Процесс криогенной адсорбции для извлечения ксенона - RU2707767C1
Код документа: RU2707767C1
Чертежи








Описание
Область применения изобретения
Изобретение по существу относится к процессу адсорбции для извлечения ксенона из потока криогенной жидкости или газа, в котором слой адсорбента вводят в контакт с ксенонсодержащим потоком жидкости или газа и избирательно адсорбируют ксенон из потока газа. Адсорбирующий слой удерживают в потоке до получения концентрации ксенона в эффлюенте, равной 70% от входной концентрации ксенона или больше, что обеспечивает глубокое исключение других компонентов потока, перед регенерацией с использованием способа циклического изменения температуры. Такая обработка адсорбирующего слоя перед регенерацией способствует получению из адсорбирующего слоя высокочистого продукта и дополнительно обеспечивает безопасное использование кислорода в качестве продувочного газа даже в тех случаях, когда в потоке подачи присутствуют углеводороды.
Предпосылки создания изобретения
Выдерживание адсорбирующего слоя в потоке до получения на выходе слоя концентрации Xe, равной 90% от входной концентрации или больше, позволяет использовать этот процесс в условиях, позволяющих безопасно применять кислород в качестве продувочной жидкости для процесса адсорбции с циклическим изменением температуры. Кроме того, при таком способе слой можно эксплуатировать в условиях давления и температуры, при которых одновременно могут адсорбироваться другие газы. Это позволяет расширять диапазон технологических условий, пригодных для процесса настоящего изобретения по сравнению с предыдущим уровнем техники.
В частности, в патенте США № 5,039,500, выданном Shino et al., например, описан процесс получения высокочистого ксенона при помощи жидкого кислорода (LOX) из основного конденсатора установки разделения воздуха. В процессе по Shino et al поток жидкого кислорода, содержащего ксенон, криптон и углеводороды, сначала газифицируют, а затем вводят в контакт с адсорбентом при предварительно выбранной температуре и давлении так, чтобы адсорбировать на адсорбенте ксенон, но не кислород, криптон или углеводороды, содержащиеся в потоке кислорода. Адсорбент регенерируют с использованием продувочного газа и нагревания. Можно дополнять основной процесс адсорбции другими типовыми процессами для повышения чистоты ксенона, включая разделительную колонну твердое вещество-газ, колонну с катализатором, колонну для удаления влаги и CO2 и т.п. Недостатком этого процесса является необходимость преобразования потока подачи жидкости из криогенной установки в поток газа перед приведением в контакт со слоем адсорбента. Более того, согласно п. 1 процесс адсорбции необходимо применять при предварительно выбранных условиях температуры и давления так, чтобы адсорбировать ксенон, но не криптон, углеводороды и кислород. Это накладывает ограничения на рабочие условия процесса. Однако из варианта осуществления 1 (колонка 3, строки 20–48) представляется, что такой подход с определением условий давления и температуры, при которых адсорбируется ксенон, но не кислород, криптон и углеводороды, был лишь частично успешным. В данном варианте осуществления силикагелевый адсорбент вводили в контакт с газифицированным потоком при -170°C, содержащим 31 м.д. ксенона, 70 м.д. криптона, 38 м.д. метана и низкие концентрации других углеводородов в кислородной матрице, вплоть до достижения прорыва ксенона. После нагревания газа до 120°C для регенерации адсорбента концентрации в эффлюенте составили 1,4% ксенона, 0,14% криптона, 0,066% углеводородов, а остальное - кислород. Тот факт, что криптон и углеводороды обогатили существенно выше их концентрации в потоке, аналогично ксенону, предполагается, что эти компоненты также адсорбировались в условиях, используемых на стадии адсорбции, что явно противоречит заявленному процессу. Таким образом, вероятно, именно из-за такой коадсорбции других компонентов, в особенности углеводородных компонентов, и их обогащения при адсорбции в последующих вариантах осуществления используют колонну с катализатором для удаления этих углеводородов и CO2, а затем колонну для удаления влаги для избавления от продуктов сгорания углеводородов на катализаторе. Напротив, в настоящем процессе адсорбции поток подачи может представлять собой жидкую или газовую фазу, а во время стадий продувки и нагревания, применяемых для извлечения продукта ксенона, концентрация криптона и углеводородов значительно меньше их концентрации в потоке. В уровне техники, представленном публикацией Shino et al, как заявлено в варианте осуществления 1, на стадии нагрева, применяемой для регенерации слоя адсорбента, концентрация метана 38 м.д. и низкие концентрации других углеводородов в потоке превращаются в 0,066% или 660 м.д. Такое обогащение является приблизительно 17-кратным относительно концентрации углеводородов в потоке.
В патенте США № 4,874,592, также выданном Shino et al, описан процесс адсорбции-десорбции, в котором ксенон концентрируют из вентилируемого потока жидкого кислорода на последовательных стадиях адсорбции и десорбции и в котором каталитическим способом удаляют углеводороды из потока газообразного ксенона, извлеченного после первой стадии адсорбции. Согласно примерам 1 и 2 и как показано на Фиг. 1 и 2 патента США № 4,874,592, прокачиваемый поток из ректификационной колонны, содержащий редкие газы, вводят в первую адсорбционную колонну, где обеспечивают возможность насыщения силикагелевого адсорбента, способного избирательно адсорбировать ксенон. Поток продукта из этой первой адсорбционной колонны собирают путем снижения давления и нагревания колонны. Поток продукта содержал смесь ксенона, криптона и углеводородов в обогащенных концентрациях, более высоких, чем в композиции потока. Для удаления углеводородов использовали типовой каталитический процесс и затем колонну для удаления диоксида углерода и воды перед применением второй адсорбционной колонны для дополнительного повышения чистоты продуктов редких газов. Как пояснено в примере 1 этого патента, вентилирование потока жидкого кислорода создает поток подачи газообразного кислорода (GOX) в адсорбционной системе. Как описано выше, процесс адсорбции настоящего изобретения совместим с подачей и жидкости, и газа, и, следовательно, не требуется стадия вентилирования жидкого кислорода. Более того, настоящий процесс осуществляют таким образом, что предотвращается обогащение углеводородов выше их концентрации в потоке подачи и, следовательно, не требуется стадия каталитического окисления, описанная в предшествующем уровне техники.
В патенте США № 6,658,894, выданном Golden et al, описан процесс извлечения по меньшей мере одного из ксенона или криптона из кислородсодержащего газа путем избирательной адсорбции ксенона и/или криптона с использованием цеолита типа X с замещениями на Li и Ag. В соответствии с примером 7, на котором показаны основные стадии процесса, описанного в публикации Golden et al, поток жидкого кислорода, содержащий 17 м.д. ксенона, 95 м.д. метана и 10 м.д. закиси азота, пропускали через слой силикагеля, в котором удаляли закись азота. Не содержащий закись азота эффлюент испаряли до 113 K и часть этого потока газа направляли в слой, содержащий цеолит типа X с замещениями литием и серебром. Прорыв метана обнаруживали в потоке через 190 минут, при этом после 1400 минут в потоке не происходило прорыва ксенона. На данном этапе стадию подачи останавливали и запускали регенерацию с использованием продувки газообразным азотом при 113 К. По данным, представленным на Фиг. 4 патента Golden et al, концентрация метана во время десорбции увеличивалась максимум до 8000–9000 м.д. Продукт-ксенон собирали путем дополнительного нагревания адсорбирующего слоя. Технологии Golden et al имеют следующие основные особенности:
- Применение цеолита типа X с замещениями как на Li, так и на Ag.
