Амидные производные тейкопланина или их фармацевтически пригодные кислотно-аддитивные соли, способ получения тейкопланиновых производных и фармацевтическая композиция - RU2078768C1
Код документа: RU2078768C1
Чертежи



Описание
Изобретение касается замещенных алкиламидов
тейкопланина, имеющих
формулу:
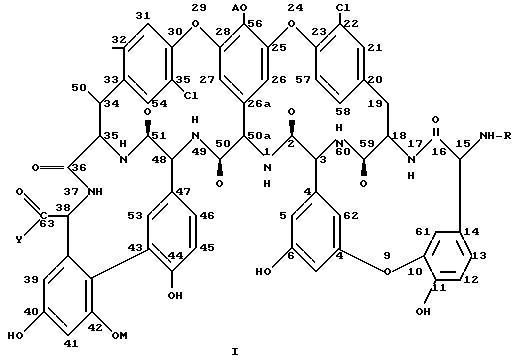
где R водород или защитная группа аминовой функциональной группы;
Y соединение формулы
-NR1-алк1-[Х- алк2]p-[T-алк3]p-[T-алк3]q-W,
где R1 водород или (C1-C4) алкил;
алк2, алк2, и алк3 каждый независимо друг от друга алкилен с линейной или разветвленной цепью с содержанием 2 10 атомов углерода;
p целое число 1 50, включая граничные значения;
q целое число 0 12, включая граничные значения;
X группы -NR2- или атом кислорода, где R2 представляет собой водород, (C1-C4)алкил, группу алк1NR3R4, где алк4 представляет собой алкилен с прямой или разветвленной цепью с содержанием 2 4 атома C, R3 представляет собой водород или (C1-C4)алкил и R4 представляет собой водород, (C1-C4)алкил или 5-6-членный циклоалкил; или R1 и R2 взятые вместе, представляют собой (C2-С4)алкиленовое звено, связывающее два атома азота, при условии, что в этом случае p равно 1;
T группа -NR5- или атом кислорода, где R5 представляет собой водород, (C1-C4)алкил; группу алк5 NR6R7, где алк5 представляет собой алкилен с прямой или разветвленной цепью с содержанием 2 4 атома C, R6 представляет собой водород или (C1-C4)алкил и R7 представляет собой водород, (C1-C4)алкил, или 5-6-членный циклоалкил; или R2 и R5 вместе друг с другом образуют (C2 -C4)алкиловое звено, соединяющее два атома азота, при условии, что в таком случае как p, так и q равны 1;
W оксигруппа, NR8N9, где R8 представляет собой H или (C1 -C6)алкил и R9 представляет собой H(1-C6)алкил, 5-6-членный циклоалкил, COOR10, где R10 представляет собой (C1-C6 )-ацилокси-(C1-C4)алкил и группу N⊕ R11R12R13 An⊖, где R11, R12 и R13 каждый независимо друг от друга представляют собой (C1-C4)алкил и An⊖ представляют собой анион, образованный из фармацевтически пригодной кислоты, при условии, что когда одновременно X представляет собой NR2, p 1 и q 0, то W не является оксигруппой;
A H или -N-[(C9-C12)алифатический ацил]-бета-D-2-доокси-2-аминоглюкопиранозил;
B водород или N-ацетил-бета-D-2-доокси-2- аминоглюкопиранозил;
M водород или альфа-D-маннопиранозил,
и их кислотно-аддитивных фармацевтически пригодных солей, при условии также, что B представляет собой водород лишь тогда, когда A и M являются одновременно атомом водорода.
Тейкопланин является международным незапатентованным названием (INN) антибиотического вещества, которое ранее называлось тейкомицином, которое получается путем культивации штамма Actinoplanes teichomyceticus vov. sp. АТСС 31121 в среде культивации, содержащей ассимилируемые источники углерода, азота и неорганических солей (см. патент США N 4239751).
Согласно процедуре, описанной в указанном патенте, антибиотический комплекс, описанный в там же, антибиотический комплекс, содержащий Тейкомицин A1, A2 и A3, извлекается из отдельного бульона брожения путем экстракции подходящим водонерастворимым органическим растворителем и осаждения из экстрагирующего растворителя согласно общепринятым процедурам. Тейкомицин A2, который является основным фактором выделенного антибиотического комплекса, затем отделяется от других путем хроматографии в колонке Sephadex® Известно (см. патент Англии N 2121401), что антибиотик Тейкомицин A2 фактически является смесью пяти близко связанных друг с другом совместно получаемых основных компонентов.
Как показали последние структурные исследования, можно представить основные компоненты 1, 2, 3, 4 и 5 тейкопланина A2 (ранее называемого Тейкомицином A2) указанной выше формулой I, в которой R представляет собой водород, Y оксигруппу, A N[(C6 -C11-)-алифатический ацил]-бета-D-2-деокси-2-амино-глюкопиранозил, B N-ацетил-бета-D-2-декси-2-амино-глюкопиранозил, а M альфа-D-маннопиранозил.
В частности, в тейкопланине A2 компонент 1 заместитель [(C10-C11)-алифатический ацил] представляет собой Z-4 деценоил, в тейкопланине A2 компонент 2 8-метил-нонаноил, в тейкопланине A2 компонент 3 деканоил, в тейкопланине A2 компонент 4 - 8-метилдеканоил, в тейкопланине A2 компонент 5 9-метилдеканоил.
Известно (см. Европейскую патентную заявку N 306645) получение тейкопланиновых соединений, в которых группа алифатической кислоты бета-D-2-деокси-2-аминоглюкопиранозилового звена представляют собой 6-метил-октансилановую группу (соединение A или RS3) или и-ионаиловую группу (соединение B или RS4).
Известны (см. статью Zanol и др. "Извлечение методом HPLC и определение структуры неосновных компонентов тейкопланина". 17-й Международный симпозиум по хроматографии. Вена. 25-30 сентября 1988 г.) два других тейкопланиновых соединения (RS1 и RS2).
Данные соединения отличаются тем, что группы алифатического ацила бета-D-2-деокси-2-аминоглюкопиранозилового звена представляют собой соответственно метил-ундеканоил (RS1) и додеканоил (RS2). Все группы сахара, в случае их присутствия, связаны с ядрами тейкопланина O-гликозидными связями.
Кроме того, найдено, что можно преобразовать тейкопланин, его чистый фактор или смесь любого из указанных факторов в любой пропорции в составляющие одно целое антибиотические продукты путем избирательного гидролиза одного или двух сахарных звеньев. Они называются антибиотиком L 17054 и антибиотиком L 17046 (см. Европейские патенты N 119575 и N 119574, соответственно).
Предпочтительными условиями гидролиза для получения антибиотика L 17054 являются следующие: 0,5 н. соляная кислота, температура 70 90oC и продолжительность процесса обычно 15 90 мин.
Антибиотик L 17054 представлен указанной выше формулой I, в которой Y представляет собой оксигруппу, R и A водород, B N-ацетил-бета-D-2-деокси-2-аминоглюколиранозил, M альфа-D-маннопиранозил, в которой сахарные звенья связаны с пептидным ядром посредством O-гликозидной связи.
Предпочтительными условиями гидролиза для получения антибиотика L 17046 являются следующие: 1 3 н. соляная кислота, температура 50 90oC, продолжительность процесса обычно 30 60 мин.
Антибиотик L 17046 представлен указанной выше формулой I, в которой Y представляет собой оксигруппу, R, A и M атомы водорода, B - N-ацетил-бета-D-2-деокси-2-амино-глюкопиранозил, где сахарное звено связано с пептидным ядром посредством O-гликозидной связи.
Известны (см. Европейскую патентную заявку N 301247) де-маннозилтейкопланиновые производные, то есть соединения указанной выше формулы I, в которой A и B имеют значения отличные от водорода, M представляет собой водород, а Y оксигруппу.
В результате полного избирательного расщепления всех сахарных звеньев тейкопланиновых соединений получается агликоновая молекула, которая называется антибиотиком L 17392 или деглюкотейкопланин, и он представлен указанной выше формулой I, в которой Y представляет собой оксигруппу, R, A, B и M каждый независимо друг от друга атом водорода (см. процесс избирательного гидролиза в Европейской патентной заявке N 146053).
Вещество, имеющее ту же структурную формулу, носит название фактора B антибиотика A 41030 (см. Европейскую патентную заявку N 0090578).
Данное вещество получается в результате микробиологического процесса, который включает брожение штамма Streptomyces virginial NRRL 12525 или Streptomyces virginial NRRL 15156 в подходящей среде, извлечение, очистку и разделение на компоненты антибиотика A 41030, антибиотического комплекса, состоящего по меньшей мере из семи факторов, включая фактор B антибиотика A 41030.
Все названные выше соединения, а именно тейкопланин, комплекс тейкопланина A2, компонент 1 тейкопланина A2, компонент 2 тейкопланина A2, компонент 3 тейкопланина A2, компонент 4 тейкопланина A2, компонент 5 тейкопланина A2, соединение A или RS3, соединение B или RS4, RS1, RS2, антибиотик L17054, антибиотик L 17046, антибиотик L 17392, производные де-маннозилтейкопланина (см. Европейскую патентную заявку N 301247) и их смеси в любой пропорции, являются исходными продуктами, пригодными для получения соответствующих алкиламидных производных согласно данному изобретению.
В описании данной патентной заявки понятие "тейкопланиновое соединение" или "тейкопланиновый исходный материал" используется для того, чтобы показать любой из указанных выше исходных материалов, а именно тейкопланин, получаемый согласно патенту США N 4239751, любую дальнейшую его очистку, комплекс тейкопланина A2, соединение указанной выше формулы I, в которой R представляет собой водород или защищающую N группу, Y оксигруппу, A - водород или -N[(C9-C12)алифатический ацил] -бета-D-2-деокси-2-аминоглюкопиранозил, B водород или N-ацетил-бета-D-2-деокси-2-аминоглюкопиранозил, M водород или альфа-D-маннопиранозил, при условии, что B может являться водородом лишь когда A и M одновременно представляют собой водород; их соль или смесь в любой пропорции.
Понятие алкил, либо как таковой, либо в комбинации с другими заместителями, включает углеводородные группы как с прямой, так и с разветвленной цепью, в частности "(C1-C6)алкил" представляет собой углеводородную цепь с прямой или разветвленной цепью с содержанием 1-6 атомов углерода, например метил, этил, пропил, 1-метилэтилбутил, 1-метилпропил, 1,1-диметилэтил, пентил, 1-метилбутил, 2-метилбутил, 1-гексил, 2-гексил, 8-гексил, 3,3 диметил-1-бутил, 4-метил-4-пентил и 3-метил -1- пентил; аналогично этому "(C1-C4)алкил" представляет собой прямую или разветвленную углеводородную цепь с содержанием 1-4 атома углерода, например алкил с содержанием 1-4 атома углерода, примеры которого перечислены выше.
Под понятием "алк1", "алк2", "алк3" имеется в виду независимо линейная или
разветвленная алкиленовая цепь с содержанием 2 10 атомов углерода, например
-CH2-CH2-,
-CH2-CH2-CH2-,
-CH2
-CH2-CH2-CH2-,
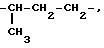
-CH2-CH2-CH2 -CH2-,
-CH2-CH2-CH2-CH2-CH2-CH2-,
-CH2-CH2-CH2-CH2-CH2- CH2-CH2-,
-CH2-CH2 -CH2-CH2-CH2-CH2- CH2-CH2-,
-CH2 -CH2-CH2-CH2-CH2- CH2 -CH2-CH2-CH2-,
-CH2-CH2-CH2-CH2 -CH2- CH2-CH2-CH2-CH2-CH2-,

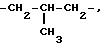
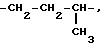
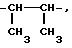
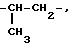

Аналогично этому "алк4" и "алк5" представляют собой независимо линейную или разветвленную алкиленовую цепь с содержанием 2 4 атома углерода, такую же, как и алкиленовая цепь с содержанием 2 4 атома углерода, примеры которой даются выше.
Предпочтительными
соединениями являются соединения формулы I,
где R водород или группа,
защищающая аминовую функциональную группу;
Y соединение формулы
-NR1-алк1
-[X-алк2]p-[T-алк]q-W,
где R1
водород или (C1-C4)алкил;
алк1, алк2 и алк3 каждый
независимо друг от друга линейная или разветвленная алкиленовая цепь с содержанием 2
4 атома углерода;
p целое число 1 12;
q целое число 0 12;
X группа -NR2- или
атом кислорода, где R2 представляет собой водород, (C1-C4)алкил, группу алк4NR3R4, в которой алк4 является линейной или
разветвленной алкиленовой цепью с содержанием 2 4 атома C, R3 представляет
собой водород или (C1-C4)алкил и R4 - водород, (C1-C4
)алкил или 5-6-членный циклоалкил; или R1 и R2 взятые вместе, образуют
(C2-C4) алкиленовое звено, связывающее два атома азота, при условии, что в данном
случае p равно 1;
Т группа -NR5 или атом кислорода, где R5
представляет собой водород, (C1-C4)алкил, группу алк5NR6R7, в которой алк5 является линейным или разветвленным алкиленом с содержанием
2 4 атома C, R6 представляет собой водород или (C1-C4)алкил, и R7 водород, (C1-C4)алкил, или 5-6-членный циклоалкил; или R2
и R5 взятые вместе, образуют (C2-C4) алкиленовое звено, связывающее
два атома азота, при условии, что в таком случае как p, так и q равны 1;
W оксигруппа,
NR8R9, где R8 представляет собой H или (C1-C6
)алкил и R9 H, (C1-C6)алкил, 5-6-членный циклоалкил, COOR10, где R10 представляет собой (C1-C6)ациклокси-(C1
-C4)алкил, и группу формулы NR11R12R13 An⊖, в которой R11, R12 и R13 каждый независимо друг от друга
представляет собой (C1-C4)алкил и An⊖ является анионом,
образованным из фармацевтически пригодной кислоты; при условии, что когда одновременно X представляет
собой NR2, p 1 и q 0, то W имеет значение отличное от оксигруппы;
А H или
-N[(C9-C12)алифатический ацил]-бета-D-2-деокси-2-аминоглюкопиранозил;
B
водород или N-ацетил-бета-D-2-деокси-2-аминоглюкопиранозил;
M водород или
альфа-D-маннопиранозил,
и их фармацевтически пригодные кислотно-аддитивные соли, при дополнительном условии,
что B представляет собой водород лишь тогда, когда А и М одновременно представляют
собой водород. Предпочтительно, когда X и/или Т представляет собой -NR2- и/или -NR5-, то
алк4 и алк5 представляют собой C2-C3 линейную
углеродную цепи.
Как описано выше, p является целым числом в интервале от 1 до 50, включая граничные значения, и q является целым числом от 0 до 12, включая граничные значения. Предпочтительно, когда X и/или Т представляют собой -NR2- и/или -NR5 -, то p и q составляют 1 12, и в то же время, когда X и Т представляют собой атомы кислорода, то p и q имеют такие значения, что p + q находится в интервале от 12 до 50.
Понятие "C5-C6 "циклоалкил", как оно используется в описании данного изобретения, охватывает группу циклопентила и циклогексила, при желании замещенную 1 3 группами низшего алкила, такого как метил и этил.
Предпочтительными соединениями являются также соединения формулы I, в которых X представляет собой группу -NR2-, где R2 является атомом водорода, (C1-C4)алкила или алк4NR3R4.
Другой группой предпочтительных соединений являются соединения формулы I, в которой p 1 и X представляет собой -NR2-, где R2 вместе с R1 образуют (C2-C3 )алкиленовое звено, связывающее атом азота. В таком случае особенно предпочтительны такие соединения, в которых алк1 представляет собой группу -CH2CH2-. Кроме того, предпочтительной группой соединений являются такие соединения, в которых алк1 представляет собой группу -CH2CH2-. Следующая предпочтительная группа соединений включает соединения формулы I, в которой p 1, q 1 и X и Т представляют собой -NR2 и -NR3-, соответственно, в которых R2 и R3 вместе друг с другом образуют (C2-C3)алкиленовое звено, связывающее атомы азота.
В данном случае особенно предпочтительны такие соединения, в которых алк2 представляет собой группу -CH2-CH2-.
Другие предпочтительные соединения представлены формулой I, в которой X и Т представляют собой атомы кислорода, p + q составляет 2 50, W представляет собой оксигруппу или NR8R9, где R8 представляет собой водород или (C1-C4)алкил и R9 водород, (C1-C4)алкил, циклопентил или циклогексил.
Другими предпочтительными соединениями являются такие соединения, в которых R8 представляет собой NR8R9, где R8 определен выше и R9 представляет собой COOR10, где R10 является (C1-C6) ацилокси-(C1-C4)алкильной группой.
В "(C1-C6)ацилокси-(C1-C4)алкиле" группа (C1
-C4алкила представляет собой метиленовое звено, при желании замещенное (C1-C3
)линейной или разветвленной алкильной цепью, например
-COOCH2
OCOCH3,

