Гликополисиалирование белков, не являющихся белками свертывания крови - RU2662807C2
Код документа: RU2662807C2
Чертежи
Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к материалам и способам для конъюгации растворимых в воде полимеров, в частности, полисиаловой кислоты, с содержащими углеводы соединениями, в частности гликопротеинами, отличными от белков свертывания крови, и к полученным конъюгатам.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Конъюгация полипептидных лекарственных средств, например, путем пегилирования или полисиалирования, защищает их от деградации в кровотоке и, таким образом, улучшает их фармакодинамические и фармакокинетические профили (Harris and Chess, Nat Rev Drug Discov. 2003; 2: 214-21; S. Jain, D. Hreczuk-Hirst, P. Laing and G. Gregoriadis, Drug Delivery Systems and Sciences, 4 (No 1): 3-9, 2004). Процесс пегилирования присоединяет повторяющиеся звенья этиленгликоля (полиэтиленгликоль (PEG)) к полипептидному лекарственному средству. Молекулы PEG имеют большой гидродинамический объем (в 5-10 раз превышающий размер глобулярных белков), являются высокорастворимыми в воде и гидратированными, нетоксичными, неиммуногенными и быстро выводятся из организма. Пегилирование молекул может приводить к увеличенной устойчивости лекарственных средств к ферментативной деградации, увеличению времени полужизни in vivo, снижению частоты дозирования, снижению иммуногенности, увеличению физической и термической стабильности, увеличению растворимости, увеличению стабильности в жидкости и снижению агрегации. Первые пегилированные лекарственные средства были одобрены FDA в ранние 1990. С тех пор FDA одобрило несколько пегилированных лекарственных средств для перорального, инъекционного и местного введения.
Сиаловые кислоты (также называемые N-ацетилнейраминовыми кислотами) и полисиаловые кислоты широко распространены в тканях животных и в меньшей степени в других видах в диапазоне от растений и грибов до дрожжей и бактерий, главным образом, в гликопротеинах и ганглиозидах.
Сокращение "PSA", используемое в настоящем документе, относится к термину "полисиаловая кислота". Аналогично, термин "mPSA", используемый в настоящем документе, относится к термину "модифицированная полисиаловая кислота".
PSA состоят из полимеров (главным образом, гомополимеров) N-ацетилнейраминовой кислоты. Вторичная аминогруппа обычно несет ацетильную группу, но вместо этого она может нести гликолильную группу. Возможные заместители на гидроксильных группах включают ацетильную, лактильную, этильную, сульфатную и фосфатную группы.

N-ацетилнейраминовая кислота
Neu5Ac
Структура сиаловой кислоты (N-ацетилнейраминовая кислота)
PSA и mPSA, главным образом, включают линейные полимеры, состоящие в основном из частей N-ацетилнейраминовой кислоты, связанных 2,8- или 2,9-гликозидными связями или их комбинациями (например, чередование 2,8- и 2,9-связей). В особенно предпочтительных PSA и mPSA гликозидные связи представляют собой α-2,8. Является пригодным, когда такие PSA и mPSA происходят из коломиновых кислот, и их обозначают в настоящем документе как "CA" и "mCA". Типичные PSA и mPSA содержат по меньшей мере 2, предпочтительно по меньшей мере 5, более предпочтительно по меньшей мере 10 и наиболее предпочтительно по меньшей мере 20 частей N-ацетилнейраминовой кислоты. Таким образом, они могут содержать от 5 до 500 частей N-ацетилнейраминовой кислоты, предпочтительно от 10 до 300 частей N-ацетилнейраминовой кислоты. PSA и CA могут представлять собой полимеры, содержащие различные группы сахаров. Они могут представлять собой сополимеры. Предпочтительно PSA и CA свободны от частей сахаров, отличных от N-ацетилнейраминовой кислоты. PSA и CA предпочтительно содержат по меньшей мере 90%, более предпочтительно по меньшей мере 95% и наиболее предпочтительно по меньшей мере 98% частей N-ацетилнейраминовой кислоты.
Когда PSA и CA содержат части, отличные от N-ацетилнейраминовой кислоты (как, например, в mPSA и mCA), они предпочтительно расположены на одном или обоих концах полимерной цепи. Такие "другие" группы могут, например, представлять собой группы, образованные из концевых групп N-ацетилнейраминовой кислоты путем окисления или восстановления.
Например, в WO -A-0187922 описаны такие mPSA и mCA, в которых невосстанавливающее концевое звено N-ацетилнейраминовой кислоты превращено в альдегидную группу путем реакции с перйодатом натрия. Кроме того, в WO 2005/016974 описаны такие mPSA и mCA, в которых восстанавливающее концевое звено N-ацетилнейраминовой кислоты подвергнуто восстановлению для восстановительного размыкания кольца на восстанавливающем концевом звене N-ацетилнейраминовой кислоты, посредством чего образуются соседняя диольная группа, с последующим окислением для превращения соседней диольной группы в альдегидную группу.
Гликопротеины, обогащенные сиаловыми кислотами, связывают селектин у человека и других организмов. Они играют важную роль в инфекциях вирусом гриппа человека. Например, сиаловая кислота может скрывать антигены маннозы на поверхности клеток-хозяев или бактерий от связывающего маннозу лектина. Это предотвращает активацию комплемента. Сиаловые кислоты также скрывают предпоследний остаток галактозы, таким образом препятствуя быстрому выведению гликопротеина посредством рецептора галактозы на клетках паренхимы печени.

Структура коломиновой кислоты (гомополимер N-ацетилнейраминовой кислоты)
CA продуцируются, среди прочих, определенными штаммами Escherichia coli, обладающими антигеном K1. CA обладают множеством физиологических функций. Они являются важными в качестве сырья для лекарственных и косметических средств.
Сравнительные исследования in vivo с полисиалированной и немодифицированной аспарагиназой показали, что полисиалирование увеличивало время полужизни фермента (Fernandes and Gregoriadis, Biochimica Biophysica Acta 1341: 26-34, 1997).
Получение конъюгатов путем образования ковалентной связи между растворимым в воде полимером и терапевтическим белком можно проводить множеством химических способов. Одним подходом для присоединения PSA к терапевтическим белкам является конъюгация полимеров через углеводные части белка. Соседние гидроксильные (OH) группы углеводов в белках можно легко окислять перйодатом натрия (NaIO4) с образованием активных альдегидных групп (Rothfus and Smith, J Biol Chem 1963; 238: 1402-10; van Lenten and Ashwell, J Biol Chem 1971; 246: 1889-94). Затем полимер можно присоединять к альдегидным группам углеводов с использованием реагентов, содержащих, например, активную гидразидную группу (Wilchek M and Bayer EA, Methods Enzymol 1987; 138: 429-42). Более поздней технологией является применение реагентов, содержащих аминооксигруппы, которые реагируют с альдегидами с образованием оксимных связей (WO 96/40662, WO 2008/025856).
Дополнительные примеры, описывающие конъюгацию PSA с терапевтическим белком, описаны в публикации US 2009/0076237, в которой описано окисление rFVIII и последующее связывание с PSA и другими растворимыми в воде полимерами (например, PEG, HES, декстран) с использованием химии гидразидов; в WO 2008/025856, в которой описано окисление различных факторов свертывания, например, rFIX, FVIII и FVIIa, и последующее связывание с полимером, например, PEG.
Недавно был описан усовершенствованный способ, включающий мягкое окисление перйодатом сиаловых кислот с образованием альдегидов с последующей реакцией с содержащим аминооксигруппу реагентом в присутствии каталитических количеств анилина (Dirksen A and Dawson PE, Bioconjugate Chem. 2008; 19,2543-8; и Zeng Y et al., Nature Methods 2009; 6: 207-9). Анилиновые катализаторы значительно ускоряют сшивание оксимов, позволяя применять очень низкие концентрации реагентов.
Несмотря на то что доступны способы конъюгации растворимых в воде полимеров с терапевтическими белками, остается потребность в разработке материалов и способов для конъюгации растворимых в воде полимеров с содержащими углеводы соединениями, отличными от белков свертывания крови, которые улучшают фармакодинамические и/или фармакокинетические свойства при одновременной минимизации затрат, ассоциированных с различными реагентами.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к материалам и способам для конъюгации растворимого в воде полимера с содержащим углевод соединением, отличным от белка свертывания крови, которое улучшает фармакодинамические и/или фармакокинетические свойства белка при одновременной минимизации затрат, связанных с различными реагентами.
В одном варианте осуществления изобретения предусмотрен способ конъюгации растворимого в воде полимера с окисленной углеводной группой содержащего углевод соединения, отличного от белка свертывания крови, включающий контактирование окисленной углеводной группы с растворимым в воде полимером в условиях, которые обеспечивают конъюгацию, где указанный растворимый в воде полимер содержит аминооксигруппу, и между окисленной углеводной группой и аминооксигруппой на растворимом в воде полимере образуется оксимная связь, или где указанный растворимый в воде полимер содержит гидразидную группу, и между окисленной углеводной группой и гидразидной группой растворимого в воде полимера образуется гидразоновая связь. Соединение может представлять собой (1) гликопротеин, отличный от белка свертывания крови, (2) ганглиозид или (3) систему для доставки лекарственного средства, содержащую углеводную группу.
Углеводную часть можно окислять с использованием специфичного к сахарам окисляющего фермента (например, галактоза- или глюкозаоксидаза) или путем инкубации с буфером, содержащим окислитель, выбранный из перйодата натрия (NaIO4), тетраацетата свинца (Pb(OAc)4) и перрутената калия (KRuO4).
Углеводную часть можно окислять по остатку сиаловой кислоты, маннозы, галактозы или глюкозы.
Растворимый в воде полимер, используемый в изобретении, может представлять собой, но не ограничиваться ими, полиэтиленгликоль (PEG), разветвленный PEG, производное PEG, PSA, mPSA, CA, mCA, гидроксиэтилцеллюлозу (HEC), декстрин, полиоксазолин, углевод, полисахариды, пуллулан, хитозан, гиалуроновую кислоту, хондроитинсульфат, дерматансульфат, крахмал, декстран, карбоксиметил-декстран, полиалкиленоксид (PAO), полиалкиленгликоль (PAG), полипропиленгликоль (PPG) полиоксазолин, полиакрилоилморфолин, поливиниловый спирт (PVA), поликарбоксилат, поливинилпирролидон, полифосфазен, полиоксазолин, сополимер полиэтилен-ангидрид малеиновой кислоты, сополимер полистирол-ангидрид малеиновой кислоты, поли(1-гидроксиметилэтиленгидроксиметилформальдегид) (PHF), фосфат 2-метакрилоилокси-2'-этилтриметиламмония (MPC).
