Оболочка с улучшенными свойствами - RU2712791C1
Код документа: RU2712791C1
Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к композиции мультимодального сополимера этилена с плотностью от 920 до 949 кг/м3 и модулем упругости при изгибе. Композиция мультимодального сополимера этилена по настоящему изобретению может быть использована в оболочке кабеля.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ
Типичный кабель, как правило, состоит из проводника, окруженного одним или более, слоем в зависимости от области применения. Например, силовой кабель имеет несколько слоев полимерных материалов, включая внутренний полупроводящий слой, за которым следует изолирующий слой и затем внешний полупроводящий слой. К этим слоям может быть добавлен один или более дополнительный вспомогательный слой(и). внешний защитный полимерный слой известен, как оболочечный слой. Другие типы кабелей представляют коммуникационные кабели, которые также содержат несколько слоев и оболочечный слой.
Одной из целей развития полиэтиленовых материалов была максимизация гибкости с сохранением при этом других механических свойств, например, высокая твердость по Шору D, для применения линейного полиэтилена низкой плотности (LLDPE) в оболочечном слое.
Поскольку внешний оболочечный слой обеспечивает внешнюю защиту кабеля, он играет важную роль в обеспечении функциональных характеристик. Безопасность, надежность и длительность функционирования являются важными ключевыми факторами, требуемыми для применения в области оболочек кабелей. С другой стороны, оболочечный слой оказывает большое влияние на общую гибкость кабеля. Следовательно, гибкость слоя оболочки является наиболее важным фактором для получения гибкого кабеля. Гибкие кабели просты в обращении и установке, в частности, при строительстве и прокладке кабелей.
Линейный полиэтилен низкой плотности (LLDPE) известен среди прочего, как материал для оболочечного слоя. Механические свойства оболочечного слоя могут быть улучшены при использовании полиэтилена, имеющего более высокую плотность, таких как полимеры из полиэтилена высокой плотности (HDPE). HDPE полимеры обеспечивают слою кабеля среди прочего улучшенную механическую прочность и абразивную износостойкость. Однако, HDPE имеет недостаток, заключающийся в ограниченной гибкости.
Оболочки из композиций мультимодального этиленового полимера хорошо известны в предшествующем уровне техники, и представлены в таких документах, как ЕР 907682 В1 и ЕР 892979 В1. В ЕР 2353169, ЕР 2351049 и ЕР 2182525 описываются композиции гибкого полиэтилена для оболочки кабеля, полученной при использовании катализатора с единым центром полимеризации на металле.
В области полимеров продолжает существовать потребность в полимерах, которые подходили бы для заданного применения, в частности, для применения в проводах и кабелях, где кабельные материалы должны отвечать высоким требованиям и строгим нормативным требованиям.
В области оболочек кабеля продолжает существовать потребность в обеспечении материала, сочетающего высокую гибкость с высокой механической прочностью.
Следовательно, объект настоящего изобретения обеспечивает бимодальную оболочку с улучшенной гибкостью по отношению к плотности, с сохранением при этом технологичности, то есть, с сохранением MFR свойств.
Дополнительно объект настоящего изобретения относится к достижению более высоких или по меньшей мере идентичных показателей твердости по Шору D (1s). Твердость по Шору D указывает на твердость материала. Этот параметр особенно важен при подземном монтаже, где кабель подвергается воздействию довольно суровых условиях.
КРАТКОЕ ОПИСАНИЕ
Определения
Используемый в описании настоящей патентной заявки термин «мультимодальный сополимер» означает сополимер, который содержит различные компоненты с различной средней молекулярной массой или различными содержаниями сомономера, или оба. Мультимодальный сополимер получают при использовании сополимеризации этилена и сомономера на двух или более стадиях полимеризации, где условия полимеризации в достаточной степени отличаются, позволяя получить различные полимеры на различных стадиях. В качестве альтернативы, мультимодальный сополимер может быть получен при проведении одностадийной полимеризации при использовании двух или более различных катализаторов или при использовании многокомпонентного катализатора, содержащего соединения по меньшей мере двух различных переходных металлов.
Используемый в описании настоящей патентной заявки термин «непрерывный процесс» означает процесс или стадию процесса, на которую сырьевые материалы подают непрерывно или периодически и с которой продукт удаляют непрерывно или периодически. Непрерывное добавление или удаление означает, что непрерывный поток поступает или выходит из процесса, стадии процесса. Непрерывное добавление или удаление означает, что во время операции процесса малые партии сырьевого материала непрерывно добавляют или малые партии продукта непрерывно удаляют из процесса или стадии процесса. Временная продолжительность цикла между партиями мала по сравнению с общим временем пребывания процесса или стадии процесса, такая как не более чем 10% общего среднего времени пребывания.
Используемый в описании настоящей патентной заявки термин «жидкая реакционная смесь» означает жидкую фазу (жидкость, газ или сверхкритическую), в которой растворены вещества, участвующие в реакции (этилен, сомономер и водород). Затем частицы, содержащие катализатор и полимер, суспендируют в жидкой реакционной смеси.
Используемый в описании настоящей патентной заявки термин «кабель» включает в объем понятия все типы проводов и кабелей, используемых в области проводов и кабелей, такие как силовые кабели и коммуникационные кабели.
Настоящее изобретение относится к композиции мультимодального сополимера этилена с плотностью от 920 до 949 кг/м3, где модуль упругости при изгибе представляет следующее уравнение:

Композиция мультимодального сополимера этилена по настоящему изобретению имеет оптимальное соотношение высокой гибкости и превосходных механических свойств.
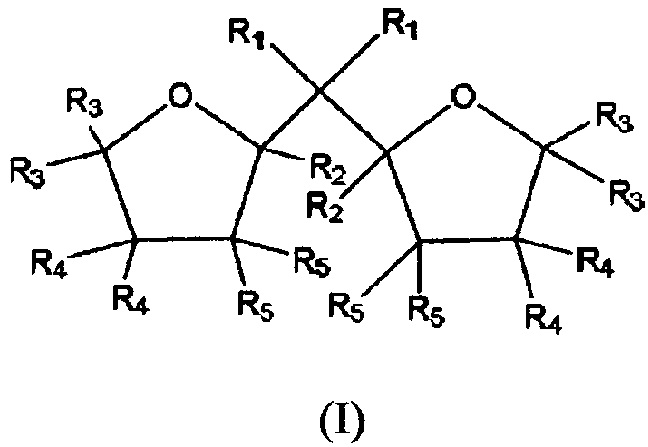
Компонент твердого катализатора, используемого при сополимеризации этилена, представляет компонент твердого катализатора Циглера-Натта для полимеризации этилена, где компонент твердого катализатора Циглера-Натта содержит магний, титан, галоид и органическое соединение внутреннего донора. Органическое соединение внутреннего донора (донор) выбирают из соединений би-(содержащих кислород в кольце) с формулой (I) или изомеры или их смеси

где в формуле (I) R1-R5 представляют идентичные или отличающиеся и могут представлять водород, линейную или разветвленную C1-С8-алкильную группу, или С3-C8-алкиленовую группу, или два или более из R1-R5 могут образовать кольцо, и два содержащих кислород кольца представляют независимо насыщенные или частично ненасыщенные или насыщенные. Используемый в описании настоящей патентной заявки термин «изомеры и смеси» означает все возможные стереоизомеры и смесь, которая может быть получена из структуры с формулой (I) в зависимости от заместителей R1-R5.
Катализатор, используемый в настоящем изобретении, содержит компонент, нанесенный на твердый MgCl2 в качестве подложки, который получают способом, включающим стадии:
a) обеспечение частиц твердого носителя аддукта MgCl2*mROH,
b) предварительной обработки частиц твердого носителя стадии а) соединением металла группы 13,
c) обработки прошедших предварительную обработку частиц твердого носителя стадии b) соединением переходного металла группы 4-6,
d) извлечение компонента твердого катализатора,
где частицы твердого носителя контактируют с органическим соединением внутреннего донора с формулой (I) или изомеры или их смеси перед обработкой частиц твердого носителя на стадии с), и где в формуле (I) или изомеры или их смеси R1-R5 представляют идентичные или отличающиеся и могут представлять водород, линейную или разветвленную C1-C8-алкильную группу, или С3-С8-алкиленовую группу, или два или более из R1-R5 могут образовать кольцо, и два, содержащих кислород кольца представляют независимо насыщенные или частично ненасыщенные или насыщенные, и где R в аддукте MgCl2*mROH представляет линейную или разветвленную алкильную группу с 1-12 атомами С, и m представляет 0-6.
Соответственно, органическое соединение внутреннего донора с формулой (I) контактирует с частицами твердого носителя перед обработкой частиц твердого носителя соединением переходного металла группы 4-6. Следовательно, органическое соединение внутреннего донора может контактировать с частицами твердого носителя перед стадией b), то есть, перед предварительной обработкой частиц твердого носителя соединением металла группы 13, или одновременно с указанной стадией предварительной обработки или после стадии b), но перед обработкой частиц твердого носителя соединением переходного металла группы 4-6.
Согласно настоящему изобретению катализатор используют согласно указанному выше в способе получения линейного полиэтилена низкой плотности при использовании многостадийного процесса.
Катализатор будет описан более подробно далее. Используемый в описании настоящей патентной заявки термин компонент катализатора Циглера-Натта (ZN) входит в объем понятия компонента катализатора, содержащего соединение переходного металла группы 4-6, соединение металла группы 13 Периодической таблицы (IUPAC, Nomenclature of Inorganic Chemistry (Номенклатура неорганической химии), 1989) и органическое соединение внутреннего донора, нанесенного на носитель на основе MgCl2.
