Гликозилированный полипептид и содержащая его фармацевтическая композиция - RU2636456C2
Код документа: RU2636456C2
Чертежи





















































































































Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к гликозилированному полипептиду и к фармацевтической композиции, содержащей указанный полипептид.
Предшествующий уровень техники
Природный человеческий интерферон-бета (ИФН-β) является гликопротеином, состоящим из 166 аминокислотных остатков. Интерферон-β принадлежит к семейству цитокинов, и, как известно, обладает иммуномодулирующим эффектом, антивирусной активностью и супрессирующим эффектом в отношении клеточной пролиферации. Далее, человеческий интерферон-β содержит три Цис в положениях 17, 31 и 141 аминокислотной последовательности, и содержит N-связанный олигосахарид типа комплекса с одинарным ветвлением на аспарагине в положении 80. Далее, известно, что он имеет дисульфидную связь на Цис в положениях 31 и 141. Интерферон-β в качестве фармацевтического средства производят с применением системы клеточной экспрессии и подразделяют на ИФН-β-1а или ИФН-β-1b, в зависимости от разницы в источнике экспрессии. В то время как ИФН-β-1а является гликопротеином, ИФН-β-1b не содержит олигосахарида. Далее известно, что ИФН-β-1а, содержащий сахарную цепь, обладает более мощным эффектом в отношении иммуногенности, антивирусной активности и противоопухолевой активности по сравнению с ИФН-β-1b.
Известно, что структура сахарной цепи, содержащаяся в гликопротеине, оказывает выраженное влияние на фармакокинетику. В частности, известно, что присутствие или отсутствие сиаловой кислоты на невосстанавливающем конце сахарной цепи оказывает влияние на увеличение времени полужизни в крови. Однако отмечалось, что предварительно биосинтезированный ИФН-β-1а имеет однородную структуру сахарной цепи в полипептиде (например, результат анализа однородности структуры сахарной цепи путем CE-TOF-MS (капиллярного электрофореза в сочетании с времяпролетной масс-спектрометрией) описан в не-патентной литературе 1). Далее, до настоящего времени не сообщалось о выделении IFN-β, имеющего по существу однородную структуру сахарной цепи, из синтезированного IFN-β или природного IFN-β. Таким образом, имеется препятствие к установлению того, что структура сахарной цепи важна для биологической активности.
В последние годы биотехнология применения системы клеточной экспрессии обеспечила производство рецептур биологически активных белков, включая интерфероны, таких как инсулин, эритропоэтин, и Г-КСФ. Сообщалось, что в этих белковых рецептурах тип гликозилирования белка обеспечивает разнообразие физических или химических свойств белка, таких как степень фолдинга или свойства конформации, стабильность, иммунная реакция, время полужизни в крови, и функция белка в биологической системе (He-патентная литература 2). Далее, с учетом критической иммуногенности, вызванной сахарной цепью нечеловеческого типа, структура сахарной цепи, добавленной к этим белковым рецептурам, предпочтительно является сахарной цепью человеческого типа.
Такие гликопротеиновые рецептуры получают посредством системы клеточной экспрессии в качестве единственного способа. Однако, как описано выше, технология такой системы клеточной экспрессии не обеспечивает контроля структуры сахарной цепи, таким образом, вызывая неоднородность в структуре сахарной цепи произведенного гликопротеина (например, Heпатентная литература 3). По этой причине имеются проблемы, такие как вариабельность качества производственных серий или неспособность оптимизации сахарной цепи. Соответственно, давно требуется способ приготовления однородного гликопротеина, в котором структура сахарной цепи легко регулируется, но в настоящее время нет сообщений о синтезе однородного гликопротеина человеческого типа, демонстрирующего биологическую активность in vivo, путем химического синтеза.
Далее, биоактивный гликопротеин, производимый посредством технологии с такой системой клеточной экспрессии, может содержать вирус или генетический материал. Далее, существует возможность контаминации таким генетическим материалом и т.д., также в случае синтеза биоактивного гликопротеина с применением сахарной цепи, приготовленной из образца биологического происхождения. Применение тепловой обработки, которая позволяет разрушить эти генетические материалы, необходимо для получения безопасной белковой рецептуры. Однако в настоящее время тепловая обработка рецептуры биоактивного гликопротеина приводит к инактивации гликопротеина, и о применении тепловой обработки не сообщалось.
Перечень цитированной литературы
Heпатентная литература
[Heпатентная литература 1] Anal Bioanall Chem (2011) 400:295-303
[Heпатентная литература 2] Nat. Biotechnol., 2006, 24, 1241-1252
[Heпатентная литература 3] J. Biotechnol, 42,117-131 (1995)
Изложение сущности изобретения
Проблемы, решаемые настоящим изобретением
Задачей, решаемой настоящим изобретением, является обеспечение гликозилированного полипептида, имеющего однородную структуру сахарной цепи, обладающего активностью интерферона-β.
Средства решения проблем
В результате повторных исследований для решения вышеуказанных проблем авторы настоящего изобретения преуспели в получении гликозилированного полипептида, имеющего однородную структуру сахарной цепи, обладающего активностью интерферона-β.
Другими словами, настоящее изобретение относится к гликозилированному полипептиду, характеризующемуся тем, что указанный гликозилированный полипептид является полипептидом, выбранным из группы, состоящей из:
(a) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1;
(b) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1, в которой одна или несколько аминокислот удалены, заменены или добавлены;
(c) аналога интерферона-β; и
(d) полипептида, имеющего 80% или более гомологии с полипептидом, состоящим из аминокислотной последовательности, представленной SEQ ID №: 1;
где полипептид имеет сахарную цепь на аминокислоте, соответствующей положению 80 в интерфероне-β и обладает активностью интерферона-β, и
указанный гликозилированный полипептид имеет по существу однородные сахарные цепи.
Таким образом, в одном варианте осуществления гликозилированный полипептид из настоящего изобретения характеризуется тем, что указанный гликозилированный полипептид приготовлен путем химического синтеза.
Далее, другой аспект настоящего изобретения относится к гликозилированному полипептиду, полученному посредством способа, включающего этап синтеза гликозилированного пептидного фрагмента и по меньшей мере двух пептидных фрагментов, и этап связывания указанного гликозилированного пептидного фрагмента и указанных по меньшей мере двух пептидных фрагментов, характеризующемуся тем, что указанный гликозилированный полипептид является полипептидом, выбранным из группы, состоящей из:
(а) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1;
(b) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1, в которой одна или несколько аминокислот удалены, заменены или добавлены;
(c) аналога интерферона-β; и
(d) полипептида, имеющего 80% или более гомологии с полипептидом, состоящим из аминокислотной последовательности, представленной SEQ ID №: 1;
где полипептид имеет сахарную цепь на аминокислоте, соответствующей положению 80 в интерфероне-β, и обладает активностью интерферона-β.
Далее, в одном варианте осуществления гликозилированного полипептида из настоящего изобретения, гликозилированный полипептид относится к гликозилированному полипептиду, полученному посредством способа, включающего этап синтеза гликозилированного пептидного фрагмента и по меньшей мере двух пептидных фрагментов, и этап связывания указанного гликозилированного пептидного фрагмента и указанных по меньшей мере двух пептидных фрагментов, характеризующемуся тем, что указанный гликозилированный полипептид является полипептидом, выбранным из группы, состоящей из:
(a) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1;
(b) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1, в которой одна или несколько аминокислот удалены, заменены или добавлены;
(c) аналога интерферона-β; и
(d) гликозилированного полипептида, имеющего 80% или более гомологии с полипептидом, состоящим из аминокислотной последовательности, представленной SEQ ID №: 1;
где полипептид имеет сахарную цепь на аминокислоте, соответствующей положению 80 в интерфероне-β, и обладает активностью интерферона-β.
Так, в одном варианте осуществления гликозилированный полипептид из настоящего изобретения характеризуется тем, что сахарная цепь в указанном гликозилированном полипептиде является связанной с аспарагином сахарной цепью.
Далее, в одном варианте осуществления гликозилированный полипептид из настоящего изобретения характеризуется тем, что сахарная цепь в указанном гликозилированном полипептиде является дисиало-сахарной цепью, представленной следующей формулой (а), или асиало-сахарной цепью, представленной следующей формулой (b).
[Химическая формула1]

[Химическая формула2]

Далее, в одном варианте осуществления гликозилированный полипептид из настоящего изобретения характеризуется тем, что Цис, соответствующий положениям 31 и 141 интерферона-β, образует дисульфидную связь в указанном гликозилированном полипептиде.
Далее, в одном варианте осуществления гликозилированный полипептид из настоящего изобретения характеризуется тем, что указанный гликозилированный полипептид подвергнут тепловой обработке.
Далее, в одном варианте осуществления гликозилированный полипептид из настоящего изобретения характеризуется тем, что указанный гликозилированный полипептид имеет чистоту 90% или более.
Далее, другой аспект настоящего изобретения относится к композиции, содержащей указанный гликозилированный полипептид, характеризующийся тем, гликозилированные полипептиды в указанной композиции являются по существу однородными.
Таким образом, в одном варианте осуществления композиция из настоящего изобретения характеризуется тем, что гликозилированные полипептиды в указанной композиции являются однородными на 90% или более.
Далее, в одном варианте композиция из настоящего изобретения характеризуется тем, что указанный гликозилированный полипептид имеет чистоту 90% или более.
Далее, композиция из настоящего изобретения характеризуется тем, что она является композицией, содержащей гликозилированный полипептид, характеризующийся тем, что указанный гликозилированный полипептид является полипептидом, выбранным из группы, состоящей из следующих полипептидов (a)-(d):
(a) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1;
(b) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1, в которой одна или несколько аминокислот удалены, заменены или добавлены;
(c) аналога интерферона-β; и
(d) гликозилированного полипептида, имеющего 80% или более гомологии с полипептидом, состоящим из аминокислотной последовательности, представленной SEQ ID №: 1;
где полипептид имеет сахарную цепь на аминокислоте, соответствующей положению 80 в интерфероне-β, и обладает активностью интерферона-β, и
гликозилированные полипептиды в указанной композиции являются по существу однородными.
Далее, другой аспект настоящего изобретения относится к фармацевтической композиции, характеризующейся тем, что она содержит (I) гликозилированный полипептид по любому из п.п. 1-7 и/или его фармацевтически пригодную соль, и (И) фармацевтически пригодный носитель.
Так, в одном варианте осуществления фармацевтическая композиция из настоящего изобретения характеризуется тем, что указанные сахарные цепи являются по существу однородными в указанном гликозилированном полипептиде.
Далее, в одном варианте осуществления фармацевтическая композиция из настоящего изобретения характеризуется тем, что сахарные цепи являются на 90% или более однородными в указанном гликозилированном полипептиде.
Далее, в одном варианте осуществления фармацевтическая композиция из настоящего изобретения характеризуется тем, что указанный гликозилированный полипептид имеет чистоту 90% или более.
Далее, в одном варианте осуществления фармацевтическая композиция из настоящего изобретения характеризуется тем, что ее применяют для лечения или профилактики интерферон-β-зависимого заболевания.
Далее, в одном варианте осуществления фармацевтическая композиция из настоящего изобретения характеризуется тем, что указанное интерферон-β-зависимое заболевание является по меньшей мере одним заболеванием, выбранным из группы, состоящей из опухоли головного мозга, включая мультиформную глиобластому, медуллобластому и астроцитому; кожной злокачественной меланомы, хронического активного гепатита В, хронического гепатита С, подострого склерозирующего панэнцефалита, компенсированного цирроза С и рассеянного склероза.
Далее, другой аспект настоящего изобретения относится к способу лечения или профилактики интерферон-β-зависимого заболевания, характеризующемуся применением эффективного количества указанного гликозилированного полипептида.
Так, в одном варианте осуществления способ из настоящего изобретения для лечения или профилактики интерферон-β-зависимого заболевания характеризуется тем, что указанное интерферон-β-зависимое заболевание является по меньшей мере одним заболеванием, выбранным из группы, состоящей из опухоли головного мозга, включая мультиформную глиобластому, медуллобластому, и астроцитому; кожной злокачественной меланомы, хронического активного гепатита В, хронического гепатита С, подострого склерозирующего панэнцефалита, компенсированного цирроза С, и рассеянного склероза.
Далее, другой аспект настоящего изобретения относится к гликозилированному полипептиду, характеризующемуся тем, что гликозилированный полипептид является полипептидом, выбранным из группы, состоящей из:
(a) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1;
(b) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1, в которой одна или несколько аминокислот удалены, заменены или добавлены;
(c) аналога интерферона-β; и
(d) полипептида, имеющего 80% или более гомологии с полипептидом, состоящим из аминокислотной последовательности, представленной SEQ ID №: 1;
где полипептид имеет сахарную цепь на аминокислоте, соответствующей положению 80 в интерфероне-β, и имеет Цис, соответствующим положениям 17, 31 и 141 в интерфероне-β, защищенную защитной группой.
Так, в одном варианте осуществления гликозилированный полипептид из настоящего изобретения характеризуется тем, что указанная сахарная цепь в указанном гликозилированном полипептиде является дисиало-сахарной цепью, имеющей карбоксигруппу сиаловой кислоты, присутствующую на защищенном невосстанавливающем конце сахарной цепи, представленной следующей формулой (с) или асиало-сахарной цепью, представленной следующей формулой (b).
[Химическая формула 3]

(где R представлено -COOBn, -COOEt, -СООМе, -COOCH2COPh, -COOCH2PhOMe, -COOCH2Ph(OMe)2, -COOCH2PhNO2, или -COOCH2Ph(NO2)2. Bn указывает бензильную группу, Et указывают этильную групп, Me указывает метальную группу, a Ph указывает фенильную группу).
[Химическая формула 4]

Далее, в одном варианте осуществления гликозилированный полипептид из настоящего изобретения характеризуется тем, что Цис, соответствующие указанным положениям 17, 31 и 141 в интерфероне-β, защищены одной или несколькими защитными группами, состоящими из Аст группы, алкоксиметильной группы, трифенилметильной группы, т-бутильной группы, бензильной группы, и этильной группы, находящейся в β-положении в указанном гликозилированном полипептиде.
Далее, другой аспект настоящего изобретения относится к способу производства гликозилированного полипептида, характеризующемуся тем, что он включает этап снятия защиты с Цис, соответствующего указанным положениям 17, 31 и 141 в интерфероне-β, защищенного защитной группой в указанном гликозилированном полипептиде, имеющем защищенный Цис.
Таким образом, один вариант осуществления способа получения гликозилированного полипептида из настоящего изобретения характеризуется тем, что он дополнительно включает этап приготовления указанного гликозилированного полипептида, имеющего Цис, соответствующий положениям 17, 31 и 141 в интерфероне-β, защищенный защитной группой, перед указанным этапом снятия защиты Цис.
Далее, в одном варианте осуществления способ получения гликозилированного полипептида из настоящего изобретения характеризуется тем, что гликозилированный полипептид подвергают тепловой обработке перед этапом удаления защитной группы с Цис.
Далее, другой аспект из настоящего изобретения относится к гликозилированному полипептиду, характеризующемуся тем, что гликозилированный полипептид является полипептидом, выбранным из группы, состоящей из:
(a) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1;
(b) полипептида, состоящего из аминокислотной последовательности, представленной SEQ ID №: 1, в которой одна или несколько аминокислот удалены, заменены или добавлены;
(c) аналога интерферона-β; и
(d) гликозилированного полипептида, имеющего 80% или более гомологии с полипептидом, состоящим из аминокислотной последовательности, представленной SEQ ID №: 1;
где полипептид имеет сахарную цепь на аминокислоте, соответствующей положению 80 в интерфероне-β, и
указанная сахарная цепь является дисиало-сахарной цепью, имеющей карбоксигруппу из сиаловой кислоты, присутствующую на невосстанавливающем конце сахарной цепи, представленной следующей формулой (с).
[Химическая формула 5]

(где R представлено -COOBn, -COOEt, -COOMe, -COOCH2COPh, -COOCH2PhOMe, -COOCH2Ph(OMe)2, -COOCH2PhNO2, или -COOCH2Ph(NO2)2. Bn указьюает бензильную группу, Et указьшает этильную группу, Me указывает метальную группу, a Ph указывает фенильную группу).
Далее, другой аспект настоящего изобретения относится к способу получения гликозилированного полипептида, характеризующемуся тем, что он включает этап удаления защитной группы из карбоксигруппы сиаловой кислоты, присутствующей на невосстанавливающем конце сахарной цепи в указанном гликозилированном полипептиде, имеющем сахарную цепь, содержащую защищенную карбоксигруппу сиаловой кислоты.
Так, в одном варианте осуществления способ получения гликозилированного полипептида из настоящего изобретения характеризуется тем, что он дополнительно включает этап приготовления указанного гликозилированного полипептида, имеющего сахарную цепь, содержащую защищенную карбоксигруппу сиаловой кислоты перед этапом снятия защиты указанной карбоксигруппы указанной сиаловой кислоты.
Далее, гликозилированный полипептид из способа получения из настоящего изобретения характеризуется тем, что он дополнительно включает этап фолдинга указанного гликозилированного полипептида.
Эффекты, достигаемые настоящим изобретением
Поскольку гликозилированный полипептид из настоящего изобретения имеет однородную структуру сахарной цепи, имеется малая вариация между сериями, и можно обеспечить гликозилированный полипептид, обладающий активностью интерферона-β, со стабильным качеством. Далее, можно сделать однородным тип связи сахарной цепи, и можно оптимизировать структуру сахарной цепи, в соответствии с заданной целью.
Далее, поскольку гликозилированный полипептид из настоящего изобретения можно подвергать тепловой обработке, можно предотвратить контаминацию вирусом или неизвестным генетическим материалом. Краткое описание чертежей
Фигура 1 является схематической диаграммой этапа сшивания гликопептидного фрагмента В, имеющего аминокислоты в положениях 68-88, и пептидного фрагмента С, имеющего аминокислоты в положениях 89-166 в аминокислотной последовательности интерферона-β в одном варианте получения гликозилированного полипептида из настоящего изобретения.
Фигура 2 является схематической диаграммой этапа сшивания пептидного фрагмента А, имеющего аминокислоты в положениях 1-67, и гликопептидного фрагмента (В+С), имеющего аминокислоты в положениях 68-166 в аминокислотной последовательности интерферона-β в одном варианте получения гликозилированного полипептида из настоящего изобретения.
Фигура 3 является схематической диаграммой этапа восстановления частного цистеина гликопептидного фрагмента (А+В+С), содержащего аминокислоты в положениях 1-166 в аминокислотной последовательности интерферона-β, приготовленного путем сшивания в аланине в одном варианте получения гликозилированного полипептида из настоящего изобретения.
Фигура 4 является схематической диаграммой, показывающей этап удаления защитной группы с цистеина, выполненного на гликопептидном фрагменте (А+В+С), содержащем аминокислоты в положениях 1-166 в аминокислотной последовательности интерферона-β, приготовленной путем сшивания после этапа восстановления в аланин в одном варианте получения гликозилированного полипептида из настоящего изобретения.
Фигура 5 демонстрирует данные ВЭЖХ (А) и масс-спектрометрии с ионизацией электрораспылением (ИЭР-МС) (В) для гликопептидного фрагмента (11), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 6 демонстрирует аминокислотную последовательность гликопептидного фрагмента (11), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 7 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (12), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 8 демонстрирует аминокислотную последовательность гликопептидного фрагмента (12), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 9 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (13), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 10 демонстрирует аминокислотную последовательность гликопептидного фрагмента (13), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 11 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (14), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 12 демонстрирует аминокислотную последовательность гликопептидного фрагмента (14), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 13 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (15), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 14 демонстрирует аминокислотную последовательность гликопептидного фрагмента (15), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 15 демонстрирует данные по чистоте гликопептидного фрагмента (15), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения по результатам ВЭЖХ.
Фигура 16 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (5), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 17 демонстрирует аминокислотную последовательность гликопептидного фрагмента (5), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 18 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (6), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 19 демонстрирует аминокислотную последовательность гликопептидного фрагмента (6), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 20 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (7), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 21 демонстрирует аминокислотную последовательность гликопептидного фрагмента (7), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 22 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (8), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 23 демонстрирует аминокислотную последовательность гликопептидного фрагмента (8), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 24 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (9), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 25 демонстрирует аминокислотную последовательность гликопептидного фрагмента (9), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 26 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (10), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 27 демонстрирует аминокислотную последовательность гликопептидного фрагмента (10), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 28 демонстрирует данные по чистоте гликопептидного фрагмента (10), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 29 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (18), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 30 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (19), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 31 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (20), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 32 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (21), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 33 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (22), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 34 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (23), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
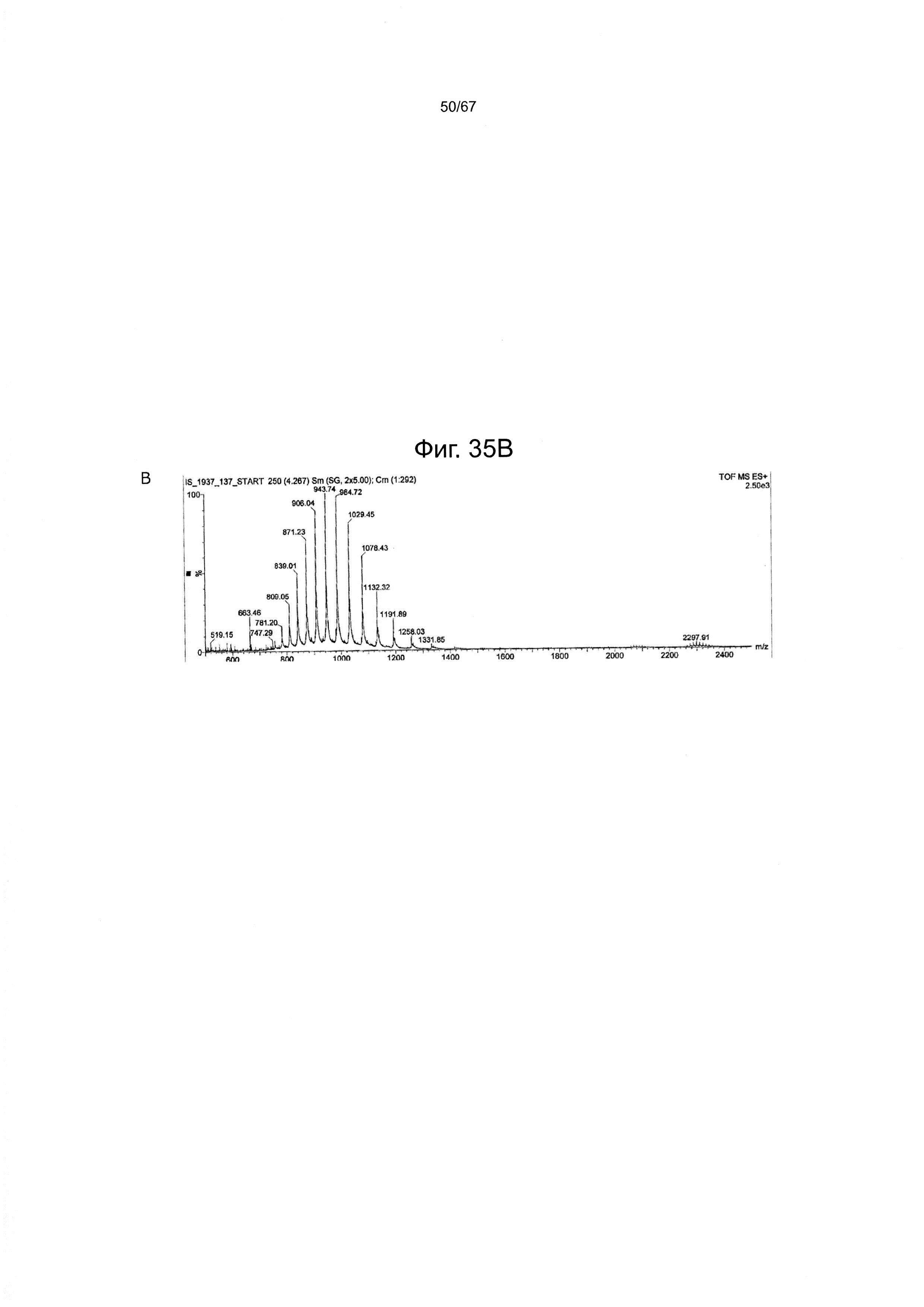
Фигура 35 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (24), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 36 демонстрирует схематическую диаграмму аминокислотной последовательности полипептида (7) с присоединенной дибензил-дисиало-сахарной цепью, имеющего Цис в положениях 17, 31 и 141, защищенные Аст группой, которая является одним вариантом осуществления гликозилированного полипептида из настоящего изобретения, и данные ВЭЖХ и ИЭР-МС анализа указанного полипептида (7) с присоединенной дибензил-дисиало-сахарной цепью до и после тепловой обработки. Фигуры 36а и 36с показывают данные ИЭР-МС до тепловой обработки, а Фигуры 36b и 36е показывают данные ИЭР-МС после тепловой обработки. Далее, Фигура 36d демонстрирует данные ВЭЖХ анализа до тепловой обработки, а Фигура 36f демонстрирует данные ВЭЖХ анализа после тепловой обработки.
Фигура 37 демонстрирует схематическую диаграмму аминокислотной последовательности полипептида (8) с присоединенной дибензил-дисиало-сахарной цепью, имеющего Цис в положениях 17, 31 и 141, защищенные Аст группой, которая является одним вариантом осуществления гликозилированного полипептида из настоящего изобретения, и данные ВЭЖХ и ИЭР-МС анализа указанного полипептида (8) с присоединенной дибензил-дисиало-сахарной цепью до и после тепловой обработки. Фигуры 37а и 37 с показывают данные ИЭР-МС до тепловой обработки, а Фигуры 37b и 37е показывают данные ИЭР-МС после тепловой обработки. Далее, Фигура 37d демонстрирует данные ВЭЖХ анализа до тепловой обработки, а Фигура 37f демонстрирует данные ВЭЖХ анализа после тепловой обработки.
Фигура 38 показывает данные анализа аффинности по отношению к рецептору ИФН- α/β 2 для химически синтезированного гликозилированного полипептида (не подвергнутого тепловой обработке), являющегося одним вариантом осуществления настоящего изобретения.
Фигура 39 показывает данные анализа аффинности по отношению к рецептору ИФН- α/β 2 для химически синтезированного гликозилированного полипептида (подвергнутого тепловой обработке), являющегося одним вариантом осуществления настоящего изобретения.
Фигура 40 демонстрирует данные фармакокинетического анализа, когда химически синтезированный гликозилированный полипептид, являющийся одним вариантом осуществления настоящего изобретения, применяли у мышей. ИФН-β Mochida, приготовленный посредством биосинтеза, использовали в качестве контроля. На Фигуре 40А показаны данные, полученные при внутривенном введении, а на 40В показаны данные, полученные при подкожном введении.
Фигура 41 демонстрирует противоопухолевую активность химически синтезированного гликозилированного полипептида, являющегося одним вариантом осуществления настоящего изобретения. В качестве контроля использовали ФБР (группа для контроля с растворителем) и ИФН-β Mochida, приготовленный посредством биосинтеза.
Фигура 42 демонстрирует данные, сравнивающие супрессорную активность в отношении клеточной пролиферации для химически синтезированного гликозилированного полипептида, который является одним вариантом осуществления настоящего изобретения, между полипептидом, прошедшим и не прошедшим тепловую обработку.
Фигура 43 является схематической диаграммой, демонстрирующей этап сшивания пептидного фрагмента В2, содержащего аминокислоты в положениях 68-75, и гликопептидного фрагмента В1, содержащего аминокислоты в положениях 76-88 в аминокислотной последовательности интерферона-β в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 44 является схематической диаграммой, демонстрирующей этап метилирования тиоловой группы (-SH) частного цистеина из гликопептидного фрагмента (В1+В2), содержащего аминокислоты в положениях 68-88 в аминокислотной последовательности интерферона-β, приготовленного путем сшивки в метилтиогруппу (-SMe) в одном варианте получения гликозилированного полипептида из настоящего изобретения.
Фигура 45 является схематической диаграммой, демонстрирующей этап интрамолекулярной ацильной транслокации, в которой цистеин, содержащий метилированную тиоловую группу (-SMe) на гликопептидном фрагменте (В1+В2), превращается в серии в одном варианте получения гликозилированного полипептида из настоящего изобретения.
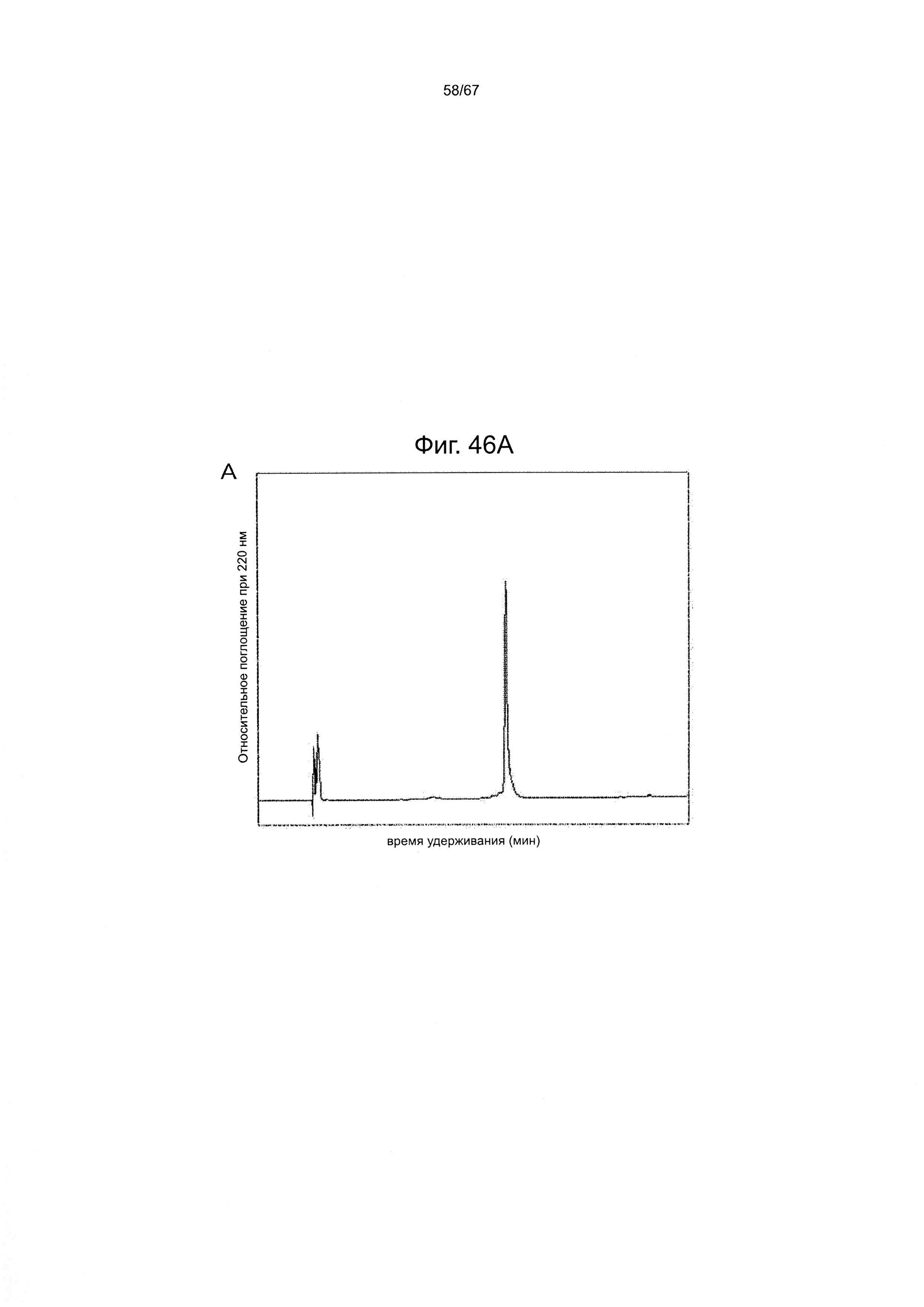
Фигура 46 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (25), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 47 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (26), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
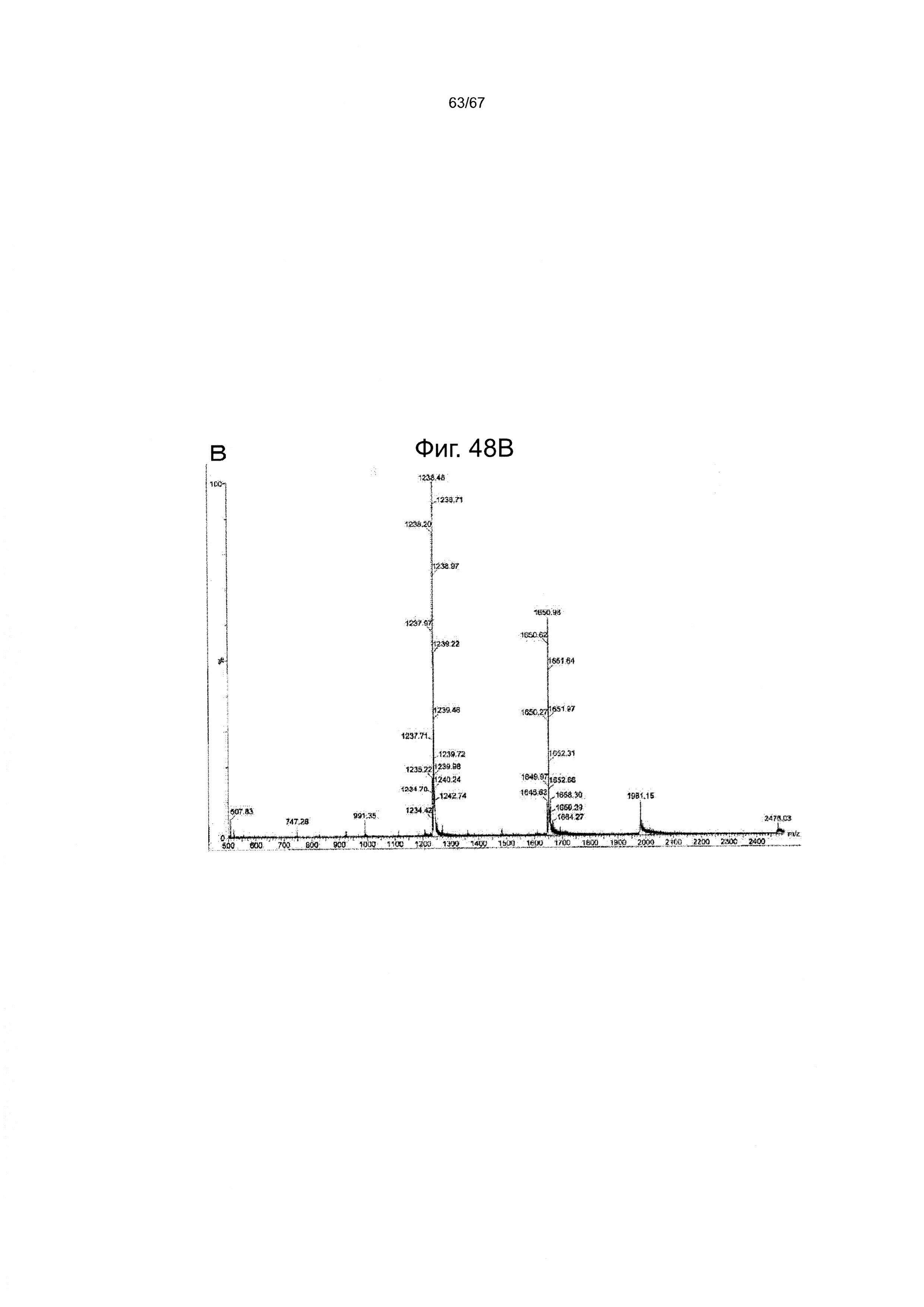
Фигура 48 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (27), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 49 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (28), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Фигура 50 демонстрирует данные ВЭЖХ (А) и ИЭР-МС (В) для гликопептидного фрагмента (29), полученного в одном варианте производства гликозилированного полипептида из настоящего изобретения.
Описание вариантов осуществления
«Интерферон-β» или «ИФН-β» означает полипептид, содержащий последовательность из 166 аминокислот, представленных SEQ ID №1. Интерферон-β содержит дисульфидную связь между цистеинами в положениях 31 и 141 в аминокислотной последовательности, указанной ниже. Далее, «интерферон-β» предпочтительно имеет аминокислотную последовательность, указанную ниже (SEQ ID №1), и соответствует интерферону-β-1а человеческого типа, имеющему сахарную цепь. Интерферон-β-1а имеет сахарную цепь, добавленную к аспарагину в положении 80 аминокислотной последовательности, указанной ниже.


