Ингибиторы вируса гепатита с - RU2298001C2
Код документа: RU2298001C2
Описание
ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение в целом относится к антивирусным соединениям и, более конкретно, к соединениям, которые ингибируют функционирование NS3 протеазы, кодируемой вирусом гепатита С (HCV, ВГС), к композициям, содержащим такие соединения, и к способам ингибирования функционирования NS3 протеазы.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
HCV является основным человеческим патогеном, поражающим, по оценкам, 170 миллионов человек, приблизительно в пять раз больше числа инфицированных вирусом иммунодефицита человека типа I. У значительной части этих HCV-инфицированных развиваются серьезные прогрессирующие заболевания печени, включая цирроз и гепатоцеллюлярный рак (Lauer, G.M.; Walker, B.D. N. Engl. J. Med, (2001), 345, 41-52).
В настоящее время для наиболее эффективной терапии HCV используют комбинацию альфа - интерферона и рибавирина, что дает длительный эффект у 40% больных (Poinard, Т. et al. Lancet (1998), 352. 1426-1432). Последние результаты клинических испытаний показывают, что пэгированный альфа-интерферон превосходит немодифицированный в качестве агента для монотерапии (Zeuzem, S. et al. N. Engl. J. Med. (2000), 343, 1666-1672). Однако, даже при использовании экспериментальных терапевтических схем, включающих комбинации пэгированного альфа-интерферона и рибаварина, у значительной части пациентов не наблюдается длительного снижения вирусной нагрузки. Таким образом, очевидна и давно ощущается необходимость в создании эффективных лекарственных средств для лечения HCV инфекции.
HCV является позитивным РНК-вирусом. На основании сравнения расшифрованной аминокислотной последовательности и большого сходства в 5' нетранслируемой области HCV (ВГС) был классифицирован как отдельный род семейства Flaviviridae. Все члены семейства Flaviviridae имеют оболочечные вирионы, которые содержат позитивный РНК-геном, кодирующий все известные вирус-специфичные белки с использованием трансляции единственной непрерывной открытой рамки считывания.
Значительная гетерогенность обнаружена в нуклеотидной и кодируемой аминокислотной последовательности по всему ВГС геному. Охарактеризовано, по меньшей мере, шесть основных генотипов и описано более 50 подтипов. Основные генотипы ВГС отличаются по их распространенности в мире, и клиническое значение генетической гетерогенности ВГС остается неясным, несмотря на многочисленные исследования возможного влияния генотипов на патогенез и терапию.
Геном ВГС представлен одноцепочечной РНК, имеющей протяженность около 9500 нуклеотидов и содержащей открытую рамку считывания (ORF), кодирующую единственный большой полипротеин, состоящий, примерно, из 3000 аминокислот. В инфицированных клетках этот полипротеин расщепляется по многим сайтам с помощью клеточных и вирусных протеаз, давая структурные и неструктурные (NS) белки. В случае ВГС на образование неструктурных белков (NS2, NS3, NS4A, NS4B, NS5A и NS5B) влияют две вирусные протеазы. Первая, и все еще плохо охарактеризованная, расщепляет на участке соединения NS2-NS3; вторая представляет собой сериновую протеазу, содержащуюся внутри N-концевой области NS3 (отсюда ее название NS3 протеаза), и опосредует все последующие расщепления по ходу транскрипции (downstream) от NS3, как в цис, в NS3-NS4A сайте расщепления, так и в транс, для остальных сайтов NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. Белок NS4A, по-видимому, выполняет множество функций, действуя как кофактор для NS3 протеазы и, возможно, способствуя локализации NS3 и других компонентов вирусной репликазы в мембране. Образование комплекса NS3 белка с NS4A, по-видимому, является необходимым для событий процессинга, повышая протеолитическую активность во всех сайтах. NS3 белок также проявляет активности нуклеозидтрифосфатазы и РНК геликазы. NS5 представляет собой РНК-зависимую РНК полимеразу, которая участвует в репликации ВГС.
Среди соединений, которые продемонстрировали эффективность при ингибировании репликации ВГС в качестве селективных ингибиторов ВГС сериновой протеазы, пептиды, описанные в Патенте США 6323180.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение охватывает соединение формулы I, включая его фармацевтически приемлемые соли, сольваты или пролекарства,

где:
(a) R1 обозначает C1-8алкил, С3-7циклоалкил или С4-10 алкилциклоалкил;
(b) m обозначает 1 или 2;
(c) n обозначает 1 или 2;
(d) R2 обозначает Н, С1-6алкил, С2-6алкенил или С3-7циклоалкил, каждый из которых, необязательно, замещен галогеном;
(e) R3 обозначает C1-8алкил, необязательно имеющий заместитель: галоген, циано, амино, С1-6диалкиламино, C6-10арил, C7-14алкиларил, С1-6алкокси, карбокси, гидрокси, арилокси, C7-14алкиларилокси, С2-6алкиловый сложный эфир, C8-15алкилариловый сложный эфир; С3-12алкенил, С3-7циклоалкил или С4-10алкилциклоалкил, где циклоалкил или алкилциклоалкил, необязательно, имеют заместитель: гидрокси, С1-6алкил, С2-6алкенил или С1-6алкокси; или R3, вместе с углеродным атомом, к которому он присоединен, образует С3-7 циклоалкильную группу, необязательно замещенную С2-6алкенилом;
(f) Y обозначает Н, фенил, замещенный нитрогруппой, пиридил, замещенный нитрогруппой, или С1-6алкил, имеющий в качестве заместителя циано, ОН или С3-7циклоалкил;
при условии, что если R4 или R5 обозначает Н, тогда Y обозначает Н;
(g) В обозначает Н, С1-6алкил, R4-(С=O)-, R4O(C=O)-, R4-N(R5)-C(=O)-, R4-N(R5)-C(=S)-, R4SO2- или R4-N(R5)-SO2-;
(h) R4 обозначает (i) C1-10алкил, необязательно имеющий заместитель: фенил, карбоксил, С1-6алканоил, 1-3 атома галогена, гидрокси, -OC(O)С1-6алкил, С1-6алкокси, амино, необязательно замещенный С1-6алкилом, амидогруппой или (низший алкил)амидогруппой; (ii) С3-7 циклоалкил, С3-7циклоалкокси или С4-10алкилциклоалкил, каждый из которых, необязательно, замещен гидрокси, карбоксилом, (С1-6алкокси)карбонилом, амино, необязательно замещенным С1-6алкиламидогруппой, или (низший алкил)амидогруппой; (iii) C6-10арил или C7-16арилалкил, каждый из которых, необязательно, замещен С1-6алкилом, галогеном, нитро, гидрокси, амидо, (низший алкил)амидо, или аминогруппой, необязательно замещенной С1-6алкилом; (iv) Het; (v) бицикло(1.1.1)пентан; или (vi) - C(O)OC1-6алкил, С2-6алкенил или С2-6алкинил;
(i) R5 обозначает Н; С1-6алкил, необязательно замещенный 1-3 атомами галогена; или С1-6алкокси, при условии, что R4 обозначает C1-10алкил;
(j) Х обозначает О, S, SO, SO2, ОСН2, СН2O или NH;
(k) R' обозначает Het; или C6-10арил или C7-10алкиларил, необязательно замещенный Ra;
и
(1) Ra обозначает С1-6алкил, С3-7 циклоалкил, С1-6алкокси, С3-7цилоалкокси, галоген-С1-6алкил, CF3, моно- или ди- галоген-С1-6алкокси, циано, галоген, тиоалкил, гидрокси, алканоил, NO2, SH, амино, С1-6алкиламино, ди (С1-6)алкиламино, ди(С1-6)алкиламид, карбоксил, (С1-6)карбоксиэфир, С1-6алкилсульфон, С1-6алкилсульфонамид, ди(С1-6)алкил(алкокси)амин, C6-10арил, C7-14алкиларил или 5-7-членный моноциклический гетероцикл;
при условии, что Х-R' не обозначает
или его фармацевтически приемлемые соль, сольват или пролекарство.
Настоящее изобретение также охватывает композиции, содержащие соединения или их фармацевтически приемлемые соли, сольваты или пролекарства и фармацевтически приемлемый носитель. В частности, настоящее изобретение включает фармацевтические композиции, пригодные для ингибирования ВГС NS3, содержащие терапевтически эффективное количество соединения по данному изобретению, или его фармацевтически приемлемой соли, фармацевтически приемлемого сольвата или пролекарства, и фармацевтически приемлемый носитель.
Кроме того, настоящее изобретение включает методы лечения больных, инфицированных ВГС, заключающиеся во введении больному терапевтически эффективного количества соединения по данному изобретению или его фармацевтически приемлемых соли, сольвата или пролекарства. Помимо этого, настоящее изобретение включает методы ингибирования ВГС NS3 протеазы путем введения больному эффективного количества соединения по данному изобретению.
Благодаря настоящему изобретению в настоящее время возможно создавать усовершенствованные лекарственные вещества, представляющие собой соединения по изобретению, которые могут быть эффективными при лечении больных, инфицированных ВГС. Конкретно, настоящее изобретение включает пептиды, которые могут ингибировать функционирование NS3 протеазы, например, в комбинации с NS4A протеазой.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Стереохимические определения и условные обозначения, применяемые в данном описании, как правило, даны по McGraw-Hill Dictionary of Chemical Terms, S.P.Parker, Ed., McGraw-Hill Book Company, New York (1984) и Stereochemistry of Organic Compounds, Eliel, E. and Wilien, S., John Wiley & Sons, Inc., New York (1994). Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активного соединения приставки D и L или R и S применяются, чтобы показать абсолютную конфигурацию молекулы относительно ее хирального(ых) центра(ов). Приставки d и l или (+) и (-) применяются для обозначения знака вращения плоскополяризованного света соединением, при этом (-) или l означают, что соединение является левовращающим, а (+) или d означают, что соединение является правовращающим. При данной химической структуре эти соединения, называемые стереоизомерами, идентичны за исключением того, что они являются зеркальными изображениями друг друга. Конкретный стереоизомер из пары зеркальных изображений может также называться энантиомером, а смесь таких изомеров часто называют энантиомерной смесью.
Номенклатура, применяемая при описании органических радикалов, например, углеводородов и замещенных углеводородов, обычно, если не указано иначе, соответствует стандартной номенклатуре, известной в технике. Комбинации групп, например, алкилалкоксиамин, если конкретно не указано иначе, включают все возможные устойчивые конфигурации. Для иллюстрации определение некоторых радикалов и комбинаций дано ниже.
Термины "рацемическая смесь" или "рацемат" относятся к эквимолярной смеси двух энантиомеров, лишенной оптической активности.
Термин "хиральный" относится к молекулам, которые не совпадают со своим зеркальным изображением при наложении друг на друга, тогда как термин "ахиральный" относится к молекулам, которые совпадают со своим зеркальным изображением при наложении друг на друга.
Термин "диастереомер" относится к стереоизомеру, который не является энантиомером, например, стереоизомеру с двумя или более центрами хиральности, и чьи молекулы не являются зеркальными изображениями друг друга. Диастереоизомеры имеют различные физические свойства, например, температуры плавления, спектральные свойства и реакционную способность. Смеси диастереоизомеров можно разделять аналитическими методами высокого разрешения, такими как электрофорез и хроматография.
Термин "энантиомеры" относится к двум стереоизомерам соединения, зеркальные изображения которых при наложении не совпадают друг с другом.
Предполагается, что термин "фармацевтически приемлемая соль" включает нетоксические соли, синтезированные из соединения, которое содержит основной или кислый фрагмент, обычными химическими методами. Как правило, такие соли можно получать реакцией свободной кислой или основной формы этих соединений со стехиометрическим количеством подходящего основания или кислоты в воде или в органическом растворителе или в их смеси; обычно предпочтительными являются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечень подходящих солей имеется в Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p.1445. Соединения по данному изобретению применимы в форме свободного основания или свободной кислоты или в форме его фармацевтически приемлемой соли. Все формы входят в объем изобретения.
Термин "терапевтически эффективное количество" означает общее количество каждого активного компонента, которое является достаточным для того, чтобы принести значительную пользу больному, например, продолжительное снижение вирусной нагрузки. При применении отдельного активного ингредиента, вводимого индивидуально, термин относится к этому индивидуальному (одному) ингредиенту. При применении в виде комбинации термин относится к объединенным количествам активных ингредиентов, которые вызывают терапевтический эффект при введении в комбинации, последовательно или одновременно.
Предполагается, что термин "соединения по изобретению" и эквивалентные ему выражения охватывают соединения Формулы I и фармацевтически приемлемые соли и сольваты, например, гидраты. Аналогично, имеется в виду, что ссылки на интермедиаты охватывают их соли и сольваты, если это возможно. Ссылки на соединение по изобретению также включают предпочтительные соединения Формулы II и III.
Термин "производное" означает химически модифицированное соединение, причем считается, что модификация является обычной для рядового специалиста-химика, такой как сложный эфир или амид кислоты, защитные группы, такие как бензильная группа для спирта или тиола, и трет-бутоксикарбонильная группа для амина.
Термин "сольват" означает физическую ассоциацию соединения по данному изобретению с одной или более молекул растворителя, органического или неорганического. Эта физическая ассоциация включает водородную связь. В некоторых примерах сольват можно выделить, например, когда одна или более молекул встроены в кристаллическую решетку кристаллического твердого вещества. "Сольват" охватывает как сольваты в растворе, так и выделяемые сольваты. Примеры сольватов включают гидраты, сольваты с этанолом, метанолом и т.п.
Термин "пролекарство" по данному описанию означает производные соединений по изобретению, которые содержат группы, отщепляемые химическим путем или в процессе метаболизма (обмена веществ), и превращаются, в процессе сольволиза или в физиологических условиях, в соединения по изобретению, фармацевтически активные in vivo. Пролекарство соединения может образовываться обычным образом за счет функциональной группы соединений, такой как амино, гидрокси или карбоксигруппы. Пролекарственная форма в виде производного часто имеет лучшую растворимость, тканевую совместимость или пролонгированное высвобождение в организме млекопитающего (см. Bungard, H., Design of Prodrugs, pp.7-9, 21-24, Elsevier, Amsterdam 1985). Пролекарства включают производные кислот, хорошо известные практическим работникам в этой области, такие, например, как сложные эфиры, получаемые реакцией исходного кислого соединения с подходящим спиртом, или амиды, получаемые реакцией исходной кислоты с подходящим амином.
Термин "пациент, больной" включает как человека, так и других млекопитающих.
Термин "фармацевтическая композиция" означает композицию, содержащую соединение по изобретению в комбинации, по меньшей мере, с одним дополнительным фармацевтическим носителем, т.е. адъювантом, эксципиентом или наполнителем, таким как разбавители, консерванты, агенты, регулирующие текучесть, агенты, способствующие измельчению, смачивающие агенты, эмульгаторы, суспендирующие агенты, подсластители, агенты, придающие вкус и залах, антибактериальные агенты, противогрибковые агенты, смазки и диспергаторы, в зависимости от природы и способа применения и лекарственных форм. Можно, например, использовать ингредиенты, перечисленные в Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA (1999).
Выражение "фармацевтически приемлемый" применяется в данном описании по отношению к тем соединениям, материалам, композициям и/или лекарственным формам, которые, по компетентному (здравому) врачебному суждению, пригодны для применения в контакте с тканями больных, не вызывая чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соизмеримых с приемлемым (разумным) соотношением риск/польза.
Термин "лечение" относится к: (i) предупреждению заболевания, нарушения или состояния у больного, который может быть предрасположен к заболеванию, нарушению и/или состоянию, но они еще не диагностированы у пациента; (ii) ингибирование (подавление) заболевания, нарушения или состояния, т.е. прекращение их развития; и (iii) ослабление заболевания, нарушения или состояния, т.е. побуждение к регрессии заболевания, нарушения и/или состояния. Термин "замещенный" по данному описанию включает замещение (замену) в одном - максимально возможном числе сайтов связывания в ядре, например, в органическом радикале, с которым связан заместитель, например, моно-, ди-, три- и тетразамещенные, если специально не указано иначе.
Термин "галоген" по данному описанию означает заместитель-галоген, выбранный из брома, хлора, фтора или йода. Термин "галогеналкил" означает алкильную группу, которая содержит один или более галоидных заместителей (атомов галогена).
Термин "алкил" по данному описанию означает ациклические линейные или разветвленные алкильные заместители и включает, например, метил, этил, пропил, бутил, трет-бутил, гексил, 1-метилэтил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил. Так, С1-6алкил относится к алкильной группе, содержащей от одного до шести атомов углерода. Термин "низший алкил" означает алкильную группу, содержащую от одного до шести, предпочтительно, от одного до четырех углеродных атомов. Термин "алкилэфир" означает алкильную группу, дополнительно содержащую сложноэфирную группу. Обычно указанное число атомов углерода, например, С2-6алкилэфир, включает все углеродные атомы в радикале.
Термин "алкенил", применяемый в данном описании, означает алкильный радикал, содержащий, по меньшей мере, одну двойную связь, например, этенил (винил), и алкил.
Термин "алкокси" по данному описанию означает алкильную группу с указанным числом углеродных атомов, присоединенную к кислородному атому. Алкокси включает, например, метокси, этокси, пропокси, 1-метилэтокси, бутокси и 1, 1-диметилэтокси. Последний радикал в уровне техники называют трет-бутокси. Термин "алкоксикарбонил" означает алкильную группу, дополнительно содержащую карбонильную группу.
Термин "циклоалкил" по данному описанию означает циклоалкильный заместитель, содержащий указанное число атомов углерода, и включает, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и спироциклические группы, такие как спироциклопропил и спироциклобутил. Термин "циклоалкокси" по данному описанию означает циклоалкильную группу, связанную с атомом кислорода, такую, например, как циклобутилокси или циклопропилокси. Термин "алкилциклоалкил" означает циклоалкильную группу, связанную с алкильной группой. Указанный интервал включает общее число атомов углерода в радикале, если специально не указано иначе. Так, С4-10алкилциклоалкил может содержать 1-7 углеродных атомов в алкильной группе и 3-9 углеродных атомов в кольце, например, циклопропилметил или циклогексилэтил.
Термин "арил" по данному описанию означает ароматический фрагмент (ароматическую группу), содержащий указанное число углеродных атомов, такой как, но без ограничения, фенил, инданил или нафтил. Например, C6-10арил относится к ароматической группе, содержащей 6-10 углеродных атомов, которая может быть в виде моноциклической или бициклической структуры. Термин "галогенарил" по данному описанию относится к арилу, имеющему в качестве заместителей один или более атомов галогена. Термины "алкиларил", "арилалкил" и "аралкил" означают арильную группу, замещенную одной или более алкильных групп. Так, C7-14алкиларильная группа может иметь 1-8 углеродных атомов в алкильной группе в случае моноциклического ароматического радикала и 1-4 углеродных атома в алкильной группе в случае конденсированного ароматического радикала. Арильные радикалы включают такие радикалы, которые замещены типичными заместителями, известными специалистам в данной области техники, например, галоген, гидрокси, карбокси, карбонил, нитро, сульфо, амино, циано, диалкиламино, галогеналкил, CF3, галогеналкокси, тиоалкил, алканоил, SH, алкиламино, алкиламид, диалкиламид, карбоксиэфир, алкилсульфонамид и алкил(алкокси)амин. Примеры алкиларильных групп включают бензил, бутилфенил и 1-нафтилметил. Термины "алкиларилокси" и "алкиларил(овый)эфир" означают алкиларильные группы, содержащие атом кислорода и сложноэфирную группу, соответственно.
Термин "карбоксиалкил" по данному описанию означает карбоксильную группу (СООН), связанную через алкильную группу по определению выше, и включает, например, масляную кислоту.
Термин "алканоил" по данному описанию означает линейные или разветвленные 1-оксоалкильные радикалы, содержащие указанное число углеродных атомов, и включает, например, формил, ацетил, 1-оксопропил (пропионил), 2-метил-1-оксопропил, 1-оксогексил и т.п.
Термин "аминоаралкил" по данному описанию означает аминогруппу, замещенную алкильной группой, такой как нижеприведенный аминоаралкил
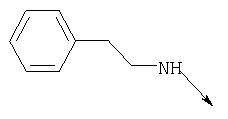
Термин "алкиламид" по данному описанию означает амид, монозамещенный алкилом, таким как

Термин "карбоксиалкил" по данному описанию означает карбоксильную группу (СООН), связанную через алкильную группу по определению выше, и включает, например, масляную кислоту.
Термин "гетероцикл", также называемый "Het", по данному описанию означает 7-12-членные бициклические гетероциклы и 5-7-членные моноциклические гетероциклы.
Предпочтительные бициклические гетероциклы представляют собой 7-12-членные конденсированные бициклические системы (оба кольца имеют общую пару атомов), содержащие 1-4 гетероатома, выбранных из азота, кислорода и серы, причем оба кольца гетероцикла являются полностью ненасыщенными. Гетероатомы азота и серы могут, необязательно, быть окисленными. Бициклический гетероцикл может содержать гетероатомы в одном или обоих циклах. Бициклический гетероцикл может также содержать заместители при любом атоме углерода в цикле, например, 1-3 заместителя. Примеры подходящих заместителей включают С1-6алкил, С3-7циклоалкил, С1-6алкокси, С3-7циклоалкокси, галоген-С1-6алкил, CF3, моно- или дигалоген-С1-6алкокси, циано, галоген, тиоалкил, гидрокси, алканоил, NO2, SH, амино, С1-6 алкиламино, ди(С1-6)алкиламино, ди(С1-6)алкиламид, карбоксил, (С1-6)карбоксиэфир, С1-6алкилсульфон, С1-6алкилсульфонамид, С1-6алкилсульфоксид, ди(С1-6)алкил(алкокси)амин, C6-10арил, C7-14алкиларил и 5-7-членный моноциклический гетероцикл. Когда два заместителя присоединены к вицинальным углеродным атомам бициклического гетероцикла, они могут соединяться с образованием цикла, например, пяти-, шести- или семичленной циклической системы, содержащей до двух гетероатомов, выбранных из кислорода и азота. Бициклический гетероцикл может быть присоединен к боковой группе, например, Х в Формуле I, по любому атому в цикле и, предпочтительно, по углероду.
Примеры бициклических гетероциклов включают, но без ограничения, следующие циклические системы:
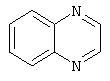


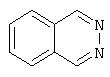





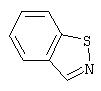

Предпочтительными моноциклическими гетероциклами являются 5-7-членные насыщенные, частично насыщенные или полностью ненасыщенные циклические системы (эта последняя подгруппа, в данном описании называемая ненасыщенной гетероароматической), содержащие в цикле от одного до четырех гетероатомов, выбранных из азота, кислорода и серы, причем гетероатомы серы и азота, необязательно, могут быть окисленными. Моноциклический гетероцикл может также содержать заместители при любом из атомов цикла, например, один-три заместителя. Примеры подходящих заместителей включают С1-6алкил, С3-7циклоалкил, С1-6алкокси, С3-7циклоалкокси, галоген-С1-6алкил, CF3, моно- или дигалоген-С1-6алкокси, циано, галоген, тиоалкил, гидрокси, алканоил, NO2, SH, амино, С1-6алкиламино, ди(С1-6)алкиламино, ди(С1-6)алкиламид, карбоксил, (С1-6 )карбоксиэфир, С1-6алкилсульфон, С1-6алкилсульфоксид, С1-6алкилсульфонамид, ди(С1-6)алкил(алкокси)амин, C6-10арил, C7-14алкиларил и 5-7-членный моноциклический гетероцикл. Моноциклический гетероцикл может быть присоединен к боковой группе, например, Х в Формуле I, по любому атому в цикле.
Примеры моноциклических гетероциклов включают, но без ограничения, следующие циклические системы:
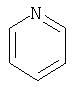
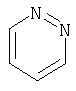


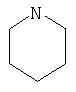
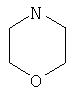
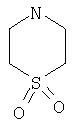

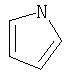



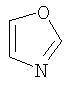



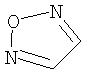
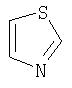

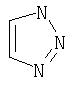




Специалист в данной области техники признает, что гетероциклы, применяемые в соединениях по данному изобретению, должны быть устойчивыми. Как правило, устойчивые соединениями являются такие соединения, которые можно синтезировать, выделить и приготовить из них препарат, используя известные специалистам в данной области техники методы, и при этом не происходит разложения соединения.
В случае применения в названиях соединений по данному изобретению обозначения "Р1', Р1, Р2, Р3 и Р4", применяемые в данном описании, картируют относительные положения аминокислотных остатков связывания ингибиторов протеазы относительно связывания субстрата природного пептида при расщеплении. Расщепление в натуральном субстрате происходит между Р1 и Р1', где положения, имеющие обозначение со штрихом (не- прим), обозначают аминокислоты, начиная от С-конца природного сайта расщепления пептида, и до N-конца; тогда как положения, имеющие обозначение со штрихом, исходят из N-конца обозначения сайта расщепления и продолжаются до С-конца. Например, Р1' относится к первому дальнему положению справа от С-конца сайта расщепления (т.е. первому положению N-конца); тогда как с Р1 начинается нумерация слева от С-конца сайта расщепления; Р2: второе положение от С-конца и т.д.) [см. Berger A & Schechter I., Transactions of the Royal Society London series (1970), B257, 249-264].
Так, в соединениях формулы I участки молекулы от "Р1' до Р4" показаны ниже:

Применяемый в данном описании термин "1-аминоциклопропилкарбоновая кислота" (Асса) относится к соединению формулы

Применяемый в данном описании термин "трет-бутилглицин" относится к соединению формулы:
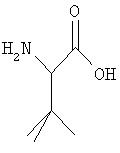
Термин "остаток" по отношению к аминокислоте или производному аминокислоты означает радикал, образующийся из соответствующей α-аминокислоты при удалении гидроксила карбоксильной группы и одного водорода α-аминокислотной группы. Например, термины Gln, Ala, Gly, Ile, Arg, Asp, Phe, Ser, Leu, Cys, Asn, Sar и Tyr представляют собой "остатки" L-глутамина, L-аланина, глицина, L-изолейцина, L-аргинина, L-аспарагиновой кислоты, L-фенилаланина, L-серина, L-лейцина, L-цистеина, L-аспарагина, саркозина и L-тирозина, соответственно.
Термин "боковая цепь" по отношению к аминокислоте или к аминокислотному остатку означает группу, присоединенную к α-углеродному атому α-аминокислоты. Например, R-группа или боковая цепь для глицина представляет собой водород, для аланина - метил, для Валина - изопропил. Для конкретных R-групп или боковых цепей α-аминокислот дается ссылка на учебник A.L.Lehninger no Biochemistry (см. главу 4).
Соединения по данному изобретению имеют структуру Формулы I:
где
(a) R1 обозначает C1-8алкил, С3-7циклоалкил или С4-10алкилциклоалкил;
(b) m обозначает 1 или 2;
(c) n обозначает 1 или 2;
(d) R2 обозначает Н, С1-6алкил, С2-6алкенил или С3-7циклоалкил, каждый из которых, необязательно, замещен галогеном;
(e) R3 обозначает C1-8алкил, необязательно имеющий заместитель: галоген, циано, амино, С1-6диалкиламино, C6-10арил, C7-14алкиларил, С1-6алкокси, карбокси, гидрокси, арилокси, C7-14алкиларилокси, С2-6алкиловый сложный эфир, C8-15алкилариловый сложный эфир; С3-12алкенил, С3-7циклоалкил или С4-10 алкилциклоалкил, где циклоалкил или алкилциклоалкил, необязательно, имеют заместитель: гидрокси, С1-6алкил, С2-6алкенил или С1-6алкокси; или R3, вместе с углеродным атомом, к которому он присоединен, образует С3-7циклоалкильную группу, необязательно замещенную С2-6алкенилом;
(f) Y обозначает Н, фенил, замещенный нитрогруппой, пиридил, замещенный нитрогруппой, или С1-6алкил, имеющий в качестве заместителя циано, ОН или С3-7циклоалкил;
при условии, что если R4 или R5 обозначает Н, тогда Y обозначает Н;
(g) В обозначает Н, С1-6алкил, R4-(С=O)-, R4O(C=O)-, R4-N(R5)-C(=O)-, R4-N(R5)-C(=S)-, R4SO2- или R4-N(R5)-SO2-;
(h) R4 обозначает (i) C1-10алкил, необязательно имеющий заместитель: фенил, карбоксил, С1-6алканоил, 1-3 атома галогена, гидрокси, -OC(O)С1-6алкил, С1-6алкокси, амино, необязательно замещенный С1-6алкилом, амидогруппой или (низший алкил)амидогруппой; (ii) С3-7циклоалкил, С3-7циклоалкокси или С4-10алкилциклоалкил, каждый из которых, необязательно, замещен гидрокси, карбоксилом, (С1-6алкокси)карбонилом, амино, необязательно замещенным С1-6алкиламидогруппой, или (низший алкил)амидогруппой; (iii) C6-10арил или C7-16 арилалкил, каждый из которых, необязательно, замещен С1-6алкилом, галогеном, нитро, гидрокси, амидо, (низший алкил)амидо, или аминогруппой, необязательно замещенной С1-6алкилом; (iv) Het; (v) бицикло(1.1.1)пентан; или (vi) - C(O)OC1-6алкил, С2-6алкенил или С2-6алкинил;
(i) R5 обозначает Н; С1-6алкил, необязательно замещенный 1-3 атомами галогена; или С1-6алкокси, при условии, что R4 обозначает C1-10алкил;
(j) Х обозначает О, S, SO, SO2, ОСН2, CH2O или NH;
(k) R' обозначает Het; или C6-10арил или C7-10алкиларил, необязательно замещенный Ra; и
(1) Ra обозначает С1-6алкил, С3-7циклоалкил, С1-6алкокси, С3-7цилоалкокси, галоген-С1-6алкил, CF3, моно- или ди-галоген-С1-6алкокси, циано, галоген, тиоалкил, гидрокси, алканоил, NO2, SH, амино, С1-6алюиламино, ди(С1-6)алкиламино, ди(С1-6)алкиламид, карбоксил, (С1-6)карбоксиэфир, С1-6алкилсульфон, С1-6алкилсульфонамид, ди(С1-6)алкил(алкокси)амин, C6-10арил, C7-14алкиларил или 5-7-членный моноциклический гетероцикл;
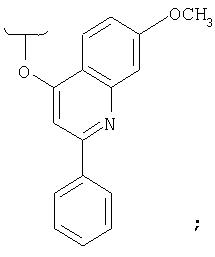
при условии, что Х-R' не обозначает
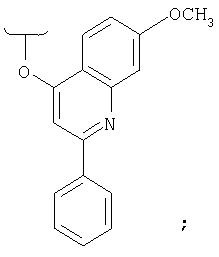
или его фармацевтически приемлемые соль, сольват или пролекарство.
Предпочтительно, R2 обозначает С2-6алкенил; R3 обозначает C1-8 алкил, необязательно замещенный С1-6алкокси, или С3-7циклоалкил; Y обозначает Н; В обозначает R4-(С=O)-, R4O(C=O)- или R4-N(R5 )-C(=O)-; R4 обозначает C1-10алкил, необязательно замещенный 1-3 атомами галогена или С1-6алкокси; или С3-7пиклоалкил или С4-10алкилциклоалкил; R5 обозначает Н; Х обозначает О или NH; и R' обозначает Het.
Заместители из каждой группы можно выбирать индивидуально и объединять в любой комбинации, которая дает устойчивое соединение по данному изобретению, также более чем один заместитель из каждой группы может быть замещен в ядре при условии, что имеются достаточно доступные сайты связывания. Например, каждый из следующих Ra заместителей, С1-6алкокси, C6арил и 5-7-членный моноциклический гетероцикл могут быть замещенными в бициклическом гетероцикле R'.
В предпочтительном аспекте соединения по данному изобретению имеют структуру Формулы II:
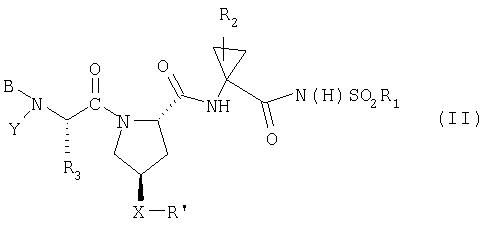
где
(a) R1 обозначает С3-7циклоалкил;
(b) R2 обозначает С1-6алкил, С2-6алкенил или С3-7циклоалкил;
(c) R3 обозначает C1-8алкил, необязательно имеющий заместитель: C6арил, С1-6алкокси, карбокси, гидрокси, арилокси, C7-14алкиларилокси, С2-6алкиловый сложный эфир, C8-15алкилариловый сложный эфир; С3-12алкенил, С3-7циклоалкил или С4-10алкилциклоалкил;
(d) Y обозначает Н;
(e) В обозначает Н, С1-6алкил, R4-(С=O)-, R4O(C=O)-, R4-N(R5)-С(=O)-, R4-N(R5)-C(=S)-, R4SO2- или R4-N(R5)-SO2-;
(f) R4 обозначает (i) C1-10алкил, необязательно имеющий заместитель: фенил, карбоксил, С1-6алканоил, 1-3 атома галогена, гидрокси, С1-6алкокси; (ii) С3-7циклоалкил, С3-7циклоалкокси или С4-10 алкилциклоалкил; или (iii) C6-10арил или C7-16арилалкил, каждый из которых, необязательно, замещен С1-6алкилом или галогеном;
(g) R5 обозначает Н; С1-6алкил, необязательно замещенный 1-3 атомами галогена;
(h) X обозначает О или NH;
(i) R' обозначает Het; или C6-10арил, необязательно замещенный Ra; и
(j) Ra обозначает С1-6алкил, С3-7циклоалкил, С1-6алкокси, галоген-С1-6алкил, галоген, амино, C6арил или 5-7-членный моноциклический гетероцикл;
при условии, что Ха-R' не обозначает
или его фармацевтически приемлемые соль, сольват или пролекарство.
В одном предпочтительном аспекте изобретения R' обозначает бициклический гетероцикл. Предпочтительно, бициклический гетероцикл содержит 1 или 2 атома азота и, необязательно, атом серы или кислорода в цикле. Предпочтительно, гетероцикл замещен, по меньшей мере, одним из заместителей: С1-6алкил, С1-6алкокси, галоген, C6арил и 5-7-членный моноциклический гетероцикл. Более предпочтительно, R' обозначает бициклический гетероцикл, содержащий 1 атом азота и замещенный метоксигруппой и, по меньшей мере, одним из заместителей: C6арил и 5-7-членный моноциклический гетероцикл.
В другом предпочтительном аспекте изобретения R' обозначает моноциклический гетероцикл. Предпочтительно, гетероцикл содержит 1 или 2 атома азота и, необязательно, атом серы или атом кислорода в цикле. Предпочтительно, гетероцикл замещен, по меньшей мере, одним из заместителей: С1-6алкил, С1-6алкокси, галоген, C6-10арил, C7-14алкиларил или 5-7-членный моноциклический гетероцикл. Более предпочтительно, R' обозначает моноциклический гетероцикл, содержащий 1 или 2 атома азота и замещенный метоксигруппой и, по меньшей мере, одним из заместителей: С6арил и 5-7-членный моноциклический гетероцикл.
В более предпочтительном варианте изобретения соединения имеют структуру, приведенную в Формуле III

где
(a) R1 обозначает С3-7циклоалкил;
(b) R2 обозначает С2-6алкенил;
(c) R3 обозначает C1-8алкил;
(d) Y обозначает Н;
(e) В обозначает R4 O(С=O)- или R4-N(R5)-C(=O)-;
(f) R4 обозначает C1-10алкил;
(g) R5 обозначает Н;
(h) R' обозначает бициклический гетероцикл, необязательно замещенный Ra, и
(i) Ra обозначает С1-6алкил, С1-6алкокси, галоген, C6арил или 5-7-членный моноциклический гетероцикл; при условии, что О-R' не обозначает
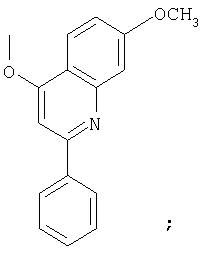
или его фармацевтически приемлемые соль, сольват или пролекарство.
Предпочтительно, R1 обозначает циклопропил или циклобутил, R2 обозначает винил, R3 обозначает трет-бутил, R4 обозначает трет-бутил и R' обозначает хинолин или изохинолин, необязательно замещенный Ra. Предпочтительно, Ra включает, по меньшей мере, один из С1-6 алкокси, C6арила и 5-7-членного моноциклического гетероцикла. В предпочтительном аспекте изобретения R1 обозначает циклопропил, R2 обозначает винил, R3 обозначает трет-бутил, R4 обозначает трет-бутил и R' обозначает изохинолин, замещенный С1-6алкокси и, по меньшей мере, одним из C6арила или 5-7-членного моноциклического гетероцикла.
Соединения по данному изобретению, благодаря наличию в них основного фрагмента, могут образовывать соли, присоединяя фармацевтически приемлемую кислоту. Соли присоединения кислот образуются из соединения Формулы I и фармацевтически приемлемой органической кислоты, включая, но без ограничения, хлористоводородную, бромистоводородную, йодистоводородную, серную, фосфорную кислоту, или органической кислоты, такой как п-толуолсульфоновая, метансульфоновая, уксусная, бензойная, лимонная, малоновая, фумаровая, малеиновая, щавелевая, янтарная, сульфаминовая или винная кислота. Таким образом, примеры таких фармацевтически приемлемых солей включают хлорид, бромид, йодид, сульфат, фосфат, метансульфонат, цитрат, ацетат, малонат, фумарат, сульфамат и тартрат.
Соли по аминогруппе могут также включать четвертичные аммониевые соли, в которых аминный азот несет подходящую органическую группу, такую как алкильная, алкенильная, алкинильная или аралкильная.
Соединения по данному изобретению, замещенные кислотной группой, могут существовать в виде солей, образованных путем присоединения основания. Такие соли присоединения основания включают соли, полученные из неорганических оснований, например, соли щелочных металлов (например, натрия и калия), соли щелочно-земельных металлов (например, кальция и магния), соли алюминия и соли аммония. Кроме того, подходящие соли присоединения основания включают соли физиологически приемлемых органических оснований, таких как триметиламин, триэтиламин, морфолин, пиридин, пиперидин, пиколин, дициклогексиламин, N,N'-дибензилэтилендиамин, 2-гидроксиэтиламин, бис-(2-гидроксиэтил)амин, три-(2-гидроксиэтил)амин, прокаин, дибензилпиперидин, N-бензил-β-фенетиламин, дегидроабиетиламин, N,N'-бисгидроабиетиламин, глюкамин, N-метилглюкамин, коллидин, хинин, хинолин, этилендиамин, орнитин, холин, N,N'-бензилфенетиламин, хлорпрокаин, диэтаноламин, диэтиламин, пиперазин, трис(гидроксиметил)аминометан и N-метилглутамин. Эти соли можно получать методами, известными специалистам в данной области техники.
Некоторые соединения по данному изобретению и их соли могут существовать в форме сольватов с водой, например, гидратов, или с органическими растворителями, такими как метанол, этанол или ацетонитрил, образуя, соответственно, "метанолат", "этанолат" или "ацетонитрилат". Настоящее изобретение включает каждый сольват и их смеси.
Помимо этого, соединения по данному изобретению, или их соли или сольваты, могут проявлять полиморфизм. Настоящее изобретение также охватывает любые такие полиморфные формы.
Соединения по данному изобретению также содержат два или более хиральных центра. Например, соединения могут включать Р1 циклопропильный элемент формулы
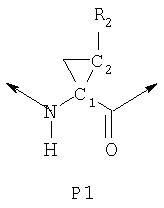
где C1 и С2, каждый, представляет собой асимметрический атом углерода в положениях 1 и 2 циклопропильного кольца. Не препятствуя другим возможным асимметрическим центрам в других сегментах соединений, присутствие этих двух асимметрических центров означает, что соединения могут существовать в виде рацемических смесей или диастереомеров, таких как диастереомеры, в которых конфигурация R2 является либо син относительно амида, либо син относительно карбонила, как показано ниже.

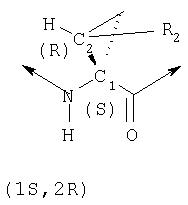


Энантиомеры можно разделять методами, известными специалистам в данной области техники, например, получая диастереомерные соли, которые можно разделять кристаллизацией, газо- жидкостной или жидкостной хроматографией, селективной реакцией одного энантиомера с энантиомер- специфичным реагентом. Следует отдавать отчет, что если нужный энантиомер в процессе разделения превращается в другую химическую частицу, тогда требуется дополнительная стадия для образования требуемой энантиомерной формы. Или же специфические энантиомеры можно синтезировать асимметрическим синтезом, используя оптически активные реагенты, субстраты, катализаторы или растворители, или асимметрическим превращением одного энантиомера в другой.
Соединения по данному изобретению могут находиться в виде пролекарства. Сложные эфиры простых алифатических или ароматических радикалов, образованные, при их наличии, из кислотных групп в боковой цепи соединений по данному изобретению, являются предпочтительными пролекарствами. В некоторых случаях желательно получать пролекарства типа двойных сложных эфиров, таких как (ацилокси)алкиловые эфиры или (алкоксикарбонил)окси)алкиловые эфиры.
Некоторые соединения по настоящему изобретению могут также существовать в различных устойчивых конформациях, которые можно разделить. Торсионная асимметрия вследствие затрудненного вращения около асимметрической одинарной связи, например, из- за стерических препятствий или деформации цикла, может способствовать разделению различных конформеров. Настоящее изобретение включает каждый конформационный изомер этих соединений и их смеси.
Некоторые соединения по данному изобретению могут существовать в цвиттерионной форме, и настоящее изобретение включает каждую цвиттерионную форму этих соединений и их смеси.
Исходные вещества, применимые для синтеза соединений по данному изобретению, известны специалистам в данной области техники и их можно легко получить, либо они являются продажными.
Соединения по данному изобретению можно получать методами, известными специалистам в данной области техники, см., например, Патент США 6323180 и Патентную заявку США 200020111313 А1. Представленные ниже методы даны в качестве иллюстрации и не претендуют на ограничение объема заявляемого изобретения. Следует знать, что может быть предпочтительным или необходимым получить такое соединение, в котором функциональная группа защищена обычной защитной группой, затем удалить защитную группу, чтобы получить соединение по данному изобретению. Подробности относительно применения защитных групп в соответствии с данным изобретением известны специалистам в данной области техники.
Соединения по данному изобретению можно синтезировать, например, по общей методике, проиллюстрированной на Схеме I (где CPG обозначает защитную группу для карбоксильной группы, a APG обозначает защитную группу для аминогруппы).
Схема I
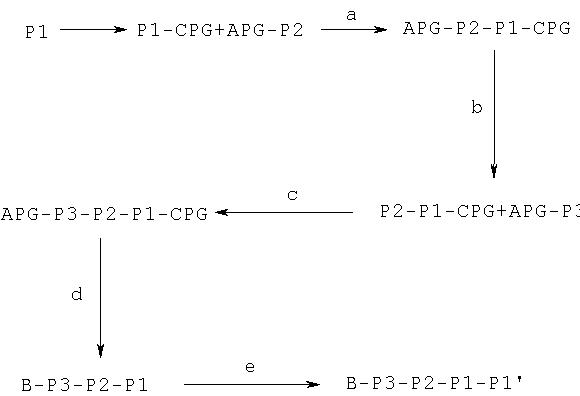
Коротко говоря, P1, P2 и Р3 могут связываться с применением общеизвестных методов пептидного связывания. Группы Р1, P2 и Р3 могут связываться друг с другом в любом порядке до тех пор, пока конечное соединение соответствует пептидам по изобретению. Например, Р3 может связываться с Р2-Р1; или Р1 может связываться с Р3-Р2.
Как правило, пептидные цепи наращивают (пептиды удлиняют), используя снятие защиты с α-аминогруппы N-концевого остатка и присоединение незащищенной карбоксильной группы к следующей соответствующим образом N-защищенной аминокислоте (сшивание, связывание) пептидным связыванием с применением описанных методов. Эти процедуры депротекции и присоединения повторяют до тех пор, пока не получают нужную последовательность. Это сшивание (связывание) можно осуществлять постадийно с составляющими аминокислотами, как изображено на Схеме I.
Связывание двух аминокислот, аминокислоты и пептида или двух пептидных фрагментов можно проводить стандартными методами связывания, таким как азидный метод, метод со смешанным ангидридом угольной- карбоновой кислот (изобутилхлорформиатный), карбодиимидный (с дициклогексилкарбодиимидом, диизопропилкарбодиимидом или водорастворимым карбодиимидом) метод, метод с активным сложным эфиром (п-нитрофениловым эфиром, имидоэфиром N-гидроксиянтарной кислоты), К-метод с реагентом Вудворда, метод с карбонилдиимидазолом, методы с фосфорными реагентами или окислительно-восстановительные методы. Некоторые из этих методов (в особенности карбодиимидный метод) можно усовершенствовать, добавляя 1-гидроксибензотриазол или 4-DMAP. Эти реакции связывания (конденсации) можно осуществлять либо в растворе (жидкой фазе), либо в твердой фазе.
Точнее, стадия сшивания (соединения, связывания) включает конденсацию (с дегидратацией) свободной карбоксильной группы одного из реагентов со свободной аминогруппой другого реагента в присутствии сшивающего (конденсирующего) агента с образованием сшитой амидной связи. Описание таких конденсирующих агентов имеется в обычных учебниках по пептидной химии, например, в М.Bodansky, "Peptide Chemistry", 2nd rev. ed., Springer-Verlag, Berlin, Germany, (1993). Примерами устойчивых конденсирующих агентов являются N, N'-дициклогексилкарбодиимид, 1-гидроксибензотриазол в присутствии N,N'-дициклогексилкарбодиимида или N-этил-N'-[(3-диметиламино)пропил]карбодиимида. Применяемым практически и полезным конденсирующим агентом является продажный гексафторфосфат (бензотриазол-1-илокси)трис-(диметиламино)фосфония, либо сам, либо в присутствии 1-гидроксибензотриазола или 4-DMAP. Другим применяемым практически и полезным конденсирующим агентом является продажный тетрафторборат 2-(1Н-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония. Еще одним применяемым практически и полезным конденсирующим агентом является продажный гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония. Реакцию конденсации проводят в инертном растворителе, например, в хлористом метилене, ацетонитриле или диметилформамиде. Избыток третичного амина, например, диизопропилэтиламина, N-метилморфолина, N-метилпирролидина или 4-DMAP, прибавляют к реакционной смеси при рН около 8. Температура реакции, как правило, составляет 0°С-50°С, а обычное время реакции от 15 мин до 24 час.
Функциональные группы составляющих аминокислот, как правило, должны быть защищены в процессе реакций конденсации во избежание образования нежелательных связей. Защитные группы, которые можно использовать, перечислены, например, в Greene, "Protective Groups in Organic Chemistry", John Wiley & Sons, New York (1981) и в "Peptides: Analysis, Synthesis, Biology", Vol.3, Academic Press, New York (1981), содержание которых вводится в данное описание в качестве ссылки.
α-Аминогруппа каждой аминокислоты, которая должна связываться с наращиваемой пептидной цепью, должна быть защищена (APG). Можно использовать любую защитную группу, известную в технике. Примеры таких групп включают: 1) ацильные группы, такие как формильная, трифторацетильная, фталильная и п-толуолсульфонильная; 2) ароматические карбаматные группы, такие как бензилоксикарбонил (Cbz или Z) и замещенные бензилоксикарбонилы, и 9-флуоренилметилоксикарбонил (Fmoc); 3) алифатические карбаматные группы, такие как трет-бутоксикарбонил (Boc), этоксикарбонил, диизопропилметоксикарбонил и аллилоксикарбонил; 4) циклоалкилкарбаматные группы, такие как циклопентилоксикарбонил и адамантилоксикарбонил; 5) алкильные группы, такие как трифенилметил и бензил; 6) триалкилсилил, такой как триметилсилил; и 7) тиолсодержащие группы, такие как фенилтиокарбонил и дитиасукциноил.
Предпочтительной α-аминозащитной группой является либо Boc, либо Fmoc. Многие производные аминокислот, соответствующим образом защищенные для пептидного синтеза, выпускаются промышленностью, α-Аминозащитную группу вновь добавляемого аминокислотного остатка отщепляют перед конденсацией (связыванием) со следующей аминокислотой. Когда используют группу Вос, выбирают методы с трифторуксусной кислотой, чистой или в хлористом метилене, или HCl в диоксане или в этилацетате. Затем нейтрализуют образовавшуюся аммониевую соль, перед конденсацией или in situ, раствором основания, таким как водные буферные растворы, или третичными аминами в хлористом метилене, или в ацетонитриле, или в диметилформамиде. Если применяют группу Fmoc, в качестве реагентов выбирают пиперидин или замещенный пиперидин в диметилформамиде, но можно использовать любой вторичный амин. Снятие защиты можно осуществлять при температуре от 0°С до комнатной температуры (rt или RT), обычно 20-22°С.
Любую аминокислоту, имеющую функциональные группы в боковой цепи, следует защищать при получении пептида с применением любой вышеописанной группы. Специалисты в данной области техники знают, что выбор и применение соответствующих защитных групп для этих функциональных групп в боковой цепи зависит от аминокислоты и наличия других защитных групп в пептиде. Выбор таких защитных групп важен потому, что группу нельзя удалять в процессе снятия защиты и конденсации α-аминогруппы.
Например, если в качестве α-аминозащитной группы применяют Boc, подходящими являются следующие защитные группы в боковой цепи: п-толуолсульфонильные (тозильные) группы можно применять для защиты аминогрупп боковых цепей аминокислот, таких как Lys и Arg; ацетамидометильную, бензильную (Bn) или трет-бутилсульфонильную группы можно применять для защиты сульфидной группы боковой цепи цистеина; (простые) бензиловые (Bn) эфиры можно применять для защиты гидроксильных групп в боковых цепях серина, треонина или гидроксипролина; и бензиловые сложноэфирные группы можно применять для защиты карбоксигруппы в боковых цепях аспарагиновой кислоты и глутаминовой кислоты.
Если для защиты α-аминогруппы выбирают Fmoc, обычно приемлемыми считаются трет-бутильные защитные группы. Например, Boc можно применять для лизина и аргинина, простой трет-бутиловый эфир - для серина, треонина и гидроксипролина, а сложный трет-бутиловый эфир - для аспарагиновой кислоты и глутаминовой кислоты. Трифенилметильный фрагмент (тритил, Trityl) можно использовать для защиты сульфидной группы в боковой цепи цистеина.
Когда наращивание пептидной цепи (удлинение пептида) завершено, все защитные группы удаляют. Если используют жидкофазный синтез, защитные группы удаляют любым способом, который диктует выбор защитной группы. Эти методы хорошо известны специалистам в данной области техники.
Далее, при получении соединений по данному изобретению можно руководствоваться нижеприведенными советами. Например, для получения соединения, содержащего R4-С(O)-, R4-S(O)2, защищенный Р3, или целый пептид, или пептидный сегмент соединяется с соответствующим ацилхлоридом или сульфонилхлоридом, соответственно, либо это соединение является промышленным соединением, либо его синтез хорошо известен в технике. Для получения соединения, содержащего R4 -С(O)-, защищенный Р3, или целый пептид, или пептидный сегмент соединяется с соответствующим хлорформиатом, либо это соединение является промышленным соединением, либо его синтез хорошо известен в технике. Для получения Boc-производных применяют (Вос)2О.
Например:

Циклопентанол обрабатывают фосгеном, получая соответствующий хлорормиат.
Хлорформиат обрабатывают заданным NH2- трипептидом в присутствии основания, такого как триэтиламин, получая циклопентилкарбамат.
При получении соединения, содержащего R4-N(R5)-С(O)- или R4-NH-C(S)-, защищенный Р3, или целый пептид, или пептидный сегмент обрабатывают фосгеном с последующей обработкой амином, как описано в SynLett. Feb 1995; (2); 142-144, или проводят реакцию с промышленным изоцианатом и подходящим основанием, таким как триэтиламин.
При получении соединения, содержащего R4-N(R5)-S(O2)-, защищенный Р3, или целый пептид, или пептидный сегмент обрабатывают либо свежеприготовленным или продажным сульфамилхлоридом с последующей обработкой амином, как описано в выложенной заявке ФРГ (1998), 84 стр. 19802350 или в Международной заявке 98/32748.
α-Карбоксильную группу С-концевого остатка обычно защищают в виде сложного эфира (CPG), который можно расщепить с образованием карбоновой кислоты. Защитные группы, которые можно применять, включают: 1) сложные алкиловые эфиры, такие как метиловый, триметилсилиловый и трет-бутиловый, 2) арилалкиловые сложные эфиры, такие как бензиловый и замещенный бензиловый, или 3) сложные эфиры, которые можно расщеплять обработкой слабым основанием или методами восстановления в мягких условиях (мягкое восстановление), такие, как трихлорэтиловый и фенациловый эфиры.
Образующаяся α-карбоновая кислота (полученная при расщеплении в результате обработки слабой кислотой, слабым основании или при мягком восстановлении) конденсируется с R1SO2 NH2 [полученным обработкой R1SO2Cl в насыщенном растворе аммиака в тетрагидрофуране] в присутствии связывающего (конденсирующего) пептид агента, такого как CDI или EDAC, в присутствии основания, такого как 4-диметиламинопиридин (4-DMAP) и/или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), с введением фрагмента Р', при этом эффективно осуществляется сборка трипептида Р1'-Р1-Р2-Р2-APG. Как правило, в этом процессе используется 1-5 эквивалентов Р' конденсирующих агентов.
Далее, если Р3 защитная группа APG удаляется и заменяется на фрагмент В описанными выше методами, а получающаяся при расщеплении α-карбоновая кислота (расщепление проводят, обрабатывая слабой кислотой, слабым основанием или методами восстановления в мягких условиях) конденсируется с R1SO2NH2 [полученным обработкой R1SO2Cl в насыщенном растворе аммиака в тетрагидрофуране или альтернативными методами] в присутствии связывающего (конденсирующего) пептид агента, такого как CDI или EDAC, в присутствии основания, такого как 4-диметиламинопиридин (4-DMAP) и/или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), с введением фрагмента Р', при этом эффективно осуществляется сборка трипептида Р1'-Р1-Р2-Р2-В. Как правило, в этом процессе используется 1-5 эквивалентов Р' конденсирующих агентов.
Соединения по данному изобретению можно получать многими методами, включая методы, описанные далее в примерах, и методы, описанные в Патенте США 6323180 и Патентной заявке США 10/001850, поданной 20 ноября 2001 г. Содержание Патента США 6323180 и Патентной заявки США 10/001850, во всей полноте вводится в данное описание в качестве ссылки.
На Схеме II, далее, показан общий способ, по которому соединения Формулы I конструируют конденсацией промежуточной трипептидкарбоновой кислоты (1) с Р' сульфонамидом. (Следует заметить, что группы R6, R7, R8, R9, R10, R11, показанные ниже, представляют собой заместители в гетероциклической системе). Указанная реакция конденсации (связывания) требует обработки карбоновой кислоты (1) конденсирующим реагентом, таким как карбонилдиимидазол, в растворителе, таком как ТТФ, который можно нагревать до кипения, с последующим прибавлением полученного производного интермедиата (1) к Р1' сульфонамиду в растворителе, таком как ТГФ или хлористый метилен в присутствии основания, такого как DBU.
Схема II

Альтернативный способ построения соединений Формулы I показан на Схеме III. Р' сульфонамидный элемент конденсируется (связывается) с Р1 элементом по способу, используемому в Схеме I. Полученный фрагмент Р1-Р1' можно затем снять защитную группу на аминоконце. В этом общем примере применяется Вое защитная группа, но специалист в данной области техники признает, что в этом процессе можно применять ряд защитных групп. Указанную Вос защитную группу можно удалить, используя, например, трифторуксусную кислоту в растворителе, таком как дихлорэтан, при этом получают депротекционированный амин в виде ТФК (TFA) соли. Указанная соль амин • ТФК может непосредственно применяться в последующей реакции конденсации или, в качестве альтернативы, соль амин • ТФК можно сначала превратить в соль амин • HCl и эту соль амин • HCl использовать в указанной реакции конденсации, показанной на Схеме III. Конденсацией указанной соли амин • HCl (3) с карбоксильным концом в присутствии конденсирующих реагентов, таких как HATU, в растворителях, таких как хлористый метилен, можно получить соединения Формулы I (4).
Схема III
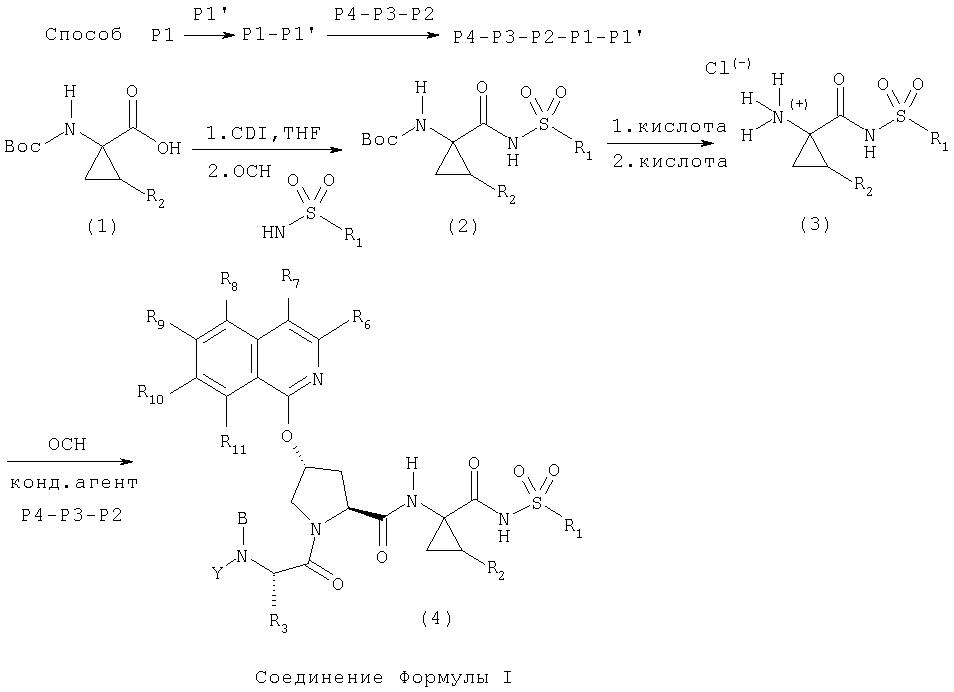
Альтернативный способ построения соединений Формулы I показан на Схеме IV. Соль - гидрохлорид Р1-Р1' концевого амина (1) конденсируется со свободной карбоксильной группой элемента Р2 в присутствии конденсирующих агентов, таких как РуВОР, в присутствии основания, такого как диизопропиламин, в растворителе, таком как хлористый метилен. Полученный интермедиат Р2-Р1-Р1' можно превратить в соединения Формулы I двухстадийным синтезом, причем на первой стадии происходит снятие защиты на Р2 аминоконце в присутствии кислоты, такой как ТФК, в растворителе, таком как хлористый метилен. Полученная соль трифторуксусной кислоты может конденсироваться с карбоксильным концом элемента Р4-Р3 в присутствии обычных конденсирующих агентов, таких как РуВор, в присутствии основания, такого как диизопропиламин, в растворителе, таком как хлористый метилен, с образованием соединений Формулы I (4).
Схема IV

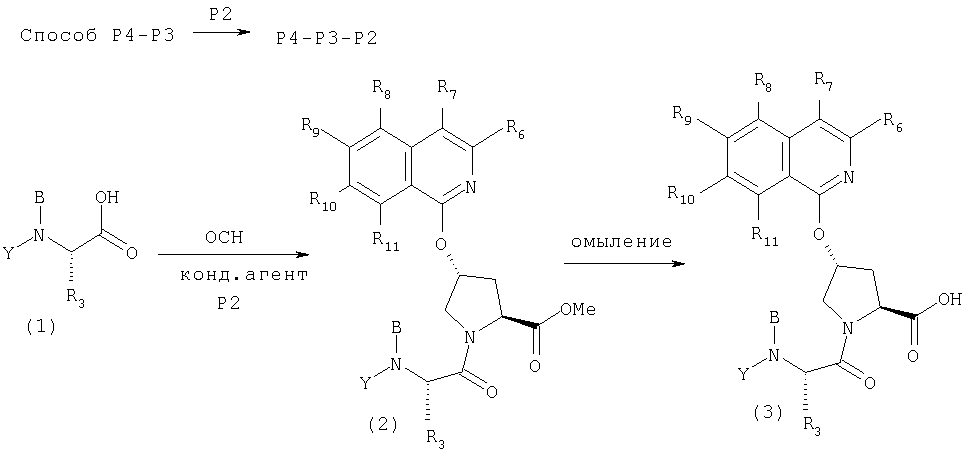
Помимо того что интермедиат Р4-Р3-Р2, используемый в вышеприведенных схемах, можно построить как описано выше, его еще можно получать так, как описано на общей Схеме V. Свободный карбоксильный конец интермедиата Р4-Р3 (1) может конденсироваться с аминоконцом Р2 элемента с образованием дипептида Р4-Р3-Р2 (2). Защитную группу с карбонильного конца интермедиата Р4-Р3-Р2 можно снять омылением сложноэфирной группы, при этом получают Р4-Р3-Р2 в виде свободной кислоты (3). Интермедиаты, подобные (3), можно превратить в соединения Формулы I, используя методы по данному описанию.
Схема V
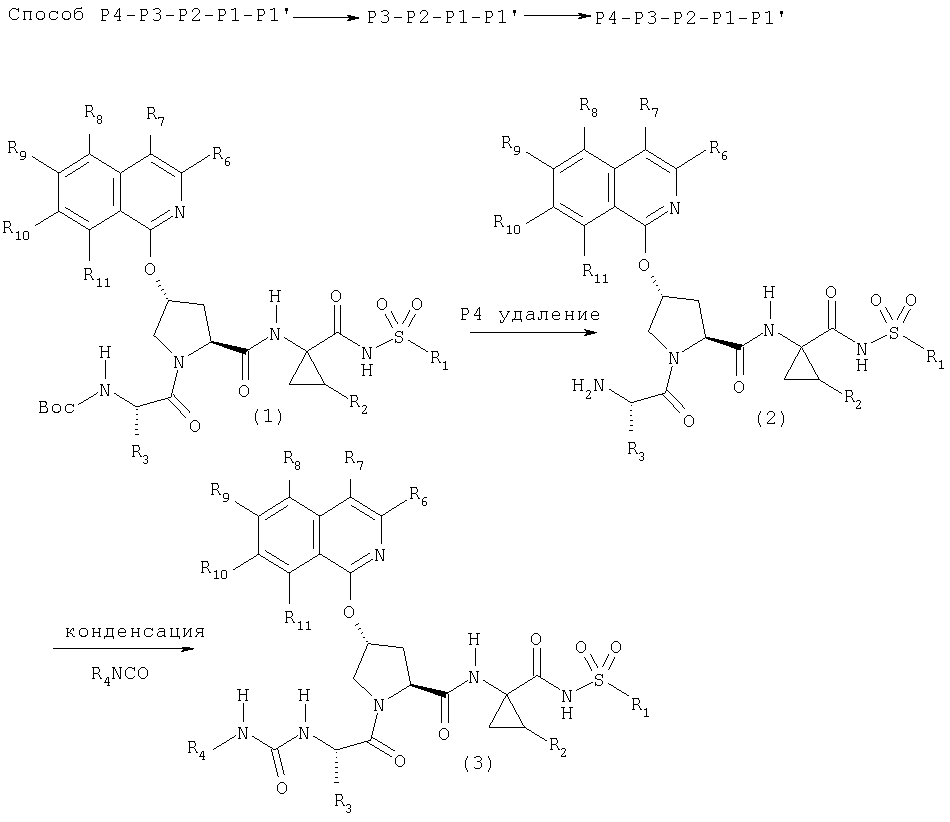
Соединения Формулы I можно также превратить в другие соединения Формулы I по данному описанию. Пример такого процесса показан на Схеме VI, где соединение Формулы I (1), содержащее группу Вос в положении Р4, превращается в соединение Формулы I (3), которое несет карбамидную группу в положении Р4. Превращение (1) в (3) можно проводить в две стадии, на первой из которых (1) превращается в амин (2) обработкой (1) кислотой, такой как ТФК, в растворителе, таком как хлористый метилен. Полученную соль амин • ТФК можно обработать изоцианатом в присутствии одного эквивалента основания, при этом получают соединение Формулы I (3), в котором фрагмент Р3 завершается карбамидной (мочевино) группой. Как указывается ранее, специалист в данной области техники признает, что интермедиат (2) можно применять в качестве исходного для получения соединений Формулы I, в которых группа Р3 завершается амидной, или сульфонамидной, или тиокарбамидной, или сульфонамидной группой. Построение указанных соединений Формулы I можно осуществлять в стандартных условиях для получения указанных Р4 функциональных групп из аминов.
Схема VI
При построении соединений Формулы I Р1' конец вводят в молекулы, используя один из общих способов, в общих чертах обрисованных выше и подробнее описанных ниже. В некоторых примерах элементы Р1', которые представляют собой циклоалкил- или алкилсульфонамиды, являются продажными или их можно получить из соответствующего алкил- или циклоалкилсульфонилхлорида обработкой указанного сульфонилхлорида аммиаком. Или же эти сульфонамиды можно синтезировать по общему методу, изображенному на Схеме VII. По этому методу продажный 3-хлорпропилсульфонилхлорид (1) превращают в соответствующий защищенный сульфонамид, например, обработкой трет-бутиламином. Затем полученный сульфонамид (2) превращают в соответствующий циклоалкилсульфонамид, обрабатывая двумя эквивалентами основания, такого как бутиллитий, в растворителе, таком как ТГФ, при низкой температуре. Защитную группу в полученном циклогексилсульфонамиде можно снять, обрабатывая его кислотой и получая при этом заданный незащищенный циклоалкилсульфонамид.
Схема VII

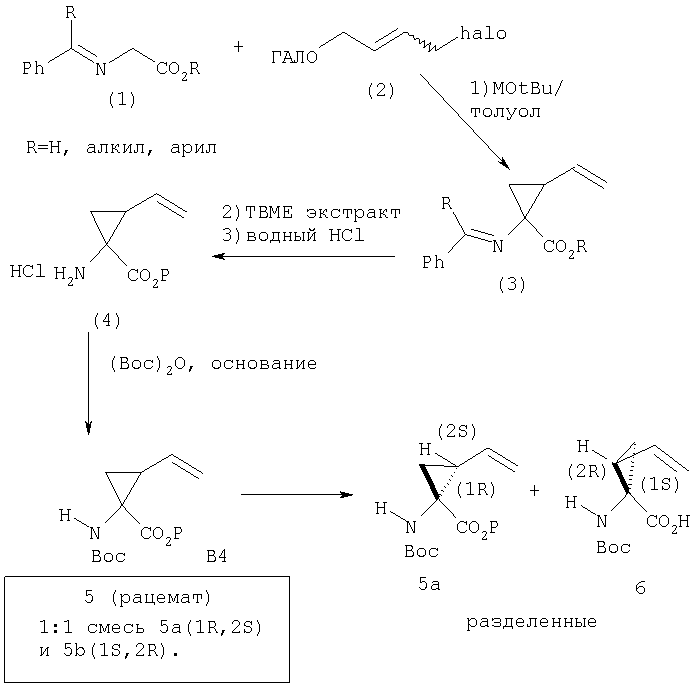
Элементы P1, применяемые для получения соединений Формулы I, в некоторых случаях являются продажными, а в иных случаях синтезируются методами по данному описанию, а затем встраиваются в соединения Формулы I методами по данному изобретению. Замещенные Р1 пиклопропиламинокислоты можно синтезировать по общему методу, показанному на Схеме VIII.
Обработка продажного или легко синтезируемого имина (1) 1,4-дигалогенбутена (2) в присутствии основания дает имин (3). Затем кислотным гидролизом 3 получают в качестве основного продукта 4, который содержит аллильный заместитель в форме син относительно карбоксильной группы. Аминогруппу соединения 4 можно защитить с помощью Вос группы, при этом получают полностью защищенную аминокислоту 5. Этот интермедиат является рацематом, который можно расщеплять ферментативным методом, при котором сложноэфирную группу соединения 5 расщепляют с помощью протеазы, получая соответствующую карбоновую кислоту. Не будучи связанными с какой-либо конкретной теорией, полагают, что эта реакция является селективной потому, что один из энантиомеров реагирует со значительно более высокой скоростью, чем его зеркальное изображение, что в результате приводит к кинетическому расщеплению промежуточного рацемата. В примерах, цитируемых в данном описании, более предпочтительным стереоизомером для интеграции в соединения Формулы I является стереоизомер 5а, стереохимия которого (1R,2S). В присутствии фермента сложноэфирная группа этого энантиомера не претерпевает расщепления и поэтому этот энантиомер 5а регенерируется из реакционной смеси. Однако, сложноэфирная группа менее предпочтительного энантиомера, 5b, имеющего стереометрию (1S,2R), претерпевает расщепление, т.е. гидролиз, при этом получается свободная кислота 6. По завершении этой реакции сложный эфир 5а можно отделить от кислоты 6 обычными методами, например, такими, как водная экстракция или хроматография.
Схема VIII
Методы получения Р2 интермедиатов и соединений Формулы I показаны на нижеприведенных Схемах. Нужно отметить, что во многих случаях реакции изображены только для одного положения интермедиата. Однако, следует понимать, что такие реакции можно использовать для получения модификаций по другим положениям этого интермедиата. Кроме того, указанные интермедиаты, условия реакции и методы, представленные в конкретных примерах, широко применимы к соединениям с другими заместителями. После представленных ниже общих схем в данном описании приводятся примеры. Как общие, так и конкретные примеры не являются ограничивающими, например, изохинолиновое ядро показано как часть общей Схемы IX, однако, этот путь представляет собой реальный способ построения соединений с другими гетероциклическими заместителями вместо изохинолинового элемента, такими как изохинолины или пиридины.
Схема IX

На Схеме IX показана конденсация N-защищенной группы С4-гидроксипролина с гетероциклом с образованием интермедиата (4) и последующее превращение указанного интермедиата в соединение Формулы I путем удлинения (наращивания, элонгации пептидной цепи) пептида по данному описанию. Следует отметить, что на первой стадии, которая представляет собой конденсацию группы С4-гидроксипролина с гетероарильным элементом, используется основание. Специалист в данной области знает, что эту конденсацию можно проводить, используя такие основания, как трет-бутоксид калия или гидрид натрия, в растворителе, таком как ДМФА, или ДМСО, или ТГФ. Эта конденсация с циклической системой изохинолина происходит по положению С1 (нумерация для циклической системы изохинолина показана на интермедиате 2 Схемы IX) и направляется хлором, который замещается в этом процессе. Следует отметить, что в этом положении можно использовать другие уходящие группы, такие как фтор, что показано на Схеме. Указанные фторированные интермедиаты (3) можно получать из соответствующих хлорированных соединений по литературным методикам, представленным в данном описании. Еще следует отметить, что положение уходящей группы (хлора или фтора) в данной циклической системе может меняться, как показано на Схеме X, где уходящая группа (фтор в данном примере) находится в положении C6 циклической системы изохинолина интермедиата (2).
Схема X
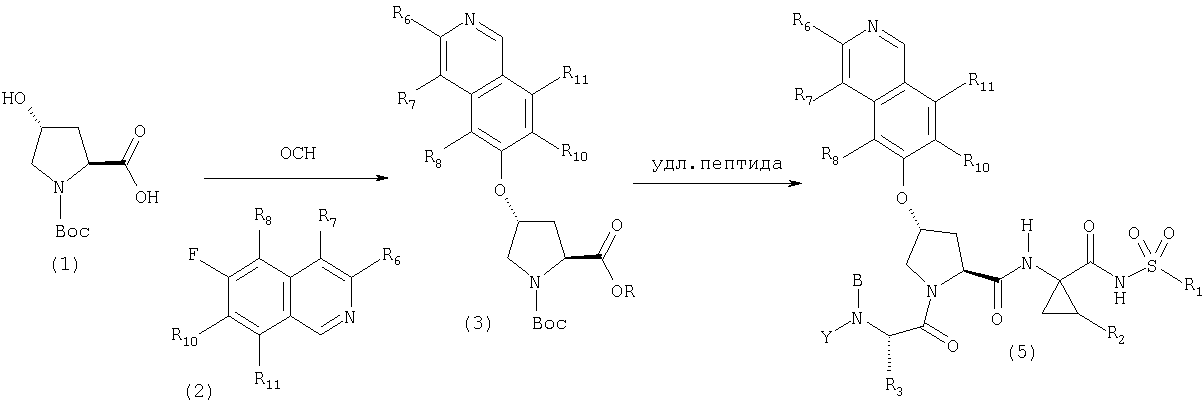
Далее следует заметить, что положение циклических гетероатомов в интермедиатах, подобных (2) на Схемах IX и X, также меняется, как показывает название гетероцикла по данному описанию. Интермедиат (2) на Схеме Х может конденсироваться с производным С4 гидроксипролина, давая элемент Р2 (3). Это C6-замещенное производное изохинолина можно превратить в соединения Формулы I методами по данному описанию.
Альтернативой вышеописанному методу конденсации С4-гидроксипролина с ароматическими и гетероароматическими группами является реакция Мицунобу, изображенная на стадии 1 Схемы XI.

На этой общей реакционной Схеме производное С4-гидроксипролина конденсируется с циклической системой хиназолина. В этой реакции используются такие реагенты, как трифенилфосфин и DEAD (диэтилазодикарбоксилат), в апротонных растворителях, таких как ТГФ или диоксан, и эту реакцию можно использовать для получения ариловых или гетероариловых эфиров. Следует отметить, что в ходе этой реакции конденсации стереохимия С4 хирального центра в производном С4-гидроксипролина изменяется, и поэтому в качестве исходного необходимо использовать производное С4-гидроксипролина с (S) стереохимией в С4 положении (как показано на Схеме XI). Стоит заметить, что в литературе описаны многочисленные модификации и усовершенствования реакции Мицунобу, вводимые в данное описание.
Во множестве примеров по данному описанию изохинолины вводятся в конечные соединения, и конкретно, в область Р2 указанных соединений. Специалист в данной области техники знает, что многие общие методы пригодны для синтеза изохинолинов. Кроме того, указанные изохинолины, полученные этими методами, можно легко ввести в конечные соединения Формулы I методами по данному описанию. В одном общем методе синтеза изохинолинов, показанном на Схеме XII,
Схема XII

Ссылка: N.Briet et al., Tetrahedron, 2002, 5761
производные коричной кислоты, изображенные в общем виде формулой (2), превращаются в 1-хлоризохинолины четырехстадийным синтезом. Указанные хлоризохинолины можно затем использовать в реакции конденсации с образованием производных С4-гидроксипролина. Превращение коричных кислот в хлорхинолины начинают с обработки коричной кислоты алкилхлорформиатом в присутствии основания. Полученный ангидрид затем обрабатывают азидом натрия, что приводит к образованию ацилазида (3), как показано на Схеме. Существуют другие методы получения ацилазидов из карбоновых кислот, например, указанную карбоновую кислоту можно обрабатывать дифенилфосфорилазидом (DPPA) в апротонном растворителе, таком как хлористый метилен, в присутствии основания. На следующей стадии реакции ацилазид (3) превращается в соответствующий изохинолон (4), как показано на Схеме. Для этого ацилазид нагревают до температуры около 190°С в высококипящем растворителе, таком как дифенилметан. Эта реакция является общей и дает замещенный изохинолон, получающийся из соответствующих производных коричной кислоты с выходами от умеренного до хорошего. Стоит отметить, что указанные производные коричной кислоты являются продажными или их можно получить из соответствующего производного бензальдегида (1) непосредственной конденсацией с малоновой кислотой или ее производными, а также по реакции Виттига. Промежуточные изохинолоны (4) на Схеме XII можно превратить в соответствующий 1-хлоризохинолин обработкой оксихлоридом фосфора. Эта реакция является общей и ее можно применять для любых изохинолонов, хинолонов или других гетероциклов по данному описанию с целью превращения гидроксизамещенных в соответствующие хлорсодержащие соединения, если существует сопряжение между указанной гидроксигруппой и атомом азота в указанных гетероциклических системах.
Другим методом синтеза циклической системы изохинолина является реакция Померанца-Фрича. Этот общий метод представлен на Схеме XIII.
Схема XIII
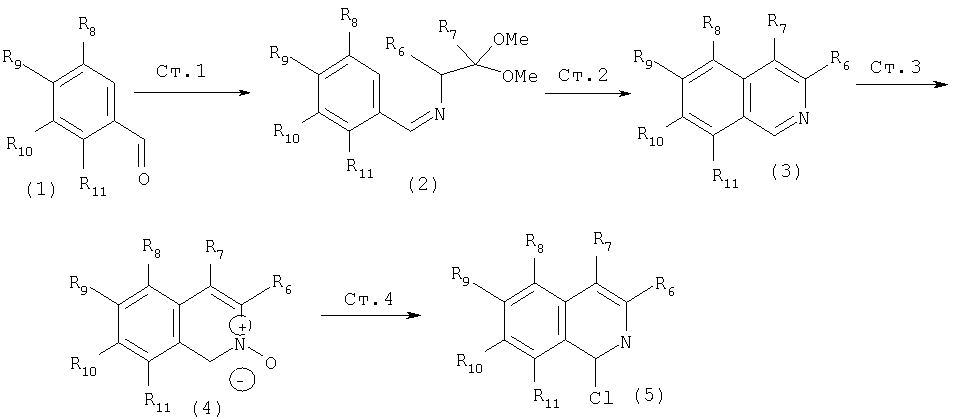
Реакция Померанца-Фрича
К.Hirao, R.Tsuchiya, Y.Yano, H.Tsue, Heterocycles 42(1) 1996, 415-422
Сначала осуществляют превращение производного бензальдегида (1) в функционализованный имин (2). Указанный имин затем превращают в циклическую систему изохинолина обработкой кислотой при повышенной температуре. Этот синтез изохинолина, изображенный на Схеме XIII, является общим, и следует отметить, что этот способ применяется практически для получения изохинолиновых интермедиатов, замещенных в положении С8 (примечание: в интермедиате (3) на Схеме XIII заместителем в С8 положении изохинолина является R11). Промежуточные изохинолины (3) можно превратить в соответствующие 1-хлоризохинолины (5) в две стадии, как показано на Схеме. На первой стадии при обработке изохинолина (3) мета-хлорбензойной кислотой в апротонном растворителе, таком как хлористый метилен, происходит образование изохинолин N-оксида (4). Интермедиат (4) можно превратить в соответствующий 1-хлоризохинолин обработкой оксихлоридом фосфора в кипящем хлороформе. Отметим, что этот двухстадийный процесс является общим и может применяться для получения хлоризохинолинов и хлорхинолинов из соответствующих изохинолинов и хинолинов, соответственно.
Другой метод синтеза циклической системы изохинолина показан на Схеме XIV.
Схема XIV

По этому методу производное орто-алкилбензамида (1) обрабатывают сильным основанием, таким как трет-бутиллитий, в растворителе, таком как ТГФ, при низкой температуре. Затем к этой реакционной смеси прибавляют нитрил, который вступает в реакцию присоединения с анионом, образующимся при депротонировании (1), что приводит к образованию (2). Эта реакция является общей и может применяться для получения замещенных изохинолинов. Интермедиат (2) на Схеме XIV можно превратить в соответствующий 1-хлоризохинолин методами по данному описанию.
Другой метод синтеза изохинолинов показан на Схеме XV. Депротонирование интермедиата (1) с применением трет-бутиллития описано выше. Однако, в данном методе указанный интермедиат "улавливается" сложным эфиром, что приводит к образованию интермедиата (2), показанного ниже. В последующей реакции кетон (2) конденсируется с ацетатом аммония при повышенной температуре, при этом образуется хинолон (3). Эта реакция является общей и может применяться для построения замещенных изохинолонов, которые затем можно превратить в соответствующие 1-хлоризохинолины по данному описанию.
Схема XV
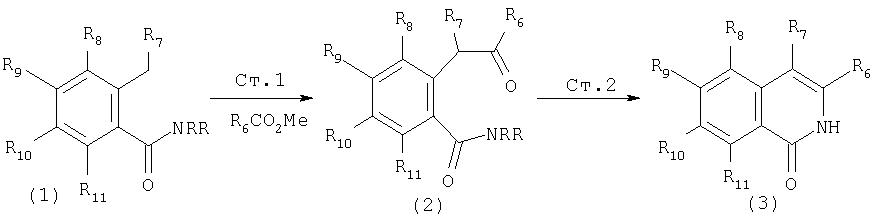
Еще один метод построения изохинолинов показан на Схеме XVI.
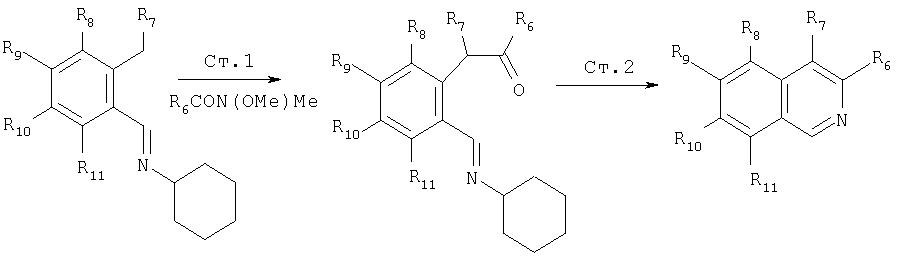
L.Flippin, J.Muchowski, JOC, 1993,2631-2632
На первой стадии этого процесса орто-алкиларилимины, такие как (1), депротонируются (вт- бутиллитий, ТГФ), а к полученному аниону прибавляют активное производное карбоновой кислоты, такое как амид Вейнреба. Полученный кетоимин (2) можно превратить в соответствующий изохинолин конденсацией с ацетатом аммония при повышенных температурах. Этот метод является общим и может применяться для синтеза замещенных изохинолинов. Указанные изохинолины можно превратить в соответствующий 1-хлоризохинолин методами по данному описанию.
Гетероциклы по данному описанию, встраиваемые в соединения Формулы I, можно дополнительно функционализовать. Очевидным для специалиста в данной области техники является то, что дополнительную функционализацию указанных гетероциклов можно проводить либо до, либо после введения этих функциональностей (гетероциклов) в соединения Формулы I. На нижеприведенных Схемах иллюстрируется это положение. Например, на Схеме XVII
Схема XVII
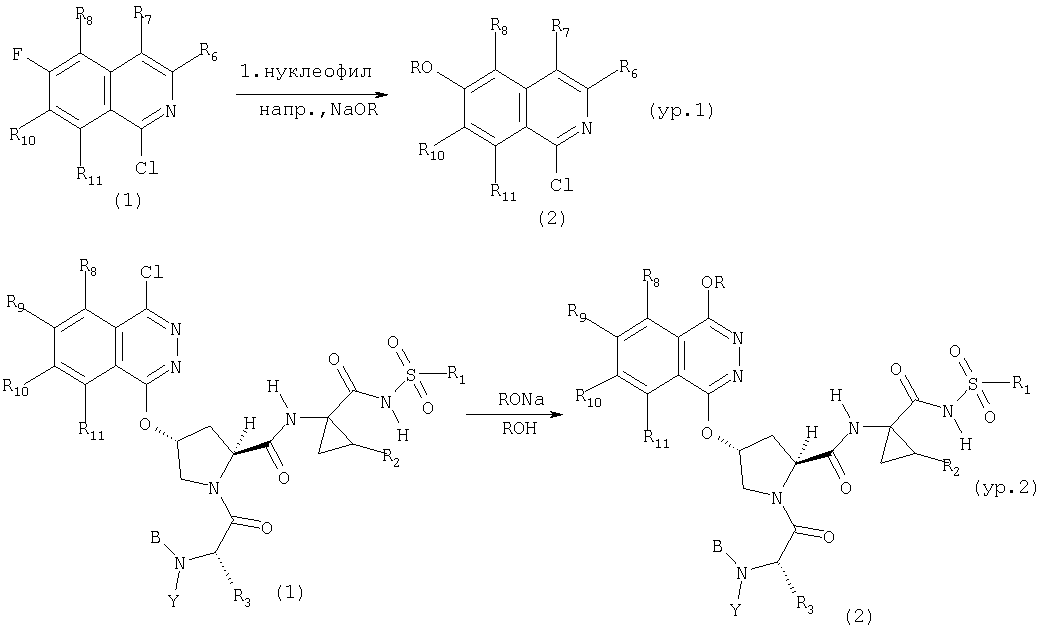
показано превращение 1-хлор-6-фторизохинолина в соответствующие 1-хлор-6-алкоксиизохинолины при обработке соединения (1) из (ур.1) алкоксидом натрия или калия в спиртовом растворителе, в котором алкоксид образуется при комнатной температуре. В некоторых случаях бывает необходимо нагревать реакционную смесь, чтобы довести реакцию до конца. Указанный хлорхинолин можно встроить в соединение Формулы I методами по данному описанию. Модификации Р2 гетероциклического элемента можно также осуществлять поле его встраивания в соединения Формулы I, как показано в (ур.2) Схемы XVII. Конкретно, соединения, такие как (1) в (ур.2), которые содержат уходящую группу в циклической системе фталазина, можно заменить на нуклеофил, такой как алкоксид, в растворителях, таких как соответствующий спирт, из которого образован алкоксид. Эти реакции можно проводить при комнатной температуре, но в некоторых случаях для завершения реакции может потребоваться нагревание реакционной смеси.
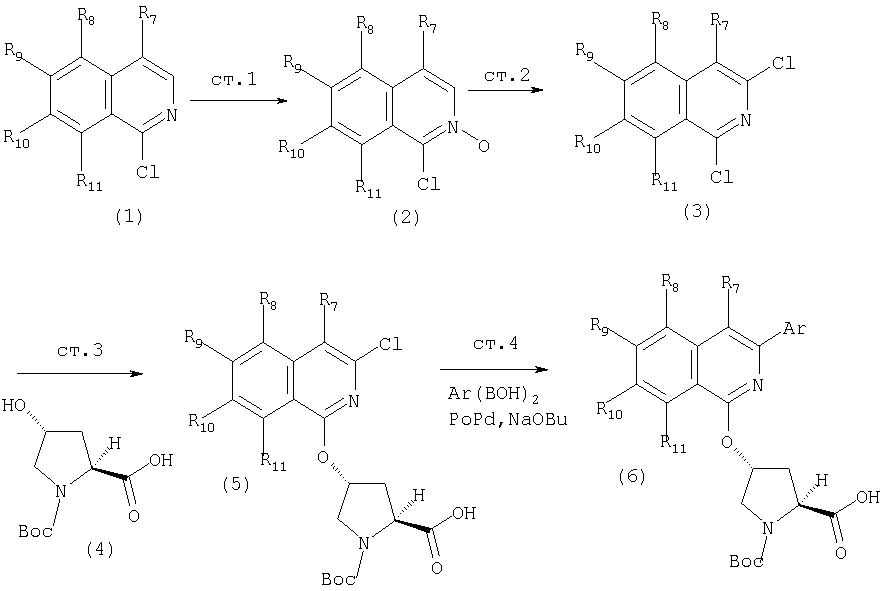
На Схеме XVIII показан общий метод модификации гетероциклов по определению в данном описании с применением реакций конденсации в присутствии палладия (опосредованных палладием). Указанную конденсацию можно применять для функционализации гетероцикла в любом положении циклической системы, получая соответствующим образом активированный или функционализованный указанный цикл, например, с помощью хлорида, как показано на Схеме. Сначала проводят реакцию 1-хлоризохинолина (1) с метахлорпербензойной кислотой, получая соответствующий N-оксид (2). Указанный интермедиат (2) можно превратить в соответствующий 1,3-дихлоризохинолин (3) обработкой оксихлоридом фосфора в кипящем хлороформе. Интермедиат (3) может конденсироваться с N-Boc-4-гидроксипролином методами по данному описанию, давая интермедиат (5), как показано на Схеме. Интермедиат (5) может сочетаться по реакции Сузуки с арилборной кислотой в присутствии палладиевого реагента и основания в растворителе, таком как ТГФ, или толуол, или ДМФА, при этом образуется С3-арилизохинолиновый интермедиат (6). В этой опосредуемой Pd реакции сочетания можно также применять гетероарилборные кислоты, при этом образуются С3-гетероарилизохинолины. Интермедиат (6) можно превратить в конечные соединения Формулы I методами по данному описанию.
Схема XVIII
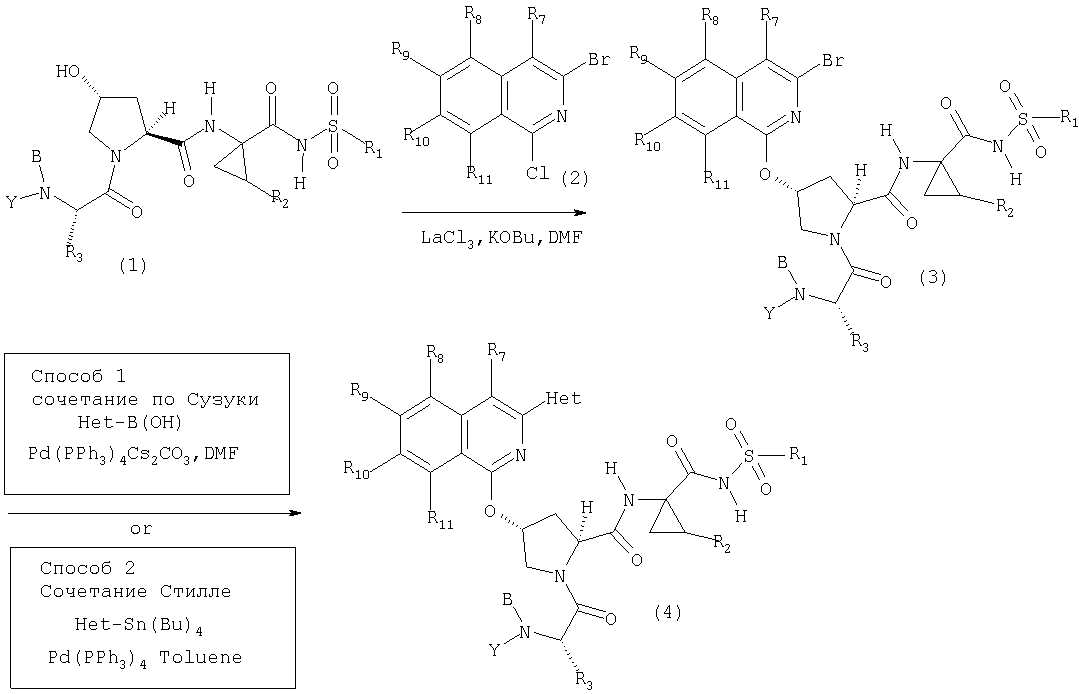
Катализируемое (опосредуемое) палладием сочетание гетероарильных систем с арильными или гетероарильными элементами можно также использовать на последних стадиях синтеза при построении соединений Формулы I, как показано на Схеме IXX. По данной Схеме промежуточный трипептидацилсульфонамид (1) сочетается (конденсируется) с 1-хлор-3-бромизохинолином (2) по вышеописанному методу замещения в гетероарильном фрагменте на алкоксид с образованием интермедиата (3). Сочетание (1) и (2) наиболее эффективно идет в присутствии катализатора, такого как хлорид лантана, по данному описанию. Циклическую систему промежуточного изохинолина (3) можно дополнительно функционализовать либо сочетанием по Сузуки (Способ 1: (3) взаимодействует с гетероарил- или арилборными кислотами в присутствии палладиевого катализатора, такого как тетра(трифенилфосфин)палладий, и основания, такого как карбонат цезия, в растворителях, таких как ДМФА), либо сочетанием по реакции Стилле (Способ 2: (3) взаимодействует с гетероарильными или арильными соединениями олова в присутствии палладиевого катализатора, такого как тетра(трифенилфосфин)палладий, в растворителях, таких как толуол).
Схема IXX
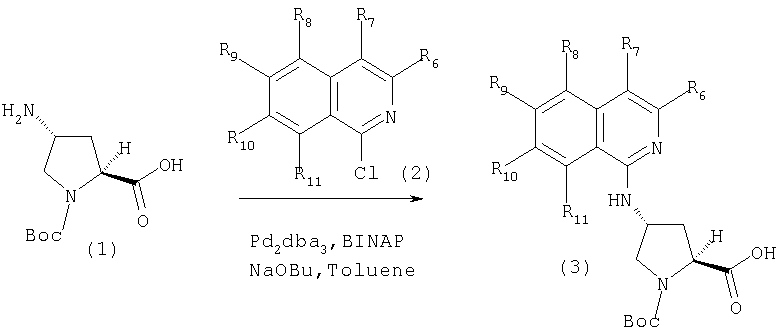
Для сочетания С4-аминопролиновых элементов с функционализованными гетероциклами можно также использовать реакции с палладием. На Схеме XX показано сочетание интермедиата (1) с функционализованным изохинолином в присутствии палладиевого катализатора и основания в растворителе, таком как толуол. Интермедиаты, подобные (3), можно превратить в соединения Формулы I методами по данному описанию.
Схема XX
Для построения циклической системы функционализованного изохинолина можно также использовать реакции [4+2] циклоприсоединения. Например, (Схема XXI) применение винилизоцианатов (1) в реакциях циклоприсоединения с бензольными предшественниками (2) дает функционализованные изохинолоны (3). Указанные изохинолины можно встраивать в соединения Формулы I, используя методы по данному описанию.
Схема XXI

Настоящее изобретение также включает композиции, содержащие соединение по данному изобретению или его фармацевтически приемлемые соль, сольват или пролекарство и фармацевтически приемлемый носитель. Фармацевтические композиции по настоящему изобретению содержат терапевтически эффективное количество соединения по изобретению, или его фармацевтически приемлемую соль, фармацевтически приемлемый сольват или фармацевтически приемлемое пролекарство, и фармацевтически приемлемый носитель, причем фармацевтически приемлемый носитель представляет собой, например, эксципиент или носитель-разбавитель.
Содержание активного ингредиента, т.е. соединение, в таких композициях, как правило, составляет 0.1-99.9 вес.% от веса композиции и очень часто составляет около 5-95 вес.%.
Фармацевтические композиции по данному изобретению можно вводить перорально, парентерально или с помощью имплантированного резервуара. Предпочтительными являются пероральное применение или инъекции. В некоторых случаях рН препарата можно корректировать с помощью фармацевтически приемлемых кислот, оснований или буферов для повышения стабильности соединения в препарате или его формы доставки. Термин "парентеральный" по данному описанию включает методы подкожной, внутрикожной, внутривенной, внутримышечной, интраартикулярной, внутрисуставной, внутригрудинной, подоболочечной и интралезиональной (внутрь поражения) инъекции или инфузии.
Фармацевтические композиции могут быть в виде стерильных препаратов для инъекции, например, в виде инъецируемой водной или масляной суспензии. Эту суспензию можно приготовить методами, известными из уровня техники, с применением диспергирующих или поверхностно-активных веществ и суспендирующих веществ. Подробности относительно получения таких соединений известны специалистам в данной области техники.
При пероральном применении фармацевтические композиции по данному изобретению могут вводиться в виде любой перорально приемлемой лекарственной формы, включая, но без ограничения, капсулы, таблетки и водные суспензии и растворы. Носители, обычно применяемые в пероральных таблетках, включают лактозу и маисовый крахмал. Обычно также добавляют смазки, такие как стеарат магния. Разбавители для перорального применения в виде капсул включают лактозу и сухой маисовый крахмал. Если водные суспензии применяются перорально, активный ингредиент соединяют с эмульгатором и суспендирующим агентом. При желании можно добавлять некоторые подсластители и/или вещества, придающие запах и вкус, и/или красители.
Другие подходящие носители для вышеуказанных композиций можно найти в стандартных фармацевтических учебниках, например, в "Remington's Pharmaceutical Sciences", 19th ed., Mack Publishing Company, Easton, Penn., 1995. Дальнейшие подробности относительно создания и приготовления подходящих форм доставки фармацевтических композиций по изобретению известны специалистам в данной области техники.
При монотерапии с целью предупреждения и лечения ВГС - опосредованного заболевания типичными являются уровни доз около 0.01-1000 миллиграмм на килограмм ("мг/кг") веса тела в день, предпочтительно, около 0.5-250 мг/кг веса тела в день соединений по изобретению. Как правило, фармацевтические композиции по данному изобретению вводят около 1-5 раз в день или же в виде непрерывного вливания. Такое введение можно применять для лечения хронических или острых состояний. Количество активного ингредиента, которое можно объединять с веществами носителя для получения разовой лекарственной формы, меняется в зависимости от хозяина, проходящего лечение, и конкретного способа применения.
Как понимает опытный специалист в данной области техники, могут требоваться дозы более низкие или более высокие, чем приведенные выше. Конкретная доза и схемы применения для любого конкретного пациента зависят от ряда факторов, включая активность конкретного применяемого соединения, возраста, веса тела, общего состояния здоровья, пола, диеты, времени применения, скорости выделения, комбинации лекарственных веществ, тяжести и течения инфекционного заболевания, предрасположенности пациента к инфекции и мнения лечащего врача. Как правило, лечение начинают с малых доз, существенно более низких, чем оптимальная доза пептида. Затем дозу повышают до тех пор, пока не будет достигнут оптимальный эффект в данных обстоятельствах. Как правило, наиболее желательно вводить соединение при уровне концентрации, который обычно дает эффективные результаты против вируса, не вызывая никаких вредных или пагубных побочных эффектов.
Если композиции по данному изобретению содержат комбинацию соединения по изобретению и один или более дополнительных терапевтических или профилактических агентов, как соединение, так и дополнительный агент обычно присутствуют в дозах около 10-100%, и, более предпочтительно, около 10-80% от дозы, которая обычно применяется по схеме монотерапии.
Если эти соединения или их фармацевтически приемлемые соли, сольваты или пролекарства готовят в виде препарата вместе с терапевтически приемлемым носителем, полученную композицию можно вводить in vivo млекопитающим, таким как человек, с целью ингибирования ВГС NS3 протеазы или с целью лечения ВГС вирусной инфекции. Такое лечение можно также осуществлять, используя соединения по данному изобретению в комбинации с агентами, которые включают, но без ограничения: иммуномодулирующие агенты, такие как интерфероны; другие антивирусные агенты, такие как рибавирин, амантадин; другие ингибиторы ВГС NS3 протеазы; ингибиторы других мишеней в жизненном цикле ВГС, таких как геликаза, полимераза, металлопротеаза или внутренний сайт прикрепления рибосом; или их комбинации. Дополнительные агенты можно объединять с соединениями по данному изобретению для создания однократной (разовой) дозы (лекарственной формы). Или же эти дополнительные агенты можно раздельно вводить млекопитающему как часть многократной дозы.
Соответственно, другой аспект данного изобретения охватывает метод ингибирования ВГС NS3 протеазной активности у пациентов с помощью введения соединения по данному изобретению, или его фармацевтически приемлемой соли, или его фармацевтически приемлемого сольвата, заместители которых имеют значение по определению выше.
В предпочтительном варианте изобретения эти методы применяются для понижения ВГС NS3 протеазной активности в организме пациента. Если фармацевтическая композиция содержит только соединение по данному изобретению в качестве активного компонента, такой метод может дополнительно включать стадию введения указанному пациенту агента, выбранного из иммуномодулирующего агента, антивирусного агента, ингибитора ВГС протеазы или ингибитора других мишеней в жизненном цикле ВГС, например, таких как геликаза, полимераза или металлопротеаза. Такой дополнительный агент можно вводить пациенту до, одновременно или после введения соединений по данному изобретению.
В другом предпочтительном аспекте эти методы применяются для ингибирования вирусной репликации у пациента. Такие методы можно применять для лечения или предупреждения ВГС заболевания.
Соединения по изобретению можно также использовать в качестве реагентов для лабораторных исследований. Соединения могут играть важную роль при проведении исследований по созданию методов анализа вирусной репликации, по оценке животных систем для анализа и по структурной биологии для дальнейшего развития знаний о механизме ВГС заболевания.
Соединения по данному изобретению можно также использовать для лечения или предупреждения вирусного загрязнения материалов и, следовательно, уменьшения риска вирусной инфекции лабораторного или медицинского персонала или пациентов, вступающих в контакт с такими материалами, например, кровью, тканями, хирургическими инструментами и одеждой, лабораторными инструментами и одеждой, и аппаратурой и материалами для сбора или переливания крови.
ПРИМЕРЫ
Приведенные далее конкретные примеры, которые иллюстрируют синтез соединений по данному изобретению, нельзя рассматривать как ограничивающие объем изобретения. Чтобы получить соединения, охватываемые данным изобретением, но специально не описанные, методы можно адаптировать к вариантам. Кроме того, немного отличающиеся варианты методов получения одинаковых соединений также будут очевидны для специалистов в данной области техники.
Если не указано иначе, процентное содержание в растворах выражает отношение веса к объему, а соотношение растворов выражается как соотношение объемов. Спектры ядерного магнитного резонанса (ЯМР) регистрируют на спектрометре Bruker 300, 400 или 500 МГц; химические сдвиги (δ) даются в миллионных долях. Флеш-хроматографию проводят на силикагеле (SiO2) методом флеш-хроматографии по Стиллу (W.C.Still et al., J. Org. Chem., (1978), 43, 2923).
Все данные жидкостной хроматографии (LC) регистрируют на жидкостном хроматографе Shimadzu LC-10AS с детектором SPB-10AV-VIS. Macc-спектрометрические (MS) данные определяют на Micromass Platform для LC методом электрораспыления (ES+).
Если не отмечено иначе, в нижеприведенных примерах каждое соединение анализируют с помощью LC/MS, используя один из семи методов в следующих условиях:
Колонки: (Метод A) - YMC ODS S7 С18 3.0×50 мм
(Метод В) - YMC ODS - A S7 С18 3.0×50 мм
(Метод С) - YMC S7 С18 3.0×50 мм
(Метод D) - YMC Xterra ODS S7 3.0×50 мм
(Метод Е) - YMC Xterra ODS S7 3.0×50 мм
(Метод F) - YMC ODS - A S7 С18 3.0×50 мм
Градиент: от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В
Время градиента: 2 мин (А, В, D, F, G); 8 мин (С, Е)
Время выдерживания: 1 мин (А, В, D, F, G); 2 мин (С, Е)
Скорость потока: 5 мл/мин
Длина волны детектора: 220 нм
Растворитель А: 10% МеОН/90% Н2 O/0.1% ТФК
Растворитель В: 10% Н2О/90% МеОН/0.1% ТФК
Сокращения, применяемые в данной заявке, включая, в частности, сокращения в иллюстрирующих нижеприведенных примерах, хорошо известны специалистам в данной области техники. Некоторые из применяемых сокращений даны ниже:
Соединения и химические интермедиаты по данному изобретению, описанные в нижеприведенных примерах, получают следующими методами. Следует заметить, что следующий далее раздел "Примеры" представлен в виде подразделов. Подразделы озаглавлены Подраздел А, Подраздел К. Номера примеров и номера соединений не идут непрерывно друг за другом по всему разделу "Примеры" заявки и, следовательно, каждый Подраздел указывает перерыв в нумерации. Нумерация внутри каждого подраздела, как правило, непрерывна. В Подразделе L описывается биологическая активность соединений. В Подразделе М описывается группа дополнительных соединений, которые можно получать методами по данному изобретению.
Подраздел А
Получение интермедиатов (промежуточных соединений):
Получение Р1 интермедиатов:
Р1 интермедиаты, описанные в этой секции, можно применять для получения соединений Формулы I методами по данному изобретению.
I Элементы Р1
1. Получение рацемического этилового эфира (1R,2S)/(1S,2R)-1 амино-2-винилциклопропанкарбоновой кислоты
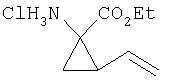
Метод А
Стадия 1
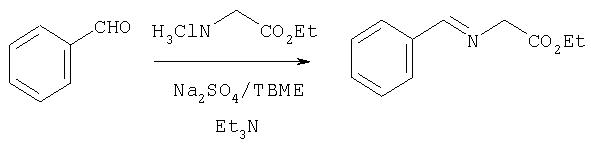
Гидрохлорид этилового эфира глицина (303.8 г, 2.16 моля) суспендируют в трет-бутилметиловом эфире (1.6 л). Прибавляют бензальдегид (231 г, 2.16 моля) и безводный сульфат натрия (154.6 г, 1.09 моля) и смесь охлаждают до 0° С в бане с ледяной водой. По каплям в течение 30 мин прибавляют триэтиламин (455 мл, 3.26 моля) и смесь перемешивают в течение 48 час при rt. Реакцию прекращают, добавляя ледяную воду (1 л), и органический слой отделяют. Водный слой экстрагируют трет-бутилметиловым эфиром (0.5 л) и объединенные органические вытяжки промывают смесью насыщенного водного раствора NaHCO3 (1 л) и рассолом (1 л.). Рассол сушат MgSO4, упаривают в вакууме, получают 392.4 г продукта - производного N-бензилимина в виде вязкого желтого масла, которое непосредственно используют на следующей стадии.1Н ЯМР (CDCl3, 300 МГц) δ 1.32 (т, J=7.1 Гц, 3Н), 4.24 (кв, J=7.1 Гц, 2Н), 4.41 (д, J=1.1 Гц, 2Н), 7.39-7.47 (м, 3Н), 7.78-7.81 (м, 2Н), 8.31 (с, 1Н).
Стадия 2

К суспензии трет-бутоксида лития (84.06 г, 1.05 моля) в сухом толуоле (1.2 л) по каплям в течение 60 мин прибавляют смесь N-бензилимина этилового эфира глицина (100.4 г, 0.526 моля) и транс-1,4-дибром-2-бутена (107.0 г, 0.500 моля) в сухом толуоле (0.6 л). По окончании прибавления к темно- красной смеси прибавляют воду (1 л) и трет-бутилметиловый эфир (ТВМЕ, 1 л). Водный слой отделяют и экстрагируют второй раз ТВМЕ (1 л). Органические вытяжки объединяют, прибавляют 1N HCl (1 л) и смесь перемешивают при комнатной температуре в течение 2 час. Органический слой отделяют и экстрагируют водой (0.8 л). Затем водные вытяжки объединяют, насыщают солью (700 г), прибавляют ТВМЕ (1 л) и смесь охлаждают до 0°С. Перемешиваемую смесь затем подщелачивают до рН 14, прибавляя по каплям 10N NaOH, органический слой отделяют, водный слой экстрагируют ТВМЕ (2×500 мл). Объединенные органические вытяжки сушат (MgSO4) и упаривают до объема 1 л. К этому раствору свободного амина прибавляют ВОС2O или ди-трет-бутилдикарбоната (131.0 г, 0.6 моля) и смесь перемешивают в течение 4 дней при rt. К реакционной смеси прибавляют дополнительное количество ди-трет-бутилдикарбоната (50 г, 0.23 моля), смесь кипятят в течение 3 час, а затем оставляют на ночь охлаждаться до комнатной температуры. Реакционную смесь сушат MgSO4 и упаривают в вакууме, получают 80 г сырого продукта. Этот остаток очищают флеш-хроматографией (2.5 кг SiO2, элюируют 1-2% MeOH/СН2Cl2), получают 57 г (53%) рацемического этилового эфира N-Boc-(1R,2S)/(1S,2R)-1 амино-2-винилциклопропанкарбоновой кислоты в виде желтого масла, твердеющего при стоянии в холодильнике:1Н ЯМР (CDCl3, 300 МГц) δ 1.26 (т, J=7.1 Гц, 3H), 1.46 (с, 9Н), 1.43-1.49 (м, 1Н), 1.76-1.82 (уш м, 1Н), 2.14 (кв, J=8.6 Гц, 1Н), 4.18 (кв, J=7.2 Гц, 2Н), 5.12 (дд, J=10.3, 1.7 Гц, 1Н), 5.25 (уш с, 1Н), 5.29 (дд, J=17.6, 1.7 Гц, 1Н), 5.77 (ддд, J=17.6, 10.3, 8.9 Гц, 1Н); MS m/z 254.16 (M-1)
Стадия 3 Получение гидрохлорида рацемического этилового эфира (1R,2S)/(1S,2R)-1 амино-2-винилциклопропанкарбоновой кислоты

Этиловый эфир N-Boc-(1R,2S)/(1S,2R)-1 амино-2-винилциклопропанкарбоновой кислоты (9.39 г, 36.8 ммоля) растворяют в 4N HCl/диоксан (90 мл, 360 ммоля) и перемешивают в течение 2 час при rt. Реакционную смесь упаривают, получают гидрохлорид этилового эфира (1R,2S)/(1S,2R)-1 амино-2-винилциклопропанкарбоновой кислоты с количественным выходом (7 г, 100%).1Н ЯМР (метанол-d4) δ 1.32 (т, J=7.1 Гц, 3H), 1.72 (дд, J=10.2, 6.6 Гц, 1Н), 1.81 (дд, J=8.3, 6.6 Гц, 1Н), 2.38 (кв, J=8.3 Гц, 1Н), 4.26-4.34 (м, 2Н), 5.24 (дд, 10.3, 1.3 Гц, 1Н), 5.40 (д, J=17.2, 1Н), 5.69-5.81 (м, 1Н).
Альтернативный способ получения гидрохлорида рацемического этилового эфира N-Boc-(1R,2S)/(1S,2R)-1 амино-2-винилциклопропанкарбоновой кислоты

К раствору трет-бутоксида калия (11.55 г, 102.9 ммоля) в ТГФ (450 мл) при -78°С прибавляют продажный N,N-дибензилимин этилового эфира глицина (25.0 г, 93.53 ммоля) в ТГФ (112 мл). Реакционную смесь нагревают до 0°С, перемешивают в течение 40 мин, а затем снова охлаждают до -78°С. К этому раствору прибавляют транс-1,4-дибром-2-бутен (20.0 г, 93.50 ммоля), смесь перемешивают в течение 1 час при 0°С и снова охлаждают до -78°С. Прибавляют трет-бутоксид калия (11.55 г, 102.9 ммоля), смесь быстро нагревают до 0°С и перемешивают еще в течение одного часа, затем упаривают в вакууме. Сырой продукт растворяют в Et2O (530 мл), прибавляют 1N раствор HCl (106 мл, 106 ммолей) и полученную двухфазную смесь перемешивают 3.5 час при rt. Слои разделяют, водный слой промывают Et2O (2х) и подщелачивают насыщенным раствором Na2HCO3. Заданный амин экстрагируют Et2O (3х) и объединенные органические вытяжки промывают рассолом, сушат (MgSO4) и упаривают в вакууме, получают свободный амин. Этот продукт обрабатывают 4N раствором HCl в диоксане (100 мл, 400 ммолей) и упаривают, получают гидрохлорид этилового эфира (1R, 2S)/(1S,2R)-1 амино-2-винилциклопропанкарбоновой кислоты в виде коричневого полужидкого вещества (выход 5.3 г, 34%), идентичного веществу, полученному по методике А, за исключением наличия небольшой примеси неидентифицированного ароматического вещества (8%).
Расщепление этилового эфира N-Boc-(1R,2S)/(1S,2R)-1 амино-2-винилциклопропанкарбоновой кислоты
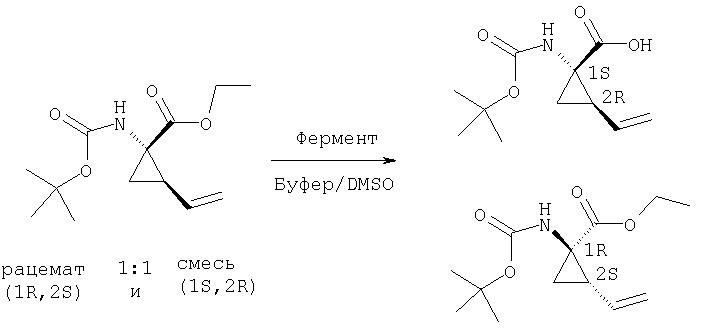
Расщепление А
К водному раствору натрийфосфатного буфера (0.1 М, 4.25 литра ("л"), рН 8), помещенному в реактор с рубашкой на 12 литров при температуре, поддерживаемой при 39°С, и при перемешивании со скоростью 300 об/мин прибавляют 511 грамм Acalase 2.4 л (около 425 мл) (Novozymes North America Inc.). Когда температура смеси достигнет 39°С, доводят рН до 8.0, добавляя 50% NaOH в воде. Затем в течение 40 мин прибавляют раствор этилового эфира N-Boc-(1R,2S)/(1S, 2R)-1 амино-2-винилциклопропанкарбоновой кислоты (85 мг) в 850 мл ДМСО. Температуру реакции поддерживают 40°С в течение 24.5 час, в течение этого времени рН смеси дважды, через 1.5 час и через 19.5 час - доводят до 8.0, прибавляя 50% NaOH в воде. Через 24.5 час определяют, что в эфире энантиомерный избыток составляет 97.2%, реакционную смесь охлаждают до комнатной температуры (26°С) и перемешивают в течение ночи (16 час), после чего в эфире энантиомерный избыток составляет 100%. Затем доводят рН реакционной смеси до 8.5, прибавляя NaOH, и полученную смесь экстрагируют МТВЕ (2×2 л). Объединенные МТВЕ вытяжки промывают 5% NaHCO3 (3×100 мл), водой (3×100 мл) и упаривают в вакууме, получают энантиомерно чистый этиловый эфир N-Boc-(1R,2S)-1 амино-2-винилциклопропанкарбоновой кислоты в виде твердого вещества светло-желтого цвета (42.55 г; чистота 97% @ 210 нм, не содержит кислоты; 100% энантиомерный избыток ("ее").
Затем водный слой после экстракции подкисляют до рН 2 с помощью 50% Н2SO4 и экстрагируют МТВЕ (2×2 л). Вытяжки МТВЕ промывают водой (3×100 мл) и упаривают, получают кислоту в виде твердого вещества желтого цвета (42.74 г; чистота: 99% @ 210 нм, не содержит эфира).

