Способ получения тетрафторсилана, метод анализа примесей в тетрафторсилане и газ на его основе - RU2274603C2
Код документа: RU2274603C2
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения тетрафторсилана, методу анализа содержания примесей в тетрафторсилане высокой чистоты и его применению.
Описания уровня техники
Тетрафторсилан (далее в тексте иногда обозначаемый как "SiF4") используется, например, в качестве сырья для получения оптических волокон, полупроводников или элементов солнечных батарей, а также для получения требуемого высокочистого продукта. Что касается способа его получения, то известен, например, способ получения SiF4 по реакции SiO2 с HF в присутствии концентрированной серной кислоты (Japanese Unexamined Patent Publication №57-135711 (JP-A-57-135711)).
Однако проблемой такого способа является образование воды в качестве побочного продукта в ходе реакции между исходным SiO2 и HF. Образовавшаяся вода может удаляться с помощью концентрированной серной кислоты, однако такое удаление оказывается неполным, и полученный SiF4 содержит значительное количество HF и гексафтордисилоксана ((SiF3)2O), полученных по реакции воды с SiF4, и, кроме этого, содержит диоксид углерода, который трудно выделить из SiF4, образование которого связывают с небольшим количеством производных углерода, содержащихся в концентрированной серной кислоте.
Кроме этого, известен способ получения SiF4 термическим разложением гексафторсиликата. Однако гексафторсиликат содержит Н2О или такие примеси, как производные кремниевой кислоты, содержащие следовые количества кислорода (например, SiO2), причем даже при удовлетворительной предварительной обработке такая примесь может реагировать с SiF4 с образованием гексафтордисилоксана в ходе осуществления термического разложения.
Известен также способ очистки SiF4, содержащего (SiF3)2 O, СО2 или HF. В том случае, когда SiF4 содержит такие газообразные примеси как (SiF3)2O, CO2 и O2 и предназначен для использования, например, в качестве сырья для производства тонкой силиконовой пленки, кислородная примесь отрицательно влияет на свойства полупроводника или волокна. В соответствие со сказанным выше, требуется SiF4 с пониженным содержанием примесей, и в равной мере необходим метод анализа следовых количеств примесей.
Что касается способа очистки SiF4, то в Japanese Unexamined Patent Publication №57-156317 (JP-A-57-156317) описывается способ очистки SiF4, содержащего (SiF3)2О в результате его контакта с адсорбентом. Однако после регенерации и дальнейшего использования такого адсорбента не всегда восстанавливается его исходная абсорбционная способность. Причина этого явления не совсем понятна, однако, считается, что она связана с разложением гексафтордисилоксана в порах адсорбента. В результате присоединения SiO2, образующейся в ходе разложения, к центрам адсорбции теряется способность адсорбента к регенерации и повторному использованию, что создает проблему, связанную с обработкой такого адсорбента, как отхода производства. Кроме этого, при недостаточном прокаливании адсорбента перед пропусканием газа протекает побочная реакция с водой с образованием гексафтордисилоксана.
Описание изобретения
Настоящее изобретение было сделано с целью устранения вышеперечисленных недостатков и состояло в разработке способа получения тетрафторсилана, метода анализа примесей в тетрафторсилане высокой чистоты и его применении.
В результате широких исследований, направленных на решение указанных выше проблем, авторы настоящего изобретения установили, что такие проблемы могут быть решены с использованием способа получения тетрафторсилана, включающего стадию (1) нагревания гексафторсиликата, стадию (2-1) реакции газообразного тетрафторсилана, содержащего гексафтордисилоксан, образовавшийся на стадии (1) с газообразным фтором, стадию (2-2) реакции газообразного тетрафторсилана, содержащего гексафтордисилоксан, образовавшийся на стадии (1) с фторидом многовалентного металла, или стадию (2-1) реакции газообразного тетрафторсилана, содержащего гексафтордисилоксан, полученный на стадии (1), с газообразным фтором, и стадию (2-3) реакции газообразного тетрафторсилана, образовавшегося на стадии (2-1) с фторидом многовалентного металла.
Кроме этого, авторы изобретения обнаружили, что указанные проблемы могут быть решены с использованием метода анализа примесей в тетрафторсилане высокой чистоты, в результате приведения тетрафторсилана, содержащего в качестве примесей газообразный Н2, газообразный O2, газообразный N2, газообразный СО, газообразный СН4 и/или газообразный CO2, в контакт с адсорбентом с целью отделения примесей от тетрафторсилана и их введения совместно с газом-носителем в газовый хроматограф с целью последующего анализа.
Настоящее изобретение было создано на базе указанного метода.
Настоящее изобретение предусматривает способ получения тетрафторсилана, включающий стадию (1) нагревания гексафторсиликата, стадию (2-1) реакции газообразного тетрафторсилана, содержащего гексафтордисилоксан, образовавшийся на стадии (1), с газообразным фтором, стадию (2-2) реакции газообразного тетрафторсилана, содержащего гексафтордисилоксан, образовавшийся на стадии (1), с фторидом многовалентного металла, или стадию (2-1) реакции газообразного тетрафторсилана, содержащего гексафтордисилоксан, полученный на стадии (1), с газообразным фтором, и стадию (2-3) реакции газообразного тетрафторсилана, образовавшегося на стадии (2-1), с форидом многовалентного металла.
Настоящее изобретение также предусматривает метод анализа примесей в тетрафторсилане высокой чистоты, заключающийся в контактировании газообразного тетрафторсилана, содержащего газообразный Н2, газообразный CO2, газообразный N2, газообразный СО, газообразный СН4 и/или CO2, с адсорбентом с целью отделения примесей от тетрафторсилана и введении примесей совместно газом-носителем в газовый хроматограф с целью анализа примесей.
Кроме этого, настоящее изобретение также предусматривает метод анализа примесей в тетрафторсилане высокой чистоты, заключающийся в введении газообразного тетрафторсилана, содержащего гексафтордисилоксан, в качестве примеси, в оптическую ячейку с окном из галогенида металла и проведении анализа на гексафтордисилоксан и/или фтористый водород методом ИК-спектрометрии.
Кроме этого, настоящее изобретение предусматривает получение газа для производства оптического волокна, содержащего тетрафторсилан, полученный описанным выше способом.
Настоящее изобретение также обеспечивает газ для производства полупроводников, содержащий газообразный тетрафторсилан, полученный описанным выше способом.
