Амидные соединения, способы получения, применение в качестве средств для лечения и профилактики заболеваний, вызываемых рнк-содержащими вирусами - RU2628800C2
Код документа: RU2628800C2
Чертежи










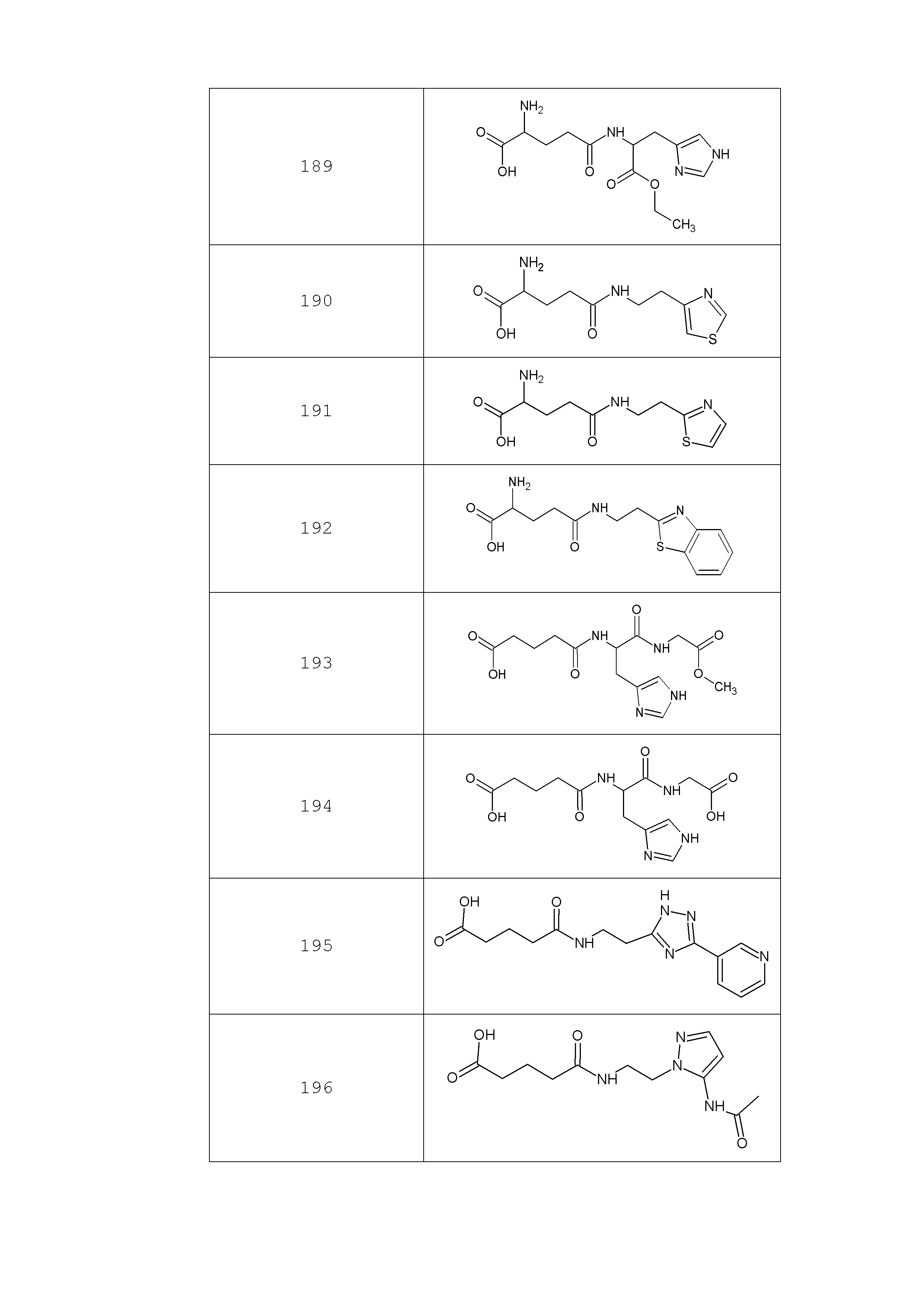



















































































Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к медицине, в частности, к применению соединений общей формулы I или их фармацевтически приемлемых солей для профилактики и лечения заболеваний, вызываемых РНК-содержащими вирусами.
Уровень техники
Вирусные инфекции являются огромной проблемой для здравоохранения. Антивирусные препараты в отношении большинства опасных и особо опасных вирусных инфекций не разработаны, а существующие нередко являются токсичными для человека или обладают недостаточной эффективностью. Большинство существующих или находящихся в разработке препаратов действуют через специфическое взаимодействие с определенными вирусными белками. Такие препараты имеют ограниченный спектр действия и способствуют быстрому появлению резистентных вариантов вирусов. В системе классификации вирусов по Балтимору к IV и V классам относятся вирусы, содержащие одноцепочечную (+) или (-) РНК. К классу IV принадлежат представители рода энтеровирусов из семейства пикорнавирусов и семейство коронавирусов, а к классу V относятся респираторно-синцитиальный вирус (РСВ) и метапневмовирус семейства парамиксовирусов.
Перечисленные группы вирусов выработали эффективную стратегию подавления антивирусных программ клетки. Столь агрессивная стратегия подавления системы клеточной противовирусной защиты приводит к высокой контагиозности и патогенности данных групп вирусов, подтверждаемой списком заболеваний, вызываемых представителями рода энтеровирусов (полиомиелит, вирусный ринит (риновирусная простуда). Среди представителей рода энтеровирусов в настоящее время наибольшую проблему составляют человеческие риновирусы. Риновирусы вызывают у человека и животных воспалительные заболевания верхних дыхательных путей, размножаясь в клетках слизистой оболочки носоглотки. Риновирусы ответственны за как минимум 80% простудных заболеваний. Помимо огромного экономического ущерба (20 млн человек/часов в США ежегодно), риновирусные инфекции вызывают большое количество осложнений, таких как синусит и воспаление среднего уха и часто обнаруживаются при вирусологическом обследовании детей, больных пневмонией. Также у детей-астматиков риновирусная инфекция является причиной обострения в 80% случаев. У взрослых, риновирусы могут вызывать как обострение астмы, так и хронической обструктивной болезни легких, хронического бронхита, муковисцидоза. Риновирусы были изолированы у больных пневмонией при иммунодефицитных состояниях.
В связи с тем, что существует более 100 антигенных разновидностей риновирусов, создание эффективной вакцины не представляется возможным (Palmenberg, A. C.; Spiro, D; Kuzmickas, R; Wang, S; Djikeng, A; Rathe, JA; Fraser-Liggett, CM; Liggett, SB (2009). "Sequencing and Analyses of All Known Human rhinovirus Genomes Reveals Structure and Evolution". Science 324 (5923): 55–9. doi: 10.1126/science. 1165557. PMID 19213880.). Кроме того, не существует эффективного химиотерапевтического средства для лечения риновирусной инфекции.
Энтеровирус типа 71 (EV71) был впервые выделен в 1970-1972 гг. от больных асептическим менингитом и больного энцефалитом в Калифорнии. Важно отметить, что в тяжелых случаях вирус приводит к развитию неврологических нарушений, таких как менингит, паралич и энцефалит. Вирус распространяется в условиях антисанитарии. При заражении вирусом EV71 поднимается температура, появляется сыпь на коже рук и ног, на ладонях, стопах, отек конечностей, возникают язвы в ротовой полости. В тяжелой форме энтеровирус может привести к летальному исходу. Как отмечается, энтеровирус-71 - один из наиболее «тяжелых» из общего числа энтеровирусов человека. Этот вирус может вызывать крупные вспышки с летальными исходами. Вакцины против энтеровируса-71 не существует, а неспецифическая терапия не разработана.
Коксакивирусная инфекция (НСХV) представляет собой большую группу заболеваний, характеризующихся выраженным клиническим полиморфизмом. Манифестация Коксакивирусной инфекции может выражаться менингитом, параличами, острыми респираторными расстройствами, пневмонией, геморрагическим конъюнктивитом, миокардитом, гепатитом, диабетом и другими синдромами. В соответствии с современной классификацией вирусов - энтеровирусы человека в составе рода Enterovirus разделены на пять видов (14): 1) полиовирус; 2) энтеровирус человека А; 3) энтеровирус человека В; 4) энтеровирус человека С; 5) энтеровирус человека D. Различные серотипы вируса Коксаки вошли в следующие виды энтеровирусов: энтеровирус человека А (Коксаки А2-8, 10, 12, 14, 16); энтеровирус человека В (Коксаки А9, Коксаки В1-6); энтеровирус человека С (Коксаки А1, 11, 13, 15, 17-22, 24).
Вирусы Коксаки, как и другие энтеровирусы человека, распространены повсеместно на земном шаре. Для стран умеренного климата характерен максимум их циркуляции в летне-осенний сезон. Вирусы обладают высокой степенью инвазивности, что обусловливает их быстрое распространение в человеческой популяции. Вирусы Коксаки часто являются причиной «внезапного» возникновения вспышек в организованных детских коллективах, больницах, наблюдается также внутрисемейное распространение инфекции. В эпидемиологии Коксакивирусной и других энтеровирусных инфекций человека важную роль играет высокий уровень изменчивости вирусного генома. Следствием этого является способность тех или иных серотипов вызывать в определенных обстоятельствах различную патологию. С другой стороны, один и тот же клинический синдром может быть обусловлен разными серотипами и разными видами энтеровирусов. В результате генетической изменчивости, селекции и быстрого распространения измененных вирусов возникают крупные вспышки заболеваний, в этиологии которых ранее данные вирусы не принимали участия, либо их циркуляция не наблюдалась в течение длительного времени.
Первичное размножение вируса Коксаки происходит в лимфоидной ткани носоглотки и кишечника. Он вызывает локальные поражения, выражающиеся симптоматикой ОРЗ, герпангины, фарингита и др. В глотке вирус определяется до 7-х суток, а с фекалиями он экскретируется 3-4 нед (при иммунодефицитах – несколько лет). Вслед за первичным размножением наступает стадия виремии, в результате которой возбудитель проникает в органы-мишени. Для вирусов Коксаки это могут быть головной и спиной мозг, мягкие мозговые оболочки, верхние дыхательные пути, легкие, сердце, печень, кожа и др. Вирусы Коксаки В могут вызывать тяжелые генерализованные патологические процессы у новорожденных. При этом в сердце, головном и спином мозге, печени, почках возникают очаги некрозов. Вирусы вызывают развитие следующих клинических синдромов: серозный менингит (Коксаки А2, 3, 4, 6, 7, 9, 10, Коксаки В1-6); острое системное заболевание детей с миокардитом и менингоэнцефалитом (Коксаки В1-5); параличи (Коксаки А1, 2, 5, 7, 8, 9, 21, Коксаки В2-5); герпангина (Коксаки А2, 3, 4, 5, 6, 8, 10); острый фарингит (Коксаки А10, 21); контагиозный насморк (Коксаки А21, 24); поражение верхних дыхательных путей и пневмония (Коксаки А9, 16, Коксаки В2-5) (16); перикардит, миокардит (Коксаки В1-5); гепатит (Коксаки А4, 9, 20, Коксаки В5); диарея новорожденных и детей младшего возраста (Коксаки А18, 20, 21, 24); острый геморрагический конъюнктивит (Коксаки А24); ящуроподобное заболевание (Коксаки А5, 10, 16); экзантема (Коксаки А4, 5, 6, 9, 16); плевродиния (Коксаки В3, 5); сыпь (Коксаки В5); лихорадка (Коксаки В1-6). Для лечения Коксакивирусной инфекции специфические химиотерапевтические препараты отсутствуют. Осуществляется патогенетическая и симптоматическая терапия, зависящая от клинической формы болезни.
К семейству Парамиксовирусов относятся представители родов респировирусов (вирус парагриппа человека 1, 2, 3, 4 и 5 типов), пневмовирусов (респираторно-синтициальный вирус) и метапневмовирусов (метапневмовирус человека).
Парамиксовирусы являются важным классом вирусов, которые связаны с респираторными заболеваниями. Респираторно- синцитиальный вирус (РСВ), как известно, является доминирующим патогеном нижних дыхательных путей во всем мире.
РСВ является важным патогеном новорожденных и детей раннего возраста и ответственен за ~ 70% тяжелых вирусных бронхиолитов и / или пневмоний, большая часть из которых характеризуется свистящим дыханием и одышкой. Данные бронхиолиты являются наиболее распространенной причиной госпитализации в зимний сезон в течение первого года жизни ребенка. Также РСВ вызывает бронхиолит, пневмонию и хроническую обструктивную болезнь легких у людей всех возрастов и в значительной степени способствует избыточной смертности в зимний сезон.
РСВ занимает лидирующее место по количеству смертельных исходов среди вирусных инфекций. Только в США более 2,4 млрд. долларов расходуется на лечение вирусных заболеваний нижнего респираторного тракта у детей. К первому году жизни 50-65% детей заражаются этим вирусом, и к двум годам заражаются почти 100% детей. К группе повышенного риска, помимо недоношенных новорожденных и пожилых относятся люди, имеющие заболевания сердечно-сосудистой, дыхательной и иммунной систем. Основываясь на опубликованных и неопубликованных данных было подсчитано, что в мире РСВ вызывает 33,8 млн случаев эпизодических острых инфекций нижнего респираторного тракта (ИНРТ), 3,4 млн тяжелых случаев ИНРТ, требующих госпитализации и 66000 - 99000 смертельных случаев среди детей в возрасте до 5 лет (Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EA, Rudan I, Weber MW, Campbell H. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet; 375:1545-55). Только в США ежегодно в лечении нуждаются 90000 недоношенных новорожденных, 125000 госпитализированных новорожденных, более 3,5 млн детей в возрасте до 2 лет и 175000 госпитализированных взрослых (Storey S. Respiratory syncytial virus market. Nat Rev Drug Discov 2010; 9:15-6.). Примерно у трети детей госпитализированных с острым бронхиолитом в первый год жизни возникает эпизодическая отдышка, и отмечается повышенная чувствительность к общим аллергенам (Schauer U, Hoffjan S, Bittscheidt J, Kochling A, Hemmis S, Bongartz S, Stephan V. RSV bronchiolitis and risk of wheeze and allergic sensitisation in the first year of life. Eur Respir J 2002; 20:1277-83). Эти симптомы могут повторяться в последующие годы (Sigurs N, Gustafsson PM, Bjarnason R, Lundberg F, Schmidt S, Sigurbergsson F, Kjellman B. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med 2005; 171:137-41). Бронхиолит может быть вызван также риновирусом, коронавирусом, вирусом гриппа, парагриппа и аденовирусом, однако РСВ является наиболее частой причиной госпитализации по поводу бронхиолита среди всех перечисленных вирусов. Адаптивный иммунитет, формирующийся в результате перенесенной РСВ инфекции как у детей (с еще незрелой иммунной системой), так и у взрослых, является краткосрочным и не обеспечивает полную противовирусную защиту. Этот факт приводит к появлению реинфекций, наблюдаемых на протяжении всей жизни. В крови новорожденных в первые месяцы жизни содержатся материнские анти-РСВ антитела, однако они не защищают ребенка.
Наиболее близко к респираторно-синцитиальному вирусу расположен метапневмовирус человека (HMPV). Этот вирус был впервые выявлен в 2001 году в Нидерландах у детей с бронхиолитом. HMPV также содержит геномную (-) ssРНК и относится к роду пневмовирусов. HMPV циркулирует по всему миру и вызывает почти универсальную инфекцию у детей. Подобно гриппу и респираторно-синцитиальному вирусу, активность HMPV наиболее высока в зимний период в умеренном климате. Большинство имеющихся данных о клинических проявлениях HMPV инфекции свидетельствуют о том, что вирус вызывает инфекции верхних дыхательных путей, бронхиолит и пневмонию. Реинфекции с HMPV происходят на протяжении всей взрослой жизни. Болезнь, как правило, протекает мягко, а у молодых взрослых часто бессимптомно. К группе повышенного риска относятся престарелые, взрослые с заболеваниями легких и с неполноценной иммунной системой. Вспышки HMPV были зарегистрированы в стационарах, при этом смертность достигала 50% среди ослабленных пожилых людей. Кроме того наблюдалось 6-12% обострений хронической обструктивной болезни легких. У реципиентов трансплантатов кроветворных стволовых клеток HMPV был связан с тяжелыми идиопатическими пневмониями.
Следует отметить, что единственным химиотерапевтическим средством, оказывающим некоторый положительный эффект при инфекциях, вызываемых (+) и (-) РНК-содержащими вирусами, является рибавирин. Однако рибавирин является относительно токсичным средством, часто вызывающим анемию. Основной его особенностью является длительное депонирование в эритроцитах. В результате, следы рибавирина обнаруживаются даже через 6 месяцев после окончания курса терапии. Упоминается также о тератогенном действии рибавирина.
Сущность изобретения
Настоящее изобретение относится к новым соединениям общей формулы I или их фармацевтически приемлемым солям для профилактики и лечения заболеваний, вызываемых РНК-содержащими вирусами, причем общая формула I имеет следующий вид:

где
R1 представляет собой
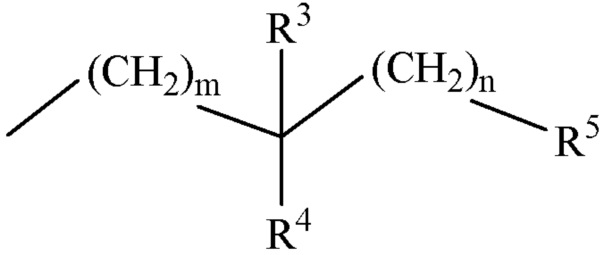
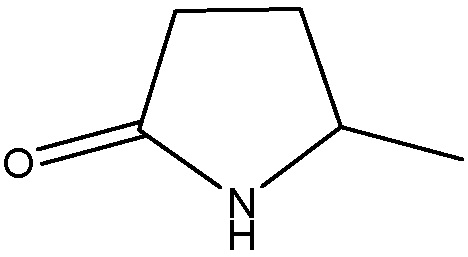


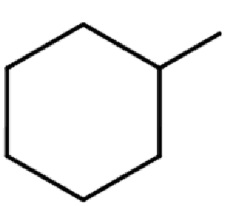
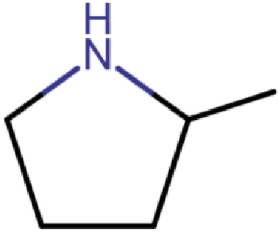



m представляет собой целое число 0 или 1;
n представляет собой целое число 0 или 1;
R2 представляет собой H или С1-С6 алкил;
R3 и R4каждый независимо представляет собой H, С1-С6 алкил, -NH2, -NHС(=O)CH3, OH и -NHC(O)CH2COOH;
R5 представляет собой -COOH, -C(O)NH2,

где R5 может быть необязательно замещен заместителем, выбранным из группы состоящей из бензила, бензил-ОС(О)-, С1-С6 алкила, OH и -NH2;
Q представляет собой
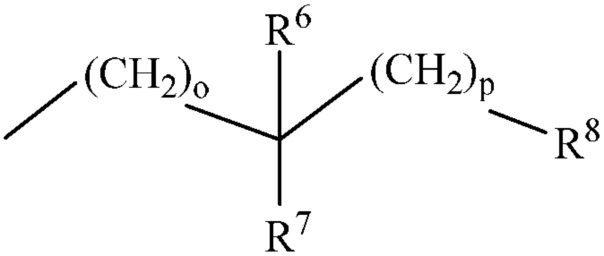
Q и R2 вместе с атомом азота, к которому они присоединены, могут образовывать цикл

Q и R1 вместе с фрагментом -C(O)N-, к которому они присоединены, могут образовывать цикл

о представляет собой целое число 0 или 2;
p представляет собой целое число от 0 до 3;
R6 и R7 каждый независимо представляет собой H, -C(O)NH2, -COOH, -CH2OH или C1-C6 алкил-NH2;
где R6 и R7 могут быть необязательно замещены C1-C6 алкилом, -CH(CH(OH)CH3)(C(O)OC2H5), -CH(CH(OH)CH3)(COOH), -CH(CH(CH3)2)(C(O)OCH3), -CH(CH(CH3)2)(C(O)NH2), -CH(CH3)C(O)OCH3, -CH(CH3)C(O)NH2, -CH(CH2CH(CH3)2)(C(O)OCH3), -CH(CH2CH(CH3)2)(C(O)ONH2), -CH(CH2OH)(COOH), -CH(CH(OH)CH3)(C(O)OCH3), -CH(CH2(OH))(C(O)OCH3), -CH2CH(OH)CH3, -(CH2)2OH, -(CH2)3OH, -CH2C(O)NH2, -CH2C(O)OCH3, -CH2COOH или -C(O)OCH3;
R8 представляет собой


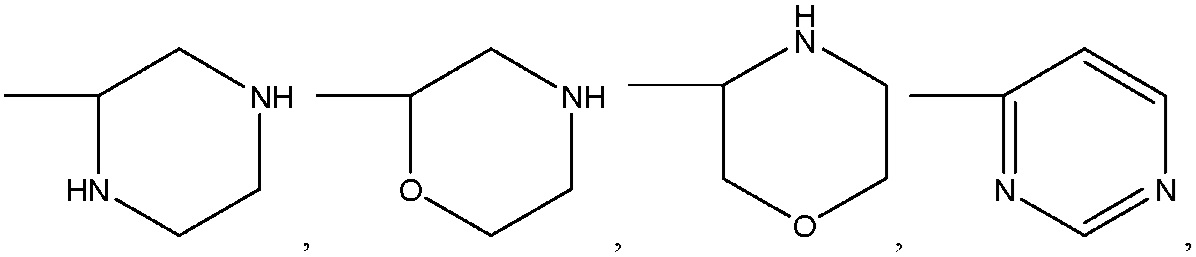
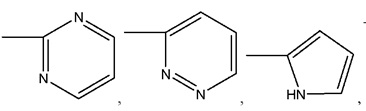
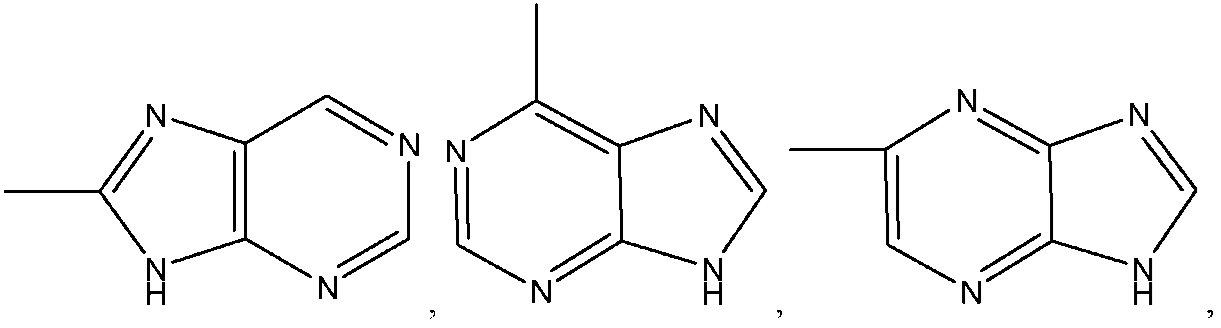



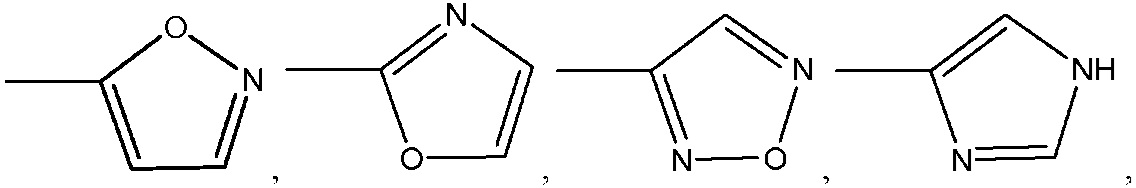
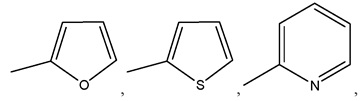
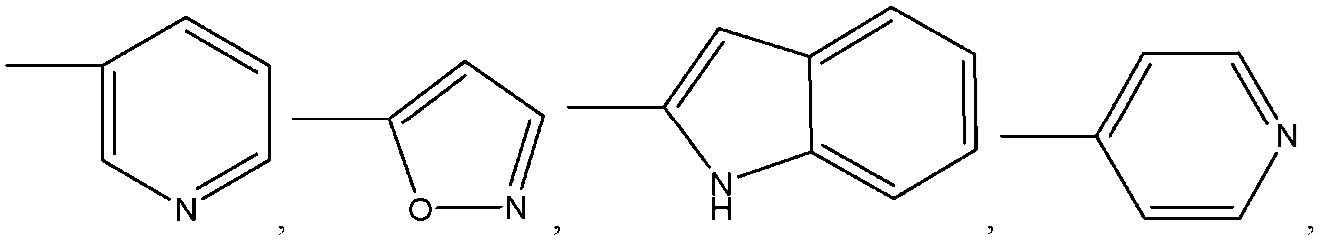

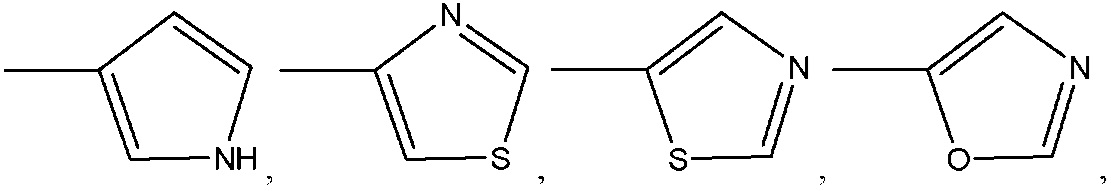






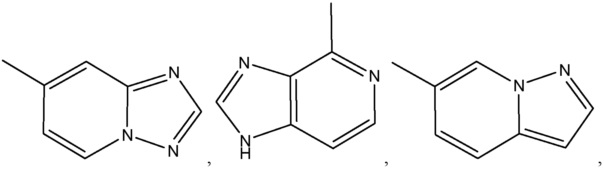



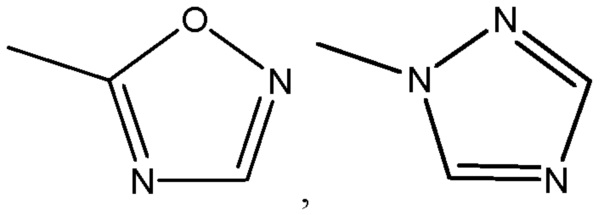
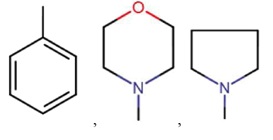
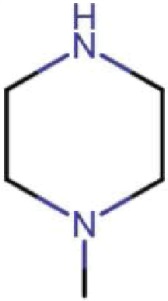
где R8 может быть необязательно замещен одним или более заместителями, выбранными из C1-C6 алкила, C1-C6 алкокси, галогена, -COOH, пиридила и –О-бензила;
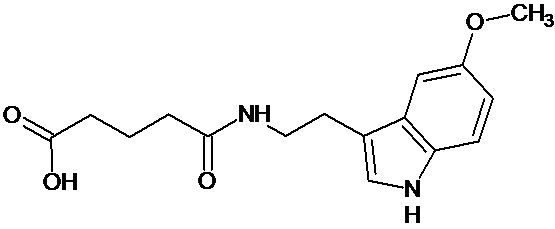
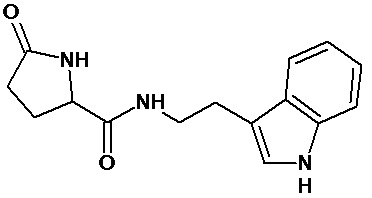
или соединение, выбранное из следующих соединений:


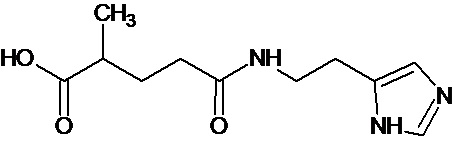

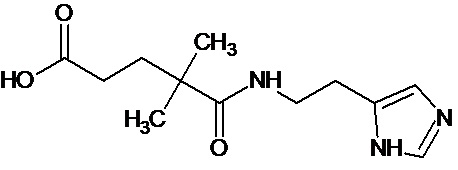

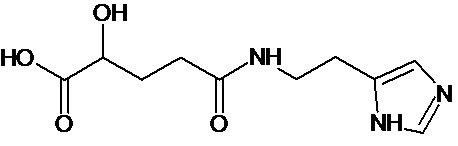
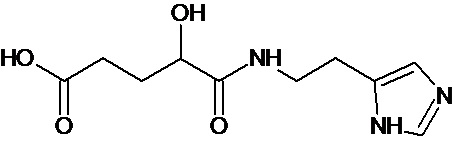
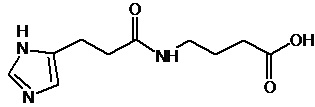


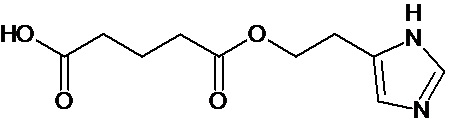

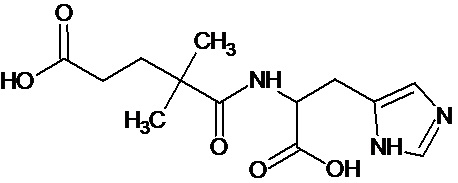



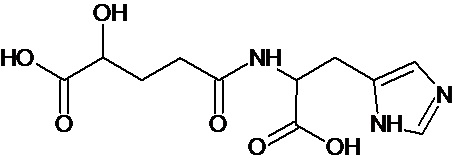
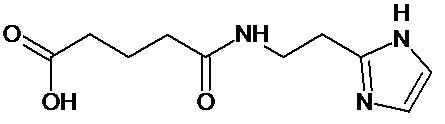

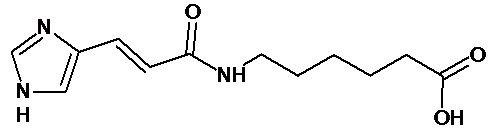

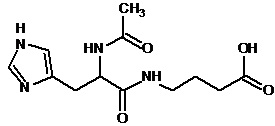
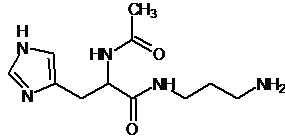
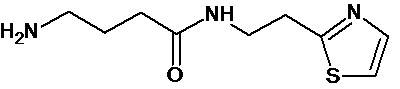

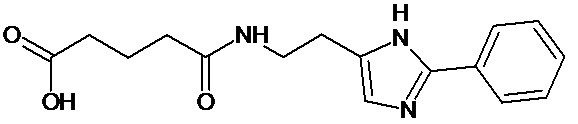

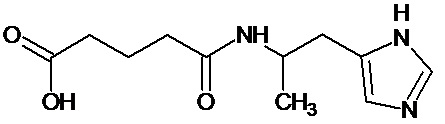
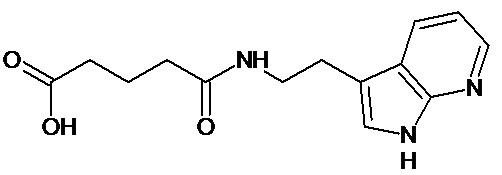

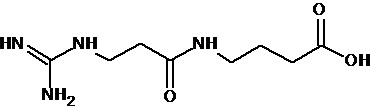
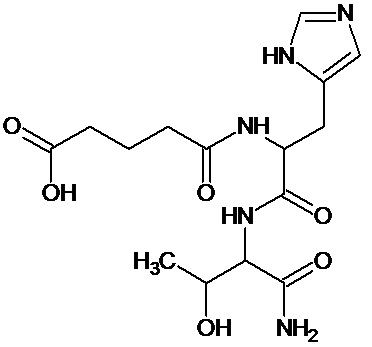
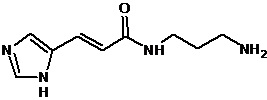
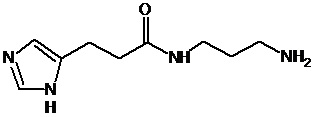
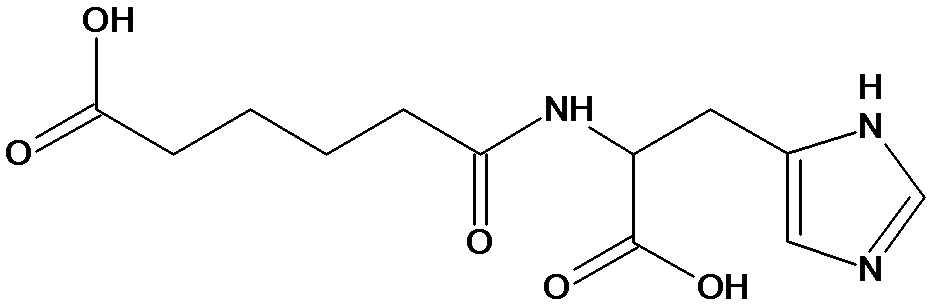

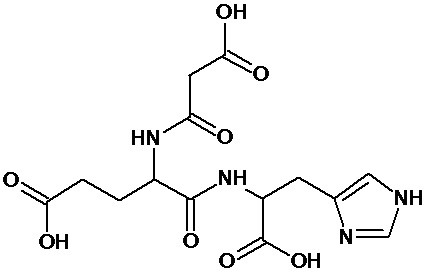

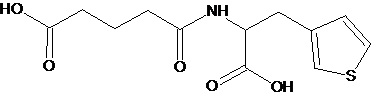


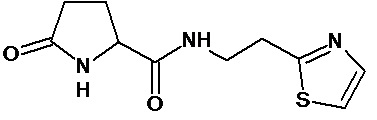



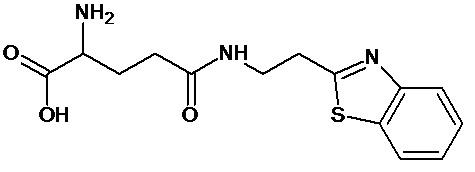
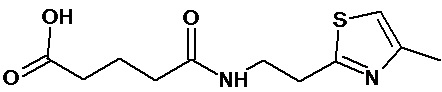
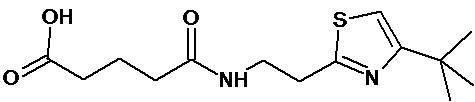


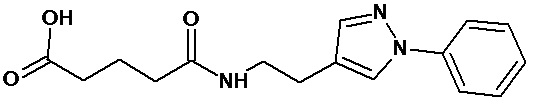



при условии, что соединение не выбрано из следующих соединений:
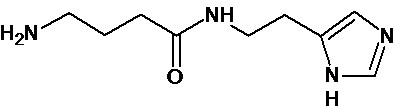




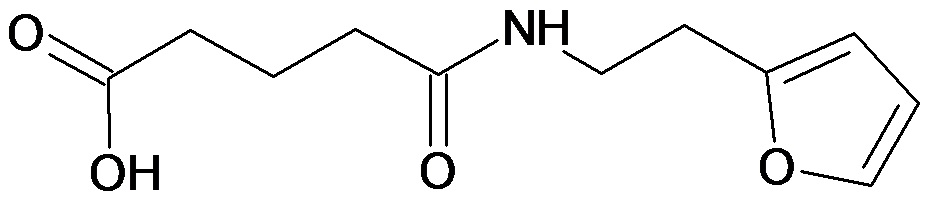


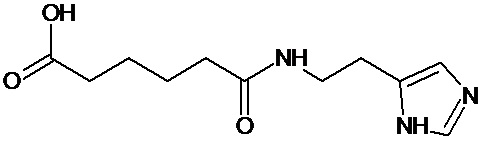



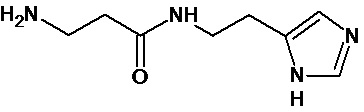
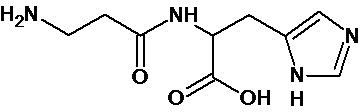


