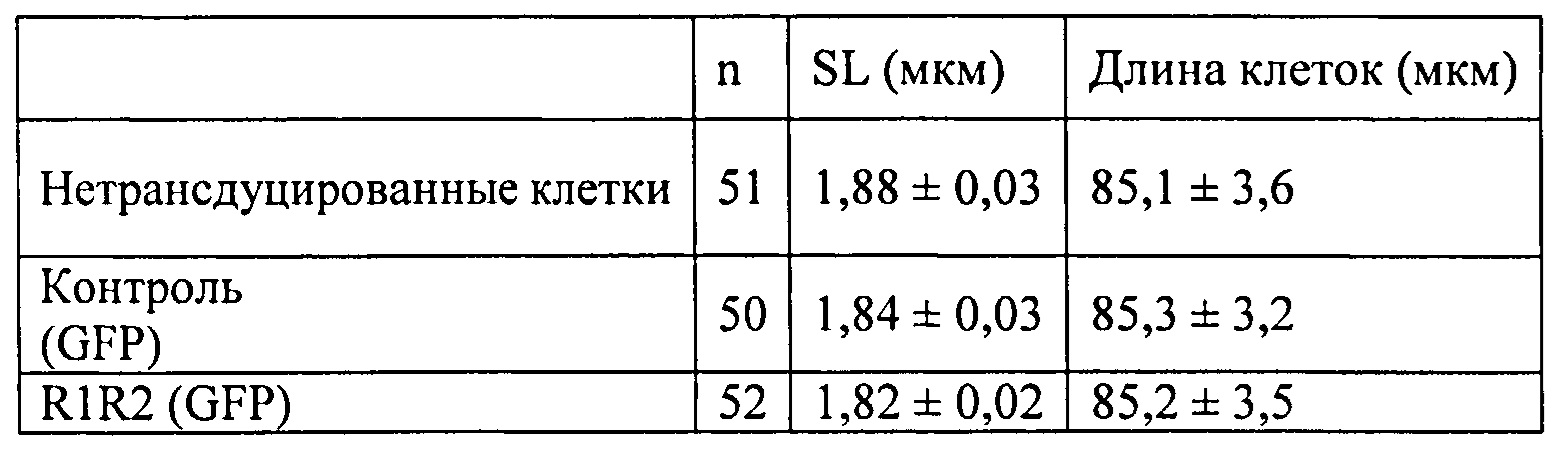
ПРИМЕР 9
[0174] Оценки сократимости. В модифицированном буфере Тирод при комнатной температуре регистрировали укорочение и расслабление клеток произвольно выбранных стимулированных кардиомиоцитов с использованием системы видеомикроскопии IonOptix. (IonOptix, Милтон, Массачусетс, США). Кальциевые токи, вызванные электрической стимуляцией, измеряли в клетках, нагруженных Fura2, с использованием оборудования IonOptix, как описано16. Флуоресценцию Fura2 измеряли с использованием спектрофотометра IonOptix (Stepper Switch), соединенного с флуоресцентным микроскопом. Испускаемую флуоресценцию Fura2 собирали с помощью 40Х объектива, пропускали через 510 нм фильтр и выявляли с помощью трубки фотоумножителя.
[0175] Эффекты гиперэкспрессии Rrm1+Rrm2 на степень и скорость стимулированного укорочения-повторного удлинения кардиомиоцитов взрослых крыс определяли путем видеодетекции длины (IonOptix). На Фигуре ба показаны типичные кривые укорочения, и на Фигуре 6b показаны типичные Ca2+-токи (Fura2 флуоресценция) для необработанных (черный), только GFP-трансдуцированных (зеленый) и Rrm1+Rrm2-трансдуцированных (красный) кардиомиоцитов. Данные для всех измерений при стимуляции 0,5 Гц суммированы в Таблица 6. Ранее был описано неблагоприятное влияние GFP на сократимость18, тогда как другие авторы не наблюдали указанного эффекта16,19. Хотя оказалось, что GFP не действует как ингибитор сокращения в настоящем исследовании, он увеличивал на 90% время расслабления, которое сопровождалось на 50% и 90% более медленным угасанием Ca2+-тока (Фигура 6c, Таблица 5). Был ли указанный эффект обусловлен повышенной активностью SERCA или более быстрой циклической активностью поперечных мостиков, приводящей к вызванной укорочением инактивации тонких филаментов и высвобождению Ca2+ от тропонина С, является объектом дальнейших исследований. Несмотря на это, кардиомиоциты, трансдуцированные Rrm1+Rrm2 (+GFP), имели значительно большую величину и скорость укорочения по сравнению с нетрансдуцированными кардиомиоцитами и только GFP-трансдуцированными контролями. Это показано на Фигуре 6 с, которая описывает % различия в скорости и степени укорочения, скорости расслабления и времени до достижения 50% и 90% расслабления. Гиперэкспрессия Rrm1+Rrm2 приводила к повышению скорости расслабления и снижению времени до достижения 50% расслабления, и оценка указанного эффекта может быть немного занижена из-за присутствия GFP. На Фигуре 6d показано % различие в свойствах Ca2+-тока, включая минимальный и максимальный уровень Ca2+ и время до достижения 50% и 90% снижения Ca2+. Не наблюдали значительного эффекта GFP или Rrm1+Rrm2+GFP на минимальный и максимальный уровень Ca2+, что указывает на то, что усиление сократимости в случае Rrm1+Rrm2, в первую очередь, было опосредовано повышенной чувствительностью миофиламентов к активации Ca2+. Примечательно, что гиперэкспрессия Rrm1+Rrm2 ускоряла повторную секвестрацию Ca2+, на что указывает сокращение времени до достижения 50% и 90% снижения, что может отчасти объяснять повышенную максимальную скорость расслабления кардиомиоцитов.
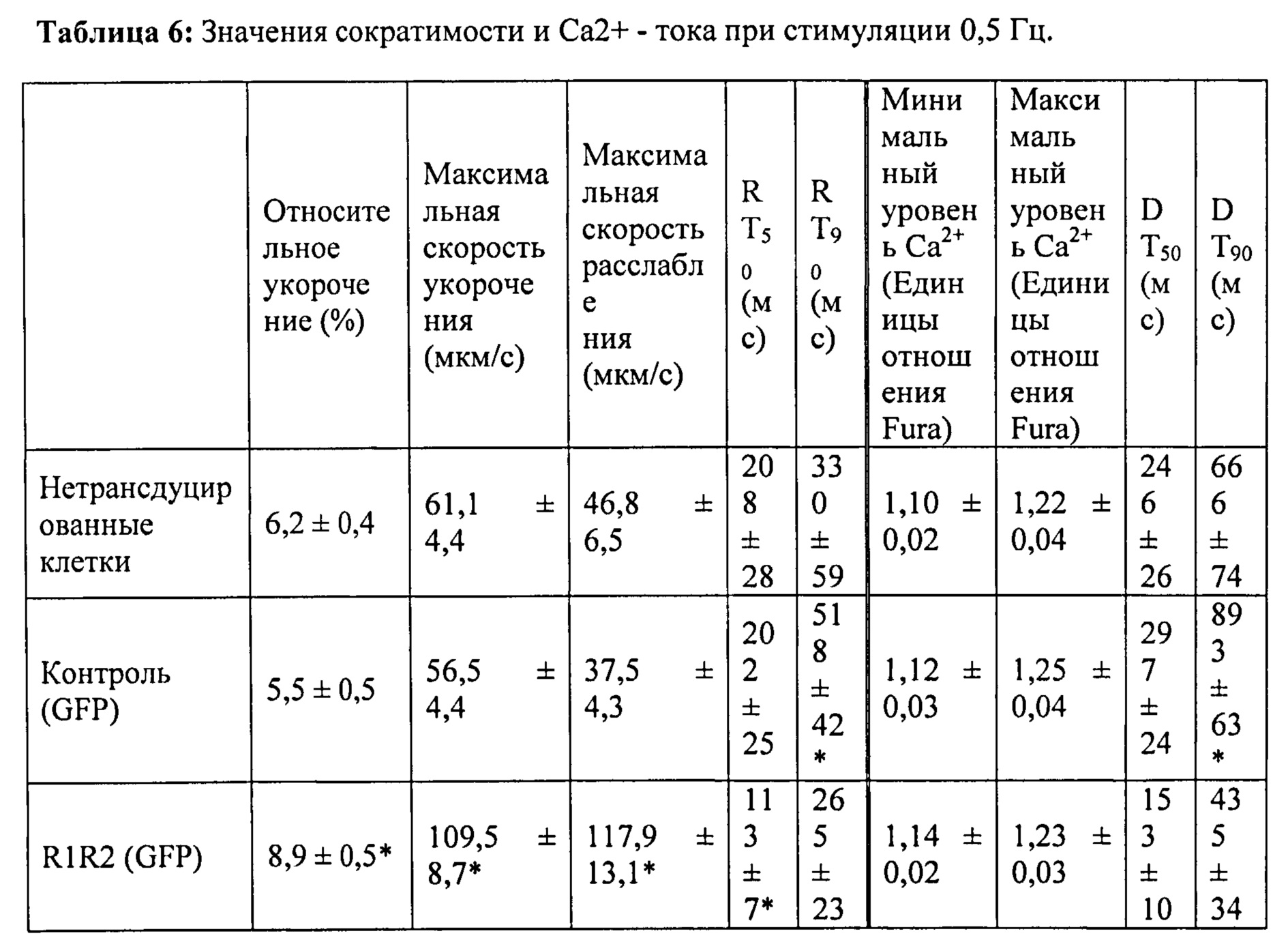

ПРИМЕР 10
[0176] Важно определить, влияет ли гиперэкспрессия Rrm1+Rrm2 на нормальный клеточный ответ на повышенные частоты стимуляции, поскольку изменения в частоте сердечных сокращений являются нормальной физиологической адаптацией к системным потребностям организма. На Фигуре 7 собраны данные о влиянии повышения частоты стимуляции (от 0,5 до 1 до 2 Гц) на относительное укорочение (7а), скорость укорочения (7b), скорость расслабления (7с) и время до достижения 90% расслабления (7с). Сократительный ответ на частоту стимуляции был одинаковым в разных группах, и Rrm1+Rrm2 трансдуцированные кардиомиоциты сохраняли усиленную функцию при всех частотах. Важно отметить, что повышенная частота сердечного ритма связана с положительным лузитропным эффектом, уменьшением времени до достижения 90% расслабления во всех группах. Наблюдалось небольшое различие между нетрансдуцированными миоцитами и трансдуцированными только GFP миоцитами, за исключением того, что время до достижения 90% расслабления было больше при 0,5 Гц у только GFP-трансдуцированных миоцитов. Также оценивали влияние частоты стимуляции на Ca2+, показанный на Фигуре 8 для минимального уровня Ca2+ (8а), максимального уровня Ca2+ (8b) и времени до достижения 50% (8с) и 90% (8d) снижения Ca2+ (DT50, DT90). Как и в случае сокращения, не наблюдали различий в характере Ca2+-тока при повышенной частоте стимуляции между нетрансдуцированными и Rrm1+Rrm2 - трансдуцированными миоцитами. Только GFP-трансдуцированные кардиомиоциты имели небольшое повышение минимального уровня Ca2+ при 2 Гц и повышение максимального уровня Ca2+ при 1 Гц и 2 Гц по сравнению с нетрансдуцированными миоцитами, но время до достижения 50% и 90% угасания было одинаковым. Как и в случае стимуляции 0,5 Гц, время до достижения 50% и 90% снижения сокращалось (более быстрое снижение) у Rrm1+Rrm2-трансдуцированных миоцитов при обеих частотах стимуляции, 1 и 2 Гц.
[0177] Поскольку наблюдалось небольшое различие между группами по минимальному и максимальному уровню Ca2+, изменения сократимости могут наилучшим образом объясняться изменениями в чувствительности миофиламентов к активации Ca2+, что показано на Фигуре 9 как эффективность сокращения, определенная в настоящей заявке как относительное укорочение кардиомиоцитов, деленная на максимальную флуоресценцию Fura2 (пик Ca2+). Кардиомиоциты, экспрессирующие Rrm1+Rrm2, имели значительно более высокую эффективность сокращения, чем нетрансдуцированные или GFP-трансдуцированные кардиомиоциты при всех частотах стимуляции. Не наблюдалось различий в эффективности сокращения между только GFP-трансдуцированными или нетрансдуцированными миоцитами, за исключением частоты стимуляции 2 Гц, что, в первую очередь, может объясняться повышенным максимальным уровнем Ca2+ у только GFP-трансдуцированных миоцитов в отсутствие повышения фракции укорочения, снижающего эффективность. Результаты по стимуляции 1 Гц и 2 Гц суммированы в Таблица 7 и Таблица 8, соответственно.
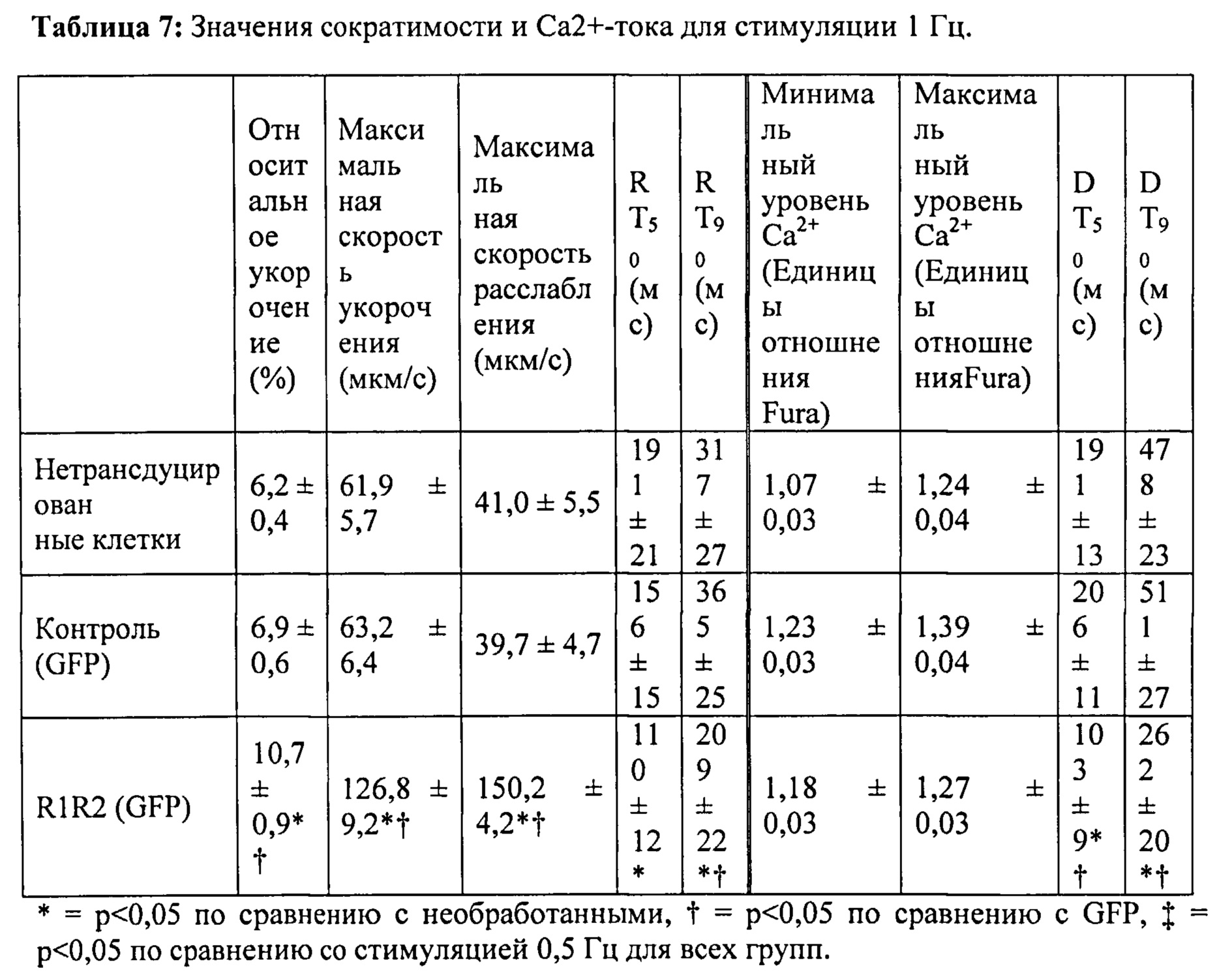
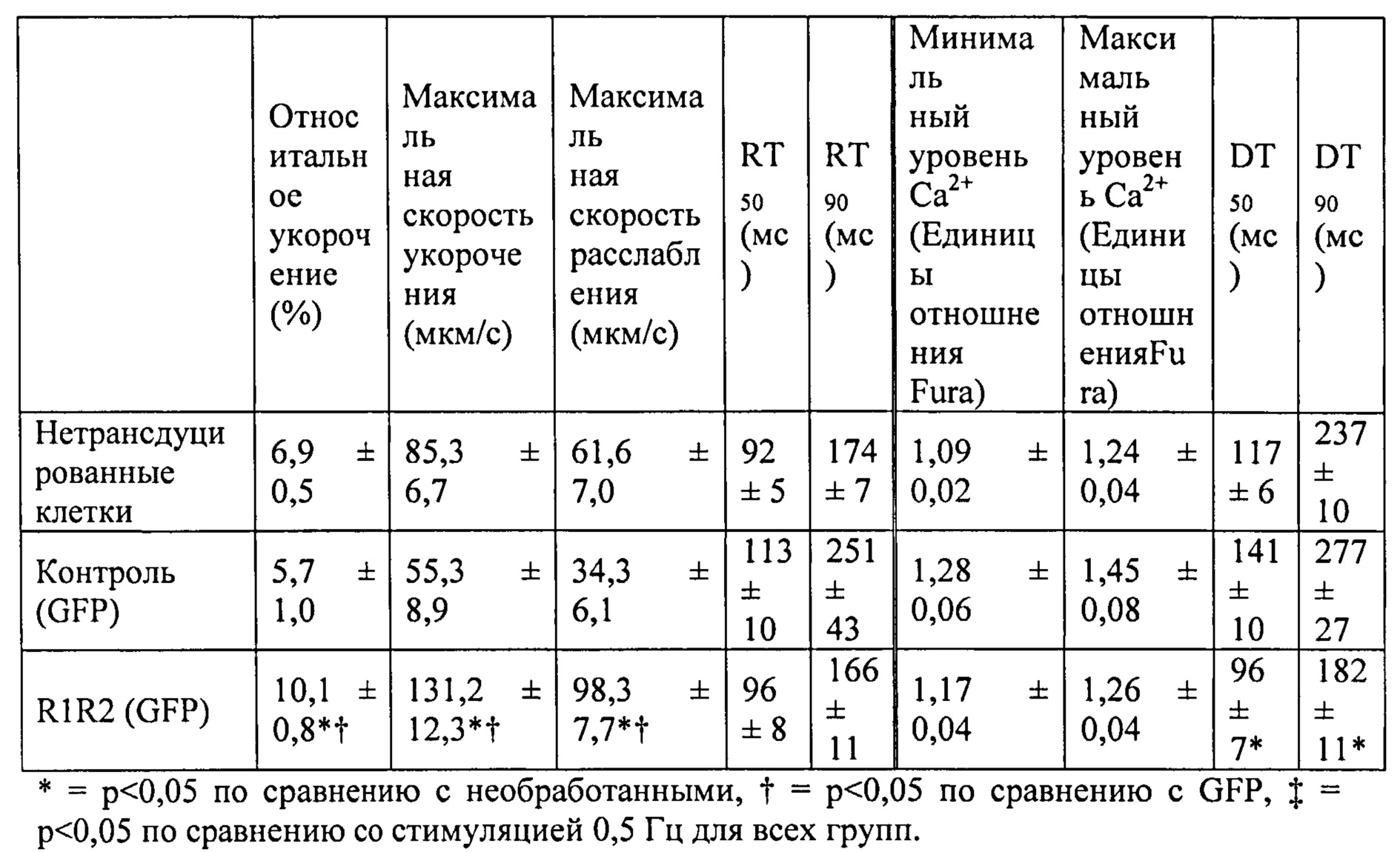
ПРИМЕР 11
[0178] Для подтверждения повышения уровня мРНК RR, белка RR и продукции дезокси-АТФ в Rrm1+Rrm2-трансдуцированных клетках, кардиомиоциты новорожденных крыс собирали и обрабатывали для проведения ПЦР в реальном времени, вестерн-блоттинга и ВЭЖХ-анализа внутриклеточной концентрации [АТФ] и [дезокси-АТФ]. Кардиомиоциты новорожденных животных использовали для достижения достаточно высокой плотности клеток для возможности точного анализа содержания нуклеотидов, поскольку известно, что внутриклеточный уровень [дезокси-АТФ] находится в пикомолярном (рМ) диапазоне. Как и в случае кардиомиоцитов взрослых животных, сократимость кардиомиоцитов новорожденных животных была значительно повышена при гиперэкспрессии R1R2. Примечательно, что поскольку кардиомиоциты новорожденных животных использовали для изучения эффектов приживления клеточного трансплантата после инфаркта миокарда14, улучшенная сократимость в указанных клетках может представлять собой другой механизм улучшения сердечной функции после инфаркта. Уровень мРНК Rrm1 и Rrm2 был значительно повышен после аденовирусной трансфекции. Параллельно с этим, на Фигурах 10а и 10b показано, что в Rrm1 и Rrm2-трансдуцированных кардиомиоцитах содержание белков Rrm1 и Rrm2 было повышено более чем в 24 раза и 46 раз, соответственно. GAPDH использовали в качестве контроля нагрузки. На Фигуре 10с показано, что Rrm1+Rrm2-трансдуцированные кардиомиоциты имели в ~10 раз повышенную клеточную концентрацию [дезокси-АТФ] по сравнению с GFP-трансдуцированными кардиомиоцитами (повышение до 0,35 нмоль/кг белка). Несмотря на то указанное повышение было значительным, поскольку [дезокси-АТФ] в норме включает менее 0,2% общего аденинтрифосфатнуклеотида, указанное повышение [дезокси-АТФ] представляло собой только ~1-1,5% общего пула адениннуклеотидов. Указанные данные предполагают, что для значительного повышения сократимости кардиомиоцитов требуется только малое количество дезокси-АТФ.
ПРИМЕР 12
[0179] Обработка данных и статистический анализ. Скорость укорочения и снижения Ca2+ оценивали с использованием нелинейного регрессионного анализа, приведенного к одной трехпараметрическому угасающей экспоненциальной функции с использованием уравнения: y=y0+ae-bx. Скорость расслабления и подъем Ca2+ оценивали путем приведения к одной трехпараметрической экспоненциальной функции с использованием уравнения: y=y0+a(1-e-bx). Расслабление и снижение Ca2+ также оценивали путем измерения времени до достижения 50% (RT50) и 90% (RT90) расслабления. Статистические различия определяли с помощью программы ANOVA с использованием критерия Стьюдента-Ньюмана-Кейлса в качестве апостериорного парного критерия (SigmaPlot 11). Различия при p-значении <0,05 считали статистически значимыми. Данные приведены как среднее ± стандартная ошибка среднего.
ПРИМЕР 13
[0180] Для изучения молекулярных механизмов регуляции сокращения и расслабления сердечными тонкими филаментами, мы сконструировали и охарактеризовали несколько вариантов cTnC. Как суммировано в Таблица 9, 1) при включении в целые cTn комплексы они связывают Ca2+(в растворе) с аффинностью (KA) в порядке: L48Q>WT>L57Q>I61Q [KA=kon/koffj (измерено методом спектроскопии стабильного состояния), 2) общие скорости диссоциации Ca2+ (koff) от cTn, (измерено с помощью спектроскопии с прерыванием потока) повышались в таком же порядке, как и КА, и 3) указанные данные хорошо коррелируют со снижением Ca2+-чувствительности (рСа50) силы при замещении вариантов в очищенных сердечных трабекулах.
