Растворимые в воде фосфонооксиметиловые эфиры затрудненных спиртов или фенолов, фармацевтические композиции на их основе, способ анестезии и способ лечения опухолевых заболеваний - RU2235727C2
Код документа: RU2235727C2
Чертежи
Описание
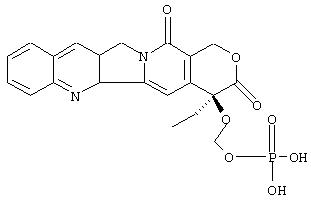
Настоящее изобретение относится к новым растворимым в воде пролекарствам из фармацевтических препаратов, содержащим алифатические или ароматические затрудненные гидроксильные группы. В частности, настоящее изобретение касается новых растворимых в воде фосфонооксиметиловых эфиров фармацевтических препаратов, содержащих стерически-затрудненный спирт или фенол, таких как камптотецин, пропофол, этопозид, витамин Е и циклоспорин А. Настоящее изобретение также относится к промежуточным продуктам, используемым для получения конечных пролекарств, а также к фармацевтическим композициям, содержащим новые соединения.
Успешная доставка пациенту фармацевтического препарата является чрезвычайно важным при лечении заболеваний. Однако применение многих испытанных в клинике лекарств с известными свойствами ограничено из-за их очень низкой растворимости в воде. Поскольку указанные лекарства имеют низкую растворимость в воде, они должны быть введены в состав с сорастворимыми фармацевтическими наполнителями, включающими поверхностно-активные вещества. Указанные поверхностно-активные вещества, как было показано, приводят к побочным эффектам у человека, что ограничивает безопасность указанных лекарств в клинических условиях и их применение для лечения ряда заболеваний.
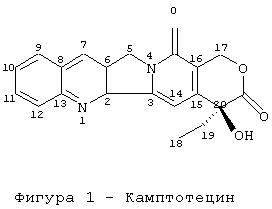
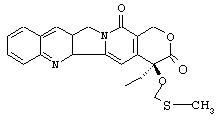
Например, камптотецин представляет собой натуральный продукт, выделенный из коры китайского дерева camptotheca, Camptotheca accuminata. Как было показано, он проявляет высокую противоопухолевую активность в опытах in vivo, проведенных на нескольких моделях животных, в отношении основных видов опухолей, таких как опухоли легкого, молочной железы, яичника, поджелудочной железы, толстой кишки и рака желудка, а также злокачественной меланомы. Камптотецин ингибирует клеточный фермент ДНК топоизомеразу I и вызывает каскад превращений, приводящих к апоптозу и программируемой смерти клетки. Топоизомераза I представляет собой жизненно важный фермент ядра клетки, ответственный за организацию и модулирование топологического строения ДНК, которое определяет способность клетки к воспроизведению, транскрибции и восстанавлению генетической информации.
Серьезным недостатком камптотецина является его крайне низкая растворимость в воде. Для того, чтобы провести биологические испытания, необходимо растворить соединение в сильном органическом растворителе (ДМСО) или изготовить лекарственный препарат в виде суспензии в Tween 80: физиологический раствор, что является нежелательным при терапевтическом лечении человека. Недавно в США были одобрены два аналога камптотецина со средней растворимостью в воде для лечения прогрессирующего рака яичников (Гикамтин, Hycamtin) и колоректального рака (Камптозар, Camptosar).
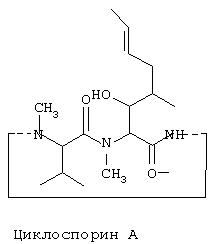
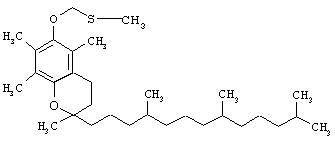
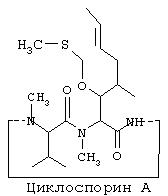
Другими лекарственными средствами, наподобие камптотецина, которые обладают теми же недостатками, являются циклоспорин A (CsA), пропофол, этопозид и Витамин Е (альфа-токоферол). Аналогично камптотецину, CsA имеет внутри своей структуры стерически затрудненный спирт, в данном случае им является вторичный спирт. CsA вводят в состав смеси Кремофор EL/этанол.
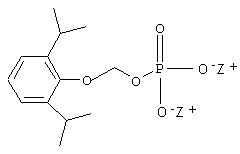
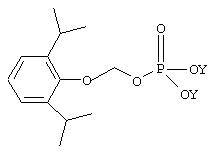
Примером стерически затрудненного, слабо растворимого в воде фенола, является пропофол, представляющий собой анестетик.
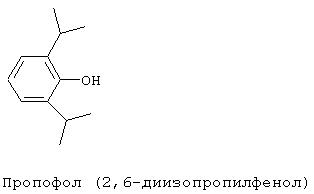
Пропофол готовят для внутривенного клинического использования in vivo в виде эмульсии масло/вода. Пропофол является не только слабо растворимым в воде, но он также вызывает боль в месте инъекции. Эту боль можно уменьшить, используя лидокаин. Из-за того, что пропофол используют в виде эмульсии, становится затруднительным и проблематичным введение в его состав других лекарственных средств, при этом изменение физических свойств состава, таких как увеличение размера капелек масла, может привести к эмболии легкого и т.д. Растворимое в воде и химически стабильное пролекарство из пропофола может обеспечить некоторые преимущества. Такой состав мог бы быть простым водным раствором, который можно было бы смешать с другими лекарственными средствами. Если пролекарство само по себе безболезненно, оно может быть более благоприятным для пациента, и наконец, не стало бы токсичности, обусловленной носителем. Другими трудно растворимыми в воде стерически затрудненными фонолами, представляющими собой противоопухолевые лекарства, являются этопозид и Витамин Е (альфа-токоферол).
Настоящее изобретение обеспечивает растворимые в воде формы лекарств, содержащие спирт или фенол, такие как камптотецин или пропофол. Что касается камптотецина, то соединения согласно настоящему изобретению представляют собой его фосфонооксиметиловые эфиры в виде свободной кислоты и их фармацевтически приемлемые соли. Растворимость в воде кислоты и солей облегчает приготовление фармацевтических составов. Все пролекарства согласно настоящему изобретению демонстрируют превосходящую растворимость в воде по сравнению с соответствующими исходными лекарствами. Методики, разработанные для соединений по настоящему изобретению, могут оказаться полезными для превращения и многих других нерастворимых в воде медицинских агентов, имеющих алифатические или ароматические затрудненные гидроксильные группы, в их растворимые в воде производные.
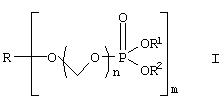
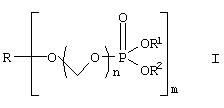
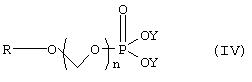
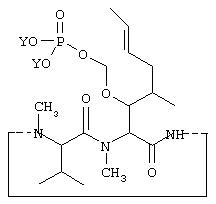
Описанное здесь изобретение включает новые композиции. Изобретение относится к растворимым в воде фосфонооксиметиловым производным фармацевтических препаратов, содержащим спирт и фенол, представленным общей формулой I:
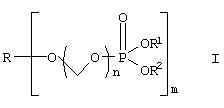
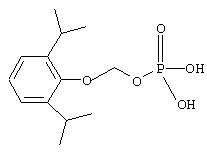
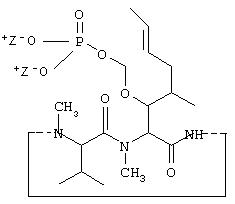
Приведенная выше формула I представляет собой производное ROH, где ROH представляет собой лекарство, содержащее спирт или фенол, такое как камптотецин, пропофол, этопозид, витамин Е и циклоспорин А. В представленной выше формуле I, n представляет собой целое число 1 или 2. Когда n имеет значение 2, ROH представляет собой предпочтительно фенолсодержащий фармацевтический препарат, такой как пропофол. Также сюда могут быть включены некоторые лекарства, для которых невозможно приготовить формы для инъекции из-за свойственной им очень низкой растворимости в воде. К ним относятся даназол, метилтестостерон, иодхинол, атовакон (danazol, methyltestosterone, iodoquinol, atovaquone). R1 представляет собой водород или ион щелочного металла, включая натрий, калий или литий или протонированный амин или протонированную аминокислоту или другой фармацевтически приемлемый катион. R2 представляет собой водород или ион щелочного металла, включая натрий, калий или литий или протонированный амин или протонированную аминокислоту или другой фармацевтически приемлемый катион. После внутривенного или перорального введения производные формулы I вновь превращаются в исходные лекарства с помощью гидролиза и/или с помощью фосфотазы.
Соответственно задачей настоящего изобретения является разработка производных нерастворимых в воде лекарств, которые демонстрируют хорошую активность и растворимость в воде.
Другой задачей настоящего изобретения является разработка фармацевтических композиций на основе указанных растворимых в воде соединений, которые содержат определенное количество соединения формулы I и фармацевтически приемлемый носитель.
Кроме того, задачей настоящего изобретения является разработка производных лекарств, имеющих хорошую стабильность при уровнях рН, пригодных для изготовления фармацевтических составов, но быстро разрушающихся in vivo под действем физиологических условий, т.е. потенциально действующих как пролекарства.
Краткое описание чертежей
Фиг.1 иллюстрирует ферментативное превращение пролекарства из пропофола в пропофол in vitro.
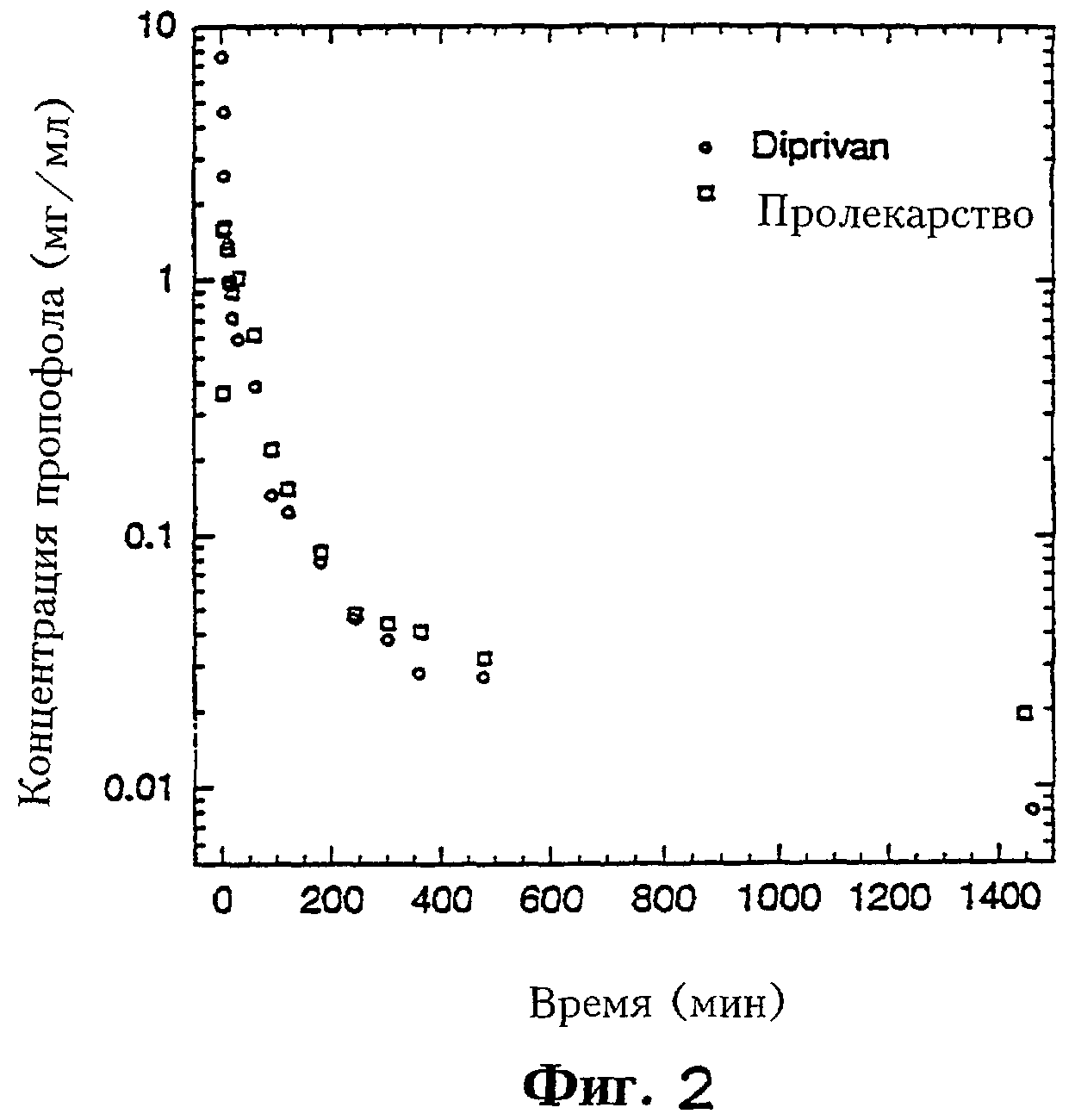
Фиг.2 иллюстрирует изменение концентрации пропофола в крови во времени, начиная с введения пролекарства из пропофола или Diprivan® в опытах на собаках.