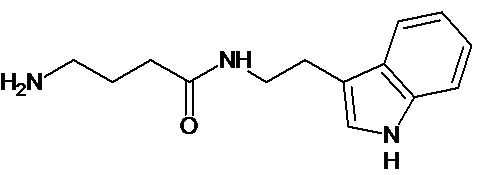


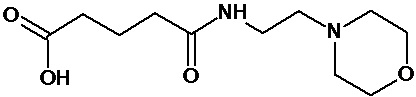



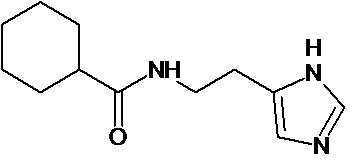



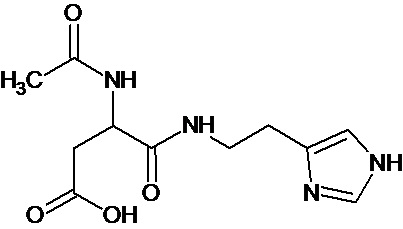
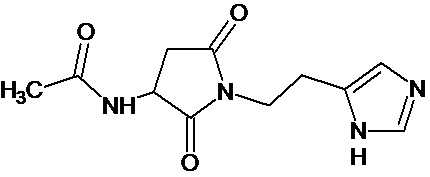
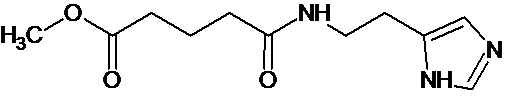
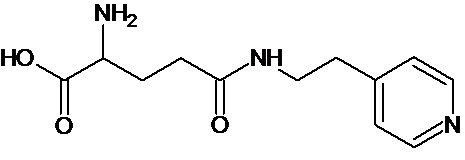
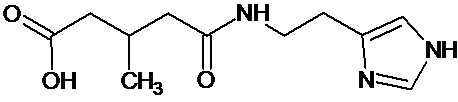




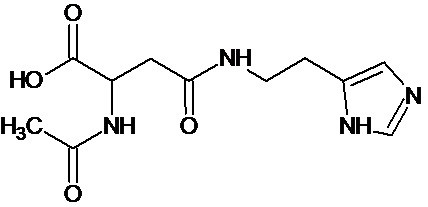



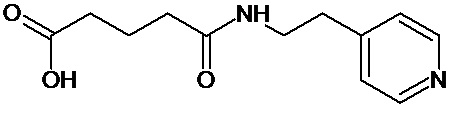


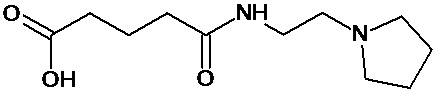
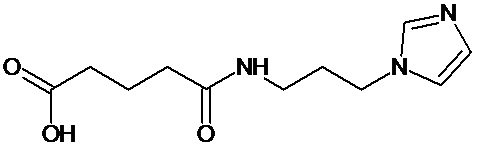


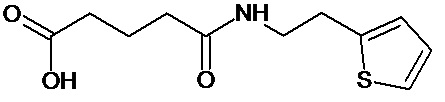
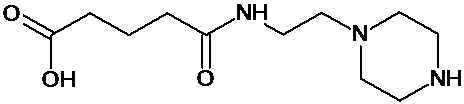
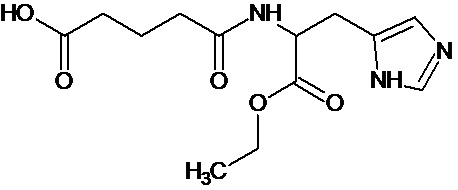


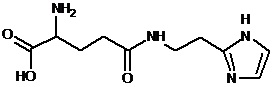
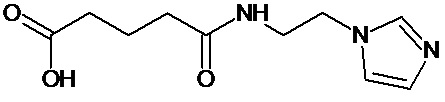

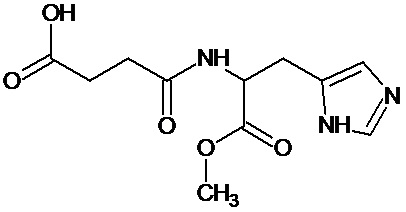


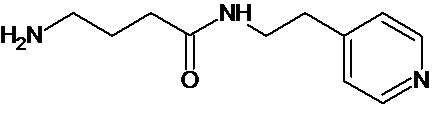
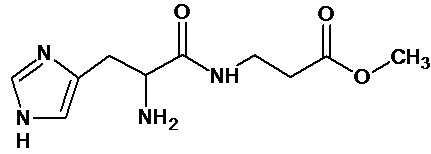
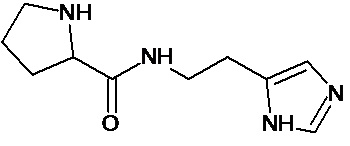
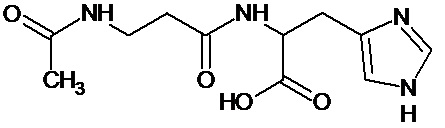

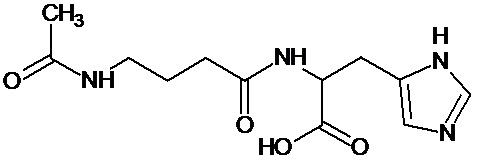
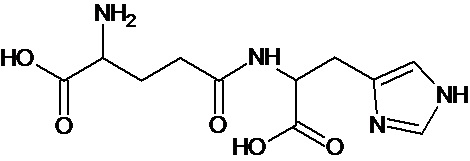

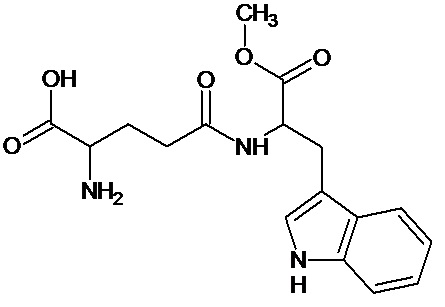

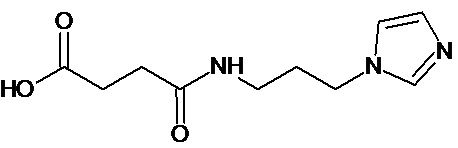


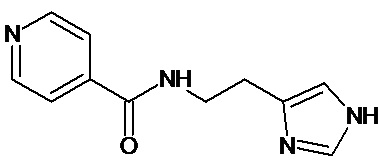


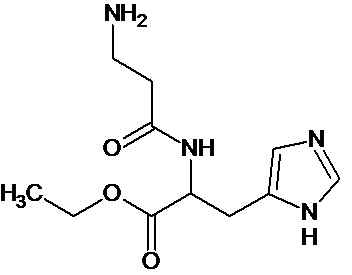
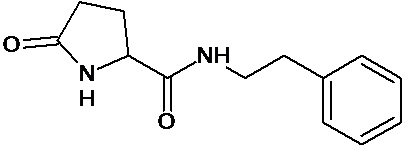
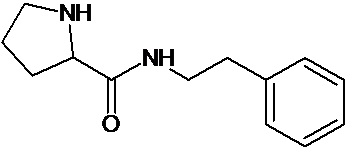



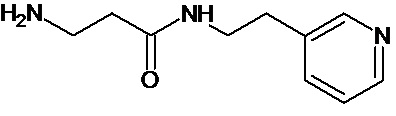
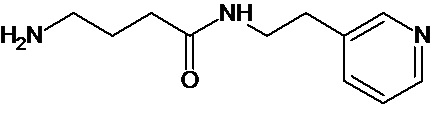
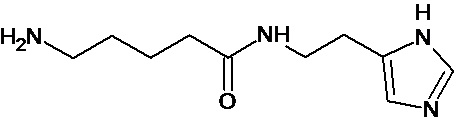



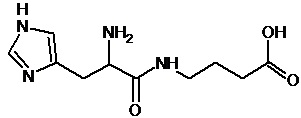


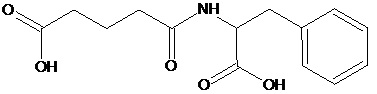



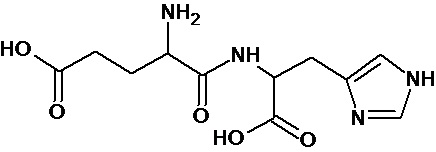


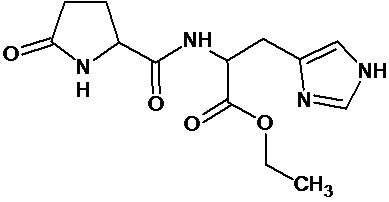


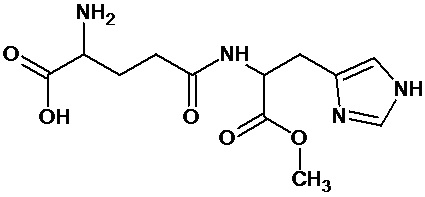


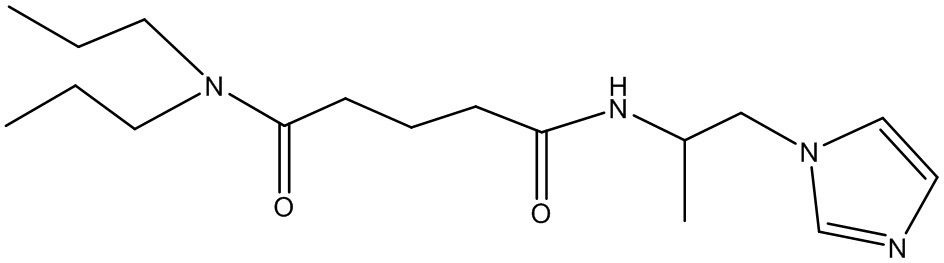

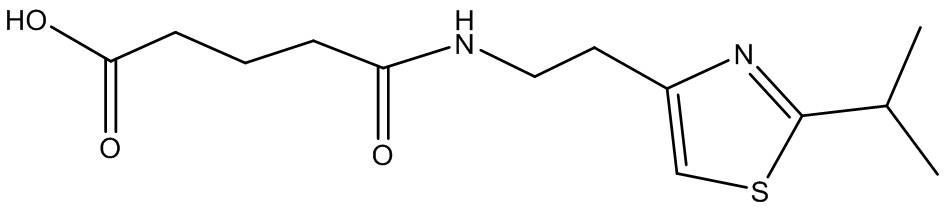


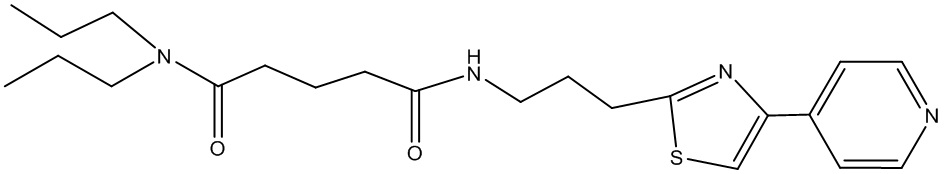


Кроме того, настоящее изобретение относится к применению ряда соединений формулы I, раскрытых ранее и необязательно подпадающих под общую структурную формулу соединений, раскрытых в публикации международной заявки WO 99/01103, по новому назначению. Более конкретно, авторами изобретения неожиданно обнаружено, что соединения общей формулы I могут использоваться в качестве нетоксичных противовирусных средств при инфекциях, вызываемых вирусами, относящимися к роду энтеровирусов, метапневмовирусов или роду пневмовирусов (без ограничения перечисленными). В частности, этими соединениями являются следующие соединения:





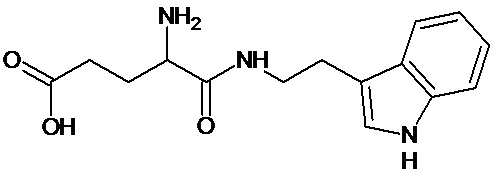

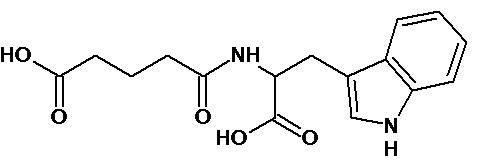
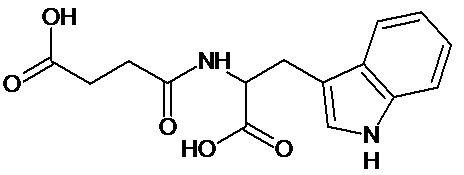

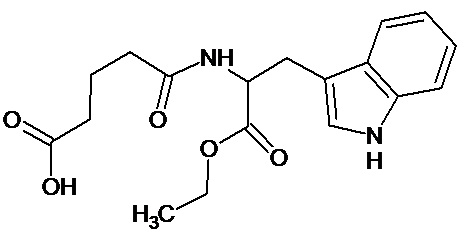

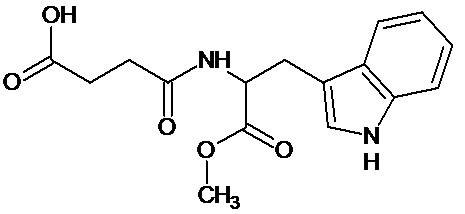
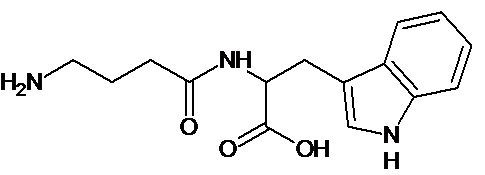



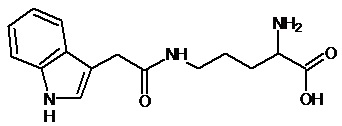
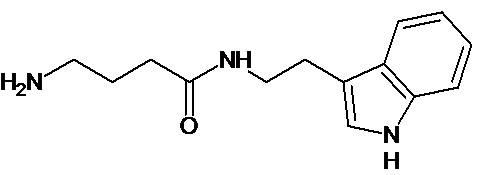


С учетом изложенного, настоящее изобретение относится к средству для лечения и/или профилактики заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов, представляющему собой соединение общей формулы I.
Изобретение далее относится к способам получения соединений общей формулы I или их фармацевтически приемлемых солей, охватывает способ профилактики и лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов, предусматривающий введение пациенту эффективного количества соединения общей формулы I или его фармацевтически приемлемой соли.
Далее, изобретение относится к фармацевтической композиции для лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов, содержащей эффективное количество соединения общей формулы I или его фармацевтически приемлемой соли.
Изобретение также относится к набору для лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов, включающему композицию по изобретению и инструкции по ее применению.
Кроме этого, изобретение относится к применению соединений общей формулы I или их фармацевтически приемлемых солей для производства лекарственного средства для лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими к роду энтеровирусов, метапневмовирусов или роду пневмовирусов.
Изобретение также включает применение соединений общей формулы I или их фармацевтически приемлемых солей для лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов.
Осуществление изобретения
Настоящее изобретение относится к соединениям общей формулы I, которое соответствует следующей формуле
или их фармацевтически приемлемым солям,
где
R1 представляет собой
m представляет собой целое число 0 или 1;
n представляет собой целое число 0 или 1;
R2 представляет собой H или С1-С6 алкил;
R3 и R4каждый независимо представляет собой H, С1-С6 алкил, -NH2, -NHС(=O)CH3, OH и -NHC(O)CH2COOH;
R5 представляет собой -COOH, -C(O)NH2,
где R5 может быть необязательно замещен заместителем, выбранным из группы состоящей из бензила, бензил-ОС(О)-, С1-С6 алкила, OH и -NH2;
Q представляет собой
Q и R2 вместе с атомом азота, к которому они присоединены, могут образовывать цикл
Q и R1 вместе с фрагментом -C(O)N-, к которому они присоединены, могут образовывать цикл
о представляет собой целое число 0 или 2;
p представляет собой целое число от 0 до 3;
R6 и R7 каждый независимо представляет собой H, -C(O)NH2, -COOH, -CH2OH или C1-C6 алкил-NH2;
где R6 и R7 могут быть необязательно замещены C1-C6 алкилом, -CH(CH(OH)CH3)(C(O)OC2H5), -CH(CH(OH)CH3)(COOH), -CH(CH(CH3)2)(C(O)OCH3), -CH(CH(CH3)2)(C(O)NH2), -CH(CH3)C(O)OCH3, -CH(CH3)C(O)NH2, -CH(CH2CH(CH3)2)(C(O)OCH3), -CH(CH2CH(CH3)2)(C(O)ONH2), -CH(CH2OH)(COOH), -CH(CH(OH)CH3)(C(O)OCH3), -CH(CH2(OH))(C(O)OCH3), -CH2CH(OH)CH3, -(CH2)2OH, -(CH2)3OH, -CH2C(O)NH2, -CH2C(O)OCH3, -CH2COOH или -C(O)OCH3;
R8 представляет собой
где R8 может быть необязательно замещен одним или более заместителями, выбранными из C1-C6 алкила, C1-C6 алкокси, галогена, -COOH, пиридила и –О-бензила;
или соединение, выбранное из следующих соединений:






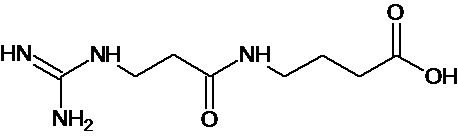




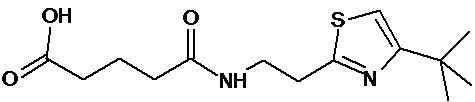
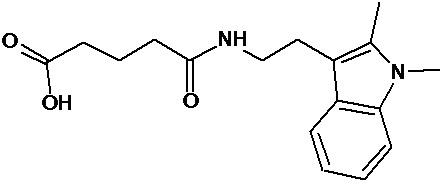
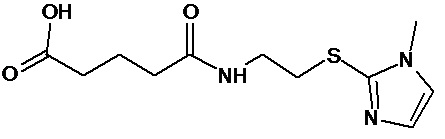
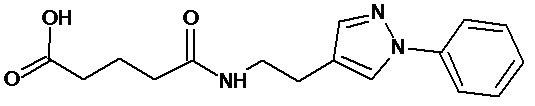
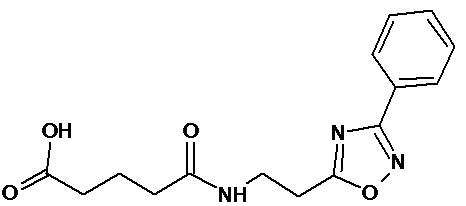

