Замедляющий окислительный агент для получения проводящих полимеров - RU2370838C9
Код документа: RU2370838C9
Чертежи

Описание
Настоящее изобретение относится к способу изготовления особых окислительных агентов, которые в смесях с исходными соединениями для получения проводящих полимеров обеспечивают большую длительность полимеризации, к изготавливаемым этим способом окислительным агентам, смесям, содержащим подобные особые (замедляющие) окислительные агенты, и их применению для изготовления конденсаторов с твердым электролитом и проводящих покрытий.
В течение последних десятилетий соединения класса π-сопряженных полимеров были предметом многочисленных публикаций, π-сопряженные полимеры называют также проводящими полимерами или синтетическими металлами.
Проводящие полимеры приобретают все большее хозяйственное значение, поскольку в отличие от металлов полимеры лучше пригодны для переработки, обладают меньшей массой и их свойства можно целенаправленно регулировать путем химического модифицирования. Примерами известных π-сопряженных полимеров являются полипирролы, политиофены, полианилины, полиацетилены, полифенилены и поли(п-фениленвинилены). Покрытия из проводящих полимеров находят самое разнообразное техническое применение. Обзор проводящих полимеров приведен в L.Groenendaal, F.Jonas, D.Freitag, H.Pielartzik & J.R.Reynolds, Adv. Mater. 12 (2000) 481-494.
Проводящие полимеры получают химическим окислительным или электрохимическим методом из соответствующих исходных соединений, например, при необходимости замещенных тиофенов, пирролов, анилинов и при необходимости олигомерных производных этих мономеров. В частности, широко распространен метод химической окислительной полимеризации, поскольку его можно технически просто реализовать на многочисленных подложках. С этой целью исходные соединения для получения проводящих полимеров полимеризуют в присутствии окислительного агента. При этом скорость полимеризации бывает настолько велика, что исходные соединения для получения проводящих полимеров и окислительный агент, как правило, приходится наносить на подложку один за другим. Однако при таком последовательном нанесении возникает проблема, состоящая в чрезвычайно большой сложности установления стехиометрического соотношения между исходными соединениями для получения проводящих полимеров и окислительным агентом. Следствием этого является неполное протекание реакции образования полимера, неполное использование исходных соединений и снижение качества проводящего покрытия и его проводимости.
Кроме того, при последовательном нанесении исходных соединений и окислительного агента возрастает количество необходимых технологических стадий, поэтому реализация последовательной технологии требует чрезвычайно больших производственных расходов. Учитывая это обстоятельство, возникло намерение совместного использования предназначенных для получения проводящих полимеров исходных соединений и окислительного агента в виде смесей с точно заданным составом компонентов.
Полимеризация в смесях окислительного агента с предназначенными для получения проводящих полимеров исходными соединениями протекает с достаточно низкими скоростями, позволяющими осуществлять ее в промышленном масштабе, лишь при пониженных температурах. Так, например, в соответствии с патентом США US 5455736 для достаточного замедления полимеризации разбавленную смесь пиррола с окислительным агентом охлаждают до низких температур. Однако использование низких температур, во-первых, является технически крайне дорогостоящим, а, во-вторых, приводит к снижению растворимости окислительного агента и чрезвычайно сильному повышению вязкости его растворов. Другой недостаток использования низких температур состоит в том, что в охлажденные растворы из окружающей среды попадает влага, что оказывает отрицательное воздействие на качество получаемых из таких растворов проводящих полимеров.
Из европейской заявки на патент ЕР-А 339340 известно о химической окислительной полимеризации 3,4-дизамещенных тиофенов. При надлежащем выборе окислительного агента указанные тиофены могут быть использованы для получения проводящих покрытий в присутствии окислительного агента также и в растворе. Однако реакция в этом случае начинается уже через несколько минут.
Из европейской заявки на патент ЕР-А 615256 известно о возможности замедления полимеризации, протекающей в смесях окислительного агента с исходными соединениями для получения проводящих полимеров, путем добавления нелетучего основания, например имидазола. Благодаря введению указанной добавки полимеризация может быть блокирована на несколько часов. Однако добавка остается в проводящем покрытии и в дальнейшем может помешать его надлежащему функционированию.
Согласно патенту США US 6001281 полимеризацию замедляют благодаря использованию двух, обладающих разными точками кипения растворителей. Более летучий растворитель подбирают таким образом, чтобы он был способен образовывать слабый комплекс с используемым в качестве окислительного агента железом(III) и, следовательно, замедлять полимеризацию. Растворитель, обладающий более высокой точкой кипения, напротив, не образует комплекса с железом(III). Для осуществления полимеризации сначала испаряют более летучий растворитель, после чего реакция протекает с высокой скоростью. Существенным недостатком предлагаемого в данном изобретении метода является необходимость сильного разбавления реакционного раствора другим растворителем. Кроме того, использование в промышленном масштабе таких растворителей, как тетрагидрофуран, является нежелательным.
Таким образом, существует потребность в окислительных агентах, которые можно было бы совместно с исходными соединениями для получения проводящих полимеров использовать для полимеризации этих соединений при удобных с точки зрения ее технического осуществления температурах и на достаточно длительное время блокировать реализуемый в промышленном масштабе процесс полимеризации без необходимости нарушения других трудоемких технологических стадий.
Учитывая вышеизложенное, в основу настоящего изобретения была положена задача подобрать и изготовить окислительные агенты, которые были бы пригодны для химической окислительной полимеризации предназначенных для получения проводящих полимеров исходных соединений, блокировали бы полимеризацию на достаточно длительное время и позволяли бы изготавливать проводящие покрытия, пригодные, например, для конденсаторов с твердым электролитом или иных сфер применения.
Другая задача настоящего изобретения состояла в том, чтобы найти пригодные окислительные агенты, которые, кроме того, были бы стабильны при хранении.
Неожиданно было обнаружено, что указанным требованиям удовлетворяют окислительные агенты, изготовленные путем обработки соли металла и органической кислоты или неорганической кислоты с органическими остатками ионитом.
Таким образом, объектом настоящего изобретения является способ изготовления окислительного агента, предназначенного для получения проводящих полимеров, отличающийся тем, что соль металла и органической кислоты или неорганической кислоты с органическими остатками обрабатывают ионитом.
Согласно настоящему изобретению возможны любые комбинации приведенных ниже общих или указанных в предпочтительных вариантах формулировок, заместителей, параметров и обозначений, а также соответствующих вариантов или предпочтительных вариантов осуществления изобретения.
В качестве ионитов могут использоваться неорганические или органические иониты, однако предпочтительными являются органические иониты.
Неорганическими анионитами являются, например, цеолиты, монтмориллониты, аттапульгиты, бентониты и другие алюмосиликаты, или кислые соли поливалентных ионов металлов, такие как фосфат циркония, вольфрамат титана, гексацианоферрат(II) никеля.
Примерами органических анионитов являются поликонденсаты, получаемые, например, из фенола и формальдегида, или полимеризаты, получаемые, например, путем сополимеризации стирола, акрилатов или метакрилатов с дивинилбензолом и снабжаемые непосредственно после получения функциональными группами. Однако возможно также использование других полимеров с соответствующими функциональными группами, например полимеров природного происхождения, таких как целлюлозы, декстраны и арагозы.
Вышеуказанные иониты приведены лишь в качестве примера и не ограничивают объема настоящего изобретения.
Иониты можно использовать в известных специалистам формах применения, например в виде шариков, гранул, порошковой смолы, введенных в ткани или волокна размолотых продуктов, в виде бумаг, покрытий или других твердых тел, в виде ионообменных мембран, жидких органических ионитов или при необходимости также магнитных ионитов. Иониты могут быть макропористыми, микропористыми или гелеобразными. Предпочтительным является использование макропористых ионитов.
В качестве ионитов предпочтительно используют аниониты. Аниониты содержат присоединенные к ним функциональные основные группы, например первичные, вторичные или третичные аминогруппы, соответственно группы четвертичного аммония. Иониты могут обладать разной основностью, которая зависит от вида и сочетания функциональных групп. Так, например, сильно щелочные иониты обычно содержат группы четвертичного аммония, в то время как слабо щелочные иониты зачастую содержат первичные, вторичные и/или третичные аминогруппы, обладающие менее выраженным щелочным характером. Однако известны также любые смешанные формы сильнощелочных и слабощелочных ионитов. Согласно настоящему изобретению пригодными предпочтительно являются слабощелочные аниониты. Они могут содержать, например, первичные, вторичные и/или третичные аминогруппы при необходимости в сочетании с группами четвертичного аммония. Особенно предпочтительными являются указанные слабощелочные иониты, содержащие в качестве функциональных групп преимущественно или исключительно третичные аминогруппы.
Иониты, а также их изготовление известны специалистам и описаны в соответствующей специальной литературе, например, в Ullmanns Encyclopädie der technischen Chemie (изд-во Chemie, Weinheim), том 13, 4-е издание, с.281-308). Однако для осуществления предлагаемого в изобретении способа пригодны также любые иониты, которые могут быть изготовлены современными методами и обладают указанными выше свойствами.
Примерами пригодных ионитов являются функционализованные третичными аминогруппами макропористые полимеры на основе стирола и дивинилбензола, выпускаемые, например, фирмой Bayer AG (Леверкузен) под торговым наименованием Lewatit®.
Согласно предлагаемому в изобретении способу иониты можно использовать без предварительной обработки. Однако возможна также предварительная обработка ионитов, например, кислотой, в частности серной кислотой, или основанием, в частности раствором едкого натра или едкого кали, например, с целью регенерации перед использованием. Используемые согласно изобретению иониты могут быть подвергнуты подобной регенерации также в том случае, если вследствие реализации предлагаемого в изобретении способа их емкость оказалась исчерпана, то есть иониты зарядились до такой степени, что уже не обладают способностью к достаточному для осуществления предлагаемого в изобретении способа ионному обмену. Благодаря регенерации иониты можно повторно использовать для осуществления предлагаемого в изобретении способа.
Обработку солей металлов ионитами осуществляют предпочтительно в присутствии одного или нескольких разных растворителей. Подобную обработку можно выполнять непрерывным или периодическим методом, например путем смешивания, перемешивания с помощью мешалки или взбалтывания с последующим разделением. В особенно предпочтительном варианте обработку солей металлов выполняют непрерывным методом. С этой целью соль металла, например, в виде раствора пропускают через содержащую ионит колонну. Однако соль металла, растворитель и ионит можно также одновременно ввести в емкость и хранить в ней в течение определенного времени, составляющего, например, от одной минуты до 72 часов. После этого ионит может быть отделен от окислительного агента, например, с помощью фильтра, мембраны или центрифуги.
В зависимости от используемого растворителя и термостойкости ионита предлагаемый в изобретении способ может быть осуществлен при температуре, например, от -20 до 120°С. Предпочтительными являются температуры, при которых способ можно легко и рентабельно реализовать в крупном масштабе, например температуры от 10 до 40°С, причем особенно предпочтительной является комнатная температура.
Количество добавляемого ионита зависит от его емкости и длительности контактирования соли металла с ионитом. При необходимости время контактирования соли металла с ионитом может быть определено в предварительных опытах. Количество ионита целесообразно подбирать таким образом, чтобы изготовленный с его использованием предлагаемый в изобретении окислительный агент обеспечивал достаточно низкую скорость полимеризации. При добавлении слишком малых количеств ионита может наступить исчерпание его емкости, прежде чем соль металла будет подвергнута достаточно полной обработке; слишком кратковременное контактирование соли металла с ионитом может привести к неполной обработке соли металла несмотря на достаточную емкость ионита. Слишком высокая емкость ионита и/или слишком длительное контактирование окислительного агента с ним могут привести к изготовлению окислительного агента, почти полностью блокирующего осуществляемую при надлежащей температуре полимеризацию. Пригодное количество добавляемого ионита при необходимости может быть определено в предварительных опытах.
Используемые иониты могут быть влагосодержащими или безводными. Согласно изобретению под влагосодержащим ионитом подразумевают прежде всего такой ионит, содержание влаги в котором составляет 1 мас.% и более. В предпочтительных вариантах осуществления изобретения используют имеющиеся в продаже иониты со стандартным влагосодержанием, составляющим, например, от 30 до 70 мас.%. Влагосодержание ионита перед обработкой соли металла при необходимости можно уменьшить, например, путем его промывки растворителем или сушки. Этот метод является особенно предпочтительным, если желательно располагать раствором окислительного агента с низким влагосодержанием.