Термин «аминокислота» в настоящей заявке применяется в самом широком значении, и включает не только природные аминокислоты, но также неприродные аминокислоты, такие как варианты и производные аминокислот. Специалисту в данной области техники понятно в свете этого широкого определения, что примеры аминокислот включают, например, природные протеиногенные L-аминокислоты; D-аминокислоты; химически модифицированные аминокислоты, такие как варианты и производные аминокислот; природные непротеиногенные аминокислоты, такие как норлейцин, β-аланин и орнитин; и химически синтезированные соединения, обладающие свойствами, хорошо известными в области характеристик аминокислот. Примеры неприродных аминокислот включают α-метиламинокислоту (такую, как α-метилаланин), D-аминокислоту, гистидино-подобную аминокислоту (такую как 2-аминогистидин, β-гидроксигистидин, гомогистидин, α-фторметилгистидин и α-метилгистидин), аминокислоту, имеющую избыток метиленов на боковой цепи («гомо» аминокислоту), и аминокислоту, в которой аминокислота с карбоксильной функциональной группой в боковой цепи замещена сульфонатной группой (такой как цистеиновая кислота). В предпочтительном аспекте аминокислота, содержащаяся в соединении из настоящего изобретения, состоит только из природных аминокислот.
Как применяется в настоящей заявке, когда одна или несколько аминокислот в аминокислотной последовательности удаляются, замещаются или добавляются, число замещенных и т.д. аминокислот не ограничивается конкретно, с тем условием, чтобы сохранялась активность интерферона-β, но означает, что, например, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 аминокислот различаются. Альтернативно, также включается случай, когда 20% или менее, или предпочтительно 10% или менее аминокислот по длине полной аминокислотной последовательности являются различными. Замещаемая или добавляемая аминокислота может быть природной аминокислотой, неприродной аминокислотой, или аналогом аминокислоты, предпочтительно природной аминокислотой.
«Пептидный фрагмент» означает пептидный фрагмент, имеющий последовательность части аминокислотной последовательности окончательного гликозилированного полипептида при получении гликозилированного пептида из настоящего изобретения. Далее, «гликозилированный пептидный фрагмент» или «гликопептидный фрагмент» в настоящей заявке означает пептидный фрагмент, содержащий гликозилированную аминокислоту в аминокислотной последовательности такого пептидного фрагмента. Гликозилированный пептид из настоящего изобретения получают путем сшивания гликозилированного пептидного фрагмента и по меньшей мере двух пептидных фрагментов.
«Аналог интерферона-β» в настоящей заявке включает полипептид, структурно подобный интерферону-β и/или полипептид, обладающий перекрывающейся структурой с интерфероном-β, например, полипептид, имеющий одну из аминокислот или несколько аминокислот из аминокислот интерферона-β, консервативно замещенных; модифицированный интерферон-β; фрагмент интерферона-β, обладающий активностью интерферона-β; и удлиненный интерферон-β, обладающий активностью интерферона-β.
«Имеющий одну из аминокислот или несколько аминокислот из аминокислот, консервативно замещенных» в настоящей заявке означает аминокислотную замену, в которой индекс гидрофильности и/или гидрофобности являются схожими у исходной аминокислоты и замещенной аминокислоты, и где не происходит явного снижения или потери активности интерферона-β до и после такого замещения.
«Модифицированный интерферон-β» в настоящей заявке означает модифицированную версию интерферона-β, включая природный вариант интерферона-β или искусственно модифицированное соединение интерферона-β. Примеры таких модификаций включают, например, алкилирование, ацилирование (такое, как ацетилирование), амидирование, карбоксилирование, образование сложного эфира, образование дисульфидной связи, гликозилирование, липидирование, фосфорилирование, гидроксилирование, и связывание с индикаторным компонентом одного или нескольких аминокислотных остатков интерферона-β.
«Удлиненный интерферон-β, обладающий активностью интерферона-β» в настоящей заявке означает пептид, содержащий одну или несколько аминокислот, добавленных к N- и/или С-концам интерферона-β, поддерживающих активность интерферона-β.
Примеры аналога интерферона-β, описанного выше, могут включать аминокислотную последовательность, известную как интерферон-β1, в котором Мет в положении 1 удален, а Цис в положении 17 замещен на Сер.
Гликозилированный полипептид из настоящего изобретения содержит полипептид, состоящий из аминокислотной последовательности, имеющей 80% или более гомологии с аминокислотной последовательностью, представленной SEQ ID №1, где гликозилированный полипептид имеет сахарную цепь на аминокислоте, соответствующей положению 80 в интерфероне-β, и обладает активностью интерферона-β. Если гликозилированный полипептид из настоящего изобретения имеет 80% или более гомологии с SEQ ID №1, он предпочтительно имеет 85% или более, 90% или более, 95% или более, или 99% или более гомологии.
«Аминокислота, соответствующая частному положению в интерфероне-β» означает в настоящей заявке аминокислоту в том же самом положении, соответствующем аминокислотной последовательности интерферона-β, до тех пор, пока нет добавления или делеции аминокислоты в гликозилированном полипептиде. Далее, если добавление или делеция аминокислоты присутствует в аминокислотной последовательности гликозилированного полипептида, это означает аминокислоту в положении с учетом сдвига аминокислотной последовательности при добавлении или делеции аминокислоты. Например, в гликозилированном пептиде, имеющем последовательность Мет1-Сер2-Тир3-Асн4- в положениях 1-4, когда одна аминокислота (Трп) добавлена между аминокислотами в положениях 2 и 3 (Мет-Сер-Трп-Тир-Асн-), то аминокислота, соответствующая аминокислоте в положении 3 (Тир) означает аминокислоту (Тир) в гликозилированном полипептиде, смещенную к С-концу путем вставки Трп.
«Сахарная цепь» в настоящей заявке означает соединение, полученное при связывании одной или нескольких единиц сахаров (моносахаридов и/или их производных). Когда происходит связывание двух или более единиц сахаров, каждая единица сахаров связывается с другой единицей путем дегидратационной конденсации с гликозидной связью между ними. Такие сахарные цепи включают, например, широкий ряд, такой как моносахариды и полисахариды, встречающиеся in vivo (глюкоза, галактоза, манноза, фукоза, ксилоза, N-ацетилглюкозамин, N-ацетилгалактозамин, сиаловая кислота, и их конъюгаты и производные), а также сахарные цепи, деградировавшие или полученные из конъюгированных биомолекул, такие как деградированные полисахариды, гликопротеины, протеогликаны, гликозаминогликаны и гликолипиды; но не ограничиваются ими. Сахарная цепь может быть линейной или разветвленной.
Далее, «сахарная цепь» в настоящей заявке также включает производное сахарной цепи, и примеры производных сахарной цепи включают сахарную цепь, в которой сахар, составляющий сахарную цепь, является, например, сахаром, имеющим карбоксигруппу (такую, как альдоновая кислота, в которой С-положение 1 окисляется с получением карбоновой кислоты (такой, как D-глюконовая кислота, которая является окисленной D-глюкозой) и уроновая кислота, в которой концевой атом С становится карбоновой кислотой (D-глюкуроновой кислотой, которая является окисленной D-глюкозой)); сахаром, имеющим аминогруппу или производное аминогруппы (таким, как D-глюкозамин или D-галактозамин); сахаром, имеющим как амино, так и карбоксигруппы (таким, как N-глюкоилнейраминовая кислота и N-ацетилмурамовая кислота); дезоксилированным сахаром (таким, как 2-дезокси- D-рибоза); сульфатированным сахаром, включающим сульфатную группу; и фосфорилированным сахаром, включающим фосфатную группу; но не ограничивается ими.
Предпочтительной сахарной цепью в настоящей заявке является сахарная цепь, которая не нарушает активности интерферона-β при добавлении к гликозилированному полипептиду.
Такая сахарная цепь в гликозилированном полипептиде из настоящего изобретения не ограничивается конкретно, и может быть сахарной цепью, существующей как гликоконъюгат in vivo (такой, как гликопептид (или гликопротеин), протеогликан и гликолипид), или она может быть сахарной цепью, не существующей в виде гликоконъюгата in vivo.
Сахарная цепь, которая существует в виде гликоконъюгата in vivo, является предпочтительной с учетом того факта, что гликозилированный полипептид из настоящего изобретения применяют in vivo. Примеры таких сахарных цепей включают N-или О-связанные сахарные цепи, которые являются сахарными цепями, связанными с пептидом (или белком) in vivo, таким как гликопептид (или гликопротеин). Предпочтительно применяется N-связанная сахарная цепь. N-связанные сахарные цепи могут включать, например, форму с высоким содержанием маннозы, комплексную форму, или гибридную форму, особо предпочтительно комплексную форму.
В одном предпочтительном аспекте настоящего изобретения сахарная цепь в гликозилированном полипептиде из настоящего изобретения является сахарной цепью комплексного типа. Сахарная цепь комплексного типа характеризуется тем, что она содержит два или более типов моносахаридов, и имеет основную структуру, показанную ниже, и лактозаминовую структуру, показанную Galβ1-4GlcNAc.
[Химическая формула 6]

Примеры предпочтительных сахарных цепей комплексного типа, используемых в настоящей заявке, включают, например, сахарную цепь, представленную следующей общей формулой:
[Химическая формула 7]
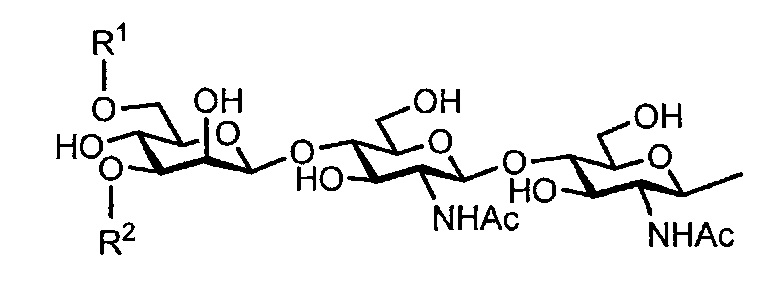
[где R1 и R2 являются идентичными или различными, и являются:
[Химическая формула 8]
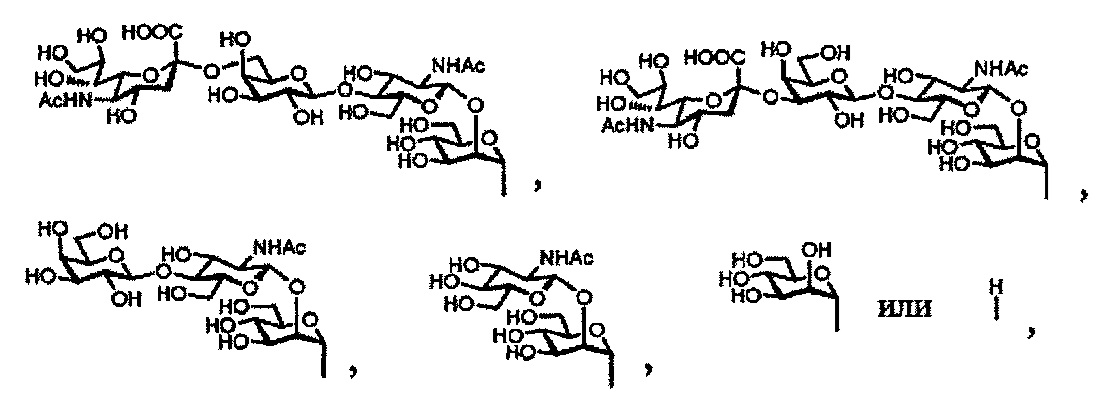
а Ас является ацетильной группой].
Далее, в настоящем изобретении сахарная цепь комплексного типа содержит сахарную цепь комплексного типа с двойным ветвлением. Сахарная цепь комплексного типа с двойным ветвлением означает цепь, имеющую сахарные цепи с одинарным ветвлением, состоящие из 0-3 сахаров в связи с каждой из двух манноз на конце основной структуры. Сахарная цепь комплексного типа с двойным ветвлением предпочтительно является, например, дисиало-сахарной цепью, показанной ниже:
[Химическая формула 9]

моносиало-сахарной цепью:
[Химическая формула 10]
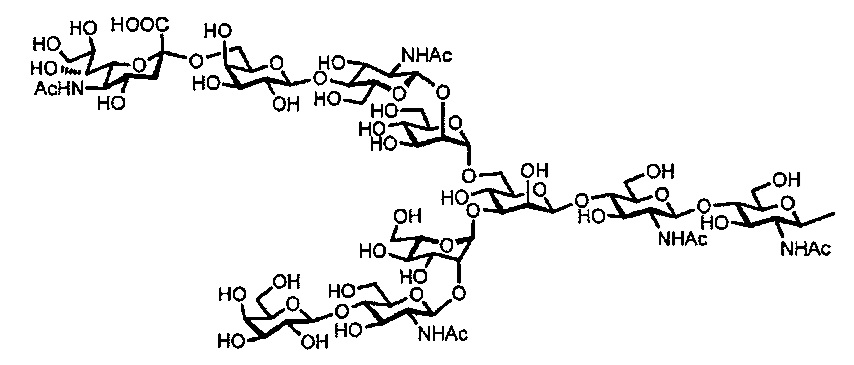
[Химическая формула 10A]

асиало-сахарной цепью:
[Химическая формула 11]
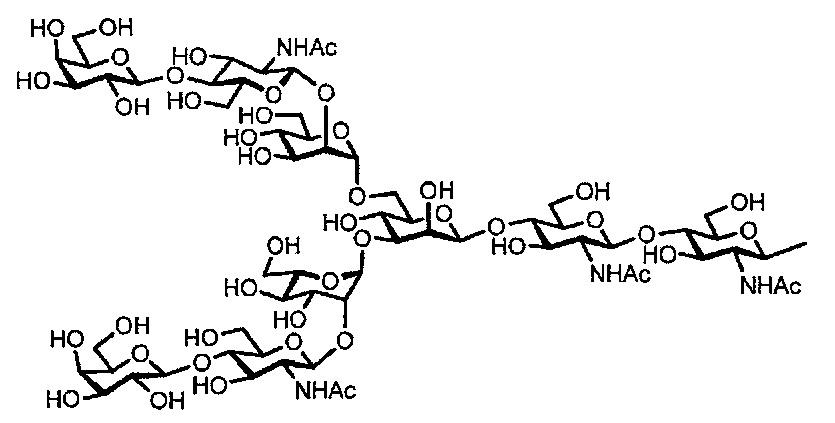
Ди-GlcNAc сахарной цепью:
[Химическая формула 12]

и диманнозной сахарной цепью:
[Химическая формула 13]

Дисиало-сахарная цепь или асиало-сахарная цепь является предпочтительной в качестве сахарной цепи комплексного типа с двойным ветвлением, наиболее предпочтительной является дисиало-сахарная цепь.
Далее, сахарная цепь комплексного типа из настоящего изобретения включает не только вышеуказанную сахарную цепь комплексного типа с двойным ветвлением (сахарную цепь комплексного типа с двумя ветвями), но также сахарную цепь комплексного типа с тройным ветвлением (сахарную цепь комплексного типа с тремя ветвями) и сахарную цепь комплексного типа с четвертичным ветвлением (сахарную цепь комплексного типа с четырьмя ветвями). Например, сахарные цепи комплексного типа с тройным или четвертичным ветвлением могут включать трисиало-сахарную цепь, представленную структурной формулой ниже:
[Химическая формула 14]

[Химическая формула 15]

и тетрасиало-сахарную цепь, представленную структурной формулой ниже:
[Химическая формула 16]

Далее, сахарные цепи комплексного типа с тройным и четвертичным ветвлением могут также включать сахарную цепь, в которой один или несколько сахаров удалены из невосстанавливающего конца этих трисиало- и тетрасиало-сахарных цепей.
Далее, сахарная цепь комплексного типа из настоящего изобретения включает цепь с добавлением фукозы. Сахарные цепи комплексного типа с добавленной фукозой могут включать фукозосодержащую сахарную цепь комплексного типа, представленную структурной формулой ниже:
[Химическая формула 17]

[Химическая формула 18]

[Химическая формула 19]

[Химическая формула 20]
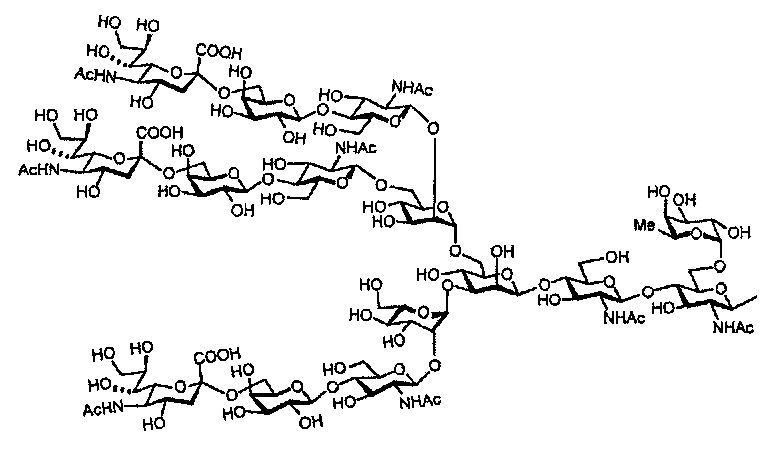
[Химическая формула 21]
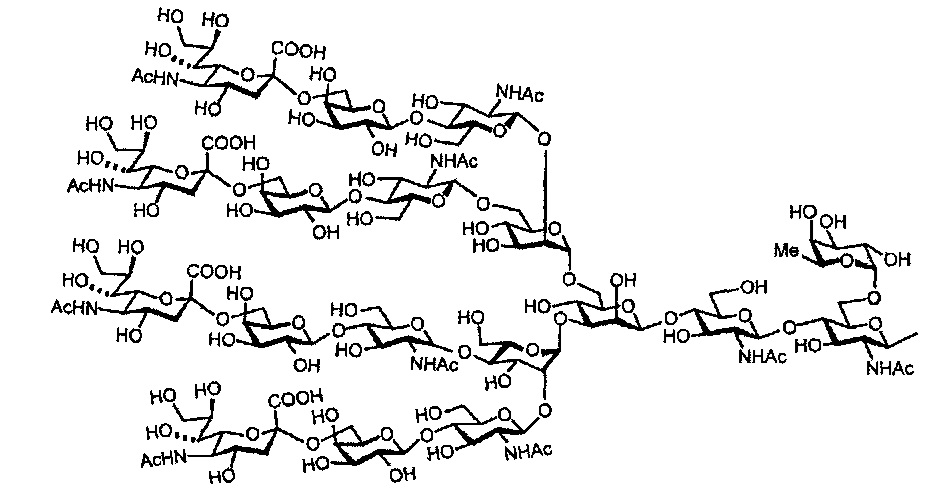
Далее, может также быть включена сахарная цепь, в которой один или несколько сахаров удалены с невосстанавливающего конца этой фукозосодержащей сахарной цепи комплексного типа.
Далее, «сахарная цепь комплексного типа с двойным ветвлением», «дисиало-сахарная цепь», «моносиало-сахарная цепь», «асиало-сахарная цепь», «ди-GlcNAc-сахарная цепь», «диманнозная сахарная цепь», «сахарная цепь комплексного типа с тройным ветвлением», «сахарная цепь комплексного типа с четвертичным ветвлением» и «фукозосодержащая сахарная цепь комплексного типа» в настоящей заявке включает не только то, что указано в вышеприведенных химических формулах, но также цепи с типом связывания, отличающимся от примеров, показанных в химических формулах, и такие сахарные цепи также предпочтительно применяются в качестве сахарной цепи из настоящего изобретения. Примеры таких сахарных цепей включают, например, дисиало- или моносиало-сахарные цепи, в которых сиаловая кислота и галактоза связаны (α2→3) связью.
Далее, сахарная цепь комплексного типа из настоящего изобретения также включает сахарную цепь, имеющую полилактозаминовую структуру или сиалил-полилактозаминовую структуру, представленную следующей формулой.
[Химическая формула 22]

(где n является целым числом от 2 до 3)
[Химическая формула 23]

(где n является целым числом от 2 до 3).
Далее, сахарная цепь с высоким содержанием маннозы, применяемая в настоящей заявке, является сахарной цепью, имеющей две или три маннозы, дополнительно связанные с основной структурой сахарной цепи комплексного типа, описанной выше. Поскольку сахарные цепи с высоким содержанием маннозы являются громоздкими, стабильность в крови может повыситься при связывании сахарной цепи с высоким содержанием маннозы с пептидом. Сахарная цепь, содержащая 5-9 манноз, такая как сахарная цепь с высоким содержанием маннозы млекопитающих, является предпочтительной, но она может быть сахарной цепью, содержащей больше манноз, такой как дрожжевая сахарная цепь с высоким содержанием маннозы. Примеры сахарных цепей с высоким содержанием маннозы, предпочтительно применяемых в настоящей заявке, могут включать, например, цепь с высоким содержанием маннозы-5 (М-%):
[Химическая формула 24]

и цепь с высоким содержанием маннозы-9 (М-9):
[Химическая формула 25]

Предпочтительные сахарные цепи могут также включать, например, сахарную цепь, имеющую идентичную структуру (сахарную цепь, в которой тип составляющего сахара и тип связывания являются идентичными) с сахарной цепью, существующей в организме человека, как гликопротеин, связанный с белком (такую, как сахарная цепь, описанная в «FEBS LETTERS Vol. 50, No. 3, Feb. 1975»), или сахарную цепь, в которой один или несколько сахаров удалены из невосстанавливающего конца цепи. В частности, могут быть включены сахарные цепи, перечисленные ниже.
[Химическая формула 26]