Дигалоид магния используют в качестве сырьевого материала для получения носителя. Твердый носитель представляет носитель, где спирт связан с координационной связью с дигалоидом Mg, предпочтительно MgCl2. MgCl2 смешивают со спиртом (ROH) и образуется твердый носитель MgCl2*mROH согласно хорошо известным методам. В качестве примеров для получения галоида магния могут быть использованы методы распылительной сушки или кристаллизации при распылении. В настоящем изобретении используют сферический или гранулированный материал носителя MgCl2*mROH с различными размерами (5-100 μм). Спирт, используемый при получении материала носителя MgCl2*mROH, представляет спирт ROH, где R представляет линейную или разветвленную алкильную группу, содержащую 1-12 атомов углерода, предпочтительно 1-8 атомов углерода, более предпочтительно 1-4 атомов углерода. Как правило, используют этанол. В MgCl2*mROH m представляет от 0 до 6, предпочтительно от 1 до 4, более предпочтительно от 2,7 до 3,3.
MgCl2*mROH представляет доступный из коммерческих источников или может быть получен при использовании методов, описанных в предшествующем уровне техники. Методы получения носителя MgCl2*mROH описаны в нескольких патентах, например, в ЕР-А-376936, ЕР-А-424049, ЕР-А-655073 и ЕР-А-614467.
Соединение металла группы 13, используемое на стадии b), предпочтительно представляет соединение алюминия.
По существу предпочтительно соединение алюминия представляет соединение алюминия с формулой Al(алкил)хХ3-х, где каждый алкил независимо представляет алкильную группу с 1-12 атомами углерода, предпочтительно 1-8 атомами углерода, более предпочтительно 1-6 атомами углерода, X представляет галоид, предпочтительно хлор, и 1<х<3. Алкильная группа может представлять линейную, разветвленную или циклическую, или смесь таких групп.
Предпочтительные соединения алюминия представляют хлориды диалкил алюминия или соединения триалкил алюминия, например, хлорид диметил алюминия, хлорид диэтил алюминия, хлорид ди-изобутил алюминия и триэтилалюминий или их смеси. Наиболее предпочтительно соединение алюминия представляет соединение триалкил алюминия, в частности соединение триэтил алюминия.
Соединение переходного металла группы 4-6 предпочтительно представляет соединение переходного металла группы 4 или соединение ванадия, более предпочтительно соединение титана. По существу предпочтительно соединение титана представляет галоид, содержащий соединение титана с формулой XyTi(OR8)4-y, где R8 представляет C1-20 алкильную, предпочтительно С2-10 и более предпочтительно С2-8 алкильную группу, X представляет галоид, предпочтительно хлор, и у представляет 1, 2, 3 или 4, предпочтительно 3 или 4 и более предпочтительно 4.
Подходящие соединения титана включают монохлориды триалкокси титана, дихлорид диалкокси титана, трихлорид алкокси титана трихлорид и тетрахлорид титана. Предпочтительно используют тетрахлорид титана.
Органическое соединение внутреннего донора выбирают из соединений бициклического простого эфира с формулой (I) или изомеров или их смесей:
где в формуле (I) R1-R5 представляют идентичные или отличающиеся и могут представлять водород, линейную или разветвленную С1-C8-алкильную группу, или С3-С8-алкиленовую группу, или два или более из R1-R5 могут образовать кольцо, и два, содержащих кислород кольца представляют независимо насыщенные или частично ненасыщенные или насыщенные.
Примерами предпочтительных линейной или разветвленной C1-C8-алкильных групп являются метальная, этильная, n-пропильная, изо-пропильная, n-бутильная, сек-бутильная, трет-бутильная, пентильная и гексильные группы.
Примерами предпочтительных C3-C8-алкиленовых групп являются пентиленовая и бутиленовая группы.
Два R1 предпочтительно представляют идентичные и являются линейными C1-С4-алкильными группами, более предпочтительно метил или этил, наиболее предпочтительно оба R1 представляют метил; или два R1 образуют кольцо с атомом углерода, к которому они присоединены, предпочтительно кольцо с 3-7 атомами углерода, более предпочтительно циклопентильное или циклогексильное кольцо.
R2-R5 представляют идентичные или отличающиеся и предпочтительно представляют Н или C1-С2-алкильные группы, или два или более из R2-R5 остатков могут образовать кольцо. В случае, когда одно или более кольцо образовано остатками R2-R5, оно более предпочтительно образовано R3 и R4 и/или R4 и R5.
Предпочтительно остатки R2-R5 не образовывают кольца, и более предпочтительно максимально два из остатков R2-R5 представляют метил, другие представляют Н. Наиболее предпочтительно все R2-R5 представляют водород.
Оба содержащие кислород кольца могут представлять насыщенные или частично ненасыщенные или ненасыщенные. Каждое частично ненасыщенное или не насыщенной содержащее кислород кольцо может иметь одну или две двойные связи. Более предпочтительно оба содержащие кислород кольца представляют насыщенные.
В наиболее предпочтительном варианте осуществления настоящего изобретения используют 2,2-ди(2-тетрагидрофурил)пропан (DTHFP) с их изомерами. DTHFP, как правило представляет 1:1 моль/моль диастереоизомерную смесь D,L-(рац)-DTHFP и мезо-DTHFP.
При получении используемого в настоящем изобретении нанесенного на носитель компонента катализатора, по существу предпочтительно органическое соединение внутреннего донора, как указано выше, добавляют в смесь катализатора перед, во время или после предварительной обработки MgCl2*mROH соединением металла группы 13, но перед его обработкой переходным металлом группы 4-6.
Следовательно, компонент твердого катализатора по настоящему изобретению может быть получен при использовании процесса, включающего стадии:
i) обеспечения твердого MgCl2*mROH, где m представляет 1-4, и R представляет линейную или разветвленную алкильную группу, содержащую 1-8 атомов С;
ii) предварительной обработки частиц твердого носителя стадии i) соединением Al;
iii) добавления органического соединения внутреннего донора с формулой (I) в прошедший предварительную обработку твердый носитель стадии ii); или
iii') одновременно со стадией ii) добавление органического соединения внутреннего донора с формулой (I) в твердый носитель;
iv) обработки прошедших предварительную обработку частиц твердого носителя со стадии iii) или iii') TiCl4 и
v) извлечения компонента твердого катализатора.
Компонент твердого катализатора по настоящему изобретению также может быть получен при использовании способа, включающего стадии:
i) обеспечения твердого носителя MgCl2*mROH, где m представляет 1-4, и R представляет линейную или разветвленную алкильную группу, содержащую 1-8 атомов С;
ii-1) добавления органического соединения внутреннего донора с формулой (I) в твердый носитель стадии i);
iii-1) предварительной обработки частиц твердого носителя стадии ii-1) соединением Al;
iv-1) обработки прошедших предварительную обработку частиц твердого носителя со стадии iii-1) TiCl4 и
v-1) извлечения компонента твердого катализатора.
Согласно любому из указанных выше способов соединение Al может быть добавлено в твердый носитель перед или после добавления органического соединения внутреннего донора или одновременно с органическим соединением внутреннего донора в носитель.
Наиболее предпочтительно в указанных выше вариантах осуществления настоящего изобретения m=2,7-3,3, ROH представляет этанол, соединение алюминия представляет соединение триалкила алюминия, такое как триэтил алюминия, и органическое соединение внутреннего донора представляет 2,2-ди(2-тетрагидрофурил)пропан, или 2,2-ди-(2-фуран)-пропан, в частности 2,2-ди(2-тетрагидрофурил)пропан или их изомеры или их смеси.
Согласно способу получения катализатора по настоящему изобретению, предварительная обработка соединением металла группы 13, предпочтительно соединением алюминия, может быть проведена добавлением раствора указанного соединения алюминия в инертный органический растворитель, предпочтительно в инертный алифатический углеводородный растворитель, например, в гептан. Способ по настоящему изобретению позволяет использование концентрированного раствора соединения алюминия. В случае, когда используют триэтилалюминий (TEA), может быть использовано от 15 до 100 масс. % раствора TEA в инертный углеводородный растворитель, предпочтительно от 25 до 100 масс. % раствора TEA в инертный алифатический углеводородный растворитель, такой как гептан. Было обнаружено, что при использовании таких более концентрированных растворов, морфология сохраняется и снижается количество отходов.
Конечный компонент твердого катализатора, как правило, имеет соотношение Мг/Ti моль/моль от 1 до 10, предпочтительно от 2 до 8, предпочтительно от 3 до 7, соотношение Al/Ti моль/моль от 0,01 до 1, предпочтительно от 0,1 до 0,5, и соотношение Cl/Ti моль/моль от 5 до 20, предпочтительно от 10 до 17.
Частицы компонента твердого катализатора по настоящему изобретению могут представлять однородные по размеру частицы без мелких частиц или агломератов.
Нанесенный на носитель компонент катализатора, как указано выше, позволяет получить полимеры с повышенной молекулярной массой. Увеличение молекулярной массы происходит не за счет производительности катализатора. Производительность остается на надлежащем высоком уровне или даже повышенном по сравнению с применением компонента катализатора аналогичного типа, но при использовании отличающегося органического соединения внутреннего донора и/или полученного добавлением органического соединения внутреннего донора во время или после стадии обработки TiCl4, или при использовании указанного органического соединения в качестве внешнего донора. Следовательно, производительность катализатора, полученного при использовании способа по настоящему изобретению, делает возможным расширение технологического окна полиэтилена, так что возможна полимеризация с обоими: с более высоким и более низким количеством водорода, с сохранением при этом хорошей производительности.