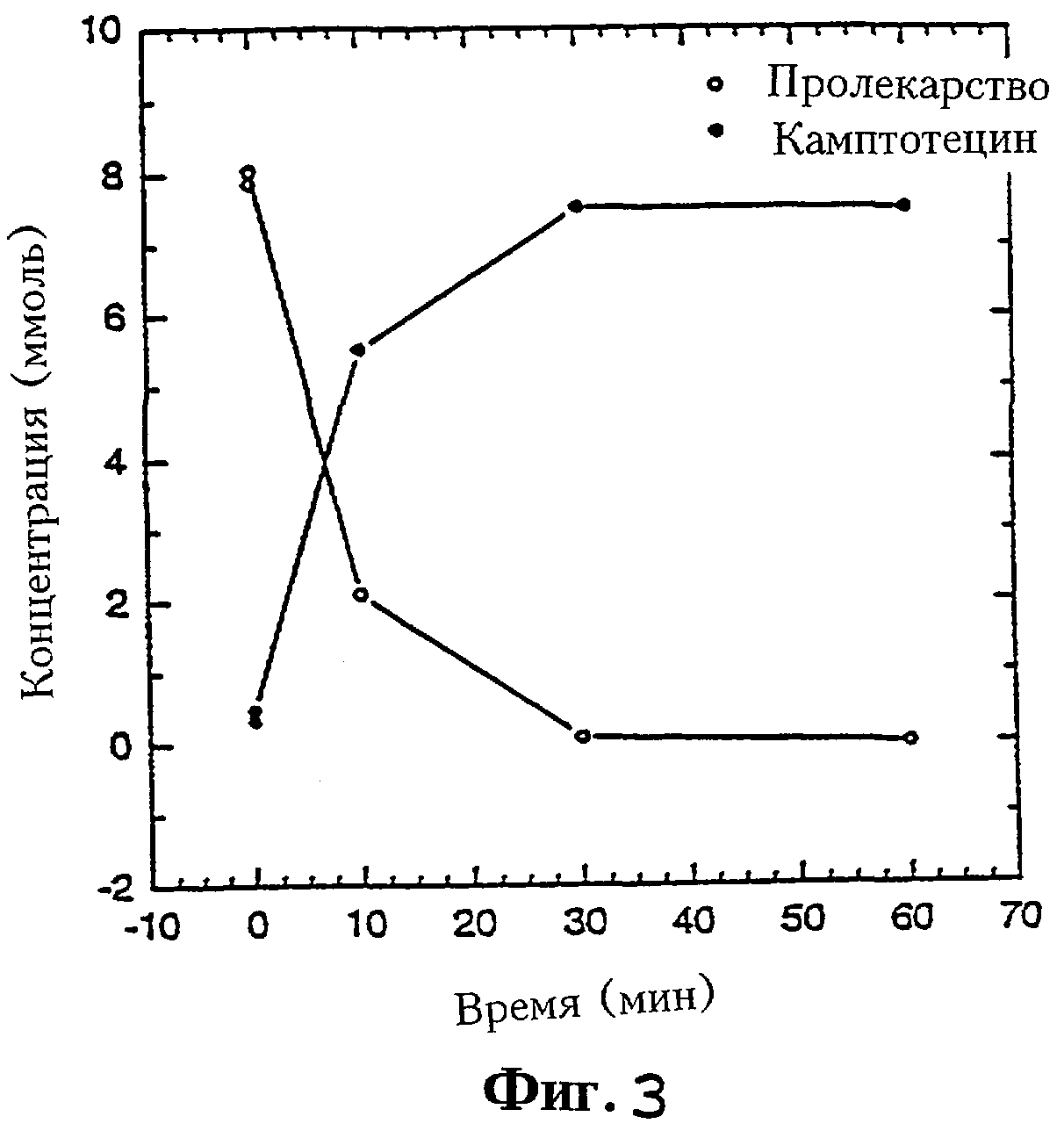
Фиг.3 иллюстрирует ферментативное превращение пролекарства из камптотецина в камптотецин в опытах in vitro.
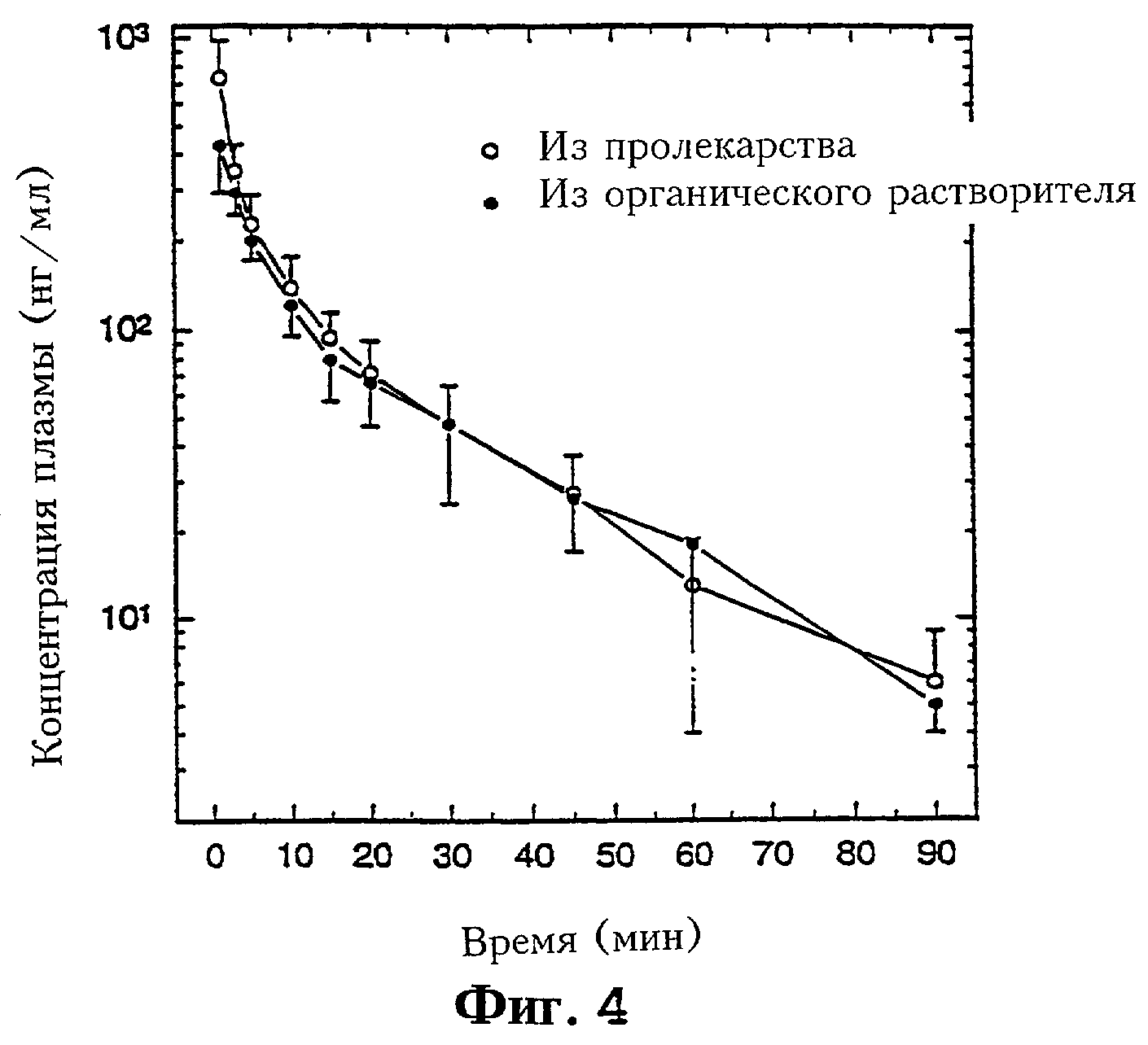
Фиг.4 иллюстрирует взаимосвязь между концентрацией камптотецина в плазме из пролекарства камптотецина и из камптотецина в органических сорастворителях в опытах на крысах.
Детальное описание изобретения
В настоящем описании, если не будет оговорено специально или не следует из контекста, использованы следующие определения.
"Фосфоно-" означает группу -Р(О)(ОН)2 и "фосфонооксиметокси" или "фосфонооксиметиловый эфир" означает, как правило, группу -ОСН2ОР(О)(ОН)2. "Метилтиометил" относится к группе –CH2SCH3. Настоящее изобретение также охватывает соединения, в которых n=2, так, что "фосфоно-ди(оксиметил) эфир" в общем означает группу –ОСН2ОСН2 ОР(О)(ОН)2.
Определение "остаток камптотецина" означает остаток, содержащий внутренний каркас камптотецина с двадцатью атомами углерода, включающий два атома азота и четыре атома кислорода, как представлено ниже, на приведенной структурной формуле с абсолютной конфигурацией.
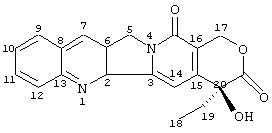
Система нумерации, приведенная выше, является системой, используемой для соответствующих производных камптотецина, и она применяется на протяжении всего описания. Например, определение С20 относится к атому углерода под номером "20".
Определение "аналог камптотецина" относится к соединению, имеющему внутренний каркас камптотецина. Должно быть понятно, что аналоги камптотецина охватывают соединения, включая, впрочем не ограничиваясь, следующие: топотекан (Topotecan), поступающий от фирмы SmithKline Beecham, иринотекан (Irinotecan) (CPT-11), поступающий от фирмы Pharmacia & Upjohn, 9-аминокамптотецин (9АС), 9-нитрокамптотецин (9NC), GI 147211 С, поступающий от фирмы Glaxo Wellcome, и DX-8951f (предшествующие шесть аналогов камптотецина в настоящее время проходят клинические испытания и они описаны в обзоре, опубликованном в Pacific West Cancer Fund автором Claire McDonald (December 1997).
Кроме того, несколько других аналогов камптотецина, также не ограничивающих круг аналогов, включены в описание путем приведения ссылок на Sawada et al., Current Pharmaceutical Design, Vol.1, No. 1, pp 113-132, а также на патенты US 5646159, 5559235, 5401747, 5364858, 5342947, 5244903, 5180722, 5122606, 5122526, 5106742, 5053512, 5049668, 4981968 и 4894456.
Несколько фармацевтических соединений, включая соответствующие производные камптотецина, содержат более одной гидроксильной группы, например 10-гидроксикамптотецин, топотекан и некоторые другие, перечисленные в приведенных выше ссылках. Должно быть понятно, что настоящее изобретение может быть применено к более, чем одной гидроксильной группе. В этом случае оно может быть осуществлено путем введения защиты для дополнительной гидроксильной группы перед образованием производного.
Определение "фосфонозащитные группы" означает остатки, которые могут быть использованы для блокирования или защиты функциональных фосфоногрупп. Предпочтительно, к таким защитным группам относятся группы, которые могут быть удалены с помощью методов, которые существенно не затрагивают остальную часть молекулы. Подходящие фосфонооксизащитные группы включают, например, бензильную (обозначаемую "Вn"), трет.-бутильную, аллильную группы.
Определение "фармацевтически приемлемая соль" означает соль металла или соль амина кислой фосфоногруппы, в которой катион не делает значительного вклада в токсичность или в биологическую активность активного вещества. Подходящие соли металлов включают литиевые, калиевые, натриевые, кальциевые, бариевые, магниевые, цинковые и алюминиевые соли. Предпочтительными солями являются соли натрия и калия.
Подходящими солями аминов являются, например, соли аммиака, трометамина, триэтаноламина, этилендиамина, глюкамина, N-метилглюкамина, глицина, лизина, орнитина, аргинина, этаноламина и это только несколько примеров. Предпочтительными солями аминов являются соли лизина, аргинина, N-метилглюкамина и трометамина.
В описании и в формуле изобретения термин -ОСН2OР(O)(ОН)2 как подразумевают, охватывает оба понятия и свободную кислоту и ее фармацевтически приемлемые соли, если из контекста не следует специально, что подразумевается только свободная кислота.
Один из аспектов настоящего изобретения обеспечивает производные фармацевтических препаратов, содержащих спирт и фенол, как показано на формуле I:
Производные формулы I могут быть получены в соответствии с последовательностью реакций, представленных на Схеме 1:
Схема 1
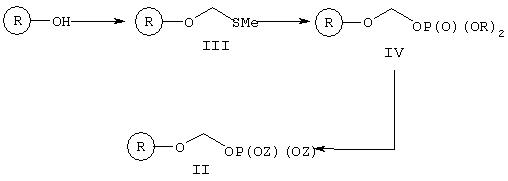
где ROH представляет собой лекарство, содержащее спирт или фенол, такой как камптотецин, пропофол, этопозид, витамин Е, циклоспорин А. Должно быть понятно, что приведенный выше путь является одним из альтернативных путей. Альтернативные пути станут очевидными при их раскрытии в описании и в примерах.
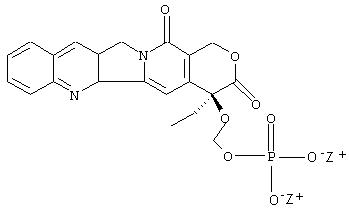
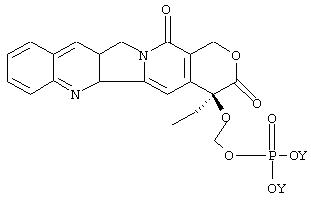
Приведенная выше схема может быть проиллюстрирована при использовании соединения камптотецина. Должно быть понятным, что эти схемы применимы и для других соединений, охватываемых формулой I и перечисленных выше, в соответствии с настоящим изобретением. Соответственно, другим аспектом настоящего изобретения является обеспечение аналогов камптотецина согласно формуле II:
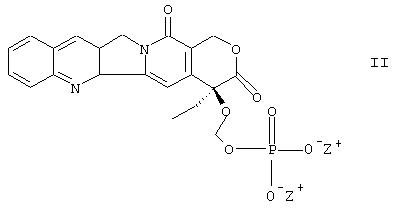
которые включают свободную кислоту, где Z представляет собой водород и их фармацевтически приемлемые соли, где Z представляет собой металл или амин.
Альтернативно формула II включает дикислоты, где Z представляет собой металл или амин в обоих случаях.
Предпочтительньми фармацевтически приемлемыми солями соединения формулы II являются щелочные соли, включая литиевые, натриевые и калиевые соли; и соли аминов, включая триэтиламиновые, триэтаноламиновые, этаноламиновые, аргининовые, лизиновые и N-метилглюкаминовые соли.
При наиболее предпочтительном воплощении изобретения производные камптотецина формулы II включают следующие соединения:
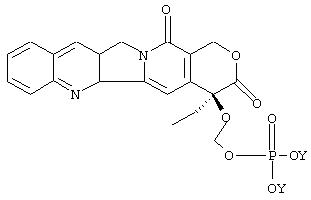
(20)-O-фосфонооксиметилкамптотецин, (20)-O-фосфонооксиметилкамптотецин моно- или динатриевая соль, (20)-O-фосфонооксиметилкамптотецин моно- или дикалиевая соль, (20)-O-фосфонооксиметилкамптотецин моно- или диаргининовая соль, (20)-O-фосфонооксиметилкамптотецин моно- или дилизиновая соль, (20)-O-фосфонооксиметилкамптотецин моно- или ди-N-метилглюкаминовая соль и (20)-O-фосфонооксиметилкамптотецин моно- или дитриэтаноламиновая соль.
Соединения формулы II могут быть получены непосредственно из камптотецина (показано как ©-ОН) в соответствии с последовательностью реакций, приведенной на схеме 2:
Схема 2
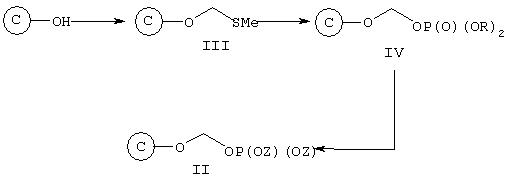
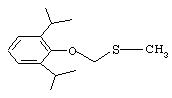
Соединение формулы III (метилтиометиловый эфир, МТМ эфир) может быть получен путем обработки камптотецина диметилсульфоксидом /уксусным ангидридом/уксусной кислотой.