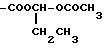
и другие.
Согласно общим определениям, изложенным выше, типичными примерами
группы
-NR1-алк1-[X-алк]p
-[T-алк3]q-W
являются следующие
-NH(CH2)2-NH(CH2)2
-NH2
-NH(CH2)2
-NH(CH2)3-NH2
-NH(CH2)2-NH(CH2)4-NH2
-NH(CH2)4-NH(CH2)2-NH2
-NH(CH2)3-NH(CH2)4-NH2
-NH(CH2)2-NH(CH2)3-NH(CH2
)2 -NH2
-NH(CH2)2-NH(CH2)4-NH(CH2)2- NH2
-NH(CH2)3-NH(CH2)4-NH(CH2)3- NH2
-NH(CH2)2-NH(CH2)3
-NH(CH2)4- NH2
-NH(CH2)4-NH(CH2)3-NH(CH2)4 -NH2
-NH(CH2)3NH(CH2)9-NH(CH2
)3 NH2
-NH(CH2)3NH(CH2)10-NH(CH2)3 NH2
-NH[(CH2)2NH]2-(CH2)2-NH2
-NH[(CH2)3NH]2-(CH2)3-NH2
-NH[(CH2)4
NH]5-(CH2)4-NH2
-NH[(CH2)5NH]3-(CH2)5
-NH2
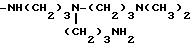

-NH(CH2)2-NH(CH2)2-NHCH3
-NH(CH2)2-NH(CH2)2-NHC2H5
-NH(CH2)2 -NH(CH2)4-NH(nC4H9)
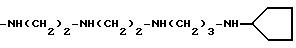


-NH(CH2)3-N[(CH2)3NH2]2

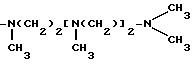
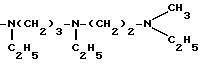
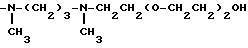
-NH(CH2)3NH(CH2)4 NHCOOCH(CH3) OCOCH3
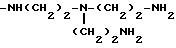


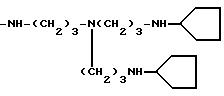

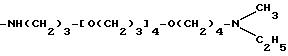
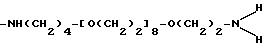
-NH-CH(CH3)CH2 -[OCH2CH2]42 OCH2 CH(CH3)NH2

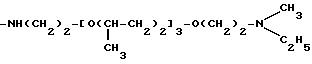
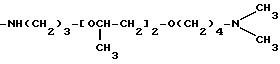

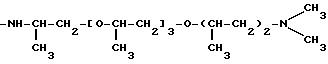


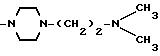



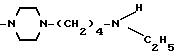

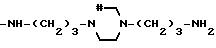
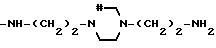
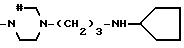