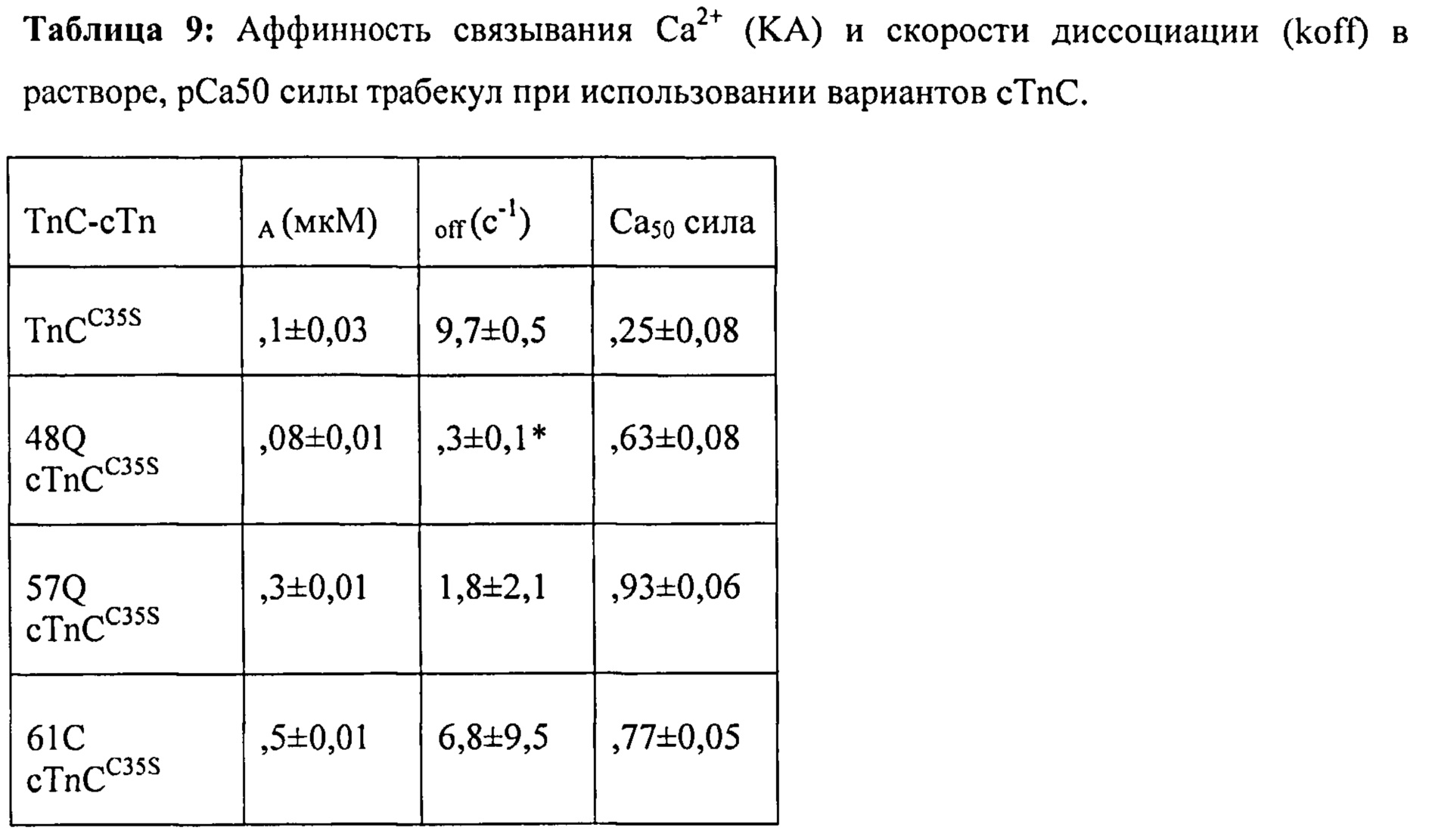
[0181] Для каждого варианта создавали аденовирусные векторы (AV) и трансдуцировали в культивируемые кардиомиоциты взрослых крыс (~250 вирусных частиц на клетку) и идентифицировали по одновременной экспрессии GFP. Данные по укорочению и внутриклеточному Ca2+-току собраны в Таблица 10. Данные (0,5 Гц стимуляция) показывают, что величина и скорость укорочения снижались в порядке L48Q>WT>L57Q>I61Q, который соответствует порядку для аффинности в растворе Ca2+ и Ca2+-чувствительности очищенной сердечной мышцы. Все варианты cTnC в AV конструкции содержали С-концевую гистидиновую метку, что позволяло проводить ПААГ-электрофорез в присутствии ДСН и Вестерн-блоттинг для количественной оценки включения в миофиламенты. Указанный уровень, как правило, варьировал от 40-60% при нагрузке AV, которую мы обычно использовали. Примечательно, что L48Q cTnC не оказывал эффект на амплитуду Ca2+-тока, что дает основания полагать, что повышенное укорочение является результатом исключительно повышенной Ca2+-чувствительности миофиламентов. Напротив, L57Q и I61Q cTnC снижали эффективность укорочения/расслабления и снижали исходную амплитуду Ca2+-тока. Одновременное снижение укорочения, амплитуды Ca2+-тока и +kFL указывает на взаимодействие миофиламентов и СПР уже через 48 часов в культуре.
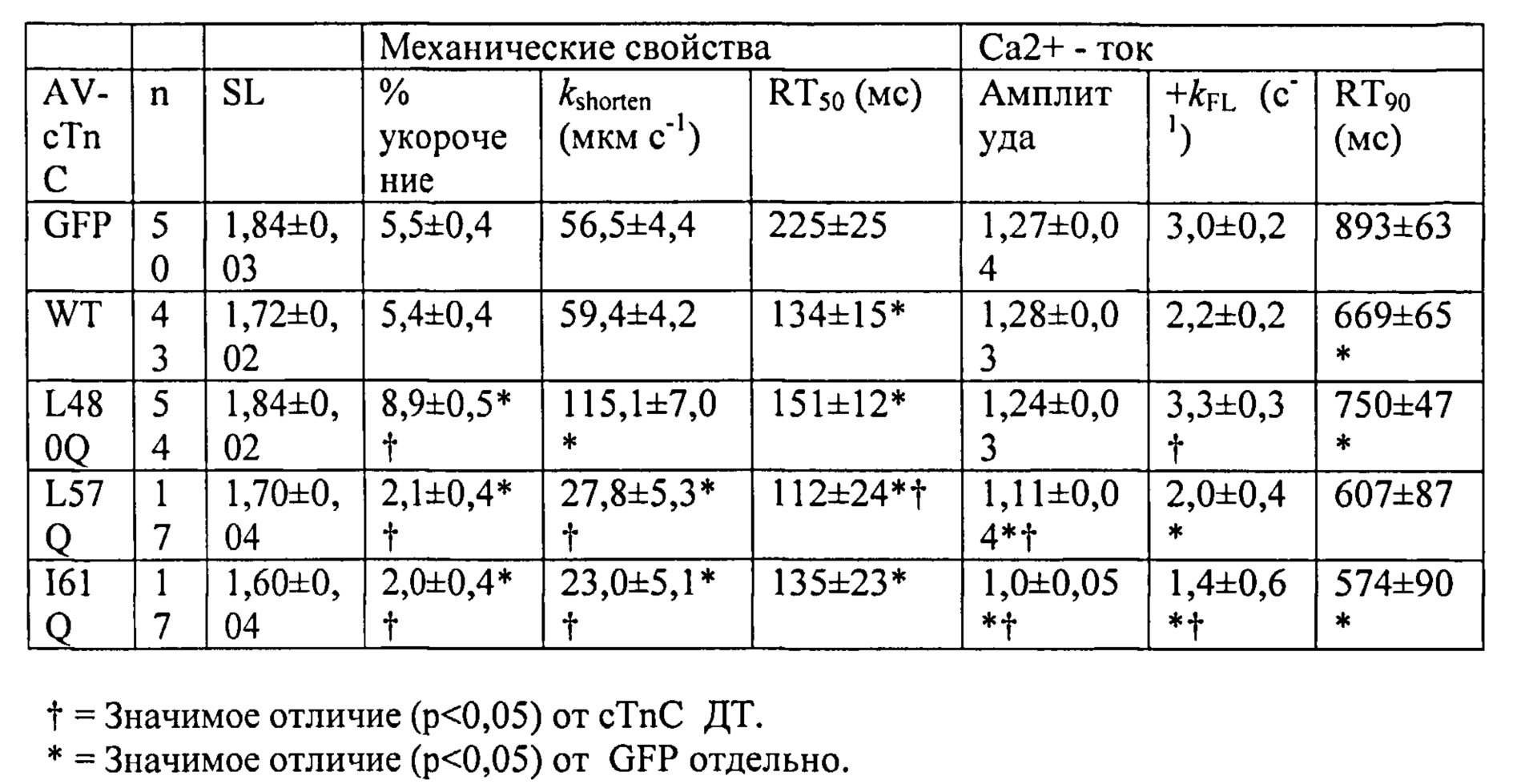
[0182] На основании данных, представленных в Таблица 9 и Таблица 10, мы определили, что варианты cTnC, которые повышают/снижают Ca2+-чувствительность силы сокращения в очищенной сердечной мышце, также повышают/снижают величину и скорость сокращения в культивируемых кардиомиоцитах. Примечательно, что как L57Q, так и I61Q cTnC снижали сократимость с сопутствующим снижением Ca2+-тока, тогда как L48Q cTnC повышал сократимость без изменения амплитуды Ca2+-тока или замедления расслабления (Таблица 10). Это происходило даже в тех случаях, когда указанные варианты только частично замещали нативные белки в тонких филаментах кардиомиоцитов, что демонстрирует, что варианты cTnC могут оказывать существенное влияние на сокращение миофиламентов и влиять на функцию СПР в отсутствие полного замещения нативного cTnC. Указанные гетерогенные популяции нативных и вариантных видов также имитируют различную пенетрантность мутаций на животных моделях и при заболевании человека, и, вероятно, будут совпадать с профилем встраивания в исследованиях на сердце in vivo.
[0183] Укорочение и Ca2+-токи были снижены в кардиомиоцитах сердец, пораженных инфарктом (4 недели) в результате длительного лигирования левой нисходящей коронарной артерии (Фигура 11). Нарушенная сократительная активность может, в первую очередь, являться результатом сниженных Ca2+-токов (Таблица 11). Важно отметить, что экспрессия L48Q cTnC восстанавливала как укорочение, так и амплитуду Ca2+-тока указанных кардиомиоцитов (Фигура 11, Таблица 11) в отличие от нормальных кардиомиоцитов, в которых усиливалось только укорочение. Указанные данные предполагать, что экспрессия L48Q cTnC влияет не только на Ca2+-чувствительность сокращения миофибрилл, но также может влиять на функцию СПР в миокарде, страдающим недостаточностью. Это, вероятно, обусловлено вклеенным в миофиламенты cTnC, а не цитозольным пулом cTnC, так как трансфекция AV-cTnC ДТ не приводила к изменению характера сократимости или Ca2+-тока (данные не представлены).
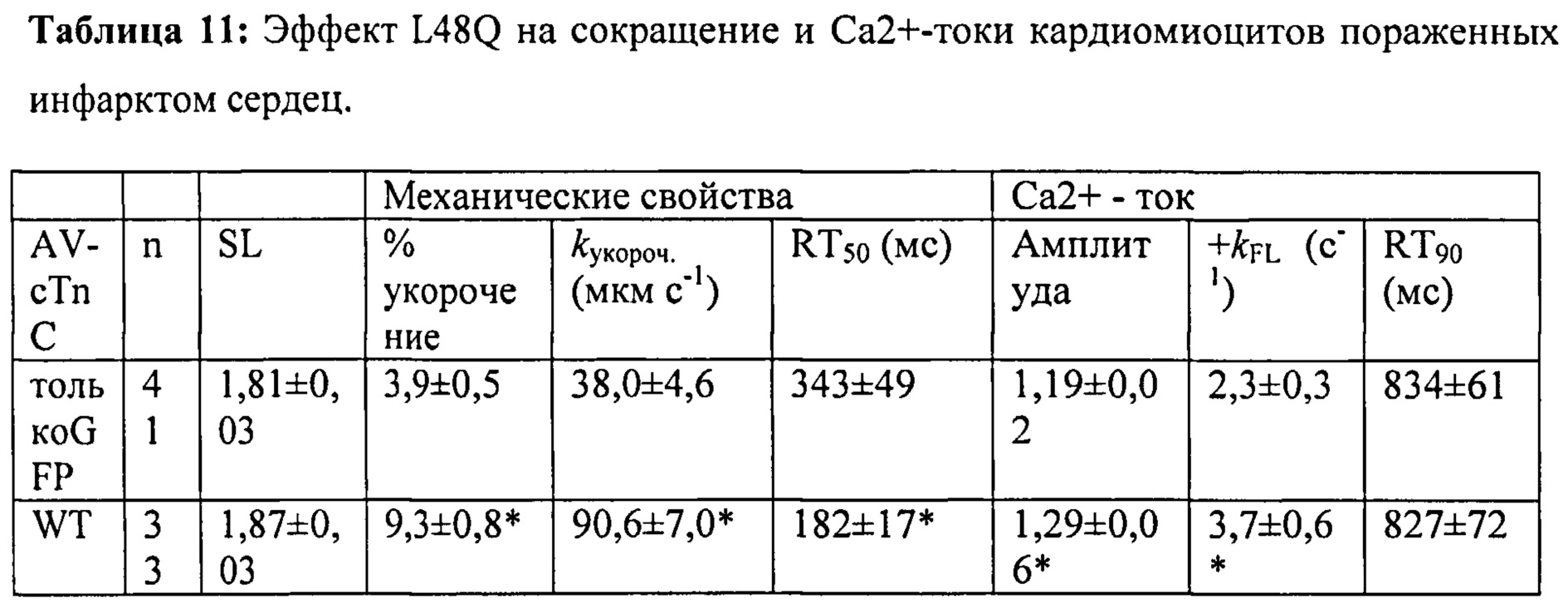
[0184] Изменения в сократимости и Ca2+-токах происходили в культуре в отсутствие стимуляции, так как взрослые кардиомиоциты были в состоянии покоя до начала анализа (48-72 часа), что дает основания полагать, что изменение Ca2+-токов может являться результатом изменений в пост-трансляционной модификации (фосфорилировании) белков СПР и/или сарколеммы, а не измененного содержания белков. Для демонстрации значимости стимуляции, мы сравнили укорочение и амплитуду Ca2+-тока у не стимулированных (в состоянии покоя) по сравнению с хронически стимулированными (1 Гц) кардиомиоцитами (Фигура 17) и обнаружили повышенное на 45% укорочение, увеличенную на 28% амплитуду Ca2+-тока и на >50% более быстрый подъем (+kFL) и угасание (kDecay) Ca2+-тока для стимулированных клеток через 30 часов после посева.
ПРИМЕР 14
[0185] Доставка гена с помощью кардиоспецифичного промотора AAV6+. Векторы rAAV6 олучают, как описано ранее

[0186] На Фигуре 12 показано первое применение AAV6-L48Q cTnC, который системно инъецировали (интраокулярно) 3 мышам, каждой в низкой (L; 0,6×1012) и высокой (Н; 1,2×10) дозе вирусных частиц. Эхокардиография показала ~20% повышение фракции выброса левого желудочка (ЛЖ) по сравнению с контролями без инъекции (UN) через две недели после инъекции, и 30-40% повышение через 3 недели. Несмотря на то, что для подтверждения указанных результатов необходимо провести имитацию инъекции и инъекцию контроля AAV6, системное введение контрольных AV-векторов не приводило к изменению функции ЛЖ. Белки миофибрилл из одной AAV6 L48Q cTnC-myc трансдуцированной мыши (и контроле без инъекции) разделяли путем ПААГ-электрофореза в присутствии ДСН, и Вестерн-блоты окрашивали антителами к cTnC. Присутствие myc-метки приводило к более медленной миграции cTnC (Фигура 13), и с помощью денситометрии было показано, что отношение cTnC-myc к нативному cTnC составляло ~40% (подобно наблюдаемому для аденовируса и трансгенных животных, ниже).
ПРИМЕР 15
[0187] Доставка гена с помощью аденовирусов (AV). AV векторы были наиболее эффективны в культуре, таким образом, для некоторых экспериментов мы будем продолжать использовать их для трансфекции варианта cTnC в кардиомиоциты. AV векторы продуцировались клетками HEK293 с репортерным белком GFP, экспрессируемым в виде отдельной кассеты в том же вирусе для идентификации трансдуцированных клеток (Фигура 14). Указанные векторы обычно обеспечивают 40-60% замещения нативного cTnC в миофибриллах культивируемых кардиомиоцитов. Указанный уровень соответствует уровню, который достигается для L48Q и I61Q cTnC-индуцибельных мышей из группы Молкентина (Molkentin, ниже).
ПРИМЕР 16
[0188] Применение трансгенных L48Q и I61Q cTnC мышей. Были получены трансгенные мыши, которые экспрессируют cTnC дикого типа (WT), L48Q или I61Q cTnC. Все конструкции используют индуцируемый кардиоспецифичный промотор тяжелой цепи а миозина (α-МНС)
[0189] Поскольку flag-эпитоп приводит к более низкой подвижности полосы cTnC (Фигура 15), отношение трансгенного cTnC к нативному cTnC определяли с помощью денситометрического метода. Отношение flag-меченного cTnC к общему cTnC представляет собой замещение в %. Линии с самой высокой экспрессией для каждой конструкции соответствовали ~40-50% замещению, подобному ожидаемому замещению для пациентов, страдающих наследственной кардиомиопатией. Кроме того, мы показали, что указанное % замещение нативного cTnC вариантом cTnC является достаточным для достижения повышенной Ca2+-чувствительности в очищенных препаратах и повышенного укорочения в интактных миоцитах. Мы подтвердим это с использованием титрационных отношений (ДТ:мутант) в очищенных трабекулах и культивируемых кардиомиоцитах для сравнения с животными моделями. Flag-антитела также доказывают сильную экспрессию трансгена у би-трансгенных мышей (DTG, ТТА+ и cTnC+), но также показывают слабую потерю индуцибельного промотора α-МНС у мышей, которые были положительными только на трансген.
ПРИМЕР 17
[0190] Применение трансгенных R92W/L cTnT мышей. Мутации, аналогичные мутациям, встречаемым у пациентов-людей, страдающих кардиомиопатией. У гетерозиготных мышей наблюдалось включение ~50% (диапазон 35-70%) варианта cTnT в миофиламенты и наблюдалась умеренная гипертрофия. Очищенная сердечная ткань, полученная от указанных мышей, имела повышенную Са2+ -чувствительность (0,3-0,4 рСа единиц) при маленькой (1,9 мкм) и большой (2,3 мкм) длине саркомера (SL)
ПРИМЕР 18
[0191] Получение пораженных инфарктом сердец. Инфаркт миокарда моделировали путем длительного лигирования левой нисходящей коронарной артерии. Это, как правило, приводит к образованию зоны инфаркта, составляющей ~30-35% площади поперечного сечения свободной стенки левого желудочка (10-15% от общей площади ЛЖ) с пониженной фракцией укорочения и повышенным расширением полости по результатам эхокардиографии (Фигура 16А-С) и потерей чувствительности к повышенной предварительной нагрузке по результатам измерений на работающем сердце (Фигура 6D).
ПРИМЕР 19
[0192] Мы будем использовать культивируемые кардиомиоциты взрослых животных, культивируемые трабекулы и препараты миофибрилл для изучения влияния вариантов cTnC на сокращение и функцию СПР. Хотя культивируемые кардиомиоциты обеспечивают подробную информацию о функции СПР во время циклов укорочения/расслабления, клетки не подвергаются внешнему растяжению, как в сердце. Культивируемые трабекулы позволяют измерять изометрическую силу и укорочение при нагрузке, наряду с анализом Ca2+-токов. Мы будем использовать маленькие тонкие трабекулы, включающие несколько слоев клеток (в глубину) для максимизации диффузии и избегания изменений сигнала из-за градиентов проникновения и диффузии флуорозонда внутри препарата. Измерения механических свойств изолированных миофибрилл позволят подробное изучение кинетики сокращения и расслабления во время максимальной и суб-максимальной активации Ca2+ и при различных (контролируемых) и поддающихся количественному определению уровнях вариантов cTnC, включенных в тонкие филаменты миофибрилл. В Таблица 12 суммированы эксперименты, проведенные для указанной цели, включая препараты, различные условии стимуляции (трансфекция AV векторами в течение 24-48 часов), условия, которые предполагается проверить, и измерения, сделанные в конце эксперимента по стимуляции.

[0193] Как показано в Таблица 12, после трансфекции культивируемых кардиомиоцитов взрослых крыс векторами AV для cTnC ДТ, L48Q, L57Q или I61Q, мы измерим сократимость и Ca2+-токи не стимулированных клеток по сравнению с клетками, подвергнутыми стимуляции. Краткосрочная стимуляция при частоте 1 Гц (0-30 мин.) обеспечит информацию по острому адаптивному ответу миофиламентов и функции СПР, ассоциированной с быстрыми адренергическими ответами, тогда как более длительная стимуляция (1-24 часов) даст информацию о более долгосрочном адаптивном ответе, включая регуляцию транскрипции (определенную на основе ПЦР в реальном времени) белков миофиламентов, СПР, сарколеммы и связанных киназ. Первоначальные эксперименты позволят нам определить, изменит ли стимуляция сокращение, характер циклической активности Ca2+ и адренергическую чувствительность. Затем мы проведем независимое изучение указанных эффектов при следующих экспериментальных условиях.
[0194] Мы будем варьировать внеклеточную концентрацию [Ca2+] ([Ca2+]ext) для определения потенциального влияния на Ca2+-индуцированное высвобождение Ca2+ (CICR), и использовать ингибиторы поперечных мостиков (СВ) миофиламентов (BDM, блеббистатин) для определения влияния механики сокращения на функцию СПР. Мы будем использовать стимуляцию α-(фенилэфрином) или β-адренергическую (изопротеренол) стимуляцию для определения того, зависит ли указанный ответ от вариантов cTnC и его взаимодействия с cTnI. Ингибиторы СВ и адренергические агонисты будут удалены до начала измерений сократимости и Ca2+-тока.
