Средство, содержащее fgf2 в качестве активного ингредиента, для лечения или профилактики астмы и хронических обструктивных заболеваний легких - RU2351356C2
Код документа: RU2351356C2
Чертежи




















Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к средству, содержащему FGF2 (фактор роста фибробластов-2, или основной фактор роста фибробластов, bFGF) в качестве активного ингредиента, для профилактики и лечения астмы и хронических обструктивных заболеваний легких (ХОЗЛ). Настоящее изобретение также относится к модели ХОЗЛ и Th1 астмы, индуцированной яичным белком (ОА) и двухцепочечной РНК (dsRNA), у мышей.
Уровень техники
В последние 20 лет распространенность астмы увеличилась почти вдвое и на сегодняшний день астмой страдает 8-10% населения планеты. Астма представляет собой хроническое воспалительное заболевание дыхательных путей и характеризуется гиперреактивностью дыхательных путей (AHR) на неспецифические раздражители и перестройкой дыхательных путей, что связано с изменением структуры и функции затрагиваемых элементов, таких как фибробласты и миофибробласты. Астму, главным образом, разделяют на бронхиальную астму и сердечную астму, но, как правило, под астмой пронимают лишь бронхиальную астму.
Одним из наиболее характерных заболеваний легких, наряду с астмой, является хроническое обструктивное заболевание легких (ХОЗЛ), которое отличается от астмы сопутствующей обструкцией дыхательных путей. ХОЗЛ занимает 4-е место среди причин смерти во всем мире и темпы роста только ХОЗЛ среди 10 наиболее значимых заболеваний увеличивается. ХОЗЛ вызывают патологические изменения в бронхиолах и паренхиме, возникающие в результате длительного воспаления в дыхательных путях и паренхиме и, таким образом, оно характеризуется облитеративным бронхиолитом и легочной эмфиземой (деструкцией паренхимы). В качестве примеров хронических обструктивных заболеваний легких можно привести обструктивный бронхит, хронический бронхиолит и эмфизему.
Лечение астмы и таких хронических обструктивных заболеваний легких основано на использовании противовоспалительных агентов или бронхорасширяющих средств. Глюкокортикоиды, модификаторы лейкотриенов и теофиллины являются характерными противовоспалительными агентами. Хотя глюкокортикоиды обладают выраженной лечебным эффектом, они влияют не на конкретную мишень, а ингибируют все иммунные и противовоспалительные ответы, что означает, что они ингибируют и необходимые иммунные ответы, и, кроме того, они обладают серьезными побочными эффектами, и, поэтому, используются в качестве ингаляционной терапии. Модификаторы лейкотриенов имеют меньше побочных эффектов, но их лечебный эффект ограничен, так как они не могут контролировать астму независимо и могут быть использованы лишь как вспомогательное средство. Применение теофиллина также ограничено из-за слабого лечебного действия и побочных эффектов.
Поэтому, необходимо создание средства для лечения астмы с выраженным действием, но с меньшим количеством побочных эффектов. Для создания таких средств необходимо полное понимание механизмов развития астмы.
Согласно общепринятой теории Т-хелперы 1 типа (Th1) или Т-хелперы 2 типа (Th2) сектерируют цитокины, которые играют важную роль в развитии астмы, конкретно, дисбаланс цитокинов Th1 и Th2 вызывает астму (гипотеза Th1/Th2) (Mosmann et al., J. Immunol., 136: 2348-57, 1986; Robinson et al., N.Engl.J.Med., 326: 298-304, 1992; Grunig et al., Science, 282: 2261-3, 1998; Richter et al., Am.J.Respir. Cell Mol.Biol., 25: 385-91, 2001). Однако, точный механизм развития астмы, индуцированной цитокинами, до сих пор не был разъяснен.
Интерлейкин-13 (IL-13), продуцируемый активированными T2-хелперными клетками, является ключевым цитокином в патогенезе астмы (Grunig et al., Science, 282: 2261-3, 1998). Подтверждением этого утверждения является обнаружение того, что гиперреактивность дыхательных путей ингибируется подавлением экспрессии IL-13 на модели аллергической астмы у животных, но вновь возникала при введении рекомбинантного IL-13 через дыхательные пути (Marsha et al., Science, 282: 2258-2261,1998).
Гистологические результаты, показанные у трансгенных по IL-13 мышей, были сходны с результатами, наблюдаемыми у больных астмой и сверхэкспрессия IL-13 вызывала воспаление дыхательных путей, увеличение секреции слизи и фиброза эпителиальных клеток (Zhu et al., J.Clin.Invest., 103:779-788,1999). Предположение о том, что IL-13 может усиливать AHR, вызывая инфильтрацию воспалительных клеток, особенно эозинофилов, остается самой известной (Hargreave et al., J. Allergy clin. Immunol., 78: 825-32, 1986). Однако на основании последних данных можно предположить, что индукция AHR может возникать в отсутствие инфильтрации эозинофилами (Venkayya et al., Am.J.Respir. Cell Mol. Biol. 26: 202-8, 2002).
Известно, что трансформирующий фактор роста β1 (TGF-β1) или эндотелиальный фактор роста сосудов (VEGF) вовлечены в патогенез астмы, индуцированной IL-13 (Lee et al., Nat.Med., 10: 1095-1103, 2004).
TGF-β1, как ключевой элемент заживления ран тканей, индуцирует фиброз тканей, который является основным патологическим изменением в перестраиваемых дыхательных путях. Конкретно, TGF-β1 преобразует фибробласты в миофибробласты, после чего миофибробласты секретируют больше коллагена, чем покоящиеся фибробласты, что приводит к перестройке дыхательных путей посредством фиброза тканей (Vignola et al., Am. J. espir. Crit. Care Med., 156: 591-599, 1997). Эта последовательность событий согласуется с данными предшествующего исследования, когда фиброз в легких трансгенных по IL-13 мышей индуцировался главным образом за счет TGF-β1-зависимого пути (Lee et al., J. Exp. Med., 194: 809-21, 2001).
В процессе фиброза ткани, TGF-β1 индуцирует секрецию фактора роста фибробластов-2 (FGF2 или основной фактор роста фибробластов, bFGF) и его рецептора-1 (FGFR-1) или рецептора FGF2 (FGFR-2). Известно, что FGF2 связан с пролиферацией эндотелиальных клеток или гладкомышечных клеток и также играет важную роль в ангиогенезе (Nugent et al., Int. J. Biochem. Cell Biol. 32: 115-20, 2000). Однако роль FGF2 в патогенезе астмы и AHR до сих пор обсуждается.
Эндотелиальный фактор роста сосудов (VEGF) является разновидностью цитокинов, которые увеличивают проникновение белков плазмы через капилляры крови, стимулируют дифференцировку и миграцию клеток и индуцируют секрецию протеаз, преобразующих клетку. VEGF также вовлечен в поддержание новых кровеносных сосудов, ингибируя апоптоз, в регуляцию иммунного ответа посредством подавления нейронального антигена и в индукцию роста и деления клеток. Авторы настоящего изобретения показали, что существует положительная обратная связь между IL-13 и VEGF в отношении иммунного ответа против антигенов и инородных веществ (Lee et al., Nat. Med., 10: 1095-1103, 2004). Тем не менее, роль FGF2 в патогенезе астмы, опосредованной VEGF, не изучена.
Интерферон-γ (IFN-γ) является еще одним ключевым цитокином, секретируемым Th1, вовлеченным в патогенез астмы. Конкретно, IFN-γ является веществом, секретируемым Т1-хелперными клетками в качестве защитного средства против патогена (Fong et al., J. Immunol., 143:2887-93,1989) и известно, что он ингибирует продукцию цитокина Th2 (Mosann et al., J. Immunol., 136: 2348-57,1986).На основании гипотезы Th1/Th2, полагают, что IFN-γ ингибирует развитие астмы, но это утверждение по прежнему противоречиво. В соответствии с предыдущими противоречивыми исследованиями, перестройка дыхательных путей, подобная той, которая наблюдается у больных астмой, происходит у трансгенных по IFN-γ мышей (Wang et al., J. Exp. Med., 192: 1587-1600, 2000) и в частности, тяжесть астмы вероятней всего связана с повышением IFN-γ (Corrogan et al., Lancet 1: 1129-32, 1988; Mognan et al., Am. J. Respir. Crit. Care Med., 161: 1790-6, 2000).
Такое противоречивое предположение согласуется с данными, что широко используемые средства для лечения астмы, такие как кортикостероиды, β2-адренергические агонисты и производные метилксантина в большей степени ингибируют иммунный ответ Th1, чем иммунный ответ Th2. Следовательно, объяснение патогенеза астмы гипотезой Th1/Th2, ограничено акцентом на важность стимуляции иммунного ответа Th2.
Тем временем, вовлечение ХОЗЛ в патогенез астмы также не было освещено. То есть развитие и прогрессирование ХОЗЛ не было объяснено, и поэтому требуется полное объяснение точного механизма вышеуказанного процесса перед созданием терапевтического средства для ХОЗЛ.
На основании результатов последних исследований на трансгенных мышах, было доказано, что IFN-γ (Wang et al., J. Exp. Med., 192: 1587-600, 2000) и IL-13 (Zheng et al., J. Clin. Invest., 106: 1081-93, 2000) вовлечены в патогенез астмы, и они являются элементами, вызывающими патологический феномен, сходный с таковым при ХОЗЛ человека. Как указано выше, цитокины в большом количестве секретируются иммунными клетками, что дает основания предположить, что иммунный ответ играет ключевую роль в патогенезе ХОЗЛ. IFN-γ и IL-13 являются важными факторами, облегчающими воспаление в дыхательных путях и паренхиме. Для заживления ран, инициированных воспалением, баланс между атакующими и защищающими факторами во время восстановления эпителиальных клеток дыхательных путей и легких является особенно важным (Lee et al., J. Exp. Med., 200: 377-89, 2004), что означает, что высокая активность атакующих факторов или слабая активность защищающих факторов может вызвать ХОЗЛ.
Авторы настоящего изобретения изучали роль FGF2 в патогенезе астмы и ХОЗЛ, опосредованных IL-13, TGF-β1, VEGF и IFN-γ, и подтвердили, что FGF2 подавляет AHR, вызванную VEGF, стимулированным IL-13, или вызванную IFN-γ, и ингибирует развитие легочной эмфиземы, инициированную воспалением дыхательных путей и паренхимы, поэтому FGF2 может эффективно использоваться для профилактики и лечения астмы и ХОЗЛ. Авторы настоящего изобретения осуществили настоящее изобретение, создав модель Th1 астмы и ХОЗЛ, индуцированных яичным белком и двухцепочечной ДНК, у животных, делая возможными проведение результативных и эффективных экспериментов для создания средств для лечения астмы и ХОЗЛ.
Описание
Техническая проблема
Задачей настоящего изобретения является создание средства, содержащего FGF2 в качестве активного ингредиента, для профилактики и лечения астмы и ХОЗЛ.
Еще одной задачей настоящего изобретения является создание модели Th1 астмы или ХОЗЛ, индуцированных аллергенами, такими как яичный белок (ОА) и двухцепочечная РНК (dsRNA), у мышей.
Техническое решение
Настоящее изобретение относится к средству для профилактики и лечения астмы, содержащему FGF2 (фактор роста фибробластов-2) в качестве активного ингредиента.
Настоящее изобретение относится к средству для профилактики и лечения астмы, для которой характерна индукция сверхэкспрессией IL-13 (интерлейкин-13).
Настоящее изобретение относится к средству для профилактики и лечения астмы, для которой характерна индукция сверхэкспрессией IFN-γ (интерферон-γ).
Настоящее изобретение относится к средству для профилактики и лечения астмы, содержащему FGF2, для ингибирования активности IL-13.
Настоящее изобретение относится к средству для профилактики и лечения астмы, содержащему FGF2, для ингибирования активности VEGF.
Настоящее изобретение относится к средству для профилактики и лечения астмы, содержащему FGF2, для подавления активности TGF-β1 (трансформирующего фактора роста-β1).
Настоящее изобретение относится к средству для профилактики и лечения ХОЗЛ, содержащему FGF2 (фактор роста фибробласта-2) в качестве активного ингредиента.
Настоящее изобретение относится к средству для профилактики и лечения ХОЗЛ, обычно индуцируемых сверхэкспрессией IFN-γ (интерферон-γ).
Настоящее изобретение относится к способу создания моделей Th1 астмы и ХОЗЛ у животных, который характеризуется непосредственным введением аллергенов, таких как яичный белок и двухцепочечная РНК, в дыхательные пути.
Настоящее изобретение относится к способу создания моделей Th1 астмы и ХОЗЛ у животных, где животным является мышь.
Настоящее изобретение относится к способу создания моделей Th1 астмы и ХОЗЛ у животных, предусматривающему следующие стадии:
(1) Сенсибилизация мышей BALB/c четырехразовым интраназальным введением 5-15 мкг полиинозиновой-полицитидиловой кислоты, двухцепочечной РНК, и 50-100 мкг яичного белка; и
(2) Сенсибилизация мышей введением 25-75 мкг яичного белка через 10 дней после первой сенсибилизации.
Настоящее изобретение относится к способу создания моделей Th1 астмы и ХОЗЛ у животных, где в вышеуказанной стадии (1) для сенсибилизации животного использовали 10 мкг двухцепочечной РНК.
Настоящее изобретение относится к способу создания моделей Th1 астмы и ХОЗЛ у животных, где в вышеуказанной стадии (1) для сенсибилизации животного использовали 75 мкг яичного белка и в вышеуказанной стадии (2) 10 дней спустя для сенсибилизации животного использовали 50 мкг яичного белка.
Настоящее изобретение относится к способу создания моделей Th1 астмы и ХОЗЛ у животных, где астма является не-эозинофильной.
Настоящее изобретение относится к модели Th1 астмы или ХОЗЛ у животных, созданной способом по настоящему изобретению.
Настоящее изобретение относится к модели Th1 астмы или ХОЗЛ у животных, где животным является мышь.
Настоящее изобретение относится к ингибитору IL-3, VEGF или TGF-β1, содержащему FGF2 (фактор роста фибробластов-2) в качестве активного ингредиента.