;
при условии, что соединение не выбрано из следующих соединений:
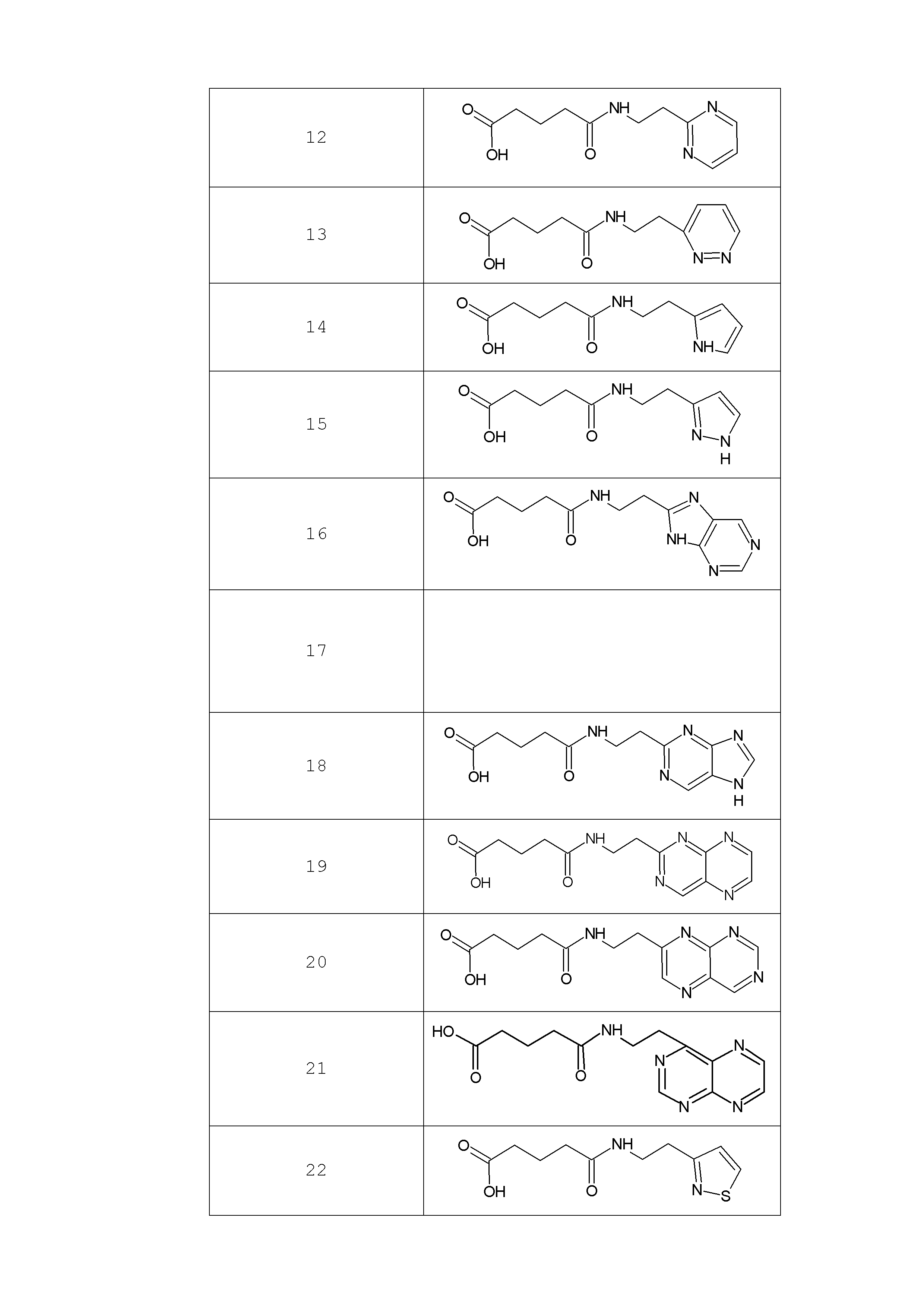
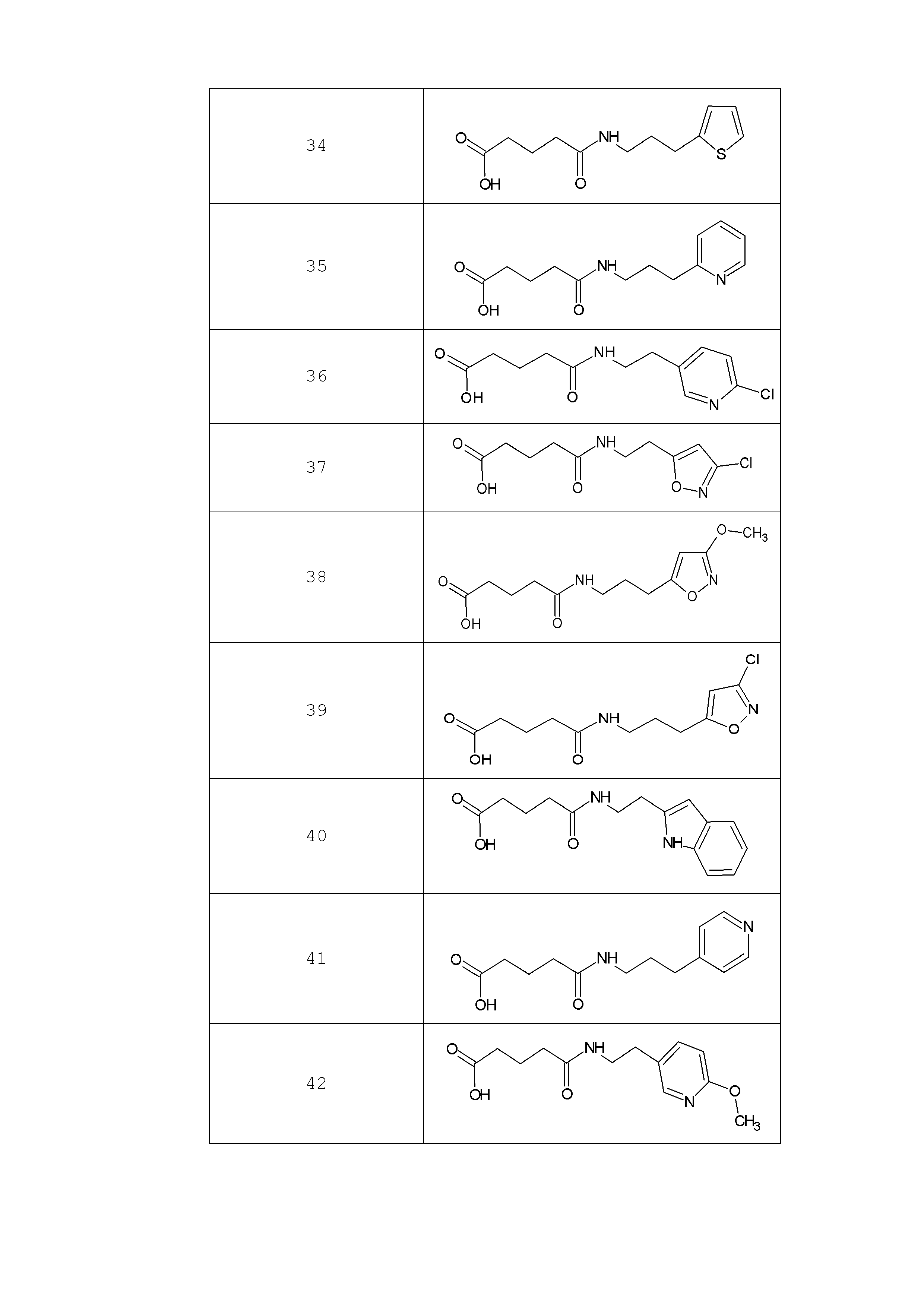
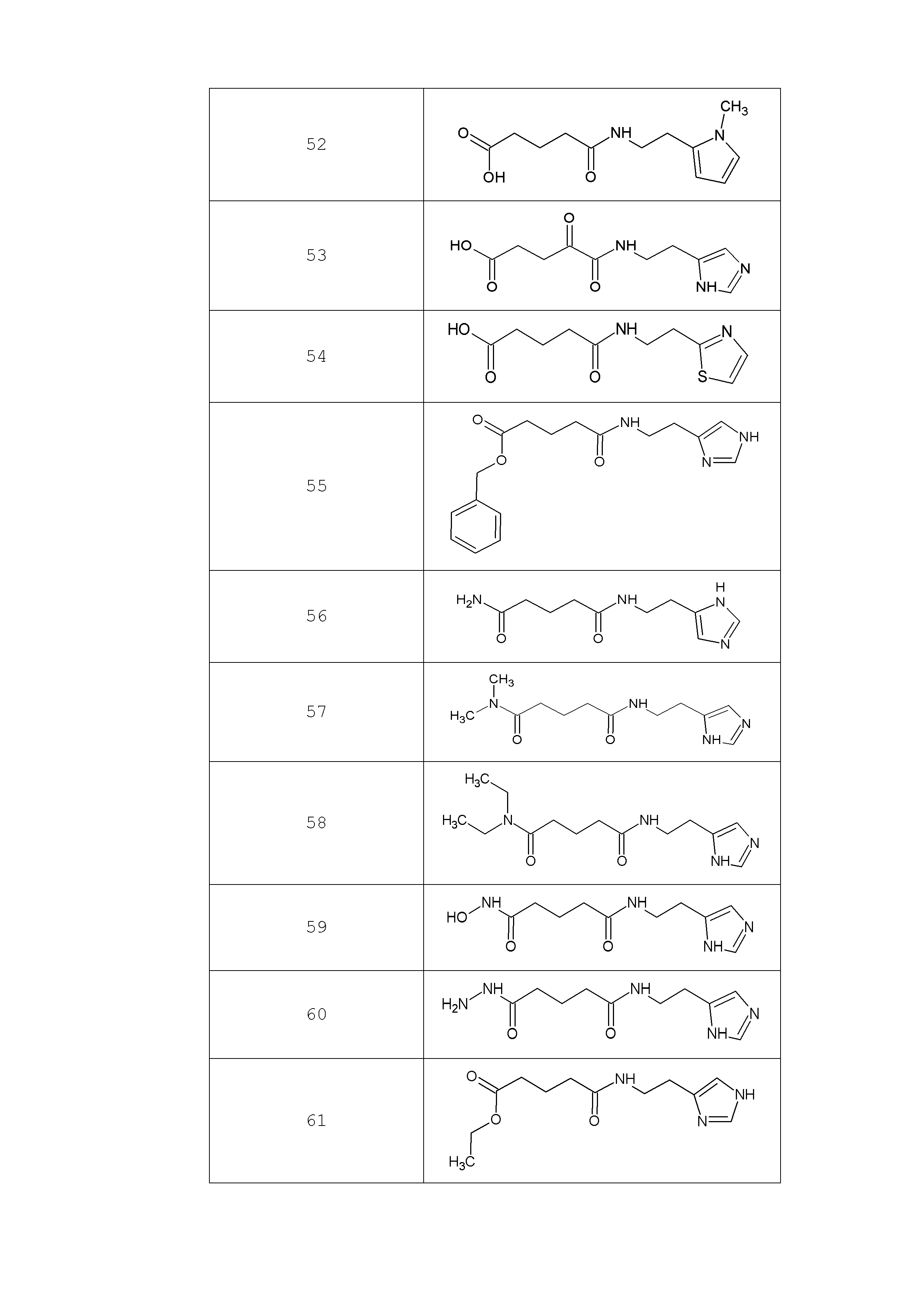
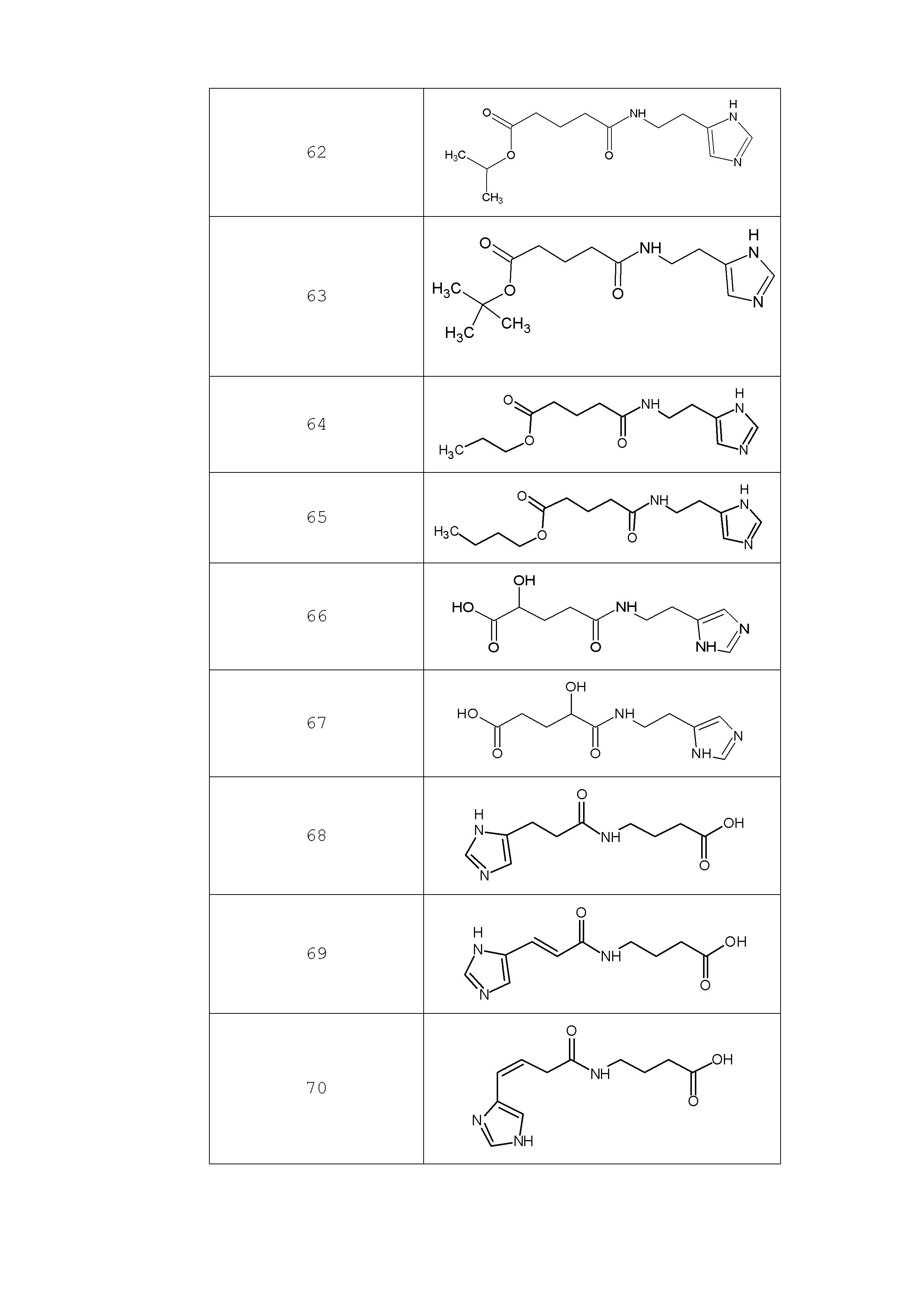
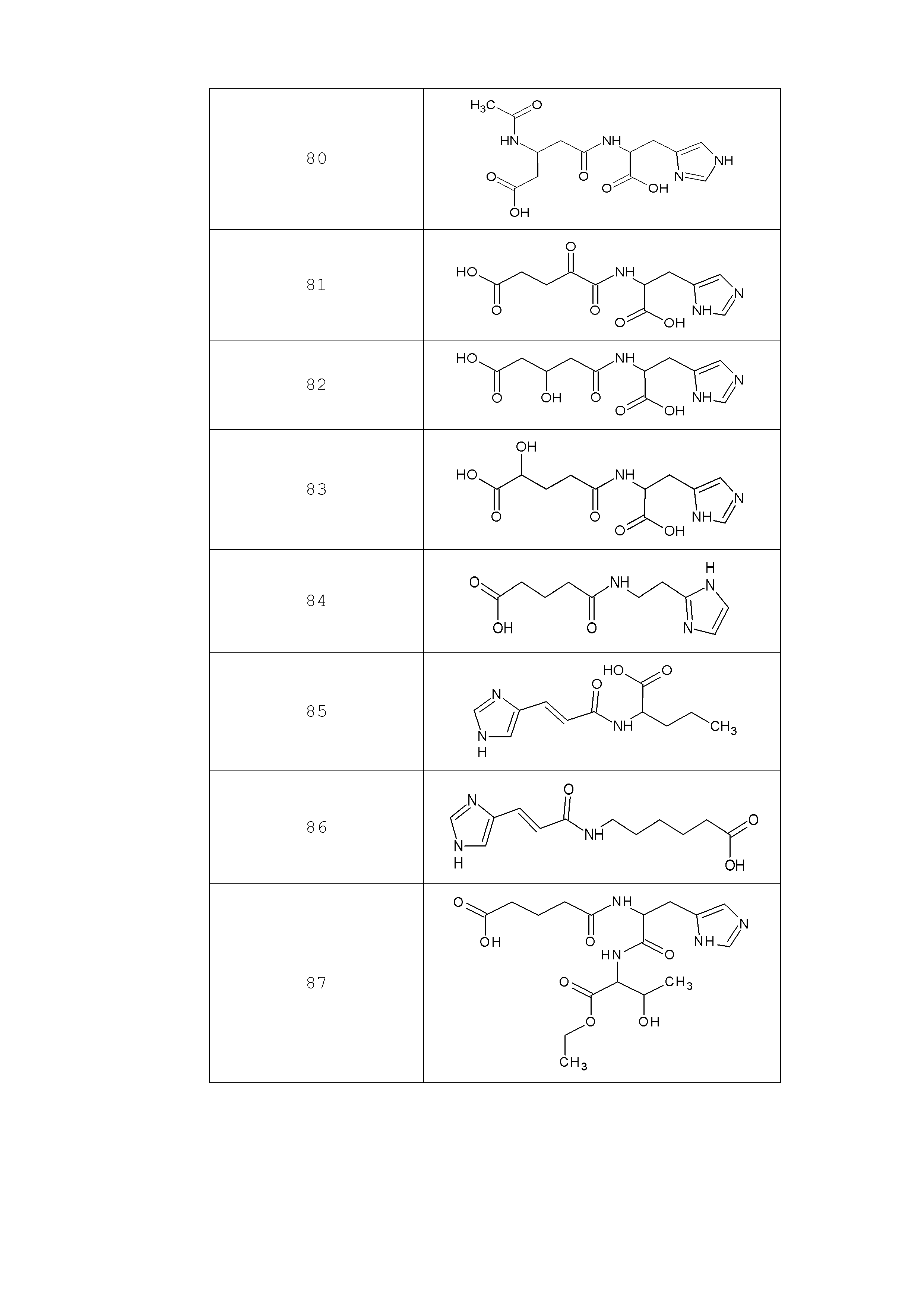
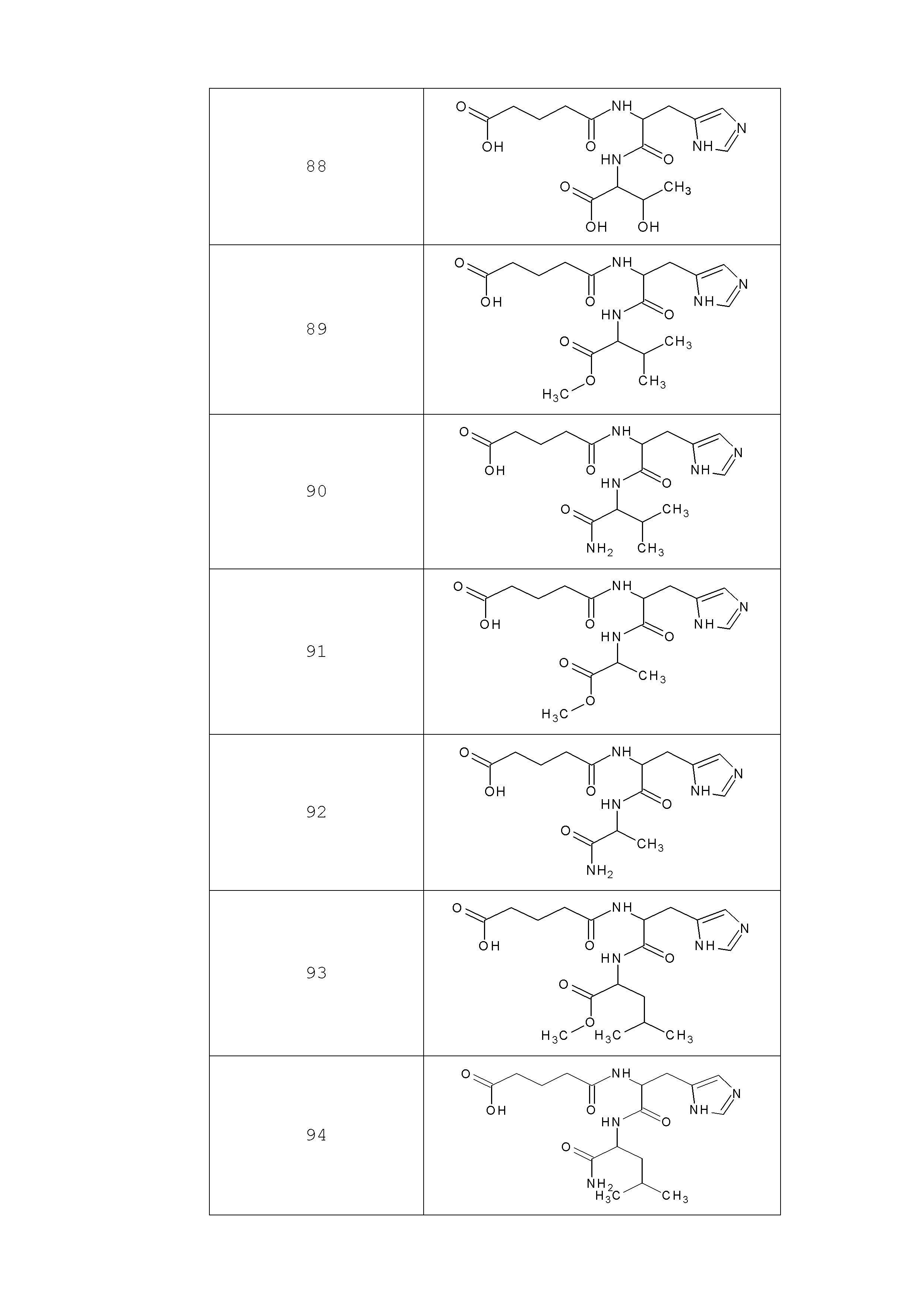
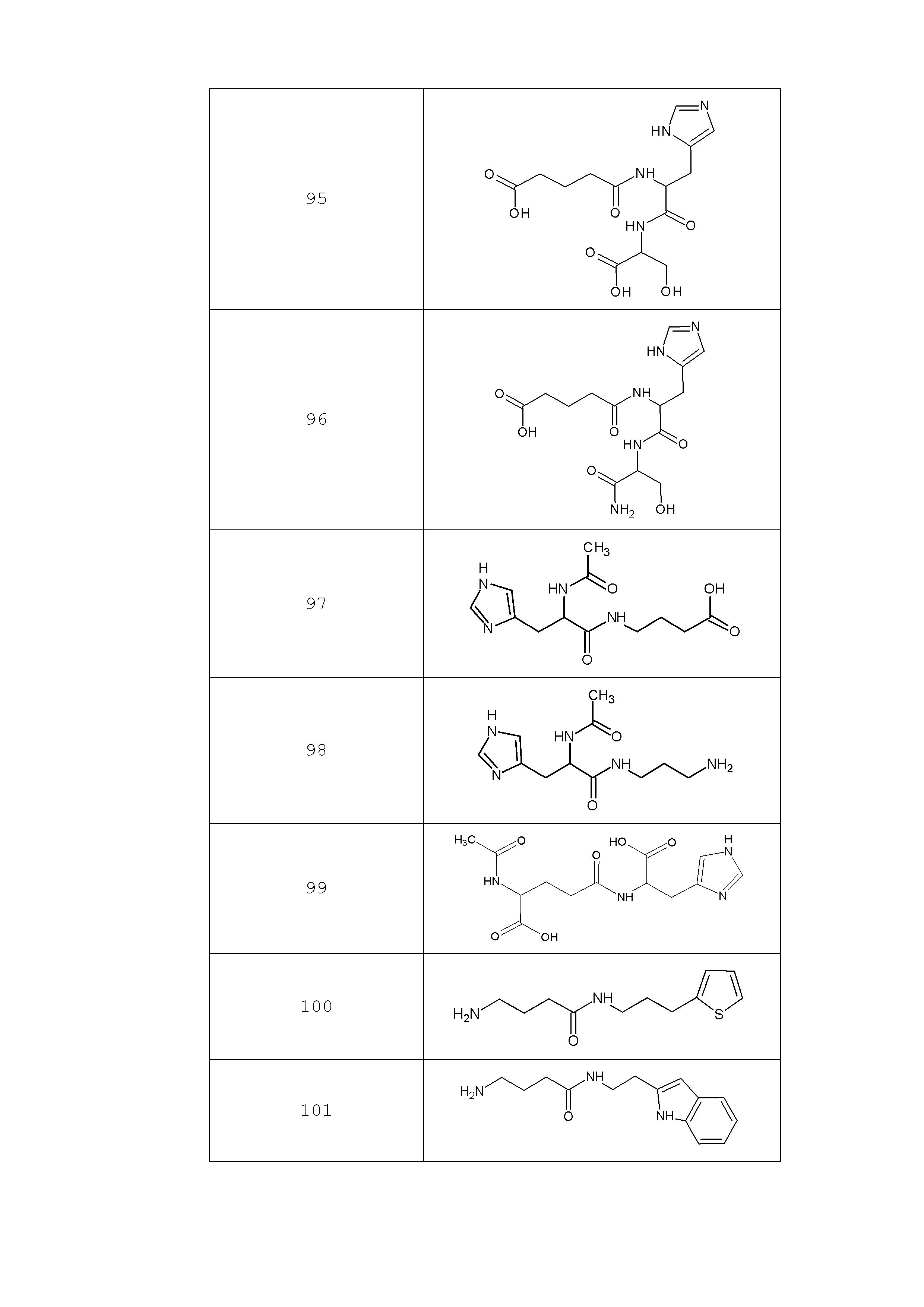
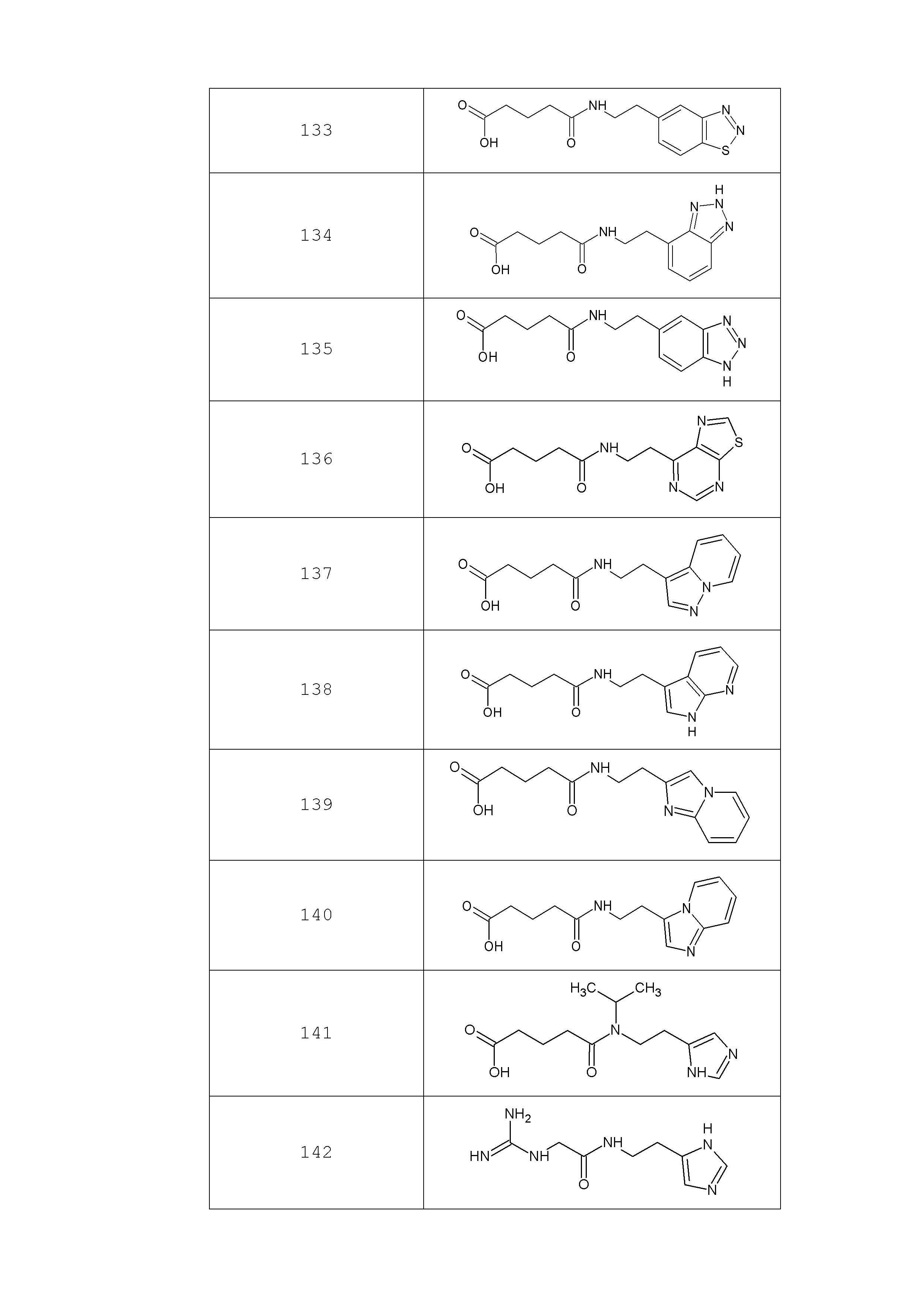
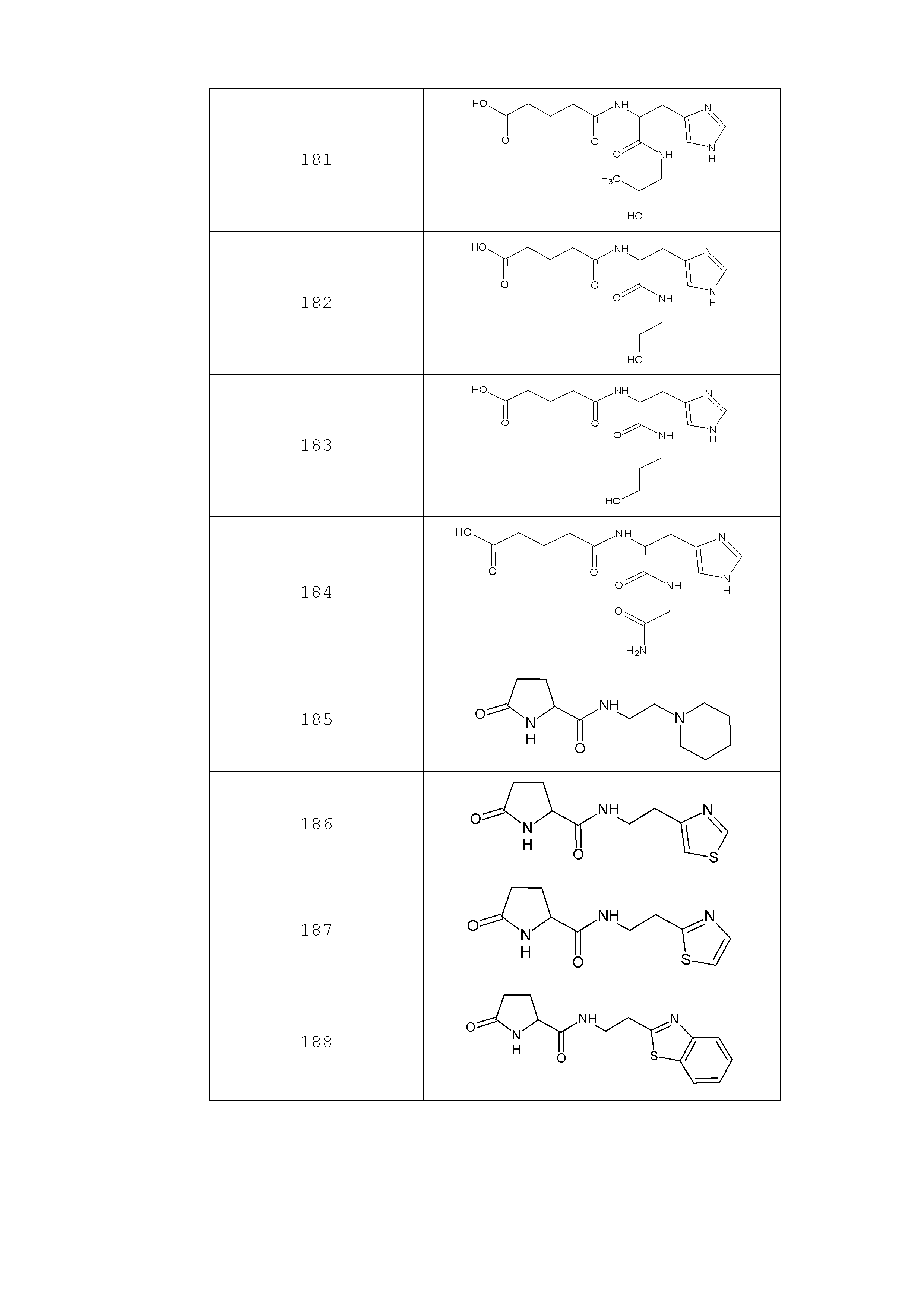
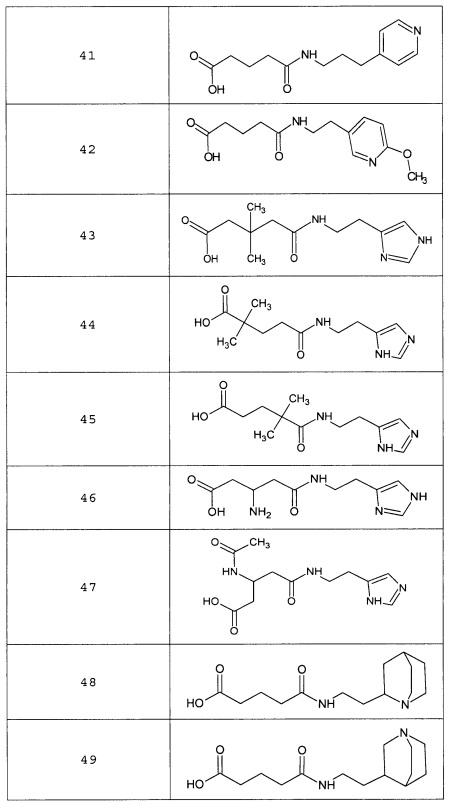
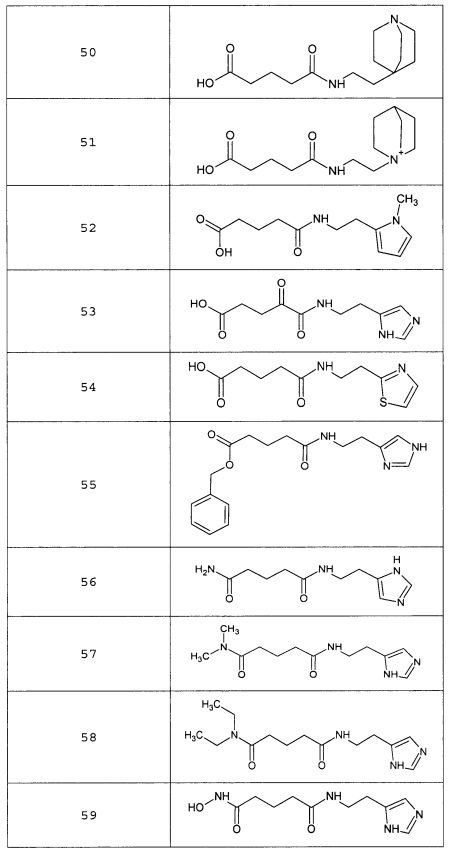
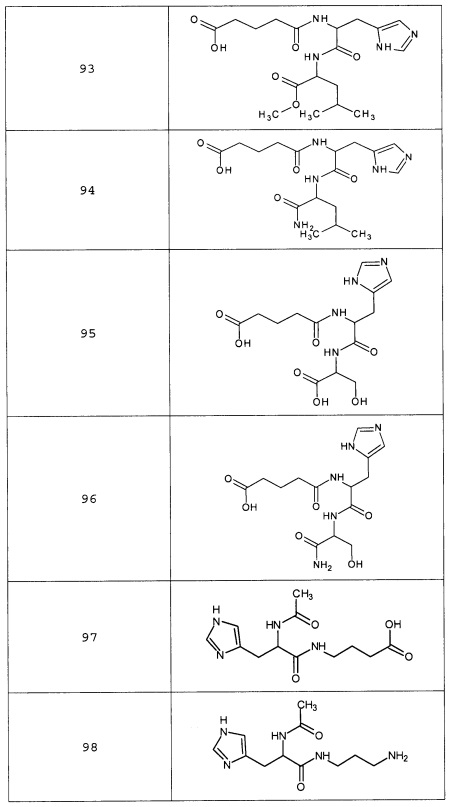
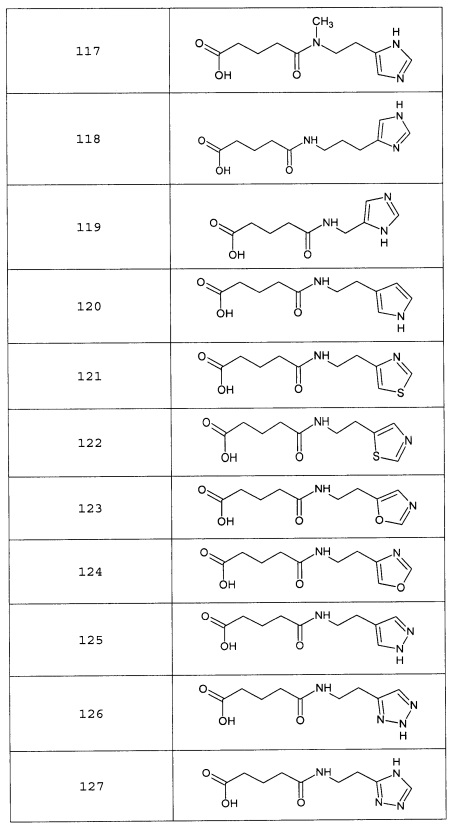
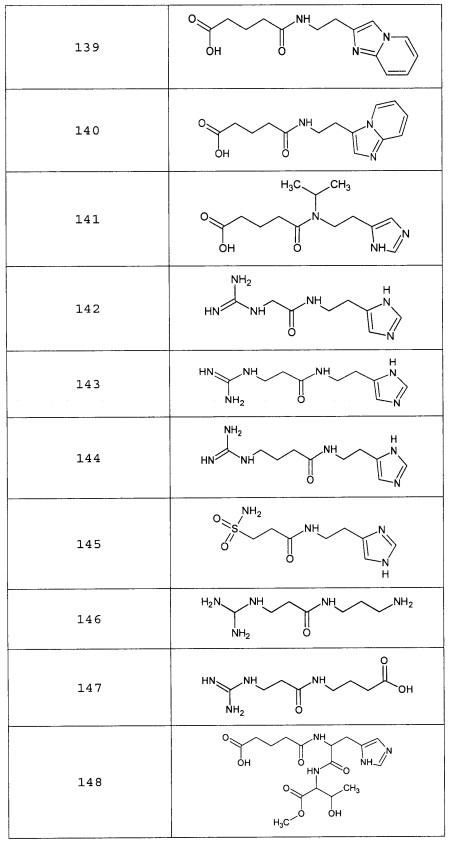
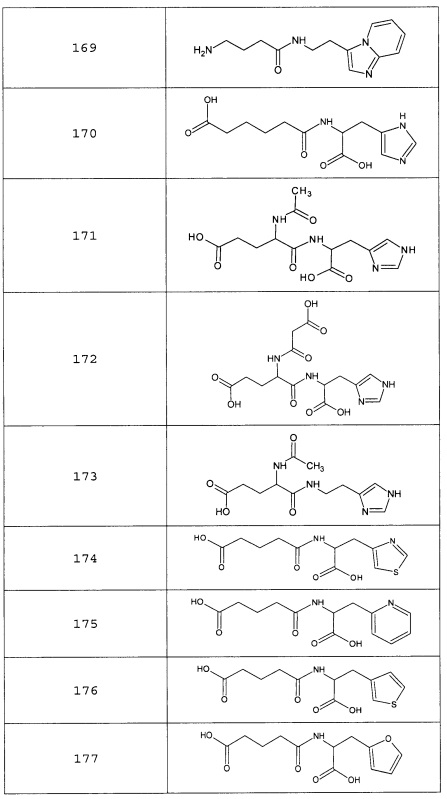
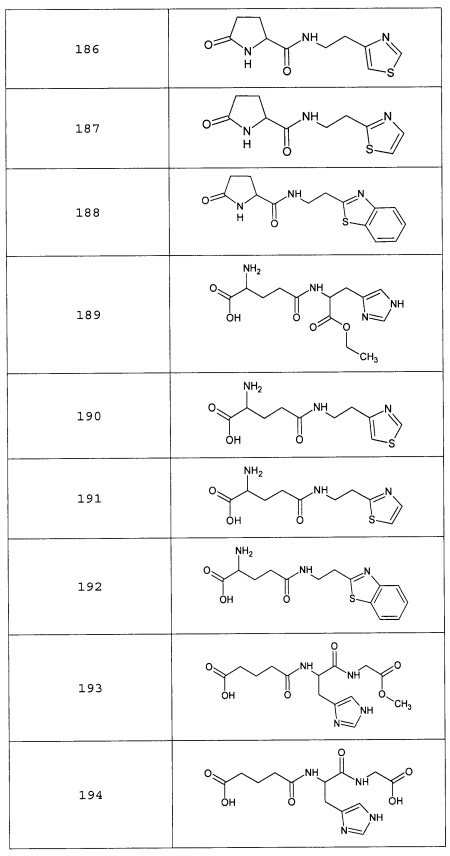
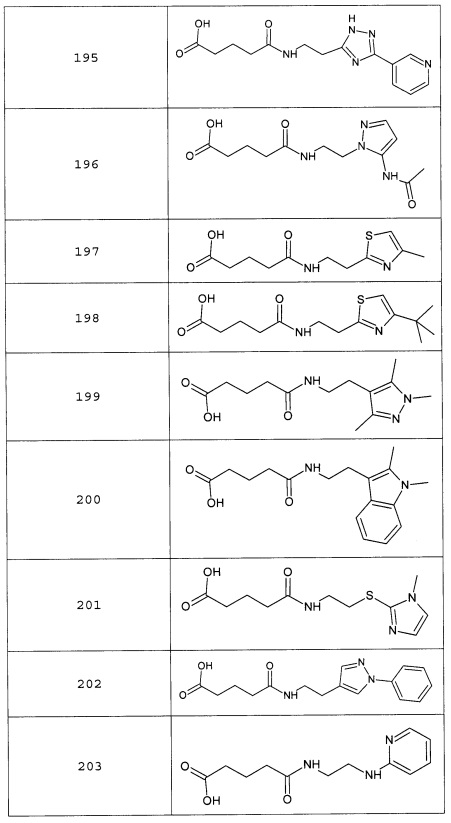
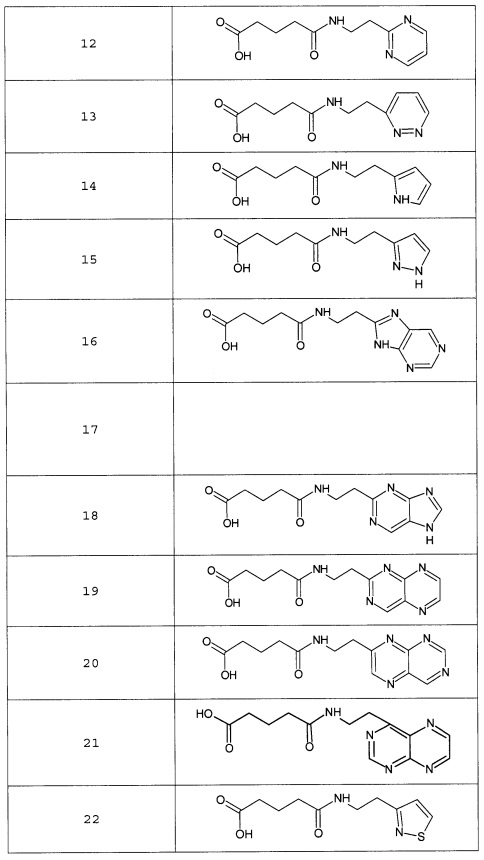
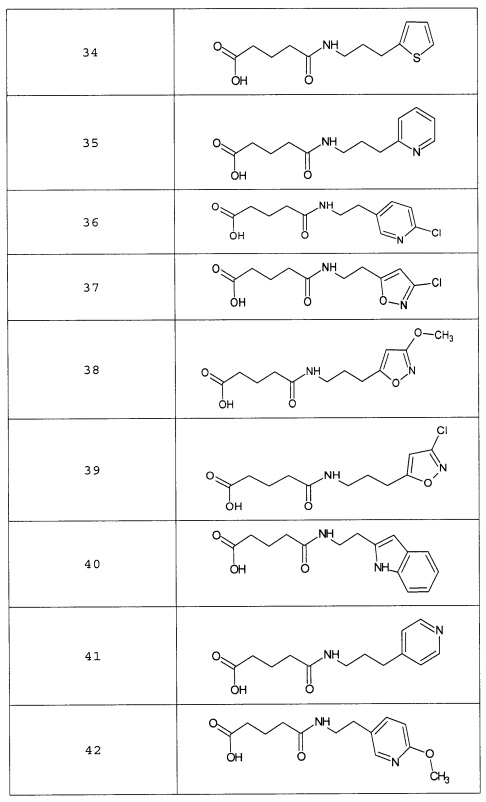
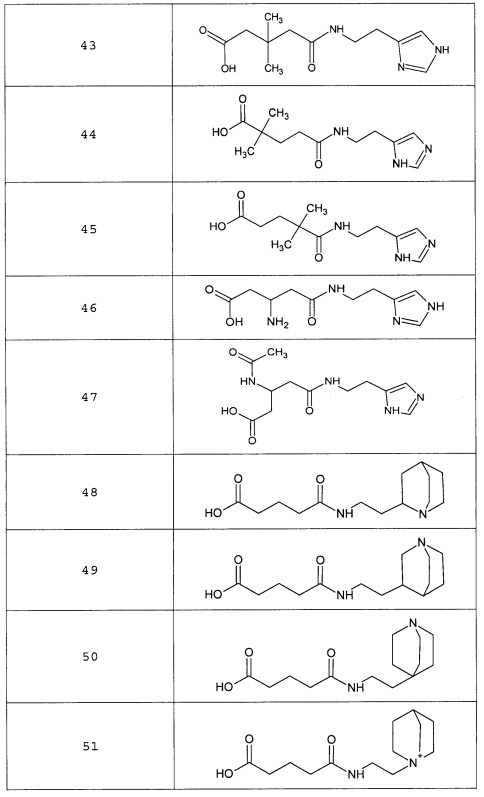
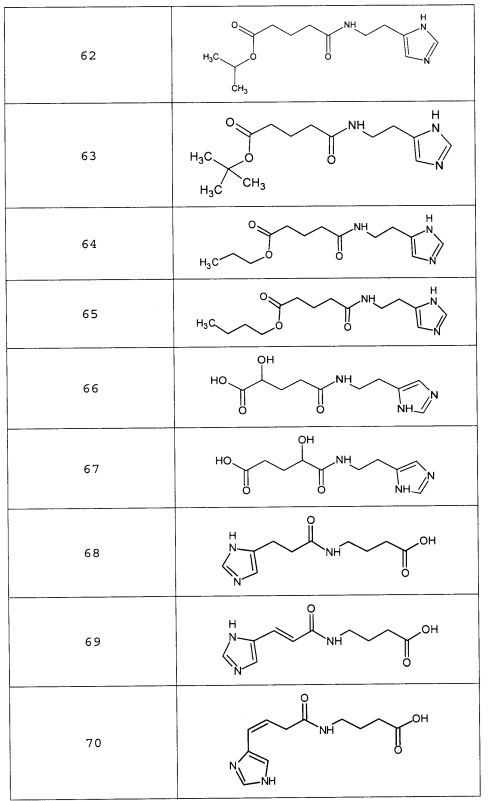
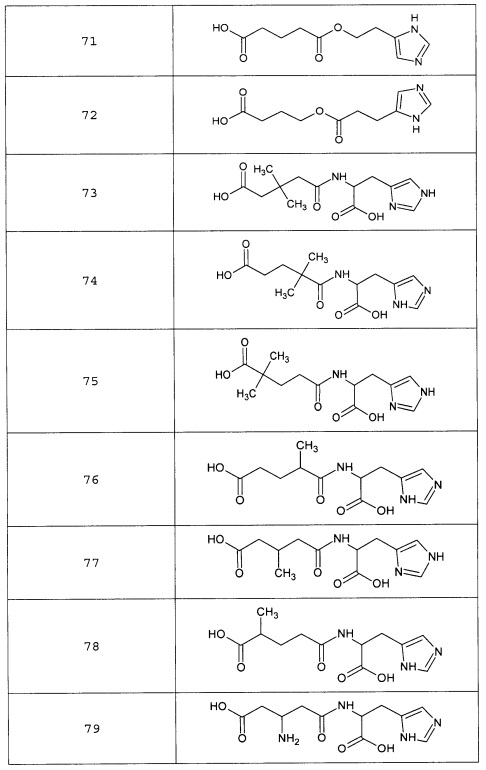
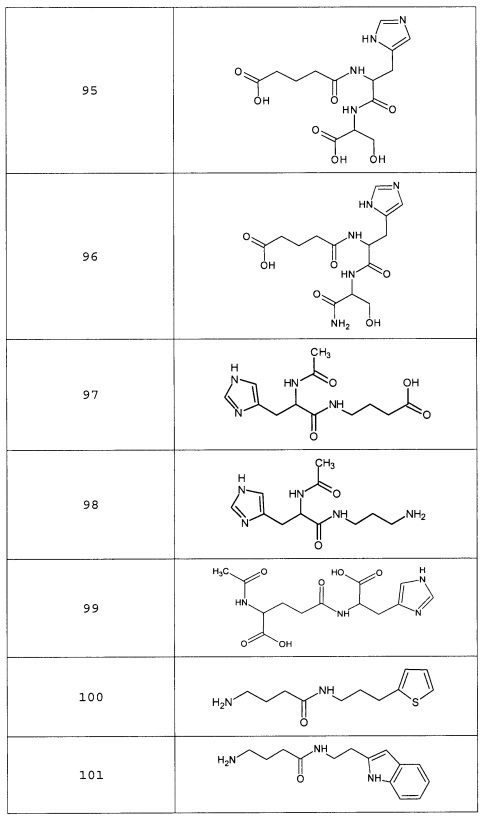
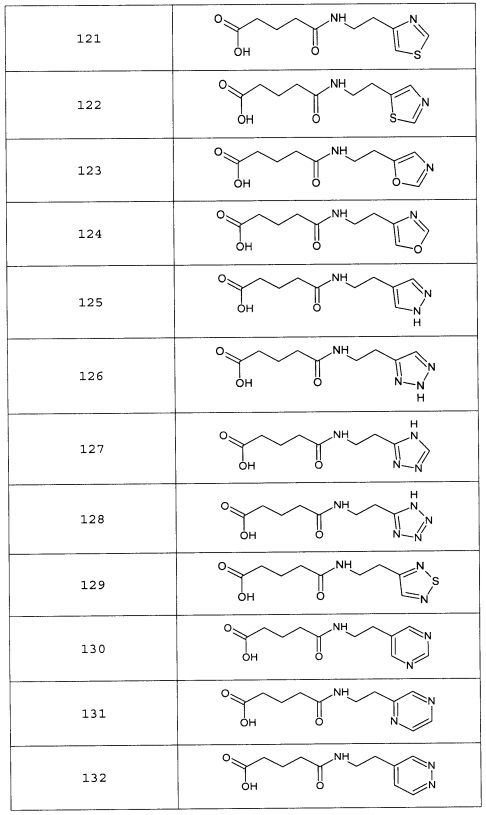
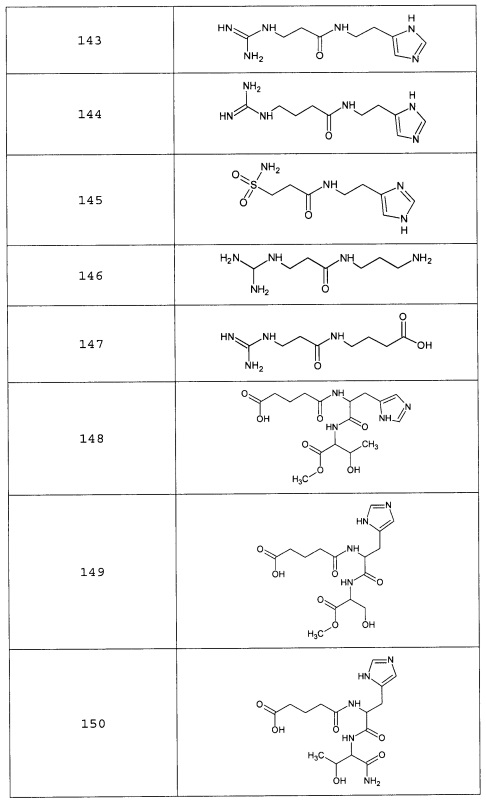
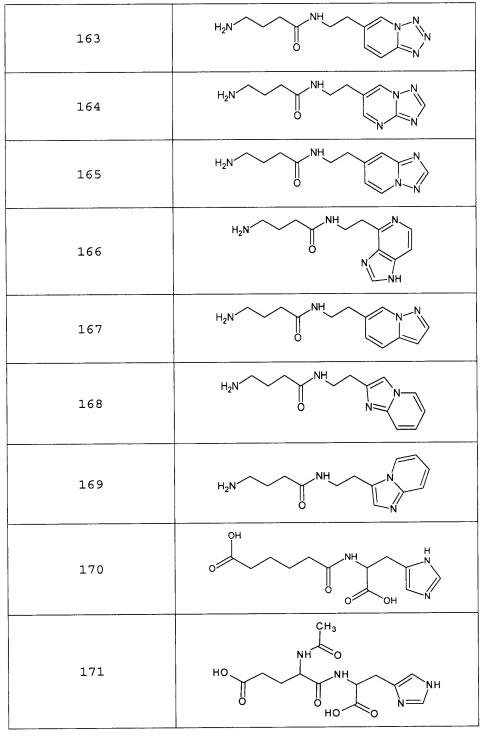
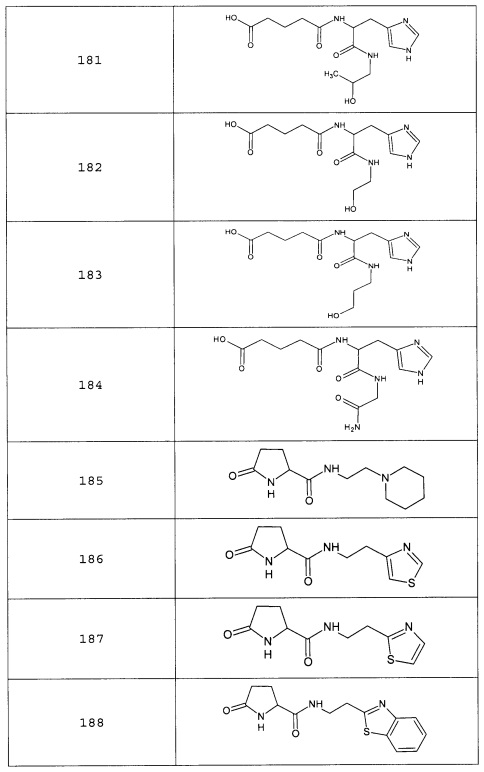
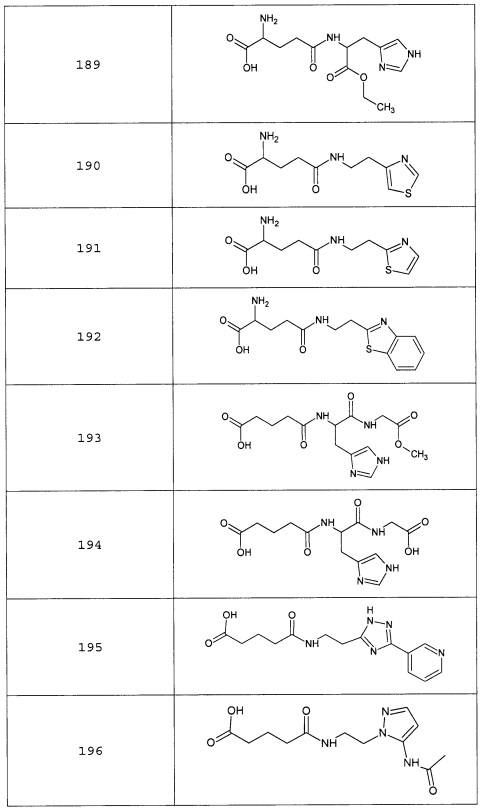
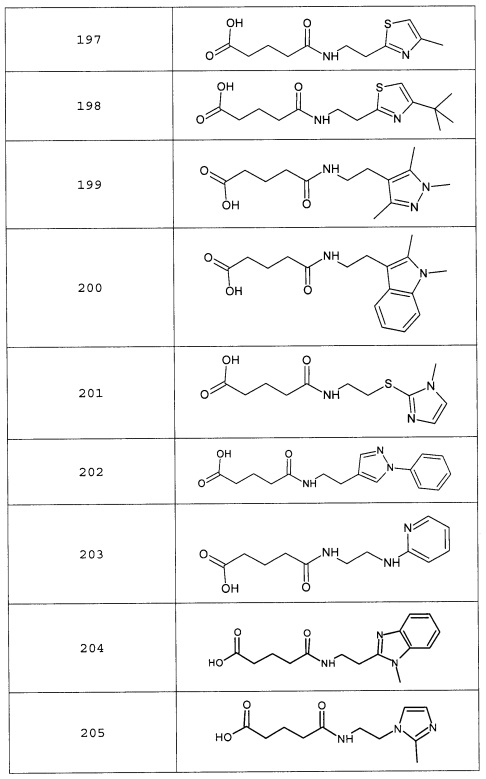
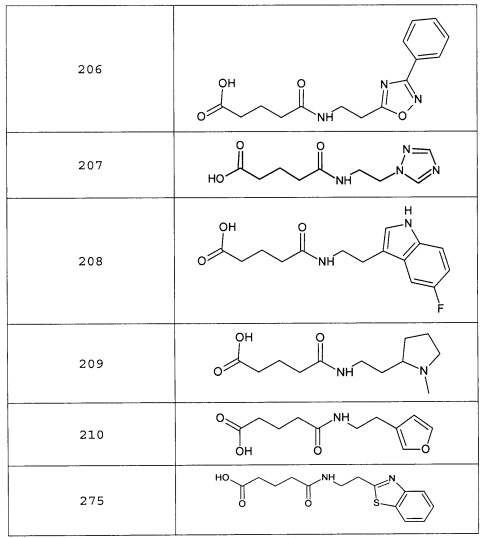
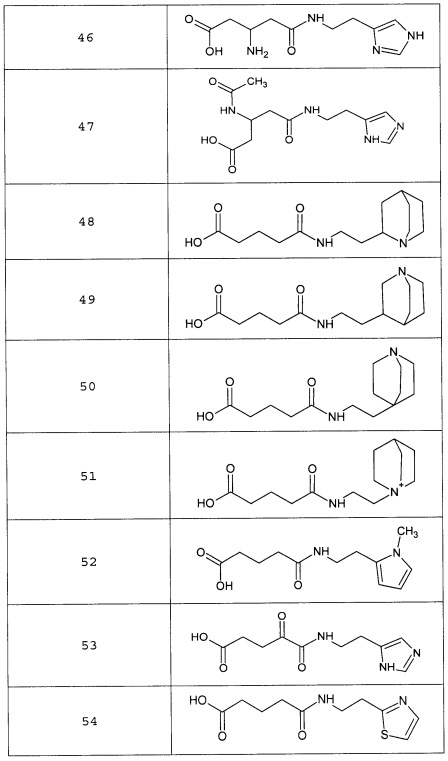
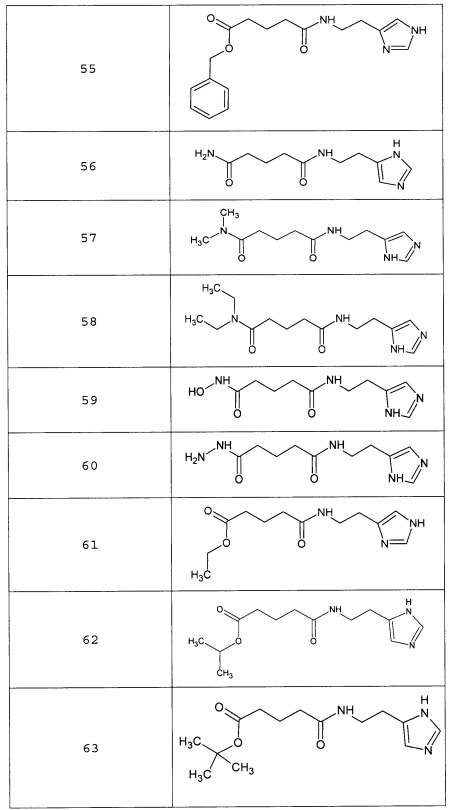
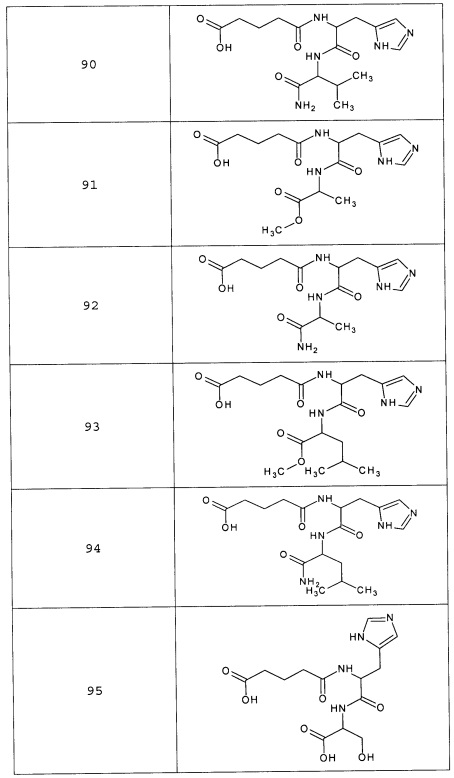
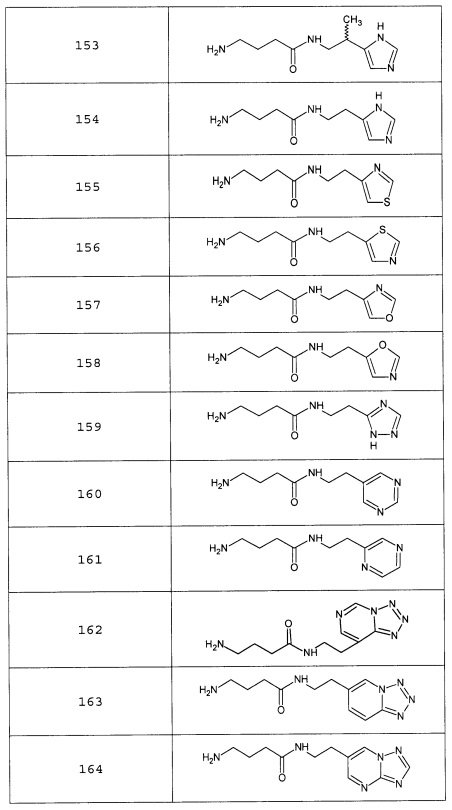
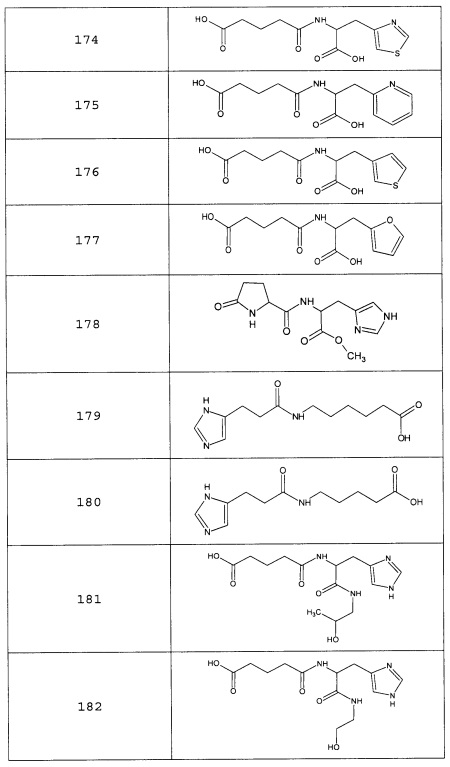
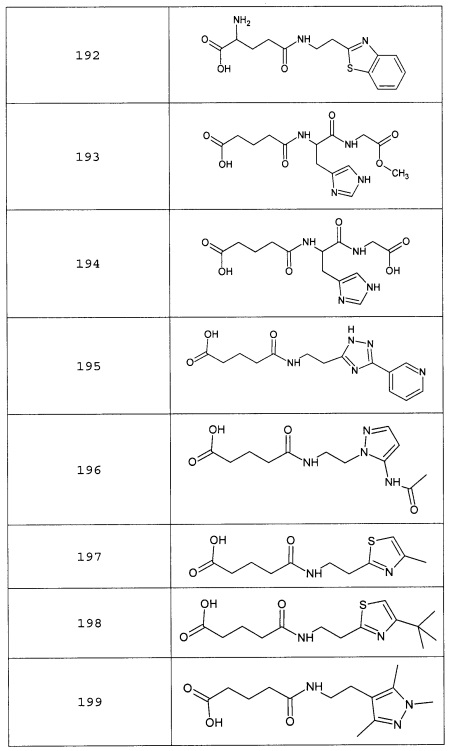
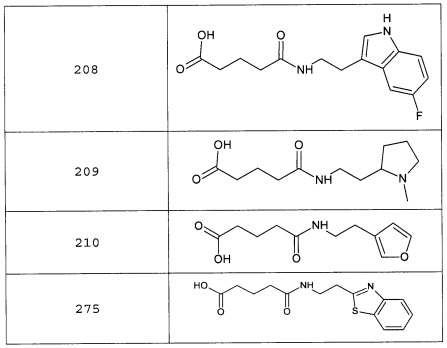
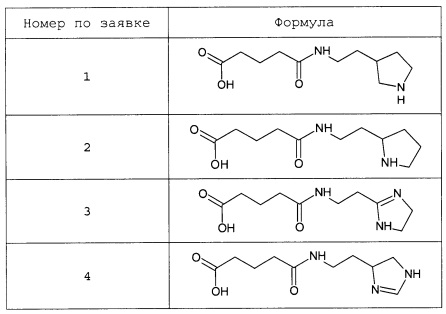
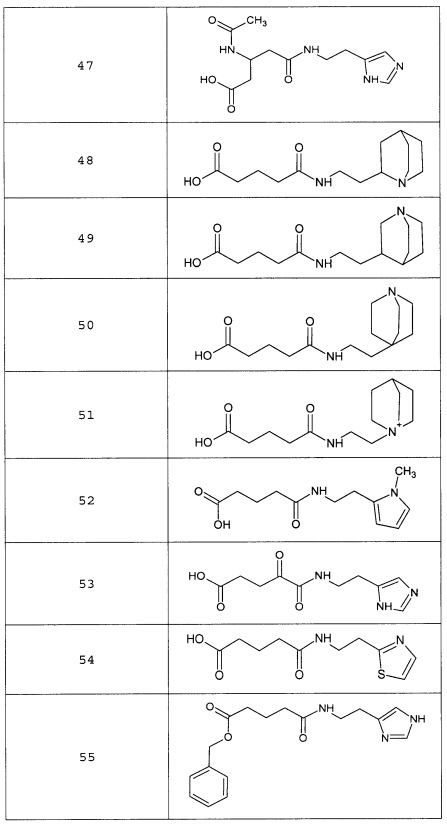
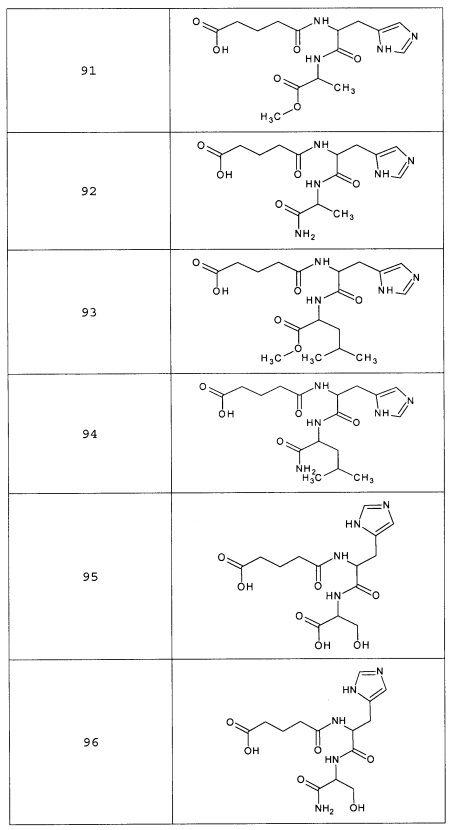
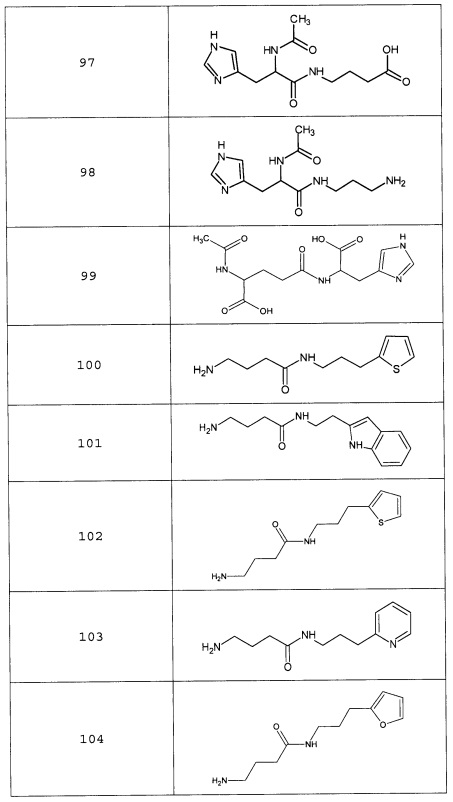
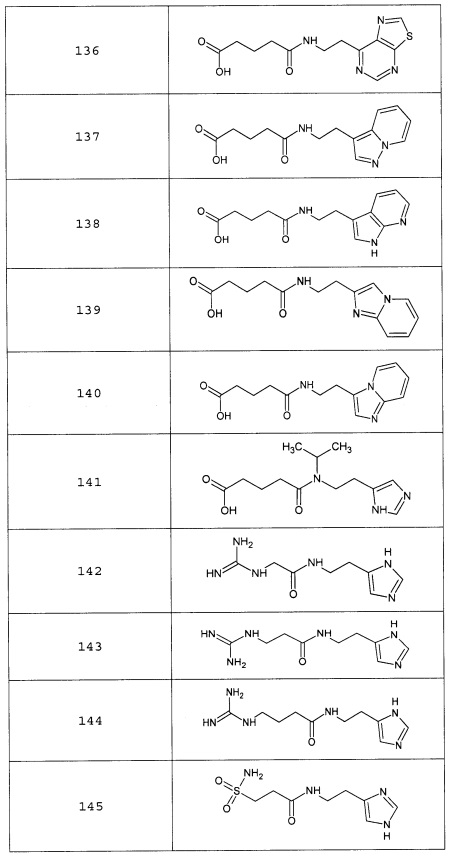
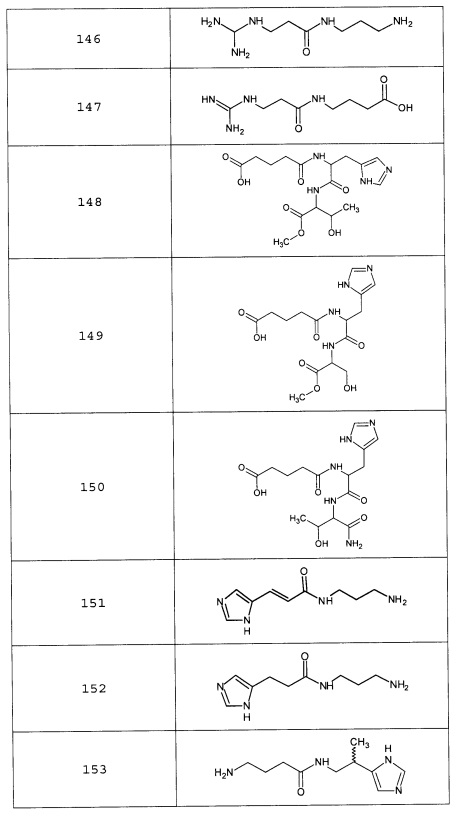
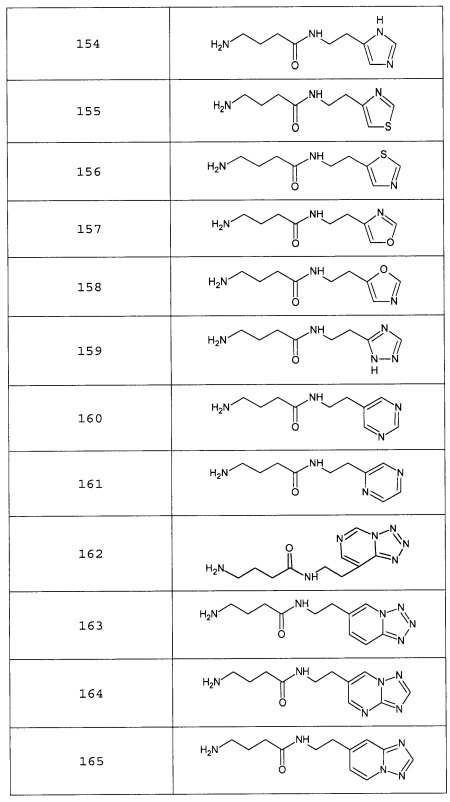
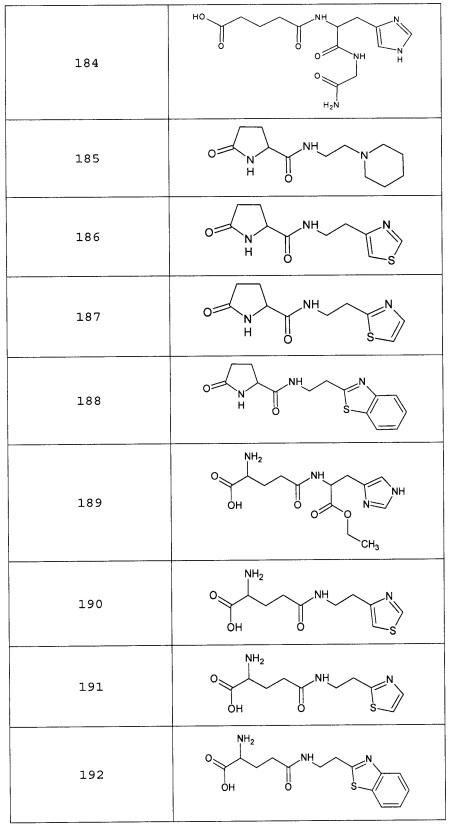
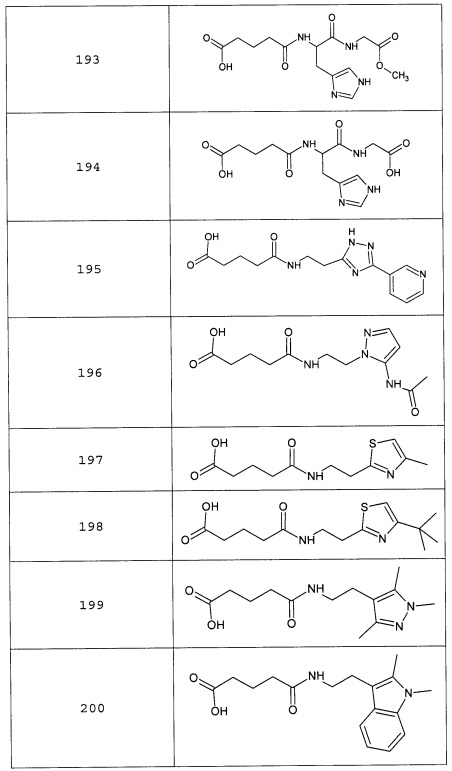
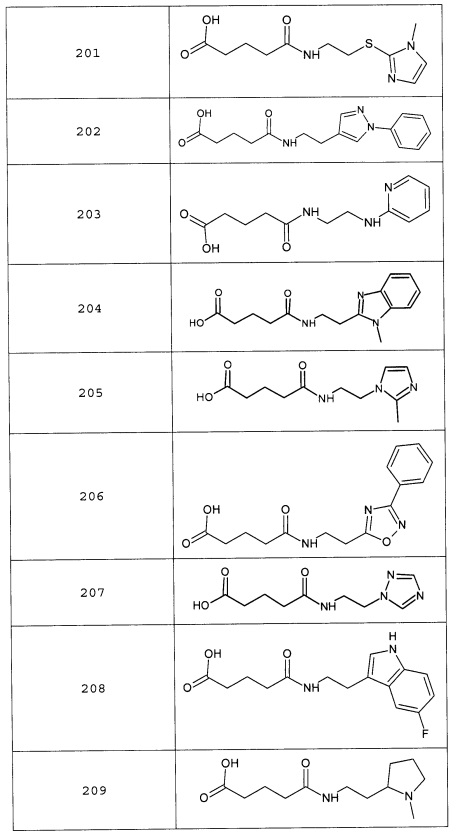
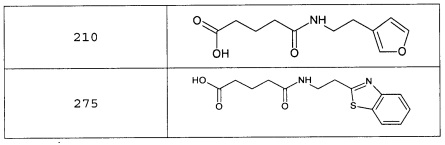
Наиболее предпочтительными соединениями настоящего изобретения являются соединения, представленные в таблице 1
Таблица 1 смотри в графической части.
Соединения общей формулы I, согласно изобретению, вводятся в твердой лекарственной форме.
Настоящее изобретение так же относится к способам получения соединений общей формулы I или их фармацевтически приемлемых солей.
В частности, настоящее изобретение относится к способу получения соединений общей формулы I, относящихся к моноамидам дикарбоновых кислот или их фармацевтически приемлемым солям, включающему взаимодействие соответствующего ангидрида с амином или дипептидом в подходящем органическом растворителе необязательно в присутствии органического основания.
Настоящее изобретение относится к способу получения соединений общей формулы I, относящихся к глутарильным производным дипептидов или их фармацевтически приемлемым солям по п. 1, включающему:
(а) синтез дипептидов исходя из ди-Boc-защищенного гистидина и соответствующей аминокислоты методом активированных пара-нитрофениловых эфиров в N,N-диметилформамиде;
(b) снятие Boc-защиты обработкой защищенного дипептида трифторуксусной кислотой; и
(с) добавление глутарового ангидрида к трифторацетатному производному дипептида в N,N-диметилформамиде в присутствии 2 эквивалентов N-метилморфолина.
Настоящее изобретение относится к способу получения соединений общей формулы I, относящихся к производным γ-аминомаслянной кислоты и соответствующего амина или их фармацевтически приемлемым солям, включающему:
(а) получение имидазолида N-Boc-γ-аминомасляной кислоты путем взаимодействия N-Boc-γ-аминомаслянной кислоты с 1,1’-карбонилдиимидозолом в среде безводного органического растворителя; и
(b) введение имидазолида N-Boc-γ-аминомасляной кислоты во взаимодействие с соответствующим амином при нагревании в среде безводного органического растворителя.
Настоящее изобретение относится к способу получения соединений общей формулы I, относящихся к производным пироглутаминовой кислоты, N-ацетил- глутаминовой килоты по α-карбоксильной группе или глутаминовой кислоты по γ-карбоксильной группе и соответствующего амина или их фармацевтически приемлемым солям методом активированных N-оксисукциимидных эфиров, включающему взаимодействие N-оксисукцинимидного эфира соответствующей кислоты с соответствующим амином в среде безводного органического растворителя при комнатной температуре.
Настоящее изобретение относится к способу получения соединений общей формулы I, относящихся к амидам, образованных 3-(4-имидазолил)акриловой кислотой и 3-(4-имидазолил)пропионовой кислотой и соответствующей аминокислотой: 2-аминопентановой кислотой, 4-аминомаслянной кислотой и 6-аминогексановой кислотой хлорангидридным методом, включающему:
(а) получение хлорангидридов соответствующих кислот с использованием преимущественно тионилхлорида,
(b) введение во взаимодействие полученного хлорангидрида без дополнительной очистки с соответствующей аминокислотой в среде безводного органического растворителя при комнатной температуре.
Изобретение далее охватывает способ профилактики и лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов, предусматривающий введение пациенту эффективного количества соединения общей формулы I или его фармацевтически приемлемой соли.
Вирус, принадлежащий роду энтеровирусов, может быть выбран из группы, включающей риновирусы, вирусы Коксаки и энтеровирус типа 71. Вирус, принадлежащий к роду пневмовирусов, представляет собой респираторно-синцитиальный вирус, а к роду метапневмовирусов – метапневмовирус человека. Доза соединения общей формулы I или его фармацевтически приемлемой соли может составлять приблизительно 0,1-30 мг/кг массы тела пациента. При этом разовая доза соединения общей формулы I может составлять приблизительно 2-300 мг. Предпочтительная длительность приема соединения общей формулы I составляет от 5 дней до 10 дней. В одном из вариантов изобретения, осуществляют профилактику или лечение обострений астмы, хронической обструктивной болезни легких, бронхита и муковисцидоза, вызванных риновирусом, респираторно-синцитиальным вирусом и метапневмовирусом.
Далее, изобретение относится к фармацевтической композиции для лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов, содержащей эффективное количество соединения общей формулы I или его фармацевтически приемлемой соли и фармацевтически приемлемые носители и наполнители. Эффективное количество соединения общей формулы I или его фармацевтически приемлемой соли, предпочтительно, составляет 0,1-30 мг/кг массы тела. При этом доза соединения общей формулы I может составлять 2-300 мг при введении 1 раз в день.
Изобретение также относится к набору для лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов, включающему композицию по изобретению и инструкции по ее применению.
Кроме этого, изобретение относится к применению соединений общей формулы I или их фармацевтически приемлемых солей для производства фармацевтической композиции для лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими к роду энтеровирусов, метапневмовирусов или роду пневмовирусов. Изобретение также включает применение соединений общей формулы I или их фармацевтически приемлемых солей для лечения заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов.
В качестве фармацевтически приемлемых солей соединений общей формулы I в настоящем изобретении могут быть использованы их соли с щелочными и щелочноземельными металлами, предпочтительно натриевая, калиевая, литиевая соли.
Кроме того, в качестве фармацевтически приемлемых солей соединений по настоящему изобретению могут быть использованы аддитивные соли органических кислот (например, формиат, ацетат, малеат, тартрат, метансульфонат, бензолсульфонат, толуолсульфонат и др.), аддитивные соли неорганических кислот (например, гидрохлорид, гидробромид, сульфат, фосфат и др.), соли с аминокислотами (например, соль аспарагиновой кислоты, соль глутаминовой кислоты и т.д.), предпочтительно хлоргидраты и ацетаты.
Соединения общей формулы I или его соли вводят в эффективном количестве, которое обеспечивает желаемый терапевтический результат.
Соединения общей формулы I или их соли могут быть введены пациенту в дозах, составляющих от 0,1 до 30 мг/кг веса тела человека в день, предпочтительно в дозах от 0,3 до 1,5 мг/кг один или более раз в день.
