Солнечные элементы, включающие в себя цепочки для аккумуляции света - RU2264677C2
Код документа: RU2264677C2
Чертежи



















































Описание
Настоящее изобретение было сделано с помощью правительственной поддержки в соответствии с грантом №DE-FG02-96ER14632 от Department of Energy и грантом №GM36238 от National Institutes of Health. Правительство США имеет определенные права на настоящее изобретение.
Область изобретения
Настоящее изобретение относится к солнечным элементам, в частности к регенеративным солнечным элементам, и к цепочкам для аккумуляции света, пригодным для использования в таких солнечных элементах.
Уровень техники
Молекулярные подходы к преобразованию солнечного света в электрическую энергию имеют богатую историю, поскольку об измеренном ″фотоэффекте″ было впервые сообщено в 1887 году в Вене (Moser, J. Montash. Chem. 1887, 8, 373.).Наиболее перспективные конструкции были разработаны в главных деталях в 1970-е годы (Gerischer, H. Photochem. Photobiol. 1972, 16, 243; Gerischer, H. Pure. Appl. Chem. 1980, 52, 2649; Gerischer, H.; Willig, F. Top. Curr. Chem. 1976, 61, 31). Два распространенных подхода изображены на фигуре 1, оба они используют молекулы, селективно поглощающие солнечный свет, называемые фотосенсибилизаторами или просто сенсибилизаторами (S) и ковалентно связанные с проводящими электродами. Поглощение света сенсибилизатором приводит к появлению возбужденного состояния, S*, которое инжектирует электрон в электрод, а затем окисляет частицу вещества в растворе. Правая часть изображает упрощенный фотоэлектросинтетический элемент. Этот элемент производит как электрическую энергию, так и химические продукты. Для работы изображенным способом в течение нескольких последних десятков лет было разработано множество молекулярных подходов, имеющих целью разложение воды на водород и кислород. В левой части изображены регенеративный элемент, который в итоге преобразует свет в электричество без каких-либо химических продуктов. В представленном регенеративном солнечном элементе имеющие место на фотоаноде реакции окисления обращаются на темновом катоде.
Принципиальная сложность с этими конструкциями солнечных элементов заключается в том, что монослой молекулярного сенсибилизатора на плоской поверхности не поглощает значительную долю падающего видимого света. Как следствие, даже если квантовые выходы переноса электрона по отношению к поглощенным фотонам являются высокими, эффективность преобразования солнечной энергии будет неприемлемо низкой из-за небольшого количества поглощаемого света. Эта проблема была замечена на ранней стадии исследования, и ее пытались обойти путем использования толстых пленок сенсибилизаторов. Эта стратегия использования толстых поглощающих слоев была безуспешной, поскольку межмолекулярное гашение возбужденных состояний в толстой пленке сенсибилизатора уменьшает выход инжекционных электронов в электрод.
Один из классов толстопленочных сенсибилизаторов предложен в так называемых органических солнечных элементах (Tang, C.W. и Albrecht, A.C. J.Chem.Phys. 1975, 63, 953-961). В данном случае пленка толщиной от 0,01 до 5 мкм, как правило, состоящая из фталоцианинов, периленов, хлорофилов, порфиринов или их смесей, осаждается на поверхности электрода и используется во влажных солнечных элементах, подобным тем, которые изображены, или в виде твердотельных устройств, где второй металл осаждается поверх органической пленки. Органический слой, как предполагается, представляет собой полупроводник с узкой запрещенной зоной и с фотопроводимостью либо n-, либо p-типа, и предлагаемые механизмы преобразования света в электрическую энергию включают в себя экситонный перенос энергии между пигментами в пленке к поверхности электрода, где имеет место перенос электронов через границу раздела. Однако важность этих предлагаемых механистических шагов не является очевидной. Повышение эффективности, которое обуславливается векторным переносом энергии между пигментами, не было убедительно продемонстрировано. Кроме того, длины диффузий экситонов, о которых сообщалось, являются короткими по сравнению с глубиной проникновения света. Соответственно большая часть света поглощается в области, где энергия не может переноситься к поверхности полупроводника. Экситоны также легко гасятся с помощью примесей или захваченного растворителя, что приводит к значительным проблемам, связанным с воспроизводимостью и сложностью изготовления. Известные в настоящее время в данной области техники органические солнечные элементы представляют собой многослойные органические пленки с ″гетеропереходами″ или легированные органические слои, которые дают ˜2%-ную эффективность при низких уровнях облучения, однако эффективность заметно падает, когда облучение приближается к интенсивности солнечного света (Forrest, S.R. et al., J.Appl.Phys. 1989, 183, 307; Schon, J.H. et al., Nature 2000, 403, 408).
Другой класс солнечных элементов на молекулярной основе представляют собой так называемые фотогальванические элементы, которые были основными устройствами для преобразования солнечной энергии на молекулярном уровне в 1940-1950-х годах (Albery, W.J. Acc.Chem.Res. 1982, 15, 142). Эти элементы отличаются от тех, которые обсуждались выше, тем, что возбужденный сенсибилизатор не подвержен переносу электрона через границу раздела. Элементы часто содержат сенсибилизаторы, заключенные в мембрану, которая дает возможность для переноса ионов и переноса заряда; мембрана физически разделяет два темновых металлических электрода и фотогенерируемых окислительно-восстановительных эквивалента. Геометрическое расположение предотвращает прямой перенос электрона в возбужденном состоянии от хромофора к электродам или обратно. Вместо этого, происходит межмолекулярное разделение заряда, и восстановительные и окислительные эквиваленты диффундируют к электродам, где имеет место перенос электрона через границу. Мембранный потенциал Нернста может генерироваться с помощью переноса электронов под действием света, происходящего в мембране. В фотоэлектросинтетических гальванических элементах могут также образовываться химические горючие материалы. Эта общая стратегия сенсибилизации электродов красителями использовалась в разнообразных вариантах в течение многих лет, но абсолютные эффективности оставались очень низкими. Albery сделал вывод, что теоретически в водном регенеративном фотогальваническом элементе может быть достигнута эффективность ˜13%. Однако реализованные к настоящему времени эффективности, как правило, составляют менее 2%.
В 1991 Gratzel и O'Regan сообщили о прорыве (O'Regan, B. et al., J.Phys.Chem. 1990, 94, 8720; O'Regan, B. and Gratzel, M. Nature 1991, 353, 737). Путем замены планарных электродов толстой пористой пленкой коллоидного полупроводника площадь поверхности для связывания сенсибилизатора увеличилась более чем в 1000 раз. Gratzel и O'Regan продемонстрировали, что монослой покрытия из сенсибилизатора, нанесенный на частицы из полупроводника, приводит к поглощению по существу всего падающего света, и эффективности преобразования энергии падающих фотонов в энергию электронов составляют единицу на индивидуальных длинах волн света в регенеративных солнечных элементах. Более того, глобальная эффективность ˜5% была реализована в условиях освещения с отношением масс воздуха в 1,5 (т.е. отношение массы атмосферы на реальном пути между наблюдателем и солнцем к массе в том случае, когда наблюдатель находится на уровне моря и при стандартном атмосферном давлении, а солнце - у него над головой); эта эффективность возросла до величины 10,69%, подтвержденной в настоящее время (Gratzel, M. в ″Future Generation Photovoltaic Technologies″ McConnell, R.D.; AIP Conference Proceedings 404, 1997, page 119). Эти солнечные элементы типа "Gratzel" уже нашли свою нишу на рынке и являются коммерчески доступными в Европе.
Эти пленки коллоидных полупроводников с высокой площадью поверхности (элементы типа «Gratzel») достигают высокого уровня поглощения, но также имеют следующие значительные недостатки: (1) для высокой эффективности требуется жидкий переход (поскольку очень нерегулярная структура поверхности делает осаждение твердотельного проводящего слоя по существу невозможным). (2) Пленки коллоидных полупроводников требуют стадий высокотемпературного отжига для понижения внутренних напряжений. Такие высокие температуры накладывают жесткие ограничения на типы проводящих подложек, которые могут быть использованы. Например, не могут быть использованы полимерные подложки, которые плавятся при температурах, более низких, чем требуемые температуры отжига. (3) Значительные потери связаны с переносом заряда через толстые пленки полупроводников. Эти потери не понижают заметно фототок, но оказывают большое воздействие на выходное напряжение, и, таким образом, мощность значительно понижается (Hagfeldt, A.; Gratzel, M. Chem. Rev. 1995, 95, 49). В соответствии с этим необходимость в новых молекулярных подходах к конструированию солнечных элементов по-прежнему остается.
Сущность изобретения
Исходя из этого настоящее изобретение предусматривает среди прочих вещей цепочку для аккумуляции света, пригодную для использования при изготовлении солнечных элементов. Цепочка для аккумуляции света содержит:
(a) первую подложку, содержащую первый электрод; и
(b) слой из стержней для аккумуляции света, электрически соединенный с первым электродом, причем каждый из стержней для аккумуляции света содержит полимер формулы I:

где:
m составляет по меньшей мере 1 и может составлять от двух, трех или четырех до 20 или более;
X1 представляет собой группу разделения заряда, имеющую возбужденное состояние с энергией, равной или более низкой, чем у Х2.
X2-Xm+1 представляют собой хромофоры.
В стержнях для аккумуляции света согласно формуле I X1 предпочтительно содержит порфириновый макроцикл, который может быть в форме двухъярусного сэндвичевого соединения. Кроме того, X2-Xm+1 также предпочтительно содержат порфириновые макроциклы.
В одном из предпочтительных вариантов воплощения стержней для аккумуляции света согласно формуле I по меньшей мере один из (например два, три, множество, большинство или все) X1-Xm+1 выбран/выбраны из группы, состоящей из хлоринов, бактериохлоринов и изобактериохлоринов.
Конкретный вариант воплощения цепочки для аккумуляции света, описанной выше, предусматривает движение дырок в направлении, противоположном направлению (переноса) энергии возбужденного состояния вдоль некоторой части или всей длины стержней для аккумуляции света, и содержит:
(a) первую подложку, содержащую первый электрод; и
(b) слой стержней для аккумуляции света, электрически соединенных с первым электродом, причем каждый из стержней для аккумуляции света содержит полимер формулы I:
где:
m составляет по меньшей мере 1 (как правило, от двух, трех или четырех до двадцати или более);
X1 представляет собой группу разделения заряда, имеющую возбужденное состояние с энергией, равной или более низкой, чем у Х2;
X2-Xm+1 представляют собой хромофоры; и
X1-Xm+1 выбраны таким образом, что при инжекции либо электрона, либо дырки из X1 в первый электрод соответствующая дырка или электрон из X1 переносится по меньшей мере к X2 и необязательно к X3, X4 и по всему пути до Xm+1. В варианте воплощения, предпочтительном в настоящее время, X1-Xm+1 выбраны таким образом, что при инжекции электрона из X1 в первый электрод соответствующая дырка из X1 переносится по меньшей мере к X2 и необязательно вплоть до Xm+1.
Цепочки для аккумуляции света обеспечивают интенсивное поглощение света и доставляют (переносят) получающееся в результате возбужденное состояние в заданное положение внутри молекулярной цепочки. Существует множество различных применений цепочек для аккумуляции света. Цепочки для аккумуляции света могут быть использованы в качестве компонентов систем для детектирования низких уровней света, в частности там, где является желательным контроль длины волны света, который собирается. Цепочки для аккумуляции света могут быть использованы в качестве входных элементов в оптоэлектронных устройствах и в качестве входного узла и системы задержки энергии в сигнальных системах на молекулярной основе. Одно из применений последних включает в себя использование во флуоресцентных сенсорах на молекулярной основе. Сенсор на молекулярной основе использует набор групп-зондов (которые связываются с анализируемым веществом), соединенных с основной молекулярной цепью, которая подвергается переносу энергии возбужденного состояния. Связывание единственного анализируемого вещества с любой из групп-зондов приводит к образованию комплекса, который может гасить возбужденное состояние, которое свободно мигрирует вдоль основной цепи (то есть экситон). Явление гашения приводит к уменьшению флуоресценции основной молекулярной цепи. Поскольку только одно присоединенное анализируемое вещество может вызвать явление гашения, чувствительность является гораздо более высокой, чем если бы присутствовало отношение 1:1 групп-зондов и флуоресцентных групп. Ранее такие флуоресцентные сенсоры на молекулярной основе использовали в основной молекулярной цепи хромофоры, поглощающие в УФ или в ближней УФ области. Цепочки для аккумуляции света, описанные здесь, являются идеально приспособленными в качестве компонентов нового класса флуоресцентных сенсоров на молекулярной основе, которые сильно поглощают (и флуоресцируют) в видимой и ближней инфракрасной области.
Особое применение цепочек для аккумуляции света, описываемых здесь, представляет собой применение в солнечных элементах. Солнечный элемент, как здесь описано, как правило, содержит:
(a) первую подложку, содержащую первый электрод;
(b) вторую подложку, содержащую второй электрод, при этом первая и вторая подложки расположены с образованием пространства между ними, и по меньшей мере один элемент из (i) первой подложки и первого электрода и (ii) второй подложки и второго электрода является прозрачным;
(c) слой стержней для аккумуляции света, электрически соединенный с первым электродом, причем каждый из стержней для аккумуляции света содержит полимер формулы I:
где:
m составляет по меньшей мере 1 (а как правило, от двух, трех или четырех до двадцати или более);
X1 представляет собой группу разделения заряда, имеющую возбужденное состояние с энергией, равной или более низкой, чем у X2;
X2-Xm+1 представляют собой хромофоры; и
X1 электрически соединена с первым электродом; и солнечный элемент дополнительно содержит
(d) электролит в пространстве между первой и второй подложками. Подвижный носитель заряда может, необязательно, быть включен в состав электролита.
В конкретном варианте воплощения приведенного выше элемента (иногда упоминаемом здесь как ″конструкция II″), солнечный элемент содержит:
(a) первую подложку, содержащую первый электрод;
(b) вторую подложку, содержащую второй электрод, при этом первая и вторая подложки расположены с образованием пространства между ними, и по меньшей мере один элемент из (i) первой подложки и первого электрода и (ii) второй подложки и второго электрода является прозрачным;
(c) слой стержней для аккумуляции света, электрически соединенный с первым электродом, причем каждый из стержней для аккумуляции света содержит полимер формулы I:
где:
m составляет по меньшей мере 1 (а как правило, от двух, трех или четырех до двадцати или более);
X1 представляет собой группу разделения заряда, имеющую возбужденное состояние с энергией, равной или более низкой, чем у X2;
X2-Xm+1 представляют собой хромофоры;
X1 электрически соединена с первым электродом; и
X1-Xm+1 выбраны таким образом, что при инжекции либо электрона, либо дырки из X1 в первый электрод соответствующая дырка или электрон из X1 переносится к X2 (и, необязательно, к X3, X4, а в некоторых случаях и по всему пути до Xm+1); солнечный элемент дополнительно содержит:
(d) электролит в пространстве между первой и второй подложками; и
(e) необязательно, но предпочтительно подвижный носитель заряда в электролите.
В варианте воплощения, предпочтительном в настоящее время, X1-Xm+1 выбраны таким образом, чтобы при инжекции электрона из X1 в первый электрод соответствующая дырка из X1 переносилась к X2 -Xm+1.
Другой конкретный вариант воплощения (иногда упоминаемый здесь как ″конструкция III″) солнечного элемента, описанного выше, содержит:
(a) первую подложку, содержащую первый электрод;
(b) вторую подложку, содержащую второй электрод, при этом первая и вторая подложки расположены с образованием пространства между ними, и по меньшей мере один элемент из (i) первой подложки и первого электрода и (ii) второй подложки и второго электрода является прозрачным;
(c) слой стержней для аккумуляции света, электрически соединенный с первым электродом, причем каждый из стержней для аккумуляции света содержит полимер формулы I:
где:
m составляет по меньшей мере 1 (а как правило, от двух, трех или четырех до двадцати или более);
X1 представляет собой группу разделения заряда, имеющую возбужденное состояние с энергией, равной или более низкой, чем у X2;
X2-Xm+1 представляют собой хромофоры;
X1 электрически соединена с первым электродом; и
Xm+1 электрически соединена со вторым электродом; солнечный элемент дополнительно содержит
(d) электролит в пространстве между первой и второй подложками.
Опять же, X1-Xm+1 могут быть выбраны таким образом, что при инжекции электронов или дырок (предпочтительно, электронов) из X1 в первый электрод соответствующая дырка или электрон из X1 переносится к X2 или, необязательно, к X3, или X4, или по всему пути до Xm+1.
Множество различных электрических устройств, содержащих солнечный элемент, описанный выше, и имеющих схемы (как правило, резистивные нагрузки), электрически присоединенные к нему, могут быть получены с солнечными элементами по настоящему изобретению, как обсуждается более подробно ниже.
Настоящее изобретение объясняется более подробно с помощью прилагаемых чертежей и описания, приведенного ниже.
Краткое описание чертежей
Фигура 1. Схемы двух распространенных молекулярных подходов для преобразования света в электрическую энергию.
Фигура 2. Общая схема линейных цепочек хромофоров (стержни для аккумуляции света).
Фигура 3. Миграция энергии вдоль стержня для аккумуляции света и использование подвижного носителя заряда для регенерации узла разделения заряда после инжекции электронов (конструкция I).
Фигура 4. Миграция энергии и миграция дырок по прыжковому механизму в противоположных направлениях (конструкция II).
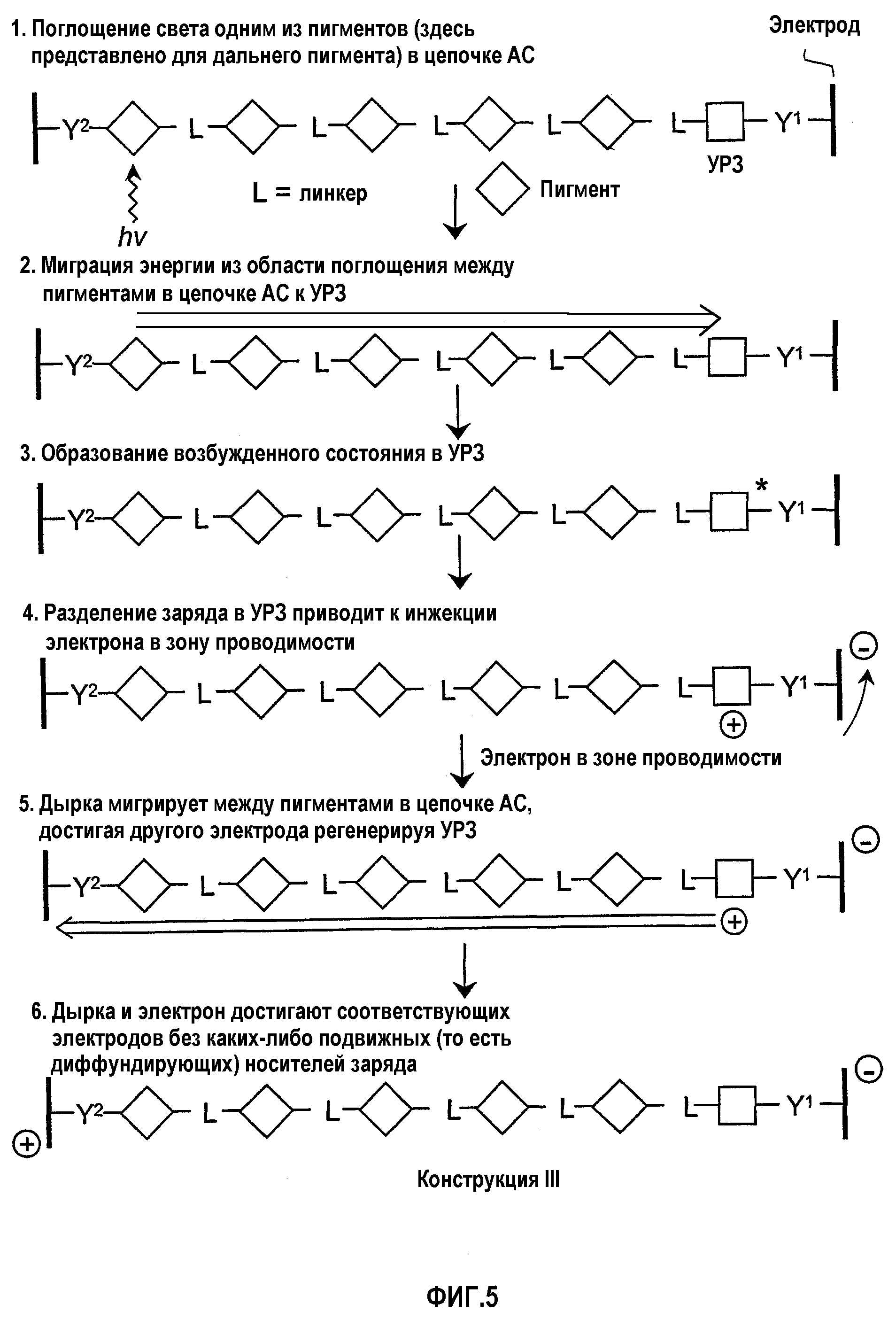
Фигура 5. Миграция энергии и миграция дырок по прыжковому механизму в противоположных направлениях в случае, когда стержни для аккумуляции света заключены между двумя электродами (конструкция III).
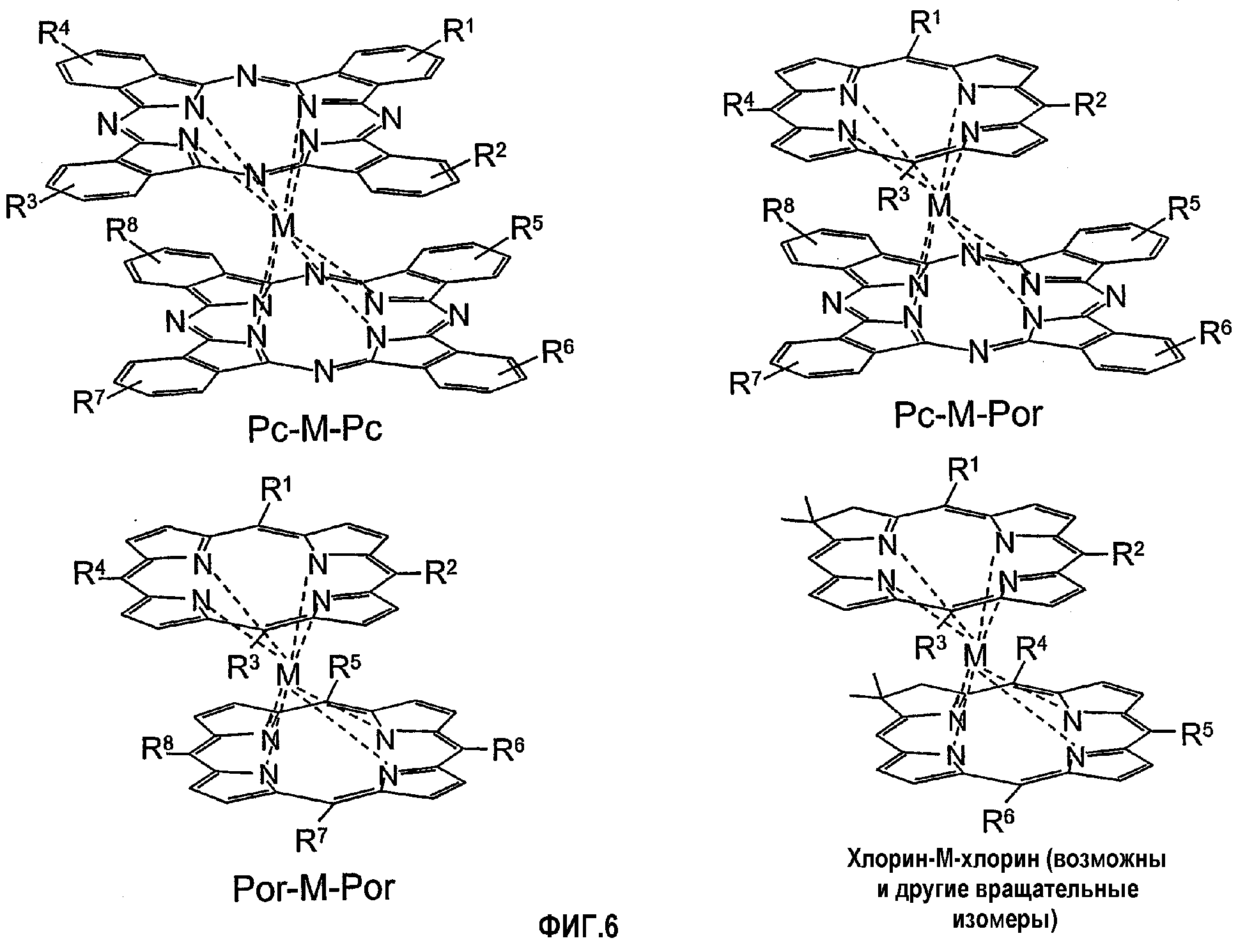
Фигура 6. Двухъярусные сэндвичевые молекулы, которые могут служить в качестве сенсибилизаторов.
Фигура 7. Механизмы сенсибилизации для полупроводника n -типа с помощью сенсибилизатора S. Здесь ECB и EVB представляют собой соответственно энергии зоны проводимости и валентной зоны полупроводника. EF представляет собой уровень Ферми полупроводника. Eo(S+/0) и Eo(S+/*) представляют собой соответственно формальные потенциалы восстановления для основного и возбужденного состояния. также изображены распределения донорных и акцепторных уровней сенсибилизатора по Gerischer.
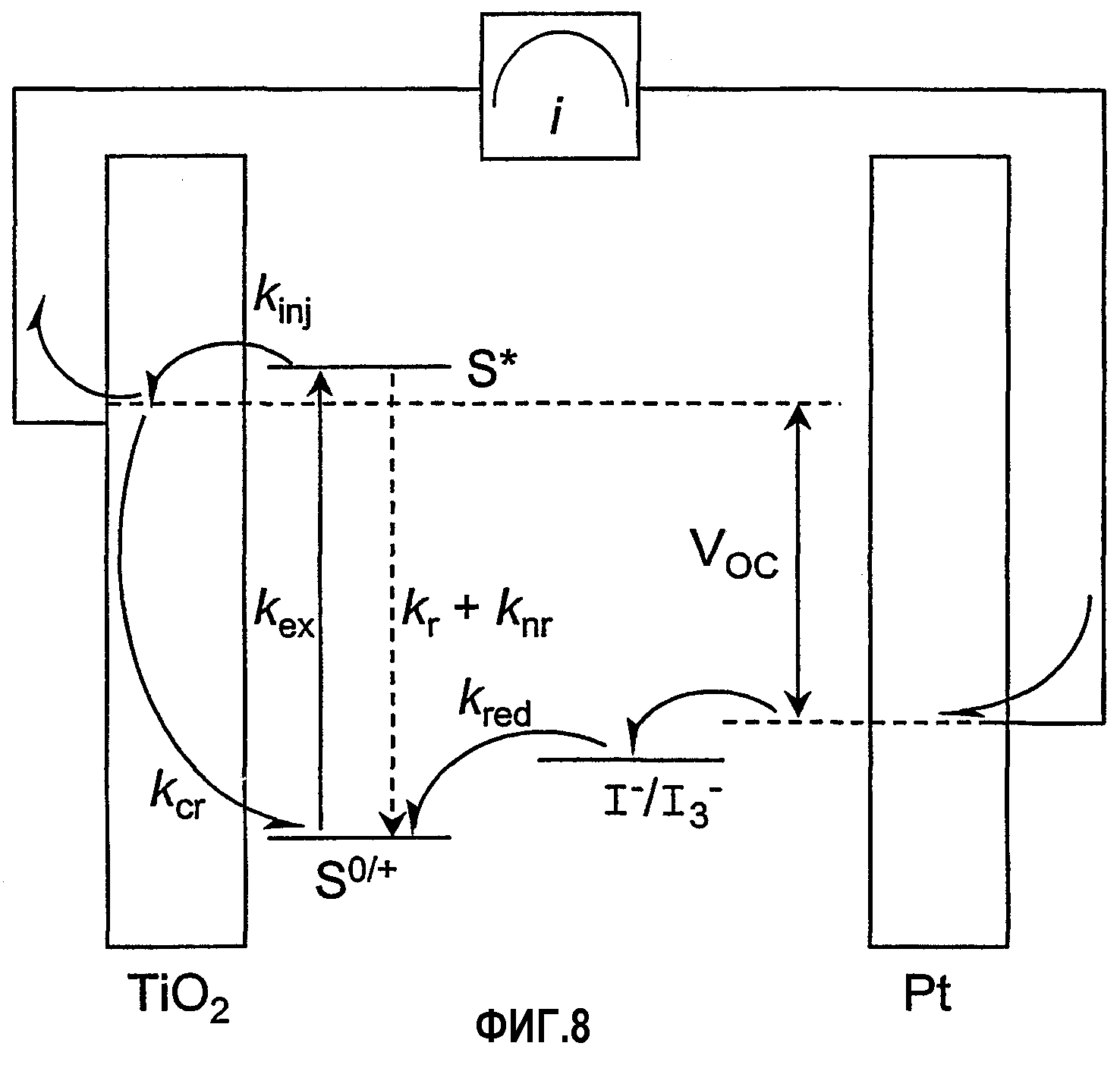
Фигура 8. Упрощенное представление механизма сенсибилизации TiO2 с помощью сенсибилизатора S. Возбуждение светом сенсибилизатора образует возбужденное состояние S*, которое инжектирует инжектируемый электрон в полупроводник с константой скорости kinj. Затем окисленный сенсибилизатор S+ регенерируется с помощью внешнего донора электронов (например, йодида) с константой скорости kred. VOC представляет собой фотопотенциал разомкнутой цепи, который представляет собой максимальную свободную энергию Гиббса, которая, в принципе, может быть получена от элемента при условиях постоянного освещения. Конкуренцию производству энергии составляет рекомбинация зарядов kcr,которая может происходить (из полупроводника) с окисленным сенсибилизатором или окисленным продуктом подвижного носителя заряда (например, трийодида).