Кроме этого, настоящее изобретение предусматривает получение газа для производства элемента солнечной батареи, содержащий тетрафторсилан, полученный описанным выше способом.
Краткое описание чертежей
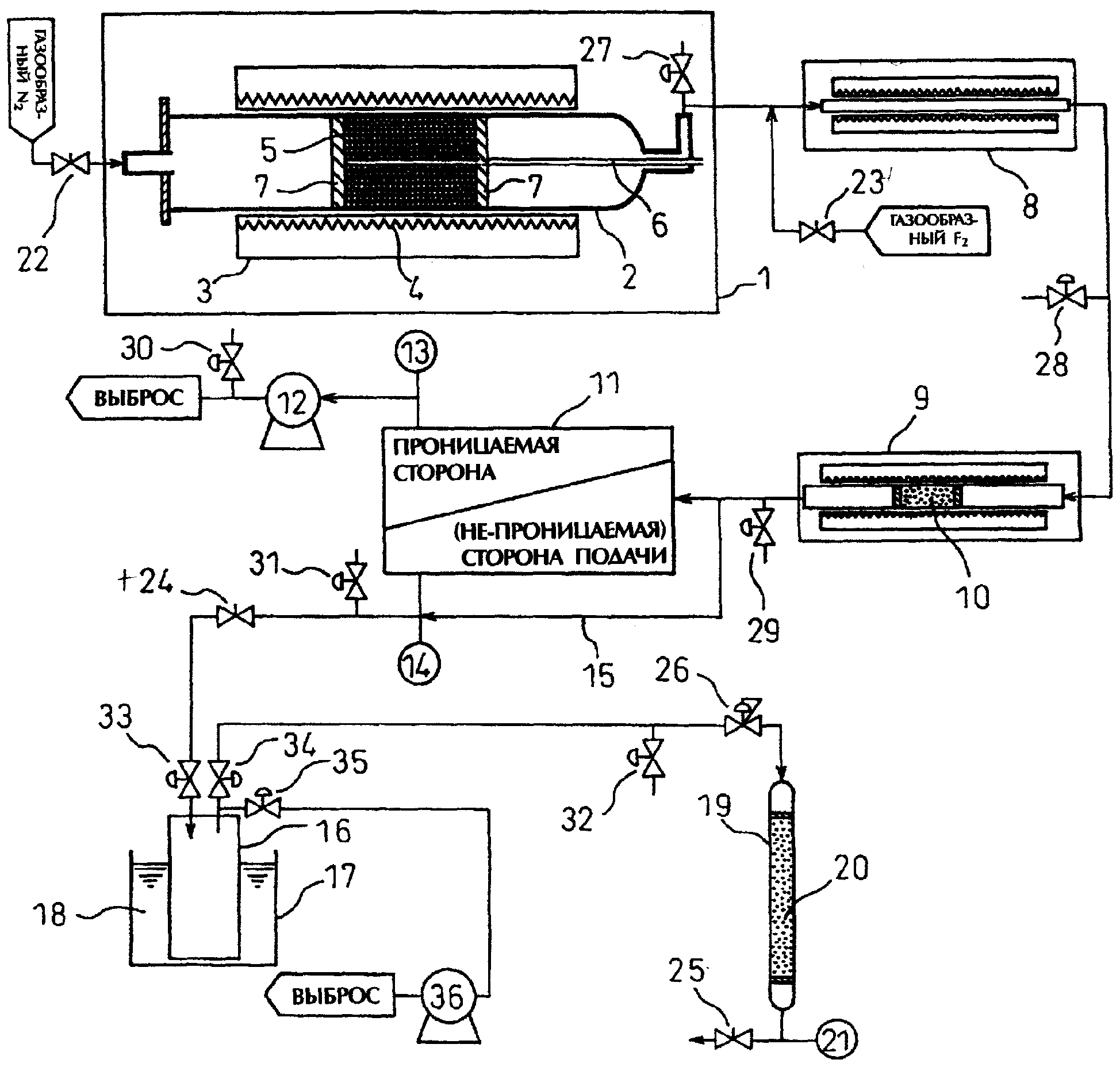
Чертеж изображает схематический вид аппарата, используемого для осуществления способа получения тетрафторсилана согласно настоящему изобретению.
На приведенном чертеже 1 обозначает реактор термического разложения, 2 - трубку для проведения реакции разложения, 3 обозначает электрическую печь, 4 - нагреватель, 5 - гексафторсиликат, 6 - термометр, 7 - фиксирующую пластину из пористого Ni, 8 обозначает реактор для получения F2 (заполненный фторидом многовалентного металла, или без него), 9 - реактор (силикон), 11 - мембранный модуль для разделения газов, 12, 36 - вакуумные насосы, 13, 14, 21 - манометры, 15 - байпас с разделительной мембраной, 16 обозначает резервуар для регенерации, 19 - абсорбционную колонну, 22-25 - вентили для регулировки скорости потока, 26 - регулятор давления, 27-32 - вентили для отбора проб и 33-35 - вентили для регенерационного резервуара.
Описание предпочтительной реализации
Ниже приводится подробное описание настоящего изобретения. Гексафторсиликат является одним из предпочтительных соединений, выбранных из группы, состоящей из гексафторсиликата щелочного металла и гексафторсиликата щелочно-земельного металла. Примерами таких соединений могут служить Li2SiF6, Na2SiF6, K2SiF6, Cs2SiF6, MgSiF6, CaSiF6, SrSiF6 и BaSiF6. Все перечисленные соединения являются недорогими и доступными промышленными продуктами, и в способе настоящего изобретения эти вещества могут использоваться по отдельности или в виде смеси из двух или более перечисленных соединений. Наиболее предпочтительным из перечисленных веществ, что обусловлено его низкой стоимостью и массовым производством, является Na2SiF6 (гексафторсиликат натрия), который получают в качестве побочного продукта в процессе производства фосфорной кислоты.
В случае использования Na2SiF6 (гексафторсиликат натрия), образующегося в процессе производства фосфорной кислоты, Na2SiF6 представляет собой кристаллический порошок с размером частиц от десятков до сотен μм, иногда содержащий 10% мас. воды. Соответственно, в способе настоящего изобретения, предназначенного для получения тетрафторсилана с использованием гексафторсиликата в качестве исходного вещества, последний предпочтительно измельчать и сушить перед проведением стадии (1). В результате измельчения гексафторсиликата площадь поверхности его кристаллов увеличивается, и это способствует сушке кристаллов.