- Доведение процесса адсорбции до точки, в которой не наблюдали прорыва ксенона.
- Десорбция в азотной атмосфере, при которой концентрация метана на выходе значительно превышает концентрацию метана в потоке подачи (95 м.д. по сравнению с 8000–9000 м.д.).
В процессе настоящего изобретения концентрации метана во время десорбции не демонстрируют такого обогащения, которое наблюдают в данных, представленных в Golden et al. Более того, настоящий процесс можно использовать в жидкой фазе, а цеолит типа Х с Li и Ag не требуется.
В патенте США № 3,971,640, выданном Golovko, описан процесс адсорбции для извлечения криптон-ксенонового концентрата из потока воздуха. В процессе Golovko поток газообразного воздуха при 90–110 K, содержащий примеси криптона, ксенона и углеводородов, пропускают через адсорбент с отверстиями пор 5–150 Å, и в течение этого времени криптон, ксенон, азот, кислород и углеводороды адсорбируются. Стадию подачи прекращают, когда на выходе из адсорбера обнаруживают криптон. В этот момент применяют десорбцию с постадийным изменением температуры, при которой температуру сначала поднимают от 90–110 K до 250–280 K, при которой ксенон, криптон, кислород, азот и углеводороды десорбируются из адсорбента, а затем слой дополнительно нагревают от температуры 250–280 K до температуры 500–650 K, причем десорбированные продукты в тот момент сбрасывают в атмосферу. В отличие от процесса, описанного в работе Golovko et al, во время стадии десорбции в процессе настоящего изобретения по существу десорбируются только ксенон и кислород, а любые дополнительные компоненты, такие как углеводороды, десорбируются в концентрациях, существенно меньших по сравнению с их концентрациями в потоке подачи. Согласно примеру 2 в публикации Golovko, измеренные при десорбции концентрации углеводородов составляют 2%, что также свидетельствует о значительной адсорбции и концентрации этих углеводородов в ходе стадии адсорбции, что отсутствует в нашем процессе. Более того, в настоящем процессе не требуется использование десорбции с многостадийными изменениями температуры вплоть до 500–650 K.
Изложение сущности изобретения
Описан процесс адсорбции для извлечения ксенона из потока криогенной жидкости или газа, в котором слой адсорбента вводят в контакт с вышеупомянутым ксенонсодержащим потоком жидкости или газа и избирательно адсорбируют ксенон из этого потока жидкости. Адсорбирующий слой доводят до по меньшей мере почти полного прорыва ксеноном для обеспечения глубокого исключения других компонентов потока, а затем выполняют регенерацию с использованием способа с циклическим изменением температуры. Такое доведение адсорбирующего слоя до почти полного прорыва ксеноном перед регенерацией позволяет получать из адсорбирующего слоя высокочистый продукт и дополнительно позволяет безопасно использовать кислород в качестве продувочного газа даже в тех случаях, когда в подаваемом потоке присутствуют углеводороды.
Подробное описание фигур
На Фиг. 1 представлена схема, иллюстрирующая стадии процесса для 1-слойного процесса адсорбции с подачей жидкости и продувкой на стадиях 1 и 2 соответственно.
На Фиг. 2 представлены профили концентрации на выходе слоя адсорбента для CH4 и Xe во время подачи (вверху) и десорбции (внизу).
На Фиг. 3 представлены профили концентрации на выходе слоя адсорбента в зависимости от времени для CH4, Xe, Kr и N2O на стадии отгонки.
На Фиг. 4a показаны профили концентрации на выходе слоя адсорбента в зависимости от времени на стадиях продувки жидким кислородом и регенерации и извлечения Xe применительно к десорбции CH4.
На Фиг. 4b показаны профили концентрации на выходе слоя адсорбента в зависимости от времени на стадиях продувки жидким кислородом и регенерации и извлечения Xe применительно к десорбции Kr.
На Фиг. 4c показаны профили концентрации на выходе слоя адсорбента в зависимости от времени на стадиях продувки жидким кислородом и регенерации и извлечения Xe применительно к десорбции Xe.
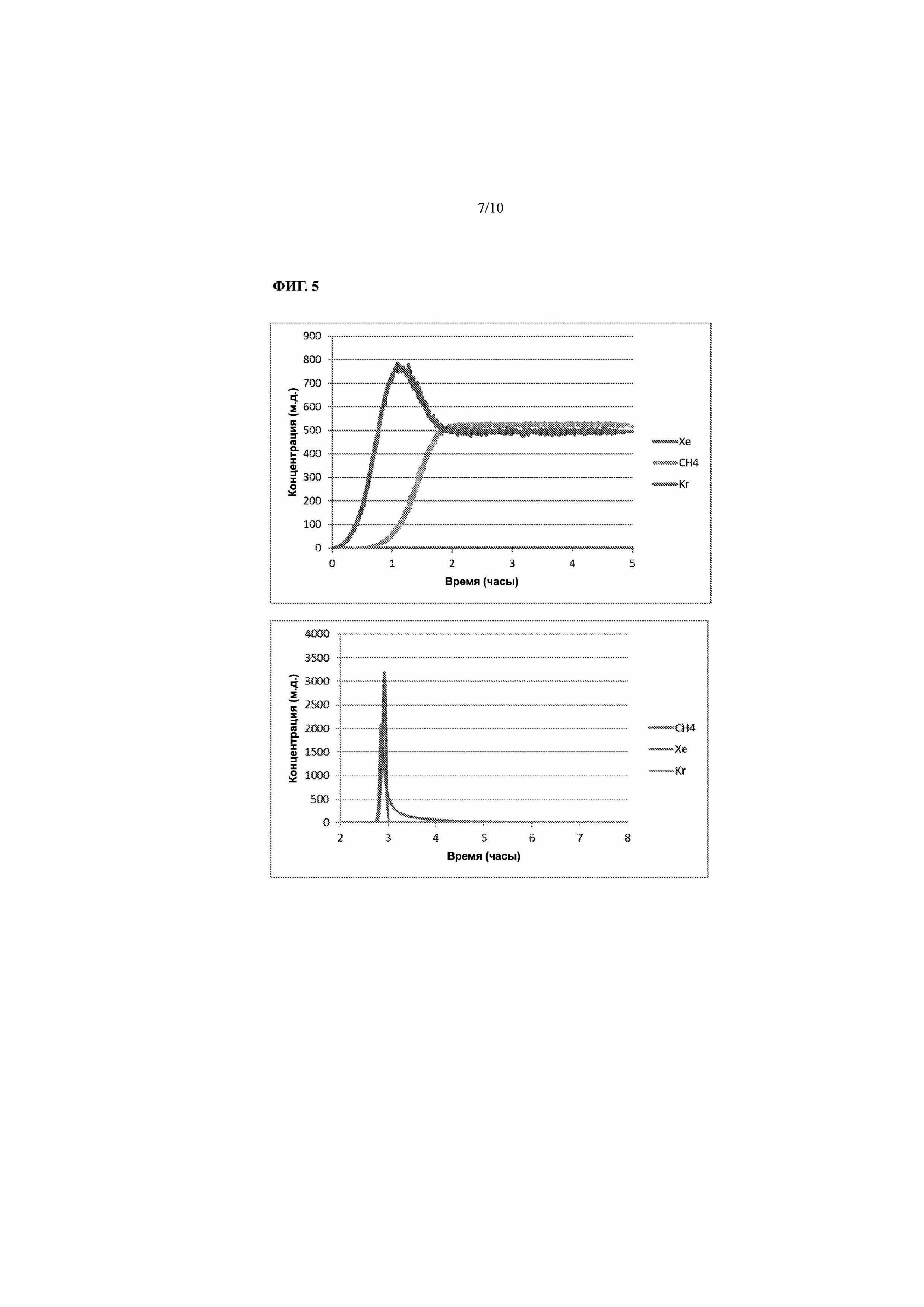
На Фиг. 5 представлены профили концентрации на выходе слоя адсорбента во время стадии отгонки газовой фазы для сравнительного примера 1, показывающие, что критерий по выходной концентрации Xe не выполнен (вверху) и что значительные количества метана, превышающие его концентрацию в потоке, десорбировали вместе с ксеноном во время нагрева слоя (внизу).
