Трансгенные животные, имеющие основные расстройства, связанные с болезнью альцгеймера - RU2373707C2
Код документа: RU2373707C2
Чертежи
























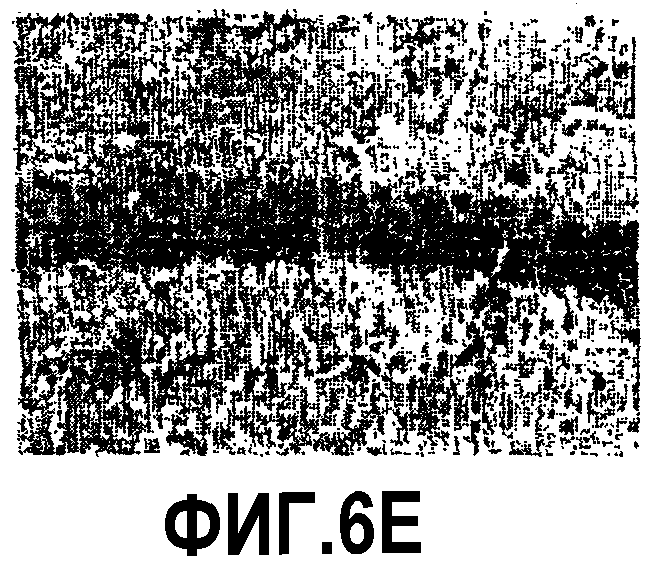



















Описание
Настоящая заявка относится к трансгенным животным моделям болезни Альцгеймера (AD). Ее объектом является, кроме того, применение этих животных.
Болезнь Альцгеймера представляет собой прогрессивное нейродегенеративное заболевание, которое затрагивает широкую пропорцию населения в возрасте. Это заболевание характеризуется в клиническом плане потерей памяти и упадком когнитивных функций, и в невропатологическом плане выраженной гибелью нейронов и присутствием в мозге внутриклеточных нейрофибриллярных отложений и внеклеточных отложений β-амилоидного пептида (Aβ), образующего амилоидные бляшки.
Амилоидные бляшки состоят в большинстве своем из пептидов Aβ из 40 или 42 остатков, которые образуются в ходе протеолиза предшественника пептида Aβ (APP). Внеклеточные отложения Aβ очень характерны для расстройств, связанных с болезнью Альцгеймера. Они представляют раннюю и неизменяемую характеристику всех форм болезни Альцгеймера, включая семейные формы (FAD). Семейные формы проявляются относительно рано (между 30 и 60 годами) и вызваны мутациями в гене APP в 5% случаев FAD с восьмью идентифицированными простыми или двойными миссенс-мутациями, в гене пресенилина 1 (PS1) в 50-70% случаев FAD с более чем 100 различными мутациями, идентифицированными до настоящего времени, и в гене пресенилина 2 в более редких случаях FAD с двумя описанными миссенс-мутациями. Показано, что мутации в этих трех генах индуцировали изменения в протеолизе APP, которые ведут к повышенному продуцированию Aβ, особенно длинной формы Aβ42, и к раннему появлению патологии и симптомов, подобных симптомам спорадических форм болезни Альцгеймера.
Животные модели, предназначенные для воспроизводства некоторых характеристик патологии болезни Альцгеймера, были уже описаны в литературе.
Речь идет, с одной стороны, о трансгенных мышах, несущих мутации в гене APP. У них развиваются патологии, подобные болезни Альцгеймера, начиная с одного года. Так, у мыши PDAPP, суперэкспрессирующей человеческий APP, несущий мутацию V717F, развиваются с возрастом отложения Aβ в мозге, но не наблюдается гибели нейронов вне расположения самих бляшек (Irizarry et al., 1997, J. Neurosc. 17(18): 7053-7059). Это явление можно назвать «эффект бляшки».
Также, мышь Tg(HuAPP695. K670N-M671L)2576, экспрессирующая человеческую изоформу APP K670N-M671L (APPSw для мутации типа Swedish), демонстрирует отложения амилоидного типа, но не обнаруживает гибели нейронов (Irizarry et al., 1997, J. Neuropathol. Exp. Neurol 56:695-973).
В исследовании Calhoun et al. (1998, Nature 395:755-756) гибель нейронов была показана в некоторых областях мозга вблизи от амилоидных бляшек у трансгенных мышей APP23 в возрасте 14-18 месяцев, экспрессирующих мутированную изоформу человеческого APP. Это наблюдение является противоречивым, так как гибель нейронов выражена слабо и происходит у животных в относительно большом возрасте и главным образом вблизи от бляшек, что могло бы соответствовать обнаруженному раньше «эффекту бляшки». Кроме того, оно почти или совсем не упоминается в недавнем комментарии, который подчеркивает, что настоящие животные модели не представляют полного подобия всем известным характеристикам патологии болезни Альцгеймера, в том числе гибели нейронов (Trojanowski, 2002, Am. J. Pathol. 160: 409-411).
С другой стороны, известно о трансгенных мышах, несущих мутации в гене PS1. У них, по-видимому, не развиваются патологии типа болезни Альцгеймера, но имеется высокое количество пептида Aβ42 (увеличение фактора 2 по отношению к PS1 дикого типа), который признан как высоко патогенным.
Кроме того, в описанных моделях трансгенных животных, несущих мутации FAD P264L или M146L в гене PS1 мыши («knock-in»), мутантный белок PS1 не экспрессируется устойчивым образом (Siman et al., 2000, J. Neurosci., 20: 8717-8726; Flood et al., 2002, Neurobiol. Aging 23: 335-348; Rozmahel et al., 2002, 23: 187-194). Эти мыши представляют также высокое количество пептида Aβ42.
В заявке WO 02/0008407 описаны такие трансгенные мыши, у которых ген, кодирующий пресенилин 1, был мутирован введением мутации P264L.
Ввиду роли белка PS1 в образовании форм Aβ42, были также получены двойные трансгенные, несущие мутации в генах APP и PS1. Как и простые трансгенные мыши, описанные выше, эти мыши демонстрируют отложения Aβ, но не демонстрируют гибели нейронов (Takeuchi et al., 2000, Am. J.Pathol. 157: 331-339).
Таким образом, существующие животные модели болезни Альцгеймера неудовлетворительны, так как с их помощью нельзя смоделировать гибель нейронов, являющуюся одной из основных характеристик нейродегенеративных заболеваний, в том числе болезни Альцгеймера.
Заявитель стремился таким образом получить животных, представляющих главные характеристики болезни Альцгеймера, в том числе гибель нейронов.
Он показал, что было возможно получать таких животных, вводя специфические мутации в ген, кодирующий белок PS1 у мышей, и скрещивая их с мышами, осуществляющими суперэкспрессию человеческого гена APP.
Первый аспект изобретения касается, таким образом, не являющегося человеком животного, имеющего, предпочтительно, в своем геноме по меньшей мере одну последовательность нуклеиновой кислоты, кодирующую пресенилин 1, имеющий по меньшей мере одну из двух мутаций, соответствующих мутациям M233T и L235P в белке PS1 мыши.
