Ионная жидкость, содержащая ион фосфония, и способ ее получения - RU2374257C2
Код документа: RU2374257C2
Чертежи
Описание
Область, к которой относится изобретение
Настоящее изобретение относится к ионной жидкости, которая находится в жидком состоянии в широком диапазоне температур от низких температур, имеющей низкую вязкость и превосходную электрохимическую стабильность, к способу ее получения и к ее использованию, включая электрические запоминающие устройства, литиевые аккумуляторные батареи, конденсаторы с двойным электрическим слоем, сенсибилизированные красителем солнечные элементы, топливные элементы и реакционные растворители.
Уровень техники
До настоящего времени сообщалось о многих ионных жидкостях, в которых имеется азотсодержащий оний-катион, такой как, типично, катион аммония. Они находятся в жидком состоянии при температуре выше 25°C, но при 25°C или ниже лишь несколько ионных жидкостей можно хранить в жидком состоянии. Кроме того, до настоящего времени сообщалось только об ионной жидкости, которая обладает высокой вязкостью при температуре, близкой к комнатной, и ее трудно использовать саму по себе в качестве электролита или растворителя (смотри патентные документы 1 и 2 и непатентные документы 1-3).
Более того, из числа ионных жидкостей, которые содержат катион, имеющий относительно низкую вязкость и температуру плавления, такой как катион имидазолиния, многие ионные жидкости трудно использовать в качестве электролита для электрических запоминающих устройств из-за недостаточной стабильности, вызванной их низкой стойкостью к восстановлению и узким окном потенциала (см. патентный документ 3 и непатентные документы 4 и 5).
Большим камнем преткновения для использования ионных жидкостей в аккумуляторных батареях, конденсаторах с двойным электрическим слоем, топливных элементах, сенсибилизированных красителем солнечных элементах, или в электролитах, растворах электролитов или добавках для электрических запоминающих устройств является то, что существует очень незначительное число ионных жидкостей, поддерживающих стабильное жидкое состояние в широком диапазоне температур от низких температур, имеющих низкую вязкость и высокую электропроводность, обладающих превосходной электрохимической стабильностью, и которые сами по себе годны к использованию.
Патентный документ 1: международная публикация № WO 02/076924 брошюрная форма
Патентный документ 2: выложенная заявка на патент Японии № 2003-331918
Патентный документ 3: выложенная заявка на патент Японии № 2001-517205
Непатентный документ 1: Hajime Matsumoto and Yoshinori Miyazaki, YOYUEN OYOBI KOONKAGAKU, Vol.44, p.7 (2001)
Непатентный документ 2: H. Matsumoto, M. Yanagida, K. Tanimoto, M. Nomura, Y. Kitagawa and Y.Miyazaki, Chem. Lett, Vol.8, p.922 (2000)
Непатентный документ 3: D.R. MacFarlane, J. Sun, J. Golding, P. Meakin and M. Forsyth, Electrochemica Acta, Vol.45, p.1271 (2000)
Непатентный документ 4: Rika Hagiwara, Electrochemistry, Vol.70, No.2, p.130 (2002)
Непатентный документ 5: Y. Katayama, S. Dan, T. Miura and T. Kishi, Journal of The Electrochemical Society, Vol.148 (2), C102-C105 (2001).
Описание изобретения
Проблемы, которые необходимо решить изобретением
Цель настоящего изобретения состоит в предложении ионной жидкости, обладающей низкой вязкостью, адекватной электропроводностью и превосходной электрохимической стабильностью, и способа получения ионной жидкости. Более того, цель настоящего изобретения заключается в предложении ионной жидкости, которую можно использовать для вышеописанных растворов электролитов, в литиевых аккумуляторных батареях, конденсаторах с двойным электрическим слоем, сенсибилизированных красителем солнечных элементах, топливных элементах, реакционных растворителях и т.п., в частности, в предложении ионной жидкости, которая стабильна в жидком состоянии при температуре, близкой к комнатной, в особенности, в предложении ионной жидкости, содержащей новый катион фосфония.
Средства решения проблем
Авторы настоящего изобретения синтезировали ряд солей, состоящих из катионных компонентов и анионных компонентов, и провели интенсивные исследования ионных жидкостей для достижения вышеуказанных целей. В результате, авторы настоящего изобретения обнаружили, что ионная жидкость, содержащая в качестве катионного компонента одну разновидность или несколько разновидностей компонентов, выбранных из группы, состоящей из органических катионов, представленных следующей ниже общей формулой (1), имеет низкую вязкость, адекватную электропроводность и превосходную электрохимическую стабильность.
[Химический продукт 1]
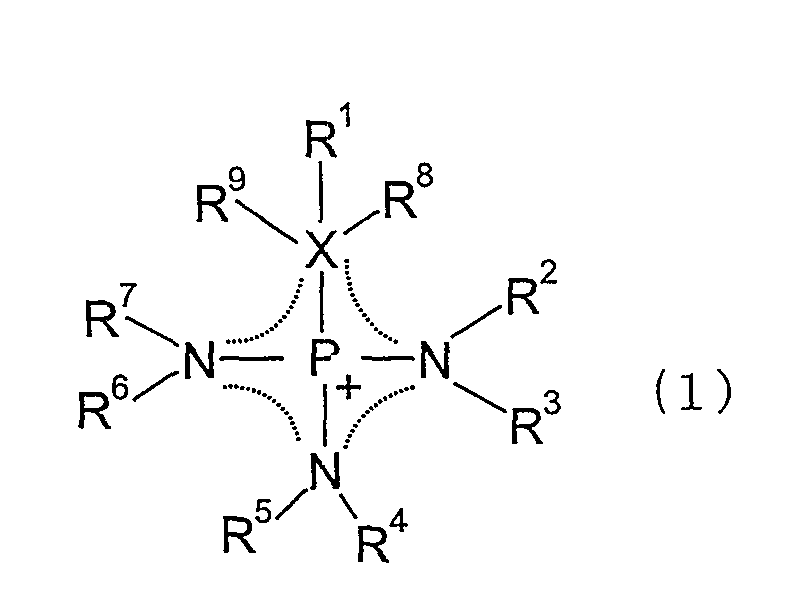
где замещающие группы R1-R9 могут быть независимо одинаковыми или отличающимися друг от друга; каждая из замещающих групп R1-R9 представляет собой атом водорода, алкильную группу с прямой или разветвленной цепью, содержащую от 1 до 30 атомов углерода, алкенильную группу с прямой или разветвленной цепью, содержащую от 2 до 30 атомов углерода, с одной или несколькими двойными связями, алкинильную группу с прямой или разветвленной цепью, содержащую от 2 до 30 атомов углерода, с одной или несколькими тройными связями, насыщенную или частично или полностью ненасыщенную циклоалкильную группу, арильную группу или гетероциклическую группу; любые атомы водорода, содержащиеся в одной замещающей группе или во множестве замещающих групп
R1-R9, могут быть частично или полностью замещены атомом галогена, или частично замещены CN группой или NO2 группой; любая одна из замещающих групп R1-R9 может образовывать циклическую структуру совместно друг с другом; любой атом углерода, содержащийся в замещающих группах R1-R9, может быть замещен атомом и/или атомной группой, выбранной из группы, состоящей из -O-, -C(O)-, -C(O)O-,
-S-, -S(O)-, -SO2-, -SO3-, -N=, -N=N-, -NH-, -NR'-, -N(R')2-, -PR'-, -P(O)R'-, -P(O)R'-O-, -O-P(O)R'-O- и -P(R')2=N-, где R' представляет собой алкильную группу с прямой или разветвленной цепью, содержащую от 1 до 10 атомов углерода, алкильную группу, частично или полностью замещенную атомом фтора, насыщенную или частично или полностью ненасыщенную циклоалкильную группу, незамещенную или замещенную фенильную группу, или незамещенную или замещенную гетероциклическую группу; X представляет собой атом серы, атом кислорода или атом углерода; R8 и R9 существуют, только когда X представляет собой атом углерода; когда X представляет собой атом углерода, X, R1, R8 и R9 могут образовывать насыщенную или частично или полностью ненасыщенную циклическую структуру совместно друг с другом; и пунктирная линия представляет сопряженную структуру.
А именно, вышеуказанные цели настоящего изобретения были достигнуты предложением ″ионной жидкости, включающей органическое вещество, представленное общей формулой (1), в качестве катионного компонента″ и ″ионной жидкости, включающей катионный компонент и анионный компонент, и катионный компонент представляет собой одну разновидность или несколько разновидностей, выбранных из группы, состоящей из катионных компонентов, представленных общей формулой (1)″.
Краткое описание чертежей
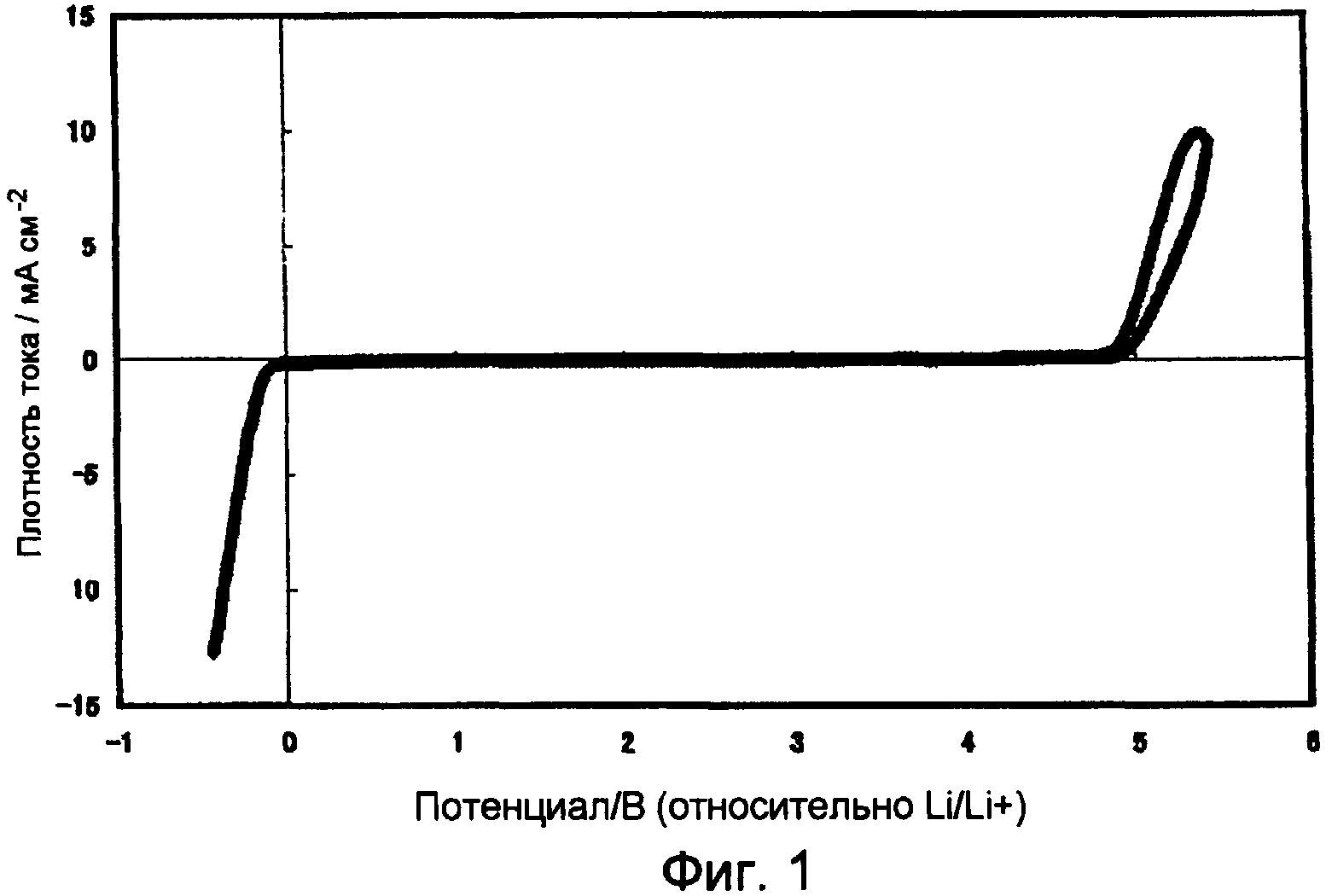
Фиг.1 представляет собой график, показывающий вольтамперную характеристику бис-трифторметансульфонилимида три(диметиламино)бутоксифосфония в примере 3.
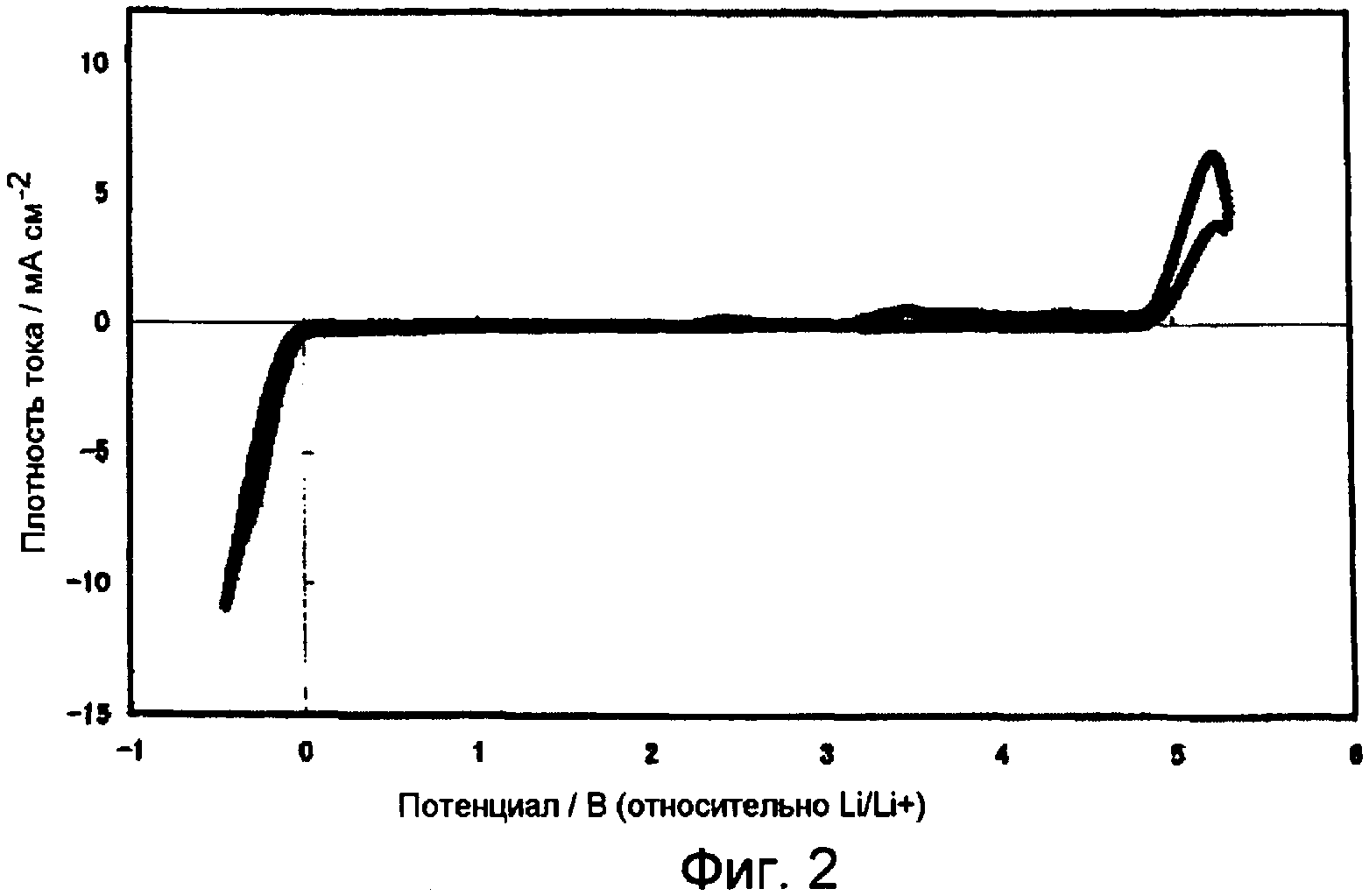
Фиг.2 представляет собой график, показывающий вольтамперную характеристику бис-трифторметансульфонилимида три(диметиламино)бутилфосфония в примере 4.