Фигура 9. Регенеративный солнечный элемент, сконструированный для функционирования, подобного тому, которое описано на фигуре 8, за исключением того, что твердотельный дырочный проводник заменяет активный при окислении-восстановлении электролит йодида/трийодида.
Фигура 10. Примеры строительных блоков, которые могут быть собраны в хромофорные цепочки.
Фигура 11. Синтетический подход (способ) к приготовлению линейных хромофорных цепочек.
Фигура 12. Рациональный синтез составляющего блока на основе димера порфирина для приготовления хромофорных цепочек.
Фигура 13. Твердофазный синтез с использованием связывания Сузуки для получения цепочек, содержащих порфирин, связанный п -фениленом.
Фигура 14. Твердофазный синтез с использованием связывания Сузуки для получения цепочек, содержащих хлорин, связанный п -фениленом.
Фигура 15. Бифункциональные составляющие блоки для использования при полимеризациях Сузуки.
Фигура 16. Рациональный синтез составляющего блока на основе бифункционального порфирина для использования при полимеризациях Сузуки.
Фигура 17. Твердофазный синтез цепочек, содержащих мезо-, мезо-связанный порфирин с присоединенной карбокси-ручкой.
Фигура 18. Твердофазный синтез цепочек, содержащих мезо-, мезо-связанный порфирин с присоединенной этиновой ручкой.
Фигура 19. Присоединение мезо-, мезо-связанной цепочки к содержащей цирконий двухъярусной сэндвичевой молекуле.
Фигура 20. Пример миграции энергии, но не миграции дырок, в хромофорной цепочке.
Фигура 21. Пример миграции энергии и миграции дырки в противоположных направлениях в хромофорной цепочке.
Фигура 22. Пример каскадной миграции энергии и миграции дырки в противоположных направлениях в хромофорной цепочке, миграция дырки происходит в определенной области цепочки.
Фигура 23. Другой пример твердофазного синтеза с использованием связывания Сузуки для получения цепочек, содержащих хлорин с этиновой ручкой, связанный п-фениленом.
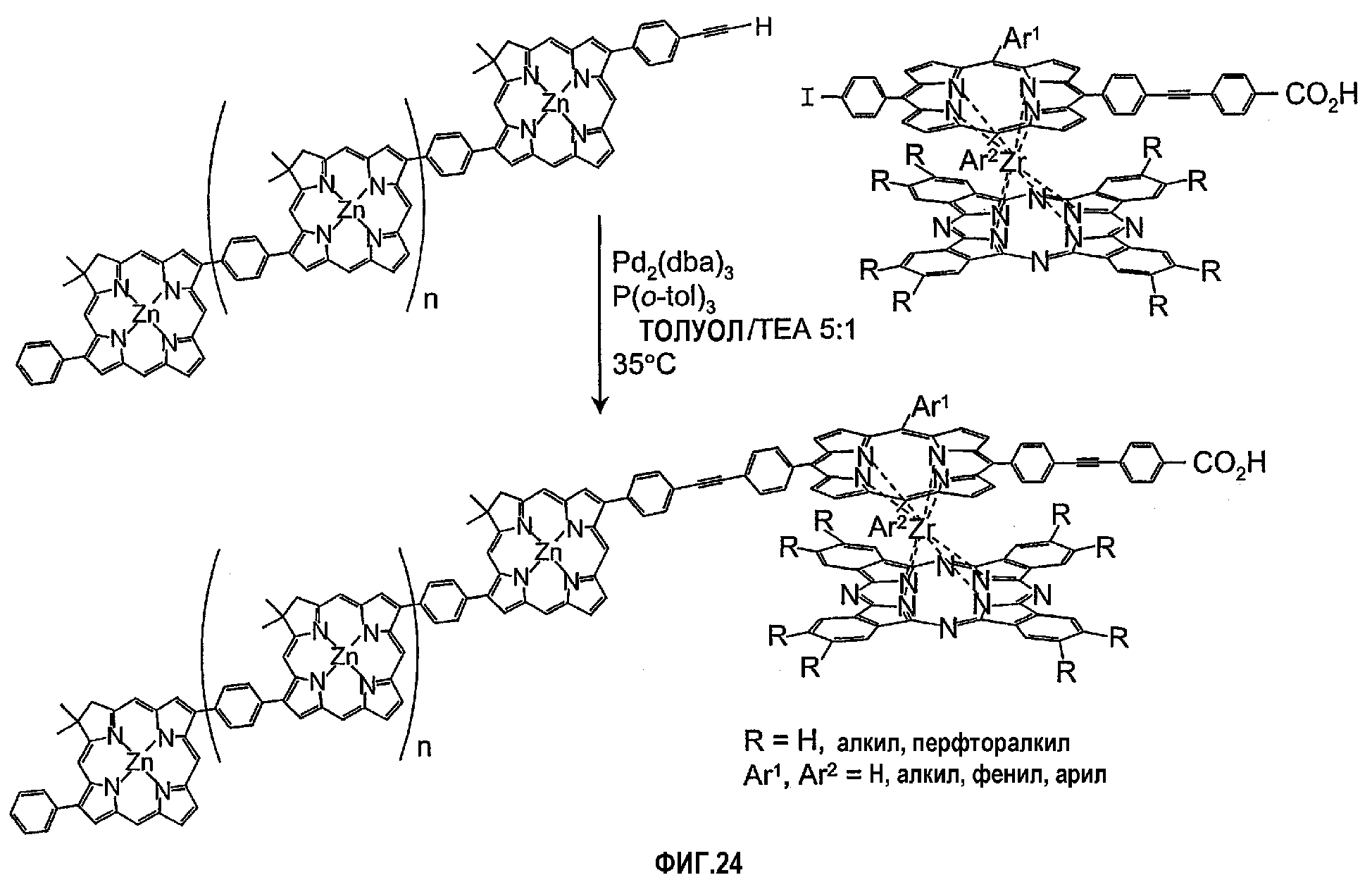
Фигура 24. Присоединение цепочки, содержащей хлорин, связанный п-фениленом, к двухъярусной сэндвичевой молекуле, содержащей цирконий.
Фигура 25. Пример обратимой миграции энергии и необратимой миграции дырки в хромофорной цепочке.
Фигура 26. Цепочка, содержащая бактериохлорин, связанный дифенилэтином.
Фигура 27. Составляющие блоки на основе хлорина, которые имеют заместители (функциональные ручки) в двух из мезо-положений, и ни одного - в β-положениях.
Фигура 28. В порфирине, имеющем самую высокую заселенную молекулярную орбиталь (HOMO) a2u, которая имеет электронную плотность преимущественно в мезо-положениях, и малую - в β-положениях, более высокие скорости (в 2,5-10 раз) наблюдаются, когда линкеры находятся в мезо, а не в β-положениях.
Фигура 29. Четыре различных составляющих блока на основе хлорина, а также номенклатура хлорина, демонстрирующая обозначения A-D колец согласно IUPAC-IUB.
Фигура 30. Ориентация переходного дипольного момента для длинноволновой полосы поглощения в хлорине со свободным основанием и металлохлорин.
Фигура 31. Парное взаимодействие составляющих блоков на основе хлорина при вхождении в ковалентно связанные цепочки.
Фигура 32. Самая высокая заселенная молекулярная орбиталь хлорина представляет собой орбиталь a2, которая имеет заметную электронную плотность на каждой из мезо- и невосстановленных β -позиций.
Фигура 33 иллюстрирует синтез составляющего блока на основе транс-хлорина с двумя β-заместителями.
Фигура 34A. Синтез новой β-замещенной Восточной половины при синтезе хлорина.
Фигура 34B. Синтез новой β-замещенной Восточной половины при синтезе хлорина, продолжающий процесс, представленный на фигуре 34A.
Фигура 35 иллюстрирует синтез новой β-замещенной Западной половины составляющего блока на основе хлорина.
Фигура 36. Другие составляющие блоки на основе хлорина, которые могут быть получены с использованием такой же стратегии синтеза, изображенной выше, и которые имеют по существу такие же физические свойства.
Фигура 37. Синтез составляющих блоков на основе транс мезо-замещенного хлорина (тип III) путем расширения процесса для приготовления хлоринов, несущих на себе соседние (цис) мезо-замещенные хлорины.
Фигура 38. Второй процесс получения составляющих блоков на основе транс мезо-замещенного хлорина (тип III).
Фигура 39. Различные составляющие блоки на основе мезо-замещенного хлорина, которые могут быть получены с помощью синтеза, описанного выше.
Фигура 40. Взаимосвязь антенных комплексов и реакционного центра для получения дырок и электронов из энергии возбуждения, стекающей с антенны.
Фигура 41. Цепочки для аккумуляции света, которые поглощают свет и подвергаются эффективному внутримолекулярному переносу энергии.
Фигура 42. Здесь представлено новое средство для удаления окислительного эквивалента из узла разделения заряда. Энергия протекает вдоль цепочки для аккумуляции света к узлу разделения заряда (УРЗ), в то время как окислительный эквивалент (дырка) протекает в обратном направлении от УРЗ к позиции в антенне, где имеют место последующие реакции с переносом электрона.
Фигура 43. Конструкция согласно фигуре 42 имеет два значительных ответвления. (1) Только два канала для доступа требуются на УРЗ: один для испускания электронов, и один тот, куда втекает энергия возбуждения, а вытекают окислительные эквиваленты (дырки).
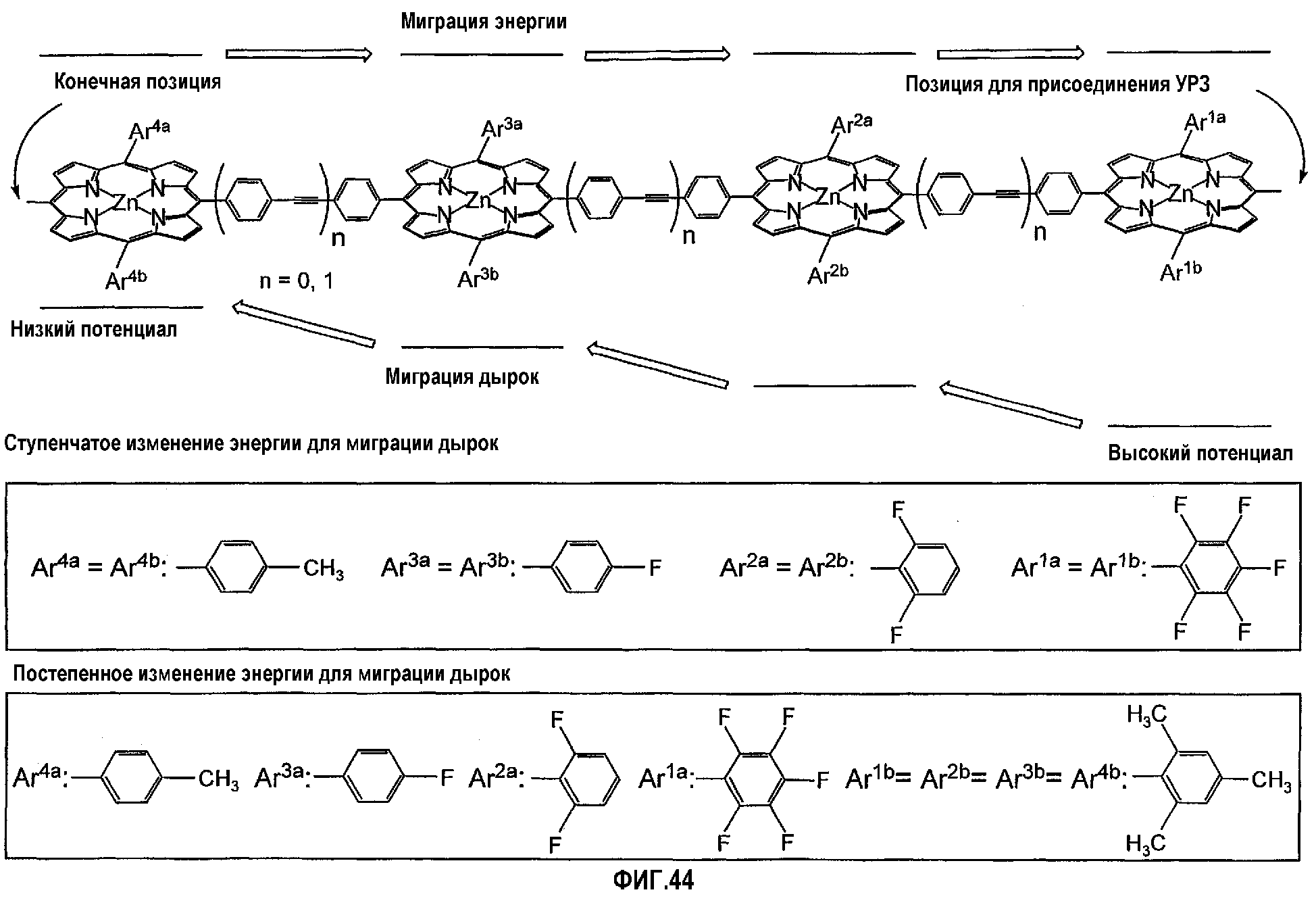
Фигура 44 иллюстрирует линейную цепочку на основе цинк- порфиринов, имеющих различные мезо-заместители.
Фигура 45 иллюстрирует линейную цепочку Mg- и Zn -порфиринов, имеющих различные мезо-заместители.
Фигура 46 иллюстрирует линейную цепочку металлохлоринов, имеющих различные мезо-заместители.
Фигура 47 иллюстрирует линейную цепочку порфиринов и хлоринов, имеющих различные мезо-заместители.
Фигура 48 иллюстрирует линейную цепочку β-замещенных хлоринов и мезо-замещенных хлоринов.
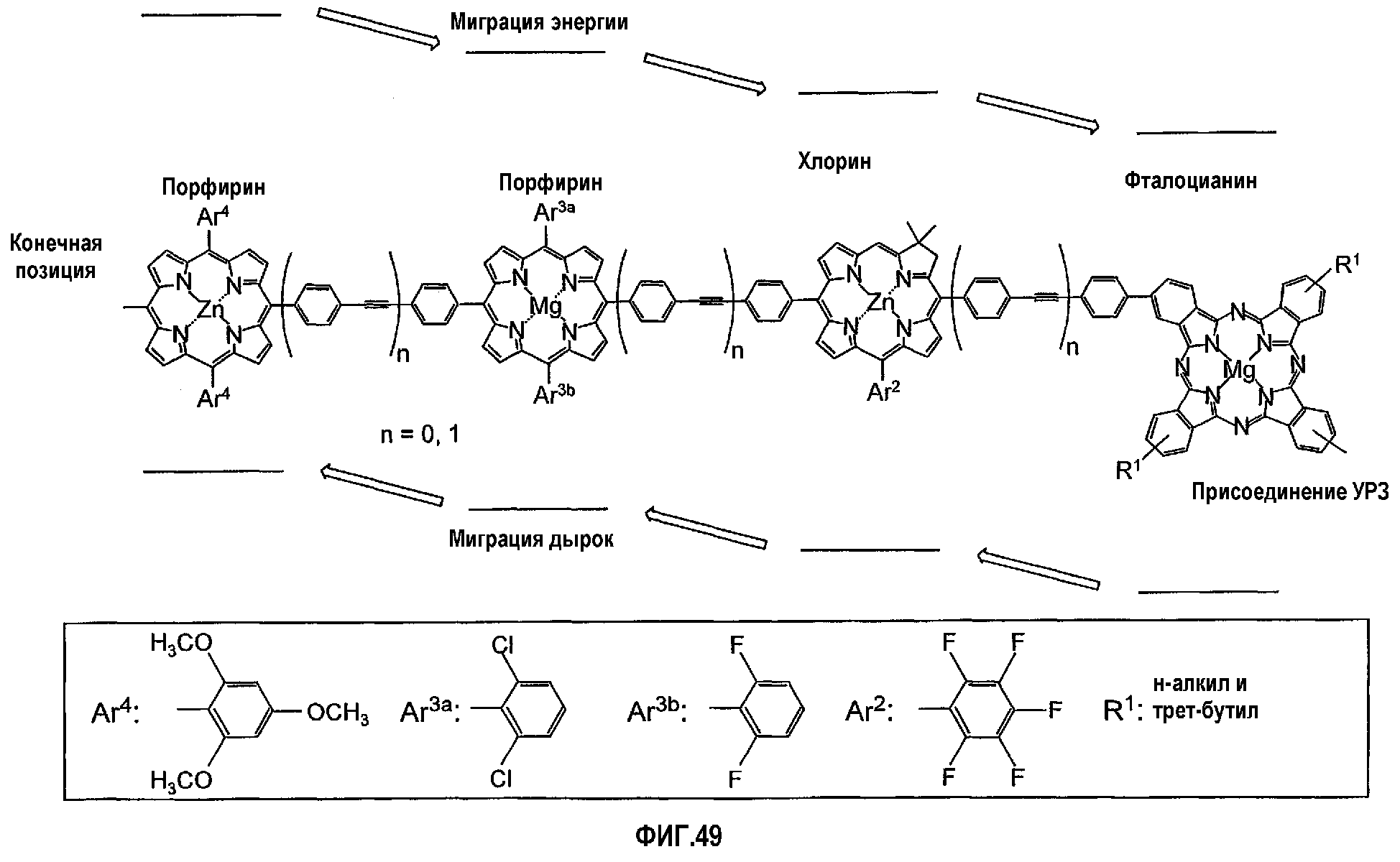
Фигура 49 иллюстрирует линейную цепочку из компонентов порфирина, хлорина и фталоцианина.
Фигура 50 иллюстрирует каскадную (катарактную, от англ. cataract) линейную цепочку, использующую домены, состоящие из множества изоэнергетических пигментов.
Фигура 51 иллюстрирует реакции, пригодные для использования при приготовлении олигомеров стержней для аккумуляции света.
Фигура 52. Полимеризация in situ, приводящая к получению стержня для аккумуляции света на поверхности (например, Au или TiO2), которая будет служить в качестве одного из электродов солнечного элемента.
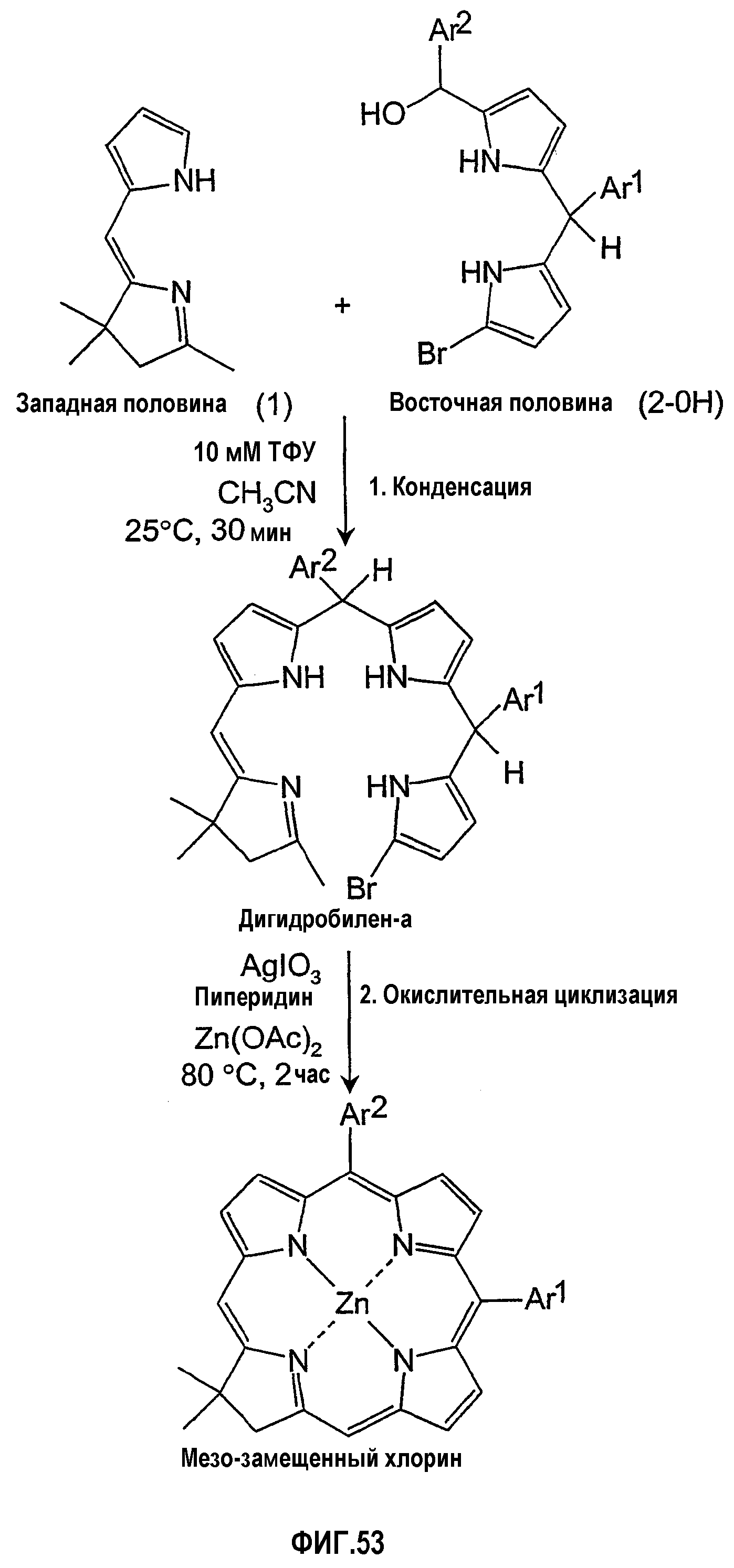
Фигура 53 иллюстрирует синтез мезо-замещенных хлоринов с помощью методик, описанных ранее.
Фигура 54 иллюстрирует синтез предшественников восточной половины (ВП) β-замещенного хлорина.
Фигура 55 дополнительно иллюстрирует синтез предшественников восточной половины β -замещенного хлорина.
Фигура 56 иллюстрирует синтез западной половины (ЗП) β -замещенного хлорина.
Фигура 57 иллюстрирует синтез β-замещенного хлорина.
Фигура 58 иллюстрирует синтез транс β-замещенного хлорина.
Подробное описание предпочтительных вариантов воплощения
Солнечные элементы, описанные здесь, требуют использования линейных хромофорных цепочек (стержней для аккумуляции света), которые обеспечивают сильное поглощение света. Кроме того, когда это желательно, описанные здесь солнечные элементы обеспечивают миграцию энергии и миграцию заряда в противоположных направлениях. Таким образом, хромофорные цепочки поглощают свет и могут проявлять внутреннее выпрямление на молекулярном уровне в потоке энергии возбужденного состояния и дырок в основном состоянии.
Без какого-либо желания ограничить настоящее изобретение можно заметить, что некоторые потенциальные преимущества солнечных элементов, описанных здесь, включают в себя следующие: они тонкие (например, стержни имеют длину не более 500 или даже 200 нанометров), легкие, портативные, гибкие, с хорошей эффективностью, твердотельные (в одном из вариантов воплощения), простые в изготовлении и имеют рациональный молекулярный дизайн. В самом деле, предполагается, что описанное здесь изобретение, сделает возможным там, где это желательно, количественное преобразование падающих фотонов в электроны на индивидуальных длинах волн света и с общими эффективностями >5% при солнечном освещении.
I. Определения
Здесь используются следующие термины и фразы:
Подложка, как здесь используется, предпочтительно представляет собой твердый материал (который может быть гибким или жестким), пригодный для использования при присоединении одной или нескольких молекул. Подложки могут быть сформированы из материалов, включая, но не ограничиваясь этим, стекло, органические полимеры, пластик, кремний, минералы (например, кварц), полупроводниковые материалы, керамика, металлы и тому подобное. Подложка может находиться в любой пригодной для использования форме, включая плоскую, планарную, искривленную, стержнеобразную и тому подобное. Подложка может быть по своей природе проводящей и служить в качестве электрода сама по себе, или электрод может быть сформирован на подложке или соединен с ней с помощью соответствующих средств (например, осаждения слоя золота или слоя проводящего оксида). Либо одна, либо обе подложки в солнечных элементах могут быть прозрачными (то есть длины волн света, которые возбуждают хромофоры, могут проходить через подложку и соответствующий электрод даже в том случае, если они визуально выглядят мутными). В цепочках для аккумуляции света подложка и электрод могут быть любого пригодного для использования типа. Одна из подложек может быть непрозрачной по отношению к длинам волн света, которые возбуждают хромофоры. Одна из подложек может быть отражающей или снабженной отражающим покрытием с тем, чтобы свет, который проходит через цепочки или стержни, отражался назад в цепочки или стержни.
Термин ″электрод″ относится к любой среде, способной переносить заряд (например, электроны) к стержню для аккумуляции света и/или от него. Предпочтительные электроды представляют собой металлы (например, золото, алюминий), неметаллы (например, проводящие оксиды, карбиды, сульфиды, селениды, теллуриды, фосфиды и арсениды, такие как сульфид кадмия, теллурид кадмия, диселенид вольфрама, арсенид галлия, фосфид галлия и тому подобное), и проводящие органические молекулы. Электроды могут быть приготовлены практически с любой 2-мерной или 3-мерной форме.
Термин ″ проводящий оксид″, как он здесь используется, относится к любому пригодному для использования проводящему оксиду, включая бинарные оксиды металлов, такие как оксид олова, оксид индия, оксид титана, оксид меди и оксид цинка, или тройные (третичные) оксиды металлов, такие как титанат стронция и титанат бария. Другие примеры пригодных для использования проводящих оксидов включают в себя, но не ограничиваются этим, оксид индия олова, диоксид титана, оксид олова, оксид галлия индия, оксид цинка и оксид цинка индия. Полупроводники на основе оксидов металлов могут быть собственными или легированными малыми количествами материалов для контроля проводимости.
Термин ″гетероциклический лиганд″, как он здесь используется, относится в целом к любой гетероциклической молекуле, состоящей из атомов углерода и содержащей по меньшей мере один, а предпочтительно, множество гетероатомов (например, N, O, S, Se, Te), причем эти гетероатомы могут быть одинаковыми или различными, и эта молекула способна к образованию координационного сэндвичевого соединения с другим гетероциклическим лигандом (который может быть таким же или иным) и металлом. Такие гетероциклические лиганды, как правило, представляют собой макроциклы, в частности, производные тетрапиррола, такие как фталоцианины, порфирины и порфиразины.
Термин ″ порфириновый макроцикл″ относится к порфирину или порфириновому производному. Такие производные включают в себя порфирины с дополнительными кольцами, орто-конденсированными или ортопери-конденсированными с порфириновым ядром; порфирины, имеющие замещение одного или нескольких атомов углерода порфиринового кольца атомом другого элемента (скелетное замещение); производные, имеющие замещение атома азота порфиринового кольца атомом другого элемента (скелетное замещение азота); производные, имеющие иные заместители, чем водород, расположенные на периферических (мезо-, β-) или внутренних атомах порфирина; производные с замещением одной или нескольких связей порфирина (гидропорфирины, например, хлорины, бактериохлорины, изобактериохлорины, декагидропорфирины, корфины, пиррокорфины и тому подобное); производные, полученные с помощью координационного связывания одного или нескольких металлов с одним или несколькими атомами порфирина (металлопорфирины); производные, имеющие один или несколько атомов, включая пиррольные и пиррометениловые блоки, вставленные в порфириновое кольцо (растянутые порфирины); производные, имеющие одну или несколько групп, удаленных из порфиринового кольца (сжатые порфирины, например, коррин, коррол) и сочетания указанных выше производных (например, фталоцианины, порфиразины, нафталоцианины, субфталоцианины и изомеры порфирина). Предпочтительные порфириновые макроциклы содержат по меньшей мере одно 5-членное кольцо.
Термин порфирин относится к циклической структуре, как правило, состоящей из четырех пиррольных колец вместе с четырьмя атомами азота и двумя атомами водорода, которые легко могут быть замещены атомами различных металлов. Типичный порфирин представляет собой гемин.
″Хлорин″ является по существу тем же термином, что и порфирин, но отличается от порфирина тем, что имеет частично насыщенное пиррольное кольцо. Основным хромофором хлорофилла, т.е. зеленого пигмента для фотосинтеза растений, является хлорин.
″Бактериохлорин″ по существу является тем же, что и порфирин, но отличается от порфирина тем, что имеет два частично насыщенных не соседствующих друг с другом (то есть транс) пиррольных кольца.
″Изобактериохлорин″ является по существу тем же, что и порфирин, но отличается от порфирина тем, что имеет два частично насыщенных соседних (то есть цис) пиррольных кольца.
Термины ″координационное сэндвичевое соединение″ или ″координационный сэндвичевый комплекс″ относятся к соединению формулы LnMn-1, где каждый L представляет собой гетероциклический лиганд, такой как порфириновый макроцикл, каждый M представляет собой металл, n равно 2 или больше, наиболее предпочтительно 2 или 3, и каждый металл располагается между парой лигандов и связывается с одним или несколькими гетероатомами (а как правило, со множеством гетероатомов, например, с 2, 3, 4, 5) в каждом лиганде (в зависимости от окисленного состояния металла). Таким образом, координационные сэндвичевые соединения не являются металлоорганическими соединениями, такими как ферроцен, в которых металл соединяется с атомами углерода. Лиганды в координационном сэндвичевом соединении, как правило, размещаются в виде пакета (то есть являются ориентированными в целом лицом друг к другу и соосно совмещенными друг с другом, хотя они могут иметь или не иметь возможность вращаться вокруг этой оси по отношению друг к другу). Смотри, например, D. Ng and J. Jiang, Chem. Soc. Rev. 26, 433-442 (1997). Координационные сэндвичевые соединения могут быть ″гомолептическими″ (где все лиганды L являются одинаковыми) или ″гетеролептическими″ (где по меньшей мере один лиганд L является иным, чем все остальные лиганды в нем).