Настоящее изобретение относится к ингибитору фиброза, воспаления дыхательных путей, AHR или перестройки дыхательных путей, содержащему FGF2 (фактор роста фибробластов-2) в качестве активного ингредиента.
Далее настоящее изобретение описано более подробно.
Настоящее изобретение относится к фармацевтической композиции для профилактики или лечения астмы, содержащей FGF2 в качестве активного ингредиента. Конкретно, настоящее изобретение относится к средству для профилактики и лечения астмы, для которой характерна индукция сверхэкспрессией IL-13 (интерлейкин-13) или IFN-γ (интерферон-γ). Для средства по настоящему изобретению характерно то, что оно ингибирует активность IL-13 (интерлейкин-13), VEGF или TGF-β1 (трансформирующий фактор роста-β1). Астму разделяют на бронхиальную астму, сердечную астму и тому подобное, но под астмой понимают просто бронхиальную астму. Астма характеризуется гиперреактивностью дыхательных путей и перестройкой дыхательных путей.
Перестройка дыхательных путей запускается повышенным иммунным ответом на аллерген, воспаление или раздражитель. При повышении иммунного ответа, Т-клетки секретируют цитокин, внутриклеточный переносчик сигнала. Секретируемый цитокин вызывает миграцию воспалительных клеток в ткани, вызывая хроническое воспаление дыхательных путей, что приводит к структурным нарушениям дыхательных путей.
Также полагают, что AHR является важным фактором для патогенеза астмы, который отличает астму от других респираторных заболеваний. AHR сопровождается гиперплазией гладкомышечных клеток дыхательных путей, сокращением и фиброзом эпителиальных клеток и легочной паренхимы, что характерно для перестройки дыхательных путей. Следовательно, воспаление дыхательных путей, AHR и перестройка дыхательных путей тесно связаны друг с другом, а именно, лечение одного из этих симптомов может приводить к неожиданному эффекту на другие симптомы и одно и то же средство может воздействовать как на воспаление дыхательных путей, так и на AHR, и перестройку дыхательных путей.
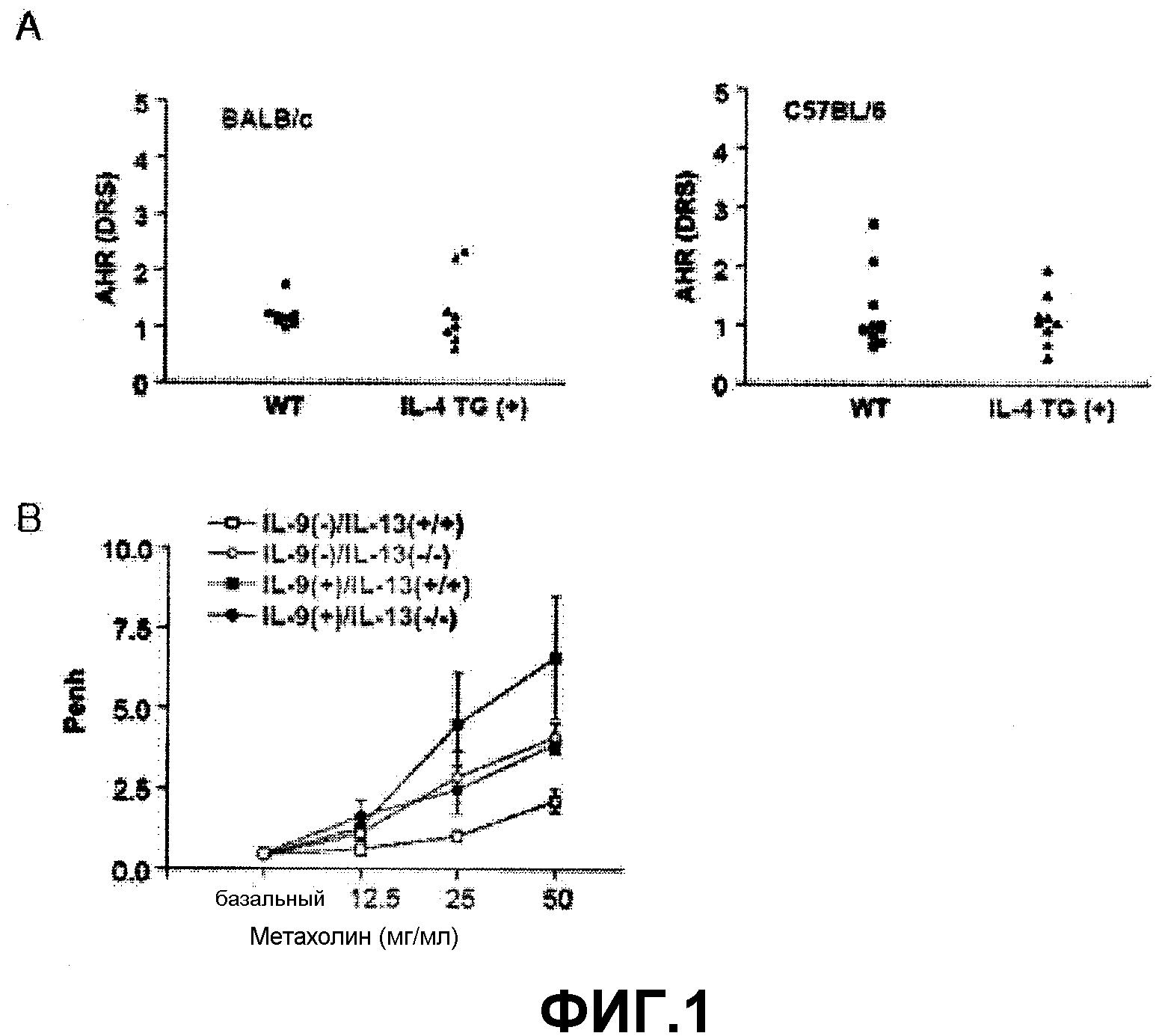
Как было показано уровень AHR у Th2 цитокин IL-4 трансгенных по Th2 цитокину IL-4 мышей (IL-4 TG(+)) был сходен с уровнем из контрольных мышей дикого типа (WT) (фиг.1А), тогда как уровень AHR у трансгенных по Th2 цитокину IL-4 мышей (IL-9(+)/IL-13(+/+)) был повышен по сравнению с уровнями у контрольных мышей дикого типа. Однако, AHR была ингибирована у мышей, нокаутированных по IL-13 (IL-9(+)/IL-13(-/-), что наводит на мысль о том, что астма, индуцированная сверхэкспрессией IL-9, была опосредована IL-13 (см. фиг.1В).
Для подтверждения вышеуказанного были созданы трансгенные по IL-13 мыши для исследования взаимосвязи между гиперреактивностью дыхательных путей и TGF-β1 и VEGF, известными как сверхэкспрессирующиеся под действием IL-13. В результате, AHR усиливалась у трансгенных по IL-13 мышей, по сравнению с контрольными мышами дикого типа (см. фиг.2).
В бронхоальвеолярном лаваже (BAL) трансгенных по IL-13 мышей, также повышались концентрации TGF-β1 и VEGF, что означает, что AHR, индуцированная IL-13, контролируется молекулами, такими как TGF-β1 и VEGF, образующимися под действием IL-13 на следующих этапах сигнального пути.
Контроль AHR, опосредованной IL-13, молекулами VEGF был подтвержден тем фактом, что AHR, опосредованная IL-13, ингибируется под действием SU1498, сигнальным блокатором рецептора 2 (см. фиг.4).
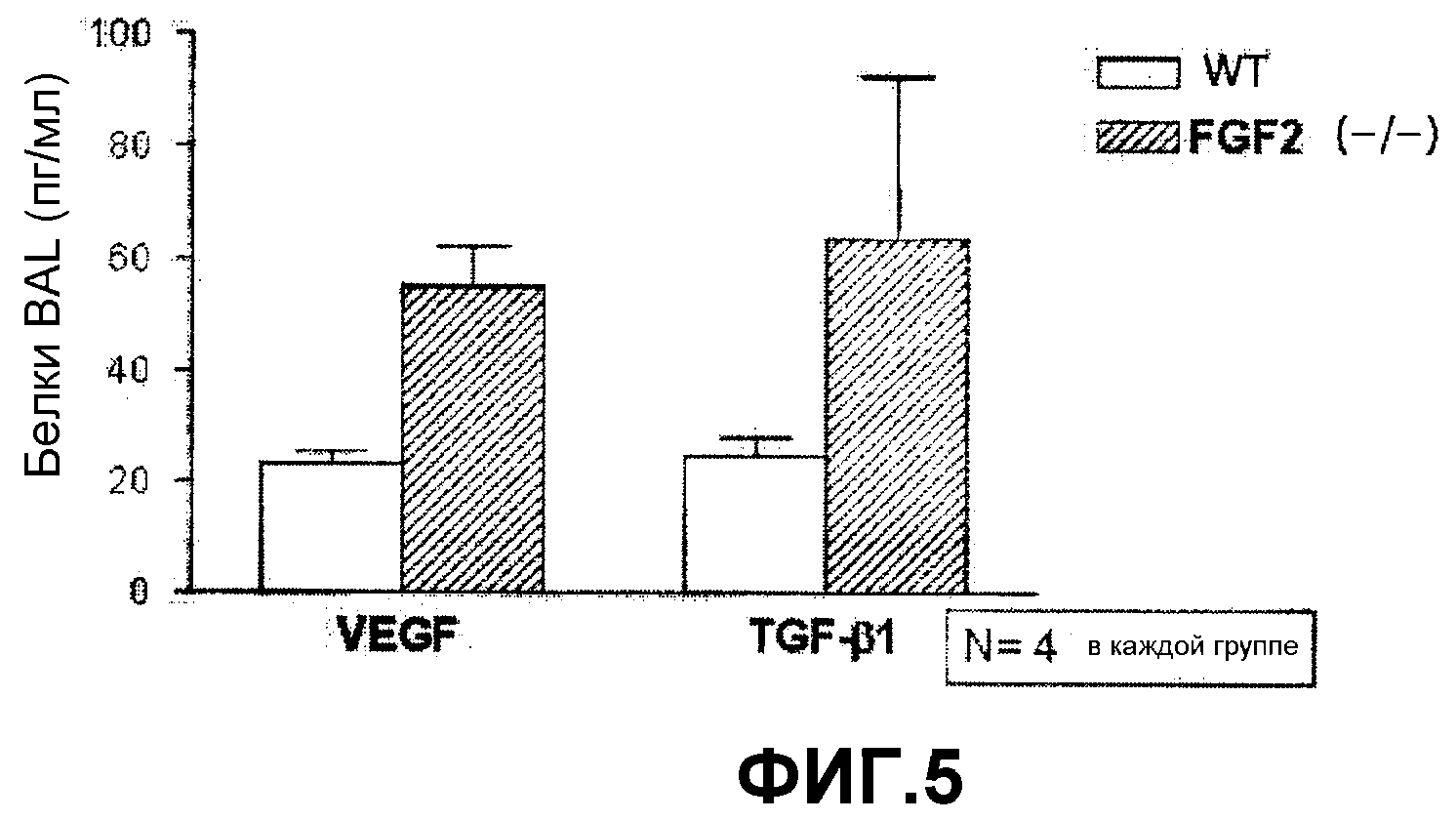
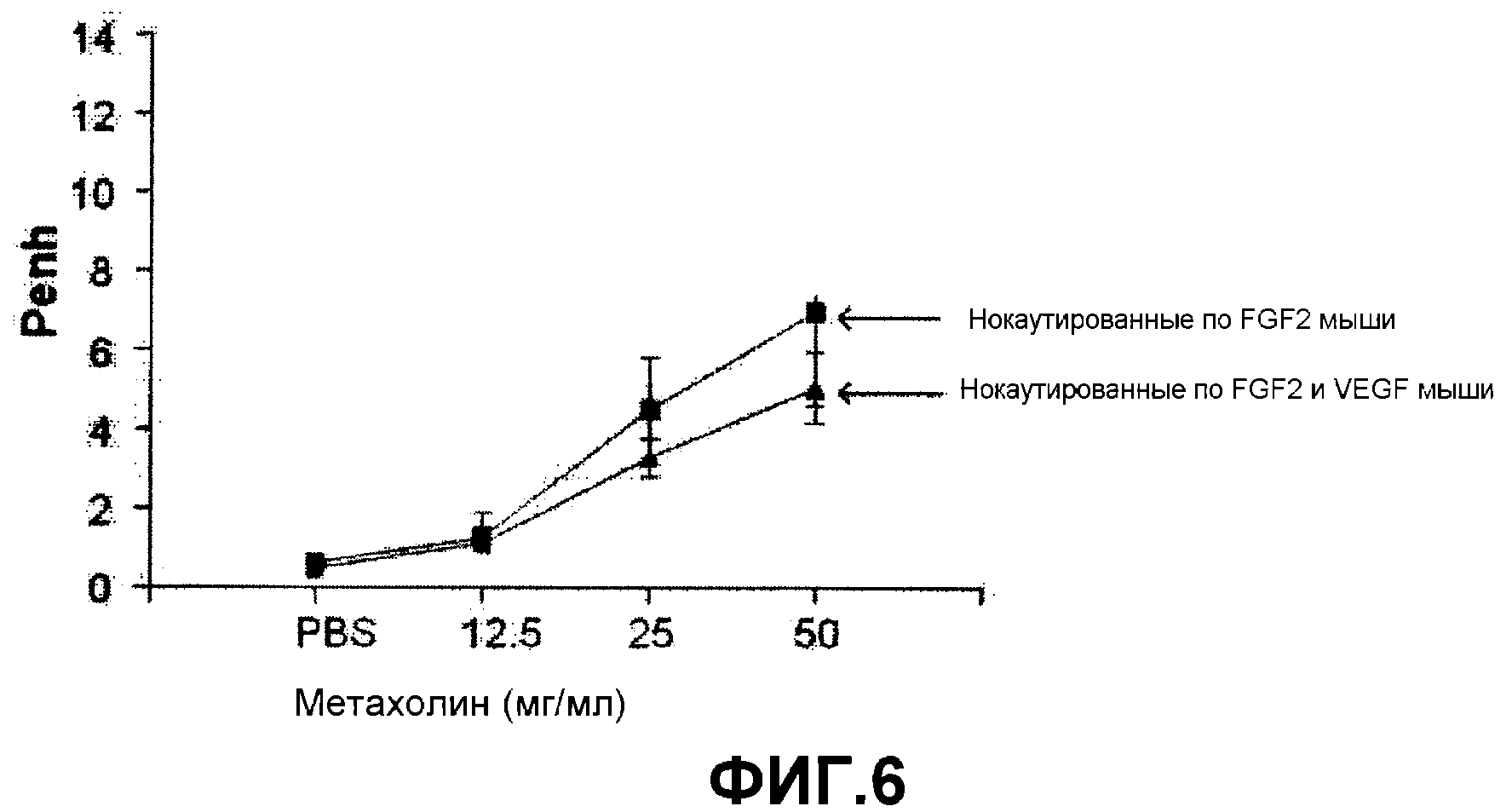
Роль FGF2 в патогенезе астмы, опосредованной IL-13, не была освещена. Таким образом, авторами настоящего изобретения была сделана попытка объяснить роль FGF2 и, наконец, подтвердить, что FGF2 является эффективным для лечения характерных симптомов астмы, индуцированной действием VEGF и TGF-β1, уровни которых контролируются IL-13. Ингибирование FGF2 приводило к повышению концентрации VEGF (см. фиг.5) на модели животных и затем вызывало AHR (см. фиг.6). Эти результаты соответствуют данным других экспериментов, в которых блокада VEGF приводила к ингибированию AHR (см. фиг.6) у мышей, дефицитных по FGF2. Фармацевтический эффект FGF2 на астму, вызванную активностью IL-13 или VEGF, подтверждается следующими данными.
