Пептидные аналоги альфа-меланоцитстимулирующего гормона - RU2496786C2
Код документа: RU2496786C2
Чертежи






































Описание
Область техники
Изобретение относится к пептидным аналогам. В частности, изобретение относится к пептидным аналогам природного альфа-меланоцитстимулирующего гормона (α-MSH), селективным в отношении рецептора меланокортина 1 (MC1R), к фармацевтическим препаратам, а также к применению этих аналогов для лечения заболеваний в медицине и ветеринарии.
Предшествующий уровень техники
Нейропептиды представляют собой небольшие биологически активные пептиды, которые широко распространены в организме и выполняют функции от нейромедиаторов до факторов роста. Многочисленные данные указывают на то, что нейропептиды обладают противовоспалительными свойствами. Одним из представителей множества нейропептидов являются меланокортиновые пептиды (меланокортины), которые связываются с рецепторами меланокортина (MC) и стимулируют их. Примером меланокортина служит α-меланоцитстимулирующий гормон (α-MSH), известный, в основном, благодаря своей способности регулировать внешнюю пигментацию; известно также, что он обладает противовоспалительными и иммуномодулирующими свойствами. Нейропептид α-MSH был обнаружен в некоторых органах и вырабатывается нейронами, гипофизом, кишечником, кожей и иммунными клетками.
Иммуномодулирующие способности α-MSH были показаны на моделях контактной гиперчувствительности, где гаптенспецифическая толерантность была вызвана инъекцией α-MSH, и при подавлении воспаления, опосредованного бактериальным эндотоксином. Было показано также, что α-MSH имеет терапевтическую активность на многих животных моделях заболеваний, таких как воспалительное заболевание кишечника, артриты и экспериментальная пересадка сердца. Другие животные модели воспаления мозга, повреждения почек и воспаления печени показали противовоспалительные эффекты, связанные с этим нейропептидом. α-MSH подавляет выработку провоспалительных цитокинов, таких как TNF-α, IL-6 и IL-1, и ингибирует хемокины, что снижает миграцию макрофагов и нейтрофилов к участкам воспаления. Оксид азота (NO) является общим медиатором для различных форм воспаления. Показано также, что синтез NO эндотоксин-стимулированными макрофагами и нейтрофилами ингибируется α-MSH. Кроме воздействия на выработку цитокинов, α-MSH снижает экспрессию MHC класса I, CD86 и CD40 моноцитов и дендритных клеток, что влияет на презентацию антигена и ко-стимуляцию. Известно также, что α-MSH усиливает образование интерлейкина 10 (IL-10) моноцитами, что, как полагают, является важным компонентом иммуносупрессивных воздействий.
Хотя молекулярные механизмы иммуномодулирующих эффектов α-MSH до конца неясны, возможный механизм действия α-MSH заключается в его способности ингибировать активацию ядерного фактора-κB в клетках. Ингибирование NF-κB приводит к подавлению выработки провоспалительных цитокинов и синтеза оксида азота макрофагами. α-MSH действует путем связывания со специфическими рецепторами, которые относятся к группе сопряженных с G-белком рецепторов с семью трансмембранными доменами. Эти рецепторы содержат рецепторы меланокортина 1 и меланокортина 3 (MCR-1 и MCR-3) на макрофагах, посредством связывания с которыми α-MSH ингибирует NF-κB. Многие из иммуномодулирующих эффектов α-MSH опосредуются также через накопление цАМФ. Связывание α-MSH с меланокортиновыми рецепторами увеличивает уровни цАМФ, что может подавлять деградацию IκB и, таким образом, ингибировать транслокацию NF-κB и выработку оксида азота.
Рецепторы MC1, с которыми связывается и которые стимулирует α-MSH, вовлечены в различные типы противовоспалительного и иммуномодулирующего ответа. Обнаружены пять типов меланокортиновых рецепторов, MC1-MC5. MC1-рецепторы обнаружены на меланоцитах, клетках меланомы, макрофагах, нейтрофилах, клетках глиомы, астроцитах, моноцитах, эндотелиальных клетках, в определенных областях мозга, яичках и яичниках. Существует постоянный интерес в получении соединений и разработке способов стимуляции MC1-рецепторов и индукции эффективного противовоспалительного и иммуномодулирующего ответа.
Сущность изобретения
Настоящее изобретение относится к по существу чистому соединению, которое селективно связывается с рецептором меланокортина 1 (MC1R); указанное соединение содержит основной тетрапептид с последовательностью His Xaa1 Arg Trp (SEQ ID NO:1) или D-Trp D-Arg Xaa2 D-His (SEQ ID NO:2); где Xaa1 представляет собой D-Cha, D-Phe или Cha, и Xaa2 представляет собой D-Cha, D-Phe или Phe; или его фармацевтически приемлемой соли. В некоторых вариантах осуществления C-концевая последовательность представляет собой D-Ser D-Ile D-Ile D-Ser D-Ser (SEQ ID NO:3).
Настоящее изобретение также относится к по существу чистому соединению, которое селективно связывается с рецептором меланокортина 1 (MC1R); указанное соединение содержит полипептид с последовательностью:
Xaa1 Xaa2 Xaa3 Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 Xaa11 Xaa12 Xaa13,
где Xaa1 представляет собой D-Val, D-Ala или D-Lys;
Xaa2 представляет собой D-Pro, D-Ala или D-Lys;
Xaa3 представляет собой D-Lys, D-Orn, D-Nle, D-Ala или D-Lys;
Xaa4 представляет собой Gly или D-Ala;
Xaa5 представляет собой D-Trp, Trp, D-3-бензотиенил-Ala, D-5-гидрокси-Trp, D-5-метокси-Trp, D-Phe или D-Ala;
Xaa6 представляет собой D-Arg, D-His или D-Ala;
Xaa7 представляет собой D-Cha, D-Phe, Phe, D-4-фтор-Phg, D-3-пиридил-Ala, D-Thi, D-Trp, D-4-нитро-Phe или D-Ala;
Xaa8 представляет собой D-His, His, D-Arg, Phe или D-Ala;
Xaa9 представляет собой D-Glu, D-Asp, D-цитруллин, D-Ser или D-Ala;
Xaa10 представляет собой D-Met, D-бутионин, D-Ile или D-Ala;
Xaa11 представляет собой D-Ser, D-Ile или D-Ala;
Xaa12 представляет собой D-Tyr, D-Ser или D-Ala;
Xaa13 представляет собой D-Ser или D-Ala;
где не более, чем один Xaa1-13 представляет собой D-Ala, за исключением, когда все Xaa1-3 представляют собой D-Ala, и не более, чем один Xaa1-3представляет собой L-аминокислоту;
или к его фармацевтически приемлемой соли.
Еще в одном из аспектов настоящее изобретение относится к по существу чистому соединению, содержащему полипептид с последовательностью:
D-Val D-Pro D-Lys Gly D-Trp D-Arg Phe D-His D-Ser D-Ile D-Ile D-Ser D-Ser (SEQ ID NO:4);
D-Val D-Pro D-Lys Gly D-Trp D-Arg D-Cha D-His D-Ser D-Ile D-Ile D-Ser D-Ser (SEQ ID NO:5);
Ser Tyr Ser Met Glu His Cha Arg Trp Gly Lys Pro Val (SEQ ID NO: 6); или
D-Val D-Pro D-Lys Gly D-Trp D-Arg D-Phe D-His D-Glu D-Met D-Ser D-Tyr D-Ser (SEQ ID NO:7);
или к его фармацевтически приемлемой соли.
В некоторых вариантах осуществления полипептиды по изобретению являются пегилированными.
В некоторых вариантах осуществления соединения по изобретению могут быть конъюгированы с биологически активной частью.
В некоторых вариантах осуществления соединения по изобретению селективно связываются с MC1R. В некоторых вариантах осуществления соединения обладают, по меньшей мере, одним из следующих свойств: способностью селективно активировать MC1R, стабильностью в плазме in vitro и устойчивостью к деградации протеазами.
В одном из аспектов изобретение относится к фармацевтической композиции, которая содержит любое по существу чистое соединение по изобретению и фармацевтически приемлемый эксципиент.
В другом аспекте изобретение относится к способу лечения аутоиммунных заболеваний или состояний у пациента, содержащему введение указанному пациенту фармацевтической композиции, которая содержит фармацевтически приемлемый эксципиент и терапевтически эффективное количество соединения по изобретению. В некоторых вариантах осуществления аутоиммунное заболевание или состояние выбрано из группы, состоящей из рассеянного склероза, сахарного диабета типа 1, апластической анемии, болезни Грейвза, целиакии, болезни Крона, волчанки, артрита, остеоартрита, аутоиммунного увеита и миастении гравис.
Еще в одном из аспектов изобретение относится к способу лечения воспаления у пациента, содержащему введение указанному пациенту фармацевтической композиции, содержащей фармацевтически приемлемый эксципиент и терапевтически эффективное количество соединения по изобретению. В некоторых вариантах осуществления воспаление связано с заболеванием, выбранным из группы, которая состоит из воспалительного заболевания кишечника, ревматоидного артрита, аллергии, атеросклероза, псориаза, гастрита и ишемической болезни сердца.
В одном из аспектов изобретение относится к способу уменьшения или замедления отторжения трансплантата у пациента, содержащему введение указанному пациенту фармацевтической композиции, которая содержит фармацевтически приемлемый эксципиент и терапевтически эффективное количество соединения по изобретению.
В другом аспекте изобретение относится к способу лечения меланомы у пациента, содержащему введение указанному пациенту фармацевтического препарата, содержащего фармацевтически приемлемый эксципиент и терапевтически эффективное количество соединения по изобретению.
Еще в одном из аспектов изобретение относится к способу лечения меланомы у пациента, содержащему введение указанному пациенту фармацевтического препарата, содержащего фармацевтически приемлемый эксципиент и терапевтически эффективное количество соединения по изобретению, которое конъюгировано с противоопухолевой полезной нагрузкой. Противоопухолевая полезная нагрузка может быть радионуклидом, радиосенсибилизирующим веществом, фотосенсибилизирующим веществом, химиотерапевтическим средством или токсином.
В дополнительном аспекте изобретение предлагает набор, содержащий фармацевтическую композицию соединения по изобретению, и, необязательно, инструкцию по применению.
Краткое описание чертежей
На фиг.1 показано уменьшение увеита под действием нативного α-MSH. На фиг.1A показаны данные мышей B10.RIII, которым ежедневно внутривенно вводили нативный α-MSH (100 мкг/мышь), когда их клинические показатели были 2-3. Увеит достоверно уменьшился по сравнению с контролем (p<0,01), не получавшими лечения. На фиг.1B показаны данные мышей B10.RIII, которым ежедневно внутрибрюшинно вводили нативный α-MSH (100 мкг/мышь) или дексаметазон (0,2 мг/кг или 2,0 мг/кг), когда их клинические показатели были 1-2 (n=5). Воспаление сетчатки уменьшилось после начала лечения (p<0,05). Звездочкой отмечены достоверные различия с контролем.
На фиг.2 проиллюстрировано улучшение увеита на поздних стадиях заболевания под действием RI α-MSH и нативного α-MSH. EAU индуцировали у мышей B10.RIII, и с 12 дня, когда у мышей развивалась поздняя стадия заболевания (показатели 3), им ежедневно внутривенно вводили нативный α-MSH, RI α-MSH или PBS в дозе 100 мкг/мышь. На фиг.2A показаны данные мышей, которым вводили нативный α-MSH или ретро-RI α-MSH и которые указывают на уменьшение глазных показателей при увеите по сравнению с контрольными мышами с PBS. На фиг.2B показаны индивидуальные максимальные глазные показатели у мышей в каждой группе на 16 день после индукции EAU (n=8). Звездочкой отмечены достоверные различия между группами (p<0,05).
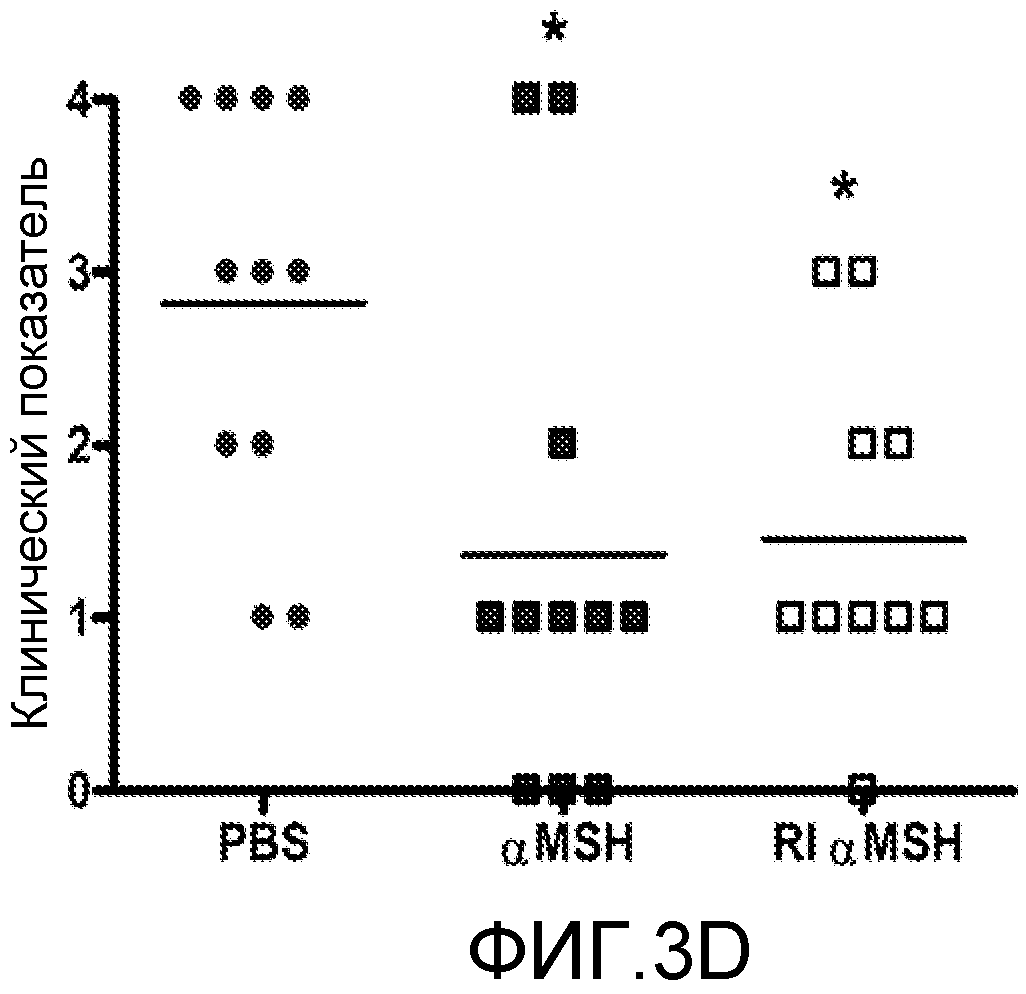
На фиг.3 показаны изображения сетчатки и индивидуальные глазные показатели животных, которым вводили α-MSH или RI α-MSH. EAU индуцировали у мышей B10.RIII, и, начиная с 13-го дня, когда мыши достигали поздней стадии заболевания, проводили ежедневное внутривенное введение нативного α-MSH, RI α-MSH или PBS в дозе 100 мкг/мышь. Показаны фундоскопические изображения сетчатки, отражающие средние величины глазных показателей каждой группы после 13-ти дней лечения (n=11). У мыши, которая получала PBS, с глазным показателем 3, были отмечены воспалительные бляшки в нескольких квадрантах глаза и васкулит рядом с оптическим нервом (фиг.3A). Мыши, которые получали α-MSH и RI α-MSH, с глазными показателями 1, показывают разрешение увеита с воспалением только вокруг оптического нерва (фиг.3B и 3C, соответственно). Сетчатки отражают среднюю величину глазного показателя для каждой группы. Индивидуальные глазные показатели мышей из каждой группы на 13-й день лечения представлены на графике. Звездочкой отмечены достоверные различия между группами (p<0,01).
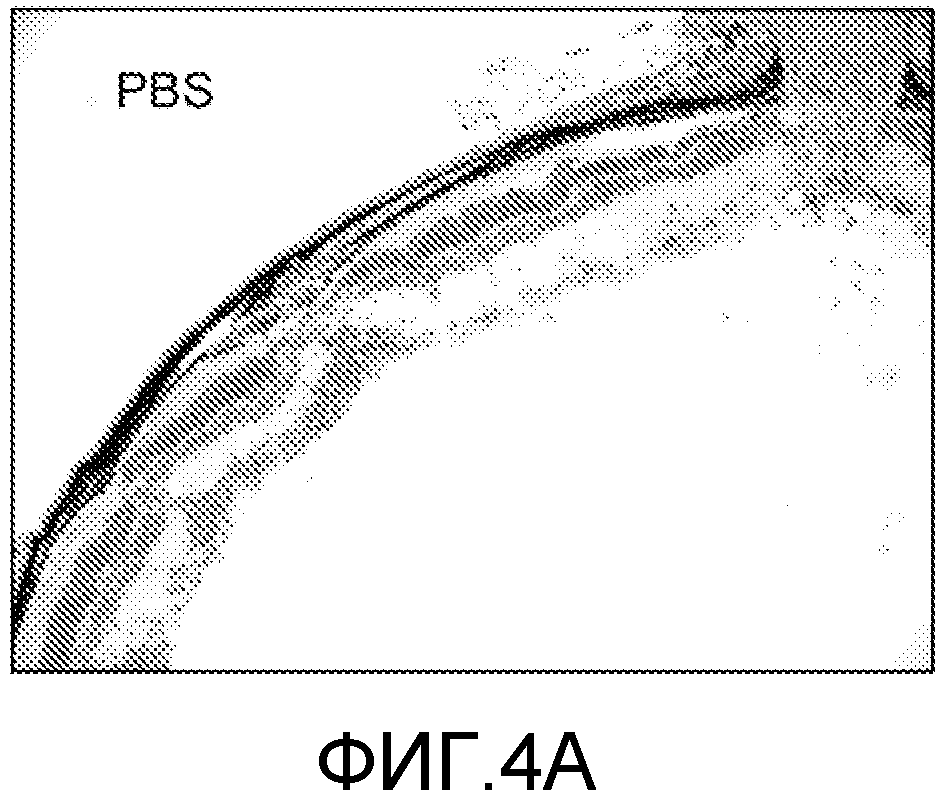
На фиг.4 проиллюстрирована гистопатология мышей с EAU, которые получали RI α-MSH и нативный α-MSH. Самкам B10.RIII мышей делали инъекцию IRBP+CFA для того, чтобы индуцировать EAU. Когда у мышей глазные показатели достигали 3, их ежедневно внутривенно вводили ретроинверсо α-MSH, нативным α-MSH или PBS в дозе 100 мкг/мышь. Группа мышей, которая получала RI α-MSH или α-MSH, показала снижение глазных воспалительных реакций (p<0,05). На фотографии показаны глаза, окрашенные гематоксилином и эозином, через 10 дней после начала лечения, и отражены средние величины глазных показателей групп мышей, которые получали PBS (фиг.4A), α-MSH (фиг.4B) и RI α-MSH (фиг.4C). Увеличение в 100 раз.
На фиг.5 показано действие на увеит ежедневной дозы ретроинверсо α-MSH, вводимой внутрибрюшинно, по сравнению с контрольным скремблированным пептидом. Мышам B10.RIII, начиная с 11 дня с начала индукции заболевания, ежедневно вводили посредством внутрибрюшинных инъекций, содержащих 100 мкг RI α-MSH или скремблированного D-аминокислотного пептидного контроля. Данные демонстрируют клинические средние глазные показатели групп мышей, которые получали RI α-MSH (n=4) и скремблированным контрольным пептидом (n=5). Мыши получали лечение в течение 13 дней. Представлены данные двух экспериментов. Звездочкой отмечены достоверные различия между группами (p<0,04).
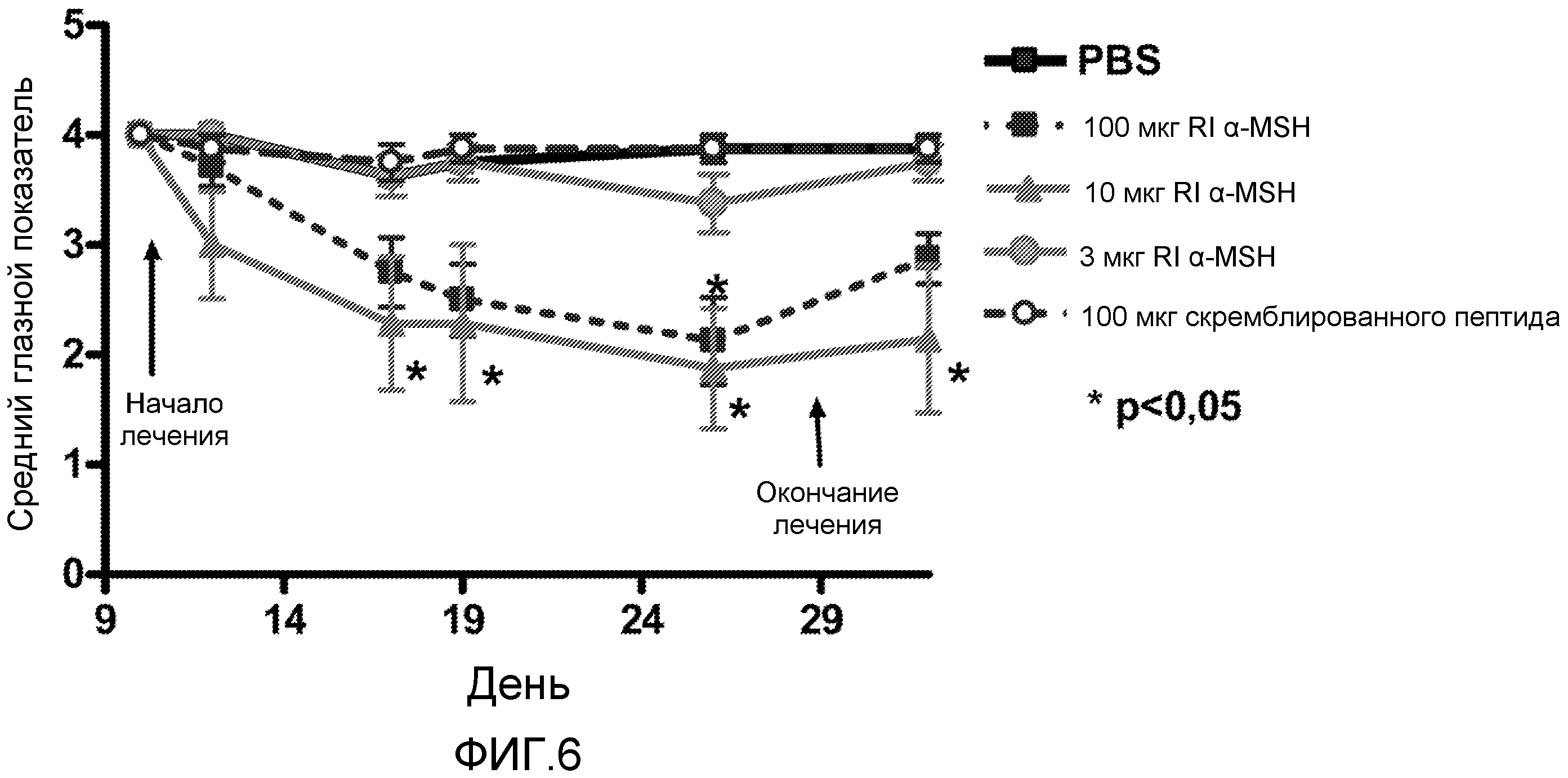
На фиг.6 проиллюстрирована эффективность RI α-MSH при лечении воспаления сетчатки. Мышам B10.RIII ежедневно вводили посредством внутрибрюшинных инъекций RI α-MSH (3, 10 или 100 мкг/мышь), если их клинические показатели были 4. Скремблированный пептидный контроль вводили инъекцией в дозе 100 мкг/мышь ежедневно. График отображает клинические показатели в динамике по времени. Наблюдали уменьшение воспаления сетчатки после начала лечения дозой 100 или 10 мкг/мышь. Ограниченный благоприятный клинический ответ наблюдали у мышей, которым вводили низкие дозы RI α-MSH (3 мкг/мышь) или PBS или контрольный скремблированный пептид (n=4). Звездочкой отмечены достоверные различия между группами (p<0,05).