Термин ″двухъярусное координационное сэндвичевое соединение″ относится к координационному сэндвичевому соединению, описанному выше, где n равно 2, и, таким образом, имеющему формулу L1-M1-L2, где каждый из L1 и L2 может быть таким же, как все другие или отличным от них. Смотри, например, J. Jiang et al., J. Porphyrins Phthalocyanines 3, 322-328 (1999).
Термин ″мультипорфириновая цепочка″ относится к дискретному количеству из двух или более ковалентно-связанных порфириновых макроциклов. Мультипорфириновые цепочки могут быть линейными, циклическими или разветвленными, но предпочтительно являются линейными. Рассматриваемые стержни для аккумуляции света предпочтительно представляют собой мультипорфириновые цепочки. Стержни для аккумуляции света или мультипорфириновые цепочки могут быть линейными (то есть все порфириновые макроциклы могут быть соединены в транс-положениях) или могут содержать один или несколько изгибов или ″изломов″ (например, путем включения одного или нескольких нелинейных линкеров в стержень для аккумуляции света или путем включения одного или нескольких цис-замещенных порфириновых макроциклов в стержень для аккумуляции света). Некоторые из порфириновых макроциклов могут дополнительно включать в себя дополнительные лиганды, в частности, порфириновые макроциклы, с образованием координационных сэндвичевых соединений, как дополнительно описано ниже. Стержни необязательно, но предпочтительно ориентируются по существу перпендикулярно по отношению к одному из электродов, а наиболее предпочтительно к обоим, т.е. первому и второму электродам.
"Хромофор" означает поглощающий свет узел, который может представлять собой узел внутри молекулы или может составлять целую молекулу. Как правило, хромофор представляет собой сопряженную систему (попеременно двойные и одинарные связи, которые могут включать в себя несвязанные электроны, но они не ограничены попеременными двойными и одинарными связями, поскольку тройные и одинарные связи, смеси попеременных тройных/двойных и одинарных связей также составляют хромофоры. Двойная или тройная связь сама по себе составляет хромофор. Гетероатомы могут быть включены в хромофор). Примеры хромофоров включают в себя циклическую сопряженную систему с 18 пи-электронами, которая придает цвет порфириновым пигментам, линейную систему из попеременных двойных и одинарных связей в зрительном пигменте ретинале или карбонильную группу в ацетоне.
"Группа разделения заряда" или "узел разделения заряда" относится к молекулярным частицам, которые при возбуждении (путем прямого поглощения или переноса энергии от другого поглотителя) передают электрон в другую часть этой же молекулы или передают электрон другой молекуле, полупроводнику или металлу. "Группа разделения заряда" или "узел разделения заряда" дают возможность для сохранения некоторой части энергии возбужденного состояния при перемещении или переносе электрона. Как правило, "группа разделения заряда" или "узел разделения заряда" располагается на том конце цепочки или стержня для аккумуляции света, с которого принимается энергия возбужденного состояния. "Группа разделения заряда" или "узел разделения заряда" облегчает или вызывает преобразование энергии возбужденного состояния в отдельный электрон проводимости, или дырку, или в пару электрон-дырка. Электрон может инжектироваться в полупроводник с помощью "группы разделения заряда" или "узла разделения заряда". Является возможным, чтобы "группа разделения заряда" или "узел разделения заряда" извлекали электрон из другой молекулы или полупроводника, тем самым создавая отрицательный заряд на "группе разделения заряда" или "узле разделения заряда" и дырку в другой молекуле или полупроводнике. Реакционный центр бактериального фотосинтеза представляет собой самый главный пример "группы разделения заряда" или "узла разделения заряда". Синтетические молекулы порфирин-хинон или порфирин-шаровая молекула (фуллерен) также функционируют, поглощая свет и используя полученную в результате энергию для переноса заряда.
Термин ″ заместитель″, как он используется здесь в формулах, в частности, обозначенный с помощью S или Sn, где n является целым числом, в предпочтительном варианте воплощения относится к группам (субузлам) с избытком или недостатком электронов, которые могут использоваться для установления окислительно-восстановительного потенциала (потенциалов) рассматриваемого соединения. Предпочтительные заместители включают в себя, но не ограничиваются этим, H, арил, фенил, циклоалкил, алкил, галоген, алкокси, алкилтио, перфторалкил, перфторарил, пиридил, циано, тиоцианато, нитро, амино, алкиламино, ацил, сульфоксил, сульфонил, амидо и карбамоил. В предпочтительных вариантах воплощения замещенная арильная группа присоединяется к порфирину или к порфириновому макроциклу, и заместители на арильной группе выбираются из группы, состоящей из арила, фенила, циклоалкила, алкила, галогена, алкокси, алкилтио, перфторалкила, перфторарила, пиридила, циано, тиоцианато, нитро, амино, алкиламино, ацила, сульфоксила, сульфонила, амидо и карбамоила. Дополнительные заместители включают в себя, но не ограничиваются этим, 4-хлорфенил, 4-трифторметилфенил и 4-метоксифенил. Предпочтительные заместители обеспечивают диапазон значений окислительно-восстановительного потенциала менее примерно 5 вольт, предпочтительно менее примерно 2 вольт, более предпочтительно менее примерно 1 вольта.
Термин ″арил″ относится к соединению, чьи молекулы имеют кольцевую структуру, характерную для бензола, нафталина, фенантрена, антрацена и тому подобное (то есть либо кольцо с 6 атомами углерода бензола, либо конденсированные кольца с 6 атомами углерода других ароматических производных). Например, арильная группа может представлять собой фенил (C6H5) или нафтил (C10H7). Можно заметить, что арильная группа, когда она действует в качестве заместителя, может и сама иметь дополнительные заместители (например, заместители, предусмотренные как Sn в различных формулах, приведенных здесь).
Термин ″алкил″ относится к парафиновой углеводородной группе, которая может быть получена из алкана путем удаления одного атома водорода из формулы. Примеры представляют собой метил (CH3-), этил (C2H5-), пропил (CH3CH2CH2-), изопропил ((CH3)2CH-).
Термин ″галоген″ относится к одному из электроотрицательных элементов группы VIIA Периодической таблицы (фтор, хлор, бром, йод, астат).
Термин ″перфторалкил″ относится к алкильной группе, где каждый атом водорода заменен атомом фтора.
Термин ″ перфторарил″ относится к арильной группе, где каждый атом водорода заменен атомом фтора.
Термин ″пиридил″ относится к арильной группе, где один узел CR заменен атомом азота.
Термин ″сульфоксил″ относится к группе состава RS(O)-, где R представляет некоторую алкильную, арильную, циклоалкильную, перфторалкильную или перфторарильную группу. Примеры включают в себя, но не ограничиваются этим, метилсульфоксил, фенилсульфоксил и тому подобное.
Термин ″сульфонил″ относится к группе состава RSO2-, где R представляет некоторую алкильную, арильную, циклоалкильную, перфторалкильную или перфторарильную группу. Примеры включают в себя, но не ограничиваются этим, метилсульфонил, фенилсульфонил, п-толуолосульфонил и тому подобное.
Термин ″карбамоил″ относится к группе состава R1(R2)NC(O)-, где R1 и R2представляют H или некоторую алкильную, арильную, циклоалкильную, перфторалкильную или перфторарильную группу. Примеры включают в себя, но не ограничиваются этим, N -этилкарбамоил, N, N-диметилкарбамоил и тому подобное.
Термин ″амидо″ относится к группе состава R1CON(R2)-, где R1 и R2представляют собой H или некоторую алкильную, арильную, циклоалкильную, перфторалкильную или перфторарильную группу. Примеры включают в себя, но не ограничиваются этим, ацетамидо, N-этилбензамидо и тому подобное.
Термин ″ацил″ относится к группе органической кислоты, в которой -OH карбоксильной группы замещен некоторым другим заместителем (RCO-). Примеры включают в себя, но не ограничиваются этим, ацетил, бензоил и тому подобное.
В предпочтительных вариантах воплощения, когда металл обозначается ″M″ или ″Mn″, где n представляет собой целое число, можно заметить, что металл может быть связан с противоионом.
Линкер представляет собой молекулу, используемую для связывания двух различных молекул, двух субузлов молекулы или молекулы и подложки. Когда все они связываются ковалентно, они образуют узлы единой молекулы.
Термин ″электрически соединенный″, когда он используется по отношению к стержню для аккумуляции света и электроду или к хромофорам, к группам разделения заряда и к электродам, относится к связи между этой группой или молекулой и связанной с ними группой или электродом, так что электроны перемещаются от среды/молекулы для хранения к электроду или от электрода к молекуле и, тем самым, изменяют окисленное состояние молекулы для хранения. Электрическое связывание может включать в себя непосредственную ковалентную связь между средой/молекулой для хранения и электродом, опосредованное ковалентное связывание (например, через линкер), непосредственное или опосредованное ионное связывание между средой/молекулой для хранения и электродом или другое связывание (например, гидрофобное связывание). Кроме того, может и не требоваться никакого реального связывания, и стержень для аккумуляции света может просто находиться в контакте с поверхностью электрода. Также не существует жесткой необходимости в контакте между электродом и стержнем для аккумуляции света, когда электрод находится достаточно близко к стержню для аккумуляции света, чтобы сделать возможным туннелирование электрона между средой/молекулой и электродом.
"Энергия возбужденного состояния" относится к энергии, хранимой в хромофоре в метастабильном состоянии после поглощения света (или переноса энергии от поглотителя). Для возбужденного синглетного (триплетного) состояния величина "энергии возбужденного состояния" определяется энергией самой короткой длины волны полосы флуоресценции (фосфоресценции). Величина "энергии возбужденного состояния" является большей или равной энергии разделенных электрона и дырки после разделения заряда.
Электролиты, используемые для осуществления настоящего изобретения, могут быть водными или неводными электролитами, включая полимерные электролиты. Электролит может содержать твердое тело или состоять из него, и в этом последнем случае солнечный элемент может быть изготовлен с исключением жидкости в пространстве между первой и второй подложками. Электролит состоит из или содержит вещество, которое повышает электрическую проводимость среды-носителя. Большинство электролитов представляют собой соли или ионные соединения. Примеры включают в себя хлорид натрия (поваренная соль), йодид лития или бромид калия в воде; тетрабутиламмоний гексафторфосфат или тетраэтиламмоний перхлорат в ацетонитриле или дихлорметане; или ионный полимер в геле.
"Подвижные носители заряда" относятся к иону, молекуле или к другим частицам, способным перемещать заряды (электроны или дырки) между двумя электродами в солнечном элементе. Примеры включают в себя хиноны в воде, расплавы солей и йодид в полимерном геле, таком как полиакрилонитрил. Примеры подвижных носителей заряда включают в себя, но не ограничиваются этим, йодид, бромид, тетраметил-1,4-фенилендиамин, тетрафенил-1,4-фенилендиамин, п-бензохинон, C60, C70, пентацен, тетратиафульвален и метилвиологен.
II. СОЛНЕЧНЫЕ ЭЛЕМЕНТЫ, СОДЕРЖАЩИЕ СТЕРЖНИ ДЛЯ
АККУМУЛЯЦИИ СВЕТА
A. Введение
Цель разработки сверхтонкого солнечного элемента с высоким коэффициентом поглощения достигается с помощью линейных цепочек хромофоров, которые служат в качестве стержней для аккумуляции света (АС). Общая конструкция линейных цепочек хромофоров изображена на фигуре 2. Пигменты (то есть молекулы, содержащие хромофоры) соединяются ковалентно с помощью линкеров с образованием линейной архитектуры. На одном конце цепочки располагается сенсибилизатор или узел разделения заряда (УРЗ). УРЗ также присоединяется к электроду с помощью линкера и функциональной группы, обозначаемой Y. На дальнем конце стержня для аккумуляции света находится конечная группа, обозначаемая Z. Конечная группа может состоять из простого алкильного или арильного заместителя или может содержать линкер для присоединения к поверхности или к противоэлектроду. При присоединении линейных молекул АС/УРЗ к электроду с помощью соединительной группы Y стержни будут ориентироваться более или менее вертикально. При этом линейная стержнеобразная архитектура делает возможным многослойное пакетирование пигментов, где каждый пигмент в стержне удерживается рядом с соседними пигментами в том же самом стержне с помощью линкера L. Структуры (модели) упаковки и расстояния между стержнями контролируются с помощью заместителей, интегрированных с пигментами. В целом они упоминаются как ″цепочки хромофоров″, причем этот термин используется взаимозаменяемо с линейными стержнями для аккумуляции света (оба термина указывают на линейную архитектуру связанных пигментов, которые эффективно поглощают свет и аккумулируют энергию (и дырки) контролируемым образом). Заметим, что термины ″сенсибилизатор″ или ″разделение заряда″ используются взаимозаменяемо; последний термин подчеркивает тот факт, что фотовозбуждаемый агент (сенсибилизатор), который инжектирует электроны в полупроводник, может состоять из множества узлов (например, из порфирина-хлорина, хлорина-хинона, бактериохлорина-шаровых молекул (фуллеренов)).
Три различных конструкции описываются для цепочек хромофоров (смотри ниже). Главным для всех конструктивных схем является тот факт, что цепочки будут аккумулировать большую долю падающего солнечного излучения. Стратегия использования монослоя молекулярных сенсибилизаторов на планарной поверхности электрода исторически была забракована, поскольку поглощается только малая доля падающего солнечного света. Описываемое изобретение концептуализирует новый молекулярный подход, в котором предварительно полученные цепочки хромофоров будут организовываться на поверхности электрода. Путем сборки цепочек перпендикулярно поверхности электрода монослойные покрытия будут приводить к значительному повышению поглощения света. Например, фталоцианины, как правило, имеют коэффициенты экстинции ˜250000 M-1см-1 в красной части видимого спектра (600-700 нм в зависимости от состояния металлирования). Монослой из таких фталоцианинов на плоской поверхности соответствует ˜10-10 моль/см2 и будет поглощать примерно 5,6% падающего света. Цепочка из 20 фталоцианинов с аддитивным поглощением (то есть без новых полос поглощения, связанных с агрегацией и/или электронными взаимодействиями), пространственно организованная таким образом, что она занимает такую же площадь поверхности, поглощала бы 68% падающего света. Если количество фталоцианинов увеличивается до 40 или фактор шероховатости поверхности электрода был равен двум, поглощалось бы 90% падающего света. Многие поверхности электродов изначально являются шероховатыми, так что монослой из цепочек в 20 хромофоров (то есть каждая цепочка состоит из 20 хромофоров) должен приводить по существу к количественному поглощению света. Этот ″выступ″ (толщина слоя) выглядит очень благоприятно в сравнении с фактором шероховатости поверхности ˜1000, необходимым для эффективной аккумуляции света, как используется в настоящее время в элементах типа Gratzel.
В конструкции I стержни АС/УРЗ присоединяются к одному из электродов через соединительную группу Y (фигура 3). Элемент включает в себя подвижные (то есть диффундирующие) носители заряда. Линейные стержни АС, обычно содержащие по 5-20 пигментов, поглощают свет. Перенос энергии возбуждения между пигментами в стержне с помощью механизмов передачи через пространство и/или через связь приводит к тому, что энергия достигает УРЗ (иллюстрируется на стадии 2, фигура 3). Затем возбужденный УРЗ инжектирует электрон в зону проводимости электрода (стадия 4). Возникшая в результате дырка остается на УРЗ и не может мигрировать в стержень АС, поскольку окислительный потенциал УРЗ является более низким, чем у непосредственно соседствующих с ней пигментов в стержне АС. Диффузия подвижного носителя заряда вблизи окисленного УРЗ приводит к переносу электрона/дырки, регенерируя УРЗ и оставляя дырку на подвижном носителе заряда. Затем дырка движется под действием диффузии подвижного носителя заряда и/или последующих процессов переноса дырки между подвижными носителями заряда до тех пор, пока не будет достигнут противоэлектрод на дальнем конце стержня АС (вблизи Z; не изображен).
В конструкции II стержни АС/УРЗ присоединяются к одному из электродов через соединительную группу Y (фигура 4). Элемент включает в себя подвижные (то есть диффундирующие) носители заряда. Все особенности являются такими же, как и в конструкции I, с тем только исключением, что дырка, образовавшаяся в УРЗ (при инжекции электрона в электрод), может мигрировать внутри линейного стержня АС. Это имеет два последствия. (1) Время жизни состояния разделения заряда увеличивается, давая существенное уменьшение процессов рекомбинации зарядов на границе раздела УРЗ-электрод. (2) Подвижные носители заряда могут получить доступ к дырке в областях, удаленных от поверхности электрода. Области, удаленные от поверхности, как ожидается, являются более доступными, тем самым облегчается перенос дырок и их миграция (посредством диффузии) к противоэлектроду.
В конструкции III стержни АС/УРЗ присоединяются к одному из электродов через соединительную группу Y (фигура 5). Противоположный конец каждого стержня присоединен к противоэлектроду. Никаких подвижных (то есть диффундирующих) носителей заряда в элементе не присутствует (хотя электролит может присутствовать). Поглощение света, миграция энергии между пигментами и перенос электрона в УРЗ происходит точно так же, как такие же процессы в конструкциях I и II. Однако дырка в УРЗ, возникающая из-за инжекции электрона в электрод, мигрирует по прыжковому механизму переноса дырок между пигментами в стержне АС, а затем переносится в противоэлектрод. Существуют несколько разновидностей этой конструкции. (1) Никаких диффундирующих носителей заряда не присутствует в элементе. (2) Только два различных канала для доступа являются необходимыми в УРЗ; один для переноса электрона в электрод и один, обеспечиваемый стержнем АС, для переноса внутрь энергии возбуждения и переноса наружу образующихся дырок. В противоположность этому, конструкция I требует трех каналов для доступа в УРЗ: один для миграции энергии внутрь, один для переноса электрона наружу и один для подвижных носителей заряда с тем, чтобы получить доступ к дырке. Отсутствие подвижных носителей заряда в конструкции III приводит к созданию твердотельного солнечного элемента.
В предыдущих исследованиях явлений аккумуляции света были созданы цепочки в форме звезд, состоящие из порфиринов и фталоцианинов (Li, J.; Lindsey, J.S.J.Org.Chem. 1999, 64, 9101 -9108; Li, J et al., J.Org.Chem. 1999, 64, 9090-9100; Li, F. et al., J.Mater.Chem. 1997, 7, 1245-1262), кластер из бор-дипирриновых красителей, окружающих порфирин (Li, F. et al J.Am.Chem.Soc. 1998, 120, 10001-10017), и линейная цепочка из четырех порфиринов и одного бор-дипиррина (Wagner, R.W.; Lindsey, J.S.J.Am.Chem.Soc. 1994, 116, 9759-9760), а также циклические цепочки (Li, J. et al., J.Am.Chem.Soc. 1999, 121, 8927-8940). Также изучалось влияние различных металлов в металлопорфиринах при модуляции скорости переноса энергии (Hascoat, P. et al., Inorg.Chem. 1999, 38, 4849-4853). Также были охарактеризованы воздействия различных линкеров на скорость переноса энергии (Hsiao, J.-S. et al., J.Am.Chem. Soc. 1996, 118, 11181-11193; Yang, S. I. et al., J.Phys.Chem. 1998, 102, 9426-9436), композицию орбиталей (Strachan, J.P. et al., J.Am.Chem. Soc. 1997, 119, 11191-11201) и место расположения линкера на порфириновом пигменте (Yang, S.I. et al., J.Am.Chem. Soc. 1999, 121, 4008-4018). Также было осуществлено моделирование миграции энергии в линейных цепочках хромофоров с целью оценки рабочих характеристик различных конструкций архитектуры молекул (Van Patten, P.G. et al., J.Phys.Chem.B 1998, 102, 4209-4216). Эти синтетические молекулы, аккумулирующие свет, сильно поглощают в видимой области и подвергаются очень эффективному переносу энергии. Как часть этих исследований, изучение свойств окисленных комплексов показало быстрый перенос дырок по прыжковому механизму между компонентами (Seth, J. et al., J.Am.Chem.Soc. 1994, 116, 10578-10592; Seth, J. et al., J.Am.Chem.Soc. 1996, 118, 11194-11207). Особенности, предложенные здесь для использования при конструировании линейных хромофорных цепочек и описанные в настоящем изобретении, подтверждаются, но не все они являются ожидаемыми исходя из всех этих предыдущих работ, составляющих уровень техники. Способы синтеза для приготовления цепочек для аккумуляции света являются достаточными для специалиста в данной области, чтобы получить молекулы, описанные здесь. В частности, существует множество способов для приготовления порфириновых составляющих блоков (a) Lindsey, J.S. et al., Tetrahedron 1994, 50, 8941-8968. (b) Lindsey, J.S. In The Porphyrin Handbook; Kadish, K.M.; Smith, K.M.; Guilard, R., Eds.; Academic Press, San Diego, CA 2000, Vol.1, pp.45-118; Cho, W.-S. et al., J.Org.Chem. 1999, 64, 7890-7901; Wagner, R.W. et al., J.Am.Chem.Soc. 1996, 118, 11166-11180; Balasubramanian, T.; Lindsey, J.S. Tetrahedron 1999, 55, 6771-6784), составляющих блоков на основе хлорина (Strachan, J.P. et al., J.Org.Chem. 2000, 65, 3160-3172), фталоцианинов (Yang, S.I. et al., J. Mater. Chem. 2000, 10, 283 -297; Tomoda, H. et al., Chem. Lett. 1980, 1277-1280; Tomoda, H. et al., Chem. Lett. 1983, 313-316) и связанных с ним хромофоров (Wagner, R.W.; Lindsey, J. S. Pure Appl. Chem. 1996, 68, 1373-1380). Способы соединения содержащих хромофоры (то есть пигмент) составляющих блоков в линейные цепочки также установлены (Wagner, R.W. et al., J.Org.Chem. 1995, 60, 5266 -5273; DiMagno, S.G. et al., J.Org.Chem. 1993, 58, 5983 -5993; Wagner, R.W. et al., Chem. Mater. 1999, 11, 2974 -2983), причем они включают в себя, но не ограничиваются этим, способы связывания, опосредуемые Pd. Были синтезированы более разработанные архитектуры, состоящие из цепочек для аккумуляции света и узлов разделения заряда, которые демонстрируют очень высокую эффективность (Kuciauskas, D. et al., J.Am.Chem. Soc. 1999, 121, 8604-8614).
B. Компоненты
Ключевыми требованиями для пигментов, предназначенных для аккумуляции света, являются интенсивное поглощение в видимой области, узкое распределение энергий возбужденного состояния (отмечаемого резкими полосами поглощения и флуоресценции), время жизни возбужденного синглетного состояния, достаточное для переноса энергии (как правило, несколько наносекунд) и совместимость с подходом к синтезу, использующим составляющие блоки и приводящим к получению линейной архитектуры. Пигменты, выбранные для использования в линейных стержнях АС, выбираются из порфиринового семейства (тетрапиррольные макроциклы). Примеры включают в себя порфирины, хлорины, бактериохлорины, тетраазапорфирины (порфиразины), фталоцианины, нафталоцианины и производные этих соединений. Порфириновые пигменты могут быть дополнены вспомогательными пигментами, такими как члены семейств периленов, ликопенов, ксантенов и дипиррометенов. Спектры поглощения таких пигментов хорошо известны специалистам в данной области техники, и их можно увидеть в различных ссылочных источниках (Du, H. et al., Photochem. Photobiol. 1998, 68, 141 -142).
Важными требованиями для линкеров, соединяющих пигменты, являются следующие. (1) Поддержка быстрых процессов переноса энергии возбужденного состояния (через связь и/или через пространство), (2) поддержка процессов переноса дырок в основном состоянии по прыжковому механизму, необходимая в некоторых случаях (конструкции II и III), и (3) достижение совместимости с подходом к синтезу, использующим составляющие блоки и приводящим к получению линейной структуры из пигментов. Выбранные линкеры для соединения пигментов в виде линейных стержней АС включают в себя 4, 4'-дифенилэтин, 4,4'-дифенилбутадиин, 4,4'-бифенил, 1,4 -фенилен, 4,4'-стильбен, 1,4-бициклооктан, 4,4'-азобензол, 4,4'-бензилиденанилин, 4,4''-терфенил и отсутствие линкера (то есть непосредственную C-C связь). Линкеры на основе п,п' -дифенилэтина и п-фенилена, как показано, поддерживают быстрый перенос энергии возбужденного состояния и процессы переноса дырок в основном состоянии по прыжковому механизму между порфириновыми молекулами.
Одно из важных требований к узлу разделения заряда (УРЗ) заключается в том, чтобы он имел энергию возбужденного состояния, равную или меньшую, чем у соседних пигментов в цепочке АС (другими словами, поглощала свет с длинами волн, равными или большими, чем пигменты в цепочке АС). Для солнечных элементов на основе полупроводников восстановительный потенциал возбужденного состояния должен быть большим, чем край (дно) зоны проводимости. Дополнительные требования к УРЗ заключаются в том, чтобы он подвергался быстрому переносу электрона в возбужденном состоянии, имел энергию, достаточную для инжекции электрона в зону проводимости электрода, и давал стабильный катион-радикал. Молекулы, выбранные для УРЗ, также выбираются из порфиринового семейства, включая порфирины, хлорины, бактериохлорины, тетраазапорфирины (порфиразины), фталоцианины, нафталоцианины и производные этих соединений. Особенно привлекательная группа производных состоит из двухъярусных сэндвичевых молекул с центральным металлом, таким как цирконий (Kim, K. et al., Inorg.Chem. 1991, 30, 2652-2656; Girolami, G.S. et al., Inorg.Chem. 1994, 33, 626-627; Girolami, G.S. et al., Angew. Chem.Int.Ed.Engl. 1996, 35, 1223-1225; Collman, J.P. et al., Inorg.Chem. 1997, 36, 5603-5608). Примеры двухъярусных сэндвичевых молекул изображены на фигуре 6. Такие двухъярусные соединения могут быть получены из любого из лигандов в семействе тетрапиррольных макроциклов.
В порфириновом семействе элекрохимический потенциал данного порфирина может быть подобран в очень широком диапазоне путем включения в состав заместителей, отбирающих электрон или высвобождающих электрон (Yang, S.I. et al., J. Porphyrins Phthalocyanines 1999, 3, 117-147). Примеры таких заместителей включают в себя арил, фенил, циклоалкил, алкил, галоген, алкокси, алкилтио, перфторалкил, перфторарил, пиридил, циано, тиоцианато, нитро, амино, N-алкиламино, ацил, сульфоксил, сульфонил, имидо, амидо и карбамоил. У мономерных порфиринов изменение элекрохимического потенциала может также быть достигнуто с помощью различных центральных металлов (Fuhrhop, J.-H.; Mauzerall, D. J.Am.Chem.Soc. 1969, 91, 4174-4181). Большое множество металлов может быть включено в порфирины. Те металлы, которые являются фотохимически активными, включают в себя Zn, Mg, Al, Sn, Cd, Au, Pd и Pt. Понятно, что некоторые металлы несут противоион. Порфирины, как правило, образуют очень стабильные катион-радикалы (Felton, R.H. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, 1978; Vol.V, pp.53 -126).
Линкеры, соединяющие УРЗ с поверхностью электрода, обеспечивают линейную архитектуру, поддерживают перенос электрона через пространство и/или через связь и имеют функциональную группу, пригодную для использования при присоединении к электроду. Примеры пригодных для использования функциональных групп включают в себя сложный эфир, карбоновую кислоту, борную кислоту, тиол, фенол, силан, гидрокси, сульфоновую кислоту, фосфоновую кислоту, алкилтиол и тому подобное. Линкеры могут состоять из 4,4'-дифенилэтина, 4,4' -дифенилбутадиина, 4, 4'-бифенила, 1,4-фенилена, 4,4'-стильбена, 1,4-бициклооктана, 4,4'-азобензола, 4,4'-бензилиденанилина, 4,4''-терфенила, 1,3-фенила, 3,4'-дифенилэтина, 3,4'-дифенилбутадиина, 3,4'-бифенила, 3, 4'-стильбена, 3,4'-азобензола, 3,4'-бензилиденанилина, 3,4''-терфенила и тому подобное.
C. Материалы
Изобретение синтезируемых хроморфорных цепочек, сконструированных для векторного переноса энергии и заряда в том случае, когда они собираются на поверхностях электродов, дает возможность для использования проводящих материалов в качестве подложек для устройств преобразования солнечной энергии. Это разнообразие материалов делает возможным конструктивную разработку солнечных элементов для конкретных применений, некоторые из которых описаны выше. Ниже описываются материалы и механизмы преобразования солнечной энергии (конструкции I-III), которые, как ожидается, дают улучшение эффективности преобразования.
1. Переходы полупроводник-хромофорная цепочка. Генерация анодного фототока представляет собой наиболее распространенный и эффективный механизм, с помощью которого солнечная энергия может аккумулироваться в полупроводниках с помощью молекулярных хромофоров (Gerischer, H. Photochem. Photobiol. 1972, 16, 243; Gerischer, H. Pure Appl.Chem. 1980, 52, 2649; Gerischer, H.; Willig, F. Top.Curr.Chem. 1976, 61, 31). Полупроводниковые материалы, такие как TiO2 (рутил или анатаз), ZnO, SrTiO3, SnO2 и In2O3,являются термодинамически стабильными и могут быть получены в виде тонких пленок, поликристаллических подложек, тонких пленок из коллоидных частиц или монокристаллов с высокой прозрачностью в видимой области. Большая запрещенная зона (>3 эВ) обеспечивает то, что прямое возбуждение полупроводника будет минимальным для случая применений на земле. Кроме того, такие материалы, как SnO2, являются коммерчески доступными на гибких полимерных подложках.
Графическое представление диаграммы типа Gerischer для повсеместно принятого механизма приведена на фигуре 7 для молекулярного сенсибилизатора S. Для изображенного случая потенциал восстановления возбужденного состояния лежит выше края (дна) зоны проводимости на величину, большую, чем энергия реорганизации [Eo (S+/*)-λ>ECB]. Это энергетическое позиционирование приводит к максимальному перекрыванию донорных уровней возбужденного состояния сенсибилизатора и непрерывной зоны проводимости полупроводника, что, в свою очередь, обеспечивает максимальную скорость переноса электрона, то есть безбарьерный перенос электрона через границу раздела. Избыток энергии инжектированного электрона диссипируется с помощью колебаний решетки (фононов) по мере того, как электрон термализуется на дне зоны проводимости. По этой причине инжекция электрона является необратимой, и в самом деле, перераспределение заселенностей возбужденного состояния никогда не наблюдается.