Животным с моделью Th2 астмы, опосредованной IL-13, вводили интраназально FGF2, с последующим изучением эффекта FGF2. В результате введения FGF2 гиперреактивность дыхательных путей на метахолин уменьшалась (см. фиг.20), уменьшалось количество воспалительных клеток в бронхоальвеолярном лаваже (BAL) (см. фиг.21) и подавлялась экспрессия IL-13 и VEGF (см. фиг.22), обоих ключевых медиаторов Th2 астмы. Результаты гистологического исследования подтвердили, что FGF2 уменьшал гипертрофию и облитерацию бронхиальной стенки почти до нормального состояния легочной ткани (см. фиг.23). Суммируя, было сделано подтверждение, что FGF2 ингибирует экспрессию VEGF и IL-13, приводя к подавлению AHR и уменьшению воспаления. Следовательно, FGF2 может эффективно использоваться для лечения астмы.
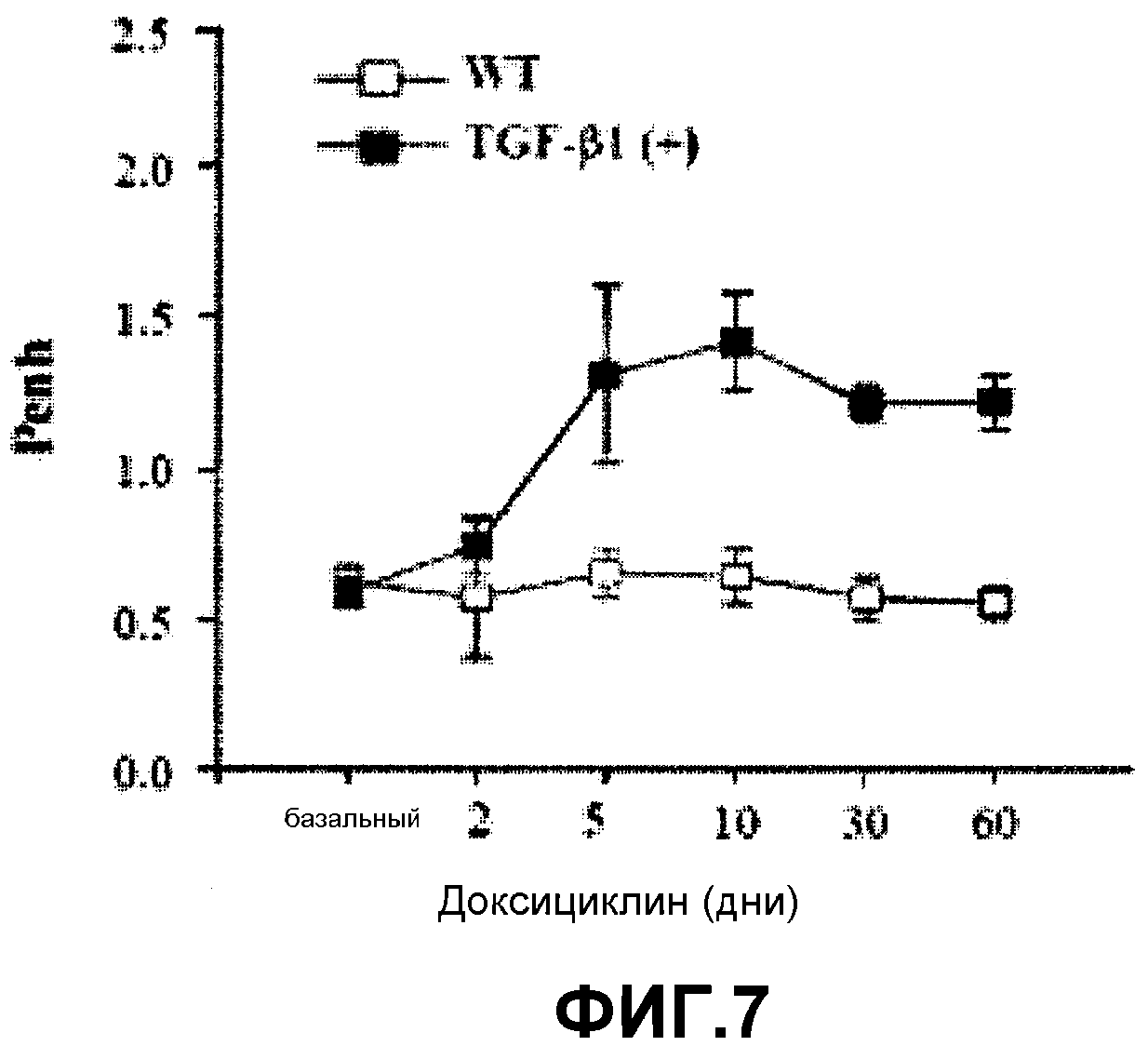
Что касается TGF-β1, молекулы, образующейся на последующих этапах сигнального пути в патогенезе IL-13 опосредованной астмы, то в предшествующих исследованиях было показано, что перестройка дыхательных путей у трансгенных по TGF-β1 мышей (Lee et al., J. Exp. Med., 200: 377-389, 2004) и бронхиальный фиброз, вызваннй IL-13, зависит от TGF-β1 (Lee et al., J. Exp. Med.,194: 809-821, 2001). TGF-β1 значительно индуцировал резистентность и облитерацию дыхательных путей у трансгенных по TGF-β1 мышей (см. фиг.7) и AHR подавлялась метахолином (см. фиг.8).
Для подтверждения взаимосвязи FGF2 и TGF-β1 при IL-13 опосредованной астме, AHR измеряли у мышей, нокаутированных по FGF2. В результате, у мышей, нокаутированных по FGF2 не было отмечено ингибирование AHR под действием TGF-β1 (см. фиг.9). Результаты показывают, что ингибирование AHR происходит не самим TGF-β1, а посредством FGF2, экспрессирующимся вместе с TGF-β1. То есть, увеличение TGF-β1 в присутствии FGF2 снижает AHR, но TGF-β1 не может подавлять повышение AHR в отсутствие FGF2.
На основании вышеуказанных результатов была подтверждена связь FGF2 c TGF-β1, как указано далее; при повреждении тканей дыхательных путей активируется иммунная система. Гладкомышечные клетки и фибробласты дыхательных путей трансформируются в миофибробласты, что приводит к развитию фиброза. В то же время, FGF2 индуцирует пролиферацию гладкомышечных клеток и фибробластов дыхательных путей и восполняет дефицит TGF-β1, а также индуцирует трансформацию миофибробластов в гладкомышечные клетки и фибробласты дыхательных путей. Суммируя, FGF2 индуцирует трансформацию миофибробластов в фибробласты, снижая количество миофибробластов, и, таким образом, FGF2 ингибирует перестройку дыхательных путей и одновременно ингибирует AHR.
Для объяснения механизма ингибирования перестройки дыхательных путей посредством FGF2, у мышей, нокаутированных по FGF2 измеряли концентрацию коллагена и AHR. В результате, количество фибробластов, секретирующих коллаген, в легких мышей, нокаутированных по FGF2, было ниже, чем у контрольных мышей дикого типа (см. фиг.10). Результат показывает, что количество фибробластов было снижено, так как FGF2 не индуцировал пролиферацию клеток, и поэтому концентрация коллагена, секретируемого этими клетками, также была понижена. Также исследовали эффект метахолина на AHR. В результате, метахолин не оказывал значительного влияния на AHR у мышей дикого типа, но резко повышал AHR у мышей, нокаутированных по FGF2 (см. фиг.9). Эти результаты показывают, что дефицит FGF2 в пути от IL-13 до TGF-β1 блокирует пролиферацию фибробластов и трансформацию миофибробластов в фибробласты, приводящую к перестройке дыхательных путей. И, кроме того, количество фибробластов, чувствительных в отношении метахолина постепенно снижалось, повышая AHR. Следовательно, FGF2 может эффективно использоваться для лечения астмы, опосредованной IL-13 и TGF-β1.
Кроме IL-13, IFN-γ, в патогенезе астмы важную роль играет цитокин Th1. Ряд предшествующих экспериментов подтвердил предположение, что Th1 цитокин, особенно IFN-γ, тесно связан с патогенезом астмы. Однако до настоящего времени не существовало модели астмы, опосредованной Th1. Этот пробел был связан с тем, что большинство исследований астмы было сфокусировано на гипотезе Th1/Th2, выделяющей важность активации Th2 в патогенезе астмы. Так, на сегодняшний день были созданы модели астмы посредством сверхэкспрессии эозинофилов или иммуноглобулина Е (IgE).
Напротив, в соответствии с последними данными, AHR может быть индуцирована вне зависимости от эозинофильного воспаления (Venkayya R., Am. J. Respir. Cell Mol Biol 2002; 26: 202-8) и количество больных неэозинофильной астмой составляет больше половины всех больных астмой (Douwes et al., Thorax, 57: 643-8, 2002). Следовательно, для изучения астмы требуется создание модели астмы типа Th1.
Поэтому, авторами настоящего изобретения была создана модель Th1 астмы или ХОЗЛ, индуцированных IFN-γ, на животных, и с помощью нее изучена роль FGF2. В результате авторами настоящего изобретения было обнаружено, что FGF2 может быть эффективно использован для лечения астмы и ХОЗЛ, опосредованных IFN-γ. Следовательно, авторы настоящего изобретения получили средство для профилактики и лечения ХОЗЛ, а также средство для профилактики и лечения астмы, содержащее FGF2 (фактор роста фибробластов-2) в качестве активного ингредиента. ХОЗЛ может быть индуцирована сверхэкспрессией IFN-γ (интерферона-γ).
Сначала была изучена взаимосвязь между IFN-γ и FGF2 при астме, индуцированной IFN-γ. Экспрессию FGF2 в легких трансгенных по IFN-γ мышей измеряли с помощью ОТ-ПЦР. В результате, экспрессия FGF2 была заметно ингибирована у трансгенных мышей, в отличие от контрольных мышей дикого типа (см. фиг.13). Результат показывает, что экспрессия FGF2 ингибируется посредством IFN-γ-сигнального пути.
Затем была изучена роль FGF2 в патогенезе воспаления дыхательных путей и AHR, индуцированной IFN-γ. У трансгенных по IFN-γ мышей (IFN-γ(+)/FGF2(+/+)) удаляли ген FGF2 и затем оценивали AHR. Также измеряли количество воспалительных клеток и уровень воспалительных цитокинов. В результате, у трансгенных по IFN-γ мышей, дефицитных по гену FGF2 (IFN-γ(+)/FGF2(+/+)) были заметно повышены AHR и воспаление (см. фиг.14А и 14В). Был сделан вывод, что повышенный уровень VEGF играет важную роль в увеличении AHR и воспалении, индуцированном дефицитом FGF2 у трансгенных по IFN-γ мышей (см. фиг.14С). Вышеуказанные результаты показывают, что FGF2 также может быть эффективно использован для лечения воспаления дыхательных путей и AHR, индуцированных IFN-γ.
Активность FGF2 исследовали на модели Th1 астмы, опосредованной IFN-γ. В результате, FGF2 снижал AHR на метахолин (см. фиг.27), и было сделано предположение, что FGF2 может быть эффективно использован для лечения IFN-γ-опосредованной астмы.
FGF2 вводили мышам с Th1 астмой и ХОЗЛ, с последующим определением у них лечебного эффекта FGF2.
Введение мышам FGF2 снижало количество воспалительных клеток в BAL (см. фиг.26) и AHR в ответ на метахолин (см. фиг.36).
Кроме AHR у мышей наблюдали апоптоз паренхиматозных клеток, характерный симптом ХОЗЛ. На AHR и апоптоз паренхиматозных клеток влияло присутствие или отсутствие FGF2. После введения FGF2, у IFN-γ-мышей снижалась не только AHR (см. фиг.27), но также деструкция паренхиматозных клеток (см. фиг.28 и 29), индуцированные IFN-γ. Введение FGF2 также снижало повреждение ткани и деструкцию альвеол или эмфизему легких, индуцированных IFN-γ (см. фиг.30 и 31).
Как объяснялось выше, на моделях астмы и ХОЗЛ, FGF2 снижал AHR и ингибировал деструкцию альвеол, что, таким образом, предполагает эффективное использование FGF2 в качестве средства для профилактики и лечения астмы и ХОЗЛ.
Средство для профилактики и лечения астмы и ХОЗЛ по настоящему изобретению, содержащее FGF2 в качестве активного ингредиента, может включать активный ингредиент в количестве 0,0001 - 50 мас.% от общей массы композиции.
Терапевтическое средство по настоящему изобретению кроме FGF2 может включать один или несколько активных ингредиентов, имеющих такую же или подобную функцию в отношении FGF2.