Измельчение кристаллического гексафторсиликата может проводиться с использованием такого устройства, как шаровая мельница, причем кристаллы измельчают до размера 100 μm и менее, предпочтительно 10 μм или менее, более предпочтительно 1 μм или менее. Затем кристаллы сушат пропусканием азота, воздуха и т.п. Температуры начала разложения могут различаться в зависимости от природы гексафторсиликата, и поэтому температуру сушки выбирают из интервала температур ниже температуры начала разложения. Так, например, в случае сушки гексафторсиликата натрия предпочтительная температура сушки кристаллов лежит в интервале от 200°С до менее 400°С, более предпочтительно, от 300°С до менее 400°С.
Цель проведения стадии сушки после измельчения кристаллов заключается в уменьшении количества таких примесей, как HF и (SiF3)2O, образующихся в качестве побочных продуктов в результате реакции SiF4, получаемого на стадии (1), с водой. Так, например, в случае использования натриевой соли в качестве гексафторсиликата SiF4 образуется по следующей ниже реакции (1), и предполагается, что в случае присутствия воды HF и (SiF3)2O образуются по реакции (2):

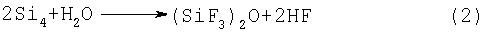
В способе получения тетрафторсилана согласно настоящему изобретению в случае присутствия HF и (SiF3)2O в SiF4 при проведении стадии (1) эти вещества могут перерабатываться на следующей стадии и не возникает каких-либо проблем, однако в результате измельчения и сушки кристаллического гексафторсиликата до проведения стадии (1) количество образующегося (SiF3)2O может быть снижено до 1/3-1/5. Причина, по которой образование (SiF3)2O не может быть исключено полностью, состоит в том, что в кристаллах остаются следовые количества воды и кислородсодержащих производных кремниевой кислоты (например, SiO2). Уменьшение количества образовавшегося количества (SiF3)2O выгодно также и в плане рентабельности процесса, поскольку в этом случае может быть уменьшено количество газообразного фтора (далее в тексте иногда используется символ "F2"), добавляемое, например, на стадии (2). Кроме этого, кристаллический гексафторсиликат предпочтительно сушить при температуре 50-200°С, что способствует последующему измельчению кристаллов.
В способе получения тетрафторсилана согласно настоящему изобретению соответствующие операции сушки и измельчения гексафторсиликата и сушки измельченных кристаллов предпочтительно проводятся до стадии (1), однако в том случае, когда используется гексафторсиликат с низким содержанием воды, указанные операции не проводятся. Вместе с тем, исчерпывающее удаление воды, содержащейся в кристаллах гексафторсиликата, является очень трудной операцией, и температура сушки имеет верхний предел, при превышении которого возникают проблемы, связанные с возможностью образования SiF4. Поэтому весьма трудно полностью предотвратить образование HF и (SiF3)2O, которое связано с наличием в кристаллах воды. Более того, кислородсодержащее производное кремниевой кислоты (например, SiO2) не может быть удалено в результате термообработки, и его наличие приводит к образованию (SiF3)2O.
Стадия (1) представляет собой нагревание гексафторсиликата с образованием SiF4 . Стадию (1) можно проводить в потоке инертного газа, например газообразного азота, или в вакууме. Предпочтительный интервал температур нагревания может быть выбран в соответствие с природой используемого гексафторсиликата. Так, например, в случае использования бариевой соли нагревание, предпочтительно, проводят при температуре в интервале 400-700°С, а в случае использования натриевой соли нагревание проводят при температуре в интервале 500-800°С.
В способе получения тетрафторсилана согласно настоящему изобретению стадию (2-1), стадию (2-2) или стадии (2-1) и (2-3) осуществляют после проведения стадии (1). Стадия (2-1) представляет собой стадию реакции между смешанным газом, содержащим SiF4 и (SiF3)2O, полученным на стадии (1), с газообразным фтором. Предпочтительная температура реакции имеет значение в интервале 100-350°С, более предпочтительно, 200-350°С. Газообразный F2 вступает в реакцию в количестве от 1 до 2 молей в расчете на моль (SiF3)2O, содержащегося в полученном SiF4. Предполагается, что реакция на стадии (2) протекает по следующему уравнению (3), согласно которому (SiF3)2O может превращаться в SiF4 и О2:

Если молярное количество газообразного F2 в 2 раза превышает количество образовавшегося (SiF3)2O, наблюдается эффект насыщения, что нежелательно в плане рентабельности процесса. Принимая во внимание коррозионную стойкость конструкционного материала реактора к воздействию газообразного F2 , предпочтительная температура реакции составляет 350°С или ниже.
Стадия (2-2) представляет собой реакцию газообразного SiF4, содержащего (SiF3)2 O, образовавшийся на стадии (1), с фторидом многовалентного металла. Стадия (2-3) представляет собой реакцию газообразного SiF4, полученного на стадии (2-1), с фторидом многовалентного металла. Реакция, протекающая на стадии (2-2) или (2-3), представляет собой разложение гексафтордисилоксана под действием фторида многовалентного металла с образованием SiF4 и O2 .
Примерами фторидов многовалентных металлов, которые могут использоваться на стадии (2-2) или (2-3), являются CoF3, MnF3, MnF4, AgF2, CeF4, PbF4 и K3NiF7. Перечисленные соединения способны активировать фтор при нагревании, и под действием активированного фтора может происходить разложение гексафтордисилоксана, протекающее по предполагаемым реакциям, представленньм уравнениями (4)-(10):

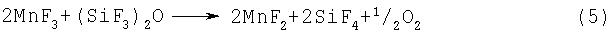
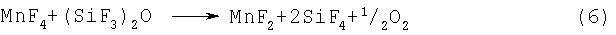
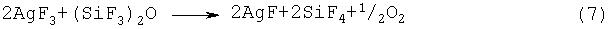
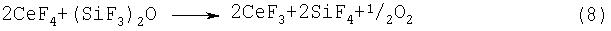


Указанные выше фториды многовалентных металлов могут использоваться по отдельности или в виде смеси.