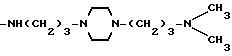
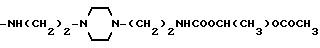
Соединения, отвечающие изобретению, проявляют антимикробное действие и находят полезное применение как полусинтетические антибактериальные агенты, действующие против грамм-положительных бактерий, а также особенно против грамм-отрицательных бактерий, и особенно против Escherichia coli и Pseudomonas aeruginosa.
Известны (см. Европейскую патентную заявку N 218099 и Международную патентную заявку N WO 88/06600) различные C63 амидные производные тейкопланинового комплекса, его отдельные компоненты и агликон и псевдоагликоны.
Соединения, отвечающие изобретению, получаются путем амидирования соответствующих производных формулы I, в которых Y представляет собой OH (то есть соответствующих карбоновых кислот).
Вещества, используемые как исходное сырье для получения соединений, отвечающих данному изобретению, могут быть либо продуктами как таковыми, либо смесями одного или нескольких продуктов.
Поскольку исходные материалы для получения соединений, отвечающих изобретению, могут использоваться в обеих указанных формах, то полученные продукты могут, в свою очередь, быть отдельными соединениями как таковыми или смесями двух или нескольких соединений указанной выше формулы I. Эти смеси соединений также охватываются изобретением и могут использоваться как таковые ввиду их биологических действий или в некоторых случаях могут быть разделены на отдельные компоненты путем известных приемов. Примеры процедур разделения, пригодных для получения отдельных компонентов из конечных смесей амидных производных тейкопланина, известны (см. Европейскую патентную заявку N 218099 и Международную патентную заявку N 88/06600).
Процедуры амидирования, описанные в указанных двух публикациях, могут
быть использованы также для получения
соединений, отвечающих данному изобретению. Данные процедуры включают конденсацию исходных карбоновых кислот, указанных выше, с избытком соответствующего амина
формулы II
NHR1
-алк1-[X-алк2]p-[T-алк3]q-W,
где R1, алк1, алк2, алк3, X,
T, p, q и W имеют те же значения,
что и выше,
в инертном органическом растворителе в присутствии конденсирующего агента.
Инертные органические растворители, используемые для реакции амидирования, это такие органические апротонные растворители, которые не оказывают неблагоприятного влияния на ход реакции и способны по меньшей мере частично солюбилизировать исходный тейкопланиновый продукт.
Примерами инертных органических растворителей являются органические амиды, сложные алкиловые эфиры, простые эфиры гликолей и многоатомных спиртов, фосфорамиды и сульфоксиды. Предпочтительными примерами инертных органических растворителей являются: диметилформамид, диметоксиэтан, гексаметилфосфорамид, диметилсульфоксид и их смеси.
Конденсирующий агент, используемый в данном изобретении, пригоден для образования амидных связей в органических соединениях, в частности при синтезе пептида.
Типичными примерами конденсирующих агентов являются (C1-C4)алкил, фенил, или гетероциклические фосфоразидаты, такие как дифенилфосфоразидат, диэтилфосфоразидат, ди(4-нитрофенил)фосфоразидат, диморфолилфосфоразидат и дифенилфосфорохлоридат. Предпочтительным конденсирующим агентом является дифенилфосфоразидат, то есть азид сложного дифенилового эфира фосфорной кислоты (DPPA). При осуществлении процесса амидирования, как указано в данном описании, аминовый реагент обычно используется в молярном избыточном количестве.
Обычно, когда аминовый реагент является недорогостоящим или довольно легко получаемым реагентом, используется 2 6-кратный молярный избыток, но предпочтителен 3-4-кратный молярный избыток.
Для осуществления процесса амидирования необходимо, чтобы амин был способен образовывать соль с карбоксильной функциональной группой тейкопланинового исходного продукта. В случае, когда амин не является достаточно сильным реагентом для образования такой соли в выбранной реакционной среде, то необходимо в реакционную смесь вводить солеобразующее основание не менее чем в равномолярном количестве с количеством исходного тейкопланина.
Использование небольшого молярного избытка аминового реагента с вводом солеобразующего основания является вполне пригодным способом, когда аминовый реагент довольно дорогостоящий и трудно получаемый продукт.
Примерами солеобразующих оснований являются третичные органические или гетероциклические амины, такие как триэтиламин, триметиамин, N-метил ирролидон или пиколин, и другие.
Конденсирующий агент обычно используется в небольшом молярном избытке, составляющим, например, 1,2 1,7-кратное количество и предпочтительно 1,5-кратное количество от тейкопланинового исходного соединения.
Кроме того, аминовый реагент может быть легко введен в реакционную среду в виде подходящей кислотно-аддитивной соли, например в виде хлоргидрата. В данном случае используется двойная молярная пропорция и предпочтительно 2- 4-кратный молярный избыток сильного основания, способного высвобождать аминовую форму из его соли. Наряду с этим, в данном случае пригодным основанием является третичный органический алифатический или гетероциклический амин, такого типа как приведено выше. Фактически, по крайней мере в некоторых случаях использование соли амина, который затем высвобождается непосредственно в ходе основного процесса указанными выше основаниями, чрезвычайно желательно, особенно когда данная соль является более стойкой, чем соответствующий свободный амин.
Температура реакции обычно изменяется в широких пределах в зависимости от типа исходного материала и условий реакции. Обычно желательно осуществлять реакцию при температурах в пределах 0-20oC.
Кроме того, время реакции также изменяется в широких пределах в зависимости от других параметров реакции. Обычно реакция конденсации завершается в течение примерно 24 48 ч.
В любом случае ход реакции регулируется посредством тонкослойной (TLC) хроматографии или жидкостной хромотографии высокого разрешения (HPLC) согласно существующим методам.
На основе результатов данных анализов специалист в данной области в состоянии оценить ход протекания реакции и решить, когда остановить реакцию и когда начать обработку реакционной массы согласно известным приемам, которые включают, например, экстракцию растворителями, осаждение путем ввода нерастворителей и т. д. в комбинации с другими общеизвестными операциями разделения и очистки, например с использованием колончатой хроматографии.
Если аминовый реагент содержит другие функциональные группы, которые не являются инертными в выбранных реакционных условиях, то эти функциональные группы должным образом защищаются посредством уже известных защитных групп.
Согласно следующему предпочтительному аспекту изобретения: соединения формулы I, в которых Y представляет собой указанную выше группу, могут быть получены путем реакции "активированного сложного эфира" карбоновой кислоты той же формулы I, в которой Y представляет собой OH, и N15-аминовая функциональная группа предпочтительно защищена, с соответствующим амином формулы II.
N15 -аминовая функциональная группа может быть защищена уже известными способами (см. например, кн. T. W.Greene. "Защитные группы в органическом синтезе". John Wiley and Sons. Нью-Йорк. 1981; M. Mc. Omie. "Защитные группы в органической химии". Plenum Press. Нью-Йорк. 1973).
Защитные группы должны быть стойкими в процессе реакции, не должны оказывать неблагоприятного влияния на реакцию амидирования и должны быть легко расщепляемыми и удаляемыми из реакционной среды по окончании реакции без изменения вновь образующейся амидной связи и общей структуры соединений, например компонентов сахара.
Типичными примерами N-защитных групп, которые могут успешно использоваться в способе данного изобретения для защиты N15-первичной аминовой функциональной группы исходного тейкопланина, и когда это нужно, аминовой функциональной группы реагента амина II, являются образующие карбамат реагенты, отличающиеся тем, что заключают в себе следующие оксикарбонильные группы: I, I диметилпропилилоксикарбонил, трет-бутилоксикарбонил, винилоксикарбонил, циннамилоксикарбонил, бензилоксикарбонил, пара-нитробензилоксикарбонил, 3, 4-диметокси-6-нитробензилоксикарбонил, 2, 4-дихлорбензилоксикарбонил, 5-бензилоксазолилметилоксикарбонил, 9 -антранилметилоксикарбонил, дифенилметилоксикарбонил, изоникотинилоксикарбонил, дифенилметилоксикарбонил, изоникотинилоксикарбонил, S-бензилоксикарбонил и другие. Другими подходящими N-защищаюшими реагентами являются альдегиды или кетоны или их производные, которые способны образовывать основания Шиффа с аминовой группой, которая должна быть защищена.
Предпочтительными примерами таких агентов, образующих основание Шиффа, являются бензальдегиды и особенно предпочтительным является 2-оксибензальдегид (салициловый альдегид).
Простым способом защиты в некоторых случаях является образование бензилиденового производного, которое может быть получено путем реакции амина с бензальдегидом в низшем спирте, таком как этанол, предпочтительно при комнатной температуре. После прекращения реакции с выбранным тейкопланиновым исходным материалом бензилиденовая защитная группа может быть удалена уже известным образом, например путем каталитической гидрогенизации, с использованием, например, палладия на углеродном носителе в качестве катализатора.
Однако в данном случае следует обратить внимание на присутствие групп, которые могут быть модифицированы путем каталитической гидрогенизации. Типичным следствием каталитической гидрогенизации амино-защищенного производного формулы I, в которой А представляет собой указанную выше группу, в которой ацилом является (Z)-4-деценоил(или содержащая его смесь), является то, что по меньшей мере частично деценоиловое соединение превращается в соответствующее деканоиловое соединение.
Как должно быть ясно для специалистов в данной области, конечный выбор конкретной защитной группы зависит от характеристик конкретного амидного производного, которое желательно. Фактически, данная амидная функциональная группа конечного соединения должна быть стойкой в условиях удаления защитной группы (или групп).
Поскольку условия удаления различных функциональных групп известны, то специалист в данной области в состоянии выбрать подходящую защитную группу.