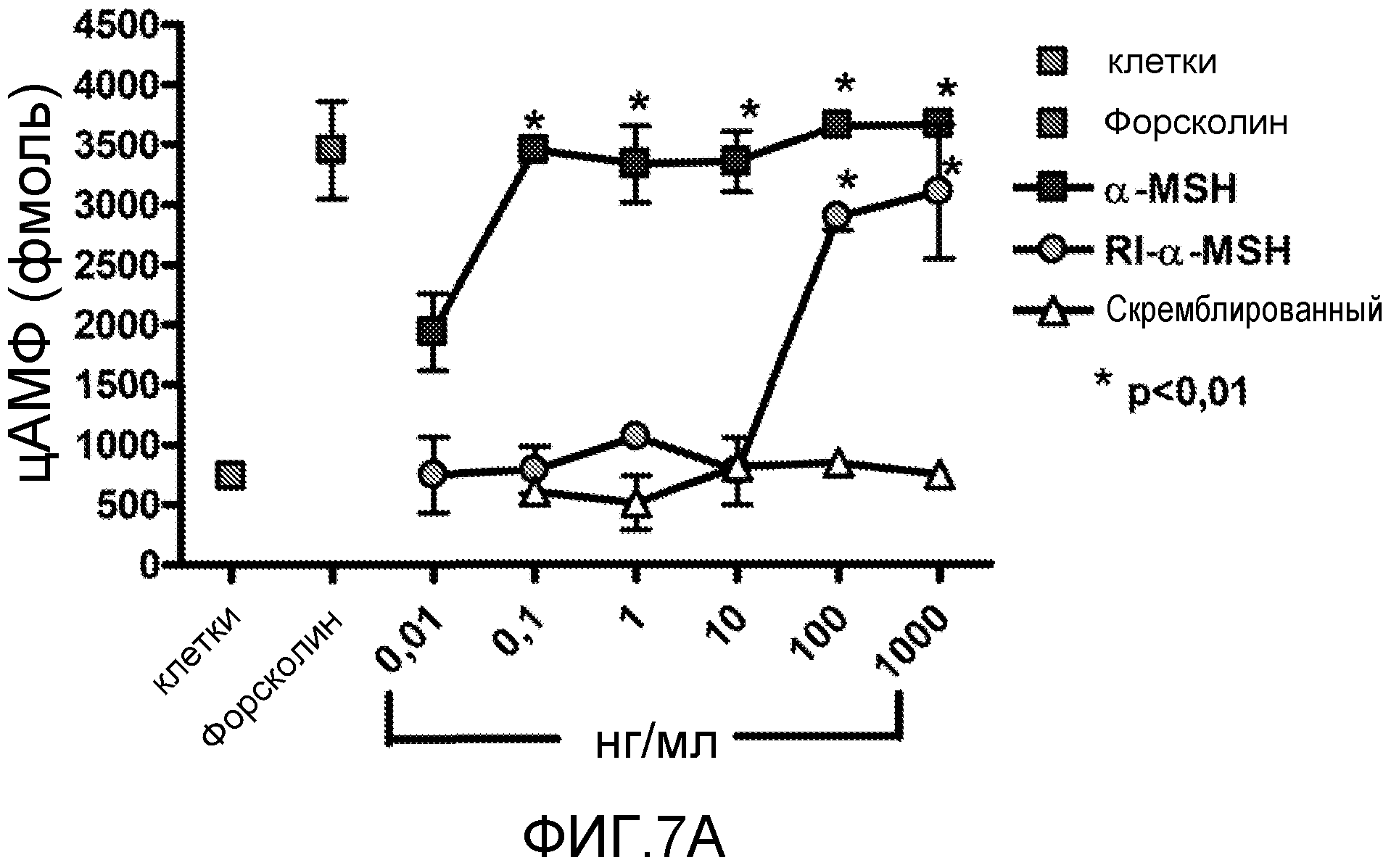
На фиг.7 показано влияние RI α-MSH на выработку цАМФ в клетках меланомы мыши. цАМФ измеряли в клетках меланомы B16-F1 после воздействия на клетки нативным α-MSH, ретроинверсо α-MSH или скремблированным контрольным пептидом в концентрациях 0,01-1000 нг/мл. На фиг.7A показано, что как нативный α-MSH, так и RI α-MSH значительно увеличивают уровни цАМФ. В качестве контроля для оценки измерения цАМФ использовали форсколин в концентрации 100 мкМ. На фиг.7B показаны данные аланинового сканирования. Аланинзамещенные пептиды RI α-MSH в концентрации 1 мкг/мл были проверены на увеличение уровней цАМФ на клеточной линии меланомы мыши B16-F1. Представлены данные двух экспериментов. Список последовательностей можно найти в таблице 1. Звездочкой отмечены достоверные различия между группами (p<0,01).
На фиг.8 проиллюстрировано течение EAE, вызванного MOG. EAE индуцировали у мышей C57BL/6 путем введения инъекций пептида MOG35-55 (200 мкг/мышь) и эмульсии CFA. Инъекцию токсина коклюша делали на 0 день и 2-й день. На 10-й день начинали ежедневное лечение α-MSH, RI α-MSH или PBS, внутрибрюшинно, дозами 100 мкг/мышь. На фиг.8A показаны клинические показатели заболевания, которые записывались ежедневно. На фиг.8B показаны индивидуальные показатели заболевания в каждой группе на 20-й день после индукции заболевания.
На фиг.9 показано снижение средних показателей EAE под влиянием RI α-MSH. EAE индуцировали у мышей C57BL/6 путем введения инъекций пептида MOG35-55 (200 мкг/мышь) и эмульсии CFA. Инъекцию токсина коклюша делали на 0 день и 2-й день. На 10-й день начинали ежедневное лечение дозами 100 мкг или 30 мкг/мышь α-MSH или RI α-MSH, 2 мг/кг дексаметазона, или PBS, внутрибрюшинно. Клинические показатели заболевания записывали ежедневно.
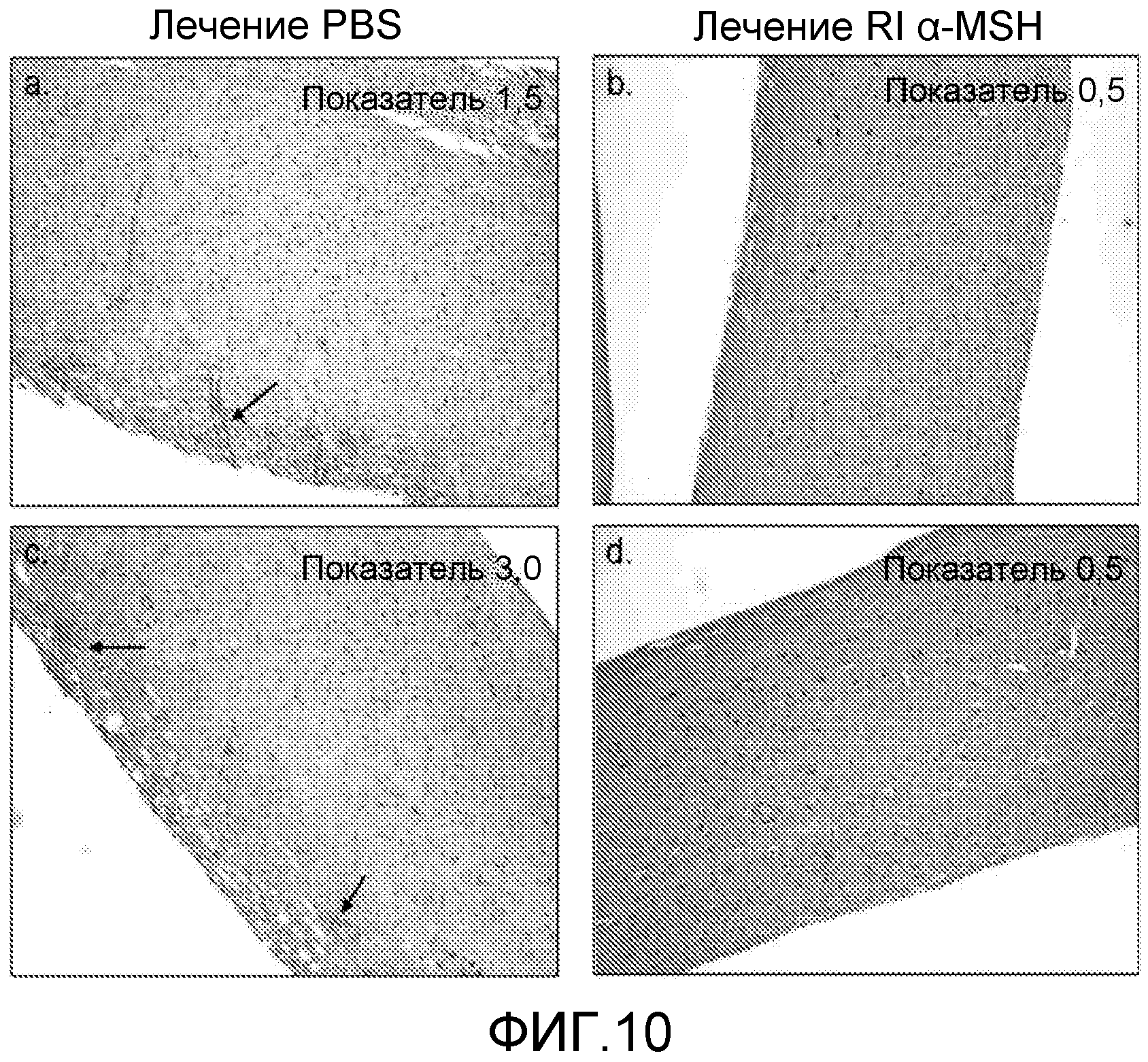
На фиг.10 показана гистология спинного мозга мышей, которым вводили RI α-MSH. EAE индуцировали у мышей C57BL/6 путем введения инъекций пептида MOG35-55 (200 мкг/мышь) и эмульсии CFA. Инъекцию токсина коклюша делали на 0 день и 2-й день. На 10-й день начинали ежедневное введение RI α-MSH в дозе 100 мкг/мышь, внутрибрюшинно. Спинной мозг собирали на 24-й день после индуцирования заболевания. Показаны две типичные мыши из каждой группы: которые получали PBS (фиг.10a и 10c) и которые получали RI α-MSH (фиг.10b и 10d). Стрелки показывают участки инфильтрации воспалительными клетками.
На фиг.11 показано количество мРНК TNFα и IL-10 в селезенке мышей, сенсибилизированных пептидом MOG, в течение стадии заболевания EAE. Мышей сенсибилизировали 200 мкг пептида MOG на 0 день и им вводили PBS, α-MSH (100 мкг) или RI α-MSH (100 мкг) ежедневно в дни 10-15. Селезенки собирали на 1 (фиг.11a и 11b), 4 (фиг.11с и 11d) и 7 (фиг.11e и 11f) дни после начала лечения и исследовали экспрессию мРНК TNFα и IL-10 с помощью количественной ПЦР. Данные показывают среднее значение для 4-х мышей в каждой из групп. Уровни РНК нормализованы по β-актину.
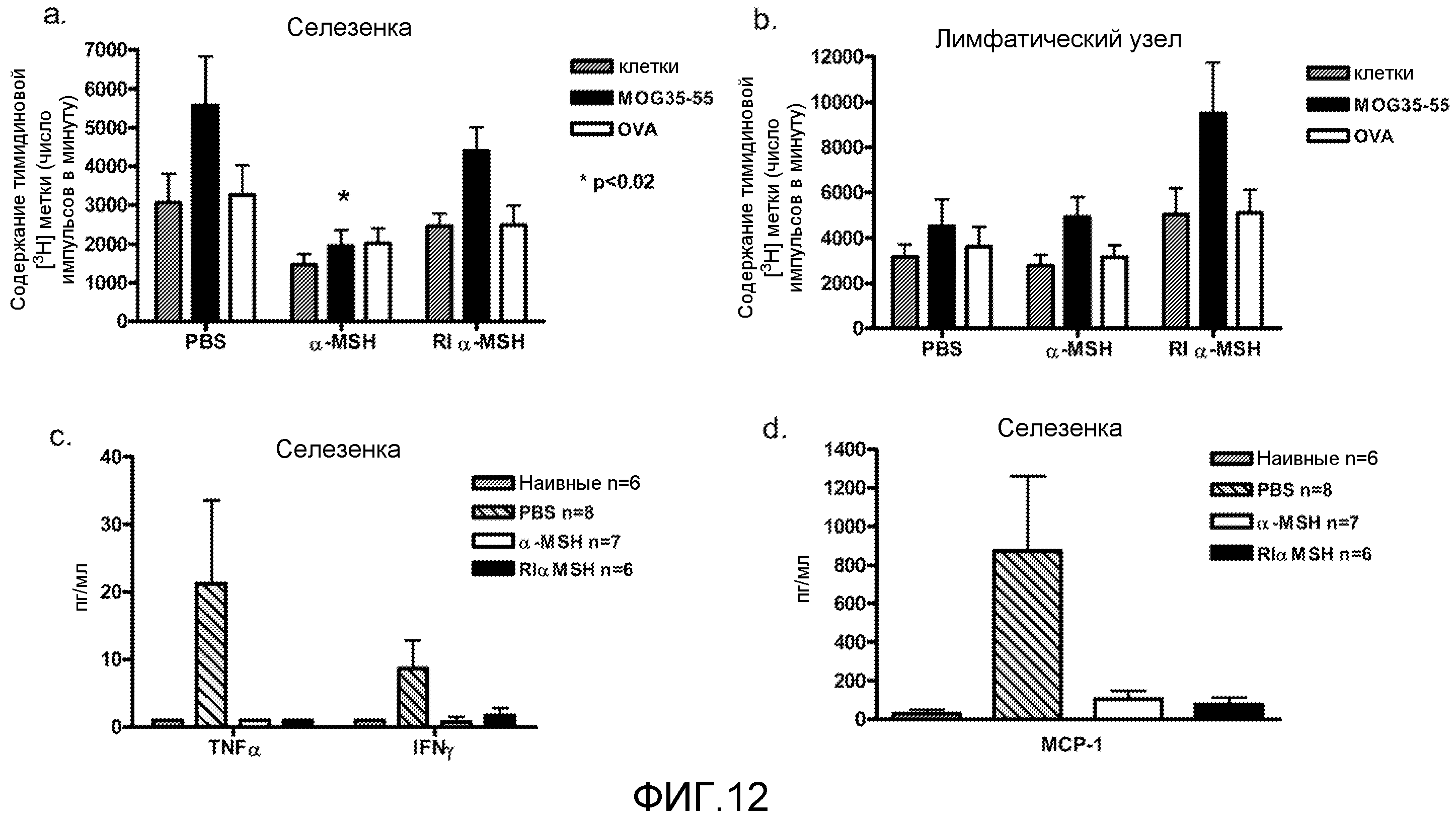
На фиг.12 проиллюстрирован вторичный иммунный ответ на пептид MOG 35-55. Мышей сенсибилизировали 200 мкг пептида MOG35-55 на 0 день. На 2-8 день мышам вводили путем инъекции внутрибрюшинно PBS, 100 мкг α-MSH или 100 мкг RI α-MSH (n=5). На 9-й день собирали клетки селезенки (фиг.12a) и лимфоузла (фиг.12b) и стимулировали 25 мкг/мл пептида MOG35-55 или пептида OVA in vitro. Клетки в культуре метили [3H]-тимидином на 3-й день. Данные представлены в виде среднего±SD. Через 24 часа собирали супернатант из культуры клеток селезенки, стимулированных пептидом MOG, и измеряли уровни цитокинов TNF-α, IFNγ (фиг.12c) и MCP-1 (фиг.12d) с помощью проточной цитометрии. Данные представлены в виде среднего±SD. Наивных мышей не сенсибилизировали пептидом MOG.
На фиг.13 проиллюстрированы цитокиновые профили в сыворотке и селезенке после введения RI α-MSH мышам, сенсибилизированных MOG. Мышей сенсибилизировали 200 мкг пептидом MOG35-55 на 0 день. На 2-8 день мышам вводили путем инъекции внутрибрюшинно PBS, 100 мкг α-MSH или 100 мкг RI α-MSH (n=5). На 9-й день собирали селезенки и сыворотки. На фиг.13a и 13b показаны уровни мРНК TNF-α и IL-10, соответственно, в селезенке, измеренные методом ПЦР в реальном времени. Данные показывают среднее значение для 4-х мышей в каждой из групп. Уровни РНК нормализованы по β-актину. Сыворотку также исследовали на уровни цитокинов методом проточной цитометрии. На фиг.13c показаны уровни TNF-α, MCP-1, IL-6 в сыворотке, и на фиг.13d показаны уровни IL-10 и IL-12 в сыворотке. Наивных мышей не сенсибилизировали пептидом MOG. Данные показаны в виде среднего±SD.
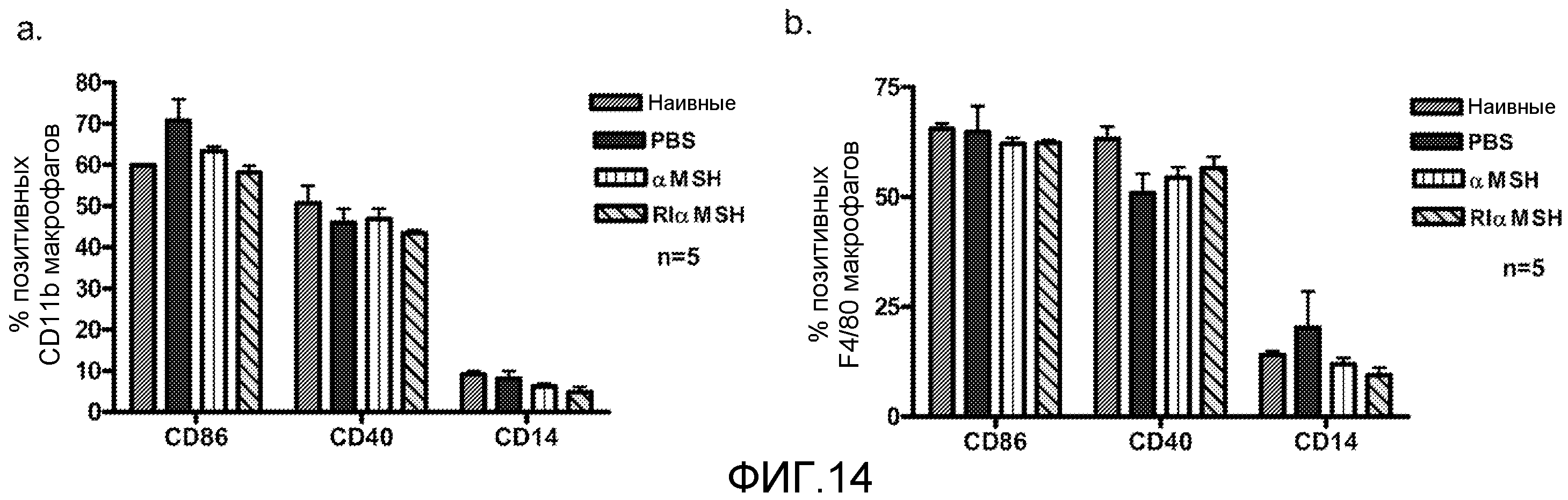
На фиг.14 показано влияние α-MSH и RI α-MSH на маркеры макрофагов. Мышей сенсибилизировали пептидом MOG in vivo и им вводили ежедневно α-MSH или RI α-MSH (100 мкг/мышь) на 1-7 день. Селезеночные макрофаги анализировали методом проточной цитометрии для измерения уровней экспрессии CD14, CD40 и CD86. Проводили селекцию клеточной популяции макрофагов по CD11b+ (фиг.14a) или F4/80+ (фиг.14b) клеткам. Данные показывают средний процент позитивных клеток в популяции макрофагов, прошедших селекцию (n=5).