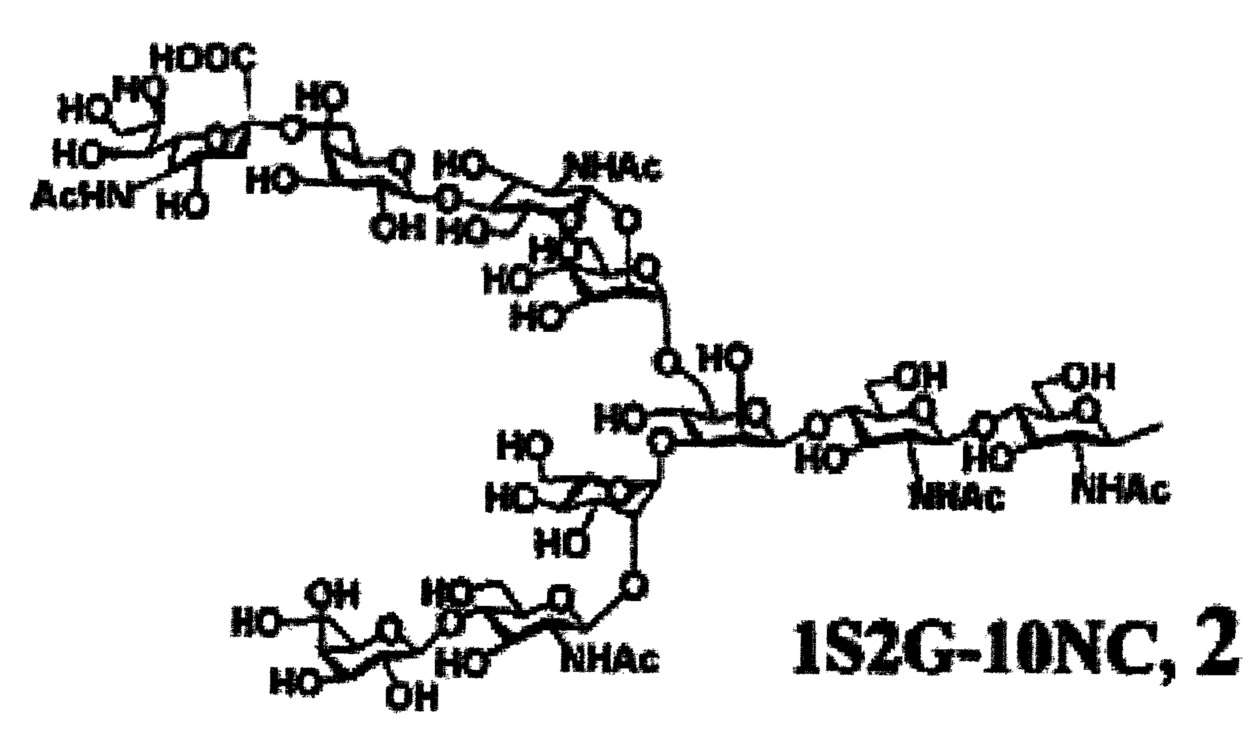

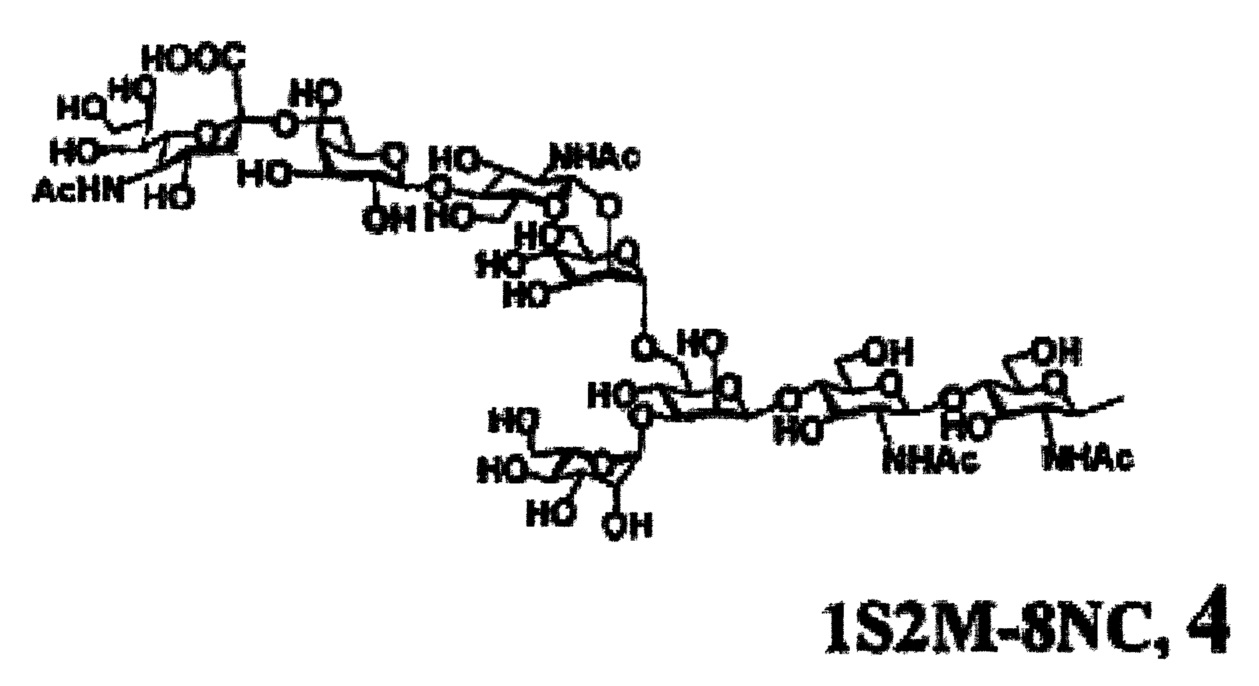


















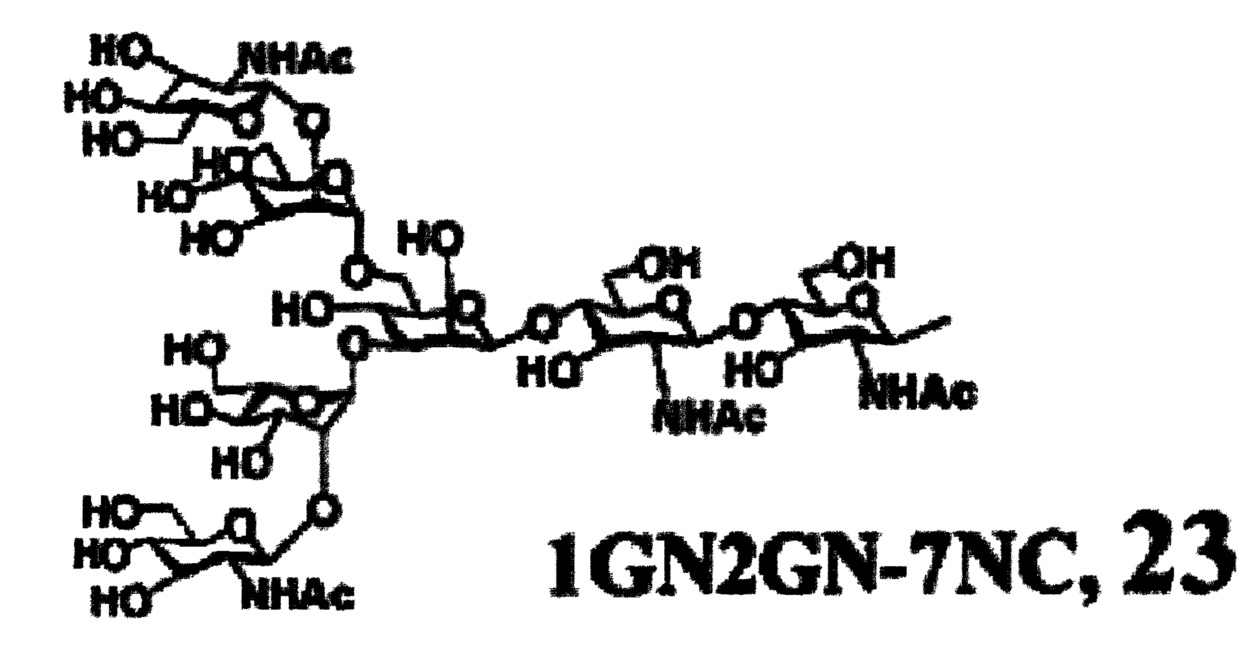

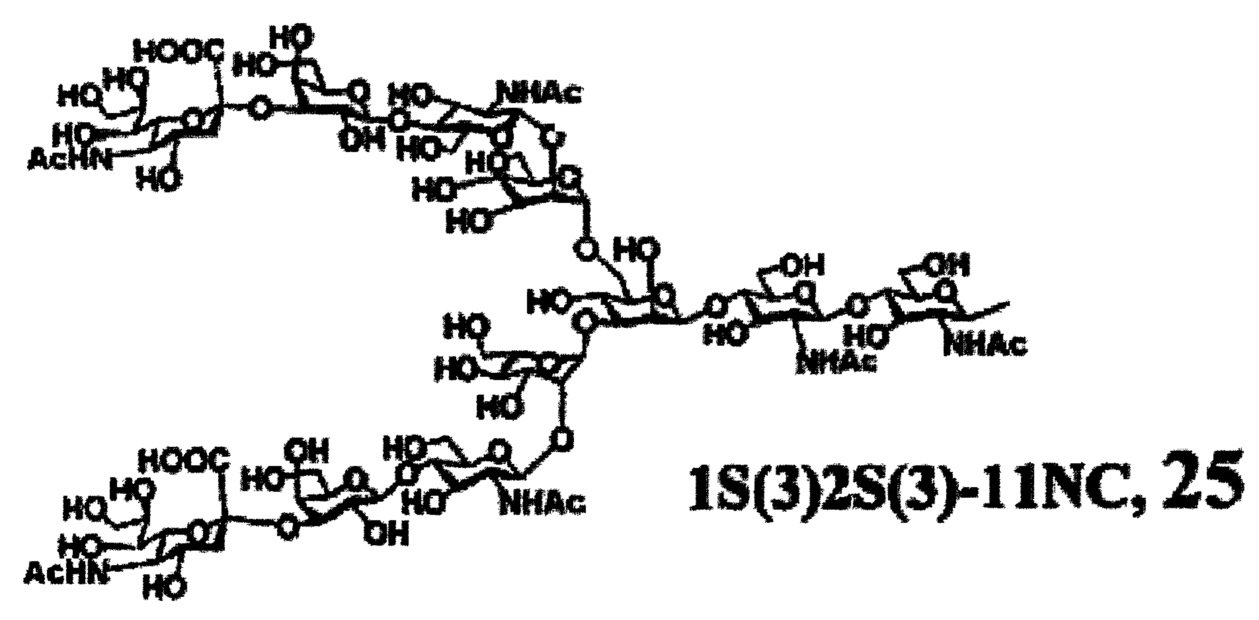





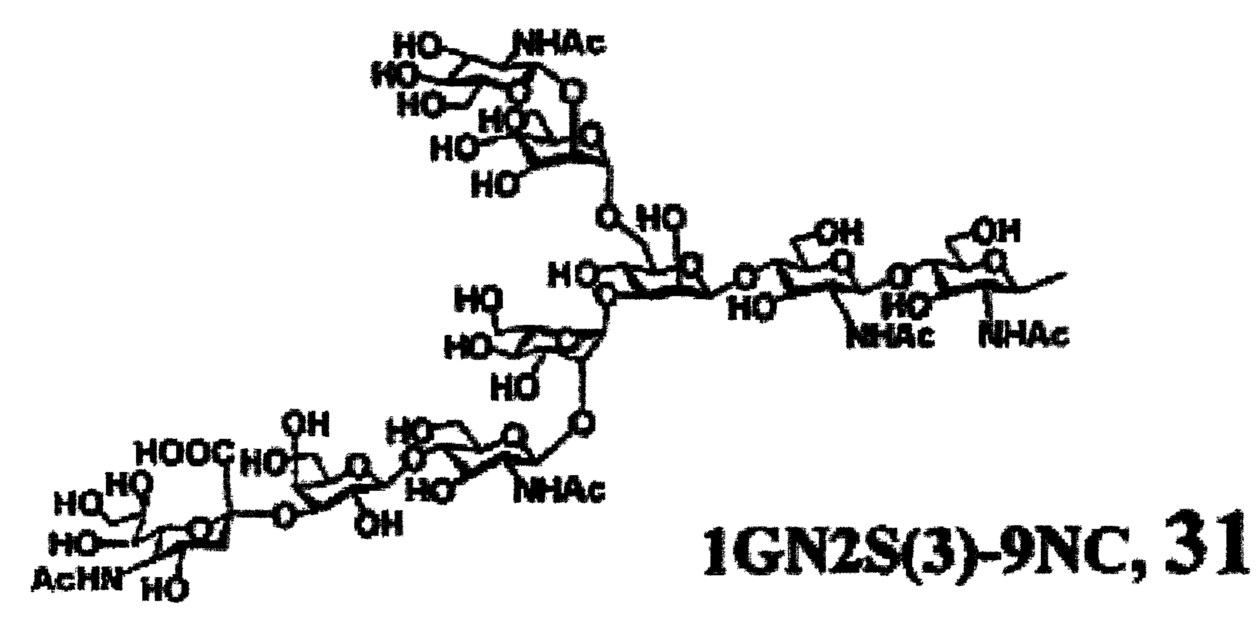


[Химическая формула 29]


В одном варианте осуществления настоящего изобретения сахарная цепь может содержать модифицированную карбоксигруппу сиаловой кислоты, помещенную на невосстанавливающем конце. Примеры такой сахарной цепи могут включать, среди сахарных цепей, перечисленных выше, сахарную цепь, содержащую сиаловую кислоту на невосстанавливающем конце, где атом водорода из карбоксигруппы (-СООН) указанной сиаловой кислоты в сахарной цепи замещен Bn, Et, Me, CH2COPh, CH2PhOMe, CH2Ph(OMe)2, CH2PhNO2, или CH2Ph(NO2)2 (т.е. карбоксигруппа сиаловой кислоты представлена -COOBn, -COOEt, -СООМе, -COOCH2COPh, -COOCH2PhOMe, -COOCH2Ph(OMe)2, -COOCH2PhNO2, или -COOCH2Ph(NO2)2).
Далее, в одном аспекте настоящего изобретения предпочтительной сахарной цепью является сахарная цепь, имеющая линейную структуру. Пример такой сахарной цепи включает олигогиалуроновую кислоту. Олигогиалуроновая кислота в настоящей заявке означает сахарную цепь, имеющую 2-32 сахара, предпочтительно 2-16 сахаров и более предпочтительно 4-8 сахаров из альтернативно связанных N-ацетилглюкозамина и глюкуроновой кислоты в линейной цепи.
Среди олигогиалуроновых кислот, применяемых в настоящем изобретении, особо предпочтительные примеры включают те, в которых единица, состоящая из N-ацетилглюкозамина и глюкуроновой кислоты, представляет 1 единицу, сахарную цепь от 2 единиц (4 сахаров) или более до 8 единиц (16 сахаров) или менее, более предпочтительно от 2 единиц (4 сахаров) до 4 единиц (8 сахаров) и наиболее предпочтительно 2 единицы (4 сахара).
Примеры гиалуроновой кислоты, предпочтительно применяемой в настоящем изобретении, включают, например, 4-сахарную олигогиалуроновую кислоту,
[Химическая формула 30]
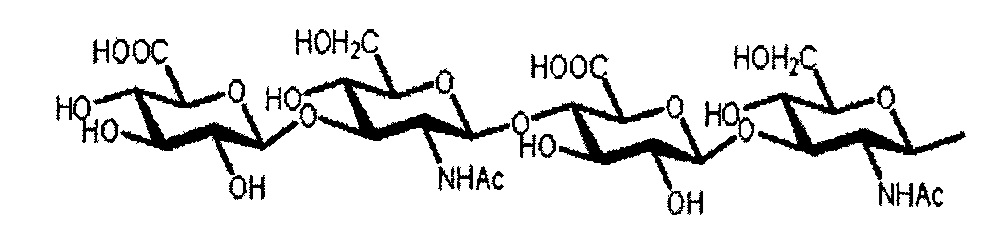
и 8-сахарную олигогиалуроновую кислоту.
[Химическая формула 31]