Ниже описывается способ получения фторида многовалентного металла применительно к случаю, когда фторид многовалентного металла представляет собой CoF3 на носителе. Так, например, Со(NO3)2·6Н2O растворяют в воде, полученный водный раствор абсорбируют на сухом Al2О3 (NST-3, производимый Nikki Kagaku K.K.), и полученный материал сушат на теплой бане до полного удаления воды. После сушки оксидом алюминия заполняют никелевую трубку и проводят прокаливание в токе N2 с целью удаления остаточных количеств воды и азотной кислоты, с получением оксида. После этого, в результате пропускания 10% F2 (разбавленного N2), проводят фторирование Со и оксида алюминия, используемого в качестве носителя.
В случае использования оксидов алюминия, титана, циркония и т.п. в качестве формирующего средства, кислород на поверхности носителя и SiF4 реагируют друг с другом с образованием гексафтордисилоксана, вследствие чего необходимо осуществлять тщательное фторирование перед пропусканием SiF4. Фторирование носителя легко осуществляется в результате пропускания нагретого газообразного фтора или HF. В результате окончательной обработки газообразным фтором перед использованием может быть получен целевой фторид многовалентного металла.
Стадии (2-2) или (2-3) предпочтительно проводят при температуре 50-350°С, более предпочтительно, при 150-350°С. При пропускании нагретого фторида многовалентного металла через смешанный газ, состоящий из гексафтордисилоксана и SiF4, происходит разложение гексафтордисилоксана с образованием SiF4 и О2. В том случае, когда линейная скорость слишком высока, зона проскока (break-through zone) удлиняется и время существования уменьшается, вследствие чего пропускание, предпочтительно, проводят с линейной скоростью 10 м/мин или меньше при обычной температуре и атмосферном давлении.
Если продолжать реакцию на стадии (2-2) или (2-3), фторид металла в высшем валентном состоянии превращается во фторид металла обычной валентности и теряет способность к фторированию, при этом на выходе из реактора детектируется гексафтордисилоксан. В этом случае можно остановить реакцию и провести повторное фторирование низшего фторида газообразньм фтором с образованием фторида металла в высшей валентности, однако с целью осуществления непрерывного режима протекания реакции также можно использовать две или более реакционных колонны, тем самым непрерывно осуществляя реакцию разложения в результате повторения циклов реакции и регенерации. Выбор времени переключения с одного режима на другой может подтверждаться данными анализа на содержание гексафтордисилоксана на выходе из реактора, проводимого методом ИК-спектрометрии с Фурье-преобразованием (FT-IR).
Стадию (2-2) или (2-3) предпочтительно проводить в присутствии газообразного фтора, и в результате пропускания нагретого газообразного фтора реакция может протекать в непрерывном режиме при регенерации фторида многовалентного металла в соответствии со следующими уравнениями (11)-(17):







Тетрафторсилан, содержащий гексафтордисилоксан в качестве примеси, смешивают с газообразным фтором в эквимолярном количестве относительно гексафтордисилоксана и пропускают через фторид многовалентного металла, в результате чего одновременно протекают реакция разложения гексафтордисилоксана под воздействием фторида многовалентного металла и регенерация фторида металла с обычной валентностью под воздействием газообразного фтора. В этом случае объемная скорость составляет 10000 ч-1 или менее, предпочтительно 5000 ч-1 или менее, более предпочтительно 1000 ч-1, при температуре окружающего воздуха и атмосферном давлении. Количество подаваемого газообразного фтора может контролироваться подачей эквимолярного количества газообразного фтора при анализе содержания гексафтордисилоксана на входе в реактор методом FT-IR.
Газообразный тетрафторсилан, полученный на стадиях (2-1), (2-2) или на стадиях (2-1) и (2-3), иногда содержит избыточное количество газообразного фтора. В соответствие со способом получения тетрафторсилана согласно настоящему изобретению, стадии (3) контактирования кремния с газообразным тетрафторсиланом, содержащим газообразный фтор, предпочтительно проводят после стадии (2-1), стадии (2-2) или стадий (2-1) и (2-3).
Стадию (3), на которой осуществляют превращение избытка газообразного фтора в SiF4, предпочтительно проводят при температуре 50°С или выше, более предпочтительно при 100°С или выше, и еще более предпочтительно при 150°С или выше. Используемый на стадии (3) кремний, предпочтительно, представляет собой кремний, поверхностные гидроксильные группы которого подвергают термообработке с использованием такого инертного газа, как азот, при температуре 400°С или выше, предпочтительно при 400-600°С.
На стадии (3) нежелательно использовать SiO2 вместо кремния, поскольку из-за реакции с HF, содержащейся в SiF4, образуется Н2О и (SiF3)2O, в соответствие со следующими схемами реакций (18) и (19):
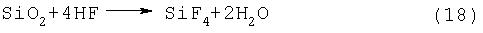
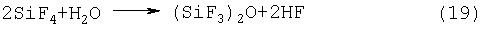
Газообразный F2 вступает в реакцию с поверхности кремния, в связи с чем, несмотря на то что размер частиц кремния и его площадь поверхности не имеют специальных ограничений, чип с размером частиц порядка нескольких мм является предпочтительным с учетом газопроницаемости, контактных характеристик или операций по заполнению. Предпочтительный кремниевый чип имеет чистоту 99,9% мас. или выше, более предпочтительно, 99,999% мас. или выше, и наиболее предпочтительно соответствует сорту полупроводниковых кремниевых плат.
Способ получения тетрафторсилана согласно настоящему изобретению предпочтительно включает стадию (4) контактирования газа, полученного на стадиях (2-1), (2-2), стадиях (2-1) и (2-3), стадиях (2-1) и (3), стадиях (2-2) и (3) или стадиях (2-1), (2-3) и (3), с газоразделительной мембраной и/или углеродными молекулярными ситами.
Газоразделительная мембрана, предпочтительно, представляет собой SiO2-ZrO2 керамическую мембрану и/или поли(4-метилпентен-1)гетерогенизированную мембрану. Углеродное молекулярное сито (молекулярно-ситовой углерод), предпочтительно, имеет размер пор порядка 5Å или менее.
SiF4, полученный по способу настоящего изобретения, может содержать примеси, образовавшиеся на соответствующих, описанных выше стадиях. Примерами примесей могут служить (SiF3)2O, H2, O2, N3 и HF. Кроме этого, могут содержаться такие примеси, как СО и CO2, образование которых связывают с небольшим количеством углерода, присутствующего в исходном гексафторсиликате. Для получения SiF4 высокой чистоты такие примеси предпочтительно отделять путем очистки.