Катализатор, используемый в процессе по настоящему изобретению, может содержать дополнительно к компоненту твердого катализатора, как указано выше, сокатализатор, который также известен, как активатор. Сокатализаторы представляют металлорганические соединения металла группы 13, как правило, соединения алюминия. Эти соединения включают галоиды алкилалюминия, предпочтительно хлориды алкилалюминия, такие как дихлорид этилалюминия, хлорид диэтилалюминия, сесквихлорид этилалюминия, хлорид диметилалюминия и аналогичное им. Они также включают соединения триалкилалюминия, такие как триметилалюминий, триэтилалюминий, три-изобутилалюминий, тригексилалюминий и три-n-октилалюминия. Также могут быть использованы другие соединения алкилалюминия, такие как изопренилалюминия. По существу предпочтительными сокатализаторами являются соединения триалкилалюминия, из которых по существу используют триэтилалюминий, триметилалюминий и триизобутилалюминий.
Катализатор по настоящему изобретению также может содержать добавку внешнего донора, такую как внешний донор. Добавки внешнего донора, которые могут быть использованы, включают, или соединения, как правило, внешние доноры типа тетрагидрофурана, силоксана или силана, и/или галоиды алкила, как известно из предшествующего уровня техники. Конечный компонент твердого катализатора, то есть, компонент твердого катализатора ZN, полученный по любому из указанных выше способов, комбинируют с соединением активатора.
Подходящими активаторами являются необязательно галогенированные (галоидзамещенные) сокатализаторы алкил алюминия с формулой (C1-С4-алкил)р-Al-Х3-р, где X представляет хлор, бром, йод или фтор; и р представляет 1, 2 или 3. С1-С4-алкильные группы могут представлять линейную или разветвленную или циклическую, или смесь таких групп. X предпочтительно представляет хлор или бром, наиболее предпочтительно X представляет хлор.
Подходящими активаторами являются, например, триметилалюминия (ТМА), триэтилалюминия (TEA) хлорид диметилалюминия (DMAC), хлорид диэтилалюминия (DEAC), хлорид диизобутилалюминия (DIBAC), дихлорид этилалюминия (EADC), дихлорид метилалюминия (MADC). Предпочтительным активатором в процессе по настоящему изобретению является триэтилалюминий.
Количество, в котором используют активатор, зависит от конкретного катализатора и активатора. Как правило, триэтилалюминий используют в таком количестве, чтобы молярное соотношение алюминия к переходному металлу, такое как Al/Ti, составляло от 1 до 1000, предпочтительно от 3 до 100 и по существу от около 5 до около 30 моль/моль.
Процесс полимеризации
Процесс полимеризации включает стадию первой полимеризации и стадию второй полимеризации. Дополнительно, процесс может включать дополнительные стадии полимеризации, например, для получения одного или более дополнительного компонента полимера или для предварительной полимеризации катализатора. Дополнительные стадии полимеризации могут предшествовать или завершать, как первую, так и вторую стадию полимеризации. Дополнительно, как первая, так и вторая стадии полимеризации могут быть разделены на две или более стадии, где либо первый гомо- или сополимер этилена, либо второй сополимер этилена получают в две или более стадии, где каждая такая стадия проводится в условиях получения, соответственно, первого гомо- или сополимера или второго сополимера.
Предварительная полимеризация
Стадиям полимеризации может предшествовать стадия предварительной полимеризации. Целью предварительной полимеризации является полимеризация малого количества полимера на катализаторе при низкой температуре и/или низкой концентрации мономера. Предварительная полимеризация позволяет улучшить производительность катализатора в суспензии и/или модифицировать свойства конечного полимера. Стадию предварительной полимеризации проводят в суспензии.
Следовательно, стадия предварительной полимеризации может быть проведена в циркуляционном реакторе. Предпочтительно предварительную полимеризацию проводят в инертном разбавителе, как правило, углеводородном разбавителе, таком как метан, этан, пропан, n-бутан, изобутан, пентан, гексан, гептан, октан и аналогичное им, или в их смесях. Предпочтительно разбавитель представляет углеводород с низкой точкой кипения с от 1 до 4 атомов углерода или смесь таких углеводородов.
Температура на стадии предварительной полимеризации, как правило, составляет от 0 до 90°C, предпочтительно от 20 до 80°C и более предпочтительно от 55 до 75°C.
Давление не является критичным показателем и, как правило, составляет от 1 до 150 бар, предпочтительно от 40 до 80 бар.
Количество мономера, как правило, составляет от около 0,1 до 1000 грамм мономера на один грамм компонента твердого катализатора, полимеризованного на стадии предварительной полимеризации. Как известно специалисту в области техники, к которой относится настоящее изобретение, частицы катализатора, полученные при непрерывной предварительной полимеризации в реакторе, не всегда содержат одинаковое количество форполимера. Наоборот, каждая частица имеет свои собственные количественные характеристики, которые зависят от времени пребывания этой частицы в реакторе предварительной полимеризации. Поскольку некоторые частицы остаются в реакторе в течение относительно длительного периода времени, а некоторые в течение относительно короткого периода времени, то также количество форполимера на различных частицах различно и некоторые отдельные частицы могут содержать количество форполимера, выходящее за указанные пределы. Однако среднее количество форполимера на катализаторе, как правило, находится в указанных выше пределах.
Молекулярная масса форполимера может контролироваться водородом, как это известно из предшествующего уровня техники. Дополнительно, для предотвращения адгезии частиц друг с другом или со стенками реактора может быть использована антистатическая добавка, как описано в WO-A-96/19503 и WO-A-96/32420.
В случае присутствия стадии предварительной полимеризации, компоненты катализатора предпочтительно все (отдельно или вместе) вводят на этой стадии предварительной полимеризации. Однако в случае, когда компонент твердого катализатора и сокатализатор могут быть введены по отдельности, то возможно введение только части сокатализатора на стадии предварительной полимеризации, а оставшуюся часть вводят на последующих стадиях полимеризации. Также в таких случаях на стадии предварительной полимеризации необходимо ввести столько сокатализатора, чтобы обеспечить достаточную реакцию полимеризации. Как правило, количество водорода и сомономера регулируют таким образом, чтобы присутствие форполимера не оказывало влияния на свойства конечного мультимодального полимера. По существу предпочтительно, чтобы скорость течения расплава форполимера была выше, чем скорость течения расплава конечного полимера, но ниже чем скорость течения расплава полимера, полученного на первой стадии полимеризации. Дополнительно, предпочтительно плотность форполимера выше, чем плотность конечного полимера. Предпочтительно плотность составляет около таковой или выше, чем плотность полимера, полученного на первой стадии полимеризации. Дополнительно, как правило, количество форполимера составляет не более чем около 5 масс. % мультимодального полимера, содержащего форполимер.
Первая стадия полимеризации
На первой стадии полимеризации получают первый гомо- или сополимер этилена. Это осуществляют введением катализатора полимеризации, необязательно с введением на стадии предварительной полимеризации или стадии, предшествующей полимеризации, как указано выше, на первой стадии полимеризации вместе с этиленом, водородом и необязательно сомономером альфа-олефина.
Водород вводят на первой стадии полимеризации для контроля MFR2 первого гомо- или сополимера этилена. Количество водорода составляет такое, что молярное соотношение водорода к этилену в жидкой реакционной смеси составляет в пределах от 200 до 50000 моль/кмоль (или моль/1000 моль), предпочтительно от 200 до 1000 моль/кмоль. В случае, когда первую стадию полимеризации проводят, как стадию суспензионной полимеризации, предпочтительно в циркуляционном реакторе, молярное соотношение водорода к этилену в жидкой реакционной смеси предпочтительно составляет от 200 до 1000 моль/кмоль, предпочтительно от 300 до 800 моль/кмоль.
Первый гомо- или сополимер этилена может представлять гомополимер. Следовательно, первый альфа-олефин не присутствует на первой стадии полимеризации. Водород присутствует в указанном выше количестве. Дополнительно, первый гомо- или сополимер этилена может представлять сополимер этилена и первого альфа-олефина. В таком случае молярное соотношение первого альфа-олефина к этилену в жидкой реакционной смеси составляет от 100 до 1000 моль/кмоль, предпочтительно от 200 до 800 моль/кмоль. Первый альфа-олефин предпочтительно выбирают из группы, состоящей из 1-бутена, 1-гексена и 4-метил-1-пентена, более предпочтительно состоящей из 1-бутена и 1-гексена. Также в этом варианте осуществления настоящего изобретения водород присутствует в указанном выше количестве.
Полученный непрерывным способом, как указано выше, первый гомо- или сополимер этилена как правило, имеет скорость течения расплава MFR2 от 100 до 1000 г/10 минут, предпочтительно от 150 до 750 г/10 минут и более предпочтительно от 200 до 600 г/10 минут. Дополнительно, первый сополимер, как правило, имеет плотность от 930 до 980 кг/м3, предпочтительно от 940 до 978 кг/м3 и наиболее предпочтительно от 945 до 976 кг/м3.
В случае, когда первый гомо- или сополимер этилена представляет сополимер этилена, предпочтительно он имеет плотность от 930 до 955 кг/м3, более предпочтительно от 940 до 953 кг/м3 и наиболее предпочтительно от 945 до 953 кг/м3.