Гибель инжектированного электрона, как ожидается, зависит от условий смещения (искривления зон) в полупроводнике. Если инжекция осуществляется, когда полупроводник находится вблизи состояния с плоскими зонами (a на фигуре 7), тогда ожидается быстрая рекомбинация с окисленным красителем, поскольку здесь не существует области электрического поля, чтобы способствовать пространственному разделению инжектированного электрона и окисленной дырки. Если полупроводник находится в условиях обеднения, инжектированный электрон отклоняется в сторону объема поверхностным электрическим полем, и рекомбинация замедляется (b на фигуре 7). Следовательно, появление тока при измерениях характеристики зависимости фототок-напряжение может быть принято в качестве грубой оценки отсутствия искривления зон. Повышенное время жизни состояния с разделенным зарядом на границах раздела молекулы-полупроводник в условиях обеднения делает возможной регенерацию сенсибилизатора с помощью внешнего донора электронов для применений в регенеративных солнечных элементах.
Упрощенный регенеративный солнечный элемент на основе молекулярного сенсибилизатора на полупроводнике n-типа изображен на фигуре 8. Возбужденный сенсибилизатор S* инжектирует электрон в полупроводник с константой скорости kinj. Молекулы окисленного красителя принимают электрон от донора (то есть подвижного носителя заряда), присутствующего в электролите, kred. Йодид представляет собой донор, изображенный на фигуре. Продукты окисления йодида восстанавливаются на темновом катоде. Суммарный процесс предоставляет возможность для генерации электрического тока светом с энергией, меньшей, чем запрещенная зона у полупроводника, причем без суммарных химических превращений. Рекомбинация заряда kcr может происходить до окисленного сенсибилизатора S+ или до окисленных частиц донора. Изображенный донор представляет собой йодид, который может быть диспергирован в воде, органическом растворителе или в полимерных гелях. Альтернативные активные при окислении-восстановлении (т.е. электрохимически активные) электролиты включают в себя Br-/Br2, хинон/гидрохинон и неорганические координационные соединения.
Могут быть использованы твердотельные элементы, которые использовали бы вместо активного при окислении-восстановлении электролита ″дырочные″ проводники, полупроводники p-типа или, возможно, металлы. Первые две альтернативы имеют прецедент, и дырочные проводники, такие как TPD (N,N-дифенил-N',N'-бис(3-метилфенил)-1,4-фенилендиамин) или OMeTAD [2,2',7,7'-тетракис(N, N-бис(п-метоксифенил)амин]-9,9'-спиробифторен, известны (Salbeck, J. et al., Synth. Met. 1997, 91, 209). В этой связи были использованы также полупроводники p-типа, такие как CuNCS (O'Regan, B.; Schwartz, D.T. Chem.Mater. 1998, 10, 1501).В обоих случаях материалы должны быть термодинамически способны к восстановлению окисленного компонента в цепочке хромофоров. Пример того, как такой элемент может работать с дырочным проводником, представлен на фигуре 9.
Путем использования хромофорных цепочек вместо одного отдельно взятого монослоя молекул сенсибилизатора S может быть реализовано значительное повышение эффективности. Например, в конструкции I сенсибилизатор S заменяется УРЗ, с которым ковалентно связывается жесткая цепочка хромофоров. Цепочка в целом простирается по нормали к поверхности электрода. Преимущество этого подхода заключается в том, что реальная площадь поверхности, занимаемая на поверхности электрода, является сравнимой с такой площадью для одного отдельно взятого сенсибилизатора S, но эффективность аккумуляции света является значительно большей. В описываемых солнечных элементах эффективность преобразования падающих фотонов в ток (IPCE) представляет собой произведение трех параметров согласно уравнению 1.
IPCE=LHE×Φinj×η (1)
Термин LHE обозначает эффективность аккумуляции света и является эквивалентом термина α по IUPAC (коэффициент поглощения), который равен доле поглощенного света (то есть (1-T), где T представляет собой коэффициент прохождения). Термин Φinj представляет собой квантовый выход инжекции электрона в электрод через границу раздела. Термин η представляет собой эффективность сбора электронов, то есть долю инжектируемых электронов, которая достигает электрической цепи. Цепочки хромофоров в конструкции I должны увеличивать LHE без уменьшения остальных параметров, и по этой причине ожидается более высокая степень преобразования солнечного света.
Применение конструкции II к сенсибилизированному полупроводниковому электроду, как ожидается, обеспечит более высокую эффективность преобразования солнечного света по сравнению с конструкцией I, поскольку «дырка» будет перемещаться к конечному хромофору в цепочке и прочь от УРЗ и полупроводникового электрода. Эта миграция дырки должна предотвратить рекомбинацию электрона в полупроводнике с ″дыркой″ в хромофорной цепочке. Кроме того, поскольку стадия регенерации будет происходить дальше от поверхности электрода, ожидается понижение рекомбинации с продуктами окисления донора. Оба этих улучшения кинетики должны повышать η, т.е. эффективность сбора электронов в уравнении 1, и поэтому ожидается более высокий фототок. Кроме того, как ожидается, возрастет фотонапряжение разомкнутой цепи VOC. В регенеративных солнечных элементах максимальная свободная энергия Гиббса, которая может быть получена, соответствует разнице энергий между уровнем Ферми полупроводника и окислительно-восстановительным потенциалом электролита VOC. При предотвращении рекомбинации инжектируемых электронов уровень Ферми повышается, а VOC увеличивается. Этот эффект может быть очень большим. Например, в элементе типа Gratzel потери на рекомбинацию до продукта окисленного йодида учитываются только как потери фототока в несколько наноампер, в то время как расходуется ˜200 мв VOC. Поскольку мощность представляет собой произведение тока и напряжения, это представляет собой значительные потери.
Может даже быть возможным исключить активные при окислении-восстановлении электролиты, дырочные проводники или переходы с полупроводником p-типа и просто использовать хромофорную цепочку для переброса ″дырки″ непосредственно в металлический противоэлектрод, как показано в конструкции III. Связанный с этим процесс, как можно себе вообразить, происходит в пленках ″органического полупроводника″, описанных выше, и является очень желательным, поскольку опосредование переноса дырок в противоэлектрод всегда требует потенциальной энергии. Гашение возбужденного состояния цепочек хромофоров металлическими поверхностями представляет собой ожидаемую проблему (смотри ниже). Однако поскольку цепочка хромофоров освещается через прозрачный полупроводник и при этом цепочка является сильно поглощающей, очень немного возбужденных состояний будет создаваться вблизи металлического противоэлектрода. Соответственно доступность линейных хромофорных цепочек может сделать возможным изготовление эффективных элементов с использованием металлических поверхностей.
2. Металл-хромофорные цепочки. Существуют два возможных процесса переноса электрона в возбужденном состоянии через границу раздела, которые могут происходить от молекулярного возбужденного состояния S*, создающегося на поверхности металла: (a) Металл принимает электрон от S* с образованием S+; или (b) металл отдает электрон S* с образованием S-. Ни один из этих процессов не наблюдался непосредственно. Эти два процесса должны конкурировать между собой, и если нет какого-либо преимущества, то в сумме никакого заряда не будет переноситься через границу раздела. Для получения стационарного фотоэлекрохимического отклика обратные для S+ (или S-) реакции с переносом электрона через границу с получением продуктов в основном состоянии также должны быть исключены. Перенос энергии от возбужденного сенсибилизатора к металлу является термодинамически выгодным и разрешается как по механизму Форстера, так и по механизму Декстера. Существуют теоретические предсказания и экспериментальные данные, описывающие гашение «с переносом энергии» молекулярных возбужденных состояний металлами.Однако эти исследования включают в себя измерения фотолюминесценции, и реальные механизмы гашения, включая перенос электрона или энергии, остаются только объектом обсуждения. Тем не менее, конкурентное гашение с переносом энергии часто используется для объяснения низких эффективностей фототока на сенсибилизированных границах раздела с металлами. Однако существует множество причин для предсказания плохих выходов сенсибилизации для металлических электродов (Gerischer, H. Photochem. Photobiol. 1972, 16, 243; Gerischer, H. Pure Appl. Chem. 1980, 52, 2649; Gerischer, H.; Willig, F. Top. Curr. Chem. 1976, 61, 31).
Цепочки хромофоров, описываемые здесь, конструируются для переноса энергии к фотоаноду, а дырок - прочь от фотоанода (конструкции II и III). Это внутреннее выпрямление должно сделать возможным предпочтительную инжекцию электрона в освещенный электрод, а также перенос дырок прочь от электрода. В этом случае обедненный слой, способствующий разделению зарядов, которое происходит на поверхности полупроводников, может и не быть необходимым, поскольку относительные скорости инжекции и рекомбинации заряда на границе раздела будут вызывать эффективное преобразование энергии. Хотя кинетический контроль динамики переноса электрона через границу раздела был и предложен ранее в качестве практической схемы для преобразования солнечного света, сообщаемые в публикациях эффективности являются очень низкими, при этом фототоки находятся в наноамперном диапазоне (Gregg, B.A.; Fox, M.A.; Bard, A.J. J.Phys.Chem. 1990, 94, 1586). В случае успеха использование металлов дало бы возможность для использования любой проводящей подложки в целях преобразования энергии. Большое множество ″прозрачных металлов″, то есть тонких металлических пленок или сеток, таких как Au, Al или Pt, на прозрачных подложках являются доступными для такого применения.
D. Синтетические подходы
Два различных подхода являются доступными для приготовления линейных стержней АС/УРЗ. Один из подходов включает в себя постадийный синтез, а другой подход включает в себя процесс полимеризации. Оба подхода используют составляющие блоки на основе пигментов, несущие на себе по меньшей мере одну, а как правило, две синтетических ручки.
Один из примеров постадийного синтетического подхода использует этин (E) и составляющие блоки на основе йод-замещенных пигментов (фигура 10). Опосредованное палладием (Pd) связывание йод-замещенного пигмента и бифункционального составляющего блока на основе пигмента, несущего на себе йодгруппу и триметилсилил-защищенный этин (TMSE), дает ковалентно-связанный димер из пигментов (фигура 11). Расщепление триметилсилил-защищенного этина с использованием тетрабутиламмоний фторида делает возможной вторую реакцию связывания, опосредованную Pd. Таким способом конструируется линейная архитектура, начиная от дальнего конца и продвигаясь в направлении ближнего конца. Конечная реакция включает в себя присоединение компонента УРЗ. Составляющий блок УРЗ несет на себе соединительную группу Y, необходимую для соединения с поверхностью электрода. Такой же способ используется для приготовления мультипорфириновых цепочек, связанных этином, как описано здесь в других местах.
Можно также представить себе большое количество составляющих блоков на основе пигментов, как отмечено здесь в других местах. В дополнение к этому, могут использоваться многомерные составляющие блоки на основе пигментов. Один из примеров включает в себя порфириновый димер, несущий на себе п -йодфенильную группу и п-[2-(триметилсилил)этинил]фенильную группу. Полученная в результате цепочка для аккумуляции света состоит из порфириновых димеров, связанных п-фениленом, которые соединены вместе с помощью п,п'-дифенилэтиновых групп. Димеры для этого подхода к составляющим блокам могут быть получены рациональным путем так, как изображено на фигуре 12.
Полимеризационный подход иллюстрируется на фигуре 13 с использованием способа связывания Сузуки для соединения составляющих блоков на основе пигментов. составляющий блок на основе пигмента, несущий на себе карбокси-группу и группу борного сложного эфира, присоединен к твердофазной смоле. В настоящее время доступно множество смол; особенно привлекательными являются смола Wang или подобная ей смола. Смола Wang представляет собой смолу на основе поперечно-сшитого полистирола, которая делает возможным легкое присоединение соединений, несущих на себе карбоновые кислоты, а отсоединение достигается путем обработки слабой кислотой в органических растворителях. Связывание Сузуки осуществляется с использованием смеси бифункциональных составляющих блоков на основе пигментов и конечного узла и одного из хорошо известных наборов условий, использующих Pd катализаторы и лиганды, которые дают высокие оборачиваемости и высокую активность. Здесь бифункциональный составляющий блок на основе пигмента несет на себе группу сложного борного эфира и йод-группу, в то время как конечный узел несет на себе только йод-группу. Полимеризация осуществляется в присутствии твердой фазы. Распределение средних длин линейных стержней контролируется с помощью отношения бифункциональных составляющих блоков на основе пигментов и конечных узлов; как правило, используется отношение 10:1 или около этого. После связывания Сузуки твердофазная смола промывается для удаления непрореагировавшего исходного материала и побочных продуктов связывания, затем желаемый продукт получается путем отщепления от смолы при стандартных условиях. Для порфириновых пигментов и кислотных условий отщепления ожидается деметаллирование металлопорфирина, и оно может быть компенсировано с помощью последующего металлирования. Затем смесь олигомеров фракционируется по размерам с помощью эксклюзионной хроматографии. Заметим, что показанный синтез происходит от узла УРЗ наружу к дальнему концу, приводя к получению линейного стержня АС/УРЗ с присоединенной функциональной группой (в данном случае, карбокси), для присоединения к электроду.
Такой же подход может быть предложен и для составляющих блоков на основе хлорина, как иллюстрируется на фигуре 14. Более широкий список, иллюстрирующий другие составляющие блоки на основе пигментов, пригодные для связывания Сузуки, изображены на фигуре 15. Два заслуживающих внимания примера включают в себя составляющий блок на основе димера пигментов и мономерный порфирин, несущий на себе два бор-дипиррометеновых красителя. Бор-дипиррометеновые красители сильно поглощают в сине-зеленой области солнечного спектра и очень эффективно передают энергию ковалентно присоединенному порфирину. Пример синтеза порфирина, несущего на себе одну йод-группу и одно производное сложного борного эфира, изображен на фигуре 16. Этот способ использует установившиеся методы для формирования дипиррометанов, ацилирование дипиррометана селективно в положениях 1,9, восстановление полученного дикетона до дипиррометан-дикарбинола и конденсирование дипиррометан-дикарбинола и дипиррометана с образованием соответствующего порфирина (Cho, W.-S. et al., J.Org.Chem. 1999, 64, 7890-7901). Последующее йодирование в единственном свободном мезо-положении дает желаемый составляющий блок, пригодный для использования при связывании Сузуки.
Полимеризация не ограничивается связыванием Сузуки. Пример мезо,мезо-связывания иллюстрируется на фигуре 17. Порфирин с одним свободным (незамещенным) мезо-положением присоединяется к твердой фазе, затем обрабатывается до получения условий мезо, мезо-связывания (AgPF6 или подобным оксидантом) в присутствии смеси порфиринов для создания стержня АС (Osuka, A.; Shimidzu, H. Angew. Chem. Int. Ed. Engl. 1997, 36, 135-137; Yoshida, N. et al., Chem. Lett. 1998, 55-56; Nakano, A. et al., Angew. Chem. Int. Ed. 1998, 37, 3023-3027; Senge, M.O.; Feng, X. Tetrahedron Lett. 1999, 40, 4165-4168). Порфирин, подвергающийся полимеризации, имеет два свободных мезо-положения, а порфирин, который служит в качестве конечных частиц, имеет только одно свободное мезо-положение. Мезо,мезо-связанные олигомеры сильно расщепляют и уширяют полосы Сорэ, что является привлекательной особенностью для достижения поглощения по всему спектру солнечного света.
Такой же полимеризационный подход может быть осуществлен для получения мезо,мезо-связанного олигомера, несущего этин на конце цепи (фигура 18). Этот синтез осуществляется с использованием нового линкера для прикрепления этинил-порфирина к твердой фазе. После отщепления замещенный этином мезо,мезо-связанный олигомер может затем подвергаться процедуре постадийного связывания для присоединения УРЗ. Пример, иллюстрирующий присоединение цирконий порфирин-фталоцианина, изображен на фигуре 19. Альтернативно, другие пигменты могут быть присоединены с помощью процедур постадийного связывания перед присоединением к УРЗ. Выбранные молекулы УРЗ включают в себя хлорины, бактериохлорины, фталоцианины, нафталоцианины или циркониевые двухъярусные сэндвичевые молекулы. Такой же синтетический подход является особенно привлекательным для использования вместе с олигомерами со связанным хлорином (смотри ниже).
Каждый из двух синтетических подходов имеет свои преимущества и недостатки. Постадийный подход дает олигомерный продукт заданной длины и делает возможным включение различных пигментов в заданных позициях. Однако постадийный подход требует достаточно большого количества манипуляций при синтезе, включая множество циклов снятия защиты и связывания, для получения желаемого продукта. полимеризационный подход быстро дает линейные олигомеры достаточной длины. Однако олигомеры получаются полидисперсными, причем синтез не дает контроля над размещением различных пигментов в цепочке. Из-за этих особенностей два этих подхода имеют различные применения.
Для конструкций I и II, где идентичные пигменты используются по всему стержню АС, может быть использован полимеризационный подход. Для конструкции III, где стержни АС/УРЗ должны быть заданной и однородной длины для размещения между электродом и противоэлектродом, должен быть использован подход постадийного синтеза. Единственное исключение может появиться в том случае, когда фракционирование по размерам может дать желаемую однородность по длине, как может происходить, когда желаемыми являются скорее короткие олигомеры, и в этом случае может быть использована полимеризация. Для конструкций I, II и III, где в стержень АС должны быть включены различные пигменты, должен быть использован постадийный подход. Также может происходить объединение постадийной процедуры и полимеризации.
Конкретные примеры порфириновых макроциклов, которые могут быть использованы в качестве лигандов для осуществления настоящего изобретения, включают в себя представленные ниже соединения формулы X, формулы XI, формулы XII, формулы XIII, формулы XIV, формулы XV, формулы XVI и формулы XVII (при этом формулы XII-XVII представляют собой различные хлорины, включая бактериохлорины и изобактериохлорины).




где:
M представляет собой металл, такой как металл, выбранный из группы, состоящей из Zn, Mg, Pt, Pd, Sn и Al, или M отсутствует (в этом случае кольцевые гетероатомы K1-K4 замещаются H,H, что требуется для выполнения условия нейтральной валентности);
K1, K2, K3, K4, K5, K6, K7 и K8 представляют собой гетероатомы, такие как гетероатомы, независимо выбранные из группы, состоящей из N, O, S, Se, Te и CH;
S1, S2, S3, S4 S5, S6, S7, S8, S9, S10, S11, S12, S13, S14, S15 и S16 представляют собой независимо выбранные заместители, которые предпочтительно обеспечивают окислительно-восстановительный потенциал менее примерно 5, 2 или даже 1 вольта. Примеры заместителей S1, S2, S3, S4 включают в себя, но не ограничиваются этим, H, арил, фенил, циклоалкил, алкил, галоген, алкокси, алкилтио, перфторалкил, перфторарил, пиридил, циано, тиоцианато, нитро, амино, алкиламино, ацил, сульфоксил, сульфонил, имидо, амидо и карбамоил.
Кроме того, каждая пара из S1 и S2, S3 и S4, S5 и S6, S7 и S8 может независимо образовывать кольцевой арен, такой как бензол, нафталин, или антрацен, который, в свою очередь, может быть незамещенным или замещенным один или несколько раз заместителем, который предпочтительно обеспечивает окислительно-восстановительный потенциал менее примерно 5, 2 или даже 1 вольта, таким как H, арил, фенил, циклоалкил, алкил, галоген, алкокси, алкилтио, перфторалкил, перфторарил, пиридил, циано, тиоцианато, нитро, амино, алкиламино, ацил, сульфоксил, сульфонил, имидо, амидо и карбамоил. Примеры таких кольцевых аренов включают в себя, но не ограничиваются этим:

(Можно понять, что все кольца являются соответствующим образом сопряженными, чтобы сохранить ароматичность конденсированных колец); здесь каждый заместитель S′ является выбранным независимо и предпочтительно обеспечивает окислительно-восстановительный потенциал менее примерно 5, 2 или даже 1 вольта. Примеры таких заместителей включают в себя, но не ограничиваются этим, H, арил, фенил, циклоалкил, алкил, галоген, алкокси, алкилтио, перфторалкил, перфторарил, пиридил, циано, тиоцианато, нитро, амино, алкиламино, ацил, сульфоксил, сульфонил, имидо, амидо и карбамоил. Конкретные примеры соединений, как описано выше, содержащих кольцевые арены, приведены ниже в формулах XX-XXIV.
В дополнение к этому, S1-S16 могут содержать связывающую группу (-Q-), ковалентно связанную с соседним порфириновым макроциклом X1-Xm+1, или связывающую группу, ковалентно связанную с указанным первым электродом. В одном из вариантов воплощения настоящего изобретения связывающие группы каждого порфиринового макроцикла ориентированы в транс-положении; в другом варианте воплощения настоящего изобретения один или несколько порфириновых макроциклов содержат связывающие группы, которые ориентированы в цис-положениях по отношению одна к другой, так что стержни для аккумуляции света содержат изгибы или изломы, или же линкер сам по себе является нелинейным или перекошенным.
Примеры порфириновых макроциклов, которые содержат кольцевые арены, как описано выше, включают в себя, но не ограничиваются этим, порфириновые макроциклы формулы XX, XXI, XXIII и XXIV, представленные ниже:


где каждый заместитель S′ является выбранным независимо и предпочтительно обеспечивает окислительно-восстановительный потенциал менее примерно 5, 2 или даже 1 вольта. Примеры таких заместителей включают в себя, но не ограничиваются этим, H, арил, фенил, циклоалкил, алкил, галоген, алкокси, алкилтио, перфторалкил, перфторарил, пиридил, циано, тиоцианато, нитро, амино, алкиламино, ацил, сульфоксил, сульфонил, имидо, амидо и карбамоил. Опять же, чтобы связать порфириновый макроцикл с подложкой или другим соединением, таким как другой порфириновый макроцикл, способами, описанными выше, порфириновый макроцикл должен содержать по меньшей мере один заместитель, а предпочтительно два заместителя S′, который является линкером (которые являются линкерами), в частности линкером, содержащим реакционно-способную группу (когда множество линкеров являются заместителями на лиганде, линкеры могут быть одинаковыми или выбранными независимо). Такие линкеры описаны выше.
Конкретные примеры координационных сэндвичевых соединений, которые могут быть использованы при осуществлении настоящего изобретения, имеют формулу XXV (для двухъярусных сэндвичевых соединений):
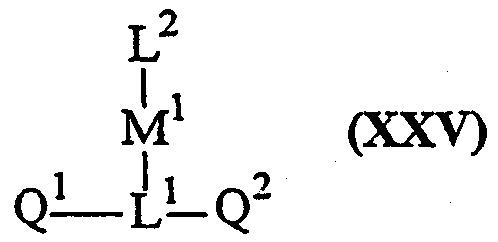
где:
M1 представляет собой металл из ряда лантаноидов, а также Y, Zr, Hf и Bi, а в ряде актиноидов Th и U (радиоактивные элементы, такие как Pm, как правило, являются менее предпочтительными);
L1 и L2 представляют собой независимо выбранные лиганды (например, порфириновые макроциклы); и
Q1 и Q2 могут присутствовать или отсутствовать, и когда присутствуют, они представляют собой независимо выбранные линкеры, описанные выше (линкер предпочтительно включает в себя защищенную или незащищенную реакционно-способную группу, такую как тио, селено или теллуро-группу). Предпочтительно присутствует по меньшей мере один элемент из Q1 или Q2.
Также можно понять, что каждый лиганд L может быть замещен одним отдельно взятым линкером Q или может быть многократно замещен линкерами Q, как объясняется более подробно ниже. Таким образом, молекула формулы XXV может быть ковалентно связанной с электродом или подложкой с помощью по меньшей мере одного из Q1 или Q2.
Каждый лиганд L может быть дополнительно замещен без отклонения от рамок соединений формулы XI выше. Например, как объясняется более подробно ниже, лиганды могут быть ковалентно связаны с другим порфириновым макроциклом, с лигандом другого координационного сэндвичевого соединения и тому подобное.
Чтобы связать порфириновый макроцикл (который может быть или не быть компонентом координационного сэндвичевого соединения) с подложкой или с другим соединением, таким как другой порфириновый макроцикл, способами, описанными выше, по меньшей мере один лиганд в порфирином макроцикле должен содержать по меньшей мере, один, а предпочтительно два заместителя S1-Sn или S′, который представляет собой линкер, в частности линкер, содержащий реакционно-способную группу (там, где множество линкеров являются заместителями на лиганде, линкеры могут быть одинаковыми или выбранными независимо). Такие линкеры обозначаются здесь как Y-Q-, где: Q представляет собой линкер, и Y представляет собой подложку, активную позицию или группу, которая может ковалентно связываться с подложкой, или активную позицию или группу, которая может ионно связываться с подложкой.
Q может представлять собой линейный линкер или перекошенный линкер, при этом линейные линкеры в настоящее время являются предпочтительными. Примеры перекошенных линкеров включают в себя, но не ограничиваются этим, 4,3′-дифенилэтин, 4,3′ -дифенилбутадиин, 4,3′-бифенил, 1,3-фенилен, 4,3′-стильбен, 4,3′-азобензол, 4,3′-бензилиденанилин и 4,3′′-терфенил. Примеры линейных линкеров включают в себя, но не ограничиваются этим, 4,4′-дифенилэтин, 4,4′-дифенилбутадиин, 4,4′-бифенил, 1,4-фенилен, 4,4′-стильбен, 1,4-бициклооктан, 4, 4′-азобензол, 4,4′ -бензилиденанилин, 4,4′′-терфенил, 3-меркаптофенил, 3 -меркаптометилфенил, 3-(2-меркаптоэтил)фенил, 3-(3-меркаптопропил)фенил, 3-(2-(4-меркаптофенил)этинил)фенил, 3 -карбоксифенил, 3-карбоксиметилфенил и тому подобное.
Y может представлять собой защищенную или незащищенную активную реакционно-способную позицию или группу на линкере, такую как тио, селено или теллуро-группу.
Таким образом, примеры линейных линкеров для Y-Q- представляют собой: 4-[2-меркаптоэтил)фенил, 4-[3-меркаптопропил)фенил, ω-алкилтиол формы HS(CH2)n-, где n=1-20, 4-карбоксифенил, 4-карбоксиметилфенил, 4-(2-карбоксиэтил)фенил, ω-алкилкарбоновую кислоту в форме HO2C(CH2)n-, где n=1-20,
4-(2-(4-карбоксифенил)этинил)фенил,
4-(2-(4-карбоксиметилфенил)этинил)фенил,
4-(2-(4-(2-карбоксиэтил)фенил)этинил)фенил,
4-(2-(4-меркаптофенил)этинил)фенил, 4-меркаптометилфенил, 4 -гидроселенофенил, 3-(2-(4-гидроселенофенил)этинил)фенил, 4 -гидротеллурофенил и 4-(2-(4-гидротеллурофенил)этинил)фенил.
Примеры перекошенных линкеров для Y-Q- представляют собой: 3-(2-(4-меркаптофенил)этинил)фенил, 3-меркаптометилфенил, 3 -гидроселенофенил, 3-(2-(4-гидроселенофенил)этинил)фенил, 3 -гидротеллурофенил и 3-(2-(4-гидротеллурофенил)этинил)фенил; и тому подобное.
Другие пригодные для использования линкеры включают в себя, но не ограничиваются этим, 2-(4-меркаптофенил)этинил, 2-(4-гидроселенофенил)этинил и 2-(4-гидротеллурофенил)этинил.
Таким образом, линкеры между соседними порфириновыми макроциклами внутри стержня для аккумуляции света или между порфириновым макроциклом и электродом, как правило, являются такими, чтобы сделать возможным сверхобмен между связываемыми хромофорами (опосредованное сообщение электронов между хромофорами, которое делает возможным или разрешает перенос и/или обмен энергии возбужденного состояния электронов и/или дырок). Примеры пригодных для использования линкеров могут в целом быть представлены формулой -Q-, где Q может быть непосредственной ковалентной связью или связывающей группой формулы:

где:
n равно 0, или 1-5, или 10;
R3 может присутствовать или отсутствовать (давая непосредственную ковалентную связь, когда R3 отсутствует и n равно 0); и
R1, R2 и R3 являются, каждый независимо, выбранными из группы, состоящей из этена, этина, арила и гетероарильных групп (например, фенила, и производных пиридина, тиофена, пиррола, фенила и тому подобное, причем эти арильные и гетероарильные группы могут быть незамещенными или замещенными один или несколько раз такими же заместителями, которые перечислены выше в связи с порфириновыми макроциклами).
Геометрия линкеров по отношению к различным хромофорам и группам разделения заряда в стержнях для аккумуляции света может изменяться. В одном из вариантов воплощения по меньшей мере один из X2-Xm+1 содержит мезо-связанный порфириновый макроцикл. В другом варианте воплощения по меньшей мере один из X2-Xm+1 содержит транс мезо-связанный порфириновый макроцикл. В другом варианте воплощения X2-Xm+1 состоят из мезо-связанных порфириновых макроциклов. В другом варианте воплощения X2-Xm+1 состоят из транс мезо-связанных порфириновых макроциклов. В другом варианте воплощения по меньшей мере один из X2-Xm+1 содержит β-связанный порфириновый макроцикл. В другом варианте воплощения по меньшей мере один из X2-Xm+1 содержит транс β -связанный порфириновый макроцикл. Еще в одном варианте воплощения X2-Xm+1 состоят из β -связанных порфириновых макроциклов. Еще в одном варианте воплощения X2 -Xm+1 состоят из транс β-связанных порфириновых макроциклов.