[0195] Для измерения укорочения и Ca2+-тока кардиомиоциты будет нагружены Fura-2-АМ, и за сокращением/расслаблением при стимуляции 0,5, 1, 2 и 3 Гц будут следить с помощью видеомикроскопии (IonOptix, Милтон, Массачусетс). Для измерений функции СПР, (Со-I), общий [Ca2+]i будут визуализировать в изолированных кардиомиоцитах, нагруженных проникающей через мембраны ацетоксиметилэфирной формой rhod-2 или fluo-4 с использованием конфокальной системы с качанием поля Nikon, соединенной с х60 линзой Nikon (NA=1,4), как описано ранее
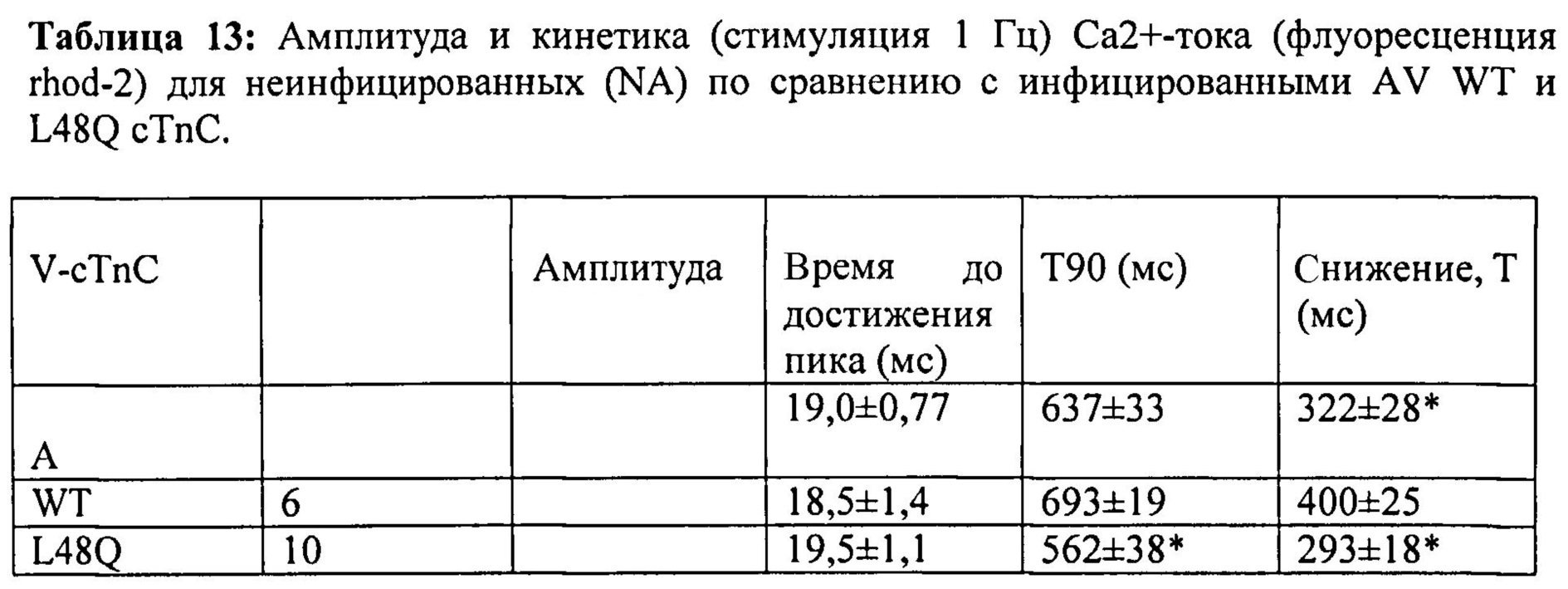
ПРИМЕР 20
[0196] Эксперименты на культивируемых и очищенных трабекулах: Мы определим, как варианты cTnC влияют на сокращение при внешней нагрузке и при различных длинах саркомеров (SL), как это происходит in vivo. Тонкие (диаметром<120 мкм, глубиной ~50 мкм) сердечные трабекулы будут культивировать, как описано Адлером и др. (Adler et al, 4
[0197] Первоначальные эксперименты позволят определить период трансфекции, который обеспечивает оптимальное включение мутанта cTnC в тонкие филаменты. Значительная продукция белка происходит в течение 24 часов трансфекции AV в культивируемых кардиомиоцитах, на что указывает сигнал GFP, и мы ожидаем получить подобные результаты на трабекулах. Сократимость и Ca2+-токи будут измерены после стимуляции при 1 Гц (Таблица 12). Препараты будут нагружены Fura-2 и помещены между силовым преобразователем и линейным двигателем в насыщенной кислородом перфузионной камере, содержащей стимулирующие электроды
[0198] Для получения подробной информации о том, как вариант cTnC влияет на сокращение миофиламентов при контролируемых субмаксимальных уровнях Ca2+, мы будем химическим путем разрушать мембрану трансдуцированных трабекул (очищать) после приложения условий стимуляции. Мы измерим рСа в зависимости от силы и скорость повторного развития силы (kTR) и укорочения при нагрузке как длинная и короткая SL. В дополнительных экспериментах очищенные трабекулы будут заменять на смеси вариантов cTnC и cTnC ДТ для определения критического уровня включения варианта (в тонкие филаменты), влияющего на сокращение. Будет проведена количественная оценка включения варианта cTnC в миофиламенты (см. ниже).
ПРИМЕР 21
[0199] Эксперименты на миофибриллах: Расслабление миофиламентов является важным компонентом нормальной сердечной функции; желудочки должны быстро расслабляться после систолы для обеспечения достаточного диастолического наполнения. Указанный важный процесс часто нарушен при патологических состояниях. Расслабление трудно оценивать на очищенных трабекулах, в которых велики диффузионные расстояния. В отличие от них, в препаратах миофибрилл малые диффузионные расстояния, которые дают возможность быстро менять растворы и измерять силу в пределах миллисекунд, что позволяет оценивать влияние вариантов cTnC на активацию и расслабление миофибрилл независимо от Ca2+-токов в интактных клетках. В раздельных экспериментах после стимуляции из измельченной сердечной мышцы, трансдуцированной вариантом cTnC, будут получены препараты миофибрилл для измерения Ca2+-зависимости скорости развития силы (kact) и kTR и скорости медленного (kREL, медленное) и быстрого (kREL, быстрое) расслабления. Примеры влияния рекомбинантных cTnC WT, L48Q и I61Q, которые замещали в сердечных миофибриллах крыс, на указанные параметры, показаны на Фигуре 19. На рисунке В показано небольшое, но значимое повышение kREL, скорости медленного расслабления и времени до 50% расслабления при использовании L48Q cTnC. Эти данный расходятся с данными, полученными на культивируемых кардиомиоцитах (Таблица 10). Очевидное различие может объясняться 1) изометрическими условиями в отличие от условий с отсутствием нагрузки, 2) различным количеством и периодом активации Ca2+ 3) различиями в % cTnC, встроенного в миофибриллы, 4) различиями в фосфорилировании миофиламентов или 5) различиями в уровне ADP, Pi или рН, которые, как известно, влияют на кинетику расслабления. В исследованиях на миофибриллах можно независимо варьировать концентрацию [АТФ], [АДФ], [Pi], [Ca2+], pH и фосфорилирование белков миофиламентов (РКА, РКС, фосфатазы и т.д.), т.е. все условия, которые варьируют в сердце и которые трудно контролировать в препаратах интактного сердца. Значительным преимуществом указанного метода является то, что развитие силы и расслабление изучают в одних и тех же условиях с получением измерений, применимых для изоволюмических (изометрических) фаз сердечной систолы (kact) и диастолы (kREL).
ПРИМЕР 22
[0200] Анализ белков: Изменения в сократительной функции, Ca2+-токах, активности в СПР Ca2+ вспышек и/или Ca2+ нагрузки при всех условиях будут сопоставлены с данными по количеству и фосфорилированию белков миофиламентов (cTnI, cTnT, MLC-2, cMyBP-С и Tm), белков СПР (PLB, RyR) и белков сарколеммы (РМСА, Ca2+ канал L-типа), полученными на клеточных культурах с высокой плотностью в соответствии с условиями эксперимента, показанными в Таблица 12. Изменения в экспрессии белков будут определены с использованием ПЦР в реальном времени и Вестерн-блоттинга. Фракции белков СПР будут получены в соответствии с опубликованными методами
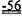
ПРИМЕР 23
[0201] Систематический набор исследований, описанных в Примерах 19-22, обеспечит подробную информацию о том, как измененное связывание Ca2+ в тонких филаментах сердца влияет на сократимость и гомеостаз Ca2+ в различных условиях, и как указанные эффекты могут опосредоваться пост-трансляционными модификациями (фосфорилированием) белков. Мы предполагаем, что существует прямая взаимосвязь между изменениями в связывании Ca2+ миофиламентами и регуляцией гомеостаза Ca2+ в кардиомиоцитах. Ожидаемые результаты и вероятные гипотезы приведены ниже:
[0202] L48Q cTnC: L48Q cTnC повышал чувствительность к Ca2+ миофиламентов (Таблица 9) и укорочение с незначительным влиянием на Ca2+-токи кардиомиоцитов в состоянии покоя (Таблица 10), тогда как стимуляция при частоте 1 Гц в течение 24 часов приводила к повышению обоих значений, а также ускоряла угасание Ca2+-тока (Фигура 17). Эти данные предполагают, что стимуляция может, в первую очередь, влиять на поведение Ca2+сарколеммы и СПР in vitro, тогда как L48Q cTnC, в первую очередь, влияет на поведение миофиламентов. Таким образом, мы ожидаем, что стимуляция будет влиять на функцию СПР (Ca2+-нагрузку, CICR и т.д.) с сопутствующим повышением силы и укорочения, возможно, путем фосфорилирования белков СПР/сарколеммы. Например, повышенная амплитуда Ca2+-тока и повторная секвестрация Ca2+ после стимуляции могли быть обусловлены повышенным фосфорилированием RyR и PLB, соответственно
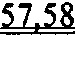
[0203] I61Q и L57Q cTnC: L57Q и I61Q cTnC снижают Ca2+-чувствительность миофиламентов, укорочение и амплитуду Ca2+-тока в кардиомиоцитах в состоянии покоя (Таблица 10). Указанные данные подтверждают сформулированное ранее предположение (предыдущий абзац) о том, что активность миофиламентов может запускать изменения в функции СПР (что также видно в данных по стимуляции). Результаты помогут определить специфические механические изменения, приводящие к снижению Ca2+-токов. Например, сниженное высвобождение Ca2+ из СПР в случае L57Q или I61Q cTnC предполагало бы сниженное фосфорилирование RyR в результате пониженной киназной активности или усиленной фосфатазной активности. В альтернативном варианте указанные варианты cTnC могут вызывать изменения в Ca2+-нагрузке СПР (с помощью сниженного фосфорилирования PLB), которые также снижали бы амплитуду Ca2+-тока. В таком случае β-адренергическая стимуляция оказывала бы большее выраженный эффект на повышение функции СПР и Ca2+-токи в I61Q или L57Q cTnC-трансдуцированных клетках по сравнению с клетками WT или L48Q cTnC-клетками. Мы ожидаем, что более высокие частоты стимуляции (2-3 Гц по сравнению со стимуляцией 0,5 Гц во время измерений) приведут к повышению сократимости путем усиления CICR и связывания Ca2+ тонкими филаментами, которое, в свою очередь, может изменить Ca2+-нагрузку и функцию СПР. Будет интересно узнать, обеспечивает ли стимуляция (1 Гц) такой же эффект. Мы ожидаем, что повышение концентрации внеклеточного [Ca2+]ext будет приводить к ослаблению эффектов L57Q или I61Q cTnC на Ca2+-токи и функцию СПР путем усиления механизма CICR.
[0204] Расслабление: Мы ожидаем, что варианты cTnC будут оказывать влияние на расслабление интактных трабекул, несмотря на то что в культивируемых кардиомиоцитах наблюдалось незначительное влияние на указанное расслабление. Это связано с тем, что сокращение в культивируемых кардиомиоцитах происходит в отсутствие нагрузки, тогда как трабекулы будут подвергаться изометрическому сокращению. Таким образом, характер связывания Ca2+ cTnC мог бы иметь более выраженный эффект во время сокращений при нагрузке, когда кинетика расслабления определяется, в первую очередь, скоростью циклической активности поперечных мостиков и отношением субстрат/продукт гидролиза АТФ, которые изменяются при ишемии
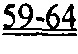
ПРИМЕР 24
[0205] Мы изучим острый и хронический эффекты варианта cTnC на сердечную функцию с использованием векторов AAV6-cTnC и трансгенных мышей. Для определения ответа на острые изменения в функции миофиламентов мы будем трансфицировать нормальных взрослых мышей вариантами AAV6-cTnC (WT, L48Q, L57Q или I61Q) с кардиоспецифичным промотором (cTnT455) посредством инъекции в хвостовую вену или посредством внутриглазной инъекции. Параллельные эксперименты будут осуществляться на трансгенных мышах L48Q cTnC и I61Q cTnC путем репрессии промотора МНС до достижения животными взрослого состояния. Дополнительные исследования будут проводиться на мышах в отсутствие репрессии указанного промотора для определения эффектов указанных вариантов cTnC на нормальное развитие и функцию сердца. Для ограничения объема задачи, мы сфокусируем исследования, в первую очередь, на взрослых мышах. Однако мы будет проводить эхокардиографическую оценку в возрасте 1, 2, 3 и 6 месяцев для выявления начала и прогрессирования любых изменений в функции. Некоторые животные будут подвергаться стрессу путем β-адренергической стимуляции с помощью изопротеренола. После проведения эхокардиографии некоторым животным будут проводить измерения гемодинамических параметров с использованием катетера Миллара (Millar), других животных усыпят, и сердца будут препарировать для проведения анализов на работающем сердце или для получения препаратов интактных или очищенных трабекул, культур кардиомиоцитов или препаратов миофибрилл.
ПРИМЕР 25
[0206] Эхокардиография. Оценку сердечной функции in vivo будут проводить неинвазивно с помощью эхокардиографии. Окологрудинные изображения по длинной оси будут использовать для определения скорости кровотока через выносящий тракт левого желудочка и площади поперечного сечения выносящего тракта (CSA) для расчета ударного объема сердца (SV=CSA × интеграл скорости кровотока (площадь аортальной скорости кровотока)) и сердечного выброса (СО=SV × частота сердечных сокращений)
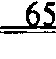
ПРИМЕР 26
[0207] Физиология in vivo. Оценку сократимости сердца в реальном времени и изменений кривой Франка-Старлинга и зависимости между давлением и объемом будут проводить с использованием миниатюрных катетеров с измерителем сопротивления/микроманометром (Millar Instruments). Гемодинамические параметры будут включать частоту сердечных сокращений, конечное диастолическое и систолическое давление, +dP/dt, -dP/dt, снижение систолической работы (SW) в зависимости от конечного диастолического объема (EDV) и константу времени угасания давления в желудочке. Указанные парламенты, демонстрирующие работу сердца, можно сопоставлять с работающим сердцем in vitro (см. ниже), измерениями динамики параметров сокращения/расслабления мышиных сердец при таких же пре- и пост-нагрузках на трабекулах и миофибриллах.
ПРИМЕР 27
[0208] Работающее сердце in vitro. После проведения эхокардиографии в конечной точке экспериментов на животных некоторые сердца будут вырезаны и помещены в аппарат работающего сердца (Experimetria) для оценки насосной функции и способности отвечать на различные пре- и пост-нагрузки (± изопротеренол), которую более трудно оценивать in vivo. Указанные сердца затем будут подготовлены для морфологического и гистологического анализа. На Фигуре 16D показан сниженный ответ (результирующая сила) на преднагрузку в пораженных инфарктом сердцах по сравнению с нормальным сердцем.
ПРИМЕР 28
[0209] Трабекулы, кардиомиоциты и миофибриллы. После проведения эхокардиографии в конечной точке эксперимента некоторые сердца будут вырезаны и желудочки будут подготовлены для получения 1) препаратов интактных или очищенных трабекул, 2) культур кардиомиоцитов или 3) препаратов миофибрилл. Экспериментальные измерения будут проводить, как описано выше.
ПРИМЕР 29
[0210] Морфологический и гистологический анализ: Некоторые препарированные сердца, включая сердца, используемые для измерений на работающем сердце in vitro, будут взяты для гравиметрического анализа, фиксированы, заделаны и окрашены (гематоксилин-эозин, трихром по Мэссону, пикросириус красный) в соответствии с общепринятым методом
ПРИМЕР 30
[0211] Системный комплекс исследований, описанных в Примерах 24-29, даст подробную информацию о краткосрочной и долгосрочной адаптации сердца к мутации, которая влияет на Ca2+-чувствительность сокращения миофиламентов in vivo, где внутренняя адаптация связана с постоянными паракринными и гормональными колебаниями. Мы ожидаем, что варианты cTnC, которые изменяют Ca2+-чувствительность миофиламентов, будут оказывать острое влияние в отличие от хронического влияния на сердечную функцию, функцию СПР и белковый профиль миофиламентов/СПР. Поскольку сердечная недостаточность связана с подавлением β-адренергической чувствительности (и стимуляцией экспрессии протеинкиназы С (РКС) и направленного фосфорилирования

[0212] Мы также будем непосредственно проверять, может ли измененная Ca2+-чувствительность миофиламентов сама по себе обуславливать функциональный или морфологический гипертрофический фенотип или фенотип дилатации желудочков. Мы предполагаем, что этого не будет происходить при относительно непродолжительной трансдукции AAV6-cTnC, но может проявляться в некоторой степени по мере старения трансгенных мышей, трансдуцированных L48Q cTnC (гипертрофия) и I61Q (дилатация). Мы ожидаем увидеть различия в профиле фосфорилирования белков СПР/сарколеммы и миофиламентов при воздействии каждого из указанных вариантов cTnC.
ПРИМЕР 31
[0213] Мы изучим влияние L48Q cTnC на пораженные инфарктом сердца и I61Q (или L57Q) cTnC на молодых и взрослых мышей с мутацией R92W/L cTnT. Пораженные инфарктом сердца. Для определения того, может ли L48Q cTnC улучшать сердечную функцию после инфаркта миокарда, мы будем трансфицировать мышей AAV6-L48Q cTnC на 3-7 день после проведения хирургического лечения инфаркта и после проведения эхокардиографии для определения степени функциональной недостаточности. Исходные эхокардиографические оценки будут проведены через 1, 2, 4 и 8 недель после трансфекции для определения наилучших временных точек для изучения улучшения функции. Мы также проведем исследование дозы вектора для определения оптимальных схем лечения и сопоставим полученные данные с измерениями по включению L48Q cTnC в миофибриллы (показано выше). Низкая доза (0,6×1012), которую инъецировали нормальным мышам (Фигура 12), была достаточно эффективной для повышения фракции выброса. У некоторых животных затем будут измерены гемодинамические параметры in situ в ходе экспериментов с использованием катетера Миллара (как описано выше). Дополнительных животных будут использовать для проведения исследований in vitro на работающем сердце, интактных и очищенных трабекулах, культивируемых кардиомиоцитах и изолированных миофибриллах. Как и в предыдущем случае, мы выявим наиболее информативные препараты и проведем на них большинство измерений для ограничения числа животных, необходимого для указанных исследований. Для определения того, предотвращает ли L48Q cTnC потерю функции левого желудочка, отдельную группу животных будут трансфицировать AAV6-L48Q cTnC до проведения хирургического лечения инфаркта. Некоторые препарированные сердца, включая сердца, используемые для проведения измерений на работающем сердце in vitro, будут взяты для гравиметрического анализа, фиксированы, заключены и окрашены для оценки признаков гипертрофии и фиброза (как описано выше). Серийные срезы пораженных инфарктом сердец будут анализировать с использованием ImageJ для количественной оценки размера инфарктной зоны и % mCherry-положительных клеток для сопоставления полученных данных с измененной функцией in vivo и in vitro после трансфекции AAV6-L48Q cTnC.
ПРИМЕР 32
[0214] Трансгенные мыши R92W/L cTnT. Мыши R92W/L cTnT будут трансдуцированы AAV6-I61Q (или L57Q) cTnC в возрасте одного месяца для определения того, приводят ли указанные варианты к ослаблению или устранению функционального фенотипа, наблюдаемого в возрасте двух месяцев, включая сниженную сердечную функцию, повышенную Ca2+-чувствительность миофиламентов и сниженную функцию СПР (связанную с фосфорилированием PLB ser16, 17
ПРИМЕР 33
[0215] Эхокардиографический анализ растяжения. Помимо возможности измерений в режиме М, современные эхокардиографические датчики имеют разрешение, достаточное для улучшенной функциональной оценки. Указанная оценка включает измерения систолического и диастолического растяжения сегментов и скорости растяжения, которые дают возможность сопоставления изменений in vivo с изменениями in vitro, обеспечивая дополнительные функциональные параметры эластичности стенки желудочка и (соответственно) движения стенок. Полученные данные будут информативны для изучения влияния вариантов cTnC на нормальные сердца, пораженные инфарктом сердца и сердца с ГКМП R92W/L. Растяжение представляет собой изменение длины по сравнению с исходным размером неподверженного стрессу миокарда путем удлинения, сжатия или укорочения
[0216] Мы ожидаем, что повышение Ca2+-чувствительности миофиламентов (L48Q cTnC) улучшит функцию пораженных инфарктом сердец и замедлит прогрессирование сердечной недостаточности. Указанный эффект может быть обусловлен снижением хронической β-адренергической стимуляции. В таком случае мы ожидаем увидеть зависимое от времени различие в балансе парасимпатического и симпатического тонуса между L48Q cTnC-трансдуцированными и неподверженными лечению пораженными инфарктом сердцами, который отражается в опосредованном α- и β-адренергической стимуляцией фосфорилировании белков.
[0217] Мы ожидаем, что трансдукция мышей R92W cTnT геном I61Q cTnC в возрасте 1 месяца будет по меньшей мере частично устранять отрицательное воздействие R92W cTnT, наблюдаемое в 2-месячном возрасте, в конечном итоге приводя к улучшению функции сердца. Указанный эффект может являться результатом изменения фосфорилирования белков СПР (таких как PLB) и Ca2+ нагрузки СПР, которое, как было показано, происходит при адаптивном ответе указанных мышей в процессе старения (58). Подобным образом, мы ожидаем, что поздняя I61Q cTnC-трансдукция (в возрасте 5-6 месяцев) R92L cTnT-мышей также будет изменять фосфорилирование белков СПР, приводя к улучшению функции и замедлению гипертрофического ответа.
ПРИМЕР 34
[0218] Было описано влияние повышенного уровня R1R2 и [дезокси-АТФ] на стимулированные сокращение культивируемых кардиомиоцитов взрослых крыс. Аденовирусные (AV) векторы. Гиперэкспрессию R1R2 индуцировали путем трансдукции кардиомиоцитов в течение ~48 часов AV векторами (~250 частиц на клетку) под контролем промотора CMV, которые экспрессируют субъединицу R1 или R2 и второй кассетой экспрессии для зеленого флуоресцентного белка (GFP). Мы также создали только GFP-экспрессирующий AV вектор в качестве контроля трансдукции/репортерного контроля. Для определения степени гиперэкспрессии R1R2 кардиомиоциты новорожденных крыс трансдуцировали AV-векторами в идентичных условиях и определяли уровень белков с помощью Вестерн-блоттинга. Уровень R1 (Фигура 23А) и R2 (Фигура 23В) был повышен в 24 раза и 46 раз, соответственно, при использовании GAPDH в качестве контроля нагрузки. ВЭЖХ-анализ показал, что указанное повышение клеточного уровня [дезокси-АТФ] было в ~10 раз (0,35 нмоль/мг белка) больше по сравнению с GFP-трансдуцированными кардиомиоцитами (Фигура 23С). Указанные данные совпадают для нескольких популяций клеток (диапазон повышения 8-12-раз) для указанной нагрузки частицами AV. В то время как указанный эффект является сильным, повышенный уровень [дезокси-АТФ] представляет собой всего только ~1-1,5% общего пула адениннуклеотида, так как [дезокси-АТФ] в норме включает >0,2% общего аденинтрифосфатнуклеотида. Указанные данные предполагают, что для значительного повышения сократимости кардиомиоцитов требуется всего небольшое количество дезокси-АТФ.
ПРИМЕР 35
[0219] Гиперэкспрессия R1R2 усиливает сократимость кардиомиоцитов. Типичные кривые (Фигура 24) показывают, что трансдукция AV-R1R2 приводила к намного большему укорочению кардиомиоцитов (А) при отсутствии повышения максимального внутриклеточного уровня Ca2+ (В; Fura-2), по результатам видеодетекции длины и флуоресцентной визуализации (IonOptix). Данные для>50 клеток (каждое условие), стимулированных при частоте 0,5 Гц (25°C), суммированы в Таблица 14, где показано, что трансфекция AV-R1R2 повышала укорочение на ~50% и скорость укорочения и расслабления на ~100% без влияния на амплитуду Са2+-тока или исходный уровень. Указанные данные предполагают, что усиление сократимости в первую очередь обусловлено эффектом на миофиламенты. Тогда как гиперэкспрессия R1R2 не изменяла величину Ca2+-тока, время до достижения 50% (DT50) и 90% (DT90) угасания сокращалось, что предполагает усиленный обратный захват Ca2+ в СПР или посредуемый Na+/Ca2+-помпой (NCX) отток Ca2+. Указанные результаты были также согласуются с результатами для стимуляции при 1 и 2 Гц.
