Композиции и способы на основе dac hyp - RU2661764C2
Код документа: RU2661764C2
Чертежи













































Описание
1. Уровень техники
Даклизумаб (DAC) представляет собой гуманизированное моноклональное IgG1антитело, которое связывается с альфа-субъединицей (CD25 или Tac) высокоаффинного рецептора человеческого интерлейкина-2 (IL-2), который экспрессируется на поверхности активированных, но не покоящихся, Т- и В-лимфоцитов. При связывании с CD25 на активированных клетках DAC блокирует образование комплекса высокоаффинного рецептора IL-2, тем самым блокируя индуцированную IL-2 пролиферацию активированных клеток.
По данным тестов прямого связывания на ФГА-бластах DAC связывается с CD25 с примерной аффинностью связывания (KD), равной 0,3 нМ, и ингибирует пролиферацию ФГА-бластов в зависимости от концентрации (Hakimi et al., 1993, J. Immunol., 151(2):1075-1085). В субоптимальной концентрации IL-2 (2,5 нг/мл) 15 нМ DAC ингибирует пролиферацию IL-2-зависимой клеточной линии Kit225/K6 на 50% (Pilsonn et al., 1997, J. Immunol., 159(3):1543-1556). В тесте оценки IL-2-зависимой пролиферации Т-клеток, индуцированной антигеном, наблюдали 50% ингибирование пролиферации под действием DAC в пределах концентраций 0,5-1 мкг/мл (3-7 нМ) (Junghans et al., 1990, Cancer Res., 50(5):1495-502).
Ранее вариант DAC был промышленно доступным для лечения острого отторжения аллотрансплантата у пациентов с трансплантированной почкой в качестве дополнительной терапии к схеме лечения иммуносупрессорами, которая включала циклоспорин и кортикостероиды Hoffman-La Roche, Inc., под торговым наименованием ZENAPAXTM. ZENAPAXTMвыпускался в виде концентрата для последующего разведения и внутривенного введения. Каждый флакон концентрата содержал 5 мл раствора, содержащего 5 мг/мл DAC, 3,6 мг/мл фосфата натрия однозамещенного моногидрата, 11 мг/мл фосфата натрия двузамещенного моногидрата, 4,6 мг/мл хлорида натрия, 0,2 мг/мл полисорбата 80 и HCl и/или NaOH, в количестве, достаточном для доведения рН до 6,9. Рекомендованная доза для взрослых и детей составляла 1,0 мг/кг при разведении рассчитанного объема 25 мг/5 мл концентрата ZENAPAXTM 50 мл 0,9% стерильного раствора хлорида натрия и внутривенном введении в периферическую или центральную вену в течение 15 мин.
Также DAC показал эффективность в лечении увеита (Nussenblatt et al., 2004, FOCIS 2004 meeting; July 18-23, Montreal, QC. Abstract 4688; Nussenblatt et al., 2003, J. Autoimmun., 21:283-293) и рассеянного склероза (см., например, Bielekova et al., 2004, Proc. Natl., Acad. Sci. USA, 101(23):8705-8708; Rose et al., 2007, Neurology, 69:785-789; патент США № 7258859) и в настоящее время является предметом проводимых клинических испытаний в лечении рассеянного склероза. Несмотря на имеющиеся сведения о том, что DAC является безопасным и эффективным, желательны высококонцентрированные жидкие композиции, имеющие длительный срок хранения, и которые удобно вводить без дополнительной формуляции или манипуляций, а также новые молекулы даклизумаба с улучшенными свойствами, такими как повышенная безопасность, по сравнению с ZENAPAXDAC.
2. Сущность изобретения
Как уже упоминалось в разделе «Уровень техники», даклизумаб представляет собой гуманизированное моноклональное IgG1антитело, которое специфически связывается с альфа-субъединицей (также относящейся далее к «CD25» или «Tac») рецептора человеческого интерлейкина-2 (IL-2), который является важным медиатором активации лимфоцитов. Ранее вариант DAC был промышленно доступным от Hoffman-La Roche, Inc., под торговым наименованием ZENAPAXTM, который проявил себя безопасным и эффективным препаратом в лечении отторжения аллотрансплантата почек, при использовании в качестве дополнительной терапии к схеме лечения иммуносупрессорами, которая включала циклоспорин и кортикостероиды (см., например, European Medicines Agency («EMEA») под наименованием ZENAPAX), и также показавшего эффективность в лечении рассеянного склероза (см., например, Bielekova et al., 2004, Proc. Natl., Acad. Sci. USA, 101(23):8705-8708; Rose et al., 2007, Neurology, 69:785-789; патент США № 7258859). Согласно EMEA ZENAPAXDAC экспрессируют в клетках GS-NS0 (клетках мышиной миеломы) и выделяют способом, включающим хроматографию на Q-сефарозе, хроматографию на S-сефарозе, диафильтрацию, хроматографию на Q-сефарозе II, ультрафильтрацию, гель-фильтрацию на S-300 и ультрафильтрацию. В настоящее время было установлено, что даклизумаб, экспрессированный в клеточной линии NS0, которая адаптирована к росту в бессывороточных, не содержащих холестерина и других продуктов животного происхождения средах, и выделенный другим способом, имеет характеристики и свойства, отличные от, и в некоторых отношениях, превосходящие даклизумаб ZENAPAX («ZENAPAXDAC»). Этот новый даклизумаб, упомянутый далее как «DAC HYP», имеет иной профиль изоформ, чем ZENAPAXDAC (по данным катионообменной хроматографии); иной профиль N-связанного гликозилирования, чем ZENAPAXDAC, хотя в клетках NS0 экспрессируются обе формы даклизумаба; и более низкой ADCC цитотоксичностью по сравнению с ZENAPAXDAC в биологических тестах.
Например, за счет гетерогенности в N- и C-концах тяжелой цепи возможно образование изоформ даклизумаба. Аминокислотная последовательность зрелой VHтяжелой цепи даклизумаба начинается в положении 20 аминокислотной последовательности, представленной на Фиг. 2 (SEQ ID NO:4). N-концевой глутамин (Q) зрелой VH тяжелой цепи (выделен жирным шрифтом, подчеркнут на Фиг. 2) способен циклизоваться, образуя пироглутамат (pE). В некоторых случаях сигнальная пептидная последовательность может быть усечена, оставляя последовательность валин-гистидин-серин (VHS), соединенную с N-концевым остатком глутамина зрелой VH тяжелой цепи. Поскольку каждая молекула даклизумаба содержит две VH тяжелой цепи, то различные N-концевые изоформы даклизумаба могут включать формы, содержащие: (1) два остатка глутамина (Q/Q); (2) один остаток глутамина и одну последовательность VHS (Q/VHS или VHS/Q); (3) две последовательности VHS (VHS/VHS); (4) один остаток глутамина и один остаток пироглутамата (Q/pE или pE/Q); (5) один остаток пироглутамата и одну последовательность VHS (pE/VHS или VHS/pE) и (6) два остатка пироглутамата (pE/pE). Также возможны различные С-концевые изоформы, которые содержат 0, 1 или 2 С-концевых остатка лизина (K) (0K, 1K или 2K), приводя к образованию сложного изоформного профиля.
Чрезвычайно удивительно, но, несмотря на то, что N-концевые глутамины VH тяжелой цепи ZENAPAX DAC полностью циклизуются с образованием пироглутамата, полной циклизации не достигается для DAC HYP. По данным катионообменной хроматографии DAC HYP отличается пиком изоформы pE/Q и пиком изоформы Q/VHS. Не желая связываться с теорией, полагают, что на данные уникальные изоформы pE/Q и Q/VHS может оказывать влияние лидерная последовательность, используемая для экспрессии DAC HYP. Следовательно, в одном аспекте настоящее изобретение относится к композициям даклизумаба, в которых изоформа pE/Q составляет 3-17%, 3-15%, 5-15%, более предпочтительно 5-12% или 7-12% от N-концевых изоформ, и/или изоформа Q/VHS составляет 1-15%, более предпочтительно 3-12% от N-концевых изоформ по данным катионообменной хроматографии.
В некоторых вариантах осуществления композиция даклизумаба отличается профилем катионообменной хроматографии, который по существу аналогичен представленному на Фиг. 18, или профилем DAC HYP, представленным на Фиг. 23.
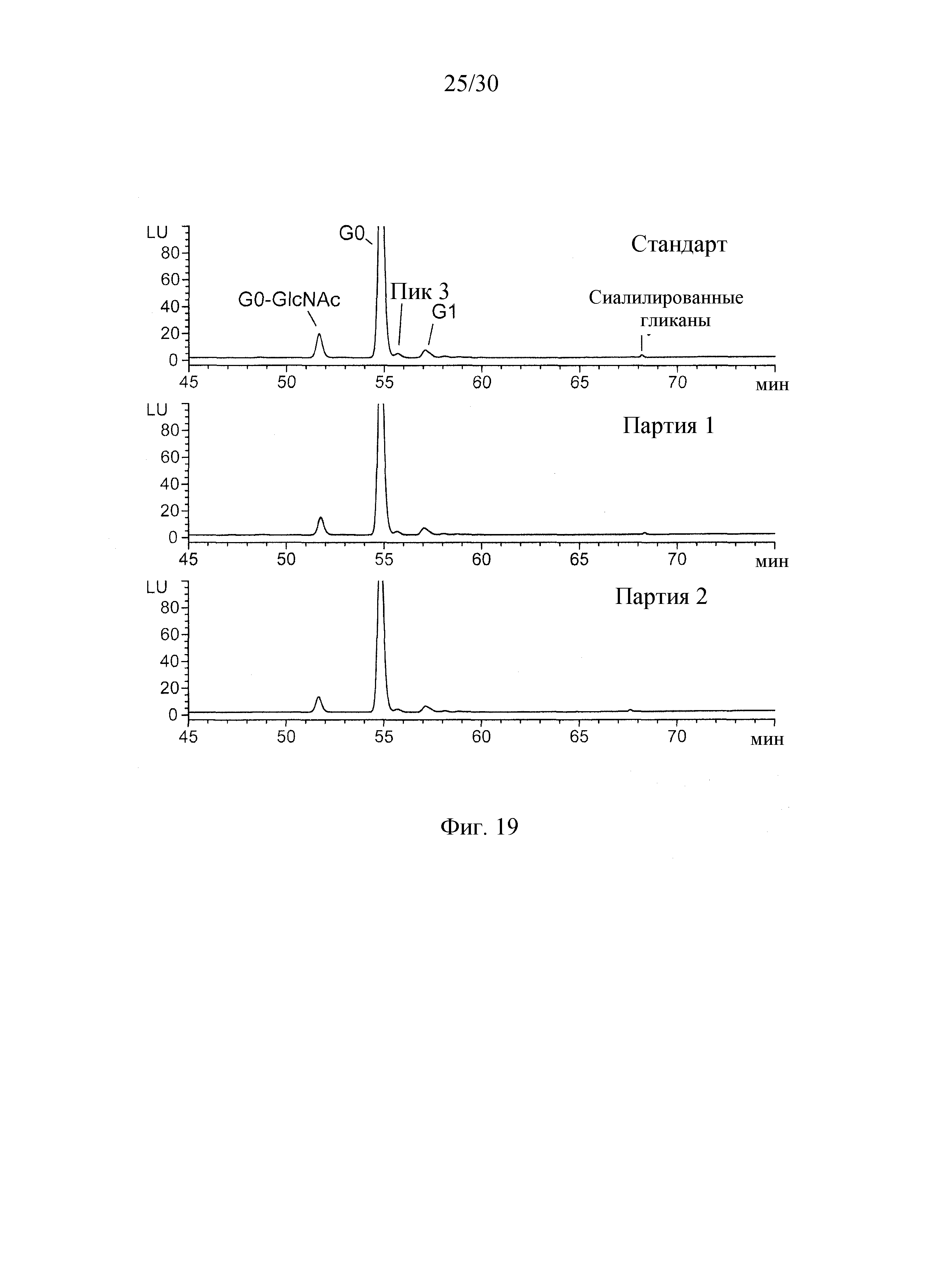
Даклизумаб содержит N-связанные олигосахариды, присоединенные к остатку Asn 296 тяжелой цепи. Когда эти N-связанные олигосахариды отщепляют с использованием амидазы PNGaseF и анализируют ВЭЖХ, то DAC HYP имеет профиль гликозилирования, отличный от ZENAPAX DAC, несмотря на тот факт, что оба продуцируются рекомбинантным путем в клеточной линии NS0. Действительно, профиль гликозилирования DAC HYP является необычно однородным. Как показано на верхней панели Фиг. 21, профиль гликозилирования ZENAPAX DAC отличается пиками, представляющими олигосахариды G0-GlcNAc, G0, G1, Man5, G2, Man6, Man7 и сиалилированный олигосахарид. На нижней панели Фиг. 21 показано, что профиль гликозилирования DAC HYP отличается двумя основными пиками, соответствующими гликоформам G0-GlcNAc и гликоформам G0, и минорным пиком, соответствующим гликоформе G1. Гликоформы G0-GlcNAc могут составлять от примерно 5% до примерно 20% AUC, как правило, от 7,2% до 14,6% AUC. Гликоформы G0 могут колебаться от 70% до 99,2% AUC, обычно от 80,9% до 99,2% AUC. Гликоформа G1 может колебаться от 1% до 9% AUC, как правило, от 1,4% до 3,8% AUC. Сиалилированные олигосахариды составляют 1,0% от общей AUC или менее.
Иммуногенность и высокая эффекторная функция могут стать проблемой для лекарственных препаратов с длительным введением. Кроме того, быстрый клиренс может приводить к снижению доступности лекарственного препарата. Как хорошо известно специалистам в данной области, различия в паттернах гликозилирования лекарственных антител могут приводить к различиям в иммуногенности. Антитела, обладающие высокооднородными паттернами гликозилирования, как это имеет место для DAC HYP, могут обеспечивать преимущественные профили иммуногенности, уровни ADCC и клиренса. Кроме того, биологические соединения, имеющие более однородные профили гликозилирования, снижают вариации «партия от партии» и могут повышать постоянство и стабильность.
Следовательно, еще в одном аспекте настоящее изобретение относится к композициям даклизумаба, которые отличаются однородным профилем N-связанного гликозилирования. В одном варианте осуществления композиция даклизумаба отличается профилем N-связанного гликозилирования, который включает примерно 5-20% гликоформ G0-GlcNAc от общей AUC, и в некоторых вариантах осуществления примерно 5-18% или примерно 7-15% (например, 7,2-14,6% или 6,9-14,7%) гликоформ G0-GlcNAc от общей AUC (и в некоторых конкретных вариантах осуществления 7,3% гликоформ G0-GlcNAc от общей AUC) и примерно 70-99,2% гликоформ G0 от общей AUC, и в некоторых вариантах осуществления примерно 75-90%, примерно 75-92% или примерно 81-88% гликоформ G0 от общей AUC (и в некоторых конкретных вариантах осуществления 86% гликоформ G0 от общей AUC) по данным ВЭЖХ. Необязательно пик G1 составляет менее чем примерно 10% от общей AUC, менее чем примерно 5%, менее чем примерно 4% или менее чем примерно 3% от общей AUC, и в некоторых вариантах осуществления его содержание находится в пределах от примерно 1% до примерно 4% (например, от 1,4% до 3,8%) или от примерно 1% до примерно 3%. Гликоформы Man5 предпочтительно составляют примерно 3% от общей AUC или менее. В других вариантах осуществления композиция даклизумаба отличается профилем N-связанных гликоформ по данным ВЭЖХ, по существу аналогичным представленному на Фиг. 19.
В некоторых аспектах композиция даклизумаба по изобретению отличается суммой двух или более пиков гликоформ. В некоторых вариантах осуществления композиции даклизумаба характеризуются (а) двумя основными пиками, соответствующими гликоформам G0-GlcNAc и гликоформам G0, которые вместе составляют примерно от 75% до 100%, от примерно 80% до примерно 100% или от примерно 85% до примерно 100% от общей AUC, и/или (b) пиками, соответствующими гликоформам Man5, Man6 и Man7, которые вместе составляют примерно 6% от общей AUC или менее, и/или (с) пиками, соответствующими гликоформам Man6 и Man7, которые вместе составляют примерно 2% от общей AUC или менее. В таких вариантах осуществления процентное содержание G0-GlcNAc, G0, G1 и/или Man5 может представлять значения, приведенные в предшествующем абзаце.
Связывающие или ингибирующие свойства DAC HYP, а также функциональная активность DAC HYP, оцененные в тесте, позволяющем определить ингибирование индуцированной IL-2 пролиферации Т-клеток, аналогичны таковым для ZENAPAX DAC. Однако очень удивительно, что DAC HYP проявляет значительно меньшую ADCC цитотоксичность по сравнению с ZENAPAX DAC, что вероятно, является результатом, по меньшей мере частично, различий в уровне нефукозилированной маннозы в паттерне гликозилирования (см. Фиг. 21). Как показано на Фиг. 22А и 22В, DAC HYP проявляет ADCC-цитотоксичность, по меньшей мере на 25% более низкую по сравнению с ZENAPAX DAC по данным клеточного теста. Как известно специалистам в данной области, пониженная ADCC-цитотоксичность DAC HYP может быть полезной для назначений, включающих длительное введение, когда не желательна гибель клеток, например, при лечении рассеянного склероза или увеита. В данных контекстах, когда терапия назначается длительно, например, при лечении рассеянного склероза и других неонкологических показаний, то терапия DAC HYP может быть безопаснее по сравнению с лечением ZENAPAX DAC.
Следовательно, в еще одном аспекте изобретение относится к композициям даклизумаба, которые характеризуются проявлением более низкой ADCC-цитотоксичности примерно на 30%, 25%, 20%, 15%, 10%, 5% или даже более в концентрации 1 мкг/мл по данным теста в условиях in vitro с использованием соотношения эффектора к клеткам-мишеням, составляющего 25:1, 40:1, 50:1 или 60:1, например, при использовании Kit225/K6 в качестве клетки-мишени и/или с использованием эффекторных клеток PBMC от 3 или более, 6 или более, 10 или более, 50 или более здоровых доноров. В конкретных вариантах осуществления изобретение относится к композициям даклизумаба, которые характеризуются проявлением ADCC-цитотоксичности в пределах 5-30%, 10-30%, 15-30%, 15-30%, 5-25%, 10-25%, 20-30%, 15-25%, 15-35% или 20-35% в концентрации 1 мкг/мл по данным теста в условиях in vitro с использованием соотношения эффектора к клеткам-мишеням, составляющего 25:1, 40:1, 50:1 или 60:1, например, при использовании Kit225/K6 в качестве клетки-мишени и/или с использованием эффекторных клеток PBMC от 3 или более, 6 или более, 10 или более, 50 или более здоровых доноров. Более низкие уровни ADCC-цитотоксичности, наблюдаемые для DAC HYP по сравнению с ZENAPAX DAC, являются удивительными с учетом того, что DAC HYP является иммуноглобулином IgG1 и не содержит мутаций рамки считывания, о которых известно, что они приводят к снижению ADCC-цитотоксичности.
Профиль безопасности DAC HYP по сравнению с ZENAPAX DAC можно дополнительно повысить при использовании бессывороточного способа с высоким выходом, который позволяет получать высокоочищенный продукт, не содержащий бычьего сывороточного альбумина (BSA). Следовательно, настоящее изобретение относится к композиции даклизумаба, которая не содержит BSA и/или является продуктом способа культивирования клеток, в котором отсутствует BSA.
Композиции даклизумаба, характеризующиеся одним или более свойствами, описанными выше (композиции DAC HYP), можно легко получить рекомбинантной экспрессией в клетках млекопитающих. Не желая связываться с какой-либо конкретной теорией способа, полагается, что одна или более уникальных характеристик и/или свойств, обсужденных выше, могут быть результатом, по меньшей мере частично, применения высокопродуктивной рекомбинантной экспресионной системы. Этого можно достичь любым способом, таким как амплификация гена с использованием DHFR, или с использованием селектируемого гена-маркера под контролем слабого промотора, предпочтительно в комбинации с сильным промотором, регулирующим экспрессию интересующего белка (предпочтительно секретируемого белка). Не желая связываться с теорией, полагают, что выбор маркеров под контролем слабого промотора облегчает идентификацию стабильных трансфектантов, в которых экспресионный вектор интегрирован в хромосомную область, которая является транскрипционно активной, с получением высоких уровней экспрессии интересующего белка. В одном варианте осуществления слабый промотор, регулирующий экспрессию селектируемого маркера, представляет собой промотор SV40 (Reddy et al., 1978, Science, 200:494-502), в котором активность одной или более энхансерных областей снижена или элиминирована посредством частичной или полной делеции, необязательно в комбинации с сильным промотором, таким как промотор CMV IE (Boshart et al., 1985, Cell 41(2):521-530), регулирующим экспрессию интересующего белка.
Следовательно, в еще одном аспекте изобретение относится к векторам, подходящим для получения рекомбинантных клеточных линий, которые стабильно экспрессируют высокие уровни даклизумаба, такого как DAC HYP, в которых экспрессия селектируемого маркера находится под контролем промотора SV40, энхансерная функция которого снижена, посредством частичной делеции одной или более энхансерных последовательностей (обозначенных dE-SV40). Конкретная промоторная последовательность dE-SV40, которую можно использовать для получения клеточных линий со стабильной экспрессией, находится в положениях 6536-6735 вектора pHAT.IgG1.rg.dE (SEQ ID NO:5), приведенного на Фиг. 3А-3D и на Фиг. 3Е (SEQ ID NO:12). Различные варианты осуществления конкретных векторов, которые можно использовать для получения клеточных линий со стабильной экспрессией, описаны в заявке на патент США 61/565419, поданной 30 ноября 2011 г., и в международной заявке PCT/US11/62720, поданной 30 ноября 2011 г., которые включены в данный документ для сведения.
В общем, векторы, пригодные для экспрессии даклизумаба, такого как DAC HYP, будут содержать один или более элементов, характерных для pHAT.IgG1.rg.dE (описанного в разделе 5.1 ниже), таких как промотор. Две цепи даклизумаба можно расположить под отдельным транскрипционным контролем, но предпочтительно в одном и том же векторе, и их кодирующие области могут представлять кДНК или геномную ДНК, содержащую интроны и экзоны. В качестве альтернативы для разделения транскрипционного контроля две цепи могут экспрессироваться в виде одного транскрипта или одной открытой рамки считывания, с их кодирующими областями, разделенными внутренним сайтом входа рибосомы или самоотщепляющейся intein последовательностью, где последовательности, кодирующие тяжелую и легкую цепи, находятся под контролем одного промотора. Приведенным в качестве примера промотором является промотор и энхансер CMV IE (в положениях 0001-0623 и 3982-4604 pHAT.IgG1.rg.dE (SEQ ID NO:5)). Дополнительные элементы включают сайты инициации транскрипции (если отсутствует в выбранном промоторе), сайты терминации транскрипции и ориджины репликации. Примеры таких элементов показаны в таблице 1, в которой приводятся компоненты pHAT.IgG1.rg.dE.
В конкретном варианте осуществления, пригодном для экспрессии обеих тяжелой и легкой цепей даклизумаба, такого как DAC HYP, из одной экзогенной нуклеиновой кислоты в клетках NS0 используется селектируемый маркер, функционирующий в клетках млекопитающих, такой как неомицинфосфотрансфераза (neor), гигромицин В-фосфотрансфераза (hygr), гигромицин В-фосфотрансфераза (Hph), пуромицин N-ацетилтрансфераза (puror), бластицидин S-дезаминаза (bsrr), ксантин-гуанинфосфорибозилтрансфераза (gpt), глутаминсинтетаза (GS) или тимидинкиназа вируса простого герпеса (HSV-tk). В предпочтительном варианте осуществления селектируемый маркер в векторе по изобретению представляет собой селектируемый маркер гуанинфосфорибозилтрансферазу E. coli под контролем промотора SV40 без энхансерной активности, кодирующей последовательности, которую можно найти в положениях 6935-7793 вектора pHAT.IgG1.rg.dE (SEQ ID NO:5), приведенного на Фиг. 3А-3D.
В еще одном аспекте изобретение относится к клеткам-хозяевам, трансфицированным векторами, пригодными для получения рекомбинантным путем даклизумаба, например, такого как DAC HYP. Клетка-хозяин может представлять любую клетку млекопитающего, включая, например, клетки яичника китайского хомячка (CHO), мышиные миеломные клетки NS0, клетки Sp2/0, клетки PER.C6, клетки Vero, клетки BHK, клетки HT1080, клетки COS7, клетки WI38, клетки CV-1/EBNA, L-клетки, клетки 3T3, клетки HEPG2, клетки MDCK и клетки 293. После трансфекции вектор может интегрировать в геном с получением клеточной линии со стабильной продукцией. Специалистам в данной области, очевидно, понятно, что нежелательно включать продукты животного происхождения в композиции, предназначенные для введения людям. Следовательно, клетки-хозяева, для которых не требуется сыворотки крови или других продуктов животного происхождения для культивирования (например, холестерина), являются предпочтительными. Клетки-хозяева, для которых требуются такие продукты животного происхождения, можно адаптировать к использованию среды без сыворотки крови и других продуктов животного происхождения. Способ адаптации мышиных миеломных клеток NS0 к росту в бессывороточной и не содержащей холестерина среде, описан Hartman et al., 2007, Biotech.&Bioeng., 96(2):294-306, и Burky et al., 2007, Biotech.&Bioeng., 96(2):281-293. Конкретный штамм клеток NS0, адаптированный к росту в бессывороточной и не содержащей холестерина среде, стабильно трансфицирован вектором, описанным выше, который можно использовать для получения DAC HYP (клон 7А11-5Н7-14-43).