Применяемые пределы MFR2 и плотности для первого гомо- или сополимера этилена известны специалисту в области техники, к которой относится настоящее изобретение. В случае, когда первой стадии полимеризации предшествует еще одна стадия полимеризации, на которой получают значительное количество полимера, указанные выше пределы MFR2 и плотности для первого гомо- или сополимера необязательно применимы для смеси полимера, содержащей полимеры, полученные на предшествующей стадии полимеризации и первой стадии полимеризации.
Первую стадию полимеризации предпочтительно проводят, как суспензионную полимеризацию. Суспензионную полимеризацию обычно проводят в инертном разбавителе, как правило, углеводородном разбавителе, таком как метан, этан, пропан, n-бутан, изобутан, пентан, гексан, гептан, октан и аналогичное им, или их смесях. Предпочтительно разбавитель представляет углеводород с низкой точкой кипения с от 1 до 4 атомов углерода или смесь таких углеводородов. По существу предпочтительный разбавитель представляет пропан, возможно содержащий незначительные количества метана, этана и/или бутана.
Содержание этилена в жидкой реакционной смеси может составлять от 1 до около 50% по молям, предпочтительно от около 2 до около 20% по молям и по существу от около 2 до около 10% по молям. Преимуществом высокой концентрации этилена является повышение производительности катализатора, а недостатком - необходимость повторной переработки большего количества этилена, чем при более низкой концентрации.
Температура на первой стадии полимеризации, как правило, составляет от 60 до 100°C, предпочтительно от 70 до 95°C. Следует избегать слишком высокой температуры для предотвращения частичного растворения полимера в разбавителе и загрязнения реактора. Давление составляет от 1 до 150 бар, предпочтительно от 40 до 80 бар.
Суспензионная полимеризация может быть проведена в любом известном реакторе, используемом для суспензионной полимеризации. Такие реакторы включают реактор непрерывного действия с механическим перемешиванием и циркуляционный реактор. По существу предпочтительно проводить полимеризацию в циркуляционном реакторе. В таких реакторах суспензия циркулирует с высокой скоростью по закрытому трубопроводу за счет циркуляционного насоса. Циркуляционные реакторы общеизвестны из предшествующего уровня техники и примеры приведены, например, в US-A-4582816, US-A-3405109, US-A-3324093, ЕР-А-479186 и US-A-5391654. Следовательно, предпочтительно проводить первую стадию полимеризации, как суспензионную полимеризацию, в одном или более циркуляционном реакторе, более предпочтительно в одном циркуляционном реакторе.
Суспензию из реактора можно отводить непрерывно или периодически. Предпочтительный способ периодического отвода заключается в использовании седиментационных колонок, где суспензия концентрируется перед выводом партии концентрированной суспензии из реактора. Использование колонок для седиментации описано, помимо прочих, в документах US-A-3374211, US-A-3242150 и ЕР-А-1310295. Непрерывный отвод описан, помимо прочих, в документах ЕР-А-891990, ЕР-А-1415999, ЕР-А-1591460 и WO-A-2007/025640. Непрерывный отвод предпочтительно сочетать с подходящим способом концентрирования, как описано в ЕР-А-1310295 и ЕР-А-1591460. Предпочтительно отводить суспензию с первой стадия полимеризации непрерывно.
В случае, когда первый гомо- или сополимер этилена представляет первый сополимер этилена, первый сомономер альфа-олефина вводят на первой стадии полимеризации для контроля плотности первого сополимера этилена. Количество сомономера, необходимое для достижения заданной плотности, зависит от типа сомономера, используемого катализатора и условий полимеризации, в частности от соотношения Н2/С2.
Содержание водорода, этилена и первого сомономера альфа-олефина может быть измерено, как известно из области техники, к которой относится настоящее изобретение, отводом потока образца из реактора или из отводимого из реактора потока, как описано в WO-A-1996035936, WO-A-1994027134 и ЕР-А-460594. Предпочтительно такой поток образца отводят на стадии снижения давления или стадии испарения, между первой и второй стадией полимеризации.
Среднее время пребывания на первой стадии полимеризации, как правило, составляет от 20 до 120 минут, предпочтительно от 20 до 70 минут. Как хорошо известно из области техники, к которой относится настоящее изобретение, среднее время пребывания τ может быть рассчитано, как:
τ=Vr/Qo,
где Vr - объем реакционного пространства (в случае циркуляционного реактора - объем реактора, в случае реактора с псевдоожиженным слоем - объем псевдоожиженного слоя), и Qo - объемная скорость потока продукта (включая полимерный продукт и жидкую реакционную смесь).
Возможно, а иногда предпочтительно, проводить первую стадию полимеризации с более чем одной стадией, например с двумя стадиями. В случае, когда первую стадию полимеризации проводят с более чем одной стадией, первый гомо- или сополимер этилена представляет смесь двух или более гомо- или сополимеров этилена. В таком случае все такие стадии следует проводить в указанных выше условиях. Дополнительно, количество первого гомо- или сополимера этилена представляет сумму количеств полимеров, полученных на всех таких стадиях.
Дополнительно, как указано выше, перед первой стадией полимеризации возможно проведение одной или более дополнительной стадии полимеризации, когда получают полимер, отличающийся от первого гомо- или сополимера этилена.
Вторая стадия полимеризации
На второй стадии полимеризации получают смесь полимера, содержащую первый гомо- или сополимер этилена и второй сополимер этилена. Это осуществляют введением частиц первого гомо- или сополимера этилена, содержащих активный катализатор, диспергированный в них, вместе с дополнительным этиленом и вторым сомономером альфа-олефина на вторую стадию полимеризации. Для контроля молекулярной масса может быть введен водород. Это приводит к образованию второго сополимера этилена на частицах, содержащих первый гомо- или сополимер этилена.
Сополимер этилена может иметь MFR5 от 0,3 до 12,0 г/10 минут. Сополимер этилена предпочтительно может иметь MFR21 от 20 до 180 г/10 минут, более предпочтительно от 25 до 100 г/10 минут. Дополнительно, предпочтительно он имеет соотношение скорости течения потока FRR21/5 от 10 до 50, более предпочтительно от 15 до 40.
Второй сомономер альфа-олефина выбирают из альфа-олефина, содержащего от 4 до 10 атомов углерода. Второй сомономер альфа-олефина может быть идентичным или отличающимся от первого сомономера альфа-олефина, в случае присутствия первого сомономера альфа-олефина. В одном предпочтительном варианте осуществления настоящего изобретения первый сомономер альфа-олефина и второй сомономер альфа-олефина представляют идентичные, такие как 1-бутен или 1-гексен, по существу предпочтительно 1-бутен. В другом предпочтительном варианте осуществления настоящего изобретения первый сомономер альфа-олефина отличается от второго сомономера альфа-олефина. Первый сомономер альфа-олефина может представлять 1-бутен, а второй сомономер альфа-олефина может представлять 1-гексен или 1-октен, более предпочтительно 1-гексен. В другом варианте осуществления настоящего изобретения первый сомономер альфа-олефина отсутствует, а второй сомономер альфа-олефина представляет 1-бутен, 1-гексен или 1-октен, или их смесь, предпочтительно 1-гексен.
Смесь полимера имеет плотность от 910 до 940 кг/м3, более предпочтительно от 915 до 930 кг/м3 и наиболее предпочтительно от 918 до 927 кг/м3.
MFR21 второго сополимера этилена не может быть измерена, поскольку второй сополимер не может быть выделен из смеси полимера.
Для достижения заданной скорости течения расплава (или молекулярной массы) смеси полимера регулируют подачу водорода. Предпочтительно подачу водорода регулируют для поддержания постоянного соотношения водорода к этилену в жидкой реакционной смеси. Фактическое соотношение зависит от катализатора наряду с типом полимеризации. Заданные свойства полимера получают при газофазной полимеризации в реакторе с псевдоожиденным слоем за счет поддержания соотношения в газовой фазе в пределах от 10 до 200 моль/кмоль, предпочтительно от 50 до 200 моль/кмоль, такое как от 100 до 175 моль/кмоль.
Второй сомономер альфа-олефина, как правило, вводят для поддержания постоянного соотношения сомономера к этилену в жидкой реакционной смеси. Соотношение сомономера к этилену, необходимое для получения полимера с заданной плотностью, зависит среди прочего от типа сомономера типа катализатора. При использовании 1-гексена в качестве сомономера заданные свойства полимера могут быть достигнуты при газофазной полимеризации в реакторе с псевдоожиженным слоем с молярным соотношением 1-гексена к этилену в газовой фазе от 50 до 400 моль/кмоль, предпочтительно от 100 до 250 моль/кмоль и по существу предпочтительно от 120 до 220 моль/кмоль.
Предпочтительно вторую стадию полимеризации проводят, как газофазную полимеризацию в псевдоожиженном слое. В газофазном реакторе с псевдоожиженным слоем олефин полимеризуют в присутствии катализатора полимеризации в восходящем газовом потоке. В реакторе, как правило, находится псевдоожиженный слой, включающий растущие полимерные частицы, содержащие активный катализатор, расположенный выше псевдоожиженного слоя.