Наилучший вариант осуществления изобретения
В качестве катионного компонента, представленного общей формулой (1), каждая из замещающих групп R1-R9 в общей формуле (1) представляет собой C1-30 алкильную группу с прямой или разветвленной цепью, насыщенную или частично или полностью ненасыщенную циклоалкильную группу, арильную группу или гетероциклическую группу. Любые атомы водорода (H), содержащиеся в одной разновидности или во множестве разновидностей данных замещающих групп R1-R9, частично или полностью замещены атомом галогена, или частично замещены CN группой или NO2. Кроме того, любой атом углерода, содержащийся в замещающих группах R1-R9, предпочтительно, замещен атомом и/или атомной группой, выбранной из группы, состоящей из -O-, -C(O)-, -C(O)O-, -S-, -S(O)-, -NR'- и -N(R')2-, где R' представляет собой C1-10 алкильную группу с прямой или разветвленной цепью, алкильную группу, частично или полностью замещенную атомом фтора, насыщенную или частично или полностью ненасыщенную циклоалкильную группу, незамещенную или замещенную фенильную группу, или незамещенную или замещенную гетероциклическую группу. Более предпочтительно, каждая группа из R1-R9 в общей формуле (1) представляет собой C1-20 алкильную группу или алкоксигруппу с прямой или разветвленной цепью (R1R9могут быть одинаковыми или отличающимися друг от друга).
Более того, Х в общей формуле (1) представляет собой атом серы, атом кислорода или атом углерода.
Анионный компонент, используемый в настоящем изобретении, представляет собой одну или несколько разновидностей, выбранных из группы, состоящей из [RSO3]-, [RfSO3]-, [(RfSO2)2N]-, [(RfSO2)3C]-, [(FSO2)3C]-, [RCH2OSO3]-, [RC(O)O]-, [RfC(O)O]-,
[CCl3C(O)O]-, [(CN)3C]-, [(CN)2CR]-, [(RO(O)C)2CR]-, [R2P(O)O]-, [RP(O)O2]2-, [(RO)2P(O)O]-, [(RO)P(O)O2]2-, [(RO)(R)P(O)O]-, [Rf2P(O)O]-, [RfP(O)O2]2-, [B(OR)4]-, [N(CF3)2]-, [N(CN)2]-, [AlCl4]-, PF6-, BF4-, SO42-, HSO4-, NO3-, F-, Cl-, Br- и I-, где каждая замещающая группа R представляет собой атом водорода, атом галогена, C1-10 алкильную группу с прямой или разветвленной цепью, C2-10 алкенильную группу с прямой или разветвленной цепью, имеющую одну или несколько двойных связей, С2-10 алкинильную группу с прямой или разветвленной цепью, имеющую одну или несколько тройных связей, или насыщенную или частично или полностью ненасыщенную циклоалкильную группу; любые атомы водорода, содержащиеся в данных замещающих группах R, могут быть частично или полностью замещены атомом галогена, или частично замещены CN группой или NO2 группой; любой атом углерода, который содержится в данных R, может быть замещен атомом и/или атомной группой, выбранными из группы, состоящей из -O-, -C(O)-, -C(O)O-, -S-, -S(O)-, -SO2-, -SO3-, -N=, -N=N-, -NR'-, -N(R')2-, -PR'-, -P(O)R'-, -P(O)R'-O-, -O-P(O)R'-O- и -P(R')2=N-, где R' представляет собой C1-10 алкильную группу с прямой или разветвленной цепью, алкильную группу, частично или полностью замещенную атомом фтора, насыщенную или частично или полностью ненасыщенную циклоалкильную группу, незамещенную или замещенную фенильную группу, или незамещенную или замещенную гетероциклическую группу; и Rf представляет собой фторсодержащую замещающую группу. Данные анионные компоненты объединяются с вышеуказанными катионными компонентами и предоставляют ионную жидкость, имеющую низкую вязкость, адекватную электропроводность и превосходную электрохимическую стабильность.
Анионный компонент, используемый в качестве противоиона катионного компонента, представленного общей формулой (1), предпочтительно, представляет собой одну или несколько разновидностей, выбранных из группы, состоящей из [RSO3]-, [RfSO3]-, [(RfSO2)2N]-, CF3SO3-, CF3COO-, PF6-, BF4-, [N(CN)2]-, [AlCl4]-, SO42-, HSO4-, NO3-, F-, Cl-, Br- и I-, и, более предпочтительно, одну или несколько разновидностей, выбранных из группы, состоящей из [RSO3]-, [RfSO3]-, [(RfSO2)2N]-, CF3SO3-, CF3COO-, [N(CN)2]-, [AlCl4]-, SO42-, HSO4- и NO3-.
Комбинация вышеуказанных катионных компонентов и данных предпочтительных анионных компонентов способна предоставить ионную жидкость, обладающую еще более предпочтительными свойствами, а именно стабильным жидким состоянием в широком диапазоне температур от низких температур, низкой вязкостью, адекватной электропроводностью и превосходной электрохимической стабильностью.
В особенно предпочтительной ионной жидкости анионный компонент, который является противоионом катионного компонента, представленного общей формулой (1), представляет собой одну или несколько разновидностей, выбранных из группы, состоящей из [RSO3]-, [RfSO3]-, [(RfSO2)2N]-, CF3SO3-, CF3COO-, PF6-, BF4-, [N(CN)2]-, [AlCl4]-, SO42-, HSO4-, NO3-, F-, Cl-, Br- и I-; и каждая из групп R1-R9 в общей формуле (1) представляет собой C1-10 алкильную группу или алкоксигруппу с прямой или разветвленной цепью (R1-R9 могут быть одинаковыми или отличными друг от друга).
В более предпочтительном случае, X в катионном компоненте, представленном общей формулой (1), является атомом серы или атомом кислорода. Ионная жидкость, замещенная данными атомами, имеет низкую температуру плавления. Еще более предпочтительной является ионная жидкость, имеющая атом кислорода в качестве X.
Когда готовят ионную жидкость, в то же время, делая упор на важность низкой вязкости, требуется выбрать конкретный катионный компонент таким образом, что R2-R7 в общей формуле (1) представляют собой C1-4 алкильные группы с прямой цепью; R8 и R9 представляют собой атомы водорода; R1 представляет собой C1-10 алкильную группу или алкоксигруппу с прямой или разветвленной цепью; X, предпочтительно, представляет собой атом серы или атом кислорода; и, особенно предпочтительно, X представляет собой атом кислорода, и в качестве анионного компонента, который является противоионом катионного компонента, требуется выбрать особое анионное соединение, предпочтительно, из (CF3SO2)2N-, PF6- или BF4-, и, особенно предпочтительно, (CF3SO2)2N-. При данных комбинациях можно получить ионную жидкость, которая показывает стабильное жидкое состояние в широком диапазоне температур от низких температур, имеющую низкую вязкость, адекватную электропроводность и превосходную электрохимическую стабильность.
Ионная жидкость по настоящему изобретению показывает превосходную электропроводность, а также имеет низкую вязкость и превосходную электрохимическую стабильность. Вследствие данных превосходных эксплуатационных характеристик ионную жидкость по настоящему изобретению используют в качестве материала для электролитов, растворов электролитов, добавок и т.п. для электрических запоминающих устройств; литиевых аккумуляторных батарей; конденсаторов с двойным электрическим слоем; топливных элементов и сенсибилизированных красителем солнечных элементов, а также используют в качестве реакционного растворителя для различных реакций. Отметим, что до настоящего времени такая ионная жидкость, которая имеет как низкую вязкость, так и электрохимическую стабильность, не была достижима. Ионная жидкость по настоящему изобретению точно удовлетворяет обоим данным свойствам.
В настоящем описании катионный компонент, представленный общей формулой (1), показывает катион фосфония, имеющий положительный заряд на атоме фосфора для удобства написания, но положительный заряд может быть делокализован по молекуле в зависимости от вида гетероатома, представленного X.
Типичный способ синтеза ионной жидкости, которая содержит катионный компонент, представленный общей формулой (1), будет указан ниже.
[Химический продукт 2]