При этом следует отметить, что конкретная доза для каждого конкретного пациента будет зависеть от многих факторов, таких как возраст, вес тела, пол, общее состояние здоровья и режим питания пациента, время и способ введения лекарственного средства, скорость его выведения из организма, а также тяжесть заболевания у данного индивида, подвергаемого лечению.
Фармацевтические композиции по настоящему изобретению содержат соединения общей формулы I или их фармацевтически приемлемую соли в количестве, эффективном для достижения желаемого результата, и могут быть приготовлены в виде стандартных лекарственных форм (например, в твердой, полутвердой или жидкой формах), содержащих соединения общей формулы I или их соли в качестве активного ингредиента в смеси с носителем или наполнителем, пригодным для внутримышечного, внутривенного, перорального, сублингвального, ингаляционного, интраназального, интраректального и трансдермального применения. Активный ингредиент может быть включен в композицию вместе с обычно используемыми нетоксичными фармацевтически приемлемыми носителями, пригодными для изготовления растворов, таблеток, пилюль, капсул, драже, суппозиториев, эмульсий, суспензий, мазей, гелей, пластырей и любых других лекарственных форм.
В качестве наполнителей могут быть использованы различные вещества, такие как сахариды, например глюкоза, лактоза или сахароза, маннит или сорбит, производные целлюлозы и/или фосфаты кальция, например, трикальций фосфат или кислый фосфат кальция; в качестве связующего компонента могут быть использованы такие, как крахмальная паста, например, кукурузный, пшеничный, рисовый, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натриевая соль карбоксиметилцеллюлозы и/или поливинилпирролидон. При необходимости могут быть использованы разрыхляющие агенты, такие как вышеупомянутые крахмалы и карбоксиметилкрахмал, поперечно сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия.
Могут быть использованы необязательные добавки, такие как агенты, регулирующие текучесть, и смазывающие агенты, такие как диоксид кремния, тальк, стеариновая кислота и ее соли, такие как стеарат магния или стеарат кальция, и/или пропиленгликоль.
В качестве добавок могут быть также использованы стабилизаторы, загустители, красители и отдушки.
В качестве мазевой основы могут быть использованы углеводородные мазевые основы, такие как вазелин белый и желтый (Vaselinum album, Vaselinum flavum), вазелиновое масло (Oleum Vaselini), мазь белая и жидкая (Unguentum album, Unguentum flavum), а в качестве добавок для придания более плотной консистенции - такие как твердый парафин и воск; абсорбтивные мазевые основы, такие как гидрофильный вазелин (Vaselinum hydrophylicum), ланолин (Lanolinum), кольдкрем (Unguentum leniens); мазевые основы, смываемые водой, такие как гидрофильная мазь (Unguentum hydrophylum); водорастворимые мазевые основы, такие как полиэтиленгликолевая мазь (Unguentum Glycolis Polyaethyleni), бентонитовые основы и другие.
В качестве основы для гелей могут быть использованы метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, оксипропилцеллюлоза, полиэтиленгликоль или полиэтиленоксид, карбопол.
В качестве основы для суппозитория могут быть использованы основы, не растворимые в воде, такие как масло какао; основы, растворимые в воде или смешиваемые с водой, такие как желатино-глицериновые или полиэтиленоксидные; комбинированные основы – мыльно-глицериновые.
При приготовлении стандартной лекарственной формы количество активного ингредиента, используемого в комбинации с носителем, может варьироваться в зависимости от реципиента, подвергающегося лечению, от конкретного способа введения лекарственного средства.
Так, например, при использовании соединений общей формулы I или их солей в виде растворов для инъекций, содержание активного агента в них составляет 0,1-5%. В качестве разбавителей могут быть использованы 0,9% раствор хлорида натрия, дистиллированная вода, раствор новокаина для инъекций, раствор Рингера, раствор глюкозы, специфические добавки для растворения. При введении в организм соединений общей формулы I или их солей в виде таблеток и суппозиториев их количество составляет 10-300 мг на стандартную лекарственную форму.
Лекарственные формы настоящего изобретения получают по стандартным методикам, таким как, например, процессы смешивания, гранулирования, формирования драже, растворения и лиофилизации.
Определения
Термин “алкил”, используемый в настоящем описании, означает насыщенный прямой или разветвленный углеводород. В некоторых вариантах осуществления, алкильные группы содержат 1-6 атомов углерода. В других вариантах осуществления, алкильные группы содержат 1-5 атомов углерода. В еще других вариантах осуществления, алкильные группы содержат 1-4 атомов углерода, и в еще других вариантах осуществления, алкильные группы содержат 1-3 атома углерода.
Термин “алкокси”, используемый в настоящем описании, относится к алкильной группе, определенной выше, присоединенной к молекуле посредством атома кислорода (“алкокси”, например, -О-алкил).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методики синтеза
Индивидуальность полученных соединений проверяли методом ТСХ на пластинках “Kieselgel 60 F254” (фирмы “Merck”, Германия). Хроматограммы проявляли хлор-тетраметилбензидиновым реактивом и реактивом Паули.
ЖХ МС-система анализа многокомпонентных смесей UPLC/MS Shimadzu 2020, включающая: хроматограф Analytical HPLC CBM-20A, насосы LC-30AD, автосамплер SIL-30AC, детекторы SPD-M20A, ELSD-LTII (evaporative light scattering detector) и масс-спектрометр LCMS-20.
Колонка - Waters ACQUITY UPLC BEH C18 1.7um 2.1x50mm; гадиент элюирования в системе растворителей: растворитель А – вода с 0,1% HCOOH, растворитель B – ацетонитрил с 0,1% HCOOH (условия А).
Колонка YMC-UltraHT Hydrosphere C18 2.0 мкм 50x2.0 мм; гадиент элюирования в системе растворителей: растворитель А – вода с 0,1% HCOOH, растворитель B – ацетонитрил с 0,1% HCOOH (условия Б).
Колонка Synergi Fusion-RP 150x2 мм, 4 мкм, 80Å; гадиент элюирования в системе растворителей: растворитель А – вода с 0,1% HCOOH, растворитель B – ацетонитрил с 0,1% HCOOH (условия В).
Колонка Shim-pack XR-ODS II 75x3 мм; гадиент элюирования в системе растворителей: растворитель А – вода с 0,1% HCOOH, растворитель B – ацетонитрил с 0,1% HCOOH (условия Г).
Аналитическую обращенно-фазовую ВЭЖХ проводили с помощью системы анализа многокомпонентных смесей органического происхождения HPLC Shimadzu, включающей: хроматограф Analytical HPLC CBM-20A, насосы LC-20AD, автосамплер SIL-20A, UV-детектор SPD-20A.
Колонка Symmetry C18 150x4,6 мм 5мкм; градиент элюирования в системе: растворитель А – водный раствор с 1-гексилсульфонатом натрия 0,0025М рН=3 , растворитель B – ацетонитрил (условия 1).
Колонка Luna C18 (2) 100A 250x4,6 мм (сер. 599779-23), градиент элюирования в системе фосфатный буферный раствор рН 3,0 – метанол (условия 2).
Колонка X-Bridge C 18, 150x4,6 мм (3,5 мкм), градиент элюирования в системе: растворитель А – водный раствор с 1-гексилсульфонатом натрия 0,0025М рН=3 , растворитель B – ацетонитрил (условия 3).
Колонка Symmetry C18 150x4,6 мм 5мкм; градиент элюирования в системе фосфатный буферный раствор рН 3,0 – метанол (условия 4).
Колонка Merk.LiChroCART 250*4mm 5mkm. LiChrospher 100RP-8E 5 mkm.C8. Serial number 1.50837.0001; градиент элюирования в системе ацетатно-аммиачный рН 7.5буфер:ацетонитрил (условия 5).
1Н-ЯМР спектры регистрировали на приборе Bruker DPX-400 (Германия).
Моноамиды дикарбоновых кислот получали путем взаимодействия соответствующего ангидрида с амином или дипептидом в органическом растворителе при разных температурных режимах в присутствии или без органического основания. В некоторых случаях использовали защищенные по NH-группе гетероцикла производные аминов. Преимущественно использовали Boc-защиту. Предпочтительными органическими растворителями для проведения реакции конденсации являются тетрагидрофуран, хлороформ, хлористый метилен, N,N-диметилформамид, дихлорметан, ацетонитрил и смесь диоксана с N,N-диметилформамидом в соотношении 3:1. Реакцию предпочтительно проводят при охлаждении до 0°С или 3-5°С, при комнатной температуре или при нагревании до 45°С или 60°С, а также при температуре кипения растворителя.
