Цитотоксические пептиды и их конъюгаты антитело-лекарственное средство - RU2586885C2
Код документа: RU2586885C2
Чертежи























































































































































Описание
Перекрестная ссылка на родственные заявки
Данная заявка испрашивает приоритеты предварительной заявки на патент США No. 61/561255, поданной 17 ноября 2011 года, и предварительной заявки на патент США No. 61/676423, поданной 27 июля 2012 года, обе из которых включены в данное описание посредством ссылки во всей их полноте.
Область изобретения
Настоящее изобретение относится к новым соединениям на основе пептидов, применимых в качестве полезных нагрузок в конъюгатах антитело-лекарственное средство (ADC), и соединениям-конструкциям полезная нагрузка-линкер, применимых в связывании с ADC. Настоящее изобретение дополнительно относится к композиции, включающей вышеупомянутые полезные нагрузки, конструкции полезная нагрузка-линкер и ADC, и способам использования этих полезных нагрузок, конструкций полезная нагрузка-линкер и ADC для лечения патологических состояний, включая рак.
Предшествующий уровень техники
Конъюгирование лекарственных средств с антителами, непосредственно или через линкеры, включает рассмотрение ряда факторов, в том числе идентичность и расположение химической группы для конъюгирования лекарственного средства, механизм высвобождения лекарственного средства, структурные элементы, обеспечивающие высвобождение лекарственного средства, и структурную модификацию для высвобождения свободного лекарственного средства. Кроме того, если лекарственное средство должно высвобождаться после интернализации антитела, механизм высвобождения лекарственного средства должен согласовываться с внутриклеточным транспортом конъюгата.
Хотя множество различных классов лекарственных средств было исследовано в отношении доставки через антитела, всего лишь несколько классов лекарственных средств проявили эффективность в качестве конъюгатов антитело-лекарственное средство, в то же время проявляя приемлемый профиль токсичности. Одним из таких классов являются ауристатины, производные природного продукта доластатина 10. Иллюстративные ауристатины включают (N-метилвалин-валин-долаизолейцин-долапролин-норэфедрин) и (N-метилвалин-валин-долаизолейцин-долапролин-фенилаланин). Однако, остается необходимость в дополнительных ауристатинах с улучшенными свойствами.
Краткое изложение сущности изобретения
Настоящее изобретение относится к цитотоксическим пентапептидам и их конъюгатам антитело-лекарственное средство, представленным формулой:

или их фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
W представляет собой


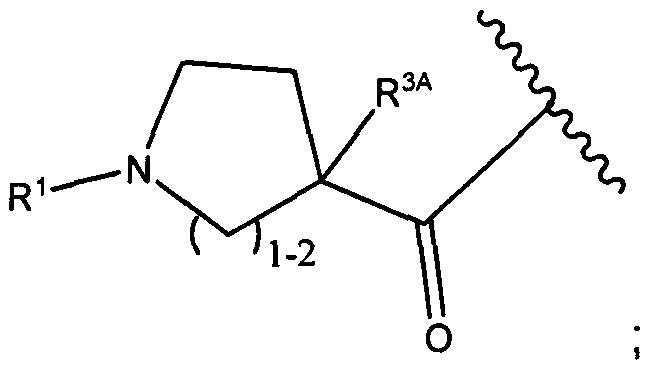
R1 представляет собой водород, C1-C8алкил или C1-C8галогеналкил, или R1 представляет собой линкер или линкер-антитело, например
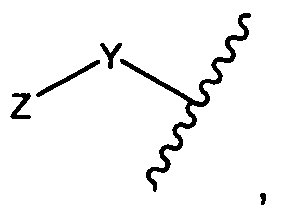
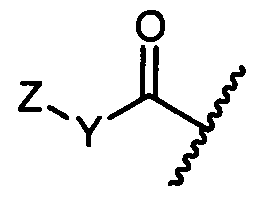

Y представляет собой -C2-C20алкилен-, -C2-C20гетероалкилен-, C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z представляет собой
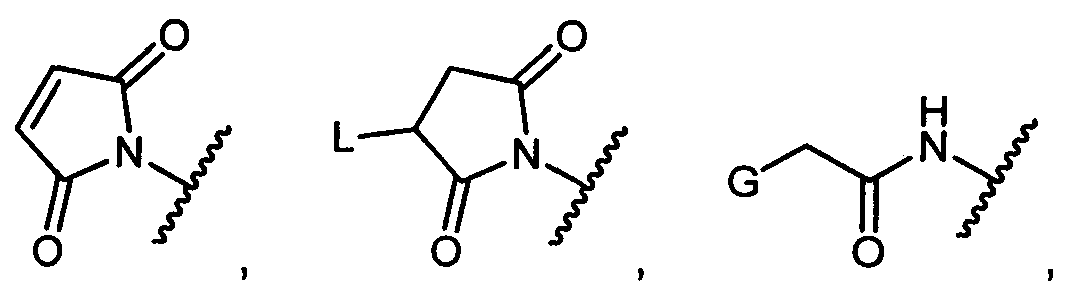


G представляет собой галоген, -OH, -SH или -S-C1-C6алкил;
L представляет собой антитело;
R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
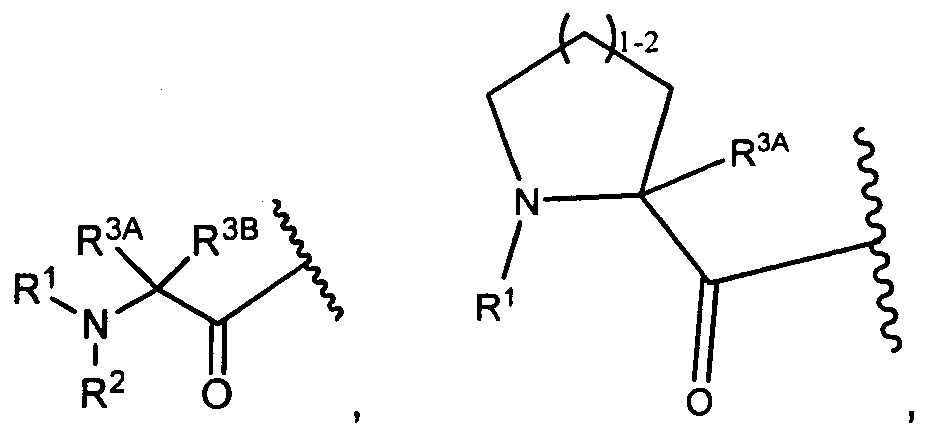
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой




или R5 представляет собой
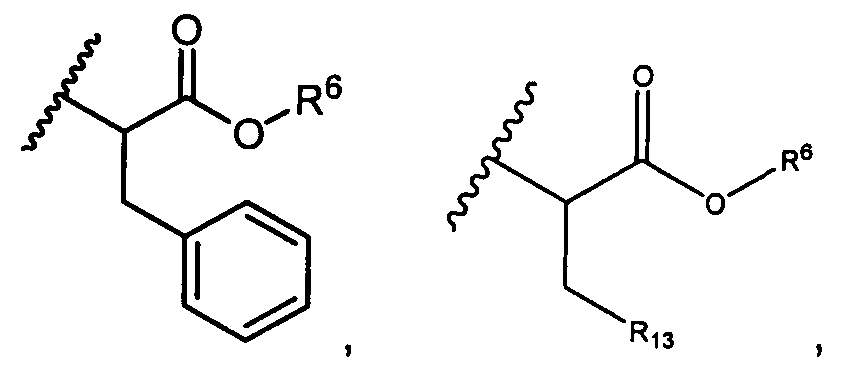


R6 представляет собой водород, -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил или -C1-C8галогеналкил;
R12 представляет собой водород, C1-C4алкил, C1-C10гетероциклил или C6-C14арил;
R13 представляет собой C1-C10гетероциклил; и
R7 представляет собой F, Cl, I, Br, NO2, CN и CF3;
h равен 1, 2, 3, 4 или 5; и
X представляет собой O или S;
при условии, что, когда R3A представляет собой водород, тогда X представляет собой S.
Настоящее изобретение относится к цитотоксическим пентапептидам и их конъюгатам антитело-лекарственное средство, представленным формулой:

или их фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
W представляет собой



R1 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил, галоген или водород; или
(2) R3A и R3B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой


или

возможно замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: C1-C8алкил, -O-(C1-C8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -OH, галоген, -N3, -NH2, -NH(R′), -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2, -S(=O)2R′ и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, C1-C8алкил и незамещенного арила;
R11 представляет собой водород, C1-C8алкил, C1-C8галогеналкил, или R11 представляет собой линкер или линкер-антитело, например



Y представляет собой C2-C20алкилен или C2-C20гетероалкилен; C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z представляет собой



G представляет собой галоген, -OH, -SH или -S-C1-C6алкил;
L представляет собой антитело;
R7 представляет собой F, Cl, I, Br, NO2, CN и CF3;
h равен 1, 2, 3, 4 или 5; и
X представляет собой О или S.
Еще один аспект изобретения относится к фармацевтической композиции, включающей эффективное количество любого из вышеупомянутых соединений и/или любого из вышеупомянутых конъюгатов антитело-лекарственное средство и фармацевтически приемлемый носитель или наполнитель.
Еще один аспект изобретения относится к способу использования эффективного количества любого из вышеупомянутых соединений и/или любого из вышеупомянутых конъюгатов антитело-лекарственное средство для лечения рака, включающему введение пациенту, нуждающемуся в этом, эффективного количества указанного соединения и/или конъюгата.
Еще один аспект изобретения относится к способу лечения рака, где указанный рак включает опухоль, метастаз или другое заболевание или расстройство, характеризующееся неконтролируемым клеточным ростом, где указанный рак выбран из группы, состоящей из рака мочевого пузыря, молочной железы, шейки матки, толстой кишки, глиомы, рака эндометрия, почки, легкого, пищевода, яичника, предстательной железы, поджелудочной железы, меланомы, рака желудка и яичек.
Краткое описание графических материалов
На Фиг. 1 показан график противоопухолевой активности четырех конъюгатов (каждый введен в дозе 1 мг/кг, Q4d×4) в виде зависимости объема опухоли от времени.
На Фиг. 2 показан график противоопухолевой активности шести конъюгатов (каждый введен в дозе 1 мг/кг, Q4d×4) в виде зависимости соотношения объем опухоли, обработанной лекарством/объем опухоли, обработанной носителем, от времени.
На Фиг. 3 показаны результаты тестирования H(C)-#D54 и H(C)-vcMMAE в дозе 1 мг/кг.
На Фиг. 4A, 4B и 4C показаны [A] результаты тестирования H(C)-#D54 и H(K)-MCC-DM1 в модели скрининга in vivo мышиного ксенотрансплантата MDA-MB-361-DYT2; [B] результаты тестирования H(C)-vcMMAE и H(C)-mcMMAF в модели скрининга in vivo мышиного ксенотрансплантата MDA-MB-361-DYT2; и [C] сравнение рассчитанного соотношения T/C для всех четырех конъюгатов. Мышей лечили по схеме q4d×4, начиная с суток 1.
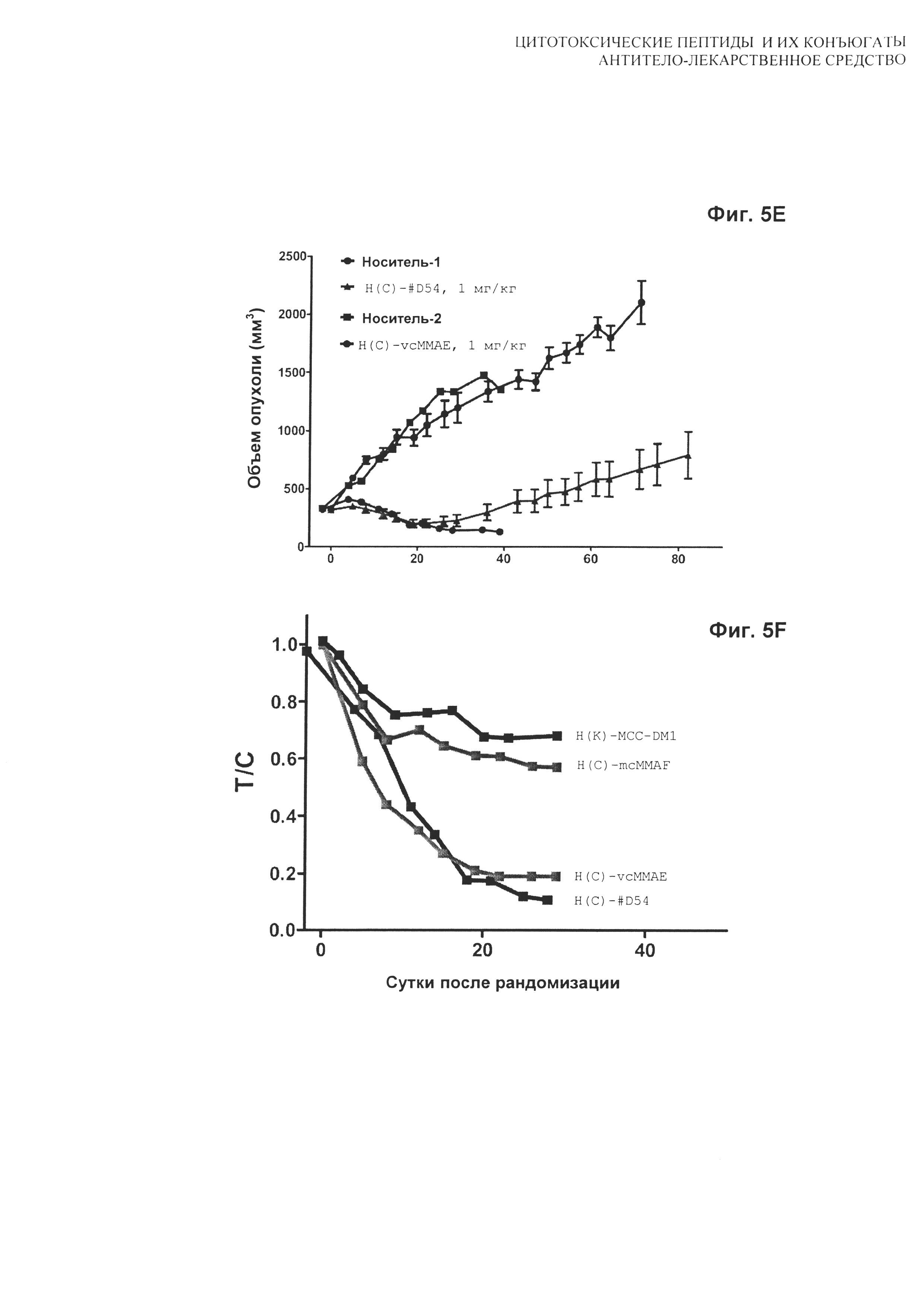
На Фиг. 5A, 5B, 5C, 5D, 5E и 5F показаны дозозависимые результаты тестирования [A] H(C)-#D54, [B] H(C)-vcMMAE, [С] H(C)-mcMMAF и [D] H(K)-MCC-DM1 в модели in vivo мышиного ксенотрансплантата N87; [E] сравнение H(C)-#D54 и H(C)-vcMMAE; и [F] сравнение T/C для всех четырех конъюгатов. Мышей лечили по схеме q4d×4, начиная с суток 1.
На Фиг. 6 показаны дозозависимые результаты тестирования H(C)-#A115 при 1 мг/кг, 3 мг/кг и 10 мг/кг в модели in vivo мышиного ксенотрансплантата N87. Мышей лечили по схеме q4d×4, начиная с суток 1.
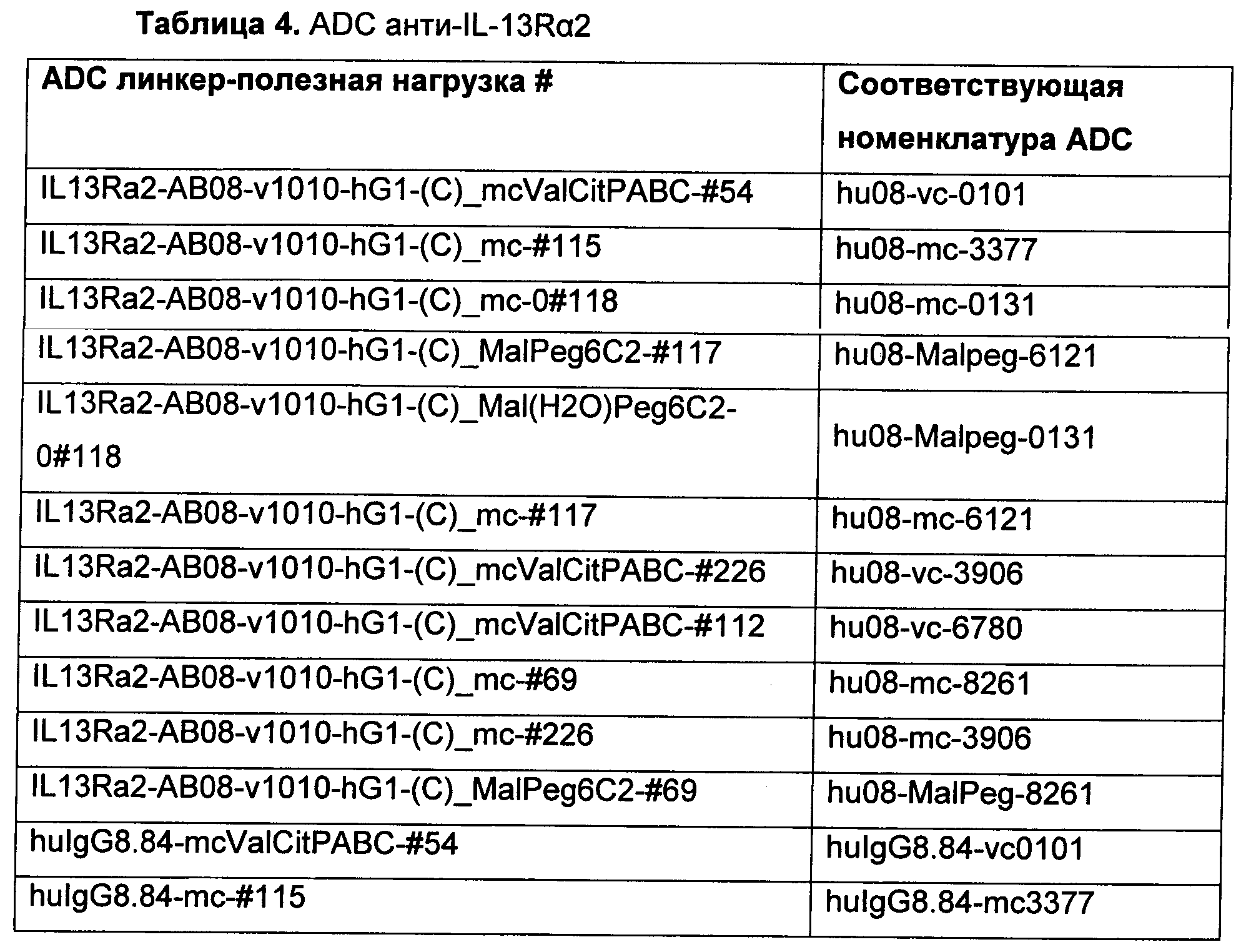
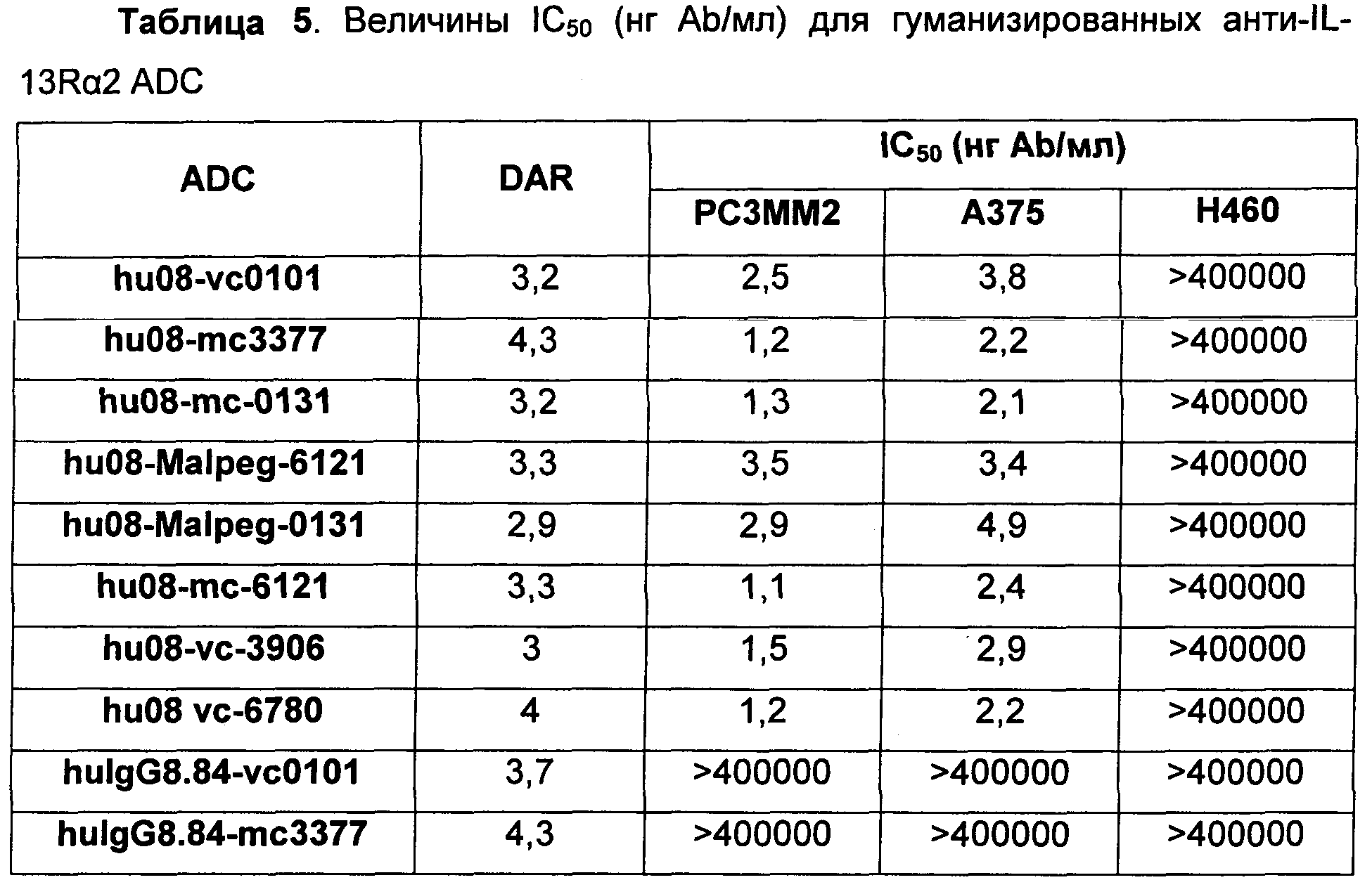
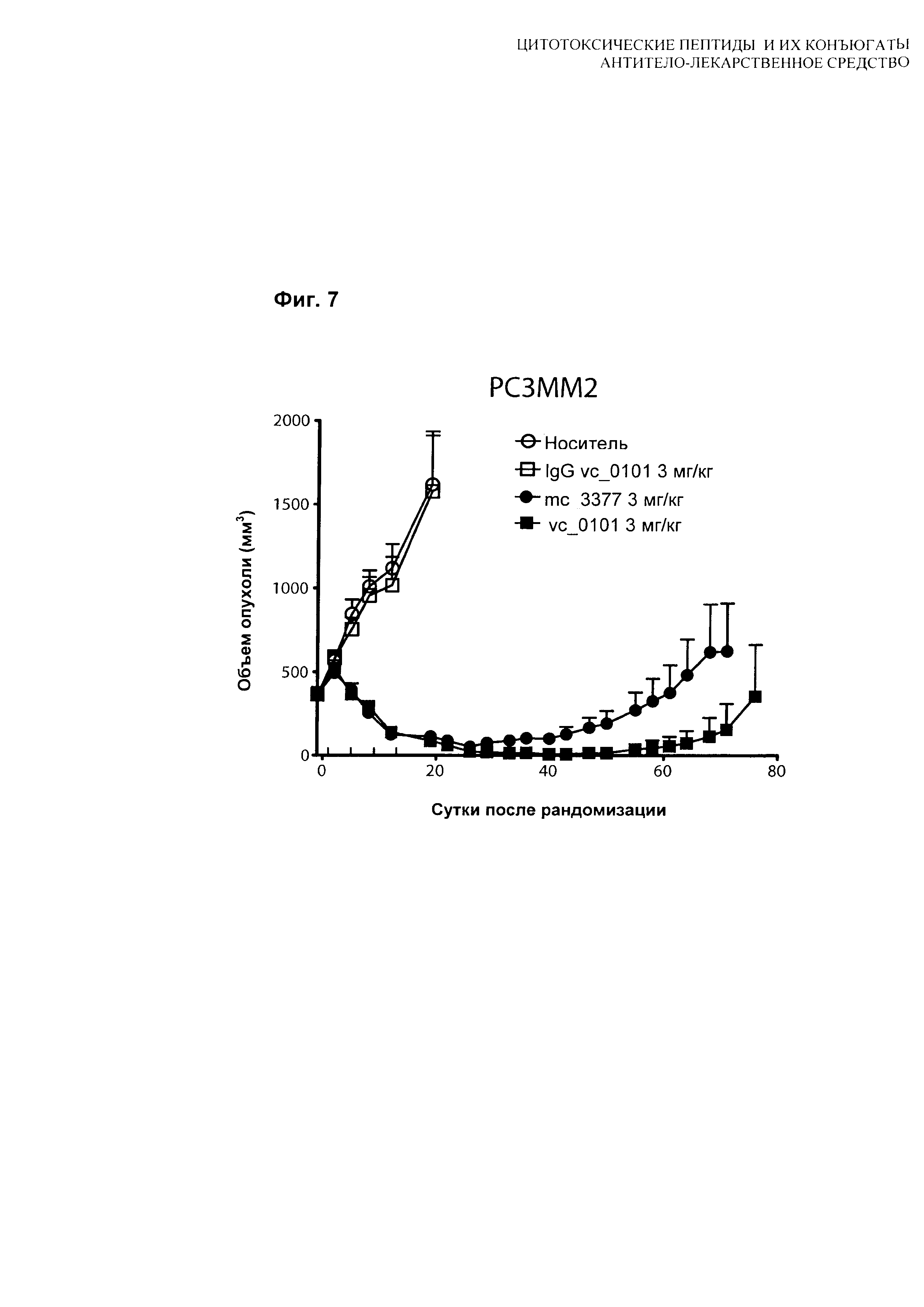
На Фиг. 7 показаны данные сравнения гуманизированного антитела hu08, конъюгированного с vc-0101 или тс-3377, тестированного в модели in vivo ксенотрансплантата с PC3MM2 клетками, клеточной линией рака простаты человека, экспрессирующей IL-13Rα2 рецептор.
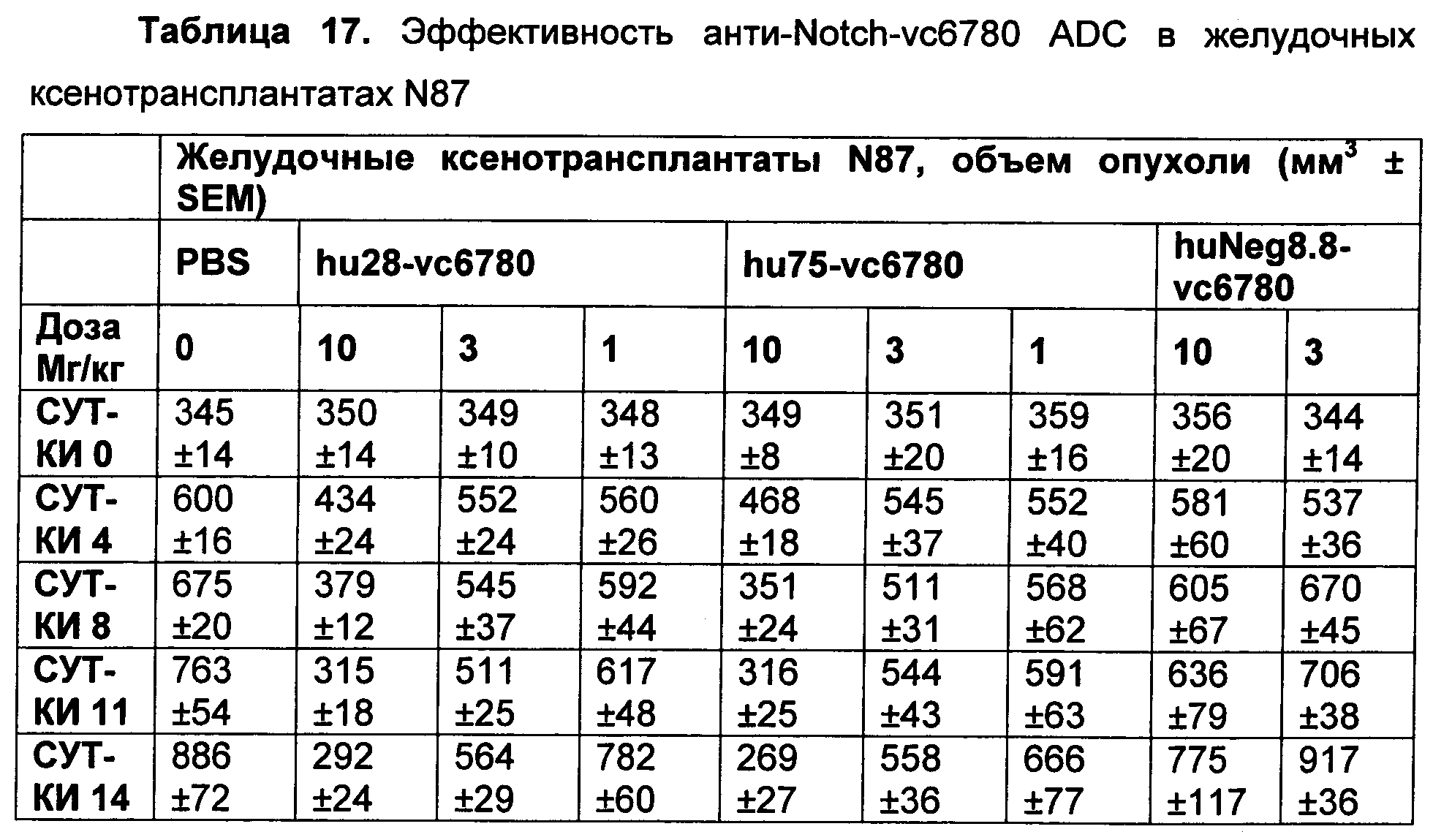
На Фиг. 8A-E показаны данные по [A] эффективности химерных крыса-человек анти-Notch ADC, введенных в дозе 5 мг/кг, в легочных ксенотрансплантатах HCC2429; [B и C] эффективности химерных крыса-человек анти-Notch ADC, введенных в дозе 5 мг/кг, в ксенотрансплантатах молочной железы MDA-MB-468; [D и E] эффективности химерных крыса-человек анти-Notch ADC, введенных в дозе 5 мг/кг, в ксенотрансплантатах желудка N87.
Подробное описание изобретения
Настоящее изобретение относится к цитотоксическим пентапептидам, к конъюгатам антитело-лекарственное средство, содержащим указанные цитотоксические пентапептиды, и к способам их использования для лечения рака и других патологических состояний. Изобретение также относится к способам использования таких соединений и/или конъюгатов in vitro, in situ и in vivo для обнаружения, диагностики или лечения клеток млекопитающих или ассоциированных патологических состояний.
Определения и сокращения
Если не указано иное, следующие термины и фразы, как их используют в данном описании, имеют следующие значения. Когда в данном описании используют товарные знаки, они включают препарат продукта, непатентованное наименование лекарственного средства и активный(е) фармацевтический(е) ингредиент(ы) указанного под товарным знаком продукта, если по контексту не указано иное.
Термин "антитело" (или "Ab") используют в данном описании в наиболее широком смысле, и он конкретно охватывает интактные моноклональные антитела, поликлональные антитела, моноспецифические антитела, мультиспецифические антитела (например биспецифические антитела) и фрагменты антител, которые проявляют желаемую биологическую активность. Интактное антитело имеет главным образом две области: вариабельную область и константную область. Вариабельная область связывается и взаимодействует с антигеном-мишенью. Вариабельная область включает гипервариабельный участок (CDR), который распознает и связывается со специфическим сайтом связывания на конкретном антигене. Константная область может распознаваться и взаимодействовать с иммунной системой (см., например, Janewayet al., 2001, Immuno. Biology, 5th Ed., Garland Publishing, New York). Антитело может быть любого типа или класса (например, IgG, IgE, IgM, IgD и IgA) или подкласса (например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2). Антитело может иметь происхождение из любого подходящего вида. В некоторых воплощениях антитело имеет человеческое или мышиное происхождение. Антитело может быть, например, человеческим, гуманизированным или химерным.
Термины "специфически связывается" и "специфическое связывание" относятся к связыванию антитела с заранее установленным антигеном. Как правило, антитело связывается с аффинностью по меньшей мере примерно 1×107 М-1 и связывается с заранее установленным антигеном с аффинностью, которая по меньшей мере в два раза больше по сравнению с его аффинностью в отношении связывания с неспецифическим антигеном (например бычьим сывороточным альбумином (BSA), казеином), отличным от заранее установленного антигена или близкородственного антигена.
Термин "моноклональное антитело", как его используют в данном описании, относится к антителу, полученному из популяции по существу гомогенных антител, то есть индивидуальные антитела, составляющие популяцию, идентичны за исключением возможных естественных мутаций, которые могут присутствовать в незначительных количествах. Моноклональные антитела являются высокоспецифичными, будучи направленными против единственного антигенного сайта. Определение "моноклональное" указывает на природу антитела как получаемого из по существу гомогенной популяции антител, и его не следует истолковывать как требующее продуцирование антитела каким-либо конкретным способом.
Термин "моноклональные антитела" специально включает "химерные" антитела, в которых часть тяжелой и/или легкой цепи идентична или гомологична соответствующей последовательности антител, имеющих происхождение из конкретных видов или принадлежащих конкретному классу или подклассу антител, в то время как остальная часть цепи(ей) идентична или гомологична соответствующим последовательностям антител, имеющих происхождение из других видов или принадлежащих другому классу или подклассу антител, а также фрагментов таких антител, при условии что они проявляют желаемую биологическую активность.
Как используют в данном описании, "H(C)-" относится к трастузумабу (товарный знак HERCEPTIN®), который означает моноклональное антитело, препятствующее связыванию HER2/neu рецептора через один из его цистеинов с соединением по изобретению. Как используют в данном описании, "H(K)-" относится к трастузумабу, который означает моноклональное антитело, препятствующее связыванию HER2/neu рецептора через один из его лизинов с соединением по изобретению.
"Интактное антитело" представляет собой антитело, которое содержит антиген-связывающую вариабельную область, а также константный домен легкой цепи (CL) и константные домены тяжелой цепи, CH1, CH2, CH3 и CH4, в зависимости от класса антител. Константные домены могут быть нативными последовательностями константных доменов (например, человеческая нативная последовательность константных доменов) или их варианты аминокислотных последовательностей.
Интактное антитело может проявлять одну или более "эффекторных функций", которые относятся к тем биологическим активностям, которые приписывают Fc-области (например, нативная последовательность Fc-области или аминокислотный вариант последовательности Fc-области) антитела. Примеры эффекторных функций антитела включают комплемент-зависимую цитотоксичность, антитело-зависимую клеточно-опосредованную цитотоксичность (ADCC) и антитело-зависимый клеточно-опосредованный фагоцитоз.
"Фрагмент антитела" содержит часть интактного антитела, предпочтительно содержащую его антиген-связывающую или вариабельную область. Примеры фрагментов антитела включают фрагменты Fab, Fab′, F(ab′)2 и Fv, диатела, триатела, тетратела, линейные антитела, одноцепочечные молекулы антитела, scFv, scFv-Fc, мультиспецифичные фрагменты антитела, образованные из фрагмента(ов) антител, фрагмент(ы), продуцируемый(е) Fab экспрессионной библиотекой, или эпитоп-связывающие фрагменты любого из вышеуказанного, которые иммуноспецифично связываются с антигеном-мишенью (например, раковым клеточным антигеном, вирусным антигеном или микробным антигеном).
Термин "вариабельный" в контексте антитела относится к определенным участкам вариабельных доменов антитела, которые сильно отличаются по последовательности и используются в связывании и специфичности каждого конкретного антитела для его конкретного антигена. Эта вариабельность сконцентрирована в трех сегментах, называемых "гипервариабельными участками" в вариабельных доменах легкой и тяжелой цепи. Более высококонсервативные участки вариабельных доменов называют каркасными областями (FR). Вариабельные домены нативных тяжелой и легкой цепей каждый содержит четыре FR, соединенные тремя гипервариабельными участками.
Термин "гипервариабельный участок" при использовании здесь относится к аминокислотным остаткам антитела, которые ответственны за связывание антигена. Гипервариабельный участок в общем содержит аминокислотные остатки из "участка, определяющего комплементарность" или "CDR" (например, остатки 24-34 (L1), 50-56 (L2) и 89-97 (L3) в вариабельном домене легкой цепи и 31-35 (H1), 50-65 (H2) и 95-102 (L3) в вариабельном домене тяжелой цепи; Kabat et al. (Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)), и/или те остатки из "гипервариабельной петли" (например, остатки 26-32 (L1), 50-52 (L2) и 91-96 (L3) в вариабельном домене легкой цепи и 26-32 (H1), 53-55 (142) и 96-101 (H3) в вариабельном домене тяжелой цепи; Chothia и Lesk, 1987, J. Mol. Biol. 196:901-917). FR остатки представляют собой остатки вариабельного домена, отличные от остатков гипервариабельной области, как определено в данном описании.
"Одноцепочечное Fv" или "scFv" фрагмент антитела содержит V.sub.H и V.sub.L домены антитела, причем эти домены присутствуют в одной полипептидной цепи. Как правило, Fv полипептид дополнительно содержит полипептидный линкер между V.sub.H и V.sub.L доменами, который дает возможность scFv образовывать желаемую структуру для связывания антигена. Для обзора scFv см. Pluckthun в The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg и Moore eds., Springer-Verlag, New York, pp. 269-315 (1994).
Термин "диатело" относится к небольшим фрагментам антитела с двумя антиген-связывающими сайтами, причем фрагменты содержат вариабельный тяжелый домен (VH), соединенный с вариабельным легким доменом (VL) в одной полипептидной цепи. При использовании линкера, который слишком короткий, чтобы обеспечить спаривание между двумя доменами на одной цепи, домены вынуждены спариваться с комплементарными доменами другой цепи и создавать два антиген-связывающих сайта. Диатела описаны более подробно, например, в EP 0404097; WO 93/11161; и Hollinger et al., 1993, Proc. Natl. Acad. Sci. USA 90:6444-6448.
"Гуманизированные" формы не являющихся человеческими (например, грызуна) антител являются химерные антитела, которые содержат минимальную последовательность, имеющую происхождение из иммуноглобулина, не являющегося человеческим. В большинстве случаев гуманизированные антитела являются человеческими иммуноглобулинами (реципиентное антитело), в которых остатки из гипервариабельного участка реципиента заменены остатками из гипервариабельного участка не являющихся человеческими видов (донорное антитело), таких как мышь, крыса, кролик или не являющийся человеческим примат, имеющих желаемую специфичность, аффинность и емкость. В некоторых случаях остатки каркасной области (FR) человеческого иммуноглобулина заменены соответствующими остатками не являющихся человеческими видов. Кроме того, гуманизированные антитела могут содержать остатки, которые не обнаруживают в реципиентном антителе или в донорном антителе. Эти модификации создают для дополнительного улучшения эффективности антитела. В общем, гуманизированное антитело содержит по существу все из по меньшей мере одного и, как правило, двух вариабельных доменов, в которых все или по существу все из гипервариабельных петель соответствуют петлям из иммуноглобулина, не являющегося человеческим, и все или по существу все из FR являются FR из последовательности человеческого иммуноглобулина. Гуманизированное антитело возможно также содержит по меньшей мере участок константной области иммуноглобулина (Fc), как правило человеческого иммуноглобулина. Для дополнительных деталей см. Jones et al., 1986, Nature 321:522-525; Riechmann et al., 1988, Nature 332:323-329; и Presta, 1992, Curr. Op. Struct. Biol. 2:593-596.
Как используют в данном описании, "выделенное" означает отделенное от других компонентов (а) природного источника, такого как растительная или животная клетка или клеточная культура, или (б) синтетической органической химической реакционной смеси. Как используют в данном описании, "очищенное" означает, что при выделении изолят содержит по меньшей мере 95%, и в другом аспекте по меньшей мере 98%, соединения (например, конъюгата) по массе изолята.
"Выделенное" антитело представляет собой антитело, которое было идентифицировано и отделено и/или выделено из компонента из его естественного окружения. Загрязняющие компоненты из его естественного окружения являются материалами, которые будут препятствовать диагностическому или терапевтическому использованию антитела, и могут включать ферменты, гормоны и другие белковые или небелковые растворенные вещества. В предпочтительных воплощениях антитело очищают (1) до более чем 95% по массе антитела, как определяют методом Лоури, и наиболее предпочтительно более чем 99% по массе, (2) до степени, достаточной для получения по меньшей мере 15 остатков N-концевой или внутренней аминокислотной последовательности, путем использования секвенатора с вращающимся стаканом, или (3) до гомогенности согласно электрофорезу на полиэтиленгликоле с додецилсульфатом натрия (SDS-PAGE) в восстанавливающих или невосстанавливающих условиях с использованием кумасси голубого или, предпочтительно, серебрянки. Выделенное антитело включает антитело in situ в рекомбинантных клетках, в виду того что по меньшей мере один компонент природного окружения антитела не будет присутствовать. Однако, обычно выделенное антитело получают по меньшей мере за одну стадию очистки.
Антитело, которое "индуцирует апоптоз", представляет собой антитело, которое индуцирует программируемую клеточную гибель, как определяют посредством связывания аннексина V, фрагментации ДНК, сжатия клетки, расширения эндоплазматического ретикулума, клеточной фрагментации и/или образования мембранных везикул (называемых апоптотическими тельцами). Клетка представляет собой опухолевую клетку, например, молочной железы, яичника, желудка, эндометрия, слюнной железы, легкого, почки, толстой кишки, щитовидной железы, поджелудочной железы или мочевого пузыря. Разные методы доступны для оценки клеточных событий, ассоциированных с апоптозом. Например, перенос фосфатидилсерина (PS) может быть измерен посредством связывания аннексина; фрагментация ДНК может быть оценена посредством электрофоретического расщепления ДНК; и конденсация ядра/хроматина наряду с фрагментацией ДНК может быть оценена посредством какого-либо увеличения в гиподиплоидных клетках.
Термин "терапевтически эффективное количество" относится к количеству лекарственного средства, эффективного для лечения заболевания или расстройства у млекопитающего. В случае рака терапевтически эффективное количество лекарственного средства может снижать количество раковых клеток; уменьшать размеры опухоли; подавлять (то есть замедлять до некоторой степени и предпочтительно останавливать) инфильтрацию раковой клетки в периферические органы; подавлять (то есть замедлять до некоторой степени и предпочтительно останавливать) метастазы опухоли; ингибировать до некоторой степени рост опухоли; и/или облегчать до некоторой степени один или более симптомов, ассоциированных с раком. До той степени, с которой лекарственное средство может ингибировать рост и/или уничтожать имеющиеся раковые клетки, оно может быть цитостатическим и/или цитотоксическим. Для терапии рака эффективность может, например, быть измерена путем оценки времени до прогрессирования заболевания (TTP) и/или определения степени ответа (RR).
Термин "существенное количество" относится к большинству, то есть более чем 50% популяции, смеси или образца.
Термин "внутриклеточный метаболит" относится к соединению, полученному в результате метаболического процесса или реакции внутри клетки на конъюгате антитело-лекарственное средство (ADC). Метаболический процесс или реакция может представлять собой ферментативный процесс, такой как протеолитическое расщепление пептидного линкера ADC. Внутриклеточные метаболиты включают, без ограничения, антитела и свободное лекарственное средство, которые могут подвергаться внутриклеточному расщеплению после поглощения, диффузии, захвата или транспорта в клетку.
Термины "внутриклеточно расщепленный" и "внутриклеточное расщепление" относятся к метаболическому процессу или реакции внутри клетки на ADC или подобном ему, посредством которых ковалентная связь, например, линкера между лекарственной группировкой и антителом разрушается с получением свободного лекарственного средства или другого метаболита конъюгата, диссоциированного от антитела внутри клетки. Расщепленные группировки ADC являются, таким образом, внутриклеточными метаболитами.
Термин "биодоступность" относится к системной доступности (то есть уровни в крови/плазме) данного количества лекарственного средства, введенного пациенту. Биодоступность представляет собой абсолютный термин, который указывает меру как времени (скорость), так и общего количества (степень) лекарственного средства, которое достигает общего кровотока из введенной лекарственной формы.
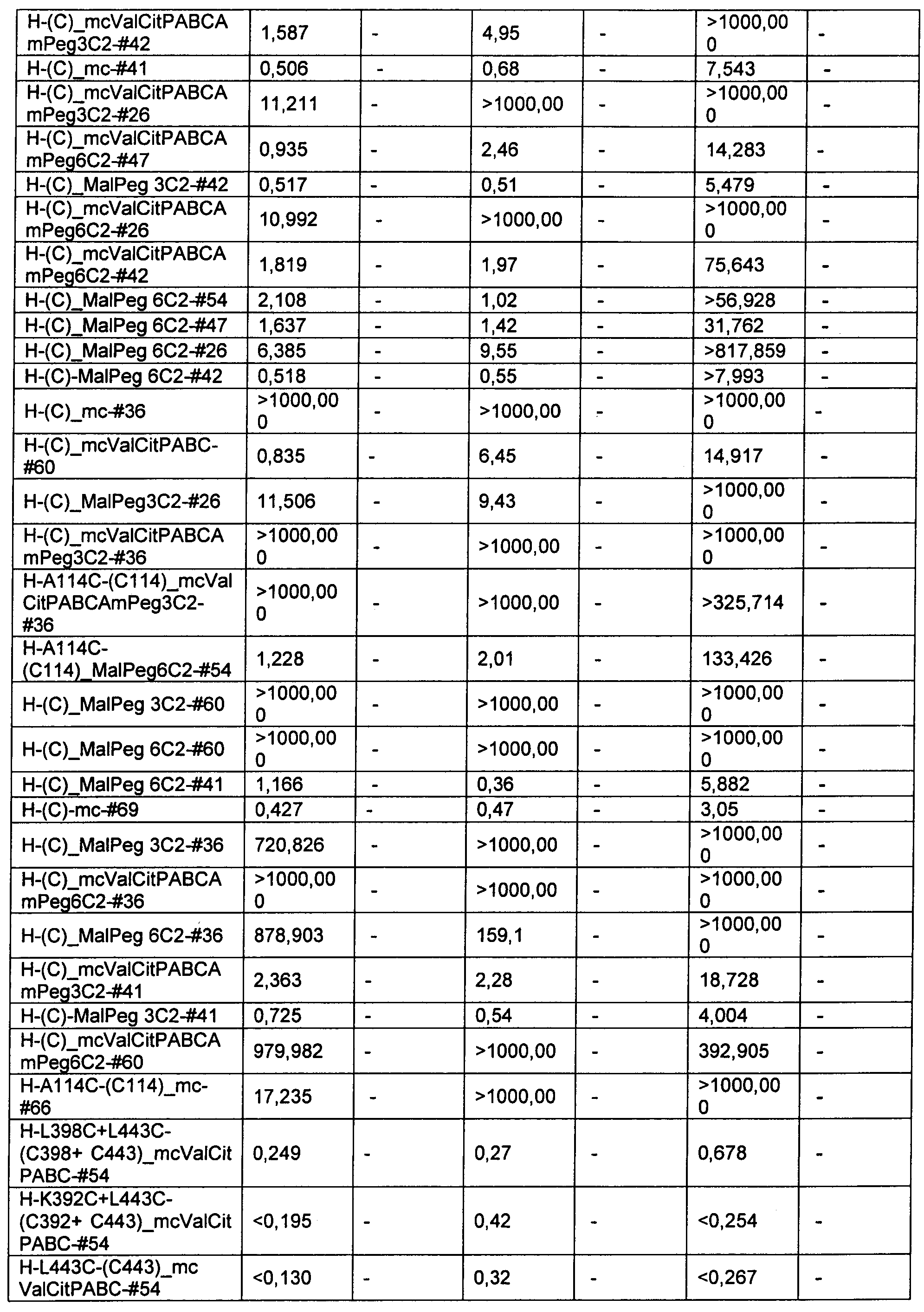
Термин "цитотоксическая активность" относится к уничтожающему клетку, цитостатическому или антипролиферативному эффекту ADC или внутриклеточного метаболита указанного ADC. Цитотоксическая активность может быть выражена в виде значения IC50, которое равно концентрации (молярной или массовой) на единицу объема, при которой выживает половина клеток.
"Расстройство" представляет собой любое состояние, при котором может быть получена польза от лечения лекарственным средством или конъюгатом антитело-лекарственное средство. Оно включает хронические и острые расстройства или заболевания, включая такие патологические состояния, которые приводят млекопитающего к рассматриваемому расстройству. Неограничивающие примеры расстройств, подлежащих лечению по настоящему изобретению, включают доброкачественные и злокачественные новообразования; лейкоз и лимфоидные новообразования, нейрональные, глиальные, астроцитальные, гипоталамические и другие эндокринные, макрофагальные, эпителиальные, стромальные и бластоцельные расстройства; а также воспалительные, ангиогенные и иммунологические расстройства.
Термины "рак" и "раковый" относятся к или описывают физиологическое состояние или расстройство у млекопитающих, которое как правило характеризуется нерегулируемым клеточным ростом. "Опухоль" содержит одну или более раковых клеток.
Примеры "пациента" включают, без ограничения, человека, крысу, мышь, морскую свинку, обезьяну, козу, корову, лошадь, собаку, кошку, птицу и домашнюю птицу. В иллюстративном воплощении пациент представляет собой человека.
Термины "лечить" или "лечение", если иное не следует по контексту, относится к терапевтическому лечению или профилактическим мерам для предотвращения обострения, где задача состоит в том, чтобы подавлять или снижать (уменьшать) нежелаемое физиологическое изменение или расстройство, такое как развитие или распространение рака. Для целей данного изобретения, благоприятные или желаемые клинические результаты включают, без ограничения, облегчение симптомов, минимизацию степени заболевания, стабилизированное (то есть не ухудшающееся) состояние заболевания, задержку или замедление прогрессирования заболевания, улучшение или облегчение болезненного состояния и ремиссию (частичную или полную), неважно может ли это быть обнаружено или нет."Лечение" может также означать продление выживания по сравнению с ожидаемым выживанием без получения лечения. Субъекты, нуждающиеся в лечении, включают тех, кто уже имеет состояние или расстройство, а также тех, у кого подозревается состояние или расстройство.
В контексте рака термин "лечение" включает любое из или все из ингибирования роста опухолевых клеток, раковых клеток или опухоли; ингибирования репликации опухолевых клеток или раковых клеток, уменьшения общего очага опухоли или снижения количества раковых клеток и улучшения одного или более симптомов, ассоциированных с заболеванием.
В контексте аутоиммунного заболевания термин "лечение" включает любое из или все из ингибирования репликации клеток, ассоциированных с аутоиммунным болезненным состоянием, включая, без ограничения, клетки, которые продуцируют аутоиммунные антитела, уменьшения очага аутоиммунного антитела и ослабления одного или более симптомов аутоиммунного заболевания.
В контексте инфекционного заболевания термин "лечение" включает любое из или все из: ингибирования роста, размножения или репликации патогена, который вызывает инфекционное заболевание, и ослабления одного или более симптомов инфекционного заболевания.
Термин "вкладыш упаковки" используют для ссылки на инструкции, включенные в коммерческие упаковки терапевтических продуктов, которые содержат информацию о показании(ях), использовании, дозировке, введении, противопоказаниях и/или предосторожностях, касающихся применения таких терапевтических продуктов.
Как используют в данном описании, термины "клетка", "клеточная линия" и "клеточная культура" используют взаимозаменяемо, и все такие обозначения включают потомство. Слова "трансформанты" и "трансформированные клетки" включают первичную клетку-субъект и культуры или потомство, происходящее от них, безотносительно количества переносов. Также следует понимать, что все потомство не может быть точно идентичным по содержанию ДНК из-за преднамеренных или случайных мутаций. Мутантное потомство, которое имеет такую же функцию или биологическую активность, как определено в отношении первоначально трансформированной клетки, также включено. Когда подразумеваются отличные обозначения, это будет ясно из контекста.
Если не указано иное, термин "алкил" сам по себе или как часть другого термина относится к прямоцепочечному или разветвленному насыщенному углеводороду, имеющему указанное количество атомов углерода (например, "C1-C8алкил" относится к алкильной группе, имеющей от 1 до 8 атомов углерода). Если количество атомов углерода не указано, алкильная группа имеет от 1 до 8 атомов углерода. Иллюстративные прямоцепочечные C1-C8алкилы включают, без ограничения, метил, этил, н-пропил, н-бутил, н-пентил, н-гексил, н-гептил и н-октил; в то время как разветвленные C1-C8алкилы включают, без ограничения, -изопропил, -втор-бутил, -изобутил, -трет-бутил, -изопентил и -2-метилбутил; ненасыщенные C2-C8алкилы включают, без ограничения, винил, аллил, 1-бутенил, 2-бутенил, изобутиленил, 1-пентенил, 2-пентенил, 3-метил-1-бутенил, 2-метил-2-бутенил, 2,3-диметил-2-бутенил, 1-гексил, 2-гексил, 3-гексил, ацетиленил, пропинил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил и 3-метил-1-бутинил.
Если не указано иное, "алкилен" сам по себе или как часть другого термина относится к насыщенному разветвленному или прямоцепочечному или циклическому углеводородному радикалу с установленным количеством атомов углерода, как правило 1-18 атомов углерода, и имеющему два моновалентных радикальных центра, полученные при удалении двух атомов водорода от одного и того же или двух разных атомов углерода исходного алкана. Типичные алкиленовые радикалы включают, без ограничения: метилен (-CH2-), 1,2-этилен (-CH2CH2-), 1,3-пропилен (-CH2CH2CH2-), 1,4-бутилен (-CH2CH2CH2CH2-) и тому подобное. "C1-C10" прямоцепочечный алкилен представляет собой прямоцепочечную насыщенную углеводородную группу формулы -(CH2)1-10-. Примеры C1-C10алкилена включают метилен, этилен, пропилен, бутилен, пентилен, гексилен, гептилен, октилен, нонилен и декален.
Если не указано иное, термин "гетероалкил" сам по себе или в комбинации с другим термином означает, если не указано иное, стабильный прямоцепочечный или разветвленный углеводород или его комбинации, полностью насыщенный или содержащий от 1 до 3 степеней ненасыщенности, состоящий из установленного количества атомов углерода и от одного до трех гетероатомов, выбранных из группы, состоящей из O, N, Si и S, и где атомы азота и серы могут возможно быть окисленными и гетероатом азота может быть возможно квартенизированным. Гетероатом(ы) O, N и S могут быть локализованы по любому внутреннему положению гетероалкильной группы. Гетероатом Si может быть локализован по любому положению гетероалкильной группы, включая положение, по которому алкильная группа присоединена к остальной части молекулы. Вплоть до двух гетероатомов могут быть расположены друг за другом.
Если не указано иное, термин "гетероалкилен" сам по себе или как часть другого заместителя означает двухвалентную группу, полученную из гетероалкила (как обсуждалось выше). Для гетероалкиленовых групп гетероатомы также могут занимать один или оба конца цепи.
Если не указано иное, "арил" сам по себе или как часть другого термина означает замещенный или незамещенный моновалентный карбоциклический ароматический углеводородный радикал из 6-20, предпочтительно 6-14, атомов углерода, полученный при удалении одного атома водорода от одного атома углерода исходной ароматической кольцевой системы. Типичные арильные группы включают, без ограничения, радикалы, полученные из бензола, замещенного бензола, нафталина, антрацена, бифенила и тому подобное. Замещенная карбоциклическуая ароматическая группа (например, арильная группа) может быть замещена одной или более, предпочтительно от 1 до 5, следующими группами: C1-C8алкил, -O-(C1-C8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -OH, галоген, -N3, -NH2, -NH(R′), -N(R′)2 и -CN; где каждый R′ независимо выбран из -H, C1-C8алкила и незамещенного арила. В некоторых воплощениях замещенная карбоциклическая ароматическая группа может дополнительно включать одно или более из следующего: -NHC(=NH)NH2, -NHCONH2, -S(=O)2R′ и -SR′. "Арилен" является соответствующей двухалентной группировкой.
"Замещенный алкил" означает алкил, в котором один или более атомов водорода каждый независимо замещен заместителем. Типичные заместители включают, без ограничения: -X, -R, -O-, -OR, -SR, -S-, -NR2, -NR3, =NR, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NRC(=O)R, -C(=O)NR2, , -SO3H, -S(=O)2R, -OS(=O)2OR, -S(=O)2NR, -S(=O)R, -OP(=O)(OR)2, -P(=O)(OR)2, , PO2H2, -AsO2H2, -C(=O)R, -C(=O)X, -C(=S)R, -CO2R, , -C(=S)OR, -C(=O)SR, -C(=S)SR, -C(=O)NR2, -C(=S)NR2 или -C(=NR)NR2, где каждый X независимо представляет собой галоген: -F, -Cl, -Br или -I; и каждый R независимо представляет собой -H, C1-C20алкил, C1-C20гетероалкил, C6-C20арил, C1-C10гетероциклил, защитную группу или группировку пролекарства. Арильные, алкиленовые и гетероалкиленовые группы, как описано выше, могут также быть просто замещенными.
Если не указано иное, "аралкил" сам по себе или как часть другого термина означает алкильную группу, как описано выше, замещенную арильную группу, как определено выше.
Если не указано иное, "C1-C10гетероциклил" сам по себе или как часть другого термина относится к моновалентной замещенной или незамещенной ароматической или неароматической моноциклической, бициклической или трициклической кольцевой системе, имеющей от 1 до 10, предпочтительно от 3 до 8, атомов углерода (также называемых кольцевыми членами) и от одного до четырех гетероатомных кольцевых членов, независимо выбранных из N, O, P или S, и полученной при удалении одного атома водорода из кольцевого атома исходной кольцевой системы. Один или более атомов N, C или S в гетероциклиле могут быть окисленными. Кольцо, которое включает гетероатом, может быть ароматическим или неароматическим. Если не указано иное, гетероциклил присоединен к его боковой группе по любому гетероатому или атому углерода, приводящему к стабильной структуре. Иллюстративные примеры C1-C10гетероциклила включают, без ограничения, тетрагидрофуранил, оксетанил, пиранил, пирролидинил, пиперидинил, пиперазинил, бензофуранил, бензотиофен, бензотиазолил, индолил, бензопиразолил, пирролил, тиофенил (тиофен), фуранил, тиазолил, имидазолил, пиразолил, триазолил, хинолинил, включая группировки, такие как 1,2,3,4-тетрагидро-хинолинил, пиримидинил, пиридинил, пиридонил, пиразинил, пиридазинил, изотиазолил, изоксазолил, тетразолил, эпоксид, оксетан и BODIPY (4,4-дифтор-4-бора-3a,4a-диаза-s-индацен) (замещенный или незамещенный). C1-C10гетероциклил может быть замещен группами в количестве вплоть до семи, включая, без ограничения, C1-C8алкил, C1-C8гетероалкил, -OR′, арил, -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(=O)2R′, -S(O)R′, галоген, -N3, -NH2, -NH(R′), -N(R′)2 и -CN; где каждый R′ независимо выбран из -H, C1-C8алкила, C1-C8гетероалкила и арила. В некоторых воплощениях замещенный гетероциклил может также включать один или более из следующих: -NHC(=NH)NH2, -NHCONH2, -S(=O)2R′ и -SR′. "Гетероцикло", "C1-C10гетероцикло" является соответствующей двухвалентной группировкой.
Если не указано иное, "гетероаралкил" сам по себе или как часть другого термина означает алкильную группу, как определено выше, замещенную ароматической гетероциклильной группой, как определено выше. Гетероаралкил является соответствующей двухвалентной группировкой.
Если не указано иное, "C3-C8карбоциклил" сам по себе или как часть другого термина представляет собой 3-, 4-, 5-, 6-, 7- или 8-членное моновалентное, замещенное или незамещенное, насыщенное или ненасыщенное неароматическое моноциклическое или бициклическое карбоциклическое кольцо, полученное при удалении одного атома водорода из кольцевого атома исходной кольцевой системы. Иллюстративные C3-C8карбоциклилы включают, без ограничения, циклопропил, циклобутил, циклопентил, циклопентадиенил, циклогексил, циклогексенил, 1,3-циклогексадиенил, 1,4-циклогексадиенил, циклогептил, 1,3-циклогептадиенил, 1,3,5-циклогептатриенил, циклооктил, циклооктадиенил, бицикло(1.1.1.)пентан, и бицикло(2.2.2.)октан. C3-C8карбоциклильная группа может быть незамещенной или замещенной группами в количестве вплоть до семи, включая, без ограничения, C1-C8алкил, C1-C8гетероалкил, -OR′, арил, -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(=O)2R′, -S(=O)R′, -OH, -галоген, -N3, -NH2, -NH(R′), -N(R′)2 и -CN; где каждый R′ независимо выбран из -H, C1-C8алкила, C1-C8гетероалкила и арила. "C3-C8карбоцикло" является соответствующей двухвалентной группировкой.
Термин "хиральный" относится к молекулам, которые имеют свойство партнера с неналагающимся зеркальным изображением, в то время как термин "ахиральный" относится к молекулам, которые являются партнерами с налагающимся зеркальным изображением.
Термин "стереооизомеры" относится к соединениям, которые имеют идентичный химический состав, но отличаются по расположению атомов или групп в пространстве.
"Диастереомер" относится к стереоизомеру с двумя или более центрами хиральности и молекулы которых не являются зеркальными отражениями друг друга. Диастереомеры имеют различные физические свойства, например, точки плавления, точки кипения, спектральные свойства и реакционноспособность. Смеси диастереомеров могут быть разделены посредством аналитических методик высокого разрешения, таких как электрофорез и хроматография.
Стереохимические определения и условные обозначения, использованные здесь, в общем следуют из S.P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms, McGraw-Hill Book Company, New York (1984); и Eliel и Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York (1994). Многие органические соединения существуют в оптически активных формах, то есть они имеют способность вращать плоскость плоскополяризованного света. При описании оптически активного соединения префиксы D и L или R и S используют для обозначения абсолютной конфигурации молекулы относительно ее хирального(ых) центра(ов). Префиксы d и I или (+) и (-) используют для обозначения знака вращения плоскополяризованного света соединением, причем (-) или I означает, что соединение является левовращающим, и соединение с префиксом (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры являются идентичными, I за исключением того, что они являются зеркальными отображениями друг друга. Конкретный стереоизомер может также называться энантиомером, и смесь таких изомеров часто называют энантиомерной смесью. Смесь 50:50 энантиомеров называют рацемической смесью или рацематом, которые могут существовать, когда в химической реакции или процессе не было никакого стереоразделения или стереоспецифичности. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомерных видов, лишенной оптической активности.
Аминокислотное "производное" включает аминокислоту, имеющую замены или модификации путем ковалентного присоединения исходной аминокислоты, такие как, например, путем алкилирования, гликозилирования, ацетилирования, фосфорилирования и тому подобное. Дополнительно включены в определение "производное", например, один или более аналогов аминокислот с замещенными связками, а также другие модификации, известные в данной области техники.
"Природная аминокислота" относится к аргинину, глутамину, фенилаланину, тирозину, триптофану, лизину, глицину, аланину, гистидину, серину, пролину, глутаминовой кислоте, аспарагиновой кислоте, треонину, цистеину, метионину, лейцину, аспарагину, изолейцину и валину, если не указано иное по контексту.
"Защитная группа" относится к группировке, которая при присоединении к реакционноспособной группе в молекуле маскирует, снижает или предотвращает ее реакционноспособность. Примеры защитных групп можно найти в T.W. Greene и P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York, 1999, и Harrison и Harrison et al., Compendium of Synthetic Organic Methods, Vols. 1-8 (John Wiley и Sons, 1971-1996), которые включены в данное описание во всей их полноте. Иллюстративные защитные группы для гидрокси включают ацильные группы, бензил и тритиловые эфиры, тетрагидропираниловые эфиры, триалкилсилиловые эфиры и аллиловые эфиры. Иллюстративные защитные группы для амино включают формил, ацетил, трифторацетил, бензил, бензилоксикарбонил (CBZ), трет-бутоксикарбонил (Boc), триметилсилил (TMS), 2-триметилсилил-этансульфонил (SES), группы тритил и замещенный тритил, аллилоксикарбонил, 9-флуоренилметилоксикарбонил (FMOC), нитро-вератрилоксикарбонил (NVOC) и тому подобное.
Примеры "защитной группы гидроксила" включают, без ограничения, метоксиметиловый эфир, 2-метоксиэтоксиметиловый эфир, тетрагидропираниловый эфир, бензиловый эфир, пара-метоксибензиловый эфир, триметилсилиловый эфир, триэтилсилиловый эфир, триизопропилсилиловый эфир, трет-бутилдиметил-силиловый эфир, трифенилметилсилиловый эфир, ацетатный сложный эфир, замещенные ацетатные сложные эфиры, пивалоат, бензоат, метансульфонат и пара-толуолсульфонат.
"Уходящая группа" относится к функциональной группе, которая может быть замещена другой функциональной группой. Такие уходящие группы хорошо известны в данной области техники, и примеры включают, без ограничения, галогенид (например хлорид, бромид, йодид), метансульфонил (мезил), пара-толуолсульфонил (тозил), трифторметилсульфонил (трифлат) и трифторметилсульфонат.
Фраза "фармацевтически приемлемая соль", как используют в данном описании, относится к фармацевтически приемлемым органическим или неорганическим солям соединения. Соединение как правило содержит по меньшей мере одну аминогруппу, и соответственно соли присоединения кислоты могут быть образованы с этой аминогруппой. Иллюстративные соли включают, без ограничения, соли сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, бисульфат, фосфат, гидрофосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, малат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, пара-толуолсульфонат и памоат (то есть 1,1′-метилен-бис-(2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может охватывать включение других молекул, таких как ацетатный ион, сукцинатный ион или другой противоион. Противоион может представлять собой любую органическую или неорганическую группировку, которая стабилизирует заряд исходного соединения. Кроме того, фармацевтически приемлемая соль может иметь более одного загруженного атома в своей структуре. В случаях множества загруженных атомов, фармацевтически приемлемая соль может иметь множество противоинов. Поэтому, фармацевтически приемлемая соль может иметь один или более загруженных атомов и/или один или более противоионов.
"Фармацевтически приемлемый сольват" или "сольват" относится к ассоциации одной или более молекул растворителя и соединения или конъюгата по изобретению. Примеры растворителей, которые образуют фармацевтически приемлемые сольваты, включают, без ограничения, воду, изопропанол, этанол, метанол, DMSO, этилацетат, уксусную кислоту и этаноламин.
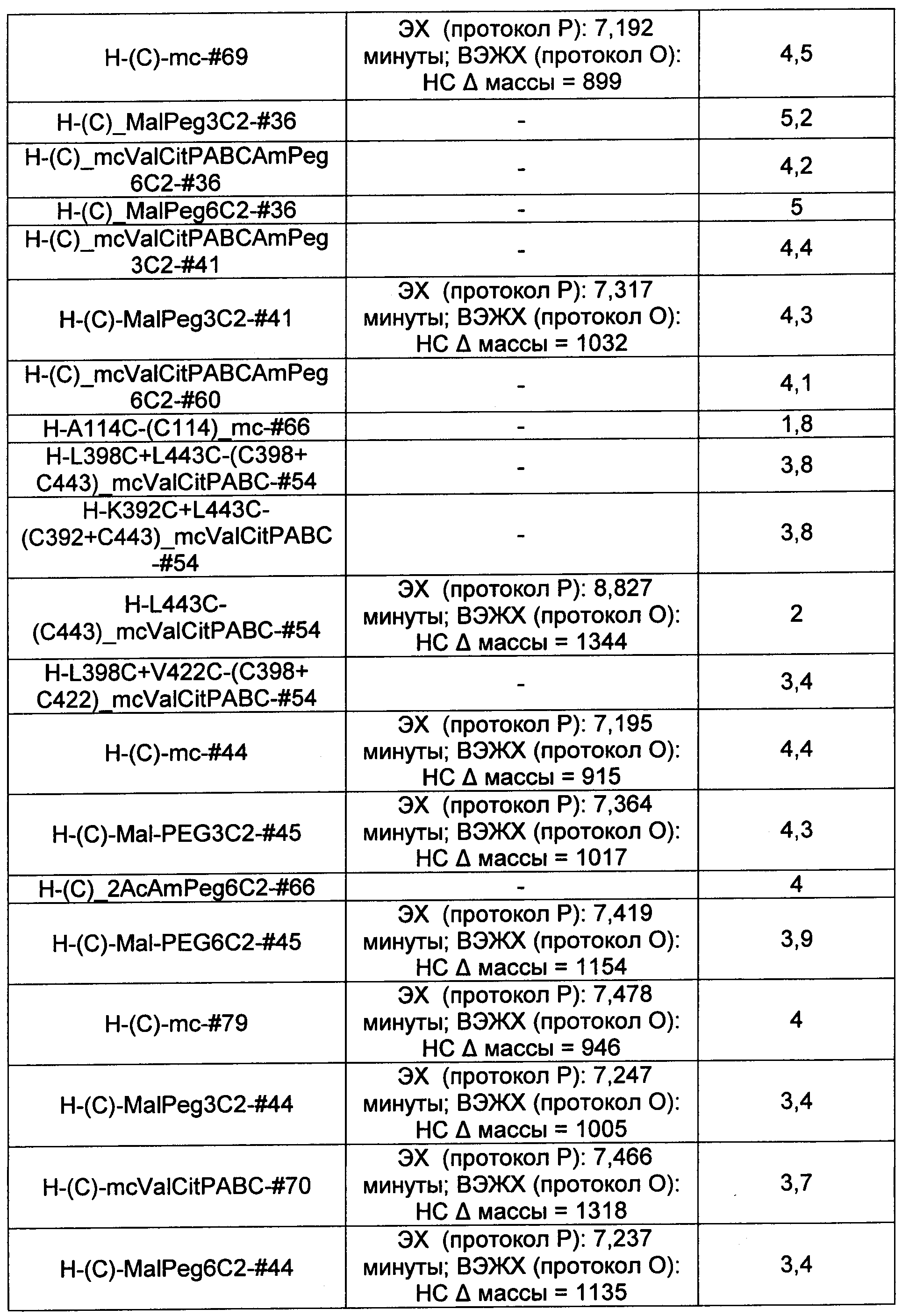
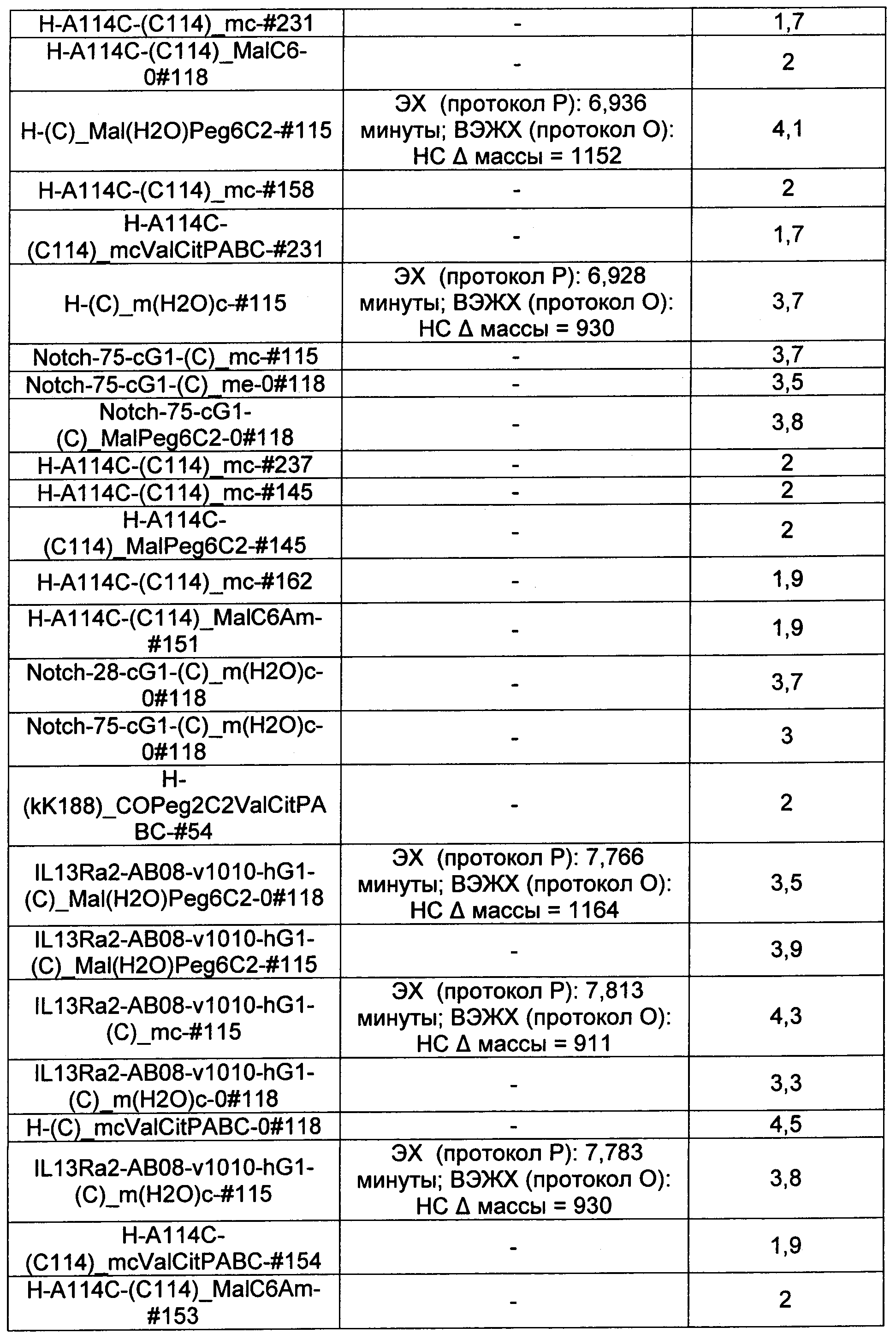
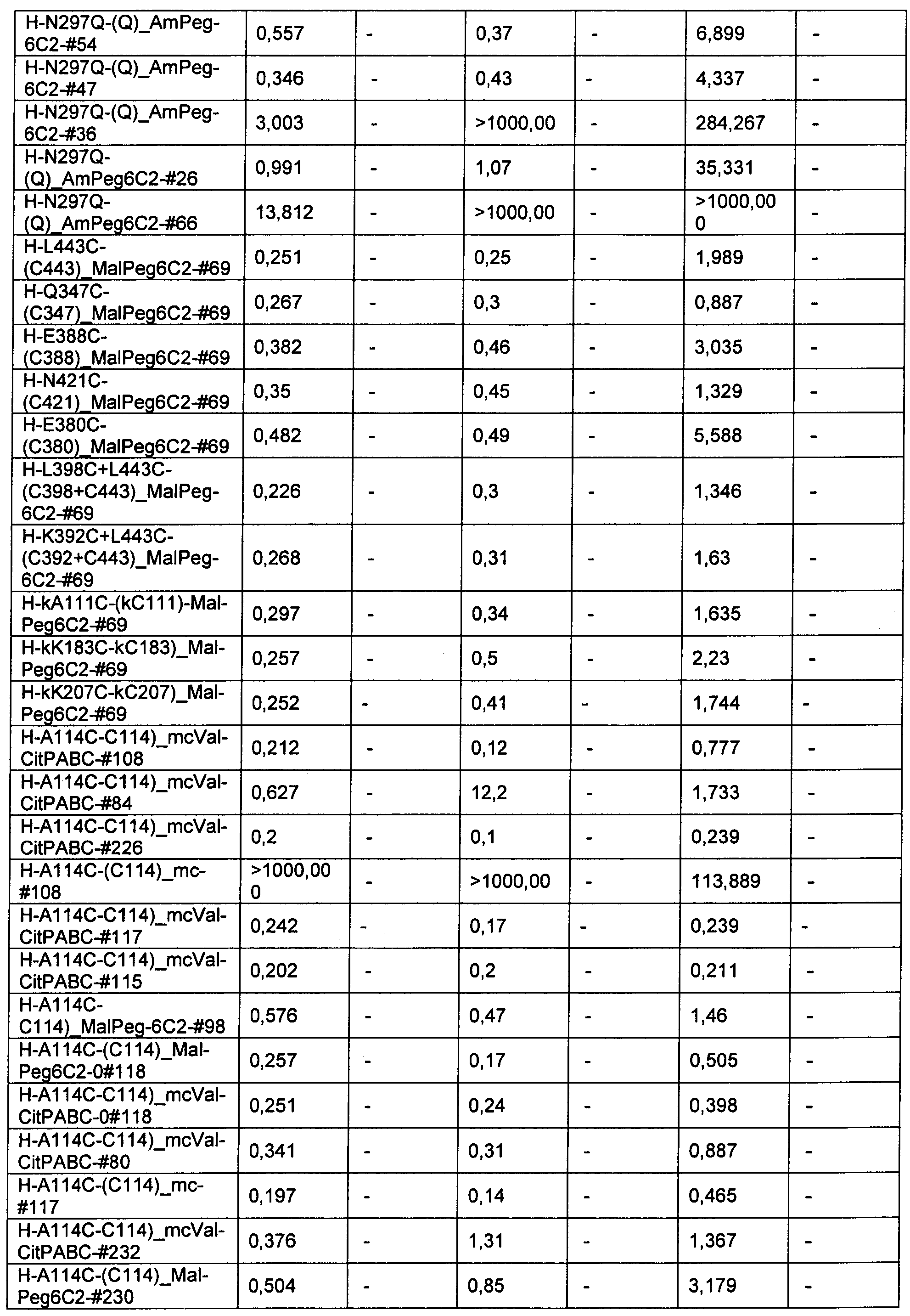
Термины "нагрузка", или "лекарственная нагрузка", или "полезная нагрузка" представляют собой или означают среднее количество полезных нагрузок ("полезная нагрузка" и "полезные нагрузки" используют в данном описании взаимозаменяемо с "лекарственным средством" и "лекарственными средствами") на антитело в ADC молекуле. Лекарственная нагрузка может находиться в диапазоне от 1 до 20 лекарственных средств на антитело. Иногда это выражают как DAR, или соотношение лекарственного средства к антителу. Композиции ADC, описанные здесь, как правило имеют соотношения DAR от 1-20 и в некоторых воплощениях от 1-8, от 2-8, от 2-6, от 2-5 и от 2-4. Типичные значения DAR равны 2, 4, 6 и 8. Среднее количество лекарственных средств на антитело, или значение DAR, может быть определено общепринятыми средствами, такими как спектроскопия в Уф/видимой области, масс-спектроскопия, твердофазный иммуноферментный анализ (ELISA) и высокоэффективная жидкостная хроматография (ВЭЖХ). Количественный показатель DAR также может быть определен. В некоторых случаях разделение, очистка и определение характеристик гомогенных ADC, имеющих конкретное значение DAR, могут быть достигнуты такими средствами, как ВЭЖХ с обращенной фазой или электрофорез. DAR может быть ограничен количеством сайтов присоединения на антитело, например, когда присоединение представляет собой цистеин-тиол, антитело может иметь только одну или несколько цистеин-тиоловых групп или может иметь только одну или несколько достаточно реакционноспособных тиоловых групп, через которые может быть присоединена единица Линкера. В некоторых воплощениях цистеин-тиол представляет собой тиоловую группу цистеинового остатка, который образует внутрицепочечную дисульфидную связь. В некоторых воплощениях цистеин-тиол представляет собой тиоловую группу цистеинового остатка, который не образует внутрицепочечную дисульфидную связь. Как правило, во время реакции конъюгирования меньше теоретического максимума группировок лекарственного средства конъюгируется с антителом. Антитело может содержать, например, много остатков лизина, которые не взаимодействуют с линкером или промежуточным линкером. Только большинство реакционноспособных групп лизина может взаимодействовать с реакционноспособным линкерным реагентом.
В общем, антитела не содержат много, если вообще содержат, свободных и реакционноспособных цистеин-тиоловых групп, которые могут быть связаны с лекарственных средством через линкер. Большинство цистеин-тиоловых остатков в антителах существуют в виде дисульфидных мостиков и должны быть восстановлены восстановителем, таким как дитиотрейтол (DTT). Антитело может быть подвергнуто денатурирующим условиям с высвобождением реакционноспособных нуклеофильных групп, таких как лизин или цистеин. Полезную нагрузку (соотношение лекарственного средства/антитела) в ADC можно контролировать несколькими разными путями, включая: (1) ограничение молярного избытка конструкции лекарственное средство-линкер относительно антитела, (2) ограничение времени или температуры конъюгирования, и (3) частичные или ограничивающие условия восстановления для модификации цистеин-тиола. Когда более одной нуклеофильной группы взаимодействуют с лекарственным средством-линкером, тогда полученный продукт представляет собой смесь ADC с распределением одной или более группировок лекарственных средств на антитело. Среднее количество лекарственного средства на антитело может быть рассчитано из смеси, например, двойным ELISA-антитело анализом, специфичным для антитела и специфичным для лекарственного средства. Индивидуальные ADC могут быть идентифицированы в смеси посредством масс-спектроскопии и разделены посредством ВЭЖХ, например хроматографии гидрофобного взаимодействия.
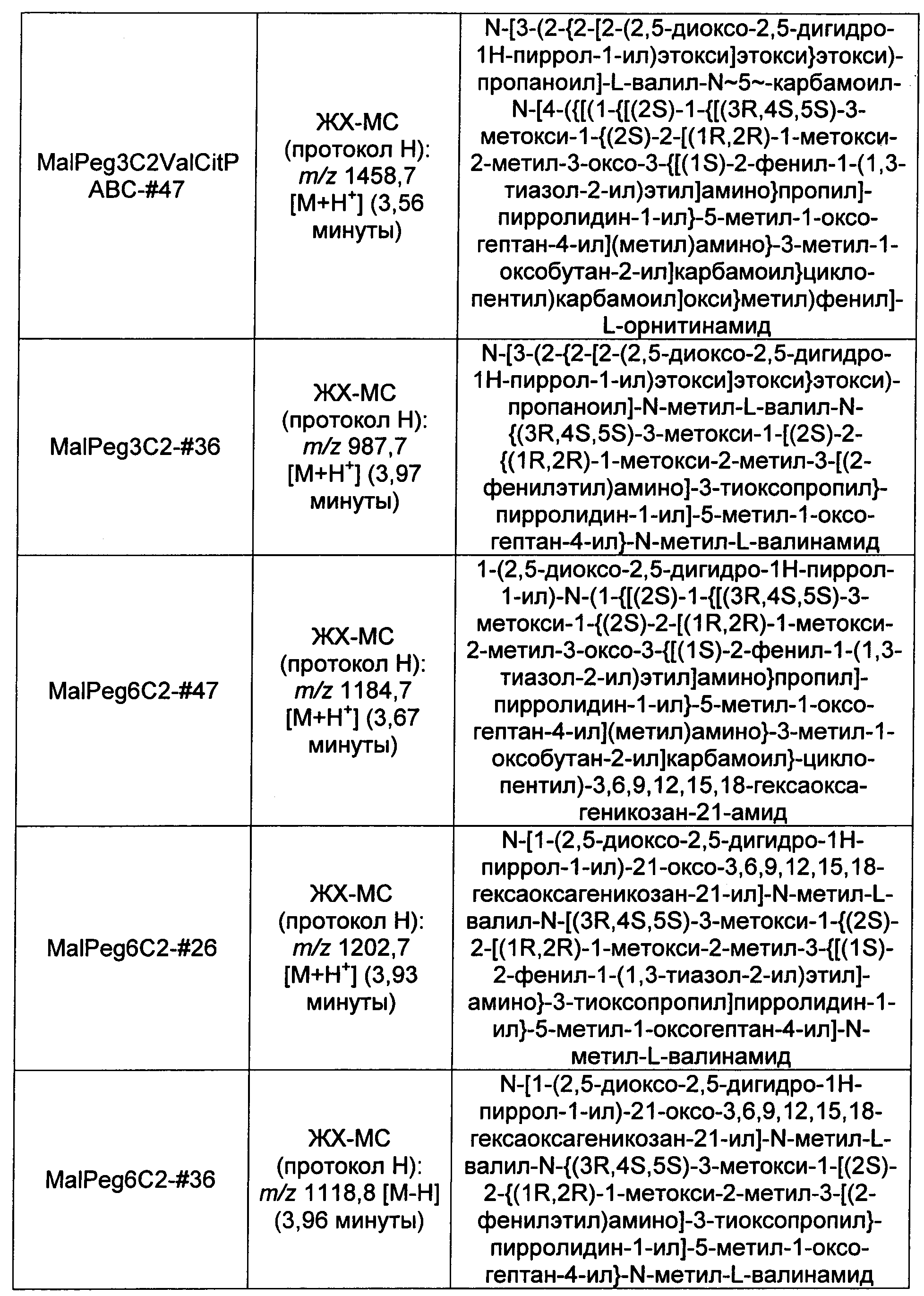
Ниже перечислены сокращения и определения, которые не могут быть иным образом определены или описаны в данной заявке: DMSO (относится к диметилсульфоксиду), МСВР (относится к масс-спектрометрии высокого разрешения), ДДМ (относится к детектированию с помощью диодной матрицы), TFA (относится к 2,2,2-трифторуксусной кислоте, или трифторуксусной кислоте), TFF (относится к тангенциальному поточному фильтрованию), EtOH (относится к этанолу), MW (относится к молекулярной массе), ВЭЖХ (относится к высокоэффективной жидкостной хроматографии), препаративная ВЭЖХ (относится к препаративной высокоэффективной жидкостной хроматографии), и т.д. (означает и так далее), тритил (означает 1,1′,1″-этан-1,1,1-триилтрибензол), THF (относится к тетрагидрофурану), NHS (относится к 1-гидрокси-2,5-пирролидиндиону), Cbz (относится к карбоксибензилу), экв. (относится к эквиваленту), n-BuLi (относится к н-бутиллитию), OAc (относится к ацетату), МеОН (относится к метанолу), i-Pr (относится к изопропилу, или пропан-2-илу), NMM (относится к 4-метилморфолину) и "-" (в таблице, означает, что на данный момент времени данные не доступны).
Соединения и его конъюгаты антитело-лекарственное средство
Один из аспектов изобретения относится к соединению формулы I:

или его фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
W представляет собой



R1 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген;
или
(2) R3A и R3B взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
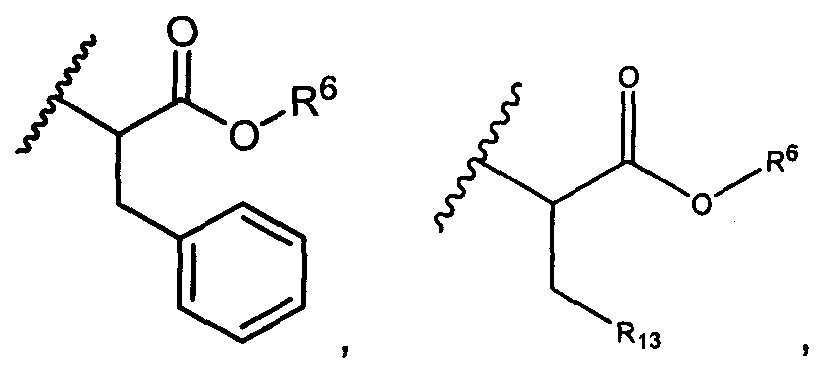
R5 представляет собой




или R5 представляет собой



R6 представляет собой водород, -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил или -C1-C8галогеналкил;
R12 представляет собой водород, C1-C4алкил, C1-C10гетероциклил или C6-C14арил;
R13 представляет собой C1-C10гетероциклил; и
X представляет собой O или S;
при условии, что, когда R3A представляет собой водород, тогда X представляет собой S.
Другой аспект изобретения относится к соединению формулы IIa:

или его фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
W представляет собой



R1 представляет собой


Y представляет собой -C2-C20алкилен-, -C2-C20гетероалкилен-; -C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z представляет собой


R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой




или R5 представляет собой
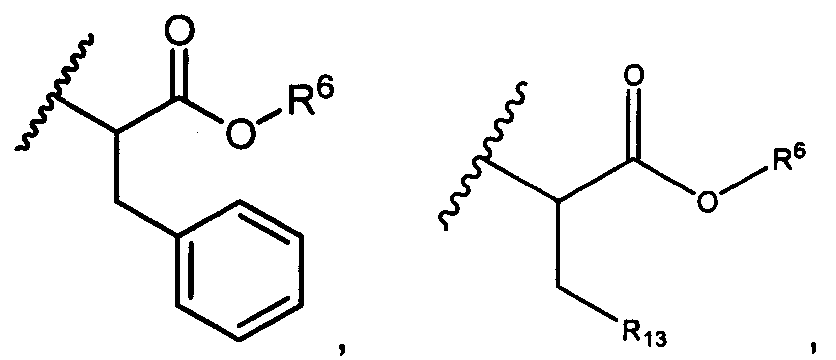


R6 представляет собой водород, -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил или -C1-C8галогеналкил;
R12 представляет собой водород, C1-C4алкил, C1-C10гетероциклил или C6-C14арил;
R13 представляет собой C1-C10гетероциклил; и
R7 в каждом случае независимо выбран из группы, состоящей из F, Cl, I, Br, NO2, CN и CF3;
R10 представляет собой водород, -C1-C10алкил, -C3-C8карбоциклил, -арил, -C1-C10гетероалкил, -C3-C8гетероцикло, -C1-C10алкилен-арил, -арилен-C1-C10алкил, -C1-C10алкилен-(C3-C8карбоцикло), -(C3-C8карбоцикло)-C1-C10алкил, -C1-C10алкилен-(C3-C8гетероцикло) и -(C3-C8гетероцикло)-C1-C10алкил, где арил в R10, содержащем арил, возможно замещен [R7]h;
h равен 1, 2, 3, 4 или 5; и
X представляет собой O или S;
при условии, что, когда R3A представляет собой водород, тогда X представляет собой S.
Другой аспект изобретения относится к соединению формулы IIIa:

или его фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
W представляет собой
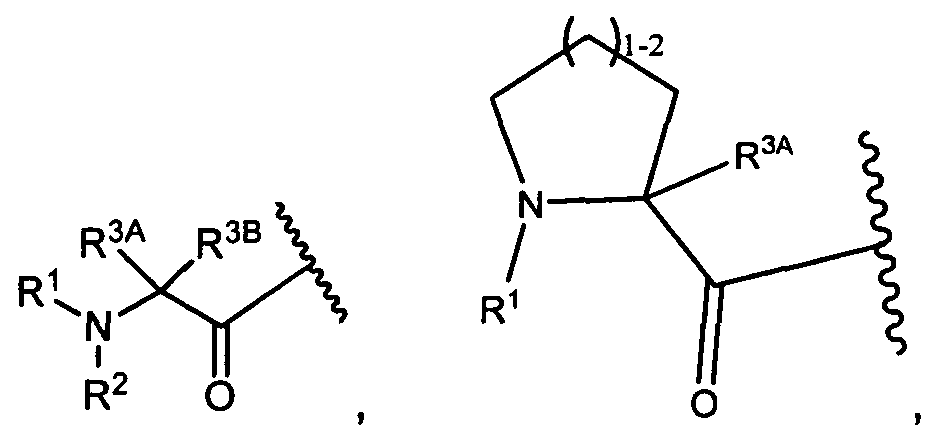


R1 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил; или
(2) R3A и R3B взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой

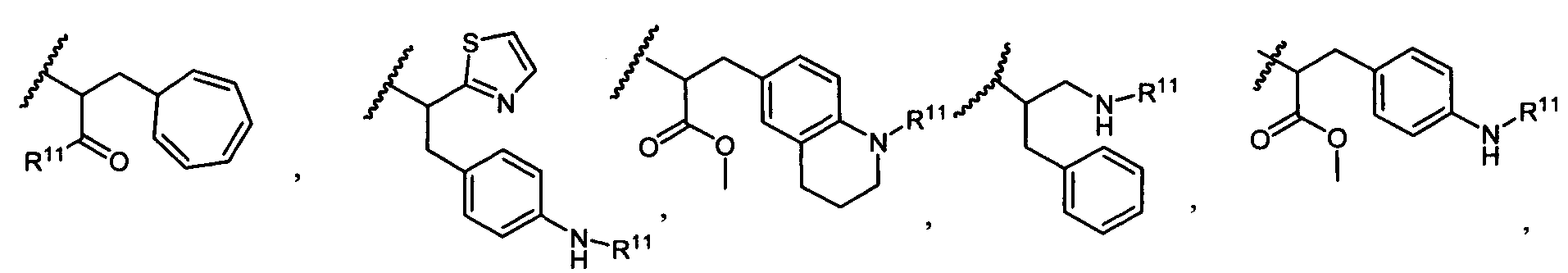
или
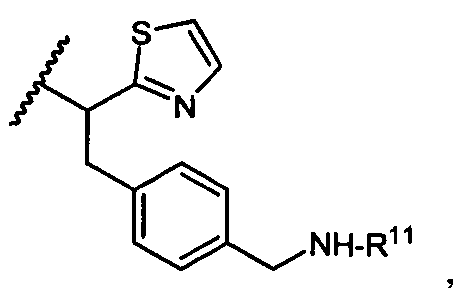
возможно замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: -C1-C8алкил, -O-(C1-C8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -OH, галоген, -N3, -NH2, -NH(R′), -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2, -S(=O)2R′ и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, C1-C8алкила и незамещенного арила;
R11 представляет собой


Y представляет собой -C2-C20алкилен-, -C2-C20гетероалкилен-, C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z представляет собой


R7 в каждом случае независимо выбран из группы, состоящей из F, Cl, I, Br, NO2, CN и CF3;
R10 представляет собой водород, -C1-C10алкил, -C3-C8карбоциклил, -арил, -C1-C10гетероалкил, -C3-C8гетероцикло, -C1-C10алкилен-арил, -арилен-C1-C10алкил, -C1-C10алкилен-(C3-C8карбоцикло), -(C3-C8карбоцикло)-C1-C10алкил, -C1-C10алкилен-(C3-C8гетероцикло) и -(C3-C8гетероцикло)-C1-C10алкил, где арил в R10, содержащем арил, возможно замещен [R7]h;
h равен 1, 2, 3, 4 или 5; и
X представляет собой O или S;
R6 представляет собой водород, C1-C8алкил или C1-C8галогеналкил; и
h равен 1, 2, 3, 4 или 5.
Другой аспект изобретения относится к соединению формулы IIb:

или его фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
W представляет собой
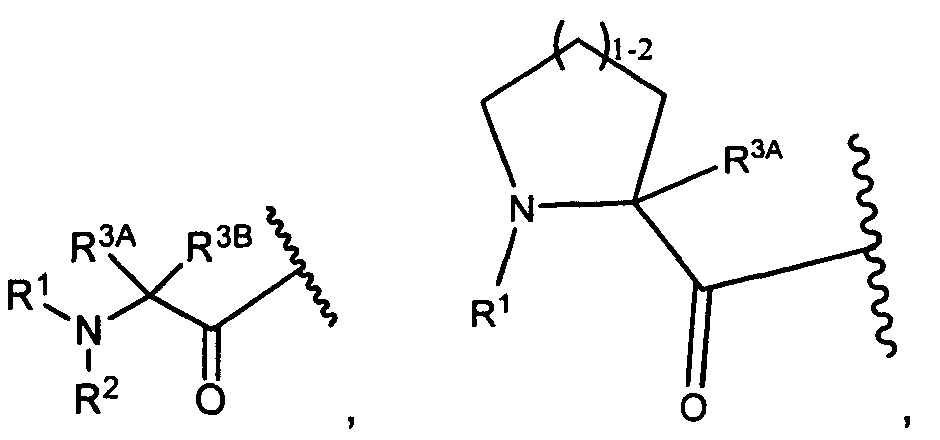
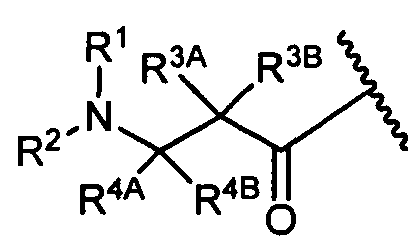

R1 представляет собой
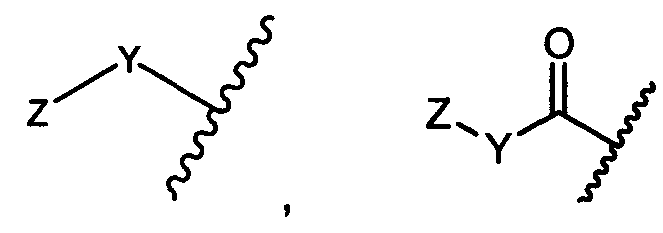

Y представляет собой -C2-C20алкилен-, -C2-C20гетероалкилен-, -C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z представляет собой


L представляет собой антитело;
R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил; или
(2) R3A и R3B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой

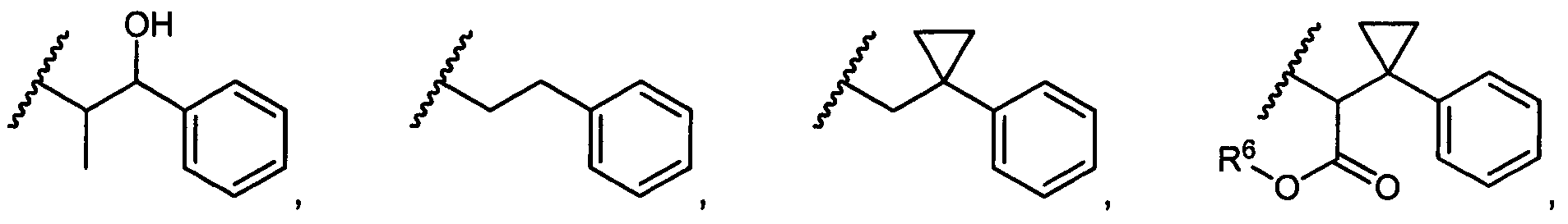


или R5 представляет собой



R6 представляет собой водород, -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил или -C1-C8галогеналкил;
R12 представляет собой водород, C1-C4алкил, C1-C10гетероциклил или C6-C14арил;
R13 представляет собой C1-C10гетероциклил; и
X представляет собой O или S;
при условии, что, когда R3A представляет собой водород, тогда X представляет собой S.
Другой аспект изобретения относится к соединению формулы IIIb:
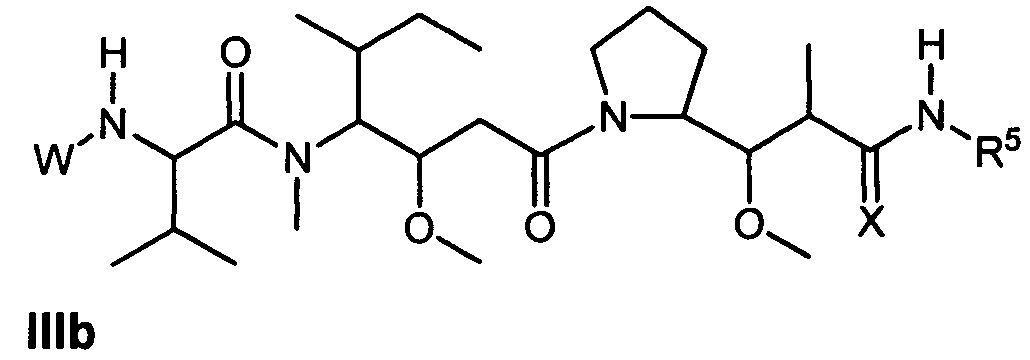
или его фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
W представляет собой


R1 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил; или
(2) R3A и R3B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой



возможно замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: -C1-C8алкил, -O-(C1-C8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -OH, галоген, -N3, -NH2, -NH(R′), -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2, -S(=O)2R′ и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, C1-C8алкила и незамещенного арила;
R11 представляет собой


Y представляет собой -C2-C20алкилен-, -C2-C20гетероалкилен-, -C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z представляет собой


L представляет собой антитело;
X представляет собой O или S.
Другой аспект изобретения относится к соединению формулы IIc:

или его фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
R1′ представляет собой


Y представляет собой -C2-C20алкилен-, -C2-C20гетероалкилен-, -C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z′ представляет собой


L представляет собой антитело;
D представляет собой -C(R4A′)(R4B′)- или отсутствует;
R2′ представляет собой водород, C1-C8алки или C1-C8галогеналкил, либо он отсутствует, когда присутствует

R3A′ и R3B′ определены как любые из следующих:
(1) R3A′ представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B′ представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил, или R3B′ представляет собой C2-C4алкилен и образует 5-7-членное кольцо, как указано посредством
(2) R3A′ и R3B′, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A′ и R4B′ определены как любые из следующих:
(1) R4A′ представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B′ представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A′ и R4B′, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой




или R5 представляет собой


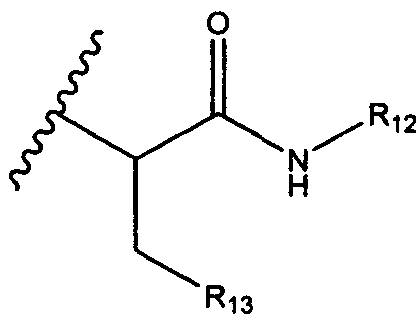
R6 представляет собой водород, -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил или -C1-C8галогеналкил;
R12 представляет собой водород, C1-C4алкил, C1-C10гетероциклил или C6-C14арил;
R13 представляет собой C1-C10гетероциклил; и
X представляет собой I или S;
при условии, что, когда R3A представляет собой водород, тогда X представляет собой S.
Другой аспект изобретения относится к соединению формулы IIIc:

или его фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
W представляет собой



R1 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил; или
(2) R3A и R3B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой


или

возможно замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующего: -C1-C8алкил, -O-(C1-C8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -OH, галоген, -N3, -NH2, -NH(R′), -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2, -S(=O)2R′ и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, C1-C8алкила и незамещенного арила;
R11′ представляет собой

Y представляет собой -C2-C20алкилен-, -C2-C20гетероалкилен-, -C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z′ представляет собой

L представляет собой антитело;
X представляет собой O или S.
Другой аспект изобретения относится к соединению формулы IId:

или его фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
L представляет собой антитело;
[линкер] представляет собой двухвалентный линкер;
D представляет собой -C(RA′)(R4B′)- или отсутствует;
R2′ представляет собой водород, C1-C8алкил или C1-C8галогеналкил, либо он отсутствует, когда присутствует

R3A′ и R3B′ определены как любые из следующих:
(1) R3A′ представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B′ представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил, или R3B′ представляет собой C2-C4алкилен и образует 5-7-членное кольцо, как указано посредством

(2) R3A′ и R3B′, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A′ и R4B′ определены как любые из следующих:
(1) R4A′ представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B′ представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A′ и R4B′, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой




или R5 представляет собой


R6 представляет собой водород, -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил или -C1-C8галогеналкил;
R12 представляет собой водород, C1-C4алкил, C1-C10гетероциклил или C6-C14арил;
R13 представляет собой C1-C10гетероциклил; и
X представляет собой O или S;
при условии, что, когда R3A представляет собой водород, тогда X представляет собой S.
Другой аспект изобретения относится к соединению формулы IIId:

или его фармацевтически приемлемым соли или сольвату, где, в каждом случае независимо,
W представляет собой



R1 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил; или
(2) R3A и R3B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой


или

возможно замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: -C1-C8алкил, -O-(C1-C8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -OH, галоген, -N3, -NH2, -NH(R′), -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2, -S(=O)2R′ и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, C1-C8алкила и незамещенного арила;
[линкер] представляет собой двухвалентный линкер;
L представляет собой антитело;
X представляет собой O или S.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где соединение представлено формулой

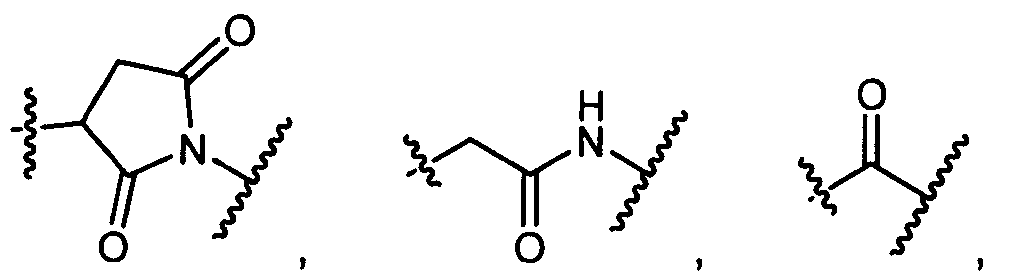
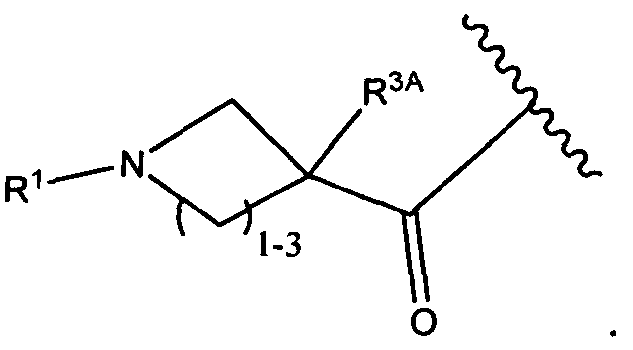
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где W представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где W представляет собой

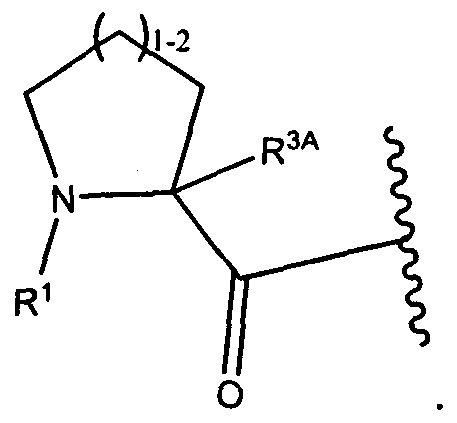
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где W представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где W представляет собой


В некоторых воплощениях изобретения W представляет собой:

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R1 представляет собой водород, C1-C8алкил или C1-C8галогеналкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R1 представляет собой водород.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R1 представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R1 представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R2 представляет собой водород.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R2 представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R2 представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R1 представляет собой водород; и R2 представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R1 представляет собой метил; и R2 представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединении и соответствующих определении, где R3A представляет собой галоген.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A представляет собой водород.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3B представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3B представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3B представляет собой изопропил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3B представляет собой C3-C8карбоциклил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3B представляет собой циклогексил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A представляет собой C1-C8алкил; и R3B представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A представляет собой метил; и R3B представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A представляет собой водород; и R3B представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A представляет собой водород; и R3B представляет собой изопропил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A и R3B взятые вместе представляют собой C2-C8алкилен или C1-C8гетероалкилен.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A и R3B взятые вместе представляют собой C2-C8алкилен.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединении и соответствующих определении, где R3A и R3B взятые вместе представляют собой -CH2CH2-.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A и R3B взятые вместе представляют собой -CH2CH2CH2-.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A и R3B взятые вместе представляют собой -CH2CH2CH2CH2-.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A и R3B взятые вместе представляют собой C1-C8гетероалкилен.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R3A и R3B взятые вместе представляют собой -CH2OCH2-.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединении и соответствующих определении, где R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и R4B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A представляет собой водород.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4B представляет собой водород.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4B представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединении и соответствующих определении, где R4B представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A представляет собой C1-C8алкил; и R4B представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A представляет собой метил; и R4B представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A представляет собой водород; и R4B представляет собой водород.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A представляет собой водород; и R4B представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединении и соответствующих определении, где R4A и R4B взятые вместе представляют собой C2-C8алкилен или C1-C8гетероалкилен.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A и R4B взятые вместе представляют собой C2-C8алкилен.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A и R4B взятые вместе представляют собой -CH2CH2-.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединении и соответствующих определении, где R4A и R4B взятые вместе представляют собой -CH2CH2CH2-.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A и R4B взятые вместе представляют собой -CH2CH2CH2CH2-.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединении и соответствующих определении, где R4A и R4B взятые вместе представляют собой C1-C8гетероалкилен.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R4A и R4B взятые вместе представляют собой -CH2OCH2-.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой
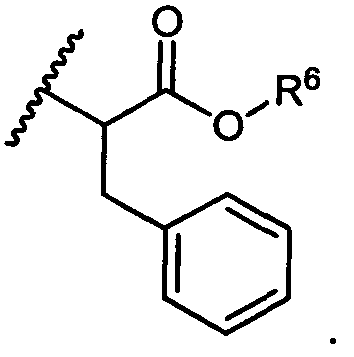
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой
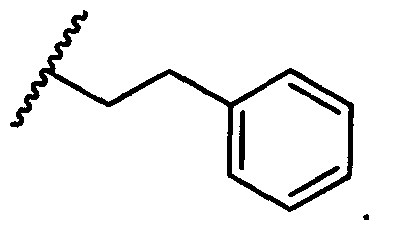
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R5 представляет собой
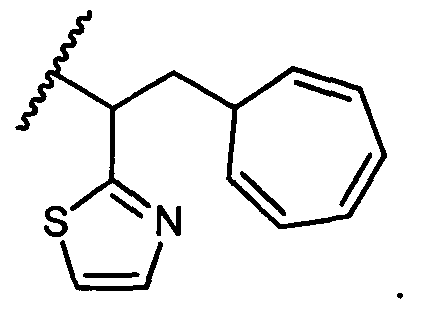
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R6 представляет собой водород.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R6 представляет собой C1-C8алкил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R6 представляет собой метил.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где X представляет собой O.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где X представляет собой S.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений или их фармацевтически приемлемых солей или сольватов и соответствующих определений, где соединение выбрано из группы, состоящей из:
N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L--валинамида;
N2-[(1-аминоциклопентил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L--валинамида;
N-метил-L-валил-N-(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-[(2-фенилэтил)амино]-3-тиоксопропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида;
N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-тиоксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-тиоксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N2-[(1-аминоциклопентил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N2-[(1-аминоциклопропил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
1-амино-N-[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]циклогексанкарбоксамида;
2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-1-валинамида;
2-метилалан ил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-1-валинамида;
2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1-фенилциклопропил)метил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L--валинамида;
N2-[(3-аминооксетан-3-ил)карбонил]-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-((3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-тиоксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N2-(3-амино-2,2-диметилпропаноил)-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N2-(3-амино-2,2-диметилпропаноил)-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида;
2-метил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[1-(бицикло[4.2.0]окта-1,3,5-триен-7-ил)-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[бицикло[4.2.0]окта-1,3,5-триен-7-ил(карбокси)метил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
2-метил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-1-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
метил-N-[(2R,3R)-3-{(2S)-1-[(3R,4S,5S)-4-{[N-(3-амино-2,2-диметилпропаноил)-L-валил](метил)амино}-3-метокси-5-метилгептаноил]-пирролидин-2-ил}-3-метокси-2-метилпропаноил]-L-фенилаланината;
N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метил-D-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-(метиламино)-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-2-амино-1-бензил-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-оксо-2-(пропиламино)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-(диэтиламино)-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-(трет-бутиламино)-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-1-валинамида;
3-метил-D-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
3-метил-L-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
L-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
D-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
1,2-диметил-L-пролил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
1,2-диметил-D-пролил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
N~2~-[2,2-диметил-3-(метиламино)пропаноил]-N-{(1S,2R)-2-метокси-4-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-амино}пропил]пирролидин-1-ил}-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида;
метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[{N-[2,2-диметил-3-(метиламино)пропаноил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноил}-пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината;
метил-N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2S)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}-пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланината;
метил-N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2R)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}-пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланината;
N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2S)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланина;
N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2R)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланина;
метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3R)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината;
метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3R)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината;
(2S)-N-[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбоксамида;
(2R)-N-[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-( циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбоксамида;
2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]-амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N~2~-{[(3R)-3-фторпирролидин-3-ил]карбонил}-N-метил-L-валинамида;
N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]-амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N~2~-{[(3S)-3-фторпирролидин-3-ил]карбонил}-N-метил-L-валинамида;
(2S)-N-[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбоксамида;
(2R)-N-[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбоксамида;
N-2-{[(3R)-3-фторпирролидин-3-ил]карбонил}-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)-этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N-2-{[(3S)-3-фторпирролидин-3-ил]карбонил}-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)-этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
1,2-диметил-D-пролил-N-(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-аминофенил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
1,2-диметил-D-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-аминофенил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
1,2-диметил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-(1,2,3,4-тетрагидрохинолин-6-ил)пропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
1,2-диметил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-аминофенил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-1-валинамида;
1,2-диметил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3R)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланина;
N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3S)-3-фторпирролидин-3-ил]-карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланина;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2R,4S)-4-карбокси-1-фенилпентан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-(бицикло[1.1.1]пент-1-иламино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(1R)-2-метокси-2-оксо-1-(1-фенилциклопропил)этил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(1S)-2-метокси-2-оксо-1-(1-фенилциклопропил)этил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(1R)-1-[(7R)-бицикло[4.2.0]окта-1,3,5-триен-7-ил]-2-метокси-2-оксоэтил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(1S)-1-[(7S)-бицикло[4.2.0]окта-1,3,5-триен-7-ил]-2-метокси-2-оксоэтил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L--валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(1S)-1-[(7R)-бицикло[4.2.0]окта-1,3,5-триен-7-ил]-2-метокси-2-оксоэтил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-1-валинамида;
N,N,2-триметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,N,2-триметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(R)-карбокси(1-фенилциклопропил)метил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
дифтор{2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(3R,4R,7S)-7-бензил-15-{2-[(3,5-диметил-1H-пиррол-2-ил-каппаN)метилиден]-2H-пиррол-5-ил-каппаN}-4-метил-5,8,13-триоксо-2-окса-6,9,12-триазапентадекан-3-ил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамидато}бора;
2-метил-D-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[{N-[(3-аминооксетан-3-ил)-карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината;
2-метилаланил-N-{(3R,4S,5S)-1-[(2S)-2-{(3R,4R,7S,12S)-7-бензил-14-[3-хлор-4-(пропан-2-илокси)фенил]-4-метил-12-[4-(8-метилимидазо[1,2-a]пиридин-2-ил)бензил]-5,8,14-триоксо-2,9-диокса-6,13-диазатетрадекан-3-ил}пирролидин-1-ил]-3-метокси-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида;
2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-{[4-(5-фтор-1,3-бензотиазол-2-ил)-2-метилфенил]амино}-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
1,2-диметил-D-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1H-индол-3-ил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S)-1-оксо-3-фенил-1-(проп-2-ен-1-илокси)пропан-2-ил]-амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-({(2S)-1-оксо-3-фенил-1-[(1H-1,2,3-триазол-4-илметил)амино]-пропан-2-ил}амино)пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S)-1-оксо-3-фенил-1-(проп-2-ин-1-иламино)пропан-2-ил]-амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1H-имидазол-4-ил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-гидроксифенил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1R)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
1,2-диметил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S)-1-оксо-3-фенил-1-(пиперазин-1-ил)пропан-2-ил]-амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида;
1,2-диметил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида; и
2-метил-D-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R1 представляет собой
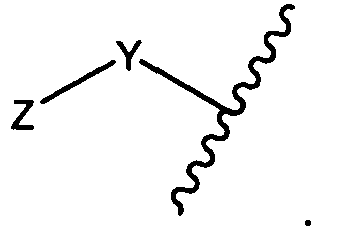
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R1 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R1 представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Y представляет собой C2-C20алкилен.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Y представляет собой -(CH2)p-; и p равен 1-10.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 1. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 2. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 3. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 4. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 5. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 6. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 7. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 8. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 9. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где p равен 10.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Y представляет собой C2-C20гетероалкилен.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Y представляет собой -(CH2CH2O)qCH2CH2-; и q равен 1-10.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 1. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 2. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 3. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 4. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 5. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 6. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 7. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 8. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 9. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где q равен 10.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R7 представляет собой F или Cl; и h равен 4 или 5.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R7 представляет собой F; и h равен 3, 4 или 5.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где R7 представляет собой F; и h равен 5.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой -NH2.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где G представляет собой Cl. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где G представляет собой Br. В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где G представляет собой I.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где соединение выбрано из группы, состоящей из соединений Таблицы 18B (см. в графической части).
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где соединение выбрано из группы, состоящей из:






В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой -NHL.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где Z представляет собой

В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где L представляет собой H(C)-.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых соединений и соответствующих определений, где L представляет собой антитело, выбранное из мышиного антитела для лечения рака шейки матки, например ореговомаба (OVAREX®); мышиного IgG2a антитела для лечения колоректального рака, например эдреколомаба (PANOREX®); химерного антитела против EGFR IgG для лечения видов рака, положительных к эпидермальному фактору роста, таких как рак головы и шеи, например цетуксимаба (ERBITUX®); гуманизированного антитела для лечения саркомы, например гуманизированного моноклонального антитела против рецептора витронектина (αvβ3) типа Vitaxin®; гуманизированного IgG1 антитела для лечения хронического миелоидного лейкоза (CLL), например алемтузумаба (САМРАТН I/H®); SMART ID10, который представляет собой гуманизированное антитело против HLA-DR для лечения неходжкинской лимфомы; 131I Lym-1 (ONCOLYM®), которое представляет собой меченное радиоактивным изотопом мышиное антитело против HLA-Dr10 для лечения неходжкинской лимфомы; гуманизированного анти-CD2 mAb для лечения лимфомы Ходжкина или неходжкинской лимфомы, например ALLOMUNE®; лабетузумаба (CEACIDE®), который представляет собой гуманизированное антитело против CEA для лечения колоректального рака; бевацизумаба (AVASTIN®), который представляет собой гуманизированное анти-VEGF-A mAb для лечения рака мозга, толстой кишки, почек или легких; ибритумомаба тиуксетана (ZEVALIN®), который представляет собой анти-CD20 моноклональное антитело для лечения неходжкинской лимфомы; офатумумаба (ARZERRA®), который представляет собой человеческое анти-CD20 моноклональное антитело для лечения хронического лимфолейкоза; панитумумаба (VECTIBIX®), который представляет собой человеческое анти-EGFR моноклональное антитело для лечения рака толстой кишки; ритуксимаба (RITUXAN®), который представляет собой анти-CD20 химерное моноклональное антитело для лечения хронического лимфолейкоза и неходжкинской лимфомы; тозитумомаба (BEXXAR®), который представляет собой анти-CD20 моноклональное антитело для лечения неходжкинской лимфомы; трастузумаба (HERCEPTIN®), который представляет собой моноклональное антитело против рецептора HER2 для лечения рака груди и желудка; ипилимумаба (YERVOY®), который представляет собой анти-CTLA4 человеческое моноклональное антитело для лечения меланомы; гемтузумаба и инотузумаба озогамицина.
В другом конкретном воплощении L включает антитела, выбранные из анти-I-13 антител, включая анти-I-13 антитела, используемые в лечении рака, например анти-IL-13Rα2 антитела.
В еще одном конкретном воплощении L включает антитела, выбранные из анти-Notch антител, включая анти-Notch антитела, используемые в лечении рака.
В некоторых воплощениях антитело L связано с линкером через серную связь или через связь сера-сера.
Другой аспект изобретения относится к конъюгату антитело-лекарственное средство, содержащему любое из вышеупомянутых соединений.
Другой аспект изобретения относится к конъюгату антитело-лекарственное средство, содержащему антитело и любое из вышеупомянутых соединений.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых конъюгатов антитело-лекарственное средство и соответствующих определений, где соединение ковалентно связано с антителом.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых конъюгатов антитело-лекарственное средство и соответствующих определений, где соединение в указанном конъюгате антитело-лекарственное средство выбрано из группы, состоящей из соединений Таблицы 18B (см. в графической части)






В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых конъюгатов антитело-лекарственное средство и соответствующих определений, где конъюгат антитело-лекарственное средство содержит между 2, 3, 4, 5, 6, 7, 8, 9 или 10 соединений по изобретению.
В некоторых воплощениях настоящее изобретение относится к любому из вышеупомянутых конъюгатов антитело-лекарственное средство и соответствующих определений, где конъюгат антитело-лекарственное средство содержит 3 или 4 соединения по изобретению.
Антительная единица (Ab)
Как отмечено выше, термин "антитело" (или "Ab") используют в данном описании в наиболее широком смысле и конкретно охватывает интактные моноклональные антитела, поликлональные антитела, моноспецифические антитела, мультиспецифические антитела (например биспецифические антитела) и фрагменты антител, которые проявляют желаемую биологическую активность. В дополнение, в то время как некоторые описанные здесь аспекты изобретения относятся к конъюгатам антитело-лекарственное средство, дополнительно предусматривается, что антительная часть конъюгата может быть заменена любым объектом, который специфически связывается или реактивно ассоциирует или образует комплексы с рецептором, антигеном или другой чувствительной группировкой, ассоциированной с данной клеточной популяцией-мишенью. Например, вместо содержания антитела, конъюгаты по изобретению могут содержать нацеливающую молекулу, которая связывается с, образует комплекс с или взаимодействует с рецептором, антигеном или другой чувствительной группировкой данной клеточной популяции для терапевтической или иной биологической модификации. Пример таких молекул включает более низкомолекулярные белки, полипептиды или пептиды, лектины, гликопротеины, не пептиды, витамины, переносящие биогенные вещества молекулы (такие как, без ограничения, трансферрин) или любые другие молекулы или вещества, связывающиеся с клеткой. В некоторых аспектах антитело или другая такая нацеливающая молекула действует для доставки лекарственного средства к конкретной клеточной популяции-мишени, с которой антитело или другая нацеливающая молекула взаимодействует.
Гетероатомы, которые могут присутствовать на антительной единице, включают серу (в одном воплощении из сульфгидрильной группы антитела), кислород (в одном воплощении из карбонильной, карбоксильной или гидроксильной группы антитела) и азот (в одном воплощении из первичной или вторичной аминогруппы антитела). Эти гетероатомы могут присутствовать на антителе в естественном состоянии этого антитела, например существующего в природе антитела, или могут быть введены в антитело посредством химической модификации.
В одном воплощении антительная единица имеет сульфгидрильную группу, и антительная единица связывается через атом серы этой сульфгидрильной группы.
В другом воплощении антитело имеет остатки лизина, которые могут взаимодействовать с активированными сложными эфирами (такие сложные эфиры включают, без ограничения, N-гидроксисукцинимидные, пентафторфенильные и пара-нитрофенильные сложные эфиры) и образовывать таким образом амидную связь, состоящую из атома азота антительной единицы и карбонила.
В еще одном воплощении антительная единица имеет один или более остатков лизина, которые могут быть химически модифицированы для включения одной или более сульфгидрильных групп. Реагенты, которые могут быть использованы для модификации лизинов включают, без ограничения, N-сукцинимидил-3-ацетилтиоацетат (SATA) и гидрохлорид 2-иминотиолана (реагент Трота).
В другом воплощении антительная единица может иметь одну или более углеводных групп, которые могут быть химически модифицированы для получения одной или более сульфгидрильных групп.
В еще одном воплощении антительная единица может иметь одну или более углеводных групп, которые могут быть окислены с получением альдегидной группы (см., например, Laguzza, et al., 1989, J. Med. Chem. 32(3):548-55). Соответствующий альдегид может образовывать связь с реакционноспособным сайтом, таким как, например, гидразин и гидроксиламин. Другие протоколы для модификации белков для присоединения или ассоциирования лекарственных средств описаны в Coligan et al., Current Protocols in Protein Science, vol. 2, John Wiley & Sons (2002) (включено в данное описание посредством ссылки).
Когда конъюгаты содержат неиммунореактивный белок, полипептидные или пептидные единицы вместо антитела, полезные неиммунореактивный белок, полипептидные или пептидные единицы включают, без ограничения, трансферрин, эпидермальные факторы роста ("EGF"), бомбезин, гастрин, гастрин-высвобождающий пептид, фактор роста тромбоцитов, интерлейкин-2 (IL-2), IL-6, трансформирующие факторы роста ("TOP"), такие как TGF-α и TGF-β, фактор роста вируса осповакцины ("VGF"), инсулин и инсулиноподобные факторы роста I и II, соматостатин, лектины и апопротеин липопротеинов низкой плотности.
Полезные поликлональные антитела представляют собой гетерогенные популяции молекул антител, имеющих происхождение из сыворотки иммунизированных животных. Полезные моноклональные антитела представляют собой гомогенные популяции антител против конкретной антигенной детерминанты (например, антигена раковой клетки, вирусного антигена, микробного антигена, белка, пептида, углевода, химического вещества, нуклеиновой кислоты или их фрагментов). Моноклональное антитело (mAb) против представляющего интерес антигена может быть получено путем использования любых методик, известных в данной области техники, которые обеспечивают продуцирование молекул антител стабильными клеточными линиями в культуре.
Полезные моноклональные антитела включают, без ограничения, человеческие моноклональные антитела, гуманизированные моноклональные антитела, фрагменты антител или химерные моноклональные антитела. Человеческие моноклональные антитела могут быть получены любым разнообразием методов, известных в данной области техники (например, Teng et al., 1983, Proc. Natl. Acad. Sci. USA. 80:7308-7312; Kozbor et al., 1983, Immunology Today 4:72-79; и Olsson et al., 1982, Meth. Enzymol. 92:3-16).
Антитело также может представлять собой биспецифическое антитело. Способы получения биспецифических антител известны в данной области техники и обсуждаются ниже.
Антитело может представлять собой функционально активный фрагмент, производное или аналог антитела, которые иммуноспецифически связываются с клетками-мишенями (например, антигенами раковой клетки, вирусными антигенами или микробными антигенами), или другими антителами, которые связываются с опухолевыми клетками или матриксом. В этом отношении, "функционально активные" означает такие фрагмент, производное или аналог, которые способны индуцировать антитела против антиидиотипических антител, которые распознают тот же самый антиген, который распознает антитело, из которого фрагмент, производное или аналог происходят. Более конкретно, в иллюстративном воплощении антигенность идиотипа молекулы иммуноглобулина может быть усилена путем делеции каркасной области и CDR последовательностей, которые являются C-концевыми к CDR последовательности, которая специфически распознает антиген. Чтобы определить какие из CDR последовательностей связываются с антигеном, синтетические пептиды, содержащие CDR последовательности, могут быть использованы в анализах связывания с антигеном посредством любой методики анализа связывания, известной в данной области техники (например, анализ BIAcore) (в отношении определения положения CDR последовательностей, см., например, Kabat et al., 1991, Sequences of Proteins of Immunological Interest, Fifth Edition, National Institute of Health, Bethesda, Md.; Kabat E et al., 1980, J. Immunology 125(3):961-969).
Другие полезные антитела включают фрагменты антител, такие как, без ограничения, F(ab′)2 фрагменты, Fab фрагменты, Fvs, одноцепочечные антитела, диатела, триатела, тетратела, scFv, scFv-FV или любая другая молекула с такой же специфичностью, как антитело.
Дополнительно, рекомбинантные антитела, такие как химерные и гуманизированные моноклональные антитела, содержащие как человеческие участки, так и участки, не являющиеся человеческими, которые могут быть получены с использованием стандартных методик рекомбинантной ДНК, представляют собой полезные антитела. Химерное антитело представляет собой молекулу, в которой различные участки имеют происхождение из различных видов животных, таких как, например, те, которые имеют вариабельную область, имеющую происхождение из константных областей мышиного моноклонального и человеческого иммуноглобулина (см., например, патент США No. 4816567 и патент США No. 4816397, которые включены в данное описание посредством ссылки во всей их полноте). Гуманизированные антитела представляют собой молекулы антител из видов, не являющихся человеческими, имеющие один или более гипервариабельных участков (CDR) из видов, не являющихся человеческими, и каркасную область из молекулы человеческого иммуноглобулина (см., например, патент США No. 5585089, который включен в данное описание посредством ссылки во всей ее полноте). Такие химерные и гуманизированные моноклональные антитела могут быть получены посредством методик рекомбинантной ДНК, известных в данной области техники, например, с использованием способов, описанных в публикации международной заявки WO 87/02671; публикациях европейских заявок No. 0184187, No. 0171496, No. 0173494; публикации международной заявки WO 86/01533; патенте США No. 4816567; публикации европейской заявки No. 012023; Berter et al., 1988, Science 240:1041-1043; Liu et al., 1987, Proc. Natl. Acad. Sci. USA 84:3439-3443; Liu et al., 1987, J. Immunol. 139:3521-3526; Sun et al., 1987, Proc. Natl. Acad. Sci. USA 84:214-218; Nishimura et al., 1987, Cancer. Res. 47:999-1005; Wood et al., 1985, Nature 314:446-449; и Shaw et al., 1988, J. Natl. Cancer Inst. 80:1553-1559; Morrison, 1985, Science 229:1202-1207; Oi et al., 1986, BioTechniques 4:214; U.S. Pat. No. 5,225,539; Jones et al., 1986, Nature 321:552-525; Verhoeyan et al., 1988, Science 239:1534; и Beidler et al., 1988, J. Immunol. 141:4053-4060; каждая из которых включена в данное описание посредством ссылки во всей их полоноте.
Полностью человеческие антитела являются особенно желательными и могут быть получены с использованием трансгенных мышей, которые неспособны к экспрессии генов тяжелых и легких цепей эндогенного иммуноглобулина, но которые могут экспрессировать гены тяжелой и легкой цепей человека. Трансгенных мышей иммунизируют обычным способом с помощью выбранного антигена, например, полноразмерным полипептидом по изобретению или его частью. Моноклональные антитела, направленные против антигена, могут быть получены с использованием традиционной гибридомной технологии. Трансгены человеческого иммуноглобулина, переносимые трансгенными мышами, перегруппировываются во время B-клеточной дифференцировки, и затем подвергаются механизму переключения классов и соматической мутации. Так, с использованием этой технологии возможно продуцирование терапевтически полезных IgG, IgA, IgM и IgE антител. Для обзора этой технологии продуцирования человеческих антител см. Lonberg и Huszar, 1995, Int. Rev. Immunol. 13:65-93. Для подробного обсуждения этой технологии продуцирования человеческих антител и человеческих моноклональных антител и протоколов продуцирования таких антител, см., например, патенты США №№5625126; 5633425; 5569825; 5661016; 5545806; каждый из которых включен в данное описание посредством ссылки во всей их полноте. Другие человеческие антитела могут быть приобретены у, например, Abgenix, Inc. (в настоящее время Amgen, Freemont, Calif.) и Medarex (Princeton, N.J.).
Полностью человеческие антитела, которые распознают выбранный эпитоп, могут быть генерированы с использованием методики, известной как "управляемая селекция". В таком подходе выбранное моноклональное антитело, не являющееся человеческим, например, мышиное антитело, используют для управления селекцией полностью человеческого антитела, распознающего тот же самый эпитоп (см., например, Jespers et al., 1994, Biotechnology 12:899-903). Человеческие антитела могут также быть получены с использованием различных методик, известных в данной области техники, включая библиотеки фагового дисплея (см., например, Hoogenboom and Winter, 1991, J. Mol. Biol. 227:381; Marks et al., 1991, J. Mol. Biol. 222:581; Quan and Carter, 2002, Rise of monoclonal antibodies as therapeutics, In Anti-IgE and Allergic Disease, Jardieu and Fick, eds., Marcel Dekker, New York, N.Y., Chapter 20, pp. 427-469).
В других воплощениях антитело представляет собой слитый белок антитела или его функционально активного фрагмента, например, в котором антитело подвергнуто слиянию через ковалентную связь (например пептидную связь) по N-концу или C-концу с аминокислотной последовательностью другого белка (или его части, предпочтительно включающей по меньшей мере 10, 20 или 50 аминокислот), который не происходит из антитела. Предпочтительно, антитело или его фрагмент ковалентно присоединены к другому белку по N-концу константной области.
Антитела включают аналоги и производные, которые являются либо модифицированными, а именно путем ковалентного присоединения любого типа молекулы, пока такое ковалентное присоединение дает возможность антителу сохранять его антиген-связывающую иммуноспецифичность. В качестве примера, но без какого-либо ограничения, производные и аналоги антител включают те, которые были дополнительно модифицированы, например, путем гликозилирования, ацетилирования, пегилирования, фосфорилирования, амидирования, дериватизации известными защитными/блокирующими группами, протеолитического расщепления, связывания с клеточной антительной единицей или другим белком, и так далее. Любая из многочисленных химических модификаций может быть осуществлена известными методиками, включая, без ограничения, специфическое химическое расщепление, ацетилирование, формилирование, метаболический синтез в присутствии туникамицина, и так далее. Дополнительно, аналог или производное могут содержать одну или более неприродных аминокислот.
Антитела могут иметь модификации (например замены, делеции или вставки) в аминокислотных остатках, которые взаимодействуют с Fc-рецепторами. В частности, антитела могут иметь модификации в аминокислотных остатках, идентифицированных как вовлеченные во взаимодействие между анти-Fc доменом и FcRn рецептором (см., например, публикацию международной заявки WO 97/34631, которая включена в данное описание посредством ссылки во всей ее полноте).
Антитела, иммуноспецифичные в отношении антигена раковой клетки, могут быть приобретены или получены любым способом, известным специалисту в данной области техники, таким как, например, химический синтез или технологии рекомбинантной экспрессии. Нуклеотидная последовательность, кодирующая антитела, иммуноспецифичные в отношении антигена раковой клетки, может быть получена, например, из базы данных GenBank или подобной ей базы данных, литературных публикаций, или традиционным клонированием и секвенированием.
В конкретном воплощении могут быть использованы известные антитела для лечения рака. Антитела, иммуноспецифичные в отношении антигена раковой клетки, могут быть приобретены или получены любым способом, известным специалисту в данной области техники, таким как, например, технологии рекомбинантной экспрессии. Нуклеотидная последовательность, кодирующая антитела, иммуноспецифичные в отношении антигена раковой клетки, может быть получена, например, из базы данных GenBank или подобной ей базы данных, литературных публикаций, или традиционным клонированием и секвенированием. Примеры антител, доступных для лечения рака, включают, без ограничения, OVAREX®, представляющее собой мышиное антитело для лечения рака шейки матки; PANOREX® (Glaxo Wellcome, NC), представляющее собой мышиное IgG2a антитело для лечения колоректального рака; цетуксимаб ERBITUX® (Imclone Systems Inc., NY), представляющий собой химерное антитело против EGFR IgG для лечения видов рака, положительных к эпидермальному фактору роста, таких как рака головы и шеи; Vitaxin® (Medlmmune, Inc., MD), представляющий собой гуманизированное антитело для лечения саркомы; CAMPATH I/H® (Leukosite, MA), представляющее собой гуманизированное IgG1 антитело для лечения хронического миелоидного лейкоза (CLL); SMART ID10 (Protein Design Labs, Inc., CA), который представляет собой гуманизированное антитело против HLA-DR для лечения неходжкинской лимфомы; ONCOLYM® (Techniclone, Inc., CA), представляющее собой меченное радиоактивным изотопом мышиное антитело против HLA-Dr10 для лечения неходжкинской лимфомы; ALLOMUNE® (BioTransplant, CA), представляющий собой гуманизированное анти-CD2 mAb для лечения лимфомы Ходжкина или неходжкинской лимфомы; CEACIDE® (Immunomedics, NJ), представляющий собой гуманизированное антитело против CEA для лечения колоректального рака; AVASTIN® (Genentech/Roche, CA), представляющий собой гуманизированное анти-VEGF-A mAb для лечения рака мозга, толстой кишки, почек или легких; ZEVALIN® (SPECTRUM Pharmaceuticals, NV), представляющий собой анти-CD20 моноклональное антитело для лечения неходжкинской лимфомы; ARZERRA® (GSK, UK), представляющий собой человеческое анти-CD20 моноклональное антитело для лечения хронического лимфолейкоза; VECTIBIX® (Amgen, CA), представляющий собой человеческое анти-EGFR моноклональное антитело для лечения рака толстой кишки; RITUXAN® (Genentech/BioGen, CA), представляющий собой анти-CD20 химерное моноклональное антитело для лечения хронического лимфолейкоза и неходжкинской лимфомы; BEXXAR® (GSK, UK), представляющий собой анти-CD20 моноклональное антитело для лечения неходжкинской лимфомы; HERCEPTIN® (Genentech, CA), представляющий собой моноклональное антитело против рецептора HER2 для лечения рака груди и желудка; YERVOY® (BMS, NJ), представляющий собой анти-CTLA4 человеческое моноклональное антитело для лечения меланомы; MYLOTARG® (Wyeth/Pfizer, NY), представляющий собой анти-CD33 гуманизированное моноклональное антитело, конъюгированное с калихеамицином для лечения острого миелоидного лейкоза; и инотузумаб озогамицин (Wyeth/Pfizer, NY), представляющий собой анти-CD22 гуманизированное моноклональное антитело, конъюгированное с калихеамицином для лечения острого миелоидного лейкоза и неходжкинской лимфомы.
В другом конкретном воплощении могут быть использованы анти-IL13 антитела, включая анти-IL13 антитела, используемые в лечении рака.
В другом конкретном воплощении могут быть использованы анти-Notch антитела, включая анти-Notch антитела, используемые в лечении рака.
В попытках обнаружить эффективные клеточные мишени для диагностики и терапии рака, исследователи пытались идентифицировать трансмембранные и другие опухолеассоциированные полипептиды, которые специфически экспрессируются на поверхности одного или более конкретных видов раковых клеток по сравнению с одной или более нормальными нераковыми клетками. Часто такие опухолеассоциированные полипептиды избыточно эскпрессируются на поверхности раковых клеток по сравнению с поверхностью нераковых клеток. Идентификация таких опухолеассоциированных антигенных полипептидов клеточной поверхности повышает способность специфически нацеливаться на раковые клетки для деструкции посредством терапий на основе антител.
Синтез соединений и их конъюгатов антитело-лекарственное средство
Соединения и конъюгаты по изобретению могут быть получены с использованием методик синтеза, изложенных ниже в разделе Примеры.
Как описано более подробно ниже, соединения и конъюгаты по изобретению могут быть получены с использованием части линкерной единицы, имеющей реакционноспособный сайт для связывания с соединением.
Линкер
Линкер (иногда обозначаемый здесь как "[линкер]") представляет собой бифункциональное соединение, которое может быть использовано для связывания лекарственного средства и антитела с образованием конъюгата антитело-лекарственное средство (ADC). Такие конъюгаты являются полезными, например, в образовании иммуноконъюгатов, направленным против опухолеассоциированных антигенов. Такие конъюгаты обеспечивают избирательную доставку цитотоксических лекарственных средств к опухолевым клеткам.
В одном воплощении линкер имеет формулу:

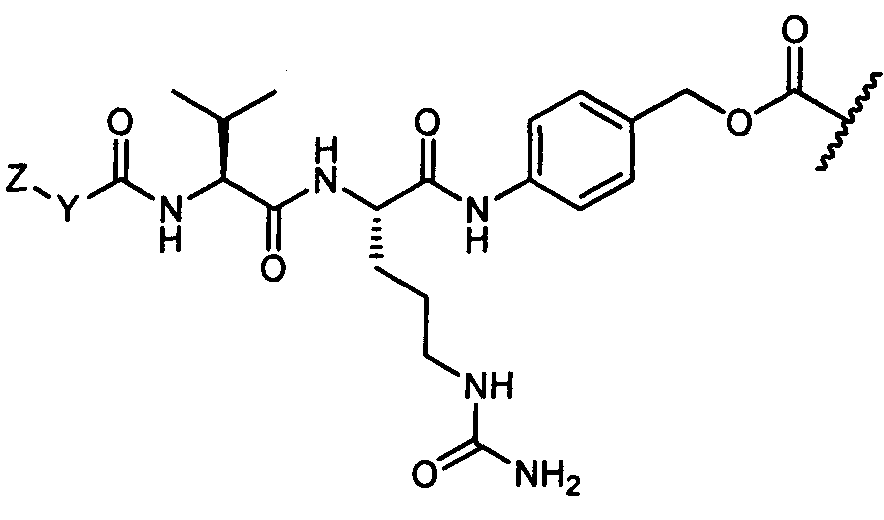
Y представляет собой C2-C20алкилен или C2-C20огетероалкилен; C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z представляет собой


R7 независимо выбран в каждом случае из группы, состоящей из F, Cl, I, Br, NO2, CN и CF3;
R10 представляет собой водород, -C1-C10алкил, -C3-C8карбоцикл, арил, -C1-C10гетероалкил, -C3-C8гетероцикло, -C1-C10алкилен-арил, -арилен-C1-C10алкил, -C1-C10алкилен-(C3-C8карбоцикло), -(C3-C8карбоцикло)-C1-C10алкил, -C1-C10алкилен-(C3-C8гетероцикло) и -(C3-C8гетероцикло)-C1-C10алкил, где арил в R10, содержащем арил, возможно замещен [R7]h; и
h равен 1, 2, 3, 4 или 5.
В ADC линкер служит для присоединения полезной нагрузки к антителу.
В одном аспекте включена вторая часть линкерной единицы, которая имеет второй реакционноспособный сайт, например электрофильную группу, которая является реакционноспособной для нуклеофильной группы, присутствующей на антительной единице (например, антителе). Полезные нуклеофильные группы на антителе включают, без ограничения, сульфгидрил, гидроксил и аминогруппы. Гетероатом нуклеофильной группы антитела является реакционноспособным для электрофильной группы на линкерной единице и образует ковалентную связь с линкерной единицей. Полезные электрофильные группы включают, без ограничения, малеимидные и галогенацетамидные группы. Электрофильная группа обеспечивает подходящий сайт для присоединения антитела.
В другом воплощении линкерная единица имеет реакционноспособный сайт, который имеет нуклеофильную группу, которая является реакционноспособной для электрофильной группы, присутствующей на антителе. Полезные электрофильные группы на антителе включают, без ограничения, альдегидные и кетонные карбонильные группы. Гетероатом нуклеофильной группы линкерной единицы может взаимодействовать с электрофильной группой на антителе и образует ковалентную связь с антителом. Полезные нуклеофильные группы на линкерной единице включают, без ограничения, гидразид, оксим, амино, гидразин, тиосемикарбазон, гидразин карбоксилат и арилгидразид. Электрофильная группа на антителе обеспечивает подходящий сайт для присоединения к линкерной единице.
Аминофункциональные группы также являются полезными реакционноспособными сайтами для линкерной единицы, поскольку они могут взаимодействовать с карбоновой кислотой или активированными сложными эфирами соединения с образованием амидной связки. Как правило, пептидные соединения по изобретению могут быть получены путем образования пептидной связи между двумя или более аминокислотами и/или пептидными фрагментами. Такие пептидные связи могут быть получены, например, согласно методике жидкофазного синтеза (см., например, Schroder и Lubke, "The Peptides", vol. 1, pp 76-136, 1965, Academic Press), который хорошо известен в области пептидной химии.
Как описано более подробно ниже, конъюгаты могут быть получены с использованием части линкера, имеющей реакционноспособный сайт для связывания с соединением по изобретению, и введением другой части линкерной единицы, имеющей реакционноспособный сайт для антитела. В одном аспекте линкерная единица имеет реакционноспособный сайт, который имеет электрофильную группу, которая является реакционноспособной с нуклеофильной группой, присутствующей на антительной единице, такой как антитело. Электрофильная группа обеспечивает подходящий сайт для присоединения антитела. Полезные нуклеофильные группы на антителе включают, без ограничения, сульфгидрильные, гидроксильные и аминогруппы. Гетероатом нуклеофильной группы антитела является реакционноспособным для электрофильной группы на Линкерной единице и образует ковалентную связь с линкерной единицей. Полезные электрофильные группы включают, без ограничения, малеимидные и галогенацетамидные группы.
В другом воплощении линкерная единица имеет реакционноспособный сайт, который имеет нуклеофильную группу, которая является реакционноспособной с электрофильной группой, присутствующей на антительной единице. Электрофильная группа на антителе обеспечивает подходящий сайт для присоединения к линкерной единице. Полезные электрофильные группы на антителе включают, без ограничения, альдегидные и кетонные карбонильные группы. Гетероатом нуклеофильной группы линкерной единицы может взаимодействовать с электрофильной группой на антителе и образует ковалентную связь с антителом. Полезные нуклеофильные группы на линкерной единице включают, без ограничения, гидразид, оксим, амино, гидразин, тиосемикарбазон, гидразин карбоксилат и арилгидразид.
Как используют в данном описании, "mc-", ранее известный как "MalC-",

Как используют в данном описании, "mcValCitPABC-", ранее известный как "MalCValCitPABC-", относится к

Как используют в данном описании, "MalPegXC2-" относится к

Как используют в данном описании, "AmPegXC2-" относится к

Как используют в данном описании, "mcValCitPABCAmPegXC2-" относится к

Как используют в данном описании, "MalPegXC2ValCitPABC-" относится к

Как используют в данном описании, "2BrAcPegXC2" относится к
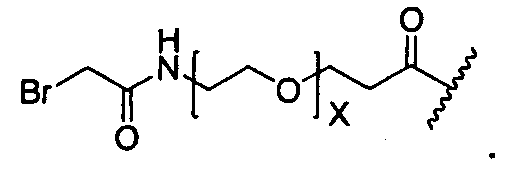
Как используют в данном описании, "mv-" относится к

Как используют в данном описании, "mb-" относится к

Как используют в данном описании, "me-" относится к

Как используют в данном описании, "MalC6-" относится к

Как используют в данном описании, "PFPCOPegXC2ValCitPABC-" означает

Как используют в данном описании, "PFPCOPegXC2AmPegYC2-" относится к

Как используют в данном описании, "PFPCOPegXC2AlaAlaAsnPABC-" относится к

Как используют в данном описании, "PFPCOPegXC2-" относится к
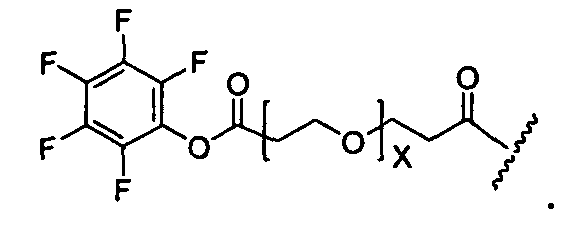
Как используют в данном описании, "PFPCOPegXC2AmPegYC2PABC-" относится к

Как используют в данном описании, "mcGly-" относится к

Как используют в данном описании, "AzCOC2Ph4AmCOPeg2C2-" относится к
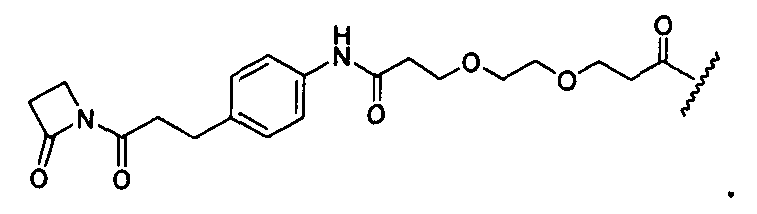
Как используют в данном описании, "AzCOC2Ph4AmPeg1C1-" относится к

Как используют в данном описании, "AcLysValCitPABC-" относится к

Конъюгирование с трансглутаминазой
В некоторых воплощениях соединение по изобретению может быть ковалентно сшито с Fc-содержащим или Fab-содержащим полипептидом, сконструированным с ацил-донорной глутамин-содержащей меткой (например, Gln-содержащими пептидными метками или Q-метками), либо эндогенный глутамин делают реакционноспособным (то есть, способность образовывать ковалентную связь в качестве ацил-донора в присутствии амина и трансглутаминазы) посредством полипептидной инженерии (например, посредством делеции, вставки, замены, мутации аминокислот или любой их комбинации на полипептиде), в присутствии трансглутаминазы, при условии, что соединение по изобретению содержит аминодонорный агент (например, небольшую молекулу, содержащую или присоединенную к реакционноспособному амину), тем самым образуя стабильную и гомогенную популяцию сконструированного Fc-содержащего полипептидного конъюгата, причем аминодонорный агент сайт-специфическим образом конъюгирован с Fc-содержащим или Fab-содержащим полипептидом через ацил-донорную глутамин-содержащую метку или экспонированный/доступный/реакционноспособный эндогенный глутамин. Например, соединения по изобретению могут быть конъюгированы как описано в публикации международной заявки No. PCT/IB2011/054899, содержание которой полностью включено в данное описание посредством ссылки. В некоторых воплощениях для облегчения конъюгирования соединения по изобретению с Fc-содержащим или Fab-содержащим полипептидом, сконструированным с ацил-донорной глутамин-содержащей меткой или эндогенным глутамином, сделанным реакционноспособным посредством полипептидной инженерии в присутствии трансглутаминазы, Z представляет собой NH2.
Конъюгирование с константной областью каппа домена легкой цепи человека
В некоторых воплощениях соединение по изобретению может быть ковалентно присоединено к боковой цепи K188 константной области каппа домена легкой цепи человека (CLк) (нумерация полноразмерной легкой цепи согласно Kabat). K188 также может называться CLк K80, если нумерацию вести только в константной области человеческой каппа, например, SEQ ID NO: 1, 2, 3 и 4).
Например, соединения по изобретению могут быть конъюгированы как описано в заявке на патент США №13/180204 или WO 2012/007896, полное содержание которых включено в данное описание посредством ссылки. В некоторых воплощениях, для облегчения конъюгирования с K188 CLк (CLк-K80), Z представляет собой

В некоторых воплощениях, для облегчения конъюгирования с K188 CLк (CLк-K80), Z представляет собой

В настоящем изобретении дополнительно предложены конъюгаты антитело-лекарственное средство, содержащие антитело или его антиген-связывающие участки, содержащие константный каппа домен, ковалентно конъюгированный с токсином по изобретению, характеризующиеся тем, что по меньшей мере один токсин по изобретению ковалентно конъюгирован с K80 SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3 или SEQ ID NO: 4 (Таблица 1, см. в графической части). В некоторых аспектах количество токсинов по изобретению, ковалентно конъюгированных с K80, может варьировать в диапазоне, нижний предел которого выбран из группы, состоящей из примерно 1,5, примерно 1,6, примерно 1,7, примерно 1,8, примерно 1,9 и примерно 2,0, и верхний предел которого выбран из группы, состоящей из примерно 2,0, примерно 2,1, примерно 2,2, примерно 2,3, примерно 2,4, примерно 2,5. В некоторых аспектах p равен примерно 2.
Конъюгирование токсина с константной областью легкой цепи антитела особенно желательно для минимизации или предотвращения любого вмешательства в связывание Fc участка антитела с Fc рецепторами (такими как FcyR и FcRn) или связывание антитела с его соответствующей мишенью. Напротив, конъюгирование соответствующего токсина с Fc участком антитела может уменьшить время полужизни антитела in vivo и/или его способность взаимодействовать с иммунной системой (эффекторная функция). Конъюгирование токсина с вариабельной областью тяжелой цепи (VH) или вариабельной областью легкой цепи (VL) антитела несет риск минимизации связывания антитела с его когнатной мишенью.
Кроме того, в то время как конъюгирование с CLк-K80 является достоверным и надежным, конъюгирование с лизинами поверхности другого антитела, каждое слегка отличной реакционноспобности и pl, может привести к гетерогенному образцу конъюгированных антител, которые могут высвобождать конъюгированные молекулы несвоевременно или нерегулярно, например во время циркуляции и до доставки эффекторной группировки к мишени посредством распознавания антитела.
В дополнение, настоящее изобретение предлагает известные полиморфизмы каппа цепи V/A по положению 45 и A/L по положению 83 (давая 3 идентифицированных человеческих константных каппа полиморфизма Km(1): V45/L83 (SEQ ID NO: 2), Km(1,2): A45/L83 (SEQ ID NO: 3) и Km(3) A45/V83 (SEQ ID NO: 4)). Вариабельность остатков по положениям 45 и 83 в SEQ ID NO: 1 может быть выбрана таким образом, чтобы обеспечивать только один, два или все три из Km(1), Km(1,2) и Km(3) полиморфизмов.
В некоторых воплощениях изобретения предложена композиция, содержащая соединение по изобретению, ковалентно конъюгированное с антителом (или его антиген-связывающим участком), где по меньшей мере примерно 50%, или по меньшей мере примерно 60%, или по меньшей мере примерно 70%, или по меньшей мере примерно 80%, или по меньшей мере примерно 90% соединения по изобретению в композиции конъюгировано с антителом или его антиген-связывающим участком по K188 CLк.
В некоторых воплощениях соединения по изобретению

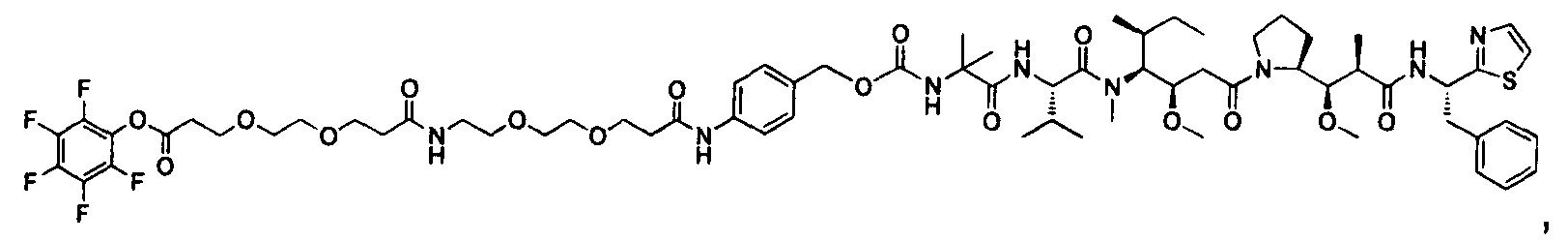


могут быть конъюгированы с объединенным сайтом каталитического антитела, такого как альдолазные антитела, или его антиген-связывающего участка. Альдолазные антитела содержат участки объединенного сайта, которые, будучи незагруженными (например, конъюгацией), катализируют реакцию альдольного присоединения между алифатическим кетонным донором и альдегидным акцептором. Содержание публикации заявки на патент США No. US 2006/205670 включено в данное описание посредством ссылки, в частности стр. 78-118, описывающие линкеры, и абзацы [0153]-[0233], описывающие антитела, их полезные фрагменты, варианты и модификации, p38C2, объединенные сайты и гипервариабельные участки (CDR), и связанная с ними технология антител (Таблица 2, см. в графической части, и репрезентативные соединения ниже):
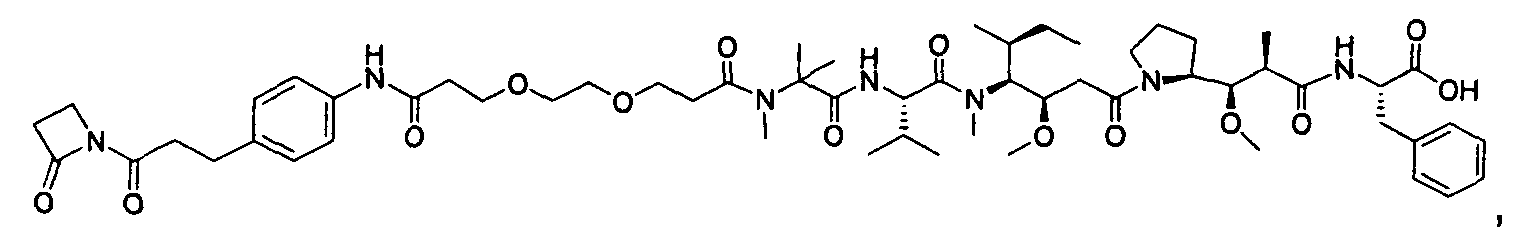

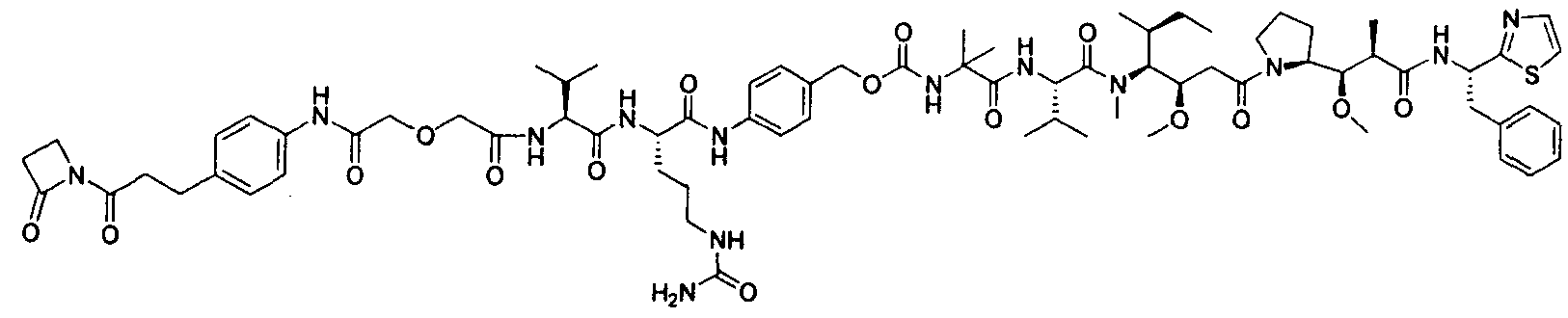

Термин "объединенный сайт" включает CDR и соседние каркасные остатки, которые вовлечены в связывание антигена.
Конъюгирование с линкерами, содержащими сукцинимиды, включая варианты с открытым кольцом
В некоторых воплощениях настоящее изобретение включает соединение по изобретению, конъюгированное через линкер на основе сукцинимида или линкер на основе сукцинимида с открытым кольцом. Стабильность связи сукцинимид-цистеин стала областью возрастающего интереса. Сукцинимиды могут быть перенесены как на in vitro, так и на in vivo экзогенные тиольные нуклеофилы, предпочтительно через реакцию ретро-Михаэля, получая малеимид, который затем подвергается действию тиола. Полагают, что гидролиз кольца приводит к виду, который устойчив к реакции ретро-Михаэля. Это делает полученный конъюгат более стабильным и потенциально более эффективным. Условия могут быть оптимизированы для принудительного открывания сукцинимидного кольца на конъюгате. Основные условия привели к легкому гидролизу кольца. Например, линкеры, содержащие полиэтиленгликольную (PEG) цепь, могут быть гидролизованы при pH 9,2 при 37°C в течение приблизительно 12 час, и линкеры, содержащие алкильную цепь, такие как "mc", могут требовать более высокую температуру и более длительную продолжительность реакции с целью завершения открытия кольца.