Терапевтическое средство по настоящему изобретению кроме вышеуказанного активного ингредиента может включать один или несколько фармацевтически приемлемых носителей для введения. Фармацевтически приемлемые носители могут быть выбраны или получены путем смешивания более одного ингредиента, выбранного из группы, состоящей из солевого раствора, стерильной воды, раствора Рингера, буферного солевого раствора, раствора декстрозы, раствора мальтодекстрозы, глицерина и этанола. Могут быть добавлены другие обычно используемые добавки, такие как антиоксидантные средства, буферный раствор, бактериостатическое средство и тому подобное. Для получения растворов для инъекций, пилюль, капсул, гранул или таблеток также могут быть добавлены разбавители, диспергирующие средства, поверхностно-активные вещества, вяжущие вещества и смазывающие вещества. Композиция по настоящему изобретению может, кроме того, быть получена в подходящих для каждого заболевания или в зависимости от ингредиентов формах, следуя способам, изложенным в Remington's Pharmaceutical Science (новейшее издание), Mack Publishing Company, Easton PA.
Терапевтическое средство по настоящему изобретению может быть введен перорально или парентерально (например, внутривенной, подкожной, интраперитонеальной, местной или интраназальной инъекцией). Парентеральное введение является предпочтительным и интраназальное введение является более предпочтительным. Эффективная доза композиции может быть определена в зависимости от массы, возраста, пола, состояния здоровья, диеты, частоты введения, способа введения, экскреции и тяжести заболевания.
Эффективная доза терапевтического средства по настоящему изобретению составляет 0,005~10 мг/кг в сутки и, предпочтительно, 0,05~1 мг/кг в сутки. Частота введения составляет один раз в сутки или, предпочтительно, несколько раз в сутки.
Терапевтическое средство по настоящему изобретению может вводиться самостоятельно или при хирургической операции, наряду с гормональной терапией, химиотерапией и регулятором биологических реакций, для профилактики и лечения астмы и ХОЗЛ.
Для исследования токсичности мышам интраназально вводили FGF2 по настоящему изобретению. В результате, оно было определено как безопасное, так как его вычисленное значение LD50 у мышей было гораздо выше, чем 1000 мг/кг.
Настоящее изобретение, кроме того, относится к способу создания модели Th1 астмы или ХОЗЛ у животных, который характеризуется непосредственным введением аллергенов, таких как яичный белок и двухцепочечная РНК, в дыхательные пути.
Способ получения предусматривает следующие стадии:
(1) Сенсибилизация мышей BALB/c четырехразовым интраназальным введением 5-15 мкг полиинозиновой-полиситидиловой кислоты, двухцепочечной РНК и 50-100 мкг яичного белка; и
(2) Сенсибилизация мышей введением 25-75 мкг яичного белка через 10 дней после первой сенсибилизации. На вышеуказанной стадии (1) количество двухцепочечной РНК для сенсибилизации предпочтительно составляет 10 мкг, а количество яичного белка составляет предпочтительно 75 мкг. На вышеуказанной стадии (2), количество яичного белка, вводимое через 10 дней для второй сенсибилизации, предпочтительно составляет 50 мкг.
Астма, упомянутая в настоящем описании, может быть не-эозинофильной.
Настоящее изобретение относится к модели Th1 астмы или ХОЗЛ у животных, полученной вышеописанным способом.
К животным могут относиться все млекопитающие, подходящие для биологических экспериментов, но мыши являются предпочтительными.
Что касается создания модели Th1 астмы или ХОЗЛ у животных, то двухцепочечная РНК (dsRNA), полученная при репликации вируса, активно индуцирует интерфероны IFN-α и IFN-γ типа 1, проявляя противовирусную активность in vivo (Guidotti et al., Annu. Rev. Immunol., 19:65-91, 2001). Интерфероны типа 1 запускают продукцию IL-12 и IFN-γ и способны индуцировать требуемый иммунный ответ, стимулируя примирование Т-клеток и созревание дендритных клеток (Londhe et al., FEBS Lett., 553: 33-8, 2003). Следовательно, авторами настоящего изобретения была создана модель астмы, индуцированной по Th1 пути после обработки dsRNA, у животных.
Последовательность и длина dsRNA, используемой для создания модели у животных, не ограничены при условии, что такое dsRNA может индуцировать Th1 астму. dsRNA может быть приобретена, предпочтительной является полиинозиновая-полицитидиловая кислота (полил:С).
У мышей с астмой, индуцированной по Th1 пути, была повышена AHR (см. фиг.15) и было повышено число лимфоцитов, нейтрофилов и макрофагов. Но число эозинофилов повышено не было. В качестве медиатора был заметно повышен только IP-10, который связан с Th1 активностью (см. фиг.16 и 17). Вышеуказанные результаты показывают, что не-эозинофильное воспаление дыхательных путей индуцируется ОА и dsRNA. Также был повышен уровень IFN-γ в бронхоальвеолярном лаваже (BAL) и уровень антиген-специфических IgG1 и IgG2 в крови (см. фиг.18 и 19). Суммируя, сенсибилизация дыхательных путей ОА и dsRNA была вызвана IFN-γ, а антиген-специфические IgG2a и антиген-специфические IgE вовлечены не были. Вышеуказанные результаты подтвердили успешное получение модели Th1 астмы у животных.
Благодаря модели на животных по настоящему изобретению также показали симптомы ХОЗЛ. А именно, размер и объем легких был увеличен, альвеолы разрушены и из-за увеличения содержания коллагена был индуцирован выраженный фиброз (см. фиг.28-фиг.31). Вышеуказанные результаты подтвердили, что мыши с Th1 астмой по настоящему изобретению также могут быть использованы в качестве модели ХОЗЛ.
Модель на животных по настоящему изобретению была подтверждена как модель Th1 или не-эозинофильной астмы, вызванной введением аллергенов (яичный белок, ОА) и двухцепочечной РНК (dsRNA) непосредственно в дыхательные пути и может эффективно использоваться для создания средства для лечения астмы и ХОЗЛ.
Описание чертежей
На фиг.1А представлена группа графиков, на которых продемонстрирована AHR, наблюдаемая у IL-4 трансгенных мышей.
На фиг.1В представлен график, на котором показана AHR, наблюдаемая у IL-9(-)/IL-13(+/+), IL-9(-)/IL-13(-/-), IL-9(+)/IL-13(+/+) и IL-9(+)/IL-13(-/-) трансгенных мышей.
На фиг.2 представлен график, на котором показано сравнение AHR трансгенных по IL-13 мышей и контрольными мышами дикого типа.
На фиг.3 представлен график, на котором показано сравнение уровней экспрессии VEGF и TGF-β1 в бронхоальвеолярном лаваже (BAL) трансгенных по IL-13 мышей и контрольными мышами дикого типа.
На фиг.4 представлен график, на котором показана AHR, наблюдаемая после введения ингибитора рецептора VEGF-2 SU 1498, у трансгенных по IL-13 мышей и у контрольных мышей дикого типа.
На фиг.5 представлен график, на котором показано сравнение уровней экспрессии VEGF и TGF-β1 в бронхоальвеолярном лаваже (BAL) мышей, нокаутированных по FGF2, и контрольными мышами дикого типа.
На фиг.6 представлен график, на котором показана AHR, наблюдаемая у мышей, нокаутированных по FGF2, или по FGF2 и VEGF.
На фиг.7 представлен график, на котором показано сравнение AHR TGF-β1 трансгенных мышей и контрольными мышами дикого типа.
На фиг.8 представлен график, на котором показаны наблюдаемые изменения AHR в соответствии со временем, у TGF-β1 трансгенных мышей и контрольных мышей дикого типа.
На фиг.9 представлен график, на котором показаны изменения AHR, наблюдаемые после введения TGF-β1, в зависимости от времени у контрольных мышей дикого типа или у мышей, нокаутированных по FGF2.
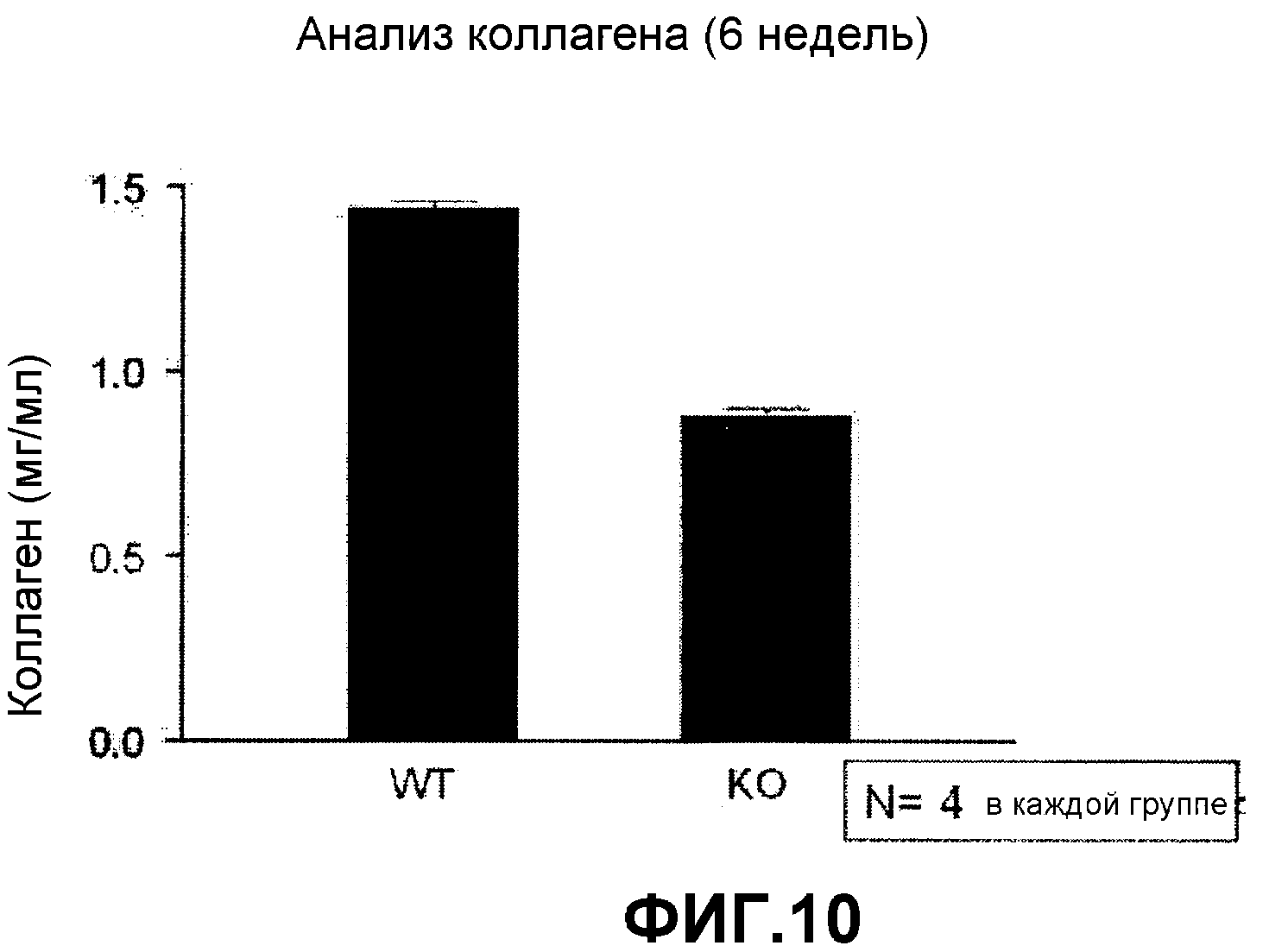
На фиг.10 представлен график, на котором показано сравнение содержания коллагена в легочной ткани мышей, нокаутированных по FGF2 и контрольными мышами дикого типа.
На фиг.11 представлен график, на котором показано сравнение AHR IFN-γ трансгенных мышей и контрольными мышами дикого типа.
На фиг.12 представлен график, на котором показаны уровни экспрессии VEGF в бронхоальвеолярном лаваже (BAL), TGF-β1 и IP-10 трансгенных по IFN-γ мышей и контрольными мышами дикого типа.
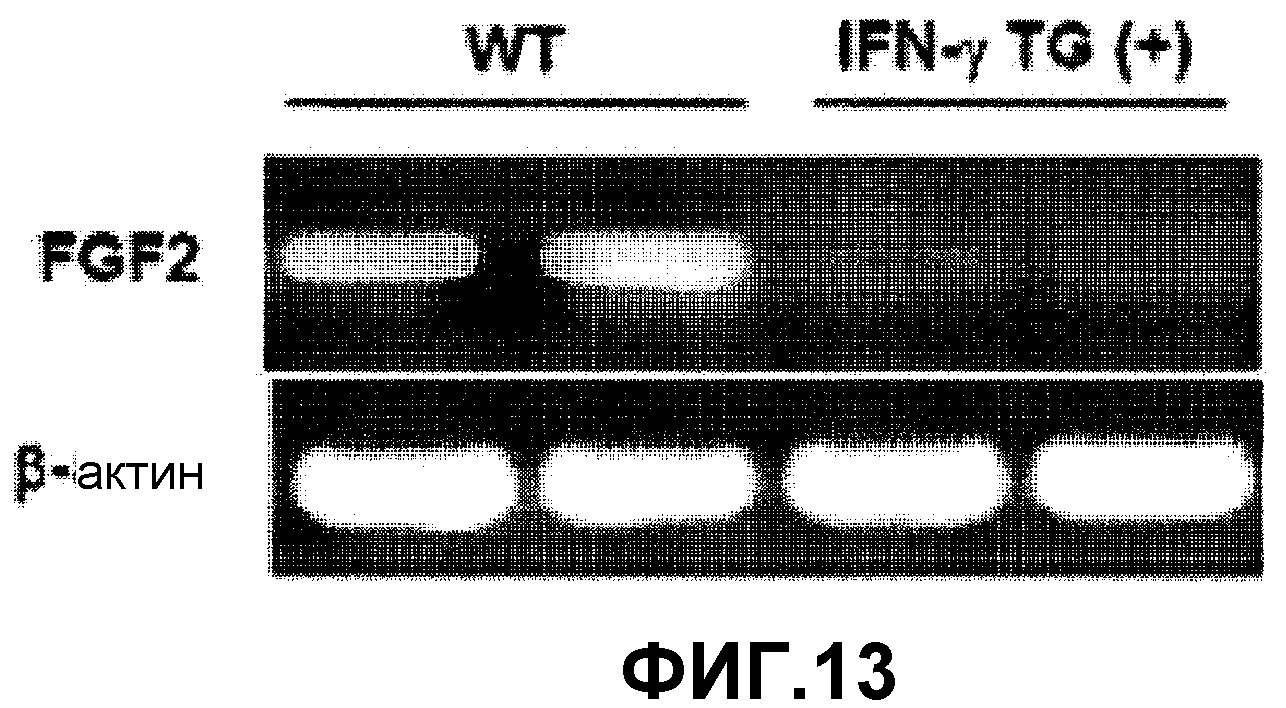
На фиг.13 представлена фотография агарозного геля, на которой видны уровни экспрессии FGF2 в ткани легких у трансгенных по IFN-γ мышей и контрольных мышей дикого типа.