Слой полимера сжижают, используя ожижающий газ, содержащий олефиновый мономер, возможно сомономер(ы), возможно регулятор роста цепи или агенты переноса цепи, такие как водород, и возможно инертный газ. Ожижающий газ вводят во входную камеру, расположенную на дне реактора. В целях обеспечения равномерного распределения газа вдоль площади поверхности поперечного сечения входной камеры, входную трубу можно снабдить приспособлением распределения потока, известным из современного уровня техники, например US-A-4933149 и ЕР-А-684871. Один или более из указанных выше компонентов может быть непрерывно добавлен в ожижающий газ для компенсации потерь, вызванных, среди прочего, реакцией или отводом продукта.
Из входной камеры газовый поток проходит вверх через решетку для ожижения в псевдоожиженный слой. Назначением решетки ожижения является равномерное разделение газового потока по поперечной площади слоя. Иногда решетки для ожижения объединяют для направления газового потока вдоль стенок реактора, как описано в документе WO-A-2005/087361. Другие типы решеток для ожижения описаны, помимо прочих, в US-A-4578879, ЕР-А-600414 и ЕР-А-721798. Обзор приведен в Gelbart и Bayens: The Design of Distributors for Gas-Fluidized Beds, Powder Technology, Vol. 42, 1985.
Ожижающий газ проходит через псевдоожиженный слой. Поверхностная скорость потока ожижающего газа должна быть выше минимальной скорости ожижения частиц, содержащихся в псевдоожиженном слое, иначе ожижение не будет происходить. С другой стороны, скорость газа должна быть ниже начальной скорости пневматического транспорта, в противном случае ожижающим газом будет захватываться весь слой. Минимальная скорость ожижения и начальная скорость пневматического транспорта может быть вычислена, если с помощью общепринятых инженерных способов установлены характеристики частиц. Обзор приведен, среди прочих, в работе Geldart: Gas Fluidization Technology, Willey & Sons, 1986.
Когда ожижающий газ контактирует со слоем, содержащим активный катализатор, реакционные компоненты газа, такие как мономеры и агенты переноса цепи, взаимодействуют в присутствии катализатора с образованием полимерного продукта. В тоже время газ нагревается благодаря тепловому эффекту реакции.
Непрореагировавший ожижижающий газ удаляется из верней части реактора и охлаждается в теплообменнике, высвобождая теплоту реакции. Газ охлаждается до температуры ниже температуры слоя в целях предотвращения нагревания слоя в результате взаимодействия. Возможно охлаждать газ до температуры частичной конденсации газа. При попадании капель жидкости в реакционную зону, они испаряются. Теплота испарения создает дополнительный вклад в остаточную теплоту реакции. Такой способ управления называется режимом конденсации, и некоторые его разновидности описаны, среди прочих, в документах WO-A-2 007/025640, US-A-4543399, ЕР-А-699213, WO-A-94/25495. Также возможно добавлять конденсирующие агенты к повторно используемому газовому потоку, как описано в ЕР-А-696293. Агенты конденсации являются компонентами, не подвергающимися полимеризации, такими как н-пентан, изопентан, н-бутан или изобутен, которые по меньшей мере частично конденсируются в холодильнике.
Далее газ сжимают и повторно направляют во входную камеру реактора. Перед подачей в реактор в ожижающий газовый поток вводят свежие реагенты в целях компенсации потерь, вызванных взаимодействием и выводом продукта. Как правило, анализируют композицию ожижающего газа, и вводят компоненты газа для сохранения постоянства композиции. Фактическую композицию определяют, исходя из желаемых свойств продукта и катализатора, используемого в полимеризации.
Катализатор можно вводить в реактор различными способами, как непрерывно, так и периодически. Среди прочих, в документах WO-A-01/05845 и ЕР-А-499759 описаны подобные способы.
Поскольку газофазный реактор является частью каскадного реактора, катализатор, как правило, распределяют среди полимерных частиц, образовавшихся в предыдущей стадии полимеризации. Полимерные частицы можно вводить в газофазный реактор способом, описанным в документах ЕР-А-1415999 и WO-A-00/26258.
Полимерный продукт можно отводить из газофазного реактора либо непрерывно, либо периодически. Можно также комбинировать эти способы. Непрерывный отвод описан, помимо прочих, в документе WO-A-00/29452. Периодический отвод описан, среди прочего, в документах US-A-4621952, ЕР-А-188125, ЕР-А-250169 и ЕР-А-579426.
Верхняя часть газофазного реактора может быть включена в так называемую зону разъединения. В такой зоне диаметр реактора увеличивается с целью снижения скорости газового потока, что позволяет частицам, уносимым из слоя ожижающим газом, возвращаться обратно в слой.
Уровень слоя можно определить различными способами, известными из современного уровня техники. Например, можно определить разницу в давлении между днищем реактора и определенным уровнем слоя вдоль всей длины реактора, уровень слоя можно рассчитать, исходя из величин разности давления. Подобный расчет приводит к получению значения уровня, усредненного по времени. Также возможно использовать ультразвуковые или радиоактивные датчики. С помощью этих способов можно определить уровень непосредственно в данный момент времени, конечно, далее эти данные можно усреднить по времени.
Также при необходимости можно вводить в газофазный реактор антистатические добавки. Подходящие антистатические добавки и способы использования их описаны, среди прочих, в документах US-A-5026795, US-A-4803251, US-A-4532311, US-A-4855370 и ЕР-А-560035. Как правило, они являются полярными соединениями и включают, среди прочего, воду, кетоны, альдегиды и спирты.
Реактор может также содержать механическую мешалку для обеспечения дополнительного перемешивания в псевдоожиженном слое. Пример подходящего устройства для перемешивания описан в документе ЕР-А-707513.
Как правило, полимеризационный реактор с псевдоожиженным слоем работает при температуре в пределах от 50 до 100°C, предпочтительно от 65 до 90°C. Подходящее давление составляет от 10 до 40 бар, предпочтительно от 15 до 30 бар.
Среднее время пребывания на второй стадии полимеризации, как правило, составляет от 40 до 240 минут, предпочтительно от 60 до 180 минут.
Как указано выше, предпочтительно проводить вторую стадию полимеризации в газовой фазе в одном или более газофазном реакторе, более предпочтительно в одном реакторе с псевдоожиженным слоем.
Смесь полимера, как правило, содержит от 25 до 57 масс. % первого гомо- или сополимер и от 43 до 75 масс. % второго сополимера. Предпочтительно смесь полимера содержит от 35 до 57 масс. % первого гомо- или сополимер этилена и от 43 до 65 масс. % второго сополимера этилена. Смесь полимера может содержать иные полимеры дополнительно к первому гомо- или сополимеру этилена и второму сополимеру этилена, но содержание первого гомо- или сополимера этилена и второго сополимера этилена должны находиться в указанных выше пределах.
Наиболее предпочтительно процесс полимеризации по настоящему изобретению проводят в каскадной последовательности, включающей по меньшей мере один циркуляционный реактор со следующим за ним по меньшей мере одним газофазным реактором.
Экструзия
После извлечения смеси полимера из реактора полимеризации ее подвергают технологической обработке на стадиях удаления остаточных углеводородов из полимера. Такие процессы технологической обработки хорошо известны из области техники, к которой относится настоящее изобретение, и могут включать стадии снижения давления, стадии продувки, стадии отгонки, стадии экстракции и аналогичное им. Также возможны комбинации различных стадий.
После удаления остаточных углеводородов полимер предпочтительно смешивают с добавками, как хорошо известно из области техники, к которой относится настоящее изобретение. Такие добавки включают антиоксиданты, стабилизаторы процесса, нейтрализаторы, лубриканты, нуклеирующие агенты, пигменты и аналогичное им.
По настоящему изобретению во время экструзии в композицию сополимера этилена может быть добавлено от 0,1 до 5 масс. % углеродной сажи, предпочтительно от 2 до 3 масс. %. Углеродная сажа предпочтительно представляет углеродную сажу типа N220.
Полимерные частицы смешивают с добавками и экструдируют с получением гранул, как известно из области техники, к которой относится настоящее изобретение. Предпочтительно на стадии экструзии используют двухшнековый экструдер со шнеками, вращающимися во встречном направлении.
Композиция полимера
Сополимер этилена может быть получен согласно указанному выше способу и согласно всем указанным вариантам осуществления настоящего изобретения. Сополимер по настоящему изобретению представляет композицию мультимодального сополимера этилена с плотностью от 920 до 949 кг/м3 и модулем упругости при изгибе, где модуль упругости при изгибе представляет следующее уравнение:
Сополимер этилена предпочтительно представляет линейный полиэтилен низкой плотности (LLDPE). Сополимер этилена предпочтительно получают проведением по меньшей мере двухстадийной полимеризация, предпочтительно с использованием непрерывного процесса.
Композиция мультимодального сополимера этилена может содержать по меньшей мере один сомономер альфа-олефина С4-С10, более предпочтительно по меньшей мере два сомономера альфа-олефина(ов), еще более предпочтительно по меньшей мере первый сомономер альфа-олефина, содержащий 1-гексен, и второй сомономер альфа-олифена, отличающийся от первого сомономера альфа-олефина. Предпочтительно сополимер этилена содержит по меньшей мере сомономеры 1-бутена и 1-гексена.
Композиция мультимодального сополимера этилена предпочтительно содержит по меньшей мере 80 масс. % сополимера этилена, более предпочтительно по меньшей мере 90 масс. %.
Сополимер этилена по настоящему изобретению может быть получен при использовании катализатора Циглера-Натта, предпочтительно органического соединения внутреннего донора с формулой соединения, представленной формулой (I):
где в формуле (I) R1-R5 представляют идентичные или отличающиеся и могут представлять водород, линейную или разветвленную C1-C8-алкильную группу, или С3-C8-алкиленовую группу, или два или более из R1-R5 могут образовать кольцо, и два, содержащих кислород кольца представляют независимо насыщенные или частично ненасыщенные или насыщенные.