Неожиданно было обнаружено, что растворы предлагаемых в изобретении окислительных агентов с низким влагосодержанием обладают стабильностью в обычных условиях хранения и транспортировки. Под обычными условиями хранения и транспортировки подразумеваются, например, давление и температура окружающей среды при транспортировке и хранении. Температура окружающей среды прежде всего зависит от географического положения и времени года и как правило не превышает, например, 30°С. Однако она может достигать также 50°С и более высоких значений.
Таким образом, в предпочтительном варианте осуществления изобретения используют иониты со столь низким влагосодержанием, чтобы влагосодержание раствора предлагаемого в изобретении окислительного агента после обработки ионитом составляло от 0 до 10 мас.%, предпочтительно от 0 до 5 мас.%, особенно предпочтительно от 0 до 2 мас.% соответственно в расчете на общую массу раствора. С этой целью перед обработкой окислительного агента ионитом содержание влаги в обладающих высоким влагосодержанием ионитах снижают, например, путем ступенчатой или непрерывной промывки безводным раствором или путем тепловой или вакуумной сушки. В качестве растворителя для промывки ионитов предпочтительно используют такой растворитель, в котором способен растворяться окислительный агент. Однако возможно также использование другого, например, более экономичного растворителя. В случае использования ионитов с высоким влагосодержанием содержание влаги в предлагаемых в изобретении окислительных агентах после обработки таким ионитом можно дополнительно понизить, например, путем сушки и последующего растворения окислительного агента в безводном растворителе или благодаря использованию влагопоглощающих средств, например молекулярных сит.
Такие, обладающие низким влагосодержание растворы предлагаемых в изобретении окислительных агентов стабильны в обычных условиях хранения и транспортировки, то есть они не образуют осадка в течение промежутка времени, достигающего нескольких месяцев. В отличие от подобных растворов растворы предлагаемых в изобретении окислительных агентов с повышенным влагосодержанием в аналогичных условиях хранения и транспортировки образуют осадок по истечении промежутка времени, который при известных обстоятельствах не превышает нескольких часов или дней. Однако для повышения стабильности при хранении растворов окислительных агентов с повышенным влагосодержанием их можно охладить до 10°С, предпочтительно до 6°С или более низких температур.
Следовательно, преимущество обладающих низким влагосодержанием растворов предлагаемых в изобретении окислительных агентов по сравнению с аналогичными растворами, обладающими повышенным влагосодержанием, состоит в том, что их транспортировка и/или хранение не требуют специального охлаждения.
Растворы предлагаемых в изобретении окислительных агентов предпочтительно содержат от 1 до 80 мас.%, особенно предпочтительно от 10 до 60 мас.%, еще более предпочтительно от 15 до 50 мас.% предлагаемого в изобретении окислительного агента.
В качестве солей металлов могут использоваться любые известные специалистам соли металлов, пригодные в качестве окислительных агентов для окислительной полимеризации тиофенов, анилинов или пирролов.
Пригодными солями металлов являются соли металлов главной или побочной группы периодической системы элементов Менделеева, причем последние в дальнейшем называют также солями переходных металлов. Предпочтительными являются соли переходных металлов. Пригодными солями переходных металлов являются, в частности, соли этих металлов и неорганической или органической кислоты, или неорганической кислоты с органическим остатком, например, соли железа(III), меди(II), xpoма(VI), церия(IV), марганца(IV), марганца(VII), рутения(III) и цинка(II).
Предпочтительными солями переходных металлов являются соли железа(III). Соли железа(III), например соли железа(III) и неорганических кислот, в частности галогениды железа(III) (например, FеСl3), или соли железа(III) и других неорганических кислот, в частности Fe(ClO4)3 или Fе2(SO4)3, и соли железа(III) и органических кислот или неорганических кислот с органическими остатками зачастую бывают недороги, легко доступны и могут быть легко переработаны.
Примерами солей железа(III) и неорганических кислот с органическими остатками являются соли кислых эфиров серной кислоты и алканолов с 1-20 атомами углерода, например лаурилсульфат железа(III).
Особенно предпочтительными солями переходных металлов являются соответствующие соли органических кислот, прежде всего соли железа(III) органических кислот.
Примерами солей железа(III) органических кислот являются соли железа(III) алкансульфокислот с 1-20 атомами углерода, таких как метановая, этановая, пропановая, бутановая или более высокомолекулярная кислота, например додекансульфокислота, алифатических перфторсульфокислот, таких как трифторметансульфокислота, перфторбутансульфокислота или перфтороктансульфокислота, алифатических карбоновых кислот с 1-20 атомами углерода, таких как 2-этилгексилкарбоновая кислота, алифатических перфторкарбоновых кислот, таких как трифторуксусная или перфтороктановая кислота, и ароматических сульфокислот, при необходимости замещенных алкильными группами с 1-20 атомами углерода, таких как бензолсульфокислота, о-толуолсульфокислота, п-толуолсульфокислота или додецилбензолсульфокислота, и циклоалкансульфокислот, таких как камфорная сульфокислота.
Могут использоваться также любые смеси указанных солей железа(III) и органических кислот.
Большим преимуществом использования солей железа(III) и органических кислот и неорганических кислот с органическими остатками является то, что такие соли не обладают коррозионным действием.
Еще более предпочтительными солями металлов являются п-толуол-сульфонат железа(III), о-толуолсульфонат железа(III) или смесь п-толуосульфоната железа(III) с о-толуолсульфонатом железа(III).
Кроме того, пригодными солями металлов являются пероксосоединения, например, пероксодисульфаты (персульфаты), в частности пероксодисульфаты аммония и щелочных металлов, такие как пероксодисульфат натрия и пероксодисульфат калия, или пербораты щелочных металлов, а также оксиды переходных металлов, например пиролюзит (оксид марганца(V)) или оксид церия(IV).
В качестве растворителей используют, в частности, следующие, инертные в реакционных условиях органические растворители: алифатические спирты, такие как метанол, этанол, изопропанол и бутанол, алифатические кетоны, такие как ацетон и метилэтилкетон, сложные эфиры алифатических карбоновых кислот, такие как этиловый эфир уксусной кислоты и бутиловый эфир уксусной кислоты, ароматические углеводороды, такие как толуол и ксилол, алифатические углеводороды, такие как гексан, гептан и циклогексан, хлорсодержащие углеводороды, такие как дихлорметан и дихлорэтан, алифатические нитрилы, такие как ацетонитрил, алифатические сульфоксиды и сульфоны, такие как диметилсульфоксид и сульфолан, амиды алифатических карбоновых кислот, такие как метилацетамид, диметилацетамид и диметилформамид, алифатические и аралифатические простые эфиры, такие как диэтиловый эфир и анизол. Кроме того, в качестве растворителя может использоваться также вода или смеси воды с указанными выше органическими растворителями. Выбранные из приведенного выше перечня растворители, вступающие в нежелательную для предлагаемого в изобретении способа реакцию с ионитом, добавляют только после обработки соли ионитом, вводя их после предварительного удаления предыдущего растворителя или в дополнение к этому растворителю.
В качестве растворителя предпочтительно используют один или несколько спиртов, воду или смесь одного или нескольких спиртов с водой. Особенно предпочтительными спиртами являются бутанол, этанол и метанол.
Изготовленный предлагаемым в изобретении способом окислительный агент после обработки ионитом может быть отделен от растворителя и при необходимости вновь растворен в том же или другом растворителе, выбранном из приведенного выше перечня.
Кроме того, объектом настоящего изобретения являются окислительные агенты или растворы окислительных агентов, которые могут быть изготовлены описанным выше предлагаемым в изобретении способом. При этом все предпочтительные варианты осуществления предлагаемого в изобретении способа и любые их комбинации относятся и к изготавливаемым этим способом окислительным агентам или их растворам. Предпочтительным объектом изобретения являются окислительные агенты или растворы окислительных агентов, изготовленные описанным выше, предлагаемым в изобретении способом.
Предлагаемые в изобретении окислительные агенты по сравнению с окислительными агентами, необработанными ионитами, при аналогичной концентрации и аналогичной реакционной температуре обеспечивают замедление, соответственно затормаживание полимеризации в реакционных смесях, состоящих из исходных соединений для получения проводящих полимеров и предлагаемого в изобретении окислительного агента. Поэтому в дальнейшем их называют также замедляющими окислительными агентами.
Скорость полимеризации в реакционных смесях при необходимости может быть дополнительно понижена (а, следовательно, тормозящий, соответственно замедляющий эффект усилен) путем разбавления и/или охлаждения этих смесей.
Кроме того, в присутствии предлагаемых в изобретении окислительных агентов проводящие покрытия при необходимости могут быть изготовлены таким же образом, как и в присутствии необработанных ионитами окислительных агентов.
Замедляющий, соответственно тормозящий эффект предлагаемых в изобретении окислительных агентов можно наблюдать простым, например, чисто визуальным методом. Для определения замедляющего эффекта можно, например, измерить время до образования первых, визуально обнаруживаемых частиц полимера. Время до появления в реакционных смесях визуально обнаруживаемых полимерных частиц предпочтительно составляет более часа, особенно предпочтительно более 10 часов и еще более предпочтительно более 20 часов.
Таким образом, объектом настоящего изобретения является применение окислительных агентов, изготовленных предлагаемым в изобретении способом, в качестве замедляющих окислительных агентов при окислительной полимеризации предназначенных для получения проводящих полимеров исходных соединений.
Под полимерами в соответствии с настоящим изобретением подразумевают любые соединения, содержащие более одной повторяющейся структурной единицы.
При этом под проводящими полимерами подразумевают соединения класса π-сопряженных полимеров, которые после окисления или восстановления обладают электрической проводимостью. В соответствии с настоящим изобретением предпочтительными проводящими полимерами следует считать π-сопряженные полимеры, которые обнаруживают электрическую проводимость после окисления. Примерами таких полимеров являются при необходимости замещенные политиофены, полипирролы и полианилины. Предпочтительными проводящими полимерами в соответствии с изобретением являются при необходимости замещенные политиофены, прежде всего при необходимости замещенные поли(3,4-этилендиокситиофены).
Под исходными соединениями для получения проводящих полимеров, ниже называемыми также просто исходными соединениями, подразумевают соответствующие таким полимерам мономеры или производные этих мономеров. Возможно также использование смеси разных исходных соединений. Пригодными мономерными исходными соединениями являются, например, при необходимости замещенные тиофены, пирролы или анилины, предпочтительно при необходимости замещенные тиофены, особенно предпочтительно при необходимости замещенные 3,4-алкилендиокситиофены.
Примером замещенных 3,4-алкилендиокситиофенов являются соединения общей формулы (I)
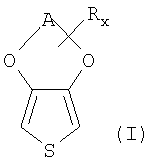
в которой
А означает при необходимости замещенный алкиленовый остаток с 1-5 атомами углерода, предпочтительно при необходимости замещенный алкиленовый остаток с 2-3 атомами углерода,
R означает линейный или разветвленный, при необходимости замещенный алкильный остаток с 1-18 атомами углерода, предпочтительно линейный или разветвленный, при необходимости замещенный алкильный остаток с 1-14 атомами углерода, при необходимости замещенный циклоалкильный остаток с 5-12 атомами углерода, при необходимости замещенный арильный остаток с 6-14 атомами углерода, при необходимости замещенный аралкильный остаток с 7-18 атомами углерода, при необходимости замещенный гидроксиалкильный остаток с 1-4 атомами углерода или гидроксильный остаток,
х означает целое число от 0 до 8, предпочтительно от 0 до 6, особенно предпочтительно 0 или 1,
причем в случае, если к остатку А присоединено несколько остатков R, они могут быть одинаковыми или разными.
Общую формулу (I) следует понимать так, что к алкиленовому остатку А может быть присоединено х заместителей R.
Еще более предпочтительными мономерными исходными соединениями являются при необходимости замещенные 3,4-этилендиокситиофены.
Примерами замещенных 3,4-этилендиокситиофенов являются соединения общей формулы (Ia)
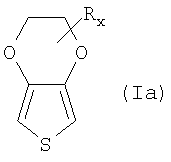
причем R и х такие, как в общей формуле (I).
Согласно изобретению под производными указанных мономерных исходных соединений подразумеваются, например, димеры или тримеры этих мономерных исходных соединений. Возможными производными указанных мономерных исходных соединений могут быть более высокомолекулярные соединения, то есть тетрамеры, пентамеры и так далее.