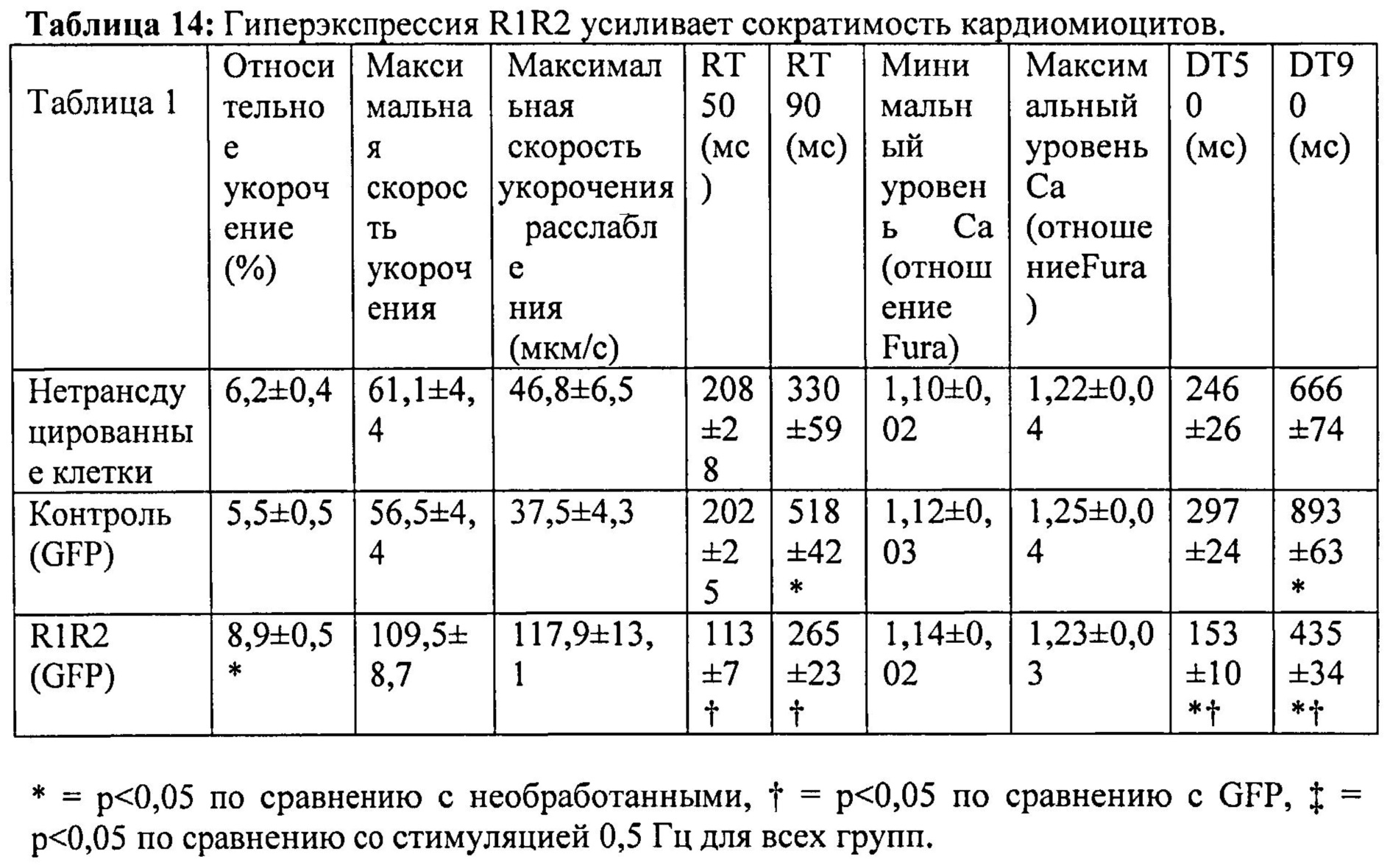
ПРИМЕР 36
[0220] Гиперэкспрессия R1R2 «восстанавливает» сократимость кардиомиоцитов пораженных инфарктом сердец. Инфаркт миокарда вызывали у взрослых крыс путем перетяжки левой нисходящей коронарной артерии. Через 4 недели кардиомиоциты взрослых крыс переводили в культуру и затем трансдуцировали AV-R1R2 или AV-GFP в течение 48 часов. Потерю функции подтверждали с помощью in vivo эхокардиографии до выделения клеток (Фигура 34). На Фигуре 25 показано, что по сравнению с контрольными клетками (не инфарктными) кардиомиоциты из пораженных инфарктом сердец имели сниженную величину укорочения (А) и скорость укорочения (В), повышенное время расслабления (С) и сниженную амплитуду Ca2+ тока (D).
[0221] Указанные эффекты восстанавливались при трансдукции AV-R1R2 (Инфаркт + R1R2). Примечательно, что гиперэкспрессия R1R2 не оказывала влияния на величину Ca2+ тока в нормальных кардиомиоцитах, но повышала величину до уровня контроля в инфарктных клетках. Указанные данные указывают на то, что гиперэкспрессия R1R2 оказывает значительное влияние на запасание и/или высвобождение Ca2+ из СПР в инфарктных клетках, и механизм, лежащий в основе указанного действия, требует более подробного изучения, которое предложено ниже.
ПРИМЕР 37
[0222] Исследования in vivo. Для определения того, как гиперэкспрессии R1R2 влияет на сердечную функцию, мы трансдуцировали С57/В16-мышей путем инъекции в хвостовую вену или прямой инъекции в сердце аденовирусами AV-R1R2(+GFP) или AV-GFP (только GFP; контроль) (4-6 животных в каждой группе). На Фиг.26А показано, что оба способа приводили к повышению фракции укорочения (FS) левого желудочка (ЛЖ) по сравнению с контролем через 4 дня после инъекции. Указанное повышение происходило благодаря сниженному внутреннему диаметру ЛЖ во время систолы без изменений во время диастолы, что предполагает более сильное сокращение желудочка (не дилатацию желудочка). На Фиг.26 В показан ЛЖ после инъекции; на увеличенном участке (справа) показана область локализованной трансдукции (стрелки), на которую указывает зеленая флуоресценция (GFP). Важно, что даже несмотря на то, что трансдукция путем прямой инъекции затрагивала небольшие участки сердца, наблюдалось значительное общее повышение функции (Фиг.26А, правый столбик).
[0223] Мы определим в ходе дополнительных исследований, имеется ли влияние на частоту сердечных сокращений и/или ударный объем сердца. Для предполагаемых исследований мы будем использовать во всех экспериментах мышей линии FVB/N, которая является исходной линией для мышей TG-R1R2. Мы также определим, происходят ли долгосрочные структурные адаптации. Результаты первоначальных исследований указывают на отсутствие выраженных морфологических различий в сердце (отношение масса сердца:масса тела и толщина и масса желудочка одинаковы), но клеточную гипертрофию, плотность миофибрилл и т.д. еще необходимо оценить.
ПРИМЕР 38
[0224] Диффузия дезокси-АТФ в миокарде может повышать эффективность трансдукции AAV6-R1R2. Удивительно, что в случае прямых инъекций AV-R1R2 наблюдалось большое повышение FS, несмотря на малую площадь трансдукции AV-R1R2 (<1% объема ЛЖ было GFP+). Эти данные позволяют предположить, что гиперэкспрессия R1R2 происходила в небольшой популяции кардиомиоцитов, и полученная в результате дезокси-АТФ диффундировала через щелевые контакты в нетрансдуцированные клетки, приводя к более глобальному повышению сократимости. Диффузия АТФ через щелевые контакты была описана другими авторами33,34. Для проверки указанного предположения мы инъецировали меченную флуоресцеином дезокси-АТФ в культивируемые чЭСК-кардиомиоциты и со временем увидели диффузию в окружающие клетки (Фиг.35А, В). Окрашенные после фиксации клетки были коннексин43-положительными, что подтверждает их соединение через щелевые контакты. Мы продолжаем описанные исследования для того, чтобы провести количественный анализ диффузии, и показать, что блокирование щелевых контактов также блокирует перенос дезокси-АТФ. Мы также сокультивировали AV-R1R2-трансдуцированные (дезокси-АТФ-донорские) кардиомиоциты, произошедшие из эмбриональных стволовых клеток человека (чЭСК-КМЦ) и нетрансдуцированные (реципиентные) чЭСК-КМЦ. Нетрансдуцированные клетки диссоциировали и сокультивировали с одной из трансдуцированных вирусом популяций. Через 48 часов после трансдукции получали изображения совместных культур и оценивали параметры сокращений с помощью видеомикроскопии (IonOptix). На Фиг.35С показано, что как у AV-R1R2-трансдуцированных миоцитов («дезокси-АТФ-донорские клетки», синяя кривая), так и у GFP-отрицательных миоцитов, которые культивировали совместно ними («WT-клетки-реципиенты дезокси-АТФ», красная кривая), наблюдалось значительно большее укорочение клеток по сравнению с AV-GFP-трансдуцированными миоцитами («контрольными донорскими клетками», зеленая кривая) или их GFP-отрицательными партнерами («контрольными клетками-реципиентами», черная кривая). Указанные данные показывают, что R1R2-гиперэкспрессирующие дезокси-АТФ-донорские клетки могут усиливать сократительные свойства соединенных с ними кардиомиоцитов-реципиентов. Данные, имеющиеся на сегодняшний день, достаточно убедительны для того, чтобы предположить, что для значительного улучшения сердечной функции может быть необязательна высокая степень R1R2-трансдукции клеток/ткани по всему сердцу.
ПРИМЕР 39
[0225] Предварительная функция сердца ex vivo. Сократительную функцию в изоволюметрических условиях регистрировали во время проведения перфузии по Лангендорффу (Langendorff) для одного WT-мышиного сердца и одного TG-R1R2-мышиного сердца. Развитие давления в ЛЖ повышалось с повышением объема ЛЖ как для сердца ДТ, так и для TG-R1R2-сердца, что указывало на нормальную чувствительность Франка-Старлинга. Однако развитие давления ЛЖ было быстрее в TG-R1R2-сердце при всех объемах ЛЖ (Фигура 28А), что подтверждало повышение насосной функции, на которое указывала повышенная FS у TG-R1R2-животных (Фигура 27). Развитие давления ЛЖ немного замедлялось в TG-RlR2-сердце в ответ на повышение уровня [Ca2+] от 2 мМ до 4 мМ (Фигура 28В), но оставалось ускоренным по сравнению с WT-сердцем на протяжении всего времени.
ПРИМЕР 40
[0226] Данные 31Р-ЯМР. Одновременно с выявлением функциональных измерений определяли содержание в миокарде высокоэнергетических фосфатов (фосфокреатина, АТФ и Pi) методом 31Р-ЯМР-спектроскопии при перфузиии (исходный уровень) и при стимуляции [Ca2+] в высокой концентрации. ЯМР-спектры (Фигура 29) показаны для сердец дикого типа, ДТ (А), и TG-R1R2 (В). В указанных спектрах нижняя кривая соответствует исходному уровню, средняя кривая советует уровню через 10 минут и верхняя кривая - через 20 минут после начала стимуляции [Ca2+] в высокой концентрации. Важно отметить, что отношение АТФ:масса сердца на исходном уровне было одинаковым для сердец ДТ и TG-R1R2 (Фигура 29С, вставка) и не менялось во время стимуляции [Ca2+] при высокой концентрации. Тогда как отношение фосфокреатина (PCr) к АТФ (Фигура 29С) было немного ниже на исходном уровне у TG-R1R2-мышей, после стимуляции [Ca2+] в высокой концентрации различий не наблюдалось. Такие результаты в первую очередь обусловлены снижением отношения PCr/АТР у мыши WT, тогда как у TG-R1R2-мыши отношение PCr/АТР сохранялось. Особо интересно, что сердце TG-R1R2 развивало намного более высокое давление ЛЖ по сравнению с сердцем WT даже через 20 минут после стимуляции [Ca2+] в высокой концентрации при одинаковом отношении PCr/АТР. Мы подтвердим и дополним указанные наблюдения данными для сильной и хронической гиперэкспрессии R1R2 и +/- гиперэкспрессии R1R2 после инфаркта миокарда.
ПРИМЕР 41
[0227] Тканеспецифичное нацеливание с использованием конструкций AAV6. Тканеспецифичность оценивали с использованием щелочной фосфатазы, экспрессия которой запускалась различными промоторами гена в конструкциях AAV6. В Таблица 15 приведено сравнение двух кардиоспецифичных промоторов (креатинкиназы 7 (СК7) и сердечного тропонина Т (cTnT455)) с неспецифичным промотором цитомегаловируса (CMV); значения приводили к значениям для СК7 в передней большеберцовой мышце (ТА). Наиболее важно отметить, что cTnT455 приводит к высокой экспрессия в сердце, но к низкой экспрессии или отсутствию экспрессии в другой ткани. Указанная специфичность сильно снижает вероятность эффектов гиперэкспрессии R1R2 на ткани, отличные от сердечной ткани.
[0228] Дезокси-АТФ не влияет на развитие силы гладкой мышцы аорты мыши. Для того, чтобы начать исследование потенциальных системных эффектов повышения уровня дезокси-АТФ, мы провели совместную работу с д-ром Франком Брозовичем (Frank Brozovich, Клиника Мейо) для определения того, влияет ли дезокси-АТФ на сокращение гладких мышц аорты мыши. На Фигуре 30А показано отсутствие отличий в последовательных сокращениях в очищенных мышечных полосках при использовании в качестве субстрата сокращения дезокси-АТФ по сравнению с АТФ. Данные нескольких экспериментов суммированы на Фигуре 30В. Кроме того, контрольные измерения показали, что дезокси-АТФ не изменяла степень фосфорилирования легкой цепи миозина, которое контролирует связывание гладкомышечного миозина с актином (данные не представлены).
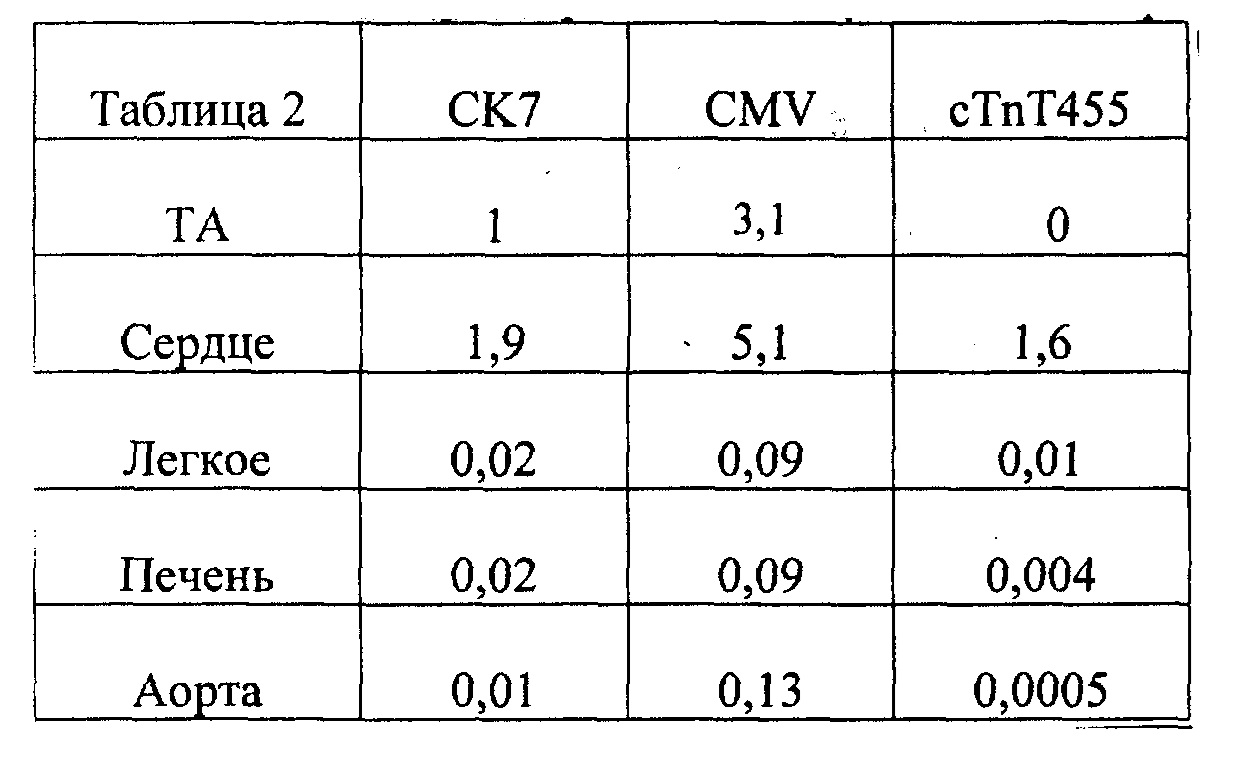
ПРИМЕР 42
[0229] AAV6-R1R2 для кардиоспецифичного нацеливанеия. Мы использовали векторы rAAV6 для острого повышения уровня R1R2 в сердце (и [дезокси-АТФ]). Векторы rAAV6 получали, как описано ранее24*. Вкратце, получали плазмиды с рекомбинантными геномами AAV, содержащие кардиоспецифичный промотор (cTnT455). R1R2-трансгенные кассеты экспрессии ко-трансдуцировали в клетки HEK293 вместе с пакующей/хелперной плазмидой pDGM6 методом CaPO4-преципитации. Векторы собирали из культуры, замораживали-оттаивали и отбирали супернатант. Для аффинной очистки использовали колонку с гепарином HiTrap (GE Healthcare, Пискатавэй, Нью-Джерси). Вирус концентрировали на градиенте сахарозы 40%), откручивали при 27,000 об./мин. (18 часов, 4°C) и повторно солюбилизировали в сбалансированном растворе Хэнкса. Векторные геномы определяли относительно плазмидных стандартов с использованием в качестве зонда олигонуклеотида участка полиаденилирования SV40, меченного на конце 32Р, с помощью Саузерн-блот-гибридизации и подтверждали с помощью количественной ПЦР.
[0230] Селективность нацеленой на сердце конструкции оценивали с использованием щелочной фосфатазы, запускаемой с помощью промоторов различных генов в конструкции AAV6. В Таблице 15 приведено сравнение двух специфичных для поперечно-исчерченных мышц промоторов - (креатинкиназы 7 (CK7) и сердечного тропонина Т (cTnT455)) - с неспецифичным промотором цитомегаловируса (CMV),
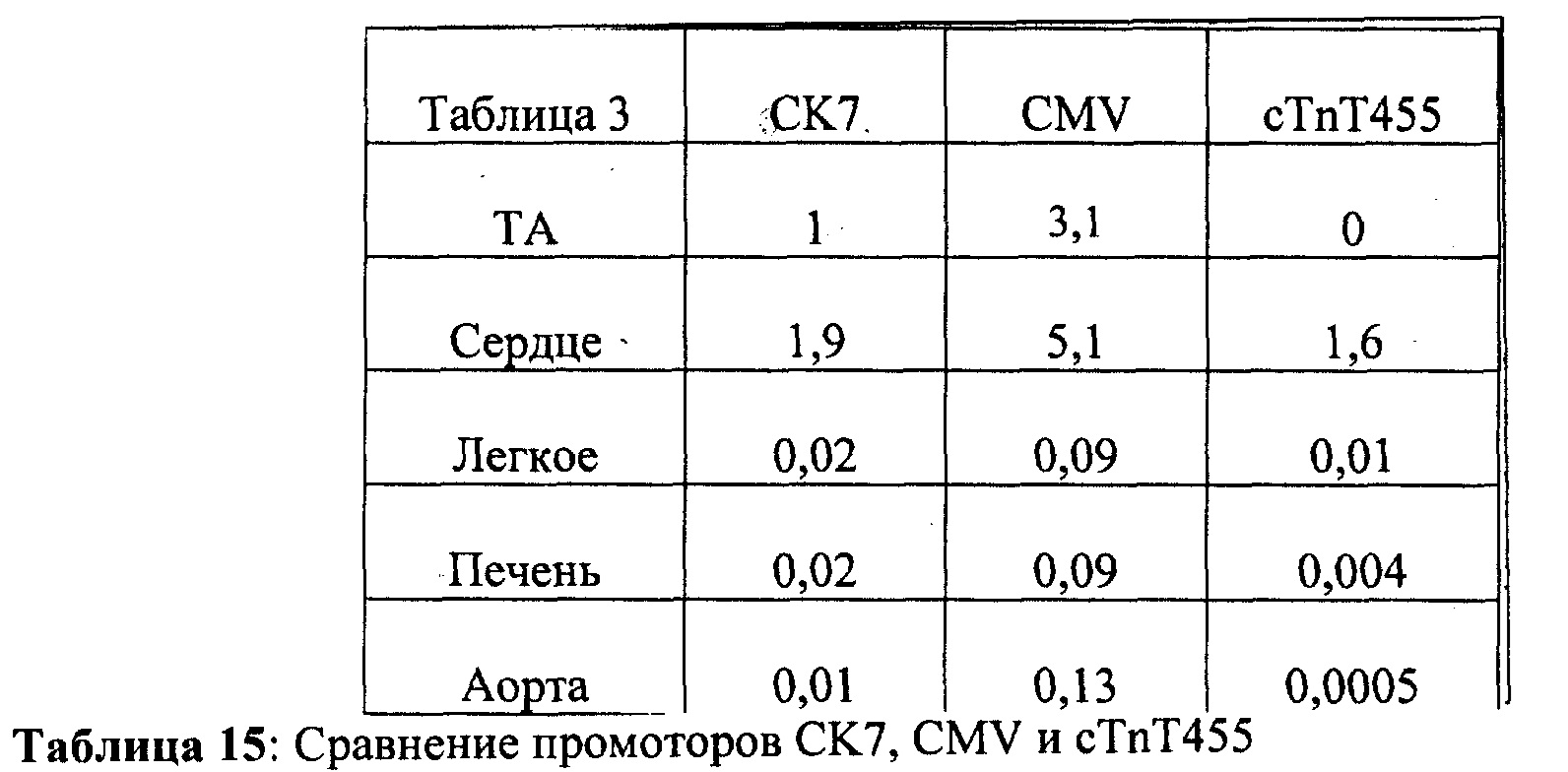
при этом значения нормировали по для СК7 в передней большеберцовой мышце (ТА). Наиболее важно отметить, что cTnT455 приводит к высокой экспрессии в сердце, но к низкой экспрессии или к отсутствию экспрессии в другой ткани, таким образом существенно снижая вероятность эффектов гиперэкспрессии R1R2 на ткани, отличные от сердечной ткани. На Фиг.36А показаны предварительные результаты Вестерн-блоттинга, подтверждающие указанные данные, согласно которым в сердечной ткани от мыши, которой инъецировали AAV6-R1R2cTnT455 (4,5 е13) наблюдалась высокая экспрессия R1 и R2 субъединицы по сравнению с контрольным сердцем мыши. Необходимо отметить, что верхние полосы являются результатом неспецифической окраски, стрелки указывают на белок R1 и R2 (идентифицированный с помощью маркеров молекулярной массы (не показаны)). Важно отметить, что экспрессия R1 и R2 в легком была крайне низкой по сравнению с сердцем и не изменялась в скелетной мышце. Эти данные, показанные на Фиг.36 для сердечной ткани, полученной от мышей, которым не проводили инъекцию (В), по сравнению с мышами, которым инъецировали AAV6-щелочную фосфатазу (темно-красный; С)36, позволяют предположить, что AAV6-R1R2cTnT455 должен был обеспечить стабильную продолжительную гиперэкспрессию R1R2. Также было показано, что стабильная экспрессия трансгена AAV6 поддерживается в течение 12+ недель у крыс37 и по меньшей мере 6+ месяцев у собак38.
