Способ лечения связанных с ifn-альфа состояний - RU2639135C2
Код документа: RU2639135C2
Чертежи




Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к иммуногенной вакцине и ее применению для лечения связанных с IFNα состояний, таких как системная красная волчанка.
УРОВЕНЬ ТЕХНИКИ
Семейство интерферонов первого типа (IFN I) включает IFNα, IFNβ IFNδ, IFN1, IFNκ, IFNτ и IFNϖ. Преобладающими формами являются IFNα, для которого у людей описаны 13 близкородственных белков , и один IFNβ. Несмотря на то, что различные формы IFN типа I могут вызывать различные биологические реакции, все IFN типа I структурно связаны (в их генах отсутствуют интроны, и они расположены на коротком плече хромосомы 9) и передают сигнал через субъединицы одного и того же рецептора (Van Boxel-Dezaire et al., Immunity 2006; 25: 361-372).
В настоящее время возрастает интерес в отношении взаимосвязи между IFN типа I и аутоиммунными нарушениями, т.к. недавно появились данные о признаках их индукции, так называемом рисунке («почерке») интерферонов, у пациентов, страдающих различными аутоиммунными заболеваниями (Baccala et al. Immunol Rev 2005; 204: 9-26). В действительности, из-за своего иммуномодулирующего действия IFN типа I, по-видимому, вовлечен в некоторые патогенные пути различных аутоиммунных состояний.
Парадигмой патогенной значимости IFN типа I в аутоиммунитете является системная красная волчанка (СКВ). СКВ представляет собой хроническое заболевание, характеризующееся полиорганной вовлеченностью вследствие аномального повреждения органов, вызванного аутоантителами, направленными на аутоантигены. СКВ имеет сложную этиологию, включающую как генетические факторы, так и факторы окружающей среды. Было показано, что уровень IFNα в сыворотке крови при СКВ коррелирует с тяжестью указанного заболевания (Dall'era et al. Ann Rheum Dis 2005; 64: 1692-7).
Синдром Шегрена (СШ), также известный как сухой синдром, представляет собой хроническое системное аутоиммунное состояние, поражающее экзокринные железы, в частности, слюнные и слезные железы. В сыворотке крови пациентов, страдающих данным заболеванием, также наблюдали повышенную активность IFNα. Наконец, было показано, что другие состояния, такие как диабет, ревматоидный артрит, склеродермия, васкулит и аутоиммунный тиреоидит, также связаны с высокими уровнями IFNα.
Недавно Sedaghat et al. также предположили, что IFN типа I может играть роль в истощении Т-клеток CD4+ у ВИЧ-положительных пациентов, т.к. они показали, что IFN типа I влияет на устойчивое состояние динамики нормальных Т-клеток CD4+ путем сдвига равновесия в сторону эффекторов Th1, которые представляют собой короткоживущие клетки, вместо долгоживущих Т-клеток памяти (Sedaghat et al. J. Virol. 2008, 82(4): 1870-1883). Это было подтверждено в публикации Mandl et al., в которой предлагается уменьшать выработку IFNα плазмацитоидными дендритными клетками для уменьшения патологической иммунной активации (Mandl et al. Nat. Med. 2008).
Кроме того, существуют данные о том, что введение IFNα обостряет сопутствующее заболевание у пациентов с псориазом, аутоиммунным тиреоидитом и рассеянным склерозом, и индуцирует подобный СКВ синдром у пациентов без аутоиммунного заболевания в анамнезе.
Следовательно, существует необходимость в агенте, ингибирующем активность IFNα.
В настоящее время пассивную иммунизацию моноклональными нейтрализующими антителами тестируют в клинических исследованиях с использованием ронтализумаба (rontalizumab) и сифалимумаба (sifalimumab) для лечения СКВ. Однако указанная терапия демонстрирует недостатки, заключающиеся в прицельном воздействии только на одну подгруппу из 13 для IFNα, и использование пассивно вводимых моноклональных антител может ограничиваться индукцией антител к лекарственному средству. Указанные антитела к лекарственному средству могут нейтрализовать или иным образом уменьшать клинический эффект указанного лекарственного средства, а также могут быть связаны с серьезными нежелательными явлениями ввиду перекрестной реактивности с аутологичными белками (De Groot et al. Trends. Immunol. 2007, 28(11)).
Таким образом, согласно настоящему изобретению предложен способ ингибирования активности IFNα in vivo путем введения терапевтически эффективного количества иммуногенного продукта, обеспечивающего активную иммунизацию, которая может нарушать иммунологическую толерантность В-клеток и образовывать высокие титры поликлональных нейтрализующих антител к IFNα, и его применение для лечения связанных с IFNα состояний.
КРАТКОЕ ОПИСАНИЕ
Одним из объектов настоящего изобретения является иммуногенный продукт, содержащий IFNα, соединенный с молекулой белка-носителя, для применения в предотвращении или лечении связанного с IFNα состояния у нуждающегося в этом субъекта, при этом терапевтически эффективное количество такого иммуногенного продукта для введения указанному субъекту составляет более 30 мкг иммуногенного продукта на одно введение, предпочтительно, по меньшей мере 60 мкг.
В одном из вариантов реализации настоящего изобретения введение указанного терапевтически эффективного количества иммуногенного продукта предотвращает появление симптомов заболевания, связанного с чрезмерной выработкой IFNα.
В другом варианте реализации настоящего изобретения введение указанного терапевтически эффективного количества иммуногенного продукта предотвращает обострение заболевания, связанного с чрезмерной выработкой IFNα.
В другом варианте реализации настоящего изобретения связанные с IFNα состояния включают системную красную волчанку, ревматоидный артрит, склеродермию, синдром Шегрена, васкулит, ВИЧ, диабет типа I, аутоиммунный тиреоидит и миозит.
В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта для введения субъекту составляет от 35 мкг до 1000 мкг иммуногенного продукта на одно введение, предпочтительно от 60 мкг до 1000 мкг.
В другом варианте реализации настоящего изобретения иммуногенный продукт вводят указанному субъекту по меньшей мере дважды в месяц.
В другом варианте реализации настоящего изобретения иммуногенный продукт дополнительно вводят указанного субъекту по меньшей мере один раз каждые три месяца.
В другом варианте реализации настоящего изобретения иммуногенный продукт дополнительно вводят субъекту, когда в образце сыворотки крови, полученном у субъекта, количество антител анти-IFNα является недетектируемым.
В другом варианте реализации настоящего изобретения указанный иммуногенный продукт является сильно инактивированным, что означает, что указанный продукт демонстрирует менее 5% противовирусной активности в условиях ТЕСТА В.
В другом варианте реализации настоящего изобретения иммуногенный продукт способен нейтрализовать противовирусную активность IFNα в условиях ТЕСТА С.
В другом варианте реализации настоящего изобретения иммуногенный продукт содержит по меньшей мере один подтип IFNα.
В другом варианте реализации настоящего изобретения указанный подтип IFNα представляет собой IFNα 2b, и молекула белка-носителя представляет собой гемоцианин фиссуреллы (KLH).
В другом варианте реализации настоящего изобретения иммуногенный продукт представляет собой вакцину, предпочтительно в форме эмульсии.
Другим объектом настоящего изобретения является единичная лекарственная форма, содержащая более 30 мкг иммуногенного продукта, содержащего IFNα, соединенный с молекулой белка-носителя, определенной выше.
Другой целью настоящего изобретения является медицинское устройство, содержащее более 30 мкг иммуногенного продукта, содержащего IFNα, соединенный с молекулой белка-носителя, определенной выше.
Другой целью настоящего изобретения является набор, содержащий по меньшей мере один пузырек, содержащий более 30 мкг, предпочтительно по меньшей мере 60 мкг иммуногенного продукта, содержащего IFNα, соединенный с молекулой белка-носителя, определенной выше, по меньшей мере один пузырек, содержащий адъювант, и средства для приведения указанного иммуногенного продукта в контакт с адъювантом и для эмульгирования смеси водного раствора с адъювантом.
В одном из вариантов реализации указанный набор согласно настоящему изобретению содержит:
- по меньшей мере один пузырек, содержащий более 30 мкг, предпочтительно по меньшей мере 60 мкг иммуногенного продукта, содержащего IFNα, соединенный с молекулой белка-носителя, согласно настоящему изобретению и средства для солюбилизирования указанного иммуногенного продукта, предпочтительно в водном растворе, или
- по меньшей мере один пузырек, содержащий раствор, предпочтительно водный раствор, содержащий более 30 мкг, предпочтительно по меньшей мере 60 мкг иммуногенного продукта, содержащего IFNα, соединенный с молекулой белка-носителя, согласно настоящему изобретению и
- по меньшей мере один пузырек, содержащий адъювант, и средства для приведения указанного раствора в контакт с адъювантом и для эмульгирования смеси раствора с адъювантом.
ОПРЕДЕЛЕНИЯ
В настоящем описании термин «интерферон α» или «IFNα» относится к белкам IFN-альфа, кодируемым функциональным геном генного локуса интерферона-альфа с идентичностью последовательности с IFN-альфа 1 75% или выше (номер GenBank NP_076918 или белок, кодируемый номером Genbank NM_024013). Примеры подтипов IFN-альфа человека включают IFN-альфа 1 (номер GenBank NP_076918), альфа 2а (номер GenBank ITF_A), альфа 2b (номер GenBank AAP20099), альфа 4 (номер GenBank NP_066546), альфа 5 (номер GenBank P01569), альфа 6 (P05013), альфа 7 (номер GenBank Р01567), альфа 8 (номер GenBank Р32881), альфа 10 (номер GenBank Р01566), альфа 14 (номер GenBank Р01570), альфа 16 (номер GenBank NP_002164), альфа 17 (номер GenBank Р01571) и альфа 21 (номер GenBank NP_002166). Примеры подтипов IFNα млекопитающих, не относящихся к человеку, могут быть найдены в Genbank, как хорошо известно специалисту в данной области техники (для ознакомления см. Pestka et al Immunological reviews 2004, 202: 8-32).
В настоящем описании термин «иммунный ответ» относится к действию, например, лимфоцитов, антигенпредставляющих клеток, фагоцитарных клеток и макромолекул, производимому вышеуказанными клетками или печенью (включая антитела, цитокины и комплемент).
В настоящем описании антитело, которое «ингибирует биологическую активность» или «нейтрализует биологическую активность» IFNα относится к антителу, ингибирующему активность цитокина по меньшей мере на 10%, 20%, 30%, 40 %, 50%, 60%, 70% или 80%, или более по сравнению с уровнем активности цитокина в отсутствии указанного антитела, например, при использовании функционального анализа, такого как анализы, описанные в разделе «Примеры».
В настоящем описании термин «молекула белка-носителя» относится к белку или пептиду длиной по меньшей мере 15 аминокислот, который при частичном ковалентном связывании с молекулой IFNα для образования гетерокомплексов, обеспечивает презентирование большого количества антигенов IFNα В-лимфоцитам.
В настоящем описании термин «субъект» включает любых млекопитающих, относящихся или не относящихся к человеку, таких как приматы, собаки, кошки, лошади, овцы…
В настоящем описании термин «пациент» относится к субъекту, пораженному связанным с IFNα состоянием.
В настоящем описании термин «эффективное количество» относится к количеству, достаточному для того, чтобы вызвать полезный или желаемый клинический результат (например, улучшение клинического состояния).
В настоящем описании термин «лечение» или «осуществление лечения» относится к клиническому вмешательству в попытке изменить естественное течение заболевания у субъекта или пациента, которого лечат, и может осуществляться либо для профилактики, либо при клинической патологии. Желаемые эффекты включают, но не ограничиваются ими, предотвращение возникновения или рецидива заболевания, облегчение симптомов, подавление, уменьшение или ингибирование каких-либо прямых или косвенных патологических последствий заболевания, снижение скорости прогрессирования заболевания, облегчение или ослабление болезненного состояния и вызов ремиссии, поддержание состояния ремиссии или улучшенный прогноз.
ПОДРОБНОЕ ОПИСАНИЕ
Хотя регуляторы IFNα встречаются в природе в организме, их способность регулировать уровни цитокинов при таких заболеваниях, как СКВ и СШ, по-видимому, недостаточна. Задачей терапевтический иммунизации против IFNα согласно настоящему изобретению является повышение уровней антител против указанного цитокинам при одновременном усилении их аффинности и нейтрализующей активности, что приводит к уменьшению избыточного количества цитокина и ингибированию его патологического действия без нарушения других метаболических и физиологических процессов.
Одной из целей настоящего изобретения является способ лечения связанного с IFNα состояния у нуждающегося в этом субъекта, включающий введение указанному субъекту терапевтически эффективного количества иммуногенного продукта, содержащего IFNα, соединенный с молекулой белка-носителя, при этом указанное терапевтически эффективное количество составляет более 30 мкг иммуногенного продукта на одно введение.
В одном из вариантов реализации настоящего изобретения указанное терапевтически эффективное количество составляет по меньшей мере 60 мкг иммуногенного продукта на одно введение.
В одном из вариантов реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 1000 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 750 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 500 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 450 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 400 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 350 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 300 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 250 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 200 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 150 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от более 30 мкг, предпочтительно более 60 мкг до 100 мкг.