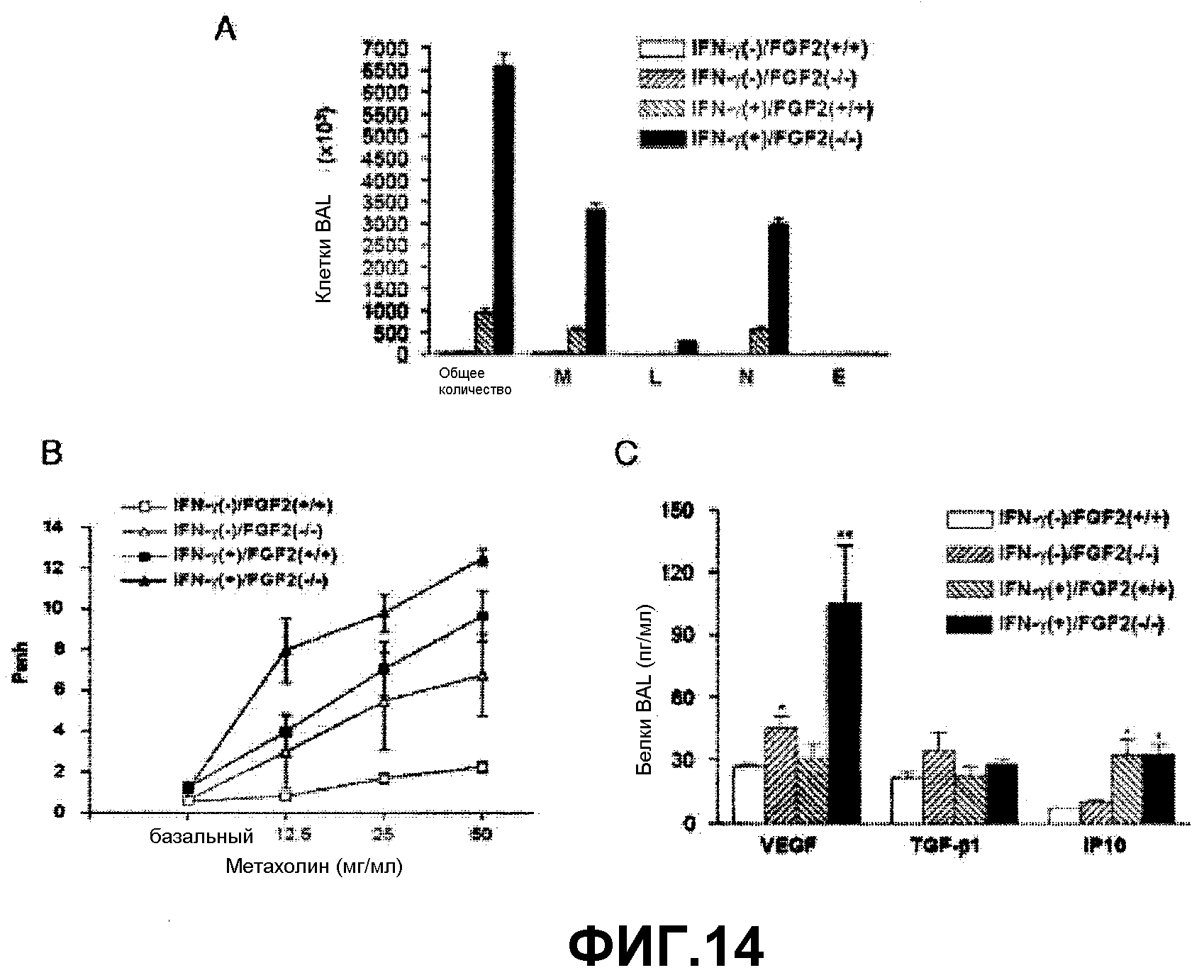
На фиг.14 представлен график, на котором показано количество всех клеток (Т), количество макрофагов (М), количество лейкоцитов (L), количество нейтрофилов (N) и количество эозинофилов (Е) в бронхоальвеолярном лаваже (BAL) у IFN-γ(-)/FGF2(+/+), IFN-γ(-)/FGF2(-/-), IFN-γ(+)/FGF2(+/+) или IFN-γ(+)/FGF2(-/-) трансгенных мышей.
На фиг.14В представлен график, на котором показана метахолин-зависимая AHR, наблюдаемая у IFN-γ(-)/FGF2(+/+), IFN-γ(-)/FGF2(-/-), IFN-γ(+)/FGF2(+/+) или IFN-γ(+)/FGF2(-/-) трансгенных мышей.
На фиг.14С представлен график, на котором показаны уровни экспрессии VEGF, TGF-β1 и IP-10 в бронхоальвеолярном лаваже у IFN-γ(-)/FGF2(+/+), IFN-γ(-)/FGF2(-/-), IFN-γ(+)/FGF2(+/+) или IFN-γ(+)/FGF2(-/-) трансгенных мышей.
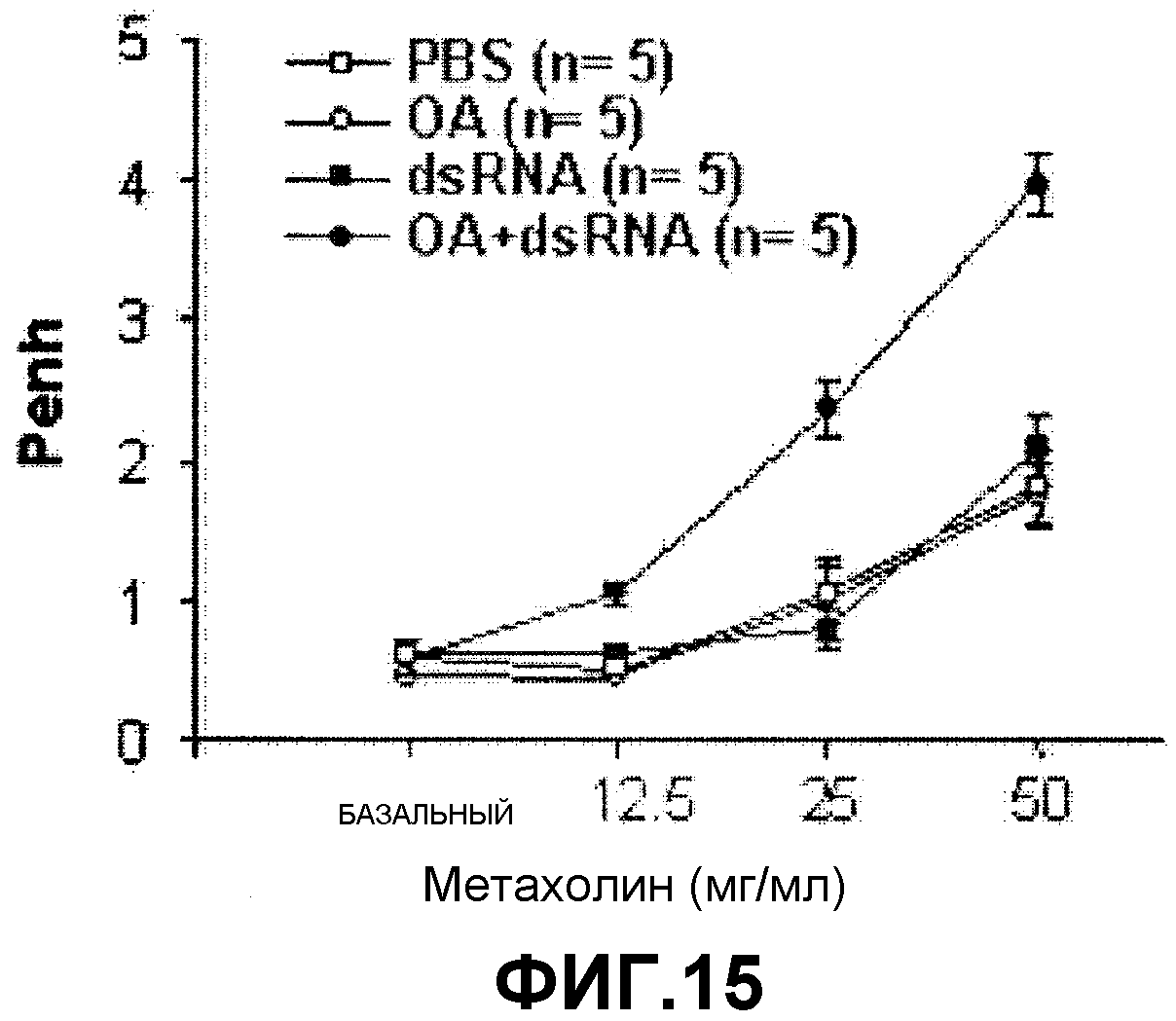
На фиг.15 представлен график, на котором показаны изменения AHR в ответ на метахолин после введения аллергена (ОА) и dsRNA по отдельности или вместе.
На фиг.16 представлен график, на котором показано количество всех клеток, количество макрофагов, количество лимфоцитов, количество нейтрофилов и количество эозинофилов в бронхоальвеолярном лаваже (BAL), измеряемое после введения аллергена (ОА) и dsRNA по отдельности или вместе.
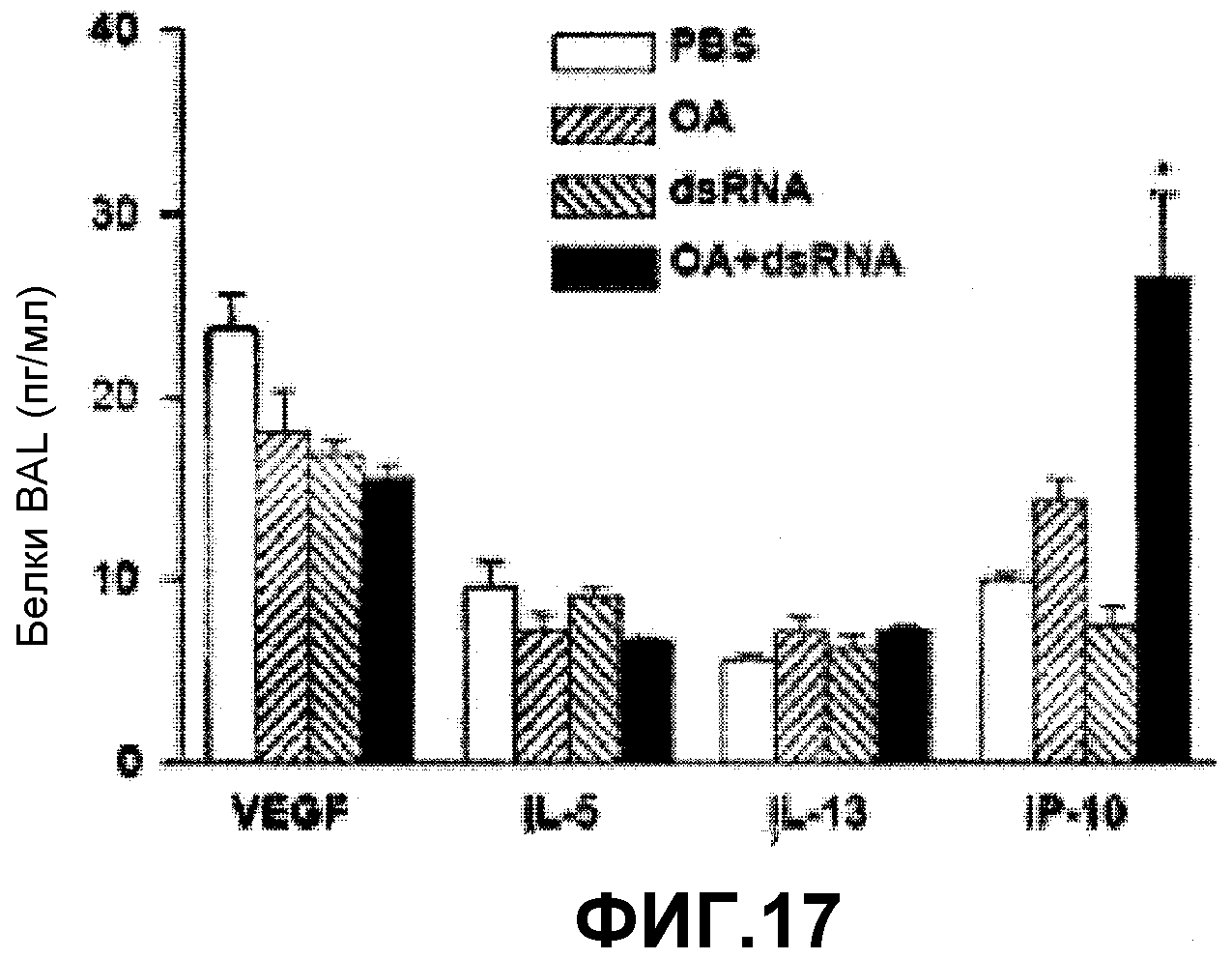
На фиг.17 представлен график, на котором показаны уровни экспрессии цитокинов (VEGF, IL-5, IL-13 и IP-10) в бронхоальвеолярном лаваже (BAL), наблюдаемые после введения аллергена (ОА) и dsRNA по отдельности или вместе.
На фиг.18 представлен график, на котором показан уровень экспрессии IFN-γ в бронхоальвеолярном лаваже (BAL), наблюдаемый после введения аллергена (ОА) и dsRNA по отдельности или вместе.