Пример принудительного гидролиза конъюгатов на основе малеимида
С целью оценки стабильности этих конъюгатов и выбора приоритетных образцов для оценки in vivo, был разработан анализ, который включает обработку конъюгатов, связанных малеимидным линкером, избытком водного глутатиона (GSH) или плазмы. Аликвоты реакционной смеси анализировали в различные точки времени для определения полезной нагрузки конъюгата, используя описанную выше методологию. Результаты (Таблица 24, см. в графической части) показывают, что связь лекарственное средство-антитело медленно расщепляется GSH-зависимым образом. Как ожидалось, скорость расщепления в высокой степени зависит от гидролиза сукцинимидного кольца. Важно то, что эти результаты, как видно, трансформируются в улучшенные фармакокинетические (PK) показатели, как измерено по увеличению площади под кривой (AUC) конъюгата и по увеличению соотношения конъюгат/Ab.
Способ оценки стабильности ADC
Образец ADC (30 мкг) в PBS смешивали с раствором глутатиона (GSH) с получением конечной концентрации GSH 0,5 мМ и концентрации белка 3 мг/мл. Контрольный образец (без GSH) готовили подобным образом из 30 мкг ADC, разбавленного до 3 мг/мл в PBS. GSH-обработанный образец ADC и контрольный образец ADC инкубировали при 37°C и отбирали пробы в моменты времени 0, 3 и 6 суток. Аликвоты восстанавливали избытком TCEP, подкисляли добавлением 0,1%-ного раствора муравьиной кислоты с 10%-ным ацетонитрилом и анализировали в отношении полезной нагрузки посредством LC/MC, как описано нише.
Анализ образца: Анализ осуществляли с использованием прибора Aglient 1100 capillary HPLC, соединенного с масс-спектрометром Waters Xevo G2 Q-TOF. Аналиты загружали на колонку Zorbax Poroshell 300SB C8 (0,5 мм × 75 мм, поддерживаемой при 80°C) с 0,1%-ной муравьиной кислотой и элюировали с использованием градиента 20-40% буфера В (80% ацетонитрила, 18% 1-пропанола, 2% воды с 0,1%-ной муравьиной кислотой) при скорости потока 20 мкл/мин в течение 5,5 минут. Масс-спектрометрическое детектирование осуществляли с ионизацией электрораспылением в режиме положительных ионов с установкой капиллярного напряжения при 3,3 кВ. Анализ данных осуществляли с использованием MaxEnt 1 функции в MassLynx и интенсивности использовали для расчета полезной нагрузки на основе предварительно описанной формулы.
Композиции и способы введения
В других воплощениях другой аспект изобретения относится к фармацевтической композиции, включающей эффективное количество соединения по изобретению и/или его конъюгата антитело-лекарственное средство и фармацевтически приемлемый носитель или наполнитель. В некоторых воплощениях композиции подходят для ветеринарного введения или введения человеку.
Фармацевтические композиции по настоящему изобретению могут быть представлены в любой форме, которая обеспечивает введение композиции пациенту. Например, композиция может быть представлена в форме твердого вещества или жидкости. Типичные пути введения включают, без ограничения, парентеральное, глазное и внутриопухолевое. Парентеральное введение включает подкожные инъекции, внутривенную, внутримышечную или внутригрудную инъекцию или инфузионные техники. В одном аспекте композиции вводят парентерально. В конкретном воплощении композиции вводят внутривенно.
Фармацевтические композиции могут быть изготовлены так, чтобы обеспечить соединению по изобретению и/или его конъюгату антитело-лекарственное средство биодоступность после введения композиции пациенту. Композиции могут принимать форму одной или более стандартных лекарственных форм, где, например, таблетка может представлять собой одноразовую стандартную форму, а контейнер соединения по изобретению и/или его конъюгата антитело-лекарственное средство в жидкой форме может содержать множество единиц дозировки.
Материалы, используемые в изготовлении фармацевтических композиций, могут быть нетоксичными в используемых количествах. Специалистам в данной области техники очевидно, что оптимальная дозировка активного(ых) ингредиента(ов) в фармацевтической композиции зависит от ряда факторов. Релевантные факторы включают, без ограничения, вид животного (например, человек), конкретную форму соединения по изобретению и/или его конъюгата антитело-лекарственное средство, путь введения и используемую композицию.
Фармацевтически приемлемый носитель или наполнитель может представлять собой твердое вещество или частицы, так что композиции находятся, например, в таблетированной или порошкообразной форме. Носитель(и) может(гут) представлять собой жидкость. В дополнение, носитель(и) может(гут) представлять собой частицы.
Композиция может быть представлена в форме жидкости, например, раствора, эмульсии или суспензии. В композиции для введения путем инъекции могут также быть включены один или более поверхностно-активных веществ, консервантов, увлажняющих агентов, диспергирующих агентов, суспендирующих агентов, буферов, стабилизаторов и изотонических агентов.
Жидкие композиции, независимо от того представляют ли они растворы, суспензии или другую подобную форму, могут также включать одно или более из следующего: стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно физиологический солевой раствор, раствор Рингера, изотонический хлорид натрия, фиксированные масла, такие как синтетические моно- или диглицериды, которые могут служить в качестве растворителя или суспендирующей среды, полиэтиленгликоли, глицерин, циклодекстрин, пропиленгликоль или другие растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатообразующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты, фосфаты или аминокислоты и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. Парентеральная композиция может быть заключена в ампулу, одноразовый шприц или многодозовый флакон, изготовленный из стекла, пластика или другого материала. Физиологический солевой раствор является примером адъюванта. Инъецируемая композиция предпочтительно является стерильной.
Количество соединения по изобретению и/или его конъюгата антитело-лекарственное средство, которое является эффективным в лечении конкретного расстройства или состояния, зависит от природы расстройства или состояния, и может быть определено путем стандартных клинических методик. В дополнение, in vitro или in vivo анализы возможно могут быть использованы, чтобы помочь идентифицировать оптимальные диапазоны дозировок. Точная доза, предназначенная для использования в композиции, зависит от пути введения и тяжести заболевания или расстройства, и будет решаться в соответствии с мнением лечащего врача и особенностей каждого пациента.
Композиции содержат эффективное количество соединения по изобретению и/или его конъюгата антитело-лекарственное средство, так чтобы получить подходящую дозировку. Как правило, такое количество составляет по меньшей мере примерно 0,01% соединения по изобретению и/или его конъюгата антитело-лекарственное средство по массе композиции. В иллюстративном воплощении фармацевтическую композицию изготавливают таким образом, чтобы единица парентеральной дозировки содержала от примерно 0,01% до примерно 2% по массе количества соединения по изобретению и/или его конъюгата антитело-лекарственное средство.
Для внутривенного введения композиция может содержать от примерно 0,01 до примерно 100 мг соединения по изобретению и/или его конъюгата антитело-лекарственное средство на кг массы тела пациента. В одном аспекте композиция может включать от примерно 1 до примерно 100 мг соединения по изобретению и/или его конъюгата антитело-лекарственное средство на кг массы тела пациента. В другом аспекте вводимое количество соединения по изобретению и/или его конъюгата антитело-лекарственное средство находится в диапазоне от примерно 0,1 до примерно 25 мг/кг массы тела.
В общем, дозировка соединения по изобретению и/или его конъюгата антитело-лекарственное средство, вводимого пациенту, как правило составляет от примерно 0,01 мг/кг до примерно 20 мг/кг массы тела пациента. В одном аспекте дозировка, вводимая пациенту, составляет от примерно 0,01 мг/кг до примерно 10 мг/кг массы тела пациента. В другом аспекте дозировка, вводимая пациенту, составляет от примерно 0,1 мг/кг до примерно 10 мг/кг массы тела пациента. В еще одном аспекте дозировка, вводимая пациенту, составляет от примерно 0,1 мг/кг до примерно 5 мг/кг массы тела пациента. В еще одном аспекте вводимая дозировка составляет от примерно 0,1 мг/кг до примерно 3 мг/кг массы тела пациента. В еще одном аспекте вводимая дозировка составляет от примерно 1 мг/кг до примерно 3 мг/кг массы тела пациента.
Соединение по изобретению и/или его конъюгат антитело-лекарственное средство могут быть введены любым общепринятым путем, например путем инфузии или болюсной инъекции. Введение может быть системным или местным. Известны различные системы доставки, например, инкапсулирование в липосомы, микрочастицы, микрокапсулы, капсулы и так далее, и они могут быть использованы для введения соединения по изобретению и/или его конъюгата антитело-лекарственное средство. В некоторых воплощениях более одного соединения по изобретению и/или его конъюгата антитело-лекарственное средство вводят пациенту.
В конкретных воплощениях может быть желательно вводить одно или более соединений по изобретению и/или их конъюгатов антитело-лекарственное средство локально в область, нуждающуюся в лечении. Этого можно достичь, в качестве примера и без ограничения, путем локальной инфузии во время хирургического вмешательства; местного нанесения, например, вместе с раневой повязкой после хирургического вмешательства; путем инъекции; посредством катетера; или с помощью имплантата, представляющего собой пористый, непористый или желатинообразный материал, включая мембраны, такие как силиконовые мембраны, или волокна. В одном воплощении введение может быть осуществлено путем прямой инъекции в место (или бывшее место), пораженное раком, опухолью или неопластической или пренеопластической тканью. В другом воплощении введение может быть осуществлено путем прямой инъекции в место (или бывшее место) проявления аутоиммунного заболевания.
В еще одном воплощении соединение по изобретению и/или его конъюгат антитело-лекарственное средство могут быть доставлены в системе контролируемого высвобождения, такой как, без ограничения, помпа, или могут быть использованы различные полимерные материалы. В еще одном воплощении система контролируемого высвобождения может быть помещена вблизи мишени соединения по изобретению и/или его конъюгата антитело-лекарственное средство, например, печени, тем самым требуя только фракцию системной дозы (см., например, Goodson, в Medical Applications of Controlled Release, выше, vol. 2, pp. 115-138 (1984)). Могут быть использованы другие системы контролируемого высвобождения, обсуждаемые в обзоре Langer (Science 249:1527-1533 (1990)).
Термин "носитель" относится к разбавителю, адъюванту или эксципиенту, с которым вводят соединение или его конъюгат антитело-лекарственное средство. Такие фармацевтические носители могут представлять собой жидкости, такие как вода и масла, в том числе из жидких углеводородов, животного, растительного или синтетического происхождения. Носители могут представлять собой солевой раствор и тому подобное. В дополнение, могут быть использованы вспомогательные, стабилизирующие и другие агенты. В одном воплощении, при введении пациенту, соединение или конъюгат и фармацевтически приемлемые носители являются стерильными. Вода является иллюстративным носителем, когда соединение или конъюгат вводят внутривенно. Солевые растворы и водные растворы декстрозы и глицерина также могут быть использованы в качестве жидких носителей, в частности для инъецируемых растворов. Композиции по настоящему изобретению при желании могут также содержать небольшие количества увлажняющих или эмульгирующих агентов, или забуферивающих pH агентов.
Композиции по настоящему изобретению могут принимать форму растворов, пеллет, порошков, препаратов непрерывного высвобождения или любую другую форму, подходящую для применения. Другие примеры подходящих фармацевтических носителей описаны в E.W. Martin "Remington′s Pharmaceutical Sciences".
В одном из воплощений соединение по изобретению и/или его конъюгат антитело-лекарственное средство изготавливают в соответствии с общепринятыми методиками в виде фармацевтической композиции, изготовленной с возможностью внутривенного введения животным, в частности людям. Как правило, носители или наполнители для внутривенного введения представляют собой стерильные изотонические водные буферные растворы. При необходимости композиции также могут включать солюбилизирующий агент. Композиции для внутривенного введения возможно могут содержать местный анестетик, такой как лигнокаин, для облегчения боли в месте инъекции. В общем, ингредиенты поставляют раздельно или в смеси в стандартной лекарственной форме, например, в виде сухого лиофилизированного порошка или свободного от воды концентрата, в герметично закрытом контейнере, таком как ампула, или саше, с указанием количества активного агента. Когда соединение по изобретению и/или его конъюгат антитело-лекарственное средство предназначены для введения посредством инфузии, они могут быть помещены, например, во флакон для инфузии, содержащий стерильную воду фармацевтической степени чистоты или солевой раствор. Когда соединение по изобретению и/или его конъюгат антитело-лекарственное средство вводят путем инъекции, ампула стерильной воды для инъекции или солевого раствора может быть предложена, так чтобы ингредиенты можно было смешать перед введением.
Композиция может включать разные материалы, которые модифицируют физическую форму твердого вещества или жидкую лекарственную форму. Например, композиция может включать материалы, которые образуют покрывающую оболочку, окружающую активные ингредиенты. Материалы, которые образуют покрывающую оболочку, как правило являются инертными и могут быть выбраны из, например, сахара, шеллака и других энетросолюбильных покрывающих агентов. Альтернативно, активные ингредиенты могут быть инкапсулированы в желатиновую капсулу.
Независимо от того является ли форма композиции по настоящему изобретению твердой или жидкой, она может включать фармакологический агент, используемый в лечении рака.
Терапевтические применения соединений и их конъюгатов антитело-лекарственное средство
Другой аспект изобретения относится к способу использования соединений по изобретению и их конъюгатов антитело-лекарственное средство для лечения рака.
Соединения по изобретению и/или их конъюгаты антитело-лекарственное средство полезны для ингибирования размножения опухолевой клетки или раковой клетки, индуцирования апоптоза в опухолевой или раковой клетке или во время лечения рака у пациента. Соединения по изобретению и/или их конъюгаты антитело-лекарственное средство могут быть использованы соответственно во множестве показаний для лечения видов рака у животного. Указанные конъюгаты могут быть использованы для доставки соединения по изобретению к опухолевой клетке или раковой клетке. Не привязываясь к какой-либо теории, в одном воплощении антитело конъюгата связывается или ассоциирует с антигеном, ассоциированным с раковой клеткой или опухолевой клеткой, и конъюгат может быть перенесен (интернализован) внутрь опухолевой клетки или раковой клетки через рецептор-опосредованный эндоцитоз или другой механизм интернализации. Антиген может быть присоединен к опухолевой клетке или раковой клетке или может быть белком внеклеточного матрикса, ассоциированным с опухолевой клеткой или раковой клеткой. В некоторых воплощениях, оказавшись в клетке, одна или более специфических пептидных последовательностей подвергаются ферментативному или гидролитическому расщеплению одной или более протеазами, ассоциированными с опухолевой клеткой или раковой клеткой, приводя к высвобождению соединения по изобретению из конъюгата. Высвобожденное соединение по изобретению затем свободно мигрирует внутри клетки и индуцирует цитотоксические или цитостатические действия. Конъюгат также может расщепляться внутриклеточной протеазой с высвобождением соединения по изобретению. В альтернативном воплощении соединение по изобретению отщепляется от конъюгата снаружи опухолевой клетки или раковой клетки, и соединение по изобретению затем проникает в клетку.
В некоторых воплощениях конъюгаты обеспечивают конъюгат-специфичное нацеливание противоопухолевого или противоракового лекарственного средства, тем самым снижая общую токсичность соединений по изобретению.
В другом воплощении антительная единица связывается с опухолевой клеткой или раковой клеткой.
В еще одном воплощении антительная единица связывается с антигеном опухолевой клетки или раковой клетки, который находится на поверхности опухолевой клетки или раковой клетки.
В еще одном воплощении антительная единица связывается с антигеном опухолевой клетки или раковой клетки, который представляет собой белок внеклеточного матрикса, ассоциированный с опухолевой клеткой или раковой клеткой.
Специфичность антительной единицы в отношении конкретной опухолевой клетки или раковой клетки может быть важна для определения тех опухолей или видов рака, которые лечатся наиболее эффективно.
Конкретные типы рака, которые можно лечить соединением по изобретению и/или его конъюгатом антитело-лекарственное средство, включают, без ограничения, карциномы мочевого пузыря, молочной железы, шейки матки, толстой кишки, эндометрия, почек, легких, пищевода, яичников, предстательной железы, поджелудочной железы, кожи, желудка и яичек, а также злокачественные новообразования крови, включая, без ограничения, лейкозы и лимфомы.
Комплексная терапия рака. Виды рака, включая, без ограничения, опухоль, метастазы, или другое заболевание или расстройство, характеризующееся неконтролируемым клеточным ростом, можно лечить или подавлять путем введения соединения по изобретению и/или его конъюгата антитело-лекарственное средство.
В других воплощениях предложены способы лечения рака, включающие введение пациенту, нуждающемуся в этом, эффективного количества соединения по изобретению и/или его конъюгата антитело-лекарственное средство и химиотерапевтического агента. В одном воплощении химиотерапевтический агент является таким, с которым лечение рака, как обнаружено, не являлось не поддающимся терапии. В другом воплощении химиотерапевтический агент является таким, с которым лечение рака, как обнаружено, не поддавалось терапии. Соединение по изобретению и/или его конъюгат антитело-лекарственное средство может быть введено пациенту, который уже подвергался хирургическому вмешательству в качестве лечения рака.
В некоторых воплощениях пациент также получает дополнительное лечение, такое как радиотерапия. В конкретном воплощении соединение по изобретению и/или его конъюгат антитело-лекарственное средство вводят одновременно с химиотерапевтическим агентом или с радиотерапией. В другом конкретном воплощении химиотерапевтический агент или радиотерапию вводят до или после введения соединения по изобретению и/или его конъюгата антитело-лекарственное средство.
Химиотерапевтический агент может быть введен на протяжении ряда сеансов. Любой один или комбинация химиотерапевтических агентов, таких как стандартное лечение химиотерапевтическим(и) агентом(ами), могут быть введены.
Дополнительно предложены способы лечения рака соединением по изобретению и/или его конъюгатом антитело-лекарственное средство в качестве альтернативы химиотерапии или радиотерапии, когда химиотерапия или радиотерапия проявляют или могут проявлять слишком сильную токсичность, например, приводят к неприемлемым или непереносимым побочным эффектам, для субъекта, подвергаемого лечению. Пациент, которого лечат, может, возможно, быть подвергнут дополнительному лечению рака, такому как хирургическое вмешательство, радиотерапия или химиотерапия, в зависимости от того, какое лечение находят приемлемым или переносимым.
Соединения по изобретению и/или его конъюгаты антитело-лекарственное средство могут также быть использованы в in vitro или ex vivo способе, таком как для лечения некоторых видов рака, включая, без ограничения, лейкозы и лимфомы, причем лечение включает аутологические трансплантаты стволовых клеток. Такое лечение может включать многостадийный процесс, при котором аутологические гематопоэтические стволовые клетки животного собирают и очищают от всех раковых клеток, причем остающуюся популяцию клеток костного мозга животного затем устраняют посредством введения высокой дозы соединения по изобретению и/или его конъюгата антитело-лекарственное средство при одновременной высокодозовой радиотерапии или без нее и трансплантат стволовых клеток внедряют обратно в организм животного. Затем проводят поддерживающую терапию, в то время как функция костного мозга восстанавливается и пациент выздоравливает.
Высвобождаемые виды соединений
Дополнительные воплощения по изобретению включают высвобождаемые химические виды соединений, внутри или вблизи раковой клетки или опухолевой клетки, как полагают, посредством ферментативного и/или гидролитического расщепления одной или более протеазой, ассоциированной с раковой клеткой или опухолевой клеткой. Такие соединения включают описанные здесь виды, а также соединения, такие как описано следующей структурой:

или их фармацевтически приемлемые соли или сольваты, где, в каждом случае независимо,
W представляет собой
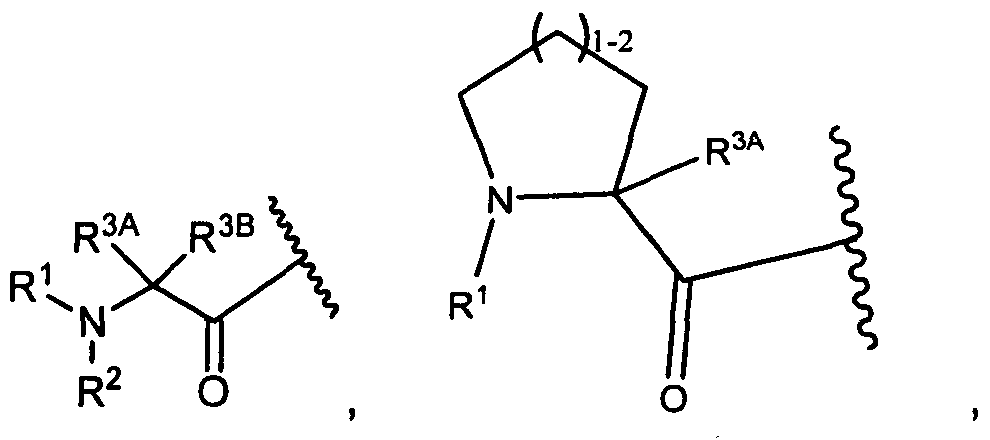

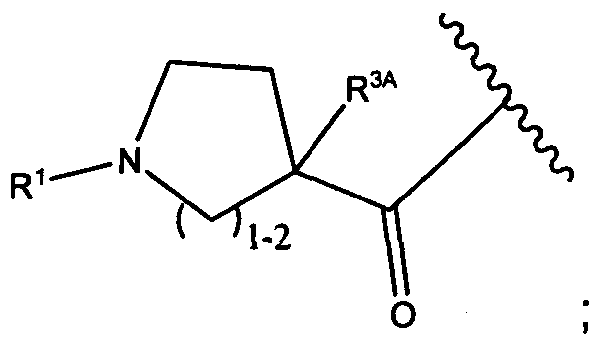
R1 представляет собой

Y представляет собой -C2-C20алкилен-, -C2-C20гетероалкилен-, C3-C8карбоцикло-, -арилен-, -C3-C8гетероцикло-, -C1-C10алкилен-арилен-, -арилен-C1-C10алкилен-, -C1-C10алкилен-(C3-C8карбоцикло)-, -(C3-C8карбоцикло)-C1-C10алкилен-, -C1-C10алкилен-(C3-C8гетероцикло)- или -(C3-C8гетероцикло)-C1-C10алкилен-;
Z″ представляет собой


Z′′′ представляет собой


G представляет собой галоген, -OH, -SH или -S-C1-C6алкил;
R2 представляет собой водород, C1-C8алкил или C1-C8галогеналкил;
R3A и R3B определены как любвые из следующих:
(1) R3A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, галоген или аралкил; и
R3B представляет собой C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил, аралкил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R4A и R4B определены как любые из следующих:
(1) R4A представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; и
R4B представляет собой водород, C1-C8алкил, C1-C8галогеналкил, C3-C8карбоциклил, C1-C10гетероциклил, арил, гетероаралкил или аралкил; или
(2) R4A и R4B взятые вместе, представляют собой C2-C8алкилен или C1-C8гетероалкилен;
R5 представляет собой
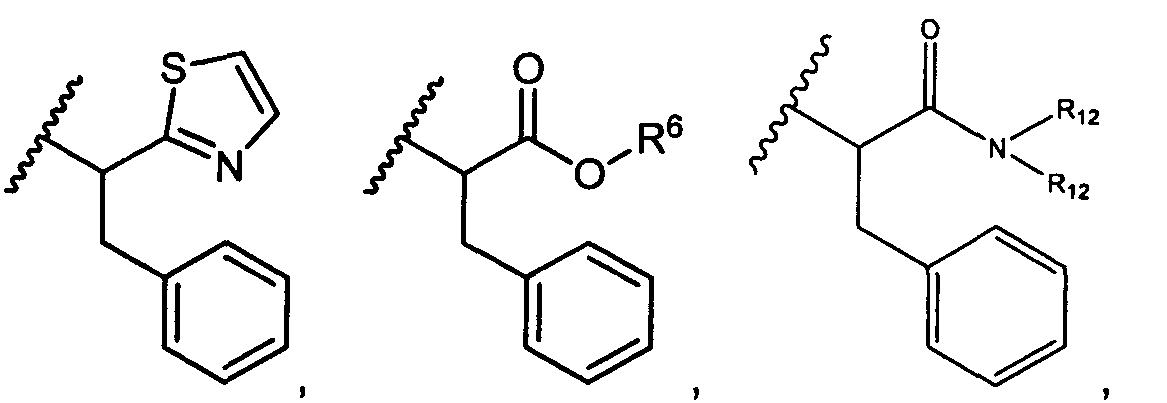



или R5 представляет собой


R6 представляет собой водород, -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил или -C1-C8галогеналкил;
R12 представляет собой водород, C1-C4алкил, C1-C10гетероциклил или C6-C14арил;
R13 представляет собой C1-C10гетероциклил; и
R7 независимо выбран для каждого случая из группы, состоящей из F, Cl, I, Br, NO2, CN и CF3;
R10 представляет собой водород, -C1-C10алкил, -C3-C8карбоциклил, -арил, -C1-C10гетероалкил, -C3-C8гетероцикло, -C1-C10алкилен-арил, -арилен-C1-C10алкил, -C1-C10алкилен-(C3-C8карбоцикло), -(C3-C8карбоцикло)-C1-C10алкил, -C1-C10алкилен-(C3-C8гетероцикло) и -(C3-C8гетероцикло)-C1-C10алкил, где арил в R10, содержащем арил, возможно замещен [R7]h;
h равен 1, 2, 3, 4 или 5; и
X представляет собой O или S.
Особый интерес представляют соединения формулы IV, имеющие структуры:



Изобретение дополнительно описано в следующих примерах, которые не предназначены ограничивать объем изобретения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Эксперименты обычно проводили в инертной атмосфере (атмосфере азота или аргона), в частности в тех случаях, когда использовали реагенты или промежуточные соединения, чувствительные к кислороду или влаге. Имеющиеся в продаже растворители и реагенты обычно использовали без дополнительной очистки, в том числе безводные растворители в подходящих случаях (обычно продукты Sure-Seal™ от Aldrich Chemical Company, Milwaukee, Wisconsin). Продукты обычно сушили под вакуумом, после чего направляли в последующие реакции или передавали для биологических исследований. Масс-спектрометрические данные получены на приборе или для жидкостной хроматографии - масс-спектрометрии (ЖХ-МС), или с использованием химической ионизации при атмосферном давлении (ХИАД), или для газовой хроматографии - масс-спектрометрии (ГХ-МС). Химические сдвиги для ядерного магнитного резонанса (ЯМР) выражены в частях на миллион (м.д., δ) со ссылкой на остаточные пики от использованных дейтерированных растворителей.
Для методик, ссылающихся на синтезы в других Примерах или Методах, можно варьировать реакционный Протокол (продолжительность реакции и температуру). В общем, реакции сопровождала тонкослойная хроматография (ТСХ) или масс-спектрометрия, и реакционные смеси в подходящих случаях подвергали обработке. Между экспериментами можно варьировать очистку: в общем, растворители и соотношения растворителей, использованные для элюентов/градиентов, выбраны для получения подходящих Rfs или времен удерживания.
Оптические вращения выполняли на поляриметре Perkin-Elmer polarimeter 343 (Serial number 9506).
МСВР осуществляли на Agilent 6220 TOF LC/MC.
Названия соединений образованы с использованием программного обеспечения ACD Labs.
Условия ВЭЖХ и ЖХ-МС, использованные для анализа
Протокол A: Колонка: Phenomenex Luna C18 (2), 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: 5% B в течение 1,5 минут, от 5% до 100% B в течение 8,5 минут, затем 100% B в течение 1 минуты; Скорость потока: 0,75 мл/минута. Температура: 25°C; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 10 мкл; Прибор: Agilent 1200 LCMS.
Протокол B: Колонка: Phenomenex Luna C18 (2), 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: 50% B в течение 1,5 минут, от 50% до 100% B в течение 6,5 минут, затем 100% B в течение 3 минут; Скорость потока: 0,75 мл/минута. Температура: 25°C; Детектирование: ДДМ 215 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 10 мкл; Прибор: Agilent 1200 LCMS.
Протокол C: Колонка: Phenomenex Luna C18 (2), 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,02% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,02% трифторуксусной кислоты в метаноле (об./об.); Градиент: от 50% до 100% В в течение 10 минут; Скорость потока: 0,75 мл/минута. Температура: не контролировали; Детектирование: ДДМ 215 нм, 254 нм; Вводимый объем: 10 мкл; Прибор: Agilent 1100 HPLC.
Протокол D: Колонка: Phenomenex Luna C18 (2), 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,02% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,02% трифторуксусной кислоты в метаноле (об./об.); Градиент: от 5% до 100% В в течение 8 минут; Скорость потока: 0,75 мл/минута. Температура: не контролировали; Детектирование: ДДМ 215 нм, 254 нм; Вводимый объем: 10 мкл; Прибор: Agilent 1100 HPLC.
Протокол E: Колонка: Phenomenex Lux Amylose-2, 250×4,6 мм, 5 мкм; Подвижная фаза A: гептан; Подвижная фаза B: этанол (денатурированный); Градиент: от 5% до 100% В в течение 10 минут; Скорость потока: 1,5 мл/минута.
Температура: не контролировали; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 150-1500 дальтон; Вводимый объем: 10 мкл; Прибор: Agilent 1100 LCMS.
Протокол F: Колонка: Waters Acquity UPLC BEH, C18, 2,1×50 мм, 1,7 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: 5% В в течение 0,1 минуты, от 5% до 95% В в течение 0,7 минуты, 95% В в течение 0,1 минуты; Скорость потока: 1,25 мл/минута. Температура: 60°C; Детектирование: 200-450 нм; MC (+) диапазон: 100-1200 дальтон; Вводимый объем: 5 мкл; Прибор: Waters Acquity.
Протокол G: Колонка: Phenomenex Luna C18 (2), 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,02% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 0% до 100% В в течение 8,5 минут; Скорость потока: 1,5 мл/минута. Температура: не контролировали; Детектирование: ДДМ 210 нм; Вводимый объем: 10 мкл; Прибор: Agilent 1100 HPLC.
Протокол H: Колонка: Phenomenex Gemini-NX, C18, 4,6×50 мм, 3 мкм, 110 А; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: от 0% до 100% В в течение 4,10 минуты, линейный, затем 100% В в течение 0,4 минуты; Скорость потока: 1,5 мл/минута. Температура: 60°C; Детектирование: ДДМ 200-450 нм; MC (+) диапазон: 100-2000 дальтон; Вводимый объем: 5 мкл; Прибор: Agilent.
Протокол I: Колонка: Atlantis T3, 75×3,0 мм, 3 мкм; Подвижная фаза A: 0,05% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: ацетонитрил; Градиент: от 5% до 95% В в течение 5,75 минуты; Скорость потока: 1,2 мл/минута. Температура: 45°C; Детектирование: ДДМ 215 нм, 230 нм, 254 нм; MC (+) диапазон: 150-1200 дальтон; Вводимый объем: 5 мкл; Прибор: Agilent 1100 LCMS.
Протокол J: Колонка: Phenomenex Luna Фенил-Гексил, 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: 5% В в течение 1,5 минут, от 5% до 100% В в течение 8,5 минут, затем 100% В в течение 1 минуты; Скорость потока: 0,75 мл/минута. Температура: 25°C; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 10 мкл; Прибор: Agilent 1200 LCMS.
Протокол K: Колонка: Symmetry-CIS, 50×2,1 мм, 3,5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B 0,1% муравьиной кислоты в метаноле (об./об.); Градиент: от 10% до 90% В в течение 6,5 минут; Скорость потока: 0,7 мл/минута. Температура: комнатная температура; Детектирование: ДДМ 215 нм; MC (+) диапазон: 100-1500 дальтон; Вводимый объем: 3 мкл; Прибор: Waters 996 PDA.
Протокол L: Колонка: XBridge C-18, 150×4,6 мм, 3,5 мкм; Подвижная фаза A: 5 мМ водный раствор ацетата аммония; Подвижная фаза B: ацетонитрил; Градиент: 10% В в течение 3 минут, затем от 10% до 80% В в течение 14 минут; Скорость потока: 0,7 мл/минута. Температура: комнатная температура; Детектирование: ДДМ 215 нм; MC (+) диапазон: 100-1500 дальтон; Вводимый объем: 3 мкл; Прибор: Waters 996 PDA.
Протокол M: Колонка: Phenomenex Luna, 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в метаноле (об./об.); Градиент: 50% В в течение 1,5 минут, от 50% до 80% В в течение 8,5 минут, затем 80% В в течение 10 минут; Скорость потока: 0,75 мл/минута. Температура: 45°C; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 90-2000 дальтон; Вводимый объем: 10 мкл; Прибор: Agilent 1200 LCMS.
Протокол N: Колонка: Phenomenex Luna C18 (2), 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,02% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 0% до 100% В в течение 23,5 минут; Скорость потока: 1,5 мл/минута. Температура: не контролировали; Детектирование: ДДМ 210 нм; Вводимый объем: 10 мкл; Прибор: Agilent 1100 HPLC
Протокол O: Колонка: Agilent Poroshell 300SB-C8, 75×2,1 мм, 2,6 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: от 20% В до 45% В в течение 4 минут; Скорость потока: 1,0 мл/минута. Температура: 60°C; Детектирование: 220 нм; MC (+) диапазон: 400-2000 Da; Вводимый объем: 10 мкл; Прибор: Agilent 1100 LC, Waters MicromassZQ MC. Деконволюцию осуществляли с использованием MaxEnt1.
Протокол P: Колонка: TSK-gel G3000SWxl, 300×7,8 мм, 10 мкм; Подвижная фаза: Фосфатно-солевой буферный раствор (PBS, 1X), pH 7,4 с 2% ацетонитрила; изократическое; Скорость потока: 1 мл/минута. Температура: комнатная температура; Вводимый объем: 5 мкл; Прибор: Agilent 1100 HPLC.
Протокол Q: Колонка: Waters Acquity UPLC HSS T3, C18, 2,1×50 мм, 1,7 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: 5% В в течение 0,1 минуты, от 5% до 95% В в течение 2,5 минут, 95% В в течение 0,35 минуты; Скорость потока: 1,25 мл/минута. Температура: 60°C; Детектирование: 200-450 нм; MC (+) диапазон: 100-2000 дальтон; Вводимый объем: 5 мкл; Прибор: Waters Acquity.
Протокол Q1: Колонка: Waters Acquity UPLC HSS T3, C18, 2,1×50 мм, 1,7 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: 5% В в течение 0,1 минуты, от 5% до 95% В в течение 1,5 минут, 95% В в течение 0,35 минуты; Скорость потока: 1,25 мл/минута. Температура: 60°C; Детектирование: 200-450 нм; MC (+) диапазон: 100-2000 дальтон; Вводимый объем: 5 мкл; Прибор: Waters Acquity.
Протокол Q2: Колонка: Xtimate C18, 2,1×30 мм, 3 мкм; Подвижная фаза A: 0,1% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 10% до 80% В в течение 0,9 минуты, 80% В в течение 0,6 минуты; 100% В в течение 0,5 минуты; Скорость потока: 1,2 мл/минута. Детектирование: ДДМ 220 нм; Температура: 25°C; Вводимый объем: 1 мкл; Прибор: Agilent.
Протокол Q3: Колонка: Xtimate C18, 2,1×30 мм, 3 мкм; Подвижная фаза A: 0,1% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент от 0% до 60% В в течение 0,9 минуты, 60% В в течение 0,6 минуты; 100% В в течение 0,5 минуты; Скорость потока: 1,2 мл/минута. Детектирование: ДДМ 220 нм; Температура: 25°C; Вводимый объем: 1 мкл; Прибор: Agilent.
Протокол R: Колонка: Phenomenex Luna, 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в метаноле (об./об.); Градиент: 5% В в течение 1,5 минут, от 5% до 100% В в течение 8,5 минут, затем 100% В в течение 1 минуты; Скорость потока: 0,75 мл/минута. Температура: 45°C; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 10 мкл; Прибор: 305 RP Agilent 1200 LCMS.
Протокол S: Колонка: Phenomenex Luna, 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,1% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: 5% В в течение 1,5 минут, от 5% до 95% В в течение 8,5 минут, затем 100% В в течение 1 минуты; Скорость потока: 1,0 мл/минута. Температура: не контролировали; Детектирование: ДДМ 210 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 10 мкл; Прибор: 305 RP Agilent 1100 HPLC.
Протокол T: Колонка: Atlantis dC18, 50×4,6 мм, 5 мкм; Подвижная фаза A: 0,05% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,05% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 5% до 95% В в течение 4,0 минут; затем удержание при 95% В в течение 1 минуты; Скорость потока: 2 мл/минута. Температура: комнатная температура; Детектирование: ДДМ 215 нм; MC (+) диапазон: 160-1000 дальтон; Вводимый объем: 3 мкл; Прибор: Waters 996 PDA.
Протокол U: Колонка: Phenomenex Luna C18 (2), 150×3,0 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: 5% В в течение 1,5 минут, от 5% до 100% В в течение 8,5 минут, затем 100% В в течение 1 минуты; Скорость потока: 0,75 мл/минута. Температура: 45°C; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 10 мкл; Прибор: Agilent 1200 LCMS.
Протокол V: Колонка: ВЭЖХ-V Ultimate XB - C18, 50×3,0 мм, 3 мкм; Подвижная фаза A: 0,225% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,225% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 30% до 90% В в течение 6 минут; Скорость потока: 1,2 мл/минута. Температура: 40°C; Детектирование: ДДМ 220 нм; Вводимый объем: 1 мкл; Прибор: SHIMADZU.
Протокол W: Колонка: ВЭЖХ-V Ultimate XB - C18, 50×3,0 мм, 3 мкм; Подвижная фаза A: 0,1% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 10% до 80% В в течение 6 минут; Скорость потока: 1,2 мл/минута. Температура: 40°C; Детектирование: ДДМ 220 нм; Вводимый объем: 3 мкл; Прибор: SHIMADZU.
Протокол X: Колонка: YMC-pack ODS-A, 150×4,6 мм, 5 мкм; Подвижная фаза A: 0,1% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 10% до 80% В в течение 6 минут; Скорость потока: 1,2 мл/минута. Детектирование: ДДМ 220 нм. Температура: 40°C; Вводимый объем: 3 мкл; Прибор: SHIMADZU.
Протокол Y: Колонка: YMC-pack ODS-A, 150×4,6 мм, 5 мкм; Подвижная фаза A: 0,1% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 0% до 95% В в течение 10 минут, затем 95% В в течение 5 минут; Скорость потока: 1,5 мл/минута; Детектирование: ДДМ 220 нм; Прибор: Agilent 1100.
Протокол Z: Колонка: Xtimate C18, 2,1×30 мкм, 3 мкм; Подвижная фаза A: 0,1% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 0% до 60% В в течение 2 минут; Скорость потока: 1,2 мл/мин. Температура: 50°C; Детектирование: 220 нм, MC (+) диапазон: 100-1000 дальтон; Вводимый объем: 1 мкл; Прибор: SHIMADZU.
Протокол AB: Колонка: Phenomenex Luna C18 (2), 150×2,0 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: от 5% до 100% В в течение 10 минут, затем 100% В в течение 2 минут; Скорость потока: 0,5 мл/минута. Температура: 25°C; Детектирование: ДДМ 210 нм, 254 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 5 мкл; Прибор: Agilent 1100 LCMS.
Протокол BB: Колонка: Phenomenex Luna C18 (2), 150×2,0 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: 5% В в течение 2,0 минут, от 5% до 100% В в течение 12 минут, и 100% В в течение 2 минут, затем от 100% до 5% В в течение 1,5 мин; Скорость потока: 0,75 мл/минута. Температура: 25°C; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 5 мкл; Прибор: Agilent.
Протокол CB: Колонка: Waters XBridge C18, 4,6×50 мм, 5 мкм; Подвижная фаза A: 0,03% гидроксида аммония в воде (об./об.); Подвижная фаза B: 0,03% гидроксида аммония в ацетонитриле (об./об.); Градиент: от 5% до 95% В в течение 4,0 минут, затем 95% В в течение 1 минуты; Скорость потока: 2 мл/минута. Температура: 25°C; Детектирование: ДДМ 215 нм, MC (+) диапазон: 160-1000 дальтон; Вводимый объем: 4 мкл; Прибор: Waters ZQ/Alliance 2795 HPLC.
Протокол DB: Колонка: Waters Atlantis dC18, 4,6×50 мм, 5 мкм; Подвижная фаза A: 0,05% трифторуксусной кислоты в воде (об./об.);
Подвижная фаза B: 0,05% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 5,0% до 95% В в течение 4,0 минут, затем 95% В в течение 1 минуты. Скорость потока: 2 мл/минута. Температура: 25°C; Детектирование: ДДМ 215 нм, MC (+) диапазон: 160-1000 дальтон; Вводимый объем: 4 мкл; Прибор: Waters ZQ/Alliance 2795 HPLC.
Протокол EB: Колонка: XBridge RP18, 2,1×50 мм, 5 мкм; Подвижная фаза A: 0,02% гидроксида аммония в воде (об./об.); Подвижная фаза B: 0,02% гидроксида аммония в ацетонитриле (об./об.); Градиент от 10% до 80% В в течение 6 минут, затем 80% в течение 2 минут; Скорость потока: 1,2 мл/минута. Детектирование: ДДМ 220 нм; Температура: 50°C.
Протокол FB: Колонка: Phenomenex Luna C18 (2), 150×2,0 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент: 5% В в течение 2,0 минут, от 5% до 100% В в течение 10 минут, и 100% В в течение 2 минут; Скорость потока: 0,50 мл/минута. Температура: 25°C; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 5 мкл; Прибор: Agilent 1200 LCMS.
В некоторых случаях в условия анализа посредством ЖХ-МС и ВЭЖХ были внесены некоторые незначительные изменения, такие как, без ограничения ими, изменение градиента или скорости потока, что обозначено символом *.
Условия ВЭЖХ, использованные для очистки
Метод A: Колонка: Phenomenex Lux Amylose-2, 250×21.2 мм, 5 мкм; Подвижная фаза A: Гептан; Подвижная фаза B: Этанол (денатурированный); Градиент: от 5% до 100% В в течение 6 мин; Скорость потока: 27 мл/минута; Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters FractionLynx.
Метод B: Колонка: Phenomenex Luna C18 (2), 150×21.2 мм, 5 мкм; Подвижная фаза A: 0,02% уксусной кислоты в воде; Подвижная фаза B: 0,02% уксусной кислоты в ацетонитриле; Градиент: 5% В в течение 1,5 минут, от 5% до 45% В в течение 8,5 минут; Скорость потока: 27 мл/минута; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters FractionLynx.
Метод C: Колонка: Phenomenex Luna C18, 100×30 мм, 10 мкм; Подвижная фаза A: 0,02% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,02% трифторуксусной кислоты в метаноле (об./об.); Градиент: от 10% до 90% В в течение 20 минут; Скорость потока: 20 мл/минута. Температура: не контролировали; Детектирование: ДДМ 210 нм, 254 нм; Вводимый объем: переменный; Прибор: Gilson.
Метод D: Колонка: Phenomenex Synergi Max-RP, 150×21,2 мм, 4 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде; Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле; Градиент: 30% В в течение 1,5 минут, от 30% до 60% В в течение 8,5 минут, от 60 до 100% В в течение 0,5 минуты, затем 100% В в течение 2 минут; Скорость потока: 27 мл/минута; Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters FractionLynx.
Метод E1: Колонка: Phenomenex Luna C18 (2), 150×21.2 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде; Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле; Градиент: 40% В в течение 1,5 минут, от 40% до 80% В в течение 8,5 минут, от 80 до 100% В в течение 0,5 минуты, затем 100% В в течение 2 минут; Скорость потока: 27 мл/минута; Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters FractionLynx ЖХ-МС.
Метод E2: Колонка: Phenomenex Luna Фенил-гексил, 150×21,2 мм, 5 мкм. Остальные Протоколы идентичны Протоколам, описанным для Метода E1.
Метод F: Колонка: Phenomenex Synergi Max-RP, 150×21,2 мм, 4 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде; Подвижная фаза B: 0,1% муравьиной кислоты в метаноле; Градиент: 44% В в течение 1,5 минут, от 44% до 77% В в течение 8,5 минут, затем 77% В в течение 10 минут; Скорость потока: 27 мл/минута; Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters FractionLynx LCMS.
Метод G: Колонка: PrincetonSFC 2-этилпиридин, 250×21,2 мм, 5 мкм; Подвижная фаза A: гептан; Подвижная фаза B: этанол (денатурированный); Градиент: 1% В в течение 1,5 минут, от 1% до 50% В в течение 8,5 минут; Скорость потока: 27 мл/минута; Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters FractionLynx LCMS.
Метод H: Колонка: Phenomenex Luna C18 (2), 150×21,2 мм, 5 мкм; Подвижная фаза A: 0,02% уксусной кислоты в воде; Подвижная фаза B: 0,02% уксусной кислоты в ацетонитриле; Градиент: 20% В в течение 1,5 минут, от 20% до 60% В в течение 10,5 минут; Скорость потока: 27 мл/минута; Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters FractionLynx LCMS.
Метод I: Колонка: Phenomenex Luna C18 (2), 150×21,2 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде; Подвижная фаза B: 0,1% муравьиной кислоты в метаноле; Градиент: 40% В в течение 1,5 минут, от 40% до 70% В в течение 8,5 минут, затем 70% В в течение 10 минут; Скорость потока: 27 мл/минута; Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters FractionLynx LCMS.
Метод J: Колонка: Phenomenex Luna C18, 100×30 мм, 5 мкм; Подвижная фаза A: 0,02% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 10% до 90% В в течение 20 минут; Скорость потока: 20 мл/минута. Температура: не контролировали; Детектирование: ДДМ 210 нм, 254 нм; Вводимый объем: переменный; Прибор: Gilson.
Метод K: Колонка: Phenomenex Luna C18 (2), 150×21,2 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде; Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле; Градиент: 20% В в течение 1,5 минут, от 20% до 50% В в течение 8,5 минут, от 50 до 100% В в течение 0,5 минуты, затем 100% В в течение 2 минут; Скорость потока: 27 мл/минута; Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters Fraction Lynx LCMS.
Метод L: Колонка: Phenomenex Luna C18 (2), 150×21,2 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде; Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле; Градиент: 30% В в течение 1,5 минут, от 30% до 50% В в течение 8,5 минут, от 50 до 100% В в течение 0,5 минуты, затем 100% В в течение 2 минут; Скорость потока: 27 мл/минута; Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters Fraction Lynx LCMS.
Метод M: Колонка: Waters Sunfire, C18, 19×100 мм, 5 мкм; Подвижная фаза A: 0,05% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,05% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: от 0 до 100% в течение 8,5 минут. Скорость потока 25 мл/минута. Детектирование: ДДМ 215 нм MC (+) диапазон: 160-1000 дальтон; Прибор: Waters FractionLynx.
Метод N: Колонка: Waters Sunfire, C18, 19×100 мм, 5 мкм; Подвижная фаза A: 0,05% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,05% муравьиной кислоты в ацетонитриле (об./об.); Градиент: от 0 до 100% в течение 8,5 минут. Скорость потока 25 мл/минута. Детектирование: ДДМ 215 нм MC (+) диапазон: 160-1000 дальтон; Прибор: Waters FractionLynx.
Метод O: Колонка: Phenomenex Luna C18, 21,2×150 мм, 5 мкм; Подвижная фаза A: 0,1% муравьиной кислоты в воде (об./об.); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об./об.); Градиент(об./об.); Градиент 20% В в течение 1,5 минут, от 20% до 40% В в течение 8,5 минут, от 40 до 100% В в течение 0,5 минуты, затем удержание 100% В в течение 1,5 минут. Скорость потока: 27 мл/минута. Детектирование: ДДМ 210-360 нм; MC (+) диапазон: 150-2000 дальтон; Прибор: Waters FractionLynx.
Метод P: Колонка: Phenomenex Gemini C18, 21,2×250 мм, 5 мкм; Подвижная фаза A: 0,225% гидроксида аммиака в воде (pH 10) (об./об.); Подвижная фаза B: 0,225% гидроксида аммиака в ацетонитриле (об./об.); Градиент: от 45% до 85% В в течение 10 минут. Скорость потока 35 мл/минута. Детектирование: ДДМ 220 нм MC (+) диапазон: 100-1200 дальтон; Прибор: Shimadzu MC Trigger.
Метод Q: Колонка: Phenomenex Synergi C18, 50×250 мм, 10 мкм; Подвижная фаза A: 0,1% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: Ацетонитрил; Градиент от 10% до 40% В в течение 25 минут. Скорость потока 100 мл/минута. Детектирование: UV/Vis (УФ/видимая область) 220 нм; Прибор: Shimadzu LC-8A.
Метод R: Колонка: Phenomenex Luna C18 (2), 250×21,2 мм, 5 мкм; Подвижная фаза A: 0,1% TFA в воде (об./об.); Подвижная фаза B: 0,1% TFA в ацетонитриле (об./об.); Градиент: от 10% до 100% в течение 30 минут; Скорость потока переменная. Температура: 25°C; Детектирование: ДДМ 215 нм, 254 нм; MC (+) диапазон: 150-2000 дальтон; Вводимый объем: 1,8 мл: Прибор: Agilent 1100 Prep HPLC.
В некоторых случаях в условия очистки были внесены некоторые незначительные изменения, такие как, без ограничения ими, изменение градиента или скорости потока, что обозначено символом *.
Общие методики
Общая методика A: Удаление Fmoc с использованием диэтиламина или пиперидина. К раствору Fmoc-содержащего соединения в дихлорметане или N,N-диметилформамиде (также называемом как DMF) добавляли равный объем диэтиламина или пиперидина. За протеканием реакции следили с помощью ЖХ-МС (или ВЭЖХ, или ТСХ). Растворители удаляли в вакууме, и в некоторых случаях остаток подвергали перегонке с гептаном от одного до четырех раз. Остаток обычно разбавляли дихлорметаном и небольшим количеством метанола, после чего окончательно концентрировали на диоксиде кремния и очищали посредством хроматографии на силикагеле, элюируя метанолом в дихлорметане (или другой подходящей смесью растворителей) с получением целевого материала (или неочищенный материал использовали как он был).
Общая методика B: Удаление Вое или расщепление сложного трет-Bu эфира с использованием трифторуксусной кислоты. К раствору Boc-содержащего соединения или соединения, содержащего сложный трет-бутиловый эфир, в дихлорметане при 0°C (или при комнатной температуре) добавляли трифторуксусную кислоту с получением соотношения трифторуксусная кислота : дихлор метан 1:4. За протеканием реакции следили с помощью ЖХ-МС (или ВЭЖХ, или ТСХ). Растворители удаляли в вакууме. Остаток три раза подвергали азеотропной перегонке с гептаном с получением целевого материала.
Общая методика C: Удаление Вое или расщепление сложного трет-бутилового эфира (также называемого как сложный трет-Bu эфир) с использованием соляной кислоты в диоксане. К раствору или Boc-содержащего соединения или соединения, содержащего сложный трет-бутиловый эфир, в диоксане (или в некоторых случаях без использования раствора, или в другом подходящем растворителе) добавляли 4 М раствор соляной кислоты в диоксане. За протеканием реакции следили с помощью ЖХ-МС (или ВЭЖХ, или ТСХ). Реакционную смесь концентрировали в вакууме, и в некоторых случаях подвергали азеотропной перегонке от одного до четырех раз со смесью гептанов.
Общая методика D: сочетание с гексафторфосфатом O-(7-азабензотриазол-1-ил)-N,N,N′,N′-тетраметилурония (HATU). К перемешиваемому раствору амина (1,0 экв.) и кислоты (1,0-2,0 экв.) в дихлорметане, N,N-диметилформамиде (также называемом как DMF) или в их смеси добавляли HATU (1,0-2,0 экв.), а затем триэтиламин (2,0-4,0 экв.) или диизопропилэтиламин (2,0-4,0 экв., также называемый как основание Хюнига). За протеканием реакции следили с помощью ЖХ-МС (или ВЭЖХ, или ТСХ); реакция обычно завершалась в пределах трех часов. Растворители удаляли в вакууме. Остаток очищали посредством хроматографии на силикагеле или посредством хроматографии с обращенной фазой или в некоторых случаях подвергали азеотропной перегонке три раза со смесью гептанов, разбавляли небольшим количеством этилацетата, после чего окончательно концентрировали на диоксиде кремния или C18-связанном диоксиде кремния и очищали посредством хроматографии на силикагеле или посредством хроматографии с обращенной фазой.
Общая методика E: сочетание с N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}-метил)фенил]-L-орнитинамидом (MalcValCitPABC-PNP). К смеси полезной нагрузке - амину (1 экв.) и N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}-метил)фенил]-L-орнитинамида (MalcValCitPABC-PNP, Eur. Pat. Appl. (1994), EP624377, 1,0-2,0 экв.) в N,N-диметилформамиде или диметилацетамиде (также называемом как DMA) добавляли пиридин (0,0-4,0 экв.), диизопропилэтиламин (0,0-4,0 экв.), 2,6-диметилпиридин (0,0-4,0 экв., также называемый как 2,6-лудитин) и гидрат 1-гидроксибензотриазола (0,01-1,1 экв., также называемый как HOBT) или 3H-[1,2,3]триазоло[4,5-b]пиридин-3-ол (0,01-1,1 экв., также называемый как HOAT). После перемешивания при 40°C - 50°C в течение 1-48 часов реакционную смесь концентрировали в вакууме и три раза подвергали азеотропной перегонке с гептаном. Неочищенный материал очищали посредством хроматографии с обращенной фазой согласно конкретному способу с получением целевого материала.
Общая методика F: конъюгирование имеющегося в продаже антитела HERCEPTIN® с линкером-полезной нагрузкой с помощью внутренних дисульфидов. Имеющееся в продаже антитело HERCEPTIN® (Genentech Inc) диализировали в фосфатно-солевом буферном растворе Дульбекко (DPBS, Lonza). Диализированное антитело восстанавливали путем добавления × эквивалентов гидрохлорида трис(2-карбоксиэтил)фосфина (TCEP, 5 мМ в дистиллированной воде) и разбавляли до конечной концентрации антитела 15 мг/мл, используя DPBS, 5 мМ 2,2′,2″,2′′′-(этан-1,2-диилдинитрило)тетра-уксусную кислоту (EDTA), pH 7,0-7,4 (Буфер A). Реакционную смесь инкубировали при 37°C в течение 1-2 часов, а затем охлаждали до комнатной температуры. Конъюгирование осуществляли путем добавления у эквивалентов линкера-полезной нагрузки (5-10 мМ в диметилацетамиде (DMA)). DMA добавляли для достижения в конечной реакционной смеси суммарного компонента органического растворителя 10-20% (об./об.), а Буфер A добавляли для достижения конечной концентрации антитела 10 мг/мл. Реакционную смесь инкубировали в течение 1-2 часов при комнатной температуре. Затем реакционную смесь подвергали буферному обмену в DPBS (pH 7,4), используя GE Healthcare Sephadex G-25 М буфер-обменные колонки согласно инструкциям производителя. Неочищенный материал очищали посредством эксклюзионной хроматографии (ЭХ) с использованием системы GE AKTA Explorer system с колонкой GE Superdex и PBS (pH 7,4) элюентом.
Общая методика G: Реакции конъюгирования проводили в верхней части устройства для центробежной ультрафильтрации, такого как Amicon Ultra 50k Ultracel filters (part #UFC805096, GE). 132 мМ исходный раствор L-цистеина приготавливали в PBS, содержащем 50 мМ EDTA. Этот раствор (50 мкл) добавляли к смеси соответствующего мутантного антитела (5 мг) в 950 мкл PBS, содержащего 50 мМ EDTA. Конечная концентрация цистеина в реакционной смеси составила 6,6 мМ. После выдерживания реакционной смеси при комнатной температуре (примерно 23°C) в течение 1,5 часов реакционную пробирку центрифугировали для концентрирования материала до приблизительно 100 мкл. Смесь разбавляли до 1 мл с помощью PBS, содержащего 50 мМ EDTA. Этот процесс повторяли 4 раза для удаления всего цистеинового восстановителя. Полученный материал разбавляли до 1 мл в PBS, содержащем 50 мМ EDTA, и обрабатывали 16 мкл 5 мМ раствора малеимидного линкера-полезной нагрузки (из Таблицы 18A, см. в графической части) в диметил-ацетамиде (DMA) (приблизительно 5 эквивалентов). После стояния при комнатной температуре (примерно 23°C) в течение 1,5 часов реакционную пробирку центрифугировали для концентрирования материала до приблизительно 100 мкл. Смесь разбавляли до 1 мл с помощью PBS. Этот процесс повторяли 2 раза для удаления избытка малеимидного реагента. Конъюгаты антитела обычно очищали посредством эксклюзионной хроматографии (ЭХ), используя систему GE AKTA Explorer system с колонкой GE Superdex200 и PBS (pH 7,4) элюентом. Нагрузку лекарственного средства на намеченный участок конъюгирования определяли, используя множество способов, в том числе масс-спектрометрию (МС), ВЭЖХ с обращенной фазой и гидрофобную хроматографию (ГИХ), как описано в другом подходящем месте. Приведенные данные (в Таблицах 19A и 19B, см. в графической части) в основном получены с помощью ЖХ-МС в условиях восстановления.
Общая методика H: 20 мМ TCEP раствор (обычно от 50 до 100 мольных эквивалентов) добавляли к антителу (обычно 5 мг) так, что конечная концентрация антитела составила 5 мг/мл в PBS, содержащем 50 мМ EDTA. После выдерживания реакционной смеси при 37°C в течение 1,5 часов антитело подвергали буферному обмену в PBS, содержащий 50 мм EDTA, используя устройство для концентрирования вращением с отсечкой 50 kD MW (промывка 3×3 мл, 10 × концентрация на цикл). В некоторых случаях также полезны альтернативные методы, такие как TFF или диализ. Полученное антитело ресуспендировали в 1 мл PBS, содержащего 50 мМ EDTA, и обрабатывали свежеприготовленным 50 мМ раствором DHA (дегидроаскорбат) в 1:1 PBS/ЕЮН (конечная концентрация DHA обычно составляет 1 мМ) и оставляли стоять при 4°C в течение ночи. Смесь антитело/DHA подвергали буферному обмену в PBS, содержащий 50 мМ EDTA, используя устройство для концентрирования вращением с отсечкой 50 kD MW (промывка 3×3 мл, 10 × концентрация на цикл). Полученное антитело ресуспендировали в 1 мл PBS, содержащего 50 мМ EDTA, и обрабатывали 10 мМ малеимидного линкера-полезной нагрузки в DMA (обычно 5-10 эквивалентов). После стояния в течение 1,5 часов, материал подвергали буферному обмену (как описано выше) в 1 мл PBS (промывки 3×3 мл, 10 × концентрация на цикл). При необходимости осуществляли очистку посредством ЭХ (как описано ранее) для удаления любого аггрегированного материала.
Общая методика I: Первоначальное конъюгирование линкера-полезной нагрузки выполняли ранее описанным способом (Общая Методика F). Полученный конъюгат антитело-лекарственное средство подвергали буферному обмену в 50 мМ боратный буфер (pH 9,2), используя устройство для ультрафильтрации (отсечка 50 kd MW). Полученный раствор нагревали до 37°C в течение 24 часов (для линкеров малеимид-Peg) или до 45°C в течение 48 часов (для малеимид-капроильных линкеров). Полученный раствор охлаждали, подвергали буферному обмену в PBS и очищали ЭХ (как описано ранее) для удаления любого аггрегированного материала. ЖХ-МС анализ материала показал, что сукцинимидное кольцо полностью раскрылось (90% или более). Примечание: в примерах, где в полезной нагрузке присутствует сложный метиловый эфир, сложный эфир в описанных условиях гидролизуется до карбоновой кислоты.
Общая методика J: Сложные пентафторфениловые эфиры конъюгировали с указанным антителом, следуя методике, ранее описанной в WO 2012007896 A1.
Общая методика K: Конъюгирование амино-алкильных линкеров осуществляли посредством фермент-опосредованного лигирования, как описано в WO 2012059882 A2.
Общая методика L. N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид (полученный аналогично #136) подвергали сочетанию с подходящей аминокислотной или аминной группировкой, используя HATU (1,0-2,0 экв.) в присутствии основания Хюнига (1,-5,0 экв.) в растворе DMF, дихлорметана или в некоторых случаях в растворе и того, и другого (или в растворе одного или более чем одного растворителя). За реакцией следили с помощью ЖХ-МС (или ТСХ, или ВЭЖХ). Реакционную смесь концентрировали в вакууме и обычно очищали посредством хроматографии на диоксиде кремния или посредством препаративной ВЭЖХ. Затем удаляли Fmoc-защиту, как описано в общей методике А, после чего осуществляли концентрирование в вакууме и очистку посредством хроматографии на диоксиде кремния или посредством препаративной ВЭЖХ.
Общая методика M. #151 подвергали сочетанию с подходящим амином, используя HATU (1,0-2,0 экв., или другой подходящий связывающий реагент) в присутствии основания Хюнига (1,0-5,0 экв.) в растворе DMF, дихлорметана или в некоторых случаях в растворе и того, и другого (или в растворе одного или более чем одного растворителя). За реакцией следили с помощью ЖХ-МС (или ТСХ, или ВЭЖХ). Реакционную смесь концентрировали в вакууме. Затем осуществляли снятие защиты Вое, как описано в общей методике B, концентрировали в вакууме и очищали посредством хроматографии на диоксиде кремния или посредством препаративной ВЭЖХ.
Общая методика N. 1-(9H-флуорен-9-ил)-3-оксо-2,7,10,13,16,19,22-гептаокса-4-азапентакозан-25-овую кислоту (или другую подходящую Fmoc-AmPegXC2-COOH) подвергали сочетанию с подходящим цитотоксическим пентапептидом (или с цитотоксическим пентапептидом, содержащем защитную группу на реакционноспособной группировке, отличной от N-конца), используя HATU (1,0-2,0 экв., или другой подходящий связывающий реагент) в присутствии основания Хюнига (1,0-5,0 экв., или другого подходящего основания) в растворе DMF, дихлорметана или в некоторых случаях в растворе и того, и другого (или в растворе одного или более чем одного растворителя). За реакцией следили с помощью ЖХ-МС (или ТСХ, или ВЭЖХ). Реакционную смесь концентрировали в вакууме. Согласно общей методике A осуществляли снятие защиты Fmoc. В некоторых случаях осуществляли второе снятие защиты для удаления защитной группы на реакционноспособной группировке на цитотоксическом пентапептиде, используя общую методику В (или другую известную из литературы подходящую методику, основанную на защитных группах). Реакционную смесь концентрировали в вакууме и очищали посредством хроматографии на диоксиде кремния или посредством препаративной ВЭЖХ.
Общая методика O. Подходящую Fmoc-AmPegXC2-COOH подвергают сочетанию с подходящим цитотоксическим пентапептидом (или с цитотоксическим пентапептидом, содержащем защитную группу на реакционноспособной группировке, отличной от N-конца) и согласно общей методике N осуществляют снятие защиты Fmoc. Реакционную смесь концентрируют в вакууме, а затем очищают посредством хроматографии на диоксиде кремния или посредством препаративной ВЭЖХ (или неочищенный материал можно использовать как он есть). Затем согласно общей методике Е собирают подходящую PABC последовательность (такую как mcValCitPABC или ее производное). В некоторых случаях, если на цитотоксической пентапептидной части молекулы присутствует защитная группа, то потом осуществляют снятие защиты, используя общую методику A или общую методику B (или другую известную из литературы подходящую методику, основанную на защитных группах). Реакционную смесь концентрируют в вакууме и очищают посредством хроматографии на диоксиде кремния или посредством препаративной ВЭЖХ.
Общая методика P. Следуют методике E, заменяя mcValCitPABC-PNP на MalPeg3C2ValCitPABC-PNP (полученный аналогично mcValCitPABC-PNP).
Общая методика Q. Подходящую Fmoc-AmPegXC2-COOH подвергают сочетанию с подходящим цитотоксическим пентапептидом (или с цитотоксическим пентапептидом, содержащем защитную группу на реакционноспособной группировке, отличной от N-конца) и осуществляют снятие защиты Fmoc, как описано в общей методике N. Реакционную смесь концентрируют в вакууме, а затем очищают посредством хроматографии на диоксиде кремния или посредством препаративной ВЭЖХ (или неочищенный материал можно использовать как он есть). К перемешиваемому раствору этого остатка в DMF при 0°C (или в некоторых случаях при немного более высокой температуре) добавляли бромуксусную кислоту (1,0-2,0 экв.), а затем основание Хюнига (1,0-5,0 экв.) и HATU (1,0-2,0 экв.) Реакционную смесь оставляли нагреваться до комнатной температуры и перемешиваться при комнатной температуре, осуществляя контроль с помощью ЖХ-МС (или ТСХ, или ВЭЖХ). Реакционную смесь концентрировали в вакууме и очищали посредством препаративной ВЭЖХ.
Общая методика R. Следуют методике E, заменяя mcValCitPABC-PNP на N-(6-{[(9H-флуорен-9-илметокси)карбонил]амино}гексаноил)-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамид (полученный аналогично mcValCitPABC-PNP). Затем осуществляли снятие защиты Fmoc (общая методика B) с последующей очисткой посредством препаративной ВЭЖХ.
Общая методика S. К перемешиваемому раствору 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаналя (1,0-3,0 экв.) в метаноле добавляли подходящий цитотоксический пентапептид (1,0 экв.), а затем муравьиную кислоту. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1-40 минут, а затем добавляли (циано-каппаС)(тригидридо)борат(1-) натрия (3,0-6,0 экв., также называемый как цианоборогидрид натрия). За реакцией следили с помощью ЖХ-МС (или ТСХ, или ВЭЖХ). В некоторых случаях добавляли дополнительное количество 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаналя (1,0-3,0 экв.). Реакционную смесь концентрировали в вакууме с последующей очисткой с использованием препаративной ВЭЖХ.
Общая методика T. 4-[3-оксо-3-(2-оксоазетидин-1-ил)пропил]анилиний получают, как описано в литературе (Bioorganic and Medicinal Chemistry Letters. 2012, vol. 22, #13, 4249-4253), который затем подвергают сочетанию с бис(пентафторфенил)-3,3′-[этан-1,2-диилбис(окси)]дипропаноатом, используя HATU в дихлорметане, а затем подвергают сочетанию с желаемым цитотоксическим пентапептидом. Затем материал очищают посредством препаративной ВЭЖХ.
Общая методика U. Fmoc-VaICitPABC-PNP подвергают сочетанию с желаемым цитотоксическим пентапептидом, следуя общей методике E, а затем следуя общей методике A удаляют Fmoc. Затем полученный остаток подвергают сочетанию с [2-оксо-2-({4-[3-оксо-3-(2-оксоазетидин-1-ил)пропил]фенил}амино)этокси]уксусной кислотой (которую получают путем сочетания 4-[3-оксо-3-(2-оксоазетидин-1-ил)пропил]анилиния с 1,4-диоксан-2,6-дионом, следуя общей методике D). Затем материал очищают посредством препаративной ВЭЖХ.
Общая методика V. Бис(пентафторфенил)-3,3′-[этан-1,2-диилбис(окси)]дипропаноат или бис(пентафторфенил)-4,7,10,13,16-пентаоксанонадекан-1,19-диоат подвергают сочетанию с желаемым цитотоксическим пентапептидом (или в некоторых случаях подвергают сочетанию с желаемым цитотоксическим пентапептидом, содержащем защитную группу на реакционноспособной группировке, отличной от N-конца), следуя общей методике D. Если присутствует защитная группа, то ее потом удаляют (используя подходящие методики, описанные в литературе). Затем материал очищают посредством препаративной ВЭЖХ.
Общая методика W. 4-({[(4-нитрофенокси)карбонил]окси}метил)фенил-1-(9H-флуорен-9-ил)-3-оксо-2,7,10-триокса-4-азатридекан-13-оат подвергают сочетанию с желаемым цитотоксическим пентапептидом, следуя общей методике E. Следуя общей методике A удаляют Fmoc. Бис(пентафторфенил)-3,3′-[этан-1,2-диилбис(окси)]дипропаноат подвергают сочетанию с этим остатком, следуя общей методике D. Затем материал очищают посредством препаративной ВЭЖХ.
Общая методика X1. N-[(9H-флуорен-9-илметокси)карбонил]-L-аланил-L-аланил-N~1~-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-N~4~-тритил-L-аспартамид подвергают сочетанию с желаемым цитотоксическим пентапептидом, следуя общей методике E. Fmoc удаляют, следуя общей методике A, а тритильную защитную группу удаляют, следуя общей методике B. Бис(пентафторфенил)-3,3′-[этан-1,2-диилбис(окси)]дипропаноат подвергают сочетанию с этим остатком, следуя общей методике D. Материал очищают посредством препаративной ВЭЖХ.
Общая методика X2. N-{3-[2-(3-этокси-3-оксопропокси)этокси]пропаноил}-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамид подвергают сочетанию с желаемым цитотоксическим пентапептидом, следуя общей методике E. Сложный этиловый эфир удаляют, используя гидроксид лития в THF и воде. Затем путем сочетания остатка с 1-гидроксипирролидин-2,5-дионом с использованием N,N′-дициклогексилкарбодиимида в THF формируют NHS-сложный эфир. Материал очищают посредством препаративной ВЭЖХ.
Общая методика X3. N-[(9H-флуорен-9-илметокси)карбонил]-D-валил-N~5~-карбамоил-N-[4-({[(2-карбоксипропан-2-ил)карбамоил]окси}метил)фенил]-L-орнитинамид подвергают сочетанию с #50 в DMSO и ацетонитриле, следуя общей методике D. Fmoc удаляют, следуя общей методике A, а затем осуществляют сочетание с бис(пентафторфенил)-3,3′-[этан-1,2-диилбис(окси)]дипропаноатом с использованием основания Хюнига в ацетонитриле. Материал очищают посредством препаративной ВЭЖХ.
Общая методика X4. N-[1-(9H-флуорен-9-ил)-3,5,12-триоксо-2,7,10-триокса-4-азадодекан-12-ил]-2-метилаланин подвергают сочетанию с #250 в ацетонитриле, следуя общей методике D. Fmoc удаляют, следуя общей методике A, а затем осуществляют сочетание с бис(пентафторфенил)-3,3′-[этан-1,2-диилбис(окси)]дипропаноатом с использованием основания Хюнига в ацетонитриле. Материал очищают посредством препаративной ВЭЖХ.
Общая методика X5. L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамид подвергают сочетанию с N~2~-ацетил-N~6~-(трет-бутоксикарбонил)-L-лизином, следуя общей методике D. Полученный остаток подвергают сочетанию с бис(4-нитрофенил)карбонатом с основанием Хюнига в DMF, а затем осуществляют сочетание с подходящим цитотоксическим пентапептидом, следуя общей методике E. Затем, следуя общей методике B, осуществляют снятие защиты в ацетонитриле. Остаток очищают посредством препаративной ВЭЖХ.
В некоторых случаях в реакционные условия были внесены незначительные изменения, такие как, без ограничения ими, касающиеся порядка добавления реагентов и реагирующих веществ и/или количества реагента или реагирующего вещества, что обозначено символом *. Более того, указанные общие методики предложены только лишь в качестве типичных и не являются ограничивающими.
Кроме предложенных выше Общих Методик релевантные ссылки, имеющие отношение к доластатину и ауристатину, включают следующие: Petit et al. J. Am. Chem. Soc. 1989, 111, 5463; Petit et al. Anti-Cancer Drug Design 1998, 13, 243 и ссылки, процитированные там; Petit et al. J. Nat. Prod. 2011, 74, 962; WO 96/33212; WO 95/09864; EP 0695758; WO 07/8848; WO 01/18032; WO 09/48967; WO 09/48967; WO 09/117531; WO 08/8603; US 7750116; US 5985837 и US 2005/9751; все из которых включены в данное описание путем ссылки во всей их полноте.
МС анализ и приготовление образцов
Для ЖХ-МС анализа образцы приготавливали, объединяя примерно 20 мкл образца (приблизительно 1 мг/мл ADC в PBS) с 20 мкл 20 мМ дитиотреитола (DTT). После выдерживания смеси при комнатной температуре в течение 5 минут, образцы анализировали в соответствии с протоколом O.
Общую нагрузку (DAR) конъюгата рассчитывали следующим образом:
Нагрузка=2*[LC1/(LC1+LC0)]+2*[HC1/(HC0+HC1+HC2+HC3)]+4*[HC2/(HC0+HC1+HC2+HC3)]+6*[HC3/(HC0+HC1+HC2+HC3)],
где указанные переменные представляют собой связанное множество: LC0 - ненагруженная легкая цепь, LC1 - однонагруженная легкая цепь, HC0 - ненагруженная тяжелая цепь, HC1 - однонагруженная тяжелая цепь, HC2 - дважды нагруженная тяжелая цепь, и HC3 - трижды нагруженная тяжелая цепь.
Использованные ЖХ-МС условия описаны в Протоколе F для времени удерживания ниже одной минуты и Протоколе H для остальных экспериментов, если не указано иное.
Получение N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (#8)