Предпочтительно, такое животное несет обе мутации.
Предпочтительно, белок PS1, несущий мутации M233T и L235P, происходит от мыши.
Особенно предпочтительно, мутантный белок пресенилин 1 является эндогенным.
Таким образом, животное согласно настоящему изобретению продуцирует предпочтительно белок, включающий последовательность SEQ ID NO:2. Предпочтительно, оно продуцирует белок, представляющий последовательность SEQ ID NO:3. Он предпочтительно заключает в своем геноме последовательность нуклеиновых кислот SEQ ID NO:1 или последовательность SEQ ID NO:8.
Последовательности SEQ ID NO:1, SEQ ID NO:2 и SEQ ID NO:8 следуют соответственно из мутаций, введенных в последовательности дикого типа SEQ ID NO:4, SEQ ID NO:5 и SEQ ID NO:9. Последовательность SEQ ID NO:5 - последовательность остатков 229-237 мышиного белка пресенилина 1 дикого типа. Последовательность SEQ ID NO:9 является последовательностью экзона 7 дикого типа гена мыши, кодирующего белок пресенилин 1, т.е. не мутантного.
Предпочтительно, животное согласно настоящему изобретению совместно экспрессирует APP, предпочтительно, человеческий APP. Такой ген может включать одну или несколько мутаций FAD. Так, мутации в гене APP могут быть одной из различных мутаций, описанных до настоящего времени в литературе. Мутации в гене APP могут быть выбраны среди мутаций "Swedish" (S), "London" (L) и "Dutch" (D), по отдельности или в сочетании.
Эти мутации хорошо описаны в литературе и характеризуются обобщенно следующими изменениями:
Также под мутацией London понимают все замены, кроме замены на изолейцин, которые расположены в положении 717 по отношению к APP770, такие как, например, мутации V717G и V717F.
Конечно, APP, который может быть использован в рамках изобретения, может быть в различных изоформах и, в частности, в формах 695, 751 и 770 или в усеченной форме, такой как, например, изоформа APP99, исключая мутацию Swedish для этой изоформы.
Предпочтительно, указанное животное включает, кроме того, предпочтительно в своем геноме последовательность нуклеиновых кислот, кодирующую весь или часть гена, кодирующего APP751. Предпочтительно, белок APP751 имеет человеческое происхождение. Он представляет предпочтительно мутации K670N и M671L (Swedich) и V717I (London).
В рамках настоящего изобретения ген APP предпочтительно находится под контролем последовательностей, обеспечивающих его сильную экспрессию в нейронах и в особенности последовательностей промоторов транскрипции, таких как экзогенный промотор. В качестве промотирующих последовательностей можно, в частности, назвать промотор HMG (Gautier et al. (1989), Nucleic Acids Res 17:20, 8389.), а также промотор PDGF (Sasahara et al. (1991), Cell 64, 217-27), промотор Thy-1 (Luthi et al. (1997, J. Neurosci 17, 4688-99) и промотор гена Приона (Scott et al. (1992), Protein Sci 1, 986-97).
Согласно особенно предпочтительному варианту осуществления изобретения животная модель включает ген APP, имеющий мутации S, D и/или L, под контролем промотора Thyl.
Таким образом, животное согласно настоящему изобретению продуцирует предпочтительно белок, включающий последовательность SEQ ID NO:7. Оно может содержать последовательность нуклеиновых кислот SEQ ID NO:6.
Предпочтительно, речь идет о трансгенной мыши, происходящей из скрещивания между трансгенной мышью ThyAPP (TG53), несущей последовательность нуклеиновой кислоты, кодирующую человеческий белок APP751SL, и трансгенной мышью, несущей последовательность нуклеиновой кислоты, кодирующую белок PS1 мыши, несущий мутации M233T и L235P.
Животные согласно настоящему изобретению воспроизводят впервые одну из наиболее важных характеристик нейродегенеративных заболеваний, которой является ранняя гибель нейронов.
Они показывают к тому же другие характеристики, традиционно описываемые для этих патологий. Животные демонстрируют ускоренное отложение амилоидных бляшек, ясно видимое с возраста 2 месяцев и особенно с 6 месяцев.
Они также демонстрируют отношение форм Aβ42 к общему Aβ, Аβ42/Aβ более приблизительно 0,9, и это наблюдается с 2,5 месяцев. Такое отношение является очень высоким по сравнению с описанным в литературе для других трансгенных мышей.
Уже видимая у мышей в возрасте 6 месяцев гибель нейронов становится ярко выраженной в 10 месяцев.
PKR (Double strand RNA-dependent Protein Kinase) представляет собой киназу, активируемую стрессом и фосфорилирующую IF2, участвующую в апоптозе.
PKR обнаруживается в гипокампе (структура, где имеет место гибель нейронов) мыши APPxPS1KI согласно изобретению в возрасте 10 месяцев. Она не обнаруживается в гипокампе трансгенных мышей APPxPS1M146L в возрасте 12 месяцев, у которых к тому же не наблюдается гибели нейронов.
Новые характеристики животных согласно настоящему изобретению делают из них исследовательские модели, более полные и типичные для расстройств, наблюдаемых у пациентов, пораженных болезнью Альцгеймера, чем описанные ранее. Эти животные особенно пригодны, таким образом, для обнаружения нейропротективных свойств соединений, предназначенных для лечения нейродегенеративных заболеваний, предпочтительно болезни Альцгеймера.
Предпочтительно, животные согласно настоящему изобретению имеют мутантные аллели ps1 в гомозиготном состоянии и APP - в гетерозиготном состоянии. Между тем, те же характеристики указанного животного могут быть описаны у животного, имеющего один мутантный аллель из двух аллелей ps1 в гетерозиготном состоянии и APP - в гетерозиготном состоянии, при, однако, менее выраженном или проявляющимся позднее фенотипе.
Другое преимущество животных согласно настоящему изобретению заключается в том, что количество мутантного белка PS1, экспрессируемого этой трансгенной мышью, эквивалентно количеству эндогенного белка PS1, в норме экспрессируемого нормальной (не трансгенной) мышью, экспрессирующей немутантный PS1. Эта характеристика делает из них выгодную исследовательскую модель - без суперэкспрессии белка PS1 - для выявления соединений, предназначенных для лечения нейродегенеративных заболеваний.
Эти соединения могут представлять собой, в частности, соединения, действующие на регуляцию гена PS1 на транскрипционном, посттранкрипционном, трансляционном, посттрансляционном уровне или на сам белок PS1, изменяя или регулируя одно или несколько из его свойств, или действуя подобным образом на партнеров взаимодействия, или на мишени белка PS1, или соединения, действующие на регуляцию APP и, более широко, любые молекулы выше сигналов, инициируемых PS1 и APP в ходе нейродегенеративного процесса.
В рамках настоящего изобретения животные предпочтительно являются млекопитающими, такими как грызуны. В частности, это может быть мышь, крыса или кролик.
Мыши и конструкции, обеспечивающие их получение, получены методами, известными специалисту.