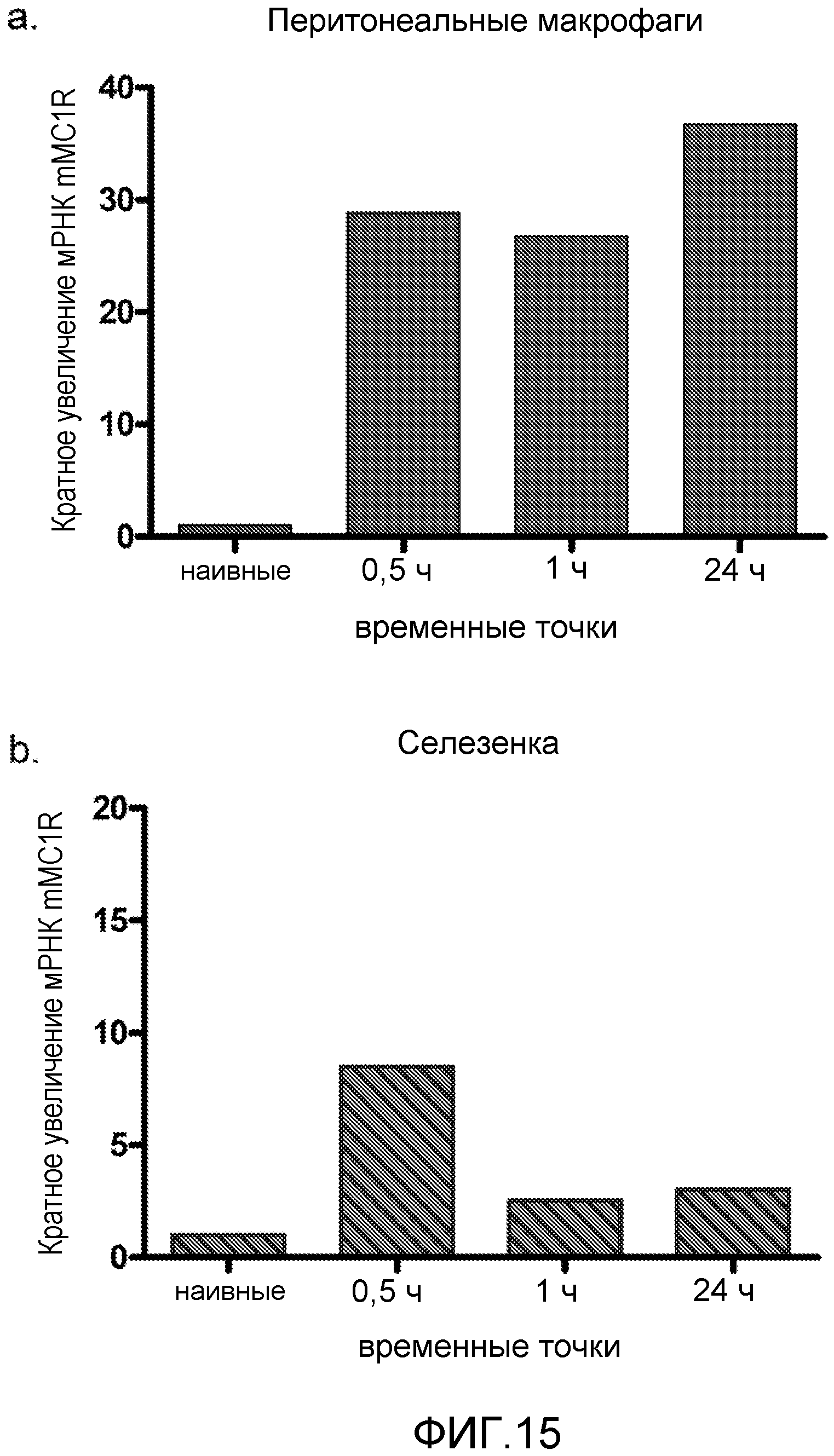
На фиг.15 показано индуцированное LPS увеличение уровней мРНК mMC1R в перитонеальных макрофагах (фиг.15a) и селезенке (фиг.15b). Мышам C57BL/6 (n=4) делали внутрибрюшинные инъекции LPS (1 мкг/мышь). Перитонеальные макрофаги и селезенки собирали через 0,5 ч, 1 ч и 24 ч. Уровни мРНК mMC1R измеряли ПЦР в реальном времени. Уровни РНК были нормализованы по 18s.
На фиг.16 показано действие MSH на модели воспаления, индуцированного LPS in vivo. Мышам C57BL/6 делали внутрибрюшинные инъекции 1 мкг LPS. Через 30 мин мышей внутрибрюшинно лечили дексаметазоном (2 мг/кг) и α-MSH (фиг.16a-16c) или аналогом RI α-MSH 891 (фиг.16d-16f). Собирали сыворотки через 2 часа после стимуляции LPS. Уровни TNF-α (фиг.16a и 16d), MCP-1 (фиг.16b и 16e) и IL-10 (фиг.16c и 16f) измеряли с помощью цитометрического анализа на гранулах методом проточной цитометрии. Данные показывают индивидуальные измерения цитокинов и среднее для каждой группы (n=6).
На фиг.17 показана стабильность RI-α-MSH и α-MSH в плазме и сыворотке. На фиг.17A показана стабильность пептидов RI-α-MSH и α-MSH в плазме и PBS при 37ºC. На фиг.17B показано время полужизни в сыворотке пептидов RI-α-MSH и α-MSH после единичной внутривенной инъекции.
На фиг.18 показаны результаты исследования связывания MSH (фиг.18A) и RI-MSH (фиг.18B) с рецепторами меланокортина 1, 3, 4 и 5.
На фиг.19 показана инверсия основного тетрапептида HfRW и ретроинверсо MSH.
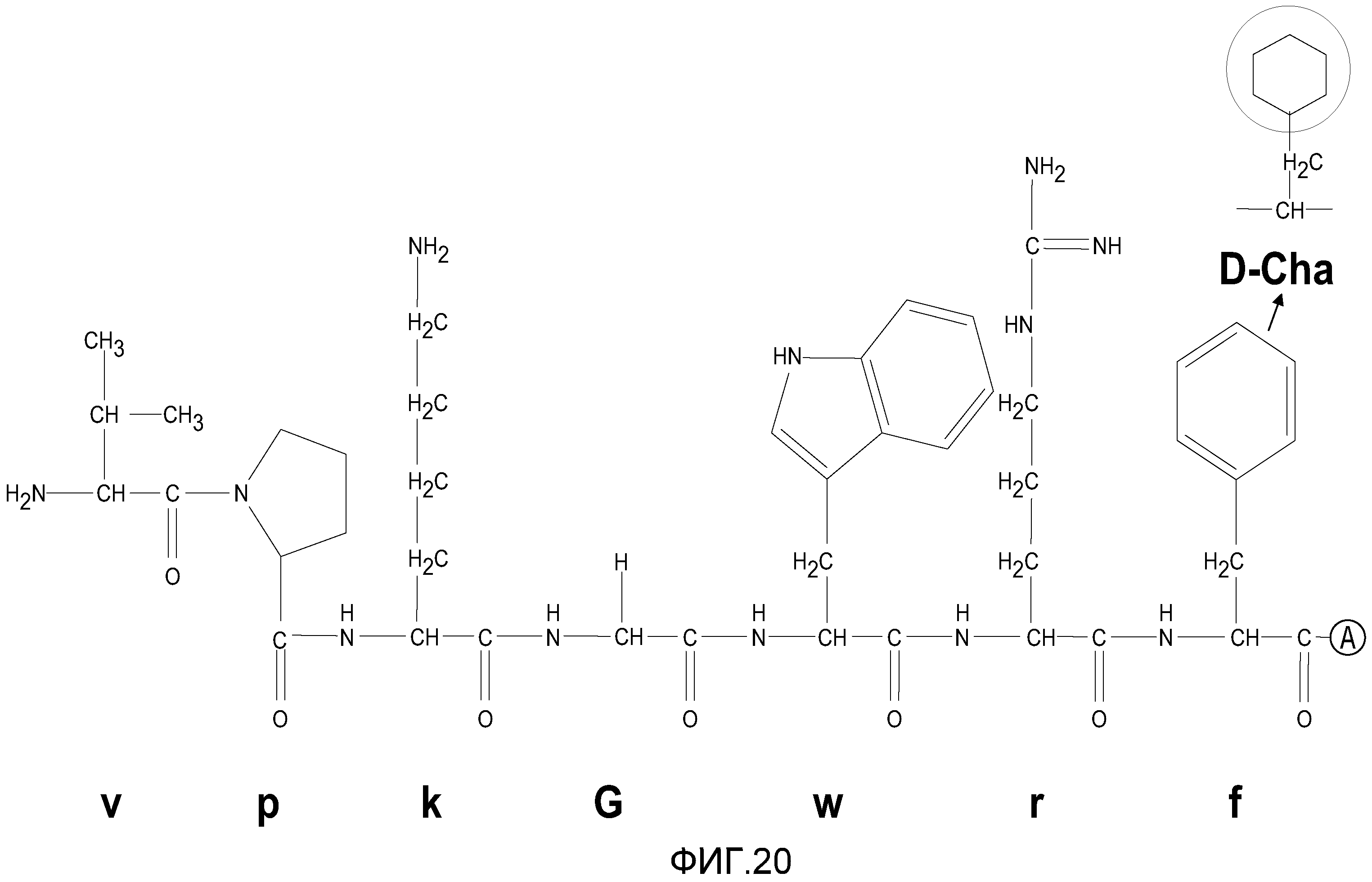
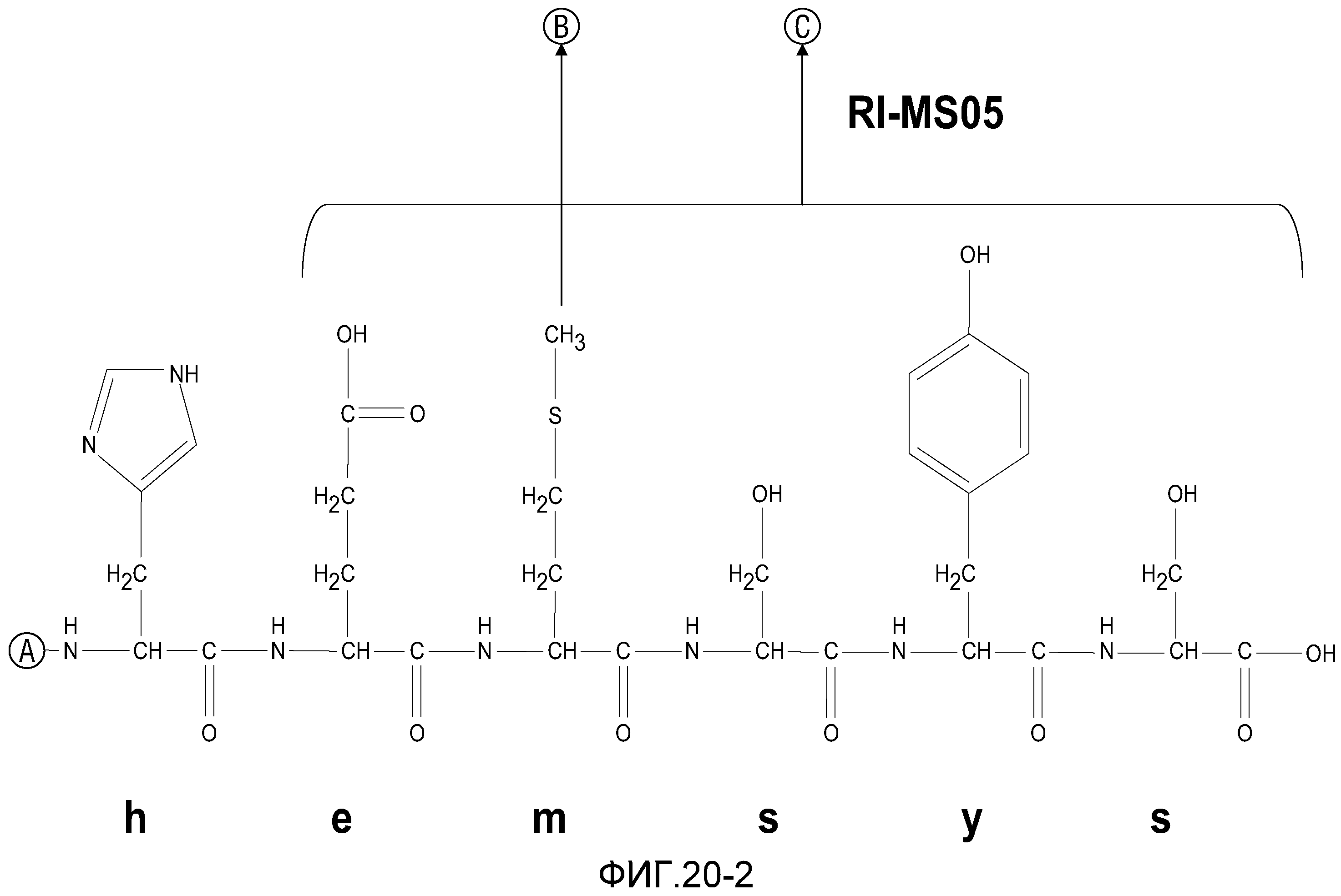
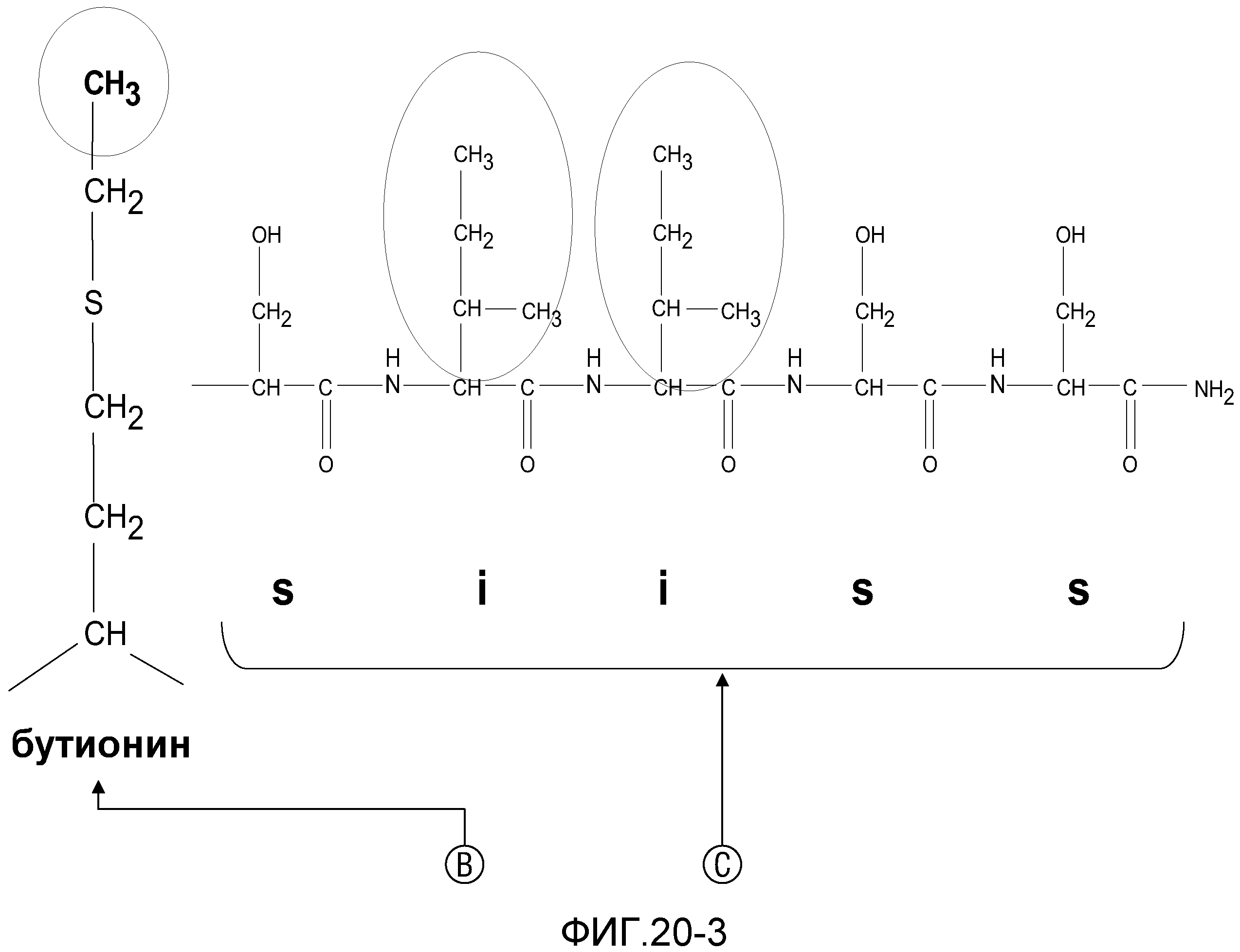
На фиг.20 проиллюстрировано замещение неприродных аминокислотных остатков в различных позициях RI-MSH.
На фиг.21 проиллюстрирован типичный анализ конкурентного связывания RI-MSH и его вариантов с MC1R.
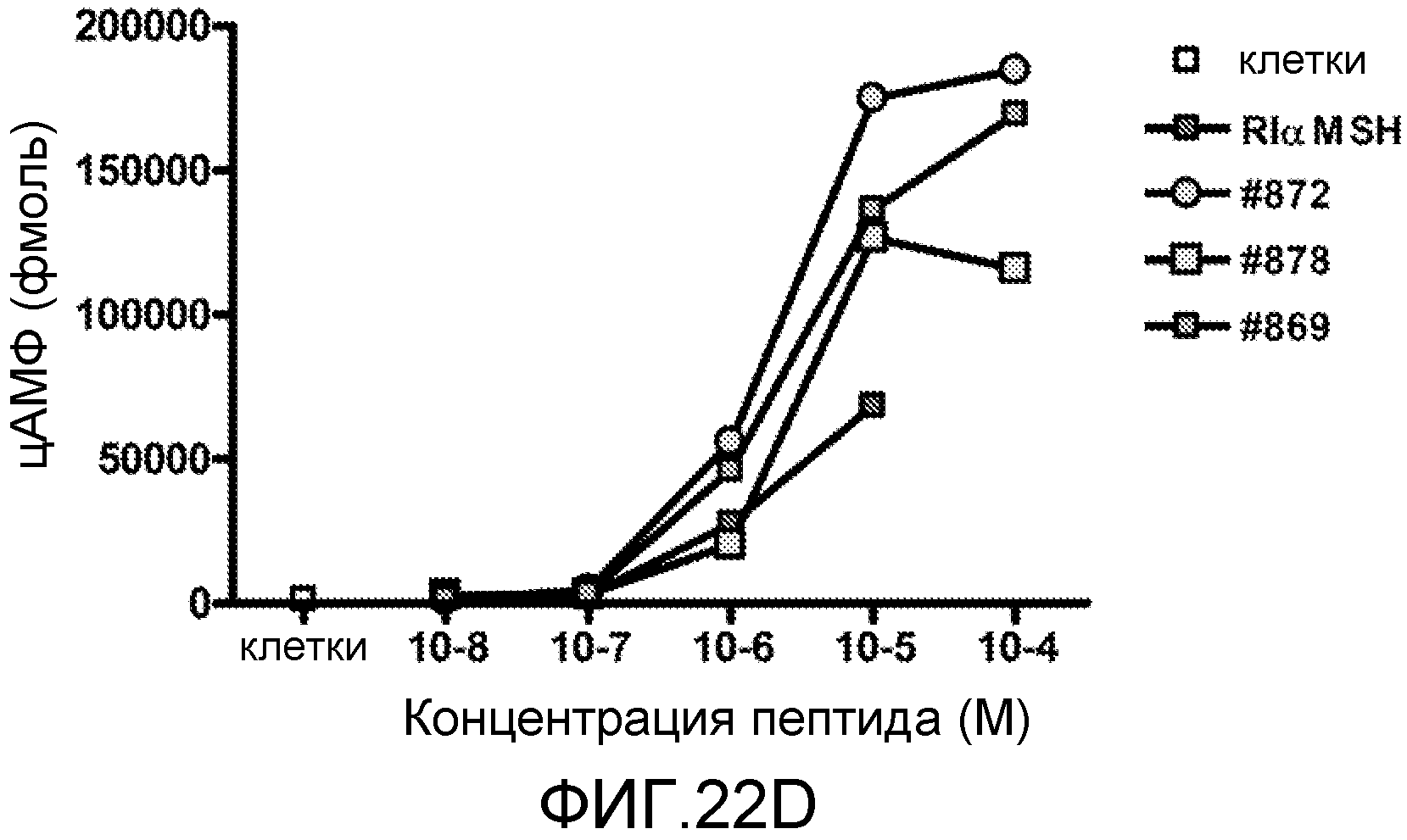
На фиг.22 проиллюстрировано влияние аналогов RI α-MSH на уровни цАМФ в клетках меланомы мыши B16 F1. Фиг.22A: аналоги 890, 891 и 892; фиг.22B: аналоги 893, 894 и 895; фиг.22C: аналоги 880, 886 и 878; и фиг.22D: аналоги 872, 878 и 869.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Белки α-MSH и MC1R и нуклеиновые в соответствии с настоящими способами не ограничиваются определенным источником или видами. Так, белки и нуклеиновые кислоты могут быть выделенными или рекомбинантными.
α-меланоцитстимулирующий гормон (α-MSH) представляет собой полипептид с последовательностью Ser Tyr Ser Met Glu His Phe Arg Trp Gly Lys Pro Val (SEQ ID NO:8) (SYSMEHFRWGKPV). В настоящее время исследуются противовоспалительные и иммуномодулирующие свойства α-MSH. α-MSH образуется при внутриклеточном протеолитическом расщеплении пропиомеланокортинового гормона (POMC). α-MSH нейропептид обнаруживается в нескольких органах и вырабатывается нейронами, гипофизом, кишечником, кожей и иммунными клетками. Сообщалось о том, что α-MSH подавляет эффекторные функции T-клеток, индуцирует регуляторные T-клетки и проявляет положительное воздействие на аутоиммунных и трансплантационных моделях.
"Конъюгат" или "конъюгированный" содержит два или более элементов, которые присоединены, соединены, спарены, образуют комплекс или другим образом ассоциированы друг с другом. Элементы могут быть присоединены друг к другу посредством ковалентных связей, ионных связей, электростатических взаимодействий, водородных связей, ван-дер-ваальсовых взаимодействий или физическими способами.
"Биологически активные" части содержат молекулу или соединение, которое вызывает или регулирует физиологический ответ. В одном из аспектов биологически активное соединение стимулирует рецепторы меланокортина, предпочтительно MC1-рецепторы.
Под "модулировать" и "модуляция" подразумевается, что активность одного или нескольких белков или белковых субъединиц усиливается или подавляется, таким образом, что экспрессия, уровень или активность больше или меньше, чем наблюдаемая в отсутствие регулятора. Например, термин "модулировать" может означать "ингибировать" или "стимулировать".
"C-концевая последовательность" относится к концу аминокислотной цепи, которая заканчивается обычно, но не обязательно, карбоксильной группой. Правило для записи пептидной последовательности состоит в том, чтобы поместить C-конец справа и писать последовательность от N- к C-концу. C-концевая последовательность может содержать от 1 до 100 аминокислот, предпочтительно, от 2 до 15 аминокислот, и даже более предпочтительно, от 3 до 10 аминокислот. C-концевая последовательность может оканчиваться карбоксильной группой, или конец может быть модифицирован хорошо известными в данной области способами содержания функциональных элементов (например, нацеливающей группы, сигнала задержки, липида и анкора).
Настоящее изобретение относится к "по существу чистому соединению". Термин "по существу чистое соединение" используют в настоящем документе для описания молекулы, такой как полипептид (например, полипептид, который связывает MC1R, или его фрагмент), который по существу свободен от других белков, липидов, углеводов, нуклеиновых кислот и других биологических материалов, с которыми он связан в природе. Например, по существу чистая молекула, такая как полипептид, может быть, по меньшей мере, на 60%, по массе сухого вещества, интересующей молекулой. Чистоту полипептидов можно определить стандартными способами, содержащими, например, электрофорез в полиакриламидном геле (например, SDS-PAGE), хроматографию на колонках (например, высокоэффективную жидкостную хроматографию (ВЭЖХ)) и анализ аминоконцевой аминокислотной последовательности.
