Способ неэлектролитической металлизации арамидных поверхностей - RU2144965C1
Код документа: RU2144965C1
Чертежи


Описание
Предпосылки создания
изобретения
Область техники, к которой относится изобретение
Данное изобретение относится к неэлектролитической металлизации арамидных волокон, в которой металл сильно приклеивается
к подложке из арамидных волокон и обеспечивает высокопроводящую поверхность. Арамид подвергается предварительной обработке перед металлизацией, включающей тщательно контролируемую выдержку в
концентрированном водном растворе азотной кислоты или в разбавленном растворе хлорсульфоновой или фторсульфоновой кислоты в органической жидкости, с последующей его промывкой, катализацией и
неэлектролитической металлизацией.
Описание известного уровня техники
Неэлектролитическая металлизация является отложением пленки металла при взаимодействии ионов металла и
агента химического восстановления в щелочном растворе. Неэлектролитическая металлизация в общем смысле является хорошо известной. Одна из трудностей в достижении успешной неэлектролитической
металлизации заключается в получении хорошей адгезии между металлизирующейся подложкой и нанесенным металлом. Хотя лишь капсулирование может быть достаточным для некоторых применений и некоторых
изделий, хорошая адгезия нанесенного металла имеет существенное значение для поверхностей волокон, потому что нанесенное металлическое покрытие должно быть достаточно надежным, чтобы выдержать
нагрузки дополнительной переработки и напряжений конечного применения.
Патент США N 5302415, выданный 12 апреля 1994 г., рассматривает способ неэлектролитической металлизации арамидных поверхностей с помощью обработки перед металлизацией, использующей 80-90% водный раствор серной кислоты.
Краткое описание изобретения
Настоящее изобретение представляет способ
металлизации арамидных волокон при увеличенных скоростях металлизации долговечным металлическим покрытием, включающий стадии: выдержки арамидных волокон в кислотном растворе 86-92 мас. % водной
азотной кислоты или 1-5 мас.% хлорсульфоновой кислоты или фторсульфоновой кислоты в органической жидкости в течение, по крайней мере, 2 с при температуре в интервале от 10 до 100oC,
нейтрализации и промывки протравленных кислотой волокон водой до тех пор, пока практически вся кислота не будет удалена, и металлизации волокон неэлектролитическим способом металлизации.
Для металлизации волокон медью процесс неэлектролитической металлизации может быть проведен при контактировании обработанных кислотой и промытых волокон с олово-палладиевым активирующим раствором, промывке волокон в воде для удаления неадгезированного металла, необязательно, погружении промытых волокон в водный ускоряющий раствор минеральной кислоты и затем погружении волокон в ванну неэлектролитической металлизации медью.
При осуществлении на практике настоящего изобретения активирующий раствор включает палладий для медного или никелевого покрытия и серебро для серебряного покрытия.
Краткое описание рисунков
Фиг. 1 является микрофотографией с увеличением х 500 волокон, неадекватно металлизированных.
Фиг. 2 является микрофотографией с увеличением х 500 волокон, обработанных в соответствии с настоящим изобретением.
Подробное описание изобретения
Давно имеется потребность в проводящих
арамидных волокнах, которые имеют долговечные металлические покрытия; и эта потребность является особенно острой для волокон, которые обладают высокой прочностью и модулем упругости.
Волокна из арамидов являются очень трудными для покрытия долговечным металлическим покрытием. Обработки и предварительные обработки поверхности арамидных волокон обычно не являются полностью удовлетворяющими.
Данное изобретение представляет способ неэлектролитической металлизации волокон из арамидов при значительно увеличенных скоростях металлизации, который в некотором отношении дает металлизированный волокнистый продукт, практически сохранивший прочность и модуль упругости, и металлическое покрытие которого является высокопроводящим и с высокой адгезией. Способ может быть осуществлен на непрерывной основе или периодически.
Под "арамидом" понимается полиамид, у которого не менее 85% амидных (-CO-NH-) связей присоединяются непосредственно к двум ароматическим кольцам. Соответствующие арамидные волокна описаны в Man-Made Fibers-Science and Technology, Volume 2, Section titled Fiber-Forming Aromatic Polyamides, p. 297, W. Black et al. Interscience Publishers, 1968. Арамидные волокна также рассматриваются в патентах США 4172938, 3869429, 3819587, 3673143, 3354127 и 3094511.