E. Примеры конструкций
Примеры конкретных молекул, которые могут быть получены в различных конструкциях, представлены на следующих схемах. Пример конструкции I изображен на фигуре 20. Zn-порфирины составляют стержень АС, а Mg-порфирин составляет УРЗ. Перенос энергии осуществляется обратимо между Zn-порфиринами, но осуществляется необратимо (в более глубокую потенциальную яму) к Mg-порфирину (Hascoat, P. et al., Inorg.Chem. 1999, 38, 4849-4853). В этой конструкции каждый из Zn- и Mg-порфиринов имеет идентичные несвязывающие мезо-заместители. При разделении заряда дырка остается на Mg-порфирине. Дырка не может переходить к Zn -порфиринам, поскольку Mg-порфирин находится при более низком потенциале, чем Zn-порфирины (Wagner, R.W. et al., J.Am.Chem.Soc. 1996, 118, 3996-3997). Окислительный потенциал УРЗ может быть подобран путем изменения индуктивного воздействия двух несвязывающих мезо-заместителей. Путем замены несвязывающих мезо-заместителей также и на Zn-порфиринах, опять же, дырку принуждают оставаться на Mg-порфирине (УРЗ). Пример конструкции II изображен на фигуре 21. Эта конструкция является подобной той, которая изображена на фигуре 19, но несвязывающие мезо-заместители на Zn- и Mg -порфиринах являются различными. Сильно отбирающие электроны заместители размещаются на Mg-порфирине (УРЗ), но не на Zn -порфиринах. Как следствие, при инжекции электрона в электрод дырка на УРЗ переносится на Zn-порфирины в цепочке АС. Перенос дырки является выгодным, поскольку Zn-порфирины находятся при более низком потенциале, чем Mg-порфирин.
Родственный пример конструкции II изображен на фигуре 22. Здесь линейный стержень АС состоит из ряда из m периленовых компонентов и ряда из n Zn-порфиринов, и УРЗ состоит из Zn -хлорина. Энергия перетекает необратимо от набора периленов к Zn-порфиринам и к Zn-хлорину. Дырка, создающаяся в УРЗ при инжекции электрона в электрод, мигрирует к Zn-порфиринам с более низким потенциалом, но не может мигрировать в ряд периленов. Этот ряд синтезируется с использованием составляющего блока на основе перилена, несущего на себе этин и йод-группу, аналогично составляющим блокам на основе порфирина.
Дополнительный пример, иллюстрирующий конструкции I и II, изображен на фигурах 23 и 24. Здесь в стержне АС используются β,β'-замещенные хлорины, а циркониевая двухъярусная порфирин-фталоцианиновая сэндвичевая молекула служит в качестве УРЗ. Циркониевые двухъярусные порфириновые сэндвичевые молекулы, как известно, являются фотохимически активными. Синтез включает в себя связывание Сузуки с образованием стержня АС из хлорина, связанного п-фениленом, который использует линкер для присоединения этина к твердой фазе. При отщеплении от твердой фазы и снятии защиты этинил-замещенный стержень АС может быть присоединен к циркониевой двухъярусной молекуле с помощью опосредованной Pd реакции этинилирования. Циркониевая двухъярусная молекула может быть получена с помощью известных способов, использующих желаемый транс-замещенный порфирин. Выбирая типы заместителей в порфириновом и фталоцианиновом компонентах двухъярусной молекулы, можно подобрать по желанию окислительный потенциал. При отсутствии высвобождающих электрон заместителей окислительный потенциал является очень низким, и дырка (образовавшаяся при инжекции электрона в электрод) остается на УРЗ (конструкция I). При заместителях, отбирающих электрон (например, когда R=F или перфторалкил; Ar1=Ar2=перфторалкил или пентафторфенил), окислительный потенциал увеличивается, и дырка мигрирует к Zn-хлоринам в стержне АС (конструкция II).
Пример конструкции III изображен на фигуре 25. Стержень АС состоит из ряда металлохлоринов. Линкер для присоединения к электроду представляет собой производное бензойной кислоты, а линкер для присоединения к противоэлектроду представляет собой тиоацетат. Такие S-ацетил защищенные тиолы подвергаются расщеплению при соприкосновении с сильным основанием или при контакте с электрохимически активными поверхностями, такими как золото (Gryko, D.T. et al., J.Org.Chem. 1999, 64, 8635-8647). Компоненты АС и УРЗ конструируются таким образом, чтобы облегчить миграцию дырки из УРЗ к дальнему концу, где происходит присоединение к противоэлектроду.
Другой тип пигмента, который может быть использован в этих конструкциях, представляет собой бактериохлорин. Бактериохлорины сильно поглощают в голубой области, подобно порфиринам, но также сильно поглощают и в ближней инфракрасной области, а также по всей видимой области (например, тетрафенилбактериохлорин, ε378нм=160000 M-1см-1, ε520нм=160000 M-1см-1,
ε740нм=130000 M-1см-1 )(Whitlock, H. W. et al. J.Am.Chem.Soc. 1969, 91, 7485). Структура линейного стержня АС, состоящего из бактериохлорина, изображена на фигуре 26.
F. сравнительное обсуждение архитектур и внутреннего выпрямления
Очень привлекательной особенностью конструкций II и III является то, что последовательность пигментов в стержнях для аккумуляции света вызывает необратимое перетекание энергии из области поглощения в УРЗ. Моделирование показывает, что такие градиенты энергии обеспечивают драматическое увеличение квантовой эффективности для энергии возбуждения, достигающей ловушки (в данном случае, узла разделения заряда) (Van Patten, P.G. et al., J.Phys.Chem. B 1998, 102, 4209-4216). Дополнительной особенностью конструкции III является то, что дырка (образующаяся в УРЗ) протекает необратимо по прыжковому механизму миграции дырок из УРЗ в противоэлектрод. Соответствующий выбор пигментов делает в стержне АС возможным необратимое протекание энергии и дырок в противоположных направлениях. Линейные стержни, которые предоставляют возможность для протекания энергии и дырок в противоположных направлениях, являются идеально пригодными для включения в сверхтонкие солнечные элементы, описываемые здесь.
Явления, описанные выше, дают возможность этим конструкциям для осуществления внутреннего выпрямления. Внутреннее выпрямление может включать в себя (i) необратимый поток энергии или электронов возбужденного состояния вдоль некоторого участка или по всей длине стержня для аккумуляции света, (ii) необратимый поток дырок вдоль некоторого участка или по всей длине стержня для аккумуляции света, или (iii) как (i), так и (ii) выше, при этом дырки и энергия или электроны движутся в противоположных направлениях.
Для необратимой миграции энергии (внутреннее выпрямление энергии или электронов), стержень для аккумуляции света должен иметь такую структуру, что E*(X1)
Для необратимой миграции дырки по прыжковому механизму (внутреннее выпрямление дырок) стержень для аккумуляции света должен иметь такую структуру, что E1/2 (X1) > E1/2(X2) > E1/2(X3)... >E1/2(Xi), где E1/2 представляет собой значение элекрохимического окислительного потенциала в некоторой средней точке для i-того хромофорного компонента X.
Хотя хромофоры стержней для аккумуляции света могут быть изоэнергетическими, условие внутреннего выпрямления, как описано выше, является предпочтительным. Однако внутреннее выпрямление не должно происходить по всему стержню для аккумуляции света, и даже не должно быть предусмотрено вблизи узла разделения заряда. Например, один или несколько изоэнергетических хромофоров может присутствовать вблизи узла разделения заряда, а внутреннее выпрямление дырок и/или энергии может происходить где-либо внутри стержня для аккумуляции света, например, в среднем или дальнем сегменте. Таким образом, термин ″внутреннее выпрямление″ энергии возбужденного состояния дырок и/или электронов с помощью стержня для аккумуляции света относится к внутреннему выпрямлению, которое происходит вдоль любого сегмента или части указанного стержня для аккумуляции света.
G. Изготовление солнечного элемента
Осаждение линейных молекул АС/УРЗ на электроде осуществляется путем взаимодействия электрода с раствором молекул АС/УРЗ, с последующей промывкой для удаления любых несвязанных частиц. Может осуществляться гомогенное или гетерогенное осаждение. То есть может осаждаться гомогенная популяция молекул или могут быть использованы смеси молекул АС/УРЗ. Одно из преимуществ последней процедуры заключается в том, что могут быть использованы молекулы, имеющие различные компоненты в цепочке АС, для перекрытия всего спектра солнечного света. Различные типы цепочек АС включают в себя, но не ограничиваются этим, следующие: цепочки все-хлорин, бактериохлорин, порфирин+фталоцианин, мезо, мезо-связанные порфирины, перилен+порфирин. Смеси этих типов цепочек АС могут быть использованы в солнечном элементе для обеспечения эффективного перекрывания всего спектра солнечного света. При таком способе не является важным, чтобы каждый стержень АС/УРЗ обеспечивал полное перекрывание всего спектра солнечного света.
Солнечные элементы, подобные фотогальваническим, могут быть изготовлены путем позиционирования узла разделения заряда вдали от поверхностей электродов. Инкорпорирование движущей силы для переноса электронов к аноду и движущей силы для переноса дырок к катоду должно приводить к эффективному преобразованию энергии. Потенциальным преимуществом такого подхода является то, что аккумуляция света и внутримолекулярный перенос заряда будет происходить на некотором расстоянии от поверхностей электродов, тем самым сводя к минимуму вредные побочные взаимодействия возбужденных состояний и электрода, такие как гашение возбужденного состояния.
III. СОСТАВЛЯЮЩИЕ БЛОКИ НА ОСНОВЕ ХЛОРИНА ДЛЯ СОЗДАНИЯ
ЦЕПОЧЕК ДЛЯ АККУМУЛЯЦИИ СВЕТА
A. Введение
Хлорины имеют три преимущества по сравнению с порфиринами при использования для сбора и использования солнечного света. (1) Хлорины сильно поглощают как в голубой области, так и в красной части всей видимой области (благодаря их зеленому цвету), эффективно преобразуя большую часть спектра солнечного света, в то время как порфирины сильно поглощают только в голубой области (благодаря их красному цвету). (2) Переходный дипольный момент при длинноволновом поглощении в хлорине является линейно поляризованным вдоль оси N-N, придавая повышенную направленность переносу энергии к соседним пигментам через пространство. В противоположность этому, в металлопорфирине переходный дипольный момент зоны длинноволнового поглощения лежит в плоскости макроцикла, эффективно локализованный вдоль обеих осей N-N (планарный осциллятор), и по этой причине наблюдается меньшая направленность переноса энергии. (3) Хлорины окисляются легче, чем порфирины, и по этой причине являются лучшими фотовосстановителями.
Недавно был описан способ синтеза, которые обеспечивает доступ к новому набору составляющих блоков на основе хлорина (Strachan, J.P. et al., J.Org.Chem. 2000, 118, 3160-3172). Составляющие блоки на основе хлорина демонстрируют типичные для хлорина спектры поглощения и флуоресценции. Составляющие блоки на основе хлорина имеют заместители (функциональные ручки) в двух из мезо-положений, и ни одного в β-положениях (фигура 27). В предыдущих исследованиях переноса энергии в мультипорфириновых цепочках было обнаружено несколько фактов, имеющих отношение к конструированию пигментов для включения в цепочки для аккумуляции света, состоящие из ковалентно-связанных пигментов: (1) процесс переноса энергии включает в себя механизмы передачи как через пространство (TS), так и через связь (TB), и наблюдаемая скорость переноса энергии представляет собой сумму этих двух процессов (Hsiao, J.-S.; et al., J.Am.Chem.Soc. 1996, 118, 11181-11193). (2) Скорость TB-переноса энергии зависит от природы молекулярных орбиталей в доноре энергии, акцепторе энергии и в линкере, соединяющем донор и акцептор (Strachan, J.P. et al., J.Am.Chem.Soc. 1997, 119, 11191-11201; Yang, S.I. et al., J.Am.Chem.Soc. 1999, 121, 4008-4018). В частности, более широкое распределение электрона (и более высокая скорость переноса) происходит при присоединении линкера в тех областях донора и акцептора, где граничащие молекулярные орбитали имеют высокую электронную плотность по сравнению с областями с низкой электронной плотностью. В качестве пояснения, в порфирине, имеющем a2u HOMO (которая имеет электронную плотность в основном в мезо-положениях и очень маленькую в β-положениях), более высокие скорости (в 2,5-10 раз) наблюдаются для линкеров в мезо -, а не в β-положениях (фигура 28). Подобным же образом, в порфирине, имеющем a1u HOMO (которая имеет электронную плотность в основном в β-положениях, и очень маленькую, если вообще имеет, в мезо-положениях), более высокие скорости наблюдаются для линкеров скорее в β-, чем в мезо-положениях.Такая разница в скоростях связывается с каждой стадией переноса энергии от пигмента к пигменту в линейных мультипигментных цепочках, и она проявляется как большое влияние на общую скорость и выход переноса энергии (Van Patten, P. G. et al., J.Phys.Chem. B 1998, 102, 4209-4216).
Факторы, которые влияют на TS-перенос энергии (механизм Форстера), хорошо известны. Двумя ключевыми определяющими факторами являются сила осциллятора для донора в возбужденном состоянии (отражается на скорости излучения для перехода в состояние самой низкой энергии) и сила осциллятора для акцептора в основном состоянии (отражается на молярном коэффициенте поглощения для перехода с самой низкой энергией). Хлорины являются идеальными кандидатами для TS-переноса энергии благодаря их большой силе осциллятора для длинноволновой области поглощения, особенно, если сравнивать с порфиринами, с их низкой силой осциллятора (то есть слабым поглощением) в красной области. Другой ключевой определяющий фактор включает в себя ориентацию соответствующих переходных дипольных моментов для донора и акцептора. Ориентационный член (κ2) для TS переноса энергии по Форстеру принимает значения 0 (ортогональные), 1 (параллельные, но не коллинеарные) и 4 (коллинеарные) в зависимости от ориентации векторов переходных дипольных моментов. Наиболее эффективный перенос энергии осуществляется при таком взаимном расположении молекул, когда переходные дипольные моменты донора и акцептора являются коллинеарными, а наименее эффективный перенос происходит при ортогональных ориентациях (Van Patten, P.G. et al., J. Phys. Chem. B 1998, 102, 4209-4216).
Предсказания относящихся к переносу энергии свойств цепочек, содержащих хлорины, могут быть подытожены. В связанных дифенилэтином мультипорфириновых цепочках (мезо-связанные, a2u HOMO) наблюдаемая скорость переноса энергии от цинк-порфирина к порфирину в виде свободного основания, как обнаружено, составляет (24 псек)-1, при этом вклад TS-переноса составляет (720 псек)-1, а TB переноса - (25 псек)-1. Для хлоринов константа скорости излучения увеличивается в ˜4 раза, а молярный коэффициент поглощения увеличивается в ≥10 раз по сравнению с порфиринами. Рассмотрение включения хлоринов в идеальной геометрии для переноса по Форстеру с одним и тем же промежуточным дифенилэтиновым линкером приводит к тому, что ориентационный член (κ2) должен составлять вплоть до ˜2 по сравнению с 1,125 для порфиринов. Суммарным результатом является ожидаемое увеличение до 100 раз скорости TS-переноса энергии хлоринов по сравнению с порфиринами. Скорость TB-переноса для хлоринов трудно оценить, но она должна попадать в диапазон, наблюдаемый для порфиринов, который находится в пределах от (25 псек)-1 до (360 псек)-1, в зависимости от плотности орбитали в области присоединения линкера. TS -перенос для хлоринов должен быть в 40-100 раз более быстрым, чем для порфиринов (то есть от (18 псек)-1 до ˜(7 псек)-1). Соответственно ожидаемая скорость переноса энергии от одного хлорина до другого должна находиться в пределах от ˜(10 псек)-1 до ˜(20 псек)-1.
При переходе от мультипорфириновых цепочек, связанных дифенилэтином, к цепочкам, связанным п-фениленом, наблюдаемая скорость переноса энергии возрастает от (24 псек)-1 до (2 псек)-1. При переходе к п-фениленовому линкеру для хлоринов более короткое расстояние должно вызывать ˜100-кратное увеличение вклада TS в скорость. В этом случае TS-механизм преобладает над скоростью TB. Соответственно скорости переноса энергии от хлорина к хлорину в цепочке, связанной п-фениленом, как ожидается, будут находиться в суб-пикосекундном диапазоне для хлоринов, замещенных как в мезо-, так и β-положениях.
Эти грубые вычисления иллюстрируют, что хлорины, как ожидается, имеют большую TS-составляющую в наблюдаемом процессе переноса энергии в мультихлориновых цепочках. Более высокие скорости переноса энергии для хлоринов по сравнению с порфиринами в сочетании с превосходными спектральными свойствами (то есть поглощения в голубой и красной областях) хлоринов по сравнению с порфиринами придают сильный импульс для конструирования цепочек для аккумуляции света, содержащих хлорин. Рассматриваются как составляющие блоки на основе мезо-замещенного хлорина, так и составляющие блоки на основе β -замещенного хлорина.
B. Молекулярный дизайн
Здесь представлены составляющие блоки на основе хлорина, сконструированные для получения эффективного переноса энергии в цепочках для аккумуляции света, содержащих хлорин. Цели представляют собой: (1) приготовление хлоринов с двумя функциональными ручками с тем, чтобы хлорины легко могли быть включены в линейные цепочки, (2) дизайн составляющих блоков на основе хлорина для получения наивысшего возможного значения ориентационного члена для TS-переноса энергии, и (3) чтобы они были соединены между собой соответствующим образом для получения наиболее широкого процесса TB-переноса энергии. Что представляют собой наилучшие области на хлорине для присоединения линкера? Четыре возможных транс-замещенных хлорина изображены на фигуре 29. Два β,β'-замещенных хлорина изображены как два хлорина, каждый из которых несет на себе два мезо-заместителя. Для оценки составляющих блоков на основе хлорина необходимо рассмотреть: (1) стерические эффекты любых заместителей, (2) ориентацию переходного дипольного момента для длинноволнового перехода, и (3) композицию пограничных молекулярных орбиталей.
Исследование четырех составляющих блоков на основе хлорина на фигуре 29 доказывает стерические ограничения в хлорине IV из-за взаимодействия мезо-заместителя, примыкающего сбоку к сдвоенным метильным группам кольца D. Остальные три хлорина I-III не имеют таких стерических взаимодействий, поэтому они превосходят IV в этом отношении.
Переходной дипольный момент для перехода в дальней красной области поляризован в хлоринах вдоль оси N-N, перпендикулярной восстановленному кольцу (кольцо D) и пересекающей кольца A и C, но не кольца B и D (фигура 30). Оценка всех четырех возможных транс-хлоринов, изображенных на фигуре 29, требует рассмотрения геометрий, получаемых при включении в ковалентно связанные цепочки. Парные взаимодействия изображены на фигуре 31, где дифенилэтиновый линкер используется для соединения хлоринов (другие линкеры, включая п-фениленовую группу, также могут быть использованы). Для мезо-связанных хлоринов κ2 принимает граничные значения 0,25 и 2,25 в зависимости от ориентации. Предполагая свободное вращение в течение времени жизни возбужденного состояния (динамическое усреднение), среднее значение κ2 составляет 1,125. Заметим, что свободное вращение ожидается вокруг цилиндрически симметричного этина, но скорость вращения может быть недостаточной для того, чтобы заставить все молекулы пройти через все конформации в течение нескольких наносекунд времени жизни возбужденного состояния. Таким образом, молекулы, находящиеся в ориентации, характеризуемой значением κ2, равным или близким к нулю, не вызовут эффективного TS-переноса энергии. β,β'-Замещенные хлорины имеют граничные значения κ2 ˜1,6, и это значение остается >1 независимо от двугранного угла вокруг этинового линкера. (Заметим, что в этом случае расстояние между центрами слегка изменяется при вращении вокруг этинового линкера.) Таким образом, β,β'-замещенные хлорины имеют чуть лучшую коллинеарность переходного дипольного момента с осью замещения (которая должна быть линейной осью мультихлориновой цепочки), чем получается для мезо-замещенных хлоринов. В итоге, составляющие блоки на основе хлорина I и II являются немного более предпочтительными по сравнению с III и IV в отношении TS -переноса энергии.
Самой высокой заселенной молекулярной орбиталью хлорина является орбиталь a2, которая помещает электронную плотность в каждой из мезо- и β-позиций (фигура 32). Соответственно является трудной оценка относительных преимуществ мезо-позиций по сравнению с β-позициями для присоединения линкера в отношении эффективного TB-переноса энергии. В отсутствие такого знания β-замещенные хлорины и мезо-замещенные хлорины, как предполагается, имеют сравнимую пригодность для использования. В любом случае поскольку расстояние, разделяющее кольца, становится очень коротким, TS-механизм будет преобладать, а TB-механизм станет вносить относительно малый вклад в наблюдаемую скорость. Главный недостаток мезо-замещенных транс-хлоринов происходит от возможной деформации макроцикла из-за стерической перенагруженности при частично насыщенном кольце. Транс-конфигурация может быть достигнута при соединении колец A и C. Сравнивая четыре возможных транс-хлорина, изображенных на схеме 4, в отношении TB-переноса энергии, можно увидеть, что мезо-замещенные хлорины (III, IV) являются худшими по сравнению с β,β'-замещенными хлоринами (I, II).
В итоге, составляющие блоки на основе хлорина I, II, и III являются пригодными для конструирования цепочек для аккумуляции света. С точки зрения синтеза составляющий блок на основе хлорина I может быть получен легче, чем II. Заметим, что предыдущий набор составляющих блоков на основе хлорина, которые были получены, обеспечивает типичные свойства поглощения хлорина, но является не пригодным для получения линейных цепочек, поскольку имеют синтетические ручки в двух соседних (цис) мезо-положениях, вместо двух транс мезо- или β-положений (фигура 27). В противоположность этому, хлорины I и II являются идеальными компонентами синтетических цепочек для аккумуляции света и должны обеспечивать высокие скорости переноса.
C. Синтез
Синтез β-замещенных составляющих блоков на основе хлорина (типа I) следует общему способу, установленному ранее для приготовления мезо-замещенных хлоринов, как обсуждалось выше. В этом способе восточная половина и западная половина подвергаются конденсации, а затем окислительной циклизации с получением хлорина. Такой же подход используется здесь для новой восточной половины и новой западной половины, каждая из который несет на себе один β-заместитель (фигура 33).
Синтез новой β-замещенной восточной половины изображен на фигурах 34A и 34B. Синтез, который построен на известной из литературы работе по разработке способа для β-замещенных составляющих блоков порфирина (Balasubramanian, T.; Lindsey, J. S. Tetrahedron 1999, 55, 6771-6784), описывается ниже в примере 1.
Синтез новой β-замещенной западной половины изображен на фигуре 35. Этот способ начинается с того же критического промежуточного соединения, которое используется для восточной половины, 2-формил-3-арилпиррола (фигура 34A). Затем западная половина приготавливается, следуя такой же последовательности реакций, которая используется для незамещенной западной половины. Последний способ находится в процессе исследования.
Составляющий блок на основе хлорина, изображенный на фигуре 33, несет на себе одну 4-(TMS-этинил)фенильную группу и одну 4-йодфенильную группу. Этот конкретный составляющий блок должен сделать возможным синтез связанных дифенилэтином, содержащих хлорин цепочек в линейной архитектуре. Другие составляющие блоки на основе хлорина, которые могут быть получены с помощью этой же стратегии синтеза и которые имеют такие же желаемые физические свойства, изображены на фигуре 36.
Синтез составляющих блоков на основе транс мезо-замещенного хлорина (тип III) получают двумя способами. Один из способов включает в себя распространение способа, установленного в последнее время, для приготовления хлоринов, несущих на себе соседние (цис) мезо-замещенные хлорины (фигура 27) (Strachan, J.P. et al., J.Org.Chem. 2000, 118, 3160). Обработка цис мезо-замещенного хлорина с помощью NBS (DiMagno, S.G. et al., J.Org.Chem. 1993, 58, 5983-5993), как ожидается, дает селективное бромирование, как изображено на фигуре 37. Альтернативно, может осуществляться йодирование с использованием йода и AgPF6 (Nakano, A. et al., Tetrahedron Lett. 1998, 39, 9489-9492). Woodward продемонстрировал, что два положения метина, присоединяемого сбоку к кольцу D, являются сильно реакционно-способными по отношению к электрофильным реагентам (Woodward, R.B.; Skaric, V.J.Am.Chem. Soc. 1961, 83, 4676-4678). Положение между кольцами A и D является стерическим затрудненным из-за парных метильных заместителей, что должно быть достаточным для получения селективного взаимодействия в положениях метина между кольцами D и C. Затем следующее далее опосредованное Pd поперечное связывание (DiMagno, S.G. et al., J.Org.Chem. 1993, 58, 5983-5993) дает желаемый транс-замещенный (Ar2, Ar3) составляющий блок на основе хлорина.
Второй способ получения транс мезо-замещенных составляющих блоков на основе хлорина (тип III) изображен на фигуре 38. приготавливают новую западную половину, которая несет на себе присоединенный к частично насыщенному кольцу заместитель, который предназначен для того, чтобы стать соответствующим мезо-заместителем.
Различные мезо-замещенные составляющие блоки на основе хлорина, которые могут быть получены таким способом и которые являются пригодными для использования при включении в синтетические цепочки для аккумуляции света, изображены на фигуре 39. Можно заметить, что заместители, которые должны использоваться в качестве арильной единицы (Ar), могут быть использованы для регулировки значения электрохимического потенциала, для целей растворимости или для контроля упаковки цепочек для аккумуляции света в самособирающихся структурах.
Для всех составляющих блоков на основе хлорина может быть использовано большое множество металлов при условии, что металлы соответствуют требованию получения фотохимически активного возбужденного состояния. Предпочтительные варианты таких металлов представляют собой Zn, Mg, Pd, Sn, и Al. Также может быть использован хлорин со свободным основанием (M=H,H). При используемом синтезе реакция образования хлорина дает цинк -хлорин, который легко деметаллируется с помощью слабой кислоты с получением хлорина со свободным основанием. Желаемый металлохлорин может затем быть получен с помощью хорошо известных реакций металлирования.
IV. ПРОТИВОПОЛОЖНО НАПРАВЛЕННЫЕ ПОТОКИ ЭНЕРГИИ ВОЗБУЖДЕННОГО СОСТОЯНИЯ И ДЫРОК В ОСНОВНОМ СОСТОЯНИИ В ЦЕПОЧКАХ ДЛЯ АККУМУЛЯЦИИ СВЕТА
A. Введение
Солнечный свет является слабым, и в этом лежит одна из главных проблем при разработке эффективных средств для использования солнечного света в виде источника энергии. Стратегия, используемая фотосинтезирующими организмами, заключается в поглощении солнечного света с помощью мультипигментных антенных комплексов, а затем в направлении полученной в результате энергии возбужденного состояния между пигментами таким образом, что возбуждение достигает реакционного центра (то есть узла разделения заряда). В реакционном центре происходит реакция разделения заряда, давая восстановительный эквивалент (электрон) и окислительный эквивалент (дырку). У растений восстановительный и окислительный эквиваленты в конечном счете используются для восстановления двуокиси углерода (с образованием углеводов) и окисления воды (с освобождением кислорода) соответственно. Таким образом, электроны протекают как от реакционного центра, так и к реакционному центру (заполняя дырку, образовавшуюся из-за переноса заряда). По этой причине реакционный центр должен иметь три канала: канал для потока энергии возбуждения от антенны, канал для испускания электронов после разделения заряда и канал для входа электронов, предназначенных для регенерации реакционного центра после разделения заряда (фигура 40). Заметим, что поток электронов внутрь и миграция дырок наружу представляют собой эквивалентные процессы, и эти термины используются как синонимы.
С точки зрения размера антенные комплексы намного превосходят реакционные центры. В то время как молекулы хлорофилла содержатся как в антенных комплексах, так и в реакционном центре, основной объем хлорофилла расположен в антенных комплексах. Например, в реакционном центре фотосинтезирующей бактерии находятся, как правило, около шести молекул хлорофилла (или соединений хлорофилла), в то время как в антенных комплексах могут присутствовать до нескольких сотен молекул хлорофилла. Антенные комплексы служат для сбора рассеянного солнечного света, а реакционные центры инициируют передачу энергии возбужденного состояния в химическое топливо посредством стабилизированного состояния с разделенным зарядом.
Генерация стабильных разделенных зарядов в реакционном центре включает в себя ряд стадий переноса электрона вдоль ряда акцепторов электронов. На каждой стадии общая обратная скорость переноса электрона (то есть рекомбинация) становится более медленной. После трех стадий отношение скоростей начальной стадии переноса вперед (kf) и скорости рекомбинации (krecomb) составляет kf/krecomb ˜ 106. Ряд быстрых переносов вперед и медленных обратных переносов дает быстрое и эффективное разделение электрона и дырки на большое расстояние.
Множество попыток было посвящено разработке синтетических антенных молекул и синтетических узлов разделения заряда (то есть эквивалента реакционного центра). В целом все антенны, полученные к настоящему времени, содержат 10 или меньше пигментов (Li, J.; Lindsey, J. S. J.Org.Chem. 1999, 64, 9101 -9108). Сконструированы и другие молекулы, которые создают малые антенны, присоединенные к узлу разделения заряда (УРЗ) (Kuciauskas, D. et al., J.Am.Chem.Soc. 1999, 121, 8604-8614). Эти молекулы продемонстрировали эффективную аккумуляцию света (то есть поглощение света и миграцию энергии) и эффективный перенос заряда. По большей части синтетические антенны, УРЗ и интегральные системы антенна-УРЗ изучались в растворе. Такие исследования могут обеспечить глубокое понимание механизмов и свойств, но редко решают проблемы, связанные с организацией сборки антенн для аккумуляции света и УРЗ, как это должно быть сделано при конструировании любой практической системы для использования солнечного света.