Согласно способу получения тетрафторсилана по настоящему изобретению, SiF4, содержащий, например, такие примеси, как О2, N3, CO, СО2 и HF, приводится в контакт с газоразделительной мембраной и/или углеродным молекулярным ситом для отделения О2, N2, CO, CO2, HF и т.п. от SiF4, в результате чего может быть получен SiF4 высокой чистоты.
Примеры используемых газоразделительных мембран включают газоразделительный мембранный модуль SiO2-ZrO2 (размер модуля: ф50×300L), выпускаемый Kyocera Corporation, и поли(4-метилпентен-1) гетерогенизированную мембрану (размер модуля: ф60×500L), выпускаемый Dai-Nippon Ink&Chemicals, Inc. Такие разделительные мембраны могут использоваться по отдельности или в комбинации.
Газоразделительные мембраны, предназначенные для использования в способе получения тетрафторсилана согласно настоящему изобретению, не ограничиваются указанными выше разделительными мембранами, при условии, что сепарационная мембрана обладает большим коэффициентом проницаемости (разделения) по SiF4 и таким примесям, как О2, N2, CO, CO2 и HF.
Углеродные молекулярные сита не ограничиваются указанным выше примером, при условии, что углеродное молекулярное сито имеет достаточно большой размер пор для адсорбции таких примесей, как O2, N2, СО, СО2 и HF и достаточно малый размер пор для абсорбции SiF4. Размер пор предпочтительно составляет 5Å или менее, поскольку такие поры абсорбируют O2, N2, CO, СО2 и HF и не адсорбируют SiF4.
Ниже описывается способ очистки газообразного SiF4 с использованием газоразделительного мембранного модуля.
Согласно способу очистки газообразного SiF4 с использованием газоразделительного мембранного модуля такой модуль вначале продувают N2 или аналогичным газом с целью удаления H2O, которая реагирует с SiF4. Очистка считается завершенной, когда при подаче газообразного N2 точка росы с проницаемой и непроницаемой стороны мембраны достигает одинакового значения. N2 не является обязательным газом, используемым для очистки, при условии, что точка росы составляет -70°С или менее.
Питающая сторона высушенного газоразделительного мембранного модуля контактирует с SiF4, содержащим такие примеси, как О2, N2, CO, СО2 и HF, и обладает селективной проницаемостью по указанным примесям, тогда как SiF4 концентрируется с непроницаемой стороны, в результате чего может быть получен SiF4 высокой чистоты. Сконцентрированный с непроницаемой стороны SiF4 может дополнительно приводится в контакт с углеродным молекулярным ситом.
Согласно способу отделения SiF4 от О2, N2, CO, СО2, HF и т.д. с использованием газоразделительного мембранного модуля, при значительном перепаде между проницаемой стороной и непроницаемой стороной, высокочистый SiF4 может быть получен с непроницаемой стороны мембраны, в связи с чем с непроницаемой стороны (сторона подачи) мембраны поддерживается давление, равное атмосферному, или более высокое давление. Кроме этого, если это желательно, то давление с проницаемой стороны мембраны может быть понижено до атмосферного давления или более низкого давления.
Ниже описывается способ очистки газообразного SiF4 с использованием углеродного молекулярного сита.
Примером используемого углеродного молекулярного сита (далее в тексте иногда используется сокращение "MSC") может служить MORSIEBON 4A (торговая марка), производимый Takeda Chemical Industries, Ltd. Согласно способу очистки с использованием адсорбента MSC загружают в резервуар и затем подвергают термической обработке в среде такого инертного газа, как N2, при температуре 100-350°С с целью удаления воды, CO2 и других адсорбированных примесей. Термообработка может проводиться в результате очистки N2 в вакууме. Термообработка считается законченной, когда подводимый и отходящий газы достигают одинаковой точки росы. N2 не является обязательным газом для проведения сушки, и может использоваться другой газ, при условии, что точка росы составляет -70°С или менее.
В результате контакта MCS с SiF4, содержащим такие примеси, как О2, N2, CO, СО2 и HF, образовавшиеся в ходе процесса настоящего изобретения, и обеспечивая абсорбцию на MSC только этих примесей, может быть получен SiF4 высокой чистоты.
Адсорбцию на MSC таких газовых примесей, как O2, N2, CO, СО2 и HF, содержащихся в SiF4, предпочтительно осуществляют в соответствие с общим методом очистки адсорбционной сепарацией, поддерживая при адсорбции низкую температуру и повышенное давление. В случае проведения адсорбции при обычных температурах давление поддерживают равным атмосферному или выше, предпочтительно 0,5 МПа или выше, более предпочтительно 1 МПа или выше. В случае абсорбции газообразных примесей при охлаждении предпочтительное давление имеет значение ниже давления сжижения SiF4.
Линейная скорость (LV, м/мин) при атмосферном давлении составляет 5 или менее, предпочтительно 2 или менее, более предпочтительно 1 или менее. Объемная скорость (SV, H-1) составляет 1000 или менее, предпочтительно 500 или менее, более предпочтительно 200 или менее.
При использовании двух абсорбционных колонн SiF4 может подвергаться непрерывной очистке в результате попеременного осуществления адсорбции и регенерации. Регенерацию можно осуществлять путем откачивания в вакууме части нагретого SiF4, находящегося в колонне адсорбционной очистки, и его подачи в колонну регенеративной десорбции, проводя продувку в направлении обратном абсорбционной очистке.
Способ получения тетрафторсилана согласно настоящему изобретению также характеризуется использованием описанного ниже аналитического метода, предназначенного для контроля протекания процесса.
Тетрафторсилан, полученный по способу настоящего изобретения, может представлять собой продукт высокой чистоты с содержанием гексафтордисилоксана в качестве примеси в количестве 1 об.ч/млн или менее. Также может быть получен тетрафторсилан высокой чистоты с содержанием гексафтордисилоксана 0,1 об.ч/млн или менее.
Ниже описывается метод анализа примесей в высокочистом тетрафторсилане настоящего изобретения. Что касается приведенных ниже численных значений, то подразумевается, что они не имеют конкретных ограничений.
Согласно настоящему изобретению, метод анализа примесей в высокочистом тетрафторсилане характеризуется тем, что осуществляют контактирование тетрафторсилана, содержащего в качестве примесей газообразный Н2, газообразный O2, газообразный N2 , газообразный СО, газообразный CH4 и/или газообразный СО2, с адсорбентом, с целью отделения примесей от тетрафторсилана и их введения совместно с газом-носителем в газовой хроматограф для их анализа.