Они могут быть получены согласно классическим трансгенным методам. В качестве примера, иллюстрируя один из способов трансгенеза, можно назвать способ электропорации генной конструкции, содержащей модифицированные гены в эмбриональных стволовых клетках мыши, и, после отбора, переноса клеток, несущих требуемое генетическое событие в бластоцисту-реципиент, такой как описывается в примерах. В этом отношении животные, мутантные по PS1, согласно изобретению получены электропорацией кассеты экспрессии, включающей нуклеиновую кислоту. Предпочтительно, эта нуклеиновая кислота представляет собой ДНК, которая может быть геномной ДНК (гДНК) или комплементарной ДНК (кДНК).
Модификация генома может следовать из изменения или модификации одного или некоторых генов "knock-in". Эта модификация может быть вызвана действием классических изменяющих или мутагенных агентов или может быть осуществлена направленным мутагенезом. В настоящем изобретении в том, что касается мутантного гена ps1, речь идет предпочтительно о гомологичной рекомбинации с помощью нацеливающего вектора, несущего предварительно мутированный с помощью направленного мутагенеза трансген, как описано в нижеследующих примерах.
Животные, экспрессирующие мутантный белок APP, получены микроинъекцией генетической конструкции в ядро зиготы.
Двойные трансгенные животные получают скрещиванием животных, мутантных по ps1, и животных, мутантных по APP.
Животные согласно настоящему изобретению могут быть использованы предпочтительно для обнаружения нейропротективных свойств соединений, предназначенных для лечения нейродегенеративных заболеваний, и преимущественно болезни Альцгеймера. Эти соединения могут быть химическими молекулами, пептидными или белковыми молекулами, антителами, химерными молекулами, а также антисмысловыми РНК или рибозимами. Выявленные соединения могут быть использованы в качестве лекарственных средств, тель-кель или в комбинации с фармацевтически приемлемым носителем для получения фармацевтической композиции. Ими могут быть, в частности, соляные растворы (фосфат мононатрия, динатрия, хлорид натрия, калия, кальция или магния и т.д., или смеси таких солей), стерильные, изотонические или сухие композиции, в частности, лиофилизованные, которые добавлением, смотря по обстоятельствам, стерилизуемой воды или физиологической сыворотки позволяют получить растворы для инъекций. Инъекции могут быть осуществлены стереотаксическим, топическим, пероральным, парентеральным, интраназальным, внутривенным, внутримышечным, подкожным, внутриглазным, чреcкожным и т.д. путем.
Другой объект изобретения касается таким образом способа выявления соединений, предназначенных для лечения нейродегенеративных заболеваний, включающего по меньшей мере следующие стадии:
- введение тестируемого соединения или смеси соединений животным согласно настоящему изобретению, и
- наблюдение эволюции одного или нескольких характерных маркеров, воспроизводящих нейропатологию, наблюдаемую у человека.
Другой объект изобретения касается способа выявления соединений, предназначенных для лечения нейродегенеративных заболеваний, включающего по меньшей мере следующие стадии:
- приведение в контакт клеток, полученных из животных согласно настоящему изобретению, с соединением или смесью соединений, и
- измерение эффекта или эффектов соединений на уровне целых клеток, в клеточных гомогенизатах или на субклеточные фракции.
Другой объект изобретения касается любого биологического продукта, происходящего из одного, из двух животных по изобретению, а также их применения для выявления соединений, предназначенных для лечения нейродегенеративных заболеваний, предпочтительно, болезни Альцгеймера. Под «биологическим продуктом» понимают, в частности, клетки, белковые экстракты, ДНК, РНК или антитела.
Таким образом, объектом настоящего изобретения являются клетки или линии клеток, происходящие от животного, такого как описано выше, в частности, эмбриональные стволовые клетки.
Объектом настоящего изобретения, кроме того, является белок PS1 мыши, несущий мутации аминокислот M в T и L в P соответственно в положениях 233 и 235. Предпочтительно, такой белок включает последовательность SEQ ID NO:2. Предпочтительно, он имеет последовательность SEQ ID NO:3.
Другой объект настоящего изобретения - нуклеиновая кислота, кодирующая белок PS1 мыши, несущий мутации аминокислот M в T и L в P соответственно в положениях 233 и 235.
Предпочтительно, такая нуклеиновая кислота согласно формуле изобретения включает последовательность SEQ ID NO:1 или последовательность SEQ ID NO:8.
Объектом настоящего изобретения, кроме того, являются последовательности, комплементарные этим нуклеиновым кислотам и векторам, включающим эти нуклеиновые кислоты или комплементарные им последовательности.
Другой аспект изобретения касается применения этих белков для выявления нейропротективных свойств соединений, предназначенных для лечения нейродегенеративных заболеваний.
Настоящее изобретение иллюстрируется следующими примерами, однако оно не ограничено только этими примерами.
В этих примерах описанные результаты доказывают преимущество мышей PS1KI и подтверждают предпочтительное использование модели PS1KIxAPP в терапевтических стратегиях, так как ее преимущество заключается в том, что она способна вопроизводить главные характеристики известных на сегодняшний день нейродегенеративных заболеваний.
Описание фигур
Фиг.1А: Схематическое представление структуры гена ps1 мыши и основных сайтов рестрикции вокруг экзона 7 дикого типа (верхняя линия) и использованного вектора нацеливания (средняя линия). Замены нуклеотидных оснований для получения мутаций кодонов М233Т и L235P в экзоне 7 (*) представлены в пунктирной рамке. Мутантный аллель PS1KI, содержащий кассету резистентности к неомицину (Neo), изображен на нижней линии. Также указано положение зонда 230 пар оснований, использованного для идентификации новорожденных.
Фиг.1B: Саузерн-блоттинг с использованием зонда 230 пар оснований, чтобы отличать аллели WT дикого типа (полоса 9,2 пар оснований) от гетерозиготных PS1KI (He, двойная полоса) и гомозиготных (Нo, полоса 7,4 пар оснований) у различных мышей.
Фиг.1C: Иммуноперенос С-концевого фрагмента PS1, показывающий, что уровни экспрессии белка PS1 не ухудшены присутствием мутаций в аллеле PS1KI.