При получении глутарильных производных дипептидов сначала осуществляли синтез дипептидов исходя из ди-Boc-защищенного гистидина и соответствующей аминокислоты методом активированных пара-нитрофениловых эфиров в N,N-диметилформамиде. Boc-защиту снимали обработкой защищенного дипептида трифторуксусной кислотой. Глутарильное производное дипептида получали добавлением глутарового ангидрида к трифторацетатному производному дипептида в N,N-диметилформамиде в присутствии 2 эквивалентов N-метилморфолина.
Производные γ-аминомаслянной кислоты и соответствующего амина синтезировали с использованием конденсирующий агентов, преимущественно с использованием 1,1'-карбонилдиимидозола. В качестве исходного соединения используют защищенное производное γ-аминомаслянной кислоты, преимущественно N-Boc-γ-аминомаслянную кислоту. При реакции N-Boc-γ-аминомаслянной кислоты с 1,1'-карбонилдиимидозолом получают активированное производное - имидазолид N-Boc-γ-аминомасляной кислоты, которое вводят взаимодействие с соответствующим амином. Обе реакции проводят в безводных органических растворителях, преимущественно в безводном ацетонитриле. Реакцию конденсации проводят при нагревании, преимущественно при 45°С.
Производные пироглутаминовой кислоты, N-ацетил- глутаминовой килоты по α-карбоксильной группе или глутаминовой кислоты по γ-карбоксильной группе и соответствующего амина получали методом активированных N-оксисукциимидных эфиров, включающему взаимодействие N-оксисукцинимидного эфира соответствующей кислоты с соответствующим амином в среде безводного органического растворителя при комнатной температуре.
Синтез амидов 3-(4-имидазолил)акриловой кислоты и 3-(4-имидазолил)пропионовой кислоты предпочтительно с 2-аминопентановой кислотой, 4-аминомаслянной кислотой и 6-аминогексановой кислотой осуществляют хлорангидридным методом. Получают хлорангидрид соответствующей кислоты с использованием преимущественно тионилхлорида, полученный хлорангидрид без дополнительной очистки вводят во взаимодействие с соответствующей аминокислотой в среде безводного органического растворителя при комнатной температуре.
Остальные соединения были синтезированы с применением стандартных методов органической химии.
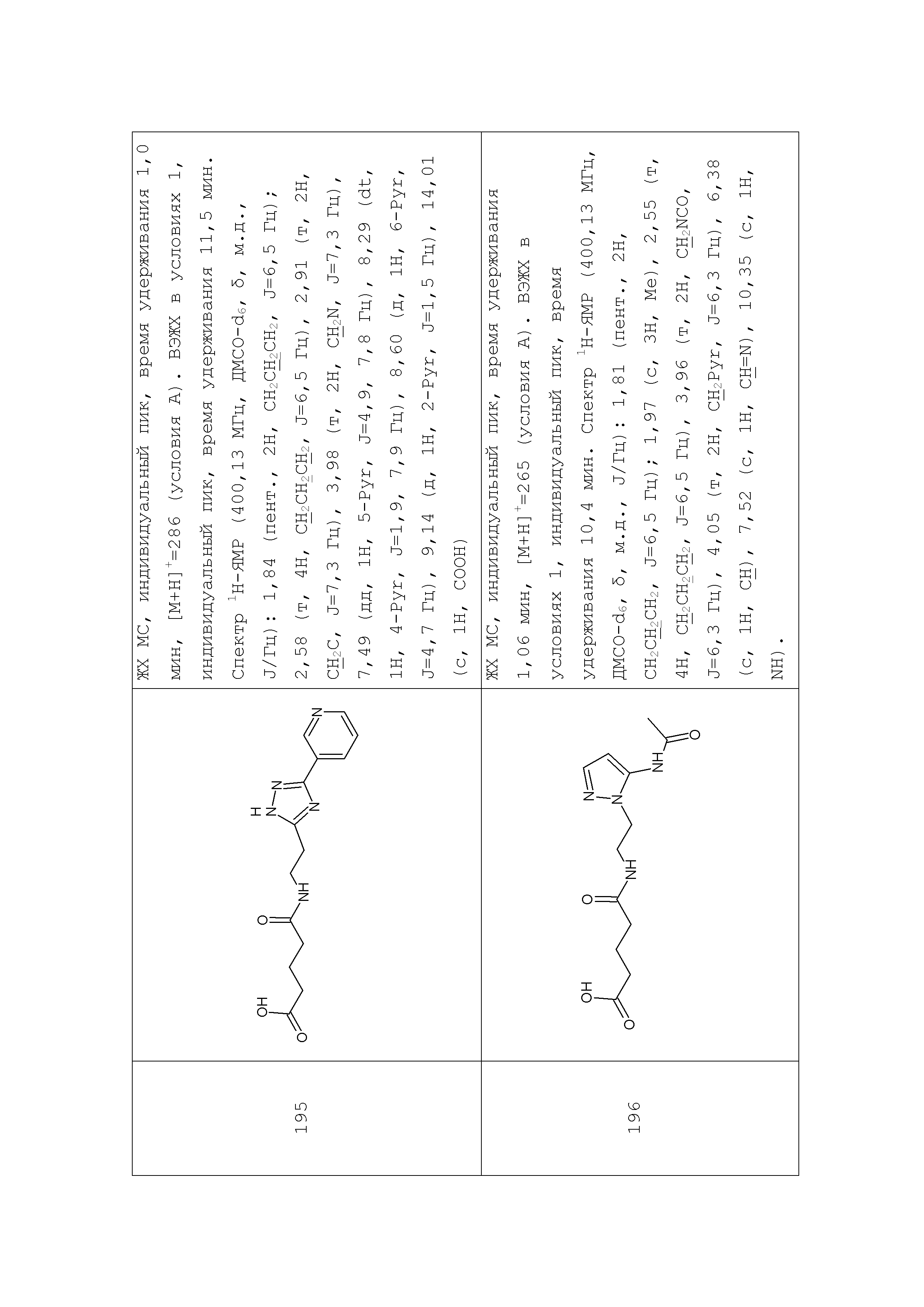
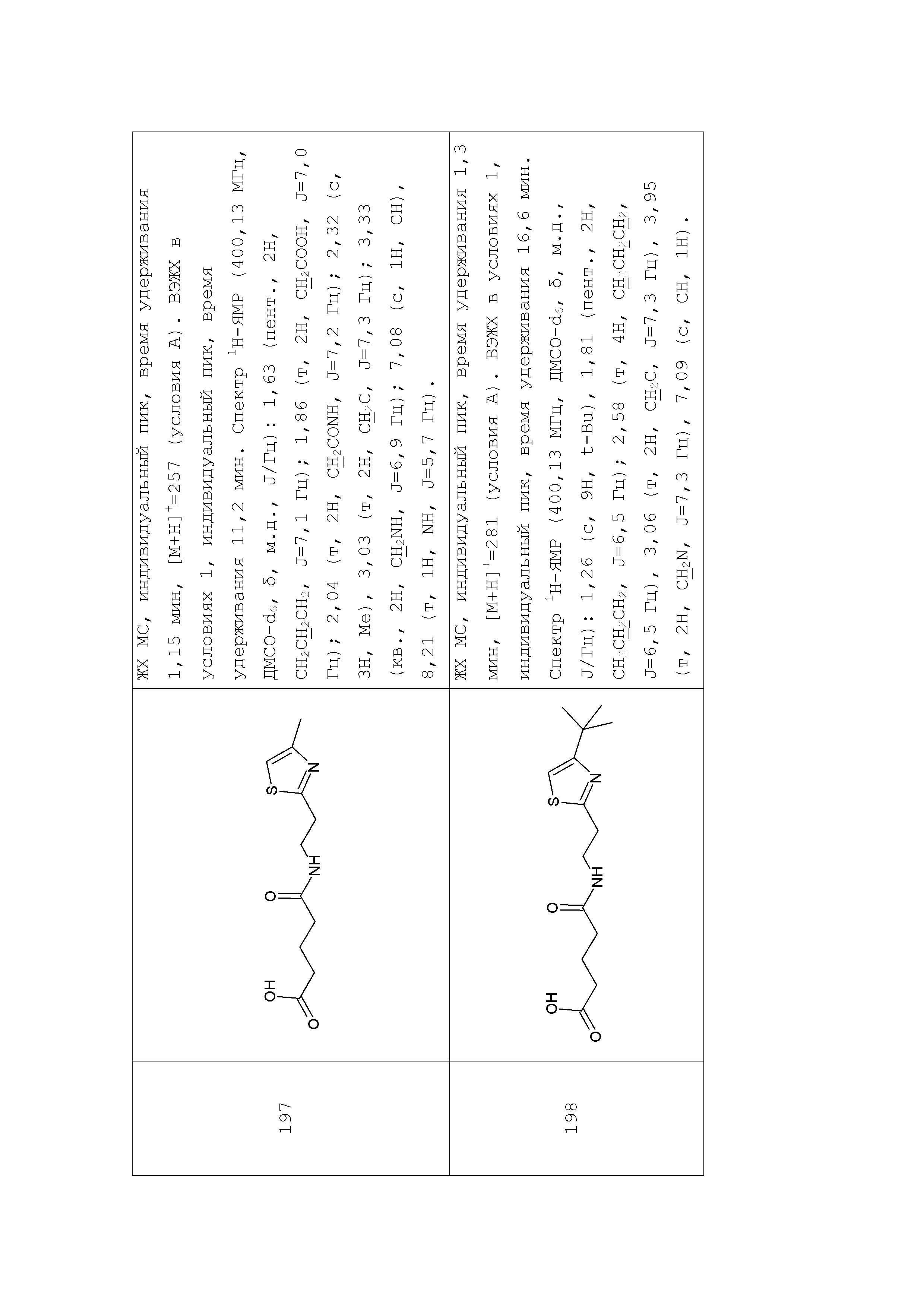
1. Синтез производных моноамидов дикарбоновых кислот на примере синтеза соединения 196
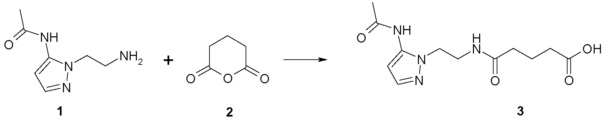
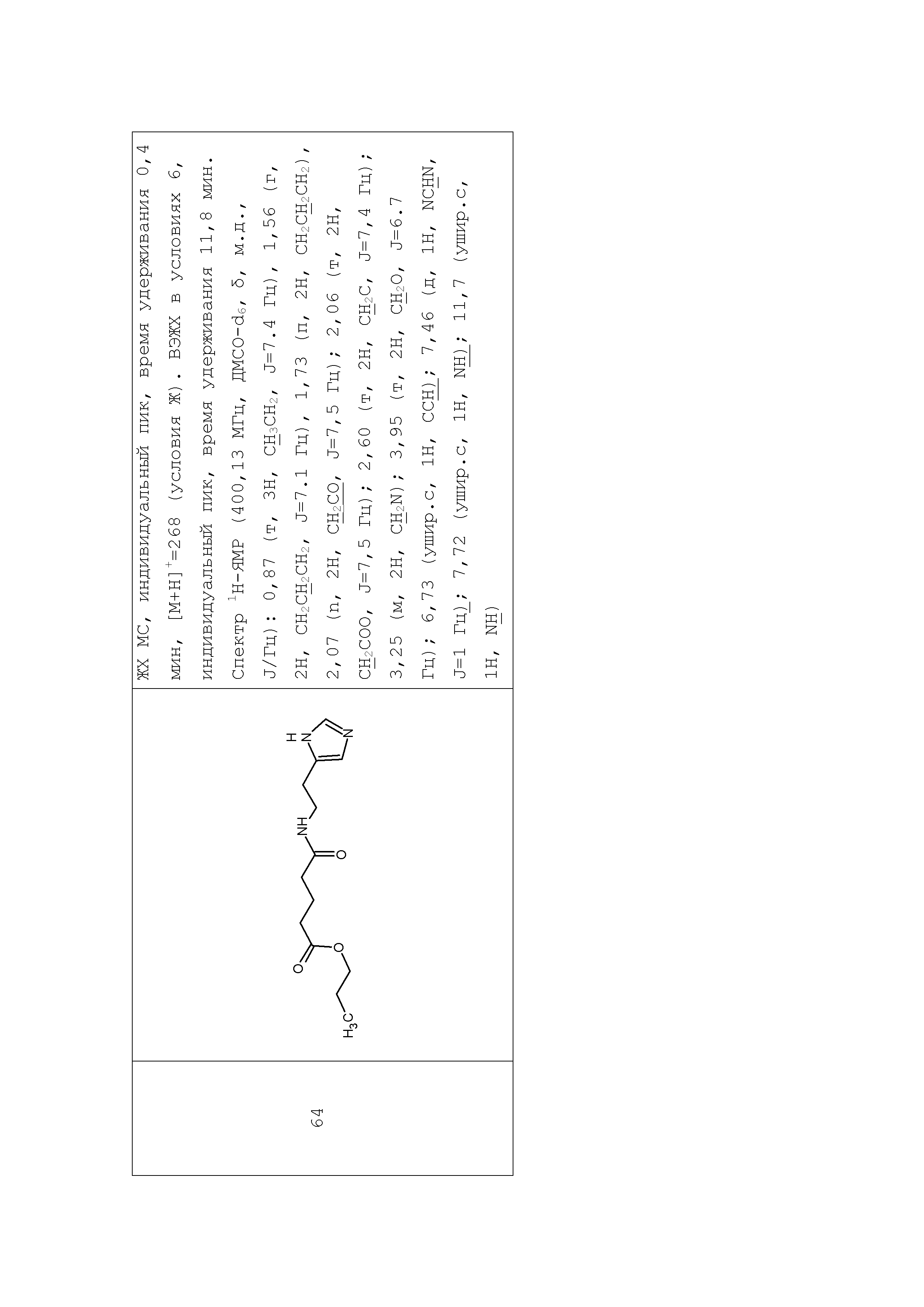
Раствор глутарового ангидрида (2) (2,850 г, 25 ммоль) в дихлорметане (25 мл) добавляли по каплям к раствору N-[1-(2-аминоэтил)-1Н-пиразол-5-ил]ацетамида дигидрохлорида (1) (3,588 г, 15 ммоль) и триэтиламина (3,440 г, 4,8 мл, 34 ммоль) в дихлорметане (50 мл) при перемешивании при комнатной температуре. Полученную реакционную смесь перемешивали при комнатной температуре в течение 6 часов до полного исчезновения исходного амина (контроль ТСХ, LCMS). Выпавший осадок соли триэтиламина отфильтровывали. Фильтрат концентрировали при пониженном давлении. Полученный остаток обрабатывали ацетоном. Образовавшийся осадок отфильтровывали, промывали ацетоном, диэтиловым эфиром и сушили на воздухе и при пониженном давлении. Соединение 3 было получено в виде белого твердого вещества (1,101 г, 26%). Rf (3) 0,52 (ДХМ/изо-пропиловый спирт, 5:1+2 капли уксусной кислоты). ЖХ МС, индивидуальный пик, время удерживания 1,06 мин, [М+Н]+=265 (условия А). ВЭЖХ в условиях 1, индивидуальный пик, время удерживания 10,4 мин. Спектр1H-ЯМР (400,13 МГц, ДМСО-d6,δ, м.д., J/Гц): 1,81 (пент., 2H, CH2CH2CH2, J=6,5 Гц); 1,97 (с, 3H, Me), 2,55 (т, 4H, CH2CH2CH2,J=6,5 Гц), 3,96 (т, 2H, CH2NCO, J=6,3 Гц), 4,05 (т, 2H, CH2Pyr, J=6,3 Гц), 6,38 (с, 1H, CH), 7,52 (с, 1H, CH=N), 10,35 (с, 1H, NH).
2. Синтез глутарильных производных дипептидов на примере синтеза соединения 94
К раствору 10 г (28,4 ммоль) паранитрофенилового эфира БОК-лейцина в 20 мл диметилформамида прибавляли 4 мл 25% водного раствора аммиака, реакционную смесь выдерживали 3 часа при комнатной температуре, упаривали досуха, остаток растирали в эфире, отфильтровывали, промывали эфиром, получили 2,5 г (10,87 ммоль) Бок-лейцинамида (R-f0,6 хлороформ:метанол:32% уксусная кислота 15:4:1). Осадок растворяли в 25 мл трифторуксусной кислоты, выдерживали 1 час, упаривали, остаток растирали в эфире, отфильтровывали, растворяли в 50 мл диметилформамида, добавляли NMM до pH-8,5, к раствору прибавляли 5,14 г (10,8 ммоль) паранитрофенилового эфира ди-БОК-L-гистидина, реакционную смесь оставляли на ночь при комнатной температуре, диметилформамид упаривали, остаток растворяли в 10 мл смеси этилацетат-гексан 8+2 мл и пропускали через колонку размерам 3×17 см заполненную суспензией силикагеля в той же смеси. Продукт элюировали этилацетатом, фракции, содержащие целевое вещество объединяли, упаривали досуха. Получили 3,95 г (9,6 ммоль) продукта с Rf-0,8 (хлороформ:метанол:32% уксусная кислота 15:4:1). Амид дипептида растворяли в 25 мл трифторуксусной кислоты, выдерживали 1 час при комнатной температуре, упаривали, остаток растирали в эфире, отфильтровывали, растворяли в 25 мл диметилформамида, добавляли NMM до рH -8,5, к полученному раствору прибавляли тремя порциями с интервалом 15-20 минут 1,14 г (10 ммоль) глутарового ангидрида, реакционную смесь выдерживали 2 часа при комнатной температуре, упаривали, к остатку прибавляли 100 мл этилацетата, оставляли на ночь, выпавший осадок отфильтровывали, растворяли в100 мл воды экстрагировали 50 мл этилацетата, водный слой упаривали до половины объема, обрабатывали активированным углем, упаривали, растворяли в 100 мл 2% уксусной кислоты, лиофилизировали. Получили 3,5 г. (91%) целевого продукта Rf-0,75 (хлороформ:метанол:32% уксусная кислота 5:3:1). ЖХ МС, индивидуальный пик, время удерживания 0,2 мин, [М+Н]+=382 (условия А). ВЭЖХ в условиях 1, индивидуальный пик, время удерживания 11,8 мин. Спектр1H-ЯМР (400,13 МГц, ДМСО-d6,δ, м.д., J/Гц): 0,82, 0,87 (д, 6H, CH2CH(CH3)2, J=6,1 Гц); 1,51 (м, 3H, CH2CH(CH3)2); 1,67 (пент., 2H, CH2CH2CH2, J=7,5 Гц); 2,15 (м, 4H, CH2CH2CH2); 2,86, 2,98 (м, 2H, CCH2CH); 4,19 (м, 1H, NCH,); 4,52 (м, 1H, CCH2CH); 7,02, 7,51 (ушир.с, 2H, NH2); 7,07 (с, 1H, CCH); 7,90 (д, 1H, NH, J=8,2 Гц); 8,08 (д, 1H, NH, J=7,9 Гц); 8,23 (с, 1H, NCHN)
3. Синтез амидов γ-аминомасляной кислоты на примере синтеза соединения 103
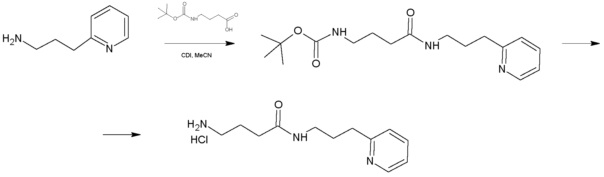
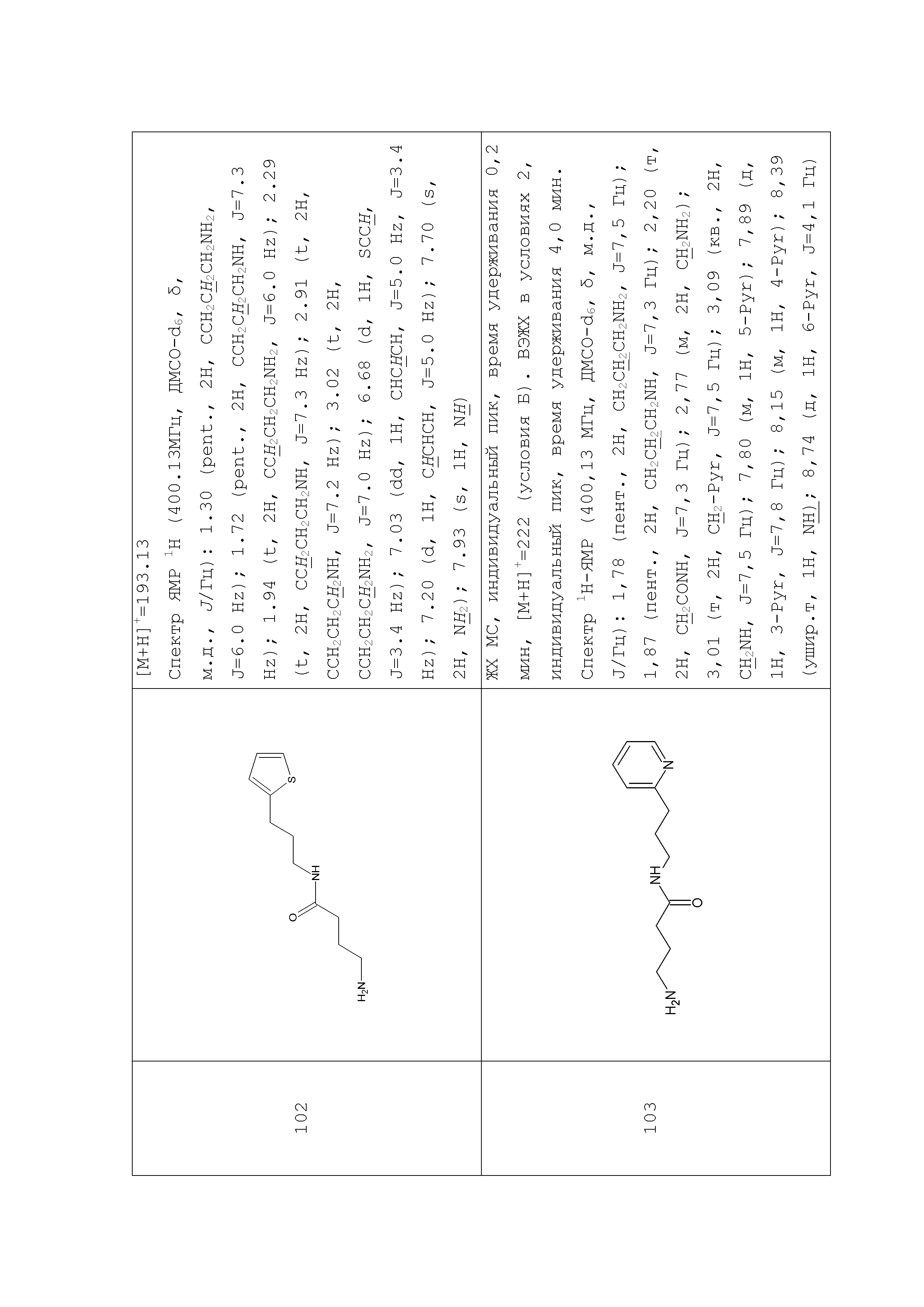
К раствору 1,36 г (0,01 моль) 2-(3-аминопропил)пиридина в 25 мл безводного ацетонитрила прибавляли раствор имидазолида N-Boc-γ-аминомасляной кислоты полученный из 2,23 (0,011 моль) N-Boc-γ-аминомасляной кислоты и 1,95 г (0,012 моль) карбонилдиимидазола в 10 мл безводного ацетонитрила. Реакционную смесь перемешивали 4 часа при 45°С, растворитель удаляли в вакууме, остаток растворяли в 300 мл эфира, промывали насыщенным водным раствором гидрокарбоната натрия (3×100 мл). Органический слой сушили над сульфатом натрия, растворитель удаляли в вакууме, остаток сушили в вакууме масляного насоса до постоянного веса. Остаток растворяли в 80 мл безводного эфира и прибавляли 35 мл 5% раствора хлористого водорода в безводном метаноле. Реакционную смесь перемешивали при комнатной температуре до исчезновения (по данным ТСХ) исходного соединения (около 4 часов), растворители удаляли в вакууме, остаток растирали с безводным эфиром, эфир декантировали после чего повторяли процедуру. Остаток под эфиром оставляли при 0ºС на 8 часов. Выпавший осадок отфильтровывали, промывали безводным эфиром (3×10 мл) и сушили в вакууме. Выход 1,62 г (63%) Rf (хлороформ - метанол 4/1) 0,48. ЖХ МС, индивидуальный пик, время удерживания 0,5 мин, [М+Н]+=222 (условия Б). ВЭЖХ в условиях 2, индивидуальный пик, время удерживания 4,00 мин. Спектр1H-ЯМР (400,13 МГц, ДМСО-d6,δ, м.д., J/Гц): 1,78 (пент., 2H, CH2CH2CH2NH2,J=7,5 Гц); 1,87 (пент., 2H, CH2CH2CH2NH, J=7,3 Гц); 2,20 (т, 2H, CH2CONH, J=7,3 Гц); 2,77 (м, 2H, CH2NH2); 3,01 (т, 2H, CH2-Pyr, J=7,5 Гц); 3,09 (кв., 2H, CH2NH, J=7,5 Гц); 7,80 (м, 1H, 5-Pyr); 7,89 (д, 1H, 3-Pyr, J=7,8 Гц); 8,15 (м, 1H, 4-Pyr); 8,39 (ушир.т, 1H, NH); 8,74 (д, 1H, 6-Pyr, J=4,1 Гц).
4. Синтез амидов пирроглутаминовой кислоты на примере синтеза соединения 178