На второй стадии процесса, представленного на схеме 2, метилтиометиловый эфир превращают в соответствующий защищенный фосфонооксиметиловый эфир (соединение формулы IV). Эту стадию осуществляют путем обработки МТМ эфира N-иодсукцинамидом и защищенным фосфатом НОР(О)(OR)2. На третьей стадии фосфоно защитные группы удаляют, что приводит к получению соединения формулы II. Например, подходящими защитной (ыми) фосфоно группой (ами) является бензил, которые может быть удален каталитическим гидрогенолизом.
Общий процесс получения соединения формулы (1) по схеме 2 более детально представлен на схеме 3.
Схема 3
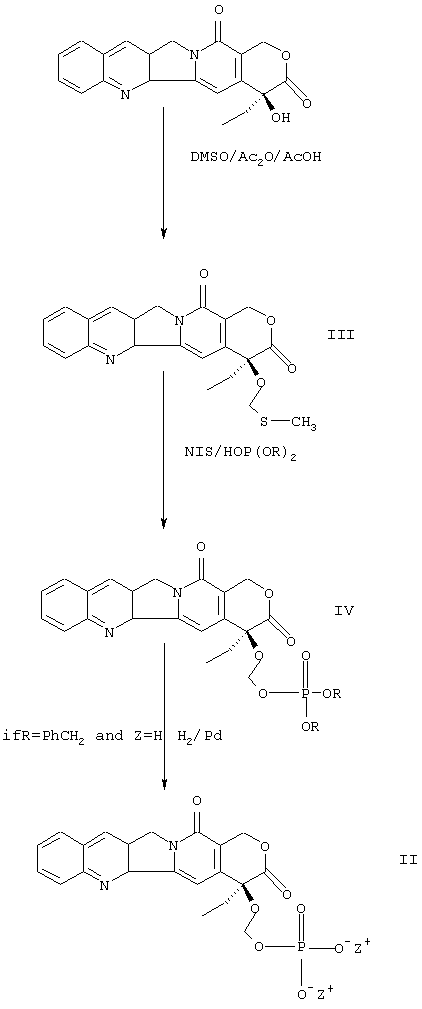
На первой стадии свободную гидроксильную группу камптотецина превращают в соответствующую метилтиоэфирную группу (-ОСН2SCН3). Такое превращение может быть осуществлено реакцией с диметилсульфоксидом в присутствии уксусного ангидрида и уксусной кислоты. Указанный способ, известный как реакция Пуммера (Pummer reaction), был успешно применен Bristol-Myers Squibb для метилтиометилирования таксола (ЕР 0604910 А1, Bioorg. Med. Chem. Lett., 6, 1837, 1996). Обычно, для получения метилтиометилового эфира реакцию проводят при комнатной температуре в течение 24-72 часов.
На второй стадии реакционной цепочки метилтиометиловый эфир превращают в соответствующий защищенный фосфонооксиметиловый эфир. Это хорошо известное превращение было успешно применено Bristol-Myers Squibb для фосфонооксиметилирования таксола (ЕР 0604910 А1, Bioorg. Med. Chem. Lett., 6, 1837, 1996). Taк, соединение формулы III обрабатывают N-иодсукцинамидом и защищенной фосфорной кислотой, такой как дибензилфосфат. Реакцию проводят в инертном органическом растворителе, таком как тетрагидрофуран и галогенированный углеводород, такой как хлористый метилен и в присутствии молекулярных сит. N-Иодсукцинимид и защищенную фосфорную кислоту используют в избытке (3-5 эквивалента) по отношению к метилтиометиловому эфиру.
На третьей стадии реакционной цепочки защитные фосфоновые группы удаляют. Снятие защиты осуществляют с помощью известных из уровня техники способов, таких как катализируемый щелочью или кислотой гидролиз, гидрогенолиз, восстановление и тому подобное. Например, каталитический гидрогенолиз может быть использован для удаления бензильных фосфонозащитных групп. Описание методик снятия защиты может быть найдено в различных справочниках, таких как T.W.Green и P.G.M.Wutz, Protective groups in organic synthesis, J.Wiley publishers. New York, NY, 1991, pp.47-67.
Основные соли соединения формулы II могут быть образованы с помощью доступных способов, включая взаимодействие соединения формулы II в виде свободной кислоты с металлическим основанием или амином. Подходящие основания металлов включают гидроксиды, карбонаты и бикарбонаты натрия, калия, лития, кальция, бария, магния, цинка и алюминия; и подходящие амины, включают триэтиламин, аммиак, лизин, аргинин, N-метилглюкамин, этаноламин, прокаин, бензатин, дибензиламин, трометамин (TRIS), хлорпрокаин, холин, диэтаноламин, триэтаноламин и тому подобное. Основные соли могут быть дополнительно очищены хроматографически с последующей лиофилизацией или кристаллизацией.
Соединения в соответствии с настоящим изобретением представляют собой фосфонооксиметиловые эфиры фармацевтических препаратов, таких как камптотецин, пропофол, этопозид, токоферол и т.д. В форме фармацевтически приемлемых солей, они демонстрируют улучшенную растворимость в воде по сравнению с исходными соединениями, таким образом позволяя получать более пригодные фармацевтические составы. Хотя это не подтверждено теоретически, однако представляется, что фосфонооксиметиловые эфиры в соответствии с настоящим изобретением представляют собой пролекарства исходных фармацевтических препаратов; фосфоно-оксоэтильный остаток, расщепляется при контакте с фосфотазой in vivo, с последующим генерированием первоначального (родительского) соединения. Как показано выше, соединения по настоящему изобретению являются эффективными фармацевтическими и терапевтическими агентами.
Например, соединения формулы II в соответствии с настоящим изобретением могут быть использованы аналогично камптотецину. Структура пролекарства камптотецина представлена выше. Таким образом онколог, являющийся специалистом в области лечения рака, будет способен определить, без дополнительных экспериментов, подходящий лечебный курс применения соединения в соответствии с настоящим изобретением. Дозировка, способ, а также график введения соединений по изобретению, не являются строго определенными и будут изменяться в зависимости от применяемого соединения. Так, соединение формулы II может быть введено любым путем, предпочтительно парентерально; доза может, например, находиться в интервале от около 0,1 до около 100 мг/кг на вес тела или от около 5 до 500 мг/м2. Соединения формулы II могут быть введены также перорально; пероральная доза может находиться в интервале от около 5 до около 500 мг/кг на вес тела. Доза, используемая на практике, может изменяться в зависимости от конкретного состава композиции, пути введения, а также конкретного места поражения, хозяина-носителя и вида опухоли, которую подвергают лечению. При определении дозировки учитываются многие факторы, которые влияют на активность лекарства, которые включают возраст, пол, диету и физические состояния пациентов.
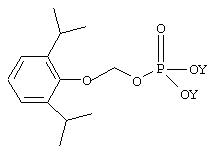
Другим примером является пролекарство профола, имеющее формулу I по настоящему изобретению. Структура пролекарства профола представлена ниже:
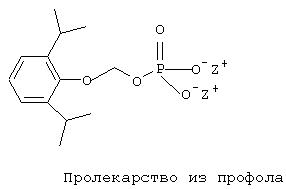
В приведенной выше формуле пролекарства из профола Z имеет те значения, что представлены в приведенной выше формуле II. Таким образом, анастезиолог, являющийся специалистом в области анастезии, способен определить, без дополнительных экспериментов, соответствующий график введения соединения по настоящему изобретению. Дозировка, способ, а также график введения соединений по изобретению не являются строго определенными и будут изменяться в зависимости от применяемого соединения. Так, соединение формулы I, такое как пролекарство из профола, может быть введено любым пригодным путем, предпочтительно парентерально; доза может, например, находиться в интервале от около 0,5 до около 10 мг/кг, вводимая в соответствии со способами ввода общего наркоза или поддержания периода общего наркоза. С другой стороны соединение формулы I может быть введено путем парентерального вливания, в этом случае дозировка может, например, находиться в интервале от 2 мкг/кг/мин до 800 мкг/кг/мин при введении согласно способам, существующим для поддержания периода общего наркоза, инициирования и поддержание MAC или ICU седативного эффекта.
Настоящее изобретение также обеспечивает фармацевтическую композицию, содержащую фармацевтически эффективное количество соединения формулы I в комбинации с одним или несколькими фармацевтически приемлемыми носителями, эксипиентами, разбавителями или адъювантами. Например, соединения в соответствии с настоящим изобретением могут быть введены в составы, изготовленные в виде таблеток, пилюль, порошков, капсул, инъекций, растворов, свечей, эмульсий, дисперсий, пищевых добавок и других пригодных форм. Они могут быть изготовлены в форме стерильных твердых составов, например, лиофилизованных, и, при желании, объединенных с другими фармацевтическими эксципиентами. Указанные твердые составы могут быть разведены стерилизованной водой, физиологическим солевым раствором или смесью воды и органического растворителя, такого как пропиленгликоль, этанол, и тому подобное или другой стерильной средой для инъекций непосредственно перед парентеральным введением.
Обычными фармацевтически приемлемыми носителями являются, например, манитол, мочевина, декстраны, лактоза, не восстановленные сахара, картофельный и маисовый крахмалы, стеарат магния, тальк, растительные масла, полиалкиленгликоли, этилцеллюлоза, поли(винилпирролидон), карбонат кальция, этилолеат, изопропилмиристат, бензилбензоат, карбонат натрия, желатин, карбонат калия, салициловая кислота. Фармацевтические композиции могут также содержать не токсичные вспомогательные вещества, такие как эмульгаторы, консерванты, смачивающие агенты и тому подобное, как например, монолаурат сорбитола, триэтаноламинолеата, полиоксиэтилен, моностеарат, глицерилтрипалмитат, диоктилнатрийсульфосукцинат, растительные масла, полиалкиленгликоли, этилцеллюлозу, поли(винилпирролидон), карбонат кальция, этилолеат, изопропилмиристат, бензилбензоат, карбонат натрия, желатин, карбонат калия, кремневую кислоту. Фармацевтические составы могут также включать нетоксичные добавки, такие как, например, эмульгирующие агенты, консерванты, смеачивающие агенты и тому подобное, например, монолаурат сорбитола, олеат триэтаноламина, моностеарат полиоксиэтилена, трипальмитат глицерина, натрий диоктилсульфосукцинат и тому подобное.
В представленной далее экспериментальной части все температуры указаны по шкале Цельсия (С), если особо не оговорено. Спектральные характеристики в соответствии с ядерным магнитньм резонансом (ЯМР) относятся к химсдвигам (δ), выраженным в частях на миллион (ррm) по отношению к тетраметилсилану (TMS), как к стандарту. Относительная площадь, определенная для различных сдвигов в спектральных данных протонного ЯМР, соответствует числу атомов водорода, содержащихся в каждой функциональной группе, входящей в молекулу. Природа химсдвигов, относящихся к мультиплетности, обозначается как уширенный синглет (бс), уширенный дуплет (бд), уширенный триплет (бт), уширенный квартет (бкв), синглет (с), мультиплет (м), дуплет (д), квартет (кв), триплет (т), дуплет дуплета (дд), дуплет триплета (дт) и дуплет квартета (дкв). Для снятия ЯМР спектров применяют такие растворители, как ацетон-d6 (дейтерированный ацетон) DMSO-d6 (пердейтериродиметилсульфоксид), D2O (дейтерированную воду), СВСl3 (дейтерохлороформ) и другие пригодные дейтерированные растворители.
В описании применяют аббревиатуры, которые широко применяют в уровне техники. Некоторые из них следующие:
MS (масс спектрометрия); HRMS (масс спектрометрия высокого разрешения); Ас (ацетил); Ph (фенил); FAB (прочная атомная бомбардировка); min (минута); h или hrs (час(сы)); NIS (N-иодосукцинимид); DMSO (диметилсульфоксид); ТГФ (тетрагидрофуран).
Приведенные далее примеры призваны проиллюстрировать синтез наиболее характерных соединений в соответствии с настоящим изобретением, однако, они не должны быть расценены как ограничивающие область изобретения никоим образом. Специалист в данной области способен перенести приведенную методику без дополнительного экспериметирования на синтез соединений, входящих в область настоящего изобретения, но не раскрытых в данном описании. Например, в приведенных примерах приведены определенные соли, однако эти соли не должны быть рассмотрены как ограничение. Иллюстрацией этому служит то, что везде повторено применение серебрянкой соли дибензилфосфата. В то же время могут быть использованы вместо серебряной соли другие соли, как соли тетераметиламмония или другие соли щелочных металлов.
ПРИМЕРЫ
1. Синтез O-Фосфонооксиметилпропофола
Ia. Синтез O-метилтиометилпропофола:
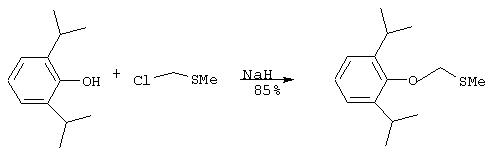
К перемешиваемой суспензии гидрида натрия (150 мг, 6,2 ммоль) в сухом НМРА (10 мл), которую выдерживают в атмосфере аргона, добавляют по каплям в течение более 15 минут пропофол (1,1 мл 97%-ный, 5,7 ммоль). Затем реакционную смесь перемешивают при комнатной температуре дополнительно 30 минут. После этого к указанной смеси добавляют по каплям хлорметилметилсульфид (550 мкл 95%-ный, 6, 2 ммоль) и затем перемешивают при комнатной температуре. Через 20 часов реакционную смесь распределяют при перемешивании между водой (10 мл) и бензолом (20 мл). Водный слой отделяют и экстрагируют бензолом (10 мл). Бензольные фракции объединяют, промывают водой (2×3 мл), сушат над сульфатом натрия и упаривают при пониженном давлении. Полученный маслянистый остаток очищают с помощью колоночной хроматографии (силикагель, гексан, затем 4:1 гексан/хлороформ), что дает 1,15 г (85% выход) названного соединения в виде бесцветного масла.
EIMS: [M+] м/з 238.
1Н ЯМР (300 МГц, CDCl3, δ): 1.24 (д, J=6.9 Гц, 12Н), 2.37 (с, 3Н). 3.37 (гепт, J=6.9 Гц, 2Н), 4.86 (с, 2Н), 7.12 (с, 3Н).13С ЯМР (75 МГц, CDCl3, δ): 15.40, 23.98, 26.68, 78.12, 124.04, 125.05, 141.74, 152.20.