Специалистам в данной области, очевидно, понятно, что базальные и питательные среды, описанные для культивирования клеток, используемых для рекомбинантного получения белка, а также другие переменные факторы, такие как схема подпитки, скорость роста, температура и уровень кислорода, могут оказывать влияние на выход и качество экспрессированного белка. Специалистам в данной области известны способы оптимизации таких условий; приведенные в качестве примера условия, описываются в разделе «Примерные варианты осуществления». Предпочтительно клетки адаптируют к росту в средах, не содержащих холестерина, сыворотки крови и других компонентов животного происхождения; в таких случаях базальная и питательная среды предпочтительно содержат определенные химические соединения, которые заменяют такие компоненты. Также было установлено, что среды, содержащие высокие концентрации глюкозы, например, 10-35 г/л глюкозы, преимущественно повышают продуктивность культивирования клеток. В конкретном варианте осуществления базальная среда содержит примерно 10-20 г/л, более предпочтительно примерно 15 г/л глюкозы, и питательная среда содержит 22-35 г/л, более предпочтительно 28 г/л глюкозы. Питательную среду можно добавлять к клеткам по возрастающей схеме подпитки, как известно в данной области, в течение периода времени, составляющего 8-15 суток, 9-13 суток или наиболее предпочтительно 10-13 суток.
Для DAC HYP, экспрессированного в штамме-продуценте 7А11-5Н7-14-43 NS0, компоненты культуральной и питательной сред и другие переменные факторы, оказывающие влияние на экспрессию и продукцию, оптимизированы. Следовательно, изобретение также относится к оптимизированным базальным средам, питательным средам, схемам подпитки и другим способам и условиям культивирования, пригодным для получения даклизумаба с высоким выходом и чистотой. Такие среды и параметры культивирования, и способы более подробно описаны в разделе 5.3.
Также было установлено, что выделение даклизумаба из клеточной культуры с использованием комбинации некоторых хроматографических стадий обеспечивает очищенный даклизумаб и лекарственную субстанцию DAC HYP, и жидкий даклизумаб и лекарственные формы даклизумаба, которые стабильны при хранении в жидкой форме в высокой концентрации, как правило, в номинальной концентрации даклизумаба и DAC HYP, составляющей, по меньшей мере, 100 мг/мл ± 10-15% и в некоторых вариантах осуществления 150 мг/мл ± 10-15% (по данным УФ-спектроскопии или показателю преломления).
Стабильные высококонцентрированные лекарственные формы даклизумаба обычно готовят заменой концентрированной композиции на обменный буфер, имеющий осмолярность в пределах примерно 267-327 мОсм/кг (например, 270-310 мОсм/кг) и рН в пределах примерно 5,8-6,2 при 25°С (например, 5,9-6,1 при 25°С), с получением промежуточной композиции и затем разведением промежуточной композиции буфером для разведения с полисорбатом с получением стабильной, высококонцентрированной жидкой композиции, содержащей примерно 100 мг/мл ± 10% даклизумаба (например, DAC HYP) и в некоторых вариантах осуществления, по меньшей мере, примерно 150 мг/мл даклизумаба (например, DAC HYP) по данным УФ-спектроскопии или показателю преломления. Буфер для разведения является таким же, что и обменный буфер, но содержит примерно 0-10% (мас./об.) полисорбата 80, и используется в таком количестве, что конечная стабильная, высококонцентрированная композиция даклизумаба содержит расчетную концентрацию (номинальную концентрацию) полисорбата 80 в пределах 0,02-0,04%, в некоторых вариантах осуществления примерно 0,03% (мас./об.). В обменный буфер и буфер для разведения можно включить различные забуферирующие агенты и наполнители для достижения осмолярности и рН в указанных пределах. Конкретный неограничивающий пример обменного буфера, пригодного для формуляции стабильного, высококонцентрированного жидкого даклизумаба и лекарственных форм DAC HYP, содержит примерно 40 мМ сукцината и примерно 100 мМ NaCl, и имеет рН примерно 6,0 при 25°С. Конкретный неограничивающий пример буфера для разведения, подходящего для применения с таким обменным буфером, содержит примерно 40 мМ сукцината, примерно 100 мМ NaCl и примерно 1% (мас./об.) полисорбата 80, и имеет рН примерно 6,0 при 25°С. Значение рН конечной композиции можно довести кислотой или основанием с получением фактического значения рН, равного примерно 6,0, при 25°С.
Стабильные, высоконцентрированные жидкие лекарственные формы даклизумаба характеризуются низкой степенью агрегации, как правило, содержат, по меньшей мере, 95% мономера и менее чем примерно 3% агрегатов, в некоторых случаях менее чем примерно 1,5% агрегатов, и обычно более чем примерно 99% мономера и менее чем примерно 0,8% агрегатов по данным эксклюзионной хроматографии. Другие характеристики высококонцентрированных жидких лекарственных форм даклизумаба более подробно приведены в разделе 5.6.
Высококонцентрированные лекарственные формы даклизумаба также отличаются длительным периодом хранения, подвергаясь не более чем 5% деградации и образованию не более чем 3% агрегатов (по данным соответственно SDS-PAGE и эксклюзионной хроматографии) в течение периода времени до 54 месяцев или более, например, в течение по меньшей мере 5 лет, при хранении при температуре 2-8°С, в течение периода времени до 9 месяцев при хранении в условиях ускоренного старения (23-27°С/60±5% относительная влажность) и в течение до 3 месяцев при хранении в стрессовых условиях (38-42°С/75±5% относительная влажность).
Как уже отмечалось выше, стабильные высококонцентрированные жидкие композиции даклизумаба можно приготовить разведением промежуточной композиции буфером для разведения с полисорбатом с получением конечной лекарственной формы даклизумаба. Следовательно, в еще одном аспекте изобретение относится к не содержащим полисорбат очищенным промежуточным композициям даклизумаба (предпочтительно DAC HYP), содержащим по меньшей мере примерно 150 мг/мл даклизумаба, в некоторых вариантах осуществления примерно 170-190 мг/мл даклизумаба, которые можно разбавить буфером для разведении с полисорбатом с получением стабильных высококонцентрированных жидких лекарственных форм даклизумаба, описанных в данном документе. В конкретном варианте осуществления концентрированные не содержащие полисорбат промежуточные композиции номинально содержат примерно 155 мг/мл или примерно 180 мг/мл даклизумаба (предпочтительно DAC HYP), примерно 40 мМ цитрата натрия и примерно 100 мМ NaCl, рН 6,0 при 25°С. В конкретном варианте осуществления концентрированные не содержащие полисорбат промежуточные композиции номинально содержат примерно 155 мг/мл или примерно 180 мг/мл даклизумаба (предпочтительно DAC HYP), примерно 40 мМ цитрата натрия и примерно 100 мМ NaCl, рН 6,0 при 25°С. Композиции даклизумаба отличаются низким содержанием агрегатов, как дополнительно описано ниже.
Было установлено, что концентрирование даклизумаба посредством ультрафильтрации приводит к образованию агрегатов, что может привести к получению высококонцентрированной лекарственной формы даклизумаба, содержащей неприемлемые количества агрегатов (например, >3%). Следовательно, предпочтительно использовать стадию «очистки» перед концентрированием лекарственной субстанции даклизумаба для удаления агрегатов. Содержание приемлемых агрегатов перед концентрированием будет зависеть от концентрации лекарственной субстанции даклизумаба, предназначенной для концентрирования, требуемой концентрации в конечной лекарственной форме даклизумаба и приемлемого уровня агрегатов в конечной лекарственной форме даклизумаба. Например, если желательно получить композицию даклизумаба с концентрацией 150 мг/мл, содержащую менее чем 3% агрегатов, и лекарственную субстанцию даклизумаба необходимо сконцентрировать в 10-30 раз (например, в 20 раз) для получения такой конечной композиции даклизумаба, то композиция даклизумаба, предназначенная для концентрирования, должна содержать <0,3% агрегатов, предпочтительно <0,2% агрегатов и предпочтительно даже более низкие количества, например, примерно 0,1% агрегатов.
Можно использовать различные известные способы для получения исходной композиции лекарственной субстанции даклизумаба, содержащей приемлемые уровни агрегатов, для концентрирования с получением концентрированной промежуточной композиции даклизумаба и конечных лекарственных форм, как описано в данном документе, включая, например, сильную катионообменную хроматографию и хроматографию гидрофобных взаимодействий. Однако неожиданнго было установлено, что слабая катионообменная хроматография снижает содержание агрегатов в композициях даклизумаба, содержащих 4-12 мг/мл даклизумаба и до 2,5% агрегатов, до очень низких уровней, как правило, 0,1% агрегатов. Применение слабой катионообменной хроматографии для удаления агрегатов является более благоприятным для окружающей среды по сравнению с хроматографией гидрофобных взаимодействий, для которой применяются растворы азотсодержащих соединений (такие как растворы сульфата аммония).
Следовательно, в еще одном аспекте изобретение относится к способам «очистки» композиций даклизумаба для удаления агрегатов таким образом, что полученная «обработанная» композиция обычно содержит примерно от 4 до 15 мг/мл даклизумаба, в которой 0,3% или менее (например, 0,2% или менее, или 0,1% или менее) находится в агрегатной форме по данным эксклюзионной хроматографии. Как правило, способ включает пропускание композиции даклизумаба, содержащей примерно 4-10 мг/мл, обычно примерно 8-9 мг/мл и предпочтительно примерно 8,5 мг/мл даклизумаба и >0,5% агрегатов, через катионообменную смолу в подходящем буфере для адсорбции даклизумаба и элюирование адсорбированного даклизумаба буфером для элюирования. Подходящие слабые катионообменные смолы включают, не ограничиваясь этим, СМ-650М (Tosoh Biosciences), CM-сефарозу, CM-HyperD. Компоненты буферов для уравновешивания, промывания и элюирования будут зависеть от используемой слабой катионообменной смолы, и они известны специалистам в данной области. Со смолой СМ-650М (Tosoh Biosciences, part Number 101392) хорошо «работает» буфер для уравновешивания и промывания, содержащий примерно 20 мМ цитрата натрия, рН 4,5, и буфер для элюирования, содержащий 20 мМ цитрата натрия и 75 мМ сульфата натрия, рН 4,5. Используемая скорость потока будет зависеть от выбора смолы и размера колонки. Для цилиндрической колонки хорошо «работает» смола CM0650M, имеющая высоту основания в пределах примерно 10-30 см (например, 17-19 см) и скорость потока примерно 50-200 см/ч (например, 90-110 см/ч, предпочтительно примерно 100 см/ч), когда хроматографию проводят при комнатной температуре или более низкой температуре, примеры значений температуры равняются 4°, 10°, 15°, 20° или 25°С. Обычные пригодные пределы температуры составляют 18-25°С (например, 18-22°С).
Для ZENAPAX EMEA способ очистки ZENAPAX DAC включает следующие двенадцать стадий:
(i) концентрирование культурального бульона;
(ii) хроматография на Q-сефарозе;
(iii) хроматография на S-сефарозе;
(iv) обработка при низком значении рН для инактивации вирусов;
(v) концентрирование/диафильтрация;
(vi) фильтрование через DV50 для удаления вирусов;
(vii) хроматография на Q-сефарозе II;
(viii) хроматография для разделения вирусов для удаления вирусов;
(ix) концентрирование ультрафильтрацией;
(x) гель-фильтрация на S-300;
(xi) концентрирование ультрафильтрацией;
(xii) заполнение флаконов в асептических условиях.
Данный способ является неэффективным и обеспечивает низкий выход при очистке. Было установлено, что более высокие выходы можно получить с использованием способа, имеющего меньшее число стадий с одновременным обеспечением более высокой степени очистки, что позволяет формулировать полученную лекарственную субстанцию даклизумаба в высококонцентрированные лекарственные формы даклизумаба, описанные выше. Следовательно, настоящее изобретение также относится к улучшенным способам выделения и/или очистки как лекарственной субстанции, так и высококонцентрированных лекарственных форм даклизумаба. В способе используется аффинная хроматография с протеином А в сочетании с сильной анионообменной хроматографией (на Q-сефарозе) и слабой катионообменной хроматографией (СМ-650М), что обеспечивает непрерывную проточную обработку без разведения промежуточного продукта способа. Улучшенный способ получения очищенной лекарственной субстанции даклизумаба включает следующие стадии:
(i) аффинная хроматография с протеином А для отделения даклизумаба от других компонентов клеточной культуры;
(ii) инактивация вирусов при низком значении рН;
(iii) сильная анионообменная хроматография на Q-сефарозе для удаления ДНК;
(iv) слабая катионообменная хроматография (СМ-650М) для уменьшения содержания агрегатов; и
(v) фильтрование для удаления вирусов.
Точные объемы, размеры колонок и рабочие параметры частично будут зависеть от масштаба очистки, как известно в данной области. Конкретные объемы, размеры колонок и рабочие параметры, пригодные для крупномасштабных очисток, описаны в разделе 5.4.
Неочищенный даклизумаб, предназначенный для очистки и необязательно формуляции с использованием вышеописанных способов, можно собрать из культуры клеток с использованием различных общепринятых способов, например, микрофильтрации, центрифугирования и глубинного фильтрования непосредственно из биореактора. Однако было установлено, что неочищенный даклизумаб можно легко собрать снижением рН клеточной культуры примерно до рН 5 при температуре ниже 15°С для флоккулирования клеток, которые можно удалить центрифугированием. В конкретном варианте осуществления неочищенный даклизумаб собирают снижением рН клеточной культуры примерно до рН 5, охлаждением культуры до температуры ниже 15°С, например, 4°С, в течение 30-90 мин и центрифугированием полученной суспензии для удаления клеток. Обычно данный способ применим для любой клеточной культуры, которая секретирует рекомбинантные белки в культуральную среду, и он не является специфическим для культур, продуцирующих даклизумаб или терапевтические антитела. Значение рН культуры можно довести с использованием разнообразных различных кислот, включая слабые или сильные органические кислоты, или слабые или сильные неорганические кислоты. Было установлено, что для культур с даклизумабом хорошо подходит лимонная кислота. Можно использовать концентрированный раствор лимонной кислоты, например, 0,5М-2М раствор, для доведения рН культуры перед сбором материала.
Очистку DAC HYP проводят с использованием трех хроматографических стадий, инактивации вирусов, фильтрования вирусов и конечной ультрафильтрации. Аффинная хроматография с протеином А является первой стадией в способе очистки, с помощью которой происходит отделение большинства примесей, связанных со способом. Для того чтобы повторно использовать колонку для аффинной хроматографии с протеином А, ее необходимо регенерировать и подвергнуть санитарной обработке. Было установлено, что водный раствор NaOH является эффективным как для регенерации, так и санитарной обработки колонки. Однако применение растворов NaOH может привести к разрушению смолы с протеином А, что повысит стоимость производства в целом. Также было установлено, что санитарная обработка смол с протеином А для аффинной хроматографии раствором, содержащим NaOH и бензиловый спирт, дает хорошие результаты и значительно повышает число возможных циклов очистки. Следовательно, изобретение относится к раствору для санитарной обработки и способу регенерации и санитарной обработки колонок и смол с протеином А для аффинной хроматографии. Обычно буфер содержит примерно от 100 до 500 мМ цитрата натрия, примерно от 10 до 30 мМ NaOH и примерно от 0,5 до 3% (об./об.) бензилового спирта, и имеет рН в пределах примерно от 10 до 13. Необязательно, буфер также может содержать другие компоненты, например, такие как соли и/или детергенты. Оба компонента, цитрат натрия и бензиловый спирт, важны для защиты смолы с протеином А от разрушения под действием NaOH и повышения микробицидной активности. В конкретных вариантах осуществления буфер для санитарной обработки протеина А содержит примерно 200 мМ цитрата натрия, примерно 20 мМ NaOH и примерно 1% (об./об.) бензилового спирта. Как описано в разделе 5.4.2, растворы для санитарной обработки, содержащие бензиловый спирт и гидроксид натрия, обладают полезными антимикробными эффектами, и их можно использовать для санитарной обработки колонок с протеином А в способах очистки для любого антитела.
Буфер для санитарной обработки можно использовать для санитарной обработки смолы с протеином А для хроматографии порционным способом, в котором смолу промывают избытком (например, 1,5-2Х объемами) буфера для санитарной обработки с последующей инкубацией в течение примерно 30-45 мин с избытком (например, 1,5-2Х объемами) буфера для санитарной обработки с последующим уравновешиванием буфером для уравновешивания или буфером для хранения. Буфер для санитарной обработки также можно использовать для санитарной обработки колонки для хроматографии с протеином А промыванием колонки избытком (например, 1,5-2Х объемами) буфера для санитарной обработки с подходящей скоростью потока (например, в пределах примерно 110-190 см/ч или 135-165 см/ч), выдерживанием колонки в условиях нулевого потока от 0 до примерно 30-40 мин, и затем промыванием колонки буфером для уравновешивания или буфером для хранения. Подходящие буферы для уравновешивания и хранения описаны в разделе 5.5.
Санитарная обработка колонок с протеином А буферами для санитарной обработки, описанными в данном документе, значительно повышает число возможных очисток, для которых можно использовать одну партию смолы. Например, в то время, когда одна партия смолы с протеином А, как правило, сохраняется только примерно 30 циклов очистки, при санитарной обработке обычными буферами на основе NaOH (например, 50 мМ NaOH, 0,5 М NaCl), то колонки с протеином А, подвергнутые санитарной обработке буферами для санитарной обработки, описанными в данном документе, можно использовать более чем 100 циклов очистки. Не желая связываться с теорией способа, полагают, что буферы для санитарной обработки, описанные в данном документе, частично защищают иммобилизованный протеин А от вызванного NaOH разрушения, тем самым увеличивая срок годности смолы. Следовательно, несмотря на то, что полагают, что усовершенствования пригодны для всех смол с протеином А, включая смолы с мутантными штаммами протеина А (например, смола MabSuRe), предназначенными для того, чтобы быть устойчивыми к деградации под действием NaOH, буферы для санитарной обработки, описанные в данном документе, являются особенно полезными при использовании для санитарной обработки смол и колонок с протеином А с применением немодифицированных иммобилизованных протеинов А или протеинов А, которые не сконструированы, чтобы быть стабильными к воздействию NaOH. Изобретение дополнительно относится к способам, включающим использованием смолы для аффинной хроматографии с протеином А для более чем 30, более чем 35 или более чем 40 циклов очистки антител, и в некоторых случаях до 50 или 100 циклов очистки белка, включающим проведение циклов очистки и промывание смолы раствором для санитарной обработки, раскрытого в данном документе.
Как уже указывалось выше, даклизумаб специфически связывается с CD25, экспрессированным на активированных, но не на покоящихся Т- и В-лимфоцитах, и блокирует связывание IL-2 с CD25, тем самым ингибируя образование комплекса высокоаффинного рецептора IL-2, и как следствие ингибирует пролиферацию активированных Т- и В-лимфоцитов. Композиции и лекарственные формы DAC, описанные в данном документе, и в частности, композиции и лекарственные формы DAC HYP, аналогично, специфически связываются с CD25 и проявляют такие же биологические свойства. Следовательно, композиции и лекарственные формы DAC, описанные в данном документе, и в частности, DAC HYP, являются пригодными для любого из тестов и терапевтических способов, описанных для даклизумаба в общем, и ZENAPAX, в частности. Следовательно, настоящее изобретение также относится к способам применения композиций и лекарственных форм DAC, описанных в данном документе, в частности, композиций и высококонцентрированной стабильной жидкой лекарственной формы DAC HYP, для ингибирования пролиферации активированных Т- и В-клеток, в применениях in vitro и in vivo в качестве терапевтического подхода для лечения заболеваний, в которых играет роль пролиферация активированных Т- и В-клеток, например, для лечения и профилактики отторжения аллотрансплантата, лечения увеита и лечения рассеянного склероза.
Как правило, способы включают контактирование активированных Т- и/или В-клеток с количеством композиции или лекарственной формы даклизумаба, описанных в данном документе, достаточным для ингибирования их пролиферации.
В отношении способов лечения, обычно способы включают введение субъекту количества композиции даклизумаба, например, композиции DAC HYP или высококонцентрированной лекарственной формы DAC, описанных в данном документе, с обеспечением терапевтического эффекта. В конкретном варианте осуществления композиции и лекарственные формы даклизумаба можно использовать для лечения рассеянного склероза, самостоятельно или в комбинации с другими средствами, такими как интерферон-бета. Композиции DAC, описанные в данном документе, можно вводить пациенту подкожно на недельной или месячной основе (например, раз в неделю, раз в две недели, дважды в месяц, раз в четыре недели или раз в месяц) в пределах доз от 75 до 300 мг (например, 75 мг, 100 мг, 125 мг, 150 мг, 175 мг, 200 мг, 225 мг, 250 мг, 275 мг или 300 мг) или в пределах от 1 мг/кг до 4 мг/кг. Композиции можно обеспечить в виде заранее заполненных шприцов, удобных для подкожного применения, предпочтительно с номинальными концентрациями даклизумаба 100 мг/мл ± 10-15% или 150 мг/мл ± 10-15%. Концентрированные композиции DAC также можно разбавить для внутривенного введения.
3. Краткое описание фигур
На Фиг. 1 приведены последовательности кДНК легкой цепи DAC HYP (SEQ ID NO:1) и транслированной аминокислотной последовательности (SEQ ID NO:2). Выделенный жирным шрифтом, подчеркнутый остаток аспартата (D) является первой аминокислотой в правильно процессированном зрелом белке; аминокислотная последовательность слева от данной остатка соответствует сигнальной последовательности.
На Фиг. 2 приведены последовательности кДНК тяжелой цепи DAC HYP (SEQ ID NO:3) и транслированной аминокислотной последовательности (SEQ ID NO:4). Выделенный жирным шрифтом, подчеркнутый остаток глутамина (Q) является первой аминокислотой в правильно процессированном зрелом белке; аминокислотная последовательность слева от данной остатка соответствует сигнальной последовательности.
На Фиг. 3A-3D представлена полная нуклеотидная последовательность вектора pHAT.IgG1.rg.dE (SEQ ID NO:5).
На Фиг. 3Е приведен конкретный вариант осуществления промотора dESV40 (SEQ ID NO:12), который можно использовать для отбора штаммов-продуцентов с высоким выходом.
На Фиг. 4А-4В приведена схематичная диаграмма вектора pHAT.IgG1.rg.dE (Фиг. 4А), который получен из pABX.gpt; вектор адаптирован к экспрессии любых генов тяжелой и легкой цепей или даже полипептида, не относящегося к антителу (Фиг. 4В).
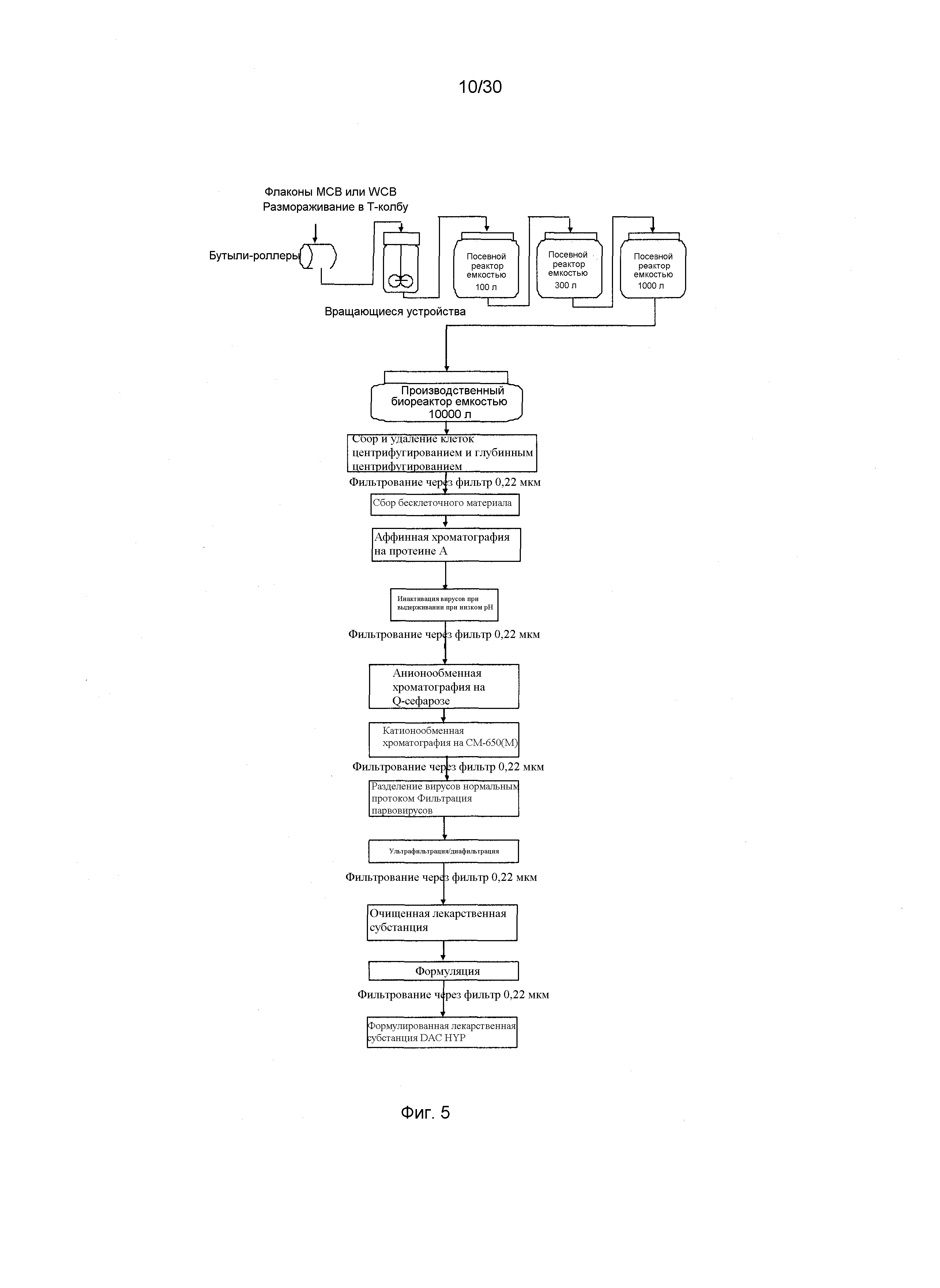
На Фиг. 5 представлен примерный способ получения DAC HYP.