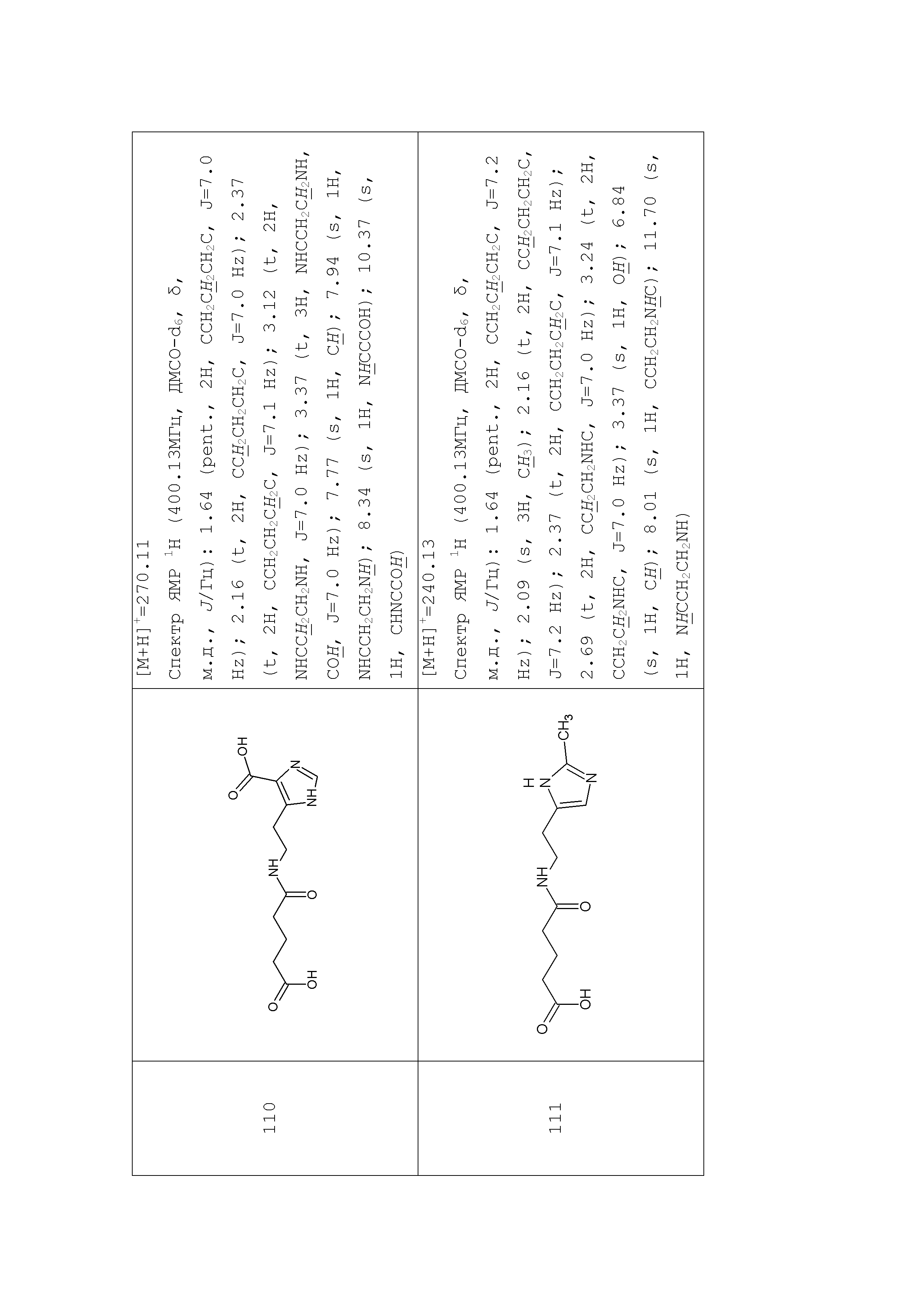
К охлажденному до 0°С раствору 3,5 г (27,1 ммоль) пироглутаминовой кислоты в 25 мл ДМФА добавляли 3,4 г (29,8 ммоль) HONSu и раствор 6,4 г (29,8 ммоль) DCC в 10 мл ДМФА. Реакционную смесь перемешивали 1 ч при 0°С и 16 ч при комнатной температуре. Осадок отделяли фильтрованием и промывали 3×10 мл этилацетатом. Растворитель из объединенных фильтратов удаляли в вакууме до образования устойчивой пены. Выход 5,8 г (95%). Rf 0,6 (хлороформ-метанол (1:1))
К раствору 2,2 г (9,7 ммоль) N-оксисукцинимидного эфира пироглутаминовой кислоты в 20 мл ДМФА добавляли 2,35 г (9,7 ммоль) дигидрохлорида гистидина и 2,87 мл (19,4 ммоль) триэтиламина. Реакционную смесь перемешивали 30 мин. при комнатной температуре. Растворитель удаляли в вакууме, остаток растворяли в 20 мл этилацетата и промывали 3×10 мл 1% лимонной кислотой, водой до нейтральной реакции и насыщенным раствором хлорида натрия. Органический слой высушивали над безводным сульфатом натрия. Растворитель удаляли в вакууме. Выход 2,36 г (80%), Rf 0,3 (хлороформ - метанол (4:1)). ЖХ МС, индивидуальный пик, время удерживания 0,23 мин. [М+Н]+=281 (условия В). ВЭЖХ в условиях 1, индивидуальный пик, время удерживания 7,0 мин. Спектр1H-ЯМР (400,13 МГц, ДМСО-d6, δ, м.д., J/Гц): 1,83-2,23 (м, 4H, CH2CH2CH), 2,93 (м, 2H, CH2CH), 3,61 (с, 3H, OCH3); 4,02 (м, 1H, CH2CH2CH); 4,50 (м, 1H, CH2CH); 6,84 (с, 1H, CCH); 7,60 (с, 1H, NCHN); 7,78 (с, 1H, NH), 8,05 (д, 1H, NH, J=7,8 Гц).
5. Синтез амидов 3-(4-имидазолил)акриловой кислоты и 3-(4-имидазолил)пропионовой кислоты на примере синтеза соединения 85