Компоненты, которые могут анализироваться методом настоящего изобретения, представляют собой следовые количества Н2, О2, N2, CO, CH4 и/или СО2. Также могут анализироваться такие компоненты, как F2, HF и (SiF3)2O.
В качестве адсорбента предпочтительно использовать активированный уголь, сферический активированный угол на основе нефтяного пека и/или углеродное молекулярное сито с размером пор порядка 6Å или более.
Согласно методу анализа настоящего изобретения, предколонку (колонка SUS с внутренним диаметром ф 3 мм и длиной 1 м), заполненную SHINCARBON-S (активированный угольный адсорбент, производимый Shimadzu Corporation) с частицами размером 60-100 меш, фиксируют на бане с постоянной температурой и выдерживают при 100°С. В такую предколонку через газовый кран вводят 1 мл SiF4, содержащего такие примеси, как Н2, О2, N2, CO, CH4, CO2, HF и (SiF3)2O. В качестве газа-носителя можно использовать гелий (Не) высокой чистоты.
В образце, переносимом Не высокой чистоты через предколонку, происходит разделение Н2, CO2, N2, СО, СН4 и CO2 и адсорбция SiF4, HF и (SiF3)2O. Такие газовые примеси, как Н2, O2, N2, CO и СН4, могут быть отделены с использованием разделительной колонки, например, с молекулярным ситом 5А (торговое наименование). В случае присутствия CO2 смесь может разделяться с использованием разделительной колонки с POLAPACK Q (торговая марка).
После этого разделенные компоненты вводят в PDD (импульсный разрядный детектор) и измеряют их концентрацию. Предел определения таких газообразных примесей, как Н2, O2, N2, CO, СН4 и CO2, составляет 0,01 об.ч/млн. Согласно методу анализа настоящего изобретения, количественный анализ может быть проведен в интервале концентраций 0,05-0,01 об.ч/млн., что позволяет анализировать SiF4 высокой чистоты.
Использование в методе анализа настоящего изобретения предколонки с упомянутым выше активированным углем обусловлено тем, что его способность к разделению образца на группу компонентов, включающую Н2, O2, N2, CO, CH4 и CO2, и группу, состоящую из основного компонента, SiF4, и примесей, HF и (SiF3)2O, значительно превосходит другие адсорбенты, например силикагель, цеолит и пористые полимерные гранулы. Предпочтительно использовать активированный уголь на основе нефтяного пека, поскольку в этом случае содержание золы (например, К2СО3) очень мало по сравнению с обычным активированным углем, и может быть достигнуто хорошее разделение основного компонента SiF4. Еще более предпочтительным адсорбентом является молекулярно ситовой уголь, на котором может быть достигнуто отличное разделение основного компонента, SiF4, по сравнению с АС и ВАС. Причиной этого, по-видимому, является хороший контроль размера пор и их распределения.
С другой стороны, адсорбированные в предколонке HF, (SiF3)2O и SiF4 могут выделяться и регенерироваться с использованием системы обратной промывки, в которой, с помощью крана, изменяется направление движения потока газа-носителя невысокой чистоты, и предколонка продувается в направлении, противоположном вводу образца. При этом температура печи, в которой находится предколонка, может быть повышена от 100 до 200°С одновременно с переключением крана, что способствует десорбции HF, (SiF3)2O и SiF4. Температура старения предколонки и разделительной колонки может иметь значение, равное обычно используемой максимальной температуре плюс 50°С.
В соответствии с настоящим изобретением метод анализа на содержание примесей в тетрафторсилане высокой чистоты заключается в введении тетрафторсилана, содержащего гексафтордисилоксан в качестве примеси, в оптическую ячейку, окно которой изготовлено из галогенида металла, и анализе гексафтордисилоксана и/или фтористого водорода методом инфракрасной спектрометрии.
В результате использования метода инфракрасной спектрометрии в анализе настоящего изобретения может быть измерена концентрация гексафтордисилоксана, содержащегося в тетрафторсилане, и концентрация фтористого водорода (HF).
При анализе (SiF3)2O, в связи с трудностью получения газообразного стандарта, содержание (SiF3)2O может быть определено, например, из соотношения оптических плотностей {(SiF3)2O/SiF4} по характеристическому ИК-поглощению (SiF3)2O при 838 см-1 и характеристическому ИК-поглощению SiF4 при 2054 см-1. В этом случае известная в технической литературе характеристика (например, Anal. Chem., 57, 104-109 (1985)), может использоваться в качестве стандарта для оптической плотности (SiF3)2O.
SiF4, содержащий (SiF3)2O, вводят в газовую ячейку с большой оптической траекторией, например, порядка 4 м или более и с использованием метода ИК-спектрометрии, определяют содержание (SiF3)2O в SiF4 в количестве до 0,1 ч/млн или менее. В качестве инфракрасного спектрометра предпочтительно использовать спектрометр с Фурье-преобразованием.
Метод анализа согласно настоящему изобретению позволяет определять низкие концентрации (SiF3)2O по поглощению в инфракрасной области спектра при длине волны 838 см-1, характеристичной для (SiF3)2O.
Кроме этого, концентрация (SiF3)2O в SiF4 может быть измерена косвенным методом в результате добавления постоянного, избыточного количества F2 к постоянному количеству SiF4, содержащего (SiF3)2O, проведения реакции между (SiF3)2O и F2 при нагревании до 300°С и определения расхода фтора.
В методе анализа согласно настоящему изобретению измерения, предпочтительно, проводят методом FT-IR, при этом, по меньшей мере, часть линии для отбора образцов, контактирующая с SiF4, изготовлена из нержавеющей стали или электрополированной нержавеющей стали, а конструкционный материал окна газовой ячейки для оптического пропускания представляет собой KCI, AgCI, KBr или CaF2. Аналогично (SiF3)2O, HF может анализироваться с концентрацией 0,1 ч/млн или менее по спектру ИК-поглощения, из оптической плотности при длине волны 4040 см-1, характеристичной для HF.
Ниже описывается применение высокочистого тетрафторсилана, полученного способом настоящего изобретения.