Стадия 1. Синтез бензил-[(2S,3S)-1-гидрокси-3-метилпентан-2-ил]метилкарбамата (#1). К раствору N-[(бензилокси)карбонил]-N-метил-L-изолейцина (52,37 г, 187,5 ммоль, 1 экв.) в тетрагидрофуране (524 мл, 0,35 М) медленно в течение 1 часа добавляли комплекс боран-тетрагидрофуран (1 М в тетрагидрофуране, 375 мл, 375 ммоль, 2 экв.) и реакционную смесь оставляли перемешиваться в течение 18 часов при комнатной температуре. Реакционную смесь охлаждали до 0°C и за 30 минут добавляли воду (30 мл). Реакционную смесь разбавляли 1 М водным раствором карбоната натрия (100 мл) и трет-бутил-метиловым эфиром (250 мл). Водный слой обратно экстрагировали трет-бутил-метиловым эфиром (100 мл). Объединенные органические слои промывали 1 М водным раствором карбоната натрия (100 мл), промывали рассолом (200 мл), сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением #1 (48,44 г, 97%-ный выход) в виде бледно-желтого масла, которое использовали на следующей стадии без дополнительной очистки.1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров: δ 7.26-7.41 (m, 5H), [5.06 (AB квартет, JAB=12,9 Гц, ΔνAB=22,8 Гц) и 5.06 (AB квартет, JAB=9,0 Гц, ΔνAB=9,0 Гц), всего 2H], [4.65 (t, J=5,3 Гц) и 4.59 (t, J=5,4 Гц), всего 1H], 3.67-3.80 (m, 1H), 3.51-3.60 (m, 1H). 3.41-3.51 (m, 1H), 2.75 и 2.71 (2 s, всего 3H), 1.49-1.64 (br m, 1H), 1.24-1.37 (br m, 1H), 0.90-1.02 (br m, 1H), 0.74-0.87 (m, 6H).
Стадия 2. Синтез бензил-метил[(2S,3S)-3-метил-1-оксопентан-2-ил]карбамата (#2). К раствору #1 (8,27 г, 31,2 ммоль, 1 экв.) в диметилсульфоксиде (41,35 мл, 0,75 М) добавляли триэтиламин (8,70 мл, 64,0 ммоль, 2,05 экв.) и смесь охлаждали до 0°C. Затем порциями добавляли комплекс триоксида серы и пиридина (10,18 г, 63,96 ммоль, 2,05 экв.), поддерживая внутреннюю температуру ниже 8°C. Реакционную смесь оставляли достигать комнатной температуры и перемешивали в течение 18 часов. Реакционную смесь вливали в воду (100 мл) и трет-бутил-метиловый эфир (100 мл). Водный слой обратно экстрагировали трет-бутил-метиловым эфиром (50 мл) и объединенные органические слои промывали рассолом (100 мл), сушили над сульфатом магния, фильтровали, концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 10% до 60% этилацетата в гептане) с получением #2 (7,14 г, 87%) в виде бесцветного масла.1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 9.61 (s, 1H), 7.26-7.42 (m, 5H), 5.01-5.13 (m, 2H), 4.04-4.12 (m, 1H), 2.86 и 2.82 (2 s, всего 3H), 1.94-2.11 (br m, 1H), 1.26-1.42 (br m, 1H).
Стадия 3. Синтез трет-бутил-(3R,4S,5S)-4-{[(бензилокси)карбонил]-(метил)амино}-3-гидрокси-5-метилгептаноата (#3). Литий-диизопропиламин получали путем добавления н-бутиллития (2,5 М раствор в тетрагидрофуране, 35,9 мл, 89,8 ммоль, 1,4 экв.) к раствору диизопропиламина (13,8 мл, 96,3 ммоль, 1,5 экв.) в тетрагидрофуране (50 мл, 1,3 М) при -78°C. Через 1 час по каплям добавляли трет-бутилацетат (15,7 мл, 116 ммоль, 1,8 экв.) и реакционную смесь перемешивали в течение дополнительных 1,5 часов, оставляя медленно нагреваться до -20°C. Реакционную смесь опять охлаждали до -78°C и добавляли раствор альдегида #2 (16,9 г, 64,2 ммоль, 1 экв.) в тетрагидрофуране (10 мл). Реакционную смесь перемешивали в течение 1,5 часов и затем гасили посредством добавления воды (100 мл). После экстракции диэтиловым эфиром (2×100 мл) объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0% до 20% ацетона в гептане) с получением #3 (8,4 г, 34%) в виде бесцветного масла. ЖХ-МС: m/z 402,4 [M+Na+], время удерживания составляет 3,91 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров: δ 7.27-7.39 (m, 5H), 5.01-5.12 (m, 2H), [4.93 (d, J=7,2 Гц) и 4.98 (br d, J=7,2 Гц), всего 1H], 4.03-4.15 (br m, 1H), 3.68-3.85 (br m, 1H), 2.65 и 2.72 (2 br s, всего 3H), 2.28-2.37 (m, 1H), 2.09-2.17 (m, 1H), 1.74-1.90 (br m, 1H), 1.41-1.51 (m, 1H), 1.39 (s, 9H), 0.92-1.01 (m, 1H), 0.77-0.92 (m, 6H).
Стадия 4. Синтез трет-бутил-(3R,4S,5S)-4-{[(бензилокси)карбонил]-(метил)амино}-3-метокси-5-метилгептаноата (#@2). К раствору #3 (8,4 г, 22 ммоль, 1 экв.) в 1,2-дихлорэтане (25 мл, 0,88 M) добавляли молекулярные сита (4 Å, 0,7 г) и протонную губку (1,8-бис(диметиламино)нафталин) (13,4 г, 59,2 ммоль, 2,7 экв.), а затем тетрафторборат триметилоксония (9,10 г, 61,6 ммоль, 2,8 экв.). После перемешивания в течение ночи реакционную смесь фильтровали через целит. Фильтрат концентрировали в вакууме и остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 40% смеси ацетон : этилацетат (1:1) в гептане) с получением #@2 (8,7 г, 68%) в виде бесцветного масла.1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров: δ 7.28-7.40 (m, 5H), 5.01-5.13 (m, 2H), 3.89-4.08 (br m, 1H), 3.70-3.82 (m, 1H), 3.18 и 3.26 (2 s, всего 3H), 2.66 и 2.71 (2 brs, всего 3H), 2.44-2.53 (m, 1H, предполагалось; частично заслонен пиком растворителя), 2.17-2.24 (m, 1H), 1.71-1.86 (br m, 1H), 1.39 и 1.39 (2 s, всего 9H), 1.31-1.40 (m, 1H), 0.94-1.08 (m, 1H), 0.76-0.91 (m, 6H).
Стадия 5. Синтез гидрохлоридной соли трет-6угип-(3R,4S,5S)-3-метокси-5-метил-4-(метиламино)гептаноата (#4). К раствору #@2 (13,37 г, 33,98 ммоль, 1 экв.) в метаноле (134 мл, 0,1 M) и концентрированной соляной кислоте (3,1 мл, 37,4 ммоль, 1,1 экв.) добавляли палладий (10%) на углероде (влажность 50%) (0,1% масс.; 1,34 г, 3,40 ммоль). Смесь гидрировали при 45 фунт-сила на кв. дюйм в течение 3 часов, затем продували азотом, фильтровали через целит и концентрировали в вакууме с получением #4 (9,20 г, 92%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 9.65 (br s, 1H), 8.97 (br s, 1H), 3.98-4.04 (m, 1H), 3.40 (s, 3H), 3.06-3.13 (br m, 1H), 2.82 (br dd, J=6, 5 Гц, 3H), 2.74-2.80 (m, 1H), 2.68 (dd, половина картины ABX, J=16,3, 4,2 Гц, 1H), 2.00-2.10 (br m, 1H), 1.73-1.84 (m, 1H), 1.46 (s, 9H), 1.38-1.45 (m, 1H), 1.13 (d, J=7,0 Гц, 3H), 0.99 (t, J=7,4 Гц, 3H).
Стадия 6. Синтез трет-бутил-(3R,4S,5S)-4-[{N-[(9H-флуорен-9-илметокси)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноата (#5). К смеси N-[(9H-флуорен-9-илметокси)карбонил]-L-валина (18,53 г, 54,60 ммоль, 1,3 экв.) и 2-хлор-4,6-диметокси-1,3,5-триазина (CDMT) (9,58 г, 54,6 ммоль, 1,3 экв.) в 2-метилтетрагидрофуране (118,00 мл, 0,34 М) добавляли N-метилморфолин (6,52 мл, 59,1 ммоль, 1,5 экв.), а затем #4 (11,80 г, 39,9 ммоль, 1 экв.). Через 3 часа реакционную смесь гасили водой (50 мл) и перемешивали в течение 15 минут. Водный слой отделяли и обратно экстрагировали 2-метилтетрагидрофураном (50 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (50 мл), сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением бесцветного масла, которое очищали посредством хроматографии на силикагеле (градиент: от 5% до 40% этилацетата в гептане) с получением #5 (26,2 г, 91%) в виде бесцветной пены. ЖХ-МС (Протокол I) m/z 581,3 [M+H+] 604,3 [M+Na+], время удерживания составляет 4,993 минуты;1H ЯМР (400 МГц, DMSO-d6), возможно, смесь ротамеров, главные характеристические сигналы: δ 7.88 (d, J=7,4 Гц, 2H), 7.71 (d, J=7,4 Гц, 2H), 7.62 (d, J=8,6 Гц, 1H), 7.41 (dd, J=7,4, 7,4 Гц, 2H), 7.27-7.34 (m, 2H), 4.13-4.32 (m, 4H), 3.70-3.82 (br m, 1H), 3.24 (s, 3H), 2.92 (br s, 3H), 2,54 (dd, J=15,7, 2,4 Гц, 1H), 2.17 (dd, J=15,4, 9,4 Гц, 1H), 1.95-2.07 (m, 1H), 1.70-1.83 (br m, 1H), 1.40 (s, 9H), 0.83-0.94 (m, 9H), 0.69 (t, J=7,2 Гц, 3H).
Стадия 7A. Синтез трет-бутил-(3R,4S,5S)-3-метокси-5-метил-4-[метил(L-валил)амино]гептаноата (#6). К раствору #5 (26 г, 42 ммоль, 1 экв.) в тетрагидрофуране (260 мл, 0,16 М) добавляли диэтиламин (22 мл) в течение 30 минуты. Реакционную смесь перемешивали в течение примерно 6 часов и затем суспензию фильтровали через целит и промывали дополнительным количеством тетрагидрофурана (25 мл). Фильтрат концентрировали в вакууме с получением бледно-желтого масла, которое перерастворяли в 2-метилтетрагидрофуране (50 мл) и опять концентрировали для обеспечения полного удаления диэтиламина. Неочищенное масло #6 (более 15,25 г) направляли на следующую стадию без дополнительной очистки.
Стадия 7B. Синтез (3R,4S,5S)-4-[{N-[(9H-флуорен-9-илметокси)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептановой кислоты (#@5). Согласно общей методике B из #5 (1,62 г, 2,79 ммоль, 1 экв.), дихлорметана (10 мл, 0,3 М) и трифторуксусной кислоты (3 мл) синтезировали #@5 (1,42 г, 97%) в виде твердого вещества, которое использовали без дополнительной очистки. ЖХ-МС m/z 525,3 [M+H+] 547,3 [M+Na+], время удерживания составляет 0,95 минуты;1H ЯМР (400 МГц, DMSO-d6), характеристические сигналы: δ 7.89 (d, J=7,6 Гц, 2H), 7.71 (d, J=7,4 Гц, 2H), 7.59 (d, J=8,8 Гц, 1H), 7.41 (dd, J=7,6, 7,4 Гц, 2H), 7.28-7.34 (m, 2H), 4.14-4.32 (m, 4H), 3.24 (s, 3H), 2.92 (br s, 3H), 2.51-2.57 (m, 1H, предполагалось; частично заслонен пиком растворителя), 2.20 (dd, J=15,9, 9,5 Гц, 1H), 1.95-2.06 (m, 1H), 1.70-1.83 (br m, 1H), 1.22-1.36 (br m, 1H), 0.84-0.93 (m, 9H), 0.70 (t, J=7,3 Гц, 3H).
Стадия 8. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-1-трет-бутокси-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#7). К смеси N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валина (19,54 г, 55,29 ммоль, 1,3 экв.) и #6 (15,25 г, 42,54 ммоль, 1 экв.) в 2-метилтетрагидрофуране (152 мл, 0,28 M) добавляли 2-хлор-4,6-диметокси-1,3,5-триазин (CDMT) (9,71 г, 55,3 ммоль, 1,3 экв.). Через 10 минут медленно добавляли N-метилморфолин (6,6 мл, 60 ммоль, 1,4 экв.), поддерживая внутреннюю температуру ниже 25°C. Реакционную смесь перемешивали в течение 4 часов, а затем гасили посредством добавления воды (50 мл). После перемешивания в течение 15 минут водный слой отделяли и обратно экстрагировали 2-метилтетрагидрофураном (50 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (100 мл), затем сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученную желтую пену очищали посредством хроматографии на силикагеле (градиент: от 5% до 35% этилацетата в гептане) с получением #7 (32 г, 97%).1H ЯМР (400 МГц, DMSO-d6, считали, что является смесью ротамеров, характеристические сигналы: δ 7.89 (d, J=7,4 Гц, 2H), 7.62 (d, J=7,4 Гц, 2H), 7.41 (br dd, J=7,4, 7,4 Гц, 2H), 7.29-7.34 (m, 2H), 4.52-4.69 (br m, 1H), 3.70-3.82 (br m, 1H), 3.22 и 3.25 (2 br s, всего 3H), 2.94 и 2.96 (2 br s, всего 3H), 2.78 и 2.81 (2 br s, всего 3H), 2.11-2.23 (m, 1H), 1.90-2.10 (m, 2H), 1.68-1.83 (br m, 1H),1.40 (s, 9H), 1.21-1.33 (br m, 1H).
Стадия 9. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (#8). К #7 (32 г, 46 ммоль, 1 экв.) в дихлорметане (160 мл, 0,29 M) по каплям в течение 10 минут добавляли трифторуксусную кислоту (17,4 мл, 231 ммоль, 5 экв.). Через 6 часов добавляли такое же количество трифторуксусной кислоты и реакцию продолжали в течение 18 часов. Реакционную смесь разбавляли толуолом (320 мл) и концентрировали в вакууме с получением #8 (35,8 г, 97%) в виде розоватого масла, которое использовали на следующей стадии без дополнительной очистки.1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: 5 7.90 (d, J=7,0 Гц, 2H), 7.62 (d, J=7,4 Гц, 2H), 7.41 (br dd, J=7,4, 7,0 Гц, 2H), 7.29-7.35 (m, 2H), 4.54-4.68 (br m, 1H), [4.09 (d, J=11 Гц) и 4.22 (d, J=10,9 Гц), всего 1H], 3.74-3.84 (br m, 1H), 3.22 и 3.24 (2 br s, всего 3H), 2.94 и 2.96 (2 br s, всего 3H), 2.78 и 2.80 (2 br s, всего 3H), 2.13-2.24 (m, 1H), 1.89-2.10 (br m, 2H), 1.70-1.81 (br m, 1H).
Получение (2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановой кислоты (#11; "Boc-Dap-кислота")
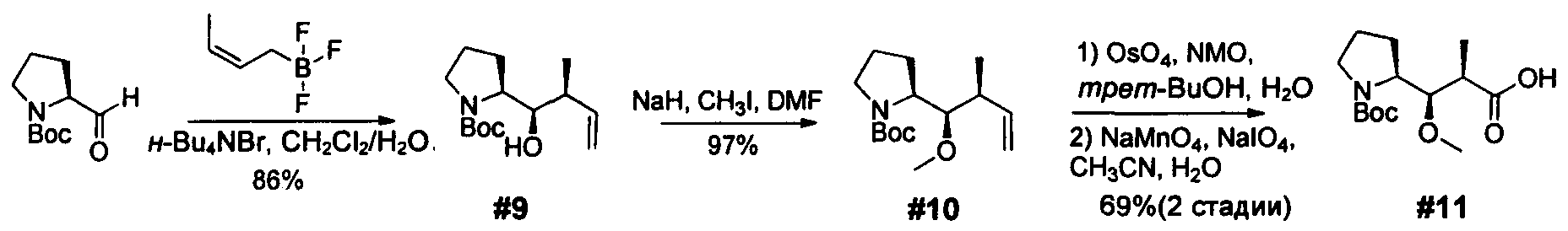
Стадия 1. Синтез трет-бутил-(2S)-2-[(1R,2S)-1-гидрокси-2-метилбут-3-ен-1-ил]пирролидин-1-карбоксилата (#9). К раствору трет-бутил-(2S)-2-формилпирролидин-1-карбоксилата (10 г, 50 ммоль, 1 экв.) в дихлорметане (120 мл, 0,42 M) добавляли (2Z)-2-бутен-1-илтрифторборат калия (9,76 г, 60,2 ммоль, 1,2 экв.), а затем бромид тетра-н-бутиламмония (3,24 г, 5,02 ммоль, 0,1 экв.) и воду (60 мл). Через 13 часов реакционную смесь разбавляли дихлорметаном (150 мл) и водой (150 мл). Водный слой отделяли и обратно экстрагировали дихлорметаном (100 мл). Объединенные органические слои промывали водным раствором хлорида натрия (5% масс., 200 мл), промывали водой (200 мл) и концентрировали в вакууме с получением #9 (примерно 13 г) в виде оранжевого масла, которое использовали без дополнительной очистки.1H ЯМР (400 МГц, CDCl3) δ 5.61-5.86 (br m, 1H), 4.97-5.09 (m, 2H), 3.80-3.98 (br m, 2H), 3.45-3.67 (br m, 1H), 3.21-3.29 (m, 1H), 2.14-2.26 (m, 1H), 1.80-2.04 (m, 3H), 1.65-1.76 (m, 1H), 1.47 (s, 9H), 1.12 (d, J=6,6 Гц, 3H).
Стадия 2. Синтез трет-бутил-(2S)-2-[(1R,2S)-1-метокси-2-метилбут-3-ен-1-ил]пирролидин-1-карбоксилата (#10). Гидрид натрия (60% в минеральном масле, 3,38 г, 84,4 ммоль, 1,1 экв.) объединяли с гексаном (40 мл) и смесь подвергали быстрому механическому перемешиванию в течение 5 минут. Твердую фазу оставляли осаждаться и удаляли гексан. Эту методику повторяли дважды для удаления минерального масла. Добавляли N,N-диметилформамид (59 мл, 1,3 М) и смесь охлаждали до 0°C; затем по каплям добавляли метилйодид (5 мл; 81 ммоль, 1,05 экв.), а потом по каплям в течение 5 минут раствор #9 (19,6 г, 76,8 ммоль, 1 экв.) в N,N-диметилформамиде (59 мл), поддерживая температуру между 0°C и 5°C. Реакционную смесь перемешивали при 0°C в течение 2 часов. Реакционную смесь гасили насыщенным водным раствором хлорида аммония (150 мл), вливали в водный раствор хлорида натрия (5% масс., 300 мл) и смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали 10%-ным водным раствором хлорида натрия (2×300 мл), промывали водой (200 мл) и концентрировали в вакууме. Полученную смесь вода - влажное масло снова подвергали концентрированию из этилацетата (150 мл) и очищали посредством хроматографии на силикагеле (градиент: от 2% до 10% этилацетата в гептане) с получением #10 (15,0 г, 73%) в виде бесцветного масла.1H ЯМР (400 МГц, CDCl3), считали, что является смесью ротамеров: δ 5.60-5.83 (m, 1H), 4.91-5.06 (m, 2H), 3.81-3.95 (br m, 1H), 3.43 (s, 3H), 3.36-3.61 (m, 2H), 3.19-3.31 (m, 1H), 2.09-2.21 (m, 1H), 1.86-2.02 (brm, 2H), 1.62-1.85 (br m, 2H), 1.47 и 1.49 (2 s, всего 9H), 1.09 (d, J=6,6 Гц, 3H).
Стадия 3. Синтез (2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановой кислоты (#11). К #10 (25,0 г, 92,8 ммоль, 1 экв.) в трет-бутаноле (100 мл, 0,93 М) сразу добавляли воду (30,00 мл), а затем N-метилморфолин-N-оксид (25,97 г, 192,1 ммоль, 2,07 экв.) и тетроксид осмия (235,93 мг, 928,04 мкмоль, 0,01 экв.). Через 12 часов смесь концентрировали в вакууме с использованием воды (20 мл) для азеотропного удаления остаточного трет-бутанола. Остаток распределяли между этилацетатом (500 мл) и водой (500 мл) с рассолом (150 мл). Водный слой снова экстрагировали этилацетатом (250 мл). Объединенные органические слои промывали водным раствором хлорида натрия (10% масс., 200 мл), промывали водой (150 мл) и концентрировали в вакууме с получением смеси вода - влажное бледно-коричневое масло, которую снова подвергали концентрированию из этилацетата (100 мл) для удаления какой-либо оставшейся воды. Этот неочищенный диол (34,76 г) использовали без дополнительной очистки.
К неочищенному диолу (34,76 г, не более 92,8 ммоль, 1 экв.) в ацетонитриле (347 мл, 0,1 М) и воде (174 мл) добавляли перманганат натрия (2,03 г, 5,73 ммоль, 0,05 экв.). Смесь охлаждали до 0°C и порциями в течение 30 минут добавляли перйодат натрия (51,46 г, 240,6 ммоль, 2,1 экв.), поддерживая внутреннюю температуру ниже 5°C. Реакционную смесь перемешивали при 0°C в течение 4 часов, а затем вливали в раствор пентагидрата тиосульфата натрия (65,40 г, 263,5 ммоль, 2,3 экв.) в воде (100 мл). Смесь фильтровали через целит и фильтрат концентрировали в вакууме. Остаток распределяли между этилацетатом (200 мл) и водой (200 мл). Водный слой обратно экстрагировали этилацетатом (250 мл) и объединенные органические слои промывали 10%-ным водным раствором лимонной кислоты. Так как целевой продукт был хорошо растворим в воде, все водные слои объединяли, обрабатывали целитом (100 г) и концентрировали в вакууме с получением не совсем белой пасты. Добавляли этилацетат (150 мл) и смесь снова подвергали концентрированию для удаления какой-либо оставшейся воды; эту операцию повторяли еще один раз. Пасту обрабатывали этилацетатом (150 мл) и помещали в вакуум при 50°C на 10 минут и фильтровали (повторяли два раза). Эти фильтраты объединяли с ранее полученным органическим слоем (после промывки лимонной кислотой), концентрировали, разбавляли этилацетатом (200 мл) и фильтровали через целит для удаления твердой фазы. Наконец, этот фильтрат концентрировали с получением #11 (22,9 г, 69% за две стадии) в виде желтоватой/коричневой пены. ЖХ-МС (Протокол I): m/z 310,1 [M+Na+], 232,1 [(M-2-метилпроп-1-ен)+H+], 188,1 [(M-Boc)+H+], время удерживания составляет 3,268 минуты;1H ЯМР (400 МГц, DMSO-d6), характеристические сигналы: δ 3.61-3.85 (br m, 2H), 3.20-3.45 (br m, 4H), 3.03-3.17 (br m, 1H), 1.59-1.93 (br m, 4H), 1.40 (br s, 9H), 1.02-1.18 (br m, 3H).
Получение соли трифторуксусной кислоты и (2R,3R)-3-метокси-2-метил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-3-[(2S)-пирролидин-2-ил]пропанамида (#19) и соли трифторуксусной кислоты и (2R,3R)-3-метокси-2-метил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-3-[(2S)-пирролидин-2-ил]пропантиоамида (#18)
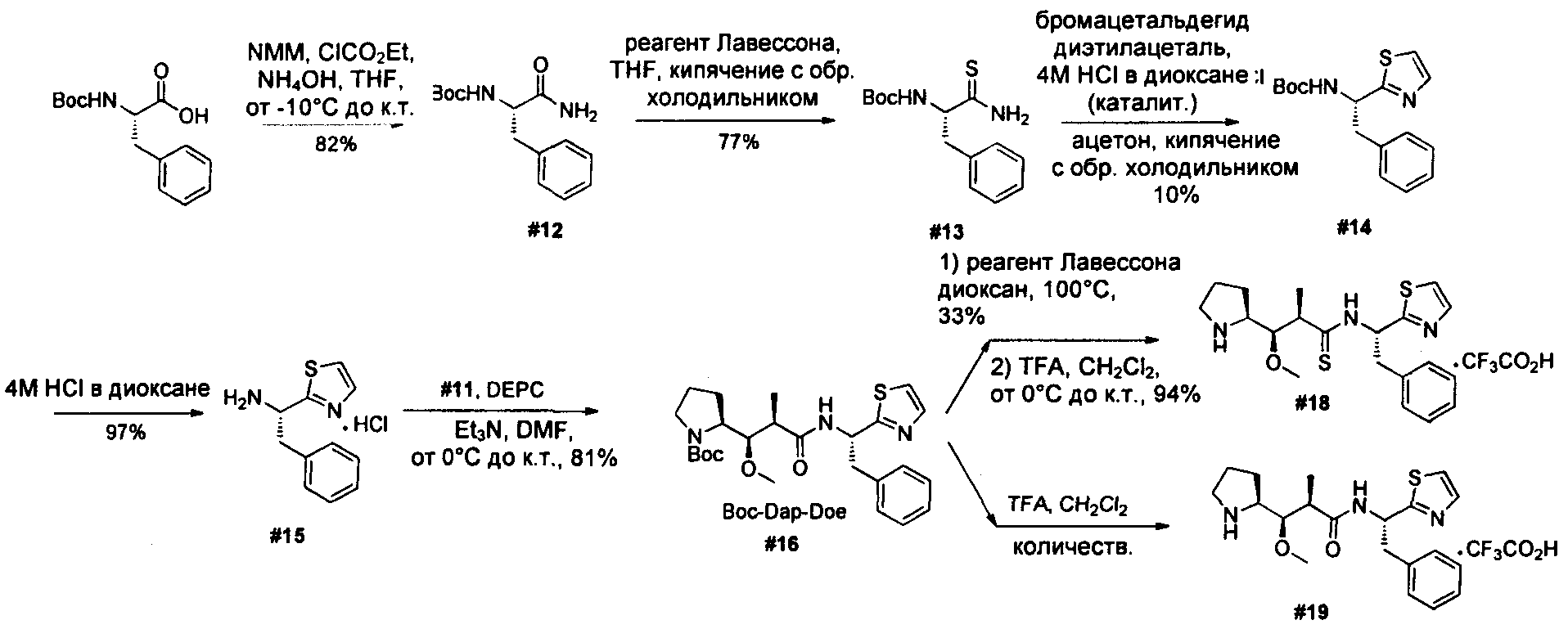
Стадия 1. Синтез Nα-(трет-бутоксикарбонил)-L-фенилаланинамида (#12). К раствору Boc-Phe-OH (30,1 г, 113 ммоль, 1 экв.) в тетрагидрофуране (378 мл, 0,3 М), охлажденному до -10°C, добавляли N-метилморфолин (13,6 мл, 124 ммоль, 1,09 экв.) и этилхлорформиат (11,8 мл, 124 ммоль, 1,09 экв.). Через 20 минут добавляли 30%-ный водный раствор гидроксида аммония (45 мл, 350 ммоль, 3,1 экв.). Смесь перемешивали при комнатной температуре в течение 18 часов, после чего концентрировали в вакууме. Остаток разбавляли этилацетатом и последовательно промывали 1 н. водным раствором бисульфата калия, водой и рассолом. Затем органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Белое твердое вещество растворяли (для этого потребовалось нагревание с перемешиванием) в этилацетате (примерно 400 мл); затем раствор оставляли охлаждаться до комнатной температуры, после чего добавляли гексан (примерно 1000 мл). Через несколько минут из реакционной смеси стало осаждаться белое вещество. Твердое вещество собирали фильтрованием, промывали гептаном (2 × примерно 150 мл) и сушили под вакуумом в течение 18 часов с получением #12 (24,50 г, 82%) в виде твердого вещества. ЖХ-МС: m/z 263,2 [M-H+], 309,2 [ ], время удерживания составляет 1,85 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, главный ротамер: δ 7.35 (br s, 1H), 7.22-7.30 (m, 5H), 7.00 (br s, 1H), 6.78 (d, J=8,6 Гц, 1H), 4.09 (ddd, J=10, 9, 4,5 Гц, 1H), 2.95 (dd, J=13,8, 4,4 Гц, 1H), 2.72 (dd, J=13,7, 10,1 Гц, 1H), 1.30 (s, 9H).
Стадия 2. Синтез трет-бутил-[(2S)-1-амино-3-фенил-1-тиоксопропан-2-ил]карбамата (#13). К раствору #12 (14,060 г, 53,192 ммоль, 1 экв.) в тетрагидрофуране (180 мл, 0,296 М) добавляли 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дитион (реагент Лавессона) (12,70 г, 31,40 ммоль, 0,59 экв.) и реакционную смесь кипятили с обратным холодильником в течение 90 минут.Реакционную смесь охлаждали до комнатной температуры и гасили посредством добавления насыщенного водного раствора бикарбоната натрия. Смесь дважды экстрагировали этилацетатом и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток растворяли в этилацетате, концентрировали в вакууме на диоксиде кремния и очищали посредством хроматографии на силикагеле (градиент: от 0% до 100% этилацетата в гептане), получая #13 (11,50 г, 77%) в виде белого твердого вещества. ЖХ-МС: m/z 279,4 [M-H+], 225,2 [(M-2-метилпроп-1-ен)+H+], 181,2 [(M-Boc)+H+];1H ЯМР (400 МГц, DMSO-d6, считали, что является смесью ротамеров, главный ротамер: δ 9.60 (br s, 1H), 9.19 (br s, 1H), 7.23-7.32 (m, 5H), 6.82 (d, J=8,8 Гц, 1H), 4.44 (ddd, J=9,4, 9,1, 4,4 Гц, 1H), 3.00 (dd, J=13,7, 4,5 Гц, 1H), 2.79 (dd, J=13,6, 9,9 Гц, 1H), 1.29 (s, 9H).
Стадия 3. Синтез трет-бутил-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамата (#14). К смеси #13 (5,65 г, 20,2 ммоль, 1 экв.) в ацетоне (101 мл, 0,2 М) добавляли бромацетальдегид-диэтилацеталь (8,76 мл, 58,2 ммоль, 2,89 экв.) и 2 капли 4 М соляной кислоты в диоксане. Смесь три раза дегазировали азотом, после чего нагревали до образования флегмы. Через 2 часа реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток растворяли в этилацетате, промывали насыщенным водным раствором бикарбоната натрия и промывали рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Полученное неочищенное оранжевое масло разбавляли этилацетатом, после чего концентрировали в вакууме на диоксиде кремния и очищали посредством хроматографии на силикагеле (градиент: от 0% до 35% этилацетата в гептане), а затем посредством хроматографии с обращенной фазой (Метод A) с получением #14 (625 мг, 10%); ВЭЖХ (Протокол E): m/z 304,5 [M+H+], 248,9 [(M-2-метилпроп-1-ен)+H+], время удерживания составляет 7,416 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, главный ротамер: δ 7.75 (d, J=3,3 Гц, 1H), 7.75 (br d, J=8,6 Гц, 1H), 7.61 (br d, J=3,1 Гц, 1H), 7.25-7.30 (m, 5H), 4.99 (ddd, J=10,5, 8,9, 4,5 Гц, 1H), 3.29-3.36 (m, 1H, предполагалось; частично заслонен сигналом воды), 2.98 (dd, J=13,8, 10,6 Гц, 1H), 1.31 (s, 9H).
Стадия 4. Синтез гидрохлоридной соли (1S)-2-фенил-1-(1,3-тиазол-2-ил)этанамина (#15). Согласно общей методике C из #14 (1,010 г, 3,318 ммоль, 1 экв.), диоксана (10 мл, 0,33 M) и 4 М раствора соляной кислоты в диоксане (20 мл, 80 ммоль, 20 экв.) синтезировали #15 (775 мг, 97%).1H ЯМР (400 МГц, DMSO-d6) δ 8.95-9.07 (br m, 3H), 7.86 (d, J=3,2 Гц, 1H), 7.73 (d, J=3,2 Гц, 1H), 7.18-7.28 (m, 3H), 7.10-7.15 (m, 2H), 4.98-5.07 (m, 1H), 3.49 (dd, J=13,3, 4,9 Гц, 1H), 3.18 (dd, J=13,4, 10,2 Гц, 1H).
Стадия 5. Синтез трет-бутил-(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-карбоксилата (#16). К раствору #11 (280 мг, 0,974 ммоль, 1 экв.) и #15 (460 мг, 1,44 ммоль, 1,48 экв.) в N,N-диметилформамиде (3 мл, 0,32 М) при 0°C добавляли диэтилфосфорилцианид (DEPC) (чистота 93%, 212 мкл, 1,30 ммоль, 1,34 экв.), а затем триэтиламин (367 мкл, 2,63 ммоль, 2,7 экв.). Через 2 часа при 0°C реакционную смесь нагревали до комнатной температуры в течение 18 часов. Затем реакционную смесь разбавляли смесью этилацетат : толуол (2:1, 30 мл) и последовательно промывали 1 М водным раствором бисульфата натрия (35 мл) и 50%-ным насыщенным водным раствором бикарбоната натрия (4×25 мл). Органический слой сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали посредством хроматографии на силикагеле (от 12% до 100% этилацетата в гептане) с получением #16 в виде светло-янтарного масла (374 мг, 81%). ЖХ-МС: m/z 474,4 [M+H+], 374,4 [(M-2-метилпроп-1-ен+H+], время удерживания составляет 3,63 минуты;1H ЯМР (400 МГц, DMSO-d6), характеристические сигналы: δ 8.66 (d, J=8,5 Гц, 1H), 7.78 (d, J=3,3 Гц, 1H), 7.64 (d, J=3,3 Гц, 1H), 7.21-7.31 (m, 4H), 7.14-7.20 (m, 1H), 5.40 (ddd, J=11,4, 8,5, 4,0 Гц, 1H), 3.23 (br s, 3H), 2.18 (dq, J=9,7, 6,7 Гц, 1H), 1.06 (d, J=6,6 Гц, 3H).
Стадия 6A. Синтез трет-бутит-(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-карбоксилата (#17). Смесь #16 (350 мг, 0,739 ммоль, 1 экв.) и 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дитиона (реагента Лавессона) (324 мг, 0,776 ммоль, 1,05 экв.) в толуоле (6 мл, 0,1 М) нагревали до 100°C. Через 10 минут смесь охлаждали до комнатной температуры. Нерастворимый материал удаляли фильтрованием и фильтрат концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 12% до 80% этилацетата в гептане), а затем посредством хроматографии с обращенной фазой (Метод E2) с получением #17 (120 мг, 33%); ВЭЖХ (Протокол J): m/z 490,2 [M+H+], время удерживания составляет 10,069 минуты; - 110 (с 0,24, МеОН);1H ЯМР (400 МГц, CD3OD), характеристические сигналы: δ 7.78 (d, J=3,3 Гц, 1H), 7.51 (d, J=3,3 Гц, 1H), 7.32-7.37 (m, 2H), 7.24-7.30 (m, 2H), 7.17-7.23 (m, 1H), 6.52-6.61 (br m, 1H), 3.62 (br dd, J=15, 4 Гц, 1H), 3.37 (s, 3H), 2.98-3.09 (br m, 1H), 2.53-2.64 (br m, 1H), 1.60-1.78 (m, 2H), 1.49 (s, 9H), 1.27 (d, J=6,5 Гц, 3H).
Стадия 6B. Синтез соли трифторуксусной кислоты и (2R,3R)-3-метокси-2-метил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-3-[(2S)-пирролидин-2-ил]пропантиоамида (#18). Согласно общей методике B из #17 (198 мг, 0,404 ммоль, 1 экв.), дихлорметана (6 мл, 0,07 M) и трифторуксусной кислоты (2 мл) синтезировали #18 (185 мг, 91%), которое использовали без дополнительной очистки. ЖХ-МС: m/z 390,1 [M+H+], время удерживания составляет 0,57 минуты;1H ЯМР (400 МГц, DMSO-d6 δ 10.91 (d, J=8,2 Гц, 1H), 9.07-9.20 (br m, 1H), 7.86-8.00 (br m, 1H), 7.83 (d, J=3,2 Гц, 1H), 7.69 (d, J=3,3 Гц, 1H), 7.27-7.36 (m, 4H), 7.21-7.26 (m, 1H), 6.33 (ddd, J=11,3, 8,3, 4,4 Гц, 1H), 3.76-3.82 (m, 1H), 3.56 (dd, J=14,6, 4,3 Гц, 1H), 3.45 (s, 3H), 3.28 (dd, J=14,6, 11,3 Гц, 1H), 3.02-3.12 (br m, 1H), 2.89-3.00 (brm, 1H), 2.72-2.89 (m, 2H), 1.69-1.83 (brm, 1H), 1.43-1.58 (m, 2H), 1.20-1.33 (m, 1H), 1.22 (d, J=6,6 Гц, 3H).
Стадия 7. Синтез соли трифторуксусной кислоты и (2R,3R)-3-метокси-2-метил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-3-[(2S)-пирролидин-2-ил]пропанамида (#19). Согласно общей методике B из #16 (607 мг, 1,28 ммоль, 1 экв.), дихлорметана (10 мл, 0,13 М) и трифторуксусной кислоты (2 мл) синтезировали #19 (640 мг, количественный выход), которое использовали на следующей стадии без дополнительной очистки.1H ЯМР (400 МГц, DMSO-d6) δ 8.96-9.07 (br m, 1H), 8.89 (d, J=8,8 Гц, 1H), 7.87-8.00 (br m, 1H), 7.80 (d, J=3,2 Гц, 1H), 7.66 (d, J=3,3 Гц, 1H), 7.28-7.34 (m, 4H), 7.20-7.27 (m, 1H), 5.43 (ddd, J=11,3, 8,6, 4,2 Гц, 1H), 3.42-3.50 (m, 2H), 3.36 (s, 3H), 3.04-3.14 (br m, 1H), 2.99 (dd, J=14,2, 11,5 Гц, 1H), 2.92-3.02 (m, 1H), 2.78-2.88 (br m, 1H), 2.34-2.42 (m, 1H), 1.73-1.84 (br m, 1H), 1.55-1.68 (m, 1H), 1.38-1.53 (m, 2H), 1.15 (d, J=6,9 Гц, 3H).
Получение гидрохлоридной соли (2R,3R)-3-метокси-2-метил-N-(2-фенилэтил)-3-[(2S)-пирролидин-2-ил]пропантиоамида (#23) и гидрохлоридной соли (2R,3R)-3-метокси-2-метил-N-(2-фенилэтил)-3-[(2S)-пирролидин-2-ил]пропанамида (#24)

Стадия 1A. Синтез трет-бутил-(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-карбоксилата {#20). К #11 (22 г, 77 ммоль, 1 экв.) в дихлорметане (383 мл, 0,2 M) и N,N-диметилформамиде (30 мл) добавляли диизопропилэтиламин (26,9 мл, 153 ммоль, 2 экв.), 2-фенилэтиламин (11,6 мл, 91,9 ммоль, 1,2 экв.) и HATU (39,0 г, 99,5 ммоль, 1,3 экв.). Реакционную смесь перемешивали в течение 18 часов, а затем концентрировали в вакууме. Остаток переносили в этилацетат (700 мл) и последовательно промывали 1 М водным раствором соляной кислоты (2×200 мл) и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Неочищенный материал переносили в дихлорметан и фильтровали. Фильтрат очищали посредством хроматографии на силикагеле (градиент: от 0% до 100% этилацетата в гептане) с получением #20 (24 г, 80%) в виде не совсем белого твердого вещества. ЖХ-МС: m/z 392,2 [M+2H+], 291,1 [(M-Boc)+H+], время удерживания составляет 0,88 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров: δ 7.80-7.89 (br m, 1H), 7.23-7.29 (m, 2H), 7.15-7.23 (m, 3H), 3.72-3.82 и 3.55-3.62 (2 br m, всего 1H), 3.45-3.55 (br m, 1H), 3.31-3.44 (br m, 2H), 3.29 (s, 3H), 3.12-3.25 (br m, 1H), 2.98-3.12 (brm, 1H), 2.71 (t, J=7,1 Гц, 2H), 2.09-2.19 (m, 1H), 1.71-1.83 (br m, 2H), 1.60-1.70 (brm, 1H), 1.49-1.60 (brm, 1H), 1.41 (s, 9H), 1.03 (d, J=6,8 Гц, 3H).
Стадия 1B. Синтез дипиридиний-1-илпентатиодифосфоната (#21). К пиридину (56 мл, 0,36 М) при 80°C добавляли пентасульфид фосфора (4,45 г, 2,19 мл, 20 ммоль, 1 экв.) и смесь кипятили с обратным холодильником (115°C) в течение 1 часа. Смесь охлаждали до комнатной температуры и продукт собирали фильтрованием с получением #21 в виде желтого твердого вещества (4,57 г, 60%); т.пл.: 165-167°C (разложение);1H ЯМР (400 МГц, DMSO-d6) δ 8.78-8.84 (m, 4H), 8.22-8.30 (m, 2H), 7.76-7.83 (m, 4H).
Стадия 2A. Синтез трет-бутил-(2S)-2-{(1R,2R)-1-метокси-2-метил-3-[(2-фенилэтил)амино]-3-тиоксопропил}пирролидин-1-карбоксилата (#22). Смесь #20 (1,200 г, 3,073 ммоль, 1 экв.) и #21 (1,40 г, 3,69 ммоль, 1,2 экв.) в ацетонитриле (15 мл, 0,20 M) подвергали воздействию микроволнового излучения при 100°C в течение 30 минут. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (150 мл) и последовательно промывали 0,5 М водным раствором соляной кислоты (100 мл) и рассолом (2×50 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 20% до 80% этилацетата в гептане) с получением #22 (670 мг, 54%) в виде белого воскоподобного твердого вещества; т.пл.: 107-109°C; ЖХ-МС: m/z 407,4 [M+H+], 351,3 [(M-2-метилпроп-1-ен)+H+], 307,3 [(M-Boc)+H+], время удерживания составляет 0,99 минуты;1H ЯМР (400 МГц, CD3CN), считали, что является смесью ротамеров: δ 8.28 (br s, 1H), 7.19-7.33 (m, 5H), 3.81-4.05 (br m, 2H), 3.60-3.81 (br m, 2H), 3.38-3.51 (br m, 1H), 3.36 (s, 3H), 3.02-3.17 (br m, 1H), 2.89-3.02 (m, 2H), 2.50-2.62 (br m, 1H), 1.71-1.85 (br m, 2H), 1.53-1.66 (br m, 2H), 1.45 (br s, 9H), 1.23 (d, J=6,7 Гц, 3H).
Стадия 3. Синтез гидрохлоридной соли (2R,3R)-3-метокси-2-метил-N-(2-фенилэтил)-3-[(2S)-пирролидин-2-ил]пропантиоамида (#23). Согласно методике C из #22 (325 мг, 0,799 ммоль, 1 экв.), диоксана (5 мл, 0,2 М) и 4 М раствора соляной кислоты в диоксане (4 мл, 16 ммоль, 20 экв.) синтезировали #23 (274 мг, количественный выход) в виде белой пены; ЖХ-МС: 308,2 [M+H+], время удерживания составляет 0,55 минуты.
Стадия 2B. Синтез гидрохлоридной соли (2R,3R)-3-метокси-2-метил-N-(2-фенилэтил)-3-[(2S)-пирролидин-2-ил]пропанамида (#24). К #20 (7,00 г, 17,9 ммоль, 1 экв.) в диоксане (50 мл, 0,36 М) и метаноле (2 мл) добавляли 4 М раствор соляной кислоты в диоксане (20 мл, 80 ммоль, 4,4 экв.). После перемешивания в течение 18 часов смесь концентрировали с получением #24 (5,86 г, количественный выход) в виде смолы, которую использовали без дополнительной очистки; ЖХ-МС: 292,2 [M+H+], время удерживания составляет 0,47 минуты.
Получение N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#26)

Стадия 1. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#25)
Согласно общей методике D из #8 (480 мг, 0,753 ммоль, 1 экв.), дихлорметана (10 мл, 0,07 М), N,N-диметилформамида (2 мл), амина #18 (401 мг, 0,941 ммоль, 1,25 экв.), HATU (372 мг, 0,979 ммоль, 1,3 экв.) и триэтиламина (367 мкл, 2,64 ммоль, 3,5 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 30% ацетона в гептане) с получением #25 (711 мг, 75%) в виде твердого вещества. ЖХ-МС: m/z 1009,7 [M+H+], время удерживания составляет 1,15 минуты; ВЭЖХ (Протокол B): m/z 505,3 [M+2H+]/2, время удерживания составляет 10,138 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.54 (br d, J=8 Гц) и 10.81 (br d, J=8 Гц), всего 1H], 7.89 (br d, J=7 Гц, 2H), [7.80 (d, J=3,3 Гц) и 7.83 (d, J=3,2 Гц), всего 1H], [7.64 (d, J=3,2 Гц) и 7.69 (d, J=3,2 Гц), всего 1H], 7.62 (br d, J=7 Гц, 2H), 7.37-7.44 (m, 2H), 7.28-7.35 (m, 4H), 7.20-7.27 (m, 2H), 7.12-7.18 (m, 1H), 6.27-6.35 и 6.40-6.48 (2 m, всего 1H), [1.14 (d, J=6,4 Гц) и 1.17 (d, J=6,3 Гц), всего 3H].
Стадия 2. Синтез N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#26). Согласно общей методике A из #25 (701 мг, 0,694 ммоль) в дихлорметане (10 мл, 0,07 М) и диэтиламине (10 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) с получением стекловидного твердого вещества. Добавляли диэтиловый эфир и гептан и смесь концентрировали в вакууме, получая #26 (501 мг, 92%) в виде белого твердого вещества. ВЭЖХ (Протокол A): m/z 787,4 [M+H+], время удерживания составляет 7,229 минуты, (чистота более 97%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.54 (brd, J=8 Гц) и 10.81 (br d, J=8. Гц), всего 1H], [7.99 (br d, J=9 Гц) и 8.00 (br d, J=9 Гц), всего 1H], [7.80 (d, J=3,3 Гц) и 7.83 (d, J=3,3 Гц), всего 1H], [7.65 (d, J=3,2 Гц) и 7.69 (d, J=3,3 Гц), всего 1H], 7.29-7.34 (m, 2H), 7.19-7.28 (m, 2H), 7.13-7.19 (m, 1H), [6.31 (ddd, J=11, 8, 4,5 Гц) и 6.45 (ddd, J=11,5, 8, 4,5 Гц), всего 1H], [4.57 (dd, J=8,9, 8,7 Гц) и 4.63 (dd, J=8,7, 8,7 Гц), всего 1H], 3.16, 3.21, 3.24 и 3.25 (4 s, всего 6H), 2.96 и 3.03 (2 br s, всего 3H), [1.14 (d, J=6,6 Гц) и 1.17 (d, J=6,4 Гц), всего 3H].
Получение N2-[(1-аминоциклопентил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#30)

Стадия 1. Синтез трет-бутил-(3R,4S,5S)-4-[{N-[(1-{[(9H-флуорен-9-илметокси)карбонил]амино}циклопентил)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноата (#27). К #6 (287 мг, 0,801 ммоль, 1 экв.) в дихлорметане (4 мл, 0,2 М) добавляли 1-{[(9H-флуорен-9-илметокси)карбонил]-амино}циклопентанкарбоновую кислоту (309 мг, 0,879 ммоль, 1,1 экв.), диизопропилэтиламин (281 мкл, 1,60 ммоль, 2 экв.) и HATU (376 мг, 0,960 ммоль, 1,2 экв.). Смесь перемешивали в течение 18 часов и разбавляли этилацетатом (15 мл). Реакционную смесь промывали 1 М водным раствором соляной кислоты (2×5 мл) и рассолом (5 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный материал очищали посредством хроматографии на силикагеле (градиент: от 0% до 60% этилацетата в гептане) с получением #27 (502 мг, 91%) в виде белой пены. ЖХ-МС: m/z 692,3 [M+H+], 714,3 [M+Na+], 636,3 [(M-2-метилпроп-1-ен)+H+], время удерживания составляет 1,13 минуты;1H ЯМР (400 МГц, DMSO-d6), характеристические сигналы: δ 7.89 (br d, J=7,4 Гц, 2H), 7.67-7.75 (m, 2H), 7.60 (br s, 1H), 7.38-7.44 (m, 2H), 7.30-7.36 (m, 2H), 7.21 (br d, J=8,8 Гц, 1H), 4.44-4.59 (m, 2H), 4.17-4.27 (m, 3H), 3.68-3.78 (br m, 1H), 3.21 (s, 3H), 2.88 (br s, 3H), 2.09-2.20 (m, 2H), 1.39 (s, 9H).
Стадия 2. Синтез (3R,4S,5S)-4-[{N-[(1-{[(9H-флуорен-9-илметокси)карбонил]амино}циклопентил)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептановой кислоты (#28). К раствору #27 (500 мг, 0,723 ммоль) в дихлорметане (7 мл, 0,1 М) добавляли трифторуксусную кислоту (3 мл). Реакционная смесь сначала стала оранжевой, затем со временем потемнела. После перемешивания в течение 18 часов растворитель удаляли в вакууме с получением #28 (460 мг, количественный выход) в виде темно-коричневого стекла, которое использовали без дополнительной очистки. ЖХ-МС: m/z 636,3 [M+H+].
Стадия 3. Синтез N2-[(1-{[(9H-флуорен-9-илметокси)карбонил]амино}-циклопентил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенип-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#29). Согласно общей методике D из #28 (50 мг, 0,079 ммоль, 1 экв.), дихлорметана (3 мл, 0,03 М), N,N-диметилформамида (0,5 мл), амина #18 (44 мг, 0,087 ммоль, 1,1 экв.), триэтиламина (33,0 мкл, 0,237 ммоль, 3 экв.) и HATU (36 мг, 0,95 ммоль, 1,2 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 30% ацетона в гептане) с получением #29 (59 мг, 67%) в виде твердого вещества. ЖХ-МС: m/z 1007,5 [M+H+], время удерживания составляет 1,11 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.54 (br d, J=8 Гц) и 10.80 (br d, J=8 Гц), всего 1H], 7.89 (br d, J=7 Гц, 2H), [7.80 (d, J=3,3 Гц) и 7.82 (d, J=3,1 Гц), всего 1H], 7.68-7.75 (m, 2H), [7.64 (d, J=3,2 Гц) и 7.68 (d, J=3,2 Гц), всего 1H], 7.38-7.44 (m, 2H), 7.27-7.36 (m, 4H), 7.12-7.25 (m, 4H), [6.30 (ddd, J=11, 8, 4,5 Гц) и 6.39-6.48 (m), всего 1H], [4.50 (br dd, J=8, 8 Гц) и 4.54-4.59 (m), всего 1H], 4.17-4.29 (m, 3H), 2.89 и 2.96 (2 br s, всего 3H), [1.13 (d, J=6,5 Гц) и 1.16 (d, J=6,4 Гц), всего 3H].
Стадия 4. Синтез N2-[(1-аминоциклопентил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#30). Согласно общей методике A из #29 (54 мг, 0,054 ммоль) в дихлорметане (6 мл, 0,9 мМ) и диэтиламине (4 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) с получением примера #30 (26 мг, 61%) в виде твердого вещества. ВЭЖХ (Протокол A): время удерживания составляет 7,233 минуты, m/z 785,4 [M+H+], (чистота более 72%).1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.54 (br d, J=8 Гц) и 10.82 (br d, J=8 Гц), всего 1H], 8.19-8.27 (m, 1H), [7.80 (d, J=3,2 Гц) и 7.83 (d, J=3,2 Гц), всего 1H], [7.65 (d, J=3,3 Гц) и 7.69 (d, J=3,3 Гц), всего 1H], 7.28-7.33 (m, 2H), 7.20-7.27 (m, 2H), 7.14-7.19 (m, 1H), [6.31 (ddd, J=11, 8, 4,5 Гц) и 6.44 (ddd, J=11, 8, 4 Гц), всего 1H], [4.53 (dd, J=9, 8 Гц) и 4.60 (dd, J=9, 7,5 Гц), всего 1H], 3.24 и 3.25 (2 s, всего 3H), 3.17 и 3.21 (2 s, всего 3H), 2.93 и 3.00 (2 br s, всего 3H), [1.14 (d, J=6,5 Гц) и 1.17 (d, J=6,5 Гц), всего 3H].
Получение 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#34)

Стадия 1. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,45,5S)-трет-бутокси-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#31). К раствору #6 (чистота 70%, 3,13 г, 6,1 ммоль, 1 экв.) в дихлорметане (40 мл, 0,15 М) добавляли N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланин (1,99 г, 6,12 ммоль, 1 экв.), диизопропилэтиламин (2,67 мл, 15,3 ммоль, 2,5 экв.) и HATU (2,79 г, 7,35 ммоль, 1,2 экв.). Реакционную смесь перемешивали в течение 18 часов, разбавляли этилацетатом, промывали 1 М водным раствором соляной кислоты и промывали рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме на диоксиде кремния. Затем материал очищали посредством хроматографии на силикагеле (градиент: от 0% до 45% этилацетата в гептане) с получением #31 (3,65 г, 90%) в виде твердого вещества. ЖХ-МС: m/z 665,5 [M+H+], 688,5 [M+Na+], 610,5 [(M-2-метилпроп-1-ен)+H+]; ВЭЖХ (Протокол C): время удерживания составляет 9,455 (чистота более 94%);1H ЯМР (400 МГц, DMSO-d6), характеристические сигналы: δ 7.89 (d, J=7,4 Гц, 2H), 7.67-7.74 (m, 2H), 7.39-7.48 (m, 3H), 7.31-7.36 (m, 2H), 7.29 (br d, J=8,8 Гц, 1H), 4.47-4.60 (br m, 1H), 4.47 (dd, J=8,6, 8,0 Гц, 1H), 4.18-4.28 (m, 3H), 3.69-3.79 (br m, 1H), 3.21 (s, 3H), 2.88 (br s, 3H), 2.15 (dd, J=15,5, 9,3 Гц, 1H), 1.91-2.01 (m, 1H), 1.67-1.81 (br m, 1H), 1.39 (s, 9H), 1.36 (br s, 3H), 1.30 (s, 3H), 0.75 (d, J=6,6 Гц, 3H), 0.66-0.73 (br m, 3H).
Стадия 2. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (#32). Согласно общей методике B из #31 (500 мг, 0,751 ммоль) в дихлорметане (7 мл, 0,1 М) и трифторуксусной кислоты (3 мл) синтезировали #32 в виде стекла (458 мг, количественный выход), которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС: m/z 611,4 [M+2H+], 632,2 [M+Na+], время удерживания составляет 0,94 минуты.
Стадия 3. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2S)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#33). Согласно общей методике D из #32 (53,0 мг, не более 0,083 ммоль, 1 экв.), дихлорметана (4 мл, 0,02 М), N,N-диметилформамида (1 мл), амина #18 (43,8 мг, 0,0870 ммоль, 1 экв.), триэтиламина (36 мкл, 0,26 ммоль, 3 экв.) и HATU (39,5 мг, 0,104 ммоль, 1,2 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 30% ацетона в гептане) с получением #33 (60 мг, 69% за две стадии). ЖХ-МС: m/z 981,4 [M+H+], время удерживания составляет 1,090 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.54 (br d, J=8 Гц) и 10.80 (br d, J=8 Гц), всего 1H], 7.86-7.91 (m, 2H), [7.80 (d, J=3,3 Гц) и 7.82 (d, J=3,3 Гц), всего 1H], 7.68-7.74 (m, 2H), [7.64 (d, J=3,2 Гц) и 7.68 (d, J=3,3 Гц), всего 1H], 7.38-7.44 (m, 2H), 7.20-7.36 (m, 6H), 7.12-7.17 (m, 1H), 6.27-6.34 и 6.40-6.47 (2 m, всего 1H), 3.22 и 3.24 (2 s, всего 3H), 3.14 и 3.18 (2 s, всего 3H), 2.90 и 2.97 (2 br s, всего 3H), 1.37 (br s, 3H), 1.31 (2 br s, всего 3H), [1.13 (d, J=6,6 Гц) и 1.16 (d, J=6,5 Гц), всего 3H].
Стадия 4. Синтез 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#34). Согласно общей методике A из #33 (55 мг, 0,055 ммоль, 1 экв.) в дихлорметане (6 мл, 0,009 М) и диэтиламине (4 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане) с получением #34 (25 мг, 60%) в виде твердого вещества. ВЭЖХ (Протокол A): m/z 759,4 [M+H+], время удерживания составляет 7,088 минуты (чистота более 75%).1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.54 (br d, J=8 Гц) и 10.81 (br d, J=8 Гц), всего 1H], 8.01-8.08 (m, 1H), [7.80 (d, J=3,1 Гц) и 7.83 (d, J=3,3 Гц), всего 1H], [7.65 (d, J=3,2 Гц) и 7.69 (d, J=3,2 Гц), всего 1H], 7.29-7.33 (m, 2H), 7.20-7.27 (m, 2H), 7.13-7.19 (m, 1H), 6.27-6.35 и 6.40-6.48 (2 m, всего 1H), [4.49 (dd, J=9, 8 Гц) и 4.56 (dd, J=9, 8 Гц), всего 1H], 3.24 и 3.25 (2 s, всего 3H), 3.17 и 3.21 (2 s, всего 3H), 2.92 и 2.99 (2 br s, всего 3H), 1.20 и 1.21 (2 s, всего 3H), 1.12 и 1.13 (2 s, всего 3H), 0.75-0.81 (m, 3H).
Получение N-метил-L-валил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-[(2-фенилэтил)амино]-3-тиоксопропил}-пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#36)

Стадия 1. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-[(2-фенилэтил)амино]-3-тиоксопропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#35). К смеси #23 (337 мг, 0,983 ммоль, 1 экв.) в дихлорметане (8 мл, 0,1 M) и N,N-диметилформамиде (1 мл) добавляли #8 (564 мг, 0,885 ммоль, 0,9 экв.), диизопропилэтиламин (383 мг, 2,95 ммоль, 3 экв.) и HATU (472 мг, 1,18 ммоль, 1,2 экв.). Через 2 часа смесь разбавляли дихлорметаном, последовательно промывали 0,1 М водной соляной кислотой и рассолом, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии с обращенной фазой (Метод G) с получением #35 (600 мг, 66%); ЖХ-МС: m/z 926,6 [M+H+], время удерживания составляет 1,16 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [9.94 (br t, J=5 Гц) и 10.16-10.23 (br m), всего 1H], 7.90 (d, J=7,2 Гц, 2H), [7.71 (br d, J=7 Гц) и 8.06 (br d, J=8 Гц), всего 1H], 7.60-7.65 (m, 2H), 7.41 (br dd, J=7, 7 Гц, 2H), 7.15-7.36 (m, 7H), 3.29 (s, 3H), 1.16-1.22 (m, 3H).
Стадия 2. Синтез N-метил-L-валил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-[(2-фенилэтил)амино]-3-тиоксопропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#36). Согласно общей методике A из #35 (465 мг, 0,502 ммоль, 1 экв.) в дихлорметане (5 мл, 0,1 М) и диэтиламине (5 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) с получением #36 (310 мг, 88%) в виде твердого вещества. ЖХ-МС: m/z 704,6 [M+H+], время удерживания составляет 0,74 минуты; МСВР: m/z, рассчитанное для C38H66N5O5S: 704,4779, найденное: 704,477 [M+H+];1H ЯМР (400 МГц, CD3OD), считали, что является смесью ротамеров, характеристические сигналы: δ 7.23-7.30 (m, 4H), 7.15-7.22 (m, 1H), [4.68 (d, J=8,6 Гц) и 4.74 (d, J=8,0 Гц), всего 1H], 3.39 и 3.40 (2 s, всего 3H), 3.12 и 3.22 (2 br s, всего 3H), [2.82 (d, J=6,0 Гц) и 2.84 (d, J=6,0 Гц), всего 1H], 2.29 и 2.30 (2 s, всего 3H), [1.27 (d, J=6,8 Гц) и 1.29 (d, J=6,6 Гц), всего 3H], [0.84 (t, J=7,4 Гц) и 0.87 (t, J=7,4 Гц), всего 3H].
Получение соли трифторуксусной кислоты и N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-тиоксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#41) и N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#42)

Стадия 1. Синтез метил-N-{(2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#37). К смеси #11 (2,7 г, 9,4 ммоль, 1 экв.) в дихлорметане (30 мл, 0,3 M) и N,N-диметилформамиде (3 мл) добавляли диизопропилэтиламин (3,30 мл, 18,8 ммоль, 2 экв.), гидрохлорид сложного метилового эфира L-фенилаланина (2,03 г, 9,40 ммоль, 1,2 экв.) и HATU (4,79 г, 12,2 ммоль, 1,3 экв.). Реакционную смесь перемешивали в течение 18 часов, а затем концентрировали в вакууме. Остаток переносили в этилацетат (100 мл) и последовательно промывали 1 М соляной кислотой (2×50 мл) и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Неочищенный материал переносили в дихлорметан и фильтровали. Фильтрат очищали посредством хроматографии на силикагеле (градиент: от 0% до 100% этилацетата в гептане) с получением #37 (2,76 г, 65%) в виде не совсем белого твердого вещества. ЖХ-МС: m/z 449,3 [M+H+], 349,2 [(M-Boc)+H+], время удерживания составляет 0,88 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 8.28 (d, J=8,2 Гц, 1H), 7.14-7.29 (m, 5H), 4.50 (ddd, J=10,9, 8,1, 4,4 Гц, 1H), 3.64 (s, 3H), 3.23 (s, 3H), 2.15-2.24 (m, 1H), 1.56-1.76 (m, 2H), 1.31-1.55 (m, 11H), 1.02 (d, J=6,6 Гц, 3H).
Стадия 2. Синтез метил-N-{(2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропантиоил}-L-фенилаланината (#38). Смесь #37 (1,52 г, 3,39 ммоль, 1 экв.) и #21 (1,68 г, 4,41 ммоль, 1,3 экв.) в ацетонитриле (12 мл, 0,28 М) подвергали воздействию микроволнового излучения при 100°C в течение 1 часа. Смесь распределяли между водой и этилацетатом. Водный слой обратно экстрагировали этилацетатом. Объединенные органические слои промывали 10%-ным водным раствором лимонной кислоты и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Материал растворяли в небольшом количестве этилацетата и концентрировали на диоксиде кремния в вакууме. После очистки посредством хроматографии на силикагеле (градиент: от 0% до 30% этилацетата в гептане) получали #38 (680 мг, 43%); ЖХ-МС: m/z 465,2 [M+H+], 487,3 [M+Na+], 365,2 [(M-Boc)+H+], время удерживания составляет 0,97 минуты; ВЭЖХ (Протокол B): 465,2 [M+H+], 487,2 [M+Na+], 365,2 [(M-Boc)+H+], время удерживания составляет 7,444 минуты (чистота более 98%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 10.23 (br d, J=7,5 Гц, 1H), 7.17-7.28 (m, 5H), 5.24 (ddd, J=11, 7,5, 4,5 Гц, 1H), 3.66 (s, 3H), 3.28 (s, 3H), 3.21 (dd, J=14,3, 4,4 Гц, 1H), 3.07 (dd, J=14,2, 11,2 Гц, 1H), 2.65-2.74 (m, 1H), 1.54-1.71 (m, 2H), 1.37 (s, 9H), 1.17 (d, J=6,4 Гц, 3H).
Стадия 3. Синтез гидрохлоридной соли метил-N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропантиоил}-L-фенилаланината (#39). Согласно общей методике C при 0°C из #38 (660 мг, 1,42 ммоль, 1 экв.), диоксана (10 мл, 0,14 M) и 4 М раствора соляной кислоты в диоксане (20 мл, 80 ммоль, 60 экв.) синтезировали #39 (590 мг) в виде не совсем белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС: m/z 365,2 [M+H+], время удерживания составляет 0,58 минуты;1H ЯМР (400 МГц, DMSO-d6) δ 10.67 (d, J=7,7 Гц, 1H), 9.42-9.54 (br m, 1H), 8.21-8.33 (br m, 1H), 7.20-7.35 (m, 5H), 5.25 (ddd, J=11,1, 7,6, 4,4 Гц, 1H), 3.76 (dd, J=8,9, 3,0 Гц, 1H), 3.68 (s, 3H), 3.39 (s, 3H), 3.24 (dd, J=14,2, 4,5 Гц, 1H), 3.13 (dd, J=14,3, 11,0 Гц, 1H), 2.93-3.09 (m, 3H), 2.85-2.93 (m, 1H), 1.72-1.84 (m, 1H), 1.36-1.60 (m, 3H), 1.22 (d, J=6,6 Гц, 3H).
Стадия 4. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#40). Согласно общей методике D из #8 (247 мг, 0,387 ммоль, 1 экв.), #39 (186 мг, не более 0,450 ммоль, 1,2 экв.), дихлорметана (10 мл, 0,04 М), N,N-диметилформамида (2 мл), HATU (176 мг, 0,464 ммоль, 1,2 экв.) и триэтиламина (189 мкл, 1,35 ммоль, 3,5 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 25% ацетона в гептане) с получением #40 (410 мг, 90% за две стадии) в виде не совсем белого твердого вещества. ЖХ-МС: m/z 984,7 [M+H+], 1006,7 [M+Na+], время удерживания составляет 1,15 минуты; ВЭЖХ (Протокол C): время удерживания составляет 9,683 минуты (чистота более 99%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.19 (br d, J=7 Гц) и 10.49 (br d, J=8 Гц), всего 1H], 7.90 (d, J=7,5 Гц, 2H), 7.60-7.65 (m, 2H), 7.38-7.45 (m, 2H), 7.29-7.35 (m, 2H), 7.14-7.28 (m, 5H), [5.20 (ddd, J=11, 7, 4 Гц) и 5.35-5.43 (m), всего 1H], 3.65 и 3.69 (2 s, всего 3H), [1.15 (d, J=6,5 Гц) и 1.18 (d, J=6,4 Гц), всего 3H].
Стадия 5A. Синтез соли трифторуксусной кислоты и N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-тиоксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#41). К раствору #40 (401 мг, 0,407 ммоль, 1 экв.) в тетрагидрофуране (10 мл, 0,03 М) добавляли раствор гидроксида лития (24,4 мг, 1,02 ммоль, 2,5 экв.) в воде (5 мл). Через 4 часа реакционную смесь концентрировали в вакууме, а затем три раза подвергали азеотропной перегонке с гептаном. Неочищенный материал растворяли в диметилсульфоксиде (7 мл) и очищали посредством хроматографии с обращенной фазой (Метод C, ввод: 7 раз по 1 мл). Соответствующие фракции концентрировали (Genevac), после чего разбавляли небольшим количеством метанола в дихлорметане. Смесь концентрировали в вакууме до стекловидного твердого вещества. Затем добавляли диэтиловый эфир, потом гептан и смесь концентрировали в вакууме с получением #41 (180 мг, 59%) в виде белого твердого вещества. ЖХ-МС: m/z 748,6 [M+H+], время удерживания составляет 0,68 минуты; ВЭЖХ (Протокол A): 748,4 [M+H+], время удерживания составляет 6,922 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 12.9 и 13.1 (2 v brs, всего 1H), [10.12 (br d, J=8 Гц) и 10.45 (br d, J=8 Гц), всего 1H], 8.75-8.90 (m, 2H), 8.62-8.73 (br m, 1H), 7.13-7.29 (m, 5H), [5.20 (ddd, J=11, 7,5, 4 Гц) и 5.40 (ddd, J=11,5, 8, 4 Гц), всего 1H], 4.55-4.73 (m, 2H), 3.23 и 3.25 (2 s, всего 3H), 3.16 и 3.18 (2 s, всего 3H), 2.97 и 3.01 (2 br s, всего 3H), 1.13-1.20 (m, 3H), 0.73-0.81 (m, 3H).
Стадия 5B. Синтез N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#42). Согласно общей методике A из #40 (561 мг, 0,570 ммоль, 1 экв.), дихлорметана (10 мл, 0,057 М) и диэтиламина (10 мл) синтезировали #42 (348 мг, 80%) в виде белого твердого вещества после хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане). ЖХ-МС: m/z 762,7 [M+H+], время удерживания составляет 0,74 минуты; ВЭЖХ (Протокол A): 762,4 [M+H+], время удерживания составляет 7,315 минуты (чистота более 95%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.20 (br d, J=7,5 Гц) и 10.50 (br d, J=8 Гц), всего 1H], 7.95-8.03 (m, 1H), 7.15-7.29 (m, 5H), [5.20 (ddd, J=11, 7,5, 5 Гц) и 5.39 (ddd, J=11, 7,5, 4 Гц, всего 1H], [4.57 (dd, J=8,8, 8,7 Гц) и 4.61 (dd, J=8,7, 8,6 Гц), всего 1H], 3.65 и 3.69 (2 s, всего 3H), 3.24 и 3.25 (2 s, всего 3H), 3.16 и 3.17 (2 s, всего 3H), 2.96 и 2.99 (2 br s, всего 3H), 2.69-2.79 (m, 1H), 2.62-2.68 (m, 1H), 2.14 и 2.15 (2 br s, всего 3H), [1.15 (d, J=6,6 Гц) и 1.18 (d, J=6,5 Гц), всего 3H], [0.75 (t, J=7,4 Гц) и 0.76 (t, J=7,3 Гц), всего 3H].
Получение гидрохлоридной соли 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-тиоксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#44) и гидрохлоридной соли 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#45)

Стадия 1. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#43). К раствору #32 (321 мг, 0,881 ммоль, 1 экв.) в дихлорметане (5 мл, 0,1 M) и N,N-диметилформамиде (1 мл) добавляли #39 (484 мг, не более 0,769 ммоль, 0,9 экв.), HATU (353 мг, 0,881 ммоль, 1 экв.) и диизопропилэтиламин (463 мкл, 2,64 ммоль, 3 экв.). После перемешивания в течение 18 часов смесь разбавляли дихлорметаном, промывали водой и рассолом, сушили над сульфатом магния, фильтровали и концентрировали на диоксиде кремния в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 30% ацетона в гептане) с получением #43 (574 мг, 68% за две стадии) в виде белого твердого вещества. ЖХ-МС: m/z 956,6 [M+H+], время удерживания составляет 4,49 минуты;1H ЯМР (400 МГц, CD3OD), считали, что является смесью ротамеров, характеристические сигналы: δ 7.80 (d, J=7,5 Гц, 2H), 7.64-7.72 (m, 2H), 7.16-7.35 (m, 7H), [5.43 (dd, J=11, 4,5 Гц) и 5.58 (dd, J=11,5, 4 Гц), всего 1H], 3.72 и 3.75 (2 s, всего 3H), 3.34 и 3.35 (2 s, всего 3H), 3.26 и 3.29 (2 s, всего 3H), 3.05 и 3.11 (2 brs, всего 3H), 1.39 и 1.40 (2 s, всего 3H), [1.24 (d, J=6,7 Гц) и 1.29 (d, J=6,4 Гц), всего 3H].
Стадия 2A. Синтез гидрохлоридной соли 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-тиоксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#44). К раствору #43 (100 мг, 0,105 ммоль, 1 экв.) в тетрагидрофуране (5 мл, 0,02 M) добавляли раствор гидроксида лития (10 мг, 0,417 ммоль, 3 экв.) в воде (3 мл). Через 3 часа реакционную смесь концентрировали в вакууме и очищали посредством хроматографии с обращенной фазой (Метод C) с получением соли трифторуксусной кислоты, которую растворяли в метаноле, обрабатывали 4 М раствором соляной кислоты в диоксане и концентрировали в вакууме с получением #44 (56 мг, 71%) в виде белого твердого вещества. ЖХ-МС: m/z 720,6 [M+H+], время удерживания составляет 0,67 минуты; ВЭЖХ (Протокол D): время удерживания составляет 8,851 минуты;1H ЯМР (400 МГц, CD3OD), считали, что является смесью ротамеров, характеристические сигналы: δ 7.17-7.31 (m, 5H), 3.34 и 3.35 (2 s, всего 3H), 3.10 и 3.16 (2 br s, всего 3H), 1.62 и 1.64 (2 s, всего 3H), 1.53 и 1.55 (2 s, всего 3H), [1.26 (d, J=6,5 Гц) и 1.30 (d, J=6,5 Гц), всего 3H], 0.84-0.91 (m, 3H).
Стадия 2B. Синтез гидрохлоридной соли 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-1-валинамида (#45). Согласно общей методике A из #43 (176 мг, 0,184 ммоль, 1 экв.), дихлорметана (4 мл, 0,05 М) и диэтиламина (4 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии с обращенной фазой (Метод C). Полученную соль трифторуксусной кислоты растворяли в метаноле, обрабатывали 4 М раствором соляной кислоты в диоксане и концентрировали в вакууме с получением #45 (100 мг, 70%) в виде белого твердого вещества. ЖХ-МС: m/z 734,6 [M+H+], время удерживания составляет 0,72 минуты;1H ЯМР (400 МГц, CD3OD), считали, что является смесью ротамеров, характеристические сигналы: δ 7.18-7.31 (m, 5H), 5.41-5.47 и 5.55-5.62 (2 m, всего 1H), 3.73 и 3.76 (2 s, всего 3H), 3.35 и 3.36 (2 s, всего 3H), 3.10 и 3.15 (2 br s, всего 3H), 1.62 и 1.64 (2 s, всего 3H), 1.53 и 1.55 (2 s, всего 3H), [1.25 (d, J=6,6 Гц) и 1.29 (d, J=6,5 Гц), всего 3H], 0.84-0.91 (m, 3H).
Получение N2-[(1-аминоциклопентил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#47)

Стадия 1. Синтез N2-[(1-{[(9H-флуорен-9-илметокси)карбонил]амино}-циклопентил)-карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#46). К раствору #19 (353 мг, 0,944 ммоль, 1 экв.) в дихлорметане (10 мл, 0,094 М) добавляли #28 (600 мг, 0,944 ммоль, 0,9 экв.), диизопропилэтиламин (498 мкл, 2,83 ммоль, 3 экв.) и HATU (444 мг, 1,13 ммоль, 1,2 экв.). После перемешивания в течение двух суток смесь концентрировали в вакууме и остаток разбавляли этилацетатом (60 мл), промывали 1 М водным раствором соляной кислоты и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток разбавляли дихлорметаном и фильтровали. Фильтрат концентрировали при пониженном давлении на диоксиде кремния и очищали посредством хроматографии на силикагеле (градиент: от 40% до 100% этилацетата в гептане) с получением #46 (644 мг, 69%) в виде белого твердого вещества. ЖХ-МС: m/z 991,8 [M+H+], время удерживания составляет 1,07 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 7.89 (br d, J=7,4 Гц, 2H), [7.77 (d, J=3,3 Гц) и 7.79 (d, J=3,3 Гц), всего 1H], 7.66-7.76 (m, 2H), [7.62 (d, J=3,3 Гц) и 7.65 (d, J=3,3 Гц), всего 1H], 7.37-7.44 (m, 2H), 7.11-7.36 (m, 7H), [5.38 (ddd, J=11, 8, 4 Гц) и 5.48-5.57 (m), всего 1H], 3.13, 3.17, 3.18 и 3.24 (4 s, всего 6H), 2.90 и 3.00 (2 br s, всего 3H), [1.05 (d, J=6,6 Гц) и 1.09 (d, J=6,8 Гц), всего 3H].
Стадия 2. Синтез N2-[(1-аминоциклопентил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#47). К смеси #46 (500 мг, 0,504 ммоль, 1 экв.) в тетрагидрофуране (8 мл, 0,06 M) добавляли диэтиламин (4 мл). После перемешивания в течение 18 часов реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) с получением #47 (374 мг, 96%) в виде белого твердого вещества. ЖХ-МС: m/z 769,6 [M+H+], время удерживания составляет 0,70 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.64 (br d, J=8,4 Гц) и 8.87 (br d, J=8,6 Гц), всего 1H], [8.22 (br d, J=9,4 Гц) и 8,26 (br d, J=9,4 Гц), всего 1H], [7.77 (d, J=3,3 Гц) и 7.80 (d, J=3,3 Гц), всего 1H], [7.63 (d, J=3,1 Гц) и 7.66 (d, J=3,3 Гц), всего 1H], 7.13-7.31 (m, 5H), [5.39 (ddd, J=11,1, 8,5, 4,2 Гц) и 5.54 (ddd, J=11,7, 8,8, 4,1 Гц), всего 1H], [4.53 (dd, J=9,2, 7,6 Гц) и 4.64 (dd, J=9,2, 6,6 Гц), всего 1H], 3.16, 3.20, 3.21 и 3.25 (4 s, всего 6H), 2.93 и 3.03 (2 br s, всего 3H), [1.05 (d, J=6,8 Гц) и 1.10 (d, J=6,6 Гц), всего 3H], 0.73-0.80 (m, 3H).
Получение N2-[(1-аминоциклопропил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#51) и 1-амино-N-[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]циклогексанкарбоксамида
(#52)

Стадия 1. Синтез соли трифторуксусной кислоты и (2R,3R)-3-метокси-2-метил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-3-[(2S)-пирролидин-2-ил]пропанамида и (3R,4S,5S)-4-[{N-[(9H-флуорен-9-илметокси)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептановой кислоты {#48). К раствору #16 (1,0 г, 2,11 ммоль, 1 экв.) и #5 (1,22 г, 2,11 ммоль, 1 экв.) в дихлорметане (20 мл, 0,1 М) при 0°C добавляли трифторуксусную кислоту (6 мл). Через 3 часа смесь концентрировали в вакууме с получением смеси #48 (1,8 г), которую использовали на следующей стадии без дополнительной очистки; ЖХ-МС (Протокол K): m/z 374,2 [M+H+], время удерживания составляет 2,093 минуты, 525,2 [M+H+], время удерживания составляет 4,875 минуты.
Стадия 2. Синтез N2-[(9H-флуорен-9-илметокси)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#49). К раствору #48 (1,8 г, не более 2,1 ммоль, 1 экв.) и диэтилцианофосфоната (DEPC) (0,51 г, 3,2 ммоль, 1,5 экв.) в 1,2-диметоксиэтане (30 мл, 0,07 М) при 0°C добавляли триэтиламин (1,47 мл, 10,6 ммоль, 5 экв.). После перемешивания при комнатной температуре в течение 2 часов смесь концентрировали в вакууме и остаток очищали посредством хроматографии на силикагеле (от 10% до 50% этилацетата в петролейном эфире) с получением #49 (0,8 г, 45%). Rf 0,6 (10% метанола в дихлорметане); ЖХ-МС (Протокол K): m/z 881,3 [M+H+], 903,3 [M+Na+], время удерживания составляет 4,837 минуты.
Стадия 3. Синтез N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#50). Согласно общей методике A из #49 (0,70 г, 0,79 ммоль, 1 экв.), дихлорметана (15 мл, 0,05 М) и диэтиламина (10 мл) синтезировали #50 (160 мг, 30%) после очистки посредством хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане). Rf 0,4 (10% метанола в дихлорметане); ЖХ-МС (Протокол K): m/z 658,3 [M+H+], 680,3 [M+Na+], время удерживания составляет 2,760 минуты.
Стадия 4A. Синтез N2-[(1-аминоциклопропил)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#51). К раствору #50 (100 мг, 0,15 ммоль, 1 экв.), гексафторфосфата бромтрис(диметиламино)фосфония (Brop, 70 мг, 0,18 ммоль, 1,2 экв.) и диизопропилэтиламина (0,08 мл, 0,45 ммоль, 3 экв.) в дихлорметане (15 мл, 0,01 М) при 0°C добавляли 1-аминоциклопропан-карбоновую кислоту (18 мг, 0,18 ммоль, 1,2 экв.). Через 2 часа смесь гасили водой и дважды экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане) с получением #51 (45 мг, 34%). Rf 0,5 (10% метанола в дихлорметане). ЖХ-МС (Протокол L): m/z 741,44 [M+H+];1H ЯМР (300 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.64 (br d, J=8 Гц) и 8.88 (br d, J=8 Гц), всего 1H], [8.16 (br d, J=9 Гц) и 8.22 (br d, J=10 Гц), всего 1H], [7.77 (d, J=3,5 Гц) и 7.79 (d, J=3,5 Гц), всего 1H], [7.63 (d, J=3,5 Гц) и 7.65 (d, J=3 Гц), всего 1H], 7.10-7.32 (m, 5H), 5.33-5.60 (m, 1H), 3.16, 3.20, 3.21 и 3.26 (4 s, всего 6H), 2.93 и 3.02 (2 br s, всего 3H), [1.05 (d, J=6,3 Гц) и 1.10 (d, J=6,3 Гц), всего 3H].
Стадия 4B. Синтез 1-амино-N-[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]циклогексанкарбоксамида (#52). К раствору #50 (120 мг, 0,18 ммоль, 1 экв.), Brop (84 мг, 0,21 ммоль, 1,2 экв.) и диизопропилэтиламина (0,1 мл, 0,54 ммоль, 3 экв.) в дихлорметане (15 мл, 0,009 M) при 0°C добавляли 1-аминоциклогексанкарбоновую кислоту (31 мг, 0,21 ммоль, 1,2 экв.). Через 2 часа смесь гасили водой и дважды экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане) с получением #52 (50 мг, 35%). Rf 0,6 (10% метанола в дихлорметане). ЖХ-МС (Протокол K): m/z 783,79 [M+H+];1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.64 (br d, J=8 Гц) и 8.87 (br d, J=9 Гц), всего 1H], 8.18-8.28 (m, 1H), [7.77 (d, J=3,5 Гц) и 7.80 (d, J=3,3 Гц), всего 1H], [7.63 (d, J=3,3 Гц) и 7.66 (d, J=3,3 Гц), всего 1H], 7.12-7.31 (m, 5H), 5.35-5,43 и 5.49-5.57 (2 m, всего 1H), [4.51 (dd, J=9, 8 Гц), и 4.61 (dd, J=9, 7 Гц), всего 1H], 3.16, 3.19, 3.21 и 3.25 (4 s, всего 6H), 2.93 и 3.02 (2 br s, всего 3H), [1.05 (d, J=6,8 Гц) и 1.10 (d, J=6,8 Гц), всего 3H].
Получение 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#54)