[0231] Таким образом, мы начали исследования для определения связи между дозой инъекции AAV6-R1R2cTnT455, динамикой и стабильностью повышенной насосной функции ЛЖ, уровнем R1R2 и [дезокси-АТФ] в ткани сердца. На Фиг.37 показан эффект 3 доз вектора, т.е. 1,5 е13, 4,5 е13 и 1,35 е14 геномов вектора AAV6-R1R2cTnT455, или физиологического раствора (контроль), которые инъецировали 3-месячным мышам (n=6 на группу), на функцию ЛЖ. Относительное укорочение (SF) ЛЖ была значительно повышенной при высокой дозе через одну неделю и при всех дозах через две недели с эквивалентными эффектами к 6 неделе. Величина SF повышалась ~25-50%, указывая на то, что эффект может достигаться при относительно низкой дозе вектора.
ПРИМЕР 43
[0232] Трансгенные мыши с гиперэкспрессией R1R2 (TG-R1R2). Мы будем использовать би-трансгенных мышей с гиперэкспрессией обеих субъединиц (Rrm1 и Rrm2) RR. На Фигура 32 показана гиперэкспрессия обеих субъединиц в сердечной мышце с денситометрическими расчетными значениями для указанных мышей TG-R1R2, которые в 33,7±7,6 (Rrm1) и в 23,7±3,4 (Rrm2) раз больше, чем соответствующие значения для мышей дикого типа (WT)25,26*. Следует отметить, что для Rrm2 верхняя полоса (*) является неспецифичной. Эндогенный белок Rrm2 не поддается выявлению в ткани WT, но в мышах TG-R1R2 он появляется в виде полосы ниже основной полосы. Тогда как еще не проводили оценку уровня дезокси-АТФ для сердечной ткани, [дезокси-АТФ] была повышена в ~10 - раз в скелетной мышце, в которой наблюдалось повышение субъединиц фермента, соответственно, в 3,3±2,1 (Rrm1) и в 35,7±11,1 (Rrm2) раз. Примечательно, что указанная величина повышения дезокси-АТФ подобна величине, определенной для кардиомиоцитов, трансдуцированных AV-R1R2 в культуре (Фигура 23). Предварительная эхокардиография указанных мышей TG-R1R2 в 6-8-месячном возрасте (измеряли в течение 3 последовательных недель) выявила среднее повышение на >50% фракции укорочения (FS) и снижение на 15%) диастолического внутреннего диаметра ЛЖ (LVIDd). Как показано на Фигуре 27, указанные различия (по сравнению с WT контролями) одинаковы по величине со значениями для предыдущих экспериментов с инъекцией AV-R1R2.
ПРИМЕР 44
[0233] Мы определим острые эффекты повышенного внутриклеточного уровня R1R2 и [дезокси-АТФ] на сердечную функцию. Мы ожидаем, что острая гиперэкспрессия R1R2 (с помощью векторов AAV6-R1R2) приведет к повышению концентрации [дезокси-АТФ] в мышиных сердцах и повышению систолической и диастолической функции. Указанные эффекты будут отражаться в 1) повышенной сократимости кардиомиоцитов и миофибрилл с более быстрым расслаблением (отчасти благодаря повышенной кинетике циклической активности поперечных мостиков), 2) повышении основного сердечного метаболизма без нарушения энергетических резервов и 3) отсутствии изменений или снижении длительности потенциала действия (благодаря усиленной секвестрации Ca2+).
[0234] Мы будем трансфицировать нормальных взрослых мышей линии FVB/N путем инъекции в хвостовую вену или путем внутриглазной инъекции векторов AAV6-R1R2 с кардиоспецифичным промотором cTnT455 (как описано выше) с использованием имитированной инъекции и инъекции вектора AAV6, содержащего только cTnT455, в качестве контролей. После инъекции каждую неделю будут проводить эхокардиографию (вплоть до 6 недель) для определения оптимальной временной точки (с максимальным эффектом) для проведения дополнительных анализов. Первоначальные исследования будут описывать сердечную функцию in vivo с помощью эхокардиографии с последующими измерениями гемодинамических показателей in situ или ex vivo с использованием сердец, перфузированных по методу Лангердорффа, для энергетического анализа и аппарата работающего сердца для оценки насосной производительности. В выбранные временные точки других мышей усыпят и их сердца будут препарировать для получения препаратов интактных или очищенных трабекул, изолированных кардиомиоцитов, препаратов миофибрилл, проведения белкового анализа и (иммуно)гистологического анализа. Указанные измерения предоставят подробную информацию о молекулярных механизмах изменений сердечной функции при сильной гиперэкспрессии R1R2.
ПРИМЕР 45
[0235] Многоуровневое исследование механических свойств целого органа. Эхокардиографию (GE Vivid7) будут проводить для всех животных. Изображения длинной окологрудинной оси будут использовать для определения скорости кровотока через выносящий тракт левого желудочка (ЛЖ) и площади поперечного сечения (CSA) выносящего тракта, для расчета ударного объема сердца (SV=CSA × интеграл скорости кровотока (площадь аортальной скорости кровотока)) и сердечного выброса (СО=SV × частота сердечных сокращений)31*. Двумерные изображения короткой оси на уровне средней части желудочка (папиллярная мышца) будут использовать для создания измерений в режиме М конечного систолического размера ЛЖ (LVESD) и конечного диастолического размера ЛЖ (LVEDD), размера передней и задней стенки (PW) ЛЖ. Указанные данные будут использовать для расчета фракции укорочения [(LVEDD-LVESD)/LVEDD × 100%] и массы стенки ЛЖ [1,05(толщина IVS+LVEDD+толщина PW)]32,33*. Некоторым животным будут проводить оценку в реальном времени сократимости сердца (закон Франка-Старлинга и зависимость давление-объем) с использованием миниатюрных катетеров с датчиком сопротивления/микроманометром (Millar Instruments). Измерения будут включать частоту сердечных сокращений, конечное диастолическое и систолическое давление, +dP/dt, -dP/dt, связанную с давлением систолическую работу (т.е., снижение систолической работы (SW) в зависимости от конечного диастолического объема (EDV)), конечное систолическое отношение между давлением и объемом и константу времени (Таи) угасания желудочкового давления. Указанные параметры можно сравнивать с измерениями для работающего сердца in vitro, измерениями для трабекул и миофибрилл в исследовании динамики сердечного сокращения/расслабления при нагрузке. Оба протокола будут осуществлять в отсутствие/присутствии добутамина для оценки потенциальных изменений адренергической чувствительности при сильной гиперэкспрессии R1R2. На других животных будут проводить измерения на работающем сердце по Нили (Neely) in vitro (Experimentria) в отсутствие потенциально мешающих гормональных колебаний для обеспечения дополнительной информации по преднагрузочной/пост-нагрузочной и адренергической чувствительности. Сердца затем будут взяты для гравиметрического анализа, фиксированы и окрашены (гематоксилин-эозин, трихром по Мэссону, пикросириус красный) в соответствии с общепринятыми методами27,68*. Срезы будут оценивать на гипертрофию, дилатацию и фиброз11,34*. Будет проведен иммуноцитохимический анализ серийных срезов сердца для оценки экспрессии R1R2 и процента трансдуцированных кардиомиоцитов для сопоставления полученных данных с функцией in vivo и in vitro.
ПРИМЕР 46
[0236] Многоуровневое исследование механических свойств на трабекулах, кардиомиоцитах и миофибриллах. В различных группах мышей после проведения эхокардиографии на конечной точке исследования сердца будут вырезаны и желудочки будут подготовлены для 1) получения интактных или очищенных трабекул, 2) культуры кардиомиоцитов или 3) препаратов миофибрилл.
[0237] Трабекулы. Мы определим, как гиперэкспрессия R1R2 влияет на сокращение интактных трабекул при внешней нагрузке при различной длине саркомеров (SL), наблюдаемой in vivo. Тонкие (диаметр <120 мкм, глубина ~50 мкм) трабекулы поместят между силовым преобразователем и линейным двигателем в насыщенной кислородом перфузионной камере, содержащей стимулирующие электроды35-37*. Изометрическую силу сокращения, кинетику сокращения и Ca2+-токи (Fura-2) и их зависимость от SL (отношение Франка-Старлинга, SL=1,9 по сравнению с 2,2 мкм) будут измерять в таких же условиях, как в случае изолированных кардиомиоцитов, со стандартным раствором Кребса-Хенселейта, 95/5% О2/СО2 при 37°C 38. Трабекулы с разрушенной мембраной будут использовать для оценки сокращения миофиламентов в стабильном состоянии с тонкой регуляцией уровня [Ca2+], [дезокси-АТФ] и [АТФ] для определения того, изменяет ли острая гиперэкспрессия R1R2 сократимость миофиламентов независимо от [дезокси-АТФ] через смену изоформ белка или изменение фосфорилирования.
[0238] Изолированные кардиомиоциты желудочков взрослых животных. Изолированные мышиные кардиомиоциты позволяют проводить высокоэффективный одновременный анализ циклов сокращения и функции СПР при различных частотах стимуляции7*. Таким образом, за Ca2+-токами (Fura-2) и укорочением/расслаблением (длина клетки и длина саркомеров) в кардиомиоцитах будут наблюдать при 0,5, 1, 2 и 3 Гц с использованием видеомикроскопии (IonOptix, Милтон, Массачусетс). Мы измерим до и после 1) различные внеклеточные уровни [Ca2+] ([Ca2+]ext), для оценки Ca2+-индуцированного высвобождения Ca2+ (CICR), 2) ингибиторы поперечных мостиков миофиламентов (BDM, блеббистатин и т.д.) для определения влияния сократительной активности на функцию СПР и 3) адренергическую стимуляцию (α-(фенилэфрин) или β (изопротеренол)) для определения присутствия изменения чувствительности (укорочение, Ca2+ высвобождение/обратный захват). Подробные способы измерения свойств потенциала действия (АР), функции CICR, Ca2+ АТФазы СПР (SERCA), NCX и Ca2+ АТФазы плазмолеммы описаны ниже.
[0239] Миофибриллы. Кинетика активации сокращения и расслабления важна для определения нормальной сердечной функции. Во время систолы быстрое развитие давления желудочков критично для эффективного изгнания крови (рабочего выхода), и во время диастолы желудочки должны быстро расслабляться для обеспечениявозможности их достаточного наполнения. Указанные свойства часто изменяются при заболевании сердца. Кинетику сокращения и расслабления миофибрилл в пределах миллисекунд сложно оценивать в очищенных трабекулах, в которых большие диффузионные расстояния замедляют уравновешивание концентрации [Ca2+] при смене раствора. Препараты миофибрилл имеют малые диффузионные расстояния, что обеспечивает возможность быстрой смены растворов и измерения силы независимо от Ca2+-токов в интактных клетках. Указанный подход использовали ранее39,40*, а новые инструменты позволяют измерять развитие силы и расслабление отдельных миофибрилл в одних и тех же условиях (Фигура 33). Указанные измерения применимы для изоволюметрических (изометрических) фаз систолы (kact) и диастолы (kREL). Важно отметить, что мы будем следить как за медленной (kREL, медленная), так и быстрой (kREL, быстрая) фазами расслабления. Считается, что медленна фаза (kREL, медленная) и ее продолжительность (tREL, медленная) отражают скорости разъединения поперечных мостиков41*, которые по результатам предшествующих исследований больше в случае дезокси-АТФ 1, 5. Таким образом, мы будем сравнивать кинетику активации и расслабления между AAV6-R1R2-трансидуцированными и контрольными сердцами с использованием АТФ или дезокси-АТФ (или различных соотношений АТФ:дезокси-АТФ). Эксперименты на миофибриллах с использованием только АТФ позволят нам оценить, изменяет ли гиперэкспрессия R1R2 сама по себе сократительные свойства миофиламентов, тогда как измерения с использованием дезокси-АТФ позволят определить вклад поперечных мостиков в более быстрое расслабление, наблюдаемое при гиперэкспрессии R1R2 в кардиомиоцитах по сравнению с контрольными клетками. Исследования на миофибриллах также позволяют независимо контролировать уровень [ADP], [Pi], [Ca2+], рН и фосфорилирования белков миофиламентов (киназы, фосфатазы), т.е. все условия, которые варьируют в сердце, коррелируют с характером метаболизма, но которые трудно контролировать в препаратах интактных сердец.
ПРИМЕР 47
[0240] Электрофизиологические измерения и взаимодействия с Ca2+. Электрофизиологические показатели будут оценивать в клетках, поддерживаемых при температуре 37°C и стимулированных при частоте между 1 и 5 Гц с использованием стандартных методов34,42-48*. АР будут зарегистрированы, как описано ранее, с использованием усилителя HEKA ЕРС-10, работающего в режиме фиксации тока. (ЕРС-10 имеет фактический контур повторителя напряжения, подобный классическому усилителю микроэлектродов49,50*). Для предотвращения диализа внутриклеточных нуклеотидов мы будем использовать метод перфорированного пэтч-клампа (который позволяет пропускать моновалентные ионы, но не молекулы больше ~200 Дальтон51*) и добавлять краситель в раствор, содержащийся в пипетке, для возможности оповещения в случае полного разрыва клетки (в соответствии с источником Strauss et al, 52*). Параметры АР, которые планируется оценить, включают частоту движения, APD (при 50, 70, и 90% реполяризации), амплитуду АР, максимальный диастолический потенциал и частоту пост-деполяризации. Для экспериментов по визуализации [Ca2+]i клетки будут нагружены Fura-2 или Fluo-4-АМ, и затем будут получены изображения с использованием систем на основе Ionoptix CCD или конфокальной системы Zeiss LSM510 МЕТА, работающей в режиме линейного сканирования. Калибровка флуоресцентных сигналов будет осуществляться с использованием способов «pseudo-ratio»53* или Fmax54*. Необходимо отметить, что в целой клетке [Ca2+]i ток состоит из притока Ca2+, последующей активации высвобождения Ca2+ из СПР (т.е., Ca2+ вспышки) и механизмов выведения Ca2+(в первую очередь NCX и SERCA). Таким образом, для понимания механизмов, лежащих в основе изменений [Ca2+]i тока, необходимо оценивать указанные потоки по отдельности. Приток Ca2+ через Ca2+ каналы L-типа (ICa) будет измерен в режиме фиксации потенциала, с использованием свободных от Na+растворов для оценки функции NCX55*. Для оценки эффектов на высвобождение Ca2+ из СПР мы будем использовать два стандартных способа: 1) одновременное измерение потенциал-зависимого ICa и [Ca2+], тока для определения коэффициента усиления электромеханического сопряжения 1 и 2) получение быстрых конфокальных изображений случаев локализованного высвобождения Ca2+ (т.е., Ca2+ вспышек)57*. Ca2+-нагрузку СПР будут измерять методом с использованием кофеина58,59*. Для анализа эффектов на функцию NCX или Ca2+ АТФазы СПР мы будем использовать стандартные ингибиторы и условия для выделения каждого потока и оценки кинетики вызванного АР и кофеином [Ca2+]i тока и токов через NCX57,60-62*.
ПРИМЕР 48
[0241] Энергетический метаболизм миокарда. Метаболизм и сокращение будут одновременно оценивать с использованием многоядерной ЯМР-спектроскопии в изолированных перфузированных сердцах, как описано ранее63,64*.
[0242] ЯМР-спектроскопия. Отсеченные мышиные сердца будут перфузировать при постоянном давлении (80 мм рт.ст.) при 37°C, как описано ранее63*. Наполненный водой баллон будет помещен в ЛЖ, конечное диастолическое давление будет доведено до 5-10 мм. рт.ст., и будет проводиться регистрация давления ЛЖ и частоты сердечных сокращений. Сократительная функция в изоволюметрических условиях будет рассчитана как резвившиеся давление ЛЖ, умноженное на частоту сердечных сокращений (произведение частоты сердечных сокращений на давление; RPP). MVO2 определяют путем измерения скорости коронарного кровотока и различий PO2 между перфузируемой жидкостью и оттоком из легочного выносящего тракта. Для достижения высокой сократительной активности концентрация [CaCl2] в перфузируемой жидкости будет повышена от 2 до 4 мМ.31Р ЯМР-спектры будут получены на исходном уровне и во время стимуляции высокой рабочей нагрузкой в течение 25 минут, затем будет проведена заморозка-фиксация с помощью зажимов Wollenberger, охлажденных жидким азотом. Относительное окисление углеродных субстратов будет определено для митохондрий путем перфузии сердец ISC-меченными субстратами с последующим замораживанием-фиксацией. 13С-ЯМР-спектроскопия и изотопный анализ будут осуществлять с использованием тканевых экстрактов, как описано ранее63,64*.
[0243] ВЭЖХ-калибровка 31Р-ЯМР-спектров. Замороженные -фиксированные ткани будут использовать для определения содержания АТФ в миокарде с помощью ВЭЖХ, как описано ранее65*. Значение содержания АТФ в миокарде, полученное с помощью ВЭЖХ, будут переводить в концентрацию [АТФ], принимая, что внутриклеточное содержание воды составляет 0,48 мл/г, и содержание белков составляет 0,15 г/г влажной ткани после поверхностного удаления воды. Значения будут использовать для калибровки площади пика АТФ31Р ЯМР-спектрах для исходного уровня для соответствующих групп. Площадь пика γ-АТР, полученная в исходных условиях, будут принимать за 100% и использовать в качестве стандартного значения для всех пиков во всех 31Р-ЯМР-спектрах.
[0244] Биогенез и функция митохондрий. Анализ митохондриального дыхания будут проводить с использованием изолированных митохондрий и пермеабилизированных миофибрилл или миоцитов с комплексом пируват/малат (Комплекс I), сукцинат (Комплекс II) и комплексом пальмитоил-карнитин в присутствии или отсутствие АДФ, разобщителей дыхания (FCCP) и различных ингибиторов66,67*. Скорость синтеза АТФ в бьющихся сердцах будут оценивать с использованием31Р-ЯМР-переноса намагниченности и сравнивать с результатами, полученными с использованием MVO263*. Плотность митохондрий будут оценивать с помощью ЕМ-изображений, также будут определять активность или экспрессию ключевых митохондриальных ферментов/белков, (цитратсинтазы, СОХ, аконитазы и VDAC). Содержание мтДНК и ее репликация, а также механизмы транскрипции, включенные в биогенез митохрндрий, будут определены, как описано68*.
[0245] Анализ миофиламентов и белков СПР. Изменения сократительной функции, Ca2+-токов, активности Ca2+-вспышек в СПР и/или Ca2+-нагрузки при всех условиях будут сравнивать с изо формами, численностью и фосфорилированием белков миофиламентов (cTnI, cTnT, MLC-2, cMyBP-С и Tm), белков СПР (PLB, RyR), белков сарколеммы (NCX, РМСА, Ca2+ канал L-типа). Изменения в уровне мРНК и экспрессии белков будут определены с использованием ПЦР в реальном времени и Вестерн-блоттинга. Фракции белков СПР будут получены в соответствии с опубликованными методами53,56. Если электрофизиологические измерения указывают на изменения, мы может оценить активность ионных каналов с помощью специфичных антител. Анализ экспрессии R1R2 будут проводить с помощью Вестерн-блоттинга (Фигура 23) или иммуноцитохмии и сопоставлять с данными конечных точек экспериментов. Специфичность промотора cTnT455 будут оценивать путем определения экспрессии R1R2 в тканях, отличных от сердечной ткани, таких как скелетные мышцы и легкое. Фосфорилирование будут анализировать с использованием флуоресцентного красителя Pro-Q Diamond (вместе с окрашиванием белков с помощью красителя Sypro Ruby) и Вестерн-блоттинга. Для оценки сайт-специфического фосфорилирования остатков серина и треонина мы можем провести масс-спектрометрический анализ.
ПРИМЕР 49
[0246] Указанные систематические исследования, описанные в Примерах 43-48, предоставят подробную многостороннюю информацию влиянии кратковременной гиперэкспрессии R1R2 и повышенного уровня [дезокси-АТФ] на сократительные свойства и Ca2+ и энергетический гомеостаз. Мы ожидаем, что относительно низкие дозы AAV6-R1R2 могут являться достаточными для усиления функции на всех уровнях механической оценки. Мы ожидаем, что у указанных мышей будет сохраняться или усиливаться сердечная β-адренергическая чувствительность. Это может происходить, если усиленная миокардиальная функция (через [дезокси-АТФ]) приводит к снижению необходимостям в β-адренергической стимуляции. Кроме того, мы ожидаем, что указанная адаптация будет более выраженной на более удаленных временных точках (3-4 недели) после пика гиперэкспрессии R1R2, а затем будут снижаться по мере угасания трансдукции. Пик адаптации может приближаться к долгосрочным изменениям, которые мы ожидаем увидеть у TG-R1R2-животных. Функциональные эффекты пост-трансляционных модификаций белков, являющихся результатом повышенного уровня [дезокси-АТФ], будут оценены с помощью клеточных/субклеточных анализов с использованием АТФ в качестве субстрата. Это могло бы привести к повышенной чувствительности миофиламентов к Ca2+ (трабекулы с разрушенной мембраной), повышенной кинетике активации и расслабления (миофибриллы) и повышенной кинетике обратного захвата (интактные трабекулы/кардиомиоциты). Однако мы не ожидаем, что отношение Франка-Старлинга будет снижаться in vivo или in vitro при гиперэкспрессии R1R2, на что указывают предварительные результаты (Фигура 28). Мы ожидаем, что гиперэкспрессия R1R2 приведет к повышению силы сокращения в интактных сердечных трабекулах, указывающему на повышение Ca2+-чувствительности и подобному повышению фракции укорочения, наблюдаемому в изолированных кардиомиоцитах. Подобным образом, мы ожидаем, что величина Ca2+-тока будет такой же, как у необработанных трабекул, но в R1R2-гиперэкспрессируюшего трабекулах может наблюдаться более быстрое снижение Ca2+ и сопутствующее повышение силы расслабления. В трабекулах с разрушенной мембраной из R1R2-гиперэкспрессируюшего миокарда может наблюдаться небольшое повышение Ca2+-чувствительности даже при использовании АТФ в качестве субстрата сокращения, которое будут отражать снижение симпатического тонуса и впоследствии приводить к снижению фосфорилирования сердечного тропонина I и миозин-связывающего белка С. Мы ожидаем увидеть отсутствие очевидных изменений во взаимодействиях с Ca2+, отличных от более быстрого угасания Ca2+-тока, подобно AV-R1R2-трансдуцированным кардиомиоцитам in vitro. Более быстрое угасание Ca2+-тока может возникать из-за 1) повышенного укорочения саркомеров, сопряженного с более быстрым отсоединением поперечных мостиков, приводящего к прогрессивному повышению диссоциации Ca2+ в тонких филаментах, делающему, таким образом, Ca2+ доступным для повторный секвестрации раньше, чем в контрольных миоцитах и 2) изменений во взаимодействующих с Ca2+ белках (отношение SERCA:фосфоламбан, фосфорилирование фосфоламбана и/или Ca2+ АТФазы плазматической мембраны) или Ca2+ АТФазных насосах при использовании дезокси-АТФ в качестве энергетического субстрата, повышающих секвестрацию Ca2+. Мы не ожидаем значительных метаболических изменений при острой гиперэкспрессии R1R2, отчасти на основании предварительных данных (Фигура 29), согласно которым содержание высокоэнергетических фосфатов в стабильном состоянии подвергается минимальному влиянию в TG-R1R2-сердце, независимо от повышенной сократительной функции.
ПРИМЕР 50
[0247] Мы определим продолжительные эффекты повышенного клеточного уровня R1R2 и [дезокси-АТФ] на сердечную функцию. Мы ожидаем, что мыши TG-R1R2 будут адаптироваться к повышенной систолической функции снижением β-адренергической стимуляции, а диастолическая функция будет поддерживаться путем адаптации механизмов взаимодействия с Ca2+. Это будет отражаться в 1) повышенной сократимости кардиомиоцитов с минимальным изменением в расслаблении, 2) такой адаптации сердечного метаболизма, которая приведет к отсудив повышения его скорости по сравнению с контролями дикого типа, 3) отсутствии изменения в электромеханическом сопряжении и 4) повышенной чувствительности к β-адренергической стимуляции.