На Фиг. 6 представлены профили концентрации на выходе слоя адсорбента во время стадии отгонки газа при комнатной температуре, стадии низкотемпературной отгонки газа (130 K), продувки и извлечения Xe и регенерации слоя.
На Фиг. 7 схематически представлены стадии процесса извлечения Kr и Xe для 1-слойного процесса адсорбции с подачей жидкости и продувкой на стадиях 1a, 1b и 2 соответственно.
На Фиг. 8 представлены профили концентрации на выходе адсорбирующего слоя в зависимости от времени для Kr и CH4 во время стадии подачи.
Подробное описание изобретения
Настоящее изобретение относится к процессу адсорбции для извлечения ксенона из потока криогенной кислородной жидкости, который можно использовать в условиях, в которых кислород можно безопасно применять в качестве продувочного газа во время регенерации, и причем процесс можно применять в условиях давления и температуры, способствующих адсорбции ксенона, а также других компонентов.
В одном варианте осуществления изобретение относится к процессу адсорбции для извлечения ксенона из потока криогенной кислородной жидкости, причем:
1. Слой адсорбента при криогенной температуре, имеющий вход и выход, приводят в контакт с потоком подачи текучей среды, содержащим кислород, ксенон и по меньшей мере один другой адсорбируемый компонент, такой как криптон и/или метан. Слой адсорбента удерживают в потоке до получения концентрации ксенона на выходе слоя, равной 70% от концентрации ксенона на входе в слой или больше; в другом варианте осуществления равной 80% от концентрации ксенона на входе в слой или больше, а в еще одном варианте осуществления равной 90% от концентрации ксенона на входе в слой или больше. Причина выдерживания слоя адсорбента в потоке до получения концентрации ксенона на выходе слоя выше определенного уровня концентрации заключается в достижении глубокого исключения всех нежелательных компонентов, в частности углеводородов, из слоя. Более конкретно, поскольку ксенон сильнее адсорбируется на адсорбенте, чем легкие углеводороды, обычно присутствующие в криогенных потоках редких газов, выдерживание слоя в потоке до получения концентрации ксенона на выходе слоя, равной 70% от концентрации ксенона на входе в слой или больше, приведет к замещению углеводородов, адсорбированных на адсорбенте, на ксенон и, следовательно, достижению более глубокого исключения углеводородов из слоя. К примерам таких углеводородов относятся, без ограничений, метан, этан, пропан, этилен и их комбинации. По очевидным причинам, связанным с безопасностью, неблагоприятным является обогащение потоков подачи текучей среды на основе кислорода сверхноминальными концентрациями C1-C3 углеводородов, что может произойти в слое адсорбента, если не будет выполнен критерий по выходной концентрации.
2. На данном этапе стадию подачи прекращают, а слой адсорбента продувают продувочным газом для удаления углеводородов и других адсорбируемых компонентов, главным образом из не обладающих избирательностью пустот в слое. Продувочный газ по существу содержит кислород, азот, аргон и/или их смеси. В одном варианте осуществления продувочный газ по существу не содержит ксенона и/или других адсорбируемых компонентов.
3. Повышение температуры слоя адсорбента до температуры, достаточной для десорбции продукта-ксенона, который можно собирать как есть и/или дополнительно обрабатывать для дополнительного повышения чистоты.
4. Охлаждение адсорбирующего слоя до криогенных температур с помощью криогенной жидкости. В одном варианте осуществления криогенная жидкость по существу не содержит ксенона и других адсорбируемых компонентов. В другом варианте осуществления криогенная жидкость содержит газообразный и/или жидкий кислород.
5. Циклическое повторение стадий 1–4.
Процесс изобретения можно применять, используя один или более адсорбирующих слоев, и его можно применять в сочетании с другими адсорбентами и/или процессами, которые могут помочь упростить подачу потока к адсорбирующему материалу для извлечения ксенона, такому как гелевая ловушка или защитный слой. К пригодным для использования адсорбентам относятся цеолиты с ионным обменом на серебро, которые имеют низкое отношение кремнезема к глинозему, < 3, и поры, достаточно большие для адсорбции ксенона. К не имеющим ограничительного характера примерам адсорбентов, пригодным для использования в процессе изобретения, относятся цеолит типа X или LSX с ионным обменом на серебро (где LSX означает вариант цеолита типа X с низким содержанием кремнезема), причем степень ионного обмена составляет по меньшей мере 80% Ag по эквивалентам; в другом варианте осуществления по меньшей мере 90% Ag по эквивалентам.
Процесс адсорбции в соответствии с изобретением обеспечивает извлечение ксенона главным образом из потока текучей среды (жидкой или газообразной фазы), без получения продукта, также обогащенного углеводородами выше и за пределами их концентрации в потоке подачи. Фактически при использовании предпочтительным способом содержание углеводородов в потоке продукта-ксенона снижено до величины, равной 50 м.д. или меньше и предпочтительно равной 1 м.д. или меньше от потока, содержащего до 2000 м.д. Если адсорбенту далеко до состояния насыщения, то есть если полный прорыв не достигнут, адсорбент будет адсорбировать следующий по эффективности компонент, к которому по нашим данным относятся углеводороды, в том числе метан. По мере попадания в слой большего количества ксенона происходит замещение и высвобождение метана из адсорбента, и его можно удалять продувкой. Доведение адсорбента до полного прорыва гарантирует отсутствие в адсорбенте емкости для углеводородов, таких как метан, и, следовательно, единственным источником этого и других углеводородов будут пустоты между данной адсорбирующей частицей и другими компонентами слоя адсорбента. В результате продувки этого пустого пространства можно эффективно удалять метан/другие углеводороды, тем самым уменьшая потенциальные риски на стадии сбора продукта-ксенона.