Аминокислота, подлежащая гликозилированию в гликозилированном полипептиде из настоящего изобретения, предпочтительно является, подобно природному интерферону-β, аминокислотой, соответствующей положению 80 в интерфероне-β. Далее, аминокислота, подлежащая гликозилированию, предпочтительно является аспарагином.
В одном предпочтительном аспекте настоящего изобретения сахарные цепи из гликозилированного полипептида из настоящего изобретения предпочтительно являются однородными. Как применяется в настоящей заявке, то, что сахарные цепи являются однородными в гликозилированном полипептиде, означает тот факт, что участки гликозилирования в пептиде, тип каждого сахара, составляющего сахарную цепь, порядок связывания между сахарами являются идентичными, когда сахарные цепи сравнивают между гликозилированными полипептидами. В частности, это означает, что структура сахарной цепи является однородной между гликозилированными полипептидами по меньшей мере на 90% или более, предпочтительно на 95%» или более и более предпочтительно 99% или более.
Далее, как применяется в настоящей заявке, то, что гликозилированные полипептиды в композиции являются однородными в композиции, содержащей гликозилированный полипептид, означает тот факт, что участки гликозилирования в пептиде, тип каждого сахара, составляющего сахарную цепь, порядок связывания сахарной цепи, тип связывания между сахарами, тип аминокислот, составляющих полипептид, и порядок аминокислотной последовательности являются идентичными, когда сахарную цепь и части полипептида являются сопоставимыми между гликозилированными полипептидами, содержащимися в указанной композиции. В частности, это означает, что структура сахарной цепи и полипептидных частей является однородной для гликозилированных полипептидов, содержащихся в композиции в количестве по меньшей мере 90% или больше, предпочтительно 95% или больше и более предпочтительно 99% или больше.
В частности, композиция и т.д., содержащая гликопептид, имеющая однородные сахарные цепи между гликопептидами, имеет постоянное качество, и в частности, является особо предпочтительной в области фармацевтического производства или количественного анализа. Долю однородных сахарных цепей или долю однородных гликозилированных полипептидов можно измерить, например, путем способа с использованием, например, ВЭЖХ, капиллярного электрофореза, ЯМР, и масс-спектрометрии.
Гликозилированный полипептид из настоящего изобретения производят путем сшивания гликозилированного пептидного фрагмента и по меньшей мере двух пептидных фрагментов.
(Пептидный фрагмент)
Как описано выше, такие гликозилированные пептидные фрагменты и пептидные фрагменты, составляющие гликозилированный полипептид из настоящего изобретения, каждый составляет часть аминокислотной последовательности интерферона-β. Другими словами, каждый фрагмент имеет аминокислотную последовательность каждой последовательности из 166 аминокислот интерферона-β, разделенного по меньшей мере на три части.
Что касается числа фрагментов и длины каждого фрагмента, предпочтительно выбрать аминокислотную последовательность каждого фрагмента с предпочтительным количеством фрагментов и предпочтительным положением аминокислот с учетом уменьшения выхода из-за удлинения при синтезе каждого фрагмента, типа аминокислоты, необходимой для связи каждого фрагмента, и снижения выхода из-за числа этапов связывания.
Длина каждого фрагмента будет варьировать в соответствии со способом приготовления фрагмента, но составляет, например, приблизительно 9-84, более предпочтительно 67-78.
Далее, что касается способа связывания каждого фрагмента, связывание можно осуществлять хорошо известным способом, таким как сшивание без тиолов, сшивание Штаудингера, сшивание с помощью сахаров и сшивание с помощью тиолов (Chem. Commu., 2011, 47, 6201-6207), (Chem. Commu., 2010, 46, 21-43), предпочтительно с помощью способа нативного химического сшивания (NCL).
Когда применяют способ нативного химического сшивания, N-конец каждого фрагмента, выбранный из аминокислотной последовательности интерферона-β, должен быть Цис. Когда выбирают иную аминокислоту, чем Цис, в качестве N-концевой аминокислоты каждого фрагмента, можно выбрать Ала, Гис, Лиз, Фен, Сер, Тре, Вал или Мет. Например, когда N-концевой аминокислотой фрагмента является Ала, можно синтезировать фрагмент, имеющий Цис, введенный на N-концевую часть вместо Ала, а змтем восстановить Цис в Ала после сшивания с другими фрагментами. Далее, когда N-концевой аминокислотой фрагмента является Фен или Вал, можно применять неприродную аминокислоту, имеющую тиоловую группу в β-положении, в качестве аминокислоты для введения с целью сшивания, а затем подвергнуть восстановительной обработке. Когда Сер или Тре выбраны в качестве N-конца фрагмента, цистеин можно ввести в качестве N-конца, а затем его можно превратить в Сер или Тре после сшивания путем метилирования тиоловой группы указанного цистеинового остатка, а затем обработки цианоген бромидом. Далее, когда Гис или Лиз выбирают в качестве N-конца фрагмента, можно получить необходимую амидную связь путем реакции сшивания с применением нуклеофильной атаки аминогруппы после введения этих аминокислот в качестве в качестве N-конца. Когда другие аминокислоты являются N-концом фрагмента, итоговый гликозилированный полипептид можно также построить путем применения способа нативного химического сшивания с хорошо известной методикой.
Альтернативно, когда цистеин отсутствует в необходимом положении аминокислотной последовательности интерферона-β, можно также ввести цистеин в необходимое положение в качестве N-конца фрагмента, с тем условием, чтобы итоговый продукт - гликозилированный полипептид обладал активностью интерферона-β. Далее, введенный цистеин можно также заменить на Ала, Сер или Тре, как указано выше, после этапа сшивания между каждым пептидным фрагментом, как необходимо.
При производстве гликозилированного полипептида из настоящего изобретения аминокислота, выбранная в качестве N-конца фрагмента, предпочтительно выбрана из Ала, Фен, Вал, Сер или Тре, с учетом числа фрагментов или выхода готового продукта, и более предпочтительно выбрана из Ала, Фен или Вал, которые можно заменить путем реакции восстановления. В качестве более специфического примера, положения 1-67 в интерфероне-β могут быть первым фрагментом, положения 68-88, имеющие Ала в положении 68 HaN-конце, могут быть вторым фрагментом, а положения 89-166, имеющие Ала в положении 89 на N-конце, могут быть третьим фрагментом. Далее, например, вышеуказанный третий фрагмент из положений 89-166, можно также производить в разделенном виде на фрагмент в положениях 89-133 и фрагмент в положениях 134-166, содержащий Ала в положении 134 на N-конце.
(Способ производства фрагмента)
Такой способ производства пептидного фрагмента можно осуществить в виде таких способов, как биосинтез, химический синтез, или синтез без применения клеток. В частности, синтез пептидного фрагмента, имеющего сахарную цепь, предпочтительно осуществляют путем химического синтеза для получения однородных сахарных цепей. Способ получения гликозилированного полипептида посредством хорошо известных способов пептидного синтеза, таких как твердофазный и жидкофазный синтез, можно применять в качестве способа производства посредством химического синтеза. Далее, когда Ала выбирают в качестве N-конца каждого фрагмента, поскольку необходимо восстановить конкретный цистеин, введенный на N-конец вместо Ала в Ала после сшивания каждого фрагмента, предпочтительно заранее защитить Цис, который не подлежит восстановлению. Соответственно, в таком случае удобно и предпочтительно получать пептидный фрагмент с помощью химического синтеза.
В качестве специфического примера способа получения гликозилированного пептидного фрагмента из настоящего изобретения, являющегося стабильным способом производства гликозилированного полипептида, имеющего однородную структуру сахарной цепи, в качестве пептидных фрагментов, можно применять способ получения гликозилированного пептидного фрагмента с использованием гликозилированного Асн в качестве гликозилированной аминокислоты, и с применением хорошо известного способа пептидного синтеза, такого как твердофазный и жидкофазный синтез. Такой способ описан, например, в Angew. Chem. Int. Ed. 2008, 47, 6851-6855. Он также описан в Международной публикации №2004/005330 (US 2005222382 (A1)), раскрытие которой настоящим включено посредством ссылки во всей полноте.
Гликозилированный полипептид можно получить, например, посредством твердофазного синтеза с применением гликозилированного аспарагина, изложение которого показано ниже.
(1) Карбоксигруппу из аминокислоты с азотом аминогруппы, защищенным липофильной защитной группой, присоединяют к смоле. В этом случае, поскольку азот аминогруппы из аминокислоты защищен липофильной защитной группой, предотвращается самоконденсация между аминокислотами, и смола реагирует с аминокислотой, обеспечивая связывание.
(2) Липофильную защитную группу полученного реактанта отсоединяют с получением свободной аминогруппы.
(3) Эту свободную аминогруппу и карбоксигруппу из любой аминокислоты, в которой азот аминогруппы защищен липофильной защитной группой, подвергают реакции амидирования.
(4) Липофильную защитную группу отсоединяют с получением свободной аминогруппы.
(5) Путем повторения вышеуказанных этапов (3) и (4) еще раз или больше, получают пептид, в котором любое число любой аминокислоты связано и соединено со смолой на одном конце и имеет свободную аминогруппу на другом конце.
(6) Наконец, путем отделения от смолы кислотой, можно получить пептид, обладающий необходимой аминокислотной последовательностью.
На этапе (1), если гликозилированный аспарагин, содержащий азот аминогруппы, защищенный липофильной защитной группой, применяют вместо аминокислоты, имеющей азот аминогруппы, защищенный липофильной защитной группой, и проводят реакцию карбоксигруппы указанной аспарагиновой части и гидроксильной группы смолы, можно получить гликозилированный пептидный фрагмент, содержащий гликозилированный аспарагин на С-конце.
Далее, после этапа (2), или после повторения этапов (3) и (4) любое число раз, т.е. один раз или больше, если применяют гликозилированный аспарагин, в котором азот аминогруппы защищен липофильной защитной группой, вместо аминокислоты, в которой азот аминогруппы защищен липофильной защитной группой на этапе (3), сахарную цепь можно добавить в любом положении. Далее, этапы (3) и (4) можно также дополнительно повторять после добавления гликозилированного аспарагина для удлинения пептида.
После связывания гликозилированной аминокислоты, если липофильную защитную группу отсоединяют с получением свободной аминогруппы, и немедленно после этого проводят этап (6), можно получить пептид, содержащий гликозилированный аспарагин на N-концевом пептиде.
Сахарные цепи, описанные выше, имеющие идентичную структуру, применяют в качестве сахарной цепи, связанной в вышеуказанном способе синтеза. Такие сахарные цепи можно получить путем любого хорошо известного способа. Примеры специфических способов, которые можно применять, включают химический синтез сахарных цепей (см., например, J. Seifert et al. Angew Chem Int. Ed. 2000, 39, p531-534) или получение цепей, отделенных от природного или искусственного источника сахарных цепей, или коммерческих цепей, но не ограничиваются ими. Гликозилированные аминокислоты, имеющие идентичную структуру в указанном способе, не ограничены, и например, выделение сахарных цепей с идентичной структурой из природного или искусственного источника сахарных цепей можно проводить путем способа, описанного, например, в WO 2004/058789. В частности, смесь, содержащую гликозилированный аспарагин (сиалилгликопептид (СГП)) выделяют из природного источника сахарных цепей, такого как куриное яйцо, с помощью способа, описанного, например, в Seko et al., Biochim Biophys Acta. 1997; 1335 (1-2):23-32; липофильную защитную группу (такую, как Fmoc) вводят в указанный гликозилированный аспарагин для получения смеси производных гликозилированного аспарагина; ее подвергают хроматографии; и сахарные цепи, имеющие различную структуру, содержащиеся в указанной смеси, можно разделить в соответствии с их структурой. Далее, гликозилированный аспарагин, имеющий частную структуру, с различными защитными группами или без них, можно получить, например, от Otsuka Chemical Co., Ltd.
Смола может быть смолой, обычно применяемой в твердофазном синтезе, и например, можно применять 2-хлоротритил-хлоридную смолу (от Merck), функционализированную хлором, амино-PEGA смолу (от Merck), функционализированную аминогруппой, NovaSyn TGT спиртовую смолу (от Merck), имеющую гидроксильную группу, смолу Ванга (от Merck), и HMPA-PEGA смолу (от Merck). Далее, может присутствовать линкер между смолой амино-PEGA и аминокислотой, и примеры таких линкеров могут включать, например, 4-гидроксиметилфеноксиуксусную кислоту (НМРА) и 4-(4-гидроксиметил-3-метоксифенокси)-бутилуксусную кислоту (НМРВ). Можно также применять смолу Н-Цис(Trt)-Тритил NovaPEG (от Merck) и т.д., в которой С-концевая аминокислота предварительно связана со смолой.
Далее, если С-конец должен быть амидирован, например, можно применять смолу Rink-Amide-PEGA (от Merck), функционализированную аминогруппой. Путем отделения пептида от этой смолы кислотой, можно амидировать С-концевую кислоту пептида.
При связывании смолы и аминокислоты, в которой азот аминогруппы защищен липофильной защитной группой, например, при использовании смолы, имеющей гидроксильную группы, или смолы, функционализированной хлором, карбоксигруппу аминокислоты связывают со смолой через сложноэфирную связь. Далее, если применяют смолу, функционализированную аминогруппой, карбоксигруппу аминокислоты связывают со смолой через амидную связь.
2-хлоротритил-хлоридная смола является предпочтительной, поскольку позволяет предотвратить рацемизацию концевого Цис при удлинении пептидной цепи при твердофазном синтезе.
Любую аминокислоту можно применять в качестве аминокислоты, и примеры включают природные аминокислоты серии (Сер), аспарагин (Асн), валин (Вал), лейцин (Лей), изолейцин (Иле), аланин (Ала), тирозин (Тир), глицин (Гли), лизин (Лиз), аргинин (Apr), гистидин (Тис), аспарагиновую кислоту (Асп), глутаминовую кислоту (Глу), глутамин (Глн), треонин (Тре), цистеин (Цис), метионин (Мет), фенилаланин (Фен), триптофан (Трп) и пролин (Про).
Примеры липофильных защитных групп могут включать, например, защитные группы на основе карбонатов и амидов, такие как 9-флуоренилметоксикарбонильная (Fmoc) группа, т-бутилоксикарбонильная (Вос) группа, бензильная группа, аллильная группа, аллилоксикарбонильная группа, и ацетильная группа. При введении липофильной защитной группы в аминогруппу, например, при введении Fmoc группы, введение можно осуществлять путем добавления 9-флюоренилметил-К-сукцинимидил-карбоната и гидрокарбоната натрия, и обеспечения реакции. Реакцию можно проводить при 0-50°С, предпочтительно при комнатной температуре, в течение примерно 1-5 часов.
В качестве аминокислоты, защищенной липофильной защитной группой, можно применять коммерческие аминокислоты. Примеры могут включать Fmoc-Cep-OH, Fmoc-Асн-ОН, Fmoc-Вал-ОН, Fmoc-Лей-ОН, Fmoc-Иле-ОН, Fmoc-Ала-ОН, Fmoc-Тир-ОН, Fmoc-Гли-ОН, Fmoc-Лиз-ОН, Fmoc-Арг-ОН, Fmoc-Гис-ОН, Fmoc-Асп-ОН, Fmoc-Глу-ОН, Fmoc-Глн-ОН, Fmoc-Tpe-OH, Fmoc-Цис-ОН, Fmoc-Мет-ОН, Fmoc-Фен-ОН, Fmoc-Τрп-OH, Fmoc-Προ-ΟΗ, Boc-Cep-OH, Вос-Асн-ОН, Вос-Вал-ОН, Вос-Лей-ОН, Вос-Иле-ОН, Вос-Ала-ОН, Вос-Тир-ОН, Вос-Глу-ОН, Вос-Лиз-ОН, Вос-Арг-ОН, Вос-Гис-ОН, Вос-Асп-ОН, Вос-Глу-ОН, Вос-Глн-ОН, Вос-Тре-ОН, Вос-Цис-ОН, Вос-Мет-ОН, Вос-Фен-ОН, Вос-Трп-ОН и Вос-Про-ОН.
Далее, аминокислота, защищенная липофильной защитной группой, в которой защитную группу вводят в боковую цепь, может включать, например, Fmoc-Ала-ОН, Fmoc-Арг(Pbf)-OH, Fmoc-AcH(Trt)-OH, Fmoc-Асп(OtBu)-OH, Fmoc-Цис(Acm)-ОН, Fmoc-Цис(tBu)-OH, Fmoc-Цис(Trt)-OH, Fmoc-Глу(OtBu)-OH, Fmoc-Глн(Trt)-ОН, Fmoc-Гли-ОН, Fmoc-Гис(Trt)-OH, Fmoc-Иле-ОН, Fmoc-Лей-ОН, Fmoc-Лиз(Вос)-ОН, Fmoc-Мет-ОН, Fmoc-Фен-ОН, Fmoc-Πpο-ΟΗ, Fmoc-Cep(tBu)-OH, Fmoc-Tpe(tBu)-OH, Fmoc-Трп(Boc)-OH, Fmoc-Тир(tBu)-OH, Fmoc-Вал-ОН, Вос-Мет-ОН и Boc-Thz-OH. Thz указывает Цис тиазолидинового типа (тиазолидин-4-карбоновую кислоту).
Если пептидный фрагмент синтезируют посредством, например, твердофазного синтеза, предпочтительно иметь аминокислоту, защищенную Вос группой на N-конце, чтобы можно было подавить побочную нуклеофильную реакцию, обусловленную аминогруппой, когда необходима тиоэтерификация после синтеза пептидного фрагмента.
Далее, если необходимо добавить линкер в аминокислотную последовательность гликозилированного полипептида, линкер можно вставить в предпочтительное положение путем использования линкера, защищенного липофильной защитной группой, вместо вышеуказанной аминокислоты, защищенной липофильной защитной группой, в процессе твердофазного синтеза.
При использовании смолы, содержащей гидроксильную группу, можно применять в качестве катализатора этерификации хорошо известные агенты для дегидратационной конденсации, например, такие как 1-мезитиленсульфонил-3-нитро-1,2,4-триазол (MSNT), дициклогексилкарбодиимид (DCC), и диизопропилкарбодиимид (DIPCDI). Далее, при использовании 2-хлоротритил-хлоридной смолы этерификацию можно проводить с таким основанием, как диизопропилэтиламин (DIPEA), триэтиламин, пиридин, и 2,4,6-коллидин. Используемая пропорция аминокислоты и агента для дегидратационной конденсации составляет 1 массовую часть первого, как правило, на 1-10 массовых частей, предпочтительно 2-5 массовых частей последнего.
Реакцию этерификации предпочтительно проводят, например, путем внесения смолы в колонку для твердофазного синтеза, промывания смолы растворителем, с последующим добавлением раствора аминокислоты. Примеры растворителей для промывания могут включать, например, диметилформамид (ДМФ), 2-пропанол и дихлорметан. Примеры растворителей для растворения аминокислоты могут включать, например, диметилсульфоксид (ДМСО), ДМФ и дихлорметан. Реакцию этерификации можно проводить при 0-50°С, предпочтительно при комнатной температуре, в течение примерно 10 минут - 30 часов, предпочтительно в течение примерно 15 минут - 24 часов.
Далее, также предпочтительно ацетилировать и кэпировать непрореагировавшие гидроксильные группы на твердой фазе к этому времени, например, уксусным ангидридом.
Отсоединение липофильной защитной группы можно выполнять, например, путем обработки основанием. Примеры оснований могут включать, например, пиперидин и морфолин. В этом случае предпочтительно проводить это в присутствии растворителя. Примеры растворителей могут включать, например, ДМСО, ДМФ и метанол.
Реакцию амидирования свободной аминогруппы с карбоксигруппой из любой аминокислоты с азотом аминогруппы, защищенным липофильной защитной группой, предпочтительно проводят в присутствии активатора и растворителя.
Примеры активаторов могут включать, например, дициклогексилкарбодиимид (ДЦК), 1-этил-3-диметиламинопропил)карбодиимид гидрохлорид (WSC/HCl), дифенилфосфорилазид (ДФФА), карбонилдиимидазол (КДИ), диэтилцианофосфонат (ДЭЦФ), бензотриазол-1-илокси-триспирролидинфосфоний (DIPCI), бензотриазол-1-илокси-триспирролидинофосфоний гексафторфосфат (РуВОР), 1-гидроксибензотриазол (HOBt), гидроксисукцинимид (HOSu), диметиламинопиридин (ДМАП), 1-гидрокси-7-азабензотриазол (HOAt), гидроксифталимид (HOPht), пентафторфенол (Pfp-OH), 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат (HBTU), 1-[бис(диметиламино)метилен]-5-хлор-1Н-бензотриазолий 3-оксид гексафторфосфат (HCTU), O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфонат (HATU), O-бензотриазол-1-ил-1,1,3,3-тетраметилуроний тетрафторборат (TBTU) и 3,4-дигидро-3-гидроди-4-окса-1,2,3-бензотриазин (DHBT).
Количество используемого активатора может составлять 1-20 эквивалентов, предпочтительно 1-10 эквивалентов и более предпочтительно 1-5 эквивалентов любой аминокислоты, в которой азот аминогруппы защищен липофильной защитной группой.
Примеры растворителей могут включать, например, ДМСО, ДМФ и дихлорметан. Реакцию можно проводить при 0-50°С, предпочтительно при комнатной температуре, в течение примерно 10-30 часов, предпочтительно в течение примерно 15 минут - 24 часов. Отсоединение липофильной защитной группы можно проводить подобно тому, что было указано выше.
Обработка кислотой является предпочтительной для отделения пептидной цепи от смолы. Примеры кислот могут включать, например, смешанный раствор трифторэтанола и уксусной кислоты (1:1), трифторуксусную кислоту (ТФУ), и фторид водорода (HF).
Пептидный фрагмент можно приготовить путем химического синтеза, такого как твердофазный синтез, как указано выше, но его можно также приготовить путем биосинтеза, хорошо известного специалистам в данной области техники. Например, необходимый пептидный фрагмент может быть экспрессирован путем введения необходимого гена в рекомбинантный вектор. Рекомбинантный вектор, применяемый в настоящей заявке, может быть любым вектором, способным трансформировать клетку-носитель, и применяют животные вирусные векторы, такие как плазмида для Е. coli, плазмида для Bacillus subtilis, и плазмиды для дрожжей, ретровирус, вирус коровьей оспы, и бакуловирус и т.д., в зависимости от клетки-акцептора. Предпочтительными являются те, которые имеют регуляторную последовательность, такую как промотор, которые могут надлежащим образом экспрессировать белок в клетке-акцепторе. Далее, клетка-акцептор может быть любой клеткой, способной экспрессировать чужеродный ген в рекомбинантном векторе, и как правило, применяют клетки Е. coli, Bacillus subtilis, дрожжей, клетки насекомых, и клетки животных.
Применяемый способ трансфекции клетки-акцептора с рекомбинантным вектором может быть способом, рутинно используемым в целом, и например, можно применять способ теплового шока, способ с хлоридом кальция, или способ электропорации для Е. coli, и способ с хлоридом лития или электропорации для дрожжей. Далее, трансформацию животной клетки можно проводить физическим способом, таким как электропорация, или химическим способом, таким как липосомный способ или способ с фосфатом кальция, или с вирусным вектором, таким как ретровирус. Далее, после введения вектора предпочтительно подтвердить, что необходимая ДНК последовательность надлежащим образом интегрирована посредством способа, хорошо известного специалистам в данной области техники. Что касается формата культивирования трансформированной клетки-акцептора, условия культивирования могут быть выбраны с учетом природы физиологии питания акцептора.
Далее, пептид, приготовленный путем биосинтеза, предпочтительно является очищенным. Способ очистки пептида может быть осуществлен путем обычной общей очистки. Например, в случае рекомбинантного белка, после культивирования бактерии или клетки, экспрессирующей рекомбинантный белок, используемый в настоящей заявке, неочищенный экстракт пептида получают путем сбора бактерии или клетки с хорошо известным способом, путем суспендирования ее в подходящем буферном растворе, разрушения бактерии или клетки, например, с помощью ультразвука, лизоцима и/или замораживания-оттаивания, а затем осаждения центрифугированием или фильтрации. Буферный раствор может содержать агент, денатурирующий белок, такой как мочевина или гуанидина гидрохлорид, или сурфактант, такой как Тритон Х-100™. Очистку экстракта таким способом, или пептида, содержащегося в надосадочной жидкости культуры, можно проводить путем хорошо известного способа очистки. Например, аффинную хроматографию, ионообменную хроматографию, фильтрацию, ультрафильтрацию, гель-фильтрацию, электрофорез, солевое осаждение и диализ можно выбрать и объединить подходящим образом для обеспечения разделения и очистки пептида.
Далее, для облегчения очистки рекомбинантного белка, можно встроить различные маркеры в вектор экспрессии. Примеры маркера, который можно применять, включают маркеры, известные специалистам в данной области техники, такие как маркер, повышающий эффективность экспрессии, или маркер, повьппающий эффективность очистки, и маркеры включают, например, тиоредоксин, GST маркер, Мус маркер, FLAG маркер и белок, связывающий мальтозу (МВР).
Далее, когда цистеин, используемый для сшивания с необходимым N-концом пептидного фрагмента, отсутствует, то цистеин также можно ввести в указанный участок. Например, можно изменить необходимый участок на цистеин в молекуле нуклеиновой кислоты, вводимой в клетку-акцептор, посредством любого способа мутагенеза, известного в данной области техники, включая ПЦР сниженной точности, сайт-специфический мутагенез, полимеразную цикличную сборку, перестановку в ДНК, мутагенез in vivo, кассетный мутагенез, рекурсивный множественный мутагенез и экспоненциальный множественный мутагенез.
Далее, когда необходима защита аминокислотной боковой цепи в пептидном фрагменте, полученном путем биосинтеза, как указано выше, защиту можно обеспечить с помощью способа, хорошо известного специалистам в данной области техники. Например, для защиты цистеина N-концевой цистеин защищают, например, формальдегидом, а тиоловую группу боковой цепи Цис остатка в пептиде можно избирательно защитить, например, S-9-флуоренилметил-тиоэфиром (Fm-SR). (Связывание каждого фрагмента).
Любой хорошо известный способ, позволяющий связать две цепи пептидных фрагментов, как описано выше, можно применять в качестве этапа связывания гликозилированного пептидного фрагмента и пептидного фрагмента, приготовленного в способе, указанном выше, пример которого может включать нативное химическое лигатирование (НХЛ, см., например, перевод с японского международной патентной заявки РСТ №2004-518621). НХЛ является способом смешивания пептидного фрагмента А, имеющего тиоэфир на С-конце, и пептидного фрагмента В, имеющего цистеин на N-конце, в буферном растворе, отсоединения тиоэфирной группы на С-конце пептида А путем нуклеофильной атаки сульфгидрильной группы цистеина на N-конце пептидного фрагмента В, и сшивания двух пептидов друг с другом посредством природной пептидной связи путем последовательной нуклеофильной атаки амина.
Соответственно, как описано выше, при использовании НХЛ на этапе сшивания, этап тиоэтерификации С-конца полипептида, располагаемого на N-конце, необходим перед этапом сшивания. Такую тиоэтерификацию можно обеспечить, например, путем активации С-концевой карбоновой кислоты, с помощью РуВОР и DIPEA, и добавления избытка алкилтиола. При использовании этого способа добавление алкилтиола предпочтительно проводят при низкой температуре от 10°С до -80°С, более предпочтительно при температуре от 0°С до -40°С, для подавления конфигурации α углерода в аминокислоте на конце фрагмента. В частности, в вышеуказанной реакции тиоэтерификации предпочтительно применять тиофенол вместо алкилтиола. Реакция тиоэтерификации при добавлении тиофенола имеет высокую скорость и может почти полностью завершить реакцию. Соответственно, побочная реакция циклизации пептидного фрагмента при реакции тиоэтерификации может быть подавлена, и таким образом, реакция тиоэтерификации является предпочтительной.
Далее, вышеуказанную тиоэтерификацию можно проводить также, например, путем способов с Fmoc или Boc, описанных в Yamamoto et al., J. Am. Chem. Soc. 2008, 130 (2), 501-510.
НХЛ проводят путем смешивания предпочтительно эквимолярных количеств полипептидной цепи из части, связывающей сахарную цепь, и полипептидной цепи из части, не связывающей сахарную цепь, в буферном растворе. В предпочтительном аспекте реакцию связывания проводят в буферном растворе с рН 6-8, и с предпочтительным рН в диапазоне 6,5-7,5. Буферный раствор может быть водным, органическим, или их смесью. Реакция связывания может дополнительно включать один или два или более типов катализаторов и/или один или два или более типов восстановителей, липидов, и других денатурирующих агентов или солюбилизаторов, и т.д. Примеры предпочтительного катализатора включают, например, тиол- и фосфинсодержащее вещество, такое как тиофенол, бензилмеркаптан, ТКЭФ, и алкилфосфин. Примеры денатурирующего агента и/или солюбилизатора включают гуанидин, водный раствор мочевины или раствор органического растворителя, такого как ТФЭ, ГФИП, ДМФ и НМП; воду, или ацетонитрил, смешанный с гуанидином и водным раствором мочевины. Скорость реакции связывания можно регулировать посредством температуры, и реакция может быть проведена при 5-55°С, предпочтительно при 15-40°С. В частности, реакцию связывания ИФН-β предпочтительно проводят в условиях 6-8М гуанидина. Например, реакция хорошо проходит в реакционной смеси с применением буферного раствора, содержащего 6М гуанидин при рН 6,8-7,8 и 1-3% тиофенола.
Если разделение продукта после этапа сшивания и исходного материала затруднено, например, например, тиоловая группа цистеина, которая была точкой сшивания, может быть преднамеренно тиоэтерифицирована путем применения избыточного количества С-концевого тиофенил-эфирного компонента с высокой реактивностью. Таким способом путем тиоэтерификации тиоловой группы пептидной боковой цепи, полученной путем сшивания, липофильность или растворимость в воде продукта можно повысить для облегчения разделения.
Если посредством этой операции производят избыточно тиоэтерифицированную тиоловую группу, ее можно легко восстановить до тиоловой группы, например, меркаптоэтансульфонатом натрия.
Когда каждый фрагмент должен иметь Ала в качестве N-конца, гликозилированный полипептид, имеющий Цис вместо Ала на N-конце, производят путем вышеуказанного способа. Соответственно, в этом случае предпочтительно возвратить аминокислоту, которая требует восстановления (Цис) на Ала, путем реакции восстановления при получении гликозилированного полипептида.
Такой способ, как радикальное восстановление и гидридное восстановление, можно применять в качестве способа восстановления Цис до Ала, особо предпочтительно радикальное восстановление.
Реакцию радикального восстановления можно проводить, например, путем добавления tBuSH, натрия 2-меркаптоэтансульфоната, или глутатиона и т.д. к растворителю, такому как ТКЭФ, содержащему пептидный фрагмент. Примеры растворителей, используемых в реакции радикального восстановления, могут включать воду, органический растворитель (такой, как ацетонитрил), или буферный раствор (такой как фосфатный буферный раствор), и специфический пример может включать Трис-карбоксиэтил-фосфиновый раствор (раствор ТКЭФ). Далее, растворитель может содержать солюбилизатор. Например, он предпочтительно содержит 6М-8М гуанидин. Далее, реакцию радикального восстановления предпочтительно проводят при рН в диапазоне 7,5-8,0, а температура реакции предпочтительно составляет 15-40°С. Когда рН ниже 7,5, реакция не проходит полностью, и таким образом, это значение не является предпочтительным. Далее, время реакции может составлять 2-6 часов. Когда она длится более 6 часов, начинается максимальное затухание, и это не является предпочтительным. В качестве примера реакции радикального восстановления, реакцию можно проводить с 7М гуанидином и 0,25М ТКЭФ в условиях рН 7,5; 20°С и в течение 4 часов.
В реакции радикального восстановления Цис, который предпочтительно не восстанавливается, может быть защищен защитной группой. В качестве такой защитной группы можно применять хорошо известные защитные группы, такие как Acm группа, алкоксиметильная группа, трифенилметильная группа, т-бутильная группа, бензильная группа, и этильная группа, в которой β положение замещено.
Далее, предпочтительно применяют контейнер для реакции, обеспечивающий небольшой мертвый объем для размещения реакционного раствора для реакции радикального восстановления для полного завершения реакции.
Далее, когда Цис, защищенный Acm группой и т.д., существует после вышеуказанного этапа восстановления, выполняют снятие защиты с Цис.
Что касается этапа снятия защиты, например, когда защитной группой для Цис является Acm группа, ее можно удалить путем реакции, например, с йодом, ацетатом ртути (II), нитратом серебра (I), или ацетатом серебра (I) в воде, метанолом, уксусной кислотой, или их смешанным раствором. Далее, когда защитной группой является алкоксиметильная группа, ее можно удалить, например, кислотным гидролизом или путем применения соли серебра или ртути. Когда защитной группой является трифенилметильная группа, ее можно удалить, например, солью ртути (II).
Вышеуказанную реакцию можно проводить, как правило, при 0-80°С, предпочтительно при 5-60°С, и более предпочтительно при 10-35°С. Время реакции предпочтительно составляет 5 минут - 24 часа в целом. Другие защитные группы можно также удалить посредством способа, хорошо известного специалистам в данной области техники. Далее, после завершения реакции, можно провести обработку, например, дитиотреитолом (ДТТ) или соляной кислотой, а затем очистить хорошо известным способом (таким как высокоэффективная жидкостная колоночная хроматография (ВЭЖХ)), если необходимо.
Далее, когда каждый фрагмент должен иметь Сер в качестве N-конца, гликозилированный полипептид, имеющий Цис вместо Сер на N-конце, производят путем вышеуказанного способа. Соответственно, в этом случае аминокислоту, которую нужно превратить в Сер (Цис), превращают в Сер после связывания при получении гликозилированного полипептида.
Такой этап превращения можно выполнить, например, путем реакции метилирования -SH группы на Цис, реакции цианирования, и последующей реакции интрамолекулярной транслокации ацила.
В частности, вышеуказанный этап превращения включает (1) этап реакции -SH группы из Цис в пептиде, содержащем Цис, с метилирующим агентом до получения -SMe группы (метилирования), и (2) этап реакции -SMe группы пептида, полученного на этапе (1), с планирующим агентом (реакция цианирования), и после реакции, превращение -Sme группы в -ОН группу через промежуточный продукт реакции путем помещения указанного пептида в основные условия (реакция интрамолекулярной транслокации ацила). Такой этап превращения описан, например, в Международной публикации 2009/017154, раскрытие которой настоящим включено посредством ссылки во всей полноте.
Примеры метилирующего агента, применяемого для реакции метилирования -SH группы Цис не ограничиваются конкретно, при условии, что он способен превратить -SH группу в -SMe группу, и он может включать, например, йодометан и метил-4-нитробензол сульфонат.
Количество используемого метилирующего агента составляет 1-1000 эквивалентов для одного цистеинового остатка из исходного материала пептида или гликопептида. Он может предпочтительно составлять 10-100 и более предпочтительно 15-30 эквивалентов. Реакцию метилирования можно проводить при 0-50°С, предпочтительно при 20-30°С, в течение примерно 10 минут - 30 часов, предпочтительно примерно 15 минут -1 часа.
Растворитель может быть буферным раствором, рН которого может быть 7-9, предпочтительно 8-9. Например, он может включать буферный раствор с рН 8,6 с 6М раствором гуанидин-гидрохлорида, 0,25 M раствора Трис-соляной кислоты, и 3,3 мМ раствора ЭДТА.
Далее, примерами планирующего агента, который можно применять с учетом стабильности, являются, например, цианоген-бромид и фенилцианат. Предпочтительно можно применять легкодоступный цианоген-бромид.
Количество используемого цианирующего агента может составлять 1-1000 эквивалентов, предпочтительно 10-100 эквивалентов, и более предпочтительно 15-30 эквивалентов на один остаток -SMe группы. Реакцию с планирующим агентом предпочтительно проводят при 0-50°С, предпочтительно при 30-40°С, в течение примерно 30 минут - 100 часов, предпочтительно примерно 12-50 часов.
Реакцию с цианирующим агентом проводят в кислых условиях, особо предпочтительно при рН 2-3. Реакцию можно проводить в кислых условиях путем применения кислого водорастворимого вещества, в частности, муравьиной кислоты, трифторуксусной кислоты, и метансульфоновой кислоты, и т.д. При ее выполнении кислое водорастворимое вещество особо предпочтительно дегазируют для предотвращения окисления атома серы. Далее, реакцию предпочтительно проводят при условии защиты от света, с учетом стабильности цианирующего агента.
В качестве растворителя можно оптимально применять водорастворимый растворитель с рН 2-3, описанный выше, например 80% раствор муравьиной кислоты, 70% раствор муравьиной кислоты и 2% раствор трифторуксусной кислоты/39% ацетонитрилсодержащий водный раствор и т.д.
Если пептид содержит метиониновый остаток в качестве аминокислоты, предпочтительно различать -SMe группу метионинового остатка и -SMe группу, полученную при реакции метилирования. Это различение можно проводить, например, путем введения защитной группы. Как применяется в настоящей заявке, метионин с введенной защитной группой (защищенный метионин) не ограничивается конкретно, при условии, что соединение не вступает в реакцию цианирования с цианирующим реагентом; например, может включать метионин сульфоксидного типа (Met(O): -CH2-CH2-S(=O)-СН3). Защищенный метиониновый остаток может быть превращен в метиониновый остаток после реакции цианирования или реакции интрамолекулярной транслокации ацила. Способ снятия защиты может быть проведен, как принято способом, хорошо известным в данной области техники.
Реакция интрамолекулярной транслокации ацила может быть проведена в более основных условиях, по сравнению с реакцией цианирования. Это обеспечивает пептид, содержащий аминокислотный остаток, имеющий -ОН группу.
Базовые условия реакции интрамолекулярной транслокации ацила могут быть кислыми или нейтральными, с тем условием, чтобы реакция проходила в более щелочных условиях, по сравнению с реакцией цианирования, более конкретно, при условии, чтобы -NH2 группа на С атоме рядом со сложноэфирной связью промежуточного продукта, полученного при реакции цианирования, не была протонированной. Что касается эффективного превращения промежуточного продукта реакции в пептид, содержащий - ОН группы, можно применять слабо или сильно основные условия.
Основные условия можно обеспечить путем добавления основного соединения, являющегося агентом для регулирования рН, хорошо известным специалисту в данной области техники (такого, как гуанидин, динатрий фосфат, Трис, натрия гидрокарбонат, гидразина гидрат, и 50 мМ водный раствор гидроксида натрия). При выполнении этого количество используемого основного компонента может составлять 0,5-1000 эквивалентов, предпочтительно 10-100 эквивалентов, и более предпочтительно 15-30 эквивалентов пептида из исходного материала.
Интрамолекулярную транслокацию ацила предпочтительно проводят при 50°С, предпочтительно при 20-30°С, в течение примерно 5 минут - 30 часов, предпочтительно в течение 5 минут - 1 часа, и более предпочтительно примерно 5 минут - 10 минут.
Прекращение реакции можно обеспечить путем снижения рН. Альтернативно, можно также непосредственно перейти к этапу очистки, например, путем ВЭЖХ, без изменения рН.
Если Тре выбран в качестве N-конца пептидного фрагмента, остаток производного треонина (Тре), содержащий -SH группу вместо Цис (или производного треоина, содержащего указанную -SH группу, защищенную дисульфидной связью и т.д.) можно ввести на N-конец пептидного фрагмента. Пептидный фрагмент, содержащий введенное производное треонина, можно подвергнуть вышеуказанной реакции метилирования, реакции цианирования, и реакции интрамолекулярной транслокации ацила после связывания, для замещения производного треонина в указанном пептидном фрагменте на треонин.
При введении сахарной цепи, содержащей сиаловую кислоту, в пептидный фрагмент, предпочтительно, чтобы применялась карбоксигруппа сиаловой кислоты на вводимой сахарной цепи, например, содержащая сиаловую кислоту сахарная цепь, защищенная, например, арильной группой, содержащей, например, бензильную группу (Bn), алкильную группу, включая, например, этильную группу (Et) или метальную группу (Me), и фенацильную группу, содержащую боковую цепь углерода, образующего кольцевую структуру, замещенную, например, дифенилметильной группой, фенацильной группой, алкокси группой, или нитро группой. В частности, например, предпочтительной является защитная группа, защищающая карбоксигруппу сиаловой кислоты, представленная COOBn, -COOEt, -СООМе, -COOCH(Ph)2, -COOCH2COPh, -COOCH2PhOMe, -COOCH2Ph(OMe)2, -COOCH2PhNO2 или -COOCH2Ph(NO2)2. Таким способом, путем защиты карбоксигруппы сиаловой кислоты бензильной группой и т.д., можно предотвратить отсоединение чувствительной к кислоте сиаловой кислоты. В частности, можно предотвратить отсоединение сиаловой кислоты от невосстанавливающего конца сахарной цепи, также при выполнении тепловой обработки, описанной ниже. Далее, введение защитной группы в карбоксигруппу сиаловой кислоты облегчает этап разделения и очистки путем ВЭЖХ и т.д. на этапе производства.
Реакцию защиты карбоксигруппы сиаловой кислоты на сахарной цепи можно провести посредством способа, хорошо известного специалисту в данной области техники. Например, удаление защитной группы с карбоксигруппы сиаловой кислоты, защищенной бензильной группой, дифенилметильной группой, или фенацильной группой можно также проводить посредством способа, хорошо известного специалисту в данной области техники. Например, реакцию снятия защиты можно проводить путем гидролиза в основных условиях, хотя они не ограничиваются конкретно следующими условиями. Реакцию снятия защиты можно проводить, как правило, при 0-50°С, предпочтительно при 0-40°С, и более предпочтительно при 0-30°С. Время реакции предпочтительно составляет, как правило, 5 минут - 5 часов. После завершения реакции ее можно нейтрализовать слабой кислотой, такой как фосфорная или уксусная кислота, а затем очистить хорошо известным способом (таким, как высокоэффективная жидкостная колоночная хроматография (ВЭЖХ), как это уместно. Этап удаления защитной группы с карбокси группы сиаловой кислоты можно проводить до или после этапа фолдинга.
Каждый этап производства интерферона-β, указанный выше, может быть в однореакторном синтезе. Например, когда интерферон-β получают путем приготовления множества фрагментов и путем множества этапов сшивания, то множество этапов сшивания могут быть однореакторным синтезом. Далее, этап восстановления цистеина необходим после соединения фрагментов, и множество этапов сшивания, также как и последующий этап восстановления цистеина, также может быть однореакторным синтезом.
Далее, этап снятия защиты цистеина и этап снятия защиты сахарной цепи сиаловой кислоты, необходимый после этапа сшивания (такой, как этап восстановления цистеина, если необходимо), также может быть однореакторным синтезом. Например, путем проведения первого этапа сшивания и второго этапа сшивания, описанного в Примерах 2 и 3 в настоящем описании в однореакторном процессе, можно повысить выход примерно в 1,6 раза.
Таким способом, проведение этапа производства в виде однореакторного процесса, является предпочтительным, поскольку можно достичь снижения времени этапа производства и облегчения путем удаления этапа ВЭЖХ, и кроме того, повысить выход.
(Этап фолдинга)
Гликозилированный пептидный фрагмент и пептидный фрагмент связывают путем вышеуказанного способа, и можно включить этап замещения необходимой аминокислоты, или этап фолдинга гликозилированного полипептида, содержащего все части, связанные после этапа снятия защиты. Различные хорошо известные способы можно применять в качестве этапа фолдинга, и они могут включать, например, диализ в буферном растворе для фолдинга, но не ограничиваются им. Буферный раствор для фолдинга не ограничен, и может содержать, например, глутатион, цистин-цистеин, и соединение, содержащее гуанидиновую группу, такое как гуанидин или его соль, и рН может составлять 6,0-9,0. Диализ можно проводить много раз, и состав или рН буферного раствора для фолдинга для каждого сеанса диализа может быть идентичным или отличающимся.
В частности, например, когда этап фолдинга проводят с тремя сеансами диализа, первый диализ может быть проведен в растворе, содержащем 8М гуанидин при рН 8,0-9,0 в течение 0,5-6 часов, второй диализ может быть проведен в растворе, содержащем 2М - 4M гуанидин при рН 8,0-9,0 в течение 6-24 часов, а третий диализ можно проводить в растворе, содержащем водный раствор 1 мМ - 20 мМ уксусной кислоты при рН 2,0-4,0 в течение 6-24 часов.
Далее, когда буферный раствор для фолдинга, применяемый на этапе фолдинга, содержит окисленный и восстановленный глутатион, предпочтительно варьировать отношении концентрации окисленного и восстановленного соединения, как уместно, в зависимости от типа сахарной цепи, добавленной к полипептиду. Например, когда сахарная цепь является дисиало-сахарной цепью, отношение окисленного глутатиона и восстановленного глутатиона, содержащихся в буферном растворе для фолдинга, предпочтительно находится в диапазоне 1:1 - 4:1, более предпочтительно 1:1 - 3:1, и особо предпочтительно 2:1. Далее, когда сахарная цепь является асиало-сахарной цепью, отношение окисленного глутатиона и восстановленного глутатиона предпочтительно находится в диапазоне 4:1 - 16:1, более предпочтительно в диапазоне 6:1 - 10:1, и особо предпочтительно 8:1.
Образование дисульфидной связи в не предназначенном для нее участке или агрегацию и т.д. можно подавить за счет исходного материала в низкой концентрации при растворении исходного материала перед фолдингом.
Подтверждение фолдинга полипептида можно обеспечить любым способом, позволяющим анализировать конформацию полипептидов, примеры которого включают способ дисульфидного картирования, определение связывания с антителом, специфичным к конформационному эпитопу, и рентгеновский анализ, но не ограничиваются ими.
(Тепловая обработка)
Далее, гликозилированный полипептид, полученный путем вышеуказанного способа, можно также подвергнуть тепловой обработке. Тепловую обработку предпочтительно проводят перед этапом фолдинга. Далее, когда сиаловая кислота присутствует на невосстанавливающем конце сахарной цепи в гликозилированном полипептиде, предпочтительно проводить тепловую обработку с карбоксигруппой сиаловой кислоты, защищенной бензильной группой, и т.д.
Тепловую обработку можно проводить, например, в буферном растворе, содержащем 6М - 10М гуанидина (Gn) при рН 3,5-7 при температуре 40°С-60°С примерно в течение 24-72 часов. Путем тепловой обработки можно достичь инактивации оболочечных вирусов, таких как ВИЧ, вирус гепатита С и вирус гепатита В, которые могут контаминировать препарат. Кроме того, гликозилированный полипептид из настоящего изобретения может сохранять активность интерферона-β даже при использовании тепловой обработки.
Далее, поскольку вероятно, что тиоловая группа цистеина обеспечивает побочную реакцию при тепловой обработке, тиоловую группу цистеина предпочтительно защищают Acm группой и т.д. на этапе тепловой обработки.
(Активность)
Гликозилированный полипептид из настоящего изобретения обладает активностью интерферона-β. «Активность интерферона-β» в настоящей заявке означает по меньшей мере один вид хорошо известных видов активности, такой как иммуномодулирующее действие, антивирусная активность и противоопухолевая активность.
Например, активность интерферона-β гликозилированного полипептида можно измерить, например, путем анализа противоопухолевой активности, описанного в Примере 12.
Анализ противоопухолевой активности можно провести, например, путем подкожного введения исследуемого гликозилированного полипептида мыши, имеющей опухоль, и определения объема опухоли со временем.
Гликозилированный полипептид из настоящего изобретения может обладать чистотой 90% или более. Далее, гликозилированный полипептид из настоящего изобретения более предпочтительно имеет чистоту 95%, 97%, 98%, 99,0%, или 99,5%, или больше.
Чистота гликозилированного полипептида в настоящей заявке означает значение выхода (г) за исключением примесей, таких как непрореагировавший материал, от общего выхода (г) при получении необходимого гликозилированного полипептида, разделенное на общий выход при получении гликозилированного полипептида, умноженное на 100. В частности, общий выход при получении гликозилированного полипептида означает выход необходимой фракции гликозилированного полипептида, полученной на этапе отделения продукта путем ВЭЖХ после этапа фолдинга. Далее, примеси и т.д. также включают гликозилированный полипептид, не обладающий идентичной структурой сахарной цепи.
В композиции, содержащей гликозилированный полипептид из настоящего изобретения, чистота гликозилированного полипептида означает чистоту гликозилированного полипептида как такового, т.е. содержащегося в композиции в виде одного ингредиента, отдельно от других ингредиентов, содержащихся в композиции.
Соответственно, композиция с высокой чистотой, содержащая гликозилированный полипептид, имеет постоянное качество, и является особо предпочтительной в областях фармацевтического производства или анализа и т.д.
Далее, чистоту гликозилированного полипептида можно измерить путем ВЭЖХ анализа. Чистота гликозилированного полипептида, измеренная посредством ВЭЖХ, может быть выражена в % от размера площади при ВЭЖХ.
(Фармацевтическая композиция)
Далее описана фармацевтическая композиция, содержащая гликозилированный полипептид из настоящего изобретения в качестве активного ингредиента.
Фармацевтическая композиция, содержащая гликозилированный полипептид из настоящего изобретения в качестве активного ингредиента, эффективна для лечения или профилактики интерферон-β-зависимых заболеваний. Как описано выше, для интерферона-β известны различные виды активности, и эти зависимые от активности заболевания также варьируют. Например, виды интерферон-β-зависимых заболеваний включают опухоль головного мозга, такую как глиобластома, медуллобластома и астроцитома, кожную злокачественную меланому, хронический активный гепатит В, хронический гепатит С, подострый склерозирующий панэнцефалит, компенсированный цирроз С, и рассеянный склероз. Фармацевтическая композиция, содержащая гликозилированный полипептид из настоящего изобретения в качестве активного ингредиента, эффективна для лечения или профилактики вышеуказанных заболеваний.
Далее, субъект для применения фармацевтической композиции, содержащей гликозилированный полипептид из настоящего изобретения в качестве активного ингредиента, означает любого индивидуума или организм, предпочтительного животного, более предпочтительно млекопитающего, и еще более предпочтительно человека.
Вышеуказанная фармацевтическая композиция составлена в форме обычной фармацевтической композиции с растворителем или вспомогательным веществом, таким как обычно применяемый наполнитель, вещество для увеличения объема, связующий агент, увлажнитель, дезинтегрант, сурфактант, и любрикант.
Примеры таких фармацевтических композиций включают, например, таблетки, пилюли, порошки, растворы, суспензии, эмульсии, гранулы, капсулы, суппозитории, ингаляционные формы, офтальмологические растворы, и инъекционные формы.
Количество гликозилированного полипептида из настоящего изобретения, содержащегося в фармацевтической композиции, не ограничивается конкретно, и может быть выбрано, как удобно, в широком диапазоне, но в целом предпочтительно составляет 1-90 масс. % гликозилированного полипептида из настоящего изобретения и более предпочтительно 1-70 масс. % от фармацевтической композиции.
Фармацевтическая композиция, содержащая гликозилированный полипептид из настоящего изобретения в качестве активного ингредиента, может дополнительно содержать другие активные ингредиенты, или может применяться в комбинации с фармацевтической композицией, содержащей другие активные ингредиенты. Далее, фармацевтическая композиция, содержащая гликозилированный полипептид из настоящего изобретения в качестве активного ингредиента, может либо содержать гликозилированный полипептид в качестве фармацевтически пригодной соли, либо дополнительно содержать один или несколько различных гликозилированньгх полипептидов из настоящего изобретения в качестве активных ингредиентов. Далее, ее можно также применять в комбинации с фармацевтической композицией, содержащей один или несколько различных гликозилированньгх полипептидов из настоящего изобретения в качестве активного ингредиента. Далее, примеры других ингредиентов, которые могут содержаться в фармацевтической композиции, могут включать фармацевтически пригодный носитель, известный специалистам в данной области техники.
Способ применения фармацевтической композиции в соответствии с настоящим изобретением не ограничивается конкретно, и применение осуществляют способом, зависящим от различных лекарственных рецептур, возраста, пола и условий заболевания пациента, и других факторов. Способ применения таблеток, пилюль, растворов, суспензий, эмульсий, гранул и капсул включает, например, пероральное применение. Далее, в случае инъекций, их можно осуществлять внутривенно, внутримышечно, внутрикожно, подкожно или интраперитонеально, по отдельности или в смеси с обычными заменителями жидкости, такими как глюкоза или аминокислота. В случае суппозиториев их применяют ректально. В случае офтальмологических растворов их вносят в ткань глаза, такую как конъюнктивальный мешок. В случае ингаляционных средств их вводят в бронхиолы или легкие.
Применяемая доза вышеуказанной фармацевтической композиции может быть выбрана, как необходимо, в зависимости от применения, пола, возраста, и степени заболевания пациента, и других условий, и например, может применяться доза 0,001-9 нмоль гликозилированного полипептида из настоящего изобретения, предпочтительно 0,01-0,2 нмоль на 1 кг массы тела.
Частота применения вышеуказанной фармацевтической композиции может быть выбрана, как необходимо, в зависимости от применения, возраста, пола, и степень заболевания пациента, и других условий, и может быть выбрана частота применения 3 раза в сутки, два раза в сутки, один раз в сутки, и менее, в соответствии с ее стабильностью в крови (такая, как один раз в неделю и один раз в месяц). Предпочтительно частота применения вышеуказанной фармацевтической композиции составляет один раз в сутки или реже.
Сахарная цепь, добавленная к гликозилированному полипептиду из настоящего изобретения, легко разрушается метаболической системой организма. Далее, в одном аспекте настоящего изобретения указанная сахарная цепь имеет структуру, представленную в связанном состоянии с гликопептидом (или гликопротеином) in vivo. Соответственно, гликозилированный полипептид из настоящего изобретения и фармацевтическая композиция, содержащая указанный пептид в качестве активного ингредиента, обладает тем преимуществом, что не оказывает побочного влияния и не обладает антигенностью даже при введении in vivo, и создает меньше проблем, связанных с потерей лекарственного эффекта из-за аллергических реакций или выработки антител.
Далее, гликозилированный полипептид из настоящего изобретения может стабильно и просто применяться в больших количествах, и очень полезен для обеспечения фармацевтических средств, обладающих устойчивым и высоким качеством.
Далее, настоящее изобретение также обеспечивает способ лечения или профилактики интерферон-β-зависимого заболевания, характеризующийся применением эффективного количества гликозилированного пептида из настоящего изобретения.
Термины, применяемые в настоящей заявке, предназначены для описания частных вариантов осуществления, и не предназначены для ограничения изобретения.
Далее, термин «включающий», как применяется в настоящей заявке, если явно не указано иное, означает наличие описанных предметов (таких как компоненты, этапы, элементы и числа), и не исключает присутствия других предметов (таких, как компоненты, этапы, элементы и числа).
Если не указано иное, все термины, используемые в настоящей заявке (включая технические и научные термины) имеют те же самые значения, которые общеприняты для специалистов в данной области технологии, к которой относится настоящее изобретение. Термины, используемые в настоящей заявке, если ясно не указано иное, должны рассматриваться как имеющие значения, соответствующие значениям, указанным в настоящей заявке и применяемым в родственных областях техники, и не должны рассматриваться как имеющие идеализированные или исключительно формальные значения.
Варианты осуществления настоящего изобретения могут быть описаны со ссылкой на схематические диаграммы. В случае схематических диаграмм, они могут быть преувеличены для более ясного представления.
Такие термины, как «первый» и «второй», применяются для обозначения различных элементов, и нужно понять, что эти элементы не ограничиваются данными терминами. Эти термины применяются исключительно с целью различения одного элемента от другого, и например, можно описать первый элемент как второй элемент, и подобным образом, описать второй элемент как первый элемент, без отделения от объема настоящего изобретения.
Настоящее изобретение далее подробно описано посредством Примеров. Однако настоящее изобретение может быть осуществлено посредством различных аспектов, и не ограничивается примерами, описанными в настоящей заявке.
Примеры
Следующие Примеры иллюстрируют способ производства 1-166 аминокислот в аминокислотной последовательности интерферона-Р, разделенной на три фрагмента из пептидного фрагмента А, имеющего аминокислоты в положениях 1-67, гликопептидного фрагмента В, имеющего аминокислоты в положениях 68-88, и пептидного фрагмента С, имеющего аминокислоты в положениях 89-166.
В частности, приведен пример способа производства, включающего следующие этапы.
(I) Этап получения пептидного фрагмента А, представленного следующей формулой (d), гликопептидного фрагмента В, представленного следующей формулой (е), и пептидного фрагмента С, представленного следующей формулой (f).
[Химическая формула 32]