Ib. Синтез O-хлорметилпропофола:
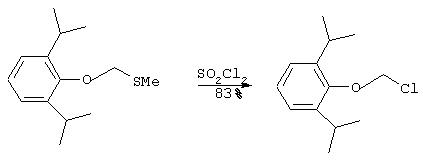
К перемешиваемому раствору O-метилтиометилпропофола (3.00 г, 12.5 ммоль) в сухом хлористом метилене (30 мл), который выдерживают в атмосфере аргона, добавляют 1М раствор SO2Cl2 в сухом хлористом метилене (12.2 мл, 12.2 ммоль) при 5°С в течение более пяти минут. Реакционную смесь перемешивают 10 минут при той же температуре и затем три часа при комнатной температуре. Растворитель упаривают при пониженном давлении и коричневый остаток в виде масла очищают с помощью флэш хроматографии на колонке (силикагель, 1:20 гексан/этилацетат), что дает 2.36 г (83% выход) названного соединения в виде желтого масла.
CIMS (NН3):[М]+м/з 226, [MH+NH3]+, м/з 244.
1ЯМР (300 МГц, CDCl3, δ): 1.22 (d, J=6.9 Гц, 12Н), 3.35 (гепт, J=6.9 Гц, 2Н), 5.76 (с, 2Н), 7.15 (м, 3Н).13С ЯМР (75 МГц, CDCl3, δ) 23.93, 26.84, 83.34, 124.34, 125.95, 141.34, 150.93.
Iс. Синтез дибензилового эфира O-фосфонооксиметилпропофола (путь-1):
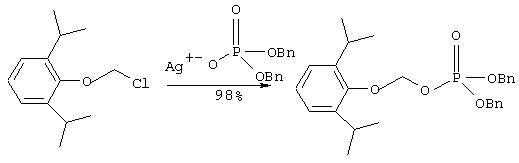
Смесь O-хлорметилпропофола (2.20 г, 9.7 ммоль), дибензилфосфата серебра (3.85 г, 10.0 ммоль) и сухого толуола (50 мл) нагревают с обратным холодильником в атмосфере аргона 45 минут. Затем смесь охлаждают до комнатной температуры и фильтруют.
После того, как растворитель упаривают в вакууме, маслянистый остаток очищают с помощью флэш хроматографии на колонке с силикагелем (9:1 гексан/этил ацетат и затем 1:1 гексан/этилацетат), что дает 4.43 г (98% выход) названного соединения в виде желтого масла.
CIMS (NН3): [МН]+, м/з 469, [МН+ NН3]+, м/з 486.
1Н ЯМР (300 МГц, CDCl3, δ): 1.17 (d, J=6.8 Гц, 12Н), 3.33 (гепт, J=6.9 Гц, 2Н), 5.00 (д, J=7.8 Гц, 2Н), 5.01 (д, J=7.8 Гц, 2Н), 5.42 (д, J=9.9 Гц, 2Н), 7.12 (м, 3Н), 7.32 (м, 10Н).13С ЯМР (75 МГц, CDCl3, δ): 23.79, 26.57, 69.15, 69.23, 94.14, 94.20, 124.07, 125.62, 127.70, 128.44, 135.42, 135.51, 141.50,151.07.
Iс. Синтез дибензилового эфира O-фосфонооксиметилпропофола (альтернативный путь 1)
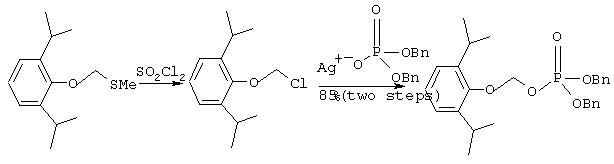
К перемешиваемому раствору О-метилтиометилпропофола (1.45 г, 6.08 ммоль) в сухом хлористом метилене (15 мл) в атмосфере аргона при 0-5°С добавляют 1М раствор SO2Cl2. в сухом хлористом метилене (6.5 мл, 6.5 ммоль) в течение более пяти минут. Реакционную смесь перемешивают 10 минут при 5°С и три часа при комнатной температуре. Затем растворитель упаривают при пониженном давлении. Оставшееся масло растворяют в толуоле (ACS-сорта, 20 мл), добавляют дибензилфосфат серебра (3.50 г, 9.1 ммоль), и полученную смесь нагревают с обратным холодильником 45 минут. Коричневую реакционную смесь охлаждают до комнатной температуры и фильтруют. Затем растворитель упаривают в вакууме, маслянистый остаток очищают с помощью колоночной хроматографии (9:1 гексан/этилацетат, затем 1:1 гексан/этилацетат), что дает 2.41 г (85% выход) названного соединения в виде желтого масла. Полученное соединение имеет тот же Rf (TCX) и1HЯMP спектр (300 МГц, СОСl3), что и достоверный образец.
Ic. Синтез дибензилового эфира О-фосфонооксиметилпропофола (альтернативный путь-2)
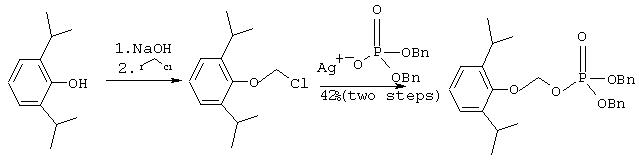
К перемешиваемой суспензии гидрида натрия (41 мг 60%-ной дисперсии в минеральном масле, 1.02 ммоль) в сухом диметоксиэтане (1.5 мл) в атмосфере аргона добавляют по каплям пропофол (200 мкл, 97%-ный, 1.04 ммоль) в течение более 5 минут и полученную смесь перемешивают дополнительно 15 минут. Полученный гомогенный раствор добавляют по каплям к перемешиваемому раствору хлориодометана (4.0 мл, 53 ммоль) в сухом диметоксиэтане (4 мл) в течение более 15 минут. Реакционную смесь перемешивают два часа, фильтруют и затем растворитель и избыток хлориодометана упаривают. Оставшееся масло растворяют в толуоле (HPLC-сорт, 10 мл). К указанному раствору добавляют дибензилфосфат серебра (400 мг, 1.04 ммоль), и полученную смесь нагревают с обратным холодильником 10 минут. Затем реакционную смесь охлаждают до комнатной температуры и фильтруют, растворитель упаривают в вакууме. Маслянистый остаток очищают с помощью флэш хроматографии на колонке с силикагелем (9:1 гексан/этилацетат и затем 1:1 гексан/этилацетат), что дает 205 мг (42% выход) названного соединения в виде желтого масла. Полученный продукт имеет тот же Rf(TCX) и1НЯМР спектр (300 МГц, СDСl3), что и достоверный образец.
В отношении приведенной выше реакции Iс (альтернативный путь - 2) возможно отметить, что должно быть понятно, что в зависимости от желаемого соединения, могут быть использованы другие реагенты. Например, когда нужно соединение формулы I n=2, хлориодометан может быть заменен на такое соединение, как Х-СН2-O-СН2-Сl, где Х является легко отщепляемой группой.
Iс. Синтез дибензилового эфира О-фосфонооксиметилпропофола (альтернативный путь - 3):
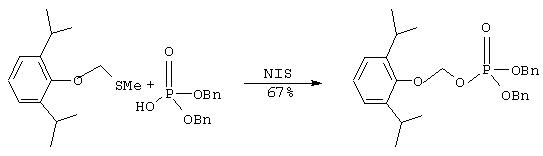
К перемешиваемому раствору о-метилтиометилпропофола (91 мг, 0.38 ммоль) в сухом хлористом метилене (2 мл) в атмосфере аргона добавляют измельченные активированные молекулярные сита 4 А(100 мг) и затем раствор дибензилфосфата (127 мг, 0.45 ммоль) и N-иодсукцинимида (102 мг, 95%-ный, 0.43 ммоль) в тетрагидрофуране (2 мл). Реакционную смесь перемешивают при комнатной температуре один час, фильтруют и разбавляют хлористым метиленом (30 мл). Полученный раствор промывают раствором тиосульфата натрия (2 мл 1М раствора), насыщенным раствором гидрокарбоната натрия (3 мл), рассолом (5 мл), сушат над смесью сульфата натрия и сульфата магния, фильтруют и концентрируют в вакууме. Маслянистый остаток очищают с помощью колоночной хроматографии на силикагеле (1:1 гексан/этилацетат), что дает 120 мг (67% выход) названного соединения в виде желтого масла. Полученный продукт имеет тот же Rf (TCX) и1НЯМР спектр (300 МГц, СDСl3), что и достоверный образец.
Iс. Синтез дибензилового эфира 0-фосфонооксиметилпропофола
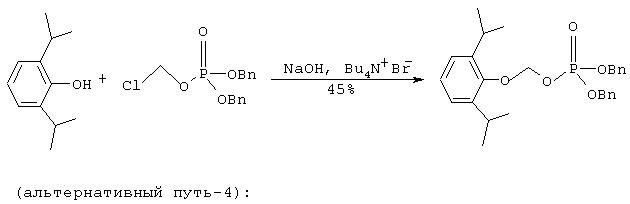
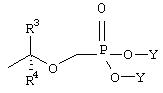
Дополнительно к приведенной выше реакции Iс (альтернативный путь - 4) должно быть отмечено, что реагент:
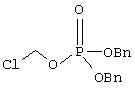
может быть вообщем представлен следующей формулой:
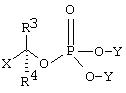
где Х представляет собой удаляемую группу, R3 и R4 каждый представляет собой атом водорода, органическую или неорганическую группу и Y является защитной группой фосфата. Примеры удаляемых групп включают хлор, бром, иод, тозилат или любую другую удаляемую группу. Примеры защитных групп для фосфатных групп включают защитные группы, которые временно блокируют реакционную способность фосфатной группы и позволяют провести селективное замещение с помощью нуклеофильной реакции замещения. Примеры таких блокирующих групп включают, но не ограничиваются, бензилом, аллилом, третичным бутилом и изопропилом, этилом и β-цианоэтилом.
Iс. Синтез дибензилового эфира O-фосфонооксиметилпропофола (альтернативный путь - 5):
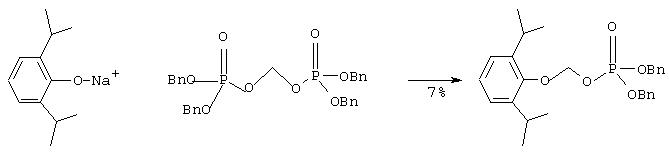
К перемешиваемой суспензии гидрида натрия (36 мг 60%-ная дисперсия в минеральном масле, 0.91 ммоль) в сухом диметоксиэтане (2 мл) в атмосфере аргона добавляют по каплям пропофол (172 мкл, 97%, 0.90 ммоль) в течение более пяти минут. Полученную смесь перемешивают при комнатной температуре дополнительно 20 минут. Затем к смеси добавляют раствор бис-(дибензилфосфоно) ацеталя формальдегида (500 мг, 0.88 ммоль) в сухом диметоксиэтане (3 мл). Реакционную смесь перемешивают при комнатной температуре 20 часов и затем при 70°С 2,5 часа. Затем смесь фильтруют и растворитель упаривают в вакууме. Маслянистый остаток очищают с помощью флэш хроматографии на колонке с силикагелем (гексан, 10:1 гексан/этил ацетат и затем 1:1 гексан/этил ацетат), что дает 29 мг (7% выход) названного соединения в виде желтого масла. Этот продукт имеет тот же Rf (TCX) и1HЯMP спектр (300 МГц, CDCl3), что и достоверный образец.
Id. Синтез O-фосфонооксиметилпропофола:
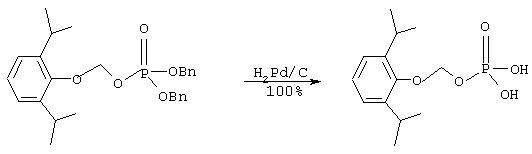
К раствору дибензилового эфира O-фосфонооксиметилпропофола (115 мг, 0.245 ммоль) в метаноле (10 мл) добавляют палладий на угле (10%, 20 мг). Смесь перемешивают в атмосфере водорода (1 атм) 1, 5 часа. Катализатор удаляют фильтрованием через целит и фильтрат упаривают при пониженном давлении, что дает 70.5 мг (100% выход) названного соединения в виде бесцветного масла, нестабильного в случае хранения при комнатной температуре.
FABMS-(GLY): [М-Н]-, м/з 287.
1Н ЯМР (300 МГц, ацетон-d6, δ): 1.19 (д, J=6.8 Гц, 12Н), 3.46 (секст, J=6.8 Гц, 2Н), 5.45 (д, J=9.7 Гц, 2Н), 7.15 с (м, 3Н).13С ЯМР (75 МГц, ацетон-d6, δ): 24.2178, 27.1496, 94.63, 94.65, 124.08, 126.30. 142.46, 152.32.
Iе. Синтез динатриевой соли O-фосфонооксиметилпропофола:
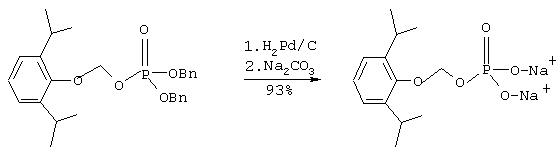
К раствору дибензилового эфира O-фосфонооксиметилпропофола (1.05 г, 2.24 ммоль) в тетрагидрофуране (100 мл) добавляют воду (5 мл) и палладий на угле (10%, 300 мг). Полученную смесь перемешивают в атмосфере водорода (1 атм) 1 час. Катализатор удаляют фильтрованием через целит, и фильтрат обрабатывают раствором гидрокарбоната натрия (263 мг в 3 мл воды, 2.12 ммоль). ТГФ упаривают при пониженном давлении и оставшийся водный раствор экстрагируют с помощью диэтилового эфира (3×3 мл). Водный слой упаривают досуха (в токе аргона или на роторном испарителе) и полученное твердое вещество сушат в течение ночи в вакууме, промывают диэтиловым эфиром (4×4 мл), гексаном (2×4 мл) и вновь сушат в вакууме, что позволяет получить 655 мг (93% выход) названного соединения в виде белого порошка. FABMS-(GLY): [M-2Na+H]-, м/з 287.
1Н ЯМР (300 МГц, D2O, δ): 1.22 (д, J=7.0 Гц, 12Н), 3.46 20 (гепт, J 6.9 Гц, 2Н), 5.27 (д, J=7.5 Гц, 2Н), 7.28 (м, 3Н).