Примерами производных замещенных 3,4-алкилендиокситиофенов являются соединения общей формулы (II)
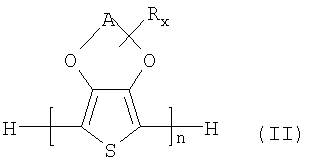
в которой
n означает целое число от 2 до 20, предпочтительно от 2 до 6, особенно предпочтительно 2 или 3, и
A, R и х такие, как в общей формуле (I).
Производные замещенных 3,4-алкилендиокситиофенов могут состоять как из одинаковых, так и из разных мономерных единиц и их можно использовать как в чистом виде, так и в виде смесей друг с другом и/или с мономерными исходными соединениями. Согласно изобретению под исходными соединениями подразумевают также окисленные или восстановленные формы этих исходных соединений, если при их полимеризации образуются проводящие полимеры, аналогичные тем, которые образуются при полимеризации указанных выше исходных соединений.
Заместителями исходных соединений, в частности тиофенов, предпочтительно 3,4-алкилендиокситиофенов, являются остатки R, аналогичные указанным для общей формулы (I).
Согласно изобретению алкиленовыми остатками с 1-5 атомами углерода А являются метилен, этилен, н-пропилен, н-бутилен или н-пентилен. Согласно изобретению алкил с 1-18 атомами углерода означает линейный или разветвленный алкильный остаток с 1-18 атомами углерода, например метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1-этилпропил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, н-гексил, н-гептил, н-октил, 2-этилгексил, н-нонил, н-децил, н-ундецил, н-додецил, н-тридецил, н-тетрадецил, н-гексадецил или н-октадецил, циклоалкил с 5-12 атомами углерода означает циклоалкильный остаток с 5-12 атомами углерода, например циклопентил, циклогексил, циклогептил, циклооктил, циклононил или циклодецил, арил с 6-14 атомами углерода означает арильный остаток с 6-14 атомами углерода, например фенил, о-толил, м-толил, п-толил, 2,3-, 2,4-, 2,5-, 2,6-, 3,4- или 3,5-ксилил, мезитил или нафтил, и аралкил с 7-18 атомами углерода означает аралкильный остаток с 7-18 атомами углерода, например бензил. Приведенный выше перечень заместителей служит для пояснения изобретения, и его не следует рассматривать как окончательный.
Заместителями остатка R могут быть многочисленные органические группы, например алкильные, циклоалкильные, арильные, галоидные, гидроксильные, эфирные, тиоэфирные, дисульфидные, сульфоксидные, сульфокислотные, сульфонатные, аминогруппы, альдегидные группы, кетогруппы, группы сложных эфиров карбоновой кислоты, карбонатные, карбоксилатные, цианогруппы, алкилсилановые, алкоксисилановые, а также карбоксиламидные группы.
Методы получения предназначенных для синтеза проводящих полимеров мономерных исходных соединений, а также их производных известны специалистам и приведены, например, в статье L.Groenendaal, F.Jonas, D.Freitag, H.Pielartzik & J.R.Reynolds, Adv. Mater. 12 (2000) 481-494 и цитированной в ней литературе.
Преимущество совместного нанесения окислительных агентов и предназначенных для получения проводящих полимеров исходных соединений наряду с технически удобными температурами состоит в значительном сокращении количества технологических операций. Другим его преимуществом является возможность установления заданного стехиометрического соотношения между реакционными партнерами. Так, например, степень превращения исходных соединений в полимер при необходимости может даже достигать почти 100%.
Кроме того, растворы или смеси изготовленных предлагаемым в изобретении способом окислительных агентов и исходных соединений особенно пригодны для создания проводящих покрытий на поверхностях пористых или гладких подложек. Благодаря равномерному распределению окислительных агентов и исходных соединений в соответствующих смесях в результате полимеризации образуются также однородные, то есть плотные (без пор или с малым содержанием пор) полимерные покрытия. В отличие от этого при последовательном нанесении окислительных агентов и исходных соединений вследствие их местного избытка или недостатка образуются пористые полимерные покрытия. В связи с высокой плотностью проводящих покрытий, изготовленных из предлагаемых в изобретении смесей, они обладают особой однородностью и высокой проводимостью.
Кроме того, благодаря использованию предлагаемого в изобретении способа доступны растворы или смеси как таковые, то есть содержащие необработанные ионитом окислительные агенты, которые пригодны для переработки в течение гораздо более длительного времени. Благодаря этому впервые предоставляется возможность использование таких смесей, соответственно растворов, в непрерывных промышленных технологических процессах.
Другим объектом настоящего изобретения являются смеси, содержащие исходные соединения для получения проводящих полимеров и один или несколько предлагаемых в изобретении окислительных агентов, а также при необходимости один или несколько растворителей, отличающиеся тем, что образование полимеров в этих смесях замедляется по сравнению со смесями, содержащими необработанные окислительные агенты.
В данном случае справедливы предпочтительные варианты, определения и примеры исходных соединений, предлагаемых в изобретении окислительных агентов и растворителей, аналогичные указанным выше.
Предлагаемые в изобретении смеси могут быть гомогенными или гетерогенными, а также однофазными или двухфазными. Под предлагаемыми в изобретении смесями предпочтительно подразумевают растворы.
Окислительные агенты и исходные соединения для получения проводящих полимеров могут быть смешаны друг с другом в виде твердых веществ и/или жидкостей. Однако предпочтительным является добавление к соответствующим смесям одного или нескольких растворителей. Пригодными, в частности, являются указанные выше растворители. Смеси могут быть изготовлены также непосредственно на подлежащей облицовке поверхности, например на оксидном слое металла или на поверхности подложки. Для этого окислительные агенты и предназначенные для получения проводящих полимеров исходные соединения наносят предпочтительно в виде растворов на подлежащие облицовке поверхности. Смесь нанесенных окислительных агентов и исходных соединений образуется позже вследствие происходящего на подлежащей облицовке поверхности перемешивания, соответственно после при необходимости осуществляемого частичного или полного испарения растворителей, причем перемешивание обусловлено диффузией окислительных агентов к исходным соединениям через соответствующую границу раздела фаз.
Предлагаемые в изобретении смеси могут содержать воду. Вода может попадать в смеси, например, вместе с предлагаемым в изобретении окислительным агентом или его раствором и/или может быть дополнительно введена в предлагаемые в изобретении смеси. Благодаря добавлению воды можно замедлить образование полимеров, протекающее в предлагаемых в изобретении смесях, то есть повысить жизнеспособность смесей. Дополнительную воду предпочтительно вводят в случае низкого влагосодержания используемых предлагаемых в изобретении окислительных агентов или их растворов. Предпочтительно добавляют от 1 до 100 мас.%, особенно предпочтительно от 1 до 60 мас.%, еще более предпочтительно от 1 до 40 мас.% воды в расчете на массу предлагаемого в изобретении окислительного агента.
Проводящие полимеры, получаемые в присутствии предлагаемых в изобретении окислительных агентов, могут быть нейтральными или катионными, однако они предпочтительно являются катионными полимерами. Определение «катионные» в данном случае относится только к тем зарядам, которыми располагает главная полимерная цепь. Эти положительные заряды должны быть уравновешены противоионами, которые в соответствии с особыми вариантами осуществления изобретения, предусматривающими замещение повторяющихся структурных единиц анионными группами, например сульфонатными или карбоксилатными группами, могут быть присоединены к полимерной цепи ковалентными связями.
При этом положительные заряды полимерной цепи могут быть уравновешены ковалентно присоединенными анионными группами частично или полностью. Если количество ковалентно присоединенных анионных групп превышает количество положительных зарядов, то несмотря на общий отрицательный заряд подобных полимеров, они согласно настоящему изобретению все равно считаются катионными полимерами, поскольку определяющее значение в данном случае имеет положительная заряженность главной полимерной цепи. Положительные заряды в общем случае не указаны в соответствующих химических формулах, поскольку точное количество и положение этих зарядов не подлежат надежному определению. Однако количество положительных зарядов составляет по меньшей мере 1 и не превышает р, причем р означает общее количество всех содержащихся в полимере, одинаковых или разных повторяющихся структурных единиц.
Для уравновешивания положительной зараженности проводящего полимера, если она уже не скомпенсирована при необходимости ковалентно присоединенными сульфонатными или карбоксилатными заместителями, то есть отрицательно заряженными остатками, проводящий полимер должен быть снабжен используемыми в качестве соответствующих противоионов анионами.
Следовательно, к смесям могут быть добавлены противоионы. Противоионами могут служить мономерные или полимерные анионы, причем последние в дальнейшем называют полианионами.
В качестве полианионов предпочтительно используют анионы полимерных карбоновых кислот, таких как полиакриловые, полиметакриловые или полималеиновые кислоты, или анионы полимерных сульфокислот, таких как полистиролсульфокислоты и поливинилсульфокислоты. Указанные поликарбоновые кислоты и полисульфокислоты могут являться также сополимерами винилкарбоновых кислот и винилсульфокислот с другими полимеризуемыми мономерами, например сложными эфирами акриловой кислоты и стиролом.
Особенно предпочтительным противоионом является анион полистиролсульфокислоты.
Молекулярная масса образующих полианионы поликислот составляет предпочтительно от 1000 до 2000000, особенно предпочтительно от 2000 до 500000. Поликислоты и их щелочные соли, например, полистиролсульфокислоты и полиакриловые кислоты, являются коммерчески доступными продуктами или могут быть синтезированы известными методами (смотри, например, Houben Weyl, Methoden der organischen Chemie, том Е 20, Makromolekulare Stoffe, часть 2, (1987), с.1141 и следующие).
В качестве мономерных анионов используют, например, анионы алкансульфокислот с 1-20 атомами углерода, таких как метансульфокислота, этансульфокислота, пропансульфокислота, бутансульфокислота или более высокомолекулярные сульфокислоты, например додекансульфокислота, анионы алифатических перфторсульфокислот, таких как трифторметансульфокислота, перфторбутансульфокислота или перфтороктансульфокислота, анионы карбоновых кислот с 1-20 атомами углерода, таких как 2-этилгексилкарбоновая кислота, анионы алифатических перфторкарбоновых кислот, таких как трифторуксусная или перфтороктановая кислота, анионы ароматических сульфокислот, при необходимости замещенных алкильными группами с 1-20 атомами углерода, таких как бензолсульфокислота, о-толуолсульфокислота, п-толуолсульфокислота или додецилбензолсульфокислота, анионы циклоалкансульфокислот, таких как камфорная сульфокислота, или тетрафторборатов, гескафторфосфатов, перхлоратов, гексафторантимонатов, гескафторарсенатов или гексахлорантимонатов.
Предпочтительными являются анионы п-толуолсульфокислоты, метансульфокислоты или камфорной сульфокислоты.
Противоионы вводят в состав смесей, например, в виде соответствующих солей щелочных металлов или свободных кислот.
Присутствующие в используемом окислительном агенте анионы предпочтительно выполняют функцию противоионов, в связи с чем отсутствует настоятельная необходимость в добавлении дополнительных противоионов.
Кроме того, к предлагаемым в изобретении смесям могут быть добавлены другие компоненты, например, одно или несколько растворимых в органических растворителях органических связующих, таких как поливинилацетат, поликарбонат, поливинилбутират, сложные эфиры полиакриловой кислоты, сложные эфиры полиметакриловой кислоты, полистирол, полиакрилонитрил, поливинилхлорид, полибутадиен, полиизопрен, простые полиэфиры, сложные полиэфиры, силиконы, сополимеры пиррола со сложными эфирами акриловой кислоты, сополимеры винилацетата со сложными эфирами акриловой кислоты и сополимеры этилена с винилацетатом, или водорастворимые связующие, такие как поливиниловые спирты, сшивающие агенты, такие как полиуретаны, соответственно полиуретановые дисперсии, полиакрилаты, полиолефиновые дисперсии, эпоксисиланы, такие как 3-глицидоксипропилтриалкоксисилан, и добавки, например поверхностно-активные вещества. Кроме того, для повышения стойкости покрытий к царапанью могут быть добавлены гидролизаты алкоксисиланов, например, на основе тетраэтоксисилана.