MFR2 композиции мультимодального сополимера этилена может составлять от 0,1 до 4 г/10 минут, предпочтительно от 0,5 до 2 г/10 минут и наиболее предпочтительно от 0,7 до 1,5 г/10 минут.
MFR5 композиции мультимодального сополимера этилена может составлять от 0,3 до 12 г/10 минут, более предпочтительно от 2 до 7 г/10 минут и наиболее предпочтительно от 2 до 5 г/10 минут.
MFR21 композиции мультимодального сополимера этилена может составлять от 20 до 180 г/10 минут, более предпочтительно от 40 до 120 г/10 минут и наиболее предпочтительно от 60 до 100 г/10 минут.
FRR21/5 композиции мультимодального сополимера этилена может составлять от 10 до 50, более предпочтительно от 15 до 40,
Твердость по Шору D (1s) композиции мультимодального сополимера этилена может составлять по меньшей мере 50, более предпочтительно по меньшей мере 53. Чем выше твердость по Шору D (1s), тем выше резистентность поверхности из композиции мультимодального сополимера этилена к механическому воздействию.
Композиция мультимодального сополимера этилена по настоящему изобретению может иметь модуль упругости при изгибе согласно следующему уравнению:

Композиция мультимодального сополимера этилена предпочтительно может иметь плотность от 930 до 949 кг/м3, предпочтительно от 930 до 940 кг/м3. Один из объектов настоящего изобретения позволяет снизить плотность с повышением, таким образом, гибкости, с сохранением при этом твердости по Шору D (1s).
Углеродная сажа может быть добавлена в композицию мультимодального сополимера этилена предпочтительно от 0,1 до 5 масс. % углерода, более предпочтительно от 0,5 до 3 масс. %, наиболее предпочтительно от 1 до 3 масс. %. Углеродная сажа предпочтительно представляет УФ углеродную сажу (UV carbon black), такую как углеродная сажа типа N220. Углеродная сажа может быть добавлена либо во время экструзии сразу же после реактора полимеризации или на отдельной стадии. Также углеродная сажа может быть добавлена в качестве мастербатча углеродной сажи непосредственно в экструдер, экструдирующий оболочку кабеля. MFR2, MFR5, MFR21 и FRR практически не изменяются при добавлении углеродной сажи.
Композиция мультимодального сополимера этилена по настоящему изобретению может дополнительно содержать традиционные добавки, такие как антиоксиданты, пигменты, УФ добавки и технологические добавки, как правило, в количестве от 0,1 масс. % до 5 масс. %.
Композиция мультимодального сополимера этилена по настоящему изобретению может быть получена по меньшей мере двухстадийной полимеризацией.
Настоящее изобретение также относится к оболочке кабеля, содержащей указанную выше композицию мультимодального сополимера этилена.
Дополнительно настоящее изобретение относится к силовому кабелю или коммуникационному кабелю, содержащему оболочку кабеля, содержащую композицию мультимодального сополимера этилена, предпочтительно к силовому кабелю, содержащему внутренний полупроводящий слой, за которым следует изолирующий слой и затем внешний полупроводящий слой. Оболочка кабеля предпочтительно состоит из композиции мультимодального сополимера этилена по настоящему изобретению.
ДЕТАЛЬНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
МЕТОДЫ ТЕСТИРОВАНИЯ
Скорость течения расплава
Скорость течения расплава (MFR) измерили согласно 1133 при температуре 190°C. Нагрузка, при которой проводят измерение, приведена как нижний индекс. Следовательно, MFR под нагрузкой 2,16 кг обозначена как MFR2. Скорость течения расплава MFR21 соответственно определяют при температуре 190°C под нагрузкой 21,6 кг, и MFR5 под нагрузкой 5 кг.
Плотность
Плотность полимера измеряют согласно ISO 1183-1:2004 Method А с использованием образцов, полученных в формах литьем под давлением согласно EN ISO 1872-2 (Feb 2007) и приводят в кг/м3.
Композиция газа в реакторе
Композиция газа в реакторе в суспензионном реакторе может быть измерена, как хорошо известно в области техники, к которой относится настоящее изобретение при испарении газа после реактора при использовании он-лайн газовой хроматографии, как описано, например, в WO-A-1996035936.
Композиция газа в реакторе в газофазном реакторе может быть проанализирована при использовании циркуляционного газа при использовании он-лайн газовой хроматографии, как хорошо известно в области техники, к которой относится настоящее изобретение.
Инструменты калибруют, как хорошо известно в области техники, к которой относится настоящее изобретение, при использовании калибровочной газовой смеси с известной композицией, близкой к таковой смеси газов, присутствующей в процессе полимеризации.
Тест на изгиб (модуль упругости при изгибе)
Модуль упругости при изгибе отражает гибкость материала. Чем выше модуль упругости при изгибе, тем ниже гибкость материала, то есть, материал сложнее деформировать заданной нагрузкой.
Тест на изгиб может быть проведен согласно методу ISO 178 с использованием образцов, полученных в формах литьем под давлением согласно EN ISO 1872-2.
Прямоугольный образец 80×4×10 мм поместили между двумя опорами. Далее на образец оказали давление приложением усилия при использовании нагружающего валика, размещенного посередине образца со скоростью 2 мм/минуту. В этом случае исследовали только модуль упругости при изгибе и, следовательно, измерили изгибающее усилие (деформация при изгибе) в пределах от 0,05% до 0,25%, по которому рассчитали модуль упругости при изгибе.
Тест на разрыв при растяжении (прочность при растяжении)
Тест на разрыв при растяжении измерили согласно ISO 527.
Скорость ползуна для тестирования на прочность при растяжении и удлинение составила 50 мм/минуту.
Образцы для тестирования получили, как описано EN ISO 1872-2, тип образца: 5А согласно ISO 527-2.
Твердость по Шору D
Твердость по Шору D (1s) определили согласно ISO868 при использовании образцов, полученных литьем в форме толщиной 4 мм. Твердость по Шору определили после 1 секунды плотного контакта прижимного устройства с тестируемым образцом. Образец получили литьем в форме согласно EN ISO 1872-2.
Количественный анализ микроструктуры с использованием ЯМР спектроскопии.
Количественную спектроскопию ядерно-магнитного резонанса (ЯМР) используют для оценки содержания сомономера в полимере.
Количественный анализ13С{1Н} ЯМР спектра записывают в состоянии расплава при использовании ЯМР спектрометра Bruker Advance III 500, работающего на частотах в пределах от 500,13 до 125,76 МГц для1Н и13С, соответственно. Весь спектр записывают при использовании13С оптимизированного 7 мм датчика измерения линейных величин под магическим углом вращения (MAS) при температуре 180°C при использовании во всей пневматике газообразного азота. Около 200 мг материала помещают в циркониевый MAS ротор с внешним диаметром 7 мм и скручивают при 4 кГц. Такая схема была выбрана главным образом в виду ее высокой чувствительности, необходимой для быстрого определения и точного количественного описания {klimke06, parkinson07, castignolles09}.
Создают стандартное одноимпульсное возбуждение при использовании NOE (ядерный эффект Оверхауза) с кратковременной задержкой повторного цикла {pollard04, klimke06} и схемой развязки RS-HEPT {fillip05, griffin07}. Всего для спектра потребовалось 1024 (1k) импульсов.
Количественные13С{1Н} ЯМР спектры обработали при использовании интегралов с определением на их основе количественных свойств. Все химические сдвиги внутренне привязаны к сигналу объемного метилена (δ+) при 30,00 частей на миллион.
Количество этилена определили при использовании интеграла сайтов метилен (δ+) при 30,00 частей на миллион с учетом количества указанных сайтов на мономер:
Е=Iδ+/2
Наличие изолированных сомономерных единиц корректируют, исходя из количества присутствующих изолированных сомономерных единиц:
Etotal(общее)=Е+(3⋅В+2⋅Н)/2
Total - общее
где В и Н определены их соответствующими сомономерами. Коррекция для последовательного и непоследовательного встраивания сомономера, при наличии, осуществляют аналогичным образом.