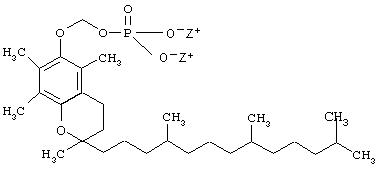
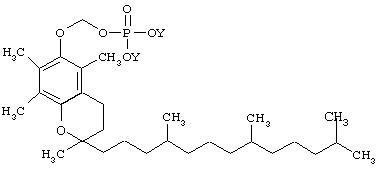
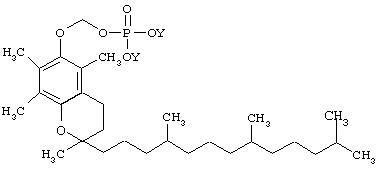
II. Синтез O-фосфонооксиметил-альфа-токоферола
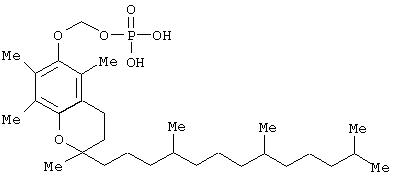
IIа. Синтез дибензилового эфира O-фосфонооксиметил-альфа-токоферола:
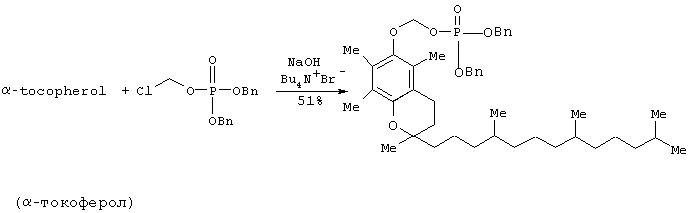
К раствору хлорметилдибензилфосфата (323 мг, 0.98 ммоль), альфа-токоферола (409 мг, 97%, 0.92 ммоль) и тетрабутиламмоний бромида (301 мг, 0.92 ммоль) в бензоле (5 мл) добавляют водный раствор гидроксида натрия (150 мг в 0.2 мл воды, 3.7 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре два часа в атмосфере аргона. Смесь затем разбавляют бензолом (10 мл), промывают водой (3×3 мл), сушат над сульфатом магния, фильтруют и упаривают при пониженном давлении. Коричневый маслянистый остаток очищают с помощью флэш хроматографии на колонке с силикагелем (10:1 гексан/этилацетат), что дает 336 мг (51% выход) названного соединения в виде желтого масла.
FABMS+(NBA): [M]+, м/з 720.
1Н ЯМР (500 МГц, CDCl3, δ): 0.85 (м, 12Н), 1.21 (с, 3Н), 1.27 (м, 24Н), 1.75 (м, 2Н), 2.06 (с, 3Н), 2.11 (с, 3Н), 2.14 (с, 3Н). 2.54 (т, J=6.8 Гц, 2Н), 4.97 (м, 4Н), 5.20 (d, J=9.3 Гц, 2Н), 7.31(м, 10Н).
IIb. Синтез O-фосфонооксиметил-альфато-коферола:
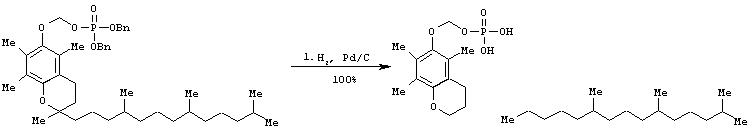
К раствору дибензилового эфира О-фосфонооксиметил-альфа-токоферола (88 мг, 0.12 ммоль) в тетрагидрофуране (10 мл) добавляют палладий на угле (10%, 15 мг). Смесь перемешивают в атмосфере водорода (1 атм) 10 минут (как показал анализ ТСХ, реакция заканчивается через 5 минут). Катализатор удаляют фильтрованием через целит, фильтрат упаривают при пониженном давлении и затем сушат в вакууме. Названное соединение получают в количестве 70 мг (100% выход) в виде коричневого масла, которое нестабильно при комнатной температуре.
FABMS+(NBA): [М]+, м/з 540, [М + Na]+, м/з 563; (NBA + Li)+: [М + Li]+, м/з 547
II c. Синтез динатриевой соли O-фосфонооксиметил-альфа-токоферола
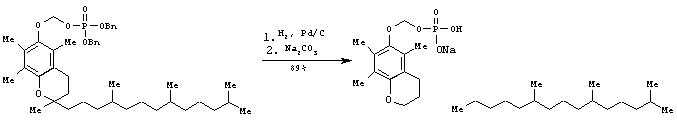
К раствору дибензилового эфира O-фосфонооксиметил-альфа-токоферола (100 мг, 0.14 ммоль) в тетерагидрофуране (10 мл) добавляют палладий на угле (10%, 18 мг). Смесь перемешивают в атмосфере водорода (1 атм) 5 минут. Катализатор удаляют фильтрованием через целит и фильтрат упаривают при комнатной температуре при пониженном давлении, затем полученный остаток растворяют в диэтиловом эфире (2 мл). Эфирный раствор обрабатывают водным раствором гидроксида натрия (11.2 мг в 100 мл воды, 0.28 ммоль) и полученную смесь перемешивают при комнатной температуре 10 минут. Эфирную фазу удаляют, а водную фазу промывают диэтиловым эфиром (3×3 мл) и затем сушат в вакууме 20 часов, что дает 73 мг (89% выход) названного соединение в виде серого твердого вещества.
FABMS+(TG/G): [МН]+, м/з 585, [М + Na]+ м/з 607
Синтез растворимых в воде производных камптотецина также будет дополнительно описан в деталях далее:
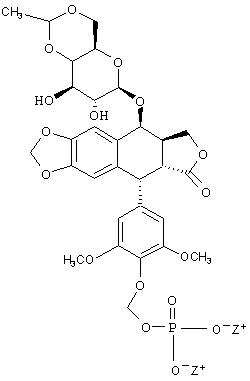
III. Синтез 20-O-Фосфонооксиметилкамтотецина
IIIa. Синтез 20-O-метилтиометилкамптотецина:
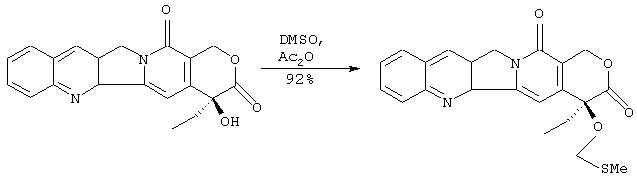
К суспензии камптотецина (5.0 г, 14.3 ммоль) в диметилсульфоксиде (250 мл) добавляют уксусный ангидрид (125 мл) и уксусную кислоту (35 мл). Гетерогенную смесь энергично перемешивают при комнатной температуре 24 часа, выливают на лед (800 мл), перемешивают 30 минут и затем экстрагируют хлористым метиленом (4×100 мл). Объединенные экстракты хлористого метилена промывают водой (2×100 мл) и сушат над сульфатом магния. Хлористый метилен удаляют при пониженном давлении, что дает коричневое твердое вещество. Твердое вещество растворяют в минимальном объеме хлористого метилена. Полученный раствор фильтруют и разбавляют 10-ти кратным избытком гексана, после чего выдерживают в течение ночи в холодильнике. Выпавшее в осадок твердое вещество отфильтровывают, промывают несколько раз гексаном и сушат, что дает 5.38 г (92% выход) названного соединения в виде светло-коричневого порошка. α-123,6°(с 0.55, СНСl3).
FABMS+(NBA): [MH]-, м/з 409.
1НЯМР (400 МГц, CDCl3, δ): 0.93 (т, J=7.2 Гц, 3Н), 2.11 (секст, J=7.6 Гц, 1Н), 2.29 (секст, J=7.6 Гц, 1Н), 2.30 (с, 3Н), 4.58 (с, 2Н), 5.33 (с, 2Н), 5.40 (д, J=17.2 Гц, 1Н), 5.62 (д, J=17.3 Гц, 1Н), 7.48 (с, 1Н), 7.69 (т, J=7.1 Гц, 1Н), 7.86 (т, J=7.1 Гц, 1Н), 7.96 (д, J=8.1 Гц, 1Н), 8.25 (д, J=8.5 Гц, 1Н), 8.42 (с, 1Н).
13С ЯМР (75 МГц, СОС13, δ): 7.76, 14.89, 33.90, 49.92, 66.68, 71.02, 76.57, 97.51, 122.63, 128.02, 128.09, 128.30, 129.71, 130.64, 131.11, 145.14, 146.10, 148.88, 152.27, 157.43, 169.34, 169.73.
IIIb. Синтез дибензилового эфира 20-О-фосфонооксиметилкамптотецина
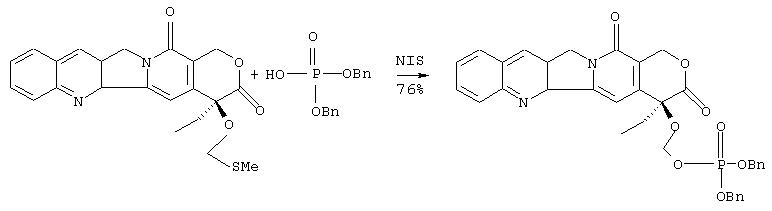
К хорошо перемешиваемой суспензии 20-O-метилтиометилкамптотецина (1.00 г, 2.44 ммоль) и измельченных активированных 4 А молекулярных сит (5 г) в тетрагидрофуране (20 мл) добавляют суспензию N-иодсукцинимида (2.00 г, 95%-ного, 8.44 ммоль) и дибензилфосфата (2.20 г, 7.83 ммоль) в хлористом метилене (12 мл). Полученную смесь энергично перемешивают при комнатной температуре 30 минут, фильтруют и разбавляют этилацетатом (300 мл). Раствор промывают водным раствором тиосульфата натрия (10%, 2×15 мл), водой (2×20 мл), рассолом (50 мл) и сушат над сульфатом магния. Смесь фильтруют и растворитель упаривают при пониженном давлении. Коричневый маслянистый остаток очищают с помощью флэш хроматографии на колонке с силикагелем (98:2 этилацетат/метанол) и сушат в вакууме в течение ночи, что дает 1,19 г (76% выход) названного соединения в виде желтой пены. α-43.1°(с 0.55, СНСl3).
FABMS+(NBA): [MH]+ м/з 639.
1Н ЯМР (400 МГц, CDCl3, δ): 0.91 (т, J=7.4 Гц, 3H), 2.09 (секст, J=7.4 Гц, 1Н), 2.26 (секст J=7.4 Гц, 1Н), 5.06 (м, 4Н), 5.28 (м, 3Н), 5.35 (д, J=17.0 Гц, 1Н), 5.48 (2xд, J=10 10.5 Гц, 1Н), 5.64 (д, J=17.3 Гц, 1Н), 7.59 (с, 1Н), 7.67 (т, J=7.0 Гц, 1Н), 7.80 (т, J=7.1 Гц, 1Н), 7.94 (д, J=8.0 Гц, 1Н), 8.13 (д,=8.5 Гц, 1Н), 8.35 (с, 1Н).
13С ЯМР (100 МГц, CDC13, δ): 7.73, 29.53, 32.49, 49.86, 66.74, 69.37, 69.44, 78.48, 88.99, 89.04, 98.09, 121.55, 127.65, 127.70, 127.90, 128.01, 128,25, 128.35, 128.36, 129.62, 130.48, 130.97, 135.45, 135.55, 145.47, 145.82, 148.76, 152.15, 157.18, 168.67.
IIIc. Синтез 20-O-фосфонооксиметилкамптотецина:
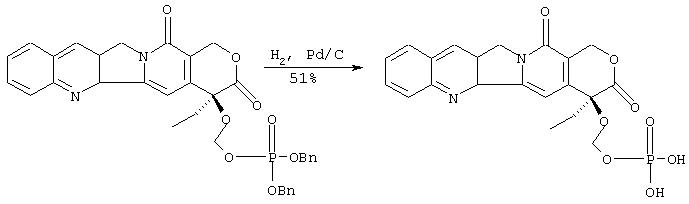
К раствору дибензилового эфира 20-O-фосфонооксиметилкамптотецина (500 мг, 0.78 ммоль) в тетрагидрофуране (100 мл) и воде (5 мл) добавляют палладий на угле (10%, 500 мг). Смесь перемешивают в атмосфере водорода (1 атм) 35 минут. Катализатор удаляют фильтрованием через целит. Целит промывают тетрагидрофураном (300 мл) и объединенные фильтраты упаривают при пониженном давлении. Полученное твердое вещество зеленого цвета промывают диэтиловым эфиром (2×20 мл), гексаном (50 мл), сушат в вакууме и затем растворяют в горячем метаноле (60 мл). Раствор фильтруют, концентрируют при пониженном давлении до остаточного объема 10 мл. После стояния при комнатной температуре в течение часа, раствор помещают в холодильник на ночь. Образовавшийся за ночь кристаллический осадок отфильтровывают и сушат в вакууме, что дает 155 мг названного соединения в виде желтого твердого вещества. Фильтрат концентрируют до объема -1 мл, который выдерживают в холодильники один час, что дает дополнительные 28 мг продукта. Общий выход: 183 мг (51%).
FABMS+(NBA): [MH]+, м/з 459, [М+Na]+, м/з 481.
1Н ЯМР (400 МГц, D2O, δ): 0.95 (т, J=7.5 Гц, 3Н), 2.25 (м. 2Н), 4.98 (д, J=5.0 Гц, 2Н), 5.14 (2xd, J=9.3 Гц, 1H), 5.22 (2xd, J=8.9 Гц, 1H), 5.48 (д, J=17.0 Гц, 1Н), 5.60 (д, J=16.9 Гц, 1H), 7.54 (с, 1H), 7.56 (т, J=7.7 Гц, 1H), 7.77 (т, J=7.2 Гц, 1H), 7.86 (д, J=8.2 Гц, 1H), 8.01 (д, J=8.5 Гц, 1H), 8.44 (с, 1H).
Химическая структура и чистота продукта также определялись с помощью1НЯМР спектроскопии его динатриевой соли, образованной из кислоты и двух мольных эквивалентов гидрокарбоната натрия в D2O.
IIIc. Синтез 20-O-фосфонооксиметилкамптотецина (альтернативный путь
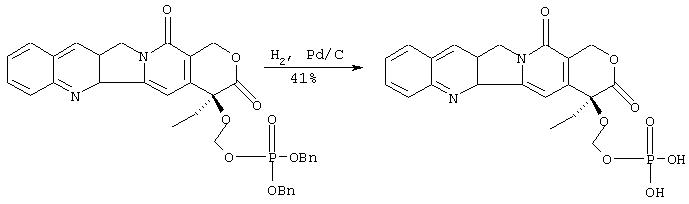
К раствору дибензилового эфира 20-O-фосфонооксиметилкамптотецина (500 мг, 0.78 ммоль) в тетрагидрофуране (100 мл) и воде (5 мл) добавляют палладия на угле (10%, 500 мг). Смесь перемешивают в атмосфере водорода (1 атм) 30 минут. Катализатор удаляют фильтрованием через целит. Целит промывают тетрагидрофураном (2×100 мл), и объединенные фильтраты обрабатывают водным раствором гидрокарбоната натрия (97 мг в 2 мл воды, 0.78 ммоль). ТГФ упаривают при пониженном давлении, водный гетерогенный остаток разбавляют водой (10 мл) и экстрагируют этилацетатом (2×3 мл). Получают желтый гомогенный раствор, который подкисляют соляной кислотой (10%) до рН=1. Полученный остаток отфильтровывают и сушат в вакууме в течение ночи, что дает 145 мг (41% выход) названного соединения в виде желтого твердого вещества.