Для окислительной полимеризации предназначенных для получения проводящих полимеров исходных соединений теоретически необходимо использовать 2,25 эквивалента окислительного агента в расчете на моль тиофена (смотри, например, J. Polym. Sci., Part A Polymer Chemistry, Vol.26, с.1287 (1988)). Возможно также использование меньшего или большего числа эквивалентов окислительного агента.
Смеси предпочтительно содержат от 1 до 30 мас.% исходных соединений для получения проводящих полимеров и от 0 до 50 мас.% связующих, сшивающих агентов и/или добавок в расчете на общую массу смеси.
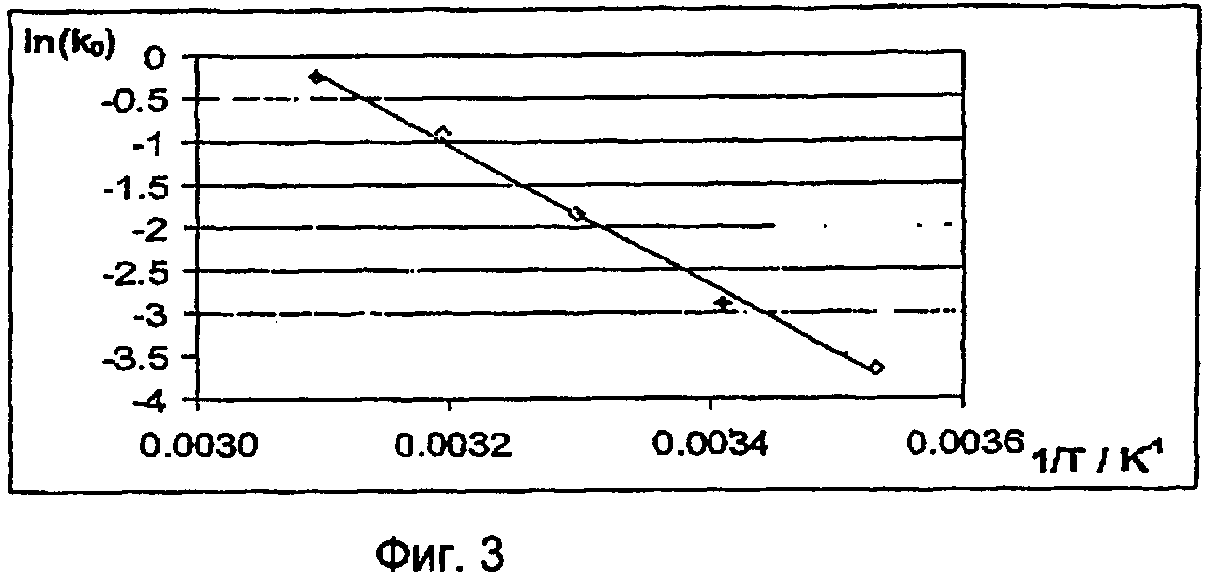
Скорость полимеризации, протекающей в смесях, содержащих исходные соединения для получения проводящих полимеров и по меньшей мере один окислительный агент, наряду с концентрацией эдуктов определяется константами скорости полимеризации. Константа скорости полимеризации k зависит от температуры в соответствии с уравнением Аррениуса:
k=ν·e-Ea/RT,
в котором ν - предэкспоненциальный множитель, Еа - энергия активации в Дж/моль, R - газовая постоянная, составляющая 8,3145 Дж·К-1·моль-1, и Т - температура в градусах Кельвина.
Энергия активации является показателем зависящей от температуры и концентрации скорости полимеризации. Чем выше энергия активации, тем медленнее протекает полимеризация и тем выше жизнеспособность смесей.
Другим объектом настоящего изобретения являются смеси, содержащие исходные соединения для получения проводящих полимеров и по меньшей мере один окислительный агент, отличающиеся тем, что энергия активации полимеризации исходных соединений составляет 75 кДж/моль или более, предпочтительно 85 кДж/моль или более, особенно предпочтительно 95 кДж/моль или более. Недостатком слишком высоких значений энергии активации может быть то, что полимеризация в этом случае начинается лишь при очень высоких температурах, которые невыгодно использовать для получения проводящих полимеров. Поэтому энергия активации предпочтительно составляет менее 200 кДж/моль, особенно предпочтительно менее 150 кДж/моль и еще более предпочтительно менее 130 кДж/моль.
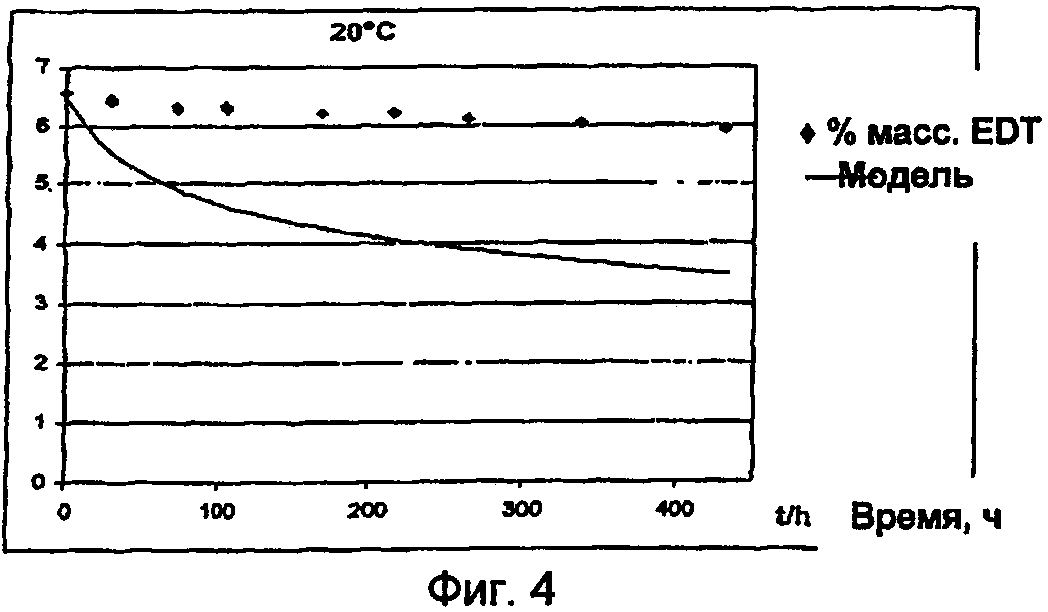
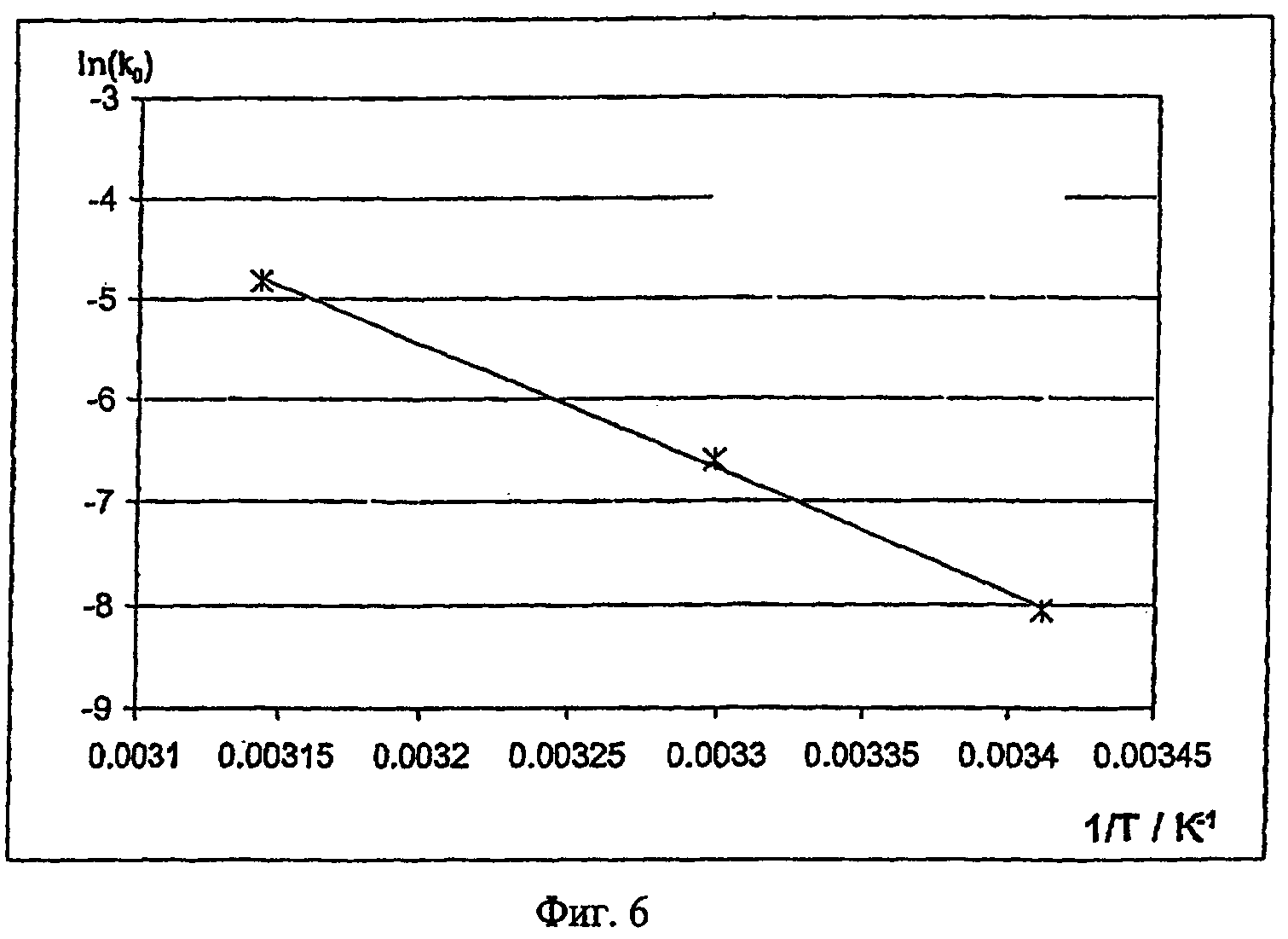
Для расчета энергии активации необходимо экспериментально определить характер изменения концентрации эдуктов (исходных соединений, окислительного агента) и продуктов полимеризации. В результате сопоставления кинетической модели отдельных реакционных стадий с характером изменения концентрации эдуктов при разных температурах получают константы скорости полимеризации при этих температурах. По зависимости константы скорости k от температуры с помощью приведенного выше уравнения можно рассчитать энергию активации полимеризации.
Методы определения энергии активации и выполнения соответствующих кинетических измерений известны специалистам и описаны, например, в F.G.Helfferich "Kinetics of homogeneous multistep reactions" (том 38 серии "Comprehensive Chemical Kinetices", изданный R.R.Compton и G.Hancock, Elsevier, Амстердам 2001). В приведенном ниже примере 11 описано определение энергии активации полимеризации, протекающей в смесях, содержащих в качестве исходного соединения 3,4-этилендиокситиофен, а в качестве окислительного агента п-толуолсульфонат железа(III).
Кроме того, предлагаемые в изобретении смеси могут содержать растворители, противоионы, связующие и/или сшивающие агенты.
В данном случае справедливы предпочтительные варианты, определения и примеры исходных соединений, противоионов, связующих, сшивающих агентов и растворителей, аналогичные указанным выше.
В качестве окислительного агента предпочтительно пригодна соль металла и органической кислоты или неорганической кислоты с органическими остатками.
В качестве таких солей могут использоваться любые известные специалистам соли металлов, пригодные в качестве окислительных агентов для окислительной полимеризации тиофенов, анилинов или пирролов.
Пригодными солями металлов являются соли металлов главной или побочной группы периодической системы элементов Менделеева, причем последние в дальнейшем называют также солями переходных металлов. Предпочтительными являются соли переходных металлов. Пригодными солями переходных металлов являются, в частности, соли этих металлов и неорганической или органической кислоты, или неорганической кислоты с органическими остатками, например соли железа(III), меди(II), хрома(VI), церия(IV), марганца(IV), марганца(VII), рутения(III) и цинка(II).
Предпочтительными солями переходных металлов являются соли железа(III). Соли железа(III), например соли железа(III) и неорганических кислот, в частности галогениды железа(III) (например, FeCl3), или соли железа(III) и других неорганических кислот, в частности Fe(ClO4)3 или Fе2(SO4)3, и соли железа(III) и органических кислот или неорганических кислот с органическими остатками часто бывают недороги, легко доступны и могут быть легко переработаны.