Наблюдали характерные сигналы, соответствующие встраиванию 1-бутена, и фракцию сомономера рассчитали, как фракцию 1-бутена в полимере относительно всех мономеров в полимере:
fBtotal=Btotal/(Etotal+Btotal+Htotal)
Total - (общее)
Количество изолированного 1-бутена, встроенного в ЕЕВЕЕ последовательности, количественно определяют при использовании интеграла *В2 сайта при 38,3 частей на миллион с учетом количества указанных сайтов на сомономер:
В=I*В2
Количество последовательно встроенного 1-бутена в ЕЕВВЕЕ последовательности количественно определяют при использовании интеграла ααВ2В2 сайта при 39,4 частей на миллион с учетом количества указанных сайтов на сомономер:
ВВ=2⋅IααВ2В2
Количество непоследовательно встроенного 1-бутена в ЕЕВЕВЕЕ последовательности количественно определяют при использовании интеграла ββВ2В2 сайта при 24,7 частей на миллион с учетом количества указанных сайтов на сомономер:
ВЕВ=2⋅IββВ2В2
Из-за перекрытия *В2 и *βВ2В2 сайтов изолированного (ЕЕВЕЕ) и непоследовательно встроенного (ЕЕВЕВЕЕ) 1-бутена, соответственно, общее количество изолированного 1-бутена корректируют, исходя из количества присутствующего непоследовательно встроенного 1-бутена:
В=I*B2-2⋅IββB2B2
Общее содержание 1-бутена рассчитали, исходя из суммы изолированного последовательно и непоследовательно встроенного 1-бутена:
Btotal=В+ВВ+ВЕВ
Total - общее
Общую молярную фракцию 1-бутена в полимере рассчитали, как следующее:
fB=Btotal/(Etotal+Btotal+Htotal)
Total - общее
Наблюдали характерные сигналы, соответствующие встраиванию 1-гексена, и фракцию сомономера рассчитали, как фракцию 1-гексена в полимере относительно всех мономеров в полимере:
fHtotal=Htotal/(Etotal+Btotal+Htotal)
Total - общее
Количество изолированного 1-гексена, встроенного в ЕЕНЕЕ последовательности, количественно определяют при использовании интеграла *В4 сайта при 39,9 частей на миллион с учетом количества указанных сайтов на сомономер:
Н=I*В4
Количество последовательно встроенного 1-гексена в ЕЕННЕЕ последовательности количественно определяют при использовании интеграла ααВ4В4 сайта при 40,5 частей на миллион с учетом количества указанных сайтов на сомономер:
НН=2⋅IααВ4В4
Количество непоследовательно встроенного 1-гексена в ЕЕНЕНЕЕ последовательности количественно определяют при использовании интеграла ββВ4В4 сайта при 24,7 частей на миллион с учетом количества указанных сайтов на сомономер:
НЕН=2⋅IββB4B4
Общую молярную фракцию 1-гексена в полимере рассчитали, как:
fH=Htotal/(Etotal+Btotal+Htotal)
Total - общее
Молярный процент введенного сомономера рассчитали по молярной фракции:
В[мол. %]=100⋅fB
Н[мол. %]=100⋅fH
Массовый процент введенного сомономера рассчитали по молярной фракции:
В [масс. %]=100⋅(fB⋅56.11)/((fB⋅56.11)+(fH⋅84.16)+((1-(fB+fH))⋅28.05))
Н [масс. %]=100⋅(fH⋅84.16)/((fB⋅56.11)+(fH⋅84.16)+((1-(fB+fH))⋅28.05))
Ссылки:
klimke06
Klimke, K., Parkinson, M., Piel, С., Kaminsky, W., Spiess, H.W., Wilhelm, M., Macromol. Chem. Phys. 2006; 207:382.
parkinson07
Parkinson, M., Klimke, K., Spiess, H.W., Wilhelm, M., Macromol. Chem. Phys. 2007; 208:2128.
pollard04
Pollard, M., Klimke, K., Graf, R., Spiess, H.W., Wilhelm, M., Sperber, O., Piel, C., Kaminsky, W., Macromolecules 2004; 37:813.
filip05
Filip, X., Tripon, С, Filip, С, J. Mag. Resn. 2005, 176, 239
griffin07
Griffin, J.M., Tripon, С., Samoson, A., Filip, C., and Brown, S.P., Mag. Res. in Chem. 2007 45, S1, S198
castignolles09
Castignolles, P., Graf, R., Parkinson, M., Wilhelm, M., Gaborieau, M., Полимер 50 (2009) 2373
busico01
Busico, V., Cipullo, R., Prog. Polym. Sci. 26 (2001) 443
busico97
Busico, V., Cipullo, R., Монет данныхсо, G., Vacatello, M., Segre, A.L., Macromoleucles 30 (1997) 6251
zhou07
Zhou, Z., Kuemmerle, R., Qiu, X., Redwine, D., Cong, R., Taha, A., Baugh, D. Winniford, В.,
J. Mag. Reson. 187 (2007) 225
busico07
Busico, V., Carbonniere, P., Cipullo, R., Pellecchia, R., Severn, J., Talarico, G., Macromol.
Rapid Commun. 2007, 28, 1128
resconi00
Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253
Материалы
Катализатор 1
Получение комплекса
В 100 литровый реактор добавили толулол (87 кг). Затем в реактор добавили Bomag А от Chemtura, (45,5 кг, 20 масс. % бутилоктил магния в гептане). Далее в реактор подали 2-этил-1-гексанол (161 кг, 99,8 масс. %) при скорости потока 24-40 кг/час. Молярное соотношение между BOMAG-A и 2-этил-1-гексанолом составило 1:1,83.
Получение компонент твердого катализатора
10 275 кг кремния (ES747JR от Crossfield, со средним размером частиц 20 μм) активировали при температуре 600°C в азоте, которым заполнили реактор для получения катализатора. Затем в реактор при комнатной температуре в течение часа добавили 411 кг 20% EADC (2,0 ммоль/г кремния) разведенного в 555 литрах. Затем температуру повысили до 35°C при перемешивании прошедшего обработку кремния в течение одного часа. Кремний сушили при температуре 50°C в течение 8,5 часов. Далее добавили 655 кг комплекса, полученного, как указано выше (2 ммоль Мг/г кремния), при 23°C в течение десяти минут. В реактор добавили 86 кг пентана при 22°C в течение десяти минут. Суспензию перемешивали в течение 8 часов при температуре 50°C. Наконец, добавили 52 кг TiCl4 в течение 0,5 часов при 45°C. Суспензию перемешивали при 40°C в течение пяти часов. Затем катализатор высушили продувкой азотом.
Катализатор 2
Получение прошедшего предварительную обработку материала подложки
Использовали реактор из нержавеющей стали с рубашкой объемом 160 дм3, снабженный спиральным перемешивающим элементом, при использовании N2 повышали давление 2,0 бар избыточного давления и понижали до 0,2 бар избыточного давления до достижения уровня O2 менее чем 3 частей на миллион. Затем емкость заполнили гептаном (20,5 кг) и 2,2-ди(тетрагидрофурил)пропаном (0,512 кг; 79 моль; DTHFP). Полученную смесь перемешивали в течение 20 минут при 40 оборотов в минуту. MgCl2⋅3EtOH носитель (6,5 кг; DTHFP/Mg=0,1 моль/моль; 27,2 моль Mg; Mg 10,18 масс. %, d10=9,5 μм, d50=17,3 μм и d90=28,5 μм, в форме гранул) добавили в реактор при перемешивании. Полученную суспензию охладили до около -20°C и в гептан добавили 33 масс. % раствор триэтилалюминий (29,4 кг, 85,0 моль Al; Al/EtOH=1,0 моль/моль) в аликвотах в течение 2,5 часов при поддержании температуры ниже 10°C. После добавления TEA реакционную смесь постепенно нагрели до 80°C в течение 2,4 часа и выдержали при этой температуре в течение дополнительных 10 минут провели отстаивание суспензии, и удалили маточный раствор через 10 μм фильтр из сетки в дне реактора в течение 15 минут. Емкость заполнили теплым толуолом (43 кг) и затем перемешали при 40 оборотах в минуту в течение 38 минут при температуре от 55 до 70°C. Суспензию отстояли в течение 10 минут при температуре от 50 до 55°C и удалили жидкость через 10 μм фильтр из сетки в дне реактора в течение 15 минут.
Получение катализатора
Емкость, содержащую прошедший предварительную обработку материал подложки, заполнили толуолом (43 кг) и затем охладили до температуры около 30°C. Добавили чистый TiCl4 (5,17 кг, 27,5 моль; Ti/Mg=1,0 моль/моль). Полученную суспензию нагрели до температуры около 90°C в течение 2 часов и выдержали при этой температуре в течение еще одного дополнительного часа при перемешивании при 40 оборотов в минуту. Суспензию отстояли в течение 10 минут при температуре около 90°C и удалили маточный раствор через 10 μм фильтр из сетки в дне реактора в течение 15 минут. Полученный твердый материал промыли дважды толуолом (43 кг каждая промывка) при ~90°C и однократно гептаном (34 кг) при ~40°C. На всех трех стадиях промывки использовали одну и ту же последовательность: добавление предварительно нагретого (90 или 40°C) растворителя, далее перемешивание при 40 оборотах в минуту в течение 30 минут, отстаивание для осаждения твердого материала в течение 10 минут, и удаление жидкость через 10 μм фильтр из сетки в дне реактора в течение 15 минут.
Полученный катализатор смешали с 20 кг белого масла и сушили в течение 4 часов при температуре 40-50°C потоком азота (2 кг/час) и вакуумом (-1 бар избыточного давления). Катализатор удалили из реактора и промыли реактор другими 20 кг масла и вывели его в тот же самый барбан. Выход сухого катализатора составил 3,60 кг (82,2% в пересчете на Mg).
Полимеризация
Примеры по настоящему изобретению
Использовали циркуляционный реактор объемом 50 дм3, работавший при температуре 60°C и давлении 58 бар. В реактор подали этилен, 1-бутен, пропановый разбавитель и водород, при этом скорость подачи этилена составила 4,0 кг/час, скорость подачи 1-бутена составила 50 г/час, скорость подачи водорода составила 10 г/час, и скорость подачи пропана составила 52 кг/час. Также в реактор ввели 3 г/час компонента твердого катализатора полимеризации, как указано выше в части «Получение катализатора», вместе с сокатализатором - триэтилалюминием, таким образом, что молярное соотношение Al/Ti составило около 15. Расчетная производительность составила 3,8 кг/час.