В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 1000 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 750 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 500 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 450 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 400 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 350 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 300 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 250 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 200 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 150 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 35 мкг до 100 мкг.
В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 1000 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 750 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 500 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 450 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 400 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 350 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 300 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 250 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 240 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 200 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 150 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 120 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 100 мкг.
В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390 мкг до 400 мкг.
В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 240 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет 60 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет 120 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет 240 мкг.
В одном из вариантов реализации терапевтически эффективное количество соответствует общему количеству белка, определенному с использованием анализа способом Бредфорда, хорошо известного в данной области техники.
В одном из вариантов реализации настоящего изобретения субъекту, которого лечат, вводят по меньшей мере дважды в месяц терапевтически эффективное количество иммуногенного продукта, описанного выше.
В другом варианте реализации настоящего изобретения субъекту, которого лечат, вводят два раза в месяц терапевтически эффективное количество иммуногенного продукта, описанного выше. В данном варианте реализации субъекту можно вводить один раз в день 0, а второй раз между днем 7 и днем 28. В другом варианте реализации субъекту можно вводить один раз в день 0, а второй раз между днем 7 и днем 21. В одном из вариантов реализации субъекту вводят один раз в день 0, а второй раз в день 28.
В другом варианте реализации настоящего изобретения субъекту, которого лечат, вводят три раза в месяц терапевтически эффективное количество иммуногенного продукта, описанного выше. В данном варианте реализации субъекту, которого лечат, можно вводить один раз в день 0, второй раз между днем 7 и днем 14, и третий раз между днем 21 и днем 28. В одном из вариантов реализации субъекту вводят один раз в день 0, второй раз в день 7 и третий раз в день 28.
В другом варианте реализации настоящего изобретения субъекту, которого лечат, вводят четыре раза в течение 3 месяцев терапевтически эффективное количество иммуногенного продукта, описанного выше. В данном варианте реализации субъекту, которого лечат, можно вводить один раз в день 0, второй раз между днем 7 и днем 14, третий раз между днем 21 и днем 28, и четвертый раз между днем 77 и днем 84. В одном из вариантов реализации субъекту вводят один раз в день 0, второй раз в день 7, третий раз в день 28 и четвертый раз в день 84.
В другом варианте реализации настоящего изобретения субъекту, которого лечат, также можно вводить один раз в три месяца терапевтически эффективное количество иммуногенного продукта, описанного выше.
В одном из вариантов реализации настоящего изобретения субъекту, которого лечат, вводят три раза в течение одного месяца, как описано выше, а затем дополнительно вводят один раз каждые три месяца терапевтически эффективное количество иммуногенного продукта, описанного выше.
В одном из вариантов реализации настоящего изобретения субъекту, которого лечат, вводят четыре раза в течение трех месяцев, как описано выше, а затем дополнительно вводят один раз в три месяца терапевтически эффективное количество иммуногенного продукта, описанного выше.
В другом варианте реализации настоящего изобретения субъекту, которого лечат, также можно вводить один раз в шесть месяцев терапевтически эффективное количество иммуногенного продукта, описанного выше.
В одном из вариантов реализации настоящего изобретения субъекту, которого лечат, вводят три раза в течение одного месяца или четыре раза в течение трех месяцев, как описано выше, а затем дополнительно вводят один раз в шесть месяцев терапевтически эффективное количество иммуногенного продукта, описанного выше.
В другом варианте реализации настоящего изобретения субъекту, которого лечат, можно дополнительно вводить один раз в год терапевтически эффективное количество иммуногенного продукта, описанного выше.
В одном из вариантов реализации настоящего изобретения субъекту, которого лечат, вводят три раза в течение одного месяца или четыре раза в течение трех месяцев, как описано выше, а затем дополнительно вводят один раз каждый год терапевтически эффективное количество иммуногенного продукта, описанного выше.
В другом варианте реализации настоящего изобретения субъекту, которого лечат, можно дополнительно вводить один раз каждые 5 лет терапевтически эффективное количество иммуногенного продукта, описанного выше.
В одном из вариантов реализации настоящего изобретения субъекту, которого лечат, вводят три раза в течение одного месяца или четыре раза в течение трех месяцев, как описано выше, а затем дополнительно вводят один раз каждые 5 лет терапевтически эффективное количество иммуногенного продукта, описанного выше.
В другом варианте реализации настоящего изобретения субъекту, которого лечат, можно дополнительно вводить один раз каждые 10 лет терапевтически эффективное количество иммуногенного продукта, описанного выше.
В одном из вариантов реализации настоящего изобретения субъекту, которого лечат, вводят три раза в течение одного месяца или четыре раза в течение трех месяцев, как описано выше, а затем дополнительно вводят один раз в 10 лет терапевтически эффективное количество иммуногенного продукта, описанного выше.
В другом варианте реализации настоящего изобретения субъекту, которого лечат, дополнительно можно вводить терапевтически эффективное количество иммуногенного продукта, описанного выше, когда количество антител к IFNα не детектируется в образце сыворотки крови, полученном у субъекта.
В одном из вариантов реализации настоящего изобретения субъекту, которого лечат, вводят три раза в течение одного месяца или четыре раза в течение трех месяцев, как описано выше, а затем дополнительно вводят терапевтически эффективное количество иммуногенного продукта, описанного выше, когда количество антител к IFNα не детектируется в образце сыворотки крови, полученном у субъекта.
Определение количества антител к IFNα в образце сыворотки крови можно осуществлять традиционными способами, известными в данной области техники, такими как твердофазный иммуноферментный анализ (ELISA) анти-IFN.
Одним из примеров осуществления такого способа является следующий:
- нанесение на 96-луночный планшет 100 нг подтипа IFNα, используемого для получения иммуногенного продукта, такого как IFNα-2b и инкубирование указанного планшета в течение ночи при 2°С-8°С,
- блокирование планшета блокирующим буфером в течение 90 минут при 37°С,
- инкубирование планшета с образцом сыворотки крови и пулом интактного образца в течение 90 минут при 37°С: образец сыворотки крови, как правило, разводят в серии разведений 1:2, начиная с разведения 200х до по меньшей мере 8 разведений,
- инкубирование планшета совместно с меченым вторичным антителом, таким как козье антитело к иммуноглобулину человека, конъюгированное с пероксидазой хрена (HRP),
- проявление комплекса с помощью субстратного раствора o-фенилендиамина дигидрохлорида (OPD). После остановки ферментативной реакции интенсивность полученного цвета определяют спектрофотометрическими способами при 492 нм. Титр анти-IFN для каждого образца выражают в виде минимального разведения,
для которого среднее значение оптической плотности (OD) выше порогового значения:
Пороговое значение = средняя OD пула интактной сыворотки × 2,08 где пороговое значение N равно 2,08.
Затем титр анти-IFN для каждого образца будет выражен в виде минимального разведения, для которого среднее значение OD выше порогового значения. Первое разведение составляет 200, пациенты считаются отрицательными, если их OD при 1/200 ниже порогового значения (Mire-Sluis et al. 2004 J. Immunol Meth. 289: 1-16).
В одном из вариантов реализации настоящего изобретения субъект, которого лечат, страдает связанным с IFNα состоянием.
В другом варианте реализации настоящего изобретения субъект, которого лечат, демонстрирует недетектируемое количество антител анти-IFNα в сыворотке крови.
[Механизм действия]
Настоящее изобретение также относится к иммуногенному продукту, который подходит для индукции иммунного ответа у млекопитающего, которому вводят указанный иммуногенный продукт, включая гуморальный иммунный ответ, при котором антитела нейтрализуют иммуносупрессивные, апоптотические или ангиогенные свойства эндогенного цитокина IFNα.
Настоящее изобретение также относится к способу индукции иммунного ответа у нуждающегося в этом млекопитающего, при этом указанный способ включает введение иммуногенного продукта, описанного выше, указанному млекопитающему. В одном из вариантов реализации указанный иммунный ответ включает гуморальный иммунный ответ, при котором индуцируются антитела, нейтрализующие иммуносупрессивные, апоптотические или ангиогенные свойства эндогенного цитокина.
В одном из вариантов реализации настоящего изобретения иммуногенный продукт является инактивированным, но иммуногенное производное цитокина IFNα химически соединено со стимулирующим Т-хелперы чужеродным белком-носителем, таким как, например KLH. Указанный иммуногенный продукт обладает способностью нарушать толерантность В-клеток, но не Т-клеток в отношении IFNα. Толерантность Т-клеток-хелперов преодолевается путем связывания IFNα с чужеродным белком-носителем.
В-клетки, специфичные в отношении IFNα, активируются после связывания антигена и эндоцитоза иммуногенного продукта, и специфичные в отношении носителя пептиды представляются через молекулы II класса главного комплекса гистосовместимости (МНС). Данный сигнал активации не является достаточным для индукции дифференцировки В-клеток в случае Т-зависимого антигена, но поскольку В-клетки процессируют аутоантигены и антигены носителя, помощь Т-клеткам может быть оказана Т-клетками, специфичными в отношении собственного белка или белка-носителя. Поскольку Т-клеточный отбор является очень строгим, не происходит специфичной активации Т-клеток для аутоантигена.
Дендритные клетки (ДК) также могут захватывать аутоантиген и молекулу-носитель, и представлять специфичные в отношении носителя пептиды через молекулы II класса МНС. Таким образом, ДК способны активировать интактные Т-клетки-хелперы, специфичные в отношении носителя. Т-клетки-хелперы, в свою очередь, способны обеспечивать специфичными в отношении носителя Т-клетками-хелперами В-клетки, специфичные в отношении аутоантигена, и представлять пептиды носителя на молекулах II класса МНС.
Т-клетки-хелперы, специфичные в отношении носителя, взаимодействуют с В-клетками, специфичными в отношении аутоантигена, что вызывает нормальный гуморальный иммунный ответ на аутоантиген.
Иммуногенный продукт главным образом используют в композициях вакцин для лечения заболевания, связанного с чрезмерной выработкой IFNα.
В частности, настоящее изобретение относится к способу лечения заболевания, связанного с чрезмерной выработкой IFNα, включающему этап введения субъекту терапевтически эффективного количества иммуногенного продукта согласно настоящему изобретению.
Настоящее изобретение также относится к способу лечения заболевания, связанного с чрезмерной выработкой IFNα, включающему введение терапевтически эффективного количества иммуногенного продукта, при этом введение иммуногенного продукта предотвращает появление симптомов указанного заболевания.
Настоящее изобретение также относится к способу лечения заболевания, связанного с чрезмерной выработкой IFNα, включающему введение терапевтически эффективного количества иммуногенного продукта, при этом введение иммуногенного продукта предотвращает обострение указанного заболевания.
Настоящее изобретение также относится к способу лечения заболевания, связанного с чрезмерной выработкой IFNα, включающему введение терапевтически эффективного количества иммуногенного продукта, при этом введение иммуногенного продукта индуцирует выработку антител, нейтрализующих активность эндогенного IFNα.
Настоящее изобретение также относится к способу лечения заболевания, связанного с чрезмерной выработкой IFNα, включающему введение терапевтически эффективного количества иммуногенного продукта, при этом введение иммуногенного продукта индуцирует нейтрализацию активности эндогенного IFNα.
Примеры заболевания, связанного с чрезмерной выработкой IFNα, включают, но не ограничиваются ими, системную красную волчанку, ревматоидный артрит, склеродермию, синдром Шегрена, васкулит, ВИЧ, диабет типа I, аутоиммунный тиреоидит и миозит.
Другая цель настоящего изобретения заключается в способе индукции выработки антител, нейтрализующих активность эндогенного IFNα, у субъекта, включающем этап введения указанному субъекту терапевтически эффективного количества иммуногенного продукта.
[Иммуногенный продукт]
Иммуногенный продукт, который применяют в соответствии с настоящим изобретением, содержит IFNα, соединенный с молекулой белка-носителя, такой как KLH, при этом указанный иммуногенный продукт является инактивированным.
Иммуногенный продукт, который применяют в соответствии с настоящим изобретением, представляет собой комплекс между по меньшей мере одним рекомбинантным подтипом IFNα и по меньшей мере одной молекулой белка-носителя, такой как, например, KLH, полученный путем конъюгации глутаральдегидом и последующей инактивации формальдегидом.
В одном из вариантов реализации настоящего изобретения молекула белка-носителя может представлять собой любую молекулу-носитель, обычно используемую в иммунологии, такую как KLH (гемоцианин фиссуреллы), овальбумин, бычий сывороточный альбумин (BSA), столбнячный анатоксин, дифтерийный анатоксин, холерный токсин В, мутантный нетоксичный дифтерийный токсин (CRM 197), белок наружной мембраны менингококка в везикулах наружной мембраны, белок наружной мембраны нетипируемой гемофильной палочки, токсин А синегнойной палочки, вирусоподобная частица (VLP)… В одном из предпочтительных вариантов реализации указанный носитель представляет собой KLH. Предпочтительно, исходный продукт KLH состоит из высокоочищенного KLH, экстрагированного из лимфы морского брюхоногого моллюска Megathura cremulata. Полученный естественным образом KLH, как правило, состоит из структуры ди-декамера, которая представляет собой нековалентный трубчатый ансамбль, состоящий из 20 субъединиц.