[Химическая формула 33]

[Химическая формула 34]
(II) Этап связывания фрагмента В и фрагмента С путем сшивания до пептидного фрагмента (В+С), и превращения цистеина тиазолидинового типа на N-конце фрагмента (В+С) в цистеин (см. Фигуру 1).
(III) Этап связывания фрагмента А и фрагмента (В+С) путем сшивания до пептидного фрагмента (А+В+С) (см. Фигуру 2).
(IV) Этап восстановления цистеина, применяемого для сшивания во фрагменте (А+В+С), в аланин (см. Фигуру 3).
(IV) Этап удаления защитной группы с цистеина (см. Фигуру 4).
В каждом фрагменте С-конец, используемый для связывания посредством сшивания, тиоэтерифицирован, а N-конец, используемый для связывания посредством сшивания, содержит цистеин. Далее, в гликопептидном фрагменте В, представленном вышеуказанной формулой (II), N-концевой боковой цистеин является цистеином тиазолидинового типа для предотвращения побочной реакции на этапе сшивания на С-конце.
N-концевой аминокислотой вышеуказанных фрагментов В и С является аланин в положениях 68 и 89 в интерфероне-β. Однако, поскольку цистеин необходим для сшивания, в вышеприведенном примере производства фрагменты синтезировали с цистеином вместо аланина на N-конце фрагментов В и С, а затем восстанавливали до аланина после сшивания.
Далее, например, фрагмент (А+В) показывает фрагмент А и фрагмент В, соединенные вместе.
Пример 1. Синтез каждого пептидного фрагмента
(1-1. Синтез пептидного фрагмента A-SPh)
Смолу Amino-PEGA (от Merck) (100 мкмоль) помещали в колонку для твердофазного синтеза, промывали в достаточной степени метиленхлоридом (ДХМ) и ДМФ, а затем добавляли ДМФ в достаточной степени для набухания. 4-гидроксиметил-3-метоксифенокси-масляную кислоту (НМРВ) (0,25 ммоль), TBTU (0,25 ммоль), и N-этилморфолин (0,25 ммоль) растворяли в ДМФ (2 мл) и помещали в колонку, и перемешивали при комнатной температуре в течение 4 часов. Смолу промывали в достаточной степени ДМФ и ДХМ, и получали смолу HMPB-PEGA, и применяли ее в качестве твердой фазы для твердофазного синтеза.
Fmoc-Фен (0,50 ммоль), MSNT (0,50 моль), и N-метилимидазол (0,375 ммоль) растворяли в ДХМ (2 мл), помещали в колонку для твердофазного синтеза, и перемешивали при 25°С в течение 4 часов.
После перемешивания смолу промывали ДХМ и ДМФ. Fmoc группу обрабатывали 20% раствором пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ, аминокислоты последовательно конденсировали посредством способа, показанного ниже, для последующего удлинения пептидной цепи.
Аминокислоту, имеющую аминогруппу, защищенную Fmoc группой, растворяли в N-метилпирролидоне (NMP) (1 мл), добавляли 0,45 M HCTU⋅HOBT/NMP (0,4 ммоль), вносили в колонку для твердофазного синтеза, и затем в колонку для твердофазного синтеза добавляли 0,9 M DIPEA/NMP (0,8 ммоль). После перемешивания при комнатной температуре в течение 20 минут, смолу промывали ДХМ и ДМФ, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. Эту операцию повторяли, и аминокислоты, защищенные Fmoc и Воc группами (0,5 ммоль), применяли для последовательной конденсации аминокислот.
Аминокислоты, защищенные Fmoc и Boc группами, Fmoc-Фен, Fmoc-Иле, Fmoc-Асн(Trt), Fmoc-Глн(Trt), Fmoc-Лей, Fmoc-Мет, Fmoc-Глу(OtBu), Fmoc-Тир(tBu), Fmoc-Иле, Fmoc-Лей-Tpe(ψMe, МеПро), Fmoc-Ала, Fmoc-Ала, Fmoc-Асп(OtBu), Fmoc-Глу(OtBu), Fmoc-Лиз(Boc), Fmoc-Глн(Trt), Fmoc-Фен, Fmoc-Глн(Trt), Fmoc-Глн(Trt), Fmoc-Лей, Fmoc-Глн(Trt), Fmoc-Лиз(Вос), Fmoc-Иле, Fmoc-Глу(OtBu), Fmoc-Глу(OtBu), Fmoc-Πpο, Fmoc-Иле, Fmoc-Асп(OtBu), Fmoc-Фен, Fmoc-AcH(Trt), Fmoc-Мет, Fmoc-Арг(Pbf), Fmoc-Асп(OtBu), Fmoc-Лиз(Вос), Fmoc-Лей, Fmoc-Цис(Acm), Fmoc-Тир(tBu), Fmoc-Глу(OtBu), Fmoc-Лей, Fmoc-Арг(Pbf), Fmoc-Гли, Fmoc-Асн(Trt), Fmoc-Лей, Fmoc-Глн(Trt), Fmoc-Трп(Boc), Fmoc-Лей, Fmoc-Лей, Fmoc-Лиз(Вос), Fmoc-Глн(Trt), Fmoc-Цис(Acm), Fmoc-Глн(Trt), Fmoc-Фен, Fmoc-Асн(Trt), Fmoc-Cep(tBu)-Cep(ψMe, МеПро), Fmoc-Арг(Pbf), Fmoc-Глн(Trt), Fmoc-Лей, Fmoc-Фен, Fmoc-Гли, Fmoc-Лей, Fmoc-Лей, Fmoc-Асн(Trt), Fmoc-Тир(tBu), Fmoc-Cep(tBu), и Вос-Мет последовательно применяли и связывали со смолой для твердофазного синтеза. В результате получали пептид из 67 остатков (1) из Фен-Иле-Асн(Trt)-Γлн(Trt)-Лей-Мет-Глу(OtBu)-Тир(tBu)-Иле-Tpe(ψMe, МеПро)-Лей-Ала-Ала-Асп(OtBu)-Глу(OtBu)-Лиз(Вос)-Глн(Trt)-Фен-Глн(Trt)-Глн(Trt)-Лей-Глн(Trt)-Лиз(Вос)-Иле-Глу(OtBu)-Глу(OtBu)-Про-Иле-Асп(OtBu)-Фен-Асн(Trt)-Мет-Арг(Pbf)-Асп(OtBu)-Лиз(Вос)-Лей-Цис(Acm)-Тир(tBu)-Глу(OtBu)-Лей-Арг(Pbf)-Гли-Асн(Trt)-Лей-Глн(Trt)-Трп(Вос)-Лей-Лей-Лиз(Вос)-Глн(Trt)-Цис(Acm)-Глн(Trt)-Фен-Асн(Τrt)-Cep(ψΜe, МеПро)-Сер(tBu)-Apr(Pbf)-Глн(Trt)-Лей-Фен-Гли-Лей-Лей-Асн(Trt)-Тир(tBu)-Cep(tBu)-Мет-BocNH (SEQ ID №2) на смоле для твердофазного синтеза.
После отмывания пептида (1), полученного выше, с ДХМ и ДМФ, добавляли смешанный раствор трифторэтанола и уксусной кислоты (1:1), пока смола не набухала в достаточной степени, а затем перемешивали ее в течение 18 часов при комнатной температуре для отсоединения пептида 1 от смолы. Промытую смолу фильтровали, и реакционный раствор концентрировали при сниженном давлении. Полученный осадок концентрировали, и получали пептид, имеющий защищенную боковую аминокислотную цепь (1) (SEQ ID №3): Фен-Иле-Асн(Trt)-Глн(Trt)-Лей-Мет-Глу(OtBu)-Тир(tBu)-Иле-Тре(ψМе, МеПро)-Лей-Ала-Ала-Асп(OtBu)-Глу(OtBu)-Лиз(Вос)-Глн(Trt)-Фен-Глн(Trt)-Глн(Trt)-Лей-Глн(Trt)-Лиз(Вос)-Иле-Глу(OtBu)-Глу(OtBu)-Про-Иле-Асп(OtBu)-Фен-Асн(Trt)-Мет-Apr(Pbf)-Асп(OtBu)-Лиз(Вос)-Лей-Цис(Acm)-Тир(tBu)-Глу(OtBu)-Лей-Арг(Pbf)-Гли-Асн(Trt)-Лей-Глн(Trt)-Трп(Вос)-Лей-Лей-Лиз(Вос)-Глн(Trt)-Цис(Acm)-Глн(Trt)-Фен-Асн(Trt)-Сер(ψМе, МеПро)-Сер(tBu)-Арг(Pbf)-Глн(Trt)-Лей-Фен-Гли-Лей-Лей-Асн(Trt)-Тир(tBu)-Сер(tBu)-Мет-BocNH.
100 мкмоль эквивалентов пептида (1), содержащего 67 аминокислотных остатков и имеющего защищенную боковую аминокислотную цепь, переносили в колбу для выделения, растворяли в ДМФ (3,0 мл), а затем охлаждали до -15°С - -20°С в атмосфере азота. Добавляли тиофенол (308 мкл, 3,0 ммоль), а затем РуВОР (260,0 мг, 0,50 ммоль) и DIPEA (85,0 мкл, 0,5 ммоль). После перемешивания при -15°С - -20°С в течение 2 часов добавляли трифторуксусную кислоту (0,1 мл), и затем постепенно восстанавливали комнатную температуру смеси. После восстановления комнатной температуры реакционный раствор концентрировали при сниженном давлении. К полученному осадку добавляли смесь трифторуксусной кислоты: воды: TIPS (=95:2,5:2,5), и перемешивали при комнатной температуре. Спустя 2 часа этот раствор вновь добавляли к диэтиловому эфиру (150 мл) для осаждения, а затем центрифугировали для удаления части раствора, с целью получения осадка, содержащего пептидный тиоэфир. Этот осадок очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=55:45 → 55:45 (5 минут) →·25:75 (35 минут) -> 5:95 (36 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте], и получали пептидный фрагмент A-SPh (1), содержащий тиофениловый эфир на С-конце (SEQ ID №4): Н2К-Мет-Сер-Тир-Асн-Лей-Лей-Гли-Фен-Лей-Глн-Арг-Сер-Сер-Асн-Фен-Глн-Цис(Аст)-Глн-Лиз-Лей-Лей-Трп-Глн-Лей-Асн-Гли-Арг-Лей-Глу-Тир-Цис(Аст)-Лей-Лиз-Асп-Арг-Мет-Асн-Фен-Асп-Иле-Рго-Глу-Глу-Иле-Лиз-Глн-Лей-Глн-Глн-Фен-Глн-Лиз-Глу-Асп-Ала-Ала-Лей-Тре-Иле-Тир-Глу-Мет-Лей-Глн-Асн-Иле-Фен-SPh. ИЭР-МС: расчет для C378H581N97O106S6: [М+4Н]4+ 2094,16; [М+5Н]5+ 1675,53; [М+6Н]6+ 1396,44; найдено 2094,06; 1675,43; 1396,17.
(1-2. Синтез присоединенного к дисиало-сахарной цепи пептидного фрагмента В-SPh)
Смолу Amino-PEGA (от Merck) (100 мкмоль) помещали в колонку для твердофазного синтеза, промывали в достаточной степени метиленхлоридом (ДХМ) и ДМФ, а затем добавляли ДМФ в достаточной степени для набухания. 4-гидроксиметил-3-метоксифенокси-масляную кислоту (НМРВ) (0,25 ммоль), TBTU (0,25 ммоль), и N-этилморфолин (0,25 ммоль) растворяли в ДМФ (2 мл) и помещали в колонку, и перемешивали при комнатной температуре в течение 4 часов. Смолу промывали в достаточной степени ДМФ и ДХМ, и получали смолу HMPB-PEGA, и применяли ее в качестве твердой фазы для твердофазного синтеза.
Fmoc-Фен (0,50 ммоль), MSNT (0,50 моль), и N-метилимидазол (0,375 ммоль) растворяли в ДХМ (2 мл), помещали в колонку для твердофазного синтеза, и перемешивали при 25°С в течение 4 часов.
После перемешивания смолу промывали ДХМ и ДМФ. Fmoc группу обрабатывали 20% раствором пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ, аминокислоты последовательно конденсировали посредством способа, показанного ниже, для последующего удлинения пептидной цепи.
Аминокислоту, имеющую аминогруппу, защищенную Fmoc группой, растворяли в N-метилпирролидоне (NMP) (1 мл), добавляли 0,45 M HCTU⋅HOBT/NMP (0,4 ммоль), вносили в колонку для твердофазного синтеза, и затем в колонку для твердофазного синтеза добавляли 0,9 M DIPEA/NMP (0,8 ммоль). После перемешивания при комнатной температуре в течение 20 минут, смолу промывали ДХМ и ДМФ, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. Эту операцию повторяли, и аминокислоты, защищенные Fmoc группой (0,5 ммоль), применяли для последовательной конденсации аминокислот.
Fmoc-Лей, Fmoc-Лей, Fmoc-Асн(Trt), Fmoc-Глу(OtBu), Fmoc-Вал, Fmoc-Иле, Fmoc-Tpe(tBu), и Fmoc-Глу(OtBu) последовательно применяли в качестве аминокислот, защищенных Fmoc группой, и получали пептид из 8 остатков Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu). Для этого пептида из 8 остатков Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ аспарагин дибензил-дисиало сахарной цепи (g) (547, 8 мг, 200 мкмоль) и DEPBT (59,9 мг, 200 мкмоль) растворяли в ДМФ/ДМСО (1:1 смешанный раствор, 3,33 мл) в центрифужной пробирке, помещали в колонку для твердофазного синтеза, добавляли DIPEA (52,3 мкл, 300 мкмоль), и перемешивали при комнатной температуре в течение 18 часов. При промывании ДМФ и ДХМ получали пептид из 9 остатков с сахарной цепью Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu)-Асн(дибензил-дисиалоолигосахарид)-FmocNH (SEQ ID №5) на твердой фазе.
[Химическая формула 35]

Для последующего удлинения гликопептидной цепи, липофильную защитную группу с пептида из 9 остатков с сахарной цепью на твердофазной смоле, полученного выше, удаляли с раствором 20% пиперидина/ДМФ (2 мл), а затем аминокислоты последовательно конденсировали способом, описанным ниже.
Аминокислоту, защищенную Fmoc группой, а также HOBt (67,6 мг, 0,50 ммоль) и DIPCI (73, 1 мкл, 0,475 ммоль) растворяли в ДМФ (6,3 мл), активировали в течение 15 минут, а затем помещали в колонку для твердофазного синтеза. После перемешивания при комнатной температуре в течение 1 часа, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 20 минут для снятия защиты. Эту операцию повторяли для последовательной конденсации аминокислот.Аминокислоты, защищенные Fmoc и Boc группами, Fmoc-Трп(Boc), Fmoc-Гли, Fmoc-Tpe(tBu), Fmoc-Cep(tBu)-Cep(ψMe, МеПро), Fmoc-Cep(tBu), Fmoc-Асп(OtBu), Fmoc-Глн(Trt), Fmoc-Арг(Pbf), Fmoc-Фен, Fmoc-Иле, and Вос-L-тиазолидин-4-карбоновую кислоту последовательно применяли и связывали со смолой для твердофазного синтеза. В результате получали гликозилированный пептидный фрагмент из 21 остатка (2) Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu)-Асн(дибензил-дисиалоолигосахарид)-Трп(Вос)-Гли-Tpe(tBu)-Cep(ψMe, МеПро)-Сер(tBu)-Сер(tBu)-Асп(OtBu)-Глн(Trt)-Арг(Pbf)-Фен-Иле-Thz-BocN (SEQ ID №6) на смоле для твердофазного синтеза.
Затем, после промывания ДХМ и ДМФ, смешанный раствор трифторэтанола и уксусной кислоты (1:1) добавляли до достаточного набухания смолы, а затем перемешивали в течение 18 часов при комнатной температуре для отсоединения гликозилированного пептидного фрагмента от смолы. Освобожденную смолу отделяли фильтрацией, и реакционный раствор концентрировали при сниженном давлении. Полученный осадок концентрировали, и получали пептид из 21 остатка с сахарной цепью (2), имеющий защищенную аминокислотную боковую цепь (SEQ ID №7): Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu)-Асн(дибензил-дисиалосахарид)-Трп(Boc)-Гли-Тре(tBu)-Сер(ψМе, МеПро)-Сер(tBu)-Сер(tBu)-Асп(OtBu)-Глн(Trt)-Арг(Pbf)-Фен-Иле-Thz-BocN.
100 мкмоль эквивалентов гликозилированного пептидного фрагмента (2), содержащего 21 аминокислотных остатков и имеющего защищенную боковую аминокислотную цепь, переносили в колбу для выделения, растворяли в ДМФ (3,0 мл), а затем охлаждали до -15°С - -20°С в атмосфере азота. Добавляли тиофенол (308 мкл, 3,0 ммоль), а затем РуВОР (260,0 мг, 0,50 ммоль) и DIPEA (85,0 мкл, 0,5 ммоль). После перемешивания при -15°С - -20°С в течение 2 часов добавляли трифторуксусную кислоту (0,1 мл), и затем постепенно восстанавливали комнатную температуру смеси. После восстановления комнатной температуры реакционный раствор концентрировали при сниженном давлении. К полученному осадку добавляли смесь трифторуксусной кислоты: воды: TIPS (=95:2,5:2,5), и перемешивали при комнатной температуре. Спустя 2 часа этот раствор вновь добавляли к диэтиловому эфиру (150 мл) для осаждения, а затем центрифугировали для удаления части раствора, с целью получения осадка, содержащего гликозилированный пептидный тиоэфир. Этот осадок очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=65:35 → 52:48 (26 минут) → 5:95 (28 минут), в линейном градиенте], и получали пептидный фрагмент с присоединенной дисиало-сахарной цепью B-SPh (2), содержащий тиофениловый эфир на С-конце (SEQ ID №8): HN-Thz-Иле-Фен-Арг-Глн-Асп-Сер-Сер-Сер-Тре-Гли-Трп-Асн(дибензил-дисиалоолигосахарид)-Глу-Тре-Иле-Вал-Глу-Асн-Лей-Лей-SPh.
ИЭР-МС: Расчет для C209H314N34O96S2: [М+3Н]3+ 1635,34; [М+4Н]4+ 1226,76; установлено 1635,04; 1226,51.
(1-3. Синтез асиало- гликозилированного пептидного фрагмента B-SPh).
Смолу Amino-PEGA (от Merck) (100 мкмоль) помещали в колонку для твердофазного синтеза, промывали в достаточной степени метиленхлоридом (ДХМ) и ДМФ, а затем добавляли ДМФ в достаточной степени для набухания. 4-гидроксиметил-3-метоксифенокси-масляную кислоту (НМРВ) (0,25 ммоль), TBTU (0,25 ммоль), и N-этилморфолин (0,25 ммоль) растворяли в ДМФ (2 мл) и помещали в колонку, и перемешивали при комнатной температуре в течение 4 часов. Смолу промывали в достаточной степени ДМФ и ДХМ, и получали смолу HMPB-PEGA, и применяли ее в качестве твердой фазы для твердофазного синтеза.
Fmoc-Фен (0,50 ммоль), MSNT (0,50 моль), и N-метилимидазол (0,375 ммоль) растворяли в ДХМ (2 мл), помещали в колонку для твердофазного синтеза, и перемешивали при 25°С в течение 4 часов.
После перемешивания смолу промывали ДХМ и ДМФ. Fmoc группу обрабатывали 20% раствором пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ, аминокислоты последовательно конденсировали посредством способа, показанного ниже, для последующего удлинения пептидной цепи.
Аминокислоту, имеющую аминогруппу, защищенную Fmoc группой, растворяли в N-метилпирролидоне (NMP) (1 мл), добавляли 0,45 M HCTU⋅HOBT/NMP (0,4 ммоль), вносили в колонку для твердофазного синтеза, и затем в колонку для твердофазного синтеза добавляли 0,9 M DIPEA/NMP (0,8 ммоль). После перемешивания при комнатной температуре в течение 20 минут, смолу промывали ДХМ и ДМФ, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. Эту операцию повторяли, и аминокислоты, защищенные Fmoc группой (0,5 ммоль), применяли для последовательной конденсации аминокислот.
В качестве аминокислот, защищенных Fmoc группой, Fmoc-Лей, Fmoc-Лей, Fmoc-AcH(Trt), Fmoc-Глу(OtBu), Fmoc-Вал, Fmoc-Иле, Fmoc-Tpe(tBu), и Pmoc-Глу(OtBu) последовательно применяли и связывали со смолой для твердофазного синтеза. В результате получали пептидный фрагмент из 8 остатков Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu) (SEQ ID №9) на смоле для твердофазного синтеза. Fmoc группу из этого пептидного фрагмента из 8 остатков обрабатывали раствором 20% пиперидина/ДМФ (1 мл) в течение 15 минут для снятия защиты. После промывания ДМФ, производное аспарагина асиало-сахарной цепи (h) (547,8 мг, 200 мкмоль) и DEPBT (59,9 мг, 200 мкмоль) растворяли в ДМФ/ДМСО (смешанный раствор 1:1; 3,33 мл) в центрифужной пробирке, помещали в колонку для твердофазного синтеза, добавляли DIPEA (52,3 мкл, 300 мкмоль) и перемешивали при комнатной температуре в течение 18 часов. Промывание ДМФ и ДХМ обеспечивало гликозилированный пептидный фрагмент из 9 остатков Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu)-Асн(Асиапоолигосахарвд)-FmocNH (SEQ ID №10) на твердой фазе.
[Химическая формула 36]