Должны быть разработаны и синтезированы цепочки для аккумуляции света, которые поглощают свет и подвергаются эффективному внутримолекулярному переносу энергии. Примеры таких молекул изображены на фигуре 41. С дифенилэтиновым линкером скорость переноса энергии от цинк-порфирина к порфирину со свободным основанием составляет (24 псек)-1. С п-фениленовым линкером скорость переноса энергии от цинк-порфирина к порфирину со свободным основанием составляет (2 псек)-1. Для зондирования роли линкера при опосредовании переноса энергии возбужденного состояния между порфиринами исследуются электрохимические свойства этих мультипорфириновых цепочек. Электрохимические потенциалы индивидуальных пигментов сохраняются при включении в цепочки. Однако дырка, образующаяся в мультипорфириновой цепочке, является делокализованной, как определяется путем анализа с помощью ЭПР. Скорость миграции дырок по прыжковому механизму между порфиринами в цепочке в жидком растворе является более высокой, чем та, которая может быть обнаружена с помощью методики ЭПР, что предполагает скорость > 107 сек-1 (Seth, J. et al., J.Am.Chem.Soc. 1994, 116, 10578-10592; Seth, J. et al., J.Am.Chem.Soc. 1996, 118, 11194-11207). Эти исследования доказывают, что существуют значительные взаимодействия электронов основного состояния в мультипорфириновых цепочках и что взаимодействие должно быть опосредовано с помощью линкера, который соединяет порфирины. Как итог, мультипорфириновые цепочки имеют желаемые особенности поглощения света и миграции энергии возбужденного состояния, которые являются главными для эффективной аккумуляции света, и в дополнение к этому обладают неожиданным свойством облегчения процессов миграции дырок по прыжковому механизму в основном состоянии при образовании окисленных комплексов.
B. Конструирование линейных цепочек
Одна из проблем при конструировании солнечных элементов включает в себя интегрирование различных компонентов, которые включают в себя антенну, УРЗ, и путей для протекания электронов от УРЗ и к нему. Здесь предложены новые средства для перемещения окислительного эквивалента прочь от узла разделения заряда. В основном антенна конструируется таким образом, что энергия протекает вдоль цепочки для аккумуляции света к узлу разделения заряда, в то время как окислительный эквивалент (дырка) протекает в обратном направлении от УРЗ к позиции в антенне, где могут иметь место последующие реакции переноса электрона (фигура 42). Эта конструкция имеет две значительных разновидности. (1) для доступа в УРЗ требуются только два канала: один для испускания электронов и один тот, где энергия возбуждения втекает, а окислительные эквиваленты (дырки) вытекают (фигура 43). Существование двух каналов вместо трех облегчает ограничения на организацию антенн для аккумуляции света вокруг УРЗ при 3-мерной упаковке. (2) Миграция дырки прочь от узла разделения заряда приводит к стабилизации состояния с разделенным зарядом. Подавляющее большинство подходов, разработанных для стабилизации состояния с разделенным зарядом в синтетических системах, сосредотачиваются на использовании ряда акцепторов электрона для перемещения электрона прочь от дырки. Сходный подход (описанный здесь) использует ряд акцепторов дырок для перемещения дырки прочь от электрона, и тем самым, для получения стабильного состояния с разделенным зарядом.
Порфириновые молекулы являются центральными в этом дизайне. Порфирины сильно поглощают свет и дают стабильные катион-радикалы при окислении. Характерной особенностью порфиринов является то, что природа заместителей, отбирающих электрон или высвобождающих электрон, присоединенных к порфирину, вызывает соответствующие изменения в элекрохимических потенциалах порфиринов, но не изменяет значительно спектр поглощения порфиринов (Seth, J.; Palaniappan, V.; Wagner, R.W.; Johnson, T.E.; Lindsey, J.S.; Bocian, D.F. J.Am.Chem.Soc. 1996, 118, 11194-11207). Следовательно, элекрохимические потенциалы порфиринов могут регулироваться без изменения уровней энергии, которые играют роль при миграции энергии. Другими словами, заместители сдвигают уровни энергии как HOMO, так и LUMO, вызывая изменения соответственно в окислительных и восстановительных потенциалах. спектр поглощения зависит от разницы между энергиями HOMO и LUMO; если как HOMO, так и LUMO сдвигаются одинаково, никакого изменения в запрещенной зоне HOMO-LUMO не наблюдается, а следовательно, спектр поглощения остается неизменным. Способность к регулировке окислительного потенциала без влияния на спектр поглощения делает возможным дизайн каскадов с переносом дырок, в то же время поддерживая связанные с переносом энергии свойства цепочек. Тщательный выбор пигментов с различными спектрами поглощения в сочетании со структурными модификациями для электрохимической регулировки дает возможность энергии и дыркам перетекать в область с меньшей потенциальной энергией в противоположных направлениях.
Следующие далее примеры иллюстрируют противоположное протекание энергии возбуждения и дырок в основном состоянии. На фигурах 44-49 диаграммы уровней энергии являются иллюстративными.
(1) Линейная цепочка цинк порфиринов, несущих на себе различные мезо-заместители (фигура 44). Все четыре порфирина имеют по существу идентичные спектры поглощения, следовательно, миграция энергии осуществляется между четырьмя изоэнергетическими порфиринами. Перенос энергии осуществляется обратимо между всеми четырьмя порфиринами. Электрохимические потенциалы расположены каскадом, при этом самый высокий потенциал находится вблизи УРЗ, а самый низший потенциал - вдали от УРЗ. По этой причине миграция дырок происходит необратимо с перемещением от высокого к низкому потенциалу.
(2) Линейная цепочка из Mg- и Zn-порфиринов, несущих на себе различные мезо-заместители (фигура 45). Mg-порфирины поглощают при несколько большей длине волны (˜5-10 нм), чем Zn -порфирины, и легче окисляются (то есть имеют более низкий потенциал), чем Zn-порфирины (Li, F. et al., J. Mater. Chem. 1997, 7, 1245-1262; Hascoat, P. et al., Inorg.Chem. 1999, 38, 4849-4853). Последовательность Zn, Zn, Mg, Mg при движении по направлению к УРЗ приводит к обратимому переносу энергии между двумя изоэнергетическими Zn-порфиринами, к необратимой миграции энергии от Zn- к Mg-порфирину и к обратимому переносу энергии между двумя изоэнергетическими Mg-порфиринами. Хотя Mg-порфирины окисляются легче, чем Zn-порфирины (с идентичными заместителями), размещение сильно отбирающих электроны заместителей на Mg-порфиринах и высвобождающих электроны заместителей на Zn-порфиринах вызывает обращение порядка следования окислительных потенциалов (Yang, S.I. et al., J. Porphyrins Phthalocyanines 1999, 3, 117-147). Для линейной цепочки из порфиринов, изображенной на фигуре 45, расположение таких заместителей создает каскад от высокого потенциала вблизи УРЗ до низкого потенциала вдали от УРЗ. Таким образом, эта система создает каскад с плавным переходом для миграции энергии и ступенчатый каскад для миграции дырок в противоположном направлении.
(3) Линейная цепочка из металлохлоринов, несущих на себе различные мезо-заместители (фигура 46). Хлорины сильно поглощают как в голубой, так и в красной областях, эффективно перекрывая большую часть спектра солнечного света. Хлорины являются членами семейства порфиринов (то есть циклического тетрапиррола). Как и у порфиринов, элекрохимические потенциалы хлоринов изменяются рациональным и предсказуемым способом в присутствии отбирающих электрон или высвобождающих электрон заместителей. Однако спектры поглощения по существу не подвержены индуктивным воздействиям заместителей. Таким образом, расположение хлоринов в линейной цепочке приводит к обратимой миграции энергии между изоэнергетическими пигментами, но к необратимому переносу дырок при перемещении прочь от УРЗ.
(4) Линейная цепочка из порфиринов и хлоринов, несущих на себе различные мезо-заместители (фигура 47). Полоса длинноволнового поглощения (а следовательно, и энергия возбужденного синглетного состояния) металлопорфирина попадает в область более коротких длин волн (более высоких энергий), чем полоса соответствующего металлохлорина. С другой стороны, хлорин легче окисляется (более низкий потенциал), чем соответствующий металлопорфирин. Энергетический каскад и каскад для дырок могут быть созданы таким образом, как изображено на фигуре 47. Сильно отбирающие электроны заместители используются вместе с хлоринами, и высвобождающие электроны заместители используются вместе с порфиринами, сдвигая хлорины к более высокому потенциалу, чем у порфиринов. Обратимый перенос энергии осуществляется между парой порфиринов, необратимый перенос осуществляется от порфирина к хлорину, и обратимый перенос осуществляется между парой хлоринов. Обратимый перенос дырок осуществляется между парой хлоринов с последующим необратимым переносом дырок от хлорина к порфирину и опять от порфирина к порфирину.
(5) Линейная цепочка из β-замещенных хлоринов и мезо-замещенных хлоринов (фигура 48). β-Замещенные хлорины, которые доступны в настоящее время, не несут на себе третьего заместителя для регулировки элекрохимического потенциала, как делают мезо-замещенные хлорины. Кроме того, β-замещенные хлорины могут плодотворно инкорпорироваться, как изображено на фигуре 48. Это расположение дает обратимую миграцию энергии и каскадный процесс переноса дырок. Заметим, что хлорины могут быть расположены с частично насыщенным кольцом, расположенным по направлению к УРЗ или от него; различными ориентациями, представленными на фигуре 48, не вызывается никаких различий в рабочих характеристиках.
(6) Линейная цепочка из порфириновых, хлориновых, и фталоцианиновых компонентов (фигура 49). Фталоцианины характеризуются очень сильным поглощением в красной области (ε˜250000 M-1см-1) и высокими окислительными потенциалами (Li, J. et al., J.Org.Chem. 1999, 64, 9090-9100; Yang, S.I. et al., J.Mater.Chem. 2000, 10, 283). Цепочка, изображенная на фигуре 49, демонстрирует каскад с последовательно изменяющимися энергиями от Zn-порфирина к Mg-порфирину, к хлорину и к фталоцианину. процесс миграции дырок осуществляется в противоположном направлении при регулировке потенциалов Mg -хлорина и Zn-порфирина с использованием мезо-заместителей.
C. Другие составы
Семейство порфиринов включает в себя различные пигменты, многие из которых могут быть использованы в линейных архитектурах для миграции энергии и дырок в противоположных направлениях. Главные члены семейства включают в себя тетраазапорфирины, гетероатом-модифицированные порфирины (например, N3O- или N3 S- вместо стандартных N4-порфиринов) (Cho, W.-S. et al., J.Org.Chem. 1999, 64, 7890-7901), коррол и многочисленные растянутые и сжатые порфирины (Van Patten, P.G. et al., J.Phys.Chem. B 1998, 102, 4209-4216).
В целом, большое множество заместителей являются доступными для регулировки элекрохимических потенциалов, включая различные арильные и алкильные группы. Галогенированные арильные группы являются особенно привлекательными для тонкой регулировки элекрохимических потенциалов.
Все цепочки, представленные на фигурах 44-49, используют дифенилэтиновые или п-фениленовые линкеры. Скорость переноса энергии при переходе от дифенилэтинового линкера к п -фениленовому линкеру увеличивается примерно в 10 раз для порфиринов (˜(2 псек)-1)5 и в 100 раз для хлоринов (суб-пикосекунды). Хотя эти линкеры являются вполне привлекательными, могут быть использованы также и другие линкеры.
D. Другие конструкции
Доступность быстрых процессов переноса энергии (то есть с помощью хлоринов или порфиринов, связанных п-фениленом) имеет важное последствие для дизайна цепочек для аккумуляции света. Моделирование воздействия скоростей переноса энергии на квантовый выход переноса энергии в ловушку, расположенную в конце линейной цепочки, показывает следующее (Van Patten, P.G. et al., J.Phys.Chem.B 1998, 102, 4209-4216): при обратимом переносе между изоэнергетическими пигментами квантовый выход уменьшается очень быстро с ростом количества пигментов. Однако квантовый выход является также очень чувствительным к скорости переноса. Повышенная скорость, даже для обратимого переноса между изоэнергетическими пигментами, замедляет падение квантового выхода с увеличением количества пигментов. Для скоростей в диапазоне от нескольких псек до долей псек линейные цепочки из разумного количества изоэнергетических пигментов (например, вплоть до 20) должны давать приемлемые квантовые выходы для возбуждения, достигающего ловушки (то есть УРЗ). Подобным же образом, при низких скоростях переноса (десятки пикосекунд) высокий квантовый выход в линейной цепочке подобных же размеров может быть получен только путем использования энергетического каскада (то есть необратимых стадий переноса энергии). В итоге линейная цепочка, состоящая из нескольких идентичных (изоэнергетических) пигментов, будет обеспечивать эффективный перенос энергии, если скорость переноса является очень высокой.
Пример цепочки, использующей множество изоэнергетических пигментов, иллюстрируется на фигуре 50. Эта цепочка включает в себя такие же пигменты на основе Zn-порфирина, Mg-порфирина и Zn -хлорина, какие используются на фигуре 49. Линкеры представляют собой исключительно п-фениленовые группы. Архитектура использует набор из n Zn-порфиринов, затем набор из n Mg-порфиринов, а затем один Zn-хлорин. Перенос энергии и дырок осуществляется обратимо между элементами данного набора, но перенос между различными наборами происходит необратимо (в сторону более низких потенциалов). Расположение пигментов напоминает тип архитектуры водопада с множеством бассейнов. Этот дизайн иллюстрирует ту концепцию, что не является необходимым иметь стадию переноса энергии к более низкому потенциалу, осуществляемую в каждом пигменте для достижения эффективного переноса энергии от позиции поглощения света к УРЗ.
Для быстрого переноса энергии идеальными выборами является использование п-фениленовых линкеров вместе с порфиринами (имеющими a2u HOMO и линкеры в мезо-положениях) (Yang, S.I. et al., J.Am.Chem.Soc. 1999, 121, 4008-4018) и/или хлоринов (имеющих линкеры в мезо- или β-положениях). Для быстрой миграции дырок по прыжковому механизму линкер должен быть соединен с позицией на пигменте, имеющей значительную электронную плотность в HOMO. в то время как перенос энергии может происходить по механизмам TS и/или TB, миграция дырок основного состояния по прыжковому механизму происходит исключительно по TB-механизму (по меньшей мере, для наблюдаемых высоких скоростей). Факторы, которые влияют на TB-миграцию энергии, являются подобными тем, которые влияют на миграцию дырок основного состояния по прыжковому механизму. Таким образом, цепочки с быстрой TB -миграцией энергии должны также давать высокие скорости миграции дырок по прыжковому механизму. Для эффективной миграции дырок по прыжковому механизму также не является важным иметь стадию миграции дырок по прыжковому механизму к более низкому потенциалу, осуществляемую на каждом пигменте, для достижения эффективного переноса от УРЗ к позиции, удаленной от него, в антенне для аккумуляции света.
Хотя в этих конструкциях энергия возбужденного состояния и дырки в основном состоянии образуются в одной и той же цепочке, при условиях быстрой динамики миграции энергии и низкого потока солнечного света вероятность одновременного присутствия возбужденного состояния и дырки является очень низкой. Соответственно гашение возбужденного состояния дырками в основном состоянии, как ожидается, является редким событием.
V. СИНТЕЗ ПОЛИМЕРОВ in situ ДЛЯ АККУМУЛЯЦИИ СВЕТА НА ЭЛЕКТРОХИМИЧЕСКИ АКТИВНЫХ ПОВЕРХНОСТЯХ
Синтез олигомеров из составляющих блоков (ВВ) на основе пигментов или стержней для аккумуляции света может происходить путем реакций нескольких различных типов. Главная проблема заключается в том, чтобы реакция, используемая для соединения составляющих блоков на основе пигментов в диадной архитектуре, также создавала бы линкер, который обеспечивает сообщение электронов между двумя пигментами. Соответственно в целом рассматривается более ограниченный набор реакций, чем тот, который существует во всей органической химии. Способы для синтеза полимерных цепочек из составляющих блоков на основе пигментов включают в себя, но не ограничиваются этим, использование следующих типов реакций (фигура 51):
связывание по Glaser (или по Eglinton) составляющих блоков на основе мономерного пигмента (с генерацией бутадиинового линкера).
связывание по Cadiot-Chodkiewicz двух различных составляющих блоков на основе пигментов (с генерацией бутадиинового линкера, соединяющего блок-сополимер).
Связывание по Sonogashira двух различных составляющих блоков на основе пигментов (с генерацией этинового линкера, соединяющего блок-сополимер).
Реакции по Heck или Виттига двух различных составляющих блоков на основе пигментов (с генерацией алкенового линкера, соединяющего блок-сополимер).
Связывание Сузуки двух различных составляющих блоков на основе пигментов (с генерацией фениленового или бифенильного линкера, соединяющего блок-сополимер).
Полимеризация составляющих блоков на основе пигментов, несущих на себе заместители, такие как две или более тиофеновых группы (с генерацией ологитиофенового линкера) или две или более пиррольных групп (с генерацией полипиррольного линкера).
Синтез олигомеров может осуществляться с использованием постадийных способов или с использованием способов полимеризации. Оба способа, как правило, требуют двух реакционно-способных групп, присоединенных к составляющему блоку на основе пигмента, для получения полимера, где составляющие блоки на основе пигмента являются интегральными компонентами основной цепи полимера. (В качестве альтернативы, менее привлекательный дизайн приводит к получению полимеров с боковыми группами, где составляющие блоки на основе пигментов присоединяются через одну связь к основной цепи полимера.) Постадийный способ синтеза, как правило, требует использования защитных групп для маскировки одного из центров реакции, и один из циклов реакции затем включает в себя связывание с последующим снятием защиты. В способе полимеризации защитные группы не используются и полимер получается в процессе, происходящем в одной колбе.
Процессы полимеризации могут иметь место в растворе или могут быть осуществлены с помощью полимера, растущего с поверхности. Полимеризация может осуществляться, начиная с твердой подложки, как в твердо-фазном синтезе пептидов или ДНК, с последующим удалением, очисткой и дальнейшей обработкой для конкретных применений. Полимеризация с получаемым полимером, присоединенным к электрохимически активной поверхности, генерирует желаемый материал для аккумуляции света in situ. Последний подход является исключительно привлекательным, устраняя потребность в обработке полимеров. Способность к исключению манипуляций с полимерами делает возможным синтез соединений, которые не проявляют достаточной растворимости в большинстве растворителей для удобства манипуляций (растворения, очистки, обработки, характеризации раствора).
Могут быть созданы полимеры, которые состоят из идентичных узлов или из различных узлов, как в блок-сополимерах или в случайных сополимерах. Альтернативно, может быть осуществлена полимеризация для создания линейной цепочки, где композиция различных составляющих блоков на основе пигментов организуется в виде градиента. Последний подход дает возможность создания энергетического каскада для протекания энергии возбужденного состояния и/или обратного протекания дырок в основном состоянии систематическим образом вдоль всей длины цепочки, как описано в других местах настоящей заявки.
Следующее далее описывает синтез in situ каскадных полимеров на электрохимически активной поверхности, такой как золото или TiO2: полимеризуемый узел (составляющий блок на основе пигмента или линкер) присоединяется к поверхности (для Au тиольная соединительная группа используется в качестве Y1; для TiO2 соединительная группа карбоновой кислоты используется в качестве Y2). Добавляется первый составляющий блок на основе пигмента (ВВ1), и связывающие реагенты добавляются для осуществления полимеризации (например, связывания по Glaser). Затем поверхность промывается для удаления связывающих реагентов (реагентов на основе меди в случае связывания по Glaser) и любых непрореагировавших ВВ1. Затем добавляют второй составляющий блок на основе пигмента (ВВ2), затем связывающие реагенты, и полимеризации дают возможность для продолжения. Такая же самая процедура для промывки осуществляется опять, а затем добавляется третий составляющий блок на основе пигмента (ВВ3), а затем связывающие реагенты, и полимеризации дают возможность для продолжения. Повторение этого процесса делает возможным систематическое выстраивание линейной цепочки из составляющих блоков на основе пигментов с монотонно изменяющимися уровнями энергии для протекания энергии возбужденного состояния и дырок в основном состоянии. Последний присоединенный мономер несет на себе один отдельно взятый реакционный центр (J-L-(ВВ)-L-Y), и соединительная группа Y используется для последующего связывания с противоположной поверхностью. Характеризация иммобилизованного на поверхности полимера достигается с помощью спектроскопии поглощения, ИК-спектроскопии, спектроскопии отражения и времяпролетной масс-спектрометрии с лазерной десорбцией.
В представленном примере (смотри фигуру 52) для поверхности из Au используется тиольная соединительная группа (X), образующая самособирающийся монослой на золоте. Такие самособирающиеся монослои являются известными для дериватизованных тиолами порфиринов (Gryko, D.T. et al., J.Org.Chem. 1999, 64, 8635-8647). Для другой поверхности, состоящей из TiO2, для присоединения используется соединительная группа карбоновой кислоты (Y). Полимеризуемые группы могут быть любого типа, описанного выше, при использовании различных именных реакций (Glaser, Sonogashira, Cadiot-Chodkiewicz, Heck, Виттиг, Сузуки и тому подобное). Конечный полимерный продукт состоит из доменов из различных составляющих блоков на основе пигментов [(ВВi)n] в виде линейной цепочки.
VI. ПРИМЕНЕНИЯ СОЛНЕЧНЫХ ЭЛЕМЕНТОВ ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Солнечные элементы по настоящему изобретению могут быть использованы во множестве различных электрических устройств. Такие устройства, как правило, содержат солнечный элемент, как описано выше, и некоторую схему (например, резистивную нагрузку), электрически соединенную с указанным солнечным элементом (например, путем создания первого электрического соединения схемы с одним из электродов солнечного элемента, и второго электрического соединения схемы с другим электродом солнечного элемента). Солнечный элемент может обеспечивать единственный источник питания для схемы, может быть вспомогательным источником, может включаться для зарядки батареи (аккумулятора) и тому подобное. Любое из множества различных электрических устройств может включать в себя солнечный элемент по настоящему изобретению, включая, но, не ограничиваясь этим, радиоприемники, телеприемники, компьютеры (такие как персональные компьютеры), процессоры, калькуляторы, телефоны, устройства для беспроволочной связи, такие как пейджеры, часы, устройства для экстренного определения местонахождения, электрические средства передвижения, источники питания для экстренных ситуаций, генераторы энергии, устройства для подсветки или лампы и другие осветительные приборы, устройства для мониторинга, устройства для проверок, детекторы излучения, устройства для создания изображений, устройства для оптической связи.
Следующие далее примеры приводятся для иллюстрации определенных аспектов настоящего изобретения и не предназначены для его ограничения.
Примеры
Рациональный синтез β-замещенных составляющих блоков на основе хлорина
В этих примерах представлен синтез β-замещенных составляющих блоков на основе хлорина. Конструируются две новые восточные половины, каждая из которых несет на себе один β-заместитель и один (не присоединяемый к соседнему составляющему блоку) мезо -заместитель, и приготавливается одна новая западная половина, которая несет на себе один β-заместитель. Эти новые предшественники используются в сочетании с предшествующей западной половиной (1) с получением трех новых хлоринов, каждый из которых несет на себе один β- и один мезо-заместитель. Также приготавливают хлорин, несущий на себе один мезо-заместитель и заместители в положениях 2 и 12. Такие составляющие блоки до сих пор не были доступны, и в сочетании с мезо-замещенными хлоринами, описанными ранее (синтез представлен на фигуре 53), должны открыть возможности для множества фундаментальных исследований, включая исследования воздействия области присоединения линкера на сообщение электронов в многочисленных архитектурах на основе хлорина.
Результаты и обсуждение
Синтез восточной половины (ВП): Синтез β-замещенной ВП начинается таким же образом, как и известный из литературы синтез β-замещенных дипиррометанов (Balasubramanian, T.; Lindsey, J.S. Tetrahedron 1999, 55, 6771-6784), но использует ряд значительных усовершенствований (фигура 54). Йодфенил-замещенный пиррол (3) легко приготавливают из 4-йодбензальдегида, моноэтилмалоната и тозилметилизоцианида. Этоксикарбонильную группу удаляют с помощью обработки NaOH в этиленгликоле при 160°C с получением 3-(4-йодфенил)пиррола (4) с выходом 91% в виде бледно-коричневых кристаллов. Примечательно, что это одностадийное декарбоксилирование является преимущественным по сравнению с двухстадийным преобразованием на подобных пиррольных соединениях (Pavri, N.R.; Trudell, M.L. J.Org.Chem. 1997, 62, 2649-2651). Формилирование по Vilsmeier-Haack 4 дает смесь двух региоизомеров (отношение ˜6:1), которые легко различаются с помощью1H ЯМР спектроскопии (смотри экспериментальный раздел). Главный изомер желательно представляет собой соединение (5) и получается в чистой форме путем перекристаллизации с выходом 62%. Защита пиррольного азота с помощью BOC группы (Tietze, L.F.; Kettschau, G.; Heitmann, K. Synthesis 1996, 851-857) дает пиррол 6 с количественным выходом. Восстановление до спирта 7 достигается путем обработки с помощью LiBH4 при низкой температуре (более долгое время реакции или более высокая температура приводят к получению перевосстановленного и лишенного защиты соединения 2-метил-3-(4-йодфенил)пиррола). Обработка спирта 7 избытком пиррола в кислых условиях приводит к получению β-замещенного, монозащищенного дипиррометана 8 с выходом 68%. Избыток пиррола является необходимым для сведения к минимуму образования трипиррометана, в то время как защита пиррольного азота является необходимой для облегчения реакции, устраняя самоконденсацию и делая возможным последующее селективное моноакрилирование. Эта методика дает β-замещенный дипиррометан как единственный региоизомер в противоположность более раним методикам, которые дают смесь двух региоизомеров (Balasubramanian, T.; Lindsey, J.S. Tetrahedron 1999, 55, 6771 -6784).
Разработаны способы для ацилирования 5-замещенных дипиррометанов, которые включают в себя получение пиррольного реагента Гриньяра с последующей обработкой с помощью хлорангидрида (Lee, C.-H.; Li, F.; Iwamoto, K.; Dadok, J.; Bothner-By, A.A.; Lindsey, J.S. Tetrahedron 1995, 51, 11645 -11672). В этом случае N-защищенный дипиррометан удерживается для селективного моноацилирования α-положения в незащищенном пиррольном узле. Обработка дипиррометана 8 2,5 эквивалентами EtMgBr в ТГФ, а затем п-толуоил хлоридом дает моноацилированный дипиррометан 9 с выходом 66% (фигура 54). Однако подобная же реакция в толуоле приводит к получению смеси моноацилированного продукта, соединения со снятой защитой и некоторых неопределенных примесей. Контрольный эксперимент включает в себя обработку дипиррометана 8 небольшим избытком EtMgBr при 0°C в ТГФ в течение 1 часа, и обычное извлечение дает исходный материал с количественным выходом, таким образом, доказывая, что BOC группа является стабильной по отношению к условиям ацилирования. Удаление BOC группы при стандартных условиях (Hasan, I. et al., J.Org.Chem. 1981, 46, 157-164) дает соединение 10. Электрофильное бромирование соединения 10 с помощью NBS (1 экв.) в ТГФ при -78°C, следуя изложенным ранее способам (избыток NBS приводит к получению значительного количества дибром соединения), дает соединение 11.
Второй β-замещенный дипиррометан приготавливают с помощью связывания по Sonogashira (Sonogashira, K. et al., Tetrahedron Lett. 1975, 4467-4470) йодфенил-замещенного соединения 10 с помощью триметилсилилацетилена. Этим способом триметилсилилэтинил дипиррометан 12 получают с количественным выходом (фигура 55). Взаимодействие соединения 12 с NBS при -78°C приводит к получению соответствующего бромдипиррометана 13 с выходом 91%.
Приготовление дипиррометана, несущего на себе заместитель в другой β-позиции, с использованием того же защищенного BOC дипиррометана 8 также является возможным при обращении порядка ацилирования и снятия защиты, что приводит к получению соединения 10. Таким образом, снятие защиты с дипиррометана 8 с помощью NaOMe/MeOH дает β-замещенный дипиррометан 14 (фигура 55). Недавно разработана процедура для селективного моноацилирования мезо-замещенных дипиррометанов с использованием EtMgBr и S -пиридил-замещенного бензотиоата (Rao, P.D.; Dhanalekshmi, S.; Littler, B.J.; Lindsey, J.S. J.Org.Chem. submitted). Применение этого способа моноацилирования к соединению 14 приводит к получению смеси из двух региоизомеров (10, 16). Попытки получения соединения 16 в виде главного продукта путем варьирования экспериментальных условий были безуспешными. Разделение двух региоизомеров является сложным и требует широкого применения колоночной флэш-хроматографии. Малый изомер 16 получают с выходом 25%. Обработка соединения 16 с помощью 1 эквивалента NBS в ТГФ при -78°C дает соединение 17 с выходом 87% в виде желтого твердого продукта. Все β -замещенные 1 -бромдипиррометаны (11, 13, 17) являются до некоторой степени нестабильными, но остаются интактными в течение нескольких недель при хранении при 0°C в атмосфере аргона.
Синтез β-замещенной западной половины. Синтез западной половины, где отсутствуют какие-либо β-заместители за исключением парной диметильной группы (1), был разработан ранее (Strachan, J.P. et al., J.Org.Chem. 2000, 65, 3160-3172). Пиррол-карбоксальдегид 5, доступный в количествах нескольких грамм, обеспечивает удобную исходную точку для синтеза новой западной половины, несущей на себе синтетическую ручку в β -положении. β-Замещенная западная половина в сочетании с β -замещенной восточной половиной сделала бы возможным синтез составляющих блоков на основе хлорина, несущих на себе два β -заместителя, расположенных на противоположных сторонах макроцикла. Применение условий реакции, используемых для получения 2-(2-нитровинил)пиррола из 2-формилпиррола (Strachan, J.P.; O'Shea, D.F.; Balasubramanian, T.; Lindsey, J.S. J.Org.Chem. 2000, 65, 3160-3172), для реакции соединения 5 приводит, в сущности, к извлечению исходного материала. После ограниченных исследований было обнаружено, что обработка соединения 5 с помощью KOAc и небольшого избытка метиламин-гидрохлорида в нитрометане (вместо метанола) в качестве растворителя при комнатной температуре в течение 2 часов (вместо 16 часов) дает желаемый продукт 18 альдол-конденсации с выходом 89% (фигура 56). Можно заметить, что более продолжительное время реакции приводит к образованию продукта дополнения по Michael нитрометана на нитровинильной группе в продукте 18, формируя 2 -(1,3-динитро-2-пропил)-3-(4-йодфенил)пиррол с выходом ˜30%. Восстановление продукта 18 с помощью NaBH4 дает соединение 19, которое подвергается дополнению по Michael вместе с мезитилоксидом в присутствии CsF при 80°C с получением продукта нитро-гексанона 20, предшественника β-замещенной западной половины. Хотя дополнение по Michael является быстрым по сравнению с образованием β-незамещенной ответной части (предшественник соединения 1), выход является несколько более низким (42% по сравнению с 65%). Обработка соединения 20 с помощью NaOMe, а затем буферным раствором TiCl3 дает β-замещенную западную половину 21 с выходом 45-50% в виде светло-зеленого твердого продукта. Выход и стабильность β-замещенной ЗП является большим, чем выход незамещенного аналога (половина 21 имеет т.пл.=141 -142°C; 1 представляет собой масло).