Получение "активированных сложных эфиров" известно (см. Fieser и Fieser. Реагенты для органического синтеза. John Wiley and Sons Inc. с. 129 130, 1967).
Примерами реагентов, образующих указанные активированные сложные эфиры, которые могут быть легко использованы в способе изобретения, являются реагенты, описанные в публикации R. Schwyzer и др. Helv. Chim. Acta 1955 г.
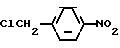
Предпочтительным реагентом данного типа является хлорацетонитрил. В данном случае сам по себе хлорацетонитрил или диметилформамид (DMF) могут использоваться как предпочтительные растворители.
Обычно инертными органическими растворителями, используемыми для получения "активированных сложных эфиров", являются органические апротонные растворители, которые не оказывают неблагоприятного влияния на ход реакции и способны по меньшей мере частично, солюбилизировать исходную карбоксикислоту.
Примерами указанных инертных органических растворителей являются органические амиды, алкиловые простые эфиры, простые эфиры гликолей и многоатомных спиртов, фосфорамиды, сульфоксиды и ароматические соединения. Предпочтительными примерами инертных органических растворителей являются диметилформамид, диметоксиэтан, гексаметилфосфорамид, диметилсульфоксид, бензол, толуол и их смеси.
Более предпочтительно, растворитель выбирается из числа следующих: ацетонитрил, диметилсульфоксид, диметилформамид. Получение активированного эфира обычно осуществляется в присутствии основания, которое не оказывает влияния на ход реакции, такого как триалкиламин, такого как триэтиламин, карбонат или бикарбонат натрия или калия. Обычно основание используется в 2 -6-молярной пропорции к исходной карбоновой кислоте тейкопланина, предпочтительно оно используется примерно в 3-кратном молярном избытке. Предпочтительным основанием является триэтиламин.
Реагент, образующий "активированный сложный эфир", используется в большом избыточном количестве относительно карбоновой кислоты исходного тейкопланина. Он обычно используется в 5-35-молярной пропорции, предпочтительно примерно в 20-30-кратном молярном избытке. Температура реакции составляет 10 60oC, предпочтительно 15 -30oC. Обычно продолжительность реакции зависит от других специфических параметров реакции и обычно составляет 3 48 ч.
В данном случае ход реакции может прослеживаться методами тонкослойной (TLC) хроматографии и жидкостной хроматографии высокого разрешения (HPLC) для определения того, когда реакция может рассматриваться как завершенная и когда могут осуществляться процедуры извлечения желаемого промежуточного продукта. Промежуточный продукт "активированный сложный эфир" может использоваться непосредственно в той же реакционной среде, где он получается, однако обычно он извлекается путем осаждения нерастворителями или путем экстракции растворителями и используется как таковой без дополнительной очистки в следующем реакционном этапе. Однако, если желательно, он может быть очищен путем колончатой хроматографии, такой как хроматография с испарительной колонкой или хроматография с колонкой с обратимой фазой.
Полученный промежуточный продукт "активированный
сложный эфир" затем реагирует с молярным избытком аминового производного формулы II
NHR1-[алк1]p-[T-алк3]q-W II
в присутствии
органического полярного растворителя при температуре 5 - 60oC, предпочтительно 10
30oC.
Органический полярный растворитель в данном случае может быть полярным протонным растворителем или апротонным растворителем.
Предпочтительными примерами органических полярных протонных растворителей являются низший (C2 -C4)алканолы, такие как этанол, н-пропанол, изо-пропанол, и-бутанол и др. или их смеси, предпочтительно используемые в сухой форме.
Предпочтительными примерами органического полярного апротонного растворителя являются N,N-диметилфомамид (DMF), гексаметилфосфорамид (HMPA) или их смеси, 1,3-диметил-3,4,5,6-тетрагидро-2(IH)-пиримидон (DMPU), диметилсульфоксид (DMSO) или диметоксиэтан (DME).
Реакция "активированного сложного эфира" с выбранным амином может осуществляться при температуре 5 60oC, но предпочтительной температурой обычно является температура 10 30oC, наиболее предпочтительно 20 - 25oC, в то время как предпочтительное молярное соотношение между промежуточным "активированным сложным эфиром" и амином II, как определено выше, составляет (1 5) (1 30), более предпочтительно (1 10) (1 20). Ход реакции можно регулировать как общепринято посредством TLC или HPLC.
Амидное производное, получаемое путем реакции амидирования, извлекается из реакционного раствора согласно общепринятым процедурам, например путем испарения растворителя или путем ввода нерастворителя. Удаление защищающей амин группы обычно осуществляется на сыром продукте, выделенном из реакционной среды амидирования.
Примеры процессов удаления указанных защитных групп из тейкопланиновых производных известны (см. например, Международную патентную заявку N WO 88/06600).
Если используются процедуры каталитической гидрогенизации, то реакция обычно осуществляется в присутствии разбавленного водного раствора сильной кислоты, предпочтительно неорганической кислоты, в органическом растворителе, смешиваемом с разбавленным водным раствором сильной кислоты. Фильтрат из данной реакционной среды затем обрабатывается для извлечения либо аддитивной соли (образуемой неорганической кислотой) амида формулы I, либо соответствующего свободного основания. Аналогичные процедуры осуществляются в том случае, когда защищающая амин группа является такой, которая может быть удалена путем обработки разбавленной неорганической кислотой, например основание Шиффа или карбонильная группа C-C алкокси, в условиях, которые не вызывают расщепления сахарных звеньев, например низкая температуре, непродолжительное время реакции.
Следующая процедура получения соединения формулы I согласно изобретению заключается в реакции N15-защищенного производного N63 амида формулы I, в которой Y представляет собой -NR1алк, XH или NR1-алк1 -[X-алк2]p-TH с реагентом формулы r-[алк]p-[T-алк3]q-W или r-[алк3]qW, соответственно, где R1, алк1, алк2, алк3, X и T имеют те же значения, что указаны выше, r представляет собой галогено, метансульфонил или тозил, в присутствии акцептора кислоты в инертном растворителе. В данных случаях p составляет предпочтительно 1 или 2, q имеет значение, отличное от нуля, предпочтительно составляет 1 или 2, X и T представляют собой предпочтительно NH или кислород, наиболее предпочтительно кислород. N15-защищенное производное N63 амида, указанное выше, получается согласно общему описанию способа получения соединений формулы I согласно изобретению.
При желании получения соединения формулы I, в которой W представляет собой -NR8R9-, где R8 определено выше, R9 является группой COOR10 и R10 (C1-C6) ацилокси-(C1-C4) алкилом, необходимо, чтобы N15-защищенное производное N63 амида, в котором W представляет собой -NHR8-, где R8 определено выше, взаимодействовало с альфа-ацилокси-алкил-пара-нитрофенилкарбонатом в присутствии безводного карбоната щелочного металла, такого как карбонат натрия.
Альфа-ацилокси-алкил-пара-нитрофенилкарбонат может быть получен известным способом (см. J. Med. Chem. 31, с. 318 322. 1988). Некоторые амиды, отвечающие изобретению, такие как амиды комплекса тейкопланина A2, отдельный его компонент или любая смесь двух или нескольких его компонентов, могут быть использованы в качестве исходного материала для получения унитарных антибиотических продуктов посредством избирательного гидролиза одной или двух молекул сахара при существовании процедуры таким образом, как уже описано в Европейских патентах N 119575 и N 119574. Альтернативный способ получения соединений формулы I, в которой A представляет собой водород, B - N-ацетил-бета-D-2-деокси-2- аминоглюкопиранозил, M альфа-D-маннопиранозил, заключается в гидролизе соответствующих амидных соединений формулы I, в которой A представляет собой -N[(C9-C12)- алифатический ацил]-бета-D-2- деокси-2-аминоглюкопиранозил, B N-ацетил-бета-D -2-деокси-2-аминоглюкопиранозил, M альфа-D-маннопиранозил (то есть карбоксамидные производные комплекса тейкопланина A2 или его отдельного компонента)(см. Европейскую патентную заявку N 146822).
Данный способ заключается в контактировании указанного выше материала с концентрированным водным раствором органической кислоты примерно при комнатной температуре, предпочтительно с водным раствором трифторуксусной кислоты концентрацией 75 95 при температуре 10 50oC.
Альтернативный способ получения соединений формулы I, в которой как A, так и M представляют собой атом водорода, B - N-ацетил-бета-D-2-деокси-2-аминоглюкопиранозил, заключается в том, соединения формулы I, в которой A представляет собой N[(C9-C12 алифатический ацил]-бета-D-2-деокси-2- аминоглюкопиранозил, B - N-ацетил-бета-D-2-деокси -2-аминоглюкопиранозил, М альфа-D-маннопиранозил, подвергается гидролизу (см. Европейскую патентную заявку N 175100).
Данный способ заключается в контактировании указанного выше исходного материала с сильной кислотой в присутствии полярного апротонного органического растворителя, выбранного из числа простых эфиров, кетонов и их смеси, которые являются жидкими продуктами, при комнатной температуре.
В последнем случае в качестве исходных материалов могут использоваться также амидные соединения формулы I, в которых A представляет собой водород, B N-ацетил-бета-D-2-деокси-2- аминоглюкопиранозил, M альфа-D-маннопиранозил, которые получаются путем гидролиза концентрированного водного раствора трифторуксусной кислоты, как описано выше.
Для извлечения кислотно-аддитивной соли величину pH реакционного раствора, полученного в результате расщепления амино-защищающей группы, обычно доводят до значения 4 7 путем ввода водного основания, например водного раствора гидрата окиси натрия, и после испарения растворителя при пониженном давлении полученный твердый продукт извлекается в форме кислотно-аддитивной соли, образуемой с сильной кислотой, которая вводится в ходе этапа удаления защиты. Этот продукт может быть дополнительно очищен общепринятыми способами, например путем колончатой хроматографии, осаждением из раствора путем ввода нерастворителей, препаративной хроматографии и другими способами. Кислотно-аддитивная соль может быть превращена в соответствующее свободное основание формулы I путем суспензирования или растворения кислотно-аддитивной соли в водном растворителе, который затем доводится до нужного значения pH, в результате чего сохраняется форма свободного основания. Данный продукт затем извлекается, например, путем экстракции органическим растворителем или превращается в другую кислотно-аддитивную соль путем добавления выбранной кислоты и осуществления процесса как описано выше.