Благодаря глубокому исключению углеводородов, достигаемому в результате настоящего процесса адсорбции, можно избежать использования сложных протоколов десорбции или использования инертных продувочных газов, таких как азот, и/или добавления других типовых операций, таких как каталитическое удаление углеводородов. Более того, имеющаяся у нас возможность обрабатывать потоки подачи, содержащие криогенный жидкий кислород, предотвращает необходимость испарения, которое при наличии углеводородов может представлять проблемы с точки зрения безопасности из-за концентрации в жидком кислороде во время испарения.
Одним из экономических преимуществ изобретения является простота этого процесса и, следовательно, меньшая потребность в капитальном оборудовании. Например, способность обрабатывать поток подачи жидкости исключает необходимость в испарителе для преобразования жидкости в газ. Способность использовать кислород в качестве продувочного газа означает уменьшение количества присоединяемого оборудования и/или дополнительных линий, например, не нужно добавлять азотную линию как в технологии Golden et al. Отсутствие углеводородов в продукте также упрощает дальнейшую обработку, и это означает, что также не требуется дополнительное капитальное оборудование, такое как каталитический окислитель и нижележащий предочиститель для удаления продуктов сгорания.
В другом варианте осуществления изобретение относится к процессу адсорбции для глубокого исключения углеводородов до уровня 50 м.д. или менее в продукте и извлечения ксенона при концентрациях ≥ 1% из криогенного потока подачи кислородной жидкости, содержащей по меньшей мере миллионные доли ксенона и углеводородов. В потоке подачи также могут присутствовать другие вещества, включая двуокись углерода, закись азота и криптон. Пример подходящего потока текучей среды содержит:
- Xe 1-200 м.д., в другом варианте осуществления – 20-180 м.д., в другом варианте осуществления - 50-150 м.д.;
- Kr 500-2000 м.д.;
- углеводороды (в виде метана) 500-2000 м.д.;
- N2O 0-100 м.д.;
- CO2 0-100 м.д.;
- Ar 0-1200 м.д.;
- миллионные доли других атмосферных газов, включая N2; и
- остальное - O2.
Вышеупомянутый богатый кислородом поток предпочтительно имеет криогенные температуры ≤ 120 K, в другом варианте осуществления ≤ 90 K, и давление до по меньшей мере 10 фунтов на кв. дюйм (изб.). Если богатый кислородом поток содержит компоненты, которые адсорбируются сильнее, чем Xe, такие как CO2 и N2O, предпочтительно по существу удалять эти вещества перед приведением потока подачи в контакт с основным адсорбирующим слоем, для чего используют гелевую ловушку, защитный слой и/или слой адсорбента в основном адсорбционном резервуаре, содержащем адсорбент для извлечения Xe. Подходящим адсорбентом для удаления этих сильно адсорбирующихся веществ является силикагель. В одном варианте осуществления силикагель имеет форму, совместимую с процессом адсорбции в уплотненном слое, например форму гранул или сфер. В другом варианте осуществления средний размер частиц формованного материала составляет по меньшей мере 0,5 мм и не более приблизительно 5 мм. В одном варианте осуществления адсорбент для извлечения Xe представляет собой цеолит с замещением на Ag, в другом варианте осуществления — цеолит AgX, причем степень обмена на Ag составляет по меньшей мере 80%, а в другом варианте осуществления — по меньшей мере 90% по эквивалентам. Цеолит AgX также полезно размещать в виде сформованных частиц, причем можно использовать сферы, экструдаты или гранулы. Средний размер частиц также преимущественно составляет по меньшей мере 0,5 мм и не более приблизительно 5 мм.
В дополнительном варианте осуществления процесс изобретения можно реализовывать на практике с использованием двух или более адсорбционных резервуаров. Адсорбционные резервуары могут быть любого известного типа, включая резервуары с вертикальным протоком, резервуары с горизонтальным протоком, резервуары с латеральным протоком или резервуары с радиальным протоком. Когда процесс изобретения осуществляют с двумя или более резервуарами, можно варьировать фазы работы слоев так, что период функционирования первого слоя начинается, когда второй слой выходит из рабочего режима так, что колебания на выходе являются минимальными. В альтернативном варианте осуществления можно так распределять циклы по фазам, что создается наложение рабочих периодов слоев.
Описание процесса адсорбции. Адсорбирующий слой для извлечения ксенона
Стадия 1. Стадия отгонки
Слой адсорбента, содержащий адсорбент, селективный к Xe, предварительно охлаждают до температуры ≤ 120 K, в другом варианте осуществления — до ≤ 90 K, криогенной кислородной жидкостью. Поток подачи также имеет температуру ≤ 120 K, в другом варианте осуществления — ≤ 90 K, давление приблизительно 10 фунтов на кв. дюйм (изб.) и содержит по меньшей мере м.д. Xe и углеводородов в кислородной основе. Адсорбент, который является селективным к Xe, постепенно насыщается Xe, пока продолжается подача потока. Стадию подачи целенаправленно продолжают до получения концентрации Xe на выходе слоя, равной по меньшей мере 70%, в другом варианте осуществления — по меньшей мере 80%, в другом варианте осуществления — по меньшей мере 90%, а в еще одном варианте осуществления — по меньшей мере 95% от концентрации на входе в слой. В этот момент подачу потока прекращают.
Стадия 2. Стадия продувки
Как только будет выполнен критерий завершения стадии 1, представляющий собой концентрацию Xe на выходе слоя, составляющую по меньшей мере 70%, в другом варианте осуществления — по меньшей мере 80%, в другом варианте осуществления — по меньшей мере 90%, а в еще одном варианте осуществления — по меньшей мере 95% от концентрации на входе в слой, слой продувают подходящим продувочным газом, выбранным из группы кислорода, азота, аргона и их смесей, при ≤ 120 К для удаления углеводородов и Xe из не имеющих избирательного характера пустот в слое адсорбента. Эту стадию продувки следует продолжать до получения ≤ 50 м.д. уровней углеводородов на выходе слоя адсорбента; в другом варианте осуществления ≤ 10 м.д., еще в одном варианте осуществления ≤ 1 м.д.
Стадия 3. Регенерация и извлечение Xe
На данной стадии температуру слоя адсорбента можно повышать от криогенных температур до по меньшей мере 250 K и вплоть до 450 K для извлечения продукта Xe с концентрацией ≥ 1%, содержащего не более 50 м.д. углеводородов, измеренных по эквиваленту метана. Температуру можно повышать с помощью продувочного газа с температурой окружающей среды или выше, а чистоту продукта Xe можно контролировать при помощи количества используемого продувочного газа с температурой окружающей среды или выше.