На фиг.19 представлен график, на котором показана продукция антиген-специфических антител (IgG1 и IgG2a) в сыворотке после введения аллергена (ОА) и dsRNA по отдельности или вместе.
На фиг.20 представлен график, на котором показана AHR, зависимая от дозы метахолина, наблюдаемая после введения рекомбинантного FGF2(rFGF2) на модели Th2 астмы у мышей и у контрольных мышей дикого типа.
На фиг.21 представлен график, на котором показано количество всех клеток, количество макрофагов, количество лимфоцитов, количество нейтрофилов и количество эозинофилов в бронхоальвеолярном лаваже (BAL), измеряемое после введения рекомбинантного FGF2 (rFGF2) мышам с Th2 астмой и у контрольных мышей дикого типа.
На фиг.22 представлен график, на котором показаны концентрации цитокинов (VEGF, IL-5, IL-13 и IP-10) в бронхоальвеолярном лаваже (BAL), наблюдаемые после введения рекомбинантного FGF2 (rFGF2) мышам с Th2 астмой и контрольным мышами дикого типа.
На фиг.23 показана фотография патологии ткани легких у мышей с Th2 астмой и у контрольных мышей дикого типа до и после введения рекомбинантного FGF2 (rFGF2).
На фиг.24 представлен график, на котором показано соотношение эозинофильного и не-эозинофильного компонента в индуцированной мокроте больного тяжелой астмой.
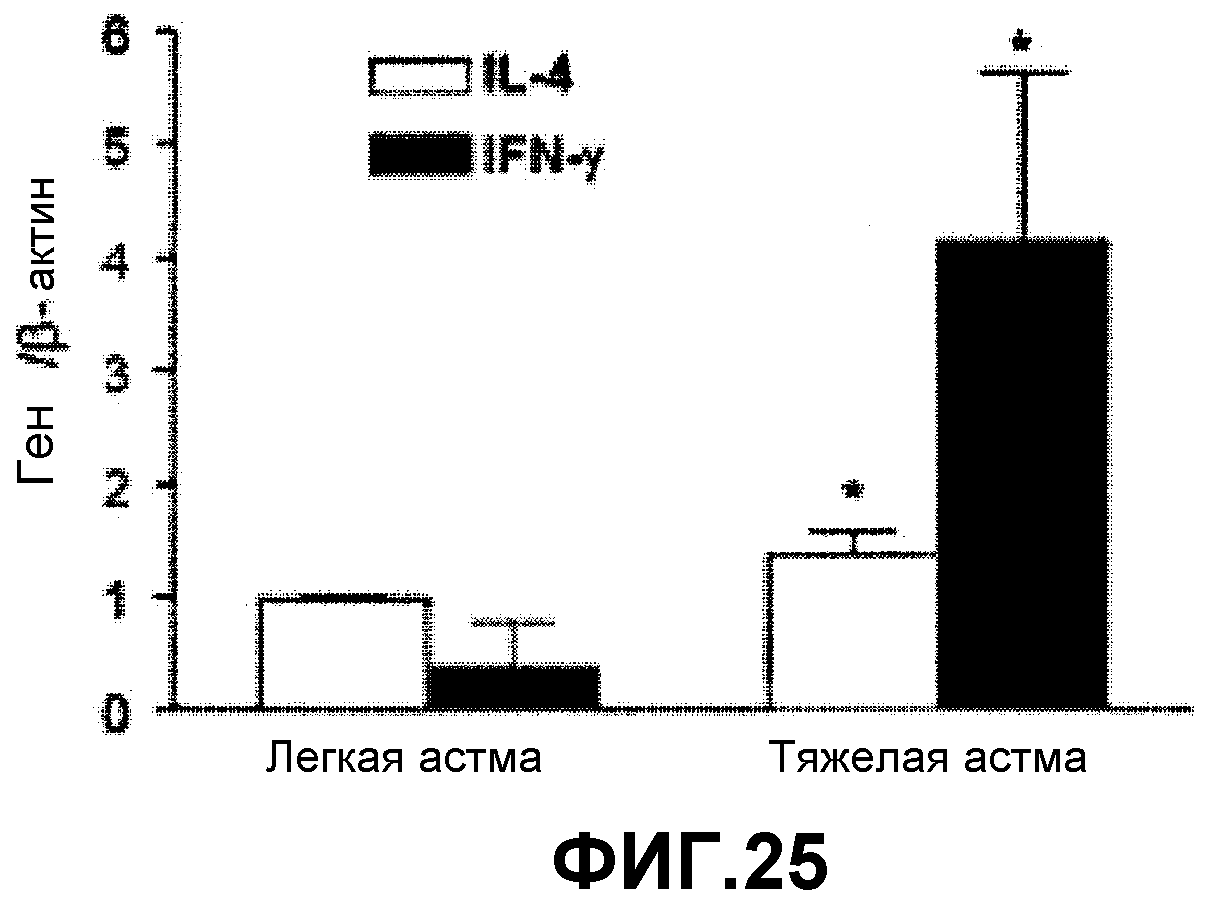
На фиг.25 представлен характер экспрессии IL-4 и IFN-γ в индуцированной мокроте больного астмой в зависимости от тяжести заболевания.
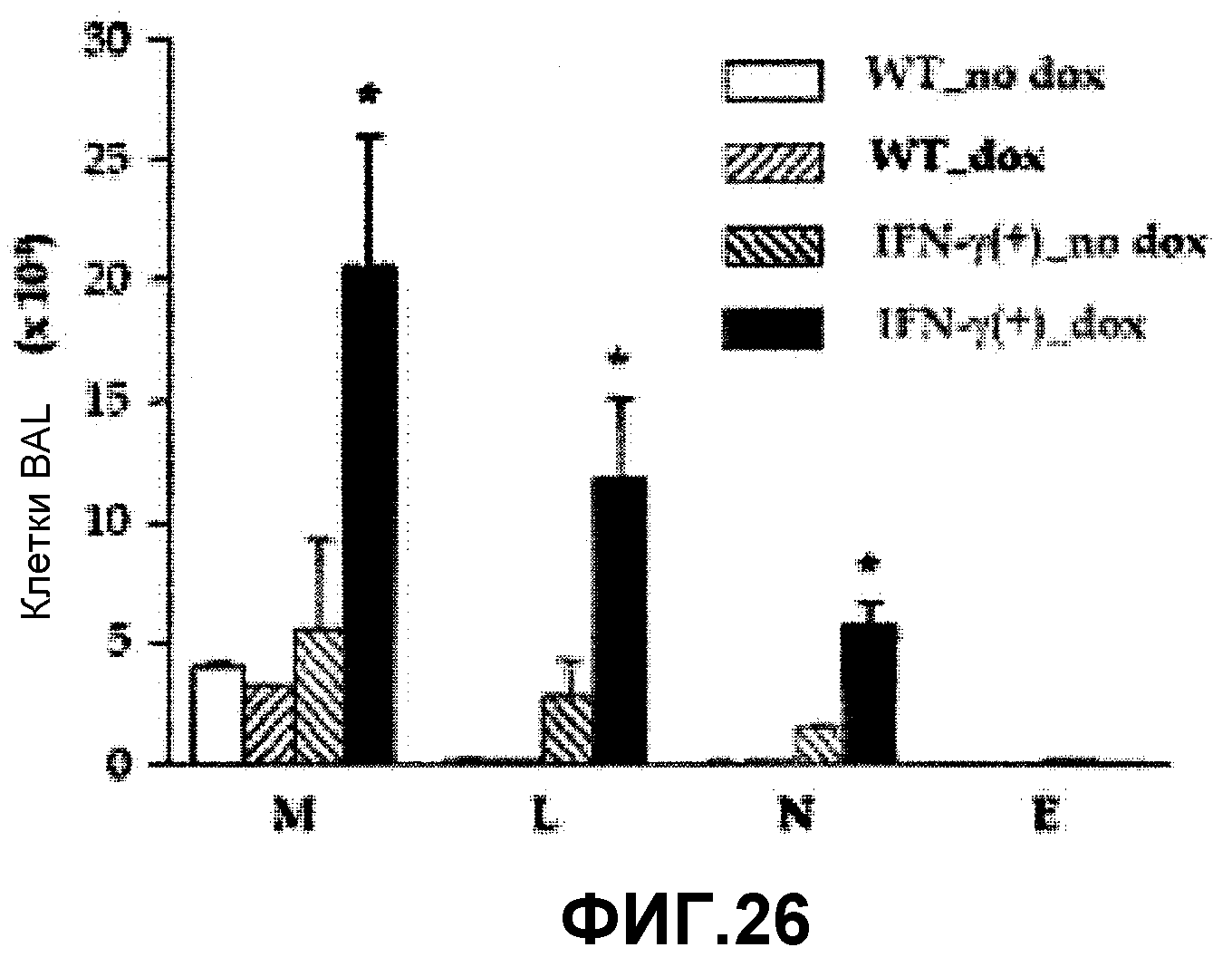
На фиг.26 представлен график, на котором показано количество всех клеток, количество макрофагов, количество лимфоцитов, количество нейтрофилов и количество эозинофилов в бронхоальвеолярном лаваже в присутствии или отсутствие индуктора сверхэкспрессии доксициклина у временно трансгенных по IFN-γ мышей и у контрольных мышей дикого типа.
На фиг.27 представлен график, на котором показана метахолин-зависимая AHR у IFN-γ(-)/FGF2(+/+), IFN-γ(-)/FGF2(-/-), IFN-γ(+)/FGF2(+/+) или IFN-γ(+)/FGF2(-/-) трансгенных мышей.
На фиг.28 представлена фотография, на которой показан размер легкого у трансгенных по IFN-γ мышей под воздействием FGF2 или в отсутствие воздействия FGF2.
На фиг.29 представлен график, на котором показано изменение объема легких трансгенных по IFN-γ мышей в зависимости от присутствия или отсутствия FGF2.
На фиг.30 представлен график, на котором показана степень фиброза легких у трансгенных по IFN-γ мышей в зависимости от присутствия или отсутствия FGF2.
На фиг.31 представлен ряд гистологических исследований ткани легких, показывающих распад паренхимы у трансгенных по IFN-γ мышей в зависимости от присутствия или отсутствия FGF2.
Вариант осуществления изобретения
Осуществленные на практике и в настоящее время предпочтительные варианты осуществления настоящего изобретения являются иллюстративными и приведены в следующих примерах.
Однако понятно, что специалист в области техники на основании описания настоящего изобретения, может осуществить модификации и уточнения, не выходя за рамки и сущности настоящего изобретения.
Пример 1. Астма, индуцированная сверхэкспрессией IL-13 и значимость VEGF и TGF-β1
Для изучения развития астмы под действием IL-13, были созданы трансгенные по IL-13 мыши и у каждой из них измеряли AHR. Также изучали эффект сверхэкспрессии IL-13 на экспрессию TGF-β1 и VEGF.
1-1. Создание трансгенных по IL-13 мышей
Трансгенных по IL-13 мышей создавали обычным способом (Zhou Zhu et al., J. Clin. Invest., 103: 779-788, 1999; Tang et al., J. Clin. Invest., 98: 2845-2853, 1996; Ray et al., J. Clin. Invest., 100: 2501-2511, 1997). Для селективной экспрессии гена-кандидата IL-13 использовали конструкцию, включающую ген-кандидат IL-13, связанный с промотором (B. Stripp and J.Whitsett, University of Cincinnati), индуцирующим экспрессию 10 кДа белка клеток Clara (СС10). Для получения индуцибельной трансгенной мыши, у которой экспрессия встроенного чужеродного гена может внешне контролироваться, получали pKS-CC10-rtTA-hGH связыванием СС10 промотора с реверсивным трансактиватором тетрациклина (rtTA) и геном гормона роста человека (hGH) (Ray et al., J. Clin. Invest., 98: 2501-2511, 1997). Плазмидную ДНК очищали на колонке Elutip-D (Schleicher and Schuell Inc, USA) и проводили диализ с микроинъекционным буфером (0,5 мМ Tris-HCl, 25 мМ ЭДТА, рН 7,5). Скрещивание мышей CBA и C57BL/6 проводили микроинъекцией интрапронуклеуса согласно литературным ссылкам, приведенным в настоящем описании, и вышеуказанную плазмидную ДНК встраивали в полученную яйцеклетку F2, что давало трансгенную мышь. Трансгенным мышам и контрольным мышам дикого типа случайным образом вводили 0,5 мг/мл доксициклина (dox) в воде и затем у каждой из них отбирали бронхоальвеолярный лаваж (BAL) для изучения уровня IL-13 и трансформации.
1-2. AHR у трансгенных по IL-13 мышей
Для подтверждения развития астмы у трансгенных по IL-13 мышей наблюдали один из наиболее характерных симптомов астмы, AHR. AHR может быть оценена на основании уклона кривой доза-эффект (DRS, Pediatric Allergy and Immunology, 14; 193, 2003) и повышенной паузы (Penh, Mckinley et al., Clinical & Experimental Immunology, 136: 224-231, 2004) согласно обычным способам, хорошо известным в этой области. Penh рассчитывается как указано далее; максимальное давление выдоха (PEP) делят на максимальное давление вдоха (PIP) и затем вычисленное значение умножают на паузу. В частности, AHR индуцировали у трансгенных мышей, полученных в примере выше (1-1), посредством распыления метахолина в течение трех минут через 24 и 48 часов после второй сенсибилизации. Максимальное давление выдоха и максимальное давление вдоха измеряли на основании общей плетизмографии каждые 10 секунд в течение 3 минут и данные усредняли (фиг.2.) На фигуре 2 показаны результаты исследования AHR у трансгенных по IL-13 мышей и у контрольных мышей дикого типа.
Как показано на фиг.2, AHR была повышена у трансгенных по IL-13 мышей по сравнению с контрольными мышами дикого типа.