В другом варианте реализации настоящего изобретения рекомбинантный подтип IFNα может представлять собой любой подтип из IFN альфа 1, альфа 2а, альфа 2b, альфа 4, альфа 5, альфа 6, альфа 7, альфа 8, альфа 10, альфа 14, альфа 16, альфа 17 и альфа 21.
Рекомбинантные подтипы IFNα могут быть получены традиционными способами, известными в данной области техники, с использованием последовательностей из Genbank, описанных выше. Например, получение рекомбинантного подтипа IFNα можно осуществлять путем культивирования клеток, содержащих вектор экспрессий, содержащий ген подтипа IFNα, а затем путем сбора телец включения и, наконец, очистки подтипа IFNα.
В одном из вариантов реализации настоящего изобретения рекомбинантный подтип IFNα представляет собой подтип IFNα 2b.
В одном из вариантов реализации настоящего изобретения иммуногенный продукт содержит по меньшей мере подтип IFNα 2b.
В одном из вариантов реализации настоящего изобретения рекомбинантный подтип IFNα находится в жидком растворе, предпочтительно буферном растворе, имеющем pH в диапазоне от 3,5, предпочтительно от 6 до 7,8.
В одном из вариантов реализации, когда субъект, которого лечат, представляет собой человека, используемый рекомбинантный IFNα является человеческим.
В одном из вариантов реализации настоящего изобретения иммуногенный продукт содержит IFNα, соединенный с молекулой белка-носителя, такой как, например KLH, при этом указанный иммуногенный продукт распознается антителом анти-IFNα.
Распознавание иммуногенного продукта антителом анти-IFNα может быть осуществлено традиционными способами, известными в данной области техники, такими как ELISA «сэндвич»-типа анти-IFNα/белок-носитель. ELISA (ТЕСТ D) проводят любыми колориметрическими способами, известными в данной области техники, такими как, например, с использованием детектирующего антитела, меченого биотином, системы амплификации поли-стрептавидин HRP и субстратного раствора о-фенилендиамина дигидрохлорида.
Одним из примеров указанного способа является следующий:
- нанесение на планшет захватывающего антитела, такого как, например, поликлональное антитело кролика анти-KLH,
- блокирование планшета блокирующим буфером (таким как 2% казеин в ФБР, например) в течение 90 минут при 37°С,
- инкубирование планшета с серией разведений иммуногенного продукта от 250 нг/мл до 8 двукратных разведений или с отрицательными контролями, такими как KLH и IFNα, в течение 90 минут при 37°С,
- инкубирование планшета совместно с детектирующим антителом, таким как, например, биотинилированное антитело анти-IFNα, в течение 90 минут при 37°С,
- инкубирование планшета совместно со стрептавидин-HRP в течение 30 минут при 37°С и проявление комплекса с помощью субстратного раствора о-фенилендиамина дигидрохлорида (OPD) в течение 30 минут. После остановки ферментативной реакции интенсивность полученного цвета определяют спектрофотометрическими способами при 490 нм.
Когда оптическая плотность лунок, содержащих иммуногенный продукт, по меньшей мере в 10 раз выше оптической плотности лунок, содержащих отрицательный контроль, специалистом в данной области техники считается, что иммуногенный продукт распознан антителом анти-IFNα и что IFNα в иммуногенном продукте соединен с KLH.
В другом варианте реализации настоящего изобретения иммуногенный продукт содержит IFNα, соединенный с молекулой белка-носителя, такой как, например, KLH, при этом указанный иммуногенный продукт является сильно иммуногенным, что означает, что указанный продукт способен индуцировать антитела анти-IFNα in vivo в условиях проводимого ниже ТЕСТА А.
Тест А осуществляют в соответствии со следующим способом:
От 0,3 до 10 мкг общего количества белка (определенных путем анализа способом Бредфорда) иммуногенного продукта вводят 6-8-недельным мышам линии Balb/с путем инъекции дважды в течение 30 дней, предпочтительно в 0 день и 21 день. Образец сыворотки крови получают до иммунизации (образец преиммунной сыворотки крови) и между 30 днем и 40 днем (образец тестируемой сыворотки крови), предпочтительно в 31 день. Проводят ELISA анти-IFNα, как объяснено выше.
Вкратце, на 96-луночные планшеты наносят 100 нг подтипа IFNα, используемого для получения иммуногенного продукта, такого как IFNα-2b, и инкубируют в течение ночи при 2°С-8°С. Затем планшет блокируют блокирующим буфером в течение 90 минут при 37°С. В лунки добавляют 100 мкл преиммунного образца в разведении 1/2500 и серии разведений от 1/2500 до 8 двукратных разведений образцов сыворотки (преиммунный и тестируемый). Наконец, в лунки добавляют меченое вторичное антитело к иммуноглобулинам мыши, такое как конъюгированное с HRP антитело, и проводят ELISA с использованием любых колориметрических способов, известных в данной области техники, таких как, например, с использованием субстратного раствора о-фенилендиамина дигидрохлорида.
Когда оптическая плотность лунок, содержащих образец тестируемой сыворотки крови, по меньшей мере в 2 раза выше оптической плотности лунок, содержащих образец преиммунной сыворотки крови, специалист в данной области техники считает, что указанный иммуногенный продукт является иммуногенным, что означает, что он индуцировал антитела анти-IFNα in vivo.
В другом варианте реализации настоящего изобретения иммуногенный продукт содержит IFNα, соединенный с молекулой белка-носителя, такой как, например, KLH, при этом IFNα является сильно инактивированным, что означает, что указанный продукт демонстрирует менее 5%, предпочтительно менее 1% противовирусной активности IFNα в условиях приведенного ниже ТЕСТА В. В одном из вариантов реализации иммуногенный продукт согласно настоящему изобретению в концентрации 500 нг/мл или больше демонстрирует менее 5%, предпочтительно менее 1% противовирусной активности IFNα в концентрации 500 нг/мл или больше в условиях ТЕСТА В.
Данный анализ основан на защитном действии IFNα от цитопатического эффекта (СРЕ) вируса везикулярного стоматита (VSV) в отношении клеток почек быка Мадин-Дарби (MDBK). Данный анализ также можно осуществлять с использованием клеток человека Нер-2С или А549 и вируса энцефаломиокардита (EMCV).
Тест В осуществляют в соответствии со следующим способом:
Иммуногенный продукт и рекомбинантный подтип IFNα, используемый для получения иммуногенного продукта (положительный контроль), разводят в количестве по меньшей мере 500 нг/мл и по меньшей мере 1000 ед/мл соответственно в базальной среде (RPMI с добавлением 2 мМ глутамина, 1 мМ пирувата натрия, 1 мМ Hepes). 50 мкл иммуногенного продукта и положительного контроля высевают в 96-луночный планшет и разводят в серии двукратных разведений в базальной среде. В каждую лунку добавляют 2 104 клеток MDBK в 50 мкл клеточной среды (RPMI с добавлением 4% FBS, 2 мМ глутамина, 1 мМ пирувата натрия и 1 мМ Hepes) и планшет инкубируют в течение ночи при 37°С, 5% CO2. Затем вирус разводят в базальной среде до по меньшей мере 10 TCID50 (доза, инфицирующая 50% культуры ткани: 10-кратное разведение для уничтожения 50% инфицированных клеток). Планшет опорожняют и добавляют 100 мкл разбавленного вируса. Затем планшет инкубируют в течение ночи при 37°С, 5% CO2.
В конце культивирования оценивают жизнеспособность клеток MDBK с использованием способов, хорошо известных в данной области техники. Одним из примеров указанных способов является следующий: в лунки добавляют раствор MTS/PMS (100 мкл MTS/5 мкл PMS; Promega G5430) 20 мкл/лунка и планшет инкубируют в течение еще 4 часов при 37°С, 5 % CO2. Затем планшет прочитывают при 490 нм на спектрофотометре.
Процент противовирусной активности рассчитывают следующим образом:
%противовирусной активности =[(ODПродукт-ODвирус)/средняя ODклетки-ODвирус)]*100
ODпродукт обозначает оптическую плотность лунки с иммуногенным продуктом или с положительным контролем (подтип IFNα).
ODвирус обозначает оптическую плотность контрольной лунки только с вирусом.
ODклетки обозначает оптическую плотность контрольной лунки с IFNα и вирусом.
Значение EC50, соответствующее количеству иммуногенного продукта, приводящему к 50% ингибированию опосредованной вирусом гибели, определяют путем интерполяции значения EC50 на ось х на графике жизнеспособность/концентрация.
Сравнение EC50 иммуногенного продукта и EC50 положительного контроля (рекомбинантный подтип IFNα, используемый для получения иммуногенного продукта) позволяет определить, демонстрирует ли иммуногенный продукт менее 5%, предпочтительно менее 1% противовирусной активности.
Может быть рассчитан фактор инактивации EC50 продукт/ECINFα: когда иммуногенный продукт демонстрирует менее 5%, предпочтительно менее 1% противовирусной активности, фактор инактивации составляет более 20, предпочтительно более 100.
В другом варианте реализации настоящего изобретения иммуногенный продукт содержит IFNα, соединенный с молекулой белка-носителя, такой как, например, KLH, при этом иммуногенный продукт способен нейтрализовать противовирусную активность IFNα в условиях приведенного ниже ТЕСТА С. В соответствии с настоящим изобретением данный анализ выполняют для оценки нейтрализующей способности сыворотки, полученной у мышей, иммунизированных иммуногенным продуктом. Нейтрализующая способность может быть оценена путем оценки жизнеспособности клеток в присутствии вируса везикулярного стоматита, реплицирующегося в клетках MDBK. Данный анализ также можно выполнять с использованием клеток человека Нер-2С и вируса EMCV.
Тест С осуществляют в соответствии со следующим способом:
От 0,3 до 10 мкг общего количества белка (определенных путем анализа способом Бредфорда) иммуногенного продукта вводят 6-8-недельным мышам линии Balb/с путем инъекции дважды в течение 30 дней, предпочтительно в 0 день и 21 день. Образец сыворотки крови получают до иммунизации (образец преиммунной сыворотки крови) и между 30 днем и 40 днем (образец тестируемой сыворотки крови), предпочтительно в 31 день.
25 мкл образцов преиммунной и тестируемой сыворотки крови высевают в 96-луночный планшет в разведении 1/200 до 8 разведений от 1/200. Положительный контроль (поликлональный анти-IFNα от PBL, Piscataway, NJ, кат. номер 31100-1), как правило, разводят для того, чтобы мог нейтрализовать активность IFNα от 3125 МЕ/лунка до 100 МЕ/лунка в базальной среде (RPMI с добавлением 2 мМ глутамина, 1 мМ пирувата натрия и 1 мМ Hepes) и 25 мкл также высевают в планшет.
В каждую лунку добавляют IFNα 25 ед./лунка (конечная концентрация) в 25 мкл базальной среды и планшет инкубируют в течение 60 минут при комнатной температуре.
В каждую лунку добавляют 20000 клеток MDBK в среде для анализа (RPMI с добавлением 4% FBS, 2 мМ глутамина, 1 мМ пирувата натрия, 1 мМ hepes) и планшет инкубируют в течение ночи при 37°С, 5% CO2.
Вирус разводят до по меньшей мере 10 TCIDso (10-кратное разведение для уничтожения 50% инфицированных клеток) в среде вируса (RPMI с добавлением 2 мМ . глутамина, 1 мМ пирувата натрия, 1 мМ hepes). Планшет опорожняют и в каждую лунку добавляют 100 мкл вируса перед инкубацией в течение 24 часов при 37°С, 5% CO2.
В конце культивирования оценивают жизнеспособность клеток MBDK с использованием способов, хорошо известных в данной области техники. Одним из примеров указанных способов является следующий: в лунки добавляют раствор MTS/PMS (100 мкл MTS/5 мкл PMS; Promega G5430) 20 мкл/лунка и планшет инкубируют в течение еще 4 часов при 37°С, 5 % CO2. Затем планшет прочитывают при 490 нм на спектрофотометре.
Относительную жизнеспособность клеток рассчитывают следующим образом:
%=[(ODобразец-ODвирус)/ODIFN+вирус)]*100
ODобразец обозначает оптическую плотность лунки с сывороткой, полученной у мыши, иммунизированной иммуногенным продуктом, или с положительным контролем (поликлональное антитело анти-IFN).
ODвирус обозначает оптическую плотность контрольной лунки только с вирусом.
ODIFN+вирус обозначает оптическую плотность контрольной лунки с IFNα и вирусом.