Стадия 4. Охлаждение слоя
После достижения температурой слоя адсорбента значения по меньшей мере 250 К и извлечения продукта Xe слой снова охлаждают до криогенных температур. Этого достигают путем остановки потока продувочного газа с температурой окружающей среды или более высокой температурой и приведения слоя адсорбента в контакт с криогенной жидкостью для охлаждения слоя до температур ≤ 120 K и предпочтительно до ≤ 90 K.
Стадии 1–4 можно применять циклически. Кроме того, данный процесс адсорбции может работать с использованием одного или более слоев адсорбента. При использовании двух адсорбирующих слоев предпочтительно использовать процесс, в котором эти два слоя адсорбента используют последовательно так, чтобы при прохождении слоем 1 стадии 1 слой 2 проходил стадии 2, 3 и 4. Особенно преимущественно использовать процесс с 2 слоями так, чтобы слой 2 прошел регенерацию в ходе стадий 2, 3 и 4 перед регистрированием в слое 1 прорыва Xe. Таким образом, в течение части стадии 1 оба слоя 1 и 2 могут работать последовательно для обеспечения выходной концентрации Xe из слоя 1, составляющей по меньшей мере 70%, в другом варианте осуществления — по меньшей мере 80%, а в еще одном варианте осуществления — по меньшей мере 90% и в еще одном варианте осуществления — по меньшей мере 95% от входной концентрации, и в то же время увеличивать общую чистоту продукта Xe с приемлемым выходом, когда эти слои последовательно соединены в течение части стадии подачи.
В одном варианте осуществления изобретения в качестве продувочного газа на стадиях 2 и 3 используют кислород, а в качестве криогенной жидкости на стадии 4 — криогенный газообразный кислород и/или жидкий кислород. Этот вариант осуществления схематически показан на Фиг. 1. Использование кислорода с температурой окружающей среды или выше в качестве продувочного газа и жидкого кислорода в качестве охлаждающей жидкости является преимущественным с точки зрения уменьшения количества вспомогательного оборудования, необходимого для процесса в целом. Использование более теплого, до 450 K, продувочного газа на стадии 3 позволяет уменьшить количество продувочного газа, необходимого для регенерации слоя, что может способствовать повышению чистоты продукта Xe, а также помогает выполнять стадию регенерации за меньшее время. Для нагревания продувочного газа до температуры выше окружающей среды при необходимости можно использовать небольшой подогреватель.
Настоящее изобретение будет проиллюстрировано ниже с помощью следующих не имеющих ограничительного характера примеров.
Примеры
Приводится всего четыре примера для сведения ключевых признаков изобретения к практике. Эти примеры иллюстрируют извлечение ксенона из различных потоков подачи, как газовой, так и жидкой фаз, а в одном варианте осуществления показывают, как можно модифицировать основной процесс адсорбции для извлечения некоторого количества криптона в дополнение к ксенону.
- Пример 1. Извлечение Xe из подаваемой жидкости с более низкой концентрацией
- Пример 2. Извлечение Xe из подаваемой жидкости с более высокой концентрацией
- Пример 3. Извлечение Xe из подаваемого газа с более высокой концентрацией
- Пример 4. Извлечение Kr и Xe из подаваемой жидкости
- Сравнительный пример. Важность критерия недоведения до прорыва Xe.
Как показывают примеры, настоящее изобретение можно реализовывать с применением при необходимости подачи газовой или жидкой фазы на стадии отгонки (стадия 1). Примеры 1 и 2 представляют собой эксперименты с жидкой фазой, а пример 3 представляет собой эксперимент с газовой фазой. Кроме того, можно адаптировать составляющий предмет изобретения процесс для обеспечения извлечения некоторого количества криптона в дополнение к ксенону (пример 4).
Пример 1. Извлечение Xe из подаваемой жидкости, содержащей 50 м.д. Xe, 500 м.д. CH4, остальное — O2.
5,568 г гранул AgLSX со средним размером частиц 0,6 мм (99% замена на Ag по эквивалентам, остальное — Na) помещали в адсорбирующий слой из нержавеющей стали (внутр. диам. 0,62 дюйма × высота 3,5 дюйма), оборудованный входным отверстием и выходным отверстием, а также термопарой, размещенной вблизи середины слоя адсорбента, для измерения температуры. Адсорбент AgLSX активировали при 350 °C в течение 4 часов в сухом азоте для уменьшения остаточного содержания влаги ≤ 0,5 мас.%, измеренного методом титрования по Карлу Фишеру. Этот активированный слой адсорбента помещали внутрь охлажденного жидким азотом криостата производства компании Oxford Instruments, в котором температуру слоя адсорбента можно контролировать с точностью ±1 °C в пределах температурного диапазона 77–300 K. Входное отверстие слоя адсорбента соединяли с магистралью, обеспечивающей пропускание через слой адсорбента потока, содержащего 50 м.д. Xe, 493 м.д. CH4, остальное — O2, либо O2 сверхвысокой степени чистоты (UHP), примененный в качестве продувочной текучей среды. Давление в данном испытательном аппарате контролировали с помощью регулятора обратного давления, установленного после слоя адсорбента на выходной линии. Если не указано иное, давление на стадии отгонки составляло 100 фунтов на кв. дюйм (изб.) и 50 фунтов на кв. дюйм (изб.) во время стадий продувки и регенерации с циклическими изменениями температуры. Для измерения состава газа, выходящего из слоя, использовали анализатор остаточных газов Omnistar (RGA) с диапазоном в 200 атомных единиц массы от компании Pffeifer Vacuum AG и с разрешением по времени приблизительно 0,1 минуты на точку данных. Данный испытательный аппарат оснащали клапанами, позволяющими пропускать поток и/или продувочные жидкости через слой адсорбента в одном или противоположных направлениях протока. Кроме того, использовали шунтирующую петлю для обеспечения возможности обхождения слоя и измерения состава потока в любой момент времени, а также для облегчения калибровки RGA. Это устройство использовали для измерения кривых прорыва для всех компонентов, отличных от O2, включая Xe и CH4, а также концентрационных профилей десорбции для всех компонентов, отличных от O2. Цель построения десорбционных кривых заключалась в определении того, отвечает ли удаление CH4 во время извлечения Xe на стадии 3 потребностям процесса (например, критерию концентрации CH4 ≤ 50 м.д.).