1-3. Эффект сверхэкспрессии IL-13 на экспрессию VEGF и TGF-β1
Приведенные далее эксперименты проводили для изучения соответствия AHR, индуцированной сверхэкспрессией IL-13, с помощью модуляторов, образующихся на последующих этапах сигнального пути IL-13, т.е. VEGF и TGF-β1. С помощью пробирки SP45 проводили катетеризацию дыхательных путей трансгенных мышей, полученных в примере выше (1-1) для получения бронхоальвеолярного лаважа (BAL), который промывали стерилизованным физиологическим раствором, содержащим 0,1% BSA и 0,05 мм ЭДТА и затем центрифугировали. В полученном супернатанте BAL, измеряли уровень VEGF и TGF-β1 с использованием набора ELISA (CalBiotech, USA) (фиг.3). На фигуре 3 представлен график, на котором показан уровень экспрессии VEGF и TGF-β1 в бронхоальвеолярном лаваже (BAL), взятом у трансгенных по IL-13 мышей и от контрольных мышей дикого типа.
Как показано на фиг.3, концентрации VEGF и TGF-β1 были повышены в BAL, полученном у трансгенных мышей с AHR, индуцированной сверхэкспрессией IL-13. Результаты показывают, что AHR, индуцированная IL-13, контролируется модуляторами TGF-β1 и VEGF, что подтверждают результаты нижеследующего примера (1-4).
1-4. Подавление AHR блокатором VEGF
Для подтверждения результатов вышеуказанного примера (1-3) в брюшную полость мыши, полученной в примере (1-1) один раз в сутки вводили, 10 мг/кг блокатора рецептора VEGF-2 SU1498 (EMD Bioscience, USA) (фиг.4). На фигуре 4 представлен график, на котором показана AHR, наблюдаемая у трансгенных по IL-13 мышей и у контрольных мышей дикого типа после введения блокатора рецептора VEGF-2 SU1498.
Как показано на фиг.4, AHR, индуцированная IL-13, подавляется блокатором рецептора VEGF-2, что указывает на то, что AHR, индуцированная IL-13, возникает в результате передачи сигнала через VEGF.
Пример 2. Роль FGF2 в патогенезе астмы, индуцированной IL-13
Для изучения роли FGF2 в патогенезе астмы, индуцированной IL-13, и действия модуляторов, образуемых на последующих этапах сигнального пути, т.е. TGF-β1 и VEGF, у мышей, нокаутированных по FGF2 (-/-), наблюдали AHR и перестройку дыхательных путей.
2-1. AHR, индуцированная дефицитом FGF2
Следующие эксперименты проводили для изучения связи FGF2 и AHR. Мышей, нокаутированных по FGF2, покупали у Jackson Lab (CA, USA). AHR (DRS) в ответ на метахолин и концентрации VEGF и TGF-β1, которые, как было показано, увеличены у трансгенных мышей, изучали в BAL по аналогии с методикой, описанной в примере (1-2) и примере (1-3) (фиг.5 и 6). На фигуре 5 представлен график, на котором показан уровень экспрессии VEGF и TGF-β1 в BAL, полученном у мышей, нокаутированных по FGF2, и у контрольных мышей дикого типа. На фигуре 6 представлен график, на котором показана AHR, наблюдаемая у мышей, нокаутированных по FGF2, и мышей, нокаутированных по FGF2, которым вводили блокатор VEGF, как описано в примере (1-4).
Как показано на фигуре 5 концентрация VEGF была увеличена у мышей, нокаутированных по FGF2, по сравнению с контрольными мышами дикого типа, что указывает на усиление AHR, что согласуется с результатами, представленными на фиг.6.
Как показано на фигуре 6, AHR была повышена у мышей, нокаутированных по FGF2, и могла быть подавлена блокатором VEGF. Результаты показывают, что AHR, индуцированная дефицитом FGF2, контролируется VEGF, а именно, введение FGF2 ингибирует экспрессию VEGF, делая его представляющим интерес кандидатом средства для профилактики и лечения астмы, опосредованной путем VEGF.
2-2. Перестройка дыхательных путей, инициированная дефицитом FGF2
Для изучения связи FGF2 с перестройкой дыхательных путей проводили следующие эксперименты, в том числе измерение пролиферации и трансформации клеток, которые сопровождались перестройкой дыхательных путей.
Мышей, нокаутированных по FGF2, покупали у Jackson Lab (CA, USA). Ткань легких получали обычным способом и анализировали концентрацию коллагена в тканях, что может быть индексом для оценки пролиферации и трансформации клеток, с использованием набора анализа Sircol Collagen (Biocolor assay, Nothern Ireland) в соответствии с инструкциями производителя (фиг.10). На фигуре 10 представлен график, на котором показана концентрация коллагена в ткани легких мышей, нокаутированных по FGF2, и контрольных мышей дикого типа. Как показано на фиг.10, концентрация коллагена была гораздо ниже у мышей, нокаутированных по FGF2, чем у контрольных мышей дикого типа.
Суммируя, в легких у мышей, нокаутированных по FGF2, снижалось количество фибробластов, секретирующих коллаген, что происходило из-за того, что фибробласты трансформировались в миофибробласты и затем мигрировали, снижая количества фибробластов. В результате, индуцировалась перестройка дыхательных путей.
На основании вышеуказанных результатов было подтверждено, что FGF2 ингибирует перестройку дыхательных путей и AHR, поэтому он может эффективно использоваться для лечения астмы.
Пример 3. Возникновение астмы под действием TGF-β1
Для изучения патогенеза астмы, опосредованной экспрессией TGF-β1, индуцированной IL-13, были созданы трансгенные по TGF-β1 мыши по аналогии с процедурой, как описано в примере (1-1), и проводили следующие эксперименты.
(3-1) Перестройка дыхательных путей, под действием TGF-β1
AHR, возникшую в результате перестройки дыхательных путей, изучали методикой, аналогичной описанной в примере выше (1-2) (фиг.7). На фигуре 7 представлен график, на котором показано сравнение AHR трансгенных по TGF-β1 мышей и контрольными мышами дикого типа в зависимости от времени. На фигуре 8 представлен график, на котором показано сравнение AHR в ответ на метахолин у трансгенных по TGF-β1 мышей и контрольных мышей дикого типа в зависимости от времени.
Как показано на фиг.7 значительная резистентность дыхательных путей индуцировалась под действием TGF-β1, что соответствует результат Penh. Как показано на фиг.8, тем не менее, AHR ингибировалась в ответ на метахолин. Такой результат показывает, что сверхэкспрессия TGF-β1 вовлечена лишь в перестройку дыхательных путей, а не в другие характерные симптомы астмы.
3-2. Роль FGF2 в ингибировании AHR под действием TGF-β1
Как объясняется в примере выше (3-1), у мышей, нокаутированных по FGF2, измеряли AHR, изучая роль FGF2 в ингибировании AHR под действием TGF-β1 (фиг.9). На фигуре 9 представлен график, на котором показана AHR, наблюдаемая у мышей, нокаутированных по FGF2, и у контрольных мышей дикого типа в зависимости от времени, после введения TGF-β1 (система R&D, USA).
Как показано на фигуре 9, ингибирование AHR под действием TGF-β1 было слабее у мышей, нокаутированных по FGF2, что указывает на то, что ингибирование AHR под действием TGF-β1 не происходит непосредственно из-за TGF-β1, а из-за FGF2, который экспрессируется совместно с TGF-β1. То есть, ингибирование AHR в результате стимуляции TGF-β1 связано с FGF2.
Пример 4. Развитие астмы, вызванное IFN-γ, и роль в FGF2
Была создана модель астмы, вызванной IFN-γ, ключевым медиатором ХОЗЛ и тяжелой астмы. С целью подтверждения роли FGF2 были проведены следующие эксперименты на трансгенных по IFN-γ мышам по аналогии с методикой, описанной в примере 1.
4-1. Увеличение AHR при сверхэкспрессии IFN-γ
Измеряли AHR у трансгенных мышей методикой, используемой в примере 1-2 выше (фиг.11). Затем измеряли уровень AHR, VEGF, TGF-β1 и белка IP-10, экспрессия которых индуцировалась IFN-γ в BAL таким же способом, который использовали в примерах 1-3 выше (фиг.12). На фигуре 11 представлен график, на котором показано сравнение AHR трансгенных по IFN-γ мышей и контрольных мышей дикого типа.
Как показано на фиг.11, у трансгенных по IFN-γ мышей AHR была однородно повышена. Как показано на фиг.12 концентрации VEGF и TGF-β1, индуцированные IL-13, экспрессируемым в Th2, не были повышены у трансгенных мышей, но был повышен белок IP-10 (интерферон-индуцибельный белок 10), который не связан с Th2. Результаты показывают, что AHR, индуцированная у трансгенных мышей в этом примере, является IFN-γ-специфичной.
4-2. Роль FGF2 в развитии AHR, индуцированной сверхэкспрессией IFN-γ
Эффект сверхэкспрессии IFN-γ на экспрессию FGF2
Для изучения роли FGF2 в развитии AHR, вызванной сверхэкспрессией IFN-γ, у трансгенных по IFN-γ мышей измеряли уровень экспрессии FGF2 (фиг.13). РНК экстрагировали из ткани легких мышей дикого типа и у трансгенных мышей обычным способом, с последующим проведением ОТ (обратной транскриптазной)-ПЦР. Вкраце, все количество РНК экстрагировали из 1 г легочной ткани, взятой от каждой мыши дикого типа и трансгенной мыши с использованием TRIzol Reagent (Life Technology, USA) в соответствии с инструкциями производителя. ОТ-ПЦР проводили с использованием выделенной РНК в качестве матрицы для синтеза кДНК с использованием набора ОТ-ПЦР (Promega, USA). ПЦР проводили с верхним праймером (5'-ACTCACATTCGAAACCCCAAAC-3') и нижним праймером (5'-CGTCAGATCGCTGGAGAC-3') с использованием 1 мкг синтезированной кДНК в качестве матрицы для амплификации FGF2-специфичной кДНК. ПЦР проводили как указано далее; предварительная денатурация при 95°С в течение 8 минут, денатурация при 95°С в течение 1 минуты, отжиг при 56°С в течение 1 минуты, полимеризация при 72°С в течение 1 минуты, 35 циклов от денатурации до полимеризации и конечной элонгации при 72°С в течение 10 минут. На фигуре 13 представлена фотография агарозного геля, на которой показана амплификация кДНК, специфичной к FGF2 легочной ткани трансгенных по IFN-γ мышей и контрольных мышей дикого типа.
Как показано на фиг.13, у трансгенных по IFN-γ мышей ингибировалась экспрессия FGF2, что указывает на то, что экспрессия FGF2 ингибируется IFN-γ.
Роль FGF2 в AHR, индуцированной сверхэкспрессией IFN-γ
Для изучения роли FGF2 в патогенезе воспаления дыхательных путей и AHR, индуцированной IFN-γ, измеряли количество воспалительных клеток и уровень белка, вовлеченного в воспаление, в BAL у мышей с различными генотипами (фиг.14). Мышей получали, как описано ранее (Zhou Zhu et al., J. Clin. Invest., 103:779-788, 1999; Tang et al., J. Clin. Invest., 98: 2845-2853, 1996; Ray et al., J. Clin. Invest., 100: 2501-2511, 1997). На фиг.14 “+” обозначает мышь с повышенной экспрессией мишеневого гена, “-” обозначает мышь, дефицитную по мишеневому гену. На фигуре 14А представлен график, на котором показано количество всех клеток (Общее количество), количество макрофагов (М), количество лимфоцитов (L), количество нейтрофилов (N) и количество эозинофилов (Е) в бронхоальвеолярном лаваже, взятом от IFN-γ(-)/FGF2(+/+), IFN-γ (-)/FGF2(-/-), IFN-γ(+)/FGF2(+/+) и IFN-γ(+)/FGF2(-/-) трансгенных мышей. На фигуре 14В представлен график, на котором показана метахолин-зависимая AHR у IFN-γ(-)/FGF2(+/+), IFN-γ (-)/FGF2(-/-), IFN-γ(+)/FGF2(+/+) и IFN-γ(+)/FGF2(-/-) трансгенных мышей. На фигуре 14С представлен график, на котором показан уровень экспрессии VEGF, TGF-β1 и IP-10 в бронхоальвеолярном лаваже (BAL) у IFN-γ(-)/FGF2(+/+), IFN-γ(-)/FGF2(-/-), IFN-γ(+)/FGF2(+/+) и IFN-γ(+)/FGF2(-/-) трансгенных мышей.
Как показано на фигуре 14, дефицит FGF2 гена у трансгенных по IFN-γ мышей приводит к повышению AHR, индуцированной IFN-γ, и увеличению плотности воспалительных клеток, ухудшая воспаление дыхательных путей. Такой результат показывает, что FGF2 может эффективно использовать для лечения астмы, индуцированной IFN-γ.
Пример 5. Ингибирование IL-13 опосредованной Th2 астмы введением FGF2
Для изучения ингибирующего эффекта белка FGF2 на IL-13 опосредованную Th2 астму проводили следующие эксперименты.
Рекомбинантный белок FGF (rFGF) покупали у Pharmacia-Upjohn Co (Италия). Для создания модели AHR у мышей, мышей BALB/c (Jackson Lab, США) дважды сенсибилизировали внутрибрюшинной инъекцией 75 мкг яичного белка (ОА) и 2 мг квасцов и через 10 дней мышей снова сенсибилизировали интраназальным введением 50 мкг яичного белка, для того, чтобы вызвать астму. Полученных мышей обозначали мышами с Th2 астмой.
Трансгенным мышам и контрольным мышам дикого типа интраназально вводили 10 мкг/мышь rFGF2 один раз в сутки в течение 4 дней или не вводили FGF2 (этой группе вводили только солевой раствор) с последующим измерением уровня AHRв ответ на метахолин (фиг.20), количество воспалительных клеток (фиг.21) и концентрации медиаторов, таких как VEGF, IL-13, IL-5 и IP-10 (фиг.22) в соответствии с методикой, описанной ранее в вышеуказанных примере (1-2) и примере (1-3).
На фигуре 20 представлен график, на котором показана AHR, зависимая от дозы метахолина, наблюдаемая у мышей с Th2 астмой и у контрольных мышей дикого типа после введения рекомбинантного FGF2. На фигуре 21 представлен график, на котором показано количество всех клеток, количество макрофагов, количество лимфоцитов, количество нейтрофилов и количество эозинофилов в бронхоальвеолярном лаваже (BAL), измеряемое после введения рекомбинантного FGF2 (rFGF2) у мышей с Th2 астмой и контрольных мышей дикого типа. На фигуре 22 представлен график, на котором показаны концентрации цитокинов (VEGF, IL-13, IL-5 и IP-10) в бронхоальвеолярном лаваже (BAL), наблюдаемые после введения рекомбинантного FGF2 (rFGF2) у мышей с Th2 астмой и у контрольных мышей дикого типа.