В одном из вариантов осуществления фраза "селективно связывается" означает, что соединение или полипептид, полученные или использованные по настоящему изобретению, предпочтительно, связываются с одним типом рецепторов, а не с другим, когда связывание происходит в присутствие смеси двух или более рецепторов (например, рецепторов меланокортина, MC1, MC2, MC3, MC4, MC5 рецепторы).
"Аминокислота" или "аминокислотная последовательность" содержит олигопептид, пептид, полипептид, или белковую последовательность, или фрагмент, часть или субъединицу любого из них, и относится к природной или синтетической молекуле. Термины "полипептид" и "белок" содержат аминокислоты, присоединенные друг к другу пептидными связями или модифицированными пептидными связями, т.е. пептидными изостерами, и могут содержать модифицированные аминокислоты, иные чем 20 закодированных в генах аминокислот. Термин "полипептид" также содержит пептиды и полипептидные фрагменты, мотивы и т.п. Заглавные однобуквенные аббревиатуры аминокислот относятся к природным L-изомерам. Строчные однобуквенные аббревиатуры аминокислот обозначают D-изомер.
Термины "полипептид", "пептид" и "белок" используют взаимозаменяемо по отношению к аминокислотным полимерам любой длины. Пептиды и полипептиды могут либо полностью состоять из синтетических искусственных аналогов аминокислот, либо представлять собой химерную молекулу, частично из природных пептидных аминокислот и частично из неприродных аналогов аминокислот. В одном из аспектов, полипептид используется в композиции, клеточной системе или способе по изобретению (например, клетка-хозяин с плазмидной экспрессией, по меньшей мере, одого фермента по изобретению). Кроме того, полипептид может относиться к соединениям, состоящим из аминокислотных полимеров, ковалентно присоединенных к другой функциональной группе (например, водорастворимая группа, нацеливающая группа, ПЭГ, неаминокислотная группа или другое терапевтическое средство).
Названия аминокислот можно сокращать с использованием следующих обозначений в скобках: пролин (Pro), валин (Val), лизин (Lys), орнитин (Orn), норлейцин (Nle), глицин (Gly), триптофан (Trp), аланин (Ala), фенилаланин (Phe), аргинин (Arg), гистидин (His), глутаминовая кислота (Glu), аспарагиновая кислота (Asp), серин (Ser), метионин (Met), изолейцин (Ile), тирозин (Tyr), циклогексилаланин (Cha), 4-фтор-D-фенилглицин (4-фтор-D-Phg), 2-тиенил-D-аланин (D-Thi).
"Лечение", "подвергать лечению", "лечить" или "терапия" в настоящем документе относится к введению млекопитающему веществ, которые способны вызывать профилактический, исцеляющий или другой положительный эффект у индивидуума. Лечение может дополнительно приводить к смягчению или улучшению заболевания или симптомов заболевания у пациента.
В настоящем документе формы единственного числа содержат ссылки на множественное число, если не указано иначе. Например, «клетка-мишень» содержит одну или несколько клеток-мишеней.
Полипептидные композиции по изобретению могут содержать любые комбинации неприродных структурных компонентов. Отдельные пептидные остатки могут быть соединены пептидными связями, другими химическими связями или посредством связывания, как например, глутаральдегид, сложные эфиры N-гидроксисукцинимида, бифункциональные малеимиды, N,N'-дициклогексилкарбодиимид (DCC) или N,N'-диизопропилкарбодиимид (DIC). Связывающие группы, которые могут быть альтернативой традиционному амидному связыванию ("пептидная связь") содержат, например, кетометилен (например, -C(=O)-CH2- для -C(=O)-NH-), аминометилен (CH2-NH), этилен, олефин (CH=CH), простой эфир (CH2-O), тиоэфир (CH2-S), тетразол, тиазол, ретроамид, тиоамид, или сложный эфир (смотрите, например, Spatola (1983) in Chemistry и Biochemistry of Aminoacids, Peptides and Proteins, Vol. 7, pp. 267-357', "Peptide Backbone Modifications", Marcel Dekker, NY, включенные в настоящий документ в качестве ссылки).
Полипептиды, используемые для осуществления на практике способа по изобретению, могут быть модифицированными любыми природными процессами, такими как посттрансляционный процессинг (например, фосфорилирование, ацилирование и т.д.), или с помощью методов химической модификации, и полученными модифицированными пептидами. Модификации могут быть сделаны в любой части полипептида, включая пептидный остов, аминокислотные боковые цепи и амино или карбокси концы. Следует понимать, что одинаковый тип модификации может присутствовать в одинаковой или различных степенях в нескольких участках в полипептиде. Также данный полипептид может иметь много типов модификации. Модификации содержат ацетилирование, ацилирование, АДФ-рибозилирование, амидирование, ковалентное присоединение флавина, ковалентное присоединение молекулы гема, ковалентное присоединение нуклеотида или нуклеотидного производного, ковалентное присоединение липида или липидного производного, ковалентное присоединение фосфатидилинозитола, образование кольца с перекрестными сшивками, формирование дисульфидной связи, деметилирование, формирование ковалентных перекрестных сшивок, образование цистеина, образование пироглутамата, формилирование, гамма-карбоксилирование, гликозилирование, образование гликофосфатидилинозитолового анкора, гидроксилирование, йодирование, метилирование, ацилирование остатком миристиловой кислоты, оксидирование, пегилирование, протеолитическое расщепление, фосфорилирование, пренилирование, селенонирование, сульфатирование, и т-РНК-опосредованное добавление аминокислот к белку, такое как аргинилирование. Смотрите, например, Creighton, T.E., Proteins - Structure и Molecular Properties 2nd Ed., W.H. Freeman и Company, New York (1993); Posttranslational Covalent Modification of Proteins, B.C. Johnson, Ed., Academic Press, New York, pp. 1-12 (1983), включенные в настоящий документ в качестве ссылки.
Дополнительные варианты осуществления соединений
В некоторых вариантах осуществления изобретение относится к по существу чистому соединению, которое селективно связывается с рецептором меланокортина 1 (MClR); указанное соединение содержит полипептид с последовательностью:
Xaa1 Xaa2 Xaa3 Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 Xaa11 Xaa12 Xaa13,
где Xaa1 представляет собой D-Val, D-Ala или D-Lys;
Xaa2 представляет собой D-Pro, D-Ala или D-Lys;
Xaa3 представляет собой D-Lys, D-Orn, D-Nle, D-Ala или D-Lys;
Xaa4 представляет собой Gly или D-Ala;
Xaa5 представляет собой D-Trp, Trp, D-3-бензотиенил-Ala, D-5-гидрокси-Trp, D-5-метокси-Trp, D-Phe или D-Ala;
Xaa6 представляет собой D-Arg, D-His или D-Ala;
Xaa7 представляет собой D-Cha, D-Phe, Phe, D-4-фтор-Phg, D-3-пиридил-Ala, D-Thi, D-Trp, D-4-нитро-Phe или D-Ala;
Xaa8 представляет собой D-His, His, D-Arg, Phe, или D-Ala;
Xaa9 представляет собой D-Glu, D-Asp, D-цитруллин, D-Ser или D-Ala;
Xaa10 представляет собой D-Met, D-бутионин, D-Ile или D-Ala;
Xaa11 представляет собой D-Ser, D-Ile или D-Ala;
Xaa12 представляет собой D-Tyr, D-Ser или D-Ala;
Xaa13 представляет собой D-Ser или D-Ala;
или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления пептидные аналоги по изобретению селективно связывают MC1R и содержат полипептид с последовательностью: Xaa1 Xaa2 Xaa3 Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 Xaa11 Xaa12 Xaa13, где Xaa1 представляет собой D-Val; Xaa2 представляет собой D-Pro; Xaa3 представляет собой D-Lys, D-Orn или D-Nle; Xaa4 представляет собой Gly; Xaa5 представляет собой D-Trp, Trp, D-3-бензотиенил-Ala, D-5-гидрокси-Trp, D-5-метокси-Trp или D-Phe; Xaa6 представляет собой D-Arg или D-His; Xaa7 представляет собой D-Cha, D-Phe, Phe, D-4-фтор-Phg, D-3-пиридил-Ala, D-Thi, D-Trp или D-4-нитро-Phe; Xaa8 представляет собой D-His, His, D-Arg, Phe или D-Ala; Xaa9 представляет собой D-Glu, D-Asp, D-цитруллин или D-Ser; Xaa10 представляет собой D-Met, D-бутионин или D-Ile; Xaa11 представляет собой D-Ser или D-Ile; Xaa12 представляет собой D-Tyr или D-Ser; Xaa13 представляет собой D-Ser, где не более, чем одна из Xaa1-13представляет собой L-аминокислоту; или его фармацевтически приемлемую соль.
В других вариантах осуществления пептидные аналоги по изобретению содержат полипептид с последовательностью: Xaa1 Xaa2 Xaa3 Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 Xaa11 Xaa12 Xaa13, где Xaa1 представляет собой D-Val; Xaa2 представляет собой D-Pro; Xaa3 представляет собой D-Lys, D-Orn или D-Nle; Xaa4 представляет собой Gly; Xaa5 представляет собой D-Trp или Trp; Xaa6 представляет собой D-Arg; Xaa7 представляет собой D-Cha, D-Phe, Phe или D-Thi; Xaa8 представляет собой D-His или His; Xaa9 представляет собой D-Glu или D-Ser; Xaa10 представляет собой D-Met, D-бутионин или D-Ile; Xaa11 представляет собой D-Ser или D-Ile; Xaa12 представляет собой D-Tyr или D-Ser; Xaa13 представляет собой D-Ser; где не более, чем одна из Xaa1-13 представляет собой L-аминокислоту; или его фармацевтически приемлемую соль.
Соединения, приведенные в настоящем документе, содержат пептидные аналоги α-меланокортинстимулирующего гормона (α-MSH).
В альтернативном варианте осуществления, пептидные аналоги состоят из D-аминокислот. В других вариантах осуществления пептиды содержат D-аминокислоты, L-аминокислоты или смесь D- и L-аминокислот. В других вариантах осуществления пептиды содержат, по меньшей мере, 40%, 50%, 60%, 70%, 80%, 90% или 100% D-аминокислот. Соединения по изобретению могут также содержать следующие неограничивающие примеры нестандартных аминокислот: D-орнитин, D-норлейцин, 3-бензотиенил-D-аланин, 5-гидрокси-D-Trp, 5-метокси-D-Trp, 4-фтор-D-фенилглицин (4-фтор-D-Phg), 3-пиридил-D-аланин, 2-тиенил-D-аланин (D-Thi), D-циклогексилаланин (D-Cha), 4-нитро-D-Phe, D-цитруллин, α-метил-D-Met и D-бутионин.
В некоторых вариантах осуществления, основной тетрапептид содержит аминокислотную последовательность His Phe Arg Trp или Trp Arg Phe His, предпочтительно в D-аминокислотной конфигурации. В другом варианте осуществления, основной тетрапептид содержит аминокислотную последовательность His D-Cha Arg Trp или Trp Arg D-Cha His, предпочтительно в D-аминокислотной конфигурации. В некоторых вариантах осуществления основной тетрапептид содержит 4 D-аминокислоты. В других вариантах осуществления, основной тетрапептид имеет, по меньшей мере, одну нестандартную аминокислоту.
Приведенные в данном документе соединения могут быть синтетическими или рекомбинантными. Соединения по изобретению могут быть получены обычными химическими способами, известными в данной области. Способы твердофазного синтеза описаны в опубликованной литературе, такой как Solid Phase Peptide Synthesis: A Practical Approach (E. Atherton, et al. 1989). Соединения по изобретению также могут быть получены обычными молекулярно-биологическими способами, известными в данной области. В рамках настоящей заявки, если не указано иначе, определения терминов и объяснения методов можно найти в любой из некоторых хорошо известных публикаций, таких как: Sambrook, J., et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press (1989); Goeddel, D., ed., Gene Expression Technology, Methods in Enzymology, 185, Academic Press, San Diego, CA (1991); "Guide to Protein Purification" in Deutshcer, M.P., ed., Methods in Enzymology, Academic Press, San Diego, CA (1989); Innis, et al., PCR Protocols: A Guide to Methods and Applications, Academic Press, San Diego, CA (1990); Freshney, R.I., Culture of Animal Cells: A Manual of Basic Technique, 2nd Ed., Alan Liss, Inc. New York, NY (1987); Murray, E.J., ed., Gene Transfer and Expression Protocols, pp. 109-128, The Humana Press Inc., Clifton, NJ and Lewin, B., Genes VI, Oxford University Press, New York (1997). Все цитируемые публикации включены в настоящий документ в качестве ссылки в полном объеме.
В некоторых вариантах осуществления пептидные аналоги по изобретению селективно связывают или активируют рецептор меланокортина-1 (MC1R). Для измерения связывания или активации MC1R может быть использован любой подходящий анализ. Например, для определения активации MC1R можно использовать индукцию цАМФ in vitro. Оценка in vitro может свидетельствовать об активации in vivo. Дополнительные варианты осуществления настоящего изобретения содержат любой полипептид, селективный для MC1-рецепторов. Выявление соединения, селективного для MCl-рецептора, может быть осуществлено подходящим методом скрининга. Неограничивающий пример анализа связывания MCl-рецептора описан в примере 4. В некоторых вариантах осуществления, предпочтительные MC1-рецепторы содержат рецептор меланокортина 1 homo sapiens (GenBank Accession No: NP_002377).
Другие варианты осуществления изобретения относятся к соединениям, которые изменяют уровни цАМФ, оксида азота (NO), TNF-α, мРНК TNF-α, мРНК IL-10, IL-10, IFNγ, IL-6, IL-12 и/или MCP-1. В некоторых вариантах осуществления соединения повышают уровни цАМФ. В других вариантах осуществления соединения нацелены на снижение или ингибирование уровней оксида азота (NO), TNF-α, мРНК TNF-α, мРНК IL-10, IL-10, IFNγ, IL-6, IL-12 и/или MCP-1. Идентичность соединений, которые изменяют вышеупомянутые уровни, можно определить методами скрининга. Для измерения вышеупомянутых уровней можно использовать подходящий анализ, известный в данной области. Неограничивающие примеры методов для определения соединений, которые демонстрируют желаемые изменения данных уровней, описаны в примерах.
В некоторых вариантах осуществления соединения по настоящему изобретению могут модулировать иммунный ответ и воспаление посредством альтернативного механизма действия, который не ограничивается механизмами, описываемыми в настоящем документе.
В некоторых вариантах осуществления пептиды по изобретению обладают улучшенной стабильностью в плазме и устойчивостью к действию протеаз. Стабильность в плазме и устойчивость к действию протеаз можно оценить любым подходящим способом. Неограничивающий пример предложен в примере 19. Оценка in vitro может указывать на активность, улучшенную устойчивость и более продолжительное время полужизни in vivo.
Способы, приведенные в настоящем документе, можно осуществлять на практике in vivo, ex vivo или in vitro.
Пегилированные пептиды
В некоторых вариантах осуществления пептиды модифицированы так, что увеличивается время полужизни пептида. В некоторых вариантах осуществления пептид пегилирован. В некоторых вариантах осуществления пегилированные пептиды относятся к пептидам, ковалентно связанным или конъюгированным с одной или несколькими цепями полимера полиэтиленгликоля (ПЭГ). Цепи полимера ПЭГ могут быть модифицированы, содержать функциональные группы или могут быть изменены другим образом. В другом варианте осуществления, полимерная цепь ПЭГ может иметь по меньшей мере одну или несколько точек ветвления. В некоторых предпочтительных вариантах осуществления полимерные цепи ПЭГ и соответствующий пегилированный пептид являются водорастворимыми, высокоподвижными в растворе, нетоксичными и неиммуногенными, без труда выходят из организма и могут иметь различное распределение в организме. В некоторых предпочтительных вариантах осуществления, фармакокинетическая природа пегилированного пептида регулируется типом цепи ПЭГ. Подходы и способы получения пегилированных пептидов известны в данной области (G. Pasuta и F.M.Veronese (2007) Polymer-drug conjugation, recent achievements и general strategies" Progress in Polymer Science 32(8-9): 933-961, которые включены в настоящий документ в качестве ссылки. Первое и второе поколение способов пегилирования белков можно найти в данной области.
Неограничивающий пример пегилирования содержит первую стадию введения подходящей функциональной группы в полимер ПЭГ с одного или обеих концов (для линейных ПЭГ). ПЭГ, которые активированы с каждого конца одной и той же активной функциональной группой называются "гомобифункциональными", а если функциональные группы различаются, то производные ПЭГ называются "гетеробифункциональными" или "гетерофункциональными". Химически активные или активированные производные полимера ПЭГ получают так, чтобы присоединить ПЭГ к желаемой молекуле. Выбор подходящей функциональной группы для производного ПЭГ основан на типе доступной реагирующей группы на молекуле, которая будет присоединена к ПЭГ. Неограничивающие примеры реагирующих аминокислот содержат лизин, цистеин, гистидин, аргинин, аспарагиновую кислоту, глутаминовую кислоту, серин, треонин и тирозин. Также можно использовать N-концевую аминогруппу и C-концевую карбоновую кислоту.
Остальные гетеробифункциональные ПЭГ для конъюгации: такие гетеробифункциональные ПЭГ весьма эффективны для связывания двух веществ, когда необходимы гидрофильные, гибкие и биосовместимые спейсеры. Предпочтительными концевыми группами для гетеробифункциональных ПЭГ являются малеимид, винилсульфоны, пиридилдисульфид, амин, карбоновые кислоты и сложные эфиры N- гидроксисукцинимида (NHS).
В некоторых вариантах осуществления изобретения, пегилированный пептид может иметь молекулярную массу в диапазоне 0,2 кДа-100 кДа. В некоторых предпочтительных вариантах осуществления изобретения, пегилированный пептид может иметь молекулярную массу в диапазоне 0,2 кДа-40 кДа. В некоторых предпочтительных вариантах осуществления изобретения, пегилированный пептид может иметь молекулярную массу в диапазоне 0,2 кДа-15 кДа. В других вариантах осуществления предпочтительная средняя молекулярная масса (в Да; как установлено размерно-эксклюзионной хроматографией) для коммерчески доступных ПЭГ, может быть выбрана из <1k, 2k, 3,5k, 5k, 10k, 20k, 30k, 40k и выше, но молекулярная масса может быть любой и зависит от желаемой фармакокинетики. Например, гетеробифункциональные ПЭГ с более низкой молекулярной массой можно использовать в качестве линкеров и для улучшения растворимости пептида. ПЭГ также может быть мульти-спейсерным, в виде вилки или разветвленным.
В некоторых вариантах осуществления пептид конъюгирован с молекулами-мишенями или действует как молекула-мишень, где молекула-мишень предпочтительно связывается с желаемым рецептором. В некоторых предпочтительных вариантах осуществления изобретения пептид конъюгирован с цитотоксическим веществом. Термин "цитотоксическое вещество" относится к веществу, которое ингибирует или предупреждает экспрессионную активность клеток, функционирование клеток и/или приводит к деструкции клеток. Термин включает радиоактивные изотопы, химиотерапевтические средства и токсины, такие как низкомолекулярные токсины или ферментативно активные токсины бактериального, грибкового, растительного или животного происхождения, включая их фрагменты и/или варианты. Неограничивающие примеры цитотоксических средств содержат майтанзин, доластатин и его аналоги, включая тазидотин и ауристатин. В других вариантах осуществления изобретения, неограничивающие примеры цитотоксических средств содержат майтанзиноиды, иттрий, висмут, рицин, А-цепь рицина, сапорин, доксорубицин, даунорубицин, таксол, бромистый этидий, митомицин, этопозид, тенопозид, винкристин, винбластин, колхицин, дигидроксиантрациндион, актиномицин, субъединица А дифтерийного токсина, укороченный Pseudomonas exotoxin (PE) A, PE40, абрин, A-цепь абрина, А-цепь модессина, альфа-сарцин, гелонин, митогеллин, рестриктоцин, феномицин, еномицин, курицин, кротин, калихимицин, ингибитор sapaonaria officinalis, и глюкокортикоид и остальные химиотерапевтические средства, а также радиоактивные изотопы такие как At211, I131, I125, Y90, Re186, Re188, Sm153, Bi212, P32 и радиоактивные изотопы Lu. Антитела могут также быть конъюгированы с ферментом, активирующим противоопухолевое пролекарство, который способен превращать пролекарство в его активную форму.
Пептид может быть связан непосредственно или опосредовано с цитотоксическим или нацеливающим агентом любым способом, известным в данной области в настоящее время. В некоторых предпочтительных вариантах осуществления изобретения, пептид присоединен к цитотоксическому средству или нацеливающему агенту с помощью одного или нескольких линкеров для создания конъюгата, при условии, что введение линкера существенно не препятствует активности, связыванию, токсичности или содержанию пептида или конъюгированного агента.
Неограничивающие примеры линкеров, содержат ионные и ковалентные связи и любую другую достаточно стабильную связь, при помощи которой нацеленный агент интернализуется клеткой, на которую нацелен конъюгированный агент. Линкерную часть выбирают в зависимости от желаемых свойств. Факторы, которые учитываются при выборе линкера, могут содержать снятие или уменьшение пространственных затруднений, происходящих из-за близости конъюгированных элементов, изменение других свойств конъюгата, таких как специфичность, токсичность, растворимость, стабильность в сыворотке и/или внутриклеточная доступность конъюгата и/или увеличение гибкости связи. Линкер может быть любым типом связи, и примеры описаны в патентах США № 7166702 и 5194425, оба которых полностью включены в настоящий документ в качестве ссылки в полном объеме.
Линкеры и связи, подходящие для химически связанных конъюгатов, содержат в качестве неограничивающих примеров, дисульфидные связи, тиоэфирные связи, пространственно-затрудненные дисульфидные связи и ковалентные связи между свободно реагирующими группами, такими как аминовые и тиоловые группы. Такие связи получают с использованием гетеробифункциональных реагентов для создания реактивных тиоловых групп на одном или обоих полипептидах и последующего реагирования тиоловых групп на одном полипептиде с реактивными тиоловыми группами или аминогруппами на другом полипептиде, к которым можно присоединить реактивные малеимидогруппы или тиоловые группы. Остальные линкеры содержат расщепляемые кислотой линкеры, такие как бисмалеимидоэтоксипропан, кислото-неустойчивые трансферриновые конъюгаты и дигидразид адипиновой кислоты, которые могли бы быть расщеплены в более кислых внутриклеточных компартментах; перекрестно-связывающие линкеры, которые расщепляются под воздействием УФ или видимого света, и линкеры, такие как различные домены, такие как CHl, CH2 и CH3, из константных областей IgG1человека (смотрите Batra et al. (1993) MoI. Immunol. 30:379-386, включенный в настоящий документ в качестве ссылки).