К источнику органического вещества, представленному общей формулой (2), по каплям добавляют алкилирующий агент (R1W) и реакцию осуществляют при предварительно определенной температуре в течение предварительно определенного времени. После того как реакционную смесь промывают диэтиловым эфиром или т.п., ее сушат под вакуумом. Алкилирующий агент (R1W) может включать диалкилсульфат, диалкилсульфонат, диалкилкарбонат, триалкилфосфат, алкилмонофторалкилсульфонат, алкилполифторалкилсульфонат, фторалкилсульфонат, алкилперфторалкилсульфонат, алкилмонофторкарбоксилат, алкилполифторкарбоксилат, алкилперфторкарбоксилат, алкилйодид, алкилбромид, алкилхлорид, серную кислоту, азотную кислоту и хлористоводородную кислоту.
Кроме того, например, ионная жидкость, имеющая различный вид аниона, может быть получена анионным обменом, как показано ниже.
[Химический продукт 3]

Здесь, ионное связывающее соединение AQ может включать, например,
LiN(CF3SO2)2, NaN(CF3SO2)2, KN(CF3SO2)2, CF3SO3Li, CF3SO3Na, CF3SO3K, CF3CH2SO3Li, CF3CH2SO3Na, CF3CH2SO3K, CF3COOLi, CF3COONa, CF3COOK, LiPF6, NaPF6, KPF6,
LiBF4, NaBF4, KBF4, LiSbF6, NaSbF6, KSbF6, NaN(CN)2, AgN(CN)2, Na2SO4, K2SO4, NaNO3 и KNO3, но оно не ограничивается данными соединениями.
В общей формуле (3) замещающие группы R1-R9 могут быть независимо одинаковыми или отличающимися друг от друга. Каждая из замещающих групп R1-R9 представляет собой атом водорода, атом галогена, C1-30 алкильную группу с прямой или разветвленной цепью, C2-30 алкенильную группу с прямой или разветвленной цепью, содержащую одну или несколько двойных связей, C2-30 алкинильную группу с прямой или разветвленной цепью, содержащую одну или несколько тройных связей, насыщенную или частично или полностью ненасыщенную циклоалкильную группу, арильную группу или гетероциклическую группу. Любые атомы водорода, содержащиеся в одной или во множестве данных замещающих групп R1-R9, могут быть частично или полностью замещены атомом галогена, или могут быть частично замещены CN группой или NO2 группой. Любые замещающие группы R1-R9 могут образовывать циклическую структуру совместно друг с другом. Любой атом углерода, содержащийся в замещающих группах R1-R9, может быть замещен атомом и/или атомной группой, выбранной из группы, состоящей из -O-, -C(O)-, -C(O)O-, -S-, -S(O)-, -SO2-, -SO3-, -N=, -N=N-, -NH-, -NR'-, -N(R')2-, -PR'-, -P(O)R'-, -P(O)R'-O-, -O-P(O)R'-O- и -P(R')2=N-, где R' представляет собой C1-10 алкильную группу с прямой или разветвленной цепью, алкильную группу, частично или полностью замещенную атомом фтора, насыщенную или частично или полностью ненасыщенную циклоалкильную группу, незамещенную или замещенную фенильную группу, или незамещенную или замещенную гетероциклическую группу. X представляет собой атом серы, атом кислорода или атом углерода. R8 и R9 существуют, только когда X представляет собой атом углерода. Когда X представляет собой атом углерода, X, R1, R8 и R9 могут образовывать насыщенную или частично или полностью ненасыщенную циклическую структуру совместно друг с другом.
Описанный выше атом галогена может включать в себя F, Cl, Br и I.
Описанная выше циклоалкильная группа может включать в себя циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил и циклодецил. Циклоалкильная группа может включать группу, которая имеет ненасыщенную связь, например, циклоалкенильную группу и циклоалкинильную группу. Циклоалкильная группа может быть частично или полностью замещена атомом галогена, или частично замещена CN группой или NO2 группой.
Описанная выше гетероциклическая группа может включать пиродинил, пиролинил, имидазолидинил, имидазолинил, пиразолидинил, пиразонил, пиперидил, пиперадинил, морфолинил или тиенил. Кроме того, данные гетероциклические группы могут содержать один или несколько заместителей, выбранных из алкильной группы, алкоксигруппы, гидроксигруппы, карбоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, тиольной группы и алкилтиольной группы, и атома галогена.
Описанная выше арильная группа может включать фенил, куменил, мезитил, толил, ксилил или т.п. Данные арильные группы могут содержать один или несколько заместителей, выбранных из алкильной группы, алкоксигруппы, гидроксигруппы, карбоксигруппы, ацильной группы, формильной группы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, тиольной группы, алкилтиольной группы и атома галогена.
Кроме того, замещающие группы R1-R9 могут включать алкоксиалкильную группу, такую как метоксиметил, метоксиэтил, этоксиметил и этоксиэтил и т.п.
Более того, в качестве гетероатома, представленного X в формуле, можно указать атом серы, атом кислорода или атом углерода. Особенно предпочтительно, здесь можно указать атом серы или атом кислорода. Посредством замещения данного атома можно получить ионную жидкость, имеющую еще более низкую температуру плавления. В качестве анионного компонента Q, который может взаимодействовать с соединением, представленным общей формулой (3), и используется в комбинации, можно перечислить вышеуказанные анионные соединения.
ПРИМЕРЫ
Настоящее изобретение далее будет подробно описано со ссылкой к следующим ниже примерам, но следует понимать, что настоящее изобретение никоим образом не ограничивается данными примерами.
Пример 1
(a) Получение метилсульфата три(диметиламино)метоксифосфония
В двухгорлую колбу в форме баклажана, оборудованную оросительным конденсатором, капельной воронкой и магнитной мешалкой, к 2,0 г (11,2 ммоль) гексаметилфосфат триамида при комнатной температуре в атмосфере азота по каплям добавляли 1,4 г (11,2 ммоль) диметилсульфата. После 15-часового перемешивания при комнатной температуре получали белую твердую соль. Соль промывали достаточным количеством эфира и сушили под вакуумом при 50°C в течение 5 ч, получая метилсульфат три(диметиламино)метоксифосфония с выходом 74%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: ацетон-d6, эталон: тетраметилсилан): δ 4,06 (д, 3H); 3,47 (с, 3Н); 2,90 (д, 18H).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 4]