В конкретных вариантах осуществления изобретения, проиллюстрированных в примерах ниже, растворимый в воде полимер представляет собой PEG или разветвленный PEG.
В следующих конкретных вариантах осуществления изобретения, проиллюстрированных в примерах ниже, растворимый в воде полимер представляет собой полисиаловую кислоту (PSA) или модифицированную PSA (mPSA). PSA или mPSA могут обладать диапазоном молекулярной массы от 350 Да до 120000 Да, от 500 Да до 100000 Да, от 1000 Да до 80000 Да, от 1500 Да до 60000 Да, от 2000 Да до 45000 Да или от 3000 Да до 35000 Да.
PSA или mPSA могут представлять собой коломиновую кислоту или модифицированную коломиновую кислоту.
В другом варианте осуществления изобретения PSA или mPSA содержат приблизительно 2-500 или 10-300 звеньев сиаловой кислоты. В другом варианте осуществления предусмотрен указанный выше способ, где окислитель представляет собой перйодат натрия (NaIO4).
Способ по изобретению может включать окисление растворимого в воде полимера с образованием альдегидной группы на концевом звене сиаловой кислоты на растворимом в воде полимере и реакцию окисленного растворимого в воде полимера с аминооксилинкером.
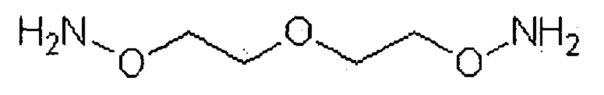
В другом варианте осуществления изобретения предусмотрен указанный выше способ, где растворимый в воде полимер получают путем реакции активированного аминооксилинкера с окисленным растворимым в воде полимером, где линкер представляет собой гомобифункциональный или гетеробифункциональный линкер. Гомобифункциональный линкер может иметь общую формулу NH2[OCH2CH2]nONH2, где n=1-50, предпочтительно 1-11, более предпочтительно 1-6. В частности, линкер может быть выбран из:
3-оксапентан-1,5-диоксиминового линкера формулы:
3,6,9-триоксаундекан-1,11-диоксиаминового линкера формулы:

PSA или mPSA можно окислять путем инкубации с окислителем с образованием концевой альдегидной группы на невосстанавливающем конце PSA.
Способ может включать окисление растворимого в воде полимера с образованием альдегидной группы на концевом звене растворимого в воде полимера, например, на концевом звене сиаловой кислоты в PSA или mPSA, и реакцию окисленного растворимого в воде полимера с аминооксилинкером. В другом варианте осуществления предусмотрен указанный выше способ, где аминооксилинкер представляет собой 3-оксапентан-1,5-диоксиамин. В родственном варианте осуществления окислитель представляет собой NaIO4.
В другом варианте осуществления изобретения предусмотрен указанный выше способ, где контактирование окисленной углеводной группы с активированным растворимым в воде полимером происходит в буфере, содержащем нуклеофильный катализатор, выбранный из группы, состоящей из анилина и производных анилина.
Гидразидная группа может быть образована на растворимом в воде полимере путем реакции окисленного растворимого в воде полимера с гидразидным линкером. Гидразидный линкер может соответственно представлять собой дигидразид адипиновой кислоты или гидразин.
В другом варианте осуществления изобретения предусмотрен указанный выше способ, кроме того, включающий стадию восстановления оксимной или гидразоновой связи в конъюгированном белке, например, путем инкубации конъюгированного белка в буфере, содержащем восстанавливающее соединение, выбранное из группы, состоящей из цианоборгидрида натрия (NaCNBH3) и аскорбиновой кислоты (витамин C). В родственном варианте осуществления восстанавливающее соединение представляет собой цианоборгидрид натрия (NaCNBH3).
В другом варианте осуществления изобретения предусмотрен конъюгированный гликопротеин, продуцированный любым из описанных выше способов. В другом варианте осуществления изобретения конъюгированный гликопротеин, отличный от белка свертывания крови, ганглиозид или система для доставки лекарственных средств содержит (a) указанный гликопротеин, ганглиозид или систему для доставки лекарственных средств; и (b) по меньшей мере один растворимый в воде аминооксиполимер, связанный с гликопротеином (a), где указанный растворимый в воде аминооксиполимер связан с гликопротеином, ганглиозидом или системой для доставки лекарственных средств через одну или несколько углеводных частей. В следующем варианте осуществления изобретения конъюгированный гликопротеин, отличный от белка свертывания крови, ганглиозид или система для доставки лекарственного средства содержит (a) указанный гликопротеин, ганглиозид или систему для доставки лекарственного средства; и (b) по меньшей мере один гидразидный растворимый в воде полимер, связанный с гликопротеином (a), где указанный гидразидный растворимый в воде полимер связан с ганглиозидом гликопротеина или системой для доставки лекарственного средства через одну или несколько углеводных частей.
ФИГУРЫ
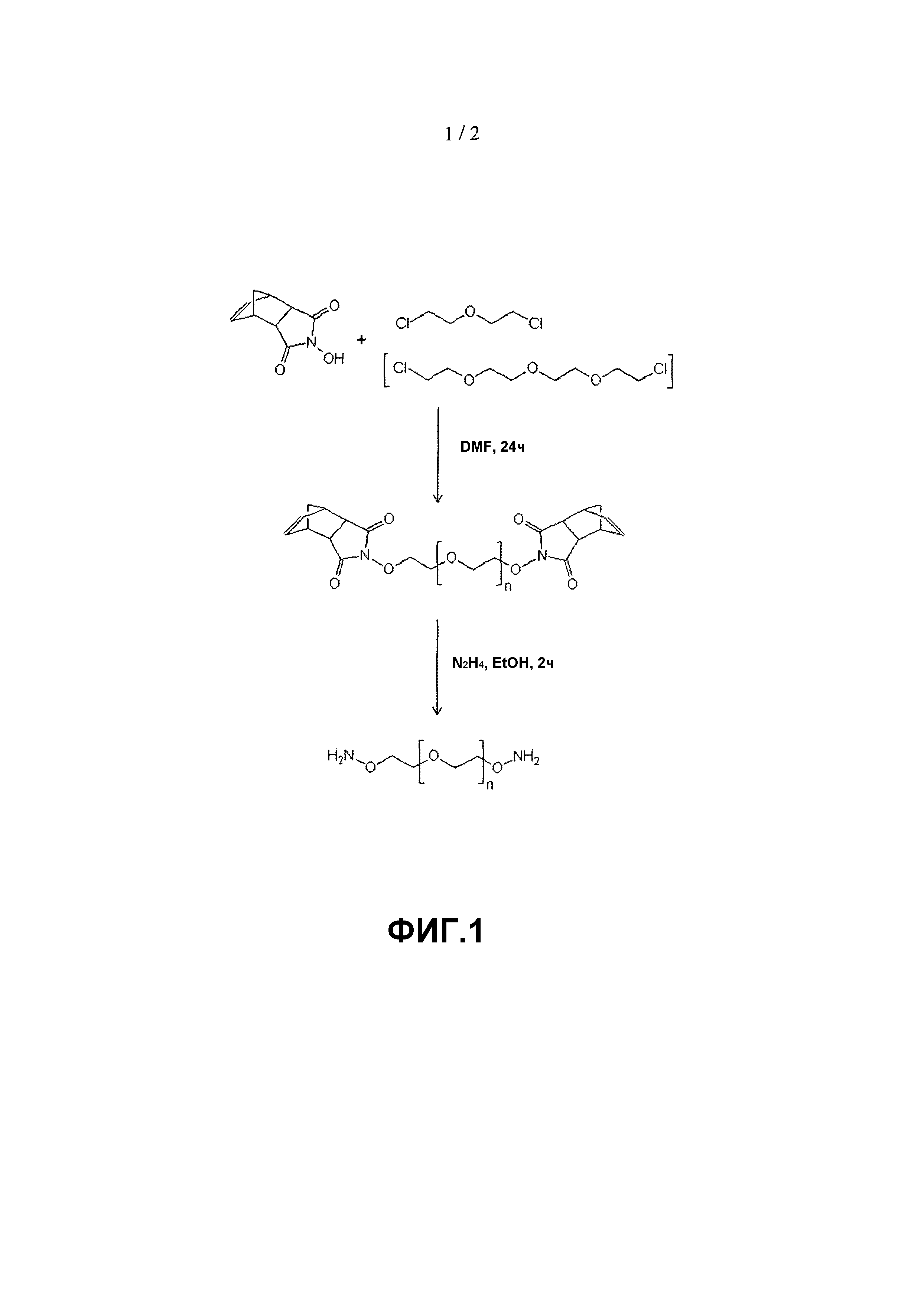
На фиг. 1 представлен синтез растворимых в воде диаминоксилинкеров: 3-окса-пентан-1,5-диоксиамина и 3,6,9-триоксаундекан-1,11-диоксиамина.