На Фиг. 6 показан мониторинг длины волны в УФ-области (280 нм), рН и электропроводность фракций продукта во время аффинной хроматографии на протеине А.
На Фиг. 7 показан мониторинг длины волны в УФ-области (280 нм), рН и электропроводности фракций продукта во время хроматографии на Q-сефарозе.
На Фиг. 8 показан мониторинг длины волны в УФ-области (280 нм), рН и электропроводности фракций продукта во время катионообменной хроматографии на СМ.
На Фиг. 9 показана схема ультрафильтрационной системы для DAC HYP.
На Фиг. 10 приведена хроматограмма 0-60 мин пептидного картирования DAC HYP. Стандартный профиль представляет препарат DAC HYP с концентрацией 100 мг/мл, и партия 1 и партия 2 соответствуют препаратам DAC HYP с концентрацией 150 мг/мл.
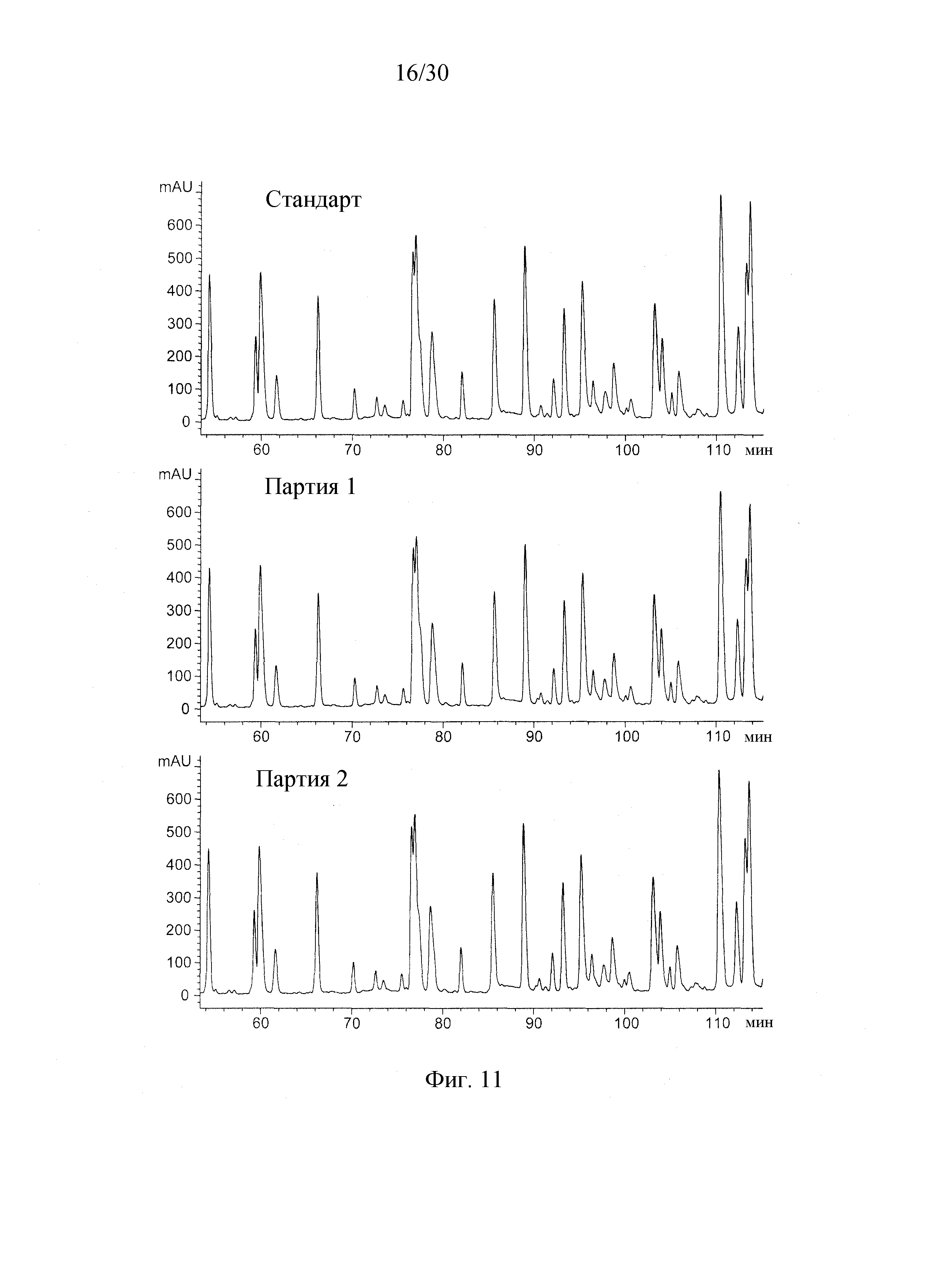
На Фиг. 11 приведена хроматограмма 55-115 мин пептидного картирования DAC HYP. Стандартный профиль представляет препарат DAC HYP с концентрацией 100 мг/мл, и партия 1 и партия 2 соответствуют препаратам DAC HYP с концентрацией 150 мг/мл.
На Фиг. 12 приведена хроматограмма 110-170 мин пептидного картирования DAC HYP. Стандартный профиль представляет препарат DAC HYP с концентрацией 100 мг/мл, и партия 1 и партия 2 соответствуют препаратам DAC HYP с концентрацией 150 мг/мл.
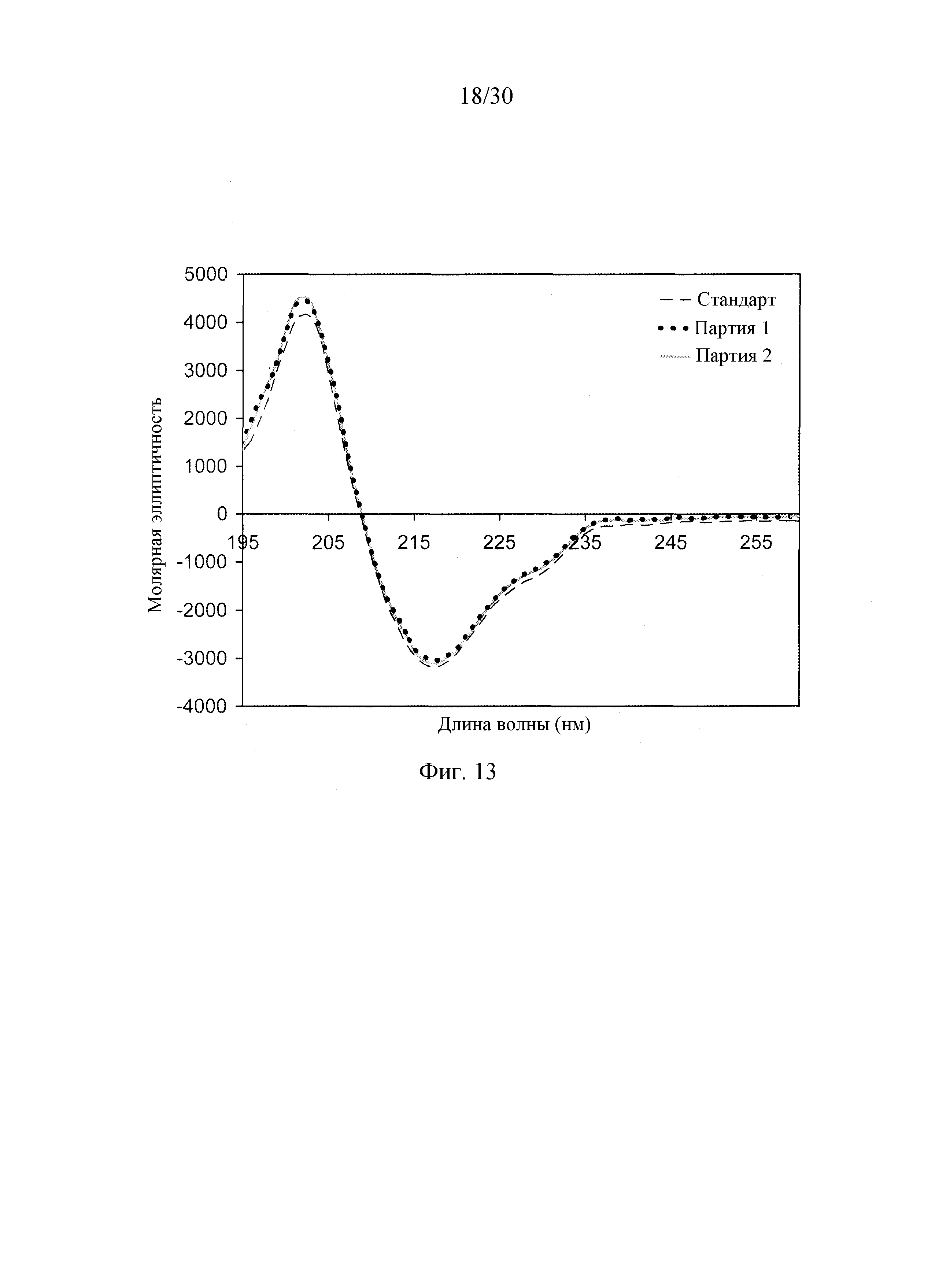
На Фиг. 13 показаны наложенные спектры кругового дихроизма серий препарата DAC HYP партии 1 и партии 2 с концентрацией 150 мг/мл. Стандарт представляет препарат DAC HYP с концентрацией 100 мг/мл.
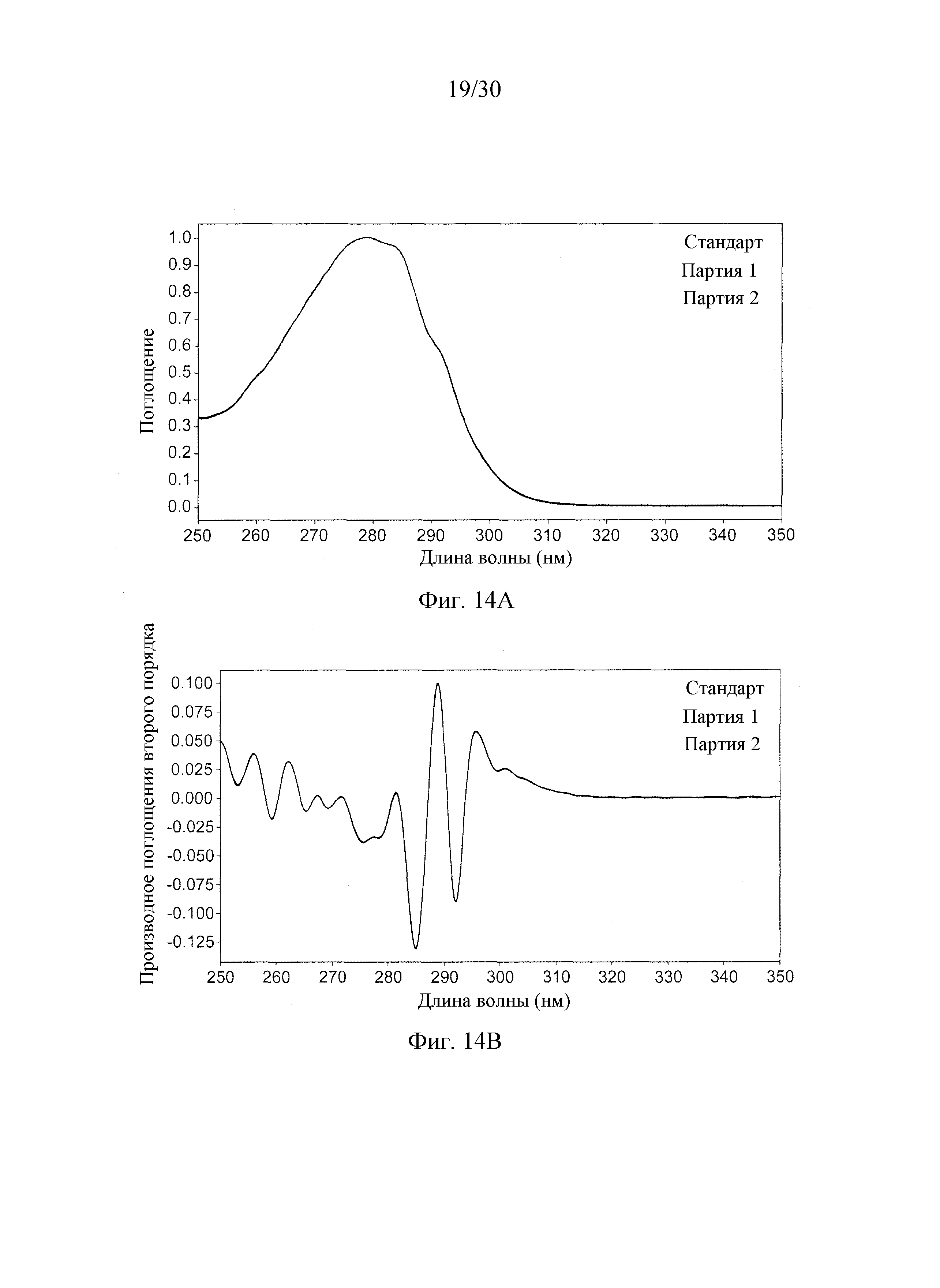
На Фиг. 14А-14В, соответственно, показаны наложенные ультрафиолетовые спектры нулевого порядка и наложенные ультрафиолетовые спектры производного второго порядка. Стандартный профиль представляет препарат DAC HYP с концентрацией 100 мг/мл, и партия 1 и партия 2 соответствуют препаратам DAC HYP с концентрацией 150 мг/мл. Все три спектра приведены на каждой из Фиг. 14А и 14В, но выглядят в виде одного спектра после наложения одного на другой.
На Фиг. 15А-15В приведены, соответственно, полномасштабные и растянутые хроматограммы эксклюзионной хроматографии. Стандартный профиль представляет препарат DAC HYP с концентрацией 100 мг/мл, и партия 1 и партия 2 соответствуют препаратам DAC HYP с концентрацией 150 мг/мл.
На Фиг. 16 приведен график агрегации DAC HYP в виде функции времени.
На Фиг. 17 показан электрофорез SDS-PAGE в редуцирующих и нередуцирующих условиях (соответственно, левая и правая панели). Стандартный профиль представляет препарат DAC HYP с концентрацией 100 мг/мл, и партия 1 и партия 2 соответствуют препаратам DAC HYP с концентрацией 150 мг/мл.
На Фиг. 18 приведены катионообменные хроматограммы DAC HYP. Стандартный профиль представляет препарат DAC HYP с концентрацией 100 мг/мл, и партия 1 и партия 2 соответствуют препаратам DAC HYP с концентрацией 150 мг/мл. Отметки пиков соответствуют различным N- и С-концевым изоформам.
На Фиг. 19 приведены хроматограммы ВЭЖХ N-связанных олигосахаридов, ферментативно отщепленных от DAC HYP. Стандартный профиль представляет препарат DAC HYP с концентрацией 100 мг/мл, и партия 1 и партия 2 соответствуют препаратам DAC HYP с концентрацией 150 мг/мл.
На Фиг. 20 приведены кривые отклика ADCC DAC HYP. Стандартный профиль представляет препарат DAC HYP с концентрацией 100 мг/мл, и партия 1 и партия 2 соответствуют препаратам DAC HYP с концентрацией 150 мг/мл.
На Фиг. 21 приведены хроматограммы ВЭЖХ N-связанных олигосахаридов, ферментативно отщепленных от DAC HYP (нижняя панель) и ZENAPAX DAC (верхняя панель), показывающие их различные профили гликозилирования.
На Фиг. 22А-22В показано сравнение ADCC-активности двух препаратов DAC HYP (именуемых Партия 3 DAC HYP и Партия 4 DAC HYP), DAC Penzberg и ZENAPAX DAC с использованием формата теста оценки ADCC с различным соотношением эффектор-к-клетке-мишени (Фиг. 22А) и формата теста оценки ADCC с различной концентрацией антитела (Фиг. 22В).
На Фиг. 23 приведено сравнение изоформ по заряду DAC HYP, DAC Penzberg и ZENAPAX DAC.
4. Подробное описание изобретения
Настоящее изобретение относится, среди прочего, к композициям DAC, обладающим специфическими свойствами, высококонцентрированным лекарственным формам DAC, пригодным для определенных путей введения, которые являются стабильными при хранении при различной температуре, векторам и клеткам-хозяевам, пригодным для получения композиций DAC, оптимизированным культуральным бульонам и условиям культивирования, пригодным для получения композиций DAC, способам очистки композиций и высококонцентрированных лекарственных форм DAC, и способам применения композиций и высококонцентрированных лекарственных форм DAC, например, для ингибирования пролиферации активированных Т- и/или В-клеток и лечения и/или профилактики заболеваний, опосредованных активированными Т- и/или В-клетками, например, таких как рассеянный склероз.
Даклизумаб (DAC) представляет собой гуманизированное моноклональное IgG1антитело, содержащее последовательность легкой цепи (VL), показанную на Фиг. 1 (положения 21-233 SEQ ID NO:2), и последовательность тяжелой цепи (VH), показанную на Фиг. 2 (положения 20-465 SEQ ID NO:4). CDR-последовательности DAC являются следующими:
VLCDR#1: S A S S S I S Y M H (SEQ ID NO:6)
VLCDR#2: T T S N L A S (SEQ ID NO:7)
VLCDR#3: H Q R S T Y P L T (SEQ ID NO:8)
VHCDR#1: S Y R M H (SEQ ID NO:9)
VHCDR#2: Y I N P S T G Y T E Y N Q K F K D (SEQ ID NO:10)
VHCDR#3: G G G V F D Y (SEQ ID NO:11)
В литературе имеются сообщения о некоторых молекулах даклизумаба, и конкретный вариант DAC ранее был промышленно доступным под торговым наименованием ZENAPAX производства Hoffman-La Roche для профилактики отторжения аллотрансплантата у пациентов с пересаженной почкой в качестве дополнения к иммунотерапии, включавшей циклоспорин и кортикостероиды. Вариант DAC продавался под торговым наименованием ZENAPAX, относящийся далее к «ZENAPAX DAC».
Другой вариант DAC, который производился в Penzberg, Германия, хотя и никогда не был промышленно доступным, он применялся в некоторых клинических испытаниях. Данный вариант относится далее к «DAC Penzberg».
Как описано в данном документе, настоящее изобретение относится, частично, к новому варианту DAC, обладающему характеристиками и свойствами, которые отличаются от, и в некоторых случаях превосходят, характеристик и свойств ZENAPAX DAC и DAC Penzberg. Следовательно, настоящее изобретение, частично, относится к композициям DAC, которые являются новыми. Новые композиции DAC отличаются одним или более признаками, более полно описанными в разделе «Заключение»:
(1) характерными pE/Q и/или Q/VHS N-концевыми изоформами;
(2) однородным профилем N-связанных олигосахаридов, отличающихся двумя основными пиками и минорным пиком;
(3) пониженной ADCC-цитотоксичностью по сравнению с ZENAPAX DAC и DAC Penzberg и
(4) низким содержанием агрегатов (<3%) при формуляции в номинальных концентрациях, таких высоких, как 150±10-15%.
Композиции DAC, обладающие одной или более данных характеристик и/или свойств, относятся в данном документе к композициям «DAC HYP». В целях приведения примеров различных аспектов и признаков изобретения, описанных в данном документе, описывается конкретная композиция DAC HYP, обладающая всеми четырьмя вышеуказанными свойствами, в качестве конкретных композиций и способов ее получения и выделения. Однако, очевидно, понятно, что композиция DAC HYP не должна обладать всеми вышеуказанными свойствами для того, чтобы попасть в объем изобретения. В конкретных вариантах осуществления DAC HYP обладает, по меньшей, двумя из характеристик (1)-(4), указанных выше (например, по меньшей мере комбинацией (1) и (2); (1) и (3); (1) и (4); (2) и (3); (2) и (4) или (3) и (4)), или по меньшей мере тремя из характеристик (1)-(4) выше (например, по меньшей мере, комбинацией (1), (2) и (3); (1), (2) и (4); (1), (3) и (4); (2), (3) и (4)). Такие композиции DAC HYP также могут содержать <3% агрегатов, <2% агрегатов и даже более низкие уровни, например, <1% агрегатов при формуляции в концентрации 100 мг ± 10-15% или даже 150 мг ± 10-15%.
Кроме того, несмотря на то, что некоторые аспекты и варианты осуществления изобретения, описанные в данном документе, иллюстрируются и приводятся в качестве примера с DAC HYP, специалисты в данной области, очевидно, понимают, что они не ограничиваются DAC HYP, и являются пригодными для композиций даклизумаба в общем, и также для IgG2, IgG3 и IgG4 анти-CD25-антител, обладающих специфическими для CD25 связывающими свойствами, аналогичными DAC, и анти-CD25-антител, подходящих для введения людям, которые не являются гуманизированными. Данные различные анти-CD25-антитела относятся в данном документе к «аналогам DAC». Такие аналоги DAC могут обычно включать шесть CDR DAC, указанных выше, но могут содержать другие CDR.
Характеристики и свойства композиций DAC HYP можно подтвердить с использованием стандартных тестов и способов. Например, профили N-концевых и С-концевых изоформ можно оценить с использованием катионообменной хроматографии при детектировании при длине волны 220 нм. В конкретном способе 100 мкл испытуемой пробы (1 мг/мл антитела, растворенного в буфере А) пропускают при комнатной температуре через колонку ProPac WCX-10 (Dionex Corporation), снабженную предколонкой ProPac WCX-10G (Dionex Corporation), с использованием следующего градиента разделения (колонку уравновешивают буфером А):
Профили N-связанного гликозилирования можно оценить отщеплением N-связанных олигосахаридов с использованием амидазы PNGase F, дериватизацией олигосахаридов флуоресцентной меткой и анализом полученной смеси с использованием нормально-фазовой ВЭЖХ с флуоресцентным детектированием. В конкретном способе дериватизированные антралиновой кислотой, отщепленные N-связанные гликаны разделяют на колонке с полимером с привитыми аминогруппами Asahipac Amino NH2P-504E (размер частиц 5 мкм, Phenomenex, ка. № CHO-2628) с использованием следующего градиентного элюирования (объем инжектирования пробы 100 мкл; колонку уравновешивают смесью 85% буфера А/15% буфера В):
Чистоту можно подтвердить с использованием электрофореза SDS-PAGE в редуцирующих условиях (минигели Precast 14% в градиенте Трис-глицин, Invitrogen Part No. 601632) и окрашиванием колоидным синим и/или эксклюзионной хроматографией с детектированием при длине волны 280 нм. В частности, 15 мкл тестируемой пробы (20 мг/мл антитела в буфере для элюирования) можно разделить при комнатной температуре на колонке 7,8×30 см TSK G3000SWXL (Toroh Biosiences, Part No. 601342), снабженной предколоночным фильтром 0,5 мкм (Upchurch, Part No: A-102X) с использованием буфера для изократического градиентного элюирования (200 мМ KPO4, 150 мМ KCl, рН 6,9) со скоростью потока 1 мл/мин.
Композиции DAC HYP и другие формуляции DAC, описанные в данном документе, такие как стабильные высококонцентрированные жидкие лекарственные формы DAC, описанные в данном документе, являются пригодными для лечения различных расстройств и состояний, которые, как полагают, опосредуются, по меньшей мере частично, активированными Т- и/или В-клетками, включая, например, отторжение аллотрансплантата и рассеянный склероз. Конкретные группы пациентов, лекарственные формы, способы введения и дозировки, и схемы, пригодные для лечения и профилактики отторжения аллотрансплантата, описаны в патенте США № 6013256 и включены в данный документ для сведения. Конкретные группы пациентов, лекарственные формы, способы введения, дозировки и схемы, пригодные для лечения и профилактики рассеянного склероза, описаны в патенте США № 7258859 и включены в данный документ для сведения. Все эти лекарственные формы, способы введения, дозировки и схемы, а также раскрытые конкретные группы пациентов и комбинированные препараты в равной степени подходят для композиций DAC HYP и, где применимо, для высококонцентрированных лекарственных форм DAC, описанных в данном документе.