К 5 мл холодного тионилхлорида небольшими порциями при интенсивном перемешивании добавляли кислоту 1 (1 г, 0,007 моль). После добавления всего количества кислоты 1 реакционную смесь перемешивали при кипении в течение 3-4 часов, затем охлаждали до комнатной температуры. Отгоняли при пониженном давлении тионилхлорид. Полученный продукт 2 тщательно промывали на фильтре Шотта абс. толуолом (3×20 мл). Выход технического продукта 2 составил 1,4 г (99%). Продукт использовали на следующей стадии без дополнительной очистки.
Суспендировали исходный хлорангидрид кислоты 2 (1,4 г, 0,009 моль) в сухом ДМФА и при перемешивании добавляли гидрохлорида амина 3 (1,34 г, 0,0098 моль) и триэтиламин (4 мл, 0,036 моль). Реакционную смесь перемешивали в течение 10 часов при комнатной температуре. По завершении реакции отфильтровывали гидрохлорид триэтиламина, растворитель удаляли в вакууме при пониженном давлении. Остаток очищали с помощью метода колоночной хроматографии на силикагеле, система растворителей хлористый метилен - метанол (15:1). Выход чистого полупродукта 4 составил 0,65 г (35%).
Растворяли исходный эфир 4 (0,65 г, 0,0026 моль) в 10 мл 50% этанола и при перемешивании добавляли КОН (0,18 г, 0,0032 моль). Перемешивание продолжали в течение 5-6 часов при комнатной температуре. По завершении реакции (ТСХ-контроль в системе хлористый метилен - метанол (10:1)), раствор фильтровали через небольшой слой целита, растворитель удаляли в вакууме. Полученную калиевую соль кислоты промывали на фильтре ацетоном, перерастворяли в абс. этаноле и высаживали целевой продукт 5 добавлением конц. HCl (1 экв). Выход продукта 5 0,55 г (90%). ЖХ МС, индивидуальный пик, время удерживания 0,2 мин, [М+Н]+=238 (условия А). ВЭЖХ в условиях 1, индивидуальный пик, время удерживания 11,8 мин.
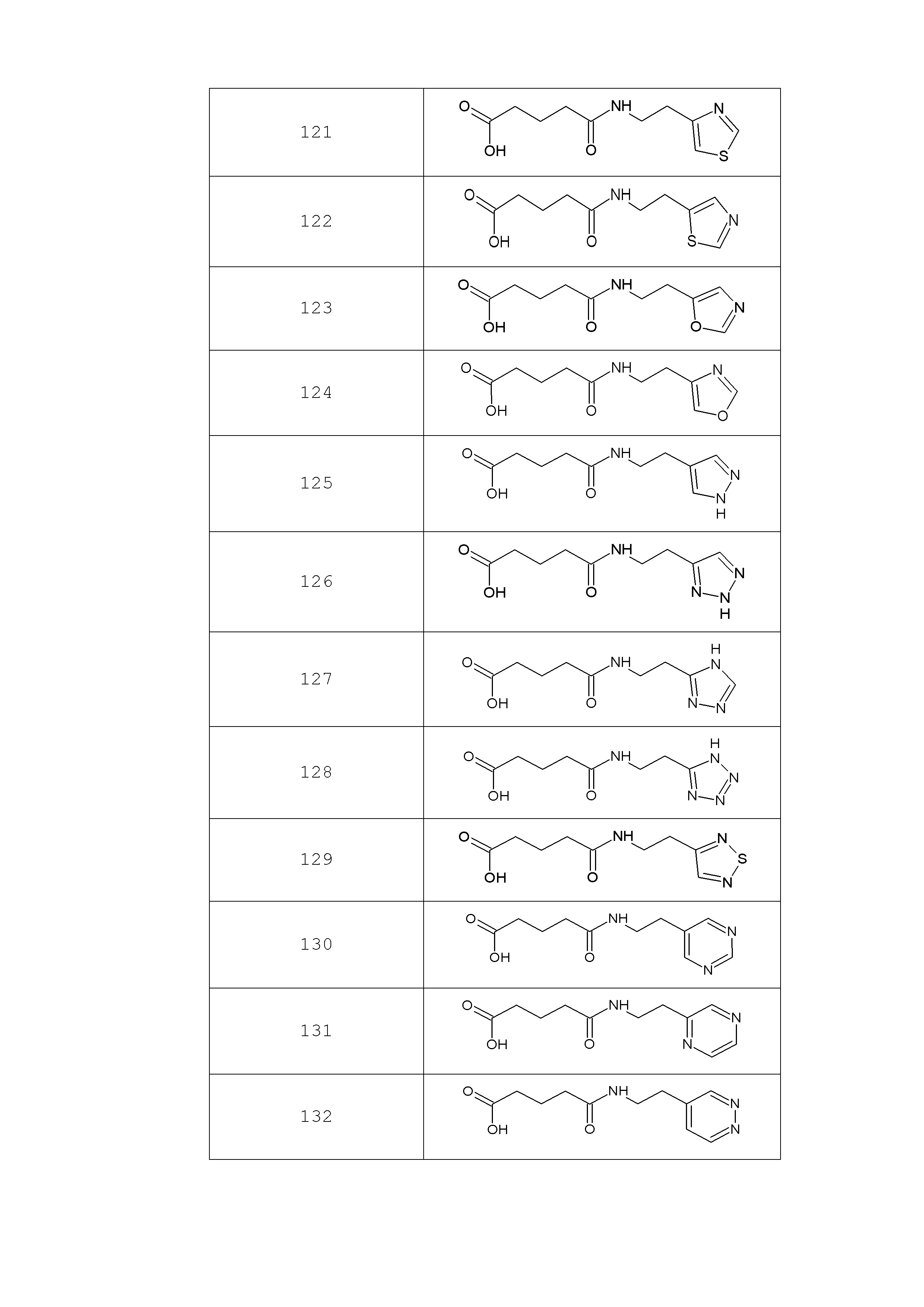
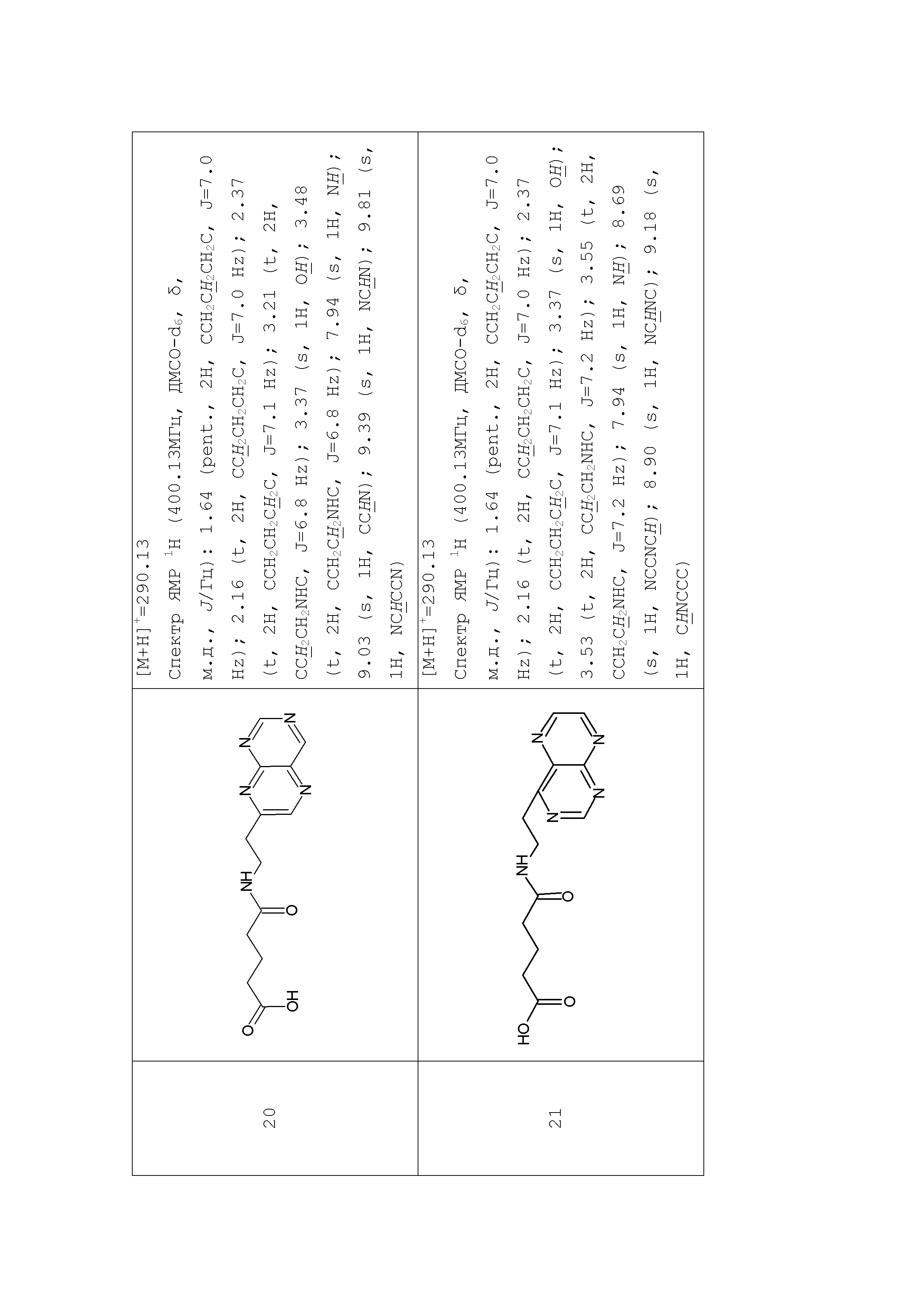
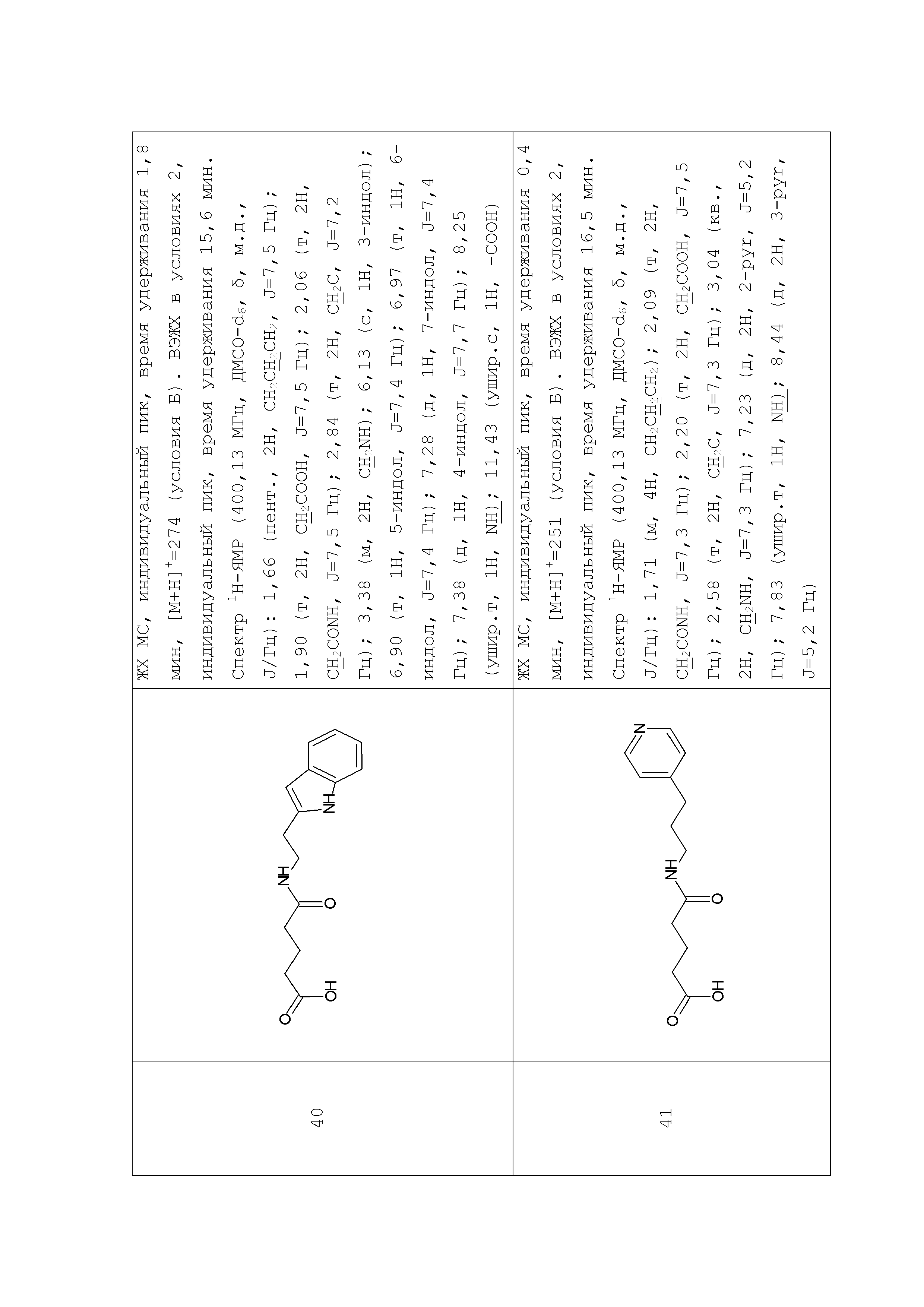
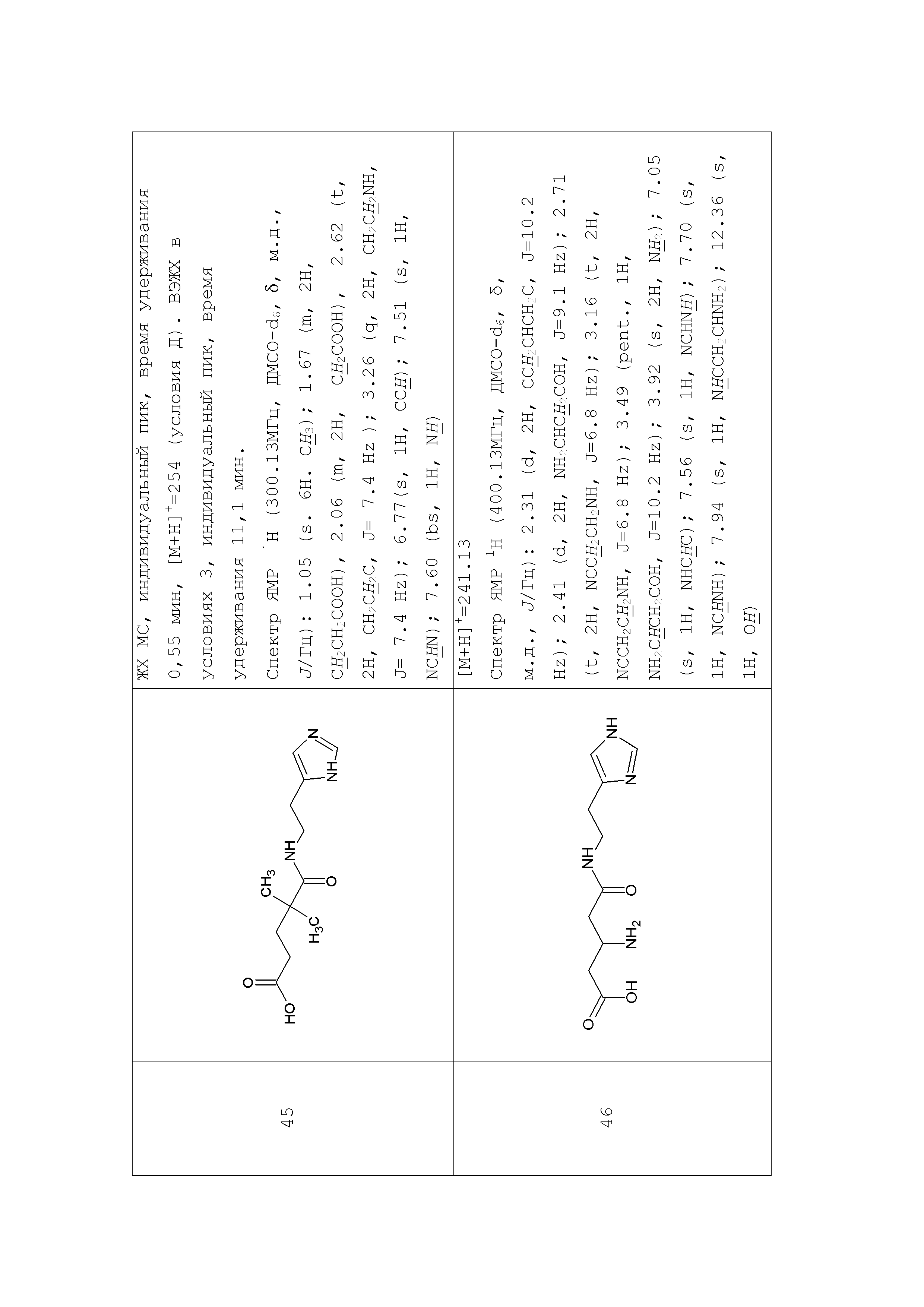
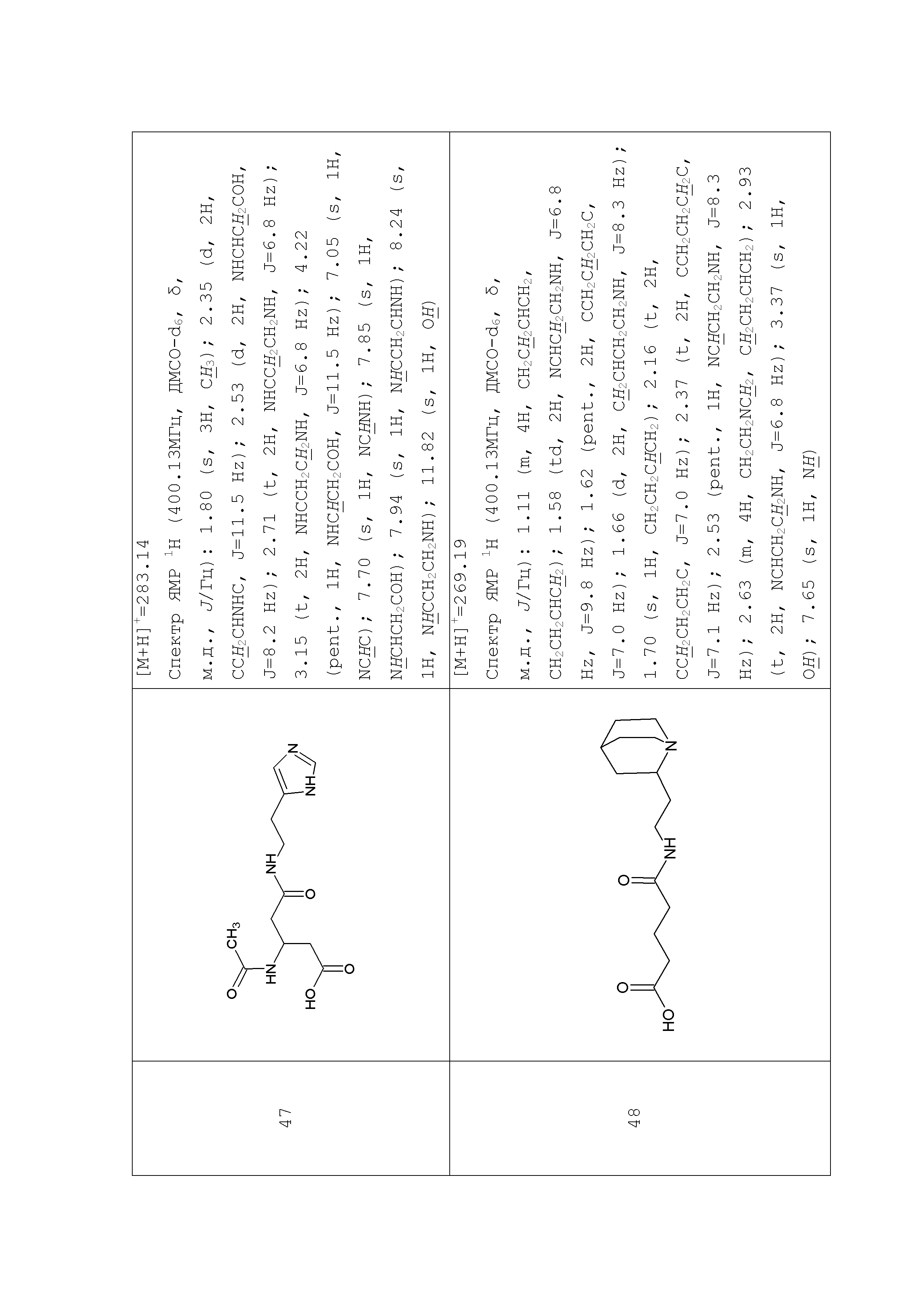
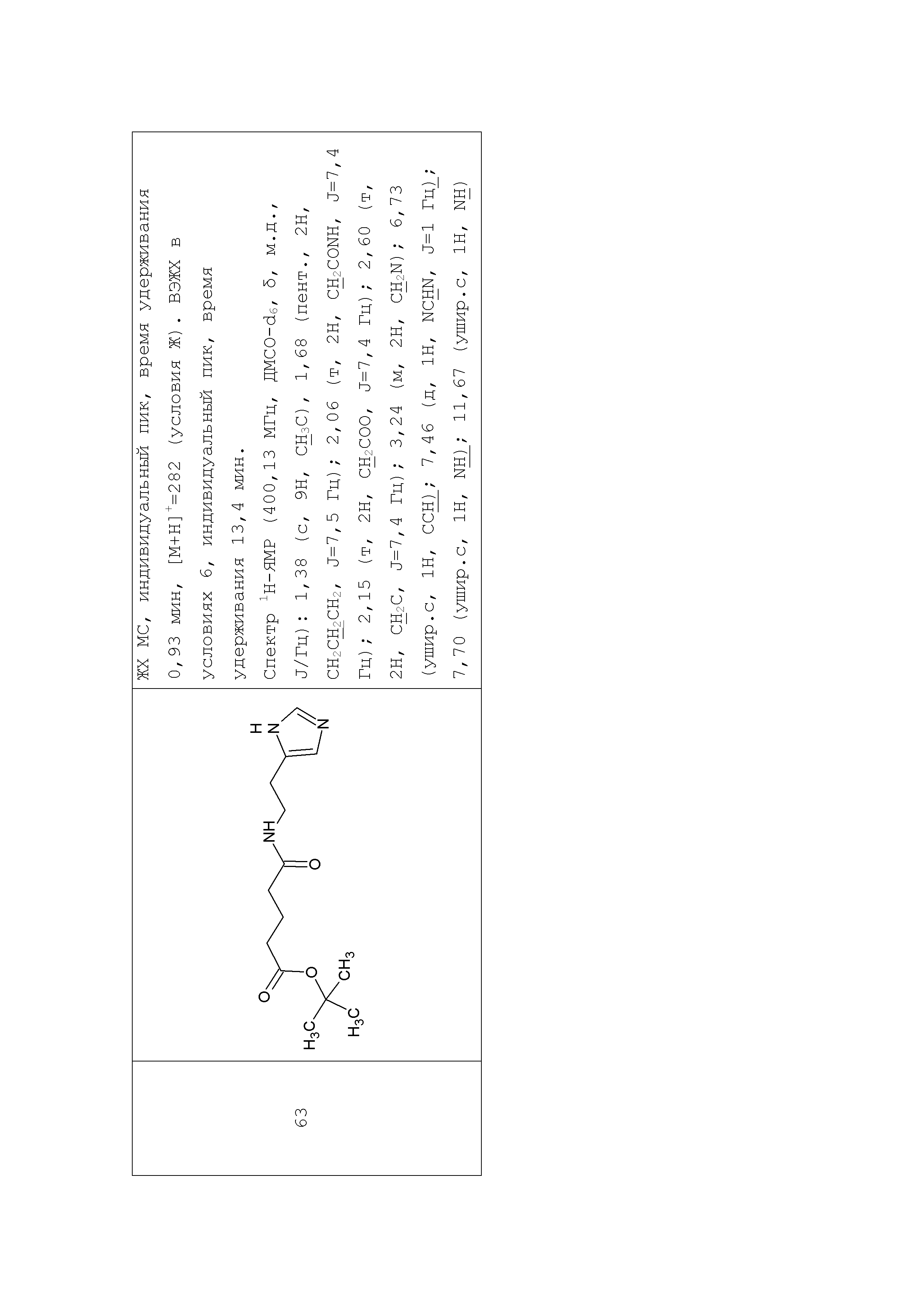
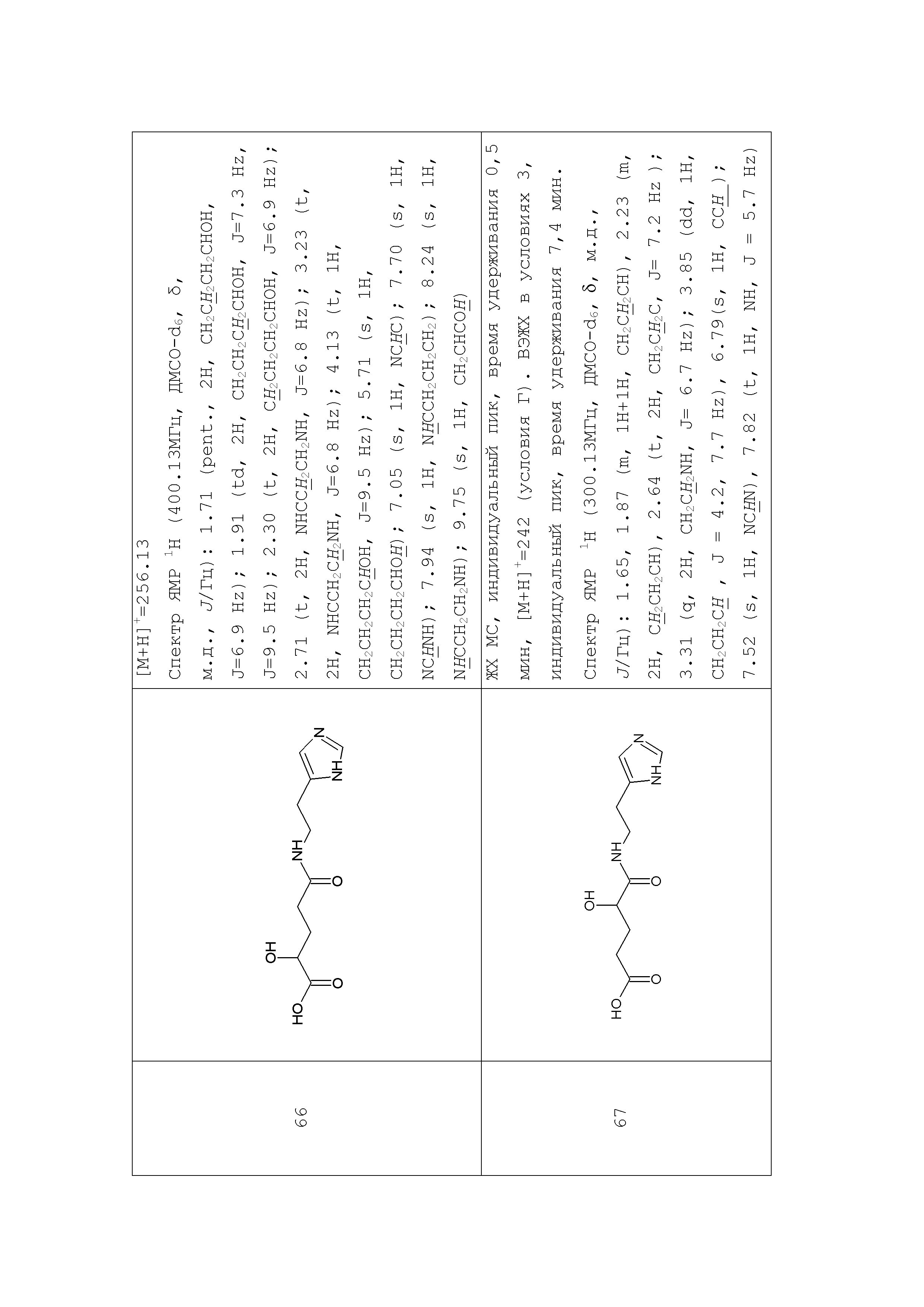
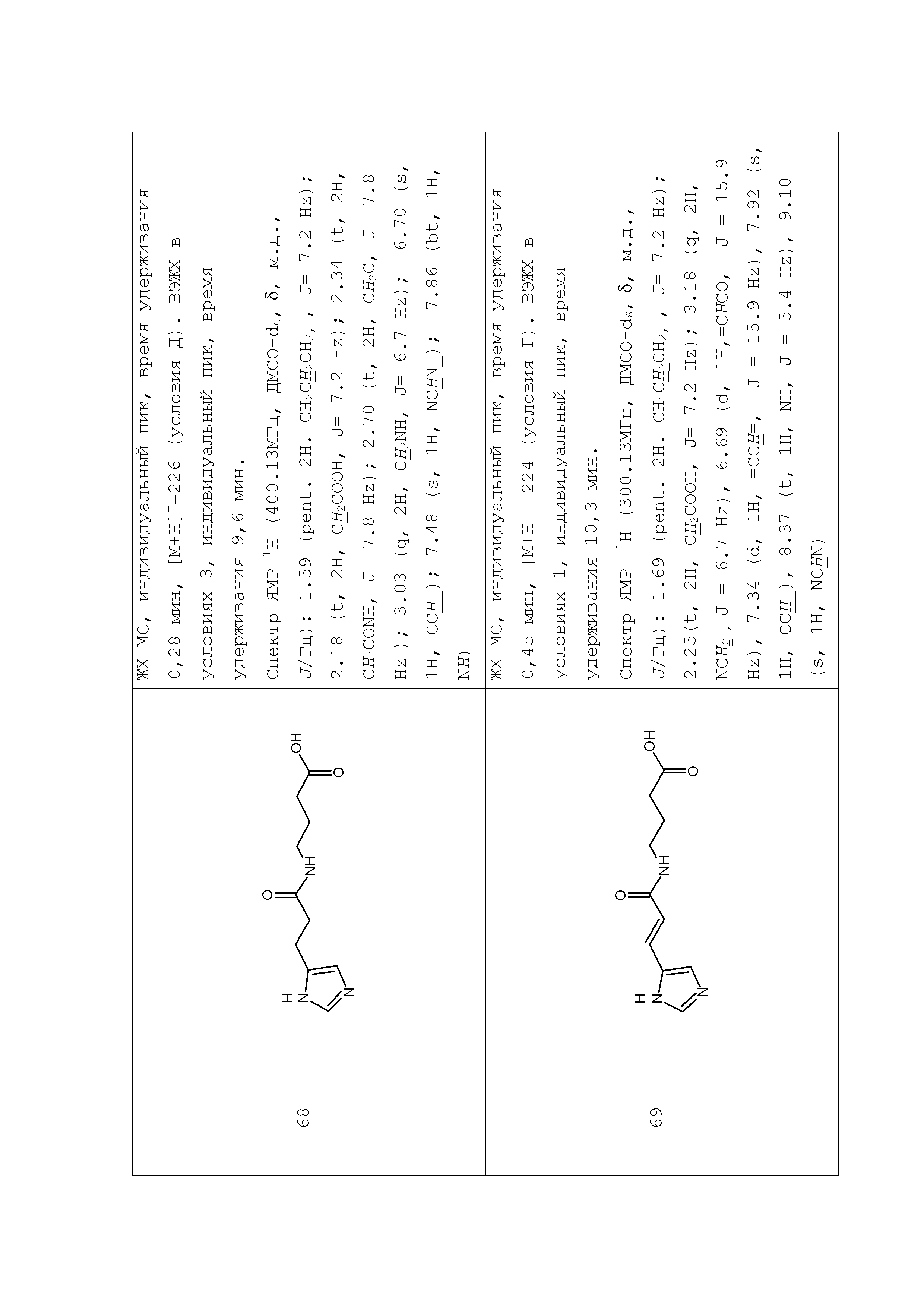
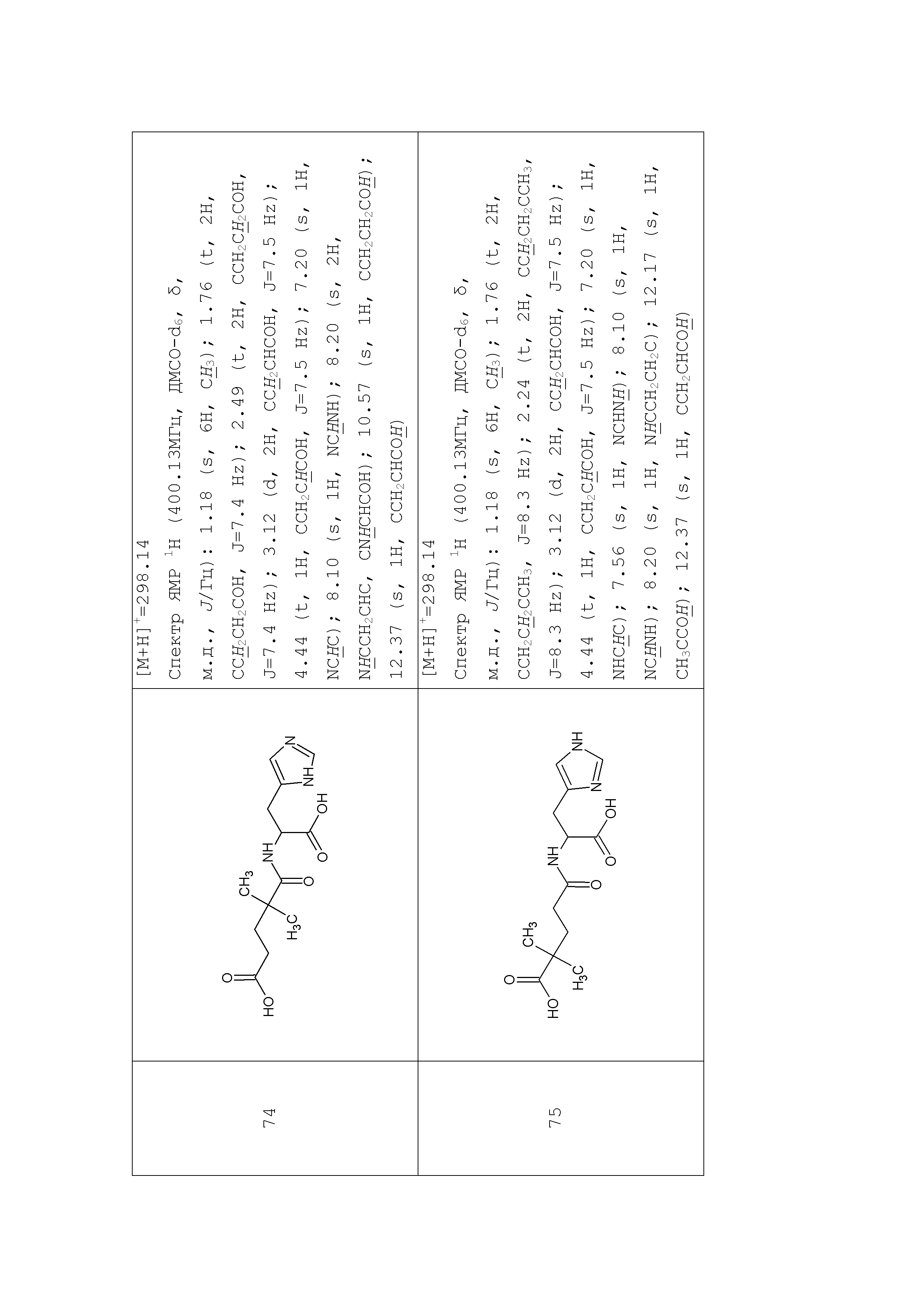
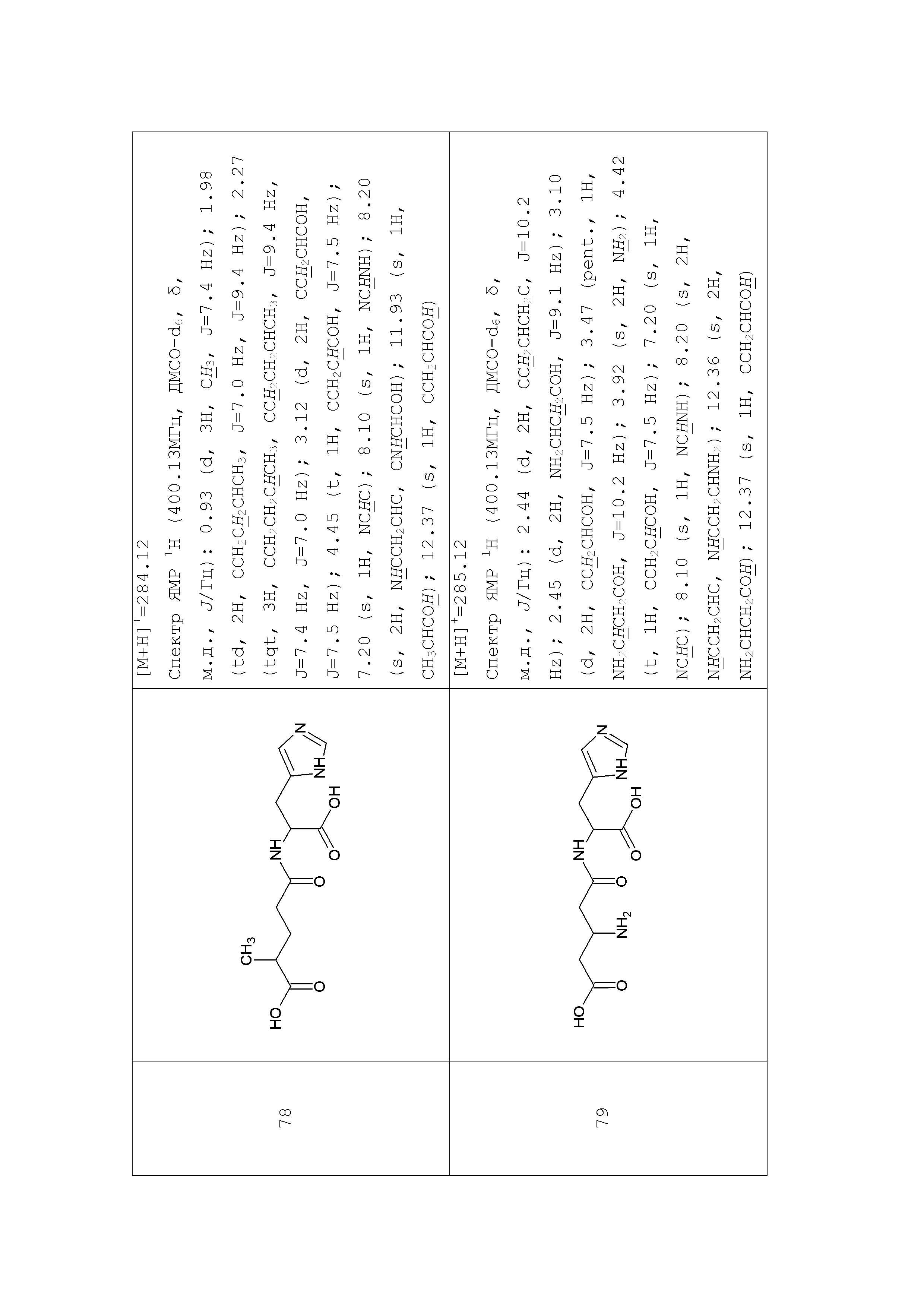
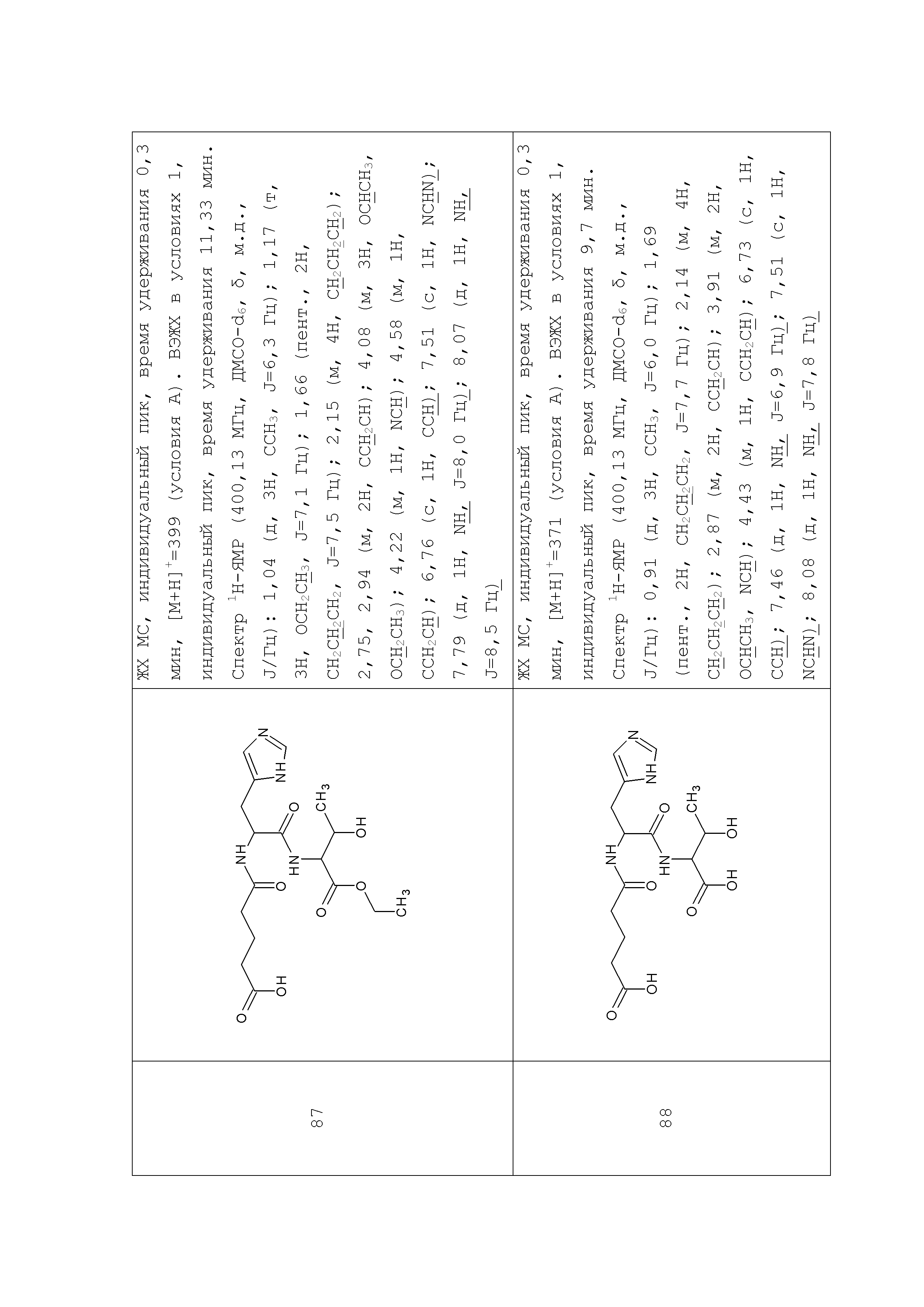
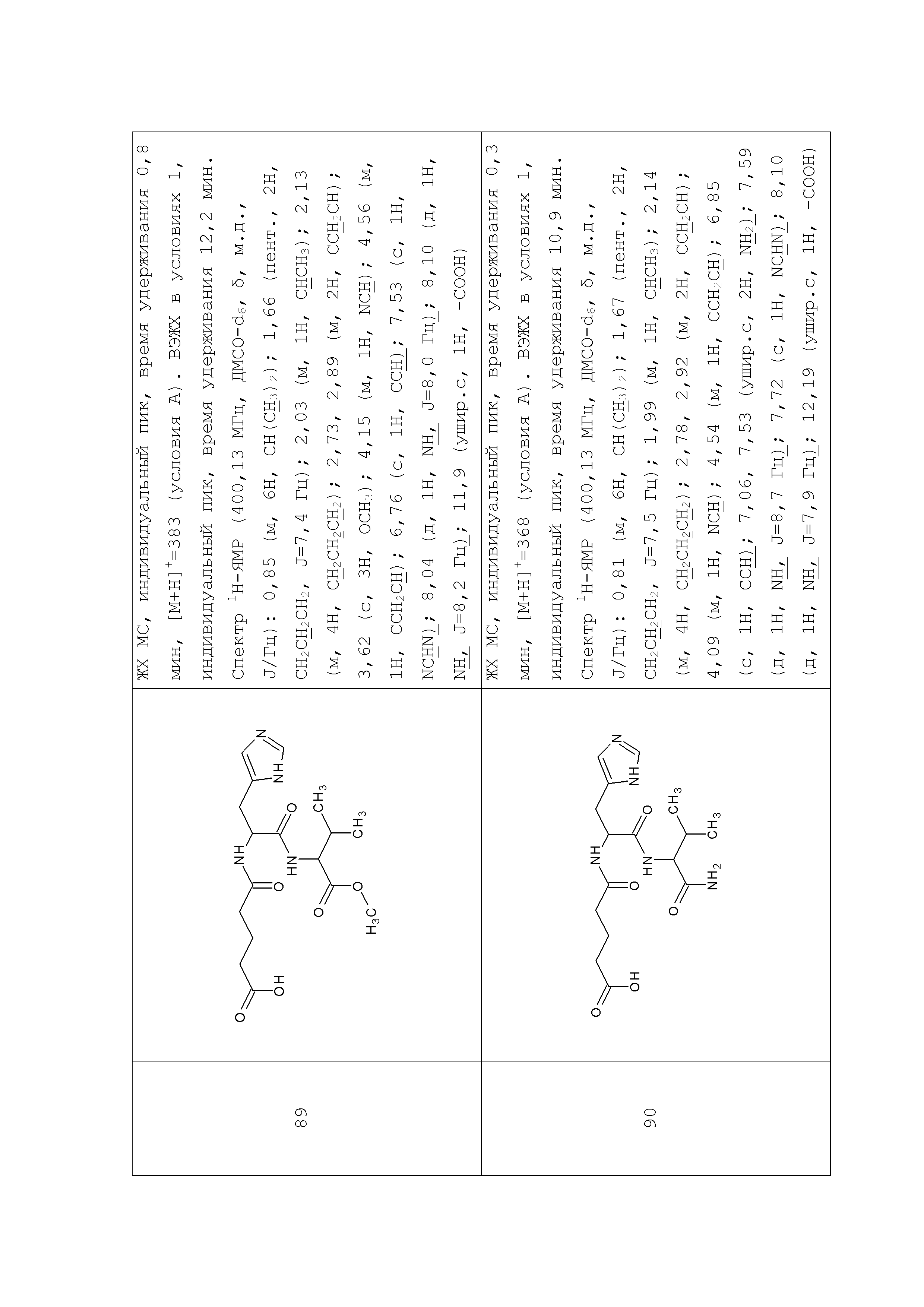
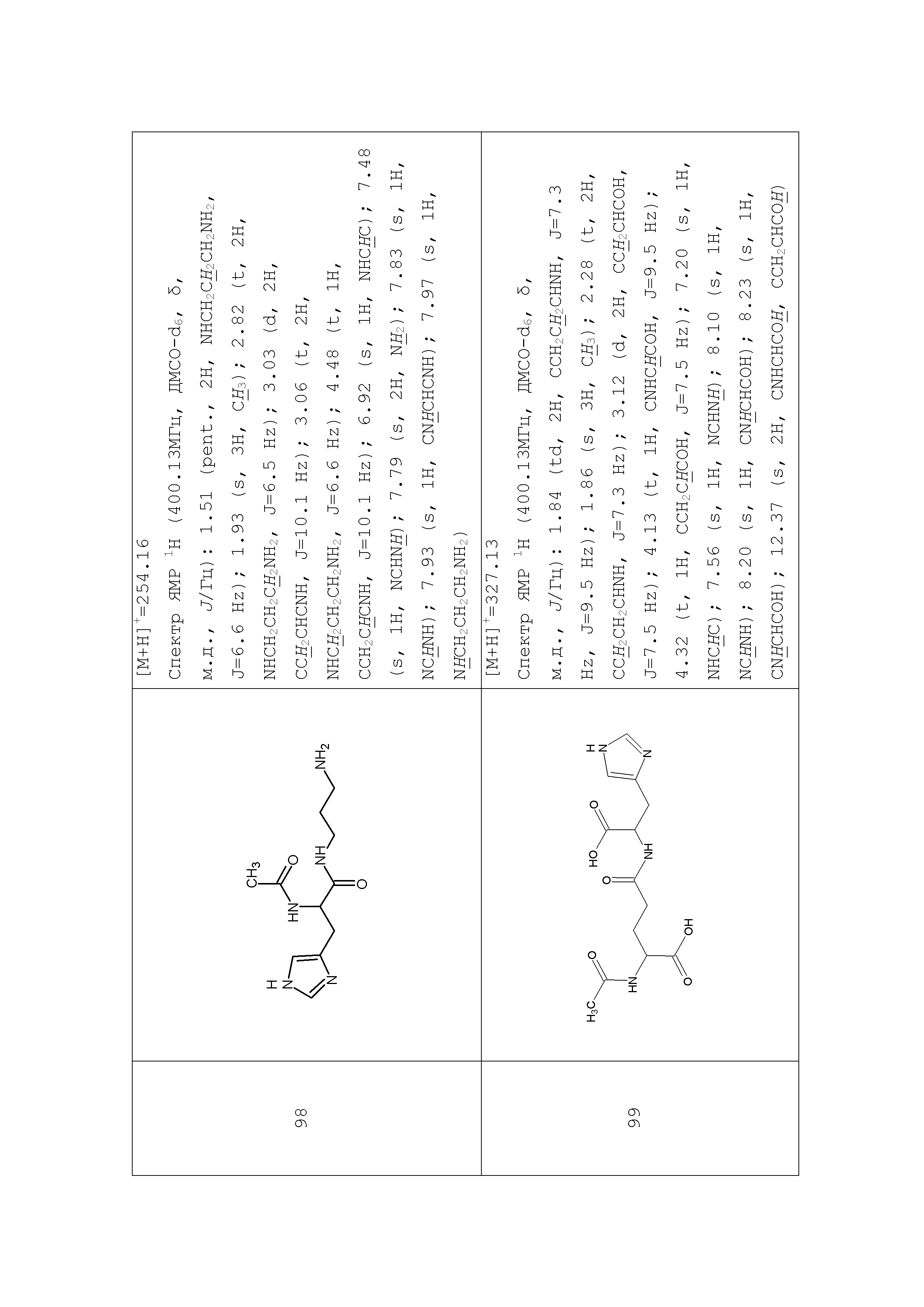
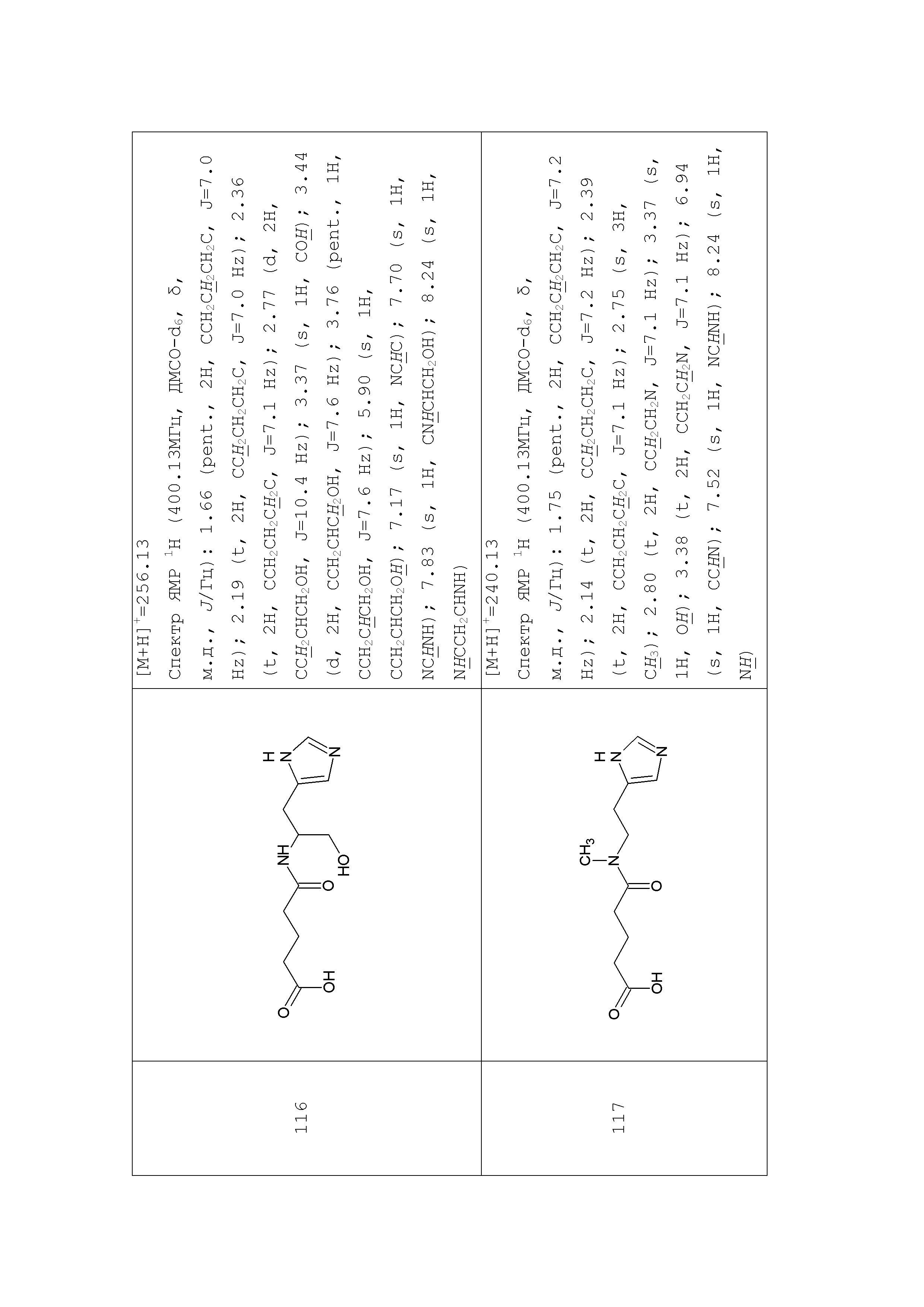
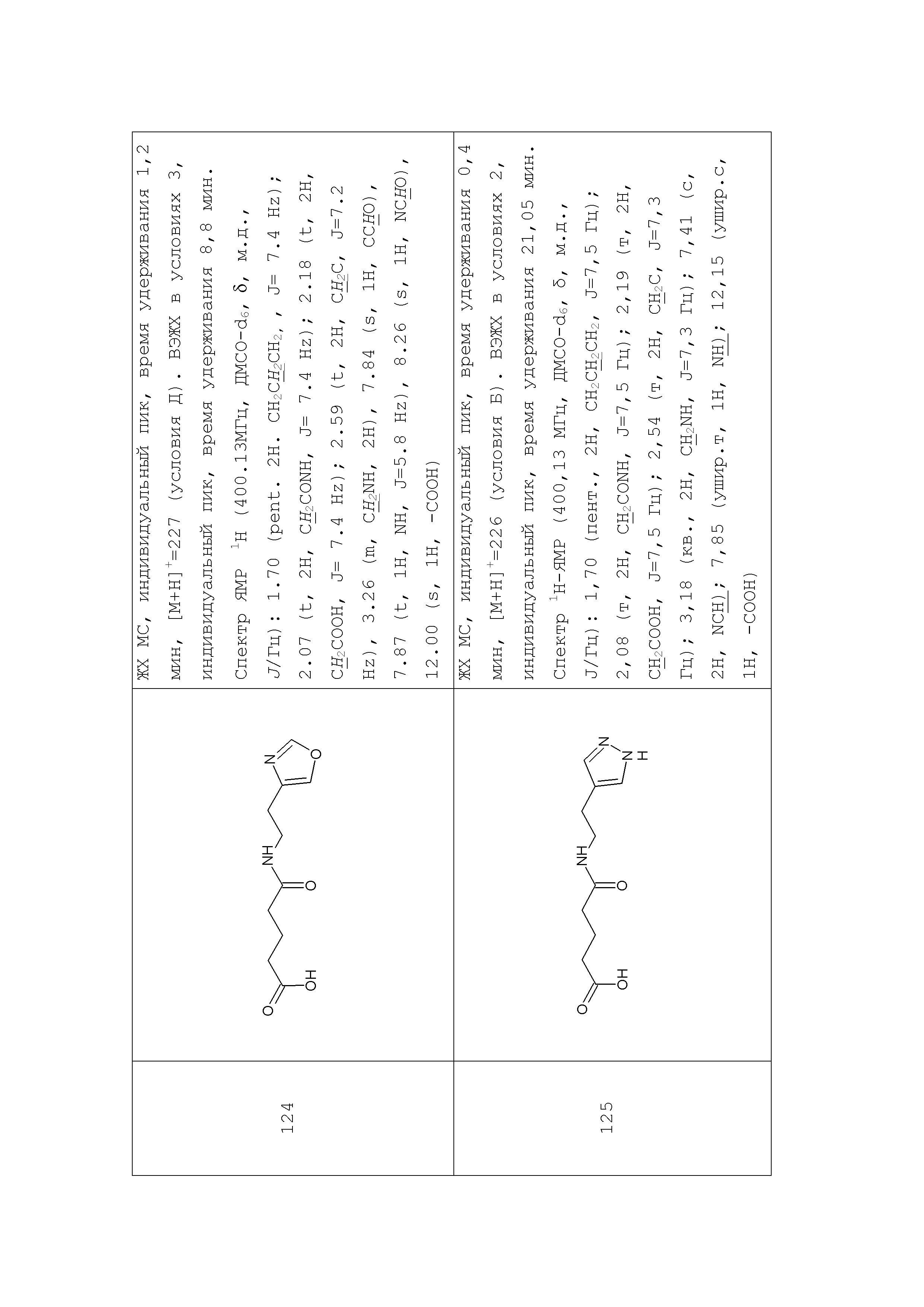
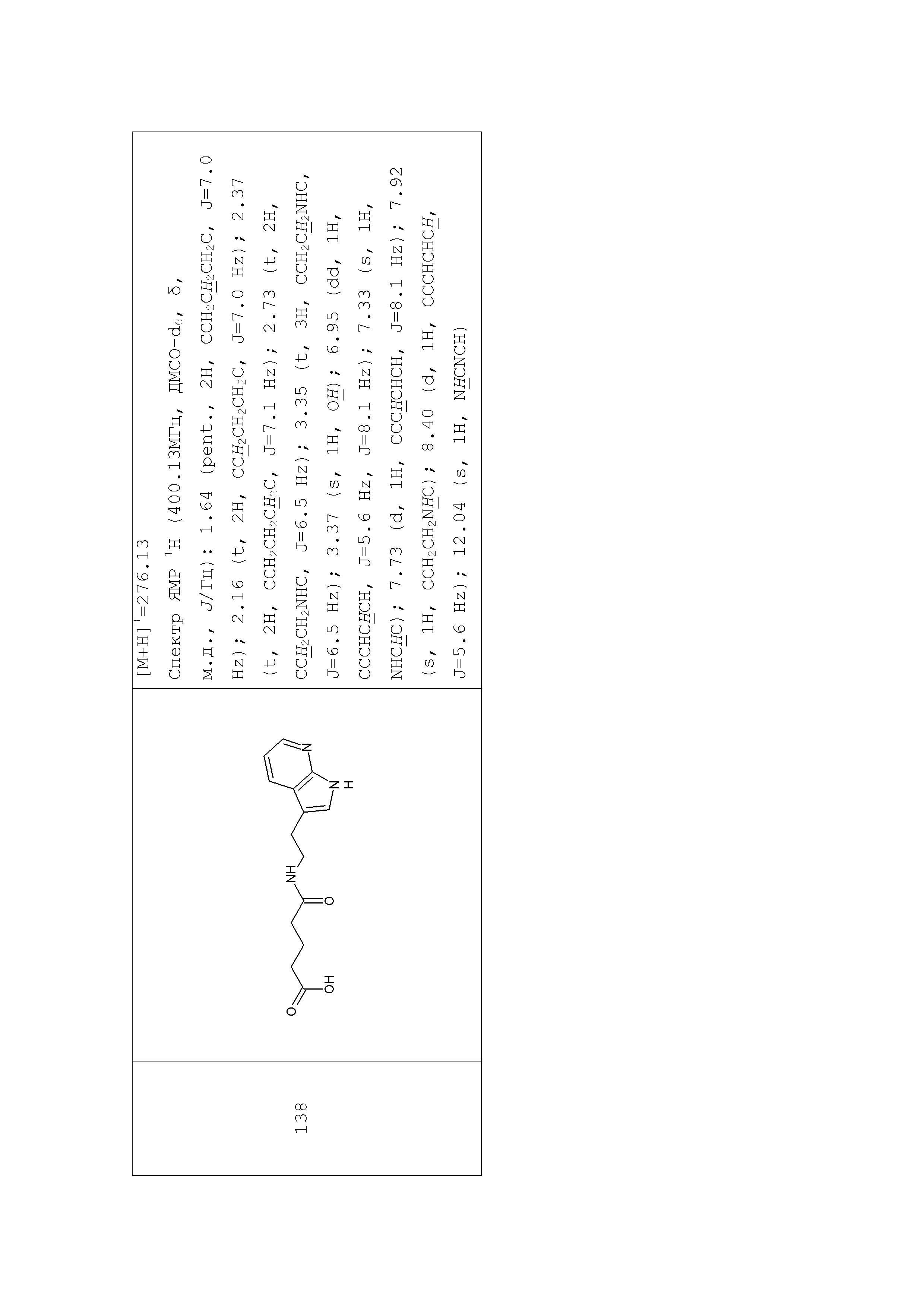
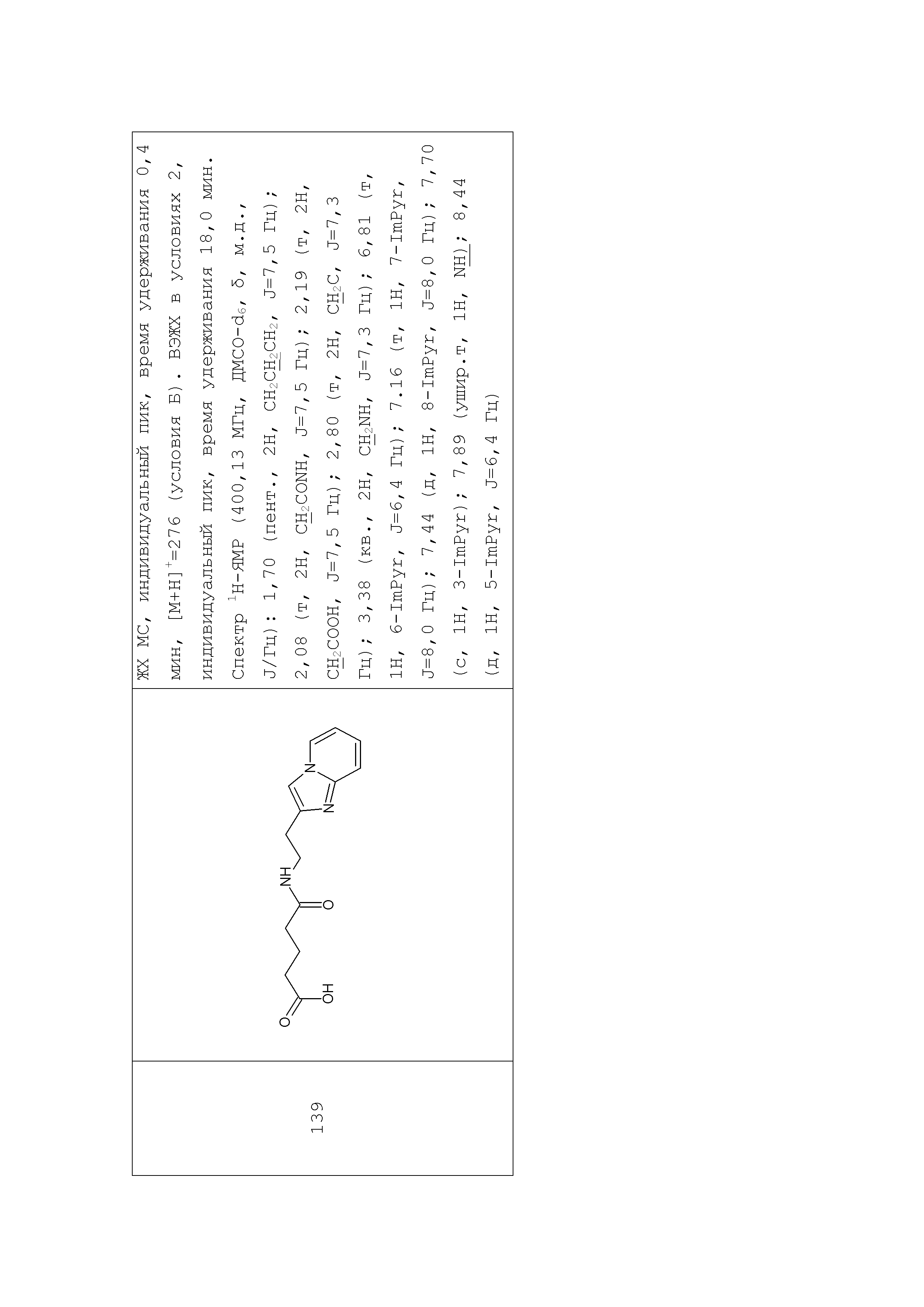
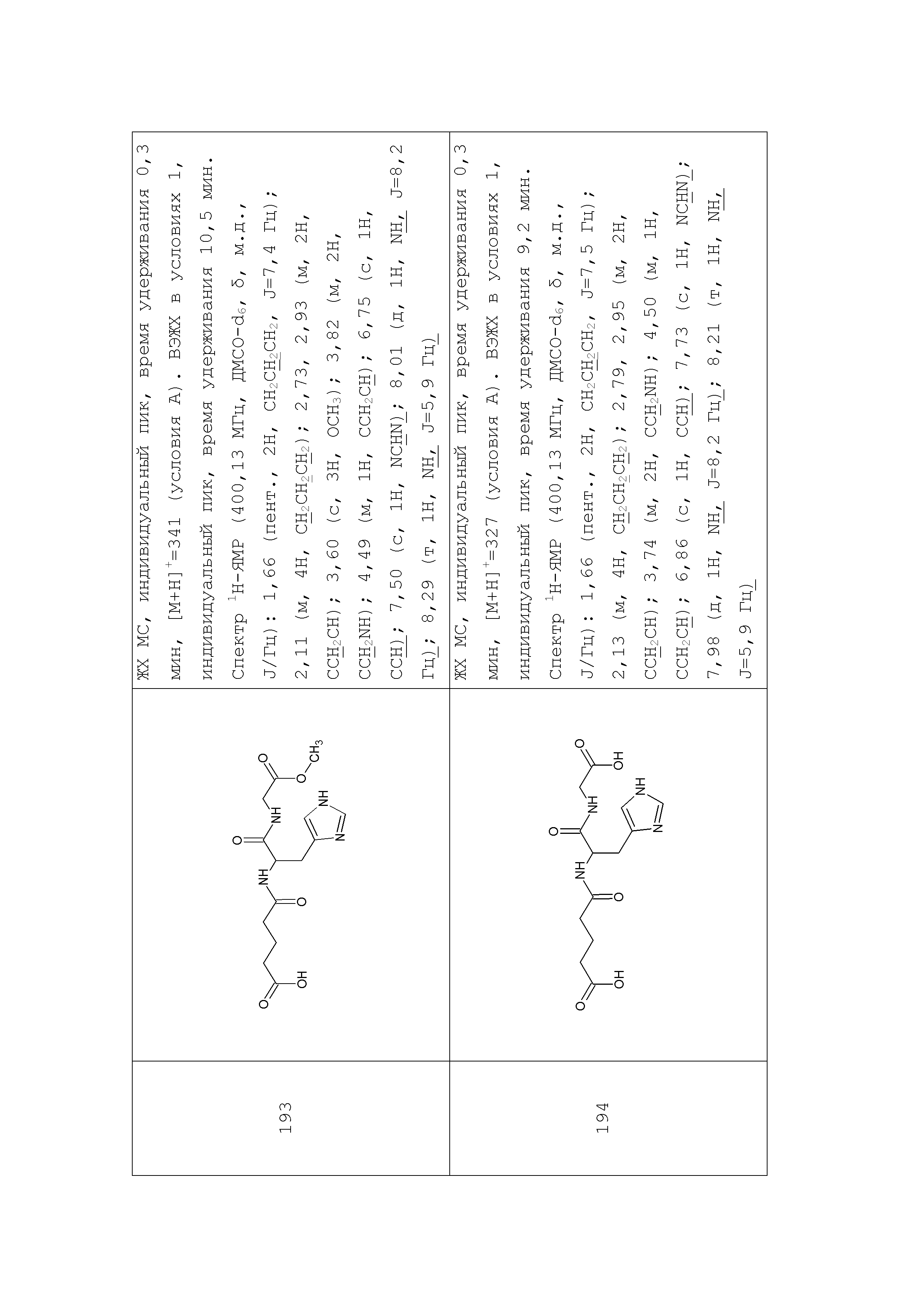
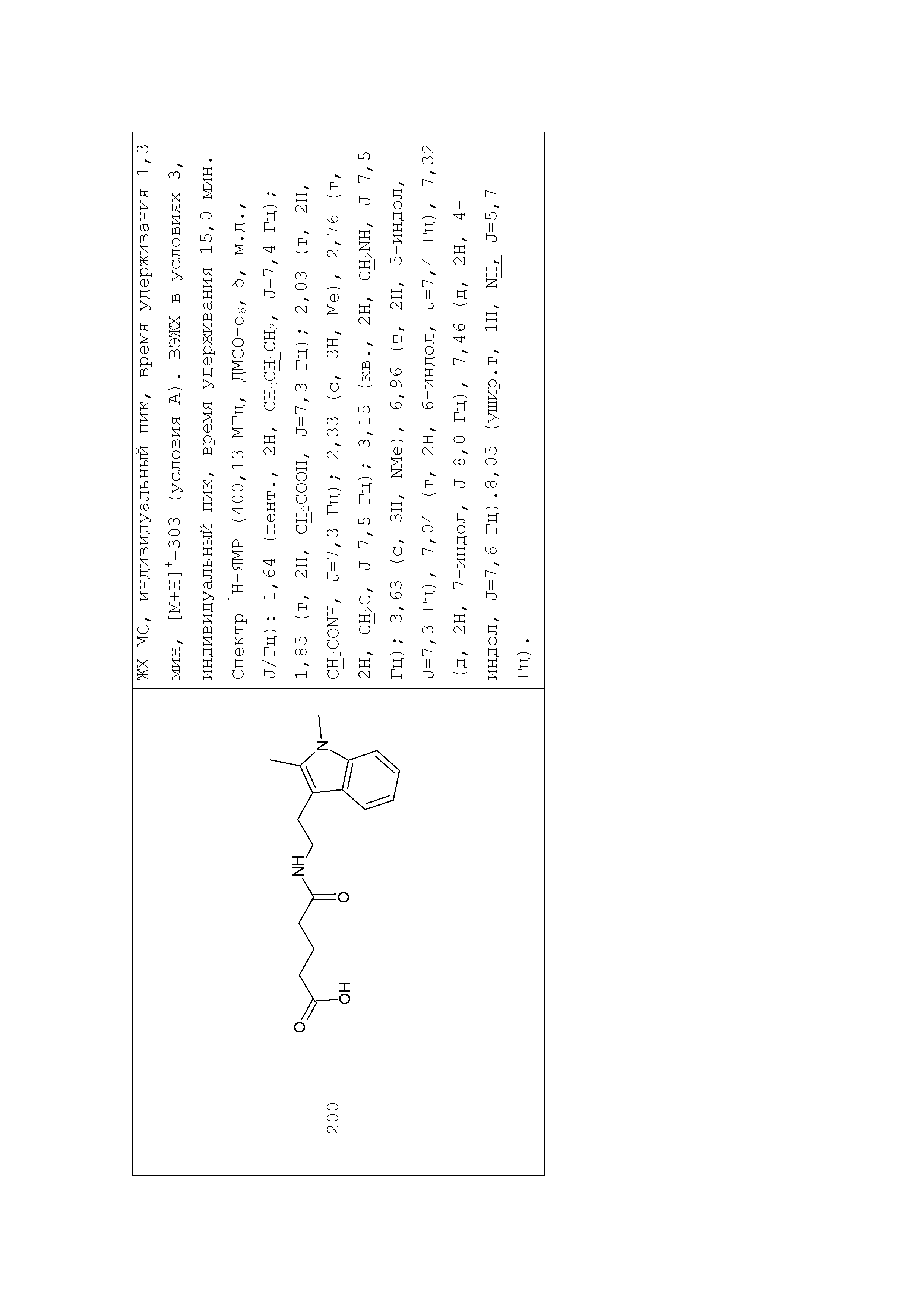
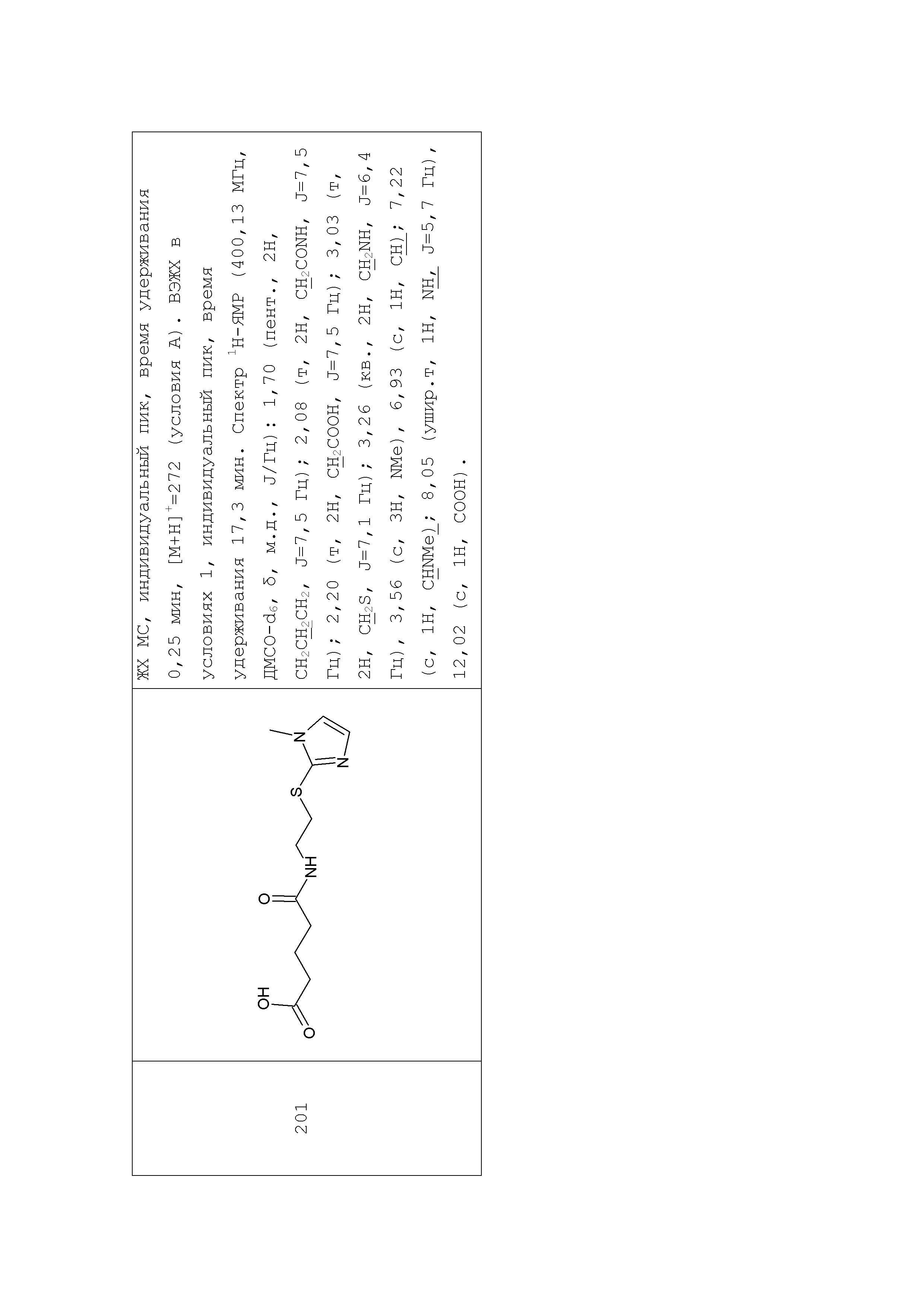
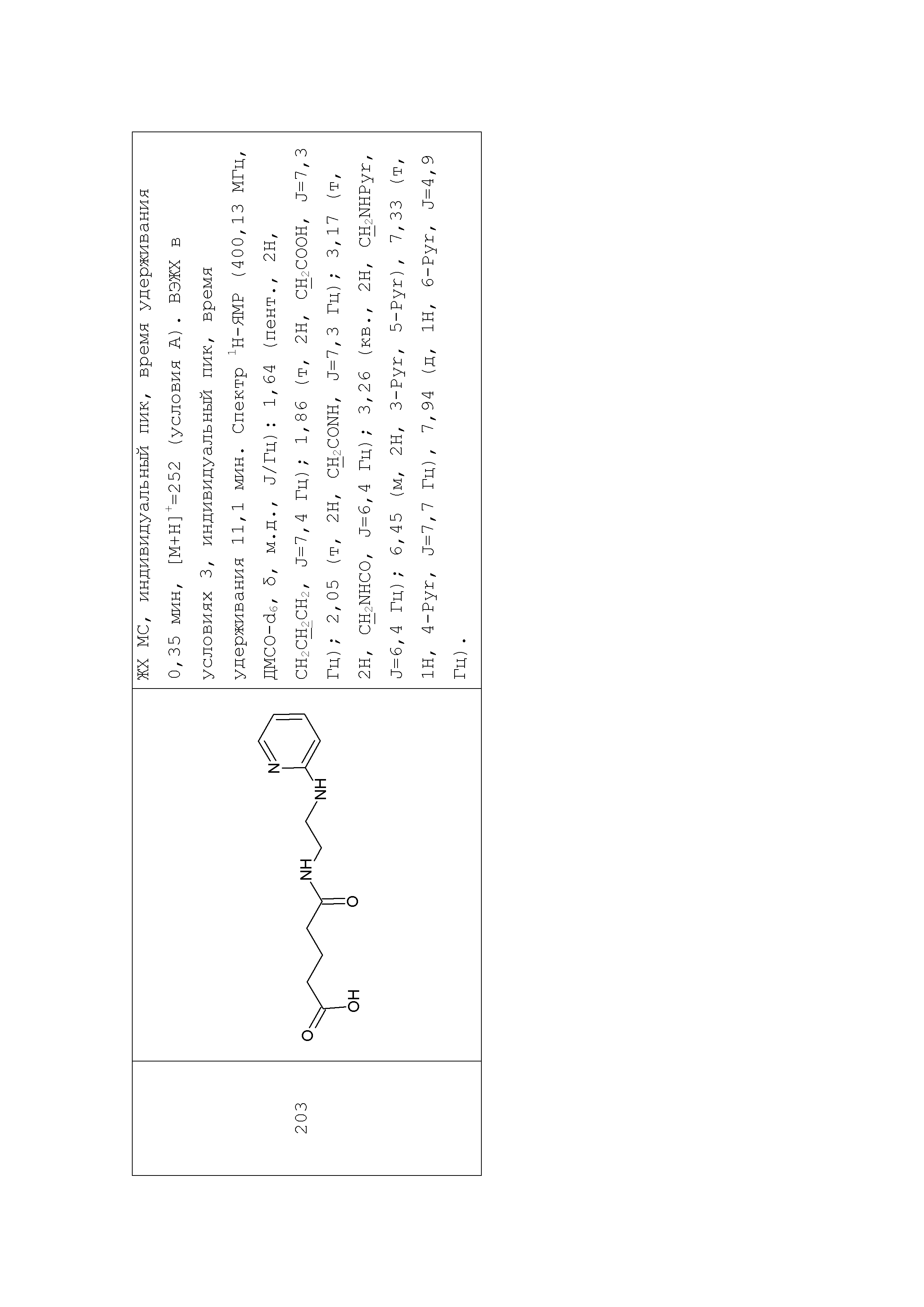
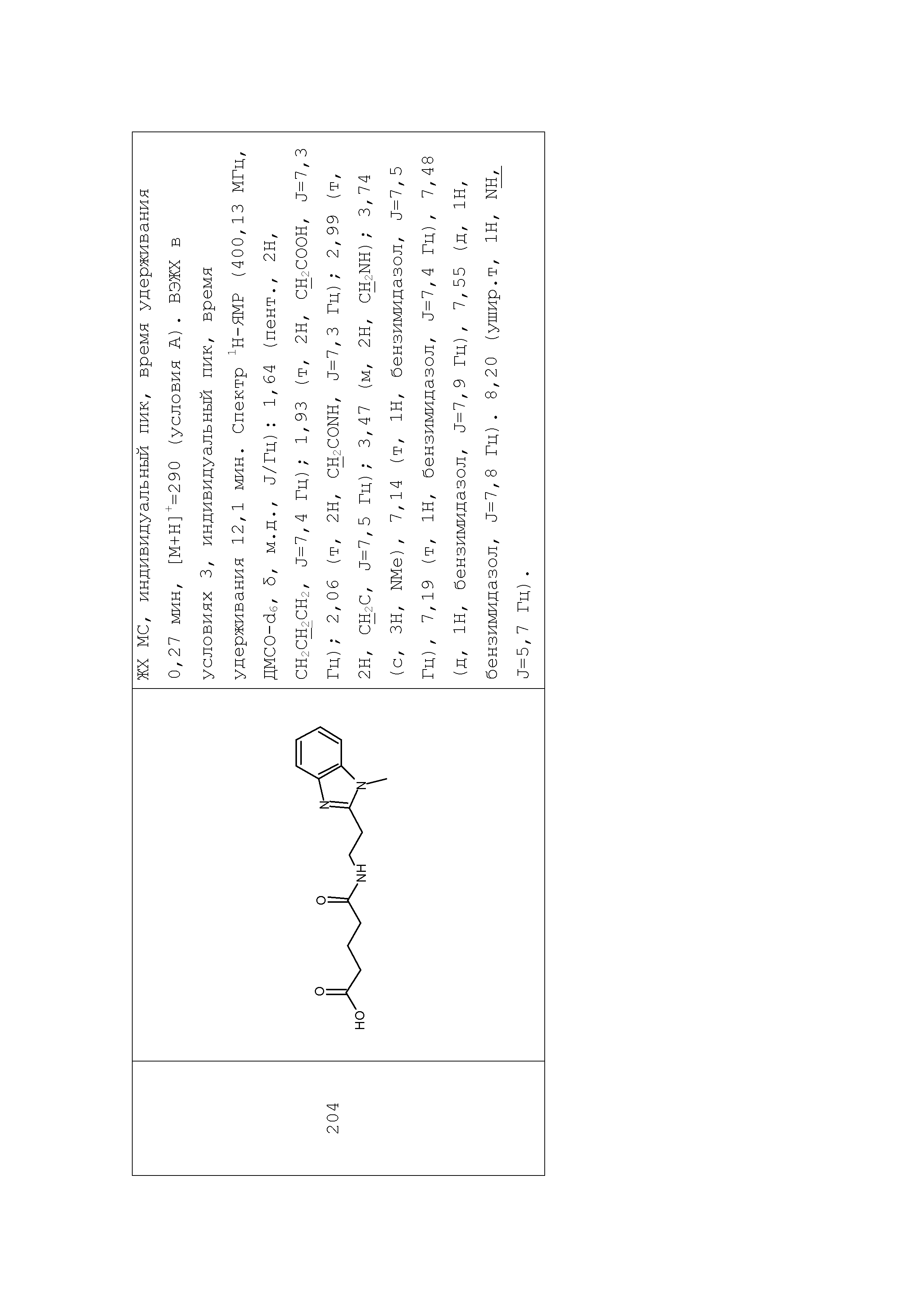
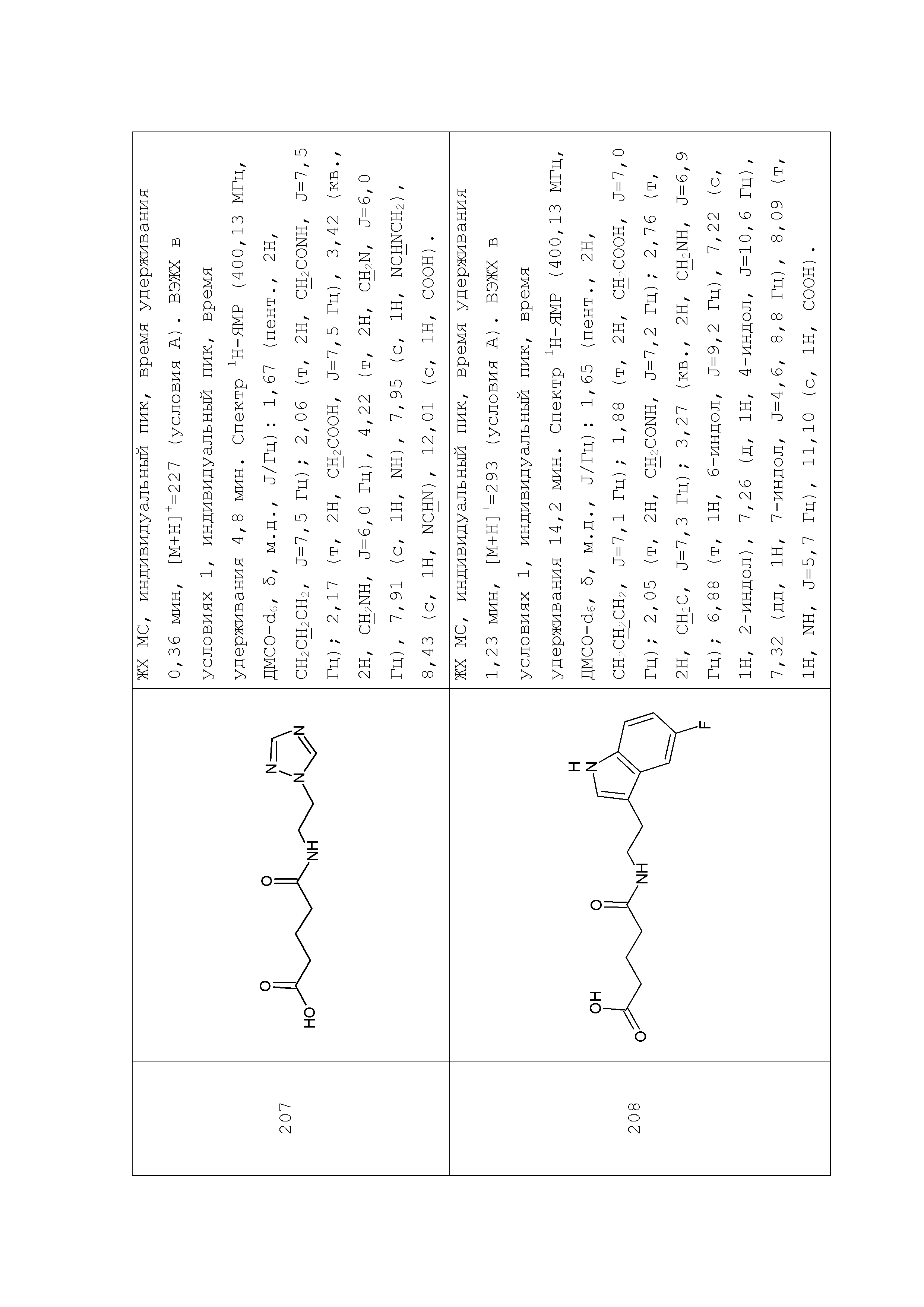
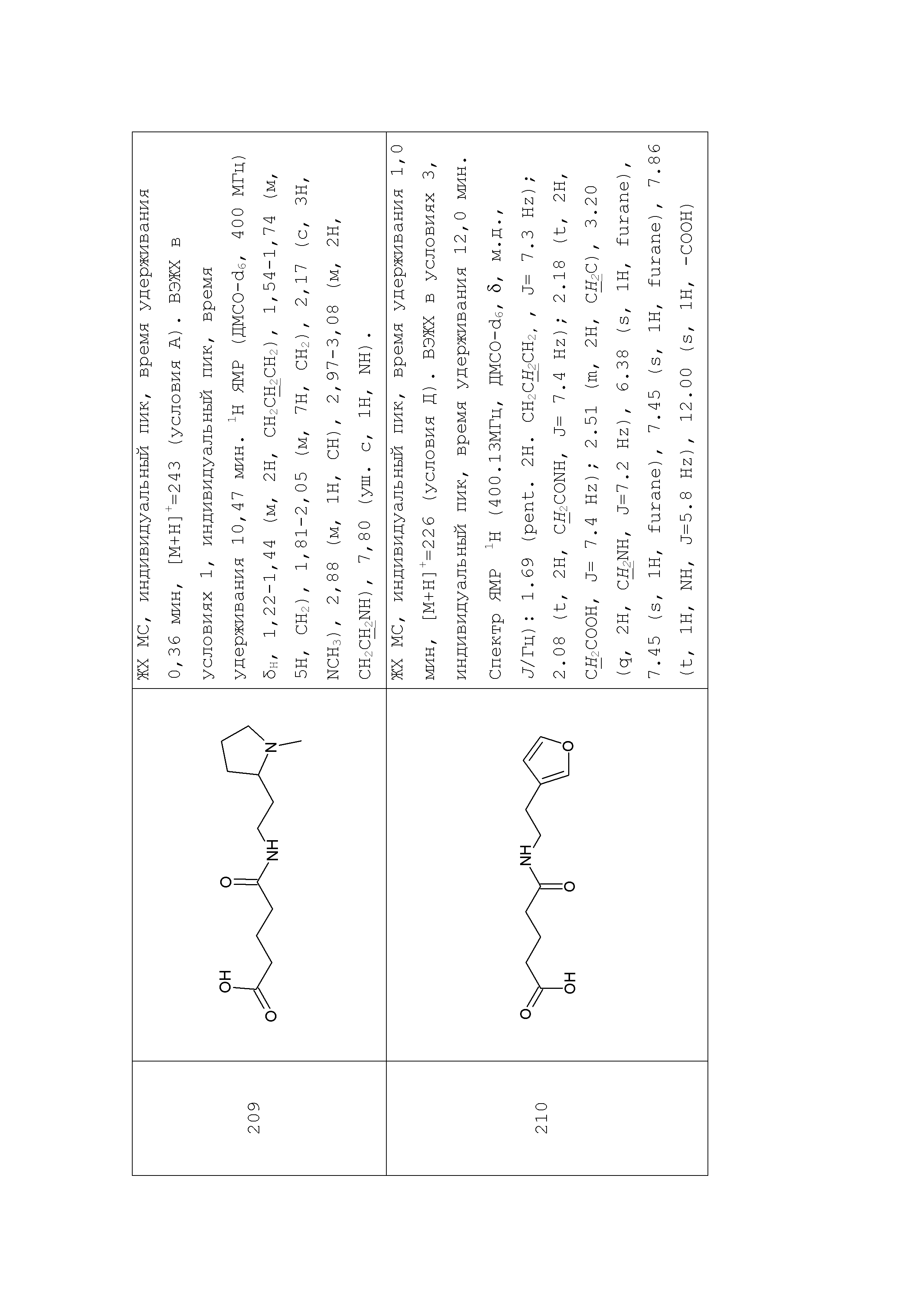
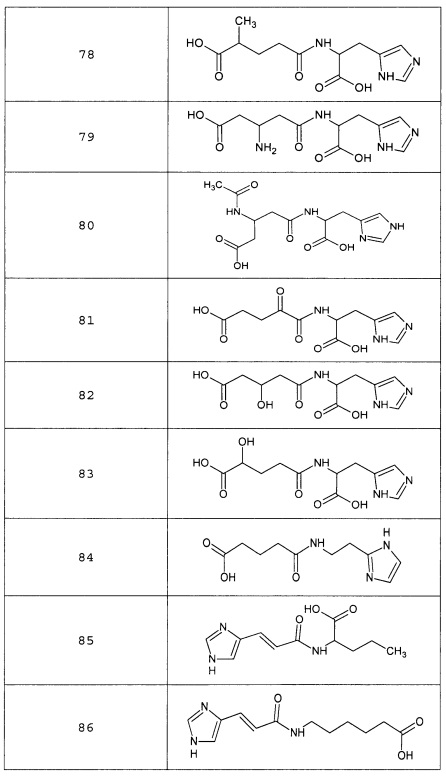
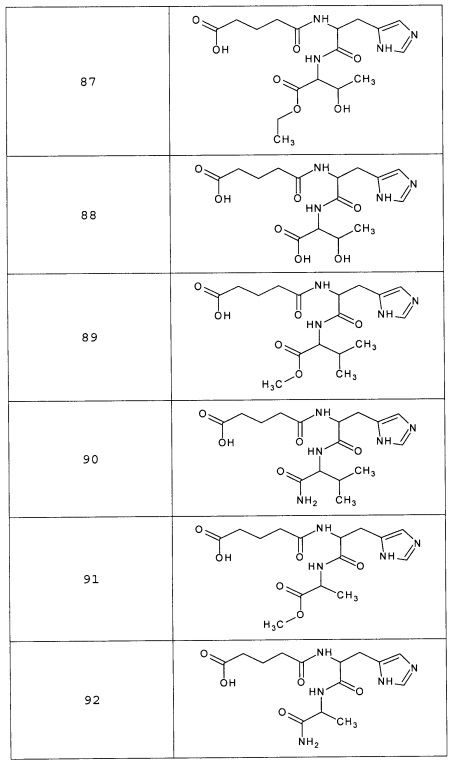
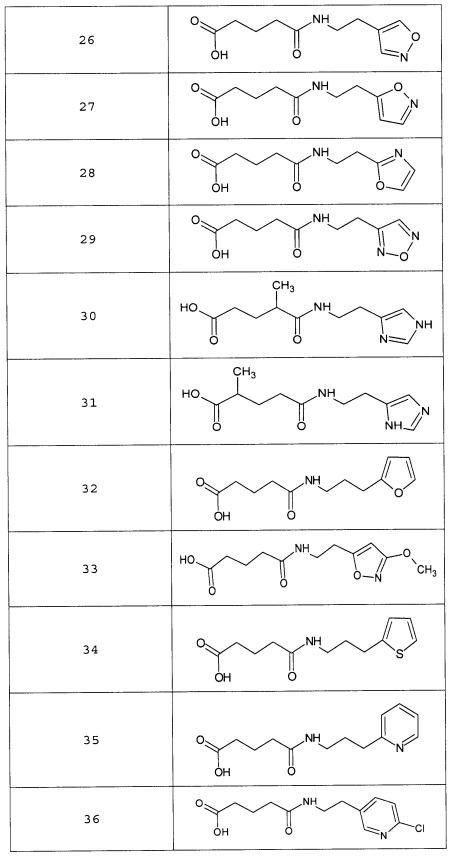
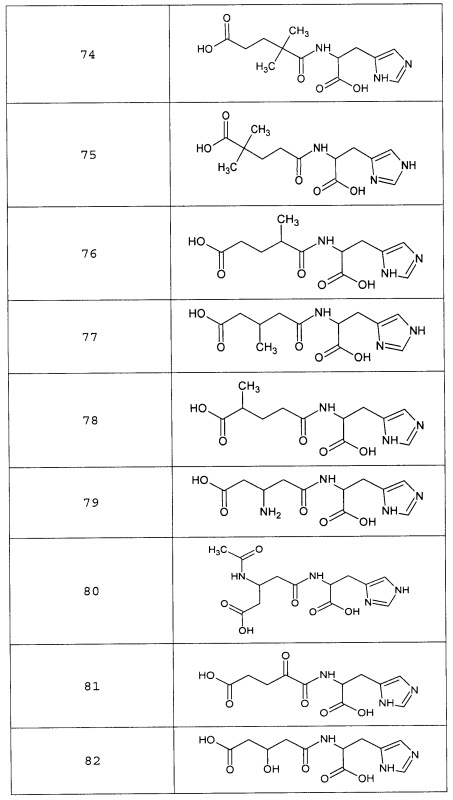
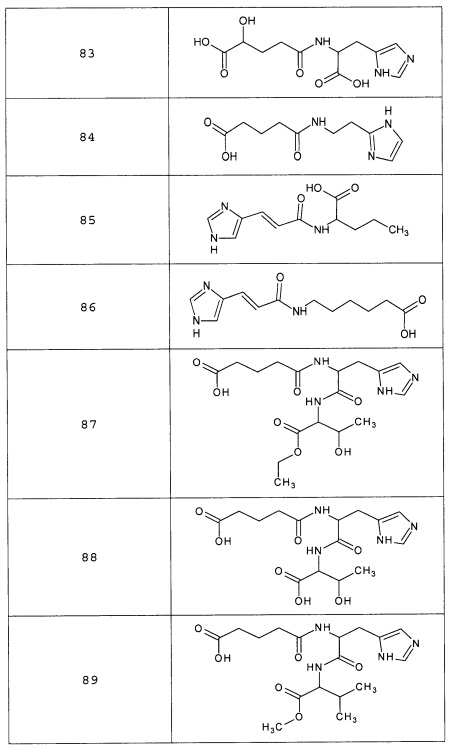
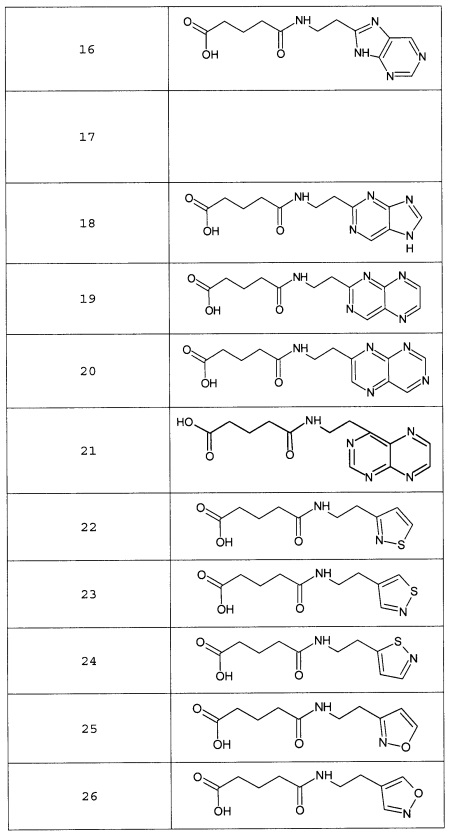
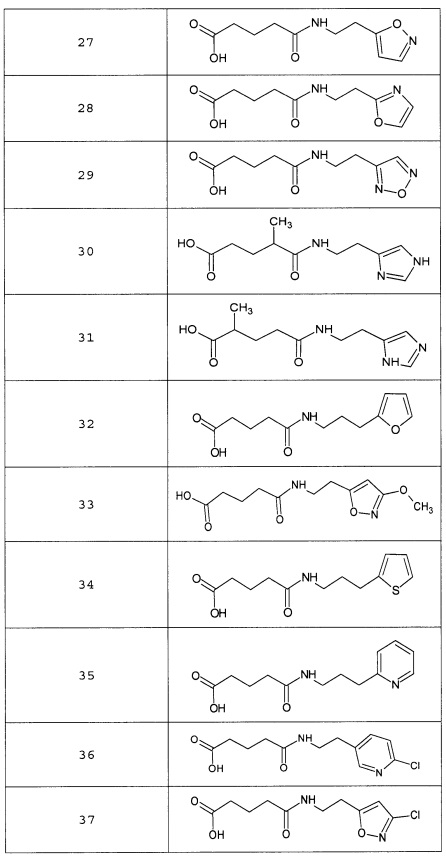
Аналогично с приведенными методиками получены следующие соединения (без ограничения перечисленными), приведенные в таблице 2:
Таблица 2 смотри в графической части.
Тесты на биологическую активность
Далее приведено детальное описание экспериментальных примеров, подтверждающих эффективность соединений общей формулы I для профилактики и лечения заболеваний в соответствии с данным изобретением, где приведенные примеры не предназначены для ограничения объема изобретения.
Пример 1
Противовирусное действие соединений общей формулы I в отношении вируса Коксаки in vivo
Для проведения исследований был использован трипсинзависимый штамм HСXV А2, предварительно адаптированный и вызывающий гибель мышей от коксаки-вирусной инфекции.
Эксперимент проводили на белых мышах самцах, весом 6-7 г. Животных инфицировали вирусом внутримышечно в объеме 0,1 мл/мышь. В опыте использовали дозу для заражения 10LD50, вызывающую летальность у мышей.
О способности соединений оказывать лечебный эффект судили по гибели инфицированных вирусом HCXV А2 мышей в опытной группе по сравнению с группой животных, не получавших лечение.
Исследуемые соединения и плацебо вводили животным перорально по лечебной схеме. В качестве плацебо мышам вводили физиологический раствор. Отрицательным контролем служили интактные животные, которые содержались в тех же условиях, что и опытные группы в отдельном помещении.
Для проведения эксперимента были сформированы группы по 14-15 мышей в каждой. Соединения вводились в дозе 30 мг/кг массы тела. Исследуемые вещества вводили перорально, 1 раз в сутки, в течение 7 дней (первое введение - через 24 часа после инфицирования). Наблюдение за животными осуществляли на протяжении 15 дней, ежедневно взвешивая и регистрируя смертность животных.
Соединения общей формулы I оказали защитное действие в отношении экспериментальной Коксаки-вирусной инфекции, снижая гибель животных и увеличивая среднюю продолжительность жизни животных. Данные по некоторым конкретным соединениям формулы I (без ограничения указанными) представлены в таблице 3.
Противовирусная активность исследованных соединений, описанная в примере, свидетельствует о том, что данные химические соединения могут быть использованы в качестве эффективных лекарственных средств при Коксаки энтеровирусной инфекции.
Таблица 3
Эффективность соединений общей формулы I в отношении Коксаки A2-вирусной инфекции на модели у мышей
Пример 2
Противовирусное действие соединений общей формулы I в отношении адаптированного к мышам РС вируса
Для определения противовирусной эффективности химических соединений в отношении РСВ на экспериментальной модели мышей in vivo использовали вирус человека hRSV, предварительно адаптированный к росту в легких мышей. Животных инфицировали вирусом в дозе 5,0 logTCID50интраназально под легким эфирным наркозом в объеме 0,05 мл/мышь. Исследуемые соединения вводили животным перорально 1 раз в сутки в течение 5 дней по лечебной схеме в дозе 30 мг/кг. Первое введение осуществляли через 24 часа после инфицирования. В качестве плацебо мышам вводили физиологический раствор. Отрицательным контролем служили интактные животные, которые содержались в отдельном помещении в тех же условиях, что и опытные группы. Экспериментальные группы содержали по 12 мышей. Препаратом сравнения служил Рибавирин в дозе 40 мг/кг.
Оценку противовирусной активности исследуемых соединений проводили по эффективности предотвращения снижения массы тела и по подавлению репродукции hRSV в легких инфицированных мышей путем измерения вирусного титра в опытных группах по сравнению с контрольной на 5-е и 7-е сутки после инфицирования.
Результаты определения массы тела животных по некоторым конкретным соединениям формулы I (без ограничения указанными) представлены в таблице 4. В группе вирусного контроля отмечали достоверное снижение массы тела мышей по сравнению с интактными животными. Противовирусная активность соединений общей формулы I проявилась в увеличении массы тела мышей по сравнению с контрольными животными.
Кроме того, терапевтическое действие соединений общей формулы I оценивали по способности подавлять репродукцию вируса hRSV в легких мышей через 5 и 7 суток после инфицирования. Титр вируса определяли титрованием 10%-ой суспензии лёгких на культуре клеток Hep-2. Результат учитывали через 2 суток инкубации при 37°С по ЦПД. Результаты определения инфекционной активности hRSV в суспензиях легких мышей в культуре клеток Hep-2 после применения исследуемых соединений и препарата сравнения представлены в таблице 5. Введение соединений общей формулы I животным приводило к снижению инфекционной активности hRSV.