Иногда после описанной выше операции может быть необходима обычная процедура обессоливания извлеченного продукта.
Так например, может с успехом использоваться колончатая хроматография с использованием полидекстрановой смолы с регулируемой пористостью (например Сефадекс LH20) или силанизированного силикагеля. После элюирования нежелаемых солей водным раствором желаемый продукт элюируется посредством линейного ингредиента или поэтапного градиента смеси воды с полярным или апротонным органическим растворителем, таким как ацетонитрил/вода с содержанием от 5 примерно до 100 ацетонитрила, и затем он извлекается путем выпаривания растворителя или лиофилизации.
Соединение формулы I в форме свободного основания может превращаться в соответствующую кислотно-аддитивную соль путем суспензирования или растворения формы свободного основания в водном растворителе и ввода небольшого молярного избытка выбранной кислоты. Полученный раствор или суспензия затем лиофилизируется с целью извлечения желаемой кислотно-аддитивной соли. В некоторых случаях вместо лиофилизации возможно извлечение конечной соли за счет осаждения путем ввода нерастворителя, смешиваемого с водой.
В случае, когда конечная соль нерастворима в органическом растворителе, в котором растворима форма свободного основания, она может извлекаться путем фильтрации из органического раствора несолевой формы после ввода стехиометрического количества небольшого молярного избытка выбранной кислоты.
Типичные и подходящие кислотно-аддитивные соли соединений формулы I включают соли, образуемые в результате стандартной реакции как с органическими, так и с неорганическими кислотами, такими как соляная кислота, бромистоводородная, серная, фосфорная, уксусная, трифторуксусная, трихлоруксусная, янтарная, лимонная, аскорбиновая, молочная, малеиновая, фумаровая, пальмитиновая, холевая, памоиновая, слизовая, камфорная, глутаровая, гликолевая, фталевая, винная, лауриновая, стеариновая, салициловая, метансульфоновая, бензолсульфоновая, сорбиновая, пикриновая, бензойная, коричная и другие кислоты.
Предпочтительными кислотно-аддитивными солями соединений, отвечающих изобретению, являются фармацевтически пригодные кислотно-аддитивные соли.
Под понятием "фармацевтически пригодные кислотно-аддитивные соли" имеются в виду соли, образуемые вместе с кислотами, которые с точки зрения биологических характеристик, процесса производства и рецептуры отвечают требованиям фармацевтической практики. Примеры кислот, пригодных для "фармацевтических кислотно-аддитивных солей" перечислены выше.
Соединения, отвечающие изобретению, имеющие форму как свободных оснований, так и их кислотно-аддитивных солей, являются полезными антибактериальными агентами, действующими как против грамм-положительных, так и против грамм-отрицательных бактерий.
Однако соединения, отвечающие данному изобретению, проявляют исключительно высокую активность действия против грамм-отрицательных бактерий, особенно против Pseudomonas aeruginosa.
Фактически, в настоящее время они являются самыми активными производными из числа тейкопланиновых антибиотиков против микроорганизмов данного вида. Эта активность особенно характерна для тех соединений, отвечающих изобретению, которые имеют деглюкотейкопланиновое ядро, но она характерна также для соединений, отвечающих изобретению, которые имеют тейкопланиновое ядро.
Антибактериальная активность соединений, отвечающих данному изобретению, может быть продемонстрирована в условиях ин витро путем стандартных двукратных испытаний разбавлением в микротитрующем устройстве с использованием питательного бульона Difco Todd-Hewitt (Strep. pyogenes и Strep. pneumonial) или питательного бульона Oxoid Iso-sensitest(Staphylococci, Strep. faecalis и грамм-отрицательные организмы). Культура питательного бульона разбавляются в достаточной степени, чтобы конечный инокулум составлял примерно 104 образующих колонии ед./мл (CFU/мл). Минимальная ингибирующая концентрация (MIC) рассматривается как наименьшая концентрация, которая не проявляет заметного роста после инкубирования в течение 18 24 ч при температуре 37oC.
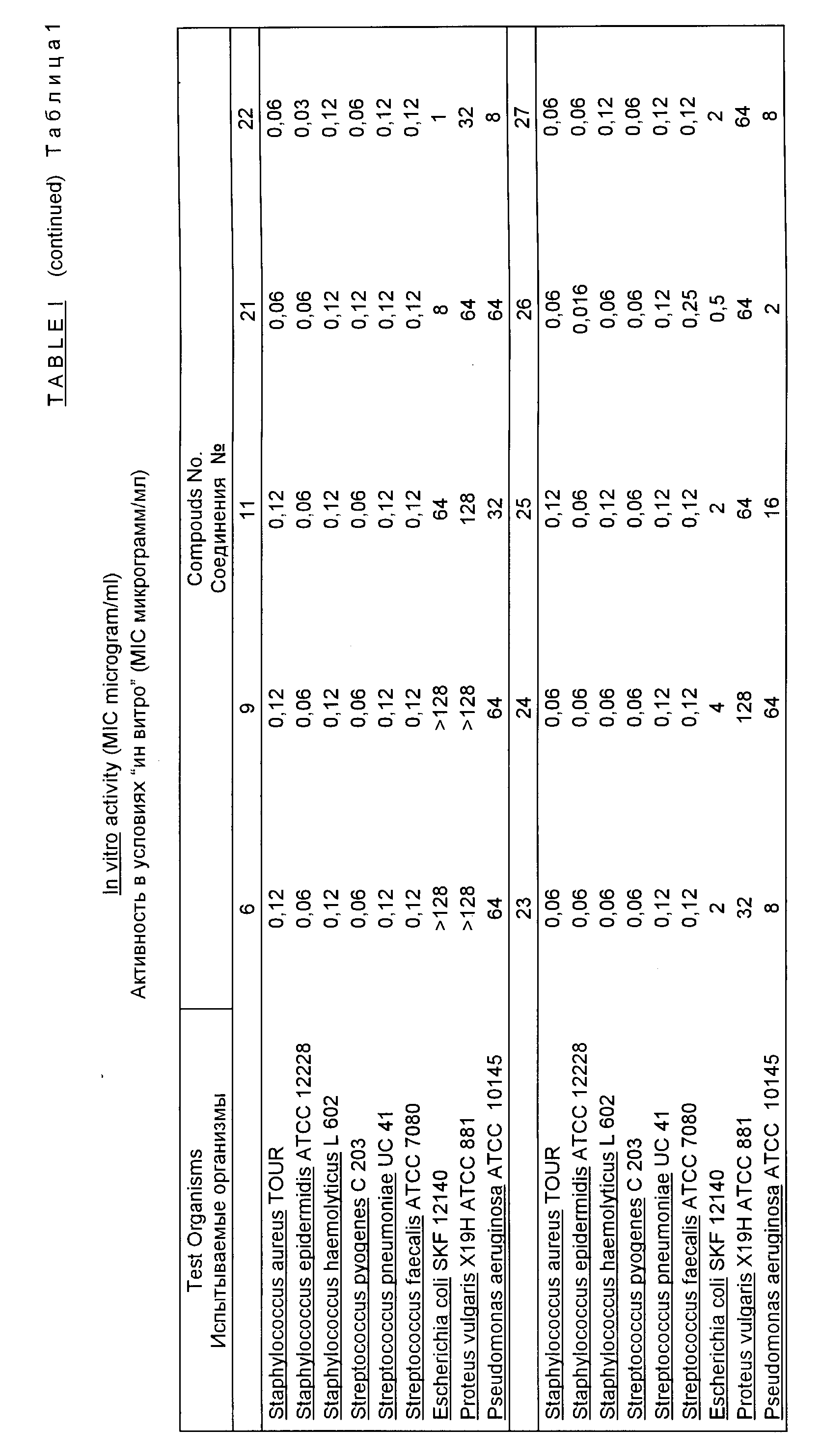
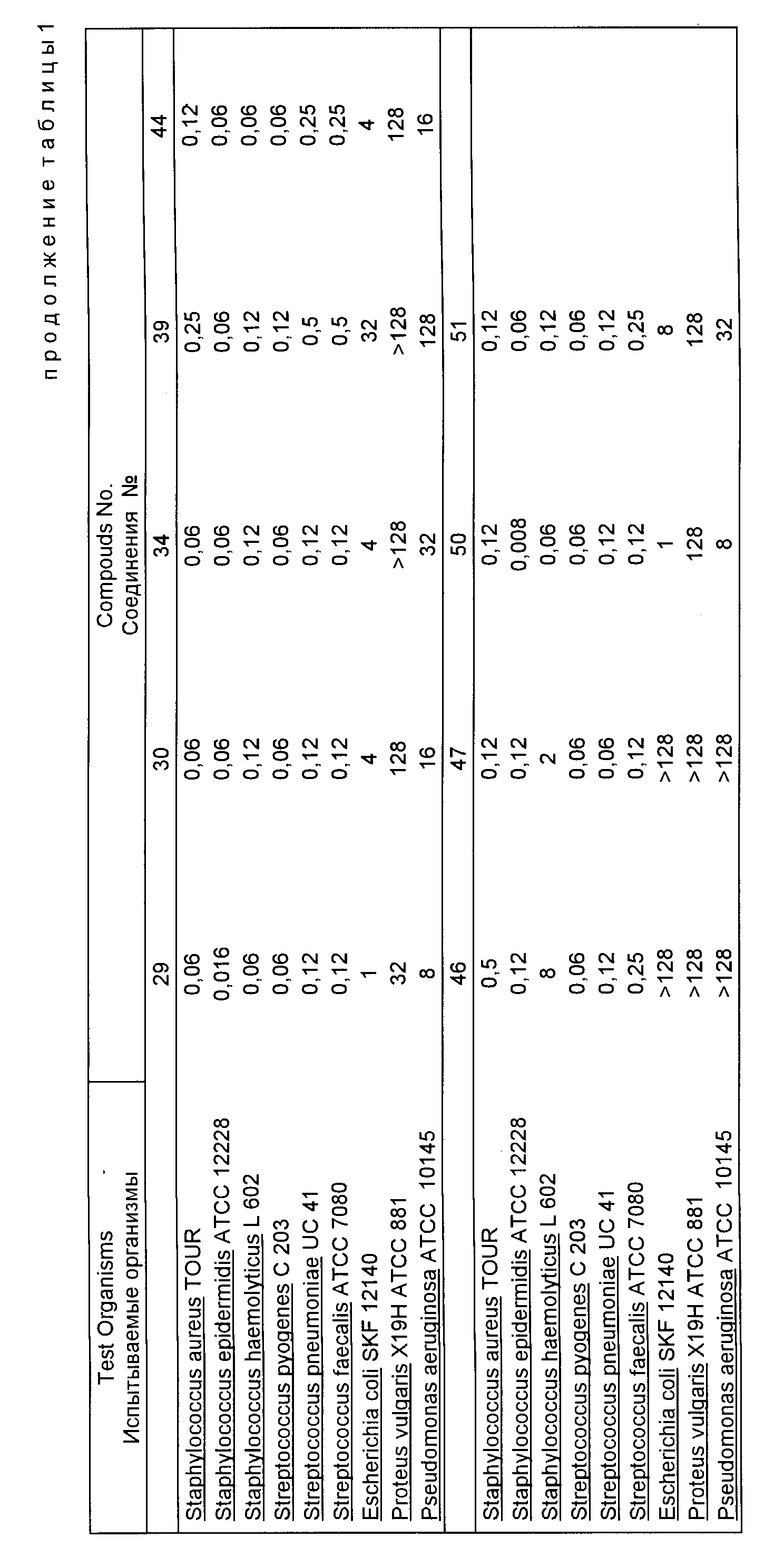
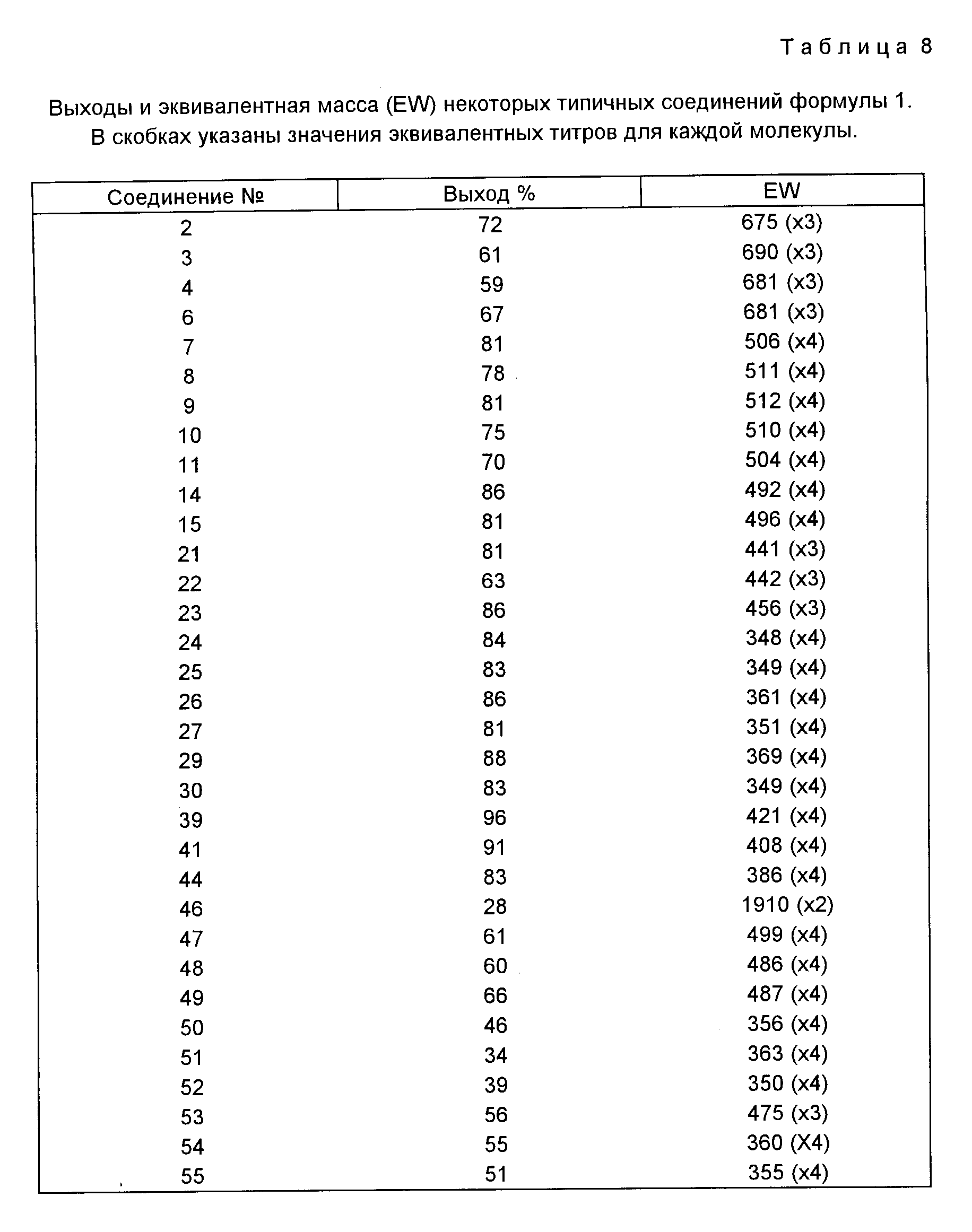
Результаты антибактериального испытания типичных соединений, отвечающих данному изобретению, суммированы в табл. 1.
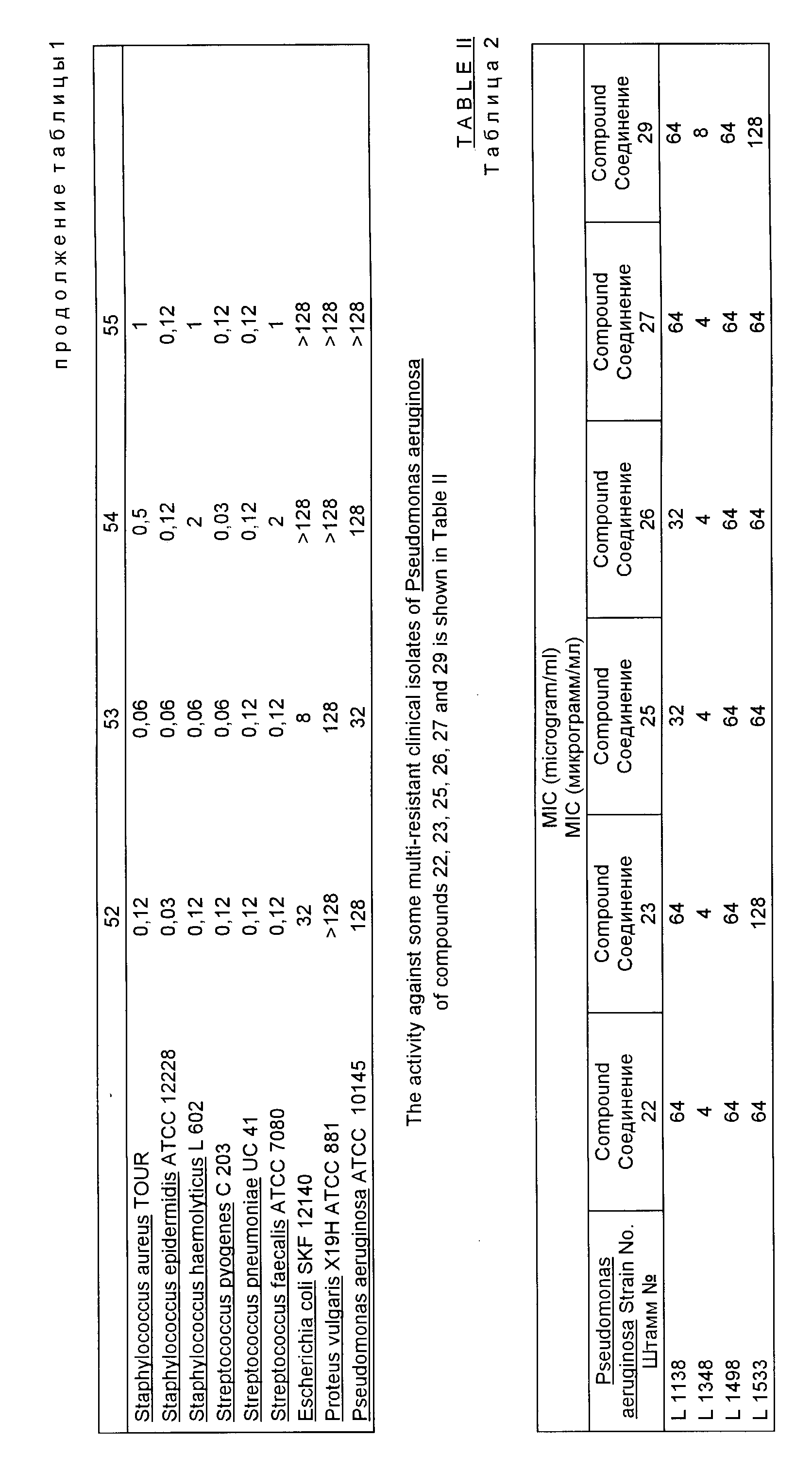
В табл. 2 показана активность соединений 22, 23, 25, 26, 27 и 29 против некоторых устойчивых ко многим антибиотикам клинических изолятов Pseudomonas aeruginosa.
Активность соединений, отвечающих изобретению, против Pseudomonas aeruginosa больше, чем активность тейкопланина и ближайших аналоговых соединений (см. Европейскую патентную заявку N 218099 и Международную патентную заявку N WO 88/06600), у которых значение MIC (мкг/мл) против того же микроорганизма никогда не бывает меньше, чем 32.
Активность соединений, отвечающих данному изобретению, против Pseudomonas aeruginosa особенно важна ввиду важности инфекционного заражения, вызванного этим штаммом.
Клиническое инфекционное заражение P. aeruginosa включает локальную инфекцию, например раны (особенно ожоги), мочевой канал, дыхательные пути, кишечник, глаза, уши, и общие инфекции (кровь, кости или септическое заражение), возникающие от мест первичной локальной инфекции у пациентов со слабой сопротивляемостью и приводящие к образованию метастатического очага в различных органах.
Прогноз для пациентов, у которых развивается септическое заражение Pseudomonas небольшой, и в некоторых публикациях сообщается об очень высокой смертности (иногда 100) (см. например, "Genetics and Biochemistry of Pseudomonas". P. H. Clarke и M. H. Richmond (гл. 2), John Wiley and Sons. 1975).
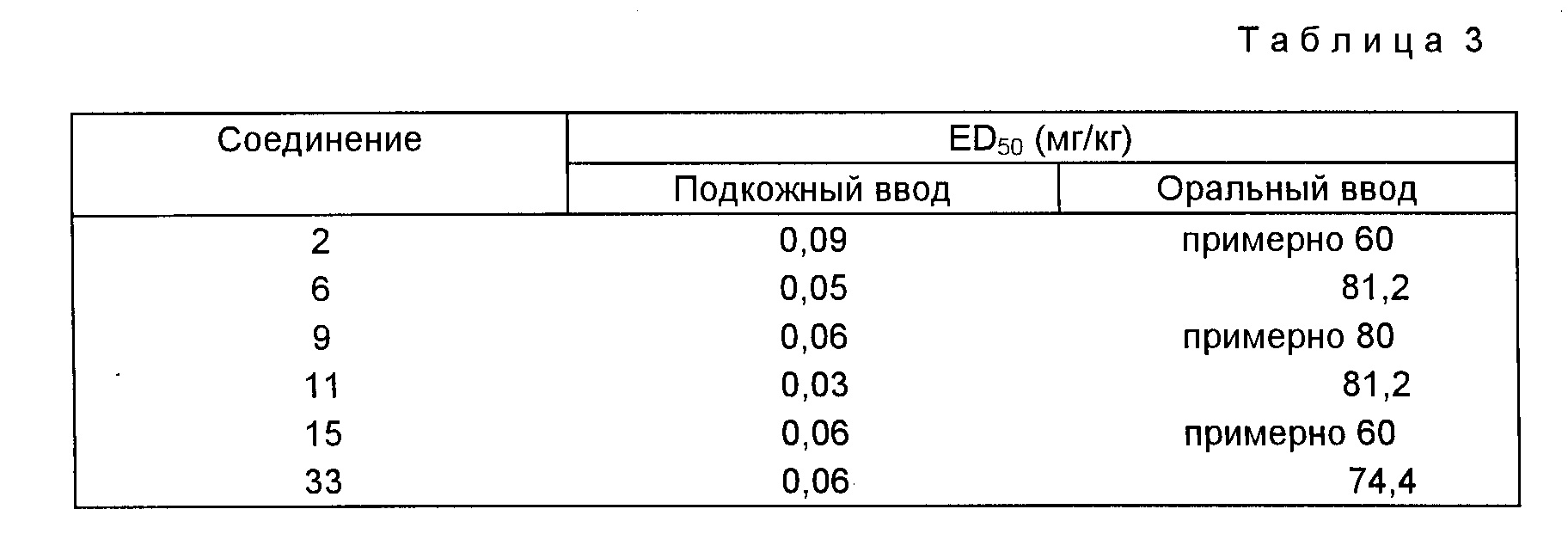
Кроме того, тейкопланиновые соединения, отвечающие изобретению, которые отличны от деглюкотейкопланина и псевдоагликонов тейкопланина, проявляют значительно более высокую активность в условиях ин виво при оральном вводе в организм по сравнению с уже известными амидными производными тейкопланина. ED50 (мг/кг) типичных соединений, отвечающих изобретению, в условиях испытаний ин виво, проведенных на мышах, септически зараженных Strep. pyogenes, полученные согласно известной процедуре (см. V. Arioli и др. Journal of Antibiotics. 1976), приводятся ниже в табл. 3.
Было найдено, что соединение 50 при испытании в условиях ин виво особенно эффективно при лечении мышей, септически зараженных E. coli, после ввода (дозой 40 мг/кг, 7/8 выжив./обработка) внутривенно и после ввода подкожно (ED50≅38 мг/кг).
Ввиду указанной выше антимикробной активности, соединения, отвечающие данному изобретению, могут использоваться как активные ингредиенты антимикробных препаратов, используемых при лечении людей и в ветеринарии для предотвращения и лечения инфекционных заболеваний, вызванных патогенными бактериями, которые чувствительны к указанным активным ингредиентам.
При таком лечении данные соединения могут использоваться как таковые или в форме смесей в любых пропорциях.
Соединения, отвечающие данному изобретению, могут вводиться в организм орально, локально или парэнтерально, причем предпочтителен парэнтеральный способ ввода. В зависимости от способа ввода в организм данные соединения могут быть приготовлены в виде различных дозированных форм. Препараты для орального ввода могут быть в форме капсул, таблеток, жидких растворов или суспензий. Как уже хорошо известно, капсулы и таблетки могут содержать наряду с активным ингредиентом обычные эксцепиенты, такие как разбавители, например лактозу, фосфат кальция, сорбит, и т. д. смазки, например стеарат магния, тальк, полиэтиленгликоль, связующие агенты, например поливинилпирролидон, желатин, сорбит, трагакант, аравийская камедь, ароматизирующие вещества и подходящие дезинтегрирующие и смачивающие вещества. Жидкие препараты, обычно в форме водных или масляных растворов или суспензий, могут содержать обычные присадки, такие как суспензирующие агенты. Для локального применения соединения, отвечающие изобретению, могут быть приготовлены в соответствующих формах, пригодных для нанесения на кожу, слизистые оболочки носа и горла или на бронхиальные ткани, и могут иметь форму кремов, мазей, жидких распыляемых растворов или ингаляторов, лепешек или препаратов, наносимых кисточкой в горло.
Следующим преимуществом соединений, отвечающих данному изобретению, является их значительно более высокая растворимость в воде при более широком диапазоне значений pH и следовательно существующие проблемы для фармацевтических композиций устраняются.
Для лечения глаз или ушей данный препарат может иметь жидкую или полужидкую форму, приготовленную в гидрофобных или гидрофильных основах, таких как мази, кремы, лосьоны, препараты для нанесения кисточкой или порошки.
Для ввода через прямую кишку соединения, отвечающие изобретению, вводятся в форме свечей в смеси с обычными носителями, такими как масло како, парафин, спермацеты или полиэтиленгликоли и их производные.
Композиции для инъекции могут иметь такие формы как суспензии, растворы или эмульсии в масле или водных носителях, и они могут содержать образующие композицию агенты, такие как суспензирующие, стабилизирующие и/или диспергирующие агенты.
Как возможный вариант, активный ингредиент может иметь порошкообразную форму для преобразования ее при подаче с соответствующим носителем, таким как стерилизованная вода.
Количество основного активного ингредиента, которое должно быть введено в организм, зависит от различных факторов, таких как размер и условия объекта, подвергаемого лечению, способ и частота ввода препарата и вызывающий заболевание агент.
Соединения, отвечающие данному изобретению, обычно эффективны при дозе их ввода в пределах примерно от 0,5 до 30 мг активного ингредиента на кг веса тела, предпочтительно в разделенной дозе, принимаемой 2 4 раза в день. Особенно желательными композициями являются такие, которые содержат примерно 20 300 мг на единичную дозу.
Примеры. Экспериментальная часть.