Стадия 1. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#53). Согласно общей методике D из #32 (2,05 г, 2,83 ммоль, 1 экв.) в дихлорметане (20 мл, 0,1 М) и N,N-диметилформамиде (3 мл), амина #19 (2,5 г, 3,4 ммоль, 1,2 экв.), HATU (1,29 г, 3,38 ммоль, 1,2 экв.) и триэтиламина (1,57 мл, 11,3 ммоль, 4 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 55% ацетона в гептане), получая #53 (2,42 г, 74%) в виде твердого вещества. ЖХ-МС: m/z 965,7 [M+H+], 987,6 [M+Na+], время удерживания составляет 1,04 минуты; ВЭЖХ (Протокол A): m/z 965,4 [M+H+], время удерживания составляет 11,344 минуты (чистота более 97%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 7.86-7.91 (m, 2H), [7.77 (d, J=3,3 Гц) и 7.79 (d, J=3,2 Гц), всего 1H], 7.67-7.74 (m, 2H), [7.63 (d, J=3,2 Гц) и 7.65 (d, J=3,2 Гц), всего 1H], 7,38-7.44 (m, 2H), 7.30-7.36 (m, 2H), 7.11-7.30 (m, 5H), [5.39 (ddd, J=11,4, 8,4, 4,1 Гц) и 5.52 (ddd, J=11,7, 8,8, 4,2 Гц), всего 1H], [4.49 (dd, J=8,6, 7,6 Гц) и 4.59 (dd, J=8,6, 6,8 Гц), всего 1H], 3.13, 3.17, 3.18 и 3.24 (4 s, всего 6H), 2.90 и 3.00 (2 br s, всего 3H), 1.31 и 1.36 (2 br s, всего 6H), [1.05 (d, J=6,7 Гц) и 1.09 (d, J=6,7 Гц), всего 3H].
Стадия 2. Синтез 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#54). Согласно общей методике A из #53 (701 мг, 0,726 ммоль) в дихлорметане (10 мл, 0,07 М) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане). Остаток разбавляли диэтиловым эфиром и гептаном и концентрировали в вакууме с получением #54 (406 мг, 75%) в виде белого твердого вещества. ЖХ-МС: m/z 743,6 [M+H+], время удерживания составляет 0,70 минуты; ВЭЖХ (Протокол A): m/z 743,4 [M+H+], время удерживания составляет 6,903 минуты (чистота более 97%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.64 (br d, J=8,5 Гц) и 8.86 (br d, J=8,7 Гц), всего 1H], [8.04 (br d, J=9,3 Гц) и 8.08 (br d, J=9,3 Гц), всего 1H], [7.77 (d, J=3,3 Гц) и 7.80 (d, J=3,2 Гц), всего 1H], [7.63 (d, J=3,3 Гц) и 7.66 (d, J=3,2 Гц), всего 1H], 7.13-7.31 (m, 5H), [5.39 (ddd, J=11, 8,5, 4 Гц) и 5.53 (ddd, J=12, 9, 4 Гц), всего 1H], [4.49 (dd, J=9, 8 Гц) и 4.60 (dd, J=9, 7 Гц), всего 1H], 3.16, 3.20, 3.21 и 3.25 (4 s, всего 6H), 2.93 и 3.02 (2 br s, всего 3H), 1.21 (s, 3H), 1.13 и 1.13 (2 s, всего 3H), [1.05 (d, J=6,7 Гц) и 1.10 (d, J=6,7 Гц), всего 3H], 0.73-0.80 (m, 3H).
Получение соли уксусной кислоты и 2-метилаланил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#56)
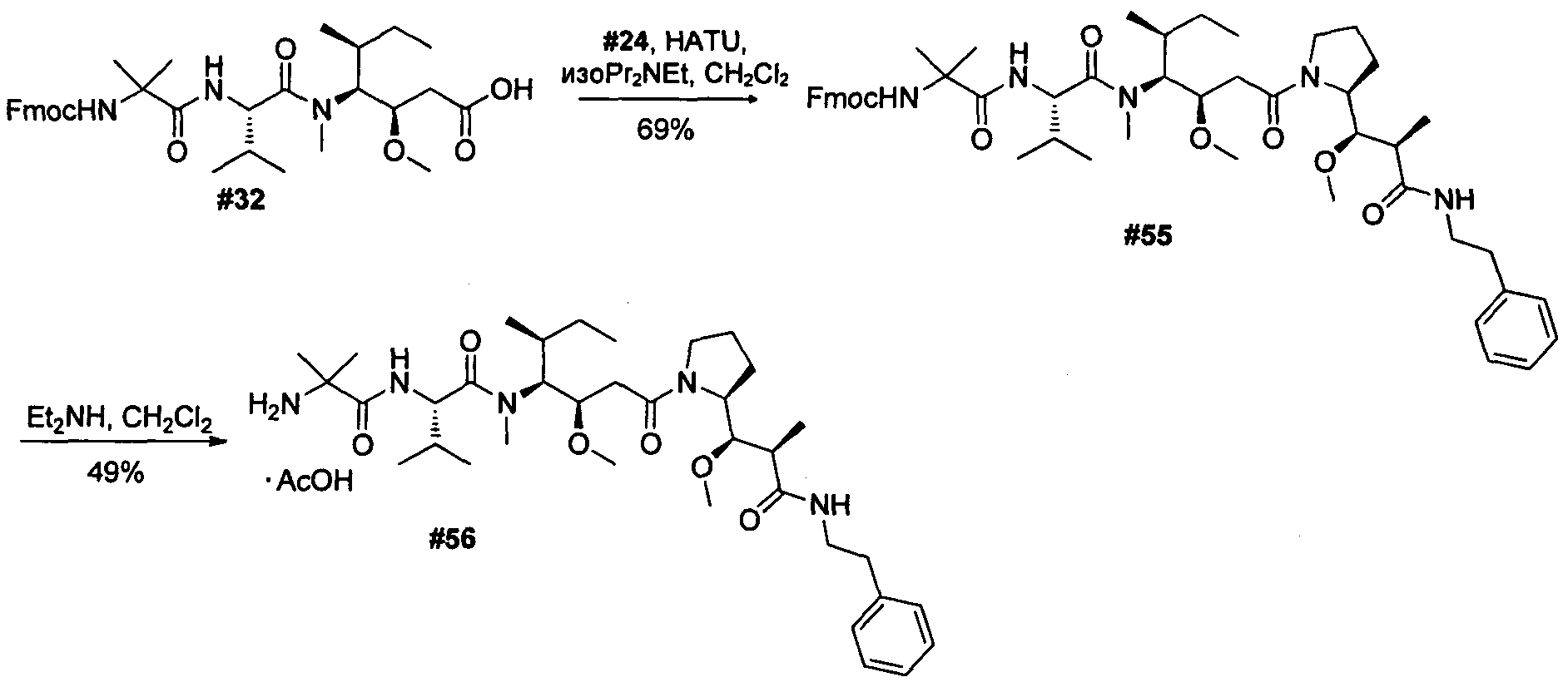
Стадия 1. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#55}. К раствору #24 (104 мг, 0,256 ммоль, 1 экв.) в дихлорметане (10 мл, 0,094 M) добавляли #32 (156 мг, 0,256 ммоль, 0,9 экв.), диизопропилэтиламин (135 мкл, 0,768 ммоль, 3 экв.) и HATU (120 мг, 0,307 ммоль, 1,2 экв.). После перемешивания в течение 18 часов смесь концентрировали в вакууме и остаток разбавляли этилацетатом (10 мл), промывали 1 М водным раствором соляной кислоты (2×5 мл) и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток разбавляли дихлорметаном и фильтровали. Фильтрат концентрировали при пониженном давлении на диоксиде кремния и очищали посредством хроматографии на силикагеле (градиент: от 0% до 100% этилацетата в гептане) с получением #55 (44 мг, 19%) в виде белого твердого вещества. ЖХ-МС: m/z 884,5 [M+2H+], время удерживания составляет 1,04 минуты.
Стадия 2. Синтез соли уксусной кислоты и 2-метилаланил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#56). К смеси #55 (44 мг, 0,050 ммоль, 1 экв.) в тетра гидрофура не (1 мл, 0,05 М) добавляли диэтиламин (0,5 мл). После перемешивания в течение 18 часов реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии с обращенной фазой (Метод B) с получением #56 (16,2 мг, 49%) в виде твердого вещества. ЖХ-МС: m/z 660,8 [M+H+], время удерживания составляет 2,23 минуты; ВЭЖХ (Протокол A): m/z 660,5 [M+H+], 682,4 [M+Na+], время удерживания составляет 6,865 минуты.
Получение 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1-фенилциклопропил)метил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#60)

Стадия 1. Синтез 1-(1-фенилциклопропил)метанамина #@1. К раствору 1-фенилциклопропанкарбонитрила (50 г, 0,34 моль, 1 экв.) в тетрагидрофуране (500 мл, 0,7 М) при 0°C добавляли алюмогидрид лития (23 г, 0,35 моль, 1,03 экв.). Реакционную смесь перемешивали при 0°C в течение одного часа, а затем при кипячении с обратным холодильником в течение одного часа. Затем реакционную смесь охлаждали и гасили водой (23 мл) и 15%-ным водным раствором гидроксида натрия (69 мл). Смесь фильтровали и концентрировали в вакууме с получением #@1 (36 г, 72%). ЖХ-МС: m/z 148,1 [M+H+], время удерживания составляет 0,86 минуты;1H ЯМР (400 МГц, CDCl3) δ 7.2-7.4 (m, 5H), 2.78 (s, 2H), 1.19 (br s, 2H), 0.72-0.84 (m, 4H).
Стадия 2. Синтез трет-бутил-(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1-фенилциклопропил)метил]амино}пропил]пирролидин-1-карбоксилата (#57). Согласно общей методике D из #11 (2,15 г, 7,48 ммоль, 1,1 экв.) в дихлорметане (20 мл, 0,3 М) и N,N-диметилформамиде (4 мл), 1-(1-фенилциклопропил)метанамина #@1 (1,001 г, 6,799 ммоль, 1 экв.), HATU (3,10 г, 8,16 ммоль, 1,2 экв.) и триэтиламина (2,84 мл, 20,4 ммоль, 3 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 100% этилацетата в гептане), получая #57 (1,93 г, 68%) в виде твердого вещества. ВЭЖХ (Протокол A при 45°C): m/z 417,3 [M+H+], время удерживания составляет 10,575 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров: δ 7.75-7.81 (m, 1H), 7.20-7.27 (m, 4H), 7.12-7.19 (m, 1H), 3.33-3.62 и 3.71-3.80 (уш. мультиплеты, всего 4H), 3.28 (s, 3H), 2.97-3.17 (br m, 2H), 2.14-2.24 (m, 1H), 1.67-1.80 (br m, 2H), 1.45-1.65 (m, 2H), 1.41 (s, 9H), 1.00 (d, J=6,6 Гц, 3H), 0.67-0.93 (m, 4H).
Стадия 3. Синтез гидрохлоридной соли (2R,3R)-3-метокси-2-метил-N-[(1-фенилцикпопропил)метил]-3-[(2S)-пирролидин-2-ил]пропанамида (#58). Согласно общей методике C из #57 (566 мг, 1,36 ммоль, 1 экв.) в диоксане (4 мл, 0,3 М) и 4 М раствора соляной кислоты в диоксане (4 мл, 16 ммоль, 11,7 экв.) синтезировали #58 (466 мг, 97%); ЖХ-МС: m/z 318,2 [M+H+], 339,2 [M+Na+], время удерживания составляет 0,56 минуты;1H ЯМР (400 МГц, DMSO-d6) δ 9.53 (br s, 1H), 8.48 (br s, 1H), 8.11 (br dd, J=5,7, 5,6 Гц, 1H), 7.23-7.30 (m, 4H), 7.14-7.21 (m, 1H), 3.58 (dd, J=7,5, 3,9 Гц, 1H), 3.50 (dd, J=13,7, 6,3 Гц, 1H), 3.34 (s, 3H), 3.21-3.29 (br m, 1H), 3.18 (dd, J=13,8, 5,0 Гц, 1H), 3.04-3.13 (br m, 2H), 2.42-2.50 (m, 1H), 1.56-1.89 (m, 4H), 1.04 (d, J=6,9 Гц, 3H), 0.71-0.91 (m, 4H).
Стадия 4. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1-фенилциклопропил)метил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#59). Согласно общей методике D из #32 (550 мг, 0,902 ммоль, 1 экв.), #58 (350 мг, 0,992 ммоль, 1,1 экв.), дихлорметана (10 мл, 0,08 M) и N,N-диметилформамида (2 мл), HATU (446 мг, 1,17 ммоль, 1,3 экв.) и триэтиламина (0,503 мл, 3,61 ммоль, 4 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 30% ацетона в гептане), получая #59 (618 мг, 69%) в виде не совсем белого твердого вещества. ЖХ-МС: m/z 908,7 [M+H+], 930,7 [M+Na+], время удерживания составляет 1,07 минуты; ВЭЖХ (Протокол B при 45°C): m/z 908,5 [M+H+], время удерживания составляет 8,721 минуты (чистота более 97%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 7.89 (d, J=7,5 Гц, 2H), 7.38-7.44 (m, 2H), 7.30-7.36 (m, 2H), [4.49 (dd, J=8,5, 7,8 Гц) и 4.59 (dd, J=8,7, 6,9 Гц), всего 1H], 4.18-4.26 (m, 3H), 3.93-4.01 (br m, 1H), 3.23 и 3.26 (2 s, всего 3H), 3.16 и 3.16 (2 s, всего 3H), 2.91 и 3.05 (2 br s, всего 3H), 1.36 и 1.37 (2 br s, всего 3H), 1.30 и 1.32 (2 br s, всего 3H), [1.00 (d, J=6,7 Гц) и 1.02 (d, J=6,6 Гц), всего 3H], 0.67-0.78 (m, 7H).
Стадия 5. Синтез 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1-фенилциклопропил)метил]амино}-пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#60). Согласно общей методике A из #59 (605 мг, 0,666 ммоль, 1 экв.), дихлорметана (10 мл, 0,067 M) и диэтиламина (10 мл) синтезировали #60 (379 мг, 83%); ВЭЖХ (Протокол A при 45°C) m/z 685,5 [M+H+], время удерживания составляет 7,072 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.03 (br d, J=9,6 Гц) и 8.07 (br d, J=9,4 Гц), всего 1H], [7.74 (br dd, J=7, 4 Гц) и 7.99 (br dd, J=5,9, 5,7 Гц), всего 1H], 7.20-7.27 (m, 4H), 7.11-7.17 (m, 1H), [4.49 (dd, J=9, 7 Гц) и 4.58 (dd, J=9, 7,5 Гц), всего 1H], 3.96-4.04 (br m, 1H), 3.24 и 3.27 (2 s, всего 3H), 3.18 и 3.19 (2 s, всего 3H), 2.93 и 3.07 (2 br s, всего 3H), 1.20 и 1.21 (2 s, всего 3H), 1.12 и 1.14 (2 s, всего 3H), [1.00 (d, J=6,7 Гц) и 1.03 (d, J=6,7 Гц), всего 3H].
Получение 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#66)

Стадия 1. Синтез циклогепта-2,4,6-триен-1-илацетонитрила (#61). К раствору безводного ацетонитрила (3,12 мл, 56,2 ммоль, 1 экв.) в тетрагидрофуране (281 мл, 0,2 М) добавляли литий-диизопропиламин (1,8 М в смеси гептан/этилбензол/тетрагидрофуран, 31,2 мл, 56,2 ммоль, 1 экв.) при -78°C. Через 20 минут при -78°C добавляли тетрафторборат тропилия (10 г, 56 ммоль, 1 экв.). Через 10 минут реакционную смесь концентрировали в вакууме и остаток разбавляли этилацетатом и промывали водой. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением коричневого масла, которое очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% этилацетата в гептане) с получением #61 (1,88 г, 25%) в виде желтого масла.1H ЯМР (400 МГц, CDCl3) δ 6.69-6.71 (m, 2H), 6.27-6.32 (m, 2H), 5.28-5.33 (m, 2H), 2.61 (d, J=7,2 Гц, 2H), 2.26-2.34 (m, 1H).
Стадия 2. Синтез 2-(циклогепта-2,4,6-триен-1-ил)этанамина (#62). К суспензии алюмогидрида лития (911 мг, 24,0 ммоль, 1,4 экв.) в безводном диэтиловом эфире (75 мл, 0,23 М) при 0°C медленно по каплям в течение 15 минут добавляли раствор #61 (2,25 г, 17,2 ммоль, 1 экв.) в диэтиловом эфире (15 мл). Реакционную смесь нагревали до комнатной температуры. Через 5 часов реакционную смесь охлаждали до 0°C и гасили посредством добавления воды (1 мл), затем фильтровали через небольшую набивку целита и промывали метанолом. Фильтрат сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением #62 (1,683 г, 73%) в виде масла золотистого цвета. ЖХ-МС: m/z 136,1 [M+H+], время удерживания составляет 0,23 минуты;1H ЯМР (400 МГц, CDCl3) δ 6.64-6.67 (m, 2H), 6.16-6.21 (m, 2H), 5.16-5.21 (m, 2H), 2.84-2.89 (m, 2H), 1.86-1.92 (m, 2H), 1.62-1.70 (m, 1H).
Стадия 3. Синтез трет-бутил-(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-карбоксилата (#63). К раствору #11 (3,57 г, 12,4 ммоль, 1 экв.) в дихлорметане (100 мл, 0,1 M) и N,N-диметилформамиде (4 мл) добавляли HATU (5,36 г, 13,7 ммоль, 1,1 экв.). Через 20 минут добавляли триэтиламин (5,20 мл, 37,3 ммоль, 3 экв.), а затем #62 (1,68 г, 12,4 ммоль, 1 экв.) и смесь перемешивали в течение 18 часов. Реакционную смесь концентрировали в вакууме и остаток переносили в этилацетат и промывали водой (50 мл). Водный слой обратно экстрагировали этилацетатом (3 раза) и объединенные органические слои сушили, фильтровали и концентрировали в вакууме с получением коричневого масла, которое очищали посредством хроматографии на силикагеле (градиент: от 0% до 100% этилацетата в гептане) с получением #63 (2,95 г, 59%-ный выход) в виде вязкого масла. ЖХ-МС: m/z 405,4 [M+H+], 427,4 [M+Na+], время удерживания составляет 0,75 минуты;1H ЯМР (400 МГц, CDCl3), считали, что является смесью ротамеров: δ 6.63-6.68 (m, 2H), 6.16-6.23 (m, 2H), 5.19 (br dd, J=9,0, 5,8 Гц, 2H), 3.51-3.63 и 3.71-3.90 (2 уш. мультиплета, всего 3H), 3.42 (s, 3H), 3.18-3.29 и 3.34-3.47 (2 уш. мультиплета, всего 3H), 2.27-2.45 (br m, 1H), 1.6-2.00 (m, 7H), 1.47 и 1.50 (2 br s, всего 9H), 1.16-1.29 (br m, 3H).
Стадия 4. Синтез гидрохлоридной соли (2R,3R)-N-[2-(циклогепта-2,4,6-триен-1-ил)этил]-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропанамида (#64)
Промежуточное соединение #63 (400 мг, 0,989 ммоль, 1 экв.) обрабатывали 4 М раствором соляной кислоты в диоксане (10 мл, 40 ммоль, 40 экв.). Через 1 час реакционную смесь концентрировали в вакууме и остаток переносили в дихлорметан и промывали 1 М раствором гидроксида натрия. Водный слой обратно экстрагировали дихлорметаном и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением #64 (301 мг, количественный выход) в виде коричневого масла, которое при стоянии стало медленно затвердевать. ЖХ-МС: m/z 305,3 [M+H+], время удерживания составляет 0,54 минуты; ВЭЖХ (Протокол G): время удерживания составляет 4,848 минуты;1H ЯМР (400 МГц, CDCl3), характеристические сигналы: δ 6.64-6.67 (m, 2H), 6.16-6.22 (m, 2H), 6.08-6.14 (br m, 1H), 5.16-5.22 (m, 2H), 3.44 (s, 3H), 3.31 (dd, J=6,3, 4,5 Гц, 1H), 2.98-3.04 (m, 1H), 2.94 (ddd, J=10,5, 7,2, 5,6 Гц, 1H), 2.81 (ddd, J=10,5, 7,7, 6,7 Гц, 1H), 2.57 (qd, J=7,1, 4,5 Гц, 1H), 1.90-1.97 (m, 2H), 1.49-1.55 (m, 1H), 1.18 (d, J=7,1 Гц, 3H).
Стадия 5. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#65). Согласно общей методике D из #32 (678 мг, 0,937 ммоль, 1 экв.) в дихлорметане (9,37 мл, 0,1 M), амина #64 (300 мг, 0,985 ммоль, 1,1 экв.), HATU (427 мг, 1,12 ммоль, 1,2 экв.) и диизопропилэтиламина (494 мкл, 2,81 ммоль, 3 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 50% ацетона в гептане), получая #65 (546 мг, 65%) в виде твердого вещества. ЖХ-МС: m/z 896,7 [M+H+], 918,7 [M+Na+], время удерживания составляет 1,06 минуты.
Стадия 6. Синтез 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#66). К раствору #65 (540 мг, 0,603 ммоль, 1 экв.) в дихлорметане (10 мл, 0,06 M) добавляли триэтиламин (10 мл) и реакционную смесь перемешивали в течение 2 часов. Смесь концентрировали в вакууме и остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) с получением #66 (347 мг, 85%) в виде бесцветного твердого вещества. ВЭЖХ (Протокол A при 45°C): m/z 674,5 [M+H+], время удерживания составляет 7,015 минуты.1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.03 (br d, J=9 Гц) и 8.05 (br d, J=9 Гц), всего 1H], [7.77 (br dd, J=5,5, 5,5 Гц) и 7.98 (br dd, J=5,5, 5,5 Гц), всего 1H], 6.54-6.65 (m, 2H), 6.10-6.19 (m, 2H), 5.11-5.19 (m, 2H), [4.48 (dd, J=9, 8 Гц) и 4.54 (dd, J=9, 7,5 Гц), всего 1H], 3.94-4.04 (br m, 1H), 3.26 и 3.29 (2 s, всего 3H), 3.17 и 3.19 (2 s, всего 3H), 2.93 и 3.06 (2 br s, всего 3H), 1.20 и 1.21 (2 s, всего 3H), 1.12 и 1.13 (2 s, всего 3H), [1.04 (d, J=6,8 Гц) и 1.07 (d, J=6,7 Гц), всего 3H].
Получение 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#69) и 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#70)

Стадия 1. Синтез гидрохлоридной соли метил-N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-фенилаланината (#67). Согласно общей методике C из #37 (2,39 г, 5,33 ммоль, 1 экв.), диоксана (10 мл, 0,53 М) и 4 М раствора соляной кислоты в диоксане (10 мл, 40 ммоль, 7,5 экв.) синтезировали #67 (2,21 г) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС: m/z 349,2 [M+H+], время удерживания составляет 0,53 минуты;1H ЯМР (400 МГц, DMSO-d6) δ 9.45-9.58 (br m, 1H), 8.63 (d, J=8,1 Гц, 1H), 8.51-8.62 (br m, 1H), 7.25-7.33 (m, 4H), 7.18-7.25 (m, 1H), 4.50 (ddd, J=10,8, 8,1, 4,5 Гц, 1H), 3.65 (s, 3H), 3.54 (dd, J=6,8, 4,5 Гц, 1H), 3.20 (s, 3H), 3.11 (dd, J=13,8, 4,5 Гц, 1H), 2.99-3.14 (br m, 3H), 2.89 (dd, J=13,8, 10,9 Гц, 1H), 2.44-2.50 (m, 1H, предполагалось; частично заслонен пиком растворителя), 1.77-1.89 (m, 1H), 1.60-1.73 (m, 2H), 1.46-1.57 (m, 1H), 1.05 (d, J=6,8 Гц, 3H).
Стадия 2. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#68). Согласно общей методике D из #32 (353 мг, 0,488 ммоль, 1 экв.) в дихлорметане (10 мл, 0,04 M), амина #67 (271 мг, не более 0,588 ммоль, 1,3 экв.), HATU (223 мг, 0,586 ммоль, 1,2 экв.) и диизопропилэтиламина (238 мкл, 1,71 ммоль, 3,5 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 40% ацетона в гептане), получая #68 (404 мг, 88% за две стадии) в виде твердого вещества. ЖХ-МС: m/z 940,7 [M+H+], 962,7 [M+Na+], время удерживания составляет 1,04 минуты; ВЭЖХ (Протокол C): время удерживания составляет 9,022 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.25 (br d, J=8 Гц) и 8.48 (br d, J=8 Гц), всего 1H], 7.89 (d, J=7,4 Гц, 2H), 7.67-7.75 (m, 2H), 7.38-7.44 (m, 2H), 7.31-7.36 (m, 2H), 7.14-7.24 (m, 5H), 4.43-4.69 (m, 3H), 4.17-4.26 (m, 3H), 3.91-3.99 (br m, 1H), 3.63 и 3.65 (2 s, всего 3H), 3.19 и 3.24 (2 s, всего 3H), 3.14 и 3.15 (2 s, всего 3H), 2.90 и 2.99 (2 br s, всего 3H), 1.36 и 1.37 (2 br s, всего 3H), 1.30 и 1.32 (2 s, всего 3H), [1.02 (d, J=6,8 Гц) и 1.06 (d, J=6,6 Гц), всего 3H].
Стадия 3A. Синтез соли трифторуксусной кислоты и 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#69). К раствору #68 (143 мг, 0,152 ммоль, 1 экв.) в тетрагидрофуране (5 мл, 0,02 M) добавляли раствор гидроксида лития (9,10 мг, 0,378 ммоль, 2,5 экв.) в воде (3 мл). Через 5 часов реакционную смесь концентрировали в вакууме, подвергали три раза азеотропной перегонке с гептаном, растворяли в диметилсульфоксиде (2,2 мл) и очищали посредством хроматографии с обращенной фазой (Метод C) с получением #69 (56 мг, 52%). ВЭЖХ (Протокол A при 45°C): 704,4 [M+H+], время удерживания составляет 6,623 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 8.08-8.22 и 8.37-8.49 (2 m, всего 5H), 7.12-7.28 (m, 5H), 3.18, 3.20 и 3.24 (3 s, всего 6H), 2.95 и 3.04 (2 br s, всего 3H), 1.52 и 1.53 (2 s, всего 3H), 1.39 и 1.41 (2 s, всего 3H), [1.02 (d, J=6,8 Гц) и 1.05 (d, J=6,6 Гц), всего 3H], 0.74-0.81 (m, 3H).
Стадия 3B. Синтез 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#70). Согласно общей методике A из #68 (240 мг, 0,255 ммоль, 1 экв.), дихлорметана (10 мл, 0,026 M) и диэтиламина (10 мл) синтезировали #70 (120 мг, 65%) в виде смеси белого твердого вещества и стекла после хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане). ВЭЖХ (Протокол A при 45°C): m/z 762,7 [M+H+], 740,4 [M+Na+], время удерживания составляет 6,903 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.26 (d, J=8,1 Гц) и 8.49 (d, J=8,3 Гц), всего 1H], [8.03 (d, J=9,5 Гц) и 8.07 (d, J=9,5 Гц), всего 1H], 7.14-7.27 (m, 5H), 3.63 и 3.67 (2 s, всего 3H), 3.16, 3.18, 3.20 и 3.25 (4 s, всего 6H), 2.92 и 3.01 (2 ors, всего 3H), 1.20 и 1.22 (2 s, всего 3H), 1.12 и 1.13 (2 s, всего 3H), [1.02 (d, J=6,8 Гц) и 1.06 (d, J=6,7 Гц), всего 3H], 0.74-0.80 (m, 3H).
Получение соли уксусной кислоты и N2-[(3-аминооксетан-3-ил)карбонил]-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#75)

Стадия 1. Синтез 3-[(трет-бутоксикарбонил)амино]оксетан-3-карбоновой кислоты (#71). К 1-аминооксетан-3-карбоновой кислоте (1,00 г, 8,54 ммоль, 1 экв.) в диоксане (15 мл, 0,5 М) добавляли раствор гидроксида натрия (1,55 г, 38,7 ммоль, 4,5 экв.) в воде (15 мл), а затем ди-трет-бутил-дикарбонат (2,09 г, 9,29 ммоль, 1,1 экв.). Образовывалось белое твердое вещество. Реакционную смесь перемешивали в течение 18 часов, а затем концентрировали в вакууме. Остаток переносили в этилацетат и промывали 1 М водным раствором соляной кислоты и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением #71 (633 мг, 38%) в виде белого твердого вещества.1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров: δ 12.93 (br s, 1H), 7.59 и 7.93 (2 br s, всего 1H), 4.71-4.78 (m, 2H), 4.47 (d, J=6,4 Гц, 2H), 1.30 и 1.38 (2 s, всего 9H).
Стадия 2. Синтез N2-[(9H-флуорен-9-илметокси)карбонил]-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#72). К #@5 (9,47 г, 18,0 ммоль, 1 экв.) и #24 (5,90 г, 18,0 ммоль, 1 экв.) в дихлорметане (250 мл, 0,072 М) добавляли диизопропилэтиламин (9,52 мл, 54,2 ммоль, 3 экв.) и HATU (8,49 г, 21,7 ммоль, 1,2 экв.). Реакционную смесь перемешивали в течение 18 часов, а затем концентрировали в вакууме. Остаток переносили в этилацетат (300 мл) и промывали 1 М водным раствором соляной кислоты (2×100 мл) и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток переносили в дихлорметан (250 мл) и фильтровали. Фильтрат концентрировали в вакууме на диоксиде кремния и очищали посредством хроматографии на силикагеле (градиент: от 0% до 50% ацетона в гептане) с получением #72 (11,61 г, 81%) в виде светло-желтого твердого вещества. ЖХ-МС: m/z 797,6 [M+H+], 819,6 [M+Na+], время удерживания составляет 1,06 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 3.26 и 3.28 (2 s, всего 3H), 3.18 и 3.20 (2 s, всего 3H), 2.95 и 3.10 (2 br s, всего 3H), 1.01-1.09 (m, 3H), 0.67-0.78 (m, 3H).
Стадия 3. Синтез N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#73). К #72 (5,16 г, 6,47 ммоль, 1 экв.) в тетрагидрофуране (10 мл, 0,65 М) добавляли диэтиламин (10 мл). Через 2 часа реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) с получением #73 (2414 мг, 65%). ЖХ-МС: m/z 576,5 [M+H+], время удерживания составляет 0,64 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 7.80-7.88 и 7.99-8.10 (2 m, всего 1H), 7.14-7.31 (m, 5H), 3.17 и 3.18 (2 s, всего 3H), 2.87 и 3.03 (2 br s, всего 3H), 1.02-1.08 (m, 3H).
Стадия 4. Синтез N2-({3-[(трет-бутоксикарбонил)амино]оксетан-3-ил}карбонил)-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#74). К #73 (100 мг, 0,174 ммоль, 1 экв.) в дихлорметане (4 мл, 0,04) и N,N-диметилформамиде (0,5 мл) добавляли #71 (45,2 мг, 0,208 ммоль, 1,2 экв.), а затем диизопропилэтиламин (92 мкл, 0,521 ммоль, 3 экв.) и HATU (102 мг, 0,260 ммоль, 1,5 экв.). Через 16 часов реакционную смесь концентрировали в вакууме и остаток переносили в этилацетат (6 мл) и промывали 1 М водным раствором соляной кислоты (2×2 мл) и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный материал очищали посредством хроматографии с обращенной фазой (Метод C) с получением #74 (140 мг), которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС: m/z 774,7 [M+H+], 796,6 [M+Na+], время удерживания составляет 0,91 минуты.
Стадия 5. Синтез соли уксусной кислоты и N2-[(3-аминооксетан-3-ил)карбонил]-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#75). К #74 (140 мг, не более 0,181 ммоль, 1 экв.) в дихлорметане (3 мл, 0,06 М) добавляли трифторуксусную кислоту (1 мл). Через 1 час реакционную смесь концентрировали в вакууме и остаток переносили в этилацетат (6 мл) и промывали насыщенным водным раствором бикарбоната натрия (2 мл) и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Половину неочищенного материала очищали посредством хроматографии с обращенной фазой (Метод B) с получением #75 (16 мг, 26%, за две стадии). ЖХ-МС: m/z 674,6 [M+H+], время удерживания составляет 0,68 минуты; ВЭЖХ (Протокол A при 45°C): m/z 674,5 [M+H+], время удерживания составляет 7,128 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 7.80-7.87 и 8.02-8.07 (2 m, всего 2H), 7.23-7.30 (m, 2H), 7.14-7.22 (m, 3H), 4.28-4.33 (m, 2H), 3.96-4.04 (br m, 1H), 3.17 и 3.19 (2 s, всего 3H), 2.96 и 3.10 (2 br s, всего 3H), [1.04 (d, J=7,0 Гц) и 1.07 (d, J=6,6 Гц), всего 3H].
Получение N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#79) и соли трифторуксусной кислоты и N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-тиоксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#80)

Стадия 1. Синтез метил-N-{(2R,3R)-3-[(2S)-1-[-{(3R,4S,5S)-4-[{N-[(9H-флуорен-9-илметокси)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропантиоил}-L-фенилаланината (#76). Согласно общей методике D из #@5 (260 мг, 0,648 ммоль, 1 экв.), #39 (340 мг, не более 0,629 ммоль, 1 экв.), дихлорметана (10 мл, 0,065 М), HATU (296 мг, 0,778 ммоль, 1,2 экв.) и диизопропилэтиламина (339 мкл, 1,94 ммоль, 3 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 40% ацетона в гептане) с получением #76 (466 мг, 83% за две стадии) в виде твердого вещества. ЖХ-МС: m/z 871,5 [M+H+], 893,5 [M+Na+], время удерживания составляет 1,10 минуты; ВЭЖХ (Протокол C): время удерживания составляет 9,249 минуты (чистота более 99%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.19 (br d, J=7,4 Гц) и 10.49 (br d, J=7,8 Гц), всего 1H], 7.89 (br d, J=7,4 Гц, 2H), 7.68-7.75 (m, 2H), 7.54-7.60 (m, 1H), 7.41 (br dd, J=7,4, 7,4 Гц, 2H), 7.28-7.36 (m, 2H), 7.15-7.28 (m, 5H), [5.20 (ddd, J=10,9, 7,3, 4,4 Гц) и 5.34-5.43 (m), всего 1H], 3.65 и 3.69 (2 s, всего 3H), 3.24 и 3.25 (2 s, всего 3H), 3.17 (br s, 3H), 2.93 и 2.98 (2 br s, всего 3H), [1.15 (d, J=6,6 Гц) и 1.18 (d, J=6,6 Гц), всего 3H].
Стадия 2. Синтез метил-N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропантиоил}-L-фенилаланината (#77). Согласно общей методике A из #76 (460 мг, 0,528 ммоль, 1 экв.), тетрагидрофурана (8 мл, 0,07 М) и диэтиламина (8 мл) синтезировали #77 (399 мг), которое использовали на следующей стадии без дополнительной очистки; ЖХ-МС m/z 649,5 [M+H+], время удерживания составляет 0,73 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы продукта: δ [10.20 (d, J=7,4 Гц) и 10.49 (d, J=7,4 Гц), всего 1H], 7.15-7.28 (m, 5H), [5.20 (ddd, J=10,9, 7,2, 4,5 Гц) и 5.34-5.42 (m), всего 1H], 3.65 и 3.68 (2 s, всего 3H), 3.24 и 3.25 (2 s, всего 3H), 3.15 и 3.15 (2 s, всего 3H), 2.83 и 2.88 (2 br s, всего 3H), [1.15 (d, J=6,6 Гц) и 1.18 (d, J=6,6 Гц), всего 3H].
Стадия 3. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#78). Согласно общей методике D из #77 (399 мг, не более 0,52 ммоль, 1 экв.), N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланина (213 мг, 0,628 ммоль, 1,2 экв.), дихлорметана (5 мл, 0,1 M), HATU (239 мг, 0,628 ммоль, 1,2 экв.) и диизопропилэтиламина (282 мкл, 1,62 ммоль, 3,1 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 50% ацетона в гептане), получая #78 (231 мг, 46% за две стадии). ЖХ-МС: m/z 970,7 [M+H+], 992,6 [M+Na+], время удерживания составляет 1,11 минуты; ВЭЖХ (Протокол C): время удерживания составляет 9,260 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.19 (d, J=7,4 Гц) и 10.47 (d, J=7,8 Гц), всего 1H], 7.89 (d, J=7,4 Гц, 2H), 7.61-7.67 (m, 2H), 7.41 (br dd, J=7,4, 7,4 Гц, 2H), 7.14-7.36 (m, 8H), [5.20 (ddd, J=11, 7, 5 Гц) и 5.38 (ddd, J=11, 8, 4 Гц), всего 1H], [4.41 (dd, J=8,6, 8,4 Гц) и 4.46 (dd, J=8,2, 8,2 Гц), всего 1H], 3.65 и 3.68 (2 s, всего 3H), 3.23 и 3.24 (2 s, всего 3H), 3.13 (br s, 3H), 2.88 и 2.93 (2 br s, всего 3H), 2.84 и 2.85 (2 s, всего 3H), 1.31 и 1.32 (2 s, всего 3H), [1.15 (d, J=6,6 Гц) и 1.18 (d, J=6,4 Гц), всего 3H].
Стадия 4A. Синтез N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#79). Согласно общей методике A из #78 (223 мг, 0,230 ммоль, 1 экв.), дихлорметана (6 мл, 0,04 M) и диэтиламина (6 мл) синтезировали #79 (146 мг, 85%) в виде белого твердого вещества после хроматографии на силикагеле (градиент: от 0% до 5% метанола в гептане, затем от 0% до 10% метанола в дихлорметане). ВЭЖХ (Протокол A при 45°C): 749,4 [M+H+], время удерживания составляет 7,315 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.20 (d, J=7,6 Гц) и 10.50 (d, J=8,0 Гц), всего 1H], 7.79-7.88 (m, 1H), 7.15-7.29 (m, 5H), [5.20 (ddd, J=11, 7, 4 Гц) и 5.38 (ddd, J=11, 8, 4 Гц), всего 1H], [4.50 (dd, J=8,8, 8,6 Гц) и 4.56 (dd, J=9, 8 Гц), всего 1H], 3.65 и 3.69 (2 s, всего 3H), 3.24 и 3.25 (2 s, всего 3H), 3.16 (brs, 3H), 2.93 и 2.97 (2 brs, всего 3H), 2.10 и 2.11 (2 s, всего 3H).
Стадия 4B. Синтез соли трифторуксусной кислоты и N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-тиоксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#80). К раствору #78 (170 мг, 0,175 ммоль, 1 экв.) в тетрагидрофуране (3 мл, 0,04 M) добавляли раствор гидроксида лития (12,6 мг, 0,525 ммоль, 3 экв.) в воде (1,5 мл). После перемешивания в течение ночи растворитель удаляли в вакууме. Остаток три раза подвергали азеотропной перегонке с гептаном. Затем остаток разбавляли диметилсульфоксидом (2,2 мл) и очищали посредством хроматографии с обращенной фазой (Метод С) с получением #80 (74 мг, 58%) в виде твердого вещества. ЖХ-МС: m/z 734,6 [M+H+], время удерживания составляет 0,69 минуты; ВЭЖХ (Протокол A при 45°C): 734,4 [M+H+], время удерживания составляет 6,903 минуты (чистота более 96%);1H ЯМР (400 МГц, DMSO-d6, считали, что является смесью ротамеров, характеристические сигналы: δ 12.9 и 13.1 (2 v br s, всего 1H), [10.12 (d, J=7,4 Гц) и 10.46 (d, J=7,8 Гц), всего 1H], 8.77-8.89 (br m, 2H), [8.47 (d, J=8,6 Гц) и 8.51 (d, J=8,6 Гц), всего 1H], 7.21-7.29 (m, 4H), 7.14-7.21 (m, 1H), [5.16-5.23 (m) и 5.38 (ddd, J=11,3, 8,2, 3,9 Гц), всего 1H], [4.51 (dd, J=9,0, 9,0 Гц) и 4.57 (dd, J=9,4, 8,6 Гц), всего 1H], 3.24 и 3.24 (2 s, всего 3H), 3.18 и 3.19 (2 s, всего 3H), 2.96 и 3.00 (2 br s, всего 3H), 1.51 и 1.53 (2 s, всего 3H), 1.40 и 1.42 (2 s, всего 3H), 1.14-1.19 (m, 3H), 0.74-0.81 (m, 3H).
Получение N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#84)
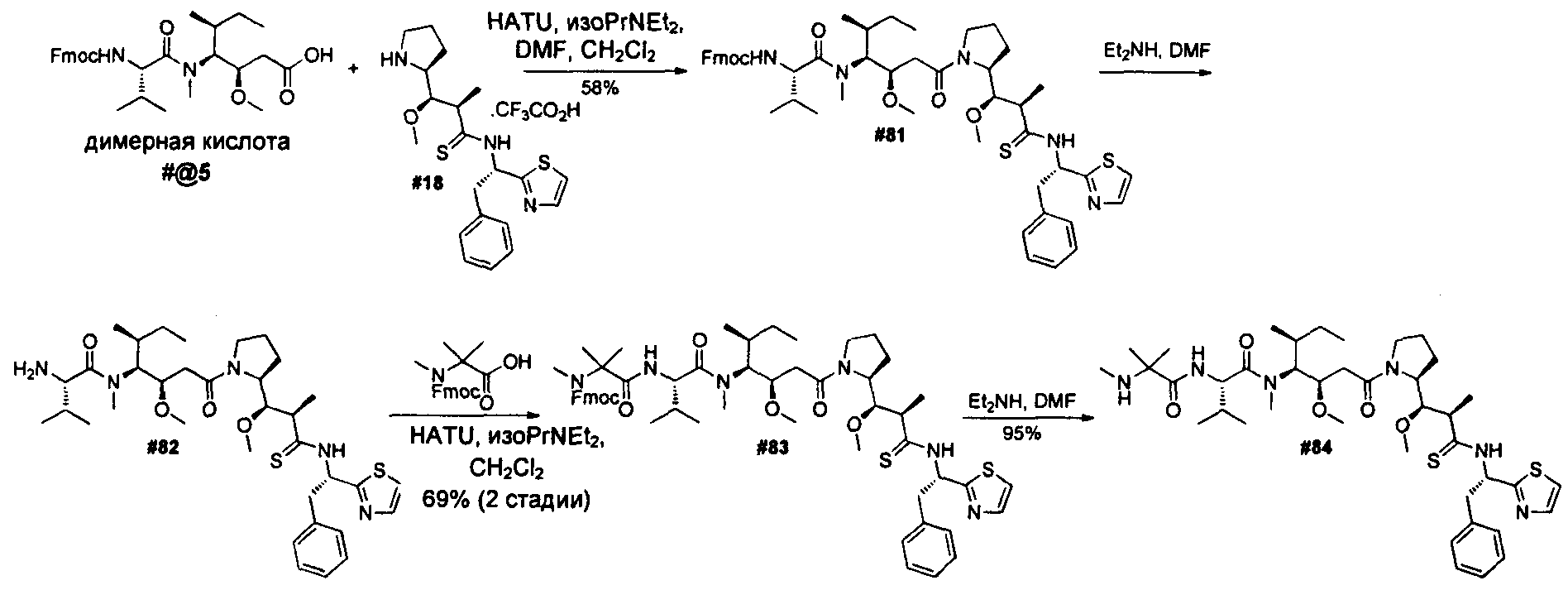
Стадия 1. Синтез N2-[(9H-флуорен-9-илметокси)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#81). Согласно общей методике D из #@5 (620 мг, 1,18 ммоль, 1 экв.), дихлорметана (10 мл, 0,1 М), амина #18 (604 мг, 1,42 ммоль, 1,2 экв.), диизопропилэтиламина (618 мкл, 3,54 ммоль, 3 экв.) и HATU (539 мг, 1,42 ммоль, 1,2 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 30% ацетона в гептане) с получением #81 (737 мг, 58%). ВЭЖХ (Протокол C): время удерживания составляет 9,235 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.54 (br d, J=8 Гц) и 10.81 (br d, J=8 Гц), всего 1H], 7.89 (d, J=7,6 Гц, 2H), [7.80 (d, J=3,3 Гц) и 7.83 (d, J=3,1 Гц), всего 1H], 7.70-7.75 (m, 2H), [7.64 (d, J=3,1 Гц) и 7.68 (d, J=3,3 Гц), всего 1H], 7.55-7.60 (m, 1H), 7.38-7.44 (m, 2H), 7.13-7.35 (m, 7H), [6.31 (ddd, J=11, 8, 4,5 Гц) и 6.40-6.48 (m), всего 1H], 3.23 и 3.24 (2 s, всего 3H), 3.17 и 3.22 (2 s, всего 3H), 2.94 и 3.01 (2 br s, всего 3H), [1.14 (d, J=6,4 Гц) и 1.17 (d, J=6,2 Гц), всего 3H].
Стадия 2. Синтез N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#82). Согласно общей методике A из #81 (733 мг, 0,818 ммоль, 1 экв.) в дихлорметане (7 мл, 0,1 М) и диэтиламине (7 мл) синтезировали #82 (670 мг), которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС: m/z674,5 [M+H+], время удерживания составляет 1,29 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы продукта: δ [10.55 (br d, J=8 Гц) и 10.84 (br d, J=8 Гц), всего 1H], [7.64 (d, J=3,1 Гц) и 7.69 (d, J=3,3 Гц), всего 1H], 7.13-7.33 (m, 5H), 6.27-6.35 и 6.38-6.47 (2 m, всего 1H), 3.23 и 3.25 (2 s, всего 3H), 3.15 и 3.19 (2 s, всего 3H), 2.84 и 2.91 (2 br s, всего 3H).
Стадия 3. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#83). Согласно общей методике D из #82 (670 мг, не более 0,818 ммоль, 1 экв.), дихлорметана (5 мл, 0,16 M), N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланина (304 мг, 0,896 ммоль, 1,1 экв.), HATU (372 мг, 0,978 ммоль, 1,2 экв.) и диизопропилэтиламина (440 мкл, 2,53 ммоль, 3,1 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 30% ацетона в гептане) с получением #83 (556 мг, 69% за две стадии). ЖХ-МС: т/г 994,7 [M+H+], время удерживания составляет 0,69 минуты; ВЭЖХ (Протокол C): время удерживания составляет 9,333 минуты (чистота более 98%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.53 (br d, J=8 Гц) и 10.80 (br d, J=8 Гц), всего 1H], 7.86-7.91 (m, 2H), [7.80 (d, J=3,3 Гц) и 7.82 (d, J=3,2 Гц), всего 1H], [7.64 (d, J=3,2 Гц) и 7.68 (d, J=3,2 Гц), всего 1H], 7.62-7.66 (m, 2H), 7.38-7.44 (m, 2H), 7.28-7.36 (m, 5H), 7.19-7.26 (m, 2H), 7.12-7.17 (m, 1H), [6.31 (ddd, J=11, 8, 4,5 Гц) и 6.44 (ddd, J=11, 8,5, 4,5 Гц), всего 1H], [4.42 (dd, J=9, 8 Гц) и 4.48 (dd, J=8, 8 Гц), всего 1H], 3.22 и 3.24 (2 s, всего 3H), 3.13 и 3.17 (2 s, всего 3H), 2.89 и 2.97 (2 br s, всего 3H), 2.84 и 2.85 (2 s, всего 3H), [1.13 (d, J=6,4 Гц) и 1.16 (d, J=6,4 Гц), всего 3H].
Стадия 4. Синтез N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-3-тиоксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#84). Согласно общей методике A из #83 (552 мг, 0,555 ммоль, 1 экв.) в дихлорметане (10 мл, 0,05 M) и диэтиламине (10 мл) синтезировали неочищенный целевой материал, который разбавляли метанолом, концентрировали в вакууме на диоксиде кремния и очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) с получением #84 (406 мг, 95%) в виде белого твердого вещества. ЖХ-МС: m/z 772,8 [M+H+], время удерживания составляет 1,35 минуты; ВЭЖХ (Протокол A): 774,4 [M+H+], время удерживания составляет 7,390 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [10.54 (br d, J=8 Гц) и 10.81 (br d, J=8 Гц), всего 1H], 7.78-7.84 (m, 2H), [7.65 (d, J=3,1 Гц) и 7.69 (d, J=3,3 Гц), всего 1H], 7.29-7.34 (m, 2H), 7.20-7.28 (m, 2H), 7.14-7.19 (m, 1H), 6.27-6.35 и 6.40-6.48 (2 m, всего 1H), [4.51 (dd, J=9, 8 Гц) и 4.57 (dd, J=9, 8 Гц), всего 1H], 3.24 и 3.25 (2 s, всего 3H), 3.16 и 3.21 (2 s, всего 3H), 2.94 и 3.00 (2 br s, всего 3H), 2.09 и 2.10 (2 s, всего 3H), 1.08 и 1.09 (2 s, всего 3H), 0.73-0.80 (m, 3H).
N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид {#83)
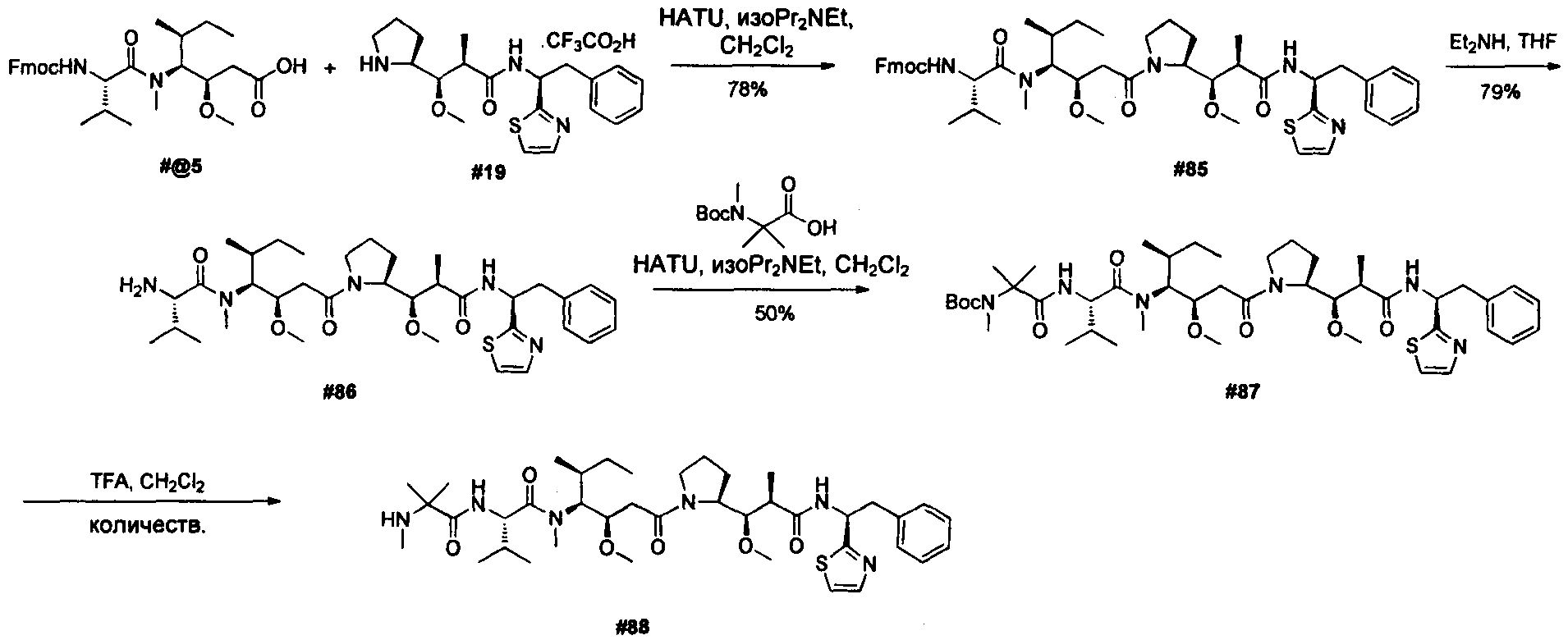
Стадия 1. Синтез N2-[(9H-флуорен-9-илметокси)карбонил]-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#85). К смеси #@5 (5,48 г, 10,4 ммоль, 1 экв.) и #19 (3,90 г, 10,4 ммоль, 1 экв.) в дихлорметане (50 мл, 0,2 M) добавляли диизопропилэтиламин (5,51 мл, 31,3 ммоль, 3 экв.), а затем HATU (4,91 г, 12,5 ммоль, 1,2 экв.). После перемешивания в течение ночи реакционную смесь концентрировали в вакууме. Остаток переносили в этилацетат (100 мл) и промывали 1 М водным раствором соляной кислоты (2×30 мл) и рассолом (30 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток переносили в дихлорметан и фильтровали; фильтрат очищали посредством хроматографии на силикагеле (градиент; от 0% до 50% ацетона в гептане) с получением #85 (7,20 г, 78%) в виде твердого вещества. ЖХ-МС: m/z 880,6 [M+H+], время удерживания составляет 1,07 минуты.
Стадия 2. Синтез N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#86). Согласно общей методике A из #85 (5,00 г, 5,68 ммоль, 1 экв.) в тетрагидрофуране (10 мл, 0,56 М) и диэтиламине (3 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) с получением #86 (2,952 г, 79%) в виде твердого вещества. ЖХ-МС: m/z 658,5 [M+H+], 680,5 [M+Na+], время удерживания составляет 0,66 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.64 (br d, J=8,4 Гц) и 8.90 (br d, J=8,8 Гц), всего 1H], [7.77 (d, J=3,3 Гц) и 7.80 (d, J=3,3 Гц), всего 1H], [7.63 (d, J=3,3 Гц) и 7.66 (d, J=3,3 Гц), всего 1H], 7.12-7.31 (m, 5H), [5.39 (ddd, J=11,2, 8,4, 4,2 Гц) и 5.54 (ddd. J=11,9, 8,9, 4,0 Гц), всего 1H], 3.15, 3.19, 3.20 и 3.26 (4 s, всего 6H), 2.86 и 2.98 (2 br s, всего 3H), [1.06 (d, J=6,6 Гц) и 1.11 (d, J=6,6 Гц), всего 3H].
Стадия 3. Синтез N-(трет-бутоксикарбонил)-N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#87). К смеси #86 (80,3 мг, 0,122 ммоль, 1 экв.) в дихлорметане (4 мл, 0,03 М) добавляли N-(трет-бутоксикарбонил)-N,2-диметилаланин (29,1 мг, 0,134 ммоль, 1,1 экв.), а затем диизопропилэтиламин (64 мкл, 0,365 ммоль, 3 экв.) и HATU (71,7 мг, 0,183 ммоль, 1,5 экв.) После перемешивания в течение ночи реакционную смесь концентрировали в вакууме. Остаток переносили в этилацетат (6 мл) и промывали 1 М водным раствором соляной кислоты (2×2 мл) и рассолом. Органический растворитель сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток переносили в дихлорметан и фильтровали; фильтрат концентрировали в вакууме на диоксиде кремния и очищали посредством хроматографии на силикагеле (градиент: от 0% до 50% ацетона в гептане) с получением #87 (58 мг, 50%) в виде белого твердого вещества. ЖХ-МС: m/z 857,7 [M+H+], 879,7 [M+Na+], время удерживания составляет 0,99 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.64 (br d, J=8 Гц) и 8.87 (br d, J=9 Гц), всего 1H], [7.77 (d, J=3,3 Гц) и 7.80 (d, J=3,3 Гц), всего 1H], [7.63 (d, J=3,2 Гц) и 7.66 (d, J=3,2 Гц), всего 1H], 7.13-7.31 (m, 5H), [6.95 (br d, J=8 Гц) и 7.06 (br d, J=8 Гц), всего 1H], 5.35-5.42 и 5.51-5.58 (2 m, всего 1H), 3.15, 3.19, 3.20 и 3.26 (4 s, всего 6H), 2.94 и 3.03 (2 br s, всего 3H), 2.83 и 2.84 (2 s, всего 3H), [1.05 (d, J=6,7 Гц) и 1.11 (d, J=6,7 Гц), всего 3H].
Стадия 4. Синтез N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фен ил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#88). К смеси #87 (58 мг, 0,068 ммоль, 1 экв.) в дихлорметане (8 мл) добавляли трифторуксусную кислоту (2 мл). После перемешивания в течение ночи реакционную смесь концентрировали в вакууме. Остаток переносили в этилацетат (10 мл), промывали насыщенным водным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением #88 (52 мг, количественный выход). ЖХ-МС 757,6 [M+H+], время удерживания составляет 0,69 минуты;1H ЯМР (400 МГц, DMSO-d6, считали, что является смесью ротамеров, характеристические сигналы: δ [8.64 (br d, J=8,6 Гц) и 8.87 (br d, J=8,6 Гц), всего 1H], 7.80-7.85 (m, 1H), [7.77 (d, J=3,3 Гц) и 7.80 (d, J=3,1 Гц), всего 1H], [7.63 (d, J=3,1 Гц) и 7.66 (d, J=3,3 Гц), всего 1H], 7.20-7.31 (m, 4H), 7.13-7.19 (m, 1H), [5.39 (ddd, J=11, 8,5, 4 Гц) и 5.49-5.56 (m), всего 1H], [4.51 (dd, J=9, 8 Гц) и 4.61 (dd, J=9, 8 Гц), всего 1H], 3.16, 3.20, 3.21 и 3.25 (4 s, всего 6H), 2.94 и 3.03 (2 br s, всего 3H), 2.10 и 2.10 (2 s, всего 3H), 1.16 (br s, 3H), 1.04-1.12 (m, 6H), 0.72-0.80 (m, 3H).
Получение соли трифторуксусной кислоты и N2-(3-амино-2,2-диметилпропаноил)-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#95)

Стадия 1. Синтез 3-{[(9H-флуорен-9-илметокси)карбонил]амино}-2,2-диметилпропановой кислоты (#93). К гидрохлоридной соли 3-амино-2,2-диметилпропановой кислоты (250 мг, 1,63 ммоль, 1 экв.) в дихлорметане (4 мл, 0,4 М) добавляли диизопропилэтиламин (859 мкл, 4,88 ммоль, 3 экв.), а затем (9H-флуорен-9-илметокси)карбонилхлорид (473 мг, 1,79 ммоль, 1,1 экв.). Реакционную смесь перемешивали в течение 18 часов, а затем концентрировали в вакууме. Остаток переносили в этилацетат (3 мл) и промывали 1 М водным раствором соляной кислоты (2×1 мл) и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и очищали посредством хроматографии на силикагеле (градиент: от 0% до 100% этилацетата в гептане) с получением #93 (250 мг, 45%) в виде масла.1H ЯМР (400 МГц, DMSO-d6) δ 12.22 (s, 1H), 7.89 (d, J=7,4 Гц, 2H), 7.72 (d, J=7,4 Гц, 2H), 7.38-7.44 (m, 2H), 7.27-7.35 (m, 3H), 4.18-4.30 (m, 3H), 3.16 (d, J=6,2 Гц, 2H), 1.05 (s, 6H).
Стадия 2. Синтез N2-(3-{[(9H-флуорен-9-илметокси)карбонил]амино}-2,2-диметилпропаноил)-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#94). К #86 (100 мг, 0,152 ммоль, 1 экв.) в дихлорметане (4 мл, 0,038 М) и N,N-диметилформамиде (0,5 мл) добавляли #93 (51,6 мг, 0,152 ммоль, 1 экв.), а затем диизопропилэтиламин (80,0 мкл, 0,457 ммоль, 3 экв.) и HATU (89,8 мг, 0,229 ммоль, 1,5 экв.). Реакционную смесь перемешивали в течение 18 часов, а затем концентрировали в вакууме. Остаток переносили в этилацетат (6 мл) и промывали 1 М водным раствором соляной кислоты (2×2 мл) и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток переносили в дихлорметан (250 мл) и фильтровали; фильтрат концентрировали в вакууме на диоксиде кремния и очищали посредством хроматографии на силикагеле (градиент: от 0% до 50% ацетона в гептане) с получением #94 (90 мг, 60%) в виде белого твердого вещества. ЖХ-МС: m/z 979,8 [M+H+], 1002,7 [M+Na+], время удерживания составляет 1,15 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы продукта: δ [8.64 (br d, J=8,6 Гц) и 8.86 (br d, J=8,6 Гц), всего 1H], 7.86-7.91 (m, 2H), [7.77 (d, J=3,3 Гц) и 7.79 (d, J=3,3 Гц), всего 1H], 7.67-7.73 (m, 2H), [7.63 (d, J=3.3 Гц) и 7.65 (d, J=3,3 Гц), всего 1H], 6.87-6.95 (m, 1H), [5.39 (ddd, J=11, 8, 4 Гц) и 5.52 (ddd, J=11,5, 9, 4 Гц), всего 1H], [4.44 (dd, J=8,4, 8,4 Гц) и 4.55 (dd, J=8,4, 8,4 Гц), всего 1H], 3.16, 3.20, 3.21 и 3.25 (4 s, всего 6H), 2.96 и 3.06 (2 br s, всего 3H), 0.69-0.77 (m, 3H).
Стадия 3. Синтез соли трифторуксусной кислоты и N2-(3-амино-2,2-диметилпропаноил)-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#95). К #94 (86 мг, 0,088 ммоль, 1 экв.) в тетрагидрофуране (2 мл, 0,04 M) добавляли диэтиламин (10 мл). После перемешивания в течение ночи реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии с обращенной фазой (Метод C) с получением #95 (55 мг, 72%). ЖХ-МС: m/z 757,5 [M+H+], время удерживания составляет 0,74 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [8.66 (br d, J=8 Гц) и 8.92 (br d, J=9 Гц), всего 1H], [7.91 (br d, J=8 Гц) и 7.97 (br d, J=9 Гц), всего 1H], [7.78 (d, J=3,3 Гц) и 7.81 (d, J=3,1 Гц), всего 1H], 7.65-7.74 (br m, 3H), [7.63 (d, J=3,3 Гц) и 7.67 (d, J=3,3 Гц), всего 1H], 7.12-7.31 (m, 5H), [5.35-5.42 (m) и 5.45-5.52 (m), всего 1H], [4.44 (dd, J=9, 9 Гц) и 4.55 (dd, J=9, 9 Гц), всего 1H], 3.17, 3.20, 3.22 и 3.25 (4 s, всего 6H), 2.96 и 3.05 (2 br s, всего 3H), 1.25 и 1.25 (2 s, всего 3H), 1.14 и 1.15 (2 s, всего 3H), [1.06 (d, J=6,6 Гц) и 1.10 (d, J=6,4 Гц), всего 3H], 0.72-0.80 (m, 3H).
Получение соли трифторуксусной кислоты и N2-(3-амино-2,2-диметилпропаноил)-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#97)

Стадия 1. Синтез N2-(3-{[(9H-флуорен-9-илметокси)карбонил]амино}-2,2-диметилпропаноил)-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#96). К #73 (100 мг, 0,174 ммоль, 1 экв.) в дихлорметане (4 мл, 0,04 М) и N,N-диметилформамиде (0,5 мл) добавляли #93 (59,1 мг, 0,174 ммоль, 1 экв.), а затем диизопропилэтиламин (92 мкл, 0,52 ммоль, 3 экв.) и HATU (102 мг, 0,260 ммоль, 1,5 экв.). Реакционную смесь перемешивали в течение 18 часов, а затем концентрировали в вакууме. Остаток переносили в этилацетат (6 мл) и промывали 1 М водным раствором соляной кислоты (2×2 мл) и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После очистки посредством хроматографии на силикагеле (градиент: от 0% до 50% ацетона в гептане) получали #96 (102 мг, 65%) в виде белого твердого вещества. ЖХ-МС: m/z 896,7 [M+H+], 918,8 [M+Na+], время удерживания составляет 1,14 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы продукта: δ 7.88 (d, J=7,4 Гц, 2H), [7.83 (br dd, J=6, 5 Гц) и 8.03 (br dd, J=6, 5 Гц), всего 1H], 7.67-7.73 (m, 2H), 7.36-7.48 (m, 3H), 7.22-7.35 (m, 4H), 7.13-7.21 (m, 3H), 6.86-6.96 (m, 1H), [4.44 (dd, J=8,6, 8,6 Гц) и 4.50 (dd, J=8,6, 8,6 Гц), всего 1H], 3.18, 3.19, 3.26 и 3.29 (4 s, всего 6H), 2.96 и 3.11 (2 br s, всего 3H), 0.70-0.77 (m, 3H).
Стадия 2. Синтез соли трифторуксусной кислоты и N2-(3-амино-2,2-диметилпропаноил)-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#97). К #96 (98 мг, 0,11 ммоль, 1 экв.) в тетрагидрофуране (2 мл, 0,04 М) добавляли диэтиламин (0,5 мл). После перемешивания в течение ночи реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии с обращенной фазой (Метод C) с получением #97 (58 мг, 68%). ЖХ-МС: m/z 674,4 [M+H+], 696,4 [M+Na+], время удерживания составляет 0,74 минуты; ВЭЖХ (Протокол A): 674,5 [M+H+], время удерживания составляет 7,072 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [7.92 (br d, J=8 Гц) и 7.97 (br d, J=8 Гц), всего 1H], [7.86 (br dd, J=6, 5 Гц) и 8.07 (br dd, J=6, 5 Гц), всего 1H], 7.64-7.74 (br m, 3H), 7.15-7.29 (m, 5H), [4.44 (dd, J=9, 9 Гц) и 4.50 (dd, J=9, 9 Гц), всего 1H], 3.26 и 3.29 (2 s, всего 3H), 3.18 и 3.20 (2 s, всего 3H), 2.96 и 3.10 (2 br s, всего 3H), 1.24 и 1.25 (2 s, всего 3H), 1.14 и 1.16 (2 s, всего 3H), 1.02-1.07 (m, 3H), 0.73-0.80 (m, 3H).
Получение соли трифторуксусной кислоты и 2-метил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида {#98)

К смеси 1-(трет-бутоксикарбонил)-2-метил-L-пролина (65,1 мг, 0,284 ммоль, 1,1 экв.) и #86 (170 мг, 0,258 ммоль, 1 экв.) в дихлорметане (5 мл, 0,03 M) добавляли HATU (0,108 мг, 0,284 ммоль, 1,1 экв.), а затем диизопропилэтиламин (139 мкл, 0,800 ммоль, 3,1 экв.). После перемешивания в течение ночи реакционную смесь охлаждали до 0°C, добавляли дихлорметан (3 мл) с последующим медленным добавлением трифторуксусной кислоты (2 мл). Реакционную смесь перемешивали при 0° в течение 5 минут, оставляли нагреваться до комнатной температуры, а затем перемешивали при комнатной температуре в течение 30 минут, после чего концентрировали в вакууме. Остаток подвергали азеотропной перегонке два раза с гептаном, разбавляли небольшим количеством дихлорметана и метанола, после чего концентрировали в вакууме на диоксиде кремния. Остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане), а затем посредством хроматографии с обращенной фазой (Метод C) с получением #98 (128 мг, 56%) в виде белого твердого вещества. ЖХ-МС: m/z 769,4 [M+H+], время удерживания составляет 1,28 минуты; ВЭЖХ (Протокол A при 45°C) m/z 769,4 [M+H+], время удерживания составляет 7,146 минуты (чистота более 98%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 9.03-9.15 (br m, 1H), 8.77-8.86 (br m, 1H), 8.69-8.76 (m, 1H), [8.66 (d, J=8,2 Гц) и 8.92 (d, J=8,6 Гц), всего 1H], [7.78 (d, J=3,1 Гц) и 7.80 (d, J=3,5 Гц), всего 1H], [7.63 (d, J=3,1 Гц) и 7.67 (d, J=3,1 Гц), всего 1H], 7.12-7.31 (m, 5H), [5.38 (ddd, J=11, 8, 4 Гц) и 5.47 (ddd, J=11, 9, 4 Гц), всего 1H], [4.46 (dd, J=9,4, 9,0 Гц) и 4.55 (dd, J=9,0, 8,6 Гц), всего 1H], 3.17, 3.20, 3.22 и 3.25 (4 s, всего 6H), 2.98 и 3.04 (2 br s, всего 3H), [1.06 (d, J=7,0 Гц) и 1.09 (d, J=6,6 Гц), всего 3H], 0.73-0.80 (m, 3H).
Получение гидрохлоридной соли метил-амино(бицикло[4.2.0]окта-1,3,5-триен-7-ил)ацетата (#102)

Стадия 1. Синтез этил-(ацетиламино)(бицикло[4.2.0]окта-1,3,5-триен-7-ил)цианоацетата (#99). Натрий (464 мг, 20,2 ммоль, 1,2 экв.) оставляли взаимодействовать с абсолютным этанолом (40 мл, 0,42 M); к полученной смеси добавляли этил-2-(ацетиламино)-2-цианоацетат (3,44 г, 20,2 ммоль, 1,2 экв.). Через 20 минут при 60°C, добавляли 7-бромбицикло[4.2.0]окта-1,3,5-триен (3,092 г, 16,89 ммоль, 1 экв.) и реакционную смесь кипятили с обратным холодильником в течение ночи, затем фильтровали и концентрировали в вакууме. Остаток разбавляли водой и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением темного масла, которое очищали посредством хроматографии на силикагеле (градиент: от 0% до 50% этилацетата в гептане) с получением #99 (4,38 г) в виде желтой смолы. ЖХ-МС: m/z 273,2 [M+H+], время удерживания составляет 2,36 минуты.
Стадия 2. Синтез (ацетиламино)(бицикло[4.2.0]окта-1,3,5-триен-7-ил)уксусной кислоты (#100). К смеси #99 (4,38 мг, менее 16,1 ммоль, 1 экв.) в метаноле (30 мл, 0,53 М) добавляли 1 н. водный раствор гидроксида натрия (38 мл, 38 ммоль, 2,4 экв.). Реакционную смесь кипятили с обратным холодильником в течение ночи, затем концентрировали в вакууме, разбавляли водой (40 мл) и подкисляли 1 н. водным раствором соляной кислоты (40 мл). Водный слой экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Полученное масло очищали посредством хроматографии на силикагеле (растворитель A: дихлорметан; растворитель B: 20% метанола в дихлорметане, содержащем 0,02% трифторуксусной кислоты; градиент: от 0% до 40% В), затем посредством сверхкритической флюидной хроматографии (колонка: Chiralpak AD-H, 250×21 мм; элюент: 85:15 диоксид углерода/метанол; скорость потока: 65 г/мин; детектирование: 210 нм; прибор: система Berger minigram preparative SFC system). Выделяли пик, элюирующийся вторым, получая #100 (600 мг, 17% за две стадии) в виде одиночного энантиомера (время удерживания составляет 3,37 минуты, чистота более 99%). ЖХ-МС: m/z 220,3 [M+H+], время удерживания составляет 2,10 минуты;1H ЯМР (400 МГц, CD3OD) δ 7.14-7.24 (m, 2H), 7.03-7.09 (m, 2H), 4.59 (d, J=8,6 Гц, 1H), 3.87 (add, J=8,5, 5,3, 2,4 Гц, 1H), 3.35 (dd, J=14,5, 5,4 Гц, 1H, предполагалось; частично заслонен пиком растворителя), 3.10 (dd, J=14,4, 2,4 Гц, 1H), 2.00 (s, 3H). Оптическое вращение: (c 0,67, метанол).
Стадия 3. Синтез гидрохлоридной соли амино(бицикло[4.2.0]окта-1,3,5-триен-7-ил)уксусной кислоты (#101). Смесь #100 (200 мг, 0,912 ммоль, 1 экв.) и 6 н. водной соляной кислоты (12,3 мл, 73,8 ммоль, 81 экв.) кипятили с обратным холодильником в течение ночи. Реакционную смесь концентрировали в вакууме с получением одиночного энантиомера #101 (195 мг) в виде не совсем желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 4. Синтез гидрохлоридной соли метил-амино(бицикло[4.2.0]окта-1,3,5-триен-7-ил)ацетата (#102). К смеси #101 (195 мг, менее 0,913 ммоль, 1 экв.) в метаноле (20 мл, 0,04 М) добавляли тионилхлорид (0,666 мл, 9,13 ммоль, 10 экв.). Через два часа кипячения с обратным холодильником реакционную смесь концентрировали в вакууме с получением одиночного энантиомера #102 (175 мг, 84% за две стадии) в виде светлоокрашенного твердого вещества. ЖХ-МС: m/z 192,3 [M+H+], время удерживания составляет 0,80 минуты; ГХ-МС: m/z 192 [M+H+], время удерживания составляет 3,206 минуты;1H ЯМР (400 МГц, CD3OD) δ 7.24-7.33 (m, 2H), 7.11-7.18 (m, 2H), 4.40 (d, J=6,9 Гц, 1H), 3.99-4.05 (m, 1H), 3.78 (s, 3H), 3.46 (dd, J=14,8, 5,4 Гц, 1H), 3.23 (dd, J=14,8, 2,5 Гц, 1H).
Получение гидрохлоридной соли (2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропановой кислоты (#103)

К смеси #11 (4,09 г, 14,2 ммоль, 1 экв.) в циклопентил метиловом эфире (10 мл, 0,14 M) добавляли 4 н. раствор хлористого водорода в диоксане (37 мл, 100 ммоль, 7 экв.). Через три часа реакционную смесь концентрировали в вакууме и три раза подвергали азеотропной перегонке с гептаном с получением #103 (1000 мг, 31%) в виде смолы, которую использовали на следующей стадии без дополнительной очистки.1H ЯМР (400 МГц, DMSO-d6) δ 9.92-10.06 (br s, 1H), 8.66-8.80 (br s, 1H), 3.89 (dd, J=5,2, 4,9 Гц, 1H), 3.43-3.53 (m, 1H), 3.39 (s, 3H), 3.06-3.17 (m, 2H), 2.66 (qd, J=7,1, 4,6 Гц, 1H), 1.71-2.03 (m, 4H), 1.11 (d, J=7,1 Гц, 3H).
Получение соли трифторуксусной кислоты и 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[1-(бицикло[4.2.0]окта-1,3,5-триен-7-ил)-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#107) и соли трифторуксусной кислоты и 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[бицикло[4.2.0]окта-1,3,5-триен-7-ил(карбокси)метил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#108)

Стадия 1. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-3-метокси-5-метил-1-оксо-1-(пентафторфенокси)-гептан-4-ил]-N-метил-L-валинамида (#104). К #32 (4,00 г, 6,56 ммоль, 1 экв.) в дихлорметане (20 мл, 0,33 М) и пиридине (1,06 мл, 13,1 ммоль, 2 экв.) по каплям добавляли пентафторфенилтрифторацетат (2,25 мл, 13,1 ммоль, 2 экв.). Реакционную смесь перемешивали в течение одного часа.
Во вторую колбу, содержащую #32 (360 мг, 0,59 ммоль) в дихлорметане (0,6 мл, 1 М) и пиридине (0,095 мл, 1,2 ммоль, 2 экв.), по каплям добавляли пентафторфенилтрифторацетат (0,203 мл, 1,18 ммоль). Эту реакционную смесь перемешивали в течение 15 минут.
Две реакционные смеси объединяли, промывали дважды 1 н. водной соляной кислотой, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Полученное желтое масло растворяли в этилацетате, адсорбировали на силикагеле и очищали посредством хроматографии на силикагеле (градиент: от 0% до 40% этилацетата в гептане) с получением #104 (4,6 г, 83%) в виде белой пены, содержащий несколько примесей. ЖХ-МС: m/z 798,3 [M+Na+], время удерживания составляет 1,23 минуты.
Стадия 2. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#105). К смеси #104 (2,00 г, менее 2,58 ммоль, 1 экв.) в дихлорметане (6 мл, 0,4 М) добавляли раствор #103 (483 мг, 2,16 ммоль, 1 экв.) в дихлорметане (2 мл), а затем диизопропилэтиламин (1,35 мл, 7,73 ммоль, 3 экв.). Реакционную смесь перемешивали в течение 16 часов, затем адсорбировали на диоксиде кремния и очищали посредством хроматографии на силикагеле (градиент: от 0% до 20% метанола в дихлорметане) с получением #105 (1,67 г, 83%) в виде белой пены. Фракции, содержащие целевой продукт с примесями (0,571 г), собирали отдельно.
Указанные выше реакцию и очистку повторяли аналогичным образом, используя #104 (2,60 г, менее 3,35 ммоль, 1 экв.), #103 (750 мг, 3,35 ммоль, 1 экв.), дихлорметан (10 мл, 0,3 М) и диизопропилэтиламин (1,35 мл, 7,73 ммоль, 2,3 экв.) и получая #105 (2,4 г, 92%) в виде желто-коричневой пены. Фракции, содержащие загрязненный продукт (1,7 г), объединяли с предыдущими загрязненными фракциями и очищали, как описано выше, с получением дополнительного количества #105 (1,30 г, количественный выход для обоих реакций за две стадии). ЖХ-МС: m/z 779,3 [M+H+], 802,3 [M+Na+], время удерживания составляет 1,05 минуты.
Стадия 3. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[1-(бицикло[4.2.0]окта-1,3,5-триен-7-ил)-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#106). К смеси #105 (225 мг, 0,289 ммоль, 1 экв.) в дихлорметане (15 мл, 0,02 М) и N,N-диметилформамиде (1 мл) добавляли HATU (136 мг, 0,347 ммоль, 1,2 экв.). Через пять минут добавляли раствор амина #102 (72,4 мг, 0,318 ммоль, 1 экв.) и диизопропиламина (203 мкл, 1,16 ммоль, 3 экв.) в дихлорметане (5 мл). Через 24 часа реакционную смесь промывали рассолом, сушили над сульфатом натрия, фильтровали, концентрировали в вакууме на силикагеле и очищали посредством хроматографии на силикагеле (градиент: от 0% до 50% ацетона в гептане) с получением одиночного энантиомера #106 (210 мг, 76%) в виде прозрачного масла. ЖХ-МС: m/z 953,1 [M+H+], время удерживания составляет 3,99 минуты;1H ЯМР (400 МГц, CDCl3), считали, что является смесью ротамеров, характеристические сигналы: 7.76 (d, J=7,5 Гц, 2H), 7.57-7.64 (m, 2H), 7.40 (dd, J=7,5, 7,4 Гц, 2H), 7.28-7.34 (m, 2H), 4.82-4.88 (m, 1H), 3.95-4.01 (m, 1H), 3.76 и 3.82 (2 s, всего 3H), 3.30, 3.31, 3.34 и 3.35 (4 s, всего 6H), [1.20 (d, J=7,0 Гц) и 1.20 (d, J=7,0 Гц), всего 3H].
Стадия 4A. Синтез соли трифторуксусной кислоты и 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[1-(бицикло[4.2.0]окта-1,3,5-триен-7-ил)-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#107). Согласно общей методике A из #106 (25 мг, 0,026 ммоль, 1 экв.) в дихлорметане (10 мл, 0,003 М) и диэтиламине (4 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии с обращенной фазой (Метод C) с получением одиночного энантиомера #107 (16 мг, 73%) в виде твердого вещества. ЖХ-МС: m/z 730,8 [M+H+], время удерживания составляет 2,13 минуты; ВЭЖХ (Протокол N): время удерживания составляет 9,889 минуты.
Стадия 4B. Синтез соли трифторуксусной кислоты и 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[бицикло[4.2.0]окта-1,3,5-триен-7-ил(карбокси)метил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#108). Одиночный энантиомер #108 (94,5 мг, 57%) синтезировали из #106 (190 мг, 0,200 ммоль) в соответствии с методикой, аналогичной описанной для синтеза #41 из #40. ЖХ-МС: m/z 716,8 [M+H+], время удерживания составляет 2,06 минуты; ВЭЖХ (Протокол N): время удерживания составляет 9,137 минуты.
Получение 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#112)