Слой адсорбента продували O2 класса UHP для удаления любых атмосферных загрязнителей перед охлаждением до 86 K и сжимали до давления 100 фунтов на кв. дюйм (изб.) с использованием UHP O2 при скорости потока эквивалента газа 1,5 ст. л./мин. Когда слой находился при этой температуре, подаваемую смесь, содержащую Xe и CH4, вводили в контакт со слоем адсорбента при таких же условиях давления, температуры и скорости потока. Через 5 часов с помощью RGA обнаружили первичный прорыв CH4. Концентрация CH4 достигла концентрации на входе через ~6,5 часов подачи. В этот момент прорыв Xe не происходил. Подачу потока продолжали и измеряли первичный прорыв Xe через 100,5 часов. Прорыв Xe до входной концентрации 50 м.д. (C/C0 = 1,0) был достигнут через 145 часов подачи. В этот момент подаваемый поток прекращали и инициировали поток UHP O2 при 0,5 ст. л./мин для очистки неселективных пустот в слое и отвода компонентов Xe и CH4. В то же время температуру слоя повышали до 130 К. После получения концентрации CH4 < 100 м.д. слой адсорбента нагревали от 130 K до 300 K со скоростью 2 К/мин, давление в слое адсорбента уменьшали до 50 фунтов на кв. дюйм (изб.), а Xe десорбировали из слоя адсорбента при продувке O2. В начале фактической десорбции Xe концентрация CH4 составляла 1 м.д. и упала до 0 м.д. через 1,2 часа. Десорбция Xe была по существу завершена через 6 часов, когда температура достигла ~300 K. Профили концентрации для стадий подачи и десорбции показаны на Фиг. 2.
Пример 2. Извлечение Xe из подаваемой жидкости, содержащей 150 м.д. Xe, 1500 м.д. Kr, 1540 м.д. CH4, 50 м.д. N2O, остальное — O2.
Слой адсорбента из примера 1 заполняли 1,83 г AgLSX (99% замена на Ag по эквивалентам, остальное — Na), средний размер частиц составлял 1,0 мм, и этот слой был заполнен ими лишь частично. Дополнительное пространство слоя заполняли по существу неадсорбирующими стеклянными гранулами, также имеющими средний размер частиц 1,0 мм. Это изменение было сделано главным образом для уменьшения времени эксперимента, которое превышало 150 часов в тесте, описанном в примере 1, в котором использовали большее количество адсорбента AgLSX. Адсорбент AgLSX перед использованием активировали, как описано в примере 1. В результате из-за присутствия N2O в подаваемом потоке газе перед слоем адсорбента AgLSX помещали гелевую ловушку, содержащую 3,54 г силикагеля класса 40 производства Grace Davison. Перед применением силикагель активировали при 200 °C в течение 4 часов для кондиционирования этого материала перед использованием. Назначение гелевой ловушки состояло в удалении как можно большего количества N2O для обеспечения функционирования более дорогого адсорбента AgLSX, как показано в примере 1, для захвата и извлечения Xe. Систему продували с помощью UHP O2 при температуре окружающей среды для удаления любых атмосферных загрязнителей из слоя адсорбента, гелевой ловушки и связанных с ними компонентов и трубопроводов. Давление устанавливали равным 100 фунтов на кв. дюйм (изб.) с помощью регулятора обратного давления. Затем с помощью жидкого N2 охлаждали гелевую ловушку до средней температуры 81,9 K. Слой адсорбента внутри криостата (как описано в примере 1) охлаждали аналогичным образом, на этот раз до средней температуры 85,6 K, по-прежнему в потоке UHP O2. После стабилизации температуры гелевой ловушки и слоя адсорбента вместо потока UHP O2 подавали Xe-содержащую смесь, имеющую следующий состав: 150 м.д. Xe, 1502 м.д. Kr, 1540 м.д. CH4, 50 м.д. N2O, остальное — O2. При стадии адсорбции скорость потока составила 0,5 ст. л./мин. Времена прорыва до C/C0 = 1 приведены в таблице 1. Профили зависимости концентрации от времени для редкого газа и загрязняющих компонентов представлены на Фиг. 3. Из этих данных очевидно, что сначала произошел прорыв Kr, затем CH4, причем прорыв обоих этих веществ произошел за время < 5 часов. С другой стороны, для полного прорыва Xe потребовалось более 52 часов. На стадии адсорбции на выходе не обнаруживали N2O.
Таблица 1. Времена прорыва для редких газов и примесей
Затем подачу смеси, содержащей редкие газы, прекращали и начинали продувку UHP O2 со скоростью 0,5 ст. л./мин и при давлении, сниженном до 50 фунтов на кв. дюйм (изб.), для выдувания из пустот и других не обладающих селективностью участков слоя адсорбента и соответствующих трубопроводов любых следов смеси, содержащей редкие газы. На стадии продувки температуру слоя адсорбента постепенно повышали до 130 K. Продолжительность продувки составляла 60 минут, после чего температуру слоя адсорбента однократно постепенно повышали до комнатной температуры для десорбции адсорбированного Xe. На стадии десорбции скорость продувки UHP O2 снижали до 0,1 ст. л./мин. Основная часть Xe десорбировалась приблизительно через 2 часа, когда температура слоя адсорбента достигла приблизительно 250 К (см. Фиг. 4a–4c).
В примерах 1 и 2 показаны результаты, которых можно добиться после выполнения стадий процесса, описанных в настоящем документе, и с использованием предпочтительного адсорбента AgX для извлечения ксенона. В этих двух примерах составы подаваемых потоков отличались в плане как концентрации ксенона, так и количества и типа примесей. Несмотря на различия в составе подаваемых потоков, тестовые испытания четко показывают, что адсорбент AgX предпочтительно адсорбирует ксеноновый компонент на стадии отгонки в жидкой фазе (стадия 1). Продувка кислородом (стадия 2) показала, что пустоты в слое и трубопроводах можно легко очищать от компонентов подаваемого потока в конце стадии 1. Нагревание слоя, как описано для стадии 3, приводит к извлечению ксенона с обогащением от низких м.д. до процентных уровней, и причем концентрации углеводородов в продукте составляли в обоих случаях ≤ 1 м.д. В этих двух экспериментах проводили конечную стадию охлаждения (стадия 4) для доведения слоя адсорбента и всех гелевых ловушек или защитных слоев, необходимых для удаления сильно адсорбирующихся загрязнителей, таких как CO2 и N2O, до криогенных температур перед началом стадии отгонки (стадия 1).
Сравнительный пример 1. Извлечение Xe из подаваемого газа, содержащего 49,7 м.д. Xe, 500 м.д. Kr, 508 м.д. CH4, 49,2 м.д. N2O, остальное — O2, причем не выполняли критерий прорыва Xe на стадии отгонки.