Изучение противовирусного действия соединений общей формулы I на модели hRSV-инфекции у мышей показало, что заявляемые соединения предотвращают снижение массы тела и снижают репродукцию вируса в легких у животных.
Пример 3
Противовирусное действие соединений общей формулы I в отношении РС вируса на модели у мышей с угнетенной иммунной системой
Противовирусную активность химических соединений в отношении респираторно-синцитиального вируса человека (штамм А2, с инфекционным титром 5×106TCID50/мл) оценивали на модели вирусной пневмонии у мышей линии Balb/c. Вирус вводили животным интраназально в объеме 50 мкл под легким эфирным наркозом. Для угнетения иммунной реакции против РС вируса за 5 дней до инфицирования животным вводили внутрибрюшинно циклофосфан в дозе 100 мг/кг. Изучаемые соединения применяли по лечебной схеме 1 раз в сутки в дозе 30 мг/кг в течение 5 дней, начиная через 24 часа после инфицирования. Активность соединений оценивали по снижению отека инфицированного респираторно-синцитиальным вирусом легкого по сравнению с контролем на пятый день после инфицирования.
Как видно из приведенных результатов по некоторым конкретным соединениям общей формулы I (без ограничения указанными) в таблице 6, инфицирование животных вирусом приводило к формированию тяжелого отека лёгких (3,15-2,05 балла из 4-х возможных). Исследуемые соединения общей формулы I оказывали нормализующее воздействие на структуру легочной ткани.
Пример 4
Противовирусное действие соединений общей формулы I на модели экспериментальной метапневмовирусной инфекции у мышей
В исследовании был использован авторский штамм HMPV, который был предварительно адаптирован к легким мышей. Изучение эффективности соединений проводили на мышах самцах линии BALB /с весом 10-12 г путем сравнения летальности леченых и контрольных животных в течение 14 дней после инфицирования. Экспериментальные группы содержали по 20 мышей. Животных инфицировали вирусом интраназально под легким эфирным наркозом в объеме 0,03 мл/мышь.
Исследуемые соединения вводили животным перорально в дозе 30 мг/кг массы тела. Животные контрольной группы получали физиологический раствор. Препараты вводили перорально 1 раз в сутки в течение 5 дней. Лечение животных начинали через 24 часа после инфицирования.
Показатель летальности для групп животных, получавших соединения общей формулы I, был снижен на 30-70%. Данные по некоторым конкретным соединениям формулы I (без ограничения указанными) представлены в таблице 7.
Пример 5
Противовирусное действие соединений общей формулы I в отношении риновируса
Для проведения исследований был использован авторский штамм hRV. Животных инфицировали вирусом интраназально под легким эфирным наркозом в объеме 0,05 мл/мышь.
Для определения эффективности соединений в отношении hRV на экспериментальной модели in vivo вирус предварительно титровали на мышах, затем проводили заражение мышей и перорально вводили исследуемые соединения. На 4-ые сутки после инфицирования проводили оценку инфекционного титра вируса титрованием легочной суспензии в культуре клеток Hela. Инфекционный титр hRV- вируса в легких животных опытной группы по сравнению с группой животных, не получавших лечение, определяли по ЦПД.
Исследуемые соединения и плацебо (физиологический раствор) вводили животным перорально 1 раз в сутки, в течение 4 дней, начиная через 12 часов после инфицирования. Соединения вводили в дозе 30 мг/кг массы тела животного. В качестве отрицательного контроля использовали интактных животных, которые содержались в отдельном помещении в тех же условиях, что и опытные группы.
Через 4 суток после инфицирования проводили оценку противовирусной активности исследуемых соединений по снижению инфекционной активности вируса, определяемой в культуре клеток Hela.
Развитие инфекционного процесса было связано с уменьшением массы тела животных в группе вирусного контроля, при этом масса тела мышей леченых исследуемыми соединениями общей формулы I была выше массы контрольных животных на 3 и 4 сутки.
Исследование веса легких мышей показало, что в процессе эксперимента вес легких инфицированных мышей превышал вес лёгких интактных животных, свидетельствуя о активно текущем инфекционном процессе. Под влиянием исследуемых соединений общей формулы I вес легких животных достоверно отличался (был ниже) от группы вирусного контроля и практически не отличался от веса легких у интактных мышей.
Результаты определения инфекционной активности hRV в суспензиях легких мышей в культуре клеток Hela после применения некоторых конкретных соединений общей формулы I (без ограничения указанными) представлены в таблице 8.
Таблица 8
Подавление репродукции hRV в легких мышей
Лечение соединениями общей формулы I приводило к снижению инфекционной активности hRV.
Изучение противовирусного действия соединений общей формулы I на модели hRV-инфекции у мышей показало, что заявляемые соединения предотвращают снижение массы тела и увеличение массы легких до значений, наблюдаемых в группе интактных животных, а также снижают репродукцию вируса в легких у животных.
Изготовление лекарственных форм
Лекарственные формы соединений общей формулы I для использования в соответствии с настоящим изобретением получают по стандартным методикам, таким как, например, процессы смешивания, гранулирования, формирование драже, растворение и лиофилизация.
Таблетированная форма
Таблетированную форму получают, используя приведенные ниже ингредиенты:
Компоненты смешивают и прессуют для образования таблеток весом 300 мг каждая.
Желатиновые капсулы
Соединение общей формулы I или его соль — 90 мг,
Лактоза (сахар молочный), крахмал картофельный, кремния диоксид коллоидный (аэросил), магния стеарат - до получения массы содержимого капсулы 220 мг
Указанные выше ингредиенты смешивают, гранулируют, гранулы помещают в твердые желатиновые капсулы в количестве 220 мг.
Суппозитории
Пример состава суппозитория:
При необходимости возможно изготовление ректальных, вагинальных и уретральных суппозиториев с соответствующими наполнителями.
Раствор для инъекций
Пример состава раствора для инъекций:
В качестве растворителя при приготовлении раствора для инъекций могут быть использованы - 0,9% раствор натрия хлорида, дистиллированная вода, раствор новокаина. Форма выпуска - ампулы, флаконы, шприц-тюбики, «insert».
Состав 1 раствора для инъекций:
Соединение общей формулы I или его соль - 100 мг
Вода дистиллированная - 5 мл
В качестве растворителя при приготовлении раствора для инъекций могут быть использованы - 0,9% раствор натрия хлорида, изотонический фосфатный буфер, НЕРЕS. Форма выпуска - ампулы, флаконы, шприц-тюбики, «insert».
Возможно изготовление различных лекарственных форм для инъекций - стерильных растворов, стерильных порошков и таблеток.
Реферат
Изобретение относится к конкретным соединениям и их фармацевтически приемлемым солям, приведенным в формуле изобретения. Соединения по изобретению предназначены для изготовления фармацевтической композиции, набора или лекарственного средства. Также изобретение относится к способу получения соединений по изобретению (вариантам). Соединения по изобретению предназначены для применения в профилактике или лечении заболеваний, вызываемых РНК-содержащими вирусами, принадлежащими роду энтеровирусов, метапневмовирусов или роду пневмовирусов. Технический результат – амидные соединения, предназначенные для лечения или профилактики заболеваний, вызываемых РНК-содержащими вирусами. 10 н. и 16 з.п. ф-лы, 8 табл., 5 пр.
Формула
















































Документы, цитированные в отчёте о поиске
Производные пептидов или их фармацевтически приемлемые соли, способ их получения, применение и фармацевтическая композиция
Комментарии