Примерами солей железа(III) и неорганических кислот с органическими остатками являются соли кислых эфиров серной кислоты и алканолов с 1-20 атомами углерода, например, лаурилсульфат железа(III).
Особенно предпочтительными солями переходных металлов являются соли этих металлов и органических кислот, прежде всего соли железа(III) и органических кислот.
Примерами солей железа(III) и органических кислот являются соли железа(III) и алкансульфокислот с 1-20 атомами углерода, таких как метановая, этановая, пропановая, бутановая или более высокомолекулярная кислота, например додекансульфокислота, алифатических перфторсульфокислот, таких как трифторметансульфокислота, перфторбутансульфокислота или перфтороктансульфокислота, алифатических карбоновых кислот с 1-20 атомами углерода, таких как 2-этилгексилкарбоновая кислота, алифатических перфторкарбоновых кислот, таких как трифторуксусная или перфтороктановая кислота, ароматических сульфокислот, при необходимости замещенных алкильными группами с 1-20 атомами углерода, таких как бензолсульфокислота, о-толуолсульфокислота, п-толуолсульфокислота или додецилбензолсульфокислота, и циклоалкансульфокислот, таких как камфорная сульфокислота.
Могут использоваться также любые смеси указанных солей железа(III) и органических кислот.
Большим преимуществом использования солей железа(III) и органических кислот и неорганических кислот с органическими остатками является то, что такие соли не обладают коррозионным действием.
Еще более предпочтительными солями металлов являются п-толуолсульфонат железа(III), о-толуолсульфонат железа(III) или смесь п-толуолсульфоната железа(III) с о-толуолсульфонатом железа(III).
Кроме того, пригодными солями металлов являются пероксосоединения, например, пероксодисульфаты (персульфаты), в частности пероксодисульфаты аммония и щелочных металлов, такие как пероксодисульфат натрия и пероксодисульфат калия, или пербораты щелочных металлов, а также оксиды переходных металлов, например, пиролюзит (оксид марганца(V)) или оксид церия(IV).
В предпочтительных вариантах предлагаемые в изобретении смеси содержат окислительный агент, который может быть изготовлен указанным выше предлагаемым в изобретении способом.
В других предпочтительных вариантах предлагаемые в изобретении смеси содержат в качестве исходных соединений для получения проводящих полимеров при необходимости замещенные тиофены, пирролы, анилины или их производные. Особенно предпочтительными являются при необходимости замещенные 3,4-этилендиокситиофены или их производные, еще более предпочтительным является 3,4-этилендиокситиофен.
В других предпочтительных вариантах предлагаемые в изобретении смеси содержат в качестве окислительного агента соль железа(III), предпочтительно п-толуолсульфонат железа(III), о-толуолсульфонат железа(III) или смесь этих солей.
В других предпочтительных вариантах в предлагаемые в изобретении смеси дополнительно вводят воду в количестве предпочтительно от 1 до 100 мас.%, особенно предпочтительно от 1 до 60 мас.%, еще более предпочтительно от 1 до 40 мас.% в расчете на массу окислительного агента.
Предлагаемые в изобретении смеси могут также образоваться на поверхности подложки in situ, например, в результате последовательного погружения подложки сначала в окислительный агент, при необходимости используемый в виде раствора, а затем в исходные соединения, при необходимости используемые в виде раствора, причем по завершении процесса погружения подложки в указанные растворы при необходимости может быть выполнена ее сушка. В этом случае смеси образуются, например, вследствие диффузии через границу раздела фаз или происходящего на поверхности подложки перемешивания разных жидкостей. Согласно изобретению такие смеси также следует считать предлагаемыми в изобретении смесями.
Используя предлагаемые в изобретении смеси, можно изготавливать электролитические конденсаторы. В принципе электролитический конденсатор изготавливают путем первоначального снабжения поверхности окисляемого металла слоем диэлектрика, то есть оксидным слоем, осуществляемого методом окисления металла, например его электрохимического окисления. Затем посредством окислительной полимеризации, согласно изобретению осуществляемой с использованием одной из указанных выше смесей, на поверхности диэлектрика химически осаждают проводящий полимер, который образует твердый электролит. Последующее нанесение обладающих высокой электропроводностью дополнительных покрытий, состоящих, например, из графита или серебра, обеспечивает возможность отвода тока. В заключение, корпус конденсатора снабжают контактами и герметизируют.
Согласно предлагаемому в изобретении способу окисляемый металл предпочтительно образует анодный корпус с развитой поверхностью, например, в виде пористого спеченного тела или пленки, которой придана шероховатость. Ниже такой корпус для краткости называют анодным корпусом.
Согласно изобретению слой состоящего из проводящего полимера твердого электролита создают на покрытом оксидным слоем анодном корпусе путем окислительной полимеризации описанных выше смесей, нанося такие смеси на оксидный слой анодного корпуса предпочтительно в виде раствора и подвергая их окислительной полимеризации, в конце которой в зависимости от активности используемого окислительного агента при необходимости осуществляют нагревание полимерного покрытия.
Таким образом, еще одним объектом настоящего изобретения является способ изготовления электролитического конденсатора, отличающийся тем, что предлагаемые в изобретении смеси наносят при необходимости в виде растворов на оксидный слой металла и подвергают химической окислительной полимеризации при температуре от -10 до 250°С до образования соответствующих полимеров.
Таким образом, другим объектом настоящего изобретения является способ изготовления электролитического конденсатора, отличающийся тем, что исходные соединения для получения проводящих полимеров и изготовленные предлагаемым в изобретении способом окислительные агенты последовательно, предпочтительно в виде растворов наносят на оксидный слой металла и подвергают химической окислительной полимеризации при температуре от -10 до 250°С до образования соответствующих полимеров.
Покрытие из проводящего полимера может быть нанесено на оксидный слой анодного корпуса непосредственно или используя промотор адгезии, например силан, и/или другой функциональный слой.
Окислительную химическую полимеризацию исходных соединений для получения проводящих полимеров в зависимости от используемого окислительного агента и желаемой длительности процесса полимеризации в общем случае осуществляют при температуре от -10 до 250°С, предпочтительно от 0 до 200°С.
Как в предлагаемые в изобретении смеси, так и в растворы можно вводить дополнительные противоионы. Пригодные противоионы являются такими, как указано выше для предлагаемых в изобретении смесей.
В предлагаемых в изобретении электролитических конденсаторах особенно предпочтительным является использование анионов мономерных алкансульфокислот, циклоалкансульфокислот или ароматических сульфокислот, поскольку содержащие такие анионы растворы способны лучше проникать в пористый анодный материал, а, следовательно, между ним и твердым электролитом может быть создана большая площадь контакта.
Кроме того, предпочтительными противоионами в предлагаемых в изобретении электролитических конденсаторах могут являться при необходимости присутствующие в окислительном агенте анионы, в связи с чем может отсутствовать настоятельная необходимость в добавлении дополнительных противоионов.
Аналогично предлагаемым в изобретении смесям растворы могут также дополнительно содержать одно или несколько связующих, сшивающих агентов и/или добавок. Пригодные связующие, сшивающие агенты и/или добавки являются такими, как указано выше для предлагаемых в изобретении смесей.
Пригодные для получения проводящих полимеров исходные соединения аналогичны указанным выше исходным соединениям.
Предлагаемые в изобретении смеси могут быть нанесены на оксидный слой анодного корпуса известными методами, например путем пропитки, налива, накапывания, разбрызгивания, напыления, нанесения раклей, намазывания или запечатывания.
При необходимости использованные растворители после нанесения смесей могут быть удалены простым испарением при комнатной температуре. Однако для обеспечения более высоких скоростей переработки более целесообразным является удаление растворителей при повышенных температурах, например при температуре от 20 до 300°С, предпочтительно от 40 до 250°С. Дополнительную тепловую обработку можно осуществлять в сочетании с удалением растворителя или по истечении определенного времени после изготовления покрытия.
Длительность тепловой обработки в зависимости от типа используемого для получения покрытия полимера составляет от 5 секунд до нескольких часов. Для тепловой обработки может использоваться также температурный профиль с переменной температурой и длительностью ее воздействия.
Тепловая обработка может быть выполнена, например, путем перемещения снабженного покрытием анодного корпуса через нагретую до желаемой температуры термокамеру с такой скоростью, чтобы обеспечить желаемое время пребывания анодного корпуса в термокамере при выбранной температуре, или путем контактирования анодного корпуса с нагретой до желаемой температуры обогреваемой плитой в течение желаемого времени воздействия температуры. Кроме того, тепловую обработку можно осуществлять, например, в одной или нескольких нагревательных печах, нагретых до разных температур.
После удаления растворителей (сушки) и при необходимости выполненной дополнительной тепловой обработки может быть целесообразным вымывание присутствующего в покрытии избыточного окислительного агента и остаточных солей, осуществляемое пригодным растворителем, предпочтительно водой или спиртами. Под остаточными солями в данном случае подразумеваются соли восстановленной формы окислительного агента и при необходимости другие присутствующие в покрытии соли.
В зависимости от типа анодного корпуса может оказаться целесообразной выполняемая предпочтительно после промывки повторная пропитка анодного корпуса смесями, благодаря чему получают более толстые полимерные покрытия.
По завершении полимеризации и предпочтительно в процессе или после промывки может оказаться целесообразным электрохимическое воссоздание оксидной пленки, цель которого состоит в исправлении возможных дефектов этой пленки и соответственно снижении величины остаточного тока готового конденсатора.
Кроме того, предлагаемый в изобретении способ характеризуется тем, что под окисляемым металлом подразумевается вентильный металл или соединение, обладающее сравнимыми с вентильным металлом свойствами.
Согласно изобретению под вентильными металлами подразумевают металлы, оксидные слои которых делают невозможным прохождение одинакового тока в обоих направлениях: при прикладывании напряжения к аноду оксидные слои вентильных металлов препятствуют прохождению тока, тогда как при прикладывании напряжения к катоду возникают слишком сильные токи, которые могут привести к разрушению оксидного слоя. К вентильным металлам относятся бериллий (Be), магний (Mg), алюминий (Al), германий (Ge), кремний (Si), олово (Sn), сурьма (Sb), висмут (Bi), титан (Ti), цирконий (Zr), гафний (Hf), ванадий (V), ниобий (Nb), тантал (Та) и вольфрам (W), а также сплав или соединение по меньшей мере одного из указанных металлов с другими элементами. Наиболее известными представителями вентильных металлов являются алюминий (Al), тантал (Та) и ниобий (Nb). Соединениями со сравнимыми свойствами являются обладающие металлической электропроводностью соединения, которые способны к окислению и оксидные слои которых обладают свойствами, аналогичными указанным выше. Так, например, хотя оксид ниобия (NbO) и обладает металлической электропроводностью, но в общем случае он не считается вентильным металлом. Однако слои оксидированного NbO обладают типичными свойствами оксидных слоев вентильных металлов, поэтому NbO или сплав или соединение NbO с другими элементами являются типичными примерами подобных соединений со сравнимыми свойствами.
Следовательно, под окисляемыми металлами подразумеваются не только сами металлы, но и их сплавы или соединения с другими элементами, если они обладают металлической электропроводностью и способностью окисляться.
Таким образом, особенно предпочтительным объектом настоящего изобретения является способ, отличающийся тем, что под вентильным металлом или соединением со сравнимыми свойствами подразумевается тантал, ниобий, алюминий, титан, цирконий, гафний, ванадий, сплав или соединение по меньшей мере одного из этих металлов с другими элементами, NbO или сплав или соединение NbO с другими элементами.
В соответствии с предлагаемым в изобретении способом окисляемый металл предпочтительно образует анодный корпус с развитой поверхностью, например, в виде пористого спеченного тела или пленки, которой придана шероховатость.
Однако предлагаемый в изобретении способ пригоден для изготовления не только электролитических конденсаторов, но и проводящих покрытий, предназначенных для использования в других областях применения.
Согласно изобретению проводящие покрытия изготавливают способом, который позволяет создавать такие покрытия путем окислительной полимеризации предлагаемых в изобретении смесей.
Таким образом, объектом настоящего изобретения является также способ изготовления токопроводящих покрытий, отличающийся тем, что предлагаемые в изобретении смеси наносят на подложку предпочтительно в виде растворов и химически полимеризуют на ней до образования проводящих полимеров при температуре от -10 до 250°С, предпочтительно от 0 до 200°С.