IIId. Синтез динатриевой соли 20 -O-фосфонооксиметилкамптотецина:
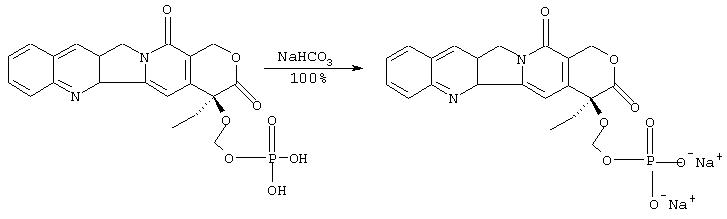
К суспензии 20-O-фосфонооксиметилкамптотецина (5 мг, 10.9 мкмоль) в оксиде дейтерия (0.5 мл) добавляют раствор гидроксида натрия в оксиде дейтерия (50 мкл от 0.44 М раствора = 22 мкмоль). Гетерогенную смесь подвергают разрушению ультразвуком в течение нескольких минут, что дает желтый гомогенный раствор названного продукта.
lHЯMP (400 МГц, D2O, через 10 мин, 96% лактон, 4% карбоксилат, δ): 1.05(т,J=7.2 Гц, 3Н), 2.27 (м, 2Н), 4.57(д, J=18.8 Гц, 1Н), 4.70 (д, J=18.9 Гц, 1Н), 5.06 (дд, J=8.3, J=5.4 Гц, 1Н), 5.18 (дд, J=7.6, J=5.5 Гц, 1Н), 5.45 (д, J=16.7 Гц, 1Н), 5.59 (д, J=16.8 Гц, 1Н), 7.34 (т, J=7.1 Гц, 1Н), 7.41 (с, 1Н), 7.60 (м, 2Н), 7.81 (д, J=8.3 Гц, 1Н), 8.17 (с, 1Н).
IIId. Синтез динатриевой соли 20-O-фосфонооксиметилкамптотецина (альтернативный путь I):
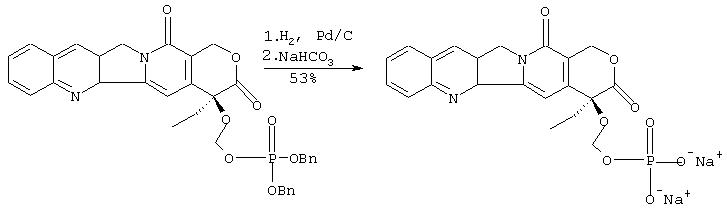
К раствору дибензилового эфира 20-O-фосфонооксиметилкамптотецина (78 мг, 0.122 ммоль) в тетрагидрофуране (10 мл) и воде (3 мл) добавляют палладий на угле (10%, 80 мг). Смесь перемешивают в атмосфере водорода (1 атм) 30 минут. Катализатор удаляют фильтрованием через целит, и фильтрат обрабатывают водным раствором гидрокарбоната натрия (20 мг в 0.5 мл воды, 0.238 ммоль). Желтый осадок отфильтровывают, промывают хлористым метиленом и сушат в вакууме, что дает 35 мг (57% выход) названного соединения (светло-коричневое твердое вещество) в виде его смеси в форме лактона (82%) и карбоксилата (18%) (по1Н ЯМР).
IIId. Синтез динатриевой соли 20-O-фосфонооксиметилкамптофецина (альтернативный путь 2):
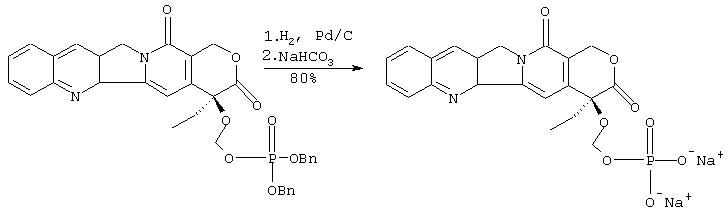
К раствору 20-O-фосфонооксиметилкамптотецина (500 мг, 0.78 ммоль) в тетрагидрофуране (100 мл) и воде (5 мл) добавляют палладий на угле (10%, 500 мг). Смесь перемешивают в атмосфере водорода (1 атм) 30 минут. Катализатор удаляют фильтрованием через целит. Целит промывают тетрагидрофураном (50 мл), и объединенные фильтраты обрабатывают водным раствором гидрокарбоната натрия (90 мг в 2 мл воды, 0.72 ммоль). Тетрагидрофуран упаривают при пониженном давлении, и остаток растворяют в воде (15 мл). Гетерогенную смеси экстрагируют этилацетатом (2×15 мл) и диэтиловым эфиром (20 мл) и полученный водный гомогенный раствор упаривают досуха в токе аргона при комнатной температуре. Остаток сушат в вакууме в течение ночи, что дает 290 мг (80% выход) названного соединения (твердое вещество оранжевого цвета) в виде смеси его в форме лактона (60%), и карбоксилата (40%) и небольшого количества побочных продуктов (определено с помощью1Н ЯМР).
IIIe. Синтез мононатриевой соли 20-O-фосфонооксиметилкамптотедина:
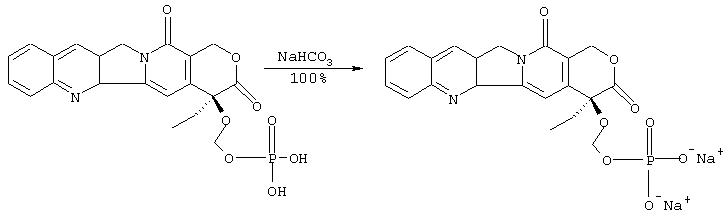
К непрерывно разрушаемой ультразвуком суспензии 20-O-фосфонооксиметилкамптотецина (5 мг, 10 ммоль) в оксиде дейтерия (0.5 мл) добавляют по каплям раствор гидроксида натрия в оксиде дейтерия пока не будет достигнута полная гомогенизация (21 мкл 0.44 М раствора = 9.2 мкмоль). Получают желтый гомогенный раствор названного соединения.
1Н ЯМР (400 МГц, D2O, δ): 1.00 (т, J=7.2 Гц, 3Н), 2.23 (м, 2Н), 4.40 (д, J=18.8 Гц, 1Н), 4.50 (д, J=18.8 Гц, 1Н), 5.10 (дд, J=9.7, J=5.9 Гц, 1Н), 5.26 (дд, J=9.0, J=6.1 Гц, 1Н), 5.39 (д, J=16.7 Гц, 1Н), 5.50 (д, J=16.7 Гц, 1Н), 7.20 (т, J=7.3 Гц, 1Н), 7.28 (с, 1Н), 7.46 (м, 2Н), 7.66 (д, J=8.4 Гц, 1Н), 8.02 (с, 1Н).
IIIf. Синтез лизиновой соли 20-O-фосфонооксиметилкамптотецина:
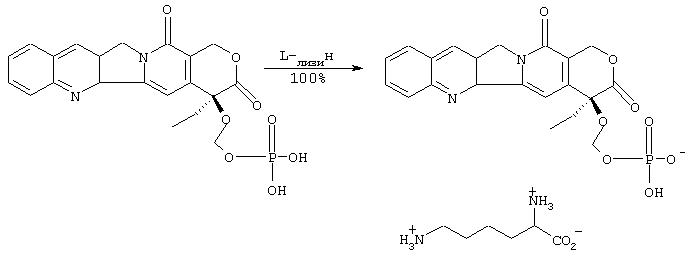
К непрерывно разрушаемой суспензии 20-O-фосфонооксиметилкамптотецина (5 мг, 10 мкмоль) в оксиде дейтерия (0.5 мл) добавляют по каплям раствор L-лизина в оксиде дейтерия (25 мкл 0.43 М раствора = 10.7 мкмоль) пока не будет достигнута полная гомогенизация. Получают желтый гомогенный раствор названного соединения.
1Н ЯМР (400 МГц, D2О, 94% лактон, 6% карбоксилат, δ): 1.02 (т, J=7.2 Гц, 1H), 1.49 (м, 2Н), 1.73 (м, 2Н), 1.88 (м, 2Н), 2.25 (м, 2Н), 3.03 (т, J=7.5 Гц, 2Н), 3.76 (т, J=6.0 10 Гц, 1Н), 4.43 (д, J=19.0 Гц, 1Н), 4.52 (д, J=18.9 Гц, 1Н), 5.11 (дд, J=9.7, J=5.8 Гц, 1Н), 5.27 (дд, J=9.2, J=5.8 Гц, 1Н), 5.41 (д, J=16.7 Гц, 1Н), 5.53 (д, J=16.7 Гц, 1Н), 7.23 (т, J=7.4 Гц, 1Н), 7.30 (с, 1H), 7.49 (м, 2Н), 7.68 (д, J=8.4 Гц, 1Н), 7.04 (с, 1Н).
IIIg. Синтез аргининовой соли 20-O-фосфонооксиметилкамптотецина
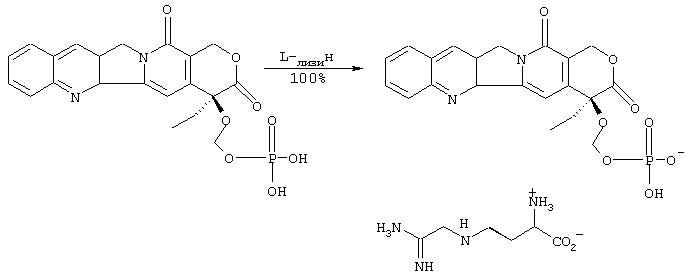
1Н ЯМР (400 МГц, D2O): 1.02 (т, J=7.1 Гц, 1H), 1.66 (м, 2Н), 1.89 (м, 2Н), 2.25 (м, 2Н), 3.20 (т, J=6.8 Гц, 2Н), 3.77 (т, J=6.0 Гц, 1H), 4.40 (д, J=19.0 Гц, 1H), 4.49 (д, J=18.8 Гц. 1H), 5.12 (дц, J=9.7, J=6.0 Гц, 1H), 5.29 (дд, J=8.8, J=6.1 Гц, 1H), 5.40 (д, J=16.7 Гц, 1H), 5.51 (д, J=16, 7 Гц, 1H), 7.20 (т, J=7.3 Гц, 1H), 7.29 (с, 1H), 7.47 (м, 2Н), 7.66 (д, J=8.3 Гц, 1H), 8.03 (с, 1H).
IIIh. Синтез N-метилглюкаминовой соли 20-O-фосфонооксиметилкамптотецина:
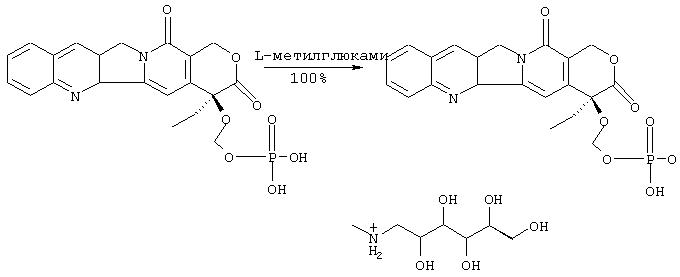
К непрерывно разрушаемой суспензии 20-O-фосфонооксиметилкамптотецина (5 мг, 10,9 мкмоль) в оксиде дейтерия (0.5 мл) добавляют по каплям раствор (D)-N-метилглюкамина в оксиде дейтерия (21 мкл 0.51 М раствора = 10.7 мкмоль) пока не будет достигнута полная гомогенизация. Получают желтый гомогенный раствор названного соединения.
1Н ЯМР (400 МГц, D2O, δ): 1.02 (т, J=7.3 Гц, 3Н), 2.25 (м, 2Н), 2.78 (с, 3Н), 3.20 (м, 2Н), 3.65 (м, 2Н), 3.80 (м, 3Н), 4.11 (м, 1Н), 4.44 (д, J=18.9 Гц, 1Н), 4.53 (д, J=19.0 Гц, 1Н), 5.12 (дд, J=9.8, J=5.9 Гц, 1Н), 5.27 (дд, J=9.2, J=5.9 Гц, 1Н), 5.41 (д, J=16.7 Гц, 1Н), 5.53 (д, J=16.7 Гц, 1Н), 7.23 (т, J=7.4 Гц, 1Н), 7.49 (м, 2Н), 7.69 (д, J=8.4 Гц, 1Н), 8.05 (с, 1Н).
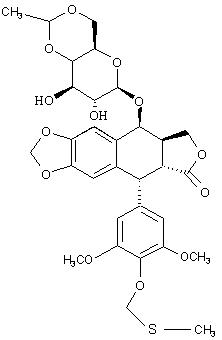
IV. Синтез 4’-O-фосфонооксиметилэтопозида:
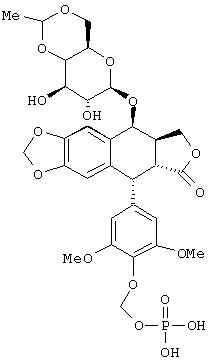
IVa. Синтез дибензилового эфира 4’-O-фосфонооксиметилэтопозида:
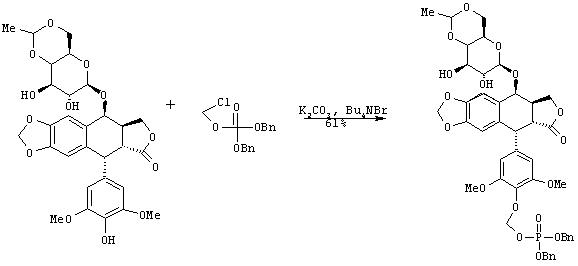
К раствору хлорметилдибензилфосфата (670 мг, 2.05 ммоль), этопозида (300 мг, 0.51 ммоль) и тетрабутиламмония бромида (164.4 мг, 0.51 ммоль) в тетрагидрофуране (0.5 мл) добавляют измельченный карбонат калия (352.4 мг, 2.55 ммоль). Полученную реакционную смесь энергично перемешивают при комнатной температуре 35 минут. Затем смесь непосредственно очищают хроматографией на колонке с силикагелем (30:1 хлористый метилен/метанол), что дает 272 мг (61% выход) названного соединения в виде белого твердого вещества с более чем 95%-ной зафиксированной трансизомерией.
FABMS+(NBA): [MH]+ м/з 879.