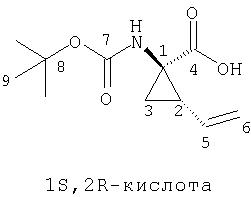
Расщепление В
К 0.5 мл 100 мМ буфера Heps•Na (pH 8.5) в лунке 24-луночного планшета (емкость: 10 мл/лунка) прибавляют 0.1 Savinase 16.0 L (протеаза из Bacillus clausii) (Novozymes North America Inc.) и раствор рацемического этилового эфира N-Вос-(1R,2S)/(1S, 2R)-1 амино-2-винилциклопропанкарбоновой кислоты (10 мг) в 0.1 мл ДМСО. Планшет закрывают (запаивают) и инкубируют при 250 об/мин и 40°С. Через 18 час энантиомерный избыток эфира составляет 44.3%, его определяют следующим образом: отбирают 0.1 мл реакционной смеси и хорошо перемешивают с 1 мл этанола; после центрифугирования 10 микролитров ("мкл") супернатанта анализируют хиральной ВЭЖХ. К оставшейся реакционной смеси прибавляют 0.1 мл ДМСО и планшет инкубируют еще в течение 3 дней при 250 об/мин и 40°С, после чего в лунку прибавляют четыре мл этанола. После центрифугирования 10 мкл супернатанта анализируют хиральной ВЭЖХ и определяют, что энантиомерный избыток составляет 100%.
Расщепление С
К 0.5 мл 100 мМ буфера Heps•Na (pH 8.5) в лунке 24-луночного планшета (емкость: 10 мл/лунка) прибавляют 0.1 Esperase 8.0 L (протеаза из Bacillus halodurans) (Novozymes North America Inc.) и раствор рацемического этилового эфира N-Вос-(1R, 2S)/(1S,2R)-1 амино-2-винилциклопропанкарбоновой кислоты (10 мг) в 0.1 мл ДМСО. Планшет закрывают (запаивают) и инкубируют при 250 об/мин и 40°С. Через 18 час энантиомерный избыток эфира составляет 39.6%, его определяют следующим образом: отбирают 0.1 мл реакционной смеси и хорошо перемешивают с 1 мл этанола; после центрифугирования 10 мкл супернатанта анализируют хиральной ВЭЖХ. К оставшейся реакционной смеси прибавляют 0.1 мл ДМСО и планшет инкубируют еще в течение 3 дней при 250 об/мин и 40°С, после чего в лунку прибавляют четыре мл этанола. После центрифугирования 10 мкл супернатанта анализируют хиральной ВЭЖХ и определяют, что энантиомерный избыток составляет 100%.
Анализ образцов проводят следующим образом:
1) Приготовление образца: Около 0.5 мл реакционной смеси хорошо перемешивают с 10 объемами EtOH. После центрифугирования 10 мкл супернатанта вкалывают (вводят) в колонку ВЭЖХ.
2) Определение конверсии:
Колонка: YMC ODS А 4.6×50 мм, S - 5 мкм
Растворитель: А, 1 мМ HCl в воде; В, MeCN
Градиент: 30% В за 1 мин; 30-45% В за 0.5 мин; 45% В за 1.5 мин; 45-30% В за 0.5 мин.
Скорость потока: 2 мл/мин
УФ детектирование: 210 нм
Время удерживания: кислота, 1.2 мин; эфир, 2.8 мин
3) Определение энантиомерного избытка для эфира:
Колонка: CHIRACEL OD-RH 4.6×150 мм, S - 5 мкм
Подвижная фаза: MeCN/50 мМ HClO4 в воде (67/33)
Скорость потока: 0.75 мл/мин
УФ детектирование: 210 нм
Время удерживания:
(1S,2R) изомер в виде кислоты: 5.2 мин;
Рацемат: 18.5 мин и 20.0 мин;
(1R,2S) изомер в виде эфира: 18.5 мин.
2. Получение этилового эфира N-Boc-(1R,2S)-1 амино-2-циклопропил-циклопропанкарбоновой кислоты

Раствор этилового эфира N-Boc-(1R,2S)-1 амино-2-винилциклопропанкарбоновой кислоты (255 мг, 1.0 ммоль) в эфире (10 мл) обрабатывают ацетатом палладия (5 мг, 0.022 ммоля). Оранжево-красный раствор помещают в атмосферу N2. По каплям в течение 1 час прибавляют избыток диазометана в эфире. Полученный раствор перемешивают при rt в течение 18 час. Избыток диазометана удаляют током азота. Полученный раствор упаривают на роторном испарителе, получая сырой продукт. Флеш-хроматография (10% EtOAc/гексан) дает 210 мг (78%) этилового эфира N-Boc-(1R,2S)-1 амино-2-циклопропилциклопропанкарбоновой кислоты в виде бесцветного масла. LC-MS (время удерживания: 2.13, аналогично методу А, исключение: время градиента 3 мин, колонка Xterra MS С18 S7 3.0×50 мм), MS m/e 270 (M++1).
3. 1-трет-Бутоксикарбониламиноциклопропанкарбоновая кислота является продажной

4. Получение гидрохлорида метилового эфира 1-аминоциклобутанкарбоновой кислоты
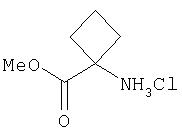
1-Аминоциклобутанкарбоновую кислоту (100 мг, 0.869 ммоля) (Tocris) растворяют в 10 мл МеОН, продувают HCl газ в течение 2 час. Реакционную смесь перемешивают в течение 18 час, а затем упаривают в вакууме, получают 144 мг желтого масла. Растирают с 10 мл эфира, получают 100 мг титульного продукта в виде белого твердого вещества.1Н ЯМР (CDCl3) δ 2.10-2.25 (м, 1Н), 2.28-2.42 (м, 1Н), 2.64-2,82 (м, 4H), 3.87 (с, 3H), 9.21 (уш. с, 3H).
5. Получение рацемического трет-бутилового эфира (1R,2R)/(1S,2S)-1 амино-2-этилциклопропанкарбоновой кислоты; изображенного ниже

Стадия 1: Получение трет-бутилового эфира 2-этилциклопропан-1,1-дикарбоновой кислоты, изображенного ниже.
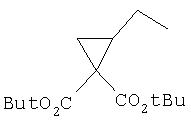
К суспензии бензилтриэтиламмонийхлорида (21.0 г, 92.2 ммоля) в 50% водном растворе NaOH (92.4 г в 185 мл Н2О) прибавляют 1,2-дибромбутан (30.0 г, 138.9 ммоля) и дитрет-бутилмалонат (20.0 г, 92.5 ммоля). Реакционную смесь энергично перемешивают в течение 18 час при rt, затем прибавляют смесь льда с водой. Сырой продукт экстрагируют CH2Cl2 (3х) и последовательно промывают водой (3х), рассолом и органические вытяжки объединяют. Органический слой сушат (MgSO4), фильтруют и упаривают в вакууме. Полученный остаток подвергают флеш - хроматографии (100 г SiO2, 3% Et2O в гексане), получают титульный продукт (18.3 г, 67.8 ммоля, выход 73%), который непосредственно используют на следующей стадии.
Стадия 2: Получение рацемического трет-бутилового эфира 2-этилциклопропан-1, 1-дикарбоновой кислоты, изображенного ниже.
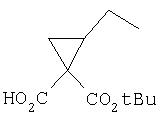
Продукт стадии 1 (18.3 г, 67.8 ммоля) прибавляют к суспензии трет-бутоксида калия (33.55 г, 299.0 ммоля) в сухом эфире (500 мл) при 0°С, а затем Н2O (1.35 мл, 75.0 ммоля) и энергично перемешивают в течение ночи при rt. Реакционную смесь выливают в смесь льда и воды и промывают эфиром (3х). Водный слой подкисляют 10% водн. лимонной кислотой при 0°С и экстрагируют EtOAc (3х). Объединенные органические вытяжки промывают водой (2х), рассолом, сушат (MgSO4) и упаривают в вакууме, получают титульный продукт в виде бледно-желтого масла (10 г, 46.8 ммоля, выход 69%).
Стадия 3: Получение трет-бутилового эфира (1R,2R)/(1S,2S)-2-этил-1-(2-триметилсилилэтоксикарбониламино)циклопропанкарбоновой кислоты, изображенного ниже.

К суспензии продукта Стадии 2 (10 г, 46.8 ммоля) и 3 г свежеактивированных молекулярных сит 4Å в сухом бензоле (160 мл) прибавляют Et3N (7.50 мл, 53.8 ммоля) и DPPA (11 мл, 10.21 ммоля). Реакционную смесь кипятят в течение 3.5 час, затем прибавляют 2-триметилсилилэтанол (13.5 мл, 94.2 ммоля) и реакционную смесь кипятят в течение ночи. Фильтруют, разбавляют Et2O, промывают 10% водным раствором лимонной кислоты, водой, насыщенным водным раствором NaHCO3, водой (2х), рассолом (2х), сушат (MgSO4) и упаривают в вакууме. Остаток суспендируют с 10 г полиизоциантаной поглотительной смолой фирмы Aldrich в 120 мл CH2Cl2, перемешивают при rt в течение ночи и получают титульный продукт (8 г, 24.3 ммоля; 52%) в виде светло-желтого масла:1Н ЯМР (CDCl3) δ 0.03 (с, 9Н), 0.97 (м, 5Н), 1.20 (уш м, 1H), 1.45 (с, 9Н), 1.40-1.70 (м, 4Н), 4.16 (м, 2Н), 5.30 (уш с, 1Н).
Стадия 4: Получение рацемического трет-бутилового эфира (1R,2R)/(1S,2S) 1-амино-2-этилциклопропанкарбоновой кислоты, изображенного ниже.

К продукту Стадии 3 (3 г, 9 ммолей) прибавляют 1.0 М раствор TBAF в ТГФ (9.3 мл, 9.3 ммоля) и смесь кипятят в течение 1.5 час, охлаждают до rt, а затем разбавляют, прибавляя 500 мл EtOAc. Раствор последовательно промывают водой (2×100 мл), рассолом (2×100 мл), сушат (MgSO4), упаривают в вакууме, получают титульный интермедиат.
II Р' элементы:
Элементы Р', получаемые как описано ниже, можно применять для получения соединений Формулы I методами по данному описанию.
1. Получение циклопропилсульфонамида:

Стадия 1: Получение N-трет-бутил-(3-хлор)пропилсульфонамида
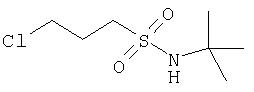
трет-Бутиламин (3.0 моля, 315.3 мл) растворяют в ТГФ (2.5 л). Раствор охлаждают до -20°С. Медленно прибавляют 3-хлорпропансульфонилхлорид (1.5 моля, 182.4 мл.). Реакционную смесь доводят до rt и перемешивают в течение 24 час. Смесь отфильтровывают и фильтрат упаривают в вакууме. Остаток растворяют в CH2Cl2 (2.0 л). Полученный раствор промывают 1N HCl (1.0 л ), водой (1.0 л), рассолом (1.0 л) и сушат Na2SO4. Раствор фильтруют и упаривают в вакууме, получают светло-желтое твердое вещество, которое кристаллизуют из гексана, получают продукт в виде белого твердого вещества (316.0 г, 99%) г.
1Н ЯМР (CDCl3) δ 1.38 (с, 9Н), 2.30-2.27 (м, 2Н), 3.22 (т, J=7.35 Гц, 2Н), 3.68 (т, J=6.2 Гц, 2Н), 4.35 (уш, 1H).
Стадия 2: Получение трет-бутиламида циклопропансульфоновой кислоты

К раствору N-трет-бутил-(3-хлор)пропилсульфонамида (2.14 г, 10.0 ммоля) в ТГФ (100 мл) при - 78°С прибавляют н-BuLi (2.5 М в гексане, 8.0 мл, 20.0 ммолей). Реакционную смесь доводят до комнатной температуры в течение 1 час. Летучие отгоняют в вакууме. Остаток распределяют между EtOAc и водой (200 мл, 200 мл). Отделенный органический слой промывают рассолом, сушат Na2SO4, фильтруют и упаривают в вакууме. Остаток перекристаллизовывают из гексана, получают заданный продукт в виде твердого вещества белого цвета (1.0 г, 56%).
1Н ЯМР (CDCl3): δ 0.98-1.00 (м, 2Н), 1.18-1.19 (м, 2Н), 1.39 (с, 9Н), 2.48-2.51 (м, 1Н), 4.19 (уш, 1Н).
Стадия 3: получение циклопропилсульфонамида

Раствор трет-бутиламида циклопропансульфоновой кислоты (110.0 г, 0.62 моля) в ТФК (500 мл) перемешивают при комнатной температуре в течение 16 час. Летучие отгоняют в вакууме. Остаток перекристаллизовывают из смеси EtOAc/гексан (60 мл/240 мл), получают нужный продукт в виде твердого вещества белого цвета (68.5 г, 91%).
1Н ЯМР (ДМСО-d6): δ 0.84-0.88 (м, 2Н), 0.95-0.98 (м, 2Н), 2.41-2.58 (м, 1Н), 6.56 (уш, 2Н).
2. Альтернативный метод получения циклоприпилсульфонамида
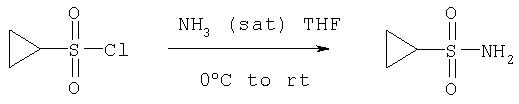
Через ТГФ (100 мл), охлажденный до 0°С, продувают газообразный аммиак до насыщения. К этому раствору прибавляют раствор 5 г (28.45 ммоля) циклопропилсульфонилхлорида (от Array Biopharma) в 50 мл ТГФ, раствор оставляют на ночь, температура при этом доходит до комнатной, и перемешивают еще в течение одного дня. Смесь упаривают до тех пор, пока не останется 1-2 мл растворителя, переносят на слой SiO2 (30 г, элюент 30-60% EtOAc/смесь гексанов), получают 3.45 г (100%) циклопропилсульфонамида в виде твердого вещества белого цвета.1Н ЯМР (Метанол-d4): δ 0.94-1.07 (м, 4Н), 2.52-2.60 (м, 1Н).13С ЯМР (Метанол-d4): δ 5.92, 33.01.
3. Получение циклобутилсульфонамида

К раствору 5.0 г (37.0 ммолей) циклобутилбромида в 30 мл безводного диэтилового эфира (Et2O), охлажденного до -78°С, прибавляют 44 мл (74.8 ммоля) 1.7 М раствора трет-бутиллития в смеси пентанов и температуру раствора медленно, в течение 1.5 час, доводят до -35°С. Эту смесь с помощью иглы медленно переносят (передавливают) в раствор 5.0 г (37.0 ммолей) свежеперегнанного сульфурилхлорида в 100 мл смеси гексанов, охлажденной до -40°С, в течение часа доводят до 0°С и осторожно упаривают в вакууме. Эту смесь снова растворяют в Et2O, промывают один раз ледяной водой, сушат (MgSO4) и осторожно упаривают. Эту смесь снова растворяют в 20 мл ТГФ, по каплям прибавляют к 500 мл насыщенного раствора NH3 в ТГФ и оставляют на ночь при перемешивании. Смесь упаривают в вакууме, сырой продукт в виде твердого вещества желтого цвета перекристаллизовывают из минимального количества СН2Cl2 в смеси гексанов с 1-2 каплями МеОН, получают 1.90 г (38%) циклобутилсульфонамида в виде твердого вещества белого цвета.1Н ЯМР (CDCl3): δ 1.95-2.06 (м, 2Н), 2.30-2.54 (м, 4Н), 3.86 (с(р?), J=8 Гц, 1Н), 4.75 (уш с, 2Н);13С ЯМР (CDCl3): δ 16.43, 23.93, 56.29. HRMS m/z (M-H)- выч. для C4H8NSO2: 134.0276, найдено 134.0282.
4. Получение циклопентилсульфонамида

Раствор 18.5 мл (37.0 ммолей) 2М циклопентилмагнийхлорида в эфире по каплям прибавляют к раствору 3.0 мл (37.0 ммолей) свежеперегнанного сульфурилхлорида (Aldrich) в 100 мл смеси гексанов, охлажденной до -78°С. Температуру смеси в течение 1 часа доводят до 0°С, а затем осторожно упаривают в вакууме. Эту смесь снова растворяют в Et2O (200 мл), промывают один раз ледяной водой (200 мл), сушат (MgSO4) и осторожно упаривают. Эту смесь снова растворяют в 35 мл ТГФ, по каплям прибавляют к 500 мл насыщенного раствора NH3 в ТГФ и оставляют на ночь при перемешивании. Смесь упаривают в вакууме, получают сырой продукт в виде твердого вещества желтого цвета, остаток фильтруют через 50 г силикагеля, используя в качестве элюента 70% EtOAc - смесь гексанов, затем раствор упаривают. Остаток перекристаллизовывают из минимального количества СН2Cl2 в смеси гексанов с 1-2 каплями МеОН, получают 2.49 г (41%) циклопентилсульфонамида в виде твердого вещества белого цвета.1Н ЯМР (CDCl3): δ 1.58-1.72 (м, 2Н), 1.74-1.88 (м, 2Н), 1.94-2.14 (м, 4Н), 3.48-3.59 (м, 1Н), 4.80 (уш с, 2Н);13С ЯМР (CDCl3): δ 25.90, 28.33, 63.54; MS m/e 148 (M-H)-.
5. Получение циклогексилсульфонамида

Раствор 18.5 мл (37.0 ммолей) 2 М циклогексилмагнийхлорида (TCI Americas) в эфире по каплям прибавляют к раствору 3.0 мл (37.0 ммолей) свежеперегнанного сульфурилхлорида в 100 мл смеси гексанов, охлажденной до -78°С. Температуру смеси в течение 1 часа доводят до 0°С, а затем осторожно упаривают в вакууме. Эту смесь снова растворяют в Et2O (200 мл), промывают один раз ледяной водой (200 мл), сушат (MgSO4) и осторожно упаривают. Эту смесь снова растворяют в 35 мл ТГФ, по каплям прибавляют к 500 мл насыщенного раствора NH3 в ТГФ и оставляют на ночь при перемешивании. Смесь упаривают в вакууме, получают сырой продукт в виде твердого вещества желтого цвета, остаток фильтруют через 50 г силикагеля, используя в качестве элюента 70% EtOAc - смесь гексанов, затем раствор упаривают. Остаток перекристаллизовывают из минимального количества СН2Cl2 в смеси гексанов с 1-2 каплями МеОН, получают 1.66 г (30%) циклогексилсульфонамида в виде твердого вещества белого цвета.1Н ЯМР (CDCl3): δ 1.11-1.37 (м, 3H), 1.43-1.56 (м, 2Н), 1.67-1.76 (м, 1Н), 1.86-1.96 (м, 2Н), 2.18-2.28 (м, 2Н), 2.91 (тт, J=12, 3.5 Гц, 1Н), 4.70 (уш с, 2Н);13С ЯМР (CDCl3): δ 25.04, 26.56, 62.74; MS m/e 162 (M-1)-.
6. Получение неопентилсульфонамида

По методике, описанной для получения циклогексилсульфонамида, из 49 мл (37 ммолей) 0.75 М неопентилмагнийхлорида (Alfa) в эфире, получают 1.52 г (27%) неопентилсульфонамида в виде белого твердого вещества.1Н ЯМР (CDCl3): δ 1.17 (с, 9Н), 3.12 (с, 2Н), 4.74 (уш с, 2Н);13С ЯМР (CDCl3): δ 29.46, 31.51, 67.38; MS m/e 150 (M-1)-.
7. Получение циклобутилкарбинилсульфонамида

Раствор 12.3 г (83 ммоля) циклобутилкарбинилбромида (Aldrich) и 13.7 г (91 ммоль) йодида натрия в 150 мл ацетона кипятят в течение ночи, а затем охлаждают до rt. Неорганические твердые вещества отфильтровывают и ацетон и циклобутилкарбинилиодид (8.41 г, 46%) отгоняют при нормальном давлении и при 150 Торр (133.3 Па) при 80°С, соответственно. Раствор 4.0 г (21.98 ммоля) циклобутилкарбинилиодида в 30 мл сухого диэтилового эфира (Et2O), охлажденный до -78°С, переносят с помощью иглы в раствор 17 мл (21.98 ммоля) 1.3 М раствора вт. бутиллития в смеси гексанов и раствор перемешивают 5 мин. В эту смесь с помощью иглы вносят раствор 3.0 г (21.98 ммоля) свежеперегнанного сульфурилхлорида в 110 мл смеси гексанов, охлажденный до -78°С, смесь в течение 1 часа доводят до rt, а затем осторожно упаривают в вакууме. Эту смесь снова растворяют в Et2O, промывают один раз ледяной водой, сушат (MgSO4) и осторожно упаривают. Эту смесь снова растворяют в 30 мл ТГФ, по каплям прибавляют к 500 мл насыщенного раствора NH3 в ТГФ и оставляют на ночь при перемешивании. Смесь упаривают в вакууме, сырой продукт в виде твердого вещества желтого цвета перекристаллизовывают из минимального количества СН2Cl2 в смеси гексанов с 1-2 каплями МеОН, получают 1.39 г (42%) циклобутилкарбинилсульфонамида в виде твердого вещества белого цвета.1Н ЯМР (CDCl3): δ 1.81-2.03 (м, 4Н), 2.14-2.28 (м, 2Н), 2.81-2.92 (м, 1Н), 3.22 (д, J=7 Гц, 2Н), 4.74 (уш с, 2Н);13С ЯМР (CDCl3): δ 19.10, 28.21, 30.64, 60.93; MS m/e 148 (M-1)-. время: 1.73, метод В. 818 (M++H) (?).
8. Получение циклопропилкарбинилсульфонамид
По методике, применяемой для получения циклобутилкарбинилсульфонамида, циклопропилкарбинилсульфонамид получают из циклопропилкарбинилбромида (Aldrich) (см. также JACS 1981, р.442-445).1Н ЯМР (CDCl3): δ 0.39-0.44 (м, 2Н), 0.67-0.76 (м, 2Н), 1.13-1.27 (м, 1Н), 3.03 (д, J=7.3 Гц, 2Н), 4.74 (уш. с, 2Н);13С ЯМР (CDCl3): δ 4.33, 5.61, 59.93; MS m/e 134 (M-1).
III Гетероциклы, применяемые в качестве исходных при построении элементов Р2 с целью последующего введения в соединения Формулы I.
1. Изохинолины

Изохинолин (1) и их замещенные аналоги можно встраивать в элементы Р2 двумя методами, коротко охарактеризованными выше и подробно представленными в данном описании. Указанные элементы Р2 (3) можно превратить в соединения Формулы I методами, аналогичными методам по данному описанию для получения сходных аналогов изохинолина.
2. Изоксазолпиридин и оксазолпиридин (1)

Изоксазольный и оксазольный гетероцикл (1) и их аналоги можно получать известными химическими реакциями и встраивать в соединения Формулы I методами по данному описанию для аналогичных изоксазолпиридиновых интермедиатов, как показано в секции В.
Подраздел В:
В Секции В для LC/MS анализа используют следующие условия:
Колонки: (Метод A): YMC ODS-A C18 S7 (4.6×33 мм)
(Метод В): YMC Xterra ODS S7 (3.0×50 мм)
(Метод С): Xterra ms C18 (4.6×33 мм)
(Метод D): YMC ODS-A C18 S3 (4.6×33 мм)
Градиент: от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В
Время градиента: 3 мин.
Время выдерживания: 1 мин.
Скорость потока: 5 мл/мин
Длина волны детектора: 220 нм
Растворитель А: 10% МеОН/90% Н2О/0.1% ТФК. Растворитель В: 90% МеОН/10% Н2O/0.1% ТФК
Следующие условия используют для преп. - ВЭЖХ разделения.
Колонки: Phenomenex - Luma 30Х100 мм, S5
Градиент: от 60% растворителя А/40% растворителя В до 0% растворителя А/100% растворителя В
Время градиента: 15 мин.
Время прекращения: 20 мин.
Скорость потока: 30 мл/мин
Длина волны детектора: 220 нм
Растворители: Растворитель А: 10% МеОН/90% Н2O/0.1% ТФК. Растворитель В: 90% МеОН/10% Н2O/0.1% ТФК
Пример 1: Получение соединения 1.
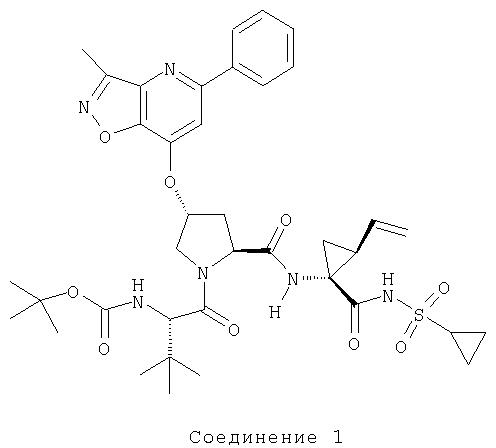
Схема 1
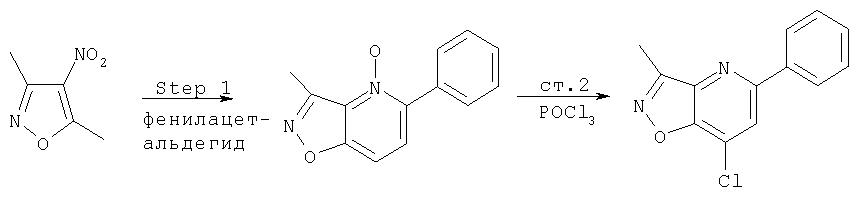
Стадия 1:
Смесь 3,5-диметил-4-нитроизоксазол (1.42 г, 10.0 ммоля), фенилацетальдегид (1.32 г, 11.0 ммоля) в пиперидине (1 мл) и этаноле (10 мл) кипятят в течение 16 час. По охлаждении до комнатной температуры продукт выпадает и его отфильтровывают. Осадок на фильтре тщательно промывают холодным этанолом, получают 1.20 г (53%) нужного продукта в виде твердого вещества белого цвета.1Н ЯМР (CDCl3): δ 2.87 (с, 3H), 7.46-7.50 (м, 3H), 7.56 (д, J=8.5 Гц, 1Н), 7.7-7.80 (м, 2Н).
LC-MS (время удерживания: 1.19 мин, метод В), MS m/z 227 (М++Н).
Стадия 2:
Раствор 3-метил-5-фенилизоксазоло[4,5-b]пиридин-4-оксид (1.00 г, 4.40 ммоля) и POCl3 (2.71 г, 17.7 ммоля) в хлороформе (10 мл) кипятят 1 час. По охлаждении до комнатной температуры к полученному раствору прибавляют хлороформ (50 мл) и промывают NaHCO3 (волн.) (две порции по 50 мл) и рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают флеш - хроматографией (4:1 гексан-EtOAc), получают 790 мг (73%) нужного продукта в виде твердого вещества белого цвета.
1Н ЯМР (CDCl3): δ 2.72 (с, 3H), 7.46-7.54 (м, 3H), 7.91 (с, 1Н), 8.00-8.03 (м, 2Н); LC-MS (время удерживания: 1.76 мин, метод В), MS m/z 245, 247 (M++H).
Схема 2
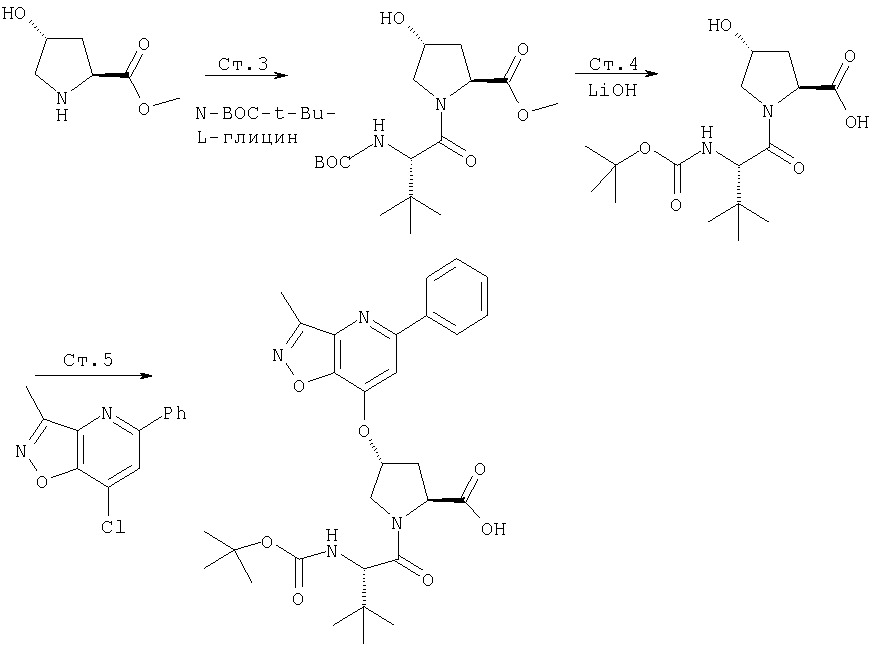
Стадия 3:
К смеси метилового 4-гидроксипирролидин-2-карбоновой кислоты (Н-Нур-ОМе HCl) (1.81 г, 10.0 ммолей), HATU (5.70 г, 15.0 ммолей) и N-BOC-трет-бутил-L-глицина (2.42 г, 105 ммолей) в СН2Cl2 (100 мл) прибавляют DIPEA (3.47 г, 31.0 ммоль) при 0°С. Перемешивают при комнатной температуре в течение 12 час, к полученному раствору прибавляют СН2Cl2 (100 мл), промывают холодной (ледяной) 5% лимонной кислотой (водн.). Органический слой промывают холодной (ледяной) 5% лимонной кислотой, 1М NaOH, рассолом, последовательно, сушат MgSO4 и фильтруют. Фильтрат упаривают в вакууме, получают 3.55 г (99%) нужного продукта в виде желтовато-белой пены. Этот продукт используют на следующей стадии без дополнительной очистки.
1Н ЯМР(CD3OD): δ 1.04 (с, 9Н), 1.43 (с, 9Н), 1.99-2.03 (м, 1H), 2.20-2.30 (м, 1Н), 3.69 (с, 3H), 3.70-3.79 (м, 2Н), 4.28 (уш, 1Н), 4.46 (уш, 1Н), 4.74-4.80 (м, 1Н);
LC-MS (время удерживания: 1.28 мин, метод В), MS m/z 359 (M++Н).
Стадия 4:
Смесь продукта Стадии 3 (3.55 г, 9.9 ммоля) в ТГФ (50 мл ), МеОН (50 мл) и LiOH моногидрата (0.83 г, 19.9 ммоля в 50 мл Н2О) перемешивают при комнатной температуре в течение ночи. После удаления летучих в вакууме, остаток растворяют в 0.1 М NaOH (100 мл). Этот водный раствор промывают эфиром (50 мл ), подкисляют 1М HCl до рН 4. Экстрагируют EtOAc (100 мл ). Органический слой промывают 5% лимонной кислотой и рассолом, служат MgSO4, упаривают досуха, получают 3.20 г (95%) нужного продукта в виде белой пены. Этот продукт используют на следующей стадии без дополнительной очистки.
1Н ЯМР (CD3OD): δ 1.02 (с, 9Н), 1.43 (с, 9Н), 2.01-2.09 (м, 1Н), 2.25-2.32 (м, 1Н), 3.70-3.85 (м, 2Н), 4.26-4.30 (м, 1Н), 4.46-4.51 (м, 2Н), 6.37-6.41 (м. 1Н); LC-MS (время удерживания: 1.14 мин, метод В), MS m/z 345 (М++Н).
Стадия 5:
К раствору продукта Стадии 4 (1.01 г, 2.93 ммоля) в ДМСО (30 мл) прибавляют трет-бутоксид калия (1.02 г, 9.08 ммоля). Полученный раствор перемешивают при комнатной температуре 1 час и прибавляют 7-хлор-3-метил-5-фенилизоксазоло[4,5-b]пиридин (0.75 г, 3.08 ммоля). Полученный раствор перемешивают 12 час. Затем прибавляют ледяную воду, подкисляют до рН 4, добавляя 1М HCl, экстрагируют EtOAc (две порции по 200 мл). Органические вытяжки промывают рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают преп. - ВЭЖХ (60% В - 100% В, 15 мин градиент), получают 305 мг (19%) нужного продукта в виде бледно-желтого твердого вещества.
1Н ЯМР (CD3OD): δ 1.02 (с, 9Н), 1.17 (с, 9Н), 2.37-2.47 (м, 1Н), 2.64 (с, 3H), 2.85-2.93 (м, 1Н), 4.00-4.08 (м, 1Н), 4.14 (уш, 1Н), 4.49-4.55 (м, 1Н), 4.62-4.71 (м, 1Н), 5.70 (м, 1Н), 7.45-7.53 (м, 3H), 7.56 (с, 1Н), 8.03-8.06 (м, 2Н);
LC-MS (время удерживания: 1.89 мин, метод В), MS m/z 553 (М+Н).
Схема 3
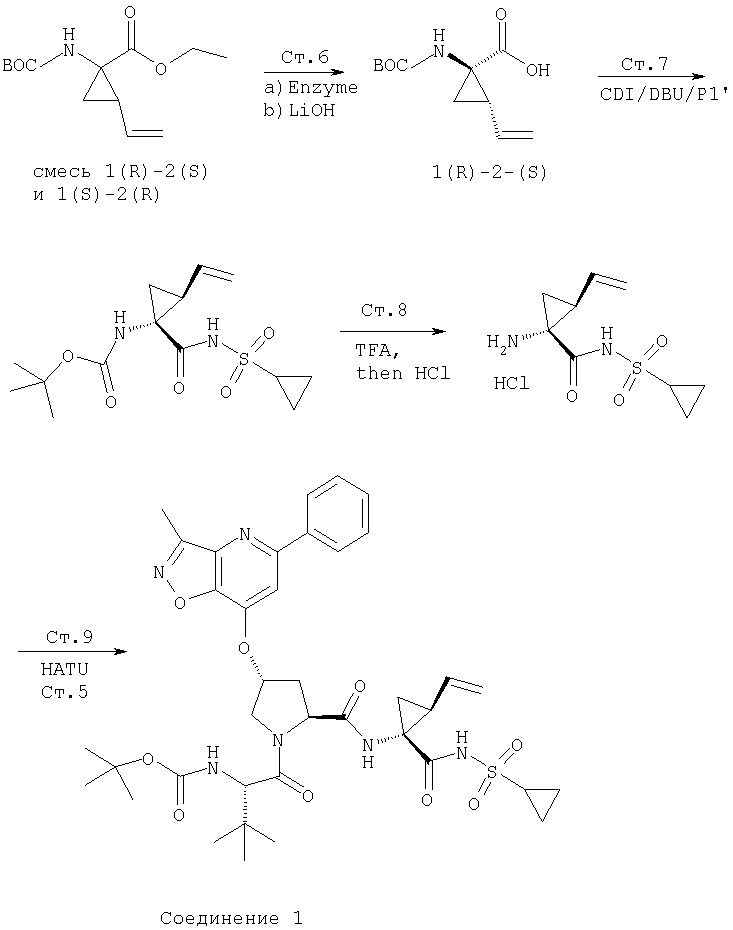
Стадия 6а
Проводится как описано в секции А.
Стадия 6b:
К раствору этилового эфира 1(R)-трет-бутоксикарбониламино-2(S)-винилциклопропанкарбоновой кислоты, продукта Стадии 6а (3.28 г, 13.2 ммоля), в ТГФ (7 мл ) и метаноле (7 мл) прибавляют суспензию LiOH (1.27 г, 53.0 ммоля) в воде (14 мл). Смесь перемешивают при комнатной температуре в течение ночи и прибавляют 1N NaOH (15 мл ) и воду (20 мл). Полученную смесь промывают EtOAc (20 мл) и органические вытяжки экстрагируют 20 мл 0.5N NaOH. Объединенные промывные воды подкисляют до рН 4, прибавляя 1N HCl и экстрагируют EtOAc (3×40 мл ). Объединенные органические вытяжки промывают рассолом и сушат (MgSO4), получают титульное соединение в виде твердого вещества белого цвета (2.62 г, 87%).1Н ЯМР (ДМСО-d6): δ 1.22-1.26 (м, 1Н), 1.37 (с, 9Н), 1.50-1.52 (м, 1Н), 2.05 (кв, J=9 Гц, 1Н), 5.04 (д, J=10 Гц, 1Н), 5.22 (д, J=17 Гц, 1Н), 5.64-5.71 (м, 1Н), 7.18, 7.53 (с, NH (ротамеры), 12.4 (уш с, 1Н));
LC-MS (время удерживания: 1.67 мин, метод В), MS m/z 228 (М+Н).
Стадия 7:
Раствор продукта Стадии 6 (2.62 г, 11.5 ммоля) и CDI (2.43 г, 15.0 ммолей) в ТГФ (40 мл) кипятят 50 мин под азотом. Раствор охлаждают до комнатной температуры и с помощью иглы переносят в раствор циклопропилсульфонамида (1.82 г, 15.0 ммолей) в ТГФ (10 мл). К полученному раствору прибавляют DBU (2.40 мл, 16.1 ммоля) и перемешивание продолжают в течение 20 час. К смеси прибавляют 1N HCl до рН 1 и ТГФ упаривают в вакууме. Суспензию экстрагируют EtOAc (2×50 мл) и объединенные органические вытяжки сушат (Na2SO4). Очистка перекристаллизацией из смеси гексанов - EtOAc (1:1) дает титульное соединение (2.4 г) в виде твердого вещества белого цвета. Маточную жидкость очищают на колонке Biotage 40S (элюент 9% ацетон в DCM), получают вторую партию титульного соединения (1.1 г). Обе партии объединяют (общий выход 92%).
1Н ЯМР (ДМСО-d6): δ 0.96-1.10 (м, 4Н), 1.22 (дд, J=5.5, 9.5 Гц, 1Н), 1.39 (с, 9Н), 1.70 (т, J=5.5 Гц, 1Н), 2.19-2.24 (м, 1Н), 2.90 (м, 1Н), 5.08 (д, J=10 Гц, 1Н), 5.23 (д, J=17 Гц, 1Н), 5.45 (м, 1Н), 6.85, 7.22 (с, NH (ротамеры);
LC-MS (время удерживания: 1.70 мин, метод В), MS m/z 331 (М++Н).
Стадия 8:
Раствор продукта Стадии 7 (3.5 г, 10.6 ммоля) в DCM (35 мл) и ТФК (32 мл) перемешивают при комнатной температуре в течение 1.5 час. Летучие удаляют в вакууме, а остаток суспендируют в 1N HCl в диэтиловом эфире (20 мл) и упаривают в вакууме. Эту процедуру повторяют один раз. Полученную смесь растирают в пентане и отфильтровывают, получая заглавное соединение в виде гигроскопического вещества не чисто белого цвета (2.60 г, 92%).
1Н ЯМР (ДМСО- d6): δ 1.01-1.15 (м, 4Н), 1.69-1.73 (м, 1Н), 1.99-2.02 (м, 1Н), 2.38 (кв, J=9 Гц, 1Н), 2.92-2.97 (м, 1Н), 5.20 (д, J=11 Гц, 1Н), 5.33 (д, J=17 Гц, 1Н), 5.52-5.59 (м, 1Н), 9.17 (уш с, 3H);
LC-MS (время удерживания: 0.24 мин, метод В), MS m/z 231 (М++Н).
Стадия 9:
К ледяной (холодной) смеси продукта Стадии 5 (70 мг, 0.13 ммоля), гидрохлорида (1-амино-2-винил-циклопропанкарбонил)амида (1R,2S)-циклопропансульфоновой кислоты, продукта Стадии 8 (37 мг, 0.14 ммоля), и HATU (72 мг, 0.19 ммоля) в DCM (2 мл) прибавляют диизопропилэтиламин (50 мг, 0.39 ммоля). Образующийся раствор оставляют на 12 час нагреваться до комнатной температуры и упаривают в вакууме. Остаток очищают в вакууме преп. - ВЭЖХ (60%В-100%В, градиент 15 мин), получают 52 мг (54%) Соединения I в виде твердого вещества сероватого цвета.
1Н ЯМР (CD3OD): δ 0.96-1.09 (м, 12Н), 1.16-1.25 (м, 10Н), 1.44-1.48 (м, 1Н), 1.87-1.91 (м, 1Н), 2.20-2.40 (м, 2Н), 2.63-2.65 (м, 4Н), 2.89-2.98 (м, 1Н), 4.08-4.20 (м, 2Н), 4.44-4.65 (м, 2Н), 5.13 (д, J=11.7 Гц, 1Н), 5.32 (д, J=15 Гц, 1Н), 5.72-5.85 (уш,2Н), 6.62 (д, J=15.0 Гц, 1Н), 7.46-7.53 (м, 3H), 7.58 (с, 1Н), 8.04-8.07 (м, 2Н);
LC-MS (время удерживания: 1.92 мин, метод В), MS m/z 765 (М++Н).
Пример 2: Получение Соединения 2.
Схема 1
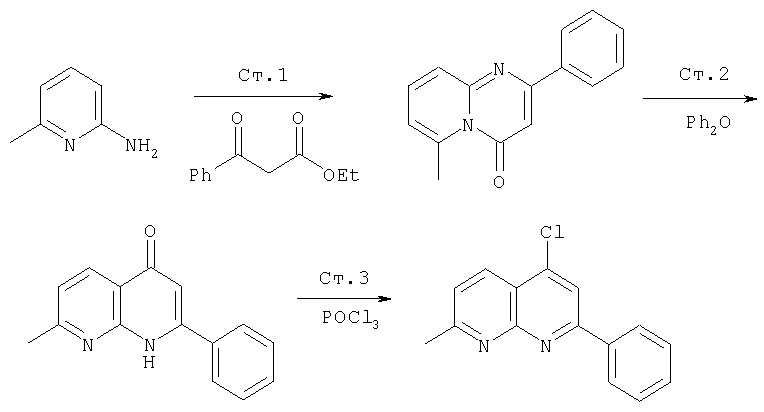
Стадия 1:
Смесь 2-амино-6-метилпиридина (1.08 г, 10.0 ммоля), этилбензоилацетата (2.30 г, 12.0 ммоля) и полифосфорной кислоты (6.00 г, 61.2 ммоля) греют при 110°С в течение 5 час. По охлаждении до комнатной температуры смесь выливают в ледяную воду (20 мл) и нейтрализуют до рН 7 с помощью 10 М NaOH. Экстрагируют CHCl3. Органический слой промывают рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают флеш - хроматографией (1:1 гексан - EtOAc), получают 510 мг (22%) заданного продукта в виде бледно- желтого вещества.
1Н ЯМР (CDCl3): δ 3.08 (с, 3H), 6.64 (д, J=7.0 Гц, 1Н), 6.71 (с, 1Н), 7.42-7.52 (м, 5Н), 8.04-8.06 (м, 2Н);
LC-MS (время удерживания: 1.21 мин, метод В), MS m/z 237 (М++Н).
Стадия 2:
Раствор 6-метил-2-фенилпиридо[1,2а]пиримидин-4-она (489 мг, 2.07 ммоля) в расплавленном диметиловом эфире (5 мл) нагревают при слабом кипении в течение 5 час. По охлаждении до комнатной температуры к образовавшейся суспензии прибавляют диэтиловый эфир (10 мл), осадок отфильтровывают. Тщательно промывают на фильтре диэтиловым эфиром, получают 450 мг (92%) нужного продукта в виде твердого вещества коричневатого цвета.
LC-MS (время удерживания: 1.25 мин, метод В), MS m/z 237 (М++Н).
Стадия 3:
Суспензию 7-метил-2-фенил-1Н-[1, 8]нафтиридин-4-она (450 мг, 1.91 ммоля) в POCl3 (10 мл) нагревают при слабом кипении в течение 3 час. Упаривают в вакууме. Остаток выливают в ледяную воду (20 мл) и нейтрализуют до рН 10, добавляя NaOH. Экстрагируют CHCl3. Органические вытяжки промывают рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают флеш-хроматографией (2:1 гексан - EtOAc), получают 450 мг (92%) нужного продукта в виде твердого вещества розового цвета.
1Н ЯМР (CD3OD): δ 2.80 (с, 3H), 7.54-7.56 (м, 3H), 7.61 (д, J=8.4 Гц, 1Н), 8.25-8.30 (м, 3H), 8.58 (д, J=8.4 Гц, 1Н);
LC-MS (время удерживания: 1.39 мин, метод В), MS m/z 255,257 (M++Н).
Схема 2
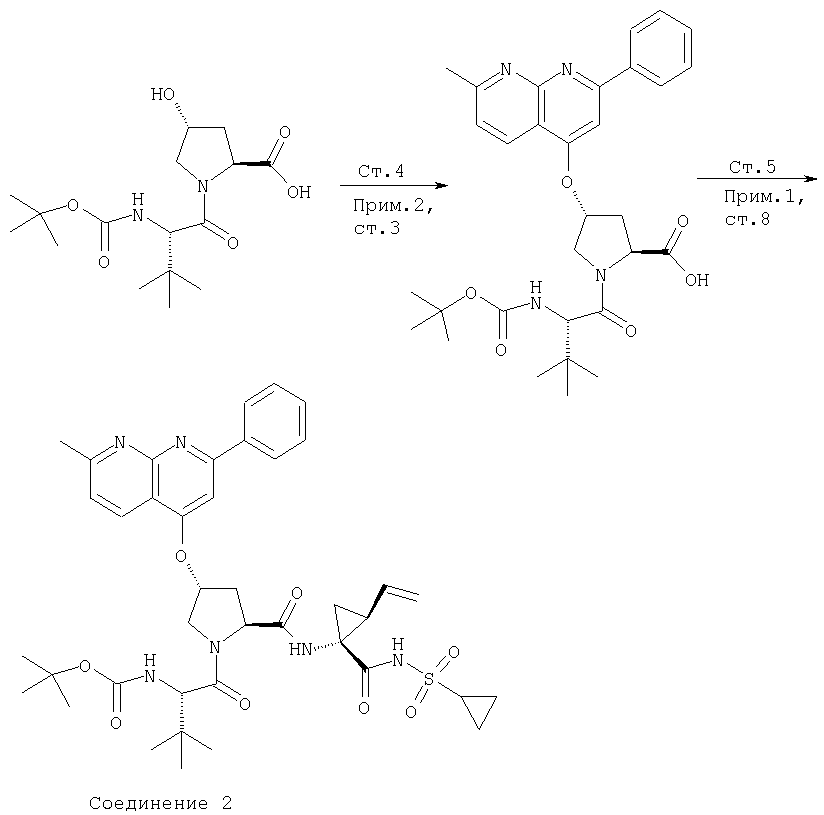
Стадия 4:
Продукт получают по методике, описанной в Примере 1, Стадия 5, за исключением того, что используют продукт 3 Стадии из Примера 2, 4-хлор-7-метил-2-фенил-[1,8]нафтиридин.
LC-MS (время удерживания: 1.55 мин, метод В), MS m/z 563 (М++Н).
Стадия 5
Соединение 2 получают по методике, описанной в Примере 1, Стадия 9, за исключением того, что используют продукт из Примера 2, Стадия 4.
1Н ЯМР (CD3OD): δ 1.01-1.10 (м, 12Н), 1.21-1.26 (м, 10Н), 1.40-1.45 (м, 1Н), 1.86-1.91 (м, 1Н), 2.20-2.29 (м, 1Н), 2.39-2.49 (м, 1Н), 2.72-2.81 (м, 1Н), 2.92-2.95 (м, 4Н), 4.10-4.16 (м, 2Н), 4.55-4.65 (м, 2Н), 5.14 (д, J=12.0 Гц, 1Н), 5.30 (д, J=15.0 Гц, 1Н), 5.67-5.82 (м, 2Н), 7.60-7.80 (м, 3H), 7.78 (д, J=8.6 Гц, 1Н), 7.87 (с, 1Н), 8.26-8.29 (м, 2Н), 8.95 (д, J=8.4 Гц, 1Н);
LC-MS (время удерживания: 1.62 мин, метод В), MS m/z 775 (M++H).
Пример 3: Получение Соединения 3.

Схема 1
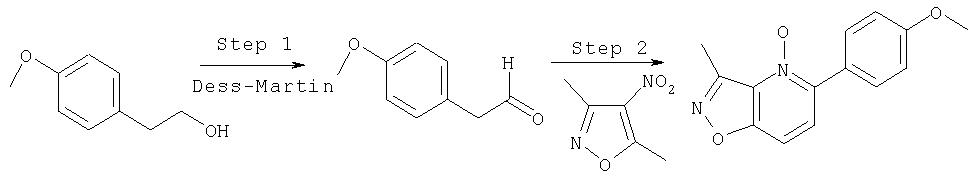
К раствору 4-метилоксифенетилового спирта (1.52 г, 10.0 ммоля) в CH2Cl2 (50 мл) при 0°С прибавляют сразу все количество реагента Десса- Мартина (4.45 г, 10.5 ммоля). Образовавшуюся смесь оставляют нагреваться до комнатной температуры в течение 1 час. Промывают нас. водным раствором Na2S2O3 и 1М NaOH, рассолом, последовательно. Сушат MgSO4, упаривают в вакууме, получают 1.50 г (100%) нужного альдегида в виде вязкого масла. Этот продукт используют без дополнительной очистки.
Стадия 2:
Раствор 3,5-диметил-4-нитроизоксазола (142 мг, 1.0 ммоль), 4-метокси-фенилацетальдегида из Примера 3, Стадия 1 (180 мг, 1.1 ммоля) в пиперидине (0.1 мл) и этаноле (2 мл) кипятят в течение 12 час. По охлаждении до комнатной температуры продукт выпадает и его отфильтровывают. Осадок на фильтре тщательно промывают холодным этанолом, получают 130 мг (51%) ожидаемого продукта в виде твердого вещества сероватого цвета.
1Н ЯМР (CDCl3) δ 2.88 (с, 3H), 3.87 (с, 3H), 7.02 (д, J=8.5 Гц, 2Н), 7.50 (д, J=9.0 Гц, 1Н), 7.57 (д, J=9.0 Гц, 1Н), 7.81 (д, J=8.5 Гц, 2Н);
LC-MS (время удерживания: 1.24 мин, метод В), MS m/z 257 (N++H).
Стадия 3:

Этот продукт получают по методике, описанной в Примере 1, Стадия 2, за исключением того, что используют продукт из Примера 3, Стадия 2.
1Н ЯМР (CDCl3) δ 2.70 (с, 3H), 3.87 (с, 3H), 7.00-7.03 (м, 2Н), 7.84 (с, 1Н), 7.96-7.98 (м, 2Н);
LC-MS (время удерживания: 1.96 мин, метод В), MS m/z 275, 277 (М++Н).
Стадия 4:
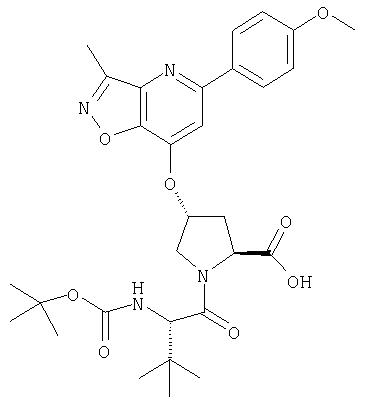
Этот продукт получают по методике, описанной в Примере 1, Стадия 5, за исключением того, что используют продукт из Примера 3, Стадия 3.
1Н ЯМР (CD3OD): δ 1.02 (с, 9Н), 1.18 (с, 9Н), 2.39-2.43 (м, 1Н), 2.63 (с, 3H), 2.75-2.80 (м, 1Н), 3.87 (с, 3H), 4.00-4.08 (м, 1Н), 4.17 (уш, 1Н), 4.49-4.55 (м, 1Н), 4.62-4.71 (м, 1Н), 5.68 (уш, 1Н), 7.05 (д, J=8.5 Гц, 2Н), 7.49 (с, 1Н), 8.00 (д, J=8.5 Гц, 2Н);
LC-MS (время удерживания: 1.89 мин, метод В), MS m/z 583 (M++H).
Стадия 5:
Соединение 3 получают по методике, описанной в Примере 1, Стадия 9, за исключением того, что используют продукт из Примера 3, Стадия 4.
1Н ЯМР (CD3OD): δ 1.01-1.09 (м, 12Н), 1.17-1.26 (м, 10Н), 1.44-1.47 (м, 1Н), 1.87-1.91 (м, 1Н), 2.20-2.40 (м, 2Н), 2.63-2.65 (м, 4Н), 2.89-2.98 (м, 1Н), 3.87 (с, 3H), 4.08-4.20 (м, 2Н), 4.44-4.65 (м, 2Н), 5.13 (д, J=11.7 Гц, 1Н), 5.32 (д, J=15.0 Гц, 1Н), 5.72-5.85 (м, 2Н), 7.05 (д, J=8.5 Гц, 2Н), 7.06 (с, 1Н), 8.01 (д, J=8.5 Гц, 2Н);
LC-MS (время удерживания: 1.96 мин, метод В), MS m/z 795 (М++Н).
Пример 4: Получение Соединения 4

Стадия 1

Этот продукт получают по методике, описанной в Примере 3, Стадии 1 и 2, за исключением того, что используют 4-фторфенетиловый спирт.
LC-MS (время удерживания: 1.18 мин, метод В), MS m/z 245 (M++H).
Стадия 2:
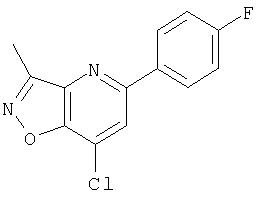
Этот продукт получают по методике, описанной в Примере 1, Стадия 2, за исключением того, что используют продукт из Примера 4, стадия 1.
1Н ЯМР (CDCl3) δ 2.71 (с, 3H), 7.17-7.20 (м, 2Н), 7.86 (с, 1Н), 8.00-8.02 (м, 2Н);
LC-MS (время удерживания: 1.71 мин, метод В), MS m/z 263, 265 (M++H).
Стадия 3

Этот продукт получают по методике, описанной в Примере 1, Стадия 5, за исключением того, что используют продукт из Примера 4, стадия 2.
LC-MS (время удерживания: 1.91 мин, метод В), MS m/z 571 (M++Н).
Стадия 4:
Этот продукт получают по методике, описанной в Примере 1, Стадия 9, за исключением того, что используют продукт из Примера 4, стадия 3.
1Н ЯМР (CD3OD): δ 1.01-1.09 (м, 12Н), 1.17-1.26 (м, 10Н), 1.44-1.47 (м, 1Н), 1.87-1.91 (м, 1Н), 2.20-2.40 (м, 2Н), 2.63-2.65 (м, 4Н), 2.89-2.98 (м, 1Н), 4.08-4.20 (м, 2Н), 4.44-4.65 (м, 2Н), 5.13 (д, J=11.7 Гц, 1Н), 5.32 (д, J=15.0 Гц, 1Н), 5.72-5.85 (м, 2Н), 7.20-7.26 (м, 2Н), 7.60 (с, 1Н), 8.09-8.14 (м, 2Н), 9.26 (уш, 1Н);
LC-MS (время удерживания: 1.91 мин, метод В), MS m/z 783 (M++Н).
Пример 5: Получение Соединения 5.
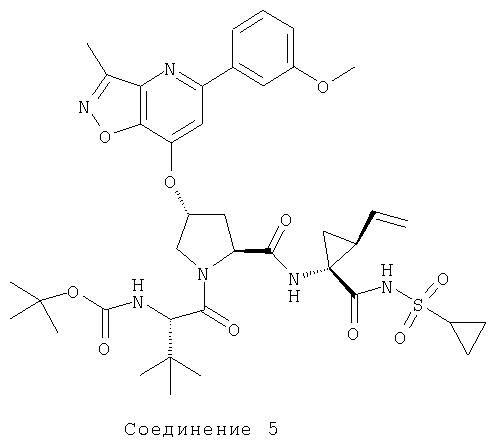
Стадия 1

Этот продукт получают по методике, описанной в Примере 3, Стадии 1 и 2, за исключением того, что используют 3-метоксифенетиловый спирт.
LC-MS (время удерживания: 1.03 мин, метод В), MS m/z 257 (М+ +Н).
Стадия 2:
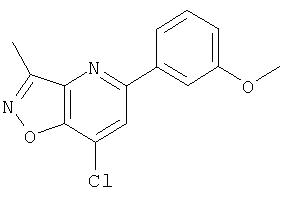
Этот продукт получают по методике, описанной в Примере 1, Стадия 2, за исключением того, что используют продукт из Примера 5, стадия 1.
1Н ЯМР (CDCl3) δ 2.72 (с, 3H), 3.90 (с, 3H), 7.00-7.02 (м, 1Н), 7.41 (т, J=8.0 Гц, 1H), 7.55 (д, J=7.5 Гц, 1Н), 7.59 (д, J=2.0 Гц. 1Н), 7.89 (с, 1Н);
LC-MS (время удерживания: 1.89 мин, метод В), MS m/z 275,277 (М++Н).
Стадия 3:
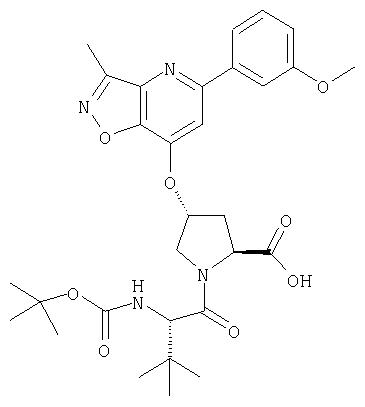
Этот продукт получают по методике, описанной в Примере 1, Стадия 5, за исключением того, что используют продукт из Примера 5, стадия 2.
1Н ЯМР (CD3OD): δ 1.02 (с, 9Н), 1.18 (с, 9Н), 2.37-2.47 (м, 1Н), 2.64 (с, 3H), 2.85-2.93 (м, 1Н), 3.88 (с, 3H), 4.00-4.08 (м, 1Н), 4.14 (уш, 1Н), 4.49-4.55 (м, 1Н), 4.62-4.71 (м, 1Н), 5.71 (уш, 1Н), 7.02-7.04 (м, 1Н), 7.40 (т, J=8.0 Гц, 1Н), 7.58-7.62 (м, 3H);
LC-MS (время удерживания: 1.90 мин, метод В), MS m/z 583 (М++Н).
Стадия 4:
Соединение 5 получают по методике, описанной в Примере 1, Стадия 9, за исключением того, что используют продукт из Примера 5, стадия 3.
1Н ЯМР (CD3OD): δ 1.01-1.09 (м, 12Н), 1.17-1.29 (м, 10Н), 1.44-1.47 (м, 1Н), 1.87-1.91 (м, 1Н), 2.20-2.40 (м, 2Н), 2.63-2.65 (м, 4Н), 2.89-2.98 (м, 1Н), 3.89 (с, 3H), 4.08-4.20 (м, 2Н), 4.44-4.65 (м, 2Н), 5.13 (д, J=11.7 Гц, 1Н), 5.32 (д, J=15.0 Гц, 1Н), 5.72-5.85 (м, 2Н), 7.02-7.05 (м, 1Н), 7.41 (т, J=8.0 Гц, 1Н), 7.55-7.61 (м, 3H);
LC-MS (время удерживания: 1.96 мин, метод В), MS m/z 795 (M++H).
Пример 6: Получение Соединения 6.

Стадия 1:

Этот продукт получают по методике, описанной в Примере 3, Стадии 1 и 2, за исключением того, что используют 2-метоксифенетиловый спирт.
LC-MS (время удерживания: 1.10 мин, метод В), MS m/z 257 (М++Н).
Стадия 2:
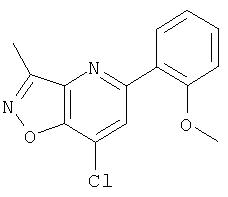
Этот продукт получают по методике, описанной в Примере 6, Стадия 2, за исключением того, что используют продукт из Примера б, стадия 1.
1Н ЯМР (CDCl3): δ 2.721 (с, 3H), 3.88 (с, 3H), 7.03 (д, J=8.0 Гц, 1Н), 7.11 (т, J=7.5 Гц, 1Н), 7.41-7.44 (м, 1Н), 7.79-7.81 (м, 1Н), 8.04 (с, 1Н);
LC- MS (время удерживания: 1.92 мин, метод В), MS m/z 275,277 (М++Н).
Стадия 3:
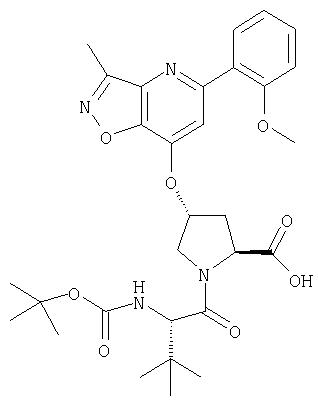
Этот продукт получают по методике, описанной в Примере 1, Стадия 5, за исключением того, что используют продукт из Примера 6, стадия 2.
1Н ЯМР (CD3OD): δ 1.02 (с, 9 Н), 1.20 (с, 9Н), 2.37-2.47 (м, 1Н), 2.63 (с, 3H), 2.85-2.93 (м, 1Н), 3.89 (с, 3H), 4.00-4.08 (м, 1Н), 4.14 (уш, 1Н), 4.49-4.55 (м, 1Н), 4.62-4.71 (м, 1Н), 5.56 (уш, 1Н), 7.09 (т, J=7.5 Гц, 1Н), 7.15 (д, J=8.5 Гц, 1Н), 7.41-7.44 (м, 1Н), 7.52 (с, 1Н), 7.67 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 1.75 мин, метод В), MS т/г 583 (М++Н).
Стадия 4:
Соединение 6 получают по методике, описанной в Примере 1, Стадия 9, за исключением того, что используют продукт из Примера 6, стадия 3.
1 Н ЯМР (CD3OD): δ 1.01-1.08 (м, 12Н), 1.17-1.26 (м, 10Н), 1.44-1.47 (м, 1Н), 1.87-1.91 (м, 1Н), 2.20-2.40 (м, 2Н), 2.63-2.65 (м, 4Н), 2.89-2.98 (м, 1Н), 3.88 (с, 3H), 4.08-4.12 (м, 1Н), 4.19 (уш, 1Н), 4.44-4.65 (м, 2Н), 5.13 (д, J=11.7 Гц, 1Н), 5.32 (д, J=15.0 Гц, 1Н), 5.59 (уш, 1Н), 5.72-5.80 (м, 1Н), 7.09 (т, J=7.5 Гц, 1Н), 7.15 (д, J=8.5 Гц, 1Н), 7.41-7.45 (м, 1Н), 7.66 (с, 1Н), 7.66-7.67 (м, 1Н);
LC-MS (время удерживания: 1.93 мин, метод В), MS m/z 795 (M++H).
Пример 7: Получение Соединения 7.

Стадия 1:
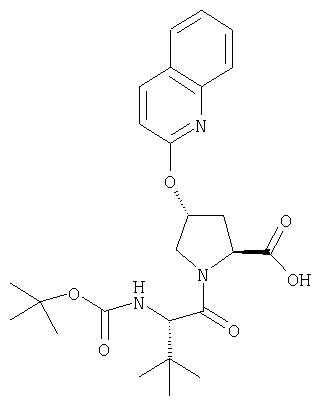
Этот продукт получают по методике, описанной в Примере 1, Стадия 5, за исключением того, что используют 2-хлорхинолин.
LC-MS (время удерживания: 1.73 мин, метод В), MS m/z 472 (М++Н).
Стадия 2:
Соединение 7 получают по методике, описанной в Примере 1, Стадия 9, за исключением того, что используют продукт из Примера 7, стадия 1.
1Н ЯМР (CD3OD): δ 1.01-1.08 (м, 12Н), 1.17-1.26 (м, 10Н), 1.44-1.47 (м, 1Н), 1.87-1.91 (м, 1Н), 2.23-2.30 (м, 2H), 2.52-2.57 (м, 1Н), 2.89-2.98 (м, 1Н), 4.10-4.14 (м, 1Н), 4.09-4.15 (м, 2H), 4.47-4.51 (m, 1Н), 5.13 (д, J=10.0 Гц, 1Н), 5.32 (д, J=17.0 Гц, 1Н), 5.73-5.78 (м, 1Н), 5.92 (уш, 1Н), 6.90-6.92 (м, 1Н), 7.42 (т, J=7.5 Гц, 1Н), 7.64 (т, J=7.5 Гц, 1Н), 7.78-7.82 (м, 2H), 8.13 (д, J=75 Гц, 1Н), 9.18 (д, 1Н);
LC-MS (время удерживания: 1.75 мин, метод В), MS m/z 684 (М+ +Н).
Пример 8: Получение Соединения 8.

Схема 1
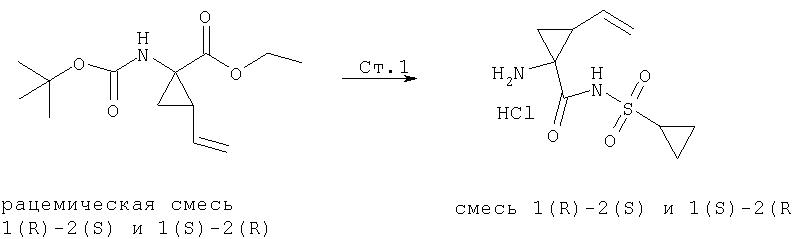
Стадия 1:
Этот продукт получают по методике, описанной в Примере 1, Стадии 6b-8, без Стадии 6а, стадии расщепления рацемата.
LC-MS (время удерживания: 0.24 мин, метод В), MS m/z 231 (М++Н).
Схема 2

Стадия 2:
К охлаждаемой льдом ("ледяной") смеси N-Boc-4-транс-гидрокси-L-пролина (1.58 г, 6.83 ммоля), гидрохлорида (1-амино-2-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (Пример 8, Стадия 1) (2.00 г, 7.52 ммоля) и HATU (3.89 г, 10.2 ммоля) в СН2 Cl2 (100 мл) прибавляют диизопропилэтиламин (4.41 г, 34.2 ммоля). Образовавшийся раствор оставляют нагреваться до комнатной температуры в течение 12 час. Прибавляют EtOAc (200 мл), промывают 5% Н3PO4 и рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают флеш - хроматографией (градиент, 2:1-1:1 гексан-ацетон), получают 1.25 г (41%) ожидаемого продукта.
1Н ЯМР (ДМСО-d6): δ 1.00-1.08 (м, 4Н), 1.34-1.40 (м, 1:2, 10Н), 1.62-1.70 (м, 1Н), 1.76-1.87 (м, 1Н), 2.02-2.21 (м, 2Н), 2.81-2.95 (м, 1Н), 3.20-3.45 (м, 2Н), 4.04-4.09 (м, 1Н), 4.26 (уш, 1Н), 5.08-5.12 (м, 1Н), 5.26 (д, J=17.1 Гц, 1Н), 5.59-5.69 (м, 1Н), 8.59, 8.87 (ротамеры,1:2,1Н), 10.48-11.15 (ротамеры, 2:1, 1Н);
LC-MS (время удерживания: 1.25 мин, метод В), MS m/z 444 (M++H).
Стадия 3:
Этот продукт получают по методике, описанной в Примере 1, Стадия 8, но используют продукт из Примера 8, Стадия 2.
LC-MS (время удерживания: 1.02 мин, метод В), MS m/z 344 (М++Н).
Стадия 4:
К ледяной смеси N-BOC-4-транс-гидрокси-L-пролина (1.58 г, 6.83 ммоля), гидрохлорида(1-амино-2-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (Пример 8, Стадия 3) (2.00 г, 7.52 ммоля) и HATU (3.89 г, 10.2 ммоля) в CH2Cl2 (100 мл) прибавляют диизопропилэтиламин (4.41 г, 34.2 ммоля). Образовавшийся раствор оставляют нагреваться до комнатной температуры в течение 12 час. Прибавляют EtOAc (200 мл), промывают 5% Н3PO4 и рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают флеш-хроматографией (градиент, 2:1-1:1 гексан-ацетон), получают 1.25 г (41%) ожидаемого продукта.
1Н ЯМР (CD3OD): δ 0.99-1.07 (м, 11Н), 1.35-1.44 (м, 13Н), 1.75-1.87 (м, 1Н), 2.09-2.22 (м, 2Н), 2.88-2.94 (м, 1Н), 3.74-3.82 (м, 2Н), 4.28-4.30 (м, 1Н), 4.33-4.38 (м, 1Н), 4.48 (уш, 1Н), 5.11-5.13 (м, 1Н), 5.30 (д, 1=15.0 Гц, 1Н), 5.70-5.78 (м, 1Н), 6.51-6.61 (м,1Н);
LC-MS (время удерживания: 1.26 мин, метод В), MS m/z 557 (M++H).
Схема 3
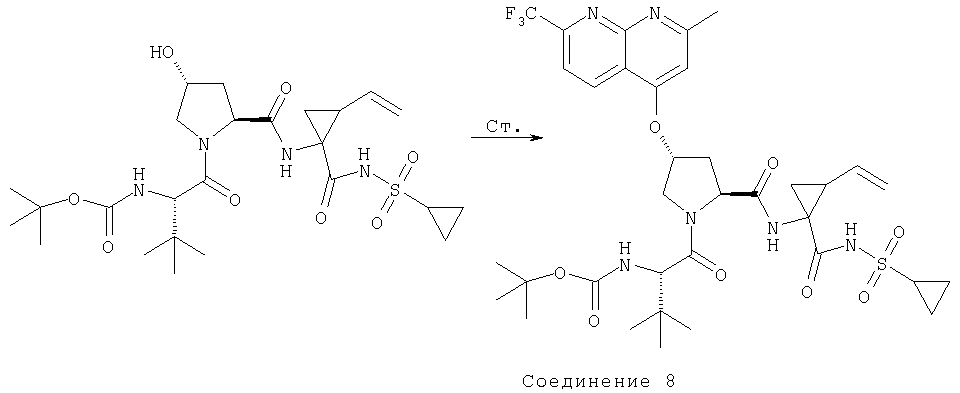
Стадия 5:
К раствору продукта из Примера 8, Стадия 4 (56 мг, 0.1 ммоля) в ДМСО (2 мл) прибавляют трет-бутоксид калия (49 мг, 0.44 ммолл). Образующийся раствор перемешивают при комнатной температуре в течение 1 час и прибавляют 4-хлор-7-метил-2-трифторметил-[1,8]нафтиридин (Р.Ferrarini et al, J Heterocyclic Chem, 1983, p1053) (30 мг, 0.12 ммоля). Полученный раствор перемешивают 12 час. Прибавляют воду со льдом, подкисляют 1М HCl до рН 4, экстрагируют EtOAc (20 мл, Х2). Органические вытяжки промывают рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают преп. ВЭЖХ, получают 16 мг (21%) Соединения 8 в виде твердого вещества розового цвета.
1Н ЯМР (CD3OD): δ 0.92-0.99 (м, 11Н), 1.01-1.04 (м, 11Н), 1.22-1.45 (м, 2Н), 1.76-1.85 (м, 1Н), 2.18-2.40 (м, 2Н), 2.76 (с, 3H), 2.86-2.97 (м, 1Н), 4.00-4.11 (м, 2Н), 4.48-4.58 (м, 2Н), 5.09-5.12 (м, 1Н), 5.28-5.31 (м, 1Н), 5.59 (уш, 1Н), 5.69-5.78 (м, 1Н), 6.39-6.48 (м, 1Н), 7.58-7.64 (м, 2Н), 8.08 (с, 1Н), 8.64-8.68 (м, 1Н), 8.85-8.91 (м, 1Н);
LC-MS (время удерживания: 1.89 мин, метод В), MS m/z 767 (M++H).
Пример 9: Получение Соединения 9.

Соединение 9 получают по методике, описанной в Примере 8, Стадия 5, но используют 7-хлор-5-этил-3-метилизоксазоло[4,5-b]пиридин (R.Nesi et al, Synth Comm. 1992, 22(16), 2349).
1Н ЯМР (CD3OD): δ 1.01-1.09 (м, 11Н), 1.21-1.25 (м, 11Н), 1.36 (т, J=7.8 Гц, 3H), 1.38-1.47 (м,.2Н), 1.80-1.90 (м,1Н), 2.20-2.31 (м,2Н), 2.59 (с, 3H), 2.90-3.00 (м, 3H), 4.01-4.18 (м, 2Н), 4.41-4.51 (м, 2Н), 5.11-5.15 (м, 1Н), 5.27-5.32 (м, 1Н), 5.58 (уш, 1Н), 5.70-5.80 (м, 1Н), 7.11 (с, 1Н), 7.72, 7.98 (1:1, 1Н), 9.00, 9.22 (1:1, 1Н);
LC-MS (время удерживания: 1.75 мин, метод В), MS m/z 717 (M++H).
Пример 10: Получение Соединения 10.

Соединение 10 получают по методике, описанной в Примере 8, Стадия 5, но используют 7-хлор-5-фенил-3-метилизоксазоло[4,5-b]пиридин (Пример 1, Стадия 2).
1Н ЯМР (CD3OD): δ 1.00-1.09 (м, 12Н), 1.16-1.25 (м, 10Н), 1.44-1.48 (м, 1Н), 1.79-1.89 (м, 1Н), 2.20-2.40 (м, 2Н), 2.64-2.66 (м, 4Н), 2.89-2.98 (м, 1Н), 4.08-4.20 (м, 2Н), 4.44-4.55 (м, 2Н), 5.11-5.16 (м, 1Н), 5.27-5.31 (м, 1Н), 5.72-5.74 (м, 2Н), 7.20-7.35 (м, 1Н), 7.46-7.51 (м, 2Н), 7.55-7.68 (м, 1Н), 8.05-8.06 (м, 2Н);
LC-MS (время удерживания: 1.97 мин, метод В), MS m/z 765 (М+ +Н).
Пример 11: Получение Соединения 11.

Схема 1

Стадия 1:
К раствору 3-метоксикоричной кислоты (11.04 г, 62 ммоля) и триэтиламина (12.52 г, 124 ммоля) в ацетоне (80 мл) при 0°С по каплям прибавляют этилхлорформиат (около 1.5 эквивалентов). Перемешивают при этой температуре 1 час, по каплям прибавляют водный раствор NaN3 (6.40 г, 100 ммоля в 35 мл Н2О) и реакционную смесь перемешивают 16 час при комнатной температуре. К смеси прибавляют воду (100 мл) и летучие отгоняют в вакууме. Полученную суспензию экстрагируют толуолом (3×50 мл) и полученные органические вытяжки сушат MgSO4. Этот высушенный раствор по каплям прибавляют к нагретому до 190°С раствору дифенилметана (50 мл) и трибутиламина (30 мл). При этом добавлении толуол отгоняется. По окончании прибавления температуру реакции поднимают до 210°С и выдерживают при ней в течение 2 час. По охлаждении выпавший продукт отфильтровывают, промывают гексаном (2×50 мл ) и сушат, получают ожидаемый продукт в виде твердого вещества белого цвета (5.53 г, 51%) (Nicolas Briet et al., Tetrahedron, 2002, 5761-5766).
LC-MS (время удерживания: 0.82 мин, метод В), MS m/z 176 (M++H).
Стадия 2:
6-Метокси-2Н-изохинолин-1-он ( 5.0 г, 28.4 ммоля) в POCl3 (10 мл) нагревают при слабом кипении в течение 3 час, упаривают в вакууме (Nicolas Briet et al., Tetrahedron, 2002, 5761-5766). Остаток выливают в лед с водой (20 мл) и нейтрализуют до рН 10, добавлял 10М раствор NaOH. Экстрагируют CHCl3. Органические вытяжки промывают рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают флеш-хроматографией (1:1 гексан - EtOAc), получают 4.41 г (80%) ожидаемого продукта в виде твердого вещества белого цвета.
1Н ЯМР (CD3OD): δ 3.98 (с, 3H), 7.34-7.38 (м, 2 Н), 7.69 (д, J=5.5 Гц, 1Н), 8.10 (д, J=6.0 Гц, 1Н), 8.23 (д, J=9.5 Гц, 1Н);
LC-MS (время удерживания: 1.42 мин, метод В), MS m/z 194 (M++H).
Схема 2
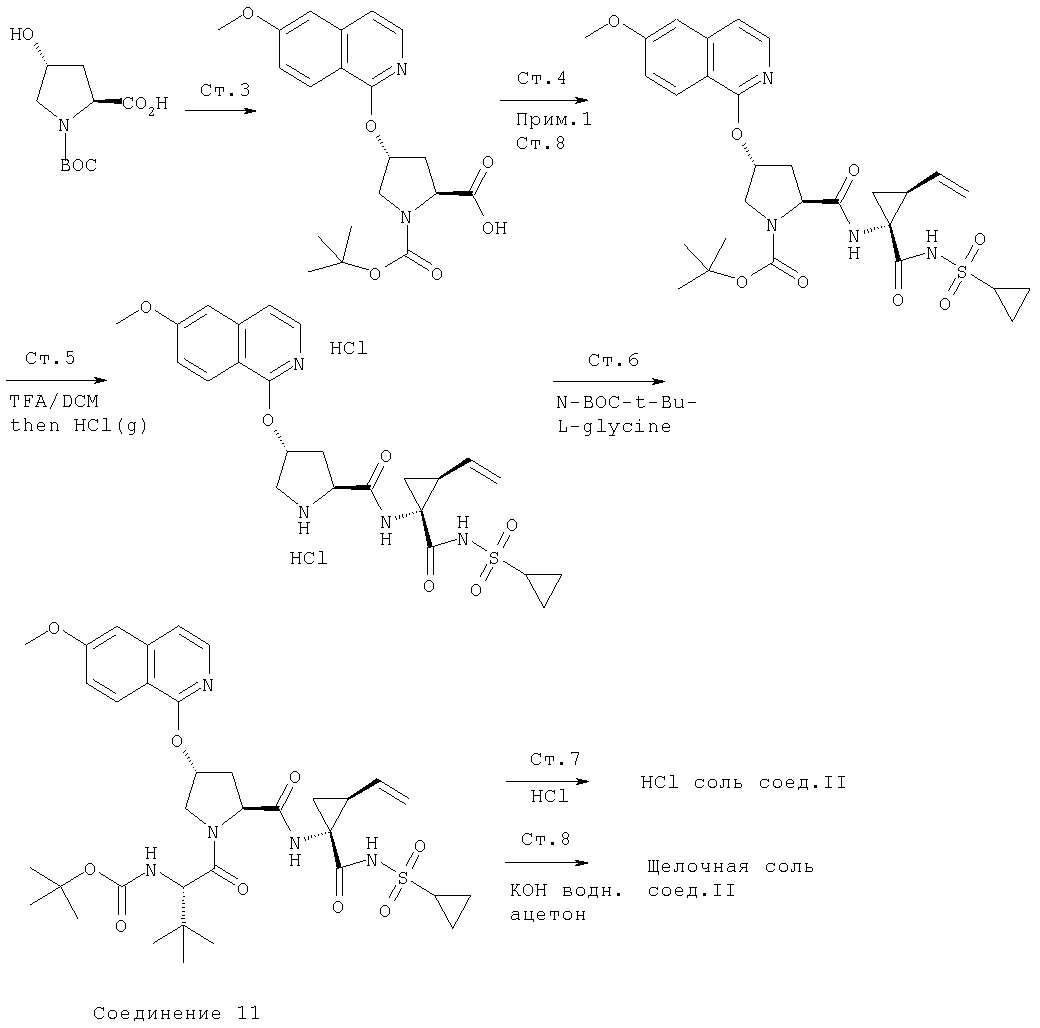
Стадия 3
К раствору N-ВОС-3-(R)-гидрокси-L-пролина (892 мг, 3.89 ммоля) в ДМСО (40 мл) при комнатной температуре сразу прибавляют все количество трет-бутоксида калия (1.34 г, 12.0 ммоля). Образующуюся суспензию перемешивают при этой температуре в течение 30 мин и охлаждают до 10°С. Прибавляют 1-хлор-6-метоксиизохинолин (Пример 11, Стадия 2) (785 мг, 4.05 ммоля) в виде твердого вещества одной порцией, и полученную смесь перемешивают при комнатной температуре в течение 12 час. Прибавляют ледяную 5% лимонную кислоту (водн.), экстрагируют EtOAc (100 мл). Водный слой снова экстрагируют EtOAc. Объединенные органические вытяжки промывают 5% лимонной кислотой (водн.) и рассолом, последовательно, сушат MgSO4, фильтруют. Фильтрат упаривают в вакууме досуха, получают 1.49 г (99%) нужного продукта в виде не чисто белой пены. Это вещество используют на следующей стадии без дополнительной очистки.
1Н ЯМР (CD3OD): δ 1.42, 1.44 (ротамеры, 9Н), 2.38-2.43 (м, 1Н), 2.66-2.72 (м, 1Н), 3.80-3.87 (м, 2Н), 3.92 (с, 3H), 4.44-4.52 (м, 1Н), 5.73 (уш, 1Н), 7.16-7.18 (м, 2Н), 7.24-7.25 (м, 1Н), 7.87-7.88 (м, 1Н), 8.07 (д, J=8.5 Гц, 1Н);
LC-MS (время удерживания: 1.62 мин, метод В), MS m/z 389 (M++H).
Стадия 4:
К смеси продукта из Примера 11, Стадия 3 (1.49 г, 3.84 ммоля), HATU (2.19 г, 5.76 ммоля) и соли (1-(R)-амино-2-(S)-винилциклопропанкарбонил)-амид HCl (Пример 1, Стадия 8) (1.12 г, 4.22 ммоля) в СН2Cl2 (50 мл) при 0°С прибавляют DIPEA (1.29 г, 11.5 ммоля) при 0° С. После перемешивания при комнатной температуре в течение 12 час к образовавшемуся раствору прибавляют СН2Cl2 (50 мл), промывают ледяной 5% лимонной кислотой (водн.). Органический слой промывают 5% лимонной кислотой (водн.) и рассолом, последовательно, сушат MgSO4 и фильтруют. Фильтрат упаривают в вакууме досуха. Остаток перекристаллизовывают из метанола, получают 1.60 г (70%) нужного продукта в виде твердого вещества белого цвета.
1Н ЯМР (CD3OD): δ 1.05-1.08 (м, 2Н), 1.16-1.20 (м, 1Н), 1.24-1.27 (м, 1Н), 1.42-1.45 (м, 10Н), 1.88 (дд, J=8.09, 5.34 Гц, 1Н), 2.24-2.30 (м, 2Н), 2.53-2.57 (м, 1Н), 2.94-2.98 (м, 1Н), 3.80 (д, J=12.5 Гц, 1Н), 3.86-3.89 (м, 1Н), 3.93 (с, 3H), 4,40-4.42 (м, 1Н), 5.13 (д, J=10.5 Гц, 1Н), 5.32 (д, J=18.0 Гц, 1Н), 5.72-5.81 (м, 2Н), 7.17-7.20 (м, 2Н), 7.26 (д, J=6.0 Гц, 1Н), 7.88 (д, J=6.0 Гц, 1Н), 8.07 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.74 мин, метод В), MS m/z 601 (М++Н).
Стадия 5:
К охлажденному льдом ("ледяному") раствору продукта из Примера 11, Стадия 4 (1.50 г, 2.50 ммоля) в СН2 Cl2 (10 мл) прибавляют ТФК (10 мл). Образующийся раствор оставляют на 2 час нагреваться до комнатной температуры. Растворитель отгоняют в вакууме. Остаток растирают с 1М HCl в эфире. Отфильтровывают, промывают эфиром, получают 1.43 г (99.8%) нужного продукта в виде гигроскопического твердого вещества белого цвета.
1Н ЯМР (CD3OD): δ 1.03-1.208 (м, 4Н), 1.26-1.31 (м, 1Н), 1.37-1.40 (м, 1Н). 1.95-1.97 (м, 1Н), 2.32-2.37 (м, 1Н), 2,42-2.48 (м, 1Н), 2.95-2.99 (м, 1Н), 3.88 (д, J=12.5 Гц, 2Н), 3.98 (с, 3H), 4.40-4.42 (м, 1Н), 5.16 (д, J=10.5 Гц, 1Н), 5.33 (д, J=18.0 Гц, 1Н), 5.62-5.69 (м, 1Н), 5.97 (уш, 1Н), 7.30-7.34 (м, 2Н), 7.47 (д, J=6.0 Гц, 1Н ), 7.90 (д, J=6.5 Гц, 1Н), 8.34 (д, J=9.0 Гц, 1Н), 9.14 (уш, 1Н);
LC-MS (время удерживания: 1.12 мин, метод В), MS m/z 501 (М++Н).
Стадия 6:
К смеси продукта из Примера 11, Стадия 5 (1,49 г, 3.84 ммоля), HATU (2.19 г, 5.76 ммоля) и N-ВОС-трет-бутил-1-глицина (1.12 г, 4.22 ммоля) в CH2Cl2 (50 мл) при 0°С прибавляют DIPEA (1.29 г, 11.5 ммоля). Перемешивают при комнатной температуре в течение 12 час, к полученному раствору прибавляют СН2Cl2 (50 мл), промывают охлажденной льдом 5% лимонной кислотой (водн). Органический слой промывают 5% лимонной кислотой (водн.) и рассолом, последовательно, сушат MgSO4 и фильтруют. Фильтрат упаривают в вакууме досуха. Остаток очищают преп. ВЭЖХ (40%В-100%В, 15 мин время градиента), получают 1.60 г (70%) Соединения 11 в виде твердого вещества белого цвета.
1Н ЯМР (CD3OD): δ 1.00-1.08 (м, 12Н), 1.23-1.25 (м, 1Н), 1.27 (с, 9Н), 1.40-1.45 (м, 1Н), 1.85-1.88 (м, 1Н), 2.20-2.30 (м, 2Н), 2.55-2.61 (м, 1Н), 2.91-2.97 (м, 1Н), 3.92 (с, 3H), 4.02-4.06 (м, 1Н), 4.21-4.24 (м, 1Н), 4.40-4.42 (м, 1Н), 4.49-4.51 (м, 1Н) 5.12 (д, J=10.5 Гц, 1Н), 5.28 (д, J=18.0 Гц, 1Н), 5.69-5.74 (м, 1Н), 5.81 (уш, 1Н), 6.60 (д, J=10.0 Гц, 1Н), 7.08-7.10 (м, 1Н), 7.18 (с. 1Н), 7.25 (д, J=6.0 Гц, 1Н), 7.88 (д, J=6.0 Гц, 1Н), 8.09 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.75 мин, метод В), MS m/z 714 (М++Н).
Анализ. Вычислено для C35H47N5O9S•0.5 Н2O: С, 58.16; Н, 6.69; N, 9.69. Найдено: С, 58.01; Н, 6.46; N, 9.55.
Стадия 7
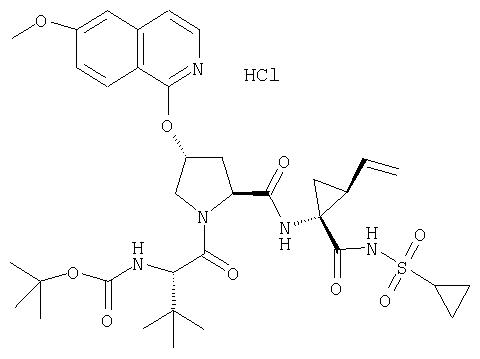
К раствору Соединения 11 (71 мг, 0.1 ммоля) в СН2Cl2 (5 мл ) при -78°С прибавляют 1М HCl в эфире (0.2 мл, 0.2 ммоля). Перемешивают при этой температуре 10 мин, летучие отгоняют в вакууме без нагревания. Остаток растирают с эфиром, фильтруют, промывают эфиром и сушат, получают 61 мг (85%) нужной HCl соли Соединения 11 в виде очень мелкодисперсного твердого вещества.
1Н ЯМР (CD3OD): δ 1.00-1.08 (м, 12Н), 1.19 (с, 9Н), 1.23-1.25 (м, 1 H), 1.40-1.45 (м, 1Н), 1.85-1.91 (м, 1Н), 2.20-2.26 (м, 1Н), 2.31-2.42 (м, 1Н), 2.65-2.78 (м, 1Н), 2.92-2.97 (м, 1Н), 4.00 (с, 3H), 4.10-4.16 (м, 2Н), 4.51-4.64 (м, 2Н), 5.13 (д, J=10.5 Гц, 1Н), 5.30 (д, J=18 Гц, 1Н), 5.69-5.79 (м, 1Н), 5.84 (уш, 1Н), 7.28 (д, J=9.3 Гц, 1Н), 7.40 (с, 1Н), 7.55 (д, J=6.3 Гц, 1Н), 7.89-7.92 (м, 1Н), 8.29 (д, J=9.0 Гц, 1Н), 9.21 (уш. 1Н);
LC-MS (время удерживания: 1.75 мин, метод В), MS m/z 714 (M++H).
Анализ. Вычислено для C35H47N5O9S•1.0 HCl: С, 56.02; H, 6.44; N,9.33; Cl, 4.72; S, 4.27. Найдено: С, 55.80; H, 6.42; N, 9.15; Cl, 4.56; S, 4.09.
Стадия 8:
Двугорлую колбу на 25 мл снабжают магнитной мешалкой, мембраной для ввода реагентов и адаптером для ввода N2. Взвешивают Соединение 11 (99.7 мг, 0.140 ммоля) и помещают в реакционную колбу. Реакционную колбу продувают и оставляют в атмосфере N2. В колбу прибавляют 850 ul ацетона, получают прозрачный раствор. К этому раствору при комнатной температуре прибавляют 780 ul 0.179 М раствора КОН (водн.), полученного растворением твердого КОН (502.8 мг, 8.97 ммоля) в 50 мл Н2O. При добавлении КОН раствор слегка нагревается, но остается прозрачным. Прозрачный раствор перемешивают при rt в течение 2 час. Продукт кристаллизуется из раствора, его отфильтровывают. Осадок на фильтре промывают холодным ацетоном, получают 42 мг (40% выход) нужного продукта в виде мелких белых игл:
1Н ЯМР (500 МГц, ДМСО-d6) δ 0.52-0.65 (м, 2Н), 0.66-0.82 (м, 2Н), 0.96 (с, 9Н), 1.21-1.24 (м, 10Н), 1.44-1.63 (м, 1H), 1.72-1.92 (м, 1H), 2.30-2.42 (м, 1H), 2.46 (д, J=7.93 Гц, 1H), 2.60-2.85 (м, 1H), 3.89 (с, 3Н), 3.93-4.05 (м, 1H), 4.07-4.24 (м, 1H), 4.41 (т, J=8.39 Гц, 1H), 4.79-4.95 (м, 1H), 4.97-5.17 (м, 1H), 5.71 (6, 1H), 5.80-6.10 (м, 1Н), 6.64 (д, J=8.54 Гц, 1H), 7.10 (д, J=8.85 Гц, 1H), 7.24-7.37 (м, 2Н), 7.90-7.96 (м, 1Н), 7.99-8.04 (б, 1H), 8.06 (д, J=9.15 Гц, 1Н).
Элементный анализ для С35Н46KN5O9S•Н2O; выч. С, 54.60; Н, 6.28; К, 5.08; N, 9.10. Найдено С, 54.88; Н, 6.23; К, 5.05; N, 9.01; MS m/e 714 (МН+).
Пример 12: Получение Соединения 12.

Соединение 12 получают по методике, описанной в Примере 11, Стадия 6, но используют N-ВОС-L-валин.
1Н ЯМР (CD3OD): δ 0.94-0.98 (м, 6Н), 1.07-1.09 (м, 3Н), 1.21-1.25 (м, 10Н), 1.40-1.43 (м, 1H), 1.88-1.89 (м, 1H), 2.05-2.09 (м, 1H), 2.22-2.35 (м, 2Н), 2.57-2.61 (м, 1H), 2.94-2.97 (м, 1Н), 3.92 (с, 3Н), 4.03-4.06 (м, 2Н), 4.47-4.55 (м, 2Н), 5.12 (д, J=10.5 Гц, 1H), 5.32 (д, J=18.1 Гц, 1H), 5.74-5.81 (м, 1Н), 5.86 (уш, 1Н), 7.10 (д, J=9.0 Гц, 1H), 7.18 (с, 1H), 7.25 (д, J=6.0 Гц, 1H), 7.88 (д, J=6.0 Гц. 1Н), 8.10 (д, J=9.0 Гц, 1H);
LC-MS (время удерживания: 1.71 мин, метод В), MS m/z 700 (М++Н).
Пример 13: Получение Соединения 13.

Соединение 13 получают по методике, описанной в Примере 11, Стадия 6, но используют N-ВОС-L-аллоизолейцин.
1 Н ЯМР (CD3OD): δ 0.89-0.96 (м, 6Н), 1.07-1.18 (м, 5Н), 1.28 (с, 9Н), 1.42-1.45 (м, 1Н), 1.50-1.54 (м, 1Н), 1.87-1.89 (м, 2Н), 2.23-2.34 (м, 2Н), 2.57-2.61 (м, 1Н), 2.92-2.95 (м, 1Н), 3.92 (с, 3H), 4.05-4.07 (м, 1Н), 4.22-4.24 (м, 1Н), 4.37-4.40 (м, 1Н), 4.54-4.56 (м, 1Н), 5.13 (д, J=10.5 Гц, 1Н), 5.32 (д, J=18.0 Гц, 1Н), 5.75-5.82 (м, 1Н), 5.86 (уш, 1Н), 7.12 (д, J=9.0 Гц, 1Н), 7.19 (с, 1Н), 7.24 (д, J=6.0 Гц, 1Н), 7.88 (д, J=6.0 Гц,1Н), 8.10 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.77 мин, метод В), MS m/z 714 (M++H).
Пример 14: Получение Соединения 14.
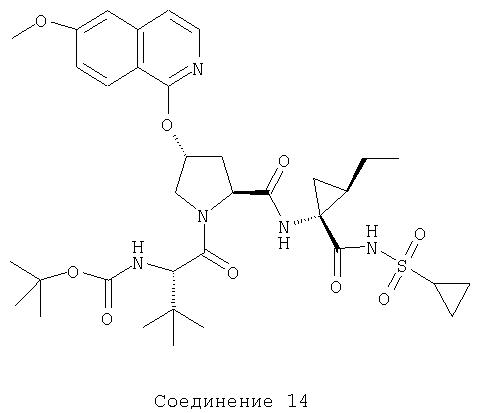
Схема 1
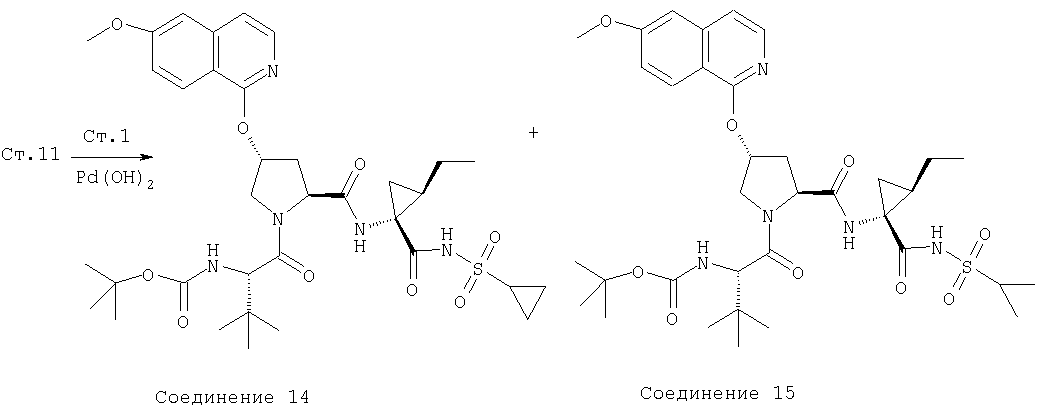
Смесь Соединения 11 (150 мг, 0.21 ммоля) и катализатора Перлманна (Pd(OH)2, 15 мг) в EtOAc (10 мл) на 20 мин помещают в шейкер Парра при давлении Н2 10 psi (68.95 кПа). Фильтруют через целит. Фильтрат упаривают в вакууме. Остаток очищают преп. ВЭЖХ, получают 67 мг (45%) Соединения 14 в виде твердого вещества белого цвета.
1Н ЯМР (CD3OD): δ 0.96-0.99 (м, 4Н), 1.04 (с, 9Н), 1.07-1.09 (м, 2Н), 1.21-1.24 (м,2Н), 1.27 (с, 9Н), 1.51-1.65 (м, 4Н), 2.25-2.27 (м, 1Н), 2.55-2.61 (м, 1Н), 2.94-2.98 (м, 1Н), 3.92 (с, 3H), 4.02-4.06 (м, 1Н), 4.21-4.24 (м, 1Н), 4.40-4.42 (м, 1Н), 4.49-4.51 (м, 1Н), 5.81 (уш, 1Н), 6.59 (д, J=10.0 Гц, 1Н), 7.08-7.10 (м, 1Н), 7.18 (д, J=1.5 Гц, 1Н), 7.24 (д, J=6.0 Гц, 1Н), 7.88 (д, J=6.0 Гц, 1Н), 8.08 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.76 мин, метод В), MS m/z 716 (М++Н).
Пример 15: Получение Соединения 15.

Соединение 15 выделяют из той же самой реакционной смеси, что и Соединение 14 с немного более продолжительным временем удерживания в качестве побочного продукта с выходом 15%.
1Н ЯМР (CD3OD): δ 0.92-1.10 (м, 17Н), 1.26-1.36 (м, 13Н), 1.64-1,72 (м, 1Н), 1.90-1.96 (м, 1Н), 2.30-2.40 (м, 1Н), 2.63-2.67 (м, 1Н), 2.96-3.00 (m, 1Н), 3.92 (с, 3H), 4.03-4.07 (м, 1Н), 4.24 (уш, 1Н), 4,40-4, 42 (м, 1Н), 4.49-4.51 (м, 1Н) 5.83 (уш, 1Н), 7.08-7.11 (м, 1Н), 7.19 (д, J=2.0 Гц. 1Н), 7.25 (д, J=6.0 Гц, 1Н), 7.89 (д, J=6.0 Гц, 1Н), 8.10 (д, J=9.0 Гц, 1Н), 8.51 (уш, 1Н);
LC-MS (время удерживания: 1.83 мин, метод В), MS m/z 718 (M++H).
Пример 16: Получение Соединения 16.
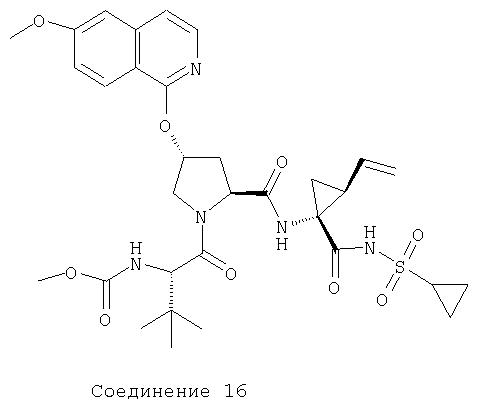
Схема 1

Стадия 1:
К раствору Соединения 11 (420 мг, 0.59 ммоля) в DCM (5 мл) при 0°С прибавляют ТФК (5 мл ). Перемешивают при этой температуре в течение 2 час, летучие удаляют в вакууме. Остаток растирают с 1М HCl в эфире (5 мл), фильтруют, промывают эфиром и сушат, получают 360 мг (89%) ожидаемой HCl соли в виде очень мелкодисперсного твердого вещества.
LC-MS (время удерживания: 1.28 мин, метод В), MS m/z 614 (М++Н).
Стадия 2:
К суспензии продукта из Примера 16, Стадия 1 (39 мг, 0.06 ммоля) и DIPEA (20 мг, 0.18 ммоля) в DCM (1 мл) при 0°С прибавляют метилхлорформиат (6.8 мг, 0.072 ммоля). Перемешивают при этой температуре 2 час, летучие отгоняют в вакууме. Остаток очищают препаративной ВЭЖХ, получают 21 мг (58%) Соединения 16 в виде белых кристаллов.
1Н ЯМР (CD3OD) δ 1.05-1.09 (м, 11Н), 1.22-1.25 (м, 2Н), 1.41-1.44 (м, 1Н), 1.86-1.89 (м, 1Н), 2.22-2.32 (м, 2Н), 2.59-2.63 (м, 1Н), 2.89-2.93 (м, 1Н), 3.48 (с, 3H), 3.92 (с, 3H), 4.06-4.10 (м, 1Н), 4.31-4.33 (м, 1Н), 4.38-4:40 (м, 1Н), 4.50-4.52 (1, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.30 (д, J=18.0 Гц, 1Н), 5.71-5.80 (м, 1Н), 5.88 (уш, 1Н), 6.95 (д, J=10.0 Гц, 1Н), 7.13-7.16 (м, 1Н), 7.19 (с, 1Н), 7.25 (д, J=6.0 Гц, 1Н), 7.88 (д, J=6.0 Гц,1Н), 8.09 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.54 мин, метод В), MS m/z 672 (М++Н).
Пример 17: Получение Соединения 17.
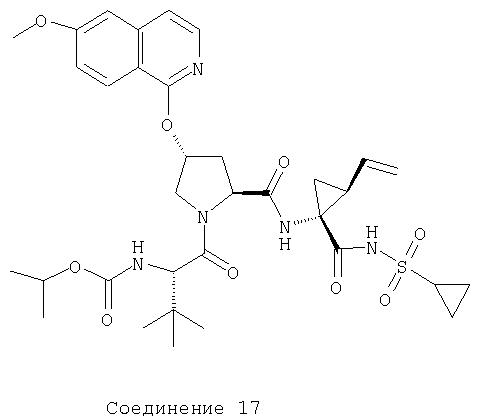
Соединение 17 получают по методике, описанной в Примере 16, Стадия 2, но используют изопропилхлорформиат.
1Н ЯМР (CD3OD) δ 1.00-1.09 (м, 15Н), 1.13-1.16 (м, 2Н), 1.24-1.26 (м, 2Н), 1.40-1.45 (м, 1Н), 1.86-1.89 (м, 1Н), 2.21-2.31 (м, 2Н), 2.55-2.61 (м, 1Н), 2.91-2.97 (м 1Н), 3 92 (с, 3H), 4.04-4.08 (м, 1Н), 4.30 (уш, 1Н), 4.40 (д, J=10Гц, 1Н), 4.49-4.54 (м, 2Н), 5.12 (д. J=10.5 Гц, 1Н), 5.29 (д, J=18.0 Гц, 1Н), 5.71-5.77 (м, 1Н), 5.84 (уш, 1Н), 6.80 (д, J=10.0 Гц, 1Н), 7.11 (д, J=9.0 Гц, 1Н), 7.19 (с, 1Н), 7.25 (д, J=6.0 Гц, 1Н), 7.88 (д, J=6.0 Гц. 1Н), 8.08 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.74 мин, метод В), MS m/z 700 (M++H).
Пример 18: Получение Соединения 18.

Соединение 18 получают по методике, описанной в Примере 16, Стадия 2, но применяют неопентилхлорформиат.
1Н ЯМР(CD3OD) δ 0.61 (уш, 1Н), 0.84 (с, 8Н), 1.05-1.09 (м, 11Н), 1.23-1.25 (м Н), 1.39-1.44 (м, 1H), 1.85-1.88 (м, 1Н), 2.20-2.30 (м, 2Н), 2.56-2.62 (м, 1Н), 2.91-2.97 (м, 1Н), 3.38 (д, J=9.0 Гц. 1Н), 3.55 (д, J=9.0 гц, 1Н), 3.92 (с, 3H), 4.02-4.06 (м, 1Н), 4.32 (д, J=9.5 Гц, 1Н), 4.41 (д, J=9.0 Гц, 1Н), 4.49-4.51 (м, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.28 (д, J=18.0 Гц, 1Н), 5.69-5.74 (м, 1Н), 5.81 (уш, 1Н), 6.90 (д, J=10.0 Гц, 1Н), 7.08-7.10 (м, 1Н), 7.19 (с, 1Н), 7.26 (д, J=6.0 Гц, 1Н), 7.88 (д, J=6.0 Гц, 1Н), 8.07 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.84 мин, метод В), MS m/z 728 (M++H).
Пример 19: Получение Соединения 19.
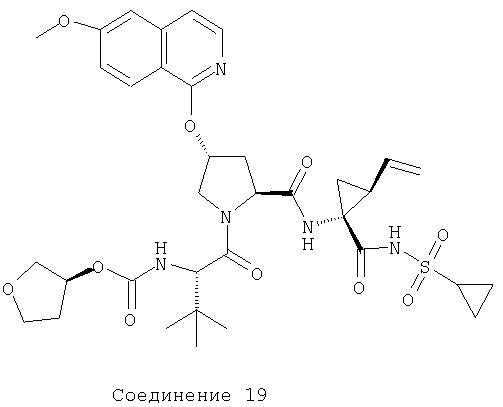
Соединение 19 получают по методике, описанной в Примере 16, Стадия 2, но используют (S)-3-фуранохлорформиат (J.Campbell, A.Good, Международная заявка WO 20020808).
1Н ЯМР (CD3OD) δ 1.03-1.08 (м, 11Н), 1.23-1.26 (м, 2Н), 1.38-1.46 (м, 1Н), 1.64-1.71 (м, 1Н), 1.85-1.90 (м, 2Н), 2.20-2.30 (м, 2Н), 2.55-2.61 (м, 1Н), 2.91-2.97 (м, 1Н), 3.66-3.72 (м, 4Н), 3.93 (с, 3H), 4.05-4.09 (м, 1Н), 4.27-4.29 (м, 1Н), 4.40-4.42 (м,1Н), 4.55-4.59 (м, 1Н), 4.75-4.77 (м, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.28 (д, J=18 Гц, 1Н), 5.73-5.80 (м, 1Н), 5.85 (уш, 1Н), 7.06 (д, J=10.0 Гц, 1Н), 7.13 (д, J=9.0 Гц, 1Н), 7.20 (с, 1Н), 7.25 (д, J=6.0 Гц, 1Н), 7.89 (д, J=6.0 Гц, 1Н), 8.07 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.52 мин, метод В), MS m/z 728 (M++Н).
Пример 20: Получение Соединения 20.
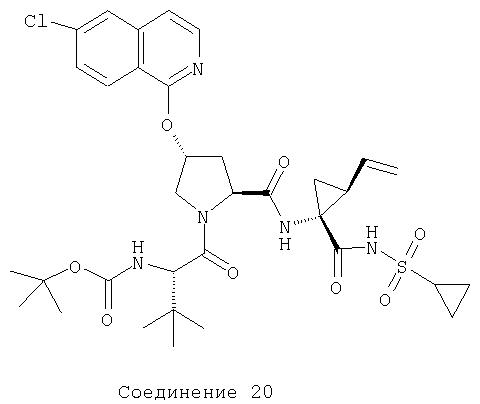
Стадия 1:

Этот продукт получают по методике, описанной в Примере 11, Стадия 2, но применяют 6-хлор-2Н-изохинолин-1-он (Nicolas Briet et al, Tetrahedron, 2002, 5761-5766).
LC-MS (время удерживания: 1.07 мин, метод В), MS m/z 180 (М++Н).
Стадия 2:

Этот продукт получают по методике, описанной в Примере 1, Стадия 5, но используют продукт из Примера 20, Стадия 1.
1Н ЯМР (CD3OD) δ 1.04 (с, 9Н), 1.20 (с, 9Н), 2.36-2.41 (м, 1Н), 2.74-2.78 (м, 1Н), 4.01-4.04 (м, 1Н), 4.19-4.21 (м, 1Н), 4.47-4.49 (м, 1Н), 4.67-4.70 (м, 1Н), 5.84 (уш, 1Н), 7.28 (д, J=6.0 Гц, 1Н), 7.47 (д, J=6.0 Гц, 1Н ), 7.84 (с, 1Н), 8.00 (д, J=6.0 Гц, 1Н), 8.20 (д. J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.88 мин, метод В), MS m/z 506 (М+ +Н).
Стадия 3:
Соединение 20 получают по методике, описанной в Примере 1, Стадия 9, но с использованием продукта из Примера 20, Стадия 2.
1Н ЯМР (CD3OD) δ 0.99-1.11 (м, 12Н), 1.20-1.26 (м, 10Н), 1.43-1.46 (м, 1Н), 1.87-1.90 (м, 1Н), 2.22-2.31 (м, 2Н), 2.60-2.64 (м, 1Н), 2.92-2.97 (м, 1Н), 4.06-4.08 (м, 1Н), 4.21-4.23 (м, 1Н), 4.45-4.47 (м, 1Н), 4.53-4.56 (м, 1Н), 5.13 (д, J=10.5 Гц, 1Н), 5.29 (д, J=18.0 Гц, 1Н), 5.72-5.80 (м, 1Н), 5.88 (уш, 1Н), 6.58 (д, J=10.0 Гц, 1Н), 7.29 (д, J=6.0 Гц, 1Н), 7.47 (д, J=9.0 Гц, 1Н), 7.86 (с, 1Н), 8.01 (д, J=6.0 Гц, 1Н), 8.18 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.94 мин, метод В), MS m/z 718 (М++Н).
Пример 21: Получение Соединения 21.