Химические линкеры и пептидные линкеры могут быть введены посредством ковалентного связывания линкера и пептида и нацеливающим или цитотоксическим средством. Гетеробифункциональные агенты, описанные ниже, можно использовать для проведения такого ковалентного связывания.
Гетеробифункциональные перекрестно-связывающие реагенты
Специалистам в данной области известно большое число гетеробифункциональных перекрестно-связывающих реагентов, которые используют для образования ковалентных связей между аминогруппами и тиоловыми группами и для введения тиоловых групп в белки (смотрите, например, the PIERCE CATALOG, Immuno Technology Catalog & Handbook, 1992-1993, который описывает препараты таких реагентов и их применение и является коммерческим источником таких реагентов; смотрите также Cumber et al. (1992) Bioconjugate Chem. 3':397-401; Thorpe et al. (1987) Cancer Res. 47:5924-5931; Gordon et al. (1987) Proc. Natl. Acad. Sci. USA 84:308-312; Walden et al. (1986) J. Mol. Cell Immunol. 2:191-197; Carlsson et al. (1978) Biochem. J. 173: 723-737; Mahan et al. (1987) Anal. Biochem. 162:163-170; Wawryznaczak et al. (1992) Br. J. Cancer 66:361-366; Fattom et al. (1992) Infection & Immun. 60:584-589). Все цитируемые источники включены в настоящий документ в качестве ссылки в полном объеме. Такие реагенты можно использовать для формирования ковалентных связей между нацеливающим агентом, хемокином и нацеленным веществом. Такие реагенты содержат в качестве неограничивающих примеров: N-сукцинимидил-3-(2-пиридилдитио)пропионат (SPDP; дисульфидный линкер); сульфосукцинимидил-6-[3-(2-пиридилдитио)пропионамидо]гексаноат (сульфо-LC-SPDP); сукцинимидилоксикарбонил-α-метилбензилтиосульфат (SMBT, пространственно-затрудненный дисульфатный линкер); сукцинимидил-6-[3-(2-пиридилдитио)пропионамидо]гексаноат (LC-SPDP); сульфосукцинимидил-4-(N-малеимидометил)циклогексан-1-карбоксилат (сульфо-SMCC); сукцинимидил-3-(2-пиридилдитио)бутират (SPDB; пространственно-затрудненный линкер с дисульфидной связью); сульфосукцинимидил-2-(7-азидо-4-метилкумарин-3-ацетамид) этил-1,3-дитиопропионат (SAED); сульфосукцинимидил-7-азидо-4-метилкумарин-3-ацетат (SAMCA); сульфосукцинимидил-6-[альфа-метил-альфа-(2-пиридилдитио)толуамидо]гексаонат (сульфо-LC-SMPT); 1,4-ди-[3'-(2'-пиридилдитио)пропионамидо]бутан (DPDPB); 4-сукцинимидилоксикарбонил-α-метил-α-(2-пиридилтио)толуол (SMPT, пространственно-затрудненный дисульфатный линкер); сульфосукцинимидил-6[α-метил-α-(2-пиридилдитио)толуамидо]гексаноат (сульфо-LC-SMPT); сложный эфир м-малеимидобензоил-N-гидроксисукцинимида (MBS); сложный эфир м-малеимидобензоил-N-гидроксисульфосукцинимида (сульфо-MBS); N-сукцинимидил(4-йодоацетил)аминобензоат (SIAB; тиоэфирный линкер); сульфосукцинимидил(4-йодоацетил)аминобензоат (сульфо-SIAB); сукцинимидил-4-(п-малеимидофенил)бутират (SMPB); сульфосукцинимидил-4-(п-малеимидофенил)бутират (сульфо-SMPB); азидобензоилгидразид (ABH).
Другие гетеробифункциональные расщепляемые перекрестно-связывающие линкеры содержат N-сукцинимидил(4-йодоацетил)-аминобензоат; сульфосукцинимидил(4-йодоацетил)-аминобензоат; 4-сукцинимидил-oxyкарбонил-a-(2-пиридилдитио)-толуол; сульфосукцинимидил-6-[a-метил-a-(пиридилдитиол)-толуамидо]гексаонат; N-сукцинимидил-3-(-2-пиридилдитио)-проприонат; сукцинимидил-6[3(-(-2-пиридилдитио)-проприонамидо]гексаонат; сульфосукцинимидил-6[3(-(-2-пиридилдитио)-проприонамидо]гексаонат; 3-(2-пиридилдитио)-пропионилгидразид, реагент Эллмана, дихлортриазиновая кислота, S-(2-тиопиридил)-L-цистеин. Другие примеры бифункциональных связывающих соединений описаны в патентах США №№ 5349066, 5618528, 4569789, 4952394 и 5137877, которые все включены в настоящий документ в качестве ссылки.
Расщепляемые кислотой, фоторазрушаемые и термочувствительные линкеры
Расщепляемые кислотой линкеры, фоторазрушаемые и термочувствительные линкеры также можно использовать, особенно в том случае, когда может быть необходимо расщепить нацеленное вещество, чтобы обеспечить ему возможность быть более легкодоступным для реакции. Расщепляемые кислотой линкеры содержат в качестве неограничивающих примеров, бисмалеимидоэтоксипропан и линкеры дигидразида адипиновой кислоты (смотрите, например, Fattom et al. (1992) Infection & Immun. 60:584-589, включенный в настоящий документ в качестве ссылки) и кислото-неустойчивые трансферриновые конъюгаты, которые содержат достаточное количество трансферрина для включения во внутриклеточный цикл обмена трансферрина (смотрите, например, Welhoner et al. (1991) J. Biol. Chem. 266:4309-4314, включенный в настоящий документ в качестве ссылки).
Фоторазрушаемые линкеры представляют собой линкеры, которые расщепляются под воздействием света (смотрите, например, Goldmacher et al. (1992) Bioconj. Chem. 3:104-107, перечень линкеров из которого включен в настоящий документ в качестве ссылки), высвобождая, таким образом, нацеленное вещество под воздействием света. Известны фоторазрушаемые линкеры, которые расщепляются под воздействием света, таким образом, высвобождая нацеленное вещество под действием света (смотрите, например, Hazum et al. (1981) in Pept., Proc. Eur. Pept. Symp., 16th, Brunfeldt, K (Ed), pp. 105-110, в котором описано применение нитробензиловой группы в качестве фоторазрушаемой защитной группы для цистеина; Yen et al. (1989) Makromol. Chem. 190:69-82, которые описывают водорастворимые фоторазрушаемые сополимеры, содержащие сополимер гидроксипропилметилакриламида, сополимер глицина, сополимер флюоресцеина и сополимер метилродамина; Goldmacher et al. (1992) Bioconj. Chem. 3:104-107, в котором описан перекрестно-связывающий линкер и реагент, который подвергается фотолитической деградации под воздействием света, близкого к УФ (350 нм); и Senter et al. (1985) Photochem. Photobiol. 42:231-237, в которых описаны нитробензилоксикарбонилхлорид перектрестно-связывающие реагенты, которые образуют фоторазрушаемые связи). Все цитируемые источники включены в настоящий документ в качестве ссылки в полном объеме. Такие линкеры могли бы использоваться, в частности, для лечения дерматологических или офтальмологических заболеваний, которые могли бы подвергаться воздействию света с использованием волоконной оптики. После введения конъюгата, глаза или кожу или другую часть тела подвергают воздействию света, что приводит к высвобождению нацеленной части из конъюгата. Такие фоторасщепляемые линкеры удобны для диагностических протоколов, когда желательно удаление нацеливающего агента для быстрого вывода из организма животного.
Остальные линкеры химических конъюгатов
Остальные линкеры, включая тритиловые линкеры, в частности, производные тритиловых групп для создания вида конъюгатов, которые обеспечивают высвобождение терапевтических средств при различных уровнях кислотности или защелоченности. Гибкость, таким образом, достигается посредством возможности предварительного выбора диапазона pH, при котором терапевтическое средство будет высвобождаться, позволяя основывать выбор линкера на известных физиологических различиях между тканями, нуждающимися в доставке терапевтического средства (смотрите, например, патент США № 5612474, включенный в настоящий документ в качестве ссылки). Например, кислотность опухолевых тканей ниже, чем нормальных.
Пептидные линкеры
Линкерные части могут быть пептидами. В конъюгате могут быть использованы пептидные линкеры. Пептидный линкер обычно имеет от приблизительно 2 до приблизительно 60 аминокислотных остатков, например, от приблизительно 5 до приблизительно 40 или от приблизительно 10 до приблизительно 30 аминокислотных остатков. Выбранная длина будет зависеть от факторов, таких как применение, для которого вводят линкер.
Линкерная часть может быть гибкой спейсерной аминокислотной последовательностью, такой как последовательности, которые известны из исследований одноцепочечных антител. Примеры таких известных линкерных групп содержат в качестве неограничивающих примеров, GGGGS, (GGGGS)n, GKSSGSGSESKS, GSTSGSGKSSEGKG, GSTSGSGKSSEGSGSTKG, GSTSGSGKSSEGKG, GSTSGSGKPGSGEGSTKG, EGKSSGSGSESKEF, SRSSG, SGSSC. Можно также использовать линкер на основе дифтерийного токсина, чувствительный к трипсину, с последовательностью AMGRSGGGCAGNRVGSSLSCGGLNLQAM.
Описаны дополнительные линкерные группы, например, в Huston et al., Proc. Natl. Acad. ScL U.S.A. 85:5879-5883, 1988; Whitlow, M., et al, Protein Engin. 6:989-995, 1993; Newton et al, Biochemistry 35:545-553, 1996; A. J. Cumber et al, Bioconj. Chem. 3:397-401, 1992; Ladurner et al, J. Mol. Biol. 273:330-337, 1997; и патенте США № 4894443; все публикации включены в настоящий документ в качестве ссылки.
Остальные линкеры содержат в качестве неограничивающих примеров: субстраты ферментов, такие как субстрат катепсина B, субстрат катепсина D, субстрат трипсина, субстрат тромбина, субстрат субтилизина, субстрат фактора Xa и субстрат энтерокиназы; линкеры, которые увеличивают растворимость, гибкость, и/или внутриклеточную расщепляемость содержат такие линкеры как (glymser)n и (sermgly)n, (смотрите, например, публикацию PCT № WO 96/06641, включенную в настоящий документ в качестве ссылки, которая предлагает примеры линкеров для применения в конъюгатах). В некоторых вариантах осуществления некоторые линкеры можно содержать для того чтобы воспользоваться преимуществами желаемых свойств каждого линкера.
Получение конъюгатов
Конъюгаты со связанными нацеленными веществами могут быть получены либо путем химической конъюгации, технологиями рекомбинантных ДНК, либо комбинированием рекомбинатной экспрессии и химической конъюгации. Пептид по изобретению и цитотоксический или нацеливающий агент могут быть связаны в любой ориентации, и в конъюгате может присутствовать более одного нацеливающего агента и/или нацеленного вещества.
В некоторых предпочтительных вариантах осуществления изобретения, цитотоксическое средство привязано к пептиду посредством гидрофильного и биосовместимого полимерного спейсера, включая короткую алкильную цепь, полисиаловую или гиалуроновую кислоту, полипептид или ПЭГ. В некоторых предпочтительных вариантах осуществления изобретения, цитотоксическое средство присоединено с помощью расщепляемого линкера, например, дисульфидной связью или пептидсодержащей последовательностью, которая расщепляется лизосомальными протеазами, такими как катепсины. В некоторых предпочтительных вариантах осуществления изобретения, спейсер присоединен либо к N-, либо к C-концу пептида.
Составы
Способы получения таких составов или композиций содержат стадию ассоциации соединения по настоящему изобретению с носителем и, необязательно, с одним или несколькими дополнительными ингредиентами. В основном, составы получают путем приведения в контакт единообразно и близко, соединения по настоящему изобретению и жидкого носителя, или мелко измельченного твердого носителя, или обоих, а затем, если необходимо, продукту придается форма.
Фармацевтические составы могут быть получены в соответствии с любым способом, известным в фармацевтическом производстве. Такие составы могут содержать подсластители, ароматизаторы, красители и консерванты. Состав может быть смешан с нетоксичными фармацевтически приемлемыми эксципиентами, которые применяются в производстве. Такой "эксципиент", как правило, относится к по существу инертному материалу, который нетоксичен и не взаимодействует с остальными компонентами композиции неблагоприятным образом. Фармацевтически приемлемые эксципиенты содержат в качестве неограничивающих примеров, жидкости, такие как вода, забуференный физиологический раствор, полиэтиленгликоль, гиалуроновая кислота, глицерин и этанол. В эксципиенты могут быть включены фармацевтически приемлемые соли, например, соли неорганических кислот, такие как трифторацетат, гидрохлориды, гидробромиды, фосфаты, сульфаты, и т.п.; и соли органических кислот, такие как ацетаты, пропионаты, малонаты, бензоаты, и т.п.
Терапевтическое средство можно вводить в виде лекарственного препарата или фармацевтической композиции, удобной для доставки. Составы могут содержать один или несколько растворителей, эмульгаторы, консерванты, буферы, эксципиенты и т.д., и могут быть предложены в таких формах, как лиофилизованные пудры, спреи, кремы, лосьоны, гели, пластыри, импланты и т.д. Фармацевтические составы для перорального введения могут быть составлены с использованием фармацевтически приемлемых носителей, хорошо известных в данной области, в соответствующих и подходящих дозах. Такие носители дают возможность лекарственным препаратам войти в состав стандартных лекарственных форм, таких как таблетки, пилюли, пудры, драже, капсулы, жидкости, пастилки, гели, сиропы, кашицы, суспензии и т.д., удобных для приема внутрь пациентом. Фармацевтические препараты для орального применения могут входить в состав в качестве твердого эксципиента, по желанию, измельчение полученной смеси и обработка смеси гранул после добавления подходящих дополнительных компонентов, если необходимо, для получения таблеток или сердцевины драже. Подходящие твердые эксципиенты представляют собой углеводные или белковые наполнители, например, сахара, включая лактозу, сахарозу, маннит, или сорбит; кукурузный, пшеничный, рисовый, картофельный крахмал или крахмал из других растений; целлюлозу, такую как метилцеллюлоза, гидроксипропилметилцеллюлоза, или натриевая карбоксиметилцеллюлоза; и камеди, включая смолу акации и трагакант; и белки, например, желатин и коллаген. Можно добавить расслоители или растворители, такие как поливинилпирролидон с перекрестными сшивками, агар, альгиновая кислота или ее соль, такая как альгинат натрия.
Водные суспензии могут содержать активное средство (например, химерный полипептид или пептидомиметик по изобретению) в смеси с эксципиентами, подходящими для производства водных суспензий. Такие эксципиенты содержат суспендирующее вещество, такое как натриевая карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, камедь трагаканта и смолу акации, и диспергенты или увлажнители, такие как природный фосфатид (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, полиоксиэтиленстеарат), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептадекаэтиленоксицетанол), продукт конденсации этиленоксида с неполным эфиром, производным жирной кислоты и гекситола (например, полиоксиэтиленсорбитмоноолеат), или продукт конденсации этиленоксида с неполным эфиром, производным жирной кислоты и ангидрида гекситола (например, полиоксиэтиленсорбитанмоноолеат). Водные суспензии могут также содержать один или несколько консервантов, таких как этил или н-пропил-н-гидроксибензоат, один или несколько красителей, один или несколько ароматизаторов и один или несколько подсластителей, таких как сахароза, аспартам или сахарин. Составы могут быть скорректированы по осмолярности.
Фармацевтические средства на масляной основе могут использоваться для введения гидрофобных активных средств по изобретению. Суспензии на масляной основе могут быть получены суспендированием активного средства (например, химерной композиции по изобретению) в растительном масле, таком как арахисовое масло, оливковое масло, сезамовое масло или кокосовое масло, или в минеральном масле, таком как жидкий парафин; или в смеси таких масел. Смотрите, например, патент США № 5716928, включенный в настоящий документ в качестве ссылки, который описывает использование необходимых масел или компонентов необходимых масел для увеличения биодоступности и снижения интер- и интраиндивидуальной вариабельности гидрофобных фармацевтических соединений, принимаемых перорально (смотрите также патент США № 5858401, включенный в настоящий документ в качестве ссылки). Масляные суспензии могут содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Можно добавлять подсластители, такие как глицерин, сорбит или сахароза, для придания препарату приятного вкуса. Такие составы можно сохранять добавлением антиоксиданта, такого как аскорбиновая кислота. Для примера масляной среды, пригодной для инъекций, смотрите Minto (1997) J. Pharmacol. Exp. Ther. 281:93-102, включенный в настоящий документ в качестве ссылки. Фармацевтические составы по изобретению могут также быть в форме эмульсий "масло в воде". Масляная фаза может быть растительным или минеральным маслом, описанным выше, или их смесью. Подходящие эмульгаторы содержат природную камедь, например, смолу акации и камедь трагаканта, природные фосфатиды, такие как соевый лецитин, сложные эфиры или неполные эфиры, производные жирных кислот и ангидридов гекситола, такие как моноолеат сорбитана, и продукты конденсации этих неполных эфиров с этиленоксидом, такие как полиоксиэтиленсорбитанмоноолеат. Эмульсия может также содержать подсластители и ароматизаторы, как и в составе сиропов и эликсиров. Такие составы могут также содержать смягчитель, консервант, или краситель.
Также следует учитывать, что композиция или медикамент, содержащий терапевтическое средство, может содержать фармацевтически приемлемый носитель, который служит в качестве стабилизатора, в частности, для пептида, белка, полинуклеотида или подобных им веществ. Примеры подходящих носителей, которые также действуют, как стабилизаторы пептидов, содержат в качестве неограничивающих примеров, фармацевтически чистые декстрозу, сахарозу, лактозу, трегалозу, маннит, сорбит, инозитол, декстран, и т.п. Остальные подходящие носители содержат, снова без ограничений, крахмал, целлюлозу, фосфаты натрия или кальция, лимонную кислоту, винную кислоту, глицин, полиэтиленгликоли (ПЭГ) с высокой молекулярной массой, и их сочетание. Возможно также использовать заряженный липид и/или детергент. Подходящие заряженные липиды содержат в качестве неограничивающих примеров, фосфатидилхолины (лецитин) и т.п. Детергенты обычно представляют собой неионное, анионное, катионное или амфотерное поверхностно-активное вещество. Примеры подходящих поверхностно-активных веществ содержат, например, поверхностно-активные вещества Тергитол® и Тритон® (Union Carbide Chemicals и Plastics, Danbury, CT), полиоксиэтиленсорбитаны, например, поверхностно-активные вещества TWEEN® (Atlas Chemical Industries, Wilmington, DE), простые эфиры полиоксиэтилена, например BRIJ, фармацевтически приемлемые сложные эфиры жирных кислот, например, лаурилсульфат и их соли (SDS), и подобные им материалы. Детальное обсуждение фармацевтически приемлемых эксципиентов, носителей, стабилизаторов и остальных вспомогательное веществ доступно в Remington's Farmaceutical Sciences (Mack Pub. Co., N. J. 1991), включенном в настоящий документ в качестве ссылки.
Фармацевтические композиции по настоящему изобретению, подходящие для парентерального введения, содержат одно или несколько соединений по изобретению в сочетании с одним или несколькими фармацевтически приемлемыми стерильными изотоническими водными или неводными растворами, дисперсиями, суспензиями или эмульсиями, или стерильные порошки, которые можно растворить в стерильных растворах для инъекций или дисперсиях непосредственно перед применением; которые могут содержать сахара, спирты, антиоксиданты, буфера, бактериостатики, растворы, которые делают состав изотоничным с кровью назначенного реципиента, или суспендирующие или загущающие компоненты.
Примеры подходящих водных и не-водных носителей, которые можно использовать в фармацевтических композициях по изобретению, содержат воду, этанол, многоатомные спирты (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.), и их подходящие смеси, растительные масла, такие как оливковое масло и пригодные для инъекций органические сложные эфиры, такие как этилолеат. Надлежащая текучесть может быть сохранена, например, с использованием покрывающих материалов, таких как лецитин, посредством сохранения требуемого размера частиц в случае рассеивания и с помощью поверхностно-активных веществ.
Такие композиции могут также содержать вспомогательные вещества, такие как консерванты, увлажнители, эмульгаторы и диспергирующие средства. Предупреждение действий микроорганизмов на соединения по настоящему изобретению можно обеспечить за счет содержания различных антибактериальных и антигрибковых агентов, например, парабена, хлоробутанола, фенола, сорбиновой кислоты и т.п. Также может быть желательным содержание в композиции изотонических агентов, таких как сахара, хлорид натрия, фосфатно-солевой буфер и т.п. Кроме того, пролонгированная абсорбция фармацевтических форм, пригодных для инъекций, может быть достигнута приблизительно содержанием агентов, которые замедляют абсорбцию, таких как моностеарат алюминия и желатин.
В некоторых случаях для того, чтобы пролонгировать действие лекарственного средства, желательно замедлить абсорбцию лекарственного средства из подкожной или внутримышечной инъекции. Этого можно достигнуть с использованием жидкой суспензии кристаллов или аморфного материала, который плохо растворяется в воде. Скорость абсорбции лекарственного средства, таким образом, зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера и формы кристалла. Альтернативно, замедленная абсорбция лекарственного средства, которое вводится парентерально, достигается растворением или суспендированием лекарственного средства в масляной среде.
В способах по изобретению, фармацевтические соединения могут быть также введены в микросферах для медленного высвобождения в организме. Например, микросферы можно вводить посредством интрадермальной инъекции лекарственного средства, которое медленно высвобождается подкожно; смотрите Rao (1995) J. Biomater ScL Полит. Ed. 7:623-645; в качестве биоразлагающихся и инъецируемых гелевых составов, смотрите, например, Gao (1995) Pharm. Res. 12:857-863 (1995); или, в виде микросфер для перорального введения, смотрите, например, Eyles (1997) J. Pharm. Pharmacol. 49:669-674. Все цитируемые источники полностью включены в настоящий документ в качестве ссылки.
Инъекции с замедленным всасыванием веществ изготавливают путем формирования матриц соединения по настоящему изобретению, микроинкапсулированных в биоразлагаемые полимеры, такие как полилактид-полигликолид. В зависимости от соотношения лекарственного средства с полимером, и природы конкретного использованного полимера, можно контролировать скорость высвобождения лекарственного средства. Примеры остальных биоразлагаемых полимеров содержат полиортоэфиры и полиангидриды. Составы инъекций с замедленным всасыванием вещества изготавливают также путем захвата лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканями тела.