(b) Получение бис-трифторметансульфонилимида три(диметиламино)метоксифосфония
3,05 г (10,0 ммоль) метилсульфата три(диметиламино)метоксифосфония, полученного в (a), растворяли в 100 мл чистой воды. После того, как примеси экстрагировали CH2Cl2, к полученному в результате водному раствору при перемешивании добавляли водный раствор 2,87 г (10,0 ммоль) бис-трифторметансульфонилимида лития в 100 мл чистой воды. После 60-минутного непрерывного перемешивания полученное в результате гидрофобное белое твердое вещество промывали водой два или три раза, экстрагировали дихлорметаном и очищали на колонке с оксидом алюминия. Экстракт концентрировали и затем сушили под вакуумом при 80°C в течение 10 ч, получая 4,50 г (выход: 95%) продукта, который представлял собой белое твердое вещество при комнатной температуре и бесцветную прозрачную жидкость при 130°C.
Соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Соединение идентифицировали как желаемое соединение, представляющее собой бис-трифторметансульфонилимид три(диметиламино)метоксифосфония. Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: ацетон-d6, эталон: тетраметилсилан): δ 4,06 (д, 3H); 2,90 (д, 18H).
19F-ЯМР (282 МГц, растворитель: ацетон-d6, эталон: CF3Cl): δ
-79,93 (с, 6F).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 5]

Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла 127°C.
Пример 2
(с) Получение этилсульфата три(диметиламино)этоксифосфония
В двухгорлую колбу в форме баклажана, оборудованную оросительным конденсатором, капельной воронкой и магнитной мешалкой, к 2,0 г (11,2 ммоль) гексаметилфосфаттриамида при комнатной температуре в атмосфере азота по каплям добавляли 2,1 г (13,4 ммоль) диэтилсульфата. После 5-дневного перемешивания при 20°C получали белую твердую соль. Соль промывали достаточным количеством эфира и сушили под вакуумом при 50°C в течение 5 ч, получая этилсульфат три(диметиламино)этоксифосфония с выходом 87%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: ацетон-d6, эталон: тетраметилсилан): δ 4,47 4,38 (м, 2H); 3,86 (кв, 2H); 2,90 (д, 18H); 1,45 (т, 3H); 1,13 (т, 3H).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 6]

(d) Получение бис-трифторметансульфонилимида три(диметиламино)этоксифосфония
3,23 г (9,7 ммоль) этилсульфата три(диметиламино)этоксифосфония, полученного в (c), растворяли в 100 мл чистой воды. После того, как примеси экстрагировали
CH2Cl2, к полученному в результате водному раствору при перемешивании добавляли водный раствор 2,8 г (9,7 ммоль) бис-трифторметансульфонилимида лития в 100 мл чистой воды. После 60-минутного непрерывного перемешивания полученное в результате гидрофобное белое твердое вещество промывали водой два или три раза, экстрагировали дихлорметаном и очищали на колонке с оксидом алюминия. Экстракт концентрировали и затем сушили под вакуумом при 80°C в течение 10 ч, получая 4,35 г (выход: 92%) продукта, который представлял собой белое твердое вещество при комнатной температуре и бесцветную прозрачную жидкость при 90°C.
Соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Соединение идентифицировали как желаемое соединение, представляющее собой бис-трифторметансульфонилимид три(диметиламино)этоксифосфония. Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: ацетон-d6, эталон: тетраметилсилан): δ 4,46 4,37 (м, 2Н), 2,90 (д, 18Н), 1,45 (т, 3Н).
19F-ЯМР (282 МГц, растворитель: ацетон-d6, эталон: CF3Cl): δ -79,91 (с, 6F).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 7]
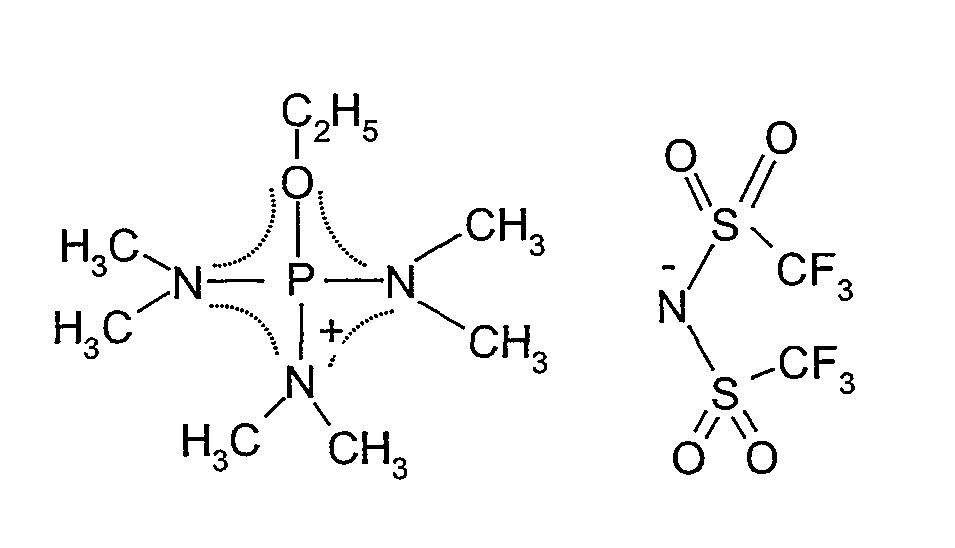
Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла 88°C.
Пример 3
(e) Получение бутилсульфата три(диметиламино)бутоксифосфония
В двухгорлую колбу в форме баклажана, оборудованную оросительным конденсатором, капельной воронкой и магнитной мешалкой, к 50,0 г (279 ммоль) гексаметилфосфаттриамида при комнатной температуре в атмосфере азота по каплям добавляли 70,4 г (335 ммоль) дибутилсульфата. После 7-дневного перемешивания при 30°C получали белую твердую соль. Соль промывали достаточным количеством эфира и сушили под вакуумом при 50°C в течение 5 ч, получая бутилсульфат три(диметиламино)бутоксифосфония с выходом 93%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: ацетон-d6, эталон: тетраметилсилан): δ 4,38 (кв, 2H); 3,82 (т, 2H); 2,90 (д, 18H); 1,80-1,73 (м, 2H); 1,55-1,30 (м, 6H); 0,96 (т, 3H); 0,90 (т, 3H).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 8]