Композиции и лекарственные формы DAC HYP, описанные в данном документе, вводят в количествах, обеспечивающих терапевтический эффект. Терапевтический эффект включает, не ограничиваясь этим, лечение лежащего в основе расстройства. Терапевтический эффект также может включать снижение или ослабление симптомов или побочных эффектов конкретного заболевания, что оценивается с использованием диагностических и других тестов. В отношении рассеянного склероза, различные способы оценки терапевтического эффекта, включая, например, применение магнитно-резонансной визуализации, для оценки очагов поражения головного мозга и/или оценки прогрессирования до инвалидизации, описаны в патенте США № 7258859, включенном в данный документ для сведения. Все эти различные тесты можно использовать для оценки терапевтического эффекта у пациентов, страдающих рассеянным склерозом.
Стабильные высококонцентрированные лекарственные формы DAC, с DAC вообще или аналогом DAC, особенно пригодны для подкожного введения при лечении хронических заболеваний, таких как рассеянный склероз. Лекарственные формы удобно вводить в виде однократной подкожной болюс-инъекции или в виде разведенного раствора для внутривенного введения. Лекарственные формы можно вводить подкожно пациенту на недельной или месячной основе (например, раз в неделю, раз в две недели, дважды в месяц, раз в четыре недели или раз в месяц) в пределах доз от 75 мг до 300 мг (например, 75 мг, 100 мг, 125 мг, 150 мг, 175 мг, 200 мг, 225 мг, 250 мг, 275 мг или 300 мг) или в пределах от 1 мг/кг до 4 мг/кг. Композиции можно обеспечить в виде заранее заполненных шприцов, удобных для подкожного применения. Разбавленные лекарственные формы можно вводить внутривенно в подходящих дозах с такой же кратностью, что при подкожном введении.
5. Примерные варианты осуществления
Различные аспекты и признаки изобретения, раскрытые в данном документе, описываются дополнительно в виде типичных вариантов осуществления. Специалистам в данной области, очевидно, понятно, что несмотря на то, что в приведенных в качестве примера вариантах осуществления используются определенные среды для клеточных культур, условия культивирования клеток, смолы для колоночной хроматографии и буферы для уравновешивания, промывания и элюирования, можно сделать обычные изменения. Кроме того, несмотря на то, что в качестве примера приводятся различные способы культивирования клеток с определенным штаммом-продуцентом (клон 7А11-5Н7-14-43), полагают, что можно успешно использовать другие штаммы-продуценты DAC с или без обычной оптимизации. Кроме того, признаки, которые описаны в ассоциации с конкретным вариантом осуществления (в разделе «Выводы» выше или в разделе «Примерные варианты осуществления», который следует), могут отличаться без существенного отрицательного влияния на требуемые свойства способов и композиций по изобретению, и более того, эти различные варианты осуществления можно комбинировать и использовать различными путями, если они не являются взаимоисключающими. Следовательно, очевидно, понятно, что приведенные в качестве примера варианты осуществления, описанные ниже, предназначены для иллюстрации и не для ограничения, и их не следует рассматривать в качестве ограничивающих формулу изобретения, которая логически вытекает из данных вариантов осуществления.
Способ получения, приведенный в качестве примера ниже, использовали для получения лекарственной субстанции DAC HYP с концентрацией 150 мг/мл. Для получения лекарственной субстанции DAC HYP с концентрацией 100 мг/мл в способ вводят небольшие изменения.
Клеточная культура, использованная для получения DAC HYP (см. раздел 5.3.) с концентрацией 100 мг/мл, не содержит эмульсии противовспенивающего агента, в то время как в клеточной культуре DAC HYP с концентрацией 150 мг/мл используется противовспенивающий агент Dow Corning Antifoam в низкой концентрации в биореакторе емкостью 10000 л для сведения до минимума пенообразования. Колонку СМ-650М (см. раздел 5.4.5) подвергают санитарной обработке буфером 0,5 М NaOH, 0,5 М сульфата натрия при получении лекарственной формы DAC HYP, имеющей конечную концентрацию антитела 100 мг/мл; сульфат натрия исключают из буфера для санитарной обработки при получении лекарственной формы DAC HYP с конечной концентрацией антитела, составляющей 150 мг/мл. Для получения DAC HYP в концентрации 100 мг/мл используют одну стадию ультрафильтрации/диафильтрации (UF/DF) в конце способа, непосредственно перед добавлением полисорбата 80 и разведения лекарственной субстанции до конечного объема (см. раздел 5.4.7), в то время как для получения лекарственной субстанции в концентрации 150 мг/мл используют две стадии UF/DF. В примерах, приведенных ниже, показан сравнительный анализ различных серий DAC HYP с концентрацией 100 и 150 мг/мл. В нескольких опытах партию DAC HYP в концентрации 150 мг/мл сравнивали с партией DAC HYP 100 мг/мл, полученной в масштабе 10000 л, относящуюся далее к «эталону серии RS0801».
5.1. Экспрессионная конструкция для DAC HYP
Гибридому, продуцирующую анти-Tac мышиное IgG2a моноклональное антитело, получали слиянием мышиной миеломной клеточной линии NS-1 со спленоцитами от мыши, иммунизированной человеческой Т-клеточной линией, полученной от пациента с Т-клеточным лейкозом (Uchiyama et al., 1981, J. Immunol., 126(4):1393-7). Анти-Tac выбрали за счет того, что оно реагирует с активированными Т-клетками, но не с покоящимися Т- или В-клетками. Затем было показано, что анти-Tac взаимодействуют с альфа-субъединицей рецептора человеческого IL-2 (Leonard et al., 1982, Nature, 300(5889):267-9).
Аминокислотные последовательности вариабельных областей легкой и тяжелой цепи мышиного анти-Tac-антитела определяли по соответствующей кДНК (Queen et al., 1989, Proc. Natl. Acad. Sci. USA, 86(24):10029-33). Аффинность связывания мышиного анти-Tac-антитела сохранялась в гуманизированной форме, как описано Queen et al. Определяющие комплементарность области (CDR) мышиного анти-Tac-антитела вначале пересаживали на акцепторную каркасную область человеческого антитела Eu. С помощью трехмерной модели были установлены ключевые остатки мышиной каркасной области, имеющие критическое значение для конформации CDR, и таким образом, для аффинности связывания, и они были заменены на человеческие аналоги в акцепторных каркасных областях. Кроме того, атипичные аминокислоты в акцепторных каркасных областях были заменены человеческими консенсусными остатками соответствующих положений для элиминации потенциальной иммуногенности.
Гены VL и VH DAC HYP конструировали в виде миниэкзонов отжигом и удлинением перекрывающихся олигонуклеотидов, как описано Queen et al. (1989). Для экспрессии DAC HYP в виде IgG1 полученные гены VL и VH клонировали в один экспрессионный вектор, как описано Cole et al. (J. Immunol., 1997, 159(7):3613-3621) и Kostelny et. al. (Int. J. Cancer, 2001, 93(4):556-565), для конструирования плазмиды pHAT.IgG1.rg.dE (см. Фиг. 3 и Фиг. 4А). Плазмида pHAT.IgG1.rg.dE содержит гены для обеих тяжелой и легкой каппа цепей даклизумаба, каждый под контролем промотора цитомегаловируса человека (CMV). Плазмида содержит ген гуанинфосфорибозилтрансферазы (gpt) E. coli в качестве селектируемого маркера. Генетические компоненты pHAT.IgG1.rg.dE приведены в таблице 1 ниже.

Промотор dESV40 охватывает положения 6536-6735 pHAT.IgG1.rg.dE (6536-6562 представляет 27 остатков энхансера А размером 72 п.н.; 6566-6629 представляет три повтора размером 21 п.н. Остатки 6536-6735 являются обратным комплементом 5172-1 и 1-133 в GenBank:J02400.1 (полный геном вируса обезьян 40)). Нуклеотидные последовательности генов легкой и тяжелой цепей DAC HYP в экспрессионном векторе были подтверждены секвенированием ДНК.
5.2. Стабильная клеточная линия для получения DAC HYP
Мышиную миеломную клеточную линию NS0 получали из Европейской коллекции клеточных культур (ECACC каталог #85110503, Salisbury, Wiltshire, Великобритания). Флакон с этими клетками NS0 размораживали в среду DMEM с добавлением 10% FBS. Клетки поддерживали во влажной атмосфере термостата при 37°С и 7,5% СО2. Затем клетки культивировали в базальной среде SFM-3 с добавлением 1 мг/мл BSA. SFM-3 представляет смесь среды DMEM и среды Хэмса F-12 с добавлением 10 мг/мл инсулина и 10 мкг/мл трансферрина. В течение примерно 3 месяцев клетки NS0 адаптировали к SFM-3 без добавлений при постепенном уменьшении количества FBS, присутствующей в культуральной среде, до ее полного отсутствия, и затем, наконец, удалением BSA на одной стадии. Полученную клеточную линию-хозяин пассажировали 15-20 раз в SFM-3 и готовили замороженный банк.
Адаптированные к SFM-3 клетки трансфицировали вектором pHAT.IgG1.rg.dE (линеаризованным с помощью фермента FspI (New England Biolabs, кат. № R0135L, серия 43)) электропорацией. Вкратце, 30-40 мкг pHAT.IgG1.rg.dE добавляли к 1×107 адаптированных клеток NS0 в фазе экспоненциального роста и дважды обрабатывали импульсом при 1,5 кВ, 25 мкФ с использованием аппарата Gene Pulser (BioRad, Richmond, CA). После электропорации клетки высевали в среду DMEM ± 10% FBS в пяти 96-луночных планшетах из расчета 20000 клеток/лунку, титр, который обеспечивал одну колонию на лунку после отбора микофенолиновой кислотой («MPA»). Как описано Hartman et al., 2007, Biotech.&Bioeng., 96(2):294-306, отбирали трансфектанты, которые стабильно интегрировали вектор, в присутствии микофенолиновой кислоты. Начиная со стабильного трансфектанта NS0, который продуцировал высокий уровень DAC HYP, проводили три последовательных цикла субклонирования методом лимитирующих разведений или флуоресцентно-активированным клеточным сортингом (FACS) в PFBM-1, содержащей 2,5 или 5% фетальной бычьей сыворотки (FBS; HyClone, Logan, UT). В каждом цикле субклонирования использовали один из лучших продуцентов для следующего цикла субклонирования. После третьего цикла субклонирования отбирали конечную продуцирующую клеточную линию (7А11-5Н7-14-43). Затем готовили посевной банк конечной продуцирующей клеточной линии замораживанием 1×107 клеток на флакон в 1 мл смеси 90% FBS/10% ДМСО (Sigma, St. Louis, MO).
5.3. Рекомбинантная продукция DAC HYP
5.3.1. Культивирование и выделение клеток
Клетки из одного флакона с посевным банком размораживают и размножают в постепенно возрастающих объемах в Т-колбах, флаконах-роллерах, вращающихся колбах и биореакторах до получения клеток в промышленном масштабе. После завершения получения производственной культуры жидкую клеточную культуру осветляют центрифугированием и глубинным фильтрованием и переносят в резервуар для сбора. Продолжительность получения производственной культуры составляет примерно 10 суток.
Культивирование и выделение клеток можно проводить с использованием разнообразных различных устройств, предназначенных для культивирования с применением обычного оборудования, известных в данной области. В еще одном примере клетки из одного флакона банка клеток размораживают и размножают в постепенно возрастающих объемах во встряхиваемых колбах и биореакторах до получения клеток в промышленном масштабе. После завершения получения производственной культуры жидкую клеточную культуру осветляют центрифугированием и глубинным фильтрованием и переносят в резервуар для сбора. Продолжительность получения производственной культуры составляет примерно 10 суток.
5.3.1.1. Приготовление посевного материала
Производственные партии инициируют размораживанием клеток из одного флакона банка клеток. Клетки переносят в Т-колбу, содержащую химически определенную среду, не содержащую белка базальную среду-2 (PFBM-2). Промышленный доступный порошок для получения PFBM-2 можно заказать в Invitrogen по запросу порошка для среды Hybridoma-SFM, приготовленного без NaCl, фенолового красного, трансферрина и инсулина, содержащего количество комплекса ЭДТА и ионов железа(III) в виде натриевой соли, который при восстановлении дает концентрацию 5 мг/л, и который содержит количества остальных компонентов, доведенные таким образом, что при восстановлении их концентрации являются такими же, как в восстановленной среде Hybridoma-SFM. Приготовленная среда PFBM-2 содержит следующие компоненты: 8 г/л приготовленного по заказу порошка; 2,45 г/л бикарбоната натрия; 3,15 г/л NaCl и 16,5 г/л D-глюкозы моногидрата (15 г/л глюкозы).
Затем клетки размножают серийным пассажем в бутылях-роллерах или вращающихся колбах каждые двое суток. Т-колбы, бутыли-роллеры и вращающиеся колбы помещают в термостат при температуре 37°С в атмосфере 7,5% СО2 для Т-колб и бутылей-роллеров и 5% СО2 для вращающихся колб.
Во вращающиеся колбы добавляют 5% СО2 в свободное пространство или продувают в культуру в зависимости от объема клеточной культуры и контролируют скорость импеллера на постоянных оборотах в минуту (об/мин). Требуемый титр посева при всех пассажах размножения посевного материала равняется 2,5×105 живых клеток/мл.
Кроме того, приготовление посевного материала можно провести с использованием способов, известных в данной области, с использованием разнообразных стандартных культуральных резервуаров, объемов и условий. Например, производственные партии можно инициировать размораживанием одного флакона с банком клеток. Клетки можно перенести во встряхиваемую колбу, содержащую химически определенную среду, не содержащую белка базальную среду-2 (PFBM-2). Промышленный доступный порошок для получения PFBM-2 можно заказать в Invitrogen по запросу порошка для среды Hybridoma-SFM, приготовленного без NaCl, фенолового красного, трансферрина и инсулина, содержащего количество комплекса ЭДТА и ионов железа(III) в виде натриевой соли, который при восстановлении дает концентрацию 5 мг/л, и который содержит количества остальных компонентов, доведенные таким образом, что при восстановлении их концентрации являются такими же, как в восстановленной среде Hybridoma-SFM. Приготовленная среда PFBM-2 содержит следующие компоненты: 8 г/л приготовленного по заказу порошка; 2,45 г/л бикарбоната натрия; 3,15 г/л NaCl и 16,5 г/л D-глюкозы моногидрата (15 г/л глюкозы). Необязательно на стадии биореактора можно добавить сульфат двухвалентной меди гептагидрата, например, в концентрации 0,04 мг/л.
Затем клетки размножают серийным пассажем во встряхиваемых колбах каждые двое суток. Встряхиваемые колбы помещают в термостат при температуре 37°С в атмосфере 7,5% СО2.
Встряхиваемые колбы встряхивают при постоянном значении оборотов в мин (об/мин) на платформе-шейкере в термостатах. Требуемая плотность посева при всех пассажах размножения инокулюма равнялась 2,2-2,5×105 живых клеток/мл.
Примерно через 14 суток после размораживания банка клеток, когда было продуцировано достаточное количество живых клеток, проводят посев в один из нескольких, как правило, в три или четыре биореактора из нержавеющей стали с перемешиваемым резервуаром. Перед применением посевной биореактор подвергают чистке на месте, обработке паром на месте и нагружают соответствующим объемом культуральной среды PFBM-2. Значение рН и содержание растворенного кислорода калибруют до того, как биореактор обрабатывают паром. В первый посевной биореактор вносят инокулят с достаточным количеством клеток для достижения первоначального титра клеток 2,2-2,5×105 живых клеток/мл. Проводят дальнейший перенос в большие объемы (как правило, в посевные биореакторы емкостью от 100 до 300 л, и затем в посевные реакторы емкостью 1000 л, или от 60 до 235 л, 950 л и 3750 л) с последующим культивированием в каждом реакторе примерно в течение двух суток и до достижения титра клеток 2,2-2,5×105живых клеток/мл. Значение рН культуры поддерживают добавлением газообразного СО2 или 1М раствора карбоната натрия (Na2CO3) при автоматическом контроле. Контролируемые рабочие условия в посевных и производственных биореакторах включают температуру, установленную на 37°С, рН 7,0 и 30% растворенного кислорода (в виде процента от насыщения воздухом). Содержимое биореакторов емкостью 100, 300 и 1000 л перемешивают соответственно при 100, 80 и 70 об/мин. В некоторых случаях контролируемые рабочие условия в посевных и производственных биореакторах включают температуру, установленную на 37°С, рН 7,0 под контролем барботирования CO2 и добавления основания, и 30% растворенного кислорода (в виде процента от насыщения воздухом). Содержимое биореакторов с большей емкостью можно перемешивать при 100, 80, 70 или 40 об/мин.
5.3.2. Биореактор для продукции клеточной культуры
Примерно через 2 суток инокулюм переносят из посевного биореактора емкостью 1000 л в производственный биореактор из нержавеющей стали с перемешивающимся резервуаром. Производственный реактор имеет рабочий объем примерно 10000 л. Перед применением биореактор подвергают чистке на месте, обработке паром на месте и загружают примерно 4000 л среды PFBM-2. Значение рН и содержание растворенного кислорода калибруют до того, как биореактор обрабатывают паром.
В другом примере инокулюм культивируют в посевном биореакторе емкостью 3750 л перед переносом в производственный биореактор из нержавеющей стали с перемешивающимся резервуаром с рабочей емкостью примерно 15000 л, который подвергают чистке на месте, обработке паром на месте и загружают примерно 4000-7000 л среды PFBM-2 перед использованием.
Требуемый титр посева в производственный биореактор находится в пределах 2,0-2,5×105 живых клеток/мл. Концентрат химически определенной безбелковой питательной среды (PFFM-3) (химически определенная концентрированная питательная среда, полученная восстановлением субкомпонентов PFFM3 1 и 2, с добавлением L-глутамина, D-глюкозы, фосфата натрия двухосновного гептагидрата, L-тирозина, фолиевой кислоты, соляной кислоты и гидроксида натрия) добавляют во время культивирования. PFFM3 содержит компоненты, приведенные в таблице 4.
Субкомпонент 1 среды PFFM3 содержит компоненты, приведенные в таблице 5, ниже.
Субкомпонент 2 среды PFFM3 содержит компоненты, приведенные в таблице 6, ниже.
Время и количество добавления PFFM3 к культуре приведено в таблице 7, ниже.
Значение рН культуры поддерживают примерно на рН 7,0, предпочтительно между 7,0 и 7,1, автоматически контролем газообразным СО2 или 1М раствором карбоната натрия (Na2CO3). Уровень кислорода падает примерно до 30% насыщения воздухом. Смесь кислород/воздух барботируют в культуру для достижения общей постоянной скорости газового потока, и растворенный кислород контролируют доведением требуемого соотношения газообразного воздуха к кислороду и повышением скорости вращения после достижения максимального соотношения кислорода к воздуху. В еще одном примере перемешивание доводят для поддержания постоянного соотношения сила/объем. Противовспениватель в виде эмульсии на основе семитикона вносят в биореактор по необходимости с учетом уровня образования пены. Периодически отбирают пробы для определения титра клеток, жизнеспособности клеток, концентрации продукта, глюкозы и лактата, растворенного О2, растворенного СО2, рН и осмолярности. Культуру в биореакторе собирают примерно через 10 суток после инокуляции. Перед сбором содержимое биореактора тестируют в виде необработанной массы.
5.3.3. Сбор и выделение клеток
Непосредственно перед сбором вначале производственный биореактор охлаждают до <15°С, затем рН доводят до 5,0±0,1 с использованием 0,5 М или 1 М, или 2 М лимонной кислоты, и выдерживают в течение примерно 30-90 или 45-60 мин для флоккуляции клеток и клеточного дебриса перед переносом в сосуд для сбора. Затем сбор с доведенным рН осветляют непрерывным центрифугированием при заранее определенных параметрах в отношении скорости резервуара и скорости потока, определенных в спецификации на партии.
Концентрат фильтруют через глубинный фильтр с последующим фильтрованием через мембрану 0,22 мкм и собирают в заранее стерилизованный резервуар. Значение рН бесклеточного собранного материала доводят примерно до 6,4 с использованием 1-2 М Трис буфера и хранят при 2-8°С для дальнейшей обработки. В некоторых случаях рН доводят в течение 12 ч после первоначального доведения рН содержимого биореактора 5,0.
5.4. Очистка DAC HYP
5.4.1. Обзор
Очистку и формуляцию DAC HYP проводят для повышения эффективности по сравнению с производственным процессом для ZENAPAX и для гарантии последовательной очистки от связанных с продуктом и способом примесей. В следующих подразделах описан способ очистки. Очистка основана на трех хроматографических методах (аффинной хроматографии на протеине А, анионообменной хроматографии на Q-сефарозе и катионообменной хроматографии на СМ-650(М)) в комбинации со стадиями инактивации вирусов при низком рН, фильтрования вирусов, ультрафильтрации/диафильтрации и формуляции. Все стадии проводятся в закрытом оборудовании. Схема способа очистки DAC HYP представлена на Фиг. 5 и описана ниже.
5.4.2. Хроматография с протеином А
Стадия хроматографии с протеином А является первой стадией очистки в последовательности нисходящих операций. Данная стадия проводится в одном или более циклов в зависимости от размера колонки, как правило, двух или трех циклах, для колонки, описанной в таблице 8А (т.е. бесклеточный собранный материал разделяют на две аликвотные порции и затем каждую порцию наносят и элюируют по отдельности с колонки с протеином А). Смола на основе рекомбинантного протеина А для аффинной хроматографии специфически связывает IgG, отделяя антитело от других компонентов клеточной культуры.
После уравновешивания колонки с протеином А буфером для уравновешивания нейтрализованный бесклеточный собранный материал пропускают через колонку для связывания антитела со смолой колонки. Буфер для уравновешивания представляет собой буфер из 20 мМ цитрата натрия, 150 мМ хлорида натрия, рН 7,0. Колонку нагружали до емкости не более 35 г антитела (белка) на литр упакованной смолы. После нанесения колонку промывают буфером для уравновешивания для удаления несвязанных и слабо связанных примесей из смолы, а также проводят промывание перед элюированием цитратным буфером для доведения концентрации цитрата и хлорида натрия на колонке. Цитратный буфер представляет 10 мМ цитрат натрия с рН 7,0. Затем связанное антитело элюируют с колонки со стадией изменения рН с использованием буфера для элюирования на основе 10 мМ цитрата натрия с рН 3.5. Обобщенные данные по условиям хроматографии на протеине А приведены в таблице 8А.
По мере элюирования продукта с колонки регистрируют поглощение элюента при длине волны 280 нм и используют для сбора фракции продукта (см. Фиг. 6).
Применение буфера для санитарной обработки, содержащего гидроксид натрия и бензиловый спирт, преимущественно приводит к гибели широкого ряда микробов, в то же время оказывая минимальное отрицательное влияние на качество смолы с протеином А. Для подтверждения этого в различные растворы для санитарной обработки вносили разные микроорганизмы и инкубировали в течение определенного периода времени. На различные временные интервалы инкубации растворы с внесенными микроорганизмами для санитарной обработки нейтрализовали и определяли титры микроорганизмов и сравнивали с контролем. Микробицидную активность выражали в log снижении количества микроорганизмов в течение периода времени. В таблице 8В показано снижение титров микроорганизмов как функции времени контакта с буфером для санитарной обработки, содержащим 20 мМ гидроксида натрия, 200 мМ цитрата натрия и 1% бензилового спирта.
В таблице 8С показано снижение титров микроорганизмов под действием различных растворов для санитарной обработки.
Приведенные выше данные свидетельствуют о том, что растворы для санитарной обработки, содержащие бензиловый спирт и гидроксид натрия, являются очень эффективными в уничтожении широкого ряда микроорганизмов, включая грамотрицательные и грамположительные бактерии, спорообразующие бактерии, дрожжи и грибы. Через 30 мин обычной санитарной обработки наблюдали не более чем 5 log10 снижение количества E. coli, Staphylococcusaureus, Pseudomonas aeruginosa и Candida albicans. Несмотря на то, что для гибели гриба (A. niger) потребовалось больше времени, в жидких клеточных культурах редко обнаруживают грибковую инфекцию. Наиболее часто выделяемыми микроорганизмами в биотехнологическом аппарате являются Bacillus, Pseudomonas и Staphylococcus. Они эффективно погибают через 30 мин контакта с раствором для санитарной обработки. В сравнении: один гидроксид натрия и бензиловый спирт не эффективны в уничтожении всех микроорганизмов. Кроме того, растворы гидроксида натрия для санитарной обработки не убивают спорообразующие Bacillus.
5.4.3. Выдерживание при низком рН для инактивации вирусов
Данная стадия предназначена для инактивации эндогенных вирусоподобных частиц и вирусов, чувствительных к низким рН. Элюат с протеина А из каждого цикла очистки на протеине А элюируют в сборный резервуар, куда добавляют 0,5 М HCl до достижения рН 3,5±0,1. Продукт переносят в резервуар для выдерживания, рН содержимого в нем проверяют другим рН-метром. Значение рН на стадии выдерживания при низком рН тщательно контролируют, поддерживая на рН 3,5±0,1 или 3,5±0,2 (например, рН 3,35-3,64) в течение 30-120 мин или 30-240 мин. Через 30-120 мин выдерживания элюат с инактивированными вирусами нейтрализуют до рН 7,8±0,1 или 7,80±0,3 (например, рН 8,05-8,34) с использованием 1 М Трис основания и затем пропускают через фильтр с размером пор 0,22 мкм в резервуар для сбора продукта. Обобщенные данные по условиям инактивации вирусов при низком рН приведены в таблице 9.