Для последующего удлинения гликопептидной цепи, липофильную защитную группу с пептида из 9 остатков с сахарной цепью на смоле для твердофазного синтеза, полученного выше, удаляли с раствором 20% пиперидина/ДМФ (2 мл), а затем аминокислоты последовательно конденсировали способом, описанным ниже. Аминокислоту, защищенную Fmoc группой, а также HOBt (67,6 мг, 0,50 ммоль) и DIPCI (73,1 мкл, 0,475 ммоль) растворяли в ДМФ (6,3 мл), активировали в течение 15 минут, а затем помещали в колонку для твердофазного синтеза. После перемешивания при комнатной температуре в течение 1 часа, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 20 минут для снятия защиты. Эту операцию повторяли для последовательной конденсации аминокислот. В качестве аминокислот, защищенных Fmoc и Boc группами, Fmoc-Трп(Boc), Fmoc-Гли, Fmoc-Tpe(tBu), Fmoc-Cep(tBu)-Сер(ψМе, МеПро), Fmoc-Cep(tBu), Fmoc-Асп(OtBu), Fmoc-Глн(Trt), Fmoc-Арг(Pbf), Fmoc-Фен, Fmoc-Иле и Вос-L-тиазолидин-4-карбоновую кислоту последовательно применяли и связывали на смоле для твердофазного синтеза. В результате получали гликозилированный пептидный фрагмент из 21 остатка (3), имеющий защищенную аминокислотную боковую цепь, Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu)-Асн(Асиалоолигосахарид)-Трп(Вос)-Гли-Тре(tBu)-Сер(ψМе, МеПро)-Cep(tBu)-Cep(tBu)-Асп(OtBu)-Глн(Trt)-Арг(Pbf)-Фен-Иле-Thz-BocN (SEQ ID №11) на смоле для твердофазного синтеза.
После промывания ДХМ и ДМФ, смешанный раствор трифторэтанола и уксусной кислоты (1:1) добавляли до достаточного набухания смолы, а затем перемешивали в течение 18 часов при комнатной температуре для отсоединения гликозилированного пептидного фрагмента (3) от смолы. Освобожденную смолу отделяли фильтрацией, и реакционный раствор концентрировали при сниженном давлении. Полученный осадок концентрировали, и получали гликозилированный пептидный фрагмент (3), имеющий защищенную аминокислотную боковую цепь (SEQ ID №12): Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Tpe(tBu)-Глу(OtBu)-Асн(Асиалоолигосахарид)-Tpπ(Boc)-Γли-Tpe(tBu)-Cep(ψMe, МеПро)-Cep(tBu)-Cep(tBu)-Асп(OtBu)-Глн(Trt)-Арг(Pbf)-Фен-Иле-Thz-BocN.
100 мкмоль эквивалентов гликозилированного пептидного фрагмента (3), содержащего 21 аминокислотных остатков и имеющего защищенную боковую аминокислотную цепь, переносили в колбу для выделения, растворяли в ДМФ (3,0 мл), а затем охлаждали до -15°С - -20°С в атмосфере азота. Добавляли тиофенол (308 мкл, 3,0 ммоль), а затем РуВОР (260,0 мг, 0,50 ммоль) и DIPEA (85,0 мкл, 0,5 ммоль). После перемешивания при -15°С - -20°С в течение 2 часов добавляли трифторуксусную кислоту (0,1 мл), и затем постепенно восстанавливали комнатную температуру смеси. После восстановления комнатной температуры реакционный раствор концентрировали при сниженном давлении. К полученному осадку добавляли смесь трифторуксусной кислоты: воды: TIPS (=95:2,5:2,5), и перемешивали при комнатной температуре. Спустя 2 часа этот раствор вновь добавляли к диэтиловому эфиру (150 мл) для осаждения, а затем центрифугировали для удаления части раствора, с целью получения осадка, содержащего гликозилированный пептидный тиоэфир. Этот полученный осадок очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=65:35 → 52:48 (26 минут) → 5:95 (28 минут), в линейном градиенте], и получали асиало-гликозилированный пептидный фрагмент с присоединенной дисиало-сахарной цепью B-SPh (3), содержащий тиофениловый эфир на С-конце (SEQ ID №13): HN-Thz-Иле-Фен-Арг-Глн-Асп-Сер-Сер-Сер-Тре-Гли-Трп-Асн(Асиалоолигосахарид)-Глу-Тре-Иле-Вал-Глу-Асн-Лей-Лей-SPh.
ИЭР-МС: Расчет для C173H268N32O80S2: [М+3Н]3+ 1381,09; [М+4Н]4+ 1036,07; установлено 1380,90; 1035,92.
(1-4. Синтез пептидного фрагмента С)
Смолу Amino-PEGA (от Merck) (100 мкмоль) помещали в колонку для твердофазного синтеза, промывали в достаточной степени метиленхлоридом (ДХМ) и ДМФ, а затем добавляли ДМФ в достаточной степени для набухания. 4-гидроксиметил-3-метоксифенокси-масляную кислоту (НМРВ) (0,25 ммоль), TBTU (0,25 ммоль), и N-этилморфолин (0,25 ммоль) растворяли в ДМФ (2 мл) и помещали в колонку, и перемешивали при комнатной температуре в течение 4 часов. Смолу промывали в достаточной степени ДМФ и ДХМ, и получали смолу HMPB-PEGA, и применяли ее в качестве твердой фазы для твердофазного синтеза.
Fmoc-Фен (0,50 ммоль), MSNT (0,50 моль), и N-метилимидазол (0,375 ммоль) растворяли в ДХМ (2 мл), помещали в колонку для твердофазного синтеза, и перемешивали при 25°С в течение 4 часов.
После перемешивания смолу промывали ДХМ и ДМФ. Fmoc группу обрабатывали 20% раствором пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ, аминокислоты последовательно конденсировали посредством способа, показанного ниже, для последующего удлинения пептидной цепи.
Аминокислоту, имеющую аминогруппу, защищенную Fmoc группой, растворяли в N-метилпирролидоне (NMP) (1 мл), добавляли 0,45 M HCTU⋅HOBT/NMP (0,4 ммоль), вносили в колонку для твердофазного синтеза, и затем в колонку для твердофазного синтеза добавляли 0,9 M DIPEA/NMP (0,8 ммоль). После перемешивания при комнатной температуре в течение 20 минут, смолу промывали ДХМ и ДМФ, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. Эту операцию повторяли, и аминокислоты, защищенные Fmoc группой (0,5 ммоль), применяли для последовательной конденсации аминокислот. В качестве аминокислот, защищенных Fmoc и Вос группами, Fmoc-AcH(Trt), Fmoc-Арг(Pbf), Fmoc-Лей, Fmoc-Тир(tBu), Fmoc-Гли, Fmoc-Tpe(tBu), Fmoc-Лей, Fmoc-Арг(Pbf), Fmoc-Асн(Trt), Fmoc-Иле, Fmoc-Фен, Fmoc-Тир(tBu), Fmoc-Фен, Fmoc-AcH(Trt), Fmoc-Арг(Pbf), Fmoc-Лей, Fmoc-Иле, Fmoc-Глу(OtBu), Fmoc-Вал, Fmoc-Арг(Pbf), Fmoc-Вал, Fmoc-Иле, Fmoc-Tpe(tBu), Fmoc-Трп(Boc), Fmoc-Ала, Fmoc-Цис(Acm), Fmoc-Гис(Trt), Fmoc-Тир(tBu)-Cep(ψMe, МеПро), Fmoc-Глу(OtBu), Fmoc-Лиз(Вос), Fmoc-Ала, Fmoc-Лиз(Вос), Fmoc-Лей, Fmoc-Тир(tBu), Fmoc-Гис(Trt), Fmoc-Лей, Fmoc-Иле, Fmoc-Арг(Pbf), Fmoc-Гли, Fmoc-Тир(tBu), Fmoc-Тир(tBu), Fmoc-Арг(Pbf), Fmoc-Лиз(Вос), Fmoc-Лей, Fmoc-Гис(Trt), Fmoc-Лей, Fmoc-Cep(tBu)-Cep(ψMe, МеПро), Fmoc-Мет, Fmoc-Лей, Fmoc-Лиз(Вос), Fmoc-Гли, Fmoc-Арг(Pbf), Fmoc-Фен-Тре(ψМе, МеПро), Fmoc-Асп(OtBu), Fmoc-Глу(OtBu), Fmoc-Лиз(Вос), Fmoc-Глу(OtBu), Fmoc-Лей, Fmoc-Лиз(Вос), Fmoc-Глу(OtBu), Fmoc-Глу(OtBu), Fmoc-Лей, Fmoc-Вал, Fmoc-Лиз(Вос)-Тре(ψМе, МеПро), Fmoc-Лей, Fmoc-Гис(Trt), Fmoc-AcH(Trt), Fmoc-Иле, Fmoc-Глн(Trt), Fmoc-Гис(Trt), Fmoc-Тир(tBu), Fmoc-Вал, Fmoc-Асн(Trt) и Вос-Цис(Trt) последовательно применяли и связывали на смоле для твердофазного синтеза. В результате получали пептидный фрагмент из 78 остатков (4) Асн(Trt)-Арг(Pbf)-Лей-Тир(tBu)-Гли-Тре(tBu)-Лей-Арг(Pbf)-Асн(Trt)-Иле-Фен-Тир(tBu)-Фен-Асн(Trt)-Арг(Pbf)-Лей-Иле-Глу(OtBu)-Вал-Арг(Pbf)-Вал-Иле-Тре(tBu)-Трп(Boc)-Ала-Цис(Acm)-Гис(Trt)-Сер(ψМе, МеПро)-Тир(tBu)-Глу(OtBu)-Лиз(Вос)-Ала-Лиз(Вос)-Лей-Тир(tBu)-Гис(Trt)-Лей-Иле-Арг(Pbf)-Гли-Тир(tBu)-Тир(tBu)-Арг(Pbf)-Лиз(Boc)-Лей-Гис(Trt)-Лей-Сер(ψМе, МеПро)-Сер(tBu)-Мет-Лей-Лиз(Вос)-Гли-Арг(Pbf)-Тре(ψМе, МеПро)-Фен-Асп(OtBu)-Глу(OtBu)-Лиз(Вос)-Глу(OtBu)-Лей-Лиз(Вос)-Глу(OtBu)-Глу(OtBu)-Лей-Вал-Тре(ψМе, МеПро)-Лиз(Вос)-Лей-Гис(Trt)-Асн(Trt)-Иле-Глн(Trt)-Гис(Trt)-Тир(tBu)-Вал-Асн(Trt)-Цис(Trt)-BocNH (SEQ ID №14) на смоле для твердофазного синтеза.
К пептидному фрагменту (4), полученному выше, добавляли трифторуксусную кислоту: воду: TIPS (=95:2,5:2,5), и перемешивали при комнатной температуре в течение 3,5 часов для отделения пептидного фрагмента (4) от смолы. Затем добавляли диэтиловый эфир (150 мл) для осаждения, а затем подвергали операции разделения посредством центрифугирования, для разделения осадка, содержащего необходимый пептидный фрагмент (4), и растворенной части. Затем, после удаления растворенной части, наличие целевого вещества подтверждали посредством ВЭЖХ, а затем очищали с ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=65:35 → 65:35 (5 минут) → 30:70 (35 минут) → 5:95 (45 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] до получения необходимого пептидного фрагмента С (4) (SEQ ID №15): H2N-Цис-Асн-Вал-Тир-Гис-Глн-Иле-Асн-Гис-Лей-Лиз-Тре-Вал-Лей-Глу-Глу-Лиз-Лей-Глу-Лиз-Глу-Асп-Фен-Тре-Арг-Гли-Лиз-Лей-Мет-Сер-Сер-Лей-Гис-Лей-Лиз-Арг-Тир-Тир-Гли-Арг-Иле-Лей-Гис-Тир-Лей-Лиз-Ала-Лиз-Глу-Тир-Сер-Гис-Цис(Acm)-Ала-Трп-Тре-Иле-Вал-Арг-Вал-Глу-Иле-Лей-Арг-Асн-Фен-Тир-Фен-Иле-Асн-Арг-Лей-Тре-Гли-Тир-Лей-Арг-Асн.
ИЭР-МС: Расчет для C441H688N124O114S3: [М+8Н]8+ 1206,89; [М+9Н]9+ 1072,90; [М+10Н]10+ 965,71; [М+11Н]11+ 804,93; найдено 1206,85; 1072,87; 965,58; 877,90; 804,90.
Пример 2. Синтез асиало-ИФН-β
Активный ИФН-β, содержащий асиало-сахарную цепь, синтезировали в целом за 5 этапов, показанных ниже.
(2-1. Первый этап: Связывание асиало-гликозилированного пептидного фрагмента В и пептидного фрагмента С).
Два типа фрагментов, асиало-гликопептидный фрагмент В из 21 остатка с тиофенил-эфирным С-концом (3) (1,8 мг) и пептидный фрагмент С из 78 остатков (4) (2,5 мг) помещали в одну колбу для выделения, растворяли в буферном растворе при рН 7,2 (0,26 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и 20 мМ раствора Трис-карбоксиэтилфосфина (ТСЕР раствора)). Затем добавляли тиофенол (7,8 мкл), и проводили реакцию при комнатной температуре. Спустя 24 часа проведение реакции подтверждали ВЭЖХ, затем к реакционному раствору добавляли 0,2 M раствор метоксамина (0,39 мл) (приготовленный путем добавления 20 мМ раствора ТСЕР к 0,2 M раствору метоксамина), и проводили реакцию при комнатной температуре. Спустя 12 часов получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий гликопептидный фрагмент, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=65:35 → 65:35 (5 минут) → 30:70 (35 минут) → 5:95 (45 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] до получения необходимого пептидного фрагмента (11), содержащего асиало-сахарную цепь в положении, соответствующем положению 80 интерферона-β (SEQ ID №16) (Фигуры 5 и 6).
ИЭР-МС: Расчет для C607Η950Ν156Ο194S4 [М+10Н]10+ 1367,52; [М+11Н]11+ 1243,29; [М+12Н]12+ 1139,77; [М+13Н]13+ 1052,17; [М+14Н]14+ 977,09; [М+15Н]15+ 912,02; [М+16Н]16+ 855,08; [М+17Н]17+ 804,84; [М+18Н]18+ 760,18; установлено 1367,64; 1243,33; 1139,80; 1052,20; 977,10; 912,03; 855,09; 804,84; 760,18.
(2-2. Второй этап: Связывание пептидного фрагмента А и асиало-гликозилированного пептидного фрагмента (В+С)).
Два типа фрагментов, пептид из 67 остатков с тиофенил-эфирным С-концом (1), полученный на вышеуказанной первом этапе (2,3 мг) и необходимый пептидный фрагмент (11), имеющий асиало-сахарную цепь в положении, соответствующем положению 80 интерферона-β (асиало-гликозилированного пептидного фрагмента (В+С)) (68-166) (1,9 мг) помещали в одну и ту же колбу для выделения, растворяли в буферном растворе с рН 7,2 (0,14 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и 20 мМ раствора ТСЕР). Затем добавляли тиофенол (4,2 мкл), и проводили реакцию при комнатной температуре. Спустя 24 часа проведение реакции подтверждали ВЭЖХ, затем добавляли натрия меркаптоэтансульфонат (1,1 мг), и проводили реакцию при комнатной температуре. Спустя 12 часов получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС.Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевое вещество, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=55:45 → 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] до получения необходимого пептидного фрагмента (12) из 166 остатков, содержащего свободную тиоловую группу на Цис в положениях 68 и 69, и имеющего асиало-сахарную цепь на Асн в положении 80 (SEQ ID №17) (Фигуры 7 и 8).
ИЭР-МС: Расчет для C979H1525N253O300S9: [М+17Н]17+ 1290,86; [М+18Н]18+ 1219,20; [М+19Н]19+ 1155,09; [М+20Н]20+ 1097,38; [М+21Н]21+ 1045,18; [М+22Н]22+ 997,71; [М+23Н]23+ 954,38; [М+24Н]24+ 914,65; [М+25Н]25+ 878,11; [М+26Н]26+ 844,37; [М+27Н]27+ 813,14; [М+28Н]28+ 784,13; [М+29Н]29+ 757,13; найдено 1219,21; 1155,11; 1097,43; 1045,23; 997,71; 954,43; 914,67; 878,11; 844,37; 813,17; 784,13; 757,16.
(Третий этап: Восстановление Цис до Ала).
Гликозилированный полипептид (12) из 166 остатков, содержащий свободную тиоловую группу на Цис в положениях 68 и 89, и имеющий асиало-сахарную цепь на Асн в положении 80, полученный на вышеуказанном втором этапе (1,0 мг), помещали в колбу для выделения, растворяли в буферном растворе при рН 7,5 (0,10 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида и 0,2 мМ раствора фосфорной кислоты), затем добавляли 0,5 M раствор Трис-этилкарбоксифосфина (раствор ТСЕР) при рН 7,5 (0,10 мл) (приготовленный из ТСЕР, 6 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и триэтиламина), tBuSH (4,2 мкл), и 0,1 M раствор VA-044 (4,2 мкл) (приготовленный путем растворения VA-044 в воде), и проводили реакцию при комнатной температуре. Спустя 4 часа подтверждали получение целевого вещества посредством ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, экстрагировали tBuSH диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевое вещество, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ /10% воды /90% ацетонитрила, А:В=55:45 → 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 минут, затем элюция с линейным градиентом] до получения гликозилированного полипептида (13) из 166 остатков, содержащего Цис в положениях 68 и 89, превращенный в Ала и имеющий асиало-сахарную цепь в Асн в положении 80 (SEQ ID №18) (Фигуры 9 и 10).
ИЭР-МС: Расчет для C979H1525N253O300S7: [М+17Н]17+ 1287,09; [М+18Н]18+ 1215,64; [М+19Н]19+ 1151,71; [М+20Н]20+ 1094,18; [М+21Н]21+ 1042,12; [М+22Н]22+ 994,80; [М+23Н]23+ 951,59; [М+24Н]24+ 911,98; [М+25Н]25+ 875,54; [М+26Н]26+ 841,91; [М+27Н]27+ 810,76; [М+28Н]28+ 781,84; [М+29Н]29+ 754,92; найдено 1287,05; 1215,71; 1151,76; 1094,23; 1042,17; 994,88; 951,64; 911,99; 875,61; 841,95; 810,77; 781,85; 754,94/
(2-4. Четвертый этап: Снятие защиты с Acm группы)
Гликозилированный полипептид (13) из 166 остатков, имеющий асиало-сахарную цепь на Асн в положении 80, полученный на вышеуказанном третьем этапе (0,3 мг), помещали в пробирку Эппендорфа, растворяли в 90% водном растворе уксусной кислоты (0,03 мл), затем добавляли ацетат серебра (0,10 мг), и проводили реакцию при комнатной температуре, защищая от света. Спустя 3,5 часа подтверждали получение целевого вещества посредством ВЭЖХ и ИЭР-МС.К реакционному раствору добавляли дитиотретитол (1,0 мг), перемешивали при комнатной температуре в течение 5 минут, а затем центрифугировали, и собирали надосадочную жидкость, удаляя осадок. Собранную надосадочную жидкость фильтровали с применением мембранного фильтра, и фильтрат, содержащий целевое вещество, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ /10% воды /90% ацетонитрила, градиент А:В=50:50 → 50:50 (5 минут) →·20:80 (35 минут) → 5:95 (35,5 минут), изократическая элюция в течение 5 минут, а затем линейный градиент] до получения гликозилированного полипептида (14), содержащего асиало-сахарную цепь на Асн в положении 80, имеющего Acm группу цистеина в положениях 17, 31 и 141 со снятой защитой (SEQ ID №19) (Фигуры 11 и 12).
ИЭР-МС: расчет для C970H1510N250O297S7: [М+17Н]17+ 1274,55; [М+18Н]18+ 1203,80; [М+19Н]19+ 1140,49; [М+20Н]20+ 1083,52; [М+21Н]21+ 1031,97; [М+22Н]22+ 985,11; [М+23Н]23+ 942,32; [М+24Н]24+ 903,10; [М+25Н]25+ 867,01; [М+26Н]26+ 833,70; [М+27Н]27+ 802,86; [М+28Н]28+ 774,23; [М+29Н]29+ 747,56; найдено 1274,62; 1203,82; 1140,53; 1083,55; 1032,01; 985,15; 942,38; 903,12; 867,05; 833,72; 802,91; 774,29; 747,55.
(2-5. Пятый этап: Этап фолдинга)
Гликозилированный полипептид (14), содержащий асиало-сахарную цепь на Асн в положении 80, полученный на вышеуказанном четвертом этапе (0,32 мг), помещали в центрифужную пробирку, растворяли в буферном растворе при рН 8,5 (3,5 мл), (приготовленном из 8 M раствора гуанидина гидрохлорида, и 0,1 мМ раствора Трис), и оставляли при комнатной температуре. Спустя 1 час раствор переносили в диализную мембрану (Spectra/Pro, MWCO: 8000). Эту диализную мембрану помещали в диализный внешний раствор А (приготовленный из 3 M раствора гуанидина гидрохлорида, 0,1 мМ раствора Трис, 8 мМ раствора восстановленного глутатиона, и 1 мМ раствора глутатиона), и диализовали при 4°С. Спустя 12 часов диализную мембрану помещали в диализный внешний раствор В (приготовленный из 1 M раствора гуанидина гидрохлорида и 0,1 мм раствора Трис), и диализовали при 4°С. Спустя 12 часов эту диализную мембрану помещали в диализный внешний раствор С (10 мМ раствор уксусной кислоты) и диализовали при 4°С. Спустя 24 часа эту диализную мембрану извлекали из внешнего диализного раствора, и внутренний раствор из диализной мембраны переносили в центрифужную пробирку. Внутренний раствор из диализной мембраны анализировали посредством ВЭЖХ, и отмечено снижение времени элюции основного пика от 17,4 минут на исходном времени поэтапной операции диализа до 11,6 минут в конце поэтапной операции диализа. Этот пик анализировали посредством ИЭР-МС, и было подтверждено снижение молекулярной массы на 2 Да с молекулярной массы перед началом операции поэтапного диализа. Затем производили прямую очистку посредством ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ /10% воды /90% ацетонитрила, градиент А:В=50:50 → 50:50 (5 минут) → 20:80 (35 минут) → 5:95 (35,5 минут), изократическая элюция в течение 5 минут, и затем с линейным градиентом] до получения гликозилированного полипептида (15) (SEQ ID №20) (Фигуры 13 и 14). Далее была определена чистота этой фракции с помощью ВЭЖХ как 99,00% (% размера площади при ВЭЖХ) (Фигура 15). Затем проводили лиофилизацию полученного гликозилированного полипептида (15). Этот лиофилизат повторно растворяли в 10 мМ водном растворе уксусной кислоты, проводили количественное определение гликопротеина с применением спектрофотометра (метод поглощения в ультрафиолетовой части спектра), и концентрацию определяли по активности образца in vitro. Далее, готовили образец для активности in vivo путем растворения 10 мМ водного раствора уксусной кислоты, содержащего гликозилированный полипептид (15) с определенной концентрацией гликопротеина, в буферном растворе, отдельно приготовленном для рецептуры (приготовленном из сывороточного альбумина человека (30 мг), хлорида натрия (24,4 мг), двузамещенного натрия фосфата гидрата (7,53 мг), и кристаллического натрия дигидрофосфата (1,41 мг)), вновь лиофилизировали, и затем растворяли в воде для инъекций, соответствующей требованиям Японской Фармакопеи.
ИЭР-МС после поэтапного диализа и после очистки с помощью ВЭЖХ: Расчет для C970H1508N250O297S7: [М+9Н]9+ 2406,37; [М+10Н]10+ 2165,83; [М+11Н]11+ 1969,03; [М+12Н]12+ 1805,03; [М+13Н]13+ 1666,25; [М+14Н]14+ 1547,31; [М+15Н]15+ 1444,22; [М+16Н]16+ 1354,02; [М+17Н]17+ 1274,43; [М+18Н]18+ 1203,68; найдено 2406,21; 2165,68; 1968,85; 1804,85; 1666,09; 1547,16; 1444,05; 1353,93; 1274,39; 1203,63.
Пример 3. Синтез дисиало-ИФН-β
Активный ИФН-β, имеющий дисиало-сахарную цепь, синтезировали в целом за 6 этапов, показанных ниже.
(3-1. Первый этап: Связывание пептидного фрагмента В, присоединенного к дибензил-дисиало-сахарной цепи, и пептидного фрагмента С)
Два типа фрагментов, пептидный фрагмент В из 21 остатка с присоединенной дибензил-дисиало-сахарной цепью, с тиофенил-эфиром на С-конце (2) (2,2 мг) и пептидный фрагмент С (4) из 78 остатков (2,5 мг) помещали в одну и ту же колбу для выделения, растворяли в буферном растворе при рН 7,3 (0,21 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0.2 мМ раствора фосфорной кислоты, и 20 мМ раствора Трис-карбоксиэтил-фосфина (раствора ТСЕР)), добавляли тиофенол (6,2 мкл), и проводили реакцию при комнатной температуре. Спустя 24 часа реакцию подтверждали с ВЭЖХ, затем к реакционному раствору добавляли 0,2 M раствора метоксамина (0,31 мл) (приготовленного путем добавления 20 мМ раствора ТСЕР к 0,2 M раствора метоксамина), и проводили реакцию при комнатной температуре. Спустя 12 часов продукцию целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий гликопептидный фрагмент, очищали посредством ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ /10% воды /90% ацетонитрила, А:В=55:45 → 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 минут, затем с линейным градиентом] до получения необходимого пептидного фрагмента (В+С) (5), содержащего, дибензил-дисиало сахарную цепь в положении, соответствующем положению 80 интерферона-β (SEQ ID №21) (Фигуры 16 и 17).
ИЭР-МС: Расчет для C643H996N158O210S4: [М+10Н]10+ 1443,10; [М+11Н]11+1312,63; [М+12Н]12+ 1203,33; [М+13Н]13+ 1110,84; [М+14Н]14+ 1031,57; [М+15Н]15+ 962,87; [М+16Н]16+ 902,75; [М+17Н]17+ 849,70; [М+18Н]18+ 802,55; [М+19Н]19+ 760,37; найдено 1443,78; 1312,66; 1203,36; 1110,84; 1031,58; 962,86; 902,76; 849,70; 802,55; 760,36.
(3-2. Второй этап: Связывание пептидного фрагмента А и присоединенного к дибензил-дисиало-сахарной цепи пептидного фрагмента (В+С))
Два типа фрагментов, пептидный фрагмент А из 67 остатков с тиофенил-эфирным С-концом (1) (2,9 мг) и пептидный фрагмент, содержащий дибензил-дисиало-сахарную цепь в положении, соответствующем положению 80 интерферона-β (фрагмент (В+С) с присоединенной дибензил-дисиало-сахарной цепью) (5), полученный на вышеуказанном первом этапе (3,3 мг), помещали в одну и ту же колбу для выделения, растворяли в буферном растворе при рН 7,2 (0,22 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида 0,2 мМ раствора фосфорной кислоты, и 20 мМ раствора ТСЕР). Затем добавляли тиофенол (7,0 мкл) и проводили реакцию при комнатной температуре. Спустя 24 часа реакцию подтверждали с помощью ВЭЖХ, добавляли натрия меркаптоэтансульфонат (2,0 мг), и проводили реакцию при комнатной температуре. Спустя 12 часов получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевое вещество, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ /10% воды /90% ацетонитрила, А:В=55:45 → 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 мин, затем в линейном градиенте] для получения гликозилированного полипептида (6) из 166 остатков, содержащего свободную тиоловую группу на Цис в положениях 68 и 89, и содержащего дибензил-дисиало-сахарную цепь на Асн в положении 80 (SEQ ID №22) (Фигуры 18 и 19).
ИЭР-МС: Расчет для C1015H1571N255O316S9: [М+16Н]16+ 1419,15; [М+17Н]17+ 1335,73; [М+18Н]18+ 1261,58; [М+19Н]19+ 1195,23; [М+20Н]20+ 1135,52; [М+21Н]21+ 1081,50; [М+22Н]22+ 1032,38; [М+23Н]23+ 987,54; [М+24Н]24+ 946,44; [М+25Н]25+ 908,62; [М+26Н]26+ 873,71; [М+27Н]27+ 841,39; [М+28Н]28+ 811,37; [М+29Н]29+ 783,43; [М+30Н]30+ 757,35; найдено 1419,12; 1335,73; 1261,55; 1195,21; 1135,48; 1081,49; 1032,35; 987,51; 946,42; 908,61; 873,70; 841,39; 811,36; 783,42; 757,36.
(3-3. Третий этап: Восстановление Цис до Ала).
Гликозилированный полипептид (6) из 166 остатков, содержащий свободную тиоловую группу на Цис в положениях 68 и 89 и имеющий дибензил-дисиало-сахарную цепь на Асн в положении 80, полученную на вышеуказанном втором этапе (4,0 мг), помещали в колбу для выделения, растворяли в буферном растворе при рН 7,5 (0,28 мл), (приготовленном из 8 M раствора гуанидина гидрохлорида и 0,2 мМ раствора фосфорной кислоты), затем добавляли 0,5 M раствор Трис-этилкарбоксифосфина (раствор ТСЕР) при рН 7,5 (0,28 мл) (приготовленный из ТСЕР, 6 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и триэтиламина), tBuSH (28 мкл) и 0,1 M раствора VA-044 (28 мкл) (приготовленного путем растворения VA-044 в воде), и проводили реакцию при комнатной температуре. Спустя 4 часа получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, tBuSH экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевое вещество, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий растворп А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ /10% воды /90% ацетонитрила, А:В=55:45 → 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 минут, затем в линейном градиенте] до получения гликозилированного полипептида (7) из 166 остатков, в котором Цис в положениях 68 и 89 превращен в Ала, и содержащего дибензил-дисиало-сахарную цепь в положении 80 (SEQ ID №23) (Фигуры 20 и 21).
ИЭР-МС: расчет для C1015H1571N255O316S7: [М+18Н]18+ 1258,02; [М+19Н]19+ 1191,86; [М+20Н]20+ 1132,32; [М+21Н]21+ 1078,44; [М+22Н]22+ 1029,47; [М+23Н]23+ 984,75; [М+24Н]24+ 943,76; [М+25Н]25+ 906,05; [М+26Н]26+ 871,24; [М+27Н]27+ 839,01; [М+28Н]28+ 809,08; [М+29Н]29+ 781,22; найдено 1258,02; 1191,86; 1132,31; 1078,39; 1029,43; 984,73; 943,76; 906,04; 871,25; 839,01; 809,06; 781,17.
(3-4. Четвертый этап: Снятие защиты с Acm группы)
Гликозилированный полипептид из 166 остатков (7), содержащий дибензил-дисиало-сахарную цепь на Асн в положении 80, полученный на вышеуказанном третьем этапе (2,9 мг), помещали в пробирку Эппендорфа, растворяли в 90% водном растворе уксусной кислоты (0,13 мл), затем добавляли ацетат серебра (0,43 мг), и проводили реакцию при комнатной температуре, защищая от света. Спустя 3,5 часа получение целевого вещества подтверждали посредством ВЭЖХ и ИЭР-МС. К реакционному раствору добавляли дитиотретитол (3,0 мг), перемешивали при комнатной температуре в течение 5 минут, а затем центрифугировали, и надосадочную жидкость собирали, отделяя от осадка. Эту собранную надосадочную жидкость фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевое вещество, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ /10% воды /90% ацетонитрила, градиент А:В=50:50 → 50:50 (5 минут) → 20:80 (35 минут) → 5:95 (35,5 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] до получения гликозилированного полипептида из 166 остатков (8), содержащего дибензил-дисиало-сахарную цепь на Асн в положении 80, содержащего лишенные защиты Acm группы в положениях 17, 31 и 141 (SEQ ID №24) (Фигуры 22 и 23).
ИЭР-МС: расчет для C1006H1556N252O313S7: [М+18Н]18+ 1246,17; [М+19Н]19+ 1180,64; [М+20Н]20+ 1121,65; [М+21Н]21+ 1068,29; [М+22Н]22+ 1019,78; [М+23Н]23+ 975,48; [М+24Н]24+ 934,88; [М+25Н]25+ 897,52; [М+26Н]26+ 863,04; [М+27Н]27+ 831,11; [М+28Н]28+ 801,47; [М+29Н]29+ 773,86; найдено 1246,16; 1180,60; 1121,62; 1068,27; 1019,74; 975,44; 934,85; 897,49; 863,00; 831,08; 801,45; 773,81.
(3-5. Пятый этап: Снятие защиты с бензильной группы).
Гликозилированный полипептид (8) из 166 остатков, содержащий дибензил-дисиало-сахарную цепь на Асн в положении 80, полученный на вышеуказанном четвертом этапе (1,9 мг), помещали в колбу для выделения, растворяли в 8 M растворе гуанидина (0,17 мл) (приготовленном из гуанидина гидрохлорида и 20 мМ ТСЕР), и затем перемешивали при комнатной температуре. После охлаждения в ледяной воде, медленно добавляли 50 мМ водный раствор гидроксида натрия (0,85 мл). После реакции в ледяной воде в течение 20 минут, проводили нейтрализацию буферным раствором (2,1 мл), (приготовленным из 8 M раствора гуанидина и 0,2 M водного раствора ацетата натрия). Получение целевого вещества в реакции подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор непосредственно очищали посредством ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ /10% воды /90% ацетонитрила, градиент А:В=50:50 → 50:50 (5 минут) → 20:80 (35 минут) → 5:95 (35,5 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] до получения гликозилированного пептида (9) из 166 остатков, содержащего дисиало-сахарную цепь на Асн в положении 80 (SEQ ID №25) (Фигуры 24 и 25).
ИЭР-МС: Расчет для C992H1544N252O313S7: [М+17Н]17+ 1308,81; [М+18Н]18+ 1236,16; [М+19Н]19+ 1171,15; [М+20Н]20+ 1112,64; [М+21Н]21+ 1059,71; [М+22Н]22+ 1011,58; [М+23Н]23+ 967,64; [М+24Н]24+ 927,37; [М+25Н]25+ 890,31; [М+26Н]26+ 856,11; [М+27Н]27+ 824,44; [М+28Н]28+ 795,03; [М+29Н]29+ 767,65; [М+30Н]30+ 742,09; найдено 1308,80; 1236,17; 1171,12; 1112,63; 1059,67; 1011,56; 967,63; 927,34; 890,29; 856,09; 824,42; 795,01; 767,63; 742,08.
(3-6. Шестой этап: Этап фолдинга).
Гликозилированный пептид (9), содержащий дисиало-сахарную цепь на Асн в положении 80, полученный на вышеуказанном пятом этапе (1,3 мг), помещали в центрифужную пробирку, растворяли в буферном растворе при рН 8,5 (13 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида и 0,1 мМ раствора Трис), и оставляли при комнатной температуре. Спустя 1 час этот раствор переносили на диализную мембрану (Spectra/Pro, MWCO: 8000). Эту диализную мембрану помещали во внешний диализный раствор А (приготовленный из 3 M раствора гуанидина гидрохлорида, 0,1 мМ раствора Трис, 2 мМ раствора восстановленного глутатиона, и 1 мМ раствора глутатиона), и диализовали при 4°С. Спустя 12 часов диализную мембрану помещали в диализный внешний раствор В (приготовленный из 1 M раствора гуанидина гидрохлорида, и 0,1 мМ раствора Трис), и диализовали при 4°С. Спустя 12 часов диализную мембрану помещали в диализный внешний раствор С (10 мМ раствор уксусной кислоты) и диализовали при 4°С. Спустя 24 часа эту диализную мембрану извлекали из внешнего диализного раствора, и внутренний раствор из диализной мембраны переносили в центрифужную пробирку. Внутренний раствор из диализной мембраны анализировали посредством ВЭЖХ, и время элюции основного пика было уменьшено от 28,0 минут на исходном времени поэтапной операции диализа до 26,1 минуты в конце поэтапной операции диализа. Этот пик анализировали посредством ИЭР-МС, и было подтверждено снижение молекулярной массы на 2 Да с молекулярной массы перед началом операции поэтапного диализа. Затем производили прямую очистку посредством ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ /10% воды /90% ацетонитрила, градиент А:В=46:54 → 46:54 (5 минут) → 34,5:65,5 (28 минут) → 5:95 (28,5 минут), изократическая элюция в течение 5 минут, и затем с линейным градиентом] до получения гликозилированного полипептида (10) (SEQ ID №26) (Фигуры 26 и 27). Далее определяли чистоту этой фракции с ВЭЖХ, она составила 99,36% (% площади при ВЭЖХ) (Фигура 28). Затем проводили лиофилизацию полученного гликозилированного полипептида (10). Этот лиофилизат повторно растворяли в 10 мМ водном растворе уксусной кислоты, и проводили количественное определение гликопротеина с помощью спектрофотометра (способ определения поглощения в ультрафиолетовой области спектра), и определяли концентрацию по активности образца in vitro.
Далее, готовили образец для активности in vivo путем растворения 10 мМ водного раствора уксусной кислоты, содержащего гликозилированный полипептид (10) с определенной концентрацией гликопротеина, в буферном растворе, отдельно приготовленном для рецептуры (приготовленном из сывороточного альбумина человека (30 мг), хлорида натрия (24,4 мг), двузамещенного натрия фосфата гидрата (7,53 мг), и кристаллического натрия дигидрофосфата (1,41 мг)), вновь лиофилизировали, и затем растворяли в воде для инъекций, соответствующей требованиям Японской Фармакопеи.
ИЭР-МС после поэтапного диализа и после ВЭЖХ очистки: расчет для C992H1542N252O313S7: [М+10Н]10+ 2224,08; [М+11Н]11+ 2021,98; [М+12Н]12+ 1853,57; [М+13Н]13+ 1711,06; [М+14Н]14+ 1588,92; [М+15Н]15+ 1483,05; [М+16Н]16+ 1390,43; [М+17Н]17+ 1308,69; [М+18Н]18+ 1236,0; найдено 2223,88; 2021,80; 1853,43; 1710,91; 1588,77; 1482,92; 1390,35; 1308,61; 1236,01.
Аминокислотная последовательность интерферона-β была получена при разделении на три фрагмента в вышеуказанных Примерах 2 и 3, но в Примерах 4-6 ниже показано получение аминокислотной последовательности интерферона-β при разделении на четыре фрагмента.
В Примерах 4-6 гликозилированный полипептид получен из четырех фрагментов в целом, путем дополнительного разделения пептидного фрагмента С, содержащего аминокислоты в положениях 89-166 в интерфероне-β, на фрагмент C1 из положений 89-134 и фрагмент С2 из положений 135-166, содержащего Ала в положении 134 на N-конце.
Пример 4. Синтез пептидного фрагмента (С1+С2)
(4-1. Синтез пептидного фрагмента C1-SPh)
Смолу Amino-PEGA (от Merck) (100 мкмоль) помещали в колонку для твердофазного синтеза, промывали в достаточной степени метиленхлоридом (ДХМ) и ДМФ, а затем добавляли ДМФ в достаточной степени для набухания. 4-гидроксиметил-3-метоксифенокси-масляную кислоту (НМРВ) (0,25 ммоль), TBTU (0,25 ммоль), и N-этилморфолин (0,25 ммоль) растворяли в ДМФ (2 мл) и помещали в колонку, и перемешивали при комнатной температуре в течение 4 часов. Смолу промывали в достаточной степени ДМФ и ДХМ, и получали смолу НМРВ-PEGA, и применяли ее в качестве твердой фазы для твердофазного синтеза.
Fmoc-Фен (0,50 ммоль), MSNT (0,50 моль), и N-метилимидазол (0,375 ммоль) растворяли в ДХМ (2 мл), помещали в колонку для твердофазного синтеза, и перемешивали при 25°С в течение 4 часов.
После перемешивания смолу промывали ДХМ и ДМФ. Fmoc группу обрабатывали 20% раствором пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ, аминокислоты последовательно конденсировали посредством способа, показанного ниже, для последующего удлинения пептидной цепи.
Аминокислоту, имеющую аминогруппу, защищенную Fmoc группой, растворяли в N-метилпирролидоне (NMP) (1 мл), добавляли 0,45 M HCTU⋅HOBT/NMP (0,4 ммоль), вносили в колонку для твердофазного синтеза, и затем в колонку для твердофазного синтеза добавляли 0,9 M DIPEA/NMP (0,8 ммоль). После перемешивания при комнатной температуре в течение 20 минут, смолу промывали ДХМ и ДМФ, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. Эту операцию повторяли, и аминокислоты, защищенные Fmoc группой (0,5 ммоль), применяли для последовательной конденсации аминокислот.
В качестве аминокислот, защищенных Fmoc или Boc группой, использовали Fmoc-Лиз(Вос), Fmoc-Лей, Fmoc-Тир(tBu), Fmoc-Гис(Trt), Fmoc-Лей, Fmoc-Иле, Fmoc-Apr(Pbf), Fmoc-Гли, Fmoc-Тир(tBu), Fmoc-Тир(tBu), Fmoc-Арг(Pbf), Fmoc-Лиз(Вос), Fmoc-Лей, Fmoc-Гис(Trt), Fmoc-Лей, Fmoc-Cep(tBu)-Cep(ψMe, MePro), Fmoc-Мет, Fmoc-Лей, Fmoc-Лиз(Вос), Fmoc-Гли, Fmoc-Арг(Pbf), Fmoc-Фен-Tpe(ψMe, MePro), Fmoc-Асп(OtBu), Fmoc-Глу(OtBu), Fmoc-Лиз(Вос), Fmoc-Глу(OtBu), Fmoc-Лей, Fmoc-Лиз(Вос), Fmoc-Глу(OtBu), Fmoc-Глу(OtBu), Fmoc-Лей, Fmoc-Вал, Fmoc-Лиз(Boc)-Tpe(ψMe, МеПро), Fmoc-Лей, Fmoc-Гис(Trt), Fmoc-Асн(Trt), Fmoc-Иле, Fmoc-Глн(Trt), Fmoc-Гис(Trt), Fmoc-Тир(tBu), Fmoc-Вал, и Fmoc-Асн(Trt), а в качестве аминокислоты, защищенной Вос группой, Boc-L-тиазолидин-4-карбоновую кислоту последовательно применяли и связывали на смоле для твердофазного синтеза. В результате получали пептидный фрагмент из 46 остатков (16) Лиз(Вос)-Лей-Тир(tBu)-Гис(Trt)-Лей-Иле-Арг(Pbf)-Гли-Тир(tBu)-Тир(tBu)-Арг(Pbf)-Лиз(Boc)-Лей-Гис(Trt)-Лей-Сер(ψМе, МеПро)-Сер(tBu)-Мет-Лей-Лиз(Вос)-Гли-Арг(Pbf)-Τpe(ψΜe, МеПро)-Фен-Асп(OtBu)-Глу(OtBu)-Лиз(Вос)-Глу(OtBu)-Лей-Лиз(Вос)-Глу(OtBu)-Глу(OtBu)-Лей-Вал-Tpe(ψMe, МеПро)-Лиз(Вос)-Лей-Гис(Trt)-Асн(Trt)-Иле-Глн(Trt)-Гис(Trt)-Тир(tBu)-Вал-AcH(Trt)-Thz-BocN (SEQ ID №27) на смоле для твердофазного синтеза.
Смолу для твердофазного синтеза, содержащую пептидный фрагмент (16), полученный выше, промывали ДХМ и ДМФ, затем добавляли смешанный раствор трифторэтанола и уксусной кислоты (1:1) до достаточного набухания смолы, и перемешивали в течение 18 часов при комнатной температуре до отсоединения пептидного фрагмента (16) от смолы. Освобожденную смолу отделяли фильтрацией, и реакционный раствор концентрировали при сниженном давлении. Полученный остаток концентрировали, и получали пептидный фрагмент (16) с защищенной боковой аминокислотной цепью (SEQ ID №28): Лиз(Вос)-Лей-Тир(tBu)-Гис(Trt)-Лей-Иле-Арг(Pbf)-Гли-Тир(tBu)-Тир(tBu)-Арг(Pbf1)-Лиз(Вос)-Лей-Гис(Trt)-Лей-Сер(ψМе, МеПро)-Сер(tBu)-Мет-Лей-Лиз(Boc)-Гли-Арг(Pbf)-Тре(ψМе, МеПро)-Фен-Асп(OtBu)-Глу(OtBu)-Лиз(Boc)-Глу(OtBu)-Лей-Лиз(Boc)-Глу(OtBu)-Глу(OtBu)-Лей-Вал-Tpe(ψMe, МеПро)-Лиз(Вос)-Лей-Гис(Trt)-Асн(Trt)-Иле-Глн(Trt)-Гис(Trt)-Тир(tBu)-Вал-Асн(Trt)-Thz-ВосМ.
100 мкмоль эквивалентов пептида (16), содержащего 46 аминокислотных остатков и имеющего защищенную боковую аминокислотную цепь, переносили в колбу для выделения, растворяли в ДМФ (3,0 мл), а затем охлаждали до -15°С - -20°С в атмосфере азота. Добавляли тиофенол (308 мкл, 3,0 ммоль), а затем РуВОР (260,0 мг, 0,50 ммоль) и DIPEA (85,0 мкл, 0,5 ммоль). После перемешивания при -15°С - -20°С в течение 2 часов добавляли трифторуксусную кислоту (0,1 мл), и затем постепенно восстанавливали комнатную температуру смеси. После восстановления комнатной температуры реакционный раствор концентрировали при сниженном давлении. К полученному осадку добавляли смесь трифторуксусной кислоты: воды: TIPS (=95:2,5:2,5), и перемешивали при комнатной температуре. Спустя 2 часа этот раствор вновь добавляли к диэтиловому эфиру (150 мл) для осаждения, а затем центрифугировали для удаления части раствора, с целью получения осадка, содержащего пептидный тиоэфир. Этот осадок очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=65:35 → 65:35 (5 минут) → 30:70 (35 минут) → 5:95 (45 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте], и получали пептидный фрагмент C1-SPh (16), содержащий тиофениловый эфир на С-конце (SEQ ID №29): HN-Thz-Асн-Вал-Тир-Гис-Глн-Иле-Асн-Гис-Лей-Лиз-Тре-Вал-Лей-Глу-Глу-Лиз-Лей-Глу-Лиз-Глу-Асп-Фен-Тре-Арг-Гли-Лиз-Лей-Мет-Сер-Сер-Лей-Гис-Лей-Лиз-Арг-Тир-Тир-Гли-Арг-Иле-Лей-Гис-Тир-Лей-Лиз-SPh.
ИЭР-МС: расчет для C263H412N72O67S3: [М+4Н]4+ 1438,68; [М+5Н]5+ 1151,14; [М+6Н]6+ 959,45; [М+7Н]7+ 822,53; [М+8Н]8+ 719,84; найдено 1438,51; 1151,00; 959,34; 822,43; 719,74.
(4-2. Синтез пептидного фрагмента С2)
Смолу Amino-PEGA (от Merck) (100 мкмоль) помещали в колонку для твердофазного синтеза, промывали в достаточной степени метиленхлоридом (ДХМ) и ДМФ, а затем добавляли ДМФ в достаточной степени для набухания. 4-гидроксиметил-3-метоксифенокси-масляную кислоту (НМРВ) (0,25 ммоль), TBTU (0,25 ммоль), и N-этилморфолин (0,25 ммоль) растворяли в ДМФ (2 мл) и помещали в колонку, и перемешивали при комнатной температуре в течение 4 часов. Смолу промывали в достаточной степени ДМФ и ДХМ, и получали смолу HMPB-PEGA, и применяли ее в качестве твердой фазы для твердофазного синтеза.
Fmoc-Фен (0,50 ммоль), MSNT (0,50 моль), и N-метилимидазол (0,375 ммоль) растворяли в ДХМ (2 мл), помещали в колонку для твердофазного синтеза, и перемешивали при 25°С в течение 4 часов.
После перемешивания смолу промывали ДХМ и ДМФ. Fmoc группу обрабатывали 20% раствором пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ, аминокислоты последовательно конденсировали посредством способа, показанного ниже, для последующего удлинения пептидной цепи.
Аминокислоту, имеющую аминогруппу, защищенную Fmoc группой, растворяли в N-метилпирролидоне (NMP) (1 мл), добавляли 0,45 M HCTU⋅HOBT/NMP (0,4 ммоль), вносили в колонку для твердофазного синтеза, и затем в колонку для твердофазного синтеза добавляли 0,9 M DIPEA/NMP (0,8 ммоль). После перемешивания при комнатной температуре в течение 20 минут, смолу промывали ДХМ и ДМФ, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. Эту операцию повторяли, и аминокислоты, защищенные Fmoc группой (0,5 ммоль), применяли для последовательной конденсации аминокислот. В качестве аминокислот, защищенных Fmoc или Boc группой, Fmoc-Асн(Trt), Fmoc-Арг(Pbf), Fmoc-Лей, Fmoc-Тир(tBu), Fmoc-Гли, Fmoc-Tpe(tBu), Fmoc-Лей, Fmoc-Арг(Pbf), Fmoc-Асн(Trt), Fmoc-Иле, Fmoc-Фен, Fmoc-Тир(tBu), Fmoc-Фен, Fmoc-Асн(Trt), Fmoc-Арг(Pbf), Fmoc-Лей, Fmoc-Иле, Fmoc-Глу(OtBu), Fmoc-Вал, Fmoc-Арг(Pbf), Fmoc-Вал, Fmoc-Иле, Fmoc-Tpe(tBu), Fmoc-Трп(Boc), Fmoc-Ала, Fmoc-Цис(Acm), Fmoc-Гис(Trt), Fmoc-Тир(tBu)-Сер(ψМе, МеПро), Fmoc-Глу(OtBu), Fmoc-Лиз(Вос), и Вос-Цис(Trt) последовательно применяли и связывали на смоле для твердофазного синтеза. В результате получали фрагмент (17) из 32 остатков Асн(Trt)-Арг(Pbf)-Лей-Тир(tBu)-Гли-Тре(tBu)-Лей-Арг(Pbf)-Асн(Trt)-Иле-Фен-Тир(tBu)-Фен-Асн(Trt)-Арг(Pbf)-Лей-Иле-Глу(OtBu)-Вал-Арг(Pbf)-Вал-Иле-Tpe(tBu)-Tpπ(Boc)-Ала-Цис(Acm)-Цис(Trt)-Cep(ψMe, МеПро)-Тир(tBu)-Глу(OtBu)-Лиз(Вос)-Цис(Trt)-BocNH (SEQ ID №30) на смоле для твердофазного синтеза.
К пептидному фрагменту (17), полученному выше, добавляли трифторуксусную кислоту: воду: TIPS (=95:2,5:2,5), и перемешивали при комнатной температуре в течение 3,5 часов для отсоединения пептидного фрагмента (17) от смолы. Затем этот раствор добавляли к диэтиловому эфиру (150 мл) для осаждения, а затем подвергали разделению посредством центрифугирования для отделения осадка, содержащего необходимый пептид, и раствора. Затем, после удаления раствора, целевое соединение подтверждали с помощью ВЭЖХ, а затем очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=65:35 → 65:35 (5 минут) → 30:70 (35 минут) → 5:95 (45 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте], для получения необходимого пептидного фрагмента С2 (17) (SEQ ID №31): Н2М-Цис-Лиз-Глу-Тир-Сер-Гис-Цис(Acm)-Ала-Трп-Тре-Иле-Вал-Арг-Вал-Глу-Иле-Лей-Арг-Асн-Фен-Тир-Фен-Иле-Асн-Арг-Лей-Тре-Гли-Тир-Лей-Арг-Асн.
ИЭР-МС: Расчет для C185H282N52O47S2: [М+3Н]3+ 1351,22; [М+4Н]4+ 1013,67; [М+5Н]5+ 811,13; найдено 1351,02; 1013,51; 811,01.
Пример 5. Связывание пептидного фрагмента C1 и пептидного фрагмента С2.
Два типа фрагментов, пептидный фрагмент из 46 остатков с тиофенил-эфирным С-концом C1-SPh (16) (8,7 мг) и пептидный фрагмент С2 (17) из 32 остатков (3,5 мг) помещали в одну и ту же колбу для выделения, растворяли в буферном растворе при рН 6,8 (1,0 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и 20 мМ раствора Трис-карбоксиэтилфосфина (ТСЕР раствора)), затем добавляли тиофенол (30 мкл), и проводили реакцию при комнатной температуре. Спустя 24 часа проведение реакции подтверждали ВЭЖХ, затем к реакционному раствору добавляли 0,2 M раствор метоксамина (1,5 мл) (приготовленный путем добавления 20 мМ раствора ТСЕР к 0,2 M раствору метоксамина), и проводили реакцию при комнатной температуре. Спустя 12 часов получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевой фрагмент, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% вода /90% ацетонитрила, градиент А:В=65:35 → 65:35 (5 минут) → 30:70 (35 минут) → 5:95 (45 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] для получения необходимого пептидного фрагмента (SEQ ID №32) (пептидный фрагмент (С1+С2)) (89-166) (Фигура 29).
ИЭР-МС: расчет для C441H688N124O114S4: [М+8Н]8+ 1210,90; [М+9Н]9+ 1076,47; [М+10Н]10+ 968,92; [М+11Н]11+ 880,93; [М+12Н]12+ 807,60; [М+13Н]13+ 745,55; [М+14Н]14+ 692,37; найдено 1210,77; 1076,46; 968,91; 880,92; 808,50; 745,47; 692,37.
Пример 6. Способ 2 синтеза асиало-ИФН-β.
(6-1. Связывание асиало-гликозилированного пептидного фрагмента В и пептидного фрагмента (С1+С2))
Два типа фрагментов, асиало-гликозилированный пептидный фрагмент В из 21 остатка с тиофенил-эфирным С-концом (3) (1,5 мг) и пептидный фрагмент (18) из 78 остатков (пептидный фрагмент С1-С2) (2,4 мг) помещали в одну и ту же колбу для выделения, растворяли в буферном растворе при рН 7,2 (0,25 мл) приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и 20 мМ раствора Трис-карбоксиэтилфосфина (ТСЕР раствора)). Затем добавляли тиофенол (7,4 мкл), и проводили реакцию при комнатной температуре. Спустя 24 часа проведение реакции подтверждали ВЭЖХ, затем к реакционному раствору добавляли 0,2 M раствор метоксамина (0,37 мл) (приготовленный путем добавления 20 мМ раствора ТСЕР к 0,2 M раствору метоксамина), и проводили реакцию при комнатной температуре. Спустя 12 часов получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий гликопептидный фрагмент, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=65:35 → 65:35 (5 минут) → 30:70 (35 минут) → 5:95 (45 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] для получения необходимого пептидного фрагмента (19), содержащий асиало-сахарную цепь в положении, соответствующем положению 80 интерферона-β (SEQ ID №33) (асиало-гликозилированньгй пептидный фрагмент (В+С1+С2)) (68 - 166) (Фигура 30).
ИЭР-МС: Расчет для C607H950N156O194S5: [М+10Н]10+ 1370,73; [М+11Н]11+ 1246,21; [М+12Н]12+ 1142,44; [М+13Н]13+ 1054,64; [М+14Н]14+ 979,38; [М+15Н]15+ 914,15; [М+16Н]16 857,08; [М+17Н]17+ 806,72; [М+18Н]18+ 761,96; [М+19Н]19+ 721,91; найдено 1370,66; 1246,14; 1142,38; 1054,56; 979,38; 914,08; 856,99; 806,65; 761,92; 721,86.
(6-2. Связывание пептидного фрагмента А и гликопептидного фрагмента В+С1+С2)
Два типа фрагментов, пептидный фрагмент из 67 остатков с тиофенил-эфирным С-концом А (1) (3,0 мг) и необходимый пептидный фрагмент (19), содержащий асиало-сахарную цепь в положении, соответствующем положению 80 интерферона-β (асиало-гликозилированного пептидного фрагмента В+С1+С2), полученного в вышеуказанном пункте 6-1 (68-166) (3,3 мг), помещали в ту же самую колбу для выделения, растворяли в буферном растворе при рН 7,2 (0,24 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и 20 мМ раствора ТСЕР). Затем добавляли тиофенол (7,2 мкл), и проводили реакцию при комнатной температуре. Спустя 24 часа проведение реакции подтверждали ВЭЖХ, затем добавляли натрия меркаптоэтансульфонат (2,1 мг), и проводили реакцию при комнатной температуре. Спустя 12 часов получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевой фрагмент, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=55:45 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] для получения гликозилированного полипептида (20) из 166 остатков, содержащего свободную тиоловую группу на Цис в положениях 68, 89 и 135, и содержащих асиало-сахарную цепь на Асн в положении 80 (SEQ ID №34) (Фигура 31).
ИЭР-МС: расчет для C979H1525N253O299S11: [М+17Н]17+ 1293,69; [М+18Н]18+ 1221,88; [М+19Н]19+ 1157,62; [М+20Н]20+ 1099,79; [М+21Н]21+ 1047,47; [М+22Н]22+ 999,90; [М+23Н]23+ 956,47; [М+24Н]24+ 916,66; [М+25Н]25+ 880,03; [М+26Н]26+ 846,22; [М+27Н]27+ 814,92; [М+28Н]28+ 785,85; найдено 1292,75; 1221,01; 1156,79; 1098,99; 1046,71; 999,19; 955,78; 916,00; 879,39; 845,63; 814,34; 785,29.
(6-3. Восстановление Цис в Ала)
Гликозилированный полипептид (20) из 166 остатков, содержащий свободную тиоловую группу на Цис в положениях 68, 89 и 135, и содержащий асиало-сахарную цепь на Асн в положении 80, полученный на вышеуказанном этапе 6-2 (1,1 мг), помещали в колбу для выделения, растворяли в буферном растворе при рН 7,5 (0,20 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты), затем добавляли 0,5 M раствор Трис-этилкарбоксифосфина (раствор ТСЕР) с рН 7,5 (0,20 мл) (приготовленный из ТСЕР, 6 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и триэтиламина), tBuSH (20 мкл), и 0,1 M раствор VA-044 (приготовленный путем растворения VA-044 в воде), и проводили реакцию при комнатной температуре. Спустя 4 часа получение целевого вещества подтверждали ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, tBuSH экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевое вещество, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=55:45 → 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] для получения гликозилированного полипептида (21) из 166 остатков, содержащего Цис в положениях 68, 89 и 135, превращенный в Ала, и имеющий асиало-сахарную цепь на Асн в положении 80 (SEQ ID №35) (Фигура 32).
ИЭР-МС: расчет для C979H1525N253O300S7: [М+17Н]17+ 1287,09; [М+18Н]18+ 1215,64; [М+19Н]19+ 1151,71; [М+20Н]20+ 1094,18; [М+21Н]21+ 1042,12; [М+22Н]22+ 994,80; [М+23Н]23+ 951,59; [М+24Н]24+ 911,98; [М+25Н]25+ 875,54; [М+26Н]26+ 841,91; [М+27Н]27+ 810,76; [М+28Н]28+ 781,84; [М+29Н]29+ 754,92; найдено 1287,11; 1215,69; 1151,72; 1094,20; 1042,16; 994,83; 951,61; 911,98; 875,57; 841,93; 810,80; 781,88; 754,88.
Гликозилированный полипептид (15) можно получить путем применения этапов удаления защиты с Acm группы и фолдинга, в условиях, подобных тем, которые описаны в 2-4, 2-5, с гликозилированным полипептидом (21), полученным в 6-3.
Пример 7. Способ 2 синтеза дисиало-ИФН-β
(7-1. Связывание пептидного фрагмента В с прикрепленной дисиало-сахарной цепью и пептидного фрагмента (С1+С2)).
Два типа фрагментов, пептидный фрагмент В из 21 остатка с присоединенной дибензил-дисиало-сахарной цепью с тиофенил-эфирным С-концом (2) (17,9 мг), и пептидный фрагмент (18) из 78 остатков (пептидный фрагмент (С1+С2)) (22,9 мг) помещали в ту же самую колбу для выделения, растворяли в буферном растворе при рН 7,2 (2,37 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и 20 мМ раствора Трис-карбоксиэтил-фосфина (раствора ТСЕР)). Затем добавляли тиофенол (71,1 мкл), и проводили реакцию при комнатной температуре. Спустя 24 часа проведение реакции подтверждали ВЭЖХ, затем к реакционному раствору добавляли 0,2 M раствор метоксамина (3,55 мл) (приготовленного путем добавления 20 мМ раствора ТСЕР к 0,2 M раствору метоксамина), и проводили реакцию при комнатной температуре. Спустя 12 часов получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС.Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий гликопептидный фрагмент, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=55:45 → 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] до получения необходимого пептидного фрагмента (22), содержащего дибензил-дисиало-сахарную цепь в положении, соответствующем положению 80 интерферона-β (SEQ ID №36) (Фигура 33).
ИЭР-МС: Расчет для C643H996N158O210S4: [М+11Н]11+ 1315,55; [М+12Н]12+ 1206,00; [М+13Н]13+ 1113,31; [М+14Н]14+ 1033,86; [М+15Н]15+ 965,00; [М+16Н]16+ 904,75; [М+17Н]17+ 851,59; [М+18Н]18+ 804,34; [М+19Н]19+ 762,06; найдено 1315,59; 1205,97; 1113,28; 1033,83; 964,98; 904,74; 851,57; 804,32; 762,02.
(7-2. Пептидный фрагмент А, и пептидный фрагмент (В1+С1+С2) с присоединенной дибензил-дисиало-сахарной цепью).
Два типа фрагментов, пептидный фрагмент из 67 остатков с тиофенил-эфирным С-концом А (1) (4,7 мг) и необходимый пептидный фрагмент (22), содержащий дибензил-дисиало-сахарную цепь в положении, соответствующем положению 80 интерферона-β, полученного в вышеуказанном пункте 7-1 (5,2 мг) помещали в одну колбу для выделения, растворяли в буферном растворе при рН 7,2 (0,35 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и 20 мМ раствора ТСЕР). Затем добавляли тиофенол (11 мкл), и проводили реакцию при комнатной температуре. Спустя 24 часа проведение реакции подтверждали ВЭЖХ, затем добавляли натрия меркаптоэтансульфонат (3,3 мг), и проводили реакцию при комнатной температуре. Спустя 12 часов получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевое вещество, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=55:45 → 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] до получения пептидного фрагмента (23) из 166 остатков, содержащего свободную тиоловую группу на Цис в положениях 68, 89 и 135, и содержащего дибензил-дисиало-сахарную цепь на Асн в положении 80 (SEQ ID №37) (Фигура 34).
ИЭР-МС: расчет для C1015H1571N255O316S10: [М+16Н]16+ 1421,16; [М+17Н]17+ 1337,62; [М+18Н]18+ 1263,36; [М+19Н]19+ 1196,92; [М+20Н]20+ 1137,13; [М+21Н]21+ 1083,02; [М+22Н]22+ 1033,84; [М+23Н]23+ 988,93; [М+24Н]24+ 947,77; [М+25Н]25+ 909,90; [М+26Н]26+ 874,94; [М+27Н]27+ 842,57; [М+28Н]28+ 812,52; [М+29Н]29+ 784,53; найдено 1421,13; 1337,57; 1263,35; 1196,90; 1137,08; 1082,99; 1033,80; 988,89; 947,73; 909,87; 874,92; 842,55; 812,51; 784,50.
(7-3. Восстановление Цис до Ала)
Пептидный фрагмент (23) из 166 остатков, содержащий свободную тиоловую группу на Цис в положениях 68, 89 и 135, и содержащий дибензил-дисиало-сахарную цепь на Асн в положении 80, полученный в вышеуказанном примере 7-2 (6,1 мг), помещали в колбу для выделения, растворяли в буферном растворе при рН 7,5 (0,27 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты), затем добавляли 0,5 M раствор Трис-этилкарбоксифосфина (раствор ТСЕР) с рН 7,5 (0,27 мл) (приготовленный из ТСЕР, 6 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и триэтиламина), tBuSH (100 мкл), и 0,1 M раствор VA-044 (100 мкл) (приготовленный путем растворения VA-044 в воде), и проводили реакцию при комнатной температуре. Спустя 4 часа получение целевого вещества подтверждали ВЭЖХ и ИЭР-МС. Реакционный раствор переносили в центрифужную пробирку, тиофенол экстрагировали диэтиловым эфиром, затем водный слой, содержащий целевое вещество, фильтровали с помощью мембранного фильтра, и фильтрат, содержащий целевое вещество, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=55:45 → 55:45 (5 минут) → 25:75 (35 минут) → 5:95 (36 минут), изократическая элюция в течение 5 минут, а затем в линейном градиенте] для получения пептидного фрагмента из 166 остатков (24), содержащего Цис в положениях 68, 89 и 135, превращенный в Ала, и содержащего дибензил-дисиало-сахарную цепь на Асн в положении 80 (SEQ ID №38) (Фигура 35).
ИЭР-МС: расчет для C1015H1571O316S7: [М+18Н]18+ 1258,02; [М+19Н]19+ 1191,86; [M+20H]20+ 1132,32; [М+21Н]21+ 1078,44; [М+22Н]22+ 1029,47; [М+23Н]23+ 984,75; [М+24Н]24+ 943,76; [М+25Н]25+ 906,05; [М+26Н]26+ 871,24; [М+27Н]27+ 839,01; [М+28Н]28+ 809,08; [М+29Н]29+ 781,22; найдено 1258,05; 1191,87; 1132,37; 1078,47; 1029,51; 984,77; 943,79; 906,05; 871,25; 839,04; 809,05; 781,24.
Гликозилированный полипептид (10) можно получить путем применения этапов снятия защиты с Acm группы, снятия защиты с бензильной группы, и фолдинга в условиях, подобных условиям на этапах, описанных в примерах 3-4, 3-5 и 3-6, для гликозилированного полипептида (24), полученного в вышеуказанном примере 7-3.
В вышеуказанных Примерах 1-3 пептидный фрагмент, содержащий Ала на N-конце, конструировали и применяли для связывания. Соответственно, в Примерах 1 -3 пептидный фрагмент, содержащий Цис, введенный в положение Ала, помещенный на N-конце указанного пептидного фрагмента синтезировали, сшивали, а затем Цис восстанавливали в Ала.
В следующем Примере конструировали пептидный фрагмент, содержащий Сер на N-конце. Другими словами, показан пример, в котором пептидный фрагмент, содержащий Цис, введенный в положение Сер, синтезировали, сшивали, а затем Цис превращали в Сер. В частности, показан пример, в котором пептидный фрагмент В, содержащий аминокислоты в положениях 68-88 в аминокислотной последовательности интерферона-β, синтезируют путем деления на фрагмент В2 из положений 68-75 и фрагмент В1 из положений 76-88, содержащий Цис на N-конце вместо Сер в положении 76, и после этапа сшивания синтетического пептидного фрагмента (B1+B2)-SR, содержащего Цис в качестве аминокислоты в положении, соответствующий положению 76 интерферона-β (далее обозначаемого как пептидный фрагмент (B1+B2)-SR (Цис-тело)) путем сшивания, и этапа превращения Цис в положение, соответствующее положению 76 интерферона-β в пептидном фрагменте (B1+B2)-SR (Цис-тело) в Сер, синтезируют пептидный фрагмент (B1+B2)-SR, содержащий Сер в качестве аминокислоты в положении, соответствующем положению 76 интерферона-β.
Другими словами, в частности, приводится пример способа производства, включающий следующие этапы.
(I) Этап получения пептидного фрагмента В1, представленного следующей формулой (i).
[Химическая формула 37]