Поток суспензии периодически отводили из реактора и направляли в циркуляционный реактор объемом 350 дм3, работавший при температуре 85°C и давлении 56 бар. В реактор добавили свежий пропан со скоростью подачи 116 кг/час, этилен и водород, таким образом, что содержание этилена в жидкой реакционной смеси составило 3,8 мол. %, и молярное соотношение водорода к этилену составило 500 моль/кмоль. Отведенный из реактора сополимер этилена имел MFR2 300 г/10 минут и плотность 972 кг/м3. Производительность составила 38 кг/час.
Суспензию периодически отводили из циркуляционного реактора и направляли в емкость для испарения, работавшую при температуре 50°C и давлении 3 бар. Из нее полимер направили в газофазный реактор с псевдоожиженным слоем, работавший под давлением 20 бар температуре 75°C. Добавили дополнительный этилен, сомономер 1-гексена, азот в качестве инертного газа и водород, таким образом, что содержание этилена в жидкой реакционной смеси составило 20 мол. %, соотношение водорода к этилену составило 150 моль/кмоль, и молярное соотношение 1-гексена к этилену составило 210 моль/кмоль. Производительность полимера в газофазном реакторе составила 55 кг/час и, следовательно, общая скорость отвода полимера из газофазного реактора составила около 97 кг/час. Полимер имел скорость течения расплава MFR5 3,0 г/10 минут и плотность 920 кг/м3. Разделение продукта (масс. % форполимера/масс. % компонента 1-ой стадии/масс. % компонента 2-ой стадии) составило 4/44/52.
Порошкообразный полимер компаундировали и гранулировали с 0,2 масс. % Irganox В 141, 6,6 масс. % мастербатча углеродной сажи на основе линейного полиэтилена низкой плотности (LLDPE) (MFR190°C/2,16 кг=2 г/10 минут), содержащего ~40% углеродной сажи типа N220, 0,1 масс. % Irganox В 225 FF и 0,05 масс. % стеарата кальция.
Процедуру по Примеру 1 по настоящему изобретению повторили при условиях, приведенных в Таблице 1.
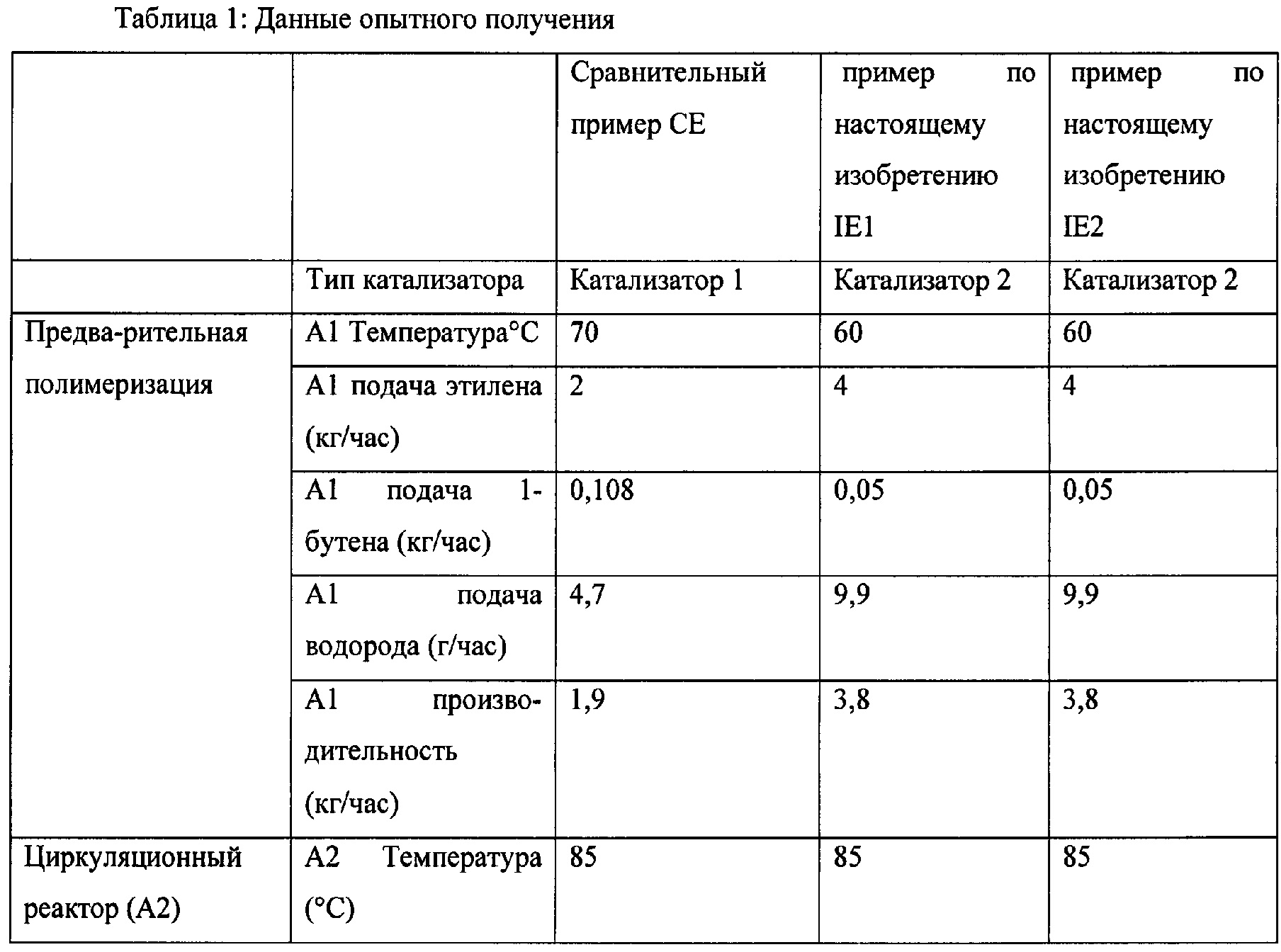

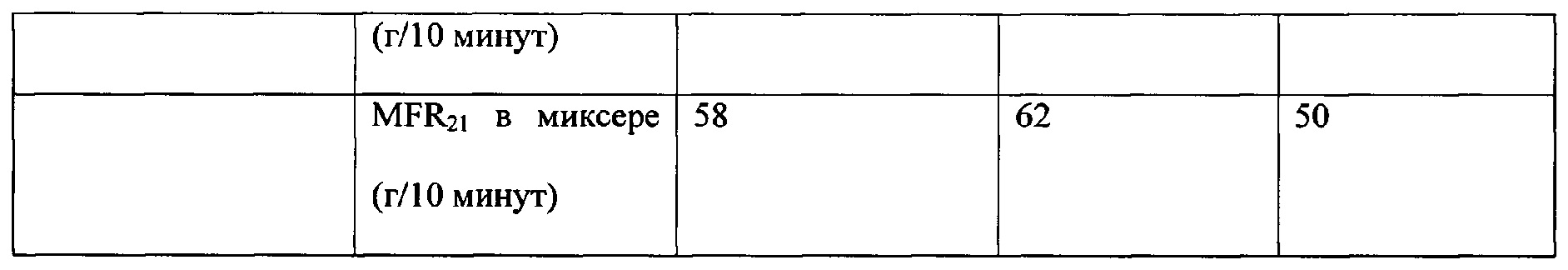
IE1 и IE2 получили при использовании катализатора 2. СЕ получили при использовании катализатора 1. СЕ получили с аналогичными технологическими свойствами, и он имел значительно более высокую гибкость по сравнению с IE1 и IE2.
IE1, IE2 и СЕ получили, как указано выше, в опытном масштабе по сравнению с коммерческим контрольным образцом LE8707, доступным от Borealis AG, полученным при промышленном масштабе производства. Сравнительный пример СЕ получили максимально близким к коммерческому контрольному образцу по выбору технологических параметров и выбору катализатора. Видно, что материал, полученный при опытном масштабе производства (СЕ), значительно менее гибкий по сравнению с коммерческим продуктом. Это указывает на то, что настоящее изобретение будет работать даже еще лучше при промышленном масштабе производства.
В Таблице 2 проводится сравнение IE1, IE2 и СЕ с LE8707. Оба, и IE1, и IE2 имеют более низкий модуль упругости при изгибе по сравнению с LE8707, но сравнимы по твердости.


Как видно из Таблицы 2, сравнительные примеры и пример по настоящему изобретению имеют аналогичную плотность. Неожиданно было обнаружено, что гибкость примера по настоящему изобретению значительно выше, чем таковая сравнительного примера, при этом твердость по Шору D повышена. Другие свойства находятся в пределах, аналогичных таковым сравнительного примера.
Хотя настоящее изобретение описано со ссылкой на различные варианты его осуществления, специалисту в области техники, к которой относится настоящее изобретение, понятно, что могут быть сделаны изменения без отклонения от объема настоящего изобретения. Подробное описание следует рассматривать, как иллюстрирующие, а приложенная формула изобретения включает в объем притязаний все эквиваленты настоящего изобретения.
Реферат
Изобретение относится к композиции мультимодального сополимера этилена, предназначенной для получения оболочки кабеля с высокой гибкостью. Композиция содержит в количестве не менее 80 мас.% сополимера этилена и по меньшей мере одного сомономера С-Сальфа-олефина, а также имеет плотность от 920 до 949 кг/ми модуль упругости при изгибе, соответствующий уравнению:Модуль упругости при изгибе <21,35·плотность - 19585.Композиция по изобретению обеспечивает получение материала, сочетающего высокую гибкость с высокой механической прочностью. 2 н. и 11 з.п. ф-лы, 2 табл., 4 пр.
Формула
Комментарии