5.4.4. Анионообменная проточная хроматография на Q-сефарозе
Стадию анионообменной хроматографии на Q-сефарозе используют для снижения содержания связанных с продуктом и способом примесей (например, нуклеиновых кислот, белков клеток-хозяев, агрегатов продукта, выщелоченного лиганда протеина А и т.д.) и для придания способу очистки дополнительной возможности элиминировать вирусы. Электропроводность и рН нагрузки выбирают таким образом, чтобы антитело протекало через колонку и отрицательно заряженные примеси, такие как белки клеток-хозяев и клеточная ДНК, связывались с положительно заряженной смолой.
Колонку для анионообменной хроматографии уравновешивают буфером для уравновешивания, состоящим из 20 мМ Трис, 20 мМ хлорида натрия, рН 7,8. Продукт с доведенным рН со стадии выдерживания при низком рН наносят на колонку до емкости не более 60 г антитела (белка) или не более 30-60 г антитела (белка) на литр упакованной смолы. После завершения нагрузки несвязанное антитело и примеси удаляют с колонки буфером для уравновешивания.
Сбор продукта проводят по поглощению элюента при длине волны 280 нм (см. Фиг. 7).
Скорость потока при санитарной обработке составляет 100 см/ч, и время выдерживания равняется 60 мин.
Обобщенные данные по условиям хроматографии на Q-сефарозе приведены в таблице 10.
Для некоторых применений объем буфера для хранения устанавливают на 3, и температуру колонки при хранении устанавливают на 5-25°С.
5.4.5. Катионообменная хроматография на СМ-650(М)
Данная стадия является последней стадией, используемой в способе для снижения микроколичеств примесей, связанных со способом и продуктом. На данной стадии в дополнение к снижению содержания агрегатов и фрагментов расщепления антитела также уменьшается количество связанных со способом примесей, таких как нуклеиновые кислоты и белки клеток-хозяев, и выщелоченный протеин А.
Колонку уравновешивают буфером для уравновешивания из 20 мМ цитрата натрия, рН 4,5. Значения рН пула элюатов с анионообменной колонки доводят до 4,5±0,1 или 4,5±0,2 (например, 4,35-4,64) с использованием 0,5 М лимонной кислоты и нагружают на колонку с требуемой емкостью нагрузки, составляющей не более чем 25 или 30 г антитела (белка) на литр упакованной смолы. После стадии связывания колонку промывают буфером для уравновешивания для удаления любых несвязанных или слабо связанных примесей со смолы. Затем связанное антитело элюируют с колонки постадийным элюированием буфером для элюирования на основе 20 мМ цитрата натрия, 75 мМ сульфата натрия, рН 4,5. Сбор пиков проводят по поглощению элюента при длине волны 280 нм (см. Фиг. 8).
Обобщенные данные по условиям хроматографии на СМ-650(М) приведены в таблице 11.
5.4.6. Нанофильтрация
Целью стадии нанофильтрации является обеспечение для способа очистки дополнительной возможности элиминировать вирусы. Удаление вирусов и вирусоподобных частиц на этой стадии достигается посредством эксклюзионного механизма. Поры фильтра сконструированы таким образом, чтобы антитело проходило через фильтр, в то время как вирусоподобные частицы и вирусы оставались на верхней стороне фильтра.
Элюат с катионообменной колонки, который был пропущен через фильтр с размером пор 0,22 или 0,1 мкм, пропускают через небольшой, задерживающий вирусы нанофильтр с последующим быстрым смывом DAC HYP буфером для формуляции с полисорбатом 80 (40 мМ сукцината, 100 мМ хлорида натрия, рН 6,0). Стадию смыва буфером применяют для извлечения антитела, которое остается на фильтре и корпусе фильтра.
Обобщенные данные по параметрам нанофильтрации приведены в таблице 12.
5.4.7. Ультрафильтрация/диафильтрация (UF/DF)
Данная стадия способа предназначена для концентрирования продукта и обмена буфера в продукте на буфер для формуляции DAC HYP без полисорбата 80. Ее проводят в режиме тангенциального потока с использованием мембраны с номинальной задерживаемой молекулярной массой 30 кДа. Две стадии ультрафильтрации/диафильтрации используют для получения композиции с концентрацией 150 мг/мл за счет предполагаемого объема продукта в конечной концентрации и относительного объема удерживания каждой системы UF.
Первую стадию проводят с использованием большой системы UF (см. Фиг. 9). Фильтрат после нанофильтрации вначале концентрируют до концентрации примерно 30 мг/мл и затем диафильтруют в обменный буфер (буфер для формуляции без полисорбата 80). Раствор диафильтрованного антитела дополнительно концентрируют примерно до 100 мг/мл, затем выделяют в концентрации примерно 55 мг/мл из системы UF/DF и переносят через фильтр с размером пор 0,22 мкм. Раствор диафильтрованного антитела также можно получить в концентрации примерно 20-60 мг/мл. Обобщенные данные по параметрам первой стадии приведены в таблице 13.
Вторая стадия проводится с использованием меньшей UF системы, но с мембраной с той же номинальной задерживаемой молекулярной массой 30 кДа. Раствор DAC HYP (обычно с концентрацией 55 мг/мл) концентрировали примерно до концентрации 180 мг/мл, извлекали из UF системы и переносили через фильтр с размером пор 0,22 мкм. UF систему промывали буфером для формуляции без полисорбата 80 и переносили через фильтр с размером пор 0,22 мкм с получением очищенной лекарственной субстанции с концентрацией примерно 170 мг/мл или примерно 150-170 мг/мл. Обобщенные данные по параметрам второй стадии приведены в таблице 14.
5.5. Формуляция DAC HYP
Конечной стадией способа является разведение очищенной лекарственной субстанции до конечной требуемой концентрации 150 или 100 мг/л±10%, т.е. конечной требуемой концентрации 150±15 мг/мл (в случае лекарственной формы с концентрацией 150 мг/мл) или 100±10 мг/мл (в случае лекарственной формы с концентрацией 100 мг/мл), в буфере, содержащем подходящую концентрацию полисорбата 80. Формуляцию проводят постадийно.
Например, вначале буфер для формуляции без полисорбата 80 (40 мМ сукцината натрия, 100 мМ хлорида натрия, рН 6,0) добавляют к очищенной лекарственной субстанции для достижения 90% требуемого объема формулированной лекарственной субстанции. Затем добавляют расчетное количество буфера для разведения с полисорбатом 80 (40 мМ сукцината, 100 мМ хлорида натрия, 1% полисорбата 80, рН 6,0) для достижения требуемой концентрации 0,03% (мас./об.) полисорбата 80 в конечной лекарственной форме. Наконец, доводят объем продукта с использованием буфера для формуляции (приготовленного из сукцината и янтарной кислоты для композиции с концентрацией 150 мг/мл и сукцината и HCl для композиции с концентрацией 100 мг/мл) без полисорбата с достижением конечной концентрации антитела 150±15 мг/мл (предпочтительно 150±8 мг/мл). Лекарственный продукт с концентрацией 100 мг/мл формулируют аналогичным способом до конечной концентрации 100±10 мг/мл (предпочтительно 100±5 мг/мл).
Формулированную лекарственную субстанцию фильтруют через фильтр с размером пор 0,22 мкм в контейнер BioProcess Container TM (мешок BPC® (или аналогичный), который помещают внутри полутвердой цилиндрической основы). Основа включает BPC с крышкой и обеспечивает защитный барьер между гибким мешком и внешней средой. Формулированную лекарственную субстанцию хранят при температуре 2-8°С в доступном для контроля охладителе для операций этапа fill/finish для лекарственных продуктов.
Обобщенные данные по условиям формуляции приведены в таблице 15.
По 1 мл лекарственного продукта разливают во флаконы или шприц. Обобщенные данные по компонентам конечных продуктов с концентрацией 150 и 100 мг/мл приведены в таблице 16 (все количества представляют номинальные значения).
5.6. Характеристика лекарственной субстанции DAC HYP
DAC HYP гликозилирован в аминокислотном положении 296 обеих субъединиц тяжелой цепи основной олигосахаридной формой, находящейся в виде центральной фукозилированной биантенной структуры без концевой галактозы.
N-конец тяжелой цепи DAC HYP находится в виде трех основных форм: 1) N-концевого глутамина (выведенный по последовательности ДНК); (2) N-концевого пироглутамина (образовавшегося в результате циклизации N-концевого глутамина), и (3) N-концевых остатков валина, гистидина и серина в дополнение к выведенному остатку N-концевого глутамина (образовавшегося в результате неполного отщепления сигнального пептида).
С-конец тяжелой цепи DAC HYP находится с и без С-концевого остатка лизина. В основной форме отсутствует С-концевой остаток лизина, что привит к образованию С-концевого глицина.
DAC HYP имеет расчетную молекулярную массу, равную 144 кДа, основанную на первичном аминокислотном составе, определенном по нуклеотидной последовательности. Соответствующая молекулярная масса восстановленной тяжелой цепи составляет 48,9 кДа и восстановленной легкой цепи составляет 23,2 кДа; в значениях этих масс не учитывается содержание углеводов и посттрансляционные модификации.
Связывание DAC HYP является высокоспецифическим для CD25, который экспрессируется на активированных, но не на покоящихся Т- и В-лимфоцитах. Связывание DAC HYP с CD25 на этих активированных клетках блокирует связывание IL-2 с CD25 и последующее образование комплекса высокоаффинного рецептора IL-2. Следовательно, блокируется пролиферация активированных клеток, индуцированная IL-2. Полагают, что наблюдаемая и потенциальная терапевтическая эффективность DAC HYP большей частью базируется на его ингибирующем эффекте в отношении пролиферации активированных аутореактивных Т-клеток. Однако DAC HYP может проявлять свое терапевтическое действие через блокирующий эффект в отношении других CD25-несущих клеточных типов, таких как эозинофилы.
Для подтверждения того, что высококонцентрированные композиции DAC HYP с содержанием 150 мг/мл были подходящими для клинических исследований, была проведена их всесторонняя физико-химическая и биологическая оценка для характеристики и сравнения двух партий лекарственной субстанции DAC HYP с концентрацией 150 мг/мл, обозначенных в дальнейшем «партия 1» и «партия 2» (или, соответственно, «партия 150-1» или «партия 150-2»), к эталону RS0801, который получен из серии DAC HYP с концентрацией 100 мг/мл при культивировании в объеме 10000 л.
Результаты свидетельствуют о том, что серии лекарственного продукта DAC HYP с концентрацией 150 мг/мл, имеющие высокую чистоту, сравнимы с сериями с концентрацией 100 мг/мл и подходят для применения в клинических испытаниях. Обобщенные данные по этим характеристикам приведены в таблице 17.
5.6.1. Цвет, внешний вид и прозрачность
Внешний вид лекарственной субстанции DAC HYP оценивали визуально, исследуя цвет и прозрачность раствора на прямом свете на черном фоне и белом фоне без увеличения. Раствор также оценивали на наличие видимых частиц. Описание типичного внешнего вида различных серий различных серий лекарственного продукта DAC HYP приведено в таблице 17.
5.6.2. Определение рН
Значение рН DAC HYP определяли согласно протоколу № 791 Фармакопеи США. Данные по пределам рН различных серий лекарственного продукта DAC HYP обобщены в таблице 17.
5.6.3. Концентрация продукта по данным УФ-спектроскопии
Концентрацию DAC HYP определяли УФ-спектроскопией. Образцы DAC HYP разводили гравиметрически буфером. Поглощение в УФ-свете каждого разбавленного раствора пробы определяли при длине волны 278 нм против буфера. Концентрацию белка в пробе рассчитывали с использованием коэффициента поглощения DAC HYP. Концентрации белка в различных сериях лекарственного продукта DAC HYP приведены в таблице 17.
5.6.4. N-концевое секвенирование
Серии DAC HYP с концентрацией 150 мг/мл оценивали с использованием N-концевого секвенирования. Пробы анализировали с использованием автоматического секвенатора на основе деградации по Эдману.
Вычисленную аминокислотную последовательность легкой цепи по 15 первым остаткам DIQMTQSPSTLSASV (SEQ ID NO:13) подтверждали для всех проб.
Большая часть тяжелой цепи DAC HYP блокируется остатком пироглутамата (pE), который не образует N-концевую последовательность тяжелой цепи. Наиболее распространенная N-концевая последовательность тяжелой цепи DAC HYP начинается с последовательности из валина, гистидина, серина (VHS), образовавшейся в результате отсутствия процессинга трех концевых остатков сигнального пептида тяжелой цепи. Четырнадцать из первых пятнадцати N-концевых остатков были подтверждены для последовательности VHS (VHSQVQLVQSGAEVK (SEQ ID NO:14)) во всех пробах. Четвертый остаток, глутамин, невозможно было определить за счет большого количества глутамина, детектированного в LC в предшествующем цикле секвенирования. Доказательство наличия тяжелой цепи с N-концевым глутамином также имело место для всех проб. Данная последовательность является результатом природного остатка N-концевого глутамина тяжелой цепи, не подвергающегося циклизации в пироглутамат. Результаты N-концевого секвенирования для серий 150 мг/кг совпадают с последовательностями, выведенными из последовательностей, кодирующих тяжелую и легкую цепи. Сравнимые результаты получали для серий продукта с концентрацией 100 мг/мл.
5.6.5. Массовый анализ тяжелой и легкой цепи
Молекулярные массы тяжелой и легкой цепей серий DAC HYP с концентрацией 150 мг/мл и эталона RS0801 определяли жидкостной хроматографией-масс-спектрометрией (LC-MC). Все серии дегликозилировали с использованием амидазы PNGaseF, восстанавливали дитиотреитолом, алкилировали йодоуксусной кислотой и разделяли хроматографией на обращенной фазе. Теоретические массы тяжелой и легкой цепей рассчитывали по белковой последовательности. Определенные массы образцов колебались в пределах 1 Да от расчетных масс, как представлено в таблице 18 ниже.
Как описано в предыдущем подразделе, известно, что две наиболее распространенные формы тяжелой цепи DAC HYP содержат остаток N-концевого пироглутамата (pE) или последовательность из валина, гистидина, серина (VHS), и отсутствует С-концевой лизин. Молекулярные массы, полученные для двух наиболее преобладающих вариантов тяжелой цепи и легкой цепи в сериях с концентрацией 150 мг/мл, были сравнимы с эталоном RS0801 и совпадали со значениями масс, выведенными для белковых последовательностей.
Вместе с результатами пептидного картирования, приведенными в следующем подразделе, результаты определения массы тяжелой и легкой цепей подтверждают наличие предполагаемой первичной последовательности легкой цепи и тяжелой цепи в сериях DAC HYP с концентрацией 150 мг/мл.
5.6.6. Пептидное картирование
Серии DAC HYP с концентрацией 150 мг/мл и эталон RS0801 (DAC HYP, полученный из серии лекарственной субстанции с концентрацией 100 мг/мл при культивировании в объеме 10000 л) оценивали с использованием пептидного картирования с обращенно-фазовой ВЭЖХ. Все серии восстанавливали дитиотреитолом, алкилировали йодоуксусной кислотой и ферментативно расщепляли трипсином. Полученные пептиды разделяли обращенно-фазовой хроматографией и детектировали по поглощению в УФ-свете при длине волны 215 нм с получением пептидных карт.
Для проверки первичной аминокислотной последовательности пептидные карты серий с концентрацией 150 мг/мл сравнивали с эталоном RS0801. Пептиды, соответствующие 90% предполагаемых остатков тяжелой и легкой цепей, ранее были идентифицированы масс-спектрометрией в пептидной карте эталона RS0801. Остатки, которые не учитывались при пептидном картировании, представляют собой единичные аминокислоты или находятся в очень полярных дипептидах, и по предположениям они не удерживаются на обращенно-фазовой колонке. Идентифицировали массы, совпадающие с пироглутаматом, глутамином и VHS-последовательностью в N-конце N-концевого пептида тяжелой цепи, в эталоне. DAC HYP содержит консенсусный сайт для N-связанного гликозилирования в Fc-области тяжелой цепи в остатке Asn296, и устанавливали массы, совпадающие со связанными сложными центральными биантенными олигосахаридами, для пептида, содержащего остаток Asn296.
Сравнение пептидных карт серий DAC HYP с концентрацией 150 мг/мл с эталоном RS0801 приведено на Фиг. 10 (от 0 до 60 мин), Фиг. 15 (от 55 до 115 мин) и Фиг. 12 (от 110 до 170 мин). Пептидные карты серий DAC HYP с концентрацией 150 мг/мл были сравнимы с эталоном RS0801, что подтверждает наличие предполагаемой первичной структуры в сериях 150 мг/мл.
5.6.7. Спектроскопия кругового дихроизма
Серии DAC HYP 150 мг/мл и эталона RS0801 анализировали спектроскопией кругового дихроизма в дальней УФ-области (far-UV CD) для оценки вторичной структуры. Перед анализом образцы разводили водой до конечной концентрации белка 0,2 мг/мл. Спектры снимали от 195 до 260 нм с ячейкой 0,1 см, и полученный сигнал преобразовывали в молярную эллиптичность после вычитания буфера.
Наложенные друг на друга спектры far-UV CD серий DAC HYP партии 1 и партии 2 и эталона RS0801 приведены на Фиг. 13. Спектры всех серий имели положительную полосу примерно при 202 нм и отрицательную полосу примерно при 217 нм. Отрицательная полоса при 217 нм является характерной для β-складчатой конформации, преобладающей конформации молекул IgG. Спектры far-UV CD DAC всех серий были аналогичными, что является подтверждением одинаковой вторичной структуры серий DAC HYP с концентрацией 150 мг/кг и референс-стандарта RS0801.
5.6.8. Ультрафиолетовая спектроскопия
Серии DAC HYP 150 мг/мл и эталон RS0801 анализировали ультрафиолетовой (УФ) спектроскопией для оценки третичной структуры. Перед анализом пробы разводили буфером для формуляции (40 мМ сукцината, 100 мМ хлорида натрия, 0,03% полисорбата 80, рН 6,0) для конечной концентрации белка 0,5 мг/мл. Спектры снимали в диапазоне волн от 250 до 350 нм с использованием кварцевой кюветы с длиной оптического пути 1 см и нормализовали к поглощению 1,0 при 280 нм.
Наложенные друг на друга УФ-спектры нулевого порядка и производные второго порядка (рассчитанные по ровным данным нулевого порядка) приведены, соответственно, на Фиг. 14А и 14В. Спектры нулевого порядка и производные второго порядка всех тестированных серий накладывались друг на друга, что подтверждает одинаковую третичную структуру серий DAC HYP 150 мг/кг и эталона RS0801.
5.6.9. Эксклюзионная хроматография
Эксклюзионную хроматографию (SEC) проводили с использованием колонки с пористым диоксидом кремния с водной подвижной фазой и при детектировании по поглощению в УФ-свете при 280 нм. В частности, 15 мкл испытуемого образца (антитело в концентрации 20 мг/мл в буфере для элюирования) анализировали при комнатной температуре на колонке 7,8×30 см TSK G3000SWXL (Tosoh Bioscinces, part No: 601342), снабженной предколоночным фильтром с размером пор 0,5 мкм (Upchurch, part No: A-102X) с использованием буфера для изократического градиентного элюирования (200 мМ KPO4, 150 мМ KCl, рН 6,9) со скоростью потока 1 мл/мин.
Как показано на Фиг. 15А (в полном масштабе) и Фиг. 15В (в растянутом масштабе), хроматографические профили DAC HYP 150 мг/мл были сравнимы с эталоном RS0801. Во всех сериях был одинаковый основной пик, соответствующий мономеру даклизумабу, и минорный пик агрегатов. Результаты по агрегатам в сериях DAC HYP с концентрацией 150 мг/мл были сравнимы с DAC HYP 100 мг/мл, как показано в таблице 19.
Серии 150 мг/мл и эталон RS0801 анализировали с использованием SEC с детектированием мультиуглового рассеяния света (SEC-MALS) для определения молекулярной массы пика агрегата. Для всех серий молекулярная масса, полученная для пика агрегата, составляла примерно 300 кДа, что совпадало с молекулярной массой димера антитела.
За образованием агрегатов DAC HYP следили в течение 18 месячного периода. Содержание агрегатов в композиции возрастало, но процент агрегатов оставался без изменений и не превышал примерно 1,5% при хранении при 5°С в течение 18 месяцев (см. Фиг. 16). Вновь полученные данные по новым партиям указывают, что примерно 1,8-1,9% агрегатов может появиться через 6 недель хранения при 5°С, но для всех тестированных проб менее чем 3% агрегатов образовывалось в течение 5 лет при хранении при 5°С.
5.6.10. Аналитическая ультрафильтрация по скорости седиментации
Мономер и агрегаты в сериях DAC HYP 150 и 100 мг/мл характеризовали с использованием аналитической ультрафильтрации по скорости седиментации (SV-AUC). Значения коэффициента седиментации и относительное содержание мономера и каждого из агрегатов приведено в таблицах 20 и 21 ниже.
Мономер был основным компонентом, который обнаруживали в каждой из серий. Коэффициент седиментации пика мономера совпадал среди серий, указывая на то, что конформация мономера является сравнимой между сериями с концентрациями 150 и 100 мг/мл. Содержание мономера в сериях с концентрацией 150 мг/мл было сравнимо с сериями с содержанием продукта 100 мг/мл.
Преобладающие виды агрегатов в каждой из серий имели значения коэффициента седиментации, соответствующие димеру антитела. Это согласуется с результатами SEC-MALS, которые указывают на то, что пик агрегата при SEC в основном состоит из димера антитела (см. предыдущий подраздел). Также отмечали наличие низких уровней более крупных видов агрегатов, имеющих коэффициенты седиментации, которые соответствовали триммеру и тетрамеру в каждой из серий по значению AUC. Содержание димера, триммера и тетрамера в сериях с 150 мг/мл было сравнимо с сериями, содержащими 100 мг/мл.
5.6.11. Количественный анализ электрофорезом SDS-PAGE в редуцирующих условиях
Чистоту препаратов определяли электрофорезом SDS-PAGE с использованием 4-20% (обычно 14%) Трис-глициновых гелей с окрашиванием коллоидным синим. Пробы анализировали в редуцирующих условиях с нагрузкой 10 мкг образца. Чистоту рассчитывали делением суммы площади полос тяжелой цепи и легкой цепи на общую площадь полос по данным денситометрии.
Как показано в таблице 22 ниже, серии с концентрацией 150 мг/мл имели высокую чистоту и были сравнимы по этому показателю с сериями 100 мг/мл.
5.6.12. Качественный анализ электрофорезом SDS-PAGE
Чистоту DAC HYP определяли электрофорезом на гелях в редуцирующих и нередуцирующих условиях. Для анализа использовали Precast 14% или 8-16% Трис-глициновые гели. Аликвотные порции из двух партий композиции DAC HYP с концентрацией 150 мг/мл сравнивали со стандартной партией, как описано ранее. Результаты анализа чистоты DAC HYP с использованием редуцированных и нередуцированных гелей приведены на Фиг. 17. Паттерн полос для серий 150 мг/мл был сравним с эталоном RS0801, с отсутствием других полос в сериях 150 мг/мл.
5.6.13. Катионообменная хроматография
Распределение изоформ по заряду в сериях DAC HYP 150 мг/мл и сериях 100 мг/мл оценивали с использованием катионообменной хроматографии (CEX). CEX проводили с использованием колонок с непористым на основе карбоксилата, слабым катионообменником при детектировании при 220 нм. 100 мкл тестируемого образца (антитело в концентрации 1 мг/мл, растворенное в буфере А) разделяли на колонке ProPac WCX-10 (Dionex Corporation), снабженной предколонкой ProPac WCX-10G (Dionex Corporation) в следующем градиенте разделения (колонку уравновешивают буфером А).
Как показано на Фиг. 18, хроматограммы CEX серий 150 мг/мл совпадали с хроматограммами эталона RS0801 без детектирования новых изоформ по заряду в сериях 150 мг/мл. Пять основных изоформ, присутствующих на хроматограммах CEX, являются результатом гетерогенности в N-конце тяжелой цепи, и они включают: (1) два остатка пироглутамата (pE/pE); (2) один остаток пироглутамата и один остаток глутамина (pE/Q); (3) один остаток пироглутамата и одну последовательность VHS (усеченный сигнальный пептид VHS, предшествующий N-концевому остатку глутамина зрелой тяжелой цепи) (pE/VHS); (4) один остаток глутамина и одну последовательность VHS (Q/VHS); (5) две последовательности VHS (VHS/VHS). Также разделялись изоформы С-концевого лизина (K), и они представлены на Фиг. 18. Каждая из N-концевых изоформ, описанных выше, может находиться в виде различных С-концевых изоформ (0, 1 или 2 K). За счет сложного состава смеси с использованием описанного метода хорошо разделяются только изоформы С-концевого лизина, и они определяются для основных изоформ pE/pE и pE/VHS.
Количественные результаты по N- и С-концевым изоформам для серий с содержанием продукта 150 и 100 мг/мл представлены, соответственно, в таблицах 23 и 24, в которых приведенный процент основан на проценте площади под кривой концентрации (AUC) определенного пика по сравнению с общей AUC всех пиков.