С арамидом могут быть использованы добавки, и, как особый случай, было установлено, что до 30 мас.% поливинилпирролидона может быть введено в поли(п-фенилентерефталамидом) в арамидные волокна, металлизируемые способом данного изобретения.
Пара-арамидами являются первичные полимеры в волокнах данного изобретения, а поли(п-фенилентерафталамид) является предпочтительным пара-амидом. Под поли(п-фенилентерефталамидом) понимается гомополимер, получающийся моль-на-моль полимеризацией п-фенилендиамина и терефталоилхлорида, а также сополимеры, получающиеся введением небольших количеств других диаминов с п-фенилендиамином и небольших количеств других хлоридов двухосновной кислоты с терефталоилхлоридом. Как правило, другие диамины и другие хлориды двухосновной кислоты могут быть использованы в количествах до примерно 10 мол.% р-фенилендиамина или терефталоилхлорида или возможно немного больших только при условии, что другие диамины и хлориды двухосновной кислоты не имеют реакционноспособных групп, которые мешают реакции полимеризации. Поли(п-фенилентерефталамид) также означает сополимеры, получающиеся от введения других ароматических диаминов и других хлоридов ароматических двухосновных кислот, таких как, например, 2,6-нафталоилхлорид или хлор- или дихлор-терефталоилхлорид, при условии только, что другие ароматические диамины и ароматические хлориды двухосновных кислот присутствуют в количествах, которые позволяют получить анизотропные прядильные "сиропы". Получение р-фенилентерефталамида описывается в патентах США N 3869429, 4308374, 4698414.
В волокнах данного изобретения также могут использоваться мета-арамиды, и поли(м-фениленизофталамид) является предпочтительным мета-арамидом. Под поли(м-фениленизофталамидом) подразумевается гомополимер, получающийся моль-на-моль полимеризацией м-фенилендиамина и изофталоилхлорида, а также сополимеры, получающиеся от введения небольших количеств других диаминов с м-фенилендиамином и небольших количеств других хлоридов двухосновных кислот с изофталоилхлоридом. Как правило, другие диамины и другие хлориды двухосновных кислот могут быть использованы в количествах до 10 мол.% м-фенилендиамина или изофталоилхлорида или возможно немного больших при условии только, что другие диамины и хлориды двухосновных кислот не имеют реакционноспособных групп, которые мешают реакции полимеризации. Поли(м-фениленизофталамид) также означает сополимеры, получающиеся от введения других ароматических диаминов и других ароматических хлоридов двухосновных кислот при условии только, что другие ароматические диамины и ароматические хлориды двухосновных кислот присутствуют в количествах, которые не ухудшают желательные характеристики арамида.
Арамидные волокна, полученные мокрым или воздушным способами прядения из вышеуказанных патентов, коагулируются в так называемую "невысыхающую" форму, в которой волокно содержит значительно более 75 мас.% воды. "Невысыхающие" волокна затем сушатся до менее примерно 20 мас.% воды для того, чтобы разрушить полимерную структуру волокна. Волокнами, пригодными для использования в способе настоящего изобретения, являются сухие волокна, имеющие влагосодержание менее 20 мас. %. Обычно волокна, используемые в способе настоящего изобретения, должны быть даже более сухими, имеющими влагосодержание около 3,5-7% воды.
На первой стадии способа данного изобретения металлизируемые арамидные волокна контактируют с кислотой предварительной обработки. Кислотой предварительной обработки, используемой при осуществлении данного изобретения, является водная азотная кислота или хлор- или фторсульфоновая кислота в органической жидкости, не реагирующей с кислотой. Было установлено, что ни водная соляная кислота, ни водная фосфорная кислота не дают приемлемые результаты при использовании в качестве кислоты предварительной обработки; было установлено, что хлор- и фторсульфоновые кислоты разлагаются в воде и должны использоваться в неводной жидкости.