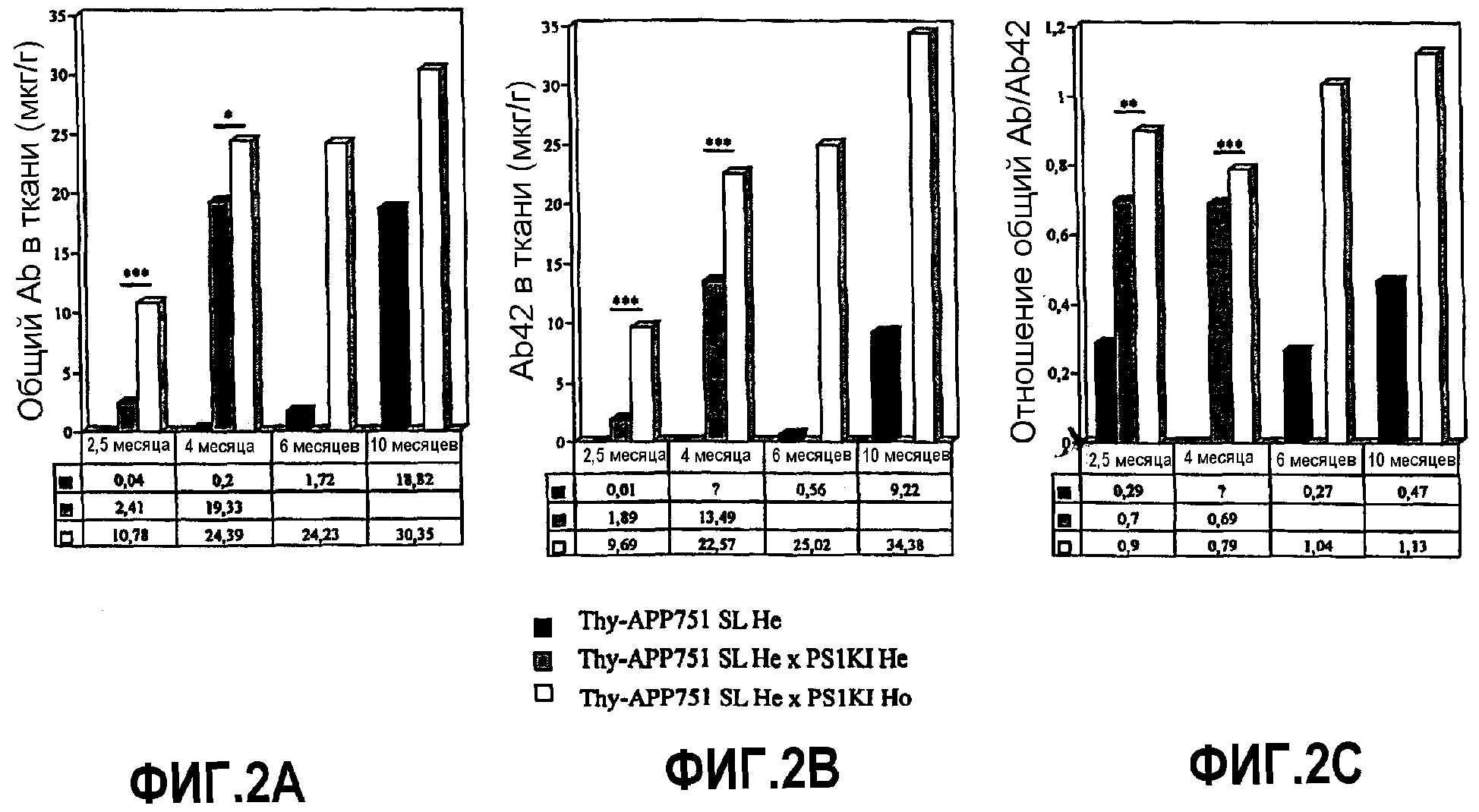
Фиг.2А, 2B и 2C: Определение количества соответственно общего Aβ42 и отношения общий Aβ/Aβ42, в возрасте 2,5, 4, 6 и 10 месяцев.
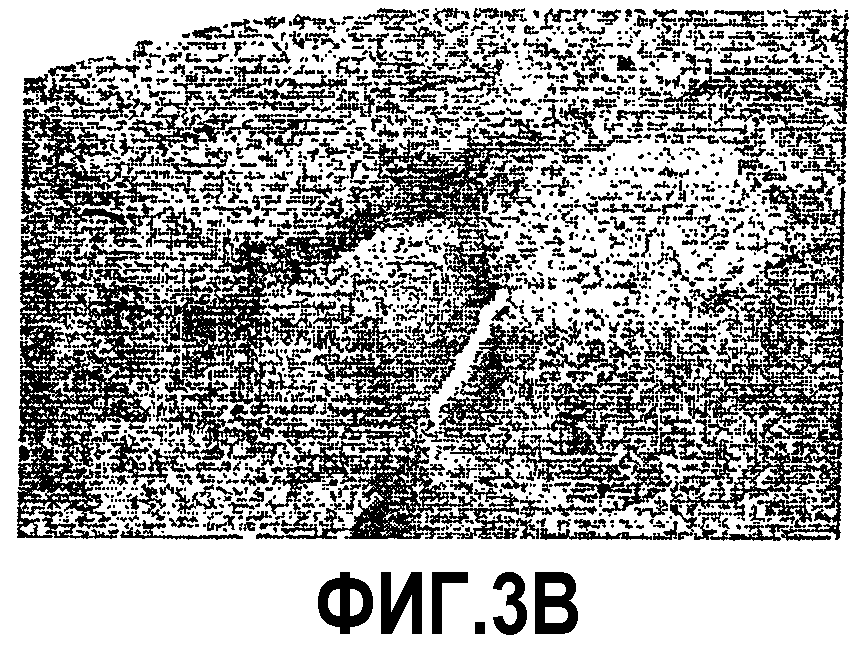
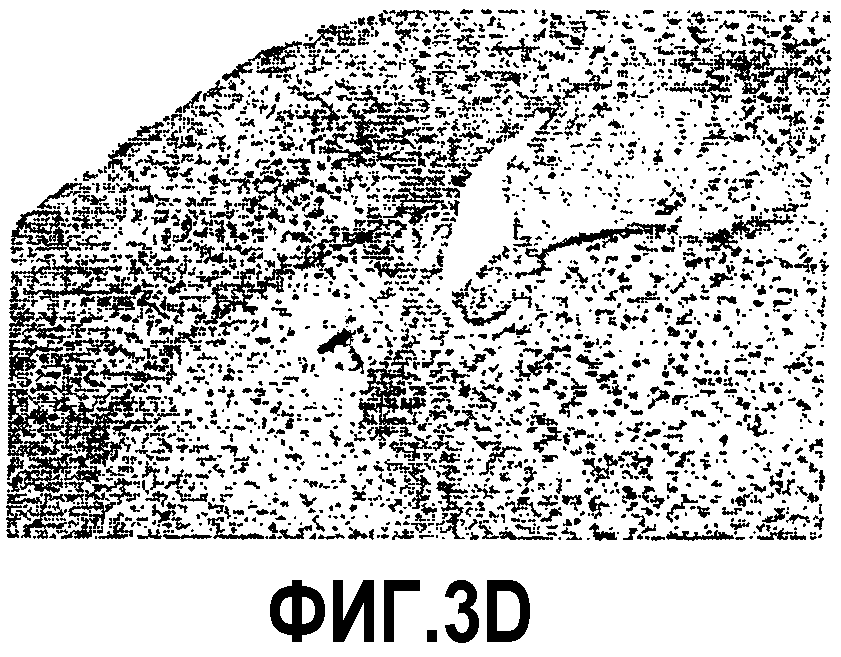
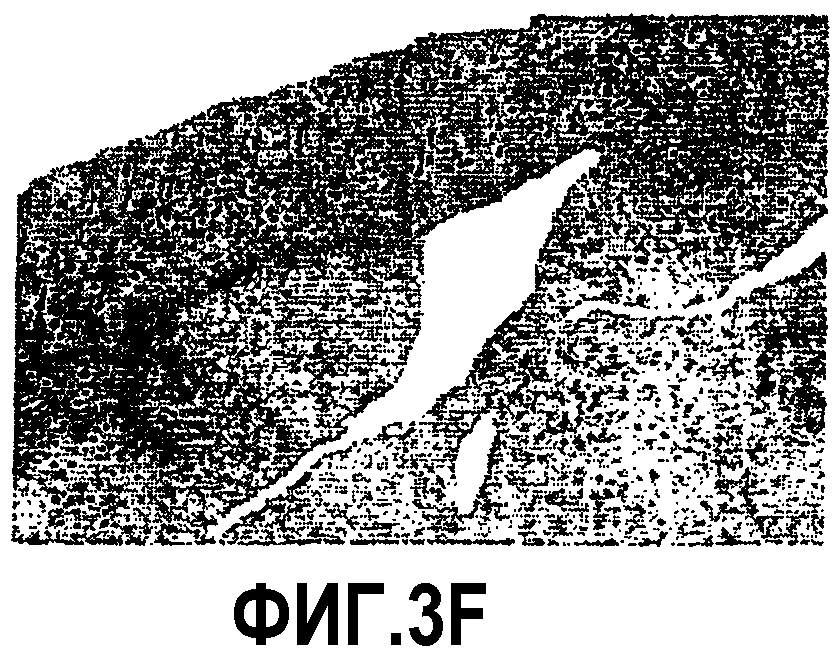
Фиг.3: Ускорение процесса отложения пептида Aβ у мышей APP751SLхPS1KI Ho. Показательная картинка регионального распределения внеклеточных отложений пептида Aβ в мозге в 6 месяцев. Изображения демонстрируют иммуномечение Aβ (Ac 4G8) у 3 мышей APP751SL (фиг.3A, 3B, 3C) и 3 мышей APP751SLxPS1KI Ho (фиг.3D, 3Е, 3F). Иммуномечение выявляет появление в возрасте 6 месяцев первых еще редких отложений в коре (Cx) и в гипокампе (Hp) мышей APP751SL. Для сравнения, у мышей APP751SLxPS1KI Ho того же возраста число отложений в этих областях значительно увеличено. Следует отметить, что у этих мышей отложения присутствуют уже в значительном количестве в таламусе (T).
Фиг.4: Прогрессия с возрастом процесса отложения пептида Aβ. Показательная картинка регионального распределения отложений Aβ в мозге в возрасте 10 месяцев. Изображения соответствуют 2 мышам APP751 SL (фиг.4A, 4B) и 2 мышам APP751SLxPS1KI Ho (фиг.4C, 4D). У мышей APP751SL иммуномечение выявляет значительное увеличение числа и размера отложений в коре (Cx) и в гипокампе (Hp) в возрасте 10 месяцев по сравнению с возрастом 6 месяцев и появление первых отложений в таламусе (см. фиг.3). Плотность и размер отложений также более значительны в 10 месяцев в коре, в гипокампе и таламусе мышей APP751SLxPS1KI Ho. Следует отметить, что у этих мышей небольшое число отложений может быть обнаружено в полосатом теле (St).
Фиг.5: Процесс гибели нейронов в CA1 у мыши APP751SLxPS1KI Ho. Показательная картинка поражения пирамидальных нейронов в гипокампе мышей APP751SLxPS1KI Ho в возрасте 10 месяцев. Изображения представляют окрашивание фиолетовым Crésyl, при слабом увеличении в гипокампе, у 2 мышей PS1KI Ho (фиг.5A, 5B), 2 мышей APP751SL (фиг.5C, 5D) и 2 мышей APP751SLxPS1KI Ho (фиг.5E, 5F). Плотность и толщина слоев пирамидальных клеток в гипокампе качественно сравнимы у мышей APP751SL и мышей PS1KI Ho в возрасте 10 месяцев. Напротив, в том же возрасте, они явно сокращены у мышей APP751SLxPS1KI Ho, в особенности в слое 1 Corne d'Ammon (CA1). Следует отметить, что число мелких клеток, окрашенных в голубой цвет (клетки глиального типа) увеличено в гипокампе мышей APP751SLxPS1KI Ho.