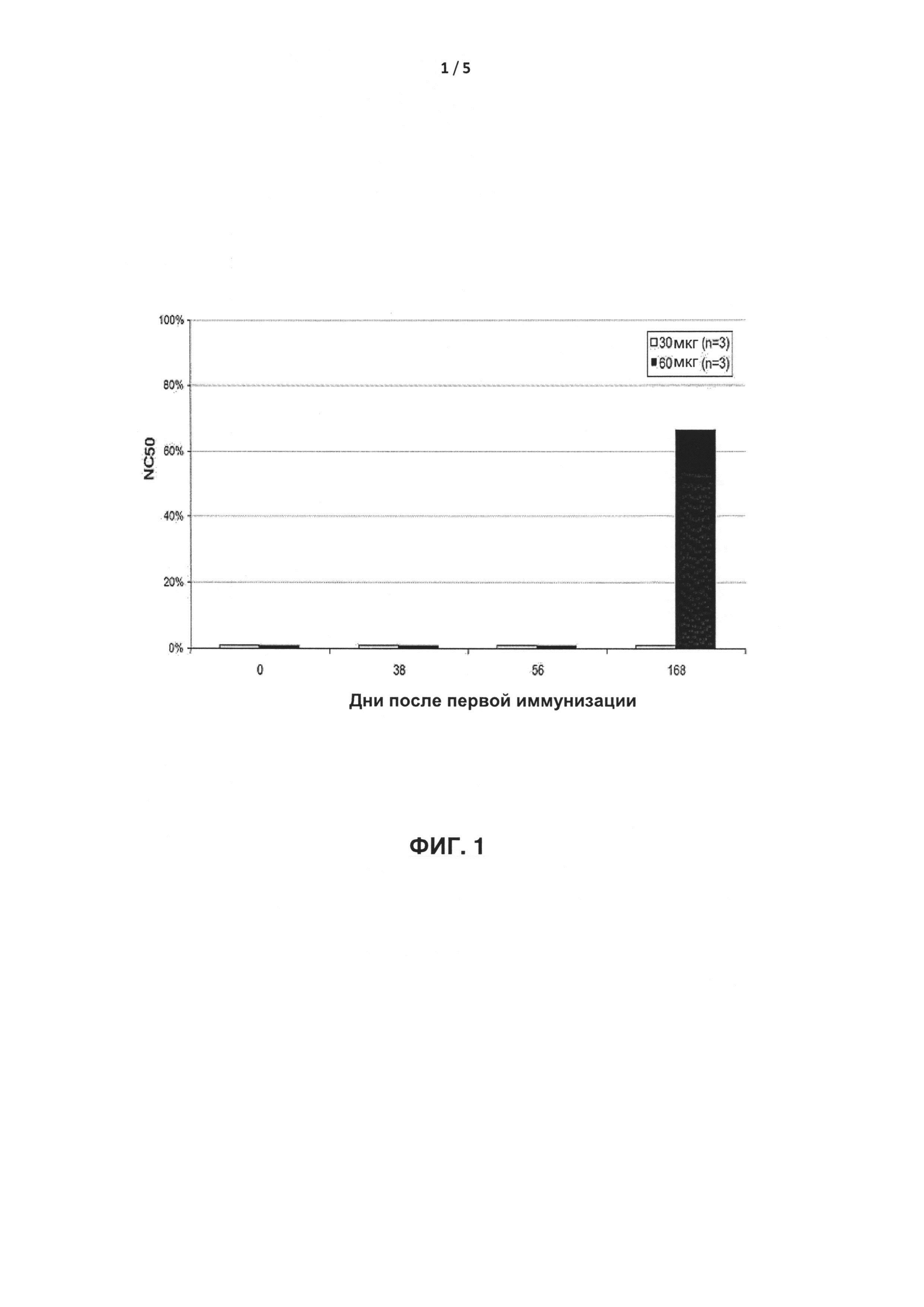
Значение NC50, соответствующее разведению сыворотки, приводящему к 50% нейтрализации опосредованной вирусом гибели, выраженное в виде фактора разведения или нейтрализующей единицы/мл, определяют путем интерполяции значения NC50 на ось x на графике жизнеспособность/концентрация.
В ТЕСТЕ С результат, демонстрирующий, что сыворотка, полученная у мыши, иммунизированной иммуногенным продуктом, не защищает клетки MBDK от гибели, означает, что иммуногенный продукт обладает способностью индуцировать антитела, направленные против IFNα, которые нейтрализуют его противовирусную активность.
В одном из вариантов реализации иммуногенный продукт содержит IFNα, соединенный с молекулой белка-носителя, такой как, например, KLH, при этом иммуногенный продукт способен нейтрализовать по меньшей мере 50% противовирусной активности IFNα в условиях ТЕСТА С. В указанном варианте реализации может быть рассчитано NC50. Если разведение сыворотки не способно нейтрализовать по меньшей мере 50% противовирусной активности IFNα в условиях ТЕСТА С, NC50 продукта не может быть рассчитано.
В одном из вариантов реализации настоящего изобретения иммуногенный продукт содержит IFNα, соединенный с молекулой белка-носителя, такой как, например, KLH, при этом соотношение IFNα/носитель по массе находится в диапазоне от 0,06 до 0,6.
В другом варианте реализации настоящего изобретения иммуногенный продукт содержит IFNα, соединенный с молекулой белка-носителя, такой как, например, KLH, при этом соотношение IFNα/носитель составляет от 0,1 до 0,5.
В другом варианте реализации настоящего изобретения иммуногенный продукт содержит IFNα, соединенный с молекулой белка-носителя, такой как, например, KLH, при этом соотношение IFNα/носитель составляет 0,3.
В другом варианте реализации настоящего изобретения иммуногенный продукт содержит IFNα, соединенный с молекулой белка-носителя, такой как, например, KLH, при этом соотношение IFNα/носитель составляет 0,05, 0,1, 0,2, 0,21, 0,22, 0,23, 0,24, 0,25, 0,26, 0,27, 0,28, 0,29, 0,3, 0,31, 0,32, 0,33, 0,34, 0,35, 0,36, 0,37, 0,38, 0,39, 0,4, 0,5.
Указанное соотношение может быть рассчитано в соответствии со способом, основанным на УФ-детектировании и детектировании флуоресценции (Тест Е), как описано в Примере 10.
[Способ получения иммуногенного продукта]
В одном из вариантов реализации настоящего изобретения киноид (kinoid) IFNα получают в соответствии со следующим способом
a) смешивание по меньшей мере одного рекомбинантного подтипа IFNα человека и по1 меньшей мере одной молекулы белка-носителя с глутаральдегидом и блокирование реакции путем добавления гасящего соединения, выбранного из (i) восстановителя и (ii) аминокислоты, выбранной из группы, состоящей из лизина и глицина, и их смеси,
b) удаление соединений, имеющих молекулярную массу менее 10 кДа или менее 8 кДа ,
c) добавление формальдегида;
d) блокирование реакции с формальдегидом путем добавления гасящего соединения, выбранного из (i) восстановителя и (ii) аминокислоты, выбранной из группы, состоящей из лизина и глицина, и их смеси,
e) сбор указанного иммуногенного продукта.
В одном из вариантов этапа а) сначала смешивают IFNα и молекулу белка-носителя, такую как, например, KLH в соответствующих количествах перед добавлением глутаральдегида.
В одном из вариантов реализации IFNα и KLH смешивают на этапе а) в молярном соотношении IFNα:субъединица KLH в диапазоне от 10:1 до 40:1. В другом варианте реализации IFNα и KLH смешивают на этапе а) в молярном соотношении IFNα:субъединица KLH в диапазоне от 15:1 до 25:1. В другом варианте реализации IFNα и KLH смешивают на этапе а) в молярном соотношении IFNα:субъединица KLH в диапазоне от 20:1 до 25:1.
В одном из вариантов этапа а) глутаральдегид используют в конечной концентрации в реакционной смеси в диапазоне от 1 мМ до 250 мМ, предпочтительно от 20 мМ до 30 мМ, более предпочтительно от 22,5 мМ до 25 мМ. В одном из вариантов этапа а) глутаральдегид инкубируют совместно с IFN α и KLH в течение периода времени в диапазоне от 15 минут до 120 минут, предпочтительно примерно 30, 35, 40, 45, 50, 60, 70, 80, 90 минут. В одном из вариантов реализации глутаральдегид добавляют в концентрации 22,5 мМ в течение примерно 45 минут. Предпочтительно, этап а) инкубации совместно с глутаральдегидом осуществляют при температуре в диапазоне от 18°С до 37°С, предпочтительно от 18°С до 27°С.
В соответствии с одним из вариантов реализации реакцию с глутаральдегидом (этап а) останавливают перед удалением соединений, имеющих молекулярную массу менее 10 кДа (этап b), путем добавления гасящего соединения, предпочтительно гасящего соединения, выбранного из (i) восстановителя и (ii) аминокислоты, выбранной из группы, состоящей из лизина и глицина, и их смеси.
Восстановитель может включать любой из восстановителей, известных в данной области техники, которые вследствие наличия восстановительных свойств обладают способностью восстанавливать оставшиеся иминные группировки, образованные при воздействии альдегидом. Восстановитель может быть выбран из группы, состоящей из борогидрида натрия, цианоборогидрид натрия.
В соответствии с одним из вариантов реализации в вариантах реализации, в которых указанное гасящее соединение представляет собой аминокислоту, указанная аминокислота включает глицин. В некоторых вариантах этапа b), в которых для блокирования реакции с глутаральдегидом используют глицин и/или лизин, выбранную аминокислоту используют в конечной концентрации в реакционной смеси в диапазоне от 0,01 М до 1 М, предпочтительно от 0,05 М до 0,5 М и наиболее предпочтительно от 0,08 М до 0,2 М, например, в концентрации 0,1 М, как показано в примерах в настоящем описании. В одном из вариантов реализации инкубацию совместно с гасящим соединением осуществляют в течение периода времени в диапазоне от 1 минуты до 120 минут, предпочтительно от 5 минут до 60 минут, например, в течение 30 минут, как показано в примерах в настоящем описании. В другом варианте реализации данный этап осуществляют при температуре в диапазоне от 18°С до 30°С, предпочтительно от 18°С до 25°С.
На этапе b) удаляют малые соединения массой менее 10 кДа, присутствующие в реакционной смеси. Данные малые соединения главным образом включают избыточный глутаральдегид и избыточные молекулы гасящего соединения, которые не прореагировали с IFN а и KLH. Этап Ь) может быть осуществлен в соответствии с любым известным способом, позволяющим удалять соединения массой менее 10 кДа; указанные способы включают диализ с помощью диализной мембраны с порогом отсечения 10 кДа или фильтрацию с использованием фильтрационной мембраны, имеющей порог отсечения 10 кДа. В качестве иллюстрации, этап b) может состоять из этапа тангенциальной поточной фильтрации с использованием фильтрационной мембраны, имеющей порог отсечения 10 кДа, как показано в примерах в настоящем описании. Ретентат фильтрации, свободный от нежелательных малых соединений, собирают в конце этапа b). При необходимости этап b) может включать предварительный этап удаления возможных агрегатов соединений, присутствующих в реакционной смеси, полученной в конце этапа b). Указанный предварительной этап может состоять из этапа обычной фильтрации для удаления агрегатов, в конечном счете, присутствующих в суспензии в жидком растворе, например, этапа фильтрации с использованием соответствующей фильтрационной мембраны, например, фильтрационной мембраны, имеющей размер пор 0,2 мкм.
В одном из вариантов этапа с) указанного способа формальдегид добавляют в конечной концентрации от 6 мМ до 650 мМ, предпочтительно от 25 мМ до 250 мМ. В одном из вариантов этапа с) указанного способа формальдегид добавляют в течение периода времени от 1 часа до 336 часов, предпочтительно от 1 часа до 144 часов. В одном из вариантов реализации формальдегид используют в конечной концентрации от 50 до 100 мМ, предпочтительно 66 мМ в течение от 20 до 50 часов, предпочтительно 40 часов.
На этапе с) инкубацию совместно с формальдегидом проводят предпочтительно при температуре в диапазоне от 30°С до 40°С, например, при 37°С, как показано в примерах в настоящем описании.
На этапе d) указанного способа реакцию с формальдегидом останавливают путем добавления гасящего соединения, предпочтительно гасящего соединения, выбранного из (i) восстановителя и (ii) аминокислоты, выбранной из группы, состоящей из лизина и глицина.
Восстановитель может включать любой из восстановителей, известных в данной области техники, которые вследствие наличия восстановительных свойств восстанавливают оставшиеся иминные группировки, образованные при воздействии альдегидом. Восстановитель может быть выбран из группы, состоящей из борогидрида натрия, цианоборогидрид натрия. В соответствии с одним из вариантов реализации в вариантах реализации, в которых указанное гасящее соединение представляет собой аминокислоту, указанная аминокислота включает глицин. В некоторых вариантах этапа b), в которых для блокирования реакции с формальдегидом используют глицин и/или лизин, выбранную аминокислоту используют в конечной концентрации в реакционной смеси в диапазоне от 0,01 М до 1,5 М, предпочтительно от 0,05 М до 1 М и наиболее предпочтительно от 0,1 М до 0,2 М, например, в концентрации 0,1 М, как показано в примерах в настоящем описании. В одном из вариантов реализации инкубацию совместно с гасящим соединением осуществляют в течение периода времени в диапазоне от 5 минут до 120 минут, предпочтительно от 10 минут до 60 минут, например, в течение 30 минут, как показано в примерах в настоящем описании. В другом варианте реализации данный этап осуществляют при температуре в диапазоне от 18°C до 30°C, предпочтительно от 18°C до 25°C.