В описанных ниже примерах исходным материалом может быть комплекс тейкопланина A (TGA), его отдельный компонент или любая смесь двух или нескольких указанных компонентов.
Типичные комплексные смеси состоят в основном из пяти компонентов, соответствующих указанной выше формуле I, в которой группы алифатического ацила бета-D-2-деокси-2-аминоглюкопиранозилового радикала, представленные как символ A, имеют соответственно следующие значения: Z-(4-)-деканоил (AC1); 8-метилнонаноил (AC2); деканоил (AC3); 8-метилдеканоил (AC4) и 9-метилдеканоил (AC5); B представляет собой N-ацетил-бета-D-2-деокси-2- аминоглюкопиранозил (AcGl4), M - альфа-D-маннопиранозил (Man), а Y OH.
Данная смесь идентифицируется как акроним TGAC1-5.
В случае, когда в качестве исходного материала используется один из компонентов указанной смеси, он идентифицируется как: TGnAC1, TGAC2, TGAC3, TGAC4 или TGAC5, в зависимости от специфического алифатического ацила остальной части указанного аминоглюкопианозилового радикала.
При использовании смеси одного или нескольких компонентов обозначение ее такое же, как в той же системе для комплекса. Так например, Акроним TGAC2-5 показывает, что это смесь компонентов 2 5, в которой компонент 1 уже больше не присутствует. Эта смесь получается, когда каталитическая гидрогенизация насыщает двойную связь компонента 1, преобразующего этот компонент в компонент 3. Акроним TGAC2, 3 означает, что это смесь компонентов 2, 3 и Акроним TGAC4,5 показывает, что это смесь компонентов 4 и 5.
Антибиотик L17392 (то есть агликон тейкопланина) представлен как Акроним DTG, в то время как псевдоагликоны L17054 и L17046 представлены соответственно как TGA3-1 и TGA3-2, и диманнозил псевдогликон (Европейская патентная заявка N 301247) представлен как DM-TGAC.
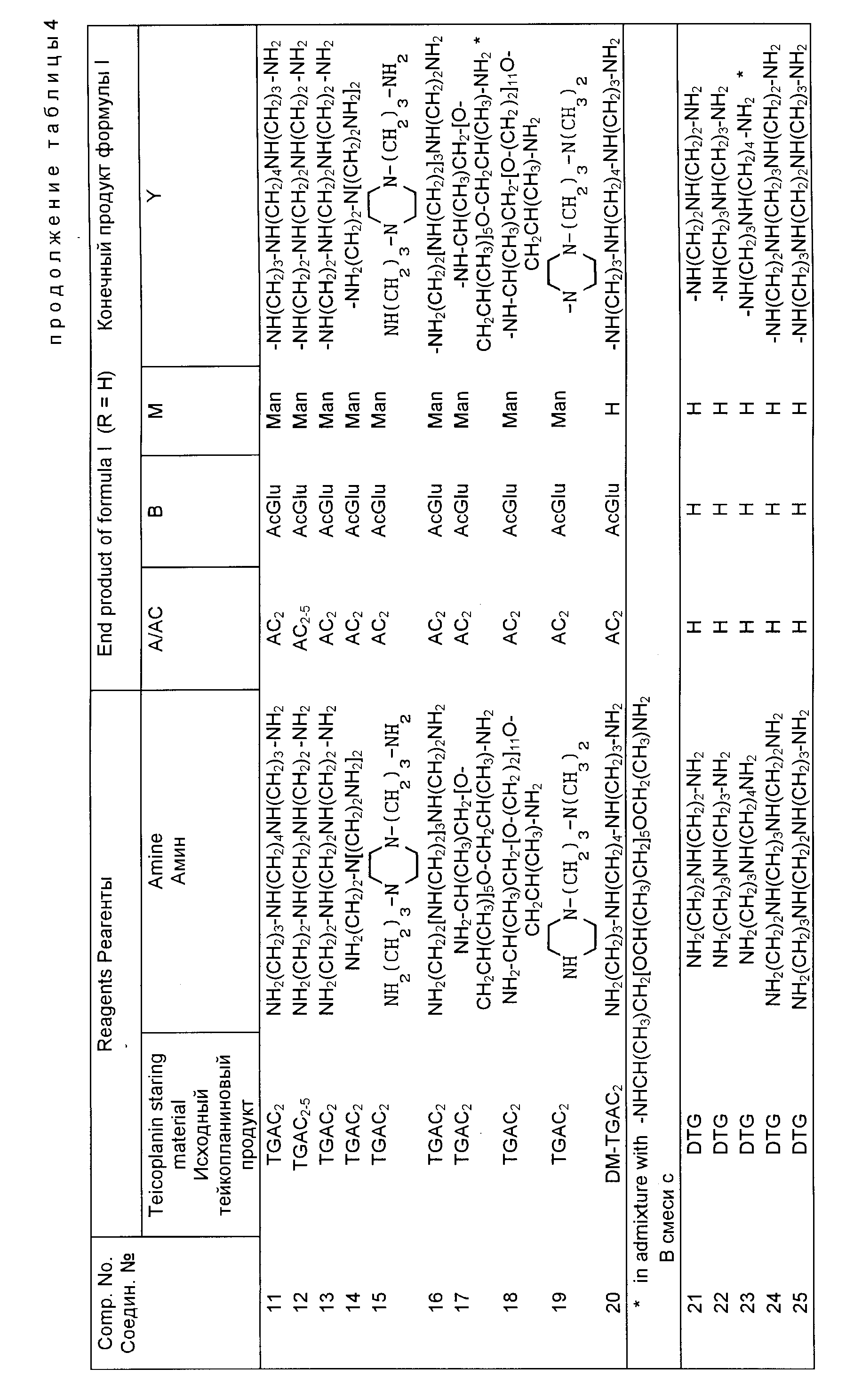
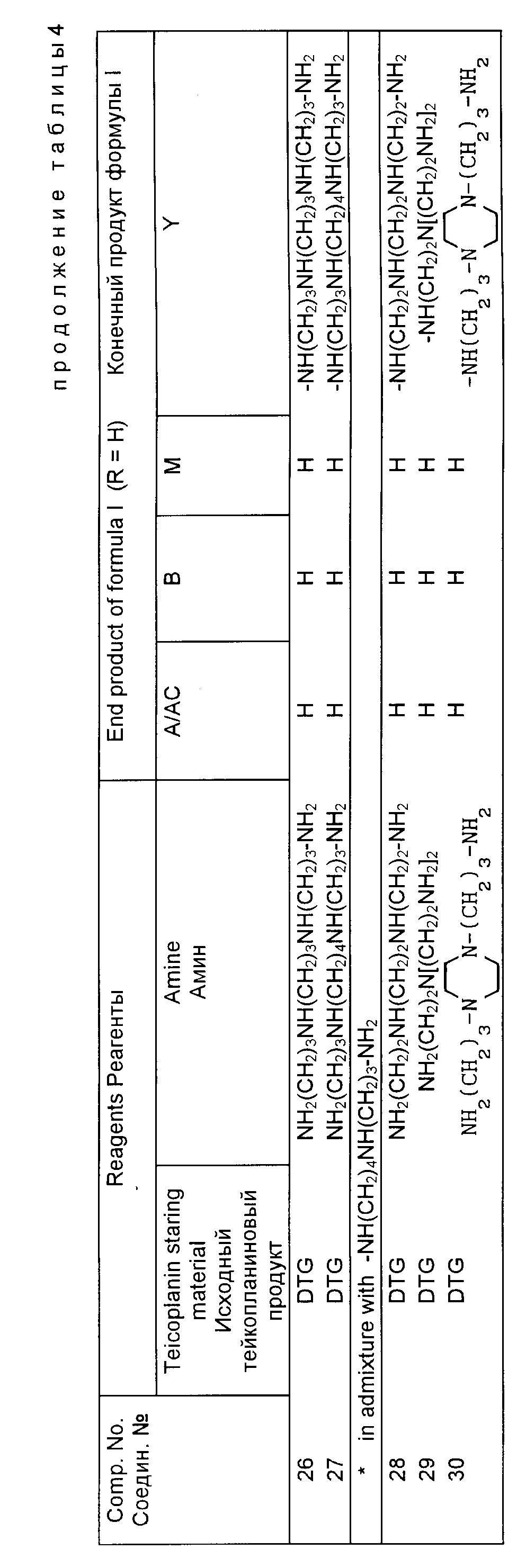
Полученные конечные продукты в приведенных табл. 4 6 определяются формулой I, приведенной выше, где символ A для алифатического ацила как заместителя бета-D-2-деокси-2-аминоглюкопиранозилового радикала (A/AC) идентифицируется с использованием обычных обозначений AC1, AC2, AC3, AC4, AC5, как разъяснялось выше. Когда получается смесь двух или более компонентов, то она обозначается по такой же системе, как указано выше.
Примеры 1 30.
В случае, когда желательны N63-карбоксиамиды смесей TGAC2-5, осуществляются следующие процедуры.
A. Получение комплекса N15-бензилоксикарбонил(CBZ)тейкопланина A2 и его отдельных компонентов 1 5.
Раствор 4,5 мл бензилхлорформата в 10 мл сухого ацетона вводится по каплям при комнатной температуре в перемешанный раствор 45 г (примерно 24 ммоль) комплекса тейкопланина A (или его отдельного компонента 1 5) и 6 мл (примерно 44 ммоль) триэтиламина (TEA) в 300 мл диметилформамида (DMF). После примерно 60 мин вводится 600 мл простого этилового эфира и образующийся осадок (примерно 59 г) извлекается путем фильтрации и повторно растворяется в 2,5 л смеси ацетон вода (1 1, об/об). Полученный раствор концентрируется при 35o C при пониженном давлении до объема примерно 1,6 л, затем он экстрагируется 1,6 л простого этилового эфира, который отделяется и удаляется.
Величина pH водного слоя доводится до 4,8 посредством ледяной уксусной кислоты и он экстрагируется 1,5 л и-бутанола. Органический слой отделяется, промывается 1,5 л воды (2 х 750 мл), затем концентрируется до объема примерно 200 мл при 45oC при пониженном давлении. При вводе этилацетата (примерно 800 мл) отделяется твердый продукт, который извлекается путем фильтрации, промывается простым этиловым эфиром (примерно 500 мл) и высушивается при комнатной температуре в вакууме в течение ночи, в результате получается 45,7 г (примерно 96) чистого конечного соединения.
В. Получение цианометилового сложного эфира N15-CBZ-Тейкопланин-A2 комплекса и отдельных его компонентов.
В перемешанный раствор 45 г (примерно 22 ммоль) N15-CBZ-тейкопланин A2 комплекса (или его отдельного компонента) в 450 мл DMF вводят при комнатной температуре 5,25 л (примерно 37 ммоль) TEA и 60 мл хлорацетонитрила. По прошествии 20 ч реакционная смесь вливается в 4,5 л этилацетата и осадок (примерно 50 г) извлекается путем фильтрации и повторно растворяется в 900 мл смеси метанол вода (1 1, об/об). Величина pH полученного раствора доводится до 5,5 посредством ледяной уксусной кислоты, затем вводится 1,1 л и-бутанола. Наибольшая часть метанола испаряется при 35oC при пониженном давлении, а в результате получается смесь (примерно 1,5 л) и-бутанола и воды, из которой отделяется органический слой, промывается 500 мл воды и концентрируется при 40oC при пониженном давлении до объема примерно 200 мл. При вводе 800 мл этилацетата отделяется твердое вещество, которое извлекается, промывается 500 мл простого этилового эфира и высушивается при 35oC в вакууме в течение ночи, в результате получается 44,2 г (выход примерно 98) чистого конечного соединения.
С. Получение N63-карбоксиамидов комплекса N15-CBZ-тейкопланина A2 и его отдельных компонентов 1 5.
Раствор 16 г (примерно 8 ммоль) N15-CBZ- тейкопланин A2 комплекса (или его отдельного компонента 1 5), цианометилового сложного эфира и большого избытка (50 100 ммоль) подходящего аминового реагента в 160 мл DMF или DMSO перемешивается при комнатной температуре в течение 60 120 мин, после чего вводится 160 мл абсолютного этанола, а затем 1,5 л этилацетата. Отделяется твердый продукт, который извлекается путем фильтрации и промывается 500 мл простого этилового эфира, затем высушивается при комнатной температуре в воздухе, в результате получается порошок (обычно выход > 85), который является достаточно чистым продуктом (как показывает жидкостная хроматография HPLC, степень чистоты > 90) для последующего этапа гидрогенизации.
D. Получение N63-карбоксиамидов комплекса тейкопланина A2 и его отдельных компонентов 2 5.
Продукт, полученный как описано выше, (5 ммоль) растворяется в 500 мл смеси метанол 0,4 н. соляная кислота (7 3, об/об) и полученный раствор гидрогенизируется при комнатных температуре и давлении в присутствии 5 Pd/C (5 г). Как только реакция завершается (как показывает HPLC) катализатор удаляется путем фильтрации через слой целита (BDH 545). Величина pH чистого фильтрата доводится до 6,5 посредством 11 н. NaOH и вводится 500 мл и н-бутанола. Полученная смесь концентрируется при 40oC при пониженном давлении до объема примерно 150 мл, затем вводится 350 мл простого этилового эфира и полученный осадок извлекается путем фильтрации. При протекании реакции в субстрате, содержащем производное, соответствующее компоненту 1 комплекса тейкопланина A2, соответствующий конечный продукт не содержит карбоксиамида компонента 1, поскольку он почти полностью преобразуется в карбоксиамид компонента 3.
Е. Очистка продуктов путем хроматографии в колонке с обратимой фазой.
Сырые продукты, полученные как описано выше, (10 г) растворяются в смеси (300 мл) ацетонитрила и воды (1 1, об/об). Затем вводится вода до тех пор, пока не образуется мутный раствор (в любом случае вводится не более 700 мл воды), ввод осуществляется сверху колонки, наполненной 500 г силанизированного силикагеля (0,06 0,2 мин; Merck Co.), полученного в той же смеси растворителей (то есть CH3CH и H2O в соотношении, рассчитанном на основе количества вводимой H2O для получения указанного выше мутного раствора), который получен в начале процесса осаждения. Колонка элюируется линейным градиентом от 10 до 80 ацетонитрила в воде с предварительным доведением величины pH до 2,2 посредством ледяной уксусной кислоты в течение 15 ч при скорости 400 мл/ч, со сбором 25 мл фракций, которые контролируются посредством HLPC. Фракции, содержащие желаемый чистый продукт, соединяются и вводятся достаточное количество и-бутанола для получения после концентрирования при 45oC в вакууме мутного сухого бутанольного раствора. При вводе трех объемов простого этилового эфира отделяется твердый продукт, который извлекается, промывается простым эфиром и высушивается при комнатной температуре в вакууме в течение ночи, в результате получается чистое конечное соединение.
Таким образом получается соединения, отвечающие данному изобретению, в форме свободных оснований (ГВ), когда группа основания, присутствующая в молекуле, является свободной аминогруппой в положении 15 комплекса тейкопланина A2 или когда дополнительная аминогруппа, введенная вместе с амидным заместителем, не является достаточно основной для образования кислотно-аддитивной соли с уксусной кислотой. Иными словами, они извлекаются в форме ацетатов. Получение соответствующих хлоргидратов, когда требуется форма кислотно-аддитивной соли, осуществляется согласно следующим процедурам.
1 ммоль амида комплекса тейкопланина A2 (или его отдельного компонента) либо в форме свободного основания, либо в форме ацетата, растворяется в 10 мл DMF. Затем вводится с одновременным перемешиванием при 5oC 10-ный молярный избыток 10 н. HCl (0,11 мл для одной аминовой функциональной группы, которая должна быть превращена в соль, 0,22 мл для двух аминовых функциональных групп, и т. д.), после чего вводится 40 мл простого этилового эфира. Образующийся осадок затем извлекается путем фильтрации, промывается простым этиловым эфиром и высушивается при комнатной температуре в вакууме в течение ночи (выход > 95).
Когда желательны N63 карбоксамиды компонента 1 комплекса тейкопланина A2 (TG AC1) (или смесь TGAC1-5), осуществляются следующие процедуры.
А'. Получение комплекса N15-трет-бутилоксикарбонил (t-BOC)-тейкопланина A2 и его отдельных компонентов 1 5.
Раствор 10 г (примерно 5 ммоль) комплекса тейкопланина A2 или его отдельного компонента 1 5, 1,2 мл (примерно 8,5 ммоль) триэтиламина (TEA) и 2,4 г (примерно 8 ммоль) трет-бутил-2,4-4, 5-трихлорфенилкарбоната в 100 мл диметилформамида перемешивается при комнатной температуре в течение 24 ч. Затем он вливается в 200 мл воды. Величина pH полученного мутного раствора доводится до 3 посредством 1 н. HCl и он экстрагируется 600 мл смеси н-бутанол этилацетат (35 65, об/об). Органический слой отделяется, промывается водой (2 х 100 мл), затем концентрируется до объема примерно 100 мл при 45oC при пониженном давлении. При вводе этилацетата (примерно 400 мл) отделяется твердый продукт, который извлекается путем фильтрации, промывается простым этиловым эфиром (примерно 200 мл) и высушивается при комнатной температуре в вакууме в течение ночи и в результате получается 10,3 г (примерно 98) чистого конечного соединения.