Фиг.6: Процесс гибели нейронов в CA1 у мышей APP751SLxPS1KI Ho. Показательная картинка нейронного поражения в CA1 в возрасте 10 месяцев с использованием других нейронных маркеров, метилового зеленого и иммуномечения BIP. Изображения представляют окрашивание метиловым зеленым, в сильном увеличении в CA1, у не мутантной мыши (фиг.6A), мыши PKS1KI Ho (фиг.6B), мыши APP751SL (фиг.6C) и мыши APP751SLxPS1KI Ho (фиг.6D). Они представляют иммуномечение BIP в сильном увеличении в CA1 у мыши PS1KI Ho (фиг.6E) и мыши APP751SLxPS1KI Ho (фиг.6F). По сравнению с нетрансгенными мышами, PS1KI Ho и APP751SL, число нейронов, окрашенных метиловым зеленым, заметно уменьшено в области CA1 мыши APP751SLxPS1KI Ho. Следует отметить наличие окрашенных клеток глиального типа в значительном количестве в паренхиме гипокампа этой двойной трансгенной мыши. Иммуномечение BIP подтверждает также значительную гибель пирамидальных нейронов в CA1 у мыши APP751SLxPS1KI Ho в возрасте 10 месяцев.
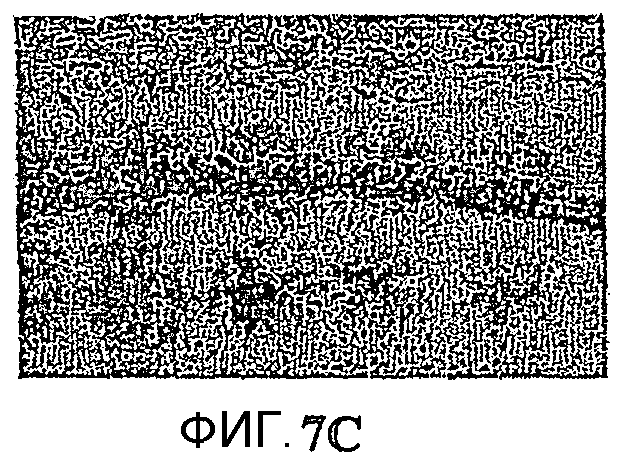
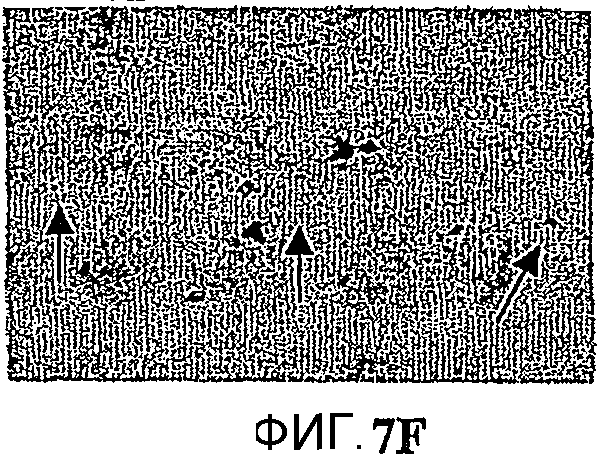
Фиг.7: Гибель нейронов в CA1 и внутриклеточное отложение пептида Aβ. Показательная картинка обоих патологических процессов, нейронное поражение и аномальная внутрицеребральная аккумуляция пептида Aβ в возрасте 10 месяцев. Изображения представляют, в сильном увеличении в CА1, иммуномечение Aβ у 2 мышей APP751 (фиг.7A, 7B) и 2 мышей APP751SLхPS1KI Ho (фиг.7Е, 7F). Они представляют, в сильном увеличении в CA1, окрашивание фиолетовым Crésyl у мыши APP751 (фиг.7C, 7D), мыши APP751SLхPS1KI Ho (фиг.7G, 7Н) и 2 мышей PS1KI Ho (фиг.7I, 7J). В возрасте 10 месяцев как у простых мышей APP751SL, так и у двойных APP751SLxPS1KI Ho, внеклеточные отложения Aβ наблюдаются в большинстве своем с одной и с другой стороны слоя нейронов в CA1. Напротив, в CA1 (характеризующейся нейронным поражением, выраженным по всей длине слоя у мыши APP751SLxPS1KI Ho (фиг.7C, 7D), иммуномечение Aβ зернистого вида (соответствующего аномальной внутринейронной аккумуляции пептида Aβ, см. стрелки) более интенсивно у мышей APP751SLxPS1KI Ho. Это справедливо также в возрасте 6 месяцев (см. фиг.8).