Стадия 1. Синтез трет-бутил-(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-карбоксилата (#109). К раствору #11 (2,00 г, 6,96 ммоль, 1 экв.) в дихлорметане (21 мл, 0,3 М) и N,N-диметилформамиде (3 мл) добавляли HATU (3270 мг, 8,35 ммоль, 1,2 экв.). Через две минуты добавляли амин (1R,2S)-(+)-норэфедрин (1,07 мг, 6,96 ммоль, 1 экв.) и триэтиламин (1,94 мл, 13,9 ммоль, 2 экв.). Через два часа реакционную смесь разбавляли этилацетатом (100 мл), промывали 1 М водным раствором соляной кислоты и рассолом, сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0% до 60% этилацетата в гептане) с получением #109 (2,18 г, 74%) в виде белого твердого вещества. ЖХ-МС: m/z 321,3 [(M-Boc)+H+], время удерживания составляет 3,14 минуты;1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 7.64 (d, J=8,6 Гц, 1H), 7.24-7.33 (m, 4H), 7.15-7.21 (m, 1H), 5.35 (br d, J=5 Гц, 1H), 4.45 (br dd, J=5, 5 Гц, 1H), 3.91-4.00 (m, 1H), 3.30-3.39 (m, 1H), 3.26 (s, 3H), 2.94-3.07 (m, 1H), 2.04-2.14 (m, 1H), 1.46-1.78 (m, 4H), 1.40 (s, 9H), 0.97-1.04 (m, 6H).
Стадия 2. Синтез соли трифторуксусной кислоты и (2R,3R)-N-[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропанамида (#110). Согласно общей методике C при 0°C из #109 (414 мг, 0,984 ммоль, 1 экв.), диоксана (5 мл, 0,2 М) и 4 М раствора хлористого водорода в диоксане (15 мл, 60 ммоль, 60 экв.) синтезировали неочищенное целевое соединение, которое очищали посредством хроматографии с обращенной фазой (Метод C) с получением #110 (120 мг, 34%) в виде вязкой жидкости. ЖХ-МС: m/z 321,1 [M+H+], время удерживания составляет 0,55 минуты;1H ЯМР (400 МГц, DMSO-d6), характеристические сигналы: δ 7.90 (d, J=8,6 Гц, 1H), 7.28-7.36 (m, 4H), 7.20-7.27 (m, 1H), 4.46 (d, J=6,2 Гц, 1H), 3.48 (dd, J=8,6, 2,3 Гц, 1H), 3.38 (s, 3H), 2.92-3.16 (m, 3H), 2.24-2.35 (m, 1H), 1.49-1.88 (m, 4H), 1.09 (d, J=6,6 Гц, 3H), 1.01 (d, J=6,6 Гц, 3H).
Стадия 3. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#111). Согласно общей методике D из #32 (140 мг, 0,230 ммоль, 1 экв.), #110 (110 мг, 0,253 ммоль, 1,1 экв.), дихлорметана (3 мл, 0,08 М), N,N-диметилформамида (0,5 мл), HATU (96,2 мг, 0,253 ммоль, 1,1 экв.) и триэтиламина (96 мкл, 0,69 ммоль, 3 экв.) синтезировали неочищенный целевой продукт, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 40% ацетона в гептане) с получением #111 (220 мг, 95%). ЖХ-МС: m/z 912,4 [M+H+], 935,4 [M+Na+], время удерживания составляет 2,15 минуты; ВЭЖХ (Протокол B): m/z 912,5 [M+H+], 934,5 [M+Na+], время удерживания составляет 10,138 минуты (чистота более 94%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ 7.89 (d, J=7,8 Гц, 2H), 7.66-7.75 (m, 2H), 7.41 (dd, J=7,4, 7,4 Гц, 2H), 7.12-7.20 (m, 1H), [5.33 (d, J=4,7 Гц) и 5.38 (d, J=4,7 Гц), всего 1H], 3.15, 3.18, 3.22 и 3.23 (4 s, всего 6H), 1.30, 1.33, 1.36 и 1.39 (4 s, всего 6H), 0.95-1.06 (m, 6H).
Стадия 4. Синтез 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#112). Согласно общей методике А из #111 (210 мг, 0,230 ммоль) в дихлорметане (5 мл, 0,05 M) и диэтиламине (5 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) с получением смеси масла и твердого вещества. Добавляли диэтиловый эфир и гептан и смесь концентрировали в вакууме, получая #112 (81 мг, 51%) в виде белого твердого вещества. ЖХ-МС: m/z 690,4 [M+H+], время удерживания составляет 1,10 минуты; ВЭЖХ (Протокол A): m/z 690,5 [M+H+], 712,4 [M+Na+], время удерживания составляет 7,229 минуты (чистота более 90%);1H ЯМР (400 МГц, DMSO-d6), считали, что является смесью ротамеров, характеристические сигналы: δ [7.62 (br d, J=8 Гц), 7.88 (br d, J=8 Гц), 8.07 (br d, J=9 Гц) и 8.11 (br d, J=9 Гц), всего 2H], 7.15-7.34 (m, 5H), [5.34 (d, J=4 Гц) и 5.41 (d, J=5 Гц), всего 1H], 3.18, 3.21, 3.23 и 3.25 (4 s, всего 6H), 2.93 и 3.08 (2 br s, всего 3H), 1.15, 1.18, 1.21 и 1.25 (4s, всего 6H).
Получение соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (115)
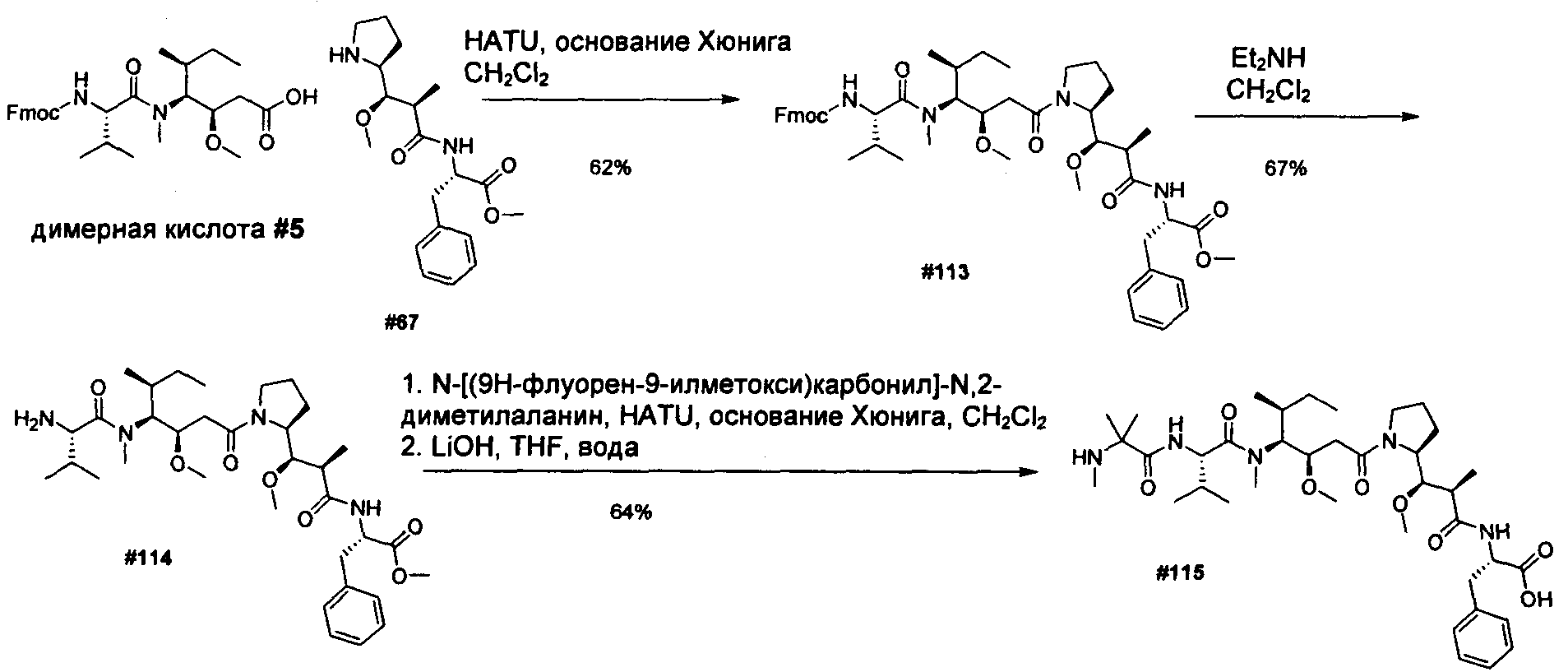
Стадия 1. Синтез метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[{N-[(9H-флуорен-9-илметокси)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#113). К перемешиваемой смеси димерной кислоты #5 (12,1 г, 23,0 мМ) и #67 (11,5 г, 23,0 мМ) в 75 мл дихлорметана под азотом добавляли HATU (10,8 г, 27,6 мМ), а затем основание Хюнига (12,1 мл, 69,0 мМ). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 15 часов. Реакционную смесь концентрировали до меньшего объема, переносили в этилацетат и промывали 1 н. HCl два раза. Затем органический слой промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Потом остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 70% ацетона в смеси гептанов), получая #113 (12,3 г, 62%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 855,3 [M+H+], 877,2 [M+Na+], время удерживания составляет 2,32 минуты; ВЭЖХ (Протокол R): m/z 855,5 [M+H+], время удерживания составляет 9,596 минуты (чистота более 97%).
Стадия 2. Синтез метил-N-{-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланината (#114). Согласно общей методике A из #113 (12 г, 14 ммоль, 1 экв.), дихлорметана (60 мл, 0,24 М) и диэтиламина (40 мл, 390 мМ) синтезировали #114 (5,9 г, 67%) в виде белого/светло-желтого твердого вещества после очистки посредством хроматографии на силикагеле (градиент: от 0% до 25% метанола в дихлорметане). ЖХ-МС (Протокол Q): m/z 633,0 [M+H+], время удерживания составляет 1,19 минуты. ВЭЖХ (Протокол A): m/z 633,5 [M+H+], время удерживания составляет 7,142 минуты (чистота более 98%).
Стадия 3. Синтез соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#115). К перемешиваемой смеси N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланина (167 мг, 0,493 мМ), #114 (260 мг, 0,411 мМ) и HATU (188 мг, 0,493 мМ) в 10 мл дихлорметана добавляли основание Хюнига (0,14 мл, 0,82 мМ). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа и 20 минут. Реакционную смесь концентрировали. К неочищенному материалу добавляли THF (9 мл) и к полученной перемешиваемой смеси добавляли гидроксид лития (49,2 мг, 2,06 мМ), растворенный в 3 мл воды. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали, а затем очищали посредством хроматографии с обращенной фазой С 18 при среднем давлении (градиент: от 5% до 45% воды в ацетонитриле с 0,02% TFA в каждой фазе), получая #115 (218 мг, 64%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 718,7 [M+H+], 740,6 [M+Na+], время удерживания составляет 1,21 минуты. ВЭЖХ (Протокол A при 45°C): m/z 718,4 [M+H+], время удерживания составляет 6,903 минуты.1H ЯМР (400 МГц, DMSO-d6) δ 8.81-8.95 (m), 8.44-8.50 (m), 8.42 (d), 8.15 (d), 7.14-7.28 (m), 4.71-4.78 (m), 4.57-4.66 (m), 4.49-4.56 (m), 4.41-4.48 (m), 3.94-4.05 (m), 3.72-3.79 (m), 3.39-3.60 (m), 2.95-3.33 (m), 2.78-2.89 (m), 2.69 (s), 2.43-2.50 (m), 2.08-2.42 (m), 1.60-1.92 (m), 1.20-1.57 (m), 0.84-1.11 (m), 0.74-0.83 (m).
Получение соли трифторуксусной кислоты и 2-метил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#117) и соли трифторуксусной кислоты и 2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#118)

Стадия 1. Синтез 1-(трет-бутоксикарбонил)-2-метил-L-пролил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]4-оксобутил}-N-метил-L-валинамида (#116). К перемешиваемому раствору #114 (1,02 г, 1,61 ммоль, 1,0 экв.) и 1-(трет-бутоксикарбонил)-2-метил-L-пролина (443 мг, 1,93 ммоль, 1,2 экв.) в 12 мл дихлорметана добавляли HATU (735 мг, 1,93 ммоль, 1,2 экв.), а затем основание Хюнига (1,12 мл, 6,45 ммоль, 4,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали, разбавляли этилацетатом, после чего промывали 0,5 н. HCl и рассолом. Затем органическую фазу сушили над сульфатом натрия, концентрировали до меньшего объема, а затем окончательно концентрировали на диоксиде кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-45% ацетона в смеси гептанов), получая #116 (1,02 г, 74%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 844,3 [M+H+], 867,2 [M+Na+], время удерживания составляет 2,15 минуты.
Стадия 2A. Синтез соли трифторуксусной кислоты и 2-метил-L-пролил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#117). К перемешиваемому раствору #116 (450 мг, 0,533 ммоль, 1,0 экв.) в 7 мл дихлорметана при 0°C добавляли TFA (3 мл, 40 ммоль, 70 экв.). Реакционную смесь оставляли перемешиваться при 0°C в течение 5 минут, а затем оставляли нагреваться до комнатной температуры при перемешивании в течение 20 минут. Реакционную смесь концентрировали, разбавляли дихлорметаном и небольшим количеством метанола, после чего окончательно концентрировали на диоксиде кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-20% метанола в этилацетате), получая #117 (396 мг, 89%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 744,5 [M+H+], 767,2 [M+Na+], время удерживания составляет 1,40 минуты; ВЭЖХ (Протокол A при 45°C): m/z 744,5 [M+H+], время удерживания составляет 7,149 минуты (чистота более 91%).1H ЯМР (400 МГц, DMSO-d6) δ 8.73-9.14 (m), 8.66 (br d), 8.50 (d), 8.22 (d), 7.12-7.25 (m), 4.67-4.74 (m), 4.41-4.63 (m), 3.93-4.00 (m), 3.73 (dd), 3.63 (d), 3.46-3.57 (m), 3.38-3.45 (m), 3.26-3.23 (m), 3.22-3.25 (m), 3.06-3.22 (m), 2.99-3.05 (m), 2.93-2.97 (m), 2.80-2.89 (m), 2.75-2.78 (m), 2.64-2.67 (m), 2.46-2.50 (m), 2.27-2.43 (m), 2.00-2.26 (m), 1.85-1.99 (m), 1.70-1.83 (m), 1.52-1.69 (m), 1.33-1.51 (m), 1.18-1.31 (m), 0.98-1.07 (m), 0.93-0.97 (m), 0.82-0.92 (m), 0.71-0.78 (m).
Стадия 2B. Синтез соли трифторуксусной кислоты и 2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#118). К перемешиваемому раствору #116 (435 мг, 0,515 ммоль) в 4 мл THF под азотом добавляли LiOH (24,7 мг, 1,03 ммоль, 2,0 экв.), растворенный в 2 мл воды. Реакционную смесь оставляли перемешиваться при комнатной температуре до тех пор, пока анализ методом ЖХ-МС не указал на омыление метилового сложного эфира. Реакционную смесь концентрировали в вакууме, а затем помещали под вакуум. Реакционную смесь разбавляли дихлорметаном и помещали под азот. К этой перемешиваемой смеси добавляли TFA (3 мл, 40,5 ммоль, 80 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 30 минут. Затем реакционную смесь концентрировали. Остаток очищали посредством хроматографии с обращенной фазой C18 при среднем давлении (градиент: от 5% до 60% ацетонитрила в воде с 0,02% TFA в каждой фазе), получая #118 (396 мг, 89%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 730,2 [M+H+], время удерживания составляет 1,18 минуты; ВЭЖХ (Протокол A при 45°C): m/z 730,5 [M+H+], время удерживания составляет 7,088 минуты (чистота более 98%).1H ЯМР (400 МГц, DMSO-d6) δ 9.04-9.13 (m), 8.75-8.87 (m), 8.70 (d), 8.38 (d), 8.11(d), 7.10-7.24 (m), 4.66-4.74 (m), 4.48-4.64 (m), 4.37-4.47 (m), 3.91-3.99 (m), 3.77 (m), 3.47-3.56 (m), 3.33-3.47 (m), 3.08-3.30 (m), 2.93-3.07 (m), 2.75-2.86 (m), 2.63-2.69 (m), 2.45-2.50 (m), 2.28-2.44 (m), 2.03-2.27 (m), 1.88-2.02 (m), 1.68-1.86 (m), 1.55-1.67 (m), 1.30-1.47 (m), 1.17-1.29 (m), 0.98-1.05 (m), 0.93-0.97 (m), 0.83-0.92 (m), 0.71-0.79 (m).
Получение 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#123)

Стадия 1. Синтез (2R,3R)-3-{(2S)-1-[(9H-флуорен-9-илметокси)карбонил]пирролидин-2-ил}-3-метокси-2-метилпропановой кислоты (#119). К перемешиваемому раствору #11 (2,4 г, 8,4 ммоль, 1,0 экв.) в 10 мл диоксана под азотом добавляли 4 M HCl в диоксане (20 мл, 80 мМ, 10 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 3 часов, после чего концентрировали в вакууме и помещали под высокий вакуум. Затем неочищенный материал растворяли в 30 мл 10%-ного Na2CO3. Потом полученный раствор добавляли к перемешиваемому раствору 1-{[(9H-флуорен-9-илметокси)карбонил]окси}пирролидин-2,5-диона (2,96 г, 8,77 ммоль, 1,05 экв.) в 30 мл DME. Реакционную смесь оставляли перемешиваться при комнатной температуре до тех пор, пока анализ методом ТСХ (20% метанол / 40% этилацетат / 40% гептаны) не указал на потребление Boc-незащищенного исходного материала. Реакционную смесь концентрировали в вакууме до меньшего объема, промывали дважды эфиром, подкисляли до pH 2, использую концентрированную HCl, а затем экстрагировали три раза раствором из 90% дихлорметана и 10% метанола. Органическую фазу промывали насыщенным бикарбонатом натрия и рассолом, после чего сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до коричневого твердого вещества #119 (3,4 г, количеств.). ЖХ-МС (Протокол Q): m/z 410,0 [M+H+], время удерживания составляет 1,81 минуты.
Стадия 2. Синтез трет-бутил-N-[(2R,3R)-3-{(2S)-1-[(9H-флуорен-9-илметокси)карбонил]пирролидин-2-ил}-3-метокси-2-метилпропаноил]-L-фенилаланината (#120). К перемешиваемому раствору гидрохлоридной соли трет-бутил-L-фенилаланината (1,67 г, 6,5 ммоль, 1,0 экв.) и #119 (5,9 г, 6,5 ммоль, 1,0 экв.) в 50 мл дихлорметана и 5 мл DMF добавляли HATU (2,9 г, 7,9 ммоль, 1,2 экв.), а затем основание Хюнига (5,6 мл, 32 ммоль, 5,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 45 минут. Реакционную смесь концентрировали, разбавляли этилацетатом, промывали 0,5 н. HCl и рассолом, после чего окончательно концентрировали на диоксиде кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-25% ацетона в гептане), получая #120 (3,14 г, 79%) в виде бело-желтого твердого вещества. ЖХ-МС (Протокол Q): m/z 613,1 [M+H+], время удерживания составляет 2,37 минуты.
Стадия 3. Синтез трет-бутил-N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-фенилаланината (#121). К перемешиваемому раствору #120 (2,87 г, 4,68 ммоль, 1,00 экв.) в 20 мл дихлорметана добавляли диэтиламин (10 мл, 95 мМ, 20,5 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. Добавляли дополнительное количество диэтиламина (10 мл, 95 ммоль, 20,5 экв.) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение еще 3 часов. Реакционную смесь концентрировали в вакууме и помещали под высокий вакуум, получая #121 (1,8 г, количеств.) в виде желто-белой смеси масла и твердого вещества. ЖХ-МС (Протокол Q): m/z 391,1 [M+H+], время удерживания составляет 1,05 минуты.
Стадия 4. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#122). К перемешиваемому раствору #121 (0,55 г, 1,0 ммоль, 1,0 экв.) в 10 мл дихлорметана и 1 мл DMF добавляли #32 (0.62 г, 1,0 ммоль, 1,0 экв.), а затем HATU (0,42 г, 1,1 ммоль, 1,1 экв.) и основание Хюнига (0,72 мл, 4,1 ммоль, 4,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение приблизительно 21 часов. Реакционную смесь концентрировали, разбавляли этилацетатом, а затем промывали 0,5 н. HCl и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали до меньшего объема, после чего окончательно концентрировали на диоксиде кремния. Затем осуществляли хроматографию на диоксиде кремния (градиент: 0%-40% ацетона в гептане), получая #122 (0,62 г, 62%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 982,3 [M+H+], время удерживания составляет 2,44 минуты.
Стадия 5. Синтез 2-метилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#123). К перемешиваемой смеси #122 (600 мг, 0,611 ммоль, 1,00 экв.) в 15 мл дихлорметана добавляли диэтиламин (5 мл, 50 ммоль, 80 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали в вакууме и смесь очищали посредством хроматографии на диоксиде кремния (градиент: 0%-40% метанола в дихлорметане), получая #123 (0,46 г, 99%) в виде твердого вещества. ЖХ-МС (Протокол Q1): m/z 760,3 [M+H+], время удерживания составляет 0,83 минуты.1H ЯМР (400 МГц, CD3OD), δ 7.14-7.30 (m), 4.70-4.78 (m), 4.56-4.64 (m), 4.05-4.19 (m), 3.87 (dd), 3.79-3.84 (m), 3.72-3.77 (m), 3.62-3.70 (m), 3.46-3.56 (m), 3.37-3.45 (m), 3.33-3.36 (m), 3.16-3.24 (m), 3.09-3.11 (m), 2.98-3.05 (m), 2.95 (d), 2.91 (d), 2.87 (d), 2.83 (d), 2.73-2.79 (m), 2.40-2.51 (m), 2.29-2.39 (m), 2.16-2.28 (m), 2.04-2.15 (m), 2.01 (s), 1.73-1.96 (m), 1.50-1.68 (m), 1.47-1.49 (m), 1.46 (s), 1.43 (s), 1.38 (s), 1.35 (d), 1.23-1.32 (m), 1.17-1.22 (m), 1.15 (d), 1.04-1.11 (m), 0.94-1.03 (m), 0.82-0.91 (m).
Получение метил-N-[(2R,3R)-3-{(2S)-1-[(3R,4S,5S)-4-{[N-(3-амино-2,2-диметилпропаноил)-L-валил](метил)амино}-3-метокси-5-метилгептаноил]пирролидин-2-ил}-3-метокси-2-метилпропаноил]-L-фенилаланината (#126)

Стадия 1. Синтез 3-{[(9H-флуорен-9-илметокси)карбонил]амино}-2,2-диметилпропановой кислоты (#124). Раствор гидрохлорида 3-амино-2,2-диметилпропановой кислоты (1,0 г, 6,5 ммоль, 1,0 экв.) в 10 мл 10%-ного Na2CO3 добавляли к раствору 1-{[(9H-флуорен-9-илметокси)карбонил]окси}-пирролидин-2,5-диона (2,3 г, 6,5 ммоль, 1,0 экв.) в 10 мл DME. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь концентрировали до меньшего объема, а затем два раза промывали эфиром. Водный слой подкисляли до pH менее 2 концентрированной HCl, а затем три раза экстрагировали раствором из 10% метанола и 90% дихлорметана. Органические фазы объединяли, после чего промывали 1 М HCl и рассолом. Органический слой сушили над сульфатом натрия и концентрировали в вакууме, получая #124 (2,2 г, 98%) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 362,0 [M+Na+], время удерживания составляет 0,89 минуты.
Стадия 2. Синтез метил-N-[(2R,3R)-3-{(2S)-1-[(3R,4S,5S)-4-{[N-(3-{[(9H-флуорен-9-илметокси)карбонил]амино}-2,2-диметилпропаноил)-L-валил](метил)амино}-3-метокси-5-метилгептаноил]пирролидин-2-ил}-3-метокси-2-метилпропаноил]-L-фенилаланината (#125). К перемешиваемому раствору #114 (200 мг, 0,316 ммоль, 1,00 экв.) в 2 мл дихлорметана добавляли #124 (107 мг, 0,316 ммоль, 1,00 экв.), а затем основание Хюнига (0,167 мл, 0,948 ммоль, 3,00 экв.) и HATU (149 мг, 0,379 ммоль, 1,20 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 12 часов. Реакционную смесь концентрировали до меньшего объема, переносили в 10 мл этилацетата и промывали два раза 5 мл 1 М HCl и один раз 5 мл рассола. Органический слой сушили над сульфатом натрия и декантировали. Органическую фазу концентрировали в вакууме и неочищенный материал переносили в дихлорметан. Осадок фильтровали. Органический слой концентрировали в вакууме и остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-50% ацетона в гептане), получая #125 (235 мг, 78%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 954,2 [M+H+], время удерживания составляет 2,28 минуты.
Стадия 3. Синтез метил-N-[(2R,3R)-3-{(2S)-1-[(3R,4S,5S)-4-{[N-(3-амино-2,2-диметилпропаноил)-L-валил](метил)амино}-3-метокси-5-метилгептаноил]пирролидин-2-ил}-3-метокси-2-метилпропаноил]-L-фенилаланината (#126). К перемешиваемому раствору #125 (235 мг, 0,246 ммоль, 1,00 экв.) в 2 мл THF добавляли диэтиламин (1 мл, 10 мМ, 40,6 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-30% метанола в этилацетате), получая #126 (101 мг, 56%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 732,2 [M+H+], время удерживания составляет 1,32 минуты.1H ЯМР (400 МГц, DMSO-d6) δ 8.51 (dd), 8.28 (d), 7.15-7.29 (m), 5.77 (s), 4.55-4.77 (m), 4.44-4.54 (m), 3.94-4.10 (m), 3.73-3.79 (m), 3.66 (d), 3.49-3.60 (m), 3.40-3.48 (m), 3.10-3.36 (m), 3.00-3.09 (m), 2.83-2.98 (m), 2.57-2.77 (m), 2.19-2.46 (m), 1.87-2.14 (m), 1.61-1.86 (m), 1.36-1.55 (m), 1.23-1.36 (m), 1.12-1.22 (m), 0.97-1.11 (m), 0.82-0.96 (m), 0.73-0.81 (m).
Получение N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#130)

Стадия 1. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(3R,4S,5S)-1-трет-бутокси-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#127). В круглодонную колбу, содержащую #6 (4,7 г, 7,9 ммоль, 1,0 экв.) и N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланин (3,2 г, 9,4 ммоль, 1,2 экв.) и магнитную мешалку, под азотом добавляли 50 мл дихлорметана, а затем HATU (3,6 г, 9,4 ммоль, 1,2 экв.) и основание Хюнига (5,5 мл, 32 ммоль, 4,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 12 часов. Реакционную смесь концентрировали до меньшего объема, переносили в этилацетат, после чего промывали 1 н. HCl и рассолом. Затем органическую фазу сушили над сульфатом натрия, фильтровали, а потом окончательно концентрировали на диоксиде кремния. Остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-30% ацетона в гептане), получая #127 (4,2 г, 78%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 680,2 [M+H+], время удерживания составляет 2,52 минуты.
Стадия 2. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (#128). К перемешиваемому раствору #127 (4,2 г, 6,1 ммоль, 1,0 экв.) в 21 мл дихлорметана под азотом добавляли TFA (7 мл, 90 ммоль, 10 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 4 часов. Реакционную смесь концентрировали в вакууме, один раз подвергали азеотропной перегонке с гептаном, а затем помещали под высокий вакуум, получая #128 в виде белого/слегка-желтого твердого вещества (3,8 г, количеств.). ЖХ-МС (Протокол Q): m/z 624,2 [M+H+], время удерживания составляет 2,01 минуты.
Стадия 3. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#129). К перемешиваемому раствору #128 (1,67 г, 3,1 ммоль, 1,0 экв.) в 20 дихлорметана и 2 мл DMF добавляли #121 (2,4 г, 3,1 ммоль, 1,0 экв.), потом HATU (1,29 г, 3,39 ммоль, 1,1 экв.), а затем основание Хюнига (2,2 мл, 12,3 ммоль, 4,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 2 часов. Реакционную смесь концентрировали, разбавляли этилацетатом, после чего промывали 0,5 н. HCl и рассолом. Органическую фазу сушили над сульфатом натрия, а затем окончательно концентрировали на диоксиде кремния. Остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-50% ацетона в смеси гептанов), получая #129 (1,9 г, 62%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 996,3 [M+H+], время удерживания составляет 2,53 минуты.
Стадия 4. Синтез N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#130). К перемешиваемому раствору #129 (823 мг, 0,826 ммоль, 1,00 экв.) в 15 мл дихлорметана добавляли диэтиламин (4 мл, 40 ммоль, 50 экв,). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 14,5 часов. Реакционную смесь концентрировали в вакууме и один раз подвергали азеотропной перегонке с гептанами. Остаток разбавляли дихлорметаном и небольшим количеством метанола, после чего концентрировали на диоксиде кремния. Остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-20% метанола в этилацетате), получая #130 (518 мг, 81%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 774,3 [M+H+], время удерживания составляет 1,48 минуты. ВЭЖХ (Протокол A при 25°C): m/z 774,5 [M+H+], время удерживания составляет 7,733 минуты (чистота более 98%).1H ЯМР (400 МГц, DMSO-d6) δ 8.36 (d), 8.14 (d), 7.81 (t), 7.14-7.25 (m), 7.01-7.07 (m), 4.87-4.94 (m), 4.78-4.85 (m), 4.67-4.76 (m), 4.46-4.65 (m), 4.29-4.40 (m), 3.93-4.03 (m), 3.70-3.81 (m), 3.49-3.60 (m), 3.38-3.47 (m), 3.29-3.36 (m), 3.15-3.28 (m), 2.98-3.13 (m), 2.94 (br s), 2.74-2.89 (m), 2.64-2.69 (m), 2.18-2.45 (m), 2.02-2.14 (m), 1.90-2.01 (m), 1.62-1.87 (m), 1.40-1.55 (m), 1.37 (d), 1.20-1.33 (m), 1.16 (d), 1.01-1.10 (m), 0.90-0.98 (m), 0.82-0.89 (m), 0.69-0.79 (m).
Получение соли трифторуксусной кислоты и 2-метил-D-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#131)

Стадия 1. Синтез соли трифторуксусной кислоты и 2-метил-D-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#131). К перемешиваемому раствору #114 (164 мг, 0,259 ммоль, 1,0 экв.) и 1-трет-бутоксикарбонил)-2-метил-D-пролина (71,3 мг, 0,311 ммоль, 1,2 экв.) в 4 мл дихлорметана добавляли HATU (118 мг, 0,311 ммоль, 1,2 экв.), а затем основание Хюнига (0,180 мл, 1,04 ммоль, 4 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 30 минут. Реакционную смесь концентрировали. Реакционную смесь переносили в 3,5 мл дихлорметана и помещали под азот. К полученному перемешиваемому раствору добавляли TFA (1,5 мл, 20 ммоль, 76 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 1 часа. Реакционную смесь концентрировали и помещали под высокий вакуум. После очистки (Метод J*) получали #131 (119 мг, 54%) в виде белого твердого вещества. ВЭЖХ (Протокол A при 45°C): m/z 744,5 [M+H+], время удерживания составляет 7,342 минуты (чистота более 98%).1H ЯМР (400 МГц, DMSO-d6) δ 9.08-9.18 (m), 8.79-8.89 (m), 8.76 (t), 8.54 (d), 8.29 (d), 7.14-7.31 (m), 4.70-4.79 (m), 4.57-4.66 (m), 4.45-4.55 (m), 3.96-4.04 (m), 3.74-3.80, 3.66 (d), 3.48-3.61 (m), 3.40-3.48 (m), 3.09-3.34 (m), 3.00-3.09 (m), 2.95-3.00 (m), 2.83-2.93 (m), 2.36-2.53 (m), 2.21-2.35 (m), 2.10-2.19 (m), 1.99-2.10 (m), 1.61-1.09 (m), 1.36-1.53 (m), 1.21-1.35 (m), 1.02-1.10 (m), 0.94-1.0 (m), 0.86-0.93 (m), 0.73-0.82 (m).
Получение соли трифторуксусной кислоты и 2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#134)
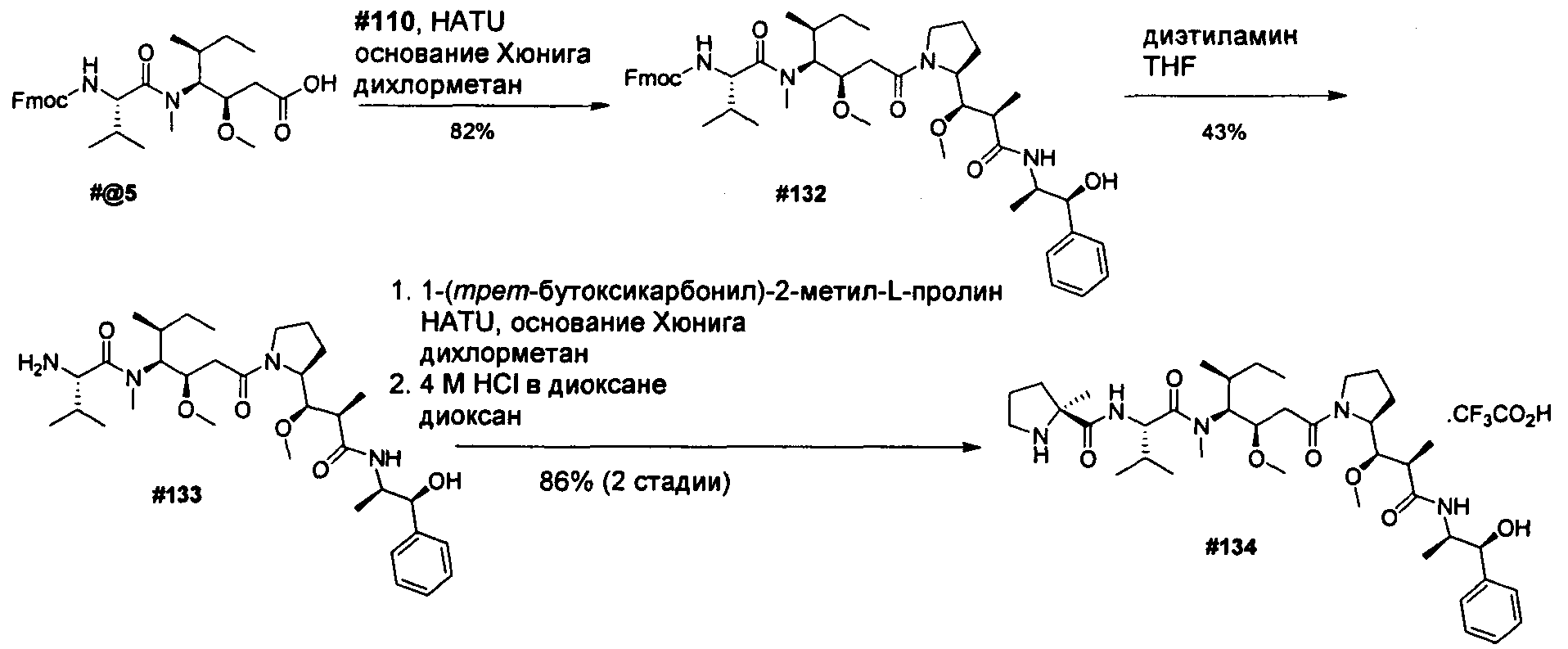
Стадия 1. Синтез N~2~-[(9H-флуорен-9-илметокси)карбонил]-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#132). В колбу, содержащую #@5 (1,14 г, 2,17 ммоль, 1,0 экв.), добавляли 10 мл дихлорметана, а затем основание Хюнига (1,15 мл, 6,52 ммоль, 3,0 экв.), HATU (1,02 г, 2,61 ммоль, 1,2 экв.) и #110 (0,776 г, 2,17 ммоль, 1,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 30 минут, а затем концентрировали в вакууме. Неочищенный материал переносили в 50 мл этилацетата, промывали два раза 25 мл 1 М HCl и один раз 25 мл рассола. Органическую фазу сушили над сульфатом натрия и декантировали. Органическую фазу концентрировали в вакууме, переносили в 30 мл дихлорметана и полученный осадок отфильтровывали. Органическую фазу концентрировали в вакууме и остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-50% ацетона в смеси гептанов), получая #132 (1,33 г, 81%) в виде твердого вещества. ЖХ-МС (Протокол Q): m/z 849,2 [M+Na+], время удерживания составляет 2,19 минуты.
Стадия 2. Синтез N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#133). К перемешиваемому раствору #132 (1,33 г, 1,60 ммоль, 1,0 экв.) в 10 мл THF добавляли диэтиламин (5 мл, 50 мМ, 31,3 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-30% метанола в этилацетате), получая #133 (418 мг, 43%) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 605,2 [M+H+], время удерживания составляет 1,48 минуты.
Стадия 3. Синтез соли трифторуксусной кислоты и 2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#134). HATU (151 мг, 0,398 ммоль, 1,2 экв.), #133 (201 мг, 0,332 ммоль, 1,0 экв.) и 1-(трет-бутоксикарбонил)-2-метил-L-пролин (91,3 мг, 0,398 мМ, 1,2 экв.) объединяли под азотом в круглодонной колбе, содержащей магнитную мешалку. Добавляли 5 мл дихлорметана, а затем основание Хюнига (0,231 мл, 1,33 ммоль, 4,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 15 часов. Затем реакционную смесь концентрировали в вакууме и помещали под высокий вакуум. Затем к остатку добавляли 4 мл диоксана, а потом 4 М HCl в диоксане (4 мл, 20 ммоль, 50 экв.). Затем реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Реакционную смесь затем концентрировали в вакууме и остаток очищали посредством хроматографии с обращенной фазой C18 при среднем давлении (градиент: от 5% до 90% ацетонитрила в воде с 0,02% TFA в каждой фазе), получая #134 (237 мг, 86%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 716,3 [M+H+], время удерживания составляет 1,16 минуты; ВЭЖХ (Протокол A при 45°C): m/z 716,5 [M+H+], время удерживания составляет 6,930 минуты (чистота более 98%).1H ЯМР (400 МГц, DMSO-d6) 9.12-9.21 (m), 8.79-8.90 (m), 8.70-8.78 (m), 7.95 (d), 7.64 (d), 7.25-7.36 (m), 7.16-7.23 (m), 4.74-4.80 (m), 4.61-4.69 (m), 4.41-4.59 (m), 3.91-4.06 (m), 3.78 (dd), 3.54-3.64 (m), 3.45-3.51 (m), 3.17-3.36 (m), 3.02-3.15 (m), 3.00 (br s), 2.40-2.48 (m), 2.24-2.35 (m), 1.91-2.21 (m), 1.68-1.90 (m), 1.61-1.68 (m), 1.48-1.59 (m), 1.22-1.35 (m), 0.97-1.09 (m), 0.84-0.97 (m), 0.74-0.83 (m).
Получение соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-(метиламино)-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#140), соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-2-амино-1-бензил-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#141), соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-оксо-2-(пропиламино)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#142), соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-(диэтиламино)-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#143) и соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-(трет-бутиламино)-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#144)


Стадия 1. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-5-метил-1-оксо-1-(пентафторфенокси)-гептан-4-ил]-N-метил-L-валинамида (#135). К раствору #128 (4,18 г, 6,70 ммоль, 1,0 экв.) в 50 мл дихлорметана добавляли пентафторфенил-3,3,3-трифторпропаноат (2,44 мл, 13,4 ммоль, 2,0 экв.), а затем пиридин (1,61 мл, 20,1 ммоль, 3,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 12 часов. Реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-70% ацетона в смеси гептанов), получая #135 (5,2 г, 98%) в виде белой пены. ЖХ-МС (Протокол Q1): m/z 812,1 [M+Na+], время удерживания составляет 1,24 минуты.
Стадия 2. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#136). К перемешиваемому раствору 4 М HCl в диоксане (10 мл, 25 ммоль, 3,7 экв.) в 10 мл диоксана добавляли #11 (2,31 г, 8,05 ммоль, 1,2 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 6 часов. Реакционную смесь концентрировали в вакууме, получая желтую смолу. К полученному остатку добавляли раствор #135 (5,3 г, 6,7 ммоль, 1,0 экв.) в 30 мл дихлорметана, а затем основание Хюнига (3,5 мл, 20 ммоль, 3 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли дихлорметаном, после чего промывали 1%-ным водным раствором HCl, а затем рассолом. Органический слой сушили над сульфатом натрия, концентрировали в вакууме и остаток очищали посредством хроматографии на диоксиде кремния (градиент: 20%-50% этилацетата в смеси гептанов, затем 93% этилацетата, 6,6% метанола и 0,4% уксусной кислоты), получая #136 (4,87 г, 92%) в виде не совсем белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 793,3 [M+H+], время удерживания составляет 1,07 минуты.
Стадия 3. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-(пентафторфенокси)пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#137). К раствору #136 (2,8 г, 3,5 ммоль, 1,0 экв.) в 30 мл дихлорметана добавляли пентафторфенил-3,3,3-трифторпропаноат (1,3 мл, 7,1 ммоль, 2,0 экв.), а затем пиридин (0,85 мл, 10,6 мМ). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-70% ацетона в гептане), получая #137 (3,1 г, 92%) в виде белого порошка. ЖХ-МС (Протокол Q1): m/z 959,2 [M+H+], время удерживания составляет 1,28 минуты.
Стадия 4. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#138). К перемешиваемому раствору #137 (493 мг, 0,514 ммоль, 1,0 экв.) в 4 мл DMF добавляли L-фенилаланин (84,9 мг, 0,514 ммоль, 1,0 экв.), а затем основание Хюнига (0,27 мл, 1,54 ммоль, 3,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 12 часов. Реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-100% этилацетата в гептане), получая #138 (200 мг, 41%) в виде белой пены. ЖХ-МС (Протокол Q1): m/z 940,3 [M+H+], время удерживания составляет 1,08 минуты.
Стадия 5. Синтез N-[(9H-флуорен-9-илметокси)карбонил]-N,2-диметилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S)-1-оксо-1-(пентафторфенокси)-3-фенилпропан-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#139). К перемешиваемому раствору #138 (200 мг, 0,213 ммоль, 1,0 экв.) в 5 мл дихлорметана добавляли пентафторфенил-3,3,3-трифторпропаноат (126 мг, 0,426 мМ, 2,0 экв.), а затем пиридин (0,051 мл, 0,64 ммоль, 3,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 12 часов. Реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии на диоксиде кремния (градиент: 0%-100% этилацетата в смеси гептанов), получая #139 (174 мг, 74%) в виде желтого масла. ЖХ-МС (Протокол Q1): m/z 1128 [M+Na+], время удерживания составляет 1,23 минуты.
Стадия 6A. Синтез соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-(метиламино)-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#140). К перемешиваемому раствору #139 (20 мг, 0,018 ммоль, 1,0 экв.) в 1 мл THF добавляли метиламин (1 М в THF, 0,18 мл, 0,18 ммоль, 10 экв.) и смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали, разбавляли DMSO и подвергали очистке (Метод J*). Фракции собирали и концентрировали в вакууме с получением #140 (4,0 мг, 30%) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 731,2 [M+H+], время удерживания составляет 0,70 минуты.1H ЯМР (400 МГц, метанол-d4) 7.30-7.41 (m), 4.71-4.78 (m), 4.58-4.69 (m), 4.04-4.15 (m), 3.86-3.98 (m), 3.73-3.78 (m), 3.61-3.70 (m), 3.50-3.58 (m), 3.32-3.47 (m), 3.23-3.26 (m), 3.17-3.22 (m), 3.07-3.15 (m), 2.95-2.98 (m), 2.76-2.91 (m), 2.68-2.75 (m), 2.63-2.66 (m), 2.43-2.51 (m), 2.22-2.28 (m), 1.99-2.11 (m), 1.74-1.96 (m), 1.21-1.31 (m), 1.17-1.20 (m), 0.92-1.10 (m), 0.79-0.89 (m).
Стадия 6B. Синтез соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-2-амино-1-бензил-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#141). Следуя такой же методике, как и для #140, используя #139 (20 мг, 0,018 ммоль, 1,0 экв.), раствор аммиака (7 М в метаноле, 0,026 мл, 0,18 ммоль, 10 экв.) и очистку (Метод J*), получали #141 (3,0 мг, 20%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 717,2 [M+H+], время удерживания составляет 0,79 минуты.1H ЯМР (400 МГц, метанол-d4) 7.22-7.30 (m), 7.14-7.21 (m), 4.57-4.4.80 (m), 4.02-4.17 (m), 3.92-3.98 (m), 3.84-3.91 (m), 3.32-3.74 (m), 3.17-3.27 (m), 3.06-3.14 (m), 2.77-3.05 (m), 2.65 (s), 2.43-2.51 (m), 2.21-2.26 (m), 1.98-2.13 (m), 1.70-1.94 (m), 1.32-1.69 (m), 1.16-1.31 (m), 0.89-1.13 (m), 0.80-0.88 (m).
Стадия 6C. Синтез соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-оксо-2-(пропиламино)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#142). Следуя такой же методике, как и для #140, используя #139 (20 мг, 0,018 ммоль, 1,0 экв.), н-пропиламин (1 М в THF, 0,18 мл, 0,18 ммоль, 10 экв.) и очистку (Метод J*), получали #142 (3,0 мг, 20%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 759,2 [M+H+], время удерживания составляет 0,74 минуты.1H ЯМР (400 МГц, метанол-d4) 7.15-7.29 (m), 4.71-4.79 (m), 4.52-4.68 (m), 4.04-4.17 (m), 3.87-3.99 (m), 3.73-3.99 (m), 3.73-3.79 (m), 3.50-3.70 (m), 3.34-3.49 (m), 3.06-3.23 (m), 2.79-2.99 (m), 2.44-2.50 (m), 2.28-2.43 (m), 2.22-2.27 (m), 1.75-2.10 (m), 1.34-1.61 (m), 1.16-1.29 (m), 0.92-1.10 (m), 0.77-0.89 (m).
Стадия 6D. Синтез соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-(диэтиламино)-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#143). Следуя такой же методике, как и для #140, используя #139 (20 мг, 0,018 ммоль, 1,0 экв.), диэтиламин (1 М в THF, 0,18 мл, 0,18 ммоль, 10 экв.) и очистку (Метод J*), получали #143 (4,0 мг, 30%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 773,3 [M+H+], время удерживания составляет 0,77 минуты.1H ЯМР (400 МГц, метанол-d4) 7.16-7.33 (m), 5.10-5.17 (m), 4.96-5.07 (m), 4.68-4.75 (m), 4.60-4.65 (m), 3.61-4.23 (m), 3.35-3.67 (m), 3.16-3.26 (m), 2.99-3.15 (m), 2.78-2.94 (m), 2.30-2.52 (m), 2.19-2.28 (m), 1.73-2.13 (m), 1.83-1.45 (m), 1.19-1.31 (m), 0.92-1.18 (m), 0.80-0.89 (m).
Стадия 6E. Синтез соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-(трет-бутиламино)-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида #144. Следуя такой же методике, как и для #140, используя #139 (20 мг, 0,018 ммоль, 1,0 экв.), трет-бутиламин (1 М в THF, 0,18 мл, 0,18 ммоль, 10 экв.) и очистку (Метод J*), получали #144 (3,4 мг, 24%) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 773,3 [M+H+], время удерживания составляет 0,74 минуты.1H ЯМР (400 МГц, DMSO-d6) δ 8.21 (d), 8.03-7.98 (m), 7.92 (d), 7.81-7.62 (m), 7.46-7.16 (m), 4.83-4.69 (m), 4.68-4.56 (m), 4.21-4.07 (m), 3.92-3.86 (m), 3.83-3.80 (m), 3.74-3.65 (m), 3.60-3.48 (m), 3.47-3.36 (m), 3.28-3.13 (m), 3.11-3.01 (m), 2.96-2.82 (m), 2.69-2.62 (m), 2.54-2.43 (m), 2.38-2.12 (m), 2.00-1.76 (m), 1.69-1.161 (m), 1.60-1.53 (m), 1.52-0.98 (m), 0.94-0.86 (m).
Получение соли трифторуксусной кислоты и N,2-диметилаланил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#145)

Стадия 1. Синтез соли трифторуксусной кислоты и N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1R,2S)-2-гидрокси-1-метил-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#145). К перемешиваемому раствору #137 (300 мг, 0,313 ммоль, 1,0 экв.) в 3 мл DMF добавляли (1S,2R)-2-амино-1-фенилпропан-1-ол (54,8 мг, 0,344 ммоль, 1,1 экв.), а затем основание Хюнига (0,164 мл, 0,939 ммоль, 3,0 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 12 часов. Затем добавляли 20%-ный раствор пиперидина в DMF (1 мл, 2,2 ммоль, 7,0 экв.) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. После очистки (Метод J*) с последующим концентрированием содержимого подходящих пробирок получали #145 (190 мг, 74%) в виде белого порошка. ЖХ-МС (Протокол Q): m/z 704,3 [M+H+], время удерживания составляет 0,67 минуты.1H ЯМР (400 МГц, CD3OD), δ 7.97 (d), 7.73 (d), 7.37-7.41 (m), 7.27-7.36 (m), 7.19-7.25 (m), 4.70-4.75 (m), 4.58-4.63 (m), 4.49-4.54 (m), 4.14-4.30 (m), 4.04-4.11 (m), 3.87 (dd), 3.63-3.77 (m), 3.51-3.58 (m), 3.46-3.49 (m), 3.38-3.43 (m), 3.25-3.37 (m), 3.15-3.23 (m), 3.11-3.14 (m), 3.01-3.02 (m), 2.59-2.64 (m), 2.52-2.55 (m), 2.44-2.52 (m), 2.41-2.43 (m), 2.07-2.26 (m), 1.73-2.0 (m), 1.65-1.73 (m), 1.59-1.65 (m), 1.51-1.59 (m), 1.32-1.46 (m), 1.23-1.26 (m), 1.08-1.21 (m), 0.94-1.07 (m), 0.83-0.92 (m).
Получение 3-метил-D-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#146), 3-метил-L-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#147), L-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#148) и D-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#149)

Стадия 1A. Синтез 3-метил-D-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#146). Раствор #114 (225 мг, 0,356 ммоль, 1,0 экв.) в 2 мл дихлорметана добавляли к раствору N-[(9H-флуорен-9-илметокси)карбонил]-3-метил-D-изовалина (126 мг, 0,356 ммоль, 1,0 экв.) в 4 мл дихлорметана. Добавляли основание Хюнига (0,188 мл, 1,07 ммоль, 3,0 экв.), а затем HATU (167 мг, 0,427 ммоль, 1,2 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали в вакууме, затем переносили в этилацетат, после чего промывали два раза 1 М HCl и один раз рассолом. Органический слой сушили над сульфатом натрия и декантировали. Органический растворитель удаляли с помощью оборудования genevac. Добавляли THF (4 мл), а затем диэтиламин (2 мл, 19 ммоль, 53,4 экв.). Реакционную смесь оставляли перемешиваться в течение примерно 12 часов. Реакционную смесь концентрировали с использованием оборудования genevac с последующей хроматографией на диоксиде кремния (градиент: 0%-30% метанола в этилацетате), получая #146 (183 мг, 69%) в виде твердого вещества. ЖХ-МС (Протокол Q): m/z 746,4 [M+H+], время удерживания составляет 1,37 минуты.1H ЯМР (400 МГц, DMSO-d6) δ 8.55 (d), 8.26-8.36 (m), 7.88-8.03 (m), 7.81 (d), 7.41-7.53 (m), 7.13-7.30 (m), 7.01 (s), 4.71-4.79 (m), 4.44-4.70 (m), 3.96-4.04 (m), 3.70-3.80 (m), 3.62-3.69 (m), 3.40-3.61 (m), 2.76-3.35 (m), 2.67-2.71 (m), 2.56-2.58 (m), 2.06-2.46 (m), 1.61-1.90 (m), 1.14-1.54 (m), 0.72-1.12 (m).
Стадия 1B. Синтез 3-метил-L-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#147). Раствор #114 (224 мг, 0,354 ммоль, 1,0 экв.) в 2 мл дихлорметана добавляли к раствору N-[(9H-флуорен-9-илметокси)карбонил]-3-метил-L-изовалина (125 мг, 0,354 ммоль, 1,0 экв.) в 4 мл дихлорметана. Добавляли основание Хюнига (0,187 мл, 1,06 ммоль, 3,0 экв.), а затем HATU (167 мг, 0,425 ммоль, 1,2 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали в вакууме, затем переносили в этилацетат, после чего промывали два раза 1 M HCl и один раз рассолом. Органический слой сушили над сульфатом натрия и декантировали. Органический растворитель удаляли с помощью оборудования genevac. Добавляли THF (4 мл), а затем диэтиламин (2 мл, 19 ммоль, 53,7 экв.). Реакционную смесь оставляли перемешиваться в течение примерно 12 часов. Реакционную смесь концентрировали с использованием оборудования genevac с последующей хроматографией на диоксиде кремния (градиент: 0%-30% метанола в этилацетате), получая #147 (216 мг, 82%) в виде твердого вещества. ЖХ-МС (Протокол Q): m/z 746,6 [M+H+], время удерживания составляет 1,29 минуты.1H ЯМР (400 МГц, DMSO-d6) δ 8.56 (m), 8.31-8.39 (m), 8.50 (d), 8.30 (br d), 7.87-8.01 (m), 7.80 (d), 7.40-7.53 (m), 7.14-7.30 (m), 4.45-4.78 (m), 3.94-4.04 (m), 3.70-3.79 (m), 3.61-3.69 (m), 3.42-3.59 (m), 2.97-3.37 (m), 2.80-2.92 (m), 2.32-2.49 (m), 2.05-2.30 (m), 1.61-1.89 (m), 1.37-1.56 (m), 1.14-1.135 (m), 0.70-1.11 (m).
Стадия 1C. Синтез L-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#148). Раствор #114 (447 мг, 0,707 ммоль, 1,0 экв.) в 2 мл дихлорметана добавляли к раствору N-[(9H-флуорен-9-илметокси)карбонил]-L-изовалина (240 мг, 0,707 ммоль, 1,0 экв.) в 4 мл дихлорметана. Добавляли основание Хюнига (0,373 мл, 2,12 ммоль, 3,0 экв.), а затем HATU (332 мг, 0,425 ммоль, 1,2 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали в вакууме, затем переносили в этилацетат, после чего промывали два раза 1 М HCl и один раз рассолом. Органический слой сушили над сульфатом натрия и декантировали. Органический растворитель удаляли с помощью оборудования genevac. Добавляли THF (4 мл), а затем диэтиламин (2 мл, 19 ммоль, 26,9 экв.). Реакционную смесь оставляли перемешиваться в течение примерно 12 часов. Реакционную смесь концентрировали с использованием оборудования genevac с последующей хроматографией на диоксиде кремния (градиент: 0%-30% метанола в этилацетате), получая #148 (182 мг, 35%) в виде твердого вещества. ЖХ-МС (Протокол Q1): m/z 732,3 [M+H+], время удерживания составляет 0,71 минуты.1H ЯМР (400 МГц, DMSO-d6) δ 8.56 (d), 8.46-8.52 (m), 8.30 (d), 8.02-8.15 (m), 7.98 (d), 7.80 (d), 7.40-7.53 (m), 7.15-7.30 (m), 4.70-4.80 (m), 4.44-4.69 (m), 3.96-4.05 (m), 3.70-3.79 (m), 3.62-3.69 (m), 3.41-3.59 (m), 2.99-3.35 (m), 2.31-2.95 (m), 2.67-2.71 (m), 2.55-2.59 (m), 2.32-2.48 (m), 2.20-2.31 (m), 1.97-2.19 (m), 1.61-1.88 (m), 1.37-1.56 (m), 1.20-1.34 (m), 1.14-1.19 (m), 1.02-1.11 (m), 0.97-1.01 (m), 0.86-0.96 (m), 0.71-0.83 (m).
Стадия 1D. Синтез D-изовалил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-бензил-2-метокси-2-оксоэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#149). Раствор #114 (447 мг, 0,707 ммоль, 1,0 экв.) в 2 мл дихлорметана добавляли к раствору N-[(9H-флуорен-9-илметокси)карбонил]-D-изовалина (240 мг, 0,707 ммоль, 1,0 экв.) в 4 мл дихлорметана. Добавляли основание Хюнига (0,373 мл, 2,12 ммоль, 3 экв.), а затем HATU (332 мг, 0,425 ммоль, 1,2 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали в вакууме, затем переносили в этилацетат, после чего промывали два раза 1 М HCl и один раз рассолом. Органический слой сушили над сульфатом натрия и декантировали. Органический растворитель удаляли с помощью оборудования genevac. Добавляли THF (4 мл), а затем диэтиламин (2 мл, 19 ммоль, 26,9 экв.). Реакционную смесь оставляли перемешиваться в течение примерно 12 часов. Реакционную смесь концентрировали с использованием оборудования genevac с последующей хроматографией на диоксиде кремния (градиент: 0%-30% метанола в этилацетате), получая #149 (154 мг, 30%) в виде твердого вещества. ЖХ-МС (Протокол Q): m/z 732,0 [M+H+], время удерживания составляет 1,24 минуты.1H ЯМР (400 МГц, DMSO-d6) δ 8.55 (d), 8.38-8.46 (m), 8.29 (d), 8.03-8.14 (m), 7.97 (d), 7.81 (d), 7.40-7.53 (m), 7.14-7.28 (m), 7.02 (s), 4.71-4.79 (m), 4.43-4.69 (m), 3.96-4.05 (m), 3.71-3.80 (m), 3.62-3.70 (m), 3.49-3.60 (m), 3.40-3.48 (m), 3.15-3.34 (m), 3.10-3.14 (m), 3.01-3.09 (m), 2.94-3.00 (m), 2.83-2.93 (m), 2.65-2.71 (m), 2.55-2.59 (m), 2.32-2.48 (m), 2.04-2.31 (m), 1.61-1.89 (m), 1.37-1.52 (m), 1.21-1.35 (m), 1.15-1.20 (m), 1.02-1.10 (m), 0.75-1.01 (m).
Получение 1,2-диметил-L-пролил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#151)

Стадия 1. Синтез 1,2-диметил-L-пролина (#150). Колбу Парра, содержащую 2-метил-L-пролин (1,0 г, 7,7 ммоль, 1,0 экв.), 40 мл метанола, формальдегид (37% масс.в воде, 2,1 мл, 77 ммоль, 10 экв.) и палладий (10% масс. на углероде, 313 мг, 2,94 ммоль, 0,38 экв.) помещали на шейкер Парра и оставляли встряхиваться при 40 фунт-сила на кв. дюйм водорода в течение примерно 12 часов. Водород удаляли и реакционную смесь фильтровали через набивку целита, которую ополаскивали раствором из 50% метанола и 50% дихлорметана. Остаток концентрировали в вакууме, получая #150 (1,1 г, 100%) в виде белого/слегка черного твердого вещества. ЖХ-МС (Протокол Q): m/z 144,0 [M+H+], время удерживания составляет 0,17 минуты.
Стадия 2. Синтез 1,2-диметил-L-пролил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#151). К перемешиваемой смеси #114 (125 мг, 0,198 ммоль, 1,0 экв.), #150 (37 мг, 0,26 ммоль, 1,3 экв.) и HATU (98 мг, 0,26 ммоль, 1,3 экв.) в 5 мл дихлорметана добавляли основание Хюнига (0,14 мл, 0,80 ммоль, 4,1 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме. К неочищенному материалу добавляли THF (6 мл). К полученной перемешиваемой смеси добавляли LiOH (14 мг, 0,59 ммоль, 3,0 экв.), растворенный в 2 мл воды. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 90 минут. Реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии с обращенной фазой C18 при среднем давлении (градиент: от 5% до 40% ацетонитрила в воде с 0,02% TFA в каждой фазе), получая #151 (147 мг, 69%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 744,3 [M+H+], время удерживания составляет 1,19 минуты; ВЭЖХ (Протокол A при 45°C): m/z 744,4 [M+H+], время удерживания составляет 6,631 минуты (чистота более 98%).1H ЯМР (400 МГц, DMSO-d6) δ 9.57-9.71 (m), 8.75 (d), 8.42 (d), 8.15 (d), 7.14-7.29 (m), 4.70-4.79 (m), 4.40-4.68 (m), 3.95-4.03 (m), 3.73-3.80 (m), 3.37-3.61 (m), 2.97-3.31 (m), 2.79-2.88 (m), 2.66-2.76 (m), 2.54-2.58 (m), 2.31-2.43 (m), 1.94-2.29 (m), 1.57-1.91 (m), 1.21-1.52 (m), 0.85-1.10 (m), 0.74-0.82 (m).
Получение 1,2-диметил-D-пролил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#153)

Стадия 1. Синтез 1,2-диметил-D-пролина (#152). В колбу Парра, содержащую 2-метил-D-пролин (432 мг, 3,34 ммоль, 1,0 экв.), формальдегид (37% масс. в воде, 1,0 мл, 37 мМ, 11 экв.), 3,5 мл метанола и 1 мл воды, добавляли палладий (10% масс. на углероде, 108 мг, 0,304 ммоль, 0,304 экв.). Колбу помещали на шейкер Парра и оставляли встряхиваться при 30 фунт-сила на кв. дюйм водорода в течение примерно 48 часов. Водород удаляли и реакционную смесь подвергали промывке через набивку целита, которую потом промывали метанолом. Органическую фазу концентрировали в вакууме, а затем подвергали азеотропной перегонке с толуолом, получая #152 (517 мг, 100%) в виде твердого вещества.1H ЯМР (400 МГц, метанол-d4): δ [3.61-3.56 (m, 1H), 3.07-2.96 (m, 1H), 2.68 (br s, 3H), 2.34-2.22 (m, 1H), 2.01-1.88 (m, 1H), 1.87-1.73 (m, 1H), 1.40 (br s, 3H)].
Стадия 2. Синтез 1,2-диметил-D-пролил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#153). К перемешиваемой смеси #114 (240 мг, 0,379 ммоль, 1,0 экв.), #152 (71 мг, 0,49 ммоль, 1,3 экв.) и HATU (188 мг, 0,49 ммоль, 1,3 экв.) в 10 мл дихлорметана добавляли основание Хюнига (0,27 мл, 4,1 мМ, 4,1 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме. К неочищенному материалу добавляли THF (6 мл). К полученной перемешиваемой смеси добавляли LiOH (36 мг, 1,5 ммоль, 4 экв.), растворенный в 2 мл воды. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме и остаток очищали посредством хроматографии с обращенной фазой C18 при среднем давлении (градиент: от 5% до 40% ацетонитрила в воде с 0,02% TFA в каждой фазе), получая #153 (220 мг, 78%) в виде белого твердого вещества. ЖХ-МС (Протокол Q): m/z 744,8 [M+H+], время удерживания составляет 1,16 минуты; ВЭЖХ (Протокол A при 45°C): m/z 744,4 [M+H+], время удерживания составляет 6,713 минуты (чистота более 98%).1H ЯМР (400 МГц, DMSO-d6) δ 9.72-9.85 (m), 8.65 (t), 8.41 (d), 8.14 (d), 7.14-7.28 (m), 4.69-4.79 (m), 4.38-4.53 (m), 3.95-4.04 (m), 3.73-3.79 (m), 3.37-3.62 (m), 3.13-3.33 (m), 2.95-3.10 (m), 2.79-2.89 (m), 2.67-2.75 (m), 2.00-2.46 (m), 1.61-1.90 (m), 1.22-1.54 (m), 1.02-1.09 (m), 0.95-1.01 (m), 0.85-0.94 (m), 0.75-0.83 (m).
Получение соли трифторуксусной кислоты и N~2~-[2,2-диметил-3-(метиламино)пропаноил]-N-{(1S,2R)-2-метокси-4-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-1-валинамида (#154)

Стадия 1. Синтез соли трифторуксусной кислоты и N~2~-[2,2-диметил-3-(метиламино)пропаноил]-N-{(1S,2R)-2-метокси-4-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]-пирролидин-1-ил}-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (#154). В сосуд, содержащий #50 (100 мг, 0,152 ммоль, 1,0 экв.) и 1 мл дихлорметана, добавляли 2,2-диметил-3-(метиламино)пропановую кислоту (36 мг, 0,152 ммоль, 1,0 экв.), а затем основание Хюнига (0,080 мл, 0,456 ммоль, 3,0 экв.) и HATU (66 мг, 0,17 ммоль, 1,1 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме, затем переносили в этилацетат, после чего промывали два раза 1 М HCl и один раз рассолом. Органический слой сушили над сульфатом натрия и декантировали. Реакционную смесь концентрировали в вакууме. Добавляли диоксан (1 мл), а затем 4 М HCl в диоксане (1,0 мл, 4,0 ммоль, 26 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 12 часов. Реакционную смесь концентрировали в вакууме. Неочищенный материал очищали посредством хроматографии с обращенной фазой C18 при среднем давлении (градиент: от 10% до 100% ацетонитрила в воде с 0,02% TFA в каждой фазе), получая #154 (55,8 мг, 41%) в виде твердого вещества. ЖХ-МС (Протокол Q): m/z 771,8 [M+H+].1H ЯМР (400 МГц, DMSO-d6) δ 8.70 (d), 8.45 (d), 7.90-8.15 (m), 7.82 (d), 7.75 (d), 7.55 (dd), 7.40 (dd), 6.90-7.10 (m), 5.10-5.30 (m), 4.45-4.55 (b), 4.30-4.45 (m), 4.20-4.30 (m), 3.75-3.90 (m), 3.50-3.60 (m), 3.15-3.40 (m), 3.05-3.15 (m), 2.85-3.05 (m), 2.60-2.85 (m), 2.25-2.40 (m), 1.80-2.25 (m), 1.70-1.80 (m), 1.20-1.60 (m), 0.80-1.10 (m), 0.05-0.80 (m).
Получение соли трифторуксусной кислоты и метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[{N-[2,2-диметил-3-(метиламино)пропаноил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#155)

Стадия 1. Синтез соли трифторуксусной кислоты и метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[{N-[2,2-диметил-3-(метиламино)пропаноил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#155). В сосуд, содержащий #114 (96,2 мг, 0,152 ммоль, 1,0 экв.) и 1 мл дихлорметана, добавляли 2,2-диметил-3-(метиламино)пропановую кислоту (36,1 мг, 0,152 ммоль, 1,0 экв.), а затем основание Хюнига (0,080 мл, 0,456 ммоль, 3,0 экв.) и HATU (66 мг, 0,17 ммоль, 1,1 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме, затем переносили в этилацетат, после чего промывали два раза 1 М HCl и один раз рассолом. Органический слой сушили над сульфатом натрия и декантировали. Реакционную смесь концентрировали в вакууме. Добавляли диоксан (1 мл), а затем 4 М HCl в диоксане (1,0 мл, 4,0 ммоль, 26 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение примерно 12 часов. Реакционную смесь концентрировали в вакууме. Неочищенный материал очищали посредством хроматографии с обращенной фазой C18 при среднем давлении (градиент: от 10% до 100% ацетонитрила в воде с 0,02% TFA в каждой фазе), получая #155 (22,2 мг, 17%).1H ЯМР (400 МГц, DMSO-d6) δ 8.55 (d), 8.22 (d), 8.15-8.35 (m), 7.90-8.05 (m) 7.10-7.25 (m) 4.70-4.80 (m), 4.55-4.65 (m), 4.45-4.52 (m), 3.93-4.00 (m), 3.72-3.78 (m), 3.60-3.70 (m), 3.50-3.60 (m), 3.40-3.50 (m), 2.80-3.30 (m), 2.45-2.60 (m), 2.00-2.45 (m), 1.60-1.80 (m), 1.35-1.50 (m), 1.10-1.35 (m).
Получение соли трифторуксусной кислоты и метил-N-{-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2S)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-1-фенилаланината (#158) и соли трифторуксусной кислоты и метил-N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2R)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланината (#159)

Стадия 1. Синтез (2S)-1-(трет-бутоксикарбонил)-2-метилпиперидин-2-карбоновой кислоты (#156) и (2R)-1-(трет-бутоксикарбонил)-2-метилпиперидин-2-карбоновой кислоты (#157). 1-(трет-Бутоксикарбонил)-2-метилпиперидин-2-карбоновую кислоту (500 мг, 2,06 ммоль, 1 экв.) разделяли посредством сверхкритической флюидной хроматографии (колонка: Chiralcel OJ-H, 250×21 мм; элюент: 90:10 диоксид углерода/этанол; скорость потока: 65 г/мин) с получением соответствующих энантиомеров. Выделяли пик, элюирующийся первым (время удерживания составляет 1,57 минуты), с получением #156 в виде смолы (140 мг, 28%) (стереохимия установлена произвольно (S энантиомер)).1H ЯМР (400 МГц, CDCl3) δ 3.83-3.90 (m, 1H), 2.93-3.01 (m, 1H), 1.87-1.97 (m, 1H), 1.67-1.77 (m, 3H), 1.48-1.66 (m, 2H), 1.46 (s, 3H), 1.44 (s, 9H). Оптическое вращение: [α]D25-21,7° (с 0,40, хлороформ). Выделяли пик, элюирующийся вторым (время удерживания составляет 2,22 минуты), с получением #157 в виде масла (255 мг, 51%) (стереохимия установлена произвольно (R энантиомер)).1H ЯМР (400 МГц, CDCl3) δ 3.83-3.90 (m, 1H), 2.93-3.01 (m, 1H), 1.87-1.97 (m, 1H), 1.67-1.77 (m, 3H), 1.48-1.66 (m, 2H), 1.46 (s, 3H), 1.44 (s, 9H). Оптическое вращение: [α]D25+30,2° (хлороформ).
Стадия 2A. Синтез соли трифторуксусной кислоты и метил-N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2S)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланината (#158). К раствору #156 (8,3 мг, 0,034 ммоль, 1 экв.) в дихлорметане (0,3 мл) и N,N-диметилформамиде (0,05 мл) добавляли N,N-диизопропилэтиламин (0,018 мл, 0,102 ммоль, 3 экв.), а затем HATU (16,1 мг, 0,041 ммоль, 1,2 экв.). Реакционную смесь перемешивали в течение 15 минут, добавляли #114 (23,4 мг, 0,037 ммоль, 1,1 экв.) и перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли дихлорметаном (2,5 мл) и добавляли 10%-ную лимонную кислоту (1,5 мл). Слои разделяли с использованием фазоразделительного картриджа, водный слой экстрагировали дихлорметаном (2×2,5 мл) и объединенные органические слои концентрировали в вакууме. Остаток растворяли в дихлорметане (4 мл) и добавляли трифторуксусную кислоту (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали в вакууме. После очистки посредством хроматографии с обращенной фазой (Метод M*) получали #158 (10,6 мг, 49%). ВЭЖХ (Протокол T): m/z 758,4 [M+H+], время удерживания составляет 2,53 минуты (чистота более 99%).1H ЯМР (400 МГц, DMSO-d6) δ 8.74-8.90 (m), 8.49-8.55 (m), 8.24 (d), 8.08-8.12 (m), 7.94-8.01 (m), 7.14-7.26 (m), 4.71-4.77 (m), 4.57-4.68 (m), 4.44-4.55 (m), 3.94-4.0 (m), 3.73-3.78 (m), 3.40-3.72 (m), 3.16-3.32 (m), 2.98-3.16 (m), 2.82-2.92 (m), 2.47-2.56 (m), 2.38-2.44 (m), 2.20-2.37 (m), 2.08-2.19 (m) 1.74-1.88 (m), 1.61-1.73 (m), 1.52-1.59 (m), 1.22-1.52 (m), 1.05 (dd), 0.94-1.00 (m), 0.85-0.93 (m), 0.74-0.79 (m).
Стадия 2B. Синтез соли трифторуксусной кислоты и метил-N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2R)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланината (#159). К раствору #157 (7,8 мг, 0,032 ммоль, 1 экв.) в дихлорметане (0,3 мл) и N,N-диметилформамиде (0,05 мл) добавляли N,N-диизопропилэтиламин (0,017 мл, 0,096 ммоль, 3 экв.), а затем HATU (14,9 мг, 0,038 ммоль, 1,2 экв.). Реакционную смесь перемешивали в течение 15 минут, добавляли #114 (22,1 мг, 0,035 ммоль, 1,1 экв.) и перемешивали при комнатной температуре в течение 3 часов, затем концентрировали в вакууме. После очистки остатка посредством хроматографии на силикагеле (градиент: от 0 до 80% ацетона в гептане) получали белое твердое вещество, которое растворяли в диоксане (0,2 мл), и добавляли 4 н. HCl в диоксане (0,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и добавляли дополнительное количество 4 н. HCl в диоксане (0,1 мл). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, концентрировали в вакууме. После очистки посредством хроматографии с обращенной фазой (Метод M*) получали #159 (6,6 мг, 33%). ВЭЖХ (Протокол T): m/z 758,4 [M+H+], время удерживания составляет 2,46 минуты (чистота составляет 89%).1H ЯМР (400 МГц, DMSO-d6) δ 8.86-8.95 (m), 8.75-8.84(m), 8.48-8.54 (m), 8.33-8.45 (m), 8.22-8.27 (m), 8.17-8.19 (m), 7.99-8.12 (m), 7.83-7.91 (m), 7.13-7.29 (m), 7.04-7.08 (m), 4.69-4.76 (m), 4.55-4.66 (m), 4.45-4.53 (m), 3.96-4.01 (m), 3.41-3.78 (m), 3.28-3.33 (m), 3.24-3.27 (m), 3.16-3.23 (m), 3.11-3.15 (m), 3.02-3.10 (m), 2.93-3.02 (m), 2.91-2.93 (m), 2.84-2.91 (m), 2.76-2.82 (m), 2.69-2.71 (m), 2.60-2.63 (m), 2.53-2.55 (m), 2.47-2.53 (m), 2.40-2.46 (m), 2.30-2.38 (m), 2.20-2.30 (m), 2.06-2.17 (m), 1.75-1.87 (m), 1.52-1.74 (m), 1.35-1.51 (m), 1.14-1.34 (m), 1.01-1.08 (m), 0.92-1.0 (m), 0.85-0.94 (m), 0.74-0.82 (m).
Получение соли трифторуксусной кислоты и N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2S)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланина (#162) и соли трифторуксусной кислоты и N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2R)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланина (#163)