Полипептид можно вводить сам по себе или в сочетании с другим терапевтическим соединением. В частности, полипептид можно вводить в сочетании с терапевтическим соединением, которое применяют для лечения меланомы, воспаления или аутоиммунного заболевания у млекопитающих. Полипептид и дополнительное терапевтическое соединение могут быть в составе одной и той же или разных композиций. Полипептид можно вводить одновременно, последовательно или раздельно с дополнительным терапевтическим соединением.
Дозировка
Фактические уровни дозировки активных ингредиентов в фармацевтических композициях по настоящему изобретению можно изменять для получения количества активного ингредиента, которое эффективно для достижения желаемого терапевтического ответа у конкретного пациента, композиции и способа введения, и не токсично для пациента. Если соединения по настоящему изобретению назначают в качестве лекарственных средств людям и животным, их можно давать самостоятельно или в составе фармацевтической композиции, содержащей, например, от 0,1 до 99% активного ингредиента, более предпочтительно, от 10 до 30%, в сочетании с фармацевтически приемлемым носителем.
Выбранный уровень дозы будет зависеть от ряда факторов, включая активность применяемого конкретного состава по настоящему изобретению, путь введения, время введения, скорость экскреции или метаболизм конкретного применяемого соединения, скорость и объем абсорбции, длительность лечения, другие лекарственные средства, составы и/или материалы, которые используются в сочетании с применяемым конкретным составом, возраст, пол, вес, состояние, здоровье в целом и медицинская история пациента, которого лечат, и другие подобные факторы, хорошо известные в медицине.
Терапевт или ветеринар со средними знаниями в данной области могут легко определить и прописать эффективное количество требуемой фармацевтической композиции. Например, терапевт или ветеринар могут начать с более низкого уровня доз соединения по изобретению, используемого в фармацевтической композиции, чем требуется для достижения желаемого терапевтического эффекта и постепенно увеличивать дозу, пока не будет достигнут желаемый эффект.
Обычно подходящая ежедневная доза соединения по изобретению будет тем количеством соединения, которое представляет собой самую низкую дозу, оказывающую терапевтический эффект. Такая эффективная доза будет, как правило, зависеть от факторов, описанных выше, например, в общем, оральная, внутривенная, интрацеребровентрикулярная и подкожные дозы соединения по настоящему изобретению для пациента, который нуждается в указанном эффекте, будут иметь неограниченный диапазон от приблизительно 1 мкг до приблизительно 5 мг на кг массы тела в час. В других вариантах осуществления доза будет иметь неограниченный диапазон от приблизительно 5 мкг до приблизительно 2,5 мг на кг массы тела в час. В дополнительных вариантах осуществления доза будет иметь неограниченный разброс от приблизительно 5 мкг до приблизительно 1 мг на кг массы тела в час.
При желании, эффективную ежедневную дозу активного соединения можно вводить в виде двух, трех, четырех, пяти, шести или более суб-доз раздельно через соответствующие интервалы на всем протяжении дня, необязательно, в стандартных лекарственных формах. В одном из вариантов осуществления соединение вводят одной дозой в сутки. В других вариантах осуществления, соединение вводят постоянно, внутривенно или другим путем. В других вариантах осуществления соединение вводят реже, чем ежедневно, например, раз в неделю или реже.
В то время как возможно вводить состав по настоящему изобретению сам по себе, предпочтительно вводить состав в виде фармацевтический состава (композиции).
Пациент, получающий такое лечение, представляет собой любое животное, которому это необходимо, включая приматов, в частности, людей, и других млекопитающих, таких как кролики, лошади, домашний скот, такой как бык, свинья, коза и овца; и домашнюю птицу и, в общем, домашних животных.
Состав по изобретению можно вводить как есть или в смеси с фармацевтически приемлемыми носителями и можно также вводить в сочетании с антимикробными агентами, такими как пенициллины, цефалоспорины, аминогликозиды и гликопептиды. Совместная терапия, таким образом, содержит последовательное, одновременное и раздельное введение активного соединения таким образом, что терапевтические эффекты первой введенной дозы не исчезают полностью к моменту, когда вводится следующая.
Возможные пути введения описанных соединений
Соединения можно вводить людям и животным для терапии любым подходящим путем введения. В настоящем документе, термин "путь" введения содержит, в качестве неограничивающих примеров, подкожную инъекцию, внутривенную инъекцию, внутриглазную инъекцию, внутрикожную инъекцию, внутримышечную инъекцию, внутрибрюшинную инъекцию, интратрахеальное введение, введение внутрь жировой ткани, внутрисуставное введение, подоболочечное введение, эпидуральное введение, ингаляцию, интраназальное введение, пероральное введение, сублингвальное введение, буккальное введение, ректальное введение, вагинальное введение, интрацистернальное введение, трансдермальное введение и местное введение, или введение путем локальной доставки (например, с помощью катетера или стента). Соединения также можно вводить или вводить совместно в лекарственных формах с медленным высвобождением.
В способах по изобретению, фармацевтические соединения можно также вводить посредством интраназального, внутриглазного, периокулярного и интравагинального путей, включая суппозитории, вдувания, пудры и аэрозольные составы (для примеров стероидные ингаляторы, смотрите Rohatagi (1995) J. Clin. Pharmacol. 35:1187-1193; Tjwa (1995) Ann. Allergy Asthma Immunol. 75:107-111, включенный в настоящий документ в качестве ссылки). Составы суппозиториев можно получить смешиванием лекарственного средства с подходящим, не раздражающим эксципиентом, который твердый при обычной температуре и жидкий при температуре тела и будет, таким образом, плавиться в теле для высвобождения лекарственного средства. Такими материалами являются масло какао и полиэтиленгликоли.
В способах по изобретению, фармацевтические соединения можно доставлять трансдермально, путем местного введения, изготовленными в виде полосок для аппликаций, растворов, суспензий, эмульсий, гелей, кремов, мазей, паст, желе, пудр и аэрозолей.
В способах по изобретению, фармацевтические соединения можно вводить парентерально, посредством внутривенного (IV) введения или введения в полости тела (например, в синовиальное пространство) или в просвет органа. Такие составы могут содержать раствор активного средства, растворенного в фармацевтически приемлемом носителе. Приемлемые среды для лекарств и растворители, которые могут быть использованы, это вода и раствор Рингера, изотонический хлорид натрия. Кроме того, можно использовать в качестве растворителя или суспендирующей среды стерильные жидкие масла. Для этой цели может быть использовано любое мягкое жирное масло, включая синтетические моно- или диглицериды.
Как описано в настоящем документе, способы по настоящему изобретению можно использовать отдельно или в сочетании с другими подходами к лечению аутоиммунных заболеваний или других состояний, описываемых в настоящем документе.
Конкретная терапевтическая комбинация (лекарственных препаратов или процедур) для использования в комбинированной схеме лечения должна учитывать совместимость желаемых лекарственных препаратов и/или процедур и желаемый терапевтический эффект, оторый необходимо достигнуть. Ясно также, что используемые методы лечения должны приводить к желаемому результату для одного и того же нарушения (например, состав по изобретению можно вводить одновременно с другим агентом, который используется для лечения того же нарушения), или они могут оказывать различное действие (например, контроль любых неблагоприятных воздействий). Как применяют в настоящем документе, дополнительные терапевтические средства, которые обычно назначают для лечения или предупреждения конкретного заболевания или состояния, известны как "подходящие для заболевания или состояния, которое лечат".
Комбинированное лечение по настоящему изобретению, как определено в настоящем документе, может быть достигнуто путем одновременного, последовательного или раздельного введения индивидуальных компонентов указанного лечения.
Терапевтическое применение
Введение полинуклеотида или регуляторного соединения (далее в данном документе "терапевтическое средство"), как описано, может производиться с профилактической или терапевтической целями. При профилактическом использовании, терапевтическое средство вводят до появления любых симптомов. Профилактическое введение терапевтического средства служит для профилактики или устранения любых симптомов. В случае терапевтического применения, терапевтическое средство используют сразу (или вскоре) после появления симптомов заболевания или нарушения. Терапевтическое введение терапевтического средства служит для смягчения любого фактического обострения симптомов.
Индивидуум, получающий лечение, может быть любым млекопитающим. В одном из аспектов, млекопитающее является человеком. В другом аспекте человек страдает аутоиммунным заболеванием или состоянием. В других аспектах, аутоиммунное заболевание или состояние связано с или выбрано из группы, состоящей из рассеянного склероза, диабета типа 1, апластической анемии, болезни Грейвза, целиакия, болезни Крона, волчанки, артрита, остеоартрита, аутоиммунного увеита, аутоиммунного энцефаломиелита и миастении гравис.
В других аспектах, у человека имеется воспалительное заболевание или состояние. В другом аспекте, воспалительное заболевание или состояние связано или выбрано из группы, состоящей из воспалительного заболевания кишечника, ревматоидного артрита, аллергии, атеросклероза, псориаза, гастрита и ишемической болезни сердца. В другом аспекте, воспаление связано со смертью мозга, предпочтительно, где уровни циркулирующего эндогенного α-MSH или α-MSH в ткани мозга снижены. Еще в одном из аспектов, терапевтическое средство используют для лечения пациента с нарушением или заболеванием, опосредованным α-MSH или MC1-рецептором.
В другом аспекте настоящее изобретение относится к стационарному лечению пациентов с меланомой. В одном из аспектов, настоящее изобретение уменьшает проявления или улучшает течение меланомы у пациентов. Еще в одном из аспектов, соединения по настоящему изобретению используют в способах детекции и получения изображений меланомы.
Наборы, содержащие вышеописанные составы
Изобретение также относится к наборам для проведения схемы лечения по изобретению. Такие наборы содержат терапевтически эффективные количества пептида, имеющего специфическую активность регулятора MC1R, в фармацевтически приемлемой форме, отдельно или в сочетании с другими агентами, в фармацевтически приемлемой форме. Предпочтительные фармацевтические формы содержат пептиды в сочетании со стерильным физиологическим раствором, раствором декстрозы, буферным раствором или другой фармацевтически приемлемой стерильной жидкостью. Альтернативно, композиция может быть лиофилизована или обезвожена. В этом случае набор может дополнительно содержать фармацевтически приемлемый раствор, предпочтительно стерильный для создания раствора для инъекций. В другом варианте осуществления, набор может дополнительно содержать иглу или шприц, предпочтительно, в стерильной упаковке, для инъекции композиции. В других вариантах осуществления набор дополнительно содержит инструкцию о способах введения композиции пациенту. Инструкция о способах может быть в виде написанного вкладыша, аудиозаписи, аудиовидеозаписи или любого другого способа инструктирования о введении композиции пациенту. В одном из вариантов осуществления набор содержит (i) первичный контейнер, содержащий пептид, имеющий специфическую MCIR-модуляторную активность; и (ii) инструкцию по применению.
Дополнительные варианты осуществления способа лечения
В некоторых вариантах осуществления изобретение относится к способу уменьшить или замедлить отторжение трансплантата у пациента, включая введение указанному пациенту фармацевтической композиции, которая содержит фармацевтически приемлемый эксципиент и эффективное количество, по меньшей мере, одного из пептидных соединений, описываемых в данном документе. В некоторых вариантах осуществления изобретение относится к способу уменьшения или замедления иммунного ответа пациента, который вызван трансплантированной тканью, клетками или органом. Неограничивающими примерами трансплантированных тканей являются аллотрансплантат при экспериментальной трансплантации сердца и клетки панкреатических островков.
Иммуносупрессивная активность на экспериментальной модели аутоиммунного энцефаломиелита (EAE)
Новые D-аминокислотные пептидные аналоги α-MSH были созданы и исследованы их иммуномодулирующие эффекты на экспериментальных моделях аутоиммунного увеита (EAU) и аутоиммунного энцефаломиелита (EAE), а также на суррогатной модели воспаления, индуцированного липополисахаридом (LPS). Лечение аналогом RI α-MSH снизило клинические показатели заболевания и возникновение заболевания на модели EAU и на мышиной модели EAE, индуцированного MOG. Кроме того, уровни экспрессии мРНК TNF-α и IL-10 в селезенке и лимфоузле мышей, примированных MOG, были снижены у мышей, которых лечили. На модели системного воспаления, индуцированного LPS, лечение α-MSH и аналогом привело к снижению уровней цитокинов в сыворотке. Эти данные указывают на то, что такие новые аналоги α-MSH обладают потенциалом для терапевтического применения при воспалении и аутоиммунном заболевании.
Материалы и способы
Пептиды. α-MSH (SYSMEHFRWGKPV) был приобретен у Bachem (King of Prussia, PA). D-аминокислотный аналог RI α-MSH (vpkgwrfhemsys), аланинзамещенный пептид RI α-MSH, (Stearyl) HfRW (820), (Ph(CH2)3CO) HfRW (819), и аналог RI α-MSH 891 [vpkGwr(D-Cha)hsiiss] были синтезированы Genzyme Corporation стандартным твердофазным методом и очищены обратнофазной ВЭЖХ. Скремблированный D-аминокислотный контрольный пептид (ksrsmgvfpeyh) был синтезирован Genzyme Corporation. Посторонний D-аминокислотный контрольный пептид (plykkiikklles) был синтезирован Genzyme Corporation.
Животные. Самки мышей B10.RIII-H2r в возрасте от пяти до шести недель были приобретены у Jackson Laboratories (Bar Harbor, ME).
Самки и самцы мышей C57BL/6 в возрасте от шести до восьми недель были приобретены у Jackson Laboratories (Bar Harbor, ME). Все протоколы для экспериментов с животными были одобрены Комитетом по биоэтике (IACUC) в Genzyme Corporation.
Индукция EAU. Самкам B10.RIII вводили подкожно в два участка (между лопатками и в область таза) суммарно 200 мкг пептида 161-180 белка, связывающего интерфоторецептор (IRBP) (New England Пептид; Fitchburg, MA), соответственно в 2 мг/мл полного адъюванта Фрейнда (CFA; Sigma, St. Louis, MO) с микобактериями туберкулеза H37Ra (Difco, Detroit, MI).
Оценка глаз. Мышам расширяли глаза с помощью одной-двух капель 1% мидриасила (Alcon; Humacao, Puerto Rico) и оставляли в затемненной комнате приблизительно на 5 мин. Мышей удерживали в руках, и сетчатки обоих глаз наблюдали с использованием непрямого офтальмоскопа с линзой 78 диоптрий. Глаза оценивались по воспалению с использованием системы оценки прогрессирования от 0 до 5. Показатель 0: нормальная сетчатка. Показатель 1: сосудистое воспаление непосредственно рядом с оптическим нервом. Показатель 2: <5 воспалительных бляшек, ограниченные одним квадрантом глаза. Показатель 3: >5 воспалительных бляшек более чем в одном квадранте глаза. Показатель 4: воспалительные бляшки прилегают друг к другу. Показатель 5: отслоение сетчатки. У мышей собирали глаза целиком и помещали в PBS. Глаза заливали в среде OCT для фиксации заморозкой. Кусочки размером 5 мкм вырезали через плоскость сосочка зрительного нерва и окрашивали гематоксилином и эозином.
Исследование связывания с рецепторами меланокортина. Профили связывания для α-MSH, RI α-MSH и скремблированного пептида измеряли для меланокортиновых рецепторов: 1, 3, 4 и 5. Связывание производили с использованием реакции конкурентного связывания с (Nle4,D-Phe7) α-MSH (Bachem). Анализ связывания был выполнен в Cerep laboratories (Paris, France) с использованием кодированных образцов.
Связывание пептидов также сопровождалось конкуренцией с125I-NDP-MSH за связывание с рецепторами меланокортина 1, 3, 4 и 5, с использованием препаратов мембран от клеточных линий HEK293. Пептиды смешивали с125I-(Nle4,D-Phe7)-α-MSH (PerkinElmer, Boston MA) в 96-луночных планшетах с коническим дном в связывающем буфере (25 мМ HEPES pH 7, 1,5 мМ CaCl2, 1 мМ MgSO4, 100 мМ NaCl, 0,2% BSA) и смешивали с препаратами мембран от трансфецированных клеточных линий HEK293 (Perkin Elmer, Boston MA), содержащими 1-10 фмоль рецептора, в течение 1 ч при 25ºC. (Nle4,D-Phe7)-NDP-MSH (Bachem) в концентрации 3 мкМ использовали в качестве положительного контроля. Смеси фильтровали через 96-луночные GFC фильтры (Millipore, Billerica, MA), промывали 3 раза связывающим буфером без BSA, сушили и считали.
Измерение цАМФ. Клетки B16-F1 (клеточная линия меланомы), полученные из American Type Culture Collection (Manassas, VA) высаживали в плоскодонные 96-луночные планшеты с плотностью 5×104/лунку и культивировали в течение ночи в среде DMEM (Cambrex, Walkers ville, MD), дополненной 2 мМ глутамином, антибиотиками (100 U/мл пенициллин и 100 U/мл стрептомицин) и 10% эмбриональная телячьей сывороткой, инактивированной нагреванием (Invitrogen, Grand Island, NY). Клетки затем обрабатывали нативным α-MSH (0,01-1000 нг/мл), ретроинверсо α-MSH (0,01-1000 нг/мл), или скремблированным D-аминокислотным контрольным пептидом (0,01-1000 нг/мл) в течение 30 мин. Клетки лизировали и измеряли уровни внутриклеточного цАМФ с помощью набора для иммуноферментного анализа (Amersham Biosciences, Piscataway, NJ). В качестве позитивного контроля использовали форсколин в концентрации 100 мкМ (Sigma, St. Louis, MO).
Индукция EAE и оценка. Самки мышей C57BL/6 были иммунизированы эмульсией пептида MOG35-55 (200 мкг/мышь; New England Пептид; Fitchburg, MA) в полном адъюванте Фрейнда (CFA; Sigma, St. Louis, MO), содержащем 0,6 мг Mycobacterium tuberculosis (Difco; Detroit, MI). Эмульсию вводили путем подкожной инъекции в двух местах в объеме 0,2 мл на мышь. Токсин Bordetella pertussis (PTX; Sigma) в PBS применяли в дозе 400 нг/животное посредством внутрибрюшинного введения и вводили в день 0 и день 3. Мышей ежедневно проверяли на паралитические симптомы EAE. Мыши были оценены по клиническим симптомам с использованием системы оценки прогрессирования от 0 до 5. Показатель 0: нет заболевания; показатель 1: вялый хвост; показатель 2: слабость задних конечностей; показатель 3: паралич задних конечностей; показатель 4: слабость/частичный паралич передних конечностей; показатель 5: смерть. Собирали спинной и головной мозг и заливали парафином для окраски гематоксилином и эозином.
Анализ пролиферации. Спленоциты и клетки лимфоузла мышей C57BL/6 культивировали в 96-луночных планшетах с плотностью 5×105 клеток/лунку. Пептид MOG35-55 или OVA 257-264 был добавлен в лунки в концентрации 25 мкг/мл. Супернатант был собран через 48 часов после стимуляции пептидом MOG. После 3-х дней в культуре клетки обрабатывали [3H]-тимидином в дозе 1 мкCi/лунку в течение 8 часов. Содержание тимидина измеряли по числу импульсов в минуту.
Анализ РНК цитокинов с помощью RT-QPCR. Общая РНК была выделена из тканей с помощью TRIzol (Invitrogen, Carlsbad, CA). 1 мкг общей РНК был обратно транскрибирован и использован в количественной ПЦР с использованием содержания SYBR green с реагентами от Applied Biosystems (Foster City, CA) на приборе ABI 7900. Стандарт кДНК был использован в каждой ПЦР для цитокинов, и полученные концентрации были нормализованы по β-актину мыши. Для получения данных были использованы следующие праймеры: TNFα мыши прямой: ggcaggtctactttggagtcattgc, и обратный: acattcgaggctccagtgaattcgg (1). Заявители использовали IL-10 мыши прямой: tgctatgctgcctgctctta, и обратный: tcatttccgataaggcttgg (2). Последовательности для β-актина мыши были: прямая: gtgggccgctctaggcaccaa, и обратная: ctctttgatgtcacgcacgatttc (3).
CBA анализ. Проточный цитометрический анализ профилей воспалительных цитокинов сыворотки мыши или клеточного супернатанта проводили с помощью Cytometric Bead Array Kit (CBA), произведенного BD Biosciences (San Jose, CA). Образцы обрабатывали по протоколу производителя.
Анализ РНК mMC1R РНК с помощью RT-QPCR. Общую РНК выделяли из тканей с помощью TRIzol (Invitrogen, Carlsbad, CA). 1 мкг общей РНК использовали для проведения обратной транскрипции и использовали для количественной ПЦР с набором Taqman с реагентами от Applied Biosystems (Foster City, CA) на приборе ABI 7900. Стандарт кДНК использовали в каждой ПЦР для mMC1R, и полученные концентрации были нормализованы по эукариотическому эндогенному контролю 18S (VIC/MGB зонд, Applied Biosystems part # 4319413E). Были использованы следующие праймеры для mMC1R: прямой: CTCTGCCTCGTCACTTTCTTTCTA, и обратный: AACATGTGGGCATACAGAATCG, и зонд: CCATGCTGGCACTCA. Праймеры были подобраны с помощью Primer Express 3,0 (Applied Biosystems, Foster City, CA).
Модель LPS. Самцам мышей C57BL/6 вводили внутрибрюшинно LPS (1 мкг/мышь). Через 30 минут мышам вводили дексаметазон (2 мг/кг; Sigma), аналог RI-α-MSH 891 или RI-α-MSH. Через 2 часа после введения LPS у мышей брали кровь на сыворотку. Анализ цитокинов проводили с помощью проточной цитометрии с набором для цитометрического анализа на гранулах (BD Biosciences).
Статистический анализ. Результаты представлены в виде среднего±SD. Каждый эксперимент повторили, по меньшей мере, дважды. Для анализа результатов использовали однофакторный дисперсионный анализ и критерий множественных сравнений Тьюки.
Следующие примеры предлагаются для того, чтобы проиллюстрировать изобретение, но не являются ограничивающими.
Пример 1
Иммуносупрессорная активность на экспериментальной модели аутоиммунного увеита (EAU)
Аутоиммунный увеит представляет собой воспаление глаза, которое ведет к потере зрения и боли. Единственными терапевтическими средствами при увеите в настоящее время являются стероиды и иммуносупрессивные лекарственные средства, которые часто оказывают серьезное токсическое действие на глаз и организм в целом. Таким образом, желательна более безопасная альтернативная терапия.
EAU представляет собой животную модель увеита человека, заболевания, которым страдают 2,3 миллиона человек в Соединенных Штатах. Иммунизация интерфоторецепторным ретиноид-связывающим белком (IRBP) или его пептидными фрагментами или ретинальным S-антигеном (S-Ag) с адъювантом может индуцировать заболевание у предрасположенных к нему линий грызунов. Заболевание содержит инфильтрацию сетчатки глаза воспалительными клетками и повреждение фоторецепторов с естественным выздоровлением без спонтанных рецидивов. Адоптивный перенос увеитогенных T-клеток сингенным реципиентам у грызунов дает основания полагать, что увеит представляет собой органспецифическое аутоиммунное заболевание, опосредованное Т-клетками, подобно многим другим сходным аутоиммунным заболеваниям, таким как рассеянный склероз, диабет типа 1 и ревматоидный артрит.