Получение хлорина. Предшествующие способы синтеза хлорина включают в себя: (1) получение бромдипиррометан-монокарбинола (2 -OH, ВП) путем восстановления карбонильной группы в предшественнике ВП, (2) кислотно-катализируемую конденсацию ВП и ЗП (1) с получением дигидробилена-a, и (3) окислительную опосредованную металлом циклизацию с получением хлорина (Strachan, J.P.; O'Shea, D.F.; Balasubramanian, T.; Lindsey, J.S. J.Org.Chem. 2000, 65, 3160-3172). Все три стадии осуществляются последовательно в один и тот же день. Та же самая процедура используется и здесь за исключением того, что условия извлечения являются иными из-за лабильной природы β-замещенных предшественников ВП (11, 13, 17) и соответствующих β-замещенных восточных половин. В типичной реакции соединение 11 обрабатывается с помощью NaBH4 в ТГФ/MeOH (4:1) при комнатной температуре в атмосфере аргона. При исчезновении исходного материала (анализ с помощью ТСХ) продукт извлекается из реакционной смеси, и карбинол 11-OH обрабатывают с помощью 1,2 эквивалента ЗП 1 при комнатной температуре в CH3CN, содержащем ТФУ. Через 25-30 минут конечный дигидробилен-a получают путем гашения реакционной смеси с помощью водного раствора NaHCO3 и извлечения CH2Cl2. Добавляют безводный толуол и по 15 молярных эквивалентов каждого из AgIO3, Zn(OAc)2 и пиперидина и смесь нагревают при 80°C в течение ˜2,5 час. Реакционная смесь медленно превращается из красной в зеленую, что указывает на образование хлорина. Фильтрация реакционной смеси через прослойку из окиси кремния с последующей колоночной хроматографией дает хлорин Zn-22 с чистотой >90%. Осаждение с помощью CH2Cl2/гексана приводит к получению чистого хлорина (Zn -22) с выходом 18% (фигура 57). Подобная же обработка восточной половины 13-OH и соединения 1 дает цинк-хлорин Zn-23 с выходом 22%. Восточная половина (17), несущая на себе β -заместитель в положении 8, взаимодействует подобным же образом с соединением 1, давая цинк-хлорин Zn-24.
Zn-хлорины 22-24 несут на себе по одному β-заместителю. Для приготовления хлорина, несущего на себе два β-заместителя, 13-OH и западная половина 21 реагируют с получением цинк хлорина Zn-25 с выходом 24% (фигура 58). Этот хлорин имеет йодфенильную группу и этинилфенильную группу в β-положении на противоположных сторонах макроцикла. Порфирины, несущие на себе йодфенильные и этинилфенильные группы в транс-ориентации, используются при постадийном синтезе линейных мультипорфириновых цепочек (Wagner, R.W.; Lindsey, J.S. J.Am.Chem.Soc. 1994, 116, 9759-9760; Wagner, R.W.; Ciringh, Y.; Clausen, P.C.; Lindsey, J.S. Chem.Mater. 1999, 11, 2974-2983; Lindsey, J.S. et al., Tetrahedron 1994, 50, 8941-8968). Аналогичные линейные мультихлориновые цепочки могут быть получены с помощью Zn-25. В каждой из этих реакций с образованием хлоринов получается только один хлориновый продукт, что указывает на отсутствие перестановок в ходе реакции. Эта методика является совершенно общей, и выходы в 18-24%, получаемые для трех β-замещенных восточных половин (11-OH, 13-OH, 17-OH) и β-замещенной западной половины (21), заметно превосходят ˜10%, получаемые для мезо-замещенных восточных половин (2-OH) и западной половины (1).
Zn-хлорины деметаллируются с получением соответствующих хлоринов со свободным основанием путем обработки с помощью ТФУ в CH2Cl2. В большинстве случаев сырой продукт является достаточно чистым для анализа, в то время как в других случаях хлорин со свободным основанием дополнительно очищается с помощью короткой колонки с окисью кремния.
Спектральные свойства хлоринов. Спектры1H ЯМР. Доступная информация в спектрах ЯМР относительно хлоринов получена в основном для природных хлоринов, которые несут на себе алкильные группы, больше всего в β-положениях.1H ЯМР спектрыβ-замещенных хлоринов со свободным основанием (22-25) и Zn-хлоринов (Zn-22-Zn-25) легко интерпретируются и подтверждают ожидаемые структуры замещения. В 22, два NH протона появляются в виде широких пиков при δ -2,15 и -1,85 м.д., и сигнал при меньшей напряженности поля появляется для одного из мезо-замещенных протонов (приписывается C-10) при δ 9,84 м.д.. Восстановленное кольцо демонстрирует синглет при δ 2,07 м.д. (парные диметильные группы) и другой синглет при δ 4,64 м.д. (кольцевая CH2), что наблюдается также в мезо-замещенных хлоринах. Другие характерные особенности включают в себя AB квартет при δ 8,85 м.д. (β-пиррольные протоны кольца A), два дублета при δ 8,64 и 8,90 м.д. (β-пиррольные протоны кольца B), и синглеты при δ 8,91 (для 2H) и 8,99 м.д. (два мезо-протона при C-15 и C-20, и один β-пиррольный протон кольца C). Значительные изменения для β-замещенного Zn-22 представляют собой отсутствие сигналов, соответствующих протонам NH, и небольшие сдвиги в сторону больших напряженностей поля от парной диметильной группы (δ 2,01 м.д.), кольцевых метиленовых протонов (δ 4,48 м.д.) и всех мезо- и β-пиррольных протонов. Подобные же тенденции наблюдаются для хлорина со свободным основанием 23 и цинк-хлорина Zn-23.
Спектр1H ЯМР хлорина 24 является несколько иным из-за различий в структуре замещения по периметру молекулы. Характерные особенности в дополнение к отличным величинам химических сдвигов для двух протонов NH включают в себя синглет при δ 8,64 м.д. (β -пиррольный протон кольца B) и сдвинутый в сторону меньших напряженностей поля сигнал при δ 9,17 м.д. в виде дублета (один из β-пиррольных протонов кольца C). Спектр1H ЯМР хлорина 25 является более простым. β-Пиррольные протоны кольца B появляются в виде двух дублетов при δ 8,62 и 8,88, а квартет AB, соответствующий β-пиррольным протонам кольца A в хлоринах 22-24, отсутствует. Оставшиеся мезо-протоны и β-пиррольные протоны дают резонанс как пять синглетов. Zn-25 показывает подобную же структуру за исключением небольшого сдвига в сторону больших напряженностей поля пиков, связанных с мезо- и β-протонами.
Отличительная особенность этого набора хлоринов заключается в том, что β-пиррольные протоны кольца B проявляются при несколько больших напряженностях поля по сравнению со всеми другими пиррольными протонами. Это указывает на то, что β -пиррольная двойная связь кольца B не участвует полностью в обобществлении электронов 18π кольца хлоринового макроцикла.
Спектры поглощения. Каждый из хлоринов со свободным основанием (22-25) демонстрирует интенсивную полосу Сорэ и характерную сильную полосу Qy. Полоса Сорэ в каждом случае демонстрирует плечо на коротких длинах волн со значительной интенсивностью, что приводит к ширине пика на уровне полумаксимума в пределах 32-35 нм для 22-25. Подобная особенность спектра наблюдается для предыдущего набора мезо-замещенных хлоринов со свободным основанием, которые исследовались. Полоса Сорэ немного сдвигается в красную сторону, поскольку заместитель перемещается из положения 8 (24) в 12 (22, 23), в 2 и 12 (25). Значительные различия в положении максимума поглощения Qy и интенсивности поглощения происходят в зависимости от позиции замещения хлорина. Положение максимума поглощения Qy находится в пределах от 637 до 655 нм и изменяется параллельно с красным сдвигом полосы Сорэ. Кроме того, наблюдается гиперхромный эффект полосы Qy, который сопровождает батохромный сдвиг. Хотя точное определение молярных коэффициентов поглощения может быть сложным, особенно при манипуляциях с малыми образцами, отношение полос Qy и Сорэ обеспечивает относительную меру изменения интенсивностей полос. Отношение полос Сорэ/Qy уменьшается от 4,3 (24) до 2,5 (25). Эти данные перечислены в таблице 1. Можно заметить, что хлорины с йодфенильной или этинилфенильной группой в положении 12 демонстрируют приблизительно одинаковые спектры поглощения. Для сравнения, мезо-замещенные хлорины со свободным основанием демонстрируют максимумы поглощения при 411-414 нм и 640-644 нм.
Каждый из цинк-хлоринов (Zn-22-Zn-25) демонстрирует интенсивную полосу Сорэ и характерную сильную полосу Qy. Полоса Сорэ в каждом случае является узкой (ширина пика на уровне полумаксимума 18-21 нм) только с очень слабым плечом в области коротких длин волн. Полоса Qy подвергается красному сдвигу от 606 нм до 628 нм, поскольку положение заместителя изменяется c 8 (Zn-24) на 12 (Zn-22, Zn-23), до 2 и 12 (Zn-25). Также наблюдается одновременное увеличение интенсивности полосы Qy. Эти результаты перечислены в таблице 1. У всех исследуемых хлоринов красный сдвиг полосы Сорэ сопровождается более выраженным красным сдвигом полосы Qy. Единственное несоответствие в этой тенденции происходит при сравнении Zn-24 и Zn-22 (или Zn-23). Первый из них имеет самую короткую длину волны для полосы Qy (606 нм), но полосу Сорэ на 415 нм, по сравнению с 615 нм и 411 нм для них же в последнем случае. Для сравнения, мезо-замещенные цинк-хлорины демонстрируют максимумы поглощения при 412 нм и 608 нм.
Флуоресцентные спектры и выходы. Подобно мезо-замещенным хлоринам, хлорины со свободным основанием 22-24 демонстрируют характерную узкую зону флуоресценции при 640 нм и более слабое испускание в области 660-720 нм. Последняя демонстрирует два хорошо различимых максимума приблизительно при 680 и 710 нм. Спектр испускания хлорина 25 со свободным основанием сдвигается к 660 нм и 726 нм. Zn-хлорины Zn-22 и Zn-23 демонстрируют каждый узкую зону флуоресценции вблизи 620 нм и слабую полосу на 676 нм, в то время как испускание Zn-24 появляется на 609 и 661 нм. Спектр испускания Zn-25 сильнее сдвинут в красную сторону, чем это наблюдается для формы свободного основания 25 (635 и 691 нм). Квантовые выходы флуоресценции определяются для тех хлоринов, у которых отсутствуют йодфенильные заместители (которые демонстрируют уменьшение выходов из-за эффекта тяжелого атома). Квантовый выход флуоресценции для хлорина 23 со свободным основанием составляет 0,25, в то время как для Zn-23 он составляет 0,11. Эти величины согласуются с величинами для других природных или синтетических хлоринов.
Выводы. Синтез хлоринов, описанный здесь, предусматривает следующие особенности: (1) контроль над положением восстановленного кольца, (2) поддержание заданного уровня гидрирования хлорина путем использования парных диметильных групп, (3) размещение синтетических ручек в заданных местах по периметру макроцикла, и (4) единственный хлориновый продукт, тем самым, облегчается очистка. Способность к включению заместителей в различных положениях (2, 5, 8, 10, или 12) по периметру хлорина открывает ряд принципиальных возможностей. При различных структурах замещения положение полосы поглощения Qy может быть изменено в пределах 637-655 нм для хлоринов со свободным основанием, и 606-628 нм для цинк хлоринов, делая возможным более широкое перекрывание спектра. Хлорин, несущий на себе синтетические ручки в положениях 2 и 12 (25), должен создавать возможность для включения составляющих блоков на основе хлорина в линейные архитектуры. Доступность семейства синтетических хлоринов, несущих на себе различные заместители в заданных положениях, должно облегчить систематические исследования воздействия заместителей и расширить рамки содержащих хлорин модельных систем.
Экспериментальный раздел
Общая часть. Спектры1H и13C ЯМР (300 МГц) получают в CDCl3, если не указано иного. Спектры поглощения (Cary 3, интервалы между точками данных 0,25 нм) и спектры флуоресценции (Spex FluoroMax, интервалы между точками данных 1 нм) измеряют рутинными способами. Хлорины анализируют в исходной форме с помощью масс-спектрометрии с лазерной десорбцией (LD-MS) в отсутствие матрицы (Fenyo, D. et al., J. Porphyrins Phthalocyanines 1997, 1, 93-99; Srinivasan, N. et al., J. Porphyrins Phthalocyanines 1999, 3, 283-291). Пиррол дистиллируют при атмосферном давлении из CaH2. Температуры плавления не корректируются. п-Йодбензальдегид получают от Karl Industries. Все другие реагенты и исходные материалы получают от Aldrich. Спектральные параметры, включая молярные коэффициенты поглощения и квантовые выходы флуоресценции (Φf), определяются таким образом, как описано ранее (Strachan, J.P. et al., J.Org.Chem. 2000, 65, 3160-3172).
Хроматография. препаративная хроматография осуществляется с использованием окиси кремния (Baker) или окиси алюминия (Fisher A540, 80-200 меш) и элюирующих растворителей на основе гексана, смешанного с этилацетатом или CH2Cl2.
Растворители. ТГФ дистиллируют из натрий бензофенон кетила по потребности. CH3CN (Fisher certified A.C.S.) дистиллируют из CaH2 и хранят над порошкообразными молекулярными ситами. Нитрометан хранят над CaCl2. CH2Cl2 дистиллируют из CaH2. Сухой метанол приготавливают следующим образом. Магниевую стружку (5 г) и йод (0,5 г) вместе с 75 мл метанола нагревают до тех пор, пока йод не исчезнет, а весь магний не преобразуется в метоксид. Добавляют вплоть до 1 л метанола и нагревают с обратным холодильником в течение минимум 2 часов перед сбором. Другие растворители используют в том виде, как они получены.
Соединения 1(Strachan, J.P. et al., J.Org.Chem. 2000, 65, 3160-3172) и 3 (Balasubramanian, T.; Lindsey, J. S. Tetrahedron 1999, 55, 6771-6784) приготавливают в соответствии с процедурами, приведенными в литературе.
3-(4-Йодфенил)пиррол (4). Следуя изложенным ранее процедурам (Balasubramanian, T.; Lindsey, J.S. Tetrahedron 1999, 55, 6771-6784), смесь 3-этоксикарбонил-4-(4-йодфенил)пиррола (7,20 г, 21,1 ммоль) и этиленгликоля (55 мл) в 100-мл колбе Кляйзена продувают аргоном в течение 10 мин, а затем добавляют порошкообразный NaOH (2,2 г, 55 ммоль). Колбу помещают на масляную баню при 120°C и температуру масляной бани повышают до 160°C. Через 2,5 час колбу охлаждают до комнатной температуры и обрабатывают 10% водным раствором NaCl (100 мл). Водный слой экстрагируют CH2Cl2, органические слои собирают, промывают 10% водным раствором NaCl, сушат (Na2SO4), концентрируют и перекристаллизовывают в этаноле с получением светло-коричневых кристаллов (5,18 г, 91%). т.пл. 164-165°C;1H ЯМР δ 6,51 (м, 1H), 6,83 (м, 1H), 7,08 (с, 1H), 7,27 (д, J=8,7 Гц, 2H), 7,63 (д, J=8,7 Гц, 2H);13C ЯМР δ 89,9, 106,3, 114,7, 119,1, 123,8, 127,0, 135,2, 137,5; ЭИ-МС наблюдается 268,9702, рассчитано 268,9702. Аналитически рассчитано для C10H8IN: C, 44,6; H, 3,0; N, 5,2. Найдено: C, 44,7; H, 3,0; N, 5,1. Синтез, начиная с 4 -йодбензальдегида (35 г), моноэтил малоната, и TosMIC, осуществляется с линейным масштабированием установленных процедур с получением 21,5 г соединения 4.
2-Формил-3-(4-йодфенил)пиррол (5). Раствор соединения 4 (5,15 г, 19,1 ммоль) в ДМФ (6,1 мл) и CH2 Cl2 (140 мл) в атмосфере аргона охлаждают до 0°C и добавляют затем POCl3 (2,11 мл, 22,6 ммоль) по каплям. Через 1 час колбу нагревают до комнатной температуры и перемешивают в течение ночи. Реакцию гасят при 0°C с помощью 2,5 M NaOH (100 мл). Смесь выливают в воду (500 мл), экстрагируют CH2Cl2, и объединенные органические слои промывают водой, насыщенным раствором соли, сушат (Na2SO4) и растворитель удаляют в вакууме.1H ЯМР спектроскопия показывает два региоизомера при отношении ˜ 6:1. Малый изомер демонстрирует сигналы при δ 7,21 и 7,39 м.д., по сравнению с сигналами при δ 6,42 и 7,14 для главного изомера. Сигнал при самой низкой напряженности поля (7,39 м.д.) приписывается протону, соседствующему с формильной группой, что осуществляется в 2 -формил-4-арил замещенном пирроле. Перекристаллизация из этилацетата дает оранжевый твердый продукт, соответствующий главному альдегиду (2,25 г). Маточную жидкость концентрируют и очищают с помощью колоночной флэш-хроматографии [окись кремния, гексан/этилацетат (3:1)]. Первая фракция соответствует главному альдегиду (1,25 г). Общий выход указанного в заголовке соединения составляет 3,50 г (62%): т.пл. 153-154°C;1H ЯМР δ 6,42 (м, 1H), 7,14 (м, 1H), 7,22 (м, 2H), 7,76 (м, 2H), 9,59 (с, 1H), 10,72 (ушир, 1H);13C ЯМР δ 93,5, 104,3, 111,4, 125,8, 128,6, 130,8, 133,1, 137,8, 179,4; FAB-MS наблюдается 296,9663, рассчитано 296,9651; Аналитически рассчитано для C10 H8INO: C, 44,5; H, 2,7; N, 4,7. Найдено: C, 44,4; H, 2,7; N, 4,6.
N-трет-Бутоксикарбонил-2-формил-3-(4-йодфенил)пиррол (6). Следуя стандартной процедуре (Tietze, L. F.; Kettschau, G.; Heitmann, K. Synthesis 1996, 851-857), образец NaH (70 мг, 1,75 ммоль, 60% дисперсия в минеральном масле) в круглодонной колбе в атмосфере аргона дважды промывают безводным пентаном (˜5 мл). Добавляют безводный ТГФ (14 мл), а затем соединение 5 (400 мг, 1,35 ммоль). После перемешивания в течение 30 мин при комнатной температуре, добавляют (BOC)2O (325 мг, 1,5 ммоль), и продолжают перемешивание в течение следующих 2 час. Реакцию гасят 50% насыщенным водным раствором NH4Cl (50 мл). Смесь экстрагируют простым эфиром, и объединенные органические слои промывают насыщенным раствором соли, сушат (Na2SO4) и фильтруют [окись кремния, гексан/этилацетат (4:1)] с получением вязкого масла (535 мг, количественный).1H ЯМР δ 1,64 (с, 9H), 6,33 (д, J=3,0 Гц, 1H), 7,30 (д, J=8,1 Гц, 2H), 7,46 (д, J=3,0 Гц, 1H), 7,72 (д, J=8,1 Гц, 2H), 10,22 (с, 1H);13C ЯМР δ 27,7, 85,8, 94,2, 113,2, 126,7, 128,5, 131,3, 132,8, 137,0, 137,4, 148,3, 181,6; FAB-MS наблюдается 397,0176, рассчитано 397,0175 (C16H16INO3).
N-трет-Бутоксикарбонил-2-гидроксиметил-3-(4-йодфенил)пиррол (7). Раствор соединения 6 (400 мг, 1,0 ммоль) в безводном ТГФ (12 мл) в атмосфере аргона охлаждают до -20 - -25°C, и по частям добавляют LiBH4 (55 мг, 2,5 ммоль). Реакцию отслеживают с помощью ТСХ (окись кремния, гексан/этилацетат (4:1)), и когда уже не детектируется никакого исходного материала (20-25 мин), реакцию гасят холодной водой (30 мл). Водный слой экстрагируют CH2Cl2 и органический слой сушат (Na2SO4), концентрируют и очищают с помощью колоночной флэш-хроматографии [окись кремния, гексан/этилацетат, содержащий 1% Et3N (3:1)] с получением смолы (330 мг, 82%).1H ЯМР δ 1,62 (с, 9H), 3,61 (ушир, 1H), 4,66 (д, J=7,2 Гц, 2H), 6,25 (д, J=3,6 Гц, 1H), 7,18 (д, J=8,1 Гц, 2H), 7,22 (д, J=3,6 Гц, 1H), 7,71 (д, J=8,1 Гц, 2H);13C ЯМР δ 27,8, 55,3, 84,7, 92,4, 111,2, 121,3, 127,9, 130,0, 130,4, 134,1, 137, 5, 149,8; FAB-MS наблюдается 399,0336, рассчитано 399,0331 (C16H18INO3).
3-(4-Йодфенил)-10-N-(трет-бутоксикарбонил)дипиррометан (8). Раствор соединения 7 (1,2 г, 3,0 ммоль) и пиррола (3,36 мл, 48 ммоль) в 1,4-диоксане (36 мл) при комнатной температуре обрабатывают 10% водным раствором HCl (6,0 мл). Реакцию отслеживают с помощью ТСХ [окись кремния, гексан/этилацетат (4:1)]. Через 4 часа добавляют насыщенный водный раствор NaHCO3 (50 мл) и воду (50 мл) и смесь экстрагируют CH2Cl2. Объединенные органические слои промывают водой, насыщенным раствором соли, сушат (Na2SO4), концентрируют и очищают с помощью флэш-хроматографии [окись кремния, гексан/этилацетат (4:1)]. Неполярный продукт выделяют в малых количествах (не характеризуется). Желаемый продукт получается в виде бледно-коричневого твердого продукта (920 мг, выходом 68%): т.пл. 128 -129°C;1H ЯМР δ 1, 57 (с, 9H), 4,18 (с, 2H), 5,87 (ушир, 1H), 6,10 (м, 1H), 6,22 (д, J=3,0 Гц, 1H), 6,64 (м, 1H), 7,16 (д, J=8,0 Гц, 2H), 7,24 (д, J=3,6 Гц, 1H), 7,71 (д, J=8,0 Гц, 2H), 8,78 (ушир, 1H);13C ЯМР δ 24,6, 27,8, 84,3, 92,1, 105,8, 107,9, 111,6, 116,3, 121,0, 126,8, 128,5, 130,4, 130,8, 135,0, 137,4, 150,0; FAB-MS наблюдается 448,0659, рассчитано 448,0648; Аналитически рассчитано для C20H21IN2O2: C, 53,6; H, 4,7; N, 6,3. Найдено: C, 54,1; H, 4,9; N, 5,9.
3-(4-Йодфенил)-9-(4-метилбензоил)-10-N-(трет-бутоксикарбонил)дипиррометан (9). Раствор соединения 8 (448 мг, 1,0 ммоль) в безводном ТГФ (15 мл) в атмосфере аргона при 0°C медленно обрабатывают EtMgBr (1 M в ТГФ, 2,5 мл, 2,5 ммоль). Смесь перемешивают в течение 30 минут при 0°C, затем медленно добавляют п-толуоил хлорид (200 мкл, 1,5 ммоль), и перемешивание продолжают в течение 1 час при 0°C. Реакцию гасят насыщенным водным раствором NH4Cl и экстрагируют CH2Cl2. Объединенные органические слои промывают водой, насыщенным раствором соли, сушат (Na2SO4), концентрируют, и продукт очищают с помощью колоночной флэш-хроматографии [окись кремния, гексан/этилацетат (4:1)]. Продукт получается в виде беловатого твердого продукта (375 мг, 66%): т.пл. 120-121°C; (из-за возможных ротамеров, данные1H ЯМР и13C ЯМР являются не очень четкими)1H ЯМР δ 1,56 (с, 9H), 2,42 (с, 3H), 4,29 (с, 2H), 5,95 (м, 1H), 6,26 (м, 1H), 6,76 (м, 1H), 7,09 (м, 2H), 7,16 (м, 1H), 7,25 (м, 2H), 7,31 (м, 1H), 7,71 (д, 8,7 Гц, 2H), 7,77 (д, J=8,1 Гц, 2H), 9,95 (ушир, 1H);13C ЯМР δ 25,2, 27,8, 31,7, 84,8, 92,3, 109,3, 111,5, 119,6, 121,5, 125,8, 126,3, 128,9, 129,0, 130,2, 130,6, 134,7, 135,8, 137,5, 138,6, 142,0, 149,7, 183,8; Аналитически рассчитано для C28 H27IN2O3: C, 59,4; H, 4,8; N, 5,0. Найдено: C, 59,4; H, 4,6; N, 5,1.
3-(4-Йодфенил)-9-(4-метилбензоил)дипиррометан (10). Следуя стандартному способу для снятия защиты с BOC-защищенных пирролов (Hasan, I. et al., J.Org.Chem. 1981, 46, 157-164), раствор соединения 9 (328 мг, 0,58 ммоль) в безводном ТГФ (4 мл) в атмосфере аргона при комнатной температуре обрабатывают с помощью метанолового раствора NaOMe (0,7 мл, приготавливают путем растворения 200 мг NaOMe в 1,0 мл MeOH). Через 20-25 мин реакцию гасят смесью гексана и воды (20 мл, 1:1) и экстрагируют этилацетатом. Объединенные органические слои промывают водой, насыщенным раствором соли, сушат (Na2SO4) и очищают с помощью колоночной флэш-хроматографии [окись кремния, гексан/этилацетат (3:1)] с получением бледно-коричневого твердого продукта (216 мг, 80%): т.пл. 185-186°C;1H ЯМР δ 2,43 (с, 3H), 4,17 (с, 2H), 6,15 (м, 1H), 6,56 (м, 1H), 6,85 (м, 1H), 7,17 (м, 2H), 7,28 (м, 2H), 7,69 (м, 2H), 7,77 (д, J=7,8 Гц, 2H), 9,43 (ушир, 1H), 10,88 (ушир, 1H);13C ЯМР δ 21,6, 25,2, 90,6, 108,6, 110,3, 117,4, 121,1, 122,3, 123,9, 129,0, 129,1, 130,0, 130,7, 135,5, 136,2, 137,4, 139,4, 142,6, 185,2; FAB-MS наблюдается 466,0561, рассчитано 466,0542; Аналитически рассчитано для C23H19IN2O: C, 59,2; H, 4,1; N, 6,0. Найдено: C, 59,3; H, 4,2; N, 5,9.
1-Бром-3-(4-йодфенил)-9-(4-метилбензоил)дипиррометан (11). Следуя изложенным ранее процедурам (Strachan, J.P. et al., J.Org.Chem. 2000, 65, 3160-3172), соединение 10 (120 мг, 0,26 ммоль) растворяют в безводном ТГФ (6 мл) и охлаждают до -78°C в атмосфере аргона. добавляют перекристаллизованный NBS (46 мг, 0,26 ммоль) и реакционную смесь перемешивают в течение 1 час (-78°C), а затем гасят смесью гексана и воды (20 мл, 1:1), и дают ей возможность нагреться до 0°C. Водную часть экстрагируют эфиром химической чистоты и объединенные органические слои сушат над K2CO3. Растворитель выпаривают в вакууме при комнатной температуре. Очистка с помощью колоночной флэш-хроматографии [окись кремния, гексан/простой эфир (2:1)] дает желтый твердый продукт (120 мг, 85%). Бромдипиррометан является нестабильным, но может храниться в течение нескольких недель при 0°C. т.пл. 160°C (разл.);1H ЯМР δ 2,44 (с, 3H), 4,09 (с, 2H), 6,12 (д, J=3,0 Гц, 1H), 6,16 (м, 1H), 6,89 (м, 1H), 7,14 (д, J=7,8 Гц, 2H), 7,30 (д, J=7,8 Гц, 2H), 7,71 (д, J=8,1 Гц, 2H), 7,80 (д, J=8,1 Гц, 2H), 10,33 (ушир, 1H), 11,59 (ушир, 1H);13C ЯМР δ 21,6, 24,9, 91,1, 97,9, 110,2, 110,5, 122,8, 123,5, 125,4, 129,2, 130,2, 130,0, 130,8, 135,2, 135,4, 137,5, 139,9, 142,8, 186,1; FAB-MS наблюдается 543,9642, рассчитано 543,9647; Аналитически рассчитано для C23H18BrIN2O: C, 50,7; H, 3,3; N, 5,1. Найдено: C, 51, 3; H, 3,5; N, 5,2.