(II) Этап получения гликопептида В2, представленного следующей формулой (j).
[Химическая формула 38]

(III) Этап получения гликопептида, представленного следующей формулой (к), путем сшивания пептида В1 и гликопептида В2, с последующим превращением конкретного цистеина на указанном пептиде в серии (см. Фигуры 43-45).
[Химическая формула 39]

Пример 8. Синтез пептидного фрагмента (B1+B2)-SR с прикрепленной дисиало-сахарной цепью.
(8-1. Синтез пептидного фрагмента В1-SR)
Матрицу HMPB-Chem (от Biotage) (0,10 ммоль) помещали в колонку для твердофазного синтеза, промывали в достаточной степени метиленхлоридом (ДХМ) и ДМФ, а затем обеспечивали достаточное набухание с ДХМ.
Fmoc-Лей (0,50 ммоль), MSNT (0,50 ммоль), и N-метилимидазол (0,375 ммоль) растворяли в ДХМ (2 мл), помещали в колонку для твердофазного синтеза, и перемешивали при 25°С в течение 4 часов.
После перемешивания смолу промывали с ДХМ и ДМФ. Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ аминокислоты последовательно конденсировали способом, показанным ниже, для последующего удлинения пептидной цепи.
Аминокислоту, имеющую аминогруппу, защищенную Fmoc группой, растворяли в N-метилпирролидоне (NMP) (1 мл), добавляли 0,45 М HCTU⋅HOBT/NMP (0,4 ммоль), вносили в колонку для твердофазного синтеза, и затем в колонку для твердофазного синтеза добавляли 0,9 М DIPEA/NMP (0,8 ммоль). После перемешивания при комнатной температуре в течение 20 минут, смолу промывали ДХМ и ДМФ, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. Эту операцию повторяли, и аминокислоты, защищенные Fmoc группой (0,5 ммоль), применяли для последовательной конденсации аминокислот.
Fmoc-Лей, Fmoc-Лей, Fmoc-AcH(Trt), Fmoc-rny(OtBu), Fmoc-Вал, Fmoc-Иле, Fmoc-Tpe(tBu) и Ртос-Глу(СНВи) последовательно применяли в качестве аминокислот, защищенных Fmoc группой, и получали пептид из 8 остатков Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu). В этом пептиде из 8 остатков обрабатывали Fmoc группой с раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ, аспарагиновое производное дибензил-дисиало-сахарной цепи (g) (547,8 мг, 200 мкмоль) и DEPBT (59,9 мг, 200 мкмоль) растворяли в ДМФ/ДМСО (смесь 1:1, 3,33 мл) в центрифужной пробирке, помещали на колонку для твердофазного синтеза, добавляли DIPEA (52,3 мкл, 300 мкмоль), и перемешивали при комнатной температуре в течение 18 часов. Промывание ДМФ и ДХМ обеспечило пептид из 9 остатков с сахарной цепью Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu)-Асн(дибензил-дисиалоолигосахарид)-FmocNH (SEQ ID №39) на твердой фазе.
[Химическая формула 40]