[0248] Для исследования хронической гиперэкспрессии R1R2, мы будем использовать модель битрансгенных мышей (TG-R1R2), которые экспрессируют обе субъединиц рибонуклеотидредуктазы на высоком уровне на протяжении всей жизни. Мы будем начинать изучение со взрослых мышей в возрасте 6 месяцев. Для определения прогрессирования изменения функции и/или адаптации в зависимости от времени за молодыми мышами будут наблюдать с помощью эхокардиографии вплоть до 6-месячного возраста. На основании полученных показателей мы определим более ранние временные точки для исследования. В указанные временные точки будут проведены все экспериментальные оценки, как описано выше. Указанные многосторонние измерения обеспечат подробные данные о молекулярных механизмах адаптации сердечной функции при хронической гиперэкспрессии R1R2, а также возможность сравнения с экспериментами, описанными выше.
[0249] Указанные исследования предоставят подробную информацию о долговременной сердечной адаптации к гиперэкспрессии R1R2 in vivo, где внутренняя адаптация связана с постоянными паракринным и гормональными колебаниями. Мы ожидаем, что гиперэкспрессия R1R2 может повышать β-адренергическую чувствительность посредством адаптации к более высокой общей сократимости, чего не происходит при кратковременной гиперэкспрессии R1R2. Мы не ожидаем увидеть снижения отношения Франка-Старлинга in vivo или in vitro при гиперэкспрессии R1R2, на основании предварительных результатов (Фигура 28) и поскольку зависимость активации сокращения от SL поддерживается при дезокси-АТФ3*. Сердечный выброс можно поддерживать снижением частоты сердечных сокращений (меньший β-адренергический стимула) для компенсации повышенного ударного объема сердца (из-за дезокси-АТФ). Мы также не ожидаем обнаружить функциональный или морфологической гипертрофический фенотип или фенотип дилатации желудочка у мышей TG-R1R2, и предварительные данные предполагают отсутсвие указанного эффекта. Мы ожидаем, что Ca2+ нагрузка СПР сохраняется, так как повышенная функция ЛЖ, в первую очередь, вызвана усиленной активностью миофиламентов. Если активность Ca2+-насоса в СПР постоянно повышена, что приводит к более быстрому снижению Ca2+-тока, то она должна приводить к более короткой продолжительности потенциала действия (APD). Мы ожидаем, что энергетические резервы (PCr, АТФ) будут сохраняться, и будет наблюдаться близкий, но повышенный ответ на стимуляцию высокой концентрацией Ca2+, и что функция митохондрий будет незначительно повышена из-за повышенного использования АТФ и дезокси-АТФ миозином. Вкратце, мы ожидаем увидеть повышенную основную функцию, но с поддержанием способности отвечать на повышенные требования, при минимальной адаптации взаимодействий с Ca2+ и энергетического гомеостаза.
ПРИМЕР 51
[0250] Мы определим влияние повышенного клеточного уровня R1R2 и [дезокси-АТФ] на сердечную функцию пораженных инфарктом сердец. Ожидается, то острая гиперэкспрессии R1R2 и повышенный уровень [дезокси-АТФ] приведет к улучшению работоспособности сердца и по меньшей мере временной остановке прогрессирования в развитие сердечной недостаточности пораженных инфарктом сердец. Многообещающие предварительные данные, согласно которым трансдукция AV-R1R2 восстанавливает сниженные сократительные свойства и характер Ca2+-тока у культивируемых кардиомиоцитов из инфарктных крысиных сердец (Фигура 25), предполагают, что это возможно. Повышение сократительной способности миофиламентов могло бы улучшить сердечную функцию, таким образом, приводя к снижению хронического β-адренергического сигналинга и РКС-опосредованного фосфорилирования, связанного с прогрессированием в сердечную недостаточность. Будет интересно узнать, приводит ли временное усиление функции (при введении одной дозы AAV6-R1R2) к адаптациям, обеспечивающим длительное улучшение. Также известно, что сердце, страдающее сердечной недостаточностью, «испытывают энергетический голод»69*. Многочисленные клинические исследования показали, что сердечная недостаточность усугубляется при энергозатратных терапиях, таких как использование агонистов адренергического рецептора, и улучшается при энергосберегающих терапиях, таких как использование ACEI и бета-блокады. Крайне важно, что повышение уровня дезокси-АТФ не влияет негативно на энергетический обмен в миокарде. Таким образом, поразительно, что предварительные данные показали отсутствие изменений в содержании высокоэнергетических фосфатов при более высокой сократительной функции. Мы определим, будет ли это работать в случае пораженных инфарктом сердец, в которых неоднократно наблюдался нарушенный энергетический метаболизм.
[0251] Мы изучим влияние острой гиперэкспрессии R1R2 на пораженные инфарктом сердца с использованием AAV6-R1R2. Инфаркт миокарда создавали путем длительного лигирования левой нисходящей коронарной артерии. У крыс это, как правило, приводит к появлению инфарктных зон размером ~20-35% от площади поперечного сечения свободной стенки ЛЖ (10-15% от общей площади ЛЖ) со пониженной фракцией укорочения, систолической и диастолической дилатацией желудочка (Фигура 34А-С) и потерей чувствительности работающих сердец к повышенной предварительной нагрузке (Фигура 34D).
[0252] Данные исследования in vivo подтверждают более ранние исследования на культивируемых кардиомиоцитах. В указанном исследовании крысам непосредственно инъецировали в сердце AAV6-R1R2 (как описано выше) на пятый день после инфаркта. Эхокардиографию проводили (вплоть до 8 недель) после трансдукции; результаты показаны на ФИГ.38. Через одну неделю после инъекции FS была улучшена, и через 2 недели FS для сердец инфаркт+вектор была такой же, как для ложнооперированных (без инфаркта) сердец. Указанное восстановление сохранялось до 8 недели, на которой эксперимент заканчивали, что демонтировало улучшение сердечной функции пораженных инфарктом сердец при введении AAV6-R1R2.
[0253] Для того, чтобы определить, помогает ли острая гиперэкспрессия R1R2 предотвращать потерю функции ЛЖ, отдельные животные будут трансдуцированы AAV6-R1R2 за 1-2 недели до проведения хирургического лечения инфаркта. Для всех экспериментов несколько мышей или крыс будут взяты для оценки гемодинамических свойств in situ с помощью экспериментов с использованием катетера Миллара (см. выше). Дополнительные животные будет использоваться для исследований in vitro на работающем сердце, интактных и очищенных трабекулах, культивируемых кардиомиоцитах и изолированных миофибриллах. Как указано выше, мы определим, какие препараты являются наиболее информативными и сфокусируем на них большинство измерений для ограничения числа животных, необходимых для указанных исследований. Некоторые препарированные сердца будут взвешены, фиксированы, заключены и окрашены, и будет проведена их оценка на гипертрофию и фиброз (как выше). Будет приведен иммуноцитохимический анализ на серийных срезах для оценки экспрессии и пенетрантности R1R2 для сопоставления с изменениями функции in vivo и in vitro.
[0254] Мы ожидаем, что гиперэкспрессия R1R2 и повышенный уровень [дезокси-АТФ] улучшат функцию сердца пораженных инфарктом сердец в выбранный для анализа момент времени. Будет улучшен ответ на стимуляцию Ca2+ в высокой концентрации, β-адренергическую стимуляцию и повышающиеся преднагрузки. Измерения на работающем сердце Нили in vitro для сердец, описанных на ФИГ.38, показали потерю чувствительности к предварительной нагрузке сердец (сердечная недостаточность), которые были поражены инфарктом (необработанные), но восстановление чувствительности к предварительной нагрузке пораженных инфарктом сердец, получавших векторы, до уровня контрольных не пораженных инфарктом сердец, что, таким образом, демонстрирует восстановление сердечной функции. См. ФИГ.39, где сила приведена в единицах г⋅см/мин. Указанный эффект может быть обусловлен снижением хронической β-адренергической стимуляции (которую можно оценить путем мониторинга плазматических гормонов), что должно было бы отражаться в многостороннем анализе в улучшении 1) Ca2+-токов, 2) сокращения миофиламентов и величины и кинетики расслабления и 3) энергетического профиля. Мы также ожидаем увидеть различие между подержанными и неподверженными лечению сердцами в α- и β-адренергически опосредуемом фосфорилировании белков кардиомиоцитов.
[0255] Несмотря на то, что настоящее изобретение было выше описано достаточно подробно с использованием иллюстраций и примеров для ясности понимания, специалисту в данной области техники очевидно, что определенные изменения и модификации могут быть реализованы на практике в пределах объема прилагаемой формулы изобретения. Кроме того, каждая ссылка, цитируемая в настоящей заявке, полностью включена в нее посредством ссылки в той же степени, как если бы каждая ссылка была конкретно включена посредством ссылки. В случае несоответствия между настоящей заявкой и ссылкой, предложенной в настоящей заявке, следует руководствоваться настоящей заявкой.
Ссылки
[0256] 1. De tombe PP. Altered contractile function in heart failure. Cardiovascular Research. 1998; 37(2): 367-380.
[0257] 2. Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005; 115(9): 2305-2315.
[0258] 3. Sabbah HN. Biologic rationale for the use of beta-blockers in the treatment of heart failure. Heart Fail Rev. 2004; 9(2): 91-97.
[0259] 4. Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006; 113(2): 305-315.
[0260] 5. Wyskovsky W, Hauptner R, Suko J. Drug-induced calcium release from heavy sarcoplasmic reticulum of skeletal muscle. Biochim. Biophys. Acta. 1988; 938: 89-96.
[0261] 6. MacGowan GA. The myofilament force-calcium relationship as a target for positive intropic therapy in congestive heart failure. Cardiovasc Drugs Ther. 2005; 19(3): 203-210.
[0262] 7. Moreno-Gonzalez A, Fredlund J, Regnier M. Cardiac troponin C (TnC) and a site I skeletal TnC mutant alter Ca2+ versus crossbridge contribution to force in rabbit skeletal fibres. J Physiol. 2005; 562(Pt3): 873-884.
[0263] 8. Tikunova SB, Davis JP. Designing calcium sensitizing mutations in the regulatory domain of cardiac troponin C.J Biol Chem. 2004; 279(34): 35341-35352.
[0264] 9. Kreutziger KL, Piroddi N, Tesi C, Poggesi C, Regnier M. Cooperative activation and tension kinetics in cardiac muscle are strongly modulated by calcium binding kinetics of troponin C. Journal of Molecular and Cellular Cardiology. 2010;In press.
[0265] 10. Santana LF, Kranias EG, Lederer WJ. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J Physiol. 1997; 503(1): 21-29.
[0266] 11. Moreno-Gonzalez A, Korte FS, Dai J, Chen KY, Ho B, Reinecke H, Murry CE, Regnier M. Cell therapy enhances function of remote non-infarcted myocardium. Journal of Molecular and Cellular Cardiology. 2009; 47(5): 603-613.
[0267] 12. Herron TJ, Vandenboom R, Fomicheva E, Mundada L, Edwards T, Metzger JM. Calcium-independent negative inotropy by beta-myosin heavy chain gene transfer in cardiac myocytes. Circ Res. 2007; 100(8): 1182-1190.
[0268] 13. Badrian B, Bogoyevitch MA. Changes in the transcriptional profile of cardiac myocytes following green fluorescent protein expression. DNA Cell Biology. 2007; 26(10): 727-736.
[0269] 14. Nishimura S, Nagai S, Sata M, Katoh M, Yamashita H, Saeki Y, Nagai R, Sugiura S. Expression of green fluorescent protein impairs the force-generating ability of isolated rat ventricular cardiomyocytes. Mol Cell Biochem. 2006; 286(1-2): 59-85.
[0270] 15. Herron TJ, Devaney E, Mundada L, Arden E, Day S, Guerrero-Serna G, Turner I, Westfall MV, Metzger JM. Ca2+-independent positive molecular inotropy for failing rabbit and human cardiac muscle by alpha-myosin motor gene transfer. Faseb J. 2010; 24(2): 415-424.
[0271] 16. Feest ER, Tu A, Luo C, Nowakowski SG, Regnier M. Effect of varying mutant troponin C content on contractile properties of striated muscle. Biophys. J. 2009; 96(3): 228a.
[0272] 17. Norman C, Rall JA, Tikunova SB, Davis JP. Modulation of the rate of cardiac muscle contraction by troponin C constructs with various calcium binding affinities. Am J Physiol Heart Circ Physiol. 2007; 293: H2580-H2587.
[0273] 18. Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrythmia in mice. Journal of Clinical Investigation. 2008; 118(12): 3893-3903.
[0274] 19. Solaro RJ, Gambassi G, Warshaw DM, Keller MR, Spurgeon HA, Beier N. Stereoselective actions of thiadiazinones on canine cardiac myocytes and myofilaments. Circ Res. 1993; 73(6): 981-990.
[0275] 20. White J, Lee JA, Shah N, Orchard CH. Differential effects of the optical isomers of EMD 53998 on contraction and cytoplasmic Ca2+ in isolated ferret cardiac muscle. Circ Res. 1993; 73(1): 61-70.
[0276] 21. Korte FS, Dai J, Weiss R, Vaziri C, Murry CE, Regnier M. Overexpression of cardiomyocyte ribonucleotide reductase (RR) increases contraction without affecting relaxation in adult cardiomyocytes from normal and infarcted hearts. Journal of Muscle Research and Cell Motility. 2010: abstract.
[0277] 22. Lim CC, Yang H, Yang M, Wang CK, Shi J, Berg EA, Pimental DR, Gwathmey JK, Hajjar RJ, Helmes M, Costello CE, Huo S, Liao R. A novel mutant cardiac troponin C disrupts molecular motions critical for calcium binding affinity and cardiomocyte contractility. Biophys. J. 2008; 94: 3577-3589.
[0278] 23. Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J. Physiol. Lond. 1999; 517(1): 143-157.
[0279] 24. Preetha N, Yiming W, Helmes M, Fukuda N, Siegfried L, Granzier H. Restoring force development by titin/connectin and assessment of Ig domain unfolding. Journal of Muscle Research and Cell Motility. 2005; 26(307-317).
[0280] 1. Ganguly PK, Pierce GN, Dhalla KS, Dhalla NS. Defective sarcoplasmic reticular calcium transport in diabetic cardiomyopathy. American Journal of Physiology. 1983; 244: E528-E535.
[0281] 2. Sabbah HN. Biologic rationale for the use of beta-blockers in the treatment of heart failure. Heart Fail Rev. 2004; 9(2): 91-97.
[0282] 3. LeWinter MM. Functional consequences of sarcomeric protein abnormalities in failing myocardium. Heart Fail Rev. 2005; 10(3): 249-257.
[0283] 4. Palmer BM. Thick filament proteins and performance in human heart failure. Heart Fail Rev. 2005; 10(3): 187-197.
[0284] 5. Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005; 115(9): 2305-2315.
[0285] 6. Regnier M, Rivera AJ, Chen Y, Chase PB. 2-deoxy-ATP enhances contractility of rat cardiac muscle. Circ. Res. 2000; 86(12): 1211-1217.
[0286] 7. Regnier M, Martin H, Barsotti RJ, Rivera AJ, Martyn DA, Clemmens E. Cross-bridge versus thin filament contributions to the level and rate of force development in cardiac muscle. Biophys J. 2004; 87(3): 1815-1824.
[0287] 8. Adhikari BB, Regnier M, Rivera AJ, Kreutziger KL, Martyn DA. Cardiac length dependence of force and force redevelopment kinetics with altered cross-bridge cycling. Biophys J. 2004; 87(3): 1784-1794.
[0288] 9. Clemmens EW, Regnier M. Skeletal regulatory proteins enhance thin filament sliding speed and force by skeletal HMM. J Muscle Res Cell Motil. 2004; 25(7): 515-525.
[0289] 10. Schoffstall B, Clark A, Chase PB. Positive inotropic effects of low dATP/ATP ratios on mechanics and kinetics of porcine cardiac muscle. Biophys J. 2006; 91(6): 2216-2226.
[0290] 11. Kashlan OB, Cooperman BS. Comprehensive model for allosteric regulation of mammalian ribonucleotide reductase: refinements and consequences. Biochemistry. 2003; 42(6): 1696-1706.
[0291] 12. Kashlan OB, Scott CP, Lear JD, Cooperman BS. A comprehensive model for the allosteric regulation of mammalian ribonucleotide reductase. Functional consequences of ATP- and dATP-induced oligomerization of the large subunit. Biochemistry. 2002; 41(2): 462-474.
[0292] 13. Santana LF, Kranias EG, Lederer WJ. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J Physiol. 1997; 503(1): 21-29.
[0293] 14. Moreno-Gonzalez A, Korte FS, Dai J, Chen KY, Ho B, Reinecke H, Murry CE, Regnier M. Cell therapy enhances function of remote non-infarcted myocardium. Journal of Molecular and Cellular Cardiology. 2009; 47(5): 603-613.
[0294] 15. He TC, Zhou S, Da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998; 95: 2509-2514.
[0295] 16. Herron TJ, Vandenboom R, Fomicheva E, Mundada L, Edwards T, Metzger JM. Calcium-independent negative inotropy by beta-myosin heavy chain gene transfer in cardiac myocytes. Circ Res. 2007; 100(8): 1182-1190.
[0296] 17. Badrian B, Bogoyevitch MA. Changes in the transcriptional profile of cardiac myocytes following green fluorescent protein expression. DNA Cell Biology. 2007; 26(10): 727-736.
[0297] 18. Nishimura S, Nagai S, Sata M, Katoh M, Yamashita H, Saeki Y, Nagai R, Sugiura S. Expression of green fluorescent protein impairs the force-generating ability of isolated rat ventricular cardiomyocytes. Mol Cell Biochem. 2006; 286(1-2): 59-85.
[0298] 19. Herron TJ, Devaney E, Mundada L, Arden E, Day S, Guerrero-Serna G, Turner I, Westfall MV, Metzger JM. Ca2+-independent positive molecular inotropy for failing rabbit and human cardiac muscle by alpha-myosin motor gene transfer. Faseb J. 2010; 24(2): 415-424.
[0299] 20. Regnier M, Rivera AJ, Chen Y, Chase PB. 2-deoxy-ATP enhances contractility of rat cardiac muscle. Circ Res. 2000; 86(12): 1211-1217.
[0300] 21. Schoffstall B, Clark A, Chase P. Positive inotropic effects of low dATP/ATP ratios on mechanics and kinetics of porcine cardiac muscle. Biophys J. 2006; 91(6): 2216-2226.
[0301] 22. Schoffstall B, Chase P. Increased intracellular [dATP] enhances cardiac contraction in embryonic chick cardiomyocytes. Journal of Cellular Biochemistry. 2008; 104(6): 2217-2227.
[0302] 23. Xu X, Page JL, Surtees JA, Liu H, Lagedrost S, Lu Y, Bronson R, Alani E, Nikitin AY, Weiss RS. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 2008; 68(8): 2652-2660.
[0303] 24. Regnier M, Lee DM, Homsher E. ATP analogs and muscle contraction: mechanics and kinetics of nucleoside triphosphate binding and hydrolysis. Biophys J. 1998; 74(6): 3044-3058.
[0304] 25. Regnier M, Homsher E. The effect of ATP analogs on posthydrolytic and force development steps in skinned skeletal muscle fibers. Biophys J. 1998; 74(6): 3059-3071.
[0305] 26. Regnier M, Martyn DA, Chase PB. Calcium regulation of tension redevelopment kinetics with 2-deoxy-ATP or low [АТФ] in rabbit skeletal muscle. Biophys J. 1998; 74(4): 2005-2015.
[0306] 27. Janssen PM, Periasamy M. Determinants of frequency-dependent contraction and relaxation of mammalian myocardium. J Mol Cell Cardiol. 2007; 43(5): 523-531.
[0307] 28. Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994; 476: 279-293.
[0308] 29. Bers DM. Cardiac excitation-contraction coupling. Nature. 2002; 415: 198-205.
[0309] 30. Luo Y, Davis JP, Smillie LB, Rall JA. Determinants of relaxation rate in rabbit skinned skeletal muscle fibres. J Physiol. 2002; 545(Pt3): 887-901.
[0310] 31. Luo Y, Davis JP, Tikunova SB, Smillie LB, Rall JA. Myofibrillar determinants of rate of relaxation in skinned skeletal muscle fibers. Adv Exp Med Biol. 2003; 538: 573-581.
[0311] 32. Piroddi N, Belus A, Scellini B, Tesi C, Giunti G, Cerbai E, Mugelli A, Poggesi C. Tension generation and relaxation in single myofibrils from human atrial and ventricular myocardium. Pflugers Arch. 2007; 454(1): 63-73.
[0312] 33. Tesi C, Piroddi N, Colomo F, Poggesi C. Relaxation kinetics following sudden Ca(2+) reduction in single myofibrils from skeletal muscle. Biophys J. 2002; 83(4): 2142-2151.
[0313] 34. Edman KAP. The velocity of unloaded shortening and its relation to sarcomere length and isometric force in vertebrate muscle fibers. J. Physiol. Lond. 1979; 291: 143-159.
[0314] 35. Hinken AC, McDonald KS. Inorganic phosphate speeds loaded shortening in rat skinned cardiac myocytes. American Journal of Physiology. 2004; 287: C500-C507.
[0315] Willott R, Gomes A, Chang A, Parvatiyar M, Pinto J, Potter J. Mutations in Troponin that cause ГКМП, ДКМП AND RCM: what can we learn about thin filament function? J Mol Cell Cardiol. 2010; 48(5): 882-92.
[0316] Parvatiyar M, Pinto J, Liang J, Potter J. Predicting cardiomyopathic phenotypes by altering Ca2+ affinity of cardiac troponin C. J Biol Chem. 2010; 285(36): 27785-97. PMCID: PMC2934646.
[0317] Sjaastad I, Wasserstrom J, Sejersted O. Heart failure — a challenge to our current concepts of excitation-contraction coupling. J Physiol. 2003; 546(Pt 1): 33-47. PMCID: PMC2342477.
[0318] DiPaola N, Sweet W, Stull L, Francis G, Schomisch Moravec C. Beta-adrenergic receptors and calcium cycling proteins in non-failing, hypertrophied and failing human hearts: transition from hypertrophy to failure. J Mol Cell Cardiol. 2001; 33(6): 1283-95.
[0319] Minamisawa S, Hoshijima M, Chu G, Ward C, Frank K, Gu Y, et al. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999; 99(3): 313-22.
[0320] Kreutziger KL, Piroddi N, Tesi C, Poggesi C, Regnier M. Cooperative activation and tension kinetics in cardiac muscle are strongly modulated by calcium binding kinetics of troponin C. Journal of Molecular and Cellular Cardiology. 2010; Accepted with Revisions.
[0321] Korte FS, Murry C, Regnier M. Targeting Myofilaments To Improve Cardiomyocyte Contractile Properties in Heart Disease. Molecular Therapy. 2009; 17(S1): 914a.
[0322] The World Health Report 2008: World Health Organization; 2008 Contract No.: Document Number|.
[0323] Tardiff J. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005; 10(3): 237-48.
[0324] Palmiter K, Tyska M, Haeberle J, Alpert N, Fananapazir L, Warshaw D. R403Q and L908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J Muscle Res Cell Motil. 2000; 21(7): 609-20.
[0325] Blanchard E, Seidman C, Seidman J, LeWinter M, Maughan D. Altered crossbridge kinetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. Circ Res. 1999; 84(4): 475-83.
[0326] Gomes A, Potter J. Molecular and cellular aspects of troponin cardiomyopathies. Ann N Y Acad Sci. 2004; 1015: 214-24.
[0327] Gomes A, Barnes J, Harada K, Potter J. Role of troponin T in disease. Mol Cell Biochem. 2004; 263(1-2): 115-29.