3-[4-(Триметилсилилэтинил)фенил]-9-(4-метилбензоил) дипиррометан (12). Образцы соединения 10 (279 мг, 0,599 ммоль), Pd2(dba)3 (42 мг, 0,046 ммоль), Ph3As (113 мг, 0,369 ммоль) и CuI (9 мг, 0,047 ммоль) добавляют в 25-мл колбу Шленка. Колбу откачивают и три раза продувают аргоном. Затем добавляют деаэрированный безводный ТГФ/Et3N (6 мл, 1:1), а затем триметилсилилацетилен (127 мкл, 0,90 ммоль). Колбу герметизируют, погружают в масляную баню (37°C), и смесь перемешивают в течение ночи. Затем добавляют CH2Cl2 (20 мл), и смесь фильтруют через прокладки из Целита, несколько раз промывают CH2Cl2, концентрируют и остаток очищают с помощью колоночной флэш-хроматографии [окись кремния, гексан/этилацетат (3:1)] с получением желтого твердого продукта (262 мг, количественно): т.пл. 126-127°C;1H ЯМР δ 0,26 (с, 9H), 2,43 (с, 3H), 4,19 (с, 2H), 6,16 (м, 1H), 6,28 (м, 1H), 6,55 (м, 1H), 6,85 (м, 1H), 7,28 (д, J=8,7 Гц, 2H), 7,38 (д, J=8,1 Гц, 2H), 7,49 (д, J=8,7 Гц, 2H), 7,77 (д, J=8,1 Гц, 2H), 9,51 (ушир, 1H), 10,96 (ушир, 1H);13C ЯМР δ 0,0, 21,5, 25,3, 105,4, 108,6, 110,3, 117,4, 119,9, 121,5, 122,3, 124,1, 127,6, 129,0, 129,1, 130,7, 132,0, 135,5, 137,0, 139,5, 142,6, 185,2; FAB-MS наблюдается 436, 1972, рассчитано 436,1971; Аналитически рассчитано для C28H28N2OSi: C, 77,0; H, 6,5; N, 6,4. Найдено: C, 76,3; H, 6,3; N, 6,3.
1-Бром-3-[4-(триметилсилилэтинил)фенил]-9-(4-метилбензоил)дипиррометан (13). Следуя процедуре для синтеза соединения 11, обработка соединения 12 (150 мг, 0,34 ммоль) с помощью NBS (60 мг, 0,34 ммоль) дает бледно-желтый твердый продукт (160 мг, 91%): т.пл. 140°C (разл.);1H ЯМР δ 0,26 (с, 9H), 2,44 (с, 3H), 4,12 (с, 2H), 6,17 (м, 2H), 6,89 (м, 1H), 7,31 (м, 4H), 7,50 (д, J=9,0 Гц, 2H), 7,80 (д, J=8,1 Гц, 2H), 10,16 (ушир, 1H), 11,42 (ушир, 1H);13C ЯМР δ 0,0, 21,5, 25,0, 94,1, 97,9, 105,2, 110,3, 110,5, 120,4, 123,3, 125,5, 127,7, 129,2, 130,7, 132,1, 135,4, 135,9, 139,7, 142,8, 185,9; FAB-MS наблюдается 514,1079, рассчитано 514,1076; Аналитически рассчитано для C28H27BrN2OSi: C, 65,2; H, 5,3; N, 5,4. Найдено: C, 65,1; H, 5,2; N, 5,3.
3-(4-Йодфенил)дипиррометан (14). Следуя процедуре снятия защиты, используемой для приготовления соединения 10, образец соединения 8 (225 мг, 0,50 ммоль) в безводном ТГФ (4 мл) в атмосфере аргона при комнатной температуре обрабатывают метаноловым раствором NaOMe (0,6 мл, приготавливают путем растворения 200 мг NaOMe в 1,0 мл MeOH). Через 15 мин реакцию гасят смесью гексана и воды (14 мл, 1:1), экстрагируют этилацетатом и объединенные органические слои промывают водой, насыщенным раствором соли, затем сушат над Na2SO4. Остаток пропускают через фильтрационную колонку с получением светло-коричневого твердого продукта (160 мг, 92%). Аналитические данные находятся в соответствии с данными литературы (Balasubramanian, T.; Lindsey, J. S. Tetrahedron 1999, 55, 6771 -6784).
3-(4-Йодфенил)-1-(4-метилбензоил)дипиррометан (16). Следуя общей процедуре моноацилирования для незащищенных дипиррометанов (Rao, P.D. et al., J.Org.Chem. 2000, 65, 1084-1092,), EtMgBr (1 M раствор в ТГФ, 2,2 мл, 2,2 ммоль) добавляют к раствору соединения 14 (385 мг, 1,1 ммоль) в безводном ТГФ (14 мл). После перемешивания в течение 10 мин колбу охлаждают до -78°C и медленно добавляют раствор пиридилового тиоэфира 15 (255 мг, 1,1 ммоль) в безводном ТГФ (3 мл). Через несколько минут охлаждающую баню удаляют, перемешивание продолжают в течение 1 час, затем смесь гасят насыщенным водным раствором NH4Cl, водой, а затем экстрагируют CH2Cl2. Объединенные органические слои промывают водой, насыщенным раствором соли, сушат (Na2SO4) и концентрируют. Два образовавшихся региоизомера очищают с помощью двух последовательных флэш-колонок [окись кремния, гексан/этилацетат (3:1)] с получением малого изомера 16 (130 мг, 25%) и главного изомера 10 (270 мг, 53%). Данные для 16: т.пл. 190°C (разл.);1H ЯМР δ 2,43 (с, 3H), 4,15 (с, 2H), 6,05 (м, 1H), 6,13 (м, 1H), 6,58 (м, 1H), 6,94 (м, 1H), 7,19 (д, J=8,7 Гц, 2H), 7,28 (д, J=8,1 Гц, 2H), 7,73 (д, J=8,7 Гц, 2H), 7,78 (д, J=8,1 Гц, 2H), 9,17 (ушир, 1H), 10,83 (ушир, 1H);13C ЯМР δ 21,6, 25,2, 91,8, 106,8, 108,3, 117,8, 121,1, 124,3, 127,2, 129,1, 129,2, 129,7, 130,2, 134,6, 135,4, 136,3, 137,6, 142,9, 185,4; FAB-MS наблюдается 466,0573, рассчитано 466,0542; Аналитически рассчитано для C23H19IN2O: C, 59,2; H, 4,1; N, 6,0. Найдено: C, 59,1; H, 4,2; N, 5,8.
9-Бром-3-(4-йодфенил)-1-(4-метилбензоил)дипиррометан (17). Следуя процедуре для синтеза соединения 11, обработка соединения 16 (186 мг, 0,400 ммоль) с помощью NBS (72 мг, 0,405 ммоль) дает бледно-желтый твердый продукт (189 мг, 87%): т.пл. 140°C (разл.);1H ЯМР δ 2,43 (с, 3H), 4,08 (с, 2H), 5,94 (м, 1H), 6,00 (м, 1H), 6,96 (д, J=2,1 Гц, 1H), 7,18 (д, J=8,7 Гц, 2H), 7,29 (д, J=8,1 Гц, 2H), 7,74 (д, J=8,7 Гц, 2H), 7,80 (д, J=8,1 Гц, 2H), 9, 80 (ушир, 1H), 11,53 (ушир, 1H); осуществляется попытка измерений13C ЯМР в CDCl3, но соединение разлагается при длительном получении данных. FAB-MS наблюдается 543,9628, рассчитано 543,9647; Аналитически рассчитано для C23H19IN2O: C, 50,7; H, 3,3; N, 5,1. Найдено: C, 51,2; H, 3,4; N, 5,0.
2-(2-транс-Нитровинил)-3-(4-йодфенил)пиррол (18). Смесь соединения 5 (1,485 г, 5,00 ммоль), KOAc (492 мг, 5,01 ммоль), метиламин-гидрохлорида (402 мг, 5,95 ммоль) и нитрометана (45 мл) в атмосфере аргона перемешивают при комнатной температуре. Смесь медленно становится оранжевой и дает красно-оранжевый осадок. Реакцию отслеживают с помощью ТСХ, и после перемешивания в течение 2 час ТСХ демонстрирует появление нового компонента и исчезновение соединения 5. (Более длительное время реакции (10 час) приводит к образованию продукта дополнения по Michael, 2-(1, 3-динитро-2-пропил)-3-(4-йодфенил)пиррола, с выходом ˜30%.) Реакцию гасят насыщенным раствором соли, экстрагируют этилацетатом и органические слои сушат (Na2SO4) и концентрируют. Остаток обрабатывают горячим этилацетатом и фильтруют, затем концентрируют и растворяют в горячем CH2Cl2 с последующим осаждением при добавлении холодного гексана и с получением оранжевого твердого продукта (1,52 г, 89%): т.пл. 217-218°C (разл.);1H ЯМР (ацетон-d6) δ 6,56 (д, J=2,1 Гц, 1H), 7,32 (д, J=8,2 Гц, 2H), 7,35 (м, 1H), 7,81 (д, J=13,5 Гц, 1H), 7,90 (д, J=8,3 Гц, 2H), 7,99 (д, J=13,4 Гц, 1H);13C ЯМР (ацетон -d6) δ 93,4, 112,5, 121,3, 127,1, 127,2, 128,4, 131,8, 132,8, 135,4, 138,9; FAB-MS наблюдается 339,9720, рассчитано 339,9709. Аналитически рассчитано для C12H9IN2O2: C, 42,4; H, 2,7; N, 8,2. Найдено: C, 41,8; H, 2,6; N, 7,9; λabs (толуол) 395 нм.
2-(2-Нитроэтил)-3-(4-йодфенил)пиррол (19). Следуя процедуре для β-незамещенного пиррола (Strachan, J.P. et al., J.Org.Chem. 2000, 65, 3160-3172), образец соединения 18 (1,36 г, 4,00 ммоль) растворяют в безводном ТГФ/MeOH (40 мл, 9:1) в атмосфере аргона при 0°C. по частям добавляют NaBH4 (605 мг, 16,00 ммоль) и перемешивание продолжают в течение 1 час при 0°C. Затем смесь перемешивают в течение 2 час при комнатной температуре. Реакционную смесь нейтрализуют уксусной кислотой (pH 7), затем добавляют воду (50 мл) и смесь экстрагируют этилацетатом. Объединенные органические слои промывают водой, насыщенным раствором соли, сушат (Na2SO4), концентрируют и очищают с помощью пропускания через короткую колонку [окись кремния, гексан/этилацетат (3:1)] с получением беловатого твердого продукта (1,2 г, 88%): т.пл. 88-89°C;1H ЯМР δ 3,41 (т, J=6,6 Гц, 2H), 4,52 (т, J=6, 6 Гц, 2H), 6,26 (с, 1H), 6,74 (с, 1H), 7,07 (д, J=8,1 Гц, 2H), 7,69 (д, J=8,1 Гц, 2H), 8,33 (ушир, 1H);13C ЯМР δ 24,0, 75,0, 91,1, 109,3, 117,8, 122,1, 122,2, 129,8, 135,7, 137,7; FAB-MS наблюдается 341,9877, рассчитано 341,9865; Аналитически рассчитано для C12H11IN2O2: C, 42,1; H, 3,2; N, 8,2. Найдено: C, 42,3; H, 3,3; N, 8,1.
1-[3-(4-Йодфенил)пирро-2-ил]-2-нитро-3,3-диметил-5-гексанон (20). Следуя процедуре для β-незамещенного пиррола (Strachan, J.P. et al., J.Org.Chem. 2000, 65, 3160-3172), смесь соединения 19 (1,03 г, 3,0 ммоль), CsF (2,28 г, 15,0 ммоль) и мезитил оксида (1,72 мл, 15,0 ммоль) в безводном ацетонитриле (22,5 мл) нагревают при 80°C в течение 2,5 - 3 час, за это время смесь превращается из бесцветной в коричневую, а затем в темно-красную. Анализ с помощью ТСХ подтверждает отсутствие исходного материала. Растворитель выпаривают в вакууме, остаток извлекают этилацетатом и фильтруют через прокладку из окиси кремния с использованием этилацетата в качестве элюента. Растворитель выпаривают в вакууме и продукт очищают с помощью гравитационной колонны [окись алюминия, гексан/этилацетат (2:1)] с последующей перекристаллизацией из CH2Cl2/гексан и получением коричневых кристаллов (550 мг, 42%): т.пл. 124-125°C;1H ЯМР δ 1,08 (с, 3H), 1,19 (с, 3H), 2,11 (с, 3H), 2,37 (д, J=17,4 Гц, 1H), 2,56 (д, J=17,4 Гц, 1H), 3,15 (м, 1H), 3,39 (м, 1H), 5,20 (м, 1H), 6,21 (м, 1H), 6,68 (м, 1H), 7,10 (м, 2H), 7,70 (м, 2H), 8, 22 (ушир, 1H);13C ЯМР δ 23,9, 24,2, 24,8, 31,6, 36,8, 51,2, 91,1, 94,2, 109,1, 117,8, 122,2, 122,4, 130,1, 135,9, 137,5, 206,7; FAB-MS наблюдается 440,0605, рассчитано 440,0597; Аналитически рассчитано для C18H21IN2O3: C, 49,1; H, 4,8; N, 6,4. Найдено: C, 49,1; H, 4,7; N, 6,3.
1,3,3-Триметил-7-(4-йодфенил)-2, 3-дигидродипирин (21). Следуя процедуре для β-незамещенного пиррола (Strachan, J.P. et al., J.Org.Chem. 2000, 65, 3160-3172), раствор соединения 20 (220 мг, 0,50 ммоль) в безводном ТГФ (5,0 мл) в атмосфере аргона обрабатывают NaOMe (135 мг, 2,5 ммоль) и смесь перемешивают в течение 1 час при комнатной температуре (первая колба). Во второй колбе объединяют TiCl3 (8,6 мас.% TiCl3 в 28 мас.% HCl, 3,8 мл, 2,5 ммоль, 5,0 молярных эквивалентов) и H2O (20 мл), NH4OAc (˜15 г) добавляют в качестве буфера для доведения pH раствора до 6,0, а затем добавляют ТГФ (5 мл). Нитронатный анион соединения 20, образовавшийся в первой колбе, переносится через канюлю в раствор TiCl3 с буфером во второй колбе. Дополнительный ТГФ (3 мл) добавляют в колбу с нитронатным анионом, и супернатант также переносится в раствор TiCl3 с буфером. Полученную смесь перемешивают при комнатной температуре в течение 6 час в атмосфере аргона. Затем смесь экстрагируют этилацетатом и объединенные органические слои промывают насыщенным водным раствором NaHCO3, водой, насыщенным раствором соли, а затем сушат (MgSO4). Растворитель удаляют при пониженном давлении при комнатной температуре. Сырой продукт пропускают через короткую колонку [окись алюминия, гексан/этилацетат (2:1)] с получением светло-зеленого твердого продукта (80-92 мг, 45 -50%): т.пл. 140-142°C;1H ЯМР δ 1,18 (с, 6H), 2,22 (с, 3H), 2,52 (с, 2H), 5,89 (с, 1H), 6,26 (м, 1H), 6,85 (м, 1H), 7,19 (м, 2H), 7,69 (м, 2H), 11,09 (ушир, 1H);13C ЯМР δ 20,7, 29,1, 29,7, 41,2, 53,7, 90,3, 102,3, 108,6, 118,5, 122,2, 127,5, 130,4, 136,8, 137,4, 161,9, 177,2; FAB-MS наблюдается 390,0595, рассчитано 390,0593 (C18H19IN2); λabs (толуол) 352 нм.
Общая процедура для получения хлорина: Zn(II)-17,18-Дигидро-18, 18-диметил-5-(4-метилфенил)-12-(4-йодфенил)порфирин (Zn-22). Следуя общей процедуре (Strachan, J.P. et al., J.Org.Chem. 2000, 65, 3160-3172), к раствору соединения 11 (110 мг, 0,20 ммоль) в 7,5 мл безводного ТГФ/MeOH (4:1) при комнатной температуре малыми порциями добавляют избыток NaBH4 (100 мг, 2,6 ммоль). Реакцию отслеживают с помощью ТСХ [окись алюминия, гексан/этилацетат (3:1)], и при ее завершении смесь гасят холодной водой (˜10 мл), затем экстрагируют дистиллированным CH2Cl2 (3Ч25 мл). Объединенные органические слои промывают насыщенным раствором соли (50 мл), сушат (K2CO3) в течение 2-3 мин и концентрируют в вакууме при комнатной температуре, чтобы оставить полученный в результате карбинол 11 -OH в ˜1-2 мл CH2Cl2. ЗП 1 (45 мг, 0,24 ммоль) растворяют в нескольких мл безводного CH3CN и объединяют с 11-OH, затем добавляют дополнительно безводный CH3CN с получением в целом 22 мл CH3CN. Раствор перемешивают при комнатной температуре в атмосфере аргона и добавляют ТФУ (20 мкл, 0,26 ммоль). Реакцию отслеживают с помощью ТСХ [окись алюминия, гексан/этилацетат (3:1)], которая через 25-30 мин демонстрирует исчезновение ВП и появление яркого пятна как раз под ЗП. Реакционную смесь гасят 10% водным раствором NaHCO3 и экстрагируют дистиллированным CH2Cl2 (3Ч25 мл). Объединенные органические слои промывают водой, насыщенным раствором соли, сушат (Na2SO4) и растворитель удаляют в вакууме при комнатной температуре. Остаток растворяют в 14 мл безводного толуола в атмосфере аргона, затем добавляют AgIO3 (848 мг, 3,0 ммоль), пиперидин (300 мкл, 3,0 ммоль) и Zn(OAc)2 (550 мг, 3,0 ммоль). Полученную смесь нагревают при 80°C в течение 2,5 час. Реакцию отслеживают с помощью ТСХ [окись кремния, гексан/CH2Cl2, (1:1); показывает одно отдельно взятое зеленое пятно] и спектроскопии поглощения (полосы при ˜410 нм и ˜610 нм). Изменение цвета реакционной смеси с красного на зеленый указывает на образование хлорина. Реакционную смесь охлаждают до комнатной температуры, затем пропускают через короткую колонку (окись кремния, CH2Cl2). Главную фракцию концентрируют и снова подвергают хроматографии [окись кремния, гексан/CH2Cl2 (2:1, затем 1:1)]. Полученный зелено-голубой твердый продукт растворяют в минимальном количестве CH2Cl2 и осаждают путем добавления гексана с получением зелено-голубого твердого продукта (25 мг, 18%).1H ЯМР δ 2,01 (с, 6H), 2,67 (с, 3H), 4,48 (с, 2H), 7,50 (д, J=7,2 Гц, 2H), 7,91 (д, J=7, 2 Гц, 2H), 7,95 (д, J=8,1 Гц, 2H), 8,09 (д, J=8,1 Гц, 2H), 8,51 (д, J=4,2 Гц, 1H), 8,67 (м, 5H), 8,78 (д, J=4,2 Гц, 1H), 9,56 (с, 1H); LD-MS наблюдается 693,78; FAB-MS наблюдается 694,0580, рассчитано 694,0572 (C35H27IN4Zn); λabs (толуол)/нм 411 (log ε=5,33, ширина пика на уровне полумаксимума = 18 нм), 616 (4,76), λem 619, 674 нм.
Замечания относительно получения хлорина: (1) Полное восстановление карбонила в предшественнике ВП до соответствующего карбинола иногда требует дополнительного NaBH4 . Восстановление должно быть завершено перед осуществлением реакции получения хлорина. (2) При извлечении ВП органические слои сушат в K2CO3 (карбинол быстро разлагается при сушке над Na2SO4 или MgSO4). Является важным не доводить раствор ВП до высыхания, поскольку ВП в высушенной форме является абсолютно лабильной. (3) ВП при извлечении и конденсационный раствор, дающий дигидробилен-a, как правило, являются либо желтыми, либо ярко-красными; эти растворы приводят к получению хлоринов с хорошим выходом. В некоторых случаях наблюдается дополнительное потемнение, в этом случае получают низкие выходы хлоринов.
Общие условия для металлирования. 17,18-Дигидро-18,18-диметил-5-(4-метилфенил)-12-(4-йодфенил)порфирин (22). К раствору Zn-22 (10 мг, 14,4 мкмоль) в безводном CH2Cl2 (5 мл) добавляют ТФУ (58 мкмоль, 0,75 ммоль). После перемешивания в течение 30 мин при комнатной температуре (мониторинг с помощью ТСХ и УФ-видимой спектроскопии) реакцию гасят 10% водным раствором NaHCO3 (20 мл) и экстрагируют CH2Cl2. Объединенные органические слои промывают водой, сушат (Na2SO4) и концентрируют. Дополнительная очистка (если необходимо) достигается с помощью хроматографии в короткой колонке [окись кремния, гексан/CH2Cl2 (1:1 затем 1:2)], с получением зеленого твердого продукта (8,0 мг, 88%).1H ЯМР δ -2,15 (ушир, 1H), -1,85 (ушир, 1H), 2,07 (с, 6H), 2,69 (с, 3H), 4,64 (с, 2H), 7,54 (д, J=7,5 Гц, 2H), 8, 04 (м, 4H), 8,16 (д, J=8,1 Гц, 2H), 8,64 (д, J=4,5 Гц, 1H), 8,85 (AB квартет, J=4,5 Гц, 2H), 8,90 (м, 3H), 8,99 (с, 1H), 9,84 (с, 1H); LD-MS наблюдается 633,88; FAB-MS наблюдается 632,1434, рассчитано 632,1437 (C35H29IN4); λabs (толуол)/нм 414 (log ε=5,13, ширина пика на уровне полумаксимума = 34 нм), 505 (4,12), 643 (4,65); λem 646, 682 нм.
Zn(II)-17,18-Дигидро-18,18-диметил-5-(4-метилфенил)-12-{4-[2-(триметилсилил)этинил]фенил}порфирин (Zn-23). Следуя общей процедуре для получения хлорина, реакция 13-OH [приготавливают из 13 (130 мг, 0,25 ммоль)] и соединения 1 (57 мг, 0,30 ммоль) дает голубой твердый продукт (36 мг, 22%).1H ЯМР δ 0,35 (с, 9H), 2,03 (с, 6H), 2,67 (с, 3H), 4, 54 (с, 2H), 7,50 (д, J=8,1 Гц, 2H), 7,86 (д, J=8,1 Гц, 2H), 7,96 (д, J=7,5 Гц, 2H), 8,16 (д, J=8,1 Гц, 2H), 8,53 (д, J=4,5 Гц, 1H), 8,60 (с, 1H), 8,68 (м, 2H), 8,73 (д, J=4,5 Гц, 1H), 8,75 (с, 1H), 8, 80 (д, J=4,5 Гц, 1H), 9,63 (с, 1H); LD-MS наблюдается 665,74; FAB-MS наблюдается 664,2007, рассчитано 664,2001; (C40H36IN4SiZn); λabs (толуол)/нм 413 (log ε=5,31, ширина пика на уровне полумаксимума = 21 нм), 618 (4,77), λem 622, 676 нм (Φf=0,11).
17,18-Дигидро-18, 18-диметил-5-(4-метилфенил)-12-{4-[2-(триметилсилил)этинил]фенил}порфирин (23). Следуя общей процедуре деметаллирования, образец Zn-23 (10 мг, 15 мкмоль) дает зеленый твердый продукт (8,0 мг, 89%).1H ЯМР δ -2,15 (ушир, 1H), -1,85 (ушир, 1H), 0,35 (с, 9H), 2,07 (с, 6H), 2,69 (с, 3H), 4,64 (с, 2H), 7,53 (д, J=7,5 Гц, 2H), 7,91 (д, J=8,1 Гц, 2H), 8,03 (д, J=8,1 Гц, 2H), 8,27 (д, J=8,1 Гц, 2H), 8,64 (д, J=4,5 Гц, 1H), 8,84 (AB квартет, J=4,5 Гц, 2H), 8,89 (м, 2H), 8,93 (с, 1H), 8,99 (с, 1H), 9,86 (с, 1H); LD-MS наблюдается 604,31; FAB-MS наблюдается 602,2880, рассчитано 602, 2866 (C40H38IN4Si); λabs (толуол)/нм 415 (log ε=4,97, ширина пика на уровне полумаксимума = 36 нм), 506 (3,96), 647 (4,49); λem 648, 685, 715 нм (Φf=0,25).
Zn(II)-17,18-Дигидро-18,18-диметил-5-(4-метилфенил)-8-(4-йодфенил)порфирин (Zn-24). Следуя общей процедуре для получения хлорина, реакция 17-OH [приготавливают из соединения 17 (110 мг, 0,20 ммоль)] и соединения 1 (45 мг, 0,24 ммоль) дает голубой твердый продукт (30 мг, 24%).1H ЯМР δ 2,03 (с, 6H), 2,67 (с, 3H), 4,51 (с, 2H), 7,50 (д, J=8,1 Гц, 2H), 7,86 (д, J=8,1 Гц, 2H), 7,97 (д, J=8,1 Гц, 2H), 8,02 (д, J=8,1 Гц, 2H), 8,54 (с, 1H), 8,60 (с, 1H), 8,69 (м, 4H), 8,97 (д, J=4,2 Гц, 1H), 9,61 (с, 1H); LD-MS наблюдается 696,39; FAB-MS наблюдается 694,0607, рассчитано 694,0572 (C35H27IN4Zn); λabs (толуол)/нм 416 (log ε=5,13, ширина пика на уровне полумаксимума = 18 нм), 607 (4,49); λem609,661 нм.
17,18-Дигидро-18,18-диметил-5-(4-метилфенил)-8-(4-йодфенил)порфирин (24). Следуя общей процедуре деметаллирования, образец Zn-24 (10 мг, 14,4 мкмоль) дает зеленый твердый продукт (7,5 мг, 83%).1H ЯМР δ -2,20 (ушир, 1H), -1,96 (ушир, 1H), 2,07 (с, 6H), 2,68 (с, 3H), 4,63 (с, 2H), 7,53 (д, J=8,1 Гц, 2H), 7,86 (д, J=8,7 Гц, 2H), 8,03 (м, 4H), 8,64 (с, 1H), 8,85 (м, 3H), 8,91 (с, 1H), 8,99 (с, 1H), 9,17 (д, J=4,5 Гц, 1H), 9,83 (с, 1H); LD-MS наблюдается 631,58; FAB-MS наблюдается 632,1454, рассчитано 632,1437 (C35H29IN4); λabs (толуол)/нм 410 (log ε=5,11, ширина пика на уровне полумаксимума = 32 нм), 504 (4,01), 638 (4,48); λem639, 679, 702 нм.
Zn(II)-17,18-Дигидро-18,18-диметил-2-(4-йодфенил)-5-(4-метилфенил)-12-{4-[2-(триметилсилил)этинил]фенил}порфирин (Zn -25). Следуя общей процедуре для получения хлорина, реакция 13-OH [приготавливают из соединения 13 (103 мг, 0,20 ммоль)] и соединения 21 (86 мг, 0,22 ммоль) дает голубой твердый продукт (42 мг, 24%).1H ЯМР δ 0,36 (с, 9H), 1,96 (с, 6H), 2,67 (с, 3H), 4,48 (с, 1H), 7,50 (д, J=7,5 Гц, 2H), 7,82 (д, J=8,7 Гц, 2H), 7,86 (д, J=8,1 Гц, 2H), 7,97 (д, J=8,1 Гц, 2H), 8,02 (д, J=8,1 Гц, 2H), 8,13 (д, J=7,8 Гц, 2H), 8,51 (д, J=4,2 Гц, 1H), 8,63 (с, 1H), 8,67 (с, 1H), 8,70 (с, 2H), 8,78 (д, J=4,2 Гц, 1H), 9,58 (с, 1H); LD-MS 866,34; FAB-MS наблюдается 866,1257, рассчитано 866,1280 (C46H39IN4SiZn); λabs (толуол)/нм 417 (log ε=5,32, ширина пика на уровне полумаксимума = 21 нм), 629 (4,90); λem 635, 691 нм.
17, 18-Дигидро-18,18-диметил-2-(4-йодфенил)-5-(4-метилфенил)-12-{4-[2-(триметилсилил)этинил]фенил}порфирин (25). Следуя общей процедуре деметаллирования, образец Zn-25 (11,0 мг, 13,7 мкмоль) дают зеленый твердый продукт (8,0 мг, 78%).1H ЯМР δ -1,95 (ушир, 1H), -1,70 (ушир, 1H), 0,36 (с, 9H), 2,0 (с, 6H), 2,68 (с, 3H), 4,60 (с, 2H), 7,53 (д, J=8,1 Гц, 2H), 7,91 (д, J=8,1 Гц, 2H), 8, 03 (д, J=8,1 Гц, 2H), 8,07 (д, J=8,1 Гц, 2H), 8,26 (д, J=8,1 Гц, 2H), 8,62 (д, J=4,2 Гц, 1H), 8,81 (с, 1H), 8,88 (д, J=4,2 Гц, 1H), 8,91 (с, 1H), 8,95 (с, 1H), 8,96 (с, 1H), 9,84 (с, 1H); LD-MS 804,02; FAB-MS наблюдается 804,2157, рассчитано 804,2145 (C46H41IN4Si); λabs (толуол)/нм 422 (log ε=5,09, ширина пика на уровне полумаксимума = 34 нм), 509 (4,08), 655 (4,68); λem 660, 726 нм.
Альтернативный подход к хлоринам, которые несут на себе парный диметиловый замок, исключают боковое сближение мезо- и β -заместителей и могут быть использованы в модельных системах, включающих в себя взаимодействие трипиррольного комплекса с пирролом, функционализированным для последующей обработки (Montfortc, F.-P.; Kutzki, O. Angew. Chem. Int. Ed. 2000, 39, 599-601; Abel, и Montfortc, F.-P. Tetrahedron Lett. 1997, 38, 1745-1748: Schmidt, W.; Montfortc, F.-P. Synlett 1997, 903-904).
Все указанное выше представляет собой иллюстрацию настоящего изобретения, и не должно рассматриваться как его ограничение. Объем настоящего изобретения определен с помощью следующей далее формулы изобретения, при этом эквиваленты формулы изобретения должны включаться в нее.
Реферат
Изобретение относится к солнечным источникам света. Технический результат изобретения: получение тонких, легких, портативных, гибких, с хорошей эффективностью - более 5% - солнечных элементов. Сущность: солнечный элемент включает в себя цепочку для аккумуляции света, которая содержит: (а) первую подложку, содержащую первый электрод; и (b) слой стержней для аккумуляции света, электрически соединенных с первым электродом. Каждый из стержней для аккумуляции света содержит полимер формулы I:


Формула



Комментарии