Другим объектом настоящего изобретения является также способ изготовления проводящих покрытий, отличающийся тем, что исходные соединения для получения проводящих полимеров и изготовленный предлагаемым в изобретении способом окислительный агент при необходимости в виде соответствующих растворов последовательно наносят на подложку и химически полимеризуют на ней до образования соответствующих проводящих полимеров при температуре от -10 до 250°С.
Типичные и предпочтительные реакционные условия, мольные соотношения, представленные в массовых процентах данные, растворители, окислительные агенты, исходные соединения для получения проводящих полимеров, а также приводимые в этой связи варианты, соответственно особенности осуществления окислительной полимеризации аналогичны указанным выше при рассмотрении изготовления электролитических катализаторов.
Наряду с указанными выше методами нанесения покрытий, используемыми для изготовления конденсаторов, в качестве метода, пригодного для нанесения смесей или растворов на плоские субстраты, предлагается также центробежный метод.
Растворы аналогично предлагаемым в изобретении смесям могут также дополнительно содержать одно или несколько связующих, сшивающих агентов и/или добавок. Пригодные связующие, сшивающие агенты и/или добавки являются такими, как указано выше для предлагаемых в изобретении смесей.
В растворы, как и в предлагаемые в изобретении смеси, также можно вводить дополнительные противоионы. Пригодными противоионами являются такие, как указано выше для предлагаемых в изобретении смесей, причем при образовании полимерных пленок полианионы обеспечивают улучшение пленкообразующих свойств, поэтому предпочтительным является использование полианионов.
После полимеризации и при необходимости осуществляемой сушки изготовленные предлагаемым в изобретении способом токопроводящие покрытия аналогично рассмотренному выше изготовлению электролитических конденсаторов могут быть подвергнуты промывке пригодными растворителями с целью удаления избыточного окислительного агента и остаточных солей.
В качестве подложки может быть использовано, например, стекло, дымчатое (гибкое) стекло или полимер.
Особенно пригодными, используемыми в качестве подложки полимерами являются поликарбонаты, сложные полиэфиры, например полиэтилентерефталат, соответственно полиэтиленнафтенат, сополикарбонаты, полисульфон, полиэфирсульфон, полиимид, полиэтилен, полипропилен или циклические полиолефины, соответственно сополимеры циклоолефинов, гидрированные полимеры или сополимеры стирола.
Пригодными полимерными подложками могут быть, например, полиэфирные и полиэфирсульфоновые пленки фирмы Sumitomo или поликарбонатные пленки фирмы Bayer AG (марки Makrofol®).
Изготовленные предлагаемым в изобретении способом проводящие покрытия могут оставаться на подложке или их можно отделять от нее.
В зависимости от сферы применения политиофеновые покрытия обладают толщиной от 1 нм до 100 мкм, предпочтительно от 10 нм до 10 мкм, особенно предпочтительно от 50 нм до 1 мкм.
Изготовленные предлагаемым в изобретении способом проводящие покрытия прекрасно пригодны для использования в качестве антистатических покрытий, прозрачных нагревательных элементов, при необходимости прозрачных электродов, покрытий с дырочной инжекцией или дырочной электропроводностью для органических светодиодов, для металлизации сквозных отверстий печатных плат или в качестве твердого электролита в электролитических конденсаторах. Проводящие покрытия предпочтительно являются прозрачными.
Проводящие покрытия можно использовать в качестве антистатической облицовки пленок и упаковок для электронных деталей, для отделки полимерных пленок, а также для облицовки мониторов. Кроме того, проводящие покрытия можно использовать в качестве материала катода в конденсаторах, в качестве прозрачных электродов, например, дисплеев (в частности, для замены электродов на основе индия и оксида олова), или в качестве электрического проводника в полимерных электронных деталях. Другими сферами возможного использования проводящих покрытий являются датчики, батареи, солнечные элементы, электрохромные окна (окна со встроенным микропроцессором) и дисплеи, а также защита от коррозии.
Кроме того, объектом настоящего изобретения является применение предлагаемых в изобретении, соответственно изготовленных предлагаемым в изобретении способом окислительных агентов, а также предлагаемых в изобретении смесей для изготовления проводящих покрытий и электролитических конденсаторов.
Приведенные ниже примеры не ограничивают объема настоящего изобретения.
Примеры
Пример 1
a) Изготовление раствора предлагаемого в изобретении окислительного агента
Стаканом для волюметрических измерений отмеривали две объемные части раствора п-толуолсульфоната железа(III) в этаноле концентрацией 40 мас.% и одну объемную часть слабощелочного макропористого анионита Lewatit® MP 62 (фирма Bayer AG). Твердый анионит отмеривали путем простой засыпки (аналогичным образом отмеривали объемные части твердых анионитов и в дальнейших примерах). Затем отмеренные объемы этанольного раствора п-толуолсульфоната железа(III) и анионита в течение 24 часов перемешивали в закрытом сосуде с помощью встряхивающего устройства. По завершении перемешивания отфильтровывали анионит.
b) Изготовление предлагаемой в изобретении смеси окислительного агента с исходными соединениями
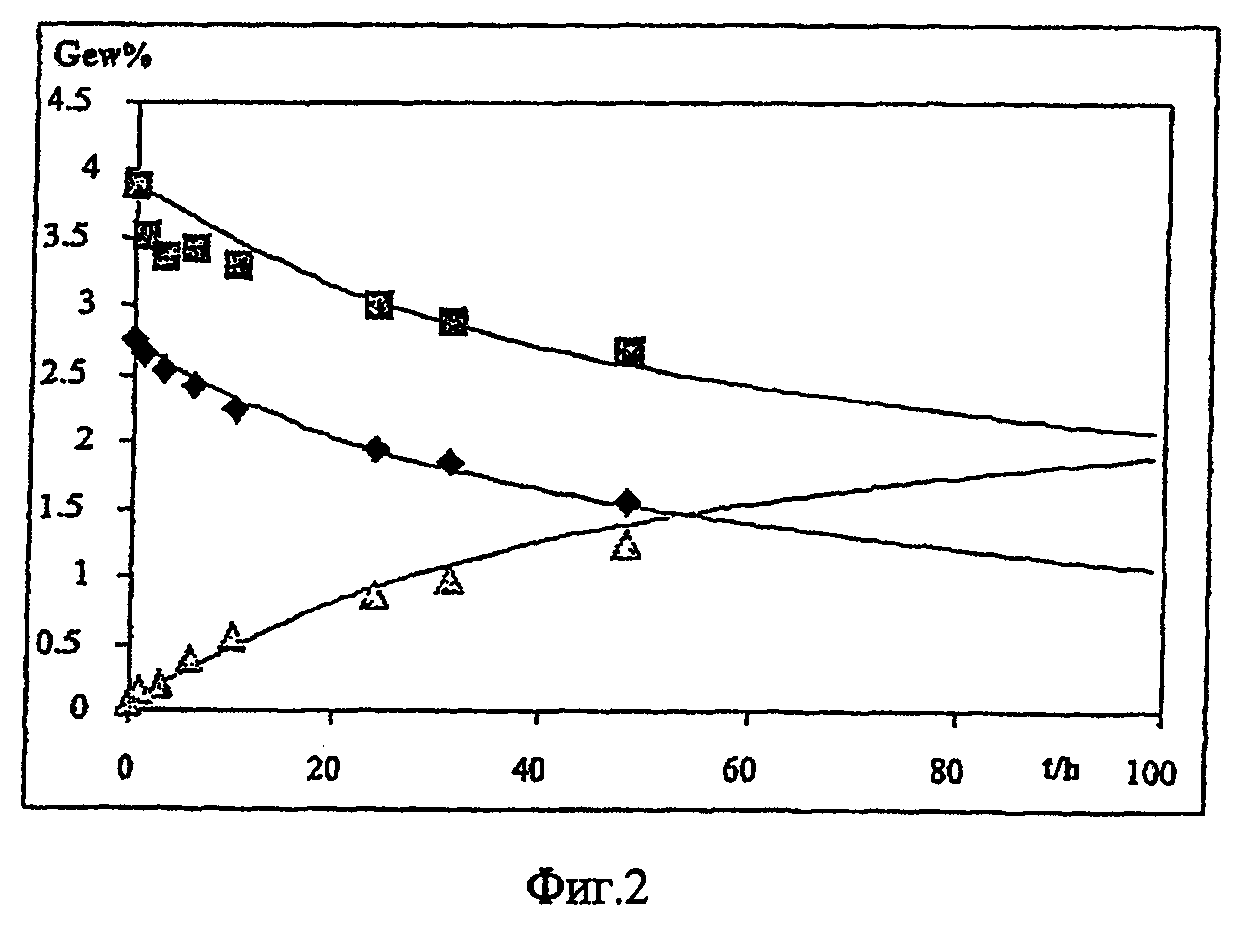
1 массовую часть 3,4-этилендиокситиофена (фирмы Baytron® M, H.C.Starck GmbH) и 20 массовых частей приготовленного в соответствии с п.а) раствора предлагаемого в изобретении окислительного агента перемешивали мешалкой и хранили полученную смесь в холодильнике при температуре около 6°С. Через регулярные промежутки времени предлагаемую в изобретении смесь в виде тонкой пленки раствора просвечивали лампой и следили за появлением частиц твердого вещества. Промежуток времени от момента изготовления смеси до появления первых визуально обнаруживаемых частиц служил мерой жизнеспособности смеси.
Жизнеспособность смеси составляла 24 часа.
Пример 2
К раствору п-толуолсульфоната железа(III) в этаноле концентрацией 40 мас.% аналогично примеру 1 добавляли разные количества анионита, после чего определяли жизнеспособность соответствующих смесей с 3,4-этилендиокситиофеном.
a) Изготовление растворов предлагаемых в изобретении окислительных агентов
Этанольный раствор п-толуолсульфоната железа(III) концентрацией 40 мас.% перемешивали в течение 7 часов со слабощелочным макропористым анионитом Lewatit® МР 62 (Bayer AG) с помощью встряхивающего устройства при объемном соотношении соответствующих компонентов 9:1, 3:1 и 2:1, после чего отфильтровывали анионит.
b) Изготовление предлагаемых в изобретении смесей окислительного агента с исходными соединениями
Получали смесь, состоящую из 1 массовой части 3,4-этилендиокситиофена (Baytron® М, Н.С.Starck GmbH) и 20 массовых частей полученного согласно п.а) раствора предлагаемого в изобретении окислительного агента, и хранили полученную смесь в холодильнике при температуре около 6°С. Жизнеспособность смеси определяли аналогично примеру 1.
c) Изготовление несоответствующей изобретению сравнительной смеси необработанного ионитом окислительного агента с исходными соединениями
Для сравнения получали смесь 1 массовой части 3,4-этилендиокситиофена (Baytron® М, H.C.Starck GmbH) с 20 массовыми частями необработанного ионитом этанольного раствора п-толуолсульфоната железа(III) концентрацией 40 мас.%, которую хранили при температуре около 6°С и также анализировали указанными выше методом (сравнительная смесь).
Были получены следующие результаты:
Предлагаемые в изобретении смеси обладали гораздо более высокой жизнеспособностью по сравнению со смесью, содержащей необработанный ионитом п-толуолсульфонат железа(III).
Пример 3
Для определения проводимости полимерных пленок, изготовленных из предлагаемых в изобретении смесей, соответствующие пленки наносили центробежным методом и полимеризовали.