Схема 1

Стадия 1:
К смеси продукта из Примера 1, Стадия 4 (3.00 г, 8.72 ммоля), HATU (4.97 г, 13.1 ммоля) и продукта из Примера 1, Стадия 8 (2.55 г, 9.59 ммоля), в СН2Cl2 (100 мл) при 0°С прибавляют DIPEA (3.02 г, 27.0 ммоля). Перемешивают при комнатной температуре в течение 12 час, к полученному раствору прибавляют CH2Cl2 (100 мл), промывают ледяной 5% лимонной кислотой (водн.). Органический слой промывают 5% лимонной кислотой (водн.) и рассолом, последовательно, сушат MgSO4 и фильтруют. Фильтрат упаривают в вакууме досуха. Остаток очищают флеш-хроматографией на колонке (элюент 1:1 гексан: ацетон), получают 3.64 г (75%) ожидаемого продукта в виде пены.
LC-MS (время удерживания: 1.41 мин, метод В), MS m/z 557 (М++Н).
Схема 2
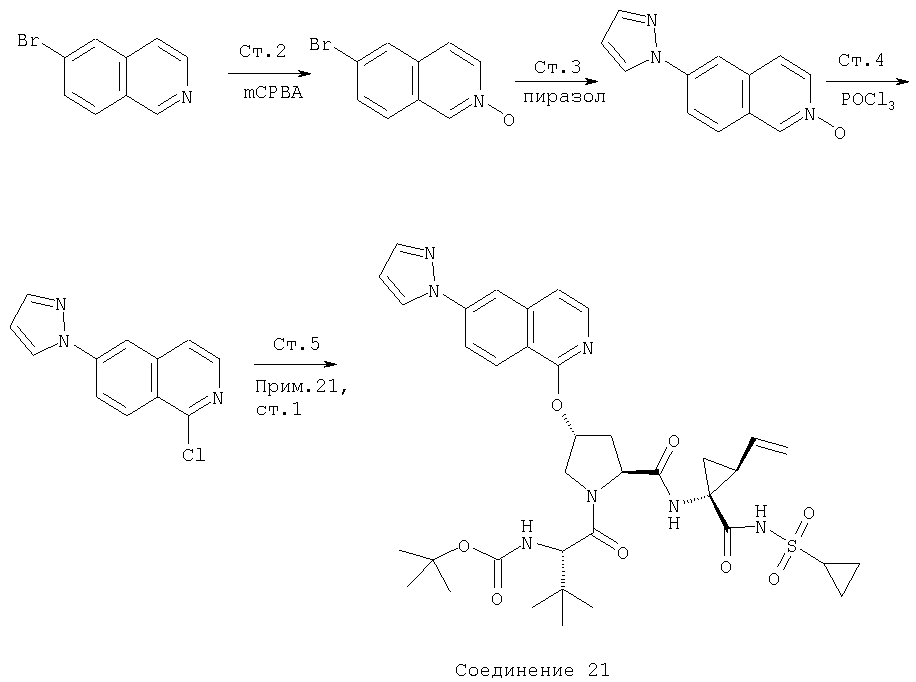
Стадия 2:
К охлажденному льдом раствору 6-бромизохинолина (4.16 г, 20 ммолей) в СН2Cl2 (100 мл) одной порцией прибавляют сухой мСРВА (9.38 г, 77% чистота, 42 ммоля). Перемешивают при комнатной температуре в течение 12 час, прибавляют СН2Cl2 (100 мл) и промывают 1М NaOH (100 мл, Х2) и рассолом. Органический слой сушат MgSO4, фильтруют, упаривают досуха, получают 3.83 г (86%) нужного продукта в виде твердого вещества белого цвета.
LC-MS (время удерживания: 0.77 мин, метод В), MS m/z 224, 226 (М++Н).
Стадия 3:
Смесь 6-бромизохинолин 2-оксида (88 мг, 0.2 ммоля), пиразола (68 мг, 1.0 ммоль), CuBr (57 мг, 0.4 ммоля) и карбоната цезия (130 мг, 0.4 ммоля) в ДМФА (2 мл) нагревают при 140°С в течение 4 час в запаянной ампуле. После фильтрования фильтрат очищают преп. ВЭЖХ, получают 41 мг (98%) ожидаемого вещества в виде твердого вещества белого цвета.
1Н ЯМР (CDCl3) δ 6.58-6.59 (м, 1Н), 7.82 (д, J=1.0 Гц, 1Н), 7.89 (д, J=7.0 Гц, 1Н), 8.02 (д, J=9.0 Гц, 1Н), 8.11 (д, J=2.5 Гц, 1Н), 8.18-8.22 (м, 2Н), 8.29 (д, J=7.0 Гц, 1Н), 9.07 (уш, 1Н);
LC-MS (время удерживания: 0.77 мин, метод В), MS m/z 212 (М++Н).
Стадия 4:
Этот продукт получают в виде не чисто белого твердого вещества по методике, описанной в Примере 11, Стадия 2, но при использовании 6-пиразолизохинолин 2-оксида.
1H ЯМР(CD3OD) δ 7.82-7.83 (м, 2Н), 8.23-8.32 (м, 4Н), 8.44-8.49 (м, 2Н);
LC-MS (время удерживания: 1.35 мин, метод В), MS m/z 230 (M++H).
Стадия 5:
К раствору продукта из Примера 21, Стадия 1 (45 мг, 0.08 ммоля) в ДМСО (2 мл) прибавляют трет-бутоксид калия (41 мг, 0.37 ммоля). Полученный раствор перемешивают при комнатной температуре в течение 30 мин и прибавляют 1-хлор-6-пиразол-1-илизохинолин (17 мг, 0.07 ммоля). Полученный раствор перемешивают 12 час. Гасят водой со льдом, подкисляют 1М HCl до рН 4, экстрагируют EtOAc (20 мл, Х2). Органические вытяжки промывают рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают преп. ВЭЖХ, получают 10 мг (16%) Соединения 21 в виде твердого вещества розового цвета.
1Н ЯМР (CD3OD) δ 1.04-1.10 (м, 12Н), 1.23-1.27 (м, 10Н), 1.43-1.47 (м, 1Н), 1.87-1.91 (м, 1Н), 2.22-2.29 (м, 2Н), 2.61-2.68 (м, 1Н), 2.92-2.98 (м, 1Н), 4.07-4.11 (м, 1Н), 4.24 (уш, 1Н), 4.46-4.60 (м, 2Н), 5.13 (д, J=10.5 Гц, 1Н), 5.29 (д, J=18 Гц, 1Н), 5.70-5.83 (м, 1Н), 5.89 (уш, 1Н), 6.59-6.61 (м, 1Н), 7.40 (д, J=10.0 Гц, 1Н), 7.80 (д, J=2.5 Гц, 1Н), 8.01 (д, J=10.0 Гц, 2Н), 8.15 (с, 1Н), 8.31 (д, J=15.0 Гц, 1Н), 8.42 (д, J=4.5 Гц, 1Н);
LC-MS (время удерживания: 1.77 мин, метод В), MS m/z 750 (М++Н).
Пример 22: Получение Соединения 22.
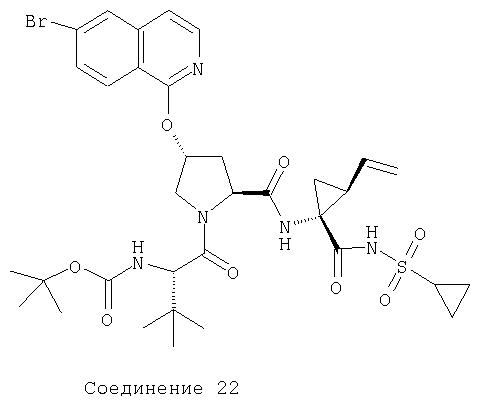
Стадия 1
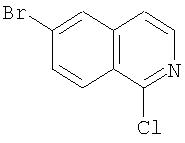
Этот продукт получают в виде не чисто белого твердого вещества по методике, описанной в Примере 11, Стадия 2, но с применением 6-бромизохинолин 2-оксида.
1Н ЯМР (CD3OD) δ 7.73 (д, J=5.5 Гц, 1H), 7.85-7.91 (м, 1Н), 8.22-8.31 (м, 3H);
LC-MS (время удерживания: 1.77 мин, метод В), MS m/z 750 (M++Н).
Стадия 2:
Соединение 22 получают в виде твердого вещества белого цвета по методике, описанной в Примере 21, Стадия 5, но с применением 1-хлор-6-бромизохинолина.
1Н ЯМР (CD3OD) δ 0.99-1.09 (м, 12Н), 1.22-1.27 (м, 10Н), 1.40-1.47 (м, 1Н), 1.86-1.91 (м, 1Н), 2.20-2.34 (м, 2Н), 2.57-2.66 (м, 1Н), 2.90-2.97 (м, 1Н), 4.05-4.09 (м, 1Н), 4.21 (уш, 1Н), 4.44-4.57 (м, 2Н), 5.13 (д, J=10.5 Гц, 1Н), 5.29 (д, J=18.0 Гц, 1Н), 5.70-5.82 (м, 1Н), 5.88 (уш, 1Н), 7.29 (д, J=9.5 Гц, 1Н), 7.60-7.63 (м, 1Н), 8.00-8.12(м, 3H);
LC-MS (время удерживания: 1.90 мин, метод В), MS m/z 762, 764 (М++Н).
Пример 23: Получение Соединения 23.
Стадия 1:
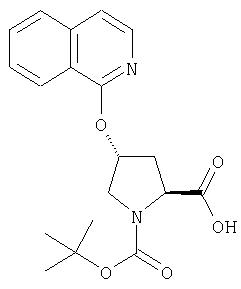
Этот продукт получают в виде белого твердого вещества по методике, описанной в Примере 11, Стадия 3, но с применением 1- хлоризохинолина.
1Н ЯМР (CD3OD) δ 1.42, 1.44 (ротамеры, 9Н), 2.39-2.44 (м, 1Н), 2.68-2.72 (м, 1Н), 3.80-3.87 (м, 2Н), 4.44-4.52 (м, 1Н), 5.78 (уш, 1Н), 7.32-7,33 (м, 1Н), 7.58 (т, J=7.8 Гц, 1Н), 7.71 (т, J=7.5 Гц, 1Н), 7.81 (д, J=8.0 Гц, 1Н), 7.95 (д, J=6.0 Гц, 1Н), 8.19 (д, J=8.0 Гц,1Н);
LC-MS (время удерживания: 1.61 мин, метод В), MS m/z 359 (M++H).
Стадия 2:

Этот продукт получают в виде не чисто белого твердого вещества по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 23, Стадия 1.
1Н ЯМР (ДМСО-d6) δ 1.00-1.09 (м, 4Н), 1.35-1.38 (м, 10Н), 1.69-1.84 (м, 1Н), 2.11-2.66 (м, 3H), 2.89-2.93 (м, 1Н), 3.62-3.89 (м, 2Н), 4.31 (т, J=8.1 Гц, 1Н), 5.12 (д, J=10.8 Гц, 1Н), 5.27 (д, J=16.8 Гц, 1Н), 5.58-5.70 (м, 1Н), 5.76 (уш, 1Н), 7.43 (4д, J=5.7 Гц, 1Н), 7.66 (т, J=7.4 Гц, 1Н), 7.79 (т, J=7.5 Гц, 1Н), 7.92 (д, J=8.1 Гц, 1Н), 8.02 (д, J=10.0 Гц, 1Н), 8.13 (д, J=8.1 Гц, 1Н), 9.02 (уш, 1Н);
LC-MS (время удерживания: 1.72 мин, метод В), MS m/z 571 (М+ +Н).
Стадия 3:

Этот продукт получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 23, Стадия 2.
LC-MS (время удерживания: 1.16 мин, метод В), MS m/z 471 (М++Н).
Стадия 4:
Соединение 23 получают в виде белого твердого вещества по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 23, Стадия 3.
1Н ЯМР (CD3OD) δ 1.00-1.09 (м, 12Н), 1.25-1.27 (м, 10Н), 1.42-1.46 (м, 1Н), 1.86-1.90 (м, 1Н), 2.22-2.34 (м, 2Н), 2.60-2.67 (м, 1Н), 2.92-2.99 (м, 1Н), 4.06-4.11 (м, 1Н), 4.26 (уш, 1Н), 4.45-4.57 (м, 2Н), 5.12 (д, J=10.2 Гц, 1Н), 5.27 (д, J=16.8 Гц, 1Н), 5.70-5.82 (м, 1Н), 5.88 (уш, 1Н), 7.32 (д, J=6.0 Гц, 1Н), 7.52 (т, J=7.4 Гц, 1Н), 7.70 (т, J=7,5 Гц, 1Н), 7.80 (д, J=8,1 Гц, 1Н), 7.97 (д, J=6 Гц, 1Н), 8.20 (д, J=8.4 Гц. 1Н), 9.18 (уш, 1Н);
LC-MS (время удерживания: 1.80 мин, метод В), MS m/z 684 (М++Н).
Пример 24: Получение Соединения 24.

Схема 1
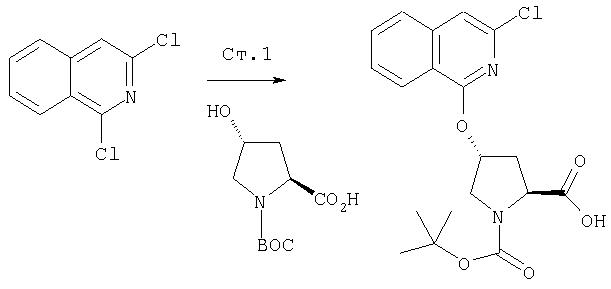
Стадия 1:
К раствору N-BOC-3-(R)-гидрокси-1-пролина (6.22 г, 26.9 ммоля) в ДМФА (250 мл) при 0°С прибавляют NaH (60%, 3.23 г, 80.8 ммоля) несколькими порциями. Полученную суспензию перемешивают при этой температуре в течение 30 мин. Одной порцией прибавляют сухой 1,3-дихлоризохинолин (5.33 г, 26.9 ммоля) и полученную смесь перемешивают при комнатной температуре в течение 12 час. Прибавляют ледяную 5% лимонную кислоту (водн), экстрагируют EtOAc (300 мл). Водную фазу снова экстрагируют EtAc. Объединенные органические вытяжки промывают 5% лимонной кислотой (водн.) и рассолом, последовательно, сушат MgSO4, фильтруют. Фильтрат упаривают в вакууме досуха, получают 10.53 г (99.8%) 1-трет-бутиловый эфир 4-(6-метоксиизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты в виде не чисто белой пены. Этот продукт используют на следующей стадии без дополнительной очистки.
1Н ЯМР (CD3OD) δ 1.43, 1.44 (ротамеры, 9Н), 2.39-2.44 (м, 1Н), 2.68-2.72 (м, 1Н), 3.80-3.90 (м, 2Н), 4.44-4.52 (м, 1Н), 5.77 (уш, 1Н), 7.39 (с, 1Н), 7.58 (т, J=7.3 Гц, 1Н), 7.71-7.78 (м, 2Н), 8.16 (д, J=7.5 Гц, 1Н);
LC-MS (время удерживания: 1.80 мин, метод В), MS m/z 392 (M++Н).
Стадия 2

Этот продукт получают по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 24, Стадия 1.
1Н ЯМР (CD3OD) δ 1.02-1.08 (м, 2Н), 1.18-1.26 (м, 2Н), 1.44-1.48 (м, 10Н), 1.84-1.91 (м, 1Н), 2.22-2.36 (м, 2Н), 2.57-2.60 (м, 1Н), 2.95-2.99 (м, 1Н), 3.81-3.93 (м, 2Н), 4.38-4.41 (м; 1Н), 5.13 (д, J=10.8 Гц, 1Н), 5.31 (д, J=16.8 Гц, 1Н), 5.75-5.82 (м, 2Н), 7.41 (с, 1Н), 7.59 (т, J=7.4 Гц, 1Н), 7.74-7.79 (м, 2Н), 8.16 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 1.82 мин, метод В), MS m/z 605 (М++Н).
Стадия 3:
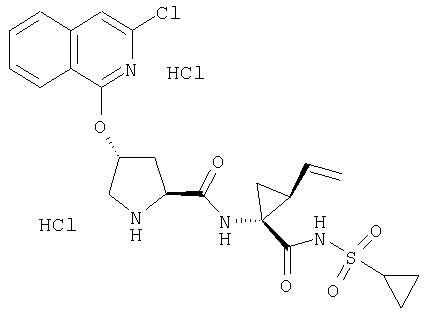
Этот продукт получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 24, Стадия 2.
LC-MS (время удерживания: 1.30 мин, метод В), MS m/z 505 (M++Н).
Стадия 4:
Этот продукт получают по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 24, Стадия 3.
1Н ЯМР (CD3OD) δ 0.99-1.09 (м, 12Н), 1.22-1.29 (м, 10Н), 1.42-1.46 (м, 1Н), 1.86-1.90 (м, 1Н), 2.21-2.34 (м, 2Н), 2.62-2.66 (м, 1Н), 2.92-2.99 (м, 1Н), 4.06-4.11 (м, 1Н), 4.26 (уш, 1Н), 4.46-4.56 (м, 2Н), 5.13 (д, J=10.5 Гц, 1Н), 5.29 (д, J=17.2 Гц, 1Н), 5.72-5.79 (м, 1Н), 5.89 (уш, 1Н), 7.40 (д, J=6.0 Гц, 1Н), 7.52 (т, J=7.4 Гц, 1Н), 7.72-7.76 (м, 2Н), 8.18 (д, J=8.5 Гц, 1Н);
LC-MS (время удерживания: 1.80 мин, метод В), MS m/z 684 (M++H).
Пример 25: Получение Соединения 25.
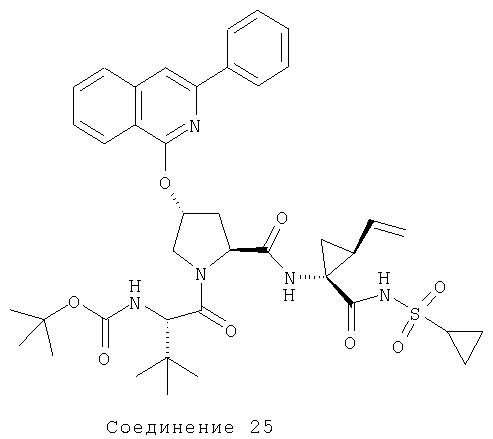
Схема 1

Стадия 1:
Смесь соединения из Примера 24, Стадия 1 (39 мг, 0.10 ммоля), фенилборной кислоты (14.6 мг, 0.12 ммоля), трет-бутоксида натрия (38 мг, 0.40 ммоля) и ((трет-Bu)2POH)2PdCl2 (POPd) (5 мг, 0.01 ммоля) в ТГФ (2 мл) кипятят 4 час. По охлаждении к полученной смеси добавляют 5% лимонную кислоту (водн.) и экстрагируют EtOAc (20 мл). Органические вытяжки промывают рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают преп. ВЭЖХ, получают 36 мг (83%) ожидаемого продукта в виде не чисто белой пены.
1Н ЯМР (CD3OD) δ 1.43, 1.45 (ротамеры, 9Н), 2.51-2.56 (м, 1Н), 2.74-2.82 (м, 1Н), 3.88-3.92 (м, 1Н), 3.98-4.01 (м, 1Н), 4.50-4.57 (м, 1Н), 5.95 (уш, 1Н), 7.36-7,39 (м, 1Н), 7.45-7.48 (м, 2Н), 7.55 (т, J=7.3 Гц, 1Н), 7.70 (т, J=7.5 Гц. 1Н), 7.84-7.89 (м, 2Н), 8.14-8.17 (м, 3Н), 9.05 (уш, 1Н);
LC-MS (время удерживания: 1.97 мин, метод В), MS m/z 435 (М++Н).
Стадия 2:
Этот продукт получают по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 25, Стадия 1.
1Н ЯМР (ДМСО-d6) δ 0.98-1.10 (м, 4Н), 1.38-1.41 (м, 10Н), 1.74-1.81 (м, 1Н), 2.18-2.34 (м, 2Н), 2.47-2.49 (м, 1Н), 2.95-2.99 (м, 1Н), 3.74-3.96 (м, 2Н), 4.34-4.37 (м, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.26 (д, J=17.8 Гц, 1Н), 5.75-5.82 (м, 1Н), 5.95 (уш, 1Н), 7.41-7.45 (м, 1Н), 7.51-7.54 (м, 2Н), 7.61-7.64 (м, 1Н), 7.78-7.82 (м, 1Н), 7.98 (д, J=9.0 Гц, 1Н), 8.06 (с, 1Н), 8.13-8.14 (м, 1Н), 8.18-8.20 (м, 2Н), 9.05 (уш, 1Н), 10.34 (уш, 1Н);
LC-MS (время удерживания: 1.99 мин, метод В), MS m/z 647 (M++H).
Стадия 3:
Этот продукт в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 25, Стадия 2.
LC-MS (время удерживания: 1.55 мин, метод В), MS m/z 547 (М++Н).
Стадия 4:
Соединение 25 в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 25, Стадия 3.
1Н ЯМР (CD3OD) δ 0.92-1.09 (м, 12Н), 1.26-1.30 (м, 10Н), 1.43-1.46 (м, 1Н), 1.87-1.90 (м, 1Н), 2.21-2.26 (м, 1Н), 2.36-2.41 (м, 1Н), 2.70-2.75 (м, 1Н), 2.93-2.97 (м, 1Н), 4.18-4.30 (м, 2Н), 4.46-4.48 (м, 1Н), 4.55-4.58 (м, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.29 (д, J=18.0 Гц, 1Н), 5.72-5.79 (м, 1Н), 6.10 (уш, 1Н), 7.37-7.40 (м, 1Н), 7.46-7.49 (м, 3Н), 7.70 (т, J=7.5 Гц, 1Н), 7.85-7.89 (м, 2Н), 8.16-8.20 (м, 3Н);
LC-MS (время удерживания: 2.08 мин, метод В), MS m/z 760 (M++Н).
Пример 26: Получение Соединения 26.
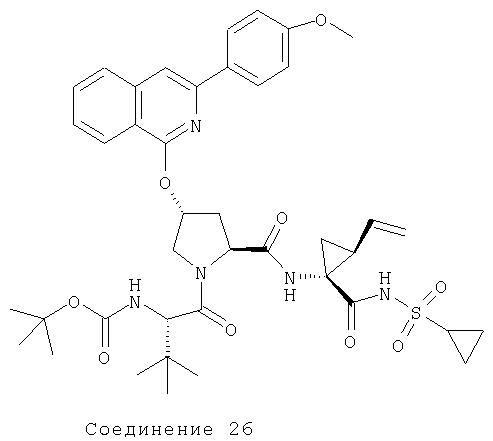
Стадия 1:
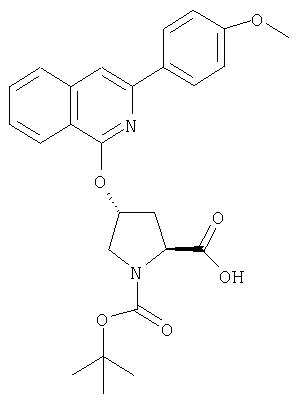
Этот продукт получают по методике, описанной в Примере 25, Стадия 1, но с применением 4-метоксифенилборной кислоты.
1Н ЯМР (CD3OD) δ 1.40, 1.45 (ротамеры, 9Н), 2.50-2.55 (м, 1H), 2.73-2.81 (м, 1Н), 3.81-3.89 (м, 4Н), 3.98-4.01 (м, 1Н), 4.50-4.57 (м, 1Н), 5.93 (уш, 1Н), 7.02 (д, J=9.0 Гц, 2Н), 7.50 (т, J=7.3 Гц, 1Н), 7.67 (т, J=7.5 Гц, 1Н), 7.73 (с, 1Н), 7.83 (д, J=8.5 Гц, 1Н), 8.09 (д, J=8.5 Гц, 2Н), 8.15 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 2.00 мин, метод В), MS m/z 465 (M++Н).
Стадия 2:
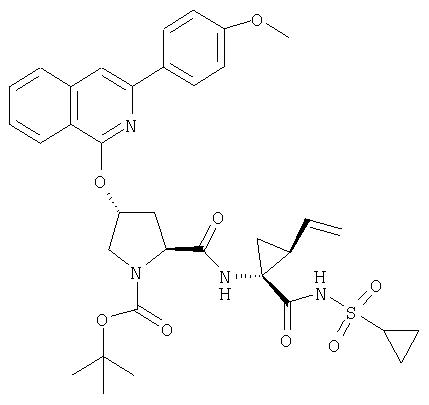
Этот продукт получают по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 26, Стадия 1.
1Н ЯМР (CD3OD) δ 1.06-1.09 (м, 2Н), 1.17-1.27 (м, 2Н), 1.42-1.47 (м, 10Н), 1.88-1.90 (м, 1Н), 2.21-2.26 (м, 1Н), 2.33-2.39 (м, 1H), 2.61-2.65 (м, 1Н), 2.95-2.99 (м, 1Н), 3.85 (с, 3Н), 3.86-3.90 (м, 1Н), 3.99-4.00 (м, 1Н), 4.43-4.45 (м, 1Н), 5.13 (д, J=10.8 Гц, 1Н), 5.31 (д, J=18.0 Гц, 1Н), 5.77-5.80 (м, 1Н), 5.99 (уш, 1Н), 7.02 (д, J=9.0 Гц, 2Н), 7.51 (т, J=7.3 Гц, 1Н), 7.68 (т, J=7.5 Гц, 1Н), 7.76 (с, 1Н), 7.84 (д, J=8.5 Гц, 1Н), 8.09 (д, J=8.5 Гц, 2Н), 8.15 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 2.02 мин, метод В), MS m/z 677 (M++H).
Стадия 3:
Этот продукт в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 26, Стадия 2.
LC-MS (время удерживания: 1.53 мин, метод В), MS m/z 577 (M++Н).
Стадия 4:
Соединение 26 получают по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 26, Стадия 3.
1Н ЯМР (CD3OD) δ 1.26-1.30 (м, 10Н), 1.44-1.46 (м, 1Н), 1.87-1.90 (м, 1Н), 2.21-2.26 (м, 1Н), 2.36-2.41 (м, 1Н), 2.70-2.75 (м, 1Н), 2.93-2.97 (м, 1Н), 3.86 (с, 3Н), 4.18-4.25 (м, 1Н), 4.30 (уш, 1Н), 4.46-4.48 (м, 1Н), 4.55-4.58 (м, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.29 (д, 7=18.0 Гц, 1Н), 5.72-5.79 (м, 1Н), 6.08 (уш, 1Н), 7.02 (д, J=9.0 Гц, 2Н), 7.44 (т, J=7.3 Гц, 1Н), 7.66 (т, J=7.5 Гц, 1Н), 7.75 (с, 1Н), 7.83 (д, J=8.5 Гц, 1Н), 8.09 (д, J=8.5 Гц, 2Н), 8.15 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 2.03 мин, метод В), MS m/z 790 (M++Н).
Пример 27: Получение Соединения 27.

Стадия 1:
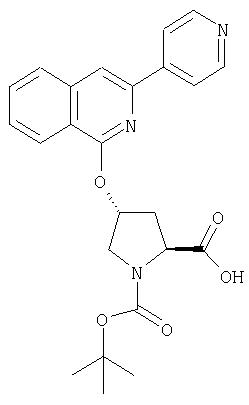
Этот продукт получают по методике, описанной в Примере 25, Стадия 1, но с применением 4-пиридилборной кислоты.
1Н ЯМР (CD3OD) δ 1,43, 1.46 (ротамеры, 96), 2.53-2.56 (м, 1Н), 2.80-2.89 (м, 1Н), 3.90-3.93 (м, 1Н), 4.00-4.05 (м, 1Н), 4.50-4.57 (м, 1Н), 6.00, 6.05 (ротамеры, 1Н), 7.80 (т, J=7.3 Гц, 1Н), 7.87 (т, J=7.3 Гц, 1Н), 7.87 (т, J=7.5 Гц, 1Н), 8.08 (д, J=8.5 Гц, 1Н), 8.32 (д, J=8.0 Гц, 1Н), 8.49 (с, 1Н), 8.84 (д, J=6.0 Гц, 2H), 8.84 (д, J=6.5 Гц, 2Н);
LC-MS (время удерживания: 1.39 мин, метод В), MS m/z 436 (М++Н).
Стадия 2
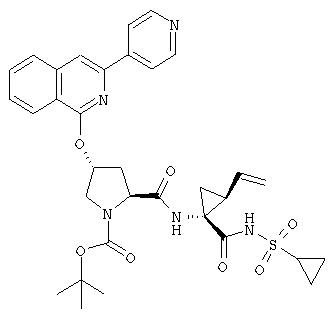
Этот продукт получают по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 27, Стадия 1.
1Н ЯМР (CD3OD) δ 1.06-1.09 (м, 2Н), 1.17-1.27 (м, 2Н), 1.42-1 46 (м, 10Н), 1.88-1.90 (м, 1Н), 2.21-2.26 (м, 1Н), 2.33-2.39 (м, 1Н), 2.61-2.65 (м, 1Н), 2.95-2.99 (м, 1Н), 3.88-3.90 (м, 1Н), 4.01-4.08 (м, 1Н), 4.43-4.45 (м, 1Н), 5.15 (д, J=10.8 Гц, 1Н), 5.32 (д, J=18.0 Гц, 1Н), 5.77-5.80 (м, 1Н), 6.10 (уш, 1Н), 7.79 (т, J=7.3 Гц, 1Н), 7.88 (т, J=7.5 Гц, 1Н), 8.08 (д, J=8.5 Гц, 1Н), 8.31 (д, J=8.0 Гц, 1Н), 8.47 (с, 1Н), 8.79 (д, J=7.0 Гц, 2Н), 8.86 (д, J=6.5 Гц, 2Н);
LC-MS (время удерживания: 1.39 мин, метод В), MS m/z 436 (М++Н).
Стадия 3:

Этот продукт в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 27, Стадия 2.
LC-MS (время удерживания: 0.96 мин, метод В), MS m/z 548 (М++Н).
Стадия 4:
Соединение 27 получают по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 27, Стадия 3.
1Н ЯМР (CD3OD) δ 0.94-1.09 (м, 12Н), 1.22-1.26 (м, 10Н), 1.44-1.49 (м, 1Н), 1.88-1.92 (м, 1Н), 2.22-2.25 (м, 1Н), 2.41-2.44 (м, 1Н), 2.70-2.75 (м, 1Н), 2.93-2.98 (м, 1Н), 4.18-4.21 (м, 1Н), 4.25 (уш, 1Н), 4.53-4.62 (м, 2Н), 5.12 (д, J=10.0 Гц, 1Н), 5.29 (д, J=20.0 Гц, 1Н), 5.72-5.77 (м, 1Н), 6.12 (уш, 1Н), 7.67 (т, J=7.3 Гц, 1Н), 7.82 (т, J=7.5 Гц, 1Н), 8.02 (д, J=8.5 Гц, 1Н), 8.29 (д, J=8.0 Гц, 1Н), 8.31 (с, 1Н), 8.55 (д, J=7.0 Гц, 2Н), 8.76 (д, J=6.5 Гц, 2Н);
LC-MS (время удерживания: 1.49 мин, метод В), MS m/z 761 (М++Н).
Пример 28: Получение Соединения 28.

Стадия 1:

Этот продукт получают по методике, описанной в Примере 25, Стадия 1, но с применением 4-N,N-диметиламинофенилборной кислоты.
LC-MS (время удерживания: 1.64 мин, метод В), MS m/z 478 (М++Н).
Стадия 2:

Этот продукт получают по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 28, Стадия 1.
LC-MS (время удерживания: 1.70 мин, метод В), MS m/z 690 (М++Н).
Стадия 3:

Этот продукт в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 28, Стадия 2.
LC-MS (время удерживания: 1.20 мин, метод В), MS m/z 590 (М++Н).
Стадия 4:
Соединение 28 получают по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 28, Стадия 3.
1Н ЯМР (ДМСО-d6) δ 0.92-1.10 (м, 13Н), 1.30 (с, 9Н), 1.35-1.38 (м, 1Н), 1.68-1.71 (м. 1Н), 2.12-3.00 (м, 2Н), 2.59-2.62 (м, 1Н), 2.91-2.95 (м, 1Н), 2.99 (с, 6Н), 3.93-4.10 (м, 2Н), 4.32-4.40 (м, 2Н), 5.09 (д, J=11.5 Гц, 1Н), 5.23 (д, J=19.0 Гц, 1Н), 5.54-5.64 (м, 1Н), 5.92 (уш, 1Н), 6.83 (д, J=9.0 Гц, 2Н), 7.42 (т, J=7.3 Гц, 1Н), 7.70 (т, J=7.5 Гц, 1Н), 7.81 (с, 1Н), 7.87 (д, J=8.5 Гц, 1Н), 8.04 (д, J=9.0 Гц, 2Н), 8.15 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 1.72 мин, метод В), MS m/z 803 (M++Н).
Пример 29: Получение Соединения 29.

Стадия 1:

Этот продукт получают по методике, описанной в Примере 25, Стадия 1, но с применением 4-цианофенилборной кислоты.
LC-MS (время удерживания: 1.87 мин, метод В), MS m/z 460 (М++Н).
Стадия 2:

Этот продукт получают по методике, описанной в Примере 11, Стация 4, но с применением продукта из Примера 29, Стадия 1.
LC-MS (время удерживания: 1.88 мин, метод В), MS m/z 672 (М++Н).
Стадия 3:

Этот продукт в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 29, Стадия 2.
LC-MS (время удерживания: 1.41 мин, метод В), MS m/z 572 (М++Н).
Стадия 4:
Соединение 29 получают в виде белого твердого вещества по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 29, Стадия 3.
1Н ЯМР (ДМСО-d6) δ 0.92-1.09 (м, 12Н), 1.25-1.26 (м, 10Н), 1.42-1.46 (м, 1Н), 1.86-1.89 (м, 1Н), 2.20-2.22 (м, 1Н), 2.33-2.34 (м, 1Н), 2.68-2.71 (м, 1Н), 2.93-2.95 (м, 1Н), 4.13-4.28 (м, 2Н), 4.49-4.60 (м, 2Н), 5.12 (д, J=10.5 Гц, 1Н), 5.28 (д, J=18.0 Гц, 1Н), 5.71-5.80 (м, 1Н), 6.09 (уш, 1Н), 7.56 (т, J=7.3 Гц, 1Н), 7.74 (т, J=7.5 Гц, 1Н), 7.83 (д, J=10.5 Гц, 2Н), 7.93 (д, J=7.5 Гц, 1H), 8.01 (с, 1Н), 8.22 (д, J=7.5 Гц, 1Н), 8.37 (д, J=10.5 Гц, 2Н);
LC-MS (время удерживания: 1.72 мин, метод В), MS m/z 803 (М++Н).
Пример 30: Получение Соединения 30.
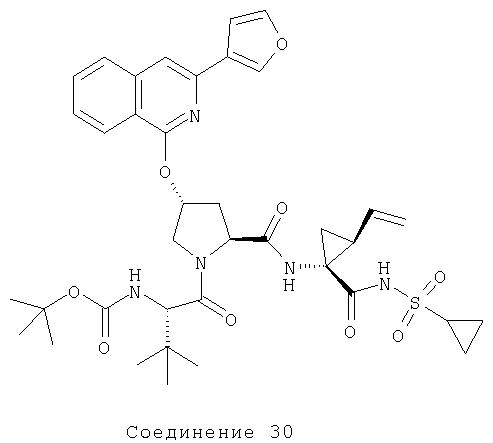
Стадия 1:
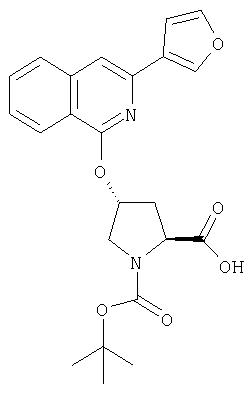
Этот продукт получают по методике, описанной в Примере 25, Стадия 1, но с применением 3-фуранборной кислоты.
LC-MS (время удерживания: 1.85 мин, метод В), MS m/z 425 (М++Н).
Стадия 2:

Этот продукт получают по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 30, Стадия 1.
LC-MS (время удерживания: 1.88 мин, метод В), MS m/z 637 (М++Н).
Стадия 3:

Этот продукт получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 30, Стадия 2.
LC-MS (время удерживания: 1.38 мин, метод В), MS m/z 537 (М++Н).
Стадия 4:
Соединение 30 получают в виде белого твердого вещества по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 30, Стадия 3.
1Н ЯМР (CD3OD) δ 0.95-1.09 (м, 12Н), 1.23-1.30 (м, 10Н), 1.43-1.46 (м, 1Н), 1.87-1.90 (м, 1Н), 2.21-2.23 (м, 1Н), 2.30-2.34 (м, 1Н), 2.64-2.70 (м, 1Н), 2.93-2.96 (м, 1Н), 4.11-4.29 (м,2Н), 4.41-4.44 (м, 1 Н), 4.54-4.56 (м, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.29 (д, J=17.5 Гц, 1Н), 5.71-5.80 (м, 1Н), 6.02 (уш, 1Н), 7.00 (с, 1Н), 7.44 (т, J=7.2 Гц, 1Н), 7.52 (с, 1Н), 7.57 (с, 1Н), 7.66 (т, J=7.0 Гц, 1Н), 7.79 (д, J=8.0 Гц, 1Н), 8.14-8.17 (м, 2Н);
LC-MS (время удерживания: 1.93 мин, метод В), MS m/z 750 (М++Н).
Пример 30: Получение Соединения 30.

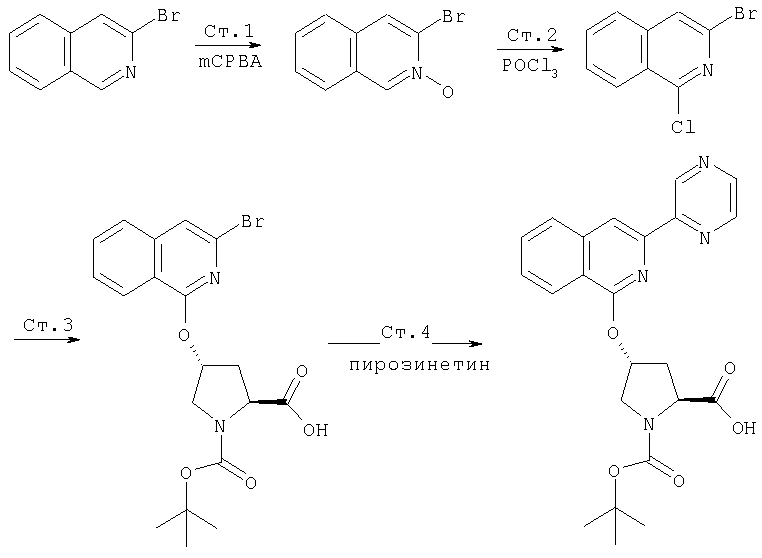
Стадия 1:
Этот продукт в виде белого твердого вещества получают по методике, описанной в Примере 21, Стадия 2, но с применением 3-бромизохинолина (Atkins et al., JOC, 1973, 400).
1Н ЯМР (CDCl3) δ 7.60-7.62 (м, 2Н), 7.71-7.73 (м, 2Н), 8.12 (с, 1Н), 8.99 (с, 1Н);
LC-MS (время удерживания: 0.78 мин, метод В), MS m/z 224, 226 (M++H).
Стадия 2:
Этот продукт в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 2, но с применением 3-бромизохинолина 2-оксида.
1Н ЯМР (CDCl3) δ 7.66-7.71 (м, 1Н), 7.74-7.76 (м, 2Н), 7.83 (с, 1Н), 8.29 (д, J=8.5 Гц, 1Н);
LC-MS (время удерживания: 1.55 мин, метод В), MS m/z 244,244 (М++Н).
Стадия 3:
Этот продукт в виде пены получают по методике, описанной в Примере 11, Стадия 3, но с применением 3-бром-1-хлоризохинолина.
1Н ЯМР (CD3 OD) δ 1.43, 1.44 (ротамеры, 9Н), 2.41-2.47 (м, 1Н), 2.69-2.72 (м, 1Н), 3.80-3.84 (м, 1H), 3.88-3.90 (м, 1Н), 4.46-4.52 (м, 1Н), 5.76 (уш, 1Н), 7.57-7.61 (м, 2Н), 7.73-7.75 (м, 2Н), 8.15 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 1.79 мин, метод В), MS m/z 437,439 (M++H).
Стадия 4:
Смесь 2-трибутилстаннилпиразина (44 мг, 0.12 ммоля), тетракис(трифенилфосфин)палладия (0) (0.12 мг, 0.01 ммоля) и продукта из Примера 31, Стадия 3 (44 мг, 0.1 ммоля) в толуоле (1 мл) кипятят в течение 3 час. Удаляют летучие в вакууме, остаток очищают преп. ВЭЖХ, получают 35 мг (80%) нужного продукта в виде твердого вещества желтого цвета.
LC-MS (время удерживания: 1.77 мин, метод В), MS m/z 437 (М++Н).
Стадия 5:

Этот продукт получают по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 31, Стадия 4.
LC-MS (время удерживания: 1.78 мин, метод В), MS m/z 649 (М++Н).
Стадия 6:
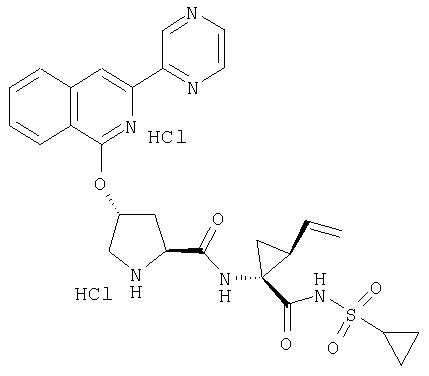
Этот продукт в виде твердого вещества белого цвета получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 31, Стадия 5.
LC-MS (время удерживания: 1.26 мин, метод В), MS m/z 549 (M++Н).
Стадия 7.
Соединение 31 получают по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 31, Стадия 6.
1Н ЯМР (CD3OD) δ 0.95-1.10 (м, 12Н), 1.24-1.27 (м, 10Н), 1.44-1.47 (м, 1Н), 1.87-1.90 (м, 1Н), 2.19-2.22 (м, 1Н), 2.38-2.44 (м, 1Н), 2.71-2.76 (м, 1Н), 2.93-2.96 (м, 1Н), 4.18-4.28 (м, 2Н), 4.50-4.61 (м, 2Н), 5.12 (д, J=10.5 Гц, 1Н), 5.29 (д, J=17.5 Гц, 1Н), 5.71-5.80 (м, 1Н), 6.12 (уш, 1Н), 7.60 (т, J=7.2 Гц, 1Н), 7.77 (т, J=7.0 Гц, 1Н), 7.97 (д, J=8.5 Гц. 1Н), 8.26 (д, J=8.5 Гц, 1Н), 8.44 (с, 1Н), 8.59 (с, 1Н), 8.70 (с, 1Н), 9.61 (с, 1Н);
LC-MS (время удерживания: 1.84 мин, метод В), MS m/z 762 (М++Н).
Пример 32: Получение Соединения 32.

Стадия 1:
Этот продукт, 2-оксиизохинолин-3-карбонитрил, в виде белого твердого вещества получают по методике, описанной в Примере 21, Стадия 2, но с применением 3-цианоизохинолина 2-оксида.
1Н ЯМР (ДМСО-d6) δ 7.74 (т, J=8.0 Гц, 1Н), 7.84 (т, J=8.2 Гц, 1Н), 7.97 (д, J=8.5 Гц, 1Н), 8.03 (д, J=8.5 Гц, 1Н), 8.85 (с, 1Н), 9.17 (с, 1Н);
LC-MS (время удерживания: 0.48 мин, метод В), MS m/z 171 (М++Н).
Стадия 2:

Этот продукт, 1-хлоризохинолин-3-карбонитрил, в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 2, но с применением 3-цианоизохинолин 2-оксида.
1Н ЯМР (CDCl3) δ 7.87-7.91 (м, 2Н), 7.92-7.94 (м, 1Н), 8.09 (с, 1Н), 8.42-8.44 (м, 1Н);
LC-MS (время удерживания: 1.22 мин, метод В), MS m/z 189 (M++H).
Стадия 3:
Этот продукт получают по методике, описанной в Примере 1, Стадия 5, но с применением 1-хлоризохинолин-3-карбонитрила.
1Н ЯМР (CD3OD) δ 1.05 (с, 9Н), 1.17 (с, 9Н), 2.34-2.40 (м, 1Н), 2.71-2.78 (м, 1Н), 4.09-4.11 (м, 1Н), 4.21 (уш, 1Н), 4.48-4.52 (м, 1Н), 4.68-4.72 (м, 1Н), 5.89 (уш, 1Н), 7.74 (т, J=7.5 Гц, 1Н), 7.86 (т, J=7.5 Гц, 1Н), 7.94-7.97 (м, 2Н), 8.31 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 1.66 мин, метод В), MS m/z 497 (M++H).
Стадия 4:
Соединение 32 получают в виде твердого вещества белого цвета по методике, описанной в Примере 1, Стадия 9, но с применением продукта из Примера 32, Стадия 3.
1Н ЯМР (CD3OD) δ 1.04-1.09 (м, 12Н), 1.20-1.27 (м, 10Н), 1.39-1.45 (м, 1Н), 1.85-1.88 (м, 1Н), 2.20-2.30 (м, 2Н), 2.63-2.71 (м, 1Н), 2.91-2.97 (м, 1Н), 4.09-4.13 (м, 1Н), 4.23 (д, J=9.3 Гц, 1Н), 4.49-4.58 (м, 2Н), 5.13 (д, J=10.5 Гц, 1Н), 5.28 (д, J=18.0 Гц, 1Н), 5.69-5.81 (м, 1Н), 5.92 (уш, 1Н), 6.60 (д, J=10.0 Гц, 1Н), 7.72 (т, J=7.5 Гц, 1Н), 7.86 (т, J=7.5 Гц, 1Н), 7.96-7.99 (м, 2Н), 8.29 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 1.75 мин, метод В), MS m/z 714 (М++Н).
Пример 33: Получение Соединения 33.
Стадия 1:

Этот продукт, 3-метилизохинолин-2-оксид, в виде белого твердого вещества получают по методике, описанной в Примере 21, Стадия 2, но с применением 3-метилизохинолина.
1Н ЯМР (CD3 OD) δ 2.64 (с, 3Н), 7.64-7.72 (м, 2Н), 7.88-7.95 (м, 2Н), 9.05 (с, 1H);
LC-MS (время удерживания: 0.61 мин, метод В), MS m/z 160 (M++Н).
Стадия 2:
Этот продукт, 1-хлор-3-метилизохинолин, в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 2, но с применением 3-метилизохинолин 2-оксида.
1Н ЯМР (CDCl3) δ 2.65 (с, 3Н), 7.25 (с, 1Н), 7.61 (т, J=7.5 Гц, 1Н), 7.69 (т, J=7.5 Гц, 1Н), 7.74 (д, J=8.0 Гц, 1Н), 8.27 (д, J=8.5 Гц, 1Н);
LC-MS (время удерживания: 1.47 мин, метод В), MS m/z 178 (М++Н).
Стадия 3:

Этот продукт, в виде белого твердого вещества, получают по методике, описанной в Примере 1, Стадия 5, но с применением 1-хлор-3-метилизохинолина.
1Н ЯМР (CD3OD) δ 1.05 (с, 9Н), 1.23 (с. 9Н), 2.51 (с, 3Н), 2.34-2.40 (м, 1Н), 2.72-2.78 (м, 1Н), 4.05-4.12 (м, 1Н), 4.26 (уш, 1Н), 4.41 (д, J=10 Гц, 1Н), 4.62-4.67 (м, 1Н), 5.90 (уш, 1Н), 7.14 (с, 1Н), 7.38 (т, J=7.5 Гц, 1Н), 7.62 (т, J=7.5 Гц, 1Н), 7.68 (д, J=8.0 Гц, 1Н), 8.14 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 1.84 мин, метод В), MS m/z 486 (М++Н).
Стадия 4:
Соединение 33 получают в виде твердого вещества белого цвета по методике, описанной в Примере 1, Стадия 9, но с применением продукта из Примера 33, Стадия 3.
1Н ЯМР (CD3OD) δ 0.99-1.09 (м, 12Н), 1.23-1.25 (м, 10Н), 1.41-1.45 (м, 1Н), 1.86-1.90 (м, 1Н), 2.21-2.31 (м, 2Н), 2.52 (с, 3Н), 2.58-2.61 (м, 1Н), 2.91-2.97 (м, 1Н), 4.08-4.12 (м, 1Н), 4.28 (уш, 1Н), 4.40 (д, J=10.0 Гц, 1Н), 4.50-4.55 (м, 1Н), 5.12 (д, J=10.0 Гц, 1Н), 5.30 (д, J=18.0 Гц, 1Н), 5.71-5.81 (м, 1Н), 5.93 (уш, 1Н), 7.13 (с, 1Н), 7.38 (т, J=7.5 Гц, 1Н), 7.62 (т, J=7.5 Гц, 1Н), 7.68 (д, J=8.0 Гц, 1Н), 8.12 (д, J=8.0 Гц, 1Н), 9.12 (уш, 1Н);
LC-MS (время удерживания: 1.85 мин, метод В), MS m/z 698 (M++Н).
Пример 34: Получение Соединения 34.

Стадия 1:
Этот продукт, 3-циклопропилизохинолин-2-оксид, в виде белого твердого вещества получают по методике, описанной в Примере 21, Стадия 2, но с применением 3-циклопропилизохинолина (L.Flippin, J.Muchowsky, J.O.C, 1993, 2631-2632).
LC-MS (время удерживания: 0.95 мин, метод В), MS m/z 186 (M++H).
Стадия 2:

Этот продукт, 1-хлор-3-циклопропилизохинолин, получают по методике, описанной в Примере 11, Стадия 2, но с применением 3-циклопропилизохинолин 2-оксида.
1Н ЯМР (CD3OD) δ 1.00-1.04 (м, 4Н), 2.11-2.18 (м, 1Н), 7.55 (с, 1Н), 7.61 (т, J=8.0 Гц, 1Н), 7.72 (т, J=8.0 Гц, 1Н), 7.83 (д, J=13.5 Гц, 1Н), 8.27 (д, J=14.5 Гц, 1Н);
LC-MS (время удерживания: 1.70 мин, метод В), MS m/z 204 (M++Н).
Стадия 3:

Этот продукт в виде твердого вещества белого цвета получают по методике, описанной в Примере 1, Стадия 5, но с применением 1-хлор-3-циклопропилизохинолина.
1Н ЯМР (CD3OD) δ 0.93-1.05 (м, 13Н), 1.29 (с, 9Н), 2.06-2.10 (м, 1Н), 2.39-2.44 (м, 1Н), 2.70-2.76 (м, 1Н), 4.05-4.12 (м, 1Н), 4.27 (уш, 1Н), 4.35 (д, J=10.0 Гц, 1Н), 4.62-4.67 (м, 1Н), 5.78 (уш, 1Н), 7.18 (с, 1Н), 7.38 (т, J=7.5 Гц, 1Н), 7.61 (т, J=7.5 Гц, 1Н), 7.66 (д, J=8.0 Гц, 1Н), 8.09 (д, J=8.0 Гц, 1Н);
LC-MS (время удерживания: 1.96 мин, метод В), MS m/z 512 (М++Н).
Стадия 4:
Соединение 34 получают в виде твердого вещества белого цвета по методике, описанной в Примере 1, Стадия 9, но с применением продукта из Примера 34, Стадия 3.
1Н ЯМР (CD3OD) δ 0.93-1.09 (м, 16Н), 1.24-1.30 (м, 10Н), 1.42-1.46 (м, 1Н), 1.87-1.90 (м, 1Н), 2.06-2.11 (м, 1Н), 2.21-2.32 (м, 2Н), 2.56-2.61 (м, 1Н), 2.92-2.97 (м, 1Н), 4.08-4.12 (м, 1Н), 4.28 (уш, 1Н), 4.32 (д. J=10.0 Гц, 1Н), 4.48-4.53 (м, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.30 (д, J=17.5 Гц, 1Н), 5.72-5.77 (м, 1Н), 5.82 (уш, 1Н), 7.18 (с, 1Н), 7.36 (т, J=7.5 Гц, 1Н), 7.60 (т, J=7.5 Гц, 1Н), 7.67 (д, J=8.0 Гц, 1Н), 8.07 (д, 7=8.0 Гц, 1Н);
LC-MS (время удерживания: 2.00 мин, метод В), MS m/z 724 (М++Н).
Пример 35: Получение Соединения 35.
Схема 1

Стадия 1:
Смесь 3-гидроксиизохинолина (725 мг, 5.0 ммолей), карбоната цезия (4.89 г, 15.0 ммолей), MeI (781 мг, 5.5 ммоля) в ДМФА (50 мл) перемешивают при комнатной температуре в течение 12 час. К смеси прибавляют EtOAc (200 мл), фильтруют, последовательно промывают водой (200 мл, Х2) и 1М NaOH (водн.), рассолом. Органические вытяжки сушат MgSO4, фильтруют, упаривают. Остаток очищают преп. ВЭЖХ, получают 120 мг (15%) нужного продукта в виде твердого вещества белого цвета.
1Н ЯМР (CDCl3) δ 4.03 (с, 3Н), 6.99 (с, 1Н), 7.36 (т, J=8.0 Гц, 1Н), 7.56 (т, J=8.2 Гц, 1Н), 7.68 (д, J=8.5 Гц, 1Н), 7.87 (J=8.5 Гц, 1Н);
LC-MS (время удерживания: 0.54 мин, метод В), MS m/z 160 (М+ +Н).
Стадия 2:
Этот продукт, 3-метоксиизохинолин-2-оксид, в виде белого твердого вещества получают по методике, описанной в Примере 21, Стадия 2, но с применением 3-метоксиизохинолина.
LC-MS (время удерживания: 0.83 мин, метод В), MS m/z 176 (М++Н).
Стадия 3:

Этот продукт, 1-хлор-3-метоксиизохинолин, в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 2, но с применением 3-метоксиизохинолин 2-оксида.
LC-MS (время удерживания: 1.62 мин, метод В), MS m/z 194 (M++H).
Стадия 4:

Этот продукт, в виде белого твердого вещества, получают по методике, описанной в Примере 1, Стадия 5, но с применением 1-хлор-3-метоксиизохинолина.
1Н ЯМР (CD3OD) δ 1.05 (с, 9Н), 1.23 (с, 9Н), 2.35-2.43 (м, 1Н), 2.72-2.79 (м, 1Н), 3.96 (с, 3Н), 4.01-4.11 (м, 1Н), 4.26 (уш, 1Н), 4.48 (д, J=10.0 Гц, 1Н), 4.62-4.67 (м, 1Н), 5.83 (уш, 1Н), 6.61 (с, 1Н), 7.25 (т, J=7.5 Гц, 1Н), 7.54 (т, J=7.5 Гц, 1Н), 7.63 (д, J=8.1 Гц, 1Н), 8.08 (д, J=8.4 Гц, 1Н);
LC-MS (время удерживания: 1.82 мин, метод В), MS m/z 502 (М+ +Н).
Стадия 5:
Соединение 35 получают в виде твердого вещества белого цвета по методике, описанной в Примере 1, Стадия 9, но с применением продукта из Примера 35, Стадия 4.
1Н ЯМР (CD3OD) δ 1.04-1.08 (м, 12Н), 1.24-1.27 (м, 10Н), 1.43-1.45 (м, 1Н), 1.86-1.89 (м, 1Н), 2.21-2.26 (м, 1Н), 2.30-2.34 (м, 1Н), 2.62-2.66 (м, 1Н), 2.91-2.97 (м, 1Н), 3.99 (с, 3Н), 4.09-4.12 (м, 1Н), 4.27-4.28 (м, 1Н), 4.46 (д, J=10.0 Гц, 1Н), 4.51-4.58 (м, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.30 (д, J=18.0 Гц, 1Н), 5.72-5.76 (м, 1Н), 5.88 (уш, 1Н), 6.62 (с, 1Н), 7.26 (т, J=7.5 Гц, 1Н), 7.55 (т, J=7.5 Гц, 1Н), 7.65 (д, J=8.0 Гц, 1Н), 8.06 (д, J=8.5 Гц, 1Н);
LC-MS (время удерживания: 1.85 мин, метод В), MS m/z 714 (М++Н).
Пример 36: Получение Соединения 36.

Схема 1
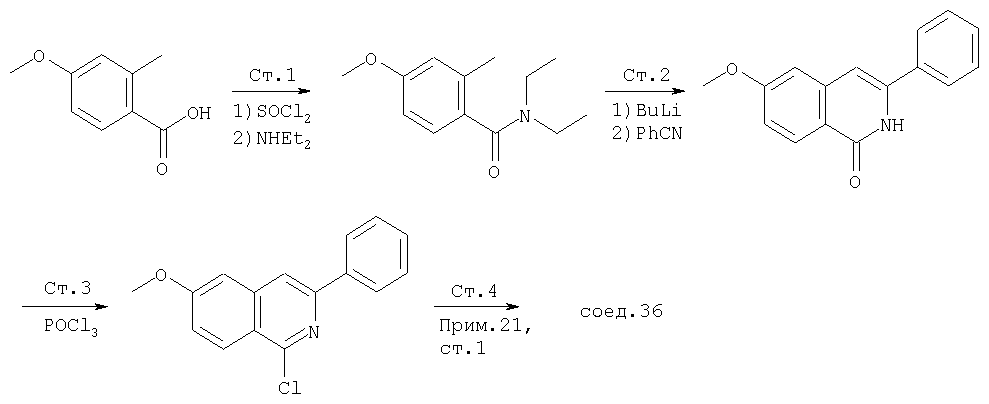
Стадия 1:
Смесь 4-метокси-2-метилбензойной кислоты (5.00 г, 30.1 ммоля) и тионилхлорида (20.0 г, 0.17 моля) кипятят 30 мин. Отгоняют летучие в вакууме. Выдерживают в вакууме масляного насоса в течение ночи, хлорангидрид кислоты в виде вязкого масла используют на следующей стадии без дополнительной очистки.
К раствору 4-метокси-2-метилбензоилхлорида в СН2Cl2 (60 мл) при 0°С по каплям прибавляют диэтиламин. Полученную смесь оставляют при перемешивании на 2 часа нагреваться до комнатной температуры. Летучие удаляют в вакууме. Остаток растирают с EtOAc (100 мл) и отфильтровывают. Фильтрат промывают 1М HCl, 1М NaOH и рассолом, сушат MgSO4. После упаривания растворителя получают 6.51 г (98%) ожидаемого продукта в виде вязкого масла.
LC-MS (время удерживания: 1.20 мин, метод В), MS m/z 222 (М++Н).
Стадия 2:
К раствору N,N-диэтил-4-метокси-2-метилбензамида (221 мг, 1.0 ммоль) в ТГФ (2 мл) при -78°С по каплям прибавляют н-BuLi (0.84 мл 2.5 М раствора в гексане, 2.10 ммоля). Полученный оранжевый раствор выдерживают при этой температуре еще в течение 30 мин, по каплям прибавляют бензонитрил (103 мг, 1.0 ммоль). Полученный раствор оставляют при перемешивании при комнатной температуре. Прибавляют 5% лимонную кислоту. Фильтруют, промывают водой, сушат. Растирают со смесью 2:1 гексан - EtOAc (5 мл), получают 205 мг (82%) нужного продукта в виде твердого вещества белого цвета.
1Н ЯМР (ДМСО-d6) δ 3.89 (с, 3Н), 6.84 (с, 1Н), 7.05-7.07 (м, 1H), 7.18 (д, J=2.5 Гц, 1Н), 7.44-7.51 (м, 3Н), 7.78 (д, J=7.0 Гц, 1Н), 8.11 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.20 мин, метод В), MS m/z, 252 (М++Н).
Стадия 3:
Этот продукт, 1-хлор-6-метокси-3-фенилизохинолин, в виде белого твердого вещества получают по методике, описанной в Примере 11, Стадия 2, но с применением 6-метокси-3-фенил-2Н-изохинолин-1-она.
1Н ЯМР (CDCl3) δ 3.97 (с, 3Н), 7.12 (д, J=2.5 Гц, 1Н), 7.23-7.26 (м, 1Н), 7.40-7.42 (м, 1Н ), 7.46-7.50 (м, 2Н), 7.89 (с, 1Н), 8.08 (д, J=7.0 Гц, 2Н), 8.21 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.90 мин, метод В), MS m/z 270, 271 (M++H).
Стадия 4:
К раствору продукта из Примера 21, Стадия 1 (320 мг, 0.57 ммоля) в ДМСО (5 мл) прибавляют трет-бутоксид калия (321 мг, 2.87 ммоля). Полученный раствор перемешивают при комнатной температуре 30 мин и прибавляют 1-хлор-6-метокси-3-фенилизохинолина (Пример 36, Стадия 3) (155 мг, 0.57 ммоля). Полученный раствор перемешивают 12 час. Прибавляют ледяную воду, подкисляют 1М HCl до рН 4, экстрагируют EtOAc (20 мл, Х2). Органические вытяжки промывают рассолом, сушат MgSO4, фильтруют, упаривают. Остаток очищают преп. ВЭЖХ (от 40%В до 100%В, 15 мин градиент), получают 289 мг (64%) Соединения 36 в виде твердого вещества белого цвета.
1Н ЯМР (CD3OD) δ 0.95-1.05 (м, 12Н), 1.24-1.32 (м, 10Н), 1.44-1.46 (м, 1Н), 1.87-1.90 (м, 1Н), 2.20-2.26 (м, 1Н), 2.30-2.36 (м, 1Н), 2.65-2.71 (м, 1Н), 2.93-2.97 (м, 1Н), 3.94 (с, 3Н), 4.12-4.28 (м, 2Н), 4.38-4.52 (м, 2Н), 5.12 (д, J=10.0 Гц, 1Н), 5.28 (д, J=17.0 Гц, 1Н), 5.69-5.74 (м, 1Н), 6.05 (уш, 1Н), 7.06-7.07 (м, 1Н), 7.26 (с, 1Н), 7.37-7.39 (м, 1Н), 7.44-7.48 (м, 2Н), 7.77 (с, 1Н). 8.07 (д, J=9.0 Гц, 1Н), 8.15 (д, J=8.5 Гц,2Н);
LC-MS (время удерживания: 2.02 мин, метод В), MS m/z 790 (М++Н).
Пример 37: Получение Соединения 37.

Схема 1

Стадия 1:
К раствору N, N-диэтил-4-метокси-2-метилбензамида (633 мг, 2.9 ммоля) в ТГФ (15 мл) при -78°С прибавляют по каплям н-BuLi (2.3 мл 2.5 М в гексане, 5.74 ммоля). Полученный красный раствор выдерживают при этой температуре еще в течение 30 мин, переносят с помощью иглы в раствор этилового эфира тиазол-2-карбоновой кислоты (A.Medici et al., Tetrahedron Lett. 1983, p2901) (450 мг, 2.9 ммоля) в ТГФ (5 мл) при -78°С. Полученный зеленый раствор перемешивают 2 час при комнатной температуре. Прибавляют нас. водн. раствор NH4Cl и экстрагируют EtOAc (50 мл). Органические вытяжки промывают нас. водн. раствором NH4Cl и рассолом, сушат, очищают флеш - хроматографией на колонке, элюент 2:1 EtOAc: гексан, получают 405 мг (45%) нужного продукта в виде вязкого масла не чисто белого цвета.
1Н ЯМР (CDCl3) δ 1.08 (т, J=7.0 Гц, 6Н), 3.22 (уш, 2Н), 3.44 (уш, 2Н), 3.79 (с, 3Н), 4.59 (с, 2Н), 6.79-6.81 (м, 1Н), 6.86 (д, J=2.5 Гц, 1Н), 7.16 (д, J=8.5 Гц, 1Н), 7,66 (д, J=3.0 Гц, 1Н), 8.00 (д, J=3.0 Гц, 1Н);
LC-MS (время удерживания: 1.30 мин, метод В), MS m/z 330 (М++Н).
Стадия 2:
Смесь N,N-диэтил-4-метокси-2-(2-оксо-2-тиазол-2-илэтил)-бензамида (405 мг, 1.22 ммоля) и NH4OAc (3.0 г, 38.9 ммоля) нагревают 1 час при 140°С в запаянной ампуле. Расплав выливают в ледяную воду, фильтруют, осадок на фильтре тщательно промывают водой. Высушенное твердое вещество (240 мг, 76%) используют на следующей стадии без дополнительной очистки.
LC-MS (время удерживания: 1.24 мин, метод В), MS m/z 259 (М++Н).
Стадия 3:
Этот продукт, 1-хлор-6-метокси-3-тиазол-2-илизохинолин, получают в виде твердого вещества белого цвета по методике, описанной в Примере 11, Стадия 2, но используют 6-метокси-3-тиазол-2-ил-2Н-изохинолин-1-он.
1Н ЯМР (CDCl3) δ 3.97 (с, 3Н), 7.16 (д, J=4.0 Гц, 1Н), 7.27-7.31 (м, 1Н), 7.46 (д, J=5.0 Гц, 1Н), 7.93 (д, J=5.5 Гц, 1Н), 8.22 (д, J=15.5 Гц, 1Н), 8.39 (с, 1Н);
LC-MS (время удерживания: 1.66 мин, метод В), MS m/z 277 (M++H).
Стадия 4:
Соединение 37 получают по методике, описанной в Примере 36, Стадия 4, но с применением 1-хлор-6-метокси-3-тиазол-2-ил-2Н-изохинолин-1-она.
1Н ЯМР (CD3OD) δ 0.97-1.09 (м, 12Н), 1.24-1.29 (м, 10Н), 1.44-1.46 (м, 1Н), 1.87-1.90 (м. 1Н), 2.20-2.26 (м, 1Н), 2.30-2.36 (м, 1Н), 2.65-2.71 (м, 1Н), 2.93-2.96 (м, 1Н), 3.96 (с, 3Н), 4.12-4.27 (м, 2Н), 4.38-4.52 (м, 2Н), 5.12 (д, J=10.5 Гц, 1Н), 5.29 (д, J=17.5 Гц, 1Н), 5.69-5.74 (м, 1Н), 5.99 (уш, 1Н), 7.14 (д, J=9.0 Гц, 1Н), 7.33 (с, 1Н), 7.66 (д, J=3.5 Гц, 1Н), 7.93 (д, J=3.0 Гц, 1Н), 8.05 (с, 1Н), 8.11 (д, J=9.0 Гц, 1Н), 9.14 (уш, 1Н);
LC-MS (время удерживания: 1.89 мин, метод В), MS m/z 797 (М++Н).
Пример 38: Получение Соединения 38.

Схема 1
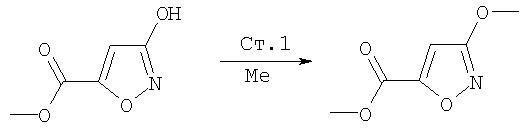
Стадия 1:
Смесь метилового эфира 3-гидроксиизоксазол-5-карбоновой кислоты (5.72 г, 0.04 моля), йодистого метила (6.82 г, 0.044 ммоля) и карбоната цезия (39.1 г, 0.12 моля) в ДМФА (200 мл) перемешивают при комнатной температуре в течение ночи. Прибавляют EtOAc (1 л), фильтруют. Фильтрат промывают последовательно водой (1 л, Х2), 1М NaOH и рассолом, сушат MgSO4, упаривают в вакууме, получают 4.80 г (76%) нужного продукта в виде твердого вещества белого цвета. Полученный продукт используют на следующей стадии без дополнительной очистки.
1Н ЯМР (CDCl3) δ 3.92 (с, 3Н), 4.00 (с, 3Н), 6.51 (с, 1Н);
LC-MS (время удерживания: 0.69 мин, метод В), MS m/z 158 (М++Н).
Стадия 2:

Этот продукт, N, N-диэтил-4-метокси-2-[2-(3-метоксиизоксазол-5-ил)-2-оксоэтил]-бензамид, получают по методике, описанной в Примере 37, Стадия 1, но при использовании метилового эфира 3-метоксиизоксазол-5-карбоновой кислоты.
LC-MS (время удерживания: 1.28 мин, метод В), MS m/z, 347 (М++Н).
Стадия 3:
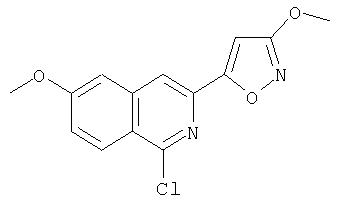
Этот продукт, N,N-диэтил-4-метокси-2-[2-(3-метоксиизоксазол-5-ил)-2-оксоэтил]-бензамид, получают по методике, описанной в Примере 37, Стадия 1, но при использовании метилового эфира 3-метоксиизоксазол-5-карбоновой кислоты.
1Н ЯМР (ДМСО-d6) δ 3.89 (с, 3Н), 3.97 (с, 3Н), 7.01 (с, 1Н), 7.14-7.16 (м, 2Н), 7.43 (с, 1Н), 8.13 (д, J=8.5 Гц, 1Н);
LC-MS (время удерживания: 1.28 мин, метод В), MS m/z 273 (М++Н).
Стадия 4:
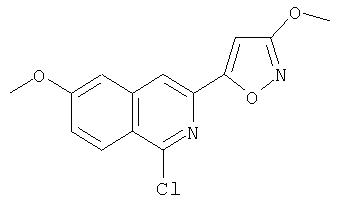
Этот продукт, 1-хлор-6-метокси-3-(3-метоксиизоксазол-5-ил)-изохинолин, получают в виде белого твердого вещества по методике, описанной в Примере 11, Стадия 2, но с использованием 6-метокси-3-(3-метоксиизоксазол-5-ил)-2Н-изохинолин-1-она.
1Н ЯМР (CDCl3) δ 3.97 (с, 3Н), 4.04 (с, 4Н), 6.60 (с, 1Н), 7.17 (д, J=2.5 Гц, 1Н), 7.31-7.33 (м, 1Н), 8.02 (с, 1Н), 8.23 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.73 мин, метод В), MS m/z 293 (M++Н).
Стадия 5:
Соединение 38 получают по методике, описанной в Примере 36, Стадия 4, но с использованием 1-хлор-6-метокси-3-(3-метоксиизоксазол-5-ил)-изохинолина.
1Н ЯМР (CDCl3) δ 0.99-1.09 (м, 12Н), 1.23-1.28 (м, 10Н), 1.44-1.46 (м, 1Н), 1.87-1.90 (м, 1Н), 2.20-2.26 (м, 1Н), 2.30-2.36 (м, 1Н), 2.65-2.71 (м, 1Н), 2.93-2.96 (м, 1Н), 3.95 (с, 3Н), 4.02 (с, 3Н), 4.13-4.14 (м, 1Н), 4.24-4.26 (м, 1Н), 4.41-4.42 (м, 1Н), 4.52-4.55 (м, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.29 (д, J=17.0 Гц, 1Н), 5.72-5.79 (м, 1Н), 5.96 (уш, 1Н), 6.60 (с, 1Н), 7.15-7.17 (м, 1Н), 7.32 (с, 1Н), 7.80 (с, 1Н), 8.10 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.95 мин, метод В), MS m/z 811 (М++Н).
Пример 39: Получение Соединения 39.
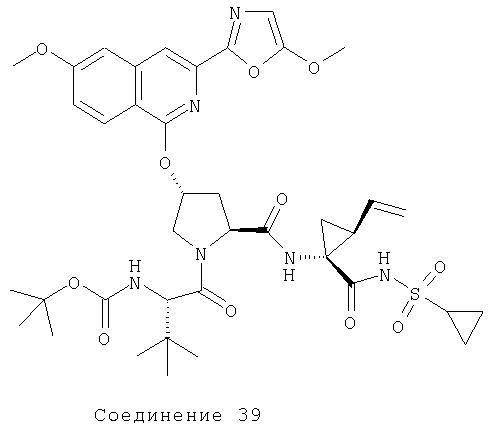
Стадия 1:

Этот продукт, N, N-диэтил-4-метокси-2-[2-(5-метоксиоксазол-2-ил)-2-оксоэтил]-бензамид, получают по методике, описанной в Примере 37, Стадия 1, но при использовании этилового эфира 5-метоксиоксазол-2-карбоновой кислоты.
LC-MS (время удерживания: 1.24 мин, метод В), MS m/z 347 (М++Н).
Стадия 2:

Этот продукт, 6-метокси-3-(5-метоксиоксазол-2-ил)-2Н-изохинолин-1-он, получают в по методике, описанной в Примере 37, Стадия 2, но с использованием N,N-диэтил-4-метокси-2-[2-(5-метоксиоксазол-2-ил)-2-оксоэтил]-бензамида.
1Н ЯМР (ДМСО-d6) δ 3.94 (с, 3Н), 4.01 (с, 3Н), 6.34 (с, 1Н), 6.99 (д, J=2.0 Гц, 1Н), 7.12-7.18 (м, 1Н), 7.25 (с, 1Н), 8.32 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1,22 мин, метод В), MS m/z 274 (М++Н).
Стадия 3:

Этот продукт, 1-хлор-6-метокси-3-(5-метоксиоксазол-2-ил)-изохинолин, получают в виде белого твердого вещества по методике, описанной в Примере 11, Стадия 2, но с использованием 6-метокси-3-(5-метоксиоксазол-2-ил)-2Н-изохинолин-1-она.
1Н ЯМР (CDCl3) δ 3.96 (с, 3Н), 4.00 (с, 3Н), 6.34 (с, 1Н), 7.12 (д, J=2.5 Гц, 1Н), 7.28-7.31 (м, 1Н), 8.13 (с, 1Н), 8.23 (д, J=9.0 Гц, 1Н);
LC-MS (время удерживания: 1.58 мин, метод В), MS m/z 291,293 (М++Н).
Стадия 4:
Соединение 39 получают по методике, описанной в Примере 36, Стадия 4, но с использованием 1-хлор-6-метокси-3-(2-метоксиоксазол-2-ил)-изохинолина.
1Н ЯМР (CD3OD) δ 0.99-1.09 (м, 12Н), 1.23-1.28 (м, 10Н), 1.44-1.46 (м, 1Н), 1.87-1.90 (м, 1Н), 2.20-2.26 (м, 1Н), 2.30-2.36 (м, 1Н), 2.65-2.71 (м, 1Н), 2.93-2.96 (м, 1Н), 3.95 (с, 3Н), 4.02 (с, 3Н), 4.13-4.14 (м, 1Н), 4.25 (уш, 1Н), 4.41-4.42 (м, 1Н), 4.52-4.55 (м, 1Н), 5.12 (д, J=10.0 Гц, 1Н), 5.29 (д, J=17.0 Гц, 1Н), 5.72-5.79 (м, 1Н), 6.07 (уш, 1Н), 6.45 (с, 1Н), 7.15-7.16 (м, 1Н), 7.29 (с, 1Н), 7.85 (с, 1Н), 8.10 (д, J=9.0 Гц, 1Н), 9.11 (уш, 1Н);
LC-MS (время удерживания: 1.75 мин, метод В), MS m/z 811 (M++H).
Пример 40: Получение Соединения 40.
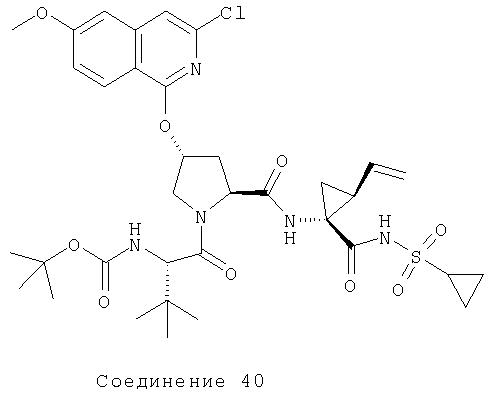
Схема 1

Стадия 1:
Этот продукт, 1-хлор-6-метоксиизохинолин 2-оксид, получают по методике, описанной в Примере 21, Стадия 2, но с использованием 1-хлор-6-метоксиизохинолина (продукт из Примера 11, Стадия 2).
1Н ЯМР (CDCl3) δ 4.00 (с, 3Н), 7.14 (д, J=2.5 Гц, 1Н), 7.41-7.43 (м, 1Н), 7.62 (д, J=7.0 Гц, 1Н), 8.15 (д, J=9.5 Гц, 1Н), 8.36 (4, J=7.0 Гц, 1Н);
LC-MS (время удерживания: 0.85 мин, метод В), MS m/z 210 (M++Н).
Стадия 2:
Этот продукт, 1,3-дихлор-6-метоксиизохинолин, получают по методике, описанной в Примере 11, Стадия 2, но с использованием 1-хлор-6-метоксиизохинолин 2-оксида.
1Н ЯМР (CDCl3) δ 3.94 (с; 3Н), 6.98 (с, 1Н), 7.25-7.26 (м, 1H), 7.52 (с; 1Н), 8.16 (д, J=9.5 Гц, 1Н);
LC-MS (время удерживания: 1.54 мин, метод В), MS m/z 228, 230 (M++Н).
Стадия 3:
Этот продукт получают по методике, описанной в Примере 24, Стадия 1, но с применением 1,3-дихлор-6-метоксиизохинолина.
1Н ЯМР (CD3OD) δ 1.43, 1.44 (ротамеры, 9Н), 2.39-2.44 (м, 1Н), 2.68-2.72 (м, 1Н), 3.80-3.90 (м, 2Н), 3.91 (с, 3Н), 4.79-4.82 (м, 1Н), 5.71 (уш, 1Н), 7.10-7.14 (м, 2Н), 7.26 (с, 1Н), 7.99-8.01 (м, 1Н);
LC-MS (время удерживания: 1.79 мин, метод В), MS m/z 422 (M++H).
Стадия 4:
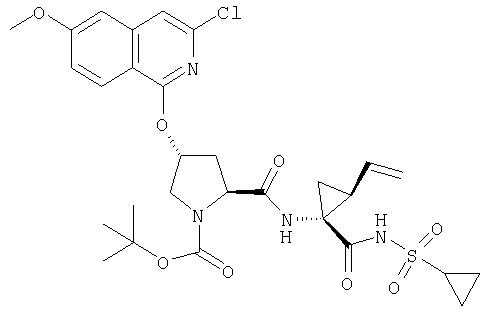
Этот продукт получают по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 40, Стадия 3.
LC-MS (время удерживания: 1.83 мин, метод В), MS, m/z 635 (М++Н).
Стадия 5:

Этот продукт в виде твердого вещества белого цвета получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 40, Стадия 4.
LC-MS (время удерживания: 1.36 мин, метод В), MS m/z 535 (M++Н).
Стадия 6:
Соединение 40 в виде твердого вещества белого цвета получают по методике, описанной в Примере 11, Стадия 6, но с использованием продукта из Примера 40, Стадия 5.
1Н ЯМР (CD3OD) δ 1.07-1.11 (м, 12Н), 1.26-1.30 (м, 10Н), 1.46-1.48 (м, 1Н), 1.87-1.91 (м, 1Н), 2.21-2.34 (м, 2Н), 2.62-2.66 (м, 1Н), 2.94-2.99 (м, 1Н), 3.95 (с, 3Н), 4.06-4.11 (м, 1Н), 4.26-4.28 (м, 1Н), 4.46-4.56 (м, 2Н), 5.15 (д, J=10,0 Гц, 1Н), 5.29 (д, J=17.0 Гц, 1Н), 5.72-5.79 (м, 1Н), 5.89 (уш, 1Н), 6.63 (д, J=9.0 Гц, 1Н), 7.08-7.09 (м, 1Н), 7.18 (с, 1Н), 7.34 (с, 1Н), 8.08 (д, J=9.5 Гц, 1Н);
LC-MS (время удерживания: 1.99 мин, метод В), MS m/z 748 (М++Н).
Пример 41: Получение Соединения 41.

Стадия 1:
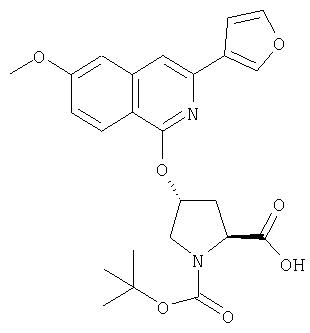
Этот продукт получают по методике, описанной в Примере 30, Стадия 1, но с применением продукта из Примера 40, Стадия 3.
LC-MS (время удерживания: 1.85 мин, метод В), MS m/z, 455 (M++Н).
Стадия 2:
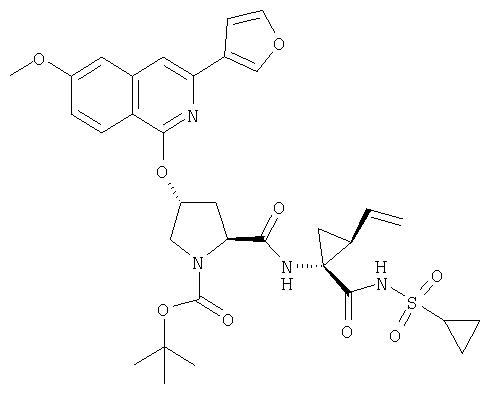
Этот продукт в виде пены получают по методике, описанной в Примере 11, Стадия 4, но с применением продукта из Примера 41, Стадия 1.
LC-MS (время удерживания: 1.88 мин, метод В), MS m/z 667 (М++Н).
Стадия 3:
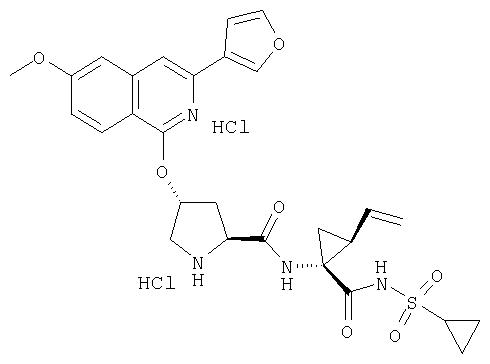
Этот продукт в виде твердого вещества белого цвета получают по методике, описанной в Примере 11, Стадия 5, но с применением продукта из Примера 41, Стадия 2.
LC-MS (время удерживания: 1.38 мин, метод В), MS m/z 567 (М++Н).
Стадия 4:
Соединение 41 в виде твердого вещества белого цвета получают по методике, описанной в Примере 11, Стадия 6, но с применением продукта из Примера 41, Стадия 3.
1Н ЯМР (CD3OD) δ 0.99-1.04 (м, 12Н), 1.22-1.31 (м, 10Н), 1.43-1.45 (м, 1Н), 1.87-1.89 (м, 1Н), 2.22-2.24 (м, 1Н), 2.30-2.34 (м, 1Н), 2.65-2.68 (м, 1Н), 2.93-2.96 (м, 1Н), 3.92 (с, 3Н), 4.11-4.14 (м, 1Н), 4.28-4.30 (м, 1Н), 4.38-4.42 (м, 1Н), 4.53-4.55 (м, 1Н), 5.12 (д, J=10.0 Гц, 1Н), 5.29 (д, J=18.0 Гц, 1Н), 5.72-5.77 (м, 1Н), 5.99 (уш, 1Н), 6.61 (д, J=5.0 Гц, 1Н), 6.98 (с, 1Н), 6.99-7.02 (м, 1Н), 7.17 (с, 1Н), 7.44 (с, 1Н), 7.57 (д, J=5.0 Гц, 1Н), 8.03 (д, J=10.0 Гц, 1Н), 8.14 (с, 1Н);
LC-MS (время удерживания: 1.92 мин, метод В), MS m/z 780 (М++Н).
Пример 42: Получение Соединения 42.
Получают по методике получения Соединения 11 из Примера 11, но вместо 3-метоксикоричной кислоты в качестве исходного для Р2 элемента используют 3-этоксикоричную кислоту.
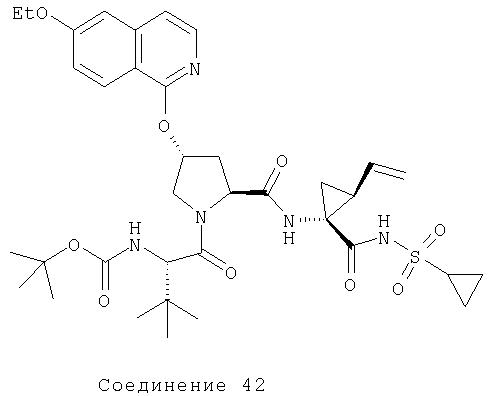
1Н ЯМР (CD3OD) δ м.д. 0.98-1.09 (м, 15Н), 1.24-1.31 (м, 10Н), 1.42-1.46 (м, 1Н), 1.85-1.90 (м, 1Н), 2.19-2.32 (м, 2Н), 2.57-2.63 (м, 1Н), 2.91-2.97 (м, 1Н), 4.03-4.09 (м, 1Н), 4.17 (кв, J=7.0 Гц, 2Н), 4.42 (д, J=11.3 Гц, 1Н), 4.49-4.54 (м, 1Н), 5.12 (д, J=17.4 Гц, 1Н), 5.72-5.78 (м, 1Н), 5.83 (с, 1Н), 7.07-7.10 (м, 1Н), 7.15 (с, 1Н), 7.22 (д, J=5.8 Гц, 1Н), 7.87 (д, J=5.8 Гц, 1Н), 8.08 (д, J=8.8 Гц, 1Н);
MS:(M+H)+ 728.
Подраздел С:
Пример 45: Получение 45.
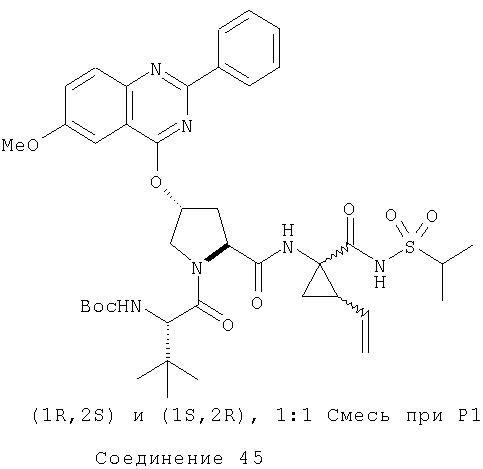
Схема 1

Стадия 1:
К раствору 2-бром-5-метоксибензойной кислоты (1.68 г, 7.27 ммоля) в ДМФА (50 мл) в колбе для работы при нормальном давлении (Chemglass) прибавляют бензамидин (1.25 г, 8.00 ммоля), К2СО3 (6.0 г, 43.6 ммоля) и медный порошок (336 мг, 1.45 ммоля). Реакционную смесь греют при 180°С в течение 1 час. Медь и избыток К2СО3 удаляют фильтрованием в вакууме и промывают МеОН. Фильтрат упаривают и полученный сырой продукт очищают флеш-хроматографией на колонке (SiO2, 5% МеОН в DCM), получают твердое вещество светло-зеленого цвета (1.55 г, выход 84%):1Н ЯМР (ДМСО-d6): δ 3.84 (с, 3Н), 7.26 (д, J=7.8 Гц, 1Н), 7.46 (уш с, 5Н), 7.57 (с, 1Н), 8.38 (ушс, 1Н);
MS m/z (МН+) 253.
Стадия 2:
К суспензии Вос-цис-гидроксипролин-ОМе (2.0 г, 8.15 ммоля) и продукта из Примера 45, Стадия 1 (2.26 г, 8.97 ммоля) в ТГФ (82 мл) при 0°С прибавляют Ph3Р и диизопропил азокарбоксилат (1.98 г, 8.97 ммоля). Перемешивают при rt в течение 17 час, к реакционной смеси прибавляют EtOAc (100 мл) и промывают Н2О (50 мл).
Водный слой отделяют и снова экстрагируют EtOAc (2×50 мл). Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают, получают вязкое масло, которое снова растворяют в минимальном количестве EtOAc и прибавляют смесь гексанов для осаждения основной части побочного продукта Ph3РО. Ph3РО отфильтровывают в вакууме, а жидкий фильтрат упаривают. Полученное вязкое масло очищают флеш-хроматографией на колонке (SiO2, 4:1 гекс.: EtOAc), получают твердый продукт белого цвета (1.76 г, выход 45%):1 Н ЯМР (60/40 ротамеры, CDCl3) δ 1.47 (с, 9Н), 2,49-2.55 (м, 1Н), 2.73-2.83 (м, 1Н), 3.80 (с, 1.8Н), 3.81 (с, 1.2Н), 3.96 (с, 3Н), 4.03-4.09 (м, 1Н), 4.54 (т, J=8.0 Гц, 0.6Н), 4.66 (с, J=7.8 Гц), 4.96-5.06 (м, 1Н), 5.97 (уш с, 0.6Н), 6.04 (уш с, 0.4Н), 7.33 (дд, J=6.1, 2.7 Гц, 1Н), 7.46-7.51 (м, 4Н), 7.91 (д, J=9.2 Гц, 1Н), 8.49 (т, J=8.5 Гц, 2Н);
13С ЯМР (ротамеры, CDCl3) δ 21.7, 22.0, 28.3, 28.4, 35.8, 36.8, 52.3, 52.4, 52.6, 55.8, 55.9, 57.9, 58.3, 74.5, 74.9, 80.6, 101.2, 101.3, 115.7, 125.8, 126.0. 128.1, 128.5, 129.7, 130.2, 137.9, 147.8, 153.8, 157.7, 158.0, 158,0, 164.8, 173.1, 173.3;
MS m/z (MH+) 480.
Схема 2
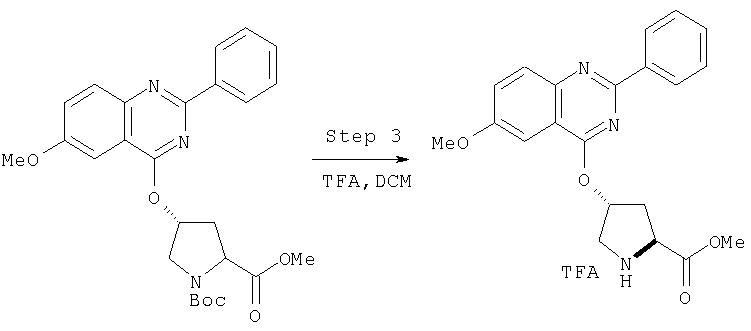


Стадия 3:
Продукт из Примера 45, Стадия 2 (760.0 мг, 1.59 ммоля) растворяют в 50% ТФК в DCM и перемешивают 2 час при rt. Растворитель упаривают и оставшееся коричневое вязкое масло сушат в вакууме в течение ночи. Продукт используют непосредственно на следующей стадии.
Стадия 4:
К раствору коричневого маслообразного продукта из Примера 45, Стадия 3 (963 мг, 1.59 ммоля) и DIPEA (1.23 г, 9.54 ммоля) в DCM (11 мл) прибавляют N-BOC L-tBuGly (440 мг, 1.90 ммоля), HBTU (902 мг, 2.38 ммоля) и HOBt (364 мг, 2.38 ммоля). Перемешивают при rt в течение 14 час, растворитель и избыток DIPEA упаривают и полученное коричневое вязкое масло очищают флеш-хроматографией на колонке (SiO2, 4:1 гекс.: EtOAc), получают твердое вещество белого цвета (0.922 мг, выход в расчете на две стадии 98%):1Н ЯМР (CDCl3/MeOD) δ 0.94 (с, 9Н), 1.15 (с, 9Н), 2.38-2.42 (м, 1Н), 2.60-2.73 (м, 1Н), 3.61 (с, 3Н), 3.83 (с, 3Н), 4.08-4.17 (м, 2Н), 4.25 (д, J=11.5 Гц, 1Н), 4.69 (т, J=8.0 Гц, 1Н), 5.99 (уш с, 1Н), 7.13 (с, 1Н), 7.38 (с, 5Н), 7.80 (д, J=9.0 Гц, 1Н), 8.32 (д, J=5.5 Гц, 1Н);13С ЯМР (CDCl3/MeOD) δ 29.6, 31.4, 31.6, 33.04, 38.2, 39.0, 55.8, 56.9, 59.2, 61.5, 62.1, 78.3, 83.1, 105.0, 119.0, 129.4, 131.5, 131.9, 132.6, 133.8, 141.2, 151.0, 161.4, 161.6, 168.2, 175.2, 175.7; MS m/z (MH+) 593.
Стадия 5:
К раствору продукта из Примера 45, Стадия 4 (409 мг, 0.69 ммоля) в ТГФ (10 мл) прибавляют 1N NaOH (2 мл). Перемешивают при rt в течение 19 час, реакционную смесь подкисляют концентрированной HCl, примерно, до рН 5 и экстрагируют DCM (3×50 мл). Объединенные органические вытяжки сушат MgSO4 и упаривают, получают твердый продукт желтого цвета (370 мг, выход 92%), который после сушки в вакууме используют непосредственно на следующей стадии:1Н ЯМР (CDCl3) δ 1.05 (с, 9Н), 1.25 (с, 9Н), 2.76-2.83 (м, 2Н), 3.94 (с, 3Н), 4.23-4.27 (м, 2Н), 4.41 (д, J=11.6 Гц, 1Н), 4.92 (т, J=7.6 Гц, 1Н), 5.20 (д, J=8.9 Гц, 1Н), 6,08 (уш с, 1Н), 7.31 (с, 1Н), 7.46-7.50 (м, 5Н), 7.93 (д, J=9.15 Гц, 1Н), 8.51 (д, J=7.3 Гц, 2Н); MS m/z (MH+) 579.
Схема 3
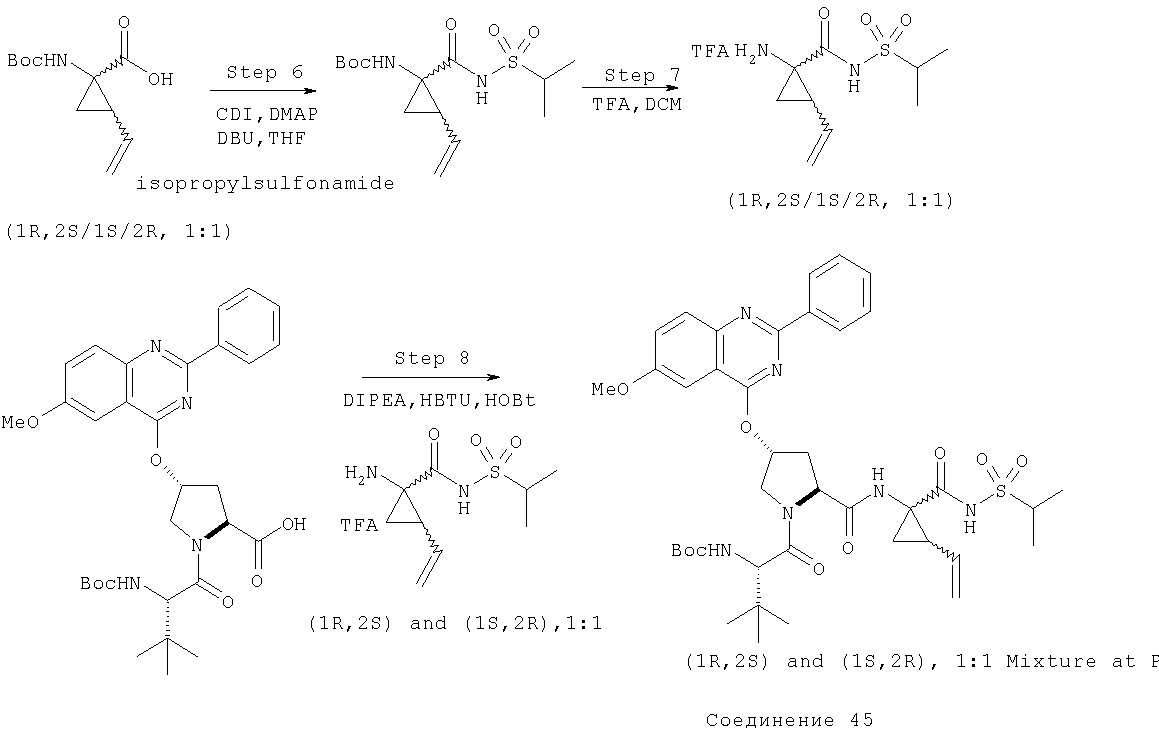
Стадия 6
К раствору N-Boc-винилциклопропанкарбоновой кислоты (1R,2S/1S,2R 1:1 смесь) (1.01 г, 4.46 ммоля) в ТГФ (20 мл) и ДМСО (2 мл) прибавляют CDI (1.08 г, 6.69 ммоля) и DMAP (817 мг, 6.69 ммоля). Перемешивают при 70°С в течение 1 час, реакционную смесь оставляют охлаждаться до комнатной температуры и обрабатывают изопропилсульфонамидом (1.1 г, 8.92 ммоля) и DBU (1.36 г, 8.92 ммоля). Реакционную смесь перемешивают при rt в течение 16 час, упаривают и очищают флеш-хроматографией на колонке (SiO2, 5% МеОН в DCM), получают коричневое вязкое масло (1.4 г, выход 98%):1Н ЯМР (Метанол-d4) δ 1.25 (м, 1Н), 1.33 (д, J=6.7 Гц, 3Н), 1.36 (д, J=6.7 Гц, 3Н), 1.45 (с, 9Н), 1.84 (дд, J=7.6, 5.2 Гц, 1Н), 2.16 (д, J=7.6 Гц, 1Н), 3.58 (уш с, 1Н), 5.08 (д, J=11.6 Гц, 1Н), 5.27 (д, J=15.6 Гц, 1Н), 5.58-5.66 (м, 1Н); MS m/z (МН+) 332.
Стадия 7:
Продукт из Примера 45, Стадия 6 (113 мг, 0.34 ммоля) обрабатывают 50% раствором трифторуксусной кислоты в DCM (10 мл) и перемешивают при rt в течение 1.4 час. Растворитель и избыток трифторуксусной кислоты удаляют в вакууме. Полученное вязкое коричневое масло сушат в вакууме (1.3 г, выход 99%) и используют без дополнительной очистки:1Н ЯМР (ДМСО-d6) δ 1.24 (д, J=6.7 Гц, 3Н), 1.26 (4, J=6.7 Гц, 3Н), 1.54 (дд, J=9.6, 6.6 Гц, 1Н), 1.99 (т, J=6.9 Гц, 1Н), 2.24 (д, J=8.5 Гц, 1Н), 3.58-3.63 (м, 1Н), 5.18 (д, J=10.4 Гц, 1Н), 5.33 (д, J=17.1 Гц, 1Н), 5.61-5.69 (м, 1Н), 8.83 (уш с, 3Н);13С ЯМР (ДМСО-d6) δ 15.2, 15.9, 16.5, 29.9, 41.6, 52.1, 116.0, 118.9, 132.0, 158.2, 167.3; MS m/z (MH+) 233.
Стадия 8:
К смеси продукта из Примера 45, Стадия 5 (117 мг, 0.338 ммоля) и DIPEA (174 мг, 1.35 ммоля) в DCM (5 мл) прибавляют HBTU (128 мг, 0.338 ммоля), HOBt (52 мг, 0.338 ммоля) и продукт из Примера 45, Стадия 7 (130 мг, 0.225 ммоля). Перемешивают при rt в течение 16 час, смесь упаривают и полученное вязкое коричневое масло очищают флеш-хроматографией на колонке (SiO2, 1:3 гекс.: EtOAc, затем 95:5 DCM:МеОН), получают не чисто белый твердый продукт (150 мг, выход 84%). Конечный продукт, Соединение 45, представляет собой смесь изомеров; вариация имеется в Р1 винилциклопропильном фрагменте молекулы (1R, 2S/1S, 2R 1:1 смесь):1Н ЯМР (Метанол-d4) δ 0.92 (уш с, 2H), 1.03 (с, 9Н), 1.17(с, 9Н), 1.27-1.38 (м, 9Н), 1.42-1.46 (м, 1Н), 1.83 (дд, J=8.1, 5.3 Гц, 0.4Н), 1.90 (дд, J=7.9, 5.5 Гц, 0.6Н), 2.24-2.31 (м, 1Н), 2.37-2.45 (м, 1Н), 2.67-2.75 (м, 1Н), 3.73-3.79 (м, 1Н), 3.90 (с, 3Н), 4.21 (дд, J=9.3, 6.0 Гц, 2Н), 4.48 (д, J=11.3 Гц, 1Н), 4.61 (кв, J=8.9 Гц, 1Н), 5.14 (т, J=9.0 Гц, 1Н), 5.33 (т, J=17.9 Гц, 1Н), 5.70-5.76 (м, 1Н), 6.06 (д, J=11.9 Гц, 1Н), 6.61 (д, J=8.9 Гц, 1Н), 7.34 (д, J=2.8 Гц, 1Н), 7.49 (уш с, 5Н), 7.87 (д, J=8.9 Гц, 1Н), 8.46 (д, J=4.3 Гц, 2H);13С ЯМР (Метанол-d4) δ 15.7, 16.1, 16.5, 16.8, 23.9, 27.1, 28.6, 35.8, 36.0, 36.2, 36.3, 36.4, 42.6, 42.8, 54.7, 54.8, 55.5, 56.4, 61.1, 61.2, 80.5, 102.9, 117.0, 118.8, 118.9, 126.8, 129.4, 129.6, 130.2, 131.5, 134.4, 139.2, 148.8, 158.0, 159.3, 159.8, 166.3, 171.1, 175.1, 184.3; MS m/z (MH+) 793.
Пример 46: Получение Соединения 46.

Соединение 46 получают по методике Стадий 1-5 и Стадии 8 из Примера 45 за исключением нижеприведенных модификаций:
Стадия 1:
Модификации: В качестве исходных веществ используют 2-бром-4,5-диметилбензойную кислоту и циклопропилкарбамидин гидрохлорид.
Продукт:
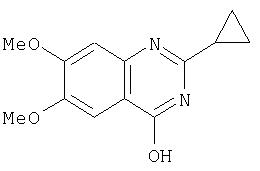
1Н ЯМР (ДМСО-d6) δ 0.97-1.01 (м, 2Н), 1.03-1.06 (м, 2H), 1.90-1.94 (м, 1Н), 3.84 (с, 3Н), 3.87 (с, 3Н), 6.93 (с, 1Н), 7.37 (с, 3Н), 12.28 (с, 1Н);13С ЯМР (ДМСО-d6) δ 9.03, 13.17, 55.47, 55.73, 104.81, 107.27, 113.26, 145.16, 147.48, 154.44, 157.21, 160.89; MS m/z (MH+) 247.
Стадия 2:
Модификации: В качестве исходного вещества вместо продукта из Примера 45, Стадия 1, используют продукт из Примера 46, Стадия 1.
Продукт:

1Н ЯМР (CDCl3) δ 1.00-1.04 (м, 2Н), 1.07-1.11 (м, 2Н), 1.43 (с, 5.4Н), 1.46 (с, 3.6Н), 2.17-2.21 (м, 1Н), 2.37-2.43 (м, 1Н), 2.62-2.69 (м, 1Н), 3.75 (с, 1.8Н), 3.78 (с, 1.2Н), 3.92 (д, J=2.8 Гц, 1Н), 4.00 (с, 3.6Н), 4.01 (с, 2.4Н), 4.48 (т, J=8.0 Гц, 0.6Н), 4.59 (т, J=7.6 Гц, 0.4Н), 5.7 (уш с, 0.6Н), 5.74 (уш с, 0.4Н), 7.18 (с, 1Н), 7.20 (с, 1Н);13С ЯМР (CDCl3) δ 9.6, 9.7, 18.1, 28.3, 28.4, 35.8, 36.7, 52.2, 52.4, 56.3, 57.8, 58.2, 74.0, 74.5, 80.5, 80.6, 101.0, 101.1, 106.3, 108.6, 148.8, 149.1, 153.8, 155.4, 164.4, 165.9, 172.9, 173.2; LC-MS m/z (МН+) 474.
Стадии 3 и 4:
В качестве исходного вещества вместо продукта из Примера 45, Стадия 2, используют продукт из Примера 46, Стадия 2.
Продукт:
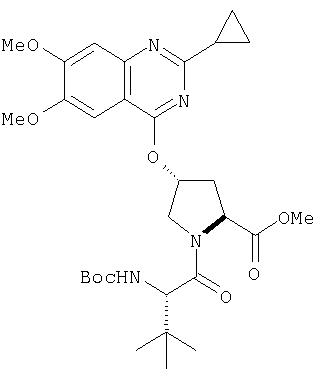
1Н ЯМР (Метанол-d4) δ 1.04 (с, 9Н), 1.08-1.21 (м, 4Н), 1.14 (с, 9Н), 2.17-2.21 (с, 1Н), 2.39-2.41 (м, 1Н), 2.74-2.77 (м, 1Н), 3.77 (с, 3Н), 3.92 (с, 3Н), 3.98 (м, 3Н), 4.09 (дд, J=11.4, 3.8 Гц, 1Н), 4.17 (д, J=8.9 Гц, 1Н), 4.42 (д, J=11.3 Гц, 1Н), 4.76 (т, J=8.2 Гц, 1Н), 5.81 (уш с, 1Н), 6.43 (д, J=8.6 Гц, 1Н), 7.14 (д, J=6.1 Гц, 1Н), 7.27 (д, J=5.8 Гц, 1Н);13С ЯМР (Метанол-d4) δ 10.0, 10.3, 18.6, 26.9, 28.5, 28.8, 35.8, 36.1, 38.9, 52.4, 54.9, 56.7, 59.6, 60.5, 76.6, 80.4, 102.7, 106.2, 109.9, 149.8, 150.7, 157.6, 166.0, 167.3, 173.5, 173.6; MS m/z (MH+) 587.
Стадия 5:
В качестве исходного вещества вместо продукта из Примера 45, Стадия 4, используют продукт из Примера 46, Стадия 4.
Продукт:

1Н ЯМР (Метанол-d4) δ 1.03 (с, 9Н), 1.13(с, 9Н), 1.20-1.23 (м, 4Н), 2.15-2.19 (м, 1Н), 2.40-2.45 (м, 1Н), 2.70-2.76 (м, 1Н), 3.90 (с, 3Н), 3.96 (с, 3Н), 4.08 (дд, J=11.4, 3.8 Гц, 1Н), 4.17 (д, J=5,8 Гц, 1Н), 4.37 (д, J=11.3 Гц, 1Н), 4.71 (т, J=8.1 Гц, 1Н), 5.77 (уш с, 1Н), 7.09 (с, 1Н), 7.20 (с, 1Н);13С1Н ЯМР (Метанол-d4) δ 10.2, 10.5, 18.6, 26.9, 28.5, 28.8, 36.0, 36.3, 54.9, 56.8, 59.7, 60.4, 76.8, 80.4, 102.6, 105.9, 109.9, 126.9, 127.9, 149.3, 150.8, 157.65, 157.8, 166.1, 167.3, 173.3, 175.1; MS m/z (MH+) 573.
Стадия 8:
В качестве исходного вещества вместо продукта из Примера 45, Стадия 5, используют продукт из Примера 46, Стадия 5.
Конечный продукт. Соединение 46, представляет собой смесь изомеров; вариация имеется в Р1 винилциклопропильном фрагменте молекулы (1R, 2S/1S, 2R 1:1 смесь).
Продукт:

1Н ЯМР (Метанол-d4) δ 1.03 (с, 9Н), 1.05-1.09 (м, 4Н), 1.16 (с, 4.5Н), 1.17 (с, 4.5Н), 1.19-1.22 (м, 1Н), 1.31 (д, J=6.7 Гц, 2Н), 1.33-1.38 (м, 7Н), 1.18-1.89 (м, 1Н), 2.15-2.20 (м, 2Н), 2.35-2.44 (м, 1Н), 3.23 (кв, J=7.4 Гц, 1Н), 3.70-3.75 (м, 1Н), 3.91 (с, 3Н), 3.98 (с, 3Н), 4.08-4.13 (м, 2Н), 4.16 (дд, J=8.9, 3.1 Гц, 1Н), 4.38 (т, J=13.1 Гц, 1Н), 4.58-4.62 (м, 1Н), 4.06 (м, 1Н), 5.29 (т, J=15.2 Гц, 1Н), 5.83 (уш с, 1Н), 7.15 (с, 1Н), 7.27 (д, J=4.3 Гц, 1Н); MS m/z (MH+) 787.
Пример 47: Получение Соединения 47.