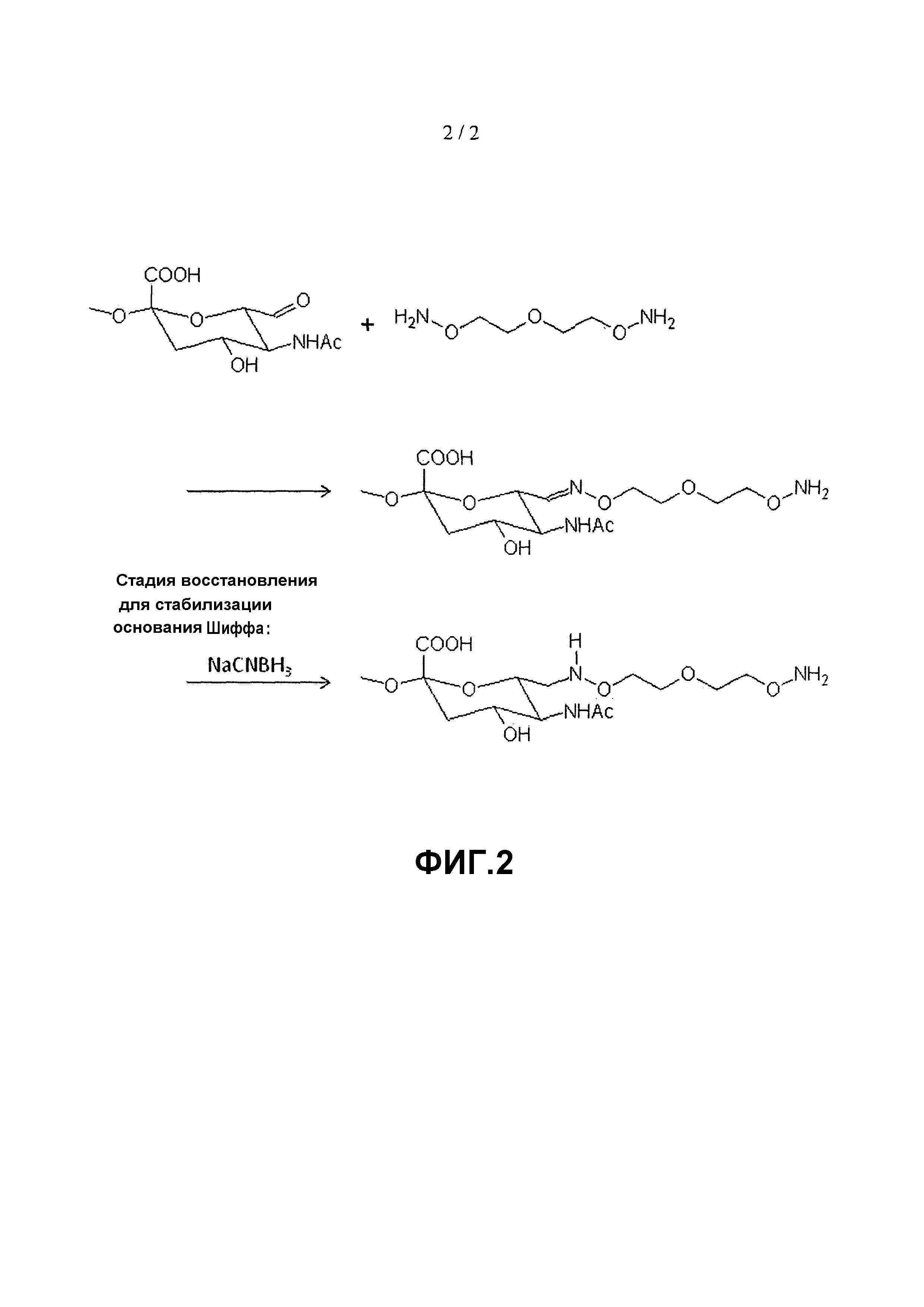
На фиг. 2 представлено получение аминоокси-PSA.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Фармакологические и иммунологические свойства углеводсодержащих соединений, таких как гликопротеины, отличные от белков свертывания крови, можно улучшать путем химической модификации и конъюгации с растворимым в воде полимером, в частности, PEG или PSA или mPSA. Свойства полученных конъюгатов, как правило, строго зависят от структуры и размера полимера. Таким образом, обычно являются предпочтительными полимеры с определенным и узким распределением размеров. PSA и mPSA, используемые в конкретных примерах, можно очищать так, чтобы получать конечный препарат PSA с узким распределением размеров.
ГЛИКОПРОТЕИНЫ
Как описано в настоящем документе, главным образом, изобретением предусматриваются гликопротеины, отличные от белков свертывания крови, включая, но не ограничиваясь ими, цитокины такие как интерлейкины, альфа-, бета- и гамма-интерфероны, колониестимулирующие факторы, включая гранулоцитарные колониестимулирующие факторы, фибробластные факторы роста, тромбоцитарные факторы роста, активирующий фосфолипазу белок (PUP), инсулин, растительные белки, такие как лектины и рицины, факторы некроза опухоли и родственные аллельные формы, растворимые формы рецепторов фактора некроза опухоли, рецепторы интерлейкинов и растворимые формы рецепторов интерлейкинов, факторы роста, тканевые факторы роста, трансформирующие факторы роста, такие как TGFα или TGFβ и эпидермальные факторы роста, гормоны, соматомедины, пигментарные гормоны, гипоталамические рилизинг-факторы, антидиуретические гормоны, пролактин, хорионический гонадотропин, фоликулостимулирующий гормон, тиреотропный гормон, тканевой активатор плазминогена и иммуноглобулины, такие как IgG, IgE, IgM, IgA и IgD, моноклональные антитела, эритропоэтин (EPO), факторы крови, отличные от белков свертывания крови, галактозидазы, α-галактозидазы, β-галактозидазы, ДНКазы, фетуин, их фрагменты и любые слитые белки, содержащие любой из упомянутых выше белков или их фрагментов вместе с терапевтическими гликопротеинами. В одном варианте осуществления гликопротеин представляет собой EPO. В следующем варианте осуществления гликопротеин представляет собой галактозидазу. В следующем варианте осуществления гликопротеин представляет собой ДНКазу. В следующем варианте осуществления гликопротеин представляет собой фетуин. Наконец, в следующем варианте осуществления гликопротеин представляет собой гранулоцитарный колониестимулирующий фактор.
Как используют в настоящем документе, "биологически активное производное" или "биологически активный вариант" включает любое производное или вариант молекулы, имеющие по существу те же функциональные и/или биологические свойства, что и указанная молекула, такие как свойства связывания, и/или ту же структурную основу, такую как пептидная основная цепь или основное полимерное звено.
"Аналог", "вариант" или "производное" представляет собой соединение, по существу сходное по структуре и имеющее ту же биологическую активность, хотя в определенных случаях в отличной степени, что и встречающаяся в природе молекула. Например, полипептидный вариант относится к полипептиду, обладающему по существу сходной структурой и имеющему ту же биологическую активность, что и эталонный полипептид. Варианты или аналоги отличаются составом их аминокислотных последовательностей по сравнению со встречающимся в природе полипептидом, из которого происходит аналог, на основании одной или нескольких мутаций, вовлекающих (i) делецию одного или нескольких аминокислотных остатков на одном или нескольких концах полипептида и/или в одной или нескольких внутренних областях встречающейся в природе полипептидной последовательности (например, фрагменты), (ii) инсерцию или добавление одной или нескольких аминокислот на одном или нескольких концах (как правило, "добавление" или "слияние") полипептида и/или в одной или нескольких внутренних областях (как правило, "инсерция") встречающейся в природе полипептидной последовательности, или (iii) замену одной или несколькими аминокислотами других аминокислот во встречающейся в природе полипептидной последовательности. В качестве примера, "производное" относится к полипептиду, обладающему той же или существенно сходной структурой по сравнению с полипептидом, который был модифицирован, например, химически.
Полипептиды вариантов или аналогов включают варианты с инсерцией, где к аминокислотной последовательности белка по изобретению добавлены один или несколько аминокислотных остатков. Инсерции могут быть расположены на любом или обоих концах белка и/или они могут быть расположены во внутренних областях аминокислотной последовательности белка. Варианты с инсерцией с дополнительными остатками на любом или обоих концах, включают, например, слитые белки и белки, включающие маркерные аминокислоты или другие аминокислотные метки. В одном аспекте молекула белка необязательно содержит N-концевой Met, особенно когда молекула экспрессируется рекомбинантно в бактериальной клетке, такой как E.coli.
В вариантах с делецией один или несколько аминокислотных остатков в белке или полипептиде, как описано в настоящем документе, удалены. Делеции могут быть проведены на одном или обоих концах белка или полипептида и/или с удалением одного или нескольких остатков в аминокислотной последовательности белка. Варианты с делецией, таким образом, включают фрагменты белковой или полипептидной последовательности.
В вариантах с заменой один или несколько аминокислотных остатков белка или полипептида удалены и заменены альтернативными остатками. В одном аспекте замены являются консервативными в природе и консервативные замены этого типа хорошо известны в данной области. Альтернативно изобретение охватывает замены, которые также являются неконсервативными. Иллюстративные консервативные замены описаны в Lehninger, [Biochemistry, 2nd Edition; WO rth Publishers, Inc., New York (1975), pp.71-77] и представлены непосредственно ниже.
Альтернативно, иллюстративные консервативные замены указаны непосредственно ниже.
ГАНГЛИОЗИДЫ
В вариантах осуществления изобретения ганглиозиды конъюгируют с растворимыми в воде полимерами, например, PEG или PSA или mPSA. Известно, что ганглиозиды обеспечивают клетки с отличающимися поверхностными маркерами, которые участвуют в распознавании клеток и межклеточной коммуникации. Они пригодны в качестве лекарственных средств.
Конъюгаты по изобретению могут содержать ганглиозид и растворимый в воде полимер, в котором ганглиозид содержит гликосфинголипид (церамид и олигосахарид) с одной или несколькими сиаловыми кислотами, присоединенными к цепи сахаров. Ганглиозиды можно классифицировать согласно тому, сколько звеньев сиаловых кислот присутствуют на молекуле. Примерами ганглиозидов являются GM1, GM2 и GM3 (моносиалоганглиозиды), GD1a, GD1b, GD2 и GD3 (дисиалоганглиозиды), GT1b (трисиалоганглиозид) и GQ1 (тетрасиалоганглиозид).
Для применения в настоящем изобретении предпочтительные ганглиозиды содержат церамид, связанный с глюкозой, которая связана с первой галактозой, которая связана с N-ацетилгалактозамином, который связан со второй галактозой. Эта вторая галактоза может быть связана с одной сиаловой кислотой. Первая галактоза может быть связана с одной, двумя, тремя или четырьмя сиаловыми кислотами. Сиаловые кислоты могут быть присоединены либо в качестве мономеров (по одной на каждой из молекул галактозы), либо в качестве олигосиаловых кислот (2-4 сиаловых кислоты) к первой галактозе.
При введении терапевтические ганглиозиды должны циркулировать в крови в течение длительных периодов. Чтобы действие ганглиозидов на ткани-мишени было более эффективным, их можно подвергать полисиалированию, например, способом по изобретению.
СИСТЕМЫ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ
В следующих вариантах осуществления изобретения системы для доставки лекарственных средств конъюгируют с растворимым в воде полимером, например, PEG или PSA или mPSA. Как правило, система для доставки лекарственного средства (DDS) представляет собой молекулярную структуру или структуру в виде частиц, которая может контролировать судьбу и эффект лекарственных средств, связанных с этой структурой. DDS можно подразделить на два основных типа. Первый тип включает макромолекулы (MDDS), например, антитела, неогликопротеины, а также синтетические полимеры, такие как поли(гидроксипропилметакриламид), полилизин и полимеризованные алкилцианоакрилаты. Связывание лекарственных средств с различными типами макромолекулярных носителей, включая моноклональные антитела, для нацеливания лекарственного средства в желаемые области, описано, например, Gregoriadis в Nature 265, 407-411 (1977). Вторым типом являются DDS в форме частиц (PDDS), которые содержат, например, наносферы или микросферы, которые содержат биодеградируемые материалы, такие как альбумин, или полубиодеградируемые материалы, такие как декстран и алкилцианоакрилатные полимеры, или носители, образованные из неионных поверхностно-активных веществ, или липосомы - для подробного описания которых см., например, Gregoriadis в NIPS, 4, 146-151 (1989).