Как показано на фиг.20, ингибирующий эффект на AHR в ответ на метахолин, был значительней у мышей с Th2 астмой, которым вводили rFGF2, по сравнению с мышами с Th2 астмой, которым не вводили rFGF2. Как показано на фиг.21, количество воспалительных клеток в BAL было значительно ниже у мышей с Th2 астмой, которым вводили rFGF2, чем у мышей с Th2 астмой, которым не вводили rFGF2. Как показано на фиг.22, концентрации IL-13 и VEGF, ключевых медиаторов Th2 астмы, были значительно снижены у мышей с Th2 астмой, которые получали rFGF2, чем у мышей с Th2 астмой, которые не получали rFGF2. При этом экспрессия IL-5 и IP-10, о которых известно, что они не связаны с Th2 астмой, не изменялась.
Также проводили гистологический анализ бронхиальной стенки мышей с астмой после введения rFGF2 (фиг.23). На фигуре 23 представлена патологическая фотография ткани легких мышей с Th2 астмой и контрольных мышей дикого типа до (А) и после (В) введения рекомбинантного FGF2 (rFGF2).
Как показано на фиг.23, как результат гистологического анализа (В) стенки бронха мышей, которым вводили rFGF2, гипертрофия и облитерация стенки бронха были снижены введением rFGF2 до нормального уровня (С).
На основании вышеприведенных результатов, было подтверждено, что FGF2 ингибирует экспрессию VEGF и IL-13, и таким образом, ингибирует AHR и воспаление дыхательных путей, опосредованных Th2. Следовательно, FGF2 можно эффективно использовать для профилактики и лечения астмы.
Пример 6. Ингибирование Th1 астмы и ХОЗЛ, опосредованных IFN-γ, введением FGF2
Для изучения ингибирующей активности FGF2 у мышей с Th1 астмой, опосредованной IFN-γ проводили следующие эксперименты
Рекомбинантный белок FGf2 покупали у Pharmacia-Upjohn Co. (Италия). Мышей с Th1 астмой, опосредованной IFN-γ, получали, как указано далее.
6-1. Создание моделей Th1 астмы и ХОЗЛ у животных с использованием яичного белка и двухцепочечной РНК
Мышей BALB/c (Jackson Lab, США) интраназально сенсибилизировали четырехразовым введением 10 мкг синтезированной dsRNA полиинозиновой-полицитидиловой кислоты (PolylC, Sigma,USA) и 75 мкг яичного белка (ОА), по отдельности или вместе. Через 10 дней мышам интраназально вводили 50 мкг ОА для индукции астмы. Полученных мышей называли мышами с Th1 астмой. Мышам отрицательного контроля вводили только фосфатный буферный раствор (PBS).
1) Подтверждение характеристик Th1 астмы
Для подтверждения индукции Th1 астмы у мышей, оценивали AHR в ответ на метахолин (фиг.15), количество воспалительных клеток в BAL (фиг.16) и концентрации медиаторов, таких как VEGF, IL-13, IL-5 и IP-10 (фиг.17) в соответствии с методикой, описанной в примере (1-2) и в примере (1-3). На фигуре 15 представлен график, на котором показано изменение AHR в ответ на метахолин после введения аллергена (ОА) и dsRNA по отдельности или вместе. На фигуре 16 представлен график, на котором показано количество всех клеток, количество макрофагов, количество лимфоцитов, количество нейтрофилов и количество эозинофилов в бронхоальвеолярном лаваже (BAL), измеренное после введения аллергена (ОА) и dsRNA по отдельности или вместе. На фигуре 17 представлен график, на котором показан уровень экспрессии цитокинов (VEGF, IL-13, IL-5 и IP-10) в бронхоальвеолярном лаваже (BAL), наблюдаемый после введения аллергена (ОА) и dsRNA по отдельности или вместе.
Как показано на фигуре 15, AHR в ответ на метахолин была повышена у мышей с астмой, индуцированной ОА и dsRNA.
Как показано на фигуре 16, количество лимфоцитов, нейтрофилов и макрофагов у мышей было повышено, а количество эозинофилов не изменялось.
Как показано на фигуре 17, у мышей был существенно повышен только медиатор IP-10. Вышеуказанные результаты показывают ОА и dsRNA индуцировали не-эозинофильное воспаление дыхательных путей.
Кроме того, для подтверждения того, что IFN-γ индуцирует вышеуказанную Th1 астму, измеряли уровни IgG1 и IgG2a IFN-γ в бронхоальвеолярном лаваже (BAL) (фиг.18 и фиг.19). На фигуре 18 представлен график, на котором показан уровень экспрессии IFN-γ в BAL, когда ОА и dsRNA вводили по отдельности или вместе. На фигуре 19 представлен график, на котором показана продукция антител, специфичных к антигену (IgG1 и IgG2a) в сыворотке, когда ОА и dsRNA вводили по отдельности или вместе.
Как показано на фигуре 18 и фигуре 19, уровень IFN-γ в BAL был по меньшей мере в три раза выше и уровни IgG1 и IgG2 в сыворотке были также повышены. Вышеуказанные результаты показывают, что астма, индуцированная OV и dsRNA, была связана с IFN-γ и антиген-специфическими IgG2a и не была связана с IgE, который вовлечен в Th2 астму.
(2) Подтверждение характеристик ХОЗЛ
Для подтверждения индукции ХОЗЛ у вышеуказанных мышей, измеряли размер и объем легких и концентрацию коллагена (фиг.28, фиг.29 и фиг.30). На фигуре 28D представлен график, на котором показан размер легких у мышей. На фигуре 29D представлен график, на котором показан объем легких у мыши. На фигуре 30D представлен график, на котором показан уровень фиброза в легких трансгенных мышей.
Как показано на фиг.28D, фиг.29D и фиг.30D размер и объем легких и концентрация коллагена была заметно повышена у мышей с Th1 астмой, что делает возможным предположить, что мыши имеют признаки ХОЗЛ. При увеличении размера и объема легких и концентрации коллагена повреждение легочной ткани, деструкция альвеол и эмфизема легких являются характерными симптомами ХОЗЛ, которые также присутствовали у мышей, как показано на фиг.31.
На фигуре 31А представлена фотография патологической ткани легких с деструкцией паренхимы у мышей с Th1 астмой. Как показано на фиг.31, в легких трансгенных по IFN-γ мышей наблюдали увеличение площади альвеол, вызванное деструкцией паренхимы, что является одним из характерных симптомов ХОЗЛ.
Увеличение размера и объема легких и апоптоза в альвеолах наряду с увеличением содержания коллагена подтверждало серьезный фиброз.
На основании вышеприведенных результатов, было подтверждено, что мышей с Th1 астмой можно эффективно использовать в качестве модели ХОЗЛ, демонстрирующей патогенез ХОЗЛ.
6-2. Ингибирование Th1 астмы введением FGF2
Нижеприведенные эксперименты проводили для изучения может ли белок FGF2 ингибировать астму у мышей с Th1 астмой, опосредованной IFN-γ. Мышей, полученных в примере выше (6-1) обозначали как экспериментальная группа мышей с Th1 астмой. Мышам с Th1 астмой, полученным в примере (6-1), и контрольным мышам дикого типа вводили рекомбинантный FGF2 в соответствии с методикой, которую использовали в примере 5. Затем измеряли AHR в ответ на метахолин в обеих группах, следуя методике, описанной в примере (1-2).
На фигуре 27 представлен график, на котором показана AHR, зависимая от дозы метахолина, наблюдаемая у мышей с Th1 астмой и у контрольных мышей дикого типа, после введения rFGF2.
Как показано на фигуре 27, AHR в ответ на метахолин была значительно ниже у мышей, которым вводили rFGF2, чем у мышей, которым не вводили rFGF2.
На фигуре 28 представлена фотография, на которой показано сравнение размера легких у мышей с Th1 астмой, получавших rFGF2 и мышами, не получавших rFGF2.
Как показано на фигуре 28, размер легких мышей, получавших rFGF2 был меньше, чем у мышей с Th1 астмой, не получавших rFGF2.
Вышеуказанные результаты показывают, что FGF2 уменьшает характерные симптомы астмы у трансгенных по IFN-γ мышей, поэтому его можно эффективно использовать для лечения астмы, индуцированной IFN-γ.
(6-3). Ингибирование ХОЗЛ введением FGF2
Нижеприведенные эксперименты проводили для изучения эффекта ингибирования FGF2 на ХОЗЛ. Рекомбинантный FGF2 вводили трансгенным мышам, полученным в примере выше (6-1), и контрольным мышам дикого типа в соответствии с методикой, описанной в примере 5. Затем измеряли размер и объем легких и концентрацию коллагена, которые являются главным показателем ХОЗЛ у мышей (фиг.28, фиг.29 и фиг.30). На фигурах 28А, В, С и D показан размер легких и нормальных мышей и мышей с ХОЗЛ, объем легких показан на графике фиг.29. На фигурах 30А, В, С и D представлены графики, на которых показан уровень фиброза легких у контрольных мышей дикого типа и мышей с ХОЗЛ после введения rFGF2 или при отсутствии его введения. На фигуре 31 представлен ряд фотографий патологической ткани легких, на которых видна деструкция паренхимы у трангенных по IFN-γ мышей в зависимости от наличия или отсутствия FGF2.
Как показано на фиг.28С и D и на фиг.29С и D, после введения rFGF2 объем легких был заметно снижен. Как показано на фиг.30С и D при введении rFGF2 мышам с ХОЗЛ концентрация коллагена также была снижена. Снижение размера и объема легких и снижение содержания коллагена означает, что rFGF2 можно эффективно использовать для лечения ХОЗЛ, как показано на фигуре 31.
На фигуре 31 представлена серия фотографий патологической ткани легких, на которых видна деструкция паренхимы у мышей с ХОЗЛ. Как показано на фиг.31А, увеличение площади альвеол, вызванное апоптозом паренхимы, характерный симптом у пациентов с ХОЗЛ, наблюдали в легких мышей, не получавших rFGF2 (В), в отличие от легких группы, получавших rFGF2 (А), что означает, что введение rFGF2 эффективно для лечения ХОЗЛ.
Вышеуказанные результаты подтвердили, что размер и объем легких, апоптоз в альвеолах и содержание коллагена вовлечены в фиброз у мышей с ХОЗЛ, и введение FGF2 эффективно для лечения таких патологических симптомов.
Пример 7. Сверхэкспрессия IFN-γ на модели астмы у человека
Нижеприведенные эксперименты проводили для изучения может ли астма у человека быть индуцирована сверхэкспрессией IFN-γ и не-эозинофильных клеток. Мокроту получали у 215 взрослых людей, больных астмой, была показана обратимая обструкция дыхательных путей и проведено измерение жизненной емкости легких с использованием спирометрии в соответствии с обычным способом.
Также на бронхи воздействовали метахолином для изучения функции легких (фиг.24). На фигуре 24 представлен график, на котором показано соотношение эозинофилов и не-эозинофилов в мокроте пациента с тяжелой астмой. Как показано на фиг.24, более чем у половины пациентов была подтверждена не-эозинофильная астма, а не-эозинофильная астма. С целью подтверждения факторов, опосредующих астму, измеряли уровень IL-4 и IFN-γ (фиг.25). На фигуре 25 представлен график экспрессии IL-4 и IFN-γ в мокроте больного с астмой в зависимости от тяжести заболевания.
Как показано на фиг.25, экспрессия IFN-γ, которая связана с Th1 астмой, была повышена у пациентов с тяжелой астмой, при этом экспрессия IL-4, которая связана с Th2 астмой, не изменялась. Результаты показывают, что пациенты с астмой, в частности с тяжелой астмой, имеют IFN-γ-опосредованную не-эозинофильную Th1 астму.
Промышленная применимость
Как объяснено выше, терапевтическое средство по настоящему изобретению, содержащее FGF2 в качестве активного ингредиента, может эффективно использоваться для профилактики и лечения фиброза, воспаления дыхательных путей, повышенной реактивности дыхательных путей, перестройки дыхательных путей, астмы и ХОЗЛ. Кроме того, модели астмы и ХОЗЛ у животных, разработанные с использованием яичного белка и двухцепочечной РНК также могут эффективно использоваться для создания терапевтического средства для лечения астмы и ХОЗЛ.
Специалисту в данной области техники понятно, что мысли и специфические варианты осуществления изобретения, концепция и конкретные варианты осуществления, описанные в вышеуказанном описании могут быть легко использованы в качестве основы для модификации или создания других вариантов осуществления изобретения для достижения целей настоящего изобретения. Специалисту в данной области техники также понятно, что такие эквивалентные варианты осуществления изобретения не выходят за рамки и сущность изобретения как указано в прилагаемой формуле изобретения.
Реферат
Изобретение относится к созданию средства для лечения или профилактики астмы или хронических обструктивных заболеваний легких (ХОЗЛ). Средство для лечения и профилактики астмы содержит FGF2 (фактор роста фибробластов-2) в качеств активного ингредиента, причем астма вызвана сверхэкспрессией IL-13 (интерлейкин-13) или сверхэкспрессией IFN-γ (интерферон-γ), где FGF2 ингибирует активность IL-13, FGF2 ингибирует активность VEGF и FGF2 ингибирует активность TGF-β1 (трансформирующий фактор роста-β1). Средство для лечения или профилактики хронического обструктивного заболевания легких (ХОЗЛ), содержащее FGF2 (фактор роста фибробластов-2) в качестве активного ингредиента, где хроническое обструктивное заболевание легких (ХОЗЛ) вызвано сверхэкспрессией IFN-γ (интерферон-γ). Ингибитор активности IL-13, содержащий FGF2 (фактор роста фибробластов-2) в качестве активного ингредиента. Ингибитор активности VEGF, содержащий FGF2 (фактор роста фибробластов-2) в качестве активного ингредиента. Ингибитор активности TGF-β1, содержащий FGF2 (фактор роста фибробластов-2) в качестве активного ингредиента. 5 н. и 3 з.п. ф-лы, 31 ил.
Формула
Документы, цитированные в отчёте о поиске
Производные феноксифенилуксусной кислоты
Комментарии