1Н ЯМР (400 МГц. СDСl3, δ): 1.41 (д, J=5.0 Гц, 3Н), 2.79 (уширенный с 1Н), 2.86 (м, 1Н), 2.97 (уширенный с, 1Н), 3.30 (дц, J=14.2, J=5.3 Гц, 1H), 3.35 (м, 2Н), 3.45 (т. J=8.5, J=8.0 Гц, 1Н), 3.59 (м, 1Н), 3.66 (с, 6Н), 3.74 (м, 1Н), 4.19 (м, 1Н), 4.20 (т, J=8.5, J=8.0 Гц, 1Н). 4.42 (дд, J=10.3, J=9.1 Гц, 1Н), 4.60 (д, J=5.2 Гц, 1Н), 4.64 (д, J=7.6 Гц, 1Н), 4.76 (кв, J=5.0 Гц, 1Н), 4.92 (д, J=3.4 Гц, 1Н), 5.03 (дд, J=7.3, J=4.3 Гц, 4Н), 5.54 (дд, J=11.7, J=5.1 Гц, 1Н), 5.59 (дд, J=11.3, J=5.1 Гц, 1Н), 5.99 (д, J=3.5 Гц, 2Н), 6.26 (с, 2Н), 6.51 (с, 1Н), 6.84 (с, 1Н), 7.33 (м, 10Н).
13С ЯМР (75 МГц, CDCl3, δ): 20.21, 37.49, 41.00, 43.78, 56.07, 66.32, 67.87, 67.97, 69.06, 69.14, 73.01, 73.29, 74.47, 79.70, 92.55, 92.62, 99.70, 101.57, 101.72, 107.89, 109.13, 110.55, 127.82, 127.97, 128.15, 128.35, 128.43. 132.40, 133.08, 135.68, 135.78, 136.49, 147.14, 148.73, 152.18, 174.90.
IVb. Синтез 4’-O-фосфонооксиметилэтопозида:
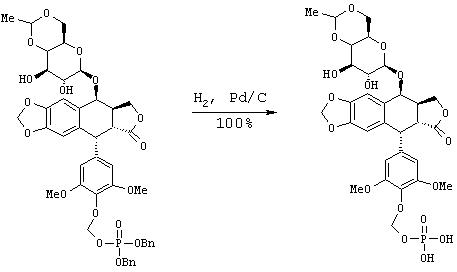
К раствору дибензилового эфира 4’-O-фocфoнooкcимeтилэтoпoзидa (20.5 мг, 0.023 ммоль) в тетрагидрофуране (2 мл) добавляют палладий на угле (10%, 5 мг). Смесь перемешивают в атмосфере водорода (1 атм) 10 минут. Катализатор удаляют фильтрованием через целит и тетрагидрофуран упаривают при пониженном давлении. Полученный остаток сушат в вакууме, что дает 16 мг (100% выход) названного соединения в виде белого твердого вещества.
FABMS+(NBA): [MH]+ м/з 699.
1Н ЯМР (400 МГц, СDСl3/DMSO-d6, δ): 1.29 (д, J=5.0 Гц, 3Н), 2.78 (м, 1Н), 3.21 (м, 2Н), 3.29 (т, J=8.6, J=7.8 Гц, 1Н), 3.37 (дд, J=14.0, J=5.3 Гц, 1Н), 3.52 (м, 2Н), 3.62 (с, 6Н), 4.09 (м, 1Н), 4.17 (т, J=8.1 Гц, 1Н), 4.38 (дд, J=8.8, J=8.7 Гц, 1Н), 4.44 (д, J=7.6 Гц, 1Н), 4.48 (д, J=5.3 Гц, 1Н), 4.66 (кв. J=5.0 Гц, 1Н), 4.88 (д, J=3.3 Гц, 1Н), 5.05 (уширенный с, 7Н), 5.40 (дд, J=10.7, J=7.8 Гц, 1Н), 5.43 (дд, J=10.4, J=7.5 Гц, 1Н), 5.89 (дд, J=8.8 Гц, 1Н), 6.18 (с, 2Н), 6.41 (с, 1Н), 6.78 (с, 1Н).
VIc. Синтез диатртевой соли 4’-O-фосфонооксиметилэтопозида
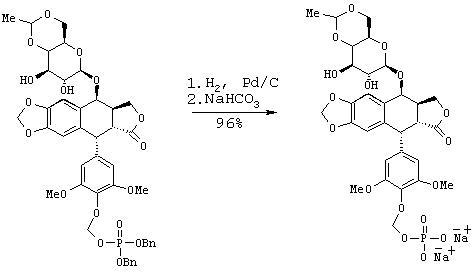
К раствору дибензилового эфира 4’-O-фocфoнooкcимeтилэтoпoзидa (200 мг, 0.227 ммоль) в тетрагидрофуране (10 мл) добавляют палладий на угле (10%, 45 мг). Смесь перемешивают в токе водорода (1 атм) 25 минут. Катализатор удаляют путем фильтрования через целит. Фильтрат упаривают при пониженном давлении, и остаток сушат в вакууме. Полученное в результате белое твердое вещество растворяют в водном растворе гидрокарбоната натрия (2.9 мл 0.136 М=0.394 ммоль). Полученную гетерогенную смесь смешивают с активированным углем, перемешивают несколько минут и затем фильтруют через фильтр с размером пор 40 мкм. Гомогенный, бесцветный фильтрат лиофилизуют, что дает 140 мг (96% выход) названного соединения в виде белого твердого вещества с более, чем на 95% сохраняемой стереохимией.
FABMS+(NBA): [МН]+, м/з 743, [М - Na +2H]+, м/з 721, [М - 2Na +3Н]+ м/з 699.
1Н ЯМР (400 МГц, D2O, δ): 1.37 (д, J=5.1 Гц, 3Н), 3.10 (м, 1Н), 3.37 (дд, J=8.9, J=8.0 Гц, 1Н), 3.48 (м, 2Н), 3.65 (м, 3Н), 3.75 (с, 6Н), 4.29 (дд, J=10.4, J=4.5 Гц. 1Н), 4.41 (т, J=8.3, J=8.0 Гц, 1Н), 4.49 (дд, J=10.5, J=8.9 Гц, 1Н), 4.68 (д, J=5.7 Гц, 1Н), 4.74 (д, J=7.8 Гц, 1Н), 4.91 (кв, J=5.0 Гц, 1Н), 5.13 (д, J=3.0 Гц, 1Н), 5.26 (2хд, J=5.3, J=3.3 Гц, 1Н), 5.28 (2хд, J=5.3, J=3.3 Гц, 1Н), 5.98 (д, J=10.5 Гц, 2Н), 6.40 (с, 2Н), 6.58 (с, 1Н), 7.00 (с, 1Н).
13С ЯМР (125 МГц, D2O, δ): 22.13, 40.74, 43.56, 46.11, 59.12, 68.70, 70.41, 72.40, 75.46, 75.95, 76.95, 82.46, 94.87, 102.88, 103.66, 104.62, 111.14. 112.82, 113.23, 130.73, 135.45, 135.74, 140.22, 149.56, 151.43, 154.94, 166.36, 181.61.
31P ЯМР (200 МГц, D2O, δ): с (2.19).
V. Синтез фосфонооксиметилирующих агентов
Va. Синтез хлорметилдибензилфосфата
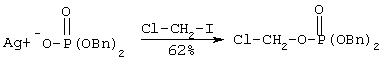
К нагреваемому с обратным холодильником раствору хлориодометана (25 г, 97%, 0.14 моль) в толуоле (HPLC-сорт, 30 мл) добавляют дибензилфосфат серебра (7.0 г, 0.018 моль) в несколько порций в течение более 20 минут. Нагревание с обратным холодильником продолжают 1 час. Затем реакционную смесь охлаждают до комнатной температуры и фильтруют. Растворитель упаривают при пониженном давлении. Маслянистый остаток очищают с помощью флэш хроматографии на колонке с силикагелем (7:3 гексан/этилацетат), что дает 3.63 г (62% выход) названного соединения в виде желтого масла.
FABMS+(NBA): [МН]+, м/з 327
1Н ЯМР (300 МГц, CDCl3, δ): 5.10 (д, J=8.0 Гц, 4Н), 5.63 (д, J=15.7 Гц, 2Н), 7.36 (с, 10Н).
13С ЯМР (75МГц, CDCI3, δ): 69.68, 69.75, 73.33, 73.42, 127.93,128.51, 128.63, 135.07.
Vb. Синтез дибензил (п-толуолсульфонметил)-фосфата:
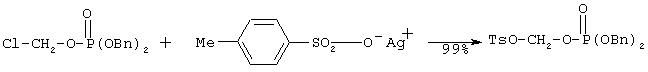
К перемешиваемому раствору п-толуолсульфоната серебра (600 мг, 2.15 ммоль) в сухом ацетонитриле (3 мл) добавляют хлорметилдибензилфосфат (150 мг, 0.46 ммоль) в атмосфере аргона. Затем реакционную смесь перемешивают 21 час при комнатной температуре. Растворитель удаляют и остаток экстрагируют эфиром (3×3 мл). Объединенные экстракты фильтруют, упаривают и сушат в вакууме, что дает 210 мг (99% выход) названного соединения в виде белого твердого вещества.
EIMS:[MH]+, м/з 463.
1Н ЯМР (300 МГц, CDCl3, δ): 2.37 (с, 3Н), 4.91 (2 х д, J=7.9 Гц, 4Н), 5.61 (д, J=14.2 Гц, 2Н), 7.29 (м, 12Н), 7.78 (д, J=8.4 Гц, 2Н).
Со ссылкой на приведенную выше реакцию Vb, с учетом разъяснений, данных в Iс выше, реагент:
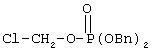
может быть вообщем представлен следующей формулой:
где все символы имеют значения, приведенные выше.
Vc. Синтез формальдегида бис(дибензилоксифосфоно)-ацеталя:
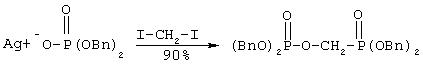
К раствору дииодометана (4 мл, 50 ммоль) в сухом толуоле (15 мл) добавляют дибензилфосфат серебра (3.0 г, 7.8 ммоль). Полученную смесь нагревают с обратным холодильником 15 минут в атмосфере аргона. Смесь затем охлаждают до комнатной температуры и фильтруют. После этого растворитель упаривают в вакууме. Маслянистый остаток очищают с помощью флэш хроматографии на колонке с силикагелем (1:1 гексан/этилацетат и затем этилацетатом) с получением желтого масла, которое затем кристаллизуют, что дает 1.97 г (90% выход) названного соединения в виде белого твердого вещества, т.пл. 39-42°С.
CIMS (NНз): [МН]+, м/з 569.
1Н ЯМР (300 МГц, СDСl3, δ): 5.03 (д, J=7.9 Гц, 8Н), 5.49 (т, J=14.3 Гц, 2Н), 7.30 (м, 20Н).
13С ЯМР (75 МГц, CDCl3, δ): 69.54, 69.61, 86.48, 127.88, 128.48, 128.55, 135.10, 135.20.
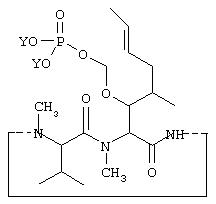
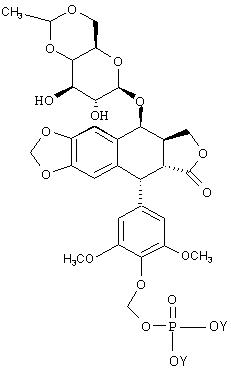
VI Синтез O-фосфонооксиметилциклоспорина А:
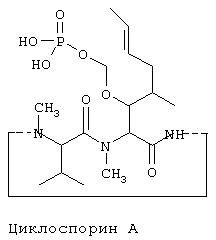
VIa. Синтез O-метилтиометилциклоспорина А:
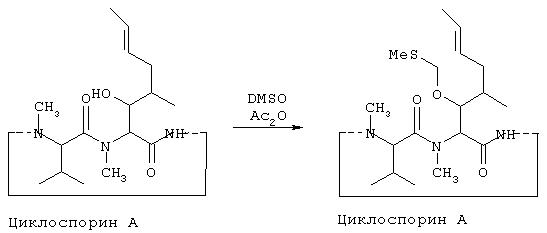
К суспензии циклоспорина А в диметилсульфоксиде (250 мл) добавляют уксусный ангидрид (125 мл) и уксусную кислоту (35 мл). Гетерогенную смесь энергично перемешивают при комнатной температуре 24 часа, выливают на лед (800 мл), перемешивают 30 минут и затем экстрагируют хлористьм метиленом (4×100 мл). Объединенные экстракты хлористого метилена промывают водой (2×100 мл) и сушат над сульфатом магния. Хлористый метилен удаляют при пониженном давлении, что приводит к образованию целевого соединения. Его затем дополнительно очищают с помощью хроматографии на силикагеле.
VIb. Синтез дибензилового эфира O-фосфонооксиметилциклоспорина А:
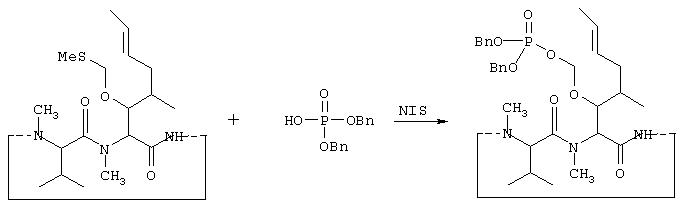
К хорошо перемешиваемой суспензии O-метилтиометилциклоспорина А и измельченных, активированных молекулярных сит, 4А (5 г) в тетрагидрофуране (20 мл) добавляют суспензию N-иодсукцинимида (2.00 г, 95%-ный, 8.44 ммоль) и дибензилфосфата (2.20 г, 7.83 ммоль) в хлористом метилене (12 мл). Полученную смесь энергично перемешивают при комнатной температуре 30 минут, фильтруют и разбавляют этилацетатом (300 мл). Раствор промывают водным раствором тиосульфата натрия (10%, 2×15 мл), водой (2×20 мл), рассолом (50 мл) и сушат над сульфатом натрия. Смесь фильтруют и растворитель упаривают при пониженном давлении. Остаток очищают с помощью флэш хроматографии на колонке с силикагелем.