Микроокружение глаза представляет собой иммунологически привилегированную область, в которой существуют механизмы для поддержания иммуносупрессии на месте для предотвращения локального воспаления. Несколько нейропептидов, которые экспрессируются нейронами ткани глаза, помогают поддерживать иммунную привилегию глаза. Одним из таких нейропептидов является α-MSH, который постоянно экспрессируется в микроокружении глаза в физиологической концентрации 30 пг/мл.
Системное введение нативного α-MSH во время индукции увеита эндотоксином у крыс уменьшает число инфильтрирующих клеток и IL-6, TNF-α, MCP-1, MIP-2 и уровни оксида азота в водянистой влаге дозозависимым образом. У мышиной модели EAU введение α-MSH на пике воспаления сетчатки снижает тяжесть заболевания. α-MSH может индуцировать егуляторные Т-клетки через рецептор меланокортина 5 (MCR-5) на T-клетках и подавлять EAU.
Была исследована эффективность лечения нативным α-MSH на мышиной модели заднего увеита. Увеит был индуцирован у мышей B10.RIII с помощью инъекции IRBP 161-180 и CFA. Заболевание начиналось примерно на десятый день после примирования. На 13-й день, когда достигались глазные показатели 2-3, мышам вводили нативный α-MSH в дозе 200 мкг/мышь, внутривенно, в течение 7 последовательных дней или оставляли их нелеченными. Поскольку профилактическое лечение нацелено, скорее на фазу индукции заболевания, чем на эффекторную фазу, лечение начинали после появления активного воспаления сетчатки. Кроме того, такая стратегия лечения лучше повторяет клиническое применение. Мыши, которых лечили α-MSH, показали снижение средних клинических глазных показателей на всем протяжении семидневного курса лечения по сравнению с нелеченными мышами (фиг.1a). Мыши, которых лечили α-MSH, показали максимальный средний глазной показатель 2,83±0,39 на 13 день по сравнению с группой нелеченных мышей, которые достигли максимального среднего глазного показателя 3,67±0,52 на 15 день.
Пример 2
Сравнение нативного α-MSH с дексаметазоном на экспериментальной модели аутоиммунного увеита B10.RIII
Современные способы терапии увеита содержат кортикостероиды и иммуносупрессирующие средства. Эффективность нативного α-MSH сравнивали с известным кортикостероидным препаратом, дексаметазоном. У мышей B10.RIII индуцировали увеит посредством инъекций IRBP161-180 и CFA. С начала заболевания, когда мыши достигали клинического глазного показателя 1, мышам ежедневно вводили внутрибрюшинно нативный α-MSH (100 мкг/мышь), дексаметазон (0,2 мг/кг) или дексаметазон 2,0 мг/кг. Мышей лечили ежедневно в течение 21 дня. Мыши, которых лечили нативным α-MSH, показали значительное уменьшение средних клинических глазных показателей на всем протяжении курса лечения по сравнению с контрольными мышами, которым вводили PBS (фиг.1B). Данные показывают, что ежедневное внутрибрюшинное введение нативного α-MSH подавляет увеит в большей степени, чем лечение дексаметазоном, как в дозе 0,2 мг/кг, так и в дозе 2,0 мг/кг.
Пример 3
Новый ретро-инверсо аналог α-MSH специфически связывается с MCR-1
Новый стабильный D-аминокислотный пептидный аналог нативного α-MSH (RI α-MSH) был синтезирован и изучен на предмет иммунномодуляторных свойств in vitro и на экспериментальной модели аутоиммунного увеита (EAU). Исследования связывания указывают, что в отличие от нативного α-MSH, RI α-MSH специфически связывается с противовоспалительным рецептором α-MSH (MCR-1), но не с другими рецепторами α-MSH (MCR-3, 4 или 5).
Было изучено связывание аналога RI α-MSH с меланокортиновыми рецепторами (MCR). Был использован анализ конкурентного связывания с применением панели MCR, содержащей MCR1, 3, 4 и 5. Связывание пептидов сопровождалось конкуренцией с125I-NDP-MSH, как описано ранее. Нативный α-MSH связался с MCR 1, 3, 4 и 5. Однако было обнаружено, что, в отличие от нативного α-MSH, RI α-MSH связался только с MCR-1, который регулирует воспалительные ответы (таблица 1). Скремблированный D-аминокислотный контрольный пептид не связался ни с каким из меланокортиновых рецепторов. Разработка аналога α-MSH со специфическим связыванием с MCR-1, и исключение связывания с MCR-3 и MCR-4, может уменьшить возможные побочные эффекты, которые нативный α-MSH создает посредством связывания с этими рецепторами.
Связывание пептидов сопровождалось конкуренцией с125I-NDP-MSH за связывание с рецепторами меланокортина 1, 3, 4 и 5 с использованием мембранных препаратов клеточных линий HEK293. Как показано на фиг.18, RI α-MSH показал высокую селективность к MC1R, с величинами Ki более чем 30 мкМ для MCR 3, 4 и 5, в то время как α-MSH показал значительное связывание со всеми четырьмя рецепторами, с менее чем 100-кратной селективностью к MC1R относительно MC3R.
Пример 4
Лечение ретроинверсо аналогом α-MSH улучшает заболевание при EAU
Иммуномодулирующие эффекты нового ретроинверсо пептидного аналога α-MSH исследовали на мышиной модели экспериментального аутоиммунного увеита (EAU) и сравнивали результаты с нативным пептидом α-MSH. Систематическая доставка RI α-MSH в начале заболевания или в течение поздней стадии заболевания кардинально и воспроизводимо улучшает увеит. Кроме того, лечение новым пептидным аналогом RI α-MSH подавляет увеит с аналогичной величиной, что и нативный пептид α-MSH. Эти данные указывают на то, что новый аналог RI α-MSH демонстрирует противовоспалительную активность и имеет терапевтическую пользу при увеите и других аутоиммунных заболеваниях и воспалении.
EAU индуцировали у самок мышей B10.RIII с помощью IRBP 161-180. Была исследована эффективность RI α-MSH и нативного α-MSH скорее в качестве лекарственного препарата, чем профилактического лечения. Мышей лечили ежедневно внутривенными инъекциями RI α-MSH (l00 мкг/мышь), нативного α-MSH или PBS, начиная с умеренной стадии увеита (глазные показатели 2). Как видно на фиг.2A, контрольные мыши, получавшие PBS, достигли максимальных глазных показателей на 16-й день, со средним глазным показателем 2,75±0,68. Однако мыши, которых лечили ежедневно аналогом RI α-MSH или нативным α-MSH, показали достоверное снижение средних клинических глазных показателей на всем протяжении курса лечения по сравнению с мышами, получавшими PBS. На 16-й день, через четыре дня после начала лечения, 5 из 8 мышей в группе, получавшей PBS, имели максимальный глазной показатель 3 или более, в то время как только 1 из 8 мышей, получавших нативный α-MSH и 0 из 8 мышей, получавших RI α-MSH, имели максимальный глазной показатель 3 или более (фиг.2B).
Пример 5
Снимки сетчатки и гистологическое исследование пациентов, которых лечили RI α-MSH, показывает супрессию заболевания на моделях EAU
Снимки сетчатки мышей, которых, начиная с поздней стадии увеита, лечили ежедневно RI α-MSH или нативным α-MSH, показали супрессию заболевания по сравнению с контрольными мышами, получавшими PBS. Фундоскопические снимки получали на 13 день после начала лечения (фиг.3). Заболевание у группы, получавшей PBS, стабилизировалось или прогрессировало со средней величиной глазного показателя равной 3, тяжелым васкулитом и воспалительными бляшками на всем протяжении глаза (фиг.3A). Однако заболевание быстро разрешалось, как у мышей, которых лечили RI α-MSH, так и у мышей, которых лечили нативным пептидом α-MSH. Снимки сетчатки демонстрируют глазные показатели равные 1, с воспалением только у оптического нерва (фиг.3B и 3C). Максимальные глазные показатели для мышей по отдельности в каждой группе показаны на фиг.3D. В группе, которую лечили PBS, 7 из 11 мышей имели глазные показатели 3 или более. Однако только у двух 2 из 11 мышей, которых лечили нативным α-MSH и RI α-MSH, глазные показатели были 3 или более.
Гистологическое исследование глаз на 10 день после начала ежедневного лечения показало, что как RI α-MSH, так и нативный α-MSH уменьшают патологию глаза (фиг.4). Мыши, которых лечили нативным α-MSH и RI α-MSH, демонстрируют нормальную архитектуру сетчатки с легким воспалением в области оптического нерва (фиг.4B и 4C). Напротив, мыши, которых лечили PBS, демонстрируют глазное воспаление и повреждение тканей. Наблюдалось воспаление в области сетчатки и оптического нерва с повреждением фоторецепторов (фиг.4A).
Пример 6
Сравнение внутрибрюшинных введений ретроинверсо α-MSH со скремблированным контрольным пептидом при EAU
Увеит индуцировали у мышей B10.RIII путем инъекций IRBP161-180 и CFA. Заявители оценивали лечение RI α-MSH, начатое на поздней стадии заболевания (глазные показатели 2-3), проводимое посредством ежедневных внутрибрюшинных инъекций, и сравнивали эффективность с контрольным скремблированным D-аминокислотным пептидом. Мышей лечили ежедневно, внутрибрюшинно, в течение 13 дней RI α-MSH или скремблированным пептидом в дозе 100 мкг/мышь. Мыши, которых лечили RI α-MSH, показали достоверное снижение (p<0,04) средних клинических глазных показателей на 15, 21 и 23 дни течения заболевания по сравнению с контрольными мышами, получавшими скремблированный пептид (фиг.5). Индивидуальные максимальные глазные показатели на 23 день после индукции заболевания выявили 75% мышей с показателями ≤1 в группе RI α-MSH, по сравнению с отсутствием мышей с показателями ≤1 в группе контрольного скремблированного пептида. Индивидуальный вес мышей записывали в четырех временных точках на всем протяжении заболевания. Все мыши в обеих группах, леченные RI α-MSH и скремблированным пептидом, сохранили свой вес или показали нормальное увеличение массы (данные не показаны). Ежедневные внутрибрюшинные дозы пептидов не привели к потере веса. Кроме того, внутрибрюшинный путь введения нативного α-MSH или RI α-MSH в дозе 100 мкг/мышь являлся эффективным при увеите, когда лечение назначали в начале заболевания, когда мыши имели клинический глазной показатель, равный 1 (данные не показаны). Таким образом, внутрибрюшинный путь введения RI α-MSH показал значительное снижение показателей заболевания по сравнению с скремблированным контрольным пептидом.
Пример 7
Введение RI α-MSH в различных дозах при модельном EAU
Заявители также изучили эффективность и оптимальную дозировку при лечении ретроинверсо α-MSH на поздней стадии тяжелого увеита. Мышам B10.RIII делали инъекции IRBP161-180 и CFA для индукции увеита. Мышей лечили ежедневно, внутрибрюшинно, PBS, контрольным скремблированным пептидом (100 мкг/мышь), или RI α-MSH в дозе 100 мкг, 10 мкг или 3 мкг/мышь. Лечение начинали на пике заболевания, глазной показатель у мышей был равен 4, что содержало воспалительные бляшки на всем протяжении заднего сегмента глаза и возможные геморрагии. У групп с PBS и контрольным скремблированным пептидом заболевание осталось тяжелым (фиг.6). Напротив, лечение RI α-MSH в дозе 100 мкг или 10 мкг/мышь быстро улучшало увеит (p≤0,05). Кроме того, дозы в 10 и 100 мкг/мышь были одинаково эффективными. Доза RI α-MSH в 3 мкг/мышь не снижала и не замедляла заболевание.
Пример 8
Действие ретроинверсо α-MSH на уровни цАМФ
Были исследованы уровни цАМФ в клетках меланомы B16-F1 после лечения нативным α-MSH, RI α-MSH, или скремблированным контрольным пептидом. Клеточная линия меланомы B16-F1 экспрессирует большое количество рецепторов MCRl (3000-4000 рецепторов/клетку) по сравнению с макрофагальными клеточными линиями, которые экспрессируют только 100-200 рецепторов/клетку. Таким образом, для изучения эффекта лечения RI α-MSH заявители выбрали клеточную линию меланомы. Клетки мышиной меланомы B16-F1 обрабатывали нативным α-MSH, RI α-MSH, или контрольным скремблированным пептидом в концентрации 1 пг/мл-1 мкг/мл. Через 30 минут клетки лизировали и измеряли внутриклеточный цАМФ с помощью ферментного иммуноанализа. Контрольная обработка клеток форсколином, который обычно используется для повышения уровня цАМФ, в дозе 100 мкМ показала увеличение цАМФ (3455,39±406,6 SD). Уровни цАМФ были достоверно повышены в зависимости от дозы в клетках, обработанных нативным α-MSH по сравнению с необработанными клетками (фиг.7A). Аналог RI α-MSH также достоверно повышал уровни цАМФ в зависимости от дозы по сравнению с необработанными клетками или контролем со скремблированным пептидом. Однако была необходима более высокая концентрация RI α-MSH (100 нг/мл), для того чтобы повысить цАМФ до уровней, сравнимых с нативным α-MSH, который увеличивал цАМФ при концентрации 10 пг/мл. Это различие в концентрации пептидов, необходимой для увеличения уровней цАМФ, может быть результатом аффинности связывания с рецептором MCR1.
Пример 9
Варианты последовательностей и действие на уровни цАМФ и связывание с MClR
MSH связывается с MC1R, MC3R, MC4R и MC5R, при этом MC1R является одной из наиболее желаемых мишеней при иммунных заболеваниях. Ретроинверсо MSH (RI-MSH) создавали для того, чтобы повысить стабильность в плазме (фиг.17) и селективность к MC1R, но его сродство к MC1R в 11 раз ниже, чем таковое у нативного пептида MSH (таблица 1). Для восстановления сродства к MC1R, заявители ввели в RI-MSH модификации, о которых было известно, что они улучшают сродство MSH к MC1R. Из трех замен в N-концевой последовательности SYSME - жирный ацил, фенилмасляная кислота и последовательность SSIIS, которые увеличивают сродство MSH к MC1R, только одна, последняя, привела к значительным улучшениям для RI-MSH. D-аланиновое сканирование аналогов RI-MSH выявило сходную связь структурной активности, как и для аланинового сканирования аналогов MSH, но стереохимия инверсионного сканирования основного MC1R-связывающего региона из 4-х остатков выявила значительные различия между MSH и RI-MSH. Кроме того, циклогексилаланиновая замена ключевого остатка фенилаланина улучшает связывание RI-MSH, но не MSH, с MC1R. Комбинирование циклогексигексилаланиновой и SSIIS замен привело к полному восстановлению сродства MSH к MC1R, в то время как сохранение ретроинверсо конфигурации является критичным для улучшенной стабильности и высокой MC1R-селективности RI-MSH.
Набор аналогов ретроинверсо MSH (RI-MSH), полученных аланиновым сканированием (пептиды 804-816), был получен и проверен на индукцию цАМФ в клетках меланомы мыши B16/F1 и меланомы человека M624. Подгруппа, составленная на основании результатов по цАМФ, затем была протестирована на связывание с MC1R. Наблюдаемые величины Ki для связывания с MC1R представлены в таблице 1.
Клетки мышиной меланомы B16-F1 обрабатывали нативным α-MSH, RI α-MSH, скремблированным контрольным пептидом, KPV или аланинзамещенными пептидами RI α-MSH в концентрации 1 мкг/мл (фиг.7B). Контрольное лечение клеток форсколином (100 мкМ) показало увеличение цАМФ (3294,82±54,53). Уровни цАМФ были значительно повышены в клетках, которые обрабатывали нативным α-MSH и RI α-MSH, по сравнению с необработанными клетками и клетками, обработанными скремблированным пептидом или KPV. Аланинзамещенные пептиды, обозначенные 810, 811 и 812, не показали увеличения активности цАМФ; наблюдаемые уровни цАМФ были равны таковым у необработанных клеток и клеток, обработанных скремблированным пептидом или KPV. Предполагают, что пептиды, обозначенные 810, 811 и 812 с аланиновыми заменами в последовательности центрального основного тетрапептида (D-Trp D-Arg D-Phe D-His область; AA 5-8), вовлечены в связывание нативного α-MSH с меланокортиновым рецептором и его биологической активностью. Аланиновые замены в N-концевой или C-концевой областях пептида RI α-MSH не влияют на накопление цАМФ в клетках меланомы. Аланиновые замены в метионине и гистидине (807 и 809) также показали снижение накопления цАМФ, хотя и не настолько значительное, как наблюдаемые в последовательности основного тетрапептида (810, 811 или 812). Замены в основном тетрапептиде (wrfh) и, в меньшей степени, метионина 4 в RI-MSH критичны для связывания с MC1R.
Другие перечисленные способы для усиления сродства MSH к MC1R продемонстрированы с использованием селекции, основанной на фаговом дисплее вариантов последовательностей MSH. Последовательность, высокоселективная к MC1R (MS05), была обнаружена путем рекомбинации пептида, выбранного из фагового дисплея и части родительской последовательности MSH, которая давала субнаномолярное сродство к MC1R. Неожиданно, ретроинверсо вариант этой последовательности показал большее сродство к MC1R (1 нМ, таблица 1, пептид 886), чем RI-MSH (4 нМ), несмотря на то, что стало известно, что MS05 имеет немного более низкое сродство к MC1R, чем MSH (Ki 0,865 нМ по сравнению с 0,557 нМ для MSH).
Замена ряда неприродных аминокислотных остатков, привносящих небольшие различия в заряде и структуре, в каждой из позиций в RI-MSH показала, что MC1R допускает только высококонсервативные замены. Все замены, кроме трех, показали меньшее или равноценное связывание. Две замены приводили к значительному увеличению сродства к MC1R: замена D-фенилаланина D-циклогексилаланином (D-Cha, пептид 869; 2,2 нМ Ki) и замена D-метионина D-бутионином (пептид 878, 2,3 нМ Ki). Была произведена замена L-циклогексилаланином, и было обнаружено, что она незначительно ингибирует связывание с MC1R (пептид 892, Ki 0,51 нМ по сравнению с 0,41 нМ для MSH). Неожиданно, комбинация бутиониновой и циклогексиаланиновой замен не привела к дальнейшему значительному увеличению сродства к MC1R (пептид 890, 1,9 нМ Ki). Однако комбинация замен, содержащая замену в ретроинверсо N-концевой последовательности MS05 (siiss) вместо C-концевой последовательности (emsys) и D-циклогексилаланин вместо D-Phe в RI-MSH, приводит к большему усилению связывания, чем каждая из замен по отдельности, указывая на синергичность эффектов. Полученный пептид (891) демонстрирует Ki для MC1R, неотличимую от MSH. Хотя эта и другие замены также создают увеличение сродства к остальным рецепторам MCR, значения Ki оставались в микромолярном диапазоне, указывая на то, что селективность RI-MSH была в значительной степени сохранена. Замены изображены на фиг.20. Репрезентативный анализ конкурентного связывания представлен на фиг.21. Смотрите таблицу 1 для полученных величин Ki.
Пример 10
Лечение RI α-MSH при EAE снижает клинические показатели заболевания
Оценивали эффект введения нативного α-MSH и аналога RI α-MSH на мышиной модели хронического прогрессирующего EAE. Самки мышей C57BL/6 были иммунизированы 200 мкг пептида MOG 35-55 в эмульсии с CFA. Токсин коклюша вводили в день 0 и день 2. Мышей осматривали ежедневно на признаки клинических симптомов и потерю веса. Мышей оценивали на симптомы паралича, начиная с восьмого дня и присваивали показатели по системе оценок от 0 до 5. Появление клинических симптомов паралича обнаруживалось приблизительно на 9-11 день у большинства мышей (фиг.8A). Ежедневное внутрибрюшинное (i.p.) лечение пептидами α-MSH или RI α-MSH в дозе 100 мкг/мышь или контрольным PBS начинали на 10 день. Мыши, которых лечили 100 мкг RI α-MSH, продемонстрировали достоверное снижение среднего клинического показателя заболевания по сравнению с контролем с PBS (фиг.8A). Достоверность p≤0,05 была достигнута на 14-22 дни для RI α-MSH, по сравнению с контролем с PBS. Однако лечение нативным пептидом α-MSH не оказало воздействия на индукцию или прогрессирование заболевания. Максимальный процент возникновения заболевания в группе мышей, получавших RI α-MSH (20%) был также снижен по сравнению с группой с PBS (80%) или группой, получавшей нативный α-MSH (75%).
Пример 11
Вариации дозировки RI α-MSH при EAE
Терапевтический эффект RI α-MSH проявился при дозе 100 мкг/мышь. Лечение пептидами α-MSH или RI α-MSH в дозе 100 мкг/мышь и 30 мкг/мышь исследовали на мышиной модели EAE, индуцированного MOG. На 10 день после иммунизации MOG начинали ежедневное внутрибрюшинное лечение. Ежедневное лечение дексаметазоном (2 мг/кг) добавили в качестве контрольной терапии. Мыши, которых лечили 100 мкг RI α-MSH, неоднократно показали снижение среднего клинического показателя заболевания по сравнению с контролем с PBS (фиг.9). Однако доза RI α-MSH 30 мкг/мышь не имела достоверного влияния на снижение средних клинических показателей. Лечение дексаметазоном также показало снижение показателей заболевания на всем протяжении течения заболевания. Хотя мыши, которых лечили нативным α-MSH, имели более низкие средние клинические показатели, чем мыши, которых лечили PBS, такие мыши не показали сходной эффективности, как мыши, которых лечили RI α-MSH или дексаметазоном. Процент возникновения заболевания у группы, которую лечили RI α-MSH, достиг максимума в 35%, дексаметазоном - 40% по сравнению с мышами, получавшими PBS, которые показали максимум возникновения заболевания - 75% (фиг.9). Эти данные указывают, что лечение пептидным аналогом RI α-MSH пептид при EAE достоверно снижает средние клинические показатели заболевания и частоту возникновения заболевания.
Пример 12
Гистология ЦНС
Спинной мозг собирали на 24 день после индукции заболевания у мышей, которых лечили PBS и RI α-MSH. Для оценки степени воспаления и количества бляшек срезы спинного мозга были окрашены гематоксилином и эозином. Патология EAE проявилась в очаговых областях инфильтрации воспалительными клетками и демиелинизации. Гистопатологическое исследование спинного мозга продемонстрировало эффективность лечения RI α-MSH на модели MOG-индуцированного EAE. Срезы от типичных мышей со средними клиническими показателями из каждой группы представлены на фиг.10a-10d. Данные выявили обширные воспалительные инфильтраты у группы мышей, получавшей PBS, по сравнению с мышами, получавшими RI α-MSH, у которых отсутствовала очаговая область воспаления.