Для последующего удлинения гликопептидной цепи, липофильную защитную группу с пептида из 9 остатков с сахарной цепью на твердофазной смоле, полученного выше, удаляли с раствором 20% пиперидина/ДМФ (2 мл), а затем аминокислоты последовательно конденсировали способом, описанным ниже.
Аминокислоту, защищенную Fmoc группой, а также HOBt (67,6 мг, 0,50 ммоль) и DIPCI (73,1 мкл, 0,475 ммоль) растворяли в ДМФ (6,3 мл), активировали в течение 15 минут, а затем помещали в колонку для твердофазного синтеза. После перемешивания при комнатной температуре в течение 1 часа, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 20 минут для снятия защиты. Эту операцию повторяли для последовательной конденсации аминокислот. В качестве аминокислот, защищенных Fmoc и Вос группами, Fmoc-Tpn(Boc), Fmoc-Гли, Fmoc-Tpe(tBu) и Вос-Цис(Trt) последовательно применяли и связывали со смолой для твердофазного синтеза. В результате получали гликозилированный пептидный фрагмент (25) из 13 остатков Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu)-Асн (дибензил-дисиалоолигосахарид)-Трп(Вос)-Гли-Тре(tBu)-Цис(Trt)-BocNH (SEQ ID №40) на смоле для твердофазного синтеза.
Затем, после промывания ДХМ и ДМФ, смешанный раствор трифторэтанола и уксусной кислоты (1:1) добавляли до достаточного набухания смолы, а затем перемешивали в течение 18 часов при комнатной температуре для отсоединения гликозилированного пептидного фрагмента от смолы. Освобожденную смолу отделяли фильтрацией, и реакционный раствор концентрировали при сниженном давлении. Полученный осадок концентрировали, и получали гликозилированный пептид (25), имеющий защищенную аминокислотную боковую цепь (SEQ ID №41): Лей-Лей-Асн(Trt)-Глу(OtBu)-Вал-Иле-Тре(tBu)-Глу(OtBu)-Асн(дибензил-дисиалоолигосахарид)-Трп(Вос)-Гли-Тре(tBu)-Цис(Trt)-BocNH.
100 мкмоль эквивалентов гликозилированного пептидного фрагмента (25), содержащего 13 аминокислотных остатков и имеющего защищенную боковую аминокислотную цепь, переносили в колбу для выделения, растворяли в ДМФ (3,0 мл), а затем охлаждали до -15°С - -20°С в атмосфере азота. Добавляли 2-меркаптоэтансульфонат (492,5 мкл, 3,0 ммоль), а затем РуВОР (260,0 мг, 0,50 ммоль) и DIPEA (85,0 мкл, 0,5 ммоль). После перемешивания при -15°С - -20°С в течение 2 часов добавляли трифторуксусную кислоту (0,1 мл), и затем постепенно восстанавливали комнатную температуру смеси. После восстановления комнатной температуры реакционный раствор концентрировали при сниженном давлении. К полученному осадку добавляли смесь трифторуксусной кислоты: воды: TIPS (=95:2,5:2,5), и перемешивали при комнатной температуре. Спустя 2 часа этот раствор вновь добавляли к диэтиловому эфиру (150 мл) для осаждения, а затем центрифугировали для удаления части раствора, с целью получения осадка, содержащего пептидный тиоэфир. Этот осадок очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=72:28 → 59:41 (26 минут), в линейном градиенте], и получали пептидный фрагмент с присоединенной дибензил-дисиалосахарной цепью B1-SR (25), содержащий алкильный тиоэфир на С-конце (SEQ ID №42): H2N-Цис-Тре-Гли-Трп-Асн(дибензил-дисиалоолигосахарид)-Глу-Тре-Иле-Вал-Глу-Асн-Лей-Лей-SR (Фигура 46). -SR указывает этилсульфонатный тиоэфир.
(8-2. Синтез пептидного фрагмента B2-SPh)
Матрицу HMPB-Chem (от Biotage) (0,10 ммоль) помещали в колонку для твердофазного синтеза, достаточно промывали метиленхлоридом (ДХМ) и ДМФ, а затем обеспечивали достаточное промывание ДХМ.
Fmoc-Cep (tBu) (0,50 ммоль), MSNT (0,50 ммоль) и N-метилимидазол (0,375 ммоль) растворяли в ДХМ (2 мл), помещали в колонку для твердофазного синтеза, и перемешивали при 25°С в течение 4 часов.
После перемешивания смолу промывали с ДХМ и ДМФ. Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. После промывания ДМФ аминокислоты последовательно конденсировали способом, показанным ниже, для последующего удлинения пептидной цепи.
Аминокислоту, имеющую аминогруппу, защищенную Fmoc группой, растворяли в N-метилпирролидоне (NMP) (1 мл), добавляли 0,45 M HCTU⋅HOBT/NMP (0,4 ммоль), вносили в колонку для твердофазного синтеза, и затем в колонку для твердофазного синтеза добавляли 0,9 M DIPEA/NMP (0,8 ммоль). После перемешивания при комнатной температуре в течение 20 минут, смолу промывали ДХМ и ДМФ, Fmoc группу обрабатывали раствором 20% пиперидина/ДМФ (2 мл) в течение 15 минут для снятия защиты. Эту операцию повторяли, и аминокислоты, защищенные Fmoc группой (0,5 ммоль), применяли для последовательной конденсации аминокислот.
Fmoc-Cep(tBu), Fmoc-Acn(OtBu), Fmoc-Глн(Trt), Fmoc-Арг(Pbf), Fmoc-Фен, Fmoc-Иле и Вос-L-тиазолидин-4-карбоновую кислоту последовательно применяли в качестве аминокислот, защищенных Fmoc группой, и связывали со смолой для твердофазного синтеза. В результате получали пептидный фрагмент (26) из 8 остатков этом пептиде из 8 остатков Сер(tBu)-Сер(tBu)-Асп(OtBu)-Глн(Trt)-Арг(Pbf)-Фен-Иле-Thz-BocN (SEQ ID №43) на смоле для твердофазного синтеза.
Затем, после промывания ДХМ и ДМФ, смешанный раствор трифторэтанола и уксусной кислоты (1:1) добавляли для достаточного замачивания смолы, и перемешивали в течение 18 часов при комнатной температуре для отсоединения пептидного фрагмента от смолы. Освобожденную смолу отделяли фильтрацией, и реакционный раствор концентрировали при сниженном давлении. Полученный остаток концентрировали, и получали пептид (26) с защищенной боковой аминокислотной цепью (SEQ ID №44): Cep(tBu)-Cep(tBu)-Асп(OtBu)-Глн(Trt)-Арг(Pbf)-Фен-Иле-Thz-BocN.
100 мкмоль эквивалентов гликозилированного пептидного фрагмента (26), содержащего 8 аминокислотных остатков и имеющего защищенную боковую аминокислотную цепь, переносили в колбу для выделения, растворяли в ДМФ (3,0 мл), а затем охлаждали до -15°С - -20°С в атмосфере азота. Добавляли 2-тиофенол (308 мкл, 3,0 ммоль), а затем РуВОР (260,0 мг, 0,50 ммоль) и DIPEA (85,0 мкл, 0,5 ммоль). После перемешивания при -15°С - -20°С в течение 2 часов добавляли трифторуксусную кислоту (0,1 мл), и затем постепенно восстанавливали комнатную температуру смеси. После восстановления комнатной температуры реакционный раствор концентрировали при сниженном давлении. К полученному осадку добавляли смесь трифторуксусной кислоты: воды: TIPS (=95:2,5:2,5), и перемешивали при комнатной температуре. Спустя 2 часа этот раствор вновь добавляли к диэтиловому эфиру (150 мл) для осаждения, а затем центрифугировали для удаления части раствора, с целью получения осадка, содержащего пептидный тиоэфир. Этот осадок очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=78:22 → 68:32 (20 минут), в линейном градиенте], для получения пептидного фрагмента B2-SPh (26), содержащего тиофениловый сложный эфир на С-конце (SEQ ID №45): HN-Thz-Иле-Фен-Арг-Глн-Асп-Сер-Сер-SPh (Фигура 47).
(8-3. Связывание пептидного фрагмента B1-SR с прикрепленной дибензил-дисиало-сахарной цепью, и пептидного фрагмента B2-SPh)
Два типа фрагментов, пептидный фрагмент B1-SR с прикрепленной дибензил-дисиало-сахарной цепью, из 13 остатков, с алкил-тиоэфирным С-концом (0,60 мг), и пептидный фрагмент B2-SPh из 8 остатков (0,16 мг) помещали в колбу для выделения, растворяли в буферном растворе при рН 7,2 (0,15 мл) (приготовленном из 8 M раствора гуанидина гидрохлорида, 0,2 мМ раствора фосфорной кислоты, и 20 мМ раствора Трис-карбоксиэтил-фосфина (раствора ТСЕР)), и проводили реакцию при комнатной температуре. Спустя 14,5 часов получение целевого вещества подтверждали с помощью ВЭЖХ и ИЭР-МС. Реакционный раствор фильтровали с помощью мембранного фильтра, и фильтрат, содержащий гликопептидный фрагмент, очищали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/ 10% вода /90% ацетонитрила, градиент А:В=78:22 → 56:44 (22 минуты), изократическая элюция в течение 5 минут, а затем в линейном градиенте] до получения необходимого гликозилированного пептидного фрагмента (B1+B2)-SR (Цис-тела) (27), содержащего дибензил-дисиало-сахарную цепь и имеющего алкил-тиоэтерифицированный С-конец (SEQ ID 346) (Фигура 48).
(8-4. Метилирование Цис)
5,3 мг (1,03 мкмоль) полученного алкил-тиоэфира пептида из 21 остатка (27) помещали в пробирку Эппендорфа, растворяли в буферном растворе при рН 8,6 (1,09 мл) (приготовленного из 8 M раствора гуанидина гидрохлорида, 0,25 M раствора Трис-соляной кислоты, и 3,3 мМ раствора ЭДТА) и затем добавляли ацетонитрил (0,36 мл) и метил-4-нитробензол-сульфонат (5,1 мг) при 25°С. Спустя 30 минут добавляли 10% раствор ТФУ (0,2 мл) при рН 4, а затем реакционный раствор очищали посредством ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=72:55 → 55:45 (34 минуты), изократическая элюция в течение 5 минут, а затем в линейном градиенте], для получения гликозилированного пептидного фрагмента (B1+B2)-SR (Цис-тела) (28), содержащего дибензил-дисиало-сахарную цепь, имеющую метилированный атом серы в цистеиновом остатке, содержавшийся в исходном материале, и содержащий тиоэтерифицированный С-концевой алкил (SEQ ID №47) (Фигура 49).
(8-5, Цианирование метилированного Цис и реакция интрамолекулярного переноса ацила после цианирования)
Полученный пептидный фрагмент (28), содержащий дибензил-дисиало-сахарную цепь, содержащую метилированный атом серы в цистеиновом остатке, помещали в пробирку Эппендорфа, растворяли в 0,1 мМ 80% растворе муравьиной кислоты (3,3 мл), и добавляли цианоген бромид 23,2 мг (219 мкмоль) при 25°С. После защиты контейнера для реакции от света, реакцию продолжали при 37°С, и спустя 26,5 часов реакционный раствор концентрировали при сниженном давлении.
Полученный осадок растворяли в буферном растворе при рН 8,0 (приготовленном из 8 M раствора гуанидина гидрохлорида и 0,2 С раствора фосфорной кислоты) (0,33 мл), а затем проводили реакцию при 37°С. Спустя 1 час добавляли раствор 20% пиперидина/ДМФ (3,3 мкл, 1% о/о), и после подтверждения завершения реакции с ВЭЖХ, проводили очистку с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=72:55 → 55:45 (34 минуты), изократическая элюция в течение 5 минут, а затем в линейном градиенте], для получения промежуточного продукта реакции гликозилированного пептидного фрагмента (В1+В2)-SR (29), содержащего 21 остаток и дибензил-дисиало-сахарную цепь, и имеющего тиоэтерифицированный алкил на С-конце (SEQ ID №48) (Фигура 50). -SR показывает этил-сульфонатный тиоэфир.
Дисиало-ИФН-β можно синтезировать, подобно Примеру 3, с применением гликозилированного полипептида, полученного в вышеприведенном пункте 8-5 (29), в качестве пептидного фрагмента В с присоединенной дибензил-дисиало-сахарной цепью в Примере 3.
Пример 9. Способ тепловой обработки
(9-1. Тепловая обработка 17.31.141Цис(Acm)-80N(дибензил-дисиалоолигосахарид)-ИФН-β)
Гликозилированный полипептид, полученный в вышеприведенном пункте 3-3 (7) (17.31.141Цис(Acm)-80N(дибензил-дисиалоолигосахарид)-ИФН-β) (3,7 мг), помещали в 10 мл колбу для выделения, и добавляли 0,2 M фосфатный буферный раствор с рН 7,0 (содержащий 8 M гуанидин) (3,3 мл; концентрация субстрата 50 мМ). Колбу для выделения закрывали стеклянной пробкой, и нагревали на масляной бане до 60°С. Спустя 10 часов восстанавливали комнатную температуру раствора, а затем проводили ВЭЖХ анализ. После подтверждения отсутствия разрушения посредством ВЭЖХ и ИЭР-МС (Фигура 36), раствор обессоливали с помощью ВЭЖХ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10% воды/90% ацетонитрила, градиент А:В=50:50 → 50:50 (5 минут) → 20:80 (35 минут) → 5:95 (35,5 минут), изократическая элюция в течение 5 минут, затем с линейным градиентом], и получали образец, подвергнутый тепловой обработке (4,0 мг).
(9-2. Тепловая обработка 80М(дибензил-дисиалоолигосахарида)-ИФН-β)
Гликозилированный полипептид, полученный в вышеприведенном пункте 3-4 (8) 80М(дибензил-дисиалоолигосахарид)-ИФН-β) (30 мкг), помещали в 0,5 мл пробирку Эппендорфа, и добавляли 0,2 M буферный раствор с рН 7,0 (содержащий 8 M гуанидин) (30 мкл; концентрация субстрата 50 мМ). Пробирку закрывали, и нагревали на масляной бане до 60°С. Спустя 10 часов восстанавливали комнатную температуру раствора, а затем проводили ВЭЖХ анализ [колонка: SHISEIDO proteonavi (С4, 5 мкм), ϕ20 × 250 мм, скорость потока: 7,0 мл/мин, элюирующий раствор А: 0,1% водный раствор ТФУ, В: 0,09% ТФУ/10%) воды/90% ацетонитрила, градиент А:В=50:50 → 50:50 (5 минут) → 20:80 (35 минут) → 5:95 (35,5 минут), изократическая элюция в течение 5 минут, и проверяли чистоту посредством ВЭЖХ и ИЭР-МС (Фигура 37).
При сравнении данных ВЭЖХ и ИЭР-МС с гликозилированным полипептидом (7), содержащим защищенный Цис, использованным в тесте нагревания в вышеприведенном пункте 9-1, полученное количество гликозилированного полипептида (8), содержащего незащищенный цистеин, снижалось из-за тепловой обработки.
Пример 10. Анализ аффинности рецепторов.
Проводили анализ аффинности рецепторов не прошедшего или прошедшего тепловую обработку химически синтезированного ИФН-β в отношении ИФН-α/β-рецептора 2.
Активность связывания не прошедшего или прошедшего тепловую обработку химически синтезированного ИФН-β в отношении ИФН-α/β-рецептора 2 измеряли с помощью ProteOn XPR36 (Bio-rad) с применением ППР (технологии поверхностного плазмонного резонанса).
В частности, 10 мкг/мл ИФН-α/β рецептора 2 (10 мМ уксуснокислого буферного раствора, рН 4,5) фиксировали на GLM чипе (Bio-rad) в соответствии с инструкциями по применению набора для связывания аминов (Bio-rad) и промывали 0,005% Твин 20/ФБР при скорости потока 30 мкг/мин. Не подвергшийся тепловой обработке полипептид (10) с прикрепленной дисиало-сахарной цепью, полученный в Примере 3, или подвергшийся тепловой обработке полипептид с прикрепленной дисиало-сахарной цепью, или прошедший тепловую обработку полипептид с прикрепленной дисиало-сахарной цепью, полученный путем обработки полипептида с прикрепленной дисиало-сахарной цепью, прошедшего тепловую обработку в вышеуказанном пункте 9-1, посредством этапа de-Acm, этапа снятия защиты с бензильной группы, и этапа фолдинга, описанных выше в пунктах 3-4 - 3-6, доводили до 0,31-25 нМ с 0,1% БСА и 2 мМ ЭДТА/ФБР, и добавляли на чип со скоростью потока 50 мкл/мин. Анализ гликозилированного полипептида (10) проводили для каждой из пяти концентраций 0,31 нМ, 0,93 нМ, 2,8 нМ, 8,3 нМ и 25 нМ. Константу связывания (ka), константу диссоциации (kd), и константу диссоциации связывания (Kd) анализировали с программным обеспечением ProteOn Manager. Результаты показаны на Фигуре 38, Фигуре 39, и в Таблице 1.
На Фигуре 38 показан график анализа аффинности связывания полипептида (10) с присоединенной дисиало-сахарной цепью, не подвергнутого тепловой обработке, с ИФН-α/β рецептором 2. Далее, на Фигуре 39 показан график анализа аффинности связывания полипептида с присоединенной дисиало-сахарной цепью, подвергнутого тепловой обработке, с ИФН-α/β рецептором 2. Как показано на Фигуре 38, Фигуре 39 и в Таблице 1, химически синтезированный гликозилированный полипептид из настоящего изобретения обладает аффинностью к рецептору, эквивалентной аффинности не подвергнутого тепловой обработке гликозилированного полипептида, даже после тепловой обработки.

Пример 11. Фармакокинетический анализ
Пример фармакокинетического анализа для внутривенного и подкожного введения гликозилированных полипептидов в соответствии с настоящим изобретением, приготовленных в вышеуказанных Примерах, показан ниже.
(11-1. Приготовление жидкости для введения и реагента)
Асиало-гликозилированный полипептид (15) (асиало-ИФН-β), приготовленный в Примере 2, и полипептид (10) с прикрепленной дисиало-сахарной цепью (дисиало-ИФН-β), приготовленный в Примере 3, применяли в качестве химически синтезированного ИФН-β. Добавляли БСА и фосфатный буферный раствор, и проводили лиофилизацию. Жидкость для введения готовили путем растворения препарата с 60 мкл воды milli-Q непосредственно перед введением, и доводили до 500,000 МЕ/мл с фосфатно-солевым буферным раствором (ФБР, Wako Pure Chemical Industries, Ltd.). Далее, ИФН-Р Mochida для инъекций, приготовленный посредством биосинтеза (Mochida Pharmaceutical Co., Ltd.), применяли в качестве контрольного ИФН-β. Полученную воду для инъекций применяли для растворения до 12 миллионов МЕ/мл, а затем доводили до 500,000 МЕ/мл с ФБР. Для ЭДТА-ФБР ФБР добавляли так, чтобы концентрация EDTA-2Na (Wako Pure Chemical Industries, Ltd.) составила 2 мМ. Химически синтезированный гликозилированный полипептид, применяемый для фармакокинетического анализа из настоящего Примера, не подвергался тепловой обработке. Далее, ИФН-β Mochida является ИФН-1а, полученным из фибробластов человека, приготовленным посредством биосинтеза.
(11-2. Применение и сбор крови)
Препарат вводили мышам (мыши BALB/c, самцы, масса тела 20,45-25,28 г) в дозе 2 миллиона МЕ/кг в сытом состоянии в орбитальную вену или дорзально подкожно с инсулиновым шприцем 29 G × 1/2 (Terumo Corporation) в объеме 4 мл/кг.75 мкл крови отбирали из орбитальной вены в капиллярную пробирку для гематокрита (HIRSHMANN LABORGERATE) с гепарином до введения, а также спустя 2, 10 и 30 минут и 1, 3, 6 и 8 часов после внутривенного введения, и до применения и спустя 10 и 30 минут и 1, 2, 4, 6 и 8 часов при подкожном введении. Кровь сразу смешивали с ЭДТА-ФБР такого же объема, как собранная кровь, и центрифугировали (15000 об/мин, 4°С, 10 минут). 90 мкл надосадочной жидкости отбирали в качестве образца плазмы. Образцы плазмы замораживали для хранения до проведения анализа. Используемые наконечники и пробирки были продуктами с низкой абсорбцией от ВМ Equipment Go., Ltd.
(11-3. Измерение концентрации в крови)
Набор для ИФА для человеческого интерферона-β (Kamakura Techno-Science, Inc.) применяли для определения концентрации гликозилированного полипептида (ИФН-β). Другими словами, готовили образцы плазмы, разбавленной в 360, 120 и 12 раз, как необходимо, с раствором для разбавления, поставляемом в наборе, для анализа образцов. В качестве эталона для получения стандартной кривой, использовали химически синтезированный ИФН-β той же серии, которую применяли для введения, и ИФН-β Mochida для инъекций, для получения 200; 100; 50; 25; 12,5; 6,25 и 3,125 МЕ/мл с раствором для разбавления, поставляемым в наборе. Пустой образец плазмы добавляли к стандартной кривой, в зависимости от разведения образца плазмы так, чтобы доставляемое количество было эквивалентным. Путем умножения полученных результатов и значения разбавления, и дальнейшего умножения на значение разбавления 2 в ЭДТА-ФБР в качестве антикоагулянтной обработки, рассчитывали концентрацию в крови. Развитие концентрация ИФН-β в плазме показано на Фигуре 40.
(11-4. Расчет фармакокинетических параметров)
Из полученного графика концентрации ИФН-β рассчитывали площадь под фармакокинетической кривой (AUC) путем формулы трапеций с применением метода анализа моментов. Далее, расчетную исходную концентрацию определяли путем метода экстраполяции для внутривенного введения (С0), и определяли максимальную концентрацию в плазме (Cmax) по полужизни в плазме (t1/2), среднему времени удержания (MRT), и действительному значению при подкожном введении. Полученные фармакокинетические параметры показаны в Таблице 2.
На Фигуре 40 видно, что гликозилированный полипептид, полученный путем химического синтеза в соответствии с настоящим изобретением, показал эквивалентную кинетику в крови, по сравнению с ИФН-β, полученным путем биосинтеза.


Пример 12. Анализ противоопухолевой активности
(12-1. Клеточная культура и приготовление реагента).
Клетки Дауди, которые являются клетками лимфомы Беркитта, применяли для анализа противоопухолевой активности. Применяли среду RPMI 1640 (Invitrogen) с добавлением 10% эмбриональной телячьей сыворотки (GIBCO), подвергнутой тепловой обработке при 56°С в течение 30 минут, и пенициллина/стрептомицина (SIGMA). Необработанные чашки (IWAKI) применяли для культивирования, культивирование проводили при 37°С и при 5% CO2 концентрации, и осуществляли посев каждые 2-3 суток.
(12-2. Способ подготовки мыши-носителя опухоли).
Клетки Дауди, культивированные на вышеуказанном этапе 12-1, собирали в пробирку и центрифугировали (1300 об/мин, 4°С, 3 минуты). Надосадочную жидкость извлекали аспиратором, добавляли HBSS (Nacalai), и суспендировали клетки. Эту обработку клеток отмыванием проводили в целом три раза. Определяли количество клеток гемоцитометром, и готовили суспензию клеток 2×108 клеток/мл с HBSS. Непосредственно перед инокуляцией клеток Дауди, добавляли Matrigel (BD) в том же самом объеме, как и клеточную суспензию, для получения 2-кратного разведения, с целью приготовления клеточной суспензии для инокуляции. Клеточную суспензию для инокуляции хранили на льду до извлечения непосредственно перед инокуляцией. Сомнопентил (Kyoritsuseiyaku Corporation), разбавленный до 5 мг/мл с ФБР, применяли в качестве анестезирующего лекарственного средства. 250-300 мкл анестетика вводили интраперитонеально ТКИД мышам (С.B-17/Icr-scid/scid Jcl мыши, самцы) (CLEA Japan, Inc.) с помощью инсулинового шприца 29 G х 1/2 (Terumo). После подтверждения анестезии, шерсть на правом боку мыши сбривали электробритвой. 100 мкл клеточной суспензии для инокуляции вводили подкожно с иглой для инъекции 26 G 1/2 (Terumo) и 1 мл стеклянным шприцем (Terumo).
(12.3. Способы анализа и оценки противоопухолевой активности)
Спустя примерно 30 суток после инокуляции клеток согласно 11-3, основную ось (мм) и второстепенную ось (мм) образованной опухолевой ткани измеряли кронциркулем (Mitsutoyo). По полученным данным определяли объем опухоли (мм3). Объем опухоли рассчитывали по следующей формуле: объем опухоли (мм3) = значение по основной оси (мм) × значение по второстепенной оси × значение по второстепенной оси × 0,5. Определяли объем опухоли по времени и строили график для оценки мощности противоопухолевой активности.
(12-4. Способы приготовления жидкости для введения и ее применения)
Асиало-гликозилированный полипептид (15), синтезированный в Примере 2, и полипептид с присоединенной дисиало-сахарной цепью (10), синтезированный в Примере 3, готовили в дозе 5 миллионов МЕ/мл в ФБР. ИФН-β Mochida для инъекций, приготовленный путем биосинтеза (Mochida Pharmaceutical Co., Ltd.). применяли в качестве контроля ИФН-β. Приготовленную воду для инъекций применяли для растворения до 12 миллионов МЕ/мл, и затем доводили до 5 миллионов МЕ/мл с ФБР. Приготовление жидкости для введения проводили непосредственно перед применением. Мышей-носителей опухоли распределяли на 4 группы с применением объема опухоли, измеренного выше в 12-3 (n = 4/группу). Объем опухоли к моменту распределения на группы составил примерно 800 мм. Применение осуществляли с приготовленной жидкостью для введения подкожно в область спины в объеме 4 мл/кг, так чтобы доза составила 20 миллионов МЕ/кг, с инсулиновым шприцем 29 G x 1/2. Для контрольной группы с введением растворителя ФБР, используемый для приготовления жидкости для введения, применяли в объеме 4 мл/кг. Разделение на группы и первое применение проводили на 0 сутки, и всего было проведено 10 подкожных введений каждый день до 9 суток.
(12-5. Оценка мощности противоопухолевой активности)
Объем опухоли рассчитывали на 3, 6, 8, 10, 13, 15 и 17 сутки со способом, показанным в 12-3, и изменением объема опухоли со временем показано на Фигуре 41.
На Фигуре 41 видно, что гликозилированный полипептид, приготовленный путем химического синтеза в соответствии с настоящим изобретением, показал эквивалентную или превосходящую противоопухолевую активность по сравнению с ИФН-β, приготовленным путем биосинтеза. В частности, гликозилированный полипептид (19) из настоящего изобретения, в котором прикрепленные дисиало-сахарные цепи являются по существу однородными, показал более высокую противоопухолевую активность, по сравнению с ИФН-β, приготовленным путем биосинтеза.
Пример 13. Оценка способности к супрессии клеточной пролиферации после тепловой обработки.
Далее оценивали способность к супрессии клеточной пролиферации подвергнутого тепловой обработке полипептида с присоединенной дисиало-сахарной цепью, полученного при воздействии на полипептид с присоединенной дисиало-сахарной цепью, подвергнутый тепловой обработке в вышеуказанном пункте 9-1, этапу де-Acm, этапу снятия защиты бензильной группы, и этапу фолдинга, описанным в вышеприведенных пунктах 3-4 - 3-6.
Специфическую оценку способности к супрессии клеточной пролиферации проводили следующим образом.
Клеточную линию лимфомы Беркитта человека, клетки Дауди, суспендировали в среде RPMI 1640, содержащей 10% эмбриональной телячьей сыворотки, 100 Ед/мл пенициллина, и 100 мкг/мл стрептомицина (10% FCS-RPMI 1640), до 1,25 × 105 клеток/мл. 1 × 104/80 мкл/ячейку клеточной суспензии помещали в 96-луночный плоскодонный планшет, далее добавляли 20 мкл/ячейку полного химическим синтезированного ИФН-β, разбавленного 10% FCS-RPMI 1640, и культивировали в СО2 инкубаторе с концентрацией СО2, доведенной до 5%, при 37°С в течение 3 суток. Способность к супрессии клеточной пролиферации измеряли с набором для подсчета клеток-8 (DOJINDO), в соответствии с инструкцией, прилагаемой к набору, с дегидрогеназной активностью митохондрий на 3 сутки культивирования в качестве индикатора.
Далее, не подвергшийся тепловой обработке полипептид (10) с присоединенной дисиало-сахарной цепью применяли в качестве контроля. Результаты показаны на Фигуре 42. Как показано на Фигуре 42, была установлена эквивалентная способность к супрессии клеточной пролиферации, по сравнению с не подвергнутым тепловой обработке гликозилированным полипептидом, для гликозилированного полипептида даже после тепловой обработки.
Реферат
Изобретение относится к области биотехнологии, конкретно к композиции для лечения или профилактики интерферон-β-зависимого заболевания, и может быть использовано в медицине.Синтетическим путем получают гликозилированный полипептид, имеющий однородную структуру сахарной цепи и обладающий активностью интерферона-β. Однородность структуры сахарной цепи и наличие сиаловой кислоты на невосстанавливающем конце сахарной цепи увеличивает время полужизни гликозилированной формы интерферона в крови и улучшает его фармакокинетические свойства. 3 н. и 9 з.п. ф-лы, 50 ил., 2 табл., 12 пр.
Формула
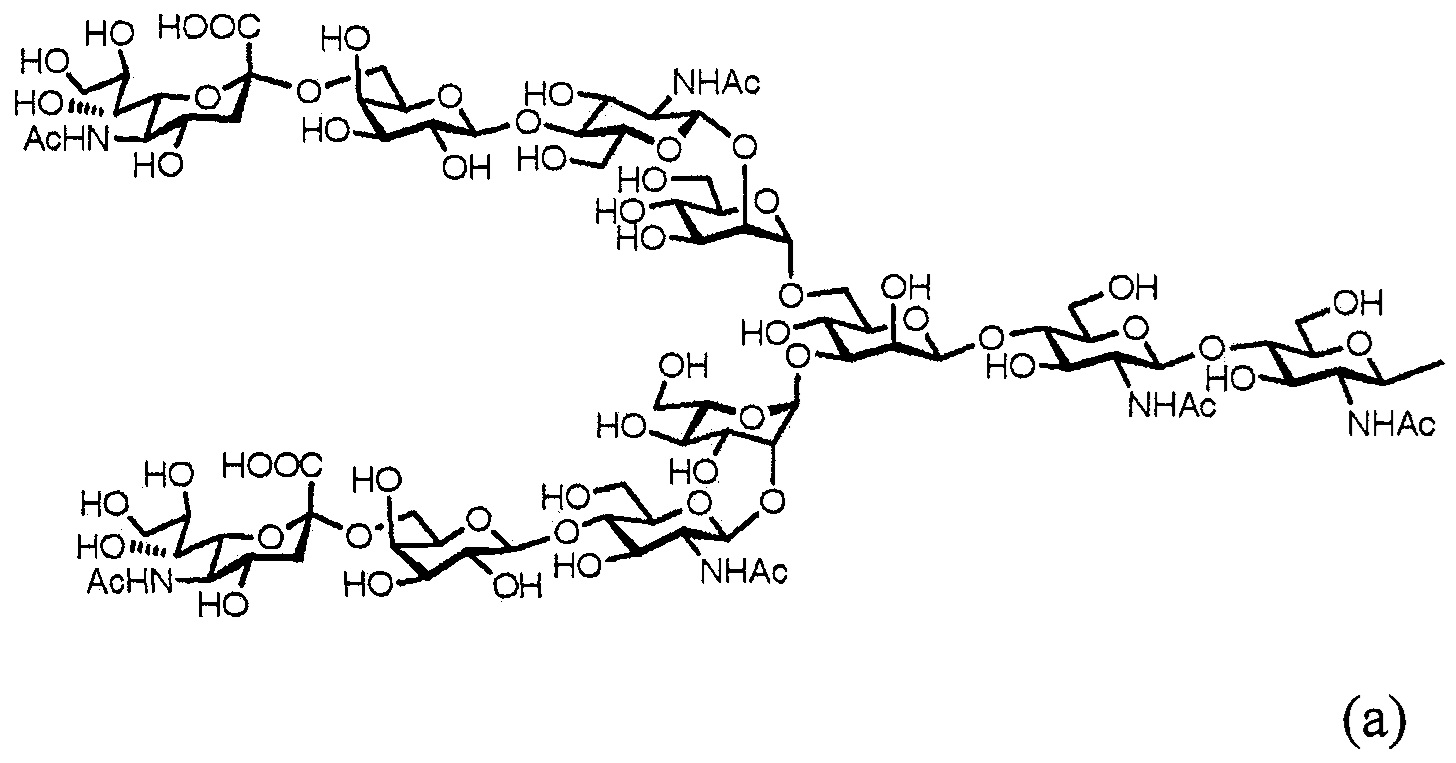

Комментарии