Фиг.8: Ранний запуск процесса гибели нейронов в CA1 у APP751SLxPS1KI Ho. Показательная картинка области CA1 гипокампа в возрасте 6 месяцев. Изображения представляют, в сильном увеличении в CA1, иммуномечение Aβ у 3 мышей APP751 (фиг.8A, 8B, 8C) и 3 мышей APP751SLхPS1KI Ho (фиг.8G, 8Н, 8I). Они представляют окрашивание фиолетовым Crésyl у мышей APP751 (фиг.8D, 8E, 8F) и мышей APP751SLxPS1KI HO (фиг.8J, 8K, 8L). В 6 месяцев область CA1 гипокампа у мыши APP751SLxPS1KI Ho характеризуется уже значительным числом внеклеточных отложений Aβ (фиг.8I), интенсивным внутриклеточным зернистым мечением Aβ (см. стрелки) и гибелью нейронов, окрашенных фиолетовым Crésyl с сопутствующим увеличением числа клеток глиального типа (фиг.8L). Для 2 других мышей APP751SLxPS1KI Ho слой нейронов CA1, окрашенный фиолетовым Crésyl слабо (фиг.8J), или вовсе не дезорганизован (фиг.8K). Следует отметить, что для этих двух мышей внутриклеточное иммуномечение Aβ кажется менее интенсивным и более диффузным (фиг.8G, 8Н), чем у 3-й мыши (фиг.8I).
ПРИМЕРЫ
Пример 1
Конструирование нацеливающего вектора,
несущего мутации M233T и L235P
Цель состояла в том, чтобы ввести две мутации в экзон 7 гена PS1 мыши с заменой остатков M233 на T и L235 на P. Оба новых кодона соответствуют мутациям, идентифицированным у пациентов с болезнью Альцгеймера с ранним началом (FAD).
Линию мышей PS1 knock-in (PS1KI) получали, используя стратегию двустадийного мутагенеза, подобную описанной в Kwok et al. (1997, Neuroreport 8; 157-42) и Champion et al. (1996, Neuroreport 7, 1582-4).
Стратегия имела целью конструирование нацеливающего вектора, несущего замены нуклеиновых кислот в кодонах 233-235 гена ps1 мыши (см. фиг.1A).
Вкратце, геномный фрагмент 17 пар оснований гена PS1 мыши был выделен путем скрининга банка геномной ДНК мыши 129SvJ, сконструированного в бактериофаге-лямбда (Stratagene, номер по каталогу 946313). Анализ путем расщепления рестрикционными ферментами, секвенирования и сравнения с доступными частичными последовательностями мышиного гена PS1 (Mitsuda et al. 1997, JBC 272, 23489-97) показал, что этот фрагмент содержит область от интрона 5 до экзона 11 гена PS1 мыши.
Субфрагмент BamHI-HindIII (9,8 тыс. пар оснований), содержащий часть интрона 5, экзон 6, интрон 6, экзон 7 и часть интрона 7, был субклонирован в плазмиде pGEM-llZf (+) (Promega, Франция) (фиг.1A). Мутагенез 2 кодонов осуществляли, используя комплект Gene Editor (Promega) на фрагменте ДНК, содержащем экзон 7, и подтверждали секвенированием нуклеотидов.
Длинное плечо (5') вектора гомологичной рекомбинации было получено клонированием фрагмента BamHI-XbaI (7 тыс. пар оснований), содержащего экзон 6. Короткое плечо (3') было получено субклонированием фрагмента XbaI-EcoRI (1,8 тыс. пар оснований), содержащего мутагенизированный экзон 7. Кассета положительного отбора (pMCI-Neo кассета) была введена в сайт XbaI, расположенный в интроне 6 в положении - 470 пар оснований в 5'-конце экзона 7 (см. фиг.1А).
Пример 2
Получение клеток, содержащих PS1KI
Нацеливающий вектор, описанный в примере 1, линеаризовывали расщеплением с NotI и электропорировали в линию эмбриональных стволовых клеток (ES) CK35, предоставленных Dr. Charles Babinet, Институт Пастера, Париж, Франция.
Клетки культивировали, как описано выше (W. Wurtz and A. Joyner, Gene Targeting: A Practical Approach by Alexandra L. Joyner (Editor). Oxford University Press; 2nd edition (February 15, 2000)).
430 клеточных клонов, способных носить гомологичную рекомбинацию, были отобраны в присутствии G418. Геномная ДНК этих клонов была проанализирована Саузерн-блоттингом, как описано раньше (Sambrook, Fritsch and Maniatis, Molecular Cloning; A Laboratory Manual, Cold String Harbor Laboratory Press,2nd edition, 1989) с использованием зонда PS1, расположенного вне области рекомбинации (фиг.1A) длинного плеча. Четыре клеточных клона, несущих целевые мутации в гене PS1, были таким образом идентифицированы. Эти клеточные клоны использовали для получения линии трансгенных мышей PS1KI.
Пример 3
Конструирование линии мышей PS1KI
Клон 18C5 инъецировали в бластоциты мышей C57B1/6.
Пять из числа полученных химерных мышей показали передачу мутантного аллеля ps1 зародышевой линии (и, таким образом, их потомству).
Исходя из этих основателей, получили линию мышей PS1KI с чистым генетическим фондом 129SV и со смешанным фондом 129SV-C57B1/6.
Наличие мутантного аллеля PS1KI в гетерозиготном (He) или гомозиготном (Ho) состоянии определяли Саузерн-блоттингом с помощью зонда ps1 230 пар оснований (фиг.1B). Эти мутантные мыши являются жизнеспособными и плодовитыми.