Предварительная обработка настоящего изобретения может осуществляться при использовании водной азотной кислоты концентрации от примерно 86 мас.% до концентрации, где имеется чрезмерная опасность для обрабатываемого материала. Пределы концентрации кислоты, конечно, зависят от температуры и длительности предварительной обработки. Предварительная обработка обычно проводится при окружающей температуре, обычно 20-40oC, и при средней продолжительности обычно 5-60 с. Если температура или продолжительность предварительной обработки увеличиваются, концентрация кислоты может быть соответственно снижена. При повышенной температуре или увеличенной продолжительности может быть эффективной азотная кислота концентрации менее 86 мас. %, а при сниженных температурах или уменьшенной продолжительности может использоваться азотная кислота концентрации более 86 мас.%. Когда используется кислота слишком низкой концентрации, предварительная обработка является неэффективной для получения высокой адгезии нанесенного металла, а когда используется кислота слишком высокой концентрации, обработанные волокна являются чрезмерно разрушенными.
Для предварительной обработки данного изобретения хлорсульфоновая кислота и фторсульфоновая кислота используются в относительно разбавленных концентрациях в органической жидкости. Органические жидкости, которые являются пригодными для использования, включают любую, с которой смешиваются кислоты и с которой кислоты не реагируют. Примеры таких жидкостей включают метиленхлорид, гексан, циклогексан и т.п. Концентрация этих галоидсульфоновых кислот для предварительной обработки данного изобретения должна составлять от примерно 1 мас.% до концентрации, где имеется чрезмерная опасность для обрабатываемых материалов, примерно 5 мас.%. Условия предварительной обработки с использованием этих галоидсульфоновых кислот обычно являются такими же, как для водной азотной кислоты.
Предварительная обработка арамидных волокон с использованием описанных выше кислот при заданных концентрациях, длительности и температурах дает чрезвычайно быстрый набор скорости металлизации, как будет показано в приведенном далее Примере. Хотя причина такого быстрого набора скорости полностью невыяснена, ясно, что обработка азотной кислотой при концентрациях 86-91% при температуре 30oC и обработка хлор- или фторсульфоновой кислотой при концентрациях 1-5% при температуре 30oC дает набор скорости металлизации арамидных волокон, который резко возрастает.
Температура ванны кислотной предварительной обработки должна быть в интервале от 10 до 100oC, предпочтительно примерно 20-40oC. Верхний температурный предел определяется вредным воздействием на прочностные свойства и сплавлением филаментной нити, тогда как нижний температурный предел является предметом целесообразности: более низкие температуры требуют неприемлемо большего времени для адекватной обработки.
Волокна, которые могут быть любой нужной толщины, контактируют с кислотой в течение не менее 2 с. При более коротких временах экспозиции трудно, в конечном счете, достигнуть достаточной глубины обработки. Более длительная экспозиция в то же время дает чрезмерное растрескивание элементарных волокон и вызывает потерю прочностных свойств. Как правило, контактирование волокон с кислотой в течение более 120 с даже при умеренных температурах приводит к разрушению волокон. Предпочтительное время контакта составляет примерно 15-40 с. Время экспозиции в кислоте может быть снижено увеличением температуры и/или увеличением концентрации кислоты. Эффективное осуществление на практике способа данного изобретения требует разумной комбинации концентрации кислоты, температуры и продолжительности травления.
Стадия кислотного травления способа данного изобретения делает поверхность волокон протравленной и обуславливает структурные изменения и микротрещины, образующиеся на поверхности волокна. На прилагаемых рисунках Фиг. 1 представляет микрофотографию волокон из поли (р-фенилентерефталамида), которые были погружены на 20 с в 85 мас.% азотную кислоту при температуре около 20oC, а Фиг. 2 представляет микрофотографию волокон из поли(р- фенилентерефталамида), которые были погружены на 5 с в 90 мас.% азотную кислоту при температуре около 20oC. Волокна на Фиг. 1 являются гладкими и не имеющими видных изменений при обработке, тогда как волокна на Фиг. 2 имеют трещины и неравномерное расслаивание по своей длине. Обработка, показанная на Фиг. 1, является неадекватной для получения металлического покрытия с высокой адгезией настоящего изобретения; а обработка, показанная на Фиг. 2, дает требуемую адгезию металла.
Важный аспект настоящего изобретения состоит в том, что предварительная обработка использует кислоты в условиях, которые действительно изменяют структуру волокон для того, чтобы достигнуть требуемой адгезии нанесенного металла. Хотя изменение поддерживается на допустимом уровне, предварительная обработка должна изменять волокна для достижения нужного результата.