(d) Получение бис-трифторметансульфонилимида три(диметиламино)бутоксифосфония
58,4 г (150 ммоль) бутилсульфата три(диметиламино)бутоксифосфония, полученного в (e), растворяли в 200 мл чистой воды. К полученному в результате водному раствору при перемешивании добавляли водный раствор 43,1 г (150 ммоль) бис-трифторметансульфонилимида лития в 150 мл чистой воды. После 2-часового непрерывного перемешивания полученную в результате гидрофобную прозрачную жидкость промывали водой пять раз и экстрагировали дихлорметаном. Экстракт концентрировали и затем сушили под вакуумом при 80°C в течение 20 ч, получая 76,9 г (выход: 99%) продукта, который представлял собой бесцветную прозрачную жидкость при комнатной температуре.
Соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Соединение идентифицировали как желаемое соединение, представляющее собой бис-трифторметансульфонилимид три(диметиламино)бутоксифосфония. Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: ацетон-d6, эталон: тетраметилсилан): δ 4,36 (кв, 2H); 2,90 (д, 18H); 1,84-1,75 (м, 2H); 1,55-1,42 (м, 2H); 0,96 (т, 3H).
19F-ЯМР (282 МГц, растворитель: ацетон-d6, эталон: CF3Cl): δ -79,92 (с, 6F).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 9]
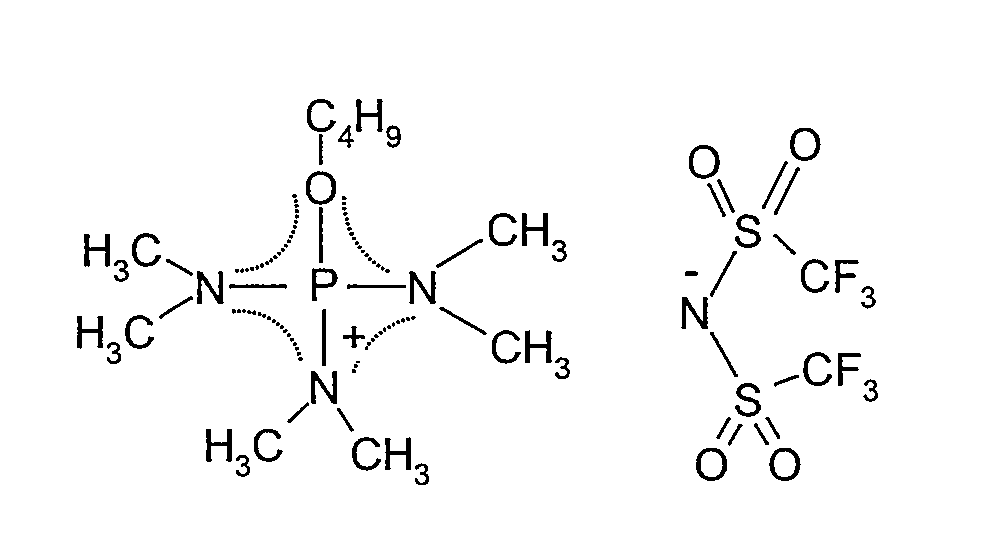
Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла -7,5°C, а температура кристаллизации составляла -67°C. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Начальная температура потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 200°C. Данные результаты показывают, что соль из настоящего примера поддерживает стабильное жидкое состояние в широком диапазоне температур от -7,5°C до 200°C.
Вязкость, измеренная вискозиметром вибрационного типа (поставляемый A&D Co., Ltd), составляла 45 мПа·с при 25°C.
Электропроводность, измеренная методом импеданса по переменному току (Electrochemical Measurement System HZ-3000, поставленная Hokuto Denko Corp.), составила 0,3 См·м-1 при 25°C.
Кроме того, циклическая вольтаммограмма, измеренная Electrochemical Measurement System HZ-3000, поставленной Hokuto Denko Corp., с использованием Pt рабочего электрода, Pt противоэлектрода и Li электрода сравнения, показало, что окно потенциала составляло от -0,1 В до 4,9 В по отношению к потенциалу Li/Li+. Вольтамперная характеристика бис-трифторметансульфонилимида три(диметиламино)бутоксифосфония показана на Фиг.1.
Пример 4
(g) Получение бутилсульфата три(диметиламино)бутилфосфония
В двухгорлую колбу в форме баклажана, оборудованную оросительным конденсатором, капельной воронкой и магнитной мешалкой, к 24,2 г (149 ммоль) триамида гексаметилфосфора при комнатной температуре в атмосфере азота по каплям добавляли 37,4 г (178 ммоль) диэтилсульфата. После 3-дневного перемешивания при комнатной температуре получали белую твердую соль. Соль промывали достаточным количеством эфира и сушили под вакуумом при 50°C в течение 5 ч, получая бутилсульфат три(диметиламино)бутилфосфония с выходом 94%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: ацетон-d6, эталон: тетраметилсилан): δ 3,83 (т, 2H); 2,85 (д, 18H); 2,73-2,63 (м, 2H); 1,70-1,33 (м, 8H); 0,97 (т, 3H); 0,90 (т, 3H).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 10]

(h) Получение бис-трифторметансульфонилимида три(диметиламино)бутилфосфония
37,4 г (100 ммоль) бутилсульфата три(диметиламино)бутилфосфония, полученного в (g), растворяли в 200 мл чистой воды. К полученному в результате водному раствору при перемешивании добавляли водный раствор 28,7 г (100 ммоль) бис-трифторметансульфонилимида лития в 150 мл чистой воды. После 2-часового непрерывного перемешивания полученную в результате гидрофобную прозрачную жидкость промывали чистой водой пять раз и экстрагировали дихлорметаном. Экстракт концентрировали и затем сушили под вакуумом при 80°C в течение 20 ч, получая 46,7 г (выход: 93%) продукта, который представлял собой бесцветную прозрачную жидкость при комнатной температуре.
Соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Соединение идентифицировали как желаемое соединение, представляющее собой бис-трифторметансульфонилимид три(диметиламино)бутилфосфония. Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: ацетон-d6, эталон: тетраметилсилан): δ 2,85 (д, 18H); 2,66-2,56 (м, 2H); 1,75-1,63 (м, 2H); 1,57-1,45 (м, 2H); 0,97 (т, 3H).
19F-ЯМР (282 МГц, растворитель: ацетон-d6, эталон: CF3Cl): δ -79,87 (с, 6F).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 11]

Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла 20,8°C, а температура кристаллизации составляла -0,6°C. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Начальная температура потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 320°C. Данные результаты показывают, что соль из настоящего примера поддерживает стабильное жидкое состояние в широком диапазоне температур от 20,8°C до 320°C.
Вязкость, измеренная вискозиметром вибрационного типа (поставляемый A&D Co., Ltd), составляла 53 мПа·с при 40°C.
Электропроводность, измеренная методом импеданса по переменному току (Electrochemical Measurement System HZ-3000, поставленная Hokuto Denko Corp.), составила 0,3 См·м-1 при 40°C.
Кроме того, циклическая вольтаммограмма, измеренная Electrochemical Measurement System HZ-3000, поставленной Hokuto Denko Corp., с использованием Pt рабочего электрода, Pt противоэлектрода и Li электрода сравнения, показала, что окно потенциала составляло от 0 В до 4,9 В по отношению к потенциалу Li/Li+. Вольтамперная характеристика бис-трифторметансульфонилимида три(диметиламино)бутилфосфония показана на Фиг.2.
Пример 5
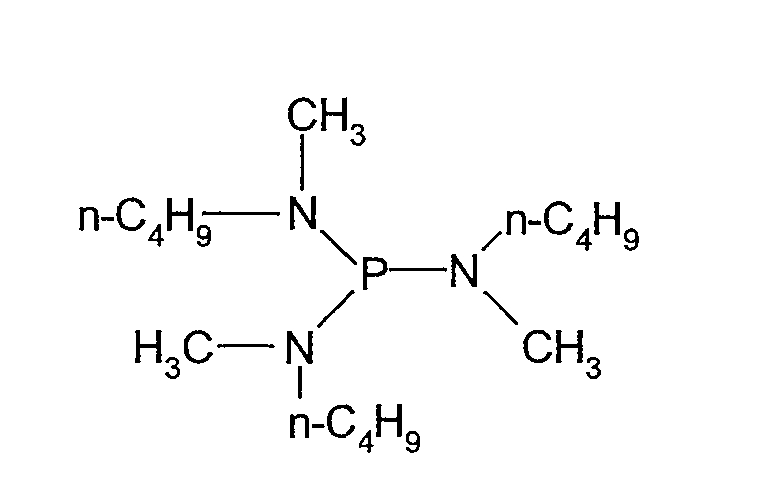
(i) Получение трис(метилбутиламино)фосфина
В 1000 мл трехгорлую колбу, оборудованную капельной воронкой и магнитной мешалкой, при комнатной температуре и в атмосфере азота добавляли 8,7 мл (0,10 моль) трихлорида фосфора и 1000 мл безводного диэтилового эфира. После охлаждения смеси на ледяной бане постепенно по каплям при перемешивании добавляли 70 мл (0,60 моль) метилбутиламина. После этого реакционную смесь перемешивали в течение 1 ч при охлаждении на ледяной бане. Реакционную смесь отфильтровали под давлением в атмосфере азота, и полученные в результате кристаллы три раза промывали безводным диэтиловым эфиром. Кристаллы очищали дистилляцией при температуре от 105°C до 118°C при пониженном давлении 0,2 кПа, получая 21,28 г трис(метилбутиламино)фосфина, который представлял собой прозрачную жидкость. Выход составлял 74%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 2,76 (м, 6H); 2,43 (д, 9H); 1,45 (м, 6H); 1,27 (м, 6H); 0,91 (т, 9H).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 120,88 (с, 1P).
Структурная формула показана ниже.
[Химический продукт 12]
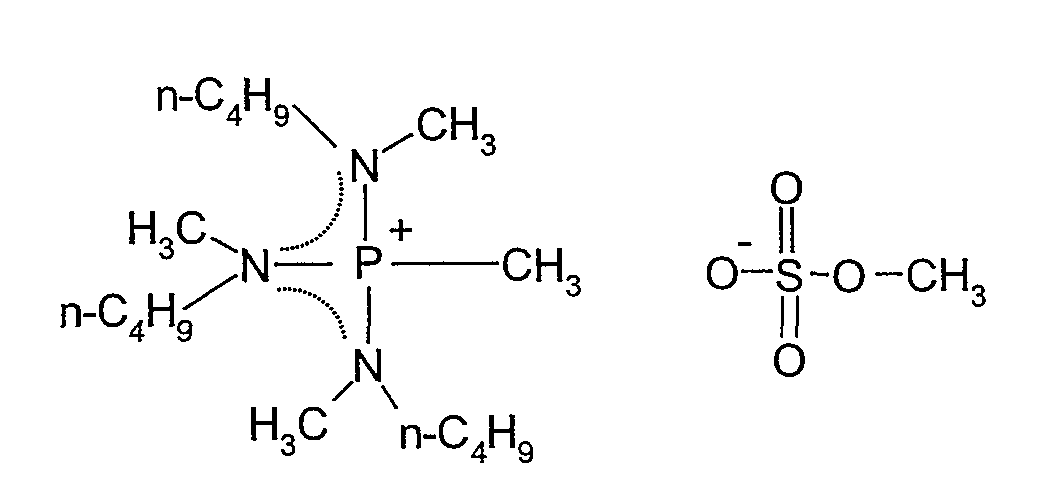
(h) Получение метилсульфата трис(метилбутиламино)метилфосфония
В 50 мл двухгорлую колбу, оборудованную магнитной мешалкой, при комнатной температуре в атмосфере азота добавляли охлажденные на ледяной бане 4,00 г (0,0138 моль) трис(метилбутиламино)фосфина, полученного в (i), и затем по каплям добавляли 1,6 мл (0,017 моль) диметилсульфата. После 12-часового перемешивания при комнатной температуре реакционную смесь три раза промывали диэтиловым эфиром и затем сушили под вакуумом при комнатной температуре, получая 4,18 г метилсульфата трис(метилбутиламино)метилфосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 73%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 3,71 (с, 3H); 2,96 (м, 6H); 2,76 (д, 9H); 2,09 (д, 3H); 1,57 (м, 6H); 1,33 (м, 6H); 0,96 (т, 9H).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): 58,79 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 13]
Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура стеклования составляла -70,4°C. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 263,5°C.
Пример 6
Получение бис-трифторметансульфонилимида трис(метилбутиламино)метилфосфония
В 100 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 1,00 г (0,0024 моль) метилсульфата трис(метилбутиламино)метилфосфония, полученного в (j), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,8 г (0,0026 моль) бис-трифторметансульфонилимида лития в 10 мл ультрачистой воды. Реакционную смесь перемешивали при комнатной температуре в течение 62 ч. Полученную в результате соль экстрагировали 20 мл CH2Cl2, и водный слой дополнительно экстрагировали 20 мл CH2Cl2. Затем органический слой три раза промывали 20 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°C, получая 0,91 г бис-трифторметансульфонилимида трис(метилбутиламино)метилфосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 65%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 2,91 (м, 6H); 2,71 (д, 9H); 1,92 (д, 3H); 1,56 (м, 6H); 1,32 (м, 6H); 0,96 (т, 9H).
19F-ЯМР (282 МГц, растворитель: CDCl3, эталон: CF3Cl): δ -78,82 (с, 6F).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 57,98 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 14]


Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла -5,5°С. Температура кристаллизации составляла -48,4°С. Температура стеклования составляла -82,9°С. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°С/мин, составляла 377,6°С.
Пример 7
(1) Получение тетрафторбората трис(метилбутиламино)метилфосфония
В 100 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 1,00 г (0,0024 моль) метилсульфата трис(метилбутиламино)метилфосфония, полученного в (j), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,3 г (0,0026 моль) тетрафторбората аммония в 10 мл ультрачистой воды. Реакционную смесь перемешивали при комнатной температуре в течение 62 ч. Полученную в результате соль экстрагировали 20 мл СН2Сl2, и водный слой дополнительно экстрагировали 20 мл СН2Сl2. Затем органический слой три раза промывали 20 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°C, получая 0,60 г тетрафторбората трис(метилбутиламино)метилфосфония, который представлял собой белое твердое вещество при комнатной температуре. Выход составлял 64%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 2,96 (м, 6H); 2,73 (д, 9H); 1,99 (д, 3H); 1,55 (м, 6H); 1,33 (м, 6H); 0,95 (т, 9H).
19F-ЯМР (282 МГц, растворитель: CDCl3, эталон: CF3Cl): δ -152,69 (д, 4F).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 58,72 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 15]

Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла 116,5°C. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 404,6°C.
Пример 8
(m) Получение гексафторфосфата трис(метилбутиламино)метилфосфония
В 100 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 1,00 г (0,0024 моль) метилсульфата трис(метилбутиламино)метилфосфония, полученного в (j), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,4 г (0,0026 моль) гексафторфосфата лития в 10 мл ультрачистой воды. Реакционную смесь перемешивали при комнатной температуре в течение 86 ч. Полученную в результате соль экстрагировали 20 мл CH2Cl2, и водный слой дополнительно экстрагировали 20 мл CH2Cl2. Затем органический слой три раза промывали 20 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°C, получая 0,48 г гексафторфосфата трис(метилбутиламино)метилфосфония, который представлял собой белое твердое вещество при комнатной температуре. Выход составлял 44%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 2,92 (м, 6H); 2,72 (д, 9H); 1,92 (д, 3H); 1,56 (м, 6H); 1,32 (м, 6H); 0,96 (т, 9H).
19F-ЯМР (282 МГц, растворитель: CDCl3, эталон: CF3Cl): δ -72,84 (d, 6F).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): 58,32 (м, 1P); -144,25 (гепт, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 16]
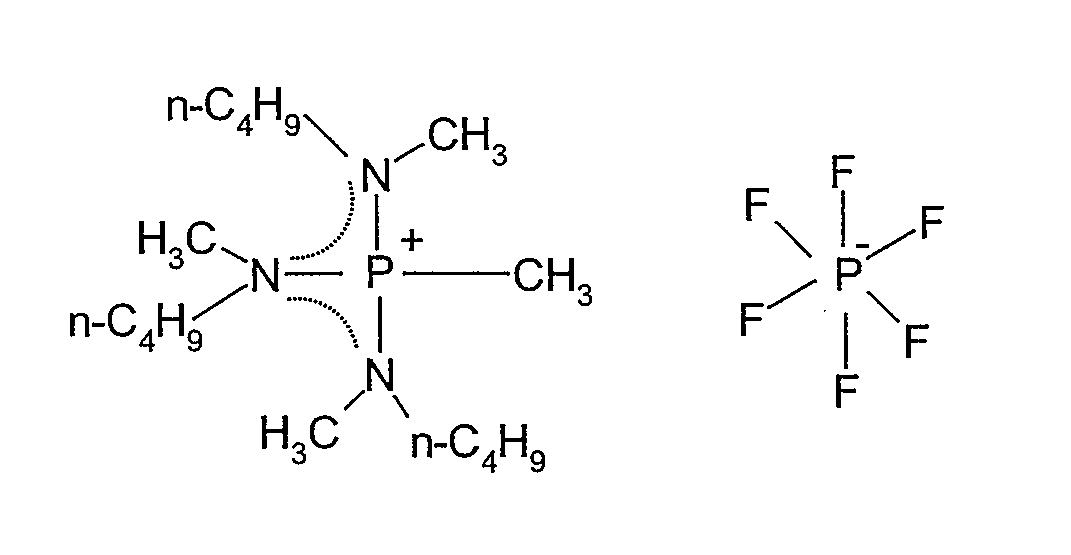
Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Не наблюдалось никакого пика, соответствующего температуре плавления. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 393,2°C.
Пример 9
(n) Получение этилсульфата трис(метилбутиламино)этилфосфония
В 50 мл двухгорлую колбу, оборудованную магнитной мешалкой, при комнатной температуре в атмосфере азота добавляли охлажденные на ледяной бане 4,00 г (0,0138 моль) трис(метилбутиламино)фосфина, полученного в (i), и затем по каплям добавляли 2,2 мл (0,017 моль) диэтилсульфата. После 37-часового перемешивания при 30°C реакционную смесь три раза промывали диэтиловым эфиром и сушили под вакуумом при комнатной температуре, получая 3,41 г этилсульфата трис(метилбутиламино)этилфосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 57%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 4,09 (м, 2H); 2,96 (м, 6H); 2,78 (д, 9H); 2,60 (м, 2H); 1,59 (м, 6H); 1,40-1,24 (м, 12H); 0,96 (т, 9H).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 61,87 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 17]

Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Не наблюдалось никакого пика, соответствующего температуре плавления. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 250,5°C.
Пример 10
(o) Получение бис-трифторметансульфонилимида трис(метилбутиламино)этилфосфония
В 100 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 1,00 г (0,0023 моль) этилсульфата трис(метилбутиламино)этилфосфония, полученного в (n), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,8 г (0,0026 моль) бис-трифторметансульфонилимида лития в 10 мл ультрачистой воды. Реакционную смесь перемешивали при комнатной температуре в течение 62 ч. Полученную в результате соль экстрагировали 20 мл CH2Cl2, и водный слой дополнительно экстрагировали 20 мл CH2Cl2. Затем органический слой три раза промывали 20 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°C, получая 0,73 г бис-трифторметансульфонилимида трис(метилбутиламино)этилфосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 53%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: СDСl3, эталон: тетраметилсилан): δ 2,92 (м, 6Н); 2,72 (д, 9Н); 2,37 (м, 2Н); 1,58 (м, 6Н); 1,39-1,20 (м, 9Н); 0,97 (т, 9Н).
19F-ЯМР (282 МГц, растворитель: СDСl3, эталон: СF3Сl): δ - 78,83 (с, 6F).
31Р-ЯМР (121 МГц, растворитель: СDСl3, эталон: трифенилфосфин): δ 61,02 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 18]

Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла -20,6°С. Температура стеклования составляла -84,6°С. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°С/мин, составляла 362,8°С.
Электропроводность, измеренная методом импеданса по переменному току (Electrochemical Measurement System HZ-3000, поставленная Hokuto Denko Corp.), составила 0,085 См·м-1 при 25°C.
Пример 11
(p) Получение тетрафторбората трис(метилбутиламино)этилфосфония
В 100 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 0,86 г (0,0019 моль) этилсульфата трис(метилбутиламино)этилфосфония, полученного в (n), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,3 г (0,0026 моль) тетрафторбората аммония в 10 мл ультрачистой воды. Реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Полученную в результате соль экстрагировали 20 мл CH2Cl2, и водный слой дополнительно экстрагировали 20 мл CH2Cl2. Затем органический слой три раза промывали 20 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°C, получая 0,65 г тетрафторбората трис(метилбутиламино)этилфосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 84%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 2,95 (м, 6H); 2,75 (д, 9H); 2,45 (м, 2H); 1,58 (м, 6H); 1,37-1,22 (м, 9H); 0,96 (т, 9H).
19F-ЯМР (282 МГц, растворитель: CDCl3, эталон: CF3Cl): δ -153,27 (д, 4F).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 61,41 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 19]
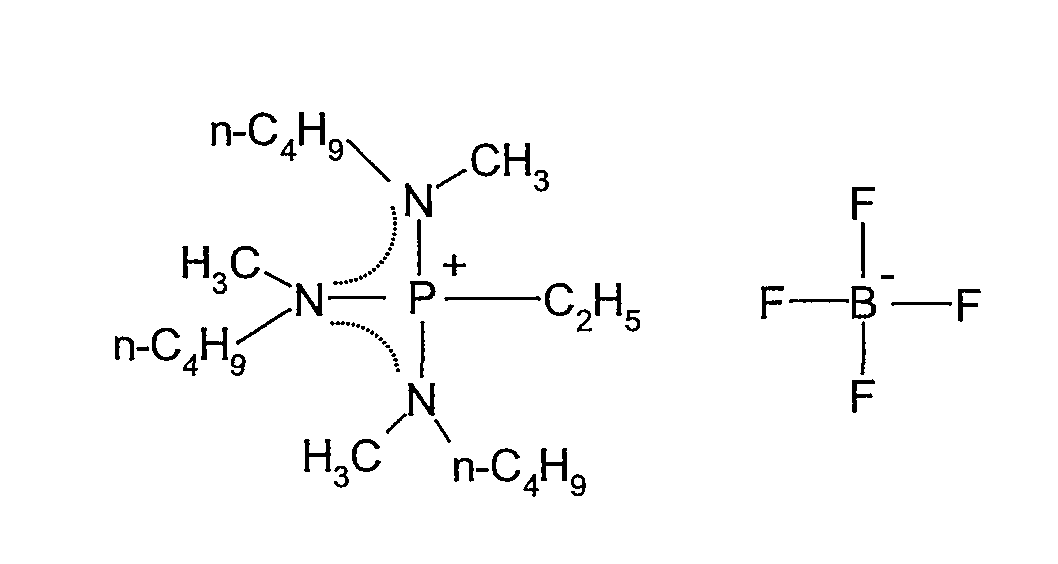
Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла 1,0°C. Температура кристаллизации составляла -32,7°C. Температура стеклования составляла -75,5°C. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 389,1°C.
Пример 12
(q) Получение гексафторфосфата трис(метилбутиламино)этилфосфония
В 100 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 1,00 г (0,0023 моль) этилсульфата трис(метилбутиламино)этилфосфония, полученного в (n), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,7 г (0,0046 моль) гексафторфосфата лития в 10 мл ультрачистой воды. Реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Полученную в результате соль экстрагировали 20 мл CH2Cl2, и водный слой дополнительно экстрагировали 20 мл CH2Cl2. Затем органический слой три раза промывали 20 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°C, получая 0,65 г гексафторфосфата трис(метилбутиламино)этилфосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 44%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 2,93 (м, 6H); 2,73 (д, 9H); 2,47 (м, 2H); 1,58 (м, 6H); 1,37-1,20 (м, 9H); 0,95 (т, 9H).
19F-ЯМР (282 МГц, растворитель: CDCl3, эталон: CF3Cl): -73,15 (д, 6F).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): 61,00 (м, 1P); -144,29 (гепт, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 20]

Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Не наблюдалось никакого пика, соответствующего температуре плавления. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 319,5°C.
Пример 13
(r) Получение н-бутилсульфата трис(метилэтиламино)н-бутилфосфония
В 50 мл двухгорлую колбу, оборудованную магнитной мешалкой, при комнатной температуре в атмосфере азота добавляли 2,33 г (0,0114 моль) трис(метилэтиламино)фосфина, полученного аналогично (i). После охлаждения на ледяной бане по каплям добавляли 2,7 мл (0,0136 моль) ди-н-бутилсульфата. Реакционную смесь перемешивали в течение 87 ч при комнатной температуре и затем 72 часа при 30°C. Затем реакционную смесь три раза промывали диэтиловым эфиром и сушили под вакуумом при комнатной температуре, получая 3,83 г н-бутилсульфата трис(метилэтиламино)н-бутилфосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 94%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): 4,04 (т, 2H); 3,11 (м, 6H); 2,77 (д, 9H); 2,48 (м, 2H); 1,67-1,37 (м, 8H); 1,24 (т, 9H); 0,99-0,88 (м, 6H).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): 59,52 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 21]

Пример 14
(s) Получение бис-трифторметансульфонилимида трис(метилэтиламино)н-бутилфосфония
В 100 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 1,00 г (0,0024 моль) н-бутилсульфата трис(метилэтиламино)н-бутилфосфония, полученного в (r), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,9 г (0,0029 моль) бис-трифторметансульфонилимида лития в 10 мл ультрачистой воды. Реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Полученную в результате соль экстрагировали 20 мл CH2Cl2, и водный слой дополнительно экстрагировали 20 мл CH2Cl2. Затем органический слой три раза промывали 20 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°С, получая 0,74 г бис-трифторметансульфонилимида трис(метилэтиламино)н-бутилфосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 57%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯMP (300 МГц, растворитель: СDСl3, эталон: тетраметилсилан): δ 3,05 (м, 6Н); 2,72 (д, 9Н); 2,28 (м, 2Н); 1,51 (м, 4Н); 1,23 (т, 9Н); 0,97 (т, 3Н).
19F-ЯМР (282 МГц, растворитель: CDCl3, эталон: СF3Сl): δ - 78,84 (с, 6F).
31Р-ЯМР (121 МГц, растворитель: СDСl3, эталон: трифенилфосфин): δ 59,02 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 22]
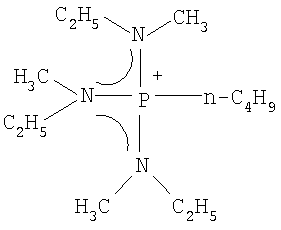
[Химический продукт 22]
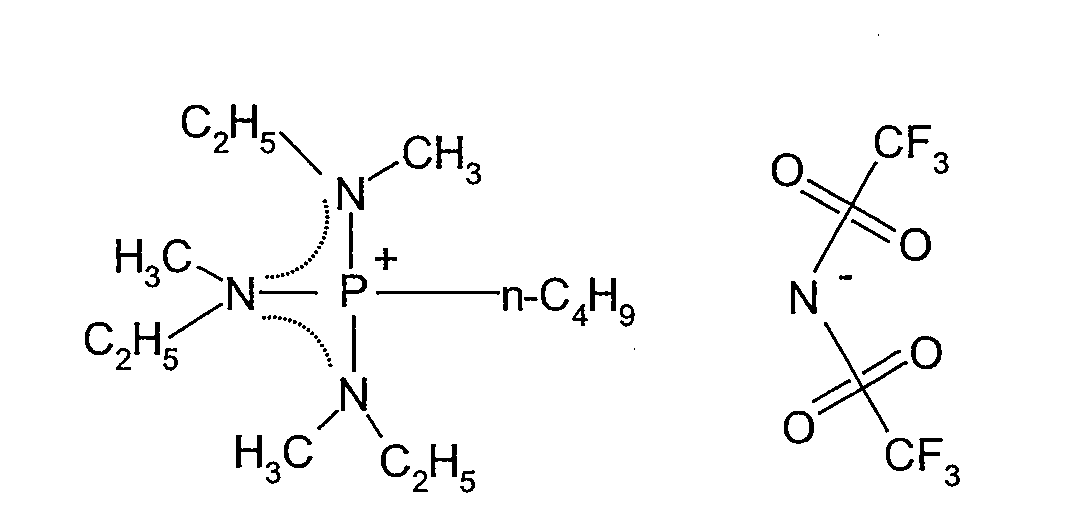
Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла -18,7°C. Температура кристаллизации составляла -47,9°C. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 393,0°C.
Пример 15
(t) Получение тетрафторбората трис(метилэтиламино)н-бутилфосфония
В 100 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 1,00 г (0,0024 моль) н-бутилсульфата трис(метилэтиламино)н-бутилфосфония, полученного в (r), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,4 г (0,0029 моль) тетрафторбората аммония в 10 мл ультрачистой воды. Реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Полученную в результате соль экстрагировали 20 мл CH2Cl2, и водный слой дополнительно экстрагировали 20 мл CH2Cl2. Затем органический слой три раза промывали 20 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°C, получая 0,87 г тетрафторбората трис(метилэтиламино)н-бутилфосфония, который представлял собой белое твердое вещество при комнатной температуре. Выход составлял 99%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 3,08 (м, 6H); 2,75 (д, 9H); 2,38 (м, 2H); 1,53 (м, 4H); 1,23 (т, 9H); 0,97 (т, 3H).
19F-ЯМР (282 МГц, растворитель: CDCl3, эталон: CF3Cl): δ -153,07 (д, 4F).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 59,40 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 23]
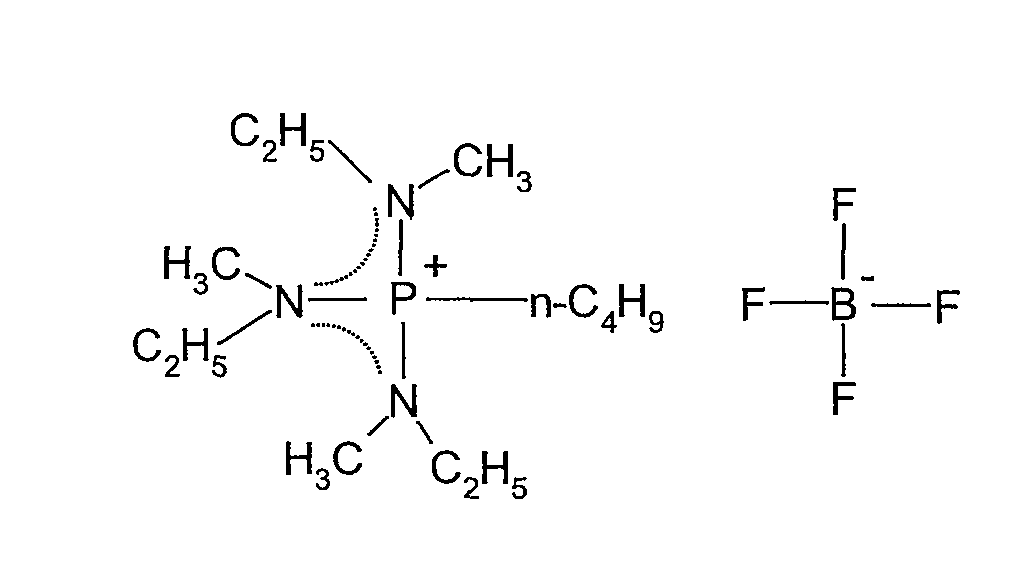
Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Не наблюдалось никакого пика, соответствующего температуре плавления. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°C/мин, составляла 333,0°C.
Пример 16
(u) Получение гексафторфосфата трис(метилэтиламино)н-бутилфосфония
В 100 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 1,00 г (0,0024 моль) н-бутилсульфата трис(метилэтиламино)н-бутилфосфония, полученного в (r), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,5 г (0,0029 моль) гексафторфосфата лития в 10 мл ультрачистой воды. Реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Полученную в результате соль экстрагировали 20 мл CH2Cl2, и водный слой дополнительно экстрагировали 20 мл CH2Cl2. Затем органический слой три раза промывали 20 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°C, получая 0,95 г гексафторфосфата трис(метилэтиламино)н-бутилфосфония, который представлял собой белое твердое вещество при комнатной температуре. Выход составлял 97%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 3,06 (м, 6H); 2,72 (д, 9H); 2,39 (м, 2H); 1,52 (м, 4H); 1,22 (т, 9H); 0,97 (т, 3H).
19F-ЯМР (282 МГц, растворитель: CDCl3, эталон: CF3Cl): δ -73,08 (д, 6F).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 59,08 (м, 1P); -144,27 (гепт, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 24]
Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). He наблюдалось никакого пика, соответствующего температуре плавления. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°С/мин, составляла 369,2°С.
Пример 17
(v) Получение оксида трис(метилбутиламино)фосфина
В 200 мл трехгорлую колбу, оборудованную капельной воронкой и магнитной мешалкой, при комнатной температуре и в атмосфере азота добавляли 1,8 мл (0,020 моль) фосфорилхлорида и 100 мл безводного дибутилового эфира. После охлаждения смеси на ледяной бане постепенно по каплям при перемешивании добавляли 21 мл (0,180 моль) метилбутиламина. Далее реакционную смесь перемешивали при 120°С в течение 36 ч, и затем реакционную смесь отфильтровывали под давлением в атмосфере азота. Полученные в результате кристаллы три раза промывали безводным дибутиловым эфиром и очищали дистилляцией при пониженном давлении 0,2 кПа и температуре от 119 до 124°C, получая 5,54 г трис(метилбутиламино)оксолина, который представлял собой прозрачную жидкость. Выход составлял 74%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 2,94 (м, 6H); 2,66 (д, 9H); 1,51 (м, 6H); 1,30 (м, 6H); 0,93 (т, 9H).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 25,26 (м, 1P).
Структурная формула показана ниже.
[Химический продукт 25]