Соединение 47 получают по методике, применяемой для получения Соединения 45 из Примера 45 (аналогичные стадии). В качестве исходных берут орто-бромбензойную кислоту, а также гидрохлорид (1R-амино-2S-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты. Соединение 47: МН+=761.
Пример 48: Получение Соединения 48.
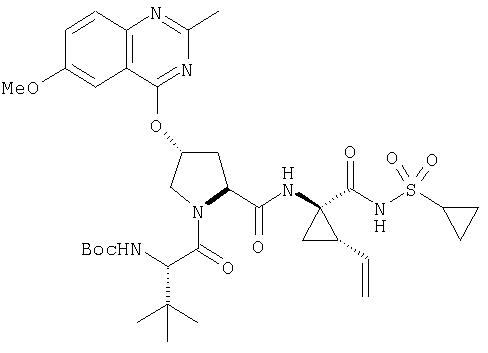
Соединение 48 получают по методике, описанной в Примере 45, Стадии 1-5 и Стадия 8, со следующими модификациями:
Стадия 1:
Модификации: В качестве исходных веществ используют ацетамидина гидрохлорид и 2-бром-5-метилбензойную кислоту.
Продукт:
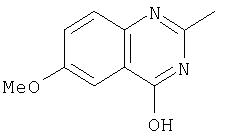
1Н ЯМР (ДМСО-d6) δ 2.31 (с, 3Н), 3.85 (с, 3Н), 7.36 (д, J=6.2 Гц, 1Н), 7.37 (с, 1Н), 7.51 (д, J=7.8 Гц, 1Н), 12.15 (с, 1Н);13С ЯМР (ДМСО-d6) δ 21.11, 55.41, 105.57, 121.22, 123.59, 128.12, 143.34, 151.68, 157.00, 161.45; LC-MS m/e (MH+) 191.
Стадия 2:
Модификации: В качестве исходного вещества вместо продукта из Примера 45, Стадия 1, используют продукт из Примера 48, Стадия 1.
Продукт:

1Н ЯМР (CDCl3) δ 1.43 (с, 5.4Н), 1.45 (с, 3.6Н), 2.38-2.45 (м, 1Н), 2.62-2.71 (с, 1Н), 2.66 (с, 1.8Н), 2.68 (с, 1.2Н), 3.77 (1.8H), 3.79 (с, 1.2Н), 3.92 (с, 3Н), 3.93-3.98 (м, 2Н), 4.49 (т, J=8.0 Гц, 0.6Н), 4.61 (т, J=7.8 Гц, 0.4Н), 5.82 (т, J=2.1 Гц. 0.6Н), 5.89 (т, J=2.3 Гц, 0.4Н), 7.26 (дд, J=4.7, 3.2 Гц, 1Н), 7.42 (дд, J=6.3, 2.8 Гц, 1Н), 7.75 (д, J=9.15 Гц, 1H);13С ЯМР (CDCl3) δ 26.1, 28.3, 28.4, 35.8, 36.7, 52.2, 52.2, 52.4, 52.5, 55.7, 55.8, 57.9, 58.2, 74.1, 74.7, 80.6, 101.0, 101.2, 114.9, 125.6, 125.9, 128.6, 147.3, 153.8, 154.5, 157.6, 157.6, 161.2, 164.6, 173.0, 173.3; LC-MS m/e (MH+) 418.
Стадии 3 и 4:
Модификации: В качестве исходного вещества вместо продукта из Примера 45, Стадия 2, используют продукт из Примера 48, Стадия 2. Продукт:

1Н ЯМР (MeOD) δ 1.03 (с, 9Н), 1.07 (с, 9Н), 2.38-2.42 (м. 1Н), 2.68 (с, 3Н), 2.80 (кв, J=7.8 Гц, 1Н), 3.76 (с, 3Н), 3.89 (с, 3Н), 4.07 (дд, J=11.9, 3.4 Гц, 1Н), 4.13 (уш с, 1Н), 4.55 (д, J=12.2 Гц, 1Н), 4.78 (т, J=8.7 Гц, 1Н), 5.93 (с, 1Н), 7.37 (д, J=2.75 Гц, 1Н), 7.48-7.51 (м, 2Н), 7.70 (д, J=5.7 Гц, 1Н);13С ЯМР (MeOD) δ 25.6, 26.9, 28.4, 28.8, 35.9, 52.8, 55.0, 56.4, 59.7, 60.6, 77.2, 80.4, 102.9, 111.6, 116.5, 127.0, 128.4, 147.5, 162.7, 166.4, 173.6. LC-MS m/e (MH+) 531.
Стадия 5:
Модификации: В качестве исходного вещества вместо продукта из Примера 45, Стадия 4, используют продукт из Примера 48, Стадия 4.
Продукт:

1Н ЯМР (MeOD) δ 1.03 (с, 9Н), 1.08 (с, 9Н), 2.41-2.46 (м, 1Н), 2.68 (с, 3Н), 2.81 (кв, J=8.1 Гц, 1Н), 3.89 (с, 3Н), 4.07 (дд, J=11.8, 3.2 Гц, 1Н), 4.18 (д, J=5.5 Гц, 1Н), 4.52 (д, J=11.9 Гц, 1Н), 4.74 (т, J=8.7 Гц. 1Н), 5.93 (уш с, 1Н), 7.37 (д, J=2,81 Гц, 1Н), 7.49 (дд, J=9.2, 2.4 Гц, 1Н), 7.71 (д, J=9.2 Гц, 1Н);13С ЯМР (MeOD) δ 25.7, 26.9, 28.5, 36.1, 55.0, 56.4, 59.7, 60.5, 77.1, 80.4, 103.0, 116.5, 127.0, 128.5, 147.7, 157.8, 159.6, 162.7, 166.4, 173.5, 174.9; LC-MS m/e (MH+) 517.
Пример 48: Получение Соединения 48

К раствору продукта из Примера 48, Стадия 5 (45.8 мг, 0.089 ммоля), гидрохлорида (1R-амино-2S-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (21.0 мг, 0.089 ммоля) и DIEA (34.5 мг, 0.267 ммоля) в DCM (1 мл) прибавляют HATU (44.0 мг, 0.116 ммоля). Перемешивают при rt в течение ночи, реакционную смесь промывают 5% водным раствором NaHCO3 (1 мл). Водный слой экстрагируют 2×2 мл DCM. Органические вытяжки промывают 5% водным раствором лимонной кислоты (1 мл), рассолом, сушат MgSO4, упаривают и очищают обращенно-фазовой преп. ВЭЖХ. В процессе очистки теряется N-BOC защитная группа во фрагменте Р3 трет-лейцина молекулы:1Н ЯМР (MeOD) δ 1.07-1.12 (м, 2Н), 1.14 (с, 2Н), 1.14-1.16 (м, 2 Н), 1.17 (с, 9Н), 1.20-1.30 (м, 3Н), 1.45 (дд, J=9.46, 5.49 Гц, 1Н), 1.56 (с, 1Н), 1.92 (дд, J=8.20, 5.60 Гц, 1Н), 2.25-2.31 (м, 1Н), 2.39-2.45 (м, 1 Н), 2.73 (м, 1Н), 2.76 (с, 3Н), 2.93-2.97 (м, 1Н), 3.94 (с, 1Н), 3.96 (с, 3Н), 4.07 (с, 1Н), 4.21 (д, J=3.97 Гц, 0.4Н), 4.23 (д, J=3.97 Гц, 0.6Н), 4.31 (м, 1Н), 4.73 (дд, J=10.38, 7.02 Гц, 1Н), 5.15 (дд, J=10.38, 1.52 Гц, 1Н), 5.32 (дд, J=1.71, 1.52 Гц, 1Н), 5.71-5.78 (м, 1Н), 6.11 (т, J=3.51 Гц, 1Н), 7.46 (д, J=2.75 Гц, 1Н), 7.67 (д, J=3.06 Гц, 0.4Н), 7.69 (д, J=3.05 Гц, 0.6Н), 7.82 (с, 06Н), 7.84 (с, 04Н),
Пример 49: Получение Соединения 49.

Соединение 49 получают по методике, описанной при получении Соединения 48, за исключением того, что в качестве исходных применяют продукт из Примера 46, Стадия 5 и гидрохлорид (1R-амино-2S-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты. В процессе очистки преп. ВЭЖХ теряется N-BOC защитная группа во фрагменте Р3 трет-лейцина молекулы:1Н ЯМР (MeOD) δ 1.09 (м, 2Н), 1.14 (д, J=3.97 Гц, 2Н), 1.17 (с, 9Н), 1.25 (м, 3Н), 1.37 (м, 3Н), 1.44 (дд, J=9.31, 5.65 Гц, 2Н), 1.57 (с, 1Н), 1.92 (дд, J=8.09, 5.65 Гц, 1Н), 2.28 (дд, J=17.70, 8.55 Гц, 1Н), 2.32 (м, 1Н), 2.68 (дд, J=14.19, 7.78 Гц. 1Н), 2.95 (м, 1Н), 3.98 (с, 3Н), 4.06 (с, 3Н), 4.08 (с, 1Н), 4.22 (д, J=2.75 Гц, 1Н), 4.70 (дд, J=9.77, 7.32 Гц, 1Н), 5.15 (дд, J=10.38, 1.53 Гц, 1Н), 5.32 (дд, J=7.40, 1.22 Гц, 1Н), 5.74 (м, 1Н), 6.04 (м, 1Н), 7.24 (с, 1Н), 7.37 (с, 1Н).
Пример 50: Получение Соединения 50.
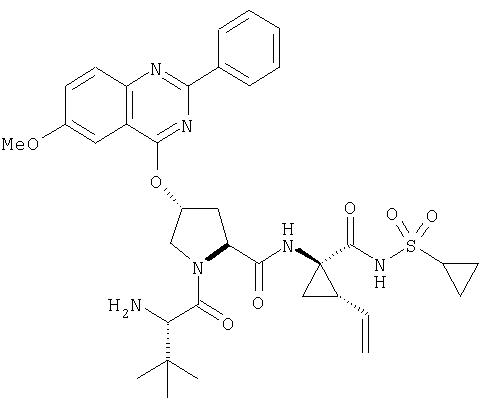
Соединение 50 получают по методике, описанной при получении Соединения 48, за исключением того, что в качестве исходных применяют продукт из Примера 45, Стадия 5, и гидрохлорид (1R-амино-2S-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты. В процессе очистки методом преп. ВЭЖХ теряется N-BOC защитная группа во фрагменте Р3 трет-лейцина молекулы:1Н ЯМР (MeOD) δ 1.10 (м, 2Н), 1.14 (с, 1Н), 1.15 (д, J=3.36 Гц, 1Н), 1.17 (д, J=3.05 Гц, 9Н), 1.22 (м, 1H), 1.27 (м, 2Н), 1.46 (дд, J=9.46, 5.49 Гц, 1Н), 1.56 (с, 1Н), 1.93 (дд, J=8.24, 5.49 Гц, 1Н), 2.29 (кв, J=8.55 Гц, 1Н), 2.48 (м, 1Н), 2.78 (дд, J=13.89, 8.09 Гц, 1Н), 2.97 (м, 1Н), 3.96 (с, 2Н), 4.07 (с, 1Н), 4.32(д, J=2.14 Гц, 2Н). 4.76 (д, J=7.02 Гц, 1Н), 4.78 (м, 1Н), 4.86 (д, J=3.05 Гц, 1Н), 5.32 (дд, J=17.09,1.22 Гц, 1Н), 5.75 (м, 1Н), 6.24 (д, J=2.44 Гц, 1Н), 7.45 (д, J=2.75 Гц, 1Н), 7.52 (м, 3Н), 7.61 (дд, J=9.16, 2.75 Гц, 1Н), 7.96 (д, J=9.16 Гц, 1Н).
Пример 51: Получение Соединения 51.

Соединение 51 получают по методике, описанной в Примере 45, Стадии 1-5 и Стадия 8, со следующими модификациями:
Стадия 1:
Модификации: В качестве исходных веществ используют трифторамидин и 2-бром-4,5-диметилбензойную кислоту.
Продукт:
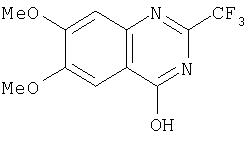
1Н ЯМР (ДМСО-d6) δ 3.92 (с, 3Н), 3.94 (с, 3Н), 7.33 (с, 1Н), 7.50 (с, 1Н), 13.40 (уш с, 1Н);13С ЯМР (ДМСО-d6) δ 55.8, 56.1, 104.9, 108.7, 150.2, 155.0; LC-MS m/e (MH+) 275.
Стадия 2:
Модификации: В качестве исходного вещества вместо продукта из Примера 45, Стадия 1, используют продукт из Примера 51, Стадия 1.
Продукт:
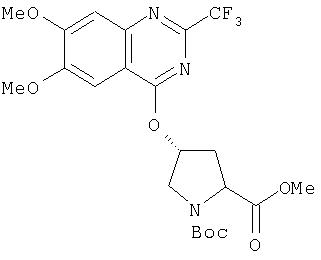
1Н ЯМР (CDCl3) δ 1.42 (с, 3.6Н), 1.44 (с, 5.4Н), 2.42-2.49 (м, 1Н), 2.67-2.73 (м, 1Н), 3.37 (с, 1.2Н), 3.78 (с, 1.8Н), 3.97 (т, J=6.5 Гц, 1Н), 4.02 (с, 2.4Н), 4.04 (с, 3.6Н), 4.48 (т, J=7.9 Гц, 0.6Н), 4.60 (т, J=7.7 Гц, 0.4Н), 5.86 (уш с, 0.6Н), 5.90 (уш с, 0.4H), 7.27-7.29 (м, 1Н), 7.38-7.44 (м, 1Н);13С ЯМР (CDCl3) δ 8.2, 28.3, 35.7, 36.7, 52.1, 52.2, 52.4, 56.5, 57.8, 58.2, 75.5, 76.0, 80.7, 100.8, 107.6, 111.0, 119.7, 148.2, 150.2, 151.4, 153.8, 154.5, 156.4, 165.1, 172.7, 173.0; LC-MS m/e (MH+) 502.
Стадии 3 и 4:
Модификации: В качестве исходного вещества вместо продукта из Примера 45, Стадия 2, используют продукт из Примера 51, Стадия 2.
Продукт:
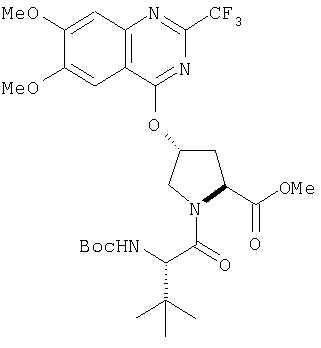
1Н ЯМР (MeOD) δ 1.03 (с, 9Н), 1.08 (с, 9Н), 2.41-2.45 (м, 1Н), 2.80-2.84 (м, 1H), 3.76 (с, 3Н), 3.96 (с, 3Н), 4.00 (с, 3Н), 4.10-4.14 (м, 2Н), 4.52 (д, J=11.6 Гц, 1Н), 4.80 (т, J=8.7 Гц, 1Н), 5.92 (уш с, 1Н), 7.35 (уш с, 2Н);13С ЯМР (MeOD) δ 26.9, 28.4, 28.8, 35.7, 36.0, 52.8, 54.8, 56.9, 59.6, 60.7, 77.9, 80.3, 102.2, 107.9, 112.4, 120.3, 149.3, 153.2, 157.8, 158.3, 173.5; LC-MS m/e (MH+) 615.
Стадия 5

1Н ЯМР (MeOD) δ 1.03 (с, 9Н), 1.09 (с, 9Н), 2.44-2.49 (м, 1Н), 2.80-2.84 (м, 1Н), 3.97 (с, 3Н), 4.01 (с, 3Н), 4.10-4.24 (м, 3Н), 4.50 (д, J=11.9 Гц, 1Н), 4.76 (т, J=7.9 Гц, 1Н), 5.93 (уш с, 1Н), 7.36 (уш с, 2Н);13С ЯМР (MeOD) δ 26.9, 28.4, 28.8, 36.0, 36.1, 54.8, 56.9, 57.0, 60.6, 77.9, 80.3, 102.3, 108.0, 112.5, 120.3, 149.3, 151.3, 153.2, 158.2, 158.3, 166.7, 173.5; LC-MS m/e (MH+) 601.
Стадия 8:
Модификации: В качестве исходного вместо продукта из Примера 45, Стадия 5 используют продукт из Примера 51, Стадия 5. Конечный продукт. Соединение 51, представляет собой смесь изомеров; вариация имеется в Р1 винилциклопропильном фрагменте молекулы (1R, 2S/1S, 2R 1:1 смесь).
Продукт:
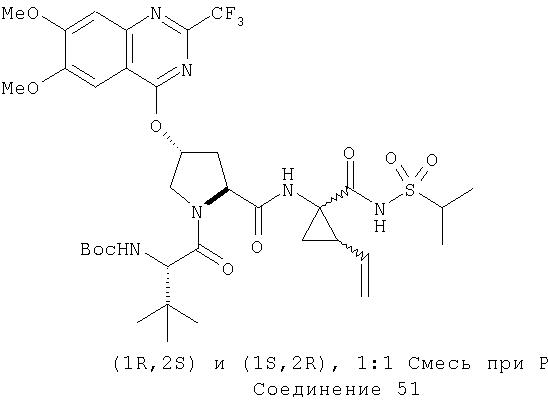
1Н ЯМР (ДМСО-d6) δ 0.23 (с, 4.5Н), 0.23 (с, 4.5Н), 0.35 (с, 4.5Н), 0.36 (с, 4.5Н), 0.45-0.59 (м, 8Н), 0.63-0.66 (м, 1Н), 1.04 (дд, J=8.2, 5.2 Гц, 1Н), 1.10 (дд, J=8.2, 5.5 Гц, 1Н), 1.47-1.53 (м, 1Н), 1.58-1.61 (м, 1Н), 1.87-1.90 (м, 1Н), 2.95-3.01 (м, 1Н), 3.17 (с, 1.5Н), 3.18 (с, 1.5Н), 3.22 (с, 3Н), 3.37 (уш с, 2Н), 3.68 (кв, J=5.9 Гц, 1Н), 3.82 (кв, J=8.6 Гц, 1Н), 4.33-4.37 (м, 1Н), 4.54 (т, J=16.5 Гц, 1Н), 4.93 (кв, J=8.9 Гц, 1Н), 5.17 (д, J=15.9 Гц, 1Н), 6.53 (с, 1Н), 6.58 (с, 1Н);13С ЯМР (ДМСО-d6) δ 12.8, 13.2, 13.7, 13.9, 19.5, 20.6, 21.1, 24.3, 25.6, 32.9, 33.1, 33.4, 33.6, 36.1, 39.7, 39.9, 51.8, 51.9, 52.4, 54.0, 54.2, 57.7, 57.7, 58.1, 58.3, 75.1, 75.3, 77.5, 84.1, 99.2, 105.2, 107.9, 109.5, 116.0, 116.1, 118.7, 123.9, 127.4, 131.5, 146.5, 148.6, 150.3, 155.1, 155.5, 163.7, 164.7, 168.2, 168.3, 170.7, 172.2; LC-MS m/e (MH+) 815.
Пример 52: Получение Соединения 52.
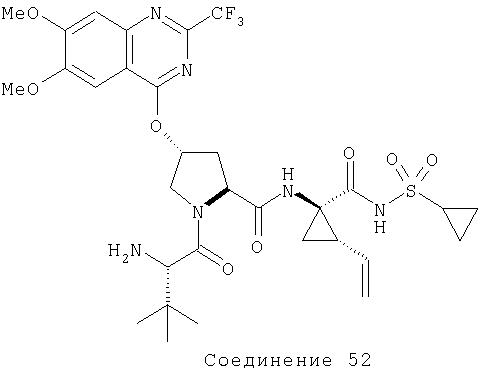
Соединение 52 получают по методике, описанной при получении Соединения 48, за исключением того, что в качестве исходных применяют продукт из Примера 51, Стадия 5, и гидрохлорид (1R-амино-2S-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты. В процессе очистки методом преп. ВЭЖХ теряется N-BOC защитная группа во фрагменте Р3 трет-лейцина молекулы:1Н ЯМР (MeOD) δ 1.11 (м, 3Н), 1.17 (с, 9Н), 1.25 (м, 3Н), 1.46 (дд, J=9.46, 5.49 Гц, 1Н), 1.92 (дд, J=8.24, 5.49 Гц, 1Н), 2.28 (кв, J=8.95 Гц, 1Н), 2.42 (м, 1Н), 2.72 (дд, J=14.19, 7.17 Гц, 1Н), 2.96 (м, 1Н), 4.01 (с, 3Н). 4.04 (м, 5Н), 4.24 (м, 2Н), 4.73 (дд, J=10.22, 7.17 Гц, 1Н), 5.15 (дд, J=10.53, 1.37 Гц, 1Н), 5.32 (д, J=17.09 Гц, 1Н), 5.75 (м, 1Н), 6.07 (с, 1Н), 7.41 (с, 1Н), 7.47 (с, 1Н).
Пример 53: Получение Соединения 53.

Схема 1

Стадия 1:
К раствору ((1R,2S)/(1S,2R), 1:1 смесь) прибавляют трифторацетат (1R-амино-2S-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (626 мг, 1.82 ммоля) в DCM (17 мл), DIPEA (555 мг, 4.29 ммоля) в DCM (17 мл), HATU (754 мг, 1.98 ммоля) и (2S, 4R) Fmoc-4-амино-1-boc-пирролидин-2-карбоновую кислоту (747 мг, 1,65 ммоля). Перемешивают при rt в течение 24 час, смесь промывают 1N HCl (10 мл), 5% водным раствором NaHCO3 (4 мл). Каждый водный слой экстрагируют DCM (25 мл). Объединенные DCM вытяжки сушат MgSO4 и упаривают. Полученное коричневое вязкое масло очищают флеш-хроматографией на колонке (SiO2, 95:5 DCM: МеОН), получают твердое вещество желтого цвета (822 мг, выход 75%):1Н ЯМР (ДМСО-d6) δ 1.04-1.09 (м, 3Н), 1.15-1.27 (м, 4Н), 1.38-1.44 (м, 7Н), 1.47 (с, 9Н), 1.84 (дд, J=8.2, 5.2 Гц, 1Н), 2.01-2.30 (м, 4Н), 2.90-2.98 (м, 1Н), 3.64-3.71 (м, 1Н), 4.16-4.22 (м, 4Н), 4.39 (уш с, 2Н), 5.13 (дд, J=10.7, 0.9 Гц, 1Н), 3.31 (д, J=17.1 Гц, 1Н), 5.72-5.79 (m, 1Н), 7.31 (т, J=7.3 Гц, 3Н), 7.38 (т, J=7.5 Гц, 3Н), 7.64 (д, J=7.02 Гц, 3Н), 7.79 (д, J=7.63 Гц, 3Н); LC-MS m/z (Na+МН+) 687.
Стадия 2
К продукту из Примера 53, Стадия 1 (500 мг, 0.752 ммоля) прибавляют 50% раствор ТФК в DCM (10 мл). Перемешивают при rt в течение 0.5 час, образовавшуюся коричневую реакционную смесь упаривают, получают твердое вещество коричневого цвета (489 мг, выход 84%):1Н ЯМР (ДМСО-d6) δ 1.03-1.19 (м, 4Н), 1.24-1.26 (м, 1Н), 1.35 (дд, J=9.5, 5.5 Гц, 1Н), 1.91-1.96 (м, 1Н), 2.22-2.30 (м, 1Н), 2.40 (уш с, 1Н), 2.93-2.98 (м, 1Н), 3.60 (уш с, 1Н), 4.21 (т, J=5.6 Гц, 2Н), 4.47 (уш с, 3Н), 5.17 (д, J=9.2 Гц, 1Н), 5.32 (д, J=17.1 Гц, 1Н), 5.64-5.67 (м, 1Н), 7.31 (т, J=7.3 Гц, 3Н), 7.39 (т, J=7.5 Гц, 3Н), 7.63 (д, J=7.3 Гц, 2Н), 7.80 (д, J=7.3 Гц, 2Н); LC-MS m/z (MH+) 565.
Схема 2

Стадия 3:
К раствору продукта из Примера 53, Стадия 2 (260 мг, 0.383 ммоля) в DCM (4 мл) прибавляют DIPEA (218 мг, 1.69 ммоля), HATU (165 мг, 0.422 ммоля) и N-BOC L-tBuGly (100 мг, 0.422 ммоля). Перемешивают при rt в течение 16 час, к реакционной смеси прибавляют Н2O (3 мл), подкисляют 1N HCI до рН 1. Водный слой экстрагируют DCM (2×15 мл). Объединенные органические вытяжки промывают 5% NaHCO3 (3 мл), рассолом (5 мл), сушат MgSO4 и упаривают. Полученное вязкое коричневое масло очищают флеш-хроматографией на колонке (SiO2, 95:5 DCM:МеОН), получают продукт в виде твердой коричневой пены (281 мг, выход 94%):1Н ЯМР (ДМСО-d6) δ 0.96-1.08 (м, 4Н), 1.05 (с, 9Н), 1.15-1.26 (м, 2Н), 1.35-1.38 (м, 5Н), 1.42 (с, 9Н), 1.85 (дд, J=9.5, 5.5 Гц, 1Н), 2.07 (уш с, 1Н), 2.22 (кв, J=8.7 Гц, 1Н), 2.92-2.95 (м, 1Н), 3.90 (уш с, 1Н), 4.20 (д, J=6.4 Гц, 3Н), 4.29-4.39 (м, 5Н), 5.13 (д, J=10.7, 1Н), 5.31 (дд, J=18.0, 5.8 Гц, 1Н), 5.70-5.77 (м, 1Н), 7.30 (т, J=7.3 Гц, 3Н), 7.39 (т, J=7.3 Гц, 4Н), 7.63 (дд, J=6.7, 2.8 Гц, 3Н), 8.80 (д, J=7.63 Гц, 3Н); LC-MS m/z (MH+) 678.
Стадия 4:
К продукту из Примера 53, Стадия 3, прибавляют 10% раствор пиперидина в ДМФА (3.3 мл). Перемешивают при rt в течение 14 час, растворитель отгоняют и полученное коричневое вязкое масло очищают флеш - хроматографией на колонке (SiO2, 95:5 DCM: МеОН), выделяют 1R,2S P1 с Rf высочайшей степени чистоты в виде бледно-желтого твердого вещества (31 мг). Другой изомер выделяют в виде смеси и не используют: LC-MS m/z (МН+) 556.
Схема 3
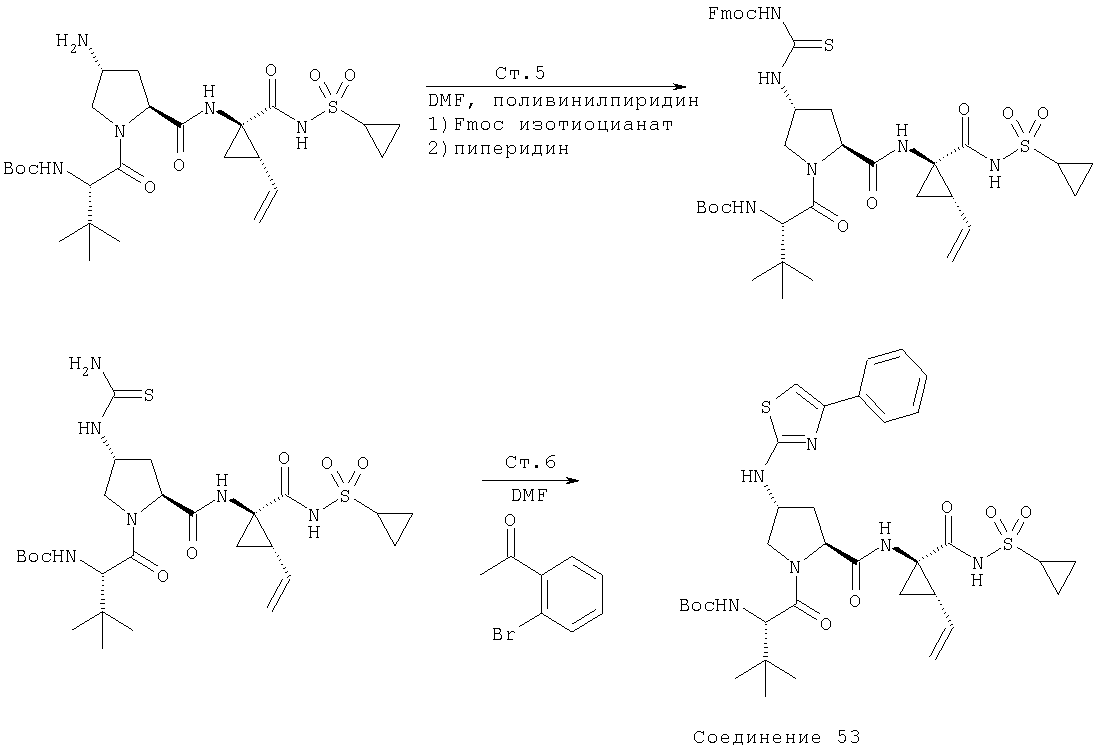
Стадии 5 и 6:
К раствору продукта из Примера 53, Стадия 4 в ДМФА (2 мл) прибавляют поливинилпиридин (13 мг) и Fmoc-изотиоцианат. Перемешивают при rt в течение 14 час, к реакционной смеси прибавляют пиперидин (172 мг, 2.02 ммоля). Перемешивают реакционную смесь при rt еще 6 час, после чего ее упаривают и сушат в вакууме в течение ночи. Сырой остаток снова растворяют в ДМФА (2 мл), прибавляют 2-бромацетофенон и перемешивают при rt еще 14h. Реакционную смесь упаривают и остаток очищают флеш-хроматографией на колонке (SiO2, 95:5 DCM:МеОН), получают Соединение 53 в виде твердого вещества светло-желтого цвета (21.9 мг, выход 50%):1Н ЯМР (ДМСО-d6) δ 0.87-0.92 (м, 1Н), 1.05 (уш с, 13Н), 1.16-1.25 (м, 4Н), 1.34-1.38 (м, 2Н), 1.42 (с, 9Н), 1.87 (т, J=6.6 Гц, 1Н), 2.22-2.25 (м, 2Н), 2.48 (т, J=10.7 Гц, 1Н), 2.93 (уш с, 1Н), 3.04 (кв, J=7.3 Гц, 1Н), 3.30-3.31 (м, 2Н), 3.43-3.49 (м, 1Н), 4.01 (д, J=10.4 Гц, 1Н), 4.07-4.12 (м, 1Н), 4.27 (т, J=9.5 Гц, 1Н), 4.44 (т, J=7.0 Гц, 1Н), 4.58 (уш с, 1Н), 5.11 (д, J=10.1 Гц, 1Н), 5.30 (дд, J=16.8, 9.6 Гц, 1Н), 5.73-5.78 (м, 1Н), 6.69 (д, J=8.2 Гц, 1Н), 6.86 (с, 1Н), 7.25 (т, J=7.3 Гц, 1Н), 7.35 (т, J=7.63 Гц, 2Н), 7.82 (д, J=8.2 Гц, 2Н); LC-MS m/z (МН+) 715.
Условия препаративной обращенно-фазовой ВЭЖХ для соединений 55-155:
Колонка Waters Xterra Prep MS C18, 5 мм (это означает размер частиц 5 микрон), 30 мм × 50 мм
Растворитель А: 10% МеОН, 90% H2O, 10 мМ NH4ОАс
Растворитель В: 90% МеОН, 10% Н2O,10 мМ NH4ОАс
Скорость потока: 50 мл/мин
Градиент: от 0% В до 100% В за 10 мин, выдерживают при 100% В в течение 4 мин
Пример 55: Получение Соединения 55.
Схема 1

Стадия 1:
К раствору Соединения 11 (1.5 г, 2.10 ммоля) в DCE (25 мл) прибавляют ТФК (TFA) (25 мл). Перемешивают при rt в течение 15 мин, реакционную смесь упаривают. Остаток - красное вязкое масло - снова растворяют в DCE (50 мл) и снова упаривают. Затем его снова растворяют в DCM (15 мл) и обрабатывают 1N раствором HCl в Et2O (25 мл). Полученную суспензию охлаждают до 0°С, фильтруют в вакууме, промывают Et2O и сушат в вакуум - шкафу, получают продукт стадии 1 в виде твердого вещества белого цвета (1.4 г, выход 97%):1Н ЯМР (CD3OD, 500 МГц) 5 1.07-11.2 (м, 3Н), 1.14 (т, J=4.12 Гц, 1Н), 1.17 (с, 9Н), 1.22 (дд, J=10.53, 4.43 Гц, 1Н), 1.21-1.27 (м, 2Н), 1.42 (дд. J=9.61, 5.34 Гц, 1Н), 1.91 (дд, J=7.93, 5.49 Гц, 1Н), 2.27 (кв, J=8.85 Гц, 1Н), 2.32-2.38 (м, 1Н), 2.70 (дд, J=13.43, 6.71 Гц, 1Н), 2.93-2.98 (м, 1Н), 3.96 (с, 3Н), 4.09 (с, 1Н), 4.14 (дд, J=12.21, 3.66 Гц, 1Н), 4.32-4.35 (м, 1Н), 4.69 (дд, J=10.53, 6.87 Гц, 1Н), 5.14 (дд, J=10.38, 1.53 Гц, 1Н), 5.31 (дд, J=17.40, 1.22 Гц, 1Н), 5.70-5.77 (м, 1Н), 5.90 (т, J=3.51 Гц, 1Н), 7.24-7.27 (м, 1Н), 7.29 (д, J=4.27 Гц, 1Н), 7.39 (т, J=4.88 Гц, 1Н), 7.90 (д, J=6.10 Гц, 1Н), 8.19 (м, 1Н), 9.22 (с, 1Н).
Стадия 2:
К раствору из смеси продукта из Примера 55, Стадия 1 (70.0 мг, 0.108 ммоля) и DIEA (41.8 мг, 0.323 ммоля) в DCM (2 мл) прибавляют уксусный ангидрид (33.0 мг, 0.323 ммоля). Перемешивают при rt в течение 14 час, растворитель отгоняют и продукт очищают обращенно - фазовой преп. ВЭЖХ, получают Соединение 55 (39.1 мг, выход 14%):1Н ЯМР (CD3OD, 500 МГц) δ 1.00-1.03 (м, 1Н), 1.06 (с, 9Н), 1.07-1.10 (м, 1Н), 1.21-1.28 (м, 2Н), 1.43 (дд, J=9.46, 5.19 Гц,1Н), 1.88 (дд, J=8.55, 5.49 Гц, 1Н), 2.23 (кв, J=8.85 Гц, 1Н), 2.27-2.32 (м, 1Н), 2.59 (дд, J=13.73, 7.02 Гц, 1Н), 2.92-2.97 (м, 1Н), 3.93 (с, 3Н), 4.12 (дд, J=11.90, 3.97 Гц, 1Н), 4.35 (д, J=11.60 Гц, 1Н), 4.51 (дд, J=10.38, 7.02 Гц, 1Н), 4.61 (дд, J=5.80, 3.05 Гц, 1Н), 4.80 (д, J=4.27 Гц, 1Н), 4.88 (д, J=3.96 Гц, 1Н), 5.12 (дд, J=10.38, 1.83 Гц, 1Н), 5.29 (дд, J=17.24, 1.37 Гц, 1Н), 5.73-5.78 (м, 1Н), 5.84 (т, J=3.66 Гц, 1Н), 7.15 (дд, J=8.85, 2.44 Гц, 1Н), 7.19 (д, J=2.44 Гц, 1Н), 7.25 (д, J=6.10 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 8.06 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.49 мин.), MS m/z 656 (МН+).
Пример 56: Получение Соединения 56.
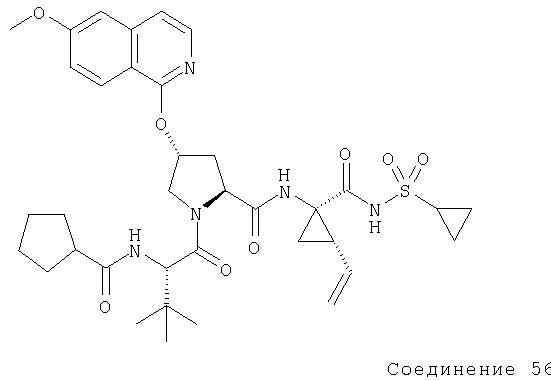
Соединение 56 получают по методике получения Соединения 55 со следующими модификациями:
Модификации: При использовании в качестве исходного циклопентанкарбонилхлорида получают Соединение 56 (18.0 мг, выход 24%):1Н ЯМР (CD3OD, 500 МГц) δ 1.00-1.03 (м, 1Н), 1.05 (с, 9Н), 1.06-1.10 (м, 2Н), 1.24-1.27 (м, 2Н), 1.25-1.61 (м, 9Н), 1.80-1.83 (м, 1Н), 1.88 (дд, J=8.24, 5.49 Гц, 1Н), 2.22-2.31 (м, 2Н), 2.58-2.65 (м, 2Н), 2.93-2.98 (м, 1Н), 3.92 (с, 3Н), 4.10 (дд, J=11.90, 3.66 Гц, 1Н), 4.35 (д, J=11.91 Гц, 1Н), 4.52 (дд, J=10.38, 7.02 Гц, 1Н), 4.65 (д, J=9.46 Гц, 1Н), 4.80 (д, J=5.49 Гц, 1Н), 4.88 (д, J=5.19 Гц, 1Н), 5.13 (дд, J=10.37, 1.83 Гц, 1Н), 5.30 (дд, J=16.80, 1.22 Гц, 1Н), 5.73-5.78 (м, 1Н), 5.84 (т, J=4.27 Гц, 1Н), 7.11 (дд, J=9.16, 2.44 Гц, 1Н), 7.19 (д, J=2.44 Гц, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 8.05 (д, J=9.16 Гц, 1Н)); LC-MS (время удерживания: 1.71 мин), MS m/z 710 (MH+).
Пример 57: Получение Соединения 57.
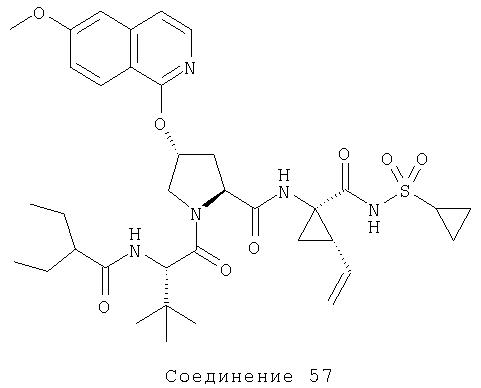
Соединение 57 получают по методике получения Соединения 55 со следующими модификациями:
Модификации: При использовании в качестве исходного 2-этилбутирилилхлорида получают Соединение 57 (20.7 мг, выход 27%):1Н ЯМР (CD3OD, 500 МГц) δ 0.66 (т, J=7.32 Гц, 3Н), 0.85 (т, J=7.32 Гц, 3Н), 1.02-1.05 (м, 1Н), 1.07 (с, 9Н), 1.10-1.12 (м, 1Н), 1.24-1.33 (м, 4Н), 1.36-1.39 (м, 1Н), 1.43 (дд, J=9.46, 5.19 Гц, 1Н), 1.48-1.51 (м, 1Н), 1.88 (дд, J=8.24, 5.19 Гц, 1Н), 2.12-2.14 (м, 1Н), 2.22 (кв, J=8.85 Гц, 1Н), 2.26-2.30 (м, 1Н), 2.59 (дд, J=13.73, 6.71 Гц, 1Н), 2.94-2.97 (м, 1Н), 3.92 (с, 3Н), 4.11 (дд, J=11.90, 3.66 Гц, 1Н), 4.40 (д, J=11.90 Гц, 1Н), 4.50 (дд, J=10.68, 7.02 Гц, 1Н), 4.75 (д, J=9.46 Гц, 1Н), 4.81 (д, J=9.16 Гц, 1Н), 4.89 (д, J=9.16 Гц, 1Н), 5.12 (дд, J=10.38, 1.53 Гц, 1Н), 5.29 (дд, J=17.09, 1.22 Гц, 1Н), 5.72-5.79 (м, 1Н), 5.85 (т, J=3.66 Гц, 1Н), 7.08 (дд, J=9.16, 2.44 Гц, 1Н), 7.19 (д, J=2.44 Гц, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 7.98 (д, J=9.16 Гц, 1Н), 8.02 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.73 мин), MS m/z 712 (MH+).
Пример 58: Получение Соединения 58.

К раствору смеси продукта из Примера 55, Стадия 1 (70.0 мг, 0.108 ммоля), DIEA (41.8 мг, 0.323 ммоля) и циклопропануксусной кислоты (16.2 мг, 0.162 ммоля) в DCM (2 мл) прибавляют HATU (61.6 мг, 0.162 ммоля). Реакционную смесь перемешивают при rt в течение ночи, промывают 5% водным NaHCO3 (1 мл). Водный слой экстрагируют DCM (2×2 мл). Объединенные органические вытяжки промывают 5% водным раствором лимонной кислоты (2 мл), рассолом, сушат MgSO4 и упаривают. Продукт очищают обращенно - фазовой препаративной ВЭЖХ, получают Соединение 58 (21.9 мг, выход 29%):1Н ЯМР (CD3OD, 500 МГц) δ 0.11-0.14 (м, 2Н), 0.43-0.47 (м, 2Н), 0.87-0.09 (м, 1Н), 1.01-1.04 (м, 1Н), 1.07 (с, 9Н), 1.09-1.12 (м, 1Н), 1.23-1.27 (м, 2Н), 1.45 (дд, J=9.46, 5.49 Гц, 1H), 1.88 (дд, J=8.24, 5.49 Гц, 1Н), 2.03 (д, J=7.32 Гц, 2Н), 2.23 (кв, J=8.75 Гц, 1Н), 2.27-2.31 (м, 1Н), 2.59 (дд, J=13.73, 7.02 Гц, 1Н), 2.92-2.96 (м,1Н), 3.93 (м, 3Н), 4.13 (дд, J=11.90, 3.97 Гц, 1Н), 4.34 (д, J=11.90 Гц, 1Н), 4.53 (дд, J=10.38, 7.02 Гц, 1Н), 4.66 (д, J=9.46 Гц, 1Н), 4.81 (д, J=6.10 Гц, 1Н), 4.89 (д, J=6.10 Гц, 1Н), 5.12 (дд, J=10.37, 1.52 Гц, 1Н), 5.30 (дд, J=17.09, 1.22 Гц, 1Н), 5.75-5.81 (м,1Н), 5.86 (с, 1Н), 7.12 (дд, J=9.16,2.44 Гц. 1Н), 7.19 (д, J=2.44 Гц, 1Н), 7.81 (д, J=9.46 Гц, 1Н), 7.89 (д, J=5.80 Гц, 1Н), 8.06 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.63 мин.), MS m/z 696 (MH+).
Пример 59: Получение Соединения 59.
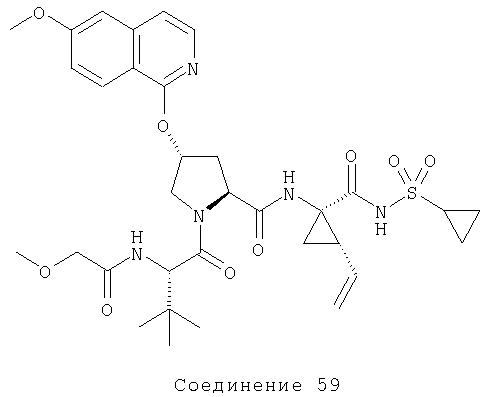
Соединение 59 получают по методике получения Соединения 58 со следующими модификациями:
Модификации: При использовании в качестве исходного вещества метоксиуксусной кислоты получают Соединение 59 (23.5 мг, выход 32%):1Н ЯМР (CD3OD, 500 МГц) δ 0.99-1.04 (м, 2Н), 1.06 (с, 9Н), 1.09-1.12 (м, 1Н), 1.22-1.27 (м, 2Н), 1.45 (дд, J=9.46, 5.49 Гц, 1Н), 1.88 (дд, J=8.24, 5.49 Гц, 1Н), 2.22 (кв. J=8.85 Гц, 1Н), 2.29-2.32 (м, 1Н), 2.60 (дд, J=13.89, 6.87 Гц, 1Н), 2.92-2.97 (м, 1Н), 3.35 (с, 3Н), 3.70 (д, J=15.26 Гц, 1Н), 3.84 (д, J=15.26 Гц, 1Н), 3.93 (с, 3Н), 4.13 (дд, J=11.90. 3.97 Гц, 1Н), 4.32 (д, J=11.60 Гц, 1Н), 4.54 (дд, J=10.38, 7.02 Гц, 1Н), 4.65 (с, 1Н), 4.81 (д, J=7.32 Гц, 1Н), 4.89 (д, J=7.32 Гц, 1Н), 5.12 (д, J=10.38 Гц, 1Н), 5.30 (д, J=16.79 Гц, 1Н), 5.74-5.81 (м, 1Н), 5.86 (т, J=3.36 Гц, 1Н), 7.14 (дд, J=9.00, 2.59 Гц, 1Н), 7.19 (д, J=2.44 Гц, 1Н), 7.26 (д, J=6.10 Гц, 1Н), 7.89 (д, J=6.10 Гц, 1Н), 8.04 (д, J=9.16 Гц, 1Н);
LC-MS (время удерживания: 1.54 мин), MS m/z 686 (MH+).
Пример 60: Получение Соединения 60.

Соединение 60 получают по методу получения Соединения 58 со следующими модификациями:
Модификации: В качестве исходного соединения берут (+)-метоксиуксусную кислоту и получают Соединение 60 (23.8 мг, выход 27%):1Н ЯМР (CD3OD, 500 МГц) δ 0.78 (д, J=7.02 Гц, 3Н), 0.83-0.88 (м, 2Н), 0.92 (т, J=7.17 Гц, 6Н), 0.94-0.98 (м, 1Н), 1.00-1.03 (м, 2Н), 1.06 (с, 9Н), 1.07-1.11(м, 1Н), 1.22-1.26 (м, 2Н), 1.31-1.36 (м, 2Н), 1.45 (дд, J=9.46, 5.49 Гц, 1Н), 1.63-1.68 (м, 2Н), 1.89 (дд, J=8.09, 5.34 Гц, 1Н), 2.01-2.04 (м, 1Н), 2.17-2.21 (м, 1Н), 2.24 (кв, J=9.00 Гц, 2Н), 2.28-2.33 (м, 1Н), 2.60 (дд, J=13.73, 7.02 Гц, 1Н), 2.93-2.98 (м, 1Н), 3.15-3.20 (м, 1Н), 3.77 (д, J=15.26 Гц, 1Н), 3.87 (д, J=1 5.26 Гц, 1Н), 3.93 (с, 3Н), 4.12 (дд, J=11.90, 3.66 Гц, 1Н), 4.32 (д, J=11.90 Гц, 1Н), 4.56 (дд, J=10.38, 7.02 Гц, 1Н), 4.65 (д, J=9.77 Гц, 1Н), 4.81 (д, J=5.80 Гц, 1Н), 4.89 (д, J=5.80 Гц, 1Н), 5.13 (дд, J=10.22, 1.68 Гц, 1Н), 5.30 (дд, J=17.09, 1.53 Гц, 1Н), 5.75-5.79 (м, 1Н), 5.85 (т, J=3.66 Гц, 1Н), 7.14 (дд, J=9.16, 2.44 Гц, 1Н), 7.19 (д, J=2.44 Гц, 1Н), 7.26 (д, J=5.80 Гц, 1Н), 7.52 (д, J=9.77 Гц, 1Н), 7.89 (д, J=5.80 Гц, 1Н), 8.03 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 2.043 мин), MS m/z 810 (MH+).
Пример 61: Получение Соединения 61.

Соединение 61 получают по методу получения Соединения 58 со следующими модификациями:
Модификации: В качестве исходного соединения берут (-)-метоксиуксусную кислоту и получают Соединение 61 (24.6 мг, выход 30%):1Н ЯМР (CD3OD, 500 МГц) δ 0.78 (д, J=7.02 Гц, 3Н), 0.82-084 (м, 1Н), 0.88 (дд, J=8.39, 3.81 Гц, 1Н), 0.91 (д, J=7.01 Гц, 3Н), 0.92 (д, J=6.41 Гц, 3Н), 0.94-0.99 (м, 2Н), 1.00-1.03 (м, 2Н), 1.06 (с, 9Н), 1.08-1.10 (м, 1Н), 1.23-1.26 (м, 2Н), 1.30-1.37 (м, 2Н), 1.44 (дд, J=9.61, 5.34 Гц, 1Н), 1.62-1.68 (м, 2Н), 1.89 (дд, J=8.24, 5.49 Гц, 1Н), 1.98-2.02 (м, 1Н), 2.13-2.16 (м, 1Н), 2.24 (кв, J=8.85 Гц, 1Н), 2.28-2.32 (м, 1Н), 2.60 (дд, J=13.73, 7.02 Гц, 1Н), 2.94-2.98 (м, 1Н), 3.08-3.13 (м, 1Н), 3.63 (д, J=15.56 Гц, 1Н), 3.93 (с, 3Н), 4.11 (дд, J=12 05, 3.81 Гц, 1Н), 4.32 (д, J=11.90 Гц, 1Н), 4.56 (дд, J=10.38, 7.02 Гц, 1Н), 4.62 (д, J=9.46 Гц, 1Н), 4.81 (д, J=6.41 Гц, 1Н), 4.89 (д, J=6.72 Гц, 1Н), 5.13 (дд, J=10.38, 1.83 Гц, 1Н), 5.30 (дд, J=17.09, 1.23 Гц, 1Н), 5.76-5.80 (м, 1Н), 5.85 (т, J=3.51 Гц, 1Н), 7.14 (дд, J=9.00, 2.59 Гц, 1Н), 7.20 (д, J=2.44 Гц, 1Н), 7.26 (д, J=5.80 Гц, 1Н), 7.52 (д, J=9.77 Гц, 1Н), 7.89 (д, J=6.10 Гц, 1Н), 8.04 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 2.05 мин), MS m/z 810 (MH+ ).
Пример 62: Получение Соединения 62.

Соединение 62 получают по методу получения Соединения 58 со следующими модификациями:
Модификации: В качестве исходного соединения берут бицикло[1.1.1]пентан-2-карбоновую кислоту и получают Соединение 62 (35.1 мг, выход 45%):1Н ЯМР (CD3OD, 500 МГц) δ 1.03 (д, J=5.49 Гц, 1Н), 1.06 (с, 9Н), 1.08-1.11 (м, 3Н), 1.23-1.29 (м, 3Н), 1.37 (дд, J=7.02, 3.36 Гц, 13Н), 1.46 (дд, J=9.46, 5.19 Гц, 1Н), 1.65 (дд, J=9.77, 2.14 Гц, 1Н), 1.69 (д, J=2.14 Гц, 1Н), 1.72 (дд, J=7.32, 3.05 Гц, 1Н), 1.88 (дд, J=8.09, 5.34 Гц, 1Н), 2.12 (дд, J=9.77, 3.05 Гц, 1Н), 2.23 (д, J=8.83 Гц, 1Н), 2.27-2.31 (м, 1Н), 2.60 (т, J=6.87 Гц, 1Н), 2.63 (д, J=1.83 Гц, 2Н), 2.68 (д, J=7.63 Гц, 1Н), 2.93-2.96 (м, 1Н), 3.23 (кв, J=7.43 Гц, 2Н), 3.70-3.75 (м, 2Н), 3.93 (с, 3Н), 4.11 (дд, J=11.90, 3.66 Гц, 1Н), 4.35 (д, J=11.90 Гц, 1Н), 4.53 (дд, J=10.53, 6.87 Гц, 1Н), 4.71 (д, J=9.46 Гц, 1Н), 4.79 (д, J=5.80 Гц, 2Н), 4.87 (д, J=5.49 Гц, 2Н), 5.13 (дд, J=10.38, 1.83 Гц, 1Н), 5.30 (дд, J=17.24, 1.37 Гц, 1Н), 5.74-5.79 (м, 1Н), 5.86 (т, J=3.20 Гц, 1Н), 7.12 (дд, J=9.16, 2.44 Гц, 1Н), 7.19 (д, J=2.44 Гц, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.72 (д, J=9.46 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.05 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.16 мин), MS m/z 708 (MH+).
Пример 63: Получение Соединения 63.

Соединение 63 получают по методу получения Соединения 58 со следующими модификациями:
Модификации: В качестве исходного соединения берут пиразин-2-карбоновую кислоту и получают Соединение 63 (42.3 мг, выход 54%):1Н ЯМР (CD3OD, 500 МГц) δ 1.00-1.04 (м, 3Н), 1.05-1.09 (м, 1Н), 1.10 (c, 9H), 1,15 (с, 1Н), 1.18-1.22 (м, 2Н), 1.43 (дд, J=9.46, 5,49 Гц, 1Н), 1.88 (дд, J=7.93, 5.49 Гц, 1Н), 2.17 (кв, J=8.65 Гц, 1Н), 2.37-2.42 (м, 1Н), 2.64 (дд, J=13.73, 7.32 Гц, 1Н), 2.91-2.95 (м, 1Н), 3.88 (с, 3Н), 3.93 (д, J=3.35 Гц, 1Н), 4.13 (дд, J=11.90, 3.36 Гц, 1Н), 4.46 (д, J=11.90 Гц, 1Н), 4.61 (дд, J=10.07, 7.32 Гц, 1Н), 4.76 (с, 1Н), 4.80 (д, J=7.63 Гц, 1Н), 4.88 (д, J=7.93 Гц, 1Н), 5.09 (д, J=10.38 Гц, 1Н), 5.27 (д, J=17.09 Гц, 1Н), 5.77-5.83 (м, 1Н), 5.85 (с, 1Н), 6.85 (дд, J=9.16, 2.44 Гц, 1Н), 7.08 (д, J=2.14 Гц, 1Н), 7.23 (д, J=5.80 Гц, 1Н), 7.87 (д, J=8.85 Гц, 1Н), 7.90 (д, J=6.10 Гц, 1Н), 8.57 (д, J=1.53 Гц, 1Н), 8.73 (д, J=2.44 Гц, 1Н), 8.81 (с, 1Н).
Пример 64: Получение Соединения 64.

К раствору смеси продукта Стадии 1, Пример 55 (70.0 мг, 0.108 ммоля) и DIEA (41.8 мг, 0.323 ммоля) в DCM (2 мл) прибавляют бензилхлорформиат (55.1 мг, 0.323 ммоля). Перемешивают при rt в течение 14 час, растворитель отгоняют и продукт очищают обращенно - фазовой препаративной ВЭЖХ, получают Соединение 64 (26.9 мг, выход 31%):1Н ЯМР (CD3OD, 500 МГц) δ 1.00 (д, J=2.14 Гц, 1Н), 1.02 (д, J=5.80 Гц, 1Н), 1.04 (с, 9Н), 1.08-1.14 (м, 1Н), 1.16 (д, J=6.71 Гц, 1Н), 1.18-1.22 (м, 2Н), 1.43 (дд, J=9.46, 5.19 Гц, 1H), 1.87 (дд, J=8.09, 5.34 Гц, 1Н), 2.17-2.22 (м, 1Н), 2.30-2.35 (м, 1Н), 2.62 (дд, J=13.73, 7.02 Гц, 1Н), 2.90-2.95 (м, 1Н), 3.88 (с, 3Н), 4.08 (дд, J=11.90, 3.66 Гц, 1Н), 4.31 (с, 1Н), 4.43 (д, J=11.60 Гц, 1Н), 4.55 (дд, J=10.07, 7.32 Гц, 1Н), 4.74 (д, J=12.21 Гц, 1Н), 4.81 (д, J=6.10 Гц, 1Н), 4.89 (д, J=5.79 Гц, 1Н), 5.10 (д, J=9.16 Гц, 1Н), 5.16 (с, 1Н), 5.28 (д, J=17.09 Гц, 1Н), 5.75-5.81 (м, 1Н), 5.83 (с, 1Н), 7.07 (дд, J=9.16, 2.44 Гц, 1Н), 7.17 (д, J=2.44 Гц, 1Н), 7.20 (д, J=7.32 Гц, 2Н), 7.25 (т, J=5.65 Гц, ЗН), 7.30-7.33 (м, 1Н), 7.34-7.37 (м, 2 Н), 7.89 (д, J=5.80 Гц, 1Н), 8.07 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.79 мин), MS m/z 748 (MH+).
Пример 65: Получение Соединения 65.
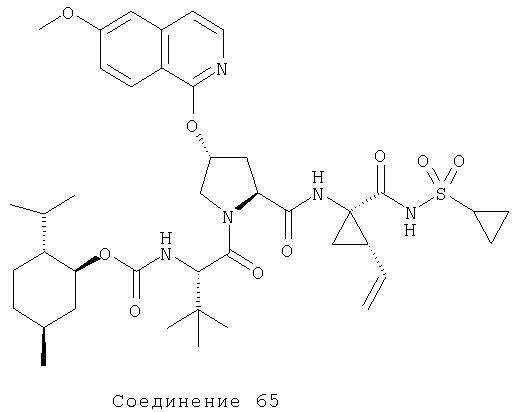
Соединение 65 получают по методу получения Соединения 64 со следующими модификациями:
Модификации: В качестве исходного соединения берут (+)-метилхлорформиат и получают Соединение 65 (28.8 мг, выход 36%):1Н ЯМР (CD3OD, 500 МГц) δ 0.72 (д, J=6.71 Гц, 3Н), 0.80 (т, J=5.80 Гц, 6Н), 0.87 (д, J=7.02 Гц, 4Н), 0.90-0.95 (м, 6Н), 0.98-1.02 (м, 5Н), 1.05 (с, 9Н), 1.07-1.12 (м. 2Н), 1.18-1.23 (м, 2Н), 1.32-1.38 (м, 3Н), 1.41 (дд, J=9.46, 5.19 Гц, 1Н), 1.46-1.48 (м, 1Н), 1.63-1.71 (м, 5Н), 1.85 (дд, J=7.93, 5.49 Гц, 1Н), 1.89-1.93 (м, 1Н), 2.00-2.03 (м, 1Н), 2.15 (кв, J=8.70 Гц, 1Н), 2.34-2.38 (м, 1Н), 2.61 (дд, J=13.73, 7.33 Гц, 1Н), 2.89-2.93 (м, 1Н), 3.73 (с, 2Н), 3.92 (с, 3Н), 4.10 (дд, J=11.60, 3.36 Гц, 1Н), 4.33 (с, 1Н), 4.41 (д, J=11.29 Гц, 1Н), 4.46-4.52 (м, 1Н), 4.54 (дд, J=9.76, 7.90 Гц. 1Н), 4.81 (д, J=5.80 Гц, 1Н), 4.89 (м, 1Н), 5.08 (д, J=11.60 Гц, 1Н), 5.26 (д, J=17.09 Гц, 1Н), 5.77-5.81 (м, 1Н), 5.83 (д, J=3.97 Гц, 1Н), 7.11 (дд, J=11.29, 1.83 Гц, 1Н), 7.18 (д, J=1.83 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 8.08 (д, J=8.85 Гц, 1Н); LC-MS (время удерживания: 2.06 мин), MS m/z 796 (MH+).
Пример 66: Получение Соединения 66.

Соединение 66 получают по методу получения Соединения 64 со следующими модификациями:
Модификации: В качестве исходного соединения берут (-)-метилхлорформиат и получают Соединение 66 (26.9 мг, выход 31%):1Н ЯМР (CD3OD, 500 МГц) δ 0.35 (д, J=6.41 Гц, 1Н), 0.51 (д, J=6.71 Гц, 2Н), 0.68 (д, J=6.71 Гц, 1Н), 0.73 (д, J=7.02 Гц, 2Н), 0.77-0.82 (м, 4Н), 0.88-0.98 (м, 10Н), 1.0-1.03 (м, 2Н), 1.05 (с, 9Н), 1.09-1.18 (м, 3Н), 1.25-1.29 (м, 1Н), 1.3-1.41 (м, 3Н), 1.60-1.71 (м, 3Н), 1.82-1.89 (м, 3Н), 2.00-2.04 (м, J=2.14 Гц, 1Н), 2.10 (кв, J=8.24 Гц, 1Н), 2.39-2.43 (м, 1Н), 2.61 (дд, J=14.04, 7.32 Гц, 1Н), 2.87-2.91 (м, 1Н), 3.73 (с, 1Н), 3.92 (с, 3Н), 4.13 (дд, J=11.75, 3.51 Гц, 1Н), 4.22-4.27 (м, 2Н), 4.30 (с, 1Н), 4.39 (д, J=11.90 Гц, 1Н), 4.48-4.55 (м, 1Н), 4.79 (д, J=5.19 Гц, 1Н), 4.87 (д, J=4.27 Гц, 1Н), 5.05 (д, J=10.07 Гц, 1Н), 5.22 (д, J=16.79 Гц, 1Н), 5.78-5.85 (м, 2Н), 7.09 (дд, J=9.16, 1.83 Гц, 1Н), 7.17 (д, J=1.83 Гц, 1Н), 7.23 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.09 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 2.05 мин), MS m/z 796 (MH+ ).
Пример 67: Получение Соединения 67.
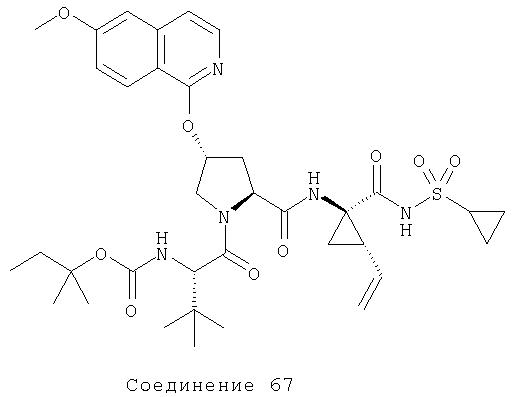
Соединение 67 получают по методу получения Соединения 64 со следующими модификациями:
Модификации: В качестве исходного соединения берут ди-трет-амилкарбонат и получают Соединение 67 (35.3 мг, выход 41%):1Н ЯМР (CD3OD, 500 МГц) δ 0.78 (т, J=7.17 Гц, 3Н), 0.89-0.95 (м, 6Н), 1.04 (с, 9Н), 1.005-1.09 (м. 3Н), 1.15 (с, 1Н), 1.22 (д, J=12.51 Гц, 6Н), 1.40 (с, 2Н), 1.42-1.46 (м, 1Н), 1.48 (с, 3Н), 1.56-1.66 (м, 2Н), 1.78 (кв, J=7.63 Гц, 1Н), 1.84 (кв, J=7.53 Гц, 1Н), 1.88 (д, J=5.80 Гц, 1Н), 2.22 (д, J=8.55 Гц, 1Н), 2.27-2.31 (м, 1Н), 2.61 (дд, J=13.73, 7.02 Гц, 1Н), 2.91-2.96 (м, 1Н), 3.92 (с, 3Н), 4.08 (д, J=12.21 Гц, 1Н), 4.26 (д, J=9.16 Гц, 1Н), 4.42 (д, J=11.29 Гц, 1Н), 4.52 (т, J=7.93 Гц, 1Н}, 5.12 (д, J=10.07 Гц, 1Н), 5.29 (д, J=17.09 Гц, 1Н), 5.73-5.80 (м, 1Н), 5.84 (с, 1Н), 6.57 (д, J=8.85 Гц, 1Н), 7.09 (д, J=8.54 Гц, 1Н), 7.18 (с, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.89 (д, J=5.80 Гц, 1Н), 8.08 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.82 мин), MS m/z 728 (МН+).
Пример 68: Получение Соединения 68.
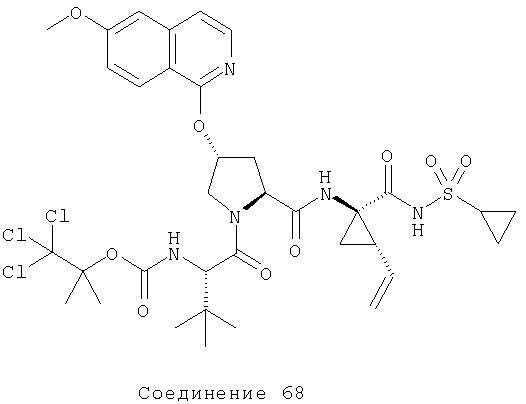
Соединение 68 получают по методу получения Соединения 64 со следующими модификациями:
Модификации: В качестве исходного соединения берут 2,2,2-трихлор-1, 1-диметилформиат и получают Соединение 68 (30.5 мг, выход 37%):1Н ЯМР (CD3OD, 500 МГц) δ 0.99 (с, 9Н), 1.04 (с, 6Н), 1.08-1.09 (м, 3Н), 1.23-1.26 (м, 3Н), 1.44 (с, 2Н), 1.46 (д, J=5.80 Гц, 1Н), 1.71 (с, 2Н), 2.23-2.33 (м, 2Н), 2.60-2.64 (м, 1Н), 2.93-2.96 (м, 1Н), 3.70 (м, 1Н), 3.71 (с, 3Н), 3.93 (с, 3Н), 4.04-4.06 (м, 2Н), 4.27 (д, J=9.16 Гц, 1Н), 4.41 (д, J=11.60 Гц, 1Н), 4.57 (д, J=10.98, 6.11 Гц, 1Н), 5.14 (д, J=12.21 Гц, 1Н), 5.32 (д, J=17.70 Гц, 1Н), 5.75-5.80 (м, 1Н), 5.84 (с, 1Н), 7.10 (дд, J=9.16, 2.44 Гц, 1Н), 7.19 (д, J=2.44 Гц, 1Н), 7.26 (д, J=6.10 Гц, 1Н), 7.90 (д, J=5.80 Гц, 1Н), 8.07 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.95 мин), MS m/z 816 (МН+).
Пример 69: Получение Соединения 69.
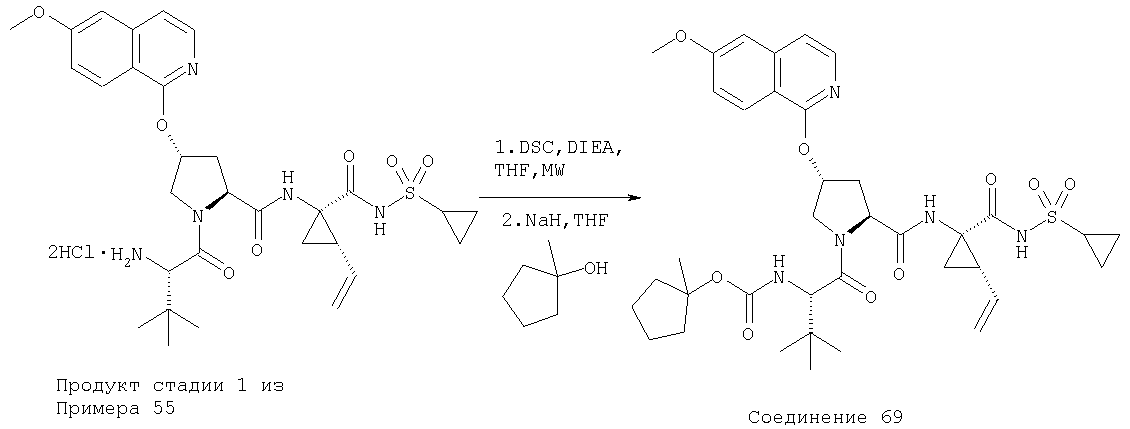
К раствору смеси продукта Стадии 1, Пример 55 (102 мг, 0.149 ммолл) и DIEA (48.2 мг, 0.373 ммоля) в ТГФ (2 мл) прибавляют N,N'-дисукцинимидилкарбонат (57.1 мг, 0.223 ммоля). Полученную суспензию облучают в микроволновой печи при 80°С в течение 15 мин. Затем прибавляют суспензию 1-метилциклопентоксида натрия, которую получают обработкой при 0°С раствора 1-метилциклопентанола (149.2 мг, 1.49 ммоля) в ТГФ (1 мл) NaH (60% в масле, 59.6 мг, 1.49 ммоля) в течение 15 мин при rt, перемешивают при rt в течение 15 мин, к реакционной смеси прибавляют насыщенный водный раствор хлористого аммония (3 мл) и экстрагируют EtOAc (10 мл). Затем органические вытяжки пропускают через колонку с целитом, упаривают и очищают обращенно - фазовой препаративной хроматографией, получают Соединение 69 (49.0 мг, выход 44%):1Н ЯМР (CD3OD, 500 МГц) δ 0.95-0.98 (м, 3Н), 0.99-1.01 (м, J=12.51 Гц, 1Н), 1.03 (с, 9Н), 1.14-1.18 (м, 2Н), 1.30 (с, 3Н), 1.40-1.47 (м, 3Н), 1.50-1.56 (м, 3Н), 1.60-1.64 (м, 1Н), 1.76-1.81 (м, 1Н), 1.83-1.85 (м, 1Н), 2.10-2.19 (м, 1Н), 2.36-2.43 (м, 1Н), 2.63 (дд, J=14.50, 7.17 Гц, 1Н), 2.86-2.90 (м, 1Н), 3.92 (с, 3Н), 4.09 (д, J=12.51 Гц, 1Н), 4.25 (д, J=1.53 Гц, 1Н), 4.43 (д, J=10.99 Гц, 1Н), 4.51-4.55 (м, 1Н), 5.06 (д, J=11.60 Гц, 1Н), 5.23 (д, J=16.78 Гц, 1Н), 5.80-5.85 (м, J=12.67, 12.67 Гц, 2Н), 7.09 (д, J=8.55 Гц, 1Н), 7.17 (с, 1Н), 7.24 (д, J=5.49 Гц, 1Н), 8.07 (д, J=9.16 Гц, 1Н);
LC-MS (время удерживания: 1.87 мин), MS m/z 740 (MH+).
Пример 70: Получение Соединения 70

Соединение 70 получают по методике получения Соединения 69 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут циклопентанол, получают Соединение 70 (85.1 мг, выход 40%).1Н ЯМР (CD3OD, 500 МГц) δ 0.98 (с, 1Н), 1.00 (д, J=4.88 Гц, 1Н), 1.03 (с, 9Н), 1.06-110 (м, 2Н), 1.24-1.29 (м, 3Н), 1.36-1.40 (м, 2Н), 1.44 (дд, J=9.31, 5.04 Гц, 2Н), 1.57-1.62 (м, 5Н), 1.69-1.73 (м, 2Н), 1.88 (дд, J=8.09, 5.65 Гц, 1Н), 2.22-2.29 (м, 2Н), 2.59-2.62 (м, 1Н), 2.92-2.96 (м, 1Н), 3.93 (с, 3Н), 4.07 (дд, J=10.99, 2.44 Гц, 1Н), 4.29 (с, 1Н), 4.42 (дд, J= 12.51, 1.53 Гц, 1Н), 4.55 (дд, J=9.77, 7.93 Гц, 1Н), 4.68-4.71 (м, 1Н), 4.81 (д, J=8.55 Гц, 1Н), 4.89 (д, J=9.46 Гц, 1Н), 5.13 (д, J=10.68 Гц, 1Н), 5.30 (д, J=16.48 Гц, 1Н), 5.73-5.78 (м, 1Н), 5.84 (с, 1Н), 7.12 (дд, J=9.15, 1.83 Гц, 1Н), 7.20 (д, J=2.14 Гц, 1Н), 7.27 (д, J=5.80 Гц, 1Н), 7.89 (д, J=5.80 Гц, 1Н), 8.09 (д, J=8.85 Гц, 1Н); LC-MS (время удерживания: 1.81 мин), MS m/z 726 (MH+).
Пример 71: Получение Соединения 71

Соединение 71 получают по методике получения Соединения 69 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут циклобутанол, получают Соединение 71 (16.2 мг, выход 39%).1Н ЯМР (CD3OD, 500 МГц) δ 0.97 (с, 3Н), 1.03 (с, 9 Н), 1.06-1.08 (м, 2Н), 1.17-1.22 (м, 3Н), 1.37-1.44 (м, 1Н), 1.82-1.85 (м, 1Н), 2.02-2.08 (м, 1Н), 2.15-2.21 (м, 1Н), 2.30-2.36 (м, 1Н), 2.49-2.54 (м, 0.4Н), 2.59-2.67 (м, 0.6Н), 2.90-2.94 (м, 1Н), 3.93 (с, 3Н), 4.09 (дд, J=8.09, 4.73 Гц, 1Н), 4.20 (д, J=10.68 Гц, 0.4Н), 4.38 (д, J=10.68 Гц, 0.6Н), 4.46 (дд, J=10.22, 7.17 Гц, 0.4Н), 4.48-4.56 (м, 1Н), 5.07-5.11 (м, 1Н), 5.26 (д, J=17.24 Гц, 0.4Н), 5.28 (д, J=17.24 Гц, 0.6Н), 5.71-5.79 (м, 1Н), 5.84 (с, 1Н), 7.07 (дд, J=8.39, 2.90 Гц, 1Н), 7.14 (д, J=2.14 Гц, 1Н), 7.20 (д, J=2.44 Гц, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.59 (д, J=5.80 Гц, 1Н), 7.88 (м, 1Н), 8.00 (д, J=9.16 Гц, 0.4Н), 8.07 (д, J=8,85 Гц, 0.6Н)); LC-MS (время удерживания: 1.25 мин), MS m/z 712 (MH+).
Пример 72: Получение Соединения 72

Соединение 72 получают по методике получения Соединения 69 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут 2-фенил-2-пропанол, получают Соединение 72 (19.0 мг, выход 42%).1Н ЯМР (CD3OD, 500 МГц) δ 0.97 (м, 1Н), 1.03 (с, 9Н), 1.06-1.09 (м, 3Н), 1.16-1.22 (м, 4Н), 1.41-1.44 (м, 1Н), 1.57 (с, 3Н), 1.86 (т, J=7.80 Гц, 1Н), 2.14-2.18 (м, 1Н), 2.30-2.35 (м, 1Н), 2.57-2.61 (м, 1Н), 2.90-2.94 (м, 1Н), 3.92 (д, J=4.27 Гц, 1Н), 3.94 (с, 3Н), 4.04 (дд, J=10.99, 3.66 Гц, 1Н), 4.18 (с, 1Н), 4.24 (д, J=10.99 Гц, 1Н), 4.52 (с, 1Н), 5.09 (д, J=10.07 Гц, 1Н), 5.26 (д, J=14.95 Гц, 1Н), 5.78-5.82 (м, 2Н), 7.07-7.12 (м, 2Н), 7.16-7.20 (м, 3Н), 7.23 (д, J=5.19 Гц, 1Н), 7.29 (д, J=7.02 Гц, 2Н), 7.84 (д, J=5.80 Гц, 1Н), 8.03 (д, J=9.46 Гц, 1Н)); LC-MS (время удерживания: 1.84 мин), MS m/z 776 (МН+).
Пример 73: Получение Соединения 73
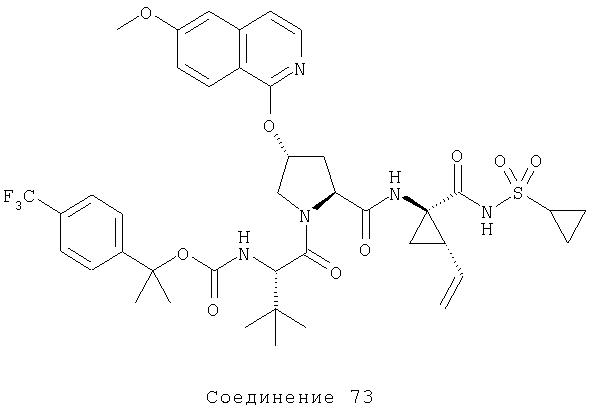
Соединение 73 получают по методике получения Соединения 69 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут 4-(трифторметил)фенилдиметилкарбинол, получают Соединение 73 (22.1 мг, выход 45%).1Н ЯМР (CD3OD, 500 МГц) δ 0.91 (с, 1Н), 0.97-1.00 (м, J=15.56 Гц, 4Н), 1.04 (с, 9Н), 1.07-1.10 (м, 2Н), 1.16-1.20 (м, 3Н), 1.30-1.31 (м, 1Н), 1.41 (дд, J=9.61, 5.34 Гц, 1Н), 1.55 (д, J=7.32 Гц, 6Н), 1.83-1.87 (м, 1Н), 2.11-2.14 (м, 1Н), 2.34-2.39 (м, 1Н), 2.57-2.62 (м, 1Н), 2.89-2.92 (м, J=11.60, 4.27 Гц, 1Н), 3.92 (с, 2Н), 3.94 (с, 3Н), 4.02-4.05 (м, 1Н), 4.17 (с, 1Н), 4.26 (д, J=11.90 Гц, 1Н), 4.53 (т, J=8.85 Гц, 1Н), 5.07 (д, J=10.07 Гц, 1Н), 5.24 (д, J=18.01 Гц, 1Н), 5.78-5.83 (м, 2Н), 7.08 (д, J=7.02 Гц, 1Н), 7.19 (с, 1Н), 7.22 (д, J=5.80 Гц, 1Н), 7.45 (дд, J=13.74, 7.63 Гц, 3Н), 7.60 (д, J=6.41 Гц, 1Н), 7.67 (д, J=7.63 Гц, 1Н), 7.84. (д, J=5.80 Гц, 1Н), 8.02 (д, J=8.54 Гц, 1Н)); LC-MS (время удерживания: 1.92 мин), MS m/z 844 (MH+).
Пример 74: Получение Соединения 74

К раствору смеси продукта стадии 1 из примера 55 (70.0 мг, 0.108 ммоля) и DIEA (41.8 мг, 0.323 ммоля) в DCM (2 мл) прибавляют трет-бутилизоцианат (32.0 мг, 0.323 ммоля). Перемешивают при rt в течение ночи, реакционную смесь упаривают и очищают обращенно - фазовой препаративной ВЭЖХ, получают Соединение 74 (42.3 мг, выход 55%):1Н ЯМР (CD3OD, 500 МГц) δ 0.96-1.00 (м, 1Н), 1.04 (с, 9Н), 1.08-1.10 (м, 3Н), 1.19 (с, 9Н), 1.22-1.31 (м, 2Н), 1.30 (м, 1Н), 1.41 (дд, J=9.46, 5.49 Гц, 1Н) 1.87 (дд, J=8.24, 5.49 Гц, 1Н), 2.20-2.29 (м, 2Н), 2.61 (дд, J=14.04, 6.72 Гц, 1Н), 2.92-2.97 (м, 1Н), 3.92 (с, 3Н), 4.08 (дд, J=11.60, 3.97 Гц, 1Н), 4.36 (с, 1Н), 4.47-4.52 (м, 2Н), 4.81 (д, J=3.36 Гц, 1Н), 4.88 (д, J=8.85 Гц, 1Н), 5.11 (дд, J=10.22, 1.68 Гц, 1Н), 5.28 (дд, J=17.09, 1.53 Гц, 1Н), 5.72-5.76 (м, 1Н), 5.85 (с, 1Н), 7.08 (дд, J=9.16, 2.44 Гц, 1Н), 7.18 (д, J=2.14 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 8.12 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.70 мин), MS m/z 713 (MH+ ).
Пример 75: Получение Соединения 75

Соединение 75 получают по методике получения Соединения 74 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут циклопентилизоцианат, получают Соединение 75 (38.5 мг, выход 49%).1Н ЯМР (CD3OD, 500 МГц) δ 0.92 (д, J=7.63 Гц, 1Н), 0.96 (с, 9Н), 0.98-1.02 (м, 1Н), 1.05 (с, 9Н), 1.07-1.10 (м, 2Н), 1.21-1.25 (м, 3Н), 1.28-1.34 (м, 1Н), 1.36-1.55 (м, 8Н), 1.58-1.65 (м, 13Н), 1.81 (м, 1Н), 1.88 (м, 6Н), 2.23 (дд, J=18.01, 8.85 Гц, 1Н), 2.29 (м, 1Н), 2.59 (дд, J=13.73, 7.02 Гц, 1Н), 2.94 (м, 1Н), 3.27 (д, J=1.83 Гц, 1Н), 3.35 (д, J=1.53 Гц, 1Н), 3.75 (м, 1Н), 3.92 (с, 3Н), 3.95 (д, J=6.41 Гц, 1Н), 3.97 (с, 1Н), 4.09 (м, 2Н), 4.40 (с, 1Н), 4.45 (д, J=11.90 Гц, 1Н), 4.52 (дд, J=10.07, 7.02 Гц, 1Н), 4.81 (д, J=7.02 Гц, 1Н), 4.89 (д, J=7.02 Гц, 1Н), 5.11 (м, 1Н), 5.29 (д, J=17.40 Гц, 1Н), 5.75 (м, 1Н), 5.85 (с, 1Н), 7.11 (дд, J=9.16, 2.44 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 7.95 (м, 1Н), 8.12 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.67 мин), MS m/z 725 (MH+).
Пример 76: Получение Соединения 76

К раствору смеси продукта стадии 1 из примера 55 (70 мг, 0.102 ммоля) и DIEA (33.0 мг, 0.255 ммоля) в ТГФ (2 мл) прибавляют N,N'-дисукцинимидилкарбонат (39.2 мг, 0.153 ммоля). Полученную суспензию облучают в СВЧ-диапазоне при 80°С в течение 15 мин. Затем к ней прибавляют трет-амиламин (88.9 мг, 1.02 ммоля). Перемешивают в течение 15 мин, реакционную смесь упаривают и очищают обращенно - фазовой препаративной ВЭЖХ, получают Соединение 76 (51 мг, 69%).1Н ЯМР (CD3OD, 500 МГц) δ 0.76 (т, J=7.48 Гц, 4Н), 0.97 (с, 1Н), 1.04 (с, 9Н), 1.13 (с, 6Н), 1 22-1.25 (м, 2Н), 1.41 (дд, J=9.61, 5.34 Гц, 1Н), 1.53 (дд, J=13.89, 7.48 Гц, 1Н), 1.58-1.62 (м, 1Н), 1.87 (дд, J=7.93, 5.49 Гц, 1Н), 2.20 (кв, J=8.65 Гц, 1Н), 2.27-2.31 (м, 1Н), 2.60 (дд, J=13.73, 7.32 Гц, 1Н), 2.92-2.96 (м, 1Н), 3.92 (с, 3Н), 4.08 (дд, J=11.75, 3.81 Гц, 1Н), 4.36 (с. 1Н), 4.46 (д, J=11.90 Гц, 1Н), 4.50 (дд, J=10.22, 7.17 Гц, 1Н), 5.10 (дд, J=10.22, 1.37 Гц, 1Н), 5.27 (дд, J=16.94, 1.07 Гц, 1Н), 5.73-5.77 (м, 1Н), 5.84 (т, J=3.51 Гц, 1Н), 7.09 (дд, J=9.16, 2.44 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 8.11 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.753 мин), MS m/z 727 (МН+).
Пример 77: Получение Соединения 77
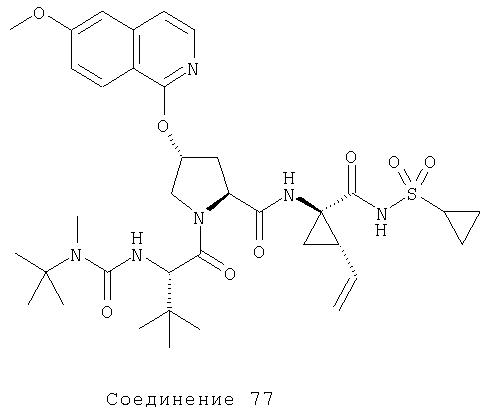
Соединение 77 получают по методике получения Соединения 76 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут трет-бутиламин, получают Соединение 77 (160.7 мг, выход 74%).1Н ЯМР (CD3OD, 500 МГц) δ 0.96-1.10 (м, 1Н), 1.06 (с, 9Н), 1.08-1.12 (м, J=5.80 Гц, 3Н), 1.26 (с, 9Н), 1.46 (дд, J=9.46, 5.19 Гц, 1Н), 1.87 (дд, J=7.93, 5.49 Гц, 1Н), 2.21 (кв, J=8.75 Гц, 1Н), 2.26-2.31 (м, 1Н), 2.57-2.62 (м, 1Н), 2.86 (с, 3Н), 2.91-2.95 (м, 1Н), 3.92 (с, 3Н), 4.09 (дд, J=11.90, 3.66 Гц, 1Н), 4.43 (с, 1Н), 4.46 (д, J=11.90 Гц, 1Н), 4.52 (дд, J=10.68, 7.02 Гц, 1Н), 5.11 (дд, J=10.22, 1.37 Гц, 1Н), 5.29 (д, J=17.09 Гц, 1Н), 5.75-5.82 (м, 1Н), 5.86 (с, 1Н), 7.09 (дд, J=9.16, 2.14 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.09 (д, J=8.85 Гц, 1Н); LC-MS (время удерживания: 1.76 мин), MS m/z 727 (MH+).
Пример 78: Получение Соединения 78
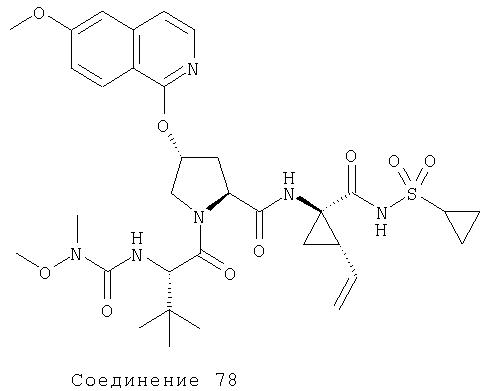
Соединение 78 получают по методике получения Соединения 76 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут N,O-диметилгидроксиламин гидрохлорид, получают Соединение 78 (62.1 мг, выход 60%).1Н ЯМР (CD3OD, 500 МГц) δ 0.99 (т, J=6.10 Гц, 1Н), 1.07 (с, 11Н), 1.22-1.26 (м, J=3.97 Гц, 2Н), 1.47 (дд, J=9.46, 5.49 Гц, 1Н), 1.88 (дд, J=8.24, 5.49 Гц, 1Н), 2.22 (д, J=8.54 Гц, 1Н), 2.30-2.33 (м, 1Н), 2.60 (дд, J=13.43, 7.02 Гц, 1Н), 2.92 (с, 3Н), 2.93-2.96 (м, 1Н), 3.66 (с, 3Н), 3.92 (с, 3Н), 4.12 (дд, J=3.66 Гц, 1Н), 4.34 (д, J=12.21 Гц, 1Н), 4.44 (д, J=9.46 Гц, 1Н), 4.54 (дд, J=10.53. 6.87 Гц, 1Н), 5.12 (д, J=10.38 Гц, 1Н), 5.30 (д, J=17.09 Гц. 1Н), 5.75-5.83 (м, 1Н), 5.86 (т, J=3.97 Гц, 1Н), 6.70 (д, J=9.77 Гц, 1Н), 7.13 (дд, J=9.16, 2.44 Гц, 1Н), 7.19 (д, J=2.44 Гц, 1Н), 7.25 (д, J=6.10 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.07 (д, J=9.16 Гц, 1Н)); LC-MS (время удерживания: 1.59 мин), MS m/z 701 (MH+).
Пример 79: Получение Соединения 79
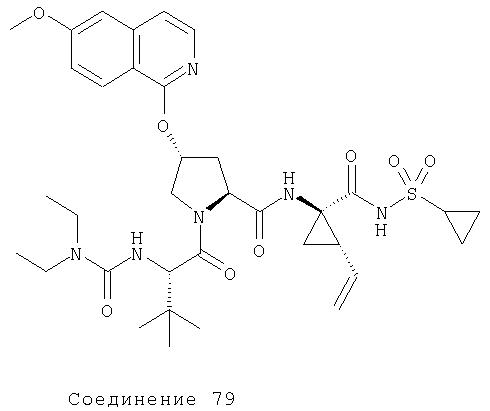
Соединение 79 получают по методике получения Соединения 76 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут диэтиламин, получают Соединение 79 (56.5 мг, выход 54%).1Н ЯМР (CD3OD, 500 МГц) δ 1.03 (кв, J=15.6 Гц, 4Н), 1.06 (д, J=1.53 Гц, 9Н), 1.05-1.10 (м, 3Н), 1.13-1.23 (м, 4Н), 1.46 (д, J=9.46, 5.19 Гц, 1Н), 1:86 (дд, J=7.93, 5.30 Гц, 1Н), 2.17 (кв, J=8.85 Гц, 1Н), 2.32-2.36 (м, 1Н), 2.60 (дд, J=14.04, 7.32 Гц, 1Н), 2.89-2.93 (м, 1Н), 3.16-3.24 (м, 4Н), 3.92 (с, 3Н), 4.14 (дд, J=11.90, 3.66 Гц, 1Н), 4.37 (д, J=11.60 Гц, 1Н), 4.51-4.55 (м, 2Н), 5.09 (д, J=10.07 Гц, 1Н), 5.27 (д, J=17.09 Гц, 1Н), 5.55 (д, J=9.46 Гц, 1Н), 5.79-5.84 (м, 1Н), 5.86 (с, 1Н), 7.11 (дд, J=8.85, 2.44 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.08 (д, J=9.16 Гц, 1Н)); LC-MS (время удерживания: 1.68 мин), MS m/z 713 (MH+).
Пример 80: Получение Соединения 80

Соединение 80 получают по методике получения Соединения 76 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут насыщенный водный раствор хлористого аммония, получают Соединение 80 (12.2 мг, выход 32%).1Н ЯМР (CD3OD, 500 МГц) δ 1.00-1.03 (м, 3Н), 1.06 (с, 9Н), 1.20-1.25 (м, 2Н), 1.42 (дд, J=9.31, 5.34 Гц, 1Н), 2.22 (д, J=9.77 Гц, 1Н), 2.29-2.35 (м, 1Н), 2.59 (дд, J=13.28, 6.87 Гц, 1Н), 2 92-2.96 (м, 1Н), 3.92 (с, 3Н), 4.14 (дд, J=11.75, 4.12 Гц, 1Н), 4.38-4.43 (м, 1Н), 4.51 (дд, J=9.92, 6.87 Гц, 1Н), 5.11 (д, J=11.90 Гц, 1Н), 5.28 (д, J=17.70 Гц, 1Н), 5.72-5.79 (м, 1Н), 5.84 (с, 1Н), 7.15 (д, J=2.44 Гц, 1Н), 7.17 (д, J=2.75 Гц, 1Н), 7.23 (д, J=5.80 Гц, 1Н), 7.87 (д, J=7.87 Гц, 1Н), 8.10 (д, J=8.85 Гц, 1Н)); LC-MS (время удерживания: 1.43 мин), MS m/z 657 (MH+).
Пример 81: Получение Соединения 81

Соединение 81 получают по методике получения Соединения 76 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут трет-октиламин, получают Соединение 81 (16.1 мг, выход 48%).1Н ЯМР (CD3OD, 500 МГц) δ 0.88 (с, 9Н), 1.00 (д, J=9.77 Гц, 5Н), 1.04 (с, 9Н), 1.17 (с, 3Н), 1.18-1.20 (м, 1Н), 1.21 (с, 3Н), 1.35 (д, J=2.44 Гц, 1Н), 1.40-1.43 (м, 1Н), 1.57 (д, J=14.95 Гц, 1Н), 1.67 (д, J=14.65 Гц, 1Н), 1.85 (дд, J=8.09, 5.34 Гц, 1Н), 2.15 (д, J=8.24 Гц, 1Н), 2.34-2.43 (м, 1Н), 2.60 (дд, J=13.73, 7.02 Гц, 1Н), 2.89-2.93 (м, 1Н), 3.92 (с, 3Н), 4.13 (дд, J=11.60, 3.97 Гц, 1Н), 4.38 (с, 1Н), 4.43 (д, J=11.90 Гц, 1Н), 4.50 (дд, J=9.77, 7.32 Гц, 1Н), 5.07 (д, J=10.38 Гц. 1Н), 5.24 (д, J=17.09 Гц, 1Н), 5.75-5.81 (м, 1Н), 5.84 (с, 1Н), 7.09 (дд, J=9.16, 2.44 Гц, 1Н), 7.17 (д, J=2.44 Гц, 1Н), 7.23 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.10 (д. J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.92 мин), MS m/z 769 (МН+).
Пример 82: Получение Соединения 82

Соединение 82 получают по методике получения Соединения 76 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут 1-(4-фторфенил)-2-метил-2-пропиламин, получают Соединение 82 (14.8 мг, выход 42%).1Н ЯМР (CD3OD, 500 МГц) δ 0.88 (с, 9Н), 1.00 (д, J=9.46 Гц, 6Н), 1.04 (с, 9Н), 1.17 (с, 3Н), 1.21 (с, 3Н), 1.32-1.37 (м, 2Н), 1.39-1.43 (м, 1Н), 1.57 (д, J=14.65 Гц, 1Н), 1.67 (д, J=14.96 Гц, 1Н), 1.82-1.86 (м, 1Н), 2.15 (т, J=9.46 Гц, 1Н), 2.33-2.43 (м, 2Н), 2.58-2.62 (дд, J=14.50, 7.78 Гц, 1Н), 2.89-2.93 (м, 1Н), 3.92 (с, 3Н), 4.12 (дд, J=11.90, 3.97 Гц, 1Н), 4.38 (с, 1Н), 4.43 (д, J=12.82 Гц, 1Н), 4.49-4.52 (м, 1Н), 5.24 (д, J=16.48 Гц, 1Н), 5.76-5.82 (м, 1Н), 5.83-5.85 (м, 1Н), 7.09 (дд, J=9.00, 2.59 Гц, 1Н), 7.17 (д, J=2.14 Гц, 1Н), 7.23 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.10 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.40 мин), MS m/z 807 (МН+).
Пример 83: Получение Соединения 83
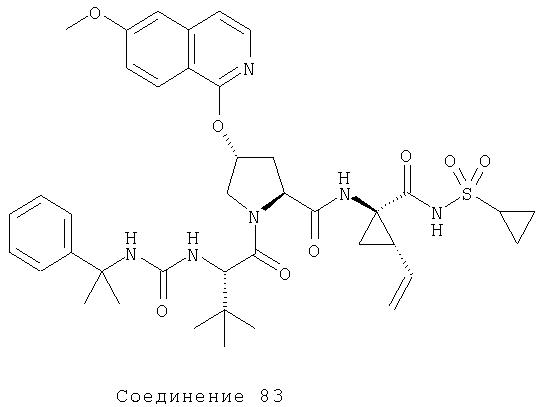
Соединение 83 получают по методике получения Соединения 76 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут кумиламин, получают Соединение 83 (64.6 мг, выход 57%).1Н ЯМР (CD3OD, 500 МГц) δ 0.87-0.91 (м, 1Н), 0.98 (д, J=9.46 Гц, 2Н), 1.01 (с, 9Н), 1.02-1.05 (м, 1Н), 1.17-1.21 (м, 3Н), 1.29 (с, 2Н), 1.40 (дд, J=9.46, 5.19 Гц, 1Н), 1.51 (д, J=3.05 Гц, 5Н), 1.85 (дд, J=8.09, 5.34 Гц, 1Н), 2.17 (кв, J=8.85 Гц, 1Н), 2.30-2.33 (м, 1Н), 2.58 (дд, J=13.58, 7.48 Гц, 1Н), 2.91-2.94 (м, 1Н), 3.92 (д, J=2.14 Гц, 1Н), 3.93 (с, 3Н), 4.05 (дд, J=11.60. 3.66 Гц, 1Н), 4.32 (д, J=9.77 Гц, 1Н), 4.51 (дд, J=9.92, 7.17 Гц, 1Н), 5.09 (дд, J=11.59, 1.52 Гц, 1Н), 5.27 (дд, J=16.71, 1.22 Гц, 1Н), 5.74-5.78 (м, 1Н), 5.82 (с, 1Н), 7.05-7.09 (м, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.19 (д, J=7.33 Гц, 1Н), 7.22 (д, J=5.80 Гц, 1Н), 7.33 (д, J=7.63 Гц, 1Н), 7.84 (д, J=5.80 Гц, 1Н); LC-MS (время удерживания: 1.76 мин), MS m/z 775 (MH+).
Пример 84: Получение Соединения 84
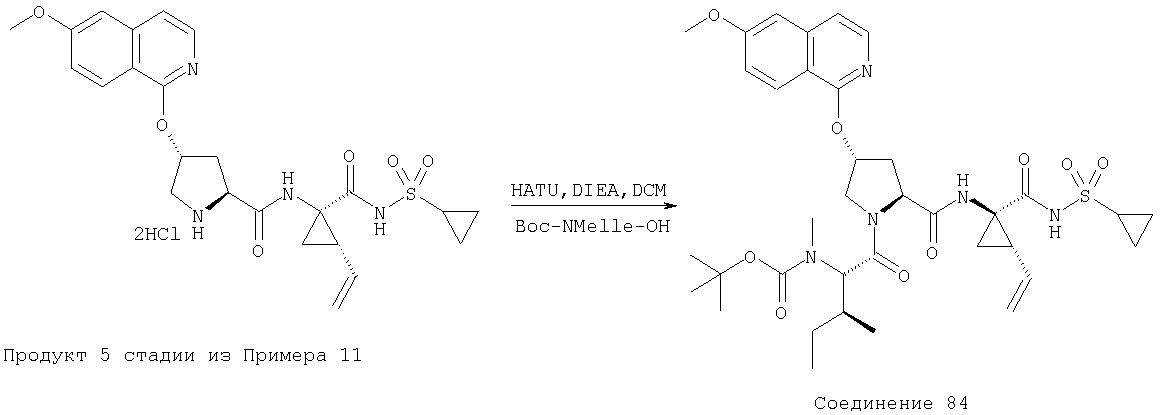
К раствору продукта стадии 5 из Примера 11 (77.0 мг, 0.136 ммоля), DIEA (70.4 мг, 0.544 ммоля) и HATU (77.5 мг, 0.204 ммоля) прибавляют Boc-MeIle-ОН (43.4 мг, 0.177 ммоля). Перемешивают при rt в течение 14 час, реакционную смесь промывают 5% водным раствором NaHCO3 (1 мл). Водный слой экстрагируют 2×2 мл DCM. Объединенные органические вытяжки промывают 5% водным раствором лимонной кислоты (2 мл), рассолом, сушат MgSO4, упаривают и очищают флеш-хроматографией на колонке (SiO2, 97:3 DCM: МеОН), получают Соединение 84 (68.4 мг, выход 69%):1Н ЯМР (CD3OD, 500 МГц) δ 0.89 (т, J=7.32 Гц, 3Н), 0.94 (дд, J=5.95, 4.43 Гц, 3Н), 1.07 (д, J=7.63 Гц, 3Н), 1.13 (с, 5Н), 1.16-1.20 (м, J=4.88 Гц, 2Н), 1.23 (с, 3Н), 1.28 (м, 1Н), 1.34-1.38 (м, 1Н), 1.41-1.47 (м, 1Н), 1.55-1.60 (м, J=7.63 Гц, 1Н), 1.87-1.91 (м, 1Н), 2.22-2.26 (м, 2Н), 2.36-2.38 (м, 1Н), 2.56-2.62 (м, 1Н), 2.81 (д, J=11.30 Гц, 2Н), 2.94-2.99 (м, 1Н), 3.92 (с, 3Н), 4.05-4.12 (м, 2Н), 4.48-4.57 (м, 2Н), 5.12 (д, =10.07 Гц, 1Н), 5.32 (м, 1Н), 5.75-5.82 (м, 1Н), 5.84-5.88 (м, 1Н), 7.09-7.13 (м, 1Н), 7.16-7.20 (м, 1Н), 7.23-7.27 (м, 1Н), 7.88 (дд, J=5.95, 2.29 Гц, 1Н), 8.04 (д, J=9.16 Гц, 0.6Н), 8.09 (д, J=9.46 Гц, 0.4Н); LC-MS (время удерживания: 1.83 мин), MS m/z 728 (MH+).
Пример 85: Получение Соединения 85
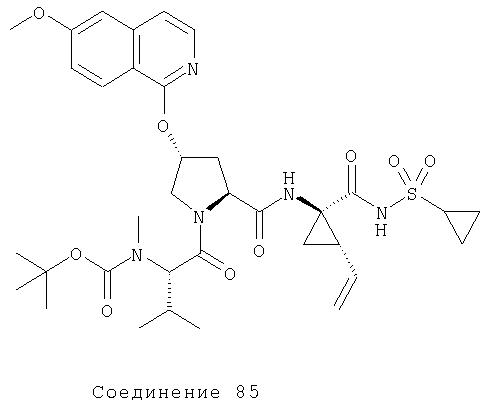
Соединение 85 получают по методике получения Соединения 84 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут Boc-MeVal-ОН, получают Соединение 85 (72.1 мг, выход 74%).1H ЯМР (CD3OD, 500 МГц) δ 0.84 (т, J=5.80 Гц, 3Н), 0.96 (д, J=6.41 Гц, 3Н), 1.08 (д, J=7.32 Гц, 2Н), 1.13 (с, 6Н), 1.16 (с, 4Н), 1.18-1.21 (м, 1Н), 1.23-1.29 (м, 1Н), 1.44 (дд, J=9.61, 5.34 Гц, 1Н), 1.88-1.92 (м, 1Н), 2.24 (д, J=10.07 Гц, 1Н), 2.32-2.39 (м, 2Н), 2.58 (дд, J=13.89, 6.26 Гц, 1Н), 2.80 (с, 3Н), 2.93-2.98 (м, 1Н), 3.93 (с, 3Н), 4.01 (дд, J=11.90, 3.36 Гц, 0.6Н), 4.12 (дд, J=11.90, 3.66 Гц, 0.4Н), 4.16 (д, J=11.29 Гц, 0.6Н), 4.38 (д, J=10.99 Гц, 0.4Н), 4.45 (д, J=10.68 Гц, 1Н), 4.47 (д, J=10.69 Гц, 0.6Н), 4.53 (дд, J=10.38, 7.02 Гц, 0.4Н), 4.58 (дд, J=10.07, 7.02 Гц, 1Н), 5.12 (д, J=4.28 Гц, 0.6Н), 5.14 (д, J=4.27 Гц, 0.4Н), 5.30 (д, J=7.32 Гц, 0.6Н), 5.34 (д, J=7.32 Гц, 0.4Н), 5.78-5.85 (м, 1Н), 5.88 (т, J=3.05 Гц, 0.6Н), 5.96 (т, J=3.97 Гц, 0.4Н), 7.13 (дд, J=9.00, 2.29 Гц, 0.6Н), 7.16 (дд, J=9.46, 2.44 Гц, 0.4Н), 7.19 (м, 1Н), 7.24 (д, J=6.10 Гц, 0.6Н), 7.26 (д, J=6.10 Гц, 0.4Н), 7.88 (д, J=5.80 Гц, 1Н), 8.02 (д, J=9.16 Гц, 0.6Н), 8.05 (д, J=9.16 Гц, 0.4Н).
Пример 86: Получение Соединения 86

Соединение 86 получают по методике получения Соединения 84 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут Boc-MeLeu-ОН, получают Соединение 86 (56.5 мг, выход 57%).1H ЯМР (CD3OD, 500 МГц) δ 0.94-0.96 (м, 6Н), 1.04-1.13 (м, 2Н), 1.17 (с, 4.5Н), 1.18 (с, 4.5Н), 1.26-1.31 (м, 1Н), 1.42 (дд, J=9.46, 5.49 Гц, 1Н), 1.46-1.51 (м, 2Н), 1.56-1.60 (м, 0.5Н), 1.69-1.72 (м, 0.5Н), 1.75-1.81 (м, 0.5Н), 1.90 (кв, J=7.50 Гц, 1Н), 2.27 (дд, J=13.89, 7.78 Гц, 1Н), 2.32-2.38 (м, 1Н), 2.58 (дд, J=14.80, 7.48 Гц, 1Н), 2.75 (с, 3Н), 2.95-2.99 (м, 1Н), 3.93 (с, 3Н), 4.03 (д, J=12.21 Гц, 1Н), 4.11-15 (м, 0.5Н), 4.28 (д, J=12.21 Гц, 1Н), 4.53 (т, J=8.50 Гц, 0.5Н), 4.59 (т, J=8.55 Гц, 0.5Н), 4.83-4.87 (м, J=6.41 Гц, 0.5Н), 4.96 (м, 0.5Н), 5.14 (дд, J=11.14, 4.73 Гц, 1Н), 5.32 (дд, J=17.70, 6.41 Гц, 1Н), 5.75-5.82 (м, 1Н). 5.90 (с, 0.5Н), 5.92 (с, 0.5Н), 7.13-7.18 (м, 1Н), 7.20 (с, 1Н), 7.25-7.27 (м, 1Н), 7.87 (т, J=4.40 Гц, 1Н), 8.05 (д, J=8.85 Гц, 1Н).
Пример 87: Получение Соединения 87
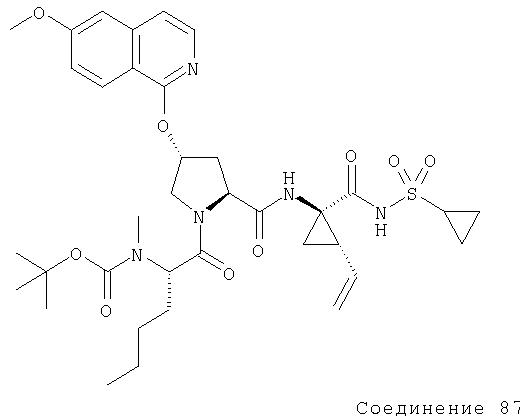
Соединение 87 получают по методике получения Соединения 84 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут Boc-MeNle-ОН, получают Соединение 87 (82.3 мг, выход 83%).1H ЯМР (CD3OD, 500 МГц) δ 0.90-0.96 (кв, J=7.53 Гц, 3Н), 1.05-1.10 (м, 2Н), 1.18 (с, 4.5Н), 1.20 (с, 4.5Н), 1.24-1.30 (м, 3Н), 1.31-1.38 (м, 1Н), 1.42 (дд, J=9.46, 5.19 Гц, 2Н), 1.72-1.81 (м, 2Н), 1.88-1.92 (м, 1Н), 2.22-2.29 (м, 1Н), 2.32-2.38 (м, 1Н), 2.58 (дд, J=13.89, 7.17 Гц, 1Н), 2.72 (с, 3Н), 2.94-2.99 (м, 1Н), 3.93 (с, 3Н), 4.02 (дд, J=9.77, 4.27 Гц, 1Н), 4.12 (дд, J=11.90, 3.35 Гц. 0.5Н), 4.24 (дд, J=11.90, 0.6 Гц, 0.5Н), 4.51-4.60 (м, 1Н), 5.14 (д, J=10.38 Гц, 1Н), 5.33 (дд, J=17.24, 4.73 Гц, 1Н), 5.75-5.82 (м, 1Н), 5.91 (с, 1Н), 7.15 (дд, J=14.34, 7.63 Гц, 1Н), 7.20 (д, J=2.44 Гц, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.87 (т, J=4.37 Гц, 1Н), 8.05 (д, J=8.85 Гц, 1Н).
Пример 88: Получение Соединения 88

Соединение 88 получают аналогично Соединению 84 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут Boc-N-Me-NVa-ОН, получают Соединение 88 (70.5 мг, выход 73%).1H ЯМР (CD3OD, 500 МГц) δ 0.96 (д, J=6.41 Гц, 3Н), 1.06-1.10 (м, 2Н), 1.18 (с, 9Н), 1.27-1.30 (м, 4Н), 1.42 (дд, J=9.46, 5.49 Гц, 1Н), 1.66-1.80 (м, 2Н), 1.88-1.92 (м, 1Н), 2.23-2.29 (м, 1Н), 2.30-2.37 (м, 1Н), 2.58 (дд, J=13.58, 7.17 Гц, 1Н), 2.73 (с, 3Н), 2.94-2.98 (м, 1Н), 3.93 (с, 3Н), 4.00-4.04 (м, 1Н), 4.12 (д, J=12.82 Гц, 0.5Н), 4.25 (д, J=12.21 Гц, 0.5Н), 4.51-4.60 (м, 1Н), 5.13 (д, J=10.68 Гц, 1Н), 5.32 (д, J=17.09 Гц, 1Н), 5.75-5.81 (м, 1Н), 5.90 (с, 1Н), 7.13-7.18 (м, 1Н), 7.20 (д, J=2.14 Гц, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.87 (с, 1Н), 8.05 (д, J=9.16 Гц, 1Н).
Пример 89: Получение Соединения 89
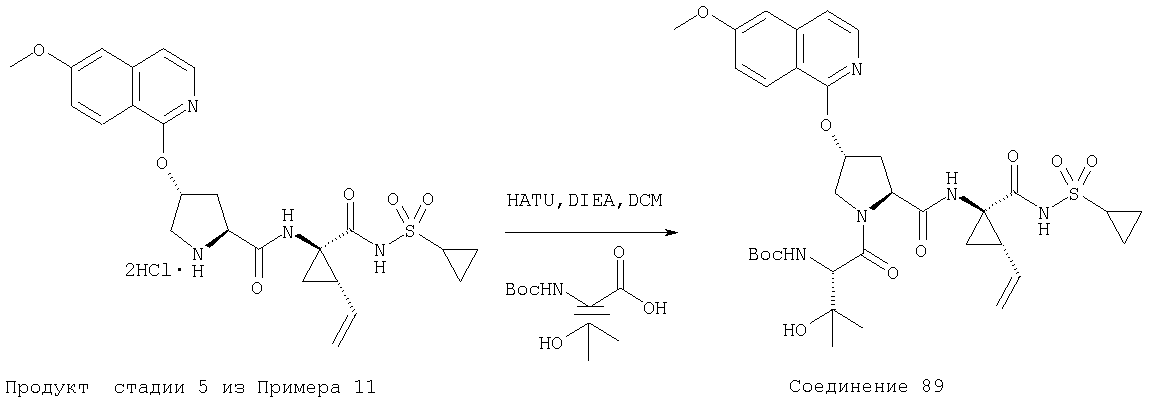
К раствору продукта стадии 5 из Примера 11 (66.0 мг, 0.123 ммоля), DIEA (63.7 мг, 0.492 ммоля) и HATU (70.0 мг, 0.184 ммоля) прибавляют 2S-трет-бутоксикарбониламино-3-гидрокси-3-метилмасляную кислоту (34.0 мг, 0.147 ммоля). Перемешивают при rt в течение 14 час, реакционную смесь промывают 5% водным раствором NaHCO3 (1 мл). Водный слой экстрагируют 2×2 мл DCM. Объединенные органические вытяжки промывают 5% водным раствором лимонной кислоты (2 мл), рассолом, сушат MgSO4, упаривают и очищают обращенно - фазовой препаративной ВЭЖХ, получают Соединение 89 (36.1 мг, выход 41%).1H ЯМР (CD3OD, 500 МГц) δ 1.07 (д, J=7.93 Гц, 2Н), 1.18 (с, 1Н), 1.20 (с, 9Н), 1.24-1.27 (м, J=11.60 Гц, 3Н), 1.30 (с, 3Н), 1.43-1.48 (м, 10Н), 1.59 (с, 1Н), 1.65 (с, 1Н), 1.87 (дд, J=8.24, 5.19 Гц, 1Н), 2.24 (кв, J=9.16 Гц, 1Н), 2.33-2.36 (м, 1Н), 2.63 (дд, J=12.97, 6.56 Гц, 1Н), 2.94-2.99 (м, 1Н), 3.92 (с, 3Н), 3.93 (с, 1Н), 4.12 (дд, J=11.60, 3.05 Гц, 1Н), 4.27-4.31 (м, 1Н), 4.54 (т, J=9.77 Гц, 1Н), 5.12 (дд, J=10.53, 1.37 Гц, 1Н), 5.30 (д, J=17.09 Гц, 1Н), 5.79-5.83 (м, 1Н), 5.85 (с, 1Н), 7.11 (дд, J=8.55, 1.83 Гц, 1Н), 7.18 (д, J=2.24 Гц, 1Н), 7.24 (д, J=5.49 Гц, 1Н), 7.88 (м, 1Н), 8.10 (д, J=8.85 Гц, 1Н); LC-MS (время удерживания: 1.637 мин), MS m/z 716 (МН+).
Пример 91: Получение Соединения 91

Соединение 91 получают аналогично Соединению 89 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут Boc-L-Thr-ОН, получают Соединение 91 (80.5 мг, выход 66%).1H ЯМР (CD3OD, 500 МГц) δ 0.93 (дд, J=8.24, 2.14 Гц, 2Н), 1.08-1.18 (м, 4Н), 1.20 (д, J=6.10 Гц, 3Н), 1.29 (с, 9Н), 1.32 (дд, J=9.61, 5.04 Гц, 1Н), 1.45 (д, J=4.27 Гц, 1Н), 1.84 (дд, J=7.63, 5.19 Гц, 1Н), 2.15 (кв, J=8.85 Гц, 1Н), 2.42-2.48 (м, 1Н), 2.64 (дд, J=14.04, 7.63 Гц, 1Н), 2.85-2.89 (м, 1Н), 3.92 (с, 3Н), 4.1-4.14 (м, 2Н), 4.30 (д, J=4.88 Гц, 1Н), 4.38 (д, J=11.60 Гц, 1Н), 4.60 (т, J=8.55 Гц, 1Н), 5.04 (дд, J=10.22, 1.68 Гц, 1Н), 5.80-5.84 (м, 2Н), 7.11 (д, J=9.16 Гц, 1Н), 7.17 (д, J=1.83 Гц, 1Н), 7.23 (д, J=5.80 Гц, 1Н), 7.87 (д, J=5.80 Гц, 1Н), 8.10 (д, J=8.85 Гц, 1Н); LC-MS (время удерживания: 1.560 мин), MS m/z 702 (MH+).
Пример 91: Получение Соединения 91
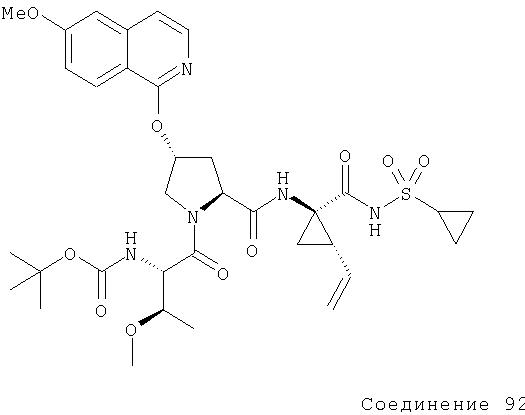
Соединение 92 получают аналогично Соединению 89 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут Boc-L-Thr(Me)-ОН, получают Соединение 92 (47.1 мг, выход 69%).1H ЯМР (CD3OD, 500 МГц) δ 0.95 (д, J=4.27 Гц, 2Н), 1.11-1.16 (м, 3Н), 1.18 (д, J=6.10 Гц, 6Н), 1.32 (с, 9Н), 1.38 (дд, J=9.31, 5.04 Гц, 1Н), 1.45 (с, 1Н), 1.85 (дд, J=7.78, 5.04 Гц, 1Н), 2.13 (д, J=9.15 Гц, 1Н), 2.46-2.51 (м, 1Н), 2.63 (дп, J=14.19, 7.78 Гц, 1Н), 2.81-2.91 (м, 1Н), 3.68-3.73 (м, 1Н), 3.92 (с, 4Н), 4.14 (д, J=12.21 Гц, 1Н), 4.35 (д, J=5.80 Гц, 1Н), 4.42 (д, J=11.29 Гц, 1Н), 4.60 (т, J=8.70 Гц, 1Н), 5.05 (д, J=10.68 Гц, 1Н), 5.24 (дд, J=16.79, 0.92 Гц, 1Н), 5.81-5.85 (м, 2Н), 7.11 (дд, J=9.16, 0.92 Гц, 1Н), 7.17 (с, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (д, 7-5.80 Гц, 1Н), 8.11 (д, J=8.55 Гц, 1Н); LC-MS (время удерживания: 1.660 мин), MS m/z 716 (MH+),
Пример 93: Получение Соединения 93

Соединение 93 получают аналогично Соединению 89 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут Boc-L-Thr(трет-Bu)-ОН, получают Соединение 93 (52.7 мг, выход 53%).1H ЯМР (CD3OD, 500 МГц) δ 1.09 (дд, J=8.24, 2.44 Гц, 2Н), 1.21-1.24 (м, 3Н), 1.28 (с, 9Н), 1.45 (с, 9Н), 1.89 (дд, J=7.93, 5.19 Гц, 1H), 2.25 (кв, J=8.34 Гц, 1Н), 2.32-2.36 (м, 1Н), 2.59 (дд, J=12.82, 6.41 Гц, 1Н), 2.95-3.00 (м, 1Н), 3.71 (с, 2Н), 3.92 (с, 3Н), 3.93-3.99 (м, 1Н), 4.10 (д, J=7.02 Гц, 1Н), 4.15 (д, J=11.90 Гц, 1Н), 4.22 (дд, J=6.10, 2.44 Гц, 2Н), 4.40 (д, J=11.60 Гц, 1Н), 4.54 (дд, J=10.01, 6.71 Гц, 1Н), 5.13 (д, J=10.38 Гц, 1Н), 5.31 (д, J=17.09 Гц, 1Н), 5.76-5.83 (м, 1Н), 5.87 (с, 1Н), 6.06 (д, J=9.46 Гц, 1Н), 6.36 (д, J=7.02 Гц, 1Н), 7.11 (д, J=8.85 Гц, 1Н), 7.18 (с, 1Н), 7.24 (д, J=5.49 Гц, 1Н), 7.88 (м, 1Н), 8.08 (д, J=9.16 Гц, 1Н).
Пример 94: Получение Соединения 94

Соединение 94 получают аналогично Соединению 89 с нижеприведенными модификациями:
Модификации: В качестве исходного вещества берут Boc-(2S,3S)-2-амино-3-метоксибутановую кислоту, получают Соединение 94 (150.2 мг, выход 80%).1H ЯМР (CD3OD, 500 МГц) δ 1.04-1.13 (м, 3Н), 1.17 (д, J=6.10 Гц, 3Н), 1.20-1.24 (м, 2Н), 1.27 (с, 9Н), 1.44-1.48 (м, 2Н), 1.86 (дд, J=7.93, 5.49 Гц, 1Н), 2.24 (кв, J=8.65 Гц, 1Н), 2.34-2.37 (м, 1Н), 2.61 (дд, J=14.19, 7.17 Гц, 1Н), 2.94-2.99 (м, 1Н), 3.66 (м, 1Н), 3.92 (с, 3Н), 4.13 (дд, J=12.36, 3.81 Гц, 1Н), 4.37 (дд, J=22.58, 10.99 Гц, 2Н), 4.54 (дд, J=10.38, 7.63 Гц, 1Н), 5.12 (д. J=10.68 Гц, 1Н), 5.31 (д, J=17.40 Гц, 1Н), 5.77-5.82 (м, 1Н), 5.85 (с, 1Н), 7.12 (д, J=9.16 Гц, 1Н), 7.18 (с, 1Н), 7.25 (д, J=6.10 Гц, 1Н), 7.89 (д, J=5.80 Гц, 1Н), 8.10 (д, J=8.85 Гц, 1Н); LC-MS (время удерживания: 1.673 мин), MS m/z 716 (MH+).
Пример 95: Получение Соединения 95
Схема 1.
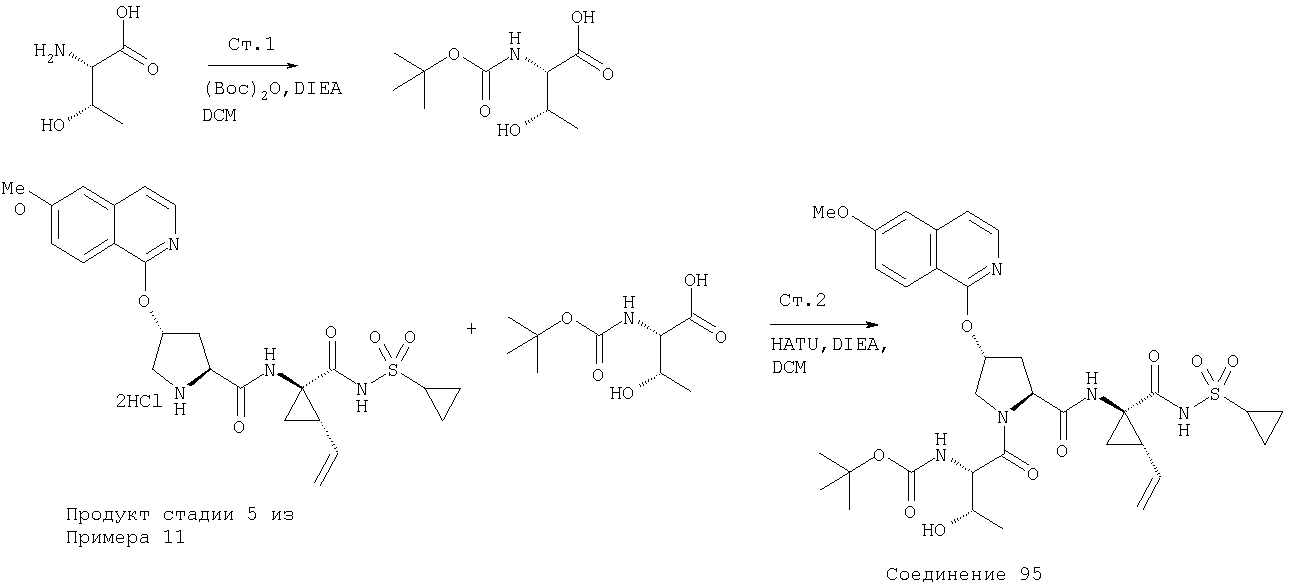
Стадия 1
К смеси Н-allo-THr-ОН (5.0 г, 41.98 ммоля) и DIEA (10.9 г, 83.96 ммоля) в DCM (150 мл) прибавляют ди-трет-бутилдикарбонат (13.7 г, 62.97 ммоля). Перемешивают при rt в течение 14 час, реакционную смесь промывают 3×100 мл DCM. Объединенные органические вытяжки сушат MgSO4 и упаривают. LC/MS показывает, что основная часть продукта остается в Н2O слое. Поэтому водный слой упаривают. Продукт очищают флеш-хроматографией на колонке (SiO2, 90:10 DCM: МеОН), получают Boc-allo-THr-ОН; LC-MS (время удерживания: 0.727 мин), MS m/z 242 (MNa+).
Стадия 2
К раствору продукта стадии 5 из Примера 11 (100.0 мг, 0.174 ммоля), DIEA (67.6 мг, 0.522 ммоля) и HATU (106.0 мг, 0.278 ммоля) прибавляют продукт вышеописанной стадии 1 (57.3 мг, 0.262 ммоля). Перемешивают при rt в течение 3 час, реакционную смесь промывают 5% водным раствором NaHCO3 (1 мл). Водный слой экстрагируют 2× 2 мл DCM. Объединенные органические вытяжки промывают 5% водным раствором лимонной кислоты (2 мл), рассолом, сушат MgSO4, упаривают и очищают обращенно-фазовой препаративной ВЭЖХ, получают Соединение 95 (39.1 мг, выход 32%):1H ЯМР (CD3OD, 500 МГц) δ 1.02 (д, J=8.55 Гц, 1Н), 1.18-1.23 (м, 3Н), 1.25 (с, 9Н), 1.38 (дд, J=9.15, 6.30 Гц, 1Н), 1.84 (дд, J=7.93, 5.19 Гц, 1Н), 2.19-2.24 (м, 1Н), 2.38-2.43 (м, 1Н), 2.65 (дд, J=14.19, 6.87 Гц, 1Н), 2.92-2.96 (м, 1Н). 3.92 (с, 3Н), 3.93 (с, 2Н), 4.16-4.19 (м, 1Н), 4.23 (д, J=8.24 Гц, 1Н), 4.44 (д, J=12.21 Гц, 1Н), 4.57-5.81 (м, 1Н), 5.09 (д, J=10.68 Гц, 1Н), 5.29 (д, J=17.09 Гц, 1Н), 5.75-5.81 (м, 1Н), 5.83-5.85 (м, 1Н), 7.11 (д, J=10.38 Гц, 1Н), 7.18 (д, J=1.83 Гц, 1Н), 7.24 (д, J=6.41 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.11 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.583 мин), MS m/z 702 (MH+).
Пример 96: Получение Соединения 96
Схема 1.