B'. Получение сложного эфира комплекса t-трет-BOC-тейкопланина A2 и его отдельного компонента 1 5.
Осуществляя процедуру таким же образом, как описано выше (см. пункт B), получают конечное соединение (с выходом примерно 98) из комплекса t-BOC-тейкопланина A2.
C'. Получение N63-карбоксамидов комплекса N63-t-BOC-тейкопланина A2 и его отдельного компонента 1 5.
Осуществляя процедуру таким же образом, как описано выше (см. пункт C), но используя диметилсульфоксид (DMSO) вместо DMF в качестве предпочтительно растворителя, получают конечные соединения из цианометилового сложного эфира N15-BOC-тейкопланинового A2 комплекса с тем же выходом (обычно > 85) и с той же степенью частоты (как показывает HPLC обычно > 90).
D'. Получение карбаксамидов комплекса тейкопланина A2 или его отдельного компонента.
Продукт (N63 карбоксамид комплекса N15-t-BOC-тейкопланина A2 или его отдельного компонента) растворяется в 40 мл сухой трифторуксусной кислоты (TFA) при 10oC. Как только образуется прозрачный раствор (примерно 2 мин) (в любом случае не более чем за 5 мин после ввода TFA) реакционная смесь разбавляется 40 мл метанола при одновременном охлаждении при 10oC. При вводе 420 мл простого этилового эфира отделяется осадок, который извлекается путем фильтрации и промывается простым этиловым эфиром (5 х 200 мл).
Очистка продуктов легко осуществляется путем растворения сырого вещества (5 г) в смеси (150 мл) ацетонитрила и воды (1 1, об/об), доведения величины pH полученного раствора до 6 посредством 1 н. NaOH и последующего разбавления водой и последующей хроматографической процедурой, как описано выше (см. пункт E).
Когда желательны N63-карбоксамиды деглюкотейкопланина (ДТС), осуществляются следующие процедуры.
A''. Получение N15- трет-бутилоксикарбонил(t-BOC)деглюкотейнопланина.
В перемешиваемый раствор 45 г (примерно 37 ммоль) антибиотика L17392 (деглюкотейнопланин) в 600 мл DMF вводят 19,3 г (примерно 65 ммоль) трет-бутил-2,4,5-трихлорфенилкарбоната и 10,2 мг (примерно 74 ммоль) TEA. Полученная реакционная смесь перемешивается при комнатной температуре в течение 24 ч, после чего она вливается в 1,5 л воды. Величина pH полученного раствора доводится до 3 посредством 1 н. соляной кислоты, затем он экстрагируется 3 л смеси этилацетата и н-бутанола (2 1, об/об). Органический слой отделяется, промывается 1 л воды, затем концентрируется при 40oC в вакууме до объема примерно 300 мл. При вводе 700 мл простого этилового эфира отделяется твердый продукт, который извлекается путем фильтрации, промывается 200 мл простого этилового эфира и высушивается при комнатной температуре в вакууме в течение ночи, в результате получается 44 г (92) чистого конечного соединения.
B''. Получение цианометилового сложного эфира N15-t-BOC-деглюкотейкопланина.
Раствор 44 г (примерно 33 ммоль) N15-t-BOC-деглюкотейкопланина, 4,7 мл (примерно 34 ммоль) TEA и 44 мл хлорацетонитрила в 440 мл DMF перемешивается при комнатной температуре в течение 20 ч, после чего вводится 1 л этилацетата и осадок извлекается путем фильтрации. Он снова растворяется (примерно 46г) в 1,5 л смеси метанол вода (1 2, об/об) и величина pH полученного раствора доводится до 5,6 посредством ледяной уксусной кислоты.
После ввода 2 л н-бутанола наибольшая часть метанола испаряется при 30oC в вакууме и органический слой отделяется, промывается 1 л воды, затем концентрируется при 35oC в вакууме до конечного объема примерно 300 мл. При вводе 700 мл этилацетата образуется твердый осадок, который извлекается путем фильтрации, промывается 500 мл простого этилового эфира, затем высушивается при комнатной температуре в вакууме в течение ночи, в результате получается 42,5 г (96) чистого конечного соединения.
C''. Получение N63 карбоксамидов N15-t-BOC-деглюкотейкопланина.
В перемешанный раствор 14 г (примерно 10 ммоль) N105-t-BOC-деглюкотейкопланина и большого избытка (100 150 ммоль) соответствующего аминового реагента в 200 мл МГ вводят 8,9 мл (примерно 150 ммоль) ледяной уксусной кислоты при комнатной температуре. Молярное количество ледяной уксусной кислоты зависит от структуры аминового реагента. Фактически, для 1 ммоль амина требуется 0,5 ммоль ледяной уксусной кислоты, когда амин не содержит дополнительных основных функциональных групп, 1 ммоль ледяной уксусной кислоты, когда амин содержит одну дополнительную основную функциональную группу, 2 ммоль, когда амин содержит две дополнительные основные фукциональные группы и т. д. Хотя присутствие уксусной кислоты необязательно для конденсации, иногда она бывает нужна для устранения боковой эпимеризации молекулы в C3 положении, которое может происходить в основных условиях процесса.
Кроме того, присутствие кислоты не оказывает влияния на скорость реакции конденсации в большинстве случаев.
После 3 6 ч (реакция, за исключением некоторых случаев, завершается в течение 3 ч) вводится 600 мл этилацетата и полученный осадок извлекается путем фильтрации, промывается 200 мл простого этилового эфира и высушивается при комнатной температуре в вакууме, в результате получается продукт, достаточно чистый для последующего этапа удаления защиты (выход > 75).
D''. Получение N63 карбоксиамидов деглюкотейкопланина.
Раствор 1 ммоль продукта, полученного как описано выше, который имеет общий титр HPLC > 85 и содержит ацетат аминового реагента как основную примесь, в 25 30 мл безводной трифторуксусной кислоты (TFA) перемешивается при комнатной температуре в течение 20 мин, затем растворитель выпаривается при 25oC при пониженном давлении. Маслянистый осадок повторно растворяется в 50 мл смеси вода ацетонитрил (6 4, об/об) и полученный раствор разбавляется водой до начала осаждения. Величина pH полученной суспензии доводится до 3,0 посредством 1 н. соляной кислоты (если это необходимо) и полученный раствор вводится сверху в колонку, содержащую 100 г силанизированного силикагеля (0,06 0,2 мин, Merck Co.) в воде.
E''. Очистка продуктов путем колончатой хроматографии с обратимой фазой.
Колонка, заполненная продуктом, как описано выше, элюируется 1 л воды, затем осуществляется элюирование линейным градиентом от 10 ацетонитрила в воде до 50 ацетонитрила в 0,01 н. соляной кислоты в течение 15 ч при скорости потока 200 мл/ч и при этом собирают фракции объемом 10 мл. Фракции, содержащие чистый продукт, сливаются, и добавляется достаточное количество н-бутанола для получения после концентрирования полученной смеси мутного сухого бутанольного раствора (30 100 мл). При вводе трех объемов простого этилового эфира отделяется твердый продукт, который извлекают путем фильтрации, промывают простым этиловым эфиром и высушивают при комнатной температуре в вакууме в течение 2 3 дней, в результате получаются конечные чистые амиды деглюкотейкопланина в форме хлоргидратов.
Соответствующие трифторацетаты получаются путем осуществления оказанной выше хроматографической процедуры очистки, но с элюированием линейным градиентом 10 60 ацетонитрила в воде и с поддержанием величины pH элюента равной 2,5 путем ввода трифторуксусной кислоты.
Используя
соответствующие
реагенты TGAC, его отдельные компоненты, DTG или DMTAC и амины формулы
-NHR1-алк1-[X-алк2]p- [T-алк3]q-W
в описанных
выше условиях получают соединения, приведенные в табл. 4.
Пример 31. Получение соединения 31 формулы I
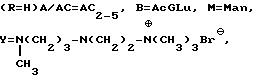
Раствор 2 г (примерно 1 ммоль) сложного цианометилового эфира, N15-CBZ-тейкопланинового A2 комплекса, полученного как описано выше, и 2 мл 1,3-диметил-1,3-пропандиамина в 20 мл DMF перемешивается при комнатной температуре в течение 2 ч, после чего вводится 20 мл абсолютного этанола, а затем 200 мл этилацетата. Отделяется твердый продукт, который извлекается путем фильтрации, промывается 50 мл простого этилового эфира и высушивается в вакууме при комнатной температуре в течение ночи и в результате получается 1,95 г чистого комплекса N15-CBZ-тейкопланина A2-1-метил-3-(метиламино)пропил-амида.
В перемешанный раствор 1,37 г (0,65 ммоль) указанного выше соединения в 100 мл сухого материала вводят при комнатной температуре 1 г (9,4 ммоль) безводного бикарбоната натрия и 2,5 г (10,1 ммоль) 2-бромэтилтриметиламмонийбромида. Реакционная смесь перемешивается при 45oC в течение трех дней, затем охлаждается до 10oC и вливается в 100 мл воды. Метанол выпаривается при 30oC при пониженном давлении и водная фаза экстрагируется 300 мл смеси n-BuOH/EtOAc (1/2 об/об). Органический слой отделяется и концентрируется при 40oC при пониженном давлении до небольшого объема (примерно 20 мл). При вводе 180 мл простого этилового эфира осажденный твердый продукт (1,12 г N15-CBZ предшественника указанного в названии соединения) извлекается путем гидрогенизации в тех же условиях, что описаны в примере 1 и в результате получается 0,45 г соединения 31.
Пример 32. Получение соединения 32 формулы I
(R=H, A/AC=H, B=H, M=H,
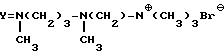
Осуществляя процедуру таким же образом, как описано в примере 31, но используя раствор 2 г сложного цианометилового эфира N15-CBZ-деглюкотейкопланина, получают соединение 32.
Пример 33. Получение соединения 33 формулы I
(R=H, A/AC=AC2-5, B=AcGlu, M=Man,
Y=-H(CH2)3NH(CH2)4
HCOOCH(CH3)OCOCH3
в смеси с
YNH(CH2)4NH (CH2)3NHCOOCH(CH3)OCOCH3).
В перемешанный раствор 2 г (0,9 ммоль) N15-CBZ-производного соединения 5, полученного как описано в примере1, в 50 мл сухого диметилформамида, вводят 1,2 г (11 ммоль) безводного карбоната натрия и 2,7 г (10 ммоль) альфа-ацетокси-этил-пара-нитрофенилкарбоната при комнатной температуре. По прошествии 3 ч реакционная смесь вливается в 500 мл этилацетата и осажденный твердый продукт извлекается, промывается 100 мл этилацетата и гидрогенизируется, как описано выше в примере 1, и в результате получается 0,57 г конечного соединения 33.
Пример 34. Получение
соединения 34 формулы I
(R=H, A/AC=H, B=H, M=H,
Y=NH(CH2)3NH(CH2)4 NHCOOCH(CH3)OCOCH3
в системе с
Y=NH(CH2)4NH(CH2)3NHCOOCH(CH3) OCOCH3)
Осуществляя процедуру таким образом, как
описано в примере 32, но используя 2 г N15-CBZ-производного соединения 23, получают 0,6 соединения 34.
Примеры 35-36. Получение соединения 35 формулы I
(R=H,
A/AC=AC2-5, B=AcGlu, M=Man,
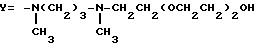
и соединения 36 формулы I
(R=H, A/AC=AC2-5, B=AcGlu, M=Man,