Лекарственные средства можно либо ковалентно связывать с, либо пассивно заключать в DDS. Например, PDDS, содержащие везикулы или липосомы из поверхностно-активного вещества, могут заключать в себе гидрофильные или гидрофобные фармацевтически активные соединения вследствие формирования соответствующей комбинации слоев поверхностно-активного вещества или липидных молекул. Фармацевтически активные соединения обычно ковалентно связывают с MDDS связью, которая может расщепляться или может не расщепляться в организме, например, до или после того, как активное соединение выполняет его функцию.
Многие из MDDS обладают присущей (например, антитела) или приобретенной (например, неогликопротеины) способностью быть распознанными клетками-мишенями или тканями-мишенями через рецепторы на их поверхности. Как правило, такие DDS захватываются специфично мишенью при инъекции. Однако специфичный захват ограничен объемом DDS, захватываемого другими, не имеющими отношения (к терапии) тканями. Причиной этому является то, что антитела и другие белки DDS (независимо от их специфичности к мишени) должны, подобно другим белкам, быть катаболизированы в конце их биологической жизни.
Синтетические полимеры, используемые в MDDS макромолекулярного типа представляют собой, например, поли(гидроксипропилметакриламид)полилизин и полимеризованные алкилцианоакрилаты. Они могут катаболизироваться в ретикулоэндотелиальной системе (RES) или другими тканями с помощью соответствующих лизосомальных ферментов. Может быть желательным снижение скорости катаболизма таких биодеградируемых DDS макромолекулярного типа различными способами, например, путем снижения захвата DDS RES или другими тканями, или путем снижения деградации лизосомальными ферментами после захватывания RES.
DDS в форме частиц (PDDS), как правило, удаляются из кровотока посредством RES. Вследствие их преимущественного удаления RES, PDDS часто используют для доставки лекарственных средств в эти ткани. Однако часто желательно, чтобы PDDS были направлены в ткани, отличные от тканей RES. Для достижения этой цели необходимо блокировать или замедлять перехват PDDS посредством RES.
DDS для применения в изобретения могут исходно не содержать гликоны. Возможно добавление или иное включение гликона в структуру DDS. Примерами таких случаев являются липосомы, включающие маннозилированный или галактозилированный липид. Эти гликолипосомы нацеливают активные средства в ткани, которые экспрессируют рецептор маннозы или галактозы, соответственно.
Когда DDS должны циркулировать в крови в течение длительных периодов, так чтобы, например, захват тканями был более эффективным (как в случае паренхимных клеток печени), их преимущественно полисиалируют способами по изобретению.
ВВЕДЕНИЕ
В одном варианте осуществления конъюгированное соединение по настоящему изобретению можно вводить путем инъекции, такой как внутривенная, внутримышечная или внутрибрюшинная инъекция. Композиции могут быть пригодны в качестве терапевтических, диагностических и/или сходных средств.
Для введения композиций, содержащих конъюгированное соединение по настоящему изобретению человеку или тестируемым животным, в одном аспекте композиции содержат один или несколько фармацевтически приемлемых носителей. Термины "фармацевтически" или "фармакологически приемлемый" относятся к молекулярным структурам и композициям, которые являются стабильными, ингибируют деградацию белка, такую как агрегация и продукты расщепления и, кроме того, не вызывают аллергических или иных неблагоприятных реакций при введении с использованием путей, хорошо известных в данной области, как описано ниже. "Фармацевтически приемлемые носители" включают любые и все клинически пригодные растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства, обеспечивающие изотоничность и замедляющие всасывание средства и т.п., включая средства, описанные выше.
Как используют в настоящем документе, "эффективное количество" включает дозу, пригодную для лечения млекопитающего, имеющего клинически установленное нарушение.
Композиции можно вводить перорально, местно, трансдермально, парентерально, с помощью ингалируемого спрея, вагинально, ректально или путем внутричерепной инъекции. Термин "парентеральный", как используют в настоящем документе, включает подкожные инъекции, внутривенную, внутримышечную, интрацистернальную инъекцию или способы инфузии. Также предусмотрено введение путем внутривенной, внутрикожной, внутримышечный, внутригрудной, внутрибрюшинной, интратекальной, ретробульбарной, внутрилегочной инъекции и/или хирургической имплантации в конкретную область. Как правило, композиции по существу не содержат пирогенов, а также других примесей, которые могут быть вредоносными для реципиента.
Однократное или многократные введения композиции можно проводить с уровнями и расписанием введения доз, выбираемыми лечащим врачом. Для профилактики или лечения заболевания соответствующая дозировка будет зависеть от типа заболевания, подлежащего лечению, как описано выше, тяжести или течения заболевания, введения лекарственного средства в профилактических или терапевтических целях, предшествующей терапии, клинического анамнеза пациента и ответа на лекарственное средство, и мнения лечащего врача.
Также настоящее изобретение относится к фармацевтической композиции, содержащей эффективное количество конъюгированного соединения или белка, как определено в настоящем документе. Фармацевтическая композиция, кроме того, может содержать фармацевтически приемлемый носитель, разбавитель, соль, буфер или эксципиент. Фармацевтическую композицию можно применять для лечения клинически установленных нарушений. Фармацевтическая композиция по изобретению может представлять собой раствор или лиофилизированный продукт. Растворы фармацевтической композиции можно подвергать любому пригодному способу лиофилизации.
В качестве дополнительного аспекта изобретение относится к наборам, которые содержат композицию по изобретению, упакованную так, чтобы облегчить ее применение для введения индивидуумам. В одном варианте осуществления такой набор включает соединение или композицию, описанные в настоящем документе (например, композицию, содержащую конъюгированный белок), упакованные в контейнер, такой как закрытая бутыль или емкость, с ярлыком, прикрепленным к контейнеру или включенным в упаковку, на котором описано использование соединения или композиции в способе применения. В одном варианте осуществления набор содержит первый контейнер, имеющий композицию, содержащую конъюгированный белок, и второй контейнер, имеющий физиологически приемлемый раствор для разбавления композиции в первом контейнере. В одном аспекте соединение или композиция упакованы в виде единичной дозированной формы. Кроме того, набор может включать устройство, пригодное для введения композиции согласно конкретному пути введения. Предпочтительно, набор содержит ярлык, на котором описано применение композиции терапевтического белка или пептида.
В одном варианте осуществления производное сохраняет полную функциональную активность исходных терапевтических соединений и обеспечивает увеличенное время полужизни in vivo по сравнению с исходными терапевтическими соединениями. В другом варианте осуществления производное сохраняет по меньшей мере 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44. 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 110, 120, 130, 140 или 150 процентов (%) биологической активности относительно исходного соединения.
СИАЛОВАЯ КИСЛОТА И PSA
Как используют в настоящем документе "части сиаловых кислот" включают мономеры или полимеры ("полисахариды") сиаловых кислот, которые растворимы в водном растворе или суспензии и имеют небольшое отрицательное влияние, такое как побочные эффекты, или не имеют его, у млекопитающих при введении конъюгата PSA-белок в фармацевтически эффективном количестве. В одном аспекте PSA и mPSA характеризуются как имеющие 1, 2, 3, 4, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400 или 500 звеньев сиаловых кислот. В некоторых аспектах различные звенья сиаловых кислот объединены в цепь.
В одном варианте осуществления изобретения часть сиаловых кислот в PSA или mPSA является высокогидрофильной, и в другом варианте осуществления все соединение является высокогидрофильным. Гидрофильность обеспечивается, главным образом, выступающими карбоксильными группами звеньев сиаловых кислот, а также гидроксильными группами. Сахаридное звено может содержать другие функциональные группы, такие как амино, гидроксильную или сульфатную группу или их комбинации. Эти группы могут присутствовать на встречающихся в природе сахаридных соединениях или могут быть внесены в производные полисахаридные соединения. PSA и mPSA, используемые в способах и конъюгатах по изобретению, можно далее охарактеризовывать, как описано выше в разделе "Уровень техники изобретения".
Встречающийся в природе полимер PSA доступен в качестве полидисперсного препарата, демонстрирующего широкое распределение размеров (например, Sigma C-5762) и высокую полидисперсность (PD). Поскольку полисахариды обычно продуцируются в бактериях, несущих в себе присущий им риск совместно очищающихся эндотоксинов, очистка длинных цепей полимеров сиаловых кислот может привести к вероятности увеличенного содержания эндотоксинов. Короткие молекулы PSA с 1-4 звеньями сиаловых кислот также можно получать синтетически (Kang SH et al., Chem Commun. 2000; 227-8; Ress D and Linhardt RJ, Current Organic Synthesis. 2004; 1:31-46), таким образом минимизируя риск высоких уровней эндотоксинов. Однако также можно изготавливать препараты PSA с узким распределением размеров и низкой полидисперсностью, которые не содержат эндотоксинов. Полисахаридные соединения, особенно пригодные для изобретения, представляют собой, в одном аспекте, соединения, продуцируемые бактериями. Некоторые из этих встречающихся в природе полисахаридов известны в качестве гликолипидов. В одном варианте осуществления полисахаридные соединения по существу не содержат концевых галактозных звеньев.
В различных вариантах осуществления соединение связано или ассоциировано с соединением PSA или mPSA в стехиометрических количествах (например, 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:7, 1:8, 1:9 или 1:10 и т.д.). В различных вариантах осуществления с соединением связаны 1-6, 7-12 или 13-20 единиц PSA и/или mPSA. В других вариантах осуществления с соединением связаны 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 или более единиц PSA и/или mPSA.
Необязательно, соединение модифицировано путем внесения участков гликозилирования (т.е. участков, отличных от природных участков гликозилирования). Такую модификацию можно проводить с использованием стандартных молекулярно-биологических способов, известных в данной области. Более того, соединение перед конъюгацией через одну или несколько углеводных групп можно гликолизировать in vivo или in vitro.