Слой адсорбента из примера 1 заполняли 1,85 г AgLSX (99% замена на Ag по эквивалентам, остальное — Na), средний размер частиц составлял 1,0 мм, и этот слой был заполнен ими лишь частично. Дополнительное пространство в слое заполняли по существу неадсорбирующими стеклянными гранулами, также имеющими средний размер частиц 1,0 мм. Адсорбент AgLSX активировали перед использованием, как описано в примере 1. В результате из-за присутствия N2O в подаваемом потоке газе перед слоем адсорбента AgLSX помещали гелевую ловушку, содержащую 2,85 г силикагеля класса 40 производства Grace Davison. Силикагель активировали перед использованием, как описано в примере 2. Назначение гелевой ловушки состояло в удалении как можно большего количества N2O, как описано в примере 2. Систему продували с помощью UHP O2 при температуре окружающей среды для удаления любых атмосферных загрязнителей из слоя адсорбента, гелевой ловушки и связанных с ними компонентов и трубопроводов. Затем с помощью жидкого N2 охлаждали гелевую ловушку до средней температуры 81,9 K. Слой адсорбента внутри криостата (как описано в примере 1) охлаждали аналогичным образом, на этот раз до средней температуры 130 K, по-прежнему в потоке UHP O2, а давление устанавливали равным 60 фунтов на кв. дюйм (изб.) при помощи регулятора обратного давления, установленного после слоя адсорбента. После стабилизации температуры гелевой ловушки и слоя адсорбента вместо потока UHP O2 подавали Xe-содержащую смесь, имеющую следующий состав: 49,7 м.д. Xe, 500 м.д. Kr, 508 м.д. CH4, 49,2 м.д. N2O, остальное — O2. При стадии адсорбции скорость потока составила 1,0 ст. л./мин. Стадию подачи продолжали в течение 5 часов, к этому времени прорыв Xe не наблюдался (см. Фиг. 5). Напротив, к этому моменту произошел полный прорыв как Kr, так и CH4. Для регенерации слоя и извлечения продукта Xe использовали следующую последовательность стадий. Сначала слой адсорбента и трубы продували UHP O2 при температуре 130 K и давлении 60 фунтов на кв. дюйм (изб.) в течение 3 минут для очистки по меньшей мере пустот в слое адсорбента и труб от загрязнителей из подаваемого потока. После такой продувки под высоким давлением давление в слое адсорбента снижали до 6 фунтов на кв. дюйм (изб.), используя регулятор обратного давления, и продолжали продувку UHP O2 при 130 K в течение дополнительных 20 минут. После такой продувки при сниженном давлении скорость потока уменьшали до 0,1 ст. л./мин и слой адсорбента продували в течение 4 минут N2 для дополнительного удаления из слоя адсорбента загрязнений, которые могли удерживаться более прочно, и, следовательно, их можно проще удалять с использованием в качестве продувочного газа более полярного N2 вместо O2. После такой краткой продувки N2 запускали продувку потоком O2 в том же направлении приблизительно в течение 90 мин для продувки слоя адсорбента и линий от N2, который создает помехи сигналу CH4 на анализаторе RGA. В этот момент газ снова переключали на O2 противоположного направления потока и температуру слоя повышали до 300 K. Скорость потока при прогревании слоя адсорбента устанавливали 0,5 ст. л./мин. Как показано на Фиг. 5, при данных условиях Xe достигал пиковой чистоты 2075 м.д., а пиковая чистота CH4 также сильно повысилась до 3180 м.д.
В этом сравнительном примере не соблюдали критерий, касающийся необходимости прорыва Xe до по меньшей мере 90% от концентрации в потоке и предпочтительно по меньшей мере 95% от концентрации в потоке. В результате этого чистота извлеченного Xe была ниже, и, что более важно, концентрация CH4 в продукте Xe превысила концентрацию в подаваемом потоке газа (3180 м.д. по сравнению с 508 м.д.).
Пример 3. Извлечение Xe из подаваемого газа, содержащего 150 м.д. Xe, 1502 м.д. Kr, 1540 м.д. CH4, 50 м.д. N2O, остальное — O2.
1,85 г адсорбента AgLSX, имеющего средний размер частиц 1,0 мм, как и ранее загружали в слой адсорбента, заполняя дополнительное пустое пространство стеклянными гранулами, также имеющими средний размер частиц 1,0 мм. Адсорбент AgLSX активировали перед использованием, как описано в примере 1. Для этого исследования также использовали гелевую ловушку с 2,85 г силикагеля того же типа, как описано в примере 2. Силикагель активировали, как описано в примере 2. Всю испытательную установку, включая гелевую ловушку, слой адсорбента и все подключенные трубопроводы, перед началом испытания продували при комнатной температуре с помощью UHP O2 для удаления любых атмосферных загрязнителей. Давление устанавливали равным 100 фунтов на кв. дюйм (изб.) с помощью регулятора обратного давления. Гелевую ловушку охлаждали до средней температуры 81,9 K все еще в потоке UHP O2. Слой адсорбента намеренно оставляли при комнатной температуре для исследования захвата Xe в условиях температуры окружающей среды. Как только защитный слой достигал нужной температуры, поток переключали на смесь с редкими газами, содержащую: 150 м.д. Xe, 1502 м.д. Kr, 1540 м.д. CH4, 50 м.д. N2O, а остальное — O2. Скорость потока устанавливали равной 0,35 ст. л./мин. При потоке комнатной температуры прорыв Xe до концентрации потока был достигнут через приблизительно через 8 часов. Затем температуру слоя адсорбента снижали от комнатной температуры до 130 K с помощью криостата, а скорость потока увеличивали до 2,5 ст. л./мин. После снижения температуры концентрация Xe на выходе из слоя адсорбента снизилась, что указывает на более высокую адсорбцию Xe при этой более низкой, но по-прежнему соответствующей газовой фазе температуре. Приблизительно через 8 часов при 130 K снова был достигнут прорыв Xe до концентрации в подаваемом потоке. В этот момент поток переключали со смеси, содержащей редкие газы, на UHP O2, давление уменьшали до 50 фунтов на кв. дюйм (изб.), а скорость потока уменьшали до 0,5 ст. л./мин для стадии продувки. Стадию продувки продолжали в течение 60 минут, после чего температуру слоя постепенно повышали с 130 K до комнатной температуры для стадии извлечения Xe и регенерации слоя. Как показано на Фиг. 6, на стадии извлечения Xe не определяли CH4, но из адсорбента было высвобождено небольшое количество N2O, вероятнее всего из-за высокой скорости потока 2,5 ст. л./мин, применяемой для уменьшения общего времени испытания.
Процесс изобретения можно также адаптировать для извлечения криптона из потока газа или жидкости, содержащей по меньшей мере Kr, Xe, CH4, а остальное — кислород. На Фиг. 7 представлен один вариант осуществления, в котором кислород используют в качестве продувочного газа на стадиях 2 и 3, а для охлаждения слоя на стадии 4 используют жидкий кислород. Один вариант процесса, который позволяет отделить криптон от этой газовой смеси, основан на адсорбенте AgX, который имеет меньшую аффинность к данному компоненту по сравнению с метаном или ксеноном. В результате прорыв не столь прочно связываемого криптона происходит первым до метана и/или других углеводородов и ксенона. Следовательно, можно извлекать часть криптона в данный период до прорыва неприемлемых количеств метала и/или других углеводородов. Процесс адсорбции для извлечения криптона и ксенона включает в себя следующие стадии.
Стадия 1a. Стадия отгонки и извлечения Kr
К слою адсорбента, содержащему адсорбент, селективный к Хе, предварительно охлажденному до ≤ 120 K, а предпочтительно до ≤ 90 K криогенной кислородной жидкостью, на вход слоя адсорбента подают поток, также имеющий температуру ≤ 120 K, а предпочтительно ≤ 90 K, и давление приблизительно 10 фунтов на кв. дюйм (изб.) и содержащий по меньшей мере миллионные доли Kr, Хе и углеводородов в кислородной основе. Адсорбент, который является селективным к Хе, обеспечивает отделение углеводородов/Kr на раннем этапе стадии отгонки, поскольку он более интенсивно адсорбирует углеводород и обеспечивает сначала прорыв слоя адсорбента криптоном. На данном этапе Kr можно извлекать до получения содержания углеводородов больше 10 м.д. на выходе слоя адсорбента, до получения концентрации углеводородов в продукте криптоне, равной 50 м.д..