Соединение 96 получают аналогично Соединению 95 с нижеприведенными модификациями:
Модификации:
Стадия 1
Гидрохлорид (2S,3S)-2-амино-3-этоксибутановой кислоты используют в качестве исходного вещества в стадии 1, получая Boc-(2S,3S)-2-амино-3-этоксибутановой кислоты; LC-MS (время удерживания: 1.067 мин), MS m/z 270 (M+Na+).
Стадия 2
Продукт стадии 1 затем аналогично реагирует с продуктом стадии 5 из Примера 11, давая Соединение 96 (55.3 мг, выход 44%):1H ЯМР (CD3OD, 500 МГц) δ 0.94 (т, J=6.87 Гц. 1Н), 0.97-1.03 (м, 2Н), 1.08-1.11 (м, 2Н), 1.13-1.15 (м, 2Н), 1.17 (м, J=6.10 Гц, 6Н), 1.29 (с, 9Н), 1.41-1.45 (м, 3Н), 1.85 (дд, J=7.48, 5.34 Гц, 1Н), 2.12-2.19 (м, 1Н), 2.43-2.49 (м, 1Н), 2.60 (дд, J=13.73, 6.80 Гц, 1Н), 2.89-2.93 (м, 1Н), 3.50-3.57 (м, 2Н), 3.73-3.78 (м, 1Н), 3.92 (с, 3Н), 4.18 (д, J=8.85 Гц, 1Н), 4.35 (д, J=12.21 Гц, 1Н), 4.39 (д, J=8.55 Гц, 1Н), 4.53 (т, J=7.78 Гц, 1Н), 5.07 (д, J=9.16 Гц, 1Н), 5.25 (д, J=18.01 Гц, 1Н), 5.82 (т, J=9.85 Гц, 1H), 5.88 (т, J=9.80 Гц, 1Н), 7.11 (д, J=5.19 Гц, 1Н), 7.18 (д, J=2.14 Гц, 1Н), 7.24 (д, J=5.49 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 8.10 (д, J=8.85 Гц, 1Н);
LC-MS (время удерживания: 1.743 мин), MS m/z 730 (MH+).
Пример 97: Получение Соединения 97
Схема 1.

Соединение 97 получают аналогично Соединению 95 с нижеприведенными модификациями:
Модификации:
Стадия 1
Н-allo-THr(трет-Bu)-ОН используют в качестве исходного вещества в стадии 1, получая Boc-(2S,3S)-1-амино-3-этоксибутановую кислоту; LC-MS (время удерживания: 1.363 мин), MS m/z 298 (M+Na+).
Стадия 2
Продукт стадии 1 затем аналогично реагирует с продуктом стадии 5 из Примера 11, давая Соединение 97 (48.2 мг, выход 37%): LC-MS (время удерживания: 1.820 мин), MS m/z 758 (МН+).
Пример 99: Получение Соединения 99

Соединение 99 получают аналогично Соединению 95 с нижеприведенными модификациями:
Модификации: В качестве исходного берут Соединение 84, получают Соединение 99 (60.3 мг, выход 98%);1H ЯМР (CD3OD, 500 МГц) δ 1.00 (кв, J=7.12 Гц, 3Н), 1.10-1.13 (м, 5Н), 1.20-1.31 (м, 3Н), 1.41 (дд, J=9.46, 5.49 Гц, 1Н), 1.61-1.68 (м, 1Н), 1.92 (дд, J=8.24, 5.49 Гц, 1Н), 2.04-2.09 (м, 1Н), 2.28 (кв, J=8.55 Гц, 1Н), 2.34-2.39 (м, 1Н), 2.57 (с, 3Н), 2.64-2.70 (м, 1Н), 2.94-2.97 (м, 1Н), 3.93 (с, 3Н), 4.07-4.14 (дд, J=12.05, 3.81 Гц, 1Н), 4.13 (д, J=6.10 Гц, 1Н), 4.18 (д, J=5.80 Гц, 1Н), 4.25 (д, J=12.21 Гц, 1Н), 4.66-4.73 (м, 1Н), 5.15 (д, J=10.68 Гц, 1Н), 5.32 (д, J=17.09 Гц, 1Н), 5.70-5.79 (м, 1Н), 5.92 (т, J=3.66 Гц, 0.4Н), 5.95 (т, J=3.66 Гц, 0.6Н), 7.17 (дд, J=9.16, 2.44 Гц, 1Н), 7.22 (д, J=2.14 Гц, 1Н), 7.28 (дд, J=5.80, 3.36 Гц, 1Н), 7.91 (дд, J=5.80, 4.27 Гц, 1Н), 8.03 (д, J=8.85 Гц, 0.6Н), 8.07 (д, J=9.16 Гц, 0.4Н); LC-MS (время удерживания: 1.33 мин), MS m/z 628 (MH+).
Пример 100: Получение Соединения 100

К раствору Соединения 94 (0.600 г, 0.838 ммоля) в DCE (3 мл) прибавляют ТФК (3 мл). Перемешивают при rt в течение 15 мин, реакционную смесь упаривают. Полученное вязкое масло снова растворяют в DCE (5 мл) и снова упаривают. Затем растворяют в DCM (2 мл ) и прибавляют 1N раствор НС1 в Et2O (10 мл). Полученную суспензию охлаждают до 0°С, фильтруют в вакууме, промывают Et2O и сушат в вакуум-шкафу, получают продукт в виде соли-бис-гидрохлорида, белого твердого вещества (527.1 г, выход 91%):1H ЯМР (CD3OD, 500 МГц) δ 1.08-1.15 (м, 2Н), 1.21 (д, J=6.71 Гц, 4Н), 1.28-1.33 (м, 1Н), 1.41 (дд, J=9.46. 5.49 Гц, 1Н), 1.91 (дд, J=8.24, 5.49 Гц, 1Н), 2.28 (кв, J=8.65 Гц, 1Н), 2.34-2.37 (м, 1Н), 2.68 (дд, J=13.12, 7.02 Гц, 1Н), 2.81 (с, 3Н), 2.93-2.98 (м, 1Н), 3.45 (с, 3Н), 3.94 (с, 3Н), 3.96-4.00 (м, 1Н), 4.16 (дд, J=11.90, 3.66 Гц, 1Н), 4.27 (д, J=11.60 Гц, 1Н), 4.59 (д, J=4.58 Гц, 1Н), 4.69 (дд, J=10.07, 7.02 Гц, 1Н), 5.14 (дд, J=10.53, 1.37 Гц, 1Н), 5.32 (д, J=17.09 Гц, 1Н), 5.70-5.77 (м, 1Н), 5.94 (т, J=3.66 Гц, 1Н), 7.19 (д, J=9.16 Гц, 1Н), 7.24 (с, 1Н), 7.32 (с, 1Н), 7.91 (д, J=5.80 Гц, 1Н), 8.09 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.213 мин), MS m/z 616 (МН+).
Пример 101: Получение Соединения 101
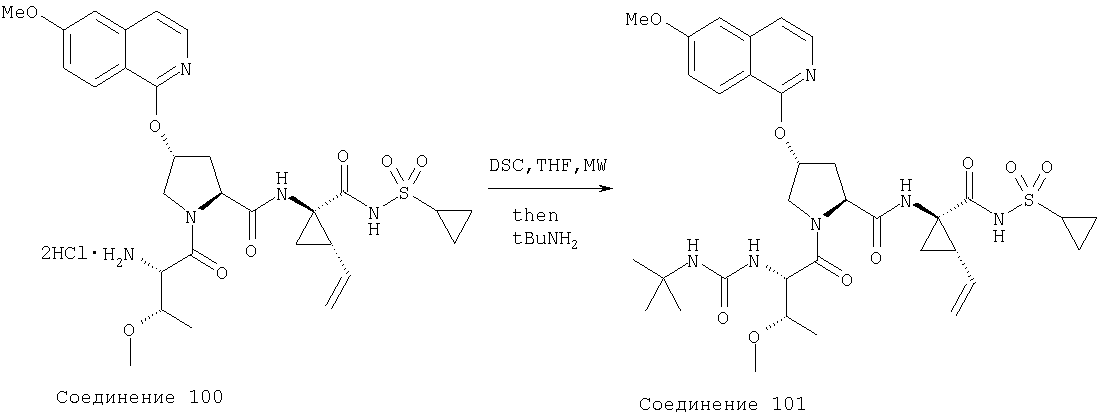
К раствору смеси Соединения 100 (80 мг, 0.116 ммоля) и DIEA (31.5 мг, 0.244 ммоля) в ТГФ (2 мл ) прибавляют N,N'-дисукцинилимидилкарбонат (44.6 мг, 0.174 ммоля). Полученную суспензию облучают в СВЧ-диапазоне при 80°С в течение 15 мин. Затем к ней прибавляют трет-амиламин (84.8 мг, 1.16 ммоля). Перемешивают при rt в течение 15 мин, реакционную смесь упаривают и очищают обращенно - фазовой препаративной ВЭЖХ, получают Соединение 101 (65 мг, 79%).1H ЯМР (CD3OD, 500 МГц) δ 1.03-1.08 (м, 3Н), 1.16 (д, J=6.41 Гц, 3Н), 1.19 (с, 9Н), 1.21-1.25 (м, 2Н), 1.27 (д, J=6.10 Гц, 1Н), 1.45 (дд, J=9.46, 5.19 Гц, 1Н), 1.85 (дд, J=8.24, 5.19 Гц, 1Н), 2.23 (кв, J=9.46 Гц, 1Н), 2.34-2.41 (м, 1Н), 2.61 (дд, J=14.34, 7.32 Гц, 1Н), 2.94-2.97 (м, 1Н), 358-3.63 (м, 1Н), 3.92 (с, 3Н), 4.15 (дд, J=12.05, 3.81 Гц, 1Н), 4.39 (д, J=11.60 Гц, 1Н), 4.55 (дд, J=9.92, 7.48 Гц, 1Н), 5.11 (д, J=10.68 Гц, 1Н), 5.29 (д, J=17.40 Гц, 1Н), 5.77-5.83 (м, 1Н), 5.86 (с, 1Н), 7.12 (дд, J=9.00, 2.29 Гц, 1Н), 7.18 (д, J=2.14 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7. 88 (д, J=5.80 Гц, 1Н), 8.10 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.617 мин), MS m/z 715 (MH+).
Пример 102: Получение Соединения 102

Соединение 102 получают аналогично Соединению 101 с нижеприведенными модификациями:
Модификации: В качестве исходного берут трет-амиламин, получают Соединение 102 (62.5 мг, выход 74%):1H ЯМР (CD3OD, 500 МГц) δ 0.77 (т, J=7.48 Гц, 2Н), 0.84 (т, J=7.48 Гц, 1Н), 1.04-1.08 (м, 2Н), 1.13 (д, J=1.22 Гц, 9Н), 1.16 (д, J=6.41 Гц, 3Н), 1.21 (с, 1Н), 1.22-1.28 (м, 2Н), 1.44 (дд, J=9.46, 5.19 Гц, 1Н), 1.52-1.57 (м, 1Н), 1.58-1.62 (м, 1Н), 1.85 (дд, J=7.93, 5.19 Гц, 1Н), 2.21-2.25 (м, 1Н), 2.34-2.39 (м, 1Н), 2.61 (дд, J=13.58, 7.17 Гц, 1Н), 2.93-2.98 (м, 1Н), 3.59-3.64 (м, 1Н), 4.15 (дд, J=11.75, 3.81 Гц, 1Н), 4.38 (д, J=12.51 Гц, 1Н), 4.50 (д, J=7.63 Гц, 1Н), 4.55 (дд, J=9.92, 7.78 Гц, 1Н), 5.11 (д, J=9.77 Гц, 1Н), 5.29 (д, J=16.79 Гц, 1Н), 5.77-5.83 (м, 1Н), 5.86 (т, J=4 73 Гц, 1Н), 7.12 (дд, J=8.85, 2.44 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.24 (д, J=6.10 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.10 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.617 мин), MS m/z 715 (МН+).
Пример 103: Получение Соединения 103
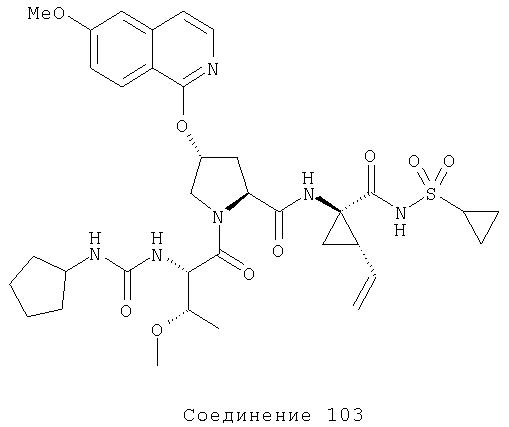
Соединение 102 получают аналогично Соединению 101 с нижеприведенными модификациями:
Модификации: В качестве исходного берут циклопентиламин, получают Соединение 103 (56.4 мг, выход 67%):1H ЯМР (CD3OD, 500 МГц) δ 1.01-1.08 (м, 2Н), 1.07 (д, J=6.10 Гц, 1Н), 1.16 (д, J=6.10 Гц, 3Н), 1.21-1.25 (м, 3Н), 1.30-1.33 (м, 1Н), 1.44 (дд, J=9.77,5.19 Гц, 1Н), 1.51-1.56 (м, 2Н), 1.60-1.65 (м, 2Н), 1.71-1.75 (м, 1Н), 1.80-1.84 (м, 1Н), 1.86 (дд, J=8.09, 5.34 Гц, 1Н), 2.20-2.25 (м, 1Н), 2.37-2.41 (м, 1Н), 2.61 (дд, J=14.04, 7.32 Гц, 1Н), 2.93-2.98 (м, 1Н), 3.60-3.65 (м, 1Н), 3.75-3.80 (м, 1Н), 3.92 (с, 3Н), 4.17 (дд, J=12.05, 3.81 Гц, 1Н), 4.37 (д, J=11.90 Гц, 1Н), 4.55-4.59 (м, 2Н), 5.10 (д, J=11.60 Гц, 1Н), 5.29 (д, J=16.48 Гц, 1Н), 5.78-5.83 (м, 1Н), 5.85 (д, J=2.44 Гц, 1Н), 7.13 (дд, J=9.16, 2.44 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.09 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.607 мин), MS m/z 727 (MH+).
Пример 104: Получение Соединения 104

К раствору смеси Соединения 100 (80 мг, 0.116 ммоля) и DIEA (31.5 мг, 0.244 ммоля) в ТГФ (2 мл) прибавляют N,N'-дисукцинилимидилкарбонат (44.6 мг, 0.174 ммоля). Полученную суспензию облучают в СВЧ-диапазоне при 80°С в течение 15 мин. Затем прибавляют суспензию циклопентоксида натрия, которую готовят, смешивая при 0°С раствор циклопентанола (110 мг, 1.28 ммоля) в ТГФ (1 мл) с NaH (60% в масле, 46.4 мг, 1.16 ммоля) в течение 15 мин при rt. Реакционную смесь перемешивают при rt 15 мин, прибавляют насыщенный водный раствор хлорида аммония (1 мл) и экстрагируют EtOAc (5 мл). Затем органические вытяжки пропускают через колонку с целитом (hydromatrix), упаривают и очищают обращенно-фазовой препаративной ВЭЖХ, получают Соединение 104 (38.2 мг, 45%):1H ЯМР (CD3OD, 500 МГц) δ 1.03-1.09 (м, 3Н), 1.16 (д, J=6.10 Гц, 3Н), 1.20-1.25 (м, 1Н), 1.25-1.30 (м, J=10.22, 5.34 Гц, 1Н), 1.40-1.45 (м, J=10.83, 3.81 Гц, 1Н), 1.46 (дд, J=9.61, 5.34 Гц, 1Н), 1.58-1.63 (м, 3Н), 1.70-1.75 (м, 2Н), 1.86 (дд, J=7.63, 5.49 Гц, 1Н), 2.22-2.26 (м, 1Н), 2.34-2.39 (м, 1Н), 2.59-2.64 (м, 1Н), 2.94-2.98 (м, 1Н), 3.67 (дд, J=7.78, 6.56 Гц, 1Н), 3.92 (с, 3Н), 4.13 (дд, J=10.83, 4.12 Гц, 1Н), 4.37-4.42 (м, 1Н), 4.56 (дд, J=10.07, 7.32 Гц, 1Н), 4.71-4.76 (м, 1Н), 5.12 (д, J=10.68 Гц, 1Н), 5.31 (д, J=16.79 Гц, 1Н), 5.80 (м, 1Н), 5.85 (с, 1Н), 7.13 (д, J=10.68 Гц, 1Н), 7.19 (д, J=1.83 Гц, 1Н), 7.25 (д, J=6.10 Гц, 1Н), 7.89 (д, J=5.80 Гц, 1Н), 8.09 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.697 мин), MS m/z 728 (MH+).
Пример 105: Получение Соединения 105

К раствору смеси Соединения 100 (80.0 мг, 0.116 ммоля) и DIEA (31.5 мг, 0.244 ммоля) в DCM (2 мл) прибавляют дитрет-амилдикарбонат (57.1 мг, 0.232 ммоля). Перемешивают при rt в течение 14 час, растворитель отгоняют и продукт очищают обращенно - фазовой препаративной ВЭЖХ, получают Соединение 105 (62.5 мг, выход 74%);1H ЯМР (CD3OD, 500 МГц) δ 0.79 (т, J=7.48 Гц, 3Н), 1.04-1.08 (м, 3Н), 1.17 (д, J=6.10 Гц, 3Н), 1.19-1.23 (с, 3Н), 1.24 (с, 3Н), 1.39-1.43 (м, 1Н), 1.46 (дд, J=9.61, 5.34 Гц, 1Н), 1.60-1.65 (м, 2Н), 1.86 (дд, J=7.93, 5.49 Гц, 1Н), 2.22 (кв, J=8.85 Гц, 1Н), 2.35-2.40 (м, 1Н), 2.61 (дд, J=14.04, 7.15 Гц, 1Н), 2.94-3.00 (м, 1Н), 3.64-4.00 (м, 1Н), 3.92 (с, 4Н), 4.14 (дд, J=11.90, 3.05 Гц, 1Н), 4.35 (д, J=7.93 Гц, 1Н), 4.40 (д, J=11.90 Гц, 1Н), 4.55 (дд, J=9.31, 7.78 Гц, 1Н), 5.11 (д, J=10.68 Гц, 1Н), 5.30 (д, J=16.79 Гц, 1Н), 5.79-5.83 (м, 1Н), 5.85 (с, 1Н), 7.12 (д, J=9.16 Гц, 1Н), 7.18 (с, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.86-7.90 (м, 1Н), 8.09 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.740 мин), MS m/z 730 (MH+).
Пример 106: Получение Соединения 106

Соединение 106 получают аналогично Соединению 105 с нижеприведенными модификациями:
Модификации: В качестве исходного берут 2,2,2-трифтор-1,1-диметилэтиловый эфир пиридин-2-илкарбоновой кислоты, получают Соединение 106 (58.1 мг, выход 65%):1H ЯМР (CD3OD, 500 МГц) δ 1.04-1.08 (м, 3Н), 1.17 (д, J=6.10 Гц, 3Н), 1.19-1.23 (м, 1Н), 1.23-1.27 (м, 1Н), 1.28 (с, 3Н), 1.46 (дд, J=9.46, 5.19 Гц, 2Н), 1.49 (с, 2Н), 1.86 (дд, J=8.09, 5.34 Гц, 1Н), 2.21 (кв, J=8.85 Гц, 1Н), 2.36-2.40 (м, 1Н), 2.62 (дд, J=13.74, 7.32 Гц, 1Н), 2.93-2.98 (м, 1Н), 3.65-3.70 (м, 1Н), 3.92 (с, 3Н), Н), 4.12 (дд, J=11.90, 3.66 Гц, 1Н), 4.31 (д, J=8.24 Гц, 1Н), 4.42 (д, J=11.90 Гц, 1Н), 4.57 (д, J=10.07, 7.32 Гц, 1Н), 5.11 (д, J=10.38 Гц, 1Н), 5.30 (д, J=16.79 Гц, 1Н), 5.78-5.83 (м, 1Н), 5.84 (с, 1Н), 7.12 (дд, J=9.00, 2.29 Гц, 1Н), 7.19 (д, J=2.14 Гц, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.89 (д, J=6.10 Гц, 1Н), 8.09 (д, J=8.85 Гц, 1Н); LC-MS (время удерживания: 1.770 мин), MS m/z 770 (МН+ ).
Пример 107: Получение Соединения 107
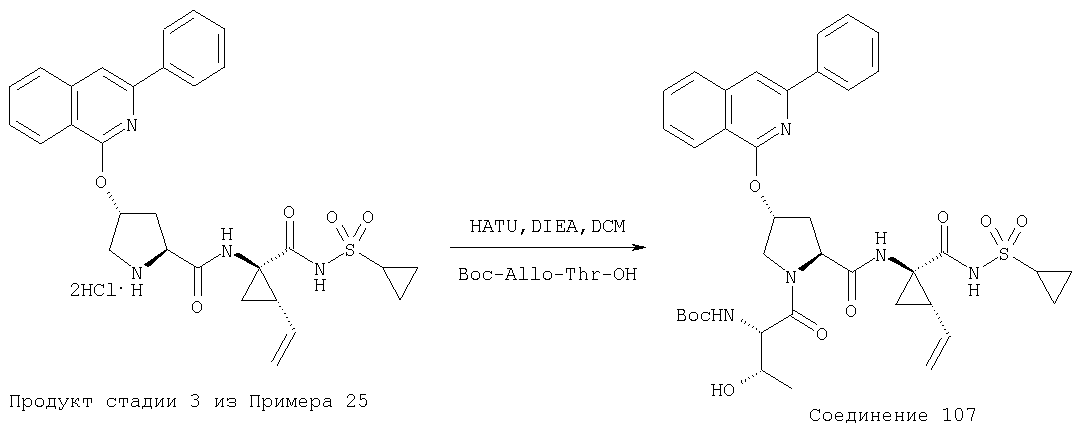
К раствору продукта стадии 3 из Примера 25 (100.0 мг, 0.116 ммоля), DIEA (62.5 мг, 0.463 ммоля) и HATU (92.0 мг, 0.242 ммоля) прибавляют Boc-allo-Thr-ОН (43.5 мг, 0.177 ммоля). Реакционную смесь перемешивают при rt в течение 3 час, промывают 5% водным раствором NaHCO3 (1 мл ). Водный слой экстрагируют 2×2 мл DCM. Объединенные органические вытяжки промывают 5% водным раствором лимонной кислоты (1 мл), рассолом, сушат MgSO4, упаривают и очищают обращенно-фазовой препаративной ВЭЖХ, получают Соединение 107 (62.5 мг, выход 52%):1H ЯМР (CD3OD, 500 МГц) δ 0.8-1.02 (м, 1Н), 1.04-1.08 (м, 2Н), 1.23-1.27 (м, 12Н), 1.42 (дд, J=9.46, 5.19 Гц, 1Н), 1.86 (т, J=6.26 Гц, 1Н), 2.23-2.27 (м, 1Н), 2.46-2.50 (м, 1Н), 2.76 (дд, J=14.04, 6.71 Гц, 1Н), 2.95-2.99 (м, 1Н), 3.94-3.98 (м, 1Н), 4.28 (д, J=7.32 Гц, 2Н), 4.52 (д, J=12.51 Гц, 1Н), 4.63 (т, J=9.00 Гц, 1Н), 5.12 (д, J=10.07 Гц, 1Н), 5.31 (д, J=16.79 Гц, 1Н), 5.77-5.83 (м, 1Н), 6.09 (с, 1Н), 7.36-7.41 (м, 1Н), 7.47 (т, J=7.17 Гц, 3Н), 7.52 (д, J=7.63 Гц, 1Н), 7.70 (т, J=7.17 Гц, 1Н), 7.85 (с, 1Н), 7.88 (д, J=8.24 Гц, 1Н), 8.17 (д, J=7.93 Гц, 2Н), 8.22 (д, J=7.63 Гц, 1Н); LC-MS (время удерживания: 1.937 мин), MS m/z 748 (MH+).
Пример 108: Получение Соединения 108

Соединение 108 получают аналогично Соединению 107 с нижеприведенными модификациями:
Модификации: В качестве исходного берут Вос-(2S, 3S)-амино-3-метоксибутановую кислоту, получают Соединение 108 (75.1 мг, выход 51%):1H ЯМР (CD3OD, 500 МГц) δ 0.80-1.02 (м, 4Н), 1.18 (д, J=6.10 Гц, 3Н), 1.28 (с, 9Н), 1.44 (дд, J=9.77, 1.30 Гц, 1Н), 1.45-1.50 (м, 1Н), 1.85-1.90 (м, 1H), 2.14-2.18 (м, 1Н), 2.55-2.59 (м, 1Н), 2.72-2.76 (м, 1Н), 2.91-2.95 (м, 1Н), 3.34 (с, 3Н), 3.65-3.69 (м, 1Н), 4.32 (д, J=10.68 Гц, 1Н), 4.40 (д, J=7.93 Гц, 1Н), 4.46 (д, J=13.12 Гц, 1Н), 4.60 (т, J=8.24 Гц, 1Н), 5.07 (д, J=9.46 Гц, 1Н), 5.26 (д, J=17.40 Гц, 1Н), 5.82-5.86 (м, 1Н), 6.08 (с, 1Н), 7.38 (дд, J=7.32, 6.10 Гц, 1Н), 7.47 (т, J=7.02 Гц, 3Н), 7.51 (д, J=5.80 Гц, 1Н), 7.69 (т, J=6.56 Гц, 1Н), 7.85 (с, 1Н), 7.88 (д, J=7.63 Гц. 1Н), 8.18 (д, J=8.24 Гц, 3Н), 8.21 (д, J=9.16 Гц, 1Н); LC-MS (время удерживания: 1.973 мин), MS m/z 762 (MH+).
Пример 109: Получение Соединения 109
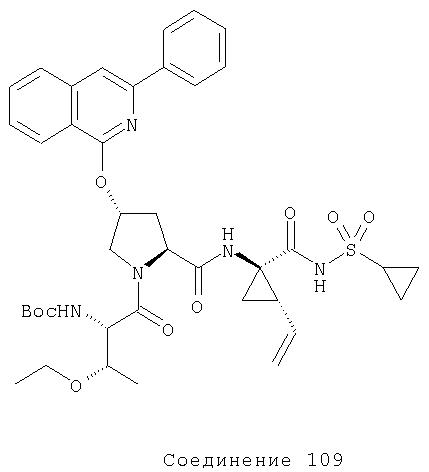
Соединение 109 получают аналогично Соединению 107 с нижеприведенными модификациями:
Модификации: В качестве исходного берут Вос-(2S,3S)-амино-3-этоксибутановую кислоту, получают Соединение 109 (57.2 мг, выход 47%);1H ЯМР (CD3OD, 500 МГц) δ 1.02-1.08 (м, 4Н), 1.17 (д, J=6.10 Гц, 6Н), 1.19 (с, 1Н), 1 9-1.24 (м,1Н), 1.23-1.27 (м, J=3.97 Гц, 1Н), 1.30 (с, 9Н), 1.44 (дд, J=9.77. 5.50 Гц, 1Н), 1.46 (с,1Н), 1.88 (дд, J=7.78, 5.95 Гц, 1Н), 2.20-1.25 (м, 1Н), 2.49-2.54 (м, 1Н), 2.69-2,73 (м, 1Н), 2.93-2.97 (м, 1Н), 3.53-3.57 (м, 2Н), 3.75-3.80 (м, 1Н), 4.34 (дд, J=11.75, 3.20 Гц, 1Н), 4.42 (т, J=8.30 Гц, 2Н), 4.57 (т, J=8.09 Гц, 1Н), 5.11 (д, J=10.38 Гц, 1Н), 5.29 (д, J=17.40 Гц, 1Н), 5.78-5.83 (м, 1Н), 6.09 (с, 1Н), 7.38 (т, J=7.32 Гц, 1Н), 7.47 (т, J=7.63 Гц, 2Н), 7.52 (д, J=7.02 Гц, 1Н), 7.70 (т, J=7.93 Гц, 1Н), 7.86 (с, 1Н), 7.88 (д, J=7.93 Гц, 1Н), 8.18 (д, J=7.32 Гц, 2Н), 8.22 (д, J=8.24 Гц, 1Н); LC-MS (время удерживания: 2.030 мин), MS m/z 776 (МН+).
Пример 110: Получение Соединения 110

Соединение 110 получают аналогично Соединению 89 с нижеприведенными модификациями:
Модификации: В качестве исходного берут Boc-L-Thr(Bn)-ОН, получают Соединение 110 (49.8 мг, выход 48%); LC-MS (время удерживания: 1.857 мин), MS m/z 792 (MH+).
Подраздел D
Пример 120: Получение Соединения 120

Схема 1

Стадия 1:
Раствор Соединения 23 (см. Пример 23) (1.50 г, 2.19 ммоля) в DCM (50 мл) и трифторуксусной кислоте (50 мл) перемешивают 3 час при rt. Смесь упаривают в вакууме, получают вязкий остаток, который затем растворяют в 1,2-дихлорэтане и снова упаривают в вакууме, получают нужную бис-соль трифторуксусной кислоты в виде не чисто белого стекловидного твердого вещества (выход количественный). Вещество используют непосредственно на следующей стадии без дополнительной очистки.
Стадия 2:
К раствору продукта из Примера 120, Стадия 1 (118 мг, 0.146 ммоля) в 1,2-дихлорэтане (3 мл) прибавляют п-толилхлорформиат (32.4 мг, 0.190 ммоля) и N,N-диизопропилэтиламин (94.5 мг, 0.731 ммоля). Смесь перемешивают при rt в течение 72 час. Промывают буферным раствором рН4 (3×3 мл) и промывные воды снова экстрагируют 1,2-дихлорэтаном (3 мл). Органические вытяжки объединяют и упаривают в вакууме. Сырой продукт растворяют в МеОН и очищают обращенно - фазовой препаративной ВЭЖХ, получают титульное соединение (Соединение 120) в виде желтого стекловидного вещества (64.2 мг, выход 61.1%):1H ЯМР (CD3OD, 500 МГц) δ 1.06-1.10 (м, 3H), 1.12 (с, 9Н), 1.24-1.28 (м, 2Н), 1.44 (дд, J=9.31, 5.34 Гц, 1Н), 1.89 (дд, J=7.93, 5.49 Гц, 1Н), 2.21-2.28 (м, 2Н), 2.31 (с, 3H), 2.62-2.66 (м, 1Н), 2.93-2.99 (м, 1Н), 4.12 (дд, J=11.90, 3.66 Гц, 1Н), 4.42 (д, J=11.60 Гц, 1Н), 4.57 (дд, J=10.22, 7.17 Гц, 1Н), 5.13 (д, J=10.38 Гц, 1Н), 5.30 (д, J=17.09 Гц, 1Н), 5.76 (ддд, J=17.09, 9.77, 9.46 Гц, 1Н), 5.87 (с, 1Н), 6.79 (д, J=8.24 Гц, 2Н), 7.07 (д, J=8.24 Гц, 2Н), 7.30 (д, J=6.10 Гц, 1Н), 7.40 (т, J=7.63 Гц, 1Н), 7.68 (т, J=7.63 Гц, 1Н), 7.79 (д, J=8.24 Гц, 1Н), 7.93 (д, J=5.80 Гц, 1Н), 8.17 (д, J=8.24 Гц, 1Н); MS m/z 718 (MH+).
Пример 121: Получение Соединения 121
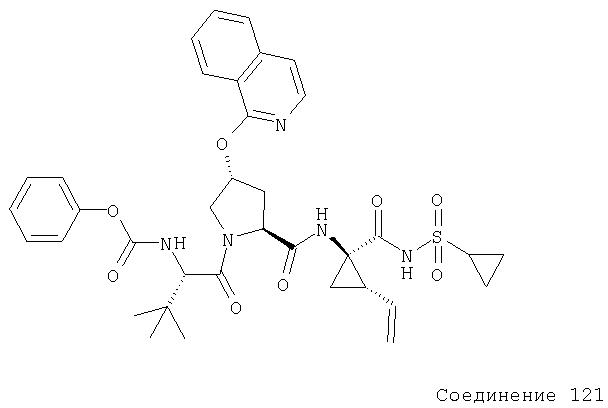
Соединение 121 получают по Схеме 1 из Примера 120 за исключением того, что на стадии 2 вместо п-толилхлорформиата берут фенилхлорформиат.
Стадия 2:
Модификации: берут 30 мг (0.19 ммоля) фенилхлорформиата, получают 89.0 мг продукта в виде желтого стекловидного вещества (выход 50%). MS m/z 704 (MH+).
Пример 122: Получение Соединения 122

Соединение 122 получают по Схеме 1 из Примера 120 за исключением того, что на стадии 2 вместо п-толилхлорформиата берут 4-фторфенилхлорформиат.
Стадия 2:
Модификации: берут 33 мг (0.19 ммоля) 4-фторфенилхлорформиата, получают 83.1 мг продукта в виде желтого стекловидного вещества (выход 78.8%). MS m/z 722 (МН+).
Пример 123: Получение Соединения 123

Соединение 123 получают по Схеме 1 из Примера 120 за исключением того, что на стадии 2 вместо п-толилхлорформиата берут 4-метоксифенилхлорформиат.
Стадия 2:
Модификации: берут 35 мг (0.19 ммоля) 4-метоксифенилхлорформиата, получают 70.2 мг продукта в виде желтого стекловидного вещества (выход 65.4%).1H ЯМР (CD3OD, 500 МГц) δ 1.06-1.10 (м, 3H), 1.11 (с, 9Н), 1.24-1.28 (м, 2Н), 1.44 (дд, J=9.46, 5.49 Гц, 1Н), 1.89 (дд, J=7 93, 5.49 Гц, 1Н), 2.24 (кв, J=8.85 Гц, 1Н), 2.31 (ддд, J=13.81, 10.30, 3.97 Гц, 1Н), 2.62-2.66 (м, 1Н), 2.94-2.98 (м, 1Н), 3.77 (с, 3H), 4.12 (дд, J=11.60, 3.66 Гц, 1Н), 4.42 (д, J=1 1.60 Гц, 1Н), 4.57 (дд, J=10.07, 7.32 Гц, 1Н), 5.13 (д, J=10.68 Гц, 1Н), 5.30 (д, J=16.79 Гц, 1Н), 5.72-5.80 (м, 1Н), 5.87 (с, 1Н), 6.80 (д, J=2.44 Гц. 4Н), 7.30 (д, J=5.80 Гц, 1Н), 7.42 (т, J=7.48 Гц, 1Н), 7.69 (т, J=7.63 Гц, 1Н), 7.80 (д, J=7.93 Гц, 1Н), 7.93 (д, J=5.80 Гц, 1Н), 8.18 (д, J=8.24 Гц, 1Н); MS m/z 734 (MH+).
Пример 124: Получение Соединения 124
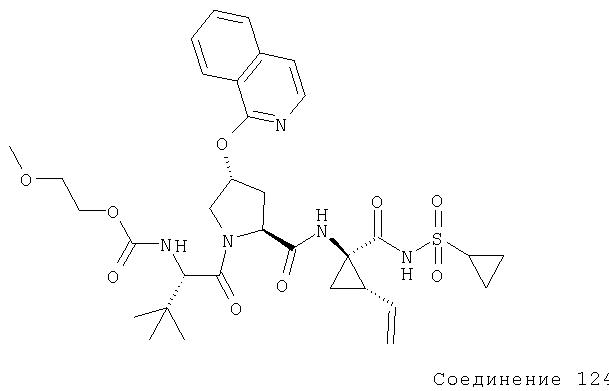
Соединение 124 получают по Схеме 1 из Примера 120 за исключением того, что на стадии 2 вместо п-толилхлорформиата берут 2-метоксиэтиловый эфир хлормуравьиной кислоты.
Стадия 2:
Модификации: берут 26 мг (0.19 ммоля) 2-метоксиэтилового эфира хлормуравьиной кислоты, получают 87.4 мг продукта в виде желтого вязкого (липкого) масла (выход 87.2%).1H ЯМР (CD3OD, 500 МГц) δ 0.96-1.02 (м, 3H), 1.05 (с, 9Н), 1.16-1.18 (м, 2Н), 1.40(дд,J=9.46,5.19Гц, 1Н), 1.85 (дд, J=7.93, 5.19 Гц, 1Н), 2.15 (кв, J=8.75 Гц, 1Н), 2.40 (ддд, J=13.89, 10.07, 4.12 Гц, 1Н), 2.65 (дд, J=13.58, 7.17 Гц, 1Н), 2.90 (ддд, J=12.89, 8.16, 4.88 Гц, 1Н), 3.27 (с, 3H), 3.36-3.44 (м, 2Н), 3.81-3.84 (м, 1Н), 3.92-3.96 (м, 1Н), 4.12 (дд, J=11.60, 3.36 Гц, 1Н), 4.44 (д, J=11.60 Гц, 1Н), 4.57 (дд, J=9.46, 7.93 Гц, 1Н), 5.07 (д, J=10.38 Гц, 1Н), 5.25 (д, J=17.09 Гц, 1Н), 5.80 (ддд, J=17.32, 9.77, 9.54 Гц, 1Н), 5.87 (с, 1Н), 7.32 (д, J=5.80 Гц, 1Н), 7.55 (т, J=7.32 Гц, 1Н), 7.70 (т, J=7.48 Гц, 1Н). 7.80 (д, J=7.93 Гц, 1Н), 7.96 (д, J=5.80 Гц, 1Н), 8.19 (д, J=8.24 Гц, 1Н); MS m/z 686 (МН+).
Пример 125: Получение Соединения 125
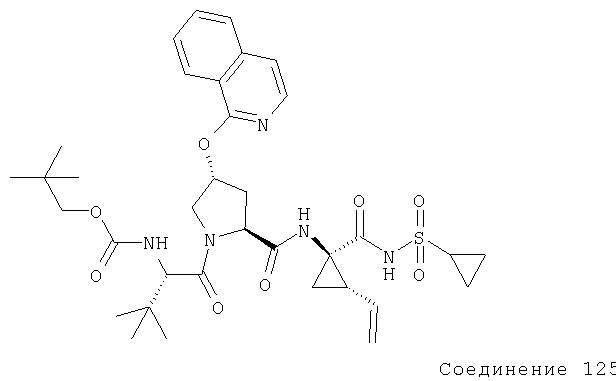
Соединение 125 получают по Схеме 1 из Примера 120 за исключением того, что на стадии 2 вместо п-толилхлорформиата берут неопентилхлорформиат.
Стадия 2:
Модификации: берут 29 мг (0.19 ммоля) неопентилхлорформиата, получают 57.4 мг продукта в виде желтого стекловидного вещества (выход 56.2%).1H ЯМР (CD3OD, 500 МГц) δ 0.83 (с, 9Н), 1.05 (д, J=2.44 Гц, 9Н), 1.07-1.09 (м, 2Н), 1.23-1.27 (м, 2Н), 1.43-1.46 (м, 1Н), 1.87-1.90 (м, 1H), 2.21-2.25 (м, 1Н), 2.29-2.33 (м, 1Н), 2.61-2.65 (м, 1Н), 2.92-2.96 (м, 1Н), 3.42 (д, J=10.07 Гц, 1Н), 3.56 (д, J=10.07 Гц, 1Н), 4.09-4.11 (м, 1Н), 4.33 (д, J=9.16 Гц, 1Н), 4.43 (д, J=11.29 Гц, 1Н), 4.54-4.57 (м, 1Н), 5.12 (д, J=10.07 Гц, 1Н), 5.30 (д, J=17.40 Гц, 1Н), 5.73-5.80 (м, 1Н), 5.88 (с, 1Н), 7.33 (д, J=5.49 Гц, 1Н), 7.53 (м, 1Н), 7.71 (т, J=6.87 Гц, 1Н), 7.81 (д, J=7.93 Гц, 1Н), 7.97 (д, J=5.80 Гц, 1Н), 8.19 (д, J=7.63 Гц, 1Н); MS m/z 698 (MH+).
Пример 126: Получение Соединения 126
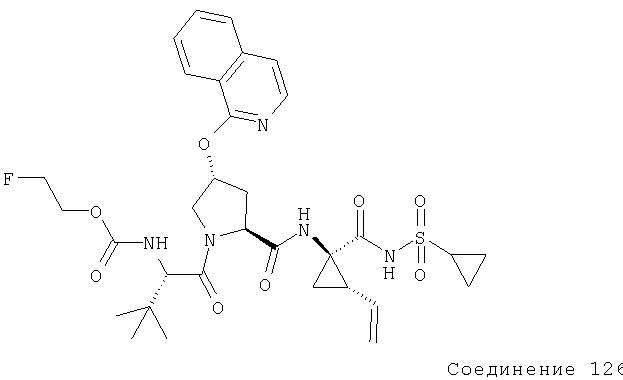
Соединение 126 получают по Схеме 1 из Примера 120 за исключением того, что на стадии 2 вместо п-толилхлорформиата берут 2-фторэтилхлорформиат.
Стадия 2:
Модификации: берут 24 мг (0.19 ммоля) 2-фторэтилхлорформиата, получают 58.9 мг продукта в виде желтого стекловидного вещества (выход 59,8%).1H ЯМР (CD3OD, 500 МГц) δ 1.05 (д, J=2.14 Гц, 9Н), 1.07-1.09 (м, 2Н), 1.22-1.27 (м, 2Н), 1.42-1.45 (м, 1Н), 1.87-1.90 (м, 1Н), 2.24 (кв, J=8.75 Гц, 1Н), 2.28-2.33 (м, 1Н), 2.63 (дд, J= 13.43, 6.41 Гц, 1Н), 2.92-2.96 (м, 1Н), 3.92-4.10 (м, 3H), 4.31-4.37 (м, 2Н), 4.42-4.46 (м, 2Н), 4.54-4.57 (м, 1Н), 5.12 (д, J=10.38 Гц, 1Н), 5.29 (д, J=17.09 Гц, 1Н), 5.71-5.79 (м, 1Н), 5.88 (с, 1Н), 7.33 (д, J=5.80 Гц, 1Н), 7.55 (т, J=7.17 Гц, 1Н), 7.71 (м, 1Н), 7.81 (д, J=7.93 Гц, 1Н), 7.96 (д, J=5.80 Гц, 1Н), 8.19 (д, J=7.63 Гц, 1Н); MS m/z 674 (MH+).
Пример 127: Получение Соединения 127

Соединение 127 получают по Схеме 1 из Примера 120 за исключением того, что на стадии 2 вместо п-толилхлорформиата берут 2-метоксифенилхлорформиат.
Стадия 2:
Модификации: берут 35 мг (0.19 ммоля) 2-метоксифенилхлорформиата, получают 97.6 мг продукта в виде желтого вязкого (липкого) масла (выход 91.1%). MS m/z 734 (MH+).
Пример 128: Получение Соединения 128

Соединение 128 получают по Схеме 1 из Примера 120 за исключением того, что на стадии 2 вместо п-толилхлорформиата берут 2-(-)-(1R)-ментилхлорформиат.
Стадия 2:
Модификации: берут 42 мг (0.19 ммоля) 2-(-)-(1R)-ментилхлорформиата, получают 69.1 мг продукта в виде белого стекловидного вещества (выход 61.7%). MS m/z 766 (МН+).
Пример 129: Получение Соединения 129

Соединение 129 получают по Схеме 1 из Примера 120 за исключением того, что на стадии 2 вместо п-толилхлорформиата берут гексилхлорформиат.
Стадия 2:
Модификации: берут 31 мг (0.19 ммоля) гексилхлорформиата, получают 66.7 мг продукта в виде желтого стекловидного вещества (выход 64.1%).1H ЯМР (CD3OD, 500 МГц) δ 0.87-0.99 (м, 5Н), 1.05 (с, 9Н), 1.07-1.09 (м, 2Н), 1.22-1.28 (м, 6Н), 1.43-1.48 (м, 3H), 1.88 (дд, J=8.24, 5.49 Гц, 1Н), 2.24 (кв, J=8.85 Гц, 1Н), 2.28-2.33 (м, 1Н), 2.63 (дд, J=14.34, 7.63 Гц, 1Н), 2.92-2.97 (м, 1Н), 3.72 (дт, J=10.61, 6.60 Гц, 1Н), 3.81-3.86 (м, 1Н), 4.10 (дд, J=11.60, 3.36 Гц, 1Н), 4.32 (д, J=8.85 Гц, 1Н), 4.43 (д, J=11.90 Гц, 1Н), 4.55 (дд, J=9.77, 7.32 Гц, 1Н), 5.13 (д, J=10.38 Гц, 1Н), 5.30 (д, J=17.09 Гц, 1Н), 5.76 (ддд, J=17.09, 10.07, 9.16 Гц, 1Н), 5.89 (с, 1Н), 7.33 (д, J=5.80 Гц, 1Н), 7.54 (т, J=7.48 Гц, 1Н), 7.69-7.72 (м, 1Н), 7.81 (д, J=8.24 Гц, 1Н), 7.97 (д, 7-6.10 Гц, 1Н), 8.20 (д, J=8.24 Гц, 1Н); MS m/z 712 (MH+).
Пример 130: Получение Соединения 130

Схема 1
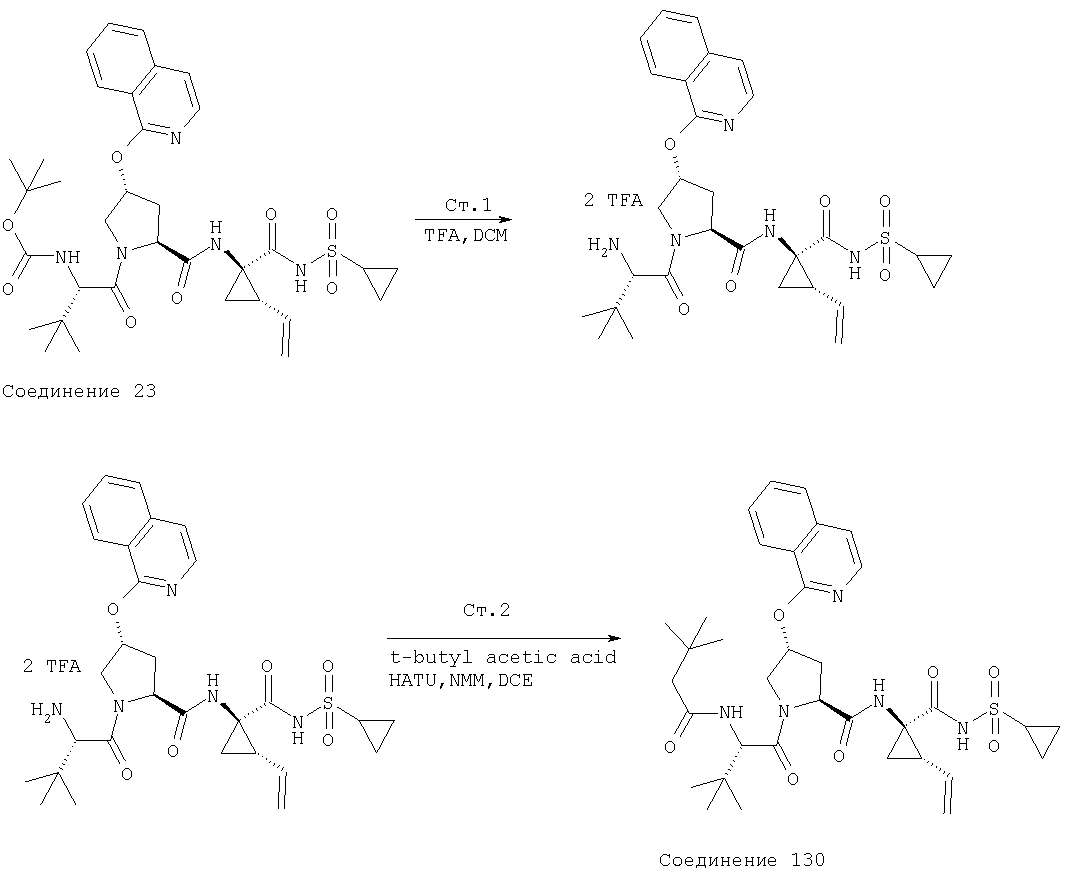
Стадия 1:
Раствор Соединения 23 (см. Пример 23) (1.50 г, 2.19 ммоля) в DCM (50 мл) и трифторуксусной кислоте (50 мл) перемешивают в течение 3 часов. Смесь упаривают в вакууме, полученный вязкий остаток растворяют в 1,2-дихлорэтане и снова упаривают в вакууме, получают нужный продукт - соль с двумя молекулами трифторуксусной кислоты (бис-соль) в виде не чисто белого стекловидного вещества (количественный выход). Вещество непосредственно используют на следующей стадии без дополнительной очистки.
Стадия 2:
Смесь продукта стадии 1 (118 мг, 0.146 ммоля), трет-бутилуксусной кислоты (22 мг, 0.19 ммоля), HATU (72 мг, 0.19 ммоля) и N-метилформалина (59 мг, 0.58 ммоля) в 1,2-дихлорэтане перемешивают в течение 24 час при rt. Реакционную смесь промывают буферньм раствором с рН 4 (3×3 мл) и промывные воды снова экстрагируют 1,2-дихлорэтаном (3 мл). Органические вытяжки объединяют и упаривают в вакууме. Сырой продукт растворяют в МеОН и очищают обращенно-фазовой препаративной ВЭЖХ, получают титульное соединение (Соединение 130) в виде стекловидного вещества слегка желтоватого цвета (43.4 мг, выход 43.5%).1H ЯМР (CD3OD) δ 0.82 (д, J=1.83 Гц, 9Н), 1.06 (д, J=2.14 Гц, 9Н), 1.07-1.10 (м, 2Н), 1.22-1.28 (м, 2Н), 1.43-1.46 (м, 1Н), 1.87-1.90 (м, 1Н), 1.99 (д, J=1.83 Гц, 2Н), 2.20-2.26 (м, 1Н), 2.27-2.33 (м, 1Н), 2.59-2.64 (м, 1Н), 2.93-2.97 (м, 1Н), 4.12-4.14 (м, 1Н), 4.42 (д, J=11.60 Гц, 1Н), 4.51-4.55 (м, 1Н), 4.67 (дд, J=9.31,1.98 Гц, 1Н), 5.11-5.14 (м, 1Н), 5.29 (д, J=17.40 Гц, 1Н), 5.72-5.80 (м, 1Н), 5.89 (д, J=1.83 Гц, 1Н), 7.32 (дд, J=5.80, 2.14 Гц, 1Н), 7.52-7.55 (м, 1Н), 7.69-7.72 (м, 1Н), 7.81 (д, J=8.24 Гц, 1Н), 7.96 (дд, J=5.80, 1.83 Гц, 1Н), 8.17 (д, J=8.24 Гц, 1Н); MS m/z 682 (MH+).
Пример 131: Получение Соединения 131
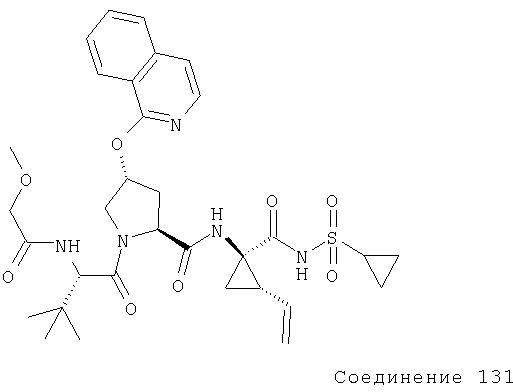
Соединение 131 получают по Схеме 1 из Примера 130 за исключением того, что на стадии 2 вместо трет-бутилуксусной кислоты берут метоксиуксусную кислоту.
Стадия 2:
Модификации: берут 17 мг (0.19 ммоля) метоксиуксусной кислоты, получают 49.9 мг продукта в виде слегка желтоватого стекловидного вещества (выход 52.0%).1H ЯМР (CD3OD, 500 МГц) δ 1.05-1.08 (м, 11Н), 1.24-1.26 (м, 2Н), 1.45 (ддд, J=9.31, 5.34, 3.66 Гц, 1Н), 1.88 (ддд, J=8.39, 5.19, 3.81 Гц, 1Н), 2.21-2.27 (м, 1Н), 2.29-2.35 (м, 1Н), 2.60-2.64 (м, 1Н), 2.91-2.97 (м, 1Н), 3.34 (д, J=3.66 Гц, 3H), 3.69 (дд, J=15.26, 3.66 Гц, 1Н), 3.81-3.85 (м, 1Н), 4.15 (дт, J=11.67, 3.62 Гц, 1Н), 4.35 (д, J=11.90 Гц, 1Н), 4.55 (ддд, J=10.30, 6.94, 3.20 Гц, 1Н), 4.66 (дд, J=9.61, 3.51 Гц, 1Н), 5.11-5.14 (м, 1Н), 5.28-5.32 (м, 1Н), 5.73-5.81 (м, 1Н), 5.90 (д, J=3.36 Гц, 1Н), 7.33 (дд, J=5.65, 3.20 Гц, 1Н), 7.54-7.58 (м, 1Н), 7.69-7.73 (м, 1Н), 7.80-7.82 (м, 1Н), 7.95-7.97 (м, 1Н), 8.15 (дд, J=8.39, 2.59 Гц, 1Н); MS m/z 656 (MH+).
Пример 132: Получение Соединения 132
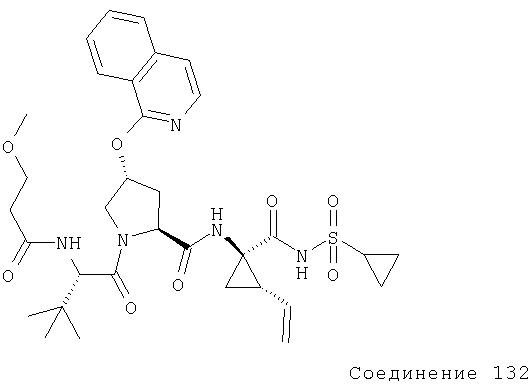
Соединение 132 получают по Схеме 1 из Примера 130 за исключением того, что на стадии 2 вместо трет-бутилуксусной кислоты берут метоксипропионовую кислоту.
Стадия 2:
Модификации: берут 20 мг (0.19 ммоля) метоксипропионовой кислоты, получают 50.0 мг продукта в виде желтого стекловидного вещества (выход 51.1%).1H ЯМР (CD3OD, 500 МГц) δ 1.06 (д, J=1.83 Гц, 9Н), 1.07-1.09 (м, 2Н), 1.23-1.27 (м, 2Н), 1.44 (ддд, J=9.38, 5.26, 1.83 Гц, 1Н), 1.87-1.90 (м, 1Н), 2.21-2.27 (м, 1Н), 2.29-2.33 (м, 2Н), 2.40-2.46 (м, 1Н), 2.59-2.64 (м, 1Н), 2.92-2.97 (м, 1Н), 3.25 (д, J=1.83 Гц, 3H), 3.45-3.54 (м, 2Н), 4.12-4.16 (м, 1Н), 4.37 (д, J=11.60 Гц, 1Н), 4.52-4.55 (м, 1Н), 4.65 (дд, J=9.16, 1.83 Гц, 1Н), 5.12 (д, J=10.38 Гц, 1Н), 5.30 (д, J=17.40 Гц, 1Н), 5.72-5.80 (м, 1Н), 5.89 (с, 1Н), 7.33 (дд, J=5.65, 1.98 Гц, 1Н), 7.54-7.58 (м, 1Н), 7.69-7.73 (м, 1Н), 7.81 (д, J=8.24 Гц, 1Н), 7.96 (дд, J=6.10, 1.83 Гц, 1Н), 8.18 (д, J=8.24 Гц, 1Н); MS m/z 670 (MH+).
Пример 133: Получение Соединения 133

Соединение 133 получают по Схеме 1 из Примера 130 за исключением того, что на стадии 2 вместо трет-бутилуксусной кислоты берут (S)-1,4-бензодиоксан-2-карбоновую кислоту.
Стадия 2:
Модификации: берут 35 мг (0.19 ммоля) (S)-1, 4-бензодиоксан-2-карбоновой кислоты, получают 54.0 мг продукта в виде слегка желтоватого стекловидного вещества (выход 49.5%).1H ЯМР (CD3OD) δ 0.83 (д, J=3.36 Гц, 9Н), 1.05-1.08 (м, 2Н), 1.20-1.25 (м, 1Н), 1.26-1.31 (м, 1Н), 1.45-1.49 (м, 1Н), 1.86-1.90 (м, 1Н), 2.21-2.25 (м, 1Н), 2.28-2.34 (м, 1Н), 2.59-2.65 (м, 1Н), 2.90-2.94 (м, 1Н), 4.12-4.17 (м, 2Н), 4.32 (д, J=11.90 Гц, 1Н), 4.35-4.39 (м, 1Н), 4.55-4.61 (м, 3H), 5.11-5.14 (м, 1Н), 5.28-5.32 (м, 1Н), 5.75-5.83 (м, 1Н), 5.90 (д, J=3.66 Гц, 1Н), 6.80-6.89 (м, 3H), 7.03-7.07 (м, 1Н), 7.32-7.34 (м, 1Н), 7.55-7.58 (м, 1Н), 7.68-7.72 (м, 1Н), 7.80-7.82 (м, 1Н), 7.96-7.98 (м, 1Н), 8.15-8.18 (м, 1Н); MS m/z 746 (MH+).
Поимер 134: Получение Соединения 134
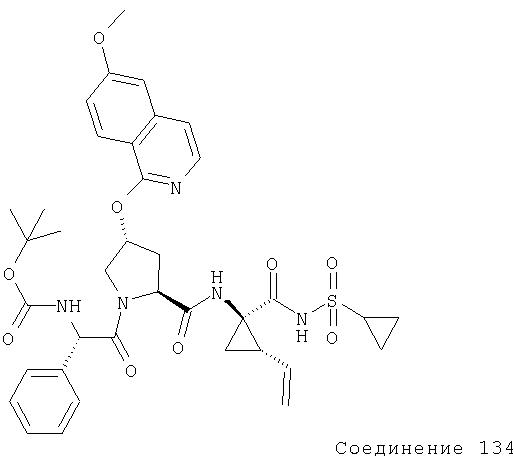
Схема 1
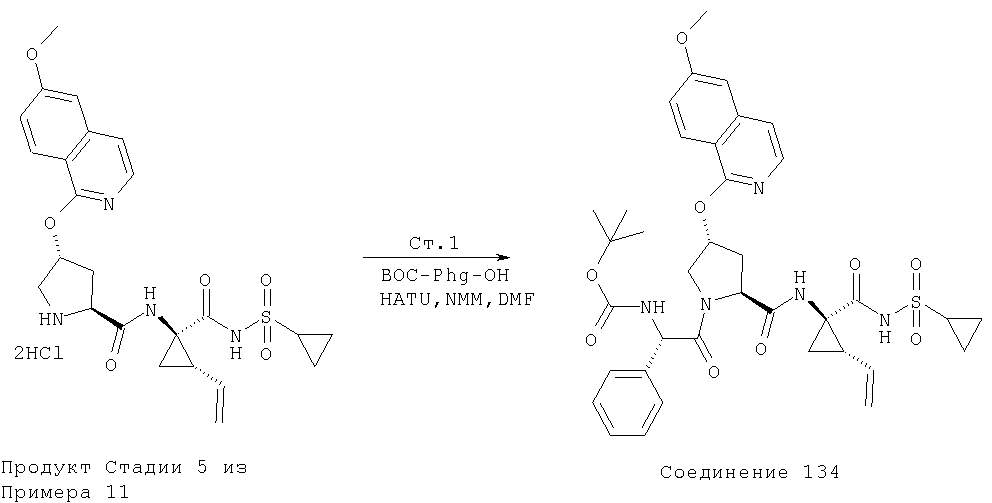
Смесь продукта из Примера 11, Стадия 5 (100 мг, 0.172 ммоля), N-α-трет-бутоксикарбонил-1-фенилглицина (45.3 мг, 0.180 ммоля), HATU (84.9 мг, 0.223 ммоля) и N-метилморфолина (87.0 мг, 0.859 ммоля) в ДМФА (1.0 мл) перемешивают при rt в течение 18 час. Смесь очищают непосредственно обращенно - фазовой препаративной ВЭЖХ, получают 29.7 мг (выход 23.6%) Соединения 134 в виде белого порошка:1H ЯМР (CD3OD) δ 0.97-1.07 (м, 2Н), 1.12-1.17 (м, 1Н), 1.22-1.32 (м, 2Н), 1.38 (с, 9Н), 1.90 (дд, J=8.09, 5.34 Гц, 1Н), 2.20-2.28 (м, 2Н), 2.54 (дд, J=13.58, 6.56 Гц, 1Н), 2.85-2.89 (м, J=8.24 Гц, 1Н), 3.50 (д, J=10.99 Гц, 1Н), 3.93 (с, 3H), 4.11 (д, J=11.60 Гц, 1Н), 4.63 (дд, J=9.46, 7.32 Гц, 1Н), 5.13 (дд, J=10.38, 1.53 Гц, 1Н), 5.32 (д, J=17.09 Гц, 1Н), 5.47 (с, 1Н), 5.74-5.84 (м, 2Н), 7.16-7.19 (м, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.32-7.43 (м, 6Н), 7.86 (д, J=5.80 Гц, 1H), 8.13 (д, J=9.16 Гц, 1Н); MS m/z 734 (МН+).
Пример 135: Получение Соединения 135

Соединение 135 получают по Схеме 1 из Примера 134 за исключением того, что на стадии 1 вместо N-α-трет-бутоксикарбонил-1 -фенилглицина используют N-α -трет-бутоксикарбонил-эритро-DL-β-метилфенилаланин. Соединение 135 получают из смеси изомеров N-α-трет-бутоксикарбонил-эритро-DL-β-метилфенилаланина и полученные два диастереоизомера разделяют обращенно-фазовой препаративной ВЭЖХ. Это соединение представляет собой один изомер, который элюируют первьм с колонки для препаративной ВЭЖХ. Точная стереохимия β -метилфенилаланинового фрагмента молекулы неизвестна.
Стадия 1:
Модификации: Берут 50.4 мг (0.180 ммоля) N-α-трет-бутоксикарбонил-эритро-DL-β -метилфенилаланина, получают 29.7 мг продукта в виде белого порошка (выход 22.7%).1H ЯМР (CD3OD) δ 1.11 (д. J=7.93 Гц, 2Н), 1.15 (д, J=6.10 Гц, 3H), 1.24-1.32 (м, 11Н), 1.44 (дд, J=9.16, 5.19 Гц, 1Н), 1.90-1.94 (м, 1Н), 2.25-2.29 (м, 1Н), 2.36 (т, J=13.28 Гц, 1Н), 2.62 (дл, J=13.58, 7.17 Гц, 1Н), 2.98-3.02 (м, 1Н), 3.20-3.24 (м, 1Н). 3.91 (с, 3H), 4.11 (дд, J=11.60, 3.05 Гц, 1Н), 4.51 (д, J=10.68 Гц, 1Н), 4.57 (дд, J=10.07, 7.32 Гц, 1Н), 4.63 (д, J=12.21 Гц, 1Н), 5.14 (д, J=10.07 Гц, 1Н), 5.32 (д, J=16.79 Гц, 1Н), 5.76-5.84 (м, 1Н), 5.88 (с, 1Н), 7.08 (дд, J=8.70, 1.68 Гц, 1Н), 7.16-7.18 (м, 2Н), 7.23-7.27 (м, 5Н), 7.89 (д, J=5.80 Гц, 1Н), 8.09 (д, J=9.46 Гц, 1Н); MS m/z 762 (MH+).
Пример 136: Получение Соединения 136

Соединение 136 получают по Схеме 1 из Примера 134, за исключением того, что на стадии 1 вместо N-α-трет-бутоксикарбонил-L-фенилглицина используют N-α-трет-бутоксикарбонил-эритро-DL-β-метилфенилаланин. Соединение 136 получают из смеси изомеров N-α -трет-бутоксикарбонил-эритро-DL-β-метилфенилаланина и полученные два диастереоизомера разделяют обращенно-фазовой препаративной ВЭЖХ. Это соединение представляет собой один изомер, который элюируют вторым с колонки для препаративной ВЭЖХ. Точная стереохимия β-метилфенилаланинового фрагмента молекулы неизвестна.
Стадия 1:
Модификации: Берут 50.4 мг (0.180 ммоля) N-α-трет-бутоксикарбонил-эритро-DL-β-метилфенилаланина, получают 26.3 мг продукта в виде белого порошка (выход 20.1%).1H ЯМР (CD3OD) δ 1.04 (с, 1Н), 1.13 (д, J=6.71 Гц, 3H), 1.12-1.17 (м, 2Н), 1.30 (с, 9Н), 1.33-1.36 (м, 1Н), 1.41 (дд, J=9.46, 5.19 Гц, 1Н), 1.87 (дд, J=7.78, 5.34 Гц, 1Н), 2.29 (кв. J=8.85 Гц, 1Н), 2.36 (ддд, J=13.81, 9.99, 4.27 Гц, 1Н), 2.54 (дд, J=13.58, 7.17 Гц, 1Н), 3.00-3.04 (м, 1Н), 3.05-3.08 (м, 1Н), 3.80 (д, J=11.90 Гц, 1Н), 3.94 (с, 3H), 4.10 (дд, J=12.05, 3.81 Гц, 1Н), 4.53-4.57 (м, 1Н), 4.59 (д, J=8.24 Гц, 1Н), 5.14 (д, J=10.38 Гц, 1Н), 5.34 (д, J=17.09 Гц, 1Н), 5.78-5.85 (м, 2Н), 6.75 (т. J=7.32 Гц, 1Н), 7.03 (т, J=7.48 Гц, 2Н), 7.12 (с, 1Н), 7.14 (с, 1Н). 7.19 (дд. J=9.31, 1.68 Гц, 1Н), 7.22 (с, 1Н), 7.28 (д, J=6.10 Гц, 1Н), 7.89 (д, J=5.80 Гц, 1Н), 8.03 (д, J=8.85 Гц, 1Н); MS m/z 762 (MH+).
Пример 137: Получение Соединения 137
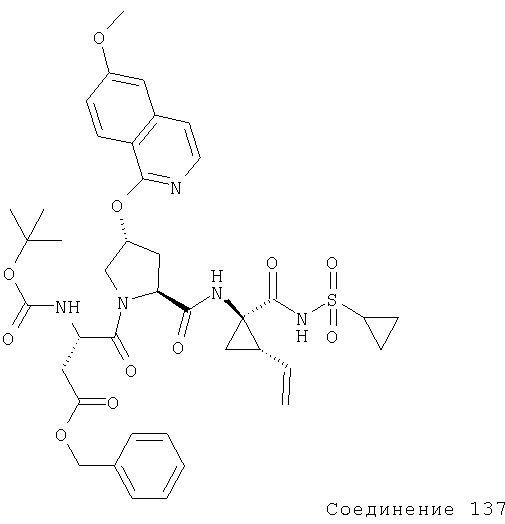
Схема 1
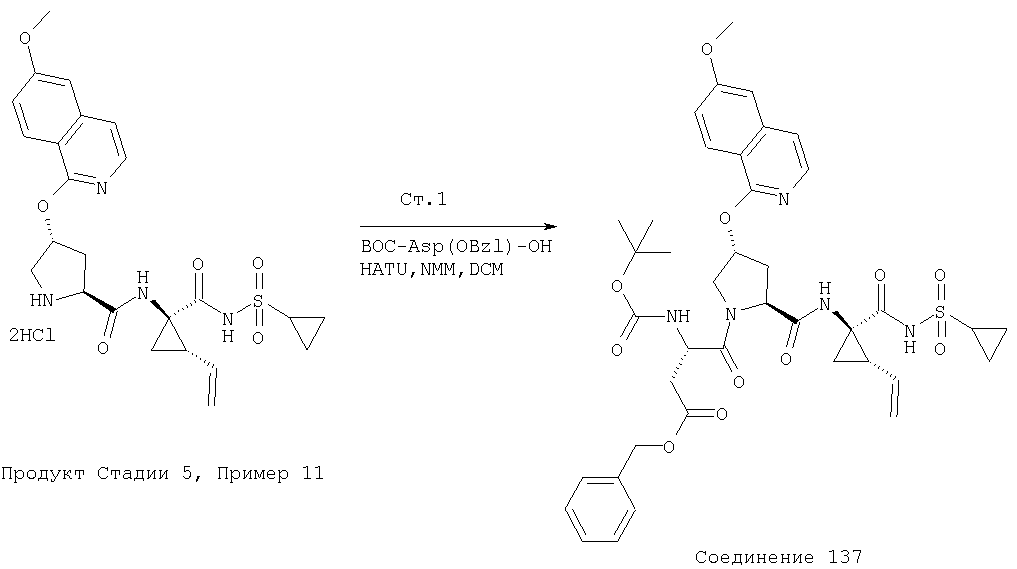
Смесь продукта Стадии 5, Пример 11 (100 мг, 0.172 ммоля), 4-бензилового эфира N-α-трет-бутоксикарбонил-L-аспарагиновой кислоты (59.5 мг, 0.180 ммоля), HATU (84.9 мг, 0.223 ммоля) и N-метилморфолина (87.0 мг, 0.859 ммоля) в DCM (3.0 мл) перемешивают при rt в течение 18 час. Реакционную смесь промывают буферным раствором с рН 4 (3×3 мл) и промывные воды снова экстрагируют DCM (3 мл). Органические вытяжки объединяют и упаривают в вакууме. Сырой продукт растворяют в МеОН и очищают обращенно - фазовой препаративной ВЭЖХ, получают Соединение 137 в виде слегка грязноватого белого стекловидного вещества (26.0 мг, выход 18.8%):1H ЯМР (CD3OD) δ 0.95-1.01 (м, 2Н), 1.16 (с, 9Н), 1.22-1.29 (м, 2Н), 1.44 (дд, J=9.46, 5.19 Гц, 1Н), 1.86 (дд, J=7.93, 5.19 Гц, 1Н), 2.26 (кв, J=8.85 Гц, 1Н), 2.32-2.37 (м, 1Н), 2.61 (дд, J=13.73, 7.32 Гц, 1Н), 2.66 (дд, J=16.48, 6.10 Гц, 1Н), 2.89 (ддд, J=12.67, 8.09, 4.88 Гц, 1Н), 3.05 (дд, J=16.63, 8.39 Гц, 1Н), 3.92(с, 3H), 4.04-4.07 (м, 1Н), 4.47 (д, J=11.90 Гц, 1Н), 4.52-4.56 (м, 1Н), 4.75 (дд, J=8.24, 6.41 Гц, 1Н), 5.12-5.14 (м, 1Н), 5.14 (с, 2Н), 5.31 (д, J=17.09 Гц, 1Н), 5.75-5.82 (м, 2Н), 7.12 (д, J=9.16 Гц, 1Н), 7.18 (д, J=2.14 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.31-7.33 (м, 1Н), 7.36 (т, J=7.32 Гц, 2Н), 7.39 (с, 1Н), 7.41 (с, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 8.10 (д, J=9.16 Гц, 1Н); MS m/z 806 (MH+).
Пример 138: Получение Соединения 138
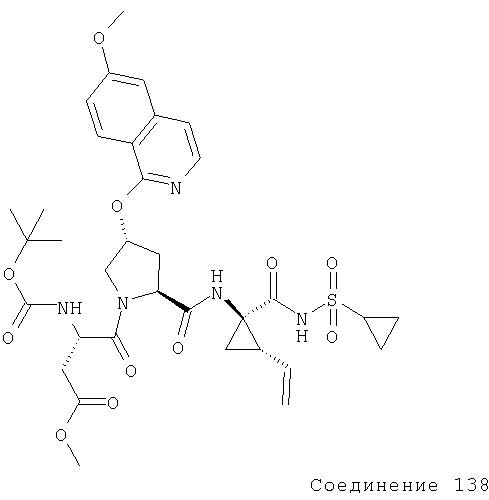
Соединение 138 получают по Схеме 1 из Примера 137 за исключением того, что на стадии 1 вместо 4-бензилового эфира N-α-трет-бутоксикарбонил-L-аспарагиновой кислоты берут 4-метиловый эфир N-трет-бутоксикарбонил-L-аспарагиновой кислоты.
Стадия 1:
Модификации: берут 45.5 мг (0.180 ммоля) 4-метилового эфира N-трет-бутоксикарбонил-L-аспарагиновой кислоты, получают 93.5 мг продукта в виде не чисто белого стекловидного вещества (выход 49.5%).1H ЯМР (CD3OD) δ 1.07-1.09 (м, 2Н), 1.17 (с, 9Н), 1.20-1.29 (м, 2Н), 1.41-1.44 (м, 1Н), 1.84-1.86 (м, 1Н), 2.26 (кв, J=8.85 Гц, 1Н), 2.33-2.38 (м, 1Н), 2.58-2.64 (м, 2Н), 2.92-3.02 (м, 2Н), 3.69 (с, 3H), 3.92 (с, 3H), 4.15 (дд, J-11.44, 2.29 Гц, 1Н), 4.49-4.56 (м, 2Н), 4.72-4.76 (м, 1Н), 5.13 (д, J=10.38 Гц, 1Н), 5.32 (д, J=17.09 Гц. 1Н), 5.74-5.82 (м, 1Н), 5.87 (с, 1Н), 7.12 (д, J=9.16 Гц, 1Н), 7.18 (с, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (дд, J=5.80, 0.92 Гц, 1Н), 8.10 (д, J=8.85 Гц, 1Н); MS m/z 730 (MH+).
Пример 139: Получение Соединения 139

Соединение 139 получают по Схеме 1 из Примера 137 за исключением того, что на стадии 1 вместо 4-бензилового эфира N-α -трет-бутоксикарбонил-L-аспарагиновой кислоты берут 4-трет-бутиловый эфир N-трет-бутоксикарбонил-L-аспарагиновой кислоты.
Стадия 1:
Модификации: берут 52.5 мг (0.180 ммоля) 4-трет-бутилового эфира N-трет-бутоксикарбонил-L-аспарагиновой кислоты, получают 125 мг продукта в виде не чисто белого стекловидного вещества (выход 99.8%).1H ЯМР (CD3 OD) δ 1.08-1.10 (м, 2Н), 1.17 (с, 9Н), 1.21-1.29 (м, 2Н), 1.46 (с, 10Н), 1.82 (дд, J=7.78, 5.34 Гц, 1Н), 2.26 (кв, J=8.75 Гц, 1Н), 2.32-2.38 (м, 1Н), 2.51 (дд, J=16.33, 7.17 Гц, 1Н), 2.63 (дд, J=14.04, 7.02 Гц, 1Н), 2.89 (дд, J=16.48, 7.63 Гц, 1Н), 2.92-2.98 (м, 1Н), 3.92 (с, 3H), 4.15 (дд, J=11.60, 3.05 Гц, 1Н), 4.50-4.57 (м, 2Н), 4.70-4.75 (м, 1Н), 5.13 (дд, J=10.38. 1.53 Гц, 1Н), 5.32 (д, J=17.40 Гц, 1Н), 5.76-5.83 (м, 1Н), 5.87 (с, 1Н), 7.13 (дд, J=8.85, 1.53 Гц, 1Н), 7.18 (д, J=2.14 Гц, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.10 (д, J=9.16 Гц, 1Н); MS m/z 772 (MH+).
Пример 140: Получение Соединения 140
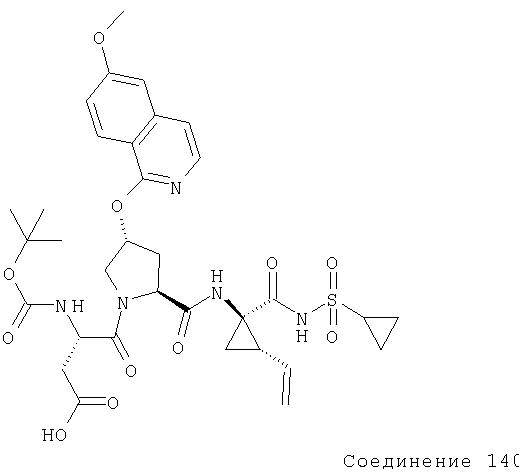
Схема 1

Соединение 138 (50.0 мг, 0.685 ммоля) растворяют в смеси ТГФ (1 мл), МеОН (1 мл) и 1.0 М водного раствора NaOH (0.137 мл, 0.137 ммоля). Через 3 час реакционную смесь нейтрализуют, прибавляя 1.0 М водный раствор HCl (0.137 мл, 0.137 ммоля). Сырую смесь упаривают в вакууме, затем прибавляют буферный раствор с рН 4 (3 мл) и DCM (3 мл) и смесь встряхивают. Слои разделяют и водный слой дополнительно экстрагируют DCM (2×1 мл). Органические вытяжки объединяют, сушат безводным MgSO4 и упаривают в вакууме, получают 48.0 мг (выход 97.9%) Соединения 140 в виде не чисто белого стекловидного твердого вещества:1H ЯМР (CD3OD) δ 2.04-1.07 (м, 2Н), 1.17 (с, 9Н), 1.22-1.25 (м, 2Н), 1.38 (дд, J=9.31, 5.34 Гц, 1Н), 1.78 (дд, J=7.78, 5.34 Гц, 1Н), 2.30 (кв, J=8.75 Гц, 1Н), 2.37-2.42 (м, 1Н), 2.48 (дд, J=15.87, 4.58 Гц, 1Н), 2.72 (дд, J=13.12, 7.63 Гц, 1Н), 2.88 (дд, J=15.72, 10.53 Гц, 1Н), 2.90-2.95 (м, 1Н), 3.92 (с, 3H), 4.21 (дд, J=11.44, 2.90 Гц, 1Н), 4.57 (т, J=8.70 Гц, 1Н), 4.62-4.65 (м, 2Н), 5.10 (д, J=10.68 Гц, 1Н), 5.34 (д, J=17.09 Гц, 1Н), 5.69-5.76 (м, 1Н), 5.83 (с, 1Н), 7.10 (д, J-9.16 Гц, 1Н), 7.17 (с, 1Н), 7.23 (д, J=6.10 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 8.12 (д, J=8.85 Гц, 1Н); MS m/z 716 (MH+).
Пример 141: Получение Соединения 141

Схема 1

Стадия 1:
Продукт Стадии 1, Пример 55 (65 мг, 0.0947 ммоля), N,N'-дисукцинимидилкарбонат (41.0 мг, 0.142 ммоля) и N,N-диизопропилэтиламин (30.6 мг, 0.237 ммоля) смешивают с сухим ТГФ (1 мл) и полученную суспензию нагревают при 80°С в микроволновом реакторе в течение 15 мин. По охлаждении до rt эту сырую смесь сразу же вводят в следующую реакцию.
Стадия 2:
К сырой реакционной смеси, полученной на Стадии 1, прибавляют смесь гидрохлорида метилового эфира L-валина (159 мг, 0.947 ммоля) и N,N-диизопропилэтиламина (122 мг, 0.947 ммоля) в сухом ТГФ (2 мл). Полученную смесь перемешивают при rt в течение 18 час. Растворитель отгоняют в вакууме, а остаток растворяют в DCM (2 мл) и промывают буферным раствором рН 4 (3×2 мл). Промывные воды объединяют и снова экстрагируют DCM (2 мл). Объединенные DCM вытяжки упаривают в вакууме, а полученный остаток растворяют в МеОН и очищают обращенно-фазовой препаративной ВЭЖХ, получают 38.1 мг (выход 52.2%) Соединения 141 в виде белого порошка.1H ЯМР (CD3OD) δ 0.83 (дд, J=6.87, 3.81 Гц. 6Н), 1.06 (с, 11Н), 1.21-1.26 (м, 2Н), 1.41 (дд, J=9.46, 5.49 Гц, 1Н), 1.87 (дд, J=8.09, 5.34 Гц, 1Н), 1.95-2.02 (м, 1Н), 2.21 (кв, J=8.85 Гц, 1Н), 2.29 (ддд, J=13.89, 9.92, 4.27 Гц, 1Н), 2.60 (дд, J=13.73, 7.02 Гц, 1Н), 2.91-2.97 (м, 1Н), 3.67 (с, 3H), 3.93 (с, 3H), 4.00 (д, J=5.49 Гц, 1Н), 4.09 (дд, J=11.90, 3.97 Гц, 1Н), 4.40-4.43 (м, 2Н), 4.52 (дд, J=10.07, 7.02 Гц, 1Н), 5.11 (дд, J=10.38, 1.53 Гц, 1Н), 5.28 (дд, J=17.09, 1.22 Гц, 1Н), 5.75 (ддд, J=17.17, 10.15, 9.00 Гц, 1Н), 5.83 (с, 1Н), 7.11 (дд,J=9.16, 2.44 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.87 (д, J=6.10 Гц, 1Н), 8.10 (д, J=8.85 Гц, 1Н); MS m/z 771 (MH+).
Пример 142: Получение Соединения 142
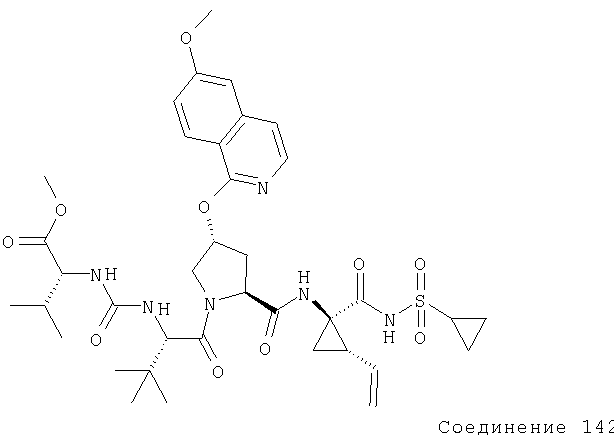
Соединение 142 получают по Схеме 1 из Примера 141 за исключением того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид метилового эфира D-валина.
Стадия 2:
Модификации: берут 159 мг (0.947 ммоля) гидрохлорида метилового эфира D-валина, получают 23.0 мг продукта в виде белого порошка (выход 31.5%):1H ЯМР (CD3OD) δ 0.88 (дд, J=13.89, 6.87 Гц, 6Н), 1.06 (с, 9Н), 1.07-1.09 (м, 2Н), 1.23-1.27 (м, 2 Н), 1.41 (дд, J=9.46, 5.49 Гц, 1Н), 1.88 (дд, J=7.93, 5.49 Гц, 1Н), 2.01-2.07 (м, 1Н), 2.23 (кв, J=9.05 Гц, 1Н), 2.31 (ддд, J=14.11, 9.99, 4.27 Гц, 1Н), 2.63 (дд, J=13.89, 7.17 Гц, 1Н), 2.93-2.98 (м, 1Н), 3.62 (с, 3H), 3.96 (с, 3H), 4.03 (д, J=5.19 Гц, 1Н), 4.09 (дд, J=11.75, 3.81 Гц, 1Н), 4.38 (с, 1Н), 4.48-4.54 (м, 2Н), 5.12 (дд, J=10.38, 1.22 Гц, 1Н), 5.29 (дд, J=17.24,1.07 Гц, 1Н), 5.70-5.77 (м, 1Н), 5.83 (с, 1Н), 7.22 (дд, J=9.00, 2.59 Гц, 1Н), 7.25 (д, 7-2.44 Гц, 1Н), 7.35 (д, J=6.10 Гц, 1Н), 7.87 (д, J=6.10 Гц, 1Н), 8.16 (д, J=9.16 Гц, 1Н); MS m/z 771 (МН+).
Пример 143: Получение Соединения 143
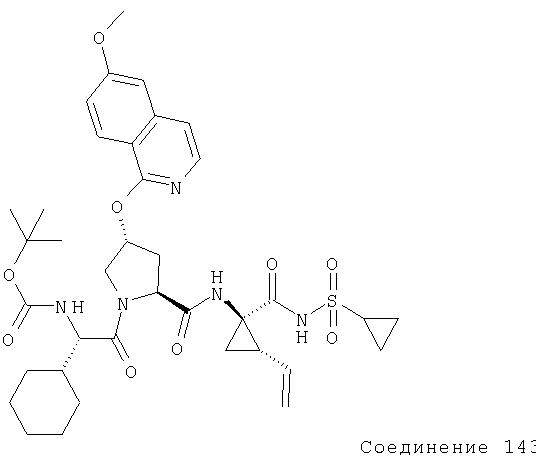
Соединение 143 получают по Схеме 1 из Примера 137 за исключением того, что на стадии 1 вместо 4-бензилового эфира N-α-трет-бутоксикарбонил-L-аспарагиновой кислоты берут N-трет-бутоксикарбонил-L-циклогексилглицин.
Стадия 1:
Модификации: берут 46.2 мг (0.180 ммоля) N-трет-бутоксикарбонил-L-циклогексилглицин, получают 93.5 мг продукта в виде белого порошка (выход 73.8%).1H ЯМР (CD3OD) δ 1.04-1.08 (дд, J=7.78, 2.29 Гц, 4Н), 1.19-1.26 (м, 4Н), 1.25 (с, 9Н), 1.41 (дд, J=9.46, 5.19 Гц, 1Н), 1.63-1.82 (м, 7Н), 1.88 (дд, J=7.93, 5.49 Гц, 1Н), 2.22 (кв, J=9.05 Гц, 1Н), 2.32-2.37 (м, 1Н), 2.59 (дд, J=13.58, 6.87 Гц, 1Н), 2.91-2.96 (м, 1Н), 3.92 (с, 3H), 4.05 (дд, J=11.75, 3.20 Гц, 1Н), 4.09 (д, J=8.85 Гц, 1Н), 4.47 (д, J=11.90 Гц, 1Н), 4.53 (дд, J=10.22, 7.17 Гц, 1Н), 5.11 (д, J=10.38 Гц, 1Н), 5.29 (д, J=16.79 Гц, 1Н), 5.79 (ддд, J=16.86, 9.92, 9.54 Гц, 1Н), 5.84 (с, 1Н), 7.10 (д, J=8.85 Гц, 1Н), 7.17 (д, J=1.53 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (д, J=6.10 Гц, 1Н), 8.09 (д, J=8.85 Гц, 1Н); MS m/z 740 (MH+).
Пример 144: Получение Соединения 144

Соединение 144 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид метилового эфира глицина.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N, N'-дисукциниимидилкарбоната, 47.0 мг (0.364 ммоля) N,N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 183 мг (1.46 ммоля) гидрохлорида метилового эфира глицина, 188 мг (1.46 ммоля) N,N-диизопропилэтиламина, получают 56.3 мг продукта в виде белого порошка (выход 52.9%):1H ЯМР (CD3OD) δ 1.21 (м, 2Н), 1.40 (дд, J=9.46, 5.49 Гц, 1Н), 1.85 (дд, J=7.78, 5.34 Гц, 1Н), 2.18 (кв, J=8.55 Гц, 1Н), 2.33 (ддд, J=13,89, 9.92, 4.27 Гц, 1Н), 2.61 (дд, J=13.73, 7.32 Гц, 1Н), 2.89-2.94 (м, 1Н), 3.65 (с, 3H), 3.69-3.77 (м, 2Н), 3.92 (с, 3H), 4.10 (дд. J=11.75, 3.81 Гц, 1Н), 4.40-4.42 (м, 2Н), 4.53 (дд, J=9.92, 7.17 Гц, 1Н), 5.08 (д, J=10.38 Гц, 1Н), 5.26 (д, J=17.09 Гц, 1Н), 5.77 (ддд, J=17.09, 10.22, 9.00 Гц, 1Н), 5.83 (с, 1Н), 7.13 (дд, J=8.85, 2.44 Гц, 1Н), 7.17 (с, 1Н), 7.23 (д, J=5.80 Гц, 1Н), 7.87 (д, J=5.80 Гц, 1Н), 8.09 (д, J=8.85 Гц, 1Н); MS m/z 729 (MH+).
Пример 145: Получение Соединения 145
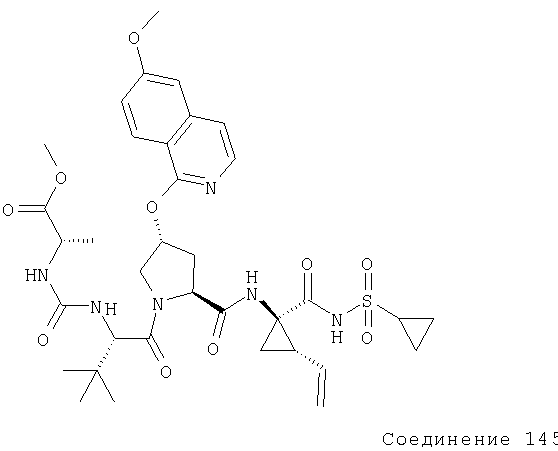
Соединение 141 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид метилового эфира L-аланина.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N,N'-дисукцинимидилкарбоната, 47.0 мг (0.364 ммоля) N,N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 203 мг (1.46 ммоля) гидрохлорида метилового эфира L-аланина, 188 мг (1.46 ммоля) N,N-диизопропилэтиламина, получают 64.3 мг продукта в виде белого порошка (выход 59.3%):1 H ЯМР (CD3OD) δ 0.97-1.02 (м, 2Н), 1.05 (м, 9Н), 1.19 (д, J=7.02 Гц, 3H), 1.18-1.22 (м, 2Н), 1.41 (дд, J=9.46, 5.19 Гц, 1Н), 1.86 (дд, J=8.09, 5.34 Гц, 1Н), 2.19 (кв, J=8.85 Гц, 1Н), 2.32 (ддд, J=13.81, 9.84, 4.43 Гц, 1Н), 2.61 (дд, J=13.73, 7.02 Гц, 1Н), 2.93 (ддд, J=12.82, 8.09, 4.73 Гц, 1Н), 3.65 (с, 3H), 3.93 (с, 3H), 3.99 (кв, J=7.22 Гц, 1Н), 4.08 (дд, J=11.75, 3.81 Гц, 1Н), 4.38 (с, 1Н), 4.42 (д, J=11.60 Гц, 1Н), 4.53 (дд. J=10.07, 7.32 Гц, 1Н), 5.09 (дд, J=10.38, 1.53 Гц, 1Н), 5.27 (дд, J=17.09, 1.22 Гц, 1Н), 5.77 (ддд, J=17.09, 10.07, 9.16 Гц, 1Н), 5.82 (с, 1Н), 7.13 (дд, J=9.16, 2.44 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.87 (д, J=5.80 Гц, 1Н), 8.10 (д, J=9.16 Гц, 1Н); MS m/z 743 (МН+).
Пример 146: Получение Соединения 146

Соединение 146 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид метилового эфира L-трет-лейцина.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N,N'-дисукцинимидилкарбоната, 47.0 мг (0.364 ммоля) N,N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 265 мг (1.46 ммоля) гидрохлорида метилового эфира L-трет-лейцина, 188 мг (1.46 ммоля) N,N-диизопропилэтиламина, получают 68.3 мг продукта в виде белого порошка (выход 59.6%):1H ЯМР (CD3OD) δ 1.06 (с, 9Н), 1.18-1.24 (м, 2Н), 1.41 (дд, J=9.46, 5.49 Гц, 1Н), 1.86 (дд, J=7.93, 5.49 Гц, 1Н), 2.19 (кв, J=8.75 Гц, 1Н), 2.30 (ддд, J=13.89, 10.07, 4.43 Гц, 1Н), 2.60 (дд, J=13,73, 7.32 Гц, 1Н), 2.91-2.96 (м, 1Н), 3.65 (с, 3H), 3.91 (с, 1Н), 3.93 (с, 3H), 4.09 (дд. J=11.60, 3.97 Гц, 1Н), 4.38 (с, 1Н), 4.43 (д, J=11.60 Гц, 1Н), 4.51 (дд, J=10.07, 7.32 Гц, 1Н), 5.10 (дд, J=10.38, 1.53 Гц, 1Н), 5.27 (дд, J=17.24, 1.37 Гц, 1Н), 5.76 (ддд, J=17.09, 10.07, 9.16 Гц, 1Н), 5.82 (с, 1Н), 7.12 (дд, J=9.16, 2.44 Гц, 1Н), 7.18 (д, J=2.14 Гц, 1Н), 7.24 (д, J=6.10 Гц, 1Н), 7.87 (д, J=5.80 Гц, 1Н), 8.11 (д, J=9.16 Гц, 1Н); MS m/z 785 (MH+).
Пример 147: Получение Соединения 147

Соединение 147 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид метилового эфира L-гистидина и количество N,N-диизопропилэтиламина на стадии 2 удваивают.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N,N'-дисукцинимидилкарбоната, 47.0 мг (0.364 ммоля) N,N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 352 мг (1.46 ммоля) гидрохлорида метилового эфира L-гистидина, 377 мг (2.91 ммоля) N,N-диизопропилэтиламина, получают 51.0 мг продукта в виде белого порошка (выход 43.2%):1H ЯМР (CD3OD) δ 1.04 (с, 11Н), 1.20-1.22 (м, 2Н), 1.41 (ддд, J=9.46, 5.34, 1.07 Гц, 1Н), 1.86-1.88 (м, 1Н), 2.23 (кв, J=8.75 Гц, 1Н), 2.28-2.33 (м, 1Н), 2.60 (дд, J=13.73, 7.02 Гц, 1Н), 2.92 (д, J=6.41 Гц, 2Н), 2.92-2.96 (м, 1Н), 3.64 (с, 3H), 3.91 (д, J=1.53 Гц, 3H), 4.04 (дд, J=11.90, 3.66 Гц, 1Н), 4.35 (с, 1Н), 4.36-4.41 (м, 2Н), 4.53 (дд, J=9.77, 7.63 Гц, 1Н), 5.10 (д, J=10.38 Гц, 1Н), 5.28 (д, J=17.40 Гц, 1Н), 5.73-5.78 (м, 1Н), 5.81 (с, 1Н), 6.82 (с, 1Н), 7.06-7.09 (м, 1Н), 7.15 (с, 1Н), 7.23 (д, J=5.80 Гц, 1Н), 7.63 (с, 1Н), 7.87 (дд, J=5.80, 1.22 Гц, 1Н), 8.08 (д, J=9.16 Гц, 1Н); MS m/z 809 (MH+).
Пример 148: Получение Соединения 148
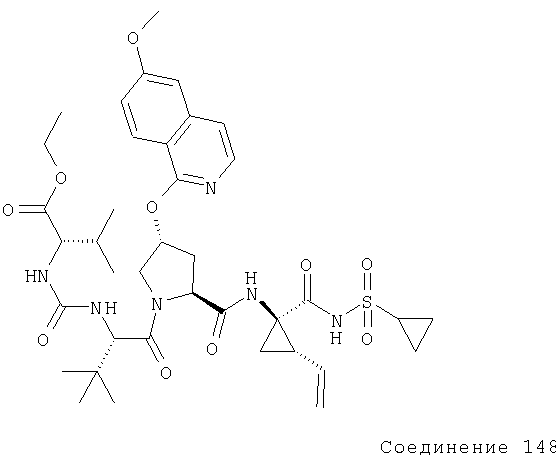
Соединение 148 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид этилового эфира L-валина.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N,N'-дисукцинимидилкарбоната, 47.0 мг (0.364 ммоля) N,N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 265 мг (1.46 ммоля) гидрохлорида этилового эфира L-валина, 188 мг (1.46 ммоля) N,N-диизопропилэтиламина, получают 69.2 мг продукта в виде белого порошка (выход 60.4%):1H ЯМР (CD3OD) δ 0.83 (дд, J=6.71, 5.19 Гц, 6Н), 1.01-1.03 (м, 2 Н), 1.06 (с, 9Н), 1.17-1.22 (м, 2Н), 1.23 (т, J=7.17 Гц, 3H), 1.40 (дд, J=9.46, 5.19 Гц, 1Н), 1.86 (дд, J=8.09, 5.34 Гц, 1Н), 1.95-2.02 (м, 1Н), 2.18 (кв, J=9.05 Гц, 1Н), 2.33 (ддд, J=13.89, 9.92, 4.27 Гц, 1Н), 2.60 (дд, J=13.89, 7.17 Гц, 1Н), 2.92 (ддд, J=12.82, 8.09, 4.73 Гц, 1Н), 3.93 (с, 3H), 3.98 (д, 7-5.19 Гц, 1Н), 4.08-4.17 (м, 3H), 4.41 (с, 1Н), 4.41-4.43 (м, 1Н), 4.52 (дд, J=10.07, 7.32 Гц, 1Н), 5.09 (дд, J=10.38, 1.53 Гц, 1Н), 5.26 (дд, J=17.09, 1.22 Гц, 1Н), 5.77 (ддд, J=17.09, 10.07. 9.16 Гц, 1Н), 5.82 (с, 1Н), 7.12 (дд, J=9.16, 2.44 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.23 (д, J=6.10 Гц, 1Н), 7.87 (д, J=6.10 Гц, 1Н), 8.10 (д, J=9.16 Гц, 1Н); MS m/z 785 (MH+).
Пример 149: Получение Соединения 149

Соединение 149 получают по Схеме 1 из Примера 137 за исключением того, что на стадии 1 вместо 4-бензилового эфира N-α-трет-бутоксикарбонил-L-аспарагиновой кислоты берут дициклогексиламиновую соль N-трет-бутоксикарбонил-L-циклопентилглицина.
Стадия 1:
Модификации: берут 76.2 мг (0.180 ммоля) дициклогексиламиновой соли N-трет-бутоксикарбонил-L-циклопентилглицина, получают 111 мг продукта в виде белого порошка (выход 89.3%).1H ЯМР (CD3OD) δ 0.98 (д, J=8.24 Гц, 2Н), 1.15-1.18 (м, 2Н), 1.24 (с, 9Н), 1.29-1.32 (м, J=18.01 Гц, 2Н), 1.38-1.40 (м, 1Н), 1.44 (дд, J=4.88, 1.53 Гц, 1Н), 1.49-1.55 (м, 2Н), 1.62-1.67 (м, 2Н), 1.74-1.80 (м, 1Н), 1.85-1.88 (м, 1Н), 2.16 (кв, J=8.75 Гц, 1Н), 2.21-2.26 (м, 1Н), 2.42 (т, J=11.90 Гц, 1Н), 2.60-2.64 (м, 1Н), 2.89-2.93 (м, 1Н), 3.92 (д, J=1.53 Гц, 3H), 4.06-4.11 (м, 2Н), 4.51-4.57 (м, 2Н), 5.07 (д, J=10.38 Гц, 1Н), 5.25 (д, J=17.09 Гц, 1Н), 5.78-5.85 (м, 2Н), 7.10 (д, J=8.85 Гц, 1Н), 7.17 (с, 1Н), 7.23 (д, J=4.27 Гц, 1Н), 7.88 (дд, J=5.95, 1.68 Гц, 1Н), 8.10 (д, J=9.16 Гц, 1Н); MS m/z 726 (MH+).
Пример 150: Получение Соединения 150
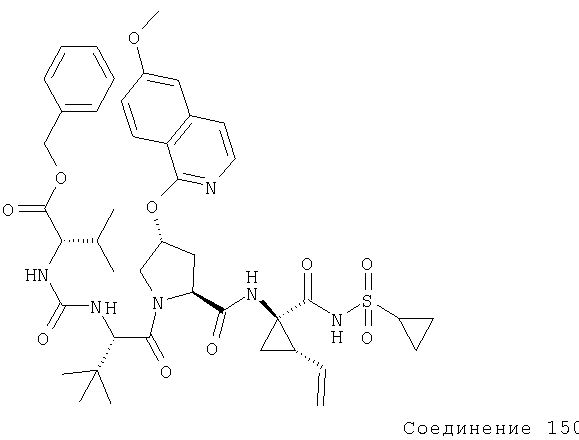
Соединение 150 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид бензилового эфира L-валина.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N,N'-дисукцинимидилкарбоната, 47.0 мг (0.364 ммоля) N,N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 356 мг (1.46 ммоля) гидрохлорида бензилового эфира L-валина, 188 мг (1.46 ммоля) N, N-диизопропилэтиламина, получают 41.0 мг продукта в виде белого порошка (выход 33.2%): MS m/z 848 (МН+).
Пример 151: Получение Соединения 151
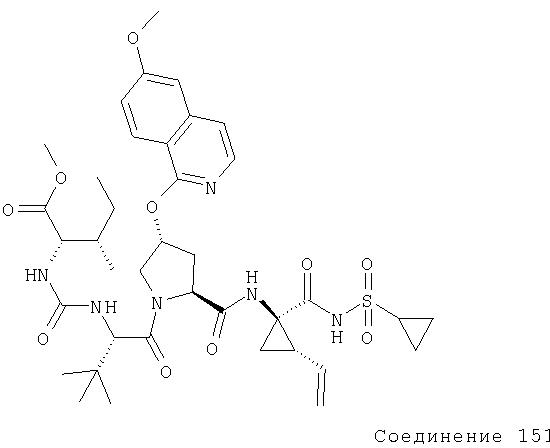
Соединение 151 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид метилового эфира L-изолейцина.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N,N'-дисукцинимидилкарбоната, 47.0 мг (0.364 ммоля) N,N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 265 мг (1.46 ммоля) гидрохлорида метилового эфира L-изолейцина, 188 мг (1.46 ммоля) N,N-диизопропилэтиламина, получают 75.5 мг продукта в виде белого порошка (выход 65.9%):1H ЯМР (CD3OD) δ 0.78-0.80 (м, 3H), 0.83-0.84 (м, 3H), 0.90-0.95 (м, 1Н), 1.01-1.03 (м, 2Н), 1.06 (д, J=3.05 Гц, 9Н), 1.17-1.21 (м, 2Н), 1.32-1.42 (м, 2Н), 1.68-1.72 (м, 1Н), 1.84-1.87 (м, 1Н), 2.14-2.20 (м, 1Н), 2.30-2.36 (м, 1Н), 2.57-2.62 (м, 1Н), 2.90-2.95 (м, 1Н), 3.66 (д, J=2.75 Гц, 3H), 3.92 (д, J=2.75 Гц, 3H), 4.05-4.12 (м, 2Н), 4.39-4.42 (м, 2Н), 4.50-4.53 (м, 1Н), 5.07-5.10 (м, 1Н), 5.23-5.28 (м, 1Н), 5.73-5.79 (м, 1Н), 5.81-5.83 (м, 1Н), 7.10-7.13 (м, 1Н), 7.17 (т, J=2.44 Гц, 1Н), 7.22-7.24 (м, 1Н), 7.85-7.87 (м, 1Н), 8.10 (дд, J=9.16, 2.75 Гц, 1Н); MS m/z 785 (MH+).
Пример 152: Получение Соединения 152
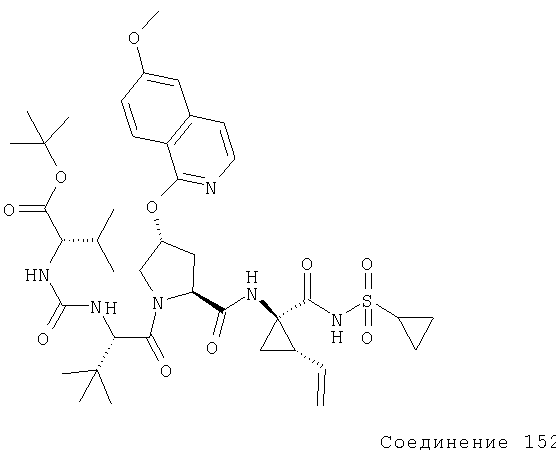
Соединение 152 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид трет-бутилового эфира L-валина.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N,N'-дисукцинимидилкарбоната, 47.0 мг (0.364 ммоля) N, N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 306 мг (1.46 ммоля) гидрохлорида трет-бутилового эфира L-валина, 188 мг (1.46 ммоля) N,N-диизопропилэтиламина, получают 93.5 мг продукта в виде белого порошка (выход 78.8%): MS m/z 814 (МН+).
Пример 153: Получение Соединения 153