5.6.14. Картирование олигосахаридов
Распределение олигосахаридов в сериях DAC HYP с содержанием продукта 150 и 100 мг/мл оценивали картированием олигосахаридов. N-связанные олигосахариды отщепляли ферментативным путем из остатка Asn296 тяжелой цепи с использованием амидазы PNGaseF. Затем олигосахариды дериватизировали флуоресцентной меткой (в данном случае антраниловой кислотой) и выделяли из антитела с использованием нейлоновой мембраны. Дериватизированные, отщепленные N-связанные гликаны разделяли при 50°С на колонке с полимером с привитыми аминогруппами Asahipac Amino NH2P-504E (размер частиц 5 мкм, Phenomenex, кат. № CHO-2628) с флуоресцентным детектированием с использованием следующего режима градиентного элюирования (объем инжекции 100 мкл; колонку уравновешивают смесью 85% буфер А/15% буфер В).
Сравнение хроматограмм серий с концентрацией 150 мг/мл с эталоном RS0801 приведено на Фиг. 19. Отмечены и приведены в таблице 25, ниже, пики олигосахаридов, составляющие, по меньшей мере, 1,0% общей площади пиков.
Все серии в основном состояли из G0 и G0-GlcNAc (G0 без G0-GlcNAc в одном плече биантенной структуры), которая является характерной для способа DAC HYP. Сиалилированные олигосахариды элюировались примерно на 68 мин, и их содержание составляет менее 1,0% во всех тестированных сериях. Не охарактеризованный олигосахарид, относящийся к пику 3, имел одинаковое содержание во всех тестированных сериях.
5.6.15. Окисление
Серии DAC HYP оценивали на потенциальное окисление метионина по определению окисленных и неокисленных триптических пептидов, присутствующих в пептидных картах. Площади пиков окисленных и неокисленных форм каждого пептида, содержащего метионин, определяли с использованием масс-спектров, полученных на ионных хроматограммах. Для каждого остатка метионина рассчитывали процент окисленного метионина делением площади пика на масс-спектре окисленного пептида на сумму площадей пиков окисленных и неокисленных пептидов.
Как представлено в таблице 26 ниже, результаты окисления метионина в сериях 150 мг/кг и пяти сериях 100 мг/кг были сравнимы.
Остатки Met251 и Met427 тяжелой цепи являются наиболее лабильными и имеют наиболее высокую степень окисления. Среди одновременно тестированных серий для оценки сравнимости уровень окисления Met251 и Met427 не превышал, соответственно, 4,8 и 1,8%.
5.6.16. Связывающая активность (связывания с CD25)
Серии DAC HYP с концентрацией 150 и 100 мг/кг оценивали на связывание с альфа-субъединицей рецептора IL-2 (CD25) с использованием ELISA в качестве показателя активности в виде фрагмента тестирования высвобождения. Растворимый CD25 иммобилизовали на микротитрационных планшетах и инкубировали с различными количествами DAC HYP. Связанный DAC HYP детектировали с использованием конъюгированного с пероксидазой хрена козьего антитела против человеческого IgG и субстрата 3,3’,5,5’-тетраметилбензидина. Строили график зависимости полученных значений поглощения от log10 концентрации DAC HYP с использованием четырехпараметрического графика и относительную активность в процентах получали с использованием анализа параллельных линий.
Результаты по лекарственной субстанции обобщены в таблице 27 ниже.
Результаты по связывающей активности серий 150 мг/кг были сравнимы с показателями для серий 100 мг/кг.
5.6.17. Поверхностный плазмонный резонанс (связывание с CD25)
Анализ поверхностного плазмонного резонанса проводили для определения константы аффинности (KD) взаимодействия связывания DAC HYP с альфа-субъединицей рецептора IL-2 (CD25).
Козье Fc-специфическое антитело против человеческого IgG иммобилизовали на поверхности чипа для захвата образцов DAC HYP, после чего вводили растворимый CD25 в различных концентрациях в двух параллелях над захваченным DAC HYP с использованием автоматического метода. Собирали данные по связыванию и корректировали с использованием проточной ячейки со стандартом и строили график с помощью программного обеспечения BIA Evaluation на основе модели Ленгмюра 1:1 с получением значений констант равновесия.
Результаты по сериям DAC HYP с концентрацией 150 мг/кг и эталона RS0801 приведены в таблице 28.
Значения константы ассоциации (ka), константы диссоциации (kd) и константы аффинности (KD) для серий с концентрацией продукта 150 мг/кг были сравнимы с эталоном RS0801.
5.6.18. Функциональная активность
Серии DAC HYP с концентрацией 150 и 100 мг/кг оценивали в отношении функциональной активности в виде фрагмента тестирования высвобождения. С помощью теста оценки функциональной активности определяют ингибирование пролиферации Т-клеток, индуцированной IL-2, по связыванию DAC HYP с альфа-субъединицей рецептора IL-2 (CD25). В присутствии IL-2 различные количества DAC HYP инкубировали с клетками KIT-225 K6 (Hori et al., 198, Blood, 70:1069-1072), которые экспрессируют рецептор IL-2. Затем детектировали ингибирование пролиферации Т-клеток под действием DAC HYP с использованием аламарового синего. Строили график зависимости полученных значений флуоресценции от log10 концентраций DAC HYP с использованием четырехпараметрического графика и определяли относительную активность с использованием анализа параллельных линий.
Результаты по функциональной активности обобщены в таблице 29.
Результаты по функциональной активности серий с концентрацией 150 мг/кг были сравнимы с показателями для серий с содержанием 100 мг/кг.
5.6.19. Антителозависимая клеточная цитотоксичность
Две серии композиций DAC HYP с концентрацией 150 мг/кг оценивали по сравнению с эталоном RS0801 DAC HYP с содержанием 100 мг/кг.
Клетки KIT-225 K6, экспрессирующие рецептор IL-2, метили51Cr и затем инкубировали с DAC HYP. Человеческие эффекторные клетки (PBMC) добавляли в различных количествах для получения разных соотношений эффектора к клеткам-мишеням (KIT-225 K6). Моноциты, несущие Fc-рецептор, взаимодействуют с Fc-доменом DAC HYP и затем вызывают требуемый лизис клеток. Степень цитотоксичности оценивали по определению высвобождения51Cr из клеток-мишеней и выражали в виде процента от максимального лизиса клеток.
PBMC от многих доноров использовали для каждой пробы. Для каждого донора процентную ADCC-активность пробы рассчитывали относительно показателя для эталона RS0801, основываясь на процентной цитотоксичности. Результаты по ADCC обобщены в таблице 30 ниже.
Кривые отклика для серий с концентрацией 150 мг/кг, эталона RS0801, положительных и отрицательных контрольных антител и контроля без антитела (для независимой от антитела клеточной цитотоксичности или AICC) приведены на Фиг. 20.
ADCC-активность серий 150 мг/кг была сравнима с эталоном RS0801.
5.6.20. Остаточный протеин А
Остаточный протеин А можно определить ELISA, в котором стандарты, контрольные пробы, контроль на планшет и тестируемые пробы разводят денатурирующим буфером и помещают в кипящую водяную баню для диссоциации протеина А и денатурации и осаждения даклизумаба. После кипячения стандарты, контроли и пробы охлаждали, центрифугировали и вносили в микротитрационный планшет, покрытый поликлональным захватывающим антителом к протеину А. Затем остаточный протеин А, присутствующий в пробах, детектировали с использованием биотинилированного антитела против протеина А вместе с стрептавидин-щелочной фосфатазой и субстратом п-нитрофенилфосфатом (PNPP). Планшет анализировали на спектрофотометрическом ридере для планшетов и строили log-log стандартную кривую, по которой определяли концентрацию протеина А. Результаты тестирования проб выражали в частях на миллион (ppm). Результаты в ppm рассчитывали делением нг/мл протеина А на концентрацию антитела в тестируемой пробе в мг/мл.
5.6.21. Содержание ДНК
Определение мышиной ДНК проводили в контрактной лаборатории с использованием количественной полимеразной цепной реакции (Q-PCR). В этом методе проводят экстракцию ДНК из пробы. Затем экстракт пробы анализируют Q-PCR с использованием мышиных специфических праймеров и зонда для амплификации специфического фрагмента повторяющегося элемента мышиной ДНК. Амплификация ДНК приводит к воспроизведению флуоресцентного сигнала, который детектируют. В пробе количественно определяют ДНК сравнением со стандартной кривой, полученной с использованием известных количеств мышиной ДНК. Результаты выражают в пикограммах ДНК на мг антитела. Обобщенные данные по среднему содержанию ДНК в различных сериях лекарственного продукта DAC HYP приведены в таблице 17.
5.6.22. Белки клетки-хозяина (HCP)
Остаточные белки клеток-хозяев в продукте определяют с использованием промышленно доступного набора. Используют аффинно-очищенное поликлональное антитело к лизату клеток NS0 для захвата и детектирования HCP NS0. Стандарт HCP получают сбором бесклеточного материала из суррогатного производственного цикла. Строят стандартную кривую с использованием рабочего стандарта HCP и готовят серийные разведения проб, содержащих HCP, в пределах стандартной кривой. Стандарты, контрольные пробы и тестируемые пробы вносят в планшет, покрытый поликлональным антителом против HCP NS0. Затем белки клеток-хозяев детектируют поликлональным антителом против HCP NS0, конъюгированным с пероксидазой хрена (HRP) вместе с субстратом 3,3’,5,5’-тетраметилбензидином (TMB). Затем планшет детектируют в спектрофотометрическом ридере для планшетов и строят четырехпараметрический график для количественного определения HCP в пробах.
Результаты анализа HCP NS0 с использованием ELISA выражают в частях на миллион (ppm). Результаты в ppm рассчитывают делением нг/мл HCP на концентрацию антитела в мг/мл. Обобщенные данные по среднему количеству HCP в различных сериях лекарственного продукта DAC HYP приведены в таблице 17.
5.6.23. Концентрация полисорбата 80
Полисорбат 80 определяют количественно спектрофотометрическим методом, который основан на образовании окрашенного кобальт-тиоцианатного комплекса с полисорбатом 80. Строят стандартную кривую с использованием серии стандартов полисорбата 80. Концентрацию полисорбата 80 в пробе определяют по стандартной кривой. Пределы концентраций полисорбата в различных сериях лекарственного продукта DAC HYP обобщены в таблице 17.
5.6.24. Осмолярность
Осмолярность определяют с использованием осмометра на основе депрессии давления пара. Перед анализом проб осмометр калибруют с использованием стандартов осмолярности, в пределы которых входят предполагаемые значения осмолярности пробы. Пределы осмолярности различных серий лекарственного продукта DAC HYP обобщены в таблице 17.
5.6.25. Выводы
Проведенные физико-химические и биологические тесты обеспечивают всестороннюю оценку лекарственных форм DAC HYP с концентрацией 150 мг/мл и DAC HYP с концентрацией 100 мг/мл.
Для всех серий DAC HYP первые 15 аминокислот тяжелой и легкой цепей, определенные N-секвенированием, пептидные карты и молекулярные массы совпадали с последовательностями генов даклизумаба.
Содержание агрегатов и распределение агрегатов по размеру во всех тестированных сериях 150 и 100 мг/мл по данным SEC-MALS и SV-AUC были сравнимы по их чистоте по данным электрофореза.
Распределение изоформ по заряду в сериях с концентрацией 150 мг/мл было аналогично этому показателю для серий 100 мг/мл с незначительными различиями в относительных количествах pE/VHS (серии 150 мг/мл = 31% pE/VHS; серии 100 мг/мл = от 34 до 42% pE/VHS) и Q/VHS (серии 150 мг/мл = от 11 до 12% Q/VHS; серии 100 мг/мл = от 4 до 9% Q/VHS) N-концевых изоформ. Характеристики DAC HYP являются следующими:
N-концевые изоформы по данным CEX:
pE/pE: 31-46%
pE/Q: 7-12%
pE/VHS: 31-42%
Q/VHS: 3-12%
VHS/VHS: 1-17%
С-концевые изоформы по данным CEX:
0K: 53-80%
1K: 14-28%
2K: 5-19%
Распределение N-связанных гликанов в DAC HYP является следующим:
G0-GlcNAc: 7,2-14,6%
G0: 80,9-88,2%
Пик 3: 1,3-1,7%
G1: 1,4-3,8%
Определенные уровни окисления DAC HYP были низкими.
DAC HYP является биологически активным по данным опытов оценки связывания с CD25 с использованием ELISA и поверхностного плазмонного резонанса. DAC HYP также характеризуется низким уровнем агрегации при хранении.
5.7. Стабильность DAC HYP
Высококонцентрированные лекарственные формы DAC HYP являются стабильными при хранении. В последующих таблицах приведены данные по стабильности серий лекарственной субстанции DAC HYP.
В таблице 31, ниже, приводятся данные по стабильности при хранении в мешках емкостью 50 мл в рекомендованных условиях (2-8°С). В таблице 32, ниже, приводятся данные по стабильности при хранении в условиях ускоренного старения в мешках емкостью 50 мл при 23-27°С. В таблице 33 ниже приводятся данные по стабильности при хранении в стрессовых условиях.
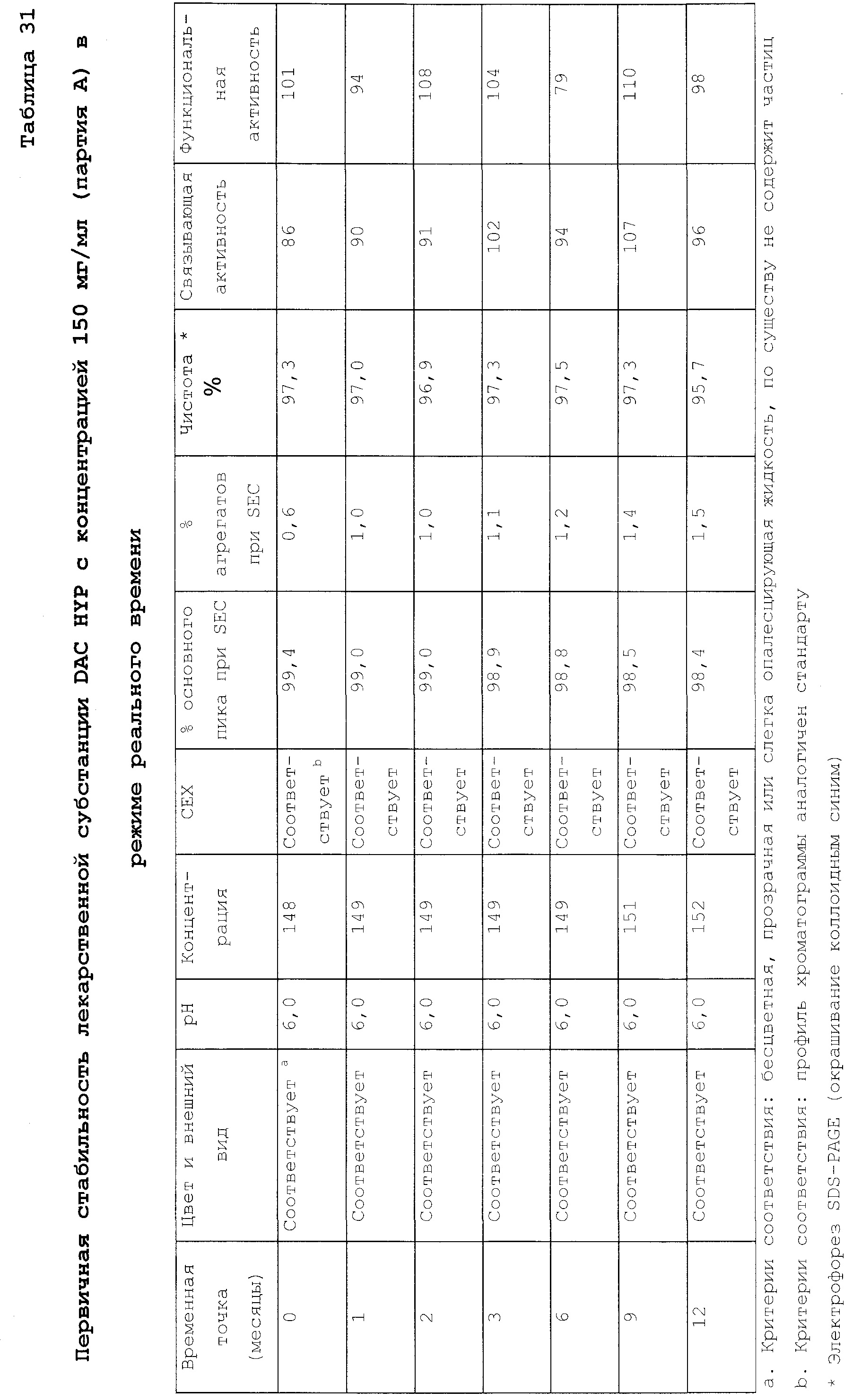


5.8. Сравнение различных форм даклизумаба
Hoffman-La Roche, Inc. («Roche») приготовил композицию даклизумаба для внутривенного введения под торговым наименованием ZENAPAXTM для лечения отторжения аллотрансплантата, испытания которого были приостановлены. DAC Penzberg представляет лекарственную форму даклизумаба для подкожного введения с концентрацией 100 мг/мл, использованную в клинических испытаниях PDL BioPharma (см. исследование CHOICE, описанное в разделе 5.9.1., ниже).
Результаты сравнения композиций DAC HYP, ZENAPAX DAC и DAC Penzberg приведены в таблице 34. В таблице буфер для формуляции представляет буфер DAC, который диафильтровали с получением конечной композиции. Следовательно, отмеченные концентрации представляют номинальные концентрации.
Различные характеристики DAC HYP сравнивали с таковыми для ZENAPAX DAC и DAC Penzberg.
Результаты сравнения по гликозилированию DAC HYP с ZENAPAX DAC приведены на Фиг. 21. Анализ типичных серий трех форм даклизумаба приведен в таблице 35.
DAC HYP также имеет достоверно более низкие уровни гликозилов маннозы (например, Man5, Man6, Man7) и более низкие уровни сиалилированных гликозилов по сравнению с ZENAPAX DAC (см., например, Фиг. 21).
Антителозависимая клеточная цитотоксичность (ADCC) является тестом in vitro, что можно использовать для оценки Fc-зависимой активности и потенциальных цитотоксических эффектов связывания антитело-мишень. С использованием мононуклеарных клеток периферической крови (PBMC) от шести здоровых доноров в качестве эффекторных клеток и клеточной линии KIT225/K6, экспрессирующей CD25, в качестве клеток-мишеней оценивали ADCC-активность различных препаратов даклизумаба в формате различных соотношений эффектора к клеткам-мишеням или в формате различных концентраций антитела.
В формате различных соотношений эффектора к клеткам-мишеням51Cr-меченные клетки KIT225/K6 (12500 клеток/лунку) предварительно инкубировали с 1 мкг/мл антитела (конечная концентрация) в течение 30 мин при 4°С в 96-луночных планшетах с V-дном в объеме 100 мкл среды для постановки теста оценки ADCC (содержащей 435 мл среды RPMI-1640; 5,0 мл L-глутамина; 50 мл инактивированной нагреванием фетальной бычьей сыворотки; 500 мкл 1000× 2-меркаптоэтанола; 5,0 мл пенициллина-стрептомицина (100×) и 5,0 мл HEPES (1 М стоковый раствор) на 500 мл); контрольные клетки инкубировали в среде для постановки теста оценки ADCC в отсутствие антитела для последующего определения антителозависимого высвобождения51Cr.
Готовили серийные разведения PBMC (эффекторы) в среде для постановки теста оценки ADCC в отдельном 96-луночном полипропиленовом планшете с получением титра клеток на 100 мкл 6,25×105 клеток; 3,13×105 клеток; 1,56×105 клеток; 7,81×104 клеток или 3,91×104 клеток. 100 мкл суспензии PBMC на лунку вносили в планшеты, содержащие51Cr-меченные клетки KIT225/K6 и антитела, с получением соотношений эффектора к мишени (E:T), равных 50:1, 25:1, 12,5:1, 6,25:1 и 3,13:1. Кроме того, 100 мкл одной среды для постановки теста оценки ADCC (без эффектора) добавляли к51Cr-меченным клеткам KIT225/K6 + mAb для определения спонтанного высвобождения51Cr. Планшеты для анализа центрифугировали при 50 RCF в течение 2 мин и инкубировали в термостате при 37°С в атмосфере с 7,5% СО2 в течение 4 ч.
За 30 мин до окончания 4-часовой инкубации в соответствующие контрольные лунки добавляли 25 мкл 8% Тритона Х-100 для определения максимального высвобождения51Cr из клеток-мишеней. После завершения 4-часовой инкубации планшеты центрифугировали при 350 RCF в течение 5 мин и 100 мкл супернатанта из каждой лунки переносили в минипробирки. Каждую минипробирку помещали в сцинтилляционный флакон и определяли радиоактивность в течение 1 мин на счетчике Beckman Gamma 5500B или аналогичном приборе.
В формате различной концентрации антитела51Cr-меченные клетки KIT225/K6 (12500 клеток/лунку; мишени) предварительно инкубировали с различными концентрациями антител mAb (5; 1; 0,2; 0,04; 0,008 и 0,0016 мкг/мл) (конечная концентрация) в течение 30 мин при 4°С в 96-луночном планшете с V-образным дном в объеме 100 мкл среды для постановки теста оценки ADCC. Контрольные клетки инкубировали с одной средой для постановки теста оценки ADCC (без mAb) для последующего определения антителонезависимого высвобождения51Cr.
Готовили серийные разведения PBMC (эффекторы) в среде для постановки теста оценки ADCC в отдельном 96-луночном полипропиленовом планшете до титра 3,13×105 клеток/100 мкл. 100 мкл суспензии PBMC на лунку вносили в планшеты, содержащие51Cr-меченные клетки KIT225/K6 + mAb с получением соотношения эффектора к мишени (E:T), равного 25:1. Кроме того, 100 мкл одной среды для постановки теста оценки ADCC (без эффектора) добавляли к51Cr-меченным клеткам KIT225/K6 + mAb для определения спонтанного высвобождения51Cr. Планшеты для анализа центрифугировали при 50 RCF в течение 2 мин и инкубировали в термостате при 37°С в атмосфере с 7,5% СО2 в течение 4 ч.
За 30 мин до окончания 4-часовой инкубации в соответствующие контрольные лунки добавляли 25 мкл 8% Тритона Х-100 для определения максимального высвобождения51Cr из клеток-мишеней. После завершения 4-часовой инкубации планшеты центрифугировали при 350 RCF в течение 5 мин и 100 мкл супернатанта из каждой лунки переносили в минипробирки. Каждую минипробирку помещали в сцинтилляционный флакон и определяли радиоактивность в течение 1 мин на счетчике Beckman Gamma 5500D или аналогичном приборе.
Результаты по оценке ADCC приведены на Фиг. 22А (формат различного соотношения эффектора к клеткам-мишеням) и на Фиг. 22В (формат различной концентрации антитела). Эти данные показывают, что максимальная ADCC-активность, достигаемая с DAC HYP, тестированным в ступенчато изменяющихся концентрациях, была примерно на 30-40% ниже по сравнению с активностью, отмеченной при тех же концентрациях ZENAPAX DAC и DAC Penzberg.
Результаты сравнения профиля изоформ DAC HYP по заряду (соответствующих N-концевым вариантам) по сравнению с ZENAPAX DAC и DAC Penzberg приведены на Фиг. 23.
5.9. Клинические испытания DAC HYP
5.9.1. Исследование CHOICE
Исследование CHOICE представляло фазу 2 рандомизированного, двойного слепого, плацебо-контролируемого испытания даклизумаба, добавленного к терапии интерфероном-бета, включавшего 230 пациентов с рецидивирующим РС. В исследовании тестировались две схемы введения 100 мг/мл DAC Penzberg (описание продукта см. выше в разделе 5.8.), который вводили подкожно: 1 мг/кг даклизумаба раз в 4 недели и 2 мг/кг даклизумаба раз в 2 недели. Результаты исследования показали, что добавление даклизумаба, вводимого в дозе 2 мг/кг раз в 2 недели в дополнение к терапии интерфероном-бета, достоверно снижало появление новых или увеличенных, контрастно усиленных гадолинием очагов через 24 недели по сравнению с терапией одним интерфероном-бета.
Результаты исследования CHOICE описаны Wynn et al., 2010, Lancet Neurol., 9(4):381-390. Лечение даклизумабом в целом хорошо переносилось. Наиболее частые нежелательные явления 3 степени тяжести были аналогичными во всех опытных группах. Нежелательные явления 3 степени тяжести наблюдали у 24% пациентов, получавших DAC/IFNβ, и у 14% пациентов, находящихся на плацебо/IFNβ. Наиболее частыми нежелательными явлениями 3 степени тяжести были инфекции, которые имели место у 7% пациентов, которых лечили DAC/IFNβ, и у 3% пациентов с плацебо/IFNβ. Отсутствовали оппортунистические инфекции или смерти, и все инфекции излечивались при применении обычных препаратов.
Результаты исследования CHOICE показали, что у пациентов с РС на фоне терапии IFNβ-a1 даклизумаб хорошо переносился и вызывал дозозависимое снижение новых или увеличенных, контрастно усиленных гадолинием (Gd+) очагов у 72% пациентов по сравнению с одним IFNβ-a1. Клиническая эффективность была связана с заметным повышением числа иммунорегуляторных природных клеток-киллеров CD56bright (NK).