[0328] Hernandez O, Szczesna-Cordary D, Knollmann B, Miller T, Bell M, Zhao J, et al. F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. J Biol Chem. 2005; 280(44): 37183-94.
[0329] Chandra M, Rundell V, Tardiff J, Leinwand L, De Tombe P, Solaro R. Ca(2+) activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. Am J Physiol Heart Circ Physiol. 2001; 280(2): H705-13.
[0330] Montgomery D, Tardiff J, Chandra M. Cardiac troponin T mutations: correlation between the type of mutation and the nature of myofilament dysfunction in transgenic mice. J Physiol. 2001; 536(Pt2): 583-92. PMCID: PMC2278862.
[0331] Miller T, Szczesna D, Housmans P, Zhao J, de Freitas F, Gomes A, et al. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J Biol Chem. 2001; 276(6): 3743-55.
[0332] Venkatraman G, Harada K, Gomes A, Kerrick W, Potter J. Different functional properties of troponin T mutants that cause dilated cardiomyopathy. J Biol Chem. 2003; 278(43): 41670-6.
[0333] Venkatraman G, Gomes A, Kerrick W, Potter J. Characterization of troponin T dilated cardiomyopathy mutations in the fetal troponin isoform. J Biol Chem. 2005; 280(18): 17584-92.
[0334] Mirza M, Marston S, Willott R, Ashley C, Mogensen J, McKenna W, et al. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J Biol Chem. 2005; 280(31): 28498-506.
[0335] Jideama N, Noland TJ, Raynor R, Blobe G, Fabbro D, Kazanietz M, et al. Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J Biol Chem. 1996; 271(38): 23277-83.
[0336] Du C, Morimoto S, Nishii K, Minakami R, Ohta M, Tadano N, et al. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Res. 2007; 101(2): 185-94.
[0337] Ahmad F, Banerjee S, Lage M, Huang X, Smith S, Saba S, et al. The role of cardiac troponin T quantity and function in cardiac development and dilated cardiomyopathy. PLoS One. 2008; 3(7): e2642. PMCID: PMC2441440.
[0338] Bristow M, Ginsburg R, Minobe W, Cubicciotti R, Sageman W, Lurie K, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982; 307(4): 205-11.
[0339] Bristow M, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, et al. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res. 1986; 59(3): 297-309.
[0340] Rockman H, Koch W, Lefkowitz R. Seven-transmembrane-spanning receptors and heart function. Nature. 2002; 415(6868): 206-12.
[0341] Moreno-Gonzalez A, Korte F, Dai J, Chen K, Ho B, Reinecke H, et al. Cell therapy enhances function of remote non-infarcted myocardium. J Mol Cell Cardiol. 2009; 47(5): 603-13.
[0342] Lewinter M, Vanburen P. Myofilament remodeling during the progression of heart failure. J Card Fail. 2002; 8(6 Suppl): S271-5.
[03431 LeWinter M. Functional consequences of sarcomeric protein abnormalities in failing myocardium. Heart Fail Rev. 2005; 10(3): 249-57.
[0344] LeWinter M, VanBuren P. Sarcomeric proteins in hypertrophied and failing myocardium: an overview. Heart Fail Rev. 2005; 10(3): 173-4.
[0345] Garvey J, Kranias E, Solaro R. Phosphorylation of C-protein, troponin I and phospholamban in isolated rabbit hearts. Biochem J. 1988; 249(3): 709-14. PMCID: PMC1148764.
[0346] Westfall M, Solaro R. Alterations in myofibrillar function and protein profiles after complete global ischemia in rat hearts. Circ Res. 1992; 70(2): 302-13.
[0347] Reiken S, Gaburjakova M, Guatimosim S, Gomez A, D'Armiento J, Burkhoff D, et al. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J Biol Chem. 2003; 278(1): 444-53.
[0348] , Obayashi M, Xiao B, Stuyvers B, Davidoff A, Mei J, Chen S, et al. Spontaneous diastolic contractions and phosphorylation of the cardiac ryanodine receptor at serine-2808 in congestive heart failure in rat. Cardiovasc Res. 2006; 69(1): 140-51.
[0349] Schwinger R, Bölck B, Münch G, Brixius K, Müller-Ehmsen J, Erdmann E. cAMP-dependent protein kinase A-stimulated sarcoplasmic reticulum function in heart failure. Ann N Y Acad Sci. 1998; 853: 240-50.
[0350] Forissier J, Carrier L, Farza H, Bonne G, Bercovici J, Richard P, et al. Codon 102 of the cardiac troponin T gene is a putative hot spot for mutations in familial hypertrophic cardiomyopathy. Circulation. 1996; 94(12): 3069-73.
[0351] Moolman J, Corfield V, Posen B, Ngumbela K, Seidman C, Brink P, et al. Sudden death due to troponin T mutations. J Am Coll Cardiol. 1997; 29(3): 549-55.
[0352] Watkins H, McKenna W, Thierfelder L, Suk H, Anan R, O'Donoghue A, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995; 332(16): 1058-64.
[0353] Blankinship M, Gregorevic P, Allen J, Harper S, Harper H, Halbert C, et al. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther. 2004; 10(4): 671-8.
[0354] Sanbe A, Gulick J, Hanks M, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003; 92(6): 609-16.
[0355] Davis J, Metzger J. Combinatorial effects of double cardiomyopathy mutant alleles in rodent myocytes: a predictive cellular model of myofilament dysregulation in disease. PLoS One. 2010; 5(2): e9140. PMCID: PMC2818843.
[0356] Navedo M, Takeda Y, Nieves-Cintrón M, Molkentin J, Santana L. Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. Am J Physiol Cell Physiol. 2010; 298(2): C211-20. PMCID: PMC2822492.
[0357] Navedo M, Nieves-Cintrón M, Amberg G, Yuan C, Votaw V, Lederer W, et al. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ Res. 2008; 102(2): e1-e11.
[0358] Rodgers B, Interlichia J, Garikipati D, Mamidi R, Chandra M, Nelson O, et al. Myostatin represses physiological hypertrophy of the heart and excitation-contraction coupling. J Physiol. 2009; 587(Pt20): 4873-86. PMCID: PMC2770153.
[0359] Rossow C, Dilly K, Yuan C, Nieves-Cintrón M, Cabarrus J, Santana L. NFATc3-dependent loss of I (to) gradient across the left ventricular wall during chronic beta adrenergic stimulation. J Mol Cell Cardiol. 2009; 46(2): 249-56.
[0360] Zhu W, Santana L, Laflamme M. Local control of excitation-contraction coupling in human embryonic stem cell-derived cardiomyocytes. PLoS One. 2009; 4(4): e5407. PMCID: PMC2671137.
[0361] Hancock W, Martyn D, Huntsman L. Ca2+ and segment length dependence of isometric force kinetics in intact ferret cardiac muscle. Circ Res. 1993; 73(4): 603-11.
[0362] Janssen P, Lehnart S, Prestle J, Lynker J, Salfeld P, Just H, et al. The trabecula culture system: a novel technique to study contractile parameters over a multiday time period. Am J Physiol. 1998; 274(5 Pt2): H1481-8.
[0363] Monasky M, Varian K, Davis J, Janssen P. Dissociation of force decline from calcium decline by preload in isolated rabbit myocardium. Pflugers Arch. 2008; 456(2): 267-76.
[0364] Varian K, Raman S, Janssen P. Measurement of myofilament calcium sensitivity at physiological temperature in intact cardiac trabeculae. Am J Physiol Heart Circ Physiol. 2006; 290(5): H2092-7.
[0365] Varian K, Janssen P. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. 2007; 292(5): H2212-9.
[0366] Huntsman L, Rondinone J, Martyn D. Force-length relations in cardiac muscle segments. Am J Physiol. 1983; 244(5): H701-7.
[0367] Guo T, Cornea R, Huke S, Camors E, Yang Y, Picht E, et al. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ Res. 2010; 106(11): 1743-52. PMCID: PMC2895429.
[0368] Gellen B, Fernandez-Velasco M, Briec F, Vinet L, LeQuang K, Rouet-Benzineb P, et al. Conditional FKBP12.6 overexpression in mouse cardiac myocytes prevents triggered ventricular tachycardia through specific alterations in excitation-contraction coupling. Circulation. 2008; 117(14): 1778-86.
[0369] Backs J, Backs T, Neef S, Kreusser M, Lehmann L, Patrick D, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA. 2009; 106(7): 2342-7. PMCID: PMC2650158.
[0370] Ling H, Zhang T, Pereira L, Means C, Cheng H, Gu Y, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009; 119(5): 1230-40. PMCID: PMC2673879.
[0371] Lygren B, Tasken K. Compartmentalized cAMP signalling is important in the regulation of Ca(2+) cycling in the heart. Biochem Soc Trans. 2006; 34(Pt4): 489-91.
[0372] Guinto P, Haim T, Dowell-Martino C, Sibinga N, Tardiff J. Temporal and mutation-specific alterations in Ca2+ homeostasis differentially determine the progression of cTnT-related cardiomyopathies in murine models. Am J Physiol Heart Circ Physiol. 2009; 297(2): H614-26. PMCID: PMC2724218.
[0373] Colomo F, Piroddi N, Poggesi C, te Kronnie G, Tesi C. Active and passive forces of isolated myofibrils from cardiac and fast skeletal muscle of the frog. J Physiol. 1997; 500(Pt2): 535-48. PMCID: PMC1159402.
[0374] Colomo F, Nencini S, Piroddi N, Poggesi C, Tesi C. Calcium dependence of the apparent rate of force generation in single striated muscle myofibrils activated by rapid solution changes. Adv Exp Med Biol. 1998; 453: 373-81; discussion 81-2.
[0375] Piroddi N, Tesi C, Pellegrino M, Tobacman L, Homsher E, Poggesi C. Contractile effects of the exchange of cardiac troponin for fast skeletal troponin in rabbit psoas single myofibrils. J Physiol. 2003; 552(Pt3): 917-31. PMCID: PMC2343446.
[0376] Piroddi N, Belus A, Scellini B, Tesi C, Giunti G, Cerbai E, et al. Tension generation and relaxation in single myofibrils from human atrial and ventricular myocardium. Pflugers Arch. 2007; 454(1): 63-73.
[0377] Belus A, Piroddi N, Scellini B, Tesi C, Amati G, Girolami F, et al. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J Physiol. 2008; 586(Pt15): 3639-44. PMCID: PMC2538824.
[0378] Stehle R, Solzin J, Iorga B, Poggesi C. Insights into the kinetics of Ca2+-regulated contraction and relaxation from myofibril studies. Pflugers Arch. 2009; 458(2): 337-57.
[0379] Zoghbi WA, Quinones MA. Determination of cardiac output by Doppler echocardiography: a critical appraisal. Herz. 1986; 11(5): 258-68.
[0380] Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V, et al. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol. 2006; 290(6): H2196-203.
[0381] Tanaka N, Dalton N, Mao L, Rockman HA, Peterson KL, Gottshall KR, et al. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation. 1996; 94(5): 1109-17.
[0382] Stevens K, Kreutziger K, Dupras S, Korte F, Regnier M, Muskheli V, et al. Physiological function and transplantation of scaffold-free and vascularized human cardiac muscle tissue. Proc Natl Acad Sci USA. 2009; 106(39): 16568-73. PMCID: PMC2746126.
[0383] Walker L, Walker J, Ambler S, Buttrick P. Stage-specific changes in myofilament protein phosphorylation following myocardial infarction in mice. J Mol Cell Cardiol. 2010; 48(6): 1180-6.
[0384] Wang J, Liu X, Arneja A, Dhalla N. Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. Can J Cardiol. 1999; 15(6): 683-90.
[0385] Wang X, Dhalla N. Modification of beta-adrenoceptor signal transduction pathway by genetic manipulation and heart failure. Mol Cell Biochem. 2000; 214(1-2): 131-55.
[0386] Korte FS, Feest ER, Razumova MV, Regnier M. Sarcomere Length Dependent Contractile Activation is Reduced in Rat Trabeculae Exchanged with cTn Containing the L48Q cTnC Variant Independently of Strong Binding Cross-Bridges. Biophysical Journal. 2010; 98(3): 356a.
[0387] He H, Javadpour M, Latif F, Tardiff J, Ingwall J. R-92L and R-92W mutations in cardiac troponin T lead to distinct energetic phenotypes in intact mouse hearts. Biophys J. 2007; 93(5): 1834-44. PMCID: PMC1948064.
[0388] Rice R, Guinto P, Dowell-Martino C, He H, Hoyer K, Krenz M, et al. Cardiac myosin heavy chain isoform exchange alters the phenotype of cTnT-related cardiomyopathies in mouse hearts. J Mol Cell Cardiol. 2010; 48(5): 979-88.
[0389] Haim T, Dowell C, Diamanti T, Scheuer J, Tardiff J. Independent FHC-related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. J Mol Cell Cardiol. 2007; 42(6): 1098-110.
[0390] Ertz-Berger B, He H, Dowell C, Factor S, Haim T, Nunez S, et al. Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proc Natl Acad Sci USA. 2005; 102(50): 18219-24. PMCID: PMC1298915.
[0391] Gilman G, Khandheria BK, Hagen ME, Abraham TP, Seward JB, Belohlavek M. Strain rate and strain: a step-by-step approach to image and data acquisition. J Am Soc Echocardiogr. 2004; 17(9): 1011-20.
[0392] Pislaru C, Abraham TP, Belohlavek M. Strain and strain rate echocardiography. Curr Opin Cardiol. 2002; 17(5): 443-54.
[0393] Becker M, Bilke E, Kuhl H, Katoh M, Kramann R, Franke A, et al. Analysis of myocardial deformation based on pixel tracking in two dimensional echocardiographic images enables quantitative assessment of regional left ventricular function. Heart. 2006; 92(8): 1102-8.
[0394] Leitman M, Lysyansky P, Sidenko S, Shir V, Peleg E, Binenbaum M, et al. Two-dimensional strain-a novel software for real-time quantitative echocardiographic assessment of myocardial function. J Am Soc Echocardiogr. 2004; 17(10): 1021-9.
[0395] 1.* Regnier M, Rivera AJ, Chen Y, Chase PB. 2-deoxy-ATP enhances contractility of rat cardiac muscle. Circ Res. 2000; 86(12): 1211-1217.
[0396] 2* Regnier M, Martin H, Barsotti RJ, Rivera AJ, Martyn DA, Clemmens E. Cross-bridge versus thin filament contributions to the level and rate of force development in cardiac muscle. Biophys J. 2004; 87(3): 1815-1824.
[0397] 3.* Adhikari BB, Regnier M, Rivera AJ, Kreutziger KL, Martyn DA. Cardiac length dependence of force and force redevelopment kinetics with altered cross-bridge cycling. Biophys J. 2004; 87(3): 1784-1794.
[0398] 4.* Regnier M, Homsher E. The effect of ATP analogs on posthydrolytic and force development steps in skinned skeletal muscle fibers. Biophys J. 1998; 74(6): 3059-3071.
[0399] 5.* Regnier M, Lee DM, Homsher E. ATP analogs and muscle contraction: mechanics and kinetics of nucleoside triphosphate binding and hydrolysis. Biophys J. 1998; 74(6): 3044-3058.
[0400] 6.* Regnier M, Martyn DA, Chase BP. Calcium regulation of tension redevelopment kinetics with 2-deoxy-ATP or low [АТФ] in rabbit skeletal muscle. Biophysical Journal. 1998; 74(4): 2005-2015.
[0401] 7.* Korte FS, Dai J, Buckley K, Feest ER, Murry CE, Regnier M. Upregulation of cardiomyocyte ribonucleotide reductase increases intracellular 2-deoxy-ATP, contractility, and relaxation. Circulation Research. 2011; In Review.
[0402] 8.* Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982; 307(4): 205-211.
[0403] 9.* Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res. 1986; 59(3): 297-309.
[0404) 10.* Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002; 415(6868): 206-212.
[0405] 11.* Moreno-Gonzalez A, Korte FS, Dai J, Chen KY, Ho B, Reinecke H, Murry CE, Regnier M. Cell therapy enhances function of remote non-infarcted myocardium. Journal of Molecular and Cellular Cardiology. 2009; 47(5): 603-613.
[0406] 12.* Lewinter MM, Vanburen P. Myofilament remodeling during the progression of heart failure. J Card Fail. 2002; 8(6 Suppl): S271-275.
[0407] 13.* LeWinter M. Functional consequences of sarcomeric protein abnormalities in failing myocardium. Heart Fail Rev. 2005; 10(3): 249-257.
[0408] 14.* LeWinter M, VanBuren P. Sarcomeric proteins in hypertrophied and failing myocardium: an overview. Heart Fail Rev. 2005; 10(3): 173-174.
[0409] 15.* Garvey JL, Kranias EG, Solaro RJ. Phosphorylation of C-protein, troponin I and phospholamban in isolated rabbit hearts. Biochem J. 1988; 249(3): 709-714.
[0410] 16.* Westfall MV, Solaro RJ. Alterations in myofibrillar function and protein profiles after complete global ischemia in rat hearts. Circ Res. 1992; 70(2): 302-313.
[0411] 17.* Reiken S, Gaburjakova M, Guatimosim S, Gomez AM, D'Armiento J, Burkhoff D, Wang J, Vassort G, Lederer WJ, Marks AR. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J Biol Chem. 2003; 278(1): 444-453.
[0412] 18.* Obayashi M, Xiao B, Stuyvers BD, Davidoff AW, Mei J, Chen SR, ter Keurs HE. Spontaneous diastolic contractions and phosphorylation of the cardiac ryanodine receptor at serine-2808 in congestive heart failure in rat. Cardiovasc Res. 2006; 69(1): 140-151.
[0413] 19.* Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005; 115(9): 2305-2315.
[0414] 20.* Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006; 113(2): 305-315.
[0415] 21.* Edes I, Kiss E, Kitada Y, Powers FM, Papp JG, Kranias EG, Solaro RJ. Effects of Levosimendan, a cardiotonic agent targeted to troponin C, on cardiac function and on phosphorylation and Ca2+ sensitivity of cardiac myofibrils and sarcoplasmic reticulum in guinea pig heart. Circ Res. 1995; 77(1): 107-113.
[0416] 22.* Wyskovsky W, Hauptner R, Suko J. Drug-induced calcium release from heavy sarcoplasmic reticulum of skeletal muscle. Biochim. Biophys. Acta. 1988; 938: 89-96.
[0417] 23.* Schoffstall B, Clark A, Chase PB. Positive inotropic effects of low dATP/ATP ratios on mechanics and kinetics of porcine cardiac muscle. Biophys J. 2006; 91(6): 2216-2226.
[0418] 24.* Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL, Miller AD, Miller DA, Chamberlain JS. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther. 2004; 10(4): 671-678.
[0419] 25.* Xu X, Page JL, Surtees JA, Liu H, Lagedrost S, Lu Y, Bronson R, Alani E, Nikitin AY, Weiss RS. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 2008; 68(8): 2652-2660.
[0420] 26.* Ylikallio E, Page JL, Xu X, Lampinen M, Bepler G, Ide T, Tyynismaa H, Weiss RS, Suomalainen A. Ribonucleotide reductase is not limiting for mitochondrial DNA copy number in mice. Nucleic Acids Research. 2010; 38(22): 8208-8218.
[0421] 27.* Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrythmia in mice. Journal of Clinical Investigation. 2008; 118(12): 3893-3903.
[0422] 28.* Solaro RJ, Gambassi G, Warshaw DM, Keller MR, Spurgeon HA, Beier N. Stereoselective actions of thiadiazinones on canine cardiac myocytes and myofilaments. Circ Res. 1993; 73(6): 981-990.
[0423] 29.* White J, Lee JA, Shah N, Orchard CH. Differential effects of the optical isomers of EMD 53998 on contraction and cytoplasmic Ca2+ in isolated ferret cardiac muscle. Circ Res. 1993; 73(1): 61-70.
[0424] 30.* Huke S, Knollmann BC. Increased myofilament Ca2+ sensitivity and arrhythmia susceptibility. Journal of Molecular and Cellular Cardiology. 2010; 48(5): 824-833.
[0425] 31.* Zoghbi WA, Quinones MA. Determination of cardiac output by Doppler echocardiography: a critical appraisal. Herz. 1986; 11(5): 258-268.
[0426] 32.* Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V, Morine KJ, Gardner TJ, Discher DE, Sweeney HL. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol. 2006; 290(6): H2196-2203.
[0427] 33.* Tanaka N, Dalton N, Mao L, Rockman HA, Peterson KL, Gottshall KR, Hunter JJ, Chien KR, Ross J, Jr. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation. 1996; 94(5): 1109-1117.
[0428] 34.* Fernandes S, Naumova AV, Zhu WZ, Laflamme MA, Gold J, Murry CE. Human embryonic stem cell-derived cardiomyocytes engraft but do not alter cardiac remodeling after chronic infarction in rats. Journal of Molecular and Cellular Cardiology. 2010; 49:941-949.
[0429] 35.* Monasky MM, Varian KD, Davis JP, Janssen PM. Dissociation of force decline from calcium decline by preload in isolated rabbit myocardium. Pflugers Arch. 2008; 456(2): 267-276.
[0430] 36.* Varian K, Raman S, Janssen P. Measurement of myofilament calcium sensitivity at physiological temperature in intact cardiac trabeculae. Am J Physiol Heart Circ Physiol. 2006; 290(5): H2092-2097.
[0431] 37.* Varian KD, Janssen PM. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. 2007; 292(5): H2212-2219.
[0432] 38.* Janssen P, Lehnart S, Prestle J, Lynker J, Salfeld P, Just H, Hasenfuss G. The trabecula culture system: a novel technique to study contractile parameters over a multiday time period. Am J Physiol. 1998; 274(5 Pt2): H1481-1488.
[0433] 39.* Kreutziger KL, Piroddi N, J.T. M, Tesi C, Poggesi C, Regnier M. Cooperative activation and tension kinetics in cardiac muscle are strongly modulated by calcium binding kinetics of troponin C. Journal of Molecular and Cellular Cardiology. 2011; 50(1): 165-174.
[0434] 40.* Kreutziger KL, Piroddi N, Scellini B, Tesi C, Poggesi C, Regnier M. Thin filament Ca2+ binding properties and regulatory unit interactions alter kinetics of tension development and relaxation in rabbit skeletal muscle. Journal of Physiology. 2008; 586(15): 3683-3700.
[0435] 41.* Belus A, Piroddi N, Tesi C. Mechanism of cross-bridge detachment in isometric force relaxation of skeletal and cardiac myofibrils. Journal of Muscle Research and Cell Motility. 2003; 24(4-6): 261-267.
[0436] 42.* Guo X, Lafiamme MA, Becker PL. Cyclic ADP-ribose does not regulate sarcoplasmic reticulum Ca2+ release in intact cardiomyocytes. Circ Res. 1996; 79: 147-151.
[0437] 43.* Lafiamme MA, Becker PL. Ca2+-induced current oscillations in rabbit ventricular myocytes. Circ Res. 1996; 78: 707-716
[0438] 44.* Lafiamme MA, Becker PL. Do beta 2-adrenergic receptors modulate Ca2+ in adult rat ventricular myocytes? Am J Physiol. 1998; 274: H1308-1314.
[0439] 45.* Zhu WZ, Santana LF, Lafiamme MA. Local control of excitation-contraction coupling in human embryonic stem cell-derived cardiomyocytes. PLoS One. 2009; 4: e5407.
[0440] 46.* Zhu WZ, Xie Y, Moyes KW, Gold JD, Askari B, Lafiamme MA. Neuregulin/erbb signaling regulates cardiac subtype specification in differentiating human embryonic stem cells. Circ Res. 2010; 107: 776-786.
[0441] 47.* Adler ED, Chen VC, Bystrup A, Kaplan AD, Giovannone S, Briley-Saebo K, Young W, Kattman S, Mani V, Lafiamme MA, Zhu WZ, Fayad Z, Keller G. The cardiomyocyte lineage is critical for optimization of stem cell therapy in a mouse model of myocardial infarction. Faseb J. 2010; 24: 1073-1081.