АМИНООКСИ-СВЯЗЬ
В одном варианте осуществления изобретения для получения конъюгатов соединения применяют реакцию гидроксиламина или производных гидроксиламина с альдегидами (например, на углеводной части с последующим окислением перйодатом натрия) с образованием группы оксима. Например, гликопротеин сначала окисляют окислителем, таким как перйодат натрия (NaIO4) (Rothfus JA et Smith EL., J Biol Chem 1963, 238, 1402-10; и Van Lenten L and Ashwell G., J Biol Chem 1971, 246, 1889-94). Окисление перйодатом, например, гликопротеинов, основано на классической реакции Малапраде, описанной в 1928 году, окислении соседних диолов перйодатом с образованием активной альдегидной группы (Malaprade L., Analytical application, Bull Soc Chim France, 1928, 43, 683-96). Дополнительными примерами такого окислителя являются тетраацетат свинца (Pb(OAc)4), ацетат марганца (MnO(Ac)3), ацетат кобальта (Co(OAc)2), ацетат таллия (TlOAc), сульфат церия (Ce(SO4)2) (US 4367309) или перрутенат калия (KRuO4) (Marko et al., J Am Chem Soc 1997,1 19, 12661-2) Под "окислителем" понимают мягко окисляющее соединение, которое способно окислять соседние диолы в углеводах, тем самым образуя активные альдегидные группы в физиологических условиях реакции.
Второй стадией является присоединение полимера, содержащего аминооксигруппу, к окисленной углеводной части, с образованием оксимной связи. В одном варианте осуществления изобретения эту стадию можно проводить в присутствии каталитических количеств нуклеофильного катализатора анилина или производных анилина (Dirksen A et Dawson PE, Bioconjugate Chem. 2008; Zeng Y et al., Nature Methods 2009; 6: 207-9). Катализ анилином значительно ускоряет сшивание оксимов, позволяя использовать очень низкие концентрации реагентов, в другом варианте осуществления изобретения оксимную связь стабилизируют восстановлением NaCNBH3 с образованием алкоксиаминовой связи.
В одном варианте осуществления изобретения стадии реакции для конъюгации PSA или mPSA с белком проводят по отдельности или последовательно (т.е. исходные материалы (например, белок, полимер и т.д.), реагенты (например, окислители, анилин и т.д.) и продукты реакции (например, окисленный углевод на белке, активированный аминооксиполимер и т.д.) разделяют между отдельными стадиями реакции).
Дополнительная информация по аминоокси-технологии может быть найдена в следующих ссылках, каждая из которых включена в полном объеме: EP 1681303A1 (HAS-модифицированный эритропоэтин); WO 2005/014024 (конъюгаты полимера и белка, связанные оксимной линкерной группой); WO 96/40662 (аминоокси-содержащие линкерные соединения и их применение в конъюгатах); WO 2008/025856 (модифицированные белки); Peri F et al., Tetrahedron 1998, 54, 12269-78; Kubler-Kielb J and Pozsgay V., J Org Chem 2005, 70, 6987-90; Lees A et al., Vaccine 2006, 24(6), 716-29; и Heredia L et al., Macromolecules 2007, 40(14), 4772-9.
Преимущества изобретения включают высокий выход конъюгата, высокое сохранение активности конъюгированного гликопротеина по сравнению с неконъюгированным белком и высокую эффективность конъюгации.
Далее изобретение проиллюстрировано с помощью следующих примеров. Примеры 1-3, 9 и 11-27 иллюстрируют конкретные варианты осуществления изобретения. Примеры 4-8 и 10 включены в качестве справочных примеров вследствие их важности для получения соответствующих конъюгатов по изобретению.
ПРИМЕРЫ
Пример 1
Получение гомобифункционального линкера NH2[OCH2CH2]2ONH2
Гомобифункциональный линкер NH2[OCH2CH2]2ONH2

(3-оксапентан-1,5-диоксиамин), содержащий две активных аминооксигруппы, синтезировали согласно Boturyn et al. (Tetrahedron 1997; 53: 5485-92) посредством двухстадийной органической реакции с использованием модифицированного синтеза первичных аминов Габриэля. На первой стадии одну молекулу 2,2-хлордиэтилового эфира подвергали реакции с двумя молекулами эндо-N-гидрокси-5-норборнен-2,3-дикарбоксиимида в диметилформамиде (DMF). Желаемый гомобифункциональный продукт получали из полученного промежуточного соединения путем гидразинолиза в этаноле. Если нет иных указаний, в примерах ниже его называют диаминоксилинкером.
Пример 2
Получение гомобифункционального линкера NH2[OCH2CH2]4ONH2
Гомобифункциональный линкер NH2[OCH2CH2]4ONH2

(3,6,9-триоксаундекан-1,11-диоксиамин), содержащий две активных аминооксигруппы, синтезировали согласно Boturyn et al. (Tetrahedron 1997;53:5485-92) посредством двухстадийной органической реакции с использованием модифицированного синтеза первичных аминов Габриэля. На первой стадии одну молекулу бис-(2-2-хлорэтокси)этилового эфира подвергали реакции с двумя молекулами эндо-N-гидрокси-5-норборнен-2,3-дикарбоксимида в DMF. Желаемый гомобифункциональный продукт получали из полученного промежуточного соединения гидразинолизом в этаноле.
Пример 3
Получение аминоокси-PSA
500 мг окисленной PSA (MМ=18,8 кДа), полученной из Serum Institute of India (Pune, Индия) растворяли в 8 мл 50 мМ натрий-ацетатного буфера, pH 5,5. Затем добавляли 100 мг 3-оксапентан-1,5-диоксиамина. После встряхивания в течение 2 ч при комнатной температуре добавляли 44 мг цианоборгидрида натрия. После встряхивания в течение дополнительных 4 ч при 4°C реакционную смесь помещали в картридж для диализа Slide-A-Lyzer (Pierce, Rockford, IL) (3,5-кДа мембрана, регенерированная целлюлоза) и подвергали диализу против PBS, pH 7,2, в течение 4 суток. Продукт замораживали при -80°C. Получение аминоокси-PSA этим способом проиллюстрировано на фигуре 2.
Пример 4
Присоединение аминоокси-PSA к rFIX и очистка конъюгата
К 12,6 мг rFIX, растворенного в 6,3 мл 50 мМ натрий-ацетатного буфера, pH 6,0, добавляли 289 мкл водного раствора перйодата натрия (10 мМ). Смесь встряхивали в темноте в течение 1 ч при 4°C и гасили в течение 15 мин при комнатной температуре путем добавления 6,5 мкл 1 M глицерина. Низкомолекулярные примеси удаляли ультрафильтрацией/диафильтрацией (UF/DF) с использованием концентраторов Vivaspin (Sartorius, Goettingen, Германия) (30-кДа мембрана, регенерированная целлюлоза). Далее к UF/DF-ретентату добавляли 43 мг аминоокси-PSA, и смесь встряхивали в течение 18 ч при 4°C. Избыток реагента PSA удаляли хроматографией гидрофобного взаимодействия (HIC). Проводимость охлажденной реакционной смеси увеличивалась до 180 мС/см, и смесь помещали в 5-мл колонку HiTrap Butyl FF (GE Healthcare, Fairfield, CT) HIC (1,6×2,5 см), предварительно уравновешенную 50 мМ HEPES, 3 М хлоридом натрия, 6,7 мМ хлоридом кальция, 0,01% Tween 80, pH 6,9. Конъюгат элюировали 2,4 объемами колонки (CV) с 50 мМ HEPES, 6,7 мМ хлоридом кальция, 0,005% Tween 80, pH 7,4 при скорости потока 5 мл/мин. Препарат аналитически охарактеризовали путем измерения количества общего белка (BCA) и хромогенной активности FIX. Для конъюгата PSA-rFIX была определена удельная активность 80,2 МЕ/мг белка (56,4% по сравнению с исходным rFIX). Результаты обобщенно представлены в таблице 1.
Пример 5
Присоединение аминоокси-PSA к rFIX в присутствии анилина в качестве нуклеофильного катализатора
К 3,0 мг rFIX, растворенного в 1,4 мл 50 мМ натрий-ацетатного буфера, pH 6,0, добавляли 14,1 мкл водного раствора перйодата натрия (10 мМ). Смесь встряхивали в темноте в течение 1 ч при 4°C и гасили в течение 15 мин при комнатной температуре добавлением 1,5 мкл 1 M глицерина. Примеси с низкой молекулярной массой удаляли эксклюзионной хроматографией (SEC) с использованием колонок для обессоливания PD-10 (GE Healthcare, Fairfield, CT). 1,2 мг окисленного rFIX, растворенного в 1,33 мл 50 мМ натрий-ацетатного буфера, pH 6,0, смешивали с 70 мкл анилина (200 мМ исходный водный раствор) и встряхивали в течение 45 мин при комнатной температуре. Затем добавляли 4,0 мг аминоокси-PSA, и смесь встряхивали в течение 2 ч при комнатной температуре и в течение других 16 ч при 4°C. Образцы отбирали через 1 ч, через 2 ч и в конце реакции через 18 ч. Далее избыток реагента PSA и свободный rFIX удаляли посредством HIC. Проводимость охлажденной реакционной смеси увеличивалась до 180 мС/см, и ее наносили в 5-мл колонку HiTrap Butyl FF (GE Healthcare, Fairfield, CT) HIC (1,6×2,5 см), предварительно уравновешенную 50 мМ HEPES, 3 M хлоридом натрия, 6,7 мМ хлоридом кальция, 0,01% Tween 80, pH 6,9. Конъюгат элюировали с помощью линейного градиента до 50 мМ HEPES, 6,7 мМ хлорида кальция, 0,005% Tween 80, pH 7,4 в 20 CV со скоростью потока 5 мл/мин.
Пример 6
Присоединение аминоокси-PSA к rFIX и восстановление с помощью NaCNBH3
К 10,5 мг rFIX, растворенного в 5,25 мл 50 мМ натрий-ацетатного буфера, pH 6,0, добавляли 53 мкл водного раствора перйодата натрия (10 мМ). Смесь встряхивали в темноте в течение 1 ч при 4°C и гасили в течение 15 мин при комнатной температуре путем добавления 5,3 мкл 1 M глицерина. Низкомолекулярные примеси удаляли ультрафильтрацией/диафильтрацией (UF/DF) с использованием концентраторов Vivaspin (Sartorius, Goettingen, Германия) (30-кДа мембрана, регенерированная целлюлоза). Далее к UF/DF-ретентату добавляли 35,9 мг аминоокси-PSA, и смесь встряхивали в течение 2 ч при комнатной температуре. Затем добавляли 53 мкл водного раствора цианоборгидрида натрия (5 M), и реакции позволяли протекать в течение дополнительных 16 ч. Затем избыток реагента PSA удаляли с помощью HIC. Проводимость охлажденной реакционной смеси увеличивалась до 180 мС/см, и смесь помещали в 5-мл колонку HiTrap Butyl FF (GE Healthcare, Fairfield, CT) HIC (1,6×2,5 см), предварительно уравновешенную 50 мМ HEPES, 3 М хлоридом натрия, 6,7 мМ хлоридом кальция, 0,01% Tween 80, pH 6,9. Конъюгат элюировали 2,4 объемами колонки (CV) с 50 мМ HEPES, 6,7 мМ хлоридом кальция, 0,005% Tween 80, pH 7,4 при скорости потока 5 мл/мин.