5.9.2. Исследование SELECT
Проводили рандомизированное, двойное-слепое, плацебо-контролируемое с отработкой доз исследование (SELECT) для определения безопасности и эффективности двух различных доз DAC HYP.
Обзор. Исследование проводилось в 76 центрах в Чешской Республике,Германии,Венгрии, Индии, Польше, России, Украине и Великобритании. Ведение каждого пациента включало наблюдение лечащего невропатолога, лечащей медсестры (или координатора исследования), невропатолога-эксперта, техника по МРТ и фармацевта (или уполномоченного лица). Использовали централизованную интерактивную систему голосового ответа для рандомизации по всем центрам. Проводили определенный протоколом анализ бесполезности после того, как 150 пациентов завершили исследование на визите на 24 неделе.
Пациенты. Соответственно критериям включения пациентов в возрасте 18-55 лет с клинически определенным рецидивирующим рассеянным склерозом (согласно критериям МкДональда 2005 #1-4; см. Polman et al., 2005, Ann. Neurol., 58:840-846), по Расширенной шкале оценки степени инвалидизации (EDSS) 0-0,50 (Kurtzke, 1983, Neurology, 33(11):1444-1452), и имеющие по меньшей мере один рецидив РС в течение 12 месяцев до рандомизации или новый Gd+ очаг поражения в головном мозге на МРТ в течение 6 недель до рандомизации, рандомизировали на лечение DAC HYP (150 или 300 мг) или плацебо в виде подкожной инъекции один раз в 4 недели в течение 52 недель. Пациенткам в детородном возрасте необходимо было принимать эффективные контрацептивы.
Пациентов исключали, если у них был первично-прогрессирующий, вторично-прогрессирующий или прогрессирующий-рецидивирующий РС, наличие злокачественного заболевания в анамнезе, тяжелые аллергические или анафилактические реакции, или известная гиперчувствительность к лекарственным препаратам, или другие медицинские состояния, которые, по мнению исследователя, могли помешать введению DAC HYP. Дополнительные критерии исключения включали предшествующее лечение DAC HYP или ZENAPAXTM, общее облучение лимфоидной системы, введение кладрибина, митоксантрона, вакцинацию Т-клетками или рецептором Т-клеток или введение любых терапевтических mAb, за исключением натализумаба или ритуксимаба. На время рандомизации пациенты не могли получать лечение циклофосфамидом или ритуксимабом в течение предшествующего года; лечение натализумабом, циклоспорином, азатиоприном, метотрексатом, иммуноглобулином внутривенно, подвергаться плазмаферезу или цитаферезу в течение предшествующих 6 месяцев; или подвергаться вакцинации живой вирусной вакциной и лечению глатирамером ацетатом, IFNβ, интерфероном-альфа за 3 месяца до рандомизации; или получать лечение кортикостероидами, 4-аминопиридином или близкими продукты в течение 30 предшествующих суток.
Характеристики групп пациентов были следующими.
Конечные точки. Основной целью данного исследования было определение того, насколько монотерапия DAC HYP снижала рецидивы РС, что оценивали по среднегодовому показателю частоты рецидивов (ARR) на неделе 52. Рецидивы определяли как новые или периодически повторявшиеся неврологические симптомы (не связанные с лихорадкой или инфекцией), продолжавшиеся >24 часов и сопровождавшиеся новыми неврологическими симптомами при обследовании неврологом-экспертом. Комитет по независимой неврологической экспертизе (INEC), состоящий из трех не информированных неврологов по РС, оценивали все подозреваемые рецидивы для установления того, насколько протокол определения рецидива РС был удовлетворительным. Только одобренные INEC рецидивы включались в основной анализ.
Вторичными целями была оценка того, насколько DAC HYP эффективен в снижении числа суммарных новых Gd+ очагов на сканах головного мозга на неделе 8, 12, 16, 20 и 24 в подгруппе пациентов; в снижении числа новых и увеличенных Т2- гиперинтенсивных очагов на неделе 52; в снижении доли пациентов с рецидивами от исходного периода до недели 52; и в улучшении качества жизни (QoL) по изменению от исходного периода по Шкале оценки влияния рассеянного склероза из 29 пунктов (MSIS-29) (Hobart et al., 2011, Brain, 124 (Pt 5):962-973) по баллам физического влияния на неделе 52. Подтвержденное прогрессирование нетрудоспособности оценивали по изменению баллов по шкале EDSS от исходного периода до недели 52 (повышение на 1,0 пункт по EDSS для исходного периода EDSS ≥1,0 или повышение на 1,5 пункта для исходного периода EDSS=0, которое сохранялось в течение 12 недель). Оценку по EDSS проводили каждые 12 недель и на неделях 20, 52, 60 и 72.
Дополнительные конечные точки QoL представляли общую оценку благополучия субъекта по EQ-визуальной аналоговой шкале (EQ-VAS) (EuroQol-a new facility for the measurement of health-related quality of life, 2011, Accessed 17.11.11, на сайте http://www.euroqol.org/) и изменению в баллах по Общему опроснику статуса здоровья EQ-5D (EuroQol-a new facility for the measurement of health-related quality of life, 2011, Accessed 17.11.11, на сайте http://www.euroqol.org/); по Краткому опроснику статуса здоровья SF-12 из 12 пунктов (Ware et al., 1996, Medical Care 34(3):220-230) и Физиологической шкале MSIS-20 на неделе 52 (Hobart et al., 2001, Brain, 124(Pt 5):962-973).
Дополнительными конечными точками на основе МРТ было число Gd+ очагов на неделе 52, объем общих, новых и вновь увеличенных Т2-гиперинтенсивных очагов на неделе 24 и 52, объем общих и вновь увеличенных Т1-гипоинтенсивных очагов «черных дыр» (определяемых как очаги, которые были изо/гипоинтенсивными по сравнению с серым веществом и которые не усиливались после введения гадолиния) на неделе 24 и 52, и процентное изменение в общем объеме головного мозга, что оценивали с использованием метода SIENA (Smith et al., 2001, J. Comput. Assis. Tomogr. 25(3):466-475).
Субпопуляции лимфоцитов определяли на многих временных точках с использованием валидированного метода FACS. NK клетки CD56bright определяются как CD3-/CD16+/CD56bright лимфоциты. Иммуногенность DAC HYP оценивали с использованием стандартного ELISA для скрининга антител к лекарственному препарату, и затем клеточный тест использовали для тестирования нейтрализующих антител для всех положительных проб.
Статистические анализы. Размер выборки, составляющий 600 пациентов, выбрали с учетом обеспечения 90% силы для детектирования 50% снижения ARR при сравнении группы с лечением DAC HYP с группой плацебо, установленного моделированием, допускающим отрицательное биноминальное распределение с 10% показателем выбывания, 5% ошибкой типа 1 и двухсторонним тестом. ARR в группе плацебо принимали за величину 0,476, основываясь на результатах недавно проведенных испытаний при RRMS. Все сообщенные р-значения были двухсторонними.
С помощью основного анализа были оценены различия в ARR между каждой группой DAC HYP и плацебо. Рецидивы, которые имели место после «спасательного» лечения с альтернативным лечением РС, были подвергнуты экспертизе. Разница была оценена с использованием модели отрицательной биноминальной регрессии, корректирующей число рецидивов в год до вхождения в исследование, к баллам по шкале EDSS в исходном периоде (EDSS ≤2,5 против EDSS>2,5) и к возрасту в исходном периоде (≤35 против >35 лет). С использованием вторичных анализов определяли различия между видами лечения с использованием отрицательной биноминальной регрессии (число Gd+ очагов в период между неделями 8 и 24; число новых и вновь увеличенных Т2-гиперинтенсивных очагов), модели пропорциональных рисков Кокса (время до первого рецидива, время до прогрессирования заболевания) и вариационный анализ (изменение баллов по шкале EDSS, объем новых и вновь увеличенных Т2-гиперинтенсивных очагов, объем новых Т1-гипоинтенсивных очагов, QoL) и модели отношения шансов (число новых Gd+ очагов на неделе 52). Определяли долю пациентов без рецидивов с использованием кривой выживания Каплана-Мейера.
Для кумулятивного числа новых Gd+ очагов в период между неделями 8 и 21, если пациент пропускал 1 или 2 последовательных сканирования или все сканы, то последнее наблюдение не в исходном периоде анализировали переносом последнего измерения вперед, или использовали среднее число очагов в каждой группе лечения, соответственно. Для других конечных точек, которые основывались на МРТ, пропущенные данные «приписывали» с использованием среднего значения по группе с тем или иным видом лечения. Относительно шкалы MSIS-29, если у пациента отсутствовало <10 пунктов, то использовали среднее значение непропущенных пунктов для «приписывания» баллов. Для пациентов с отсутствием ≥10 пунктов и других показателей QoL, использовали произвольный наклон и интерпретационную модель для установления пропущенных данных.
Для статистического анализа конечных точек по эффективности использовали отдельные сравнения данных по группе с DAC HYP в дозе 300 мг против плацебо и группе DAC HYP с дозой 150 мг против плацебо. Использовали последовательный анализ с закрытым завершением для контроля общей ошибки типа I в результате множественных сравнений.
Анализ эффективности проводили на ITT-популяции (intent-to-treat), которая включала всех рандомизированных пациентов. Однако 21 пациент из одного исследовательского центра был предположительно исключен из ITT-популяции до завершения исследования в результате неправильного введения препаратов в центре, что установили еще до завершения исследования (все пациенты в центре получали активное лечение). В анализе чувствительности все анализы основной и вторичной эффективности повторяли при включении всех рандомизированных пациентов. Все анализы безопасности основывались на безопасности для популяции, которую определяли для всех пациентов, получивших по меньшей мере одну дозу исследуемого препарата и которые прошли по меньшей мере одно обследование после рандомизации.
Заранее запланированный анализ бесполезности проводили после того, как 150 субъектов завершили визит на неделе 24 для обеспечения возможности остановить, если гипотетические эффекты DAC HYP не были очевидными. Поскольку эффективность может меняться по ходу исследования, то не планировалось рано остановить исследование для доказательства преимущества на время анализа бесполезности. Бесполезность оценивали установлением по отдельности условной силы для кумулятивного числа новых Gd+ очагов в период между неделей 8 и 24 и конечной точкой по ARR для группы с каждой дозой. Комитет по мониторингу безопасности рецензировал данные на время анализа и, основываясь на полном соответствии данных и оценке соотношения риск/польза, рекомендовал продолжить исследование.
Выводы по полученным результатам
Отобранных участников испытания рандомизировали в период с 2/15/2008 по 5/14/2010. Показатели в исходном периоде были аналогичными по всем трем группам лечения, хотя у пациентов в группе с DAC HYP в дозе 150 мг имелась тенденция к наличию большего количества Т2 и Gd+ очагов по сравнению с группой DAC HYP с дозировкой 300 мг. Из всех рандомизированных пациентов в целом 577 (93%) завершили лечение с аналогичными относительными количествами пациентов в группах DAC HYP и плацебо.
Подробные результаты
Клиническая эффективность
ARR на неделе 52 (основная конечная точка) был ниже у пациентов, рандомизированных к лечению DAC HYP в дозе 150 мг (0,21) или 300 мг (0,23), по сравнению с плацебо (0,46; таблица 36), что означает 54% снижение для DAC HYP в дозе 150 мг по сравнению с плацебо (95% ДИ, от 31 до 91%, р<0,0001) и 50% снижение для DAC HYP в дозе 300 мг по сравнению с плацебо (95% ДИ, от 26 до 66%, р=0,0002; таблица 36). В течение 52 недель доля пациентов с рецидивами снизилась в группе с DAC HYP в дозе 150 мг (19%) и DAC HYP в дозе 300 мг (20%) по сравнению с 36% в группе с плацебо (р≤0,001 для обоих сравнений) (таблица 36). По сравнению с плацебо риск непрерывного прогрессирования нетрудоспособности за 3 месяца на неделе 52 снизился на 57% (соотношение рисков = 0,43; 95% ДИ, от 0,21 до 0,88; р=0,021) в группе DAC HYP с дозой 150 мг и на 43% (соотношение рисков = 0,57; 95% ДИ, от 0,30 до 1,09; р=0,091) в группе DAC HYP с дозой 300 мг.
Наблюдали относительное улучшение по 4,0 пунктам шкалы MSIS-29 на неделе 52 в группе пациентов, получавших DAC HYP в дозе 150 мг, против плацебо, с менее выраженным изменением в группе пациентов, получавших DAC HYP в дозе 300 мг (р<0,0008 и р=0,1284 против плацебо, соответственно; таблица 36). Также наблюдали аналогичные изменения по другим показателям связанного со здоровьем качества жизни, включая физическую и физиологическую функцию, и в общем статусе здоровья (таблица 36).
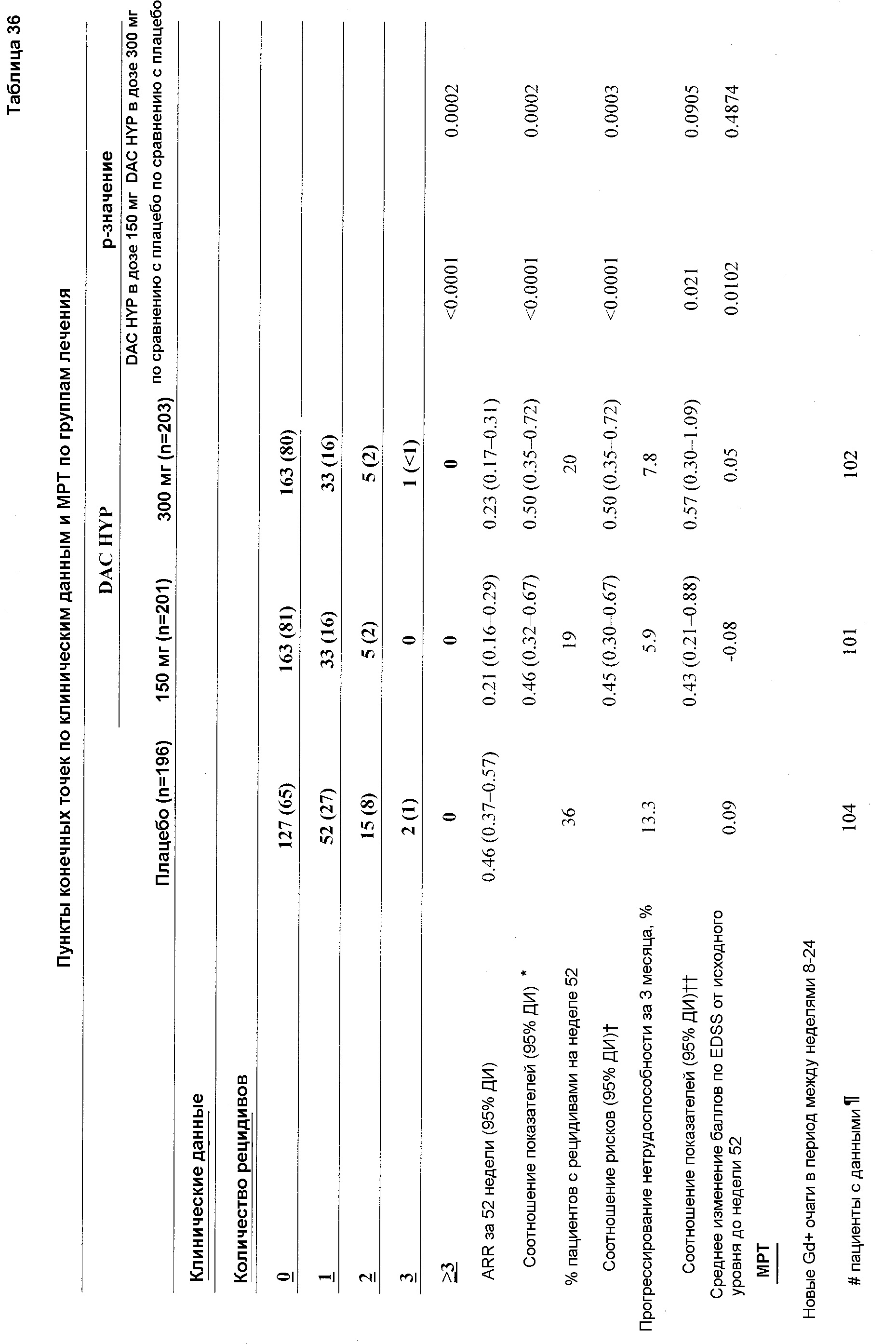



МРТ. DAC HYP приводил к снижению активности очагов РС по данным МРТ в обеих испытуемых популяциях и в подгруппе с ежемесячно проводимым МРТ в период между неделями 8 и 24 (таблица 36). В противоположность клиническим конечным точкам результаты по баллам эффективности были незначительно выше в группе с дозой 300 мг по сравнению с дозой 150 мг даже после корректировки потенциальных диспропорций в исходном периоде. Результаты продольного анализа показали, что активность Gd+ очагов была выше в группе с дозой 150 мг по сравнению с группой с дозой 300 мг в первые несколько месяцев лечения, но данные сравнялись к неделе 52 (таблица 36). Анализ чувствительности, который включал 21 пациента, исключенного из исследования, давал аналогичные результаты для всех анализов эффективности.
Безопасность. Нежелательные явления (AE) имели место у аналогичного относительного числа пациентов в группе, получавшей DAC HYP в дозе 150 мг (73%), DAC HYP в дозе 300 мг (76%) и плацебо (79%) (таблица 37). Серьезные AE имели место у 26% пациентов, находившихся на плацебо, 15% в группе с DAC HYP в дозе 150 мг и 17% в группе с DAC HYP в дозе 300 мг. Исключая рецидивы РС, SAE имели место у 6, 7 и 9% пациентов в каждой группе (таблица 37). AE, которые имели место у >5% пациентов, получавших DAC HYP, приведены в таблице 37. Частота серьезных инфекций составляла 2% у пациентов, которых лечили DAC HYP, против 0% в группе с плацебо. Среди 7 пациентов, у которых отмечали тяжелую инфекцию во время введения препарата, 1 пациент вынужден был прервать лечение за счет тяжелой инфекции, и 6 пациентов возобновили лечение после излечения инфекции. Частота кожных реакций составляла 18% у пациентов с DAC HYP в дозе 150 мг, 22% с DAC HYP в дозе 300 мг и 13% в группе плацебо (таблица 37). Серьезные кожные реакции имели место у 1% пациентов, которых лечили DAC HYP. Один пациент, получавший DAC HYP, который выздоравливал от тяжелой сыпи, умер в результате осложнения псоас-абсцесса. При вскрытии обнаружили псоас-абсцесс, ранее не диагностированный, который затронул брюшную артерию и это привело к локальному тромбозу и острому ишемическому колиту. Во время испытания было обнаружено пять злокачественных заболеваний: два случая рака шейки матки (по 1 пациентке в группе с плацебо и DAC HYP в дозе 150 мг); в одном случае новообразование в щитовидной железе в группе с DAC HYP в дозе 150 мг оказалось несерьезным узелком щитовидной железы; и два случая меланомы в группе с DAC HYP в дозе 300 мг. Меланому лечили локальным иссечением без сообщения о рецидиве.


Лабораторные анализы. У пациентов, которых лечили DAC HYP, имело место повышение общего числа NK-клеток (клеток/см3) по сравнению с плацебо на неделе 52 (среднее значение: 42,0 (DAC HYP в дозе 150 мг); 46,5 (DAC HYP в дозе 300 мг) по сравнению с -4,5 для плацебо; р=<0,001). Повышение числа клеток NK было связано с избирательным повышением числа клеток NK CD56bright со среднего значения 7,77 в исходном периоде до 44,84 в конце лечения. В противоположность имели место только незначительные в количестве клеток NK CD56bright(изменения среднего значения от 122,68 до 123,70). Повышение числа клеток NK CD56brightбыло очевидным на первой временной точке после исходного периода (неделя 4) в обеих группах DAC HYP против плацебо (р<0,0001). Количество клеток NK CD56bright возросло со среднего значения 0,6% лимфоцитов в исходном периоде до 2,8% на неделе 52. В противоположность у пациентов, получавших лечение DAC HYP, имело место небольшое снижение В-клеток и общего числа лимфоцитов (таблица 38). Число клеток CD4+ и CD8+ претерпело снижение примерно на 7-10% на неделе 52 у пациентов, которых лечили DAC HYP, и соотношение CD4+/CD8+ оставалось без изменений в течение всего периода лечения.

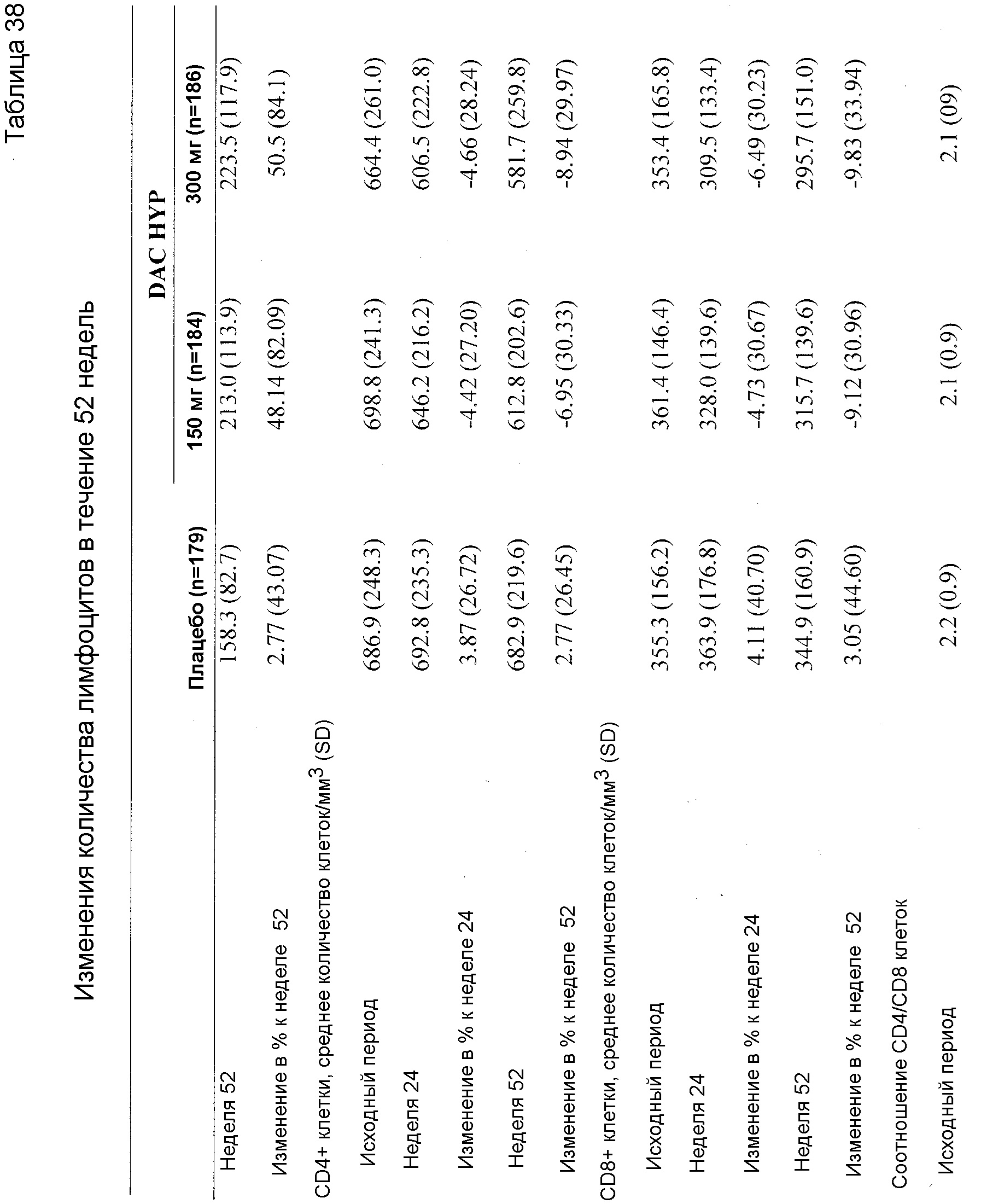

Результаты тестов оценки функции печени (LFT) выше 5X ULN имели место у 4% пациентов в группе с DAC HYP и <1% с плацебо. Данные отклонения, как правило, отмечали в позднем периоде лечения (среднее начало + сутки 308), и они проходили в течение среднего времени, составляющего 62 суток. Из 17 пациентов, получавших DAC HYP, с >5X ULN 6 продолжали или продолжили лечение DAC HYP по меньшей мере через 6 месяцев после восстановления, все без рецидива во время этого периода. У двух пациентов повышение LFT было связано с инфекциями (в одном случае это был гепатит В и в одном случае инфекция, вызванная цитомегаловирусом).
Иммуногенность. На неделе 24 нейтрализующие антитела к DAC HYP были детектированы у 6 (2%) пациентов, получавших лечение DAC HYP (5 пациентов в группе с дозой 150 мг и 1 пациент в группе с дозой 300 мг). У некоторых пациентов появление этих антител носило временный характер, и на неделе 52 нейтрализующие антитела к DAC HYP имели место только у 1 субъекта из каждой группы с DAC HYP.