Пример 4
Определение количества PS1 в линии PS1KI
После эвтаназии мозг мышей отбирали и взвешивали. Одно полушарие было сохранено для иммуногистохимии (пост-фиксация), а другое было заморожено, затем гомогенизировано индивидуально на льду при помощи Potter в 2 мл буферного раствора: 0,32 M сахарозы, Tris-HCl 4 мМ, pH 7,4, содержащего коктейль ингибиторов протеаз (CompleteТМ, Roche Diagnostics). Концентрация в белке была определена методом BCA (Pierce). Гомогенат был сохранен при -80°C.
Для обнаружения PS1 25 мкг белкового экстракта мозга инкубировали при 56°C в течение 20 мин в депозитном буфере Laemmli, содержащем мочевину 8M и дитиотреитол 50 мМ. Белки фракционировали электрофорезом на геле NuPAGE 4-12% Бис-Трис полиакриламид (SDS-PAGE) в буфере MES (2-(N-морфолино)этансульфоновая кислота). После переноса белков на нитроцеллюлозный фильтр (Amersham, Франция) фильтр нагревали в PBS в течение 5 мин для увеличения чувствительности и сразу насыщали 5% (вес./об.) порошковым обезжиренным молоком в буфере PBST (PBS, 0,05% (об./об.) Tween 20)) в течение 1 ч и инкубировали в течение ночи при 4°C с первичным антителом в единственном буфере PBST. Связывание антитела было обнаружено с антителом-анти IgG (анти-мышь), конъюгированным с пероксидазой хрена (Amersham, Франция) в разведении 1/10000 в PBST, с последующим использованием системы детекции хемилюминесценцией (Amersham, Франция) согласно инструкциям производителя. Для обнаружения PS1 первичное антитело MAB1563 (Chemicon, США) использовали в разведении 1/10000. Для полуколичественного анализа, люминесцентные сигналы были оцифрованы с помощью камеры CCD GeneGnome 16 битов (Syngene, Кембридж, Англия) и подвергнуты анализу с программным обеспечением Genetools (Syngene). Линейность сигнала была проверена благодаря стандартным кривым, установленным с образцами от 2,5 до 10 мкг гомогената на полосу.
Этот анализ иммунопереносом позволил определить, что уровни экспрессии С-концевого фрагмента мутантного PS1 остались нормальными и не снижены у мыши PS1KI233/235 (фиг.1C).
Пример 5
Получение линии PS1KIxAPP скрещиванием линий PS1KI и APP
Мыши PS1KI (описанные в примерах 1-4) были скрещены с линией трансгенных мышей, суперэкспрессирующих человеческую форму кДНК APP751, несущую мутации FAD Swedish (мутация K670N; M671L) и London (V717I) под контролем промотора Thy-1. Мыши, суперэкспрессирующие человеческую форму кДНК APP751, несущую мутации, были получены, как описано в заявке на патент WO 01/20977.
Во всех следующих опытах были использованы мыши с одним и тем же генетическим фондом для минимизации эффектов, связанных с изменениями генетического фонда.
Пример 6
Определение количества общего амилоидного пептида Аβ и Аβ42 методом иммуноэлектрохемилюминесценции
Для определения количества общей популяции Аβ в мозге (растворимые формы и агрегированные или нерастворимые формы) аликвоты гомогената мозга обрабатывали 2 объемами раствора 9M гидрохлората гуанидина (GH) в 50 мМ Tris pH7,4. Гомогенаты перемешивали в течение 1 ч с 3 периодами обработки ультразвуком по 15 мин с последующим центрифугированием при 50000×g при 4°C в течение 2 ч. Экстракты гуанидина разводили в степени 1/20 в 20 мМ буфера Tris-HCl, pH 7,6, 150 мМ NaCl, 0,5% BSA (вес./об.) и 0,05% Tween 20 (вес./об.). Концентрация пептида Aβ во фракциях была определена затем с помощью иммуноэлектрохемилюминесценции (Yang et al., 1994, Biotechnology (NY) 12(2), 193-194)) при помощи 2 моноклональных мышиных антител антипептид Aβ (4G8 и 6E10) и считывающего устройства Origen M8 Analyzer (IGEN Europe Inc. Оксфорд) согласно прописи, измененной согласно Khorkova et al. (J. Neurosci. Methods 82, 159-166 (1998)).
Моноклональное антитело 4G8 (Senetek PLC), распознающее эпитоп из остатков 17-24 пептида Aβ, рутенилировали сложным эфиром TAG-NHS согласно прописи поставщика (IGEN Europe Inc., Оксфорд). Ru-4G8 и биотинилированное антитело 6E10, эпитоп 1-10 пептида Aβ (Senetek PLC) вводили в присутствии растворимой фракции мозга и определяли количество трехчастных комплексов Ru-4G8/Аβ/6E10-biot с помощью считывающего устройства Origen. Гамму синтетического пептида Aβ (Bachem) использовали для калибровки каждого опыта. Количество пептида Aβ рассчитывали в нанограммах на грамм начального веса мозговой ткани.
Для измерения специфических форм пептида Aβ, заканчивающихся в положении 42 (Aβ42), антитело 6E10 заменяли моноклональным антителом 22F9, которое фиксируется специфически на C-конце Aβ42 (Wirths et al., 2002, Brain Pathol. 12, 275-286).
В заключение, присутствие гена ps1 knock-in (PS1-KI) приводит к
- ускорению аккумуляции Aβ (фиг.2A) и Aβ42 (фиг.2B) в мозге с эффектом, еще более выраженным, когда аллель PS1KI присутствует в гомозиготном состоянии (эффект ген-дозировка). Эффект PS1KI (Ho) выражен сильнее, чем в случае трансгенной мыши, суперэкспрессирующей PS1M146L, описанной ранее в заявке WO 01/20977;
- массовому увеличению пропорции пептида Aβ, имеющего конец β42, который представляет подавляющее большинство Aβ, когда мутация PS1KI присутствует в гомозиготном состоянии, как показывает фиг.2C (отношение Aβ42/Aβ общий составляет 0,92, в возрасте 2,5 месяца, против 0,25 в отсутствие PS1KI и промежуточная величина 0,70 в присутствии единственного аллеля PS1KI: эффект ген-дозировка). В литературе считается, что виды пептида Aβ, заканчивающиеся концом β42, представляют наиболее патологические формы пептида. Линия PS1KIxAPP представляет таким образом модель, особенно обогащенную патологическими формами.
Пример 7
Анализ отложений пептида Aβ с помощью иммуногистохимии
Для испытаний иммуногистохимии/гистологии после отбора и затем постфиксации в параформальдегиде (4%) полушария мозга подвергали криопротекции в течение 1 ночи при 4°C в буфере фосфата натрия 0,2 M (NaH2PО4, 2H2О/Na2HPO4, 12H2О, pH 7,4), содержащем сахарозу 20% (вес./об.). Затем их замораживали в течение 1 мин в изопентане, имеющем температуру -30°C в сухом льду. Срезы толщиной 25 мкм, полученные в криостате, термостатируемом при -30°C (LEICA CM3000), помещают в конечном счете в буфер PBS 0,02M, затем сохраняют при 4°C.