В соответствии с одним из вариантов указанного способа, непосредственно перед сбором на этапе е) специалист в данной области техники может осуществлять удаление веществ, имеющих молекулярную массу менее 100 кДа, любым способом, известным в данной области техники для удаления веществ, имеющих молекулярную массу более 100 кДа, из жидкого раствора. В первом варианте реализации используемый способ представляет собой этап фильтрации, который осуществляют путем использования фильтрационной мембраны, имеющей порог отсечения по меньшей мере 100 кДа, который включает этап ультрафильтрации или этап тангенциальной фильтрации. Во втором варианте реализации используемый способ состоит из этапа тангенциальной фильтрации с использованием фильтрационной мембраны, имеющей порог отсечения по меньшей мере 100 кДа. В другом варианте реализации непосредственно перед сбором на этапе е) можно осуществлять удаление веществ, имеющих молекулярную массу менее 300 кДа, путем использования фильтрационной мембраны, имеющей порог отсечения по меньшей мере 300 кДа.
[Композиция, эмульсия и вакцина, содержащая такую эмульсию]
Настоящее изобретение относится к композиции, содержащей иммуногенный продукт, описанный выше. Настоящее изобретение также относится к составу продукта согласно настоящему изобретению, при этом указанный продукт находится в эмульсии. Предпочтительно, композиция вакцины согласно настоящему изобретению содержит или состоит из указанной эмульсии. Указанная эмульсия содержит иммуногенный продукт согласно настоящему изобретению, масло и поверхностно-активное вещество или смесь по меньшей мере одного масла и по меньшей мере одного поверхностно-активного вещества. Предпочтительно, масло или смесь масло/поверхностно-активное вещество представляет собой фармацевтически приемлемый наполнитель. Более предпочтительно, смесь масла и поверхностно-активного вещества представляет собой адъювант, еще более предпочтительно иммуноадъювант. Предпочтительным адъювантом является ISA 51. Другим примером иммуноадъюванта, который может быть использован, является SWE (эмульсия масло-в-воде на основе сквалена). Другим примером иммуноадъюванта, который может быть использован, является SWE-a (эмульсия масло-в-воде на основе сквалана). Эмульсия согласно настоящему изобретению может представлять собой эмульсию вода-в-масле или эмульсию масло-в-воде.
В другом варианте реализации количество иммуногенного продукта согласно настоящему изобретению составляет более 0,01% (масс/масс.) и менее 1% (масс/масс.) от общей массы указанной эмульсии.
[Адъюванты]
Эмульсия или композиция вакцины согласно настоящему изобретению может содержать адъювант, в частности, иммуноадъюванты. В одном из вариантов реализации количество адъюванта находится в диапазоне от 0,00001% (масс/масс.) до 1%, предпочтительно от 0,0001 до 0,1%, более предпочтительно от 0,001 до 0,01% (масс/масс.) от общей массы композиции вакцины.
В композиции вакцины, указанной выше, может быть использован любой подходящий адъювант, известный специалисту в данной области техники, включая адъюванты на основе масла, такие как, например, неполный адъювант Фрейнда, адъюванты на основе миколата (например, димиколат трегалозы), бактериальный липополисахарид (ЛПС), пептидогликаны (т.е. муреины, мукопептиды или гликопротеины, такие как N-Opaca, мурамилдипептид [МДП] или аналоги МДП), монофосфориллипид A (MPL), протеогликаны (например, экстрагированные из Палочки Фридлендера (Klebsiella pneumoniae)), стрептококковые препараты (например, OK432), Биостим.ТМ. (Biostim.TM.) (например, 01 K2), «Iscoms» из EP 109942, EP 180564 и EP 231039, гидроксид алюминия, сапонин, DEAE-декстран, нейтральные масла (такие как миглиол), растительные масла (такие как арахисовое масло), липосомы, полиолы Плюроник.RTM. (Pluronic.RTM.), систему адъювантов Ribi (см., например, GB-A-2 189 141) или интерлейкины, в частности, те, которые стимулируют иммунитет, опосредованный клетками. Альтернативный адъювант, состоящий из экстрактов Amycolata, рода бактерий в порядке Актиномицеты (Actinomycetales), был описан в патенте США №4877612. В качестве альтернативы, также можно использовать SWE (сквален, 3,9%, спан (span) 0,47%, твин (tween) 80 0,47% в цитратном буфере) и SWE-a (сквалан 3,9%, спан 0,47% твин 80 0,47% в цитратном буфере). Кроме того, коммерчески доступны запатентованные смеси адъювантов. Используемый адъювант, отчасти, будет зависеть от организма-реципиента. Количество адъюванта для введения будет зависеть от типа и размера животного. Оптимальные дозы могут быть легко определены обычными способами.
Масляные адъюванты, подходящие для применения в эмульсиях вода-в-масле, могут включать минеральные масла и/или метаболизируемые масла. Минеральные масла могут быть выбраны из Байола® (Bayol®), Маркола® (Marcol.®) и Дракеола (Drakeol), включая Дракеол® 6VR (SEPPIC, Франция).®. Метаболизируемые масла могут быть выбраны из масла SP масла (описанного ниже), Эмульсигена (Emulsigen) (MPV Laboratories, Ralston, NZ), Монтанида (Montanide) 264,266,26 (Seppic SA, Париж, Франция), а также растительных масел, таких как арахисовое масло и соевое масло, масел животного происхождения, таких как рыбий жир сквалан и сквален, и токоферол и его производные.
Кроме того, адъювант может содержать один или более смачивающих или диспергирующих агентов в количествах от примерно 0,1 до 25%, более предпочтительно от примерно 1 до 10% и еще более предпочтительно примерно от 1 до 3% от объема адъюванта. Наиболее предпочтительными в качестве смачивающих или диспергирующих агентов являются неионные поверхностно-активные вещества. Подходящие неионные поверхностно-активные вещества включают блок-сополимеры полиоксиэтилен/полиоксипропилен, особенно те, которые продаются под торговым знаком Плюроник® и поставляемые BASF Corporation (Mt. Olive, N.J.). Другие подходящие неионные поверхностно-активные вещества включают полиоксиэтиленовые сложные эфиры, такие как полиоксиэтиленсорбитанмоноолеат, доступный под торговым знаком Твин 80® или маннид моноолеат. Может быть желательным включение более одного, например, по меньшей мере двух смачивающих или диспергирующих агентов в адъювант в виде части композиции вакцины согласно настоящему изобретению.
Подходящие адъюванты могут содержать, но не ограничиваются ими, поверхностно-активные вещества, известные специалисту в данной области техники, такие как, например, гексадециламин, октадециламин, лизолецитин, диметилдиоктадециламмония бромид, N,N-диоктадецил-N-N'-бис(2-гидроксиэтил-пропандиамин), метоксигексадецилглицерин и полиолы плюроники; полианионы, например, пиран, сульфат декстрана, полиинозиновая-полицитидиловая кислота (poly IC), полиакриловая кислота, карбопол; пептиды, например, мурамилдипептид, диметилглицин, тафтсин, масляные эмульсии, квасцы и их смеси. Другие возможные адъюванты содержат пептидные субъединицы В термолабильного токсина E. coli, или холерного токсина. McGhee, J.R., et al., "On vaccine development," Sem. Hematol., 30: 3-15 (1993).
[Дополнительные поверхностно-активные вещества]
В вариантах композиции вакцины согласно настоящему изобретению, содержащей эмульсию, указанная композиция вакцины предпочтительно также содержит в дополнение к комбинации иммуногенного продукта и одного или более маслянистых иммуноадъювантных веществ, одно или более поверхностно-активных веществ. Типичные варианты поверхностно-активных веществ включают маннид моноолеат, такой как Монтанид® 80, продаваемый Арлацел (Arlacel) (SEPPIC, Франция).
В одном из вариантов реализации количество поверхностно-активного вещества находится в диапазоне от 0,00001% (масс/масс.) до 1%, предпочтительно от 0,0001 до 0,1%, более предпочтительно от 0,001 до 0,01% (масс/масс.) от общей массы композиции вакцины.
[Лиофилизированные продукты]
В соответствии с одним из вариантов реализации и для целей хранения продукт или композиция вакцины согласно настоящему изобретению могут быть лиофилизированы. Таким образом, композиции вакцин могут быть представлены в высушенной способом сублимации (лиофилизированной) форме. В указанном варианте реализации иммуногенный продукт согласно настоящему изобретению комбинируют с одним или более вспомогательными веществами для лиофилизации. Различные вспомогательные вещества для лиофилизации хорошо известны специалисту в данной области техники. Лиофилизация вспомогательных веществ включает сахара, такие как лактоза и маннит.
В таком варианте реализации, в котором композиция вакцины состоит из лиофилизированной композиции для применения в виде жидкой эмульсии, содержащей поверхностно-активное вещество, композиция вакцины предпочтительно содержит количество иммуногенного продукта согласно настоящему изобретению, составляющее более 0,1% (масс./масс.) и менее 10% (масс./масс.) от общей массы указанной композиции вакцины.
[Стабилизаторы]
В некоторых вариантах реализации вакцина может быть смешана со стабилизаторами, например, для защиты склонных к распаду белков от распада, для увеличения срока годности вакцины или для повышения эффективности сушки способом сублимации. Подходящими стабилизаторами являются, в том числе SPGA (Bovarnik et al; J. Bacteriology 59: 509 (1950)), углеводы, например, сорбит, маннит, трегалоза, крахмал, сахароза, декстран или глюкоза, белки, такие как альбумин или казеин, или продукты их распада, смеси аминокислот, таких как лизин или глицин, и буферы, такие как фосфаты щелочных металлов.
[Путь введения]
Композиции вакцины согласно настоящему изобретению могут быть введены иммунизируемому субъекту любым традиционным способом, в том числе путем инъекции, например, внутрикожной, внутримышечной, интраперитонеальной или подкожной инъекции; или путем местной доставки, такой как, например, трансдермальная доставка. Лечение может состоять из одной дозы или нескольких доз в течение определенного периода времени.
[Формы дозирования]
Формы, подходящие для применения путем инъекций, могут включать стерильные растворы или дисперсии и стерильные порошки для немедленного приготовления стерильных растворов или дисперсий для инъекций. Предотвращение загрязнения микроорганизмами может быть обеспечено путем добавления в композицию вакцины консервантов, таких как различные антибактериальные и противогрибковые агенты, например, парабены, хлорбутанол, фенол, сорбиновая кислота, тимеросал и т.п. Во многих случаях может быть предпочтительным включение изотонических агентов, например, Сахаров или хлорида натрия, для уменьшения боли во время инъекции. Пролонгированная абсорбция композиций для инъекций может быть обеспечена применением в указанных композициях агентов, замедляющих абсорбцию, например, моностеарата алюминия и желатина.
В соответствии с одним из вариантов реализации лиофилизированную композицию вакцины согласно настоящему изобретению солюбилизируют в воде для инъекции и осторожно перемешивают; затем добавляют иммуноадъювант, предпочтительно ISA 51; смесь осторожно перемешивают для эмульгирования и загружают в подходящий шприц. Таким образом, настоящее изобретение также относится к медицинскому устройству, включая шприц, заполненный или предварительно заполненный композицией вакцины согласно настоящему изобретению. В идеальном варианте эмульсию готовят непосредственно перед применением. Однако шприц, содержащий эмульсию, можно хранить менее 10 часов при 2-8°С. В данном случае эмульсии необходимо дать нагреться перед инъекцией потерев в руках.
[Диапазон единичных доз]
Другой целью настоящего изобретения является единица дозирования, содержащая количество иммуногенного продукта в диапазоне от более 30 мкг до 1000 мкг. В другом варианте реализации указанная единица дозирования содержит количество иммуногенного продукта в диапазоне от 35 мкг до 1000 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 35 мкг до 750 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 35 мкг до 500 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 35 мкг до 450 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 35 мкг до 400 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 35 мкг до 350 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 35 мкг до 300 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 35 мкг до 250 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 1000 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 750 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 500 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 450 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 400 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 350 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 300 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 250 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 240 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 200 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 150 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 120 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 100 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 35, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180,190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390 до 400 мкг.
В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 60 мкг до 240 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта в диапазоне от 60 мкг до 120 мкг.
В другом варианте реализации единица дозирования содержит количество иммуногенного продукта, составляющее 60 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта, составляющее 120 мкг. В другом варианте реализации единица дозирования содержит количество иммуногенного продукта, составляющее 240 мкг. [Набор и медицинское устройство]
Настоящее изобретение также относится к набору, содержащему:
1 пузырек (Флакон номер 1), содержащий иммуногенный продукт согласнонастоящему изобретению, как правило, объемом 3 мл;
1 пузырек (Флакон номер 2), содержащий адъювант, предпочтительно ISA51; данный пузырек может содержать 3 мл адъюванта и может представлять собой емкость объемом 8 мл; 1 шприц, как правило, 1 мл Braun Injekt-F®;
1 иглу (Игла номер 1) для приготовления эмульсии; данная игла предпочтительно представляет собой иглу 20G;
1 иглу (Игла номер 2) для инъекции, предпочтительно внутримышечной инъекции; данная игла предпочтительно представляет собой иглу 23G.
Настоящее изобретение также относится к способу приготовления вакцины из набора, включающему:
(1) набирание 0,4 мл адъюванта из Флакона номер 2. Выпускание содержимого данного шприца во Флакон номер 1, содержащий 0,4 мл иммуногенного продукта. (4) набирание и выпускание всего содержимого данного пузырька достаточное количество раз для эмульгирования содержимого, как правило, 30 раз и, наконец, набирание всей эмульсии.