Протравленные кислотой волокна из поли (п-фенилентерефталамида) хорошо промывают водой для удаления практически всей кислоты. Необязательно, волокно может быть нейтрализовано основанием, таким как раствор бикарбоната натрия, который может добавляться к промывочной воде или использоваться на отдельной стадии. Также можно сушить протравленные кислотой волокна до стадии металлизации.
Сущность данного изобретения состоит в открытии того, что арамидные волокна, обработанные кислотой, как указано здесь, могут дать улучшенный металлизированный волокнистый продукт. Как правило, общеизвестные способы неэлектролитической металлизации могут быть использованы для металлизации арамидных волокон после кислотной обработки в соответствии с настоящим изобретением.
Например, для способа металлизации медью водный активирующий раствор получается при использовании катионов палладия и олова в качестве катализатора активации. Металлизируемые протравленные кислотой и промытые волокна из поли (п-фенилентерефталамида) погружаются в раствор и перемешиваются для обеспечения активации поверхности волокон. Затем волокна, при необходимости, удаляются из активирующего раствора и прополаскиваются и, при необходимости, могут быть перенесены в ванну ускорителя с разбавленной минеральной кислотой.
Волокна затем помещаются в или пропускаются через ванну металлизации с ионами меди и формальдегидом, где ионы меди образуют комплекс с удержанием их в растворе, например, с тетранатриевой солью этилендиаминтетрауксусной кислоты (ЭДТА).
При осуществлении данного изобретения могут быть использованы ванны, имеющие широкий интервал концентраций металла. Предпочтительные ванны металлизации содержат примерно 1-5 г/л меди. В описанных здесь опытах наиболее предпочтительными являются ванны с 1-3 г/л меди.
Ванна металлизации с погруженными в нее волокнами умеренно перемешивается в течение 10-20 мин для обеспечения адекватного нарастания. При снижении скорости добавляется формальдегид, pH-корректирующий щелочной раствор и раствор с ионами меди. Добавления выполняются непрерывно или периодически. Металлизированный материал затем может быть промыт и высушен. В качестве восстанавливающих агентов вместо формальдегида могут быть использованы другие материалы. Пригодными восстановителями являются гипофосфит, гидразин, гидрид бора и т.п.
Все вышеуказанные стадии могут быть проведены в различных ваннах при температурах от 10 до 60oC, предпочтительно 20-40oC.
Например, в способе металлизации серебром протравленные кислотой волокна сначала погружают в водный раствор восстанавливающего агента, такого как SnCl2/HCl. Погруженные в SnCl2/HCl волокна промываются тщательно водой для удаления избытка и неадгезированных ионов двухвалентного олова и затем переносятся в водяную ванну, к которой добавляется металлкомплексообразующий раствор нитрата серебра и аммиака при pH ванны 8-9,5. В процессе погружения в металлкомплексообразующую ванну последняя перемешивается для обеспечения того, чтобы поглощенные ионы двухвалентного олова восстанавливали ионы серебра до серебра с предпочтительным отложением на сереброактивированной полимерной поверхности. В типичном способе мольное соотношение формальдегид: серебро составляет от 1,1:1 до 2:1. Количество нитрата серебра регулируется для обеспечения нужной массы восстановленного серебра как функция металлизируемого волокнистого материала. Металлизированные серебром волокна промываются и сушатся.
Вместо серебра или меди никель или кобальт, или подобное также может наноситься на протравленные кислотой волокна с собственной комбинацией активирующего раствора, раствора восстанавливающего агента и раствора металлизации.
Процессы металлизации могут проводится на протравленных кислотой волокнах, которые высушиваются или остаются влажными после стадии травления кислотой. В случае металлизации медью на качество металлизации относительно незаметно влияние сушки волокон после контакта с кислотой. Однако процесс серебрения дает выход нанесенного серебра с наименьшим сопротивлением, когда волокна сначала сушатся при температуре примерно 15-80oC, предпочтительно 15-20oC. Когда металлизированные серебром волокна сушатся при средней температуре, заметно меньшее проникновение металлического серебра в волокнистую структуру, чем для невысохших волокон, и отмечается лучшая сплошность серебряного покрытия, чем реализуется для волокон, сушившихся при более высоких температурах.