Стадия 1b. Стадия отгонки
После получения концентрации углеводородов на выходе свыше 10 м.д. происходит завершение извлечения Kr, и стадия отгонки продолжается до постепенного насыщения селективного для Хе адсорбента AgX ксеноном по мере продолжения пропускания потока подачи. Стадию подачи целенаправленно продолжают до получения концентрации Хе на выходе слоя, равной по меньшей мере 90% и предпочтительно по меньшей мере 95% от концентрации Хе на входе в слой. В этот момент подачу потока прекращают.
Стадия 2. Стадия продувки жидким кислородом
После выполнения критерия завершения стадии 1b, представляющего собой концентрацию Хе на выходе слоя, составляющую по меньшей мере 90% от концентрации на входе в слой, слой следует продувать продувочным газом, выбранным из группы кислорода, азота, аргона и их смесей, при ≤ 120 K для удаления углеводородов и Хе из не имеющих избирательного характера пустот в слое адсорбента. Эту стадию продувки следует продолжать до получения уровней углеводородов на выходе слоя адсорбента ≤ 50 м.д., предпочтительно ≤ 10 м.д. и наиболее предпочтительно ≤ 1 м.д.
Стадия 3. Регенерация и извлечение Хе
На данном этапе температуру слоя адсорбента можно повышать от криогенных температур до по меньшей мере 250 K для извлечения продукта Хе в процентных концентрациях, содержащего не более 50 м.д. углеводородов, измеренных по эквиваленту метана. Температуру можно повышать при помощи газообразного кислорода с температурой окружающей среды или более высокой (до 450 K), а чистоту продукта Хе можно контролировать по количеству используемого продувочного газа с температурой окружающей среды.
Стадия 4. Охлаждение слоя
После достижения температурой слоя адсорбента значения по меньшей мере 250 К и извлечения продукта Xe слой необходимо снова охлаждать до криогенных температур. Этого достигают путем остановки потока продувочного газа с температурой окружающей среды или более высокой температурой и приведения слоя адсорбента в контакт с криогенной жидкостью для охлаждения слоя до температур ≤ 120 K и предпочтительно до ≤ 90 K.
Стадии 1–4 можно применять циклически. Кроме того, данный процесс адсорбции может работать с использованием одного или более слоев адсорбента. При использовании двух адсорбирующих слоев предпочтительно использовать процесс, в котором эти два слоя адсорбента используют не в одной фазе друг с другом, причем когда слой 1 находится на стадии 1а и 1b, слой 2 проходит через стадии 2, 3 и 4. Особенно преимущественно использовать процесс с 2 слоями так, чтобы слой 2 прошел регенерацию в ходе стадий 2, 3 и 4 перед регистрированием в слое 1 прорыва Xe. Таким образом, в течение части стадии 1a и 1b оба слоя 1 и 2 могут работать последовательно и обеспечивать концентрацию Xe на выходе из слоя 1 более 95% от концентрации на входе и в то же время увеличивать общий выход Xe и Kr. Предпочтительный вариант осуществления, как показано на Фиг. 7, заключается в применении кислорода в качестве продувочного газа на стадиях 2 и 3 и жидкого кислорода в качестве охлаждающей жидкости на стадии 4.
Пример 4. Извлечение Kr из подаваемой жидкости, содержащей 150 м.д. Xe, 1500 м.д. Kr, 1540 м.д. CH4, 50 м.д. N2O, остальное — O2.
Слой адсорбента из примера 1 заполняли 1,83 г AgLSX (99% замена на Ag по эквивалентам, остальное — Na), средний размер частиц составлял 1,0 мм, и этот слой был заполнен ими лишь частично. Дополнительное пространство в слое заполняли по существу неадсорбирующими стеклянными гранулами, также имеющими средний размер частиц 1,0 мм. Адсорбент AgLSX активировали перед использованием, как описано в примере 1. В результате из-за присутствия N2O в подаваемом потоке газе перед слоем адсорбента AgLSX помещали гелевую ловушку, содержащую 3,54 г силикагеля класса 40 производства Grace Davison. Этот материал активировали перед использованием, как описано в примере 2. Систему продували с помощью UHP O2 при температуре окружающей среды для удаления любых атмосферных загрязнителей из слоя адсорбента, гелевой ловушки и связанных с ними компонентов и трубопроводов. Давление устанавливали равным 100 фунтов на кв. дюйм (изб.) с помощью регулятора обратного давления. Затем с помощью жидкого N2 охлаждали гелевую ловушку до средней температуры 81,9 K. Слой адсорбента внутри криостата (как описано в примере 1) охлаждали аналогичным образом, на этот раз до средней температуры 85,6 K, по-прежнему в потоке UHP O2. После стабилизации температуры гелевой ловушки и слоя адсорбента вместо потока UHP O2 подавали Xe-содержащую смесь, имеющую следующий состав: 150 м.д. Xe, 1500 м.д. Kr, 1540 м.д. CH4, 50 м.д. N2O, остальное — O2. При стадии адсорбции скорость потока составила 0,5 ст. л./мин. Кривые прорыва для Kr, CH4 и Xe приведены на Фиг. 8. Из данных на этой фигуре видно, что прорыв Kr происходит до прорыва CH4, что позволяет извлечь Kr до возникновения прорыва CH4.
Реферат
Изобретение относится к процессу адсорбции для извлечения ксенона из потока криогенной жидкости или газа, в котором слой адсорбента вводят в контакт с ксенонсодержащим потоком жидкости или газа. Способ включает: i) подачу потока подачи при криогенных температурах на вход адсорбционного резервуара, причем указанный слой адсорбента содержит адсорбент, селективный к ксенону; ii) выдерживание указанного слоя адсорбента в потоке до получения концентрации ксенона на выходе указанного слоя, равной 70% от концентрации ксенона на входе в указанный слой адсорбента или выше; iii) завершение подачи в адсорбирующий слой и продувку продувочным газом; iv) повышение температуры указанного слоя адсорбента до температуры, обеспечивающей десорбцию всего указанного ксенона; v) извлечение продукта ксенона, десорбированного из указанного слоя адсорбента; vi) охлаждение указанного слоя адсорбента до криогенных температур с помощью криогенной жидкости и повторение стадий i)-vi) циклическим образом. При этом указанный селективный к ксенону адсорбент представляет собой цеолит типа X с ионным обменом на серебро, причем степень ионного обмена составляет 80% Ag по эквивалентам. Технический результат заключается в получении высокочистого продукта и безопасном использовании кислорода в качестве продувочного газа. 22 з.п. ф-лы, 10 ил., 1 табл., 4 пр.
Комментарии