VIc Синтез O-фосфонооксиметилциклоспорина А:
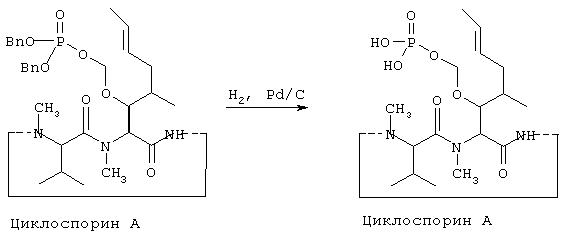
К раствору дибензилового эфира O-фосфонооксиметилциклоспорина А в тетрагидрофуране (100 мл) и воде (5 мл) добавляют палладий на угле (10%, 500 мг). Смесь перемешивают в атмосфере водорода (1 атм) 35 минут. Катализатор удаляют фильтрованием через целит. Затем целит промывают тетерагидрофураном (300 мл) и объединенные экстракты упаривают при пониженном давлении. Полученное твердое вещество промывают эфиром (2×20 мл), гексаном (50 мл), сушат в вакууме, и затем растворяют в горячем метаноле (60 мл). Раствор фильтруют, концентрируют при пониженном давлении до объема -10 мл. После стояния при комнатной температуре в течение одного часа, раствор помещают в холодильник на ночь. Кристаллический осадок, образовавшийся ночью, отфильтровывают и сушат в вакууме, что приводит к названному соединению в виде твердого вещества. Фильтрат концентрируют до объема - I мл и выдерживают в холодильнике один час, что дает дополнительное количество продукта.
БИОЛОГИЧЕСКИЕ ИСПЫТАНИЯ
Соединения по настоящему изобретению являются новыми фармацевтическими агентами; характерные соединения формулы I были испытаны как в опытах по исследованию их превращения in vitro, так и in vivo. Во всех указанных исследованиях пролекарства превращались в соответствующие фармацевтически активные исходные соединения.
(1) Оценка растворимости пролекарства пропофола в воде
Растворимость в воде пролекарства пропофола оценивается приблизительно как 500 мг/мл, исходя из данных HPLC (высокоэффективной жидкостной хроматографии, ВЭЖХ) анализа насыщенного водного раствора.
(2) Превращение пролекарства пропофола в профол in vitro
Превращение in vitro пролекарства пропофола в пропофол осуществляют при использовании щелочной фосфатазы в среде глицинового буфера с рН 10.4. Готовят 25 мл раствора при 100 мкг/мл пролекарства из пропофола в глициновом буфере. Один мл сохраняют для определения нулевой отметки во времени, оставшиеся 24 мл помещают на водяную баню при 37°С. К 24 мл раствора пролекарства из пропофола добавляют 960 мкл раствора щелочной фосфатазы в глициновом буфере с концентрацией 0.1 мг/мл, перемешивают и возвращают на водяную баню. Образцы объемом 1.5 мл отбирают через 5, 10, 20, 30, 40, 60, 90, 120, 180, 240, 300 и 360 минут. К каждому образцу сразу добавляют 10 мкл ледяной уксусной кислоты, чтобы прекратить ферментативную реакцию. Образцы анализируют с помощью ВЭЖХ, что позволяет определить концентрацию пропофола и пролекарства пропофола. Результаты превращения in vitro представлены на фиг. 1. Эти результаты показывают, что пролекарство пропофола является субстратом для щелочной фосфатазы.
(3) Оценка наивысшей токсичности на крысах.
Пролекарство пропофола готовят для внутривенной инъекции с концентрацией 68 мг/мл в 0.9% натрий хлорной инъекции, USP. Указанная концентрация эквивалентна 36 мг/мл пропофола. Перед введением, раствор пролекарства пропофола фильтруют через 0.22 мкм найлоновую мембрану.
Исследования пролекарства пропофола на крысах проводят с использованием двух мужских особей, Harlen Sprague-Dawley, крыс весом 820 и 650 г. Крысе, весом 820 г, вводят 200 мкл пролекарства пропофола внутривенно (эквивалент 9 мг/кг пропофола), в хвостовую вену. Образец крови берут из хвостовой вены приблизительно через 12 минут (с помощью гепаринизированного шприца). Крысе весом 650 г получают дозу средне седативного Metaphane® перед введением состава с пролекарством пропофола. Крысе весом 650 г инъецируют 125 мкл состава с пролекарством пропофола в хвостовую вену и образец крови из хвостовой вены отбирают приблизительно через шесть минут с помощью гепаринизированного шприца. Образцы крови от обеих крыс анализируют для обнаружения пропофола с помощью ВЭЖХ. Результаты инъекции пролекарства пропофола у обеих крыс были одинаковыми. Обе крысы стали неподвижными через несколько минут, но ни одна не потеряла установочного рефлекса. Основываясь на визуальных наблюдениях, можно сказать, что крысы полностью восстановились после инъекций пролекарства пропофола. Кровь, взятая у обеих крыс, показала наличие пропофола при анализе ВЭЖХ. Крысы не проявляли признаков дискомфорта из-за пролекарства пропофола.
(4) Фармакокинетические исследования на собаках
Фармакокинетическое исследование, включающее Diprivan® или пролекарство пропофола, осуществляют на собаке с существенным периодом вымывания между опытами. Концентрации веществ в крови определяют, используя ВЭЖХ с флуоресцентным детектором, в то время как активность мозга изучают при помощи двух отводов для электроэнцефалографии (EEG). Прежде чем вводить собаке дозу, ей накладывают повязку на глаза, уши затыкают ватой и ноги привязывают, чтобы свести на нет ее движения, а также другие внешние воздействия, так, чтобы наиболее эффективно провести наблюдение за воздействием пропофола на электроэнцефалограмму мозга собаки.
Определение концентрации пропофола в крови в зависимости от времени проводят на гончей собаке весом 13 кг. Приблизительно 8 мл крови забирают перед инъекцией, чтобы использовать ее при построении стандартной кривой и в качестве нулевого временного уровня крови. Собака получает Diprivan или состав пролекарства в объеме, эквивалентном 7 мг/кг пропофола на инъекцию в головную вену.
Забирают образцы крови объемом 2 мл из головной (но не из той вены, в которую введена инъекция), яремной или подкожной вены ноги (с помощью гепаринизированного шприца) через 1, 3, 5, 10, 15, 20 и 30 минут после инъекции. Образцы крови также забирают через 60, 90, 120, 180, 240, 300, 360, 480 и 1440 минут. Образцы крови экстрагируют, чтобы немедленно удалить пропофол после того, как их взяли у собаки. Собаку заставляют голодать приблизительно 20 часов, прежде, чем вводят Diprivan® или состав пролекарства пропофола. Через 120 минут берут образец крови и собаке позволяют пить воду. Пищу собаке дают после того, как получен образец крови, соответствующий 480 минутам. Регулярной диетой собаки является диета Hills’ Science Maintenance. Собака находится в цикле свет/тьма, в котором 12 часов в день бывает свет.
Концентрацию пропофола в образцах крови определяют, используя ВЭЖХ с флуоресцентным детектором. Результаты представлены на фиг 2. Использование методов экстракции крови и ВЭЖХ основывается на работах Plummer (1987) с минимальными модификациями. Приготовление образца и процесс исследования осуществляют следующим образом.
К образцу крови 1 мл добавляют 10 мкл внутреннего стандарта тимола (20 мг/мл) и 1 мл фосфатного буфера (0.1 М, рН 7.2), проводя перемешивание после каждого прибавления. Затем добавляют пять мл циклогексана и образцы перемешивают при 75 об/мин 20-30 минут. Органический слой отделяют путем центрифугирования в течение 1 минуты приблизительно со скоростью 2000 об/мин. Приблизительно 4.5 мл органического слоя помещают в пробирку, содержащую 50 мкл разбавленного раствора гидроксида тетраметиламмония (ТМАН) приблизительно до 1.8% (вес/объем). Растворитель упаривают досуха в токе азота и вновь разбавляют 200 мкл подвижной фазы А. Образцы центрифугируют при 15000 об/мин 30 секунд, чтобы удалить любые частицы, и супернатант инжектируют в прибор для ВЭЖХ. Образцы для стандартной кривой готовят путем смешивания 1 мл аликвот первоначальной крови, с пропофолом в концентрациях 5, 1, 0.5, 0.1 и 0.01 мкг/мл. Эти стандарты обрабатывают так же как и образцы.
Система ВЭЖХ включает следующие компоненты Shimadzu: LC-10AT насосы, SCL-10А системный контролер, RF 353 флуоресцентный детектор и SIL-10A автопробоотборник. Параметры ВЭЖХ следующие: возбуждение при 275 нм и эмиссия при 320 нм; скорость потока 1 мл/мин; объем инъекции составляет 3-30 мкл в зависимости от концентрации пропофола. Колонка ВЭЖХ представляет собой колонку Zorbax RX-C18, 15 см × 4.6 нм внутренний диаметр, размер частиц 5 мкм. Подвижная фаза А 60:40 (об/об) ацетонитрил: 25 мМ фосфат, 15 мМ ТВАР Буфер рН 7.1. Подвижная фаза В 80:10:10 (об/об/об) ацетонитрил:вода:ТГФ. Подвижную фазу В используют для очистки колонки после элюирования тимола и пропофола при использовании подвижной фазы А (4.2 и 7,4 минут соответственно).
Собака проявляла признаки наркоза после инъекции обоих составов, как показали визуальные наблюдения и EEG диаграммы. Собака пришла в себя после наркоза, вызванного обоими составами, через 20-30 минут. Уровни пропофола в крови в результате инъекции пролекарства пропофола приближаются к тем, которые возникают при инъекции Diprivan®.
(5) Оценка растворимости пролекарства камптотецина в воде
Растворимость пролекарства камптотецина выше, чем 50 мг/мл, судя по визуальному и ВЭЖХ анализам.
(6) Ферментативное изучение пролекарства камптотецина (p-cpt)
16 мкг/мл p-cpt разрушают с помощью кислотной фосфатазы (0.02 единиц/мл p-cpt раствора). В качестве среды используют 0,09 М цитратный буфер, рН 4.8 и температуру 37°С. Превращение p-cpt в камптотецин фиксируют с помощью ВЭЖХ.
Параметры ВЭЖХ:
МР: 24% калий фосфатный буфер рН 4, 76% ацетонитрил
Колонка: Zorbax RX-C18, 15 cm × 4.6 мм внутренний диаметр, 5 мкм размер частиц
Определение: 370 нм UV
Скорость потока: 1 мл/мин
Кислотная фосфатаза из простаты быка (сигма). Результаты представлены на фиг. 3. Результаты демонстрируют, что пролекарство камптотецина является субстратом для кислотной фосфатазы.
(7) Фармакокинетические исследования пролекарства камптотецина при использовании крыс
Были проведены фармакокинетические эксперименты, включающие введение доз состава пролекарства камптотецина и камптотецина мужским особям крыс Sprague-Dawley. Два состава пролекарства камптотецина, которые были подвергнуты исследованию, состояли из пролекарства, растворенного в 15 мМ фосфате, рН 4.0 и камптотецина, растворенного в органических со-растворителях. Сущность фармакокинетических экспериментов такова:
Объем состава пролекарства камптотецина или состава камптотецина готовят с такой концентрацией, чтобы крысе можно было ввести дозу эквивалентную 1 мг камптотецина на кг веса. Состав вводят крысе, используя канюлю, постоянно находящуюся в левой яремной вене крысы.
Образцы крови забирают через канюлю постоянно находящую, на правой яремной вене крысы. Обе канюли перед использованием промывают гепариновым солевым раствором, и они содержат гепариновый солевой раствор на протяжении эксперимента.
Крысам дают наркоз с помощью пентобарбитала натрия перед тем, как вставить яремные канюли, и поддерживают наркоз с помощью пентабарбитала натрия на протяжении всего исследования. Во время опыта крыс помещают на мягкую прокладку, нагреваемую до 37°С и трахеотомируют. Забирают образцы крови приблизительно по 150 мкл до введения дозы и через 1, 3, 5, 10, 15, 20, 30, 45, 60 и 90 минут после введения крысам составов.
Образцы крови помещают в микроцентрифужные пробирки и центрифугируют 20 секунд приблизительно при 15000 об/мин.
В другие микроцентрифужные пробирки помещают 50 мкл аликвоты плазмы из каждого образца крови. К плазме добавляют 150 мкл аликвоту охлажденного ацетонитрила и препарат встряхивают 5 секунд. Затем добавляют 450 мкл аликвоту охлажденного фосфата натрия (0.1 М, рН 7.2). Содержимое микроцентрифужных пробирок встряхивают 5 секунд и центрифугируют 20 секунд при приблизительно 15000 об/мин. Супернатант помещают в пробоотборник образцов ВЭЖХ при 4°С и анализируют (инъекции по 50 мкл).
Система ВЭЖХ включает следующие Shimadzu компоненты: LC-10AT насос, SCL-10А системный контролер, RF 535 флуоресцентный детектор, SIL-10A пробоотборник образцов (находящийся при 4°С), и СТО-10А нагревательную камеру (находится при температуре 30°С).
Параметры ВЭЖХ следующие: возбуждение при 370 нм и эмиссия при 435 нм; скорость потока 2 мл/мин; Колонка ВЭЖХ представляет собой колонку Hypersil ODS, 15 см × 4.6 нм внутренний диаметр, размер частиц 5 мкм. Подвижная фаза состояла из 75% 25 мМ фосфата натрия, рН 6.5/25% ацетонитрил (об/об) с 25 мМ дигидрофосфатом тетрабутиламмония, добавляемого в качестве снижающего ионизированность реагента.
Как можно видеть на фиг. 4, пролекарство обеспечивает уровни камптотецина в плазме крови, которые эквивалентны уровням, достигаемым при непосредственной инъекции камптотецина в органических сорастворителях. На чертеже изображено среднее значение со стандартным отклонением для пяти крыс, которым вводили пролекарство и шести крыс, которым вводили камптотецин.
Реферат
Изобретение относится к новым растворимым в воде фосфонооксиметиловым эфирам затрудненных спиртов и фенолов. Описываются фосфонооксиметиловые эфиры формулы I:
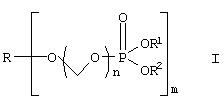
где R-О- представляет собой остаток фармацевтического соединения, содержащего спирт или фенол, за исключением таксола и производных таксола, R1 является водородом или ионом щелочного металла, или протонированным амином, или протонированной аминокислотой, R2 является водородом или ионом щелочного металла, или протонированным амином, или протонированной аминокислотой, n является целым числом 1 или 2; m является целым числом и имеет значение по крайней мере 1; и их фармацевтически приемлемые соли. Также описываются промежуточные соединения и способы их получения, фармацевтическая композиция, обладающая анастезирующим действием, фармацевтическая композиция, обладающая противоопухолевой активностью, способ анастезии и способ лечения опухолевых заболеваний. Технический результат – настоящее изобретение обеспечивает растворимые в воде формы лекарств, содержащие спирт и фенол. 11 н. и 9 з.п. ф-лы, 4 ил.

Формула
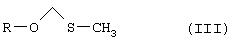

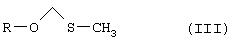
Комментарии