а) Изготовление раствора предлагаемого в изобретении окислительного агента
Бутанольный раствор п-толуолсульфоната железа(III) концентрацией 40 мас.% смешивали аналогично примеру 1 со слабощелочным макропористым анионитом Lewatit® MP 62 (Bayer AG) в объемном соотношении 2:1 и смесь выдерживали в течение 64 часов. Затем отфильтровывали анионит.
b) Изготовление предлагаемой в изобретении смеси окислительного агента с исходными соединениями, а также проводящего покрытия
Получали смесь, состоящую из 1 массовой части 3,4-этилендиокситиофена (Baytron® М, H.C.Starck GmbH) и 20 массовых частей изготовленного в соответствии с п.а) раствора предлагаемого в изобретении окислительного агента, и часть полученной смеси в течение 5 секунд наносили на предметное стекло (26 мм × 26 мм × 1 мм) посредством устройства для нанесения покрытий методом центрифугирования (Chemical Technology KW-4A) при частоте вращения 2000 мин-1. Образец в течение 60 минут сушили при 20°С, после чего в течение 15 минут промывали в стеклянной чашке метанолом. Затем образец вновь сушили в течение 15 минут при 50°С и посредством универсального электроизмерительного прибора Keithley 199 выполняли четырехточечное измерение поверхностного электрического сопротивления. Толщину покрытия определяли посредством измерителя профиля поверхностей Tencor Alpha Step 500. Зная поверхностное сопротивление и толщину покрытия, вычисляли его удельную проводимость. Оставшуюся смесь хранили в холодильнике при температуре около 6°С и использовали для определения жизнеспособности, как описано в примере 1.
c) Изготовление несоответствующей изобретению сравнительной смеси необработанного ионитом окислительного агента с исходными соединениями
Для сравнения получали смесь 1 массовой части 3,4-этилендиокситиофена (Baytron® M, H.C.Starck GmbH) с 20 массовыми частями необработанного ионитом бутанольного раствора п-толуолсульфоната железа(III) концентрацией 40 мас.%, которую хранили при 6°С и также анализировали указанными выше методом (сравнительная смесь).
Были получены следующие результаты:
Предлагаемые в изобретении смеси обладали гораздо более высокой жизнеспособностью по сравнению со смесью, содержащей необработанный ионитом окислительный агент. Вместе с тем покрытие из предлагаемой в изобретении смеси обладало значительно более высокой проводимостью и значительно меньшим поверхностным сопротивлением, чем изготовленный из сравнительной смеси образец.
Пример 4
Определяли жизнеспособность предлагаемой в изобретении смеси по сравнению со смесью из необработанной ионитом соли металла с добавлением основания.
а) Изготовление раствора предлагаемого в изобретении окислительного агента
Бутанольный раствор п-толуолсульфоната железа(III) концентрацией 40 мас.% смешивали аналогично примеру 1 со слабощелочным макропористым анионитом Lewatit® MP 62 (Bayer AG) в объемном соотношении 2:1 и смесь выдерживали в течение 64 часов. Затем отфильтровывали анионит.
b) Изготовление предлагаемой в изобретении смеси окислительного агента с исходными соединениями
Получали смесь, состоящую из 1 массовой части 3,4-этилендиокситиофена (Baytron® M, H.C.Starck GmbH) и 20 массовых частей изготовленного по п.а) раствора предлагаемого в изобретении окислительного агента. Смесь хранили в холодильнике при температуре около 6°С и определяли ее жизнеспособность, как указано в примере 1.
с) Изготовление несоответствующей изобретению сравнительной смеси необработанного ионитом окислительного агента с исходными соединениями
Для сравнения готовили смесь 1 массовой части 3,4-этилендиокситиофена (Baytron® M, H.C.Starck GmbH) с 20 массовыми частями необработанного ионитом бутанольного раствора п-толуолсульфоната железа(III) концентрацией 40 мас.% и 0,75 массовой частью имидазола, которую хранили при 6°С и также анализировали указанным выше методом (сравнительная смесь).
Были получены следующие показатели жизнеспособности:
Из обеих указанных смесей удалось изготовить полимерные пленки путем нанесения на стеклянную пластину и последующей сушки при 60°С. Однако из сравнительных смесей с добавлением увеличенного количества имидазола полимерные пленки не могли быть изготовлены и при повышении температуры до 150°С.
Жизнеспособность предлагаемой в изобретении смеси значительно выше по сравнению со смесью, содержащей необработанный ионитом окислительный агент и добавку основания (имидазола).
Пример 5
Определяли жизнеспособность предлагаемой в изобретении смеси, содержащей два разных раствора окислительного агента, изготовленного предлагаемым в изобретении способом.
a) Изготовление двух растворов предлагаемого в изобретении окислительного агента
Этанольный раствор п-толуолсульфоната железа(III) концентрацией 40 мас.% перемешивали в течение 7 часов посредством встряхивающего устройства со слабощелочным макропористым анионитом Lewatit® MP 62 (Bayer AG) при объемном соотношении компонентов 1:1, после чего отфильтровывали анионит (получали раствор 1).
Аналогичным образом путем осуществляемого в течение 7 часов с помощью встряхивающего устройства перемешивания этанольного раствора п-толуолсульфоната железа(III) концентрацией 40 мас.% со слабощелочным макропористым анионитом Lewatit® MP 62 (Bayer AG) при объемном соотношении компонентов 2:1 и последующего отфильтровывания анионита получали раствор 2.
b) Изготовление предлагаемой в изобретении смеси окислительного агента с исходными соединениями
Получали предлагаемую в изобретении смесь, состоящую из 1 массовой части 3,4-этилендиокситиофена (Baytron® М, Н.С.Starck GmbH), 10 массовых частей раствора 1 и 10 массовых частей раствора 2, и хранили ее в холодильнике при температуре около 6°С. Жизнеспособность смеси определяли аналогично примеру 1.
Жизнеспособность составляла 96 часов.
Как следует из данного примера, жизнеспособность смеси можно регулировать также путем смешивания по-разному изготовленных предлагаемых в изобретении окислительных агентов.
Пример 6
Определяли жизнеспособность предлагаемой в изобретении смеси при ее хранении при низкой температуре.
а) Изготовление раствора предлагаемого в изобретении окислительного агента Аналогично примеру 1 этанольный раствор п-толуолсульфоната железа(III) концентрацией 40 мас.% перемешивали в течение 7 часов посредством встряхивающего устройства со слабощелочным макропористым анионитом Lewatit® MP 62 (Bayer AG) в объемном соотношении 2:1, после чего отфильтровывали анионит.
b) Изготовление предлагаемой в изобретении смеси окислительного агента с исходными соединениями
Получали смесь, состоящую из 1 массовой части 3,4-этилендиокситиофена (Baytron® M, H.C. Starck GmbH) и 20 массовых частей изготовленного согласно п.а) раствора предлагаемого в изобретении окислительного агента, и хранили ее в холодильнике при температуре около -15°С. Смесь оставалась при этой температуре в жидком состоянии. Жизнеспособность смеси определяли аналогично примеру 1.
с) Изготовление несоответствующей изобретению сравнительной смеси необработанного ионитом окислительного агента с исходными соединениями
Для сравнения получали смесь 1 массовой части 3,4-этилендиокситиофена (Baytron® M, H.C.Starck GmbH) с 20 массовыми частями необработанного ионитом этанольного раствора п-толуолсульфоната железа(III) концентрацией 40 мас.%, которую хранили при -15°С и также исследовали указанным выше методом (сравнительная смесь).
Получены следующие результаты (для сравнения приведена жизнеспособность соответствующей смеси из примера 2 при ее хранении при 6°С).
Как показывает сравнение жизнеспособности смесей при разных температурах их хранения, она может быть существенно повышена благодаря охлаждению смесей до низких температур, что относится как к предлагаемой в изобретении, так и сравнительной смеси. Однако предлагаемые в изобретении смеси и при пониженных температурах обладают более высокой жизнеспособностью по сравнению со смесями, содержащими необработанный ионообменной смолой толуолсульфонат железа(III).
Пример 7
Определяли жизнеспособность предлагаемых в изобретении смесей, содержащих предлагаемые в изобретении окислительные агенты, которые были подвергнуты обработке разными ионитами.
а) Изготовление растворов предлагаемого в изобретении окислительного агента Аналогично примеру 1 бутанольный раствор п-толуолсульфоната железа(III) концентрацией 40 мас.% перемешивали со слабощелочным макропористым анионитом Lewatit® МР 62 (Bayer AG) в объемном соотношении 2:1 и выдерживали полученную смесь в течение 24 часов. Затем отфильтровывали анионит. Предлагаемые в изобретении окислительные агенты были изготовлены аналогичным образом путем обработки раствора п-толуолсульфоната железа(III) макропористым анионитом Lewatit® MP 64 (Bayer AG) со средней основностью или сильнощелочным макропористым анионитом Lewatit® MP 600 WS (Bayer AG).
b) Изготовление предлагаемых в изобретении смесей окислительного агента с исходными соединениями
Получали смесь, состоящую из 1 массовой части 3,4-этилендиокситиофена (Baytron® M, H.C.Starck GmbH) и 20 массовых частей изготовленного по п.а) раствора предлагаемого в изобретении окислительного агента, и хранили ее в холодильнике при температуре около 6°С. Жизнеспособность смеси определяли аналогично примеру 1.
Аналогичным образом получали смеси, содержащие раствор окислительного агента, изготовленный путем выполненной по п.а) обработки макропористым анионитом Lewatit® MP 64 (Bayer AG) со средней основностью или сильнощелочным макропористым анионитом Lewatit® MP 600 WS (Bayer AG), хранили полученные смеси при 6°С и определяли жизнеспособность образцов, как описано выше.
с) Изготовление несоответствующей изобретению сравнительной смеси необработанного ионитом окислительного агента с исходными соединениями
Для сравнения получали смесь 1 массовой части 3,4-этилендиокситиофена (Baytron® M, H.C.Starck GmbH) с 20 массовыми частями необработанного ионитом бутанольного раствора п-толуолсульфоната железа(III) концентрацией 40 мас.%, которую хранили при 6°С и также исследовали указанными выше методом (сравнительная смесь).
Были получены следующие результаты:
Пример 8
Получали раствор предлагаемого в изобретении окислительного агента со средним влагосодержанием и высокой стабильностью при хранении.
а) Изготовление раствора предлагаемого в изобретении окислительного агента с низким влагосодержанием
Смешивали 1 л слабоосновного макропористого анионита Lewatit® МР 62 (Bayer AG) с 2 л абсолютного этанола и перемешивали их в течение 6 часов. Затем ионообменную смолу отфильтровывали и вновь трижды кондиционировали, как описано выше.
Затем этанольный раствор п-толуолсульфоната железа(III) концентрацией 40 мас.% перемешивали аналогично примеру 1 в течение 7 часов посредством встряхивающего устройства со слабоосновным макропористым анионитом Lewatit® МР 62 (Bayer AG) в объемном соотношении 2:1 (в расчете на объем ионита до обработки этанолом), после чего отфильтровывали анионит.
b) Изготовление раствора предлагаемого в изобретении окислительного агента без предварительной обработки ионита
Аналогично примеру 1 этанольный раствор п-толуолсульфоната железа(III) концентрацией 40 мас.% перемешивали в течение 7 часов посредством встряхивающего устройства со слабощелочным макропористым анионитом Lewatit® МР 62 (Bayer AG) в объемном соотношении 2:1, после чего отфильтровывали анионит.
Раствор предлагаемого в изобретении окислительного агента, изготовленного по п.а), обладал влагосодержанием 1,1 мас.% (в расчете на общую массу раствора) и не выпадал в осадок при хранении при комнатной температуре (20°С) в течение трех месяцев.
Влагосодержание предлагаемого в изобретении окислительного агента, изготовленного по п.b), составляло 12,8 мас.% (в расчете на общую массу раствора). Выпадение хранящегося при комнатной температуре раствора в осадок наступало по истечении одной недели, а при хранении в холодильнике при температуре около 6°С спустя два месяца.
Пример 9
Определяли жизнеспособность предлагаемой в изобретении смеси, содержащей предлагаемый в изобретении окислительный агент с пониженным влагосодержанием.
a) Изготовление предлагаемой в изобретении смеси окислительного агента с исходными соединениями
Получали смесь, состоящую из 1 массовой части 3,4-этилендиокситиофена (Baytron М, Н.С.Starck GmbH) и 20 массовых частей изготовленного согласно примеру 8а) раствора предлагаемого в изобретении окислительного агента. Смесь хранили в холодильнике при температуре около 6°С и определяли ее жизнеспособность, как описано в примере 1.
b) Изготовление предлагаемой в изобретении смеси окислительного агента и исходных соединений с добавлением воды
Получали смесь, состоящую из 1 массовой части 3,4-этилендиокситиофена (Baytron® М, Н.С.Starck GmbH), 20 массовых частей изготовленного согласно примеру 8а) раствора предлагаемого в изобретении окислительного агента и 2 частей воды. Смесь хранили в холодильнике при температуре около 6°С и определяли ее жизнеспособность, как описано в примере 1.
Жизнеспособность предлагаемой в изобретении смеси по п.а) составляла 2,5 часа, а предлагаемой в изобретении смеси по п.b) 30 часов.