В перемешанную суспензию 5,3 г (примерно 2,5 ммоля) N15 -CBZ-тейкопланина А2 в 560 мл метанола вводят 1-метил-3(метиламино)пропил-амид, полученный как описано выше в примере 31), 17 мл соответствующего хлорэтокси-оксиэтилового реагента формулы ClCH2CH2(OCH2CH2)2OH и ClCH2CH2OCH2CH2OH, соответственно, и 1,86 г (13,5 ммоль) карбоната калия при комнатной температуре. После перемешивания при 45oC в течение 3 ч реакционная смесь охлаждается до 15oC и величина pH ее доводится до 6 посредством 1 н. HCl.
Метанол выпаривается при 30oC при пониженном давлении и кислотный остаточный продукт гидрогенизируется, как описано в примере 1, и в результате получается 1,9 г соединения 35 или 0,97 г соединения 36.
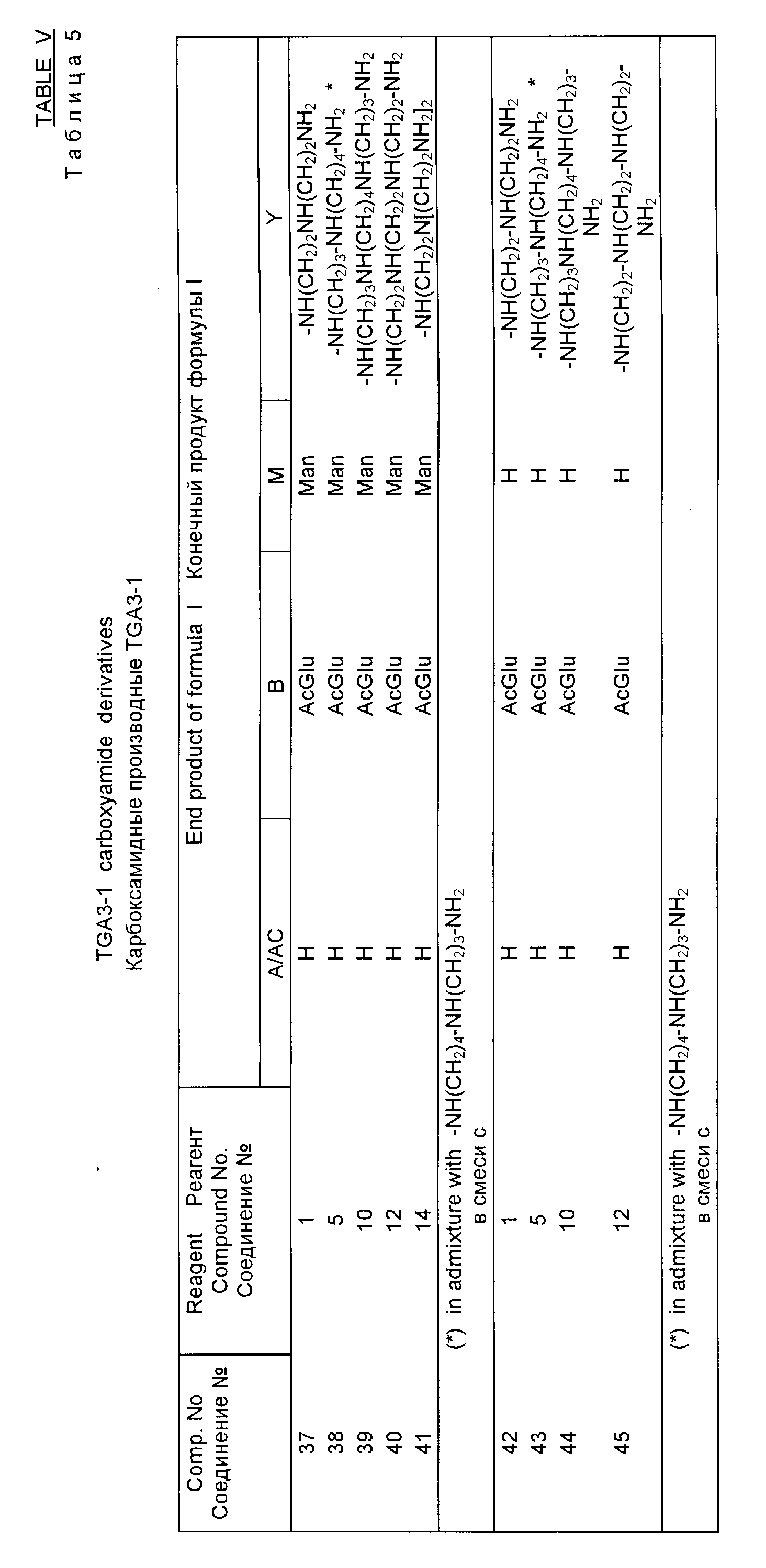
Примеры 37-41. Получение амидных производных ТСАЗ-1.
Раствор 4 г (примерно 2 ммол) подходящего амидного производного комплекса тейкопланина A2 или его отдельного компонента, полученного как описано выше и представленного в табл. 5, в 100 мл 90-ной водной трифторуксусной кислоты перемешивается при комнатной температуре в течение 2 ч, после этого растворители выпариваются и маслянистый остаточный продукт повторно растворяется в 200 мл H2O. После доведения величины pH до 8 полученный раствор вводится в колонку, наполненную 400 г силанизированного силикагеля в H2O. Осуществляется хроматография, как описано выше в примере 1, и в результате получается конечное желаемое соединение.
Примеры 42-45. Получение производных TGA3-2 амида.
Суспензия 4 г (примерно 2 ммоль) соответствующего амидного производного тейкопланинового соединения, полученного как описано выше и представленного ниже в табл. 6, в 80 мм 1, 2-диметоксиэтана (ДМЕ), перемешивается при комнатной температуре в течение 2 дней с одновременным пропусканием пузырьков HCl, после чего нерастворимое твердое вещество извлекается путем фильтрации. В результате очистки путем хроматографии в колонке, как описано выше (пример 1), получаются конечные желаемые соединения.
Примеры 46 55.
A'''. Общая процедура (с использованием дифенилфосфоразида).
В перемешанный раствор 6 ммоль тейкопланина A2 или его отдельного компонента (или смеси его компонентов в любой пропорции)) или N15-трет-бутилоксикарбонил (t-BOC)деглюкотейкопланина в 60 мл диметилсульфоксида (DMSO) вводят 30 ммоль подходящего промежуточного амина (полученного как описано ниже) и 10 ммоль дифенилфосфоразидата (DPPA) при температуре 0 5oC. После перемешивания при комнатной температуре в течение ночи вводят 240 мл этилацетата и осажденный твердый продукт извлекается и очищается в хроматографической колонке с обратимой фазой, как было описано ранее (метод Е), в результате чего получаются чистые TGAC амиды или N15 -t-ВОС-деглюкотейпланинамиды (ВОС-ДТС-амиды).
В случае амидов ВОС-ДТС или амидов, содержащих ВОС-защитные группы на амидном звене, ВОС-защитные группы удаляются путем растворения 1 ммоль этих соединений в 30 мл безводной трифторуксусной кислоты при комнатной температуре и с осуществлением процедуры таким же образом, как было описано ранее (например метод D'' для получения N63-карбоксамидов DTG).
B'''. Получение промежуточных аминов соединений 46 55.
1. Диамин-O, O'-бис(2-аминопропил)полиэтиленгликоль 1900 (Jeffamine TM 2001) был получен от Fluka Chemie AG (промежуточный амин соединения 46).
2. Для промежуточного взаимодействия аминов соединений 47 52 предварительно был получен общий промежуточный
ди-(3-ВОС-аминопропил)амин
формулы
BOC-NH-(CH2)3-NH-(CH2)3-NH-BOC
нижеследующим образом. Раствор 142 г
2-(трет-бутоксикарбонилоксиимино-2-фенила-цетонитрила (BOC-ON,
Aldrich-Chemie) в 300 мл тетрагидрофурана (THF) вводится по каплям при 10oC в перемешанный раствор 42 мл
бис-(3-аминопропил)амина (Fluka Chemie AG) в 400 мл ТНГ. После 16 ч при комнатной
температуре растворитель выпаривается и маслянистый остаточный продукт растворяется в 1 л этилацетата. Полученный
раствор промывается 1 н. NaOH (200 мл) и затем водой (2 х 300 мл), после этого он
смешивается с 0,01 н. HCl (2 х 500 мл). Величина pH водной фазы доводится до 8 посредством 1 н. NaOH и она
экстрагируется 500 мл н-бутанола. Органический слой отделяется, промывается 250 мл воды и
затем концентрируется до конечного объема примерно 70 мл. При выдержке в течение ночи при 6oC
образуются кристаллы, которые извлекаются путем фильтрации и в результате получается 75 г
чистого конечного соединения в виде свободного основания.
1Н ЯМР: 2,93, 2,44 1,47 (CH2), 1,38 (N-ВОС) 6,68 (NH).
3. N', N''-ди-t-ВОС-трис-(3-аминопропил)амин (для производных 47 50).
В перемешанный раствор 45 г указанного выше ди-t-ВОС промежуточного триада в 500 мл абсолютного этанола вводят 21 мл 3-бром-пропионитрила и 25 г карбоната калия при комнатной температуре. Реакционная смесь перемешивается в течение ночи, затем она фильтруется и концентрируется до конечного объема примерно 100 мл, после чего она разбавляется 800 мл воды. Полученный раствор (pH 8) экстрагируется этилацетатом (2 х 800). Органический слой отделяется и промывается водой (2 х 200 мл), после чего он концентрируется до конечного объема примерно 100 мл. При выдержке при 6oC в течение ночи отделяется кристаллический твердый продукт, который извлекается путем фильтрации и в результате получается 34 г ди-(3-t-ВОС-аминопропил)амино-1-пропионитрила.
1Н ЯМР: 2,94, 2,63, 2,54, 2,37, 1,47, (CH2, 1,36 (N-ВОС), 6,73 (H).
Данный продукт растворяется в 200 мл этанольного раствора, содержащего 5 г NaOH. В полученный раствор вводится 4 г никеля Рэноя, как активного катализатора (Aldrich-Chemie) и суспензия гидрогенизируется при давлении 2,5 атм. в течение 10 ч. Катализатор отфильтровывается и растворитель выпаривается. Маслянистый остаточный продукт растворяется в 500 мл этилацетата и полученный раствор промывается водой (2 х 100 мл), после чего органический растворитель выпаривается и в результате получается примерно 34 г конечного желаемого соединения.
1Н ЯМР: 2,93, 2,54, 2,32, 1,49 (CH2), 1,41 (N-ВОС); 6,77 (NH).
4. 2-(3-аминопропил)-3-(3,3-диметиламинопропил)-амино-1-пропиламино (для производного 51).
В перемешанный раствор 19 г хлоргидрата 3, 3- диметил-амино-1-пропилхлорида в 400 мл абсолютного этанола вводят при комнатной температуре 20 г ди-t-ВОС промежуточного триамина и 28 г карбоната калия, после чего вводят 3 г иодида калия. Реакционная смесь нагревается с обратным холодильником в течение 6 ч, затем она фильтруется и растворитель выпаривается. Остаточный продукт повторно растворяется в 400 мл воды и полученный раствор экстрагируется 600 мл этилацетата. Органический слой отделяется, промывается водой (2 х 200 мл) и затем растворитель выпаривается и в результате получается маслянистый остаточный продукт (8,7 г), ди-t-ВОС производное конечного соединения, достаточное чистое для последующего этапа.
1Н ЯМР: 2,91 2,42, 2,31, 2,16, 1,47 (CH2), 1,36 (N-ВОС), 2,09 (NCH3).
Раствор данного продукта в 30 мл метиленхлорида обрабатывается 30 мл сухой трифторуксусной кислоты при комнатной температуре в течение 2 ч, после чего растворители выпариваются. Маслянистый остаточный продукт растворяется в 40 мл абсолютного этанола и сухой HCl пропускается в виде пузырьков при комнатной температуре до тех пор, пока не произойдет полное осаждение продукта. После фильтрации получается 3,8 г конечного соединения в форме тетра-хлоргидрата.
1Н ЯМР: 3,2 2,91 (6-CH2); 2,13 1,90 (3-CH2); 2,73 (NCH3).
Для концентрации с ВОС-DTG используется свободное основание, которое получается путем растворения тетра-хлоргидрата (10 ммоль) в 1 н. NaOH (40 мл), после чего осуществляется выпаривание досуха образующегося раствора. Остаточный продукт затем суспензируется в метиленхлориде (100 мл) и нерастворимое вещество отфильтровывается. Растворитель выпаривается и маслянистый остаточный продукт используется как таковой без дополнительной очистки.
5. 3-(3-аминопропил)-3-(2,2-диэтиламиноэтил)-амино-1-пропиламин (для производного 52).
Осуществляя процедуру точно таким же образом, как описано выше, но используя хлоргидрат 2, 2-диэтиламино-1-этилхлорида (21 г) для реакции с ди-t-ВОС промежуточным триамином (20 г) сначала получают 11 г ди-t-ВОС производного конечного соединения. Затем защитные группы удаляются путем аналогичной обработки трифторуксусной кислотой в растворе метиленхлорида. В конечном итоге получается свободное основание (в виде масла), как описано выше, из которого получается конечное соединение (8,2 г).
1Н ЯМР: 2,6 2,3 (8-CH2), 1,42 (2-CH2), 0,92 (2-CH3).
Ход этих реакций и гомогенность конечных полиаминов проверяется методом TLC на предварительно покрытых силикагелем 60 F254 (Merck Co.) пластинах с использованием смеси метиленхлорид метанол (9 1, об/об), содержащей 1 гидроокиси аммония как подвижной фазы. Пятна проявляются с помощью иода.
6. 4-(3,3-диметиламинопропил)пиперазин (для производного 53).
В перемешанный раствор 15,8 г 3, 3-диметиламино-1-пропилхлорида в 300 мл абсолютного этанола вводят 9 мл 1 -бензилпиперазина и 14 г карбоната калия. Реакционная смесь перемешивается при нагревании с обратным холодильником в течение 6 ч, после чего она охлаждается при комнатной температуре и фильтруется. Растворитель выпаривается и маслянистый остаточный продукт растворяется в 300 мл воды. Полученный раствор экстрагируется метиленхлоридом (2 х 200 мл). Органический слой отделяется, промывается 200 мл воды и затем растворитель выпаривается. Маслянистый остаточный продукт (9 г) растворяется в 300 мл 95-ного этанола и гидрогенизируется (25oC, 1 атм.) над 3 г 10 Pd/C. В течение 6 ч абсорбируется примерно 1 л H2. Катализатор отфильтровывается и сухой HCl пропускается в виде пузырьков в прозрачный фильтрат. Отделяется твердый продукт, который извлекается, промывается абсолютным этанолом и высушивается в вакууме при комнатной температуре в течение ночи, и в результате получается 7 г чистого конечного соединения в виде три-хлоргидрата.
1Н ЯМР: 2,79, 2,53, 2,30 (CH2 пиперазин), 2,79, 2,23, 2,15 (CH2 диметиламинопропил), 2,10 (NCH3).
Свободное основание получается путем растворения данного трихлорида (6 г) в 2 н. NaOH (30 мл) с последующей экстракцией метиленхлоридом (170 мл) и выпариванием органического растворителя. Полученный в результате маслянистый остаточный продукт используется без дальнейшей очистки в процессе получения соединения 53.
7. N, N'-Бис(3-аминопропил)нонан-1,5-диамин и N, N'-Бис(3-аминопропил)декан-1,5-диамин.
Данные производные являются известными соединениями и получаются согласно известному способу (см. Israck M. J. Rosenfield S. S. Modest E. J. J. Med. Chem. 1964, 7, 710) путем моно- и ди-цианоэтилирования соответствующих альфа-, омега-алкилендиаминов с последующим каталитическим восстановлением нитрилов обычно в мягких условиях.
Для соединений, получаемых согласно процедуре A''' (46 55), следует обратиться к табл. 6.
HPLC анализа осуществляется с использованием насоса модели Вариан 5000 LC, снабженным инжектором модели Rheodyne 7125 и УФ-детектором на 254 нм.
Колонки: первая колонка (1,9 см)
Hibor hichro Cart. 25 4 (Merck) с
первым наполнителем Zichrozorb RP-8 (20 30 мкм) и последующая колонка Hibar RT 250-4 (Merck) с первым наполнителем Lichrosorb RP-8 (10 мкм)
Элементы: А, 0,2
водный HCOONH4; B,
CH3CH.
Скорость потока: 2 л/мин.
Инжекция: 20 мкл.
Элюирование: линейный градиент 20 60 B и A в течение 30 мин. Время удерживания в колонке некоторых типичных соединений представлено в табл. 7.
Кислотно-основное титрование. Продукты растворяют в MCS (метилцеллозоль) H2O (4 1, об/об), затем вводится избыток 0,01 М HCl в той же смеси растворителей и полученные растворы титруются 0,01 н.NaOH. Эквивалентная масса некоторых типичных соединений представлена в табл. 8.
1H ЯМР: спектр1Н ЯМР при 500 МГц регистрируется в температурном диапазоне 20 30oC и спектрометре Bruker AM 500 в DMSO-D6 с использованием тетраметилсилана (TMS) в качестве внутреннего стандарта (дельта 0,00 ч./млн.).
В табл. 7 представлено время удерживания (tR), определяемое как описано выше, для некоторых типичных соединений изобретения.
Далее приводятся значения данных1Н ЯМР некоторых типичных соединений, зарегистрированные в DMSO-D6 с использованием тетраметилсилана как внутреннего стандарта (дельта 0,001 ч./млн.).
Соединение 2. 3,61, 2,95 (CH2 боковая цепь); 0,83, 1,18, 1,46, 2,02 (ацильная цепь); 4,18 5,62 (пептидные CH); 1,89 (ацетилглюкозамин); 6,18 8,45 (ароматические протоны и пептидные NH).
Соединение 4. 3,45, 2,82, 2,63, 2,08, 1,66 (CH2 боковая цепь); 0,87, 1,23, 1,45, 2,01 (ацильная цепь); 1,83 (ацетилглюкозамин); 4,15 5,71 (пептидные CH); 6,26 8,56 (ароматические протоны и пептидные NH).
Соединение 6. 3,52, 2,73, 2,581,91, 1,56 (CH2 боковая цепь); 0,84, 1, 15, 1,46, 2,02 (ацильная цепь); 1,82 (ацетилглюкозамин); 3,42 (манноза); 4,15 5,69 (пептидные CH); 6,29 8,53 (ароматические протоны и пептидные NH).
Соединение 7. 3,68 3,12, 2,98, 2, 05 (CH2 боковая цепь): 0,84, 1,16, 1,44, 2,01 (ацильная цепь); 1,89 (ацетилглюкозамин), 3,42 (манноза), 4,13 5,58 (пептидные CH); 6,21 8,53 (ароматические и пептидные NH).
Соединение 8. 3,68, 2,92, 1,98 (CH2 боковая цепь); 0,82, 1,25, 1,43, 2,01 (ацильная цепь); 1,87 (ацетилглюкозамин); 4,17 5,65 (пептидные CH); 6,26 8,57 (ароматические протоны и пептидные NH).
Соединение 9. 3,48, 2,98, 1,98 (CH2 боковая цепь), 0,82, 1,12, 1,43, 2,01 (ацильная цепь); 1,86 (ацетилглюкозамин); 4,16 5,62 (пептидные CH); 6,18 8,58 (ароматические протоны и пептидные NH).
Соединение 11. 3,71 2,93, 1,98, 1,73 (CH-боковая цепь); 0,83, 1,23, 1,47, 2,02 (ацильная цепь); 1,89 (ацетилглюкозамин); 4,13 5,58 (пептидные CH); 6,21 8,62 (ароматические протоны и пептидные NH).
Соединение 15. 3,52, 3,13 (CH2 пиперазин); 3,42 2,07 (CH2 боковая цепь); 0,83, 1,16, 1,45, 2,02 (ацильная цепь); 1,88 (ацетилглюкозамин); 4,16 5,32 (пептидные CH); 6,18 8,53 (ароматические протоны и пептидные NH).
Соединение 21. 3,52, 3,04, 2,81 (CH2); 4,15 5,62 (пептидные CH); 6,18 8,62 (ароматические протоны, пептидные NH).
Соединение 22. 3,17, 2,93, 2,83, 1,87, 1,80 (CH2 боковая цепь); 4,15 5,61 (пептидные CH); 6,18 8,53 (ароматические протоны и пептидные NH).
Соединение 23. 3,42, 2,98 2,71, 1,72 1,61 (CH2 спермидин); 5,15 5,63 (пептидные CH); 6,19 8,43 (ароматические протоны и пептидные NH).
Соединение 24. 3,38, 3,12, 2,98 (CH2-N); 1,89 (CH2); 4,17 - 5,58 (пептидные CH); 6,18 8,48 (ароматические протоны, пептидные NH).
Соединение 25. 3,34, 3,08, 2, 84, 1,78 (CH2 боковая цепь); 4,16 - 5,58 (пептидные CH); 8,31 8,48 (ароматические протоны и пептидные NH).
Соединение 26. 3,36 2,98, 2,87 (CH2-N); 1,78, 1,77 (CH2); 4,15 5,62 (пептидные CH); 6,18 8,42 (ароматические протоны, пептидные NH).
Соединение 27. 3,12, 3,03 2,82, 1,88, 1,81, 1,65 (CH2 боковая цепь); 4,15 5,62 (пептидные CH); 6,19 8,41 (ароматические протоны и пептидные NH).
Соединение 29. 3,48 3,12, 2,64 (CH2-N); 4,18 5,61 (пептидные CH); 6,21 8,70 (ароматические протоны и пептидные NH).
Соединение 30. 3,49, 3,11, 2,95, 2,85, 1,85 (CH2 боковая цепь); 4,18 5,81 (пептидные CHN); 6,21 8,56 (ароматические протоны, пептидные NH).
Соединение 31. 3,01 (CH3); 2,95, 2,21, 1,68 (CH2 боковая цепь); 2,28, 2,06 (NCH3); 0,83, 1,21, 1,46 (ацильная цепь); 1,86 (ацетилглюкозамин); 3,48 (мнноза); 4,17 5,82 (пептидные CH); 6,14 8,62 (ароматические протоны и пептидные NH).
Соединение 34. 3,03 2,81, 1,82, 1,62, 1,43 (CH2-боковая цепь); 2,01 (CH3-CH); 1,23 (CH3); 4,13 5,62 (пептидные CH); 6,19 - 8,47 (ароматические протоны и пептидные NH).
Соединение 35. 5,25 (CH2-O); 4,51 (CH2OH); 2,06 (NCH3); 0,81, 1,23, 1,45, 2,02 (ацильная цепь); 1,87 (ацетилглюкозамин); 4,12 5,69 (пептидные CH); 6,14 8,48 (ароматические протоны и пептидные NH).
Соединение 36. 5,22 (CH2O); 3,72, 3,82 (CH2 боковая цепь); 1,93 (NCH2); 0,83, 1,14, 1,43, 2,01 (ацильная цепь); 1,87 (ацетилглюкозамин); 4,18 5,72 (пептидные CH); 6,31 8,45 (ароматические протоны и пептидные NH).
Соединение 44. 3,68, 3,34, 2,95, 1,99, 1,84 (Спермин); 1,87 (ацетилглюкозамин); 4,18 5,61 (пептидные CH); 6,19 8,56 (ароматические протоны и пептидные NH).
Соединение 39. 3,71, 3,372,98, 1,98, 1,82 (спермин); 1,86 (ацетилглюкозамин); 3,44 (манноза); 4,16 5,58 (пептидные CH); 6,29 8,54 (ароматические протоны и пептидные NH).
Соединение 46. 3,41, 3,23, 3,16, 2,94 (CH2 боковая цепь); 1,121,23, (CH3 боковая цепь); 0,81, 1,151,46, 2,00 (ацильная цепь); 1,86 (ацетилглюкозамин); 4,16 5,59 (пептидные CH); 6,23 8,01 (ароматические протоны, пептидные NH).
Соединение 47. 3,45, 3,09, 2, 35, 1,84 (CH2 боковая цепь); 0,82, 1,14, 1,46, 2,03 (ацильная цепь); 1,68 (ацетилглюкозамин); 4,12 5,62 (пептидные CH); 6,23 8,43 (ароматические протоны, пептидные NH).
Соединение 50. 3,43, 3,24, 2,85, 1,84 (CH2 боковая цепь); 4,12 - 5,62 (пептидные CH); 6,12 8,51 (ароматические протоны; пептидные NH).
Соединение 51. 3,45, 3,39, 3,05, 2, 91, 2,12, 1,98 (CH2 боковая цепь); 2,74 (NCH3); 4,13 5,59 (пептидные CH); 6,18 8,61 (ароматические протоны, пептидные NH).
Соединение 52. 3,48, 3,12, 1,89 (CH3-боковая цепь); 1,18 (2CH3-этил); 4,12 5,61 (пептидные CH); 6,20 8,52 (ароматические протоны, пептидные NH).
Соединение 53. 3,42, 3,39, 3,30, 1,98 (CH2-p p, CH2-пропил); 2,75 (NCH3); 4,05 5,63 (пептидные CH); 6,32 - 8,52 (ароматические протоны, пептидные NH).
Соединение 55. 3,32, 2,98, 2,76, 1,86, 1,52, 1,24 (CH2 боковая цепь), 4,13, 5,62 (пептидные CH), 6,31 8,42 (ароматические протоны, пептидные NH).
Соединение 54. 3,37, 3,34, 3,07, 2,86, 2,78, 1,75, 1,59, 1,26, (CH2-боковая цепь), 4,14 5,61 (пептидные CH), 6,21 8,34 (ароматические протоны, пептидные NH).
Пример на фармацевтическую композицию, мг:
Соединение N 50 200
NaCl
24
Вода 3,
Реферат
Использование: в качестве лекарственных соединений против грамм-положительных и грамм-отрицательных бактерий. Сущность: продукт - C63-амидные производные тейкопланина, которые получают реакцией сложного, предпочтительно ацетоэфира тейкопланина с соответствующим ди- или полиалкиламином. 3 с. и 18 з. п. ф-лы, 8 табл.
Формула
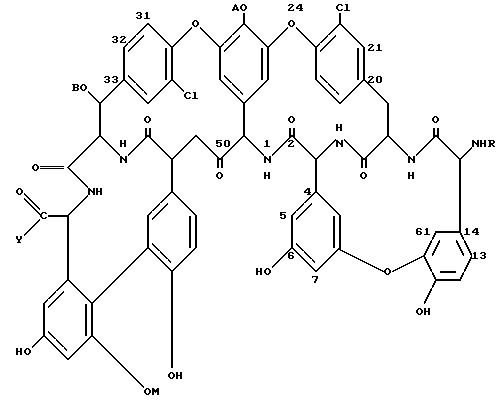
где R водород или защитная группа аминовой функции;
Y радикал -NR1-alK1-[X-alK2]p - [T-alK3]q-W, где R1 водород, C1 C4- алкил, alK1 alK3 каждый независимо друг от друга нормальный или разветвленный C2 C10-алкилен, p 1 42 целое число, q 0 2 целое число, Х группа -NR2 или кислород, R2 водород, C1 C4- алкил, группа alK4NR3R4, где alK4 нормальный или разветвленный алкилен C2 C4, R3 водород, C1 C4-алкил, R4 водород, C1 C2-алкил, или R1 и R2, взятые вместе, образуют C2 C4-алкилен, связывающий два атома азота, при условии, что p 1; Т кислород, группа NR5, где R5 водород, C1 C4-алкил, или R2 и R5, вместе взятые, образуют C2 C4-алкилен, связывающий два атома азота, при условии, что p q 1, W оксигруппа, NR8R9, где R8 водород, C1 C6-алкил, R9 водород, C1 - C6-алкил, COOR10, где R10 C1 - C6-ацилокси- C1 C4-алкил или группа N+R11R12R13Anθ, где каждый R11, R12 и R13 C1 C4-алкил, Anθ - анион, образованный от фармацевтически приемлемой кислоты, при условии, что когда Х -NR2- и одновременно с этим p 1 и q 0, то W имеет значение, отличное от оксигруппы;
A водород или N[(C9-C12)]алифатический ацил] -бета-D-2-диокси-2-аминоглюкопиранозил;
B водород, N-ацетил-бета-D-2-деокси-2-аминоглюкопиранозил;
M водород или альфа-маннопиранозил,
или их фармацевтически пригодные кислотно-аддитивные соли при условии, что B водород в случае, когда A и М одновременно водород.
NHR1-alk-[X-alk2]p-[T-alk3]q-W,
где R1, alk1 alk3, X, T, p, q и W имеют значения, указанные в п.1.
Комментарии