[0442] 48.* Xu C, Police S, Hassanipour M, Li Y, Chen Y, Priest C, O'Sullivan C, Lafiamme MA, Zhu WZ, Van Biber B, Hegerova L, Yang J, Delavan-Boorsma K, Davies A, Lebkowski J, Gold JD. Efficient generation and cryopreservation of cardiomyocytes derived from human embryonic stem cells. Regenerative Medicine. 2011; 6: 53-66.
[0443] 49.* Magistretti J, Mantegazza M, de Curtis M, Wanke E. Modalities of distortion of physiological voltage signals by patch-clamp amplifiers: a modeling study. Biophys J. 1998; 74(2 Pt1): 831-842.
[0444] 50.* Magistretti J, Mantegazza M, Guatteo E, Wanke E. Action potentials recorded with patch-clamp amplifiers: are they genuine? Trends Neurosci. 1996; 19(12): 530-534.
[0445] 51.* Akaike N, Harata N. Nystatin perforated patch recording and its applications to analyses of intracellular mechanisms. Jpn J Physiol. 1994; 44(5): 433-473.
[0446] 52.* Strauss U, Herbrik M, Mix E, Schubert R, Rolfs A. Whole-cell patch-clamp: true perforated or spontaneous conventional recordings? Pflugers Arch. 2001; 442(4): 634-638.
[0447] 53.* Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993; 262(5134): 740-744.
[0448] 54.* Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J. 2000; 78(5): 2655-2667.
[0449] 55.* Santana LF, Cheng H, Gomez AM, Cannell MB, Lederer WJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res. 1996; 78(1): 166-171.
[0450] 56.* Guo X, Laflamme MA, Becker PL. Cyclic ADP-ribose does not regulate sarcoplasmic reticulum Ca2+ release in intact cardiac myocytes. Circ Res. 1996; 79(1): 147-151.
[0451] 57.* Zhu WZ, Santana LF, Laflamme MA. Local control of excitation-contraction coupling in human embryonic stem cell-derived cardiomyocytes. PLoS ONE. 2009; 4(4): e5407.
[0452] 58.* Santana LF, Kranias EG, Lederer WJ. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J Physiol. 1997; 503(Pt1): 21-29.
[0453] 59.* Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol. 1992; 453: 591-608.
[0454] 60.* Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994; 476(2): 279-293.
[0455] 61.* Terracciano CM, Philipson KD, MacLeod KT. Overexpression of the Na(+)/Ca(2+) exchanger and inhibition of the sarcoplasmic reticulum Ca(2+)-ATPase in ventricular myocytes from transgenic mice. Cardiovasc Res. 2001; 49(1): 38-47.
[0456] 62.* Dilly KW, Rossow CF, Votaw VS, Meabon JS, Cabarrus JL, Santana LF. Mechanisms underlying variations in excitation-contraction coupling across the mouse left ventricular free wall. J Physiol. 2006; 572(Pt 1): 227-241.
[0457] 63.* Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005; 112(15): 2339-2346.
[0458] 64.* Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009; 119(21): 2818-2828.
[0459] 65.* Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, Goodyear LJ, Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J. Biol. Chem. 2003; 278(31): 28372-28377.
[0460] 66.* Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008; 3(6): 965-976.
[0461] 67.* Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005; 112(17): 2686-2695.
[0462] 68.* Karamanlidis G, Nascimben L, Couper GS, Shekar PS, Del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010;106(9):1541-1548.
[0463] 69.* Neubauer S. The failing heart-an engine out of fuel. N Engl J Med. 2007; 256(11): 1140-1151.
1. Regnier M, Rivera AJ, Chen Y, Chase PB. 2-deoxy-ATP enhances contractility of rat cardiac muscle. Circ Res. 2000; 86(12): 1211-1217.
2. Regnier M, Martin H, Barsotti RJ, Rivera AJ, Martyn DA, Clemmens E. Cross-bridge versus thin filament contributions to the level and rate of force development in cardiac muscle. Biophys J. 2004; 87(3): 1815-1824.
3. Adhikari BB, Regnier M, Rivera AJ, Kreutziger KL, Martyn DA. Cardiac length dependence of force and force redevelopment kinetics with altered cross-bridge cycling. Biophys J. 2004; 87(3): 1784-1794.
4. Regnier M, Homsher E. The effect of ATP analogs on posthydrolytic and force development steps in skinned skeletal muscle fibers. Biophys J. 1998; 74(6): 3059-3071.
5. Regnier M, Lee DM, Homsher E. ATP analogs and muscle contraction: mechanics and kinetics of nucleoside triphosphate binding and hydrolysis. Biophys J. 1998; 74(6): 3044-3058.
6. Regnier M, Martyn DA, Chase BP. Calcium regulation of tension redevelopment kinetics with 2-deoxy-ATP or low [АТФ] in rabbit skeletal muscle. Biophysical Journal. 1998; 74(4): 2005-2015.
7. Korte FS, Dai J, Buckley K, Feest ER, Adamek N, Geeves MA, Murry CE, Regnier M. Upregulation of cardiomyocyte ribonucleotide reductase increases intracellular 2 deoxy-ATP, contractility, and relaxation. J Mol Cell Cardiol. 2011; 51(6): 894-901.
8. Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982; 307(4): 205-211.
9. Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res. 1986; 59(3): 297-309.
10. Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002; 415(6868): 206-212.
11. Moreno-Gonzalez A, Korte FS, Dai J, Chen KY, Ho B, Reinecke H, Murry CE, Regnier M. Cell therapy enhances function of remote non-infarcted myocardium. Journal of Molecular and Cellular Cardiology. 2009; 47(5): 603-613.
12. Lewinter MM, Vanburen P. Myofilament remodeling during the progression of heart failure. J Card Fail. 2002; 8(6 Suppl): S271-275.
13. LeWinter M. Functional consequences of sarcomeric protein abnormalities in failing myocardium. Heart Fail Rev. 2005; 10(3): 249-257.
14. LeWinter M, VanBuren P. Sarcomeric proteins in hypertrophied and failing myocardium: an overview. Heart Fail Rev. 2005; 10(3): 173-174.
15. Garvey JL, Kranias EG, Solaro RJ. Phosphorylation of C-protein, troponin I and phospholamban in isolated rabbit hearts. Biochem J. 1988; 249(3): 709-714.
16. Westfall MV, Solaro RJ. Alterations in myofibrillar function and protein profiles after complete global ischemia in rat hearts. Circ Res. 1992; 70(2): 302-313.
17. Reiken S, Gaburjakova M, Guatimosim S, Gomez AM, D'Armiento J, Burkhoff D, Wang J, Vassort G, Lederer WJ, Marks AR. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J Biol Chem. 2003; 278(1): 444-453.
18. Obayashi M, Xiao B, Stuyvers BD, Davidoff AW, Mei J, Chen SR, ter Keurs HE. Spontaneous diastolic contractions and phosphorylation of the cardiac ryanodine receptor at serine-2808 in congestive heart failure in rat. Cardiovasc Res. 2006; 69(1): 140-151.
19. del Monte F, Harding S, Schmidt U, Matsui T, Kang Z, Matsui T, Guerrero J, Gwathmey J, Rosenzweig A, Hajjar R. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 2000; 100: 2308-2311.
20. Miyamoto M, del Monte F, Schmidt U, DiSalvo T, Kang Z, Dec G, Gwathmey J, Rosenzweig A, Hajjar R. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proceedings of the National Academy of Sciences USA. 2000; 97: 793-798.
21. Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006; 113(2): 305-315.
22. Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005; 115(9): 2305-2315.
23. Edes I, Kiss E, Kitada Y, Powers FM, Papp JG, Kranias EG, Solaro RJ. Effects of Levosimendan, a cardiotonic agent targeted to troponin C, on cardiac function and on phosphorylation and Ca2+ sensitivity of cardiac myofibrils and sarcoplasmic reticulum in guinea pig heart. Circ Res. 1995; 77(1): 107-113.
24. Wyskovsky W, Hauptner R, Suko J. Drug-induced calcium release from heavy sarcoplasmic reticulum of skeletal muscle. Biochim. Biophys. Acta. 1988; 938: 89-96.
25. Xu X, Page JL, Surtees JA, Liu H, Lagedrost S, Lu Y, Bronson R, Alani E, Nikitin AY, Weiss RS. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 2008; 68(8): 2652-2660.
26. Ylikallio E, Page JL, Xu X, Lampinen M, Bepler G, Ide T, Tyynismaa H, Weiss RS, Suomalainen A. Ribonucleotide reductase is not limiting for mitochondrial DNA copy number in mice. Nucleic Acids Research. 2010; 38(22): 8208-8218.
27. Korte FS, Dai J, Buckley K, Feest ER, Murry CE, Regnier M. Upregulation of cardiomyocyte ribonucleotide reductase increases intracellular 2-deoxy-ATP, contractility, and relaxation. Circulation Research. 2011; In Review.
28. McDonald KS, Moss RL. Strongly binding myosin cross-bridges regulate loaded shortening and power output in cardiac myocytes. Circulation Research. 2000; 87: 768-773.
29. Dobesh DP, Konhilas JP, de Tombe PP. Cooperative activation in cardiac muscle: impact of sarcomere length. Am J Physiol Heart Circ Physiol. 2002; 282(3): H1055-1062.
30. Kinoshita Y, Nishigaki K. Unexpectedly general replaceability of ATP in ATP-requiring enzymes. J Biochem. 1997; 122(1): 205-211.
31. Trumble WR, Sutko JL, Reeves JP. Cardiac sarcolemmal and sarcoplasmic reticulum membrane vesicles exhibit distinctive (Ca-Mg)-ATPase substrate specificities. J Biol Chem. 1981; 256(14): 7101-7104.
32. Baker AJ. Refueling the heart: Using 2-deoxy-ATP to enhance cardiac contractility. J Mol Cell Cardiol. 2011; 51(6): 883-884.
33. Goldberg GS, Moreno AP, Lampe PD. Gap junctions between cells expressing connexin 43 or 32 show inverse permselectivity to adenosine and ATP. J Biol Chem. 2002; 277(39): 36725-36730.
34. Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, Nedergaard M. Connexin 43 hemichannels are permeable to ATP. J Neurosci. 2008; 28(18): 4702-4711.
35. Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL, Miller AD, Miller DA, Chamberlain JS. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther. 2004; 10(4): 671-678.
36. Odom GL, Gregorevic P, Chamberlain JS. Viral-mediated gene therapy for the muscular dystrophies: successes, limitations and recent advances. Biochim Biophys Acta. 2007; 1772(2): 243-262.
37. Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, Koch WJ. Myocardial adeno-associated virus serotype 6-betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009; 119(1): 89-98.
38. Wang Z, Allen JM, Riddell SR, Gregorevic P, Storb R, Tapscott SJ, Chamberlain JS, Kuhr CS. Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum Gene Ther. 2007; 18(1): 18-26.
39. Caras IW, Martin DW, Jr. Molecular cloning of the cDNA for a mutant mouse ribonucleotide reductase Ml that produces a dominant mutator phenotype in mammalian cells. Mol Cell Biol. 1988; 8(7): 2698-2704.
40. Gillis TE, Martyn DA, Rivera AJ, Regnier M. Investigation of thin filament near-neighbour regulatory unit interactions during force development in skinned cardiac and skeletal muscle. J Physiol. 2007; 580(Pt.2): 561-576.
41. Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrythmia in mice. Journal of Clinical Investigation. 2008; 118(12): 3893-3903.
42. Solaro RJ, Gambassi G, Warshaw DM, Keller MR, Spurgeon HA, Beier N. Stereoselective actions of thiadiazinones on canine cardiac myocytes and myofilaments. Circ Res. 1993; 73(6): 981-990.
43. White J, Lee JA, Shah N, Orchard CH. Differential effects of the optical isomers of EMD 53998 on contraction and cytoplasmic Ca2+ in isolated ferret cardiac muscle. CircRes. 1993; 73(1): 61-70.
44. Huke S, Knollmann BC. Increased myofilament Ca2+ sensitivity and arrhythmia susceptibility. Journal of Molecular and Cellular Cardiology. 2010; 48(5): 824-833.
45. Zoghbi WA, Quinones MA. Determination of cardiac output by Doppler echocardiography: a critical appraisal. Herz. 1986; 11(5): 258-268.
46. Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V, Morine KJ, Gardner TJ, Discher DE, Sweeney HL. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol. 2006; 290(6): H2196-2203.
47. Tanaka N, Dalton N, Mao L, Rockman HA, Peterson KL, Gottshall KR, Hunter JJ, Chien KR, Ross J, Jr. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation. 1996; 94(5): 1109-1117.
48. Monasky MM, Varian KD, Davis JP, Janssen PM. Dissociation of force decline from calcium decline by preload in isolated rabbit myocardium. Pflugers Arch. 2008; 456(2): 267-276.
49. Varian K, Raman S, Janssen P. Measurement of myofilament calcium sensitivity at physiological temperature in intact cardiac trabeculae. Am J Physiol Heart Circ Physiol. 2006; 290(5): H2092-2097.
50. Varian KD, Janssen PM. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. 2007; 292(5): H2212-2219.
51. Janssen P, Lehnart S, Prestle J, Lynker J, Salfeld P, Just H, Hasenfuss G. The trabecula culture system: a novel technique to study contractile parameters over a multiday time period. Am J Physiol. 1998; 274(5 Pt2): H1481-1488.
52. Kreutziger KL, Piroddi N, J.T. M, Tesi C, Poggesi C, Regnier M. Cooperative activation and tension kinetics in cardiac muscle are strongly modulated by calcium binding kinetics of troponin C. Journal of Molecular and Cellular Cardiology. 2011; 50(1): 165-174.
53. Kreutziger KL, Piroddi N, Scellini B, Tesi C, Poggesi C, Regnier M. Thin filament Ca2+ binding properties and regulatory unit interactions alter kinetics of tension development and relaxation in rabbit skeletal muscle. Journal of Physiology. 2008; 586(15): 3683-3700.
54. Belus A, Piroddi N, Tesi C. Mechanism of cross-bridge detachment in isometric force relaxation of skeletal and cardiac myofibrils. Journal of Muscle Research and Cell Motility. 2003; 24(4-6): 261-267.
55. Guo X, Laflamme MA, Becker PL. Cyclic ADP-ribose does not regulate sarcoplasmic reticulum Ca2+ release in intact cardiomyocytes. Circ Res. 1996; 79: 147-151.
56. Laflamme MA, Becker PL. Ca2+-induced current oscillations in rabbit ventricular myocytes. Circ Res. 1996; 78: 707-716
57. Laflamme MA, Becker PL. Do beta 2-adrenergic receptors modulate Ca2+ in adult rat ventricular myocytes? Am J Physiol. 1998; 274: H1308-1314.
58. Zhu WZ, Santana LF, Laflamme MA. Local control of excitation-contraction coupling in human embryonic stem cell-derived cardiomyocytes. PLoS One. 2009; 4: e5407.
59. Zhu WZ, Xie Y, Moyes KW, Gold JD, Askari B, Laflamme MA. Neuregulin/erbb signaling regulates cardiac subtype specification in differentiating human embryonic stem cells. Circ Res. 2010; 107: 776-786.
60. Adler ED, Chen VC, Bystrup A, Kaplan AD, Giovannone S, Briley-Saebo K, Young W, Kattman S, Mani V, Laflamme MA, Zhu WZ, Fayad Z, Keller G. The cardiomyocyte lineage is critical for optimization of stem cell therapy in a mouse model of myocardial infarction. FasebJ. 2010; 24: 1073-1081.
61. Fernandes S, Naumova AV, Zhu WZ, Lafiamme MA, Gold J, Murry CE. Human embryonic stem cell-derived cardiomyocytes engraft but do not alter cardiac remodeling after chronic infarction in rats. Journal of Molecular and Cellular Cardiology. 2010; 49: 941-949.
62. Xu C, Police S, Hassanipour M, Li Y, Chen Y, Priest C, O'Sullivan C, Lafiamme MA, Zhu WZ, Van Biber B, Hegerova L, Yang J, Delavan-Boorsma K, Davies A, Lebkowski J, Gold JD. Efficient generation and cryopreservation of cardiomyocytes derived from human embryonic stem cells. Regenerative Medicine. 2011; 6: 53-66.
63. Magistretti J, Mantegazza M, de Curtis M, Wanke E. Modalities of distortion of physiological voltage signals by patch-clamp amplifiers: a modeling study. Biophys J. 1998; 74(2 Pt1): 831-842.
64. Magistretti J, Mantegazza M, Guatteo E, Wanke E. Action potentials recorded with patch-clamp amplifiers: are they genuine? Trends Neurosci. 1996; 19(12): 530-534.
65. Akaike N, Harata N. Nystatin perforated patch recording and its applications to analyses of intracellular mechanisms. Jpn J Physiol. 1994; 44(5): 433-473.
66. Strauss U, Herbrik M, Mix E, Schubert R, Rolfs A. Whole-cell patch-clamp: true perforated or spontaneous conventional recordings? Pflugers Arch. 2001; 442(4): 634-638.
67. Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993; 262(5134): 740-744.
68. Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J. 2000; 78(5): 2655-2667.
69. Santana LF, Cheng H, Gomez AM, Cannell MB, Lederer WJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res. 1996; 78(1): 166-171.
70. Guo X, Lafiamme MA, Becker PL. Cyclic ADP-ribose does not regulate sarcoplasmic reticulum Ca2+ release in intact cardiac myocytes. Circ Res. 1996; 79(1): 147-151.
71. Zhu WZ, Santana LF, Lafiamme MA. Local control of excitation-contraction coupling in human embryonic stem cell-derived cardiomyocytes. PLoS ONE. 2009; 4(4): e5407.
72. Santana LF, Kranias EG, Lederer WJ. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J Physiol. 1997; 503(Pt1): 21-29.
73. Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol. 1992; 453: 591-608.
74. Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994; 476(2): 279-293.
75. Terracciano CM, Philipson KD, MacLeod KT. Overexpression of the Na(+)/Ca(2+) exchanger and inhibition of the sarcoplasmic reticulum Ca(2+)-ATPase in ventricular myocytes from transgenic mice. Cardiovasc Res. 2001; 49(1): 38-47.
76. Dilly KW, Rossow CF, Votaw VS, Meabon JS, Cabarrus JL, Santana LF. Mechanisms underlying variations in excitation-contraction coupling across the mouse left ventricular free wall. J Physiol. 2006; 572(Pt1): 227-241.
77. Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005; 112(15): 2339-2346.
78. Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009; 119(21): 2818-2828.
79. Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, Goodyear LJ, Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J. Biol. Chem. 2003;278(31):28372-28377.
80. Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008; 3(6): 965-976.
81. Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005; 112(17): 2686-2695.
82. Karamanlidis G, Nascimben L, Couper GS, Shekar PS, Del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010; 106(9): 1541-1548.
83. Williams CD, Regnier M, Daniel TL. Axial and radial forces of cross-bridges depend on lattice spacing. PLoS Comput Biol. 2010; 6(12): e1001018.
84. Campbell SG, Lionetti FV, Campbell KS, McCulloch AD. Coupling of adjacent tropomyosins enhances cross-bridge-mediated cooperative activation in a markov model of the cardiac thin filament. Biophys J. 2010; 98(10): 2254-2264.
85. Campbell SG, Howard E, Aguado-Sierra J, Coppola BA, Omens JH, Mulligan LJ, McCulloch AD, Kerckhoffs RC. Effect of transmurally heterogeneous myocyte excitation-contraction coupling on canine left ventricular electromechanics. Exp Physiol 2009; 94(5): 541-552.
86. Campbell SG, McCulloch AD. Multi-scale computational models of familial hypertrophic cardiomyopathy: genotype to phenotype. J R Soc Interface. 2011; 8(64): 1550-1561.
87. Chuang JS, Zemljic-Harpf A, Ross RS, Frank LR, McCulloch AD, Omens JH. Determination of three-dimensional ventricular strain distributions in gene-targeted mice using tagged MRI. Magn Reson Med. 2010; 64(5): 1281-1288.
88. Kerckhoffs RC, Campbell SG, Flaim SN, Howard EJ, Sierra-Aguado J, Mulligan LJ, McCulloch AD. Multi-scale modeling of excitation-contraction coupling in the normal and failing heart. ConfProc IEEE Eng Med Biol Soc. 2009; 2009: 4281-4282.
89. Beeri R, Chaput M, Guerrero JL, Kawase Y, Yosefy C, Abedat S, Karakikes I, Morel C, Tisosky A, Sullivan S, Handschumacher MD, Gilon D, Vlahakes GJ, Hajjar RJ, Levine RA. Gene delivery of sarcoplasmic reticulum calcium ATPase inhibits ventricular remodeling in ischemic mitral regurgitation. Circ Heart Fail. 3(5): 627-634.
90. Bott-Flugel L, Weig HJ, Knodler M, Stadele C, Moretti A, Laugwitz KL, Seyfarth M. Gene transfer of the pancaspase inhibitor P35 reduces myocardial infarct size and improves cardiac function. J Mol Med (Berl). 2005; 83(7): 526-534.
91. Prasad KM, Smith RS, Xu Y, French BA. A single direct injection into the left ventricular wall of an adeno-associated virus 9 (AAV9) vector expressing extracellular superoxide dismutase from the cardiac troponin-T promoter protects against myocardial infarction. J Gene Med. 2011; 13(6): 333-341.
92. Lafiamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, O'Sullivan C, Collins L, Chen Y, Minami E, Gill EA, Ueno S, Yuan C, Gold J, Murry CE. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007; 25(9): 1015-1024.
93. Lafiamme MA, Zbinden S, Epstein SE, E. MC. Cell-based therapy for myocardial ischemia and infarction: pathophysiological mechanisms. Annu Rev Pathol Mech Dis. 2007; 2: 307-339.
94. Nussbaum J, Minami E, Lafiamme MA, Virag JA, Ware CB, Masino A, Muskheli V, Pabon L, Reinecke H, Murry CE. Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. Faseb J. 2007; 21(7): 1345-1357.
95. Neubauer S. The failing heart~an engine out of fuel. N Engl J Med. 2007; 256(11): 1140-1151.
Gregorevic P, Blankinship MJ, Allen J, Crawford RW, Meuse L, Miller D, Russell DW and Chamberlain JS: Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nature Med 2004; 10: 828-834.
Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert C, Miller AD and Chamberlain JS. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther 2004; 10: 671-678.
Blankinship M, Gregorevic P and Chamberlain JS: Gene therapy strategies for Duchenne muscular dystrophy utilizing recombinant adeno-associated virus vectors. Mol Ther 2006; 13: 241-249.
Salva MZ, Himeda CL, Tai PW, Nishiuchi E, Gregorevic P, Allen JM, Finn EE, Nguyen QG, Blankinship MJ, Meuse L, Chamberlain JS and Hauschka SD: Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther 2007; 15: 320-329.
Реферат
Изобретение относится к клеточным технологиям. Описан способ улучшения сердечной функции у млекопитающего, включающий: трансплантацию кардиомиоцитов в миокард указанного млекопитающего, причем указанные кардиомиоциты содержат первый вектор экспрессии, содержащий первую последовательность нуклеиновой кислоты, кодирующую субъединицу R1 рибонуклеотидредуктазы, и второй вектор экспрессии, содержащий вторую последовательность нуклеиновой кислоты, кодирующую субъединицу R2 рибонуклеотидредуктазы, где указанные первая и вторая последовательности нуклеиновой кислоты функционально связаны с промотором, который индуцирует гиперэкспрессию R1 и R2, что обеспечивает образование дезокси-АТФ и улучшение сердечной функции у млекопитающего. Изобретение расширяет возможности лечения сердечной недостаточности. 2 н. и 25 з.п. ф-лы, 40 ил., 15 табл., 51 пр.
Формула
Документы, цитированные в отчёте о поиске
Способ пролиферации кардиомиоцитов
Комментарии