Пример 7
Присоединение аминоокси-PSA (линкер: NH2[OCH2CH2]4ONH2) к rFIX и очистка конъюгата
К 5,6 мг rFIX, растворенного в 2,8 мл 50 мМ натрий-ацетатного буфера, pH 6,0, добавляли 102 мкл водного раствора перйодата натрия (10 мМ). Смесь встряхивали в темноте в течение 1 ч при 4°C и гасили в течение 15 мин при комнатной температуре добавлением 2,9 мкл 1 M глицерина. Низкомолекулярные примеси удаляли ультрафильтрацией/диафильтрацией (UF/DF) с использованием концентраторов Vivaspin (Sartorius, Goettingen, Германия) (30-кДа мембрана, регенерированная целлюлоза). Далее к UF/DF-ретентату добавляли 19 мг аминоокси-PSA, и смесь встряхивали в течение 18 ч при 4°С. Избыток реагента PSA удаляли с помощью HIC. Проводимость охлажденной реакционной смеси увеличивалась до 180 мС/см, и смесь помещали в 5-мл колонку HiTrap Butyl FF (GE Healthcare, Fairfield, CT) HIC (1,6×2,5 см), предварительно уравновешенную 50 мМ HEPES, 3 М хлоридом натрия, 6,7 мМ хлоридом кальция, 0,01% Tween 80, pH 6,9. Конъюгат элюировали 2,4 CV с 50 мМ HEPES, 6,7 мМ хлоридом кальция, 0,005% Tween 80, pH 7,4 при скорости потока 5 мл/мин.
Пример 8
Присоединение аминоокси-PSA к rFVIII
К 11 мг rFVIII, растворенного в 11 мл буфера Hepes, pH 6 (50 мМ Hepes, 5 мМ CaCl2, 150 мМ NaCl, 0,01% Tween) добавляли 57 мкл 10 мМ перйодата натрия. Смесь встряхивали в темноте в течение 30 мин при 4°C и гасили в течение 30 мин при 4°C добавлением 107 мкл 1 M водного раствора глицерина. Затем добавляли 19,8 мг аминоокси-PSA (18,8 кДа), и смесь встряхивали в течение ночи при 4°C. Ионную силу увеличивали добавлением буфера, содержащего 8 M ацетат аммония (8 M ацетат аммония, 50 мМ Hepes, 5 мМ CaCl2, 350 мМ NaCl, 0,01% Tween 80, pH 6,9), с получением конечной концентрации ацетата аммония 2,5 M. Далее реакционную смесь наносили на колонку HiTrap Butyl FF (GE Healthcare, Fairfield, CT), которая была уравновешена буфером для уравновешивания (2,5 M ацетат аммония, 50 мМ Hepes, 5 мМ CaCl2, 350 мМ NaCl, 0,01% Tween 80, pH 6,9). Продукт элюировали буфером для элюирования (50 мМ Hepes, 5 мМ CaCl2, 0,01% Tween 80, pH 7,4), и элюат концентрировали фильтрованием на центрифуге с использованием устройства Vivaspin (Sartorius, Goettingen, Germany) с 30000 MWCO.
Пример 9
Получение гомобифункционального линкера NH2[OCH2CH2]6ONH2
Гомобифункциональный линкер NH2[OCH2CH2]6ONH2

(3,6,9,12,15-пентаоксагептадекан-1,17-диоксиамин), содержащий две активных аминооксигруппы, синтезировали согласно Boturyn et al. (Tetrahedron 1997; 53: 5485-92) с помощью двухстадийной органической реакции с использованием модифицированного синтеза первичных аминов Габриэля. На первой стадии одну молекулу дихлорида гексаэтиленгликоля подвергали реакции с двумя молекулами эндо-N-гидрокси-5-норборнен-2,3-дикарбоксимида в DMF. Желаемый гомобифункциональный продукт получали из полученного промежуточного соединения гидразинолизом в этаноле.
Пример 10
Полисиалирование rFIX с использованием малеимидо/аминоокси-линкерой системы
A. Получение модифицирующего реагента
Аминоокси-PSA реагент получают с использованием малеимидо/аминоокси-линкерной системы (Toyokuni et al., Bioconjugate Chem 2003; 14, 1253-9). PSA-SH (20 кДа), содержащей свободную концевую SH-группу, и с использованием двухстадийного процесса: a) получение PSA-NH2 восстановительным аминированием окисленной PSA с помощью NH4Cl согласно WO 05016973A1, и b) внесение сульфгидрильной группы путем реакции концевой первичной аминогруппы с 2-иминотиоланом (реагент Трота/Pierce, Rockford, IL), как описано в US 7645860. PSA-SH присоединяют к малеимидогруппе линкера при pH 7,5 в буфере PBS с использованием 10-кратного молярного избытка линкера и концентрации PSA-SH 50 мг/мл. Реакционную смесь инкубируют в течение 2 часов при осторожном встряхивании при комнатной температуре. Затем избыток линкерного реагента удаляют, и буфер для аминоокси-PSA заменяют на буфер для окисления (50 мМ фосфат натрия, pH 6,0) путем диафильтрации. Буфер заменяют 25 раз с использованием регенерированной целлюлозной мембраны Pellicon XL5kD (Millipore, Billerica, MA).
B. Модификация rFIX после предшествующего окисления с помощью NaIO4
rFIX окисляли в 50-мМ натрий-фосфатном буфере, pH 6,0, с использованием 100-мкМ перйодата натрия в буфере. Смесь встряхивали в темноте в течение 1 ч при 4°C и гасили в течение 15 мин при комнатной температуре добавлением глицерина до конечной концентрации 5 мМ. Низкомолекулярные примеси удаляли с помощью эксклюзионной хроматографии (SEC) с использованием колонок для обессоливания PD-10 (GE Healthcare, Fairfield, CT). Затем к окисленному rFIX добавляли анилин до конечной концентрации 10 мМ и смешивали с реагентом аминоокси-PSA для обеспечения 5-кратного молярного избытка PSA. Реакционную смесь инкубировали в течение 2 часов при осторожном встряхивании в темноте при комнатной температуре.
C. Очистка конъюгатов
Избыток свободного реагента PSA и свободный rFIX удаляют с помощью HIC. Проводимость реакционной смеси увеличивается до 180 мС/см, и ее наносят на колонку, заполненную 48 мл Butyl-Sepharose FF (GE Healthcare, Fairfield, CT), предварительно уравновешенной 50 мМ Hepes, 3 M хлоридом натрия, 6,7 мМ хлоридом кальция, 0,01% Tween 80, pH 6,9. Затем конъюгат элюируют линейным градиентом 60% буфера для элюирования (50 мМ Hepes, 6,7 мМ хлорид кальция, pH 7,4) в 40 CV. Наконец, содержащие PSA-rFIX фракции собирают и подвергают UF/DF с использованием 30-кДа мембраны, изготовленной из регенерированной целлюлозы (Millipore). Препарат аналитически охарактеризовывают путем измерения общего количества белка (BCA) и хромогенной активности FIX. Для конъюгатов PSA-rFIX, полученных с обоими вариантами, определили удельную активность >50% по сравнению с исходным rFIX.
Пример 11
Получение реагента аминоокси-PSA
Реагент аминоокси-PSA получали согласно примеру 3. Конечный продукт подвергали диафильтрации против буфера, pH 7,2 (50 мМ Hepes) с использованием 5-кДа мембраны (регенерированная целлюлоза, Millipore), замораживали при -80°C и лиофилизировали. После лиофилизации реагент растворяли в соответствующем объеме воды и использовали для получения конъюгатов PSA-белок путем модификации углеводов.
Пример 12
Подробное описание синтеза реагента аминоокси-PSA
3-Оксапентан-1,5-диоксиамин синтезировали согласно Botyryn et al. (Tetrahedron 1997; 53:5485-92) с помощью двухстадийного органического синтеза, описанного в примере 1.
Стадия 1:
К раствору эндо-N-гидрокси-5-норборнен-2,3-дикарбоксиимида (59,0 г; 1,00 экв.) в 700 мл безводного N,N-диметилформамида добавляли безводный K2CO3 (45,51 г; 1,00 экв.) и 2,2-дихлордиэтиловый эфир (15,84 мл; 0,41 экв.). Реакционную смесь перемешивали в течение 22 часов при 50°C. Смесь упаривали до сухого состояния при пониженном давлении. Остаток суспендировали в 2 л дихлорметана и экстрагировали два раза насыщенным водным раствором NaCl (каждый раз по 1 л). Слой дихлорметана сушили над Na2SO4, а затем упаривали до сухого состояния при пониженном давлении и сушили в высоком вакууме с получением 64,5 г 3-оксапентан-1,5-диоксиэндо-2',3'-дикарбоксидиимиднорборнена в виде беловато-желтого твердого вещества (промежуточное соединение 1).
Стадия 2:
К раствору промежуточного соединения 1 (64,25 г; 1,00 экв.) в 800 мл безводного этанола добавляли 31,0 мл гидрата гидразина (4,26 экв.). Затем реакционную смесь кипятили с обратным холодильником в течение 2 часов. Смесь концентрировали до половины исходного объема путем выпаривания растворителя при пониженном давлении. Появляющийся осадок отфильтровывали. Оставшийся слой этанола упаривали до сухого состояния при пониженном давлении. Остаток, содержавший неочищенный продукт 3-оксапентан-1,5-диоксиамин, сушили в вакууме с получением 46,3 г. Неочищенный продукт далее очищали колоночной хроматографией (Silicagel 60; изократическое элюирование смесью дихлорметан/метанол, 9+1) с получением 11,7 г чистого конечного продукта 3-оксапентан-1,5-диоксиамина.