Пример 10
Изготовление конденсаторов с использованием предлагаемых в изобретении окислительных агентов
Танталовый порошок с удельной емкостью 50000 мкФ·В/г прессовали и спекали в столбики, представляющие собой пористые цилиндрические тела диаметром 2,5 мм и высотой 1,9 мм. Столбики (аноды) подвергали анодному окислению в фосфорнокислом электролите при напряжении 30В.
Этанольный раствор п-толуолсульфоната железа(III) концентрацией 40 мас.% перемешивали аналогично примеру 1 в течение 7 часов посредством встряхивающего устройства со слабощелочным макропористым анионитом Lewatit® MP 62 (Bayer AG) в объемном соотношении 2:1, после чего отфильтровывали анионит.
Получали смесь, состоящую из 1 массовой части 3,4-этилендиокситиофена (Baytron® M, H.C.Starck GmbH) и 20 массовых частей изготовленного по п.а) раствора предлагаемого в изобретении окислительного агента.
Предлагаемую в изобретении смесь использовали для пропитки анодных столбиков. Анодные столбики пропитывали в этой смеси, а затем подвергали последовательно осуществляемой 15-минутной сушке при комнатной температуре, 15-минутной сушке при 50°С и 15-минутной сушке при 150°С. В результате тепловой обработки происходила полимеризация смеси в столбиках. Затем столбики в течение 30 минут промывали метанолом. Указанную пропитку и промывку повторяли еще два раза. В заключение на столбики наносили покрытие из графита или серебра.
Аналогичным образом обрабатывали новую партию столбиков, используя аналогичную предлагаемую в изобретении смесь, которую хранили при 6°С в течение 24 часов после приготовления.
Другую партию новых столбиков обрабатывали предлагаемой в изобретении смесью, которую хранили при 6°С в течение 72 часов после приготовления и перед обработкой подвергали фильтрованию.
Изготовленные конденсаторы обладали следующими электрическими характеристиками:
Емкость конденсаторов определяли при частоте 120 Гц, эквивалентное сопротивление при частоте 100 кГц, используя мост для измерения индуктивности, емкости и активности сопротивлений (Agilent 4284A). Как следует из приведенных в таблице данных, отсутствует существенная разница в электрических характеристиках конденсаторов.
Пример 11
Определение энергии активации для предлагаемых в изобретении и несоответствующих изобретению смесей
Ниже описывается определение энергии активации полимеризации, протекающей в смесях, содержащих 3,4-этилендиокситиофен в качестве исходного соединения и п-толуолсульфонат железа(III) в качестве окислительного агента. При этом сравнивали смеси, содержащие необработанный п-толуолсульфонат железа(III) и п-толуолсульфонат железа(III), обработанный согласно изобретению ионитом.
Кинетическая модель полимеризации 3,4-этилендиокситиофена в присутствии п-толуолсульфоната железа(III)
За взаимодействием 3,4-этилендиокситиофена с п-толуолсульфонатом железа(III) следили по изменению концентрации 3,4-этилендиокситиофена, железа(III) и железа(II) в растворах реакционной смеси. Поскольку поли(3,4-этилендиокситиофен) как продукт полимеризации не обладает растворимостью и выпадает из раствора в осадок, непосредственное наблюдение за изменением его концентрации не представлялось возможным.
Для определения изменения концентрации получали реакционные смеси, состоящие из 3,4-этилендиокситиофена и спиртовых растворов п-толуолсульфоната железа(III), которые хранили при перемешивании в термостатируемых герметичных емкостях. Через регулярные промежутки времени из емкостей отбирали образцы и определяли содержание в них 3,4-этилендиокситиофена, железа(III) и железа(II). Концентрацию 3,4-этилендиокситиофена определяли методом высокоэффективной жидкостной хроматографии. Концентрацию железа(II) и железа(III) определяли фотометрически.
На фиг.1 показана схема окислительной полимеризации 3,4-этилендиокситиофена в присутствии п-толуолсульфоната железа(III), сопровождаемой образованием проводящего поли(3,4-этилендиокситиофена).
Фиг.1 - схема окислительной полимеризации 3,4-этилендиокситиофена в присутствии п-толуолсульфоната железа(III) с образованием проводящего поли(3,4-этилендиокситиофена).
Окислительная полимеризация включает следующие отдельные реакционные стадии.
Окисление мономера
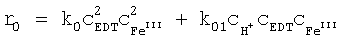
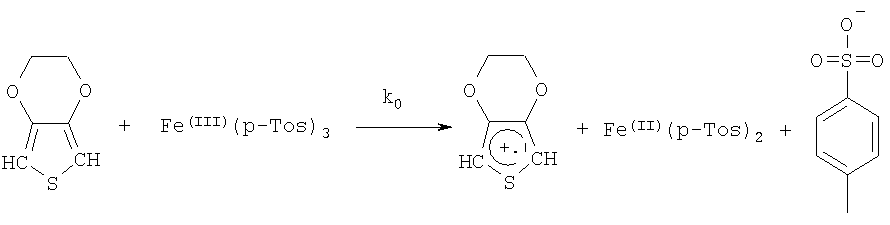
EDT - 3,4-этилендиокситиофен;
p-Tos - п-толуолсульфонат;
k01 - константа катализа высвобождающейся при окислении кислотой.
Окисление концевых групп
p-Tos - п-толуолсульфонат;
k11 - константа катализа высвобождающейся при окислении кислотой.
Рост полимерных цепей путем соединения радикалов (катионов) и депротонирования
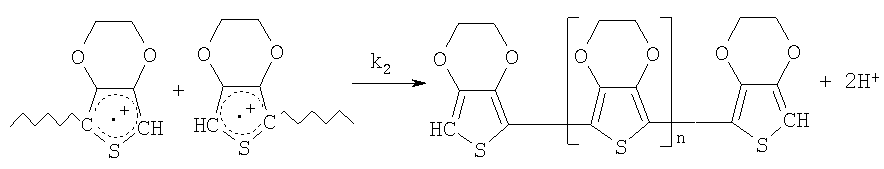
Окисление полимера
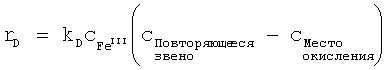
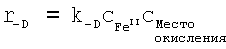
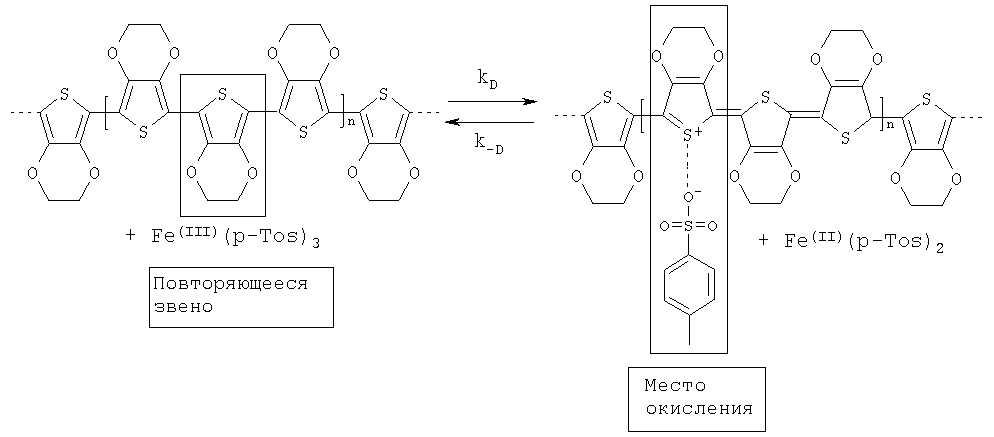
В нижеследующей таблице приведены экспериментально определенные константы скорости отдельных реакций, протекающих при окислительной полимеризации 3,4-этилендиокситиофена в присутствии п-толуолсульфоната железа(III) при 30°С, приводящей к образованию окисленного поли(3,4-этилендиокситиофена).
Определяющей скорость полимеризации реакционной стадией является окисление мономера (k0).
Приведенные в таблице константы позволяют весьма точно описать изменение во времени концентрации 3,4-этилендиокситиофена, железа(III) и железа(II) для разных исходных концентраций. На приведенном на фиг.2 примере экспериментально определенное изменение концентрации каждого из реагентов во времени сопоставлено с соответствующей кинетической моделью.
Фиг.2 - сравнение экспериментально определенного при 30°С изменения концентрации 3,4-этилендиокситиофена (■ квадрат), железа(III) (♦ ромб) и железа(II) (▲ треугольник) с соответствующей кинетической моделью (сплошные линии).
Определение энергии активации полимеризации 3,4-этилендиокситиофена в присутствии необработанного п-толуолсульфоната железа(III)
Для вычисления энергии активации определяли изменение концентрации при разных температурах полимеризации (10°С, 20°С, 30°С, 40°С, 50°С) и соответствующие значения определяющей скорость полимеризации константы k0. Согласно построенной в координатах Аррениуса графической зависимости (фиг.3) энергия активации полимеризации составила 67 кДж/моль, предэкспоненциальный множитель 5,2·1010 л3·моль-3·ч-1.
Фиг.3 - зависимость константы скорости k0 от температуры в координатах Аррениуса (символы ♦ соответствуют экспериментальным данным, линия - кинетической модели).
Определение энергии активации полимеризации 3,4-этилендиокситиофена в предлагаемых в изобретении смесях
Получали предлагаемую в изобретении смесь, состоящую из 6,3 массовых частей 3,4-этилендиокситиофена (Baytron® M, H.C.Starck GmbH), 85,2 массовых частей изготовленного согласно примеру 8а) раствора предлагаемого в изобретении окислительного агента и 8,5 массовых частей воды.
Скорость полимеризации в присутствии предлагаемого в изобретении окислительного агента была значительно ниже, чем в присутствии необработанного окислительного агента. На фиг.4 показано изменение концентрации 3,4-этилендиокситиофена в предлагаемой в изобретении смеси по сравнению с результатом моделирования, выполненного с использованием констант реакции для необработанного окислительного агента.
Фиг.4 - экспериментально определенное изменение концентрации 3,4-этилендиокситиофена (символы ♦) при полимеризации при 20°С в предлагаемых в изобретении смесях по сравнению с результатом моделирования для необработанного окислительного агента (сплошная линия).
Для согласования имеющейся кинетической модели с экспериментальными данными константы скорости реакций окисления (k0, k01, k1, k11) были пересчитаны с использованием одинакового поправочного коэффициента. Это позволило учесть более низкую скорость реакций окисления в экспериментальных условиях. Все прочие параметры оставляли без изменения. Как показано на фиг.5, экспериментально определенные изменения концентрации реагентов во времени могут быть очень точно описаны с помощью соответствующих кинетических моделей.
Фиг.5 - экспериментально определенное при 20°С изменение концентраций 3,4-этилендиокситиофена, железа(III) и железа(II) (символы) и результаты соответствующего моделирования (линии).
Для расчета энергии активации определяли изменение концентрации при разных температурах полимеризации (20°С, 30°С, 45°С) и соответствующие значения определяющей скорость полимеризации константы k0. Графическая зависимость в координатах Аррениуса показана на фиг.6. Энергия активация составила 100 кДж/моль. Предэкспоненциальный множитель составил 2,4·1014 л1·моль-1·ч-1.
Фиг.6 - зависимость константы скорости окисления мономера k0 от температуры в координатах Аррениуса (символы соответствуют экспериментальным данным, линия - кинетической модели).
В связи с более высокой энергией активации полимеризация в предлагаемой в изобретении смеси протекает гораздо медленнее, чем в смесях, содержащих необработанные окислительные агенты.
Реферат
Изобретение относится к области электротехники, в частности к способу изготовления особых окислительных агентов, которые в смесях с исходными соединениями предназначены для получения проводящих полимеров и обеспечивают большую длительность полимеризации с повышением качества покрытия, а также к смесям, содержащим подобные окислительные агенты, и их применению для изготовления конденсаторов с твердым электролитом и проводящих покрытий. Окислительный агент изготавливают путем обработки соли металла и органической кислоты или неорганической кислоты с органическими остатками ионитом до его применения в окислительной полимеризации. 8 н. и 26 з.п. ф-лы, 6 ил.
Формула
Документы, цитированные в отчёте о поиске
Электропроводный пигментный композиционный материал
Комментарии