Перед инъекцией Иглу номер 1 предпочтительно заменяют на Иглу номер 2 и из шприца выпускают воздух.
В одном из вариантов реализации указанный набор содержит:
1 пузырек (Флакон номер 1), содержащий 0,4 мл иммуногенного продукта согласно
настоящему изобретению;
1 пузырек (Флакон номер 2), содержащий по меньшей мере 0,4 мл адъюванта,
предпочтительно ISA51;
- 1 шприц, как правило, 1 мл Braun Injekt-F®;
- 1 иглу (Игла номер 1) для приготовления эмульсии; данная игла предпочтительно представляет собой иглу 20G;
- 1 иглу (Игла номер 2) для инъекции, данная игла предпочтительно представляет собой иглу 23G.
В другом варианте реализации иммуногенный продукт находится в лиофилизированной форме. Следовательно, указанный набор содержит:
- 1 пузырек (Флакон номер 1), содержащий лиофилизированный продукт согласно настоящему изобретению, как правило, объемом 3 мл;
- 1 пузырек (Флакон номер 2), содержащий воду для инъекции, как правило, объемом 2 мл;
- 1 пузырек (Флакон номер 3), содержащий адъювант, предпочтительно ISA51; данный пузырек может содержать 3 мл адъюванта и может представлять собой емкость объемом 8 мл; 1 шприц, как правило, 1 мл Braun Injekt-F®;
- 1 иглу (Игла номер 1) для приготовления эмульсии; данная игла предпочтительно представляет собой иглу 20G;
- 1 иглу (Игла номер 2) для инъекции, предпочтительно внутримышечной инъекции; данная игла предпочтительно представляет собой иглу 23G.
Настоящее изобретение также относится к способу приготовления вакцины из набора, включающему:
(1) впрыскивание воды для инъекций из Флакона номер 2 во Флакон номер 1 путем использования шприца, соединенного с Иглой номер 1;
(2) осторожное вращение Флакона номер 1 в течение 1-5 минут до полной солюбилизации препарата;
(3) с помощью того же шприца и иглы набирание адъюванта из Флакона номер 3. Выпускание содержимого данного шприца во Флакон номер 1
(4) набирание и выпускание всего содержимого данного пузырька достаточное количество раз для эмульгирования содержимого, как правило, 30 раз и, наконец, набирание всей эмульсии.
Настоящее изобретение также относится к медицинскому устройству, представляющему собой шприц, заполненный или предварительно заполненный композицией, эмульсией или вакциной согласно настоящему изобретению.
В одном из вариантов реализации указанный шприц представляет собой двухкамерный шприц, в котором одна камера содержит раствор с иммуногенным продуктом согласно настоящему изобретению, а другая камера содержит адъювант.
Настоящее изобретение также относится к медицинскому устройству, содержащему пузырек или карпулу, предварительно заполненные продуктом согласно настоящему изобретению или композицией вакцины согласно настоящему изобретению.
В одном из вариантов реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от более 30 мкг до 1000 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 35 мкг до 1000 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 35 мкг до 750 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 35 мкг до 500 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 35 мкг до 450 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 35 мкг до 400 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 35 мкг до 350 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 35 мкг до 360 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 35 мкг до 250 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 1000 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 750 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 500 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 450 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 400 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 350 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 300 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 250 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 240 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 200 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 150 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 120 мкг. В другом варианте реализации настоящего изобретения терапевтически эффективное количество иммуногенного продукта на одно введение составляет от 60 мкг до 100 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 35, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160,170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390 до 400 мкг.
В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 60 мкг до 240 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта в диапазоне от 60 мкг до 120 мкг.
В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта, составляющее 60 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта, составляющее 120 мкг. В другом варианте реализации медицинское устройство содержит количество иммуногенного продукта, составляющее 240 мкг.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1: Процент образцов сыворотки крови иммунизированных пациентов, демонстрирующих нейтрализующую активность в отношении IFNα в ходе промежуточного отчета.
Фиг.2: Дифференциальная эволюция генов, индуцированных IFN, у пациентов, получавших лечение, по сравнению с пациентами, получавшими плацебо. Из 11 пациентов, демонстрирующих повышенные уровни экспрессии генов, индуцированных IFN, в исходном состоянии, 8 получали лечение иммуногенным продуктом и 3 получали инъекции плацебо. Уровни 250 генов, индуцированных IFN, демонстрирующих самые высокие уровни сверхэкспрессии у пациентов с СКВ, оценивали с использованием микроматриц высокой плотности. Результаты представлены в виде среднего log2 (уровень экспрессии в V1) - log2 (уровень экспрессии в V6). Значение p рассчитывали с использованием t-критерия Стьюдента.
Фиг.3: Титры IFN-связывающих антител у пациентов, получающих лечение, с положительным или отрицательным IFN-признаком в исходном состоянии по сравнению с пациентами, получающими плацебо. Звездочки показывают значения p<0,05.
Фиг.4: Дифференциальная эволюция генов, индуцированных IFN, у пациентов, получающих лечение, с положительным или отрицательным IFN-признаком в исходном состоянии по сравнению с пациентами, получающими плацебо, между V10 и V0 или между V11 и V0. Результаты представлены в виде среднего Дельта Log (экспрессия генов). Звездочки показывают значения p<0,05.
Фиг.5: Изменение значений С3 в сыворотке крови у пациентов, получающих лечение, с положительным IFN-признаком в исходном состоянии и у пациентов, получающих плацебо. Звездочки показывают значения p<0,04.
ПРИМЕРЫ
ПРИМЕР 1: Получение иммуногенного продукта
Гемоцианин фиссуреллы (KLH) экстрагировали из лимфы морского брюхоногого моллюска Megathura cremulata, а затем очищали в условиях надлежащей производственной практики (GMP). Результаты анализов для определения стабильности, выполненных в условиях хранения при температуре 2-8°С, показали, что срок годности очищенного KLH составляет 36 месяцев при 2-8°С.
Рекомбинантный IFNα 2b человека получали в Е. coli в условиях GMP.
Партии продукта согласно настоящему изобретению в масштабе 350 мг IFNα получали с использованием способа изготовления, представленного ниже.
a) Конъюгация глутаральдегидом
Фильтрованный KLH добавляют к раствору IFNα 2b (IFNα 2b в 70 мМ гидрофосфате натрия pH 7,8) с соотношением IFNα:KLH 20:1 (соответствующем молярному соотношению 20 мономеров IFNα на 1 субъединицу KLH) на основе УФ-измерения концентрации.
Конъюгацию осуществляют с помощью глутаральдегида (добавляемого до достижения конечной концентрации 22,5 мМ в реакционной среде) и бората pH 9 (добавляемого до достижения конечной концентрации 28,5 мМ в реакционной среде) до получения pH, равного 8,5.
Затем данный раствор при pH 8,5 смешивают в течение 45 минут при 23±2°С.
b) Гашение глицином
Реакционную смесь гасят глицином 0,1 М в течение 30 минут.
c) Первая тангенциальная поточная фильтрация (TFF 1)
Первую TFF осуществляют с помощью системы TFF Pall Minim II и полиэфирсульфоновой мембраны 0,02 м2 с порогом отсечения по молекулярной массе 10 кДа, дезинфицированной 0,5 М NaOH и уравновешенной рабочим буфером (70 мМ гидрофосфат натрия pH 7,8).
Затем гашеный раствор осветляют путем 0,22 мкм-фильтрации. Промежуточный раствор дважды разводят в рабочем буфере, а затем подвергают диафильтрации путем тангенциальной поточной фильтрации (TFF) и с использованием 12 объемов рабочего буфера. Собирают ретентат и хранят в течение менее 20 часов.
d) Инактивация формальдегидом
Формальдегид добавляют к ретентату с достижением конечной концентрации 66,6 мМ с использованием перистальтического насоса. Реакцию инактивации проводят в течение 40 часов в инкубаторе с температурой, установленной на 37±2°С, с ежедневным перемешиванием раствора магнитной мешалкой.
e) Гашение глицином
Затем реакцию гасят 0,1 М глицина в течение 30 минут.
f) Вторая тангенциальная фильтрация (TFF 2)
Вторую TFF осуществляют с помощью системы TFF Pall Minim II и полиэфирсульфоновой мембраны 0,02 м2 с порогом отсечения по молекулярной массе 100 кДа, дезинфицированной 0,5 М NaOH и уравновешенной буфером, используемым для приготовления состава (70 мМ гидрофосфат натрия pH 7,8).
Гашеный раствор осветляют путем 0,2 мкм-фильтрации. Промежуточный раствор концентрируют до исходного тангенциального объема ≈900 мл, а затем фильтруют путем TFF 12 объемами буфера, используемого для приготовления состава (70 мМ фосфатный буфер) с удалением с низкомолекулярных гомополимеров IFNα и неактивных реагентов. Собирают ретентат, а затем разводят до теоретической концентрации 300 мкг/мл на основе определения концентрации путем анализа способом Бредфорда, а затем подвергают 0,2 мкм-фильтрации с получением иммуногенного продукта согласно настоящему изобретению.
ПРИМЕР 2: антигенность продукта
ELISA «сэндвич»-типа анти-IFNα/KLH осуществляли следующим образом. Вкратце, на 96-луночный планшет наносили захватывающее антитело: поликлональное антитело кролика анти-KLH и блокировали блокирующим буфером (таким как 2% казеин в ФБР, например) в течение 90 минут при 37°С. Планшет инкубировали в течение 90 минут при 37°С с серией разведений иммуногенного продукта от 250 нг/мл до 8 двукратных разведений или с отрицательными контролями, такими как KLH и IFNα. Затем добавляли детектирующее антитело, такое как, например, биотинилированное антитело анти-IFNα, в течение 90 минут. Наконец, планшет инкубировали совместно со стрептавидин-HRP в течение 30 минут при 37°С и комплекс проявляли с помощью субстратного раствора о-фенилендиамина дигидрохлорида (OPD) в течение 30 минут. После остановки ферментативной реакции интенсивность полученного цвета определяли спектрофотометрическими способами при 490 нм.
Данный тест подтвердил, что указанный продукт содержит IFNα, который является антигенным, т.е. распознается антителом анти-IFNα, и что указанный IFNα соединен с KLH.
ПРИМЕР 3: Иммуногенность продукта (ТЕСТ А)
4 мкг общего количества белка продукта, определенных путем анализа способом Бредфорда, вводили 7 6-8-недельным мышам линии Balb/с путем инъекции в 0 день и 21 день.
В 31 день у мышей забирали образцы крови и собирали сыворотки крови.
ELISA анти-IFNα осуществляли на преиммунных и собранных сыворотках крови следующим образом:
- на 96-луночный планшет наносили 100 нг IFNα-2b и инкубировали в течение ночи при 2°С-8°С;
- добавляли блокирующий буфер в течение 90 минут при 37°С,
- добавляли иммуногенный продукт в разведении 1/2500 до по меньшей мере 8 двукратных разведений и планшет инкубировали в течение 90 минут при 37°С,
- планшет инкубировали совместно с меченым антителом к иммуноглобулинам мыши, таким как антитело, конъюгированное с HRP, в течение 90 минут при 37°С,
- проводили ELISA с помощью субстратного раствора о-фенилендиамина дигидрохлорида (OPD). После остановки ферментативной реакции интенсивность полученного цвета определяли спектрофотометрическими способами при 490 нм. Данный тест показал, что у 7 мышей иммунизация иммуногенным продуктом приводила к наличию титров антител анти-IFNα.
ПРИМЕР 4: Остаточная активность продукта (ТЕСТ В)
Данный анализ был основан на защитном действии IFNα от цитопатического эффекта (СРЕ) вируса везикулярного стоматита (VSV) в отношении клеток почек быка Мадин-Дарби (MDBK).
Иммуногенный продукт и рекомбинантный IFNα 2b, используемый для получения иммуногенного продукта (положительный контроль), разводили в количестве по меньшей мере 500 нг/мл и по меньшей мере 1000 ед/мл соответственно в базальной среде (RPMI с добавлением 2 мМ глутамина, 1 мМ пирувата натрия, 1 мМ Hepes). 50 мкл иммуногенного продукта и положительного контроля высевали в 96-луночный планшет и разводили в серии двукратных разведений в базальной среде. В каждую лунку добавляли 2 104 клеток MDBK в 50 мкл клеточной среды (RPMI с добавлением 4% FBS, 2 мМ глутамина, 1 мМ пирувата натрия и 1 мМ Hepes) и планшет инкубировали в течение ночи при 37°С, 5% CO2. Затем вирус разводили в базальной среде до по меньшей мере 10 TCID50 (доза, инфицирующая 50% культуры ткани: 10-кратное разведение для уничтожения 50% инфицированных клеток). Планшет опорожняли и добавляли 100 мкл разбавленного вируса. Затем планшет инкубировали в течение ночи при 37°С, 5% CO2.
В конце культивирования в лунки добавляли раствор MTS/PMS (100 мкл MTS/5 мкл PMS; Promega G5430) 20 мкл/лунка и планшет инкубировали в течение еще 4 часов при 37°С, 5 % CO2. Затем планшет прочитывали при 490 нм на спектрофотометре.