Методы испытаний
Электросопротивление
Элемент сопротивления
конструируется установкой медных
электродов длиной 25,4 мм параллельно и на 25,4 мм в сторону на плоском блоке непроводника, такого как полиэтилен. Электроды соединяются с омметром, таким как
мультиметр Кейтли 173А, и сопротивление
ткани определяется при прижатии элемента к ткани, расположенной на плоской непроводящей поверхности. Сопротивление фиксируется в омах на квадрат.
Описание предпочтительных
вариантов
Получение металлизированных волокон.
В приведенном ниже Примере используется следующая методика.
Металлизируемые волокна либо сначала обрабатываются кислотой предварительной обработки и затем переплетаются в небольшие тканевые рукава, либо сначала переплетаются, а затем обрабатываются кислотой предварительной обработки. Конечно, волокна для сравнительных примеров не подвергаются кислотной обработке или обрабатываются кислотой за пределами интервалов концентраций или условий обработки, требуемых данным изобретением. Используется трикотажная машина марки КОМЕТ фирмы Скотт энд Уильяме, Лакония, Нью-Гемпитр, США, имеющая головку диаметром 8,89 см (3,5 дюйма); получается ткань, состоящая из шести рядов (стежки параллельны оси цилиндра) и пяти петельных столбиков (стежки перпендикулярны оси цилиндра).
Каждый из образцов трикотажной ткани затем подвергается неэлектролитической металлизации
медью с
использованием серийно выпускаемых химических препаратов следующим образом:
(a) контактирование тканей в течение примерно 10 минут при температуре около 40oC с водным
активирующим
раствором минеральной кислоты, хлоридом двухвалентного олова и палладия, например раствором 60 мл "Кататрозит" 44 фирмы Шиплей Ко. , и с водным раствором хлорида натрия и олова, например
раствором
540 г "Катапреп" 404 фирмы Шиплей Ко. в 1700 мл воды с получением палладий-оловосодержащего комплекса для активации поверхности волокон;
(b) промывка пряжи в течение примерно 5
минут с двумя
переменами воды при температуре около 25oC,
(с) погружение пряжи примерно на 20 минут при температуре около 40oC в ванну водной металлизации, содержащую,
например, 240
мл "Циркупозит" 3350М фирмы Шиплей и Ко. , 84 мл "Циркупозит" 3350А фирмы Шиплей и Ко., 200 мл "Циркупозит" 3350В фирмы Шиплей и Ко. и 1476 мл воды;
(d) промывка пряжи в течение
примерно 7
мин двумя переменами воды при температуре около 25oC и
(е) сушка пряжи до утра в вакуумном термошкафу при температуре около 20oC.
"Циркупозит® 3350M" является товарным знаком фирмы Шиплей Ко. для раствора 25 мас. % этилендиаминтетрауксусной кислоты и 75 мас.% воды и безопасных материалов.
"Циркупозит® 3350A" является товарным знаком фирмы Шиплей Ко. для раствора 7 мас. % формальдегида, 10 мас.% сульфата меди, 3 мас.% соляной кислоты и 80 мас.% воды и безопасных материалов.
"Циркупозит® 3350B" является товарным знаком фирмы Шиплей Ко. для раствора 5 мас.% гидроксида натрия, 95 мас.% воды и безопасных материалов.
Для целей этих примеров проводится анализ металлизированных волокон на металлическую медь для определения количества меди, отложенной в процессе металлизации.
Пример 1 и
Сравнительные примеры
1-4
В этих примерах исследуется влияние азотной кислоты в качестве кислоты предварительной обработки. В этих примерах обрабатываемые волокна используются в виде
арамидной пряжи 400 денье,
имеющей 1,5 денье на филамент (445 дтекс, имеющей 1,7 дтекс на филамент), выполненной из поли(р-фенилентерефталамида) и поставляемой фирмой Е.И. дю Пон де Немур энд Компани
под торговой маркой
"КЕВЛАР"29.
Пряжа из волокон обрабатывается погружением в азотную кислоту при температуре около 20oC при концентрациях и с продолжительностью, указанных в Таблице 1; затем тщательно промывается водой и погружается в 8 мас.% раствор бикарбоната натрия на 5 мин перед промывкой в воде снова в течение полутора часов. Протравленная пряжа сушится на воздухе и из нее изготавливаются трубки, которые металлизируются в соответствии с вышеописанной методикой.