Соединение 153 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут (S)-(+)-1-метокси-2-пропиламин.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N,N'-дисукцинимидилкарбоната, 47.0 мг (0.364 ммоля) N,N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 130 мг (1.46 ммоля) (S)-(+)-1-метокси-2-пропиламина, 188 мг (1.46 ммоля) N,N-диизопропилэтиламина, получают 50.3 мг продукта в виде белого порошка (выход 47.3%):1H ЯМР (CD3OD) δ 0.96 (д, J=7.02 Гц, 3H), 0.99-1.01 (м, 2Н), 1.04 (s, 9 Н), 1.15-1 18 (m, 2 H), 1.39 (дд, J=9.61, 5.34 Гц, 1Н), 1.84 (дд, J=7.93, 5.19 Гц, 1Н), 1.93 (с, 3H), 2.15 (кв, J=9.05 Гц, 1Н), 2.37 (ддд, J=13.96, 9.84, 4.58 Гц, 1Н), 2.61 (дд, J=14.04, 7.32 Гц, 1Н), 2.90 (ддд, J=12.89, 8.16, 4.88 Гц, 1Н), 3.19-3.28 (м, 2Н), 3.64-3.67 (м, 1Н), 3.92 (с, 3H), 4.12 (дд, J=11.60, 3.97 Гц, 1Н), 4.40 (с, 1Н), 4.44 (д, J=11.90 Гц, 1 Н), 4.53 (дд, J=9.77, 7.32 Гц, 1Н), 5.07 (дд, J=10.22, 1.68 Гц, 1Н), 5.24 (дд, J=17.24, 1.37 Гц, 1Н), 5.76-5.81 (м, 1Н), 5.83 (с, 1Н), 7.10 (дд, J=9.16. 2.44 Гц, 1Н), 7.17 (д, J=2.44 Гц, 1Н), 7.23 (д, J=5.80 Гц, 1Н), 7.87 (д, J=5.80 Гц, 1Н), 8.10 (д, J=9.16 Гц, 1Н); MS m/z 729 (MH+).
Пример 154: Получение Соединения 154
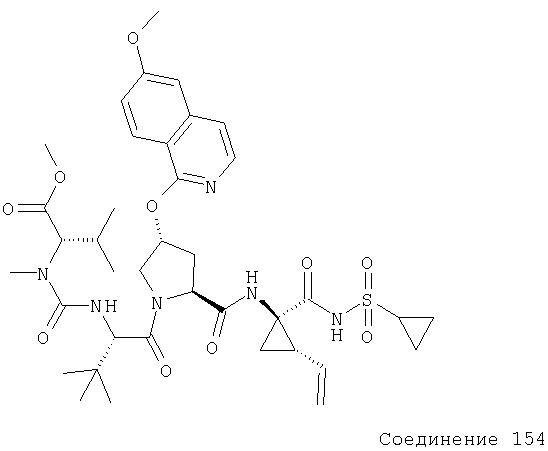
Соединение 154 получают по Схеме 1 из Примера 141 за исключением увеличения масштаба и того, что на Стадии 2 вместо гидрохлорида метилового эфира L-валина берут гидрохлорид метилового эфира N-метил L-валина.
Стадия 1:
Модификации: берут 100 мг (0.146 ммоля) продукта из Примера 55, Стадия 1; 62.2 мг (0.219 ммоля) N,N'-дисукцинимидилкарбоната, 47.0 мг (0.364 ммоля) N,N-диизопропилэтиламина.
Стадия 2:
Модификации: берут 265 мг (1.46 ммоля) гидрохлорида метилового эфира N-метил L-валина, 188 мг (1.46 ммоля) N,N-диизопропилэтиламина, получают 68.2 мг продукта в виде белого порошка (выход 59.5%):1H ЯМР (CD3OD) δ 0.70 (дд, J=6.71, 2.14 Гц, 3H), 0.89 (дд, J=6.41, 2.44 Гц, 3H), 0.96-0.98 (м, 1Н), 1.02-1.04 (м, 2Н), 1.07 (д, J=2.14 Гц, 9Н), 1.18-1.22 (м, 2Н), 1.43-1.47 (м, 1Н), 1.84-1.87 (м, 1Н), 2.11-2.19 (м, 2Н), 2.31-2.37 (м, 1Н), 2.58-2.63 (м, 1Н), 2.87 (д, J=2.44 Гц, 3H), 2.90-2.94 (м, 1Н), 3.65 (д, J=2.14 Гц, 3H), 3.92 (д, J=2.14 Гц, 3H), 4.10-4.14 (м, 1Н), 4.25 (дд, J=10.07, 1.22 Гц, 1Н), 4.48 (д, J=2.44 Гц, 1Н), 4.50-4.54 (м, 1Н), 5.08-5.10 (м, 1Н), 5.25-5.28 (дд, J=17.09,1.53 Гц, 1Н), 5.78-5.85 (м, 2Н), 7.10-7.13 (м, 1Н), 7.18-7.19 (м, 1Н), 7.24 (дд, J=5.95, 2.59 Гц, 1Н), 7.87-7.89 (м, 1Н), 8.10 (дд, J=9.00, 2.59 Гц, 1Н); MS m/z 785 (MH+).
Пример 155: Получение Соединения 155

Схема 1

Стадия 1;
К раствору продукта Стадии 1, Пример 55 (100 мг, 0.146 ммоля) в сухом ТГФ (2 мл) прибавляют 2,2,2-трифтор-1,1-диметилэтиловый эфир пиридин-2-илкарбоновой кислоты (44.0 мг, 0.175 ммоля) и N-метилморфолин (59 мг, 0.58 ммоля). Смесь перемешивают при rt в течение 24 час. Промывают, упаривают в вакууме и остаток растворяют в DCM (2 мл). Раствор промывают буферным раствором рН 4 (3×3 мл) и промывные воды снова экстрагируют DCM (3 мл). Органические вытяжки растворяют затем в МеОН и очищают обращенно - фазовой препаративной ВЭЖХ, получают Соединение 155 в виде белого порошка (38.5 мг, выход 34.3%):1H ЯМР (CD3OD) δ 1.04 (с, 11Н), 1.19-1.22 (м, 2Н), 1.23 (м, 3H), 1.43 (дд, J=9.31, 5.34 Гц, 1Н), 1.46 (с, 3H), 1.87 (дд, J=7.93, 5.49 Гц, 1Н), 2.19 (кв, J=8.85 Гц, 1Н), 2.34 (м, 1Н), 2.62 (дд, J=13.73, 7.02 Гц, 1Н), 2.92 (ддд, J=12.67, 8.09, 4.88 Гц, 1Н), 3.92 (с, 3H), 4.06 (дд, J=11.90, 3.36 Гц, 1Н), 4.23 (с, 1Н), 4.43 (д, J=11.60 Гц, 1Н), 4.56 (дд, J=10.38, 7.32 Гц, ГН), 5.10 (д, J=10.38 Гц, 1Н), 5.27 (д, J=17.09 Гц, 1Н), 5.75-5.80 (м, 1Н), 5.82 (с, 1Н), 7.10(дд, J=9.16, 2.44 Гц, 1Н), 7.18 (д, J=2.44 Гц, 1Н), 7.25 (д, J=6.10 Гц, 1Н), 7.89 (д, J=5.80 Гц, 1Н), 8.07 (д, J=9.16 Гц, 1Н); MS m/z 768 (MH+).
Подраздел Е
Условия LC-MS для Секции Е
"метод А" колонка 3.0Х50 мм Xterra @ 4 мин градиент и скорость потока 4 мл/мин
"метод В" колонка 3.0Х50 мм Xterra @ 3 мин градиент и скорость потока 4 мл/мин
"метод С" колонка 4.6Х50 мм Xterra @ 4 мин градиент и скорость потока 4 мл/мин
"метод D" колонка 4.6Х50 мм Xterra @ 3 мин градиент и скорость потока 4 мл/мин
Пример 180: Получение Соединения 180
Общая схема синтеза

Соединения 180-183 получают по общей схеме синтеза, изображенной выше. Эти отдельные реакции подробно описаны в другом месте. За исключением первой стадии алкилирования, где в случае трет-бутоксид калия в ТГФ (от Aldrich Chemicals) в ДМФА предлагается более удобная методика обработки: основную часть растворителя ДМФА отмывают водой по завершении алкилирования.
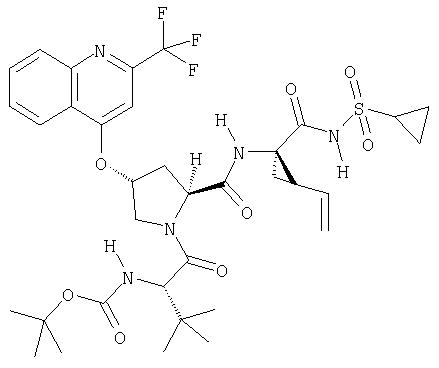
Так Соединение 180: BOCNH-P3(L-трет-BuGly)-P2[(4R)-(2-трифтометилхинолин-4-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан: вещество получают в виде белой пены с выходом 61%. LC/MS Rt- мин (MNa+) [метод А]: 3.35 (774).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 13Н), 1.21 (с, 9Н), 1.42 (дд, J=9.17, 5.26 Гц, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.21 (м, 1Н), 2.33 (м, 1Н), 2.64 (дд, J=13.94, 6.60 Гц, 1Н), 2.93 (м, 1Н), 4.09 (дд, J=11.49, 2.69 Гц, 1Н), 4.21 (с, 1Н), 4.52 (м, 1 Н), 4.56 (д, J=12.23 Гц, 1Н), 5.10 (дд, J=10.39, 1.59 Гц, 1Н), 5.27 (д, J=16.87 Гц, 1Н), 5.58 (с, 1Н), 5.72 (м, 1Н), 7.36 (с, 1Н), 7.61 (т, J=7.70 Гц, 1Н), 7.83 (т, J=7.34 Гц, 1Н), 8.07 (д, J=8.56 Гц, 1Н), 8.26 (д, J=8.56 Гц, 1Н).
Пример 181: Получение Соединения 181

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(2,8-бистрифтометилхинолин-4-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан: вещество получают в виде белой пены с выходом 52%. LC/MS Rt- мин (MNa+) [метод А]: 3.60 (843).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 11Н), 1.16 (с,9Н), 1.22 (м, 2Н), 1.42 (дд, J=9.29, 5.38 Гц, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.21 (кв, J=8.97 Гц, 1Н), 2.33 (м, 1Н), 2.65 (дд, J=13.94, 6.85 Гц, 1Н), 2.93 (м, 1Н), 4.07 (дд, J=11.98, 2.69 Гц, 1Н), 4.17 (с, 1Н), 4.52 (дд, J=10.52, 6.85 Гц, 1Н), 4.58 (д, J=11.98 Гц, 1Н), 5.10 (д, J=10.27 Гц, 1Н), 5.27 (д, J=17.12 Гц, 1Н), 5.60 (с, 1Н), 5.72 (м, 1Н), 7.46 (с, 1Н), 7.69 (т, J=7.83 Гц, 1Н), 8.18 (д, J=7.34 Гц, 1Н), 8.50 (д, J=8.31 Гц, 1Н).
Пример 182: Получение Соединения 182
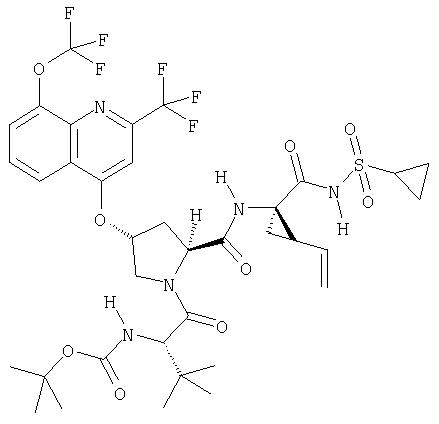
BOCNH-P3(L-трет-BuGly)-Р2[(4R)-(2-трифторметил, 8-трифтометоксихинолин-4-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан: вещество получают в виде белой пены с выходом 99%. LC/MS Rt- мин (MNa+) [метод А]: 3.62 (858).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 11Н), 1.21 (м, 11Н), 1.42 (дд, J=9.05, 5.14 Гц, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.21 (кв, J=8.64 Гц, 1Н), 2.33 (м, 1Н), 2.64 (дд, J=13.94, 6.60 Гц, 1Н), 2.93 (м, 1Н), 4.08 (дд, J=11.98, 2.69 Гц, 1Н), 4.18 (с, 1Н), 4.51 (дд, J=10.52, 6.85 Гц, 1Н), 4.57 (д, J=12.23 Гц, 1Н), 5.10 (дд, J=10.52, 1.22 Гц, 1Н), 5.27 (д, J=17.12 Гц, 1Н), 5.59 (с, 1Н), 5.72 (м, 1Н), 7.44 (с, 1Н), 7.63 (т, J=8.07 Гц, 1Н), 7.78 (д, J=7.58 Гц, 1Н), 8.24 (д, J=8.56 Гц, 1Н).
Пример 183: Получение Соединения 183

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(2-трифторметил, 8-хлорхинолин-4-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан: вещество получают в виде белой пены с выходом 64%. LC/MS Rt- мин (MNa+) [метод А]: 3.52 (808).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.04 (м, 11Н), 1.21 (м, 11Н), 1.41 (дд, J=9.41, 5.50 Гц, 1Н), 1.86 (дд, J=8.07, 5.62 Гц, 1Н), 2.20(кв, J=8.80 Hz, 1Н), 2.32 (м, 1Н), 2.63 (дд, J=13.82, 6.72 Гц, 1Н), 2.92 (м, 1Н), 4.07 (дд, J=12.10, 2.81 Гц, 1Н), 4.19 (с, 1Н), 4.50 (дд, J=10.52, 6.85 Гц, 1Н), 4.56 (д, J=11.98 Гц, 1Н), 5.10 (дд, J=10.27, 1 47 Гц, 1Н), 5.26 (д, J=17.12 Гц, 1Н), 5.57 (с, 1Н), 5.72 (м, 1Н), 7.41 (с; 1Н), 7.52 (т, J=8.07 Гц, 1Н), 7.93 (д, J=7.58 Гц, 1Н), 8.19 (д, J=8.56 Гц, 1Н).
Пример 184: Общая методика алкилирования трипептидом (Соединение 184) и Р2*
Общая схема-Получение Соединения 184 из Примера 184
Получение трипептидного компонента. Пример 184, осуществляют последовательным сочетанием с амидом с применением HATU в качестве конденсирующего агента. Ясно, что в нижеприведенной схеме могут применяться многие стандартные конденсирующие агенты.
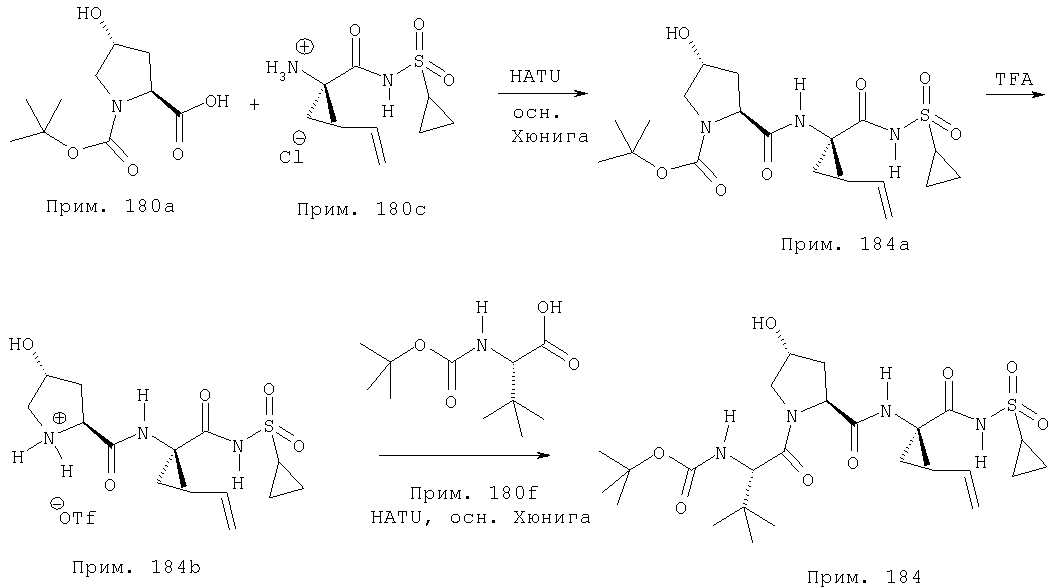
Получение интермедиата 184а:
К смеси HATU (820 мг, 2.2 ммоля), Соединения из Примера 180а (Boc-4R-гидроксипролина, 417 мг, 1.8 ммоля) и Соединения из Примера 180с (гидрохлорида (1(R)-амино-2(S)-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты, 490 мг, 1.8 ммоля) в высушенной пламенем колбе при комнатной температуре прибавляют сухой СН2Cl2 (8 мл). Смесь хранят в атмосфере сухого N2 прежде, чем охладить до -78°С. Медленно, в течение 5 мин прибавляют основание Хюнига (диизопропилэтиламин, 625 мкл, 3.6 ммоля) и смесь становится ярко-оранжевой суспензией. Перемешивание продолжают в течение часа, температура при этом поднимается до комнатной. LC/MS показывает полное превращение в нужный продукт 184а. Сырую реакционную смесь обрабатывают как обычно, промывают тремя порциями (5 мл) воды, органический остаток экстрагируют этилацетатом (3×5 мл). Сырой продукт получают, отгоняя органические растворители в вакууме. Вещество используют на следующей стадии без дополнительной очистки.
Получение интермедиата из Примера 184b:
Высушенное сухое вещество, полученное на предыдущей стадии, растворяют в 9 мл СН2Cl2 при комнатной температуре. К этому раствору прибавляют 3 мл трифторуксусной кислоты, образуется бледно-желтоватый раствор. Перемешивание продолжают 2 час при комнатной температуре. LC/MS показывает отсутствие исходного 184а, тогда как сигнал нужного продукта, 184b, является основньм наряду с сигналом, соответствующим побочному продукту, образованному из HATU на предыдущей стадии. Растворители упаривают, а твердый остаток используют на следующей стадии непосредственно после очистки.
Получение трипептида, Соединения 184:

BOCNH-P3(L-трет-BuGly)-P2[(4R)-гидрокси-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропан [Notebook 46877-128]
Сырой продукт предыдущей стадии (Пример 184b, 1.8 ммоля) смешивают с HATU (700 мг, 1.8 ммоля) и BOC-L-трет-лейцином (Пример 180f, Fluka Chemicals, 420 мг, 1.8 ммоля) в CH2 Cl2 (10 мл) при комнатной температуре. К этой суспензии прибавляют основание Хюнига (1 мл, избыток), образуется менее вязкая оранжевая суспензия. LC/MS показывает частичное превращение в соединение 184. Полное превращение в нужный продукт 184 происходит после перемешивания в течение двух дней при комнатной температуре. Сырую реакционную смесь упаривают досуха. Остаток растворяют в этилацетате. Основную часть оставшегося HATU удаляют экстракцией половинным количеством насыщенного свежеприготовленного раствора бикарбоната натрия. Последние следы HATU-остатка (1-гидрокси-7-азабензотриазола) удаляют, промывая деионизированной водой. Упариванием растворителей получают 990 мг (98%) нужного продукта в виде белой пены. Это вещество пригодно для непосредственного последующего алкилирования электрофилами, такими как хинолины и изохинолины без дополнительной очистки. LC/MS Rt- мин (MNa+) [метод А]: 3.52 (808).1 H ЯМР (400 МГц, CD3OD) δ м.д. 0.95 (с, 2Н), 0.99 (с, 9Н), 1.05 (м, 2Н), 1.21 (м, 1Н), 1.39 (м, 9Н), 1.84 (дд, J=8.31, 5.38 Гц, 1Н), 1.96 (м, 1Н), 2.11 (м, 1Н), 2.20 (м, 1Н), 2.91 (м, 1Н), 3.80 (м, 2Н), 4.28 (д, J=9.78 Гц, 1Н), 4.35 (дд, J=9.90, 6.97 Гц, 1Н), 4.47 (с, 1Н), 5.11 (м, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.75 (м, 1Н).
Алкилирование трипептида (соединение 184) электрофилами:
В высушенную на пламени круглодонную колбу помещают Соединение 184 (0.5-1.0 ммолъ), замещенный 4-хлорхинолин (1.0 эквивалент) и хлорид лантана (безводные гранулы LaCl3, Aldrich, М.в. 245 г/моль; 1.0 эквивалент. Замечание: найдено, что такая добавка помогает в некоторых случаях, особенно в случае менее реакционноспособных электрофилов. Этот реагент может иногда не применяться, если электрофилы достаточно реакционноспособны в анионном алкилировании) в 2 мл сухого ДМФА. Неорганическая соль лишь слабо растворима в ДМФА при комнатной температуре. Смесь охлаждают до -78°С (баня сухой лед/ацетон) при перемешивании под азотом. К этой охлажденной смеси прибавляют раствор трет-бутоксида калия в ТГФ (1.0 М, Aldrich, 5.5 эквивалентов) и цвет смеси изменяется от бесцветного на бледно-желтоватый или зеленоватый. Перемешивают при -78°С, время перемешивания зависит от реакционноспособности 4-хлорхинолина (от нескольких часов при -78°С до времени в течение ночи при комнатной температуре). Найдено, что неорганическая соль также превращается в конце в тонкую эмульсию. Прибавляют полунасыщенный свежеприготовленный водный раствор NH4Cl (2 мл). Органические вещества экстрагируют этилацетатом (10 мл × 3). Органические вытяжки объединяют, промывают снова деионизированной водой (10 мл × 2). Упариванием органической фракции получают сырую смесь, обогащенную нужным продуктом, по определению LC/MS. Нужный продукт выделяют препаративной ВЭЖХ в стандартных условиях разделения (как правило: 3.0Х50 мм Xterra @ 4 мин градиент и скорость потока 4 мл/мин), получают аналитически чистый нужный продукт. Алкилирование группы 1-галоизохинолинов проводят точно таким же способом.
Пример 185: Получение Соединения 185
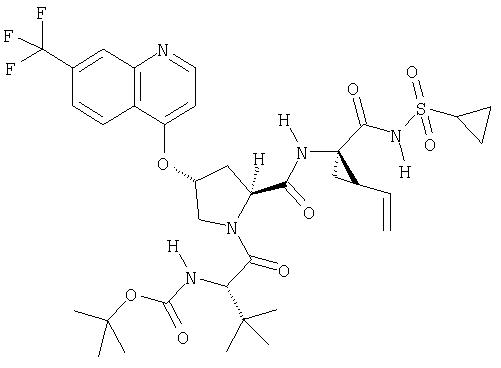
После общей процедуры алкилирования трипептида, описанной в Примере 184, получают BOCNH-P3(L-трет-BuGly)-P2[(4R)-(7-трифторметилхинолин-4-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан: продукт получают в виде белой пены с выходом 50%. LC/MS Rt- мин (MNa+) [метод В]: 2.32 (752).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (с, 9Н), 1.06 (м, 11Н), 1.22 (м, 2Н), 1.43 (дд, J=9.41, 5.26 Гц, 1Н), 1.88 (дд, J=8.19, 5.50 Гц, 1Н), 2.23 (кв, J=8.80 Гц, 1Н), 2.42 (м, 1Н), 2.75 (дд, J=14.06, 6.48 Гц, 1Н), 2.93 (м, 1Н), 4.10 (м, 2Н), 4.61 (м, 2Н), 5.12 (дд, J=10.39, 1.59 Гц, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.72 (м, 2Н), 7.61 (д, J=6.36 Гц, 1Н), 7.96 (д, J=8.80 Гц, 1Н), 8.38 (с, 1Н), 8.59 (д, J=8.56 Гц, 1Н), 9.14 (д, J=6 36 Гц, 1Н).
Пример 186: Получение Соединения 186

После общей процедуры алкилирования трипептида, описанной в Примере 184, получают BOCNH-P3(L-трет-BuGly)-P2[(4R)-(8-трифторметилхинолин-4-оксо)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропан: нужный продукт получают в виде белой пены с выходом 50%. LC/MS Rt- мин (MH+) [метод В]: 2.48 (752).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (с, 9Н), 1.05 (м, 2Н), 1.13 (с, 9Н), 1.23 (м, 2Н), 1.42 (дд, J=8.68, 5.50 Гц, 1H), 1.87 (дд, J=8.07, 5.38 Гц, 1H), 2.21 (кв, J=8.80 Гц, 1H), 2.36 (м, 1Н), 2.69 (дд, J=14.06, 6.97 Гц, 1H), 2.93 (м, 1Н), 4.08 (дд, J=11.98, 2.93 Гц, 1Н), 4.15 (с, 1Н), 4.54 (дд, J=10.52, 7.09 Гц, 1Н), 4.60 (д, J=12.47 Гц, 1Н), 5.11 (дд, J=10.52, 1.71 Гц, 1Н), 5.28 (д, J=15.90 Гц, 1Н), 5.58 (с, 1Н), 5.72 (м, 1Н), 7.32 (д, J=5.87 Гц, 1Н), 7.69 (т, J=7.95 Гц, 1Н), 8.22 (д, J=7.09 Гц, 1Н), 8.55 (д, J=8.07 Гц, 1Н), 8.88 (д, J=5.62 Гц, 1Н).
Получение изохинолиновых интермедиатов для 6-F, 6-этил, 6-изопропил и 6-трет-бутилизохинолин Р2* строительных блоков.
Как правило, 6-фтор и 6-алкилизохинолинов, применяемых в нижеприведенных экспериментах, получают по реакции Померанца-Фрича (Типичная методика: Получение оптически активного 8, 8-дизамещенного 1,1-биизохинолина, К.Hirao, R.Tsuchiya, Y.Yano, H.Tsue, Heterocycles 42(1) 1996, 415-422), как описано ниже. Продукты превращают в 1-хлорпроизводные через N-оксидные интермедиаты, как описано в другом месте.
Общая схема синтеза

Реагенты и условия реакций: (а) кипячение в бензоле, азеотропная отгонка воды; (b) первая стадия: этилхлорформиат, триметилфосфит в ТГФ, вторая стадия: тетрахлорид титана в хлороорме; (с) МСРВВА в CH2 Cl2; (d) POCl3 в бензоле
Пример 187: Получение Соединения 187
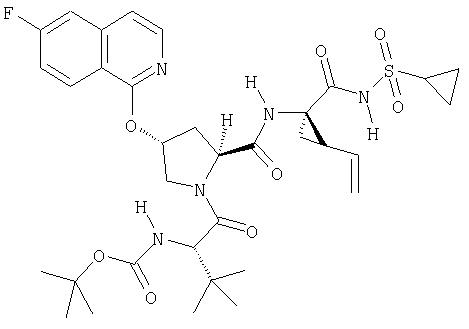
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-фторизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают в виде белой пены с выходом 12%. LC/MS Rt- мин (MNa+) [метод С]: 3.81 (724).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 13Н), 1.22 (с, 9Н), 1.42 (м, 1Н), 1.86 (м, 1Н), 2.21 (м, 2Н), 2.61 (дд, J=13.69, 6.60 Гц, 1Н), 2.93 (м, 1Н), 4.05 (д, J=13.69 Гц, 1Н), 4.21 (с, 1Н), 4.49 (м, 2Н), 5.11 (д, J=10.03 Гц, 1Н), 5.28 (д, J=17.61 Гц, 1Н), 5.72 (м, 1Н), 5.86 (д, J=4.40 Гц, 1Н). 7.31 (м, 2Н), 7.48 (д, J=8.31 Гц, 1Н), 7.97 (д, J=6.36 Гц, 1Н), 8.26 (д, J=6.11 Гц, 1Н).
Пример 188: Получение Соединения 188

Описанное выше алкилирование дает в качестве основного продукта 1-хлоризохинолин: BOCNH-P3(L-трет-BuGly)-P2[(4R)-(1-хлоризохинолин-6-оксо)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропан получают в виде белой пены с выходом 40.2%. LC/MS Rt- мин (МН+) [метод С]: 3.81 (718).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.25 (м, 2Н), 2.54 (дд, J=12.96, 6.60 Гц, 1Н), 2.92 (м, 1Н). 4.06 (дд, J=11.98, 2.69 Гц, 1Н), 4.20 (с, 1Н), 4.31 (д, J=11.74 Гц, 1H), 4.45 (дд, J=9.78, 7.58 Гц, 1Н), 5.11 (дд, J=10.27, 1.71 Гц, 1Н), 5.28 (дд, J=17.36, 1.47 Гц, 1Н), 5.36 (с, 1Н), 5.74 (м, 1Н), 7.35 (д, J=9.29 Гц, 1Н), 7.40 (с, 1Н), 7.70 (д, J=5.87 Гц, 1Н), 8.13 (д, J=5.87 Гц, 1Н), 8.25 (д, J=9.29 Гц, 1Н).
Пример 189: Получение Соединения 189

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-этилизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2 -циклопропан получают в виде белой пены в 4.6 мг желтого твердого вещества (выход 4.2%). LC/MS Rt- мин (МН+) [метод В]: 2.70 (712).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.07 (м, 2Н), 1.22 (м, 11Н), 1.28 (т, J=7.91 Гц, 3H), 1.41 (м, 1Н), 1.85 (м, 1Н), 2.25 (м, 2Н), 2.60 (дд, J=13.69, 6.85 Гц, 1Н), 2.80 (кв, J=7.66 Гц, 2Н), 2.93 (м, 1Н), 4.02 (д, J=31.06 Гц, 1Н), 4.23 (с, 1Н), 4.42 (м, 1Н), 4.54 (м, 1Н), 5.10 (д, J=10.27 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.74 (м, 1Н), 5.84 (с, 1Н), 7.25 (д, J=5.87 Гц, 1Н), 7.38 (д, J=8.56 Гц, 1Н), 7.59 (с, 1Н), 7.90 (д, J=6.24 Гц, 1Н), 8.09 (д, J=8.56 Гц, 1Н).
Пример 190: Получение Соединения 190
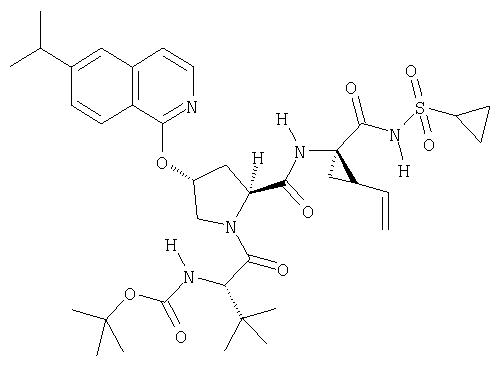
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-изопропилизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают в виде белой пены с выходом 69%. LC/MS Rt- мин (MNa+) [метод В]: 2.76 (749).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 13Н), 1.20 (м, 9Н), 1.31 (д, J=6.85 Гц, 6Н), 1.42 (м, 1Н), 1.86 (дд, J=8.07, 5.62 Гц, 1Н), 2.26 (м, 2Н), 2.62 (дд, J=13.69, 6.85 Гц, 1Н), 2.93 (м, 1Н), 3.07 (м, 1Н), 4.06 (м, 1Н), 4.21 (с, 1Н), 4.52 (м, 2Н), 5.11 (д, J=10.27 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.74 (м, 1Н), 5.84 (с, 1Н), 7.32 (д, J=6.11 Гц, 1Н), 7,46 (д, J=8.56 Гц, 1Н), 7.64 (с, 1Н), 7.90 (д, J=6.11 Гц, 1Н), 8.13 (д, J=8.56 Гц, 1Н).
Пример 191: Получение Соединения 191
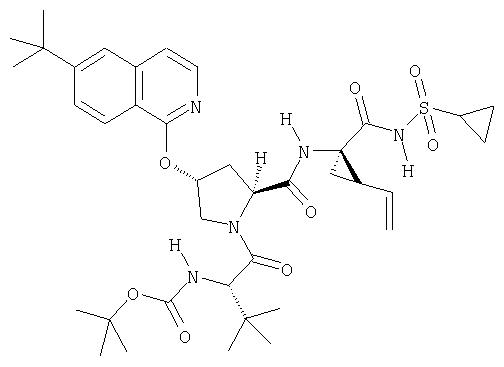
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-трет-бутилизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают в виде белой пены с выходом 81%. LC/MS Rt- мин (МН+) [метод В]: 2.84 (740).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.06 (м, 2Н), 1.18 (с, 9Н), 1.22 (м, 2Н), 1.39 (с, 9Н), 1.43 (м, 1Н), 1.87 (дд, J=8.19, 5.50 Гц, 1.Н), 2.27 (м, 2Н), 2.63 (дд, J=13.57, 6.97 Гц, 1Н), 2.93 (м, 1Н), 4.06 (м, 1Н), 4.20 (с, 1Н), 4.52 (м, 2Н), 5.10 (д, J=11.49 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.73 (м, 1Н), 5.84 (с, 1Н), 7.36 (д, J=6.11 Гц, 1Н), 7.66 (дд, J=8.80, 1.22 Гц, 1Н), 7.78 (с, 1Н), 7.91 (д, J=5.87 Гц, 1Н), 8.15 (д, J=8.80 Гц, 1Н).
Получение 6-изопропоксил- и 6-трет-бутоксилизохинолиновых интермедиатов:
Некоторые 6-алкокси-1-хлоризохинолины получают непосредственным замещением 6-фтор-1-хлоризохинолина на соответствующий алкоксид-ион, например, при действии трет-бутоксида калия (53%) или изопропоксида натрия (54%).
Общая схема синтеза

R = алкоксид-анионы, например, трет-Bu, изо-Pr
Реакцию ароматического нуклеофильного замещения в 6-фтор-1-хлоризохинолине осуществляют под действием изопропоксида натрия и трет-бутоксида в ДМФА, получают соответствующий 6-изопропокси - (54%):1H ЯМР (400 МГц, хлороформ-d) δ м.д. 1.43 (д, J=6.11 Гц, 6Н), 4.76 (м, J=6.11 Гц, 1Н), 7.08 (д, J=2.45 Гц, 1Н), 7.29 (дд, J=9.29, 2.45 Гц, 1Н), 7.50 (д, J=5.62 Гц, 1Н), 8.18 (д, J=5.87 Гц, 1Н), 8.24 (д, J=9,29 Гц, 1Н) и 6-трет-бутокси-1-хлоризохинолин (55%):1H ЯМР (400 МГц, хлороформ-d) δ м.д. 1.48 (с, 9Н), 7.31 (м, 2Н), 7.47 (д, J=5.62 Гц, 1Н), 8.18 (д, J=5.62 Гц, 1Н), 8.21 (д, J=9.78 Гц, 1Н), соответственно, в качестве основного продукта. Эти 6-алкокси-1-хлоризохинолины алкилируют трипептидом, как описано в Примере 184, получая нужные продукты, изображенные ниже.
Пример 192; Получение Соединения 192

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-изопропоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают в виде белой пены с выходом 59%. LC/MS Rt- мин (MH+) [метод С]: 3.87 (742).1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99 (с, 11Н), 1.06 (м, 2Н), 1.21 (с, 9Н), 1.37 (д, J=5.87 Гц, 6Н), 1.42 (м, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.25 (м, 2Н), 2.61 (дд, J=13,69, 6.85 Гц, 1Н), 2.92 (м, 1Н), 4.04 (дд, J=11.86, 3.30 Гц, 1Н), 4.21 (уш. с, 1Н), 4.49 (м, 2Н), 4.78 (д, J=5.87 Гц, 1Н), 5.10 (дд, J=10.39, 1.35 Гц, 1Н), 5.28 (д, J=16.87 Гц, 1Н), 5.73 (м, 1Н), 5.79 (д, J=11.25 Гц, 1Н), 7.07 (дд, J=9.05, 1.96 Гц, 1Н), 7.18 (д, J=2.20 Гц, 1Н), 7.26 (д, J=6.11 Гц, 1Н), 7.85 (д, J=6.11 Гц, 1Н), 8.10 (д, J=9.05 Гц, 1Н).
Пример 193: Получение Соединения 193

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-трет-бутоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2 -циклопропан получают в виде белой пены с выходом 39%. LC/MS Rt- мин (MH+) [метод С]: 3.99 (756).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.03 (уш с, 13Н), 1.19 (с, 9Н), 1.42 (м, 10Н), 1.86 (дд, J=7.83, 5.62 Гц, 1Н), 2.24 (м, 2Н) 2.60 (дд, J=13.69, 6.85 Гц, 1Н), 2.93 (м, 1Н), 4.04 (дд, J=11.49, 2.93 Гц. 1Н), 4.23 (с, 1Н), 4.49 (м, 2Н), 5.10 (д, J=11.25 Гц, 1Н), 5.27 (д. J=16.87 Гц, 1Н), 5.73 (м, 1Н), 5.81 (с, 1Н), 7.13 (дд, J=8.80, 1.47 Гц, 1Н), 7.25 (д, J=5.87 Гц, 1Н), 7.33 (д, J=2.20 Гц, 1Н), 7.87 (д, J=6.11 Гц, 1Н), 8.10 (д, J=9.05 Гц, 1Н).
Получение фталазиновых Р2* производных:
Как правило, как 1-хлорфталазин, так и 1,4-дихлорфталазин алкилируются легко, давая нужный продукт. Однако, продажные 1-хлорфталазин и 1,4-дихлорфталазин часто имеют в качестве примесей некоторые продукты гидролиза. Предварительная обработка POCl3 и непосредственно сразу после этого алкилирование дает более приемлемые результаты.
Общая схема синтеза

Условия реакции: (а) POCl3 в DCE; (b) Алкилирование трипептидом; (с) натриевые производные имидазола (R=СН), триазола (R=N).
Пример 194: Получение Соединения 194

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(фталазин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2 -циклопропан получают в виде белой пены с выходом 41%. LC/MS Rt- мин (MNa+) [метод В]: 2.07 (707).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.03 (м, 9Н), 1.06 (м, 4 Н), 1.14 (с, 9Н), 1.20 (м, 1Н), 1.43 (м, 1Н), 1.87 (дд, J=8.07, 5.62 Гц, 1Н), 2.24 (кв, J=8.80 Гц, 1Н), 2.38 (м, 1Н), 2.76 (дд, J=14.18, 7.09 Гц, 1Н), 2.92 (м, 1Н), 4.11 (м,2Н), 4.62 (м, 1Н), 5.11 (дд, J=10.27, 1.71 Гц, 1Н), 5.29 (дд, J=17.12, 1.22 Гц, 1Н), 5.72 (м, 1Н), 5.96 (с, 1Н), 8.26 (м, 2Н), 8.46 (м, 2Н), 9.84 (с, 1Н).
Пример 195: Получение Соединения 195

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-хлорфталазин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают в виде белой пены с выходом 23%. LC/MS Rt- мин (MNa+) [метод С]: 3.52 (742).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 11Н), 1.06 (м, 2Н), 1.14 (с, 9Н), 1.22 (м, 1Н), 1.43 (м, 1Н), 1.87 (м, 1Н), 2.21 (м, 1Н), 2.35 (м, J=10.27 Гц, 1Н), 2.70 (м, 1Н), 2.93 (м, 1Н), 4.05 (д, J=3.42 Гц, 1Н), 4.58 (м, 2Н), 5.11 (дд, J=10.39, 1.10 Гц, 1Н), 5.28 (д, J=17.36 Гц, 1Н), 5.73 (м, 1Н), 5.93 (с, 1Н), 7.99 (м, 1Н), 8.07 (т, J=7.70 Гц, 1Н), 8.26 (дд, J=8.19, 2.32 Гц, 2Н).
Получение 4-(имидазо-1-ил)фталазин и 4-(1,2,4-триазо-1-ил)фталазин Р2* производных:
Полученный выше продукт, Соединение 195, BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-хлорфталазин-1-оксо)-S-пролин]-P1(LR,2S винил Асса)-CONHSO2-циклопропан вступает в реакцию замещения с анионами типичных азолов, таких как имидазол и триазол, давая 4-азолзамещенные производные фталазина, показанные ниже:
Пример 196: Получение Соединения 196

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-(имидазо-1-ил)фталазин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают в виде белой пены с выходом 23%. LC/MS Rt- мин (MNa+) [метод В]: 1.94 (773).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.03 (с, 9Н), 1.07 (м, 2Н), 1.20 (м, 9Н), 1.24 (м, 1Н), 1.41 (м, 2Н), 1.88 (дд, J=8.19, 5.50 Гц, 1Н), 2.23 (м, 1Н), 2.41 (м, 1Н), 2.75 (м, J=14.92 Гц, 1Н), 2.93 (м, 1Н), 4.14 (м, 2Н), 4.60 (дд, J=10.15, 6.97 Гц, 2Н), 5.12 (дд, J=10.27, 1.47 Гц, 1Н), 5.29 (дд, J=17.24, 1.35 Гц, 1Н), 5.74 (м, 1Н), 6.09 (с, 1Н), 7.85 (с, 1Н), 7.94 (м, 1Н), 8.10 (м, 2Н), 8.43 (м, 1Н), 9.17 (с, 1Н), 9.46 (с, 1Н).
В процессе реакции замещения также выделяют небольшое количество побочного де-ВОС продукта (Соединение 197):
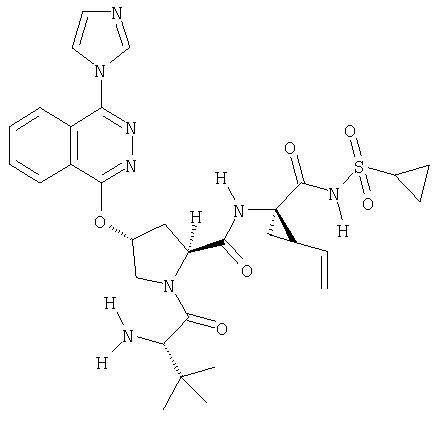
NH2-P3(L-трет-BuGly)-P2[(4R)-(4-(имидазо-1-ил)фталазин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают в виде белой пены с выходом 17%. LC/MS Rt- мин (МН+) [метод В]: 1.22 (651).1H ЯМР (400 МГц, CD3OD) δ м.д. 1.16 (с, 9Н), 1.23 (м, 2Н), 1.42 (дд, J=9.41, 5.50 Гц, 1Н), 1.89 (м, 1Н), 2.26 (кв, J=8.97 Гц, 1Н), 2.43 (м, 1Н), 2.78 (дд, J=14.06, 7.21 Гц, 1Н), 2.93 (м, 1Н), 3.74 (м, 1Н), 4.11 (с, 1Н), 4.22 (дд, J=12.23, 3.91 Гц, 1Н), 4.47 (м, 2Н), 4.71 (дд, J=10.27, 7.09 Гц, 1Н), 5.12 (м, 1Н), 5.29 (д, J=17.36 Гц, 1Н), 5.71 (м, 1Н), 6.13 (т, J=3.67 Гц, 1Н), 7.87 (с, 1Н), 7.97 (м, 1Н), 8.14 (м, 3H), 8.41 (м, 1Н), 9.47 (с, 1Н).
Пример 198: Получение Соединения 198
Это осуществляют реакцией замещения 4-хлорфталазина (Соединение 195) с натриевой солью 1,2,4-триазола в ДМФА при 55-65°С.

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-(1,2,4-триазо-1-ил)фталазин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2 -циклопропан получают в виде белой пены с выходом 62%. LC/MS Rt- мин (MNa+) [метод С]: 3.35 (774).1H ЯМР (500 МГц, CD3OD) δ м.д. 0.97 (с, 9Н), 1.01 (м, 2Н), 1.10 (с, 9Н), 1.16 (м, 1Н), 1.37 (м, 2Н), 1.82 (м, 1Н), 2.18 (д, J=8.55 Гц, 1Н), 2.32 (м, 1Н), 2.69 (дд, J=13.58, 6.87 Гц, 1Н), 2.88 (уш с, 1Н), 4.06 (д, J=11.60 Гц, 1Н), 4.13 (с, 1Н), 4.53 (м, J=9.16 Гц, 1Н), 4.61 (д, Гц, 1Н), 5.06 (д, J=10.07 Гц. 1Н), 5.23 (д, J=17.40 Гц, 1Н), 5.68 (м, 1Н), 5.98 (с, 1Н), 7.97 (м, 2Н), 8.30 (м, J=11.80 Гц, 2Н), 8.44 (д, J=7.63 Гц, 1Н), 9.14 (с, 1Н).
Получение 4-гидрокси-и 4-алкоксифталазин Р2* производных
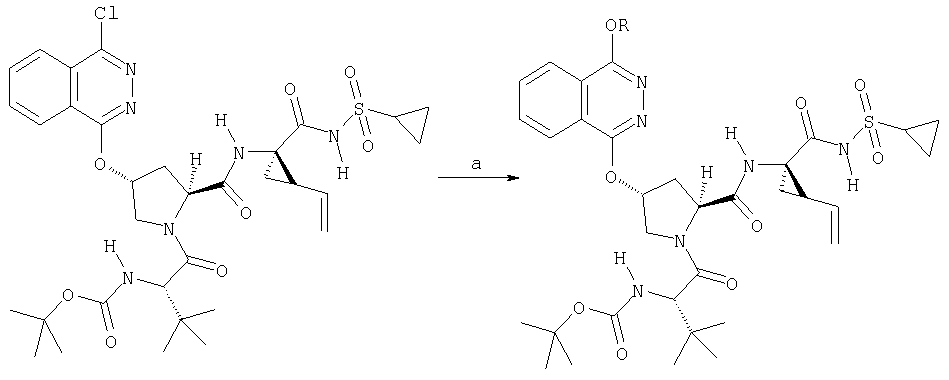
Условия реакции: (а) алкоксиды натрия, такие как метоксид, этоксид и изопропоксид.
Пример 199: Получение Соединения 199
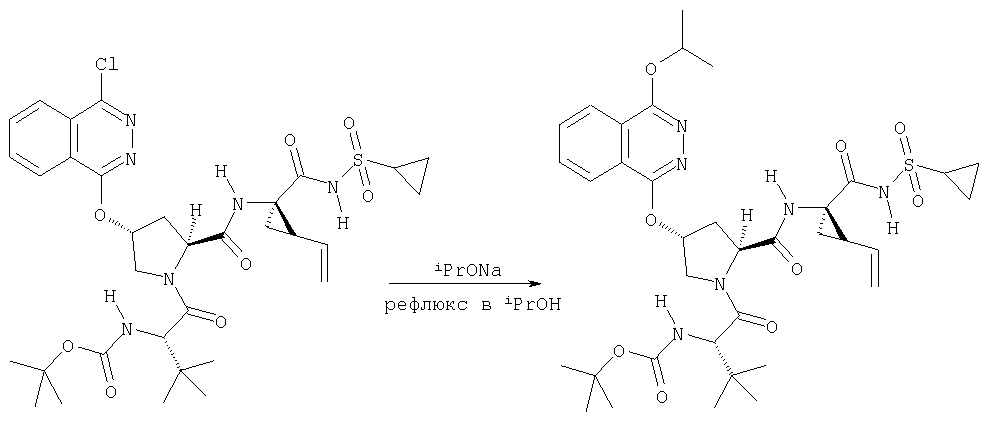
4-Хлорфталазин (Пример 195) растворяют в изопропиловом спирте при комнатной температуре и прибавляют 1.0 экв изопропоксида натрия, полученную суспензию кипятят. Получают нужный продукт, 4.5 мг (выход 20%) получают в виде твердого вещества желтого цвета. LC/MS Rt- мин (МН+) [метод В]: 2.68 (743).1 H ЯМР (400 МГц. CD3OD) δ м.д. 1.02 (м, 11Н), 1.20 (м, 11Н), 1.42 (м, 1Н), 1.49 (д, J=6 Гц, 6Н), 1.87 (дд, J=7.95, 5.50 Гц, 1Н), 2.21 (м, 1Н), 2.23 (м, 1Н), 2.66 (м, 1Н), 2.93 (м, 1Н), 4.08 (кв, J=7.09 Гц, 1Н), 4.19 (с, 1Н), 4.55 (м, 2Н), 5.11 (д, J=10.27 Гц, 1Н), 5.28 (д, J=17.61 Гц, 1Н), 5.48 (м, 1Н), 5.70 (д, J=10.03 Гц, 1Н), 5.81 (м, 1Н), 7.90 (м, 2Н), 8.16 (м, 2Н).
Пример 200: Получение Соединения 200
Аналогичным образом получают 4-этоксипроизводное: BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-этоксифталазин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан.
Получают 4.0 мг твердого вещества желтого цвета (16%). LC/MS Rt- мин (МН+): 2.52 (729) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.06 (м, 2Н), 1.16 (с, 9Н), 1.24 (м, 2Н), 1.43 (дд, J=9.78, 5.14 Гц, 1Н), 1.53 (т, J=6.97 Гц, 3H), 1.87 (дд, J=8.19, 5.50 Гц, 1Н), 2.22 (кв, J=8.97 Гц, 1Н), 2.32 (м, 1Н), 2.67 (м, 1Н), 2.93 (м, 1Н), 4.06 (д, J=8.56 Гц, 1Н), 4.19 (с, 1Н), 4.56 (м, 4Н), 5.10 (м, 1Н), 5.30 (м, 1Н), 5.74 (м, 1Н), 5.82 (с, 1Н), 7.94 (м, 2Н), 8.16 (д, J=7.83 Гц, 1Н), 8.21 (м, 1Н).
Пример 201: Получение Соединения 201
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-метоксифталазин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают с выходом 30.2%. LC/MS Rt- мин (MH+): 2.42 (715) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.96 (с, 9Н), 1.07 (м,2Н), 1.20 (м, 11Н), 1.43 (дд, J=9.29, 5.38 Гц, 1Н), 1.87 (дд, J=8.07, 5.62 Гц, 1Н), 2.22 (кв, J=8.80 Гц, 1Н), 2.31 (м, 1Н), 2.66 (д, J=8.07 Гц, 1Н), 2.93 (м, 1Н), 4.06 (дд, J=11.98, 3.18 Гц, 1Н), 4.19 (д, J=3.42 Гц, 4Н), 4.54 (м, 2Н), 5.11 (м, 1Н), 5.28 (д, J=17.36 Гц, 1Н), 5.73 (м, 1Н), 5.83 (с, 1Н), 7.95 (м, 2Н), 8.19 (м, 2Н).
Пример 202: Получение Соединения 202
Попытка провести реакцию замещения BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-хлорфталазин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропана (Пример 195) с натриевой солью тетразола дает в основном 4-гидрокси, продукт гидролиза.

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-гидроксифталазин-1-оксо)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропан получают с выходом 44.2% в виде твердого вещества бледно-кремового цвета. LC/MS Rt- мин (МН+): 2.18 (701) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.05 (м, 2Н), 1.23 (м, 11Н), 1.42 (дд, J=9.29, 5.38 Гц, 1Н), 1.87 (дд, J=8.07, 5.38 Гц, 1Н), 2.21 (м, 2Н), 2.63 (м, 1Н), 2.93 (м, 1Н), 4.00 (с, 1Н), 4.20 (с, 1Н), 4.50 (м, 2Н), 5.11 (дд, J=10.27, 1.47 Гц, 1Н), 5.29 (д, J=16.87 Гц, 1Н), 5.59 (с, 1Н), 5.73 (м, 1Н), 7.86 (дд, J=5.75, 3.30 Гц, 2Н), 8.01 (дд, J=5.87, 3.42 Гц, 1Н), 8.29 (дд, J=5.87, 3.42 Гц, 1Н).
Получение 5,6-дизамещенных изохинолин Р2* производных по методике алкилирования
Общая схема синтеза
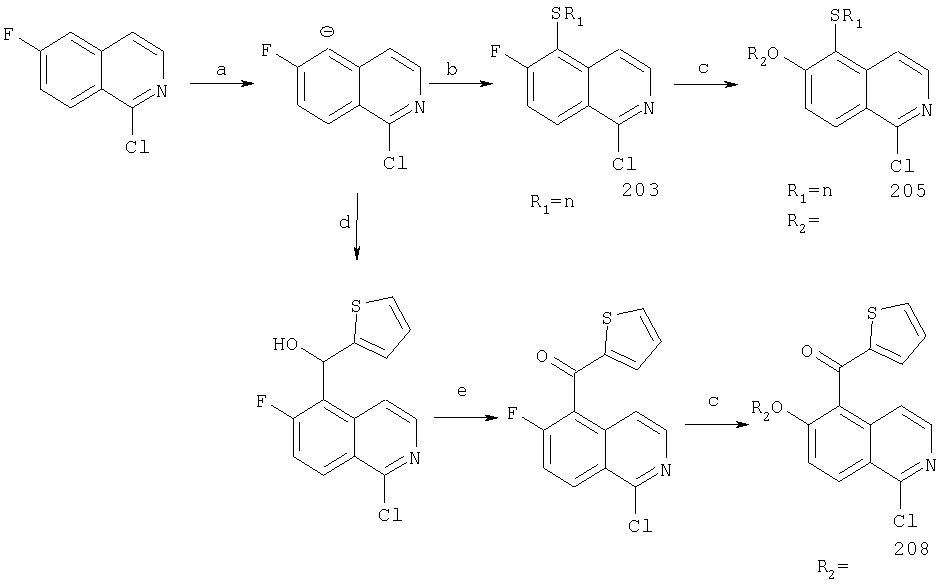
Условия реакции: (a) LDA в ТГФ; (b) Алкилсульфид, такой как (н-PrS)2; (с) Алкоксид натрия, такой как MeONa; (d) Тиофен-2-карбоксальдегид; (e) MnO2 в бензоле.
Пример 203: Получение 1-хлор-5-пропилтио-6-фторизохинолина

К охлажденному (-78°С) раствору 1-хлор-6-фторизохинолина (59 мг, 0.32 ммоля) в 2 мл ТГФ прибавляют раствор LDA в циклогексане (1.5 молярный, 0.23 мл, 0.35 ммоля). Оранжевый раствор перемешивают 2 час, прибавляют н-пропилдисульфид (60 мкл, чистое вещество, избыток). Реакционную смесь оставляют на 30 мин доходить до комнатной температуры. Прибавляют полунасыщенный раствор NH4Cl, органический остаток экстрагируют этилацетатом. LC-MS анализ показывает, примерно, 50%-ную конверсию в заданный продукт наряду с, в основном, исходным веществом. Нужный продукт очищают на короткой колонке (4 см × 2 см, силикагель типа-Н), элюируя 5%-ным раствором эфира в смеси гексанов, получают 29 мг (выход 36%) продукта. LC/MS Rt- мин (МН+) [метод С]: 3.79 (256).1H ЯМР (400 МГц, хлороформ-d) δ м.д. 0.96 (т, J=7.34 Гц, 3H), 1.52 (м, 2Н), 2.86 (м, 2Н), 7.45 (дд, J=9.29, 8.56 Гц, 1Н), 8.34 (д, J=0.73 Гц, 2H), 8.37 (м, 1Н). Это соединение алкилируют трипептидом по методике, описанной в Примере 184, получая нижеприведенное соединение:
Пример 204: Получение Соединения 204
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(1-хлор-5-пропилтиоизохинолин-6-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан
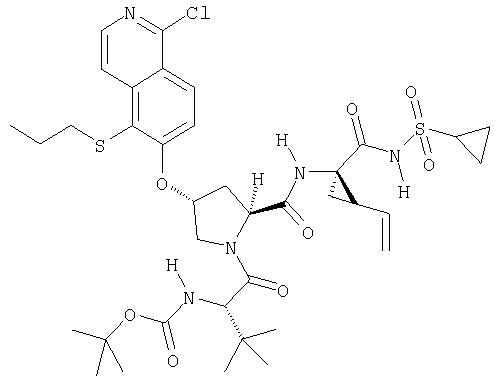
По общей методике получают 4.6 мг (3.2%) твердого вещества желтого цвета. LC/MS Rt- мин (МН+): 2.73 (792) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.93 (т, J=7.34 Гц, 3H), 0.97 (с, 9Н), 1.08 (м, 2H), 1.24 (м, 11Н), 1.43 (м, 3H), 1.86 (м, 1Н), 2.24 (м, 2H), 2.56 (м, 1Н), 2.78 (кв, J=7.09 Гц, 2H), 2.92 (м, 1Н), 4.01 (д, J=9.29 Гц, 1Н), 4.22 (с, 1Н), 4.29 (с, 1Н), 4.59 (д, J=6.85 Гц, 1Н), 5.11 (д, J=10.76 Гц, 1Н), 5.28 (д, J=17.36 Гц, 1Н), 5.49 (с, 1Н), 5.74 (м. 1Н), 7.66 (д, J=9.29 Гц, 1Н), 8.18 (д, J=6.11 Гц, 1Н), 8.41 (м, 2H).
Пример 205: Получение 5-пропилтио-6-этоксиизохинолин Р2* производных
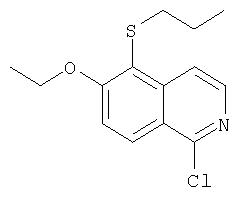
Нижеприведенная методика равно применима к другим 5-алкилтио-6 алкоксиизохинолинам при изменении указанных здесь реагентов. К раствору 1-хлор-6-фторизохинолина (88 мг, 0.48 ммоля) в 2.0 мл ТГФ в атмосфере азота при -78°С прибавляют LDA (1.5 молярный раствор в циклогексане, 0.42 мл, 0.63 ммоля), при этом образуется темно-коричневый раствор. Перемешивают при -78°С в течение 30 мин и добавляют чистый н-пропилдисульфид (85 мкл, избыток). Реакционную смесь в течение 30 мин доводят до комнатной температуры. Прибавляют полунасыщенный раствор NH4Cl, органические остатки экстрагируют этилацетатом. Органические вытяжки объединяют и сушат в вакууме до 50 микрон (Hg=0.06 мбар). Сырой продукт растворяют в 2 мл ТГФ, охлаждают до -78°С, прибавляют избыток этоксида калия (60 мг). Промежуточный изохинолин под конец очищают на колонке с силикагелем (тип-Н, Merck), элюент эфир-смесь гексанов, получают 32.2 мг (24%) чистого соединения. Метод LC/MS показал 1-хлор-5 пропилтио-6-этоксиизохинолин при Rt- мин (МН+): 3.77 (282) [метод С].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 0.94 (т, J=7.34 Гц, 3H), 1.46 (м, 2Н), 1.55 (т, J=6.97 Гц, 3H), 2.83 (т, J=7.21 Гц, 2Н), 4.32 (кв, J=6.85 Гц, 2Н), 7.36 (д, J=9.29 Гц, 1Н), 8.22 (д, J=6.11 Гц, 1Н), 8.32 (д, J=9.29 Гц, 1Н), 8.35 (д, J=6.11 Гц, 1Н). По общей методике алкилирования трипептидом (Пример 184) этот 1-хлор-5-пропилтио-6-этоксиизохинолин алкилируют трипептидом (соединение 184), получают 40.7 мг (44.8%) нужного продукта, показанного ниже.
Пример 206: Получение Соединения 206
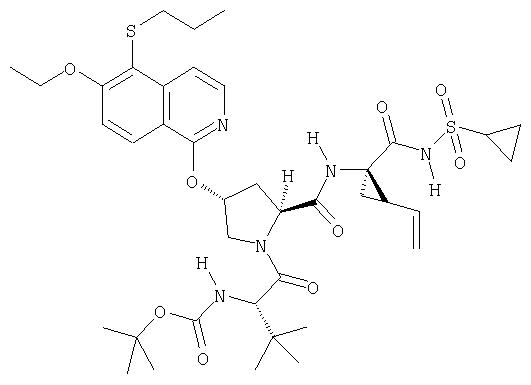
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-этокси-5-пропилтиоизохинолин-1-оксо)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропан: LC/MS Rt- мин (MH+): 2.93 (803) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.93 (т, J=7.34 Гц, 3H), 1.01 (с, 9 Н), 1.07 (м, 2Н), 1.21 (м, 11Н), 1.41 (м, 3H), 1.48 (т, J=6.85 Гц, 3H), 1.86 (дд, J=8.07, 5.62 Гц, 1Н), 2.25 (м, 2Н), 2.60 (дд, J=13.69, 6.85 Гц, 1Н), 2.81 (кв, J=6.97 Гц, 2Н), 2.93 (м, 1Н), 4.05 (м, 1 Н), 4.21 (с, 1Н), 4.27 (кв, J=7.09 Гц, 2Н), 4.43 (д, J= 11.74 Гц, 1Н), 4.52 (м, 1Н), 5.10 (д, J=10.76 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.74 (м, 1Н), 5.82 (с, 1Н), 7.30 (д, J=9.05 Гц, 1Н), 7.92 (д, J=6.36 Гц, 1Н), 7.97 (м, 1Н), 8.22 (д, J=9.05 Гц, 1Н).
Пример 207: Получение Соединения 207
Аналогичным образом эта методика применяется для получения BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-метокси-5-метилтиоизохинолин-1-оксо)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропана.

К раствору 100 мг 1-хлор-6-фторизохинолина (0.55 ммоля) в 2 мл сухого ТГФ при -78°С прибавляют LDA в ТГФ (1.3 экв). Образуется темно-коричневый раствор, затем прибавляют дисульфид и цвет раствора меняется на зеленоватый, затем светло-коричневый. К реакционной смеси прибавляют 2 мл воды и 2 мл NH4Cl, экстрагируют этилацетатом, сушат сульфатом натрия. Растворитель упаривают в вакууме, полученный остаток используют без очистки (в сыром виде). LC/MS Rt- мин (МН+): 2.23 (228) [метод В]. Сырой продукт снова растворяют в 2 мл сухого ТГФ при -78°С и прибавляют 1.3 экв. КОМе, затем реакционную смесь оставляют на ночь при перемешивании, при этом ее температура доходит до комнатной. К реакционной смеси прибавляют этилацетат и промывают рассолом, сушат сульфатом натрия. Получают 104 мг (79%). LC/MS Rt- мин (MH+): 2.04 (240) [метод В]. Интермедиат, 1-хлор-5-метилтио-6-метоксиизохинолин, вступает в реакцию алкилирования трипептидом по методике, описанной выше. По общей методике получают 70 мг (42.7%) твердого вещества желтого цвета. LC/MS Rt- мин (MH+): 2.65 (760) [метод В].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 0.94 (м, 11Н), 1.17 (с, 9Н), 1.26 (м, 2Н), 139 (м, 1Н), 1.83 (дд, J=8.07, 5.62 Гц, 1Н), 2.01 (м, 2Н), 2.23 (с, 3H), 2.45 (м, 1Н), 2.79 (м, 1Н), 3.94 (с, 3H), 3.97 (д, J=3.91 Гц, 1Н), 4.15 (с, 1Н), 4.25 (д, J=11.74 Гц, 1Н), 4.36 (дд, J=9.66, 7.21 Гц, 1Н), 4.99 (д, J=10.27 Гц, 1Н), 5.12 (д, J=16.87 Гц, 1Н), 5.69 (м, 1Н), 5.74 (с, 1Н), 7.08 (д, J=9.05 Гц, 1Н), 7.83 (м, 2Н), 8.06 (д, J=9.05 Гц, 1Н).
Пример 208: Получение 1-хлор-6-метоксиизохинолин-5-илтиофен-2-илметанона
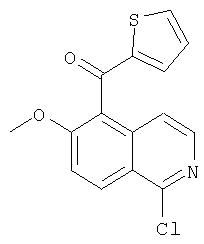
По той же методике LDA-депротонирования (получение Соединения из Примера 203) 1-хлор-6-фторизохинолина, описанной ранее, к первоначально образовавшемуся аниону прибавляют 2-тиофенкарбоксальдегид, получают 1-хлор-6-фторизохинолин-5-илтиофен-2-илметанол. Вещество окисляют до 1-хлор-6-фторизохинолин-5-илтиофен-2-илметанона с помощью MnO2 в бензоле, общий выход после хроматографической очистки 49.6%. LC/MS Rt- мин (МН+): 2.98 (292) [метод В].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 7.12 (дд, J=4.89, 3.91 Гц, 1Н), 7.40 (м, 1H), 7.53 (м, 1Н), 7.56 (дд, J=5.87, 0.73 Гц, 1Н), 7.82 (дд, J=5.01, 1.10 Гц, 1Н), 8.27 (д, J=5.87 Гц, 1Н), 8.54 (ддд, J=9.29, 5.38, 0.73 Гц, 1Н). Само нуклеофильное ароматическое замещение атома фтора осуществляют в растворе избытка метоксида калия, получая в качестве основного продукта 1-хлор-6-метоксиизохинолин-5-илтиофен-2-илметанон наряду с 25-33% 1, 6-диметоксиизохинолин-5-илтиофен-2-илметаноном. Сырой продукт (77%) используют на стадии алкилирования трипептидом без дополнительной очистки.
Пример 209: Получение Соединения 209

По общей методике алкилирования трипептидом (Пример 184) получают 35.3 мг (26.5%) BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-метокси-5-(тиофен-2-карбонил)-изохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан в виде бледно окрашенного твердого вещества. LC/MS Rt- мин (MH+): 2.54 (825) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (с, 9Н), 1.06 (м, 2Н), 1.22 (м, 2Н), 1.26 (с, 9Н), 1.43 (м, 1Н), 1.87 (дд, J=7.95, 5.50 Гц, 1Н), 2.28 (м, 2Н). 2.62 (дд, J=13.82, 6.97 Гц, 1Н), 2.93 (м, 1Н), 3.90 (с, 3H), 4.07 (дд, J=11.62, 3.06 Гц, 1Н), 4.23 (с, 1Н), 4.43 (м, 1Н), 4.55 (дд, J=9.78, 7.34 Гц, 1Н), 5.09 (м, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.74 (м, 1Н), 5.86 (с, 1H), 6.93 (д, J=6.11 Гц, 1Н), 7.11 (м, 1Н), 7.32 (дд, J=3.91, 0.98 Гц, 1Н), 7.46 (д, J=9.29 Гц, 1Н), 7.86 (с, J=6.72 Гц, 1Н), 7.91 (дд, J=4.89, 1.22 Гц, 1Н), 8.39 (д, J=9.29 Гц, 1Н).
Получение Р2* с помощью производных коричной кислоты. Общая методика, изображенная ниже, подробно описана в другом месте.
Общая схема синтеза

Пример 210: Получение Соединения 210
10.0 г мета-толилакриловой кислоты (61.7 ммоля) суспендируют в 50 мл бензола, прибавляют 12.6 мл DPPA (0.95 экв), а затем 10.3 мл триэтиламина (1.2 экв). Полученный раствор перемешивают при комнатной температуре в течение 1 час. Отгоняют летучие в вакууме и мета-толилакрилоилазид очищают флеш-хроматографией, получают 11.5 г чистого соединения (выход количественный). Это вещество в 100 мл дифенилметана по каплям в течение 1 часа прибавляют в 100 мл предварительно нагретого до 200°С дифенилметана. Полученный раствор выдерживают при этой температуре еще в течение 4 часов, затем охлаждают до комнатной температуры. Образуется белый осадок, который отфильтровывают. Твердое вещество трижды промывают гексаном и сушат. К фильтрату прибавляют 200 мл смеси гексанов, раствор оставляют на ночь для выделения второй порции. Обе порции осадка объединяют, получают 4.2 г 6-метилизохинолин-1-ол (50%). LC/MS Rt- мин (MH+): 1.31 (160) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 2.49 (с, 3H), 6.61 (д, J=7.32 Гц, 7.13 (д, J=7.02 Гц, 1Н), 7.36 (д, J=8.24 Гц, 1Н), 7.45 (с, 1Н), 8.18 (д, J=8.24 Гц, 1Н). Продукт суспендируют в 15 мл POCl3 и кипятят в течение 3 час. После удаления POCl3 в вакууме остаток распределяют между EtOAc (1 л) и холодным водным раствором NaOH (полученным из 200 мл 1.0 N NaOH и 20 мл 10.0 N NaOH) и перемешивают в течение 15 мин. Органический слой промывают водой (2×200 мл), рассолом (200 мл), сушат (MgSO4) и упаривают в вакууме, получают 1-хлор-6-метилизохинолин (67.4%). LC/MS Rt- мин (MH+): 1.92 (178) [метод В].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 2.53 (с, 3H), 7.47 (д, J=6.11 Гц, 2Н), 7.56 (с, 1Н), 8.18 (м, 2Н). Конечное алкилирование 1-хлор-6-метилизохинолина трипептидом проводят по методике, описанной ранее (Пример 184).

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-метилизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан: вещество получают в виде белой пены с выходом 18%. LC/MS Rt- мин (MNa+): 2.64 (720) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 13Н), 1.23 (м, 9Н), 1.42 (м, 1Н), 1.86 (дд, J=7.95, 5.50 Гц, 1Н), 2.25 (м, 2Н), 2.49 (с, 3H), 2.61 (дд, J=13.82, 6.48 Гц, 1Н), 2.93 (м, 1Н), 4.05 (дд, J=11.86, 3.30 Гц, 1Н), 4.23 (с, 1Н), 4.43 (д, J-11.49 Гц, 1Н), 4.52 (м, 1Н), 5.10 (д, J=11.49 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.74 (м, 1Н), 5.83 (с, 1Н), 7.24 (д, J=5.87 Гц, 1Н), 7.35 (д, J=8.07 Гц, 1Н), 7.58 (с, 1Н), 7.89 (д, J=5.87 Гц, 1Н), 8.07 (д, J=8.56 Гц, 1Н).
Пример 211: Получение Соединения 211

По общей методике, описанной выше, получают 51.0 мг (64.9%) BOCNH-P3(L-трет-BuGly)-P2[(4R)-(1,3-диокса-7-азациклопента[α]нафталин-6-ол)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циююпропана в виде бледно окрашенного твердого вещества. LC/MS Rt- мин (МН+): 2.57 (728) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99 (с, 9Н), 1.07 (м, 2Н), 1.18 (м, 11 Н), 1.42 (м, 1Н), 1.86 (дд, J=8.07, 5.62 Гц, 1Н), 2.23 (м, 2Н), 2.59 (дд, J=13.69, 6.85 Гц, 1Н), 2.93 (м, 1Н), 4.04 (дд, J=10.27 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.73 (м, 1Н), 5.82 (с, 1Н), 6.18 (с, 2Н), 7.13 (д, J=8.56 Гц, 1Н), 7.19 (д, J=6.11 Гц, 1Н), 7.81 (д, J=8.56 Гц, 1Н), 7.85 (д, J=6.11 Гц, 1Н).
Получение 5,6,7-тризамещенных изохинолин Р2* производных:

Условия реакции: (a) LDA в ТГФ; (b) Т-фторбензолсульфонимид (NFSI) для R=F или диметилсульфид (MeS)2 для R = SMe.
Полученный ранее 5,6-метилендиокси-1-хлоризохинолин депротонируют непосредственно в присутствии сильного основания, такого как LDA, получая соответствующий 7-анион, без взаимодействия с функциональной группой 1-хлор. К 7-аниону прибавляют электрофилы, такие как NFSI (N-фторбензолсульфонимид) и диметилсульфид, получают соответствующую 7-замещенную циклическую систему изохинолина.
Пример 212: Получение Соединения 212
Стадия 1:
Получение 5,6-метилендиокси-7-фтор-1-хлоризохинолина. К раствору 5, 6-метилендиокси-1-хлор-изохинолина (126 мг, 0.61 ммоля) в 4 мл ТГФ под азотом при -78°С прибавляют раствор LDA в циклогексане (1.5 молярный, 0.65 мл, 0.98 ммоля). Светлый коричневатый раствор перемешивают 15 мин и прибавляют N-фторбензолсульфонимид (NFSI, 0.3 г, 1.5 эквивалента). ТСХ показывает появление нового пятна помимо неизмененного исходного. Обработка водой с последующей экстракцией этилацетатом дает сырой продукт в виде масла, который очищают препаративной ВЭЖХ, получают 52 мг (38%). LC/MS Rt- мин (МН+): 3.09 (226) [метод С].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 6.33 (с, 2Н), 7.52 (д, J=5.87 Гц, 1Н), 7.71 (д, J=10.51 Гц, 1Н), 8.16 (д, J=5.87 Гц, 1Н).
Стадия 2
Алкилирование 5, 6-метилендиокси-7-фтор-1-хлоризохинолина трипептидом проводят как описано ранее (Пример 184), получают основной продукт в результате замещения фтора.
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-хлор-1,3-диокса-7-азациклопента[α]нафталин-4-окса)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропан, изображенный ниже,

По общей методике получают 24.3 мг (24.3%) твердого вещества желтого цвета. LC/MS Rt- мин (MH+): 2.54 (763) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00 (с, 9Н), 1.06 (м, 2Н), 1.20 (м, 2Н), 1.29 (с, 9Н), 1.42 (дд, J=9.41, 5.26 Гц, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.24 (м, 2Н), 2.54 (дд, J=13.57, 6.48 Гц, 1Н), 2.92 (м, 1Н), 4.04 (дд, J=12.10, 2.81 Гц, 1Н), 4.20 (д, J=7.34 Гц, 1Н), 4.33 (д, J=12.23 Гц, 1Н), 4.47 (дд, J=10.52, 6.85 Гц, 1Н), 5.10 (дд, J=10.39, 1.59 Гц, 1Н), 5.28 (дд, 7-17.12, 1.22 Гц, 1Н), 5.46 (д, J=5.87 Гц, 1Н), 5.74 (м, 1Н), 6.29 (м, 2Н), 7.40 (с, 1Н), 7.56 (м, 1Н), 8.01 (д, J=5.62 Гц, 1Н).
Пример 213: Получение Соединения 213
К раствору 5,6-метилендиокси-1-хлор-изохинолина (84 мг, 0.41 ммоля) в 4 мл ТГФ под азотом при -78°С прибавляют раствор LDA в циклогексане (1.5 молярный, 0.60 мл, 0.9 ммоля). Светлый коричневатый раствор перемешивают 15 мин при -78°С и прибавляют метилдисульфид (50 мкл чистого реагента, 1.4 эквивалента). ТСХ показывает появление нового пятна помимо неизмененного исходного. Обработка водой с последующей экстракцией этилацетатом дает сырой продукт в виде масла, который очищают препаративной ВЭЖХ, получают 51 мг (49%). LC/MS Rt- мин (МН+): 3.39 (254) [метод С].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 2.64 (с, 3H), 6.29 (с, 2Н), 7.49 (д, J=4.89 Гц, 1Н), 7.71 (с, 1Н), 8.11 (д, J=5.87 Гц, 1Н). Алкилирование 5,6-метилендиокси-7-метилтио-1-хлоризохинолина трипептидом проводят как описано ранее (Пример 184), получают основной продукт, изображенный ниже: BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-метилтио-1,3-диокса-7-азациклопента[α]нафталин-6-илокси)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан

По общей методике получают 59.6 мг (42.2%) твердого вещества желтого цвета. LC/MS Rt- мин (MH+): 2.70 (774) [метод В].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 1.02 (м, 11Н), 1.16 (с, 9Н), 1.32 (с, 2Н), 1.45 (м, 1Н), 1.94 (м, 1Н), 2.12 (д, J=8.56 Гц, 1Н), 2.56 (с, 3H), 2.62 (м, 2Н), 2.90 (д, J=4.40 Гц, 1Н), 4.15 (д, J=7.83 Гц, 2Н), 4.48 (д, J=12.47 Гц, 1Н), 4.62 (т, J=7.83 Гц, 1Н), 5.13 (д, J=10.52 Гц, 1Н), 5.26 (д, J=17.12 Гц, 1Н), 5.74 (д, J=16.38 Гц, 1Н), 5.95 (с, 1Н), 6.28 (с, 2Н), 7.41 (с, 1Н), 7.59 (с, 1Н), 7.85 (д, J=6.11 Гц, 1Н).
Получение 3,4-дизамещенных изохинолин Р2* производных
Пример 215: Получение Соединения 215
BOCNH-P3(L-трет-BuGly)-P2[(1R)-(2, 3-дигидро-1Н-4-азациклопента[α]нафталин-5-илокси)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан, изображенный ниже, получают по следующей схеме:
Общая схема синтеза изохинолиновой компоненты соединения 214

Примечания:
Синтез нового 1-фтор Р2* успешно осуществлен по методикам, описанным в цитируемых ниже ссылках:
(1) Rigby, James H.; Holsworth, Daniel D.; James, Kelly. Vinyl Isocyanates In Synthesis. [4+2] Cycloaddition Reactions With Benzyne Addends. Journal Of Organic Chemistry (1989), 54(17), 4019-20
(2) Uchibori, Y.; Umeno, M.; Yoshiokai, H.; Heterocycles, 1992, 34(8), 1507-1510
Пример 214: Получение Соединения 214, 5-хлор-2,3-дигидро-1Н-4-азациклопента[α]нафталина и Соединения 215 из Примера 215

2,3-Дигидро-1Н-4-азациклопента[α]нафталин-5-ол получают по методу Rigby, описанному в ссылке 1, цитированной выше. С применением POCl3, как описано в другом месте, получают 430 мг (59.8%) Соединения 214. LC/MS Rt- мин (МН+): 2.29 (204) [метод В].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 2.28 (м, 2Н), 3.19 (кв, J=7.74 Гц, 4Н), 7.58 (м, 1Н), 7.71 (м, 2Н), 8.32. (д, J=8.56 Гц, 1Н). Хлорид является достаточно реакционноспособным, чтобы его можно было алкилировать трипептидом по методике в Примере 184, получая нужный продукт, Соединение 215. Однако общий выход можно удвоить, если хлорид заместить на фторид по методу Uchibori, описанному в ссылке 2. Так, выделено 17.0 мг (23.6%) Соединения 215 в виде бледно-желтого твердого вещества. LC/MS Rt- мин (MH+): 2.80 (724) [метод В].1H ЯМР (500 МГц, CD3OD) δ м.д. 1.03 (с, 9Н), 1.09 (м, 2Н), 1.24 (м, 11Н), 1.44 (дд, J=8.24, 5.49 Гц, 1Н), 1.88 (дд, J=7.93, 5.49 Гц, 1Н), 2.25 (м, 4Н), 2.63 (дд, J=13.73, 7.02 Гц, 1Н), 2.94 (м, 1Н), 3.05 (м, 2Н), 3.10 (м, 2Н), 4.08 (дд,J=11.60, 2.75 Гц, 1Н), 4.24 (д, J=20.45 Гц, 1Н), 4.45 (д, J=11.90 Гц, 1Н), 4.54 (дд, J=9.46, 7.63 Гц, 1Н), 5.11 (м, 1Н), 5.30 (д, J=17.09 Гц, 1Н), 5.75 (м, 1Н), 5.87 (с, 1Н), 7.44 (т, J=7.02 Гц, 1Н), 7.69 (м, 2Н), 8.18 (д, J=8.24 Гц, 1Н).
Получение 3,4-дигидрофуранил- и фуранилизохинолин Р2* компонентов, Примеры 217 и 218
Общая схема синтеза

Пример 217: Получение Соединения 217, 5-хлор-2, 3-дигидро-1-окса-4-азациклопента[α]нафталина и Соединения 218, 5-хлор-1-окса-4-азациклопента[α]нафталина
Этот синтез проводят по методикам, описанным, частично, в цитируемых ниже ссылках:
(1) Hojo, Masaru; Masuda, Rioichi; Sakaguchi, Syuhei; Takagawa, Makoto, Synthesis (1986), (12), 1016-17
(2) Rigby, James H.; Holsworth, Daniel D.; James, Kelly. Vinyl Isocyanates In. Synthesis. [4+2] Cycloaddition Reactions With Benzyne Addends. Journal Of Organic Chemistry (1989), 54(17), 4019-20
(3) Uchibori, Y.; Umeno, М.; Yoshiokai, H.; Heterocycles, 1992, 34(8), 1507-1510
Как 2,3-дигидро-1-окса-4-азациклопента[α]нафталин-5-ол, так и 1-окса-4-азациклопента[α]нафталин-5-ол получают совместно по цитированным выше методикам (ссылки 1 и 2). Превращение этой пары в их хлорпроизводные проводят как обычно, с помощью POCl3: К неочищенным гидроксисоединениям (около 2 г, бледно-желтое масло) прибавляют 15 мл POCl3 и смесь кипятят 3 часа. После удаления POCl3 в вакууме остаток перемешивают с EtOAc (1 л) и холодным водным раствором NaOH (220 мл, 1.0 N) в течение 15 мин. Органический слой отделяют, промывают водой (2×200 мл), рассолом (200 мл)и сушат MgSO4, упаривают в вакууме, получают 300 мг (13.2%) Соединения из Примера 217, 5-хлор-2,3-дигидро-1-окса-4-азациклопента[α]нафталина и 100 мг (4.4%) Соединения из Примера 218, 5-хлор-1-окса-4-азациклопента[α]нафталина в виде жидкостей светло-коричневого цвета после очистки хроматографией на силикагеле. Соединение 217: LC/MS Rt- мин (МН+): 2.05 (206) [метод В].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 3.46 (т, J=9.05 Гц, 2Н), 4.82 (т, J=9.17 Гц, 2Н), 7.58 (м,1Н), 7.66 (м, 1Н), 7.85 (д, J=8.31 Гц, 1Н), 8.21 (д, J=8.56 Гц, 1Н). Соединение 218: LC/MS Rt- мин (МН+): 2.16 (204) [метод В].1 H ЯМР (400 МГц, хлороформ-d) δ м.д. 7.15 (д, J=2.20 Гц, 1Н), 7.70 (м, 1Н), 7.89 (м, 2Н), 8.27 (д, J=8.31 Гц,1Н), 8,44 (д, J=8.80 Гц, 1Н).
Получение 5-фтор-2, 3-дигидро-1-окса-4-азациклопента[α]нафталина конечных продуктов Р2* сочетания (конденсации).
Обмен хлор/фтор проводят по цитированному выше методу (ссылка 3). Так, 5-хлор-2, 3-дигидро-1-окса-4-азациклопента [α]нафталин-5-ол (Пример 217) суспендируют в 1.5 мл Bu4PHF2 и облучают в микроволновом реакторе (Реактор Смита) в течение 2 час при 120°С. После обработки водой и очистки на колонке получают 22 мг (26.9%) фторсодержащего продукта. LC/MS Rt- мин (МН+): 1.91 (190) [метод В]. Производное фурана (Пример 218), 5-хлор-1-окса-4-азациклопента [α]нафталин, в достаточной степени реакционноспособен и непосредственно алкилируется трипептидом без активации превращением во фторид.
Пример 219: Получение Соединения 219

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(2, 3-дигидро-1-окса-4-азациклопента[α]нафталин-5-илокси)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан. По общей методике алкилирования получают 22 мг (26.3%) желтого твердого вещества.
LC/MS Rt- мин (МН+): 2.65 (726) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99 (с, 9Н), 1.07 (м, 2Н), 1.20 (м, 11Н), 1.40 (м, 1Н), 1.86 (дд, J=8.07, 5.62 Гц, 1Н), 2.21 (дд, J=17.48, 8.93 Гц, 2Н), 2.60 (дд, J=13.45, 6.85 Гц, 1Н), 2.93 (м, 1Н), 3.34 (м, 2Н), 4.04 (дд, J=11.74, 3.18 Гц, 1Н), 4.24 (с, 1Н), 4.41 (д, J=11.49 Гц, 1Н), 4.51 (м, 1Н), 4.74 (т, J=9.05 Гц, 2Н), 5.11 (д, J=10.27 Гц, 1Н), 5.28 (д, J=17.36 Гц, 1Н), 5.73 (м, 1Н), 5.78 (с, 1Н), 7.43 (м, 1Н), 7.65 (т, J=7.46 Гц, 1Н), 7.74 (д, J=8.31 Гц, 1Н), 8.12 (д, J=8.56 Гц, 1Н).
Пример 220: Получение Соединения 220
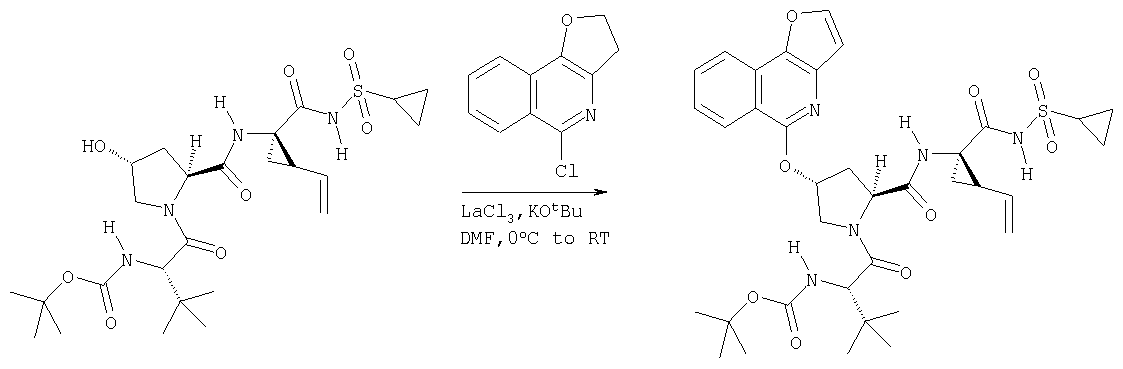
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(1-окса-4-азациклопента[α]нафталин-5-илокси)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропан. По общей методике алкилирования получают 13 мг (20%) желтого твердого вещества. LC/MS Rt- мин (MH+): 2.70 (724) [метод В].1H ЯМР (500 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.09 (м, 2Н), 1.22 (с, 9Н), 1.27 (м, 2Н), 1.46 (м, 1Н), 1.89 (дд, J=7.78, 5.65 Гц, 1Н), 2.24 (д, J=8.55 Гц, 1Н), 2.33 (т, J=9.92 Гц, 1Н), 2.68 (дд, J=13.73, 7.02 Гц, 1Н), 2.95 (м, 1Н), 4.14 (м, 1Н), 4.26 (с, 1Н), 4.50 (д, J=11.90 Гц, 1Н), 4.57 (д, J=17.09 Гц, 1Н), 5.12 (д, J=10.07 Гц, 1Н), 5.30 (д, J=17.40 Гц, 1Н), 5.75 (м, 1Н), 5.93 (с, 1Н). 6.97 (д, J=2.14 Гц, 1Н), 7.51 (т, J=7.32 Гц, 1Н), 7.81 (т, J=7,48 Гц, 1Н), 7.92 (с, 1Н), 8.13 (с, J=7.94 Гц, 1Н), 8.28 (д, J=8.24 Гц, 1Н).
Получение 3-галоген- и 3-гетероарил-4-алкокси- и 4-гидроксиизохинолин Р2* производных
Общая схема синтеза

Условия реакций: (1) МеОК в DMPU; (2) NBS в дихлорэтане; (3) МСРВА в СН2Cl2; (4) POCl3 в дихлорэтане; (5) BBr3 в CH2Cl2; (6) SEM-хлорид и основание Хюнига в СН2Cl2
4-Метоксиизохинолин (Пример 222а) получают из 1-бромизохинолина по новой удобной методике, используя обычное лабораторное оборудование и обычные реагенты. Региоселективное NBS-бромирование дает 3-бром-4-метоксиизохинолин (Пример 222b) с хорошими выходами. Окисление с помощью МСРВА протекает без осложнений и дает соответствующий N-оксид (Пример 222с), который изомеризуется в 1-хлор-3-бром-4-метоксиизохинолин (Пример 222d) по обычной методике с POCl3. 4-Метоксиизохинолин алкилируют трипептидом, получают соответствующее 3-бром-4-метокси-Р2* производное, пригодное для сочетания Стилле и Сузуки. Или же 4-метоксиизохинолин деметилируют в ВВг3 с образованием 4-гидрокси-3-бром-1-хлоризохинолина (Пример 222е). 4-Гидроксигруппу снова защищают с помощью SEM-хлорида, получая 4-SEM защищенный интермедиат, Пример 222. 4-Гидроксисоединение регенерируют по окончании реакции сочетания, снимая защитную группу в присутствии либо кислоты, либо фторида.
Пример 222d: Получение 1-хлор-3-бром-4-метоксиизохинолина
Стадия 1:
К раствору продажного 4-бромизохинолина (15 г, 73 ммоля) в 200 мл диметил-3,4,5, 6-тетрагидро-2(1Н)-пиримидинона (DMPU, Aldrich) прибавляют твердый метоксид калия (5.6 г, 80 ммолей). Реакционный сосуд помещают в масляную баню при 105°С на 20 мин. Цвет смеси после нагревания быстро меняется от начального очень бледного до темного зеленовато-коричневатого. Масляную баню отставляют и к реакционной смеси прибавляют воду, многократно экстрагируют эфиром (порциями). ТСХ показывает два новых пятна (элюент 1:1 об/об смесь гексанов и этилацетата) примерно равного размера. Деление проводят на колонке с силикагелем (Merck, тип-Н), элюируют смесью гексанов с последующим постепенньм прибавлением эфира в подвижную фазу. Нужный продукт, 4-метоксиизохинолин, (4.1 г, 35.3%) выделяют после упаривания растворителей. Выделяют также другой продукт как побочный продукт восстановления изохинолина. Идентичность побочного продукта подтверждена сравнением ЯМР-спектров со спектрами заведомого вещества. Пример 222а: LC/MS Rt- мин (MH+): 1.16 (160) [метод В].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 4.07 (с, 3H), 7.61 (м, 1Н), 7.69 (м, 1Н), 7.93 (д, J=8.07 Гц, 1Н), 8.08 (с, 1Н), 8.19 (д, J=8.56 Гц, 1Н), 8.89 (с, 1Н). [Примечание: это соединение было получено ранее, см. Zoltewicz, John A.; Oestreich, Terence М.; Sale, Alan A, Journal of The American Chemical Society (1975), 97(20), 5889-96 в "Автоклаве из монеля", и позднее на основе фокусированной микроволновой технологии в Cherng, Yie-Jia, Tetrahedron (2002), 58(6), 1125-1129. Настоящая методика не требует ни специальной аппаратуры для работы под высоким давлением, ни микроволнового оборудования для проведения реакции в препаративных масштабах.
Стадия 2:
Вещество (Пример 222а) бромируют с помощью NBS, так, к 4-метоксиизохинолину (Пример 222а, 2.1 г, 13.2 ммоля) в 1,2-дихлорэтане (DCE, 150 мл) при 70°С прибавляют N-бромсукцинимид (NBS, 1.5 г, 8.4 ммоля, 0.6Х), через час при этой температуре прибавляют вторую порцию NBS (1.5 г). Темно-коричневую смесь перемешивают еще 1 час и прибавляют третью порцию NBS (1.0 г). Бромирование контролируют по LC-MS и ведут до исчезновения исходного. Неочищенную реакционную смесь упаривают досуха и нужный продукт отфильтровывают на короткой колонке с силикагелем (Тип-Н, Merck, диаметр 3 см, высота 1.5 см), элюируя сначала неразбавленной смесью гексанов с последующим постепенным увеличением количества добавляемого эфира. Нужный продукт (Пример 222b) выделяют в виде маслообразного вещества (1.7 г, 54%). LC/MS Rt- мин (MH+): 2.65 (238) [метод С].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 4.04 (с, 3H), 7.64 (т, J=7.58 Гц, 1Н), 7.76 (т, J=7.09 Гц, 1Н), 7.99 (д, J=8.31 Гц, 1Н), 8.11 (д, J=8.31 Гц, 1Н), 8.85 (с, 1Н). [3-Бром-4-метоксиизохинолин ранее был получен другим методом, см.: Finkentey, Christel; Langhals, Elke; Langhals, Heinz. Chemische Berichte (1983), 116(6), 2394-7. ЯМР-спектр продукта идентичен описанному в литературе].
Стадия 3:
Продукт бромирования с помощью NBS окисляют МСРВА в хлористом метилене при комнатной температуре. Так, МСРВА (1.80 г, чистота 77%, 8.0 ммолей) прибавляют к раствору 3-бром-4-метоксиизохинолина (Пример 222b, 1.65 г, 6.9 ммоля) в 35 мл СН2Cl2. Раствор перемешивают 4 час, образуется белая суспензия. К смеси прибавляют раствор бикарбоната натрия (5% свежеприготовленный, 20 мл), органические вещества экстрагируют CH2Cl2 (10×25 мл). Многократная экстракция органическим растворителем необходима, чтобы экстрагировать в какой-то степени растворимый в воде N-оксид. Сырой продукт, полученный после упаривания растворителей, очищают далее фильтрованием через силикагель, получают 1.36 г (5.4 ммоля, 78%) N-оксида (Пример 222с) в виде воскообразного вещества. LC/MS Rt- мин (MH+): 1.79 (254) [метод С].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 4.07 (с, 3H), 7.63 (м, 2Н), 7.72 (м, 1Н), 8.00 (м, 1Н), 8.86 (с, 1Н).
Стадия 4:
Конечную перегруппировку N-оксида проводят как обычно в POCl3 по методике, описанной в другом месте. Выход Соединения из Примера 222d практически количественный. LC/MS Rt- мин (МН+): 2.69 (272) [метод D].1H ЯМР в виде HCl соли (400 МГц, хлороформ-d) δ м.д. 4.07 (с, 3H), 7.81 (м, 1Н), 7.92 (м, 1Н), 8.17 (д, J=8.31 Гц, 1Н), 8.34 (д, J=8.31 Гц, 1Н).1H ЯМР в виде свободного основания (400 МГц, хлороформ-d) δ м.д. 4.03 (с, 3H), 7.72 (м, 1Н), 7.81 (м, 1Н), 8.12 (д, J=8.56 Гц, 1Н), 8.28 (д, J=8.56,1Н).
Пример 223: Получение Соединения 223
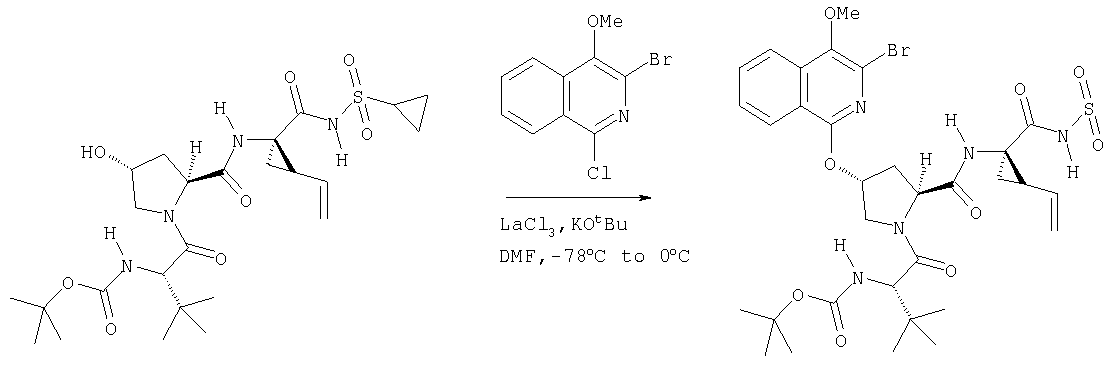
Свободное основание (Пример 222d), полученное на предыдущей стадии, алкилируют фрагментом трипептида по методике алкилирования (Пример 184), описанной в другом месте, получают 79% ожидаемого продукта в виде белого, как бумага, твердого вещества. LC/MS Rt- мин (MNa+): 3.91 (814) [метод С].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (с, 9Н), 1.06 (дд, J=8.07, 1.47 Гц, 2Н), 1.22 (м, 11Н), 1.42 (дд, J=9.78, 5.14 Гц, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.22 (дд, J=18.10, 9.29 Гц, 1Н), 2.28 (м, 1Н), 2.61 (дд, J=13.57, 6.97 Гц, 1Н), 2.93 (м, 1Н), 3.92 (с, 3H), 4.06 (дд, J=11.86, 2.81 Гц, 1Н), 4.22 (с, 1Н), 4.43 (д, J=11.49 Гц, 1Н), 4.51 (м, 1Н), 5.10 (д, J=10.52 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.74 (м, 1Н), 5.81 (с, 1Н), 7.56 (т, J=7.58 Гц, 1Н), 7.78 (т, J=7.58 Гц, 1Н), 8.00 (д, J=8.31 Гц, 1Н), 8.16 (д, J=8.56 Гц, 1Н).
Примеры 224 и 225: Получение Соединений 224 и 225
4-Метоксигруппу в 1-хлор-3-бром-4-метоксихинолине (Пример 222d), описанном ранее, превращают в α-триметилсилилэтоксиметильную (SEM) группу по следующей методике. 1-Хлор-3-бром-4-метоксихинолин (Пример 222d) деметилируют под действием BBr3 (0.2-0.3 молярная концентрация-конечная концентрация BBr3 в реакционной смеси) при комнатной температуре в течение 12 час. Найдено, что высокая концентрация ВВr3 необходима и эффективна при проведении такого деметилирования. К неочищенной реакционной смеси прибавляют 50 объемов сухого метанола и упаривают досуха. Деметилирование проходит практически количественно. Пример 222е: LC/MS Rt- мин (МН+): 2.32 (258) [метод D].1H ЯМР в виде HCl соли (400 МГц, хлороформ-d) δ м.д. 5.83 (уш. с, 1Н), 7.73 (т. J=7.70 Гц, 1Н), 7.79 (т, J=7.58 Гц, 1Н), 8.22 (м, 2Н). 4-Гидрокси-3-бром-1-хлоризохинолин (Пример 222е) снова защищают с помощью 2-(триметилсилил)этоксиметилхлорида (SEM-Cl). Неочищенное свободное основание предыдущего препарата сушат до 40 микрон (Hg) при комнатной температуре и снова защищают с помощью SEM-хлорида. К раствору 4-гидроксисоединения (Пример 222е, 1.33 г, 5.2 ммоля) в хлористом метилене (50 мл) при 0°С последовательно прибавляют диизопропилэтиламин (2 мл, 11.5 ммоля) и SEM-хлорид (1.8 мл, 10 ммолей). Смесь перемешивают 10 мин и промывают свежеприготовленным раствором NaHCO3 (5%, 100 мл). Органические вещества экстрагируют несколькими порциями хлористого метилена, объединенные органические вытяжки снова промывают 20 мл деионизированной воды и упаривают в вакууме. Введение защитной SEM-группы осуществляется практически количественно.
Пример 222: LC/MS Rt- мин (МН+): 3.40 (410) [метод D].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 0.03 (с, 9Н), 0.99 (м, 2Н), 3.98 (м, 2Н), 5.33 (с, 2Н), 7.72 (м, 1Н), 7.80 (м, 1Н), 8.17 (д, J=8.56 Гц, 1Н), 8.27 (д, J=8.07 Гц, 1Н). Алкилирование 4-SEM-защищенного изохинолина трипептидом: Соединения 224 и 225 получают по той же самой реакции алкилирования. Наиболее вероятно, что 4-гидроксисоединение (Соединение 224) получают в результате очистки с помощью препаративной ВЭЖХ вследствие присутствия ТФК.