Рассчитывали процент противовирусной активности иммуногенного продукта, и для двух партий тестируемого продукта противовирусная активность составляла менее 1% противовирусной активности IFNα.
ПРИМЕР 5: Нейтрализующая способность продукта (ТЕСТ С)
Нейтрализующую способность продукта оценивали путем оценки жизнеспособности клеток в присутствии вируса везикулярного стоматита, реплицирующегося в клетках MDBK.
4 мкг общегоколичства белка (определенных путем анализа способом Бредфорда) иммуногенного продукта вводили 6-8-недельным мышам линии Balb/с путем инъекции в 0 день и 21 день. Образец сыворотки крови получали до иммунизации (образец преиммунной сыворотки крови) и в 31 день (образец тестируемой сыворотки крови).
25 мкл образцов преиммунной и тестируемой сыворотки крови высевали в 96-луночный планшет в разведении 1/200 до 8 разведений от 1/200. Положительный контроль (поликлональный анти-IFNα от PBL, Piscataway, NJ, кат. номер 31100-1) разводили от 3125 МЕ/лунка до 100 МЕ/лунка в базальной среде (RPMI с добавлением 2 мМ глутамина, 1 мМ пирувата натрия и 1 мМ Hepes) и 25 мкл также высевали в планшет.
В каждую лунку добавляли IFNα 25 ед/лунка (конечная концентрация) в 25 мкл базальной среды и планшет инкубировали в течение 60 минут при комнатной температуре.
В каждую лунку добавляли 20000 клеток MDBK в среде для анализа (RPMI с добавлением 4% FBS, 2 мМ глутамина, 1 мМ пирувата натрия, 1 мМ hepes) и планшет инкубировали в течение ночи при 37°С, 5% CO2.
Вирус разводили до по меньшей мере 10 TCID50 (10-кратное разведение для уничтожения 50% инфицированных клеток) в среде вируса (RPMI с добавлением 2 мМ глутамина, 1 мМ пирувата натрия, 1 мМ hepes). Планшет опорожняли и в каждую лунку добавляли 100 мкл вируса перед инкубацией в течение 24 часов при 37°С, 5% CO2.
В конце культивирования в лунки добавляли раствор MTS/PMS (100 мкл MTS/5 мкл PMS; Promega G5430) 20 мкл/лунка и планшет инкубировали в течение еще 4 часов при 37°С, 5% CO2. Затем планшет прочитывали при 490 нм на спектрофотометре.
NC рассчитывали для всех 7 тестируемых образцов: средняя NC=253789 МЕ/мл (SEM=172526), что демонстрирует, что все сыворотки крови содержали антитела анти-IFNα, способные нейтрализовать противовирусную активность IFNα.
ПРИМЕР 6: Примеры композиций и вакцины, содержащей иммуногенный продукт
В Таблице 1 описана одна из типичных композиций, содержащих иммуногенный продукт.

В Таблице 2 описана одна из типичных вакцин, содержащих иммуногенный продукт.

ПРИМЕР 7: Клиническое исследование
Клиническое исследование проводили с использованием композиции вакцины, описанной в Таблице 2.
Дизайн исследования:
Осуществляли 3 или 4 введения продукта в 0 день, 7 день и 28 день или в 0 день, 7 день, 28 день и 84 день взрослым, подверженным СКВ.
Тестировали следующие дозы продукта: 30 мкг, 60 мкг, 120 мкг и 240 мкг. Популяция исследования:
28 пациентов мужского или женского пола в возрасте от 18 до 50 лет с СКВ со степенью выраженности от мягкой до умеренной (индекс активности СКВ (SLEDAI) 4-10), активным заболеванием несмотря на получение лечения. Нормальный контрольный признак гена интерферона устанавливали у 48 здоровых добровольцев. Мононуклеарные клетки периферической крови (МКПК) 18 из 48 здоровых добровольцев стимулировали in vitro интерферонами типа I для идентификации признака интерферона на матрицах высокой плотности. Признак СКВ устанавливали путем сравнения признаков здоровых добровольцев и пациентов с СКВ на исходном уровне.
Промежуточный анализ осуществляли у пациентов, включенных в первые три группы, т.е. получивших 30, 60 или 120 мкг дозы или плацебо.


Результаты
Безопасность и переносимость вакцины
В качестве связанных серьезных нежелательных явлений (SAEs) сообщали о двух обострениях волчанки. Первое произошло в группе плацебо. Второе произошло после первой инъекции 240 мкг IFN-K у пациента, который самопроизвольно прекратил терапию кортикостероидами через два дня после инъекции. Данное резкое прекращение лечения кортикостероидами, вероятно, участвовало в возникновении обострения. Регулярные промежуточные анализы безопасности проводил независимый комитет по безопасности. Не обнаруживали клинически значимого изменения лабораторных показателей (гематология, биохимия, моча).
Иммуногенность вакцины
Титры антител анти-IFNα измеряли путем ELISA в образцах сыворотки крови, полученных у пациентов.
ELISA анти-IFNα проводили, как описано выше.
Результаты показывают, что титры антител анти-IFNα детектировали во всех группах, получавших лечение иммуногенным продуктом, начиная с 28 дня. Нейтрализующая активность вакцины
Нейтрализующую активность оценивали in vitro с использованием следующего способа:
50 мкл образцов сыворотки крови, полученных из сывороток крови пациентов, высевали в 96-луночный планшет в разведении 1/200 до 8 разведений от 1/200.
Положительный контроль (поликлональный анти-IFN от PBL Piscataway, NJ, 31100-1) разводили от 100 нг/лунка до 3,125 нг/лунка и 50 мкл добавляли в планшет. Разведения осуществляли в базальной среде (RPMI с добавлением 2 мМ глутамина, 1 мМ пирувата натрия и 1 мМ hepes).
В каждую лунку добавляли IFNα 2b 10 ед/лунка (конечная концентрация) и планшет инкубировали в течение 60 минут при комнатной температуре.
В каждую лунку добавляли 30000 клеток MDBK в среде для анализа (RPMI с добавлением 4% FBS, 2 мМ глутамина, 1 мМ пирувата натрия, 1 мМ hepes) и планшет инкубировали в течение ночи при 37°С, 5% CO2.
Вирус разводили до по меньшей мере 10 TCID50 (10-кратное разведение для уничтожения 50% инфицированных клеток) в среде вируса (RPMI с добавлением 2 мМ глутамина, 1 мМ пирувата натрия, 1 мМ hepes). Планшет опорожняли и в каждую лунку добавляли 100 мкл вируса перед инкубацией в течение 24 часов при 37°С, 5% CO2.
В конце культивирования в лунки добавляли раствор MTS/PMS (100 мкл MTS/5 мкл PMS; Promega G5430) 20 мкл/лунка и планшет инкубировали в течение еще 4 часов при 37°С, 5% CO2. Затем планшет прочитывали при 490 нм на спектрофотометре.
Результаты промежуточного отчета показали, что ни в одной из сывороток крови у пациентов, получавших лечение 30 мкг иммуногенного продукта, не присутствовали антитела анти-IFNα, обладающие нейтрализующей способностью, в 168 день после иммунизации, тогда как в сыворотках крови у пациентов, получавших лечение 60 мкг иммуногенного продукта, присутствовали антитела анти-IFNα, обладающие нейтрализующей способностью, в 168 день (Фиг.1).
Более того, результаты конечного отчета показали, что нейтрализующую активность детектировали у 50% субъектов, получавших лечение 60 мкг или 120 мкг иммуногенного продукта, и у 80% субъектов, получавших лечение 240 мкг иммуногенного продукта (Таблица 5).

Данные результаты показали, что лечение более 30 мкг иммуногенного продукта необходимо для нейтрализации IFNα in vivo.
ПРИМЕР 8: Транскриптомный анализ
МКПК собирали в некоторые моменты времени до и после инъекции иммуногенного продукта. Для данного промежуточного анализа экстрагировали тотальную РНК в образцах V1 (0 день) и V6 (38 день после первой инъекции), обозначенных в соответствии со стандартным протоколом Affymetrix, и гибридизовали на матрицах Genechip HGU133 Plus 2.0. Статистический анализ выполняли на GeneSpring после нормализации образцов RMA (робастный анализ микроматриц).
Алгоритмы неконтролируемой кластеризации выполняли на исходных образцах и группировали пациентов на две категории: пациенты с (n=11) и пациенты без (n=7) наличия «признака интерферона» на исходном уровне, т.е. на спонтанной сверхэкспрессии генов, индуцированных интерфероном типа I (гены, индуцированные IFN, идентифицировали экспериментально на основе анализов микроматриц контрольных МКПК, стимулированных IFN). Не удивительно, что титры двухцепочечной ДНК (дцДНК) были значительно выше у пациентов с признаком (среднее значение +/-SEM: 131,1. +/-50,1 МЕ/мл) по сравнению с пациентами без указанного признака (среднее значение +/-SEM: 44,7 +/-33,3, p=0,006 по критерию Манна-Уитни). Измеримые антитела анти-IFNα обнаруживали в 8 образцах 8 пациентов при последующем наблюдении с признаком IFN на исходном уровне, которые получали иммуногенный продукт, тогда как это имело место только у 2 из 6 пациентов без признака IFN, получавших лечение иммуногенным продуктом, и ни у одного из 4 индивидуумов, получавших лечение плацебо (p=0,002 по критерию хи-квадрат). Из 11 пациентов с исходным IFN-признаком 2 получали дозу 30 мкг, 1 получал дозу 60 мкг, 5 получали дозу 120 мкг и 3 лечили инъекцией плацебо. Изменения, наблюдаемые в экспрессии генов, индуцированных IFN, между V1 и V6, значительно отличались у пациентов, получавших лечение иммуногенным продуктом по сравнению с пациентами, получавшими лечение плацебо (Фиг.2).
Данный результат позволяет предположить, что иммуногенный продукт влияет на экспрессию генов, индуцированных IFN, in vivo.
ПРИМЕР 9: признак IFN и ответ на лечение иммуногенным продуктом
«Признак интерферона» на исходном уровне измеряли у 28 пациентов с СКВ из Примера 7 со степенью выраженности от мягкой до умеренной. Признак интерферона (IFN-признак) соответствует показателю, рассчитанному с использованием 21 гена, индуцированного IFN, и был описан в Yao et al, Arthritis & Rheumatism, 2009, 60 (6): 1785-1796. Измерение признака интерферона проводили, как описано в Примере 8.
Из 28 пациентов с СКВ из Примера 7, 19 демонстрировали положительный признак интерферона и 9 - отрицательный признак интерферона на исходном уровне.
Признак интерферона и активность заболевания СКВ
Измеряли титры антител к дцДНК и уровни СЗ в сыворотке крови в качестве показателей активности заболевания в обеих группах пациентов.
Титры антител к дцДНК определяли с использованием набора DPC Anti-DNA (PIKADD-4) от Diagnostic Products Corporation.
Уровни С3 в сыворотке крови определяли с использованием набора Complement С3 (набор # 446450) от Beckman Coulter.

Как показано в Таблице 6 выше, пациенты с СКВ с положительным признаком интерферона на исходном уровне демонстрируют биологические показатели более высокой активности заболевания.
Признак интерферона и ответ на лечение иммуногенным продуктом согласно настоящему изобретению
Эффекты лечения иммуногенным продуктом согласно настоящему изобретению, описанного в Примере 7, сравнивали у пациентов с СКВ с положительным и отрицательным IFN-признаком на исходном уровне.
Ответ анти-IFN-альфа
Титры IFN-связывающих антител измеряли, как описано в Примере 7, в V6 (38 день), V10 (112 день) и V11 (168 день после иммунизации).
Результаты показали, что пациенты с СКВ с положительным признаком интерферона продуцируют в десять раз больше IFN-связывающих антител в ответ на иммуногенный продукт согласно настоящему изобретению, чем пациенты с СКВ с отрицательным признаком интерферона на исходном уровне (Фиг.3).
Гены, индуцированные IFN
Эволюция экспрессии генов, индуцированных IFN, между V0 и V10 или V11 измеряли у получавших лечение пациентов с положительным или отрицательным признаком IFN на исходном уровне и у пациентов, получавших лечение плацебо.
Результаты показали, что по сравненшо с пациентами, получавшими плацебо, и пациентами с отрицательным IFN-признаком, влияние терапии иммуногенным продуктом согласно настоящему изобретению на гены, индуцированные IFN, сильно и значительно отличалось d V10 и V11 у пациентов с СКВ с положительным IFN-признаком (Фиг.4).
Комплемент С3
Значения С3 в сыворотке крови измеряли у пациентов, получавших лечение, и у пациентов, получавших плацебо, в V-1 (за 30 дней до иммунизации), V0 (день иммунизации), V7 (56 день), V10 (112 день) и V11 (138 день после иммунизации), как описано выше.
Результаты показали, что происходит значительное увеличение уровней С3 у пациентов, получающих лечение, по сравнению с пациентами, получающими плацебо. Более того, происходит значительное увеличение уровней С3 у пациентов с СКВ с положительным IFN-признаком, которых лечат иммуногенным продуктом согласно настоящему изобретению (Фиг.5).