Пример 13
Получение полимера аминоокси-PSA
1,3 г окисленной коломиновой кислоты (23 кДа) растворяли в 18 мл 50 мМ ацетата натрия pH 5,5±0,02. 20-Кратный молярный избыток 1,11-диамино-3,6,9-триоксаундекана (также называемого 3,6,9-триоксаундекан-1,11-диоксиамином) растворяли в минимальном количестве 50 мМ ацетата натрия (pH 5,5±0,02) и добавляли к раствору PSA. Конечная концентрация коломиновой кислоты составляла 62,5 мг/мл. Эту реакционную смесь инкубировали в течение 2±0,1 ч при 22±1,0°C на мягком смесителе (22 колебания в минуту). После этого к указанной выше реакционной смеси добавляли 0,65 мл раствора NaCNBH3 в концентрации 160 мг/мл до конечной концентрации 5,00 мг/мл. Его инкубировали в течение 3,0±0,20 часов при 4,0±1,0°C на устройстве для встряхивания (22 колебания в минуту) в воздухонепроницаемом не содержащем эндотоксинов контейнере с достаточным количеством свободного пространства для перемешивания. Для очистки образец разбавляли 2 мМ триэтаноламином, pH 8,0±0,02, до конечной концентрации коломиновой кислоты 20 мг/мл. Реакционную смесь обессоливали для удаления избытка 1,11-диамино-3,6,9-триоксаундекана, NaCNBH3 и побочных продуктов реакции. После этого проводили обессоливание на колонке Sephadex G25 с использованием 20 мМ триэтаноламинового буфера (pH 8,0±0,02). pH обессоленного образца доводили до pH 7,8-8,0 и подвергали ультрафильтрации/диафильтрации посредством 20 мМ TEA pH 8,0 один раз и 2 мМ триэтаноламина (TEA) pH 8,0 два раза. Образец лиофилизировали и хранили при -80°C.
Альтернативно, очистку проводили в присутствии высокого содержания соли в ходе стадий обессоливания и ультрафильтрации/диафильтрации (UF/DF). Также использовали анионообменную хроматографию в высоком содержании соли для получения высокочистого аминоокси-PSA. По аналогии синтезировали аминоокси-PSA с различной молекулярной массой.
Пример 14
Присоединение диаминоокси-(3,6,9-триоксаундекан-1,11-диоксиамин)-PSA к β-галактозидазе
Для окисления β-галактозидазы (β-Gal) использовали различные концентрации NaIO4 (в диапазоне от 0,157 мМ до 2 мМ). 0,5 мг β-Gal окисляли при кислом pH 5,75 при 4°C в течение 30 минут в темноте. Окисление останавливали добавлением NaHSO3 до конечной концентрации 5 мМ. Реакцию конъюгации проводили с использованием окисленного β-Gal с полимером диаминоокси-PSA (22 кДа). Конечная концентрация полимера в реакционной смеси составляла 1,25 мМ, в то время как концентрация β-Gal находилась в диапазоне от 0,125 мг/мл до 0,76 мг/мл. Все реакции проводили при pH 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C, и образцы собирали с временными интервалами 1, 2 и 24 часов. Конъюгаты охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгата при SDS PAGE наблюдали сдвиг полосы, и он также подтверждался вестерн-блоттингом.
Исходя из наилучших условий реакций, 1,9 мг β-Gal окисляли 1,5 мМ NaIO4 в течение 30 минут при 4°C, а затем окисление останавливали добавлением NaHSO3 до конечной концентрации 5 мМ. Реакцию конъюгации проводили с использованием окисленного β-Gal с полимером диаминоокси-PSA. Конечные концентрации полимера и белка в реакционной смеси составляли 1,25 мМ и 0,76 мг/мл, соответственно. Конечное значение pH реакционной смеси составляло приблизительно 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C в течение 2 часов. Очищенные и неочищенные конъюгаты охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгата при SDS PAGE наблюдали сдвиг полосы, и он также подтверждался вестерн-блоттингом с использованием антитела против PSA. Активность конъюгатов PSA-βGal in vitro была сравнима с исходным белком при использовании универсального набора для βGal анализа (All in one βGal assay kit) (Pierce). В сравнимых конъюгатах наблюдали менее чем 50% активность при использовании химии альдегидных линкеров. Кроме того, процесс в целом масштабировали вплоть до 3 раз.
Пример 15
Присоединение диаминоокси-PSA к фетуину
Фетуин окисляли 10 мМ NaIO4 в течение 60 минут при 4°C в темноте, и окисление останавливали добавлением NaHSO3 до конечной концентрации 10 мМ. Реакцию конъюгации проводили с использованием окисленного фетуина с полимером диаминоокси-PSA (23 кДа). Конечная концентрация полимера в реакционной смеси составляла 2,5 мМ при pH 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Конечная концентрация белка в реакционной смеси составляла 0,714 мг/мл, и реакцию проводили при 4°C в течение 2 часов. Эти конъюгаты охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгатов при SDS PAGE наблюдали сдвиг полосы, и он также подтверждался вестерн-блоттингом.
Для масштабирования реакции 5 мг фетуина окисляли 10 мМ NaIO4 в течение 60 минут при 4°C в темноте, а затем окисление останавливали добавлением NaHSO3 до конечной концентрации 10 мМ. Реакцию конъюгации проводили с использованием окисленного фетуина с полимером диаминоокси-PSA (23 кДа). Конечная концентрация полимера в реакционной смеси составляла 2,5 мМ при pH 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C, и через 2 часа отбирали образец. Очищенные и неочищенные конъюгаты охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгатов при SDS PAGE наблюдали сдвиг полосы, и он также подтверждался вестерн-блоттингом.
Пример 16
Присоединение диаминоокси-PSA к фетуину с анилином для действия в качестве нуклеофильного катализатора
0,2 мг фетуина окисляли 10 мМ NaIO4 в течение 30 минут при 4°C в темноте, а затем окисление останавливали добавлением NaHSO3 до конечной концентрации 5 мМ. Реакцию конъюгации проводили с использованием окисленного фетуина с полимером диаминоокси-PSA (23 кДа). Конечная концентрация полимера в реакционной смеси составляла 1,25 мМ. Конечное значение pH реакционной смеси составляло 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Конечная концентрация белка в реакционной смеси составляла 0,125 мг/мл. К 1,6 мл реакционной смеси добавляли 84,21 мкл 200 мМ раствора анилина. Реакцию проводили при 4°C в течение ночи.
Пример 17
Присоединение диаминоокси-PSA к эритропоэтину (ΕΡΟ)
0,2 мг EPO окисляли с помощью 10 мМ NaIO4 в течение 30 минут при 4°C. Окисление останавливали добавлением NaHSO3 до конечной концентрации 5 мМ. Реакцию конъюгации проводили с использованием окисленного EPO с диаминоокси-полимером размером 23 кДа. Конечная концентрация полимера в реакционной смеси составляла 1,25 мМ. Конечная концентрация EPO в реакционной смеси составляла 0,125 мг/мл. Конечное значение pH реакционной смеси составляло приблизительно 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C в течение 24 часов. Неочищенный конъюгат охарактеризовывали с использованием SDS PAGE. Для конъюгатов при SDS PAGE наблюдали сдвиг полосы.
Пример 18
Присоединение диаминоокси-PSA к EPO с анилином для действия в качестве нуклеофильного катализатора
0,2 мг EPO окисляли 10 мМ NaIO4 в течение 30 минут при 4°C. Окисление останавливали добавлением NaHSO3 до конечной концентрации 5 мМ. Реакцию конъюгации проводили с использованием окисленного EPO с полимером диаминоокси-PSA (22 кДа). Конечная концентрация полимера в реакционной смеси составляла 1,25 мМ. Конечное значение pH реакционной смеси составляло 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Конечная концентрация белка в реакционной смеси составляла 0,125 мг/мл. К 1,6 мл реакционной смеси добавляли 84,21 мкл 200 мМ раствора анилина. Реакцию проводили при 4°C в течение ночи. Конъюгаты охарактеризовывали с использованием SDS PAGE. Для конъюгатов при SDS PAGE наблюдали сдвиг полосы. В отношении активности конъюгатов не наблюдали неблагоприятного эффекта.
Пример 19
Присоединение диаминоокси-PSA к ДНКазе
Для гликополисиалирования ДНКазы для реакции конъюгации использовали ДНКазу бычьей поджелудочной железы. Этот источник ДНКазы поставляли в качестве лиофилизированного порошка, который хранили при -20°C. Перед реакцией этот лиофилизированный порошок растворяли в натрий-ацетатном буфере (pH 5,75). Полимер, использованный для гликополисиалирования, имеет массу в диапазоне от 10 кДа до 22 кДа. Для окисления гликонной части на ДНКазе, в качестве окислителя использовали NaIO4 до конечной концентрации 1 мМ. ДНКазу окисляли при кислом pH 5,75 при 4°C в течение 30 минут. Окисление останавливали добавлением NaHSO3 до конечной концентрации 2 мМ. После завершения окисления реакцию конъюгации проводили добавлением полимера диаминоокси-PSA до конечной концентрации 1,25 мМ. К реакционной смеси добавляли NaCNBH3 до конечной концентрации 50 мМ или 3,17 мг/мл, и полисиалирование ДНКазы проводили при 4,0±1,0°C в течение по меньшей мере 2 часов. Реакцию останавливали 25-кратным молярным избытком Tris относительно полимера. Конъюгаты охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгата при SDS PAGE наблюдали сдвиг полосы, и положительный результат получали с помощью вестерн-блоттинга. Активность измеряли в качестве 95% (по сравнению с менее чем 50%, наблюдаемыми в сравнимых конъюгатах, полученных с использованием химии альдегидных линкеров).
Пример 20
Присоединение диаминоокси (3-оксапентан-1,5-диоксиаминового линкера)-PSA к β-галактозидазе
Для окисления β-галактозидазы использовали NaIO4 в концентрации 2 мМ. 3 мг β-галактозидазы окисляли при кислом pH, равном 5,75, при 4°C в течение 30 минут, а затем окисление останавливали добавлением NaHSO3 до конечной концентрации 2 мМ. Реакцию конъюгации проводили с использованием окисленной β-галактозидазы с полимером диаминоокси-PSA (23 кДа). Конечная концентрация полимера в реакционной смеси составляла 1,5 мМ. Конечная концентрация β-галактозидазы в реакционной смеси составляла 0,867 мг/мл. Конечное значение pH реакционной смеси составляло приблизительно 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C в течение 2 часов. Конъюгаты охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгата при SDS PAGE наблюдали сдвиг полосы, и положительный результат получали с помощью вестерн-блоттинга.
Пример 21
Получение гидразид-коломиновой кислоты
Авторы настоящего изобретения использовали следующий протокол для получения PSA-гидразида (коломиновая кислота-гидразид) с использованием дигидразида адипиновой кислоты. Аналогичные способы использовали для получения других PSA-гидразидов.