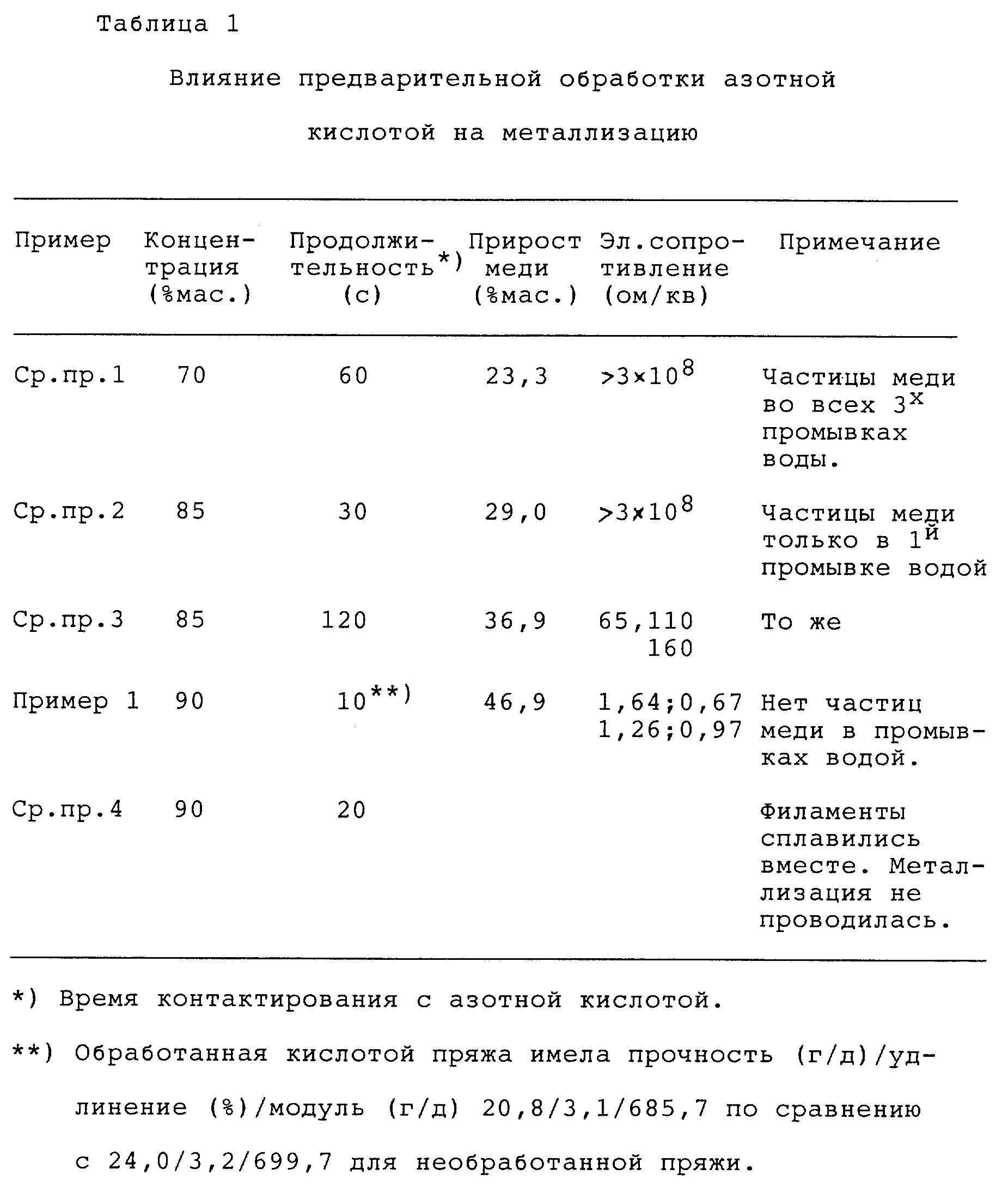
Приведенные в Таблице 1 данные по приросту меди (выраженному в мас.% металлизированного волокна) и электросопротивлению показывают, что азотная кислота при 20oC является эффективной при концентрациях выше 85 мас.% для предварительной обработки волокон для обеспечения металлизации медью. Концентрация азотной кислоты 85 мас.% является эффективной при немного более высоких температурах предварительной обработки до примерно 50oC; а 86 мас.% может быть использована эффективно при 20oC. Данные Таблицы 1 также показывают, что предварительная обработка азотной кислотой адекватной концентрации и соответствующей длительности дает металлическую пленку с высокой адгезией, на что указывает отсутствие медных частиц при визуальном анализе промывных вод металлизации. Присутствие медных частиц в промывных водах металлизации принимается за показатель плохой адгезии меди к волокнистой подложке: чем больше частиц, тем хуже адгезия.