(w) Получение этилсульфата трис(метилбутиламино)этилфосфония
В 50 мл двухгорлую колбу, оборудованную магнитной мешалкой, при комнатной температуре в атмосфере азота добавляли 2,26 г (0,0074 моль) трис(метилбутиламино)оксолина, полученного в (v), и затем по каплям добавляли 1,2 мл (0,0089 моль) диэтилсульфата. Смесь перемешивали при 30°C в течение 69 ч, затем три раза промывали диэтиловым эфиром и сушили под вакуумом при комнатной температуре, получая 0,65 г этилсульфата трис(метилбутиламино)этоксифосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 19%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 4,36 (м, 2H); 4,10 (кв., 2H); 3,02 (м, 6H); 2,84 (д, 9H); 1,58 (м, 6H); 1,45 (т, 3H); 1,40-1,26 (м, 9H); 0,96 (т, 9H).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 35,87 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру).
[Химический продукт 26]

Пример 18
(x) Получение бис-трифторметансульфонилимида трис(метилбутиламино)этоксифосфония
В 50 мл колбу в форме баклажана, оборудованную магнитной мешалкой, добавляли 0,65 г (0,0014 моль) этилсульфата трис(метилбутиламино)этоксифосфония, полученного в (w), и 10 мл ультрачистой воды, и затем при перемешивании добавляли водный раствор 0,5 г (0,0015 моль) бис-трифторметансульфонилимида лития в 10 мл ультрачистой воды. Реакционную смесь перемешивали при 30°C в течение 62 ч. Полученную в результате соль экстрагировали 20 мл CH2Cl2, и водный слой дополнительно экстрагировали 20 мл CH2Cl2. Затем органический слой три раза промывали 40 мл ультрачистой воды, экстракт концентрировали на роторном испарителе, три раза промывали диэтиловым эфиром и затем сушили под вакуумом при 80°C, получая 0,8 г бис-трифторметансульфонилимида трис(метилбутиламино)этоксифосфония, который представлял собой прозрачную жидкость при комнатной температуре. Выход составлял 93%.
Полученное в результате соединение идентифицировали спектрометром ядерного магнитного резонанса (BRUKER Ultra Shield 300 NMR спектрометр, поставляемый BRUKER Corp.). Спектральные данные показаны ниже.
1H-ЯМР (300 МГц, растворитель: CDCl3, эталон: тетраметилсилан): δ 4,23 (м, 2H); 2,98 (м, 6H); 2,77 (д, 9H); 1,58 (м, 6H); 1,46-1,27 (м, 9H); 0,96 (т, 9H).
19F-ЯМР (282 МГц, растворитель: CDCl3, эталон: CF3Cl): δ -78,83 (с, 6F).
31P-ЯМР (121 МГц, растворитель: CDCl3, эталон: трифенилфосфин): δ 35,83 (м, 1P).
Структурная формула показана ниже (пунктирная линия в формуле представляет сопряженную структуру)
[Химический продукт 27]

Температуру плавления измеряли сканирующим дифференциальным калориметром (DSC8230, поставляемый Shimadzu Corp.). Температура плавления составляла -19,9°С. Температура кристаллизации составляла -55,8°С. Температура стеклования составляла -85,9°С. Температуру термического разложения измеряли термогравиметрическим анализатором (TG8120, поставляемый Rigaku Corp.). Температура 5% потери массы, измеренная при скорости повышения температуры 10°С/мин, составляла 208,6°С.
Промышленная применимость
Настоящее изобретение предлагает ионную жидкость, которая показывает стабильное жидкое состояние в широком диапазоне температур от низких температур, и имеет низкую вязкость, адекватную электропроводность и превосходную электрохимическую стабильность.
Ионную жидкость по настоящему изобретению можно использовать в таких областях применения, как литиевые аккумуляторные батареи, конденсаторы с двойным электрическим слоем, топливные элементы, сенсибилизированные красителем солнечные элементы, электролиты, растворы электролитов или добавки для электрических запоминающих устройств, реакционные растворители и т.п.
Реферат
Настоящее изобретение относится к ионным жидкостям на основе катиона, представленного формулой (I): ! ! где замещающие группы R1-R9 выбраны из водорода, алкила; любой атом углерода в R1-R9 может быть замещен группой -O-, -С(O)-, -С(O)O-, -S-, -S(O)-, -SO2- или -SO3-; Х представляет собой S, О или С; R8 и R9 существуют, только когда Х представляет собой углерод; анион выбран из групп [RSO3]-, [RfSO3]-, [(RfSO2)2N]-, [(FSO2)3C]-, [RCH2OSO3]-, [RC(O)O]-, [RfC(O)O]-, [CCl3C(O)O]-, [(CN)3C]-, [(CN)2CR]-, [(RO(O)C)2CR]-, [B(OR)4]-, [N(CF3)2]-, [N(CN)2]-, [AlCl4]-, PF6 -, BF4 -, SO4 2-, HSO4 -, ! NO3 -; где R представляет собой водород, галоген, алкил, алкенил, алкинил, циклоалкил, Rf представляет собой фторсодержащую замещающую группу. Технический результат - получение новых ионных жидкостей с улучшенными электрохимическими свойствами. 14 з.п.ф-лы, 2 ил.
Формула
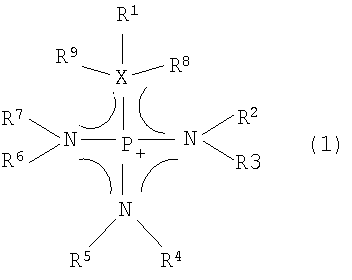
где замещающие группы R1-R9 могут быть независимо одинаковыми или отличающимися друг от друга; каждая из замещающих групп R1-R9представляет собой атом водорода или алкильную группу с прямой или разветвленной цепью, содержащую от 1 до 30 атомов углерода; любой атом углерода, содержащийся в замещающих группах R1-R9, может быть замещен атомом и/или атомной группой, выбранной из группы, состоящей из -O-, -С(O)-, -С(O)O-, -S-, -S(O)-, -SO2-, -SO3-;
Х представляет собой атом серы, атом кислорода или атом углерода; R8 и R9существуют, только когда Х представляет собой атом углерода и пунктирная линия представляет сопряженную структуру;
анионный компонент представляет собой одну или несколько разновидностей, выбранных из группы, состоящей из [RSO3]-, [RfSO3]-, [(RfSO2)2N]-, [(FSO2)3C]-, [RCH2OSO3]-, [RC(O)O]-, [RfC(O)O]-, [CCl3C(O)O]-, [(CN)3C]-, [(CN)2CR]-, [(RO(O)C)2CR]-, [B(OR)4]-, [N(CF3)2]-, [N(CN)2]-, [AlCl4]-, PF6-, BF4-, SO42-, HSO4-, NO3-; где каждая замещающая группа R представляет собой атом водорода, атом галогена, алкильную группу с прямой или разветвленной цепью, содержащую от 1 до 10 атомов углерода, алкенильную группу с прямой или разветвленной цепью, содержащую от 2 до 10 атомов углерода, с одной или несколькими двойными связями, алкинильную группу с прямой или разветвленной цепью, содержащую от 2 до 10 атомов углерода, с одной или несколькими тройными связями, насыщенную или частично или полностью ненасыщенную циклоалкильную группу; любые атомы водорода, содержащиеся в замещающих группах R, могут быть частично или полностью замещены атомом галогена; любой атом углерода, содержащийся в замещающих группах R, может быть замещен атомом и/или атомной группой, выбранной из группы, состоящей из -O-, -С(O)-, -С(O)O-, -S-, -S(O)-, -SO2- и -SO3-; и Rf представляет собой фторсодержащую замещающую группу.
Комментарии