Заключение. Антагонистическое воздействие на CD25 при ежемесячном подкожном введении DAC HYP в виде монотерапии показало значительные и клинически значимые эффекты в течение 1 года в отношении активности РС, о чем судили, например, по снижению показателя частоты рецидивов, числа новых определяемых на МРТ очагов и прогрессированию инвалидизации в подавляющей популяции не подвергавшихся воздействию пациентов с РС.
6. Конкретные варианты осуществления, включение для сведения
Различные аспекты настоящего изобретения описаны в вариантах осуществления, приведенных ниже в последующих пронумерованных пунктах.
1. Модифицированная клетка NS0, которая адаптирована к росту в бессывороточных и не содержащих холестерина средах и которая сконструирована для экспрессии рекомбинантного белка, где указанная клетка способна достигать объемного выхода, превышающего 100 мг/л/сутки рекомбинантного белка, в культуре объемом 100 л в 10-суточном способе с подпиткой при культивировании в бессывороточной и не содержащей холестерина среде.
2. Модифицированная клетка NS0 по варианту осуществления 1, которая способна достигать объемного выхода, превышающего 100 мг/л/сутки рекомбинантного белка, в культуре объемом 1000 л в 10-суточном способе с подпиткой при культивировании в средах, не содержащих холестерина и компонентов животного происхождения.
3. Модифицированная клетка NS0 по варианту осуществления 1, которая способна достигать объемного выхода, превышающего 100 мг/л/сутки рекомбинантного белка, в культуре объемом 16000 л в 10-суточном способе с подпиткой при культивировании в средах, не содержащих холестерина и компонентов животного происхождения.
4. Модифицированная клетка NS0 по одному из вариантов осуществления 1-3, к которой добавляют питательную среду по следующей схеме, где добавленный объем представляет процент от первоначального объема клеточной культуры:
5. Модифицированная клетка NS0 по варианту осуществления 1, которая способна достигать объемного выхода, превышающего 200 мг/л/сутки рекомбинантного белка, в культуре объемом по меньшей мере 100 л в 13-суточном способе с подпиткой.
6. Модифицированная клетка NS0 по варианту осуществления 5, которая способна достигать объемного выхода, превышающего 200 мг/л/сутки рекомбинантного белка, в культуре объемом 1000 л в 13-суточном способе с подпиткой при культивировании в средах, не содержащих холестерина.
7. Модифицированная клетка NS0 по варианту осуществления 5, которая способна достигать объемного выхода, превышающего 100 мг/л/сутки рекомбинантного белка, в культуре объемом 16000 л в 10-суточном способе с подпиткой при культивировании в бессывороточных и не содержащих холестерина средах.
8. Модифицированная клетка NS0 по варианту осуществления 1, которую стабильно трансфицируют нуклеиновой кислотой, пригодной для экспрессии моноклонального анти-CD25-антитела.
9. Модифицированная клетка NS0 по варианту осуществления 8, где моноклональное анти-CD25-антитело содержит VL легкой цепи, соответствующую по последовательности положениям 21-233 в SEQ ID NO:2, и VH тяжелой цепи, соответствующую по последовательности положениям 20-465 в SEQ ID NO:4.
10. Модифицированная клетка NS0 по варианту осуществления 1, которую трансформируют вектором pAbX.gpt.
11. Модифицированная клетка NS0 по варианту осуществления 1, которую трансформируют вектором pHAT.IgG1.rg.dE.
12. Модифицированная клетка NS0 по варианту осуществления 1, которая обозначена как клон 7А11-5Н7-14-43.
13. Способ получения рекомбинантного белка, включающий культивирование модифицированной клетки NS0 по одному из вариантов осуществления 1-12.
14. Способ по варианту осуществления 13, в котором модифицированную клетку NS0 культивируют в условиях, которые приводят к продукции по меньшей мере 100 мг/л/сутки рекомбинантного белка в 100 л, 1000 л или 16000 л культуры в 10-суточном способе с подпиткой, или по меньшей мере 200 мг/л/сутки рекомбинантного белка в 100 л, 1000 л или 16000 л культуры в 13-суточном способе с подпиткой.
15. Способ по вариантам осуществления 13 или 14, в котором модифицированную клетку NS0 культивируют в отсутствие сыворотки крови и холестерина.
16. Способ по варианту осуществления 15, в котором модифицированную клетку NS0 культивируют в отсутствие трополона и гидрокортизона.
17. Способ по вариантам осуществления 13 или 14, в котором модифицированную клетку NS0 культивируют в базальной и/или питательной среде, содержащей 10-35 г/л глюкозы.
18. Способ по варианту осуществления 17, в котором модифицированную клетку NS0 культивируют в базальной среде, содержащей 15 г/л глюкозы, и/или питательной среде, содержащей 28 г/л глюкозы.
19. Способ по варианту осуществления 18, в котором базальная среда состоит из компонентов PFBM2±10%.
20. Способ по варианту осуществления 18, в котором питательная среда состоит из компонентов PFFM3±10%.
21. Способ по вариантам осуществления 19 или 20, в котором клетку культивируют в базальной среде в течение 1-3 суток и затем в питательной среде в течение 10-13 суток.
22. Способ по варианту осуществления 18, в котором питательную среду добавляют по схеме, приведенной в таблице 7, ±10%.
23. Вектор, пригодный для рекомбинантной экспрессии интересующего белка, содержащий слабый промотор, регулирующий экспрессию селектируемого маркера, функционирующего в клетках млекопитающих, и сильный промотор, регулирующий экспрессию интересующего белка.
24. Вектор по варианту осуществления 23, где интересующий белок является терапевтическим антителом.
25. Вектор по варианту осуществления 24, где терапевтическое антитело представляет собой анти-CD25-антитело.
26. Вектор по варианту осуществления 25, где анти-CD25-антитело содержит CDR даклизумаба.
27. Вектор по варианту осуществления 26, где анти-CD25-антитело представляет собой даклизумаб.
28. Способ получения клетки-хозяина млекопитающих, которая имеет высокий объемный выход интересующего белка, включающий трансфекцию клетки вектором по любому из вариантов осуществления 23-27 и отбор клетки, которая способна продуцировать по меньшей мере 100 мг/л/сутки рекомбинантного белка в 100 л, 1000 л или 16000 л культуры в 10-суточном способе с подпиткой, или по меньшей мере 200 мг/л/сутки рекомбинантного белка в 100 л, 1000 л или 16000 л культуры в 13-суточном способе с подпиткой.
29. Композиция, содержащая даклизумаб, в которой даклизумаб характеризуется присутствием N-связанной изоформы тяжелой цепи pE/Q и/или N-концевой изоформы тяжелой цепи Q/VHS.
30. Композиция по варианту осуществления 29, в которой N-концевая изоформа тяжелой цепи pE/Q составляет примерно 6-15% даклизумаба.
31. Композиция по варианту осуществления 29, в которой N-концевая изоформа тяжелой цепи pE/Q составляет примерно 7-12% даклизумаба.
32. Композиция по любому из вариантов осуществления 29-31, в которой N-концевая изоформа тяжелой цепи Q/VHS составляет примерно 1-15% даклизумаба.
33. Композиция по варианту осуществления 32, в которой N-концевая изоформа тяжелой цепи Q/VHS составляет примерно 3-12% даклизумаба.
34. Композиция по варианту осуществления 29, в которой тяжелая цепь даклизумаба находится в следующих N-концевых изоформах:
35. Композиция по варианту осуществления 29, в которой тяжелая цепь даклизумаба находится в следующих N-концевых изоформах:
36. Композиция по варианту осуществления 29, в которой даклизумаб характеризуется профилем изоформ при катионообменной хроматографии, по существу аналогичным представленному на Фиг. 18.
37. Композиция по варианту осуществления 29, в которой даклизумаб представляет собой DAC HYP.
38. Композиция, содержащая даклизумаб, в которой даклизумаб характеризуется профилем N-связанного гликозилирования по данным ВЭЖХ, содержащим два основных пика, один соответствующий олигосахариду G0-GlcNAc, и один, соответствующий олигосахариду G0, где объединенные AUC данных двух пиков составляют примерно 88-99,5% от общей AUC всех пиков.
39. Композиция по варианту осуществления 38, в которой AUC пика G0-GlcNAc составляет примерно 5-18% от общей AUC всех пиков, и AUC пика G0 составляет примерно 75-92% от общей AUC всех пиков.
40. Композиция по варианту осуществления 39, в которой AUC пика G0-GlcNAc составляет примерно 6-16% от общей AUC всех пиков, и AUC пика G0 составляет примерно 78-90% от общей AUC всех пиков.
41. Композиция по любому из вариантов осуществления 38-40, в которой профиль N-связанного гликозилирования имеет менее чем примерно 3% Man5.
42. Композиция по варианту осуществления 41, в которой профиль N-связанного гликозилирования имеет менее чем примерно 0,5% G2, Man6 и/или Man7.
43. Композиция по варианту осуществления 38, в которой профиль N-связанного гликозилирования содержит третий пик, соответствующий сиалилированным олигосахаридам, и AUC пика сиалилированных олигосахаридов составляет 1% или менее от общей AUC всех пиков.
44. Композиция по варианту осуществления 38, в которой профиль N-связанного гликозилирования содержит третий пик, соответствующий олигосахариду G1, и AUC пика G1 составляет примерно 1-5% от общей AUC всех пиков.
45. Композиция по варианту осуществления 44, в которой AUC пика G1 составляет примерно 1-2% от общей AUC всех пиков.
46. Композиция по варианту осуществления 38, в которой даклизумаб имеет профиль N-связанного гликозилирования по данным ВЭЖХ, по существу аналогичный представленному на Фиг. 19 или представленному на нижней панели Фиг. 21.
47. Композиция по варианту осуществления 38, в которой даклизумаб представляет собой DAC HYP.
48. Композиция, содержащая даклизумаб, который проявляет менее 35% среднюю ADCC-цитотоксичность по данным клеточного теста в условиях in vitro с использованием эффекторных клеток по меньшей мере от 3 здоровых доноров и клеток Kit 225 K6 в качестве клеток-мишеней, в концентрации даклизумаба 1 мкг/мл и при соотношении эффектора к клеткам-мишеням, равным примерно 25:1.
49. Композиция по варианту осуществления 48, где даклизумаб проявляет 10-30% среднюю ADCC-цитотоксичность в указанном тесте.
50. Композиция по вариантам осуществления 48 или 49, где в указанном тесте используются эффекторные клетки по меньшей мере от 6 здоровых доноров.
51. Композиция по вариантам осуществления 48 или 49, где в указанном тесте используются эффекторные клетки по меньшей мере от 10 здоровых доноров.
52. Композиция по любому из вариантов осуществления 48-51, в которой даклизумаб представляет собой DAC HYP.
53. Композиция, пригодная для получения лекарственной формы даклизумаба, содержащая примерно 150-190 мг/мл даклизумаба и такие количества наполнителей, что разведение композиции буфером для разведения дает разбавленную композицию, которая содержит примерно 85-165 мг/мл даклизумаба и имеет осмолярность в пределах примерно 267-327 мОсм/кг и рН в пределах примерно 5,8-6,2 при 25°С, и в которой по меньшей мере примерно 95% даклизумаба находится в мономерной форме по данным эксклюзионной хроматографии.
54. Композиция по варианту осуществления 53, которая содержит такие количества наполнителей, что при разведении композиции буфером для разведения разбавленная композиция содержит примерно 85-115 мг/мл даклизумаба.
55. Композиция по варианту осуществления 53, которая содержит такие количества наполнителей, что при разведении композиции буфером для разведения разбавленная композиция содержит примерно 150±15 мг/мл даклизумаба.
56. Композиция, содержащая примерно от 4 до 15 мг/мл даклизумаба, где 0,1% или менее даклизумаба находится в агрегатной форме.
57. Композиция по варианту осуществления 56, которую получают очисткой композиции даклизумаба, содержащей примерно от 4 до 15 мг/мл даклизумаба, где до 2,5% даклизумаба находится в агрегатной форме, с использованием колоночной хроматографии на слабой катионообменной смоле.
58. Композиция по варианту осуществления 57, где слабая катионообменная смола представляет собой СМ-650М.
59. Композиция по варианту осуществления 58, где смолу СМ-650М уравновешивают буфером для уравновешивания, содержащим примерно 20 мМ цитрата натрия, рН 4,4-4,6, и даклизумаб элюируется буфером для элюирования, содержащим примерно 20 мМ цитрата натрия и примерно 75 мМ сульфата натрия, рН 4,4-4,6.
60. Композиция по варианту осуществления 59, где хроматографию проводят на цилиндрической колонке с использованием основания смолы с высотой примерно 10-30 см или примерно 17-19 см, и даклизумаб элюируется при температуре в пределах примерно 4-22°С или примерно 18-22°С, и со скоростью потока в пределах примерно 50-200 см/ч или примерно 90-110 см/ч.
61. Композиция, подходящая для введения людям, содержащая примерно 85-165 мг/мл даклизумаба; и примерно 0,02-0,04% (мас./об.) полисорбата 80, где композиция имеет осмолярность в пределах примерно 267-327 мОсм/кг и рН в пределах примерно 5,8-6,2 при 25°С, и по меньшей мере примерно 95% даклизумаба находится в мономерной форме по данным эксклюзионной хроматографии.
62. Композиция по варианту осуществления 61, в которой по меньшей мере примерно 99% даклизумаба находится в мономерной форме по данным эксклюзионной хроматографии.
63. Композиция по варианту осуществления 61, которая содержит примерно 85-115 мг/мл даклизумаба.
64. Композиция по варианту осуществления 63, которая в основном состоит из примерно 100 мг/мл даклизумаба, примерно 40 мМ сукцината натрия, примерно 100 мМ хлорида натрия и примерно 0,03% (мас./об.) полисорбата 80, и имеет рН примерно 6,0 при 25°С.
65. Композиция по варианту осуществления 61, которая содержит примерно 135-165 мг/мл даклизумаба.
66. Композиция по варианту осуществления 65, которая в основном состоит из примерно 150 мг/мл даклизумаба, примерно 40 мМ сукцината натрия, примерно 100 мМ хлорида натрия и примерно 0,03% (мас./об.) полисорбата 80, и имеет рН примерно 6,0 при 25°С.
67. Композиция по варианту осуществления 65, которую получают способом, включающим стадии концентрирования композиции даклизумаба, содержащей примерно 4-15 мг/мл даклизумаба, посредством ультрафильтрации в подходящем буфере с достижением концентрации даклизумаба в пределах примерно 85-180 мг/мл и необязательно разведения концентрированной композиции буфером для разведения.
68. Фармацевтическая композиция, подходящая для подкожного введения, содержащая примерно 85-165 мг/мл даклизумаба, в которой процент даклизумаба в агрегатной форме не превышает примерно 3% после хранения в течение 12 месяцев при температуре в пределах примерно 2-8°С.
69. Фармацевтическая композиция по варианту осуществления 68, которая содержит примерно 85-115 мг/мл даклизумаба.
70. Фармацевтическая композиция по варианту осуществления 68, которая содержит примерно 135-165 мг/мл даклизумаба.
71. Фармацевтическая композиция по вариантам осуществления 69 или 70, в которой процент даклизумаба в агрегатной форме не превышает примерно 2% после хранения в течение 12 месяцев при температуре в пределах примерно 2-8°С.
72. Фармацевтическая композиция по вариантам осуществления 69 или 70, в которой процент даклизумаба в агрегатной форме не превышает примерно 3% после хранения в течение 18 месяцев при температуре в пределах примерно 2-8°С.
73. Способ сбора рекомбинантного белка из клеточной культуры, включающий стадии:
(i) доведения рН клеточной культуры, которая экспрессирует и секретирует рекомбинантный белок, до рН в пределах примерно 4,5-5,5;
(ii) инкубации клеточной культуры с доведенным рН в течение примерно 30-90 мин при температуре в пределах примерно 4-15°С; и
(iii) центрифугирования инкубированной клеточной культуры с доведенным рН для удаления клеточного дебриса.
74. Способ получения очищенной композиции даклизумаба, включающий стадии:
(i) адсорбции даклизумаба из неочищенного препарата даклизумаба на смолу для аффинной хроматографии;
(ii) промывания смолы для аффинной хроматографии буфером для промывания для удаления загрязняющих примесей;
(iii) элюирования адсорбированного даклизумаба буфером для элюирования;
(iv) инактивации вирусов в элюате доведением рН примерно до 3-4 и инкубации элюата с доведенным рН при определенной температуре в течение периода времени, достаточного для инактивации вирусов;
(v) нейтрализации элюата с инактивированными вирусами до рН в пределах примерно 7,7-7,9 (при 25°С);
(vi) пропускания нейтрализованного элюата через смолу для сильной анионообменной хроматографии;
(vii) адсорбции даклизумаба элюата со стадии (vi) на смоле для слабой катионообменной хроматографии; и
(viii) элюирования адсорбированного даклизумаба со смолы для слабой катионообменной хроматографии.
75. Способ по варианту осуществления 74, в котором неочищенный препарат даклизумаб собирают из клеточной культуры.
76. Способ по варианту осуществления 74, в котором неочищенный препарат даклизумаб получают культивированием клетки-хозяина 7А11-5Н7-14-43 в условиях, в которых даклизумаб секретируется в культуральную среду, и сбором секретированного даклизумаба.
77. Способ по варианту осуществления 76, в котором даклизумаб собирают с использованием способа по варианту осуществления 73.
78. Способ по варианту осуществления 74, который дополнительно включает стадии:
(ix) фильтрования элюированной композиции даклизумаба со стадии (viii) для удаления вирусов; и
(x) концентрирования профильтрованного раствора с использованием ультрафильтрации с получением очищенной композиции даклизумаба, содержащей примерно 85-180 мг/мл даклизумаба.
79. Способ по варианту осуществления 78, который дополнительно включает стадию разведения очищенной композиции даклизумаба буфером для разведения с получением композиции, содержащей примерно 85-165 мг/мл даклизумаба и примерно 0,02-0,04% (мас./об.) полисорбата 80, где композиция имеет осмолярность в пределах примерно 267-327 мОсм/кг и рН в пределах примерно 5,8-6,2 при 25°С, и по меньшей мере примерно 95% даклизумаба находится в мономерной форме по данным эксклюзионной хроматографии.
80. Способ по варианту осуществления 79, в котором полученная композиция содержит менее чем примерно 50 ppm белков клеток-хозяев из рекомбинантного источника даклизумаба, менее чем примерно 10 ppm протеина А, и не более чем 3% даклизумаба в композиции находится в агрегатной форме.
81. Базальная среда PFBM2.
82. Питательная среда PFFM3.
83. Композиция даклизумаба, которую получают способом, включающим стадию культивирования клетки-хозяина по любому из вариантов осуществления 1-12 в условиях, в которых даклизумаб секретируется в культуральную среду.
84. Композиция даклизумаба по варианту осуществления 83, где способ дополнительно включает стадию выделения секретированного даклизумаба из клеточной культуральной среды.
85. Буфер, пригодный для санитарной обработки смолы с протеином А для аффинной хроматографии, содержащий примерно 100-500 мМ цитрата натрия, примерно 10-30 мМ NaOH и примерно 0,5-3% (об./об.) бензилового спирта.
86. Способ санитарной обработки колонки с протеином А для аффинной хроматографии, включающий промывание колонки буфером для санитарной обработки по варианту осуществления 85 со скоростью потока и в течение периода времени, достаточных для санитарной обработки колонки.
87. Способ по варианту осуществления 86, в котором колонку промывают примерно 1,8 объемами колонки буфера для санитарной обработки со скоростью потока 150 см/ч, промытую колонку инкубируют без протока в течение примерно 30-45 мин и затем уравновешивают буфером для уравновешивания.
88. Способ по варианту осуществления 87, в котором буфер для уравновешивания содержит примерно 20 мМ цитрата натрия и 150 мМ NaCl, и имеет рН примерно 7 (при 25°С).
89. Способ лечения пациента, страдающего рассеянным склерозом, включающий введение пациенту количества композиции DAC HYP, достаточного для обеспечения терапевтического эффекта.
90. Способ по варианту осуществления 89, в котором композицию DAC HYP вводят внутривенно.
91. Способ по варианту осуществления 90, в котором композицию DAC HYP вводят в количестве, соответствующем примерно 0,8-0,9 мг/кг DAC HYP.
92. Способ по варианту осуществления 91, в котором композицию DAC HYP вводят в количестве, соответствующем примерно 1 мг/кг DAC HYP.
93. Способ по любому из вариантов осуществления 89-92, в котором DAC HYP вводят один раз в неделю в течение по меньшей мере 6 недель, по меньшей мере 12 недель, по меньшей мере 24 недель.
94. Способ по любому из вариантов осуществления 89-93, в котором DAC HYP вводят в виде монотерапии.
95. Способ по варианту осуществления 94, в котором пациент не отвечал на предшествующую терапию интерфероном-бета или прервал предшествующее лечение интерфероном-бета.
96. Способ по любому из вариантов осуществления 89-93, в котором DAC HYP вводят дополнительно к интерферону-бета.
97. Способ по варианту осуществления 89, в котором композицию DAC HYP вводят подкожно.
98. Способ по варианту осуществления 97, в котором композицию DAC HYP вводят в количестве, соответствующем примерно 1 мг/кг DAC HYP.
99. Способ по варианту осуществления 98, в котором композицию DAC HYP вводят один раз в две недели.
100. Способ по варианту осуществления 99, в котором композицию DAC HYP вводят в целом в течение примерно 24 недель.
101. Способ по варианту осуществления 97, в котором композицию DAC HYP вводят в количестве, соответствующем примерно 2 мг/кг DAC HYP.
102. Способ по варианту осуществления 101, в котором композицию DAC HYP вводят один раз в 4 недели.
103. Способ по варианту осуществления 102, в котором композицию DAC HYP вводят в целом в течение примерно 24 недель.
104. Способ по варианту осуществления 103, в котором композицию DAC HYP вводят в количестве, соответствующем от 75 до 300 мг DAC HYP.
105. Способ по варианту осуществления 104, в котором композицию DAC HYP вводят в количестве, соответствующем 150 мг.
106. Способ по варианту осуществления 104, в котором композицию DAC HYP вводят в количестве, соответствующем 300 мг.
107. Способ по любому из вариантов осуществления 103-106, в котором композицию DAC HYP вводят один раз в 4 недели.
108. Способ по варианту осуществления 107, в котором композицию DAC HYP вводят в целом в течение по меньшей мере 48 недель.
109. Способ по любому из вариантов осуществления 103-108, в котором DAC HYP вводят в виде монотерапии.
110. Способ по варианту осуществления 109, в котором пациент не отвечал на предшествующую терапию интерфероном-бета или прервал предшествующее лечение интерфероном-бета.
111. Способ по любому из вариантов осуществления 103-108, в котором DAC HYP вводят дополнительно к интерферону-бета.
112. Рекомбинантный белок из клеточной культуры, полученный или который может быть получен способом, включающим стадии:
(i) доведения рН клеточной культуры, которая экспрессирует и секретирует рекомбинантный белок, до рН в пределах примерно 4,5-5,5;
(ii) инкубации клеточной культуры с доведенным рН в течение примерно 30-90 мин при температуре в пределах примерно 4-15°С; и
(iii) центрифугирования инкубированной клеточной культуры с доведенным рН для удаления клеточного дебриса.
113. Очищенная композиция даклизумаба, полученная или которая может быть получена способом, включающим стадии:
(i) адсорбции даклизумаба из неочищенного препарата даклизумаба на смолу для аффинной хроматографии;
(ii) промывания смолы для аффинной хроматографии буфером для промывания для удаления загрязняющих примесей;
(iii) элюирования адсорбированного даклизумаба буфером для элюирования;
(iv) инактивации вирусов в элюате доведением рН до рН в пределах примерно 3-4 и инкубации элюата с доведенным рН при определенной температуре в течение периода времени, достаточного для инактивации вирусов;
(v) нейтрализации элюата с инактивированными вирусами до рН в пределах примерно 7,7-7,9 (при 25°С);
(vi) пропускания нейтрализованного элюата через смолу для сильной анионообменной хроматографии;
(vii) адсорбции даклизумаба элюата со стадии (vi) на смоле для слабой катионообменной хроматографии; и
(viii) элюирования адсорбированного даклизумаба со смолы для слабой катионообменной хроматографии.
114. Очищенная композиция даклизумаба, полученная или которая может быть получена способом согласно любому из вариантов осуществления по пп. 74-80.
Все публикации, патенты, заявки на патент и другие документы, цитированные в данной заявке, включены в полном объеме в данный документ для сведения для всех целей в той степени, как если бы каждая отдельная публикация, патент, заявка на патент и другой документ указывались по отдельности для включения для сведения для всех целей.
Несмотря на то, что были показаны и описаны различные конкретные варианты осуществления, очевидно, понятно, что могут быть сделаны различные изменения, не отступая от сущности и объема изобретения.
Реферат
Настоящее изобретение относится к области биотехнологии, конкретно к композиции даклизумаба, и может быть использовано в медицине. Полученная композиция препарата даклизумаба подходит для подкожного введения и может быть применена при лечении индивидов, страдающих от рассеянного склероза. Заявленное изобретение отличается от известных аналогов иными профилями изоформ и N-связанного гликозилирования и проявляет более низкую ADCC цитотоксичность. 2 н. и 6 з.п. ф-лы, 38 табл., 9 пр., 23 ил.
Комментарии