Сравнительный пример 5.
В данном примере исследуется влияние фосфорной кислоты в качестве кислоты предварительной обработки.
Та же арамидная пряжа, которая использовалась в предыдущих примерах, обрабатывается водной фосфорной кислотой концентрацией около 87 мас.% в течение 60 с по методике, описанной в Примере 1. Обработанная кислотой пряжа нейтрализуется, промывается, перерабатывается в небольшие тканевые рукава и металлизируется медью согласно описанной выше методике. Прирост меди на тканевых рукавах оказывается равным только 23,3%. Тканевый рукав покрывается медью неравномерно, и электросопротивление ткани равняется более 3•108 Ом/кв. Данный пример показывает, что предварительная обработка арамидной пряжи высококонцентрированной фосфорной кислотой не способствует металлизации медью по сравнению с воздействием предварительной обработки азотной кислотой примерно равной концентрации.
Примеры 2-4 и Сравнительные примеры 6 и 7
В этих примерах исследуется влияние
хлорсульфоновой кислоты в качестве кислоты предварительной обработки.
Арамидная пряжа из предыдущих примеров перерабатывается в небольшие тканевые рукава в соответствии с вышеописанной методикой; и пряжа предварительно обрабатывается в виде этого рукава. Условия предварительной обработки рукавов перед металлизацией приводятся в Таблице 2. Данные показывают, что 2 мас. % хлорсульфоновая кислота (ClSO3H) в любом из метиленхлорида, гексана или циклогексана оказывает разное влияние на прирост меди и электросопротивление. При использовании предварительной обработки хлорсульфоновой кислотой не наблюдается медных частиц в промывных водах после металлизации.
Хлорсульфоновая кислота может быть использована в качестве эффективной предварительной обработки при такой низкой концентрации, как 1 мас.% и в любой органической жидкости, которая смешивается, но не взаимодействует с кислотой. Температура предварительной обработки обычно составляет около 20oC с активностью, увеличенной при увеличении температуры, а продолжительность предварительной обработки обычно составляет менее 60 с. При концентрации кислоты более 5 мас.% арамидные волокна могут значительно ухудшить прочностные свойства при предварительной обработке при слишком высокой температуре или для слишком большой продолжительности.
Фторсульфоновая кислота используется для предварительной обработки таким же образом и в тех же условиях, что и хлорсульфоновая кислота.
Сравнительный пример 8
В данном
примере
исследуется влияние соляной кислоты в качестве кислоты предварительной обработки.
Арамидная пряжа из предыдущих примеров перерабатывается в небольшой тканевый рукав описанным выше способом. Пряжа предварительно обрабатывается в виде этого рукава водной соляной кислотой концентрации примерно 38 мас.% в течение 60 минут при температуре около 20oC. Обработанный кислотой рукав затем нейтрализуется, промывается, сушится на воздухе и металлизируется медью по способу, описанному в Примере 1. Прирост меди на тканевом рукаве равняется только 26 мас.%, а электросопротивление равняется более 3•108 Ом/кв.
Реферат
Описывается способ неэлектролитической металлизации арамидных волокон с нанесением долговечного металлического покрытия, включающий контактирование арамидных волокон с кислотным раствором в течение не менее 2 с при температуре в интервале 10 - 100°С, промывку водой до практического удаления всей кислоты, контактирование волокон с активирующим раствором, промывку и погружение в раствор катионов наносимого металла, отличающийся тем, что в качестве кислотного раствора используют раствор, выбранный из группы, включающей 86-91 мас.%-ную водную азотную кислоту, 1-5 мас.%-ную хлорсульфоновую кислоту в органической жидкости и 1-5 мас.%-ную фторсульфоновую кислоту в органической жидкости. Технический результат - увеличение скорости металлизации с сохранением волокнистого продукта, практически сохранившего прочность и модуль упругости, металлическое покрытие которого является высокопроводящим и с высокой адгезией. 3 з.п. ф-лы, 2 табл., 2 ил.

Комментарии