Пример 13
Измерение TNF-α и IL-10 в селезенке во время прогрессирования заболевания
Был исследован механизм действия эффектов лечения RI α-MSH при EAE. Известно, что нативный α-MSH воздействует на моноциты/макрофаги за счет снижения уровня TNF-α и повышения IL-10. Уровни мРНК TNF-α и IL-10 в селезенке мышей, примированных MOG, которых лечили RI α-MSH или нативным α-MSH, оценивали с помощью количественной ПЦР. Мыши были иммунизированы MOG p35-55, ежедневное лечение RI α-MSH или нативным α-MSH началось на 10 день. Образцы селезенки брали у мышей на 1, 4 и 7 дни после начала ежедневного лечения. Данные показывают, что лечение RI α-MSH и α-MSH достоверно понижает (p≤0,001) TNF-α по сравнению с контролем с PBS после 7 дней ежедневного лечения (фиг.11e). Однако не было никаких достоверных изменений уровня TNF-α в предыдущих временных точках (день 1 и день 4; фиг.11a и 11с). Уровни мРНК IL-10 были также снижены к седьмому дню, как в RI α-MSH, так и в α-MSH группах по сравнению с контролем с PBS (фиг.11f). Однако никаких различий в уровнях мРНК IL-10 не было обнаружено в более ранних временных точках (фиг.11b и 11d). Хотя лечение нативным α-MSH не уменьшает средние клинические показатели заболевания или процент возникновения заболевания, в этом исследовании лечение α-MSH уменьшало уровни мРНК в селезенке, как TNF-α, так и IL-10 по сравнению с контролем с PBS к 17 дню прогрессирования заболевания. Аналог RI α-MSH способен снижать уровни мРНК TNF-α в селезенке после ежедневного лечения.
Пример 14
Влияние RI α-MSH на вторичный иммунный ответ к пептиду MOG
Были исследованы эффекты лечения мышей α-MSH или RI α-MSH in vivo на вторичный иммунный ответ к пептиду MOG 35-55. Мыши были примированы пептидом MOG35-55, и со 2 по 8 день мышей лечили PBS, или 100 мкг α-MSH, или 100 мкг RI α-MSH. На 9 день селезенку и дренирующие лимфоузлы собрали и проанализировали на вторичные иммунные ответы к пептиду MOG35-55 in vitro методом инкорпорации [3H]-тимидина. Данные показывают значительное снижение пролиферативного ответа клеток селезенки на пептид MOG35-55 в группе мышей, которых лечили α-MSH по сравнению с группой, получавшей PBS (фиг.12a). Группа, которую лечили RI α-MSH, показала слабое уменьшение вторичного иммунного ответа к пептиду MOG35-55. Однако вторичный иммунный ответ к пептиду MOG в клеточной популяции лимфоузла не показал достоверного различия в степени ответа между группами, которых лечили PBS, α-MSH и RI α-MSH (фиг.12b).
Был собран клеточный супернатант из клеток селезенки, которые были стимулированы пептидом MOG35-55 in vitro от каждой из леченных in vivo групп (наивные, PBS, α-MSH, RI α-MSH). Уровни цитокинов измеряли с использованием цитометрического анализа на гранулах методом проточной цитометрии. Данные показывают снижение уровней TNF-α, IFNγ, IL-6 и MCP-1, как в группе, которую лечили α-MSH, так и в группе с RI α-MSH по сравнению с группой, получавшей PBS (фиг.12c и 12d). Уровни цитокинов в супернатанте клеток селезенки в обеих, α-MSH и RI α-MSH, группах были сходны с уровнями цитокинов из клеток селезенки непримированных мышей.
Таким образом, лечение пептидом RI α-MSH оказывало влияние на цитокиновый вторичный иммунный ответ к пептиду MOG, но не влияло на Т-клеточный пролиферативный ответ.
Пример 15
Эффекты лечения мышей α-MSH или RI α-MSH в течение примирующей фазы EAE
Мыши были иммунизированы пептидом MOG 35-55, и со 2 по 8 день их лечили ежедневно контрольным PBS, α-MSH или RI α-MSH. Селезенки от отдельных мышей собирали на 9 день и оценивали экспрессию мРНК цитокинов с использованием ПЦР в реальном времени. Фиг.13 показывает уровни экспрессии мРНК TNF-α и IL-10 в селезенке мышей, которых лечили PBS, α-MSH или RI α-MSH в примирующей фазе заболевания. Наивные мыши, которые не были иммунизированы пептидом MOG, были использованы для оценки базовых уровней мРНК TNF-α и IL-10. Лечение α-MSH или RI α-MSH снижало уровни мРНК IL-10 и TNF-α по сравнению с контролем с PBS.
Образцы сыворотки крови были также собраны на 9 день исследования для оценки уровней цитокинов: TNF-α, MCP-1, IL-12, IL-10 и IL-6 (фиг.13c и 13d). Данные показывают снижение MCP-1 и IL-6 в обеих группах, с α-MSH и RI α-MSH, по сравнению с контрольной группой, получавшей PBS. MCP-1 был значительно снижен (p<0,01) в группе, которую лечили RI α-MSH по сравнению с контролем с PBS. Дополнительно, группа с RI α-MSH продемонстрировала снижение TNF-α и IL-12 по сравнению с группой с PBS. Не было никаких различий в сывороточных уровнях IL-10 между тремя исследованными группами.
Пример 16
RI α-MSH не влияет на макрофагальные маркеры
Исследовали уровни поверхностной экспрессии CD86, CD40 и CD14 на селезеночных макрофагах CD11b+ и F4/80+ методом проточной цитометрии у мышей, иммунизированных MOG 35-55, через 7 дней ежедневного приема доз α-MSH или RI α-MSH. Данные показывают отсутствие различий в уровнях экспрессии CD86, CD40 или CD14 в клеточной популяции селезеночных макрофагов CD11b+ или F4/80+ у мышей, которым вводили α-MSH или RI α-MSH (фиг.14). Похожие результаты были получены для моноцитов крови, которые были исследованы на уровни экспрессии CD86, CD40 и CD14 (данные не показаны).
Пример 17
Результаты на мышиной модели воспаления, индуцированного LPS
Сообщалось, что нативный α-MSH подавляет воспаление за счет снижения выработки TNF-α посредством стимуляции рецепторов меланокортина (MCR), в частности, рецептора меланокортина 1. LPS стимулирует воспалительные медиаторы, такие как TNF-α, MCP-1, IL-6 и IFNγ, и используется в качестве модели острого воспаления. Было исследовано, может ли аналог α-MSH подавлять воспалительные цитокины на мышиной модели воспаления, индуцированного LPS. Были определены уровни экспрессии MC1R в селезенке и перитонеальных макрофагах после введения LPS. Мышам C57BL/6 делали инъекции LPS, внутрибрюшинно, и забирали селезенку и перитонеальные макрофаги для анализа в нескольких временных точках после инъекции LPS. Данные не показывают заметных различий в уровнях экспрессии мРНК MC1R в любой из временных точек в перитонеальных макрофагах (фиг.15a). Однако введение LPS все же увеличивало уровни мРНК MC1R по сравнению с наивными мышами, которых не стимулировали LPS. В селезенке уровни мРНК MC1R были увеличены 30 минут спустя инъекции LPS и затем снизились через час после инъекции (фиг.15b). LPS сходно увеличивает уровни мРНК MC1R в селезенке по сравнению с базовыми уровнями у наивных мышей.
Данные, демонстрирующие экспрессию мРНК MClR в селезенке и перитонеальных макрофагах, указывают, что эти 30 минут после стимуляции LPS могли бы быть оптимальной временной точкой для лечения α-MSH или аналогом RI α-MSH. Мышам C57BL/6 делали инъекции LPS, внутрибрюшинно, и через 30 минут вводили внутрибрюшинно нативный α-MSH или аналог RI α-MSH 891 в дозах в диапазоне от 5 мг/кг до 0,156 мг/кг. Аналог RI α-MSH 891 представляет собой аналог RI α-MSH, в котором d-Cha и D-аминокислоты hsiiss были добавлены к последовательности основного пептида. Показано, что аналог RI α-MSH 891 имеет повышенную аффинность связывания с мышиным MC1R и повышенный потенциал для стимуляции цАМФ (данные не показаны). Результаты показывают значительное снижение уровней TNF-α и IL-10 в группе, которую лечили нативным α-MSH по сравнению с контролем с PBS (фиг.16a и 16c). Лечение нативным α-MSH не влияло на MCP-1 (фиг.16b). Лечение мышей, стимулированных LPS, аналогом RI α-MSH 891, показало сходные результаты, со значительным снижением TNF-α и IL-10 и отсутствием действия на уровни MCP-1 по сравнению с контролем с инертным веществом (фиг.16d-16f). Более низкие дозы как нативного α-MSH, так и аналога RI α-MSH 891 показали наибольшую супрессию воспалительных цитокинов. Положительный контроль с дексаметазоном показал постоянную супрессию уровней TNF-α и MCP-1.
Пример 18
Стабильность пептидов в плазме
Время полужизни нативного α-MSH в сыворотке оценивают приблизительно в 10 минут. Несмотря на такое ограниченное время полужизни, пептид все же способен вызывать мощную противовоспалительную активность. Однако необходимы более стабильные аналоги α-MSH для того, чтобы увеличить силу противовоспалительных действий и в дальнейшем разработать терапевтическое средство. D-аминокислотный аналог нативного α-MSH (обозначаемый ретроинверсо α-MSH или RI α-MSH) был синтезирован и является более стабильным, чем нативный α-MSH. D-аминокислотная форма пептидов более устойчива к протеолизу и метаболизируется с меньшей скоростью, чем L-аминокислотная форма пептидов.
Стабильность RI-α-MSH определяли путем инкубации в плазме или PBS in vitro при 37ºC в течение 24 часов. Отбирали аликвоты и осаждали пептиды двумя объемами ацетонитрила. α-MSH был использован в качестве контроля, и брадикинин - в качестве внутреннего стандарта. После центрифугирования образцы были заморожены при -80ºC до LC/MS/MS (MRM) анализа с использованием разделения на колонках C18 и метода позитивной ионизации электрораспылением. Как показано на фиг.17a, время полужизни MSH в плазме - приблизительно 3 часа, но он был стабилен в PBS. Однако RI-α-MSH был стабилен, как в PBS, так и в плазме, не показывая заметной деградации спустя 24 часа.
Фармакокинетические (PK) профили для α-MSH и RI-α-MSH показывают, что RI-α-MSH имеет более долгое время полужизни в сыворотке, чем нативный α-MSH (фиг.17b). Мыши, получившие единичную дозу 100 мкг RI-α-MSH или α-MSH, имели доступные для измерения сывороточные уровни RI-α-MSH спустя 24 часа, но не было выявляемых уровней α-MSH через 120 минут после введения (n=5).
Пример 19
Связывающие эффекты ретроинверсо пептидов
Основного тетрапептида MSH, содержащего D-Phe (HfRW), достаточно для получения значительного связывания с MC1R (20-50 нМ Ki). Однако наблюдали незначительное связывание при использовании ретроинверсо версии того же тетрапептида (wrFh, 883, Ki>30 мкМ, таблица 1), что указывает на то, что такой ретроинверсо пептид не может полностью контактировать с рецептором тем же способом, что и HfRW. Добавление жирных ацильных групп к N-концу HfRW селективно улучшает его связывание с MC1R, давая аффинность, сравнимую с MSH. Это наблюдали для стеарил-HfRW пептида и MC1R (1,3 нМ, пептид 820). Добавление диаминогексанстеариловой группы к ретроинверсо wrFh (пептид 882) улучшает связывание с MC1R (120 нМ, >250 раз) в той же степени, как и добавление стеариновой кислоты к N-концу HfRW (80-кратно), но не достигает сродства, наблюдаемого для RI-MSH (4,1 нМ), снова указывая на то, что в связывании RI-MSH с MC1R играют большую роль другие остатки RI-MSH, чем для MSH.
Пример 20
Стереохимия влияет на связывание ретроинверсо пептидов
Стереохимия остатков в основном ретроинверсо тетрапептиде имеет влияние на связывание с MC1R, существенно отличающееся от L-формы MSH. Как показано на фиг.19, инверсия остатков основного тетрапептида в RI-MSH в L-форму (пептиды 884, 893-895) снижает сродство пептида к MC1R. Поразительно и неожиданно, инверсия D-Phe в L-Phe в RI последовательности приводит к 20-кратному снижению связывания, в то время как сообщают, что соответствующая замена в HFRW (L-Phe на D-Phe) улучшает связывание в 400 раз. Было обнаружено для остальных остатков,что стереохимическая инверсия имеет меньший эффект для RI-MSH, чем для основного пептида HfRW, снова указывая на то, что относительная важность каждого остатка в основном тетрапептиде ниже в RI-MSH, чем в тетрапептиде HfRW.
Пример 21
Эффекты блокирования концевых групп ретроинверсо MSH
Другой возможный путь для достижения более высокого сродства к MC1R основан на наблюдении, что блокирование концевых групп пептида HfRW фенилмасляной кислотой селективно и сильно повышает сродство HfRW к MC1R (Ki=6 пМ). Однако не наблюдали никакого повышения аффинности укороченного RI-MSH, несущего C-концевой фенилпропиламид (пептид 847), с Ki=150 нМ, которую наблюдали как для пептида, укороченного на остаток гистидина (пептид 847int), так и для его аминопропилфенилового аддукта (пептид 847, таблица 1), что демонстрирует, что для RI-MSH, связывание изменяется таким образом, который не позволяет одновременного взаимодействия тетрапептидной последовательности и предполагаемого ароматического сайта взаимодействия рядом с гистидин-связывающим элементом в MC1R.
Пример 22
Синтез токсинового конъюгата ретроинверсо MSH
Модифицированную версию пептида 891 (таблица 1), в которой к N-концу добавлен цистеин, получали посредством Fmoc-химии. Конъюгат монометилауристатина E (MMAE), содержащий чувствительный к протеазам валин-цитруллиновый линкер с малеимидо-капроиловой частью для связывания с тиолами, синтезировали согласно Doronina et al. (2003) Nature Biotechnol. 21:778-784, включенной в настоящий документ в качестве ссылки. Пептид и конъюгат MMAE инкубировали в растворе 25 мМ фосфата натрия, 2 мМ ЭДТА pH 7 в течение 14 часов при 25ºC и очищали продукт с помощью C18 обратнофазовой ВЭЖХ (ОФ-ВЭЖХ).
Пример 23
Действие аналогов RI α-MSH на цАМФ в клеточных линиях меланомы мыши
Как нативный α-MSH, так и RI α-MSH повышают уровни цАМФ в клетках меланомы мыши. Были созданы пептидные аналоги RI α-MSH для того, чтобы определить, будут ли модификации в последовательности основного пептида RI α-MSH повышать уровни цАМФ по сравнению с пептидом RI α-MSH. Дозозависимый эксперимент проводили для созданных аналогов RI α-MSH с анализом цАМФ с использованием клеток мышиной меланомы B16-F1.
Клеточные культуры in vitro: Клетки меланомы мыши B16-F1 культивировали в 96-луночных планшетах с плотностью 5×104 клеток/лунку в течение ночи в среде с L-глутамином, пенициллином/стрептавидином и FBS. Удаляли среду и добавляли к клеткам новую среду с IBMX на 1 час. Клетки затем обрабатывали RI α-MSH или пептидными аналогами RI α-MSH (890, 891, 892, 893, 894 или 895). Через 30 минут клетки лизировали с использованием набора для анализа цАМФ, и супернатант использовали для анализа. В качестве положительных контролей использовали форсколин в концентрации 10 мкМ.
Уровни цАМФ: Внутриклеточный цАМФ измеряли с помощью cAMP competition Assay Kit (Amersham Biosciences). Все образцы клеточных лизатов для анализа были разведены 1:100.
Пептиды
869 vpkGwr(d-Cha)hemsys
872 vpkGwrfremsys
880 vpkGwrFhsiiss
878 vpkGwrfhe(d-бутионин)sys
886 RI-MS05 vpkgwrfhsiiss
890 vpkGwr(d-Cha)he(d-бутионин)sys
891 vpkGwr(d-Cha)hsiiss
892 SYSMEH(Cha)RWGKPV
893 vpkGWrfhemsys
894 vpkGwRfhemsys
895 vpkGwrfHemsys
Результаты: Клетки меланомы мыши B16-F1 обрабатывали RI α-MSH или аналогами RI α-MSH (869, 872, 880, 878, 886 и 890-895) в диапазоне концентраций от 10-4 до 10-11 M. Уровни цАМФ от клеток, обработанных RI α-MSH или аналогами RI α-MSH. Большинство аналогов RI α-MSH показали повышенные значения EC50 по сравнению с RI α-MSH, за исключением аналога 894. Аналоги 891, 892 и 886 показали наибольшее увеличение значений EC50 при анализе цАМФ по сравнению с пептидом RI α-MSH. Таким образом, аналоги RI α-MSH показали улучшенный дозозависимый эффект и величины EC50 по сравнению с пептидом RI α-MSH. Смотрите графические данные на фиг.22 и данные EC50 в таблице 2.
Реферат
Изобретение относится к стабильным пептидным аналогам альфа-меланоцитстимулирующего гормона (α-MSH), которые имеют сродство к рецептору меланокортина 1 (MC1R), фармацевтическим препаратам пептидных аналогов α-MSH, а также к способам применения этих аналогов для лечения заболеваний в медицине и в ветеринарии, связанных с MC1R. 11 н. и 5 з.п. ф-лы, 22 ил., 2 табл., 23 пр.
Формула
His Xaa1 Arg Trp (SEQ ID NO:1) или D-Trp D-Arg Xaa2 D-His (SEQ ID NO:2),
где Xaa1 представляет собой D-Cha или Cha, и
Xaa1 представляет собой D-Cha;
или его фармацевтически приемлемая соль,
и, необязательно, где указанный полипептид содержит C-концевой полипептид с последовательностью: D-Ser D-Ile D-Ile D-Ser D-Ser (SEQ ID NO:3).
Xaa1 Xaa2 Xaa3 Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 Xaa11 Xaa12 Xaa13,
где Xaa1 представляет собой D-Val, D-Ala или D-Lys;
Xaa2 представляет собой D-Pro, D-Ala или D-Lys;
Xaa3 представляет собой D-Lys, D-Orn, D-Nle, D-Ala или D-Lys;
Xaa4 представляет собой Gly или D-Ala;
Xaa5 представляет собой D-Trp, Trp, D-3-бензотиенил-Ala, D-5-гидрокси-Trp, D-5-метокси-Trp, D-Phe или D-Ala;
Xaa6 представляет собой D-Arg, D-His или D-Ala;
Xaa7 представляет собой D-Cha, D-Phe, Phe, D-4-фтор-Phg, D-3-пиридил-Ala, D-Thi, D-Trp, D-4-нитро-Phe или D-Ala;
Xaa8 представляет собой D-His, His, D-Arg, Phe или D-Ala;
Xaa9 представляет собой D-Glu, D-Asp, D-цитруллин, D-Ser или D-Ala;
Xaa10 представляет собой D-Met, D-бутионин, D-Ile или D-Ala;
Xaa11 представляет собой D-Ser, D-Ile или D-Ala;
Xaa12 представляет собой D-Tyr, D-Ser или D-Ala; и
Xaa13 представляет собой D-Ser или D-Ala;
где не более чем один Xaa1-13 представляет собой D-Ala, исключая случай, когда все Xaa1-3 представляют собой D-Ala, и не более чем один Xaa1-13 представляет собой L-аминокислоту;
или его фармацевтически приемлемая соль.
Xaa1 Xaa2 Xaa3 Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Хаа9 Xaa10 Xaa11 Хаа12 Xaa13,
где Xaa1 представляет собой D-Val;
Xaa2 представляет собой D-Pro;
Xaa3 представляет собой D-Lys, D-Orn или D-Nle;
Xaa4 представляет собой Gly;
Xaa5 представляет собой D-Trp, Trp, D-3-бензотиенил-Ala, D-5-гидрокси-Trp, D-5-метокси-Trp или D-Phe;
Xaa6 представляет собой D-Arg или D-His;
Xaa7 представляет собой D-Cha, D-Phe, Phe, D-4-фтор-Phg, D-3-пиридил-Ala, D-Thi, D-Trp или D-4-нитро-Phe;
Xaa8 представляет собой D-His, His, D-Arg, Phe или D-Ala;
Xaa9 представляет собой D-Glu, D-Asp, D-цитруллин или D-Ser;
Xaa10 представляет собой D-Met, D-бутионин или D-Ile;
Xaa11 представляет собой D-Ser или D-Ile;
Xaa12 представляет собой D-Tyr или D-Ser; и
Xaa13 представляет собой D-Ser;
где не более чем один Xaa1-13 представляет собой L-аминокислоту; или его фармацевтически приемлемая соль.
Xaa1 Xaa2 Xaa3 Xaa4 Xaa5 Xaa6 Xaa7 Xaa8Xaa9Xaa10 Xaa11 Xaa12 Xaa13,
где Xaa1 представляет собой D-Val;
Xaa2 представляет собой D-Pro;
Xaa3 представляет собой D-Lys, D-Orn или D-NIe;
Xaa4 представляет собой Gly;
Xaa5 представляет собой D-Trp или Trp;
Xaa6 представляет собой D-Arg;
Xaa7 представляет собой D-Cha, D-Phe, Phe или D-Thi;
Xaa8 представляет собой D-His или His;
Xaa9представляет собой D-Glu или D-Ser;
Xaa10 представляет собой D-Met, D-бутионин или D-Ile;
Xaa11 представляет собой D-Ser или D-Ile;
Xaa12 представляет собой D-Tyr или D-Ser; и
Xaa13 представляет собой D-Ser;
где не более чем один Xaa1-13 представляет собой L-аминокислоту; или его фармацевтически приемлемая соль.
D-Val D-Pro D-Lys Gly D-Trp D-Arg Phe D-His D-Ser D-Ile D-Ile D-Ser D-Ser (SEQ ID NO:4);
D-Val D-Pro D-Lys Gly D-Trp D-Arg D-Cha D-His D-Ser D-Ile D-Ile D-Ser D-Ser (SEQ ID NO:5);
Ser Tyr Ser Met Glu His Cha Arg Trp Gly Lys Pro Val (SEQ ID NO:6); или D-Val D-Pro D-Lys Gly D-Trp D-Arg D-Phe D-His D-Glu D-Met D-Ser D-Tyr D-Ser (SEQ ID NO:7);
или его фармацевтически приемлемая соль.
способностью селективно активировать MC1R; стабильностью в плазме in vitro; или устойчивостью к деградации протеазами.
Документы, цитированные в отчёте о поиске
Лиганды рецепторов меланокортинов
Комментарии