Стадия 1A. Синтез метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(2S)-1-(трет-бутоксикарбонил)-2-метилпиперидин-2-ил]карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#160). К раствору #156 (106 мг, 0,436 ммоль, 1 экв.) в дихлорметане (3 мл) и N,N-диметилформамиде (0,5 мл) добавляли диизопропилэтиламин (0,228 мл, 1,31 ммоль, 3 экв.), а затем HATU (205 мг, 0,523 ммоль, 1,2 экв.). Реакционную смесь перемешивали в течение 15 минут, добавляли #114 (276 мг, 0,436 ммоль, 1 экв.) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли дихлорметаном (10 мл) и промывали 10%-ной лимонной кислотой (3×5 мл). Органический слой сушили над сульфатом натрия, фильтровали и фильтрат концентрировали в вакууме. После очистки остатка посредством хроматографии на силикагеле (градиент: от 0 до 80% ацетона в гептане) получали #160 (145 мг, 39%). ЖХ-МС (Протокол Q1): m/z 858,8 [M+H+], время удерживания составляет 1,12 минуты.
Стадия 1B. Синтез метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(2R)-1-(трет-бутоксикарбонил)-2-метилпиперидин-2-ил]карбонил}-1-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#161). К раствору #157 (109 мг, 0,448 ммоль, 1 экв.) в дихлорметане (3 мл) и N,N-диметилформамиде (0,5 мл) добавляли диизопропилэтиламин (0,234 мл, 1,34 ммоль, 3 экв.), а затем HATU (205 мг, 0,538 ммоль, 1,2 экв.). Реакционную смесь перемешивали в течение 15 минут и добавляли #114 (284 мг, 0,448 ммоль, 1 экв.). После перемешивания при комнатной температуре в течение 2 часов смесь разбавляли дихлорметаном (10 мл), промывали 10%-ной лимонной кислотой (3×5 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После очистки остатка посредством хроматографии на силикагеле (градиент: от 0 до 100% ацетона в гептане) получали #161 (185 мг, 48%). ЖХ-МС (Протокол Q): m/z 858,3 [M+H+], время удерживания составляет 2,25 минуты.
Стадия 2A. Синтез соли трифторуксусной кислоты и N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2S)-2-метилпиперидин-2-ил]карбонил}-1-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланина (#162). К раствору #160 (145 мг, 0,169 ммоль, 1 экв.) в тетрагидрофуране (1,25 мл) добавляли гидроксид лития (8 мг, 0,338 ммоль, 2 экв.), растворенный в воде (0,75 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и упаривали досуха в вакууме. Остаток растворяли в дихлорметане (2,5 мл) и добавляли трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали в течение 30 минут, концентрировали в вакууме и очищали посредством хроматографии с обращенной фазой C18 при среднем давлении (градиент: от 0% до 100% ацетонитрила в воде с 0,02% TFA в каждой фазе) с получением указанного в заголовке соединения #162 (145 мг, количественный выход) в виде белого твердого вещества. ВЭЖХ (Протокол U): m/z 744,5 [M+H+], время удерживания составляет 7,121 минуты (чистота составляет 98%).1H ЯМР (400 МГц, DMSO-d6) δ 8.76-8.96 (m), 8.52-8.58 (m), 8.38-8.43 (m), 8.11-8.16 (m), 7.27-7.30 (m), 7.12-7.27 (m), 7.01-7.05 (m), 4.71-4.79 (m), 4.48-4.67 (m), 4.39-4.47 (m), 3.79-4.22 (m), 3.71-3.78 (m), 3.38-3.57 (m), 3.22-3.30 (m), 3.14-3.23 (m), 3.07-3.13 (m), 2.96-3.06 (m), 2.76-2.87 (m), 2.66-2.68 (m), 2.47-2.57 (m), 2.42-2.44 (m), 2.06-2.40 (m), 1.73-1.89 (m), 1.51-1.72 (m), 1.36-1.49 (m), 1.20-1.35 (m), 1.00-1.09 (m), 0.95-0.99 (m), 0.83-0.94 (m), 0.73-0.80 (m).
Стадия 2B. Синтез соли трифторуксусной кислоты и N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(N-{[(2R)-2-метилпиперидин-2-ил]карбонил}-L-валил)амино]гептаноил}пирролидин-2-ил]-2-метилпропаноил}-L-фенилаланина (#163). Соединение #161 (185 мг, 0,216 ммоль, 1 экв.) превращали в неочищенное указанное в заголовке соединение #163, используя методику, описанную для получения #162. Неочищенный материал очищали посредством хроматографии с обращенной фазой C18 при среднем давлении (градиент: от 0% до 85% ацетонитрила в воде с 0,02% TFA в каждой фазе) с получением #163 (127 мг, 68%) в виде белого твердого вещества. ВЭЖХ (Протокол U): m/z 744,5 [M+H+], время удерживания составляет 7,077 минуты (чистота составляет 98%).1H ЯМР (400 МГц, DMSO-d6) δ 8.79-8.99 (m), 8.36-8.49 (m), 8.12-8.17 (m), 7.31-7.34 (m), 7.11-7.27 (m), 7.05-7.09 (m), 4.71-4.77 (m), 4.54-4.68 (m), 4.40-4.53 (m), 3.88-4.39 (m), 3.71-3.77 (m), 3.39-3.58 (m), 3.22-3.32 (m), 3.10-3.22 (m), 3.04-3.09 (m), 2.92-3.03 (m), 2.77-2.88 (m), 2.68-2.71 (m), 2.47-2.57 (m), 2.43-2.45 (m), 2.30-2.42 (m), 2.03-2.29 (m), 1.74-1.88 (m), 1.52-1.73 (m), 1.37-1.51 (m), 1.17-1.37 (m), 1.00-1.07 (m), 0.95-0.99 (m), 0.84-0.93 (m), 0.73-0.81 (m).
Получение соли трифторуксусной кислоты и метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3R)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#172) и соли трифторуксусной кислоты и метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3R)-фрторпирролидин-3-ил]карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#173)

Стадия 1. Синтез метил-(3R)-1-бензил-3-фторпирролидин-3-карбоксилата (#164) и метил-(3S)-1-бензил-3-фторпирролидин-3-карбоксилата (#165). Известный метил-1-бензил-3-фторпирролидин-3-карбоксилат (3900 мг, 16,4 ммоль, 1 экв.) разделяли посредством сверхкритической флюидной хроматографии (колонка: Chiralpak 1C, 250×21 мм; элюент: 95:5 диоксид углерода/пропанол; скорость потока: 65 г/мин) с получением соответствующих энантиомеров. Выделяли пик, элюирующийся первым (время удерживания составляет 3,37 минуты), с получением #164 (1720 мг, 36%) в виде одиночного энантиомера (стереохимия установлена произвольно (R энантиомер)).1H ЯМР (400 МГц, TMS-CDCl3; δ 7.17-7.30 (m, 5H), 3.74 (s, 3H), 3.65 (d, J=12,9 Гц, 1H), 3.63 (d, J=12,9 Гц, 1H), 2.86-3.03 (m, 3H), 2.61 (q, J=8,0 Гц, 1H), 2.34-2.46 (m, 1H), 2.13-2.26 (m, 1H). Оптическое вращение: [α]D25+24,7° (хлороформ). Выделяли пик, элюирующийся вторым (время удерживания составляет 3,91 минуты), с получением #165 (1600 мг, 33%) в виде одиночного энантиомера (стереохимия установлена произвольно (S энантиомер)). 1H ЯМР (400 МГц, CDCl3; (CH3)4Si), δ 7.17-7.30 (m, 5H), 3.74 (s, 3H), 3.65 (d, J=12,9 Гц, 1H), 3.63 (d, J=12,9 Гц, 1H), 2.86-3.03 (m, 3H), 2.61 (q, J=8,0 Гц, 1H), 2.34-2.46 (m, 1H), 2.13-2.26 (m, 1H). Оптическое вращение: [α]D25-23,3° (хлороформ).
Стадия 2A. Синтез 1-трет-бутил-3-метил-(3R)-3-фторпирролидин-1,3-дикарбоксилата (#166). К раствору, содержащему #164 (355 мг, 1,50 ммоль, 1 экв.) и ди-трет-бутил-карбонат (400 мг, 1,8 ммоль, 1,2 экв.) в метаноле (15,5 мл), добавляли 10% Pd/C (70 мг). Реакционную смесь гидрировали при 45 фунт-сила на кв. дюйм в шейкере Парра в течение 22 часов, фильтровали через целит, фильтрат концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0 до 30% этилацетата в гептане) с получением #166 в виде прозрачного масла (272 мг, 74%).1H ЯМР (400 МГц, CDCl3) δ 3.87 (s, 3H), 3.85-3.66 (m, 3H), 3.56 (m, 1H), 2.53-2.28 (m, 2H), 1.51 (s, 9H).
Стадия 2B. Синтез 1-трет-бутил-3-метил-(3S)-3-фторпирролидин-1,3-дикарбоксилата (#167). Соединение #165 (362 мг, 1,53 ммоль, 1 экв.) превращали в #167 с 63%-ным выходом, используя способ, описанный выше для #164.1H ЯМР (400 МГц, CDCl3) δ 3.87 (s, 3H), 3.85-3.66 (m, 3H), 3.56 (m, 1H), 2.53-2.28 (m, 2H), 1.51 (s, 9H).
Стадия 3A. Синтез (3R)-1-(трет-бутоксикарбонил)-3-фторпирролидин-3-карбоновой кислоты (#168). К раствору #166 (272 мг, 1,10 ммоль, 1 экв.), растворенного в метаноле (2,96 мл), добавляли водный раствор гидроксида натрия (2,5 M, 0,88 мл) и реакционную смесь перемешивали при комнатной температуре в течение 3,5 часов. Реакционную смесь гасили 10%-ной водной лимонной кислотой (5 мл), добавляли этилацетат (100 мл) и слои разделяли. Органический слой промывали 10%-ной лимонной кислотой, водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением #168 в виде белого твердого вещества (253 мг, 99%).1H ЯМР (400 МГц, CDCl3) δ 3.96-3.69 (m, 3H), 3.59 (m, 1H), 2.59-2.33 (m, 2H), 1.51 (s, 9H). ЖХ-МС (Протокол Q1): m/z 232,1 [M-H+], время удерживания составляет 0,67 минуты. Время удерживания при хиральной ВЭЖХ: 3,39 мин (чистота составляет 99%) (колонка: Chiralpak AD-H, 4,6 мм × 25 см, подвижная фаза 5-60% CO2/метанол, скорость потока 3,0 мл/мин); оптическое вращение: [α]D25 4,8 (c=0,52, MeOH).
Стадия 3B. Синтез (3S)-1-(трет-бутоксикарбонил)-3-фторпирролидин-3-карбоновой кислоты (#169). К раствору #167 (238 мг, 0,963 ммоль, 1 экв.), растворенного в метаноле (2,6 мл), добавляли водный раствор гидроксида натрия (2,5 M, 0,88 мл) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Затем реакционную смесь гасили 10%-ной водной лимонной кислотой (5 мл), добавляли этилацетат (100 мл) и слои разделяли. Органический слой промывали 10%-ной лимонной кислотой, водой и рассолом, затем сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением #169 в виде белого твердого вещества (221 мг, 99%).1H ЯМР (400 МГц, CDCl3) δ 3.96-3.69 (m, 3H), 3.59 (m, 1H), 2.59-2.33 (m, 2H), 1.51 (s, 9H). ЖХ-МС (Протокол Q1): m/z 232,1 [M-H+], время удерживания составляет 0,67 минуты. Время удерживания при хиральной ВЭЖХ: 3,95 мин (чистота составляет 98%) (колонка: Chiralpak AD-H, 4,6 мм × 25 см, подвижная фаза 5-60% CO2/метанол, скорость потока 3,0 мл/мин); оптическое вращение: [α]D25-3,6 (c=0,55, MeOH).
Стадия 4A. Синтез (3R)-1-(трет-бутоксикарбонил)-3-фторпирролидин-3-карбоксилата лития (#170). К раствору #168 (50 мг, 0,21 ммоль, 1 экв.) в метаноле (0,2 мл) добавляли раствор гидроксида лития (9,2 мг, 0,38 ммоль, 1,8 экв.), растворенного в воде (0,1 мл). Затем добавляли тетрагидрофуран (0,3 мл) и реакционную смесь перемешивали при 45°C в течение 18 часов. Реакционную смесь концентрировали в вакууме и материал подвергали азеотропной перегонке (3X) с толуолом (2 мл), получая #170 (51 мг, 100%) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 4B. Синтез (3S)-1-(тре-бутоксикарбонил)-3-фторпирролидин-3-карбоксилата лития (#171). К раствору #169 (75 мг, 0,32 ммоль, 1 экв.) в метаноле (0,3 мл) добавляли раствор гидроксида лития (13,8 мг, 0,572 ммоль, 1,8 экв.) в воде (0,4 мл). Затем добавляли тетрагидрофуран (0,45 мл) и реакционную смесь перемешивали при 45°C в течение 18 часов. Реакционную смесь концентрировали в вакууме и материал подвергали азеотропной перегонке (3X) с толуолом (4 мл), получая #171 (77 мг, 100%) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 5A. Синтез соли трифторуксусной кислоты и метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3R)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#172). К суспензии #168 (36,9 мг, 0,158 ммоль, 1 экв.) и #114 (100 мг, 0,158 ммоль, 1,0 экв.) в N,N-диметилформамиде (0,8 мл) и дихлорметане (3,6 мл) добавляли N,N-диизопропилэтиламин (0,083 мл, 0,474 ммоль, 3 экв.), а затем HATU (60,7 мг, 0,158 ммоль, 1,0 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли этилацетатом и последовательно промывали водой, 10%-ной водной лимонной кислотой (масс./об.) и рассолом. Органический слой сушили над сульфатом натрия и концентрировали в вакууме с получением 220 мг (164% от теоретического значения) неочищенного промежуточного соединения. Часть этого неочищенного промежуточного соединения (50 мг, 23%) растворяли в дихлорметане (1,5 мл) и добавляли трифторуксусную кислоту (0,4 мл). Смесь перемешивали при комнатной температуре в течение 2 часов и упаривали досуха в вакууме. После очистки посредством хроматографии с обращенной фазой (Метод M*) получали #172 (15,8 мг, 51%). ЖХ-МС (Протокол Q): m/z 748,9 [M+H+], время удерживания составляет 1,29 минуты.1H ЯМР (DMSO-d6) δ 9.16-9.43 (m), 8.48-8.53 (m), 8.40-8.44 (m), 8.34-8.39 (m), 8.22-8.30 (m), 8.09-8.16 (m), 7.87-7.91 (m), 7.77-7.83 (m), 7.12-7.24 (m), 4.56-4.72 (m), 4.41-4.54 (m), 3.92-4.00 (m), 3.70-3.75 (m), 3.39-3.66 (m), 3.20-3.25 (m), 3.12-3.20 (m), 2.97-3.11 (m), 2.94 (ors), 2.75-2.89 (m), 2.63-2.69 (m), 2.47-2.54 (m), 2.29-2.45 (m), 2.15-2.27 (m), 2.02-2.15 (m), 1.57-1.87 (m), 1.33-1.51 (m), 1.19-1.30 (m),1.02 (dd), 0.82-0.97 (m), 0.70-0.79 (m).
Стадия 5B. Синтез соли трифторуксусной кислоты и метил-N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3R)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланината (#173). К суспензии #169 (36,9 мг, 0,158 ммоль, 1 экв.) и #114 (100,0 мг, 0,158 ммоль, 1 экв.) в N,N-диметилформамиде (0,8 мл) и дихлорметане (3,6 мл) добавляли N,N-диизопропилэтиламин (0,083 мл, 0,474 ммоль, 3 экв.), а затем HATU (60,7 мг, 0,158 ммоль, 1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 14 часов, разбавляли этилацетатом и последовательно промывали водой, 10%-ной водной лимонной кислотой (масс./об.) и рассолом. Органический слой сушили над сульфатом натрия и концентрировали в вакууме с получением 180 мг (134% от теоретического значения) неочищенного промежуточного соединения. Часть этого неочищенного промежуточного соединения (50 мг, 27%) растворяли в дихлорметане (1,5 мл), добавляли трифторуксусную кислоту (0,4 мл) и смесь перемешивали при комнатной температуре в течение 2 часов и упаривали досуха в вакууме. После очистки посредством хроматографии с обращенной фазой (Метод M*) получали #173 (17,6 мг, 45%). ЖХ-МС (Протокол Q): m/z 748,9 [M+H+], время удерживания составляет 1,29 минуты.1H ЯМР (DMSO-d6) δ 9.35-9.50 (m), 9.22-9.34 (m), 8.47-8.52 (m), 8.39-8.45 (m), 8.30-8.37 (m), 8.22-8.24 (m), 8.09-8.13 (m), 7.80-7.85 (m), 7.67-7.72 (m), 7.09-7.24 (m), 6.97-6.98 (m), 4.65-4.72 (m), 4.55-4.64 (m), 4.41-4.50 (m), 3.92-3.99 (m), 3.42-3.75 (m), 3.32-3.39 (m), 3.25-3.31 (m), 3.21-3.24 (m), 3.11-3.20 (m), 3.06-3.11 (M), 2.97-3.05 (m), 2.95 (br s), 2.83-2.88 (m), 2.76-2.82 (m), 2.65-2.70 (m), 2.44-2.54 (m), 2.15-2.42 (m), 2.02-2.14 (m), 1.57-1.84 (m), 1.33-1.48 (m), 1.19-1.30 (m), 1.02 (dd), 0.92-0.97 (m), 0.82-0.91 (m), 0.70-0.77 (m).
Получение гидрохлоридной соли (2S)-N-[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-(метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбокс-амида (#178) и гидрохлоридной соли (2R)-N-[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-(метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбокс-амида (#180)

Стадия 1. Синтез пентафторфенил-(3R,4S,5S)-4-[{N-[(9H-флуорен-9-илметокси)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноата (#174). К раствору #@5 (19,43 г, 37,03 ммоль, 1 экв.) в дихлорметане (100 мл) и пиридине (5,86 г, 74,1 ммоль, 2 экв.) добавляли пентафтор-фенилтрифторацетат (20,7 г, 74,1 ммоль, 2 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0 до 52% этилацетата в гептане) с получением #174 (23,58 г, 92%) в виде желтого масла. ЖХ-МС (Протокол Q1): m/z 691,2 [M+H+], время удерживания составляет 1,23 минуты.
Стадия 2. Синтез N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N~2~-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валинамида (#175). К раствору #174 (706 мг, 1,02 ммоль, 1 экв.) и #64 (311 мг, 1,02 ммоль, 1 экв.) в дихлорметане (3 мл) добавляли N,N-диизопропилэтиламин (400 мг, 3,07 ммоль, 3 экв.). После 18 часов перемешивания при комнатной температуре реакционную смесь концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0 до 100% этилацетата в гептане) с получением #175 (560 мг, 68%) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 611,8 [M+H+], время удерживания составляет 1,15 минуты.
Стадия 3. Синтез N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#176). Согласно общей процедуре A из #175 (560 мг, 0,690 ммоль, 1 экв.) в дихлорметане (9 мл) и N,N-диэтиламине (6,0 мл) синтезировали неочищенное целевое соединение, которое очищали посредством хроматографии на силикагеле (градиент: от 0 до 50% метанола в дихлорметане) с получением #176 (351 мг, 87%) в виде желтого масла. ЖХ-МС (Протокол Q1): m/z 589,5 [M+H+], время удерживания составляет 0,72 минуты.
Стадия 4A. Синтез трет-бутил-(2S)-2-{[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил](метил)-амино}-3-метил-1-оксобутан-2-ил]карбамоил}-2-метилпиперидин-1-карбоксилата (#177). Согласно общей методике D из #176 (100 мг, 0,170 ммоль, 1 экв.), #156 (53,8 мг, 0,221 ммоль, 1,3 экв.), дихлорметана (4,5 мл), HATU (84,9 мг, 0,221 ммоль, 1,3 экв.) и N,N-диизопропилэтиламина (0,123 мл, 0,697 ммоль, 4,1 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0 до 100% этилацетата в гептане) с получением #177 (145 мг, предполагается количественный выход) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 814,7 [M+H+], время удерживания составляет 1,14 минуты.
Стадия 4B. Синтез трет-бутил-(2R)-2-{[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-(метил)-амино}-3-метил-1-оксобутан-2-ил]карбамоил}-2-метилпиперидин-1-карбоксилата (#179). Согласно общей методике D из #176 (100 мг, 0,170 ммоль, 1 экв.), #157 (53,8 мг, 0,221 ммоль, 1,3 экв.), дихлорметана (4,5 мл), HATU (84,9 мг, 0,221 ммоль, 1,3 экв.) и N,N-диизопропилэтиламина (0,123 мл, 0,697 ммоль, 4,1 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0 до 100% этилацетата в гептане) с получением #179 (155 мг, предполагается количественный выход) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 814,7 [M+H+], время удерживания составляет 1,14 минуты.
Стадия 5A. Синтез гидрохлоридной соли (2S)-N-[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-(метил)-амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбоксамида (#178). Согласно общей методике C из #177 (143 мг, 0,176 ммоль, 1 экв.) и 4 М раствора соляной кислоты в диоксане (2,0 мл) синтезировали целевой материал в виде смолы (145 мг). Порцию этого неочищенного остатка (25 мг) подвергали азеотропной перегонке со смесью метанол/ацетонитрил с получением #178 (20 мг, 89%-ный выход) в виде белого твердого вещества.1H ЯМР (400 МГц, DMSO-d6), δ 8.96-9.07 (m), 8.79-8.96 (m), 8.58 (d), 8.02-8.08 (m), 7.77-7.83 (m), 7.25-7.31 (m), 7.19-7.24 (m), 6.56-6.67 (m), 6.12-6.21 (m), 5.13-5.22 (m), 4.72-4.81 (m), 4.63-4.70 (m), 4.50-4.59 (m), 4.07-4.16 (m), 3.98-4.05 (m), 3.80-3.86 (m), 3.55-3.76 (m), 3.46-3.54 (m), 3.38 -3.44 (m), 3.26-3.35 (m), 3.18-3.24 (m), 3.05-3.18 (m), 2.98-3.04 (m), 2.40-2.55 (m), 2.27-2.35 (m), 2.08-2.27 (m), 1.74-1.98 (m), 1.50-1.74 (m), 1.20-1.46 (m), 0.73-1.16 (m). ЖХ-МС (Протокол Q1): m/z 714,6 [M+H+], время удерживания составляет 0,76 минуты. ВЭЖХ (Протокол U): m/z 714,5 [M+H+] время удерживания составляет 7,124 минуты (чистота составляет 91%).
Стадия 5B. Синтез гидрохлоридной соли (2R)-N-[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил](метил)-амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбокс-амида (#180). Согласно общей методике С из #179 (162 мг, 0,199 ммоль, 1 экв.) и 4 М раствора соляной кислоты в диоксане (2,0 мл) синтезировали целевой материал в виде смолы (155 мг). Порцию этой смолы (25 мг) подвергали азеотропной перегонке со смесью 1/1 метанол/ацетонитрил с получением #180 (20 мг, 83%) в виде твердого вещества.1H ЯМР (400 МГц, DMSO-d6), δ 9.02-9.13 (m), 8.83-8.93 (m), 8.39-8.46 (m), 8.00-8.06 (m), 7.78 (t), 7.24-7.30 (m), 7.16-7.21 (m), 6.54-6.65 (m), 6.09-6.19 (m), 5.11-5.18 (m), 4.69-4.78 (m), 4.59-4.68 (m), 4.46-4.56 (m), 4.08-4.13 (m), 3.95-4.03 (m), 3.77-3.85 (m), 3.54-3.73 (m), 3.43-3.53 (m), 3.37-3.42 (m), 3.24-3.33 (m), 3.16-3.22 (m), 3.03-3.15 (m), 2.99-3.02 (m), 2.89-2.98 (m), 2.65-2.76 (m), 2.41-2.54 (m), 2.15-2.39 (m), 2.07-2.15 (m), 1.51-1.94 (m), 1.49 (d), 1.38 (t), 1.20-1.32 (m), 1.02-1.09 (m), 0.84-0.97 (m), 0.73-0.81 (m). ЖХ-МС (Протокол Q1): m/z 714,6 [M+H+], время удерживания составляет 0,76; ВЭЖХ (Протокол U): m/z 714,4 [M+H+], время удерживания составляет 7,409 минуты (чистота составляет 90%).
Получение соли муравьиной кислоты и 2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#182), соли трифторуксусной кислоты и N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N~2~-{[(3R)-3-фторпирролидин-3-ил]карбонил}-N-метил-L-валинамида (#184) и соли трифторуксусной кислоты и N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N~2~-{[(3S)-3-фторпирролидин-3-ил]карбонил}-N-метил-L-валинамида (#186)

Стадия 1A. Синтез 1-(трет-бутоксикарбонил)-2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#181). Согласно общей методике D из #176 (100 мг, 0,170 ммоль, 1 экв.), (S)-1-(трет-бутоксикарбонил)-2-метил-L-пролина (50,7 мг, 0,221 ммоль, 1,3 экв.), дихлорметана (4,3 мл), HATU (84,9 мг, 0,221 ммоль, 1,3 экв.) и N,N-диизопропилэтиламина (0,123 мл, 0,697 ммоль, 4,1 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0 до 100% этилацетата в гептане) с получением #181 (142 мг, предполагался количественный выход). ЖХ-МС (Протокол Q1): m/z 800,6 [M+H+], время удерживания составляет 1,11 минуты.
Стадия 1B. Синтез трет-бутил-(3R)-3-{[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил](метил)-амино}-3-метил-1-оксобутан-2-ил]карбамоил}-3-фторпирролидин-1-карбокси-лата (#183). К раствору #170 (18,2 мг, 0,076 ммоль, 1 экв.) в дихлорметане (1,8 мл) и N,N-диметилформамиде (0,3 мл) добавляли N,N-диизопропилэтиламин (0,040 мл, 0,228 ммоль, 3 экв.), а затем HATU (29,2 мг, 0,076 ммоль, 1 экв.). После перемешивания в течение 10 минуты при комнатной температуре добавляли #176 (45 мг, 0,076 ммоль, 1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и добавляли дополнительное количество HATU (29 мг, 0,076 ммоль, 1 экв.). Спустя 8 часов реакционную смесь концентрировали в вакууме с получением #183 (61,0 мг, количественно), который использовали на следующей стадии без дополнительной очистки. ЖХ-МС (Протокол Q1): m/z 826,6 [M+Na+], время удерживания составляет 1,05 минуты.
Стадия 1С. Синтез трет-бутил-(3S)-3-{[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил](метил)-амино}-3-метил-1-оксобутан-2-ил]карбамоил}-3-фторциклопентанкарбоксилата (#185). К раствору #171 (24 мг, 0,1 ммоль, 1 экв.) в дихлорметане (2,35 мл) и N,N-диметилформамиде (0,33 мл) добавляли N,N-диизопропилэтиламин (0,053 мл, 0,300 ммоль, 3 экв.), а затем HATU (38,4 мг, 0,100 ммоль, 1 экв.). После перемешивания при комнатной температуре в течение 10 минуты добавляли #176 (58,9 мг, 0,1 ммоль, 1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и добавляли дополнительное количество HATU (38,4 мг, 0,100 ммоль, 1 экв.) и N,N-диметилформамида (0,2 мл) и перемешивали в течение дополнительных 9 часов. Реакционную смесь концентрировали в вакууме с получением #185 (80 мг, количественно), который использовали на следующей стадии без дополнительной очистки. ЖХ-МС (Протокол Q1): m/z 804,6 [M+H+], время удерживания составляет 1,05 минуты.
Стадия 2A. Синтез соли муравьиной кислоты и 2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксо-гептан-4-ил]-N-метил-L-валинамида (#182). Согласно общей методике C из #181 (136 мг, 0,170 ммоль, 1 экв.) и 4 М раствора соляной кислоты в диоксане (2 мл) синтезировали целевой материал в виде смолы (142 мг). Порцию этого неочищенного остатка (20 мг, 14%) подвергали азеотропной перегонке со смесью 1/1 метанол/ацетонитрил и затем очищали посредством хроматографии с обращенной фазой (Метод O) с получением #182 (10 мг, 57% за две стадии) в виде твердого вещества. ВЭЖХ (Протокол U): m/z 700,4 [M+H+], время удерживания составляет 7,106 минуты (чистота более 90%).1H ЯМР (400 МГц, DMSO-d6), δ 8.25-8.39 (m), 8.20-8.25 (m), 7.96-7.99 (m), 7.74-7.77 (m), 6.55-6.63 (m), 6.10-6.18 (m), 5.11-5.18 (m), 4.66-4.72 (m), 4.51-4.61 (m), 4.46-4.50 (m), 3.96-4.01 (m), 3.37-3.86 (m), 3.20-3.36 (m), 3.11-3.19 (m), 3.03-3.11 (m), 2.98-3.03 (m), 2.90-2.96 (m), 2.77-2.79 (m), 2.65-2.73 (m), 2.57-2.63 (m), 2.47-2.56 (m), 2.36-2.46 (m), 2.26-2.32 (m), 2.14-2.25 (m), 2.03-2.10 (m), 1.92-2.03 (m), 1.72-1.92 (m), 1.64-1.72 (m), 1.60-1.64 (m), 1.50-1.59 (m), 1.39-1.48 (m), 1.23-1.32 (m), 1.18-1.22 (m), 1.12-1,14 (m), 1.01-1.09 (m), 0.83-1.00 (m), 0.74-0.83 (m), 0.70- 0.74 (m).
Стадия 2B. Синтез соли трифторуксусной кислоты и N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N~2~-{[(3R)-3-фторпирролидин-3-ил]карбонил}-N-метил-L-валинамида (#184). Согласно общей методике C из #183 (61,1 мг, 0,076 ммоль, 1 экв.), дихлорметана (0,3 мл) и 4 М раствора соляной кислоты в диоксане (0,9 мл) синтезировали неочищенный целевой материал (140 мг). Порцию этого неочищенного материала (98 мг, 69%) очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #184 (7,6 мг, 20% за две стадии). ВЭЖХ (Протокол T): m/z 704,5 [M+H+], время удерживания составляет 2,50 минуты (чистота составляет 84%).1H ЯМР (400 МГц, DMSO-d6), δ 8.67-8.71 (m), 8.44-8.48 (m), 8.27-8.33 (m), 8.22-8.27 (m), 7.97-8.03 (m), 7.84-7.89 (m), 7.74-7.81 (m), 7.43-7.48 (m), 7.23-7.29 (m), 7.17-7.21 (m), 6.55-6.66 (m), 6.10-6.19 (m), 5.10-5.19 (m), 4.66-4.75 (m), 4.51-4.63 (m), 3.96-4.04 (m), 3.78-3.85 (m), 3.65-3.73 (m), 3.46-3.62 (m), 3.37-3.45 (m), 3.24-3.37 (m), 3.21-3.24 (m), 3.13-3.21 (m), 3.02-3.13 (m), 2.95-3.00 (m), 2.80-2.82 (m), 2.66-2.71 (m), 2.47-2.57 (m), 2.24-2.46 (m), 2.09-2.24 (m), 1.95-2.05 (m), 1.49-1.93 (m), 1.22-1.34 (m), 1.02-1.09 (m), 0.83-1.01 (m), 0.74-0.82 (m).
Стадия 2C. Синтез соли трифторуксусной кислоты и N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(циклогепта-2,4,6-триен-1-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N~2~-{[(3S)-3-фторпирролидин-3-ил]карбонил}-N-метил-L-валинамида (#186). Согласно общей методике C из #185 (80,3 мг, 0,1 ммоль, 1 экв.), дихлорметана (0,4 мл) и 4 М раствора соляной кислоты в диоксане (1,2 мл) синтезировали неочищенный целевой материал, порцию которого (94 мг, 53%) очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #186 (11,8 мг, 32% за две стадии).1H ЯМР (400 МГц, DMSO-d6), δ 8.85-9.07 (m), 8.29-8.41(m), 7.99-8.04 (m), 7.76-7.84 (m), 6.56-6.68 (m), 6.12-6.21 (m), 5.12-5.21 (m), 4.87-4.99 (m), 4.69-4.79 (m), 4.49-4.67 (m), 3.98-4.06 (m), 3.80-3.87 (m), 3.64-3.76 (m), 3.55-3.64 (m), 3.47-3.54 (m), 3.39-3.46 (m), 3.26-3.39 (m), 3.22-3.25 (m), 3.18-3.22 (m), 3.06-3.14 (m), 2.98-3.01 (m), 2.55-2.57 (m), 2.42-2.49 (m), 2.11-2.38 (m), 2.09 (s), 1.78-1.97 (m), 1.72-1.77 (m), 1.51-1.71 (m), 1.24-1.36 (m), 1.07 (dd), 0.83-1.03 (m), 0.75-0.82 (m). ВЭЖХ (Протокол T): m/z 704,5 [M+H+], время удерживания составляет 2,48 минуты (чистота составляет 100%).
Получение формиатной соли (2S)-N-[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-(метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбокс-амида (#188), формиатной соли (2R)-N-[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-(метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбокс-амида (#190), соли трифторуксусной кислоты и N~2~-{[(3R)-3-фторпирролидин-3-ил]карбонил}-N-(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#192) и соли трифторуксусной кислоты и N~2~4[(3S)-3-фторпирролидин-3-ил]карбонил}-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валин-амида(#194)

Стадия 1A. Синтез трет-бутил-(2S)-2-{[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)-этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)-амино}-3-метил-1-оксобутан-2-ил]карбамоил}-2-метилпиперидин-1-карбоксилата (#187). Согласно общей методике D из #86 (280 мг, 0,4 ммоль, 1 экв.), #156 (100 мг, 0,4 ммоль, 1 экв.), дихлорметана (5 мл), HATU (182 мг, 0,48 ммоль, 1,2 экв.) и N,N-диизопропилэтиламина (100 мг, 0,8 ммоль, 2 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0,01 до 0,05% метанола в дихлорметане) с получением #187 (220 мг, 62%) в виде белого твердого вещества. ВЭЖХ (Протокол V): m/z 883,57 [M+H+], время удерживания составляет 3,23 минуты (чистота составляет 95%).
Стадия 1B. Синтез трет-бутил-(2R)-2-{[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)-этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)-амино}-3-метил-1-оксобутан-2-ил]карбамоил}-2-метилпиперидин-1-карбоксилата (#189). Согласно общей методике D из #86 (400 мг, 0,6 ммоль, 1 экв.), #157 (146 мг, 0,6 ммоль, 1 экв.), дихлорметана (10 мл), HATU (259 мг, 0,72 ммоль, 1,2 экв.) и N,N-диизопропилэтиламина (158 мг, 1,2 ммоль, 2 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 0,01 до 0,05% метанола в дихлорметане) с получением #189 (220 мг, 37%) в виде белого твердого вещества. ВЭЖХ (Протокол W): m/z 883,7 [M+H+], время удерживания составляет 4,12 минуты (чистота составляет 95%).
Стадия 1C. Синтез трет-бутил-(3R)-3-фтор-3-{[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]карбамоил}пирролидин-1-карбоксилата (#191). Согласно общей методике D из #86 (300 мг, 0,45 ммоль, 1 экв.), #168 (106 мг, 0,45 ммоль, 1 экв.), дихлорметана (10 мл), HATU (194 мг, 0,54 ммоль, 1,2 экв.) и диизопропилэтиламина (117 мг, 0,9 ммоль, 2 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии с обращенной фазой (Метод P) с получением #191 (159 мг, 40%) в виде белого твердого вещества. ВЭЖХ (Протокол X): m/z 873,4 [M+H+], время удерживания составляет 3,32 минуты (чистота составляет 99%).
Стадия 1D. Синтез трет-бутил-(3S)-3-фтор-3-{[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-(метил)амино}-3-метил-1-оксобутан-2-ил]карбамоил}пирролидин-1-карбокси-лата (#193). Согласно общей методике D из #86 (300 мг, 0,45 ммоль, 1 экв.), #169 (106 мг, 0,45 ммоль, 1 экв.), дихлорметана (10 мл), HATU (194 мг, 0,54 ммоль, 1,2 экв.) и диизопропилэтиламина (117 мг, 0,9 ммоль, 2 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии с обращенной фазой (Метод P) с получением #193 (149 мг, 37%) в виде белого твердого вещества. ВЭЖХ (Протокол X): m/z 873,4 [M+H+], время удерживания составляет 3,34 минуты (чистота составляет 98%).
Стадия 2A. Синтез соли муравьиной кислоты и (2S)-N-[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбоксамида (#188). Согласно общей методике C из #187 (20 мг, 0,023 ммоль, 1 экв.), дихлорметана (0,1 мл), ацетонитрила (0,1 мл) и 4 М раствора соляной кислоты в диоксане (0,26 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии с обращенной фазой (Метод N*) с получением #188 (11,6 мг, 61%,); ВЭЖХ (Протокол T): m/z 783,8 [M+H+], время удерживания составляет 2,53 минуты (чистота составляет 96%).1H ЯМР (400 МГц, DMSO-d6), δ 8.82-8.87 (m), 8.60-8.63 (m), 8.26-8.29 (m), 7.84-7.90 (m), 7.75-7.81 (m), 7.60-7.67 (m), 7.21-7.31 (m), 7.14-7.19 (m), 5.49-5.55 (m), 5.37-5.42 (m), 4.69-4.75 (m), 4.59-4.65 (m), 4.50-4.56 (m), 3.95-4.01 (m), 3.77-3.82 (m), 3.54-3.61 (m), 3.47-3.53 (m), 3.24-3.45 (m), 3.14-3.23 (m), 3.03-3.08 (m), 2.96-3.03 (m), 2.78-2.80 (m), 2.64-2.73 (m), 2.60-2.62 (m), 2.47-2.56 (m), 2.31-2.45 (m), 2.13-2.28 (m), 2.00-2.07 (m), 1.92-1.99 (m), 1.71-1.86 (m), 1.58-1.70 (m), 1.50-1.56 (m), 1.38-1.49 (m), 1.15-1.36 (m), 1.08-1.14 (m), 1.03-1.07 (m), 0.90-1.02 (m), 0.83-0.90 (m), 0.73-0.80 (m), 0.70-0.73 (m).
Стадия 2B. Синтез соли муравьиной кислоты и (2R)-N-[(2S)-1-{[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил](метил)амино}-3-метил-1-оксобутан-2-ил]-2-метилпиперидин-2-карбоксамида (#190). Согласно общей методике C из #189 (20 мг, 0,022 ммоль, 1 экв.), дихлорметана (0,1 мл), ацетонитрила (0,1 мл) и 4 М раствора соляной кислоты в диоксане (0,26 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии с обращенной фазой (Метод N*) с получением #190 (13,2 мг, 73%,). ВЭЖХ (Протокол T): m/z 783,7 [M+H+], время удерживания составляет 2,5 минуты (чистота составляет 100%).1H ЯМР (400 МГц, DMSO-d6), δ 8.83-8.86 (m), 8.60-8.62 (m), 8.24-8.27 (m), 7.83-7.89 (m), 7.78-7.80 (m), 7.75-7.77 (m), 7.64-7.66 (m), 7.60-7.63 (m), 7.20-7.31 (m), 7.13-7.19 (m), 5.49-5.55 (m), 5.36-5.42 (m), 4.65-4.74 (m), 4.60-4.65 (m), 4.50-4.56 (m), 3.95-4.01 (m), 3.76-3.81 (m), 3.53-3.62 (m), 3.47-3.52 (m), 3.22-3.45 (m), 3.14-3.21 (m), 2.97-3.05 (m), 2.93-2.96 (m), 2.79-2.86 (m), 2.76-2.78 (m), 2.65-2.68 (m), 2.48-2.56 (m), 2.37-2.43 (m), 2.29-2.35 (m), 2.17-2.27 (m), 2.04-2.11 (m), 1.93-2.00 (m), 1.70-1.85 (m), 1.53-1.69 (m), 1.36-1.48 (m), 1.28-1.36 (m), 1.13-1.26 (m), 1.08-1.12 (m), 0.98-1.07 (m), 0.91-0.97 (m), 0.84-0.91 (m), 0.73-0.78 (m).
Стадия 2C. Синтез соли трифторуксусной кислоты и N~2~-{[(3R)-3-фторпирролидин-3-ил]карбонил}-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#192). Согласно общей методике C из #191 (10 мг, 0,011 ммоль, 1 экв.), дихлорметана (0,1 мл), ацетонитрила (0,1 мл) и 4 М раствора соляной кислоты в диоксане (0,13 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #192 (5,1 мг, 60%). ВЭЖХ (Протокол T): m/z 773,5 [M+H+], время удерживания составляет 2,43 минуты (чистота составляет 100%).1H ЯМР (400 МГц, DMSO-d6) δ. 9.32-9.44 (m), 9.17-9.21 (m), 8.91 (d), 8.63-8.69 (m), 8.38-8.43 (m), 8.22-8.27 (m), 7.80 (dd), 7.66 (dd), 7.14-7.33 (m), 5.49-5.57 (m), 5.37-5.45 (m), 4.10-4.78 (m), 3.97-4.06 (m), 3.77-3.83 (m), 3.33-3.65 (m), 3.15-3.29 (m), 2.95-3.09 (m), 2.82-2.83 (m), 2.67-2.71 (m), 2.55-2.57 (m), 2.32-2.54 (m), 2.10-2.31 (m), 2.09 (s), 1.72-1.90(m), 1.57-1.72 (m). 1.38-1.50 (m), 1.15-1.38 (m), 1.09 (dd), 0.85-1.0 (m), 0.75-0.83 (m).
Стадия 2D. Синтез соли трифторуксусной кислоты и N~2~-{[(3S)-3-фторпирролидин-3-ил]карбонил}-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}-пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#194). Согласно общей методике C из #193 (10 мг, 0,011 ммоль, 1 экв.), дихлорметана (0,1 мл), ацетонитрила (0,1 мл) и 4 М раствора соляной кислоты в диоксане (0,13 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #194 (9 мг, 93%). ВЭЖХ (Протокол T): m/z 773,8 [M+H+], время удерживания составляет 2,42 минуты (чистота составляет 100%).1H ЯМР (400 МГц, DMSO-d6) δ 9.39-9.52 (m), 9.21-9.35 (m), 8.90 (d), 8.63-8.69 (m), 8.42-8.48 (m), 8.29-8.34 (m), 7.80 (dd), 7.66 (dd), 7.22-7.33 (m), 7.13-7.21 (m), 5.47-5.57 (m), 5.36-5.44 (m), 4.43-4.93 (m), 3.97-4.05 (m), 3.64-3.83 (m), 3.32-3.61 (m), 3.15-3.29 (m), 3.06-3.09 (m), 2.95-3.05 (m), 2.89-2.95 (m), 2.82-2.84 (m), 2.67-2.72 (m), 2.54-2.56 (m), 2.48-2.53 (m), 2.28-2.48 (m), 2.11-2.28 (m), 2.09 (s), 1.57-1.72 (m), 1.38-1.47 (m), 1.15-1.37 (m), 1.09 (dd), 0.85-0.99 (m), 0.78 (t).
Получение формиатной соли 1,2-диметил-D-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-аминофенил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#200) и формиатной соли 1,2-диметил-D-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-амино-фенил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#201)

Стадия 1. Синтез 1,2-диметил-D-пролил-N-[(3R,4S,5S)-1-трет-бутокси-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#195). Согласно общей методике D из #6 (7,75 г, 21,6 ммоль, 1 экв.), #152 (3,88 г, 21,6 ммоль, 1 экв.), дихлорметана (100 мл), HATU (9,8 г, 25,9 ммоль, 1,2 экв.) и диизопропилэтиламина (11,1 г, 86,4 ммоль, 4 экв.) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 20 до 55% этилацетата в петролейном эфире) с получением #195 (11,1 г, количественный выход) в виде желтого масла.
Стадия 2. Синтез 1,2-диметил-D-пролил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (#196). Согласно общей методике B из #195 (11,1 г, 21,6 ммоль, 1 экв.), дихлорметана (100 мл) и трифторуксусной кислоты (40 мл) синтезировали неочищенный целевой материал с получением #196 (10,1 г, количественный выход), который использовали на следующей стадии без дополнительной очистки. ЖХ-МС (Протокол Z): m/z 428,5 [M+H+], время удерживания составляет 0,9 минуты.
Стадия 3. Синтез 1,2-диметил-D-пролил-N-[(3R,4S,5S)-3-метокси-5-метил-1-оксо-1-(пентафторфенокси)гептан-4-ил]-N-метил-L-валинамида (#197). К охлажденному раствору (0°C) #196 (4,0 г, 9,4 ммоль, 1 экв.) в дихлорметане (40 мл) добавляли по каплям пиридин (2,95 г, 37,6 ммоль, 4 экв.), а затем раствор пентафторфенилтрифторацетата (3,9 г, 13,6 ммоль, 1,4 экв.) в дихлорметане (5 мл). Смесь перемешивали при комнатной температуре в течение одного часа и растворитель концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 1 до 10% метанола в дихлорметане) с получением соединения #197 (4,5 г, 81,2% (за три стадии) в виде белого твердого вещества.
Стадия 4. Синтез соли трифторуксусной кислоты и 1,2-диметил-D-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]-пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#198). К охлажденному раствору (0°C) #197 (4,0 г, 7,4 ммоль, 1 экв.) в дихлорметане (25 мл) добавляли по каплям диизопропилэтиламин (3,4 г, 26,3 ммоль, 3,5 экв.), а затем раствор #103 (2,3 г, 7,6 ммоль, 1,02 экв.) в дихлорметане (15 мл). После завершения добавления смесь перемешивали при комнатной температуре в течение 16 часов и растворитель удаляли в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 1 до 10% метанола в дихлорметане) с последующей второй очисткой посредством хроматографии с обращенной фазой (Метод Q) с получением #198 (1,57 г, 57,5%) в виде белого твердого вещества. ВЭЖХ (Протокол X): m/z 597,49 [M+H+], время удерживания составляет 8,879 минуты (чистота составляет 98%). Хиральная ВЭЖХ, время удерживания: 3,328 мин (чистота составляет 98%), колонка: Chiralcel OJ-H, 250×4,6 мм, 5 мкм; подвижная фаза: метанол (0,05% диэтиламина) в CO2 от 5% до 40% за 15 минуты; скорость потока: 2,35 мл/минуту.
Стадия 5. Синтез 1,2-диметил-D-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-(пентафторфенокси)пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#199). К раствору #198 (280 мг, 0,394 ммоль, 1 экв.) в дихлорметане (2 мл) добавляли пиридин (75 мг, 0,94 ммоль, 2,4 экв.), а затем раствор пентафторфенилтрифторацетата (268 мг, 0,94 ммоль, 2,4 экв.) в дихлорметане (1,5 мл). Смесь перемешивали при комнатной температуре в течение 2,5 часов и растворитель концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 1 до 10% метанола в дихлорметане) с получением соединения #199 (279 мг, 39%) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 763,5 [M+H+], время удерживания составляет 0,93 минуты.
Стадия 6A. Синтез соли трифторуксусной кислоты и 1,2-диметил-D-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-(1,2,3,4-тетрагидро-хинолин-6-ил)пропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#200). К смеси #199 (25 мг, 0,033 ммоль, 1 экв.) и #215 (7,7 мг, 0,033 ммоль, 1 экв.) в дихлорметане (1,5 мл) добавляли N,N-диизопропилэтиламин (30,2 мг, 2,31 ммоль, 7 экв.). Реакционную смесь перемешивали в течение 5 минуты и добавляли N,N-диметилформамид (0,5 мл). После перемешивания в течение 2 ½ часов добавляли дополнительное количество N,N-диизопропилэтиламина (30,2 мг, 2,31 ммоль, 7 экв.). Спустя 3 ½ часа добавляли еще дополнительное количество N,N-диметилформамида (0,75 мл) и смесь перемешивали в течение 18 часов. Добавляли дополнительное количество N,N-диизопропилэтиламина (15,1 мг, 1,15 ммоль, 3,6 экв.) и N,N-диметилформамида (0,25 мл) и реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Реакционную смесь концентрировали в вакууме и неочищенный продукт очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #200 (12,9 мг, 48%). ВЭЖХ (Протокол T): m/z 407,7, двойной заряд [2+], время удерживания составляет 1,69 минуты (чистота составляет 100%).1H ЯМР (400 МГц, DMSO-d6) δ 9.72-9.82 (m). 8.61-8.67 (m), 8.42-8.48 (m), 8.19-8.24 (m), 7.25-7.27 (m), 7.12-7.14 (m), 6.94-7.01 (m), 6.88-6.94 (m), 6.78-6.84 (m), 6.67-6.74 (m), 6.57-6.64 (m), 4.69-4.77 (m), 4.60-4.68 (m), 4.53-4.60 (m), 4.46-4.53 (m), 4.37-4.45 (m), 3.97-4.05 (m), 3.76-3.81 (m), 3.62-3.67 (m), 3.53-3.62 (m), 3.44-3.52 (m), 3.32-3.38 (m), 3.27-3.32 (m), 3.22-3.27 (m), 3.15-3.22 (m), 3.06-3.14 (m), 2.97-3.01 (m), 2.92-2.96 (m), 2.74-2.83 (m), 2.61-2.74 (m), 2.57-2.61 (m), 2.48-2.56 (m), 2.37-2.46 (m), 2.25-2.36 (m), 2.00-2.20 (m), 1.67-1.91 (m), 1.47-1.60 (m), 1.37-1.47 (m), 1.24-1.35 (m), 1.03-1.10 (m), 0.95-1.00 (m), 0.88-0.94 (m), 0.76-0.83 (m).
Стадия 6B. Синтез соли трифторуксусной кислоты и 1,2-диметил-D-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-аминофенил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#201). К смеси #199 (25 мг, 0,033 ммоль, 1 экв.) и имеющегося в продаже метилового эфира 4-амино-L-фенилаланина (8,8 мг, 0,033 ммоль, 1 экв.) в дихлорметане (1,5 мл) добавляли N,N-диизопропилэтиламин (30,2 мг, 2,31 ммоль, 7 экв.). Реакционную смесь перемешивали в течение 5 минуты и добавляли N,N-диметилформамид (0,5 мл). Спустя 4 часа добавляли дополнительное количество N,N-диизопропилэтиламина (37,75 мг, 2,88 ммоль, 8,25 экв.) и смесь перемешивали в течение 50 минуты. Добавляли дополнительное количество N,N-диметилформамида (0,75 мл) и реакционную смесь перемешивали в течение 66 часов, концентрировали в вакууме и неочищенный продукт очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #201 (14,3 мг, 56%); ВЭЖХ (Протокол T): m/z 387,2, двойной заряд [2+], время удерживания составляет 1,50 минуты (чистота составляет 100%);1H ЯМР (400 МГц, DMSO-d6) δ 9.68-9.84 (m), 8.57-8.66 (m), 8.46-8.51 (m), 8.23-8.29 (m), 7.18-7.28 (m), 7.11-7.16 (m), 7.03-7.08 (m), 6.97-7.02 (m), 4.67-4.75 (m), 4.58-4.66 (m), 4.34-4.57 (m), 3.95-4.03 (m), 3.85-3.90 (m), 3.73-3.81 (m), 3.66-3.72 (m), 3.57-3.66 (m), 3.50-3.57 (m), 3.42-3.49 (m), 3.32-3.38 (m), 3.14-3.30 (m), 2.95-3.11 (m), 2.84-2.94 (m), 2.78-2.81 (m), 2.63-2.74 (m), 2.46-2.57 (m), 2.34-2.45 (m), 2.19-2.34 (m), 1.97-2.19 (m), 1.67-1.90 (m), 1.45-1.62 (m), 1.34-1.41 (m), 1.21-1.34 (m), 1.01-1.10 (m), 0.82-0.99 (m), 0.72-0.81 (m).
Получение формиатной соли 1,2-диметил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-(1,2,3,4-тетрагидрохинолин-6-ил)пропан-2-ил]амино}-2-метил-3-оксопропил]-пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#207), формиатной соли 1,2-диметил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-аминофенил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#208) и соли трифторуксусной кислоты и 1,2-диметил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#209)

Стадия 1. Синтез 1,2-диметил-L-пролил-N-[(3R,4S,5S)-1-трет-бутокси-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#202). Согласно общей методике D из #6 (4,3 г, 12 ммоль, 1 экв.), #150 (2,15 г, 12 ммоль, 1 экв.), дихлорметана (50 мл), HATU (5,46 г, 14 ммоль, 1,2 экв.) и диизопропилэтиламина (8,17 мл) синтезировали неочищенный целевой материал, который очищали посредством хроматографии на силикагеле (градиент: от 20 до 55% этилацетата в петролейном эфире) с получением #202 (5,2 г, 89%) в виде желтого масла.
Стадия 2. Синтез 1,2-диметил-L-пролил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (#203). Согласно общей методике В из #202 (5,2 г, 10,77 ммоль, 1 экв.), дихлорметана (45 мл) и трифторуксусной кислоты (20 мл) синтезировали неочищенный целевой материал с получением #203 (7 г, количественный выход), который использовали на следующей стадии без дополнительной очистки.
Стадия 3. Синтез 1,2-диметил-L-пролил-N-[(3R,4S,5S)-3-метокси-5-метил-1-оксо-1-(пентафторфенокси)гептан-4-ил]-N-метил-L-валинамида (#204). К охлажденному раствору (0°C) #203 (7,0 г, 10,77 ммоль, 1 экв.) в дихлорметане (15 мл) добавляли по каплям пиридин (3,41 г, 43,08 ммоль, 4 экв.), а затем раствор пентафторфенилтрифторацетата (6,03 г, 21,54 ммоль, 2 экв.) в дихлорметане (7 мл). Смесь перемешивали при комнатной температуре в течение одного часа и растворитель концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 1 до 10% метанола в дихлорметане) с получением соединения #204 (8 г, 82% за две стадии) в виде желтого твердого вещества.
Стадия 4. Синтез 1,2-диметил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#205). К охлажденному (0°C) раствору #204 (8,0 г, 10,77 ммоль, 1 экв.) в дихлорметане (25 мл) добавляли по каплям диизопропилэтиламин (5,6 г, 43,08 ммоль, 4 экв.), а затем раствор #103 (3,22 г, 10,77 ммоль, 1 экв.) в дихлорметане (15 мл). После завершения добавления смесь перемешивали при комнатной температуре в течение 16 часов и растворитель удаляли в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 1 до 10% метанола в дихлорметане) с получением #205 (2,2 г, 33%) в виде желтого твердого вещества. ВЭЖХ (Протокол X): m/z 597,42 [M+H+], время удерживания составляет 8,729 минуты (чистота более 97%), хиральная ВЭЖХ, время удерживания: 2,87 мин (чистота составляет 89%), колонка: Chiralcel OD-3, 150×4,6 мм, 3 мкм; подвижная фаза: этанол (0,05% диэтиламина) в CO2 от 5% до 40% за 12 минуты; скорость потока: 2,5 мл/минуту.
Стадия 5. Синтез 1,2-диметил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-(пентафторфенокси)пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#206). К раствору #198 (0,28 г, 0,47 ммоль, 1 экв.) в дихлорметане (2 мл) добавляли пиридин (75 мг, 0,94 ммоль, 2 экв.), а затем раствор пентафторфенилтрифторацетата (268 мг, 0,94 ммоль, 2 экв.) в дихлорметане (1,5 мл). Смесь перемешивали при комнатной температуре в течение 2,5 часов и растворитель концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (градиент: от 1 до 10% метанола в дихлорметане) с получением соединения #206 (348 мг, 97%) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 763,5 [M+H+], время удерживания составляет 0,9 минуты.
Стадия 6A. Синтез соли трифторуксусной кислоты и 1,2-диметил-1-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-(1,2,3,4-тетрагидро-хинолин-6-ил)пропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валин-амида (#207). Указанное в заголовке соединение получали из #206 (25 мг, 0,033 ммоль, 1 экв.) и #215 (7,7 мг, 0,033 ммоль, 1 экв.), используя способ, описанный выше для получения #200. Неочищенный продукт очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #207 (11,7 мг, 44%). ВЭЖХ (Протокол T): m/z 407,6, двойной заряд [2+], время удерживания составляет 1,59 минуты (чистота составляет 100%).1H ЯМР (DMSO-d6) δ 9.57-9.69 (m), 8.68-8.76 (m), 8.42-8.47 (m), 8.23-8.29 (m), 8.18-8.23 (m), 7.24-7.27 (m), 6.95-7.01 (m), 6.89-6.94 (m), 6.80-6.86 (m), 6.70-6.78 (m), 6.60-6.67 (m), 4.69-4.77 (m), 4.60-4.68 (m), 4.46-4.60 (m), 4.34-4.46 (m), 3.95-4.03 (m), 3.87-3.91 (m), 3.79-3.85 (m), 3.74-3.79 (m), 3.60-3.66 (m), 3.49-3.60 (m), 3.41-3.49 (m), 3.12-3.35 (m), 3.04-3.12 (m), 2.89-3.04 (m), 2.68-2.83 (m), 2.61-2.67 (m), 2.45-2.55 (m), 2.34-2.44 (m), 2.08-2.33 (m), 2.05-2.08 (m), 1.92-2.05 (m), 1.75-1.91 (m), 1.65-1.75 (m), 1.59-1.64 (m), 1.34-1.58 (m), 1.20-1.31 (m), 1.01-1.09 (m), 0.94-0.99 (m), 0.84-0.93 (m), 0.80-0.83 (m), 0.72-0.80 (m).
Стадия 6B. Синтез соли трифторуксусной кислоты и 1,2-диметил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3{[(2S)-3-(4-аминофенил)-1-метокси-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#208). К смеси #206 (25,0 мг, 0,033 ммоль, 1 экв.) и имеющегося в продаже метилового эфира 4-амино-L-фенилаланина (8,8 мг, 0,033 ммоль, 1 экв.) в дихлорметане (1,5 мл) добавляли N,N-диизопропилэтиламин (30,2 мг, 2,31 ммоль, 7 экв.). Реакционную смесь перемешивали в течение 5 минуты и добавляли N,N-диметилформамид (0,5 мл). Спустя 2 ½ часа добавляли дополнительное количество N,N-диизопропилэтиламина (30,2 мг, 2,31 ммоль, 7 экв.). После перемешивания в течение 3 1/2 часов добавляли дополнительное количество N,N-диметилформамида (0,75 мл) и реакционную смесь перемешивали в течение 66 часов, концентрировали в вакууме и неочищенный продукт очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #208 (13 мг, 51%); ВЭЖХ (Протокол T): m/z 387,2, двойной заряд [2+], время удерживания составляет 1,58 минуты (чистота составляет 100%);1H ЯМР (400 МГц, DMSO-d6) δ 9.54-9.69 (m), 8.68-8.75 (m), 8.46-8.50 (m), 8.33-8.37 (m), 8.22-8.31 (m), 8.09-8.14 (m), 7.17-7.27 (m), 7.07-7.16 (m), 6.99-7.05 (m), 6.92-6.99 (m), 4.69-4.75 (m), 4.60-4.68 (m), 4.42-4.59 (m), 4.34-4.42 (m), 3.95-4.03 (m), 3.85-3.90 (m), 3.74-3.80 (m), 3.65-3.72 (m), 3.62-3.65 (m), 3.42-3.62 (m), 3.31-3.36 (m), 3.24-3.30 (m), 3.11-3.24 (m), 3.03-3.11 (m), 2.96-3.03 (m), 2.81-2.92 (m), 2.65-2.76 (m), 2.43-2.55 (m), 2.34-2.43 (m), 2.06-2.33 (m), 1.93-2.05 (m), 1.75-1.89 (m), 1.66-1.74 (m), 1.58-1.65 (m), 1.47-1.57 (m), 1.34-1.43 (m), 1.20-1.32 (m), 1.10-1.14 (m), 1.00-1.09 (m), 0.83-0.99 (m), 0.72-0.81 (m).
Стадия 6C. Синтез соли трифторуксусной кислоты и 1,2-диметил-L-пролил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (#209). Указанное в заголовке соединение получали из #206 (25,0 мг, 0,033 ммоль, 1 экв.) и гидрохлорида метилового эфира L-фенилаланина (7,1 мг, 0,033 ммоль, 1 экв.), используя способ, описанный выше для получения #200. Неочищенный продукт очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #209 (10,3 мг, 41%). ВЭЖХ (Протокол T): m/z 758,7 [M+H+], время удерживания составляет 1,787 минуты (чистота составляет 100%).1H ЯМР (DMSO-d6) δ 9.57-9.70 (m), 8.70-8.75 (m), 8.50-8.56 (m), 8.32-8.42 (m), 8.24-8.26 (m), 8.10-8.15 (m), 7.14-7.27 (m), 7.12-7.13 (m), 6.98-7.00 (m), 4.69-4.77 (m), 4.55-4.67 (m), 4.43-4.53 (m), 3.94-4.02 (m), 3.73-3.78 (m), 3.62-3.68 (m), 3.48-3.60 (m), 3.39-3.48 (m), 3.23-3.33 (m), 3.13-3.22 (m), 3.08-3.13 (m), 3.02-3.08 (m), 2.96-3.01 (m), 2.81-2.94 (m), 2.76-2.80 (m), 2.66-2.75 (m), 2.62-2.66 (m), 2.46-2.55 (m), 2.31-2.45 (m), 2.09-2.29 (m), 2.05-2.09 (m), 1.93-2.04 (m), 1.74-1.88 (m), 1.65-1.74 (m), 1.59-1.65 (m), 1.36-1.52 (m), 1.21-1.35 (m), 1.01-1.08 (m), 0.94-1.00 (m), 0.83-0.94 (m), 0.73-0.81 (m).
Получение метил-(2S)-2-амино-3-(1,2,3,4-тетрагидрохинолин-6-ил)пропаноата
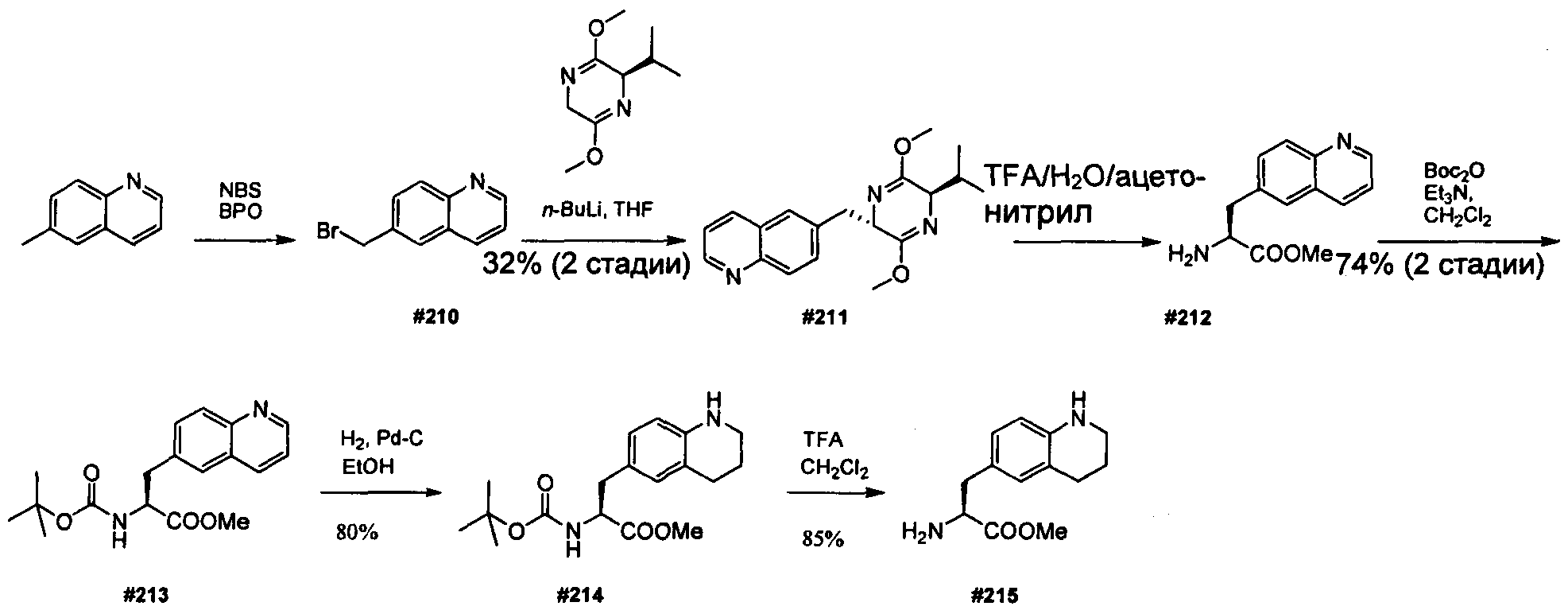
Стадия 1. Синтез 6-(бромметил)хинолина (#210). Раствор 6-метилхинолина (5 г, 35 ммоль, 1 экв.), N-бромсукцинимида (8,1 г, 45,5 ммоль, 1,3 экв.) и бензоилпероксида (840 мг, 3,5 ммоль, 0,1 экв.) в четыреххлористом углероде (100 мл) перемешивали при кипячении с обратным холодильником в течение 3 часов и затем охлаждали до комнатной температуры. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Остаток растворяли в тетрагидрофуране (100 мл) и фильтровали. Фильтрат использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2. Синтез 6-{[(2S,5R)-3,6-диметокси-5-(пропан-2-ил)-2,5-дигидропиразин-2-ил]метил}хинолина (#211). К охлажденному раствору (-70°C) (2R)-3,6-диметокси-2-(пропан-2-ил)-2,5-дигидропиразина (25,8 г, 140 ммоль, 2 экв.) в тетрагидрофуране (200 мл) добавляли по каплям н-бутиллитий (2,5 M, 64,4 мл, 161 ммоль, 2,3 экв.) и затем перемешивали в течение 30 минуты. По каплям при -65°C добавляли раствор #210 (15,4 г, 70 ммоль, 1 экв.) в тетрагидрофуране (150 мл) и затем раствор перемешивали в течение 2 часов при этой температуре. Реакционную смесь гасили насыщенным водным хлоридом аммония (100 мл) и экстрагировали этилацетатом (100 мл). Органическую фазу сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии на диоксиде кремния (градиент: от 10 до 16% этилацетата в петролейном эфире) с получением #211 (7,3 г, 32% за две стадии) в виде желтого твердого вещества. ЖХ-МС (Протокол Z): m/z 326,2 [M+H+], время удерживания составляет 0,88 минуты.
Стадия 2. Синтез метил-(2S)-2-амино-3-(хинолин-6-ил)пропаноата (#212). К раствору #211 (7,3 г, 22,5 ммоль, 1 экв.) в воде (25 мл) и ацетонитриле (80 мл) при 0°C добавляли трифторуксусную кислоту (9 мл) и раствор перемешивали при 10°C в течение ночи. Органический слой удаляли в вакууме и оставшийся водный слой подщелачивали до pH 9 с помощью карбоната натрия и использовали непосредственно на следующей стадии.
Стадия 4. Синтез метил-(2S)-2-[(трет-бутоксикарбонил)амино]-3-(хинолин-6-ил)пропаноата (#213). К раствору #212 (5,2 г, 22,5 ммоль, 1 экв.) и триэтиламина (9,1 г, 90 ммоль, 4 экв.) в смешанном растворителе из метанола (30 мл) и воды (50 мл) при 0°C добавляли ди-трет-бутилдикарбонат (17,5 г, 78,75 ммоль, 3,5 экв.) и затем раствор перемешивали при 10°C в течение ночи. Реакционную смесь фильтровали и осадок на фильтре промывали метанолом (20 мл × 2). Фильтрат экстрагировали этилацетатом (50 мл × 2) и органическую фазу концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии на диоксиде кремния (градиент: от 25 до 50% этилацетата в петролейном эфире) с получением #213 (5,5 г, 74% за две стадии) в виде желтого масла. ЖХ-МС (Протокол Z): m/z 331,2 [M+H+], время удерживания составляет 0,76 минуты.
Стадия 5. Синтез метил-(2S)-2-[(трет-бутоксикарбонил)амино]-3-(1,2,3,4-тетрагидрохинолин-6-ил)пропаноата (#214). Суспензию #213 (1,5 г, 4,55 ммоль, 1 экв.) и палладия на углероде (150 мг) в этаноле (20 мл) перемешивали при 50°C под давлением 30 фунт-сила на кв. дюйм водорода в течение ночи. Реакционную смесь фильтровали через набивку целита и фильтрат концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии на диоксиде кремния (градиент: 40% этилацетата в петролейном эфире) с получением #214 (1,2 г, 80%) в виде масла.1H ЯМР (400 МГц, CDCl3): δ 6.70 (d, 2H), 6.40 (m, 1H), 4.95 (m, 1H), 4.49 (m, 1H), 3.77 (s, 3H), 3.28 (m, 2H), 2.97 (m, 2H), 2.71 (m, 2H), 1.95 (m, 2H), 1.26 (s, 9H), ВЭЖХ (Протокол Y): m/z 357,0 [M+Na+], время удерживания составляет 5,304 минуты (чистота более 98%). Хиральная ВЭЖХ, время удерживания: 4,64 мин (чистота составляет 98%) (колонка: Chiralcel OJ-H, 150×4,6 мм, 5 мкм; подвижная фаза: этанол (0,05% диэтиламина) в CO2 от 5% до 40% за 15 минуты; скорость потока: 2,5 мл/минуту.
Стадия 6. Синтез метил-(2S)-2-амино-3-(1,2,3,4-тетрагидрохинолин-6-ил)пропаноата (#215). К раствору #214 (750 мг, 2,25 ммоль, 1 экв.) в дихлорметане (20 мл) при 0°C добавляли по каплям трифторуксусную кислоту (2 мл) и затем раствор перемешивали при 20°C в течение ночи. Реакционную смесь концентрировали в вакууме и остаток растворяли в воде (20 мл). Раствор подщелачивали с помощью карбоната натрия и экстрагировали смесью этилацетат/тетрагидрофуран (30 мл × 3). Органическую фазу сушили над сульфатом натрия и концентрировали в вакууме с получением #215 (450 мг, 85%) в виде желтого масла.1H ЯМР (400 МГц, CDCl3): δ 6.70 (d, 2H), 6.40 (m, 1H), 3.73 (s, 3H), 3.67 (m, 1H), 3.30 (m, 2H), 2.96 (m, 1H), 2.75 (m, 3H), 1.96 (m, 2H), 1.50 (br, 2H), 1.26 (br, 1H). ВЭЖХ (Протокол Y): m/z 235,14 [M+H+] время удерживания составляет 4,35 минуты (чистота более 96%). Хиральная ВЭЖХ, время удерживания: 5,71 мин (чистота составляет 98%). (Колонка: Chiralcel OJ-Н, 150×4,6 мм, 5 мкм; подвижная фаза: этанол (0,05% диэтиламина) в CO2 от 5% до 40% за 15 минуты; скорость потока: 2,5 мл/минуту.
Получение соли трифторуксусной кислоты и N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S-4-[(N-{[(3R)-3-фторпирролидин-3-ил]карбонил}-L-валил)-(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланина (#217) и соли трифторуксусной кислоты и N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3S)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}-пирролидин-2-ил]-3-метокси-2-метилпропаноил}-L-фенилаланина (#219)

Стадия 1A. Синтез метилового эфира N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3R)-1-(трет-бутоксикарбонил)-3-фторпирролидин-3-ил]карбонил}-L-валил)-(метил)-амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метил-пропаноил}-L-фенилаланина (#216). К раствору #168 (36,9 мг, 0,158 ммоль, 1 экв.) и #114 (100 мг, 0,158 ммоль, 1 экв.) в дихлорметане (3,6 мл) и N,N-Диметилформамиде (0,8 мл) добавляли диизопропилэтиламин (0,083 мл, 0,474 ммоль, 3 экв.), а затем HATU (60,7 мг, 0,158 ммоль, 1 экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 18 часов, разбавляли этилацетатом (25 мл), промывали водой (1X), 10%-ной лимонной кислотой (1X) и рассолом (1X). Органический слой сушили над сульфатом натрия, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного #216 (220 мг, 164% от теоретического значения), который использовали на следующей стадии без дополнительной очистки. ВЭЖХ (Протокол Q): m/z 848,6 [M+H+], время удерживания составляет 2,10 минуты.
Стадия 1B. Синтез метилового эфира N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3S)-1-(трет-бутоксикарбонил)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)-амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метил-пропаноил}-1-фенилаланина (#218). К раствору #169 (36,9 мг, 0,158 ммоль, 1 экв.) и #114 (100 мг, 0,158 ммоль, 1 экв.) в дихлорметане (3,6 мл) и N,N-диметилформамиде (0,8 мл) добавляли диизопропилэтиламин (0,083 мл, 0,474 ммоль, 3 экв.), а затем HATU (60,7 мг, 0,158 ммоль, 1экв.). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 18 часов, разбавляли этилацетатом (25 мл), промывали водой (1X), 10%-ной лимонной кислотой (1X) и рассолом (1X). Органический слой сушили над сульфатом натрия, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного #218 (180 мг, 134% от теретического значения), который использовали на следующей стадии без дополнительной очистки. ВЭЖХ (Протокол Q): m/z 848,6 [M+H+], время удерживания составляет 2,10 минуты.
Стадия 2A. Синтез соли трифторуксусной кислоты и N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3R)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)-амино]-3-метокси-5-метил-гептаноил}пирролидин-2-ил]-3-метокси-2-метил-пропаноил}-L-фенилаланина (#217). К раствору неочищенного #216 (134 мг) в тетрагидрофуране (4 мл) добавляли гидроксид лития (1 М, 0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и концентрировали в вакууме. Остаток растворяли в дихлорметане (2 мл) и добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали в течение 4 часов и концентрировали в вакууме. Неочищенный материал очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #217 (60 мг, 86% за две стадии) в виде смолы. ЖХ-МС (Протокол Q): m/z 734,93 [M+H+], время удерживания составляет 1,19 минуты.1H ЯМР (DMSO-d6) δ 12.62-12.83 (m), 9.30-9.43 (m), 9.17-9.28 (m), 8.34-8.41 (m), 8.22-8.31 (m), 8.08-8.15 (m), 7.87-7.93 (m), 7.76-7.81 (m), 7.11-7.23 (m), 4.93-4.99 (m), 4.81-4.88 (m), 4.55-4.71 (m), 4.48-4.54 (m), 4.37-4.45 (m), 3.92-3.99 (m), 3.69-3.75 (m), 3.31-3.65 (m), 3.25-3.30 (m), 3.20-3.24 (m), 3.12-3.19 (m), 3.08-3.10 (m), 2.97-3.07 (m), 2.92-2.97 (m), 2.75-2.84 (m), 2.64-2.70 (m), 2.43-2.57 (m), 2.28-2.43 (m), 2.15-2.26 (m), 2.02-2.15 (m), 1.56-1.87 (m), 1.31-1.48 (m), 1.05-1.30 (m), 0.97-1.06 (m), 0.82-0.97 (m), 0.71-0.79 (m).
Стадия 2B. Синтез соли трифторуксусной кислоты и N-{(2R,3R)-3-[(2S)-1-{(3R,4S,5S)-4-[(N-{[(3S)-3-фторпирролидин-3-ил]карбонил}-L-валил)(метил)-амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-3-метокси-2-метил-пропаноил}-L-фенилаланина (#219). К раствору неочищенного #218 (100 мг) в тетрагидрофуране (4 мл) добавляли 1,0 М гидроксид лития в воде (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и затем концентрировали в вакууме. Неочищенный материал растворяли в дихлорметане (2 мл) и добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали в течение 4 часов и затем концентрировали в вакууме. Неочищенный материал очищали посредством хроматографии с обращенной фазой (Метод M*) с получением #219 в виде смолы (60 мг, 91% за две стадии). ЖХ-МС (Протокол Q): m/z 734,97 [M+H+], время удерживания составляет 1,14 минуты.1H ЯМР (400 МГц, DMSO-d6), δ 12.62-12.85 (m), 9.32-9.43 (m), 9.13-9.26 (m), 8.39-8.46 (m), 8.30-8.39 (m), 8.25-8.29 (m), 8.08-8.13 (m), 7.79-7.85 (m), 7.67-7.72 (m), 7.10-7.23 (m), 4.94-5.01 (m), 4.83-4.89 (m), 4.64-4.73 (m), 4.56-4.63 (m), 4.44-4.50 (m), 4.37-4.44 (m), 3.92-3.99 (m), 3.60-3.74 (m), 3.24-3.55 (m), 3.11-3.24 (m), 3.07-3.10 (m), 3.02-3.06 (m), 2.98-3.02 (m), 2.93-2.97 (m), 2.75-2.85 (m), 2.68-2.69 (m), 2.63-2.67 (m), 2.45-2.55 (m), 2.26-2.44 (m), 2.15-2.25 (m), 2.03-2.14 (m), 1.55-1.87 (m), 1.31-1.47 (m), 1.15-1.31 (m), 0.98-1.05 (m), 0.91-0.98 (m), 0.82-0.91 (m), 0.71-0.78 (m).
Получение 2-метилаланил-N-{(3R,4S,5S)-1-[(2S)-2-{(3R,4R,7S)-7-бензил-4-метил-18-[(4S,5R)-5-метил-2-оксоимидазолидин-4-ил]-5,8,13-триоксо-2-окса-6,9,12-триазаоктадекан-3-ил}пирролидин-1-ил]-3-метокси-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#257)