Пример 10: определение соотношения IFNα/KLH в продукте согласно настоящему изобретению
Для оценки количества IFNα и KLH в продукте согласно настоящему изобретению разрабатывали способ количественного определения на основе УФ-детектирования и детектирования флуоресценции. Продукты согласно настоящему изобретению изготавливали с двумя флуоресцентными метками, каждая из которых специфична в отношении IFNα или KLH. После анализа путем эксклюзионной хроматография (SEC), количество IFNα и KLH определяли по интегрированию УФ-сигнала при 220 нм и сигнала флуоресценции (флуоресцентный детектор (FLD)), специфичного в отношении метки IFNα или метки KLH. Данный способ позволял рассчитать соотношение массы IFNα/KLH.
a) Мечение исходных веществ:
Флуоресцентные метки связывали на сульфгидрильных группах для сохранения аминогрупп, используемых во время получения продукта.
Мечение проводили в 70 мМ натрий-фосфатном буфере рН7 при комнатной температуре в течение 3 часов. KLH метили 200 молярными эквивалентами Atto565-малеимида (18507, Sigma), a IFNα - 100 молярными эквивалентами флуоресцеин-малеимида (46130, Pierce). Затем меченые белки (KLH-atto565 и IFN-Флуоресцеин) фильтровали на колонке Zeba (порог отсечения 7кДа, Thermo Scientific, 89893), кондиционированной 70 мМ фосфатным буфером рН 7,8 для удаления непрореагировавших меток.
b) Изготовление продукта:
Затем меченые исходные вещества использовали для получения меченых продуктов тем же способом, что и в Примере 1, с диализной фильтрацией вместо тангенциальной поточной фильтрации.
c) Получение стандартов гомополимеров KLH и IFNα
Для количественного аналитического способа получали стандарты гомополимеров.
Меченый стандарт гомополимеров IFNα получали тем же способом, что и в Примере 1, но с использованием 70 мМ фосфатного буфера pH 7,8 вместо KLH и диализной фильтрации вместо тангенциальной поточной фильтрации.
Меченый стандарт гомополимеров KLH получали тем же способом, что и в Примере 1, но с использованием 70 мМ фосфатного буфера pH 7,8 вместо IFNα и диализной фильтрации вместо тангенциальной поточной фильтрации.
d) Способ анализа путем эксклюзионной хроматографии
Затем партии анализировали путем SEC с УФ-детектированием и детектированием специфической флуоресценции.
60 мкл образца вводили в соединенные последовательно колонки SEC5 (1000А°) SEC3 (300А°) (Agilent, 5190-2536, 5190-2511), элюирование проводили с использованием ФБР в течение 35 минут с УФ-детектированием при 220 нм и детектированием специфической флуоресценции (для IFNα-Флуоресцеина или KLH-Atto565), как описано в Таблице 7.
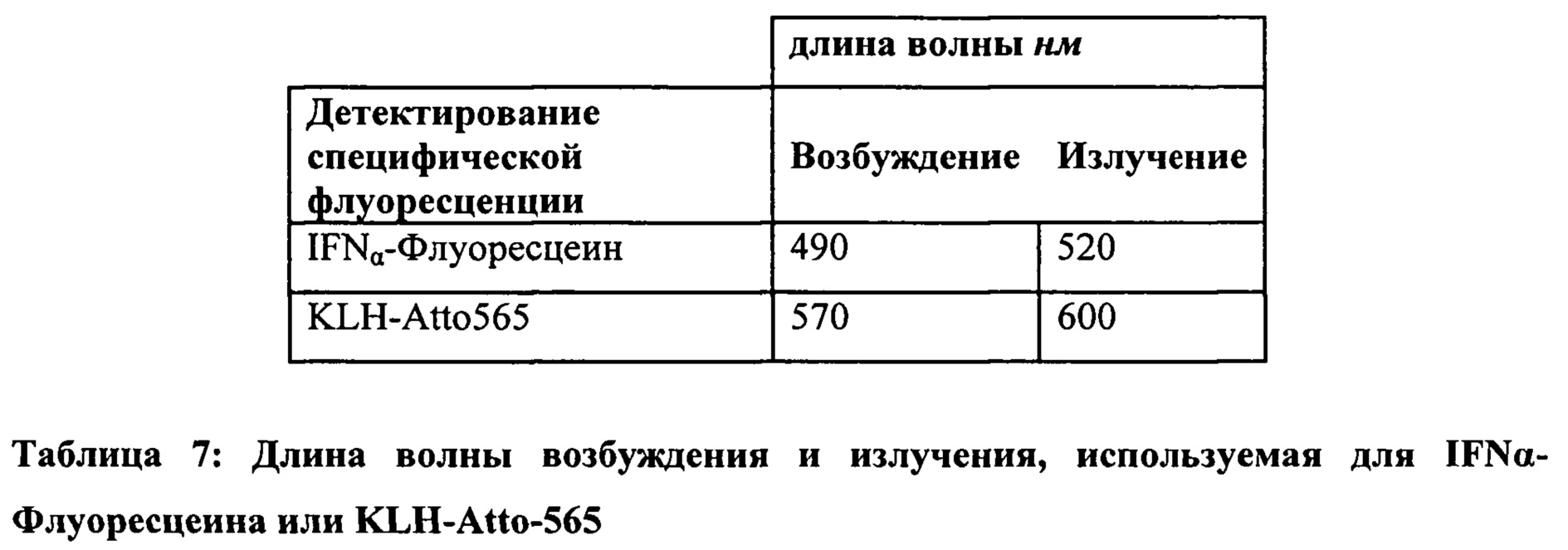
УФ-сигналы и сигналы флуоресценции (FLD) рассчитывали путем интеграции площади под пиками хроматограммы между 0 до 20 минутами.
Для проверки данного способа проводили предварительные эксперименты для демонстрации:
- Специфичности сигнала флуоресценции (не наблюдали наложения сигналов между двумя мечеными белками),
- Для каждой изготовленной партии (продукт согласно настоящему изобретению, меченые гомополимеры KLH и IFNα) получали схожие УФ-профили путем эксклюзионной ВЭЖХ,
- Не наблюдали гашения сигнала флуоресценции в результате изготовления,
- Сигналы FLD были линейными и пропорциональными УФ-сигналам. УФ-вклад меченого IFNα в получаемый меченый киноид измеряли в соответствии с кривой Площадь по FLDIFNα-Флуоресцеин=f(Площадь по УФ) меченого стандарта гомополимеров IFNα.
УФ-вклад меченого KLH в получаемый меченый киноид измеряли в соответствии с кривой Площадь по FLDKLH-atto565=f(Площадь по УФ) меченого стандарта гомополимеров KLH.
Поскольку, УФ-площадь, как было проверено, является линейной функцией концентрации белка, данный способ позволял оценивать массовый процент меченого IFNα в итоговом полученном меченом киноиде.
е) Анализ партий
Изготавливали 3 партии меченых киноидов и анализировали их данным способом.
На основе пропорциональности УФ-сигнала и концентрации, и FLD и УФ-сигнала, рассчитывали соотношение между количеством IFNα и KLH (mIFNα/mKLH) для указанных трех партий (Таблица 8).

Находили среднее соотношение mIFNα/mKLH, составляющее 0,28, с относительным стандартным отклонением <15%.
Пример 11: Продуцируемые титры антител анти-mIFNα и нейтрализующая способность, когда иммуногенный продукт согласно настоящему изобретению вводят путем инъекции в виде эмульсии с SWE или SWE-a
Получение muIFN-K:
Вкратце, смешивали IFNαA мыши (PBL Biomedical Laboratories) и нативный KLH (Sigma) в соотношении 50:1, и воздействовали 22,5 мМ глутаральдегидом в течение 45 минут. После диализа против фосфатного буферного раствора (ФБР) с удалением избытка глутаральдегида, раствор инкубировали с 66 мМ формальдегидом в течение 48 часов при 37°С. После гашения глицином (0,1 М конечный) и последующего проведения диализа против ФБР с использованием мембраны с порогом отсечения 10 кДа, препарат стерилизовали путем фильтрации с использованием 0,22-мкм мембраны и хранили при 4°С.
Протокол иммунизации:
Мышей дважды иммунизировали внутримышечно в 0 день и 21 день mIFN-K (10 мкг на инъекцию) в виде эмульсии 1 к 1 с адъювантом SE или SE-a (конечный объем 100 мкл).
Определение титров антител анти-muIFNα и анти-KLH путем ELISA Сыворотки крови анализировали на предмет антител к muIFNα или KLH путем ELISA. Вкратце, на 96-луночные планшеты Maxisorp (Nunc) наносили muIFNαA 100 нг/лунка (PBL Biomedical Laboratories) для детектирования антител анти-muIFNα или нативный KLH (Sigma) для детектирования антител анти-KLH.
Двукратное серийное разведение образцов сыворотки крови (от 1:100 до 1:51,200) добавляли в лунки. «Холостые» лунки получали 100 мкл буфера для разведения. Через 1,5 часа при 37°С антитела детектировали с помощью 100 мкл антитела к иммуноглобулину G (IgG) мыши, конъюгированного с пероксидазой хрена, и О-фенилендиамина, колориметрического субстрата для пероксидазы хрена. Пул сывороток крови мышей линии Balb/c, иммунизированных muIFN-K, использовали в качестве положительного контроля. Оптическую плотность (OD) регистрировали при длине волны 490 нм. Анализы ELISA проводили в двух повторностях. В каждом планшете две лунки оставляли для холостых проб; их среднее значение вычитали из всех лунок.
Титры антител рассчитывали путем интерполяции максимальной OD (ODmax)/2 на оси х. Используемым уравнением было y=ax+b для прямой, проходящей через две точки вокруг ODmax/2.
Определение нейтрализующей способности антител анти-muIFNa, индуцированных после иммунизации IFN-K
Нейтрализующую способность определяли с использованием классического противовирусного цитопатического анализа (EMCV/L929). В данном анализе титр противовирусной активности muIFNα определяют относительно его способности ингибировать летальный эффект вируса энцефаломиокардита (EMCV) в клетках L-929 мышей (АТСС).
Вкратце, 25 мкл разведенных образцов сыворотки крови (или контрольного антитела) добавляли в 96-луночные культуральные планшеты (Nunc) в двукратных серийных разведениях (от 1:200 до 1:6400). Коммерческое поликлональное антитело кролика анти-muIFNα (от PBL, кат. номер: 32100-1) использовали в качестве положительного контроля. После инкубации совместно с muIFNα 25 МЕ/лунка в течение 1 часа при комнатной температуре высевали клетки L-929 по 20×103 на лунку и инкубировали при 37°С. После роста в течение ночи планшеты промывали ФБР и в каждую лунку добавляли раствор EMCV 100 мкл/лунка (в 100 раз больше дозы, необходимой для уничтожения 50% клеток). Планшеты инкубировали в течение 48 часов при 37°С. Наконец, добавляли раствор MTS/PMS (3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий, внутренняя
соль/феназинметосульфат) (Promega) 20 мкл на лунку и планшеты инкубировали в течение 4 часов при 37°С, 5% CO2 в увлажненном инкубаторе (защищенном от света). Затем для каждой лунки измеряли OD при 490 нм. OD холостых лунок (лунки со 100 мкл только культуральной среды) вычитали из OD образца.
Нейтрализующую способность каждого образца рассчитывали следующим образом:
Нейтрализующая способность (%)=100×[(OD тест-OD вирус)/(OD клетки)], где OD тест представляет собой OD для тестируемого образца (клетки + IFNα + сыворотка + вирус)
ODвирус представляет собой OD для вирусного контроля (клетки +вирус) ODклетки представляет собой OD для 20000 клеток/лунка (клетки + IFNα + вирус). Нейтрализующую способность наносили на график в виде функции разведения сыворотки крови. Титр (число разведения сыворотки крови), нейтрализующий 50% значения активности IFNα, определяли путем интерполяции на линейной части кривой.
Результаты показали, что титры анти-muIFNα и титры анти-KLH присутствовали в сыворотках крови мышей, собранных на 31 день после первой инъекции muIFN-K, эмульгированного в SWE или SWE-а; и что антитела анти-muIFNα обладали нейтрализующей способностью (NC50 > 200).
Реферат
Группа изобретений относится к медицине, а именно к иммунологии, и может быть использована для применения иммуногенного продукта при лечении состояния, связанного с IFNα. Иммуногенный продукт, содержащий IFNα, соединенный с молекулой гемоцианина фиссуреллы (KLH), применяют в терапевтически эффективном количестве, которое вводят указанному субъекту за одно введение и которое составляет более 30 мкг, причем отношение IFNα к KLH по массе составляет от 0,06 до 0,6. Группа изобретений относится также к единичной лекарственной форме, содержащей более 30 мкг иммуногенного продукта, а также к набору для лечения связанного с IFNα состояния у нуждающегося в этом субъекта. Использование данной группы изобретений позволяет нейтрализовать IFNα in vivo при применении более 30 мкг иммуногенного продукта, а также увеличивает продукцию IFN-связывающих антител у пациентов в ответ на вышеуказанный иммуногенный продукт. 3 н. и 12 з.п. ф-лы, 11 пр., 8 табл., 5 ил.
Комментарии