1. Растворить 1 г активированной коломиновой кислоты в ~10 мл 20 мМ ацетата натрия pH 5,5±0,02. Конечная концентрация коломиновой кислоты должна составлять 62,5 мг/мл.
2. Растворить 25-кратный молярный избыток (относительно окисленной коломиновой кислоты "CAO") дигидразида адипиновой кислоты (ММ=174,2 г) в минимальном количестве 20 мМ ацетата натрия (pH 5,5±0,02) и добавить к раствору из 1.
3. Количество дигидразида адипиновой кислоты, подлежащее применению = (масса CAO в граммах × 25 × ММ дигидразида адипиновой кислоты в г)/ММ CAO в дальтонах = (1×25×174,2)/15×103=0,290 г.
4. После добавления раствора дигидразида адипиновой кислоты довести объем коломиновой кислоты ацетатом натрия до конечной концентрации 62,5 мг/мл. Таким образом, общий объем реакции составляет 16 мл.
5. Инкубировать реакционную смесь в течение 2±0,1 ч при 22,0±1,0°C на устройстве для встряхивания (22 колебаний в минуту).
6. Приготовить концентрированный раствор NaCNBH3 (165 мг/мл) и добавить 0,5 мл к раствору из 1 так, чтобы конечная концентрация этого раствора в конечной реакционной смеси составила 5,0 мг/мл. Инкубировать реакционную смесь в течение 3,0±0,20 часов при 4,0±1,0°C на устройстве для встряхивания (22 колебаний в минуту).
7. Поддерживать реакционную смесь в не содержащем эндотоксинов воздухонепроницаемом контейнере с избытком свободного пространства 50 мл для надлежащего перемешивания (должно быть достаточно пространства, чтобы реакционная смесь не касалась крышки контейнера).
8. После реакции в течение 3 часов при 4°C разбавить образец 2 мМ триэтаноламином (довести объем до 50 мл) при pH 8,0±0,02 для получения конечной концентрации коломиновой кислоты 20 мг/мл.
9. Обессолить реакционную смесь для удаления избытка необработанного дигидразида адипиновой кислоты, NaCNBH3 и т.д. из полимера. Это можно осуществлять с помощью GPC (с использованием матрицы носителя XK 50 Sephadex G-25; ≤1,8 мг CA/мл матрицы; высота слоя 35 см; объем колонки = 687 мл) путем наблюдения при УФ 224 нм и проводимости. Обессоливание проводят с помощью 20 мМ триэтаноламинового (pH 8,0±0,02) буфера.
10. После обессоливания коломиновую кислоту-гидразид подвергают 1 циклу ультрафильтрации, 1 цикл диафильтрации с использованием 20 мМ TEA, pH 8,0±0,02 и по меньшей мере 3 циклов диафильтрации с использованием 2 мМ TEA, pH 8,0±0,02. Это можно осуществлять с использованием 3-кДа кассет vivaflow.
11. Довести pH обессоленного образца до pH 7,8-8,0. Необязательно высушить сублимацией образец, а затем поддерживать его для вторичного высушивания для удаления избытка влаги.
Пример 22
Присоединение гидразид-PSA к эритропоэтину
Для окисления эритропоэтина (EPO) использовали NaIO4 в концентрации 10 мМ. EPO (1 мг) окисляли при pH 5,75 при 4°C в течение 30 минут, затем окисление останавливали добавлением NaHSO3 до конечной концентрации 5 мМ. Реакцию конъюгации проводили с использованием окисленного EPO с полимером гидразид-PSA. Молекулярная масса гидразид-PSA, использованного для конъюгации, составляла 24,34 кДа. Конечная концентрация гидразид-PSA в реакционной смеси составила 1,25 мМ. Конечная концентрация EPO в реакционной смеси составила 0,125 мг/мл. Конечное значение pH реакционной смеси составило приблизительно 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C в течение 24 часов. Конъюгаты охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгата при SDS PAGE наблюдали сдвиг полосы, и положительный результат получали с помощью вестерн-блоттинга.
Пример 23
Присоединение гидразид-PSA к β-галактозидазе
β-Галактозидазу (0,5-4,5 мг) окисляли с помощью от 0,625 до 2 мМ NaIO4 в течение 30 минут при 4°C. Окисление останавливали добавлением NaHSO3 до конечной концентрации 5 мМ. Реакцию конъюгации проводили с использованием окисленной β-галактозидазы с гидразид-PSA в диапазоне от 24,34 кДа до 27,9 кДа. Конечная концентрация гидразид-PSA в реакционной смеси составила 1,25 мМ. Конечная концентрация β-галактозидазы в реакционной смеси находилась в диапазоне от 0,125 мг/мл до 0,76 мг/мл. Конечное значение pH реакционной смеси должно составлять приблизительно 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C, и образцы собирали через 1, 2 и 24 часа. Очищенный и неочищенный конъюгат охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгата при SDS PAGE наблюдали сдвиг полосы, и положительный результат получали с помощью вестерн-блоттинга. Измеренная активность составила 84%. В сравнимых конъюгатах, полученных с использованием химии альдегидных линкеров, наблюдали менее чем 50% активность.
Пример 24
Присоединение гидразид-PSA к фетуину
Фетуин (0,25 мг) окисляли с помощью NaIO4 (5 или 10 мМ) в течение 30 или 60 минут при 4°C. Окисление останавливали добавлением NaHSO3 до конечной концентрации 5 или 10 мМ соответствующим образом для соответствия концентрации NaIO4, использованного для окисления. Реакцию конъюгации проводили с использованием окисленного фетуина с полимером дигидразид адипиновой кислоты-PSA. Конечная концентрация полимера в реакционной смеси составила от 1,25 до 2,5 мМ. Конечное значение pH реакционной смеси должно составлять приблизительно 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C в течение от 1 до 4 часов. Конъюгаты охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгата при SDS PAGE наблюдали сдвиг полосы, и положительный результат получали с помощью вестерн-блоттинга.
Проводили масштабированную реакцию для 5 мг фетуина с последующей очисткой полученного конъюгата. 5 мг фетуина окисляли 10 мМ NaIO4 в течение 60 минут при 4°C, а затем окисление останавливали добавлением NaHSO3 до конечной концентрации 10 мМ. Реакцию конъюгации проводили с использованием окисленного фетуина с полимером адипиновая кислота дигидразид-PSA. Конечная концентрация полимера в реакционной смеси составляла 2,5 мМ. Конечное значение pH реакционной смеси составляло приблизительно 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C, и образцы собирали через 2 часа. Очищенный и неочищенный конъюгат охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгата при SDS PAGE наблюдали сдвиг полосы, и положительный результат получали с помощью вестерн-блоттинга.
Пример 25
Присоединение гидразид-PSA к ДНКазе
ДНКазу окисляли с помощью NaIO4 в конечной концентрации в диапазоне от 0,2 мМ до 2 мМ в течение 30 минут при 4°C. Окисление останавливали добавлением NaHSO3 до конечной концентрации 2 и 5 мМ, в зависимости от концентрации NaIO4, использованной для окисления. Гликополисиалирование окисленной ДНКазы проводили добавлением к окисленной ДНКазе полимера гидразид-PSA до конечной концентрации 1,25 мМ. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл, и гликополисиалирование ДНКазы проводили при 4°С в течение периода времени в диапазоне от 1 часа до 2 часов. Реакцию останавливали 25-кратным молярным избытком Tris относительно полимера. Конъюгаты охарактеризовывали с использованием SDS PAGE и вестерн-блоттинга. Для конъюгатов при SDS PAGE наблюдали сдвиг полосы, и положительный результат получали с помощью вестерн-блоттинга. Измеренная активность составила 49%.
Пример 26
Пегилирование β-галактозидазы с использованием аминооксилинкера (3-оксапентан-1,5-диоксиамин)
β-Галактозидазу (1 мг) окисляли 1,5 мМ NaIO4 в течение 30 минут при 4°C. Окисление останавливали добавлением NaHSO3 до конечной концентрации 1,5 мМ.
Реакцию конъюгации проводили с использованием окисленной β-галактозидазы с полимером диаминоокси-PEG (20 кДа). Конечная концентрация полимера в реакционной смеси составила 1,25 мМ. Конечная концентрация β-галактозидазы в реакционной смеси составила 1 мг/мл. Конечное значение pH реакционной смеси должно составлять приблизительно 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Реакцию проводили при 4°C в течение 2 часов. Неочищенный конъюгат охарактеризовывали с использованием SDS PAGE, и для конъюгата при SDS PAGE наблюдали сдвиг полосы. Измеренная активность составила 59%.
Пример 27
Пегилирование эритропоэтина с использованием аминооксилинкера
Эритропоэтин (EPO; 0,2 мг) окисляли 5 или 10 мМ NaIO4 в 50 мМ ацетате натрия при pH 5,75 в течение 45 минут при 4°C, а затем окисление останавливали добавлением NaHSO3 до конечной концентрации 5 или 10 мМ (для соответствия концентрации NaIO4, использованной для окисления). Реакцию конъюгации проводили с использованием окисленного EPO с полимером диаминоокси-PEG (20 кДа). Конечная концентрация полимера в реакционной смеси составила 1,5 мМ. Конечное значение pH реакционной смеси должно было составлять 5,75. К реакционной смеси добавляли цианоборгидрид натрия до концентрации 50 мМ или 3,17 мг/мл. Конечная концентрация белка в реакционной смеси составила 0,4 мг/мл. Реакцию конъюгации проводили в течение ночи при 4°C.
Таким образом, изобретение относится к конъюгатам соединений, отличных от белков свертывания крови, с растворимыми в воде полимерами, в частности PSA и mPSA.
Реферат
Изобретение относится к способу конъюгации полисиаловой кислоты (PSA) или модифицированной PSA (mPSA) с окисленной углеводной частью гликопротеина, отличного от белка свертывания крови, содержащего углеводную группу, посредством контактирования окисленной углеводной части с PSA или mPSA в условиях, которые обеспечивают конъюгацию, где указанная PSA или mPSA содержат гидразидную группу, и между окисленной углеводной частью на гликопротеине и гидразидной группой на PSA или mPSA образуется гидразоновая связь, где модифицированная PSA представляет собой PSA, содержащую группу, образованную из концевой группы N-ацетилнейраминовой кислоты путем окисления или восстановления. Способ позволяет получить высокий выход конъюгата, высокое сохранение активности конъюгированного гликопротеина по сравнению с неконъюгированным белком и высокую эффективность конъюгации. 3 н. и 11 з.п. ф-лы, 2 ил., 1 табл., 27 пр.
Формула
Документы, цитированные в отчёте о поиске
N-концевое полисиалилирование
Комментарии