Стадия 1. Синтез 9H-флуорен-9-илметил-[(2S)-1-({2-[(трет-бутоксикарбонил)амино]этил}амино)-1-оксо-3-фенилпропан-2-ил]карбамата (#253). В соответствии с общей методикой D, используя N-[(9H-флуорен-9-илметокси)карбонил]-L-фенилаланин (500 мг, 1,29 ммоль, 1,0 экв.), трет-бутил-(2-аминоэтил)карбамат (207 мг, 1,29 ммоль, 1,0 экв.), HATU (620 мг, 1,55 ммоль, 1,2 экв.) и основание Хюнига (0,452 мл, 2,58 ммоль, 2,0 экв.) в 6 мл DMF, получали #253 в виде белого твердого вещества (620 мг, 91%) после концентрирования растворителя и перекристаллизации с использованием этилацетата. ЖХ-МС (Протокол Q1): m/z 552,3 [M+Na+], время удерживания составляет 1,01 минуты.
Стадия 2. Синтез N-альфа-[(9H-флуорен-9-илметокси)карбонил]-N-[2-({6-[(4S,5R)-5-метил-2-оксоимидазолидин-4-ил]гексаноил}амино)этил]-L-фенил-аланинамида (#254). Защиту в виде Boc удаляли с использованием общей методики C, используя #251 (88 мг, 0,17 ммоль, 1,0 экв.) и 4 М HCl (2,0 мл, 8,0 ммоль, 48 экв.) с последующим концентрированием в вакууме. Затем проводили реакцию сочетания согласно общей методике D, используя неочищенный остаток, 6-[(4S,5R)-5-метил-2-оксоимидазолидин-4-ил]гексановую кислоту (35,6 мг, 0,166 ммоль, 1,0 экв.), HATU (73,2 мг, 0,18 ммоль, 1,1 экв.) и основание Хюнига (0,087 мл, 0,50 ммоль, 3,0 экв.) в 2 мл DMF с последующей очисткой (Метод J), получая #254 (35 мг, 34%) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 626,3 [M+H+], время удерживания составляет 0,86 минуты.
Стадия 3. Синтез N-[2-({6-[(4S,5R)-5-метил-2-оксоимидазолидин-4-ил]гексаноил}амино)этил]-L-фенилаланинамида (#255). В соответствии с общей методикой A, используя #254 (35 мг, 0,056 ммоль, 1,0 экв.) и пиперидин (0,10 мл, 1,0 ммоль, 20 экв.) в 0,5 мл DMF, с последующей очисткой с помощью хроматографии на диоксиде кремния (0-30% метанола в дихлорметане) получают #253 (19 мг, 84%). ЖХ-МС (Протокол Q1): m/z 404,2 [M+H+], время удерживания составляет 0,48 минуты.
Стадия 4. Синтез N-[(9H-флуоренил-9-илметокси)карбонил]-2-метилаланил-N-{(3R,4S,5S)-1-[(2S)-2-{(3R,4R,7S)-7-бензил-4-метил-18-[(4S,5R)-5-метил-2-оксоимидазолидин-4-ил]-5,8,13-триоксо-2-окса-6,9,12-триаза-октадекан-3-ил}пирролидин-1-ил]-3-метокси-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамид (#256). В соответствии с общей методикой D, используя #105 (36,6 мг, 0,047 ммоль, 1,0 экв.), #255 (19 мг, 0,047 ммоль, 1,0 экв.), HATU (22,4 мг, 0,056 ммоль, 1,2 экв.) и основание Хюнига (0,025 мл, 0,141 ммоль) в 1,5 мл DMF с последующей очисткой (Метод J), получали #256 (15 мг, 27%) в виде белого твердого вещества. ЖХ-МС (Протокол Q1): m/z 1164,8 [M+H+], время удерживания составляет 0,99 минуты.
Стадия 5. Синтез 2-метилаланил-N-{(3R,4S,5S)-1-[(2S)-2-{(3R,4R,7S)-7-бензил-4-метил-18-[(4S,5R)-5-метил-2-оксоимидазол идин-4-ил]-5,8,13-триоксо-2-окса-6,9,12-триазаоктадекан-3-ил}пирролидин-1-ил]-3-метокси-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида (#257). В соответствии с общей методикой A, используя #256 (5 мг, 0,004 ммоль, 1,0 экв.) и пиперидин (0,02 мл, 0,2 ммоль, 50 экв.) в 0,7 мл DMF с последующей очисткой (Метод J), получали #257 (2 мг, 50%) в виде бесцветного стекла. ЖХ-МС (Протокол Q1): m/z 1164,8 [M+H+], время удерживания составляет 0,99 минуты.1H ЯМР (400 МГц, DMSO-d6), δ 8.44-8.52 (m), 8.06-8.20 (m), 7.96-8.01 (m), 7.69-7.83 (m), 7.20-7.28 (m), 7.11-7.19 (m), 3.38-3.83 (m), 3.19-3.26 (m), 3.03-3.12 (m), 2.98 (s), 2.91 (s), 2.75 (s), 2.65-2.70 (m), 2.01-2.36 (m), 1.65-1.87 (m), 1.39-1.57 (m), 1.13-1.37 (m), 1.04-1.08 (m), 0.74-1.01 (m).
Получение N-[5-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)пентаноил]-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (mv#115)
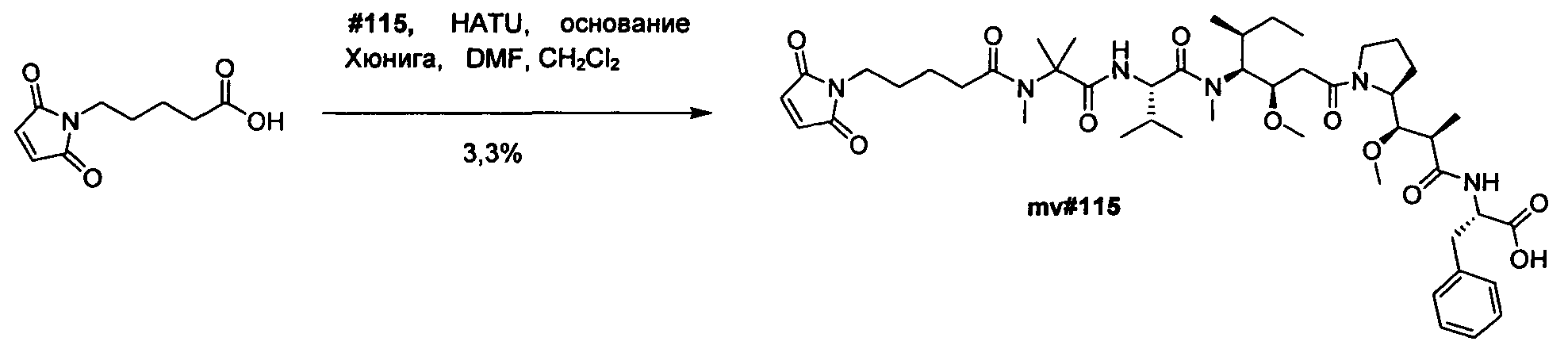
Стадия 1. Получение N-[5-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)пентаноил]-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (mv#115). К перемешиваемому раствору 5-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)пентановой кислоты (12 мг, 0,061 мМ) в 0,4 мл дихлорметана и 0,1 мл DMF добавляли HATU (23,2 мг, 0,061 мМ), а затем основание Хюнига (0,033 мл, 0,188 мМ). Реакционную смесь оставляли перемешиваться в течение 5 минут, после чего добавляли #115 (39 мг, 0,047 мМ) в виде раствора в 0,4 мл дихлорметана и 0,1 мл DMF. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 3 часов и 15 минуты, после чего гасили посредством добавления воды, содержащей небольшое количество TFA. Реакционную смесь затем концентрировали. Неочищенный материал растворяли в DMSO и очищали посредством хроматографии с обращенной фазой (Метод J). Соответствующие фракции концентрировали (Genevac). Материал затем дополнительно очищали посредством хроматографии с обращенной фазой (Метод K), а соответствующие фракции концентрировали (Genevac). Материал затем переносили в небольшой сосуд, используя дихлорметан и метанол, после чего концентрировали (Genevac) с получением mv#115 (1,4 мг, 3,3%) в виде смеси масло/твердое вещество. ВЭЖХ (Протокол A при 45°C): m/z 897,5 [M+H+], время удерживания составляет 9,149 минуты (чистота более 97%).
Получение N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (mc#115)

Стадия 1. Синтез 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноилхлорида (#248). К перемешиваемому раствору 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексановой кислоты (3,15 г, 14,9 мМ) в 15 мл дихлорметана добавляли оксалилхлорид (1,61 мл, 17,9 мМ), а затем одну каплю DMF. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение трех часов. Реакционную смесь концентрировали в вакууме. Остаток растворяли в растворе один к одному гептана и дихлорметана и затем концентрировали в вакууме. Этот процесс повторяли еще два раза с получением твердого вещества #248 (3,43 г, 100%).1H ЯМР (400 МГц, DMSO-d6): δ [7.02 (s, 2H), 3.43 (m, 2H), 2.53 (m, 1H), CH 2.18 (m, 1H), 1.54 (m, 4H), 1.26 (m, 2H)].
Стадия 2. Синтез N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-N,2-диметилаланина (#249). К перемешиваемому раствору #248 (600 мг, 2,61 мМ) в 10 мл дихлорметана добавляли N,2-диметилаланин (306 мг, 2,61 мМ), а затем триэтиламин (1,09 мл, 7,84 мМ). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение трех часов. К реакционной смеси добавляли дихлорметан и органический слой промывали три раза водой и два раза рассолом. Органический слой отделяли и затем сушили над сульфатом натрия, после чего концентрировали в вакууме. Неочищенный остаток очищали хроматографией на диоксиде кремния (0-30% метанола в дихлорметане), который предварительно был нейтрализован триэтиламином, с получением белого твердого вещества #249 (127 мг, 16%). ЖХ-МС (Протокол Q): m/z 309,0 [M-H-], время удерживания составляет 0,96 минуты.
Стадия 3. Синтез N-{(2R,3R)-3-метокси-3-[(2S)-1-{(3R,4S,5S)-3-метокси-5-метил-4-[метил(L-валил)амино]гептаноил}пирролидин-2-ил]-2-метил-пропаноил}-L-фенилаланина (#250). К перемешиваемому раствору #113 (2,10 г, 2,46 мМ) в 10 мл THF добавляли гидроксид лития (228 мг, 5,16 мМ), а затем 3 мл воды. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. Реакционную смесь подкисляли посредством добавления 1 М HCl и затем концентрировали в вакууме. Полученное белое твердое вещество переносили в 20 мл ацетонитрила и 5 мл воды. Водный слой удаляли и органический слой промывали один раз водой. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Затем добавляли этилацетат (20 мл) и неочищенное твердое вещество оставляли перемешиваться в течение 30 минуты, после чего отфильтровывали с получением белого твердого вещества #250 (1,42 г, 94%). ЖХ-МС (Протокол Q): m/z 619,5 [M+H+], время удерживания составляет 1,10 минуты. ВЭЖХ (Протокол A при 45°C) m/z 619,4 [M+H+], время удерживания составляет 6,732 минуты.
Стадия 4. Синтез N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (mc#115). К перемешиваемому раствору #249 (382 мг, 1,23 мМ) в 5 мл дихлорметана добавляли HATU (482 мг, 1,23 мМ), а затем триэтиламин (0,52 мл, 1,23 мМ). Реакционную смесь оставляли перемешиваться в течение 1 часа при комнатной температуре, а затем добавляли #250 (762 мг, 1,23 мМ). Реакционную смесь оставляли перемешиваться в течение 3 часов. Реакционную смесь концентрировали в вакууме. В результате очистки с обращенной фазой (Метод L) с последующей лиофилизацией получали белое твердое вещество mc#120 (124 мг, 11%). ВЭЖХ (Протокол A при 45°C;) m/z 911,5 [M+H+], время удерживания составляет 9,676 минуты.
Получение N-[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутаноил]-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (mb#115)

Стадия 1. Синтез N-[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутаноил]-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (mb#115). Перемешиваемый раствор 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутановой кислоты (1,2 эквивалента), HATU (1,2 эквивалента) и основания Хюнига (3 эквивалента) в DMF и дихлорметане оставляют перемешиваться в течение 30 минуты. Затем добавляют соединение #115 (1 эквивалент) в виде раствора в дихлорметане и DMF. За ходом реакции следят посредством ЖХ-МС. Реакционную смесь концентрируют и очистку завершают с помощью Isco хроматографии среднего давления с обращенной фазой (градиент: 5%-100% воды в ацетонитриле).
Получение N-[7-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гептаноил]-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (me#115)
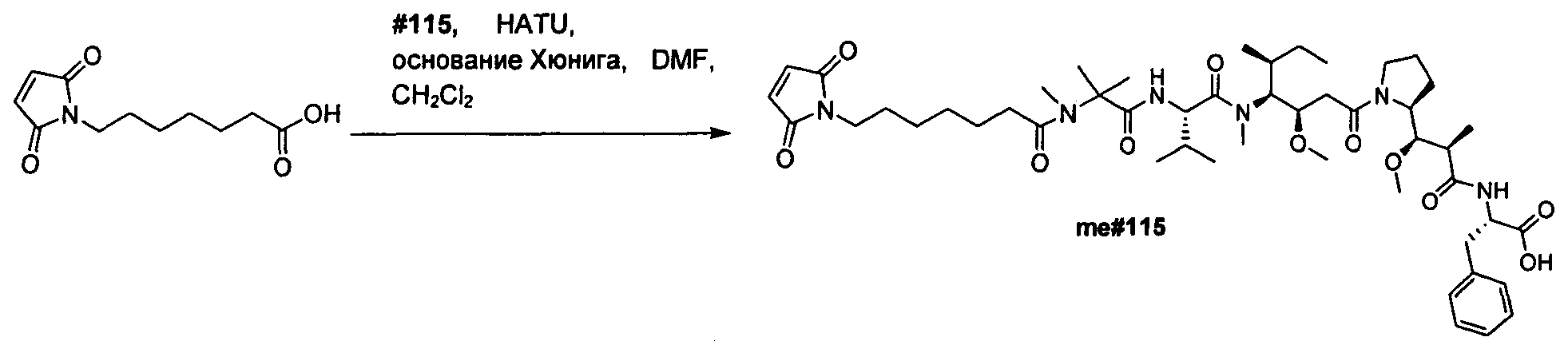
Стадия 1. Синтез N-[7-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гептаноил]-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (me#115). Перемешиваемый раствор 7-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гептановой кислоты (1,2 эквивалента), HATU (1,2 эквивалента) и основания Хюнига (3 эквивалента) в DMF и дихлорметане оставляют перемешиваться в течение 30 минуты. Затем добавляют соединение #115 (1 эквивалент) в виде раствора в дихлорметане и DMF. За ходом реакции следят посредством ЖХ-МС. Реакционную смесь концентрируют и очистку завершают с помощью Isco хроматографии среднего давления с обращенной фазой (градиент: 5%-100% воды в ацетонитриле).
Получение N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N~5~-карбамоил-N-(4-{(8S,11S,12R)-12-(2-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]-пирролидин-1-ил}-2-оксоэтил)-8-изопропил-4,5,5,10-тетраметил-11-[(1S)-1-метилпропил]-3,6,9-триоксо-2,13-диокса-4,7,10-триазатетрадец-1-ил}фенил)-L-орнитинамида (mcValCitPABC#115)

Стадия 1. Синтез N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N~5~-карбамоил-N-(4-{(8S,11S,12R)-12-(2-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-8-изопропил-4,5,5,10-тетраметил-11-[(1S)-1-метилпропил]-3,6,9-триоксо-2,13-диокса-4,7,10-триазатетрадец-1-ил}фенил)-L-орнитинамида (mcValCitPABC#115). Готовят раствор mcCValCitPABC (линкер # D, 1 эквивалент) и #115 (1 эквивалент) в DMF. Добавляют основание Хюнига (4 эквивалента), 2,6-лутидин (4 эквивалента) и HOAT (0,2 эквивалента). За ходом реакции следят посредством ЖХ-МС. Реакционную смесь концентрируют и очистку завершают с помощью Isco хроматографии среднего давления с обращенной фазой (градиент: 5%-100% воды в ацетонитриле).
Получение N-(21-амино-4,7,10,13,16,19-гексаоксагеникозан-1-оил)-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (AmPeg6C2#115)

Стадия 1. Синтез N-(21-амино-4,7,10,13,16,19-гексаоксагеникозан-1-оил)-N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамида (AmPeg6C2#115). Раствор 1-(9H-флуорен-9-ил)-3-оксо-2,7,10,13,16,19,22-гептаокса-4-азапентакозан-25-овой кислоты (1 эквивалент), HATU (1 эквивалент) и основания Хюнига (3 эквивалента) оставляют перемешиваться в течение 30 минуты. Добавляют соединение #115 в виде раствора в DMF. За ходом реакции следят с помощью ЖХ-МС. Когда реакция сочетания близка к завершению, добавляют пиперидин (5 эквивалентов). За снятием защиты в виде Fmoc следят с помощью ЖХ-МС. Реакционную смесь концентрируют и очистку завершают с помощью Isco хроматографии среднего давления с обращенной фазой (градиент: 5%-100% воды в ацетонитриле).
Получение 1,2-диметил-D-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(2S)-3-[4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-глицил}амино)фенил]-1-метокси-1-оксопропан-2-ил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида mcGly#201

Стадия 1: Синтез метилового эфира N-(трет-бутоксикарбонил)-4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицил}амино)-L-фенил-аланина (#251). К раствору метилового эфира 4-амино-N-(трет-бутоксикарбонил)-L-фенилаланина (4,1 г, 15,3 ммоль, 1 экв.) в сухом N,N-диметилформамиде (70 мл) при 0°C добавляли N,N′-дициклогексилкарбодиимид (2,9 г, 15,3 ммоль, 1 экв.). Смесь перемешивали при 0°C в течение 30 минуты. При 0°C добавляли раствор 2-(6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамидо)уксусной кислоты (3 г, 10,2 ммоль, 0,66 экв.) в сухом N,N-диметилформамиде (20 мл). Смесь перемешивали при комнатной температуре в течение 3 суток. Смесь фильтровали. Фильтрат вливали в ледяную воду (200 мл) и экстрагировали EtOAc (200 мл × 3). Экстракт промывали рассолом (200 мл), сушили над Na2SO4 и концентрировали в вакууме с получением #251 (1,8 г, выход 32,3%) в виде светло-желтого твердого вещества. ВЭЖХ (Протокол Q2) [M+Na+] 567,3, время удерживания составляет 1,02 мин.
Стадия 2: Синтез метилового эфира 4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]глицил}амино)-L-фенилаланина (#252). К раствору #251 (800 мг, 1,47 ммоль, 1 экв.) в дихлорметане (16 мл) при 0°C добавляли TFA (4,8 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Смесь концентрировали в вакууме. Остаток растворяли в воде и фильтровали. Фильтрат лиофилизировали с получением #252 (800 мг, 97,5%) в виде белого твердого вещества. ВЭЖХ (Протокол Q3) [M+H+] 445,4, время удерживания составляет 0,90 мин.
Стадия 3: Синтез 1,2-диметил-D-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(2S)-3-[4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-глицил}амино)фенил]-1-метокси-1-оксопропан-2-ил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (mcGly#201). К раствору #198 (94 мг, 0,13 ммоль, 1 экв.) и #252 (60,3 мг, 0,18 ммоль, 1,4 экв.) в N,N-диметилформамиде (2 мл) добавляли HATU (64,2 мг, 0,13 ммоль, 1 экв.), а затем N,N-диизопропилэтиламин (66 мг, 0,52 ммоль). Раствор перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь нейтрализовали с помощью водн. лимонной кислоты и концентрировали с получением неочищенного продукта, который очищали посредством хроматографии на силикагеле (элюировали смесью MeOH/DCM от 1% до 7%), затем очищали снова посредством препаративной ТСХ (смесь метанол : дихлорметан, 1:10) с получением mcGly#201 (25 мг, 16,2%) в виде белого твердого вещества: ИЭР-МС: m/z 1023,59 [M+H+], ВЭЖХ (Протокол EB) время удерживания составляет 4,0 минуты (чистота составляет 96%).1H ЯМР (DMSO-d6) δ 9.88 (d, 1H), 8.48 (d, 0.5H), 8.24 (d, 0.5H), 8.11 (m, 1H), 7.82 (m, 1H), 7.47 (d, 2H), 7.15 (m, 2H), 7.01 (s, 2H), 4.67 (m, 3H), 3.96 (m, 4H), 3.65 (m, 4H), 3.40 (m, 4H), 3.27 (m, 7H), 3.16 (m, 5H), 2.24 (m, 8H), 1.50 (m, 11H), 1.19 (m, 21H).
Получение 1,2-диметил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-{[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексил]амино}-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (MalC6Am#151)

Стадия 1. Синтез 1,2-диметил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-{[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексил]амино}-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (MalC6Am#151). В соответствии с общей методикой D, используя #151 (20 мг, 0,023 ммоль, 1,0 экв.), 1-(6-аминогексил)-1H-пиррол-2,5-дион (7,0 мг, 0,030 ммоль, 1,3 экв.), HATU (11,4 мг, 0,030 ммоль, 1,3 экв.) и основание Хюнига (0,016 мл, 0,092 ммоль, 1,3 экв.) в 2 мл дихлорметана и 0,2 мл DMF, с последующей очисткой с помощью хроматографии среднего давления с обращенной фазой C18 (градиент: от 5% до 80% ацетонитрила в воде с 0,02% TFA в каждой фазе) получали MalC6Am#151 (18,4 мг, 86%) в виде прозрачной смеси масло/твердое вещество. ЖХ-МС (Протокол Q): m/z 922,3 [M+H+], время удерживания составляет 1,43 минуты; ВЭЖХ (Протокол A при 45°C): m/z 922,4 [M+H+], время удерживания составляет 7,203 минуты.
Получение N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N-(4-{(6S,9R,10R)-6-бензил-10-[(2S)-1-{(3R,4S,5S)-4-[(1,2-диметил-L-пролил-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}пирролидин-2-ил]-9-метил-3,8-диоксо-2,11-диокса-4,7-диазадодец-1-ил}фенил)-N~5~-карбамоил-L-орнитинамида (mcValCitPABC#246)
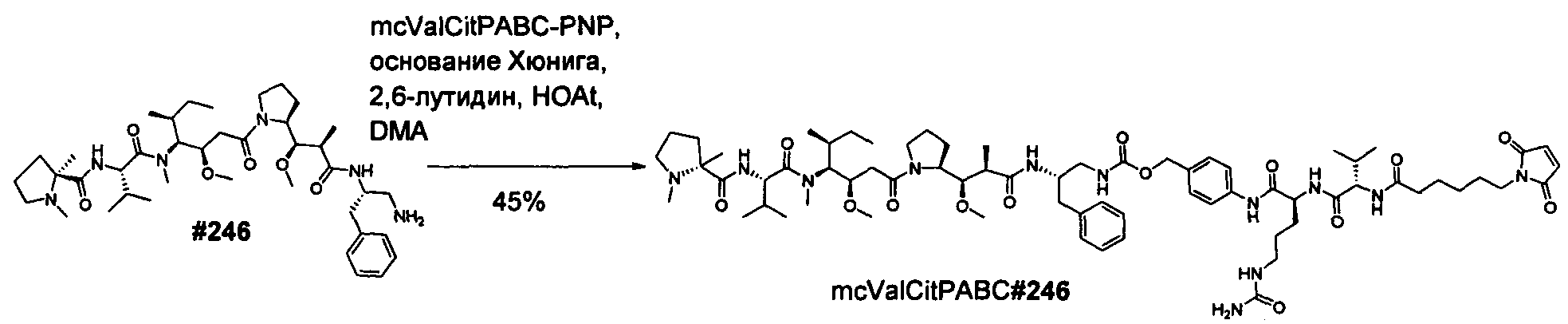
Стадия 1. Синтез N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N-(4-{(6S,9R,10R)-6-бензил-10-[(2S)-1-{(3R,4S,5S)-4-[(1,2-диметил-L-пролил-L-валил)(метил)амино]-3-метокси-5-метилгептаноил}-пирролидин-2-ил]-9-метил-3,8-диоксо-2,11-диокса-4,7-диазадодец-1-ил}фенил)-N~5~-карбамоил-L-орнитинамида (mcValCitPABC#246). В соответствии с общей методикой E, используя #246 (29,2 мг, 0,035 ммоль, 1,0 экв.), mcValCitPABC-PNP (28,8 мг, 0,039 ммоль, 1,1 экв.), 2,6-лутидин (0,016 мл, 0,14 ммоль, 4,0 экв.), основание Хюнига (0,025 мл, 0,14 ммоль, 4,0 экв.) и HOAT (4,8 мг, 0,035 ммоль, 1,0 экв.) в 2,0 мл DMA, с последующей очисткой с помощью хроматографии среднего давления с обращенной фазой C18 (градиент: от 5% до 50% ацетонитрила в воде с 0,02% TFA в каждой фазе) получали mcValCitPABC#246 (21 мг, 45%) в виде прозрачной смеси масло/твердое вещество. ЖХ-МС (Протокол Q): m/z 1327,9 [M+H+], время удерживания составляет 1,36 минуты.
Исследования HERCEPTIN® in vitro и in vivo
Следует отметить, что для следующих исследований HERCEPTIN® в отсутствие конъюгированных цитотоксических агентов не показывает существенной in vitro активности или in vivo эффективности при эквивалентных концентрациях антитела.
Методика клеточного анализа in vitro
Целевые экспрессирующие (BT474 (рак молочной железы), N87 (рак желудка), HCC1954 (рак молочной железы), MDA-MB-361-DYT2 (рак молочной железы)) или неэкспрессирующие (MDA-MB-468) клетки высевали в 96-луночные планшеты для культур клеток за 24 часа до обработки. Клетки обрабатывали 3-кратными серийными разведениями конъюгатов антитело-лекарственное средство или свободными соединениями (то есть без антитела, сконъюгированного с лекарственным средством) в двух повторностях в 10 концентрациях. Жизнеспособность клеток определяли с помощью анализа CellTiter 96® AQueous One Solution Cell Proliferation MTS (Promega, Madison WI) через 96 часов после обработки. Относительную клеточную жизнеспособность определяли как процент от необработанного контроля. Величины IC50 рассчитывали, используя четырехпараметрическую логистическую модель #203 с XLfit v 4.2 (IDBS, Guildford, Surry, UK). Результаты представлены в Таблицах 20, 21A и 21B (см. в графической части).
In vivo модель опухолевого ксенотрансплантата MDAMB-361 DYT2
Исследования эффективности in vivo конъюгатов антитело-лекарственное средство проводили с клеточной линией Her2+MDAMB-361. Для исследований эффективности 10 миллионов опухолевых клеток в 50%-ном матригеле имплантировали подкожно голым бестимусным облученным мышам в возрасте 6-8 недель. Когда размеры опухоли достигали размера от 250 до 350 мм3, посредством болюсной инъекции в хвостовую вену вводили лекарственные средства или носитель. Мышам инъецировали 1 мг/кг конъюгатов антитело-лекарственное средство с обработкой четыре раза каждые четверо суток (Q4d×4). Объем опухоли измеряют дважды в неделю в течение первых 50 суток и затем один раз каждую неделю с помощью устройства Caliper и рассчитывают по следующей формуле:
Объем опухоли = (длина × ширина2) / 2.
Результаты сравнивали по ходу исследования посредством нормализации регрессии опухоли обработанных лекарственным средством мышей путем деления объема опухоли на объем опухоли, обработанной носителем (T/C).
Шесть соединений были протестированы в трех разных исследованиях ксенотранстплантата MDA-MB-361-DYT2 для определения их противоопухолевой активности. Результаты репрезентативного исследования четырех из этих соединений демонстрируют значительную регрессию опухоли относительно мышей, обработанных носителем, на протяжении периода наблюдений 50 суток (Фиг. 1). Для сравнения результатов соединений в трех исследованиях противоопухолевую активность нормализовали путем деления объема опухоли, обработанной лекарственным средством, на объем опухоли, обработанной носителем (T/C). Представление шести величин T/C (Фиг. 2) демонстрирует, что каждое из шести соединений вызывает полную (или почти полную) регрессию опухоли на протяжении периода наблюдений, который продолжался вплоть до 107 суток для одного из исследований.
Результаты тестирования H(C)-#D54, H(C)-vcMMAE, H(C)-mcMMAF и H(K)-MCC-DM1 в исследованиях ксенотрансплантата MDA-MB-361-DYT2 представлены на Фиг. 4. График зависимости объема опухоли в группе, подвергавшейся обработке, по сравнению с контрольной группой (T/C) обеспечивает возможность сравнения между конъюгатами (см. Фиг. 5C). Эти результаты демонстрируют, что в этой модели H(C)-#D54 проявляет эффективность, эквивалентную эффективности конъюгатов HERCEPTIN® с H(C)-vcMMAE, H(C)-mcMMAF и превышающую H(K)-MCC-DM1.
In vivo модель опухолевого ксенотрансплантата N87 (HERCEPTIN®)
In vivo исследования эффективности конъюгатов антитело-лекарственное средство выполняли с моделями ксентрансплантатов, эксперессирующих мишень, используя клеточные линии N87. Для исследования эффективности 7,5 миллионов опухолевых клеток в 50%-ном матригеле имплантируют подкожно голым бестимусным мышам в возрасте 6-8 недель до того, как размеры опухоли достигнут размера между 250 и 350 мм3. Дозирование осуществляют посредством болюсной инъекции в хвостовую вену. В зависимости от опухолевого ответа на обработку животным инъецируют 1-10 мг/кг конъюгатов антитело-лекарственное средство при обработке четыре раза каждые четверо суток. У всех экспериментальных животных еженедельно осуществляют мониторинг изменения массы тела. Объем опухоли измеряют дважды в неделю в течение первых 50 суток и затем один раз каждую неделю с помощью устройства Caliper и рассчитывают согласно следующей формуле:
Объем опухоли = (длина × ширина2) / 2.
Животных гуманно умерщвляют до того, как их размеры опухоли достигают 2500 мм3. За размером опухоли наблюдают в отношении снижения после первой недели обработки. За животными может осуществляться постоянный мониторинг в отношении повторного роста опухоли после того, как обработка прекращена.
Результаты тестирования H(C)-#D54, H(C)-vcMMAE, H(C)-mcMMAF и H(K)-MCC-DM1 в модели скрининга in vivo N87 ксенотрансплантата у мышей показаны на Фиг. 3 и 5. Эти результаты демонстрируют, что в этой модели H(C)-#D54 является лучшим/сходным с конъюгатом H(C)-vcMMAE и более эффективным, чем конъюгаты H(C)-mcMMAF и H(K)-MCC-DM1.
Фармакокинетика и токсикокинетика
Фармакокинетику у мышей и токсикокинетику у крыс определяли из исследований фармакокинетики однократной дозы у мышей и токсикологии у крыс (смотри Таблицы 22 и 23 в графической части). Фармакокинетику у мышей и токсикокинетику у крыс определяли из исследований фармакокинетики однократной дозы у мышей и токсикологии у крыс. Фармакокинетику у мышей определяли из образцов, полученных от голых бестимусных мышей, которым вводили однократную дозу 3 мг/кг. Образцы собирали в течение промежутка времени вплоть до 336 ч. Токсикокинетику у крыс определяли у крыс (Sprague-Dawley (Crl:CD (SD))), которым осуществляли однократное введение H(C)-vc-MMAE или H(C)-#D54 в дозах 3, 10, и 30 мг/кг, или вводили H(C)-mc-MMAD или Н(С)-mc-MMAF в дозе 10, 30, и 100 мг/кг. Образцы собирали в течение промежутка времени вплоть до 336 часов. Концентрации в кровотоке общего антитела и ADC измеряли с помощью анализов ELISA (твердофазный иммуноферментный анализ). Площадь под кривой (AUC) рассчитывали для общего антитела и ADC для каждого ADC. Также рассчитывали AUC соотношения ADC к антителу.
Экспозиция H(C)-#D54 общего антитела и ADC были больше, чем наблюдавшиеся для H(C)-vc-MMAE у мышей в дозе 3 мг/кг и при всех дозах, оценивавшихся у крыс. Соотношение AUC для ADC и Ab для H(C)-#D54 также было больше, чем наблюдавшееся для H(C)-vc-MMAE. Эти результаты свидетельствуют о том, что H(C)-#D54 имеет большую экспозицию, и что ADC и/или конструкция линкер-полезная нагрузка потенциально более стабильны, чем H(C)-vc-MMAE.
Токсичность
Целевую независимую токсичность #D54 и компараторных конструкций линкер-полезная нагрузка (mcValCitPABC-MMAD и mcValCitPABC-MMAE), конъюгированных с моноклональным антителом, не являющимся перекрестно реактивным (IgG1), оценивали в исследовании токсичности однократной дозы у крыс с двухнедельным периодом наблюдения. Дозы конъюгатов антитело-лекарственное средство (ADC) составляли 0, 3, 10 и 30 мг/кг при n, равном 5 самцов/группу, а нагрузка конструкции линкер-полезная нагрузка была сходной среди конъюгатов (3,8, 3,2 и 4, соответственно). Эти исследования включали по меньшей мере ежедневные клинические наблюдения, еженедельное измерение массы тела, клиническую патологию (конец жизни) и некропсию (сутки 15-17) с микроскопической оценкой 9 или более тканей и любых заметных повреждений.
Гибель с сопутствующими изменениями массы тела и признаками заболевания наблюдали при дозе 30 мг/кг для всех конъюгатов и при дозе 10 мг/кг для конъюгата MMAD. В выживших группах не было клинических проявлений или изменений массы тела.
Целевые органы коъюгатов идентифицировали посредством микроскопической оценки в группах доз выживания следующим образом. Конъюгат при 10 мг/кг имел дебрис в просвете эпидидимиса (5/5, от минимального до среднего), воспаление в основании сердца (1/5 крыс, минимальное) и усиленный митоз в роговице (1/5 крыс, минимальный). Не было гистологических проявлений для конъюгата при 3 мг/кг. Для MMAD конъюгата в группе дозы выживания при 3 мг/кг имелись изменения в костном мозге и связанные с ним и в семеннике и эпидидимисе. Для MMAE конъюгата при 10 мг/кг имелись изменения в костном мозге, почке, печени и эпидидимисе. При дозе 3 мг/кг для этого конъюгата имелись изменения в почке и усиленный митоз в печени. Таким образом, в исследованиях сходного дизайна и в группах выживания конъюгат не имел проявлений в костном мозге, видимых для компараторных конъюгатов, и также не имел проявлений в почке, видимых в случае одного из компараторов.
В целом, максимальная переносимая доза (MTD) конъюгата и MMAE конъюгата составляла 10 мг/кг, а MTD для MMAD конъюгата составляла 3 мг/кг.Ненаблюдаемый уровень побочных эффектов (NOAEL) конъюгата составлял 3 мг/кг, тогда как NOAEL компараторных конъюгатов линкер-полезная нагрузка составял менее 3 мг/кг. Это исследование продемонстрировало, как токсикологический профиль для #D54 соотносится с рядом соединений, раскрытых в уровне техники.
In vitro и in vivo исследования ADC для анти-IL-13Rα2
Анти-IL-13Rα2 антитела и ADC
Гуманизированное антитело hu08 специфически связывается с рецептором IL-13Rα2. Аминокислотные и нуклеотидные последовательности для hu08 показаны в Таблице 3 (см. в графической части). CDR по Кабат (Kabat) подчеркнуты.
Гуманизированное анти-IL-13Rα2 антитело hu08 конъюгировали с различными комбинациями линкер-полезная нагрузка по настоящему изобретению, как представлено в Таблице 4 (см. в графической части). Конъюгаты антитело-лекарственное средство получали согласно способам по настоящему изобретению.
In vitro анализ цитотоксичности с анти-IL-13Rα2 ADC
Клеточные линии, экспрессирующие антиген IL-13Rα2, и клеточную линию в качестве негативного контроля культивировали с возрастающими концентрациями анти-IL-13Rα2 ADC, содержащих антитело hu08, конъюгированное с различными конструкциями линкер-полезная нагрузка по настоящему изобретению. Через четверо суток оценивали жазнеспособность каждой культуры. Величины IC50 рассчитывали с помощью логистической нелинейной регрессии и представляют как нг Ab/мл.
Данные свидетельствуют о том, что анти-1L-13Rα2 антитело hu08v1.0/1.0, конъюгированное с шестью различными полезными нагрузками ауристатина, эффективно против обеих тестировавшихся IL-13Rα2 позитивных клеточных линий (PC3MM2), имея IC50 в диапазоне от 1,1 до 4,9 нг Ab/мл или 7,3-32,7 пМ (Таблица 5, см. в графической части). Все ADC не были активны против IL-13Rα2 негативной клеточной линии, H460, а не связывающие IL-13Rα2 контрольные ADC, hulgG8.84-vc0101 и hulgG8.84-mc3377, были неактивны против всех протестированных клеточных линий.
In vivo модели подкожных ксенотрансплантатов с анти-IL-13Rα2 ADC
Гуманизированное антитело hu08 специфически связывается с рецептором IL-13Rα2. hu08 ADC с одиннадцатью различными комбинациями линкер-полезная нагрузка тестировали в in vivo модели ксенотрансплантата. Самкам бестимусных (голых) мышей подкожно инъецировали PC3MM2. Мышам с установленными опухолями размером приблизительно от 0,1 до 0,3 г (n=8-10 мышей/группу обработки) внутривенно вводили q4d×4 нормальный физиологический раствор (носитель), hu08v1.0/1.0 ADC с конструкциями линкер-полезная нагрузка vc-0101, vc-6780, vc-3906, mc-8261, mc-0131, mc-6121, mc-3377, MalPeg-8261, MalPeg-0131, MalPeg-6121 или MalPeg-3906, и несвязывающее Ab (hulgG8.84), конъюгированное с vc-0101 или mc-3377, в дозе 2 или 3 мг Ab/кг. ADC дозировали на основании содержания Ab. Опухоли измеряли по меньшей мере один раз в неделю и их размер рассчитывают как мм3 = 0,5 × (ширина опухоли2) × (длина опухоли).
Результаты эффективности in vivo, представленные в Таблице 6 (см. в графической части), показывают диапазон противоопухолевой активности с разными протестированными ADC. Относительный порядок активности является следующим: hu08-vc-0101>hu08-vc-6780>hu08-mc-0131>hu08-mc-6121>hu08-mc-3906>hu08-MalPeg-0131>hu08-MalPeg-6121>hu08-MalPeg-3906>>hu08-mc-8261. При дозовом уровне 3 мг/кг как hu08-vc-0101, так и hu08-mc-3377 демонстрировали противоопухолевую активность, тогда как несвязывающее Ab (hulgG8.84), конъюгированное с vc-0101 или mc-3377, не обладало активностью и было аналогично контрольному носителю.
Исследования анти-Notch ADC in vitro и in vivo
Анти-Notch антитела и ADC
Гуманизированные антитела, hu28 и hu75, и химерные антитела крыса-человек, ch28 и ch75, специфически связываются с Notch рецептором. Аминокислотные и нуклеотидные последовательности для hu28 и hu75 представлены в Таблице 7 (см. в графической части). CDR по Кабат подчеркнуты.
Гуманизированные анти-Notch антитела, hu28 и hu75, и химерные крыса-человек анти-Notch антитела, ch28 и ch75, конъюгировали с различными комбинациями линкер-полезная нагрузка по настоящему изобретению, как представлено в Таблице 8 (см. в графической части). Конъюгаты антитело-лекарственное средство получали в соответствии со способами по настоящему изобретению.
In vitro анализы цитотоксичности с анти-Notch ADC
Эффекты анти-Notch ADC оценивали на 1) клеточных линиях, эндогенно экспрессирующих белок Notch: HCC2429 (рак легкого), OVCAR3 (рак семенника) и MDA-MB-468 (рак молочной железы), 2) клеточных линиях, сконструированных таким образом, чтобы сверхэкспрессировать белок Notch: MDA-MB-468/hNotch и U20S/DNotch, и 3) клеточной линии, являющейся негативным контролем (SW900), с использованием индикатора клеточной жизнеспособности MTS (Promega, Madison, WI). Эти клеточные линии культивировали с возрастающими концентрациями анти-Notch ADC, содержащих гуманизированные анти-Notch антитела, hu28 и hu75, и химерные анти-Notch антитела крыса-человек, ch28 и ch75, конъюгированные с разными комбинациями линкер-полезная нагрузка по настоящему изобретению. В качестве контроля специфичности для анти-Notch-ADC на тех же клеточных линиях также тестировали не направленные против мишени контроль-ADC (huNeg8.8-ADCs или ch2H6-ADCs). Спустя четверо суток оценивали жизнеспособность каждой культуры. Величины IC50 рассчитывали посредством логистической нелинейной регрессии и представляли их как нг Ab/мл. Также представляют соотношение лекарственное средство: антитело (DAR).
В Таблице 9 (см. в графической части) представлены показатели IC50 (нг Ab/мл) для обработок гуманизированными анти-Notch ADC. HCC2429 и MDA-MB-468/hNotch клеточные линии имели два индивидуальных повтора. Данные демонстрируют, что гуманизированные анти-Notch ADC с разными конструкциями линкер-полезная нагрузка были активными и индуцировали гибель клеток в Notch экспрессирующих и сверхэкспрессирующих раковых клеточных линиях HCC2429, OVCAR3, MDA-MB-468, MDA-MB-468/hNotch, U20S/hNotch, но не в клеточной линии негативного контроля SW900, у которой отсутствует экспрессия Notch. У не направленных против мишени контроль-ADC активность либо отсутствовала (LP) и поэтому величины IC50 не генерировались как указано, либо они обладали минимальной активностью в самых высоких тестировавшихся дозах. Анти-Notch ADC, имеющие величины IC50, равные величинам IC50 для контроль-ADC, или близкие к ним, рассматривались как не обладающие активностью in vitro и указаны как LP.
В Таблице 10 (см. в графической части) показаны величны IC50 (нг Ab/мл) для обработок химерными крыса-человек анти-Notch ADC. Для экспериментов с 2-4 индивидуальными повторами рассчитывали средние величины IC50 вместе со стандартной ошибкой среднего (S.E.M.). Данные свидетельствуют о том, что химерные крыса-человек анти-Notch ADC с разными конструкциями линкер-нагрузка были акивными и индуцировали гибель клеток в Notch экспрессирующих и сверх-экспрессирующих раковых клеточных линиях HCC2429, OVCAR3, MDA-MB-468, MDA-MB-468/hNotch, U2OS/hNotch. У не направленные против мишени контроль-ADC активность либо отсутствовала (LP) и поэтому величины IC50 не генерировались как указано, либо они обладали минимальной активностью в самых высоких тестировавшихся дозах. Анти-Notch ADC, имеющие величины IC50, равные величинам IC50 для контроль-ADC, или близкие к ним, рассматривались как не обладающие активностью in vitro и указаны как LP.
In vivo модели человеческого опухолевого ксенотрансплантата с анти-Notch ADC
Гуманизированные анти-Notch антитела, hu28 и hu75, и химерные крыса-человек анти-Notch антитела, ch28 и ch75, конъюгировали с разными комбинациями линкер-полезная нагрузка и тестировали в моделях ксенотрансплантатов 37622A1 немелкоклеточного рака легкого (NSCLC), HCC2429 рака легкого, MDA-MB-468 рака молочной железы и N87 рака желудка. Для каждой описанной ниже модели первую дозу давали в Сутки 0. Опухоли измеряли по меньшей мере один раз в неделю и их объем рассчитывали по формуле:
объем опухоли (мм3) = 0,5 × (ширина опухоли2)(длина опухоли).
Рассчитывали средние объемы опухоли (±S.E.M.) для каждой обрабатываемой группы, имеющей максимум 10 и минимум 6 подлежащих включению животных.
A. Ксенотрансплантаты 37622A1 NSCLC
Эффекты анти-Notch ADC оценивали у иммунодефицитных мышей на in vivo рост человеческих опухолевых ксенотрансплантатов, которые развивались из фрагментов свежеиссеченных опухолей 37622A1 NSCLC, полученных в соответствии с надлежащими процедурами согласия (Asterand). Полученные от пациентов ксенотрансплантаты 37622A1 NSCLC подкожно пассировали in vivo как фрагменты от животного к животному у самок голых бестимусных (Nu/Nu) мышей. Когда опухоли достигали объема от 150 до 300 мм3, их исследовали для того, чтобы убедиться в однородности размера опухолей среди разных обрабатываемых групп. В модели 37622A1 NSCLC дозировали внутривенно четыре раза каждые четверо суток (Q4d×4) носитель PBS, гуманизированные анти-Notch ADC, контрольные DuNeg-8.8 ADC и цисплатин в дозах, приведенных в Таблице 11 (см. в графической части).
Цисплатин представляет собой противораковый агент на основе платины, используемый в лечении рака и считающийся стандартной терапией. Цисплатин перекрестно связывает ДНК, тем самым индуцируя апоптоз и ингибирование клеточного роста. Данные демонстрируют, что анти-Notch ADC hu28-vc0101, hu28-vc6780, hu75-vc0101 и hu75-vc6780 ингибировали рост ксенотрансплантатов 37622A1 NSCLC. Кроме того, данные показывают, что анти-Notch ADC ингибировали опухолевый рост более эффективно, чем контрольные huNeg8.8-ADC. Кроме того, данные показывают, что анти-Notch ADC ингибировали опухолевый рост более эффективно, чем цисплатин, указывая на большую эффективность, чем у химиотерапевтического лекарственного средства стандартной терапии на основе платины.
B. Легочные ксенотрансплантаты HCC2429
Аналогичные in vivo эксперименты выполняли с клеточной линией рака легкого HCC2429, как описано выше. Для получения ксенотрансплантатов самкам голых бестимусных (Nu/Nu) мышей имплантировали подкожно 3,5×106 клеток HCC2429 в 50%-ном матригеле (BD Biosciences). Когда опухоли достигали размера от 200 до 400 мм3, их исследовали для того, чтобы убедиться в однородности размера опухолей среди разных обрабатываемых групп. В модели рака легкого HCC2429 дозировали внутривенно четыре раза каждые четверо суток (Q4d×4) носитель PBS, гуманизированные анти-Notch ADC и контрольные huNeg-8.8 ADC в дозах, приведенных в Таблицах 12 и 13 (см. в графической части). Данные демонстрируют, что анти-Notch ADC hu28-vc0101, hu28-vc6780, hu75-vc0101 и hu75-vc6780 ингибировали рост легочных ксенотрансплантатов HCC2429 дозо-зависимым образом. Далее, данные показывают, что анти-Notch ADC ингибировали опухолевый рост более эффективно, чем контрольные huNeg8.8-ADC в дозах 1 и 3 мг/кг для анти-Notch ADC с vc0101 конструкциями линкер-полезная нагрузка и в дозах 3 и 10 мг/кг для анти-Notch ADC с vc6780 конструкциями линкер-полезная нагрузка. Кроме того, данные демонстрируют, что доза 3 мг/кг hu28-vc0101 была более эффективна, чем доза 10 мг/кг hu28-vc6780.
В легочной модели HCC2429 также дозировали внутривенно Q4d×4 носитель PBS, химерные крыса-человек анти-Notch ADC и контрольные huNeg-8.8 ADC в дозе 5 мг/кг, как представлено на Фиг. 8A. Данные демонстрируют, что анти-Notch ADC с нерасщепляемыми (те) и расщепляемыми (vc) линкерами и разными комбинациями полезной нагрузки ибировали рост легочных ксенотрансплантатов HCC2429. Далее, данные показывают, что химерные крыса-человек анти-Notch ADC ингибировали опухолевый рост более эффективно, чем контрольные huNeg8.8-ADC. Кроме того, данные демонтрируют, что химерные крыса-человек анти-Notch ADC с vc0101 конструкциями линкер-полезная нагрузка были более эффективными, чем другие тестировавшиеся анти-Notch ADC.
C. Ксенотрансплантаты молочной железы MDA-MB-468 Аналогичные эксперименты in vivo проводили с клеточной линией рака молочной железы MDA-MB-468, как описано выше. Клетки MDA-MB-468 классифицируют как утроенно-негативный рак молочной железы (TNBC) базально-подобного субтипа, поскольку у них отсутствует экспрессия рецептора эстрогена, рецептора прогестерона и рецептора 2 человеческого эпидермального фактора роста (HER2) (Lehmann, BD, et al, J Clin Invest. 2011; 121(7):2750-2767). Для получения ксенотрансплантатов самкам мышей SCID Hairless Outbred (SHO) в жировое тело молочной железы ортотопически имплантировали 50%-ный матригель, содержащий 10×106 клеток MDA-MB-468 (BD Biosciences). Когда опухоли достигали объема от 250 до 450 мм3, опухоли исследовали для того, чтобы удостовериться в однородности массы опухоли среди разных обработываемых групп. В модели молочной железы MDA-MB-468 внутривенно дозировали Q4d×4 носитель PBS, гуманизированные анти-Notch ADC и контрольные huNeg-8.8 ADC в дозах, представленных в Таблицах 14 и 15 (см. в графической части). Данные демонтрируют, что анти-Notch ADC hu28-vc0101, hu28-vc6780, hu75-vc0101 и hu75-vc6780 ингибировали рост ксенотрансплантатов молочной железы MDA-MB-468 дозо-зависимым образом. Далее, данные показывают, что анти-Notch ADC ингибировали опухолевый рост более эффективно, чем контрольные huNeg8.8-ADC, в дозах 1 и 3 мг/кг для ADC с vc0101 конструкциями линкер-полезная нагрузка и в дозах 1, 3 и 10 мг/кг для ADC с vc6780 конструкциями линкер-полезная нагрузка. Кроме того, данные демонтрируют, что доза 1 мг/кг анти-Notch ADC с vc0101 конструкциями линкер-полезная нагрузка была более эффективна, чем доза 3 мг/кг анти-Notch ADC с vc6780 конструкциями линкер-полезная нагрузка.
В модели молочной железы MDA-MB-468 также дозировали внутривенно Q4d×4 носитель PBS, химерные крыса-человек анти-Notch ADC и контрольные huNeg-8.8 ADC в дозе 5 мг/кг, как представлено на Фиг. 8B и 8C. Данные демонстрируют, что химерные крыса-человек анти-Notch ADC с нерасщепляемыми (mc) и расщепляемыми (vc) линкерами и разными комбинациями полезной нагрузки ингибировали рост ксенотрансплантатов молочной железы MDA-MB-468. Далее, данные показывают, что химерные крыса-человек анти-Notch ADC ингибировали опухолевый рост более эффективно, чем контрольные huNeg8.8-ADC. Далее, данные демонстрируют, что химерные крыса-человек анти-Notch ADC с vc0101 конструкциями линкер-полезная нагрузка были более эффективны, чем другие тестировавшиеся химерные крыса-человек анти-Notch ADC.
D. Желудочные ксенотрансплантаты N87
Аналогичные in vivo эксперименты проводили с клеточной линией рака желудка N87, как описано выше. Для получения ксенотрансплантатов самкам голых бестимусных (Nu/Nu) мышей имплантировали подкожно 7,5×106 клеток N87 в 50%-ном матригеле (BD Biosciences). Когда опухоли достигали объема от 250 до 450 мм3, опухоли исследовали для того, чтобы удостовериться в однородности массы опухоли среди разных обрабатываемых групп. В желудочной модели N87 дозировали внутривенно Q4d×4 носитель PBS, гуманизированные анти-Notch ADC, контрольные huNeg-8.8 ADC и цисплатин в дозах, представленных в Таблицах 16 и 17 (см. в графической части). Данные демонстрируют, что анти-NotchADC hu28-vc0101, hu28-vc6780, hu75-vc0101 и hu75-vc6780 ингибировали рост желудочных ксенотрансплантатов N87 дозо-зависимым образом. Далее, данные показывают, что анти-Notch ADC ингибировали опухолевый рост более эффективно, чем контрольные huNeg8.8-ADC, в дозах 1, 3, 5 мг/кг для ADC с vc0101 конструкциями линкер-полезная нагрузка и в дозах 3 и 10 мг/кг для ADC с vc6780 конструкциями линкер-полезная нагрузка. Кроме того, данные демонстрируют, что ADC с vc0101 конструкциями линкер-полезная нагрузка в общем были более эффктивны, чем стандартная терапия цисплатином и ADC с vc6780 конструкциями линкер-полезная нагрузка.
В желудочной модели N87 также дозировали внутривенно Q4d×4 носитель PBS, химерные крыса-человек анти-Notch ADC и контрольные huNeg-8.8 ADC в дозе 5 мг/кг, как представлено на Фиг. 8D. Данные демонстрируют, что химерные крыса-человек анти-Notch ADC с нерасщепляемыми (mc) и расщепляемыми (vc) линкерами и разными комбинациями полезной нагрузки ингибировали рост желудочных ксенотрансплантатов N87. Далее, данные показывают, что химерные крыса-человек анти-Notch ADC ингибировали опухолевый рост более эффективно, чем контрольные huNeg8.8-ADC. Кроме того, данные демонстрируют, что химерные крыса-человек анти-Notch ADC с vc0101 конструкциями линкер-полезная нагрузка были более эффективны, чем другие тестировавшиеся анти-Notch ADC.
В желудочной модели N87 также дозировали внутривенно Q4d×4 носитель PBS и химерные крыса-человек анти-Notch ADC ch28-mc0131, ch75-mc0131, ch28-m(H2O)c-0131 и ch75-m(H2O)c-0131 в дозе 5 мг/кг, как представлено на Фиг. 8E. Данные демонстрируют, что химерные крыса-человек анти-Notch ADC, имеющие mc0131 и m(H2O)c-0131 конструкции линкер-полезная нагрузка, ингибировали рост желудочных ксенотрансплантатов N87. Далее, данные демонстрируют, что химерные крыса-человек анти-Notch ADC, имеющие m(H2O)c-0131 конструкции линкер-полезная нагрузка, были более эффективными, чем химерные крыса-человек анти-Notch ADC, имеющие mc0131 конструкции линкер-полезная нагрузка.
Реферат
Изобретение относится к цитотоксическим пентапептидам формулы (I), к их конъюгатам антитело-лекарственное средство и фармацевтическим композициям. Соединения обладают противоопухолевой активностью и предназначены для лечения рака. 13 н. и 15 з.п. ф-лы, 8 ил., 29 табл.
Формула

или его фармацевтически приемлемая соль, где, в каждом случае независимо,
W представляет собой




R1 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R2 представляет собой водород, С1-С8алкил или C1-С8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; и
R3B представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой С2-С8алкилен или С2-С8гетероалкилен, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S;
R4A и R4B определены как любые из следующих:
R4A представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил; и
R4B представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил;
R5 представляет собой
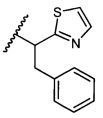










C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, С3-С8карбоциклил и С6-С14арил, возможно, замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: -С1-С8алкил, -С1-С8алкил-N(R′)2, -С1-С8алкил-С(O)R′, -С1-С8алкил-С(O)OR′, -O-(С1-С8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -ОН, галоген, -N3, -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2 и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, С1-С8алкила и незамещенного С6-С14арила, или два R′ могут вместе с азотом, к которому они присоединены, образовывать C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S;
или R5 представляет собой




R6 представляет собой водород, -С1-С8алкил, -С2-С8алкенил, -С2-С8алкинил или -С1-С8галогеналкил;
R12 представляет собой водород, С1-С4алкил, C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, или С6-С14арил;
R13 представляет собой С1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S; и
X представляет собой О.

или его фармацевтически приемлемая соль, где, в каждом случае независимо,
W представляет собой


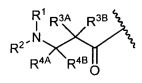

R1 представляет собой



Y представляет собой -С2-С20алкилен-, -С2-С20гетероалкилен-, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S, -С3-С8карбоцикло-, -С6-С14арилен-, -С3-С8гетероцикло-, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, -С1-С10алкилен-С6-С14арилен-, -С6-С14арилен-С1-С10алкилен-, -С1-С10алкилен-(С3-С8карбоцикло)-, -(С3-С8карбоцикло)-С1-С10алкилен-, -С1-С10алкилен-(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)- или -(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)-С1-С10алкилен-;
Z представляет собой

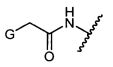
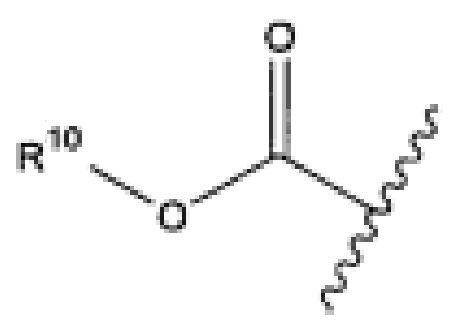

G представляет собой галоген, -ОН, -SH или -S-С1-С6алкил;
R2 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; и
R3B представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой С2-С8алкилен или С2-С8гетероалкилен, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S;
R4A и R4B определены как любые из следующих:
R4A представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил; и
R4B представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил;
R5 представляет собой










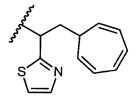
C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, С3-С8карбоциклил и С6-С14арил, возможно, замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: -С1-С8алкил, -C1-С8алкил-N(R′)2, -С1-С8алкил-С(O)R′, -С1-С8алкил-С(O)OR′, -O-(С1-С8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -ОН, галоген, -N3, -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2 и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, С1-С8алкила и незамещенного С6-С14арила, или два R′ могут вместе с азотом, к которому они присоединены, образовывать C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S;
или R5 представляет собой

R6 представляет собой водород, -С1-С8алкил, -С2-С8алкенил, -С2-С8алкинил или -С1-С8галогеналкил;
R12 представляет собой водород, С1-С4алкил, C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, или С6-С14арил;
R13 представляет собой C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S; и
R7 независимо выбран для каждого случая из группы, состоящей из F, Cl, I, Br, NO2, CN и CF3;
R10 представляет собой водород, -С1-С10алкил, -С3-С8карбоциклил, -С6-С14арил, -C1-С10гетероалкил, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S, -С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, -С1-С10алкилен-С6-С14арил, -С6-С14арилен-С1-С10алкил, -С1-С10алкилен-(С3-С8карбоцикло), -(С3-С8карбоцикло)-С1-С10алкил, -С1-С10алкилен-(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S) и -(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)-С1-С10алкил, где С6-С14арил в R10, содержащем арил, возможно, замещен [R7]h;
h равен 1, 2, 3, 4 или 5; и
X представляет собой О.

или его фармацевтически приемлемая соль, где, в каждом случае независимо,
W представляет собой
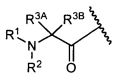



R1 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R2 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; и
R3B представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой С2-С8алкилен или С2-С8гетероалкилен, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S;
R4A и R4B определены как любые из следующих:
R4A представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил; и
R4B представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил;
R5 представляет собой


или

возможно, замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: -С1-С8алкил, -O-(С1-С8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -ОН, галоген, -N3, -NH2, -NH(R′), -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, С1-С8алкила и незамещенного С6-С14арила;
R11 представляет собой



Y представляет собой -С2-С20алкилен-, -С2-С20гетероалкилен-, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S, С3-С8карбоцикло-, -С6-С14арилен-, -С3-С8гетероцикло-, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, -С1-С10алкилен-С6-С14арилен-, -С6-С14арилен-С1-С10алкилен-, -С1-С10алкилен-(С3-С8карбоцикло)-, -(С3-С8карбоцикло)-С1-С10алкилен-, -С1-С10алкилен-(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)- или -(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)-С1-С10алкилен-;
Z представляет собой
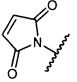




G представляет собой галоген, -ОН, -SH или -S-C1-С6алкил;
R7 в каждом случае независимо выбран из группы, состоящей из F, Cl, I, Br, NO2, CN и CF3;
h равен 1, 2, 3, 4 или 5; и
X представляет собой О.

или его фармацевтически приемлемая соль, где, в каждом случае независимо,
W представляет собой


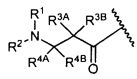
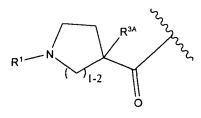
R1 представляет собой



Y представляет собой -С2-С20алкилен-, -С2-С20гетероалкилен-, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S, -С3-С8карбоцикло-, -С6-С14арилен-, -С3-С8гетероцикло-, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, -С1-С10алкилен-С6-С14арилен-, -С6-С14арилен-С1-С10алкилен-, -С1-С10алкилен-(С3-С8карбоцикло)-, -(С3-С8карбоцикло)-С1-С10алкилен-, -С1-С10алкилен-(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)- или -(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)-С1-С10алкилен-;
Z представляет собой



L представляет собой антитело;
R2 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой C1-С8алкил, насыщенный С3-С8карбоциклил или галоген; и
R3B представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой С2-С8алкилен или С2-С8гетероалкилен, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S;
R4A и R4B определены как любые из следующих:
R4A представляет собой водород, C1-С8алкил или насыщенный С3-С8карбоциклил; и
R4B представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил;
R5 представляет собой

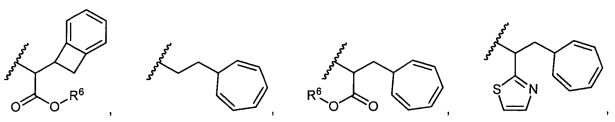
C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, С3-С8карбоциклил и С6-С14арил, возможно, замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: -С1-С8алкил, -С1-С8алкил-N(R′)2, -С1-С8алкил-С(O)R′, -С1-С8алкил-С(O)OR′, -O-(С1-С8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -ОН, галоген, -N3, -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2 и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, С1-С8алкила и незамещенного С6-С14арила, или два R′ могут вместе с азотом, к которому они присоединены, образовывать C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S;
или R5 представляет собой

R6 представляет собой водород, -С1-С8алкил, -С2-С8алкенил, -С2-С8алкинил или -С1-С8галогеналкил;
R12 представляет собой водород, С1-С4алкил, C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, или С6-С14арил;
R13 представляет собой C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S; и
X представляет собой О.

или его фармацевтически приемлемая соль, где, в каждом случае независимо,
W представляет собой

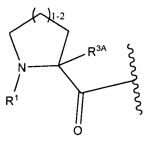
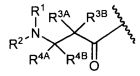

R1 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R2 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; и
R3B представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой С2-С8алкилен или С2-С8гетероалкилен, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S;
R4A и R4B определены как любые из следующих:
R4A представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил; и
R4B представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил;
R5 представляет собой

или

возможно, замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: C1-С8алкил, -O-(С1-С8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -ОН, галоген, -N3, -NH2, -NH(R′), -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, С1-С8алкила и незамещенного С6-С14арила;
R11 представляет собой



Y представляет собой -С2-С20алкилен-, -С2-С20гетероалкилен-, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S, -С3-С8карбоцикло-, -С6-С14арилен-, -С3-С8гетероцикло-, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, -С1-С10алкилен-С6-С14арилен-, -С6-С14арилен-С1-С10алкилен-, -С1-С10алкилен-(С3-С8карбоцикло)-, -(С3-С8карбоцикло)-С1-С10алкилен-, -С1-С10алкилен-(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)- или -(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)-С1-С10алкилен-;
Z представляет собой




L представляет собой антитело;
X представляет собой О.

или его фармацевтически приемлемая соль, где, в каждом случае независимо,
R1′ представляет собой


Y представляет собой -С2-С20алкилен-, -С2-С20гетероалкилен-, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S, -С3-С8карбоцикло-, -С6-С14арилен-, -С3-С8гетероцикло-, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, -С1-С10алкилен-С6-С14арилен-, -С6-С14арилен-С1-С10алкилен-, -С1-С10алкилен-(С3-С8карбоцикло)-, -(С3-С8карбоцикло)-С1-С10алкилен-, -С1-С10алкилен-(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)- или -(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)-С1-С10алкилен-;
Z′ представляет собой



L представляет собой антитело;
D представляет собой -C(R4A′)(R4B′)- или отсутствует;
R2′ представляет собой водород, С1-С8алкил или С1-С8галогеналкил, либо он отсутствует, когда присутствует

R3A′ и R3B′ определены как любые из следующих:
(1) R3A′ представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; и
R3B′ представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген, или R3B′ представляет собой С2-С4алкилен и образует 5-7-членное кольцо, как указано посредством

(2) R3A′ и R3B′, взятые вместе, представляют собой С2-С8алкилен или С2-С8гетероалкилен, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S;
R4A′ и R4B′ определены как любые из следующих:
R4A′ представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил; и
R4B′ представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил;
R5 представляет собой



C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, С3-С8карбоциклил и С6-С14арил, возможно, замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: -С1-С8алкил, -C1-С8алкил-N(R′)2, -С1-С8алкил-С(O)R′, -С1-С8алкил-С(O)OR′, -O-(С1-С8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -ОН, галоген, -N3, -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2 и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, С1-С8алкила и незамещенного С6-С14арила, или два R′ могут вместе с азотом, к которому они присоединены, образовывать C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S;
или R5 представляет собой

R6 представляет собой водород, -C1-С8алкил, -С2-С8алкенил, -С2-С8алкинил или -С1-С8галогеналкил;
R12 представляет собой водород, С1-С4алкил, C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, или С6-С14арил;
R13 представляет собой C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S; и
X представляет собой О.

или его фармацевтически приемлемая соль, где, в каждом случае независимо,
W представляет собой

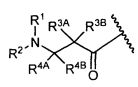

R1 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R2 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; и
R3B представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой С2-С8алкилен или С2-С8гетероалкилен, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S;
R4A и R4B определены как любые из следующих:
R4A представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил; и
R4B представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил;
R5 представляет собой
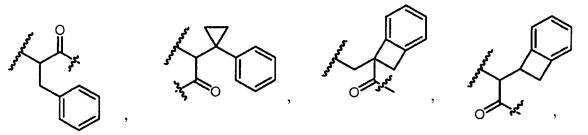

или

возможно, замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: С1-С8алкил, -О-(С1-С8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -ОН, галоген, -N3, -NH2, -NH(R′), -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, С1-С8алкила и незамещенного С6-С14арила;
R11′ представляет собой


Y представляет собой -С2-С20алкилен-, -С2-С20гетероалкилен-, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S, -С3-С8карбоцикло-, -С6-С14арилен-, -С3-С8гетероцикло-, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, -С1-С10алкилен-С6-С14арилен-, -С6-С14арилен-С1-С10алкилен-, -С1-С10алкилен-(С3-С8карбоцикло)-, -(С3-С8карбоцикло)-С1-С10алкилен-, -С1-С10алкилен-(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)- или -(С3-С8гетероцикло, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S)-С1-С10алкилен-;
Z′ представляет собой

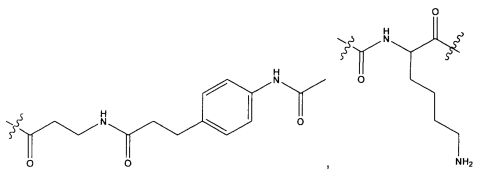
L представляет собой антитело;
X представляет собой О.

или его фармацевтически приемлемая соль, где, в каждом случае независимо,
L представляет собой антитело;
[линкер] представляет собой двухвалентный линкер;
D представляет собой -C(R4A′)(R4B′)- или отсутствует;
R2′ представляет собой водород, С1-С8алкил или C1-С8галогеналкил, либо он отсутствует, когда присутствует

R3A′ и R3B′ определены как любые из следующих:
(1) R3A′ представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; и
R3B′ представляет собой C1-С8алкил, насыщенный С3-С8карбоциклил или галоген, или R3B′ представляет собой С2-С4алкилен и образует 5-7-членное кольцо, как указано посредством

(2) R3A′ и R3B′, взятые вместе, представляют собой С2-С8алкилен или С2-С8гетероалкилен, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S;
R4A′ и R4B′ определены как любые из следующих:
R4A′ представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил; и
R4B′ представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил;
R5 представляет собой



C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, С3-С8карбоциклил и С6-С14арил, возможно, замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: -С1-С8алкил, -С1-С8алкил-N(R′)2, -С1-С8алкил-С(O)R′, -С1-С8алкил-С(O)OR′, -O-(С1-С8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -ОН, галоген, -N3, -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2 и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, С1-С8алкила и незамещенного С6-С14арила, или два R′ могут вместе с азотом, к которому они присоединены, образовывать C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S;
или R5 представляет собой
R6 представляет собой водород, -С1-С8алкил, -С2-С8алкенил, -С2-С8алкинил или -С1-С8галогеналкил;
R12 представляет собой водород, С1-С4алкил, C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S, или С6-С14арил;
R13 представляет собой C1-С10гетероциклил, имеющий от 1 до 4 гетероатомов, выбранных из N, О и S; и
X представляет собой О.

или его фармацевтически приемлемая соль, где, в каждом случае независимо,
W представляет собой



R1 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R2 представляет собой водород, С1-С8алкил или С1-С8галогеналкил;
R3A и R3B определены как любые из следующих:
(1) R3A представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; и
R3B представляет собой С1-С8алкил, насыщенный С3-С8карбоциклил или галоген; или
(2) R3A и R3B, взятые вместе, представляют собой С2-С8алкилен или С2-С8гетероалкилен, имеющий от 1 до 3 гетероатомов, выбранных из О, N и S;
R4A и R4B определены как любые из следующих:
R4A представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил; и
R4B представляет собой водород, С1-С8алкил или насыщенный С3-С8карбоциклил;
R5 представляет собой

или

возможно, замещенные 1, 2, 3, 4 или 5 группами, независимо выбранными из группы, состоящей из следующих: C1-С8алкил, -О-(С1-С8алкил), -C(O)R′, -OC(O)R′, -C(O)OR′, -C(O)NH2, -C(O)NHR′, -C(O)N(R′)2, -NHC(O)R′, -S(O)2R′, -S(O)R′, -ОН, галоген, -N3, -NH2, -NH(R′), -N(R′)2, -CN, -NHC(=NH)NH2, -NHCONH2и -SR′, где каждый R′ независимо выбран из группы, состоящей из водорода, С1-С8алкила и незамещенного С6-С14арила;
[линкер] представляет собой двухвалентный линкер;
L представляет собой антитело;
X представляет собой О.
W представляет собой























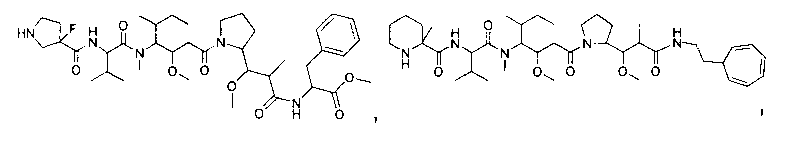











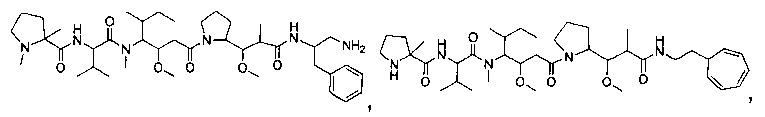


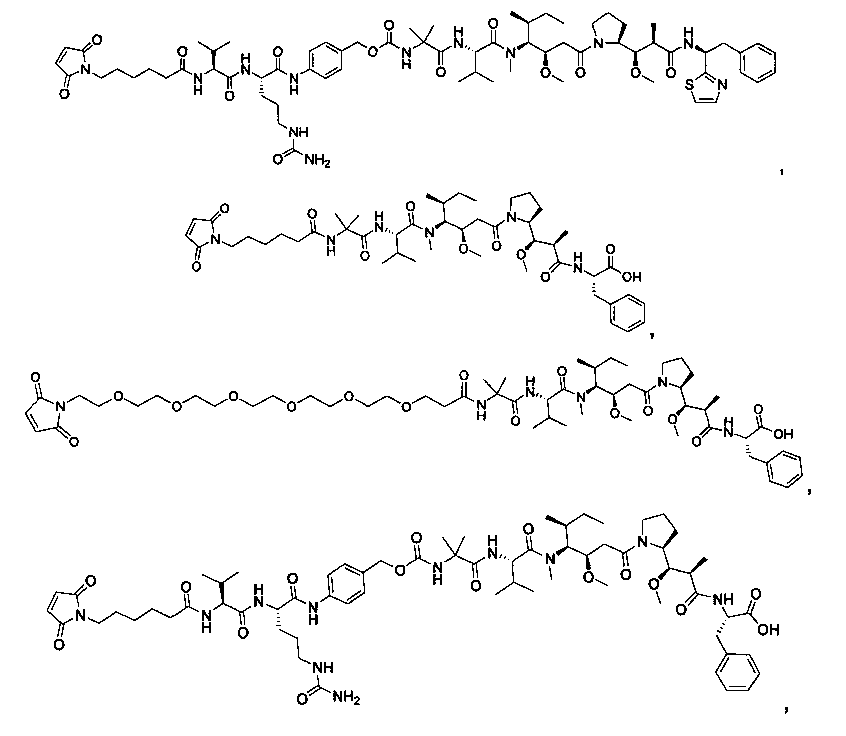
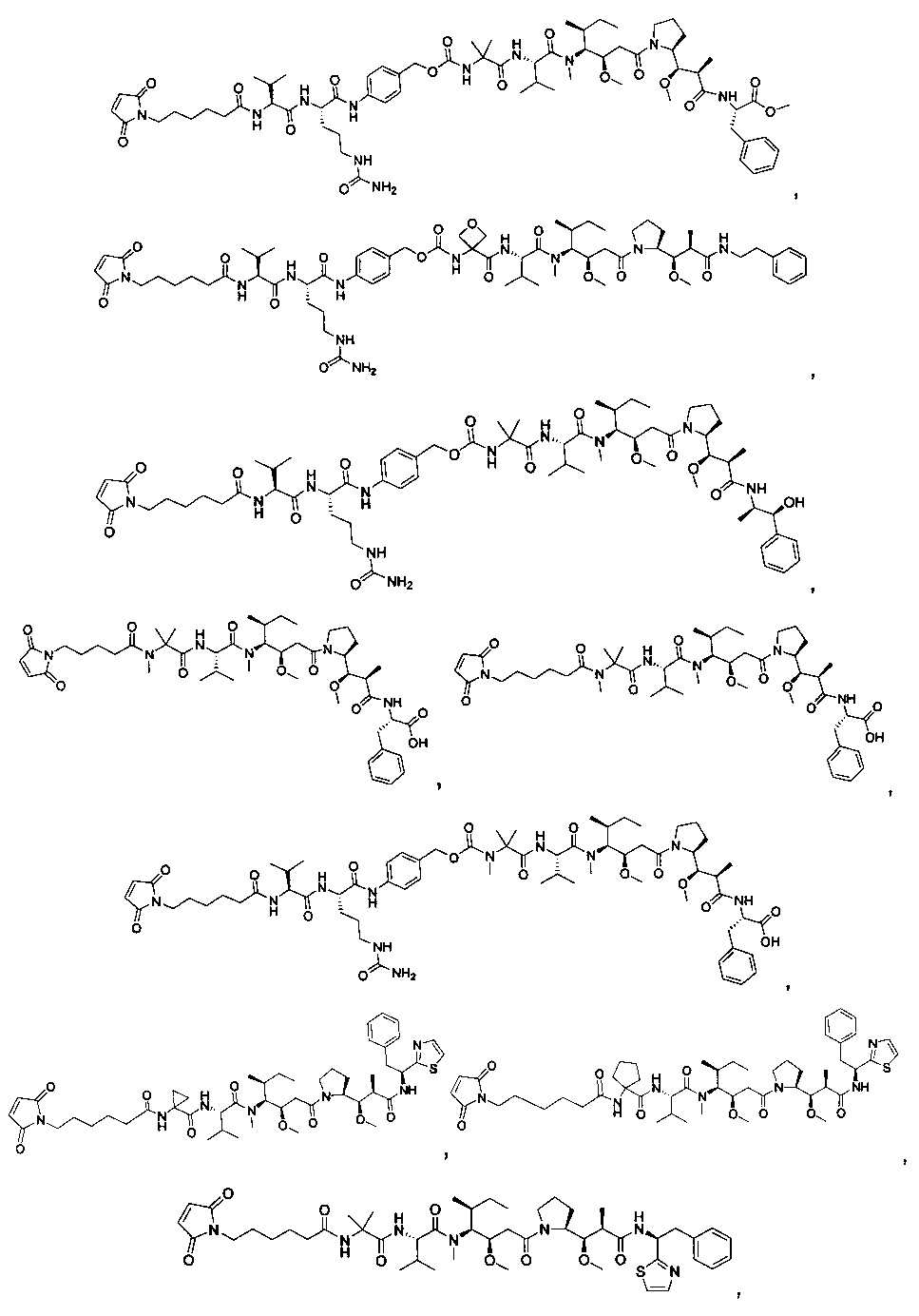







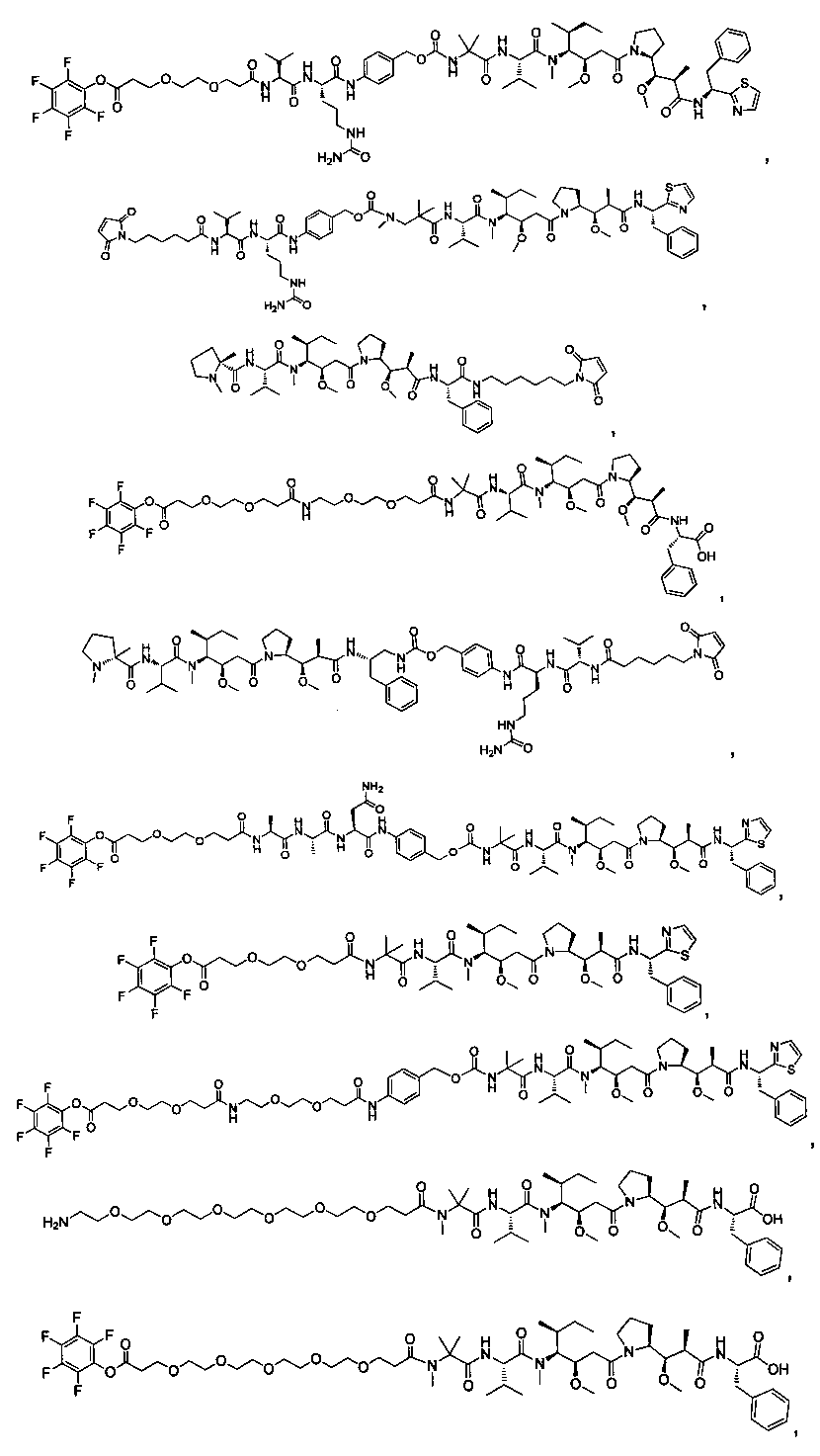

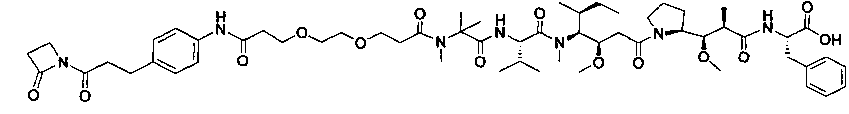








Комментарии