С повышением степени интеграции транзисторов, повлекшей за собой усовершенствование полупроводниковых приборов, стало возможным значительное увеличение интегральной плотности или скорости переключения индивидуальных транзисторов. Однако задержка при распространении, связанная с качеством электропроводки, сводит на нет увеличение скорости работы транзистора. Задержка, связанная с электропроводкой, становится особенно серьезной проблемой при уширении линии до 0,25 μм и более. Для решения такой проблемы алюминиевую проводку заменяют на медную электропроводку с низким сопротивлением и применяют низкодиэлектрическую межслоевую изолирующую пленку с целью уменьшения емкости между проводками. Примером низкодиэлектрического материала для линий шириной 0,25-0,18 или 0,13 μм может служить SiOF (оксидная пленка, допированная фтором, ε: около 3,5), полученная методом CVD в HDP (высокая плотность) плазме. Поскольку в настоящее время продолжается разработка новых способов с использованием SiOF для межслоевой изолирующей пленки и алюминиевых сплавов для электропроводки, высокочистый SiF4 настоящего изобретения может найти применение в качестве допирующего материала.
Стекло для оптических волокон содержит внутреннюю часть и покрытую часть. Внутренняя часть должна иметь более высокий показатель преломления, чем периферическая покрытая часть, с тем чтобы облегчить прохождение света в центральную часть. Увеличение показателя преломления может быть достигнуто путем введения таких легирующих добавок, как Ge, AI, Ti и т.п. Однако при этом следует учитывать побочный эффект увеличения светорассеяния на присадках, вследствие чего происходит снижение эффективности светопропускания. При добавлении фтора в покрытие величина показателя преломления может уменьшаться до значений ниже показателя преломления чистого кварца, и, вследствие этого, чистый кварц или кварц с пониженным содержанием легирующих добавок может использоваться в центральной части с целью повышения эффективности светопропускания. Фтор в атмосфере Не добавляют в ходе термообработки мелкозернистого стеклянного материала (SiO2) в атмосфере SiF4, и, таким образом, высокочистый SiF4 настоящего изобретения может использоваться в качестве газа, предназначенного для получения оптического волокна.
Настоящее изобретение дополнительно иллюстрируется приведенными ниже примерами, не ограничивающими область изобретения.
Пример 1.
Гексафторсиликат натрия (Na2SiF6) 5 со средним размером частиц около 70 μм, чистотой 89% мас. или более (содержание воды: 10% мас. или менее), полученный в качестве побочного продукта в процессе производства фосфорной кислоты, высушивали горячим воздухом при 120°С и 1500 г полученного материала загружали в центральную часть трубки 2 для проведения реакции разложения (внутренний диаметр: 90 мм, длина: 1500 мм, конструкционный материал: никель) в реакторе 1 термического разложения, показанном на чертеже, и оба конца трубки закрывали пластиной 7 из пористого Ni. Затем при поддерживании температуры Na2SiF6 ниже 400°С с помощью электропечи 3 (длина: 1000 мм), открывая клапан 22, пропускали газообразный N2 (точка росы: -70°С или менее) со скоростью 1000 мл/мин, и после подтверждения того, что концентрация HF в отходящем газе снизилась до 1 ч/млн или менее, сушку Na2SiF6 завершали.
После этого скорость потока N2 устанавливали равной 200 мл/мин и температуру электропечи 3 повышали до 700°С и поддерживали на этом значении. В результате образовывался SiF4 с концентрацией около 30% об. Образцы полученного газа отбирали с помощью вентиля 27 и определяли концентрации газовых примесей. Полученные результаты представлены в Таблице 1. Из представленных результатов можно видеть, что в продукте содержится 8560 ч/млн (SiF3)2O.
Пример 2
Газообразный SiF4 получали по методике, описанной в примере 1, за исключением того, что сухие кристаллы гексафторсиликата натрия, используемые в Примере 1, истирали с помощью измельчителя, и полученным порошком с размером частиц около 1 μм заполняли трубку 2 для проведения реакции разложения. Образцы образовавшегося газа отбирали с помощью вентиля 27 и определяли концентрации газовых примесей. Полученные результаты представлены в Таблице 1. Из полученных результатов следует, что проведение измельчения приводит к уменьшению концентрации (SiF3)2O.
Пример 3
Газообразный SiF4, полученный в Примере 2, вводили в реактор 8 для обработки F2 (конструкционный материал реакторной трубки: никель, внутренний диаметр: 8 мм, длина: 1000 мм), показанный на чертеже, через вентиль 23 вводили 3 мл 100% газообразного фтора, и при 300°С протекала реакция между (SiF3)2O, содержащимся в SiF4, и F2. Образец образовавшегося газа отбирали через вентиль 28 и анализировали. Полученные результаты представлены в Таблице 1. Из полученных результатов можно видеть, что концентрация (SiF3)2O уменьшается до значения менее 0,1 об.ч/млн.
Пример 4
В реакционную трубку (конструкционный материал: никель) реактора 9, показанного на чертеже, загружали 60 мл кремниевой крошки размером 8-10 меш, которую обрабатывали в течение 3 часов при 500°С, пропуская N2 (точка росы: -70°С или менее) со скоростью 300 мл/мин. Затем температуру реакционной трубки, заполненной кремниевой крошкой, устанавливали равной 150°С и вводили газ, полученный в Примере 3, с целью его реакции с избытком F2 и кремнием. Образцы образовавшегося газа отбирали через вентиль 29 и анализировали. Полученные результаты представлены в Таблице 1. Из представленных данных видно, что концентрация газообразного фтора уменьшена до значения менее 0,1 об.ч/млн.
Пример 5
Через газоразделительный мембранный модуль 11 (SiO2-ZrO2 мембрана, производимая Kyocera Corporation), показанный на чертеже, со скоростью 2-3 л/мин пропускали N2 (точка росы: -70°С или менее) и проводили его осушку до достижения одинаковой точки росы на входе и выходе из модуля. Со стороны подачи в сухом газоразделительном мембранном модуле 11, при атмосферном давлении, вводили газ, полученный в Примере 4, и в результате сброса давления с газопроницаемой стороны с помощью вакуумного насоса 11 такие газовые примеси, как N2, отделялись с проницаемой стороны. Образцы газа с проницаемой стороны отбирали через вентиль 30, а газ с непроницаемой стороны мембраны отбирали через вентиль 31. Полученные образцы анализировали. Результаты анализа представлены в Таблице 1. Из полученных результатов можно видеть, что большая часть газовых примесей может удаляться с проницаемой стороны мембраны.
SiF4 с непроницаемой стороны мембраны вымораживали в регенерационный резервуар 16, охлажденный до температуры -120°С с помощью жидкого N2, через вентили 24 и 33, регулируя при этом давление.