На этих срезах осуществляют иммуноферментную детекцию пептида Aβ с помощью системы детекции, предполагающей образование комплексов авидин-биотин-пероксидазы (АВС), в которых пероксидаза хрена, соединенная с авидином, биотинилирована. Коротко, после инкубации 30 мин в буфере блокировки (нормальная козья сыворотка (Chemicon) в 10% PBS, содержащем 0,1% тритона (Sigma), срезы помещают в присутствии раствора H2О2 0,3% для того, чтобы удалить эндопероксидазы, присутствующие в ткани. Затем эти срезы инкубируют в растворе первичного антитела, содержащем 0,3% тритона и 2% нормальной сыворотки (в течение ночи при 4°C). Используемое первичное анти-Aβ антитело (4G8, Senetek) (моноклональное антитело, направленное к остаткам 17-24 пептида Aβ) биотинилировано. После промываний срезы помещают непосредственно в присутствии комплекса АВС, в течение 1 часа согласно инструкциям производителя (Kit ABC Vectastin, Laboratories Vector, Burlingame, СА). 3-3'-Диаминобензидин был использован как хромоген для пероксидазного фермента.
Таким образом, ускорение аномальной аккумуляции пептида Aβ в мозге двойных трансгенных мышей APP751SLxPS1KI Ho, обнаруженное раньше биохимическими тестами на гомогенатах полушарий мозга, было подтверждено иммуногистохимией. Действительно, микроскопический анализ иммуномечения Aβ, полученного на срезах полушарий мозга, доказал существование ускоренного процесса отложения пептида Aβ в паренхиме мозга этих мышей. Действительно, в то время как первые отложения появляются в коре и в гипокампе к 6 месяцам возраста у мышей APP751SL (фиг.3), они могут быть обнаруженными с возраста 2 месяцев у двойных APP751SLxPS1KI в гомозиготном состоянии. По сравнению с простыми трансгенными APP751SL, у двойных трансгенных (APP751SLxPS1KI Ho) в возрасте 6 месяцев плотность отложений Aβ значительно больше в гипокампе и коре. Кроме того, распределение отложений является более широким, в частности, отложения уже обнаруживаются в таламусе, а также в мосту (фиг.3).
С возрастом, в частности в возрасте 10 месяцев, плотность, а также размер отложений увеличиваются в мозге простых трансгенных мышей APP751SL (фиг.4). Распределение этих отложений также шире, так как они присутствуют в таламусе. У двойных трансгенных APP751SLxPS1KI Ho в возрасте 10 месяцев подобная прогрессия процесса отложения пептида Aβ наблюдается в гипокампе, коре, таламусе и мосту. Первые отложения могут быть обнаружены в ограниченном количестве в полосатом теле (фиг.4). Напротив, мозжечок остается не затронут процессом отложения Aβ. Следует отметить, что в мозге мышей PS1KI Ho в возрасте 10 месяцев (n=4) никакого отложения пептида Aβ не обнаружено.
Пример 8
Анализ гибели нейронов с помощью гистологии и иммуногистохимии
Присутствие очень значительной пропорции патологического пептида Aβ42 повлекло за собой анализ того, развивается ли с возрастом в линии APP751SLxPS1KI Ho, помимо ускорения процесса отложения пептида Aβ, гибель нейронов. Для этого осуществляли 3 типа окрашиваний, позволяющих визуально отобразить исчезновение нейронных клеток на срезах мозговой ткани: a) гистология в фиолетовом crésyl, который окрашивает тела Nissl (органеллы цитоплазмы, ассоциированные с рибосомами шероховатого эндоплазматического ретикулума) и позволяет обнаружить на срезах мозга все нейронные и глиальные клетки; b) гистология в метиловом зеленом, который окрашивает ДНК всех клеток; c) иммуногистохимия в BIP, которая обнаруживает экспрессию в клетках шаперонного белка эндоплазматического ретикулума.
Для окрашивания в фиолетовом crésyl срезы мозговой ткани помещали на желатинированные пластины, затем инкубировали 10 минут в растворе фиолетового crésyl (C 1791, Sigma) 0,5% в дистиллированной воде. После промывания в кислой среде срезы обезвоживали.
Для окрашивания метиловым зеленым срезы поместили на желатинированные пластины, затем инкубировали 10 минут в растворе метилового зеленого (M5015 сигма) 1% в дистиллированной воде, промывали, затем обезвоживали.
Для иммуногистохимии BIP (поликлональное антитело, SPA-826, Stressgen), протокол идентичен использованному для иммуногистохимии пептида Aβ (см. выше), исключая дополнительную инкубацию (1 ч, при температуре окружающей среды) срезов в растворе вторичного биотинилированного антитела (антитело анти-IgG кролика, полученного у козы, Vector) до их инкубации в комплексе ABC.
Микроскопический анализ выявил через использование различных гистологических/иммуногистохимических маркеров уменьшение толщины слоя пирамидальных клеток гипокампа, в частности CA1, в мозге мышей APP751SLxPS1KI Ho (n=3/3) (фиг.5 и 6). Это уменьшение указывает на наличие процесса гибели нейронов, уже имеющего место в возрасте 10 месяцев. В 6 месяцев гибель нейронов имеет место в мозге 1/3 мышей, что позволяет предположить ранний запуск нейротоксического процесса (фиг.8). Анализ параллельно в гипокампе и, в частности в CA1, 2 патологических процессов, аномальной аккумуляции пептида Aβ в мозге и нейронного поражения, внушает мысль о более вероятной роли в нейротоксическом процессе внутриклеточной аккумуляции Aβ (явление, уже описанное у мышей thy1APP751SLxPS1 M146L) по сравнению с его аккумуляцией в виде внеклеточных отложений (фиг.7). Действительно, в нейронах, еще присутствующих в СА1, наблюдается аномально высокая экспрессия пептида Aβ. Кроме того, нейронное поражение в CA1 ясно наблюдается в областях, лишенных внеклеточных отложений. Следует отметить существование вероятного эффекта ген-дозировка в процессе гибели нейронов в CA1. Нейронное поражение действительно было обнаружено у мышей APP751SLxPS1KI в очень большом возрасте (>15 месяцев), имеющих только один аллель PS1KI.
Реферат
Изобретение относится к области генетической инженерии и медицины. Раскрыто животное, не являющееся человеком, имеющее последовательность нуклеиновой кислоты, кодирующую пресенилин 1, несущий мутации, соответствующие мутациям М233Т и L235P в белке PS1 мыши. Также животное содержит последовательность нуклеиновой кислоты, кодирующую весь ген или часть гена, кодирующего АРР. При этом, белок АРР представляет собой АРР751, происходит от человека и несет мутации Swedish и London. Животное предназначено для использования для борьбы с болезнью Альцгеймера. Раскрыты также белок PS1 и нуклеиновая кислота, кодирующая его. Изобретение может быть использовано в медицине для выявления соединений, предназначенных для лечения болезни Альцгеймера. 9 н. и 11 з.п. ф-лы, 50 ил., 1 табл.
Комментарии