Пример 6
В абсорбционную колонну 19 (внутренний диаметр: 16 мм, длина: 1000 мм), показанную на чертеже, загружали 100 мл MSC (MORSIEBON 4A, производимый Takeda Chemical Industries, Ltd.) и систему сушили, пропуская N2 (точка росы: -70°С или менее) при 500°С со скоростью 300 мл/мин до достижения одинаковой точки росы на входе и выходе из системы. После охлаждения и продувки Не SiF4, регенерированный в регенерационном резервуаре 16 в Примере 5, переводили в газообразное состояние при обычной температуре и вводили в абсорбционную колонну 19. В ходе этой операции устанавливали давление 0,9 МПа, а объемную скорость газообразного SiF4 устанавливали равной 350 мл/мин с использованием вентиля 25 для регулирования скорости потока, регулятора давления 26 и манометра 21. Газ, выходящий из абсорбционной колонны 19, анализировали на присутствие газообразных примесей, используя описанную выше методику; полученные результаты представлены в Таблице 1. Из приведенных результатов можно видеть, что измеренные концентрации всех примесей составляли менее 0,1 об.ч/млн.
Пример 7
Газ, полученный в Примере 4, вымораживали в регенерационный резервуар 16, охлажденный до -120°С с помощью жидкого азота, через вентили 24 и 33, регулируя давление с использованием байпасной линии 15 разделительной мембраны, изображенной на чертеже. Затем, в результате газификации при обычной температуре, SiF4, собранный в регенерационном резервуаре 16, вводили в абсорбционную колонну 19, проводя обработку тем же способом и при тех же условиях, что описаны в Примере 6. Газ на входе в абсорбционную колонну 19 и газ, отходящий из абсорбционной колонны 19, анализировали, полученные при этом результаты представлены в Таблице 1. Из этих результатов видно, что измеренные концентрации всех примесей на выходе из реактора составляли менее 0,1 об.ч/млн.
Пример 8
Приготовление фторида многовалентного металла, нанесенного на подложку: 10% CoF3/Al2О3
В 200 мл воды растворяли 26,4 г (0,0091 моля) Со(NO3)2·6Н2O [квалификация: особочистый рагент]. Полученный водный раствор абсорбировали на 100,2 г сухого Al2О3 (NST-3, производимый Nikki Kagaku K.K.) и полученный материал сушили на теплой бане до нулевого содержания воды. После сушки оксид алюминия загружали в реакционную трубку (конструкционный материал: никель) реактора 8, показанного на чертеже, и прокаливали в течение 12 часов при 400 С в токе N3 (400 мл/мин), в результате чего удаляли остаточные количества воды и азотной кислоты с получением оксида Со. После этого при 250°С, со скоростью 1000 мл/мин, пропускали 10% газообразный F2 (разбавление N2) для осуществления фторирования оксида алюминия и Со. Фторирование проводили до одинаковой концентрации фтора на входе и на выходе из реактора. Концентрацию фтора измеряли путем пропускания газа, подлежащего анализу, через 5% водный раствор KI и титрования выделившегося иода 0,1N водным раствором Na2S2O3.
Используя 100 мл полученного выше фторида многовалентного металла, осуществляли разложение гексафтордисилоксана. Газообразный SiF4, полученный в Примере 2, вводили в реактор 8 (конструкционный материал реакционной трубки: никель, внутренний диаметр: 8 мм, длина: 1000 мм), показанный на чертеже, и (SiF3)2O, содержащийся в SiF4, вступал в реакцию с фторидом многовалентного металла (CoF3) при 200°С. Образцы образовавшегося газа отбирали через вентиль 28 и анализировали. Полученные результаты представлены в Таблице 1. Из представленных результатов можно видеть, что количество (SiF3)2 O понижено до значения менее 0,1 об.ч/млн. Анализ на выходе из реактора продолжали и определяли содержание (SiF3)2O.
Пример 9
Использовали 100 мл фторида многовалентного металла, полученного в Примере 8. Газообразный SiF4, полученный в Примере 1, вводили в реактор 8 (конструкционный материал реакционной трубки: никель, внутренний диаметр: 8 мм, длина: 1000 мм), показанный на чертеже, через вентиль 23 вводили 2,3 мл 100% газообразного фтора и в результате реакции между (SiF3)2O, содержащимся в SiF4, и фторидом многовалентного металла при 250°С, происходила его регенерация под действием F2. Образцы полученного газа отбирали через вентиль 28 и анализировали. Полученные результаты представлены в Таблице 1. Из этих результатов видно, что концентрация (SiF3)2O снижена, примерно, до 500 об.ч/млн. Анализ газа на выходе из реактора продолжали, однако газообразный фтор обнаружен не был, а концентрация (SiF3)2O не менялась.
Промышленная применимость
В соответствие с приведенной выше информацией согласно настоящему изобретению может быть получен SiF4, не содержащий (SiF3)2O. Кроме этого, примесные компоненты могут анализироваться в количествах до 0,1 ч/млн или менее, и может быть получен высокочистый SiF4, требующийся для производства компонентов электронной техники, в частности для производства элемента солнечной батареи, полупроводников и оптического волокна.
Реферат
Изобретение относится к химической технологии, а именно к способу получения тетрафторсилана и газу на его основе. Способ получения тетрафторсилана включает стадию (1) нагревания гексафторсиликата, стадию (2-1) реакции газообразного тетрафторсилана, содержащего гексафтордисилоксан, образовавшийся на стадии (1) с газообразным фтором. Далее следует стадия (2-2) реакции газообразного тетрафторсилана, содержащего гексафтордисилоксан, образовавшийся на стадии (1) с фтористым соединением многовалентного металла. В качестве альтернативы стадии (2-2) присутствует стадия (2-1) реакции газообразного тетрафторсилана, содержащего гексафтордисилоксан, образовавшийся на стадии (1) с газообразным фтором. Потом следует стадия (2-3) реакции газообразного тетрафторсилана, полученного на стадии (2-1) с фтористым соединением многовалентного металла. По вышеуказанному способу получают высокочистый тетрафторсилан, содержащий гексафтордисилоксан в количестве 0,1 об.ч/млн или менее. Газообразный тетрафторсилан, полученный вышеуказанным способом, используют при производстве оптического волокна, полупроводников, элементов солнечной батареи. Технический результат состоит в получении тетрафторсилана с пониженным содержанием примесей. 5 н. и 19 з.п. ф-лы, 1 табл., 1 ил.
Комментарии