Пример 224 (15.4%): LC/MS Rt- мин (MNa+): 2.87 (800) [метод D].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (с, 9Н), 1.06 (д, J=8.31 Гц, 2Н), 1.25 (с, 9Н), 1.42 (с, 2Н), 1.86 (м, 1Н), 2.23 (м, 2Н), 2.60 (дд, J=13.21, 7.34 Гц, 1Н), 2.93 (м, 1Н), 4.06 (д, J=11.00 Гц, 1Н), 4.24 (м, 2Н), 4.38 (д, J=11.98 Гц, 1Н), 4.49 (дд, J=9.78, 7.09 Гц, 1Н), 5.10 (д, J=10.03 Гц, 1Н), 5.28 (д, J=17.36 Гц, 1Н), 5.72 (м, 1Н), 5.76 (с, 1Н), 7.52 (т, J=7.46 Гц, 1Н), 7.71 (т, J=7.09 Гц, 1Н), 8.09 (д, J=4.40 Гц, 1Н), 8.11 (д, J=4.1-6 Гц, 1Н).
Пример 225 (8.0%): LC/MS Rt- мин (MNa+): 3.46 (808) [метод D].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.01 (с, 9Н), 0.96 (м, 2Н), 1.02 (с, 11Н), 1.06 (д, J=6.60 Гц. 2Н), 1.24 (с, 9Н), 1.42 (м, 1Н), 1.86 (дд, J=7.83, 5.38 Гц, 1Н), 2.25 (м, 2Н), 2.62 (дд, J=13.69, 7.34 Гц, 1Н), 2.93 (м, 1Н), 3.97 (м, 2Н), 4.07 (дд, J=10.88, 3.55 Гц, 1Н), 4.23 (с, 1Н), 4.43 (д, J=11.25 Гц, 1Н), 4.50 (м, 1Н), 5.10 (д, J=10.76 Гц, 1Н), 5.25 (м, 3H), 5.74 (м, 1Н), 5.82 (с, 1Н), 7.57 (м, 1Н), 7.77 (т, J=7.83 Гц, 1Н), 8.06 (д, J=8.56 Гц, 1Н), 8.16 (д, J=8.31 Гц, 1H).
Получение 4Н-[1,3]диоксино[5,4]изохинолин Р2* производных
Общая схема синтеза

Условия реакций: (1) МеОК в DMPU; (2) МСРВА в СН2Cl2; (3) POCl3 в DCE; (4) BBr3 в CH2Cl2; (6) раствор НСНО в 40% H2SO4 по методу, описанному в статьях "Синтез 1,3-оксазино[5,6-с]изохинолинов и родственных соединений" (Miyoko Toyama and Hirotaka Otomasu, Chem. Pharm. Bull. 33(12), 5543-5546, 1985; (6) Фторирование по Uchibori, Y.; Umeno, M.; Yoshiokai, H.; Heterocycles, 1992, 34 (8), 1507-1510
Пример 227: Получение Соединения 227

6-Хлор-1,3-оксазино[5, 6-с]изохинолин получают по методике Miyoko Toyama и Hirotaka Otomasu с 1-хлор-4-гидроксихинолином в качестве исходного. Исходное, 1-хлор-4-гидроксиизохинолин (Пример 226с), получают по вышеприведенной схеме. Окисление 1-хлор-4-гидроксиизохинолина с помощью МСРВА (Пример 222а) проводят как обычно, выход соответствующего N-оксида 79.1% (Пример 226а). Это соединение сразу же превращают в 1-хлорпроизводное реакцией с POCl3, получают хлорид (Пример 226b) практически с количественным выходом. Неочищенный 1-хлор-4-гидроксихинолин деметилируют с помощью ВВr3 при комнатной температуре; получают соответствующий 1-хлор-4-гидроксиизохинолин (Пример 226с) в результате обработки сырой реакционной смеси после реакции с BBr3 сухим метанолом при комнатной температуре с последующим упариванием, чтобы избавиться от избытка остаточных боратов. Реакцией Miyoko Toyama и Hirotaka Otomasu из 300 мг 4-метоксиизохинолина в 4 стадии получают 266 мг 6-хлор-1, 3-оксазино[5,6-с]изохинолина (Пример 226d, общий выход 62.3%). LC/MS Rt- мин ([М-HCHO]H+): 2.45 (192) [метод D].1H ЯМР (400 МГц, хлороформ-d) δ м.д. 5.02 (с, 2Н), 5.41 (с, 2Н), 7.68 (м, 1H), 7.77 (ддд, J=8.25, 6.91, 1.22 Гц, 1Н), 8.10 (д, J=8.31 Гц, 1Н), 8.26 (д, J=8.56 Гц, 1Н).
Оказалось, что в реакции алкилирования по методике из Примера 184 хлорид является неактивным. Соответствующий 6-фтор-1,3-оксазино[5,6-с]изохинолин (Пример 226) получают по цитированной ранее методике [Uchibori, Y.; Umeno, М.; Yoshiokai, H.; Heterocycles, 1992, 34(8), 1507-1510]. Реакцию не доводят до конца и получают сырую реакционную смесь с соотношением 1:2.4 (Cl:F). Без дополнительной очистки смесь хлорид/фторид алкилируют по методике из Примера 184, после очистки препаративной ВЭЖХ получают 66 мг (50.0%) BOCNH-P3(L-трет-BuGly)-P2[(4R)-(1,3-оксазино[5,6-с]изохинолин-6-оксо)-S-пролин]-P1(LR,2S винил Асса)-CONHSO2-циклопропана. LC/MS Rt- мин (MNa+): 3.03 (764) [метод D].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.06 (дд, J=8.07, 1.96 Гц, 2Н), 1.22 (с, 10Н), 1.34 (д, J=6.11 Гц, 1Н), 1.42 (м, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.23 (м, 2Н), 2.59 (дд, J=13.82, 6.97 Гц, 1Н), 2.93 (м, 1Н), 4.03 (дд, J=11.86, 3.06 Гц, 1Н), 4.23 (с, 1Н), 4.41 (д, J=11.98 Гц, 1Н), 4.50 (дд, J=9.66, 6.97 Гц, 1Н), 4.87 (м, 2Н), 5.11 (д, J=10.52 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.34 (с, 2Н), 5.74 (м, 2Н), 7.51 (т, J=7.46 Гц, 1Н), 7.70 (т, J=7.58 Гц, 1Н), 7.95 (д, J=8.31 Гц, 1Н), 8.12 (д, J=8.31 Гц, 1Н).
Получение 4-метокси-3-гетероарил-и 3-азоилизохинолин Р2* производных по реакциям сочетания Сузуки и Стилле
Приведенные ниже методики сочетания оказались (обще)применимыми для бромпроизводного из Примера 223. Ясно, что аналогичная методика равно применима к иным комбинациям конденсирующих агентов (реагентов для сочетания) и катализаторов, нежели бор и олово.
Пример 229: Получение Соединения 229
Синтез BOCNH-P3(L-трет-BuGly)-P2[(4R)-(1-фуран-3-ил-4-метоксиизохинолин-1-оксо)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропана по реакции Сузуки показан ниже:
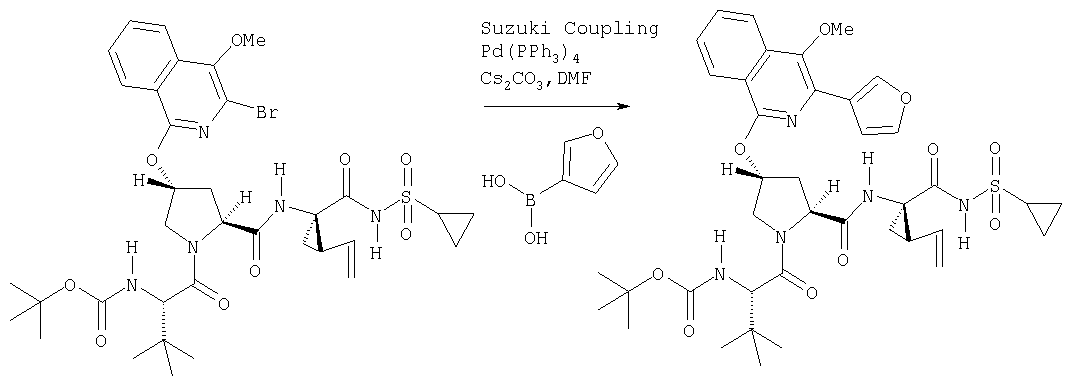
22 мг (0.028 ммоля) соединения из Примера 223 растворяют в 1 мл ДМФА, прибавляют 9.4 мг продажной бороновой кислоты (3 экв), 3 мг катализатора (10 ммольн.%) и 18 мг карбоната цезия. Смесь дважды откачивают под вакуумом, а затем нагревают при 110°С в течение 3 час. Полученный продукт очищают препаративной ВЭЖХ, получают 13.6 мг (64%) желтого твердого вещества. LC/MS Rt- мин (МН+): 2.85 (780) [метод В].1H ЯМР (500 МГц, CD3OD) δ м.д. 1.09 (м, 11Н), 1.26 (м, 12Н), 1.68 (м, 1Н), 2.27 (с, 1Н), 2.64 (м, 2Н), 2.97 (м, 1Н), 3.86 (с, 3H), 4.15 (д, J=10.38 Гц, 1Н), 4.28 (с, 1Н), 4.43 (д, J=10.99 Гц, 1Н), 4.56 (м, 1Н), 5.11 (м, 2Н), 5.63 (м, 1Н), 5.99 (с, 1Н), 7.20 (с, 1Н), 7.51 (м, 1Н), 7.61 (м, 1Н), 7.75 (т, J=7.17 Гц, 1Н), 8.03 (д, J=8.24 Гц, 1Н), 8.16 (д, J=8.24 Гц, 1Н), 8.27 (с, 1Н).
Пример 230: Получение Соединения 230
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(3-фуран-2-ил-4-метоксиизохинолин-1-оксо)-S-пролин]-P1(LR,2S винил Асса)-CONHSO2-циклопропан получают по реакции Стилле, изображенной ниже:
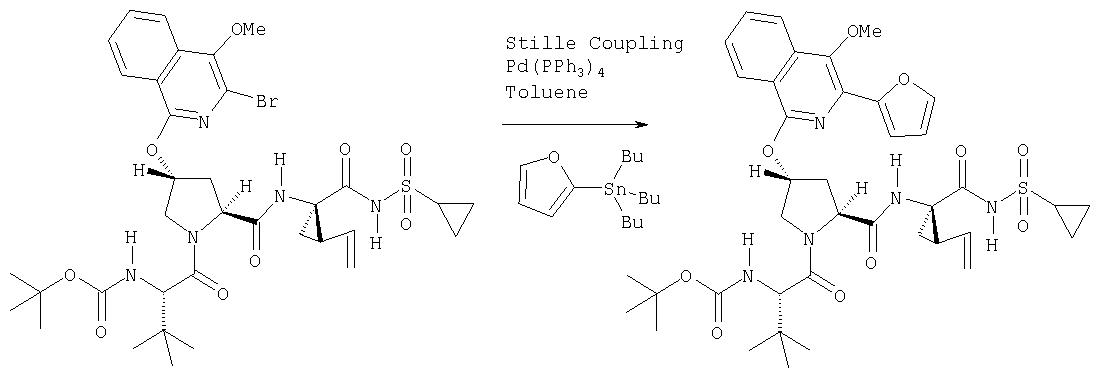
40 мг (0.5 ммоля) соединения из Примера 223, 4 мг катализатора (5 мольн.%) и 100 мкл (4 экв) продажного оловосодержащего реагента растворяют в 1 мл толуола, смесь дважды откачивают, а затем греют при 90°С в течение ночи. После разделения методом препаративной ВЭЖХ получают 19.6 мг (50.0%) твердого вещества зеленоватого цвета. LC/MS Rt- мин (МН+): 2.76 (780) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.94 (м, 2Н), 0.98 (с, 9Н), 1.09(м,2Н), 1.25 (с, 9Н), 1.39 (м, 1Н), 1.60 (м, 1Н), 2.35 (м, 1Н). 2.48 (м, 1Н), 2.74 (м, 1Н), 2.95 (м, 1Н), 3.87 (с, 3H), 4.14 (м, 1Н), 4.22 (д, J=4.16 Гц, 1Н), 4.41 (с, 1Н), 4.69 (м, 1Н), 5.26 (м, 1Н), 5.35 (м, 1Н), 5.93 (с, 1Н), 6.03 (м, 1Н), 6.61 (м, 1Н), 7.16 (д, J=3.18 Гц, 1Н), 7.50 (д, J=7.58 Гц, 1Н), 7.67 (с, 1Н), 7.73 (т, J=7.34 Гц, 1Н), 8.04 (м, 1Н), 8.17 (д, J=8.31 Гц, 1Н).
Пример 231: Получение Соединения 231
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(3-пиразин-2-ил-4-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают по реакции Стилле с выходом 7.1%. LC/MS Rt- мин (MH+): 2.51 (792) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.98 (м, 9Н), 1.11 (м, 2Н), 1.19 (с, 9Н), 1.27 (м, 2Н), 1.42 (м, 1Н), 2.37 (м, 1Н), 2.48 (м, 2Н), 2.81 (м, 1Н), 2.97 (м, 1Н), 3.83 (с, 3H), 4.07 (с, 1Н), 4.20 (д, J=4.16 Гц, 1Н), 4.54 (д, J=11.49 Гц, 1Н), 4.72 (м, 1Н), 5.27 (м, 1Н), 5.39 (м, 1Н), 5.96 (с, 1Н), 6.04 (м, 1Н), 7.63 (с, 1Н), 7.83 (с, 1Н), 8.17 (с, 1Н), 8.26 (с, 1Н), 8.60 (д, J=2.20 Гц, 1Н), 8.76 (д, J=2.20 Гц, 1Н), 9.33 (с, 1Н).
Пример 232: Получение Соединения 232

BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-метокси-3-тиазол-2-илизохинолин-1-оксо)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2 -циклопропан получают аналогично по реакции сочетания Стилле с выходом 32.2%. LC/MS Rt- мин (МН+): 2.42 (797) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.03 (с, 9Н), 1.07 (м, 2Н), 1.13 (с, 9Н), 1.22 (м, 2Н). 1.43 (дд, J=9.78, 5.14 Гц, 1Н), 1.88 (дд, J=8.07, 5.38 Гц, 1Н), 2.23 (кв, J=8.97 Гц, 1Н), 2.36 (м, 1Н), 2.67 (м, 1Н), 2.94 (м, 1Н), 4.10 (с, 3H), 4.15 (м, 1Н), 4.18 (с, 1Н), 4.53 (д, J=25.92 Гц, 1Н), 4.59 (дд, J=10.27, 7.09 Гц, 1Н), 5.12 (м, 1Н), 5.29 (д, J=17.36 Гц, 1Н), 5.73 (м, 1Н), 6.09 (с, 1Н), 7.74 (т, J=7.58 Гц, 1Н), 7.91 (т, J=7.70 Гц, 1Н), 8.00 (д, J=3.42 Гц, 1Н), 8.18 (д, J=3.18 Гц, 1Н), 8.22 (д, J=8.31 Гц, 1Н), 8.29 (д, J=8.31 Гц, 1Н).
Пример 233: Получение Соединения 233

По обычной методике алкилирования трипептидом продажного 4-хлорфуро[3,2-с]пиридина получают 5.7 мг (8.1%) твердого вещества желтого цвета (8.2%). LC/MS Rt- мин (МН+): 2.32 (674) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.07 (м, 2Н), 1.21 (м, 11Н), 1.41 (м, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.22 (дд, J=17.61, 9.05 Гц, 2Н), 2.54 (дд, J=13.69, 7.09 Гц, 1Н), 2.92 (м, 1Н), 4.06 (м, 1Н), 4.21 (м, 1Н), 4.32 (с, 1Н), 4.49 (м, 1Н), 5.11 (дд, J=10.27, 1.47 Гц, 1Н), 5.29 (дд, J=17.36, 1.22 Гц, 1Н), 5.74 (м, 1Н), 5.81 (с, 1Н), 6.83 (д, J=1.22 Гц, 1Н), 7.19 (д, J=5.87 Гц, 1Н), 7.76 (д, J=1.22 Гц, 1Н), 7.97 (д, J=5.87 Гц, 1Н).
Пример 235: Получение Соединения 235

По обычной методике алкилирования трипептидом продажного 4-хлортиено[3,2-с]пиридина получают 20.0 мг (28.1%) твердого вещества желтого цвета. LC/MS Rt- мин (MH+): 2.50 (690) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.06 (м, 2Н), 1.21 (м, 11Н), 1.42 (м, 1Н), 1.86 (дд, J=8.19, 5.50 Гц, 1Н), 2.24 (м, 2Н), 2.57 (дд, J=13.69, 6.85 Гц, 1Н), 2.93 (м, 1Н), 4.05 (дд, J=11.98, 3.18 Гц, 1Н), 4.22 (с, 1Н), 4.39 (д, J=11.74 Гц, 1Н), 4.50 (дд, J=9.90, 7.21 Гц, 1Н), 5.10 (дд, J=10.39, 1.34 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.73 (м, 1Н), 5.81 (с, 1Н), 7.45 (д, J=5.62 Гц, 1Н), 7.53 (м, 2Н), 7.94 (д, J=5.87 Гц, 1Н).
Пример 236: Получение Соединения 236

По обычной методике алкилирования трипептидом продажного 3,5-дихлор-1,2,4-тиадиазола получают 8.0 мг (11.9%) твердого вещества желтого цвета. LC/MS Rt- мин (MNa+ ): 2.37 (697) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00 (с, 9Н), 1.06 (м, 2Н), 1.22 (м, 2Н), 1.36 (с, 9Н), 1.42 (м, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.25 (м, 2Н), 2.60 (дд, J=14.18, 6.85 Гц, 1Н), 2.92 (м, 1Н), 4.03 (дд, J=12.47, 3.18 Гц, 1Н), 4.17 (с, 1Н), 4.42 (м, 2Н), 5.11 (дд, J=10.27, 1.71 Гц, 1Н), 5.29 (дд, J=17.12, 1.47 Гц, 1Н), 5.68 (с, 1Н), 5.74 (м, 1Н).
Пример 237: Получение Соединения 237
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(хиноксалин-2-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан, изображенный ниже,

получают (113.0 мг, 19.2%) по общей методике алкилирования трипептидом продажного 2-хлорхиноксалина. LC/MS Rt- мин (MNa+): 2.48 (707) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.06 (м, 2Н), 1.22 (м, 11Н), 1.42 (м, 1Н), 1.87 (дд, J=8.19, 5.50 Гц, 1Н), 2.24 (м, 1Н), 2.31 (м, 1Н), 2.57 (дд, J=13.57, 6.97 Гц, 1Н), 2.93 (м,1Н), 4.09 (дд, J=11.98, 3.18 Гц, 1Н), 4.17 (с, 1Н), 4.38 (д, J=11.74 Гц, 1Н), 4.50 (дд, J=10.27, 7.09 Гц, 1Н), 5.11 (дд, J=10.27, 1.71 Гц, 1Н), 5.29 (дд, J=17.12, 1.47 Гц, 1Н), 5.74 (м, 1Н), 5.87 (с, 1Н), 7.62 (т, J=7.46 Гц, 1Н), 7.73 (т, J=7.70 Гц, 1Н), 7.87 (м, 1Н), 7.96 (д, J=8.31 Гц, 1Н), 8.42 (с, 1Н).
Пример 238: Получение Соединения 238
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(2-трифор-6-фторхинолин-4-оксо)-S-пролин]-Р1(1R,2S винил Асса)-CONHSO2-циклопропан, изображенный ниже,
получают (17.0 мг, 23.2%) по общей методике алкилирования трипептидом продажного 2-трифторметил-4-хлор-6-фтор-хинолина. LC/MS Rt- мин (MNa+): 2.66 (792) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (с, 9Н), 1.06 (м, 2Н), 1.17 (с, 9Н), 1.23 (м, 2Н), 1.42 (м, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.21 (кв, J=8.64 Гц, 1Н), 2.32 (м, 1Н), 2.63 (дд, J=13.94, 6.85 Гц, 1Н), 2.93 (м, 1Н), 4.08 (м, 1Н), 4.18 (с, 1Н), 4.53 (м, 2Н), 5.10 (м, 1Н), 5.27 (д, J=17.12 Гц, 1Н), 5.58 (с, 1Н), 5.72 (м, 1Н), 7.39 (с, 1Н), 7.65 (м, 1Н), 7.83 (дд, J=9.29, 2.69 Гц, 1Н), 8.12 (дд, J=9.29, 5.14 Гц, 1Н).
Пример 239: Получение Соединения 239
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-фторхинолин-4-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан, изображенный ниже,

получают (26.0 мг, 39.0%) в виде желтого твердого вещества по общей методике алкилирования трипептидом с продажным 4-хлор-6-фторхинолином в качестве исходного. LC/MS Rt- мин (MNa+): 1.98 (702) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.04 (м, 11Н), 1.14 (s, 9Н) 1.23 (m, 2H), 1.42 (м, 1Н) 1.87 (дд, J=8.07, 5.38 Гц, 1Н), 2.23 (кв, J=8.80 Гц, 1Н), 2.41 (м, 1Н), 2.75 (дд. J=14.43, 6.85 Гц, 1Н), 2.93 (м, 1Н), 4.09 (с, 1Н), 4.12 (д, J=2.69 Гц, 1Н), 4.61 (м, 2Н), 5.11 (дд, J=10.39. 1.59 Гц, 1Н), 5.28 (дд, J=17.24, 1.34 Гц, 1Н), 5.70 (м, 1Н), 5.75 (с, 1Н), 7.62 (д, J=6.60 Гц, 1Н), 7.93 (м, 1Н), 8.06 (дд, J=8.68, 2.57 Гц, 1Н), 8.20 (дд, J=9.29, 4.40 Гц, 1Н), 9.06 (д, J=6.60 Гц, 1Н). Препаративной ВЭЖХ выделяют также небольшое количество побочного продукта, получающееся в этой реакции при замещении F.
Пример 240: Выделение Соединения 240
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-хлорхинолин-6-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2 -циклопропан, изображенный ниже,
получают (8.0 мг, 11.7%) как побочный продукт в виде твердого вещества желтого цвета. LC/MS Rt- мин (MNa+): 2.240 (740) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00 (с, 9Н), 1.06 (м, 2Н), 1.23 (м, 11Н), 1.42 (м, 1Н), 1.86 (дд, J=8.19, 5.50 Гц, 1Н), 2.22 (м, 1Н), 2.29 (м, 1Н), 2.55 (м, 1Н), 2.92 (м, 1Н), 4.09 (м, 1Н), 4.21 (с, 1Н), 4.30 (м, 1Н), 4.46 (дд, J=10.27, 6.85 Гц, 1Н), 5.11 (дд, J=10.27, 1.47 Гц, 1Н), 5.28 (дд, J=17.36, 1.47 Гц, 1Н), 5.43 (с, 1Н), 5.74 (м, 1Н), 7.60 (дд, J=9.29, 2.45 Гц, 1Н), 7.64 (д, J=2.45 Гц, 1Н), 7.81 (д, J=4.89 Гц, 1Н), 8.06 (д, J=9.29 Гц, 1Н), 8.72 (д, J=5.14 Гц, 1Н).
Пример 241: Получение Соединения 241
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(8-фторхинолин-4-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2 -циклопропан, изображенный ниже,

получают в виде твердого вещества желтого цвета (10.3 мг, 14.7%) по общей методике алкилирования трипептидом с продажным 4-хлор-8-фторхинолином в качестве исходного. LC/MS Rt- мин (MNa+): 1.95 (702) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.98 (с, 9Н), 1.06 (м, 2Н), 1.14(с, 9Н), 1.22 (м, 2Н), 1.42 (м, 1Н), 1.87 (дд, J=8.07, 5.62 Гц, 1Н), 2.22 (кв, J=8.72 Гц, 1Н), 2.41 (м, 1Н), 2.74 (дд, J=14.06, 6.97 Гц, 1Н), 2.93 (м, 1Н), 4.11 (м, 2Н), 4.57 (дд, J=10.39, 6.97 Гц, 1Н), 4.66 (д, J=12.23 Гц, 1Н), 5.11 (дд, J=10.27, 1.22 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.71 (м, 2Н), 7.59 (д, J=6.36 Гц, 1Н), 7.75 (м, 1Н), 7.86 (м, 1Н), 8.23 (д, J=8.56 Гц, 1Н), 9.02 (д, J=6.36 Гц, 1Н). Препаративной ВЭЖХ выделяют также побочный продукт. Производное 4-хлор -8-оксохинолина получается в результате замещения F вместо хлора в качестве уходящей группы.
Пример 242: Выделение Соединения 242
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(4-хлорхинолин-8-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан, изображенный ниже,

получают (9.0 мг, 13.2%) как побочный продукт в виде твердого вещества желтого цвета. LC/MS Rt- мин (МН+): 2.37 (718) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00 (с, 9Н), 1.06 (м, 2Н), 1.12 (с, 9Н), 1.23 (м, 2Н), 1.43 (дд, J=9.41, 5.50 Гц, 1Н), 1.87 (дд, J=8.19, 5.50 Гц, 1Н), 2.25 (м, 1Н), 2.35 (м, 1Н), 2.67 (дд, J=13.94, 7.09 Гц, 1H), 2.93 (м, 1Н), 4.10 (м, 1Н), 4.13 (с, 1Н), 4.43 (д, J=11.98 Гц, 1Н), 4.65 (дд, J=10.03, 7.09 Гц, 1Н), 5.12 (дд, J=10.27, 1.47 Гц, 1Н), 5.30 (дд, J=17.12, 1.22 Гц, 1Н), 5.51 (с, 1Н), 5.75 (м, 1Н), 7.61 (д, J=7.83 Гц, 1Н), 7.88 (т, J=8.19 Гц, 1Н), 8.04 (м, 2Н), 8.91 (д, J=5.38 Гц, 1Н).
Пример 243: Получение Соединения 243
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(3-гидроксихиноксалин-2-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан, изображенный ниже,
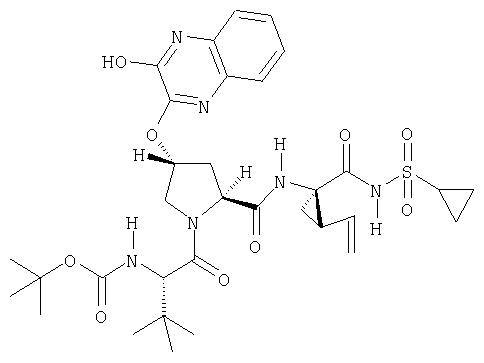
бледно-желтое твердое вещество, получают (8.0 мг, 11.4%) после спонтанного гидролиза продукта моноалкилирования, полученного по общей методике алкилирования трипептидом продажного 2,3-дихлор-хиноксалина. LC/MS Rt- мин (MNa+): 2.42 (723) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99 (с, 9Н), 1.05 (м, 2Н), 1.24 (с, 9Н), 1.40 (м, 3H), 1.86 (м, 1Н), 2.24 (м, 2Н), 2.53 (м, 1H), 2.92 (м, 1Н), 4.06 (м, 1Н), 4.16 (с, 1Н), 4.40 (м, 1Н), 4.55 (дд, J=10.39, 6.97 Гц, 1Н), 5.11 (м, 1Н), 5.29 (м, 1Н), 5.73 (м, 1Н), 5.78 (с, 1Н), 7.15 (с, 1Н), 7.26 (м, 2Н), 7.36 (т, J=7.83 Гц, 1Н), 7.61(д, J=8.07 Гц, 1Н).
Пример 244: Получение Соединения 244
BOCNH-P3(L-трет-BuGly)-P2[(4R)-(6-карбоновой кислоты диметиламидизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан получают, используя комбинацию схемы "Pd0 сочетания" и постадийную процедуру с 6-бром-1-хлоризохинолином в качестве исходного.

LC/MS Rt- мин (MNa+): 2.34 (777) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 0.98 (м, 11Н), 1.23 (м, 11Н), 1.35 (м, 1Н), 1.91 (м, 1Н), 2.29 (м, 2Н), 2.47 (м, 1Н), 2.58 (м, 1Н), 2.97 (с, 3H), 3.11 (с, 3H), 4.09 (м, 1Н), 4.24 (с, 1Н), 4.44 (м, 1Н), 4.61 (м, 1Н), 5.16 (м, 2Н), 5.57 (м, 1Н), 5.90 (с, 1Н), 7.38 (д, J=5.87 Гц, 1Н), 7.50 (д, J=8.07 Гц, 1Н), 7.86 (с, 1Н), 8.03 (д, J=5.87 Гц, 1Н), 8.27 (д, J=8.56 Гц, 1Н),
Пример 245: Получение Соединения 245
При проведении катализируемой Pd0 реакции сочетания Стилле (Пример 230) в качестве минорного выделяют побочный продукт, который идентифицируют как BOCNH-P3(L-трет-BuGly)-P2[(4R)-(3-хлор-4-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S винил Асса)-CONHSO2-циклопропан, изображенный ниже

LC/MS Rt- мин (MNa+): 2.62 (770) [метод В].1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.08 (м, 2Н), 1.18 (с, 9Н), 1.27 (м, 2Н), 1.37 (м, 1H), 1.62 (м, 1Н), 2.36 (м, 2Н), 2.73 (м, 1Н), 2.97 (м, 1Н), 3.92 (с, 3H), 4.02 (м, 1Н), 4.18 (с, 1Н), 4.48 (м, 1Н), 4.66 (м, 1Н), 5.30 (м, 2Н), 5.78 (с, 1Н), 6.04 (м, 1Н), 7.53 (т, J=7.70 Гц. 1Н), 7.77 (т, J=7.58 Гц, 1Н), 8.00 (д, J=8.56 Гц, 1Н), 8.19 (д, J=8.07 Гц, 1Н).
Подраздел F
Пример 250: Получение Соединения 250
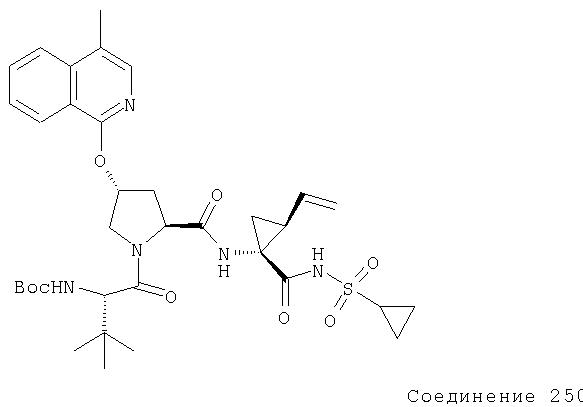
Схема 1
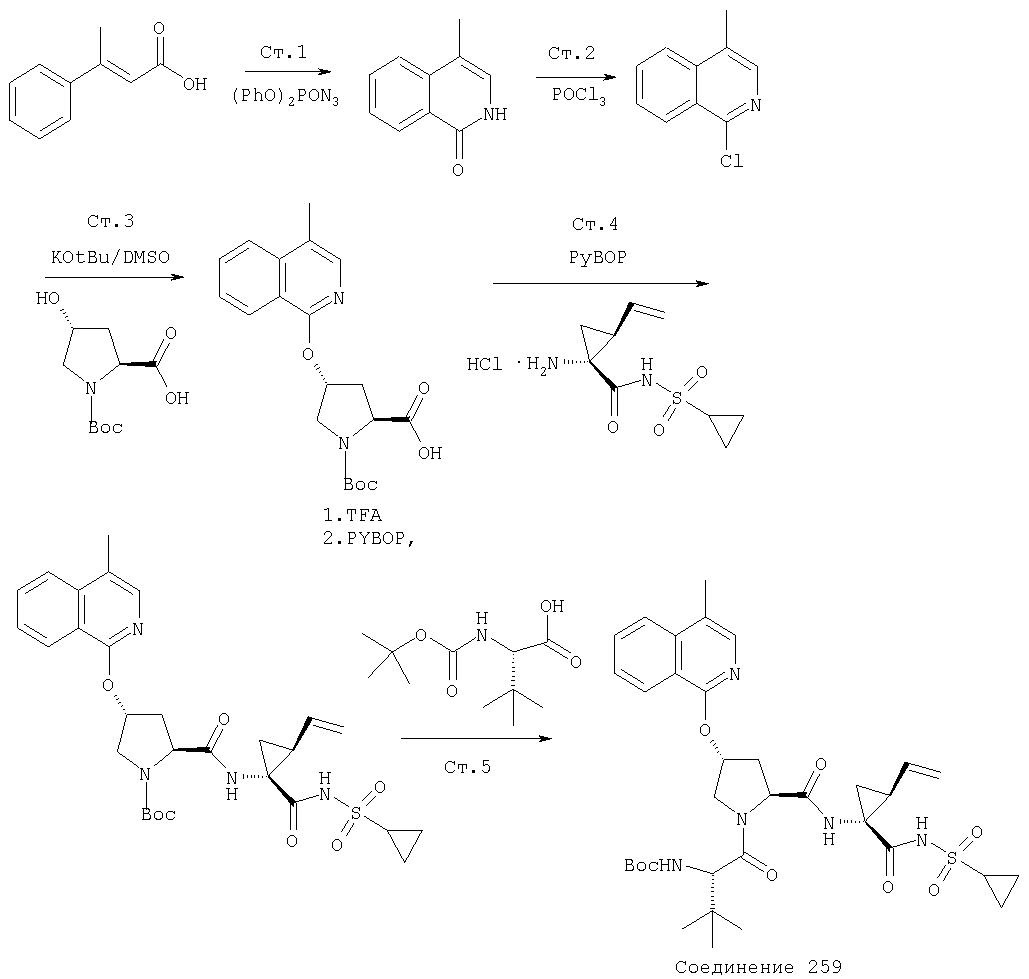
Стадия 1:
Раствор 3-фенилбут-2-еновой кислоты (16.2 г), дифенилфософорилазида (27.5 г) и триэтиламина (10.1 г) в бензоле (100 мл) перемешивают 1 час. Фильтруют через слой силикагеля, промывают бензолом и упаривают, остаток растворяют в дифенилметане (80 мл) и кипятят 3 час. Охлаждают до rt, осадок отфильтровывают, промывают бензолом и сушат, получают 10 г (63%) нужного продукта в виде твердого вещества.1H ЯМР (400 МГц, CD3OD) δ м.д. 2.30 (с, 3H), 7.00 (с, 1Н), 7.54 (м, 1Н), 7.77 (м, 2Н), 8.33 (д, J=7.34 Гц, 1Н).
Стадия 2:
Раствор 4-метил-2Н-изохинолин-1-она (4.8 г) в POCl3 (50 мл) кипятят 3 час. После охлаждения и упаривания остаток подщелачивают, добавляя 5 N NaOH, и экстрагируют СН2Cl2. Органические вытяжки промывают рассолом и сушат MgSO4. После упаривания, очистки флеш-хроматографией на Biotage с 5% раствором этилацетата в смеси гексанов в качестве элюента получают 4.8 г (90%) нужного продукта в виде твердого вещества.1H ЯМР (400 МГц, CDCl3) δ м.д. 2.59 (с, 3H), 7.68 (т, J=7.70 Гц, 1Н), 7.78 (м, 1Н), 7.94 (д, J=8.31 Гц, 1Н), 8.11 (с, 1Н), 8.35 (д, J=8.31 Гц, 1Н).
Стадия 3:
Раствор Boc-Hyp-ОН (231 мг) и трет-ВиОК (336 мг) в ДМСО (10 мл) перемешивают 0.5 час. К раствору прибавляют 1-хлор-4-метилизохинолин (178 мг) и полученную смесь перемешивают 1 день. К реакционной смеси прибавляют 5% лимонную кислоту и экстрагируют этилацетатом. Органические вытяжки промывают рассолом и сушат MgSO4. После упаривания получают 350 мг (94%) нужного продукта, который используют на следующей стадии без дополнительной очистки.1H ЯМР (400 МГц, CD3OD) δ м.д. 1.39,1.43 (2с, 9Н, ротамеры), 2.40 (дд, J=17.97, 4.52 Гц, 1Н), 2.48 (с, 3H), 2.68 (м, 1Н), 3.84 (м, 2Н), 4.46 (м, 1Н), 5.71 (с, 1Н), 7.58 (т, J=7.70 Гц, 1Н), 7.75 (м, 2Н), 7.91 (д, J=8.31 Гц, 1Н), 8.19 (м, 1Н); MS: (M+Na)+ 396.
Стадия 4:
Раствор 1-трет-бутилового эфира 4-(4-метилизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты (74 мг), гидрохлорида (1(R)-амино-2(S)-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (59 мг), РуВОР (114 мг) и изо-Pr2NEt (0.2 мл ) в СН2Cl2 (2 мл) перемешивают 2 час. Очистка флеш-хроматографией на системе Biotage с 5% МеОН в этилацетате дает 105 мг (90%) нужного продукта.1H ЯМР (400 МГц, CD3OD) δ м.д. 1.18 (м, 5Н), 1.39 (с, 9Н), 1.87 (дд, J=8.2, 5.3 Гц. 1Н), 2.28 (м, 2Н), 2.54 (м, 4Н), 2.95 (м, 1Н), 3.86 (м, 2Н), 4.40 (дд, J=9.8, 6.9 Гц, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.31 (д, J=17.6 Гц, 1Н), 5.79 (м, 2Н), 7.60 (т, J=7.5 Гц, 1Н), 7.78 (м, 2Н), 7.93 (д, J=8.3 Гц, 1Н), 8.20 (д, J=8.1 Гц, 1Н); MS: (M+Na)+607.
Стадия 5:
Раствор трет-бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(4-метилизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты (100 мг) и ТФК (3 мл) в CH2Cl2 (3 мл) перемешивают 1 час. После упаривания остаток растворяют в СН2Cl2 (2 мл) и прибавляют Boc-1-трет-лейцин (40 мг), РуВОР (104 мг) и изо-Pr2NEt (0.2 мл). Смесь перемешивают 1 час. После обработки очистка с помощью преп-ВЭЖХ дает 60 мг (52%) нужного продукта, Соединения 250, в виде твердого вещества.1H ЯМР (400 МГц, CD3OD) δ м.д. 1.04 (м, 12Н), 1.26 (м, 10Н), 1.44 (дд, J=9.5, 5.1 Гц, 1Н), 1.88 (дд, J=8.1, 5.4 Гц, 1Н), 2.26 (м, 2Н), 2.49 (с, 3H), 2.62 (дд, J=13.7, 7.1 Гц, 1Н), 2.94 (м, 1Н), 4.06 (дд, J=12.0, 3.4 Гц, 1Н), 4.25 (м, 1Н), 4.45 (д, J=11.3 Гц, 1Н), 4.53 (дд, J-10.3, 6.6 Гц, 1Н), 5.12 (д, J=10.0 Гц, 1Н), 5.29 (д, J=17.1 Гц, 1Н), 5.77 (м, 2Н), 6.63 (д, J=8.6 Гц, 1Н), 7.53 (т, J=7.8 Гц, 1Н), 7.76 (т, J=8.1 Гц, 1Н), 7.80 (с, 1Н), 7.91 (д, J=8.1 Гц, 1Н), 8.22 (д, J=8.3 Гц, 1-Н);
MS: (M+Na)+ 720.
Пример 251: Получение Соединения 251
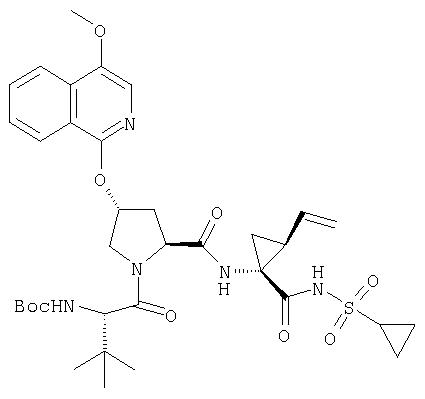
Соединение 251 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 3-метокси-3-фенилакриловую кислоту.
Стадия 1:
Модификации: используют 15 г 3-метокси-3-фенилакриловой кислоты, получают 250 мг продукта (выход 2%).
Продукт:
1H ЯМР (400 МГц, CD3COCD3) δ м.д. 3.85 (с, 3H), 6.96 (с, 1Н), 7.54 (м, 1Н), 7.71 (м, 1Н), 7.86 (д, J=8.07 Гц, 1Н), 8.31 (д, J=8.07 Гц, 1Н).
Стадия 2
Модификации: берут 200 мг 4-метокси-2Н-изохинолин-1-она, получают 150 мг продукта (выход 68%).
Продукт:
1H ЯМР (400 МГц, CD3Cl3) δ м.д. 4.05 (с, 2Н), 7.71 (м, 1H), 7.72 (м, 2Н), 7.80 (с, 1Н), 8.23 (дд, J=18.71, 7.70 Гц, 2Н).
Стадия 3:
Модификации: берут 122 мг 1-хлор-4-метоксиизохинолина, получают 218 мг продукта (выход 89%).
Продукт:

MS:(M+Na)+411
Стадия 4:
Модификации: берут 194 мг трет-бутилового эфира 4-(4-метоксиизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 298 мг (99%) продукта.
Продукт:

1H ЯМР (400 МГц, CD3OD) δ м.д. 1.17 (м, 5Н), 1.42 (с, 9Н), 1.87 (дд, J=3.2, 5.5 Гц, 1Н), 2.27 (м, 2Н), 2.54 (дд, J=13.3, 6.2 Гц, 1Н), 2.95 (м, 1Н), 3.85 (м, 2Н), 4.00 (с, 3H), 4.39 (дд, J=9.8, 6.9 Гц. 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.31 (д, J=17.1 Гц, 1Н), 5.76 (м, 2Н), 7.52 (с, 1Н), 7.62 (т, J=7.6 Гц, 1Н), 7.74 (т, J=7.2 Гц, 1Н), 8.12 (т, J=8.3 Гц, 2Н).
Стадия 5:
Модификации: берут 190 мг трет-бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(4-метилизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 270 мг (51%) продукта.
Продукт:

Данные:1H ЯМР (500 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.26 (м, 10Н), 1.43 (дд, J=8.6, 4.6 Гц, 1Н), 1.88 (дд; J=7.9, 5.5 Гц, 1Н), 2.24 (м, 2Н), 2.61 (дд, J=13.6, 6.9 Гц. 1Н), 2.94 (м, 1Н), 4.00 (с, 3H), 4.06 (дд, J=11.3, 3.1 Гц, 1Н), 4.25 (д, J=8.9 Гц, 1Н), 4.43 (д, 7-11.3 Гц, 1Н), 4.52 (м, 1Н), 5.12 (д, J=10.1 Гц, 1Н), 5.29 (д, J=17.1 Гц, 1Н), 5.75 (м, 2Н), 6.60 (д, J=8.6 Гц, 1Н), 7.55 (м, 2Н), 7.71 (т, J=7.3 Гц, 1Н), 8.09 (д, J=8.2 Гц, 1Н), 8.14 (д, J=8.2 Гц, 1Н); MS: (M+Na)+ 736.
Пример 252: Получение Соединения 252
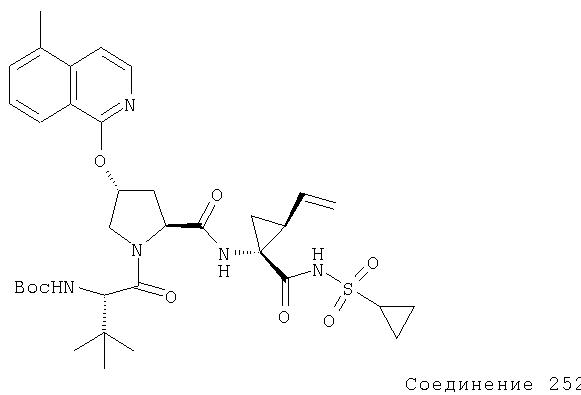
Соединение 252 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 2-метилкоричную кислоту.
Стадия 1:
Модификации; берут 20 г 2-метилкоричной кислоты, получают 14.3 г (72%) продукта
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 2.54 (с, 1Н), 6.69 (д. J=7.3 Гц, 1Н), 7.23 (д. J=Гц, 1Н), 7.39 (т, J=7.8 Гц, 1Н), 7.50 (д, J=7.1 Гц), 8.30 (д, J=8.1 Гц, 1Н), 11.62 (с, 1Н); MS: (М+Н)+ 160.
Стадия 2:
Модификации: берут 14.4 г 5-метил-1Н-изохинолин-1-она, получают 10.6 г продукта (выход 66%).
Продукт:
Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 2.67 (с, 3H), 7.55 (м, 2Н), 7.70 (дд, J=5.9, 1.0 Гц, 1Н), 8.19 (м, 1Н), 8.28 (д, Гц, 1Н); MS: (M+H)+ 178.
Стадия 3:
Модификации: берут 533 мг 1-хлор-5-метилизохинолина, получают 1116 мг продукта (выход 100%).
Продукт:

Данные: MS: (М+Н)+ 373.
Стадия 4:
Модификации: берут 372 мг трет-бутилового эфира 4-(5-метилизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 551 мг (94%) продукта.
Продукт:
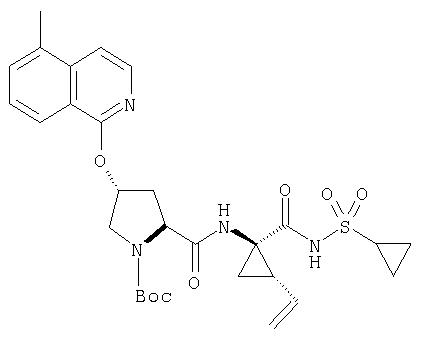
Данные: MS: (M+Na)+ 607.
Стадия 5:
Модификации: берут 551 мг трет- бутилового эфира 2- (1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(5-метилизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 274 мг (44%) продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00 (м, 12Н,) 1.23 (м, 10Н), 1.44 (м, 1Н), 1.87 (дд, J=8.1, 5.4 Гц, 1Н), 2.26 (м, 2Н), 2.62 (м, 4Н), 2.94 (м, 1Н), 4.07 (дд, J=11.9, 3.3 Гц, 1Н), 4.25 (д, J=9.5 Гц, 1Н), 4.46 (д, J=11.5 Гц, 1Н), 4.53 (дд, J=10.3, 7.1 Гц, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.29 (д, J=16.9 Гц, 1Н), 5.75 (м, 1Н), 5.86 (с, 1Н), 6.62 (д, J=9.3 Гц, 1Н), 7.39 (т, J=7.7 Гц, 1Н), 7.44 (д, J=5.9 Гц, 1Н), 7.53 (д, J=7.1 Гц, 1Н), 8.00 (д, J=6.1 Гц, 1Н), 8.06 (д, J=8.3 Гц, 1Н); MS: (M+H)+ 373.
Пример 253: Получение Соединения 253

Соединение 253 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 2-метоксикоричную кислоту.
Стадия 1:
Модификации; берут 10 г 2-метоксикоричной кислоты, получают 5.3 г (52%) продукта
Продукт:
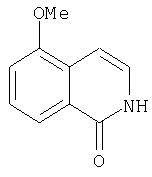
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 3.95 (с, 3H), 6.94 (д, J=7.3 Гц, 1Н), 7.08 (д, J=8.1 Гц, 1Н), 7.14 (д, J=7.3 Гц, 1Н), 7.43 (т, J=8.1 Гц, 1Н), 7.99 (д, J=8.1 Гц, 1Н), 7.99 (д, J=8.1 Гц, 1Н), 10.92 (с, 1Н); MS: (M+H)+ 176.
Стадия 2:
Модификации: берут 5.3 г 5-метокси-1Н-изохинолин-1-она, получают 5.38 г продукта (выход 92%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.01 (с, 3H), 7.04 (д, J=7.8 Гц, 1Н), 7.57 (т, J=8.1 Гц, 1Н), 7.88 (д, J=8.6 Гц, 1Н), 7.97 (д, J=5.9 Гц, 1Н), 8.25 (д, J=5.9 Гц, 1Н); MS: (M+H)+ 194.
Стадия 3:
Модификации: берут 581 мг 1-хлор-5-метоксиизохинолина, получают 1163 мг продукта (выход 100%).
Продукт:

Данные: MS: (M+H)+ 389.
Стадия 4:
Модификации: берут 117 мг 1-трет-бутилового эфира 4-(5-метоксиизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 180 мг (100%) продукта.
Продукт:

Данные: MS: (М+Н)+ 601.
Стадия 5:
Модификации: берут 177 мг трет- бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(5-метоксиизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 63 мг (44%) продукта.
Продукт:
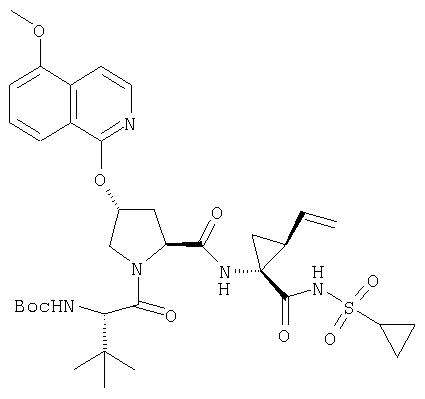
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00 (м, 12Н), 1.21 (м, 10Н), 1.38 (м, 1Н), 1.82 (дд, J=8.1, 5.6 Гц, 1Н), 2.20 (м, 2Н), 2.56 (дд, J=13.6, 6.7 Гц, 1Н), 2.88 (м, 1Н), 3.08 (м, 2Н), 3.93 (с, 3H), 4.01 (дд, J=11.9, 3.3 Гц, 1Н), 4.20 (д, J=9.1 Гц, 1Н), 4.39 (д, J=12.2 Гц, 1Н), 4.47 (дд, J=9.7, 7.0 Гц, 1Н), 5.06 (д, J=10.0 Гц, 1Н). 5.23 (д, J=16.9 Гц, 1Н), 5.70 (м, 1Н), 5.79 (с, 1Н), 6.55 (д, J=9.5 Гц, 1Н), 7.08 (д, J=7.6 Гц, 1Н), 7.37 (т, J=8.0 Гц, 1Н), 7.54 (д, J=5.9 Гц, 1Н), 7.68 (д, J=8.3 Гц, 1Н), 7.89 (д, J=5.9 Гц, 1Н); MS: (M+H)+ 714.
Пример 254: Получение Соединения 254
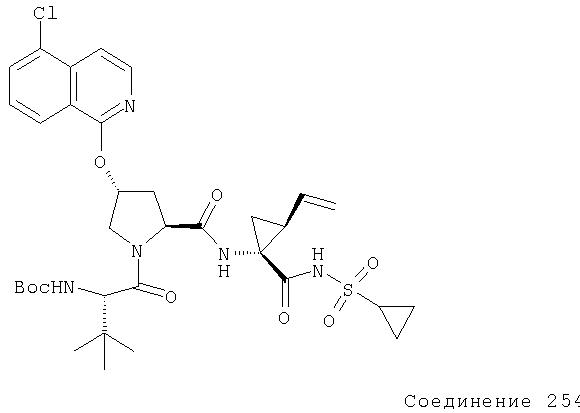
Соединение 254 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 2-хлоркоричную кислоту.
Стадия 1:
Модификации; берут 25 г 2-хлоркоричной кислоты, получают 14.6 г (59%) продукта
Продукт:
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 7.22 (д, J=7.3 Гц, 1Н), 7.42 (т, J=7.8 Гц, 1Н), 7.73 (д, J=7.8 Гц, 1Н), 8.34 (д, J=8.1 Гц, 1Н), 10.61 (с, 1Н); MS: (M+H)+ 180.
Стадия 2:
Модификации: берут 14.2 г 5-хлор-2Н-изохинолин-1-она, получают 8.28 г продукта (выход 53%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 7.60 (дд, J=8.6, 7.6 Гц, 1Н), 7.83 (м, 1Н), 8.00 (д, J=5.9 Гц, 1Н), 8.29 (дт, J=8.9, 1.0 Гц, 1Н), 8.38 (д, J=5.9 Гц, 1Н); MS: (M+H)+ 198.
Стадия 3:
Модификации: берут 594 мг 1,5-дихлоризохинолина, получают 1174 мг продукта (выход 100%).
Продукт:

Данные: MS: (М+Н)+ 393.
Стадия 4:
Модификации: берут 118 мг 1-трет-бутилового эфира 4-(5-хлоризохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 154 мг (85%) продукта.
Продукт:
Данные: MS: (M+H)+ 605.
Стадия 5:
Модификации: берут 150 мг трет- бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(5-хлоризохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 91 мг (51%) продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 0.97 (м, 12Н), 1.17 (м, 10Н), 1.38 (дд, J=9.4, 5.3 Гц, 1Н), 1.82 (дд, J=8.0, 5.5 Гц, 1Н), 2.21 (м, 2Н), 2.58 (дд, J=13.8, 7.0 Гц, 1Н), 2.88 (м, 1Н), 4.01 (дд, J=11.9, 2.8 Гц, 1Н), 4.16 (д, J=9.3 Гц, 1Н), 4.47 (м, 2Н), 5.06 (д, J=10.3 Гц, 1Н), 5.24 (д, J=16.9 Гц, 1Н), 5.70 (м, 1Н), 5.82 (с, 1Н), 6.52 (д, J=9.3 Гц, 1Н), 7.42 (т, J=8.0 Гц, 1Н), 7.57 (д, J=6.1 Гц, 1Н), 7.76 (д, J=7.6 Гц, 1Н), 8.05 (д, J=6.1 Гц, 1Н). 8.13 (д, J=8.3 Гц, 1Н); MS: (M+H)+ 718.
Пример 255: Получение Соединения 255

Соединение 255 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 2-фторкоричную кислоту.
Стадия 1:
Модификации; берут 16.6 г 2-фторкоричной кислоты, получают 8.55 г (51%) продукта
Продукт:

Данные:1H ЯМР (400 МГц, CD3COCD3) δ м.д. 6.62 (д, J=7.3 Гц, 1Н), 7.32 (д, J=7.3 Гц, 1Н), 7.47 (м, 2Н), 8.09 (м, 1Н).
Стадия 2:
Модификации: берут 8.4 г 5-фтор-1Н-изохинолин-1-она, получают 7.5 г продукта (выход 80%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 7.43 (ддд, J=9.7, 7.8, 0.9 Гц, 1Н), 7.62 (тд, J=8.2, 5.4 Гц, 1Н), 7.84 (д, J=5.6 Гц, 1Н), 8.14 (д, J=8.6 Гц, 1Н), 8.33 (д, J=5.9 Гц, 1Н); MS: (M+H)+ 182.
Стадия 3:
Модификации: берут 203 мг 1-хлор-5-фторизохинолина, получают 384 мг продукта (выход 90%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3SOCD3) δ м.д. 1.34, 1.36 (2с, 9Н, ротамеры), 2.35 (м, 1Н), 2.61 (м, 1Н), 3.65 (д, J=12.23 Гц, 1Н), 3.80 (м, 1Н), 4.35 (м, 1Н), 5.70 (с, 1Н), 7.48 (д, J=6.11 Гц, 1Н), 7.63 (м, 2Н), 7.99 (м, 1Н), 8.10 (д, J=5.87 Гц, 1Н); MS: (M+Na)+ 399.
Стадия 4:
Модификации: берут 76 мг 1-трет-бутилового эфира 4-(5-фторизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 116 мг (99%) продукта.
Продукт:

Стадия 5:
Модификации: берут 110 мг трет-бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(5-фторизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 39 мг (30%) продукта.
Продукт:
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 12Н). 1.25 (м, 10Н), 1.44 (дд, J=9.5, 5.4 Гц, 1Н), 1.88 (дд, J=8.1, 5.4 Гц, 1Н), 2.28 (м, 2Н), 2.63 (дд, J=13.8, 7.0 Гц, 1Н), 2.94 (м, 1Н), 4.07 (дд, J=11.9, 3.1 Гц, 1Н), 4.23 (д, J=9.3 Гц, 1Н), 4.52 (м, 2Н), 5.12 (дд, J=10.3, 1.5 Гц, 1Н), 5.29 (д, J=17.4 Гц, 1Н), 5.75 (м, 1Н), 5.89 (с, 1Н), 6.59 (д, J=9.1 Гц, 1Н), 7.47 (м, 3H), 8.02 (д, J=8.1 Гц, 1Н), 8.06 (д, J=6.1 Гц, 1Н); MS: (M+Na)+ 724.
Пример 256: Получение Соединения 256

Соединение 256 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 2-дифторметоксикоричную кислоту.
Стадия 1:
Модификации; берут 10.7 г 2-дифторметоксикоричной кислоты, получают 2 г (18%) продукта
Продукт:
Данные:1H ЯМР (400 МГц, CD3SOCD3) δ м.д. 6.06 (м, 2Н), 6.42 (м, 2Н), 6.71 (с, 2Н), 7.36 (с, 1Н); MS: (М+H)+ 212.
Стадия 2:
Модификации: берут 300 мг 5-дифторметокси-1Н-изохинолин-1-она, получают 300 мг продукта (выход 92%).
Продукт:
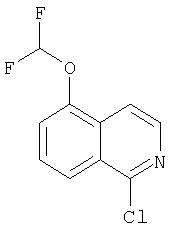
Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 6.70 (т, J=72.8 7(?) Гц, 1Н), 7.48 (м, 1Н), 7.64 (м, 1Н), 7.92 (д, J=5.87 Гц, 1Н), 8.21 (д, J=8.56 Гц, 1Н), 8.35 (д, J=5.62 Гц, 1Н).
Стадия 3:
Модификации: берут 230 мг 1-хлор-5-дифторметоксиизохинолина, получают 360 мг продукта (выход 96%).
Продукт:
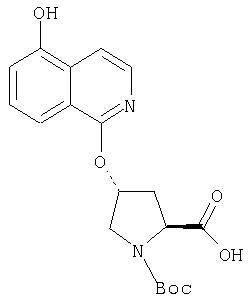
Стадия 4:
Модификации: берут 37 мг 1-трет-бутилового эфира 4-(5-гидроксиизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 57 мг (99%) продукта.
Продукт:

Стадия 5:
Модификации: берут 57 мг трет-бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(5-гидроксиизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 10 мг (15%) продукта.
Продукт:
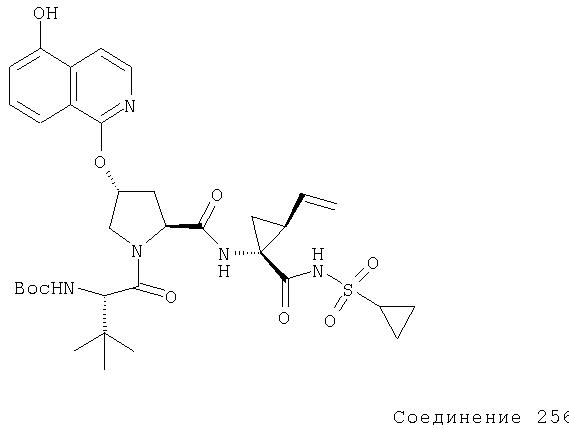
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 0.93 (м, 4Н), 1.13 (с, 9Н), 1.31 (м, 1Н), 1.49 (с, 9Н), 1.89 (дд, J=7.8, 5.4 Гц, 1H), 2.16 (кв, J=8.8 Гц, 1Н), 2.40 (м, 1Н), 2.81 (м, 1Н), 2.90 (м, 1Н), 3.76 (м, 2Н), 4.30 (м, 1Н), 4.59 (дд, J=10.2, 7.7 Гц, 1Н), 5.07 (дд, J=10.3, 1.7 Гц, 1Н), 5.26 (дд, J=17.2, 1.3 Гц, 1Н), 5.77 (дт, J=17.2, 9.6 Гц, 1Н), 5.93 (с, 1Н), 7.24 (д, J=8.6 Гц, 1Н), 7.51 (м, 2Н), 7.63 (т, J=8.0 Гц, 1Н), 7.98 (д, J=6.1 Гц, 1Н), 8.24 (д, J=8.3 Гц, 1Н); MS: (M+H)+ 700.
Пример 257: Получение Соединения 257
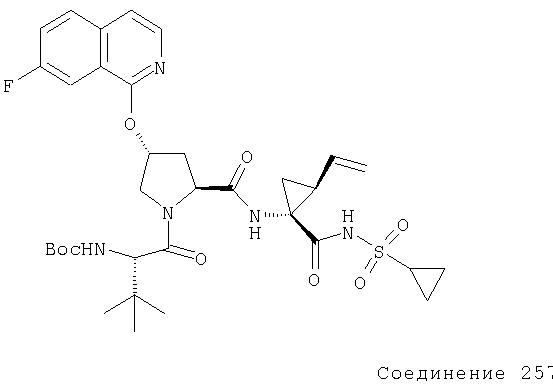
Соединение 257 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 4-фторкоричную кислоту.
Стадия 1:
Модификации: берут 16.6 г 4-фторкоричной кислоты, получают 8.2 г (49%) продукта
Продукт:
Данные:1H ЯМР.(400 МГц, CD3COCD3) δ м.д. 6.57 (д, J=7.09 Гц, 1Н), 7.21 (д, J=7.09 Гц, 1Н), 7.50 (м, 1Н), 7.72 (дд, J=8.68, 5.26 Гц, 1Н), 7.90 (дд, J=9.54, 2.93 Гц, 1Н).
Стадия 2:
Модификации: берут 8.15 г 7-фтор-2Н-изохинолин-1-она, получают 7.6 мг продукта (выход 84%).
Продукт:
Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 7.52 (тд, J=8.6, 2.6 Гц, 1Н), 7.59 (д, J=5.6 Гц, 1Н), 7.86 (дд, J=9.1,5.4 Гц, 1Н), 7.95 (дд, J=9.5, 2.5 Гц, 1Н), 8.26 (д, J=5.6 Гц, 1Н); MS:(M+H)+ 182.
Стадия 3:
Модификации: берут 191 мг 1-хлор-7-фторизохинолина, получают 350 мг продукта (выход 93%).
Продукт:

Данные: MS: (M+Na)+ 399.
Стадия 4:
Модификации: берут 75 мг 1-трет-бутилового эфира 4-(7-фторизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 100 мг (85%) продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.16 (м, 4Н), 1.41 (м, 10Н), 1.88 (дд, J=8.1, 5.4 Гц, 1Н), 2.28 (м, 2Н), 2.56 (м, 1Н,), 2.94 (м, 1Н), 3.87 (м, 2Н), 4.41 (дд, J=9.7, 7.0 Гц, 1Н). 5.12 (д, J=10.8 Гц, 1Н), 5.31 (д, J=17.1 Гц, 1Н), 5.78 (м, 2Н), 7.36 (д, J=5.9 Гц, 1Н), 7.54 (м, 1Н), 7.78 (дд, J=9.3, 2.5 Гц, 1Н), 7.90 (дд, J=9.1, 5.1 Гц, 1Н), 7.96 (д, J=5.9 Гц, 1Н); MS: (M+Na)+ 611.
Стадия 5:
Модификации: берут 95 мг трет-бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(7-фторизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 55 мг (44%) продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 12Н), 1.22 (м, 10Н), 1.44 (дд, J=9.3, 5.4 Гц, 1Н), 1.88 (дд, J=8.2, 5.5 Гц, 1Н), 2.27 (м, 2Н), 2.63 (дд, J=13.8, 7.0 Гц, 1Н), 2.94 (м, 1Н), 4.07 (дд, J=11.5, 3.2 Гц, 1Н), 4.22 (д, J=9.5 Гц, 1Н), 4.47 (д, J=11.7 Гц, 1Н), 4.55 (дд, J=10.6, 7.5 Гц, 1Н), 5.12 (д, J=10.3 Гц, 1Н), 5.29 (д, J=17.1 Гц, 1Н), 5.75 (м, 1Н), 5.87 (с, 1Н), 6.61 (д, J=9.5 Гц, 1Н), 7.36 (д, J=5.9 Гц, 1Н), 7.52 (тд, J=8.9, 2.5 Гц, 1Н), 7.79 (дд, J=9.4, 2.6 Гц, 1Н), 7.88 (дд, J=8.7, 5.5 Гц, 1Н), 7.96 (д, J=5.9 Гц, 1Н); MS: (M+Na)+ 724.
Пример 258: Получение Соединения 258

Соединение 258 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 4-хлоркоричную кислоту.
Стадия 1:
Модификации: берут 9.13 г 4-фторкоричной кислоты, получают 4 г (44%) продукта
Продукт:
Данные:1H ЯМР (400 МГц, CD3SCOCD3) δ м.д. 6.58 (д, J=7.1 Гц, 1Н), 7.20 (дд, J=7.1, 5.9 Гц, 1Н), 7.72 (м, 2Н), 8.10 (м, 1Н).
Стадия 2:
Модификации: берут 3.5 г 7-хлор-2Н-изохинолин-1-она, получают 2.8 мг продукта (выход 72%).
Продукт:

Данные:1H ЯМР (500 МГц, CDCl3) δ м.д. 7.59 (д, J=5.5 Гц, 1Н), 7.69 (дд, J=8.9, 2.1 Гц, 1Н), 7.80 (д, J=8.6 Гц, 1Н), 8.29 (д, J=5.5 Гц, 1Н), 8.34 (с, 1Н); MS: (М+Н)+ 198.
Стадия 3:
Модификации: берут 208 мг 1,7-дихлоризохинолина, получают 350 мг продукта (выход 89%).
Продукт:

Данные: MS: (M+Na)+ 415.
Стадия 4:
Модификации: берут 79 мг 1-трет-бутилового эфира 4-(7-хлоризохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 119 мг (99%) продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3CD) δ м.д. 1.17 (м, 4Н), 1.43 (м, 10Н), 1.88 (дд, J=8.31 5.4 Гц, 1Н), 2.29 (м, 2Н), 2.57 (дд, J=13.7, 6.9 Гц, 1Н), 2.95 (м, 1Н), 3.87 (м, 2Н), 4.42 (дд, J=9.9, 6.9 Гц, 1Н), 5.13 (д, J=10.3 Гц, 1Н), 5.31 (дд, J=17.1, 1.2 Гц, 1Н), 5.78 (м, 2Н), 7.35 (д, J=5.9 Гц, 1Н), 7.69 (дд, J=8.7, 2.1 Гц, 1Н), 7.84 (д, J=8.8 Гц, 1Н), 7.99 (д, J=5.9 Гц, 1Н), 8.12 (д, J=1.7 Гц, 1Н); MS: (M+Na)+ 627.
Стадия 5:
Модификации: берут 115 мг трет-бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(7-хлоризохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 36 мг (25%) продукта.
Продукт:

Данные: MS: (M+Na)+ 740.
Пример 259: Получение Соединения 259

Соединение 259 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 4-метилкоричную кислоту.
Стадия 1:
Модификации: берут 25 г 4-метилкоричной кислоты, получают 15.3 г (62%) продукта
Продукт:

Данные:1 H ЯМР (400 МГц, CD3OD) δ м.д. 2.50 (с, 3H), 6.54 (д, J=7.1 Гц, 1Н), 7.13 (д, J=7.1 Гц, 1Н), 7.49 (м, 2Н), 8.22 (с, 1Н), 11.49 (с, 1Н); MS: (M+H)+ 160.
Стадия 2:
Модификации: берут 15.3 г 7-метил-2Н-изохинолин-1-она, получают 5.15 г продукта (выход 30%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 2.58 (с, 3H), 7.56 (м, 2Н), 7.73 (д, J=8.3 Гц, 1Н), 8.09 (с, 1Н), 8.20 (д, J=5.6 Гц, 1Н); MS: (M+H)+ 178.
Стадия 3:
Модификации: берут 205 мг 1-хлор-7-метилизохинолина, получают 350 мг продукта (выход 89%).
Продукт:
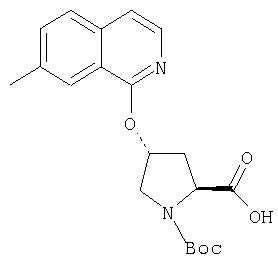
Данные: MS: (M+H)+ 373.
Стадия 4:
Модификации: берут 75 мг 1-трет-бутилового эфира 4-(7-метилизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 107 мг (95%) продукта.
Продукт:

Данные: MS: (M+Na)+ 607.
Стадия 5:
Модификации: берут 107 мг трет- бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(7-метилизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 53 мг (41%) продукта.
Продукт:
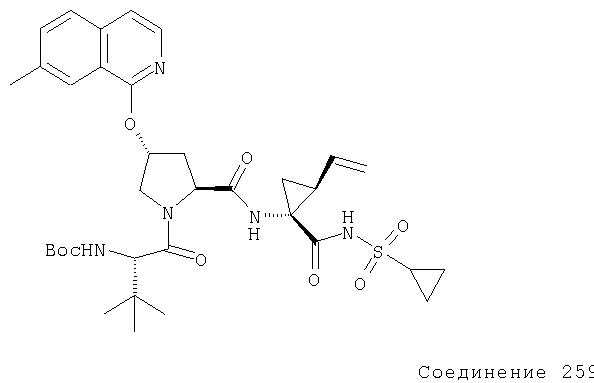
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (м, 12Н), 1.18 (с, 9Н), 1.24 (м, 1Н), 1.45 (дд, J=9.4, 5.5 Гц, 1Н), 1.88 (дд, J=8.2, 5.5 Гц, 1Н), 2.28 (м, 2Н), 2.50 (с, 3H), 2.61 (дд, J=13.8, 6.7 Гц, 1Н), 3.34 (с, 1Н), 4.09 (дд, J=11.7, 3.2 Гц, 1Н), 4.23 (с, 1Н), 4.42 (д, J=12.0 Гц, 1Н), 4.57 (дд, J=10.0,7.1 Гц, 1Н). 5.12 (дд, J=10.3, 1.5 Гц, 1Н), 5.30 (д, J=17.1 Гц, 1Н), 5.76 (м, 1Н), 5.87 (с, 1Н), 7.28 (д, J=5.9 Гц, 1Н), 7.55 (д, J=8.3 Гц, 1Н), 7.71 (д, J=8.3 Гц, 1Н), 7.89 (д, J=5.9 Гц, 1Н), 7.93 (с, 1Н); MS: (М+H)+ 698.
Пример 260: Получение Соединения 260

Соединение 260 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 4-метоксикоричную кислоту.
Стадия 1:
Модификации: берут 33 г 4-метоксикоричной кислоты, получают 7 г (33%) продукта
Продукт:

Данные:1H ЯМР (500 МГц, CD3COCD3) δ м.д. 3.90 (с, 3H), 6.49 (д, J=7.0 Гц, 1Н), 7.10 (д, J=7.3 Гц, 1Н), 7.28 (дд, J=8.6, 2.8 Гц, 1Н), 7.57 (д, J=8.9 Гц, 1Н), 7.71 (д, J=2.8 Гц, 1Н).
Стадия 2:
Модификации: берут 4 г 7-метокси-2Н-изохинолин-1-она, получают 3 г продукта (выход 68%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 3.98 (с, 3H), 7.38 (дд, J=8.9, 2.6 Гц, 1Н), 7.52 (м, 2Н), 7.73 (д, J=8.8 Гц, 1Н), 8.16 (д, J=5.4 Гц, 1Н).
Стадия 3:
Модификации: берут 533 мг 1-хлор-7-метоксиизохинолина, получают 1115 мг продукта (выход 100%).
Продукт:
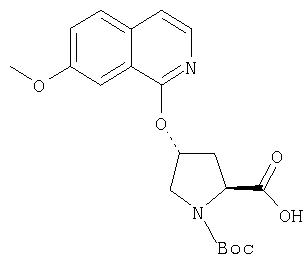
Стадия 4:
Модификации: берут 78 мг 1-трет-бутилового эфира 4-(7-метоксиизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 108 мг (99%) продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.17 (м, 4Н), 1.40 (м, 1Н), 1.43 (с, 9Н), 1.85 (дд, J=8.1, 5.4 Гц, 1Н), 2.21 (м, 2Н), 2.51 (дд, J=13.7, 6.6 Гц, 1Н), 2.93 (с, 1Н), 3.80 (м, 2Н), 3.94 (с, 3H), 4.41 (дд, J=10.0, 6.6 Гц, 1Н), 4.57 (с, 1Н), 5.11 (д, J=11.3 Гц, 1Н), 5.29 (д, J=17.1 Гц, 1Н), 5.77 (м, 2Н), 7.01 (д, J=7.8 Гц, 1Н), 7.22 (д, J=5.6 Гц, 1Н), 7.32 (д, J=8.1 Гц, 1Н), 7.58 (т, J=8.0 Гц, 1Н), 7.87 (д, J=5.9 Гц, 1Н); MS: (M+H)+ 601.
Стадия 5:
Модификации: берут 100 мг трет-бутилового эфира 2-1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(7-метоксиизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 30 мг (25%) продукта.
Продукт:

Данные:1H ЯМР (500 МГц, CD3SOCD3) δ м.д. 0.90 (м, 2Н), 0.95 (с, 9Н), 1.05 (м, 1Н), 1.12 (с, 9Н), 1.35 (м, 2Н), 1.70 (м, 1Н), 2.18 (м, 1Н), 2.92 (м, 1Н), 3.86 (с, 3H), 4.00 (м, 2Н), 4.27 (д, J=12.0 Гц, 1Н), 4.45 (т, J=8.6 Гц, 1Н). 5.09 (д, J=10.8 Гц, 1Н), 5.23 (д, J=16.9 Гц, 1Н), 5.62 (м, 1Н), 5.79 (с, 1Н), 6.55 (д, J=8.1 Гц, 1Н), 7.35 (д, J=6.6 Гц, 1Н), 7.39 (д, J=2.5 Гц, 1Н), 7.43 (дд, J=8.8, 2.2 Гц. 1Н), 7.84 (д, J=8.8 Гц, 1Н), 7.88 (д, J=5.9 Гц, 1Н); MS: (M+H)+ 714.
Примеры 261 и 262: Получение Соединений 261 и 262


Соединения 261 и 262 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 4-фтор-3-метоксикоричную кислоту.
Стадия 1:
Модификации: берут 19.6 г 4-фтор-3-метоксикоричной кислоты, получают 9.5 г (48%) продукта
Продукт:
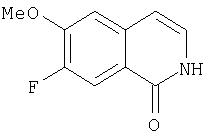
Данные:1H ЯМР (500 МГц, CD3COCD3) δ м.д. 4.00 (с, 1Н), 6.49 (д, J=7.34 Гц, 1Н), 7.19 (д, J=7.09 Гц, 1Н), 7.29 (д, J=8.07 Гц, 1Н), 7.86 (д, J=11.74 Гц, 1Н).
Стадия 2:
Модификации: берут 9 г 7-фтор-6-метокси-2Н-изохинолин-1-она, получают 7 г продукта (выход 70%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.04 (с, 3H), 7.17 (д, J=8 07 Гц, 1Н), 7.48 (д, J=5.62 Гц, 1Н), 7.94 (д, J=11.49 Гц, 1Н), 8.20 (д, J=5.62 Гц, 1Н).
Стадия 3:
Модификации: берут 222 мг 1-хлор-7-фтор-6-метоксиизохинолина, получают 406 мг продукта.
Продукты:


Стадия 4:
Модификации: берут 400 мг 1-трет-бутилового эфира 4-(7-фтор-6-метоксиизохинолин-1-илокси)-пирролидин-1, 2-дикарбоновой кислоты 1-трет-бутилового эфира 4-(7-хлор-6-метоксиизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 700 мг продуктов.
Продукты:

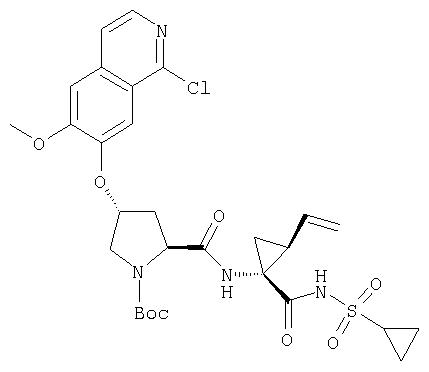
Стадия 5:
Модификации: берут 700 мг трет-бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(7-фтор-6-метоксиизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 79 мг Соединения 261 и 80 мг Соединения 262.
Продукты:
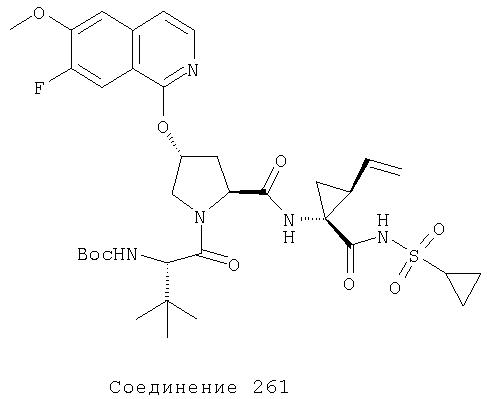

Данные для Соединения 261:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.07 (м, 12Н), 1.25 (м, 10Н), 1.44 (м, 1Н), 1.88 (дд, J=8.1, 5.6 Гц, 1Н), 2.25 (м, 2Н), 2.60 (дд, J=13.7, 6.9 Гц, 1Н), 2.94 (м, 1Н), 4.02 (м, 4Н), 4.22 (с. 1Н), 4.43 (д, J=12.2 Гц, 1Н), 4.53 (дд, J=10.3, 6.6 Гц. 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.30 (д, J=16.6 Гц, 1Н), 5.75 (м, 1Н), 5.84 (с, 1Н), 7.28 (д, J=5.9 Гц, 1Н), 7.37 (д, J=8.1 Гц, 1Н), 7.75 (д, J=11.7 Гц, 1Н), 7.91 (д, J=5.9 Гц, 1Н); MS: (M+Na)+ 754.
Данные для Соединения 262:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.07 (м, 12Н), 1.25 (м, 10Н), 1.44 (м, J=9.41, 5.20 Гц, 1Н), 1.87 (дд, J=8.3110 5.38 Гц, 1Н), 2.24 (кв, J=8.72 Гц, 2Н), 2.57 (дд, J=13.82, 7.21 Гц, 1Н), 2.94 (м, 1Н), 3.97 (д, J=5.14 Гц, 3H), 4.09 (м, J=11.00 Гц, 1Н), 4.24 (с, 1Н), 4.32 (м, 1Н), 4.50 (м, J=16.87 Гц, 1Н), 5.12 (дд, J=10.52. 1.71 Гц, 1Н), 5.30 (дд, J=17.12,1.47 Гц, 1Н), 5.38 (с, 1Н), 5.76 (м, 1Н), 7.39 (с, 1Н), 7.63 (с, 1Н), 7.66 (д, J=5.87 Гц, 1Н), 8.07 (д, J=5.62 Гц, 1Н); MS: (M+H)+ 732.
Пример 263: Получение Соединения 263

Соединение 263 получают по Схеме 1 из Примера 250 за исключением стадии 1 и стадии 2.
Стадия 3:
Модификации: берут 176 мг 1-хлор-8-метилизохинолина, получают 370 мг (100%) продукта.
Продукт:
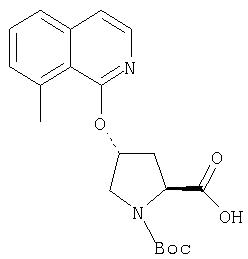
Стадия 4:
Модификации: берут 149 мг 1-трет-бутилового эфира 4-(8-метилизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 230 мг (99%) продукта.
Продукт:
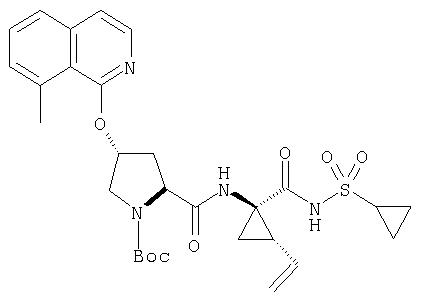
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.13 (м, 4Н), 1.42 (м, 10Н), 1.87 (дд, J=8.2, 5.3 Гц, 1Н), 2.25 (м, 2Н), 2.58 (дд, J=13.9, 6.9 Гц, 1Н), 2.83 (с, 3H), 2.96 (м, 1Н), 3.85 (м, 2Н), 4.38 (м, J=10.2, 6.7 Гц, 1Н), 5.12 (дд, J=10.4, 1.6 Гц, 1Н), 5.30 (дд, J=17.1, 1.2 Гц, 1Н), 5.76 (м, 2Н), 7.28 (д, J=5.9 Гц, 1Н), 7.36 (д, J=6.9 Гц, 1Н), 7.53 (т, J=7.7 Гц, 1Н), 7.62 (м, 1Н), 7.88 (д, J=5.6 Гц, 1Н); MS: (M+Na)+ 607.
Стадия 5:
Модификации: берут 220 мг трет-бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(8-метилизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 90 мг (35%) продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 12Н), 1.24 (м, 10Н), 1.44 (дд, J=9.3, 5.4 Гц, 1Н), 1.87 (дд, J=8.1, 5.4 Гц, 1Н), 2.25 (м, 2Н), 2.60 (дд, J=13.9, 7.3 Гц, 1Н), 2.77 (с, 3H), 2.94 (м, 1Н), 4.04 (дд, J=11.9, 3.1 Гц, 1Н), 4.27 (д, J=9.5 Гц, 1Н), 4.42 (д, J=12.0 Гц, 1Н), 4.51 (дд, J=10.8, 7.1 Гц, 1Н), 5.12 (д, J=10.3 Гц, 1Н), 5.28 (д, J=17.1 Гц, 1Н), 5.75 (м, 1Н), 5.95 (с, 1Н), 6.63 (д, J=9.1 Гц, 1Н), 7.28 (м, 2Н), 7.50 (т, J=7.7 Гц, 1Н), 7.60 (д, J=7.8 Гц, 1Н), 7.89 (д, J=5.6 Гц, 1Н); MS: (М+Na)+ 720.
Пример 264: Получение Соединения 264

Соединение 264 получают по Схеме 1 из Примера 250 за исключением стадии 1 и стадии 2.
Стадия 3:
Модификации: берут 203 мг 1-хлор-8-метоксиизохинолина, получают 340 мг (85%) продукта.
Продукт:
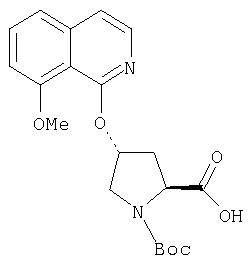
Данные:1H ЯМР (400 МГц, CD3SOCD3) δ м.д. 1.34, 1.36 (2с, 9Н, ротамеры), 2.26 (м, 1Н), 2.49 (м, 1Н), 3.67 (м, 2Н), 3.86 (с, 3H), 4.31 (м, 1Н), 5.67 (уш с, 1Н), 7.04 (д, J=7.8 Гц, 1Н), 7.30 (д, J=5.9 Гц, 1Н), 7.38 (д, J=8.1 Гц, 1Н), 7.62 (т, J=8.0 Гц, 1Н), 7.93 (д, J=5.6 Гц, 1Н), 12.64 (с, 1Н); MS. (M+Na)+ 411.
Стадия 4:
Модификации: берут 78 мг 1-трет-бутилового эфира 4-(8-метоксиизохинолин-1-илокси)-пирролидин-1,2-дикарбоновой кислоты, получают 115 мг (96%) продукта.
Продукт:

Стадия 5:
Модификации: берут 110 мг трет-бутилового эфира 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-(8-метоксиизохинолин-1-илокси)-пирролидин-1-карбоновой кислоты, получают 45 мг (34%) продукта.
Продукт:

Данные: MS. (М+H)+ 714.
Примеры 265 и 266: Получение Соединений 265 и 266

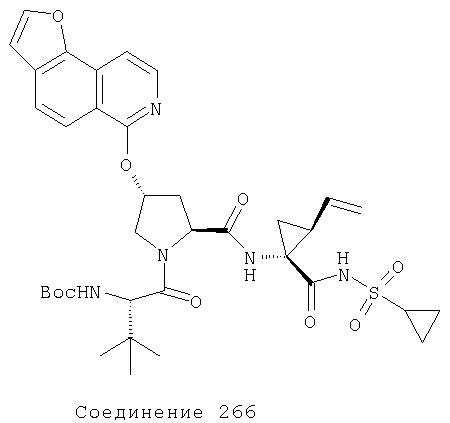
Соединения 265 и 266 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 3-(2,3-дигидробензофуран-7-ил)акриловую кислоту.
Стадия 1:
Модификации: берут 3.8 г 3-(2,3-дигидробензофуран-7-ил)акриловой кислоты, получают 2 г (53%) продукта
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 3.37 (т, J=9.05 Гц, 1Н), 4.73 (т, J=9.05 Гц, 2Н), 6.67 (д, J=7.09 Гц, 1Н), 7.10 (д, J=7.Q9 Гц, 1Н), 7.37 (д, J=8.07 Гц, 1Н), 7.81 (д, J=8.07 Гц, 1Н); MS: (М+Н)+ 188.
Стадия 2:
Модификации: берут 1.87 г 2,3-дигидро-7Н-фуро[2,3-f]изохинолин-6-она, получают 1.84 г продукта (выход 90%).
Продукт:
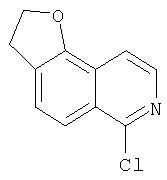
Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 3.43 (т, J=9.05 Гц, 2Н), 4.82 (т, J=9.05 Гц, 2Н), 7.52 (д, J=8.56 Гц, 1Н), 7.66 (д, J=5.62 Гц, 1Н), 7.84 (д, J=8.31 Гц, 1Н), 8.19 (д, J=5.62 Гц, 1Н); MS (M+H)+ 206.
Стадия 3:
Модификации: берут 206 мг 6-хлор-2,3-дигидро-7Н-фуро[2,3-f]изохинолина, получают 300 мг смеси продуктов.
Продукты:
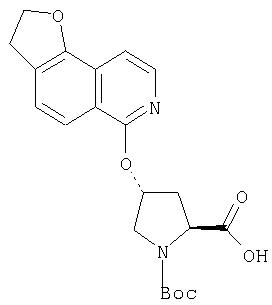
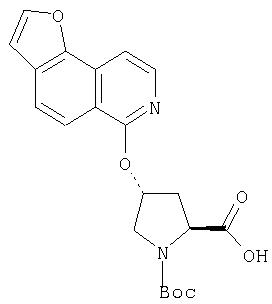
Стадия 4:
Модификации: берут 240 мг смеси продуктов стадии 3, получают 350 мг смеси продуктов.
Продукты:
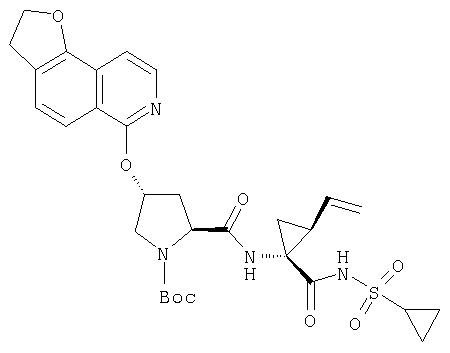

Стадия 5:
Модификации: берут 331 мг смеси продуктов стадии 4, получают 240 мг Соединения 265 и 24 мг Соединения 266.
Продукты:
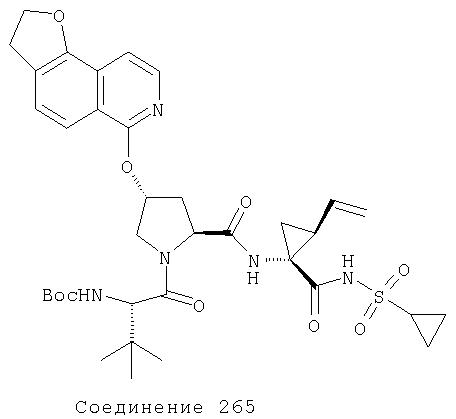

Данные для Соединения 265:1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99 (м, 12Н), 1.16 (м, 10Н), 1.36 (м, 1Н), 1.81 (дд, J=8.07, 5.62 Гц, 1Н), 2.18 (м, 2Н), 2.54 (дд, J=13.69, 6.85 Гц, 1Н), 2.87 (м, 1Н), 3.31 (т, J=9.05 Гц, 2Н), 4.01 (м, 1Н), 4.18 (с, 1Н), 4.36 (д, J=11.74 Гц, 1Н), 4.46 (дд, J=10.15, 7.21 Гц, 1Н), 4.70 (м, 2Н), 5.05 (д, J=10.27 Гц, 1Н), 5.23 (д, J=16.87 Гц, 1Н), 5.70 (м, 2Н), 7.23 (д, J=5.87 Гц, 1Н), 7.31 (д, J=8.31 Гц, 1Н), 7.63 (д, J=8.31 Гц, 1Н), 7.82 (д, J=5.87 Гц, 1Н); MS (M+H)+ 726.
Данные для Соединения 266:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.24 (м, 10Н), 1.44 (дд, J=10.03, 5.14 Гц, 1Н), 1.88 (дд, J=7.83, 5.38 Гц, 1Н), 2.27 (м, 2Н), 2.65 (дд, J=12.96, 6.36 Гц, 1Н), 2.94 (м, 1Н), 4.08 (дд, J=12.35, 3.30 Гц, 1Н), 4.25 (с, 1Н), 454 (м, 2Н), 5.12 (д, J=10.27 Гц, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.75 (м, 1Н), 5.91 (с, 1Н), 7.05 (д, J=1.96 Гц, 1Н), 7.72 (м, 2Н), 8.02 (м, 2Н), 8.11 (д, J=5.87 Гц, 1Н), 9.19 (с, 1Н); MS: (M+H)+ 724.
Примеры 267 и 268: Получение Соединений 267 и 268
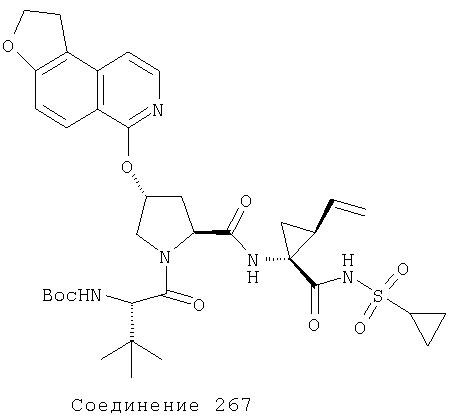

Соединения 267 и 268 получают по Схеме 1 из Примера 250 за исключением того, что на стадии 1 вместо 3-фенилбут-2-еновой кислоты берут 3-(2,3-дигидробензофуран-4-ил)акриловую кислоту.
Стадия 1:
Модификации: берут 1.14 г 3-(2,3-дигидробензофуран-4-ил)акриловой кислоты, получают 600 мг (52%) продукта
Продукт:
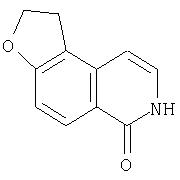
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 3.35 (т, J=8.93 Гц, 2 Н), 4.74 (т, J=8.93 Гц, 2Н), 6.49 (д, J=7.09 Гц, 1Н), 6.95 (д, J=8.56 Гц, 1Н), 7.25 (д, J=7.09 Гц, 1Н), 8.13 (д, J=8.80 Гц, 1Н); MS (М+H)+ 188.
Стадия 2:
Модификации: берут 560 мг 1,7-дигидро-2Н-фуро[3,2-f]изохинолин-6-она, получают 380 мг продукта (выход 48%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 3.47 (т, J=9.05 Гц, 2Н), 4.84 (т, J=9.05 Гц, 2Н), 7.24 (д, J=8.56 Гц, 1Н), 7.33 (д, J=5.87 Гц, 1Н), 8.20 (м, 2Н); MS (M+H)+ 206.
Стадия 3:
Модификации: берут 206 мг 6-хлор-1,2-дигидрофуро[3,2-f]изохинолина, получают 390 мг смеси продуктов.
Продукты:
Стадия 4:
Модификации: берут 216 мг смеси продуктов стадии 3, получают 330 мг смеси продуктов.
Продукты:

Стадия 5:
Модификации: берут 330 мг смеси продуктов стадии 4, получают 140 мг Соединения 267 и 25 мг Соединения 268.
Продукты:

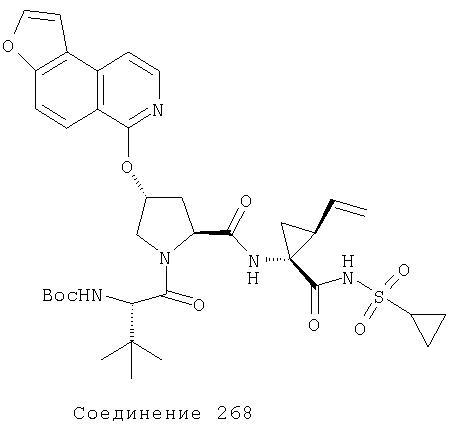
Данные для Соединения 267:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.07 (м, 12Н), 1.24 (м, 10H), 1.43 (м, 1Н), 1.88 (дд, J=8.07, 5.38 Гц, 1H), 2.26 (м, 2Н), 2.61 (дд, J=13.69, 7.09 Гц, 1Н), 2.94 (м, 1Н), 3.42 (т, J=9.05 Гц, 2Н), 4.05 (дд, J=11.86, 3.55 Гц, 1Н), 4.24 (с, 1Н), 4.50 (м, 2Н), 4.77 (т, J=8.93 Гц, 2Н), 5.12 (м, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.76 (м, 2Н), 7.03 (д, J=8.80 Гц, 1Н), 7.12 (д, J=6.11 Гц, 1Н), 7.91 (д, J=5.87 Гц, 1Н), 8.06 (д, J=8.80 Гц, 1Н); MS: (M+H)+ 726.
Данные для Соединения 268:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.19 (с, 9Н), 1.26 (м, 1Н), 1.44 (м, 1Н), 1.88 (дд, J=8.07, 5.62 Гц, 1Н), 2.24 (д, J=8.56 Гц, 2Н), 2.64 (м, 1Н), 2.95 (м, 1Н), 4.07 (м, J=3.42 Гц, 1Н), 4.24 (с, 1Н), 4.54 (м, 2Н), 5.12 (д, J=10.52 Гц, 1H), 5.30 (д, J=17.12 Гц, 1H), 5.76 (м, 1H), 5.91 (с, 1H), 7.39 (д, J=1.47 Гц, 1H), 7.68 (м, 2Н), 7.96 (д, J=1.96 Гц, 1H), 8.12 (м, 2Н); MS: (M+H)+724.
Пример 269: Получение Соединения 269

Схема 2

Стадия 1:
Раствор 2-трифторметоксикоричной кислоты (11.6 г), дифенилфосфорилазида (13.75 г) и триэтиламина (7.07 г) в бензоле (50 мл) перемешивают в течение 1 час. Фильтруют через слой силикагеля, промывают бензолом, упаривают, остаток растворяют в дифенилметане (80 мл) и кипятят 3 час. По охлаждении до rt осадок отфильтровывают, промывают бензолом и сушат, получают 5.1 г (44%) нужного продукта в виде твердого вещества.1H ЯМР (400 МГц, CD3OD) δ м.д. 6.79 (д, J=7.3 Гц, 1H), 7.29 (д, J=7.3 Гц, 1H), 7.57 (т, J=8.1 Гц, 1H), 7.70 (д, J=7.8 Гц, 1H), 8.30 (д, J=8.1 Гц, 1H); MS: (M+H)+ 230.
Стадия 2:
Раствор 5-трифторметокси-2Н-изохинолин-1-она (4.58 г) в POCl3 (50 мл) кипятят 3 час. После охлаждения и упаривания остаток подщелачивают, прибавляя 5N NaOH, и экстрагируют СН2Cl2. Органические вытяжки сушат MgSO4. Упаривают, очищают флеш-хроматографией на системе Biotage, элюируя 5% этилацетатом в смеси гексанов, получают 4.347 г (88%) нужного продукта в виде твердого вещества.1H ЯМР (400 МГц, CDCl3) δ м.д. 7.66 (м, 2Н), 7.87 (д, J=5.9 Гц, 1Н), 8.31 (м, 1Н), 8.37 (д, J=5.9 Гц, 1Н); MS: (М+H)+ 248.
Стадия 3:
К суспензии трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты (56 мг), 1-хлор-5-трифторметоксиизохинолина (25 мг) и LaCl3 (25 мг) в ДМФА (1 мл) при -78°С прибавляют трет-BuOK (0.5 мл, 1М в ТГФ) и доводят до rt. Перемешивают 30 мин, к реакционной смеси прибавляют насыщенный раствор NH4 Cl и экстрагируют этилацетатом. Упаривают, очищают преп. ВЭЖХ, получают 35 мг (46%) нужного Соединения 269 в виде твердого вещества.1H ЯМР (400 МГц, CD3OD) δ м.д. 1.03 (м, 12Н), 1.24 (м, 10H), 1.44 (дд, J=9.7, 5.3 Гц, 1Н), 1.88 (дд, J=8.1, 5.6 Гц, 1Н), 2.28 (м, 2Н), 2.64 (дд, J=13.7, 7.1 Гц, 1Н), 2.94 (м, 1Н), 4.09 (м, 1Н), 4.21 (д, J=9.3 Гц, 1Н), 4.53 (м, 2Н). 5.12 (д, J=11.5 Гц, 1Н), 5.30 (д, J=17.1 Гц, 1Н), 5.75 (м, 1Н), 5.92 (м, 1Н), 6.60 (д, J=9.5 Гц, 1Н), 7.49 (д, J=6.1 Гц, 1Н), 7.60 (м, 1Н), 7.69 (д, J=7.3 Гц, 1Н), 8.11 (д, J=6.1 Гц, 1Н), 8.22 (д, J=8.3 Гц, 1Н); MS: (M+Na)+ 790.
Пример 270: Получение Соединения 270

Соединение 270 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 2-трифторметилкоричную кислоту.
Стадия 1:
Модификации: берут 10 г 2-трифторметилкоричной кислоты, получают 5 г (50%) продукта
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 6.83 (м, 1Н), 7.33 (д, J=7.58 Гц, 1Н), 7.63 (т, J=7.83 Гц, 1Н), 8.09 (д, J=7.58 Гц, 1Н), 8.57 (д, J=8.07 Гц, 1Н).
Стадия 2:
Модификации: берут 4.4 г 5-трифторметил-2Н-изохинолин-1-она, получают 3.5 г продукта (выход 73%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 7.75 (т, J=7.95 Гц, 1Н,), 7.90 (м, 1Н), 8.12 (д, J=7.34 Гц, 1Н), 8.41 (д, J=6.11 Гц, 1Н), 8.60 (д, J=8.56 Гц, 1Н).
Стадия 3:
Модификации: берут 46 мг 1-хлор-5-трифторметилизохинолина и 111 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2, 2-диметилпропил}-карбаминовой кислоты, получают 70 мг продукта (выход 47%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.23 (м, 10H), 1.44 (дд, J=9.54, 5.38 Гц, 1Н), 1.88 (дд, J=8.07, 5.38 Гц, 1Н), 2.28 (м, 2Н), 2.65 (дд, J=13.82, 6.97 Гц, 1Н), 2.94 (м, 1Н), 4.07 (м, 1Н), 4.20 (м, 1Н), 4.56 (м, 2Н), 5.12 (м, 1Н), 5.30 (д, J=17.12 Гц, 1Н), 5.75 (м, 1Н), 5.90 (с, 1Н), 6.59 (д, J=9.05 Гц, 1Н), 7.53 (д, J=4.40 Гц, 1Н), 7.65 (т, J=7.83 Гц, 1Н), 8.12 (д, J=7.09 Гц, 1Н), 8.15 (д, J=6.36 Гц, 1Н), 8.50 (д, J=8.31 Гц, 1Н); MS: (M+Na)+ 774.
Пример 271: Получение Соединения 271

Соединение 271 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 2-хлоркоричную кислоту.
Стадия 1:
Модификации: берут 7 г 2-хлоркоричной кислоты, получают 5 г (71%) продукта
Продукт:
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 3.02 (м, 4Н), 3.91 (м, 4Н), 6.97 (д, J=7.34 Гц, 1Н), 7.18 (д, J=7.34 Гц, 1Н), 7.44 (м, 2Н), 8.02 (д, J=7.83 Гц, 1Н); MS (М+H)+ 231.
Стадия 2:
Модификации: берут 2.2 г 5-морфолин-4-ил-2Н-изохинолин-1-она, получают 2.1 г продукта (выход 87%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 3.09 (м, 4Н), 3.97 (м, 4Н), 7.32 (д, J=7.58 Гц, 1Н), 7.60 (м, 1Н), 7.91 (д, J=5.87 Гц, 1Н), 8.06 (д, J=8.56 Гц, 1Н), 8.26 (д, J=5.87 Гц, 1Н).
Стадия 3:
Модификации: берут 50 мг 1-хлор-5-морфолин-4-илизохинолина и 111 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2, 2-диметилпропил}-карбаминовой кислоты, получают 40 мг продукта (выход 26%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.07 (м, 12Н), 1.26 (м, 10H), 1.44 (д, J=7.93 Гц, 1Н), 1.88 (дд, J=7.93, 5.19 Гц, 1Н), 2.25 (м, 2Н), 2.62 (дд, J=13.73, 7.02 Гц, 1Н), 2.94 (м, 1Н), 3.06 (д, J=3.97 Гц, 4Н), 3.94 (м, 4Н), 4.07 (д, J=14.04 Гц, 1Н), 4.25 (с, 1Н), 4.45 (д, J=12.21 Гц, 1Н), 4.52 (м, 1Н), 5.12 (д, J=9.46 Гц, 1Н), 5.29 (д, J=16.79 Гц, 1Н), 5.75 (м, 1Н), 5.85 (с, 1Н), 7.34 (д, J=7.32 Гц, 1Н), 7.45 (с, J=7.78 Гц, 1Н), 7.59 (д, J=6.10 Гц, 1Н), 7.91 (д, J=7.63 Гц, 1Н), 7.97 (д, J=5.80 Гц, 1Н).
Пример 272: Получение Соединения 272

Соединение 272 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 2,3-диметоксикоричную кислоту.
Стадия 1:
Модификации: берут 10.4 г 2,3-диметоксикоричной кислоты, получают 5 г (40%) продукта
Продукт:
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 3.86 (с, 3H), 3.96 (с, 3H). 6.82 (д, J=7.2 Гц, 1Н), 7.10 (д, J=7.2 Гц, 1Н), 7.28 (д, J=8.8 Гц, 1Н), 8.07 (д, J=8.8 Гц, 1Н); MS: (М+H)+ 206.
Стадия 2:
Модификации: берут 4.1 г 5,6-диметокси-2Н-изохинолин-1-она, получают 4.03 г продукта (выход 90%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 3.97 (с, 3H), 4.05 (с, 3H), 7.65 (д, J=9.29 Гц, 1Н), 7.90 (дд, J=5.87, 0.98 Гц, 1Н), 8.12 (м, 2Н).
Стадия 3:
Модификации: берут 22 мг 1-хлор-5,6-диметоксиизохинолина и 56 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 31 мг продукта (выход 42%).
Продукт:

Данные:1H ЯМР (500 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.26 (м, 10H), 1.44 (с, 1Н), 1.88 (д, J=7.32 Гц, 1Н), 2.24 (с, 2Н), 2.60 (м, 1Н), 2.94 (м, 1Н), 3.92 (с, 3H), 3.99 (с, 3H), 4.06 (д, J=11.90 Гц, 1Н), 4.23 (с, 1Н), 4.43 (д, J=10.68 Гц, 1Н), 4.53 (м, 1Н), 5.12 (д, J=10.38 Гц, 1Н), 5.30 (д, J=17.40 Гц, 1Н), 5.77 (м, 2Н), 7.35 (д, J=9.16, 1Н), 7.46 (д, J=5.80 Гц, 1Н), 7.89 (д, J=5.80 Гц, 1Н), 7.97 (д, J=8.85 Гц, 1Н).
Получение Соединения 273
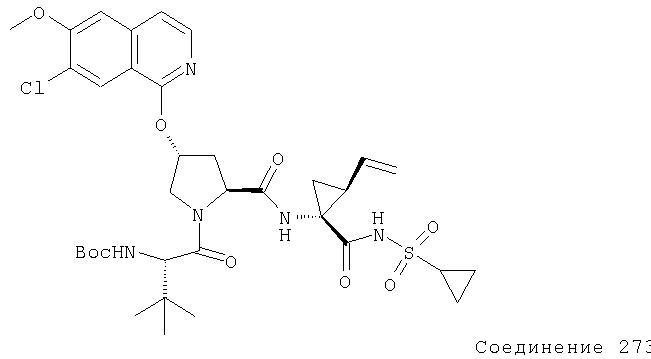
Соединение 273 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 4-хлор-3-метоксикоричную кислоту.
Стадия 1:
Модификации: берут 2.5 г 4-хлор-3-метоксикоричной кислоты, получают 1.2 г (48%) продукта
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 4.00 (с, 3H), 6.64 (д, J=7.09 Гц, 1Н), 7.15 (д, J=7.34 Гц, 1Н), 7.21 (с, 1Н), 8.22 (с, 1Н).
Стадия 2:
Модификации: берут 1.05 г 7-хлор-6-метокси-2Н-изохинолин-1-она, получают 0.8 г продукта (выход 70%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.05 (с, 3H), 7.13 (с, 1Н), 7.48 (д, J=5.38 Гц, 1Н), 8.21 (д, J=5.62 Гц, 1Н), 8.34 (с, 1Н); MS: (M+H)+ 229.
Стадия 3:
Модификации: берут 44 мг 1, 7-дихлор-6-метоксиизохинолина и 113 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2, 2-диметилпропил}-карбаминовой кислоты, получают 25 мг продукта (выход 17%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.07 (м, 12Н), 1.24 (м, 10H), 1.44 (дд, J=9.54, 5.38 Гц, 1Н), 1.88 (дд, J=8.07, 5.38 Гц, 1Н), 2.26 (м, 1Н,), 2.60 (м, J=13.69, 6.85 Гц, 1Н), 2.94 (м, 2Н), 3.98 (с, 3H), 4.06 (м, 1Н), 4.20 (м, 1Н), 4.42 (д, J=12.23 Гц, 1Н), 4.57 (м, 1Н), 5.12 (д, J=11.74 Гц, 1Н), 5.30 (д, J=17.36 Гц, 1Н), 5.76 (м, 1Н), 5.86(с, 1Н), 7.28 (д, J=5.62 Гц, 1Н), 7.33 (с, 1Н), 7.92 (д, J=5.87 Гц, 1Н), 8.09 (с, 1Н); MS: (M+H)+ 749.
Пример 274: Получение Соединения 274
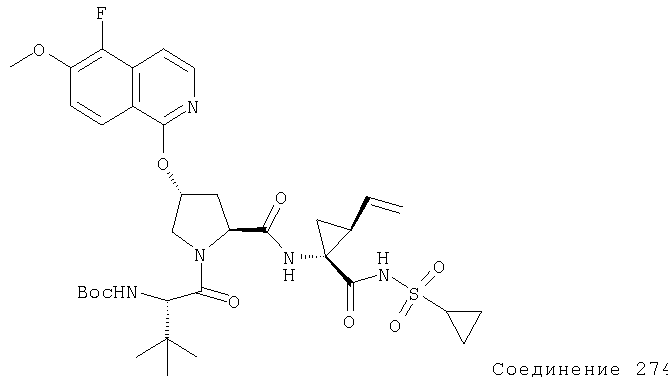
Соединение 274 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 2-фтор-3-метоксикоричную кислоту.
Стадия 1:
Модификации: берут 3.92 г 2-фтор-3-метоксикоричной кислоты, получают 2.4 г (61%) продукта
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 4.00 (ч, 3H), 6.72 (м, 1Н), 7.16 (д, J=7.34 Гц, 1Н), 7.35 (т, J=8.44 Гц, 1Н), 8.09 (д, J=8.80 Гц, 1Н).
Стадия 2:
Модификации: берут 1.93 г 5-фтор-6-метокси-2Н-изохинолин-1-она, получают 1.688 г продукта (выход 80%).
Продукт:
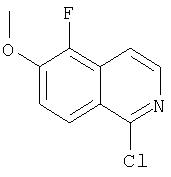
Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.08 (с, 3H), 7.44 (дд, J=9.29, 7.83 Гц, 1Н), 7.75 (д, J=5.87 Гц, 1Н), 8.12 (д, J=9.29 Гц, 1Н), 8.22 (д, J=5.87 Гц, 1Н); MS: (M+H)+ 212.
Стадия 3:
Модификации: берут 41 мг 1-хлор-5-фтор-6-метоксиизохинолина и 133 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 70 мг продукта (выход 48%).
Продукт:
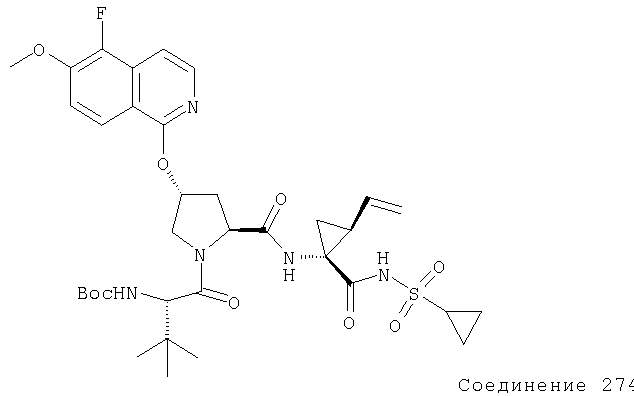
Данные:1H ЯМР (400 МГц, CDOD) δ м.д. 1.06 (м, 13Н), 1.21 (с, 9Н), 1.44 (дд, J=9.78, 5.38 Гц, 1Н), 1.88 (дд, J=8.19, 5.50 Гц, 1Н), 2.24 (д, J=9.29 Гц, 2Н), 2.62 (д, J=13.94 Гц, 1Н), 2.94 (м, 1Н), 4.05 (м, 4Н), 4.22 (д, J=9.29 Гц, 1Н), 4.45 (м, 1Н), 4.54 (дд, J=9.66, 7.21 Гц, 1Н), 5.12 (д, J=10.52 Гц, 1Н), 5.30 (д, J=16.87 Гц, 1Н), 5.76 (м, 1Н), 5.86 (с, 1Н), 7.39 (м, 2Н), 7.95 (д, J=6.11 Гц, 1Н), 8.00 (д, J=9.29 Гц, 1Н).
Пример 275: Получение Соединения 275
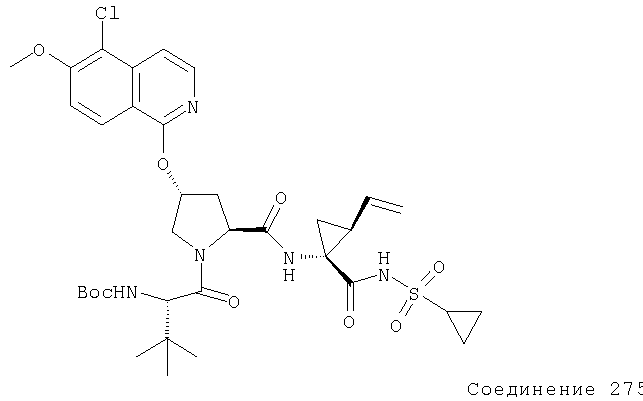
Соединение 275 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 2-хлор-3-метоксикоричную кислоту.
Стадия 1:
Модификации: берут 658 мг 2-хлор-3-метоксикоричной кислоты, получают 360 мг (54%) продукта
Продукт:
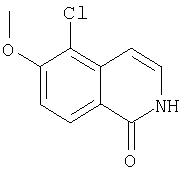
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 4.02 (с, 3H), 6.91 (д, J=7.34 Гц, 1Н), 7.23 (д, J=7.58 Гц, 1Н), 7.35 (д, J=9.05 Гц, 1Н), 8.27 (д, J=9.05 Гц, 1Н).
Стадия 2:
Модификации: берут 350 мг 5-хлор-6-метокси-2Н-изохинолин-1-она, получают 300 мг продукта (выход 80%).
Продукт:
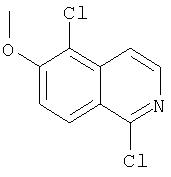
Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.09 (с, 3H), 7.43 (д, J=9.29 Гц, 1Н), 7.93 (д, J=6.11 Гц, 1Н), 8.30 (м, 2Н); MS (М+Н)+ 229.
Стадия 3:
Модификации: берут 68 мг 1, 5-дихлор-6-метоксиизохинолина и 167 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2, 2-диметилпропил}-карбаминовой кислоты, получают 130 мг продукта (выход 60%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.25 (м, 10H), 1.46 (д, J=5.62 Гц, 1Н), 1.88 (дд, J=8.07, 5.62 Гц, 1Н), 2.27 (м, 2Н), 2.62 (м, 1Н), 2.94 (м, 1Н), 4.05 (м, 4Н), 4.22 (д, J=9.05 Гц, 1Н), 4.46 (д, J=11.49 Гц, 1Н), 4.54 (дд, J=9.78, 6.36 Гц, 1Н), 5.13 (д, J=10.52 Гц, 1Н), 5.30 (д, J=15.89 Гц, 1Н), 5.76 (м, 1Н), 5.86 (с, 1Н), 7.40 (д, J=9.29 Гц, 1Н), 7.55 (д, J=6.36 Гц, 1Н), 8.01 (д, J=6.36 Гц, 1Н), 8.20 (д, J=9.29 Гц, 1H); MS: (M+H)+ 749.
Пример 276: Получение Соединения 276

Соединение 276 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-хлор-2-метоксикоричную кислоту.
Стадия 1:
Модификации: берут 4.24 г 3-хлор-2-метоксикоричной кислоты, получают 2.4 г (57%) продукта
Продукт:
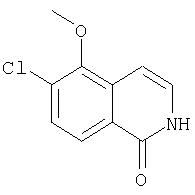
Данные:1 H ЯМР (400 МГц, CD3OD) δ м.д. 3.93 (с, 1Н), 6.85 (д, J=7.34 Гц, 1Н), 7.24 (д, J=7.34 Гц, 1Н), 7.52 (д, J=8.80 Гц, 1Н), 8.03 (д, J=8.80 Гц, 1Н); MS: (M+H)+ 210.
Стадия 2:
Модификации: берут 2.09 г 6-хлор-5-метокси-2Н-изохинолин-1-она, получают 1.9 г продукта (выход 83%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.03 (с, 2Н), 7.63 (д, J=9.05 Гц, 1Н), 7.86 (д, J=5.14 Гц, 1Н), 8.06 (д, J=9.05 Гц, 1Н), 8.32 (д, J=5.62 Гц, 1Н); MS: (М+Н)+ 229.
Стадия 3:
Модификации: берут 91 мг 1, 6-дихлор-5-метоксиизохинолина и 226 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2, 2-диметилпропил}-карбаминовой кислоты, получают 114 мг продукта (выход 38%).
Продукт:
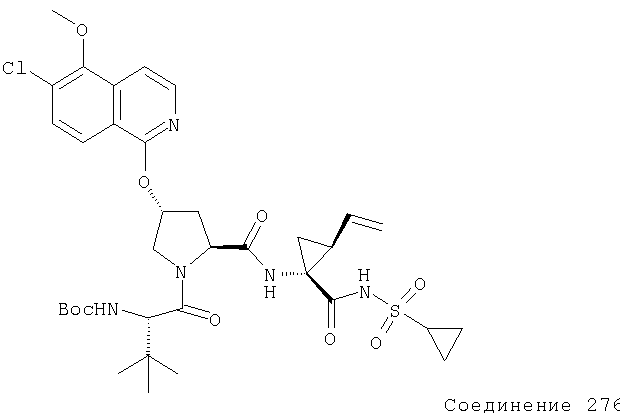
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.23 (м, 10H), 1.44 (т, J=6.72 Гц, 1Н), 1.88 (дд, J=7.95, 5.26 Гц, 1Н), 2.25 (м, 2Н), 2.62 (дд, J=13.33, 6.48 Гц, 1Н), 2.94 (м, 1Н), 3.98 (с, 3H), 4.03 (м, 1Н), 4.20 (м, 1Н), 4.51 (м, 2Н), 5 12 (д, J=10.52 Гц, 1Н), 5.32 (с, 1Н), 5.75 (м, 1Н), 5.87 (с, 1Н), 7.50 (м, 2Н), 7.95 (м, J=8.80 Гц, 1Н), 8.06 (д, J=5.87 Гц, 1Н); MS (MH+) 749.
Пример 277: Получение Соединения 277
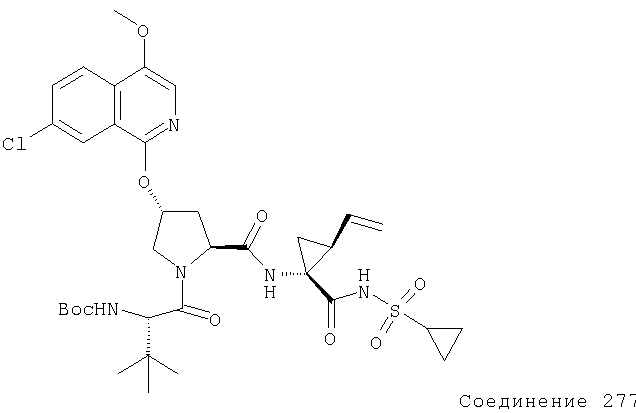
Соединение 277 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-(4-хлорфенил)-3-метоксиакриловую кислоту.
Стадия 1:
Модификации: берут 4.24 г 3-(4-хлорфенил)-3-метоксиакриловой кислоты, получают 130 мг (3%) продукта
Продукт:
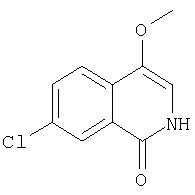
Данные:1Н ЯМР (400 МГц, CD3OD) δ м.д. 3.96 (с, 3Н), 7.19 (дд, J=8.80, 2.45 Гц, 1Н), 7.28 (д, J=2.45 Гц, 1Н), 7.34 (с, 1Н), 8.25 (д, J=9.05 Гц, 1Н); MS: (М+Н)+ 210.
Стадия 2:
Модификации: берут 105 мг 7-хлор-4-метокси-2Н-изохинолин-1-она, получают 60 мг продукта (выход 71%).
Продукт:

Данные:1Н ЯМР (400 МГц, CDCl3) δ м.д. 4.05 (с, 3Н), 7.67 (дд, J=8.80, 1.90 Гц, 1Н), 7.80 (с, 1Н), 8.16(д, J=9.05 Гц, 1Н), 8.24 (д, J=1.96 Гц, 1Н); MS: (M+H)+ 229.
Стадия 3:
Модификации: берут 46 мг 1,7-дихлор-4-метоксиизохинолина и 113 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 58 мг продукта (выход 31%).
Продукт:
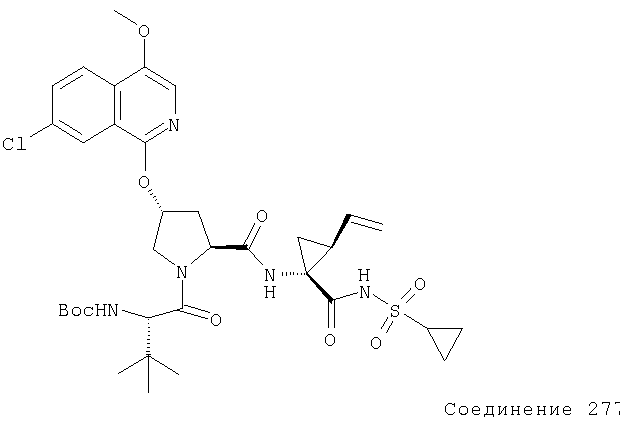
Данные:1Н ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 11Н), 1.16 (с, 9Н), 1.24 (м, 2Н), 1.44 (дд, J=9.54,5.38 Гц, 1Н), 1.88 (дд, J=8.07, 5.62 Гц, 1Н), 2.28 (м, 2Н), 2.59 (дд, J=13.69, 6.85 Гц, 1Н), 2.94 (м, 1Н), 4.00 (с, 3Н), 4.05 (д, J=11.74 Гц, 1Н), 4.19 (с, 1Н), 4.43 (д, J=1.49 Гц, 1Н). 4.56 (дд, J=10.03, 6.85 Гц, 1Н), 5.12 (д, J=11.49 Гц, 1Н), 5.30 (д, J=17.12 Гц, 1Н), 5.76 (м, 2Н), 7.57 (с, 1Н), 7.67 (д, J=8.56 Гц, 1Н), 8.04 (с, 1Н), 8.08 (д, J=8.80 Гц, 1Н); MS: (M+H)+ 748.
Пример 278: Получение Соединения 278
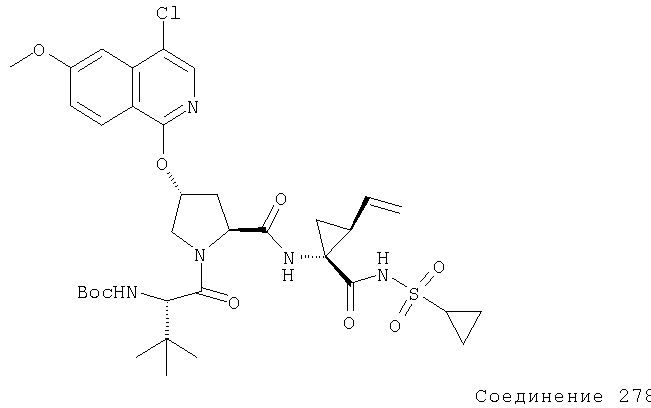
Соединение 278 получают по Схеме 2 из Примера 269 за исключением Стадии 1.
Стадия 1:
Модификации: Смесь 6-метокси-2Н-изохинолин-1-она (700 мг) и NCS (532 мг) в MeCN (10 мл) кипятят 3 час. Фильтрованием получают 600 мг (72%) нужного продукта в виде твердого вещества.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 3.96 (с, 1Н), 7.19 (дд, J=8.80,2.45 Гц, 1Н), 7.28 (д, J=2.45 Гц, 1Н), 7.34 (с, 1Н), 8.25 (д, J=9.05 Гц, 1Н); MS: (M+H)+ 210.
Стадия 2:
Модификации: берут 500 мг 4-хлор-4-метокси-2Н-изохинолин-1-она, получают 400 мг продукта.
Продукт:
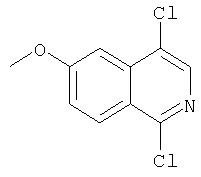
Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.01 (с, 3H), 7.35 (д, J=2.45 Гц, 1Н), 7.41 (д, J=2.45 Гц, 1Н), 8.24 (д, J=9.29 Гц, 1Н), 8.27 (с, 1Н); MS: (M+H)+ 229.
Стадия 3:
Модификации: берут 42 мг 1,4-дихлор-6-метоксиизохинолина и 117 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 70 мг продукта (выход 47%).
Продукт:
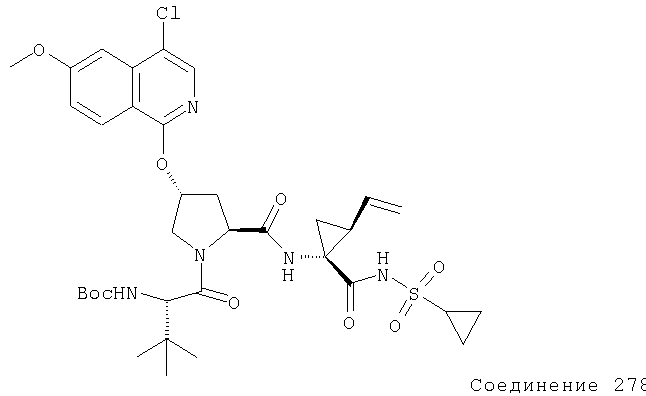
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 12Н), 1.25 (м, 10H), 1.44 (м, 1Н), 1.88 (дд, J=8.07, 5.62 Гц, 1Н), 2.24 (м, 2Н), 2.61 (дд, J=13.82, 6.72 Гц, 1Н), 2.94 (м, 1Н), 3.97 (с, 3H), 4.04 (дд, J=11.74,2.69 Гц, 1Н), 4.21 (с, 1Н), 4.49 (м, 2Н), 5.12 (д, J=10.52 Гц, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.75 (м, 2Н), 7.19 (д, J=8.80 Гц, 1Н), 7.37 (с, 1Н), 8.00 (с, 1Н), 8.13 (д, J=9.05 Гц, 1Н); MS: (M+H)+ 749.
Пример 279: Получение Соединения 279
Соединение 279 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-(3-метоксифенил)-акриловую кислоту.
Стадия 1:
Модификации: берут 4.24 г 3-(3-метоксифенил)-акриловой кислоты, получают 400 мг (10%) продукта
Продукт:

Стадия 2:
Модификации: берут 500 мг 4, 6-диметокси-2Н-изохинолин-1-она, получают 300 мг продукта (выход 69%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 3.97 (с, 3H), 4.05 (с, 3H), 7.31 (дд, J=9.17, 2.57 Гц, 1Н), 7.45 (д, J=2.69 Гц, 1Н), 7.75 (с, 1Н), 8.16 (д, J=9.29 Гц, 1Н); MS: (M+H)+ 224.
Стадия 3:
Модификации: берут 89 мг 1-хлор-4,6-диметоксиизохинолина и 223 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 160 мг продукта (выход 54%).
Продукт:
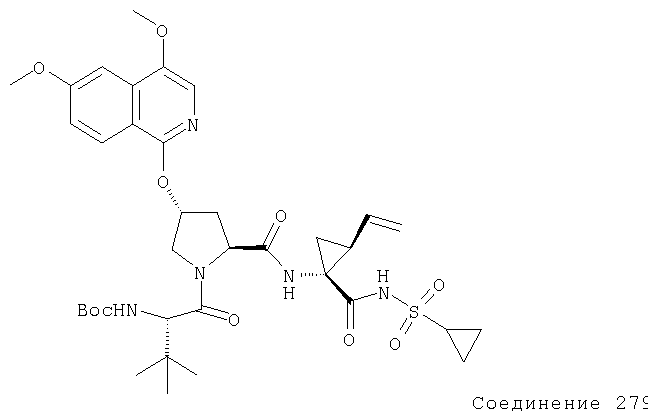
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.07 (м, 12Н), 1.21 (м, 10H), 1.43 (м, 1Н), 1.87 (дд, J=8.07, 5.62 Гц, 1Н), 2.24 (м, 2Н), 2.58 (дд, J=13.57, 6.97 Гц, 1Н), 2.94 (м, 1Н), 3.92 (с, 3H), 3.99 (с, 3H), 4.04 (дд, J=11.74, 2.93 Гц, 1Н), 4.24 (с, 1Н), 4.39 (д, J=11.98 Гц, 1Н), 4.50 (м, 1Н), 5.12 (д, J=10.52 Гц, 1Н), 5.29 (д, J=16.87 Гц, 1Н), 5.75 (м, 2Н), 7.12 (д, J=9.05 Гц, 1Н), 7.40 (д, J=2.20 Гц, 1Н), 7.48 (с, 1Н), 8.04 (д, J=9.05 Гц, 1Н); MS: (М+Н)+ 744.
Пример 280: Получение Соединения 280
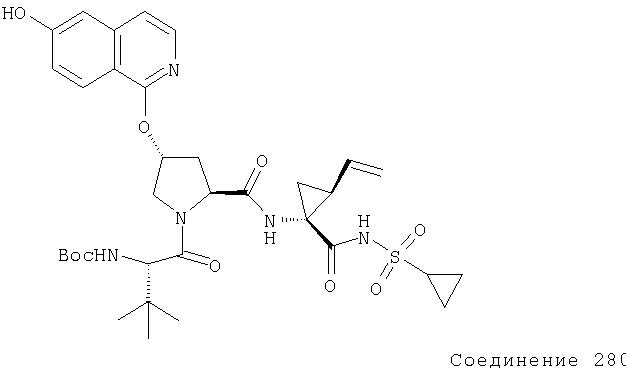
Соединение 280 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-(3-дифторметоксифенил)-акриловую кислоту.
Стадия 1:
Модификации: берут 4.28 г 3-(3-дифторметоксифенил)-акриловой кислоты, получают 3.1 мг (72%) продукта
Продукт:

Данные: MS: (М+Н)+ 212.
Стадия 2:
Модификации: берут 2 г 6-дифторметокси-2Н-изохинолин-1-она, получают 1.5 г продукта (выход 61%).
Продукт:
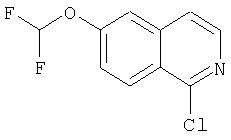
Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 6.69 (т, J=72.75 Гц, 1Н), 7.49 (м, 2Н), 8.28 (д, J=5.62 Гц, 1Н), 8.36 (д, J=9.05 Гц, 1Н); MS: (M+H)+ 230.
Стадия 3:
Модификации: берут 46 мг 1-хлор-6-дифторметоксиизохинолина и 113 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 8 мг продукта (выход 5%).
Продукт:
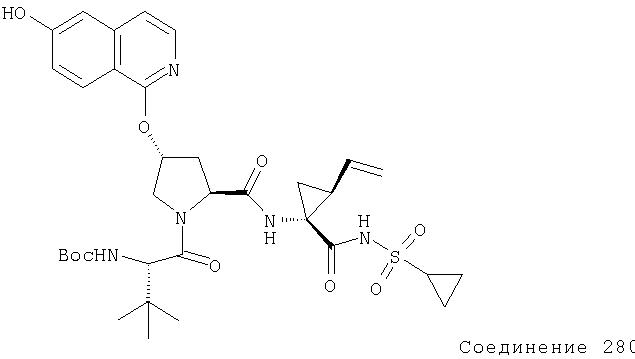
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 12Н), 1.23 (м, 10H), 1.44 (м, 2Н), 1.88 (дд, J=8.19. 5.50 Гц, 1Н), 2.30 (м, 2Н), 2.67 (д, J=13.94 Гц, 1Н), 2.93 (м, 1Н), 4.07 (д, J=10.27 Гц, 1Н), 4.21 (с, 1Н), 4.53 (д, J=6.85 Гц, 2Н), 5.13 (м, 1Н), 5.31 (с, 1Н), 5.76 (д, J=47.93 Гц, 2Н), 7.11 (м, 2Н), 7.26 (д, J=6.11 Гц, 1Н), 7.81 (д, J=6.11 Гц, 1Н), 8.16 (м, 1Н); MS: (M+H)+ 700.
Пример 281: Получение Соединения 281

Соединение 281 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-хлор-3-фенилакриловую кислоту.
Стадия 1:
Модификации: берут 11 г 3-хлор-3-фенилакриловой кислоты, получают 3.1 мг (29%) продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 7.34 (с, 1Н), 7.52 (т, J=7.58 Гц, 1Н), 7.77 (т, J=7.46 Гц, 1Н), 7.90 (д, J=8.07 Гц, 1Н), 8.39 (д, J=8.07 Гц, 1Н), 11.37 (с, 1Н); MS: (М+Н)+ 180.
Стадия 2:
Модификации: берут 3.1 г 4-хлор-2Н-изохинолин-1-она, получают 2.3 г продукта (выход 66%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 7.77 (ддд, J=8.31, 7.09, 1.22 Гц, 1Н), 7.88 (ддд, J=8.31, 7.09, 1.22 Гц, 1Н), 8.23 (д, J=8.31 Гц, 1Н), 8.34 (с, 1Н), 8.36 (д, J=8.56 Гц, 1Н); MS: (M+H)+ 198.
Стадия 3:
Модификации: берут 20 мг 1,4-дихлоризохинолина и 56 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 33 мг продукта (выход 30%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.24 (м, 10H), 1.44 (дд, J=9.41, 5.26 Гц, 1Н), 1.88 (дд, J=7.83, 5.62 Гц, 1Н), 2.27 (м, 2Н), 2.63 (дд, J=13.82, 6.97 Гц, 1Н), 2.94 (м, 1Н), 4.06 (дд, J=11.49, 2.45 Гц, 1Н), 4.22 (д, J=9.29 Гц, 1Н), 4.53 (м. 2Н), 5.12 (д, J=10.76 Гц, 1Н), 5.29.(д, J=17.12 Гц, 1Н), 5.75 (м, 1Н), 5.85 (с, 1Н), 6.60 (д, J=8.80 Гц, 1Н), 7.63 (т, J=7.58 Гц, 1Н), 7.86 (т, J=7.70 Гц, 1Н), 8.06 (с, 1Н), 8.11 (д, J=8.56 Гц, 1Н), 8.25 (д, J=8.31 Гц, 1Н); MS: (M+H)+ 718.
Пример 282: Получение Соединения 282

Соединение 282 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-трифторметоксикоричную кислоту.
Стадия 1:
Модификации: берут 20 г 3-трифторметоксикоричной кислоты, получают 2 г (8%) продукта.
Продукт:

Данные: MS: (M+H)+ 230.
Стадия 2:
Модификации: берут 2 г 6-трифторметокси-2Н-изохинолин-1-она, получают 0.7 г продукта (выход 33%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 7.51 (д, J=9.29 Гц, 1Н), 7.59 (д, J=5.62 Гц, 1Н), 7.64 (с, 1Н), 8.31 (д, J=5.62 Гц, 1Н), 8.40 (д, J=9.05 Гц, 1Н); MS: (M+H)+ 248.
Стадия 3:
Модификации: берут 50 мг 1-хлор-6-трифторметоксиизохинолина и 113 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 42 мг продукта (выход 27%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 12Н), 1.24 (м, 10H), 1.44 (дд, J=9.17, 5.50 Гц, 1Н), 1.88 (дд, J=8.07, 5.62 Гц, 1Н), 2.28 (м, 2Н), 2.63 (дд, J=13.45, 7.09 Гц, 1Н), 2.94 (м, 1Н), 4.06 (дд, J=11.25, 2.45 Гц, 1Н), 4.21 (с, 1Н), 4.53 (м, 2Н), 5.13 (д, J=10.52 Гц, 1Н), 5.30 (д, J=17.12 Гц, 1Н), 5.75 (м, 1Н), 5.89 (с, 1Н), 7.39 (м, 2Н), 7.72 (с, 1Н), 8.05 (д, J=5.87 Гц, 1Н), 8.31 (д, J=9.05 Гц, 1Н), 9.18 (с, 1Н); MS: (M+H)+ 768.
Пример 283: Получение Соединения 283

Соединение 283 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-(4-фторфенил)-3-метоксиакриловую кислоту.
Стадия 1:
Модификации: берут 3.82 г 3-(4-фторфенил)-3-метоксиакриловой кислоты, получают 198 мг (5%) продукта.
Продукт:
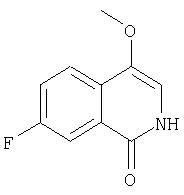
Данные: MS: (M+H)+ 194.
Стадия 2:
Модификации: берут 193 мг 7-фтор-4-метокси-2Н-изохинолин-1-она, получают 199 мг продукта (выход 94%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.05 (с, 3H), 7.49 (м, 1Н), 7.78 (с, 1Н). 7.86 (дд, J=9.66,2.57 Гц, 1Н), 8.23 (дд, J=9.29, 5.38 Гц, 1Н); MS: (М+Н)+ 212.
Стадия 3:
Модификации: берут 42 мг 1-хлор-7-фтор-4-метоксиизохинолина и 112 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 40 мг продукта (выход 14%).
Продукт:

Пример 284: Получение Соединения 284
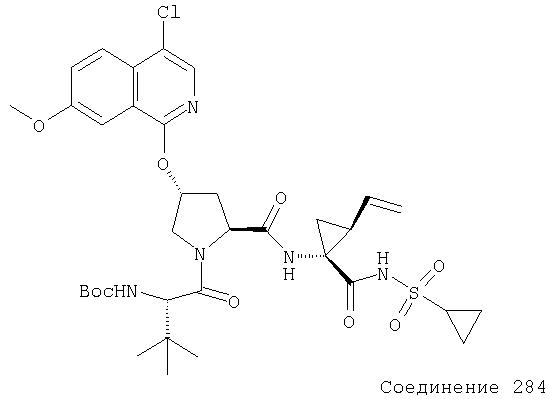
Соединение 284 получают по Схеме 2 из Примера 269 за исключением Стадии 1.
Стадия 1:
Модификации: Смесь 7-метокси-2Н-изохинолин-1-она (876 мг) и NCS (665 мг) в MeCN (10 мл) кипятят 3 час. Фильтрованием получают 500 мг (47%) нужного продукта в виде твердого вещества.
Продукт:
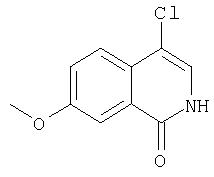
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 4.00 (с, 3H), 7.58 (м, 2Н), 8.14 (д, J=10.03 Гц, 1Н), 8.17 (с, 1Н).
Стадия 2:
Модификации: берут 418 мг 4-хлор-7-метокси-2Н-изохинолин-1-она, получают 410 мг продукта (выход 90%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.00 (с, 3H), 7.49 (дд, J=9.16, 2.44 Гц, 1Н), 7.55 (д, J=2.44 Гц, 1Н), 8.12 (д, J=9.16 Гц, 1Н), 8.21 (с, 1Н); MS: (M+H)+ 229.
Стадия 3:
Модификации: берут 42 мг 1,4-дихлор-7-метоксиизохинолина и 117 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 50 мг продукта (выход 33%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (м, 20Н), 1.24 (м, 2Н), 1.44 (м, 1Н), 1.89 (дд, J=8.19, 5.50 Гц, 1Н), 2.28 (м, 2Н), 2.62 (дд, J=13.69, 6.85 Гц, 1Н), 2.94 (м, 1Н), 3.92 (с, 3H), 4.07 (дд, J=11.98, 3.42 Гц, 1Н), 4.19 (м, 1Н), 4.44 (д, J=11.74 Гц, 1Н), 4.58 (дд, J=10.27, 7.09 Гц, 1Н), 5.12 (м, 1Н), 5.31 (д, J=17.12 Гц, 1Н), 5.78 (м. 2Н), 7.49 (м, 2Н), 7.91 (с, 1Н), 8.02 (м, 1Н); MS: (M+H)+ 749.
Пример 285: Получение Соединения 285
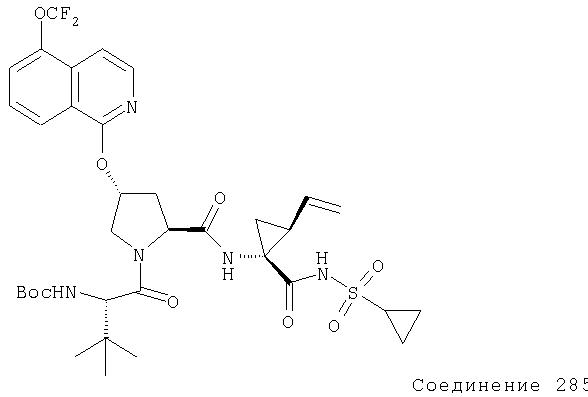
Соединение 285 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут другой реагент.
Стадия 1 и Стадия 2:
См. Соединение 256.
Стадия 3:
Модификации: берут 46 мг 1-хлор-5-дифторметоксиизохинолина и 111 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 40 мг продукта (выход 27%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.25 (м, 10H), 1.44 (м, 1Н, 1.89 (м, 1Н), 2.22 (м, 2Н), 2.62 (м, 1Н), 2,94 (м, 1Н), 4.08 (м, 1Н), 4.23 (д, J=9.54 Гц, 1Н), 4.52 (м, 2Н), 5.12 (д, J=10.76 Гц, 1Н), 5.29 (д, J=17.61 Гц, 1Н), 5.75 (м, J=10.03 Гц, 1Н), 5.88 (с, 1Н), 6.60 (с, 1Н), 7.02 (т, J=73.48 Гц, 1Н), 7.52 (м, 3H), 8.07 (м(?), J=5.75 Гц, 2Н); MS: (M+Na)+ 772.
Пример 286: Получение Соединения 286

Соединение 286 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-(2,3-дигидробензо[1,4]диоксин-5-ил)-акриловую кислоту.
Стадия 1:
Модификации: берут 4.12 г 3-(2,3-лигидробензо[1,4]диоксин-5-ил)-акриловой кислоты, получают 2.2 мг (53%) продукта.
Продукт:
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 4.37 (м, 4Н), 6.83 (д, J=7.09 Гц, 1Н), 7.02 (д, J=8.80 Гц, 1Н), 7.12 (д, J=7,34 Гц, 1Н), 7.79 (д, J=8.80 Гц, 1Н); MS: (М+H)+ 204.
Стадия 2:
Модификации: берут 2.05 г 2,3-дигидро-7H-1,4-диокса-7-азафенантрен-8-она, получают 1.5 г продукта (выход 68%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 4.42 (м, 4Н), 7.24 (д, J=9.05 Гц, 1Н), 7.77 (д, J=5.87 Гц, 1Н), 7.84 (д, J=9.05 Гц, 1Н), 8.18 (д, J=5.87 Гц, 1Н); MS: (M+H)+ 222.
Стадия 3:
Модификации: берут 88 мг 8-хлор-2,3-дигидро-7Н-1,4-диокса-7-азафенантрена и 223 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 140 мг продукта (выход 47%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.24 (м, 10H), 1.43 (дд, J=9.05, 5.14 Гц, 1Н), 1.87 (м, 1Н), 2.22 (д, J=9.29 Гц, 2Н), 2.60 (дд, J=13.45, 7.09 Гц, 1Н), 2.94 (м, 1Н), 4.05 (дд, J=11.62, 2.81 Гц, 1Н), 4 24 (с, 1Н), 4.44 (м, 6Н), 5.13 (д, J=17.36 Гц, 1Н), 5.29 (д, J=17.36 Гц, 1Н), 5.75 (м, 2Н), 7.04 (д, J=9.29 Гц, 1Н), 7.44 (д, J=5.87 Гц, 1Н), 7.69 (д, J=9.05 Гц, 1Н), 7.88 (д, J=6.11 Гц, 1Н); MS: (M+H)+ 742.
Пример 287: Получение Соединения 287

Соединение 287 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-(2,2-дифторбензо[1,3]диоксол-4-ил)-акриловую кислоту.
Стадия 1:
Модификации: берут 4.56 г 3-(2,2-дифторбензо[1,3]диоксол-4-ил)-акриловой кислоты, получают 2.2 мг (55%) продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 6.63 (д, J=7.09 Гц, 1Н), 7.29 (д, J=7.34 Гц, 1Н), 7.40 (д, J=8.80 Гц, 1Н), 8.19 (д, J=8.80 Гц, 1Н); MS: (M+H)+ 226.
Стадия 2:
Модификации: берут 2.2 г 2,2-дифтор-7Н-1, 3-диокса-7-азациклопента[а]нафталин-6-она, получают 2.1 г продукта (выход 87%).
Продукт:

Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 7.51 (д, J=9.29 Гц, 1Н), 7.65 (д, J=5.87 Гц, 1Н), 8.22 (д, J=9.05 Гц, 1Н), 8.32 (д, J=5.87 Гц, 1Н); MS: (M+H)+ 244.
Стадия 3:
Модификации: берут 48 мг 6-хлор-2,2-дифтор-1,3-диокса-7-азациклопента[а]нафталина и 113 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 40 мг продукта (выход 27%).
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (с, 12Н), 1.24 (м, 10H), 1.43 (м, 1Н), 1.88 (дд, J=8.07, 5.38 Гц, 1Н), 2.32 (д, J=3.67 Гц, 2Н), 2.64 (д, J=13.45 Гц, 1Н), 2.95 (м,1Н). 4.05 (д, J=11.49 Гц, 1Н), 4.19 (д, J=9.29 Гц, 1Н), 4.53 (м, 2Н), 5.12 (д, J=9.78 Гц, 1Н), 5.32 (с, 1Н), 5.77 (м, 2Н), 7.34 (д, J=5.87 Гц, 1Н), 7.46 (д. J=9.05 Гц, 1Н), 8.11 (м, 2Н): MS: (M+H)+ 764.
Пример 288: Получение Соединения 288

Соединение 288 получают по Схеме 2 из Примера 269 за исключением того, что на стадии 1 вместо 2-трифторметилкоричной кислоты берут 3-(2,2-дифторбензо[1,3]диоксол-5-ил)-акриловую кислоту.
Стадия 1:
Модификации: берут 1 г 3-(2,2-дифторбензо[1,3]диоксол-5-ил)-акриловой кислоты, получают 0.55 г продукта.
Продукт:

Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 6.69 (д, J=7.09 Гц, 1Н), 7.19 (д, J=7.09 Гц, 1Н), 7.47 (с, 1Н), 7.98 (с, 1Н); MS: (M+H)+ 226.
Стадия 2:
Модификации: берут 0.5 г 2,2-дифтор-6H-[1,3]-диоксоло[4,5-g]изохинолин-5-она, получают 0.4 г продукта.
Продукт:
Данные:1H ЯМР (400 МГц, CDCl3) δ м.д. 7.41 (с, 1Н), 7.57 (д, J=5.49 Гц, 1Н), 7.94 (с, 1Н), 8.27 (д, J=5.80 Гц, 1Н); MS (M+H)+ 244.
Стадия 3:
Модификации: берут 48 мг 5-хлор-2,2-дифтор-[1,3]-диоксоло[4, 5-g]изохинолина и 112 мг трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты, получают 30 мг продукта.
Продукт:
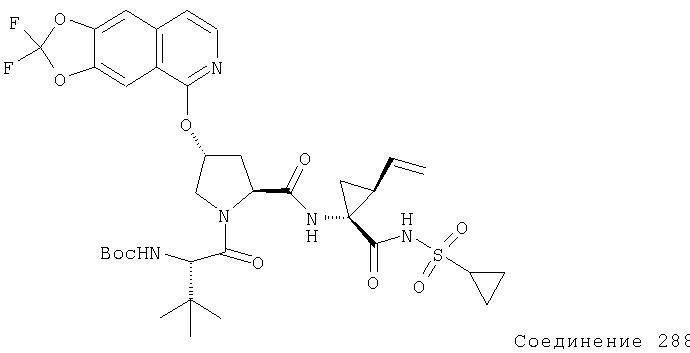
Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.06 (м, 12Н), 1.25 (м, 10H), 1,42 (м, 1Н), 1.87 (дд, J=8.07, 5.62 Гц, 1Н), 2.26 (м, 2Н), 2.61 (дд, J=13 57, 6.97 Гц, 1Н), 2.93 (м, 1Н), 4.07 (дд, J=11.86, 2.81 Гц, 1Н), 4.22 (м, 1Н), 4.40 (д, J=11.98 Гц, 1Н), 4 52 (м, 1Н), 5.11 (д, J=10.52 Гц, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.37 (с, 1Н), 5.74 (м, 1Н), 7.39 (с, 1Н), 7.56 (с, 1Н), 7.63 (д, J=5.62 Гц, 1Н), 7.99 (д, J=5.62 Гц, 1Н).
Пример 289: Получение Соединения 289
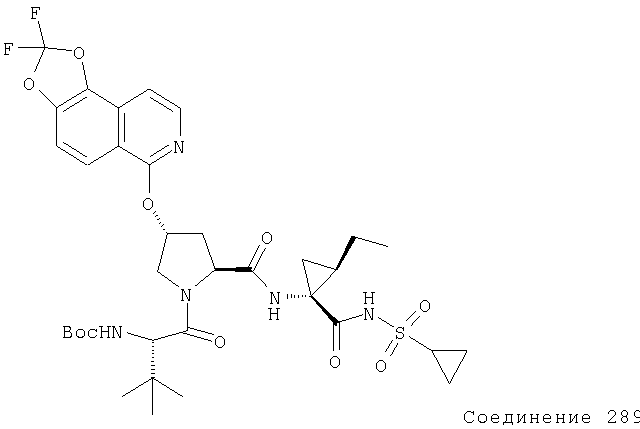
Схема 3
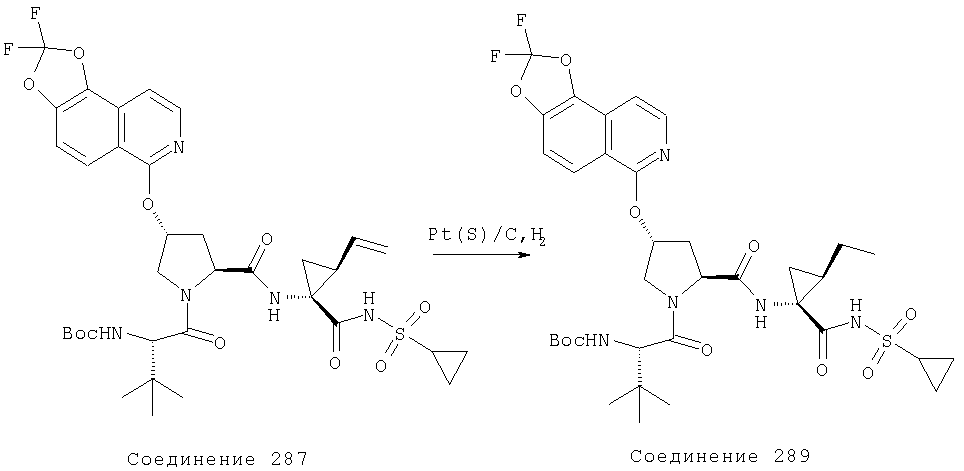
Суспензию Соединения 287 (15 мг) и Pt(S)/C (5%, 5 мг) в этилацетате (5 мл) гидрируют при 10 psi (68.95 кПа) в течение 30 мин. После фильтрования при упаривании получают 15 мг (выход количественный) Соединения 289 в виде твердого вещества.1H ЯМР (400 МГц, CD3OD) δ м.д. 1.09 (м, 26Н), 1.57 (м, 4Н), 2.30 (м, 1Н), 2.61 (м, J=13.82, 7.21 Гц, 1Н), 2.96 (м, 1Н), 4.05 (м, J=13.94 Гц, 1Н), 4.19 (д, J=9.54 Гц, 1Н), 4.53 (м, 2Н), 5.89 (с, 1Н), 7.34 (д, J=5.87 Гц, 1Н), 7.46 (д, J=9.05 Гц, 1Н), 8.09 (д, J=5.87 Гц, 1Н), 8.12 (д, J=8.80 Гц, 1Н); MS: (M+H)+ 766.
Пример 290: Получение Соединения 290

Соединение 290 (15 мг, 100%) получают по Схеме 3 из Примера 289 из 15 мг Соединения 286. Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (м, 14Н), 1.23 (м, 12Н), 1.58 (м, 4Н), 2.25 (м, 1Н), 2.58 (дд, J=13.82, 7.21 Гц, 1Н), 2.96 (м. 1Н), 4.05 (м, J=11.25, 2.93 Гц, 1Н), 4.25 (д, J=9.54 Гц, 1Н), 4.39 (м, 5Н), 4,52 (м, J=10.03, 7.34 Гц, 1Н), 5.81 (с, 1Н), 7.03 (д, J=9.05 Гц, 1Н), 7.43 (д, J=6.11 Гц, 1Н), 7.69 (д, J=9.05 Гц, 1Н), 7.88 (д, J=6.11 Гц, 1Н); MS: (M+H)+ 744.
Пример 291: Получение Соединения 291

Соединение 291 (28 мг, 100%) получают по Схеме 3 из Примера 289 из 28 г Соединения 251. Данные:1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (м, 15Н), 1.26 (м, 11Н), 1.37 (м, 1Н), 1.58 (м, 3H), 2.25 (м, 1Н), 2.58 (дд, J=743.6, 7.0 Гц, 1Н), 2.96 (м, 1Н), 3.99 (с, 3H), 4.06 (м, 1Н), 4.25 (м, 1Н), 4.44 (м, 1Н), 4.53 (дд, J=10.3, 7.6 Гц, 1Н), 5.78 (с, 1Н), 6.64 (д, J=9.8 Гц, 1Н), 7.55 (м, 2Н), 7.71 (т, J=7.3 Гц, 1Н), 8.09 (д, J=8.6 Гц, 1Н), 8.14 (д, J=8.1 Гц, 1Н); MS: (М+H)+ 738.
Пример 292: Получение Соединения 292
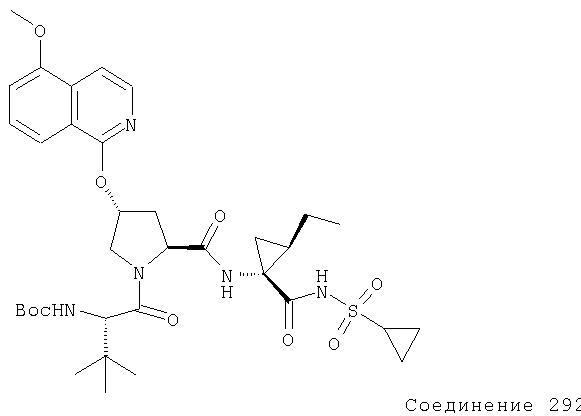
Соединение 291 (16 мг, 84%) получают по Схеме 3 из Примера 289 из 19 мг Соединения 253.1H ЯМР (400 МГц, CD3OD) δ м.д. 0.90 (м, 15Н), 1.15 (м, 12Н), 1.48 (м, 3H), 2.18 (м, 1Н), 2.51 (дд, J=13.7, 6.9 Гц, 1Н), 2.88 (м, 1Н), 3.90 (с, 3H), 3.98 (дд, J=11.6, 3.1 Гц, 1Н), 4.18 (д, J=9.5 Гц, 1Н), 4.36 (д, J=11.0 Гц, 1Н), 4.45 (дд, J=10.2, 7.2 Гц, 1Н), 5.76 (с, 1Н), 6.56 (д, J=9.3 Гц, 1Н), 7.05 (д, J=7.6 Гц, 1Н), 7.34 (т, J=8.1 Гц, 1Н), 7.51 (д, J=5.9 Гц, 1Н), 7.65 (д, J=8.3 Гц, 1Н), 7.86 (д, J=6.1 Гц, 1Н); MS: (M+Na)+738.
Пример 293: Получение Соединения 293

Соединение 293 (7 мг, 35%) получают по Схеме 3 из Примера 289 из 20 мг Соединения 252.1H ЯМР (400 МГц, CD3OD) δ м.д. 1.04 (м, 15Н), 1.27 (м, 12Н), 1.58 (м, 3H), 2.27 (м, 1Н), 2.60 (м, 4Н), 2.96 (м, 1Н), 4.07 (дд, J=11.7, 2.9 Гц, 1Н), 4.25 (с, 1Н), 4.46 (д, J=12.0 Гц, 1Н), 4.54 (дд, J=10.0, 7.6 Hz, 1H), 5.85 (s, 1H), 7.39 (t, J=7.7 Hz, 1H), 7.44 (d, J=5.9 Hz, 1H), 7.53 (d, J=6.9 Hz, 1H), 8.00 (d, J=6.1 Hz, 1H), 8.06 (d, J=8.6 Hz, 1H); MS: (M+H)+ 700.
Пример 294: Получение Соединения 294

Соединение 294 (14 мг, 78%) получают по Схеме 3 из Примера 289 из 18 мг Соединения 254.1H ЯМР (400 МГц, CD3OD) δ м.д. 0.94 (м, 15Н), 1.13 (м, 10H), 1.20 (м, 2Н), 1.50 (м, 3H), 2.21 (м, 1Н), 2.53 (дд, J=13.8, 7.0 Гц, 1Н), 2.88 (м, 1Н), 3.99 (дд, J=11.4, 2.6 Гц, 1Н), 4.14 (д, J=9.3 Гц, 1Н), 4.45 (м, 2Н), 5.79 (с, 1Н), 6.53 (д, J=9.1 Гц, 1Н), 7.40 (т, J=8.0 Гц, 1Н), 7.54 (д, J=5.9 Гц, 1Н), 7.73 (д, J=7.3 Гц, 1Н), 8.02 (д, J=6.1 Гц, 1Н), 8.10 (д, J=8.6 Гц, 1Н); MS: (M+Na)+ 742.
Пример 295: Получение Соединения 295
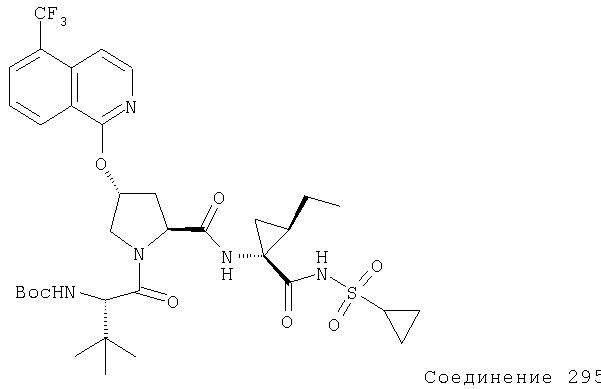
Соединение 295 (30 мг, 100%) получают по Схеме 3 из Примера 289 из 30 мг Соединения 270. MS: (M+Na)+ 776.
Пример 296: Получение Соединения 296

Соединение 296 (6.3 мг, 33%) получают по Схеме 3 из Примера 289 из 20 мг Соединения 259.1H ЯМР (400 МГц, CD3OD) δ м.д. 1,04 (м, 15Н), 1.24 (м, 12Н), 1.59 (м, 3H), 2.29 (м, 1Н), 2.50 (с, 3H). 2,60 (дд, J=13.69, 6.85 Гц, 1Н), 2.97 (м, 1Н), 4.09 (дд, J=11.74, 2.93 Гц, 1Н), 4.22 (с, 1Н), 4.43 (д, J=11.74 Гц, 1Н), 4.59 (дд, J=1 0.27, 6.85 Гц, 1Н), 5.87 (с, 1Н), 7.30 (д, J=5.87 Гц, 1Н), 7.57 (дд, J=8.31, 1.47 Гц, 1Н), 7.72 (д, J=8.31 Гц, 1Н), 7.89 (д, J=5.87 Гц, 1Н), 7.94 (с, 1Н); MS: (M+H)+ 700.
Пример 297: Получение Соединения 297
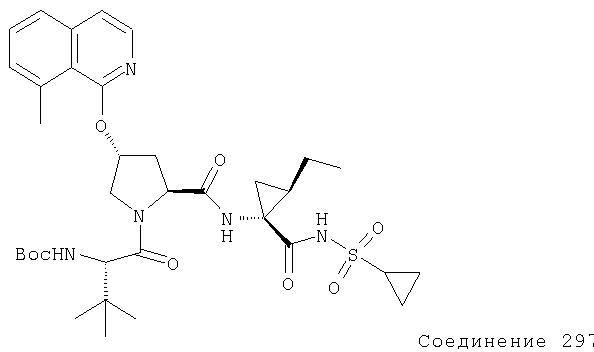
Соединение 297 (40 мг, 100) получают по Схеме 3 из Примера 289 из 40 мг Соединения 263.1H ЯМР (400 МГц, CD3OD) δ м.д. 0.98 (м, 13Н), 1.07 (м, 2Н), 1.27 (м, 12Н), 1.57 (м, 3H), 2.27 (м, 1Н), 2.58 (дд, J=14.7, 7.1 Гц, 1Н), 2.77 (с, 3H), 2.96 (м, 1Н), 4.04 (м, 1Н), 4.27 (м, 1Н), 4.42 (д, J=11.5 Гц, 1Н), 4.55 (д, J=10.6, 7.0 Гц, 1Н), 5.94 (с, 1Н), 6.65 (д, J=9.5 Гц, 1Н), 7.28 (м, 2Н), 7.50 (т, J=7.6 Гц, 1Н), 7.60 (д, J=7.6 Гц, 1Н), 7.89 (д, J=5.6 Гц, 1Н); MS: (M+Na)+ 722.
Пример 298: Получение Соединения 298
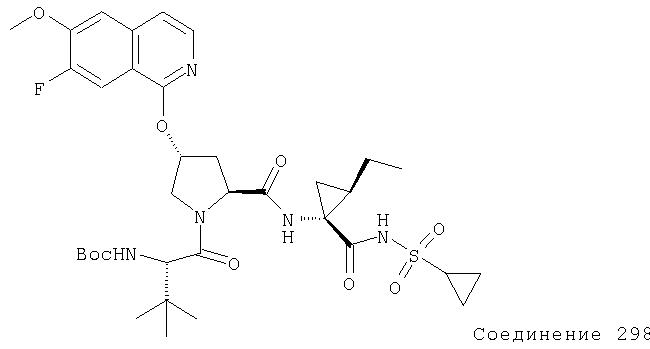
Соединение 298 (29 мг, 100%) получают по Схеме 3 из Примера 289 из 29 мг Соединения 261.1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99 (м, 15Н), 1.25 (м, 12Н), 1.60 (м, 3H), 2.28 (м, 1Н), 2.58 (м, 1Н), 2.96 (м, 1Н), 4.02 (м, 4Н), 4.22 (дд, J=8.8, 4.4 Гц, 1Н), 4.48 (м, 2Н), 5.83 (с, 1Н), 7.27 (д, J=5.6 Гц, 1Н), 7.36 (д, J=8.8 Гц, 1Н), 7.75 (д, J=11.3 Гц, 1Н), 7.90 (д, J=5.9 Гц, 1Н); MS: (М+Na)+ 756.
Пример 299: Получение Соединения 299
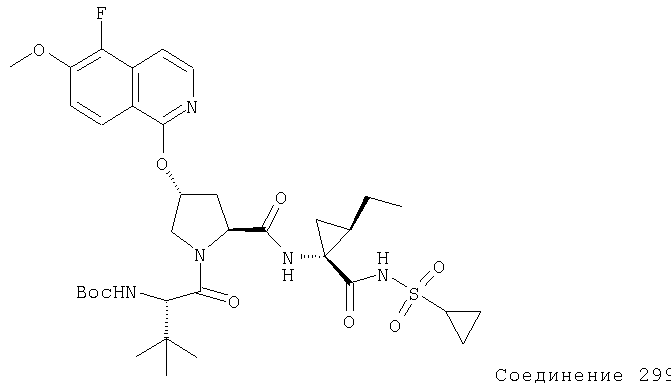
Соединение 299 (34 мг, 99%) получают по Схеме 3 из Примера 289 из 35 мг Соединения 274.1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (м, 16Н), 1.28 (м, 12Н), 1.58 (м, 2Н), 2.27 (с, 1Н), 2.59 (дд, J=13.82, 6.97 Гц, 1Н), 2.96 (м, 1Н), 4.02 (с, 3H), 4.07 (м, 1Н), 4.21 (м, 1Н), 4.44 (м, J=11.98 Гц, 1Н), 4.55 (д, J=10.27 Гц, 1Н), 5.85 (с, 1Н), 7.39 (м, 2Н), 7.95 (д, J=6.11 Гц, 1Н), 8.00 (д, J=9.29 Гц, 1Н); MS: (M+Na)+ 756.
Пример 300: Получение Соединения 300

Соединение 300 (30 мг, 100%) получают по Схеме 3 из Примера 289 из 30 мг Соединения 262.1H ЯМР (400 МГц, CD3OD) δ м.д. 1.27 (м, 30Н), 2.25 (с, 1Н), 2.54 (с, 1Н), 2.96 (м, 1Н), 3.97 (с, 3H), 4.20 (м, 3H), 4.51 (м, J=10.52, 6.85 Гц, 1Н), 5.37 (с, 1Н), 7.38 (с, 1Н), 7.62 (с, 1Н), 7.65 (д, J=5.38 Гц, 1Н), 8.06 (д, J=5.62 Гц, 1Н); MS: (M+Na)+ 773.
Подраздел G:
Условия осуществления метода LC/MS в Секции G суть следующие:
Колонка 4.6Х50 мм Xterra, градиент 3 мин и скорость потока 4 мл/мин
Схема 1: (Общая схема)

Схема 2: (Общая схема)
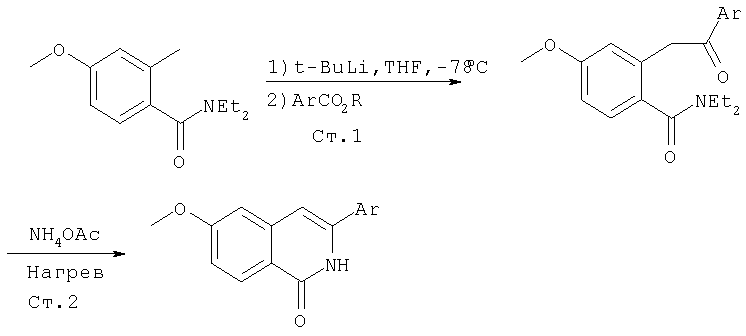
Схема 3: (Общая схема)
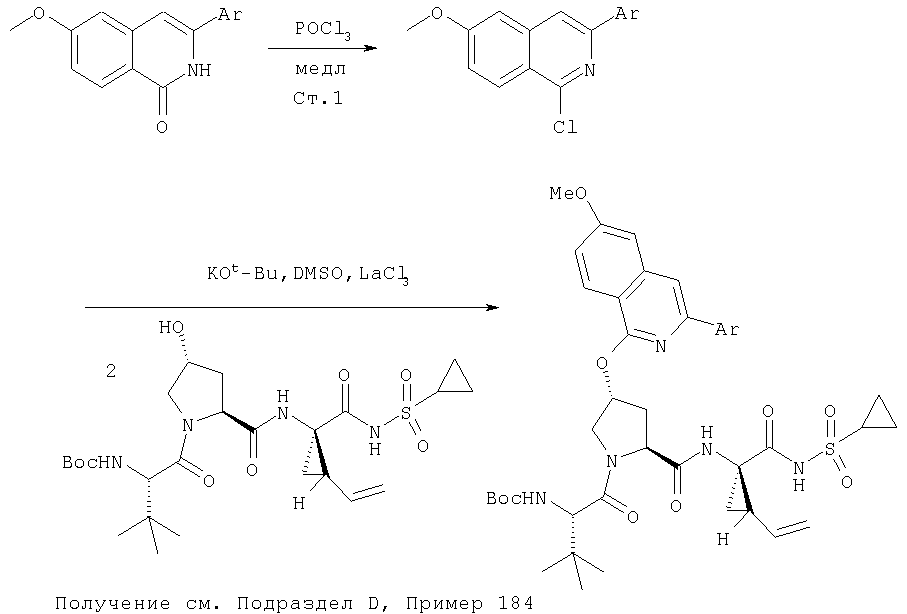
Схема 4: (Общая Схема)

Пример 320: Получение Соединения 320

Соединение 320 получают по приведенным выше Схемам 1 и 3.
Стадия 1 (Схема 1):

К раствору N,N-диэтил-4-метокси-2-метилбензамида (332 мг, 1.5 ммоля) в ТГФ (15 мл) при -78°С прибавляют трет-BuLi (1.7 М раствор в пентане, 1.3 мл, 2.25 ммоля). Образовавшийся красный раствор перемешивают при -78°С в течение 10 мин, затем прибавляют 2-цианопиридин (156 мг, 1.5 ммоля). Температуру реакционной смеси доводят до комнатной и перемешивают в течение ночи. Прибавляют насыщенный раствор NH4Cl и дважды экстрагируют этилацетатом. Объединенные органические вытяжки сушат (MgSO4) и упаривают. Сырой продукт очищают препаративной ВЭЖХ, получают соль ТФК в виде твердого вещества желтоватого цвета (85 мг, выход 15%)
1H ЯМР (400 МГц, CD3OD) δ м.д. 3.91 (м, 3H), 7.09 (дд, J=9.05, 2.45 Гц, 1Н), 7.17 (д, J=2.45 Гц, 1Н), 7.37 (с, 1Н), 7.42 (м, 1Н), 7.92 (м, 1Н), 8.08 (д, J=8.07 Гц, 1Н), 8.18 (д, J=9.05 Гц, 1Н), 8.65 (д, J=4.89 Гц, 1Н).
LC-MS (время удерживания: 2.14 мин), MS m/z 253 (MH+).
Стадия 2 (Схема 3, Стадия 1):
ТФК соль 6-метокси-3-пиридин-2-ил-2Н-изохинолин-1-она (85 мг, 0.232 ммоля) кипятят с POCl3 (3.0 мл) в течение 2 дней. POCl3 отгоняют и к остатку прибавляют лед. Затем нейтрализуют, прибавляя 10 N раствор NaOH, и получают чистое твердое вещество коричневого (бурого) цвета (62 мг, выход 99%).
LC-MS (время удерживания: 2.063 мин), MS m/z 271 (МН+).
Стадия 3 (Схема 3, Стадия 2):
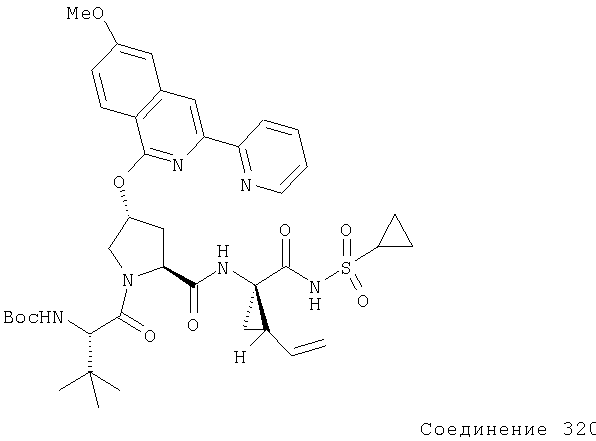
К раствору трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты (82 мг, 0.148 ммоля) и LaCl3 (36 мг, 0.148 ммоля) в ДМФА (1.5 мл), при -78°С прибавляют трет-бутоксид калия (1.0 М раствор в ТГФ, 0.74 мл, 0.74 ммоля). Реакционную смесь перемешивают 1 час, затем прибавляют 1-хлор-6-метокси-3-пиридин-2-илизохинолин (40 мг, 0.148 ммоля). Доводят до rt и перемешивают в течение ночи. Затем прибавляют воду и фильтруют. Фильтрат упаривают и остаток очищают препаративной ВЭЖХ, продукт получают в виде не чисто белого твердого вещества (Соединение 320) (23 мг, выход 20%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.87-1.08 (м, 11Н), 1.20-1.30 (м, 11Н), 1.43 (м, 1Н), 1.87 (м, 1Н), 2.22 (м, 1Н), 2.35 (м, 1Н), 2.69 (м, 1Н), 2.93 (м, 1Н), 3.94 (с, 3H), 4.16 (м, 1Н), 4.27 (м, 1Н), 4.45 (м, 1Н), 4.56 (м, 1Н), 5.10 (д, J=11.3 Гц, 1Н), 5.27 (д, J=15.9 Гц, 1Н), 5.74 (м, 1Н), 6.07 (с, 1Н), 7.12 (д, J=7.33 Гц, 1Н), 7.31 (д, J=1.96 Гц, 1Н), 7.40 (м, 1Н), 7.94 (дд, J=7.8 Гц, 1.5 Гц, 1Н), 8.11 (д, J=9.29 Гц, 1Н), 8.22 (с, 1Н), 8.45 (д, J=8.07 Гц, 1Н), 8.62 (м, 1Н). LC-MS (время удерживания: 2.393 мин), MS m/z 791 (MH+).
Пример 321: Получение Соединения 321
Схема 5:

Соединение 321 получают по приведенным выше Схемам 2 и 3, причем на стадии 1 Схемы 2 используют этиловый эфир 2-диэтиламинотиазол-4-карбоновой кислоты.
LC-MS (время удерживания: 2.76 мин), m/z 868 (МН+).
Пример 322: Получение Соединения 322

Соединение 322 получают по методике из Примера 321, за исключением того, что на Стадии 1 Схемы 2 вместо этилового эфира 2-диэтиламинотиазол-4-карбоновой кислоты используют этиловый эфир 2-диметиламинотиазол-4-карбоновой кислоты (полученный по Схеме 5, за исключением того, что вместо этилтиомочевины и этилиодида берут метилтиомочевину и метилиодид).
LC-MS (время удерживания: 2.56 мин), m/z 840 (МН+).
Пример 323: Получение Соединения 323
Соединение 323 получают по методике Стадии 3 из Примера 320, за исключением того, что вместо 1-хлор-6-метокси-3-пиридин-2-илизохинолина берут 1-хлор-6-метоксибензо[d]изоксазол.
Пример 324: Получение Соединения 324

Соединение 324 получают по методике Стадии 3 из Примера 320, за исключением того, что вместо 1-хлор-6-метокси-3-пиридин-2-илизохинолина берут 3-хлорбензо[d]изотиазол.
LC-MS (время удерживания: 1.83 мин), m/z 688 (MH+).
Пример 325: Получение Соединения 325

Соединение 325 получают по вышеприведенным Схемам 1 и 3.
Стадия 1 (Схема 1):
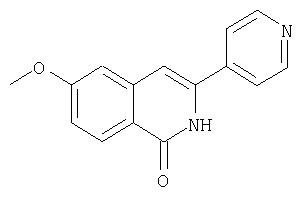
К раствору N, N-диэтил-4-метокси-2-метилбензамида (332 мг, 1.5 ммоля) в ТГФ (15 мл) при -78°С прибавляют трет-BuLi (1.7 М раствор в пентане, 1.3 мл, 2.25 ммоля). Образовавшийся красный раствор перемешивают при -78°С в течение 10 мин, затем прибавляют 4-цианопиридин (164 мг, 1.575 ммоля). Температуру реакционной смеси доводят до комнатной и перемешивают в течение ночи. Прибавляют насыщенный раствор NH4Cl, выпадает желтый осадок, который представляет собой чистый продукт (145 мг, выход 38%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 3.91 (с, 3H), 7.18 (дд, J=8.8 Гц, 2.8 Гц, 1Н), 7.26 (м, 2Н), 8.06 (д, J=6.0 Гц, 2Н), 8.16 (д, J=8.8 Гц, 1Н), 8.84 (д, J=6.0 Гц, 2Н).
LC-MS (время удерживания: 1.300 мин), MS m/z 253 (МН+).
Стадия 2 (Схема 3, Стадия I):

6-Метокси-3-пиридин-4-ил-2Н-изохинолин-1-он (134 мг, 0.531 ммоля) кипятят с POCl3 (6.0 мл) в течение 5 дней. POCl3 отгоняют и к остатку прибавляют лед. Затем нейтрализуют, прибавляя насыщенный раствор NaHCO3, и получают чистое твердое вещество коричневого (бурого) цвета (62 мг, выход 99%) (125 мг, выход 87%).
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 3.99 (с, 3H), 7 53 (дд, J=9.04 Гц, 2.44 Гц, 1Н), 7.59 (д, J=2.69 Гц, 1Н), 8.26 (д, J=9.05 Гц, 1Н), 8.30 (д, J=5.38 Гц, 2Н), 8.73 (с, 1Н), 8.85 (д, J=6.36 Гц, 2Н).
LC-MS (время удерживания: 2.027 мин), MS m/z 271 (МН+).
Стадия 3 (Схема 3, Стадия 2):

К раствору трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты (83.5 мг, 0.15 ммоля) и LaCl3 (36.8 мг, 0.15 ммоля) в ДМФА (1.5 мл), при -78°С прибавляют трет-бутоксид калия (1.0 М раствор в ТГФ, 0.75 мл, 0.75 ммоля). Реакционную смесь перемешивают 1 час, затем прибавляют 1-хлор-6-метокси-3-пиридин-4-илизохинолин (40.6 мг, 0.15 ммоля). Доводят до rt и перемешивают в течение ночи. Затем прибавляют воду и фильтруют. Фильтрат упаривают и остаток очищают препаративной ВЭЖХ, продукт получают в виде не чисто белого твердого вещества (Соединение 325) (1.6 мг, выход 1.5%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.90 (м, 2Н), 1.02 (с, 9Н), 1.17-1.31 (м, 11Н), 1.42 (м, 1Н), 1.87 (м, 1H), 2.23 (м, 1Н), 2.35 (м, 1Н), 2.68 (м, 1Н), 2.93 (м, 1Н), 3.95 (с, 3H), 4.15 (м, 1Н), 4.25 (м, 1Н), 4.45 (м, 1Н), 4.56 (м, 1Н), 5.10 (д, J=10.76 Гц, 1Н), 5.27 (д, J=17.61 Гц, 1Н), 5.74 (м, 1Н), 6.06 (с, 1Н), 7.14 (д, J=8.07 Гц, 1Н), 7.34 (с, 1Н), 8.01 (с, 1Н), 8.12 (д, J=8.81 Гц, 1Н), 8.19 (д, J=6.12 Гц, 2Н), 8.61 (д, J=5.63 Гц, 2Н).
LC-MS (время удерживания: 2.523 мин), MS m/z 791 (МН+).
Пример 326; Получение Соединения 326

Соединение 326 получают по вышеприведенным Схемам 1 и 4.
Стадия 1 (Схема 1):

К раствору N,N-диэтил-4-метокси-2-метилбензамида (332 мг, 1.5 ммоля) в ТГФ (15 мл) при -78° С прибавляют трет-BuLi (1.7 М раствор в пентане, 1.3 мл, 2.25 ммоля). Образовавшийся красный раствор перемешивают при -78°С в течение 10 мин, затем прибавляют 4-диметиламинобензонитрил (219 мг, 1.5 ммоля). Температуру реакционной смеси доводят до комнатной и перемешивают в течение ночи. Прибавляют насыщенный раствор NH4Cl, выпадает желтый осадок, который отфильтровывают и растирают с эфиром, получают не чисто белое твердое вещество, являющееся чистым продуктом (247 мг, выход 56%).
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 2.97 (с, 6Н), 3.87 (с, 3H), 6.72 (с, 1Н), 6.78 (д, J=8.80 Гц, 2Н), 6.97 (дд, J=8.80,2.45 Гц, 1Н), 7.10 (д, J=2.45 Гц, 1Н), 7.65 (д, J=8.80 Гц, 2Н), 8.05 (д, J=8.80 Гц, 1Н), 11.11 (с, 1Н).
LC-MS (время удерживания: 2.023 мин), MS m/z 295 (МН+).
Стадия 2 (Схема 4, Стадия 1):
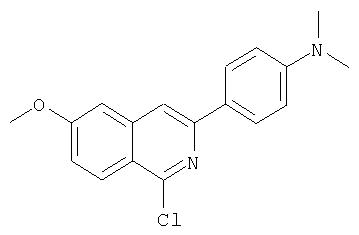
3-(4-Диметиламинофенил)-6-метокси-2Н-изохинолин-1-он (245 мг, 0.83 ммоля) кипятят с POCl3 (10.0 мл) в течение 2 дней. POCl3 отгоняют и к остатку прибавляют лед. Затем нейтрализуют, прибавляя 10N раствор NaOH, и дважды экстрагируют этилацетатом. Объединенные органические вытяжки сушат (MgSO4). После упаривания получают продукт - твердое вещество оранжевого цвета (215 мг, выход 83%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 3.01 (с, 6Н), 3.96 (с, 3H), 6.88 (д, J=9.05 Гц, 2Н), 7.20 (дд, J=9.17, 2.57 Гц, 1Н), 7.28 (д, J=2.45 Гц, 1Н), 7.94 (с, 1Н), 7.96 (д, J=9.05 Гц, 2Н), 8.13 (д, J=9.29 Гц, 1Н).
LC-MS (время удерживания: 2.543 мин), MS m/z 313 (MH+).
Стадия 3 (Схема 4, Стадия 2):

Смесь [4-(1-хлор-6-метоксиизохинолин-3-ил)-фенил]-диметиламина (110 мг, 0.35 ммоля) и тетрабутилфосфоний гидродифторида (0.5 г) в течение 20 мин нагревают в микроволновом реакторе Смита при 140°С. Затем прибавляют воду и экстрагируют этилацетатом. Органический слой отделяют, промывают водой и сушат (MgSO4). Упариванием растворителя получают продукт - твердое вещество коричневатого цвета (85 мг, выход 82%).
LC-MS (время удерживания: 2.320 мин), MS m/z 297 (МН+).
Стадия 4 (Схема 4, Стадия 3):

К раствору трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты (111 мг, 0.2 ммоля) и LaCl3 (49 мг, 0.2 ммоля) в ДМФА (2.0 мл) при -78°С прибавляют трет-бутоксид калия (1.0 М раствор в ТГФ, 1.0 мл, 1.0 ммоля). Реакционную смесь перемешивают 1 час, затем прибавляют 4-(1-фтор-6-метокси-илизохинолин-3-ил)-фенил]-диметиламин (59 мг, 0.2 ммоля). Доводят до rt и перемешивают в течение ночи. Затем прибавляют воду и фильтруют. Фильтрат упаривают и остаток очищают препаративной ВЭЖХ, продукт получают в виде желтоватого твердого вещества (Соединение 326) (17.6 мг, выход 11%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.97-1.08 (м, 11Н), 1.23 (м, 2Н), 1.31 (с, 9Н), 1.44 (м, 1Н), 1.87 (м, 1Н), 2.22 (м, 1Н), 2.34 (м, 1Н), 2.68 (м, 1Н), 2.93 (м, 1Н), 2.99 (м, 6Н), 3.91 (с, 3H), 4.17 (м, 1Н), 4.29 (м, 1Н), 4.39 (м, 1Н), 4.52 (м, 1Н), 5.10 (д, J=10.76 Гц, 1Н), 5.27 (д, J=17.11 Гц, 1Н), 5.74 (м, 1Н), 6.03 (с, 1Н), 6.83 (м, 2Н), 6.95 (м, 1Н), 7.16(с, 1Н), 7.59 (с, 1Н), 8.01 (м, 3H).
LC-MS (время удерживания: 2.850 мин), MS m/z 834 (MH+).
Пример 327: Получение Соединения 327

Соединение 327 получают по нижеприведенным Схемам 1 и 4.
Стадия 1 (Схема 1):

К раствору N,N-диэтил-4-метокси-2-метилбензамида (332 мг, 1.5 ммоля) в ТГФ (15 мл) при -78°С прибавляют трет-BuLi (1.7 М раствор в пентане, 1.3 мл, 2.25 ммоля). Образовавшийся красный раствор перемешивают при -78°С в течение 10 мин, затем прибавляют 4-диэтиламинобензонитрил (261 мг, 1.5 ммоля). Температуру реакционной смеси доводят до комнатной и перемешивают в течение ночи. Прибавляют насыщенный раствор NH4Cl, выпадает желтый осадок, который отфильтровывают, желтое твердое вещество, являющееся чистым продуктом (215 мг, выход 44%).
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 1.12 (м, 6Н), 3.39 (м, 4Н), 3.87 (с, 3H), 6.69 (с, 1Н), 6.72 (д, J=9.05 Гц, 2Н), 6.96 (дд, J=8.80, 2.45 Гц, 1Н), 7.09 (д, J=2.45 Гц, 1Н), 7.61 (д, J=9.05 Гц, 2Н), 8.04 (д, J=8.80 Гц, 1Н), 11.06 (с, 1Н).
LC-MS (время удерживания: 1.883 мин), MS m/z 323 (МН+).
Стадия 2 (Схема 4, Стадия 1):

3-(4-Диэтиламинофенил)-6-метокси-2Н-изохинолин-1-он (207 мг, 0.642 ммоля) кипятят с POCl3 (8.0 мл) в течение 1 дня. POCl3 отгоняют и к остатку прибавляют лед. Затем нейтрализуют, прибавляя насыщенный раствор NaHCO3, и дважды экстрагируют этилацетатом. Объединенные органические вытяжки сушат (MgSO4). После упаривания растворителя получают продукт - твердое вещество коричневатого цвета (180 мг, выход 82%).
LC-MS (время удерживания: 2.397 мин), MS m/z 341 (МН+).
Стадия 3 (Схема 4, Стадия 2):

Смесь [4-(1-хлор-6-метоксиизохинолин-3-ил)-фенил]-диметиламина (90 мг, 0.264 ммоля) и тетрабутилфосфоний гидродифторида (0.5 г) в течение 20 мин нагревают в микроволновом реакторе Смита при 140°С. Затем прибавляют воду и экстрагируют этилацетатом. Органический слой отделяют, промывают водой и сушат (MgSO4). Упариванием растворителя получают продукт в виде желтоватого масла (70 мг, выход 82%).
LC-MS (время удерживания: 2.254 мин), MS m/z 325 (МН+).
Стадия 4 (Схема 4, Стадия 3):
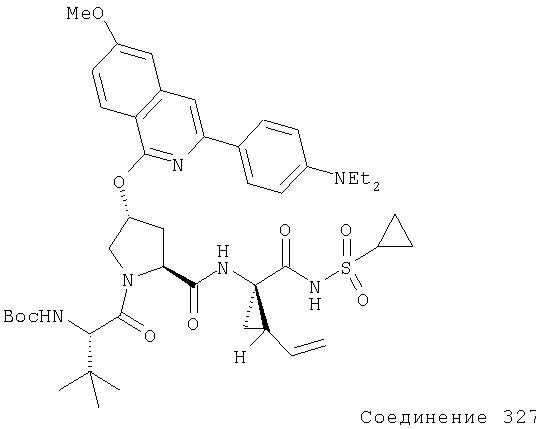
К раствору трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты (100 мг, 0.18 ммоля) и LaCl3 (66 мг, 0.27 ммоля) в ДМФА (2.0 мл) при -78°С прибавляют трет-бутоксид калия (1.0 М раствор в ТГФ, 0.9 мл, 0.9 ммоля). Реакционную смесь перемешивают 1 час, затем прибавляют 4-(1-фтор-6-метоксиизохинолин-3-ил)-фенил]-диэтиламин (70 мг, 0.216 ммоля). Доводят до rt и перемешивают в течение ночи. Затем прибавляют воду и фильтруют. Фильтрат упаривают и остаток очищают препаративной ВЭЖХ, продукт получают в виде твердого вещества белого цвета (Соединение 327) (18 мг, выход 12%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.95-1.07 (м, 11Н), 1.18 (м, 6Н), 1.25-1.38 (м, 11Н), 1.58 (м, 1Н), 1.85 (м, 1Н), 2.19 (м, 1Н), 2.34 (м, 1Н), 2.68 (м, 1Н), 2.92 (м, 1Н), 3.42 (м, 4Н), 3.90 (с, 3H), 4.16 (м, 1Н), 4.28 (м, 1Н), 4.37 (м, 1Н), 4.53 (м, 1Н), 5.07 (д, J=11.0 Гц, 1Н), 5.25 (д, J=l7.36 Гц, 1Н), 5.74 (м, 1Н), 5.99 (с, 1Н), 6.77 (д, J=8.8 Гц, 2Н), 6.94 (д, J=9.05 Гц, 1Н), 7.14 (с, 1Н), 7.56 (с, 1Н), 7.95-8.02 (м, 3H).
Пример 328: Получение Соединения 328

Соединение 328 получают по вышеприведенным Схемам 2 и 3.
Стадия 1 (Схема 2, Стадия 1):
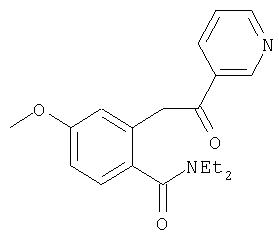
К раствору N,N-диэтил-4-метокси-2-метилбензамида (332 мг, 1.5 ммоля) в ТГФ (15 мл) при -78°С прибавляют трет-BuLi (1.7 М раствор в пентане, 2.12 мл, 3.6 ммоля). Образовавшийся красный раствор перемешивают при -78°С в течение 10 мин, затем прибавляют метилникотинат (206 мг, 1.5 ммоля). Реакционную смесь перемешивают при -78°С в течение 2 час. Прибавляют насыщенный раствор NH4Cl и дважды экстрагируют этилацетатом. Объединенные органические вытяжки сушат (MgSO4) и упаривают. Сырой продукт очищают препаративной ВЭЖХ, получают ТФК соль в виде вязкого желтоватого масла (124 мг, выход 19%).
LC-MS (время удерживания: 1.740 мин), MS m/z 349 (M+Na+).
Стадия 2 (Схема 2, Стадия 2):
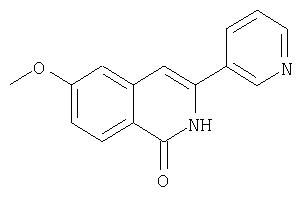
N,N-Диэтил-4-метокси-2-(2-оксо-2-пиридин-3-илэтил)-бензамид (120 мг, 0.272 ммоля) нагревают с ацетатом аммония (1 г) в течение 3 час. Затем реакционную смесь охлаждают и прибавляют воду. Экстрагируют этилацетатом и органические вытяжки сушат (MgSO4), упаривают, получают продукт в виде твердого вещества желтоватого цвета (65 мг, выход 95%).
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 3.89 (с, 3H), 6.93 (с, 1Н), 7.10 (дд, J=8.80, 2.45 Гц, 1Н), 7.19 (д, J=2.45 Гц, 1Н), 7.52 (дд, J=7.46, 4.77 Гц, 1Н), 8.15 (м, 2Н), 8.64 (дд, J=4.89, 1.47 Гц, 1Н), 8.96 (д, J=1.71 Гц, 1Н), 11.51 (с, 1Н).
LC-MS (время удерживания: 1.377 мин), MS m/z 253 (МН+).
Стадия 3 (Схема 3, Стадия I):

6-Метокси-3-пиридин-3-ил-2Н-изохинолин-1-он (65 мг, 0.258 ммоля) кипятят с POCl3 (2.5 мл) в течение 7 дней. POCl3 отгоняют и к остатку прибавляют лед. Затем нейтрализуют, прибавляя 10N раствор NaOH, и дважды экстрагируют этилацетатом. Объединенные органические вытяжки сушат (MgSO4). После упаривания получают продукт - твердое вещество желтого цвета (27 мг, выход 39%).
LC-MS (время удерживания: 2.090 мин), MS m/z 271 (МН+).
Стадия 4 (Схема 3, Стадия 2):

К раствору трет-бутилового эфира{1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты (56 мг, 0.10 ммоля) и LaCl3 (25 мг, 0.10 ммоля) в ДМФА (1.5 мл) при -78°С прибавляют трет-бутоксид калия (1.0 М раствор в ТГФ, 0.5 мл, 0.5 ммоля). Реакционную смесь перемешивают 1 час, затем прибавляют 1-хлор-6-метокси-3-пиридин-3-илизохинолин (27 мг, 0.10 ммоля). Доводят до rt и перемешивают в течение ночи. Затем прибавляют воду и фильтруют. Фильтрат упаривают и остаток очищают препаративной ВЭЖХ, продукт получают в виде твердого вещества белого цвета (Соединение 328) (17 мг, выход 21%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.95 (м, 2), 1.02 (с, 9Н), 1.20-1.30 (м, 11Н), 1.41 (m, 1Н), 1.86 (м, 1Н), 2.21 (м, 1Н), 2.35 (м, 1Н), 2.67 (м, 1Н), 2.93 (м, 1Н), 3.93 (с, 3H), 4.14 (м, 1Н), 4.26 (м, 1Н), 4.47 (д, J=11.99 Гц, 1Н), 4.55 (м, 1Н), 5.09 (д, J=10.02 Гц, 1Н), 5.26 (д, J=17.85 Гц, 1Н), 5.74 (м, 1Н), 6.07 (с, 1Н), 7.09 (м, 1Н), 7.29 (д, J=1.96 Гц, 1Н), 7.53 (м, 1Н), 7.86 (с, 1Н), 8.09 (д, J=9.05 Гц, 1Н), 8.50-8.58 (м, 2Н), 9.28 (с, 1Н).
LC-MS (время удерживания: 2.453 мин), MS m/z 791 (МН+).
Пример 329: Получение Соединения 329
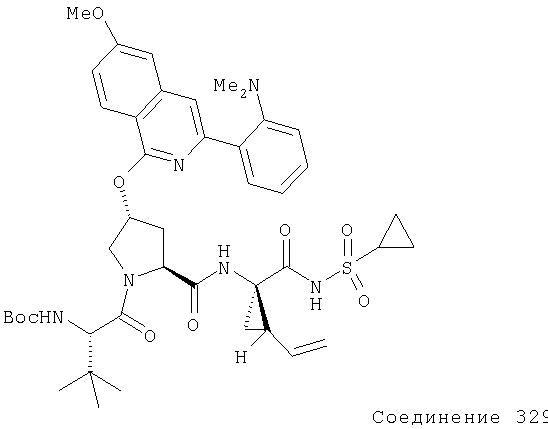
Соединение 329 получают по вышеприведенным Схемам 2 и 4.
Стадия 1 (Схема 2, Стадия 1):
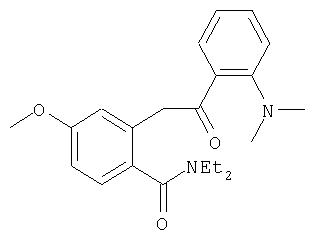
К раствору N,N-диэтил-4-метокси-2-метилбензамида (332 мг, 1.5 ммоля) в ТГФ (15 мл) при -78° С прибавляют трет-BuLi (1.7 М раствор в пентане, 2.2 мл, 3.75 ммоля). Образовавшийся красный раствор перемешивают при -78°С в течение 10 мин, затем прибавляют метиловый эфир N, N-диметилантраниловой кислоты (269 мг, 1.5 ммоля). Реакционную смесь перемешивают при -78°С в течение 2 час. Прибавляют насыщенный раствор NH4Cl и дважды экстрагируют этилацетатом. Объединенные органические вытяжки сушат (MgSO4) и упаривают. Сырой продукт очищают препаративной ВЭЖХ, получают продукт в виде вязкого желтоватого масла (256 мг, выход 46%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99-1.13 (м, 6Н), 3.23-3.31 (м, 8Н), 3.39 (м, 2Н), 3.82 (с, 3H), 4.35 (с, 2Н), 6.91 (дд, J=8.44, 2.57 Гц, 1Н), 6.99 (д, J=2.45 Гц, 1Н), 7.22 (д, J=8.56 Гц, 1Н), 7.69 (т, J=7.70 Гц, 1Н), 7.84 (м, 1Н), 7.96 (д, J=8.31 Гц, 1Н), 8.18(д, J=7.83 Гц, 1Н).
LC-MS (время удерживания: 1.557 мин), MS m/z 369 (МН+).
Стадия 2 (Схема 2, Стадия 2):
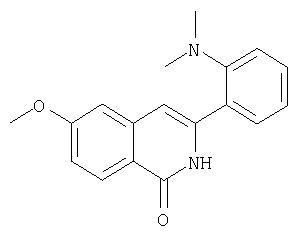
2-[2-(2-Диметиламинофенил)-2-оксоэтил]-N,N-диэтил-4-метоксибензамид (250 мг, 0.678 ммоля) нагревают с ацетатом аммония (1.5 г) в течение 2 час. Затем реакционную смесь охлаждают и прибавляют воду. Экстрагируют этилацетатом и органические вытяжки сушат (MgSO4), упаривают, получают продукт в виде твердого вещества желтоватого цвета (125 мг, выход 63%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 2.95 (с, 6Н), 3.92 (с, 3H), 6.92 (с, 1Н), 7.12 (дд, J=8.80, 2.45 Гц, 1Н), 7.16 (д, J=2.45 Гц, 1Н), 7.35 (м, 1Н), 7.55 (м, 2Н), 7.63 (д, J=7.83 Гц. 1Н), 8.20 (д, J=9.05 Гц, 1Н).
LC-MS (время удерживания: 2.097 мин), MS m/z 295 (МН+).
Стадия 3 (Схема 4, Стадия 1):
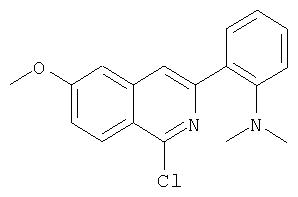
3-(2-Диметиламинофенил)-6-метокси-2Н-изохинолин-1-он (125 мг, 0.425 ммоля) кипятят с POCl3 (4.0 мл) в один день. POCl3 отгоняют и к остатку прибавляют лед. Затем нейтрализуют, прибавляя 10N раствор NaOH, и дважды экстрагируют этилацетатом. Объединенные органические вытяжки сушат (MgSO4 ). После упаривания получают продукт - твердое вещество коричневатого цвета (82 мг, выход 62%).
LC-MS (время удерживания: 2.040 мин), MS m/z 313 (MH+).
Стадия 4 (Схема 4, Стадия 2):
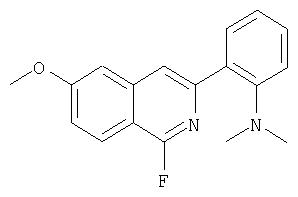
Смесь [2-(1-хлор-6-метоксиизохинолин-3-ил)-фенил]-диметиламина (82 мг, 0.262 ммоля) и тетрабутилфосфоний гидродифторида (1.0 г) в течение 20 мин. нагревают в микроволновом реакторе Смита при 140°С. Затем прибавляют воду и экстрагируют этилацетатом. Органический слой отделяют, промывают водой и сушат (MgSO4). Упариванием растворителя получают сырой продукт, который очищают препаративной ВЭЖХ. Продукт получают в виде желтоватого масла (85 мг).
1H ЯМР (400 МГц, CD3OD) δ м.д. 3.41 (с, 6Н), 4.00 (с, 3H), 7.42 (дд, J=9.05, 2.45 Гц, 1Н), 7.53 (с, 1Н), 7.71 (м, 2Н), 7.99 (м, 1Н), 8.16 (м, 2Н), 8.31 (с, 1Н).
LC-MS (время удерживания: 1.873 мин), MS m/z 297 (МН+).
Стадия 5 (Схема 4, Стадия 3):

К раствору трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты (56 мг, 0.1 ммоля) и LaCl3 (25 мг, 0.1 ммоля) в ДМФА (1.0 мл) при -78°С прибавляют трет-бутоксид калия (1.0 М раствор в ТГФ, 0.5 мл, 0.5 ммоля). Реакционную смесь перемешивают 1 час, затем прибавляют [2-(1-фтор-6-метоксиизохинолин-3-ил)-фенил]-диметиламин (30 мг, 0.1 ммоля). Доводят до rt и перемешивают в течение ночи. Затем прибавляют воду и фильтруют. Фильтрат упаривают и остаток очищают препаративной ВЭЖХ, продукт получают в виде твердого вещества белого цвета (Соединение 329) (4.0 мг, выход 5%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.98-1.08 (м, 11Н), 1.16-1.32 (м, 11Н), 1.40 (м, 1Н), 1.85 (м, 1Н), 2.16-2.32 (м, 2Н), 2.60-2.71 (м, 7Н), 2.92 (м, 1Н), 3.91 (с, 3H), 4.08 (м, 1Н), 4.26 (м, 1Н), 4.45 (м, 1Н), 4.55 (м, 1Н), 5.10 (д, J=10.27 Гц, 1Н), 5.28 (д, J=18.09 Гц, 1Н), 5.74 (м, 1Н). 5.89 (с, 1Н), 7.05 (д, J=6.85 Гц, 1Н), 7.10-7.20 (м, 2Н), 7.29 (м, 1Н), 7.63 (д, J=7.58 Гц, 1Н), 7.78 (с, 1Н), 8 07 (д, J=8.56 Гц, 1Н).
LC-MS (время удерживания: 2.550 мин), MS m/z 834 (MH+).
Пример 330: Получение Соединения 330

Соединение 330 получают по вышеприведенным Схемам 2 и 4.
Стадия 1 (Схема 2, Стадия 1):

К раствору N,N-диэтил-4-метокси-2-метилбензамида (332 мг, 1.5 ммоля) в ТГФ (15 мл) при -78°С прибавляют трет-BuLi (1.7 М раствор в пентане, 2.2 мл, 3.75 ммоля). Образовавшийся красный раствор перемешивают при -78°С в течение 10 мин, затем прибавляют метиловый эфир (3-диметиламино)бензойной кислоты (269 мг, 1.5 ммоля). Реакционную смесь перемешивают при -78°С в течение 2 час. Прибавляют насыщенный раствор NH4Cl и дважды экстрагируют этилацетатом. Объединенные органические вытяжки сушат (MgSO4) и упаривают. Сырой продукт очищают препаративной ВЭЖХ, получают ТФК соль в виде вязкого желтоватого масла (245 мг, выход 33%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (т, J=6.85 Гц, 3H), 1.09 (м, 3H), 3.11 (с, 6Н), 3.21 (м, 2Н), 3.40 (м, 2Н), 3.79 (с, 3H), 4.39 (с, 2Н), 6.84-6.91 (м, 2Н), 7.19 (д, J=8.32 Гц, 1Н), 7.35 (м, 1H), 7.49 (т, J=8.07 Гц, 1Н), 7.66-7.71 (м, 2Н).
LC-MS (время удерживания: 1.930 мин), MS m/z 369 (МН+).
Стадия 2 (Схема 2, Стадия 2):
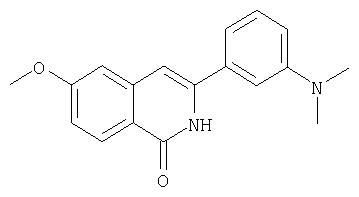
2-[2-(3-Диметиламинофенил)-2-оксоэтил]-N,N-диэтил-4-метоксибензамвд (240 мг, 0.497 ммоля) нагревают с ацетатом аммония (2.0 г) в течение 2.5 час. Затем реакционную смесь охлаждают и прибавляют воду. Получают продукт в виде твердого вещества коричневатого цвета (95 мг, выход 65%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 2.98 (с, 6Н), 3.88 (с, 3H), 6.74-6.87 (м, 2Н), 7.01-7.07 (м, 3H), 7.18 (д, J=2.44 Гц, 1Н), 7.28 (т, J=7.82 Гц, 1Н), 8.10 (д, J=8.80 Гц, 1Н).
LC-MS (время удерживания: 1.773 мин), MS m/z 295 (МН+).
Стадия 3 (Схема 4, Стадия 1):
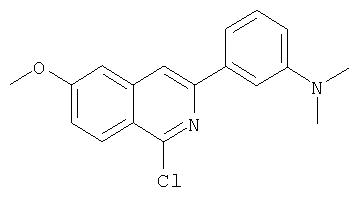
3-(3-Диметиламинофенил)-6-метокси-2Н-изохинолин-1-он (92 мг, 0.312 ммоля) кипятят с POCl3 (3.0 мл) в течение двух дней. POCl3 отгоняют и к остатку прибавляют лед. Затем нейтрализуют, прибавляя насыщенный раствор NaHCO3, и дважды экстрагируют этилацетатом. Объединенные органические вытяжки сушат (MgSO4). После упаривания растворителя получают продукт - вязкое масло коричневатого цвета (72 мг, выход 74%).
LC-MS (время удерживания: 2.297 мин), MS m/z 313 (MH+).
Стадия 4 (Схема 4, Стадия 2):

Смесь [3-(1-хлор-6-метоксиизохинолин-3-ил)-фенил]-диметиламина (72 мг, 0.23 ммоля) и тетрабутилфосфоний гидродифторида (0.5 г) в течение 20 мин нагревают в микроволновом реакторе Смита при 140°С. Затем прибавляют воду и экстрагируют этилацетатом. Органический слой отделяют, промывают водой и сушат (MgSO4). Упариванием растворителя получают продукт в виде коричневатого масла (58 мг, выход 85%).
LC-MS (время удерживания: 2.193 мин), MS m/z 297 (МН+).
Стадия 5 (Схема 4, Стадия 3):

К раствору трет-бутилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты (86 мг, 0.155 ммоля) и LaCl3 (57 мг, 0.233 ммоля) в ДМФА (1.5 мл) при -78°С прибавляют трет-бутоксид калия (1.0 М раствор в ТГФ, 0.5 мл, 0.5 ммоля). Реакционную смесь перемешивают 1 час, затем прибавляют [3-(1-фтор-6-метоксиизохинолин-3-ил)-фенил]-диметиламин (55 мг, 0.185 ммоля). Доводят до rt и перемешивают в течение ночи. Затем прибавляют воду и фильтруют. Фильтрат упаривают и остаток очищают препаративной ВЭЖХ, продукт получают в виде твердого вещества не чисто белого цвета (Соединение 330) (8.0 мг, выход 6%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99-1.09 (м, 11Н), 1.23 (м, 2Н), 1.29 (с, 9Н), 1.42 (м, 1Н), 1.86 (м, 1Н), 2.21 (м, 1Н), 2.33 (м, 1Н), 2.70 (м, 1Н), 2.93 (м, 1Н), 3.00 (с, 6Н), 3.92 (с, 3H), 4.14 (м, 1Н), 4.29 (м, 1Н), 4.44-4.57 (м, 2Н), 5.10 (д, J=11.00 Гц, 1Н), 5.27 (д, J=16.87 Гц, 1Н), 5.74 (м, 1Н), 6.01 (с, 1Н), 6.63 (д, J=8.80 Гц, 1Н), 7.03 (д, J=6.85 Гц, 1Н), 7.24 (с, 1Н), 7.28 (т, J=8.07 Гц, 1Н), 7.45 (д, J=7.82 Гц, 1Н), 7.59 (с, 1Н), 7.72 (с, 1Н), 8.05 (д, J=8.80 Гц, 1Н).
LC-MS (время удерживания: 2.707 мин), MS m/z 834 (MH+).
Пример 331: Получение Соединения 331

Соединение 331 получают методами по данному описанию.
Пример 334: Получение Соединения 334

Соединение 334 получают следующим методом:
Стадия 1:
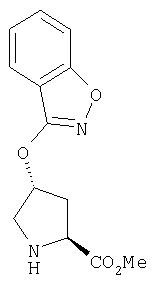
К раствору Boc-cis-HYP-OMe (122.6 мг, 0.5 ммоля) в ТГФ (15 мл) при 0°С прибавляют трифенилфосфин (196.7 мг, 0.75 ммоля) и бензо[d]изоксазол-3-ол (81 мг, 0.6 ммоля). Затем прибавляют DEAD (0.118 мл, 0.75 ммоля). Температуру реакционной смеси доводят до комнатной и перемешивают 3 час. Затем упаривают растворитель и остаток очищают препаративной ВЭЖХ, получая бесцветное вязкое масло (117 мг, выход 54%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.41 (м, 9Н), 2.38 (м, 1Н), 2.75 (м, 1H), 3.75 (м, 3H), 3.8-1 (м, 1Н), 3.90 (м, 1Н), 4.47 (м, 1Н), 5.44 (м, 1Н), 7.31 (т, J=7.46 Гц, 1Н), 7.47 (д, J=8.56 Гц, 1Н), 7.59 (т, J=7.83 Гц, 1Н), 7.66 (д, J=8.07 Гц, 1Н).
LC-MS (время удерживания: 2.65 мин), MS m/z 363 (МН+).
Затем часть продукта реакции конденсации (сочетания) (85 мг, 0.235 ммоля) растворяют в 4N растворе HCl (1.5 мл) и перемешивают 3 час. Упариванием растворителя получают HCl соль в виде желтеющего масла (85 мг, выход >100%).
LC-MS (время удерживания: 1.327 мин), MS m/z 263 (MH+).
Стадия 2:
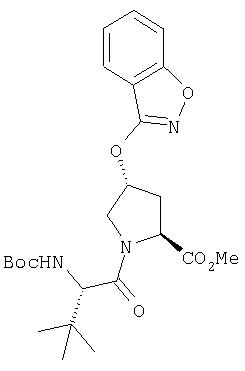
К раствору гидрохлорида метилового эфира 4-(Бензо[d]изоксазол-3-илокси)-пирролидин-2-карбоновой кислоты (85 мг, 0.285 ммоля) в CH3CN (10 мл) прибавляют N-boc-L-трет-лейцин (99 мг, 0.427 ммоля), DIEA (0.25 мл, 1.425 ммоля) и конденсирующий агент HOBt (65 мг, 0.427 ммоля) и HBTU (162 мг, 0.427 ммол). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Затем очищают на колонке для препаративной ВЭЖХ, получают продукт в виде бесцветного вязкого масла (63 мг, выход 46%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.17 (с, 9Н), 2.34 (м, 1Н), 2.78 (дд, J=14.13, 7.83 Гц, 1Н), 3.72 (с, 3H), 4.00 (дд, J=12.22, 3.42 Гц, 1Н), 4.19 (с, 1Н), 4.57 (д, J=12.23 Гц, 1Н), 4.68 (м, 1Н), 5.51 (м, 1Н), 7.27 (м, 1Н), 7.47 (д, J=8.56 Гц, 1Н), 7.57 (м, 1Н), 7.63 (д, J=8.07 Гц, 1Н).
LC-MS (время удерживания: 2.737 мин), MS m/z 498 (M+Na+).
Стадия 3:

К раствору метилового эфира 4-(Бензо[d]изоксазол-3-илокси)-1-(2-трет-бутилоксикарбониламино-3, 3-диметилбутирил)-пирролидин-2-карбоновой кислоты (63 мг, 0.132 ммоля) в смеси ТГФ (3.5 мл), метанола (2.0 мл) и воды (0.5 мл) прибавляют моногидрат гидроксида лития (83 мг, 1.89 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем ее подкисляют 1N раствором HCl до рН 3-5 и упаривают. Экстрагируют этилацетатом (2×30 мл) и объединенные органические вытяжки сушат (MgSO4). Упариванием растворителя получают желтоватое масло (61 мг, выход 100%).
К раствору вышеуказанного соединения (61 мг, 0.132 ммоля) в СН3CN (8 мл) прибавляют гидрохлорид (1R,2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (42 мг, 0.158 ммоля), DIEA (0.115 мл, 0.66 ммоля) и конденсирующий агент HOBt (30 мг, 0.198 ммоля) и HBTU (75 мг, 0.198 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ и получают в качестве конечного продукта желтую пленку (Соединение 334) (24 мг, выход 27%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01 (с, 9Н), 1.05 (м, 2Н), 1.12-1.26 (м, 11Н), 1.43 (м, 1Н), 1.86 (дд, J=8.07, 5.38 Гц, 1Н), 2.17-2.33 (м, 2Н), 2.67 (дд, J=12.96, 5.87 Гц, 1Н), 2.93 (м, 1Н), 4.05 (м, 1Н), 4.22 (м, 1Н), 4.49 (м, 2Н), 5.11 (д, J=10.21 Гц, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.55 (с, 1Н), 5.74 (м, 1Н), 7.29 (м, 1Н), 7.48 (д, J=8.32 Гц, 1Н), 7.54-7.64 (м, 2Н).
LC-MS (время удерживания: 2.767 мин), MS m/z 696 (M+Na+).
Пример 335: Получение Соединения 335
Схема 1:

Схема 2:


Стадия 1: (Схема 1, Стадия 1)
К раствору (2S,4R)-4-гидроксипирролидин-1,2-дикарбоновой кислоты (0.25 г, 1.08 ммоля) в СН3CN (10 мл) прибавляют гидрохлорид (1R,2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил))-карбаминовой кислоты (0.346 г, 1.30 ммоля), DIEA (0.94 мл, 5.41 ммоля) и конденсирующий агент HOBt (0.240 г, 1.62 ммоля) и HBTU (0.615 г, 1.62 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Получают в качестве конечного продукта желтое масло. Очищают на колонке для препаративной ВЭЖХ, получают бесцветное вязкое масло, которое затем растворяют в 4N HCl в диоксане. Реакционную смесь перемешивают при rt в течение ночи. Упариванием растворителя получают твердое вещество белого цвета в качестве продукта (200 мг, выход 49%).
LC-MS (время удерживания: 0.647 мин), MS m/z 344 (МН+).
Стадия 2: (Схема 1, Стадия 2)
К раствору вышеуказанного соединения (200 мг, 0.527 ммоля) в CH3CN (10 мл) прибавляют 2-метоксикарбониламино-3,3-диметилмасляную кислоту (0.15 г, 0.79 ммоля), DIEA (0.46 мл, 2.63 ммоля) и конденсирующий агент HOBt (0.121 г, 0.79 ммоля) и HBTU (0.30 г, 0.79 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают, получают желтоватое масло. Его очищают на колонке для препаративной ВЭЖХ и конечный продукт получают в виде твердого вещества белого цвета (интермедиат 2) (145 мг, выход 54%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 0.99-1.10 (м, 11Н), 1.24 (м, 2Н), 1.41 (дд, J=9.5, 5.5 Гц, 1Н), 1.87 (дд, J=7.9, 5.5 Гц, 1Н), 1.97 (м, 1Н), 2.13 (м, 1Н), 2.24 (м, 1Н), 2.93 (м, 1Н), 3.65 (с, 3H), 3.77-3.88 (м, 2Н), 4.33-4.39 (м, 2Н), 4.49 (м, уш, 1Н), 5.13 (д, J=10.4 Гц, 1Н), 5.31 (д, J=17.1 Гц, 1Н), 5.76 (м, 1Н).
LC-MS (время удерживания: 1.590 мин), MS m/z 515 (MH+).
Стадия 3: (Схема 2)
К раствору 2,4-дихлорпиримидина (149 мг, 1 ммоль) в ТГФ (5 мл) прибавляют тетракис(трифенилфосфин)палладий (23 мг, 2 мольн.%) и 0.5М раствор фенилцинкбромида (2.1 мл, 1.05 ммоля) в ТГФ. Реакционную смесь перемешивают при 50°С в течение ночи. Затем добавляют насыщенный раствор хлористого аммония и дважды экстрагируют EtOAc. Объединенные органические вытяжки промывают водой и сушат (MgSO4). Упариванием растворителя получают желтый остаток, который очищают препаративной ВЭЖХ, получая желтоватое масло - 2-хлор-4-фенилпиримидин.
К раствору интермедиата 2 (20 мг, 0.039 ммоля) в ДМФА (3 мл) при 0°С прибавляют NaH (3.9 мг 60% дисперсии в минеральном масле, 0.0975 ммоля). Реакционную смесь доводят до комнатной температуры и перемешивают при rt в течение 1 час. Затем прибавляют полученный выше 2-хлор-4-фенилпиримидин (18 мг сырого продукта). Реакционную смесь перемешивают при rt в течение ночи. Затем прибавляют воду и экстрагируют EtOAc. Органический слой отделяют, промывают рассолом и сушат (MgSO4). Упариванием растворителя получают желтоватое масло, которое затем очищают препаративной ВЭЖХ, получают конечный продукт (Соединение 335), представляющий собой ТФК соль, в виде вязкого бесцветного масла (5.5 мг, выход 18%).
1H ЯМР (300 МГц, CD3OD) δ м.д. 0.92-1.12 (м, 11Н), 1.25 (м, 2Н), 1.44 (дд, J=9.2, 5.5 Гц, 1Н), 1.89 (дд, J=8.1, 5.5 Гц, 1Н), 2.17-2.37 (м, 2Н), 2.57 (м, 1Н), 2.95 (м, 1Н), 3.52 (с, 3H), 4.14 (м, 1Н), 4.24-4.38 (м, 2Н), 4.51 (м, 1Н), 5.13 (д, J=10.2 Гц, 1Н), 5.31 (д, J=17.2 Гц, 1Н), 5.77 (м, 1Н), 5.86 (с, 1Н), 7.48-7.60 (м, 3H), 7.66 (д, J=5.3 Гц, 1Н), 8.18 (м, 2Н), 8.60 (д, J=5.1 Гц, 1Н).
LC-MS (время удерживания: 1.947 мин), MS m/z 669 (MH+).
Пример 336: Получение Соединения 336

Стадия 1:
К раствору 2,4-дихлорпиримидина (149 мг, 1 ммоль) в ТГФ (5 мл) прибавляют тетракис(трифенилфосфин)палладий (56 мг, 5 мольн.%) и 0.5М раствор пиридинилцинкбромида (2.4 мл, 1.2 ммоля) в ТГФ. Реакционную смесь перемешивают при 50°С в течение ночи. Затем добавляют насыщенный раствор хлористого аммония и дважды экстрагируют EtOAc. Объединенные органические вытяжки промывают водой и сушат (MgSO4). Упариванием растворителя получают желтый остаток, который очищают препаративной ВЭЖХ, получая продукт - желтоватое масло.
1H ЯМР (500 МГц, CD3OD) δ м.д. 7.61 (м, 1Н), 8.07 (м, 1Н), 8.36 (д, J=5.19 Гц, 1Н), 8.50 (д, J=7.94 Гц, 1Н), 8.75 (д, J=3.97 Гц, 1Н), 8.82 (д, J=5.19 Гц. 1Н).
LC-MS (время удерживания: 1.440 мин), MS m/z 192 (МН+).
Стадия 2:
К раствору интермедиата 2 из Примера 335 (15 мг, 0.029 ммоля) в ДМФА (3 мл) при 0°С прибавляют NaH (1.75 мг 60% дисперсии в минеральном масле, 0.0728 ммоля). Реакционную смесь доводят до комнатной температуры и перемешивают при rt в течение 1 час. Затем прибавляют полученный выше 2-хлор-4-пиридин-2-илпиримидин (9.5 мг, 0.0311 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем прибавляют воду и экстрагируют EtOAc. Органический слой отделяют, промывают рассолом и сушат (MgSO4). Упариванием растворителя получают желтоватое масло, которое затем очищают препаративной ВЭЖХ, получают конечный продукт (Соединение 335), представляющий собой ТФК соль, в виде желтоватой пленки (5.5 мг, выход 18%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 1.03 (с, 9Н), 1.08 (м, 2Н), 1.24 (м, 2Н), 1.43 (дд, J=9.77, 5.50 Гц, 1Н), 1.89 (м, 1Н), 2.24 (м, 1Н), 2.31 (м, 1Н), 2.57 (м, 1Н), 2.95 (м, 1Н), 3.50 (с, 3H), 4.13 (м, 1Н), 4.29 (м, 1Н), 4.36 (д, J=11.91 Гц, 1Н), 4.52 (м, 1Н), 5.13 (д, J=10.08 Гц, 1Н), 5.31 (д, J=16.79 Гц, 1Н), 5.76 (м, 1Н), 5.88 (м, 1Н), 7.64 (м, 1Н), 8.06-8.13 (м, 2Н), 8.54 (д, J=7.93 Гц, 1Н), 8.73-8.76 (м, 2Н).
LC-MS (время удерживания: 1.787 мин), MS m/z 670 (MH+).
Пример 337: Получение Соединения 337

Стадия 1:
К раствору 2,4-дихлорпиримидина (149 мг, 1 ммоль) в ДМФА (5 мл) прибавляют дихлорбис(трифенилфосфин)палладий (II) (35 мг, 5 мольн.%) и 2-(трибутилстаннил)тиофен (0.38 мл, 1.2 ммоля). Реакционную смесь греют при 70°С в течение 3 час. Затем добавляют насыщенный раствор KF в метаноле (20 мл) и перемешивают при rt в течение 4 час. Реакционную смесь упаривают с небольшим количеством силикагеля, остаток фильтруют через бумажный фильтр и промывают EtOAc. Затем фильтрат упаривают, остаток очищают препаративной ВЭЖХ, получая продукт в виде не чисто белого твердого вещества (110 мг, выход 35%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 7.20 (дд, J=5.01, 3.79 Гц, 1Н), 7.74 (дд, J=5.01, 1.10 Гц, 1Н), 7.77 (д, J=5.38 Гц, 1Н), 7.98 (дд, J=3.79, 1.10 Гц, 1Н), 8.55 (д, J=5.38 Гц, 1Н).
LC-MS (время удерживания: 1.453 мин), MS m/z 197 (MH+).
Стадия 2:
К раствору интермедиата 2 из Примера 335 (20 мг, 0.039 ммоля) в ДМФА (3 мл) при 0°С прибавляют NaH (7.8 мг 60% дисперсии в минеральном масле, 0.195 ммоля). Реакционную смесь доводят до комнатной температуры и перемешивают при rt в течение 1 час. Затем прибавляют полученный выше 2-хлор-4-тиофен-2-илпиримидин (16.9 мг, 0.0544 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем прибавляют воду и экстрагируют EtOAc. Органический слой отделяют, промывают рассолом и сушат (MgSO4). Упариванием растворителя получают желтоватое масло, которое затем очищают препаративной ВЭЖХ, получают два продукта (Соединение 337 и интермедиат 3).
Соединение 337: (желтоватая пленка, 3.0 мг, выход 11%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 0.98-1.07 (м, 11Н), 1.22 (м, 2Н), 1.41 (дд, J=9.54, 5.62 Гц, 1Н), 1.86 (дд, J=8.32, 5.63 Гц, 1Н), 2.19-2.31 (м, 2Н), 2.52 (м, 1Н), 2.92 (м, 1Н), 3.50 (с, 3H), 4.09 (м, 1Н), 4.25-4.32 (м, 2Н), 4.47 (дд, J=10.03, 7.34 Гц, 1Н), 5.11 (дд, J=10.27, 1.71 Гц, 1Н), 5.28 (дд, J=17.11, 1.46 Гц, 1Н), 5.69-5.79 (м, 2Н), 7.20 (дд, J=4.89, 3.66 Гц, 1Н), 7.51 (д, J=5.38 Гц, 1Н), 7.70 (д, J=4.89 Гц, 1Н), 7.95 (д, J=3.67 Гц, 1Н), 8.54 (д, J=5.14 Гц, 1Н).
LC-MS (время удерживания: 1.787 мин), MS m/z 696 (M+Na+).
Интермедиат 3: (10 мг, выход 35%).
LC-MS (время удерживания: 1.477 мин), MS m/z 617 (MH+ ).
Пример 338: Получение Соединения 338

К раствору (1-циклопропансульфониламинокарбонил-2-винилциклопропиламида 1-(2-амино-3,3-диметилбутирил)-4-(4-тиофен-2-ил-пиримидин-2-илокси)-пирролидин-2-карбоновой кислоты (10 мг, 0.0137 ммоля) в СН3 CN (5 мл) прибавляют циклопропилуксусную кислоту (2.1 мг, 0.0205 ммоля), DIEA (0.012 мл, 0.0742 ммоля) и конденсирующий агент HOBt (3.1 г, 0.0205 ммоля) и HBTU (7.8 мг, 0.0205 ммол). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Получают желтоватое масло. Его очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль (Соединение 338) в виде желтоватой пленки (4.6 мг, выход 41%).
lH ЯМР(400 МГц, CD3OD) δ м.д. 0.12 (m, 2Н), 0.48 (м, 2Н), 0.90 (м, 1Н), 1.01-1.09 (м, 11Н), 1.23 (м,2Н), 1.43 (дд, J=9.29, 5.38 Гц, 1Н), 1.87 (дд, J=8.31, 5.62 Гц, 1Н), 2.06 (м, 2Н), 2.19-2.31 (м, 2Н), 2.52 (дд, J=13.45, 6.85 Гц, 1Н), 2.93 (м, 1Н), 4.12 (дд, J=11,98, 3,91 Гц, 1Н), 4.27 (д, J=11.74 Гц, 1Н), 4.47 (дд, J=10.27, 6.85 Гц, 1Н), 4.63 (с, 1Н), 5.11 (дд, J=10.27, 1.47 Гц, 1Н), 5.28 (дд, J=17.12, 1.47 Гц, 1Н), 5.71-5.80 (м, 2Н), 7.20 (дд, J=4.89, 3.67 Гц, 1Н), 7.51 (д, J=5.38 Гц, 1Н), 7.70 (д, J=5.20 Гц, 1Н), 7.95 (д, J=3.67 Гц. 1Н), 8.48 (д, J=5.13 Гц, 1Н).
LC-MS (время удерживания: 1.833 мин), MS m/z 699 (MH+).
Пример 339: Получение Соединения 339

Соединение 339 получают по Схемам из Примера 337 и Примера 338 за исключением того, что на Стадии 1 вместо 1 2-(трибутилстаннил)тиофена из Примера 337 используют 2-(трибутилстаннил)фуран.
Стадия 1:
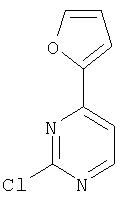
К раствору 2,4-дихлорпиримидина (149 мг, 1 ммоль) в ДМФА (5 мл) прибавляют дихлорбис(трифенилфосфин)палладий (II) (35 мг, 5 мольн.%) и 2-(трибутилстаннил)фуран (0.35 мл, 1.2 ммоля). Реакционную смесь греют при 70°С в течение 3 час. Затем добавляют насыщенный раствор KF в метаноле (20 мл) и перемешивают при rt в течение 4 час. Реакционную смесь упаривают с небольшим количеством силикагеля, остаток фильтруют через бумажный фильтр и промывают EtOAc. Затем фильтрат упаривают, остаток очищают препаративной ВЭЖХ, получая продукт в виде коричневатого твердого вещества (80 мг, выход 27%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 6.68 (дд, J=3.67, 1.71 Гц, 1Н), 7.42 (д, J=3.67 Гц, 1Н), 7.67 (д, J=5.13 Гц, 1Н), 7.30 (д, J=1.71 Гц, 1Н), 8.62 (д, J=5.14 Гц, 1Н).
LC-MS (время удерживания: 1.233 мин), MS m/z 181 (МН+).
Стадия 2:

К раствору метилового эфира {1-[2-(1-циклопропансульфониламинокарбонил-2-винилциклопропилкарбамоил)-4-гидроксиоксипирролидин-1-карбонил]-2,2-диметилпропил}-карбаминовой кислоты (20 мг, 0.0544 ммоля) в ДМФА (3 мл) при 0°С прибавляют NaH (7.8 мг 60% дисперсии в минеральном масле, 0.195 ммоля). Реакционную смесь доводят до комнатной температуры и перемешивают при rt в течение 1 час. Затем прибавляют полученный выше 2-хлор-4-фуран-2-илпиримидин (16.0 мг, 0.0544 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем прибавляют воду и экстрагируют EtOAc. Органический слой отделяют, промывают рассолом и сушат (MgSO4). Упариванием растворителя получают желтоватое масло, которое затем очищают препаративной ВЭЖХ, получают продукт реакции сочетания, не содержащий Boc-группы (3 мг, выход 11%).
Стадия 3:

К раствору (1-циклопропансульфониламинокарбонил-2-винилциклопропил-амида 1-(2-амино-3,3-диметилбутирил)-4-(4-фуран-2-ил-пиримидин-2-илокси)-пирролидин-2-карбоновой кислоты (3 мг, 0.0042 ммоля) в CH3CN (5 мл) прибавляют циклопропилуксусную кислоту (0.6 мг, 0.0063 ммоля), DIEA (0.004 мл, 0.021 ммоля) и конденсирующий агент HOBt (1.0 г, 0.063 ммоля) и HBTU (2.4 мг, 0.0063 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Получают желтое масло. Его очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль (Соединение 339) в виде желтоватой пленки (1.0 мг, выход 30%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.12 (м, 2Н), 0.48 (м, 2Н), 0.90 (м, 1Н), 0.99-1.09 (м, 11Н), 1.23 (м, 2Н), 1.43 (дд, J=9.05, 5.31 Гц, 1Н), 1.87 (м, 1Н), 2.05 (м, 2Н), 2.19-2.29 (м, 2Н), 2.50 (м, 1Н), 2.93 (м, 1Н), 4.10 (дд, J=12.23, 3.91 Гц, 1Н), 4.25 (д, J=11.99 Гц, 1Н), 4.47 (дд, J=10.52, 7.09 Гц, 1Н), 4.63 (с, 1Н), 5.11 (дд, J=10.52, 1.71 Гц, 1Н), 5.29 (дд, J=17.12, 1.47 Гц, 1Н), 5.71-5.79 (м, 2Н), 6.65 (дд, J=3.67, 1.96 Гц, 1Н). 7.38 (д, J=3.67 Гц, 1Н), 7.40 (д, J=5.38 Гц, 1Н), 7.76 (м, 1Н), 8.54 (д, J=5.38 Гц, 1Н).
LC-MS (время удерживания: 1.790 мин), MS m/z 683 (MH+).
Пример 340: Получение Соединения 340


Стадия 1:
К раствору 2,4-дихлорпиримидина (149 мг, 1 ммоль) в ДМФА (5 мл) прибавляют дихлорбис(трифенилфосфин)палладий (II) (35 мг, 5 мольн.%) и 2-(трибутилстаннил)тиазол (412 мг, 1.1 ммоля). Реакционную смесь греют при 80°С в течение 3 час. Затем добавляют насыщенный раствор KF в метаноле (20 мл) и перемешивают при rt в течение 4 час. Реакционную смесь упаривают с небольшим количеством силикагеля, остаток фильтруют через бумажный фильтр и промывают EtOAc. Затем фильтрат упаривают, остаток очищают препаративной ВЭЖХ, получая продукт в виде коричневатого твердого вещества (9 мг, выход 3%).
LC-MS (время удерживания: 1.320 мин), MS m/z 198 (MH+).
Стадия 2:
К раствору гидрохлорида (1-циклопропансульфониламинокарбонил-2-винилциклопропил-амида 1-[2-(2-циклопропилацетиламино)-3, 3-диметилбутирил]-4-гидроксипирролидин-2-карбоновой кислоты (12.5 мг, 0.0232 ммоля) в ДМФА (3 мл) при 0°С прибавляют NaH (3.7 мг 60% дисперсии в минеральном масле, 0.0928 ммоля). Реакционную смесь доводят до комнатной температуры и перемешивают при rt в течение 1 час. Затем прибавляют 2-хлор-4-тиазол-2-илпиримидин (9.0 мг, 0.0289 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем прибавляют воду и экстрагируют EtOAc. Органический слой отделяют, промывают рассолом и сушат (MgSO4). Упариванием растворителя получают сырой продукт, который затем очищают препаративной ВЭЖХ, получают продукт в виде твердого вещества белого цвета (Соединение 340) (2.83 мг, выход 17%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.12 (м, 2Н), 0.47 (м, 2Н), 0.89 (м, 1Н), 1.00-1.09 (м, 11Н), 1.22 (м, 2Н), 1.44 (дд, J=9.54, 5.38 Гц, 1Н), 1.87 (дд, J=8.07, 5.38 Гц, 1Н), 2.06 (м, 2Н), 2.20-2.32 (м, 2Н), 2.52 (дд, J=13.70, 6.85 Гц, 1Н), 2.93 (м, 1Н), 4.13 (дд, J=11.98, 3.91 Гц, 1Н), 4.30 (д, J=11.98 Гц, 1Н), 4.48 (дд, J=10.51, 7.09 Гц, 1Н), 4.63 (д, J=9.54 Гц, 1Н), 5.11 (д, J=10.51 Гц, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.73-5.80 (м, 2Н), 7.81 (д, J=5.14 Гц. 1Н), 7.84 (д, J=3.18 Гц, 1Н), 8.03 (д, J=2.93 Гц, 1Н), 8.68 (д, J=5.13 Гц, 1Н).
LC-MS (время удерживания: 1.710 мин), MS m/z 700 (MH+).
Пример 341: Получение Соединения 341
Схема 1:

Схема 2:

Стадия 1 (Схема 1, Стадия 1):
К раствору Boc-HYP-ОН (1.0 мг, 4.324 ммоля) в ДМФА (20 мл) при 0°С прибавляют NaH (0.38 мг 60% дисперсии в минеральном масле, 9.513 ммоля). Реакционную смесь доводят до комнатной температуры и перемешивают при rt в течение 1 час. Затем прибавляют 2,4-дихлорпиримидин (0.709 мг, 0.0289 ммоля). Реакционную смесь доводят до rt и перемешивают при этой температуре в течение ночи. Затем прибавляют 1N HCl и экстрагируют EtOAc. Органический слой отделяют, промывают рассолом и сушат (MgSO4). Упариванием растворителя получают сырой продукт, который затем очищают препаративной ВЭЖХ, получают продукт - бесцветное масло (0,4 г, выход 27%).
1H ЯМР (300 МГц, CD3OD) δ м.д. 1.13 (м, 9Н), 2.37 (м, 1Н), 2.62 (м, 1Н), 3.70-3.84 (м, 2Н), 4.38 (м, 1Н), 5.65 (м, 1Н), 6.88 (д, J=5.86 Гц, 1Н), 8.37 (д, J=5.86 Гц, 1Н).
LC-MS (время удерживания: 1.370 мин), MS m/z 344 (МН+).
Стадия 2: (Схема 1, Стадия 2)
К раствору 1-трет-бутилового эфира (1S,4R) 4-(2-хлорпиримидин-4-илокси)-пирролидин-1,2-дикарбоновой кислоты (0.34 г, 0.99 ммоля) в СН3CN (20 мл) прибавляют (1R,2S)/(1S, 2R) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовую кислоту (0.511 г, 1.48 ммоля), DIEA (0.86 мл, 4.95 ммоля) и ковденсирующий агент HOBt (0.226 г, 1.48 ммоля) и HBTU (0.561 г, 1.48 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Затем очищают на колонке для препаративной ВЭЖХ, получают твердое вещество желтого цвета (интермедиат 4) (0.33 г, выход 41%).
1H ЯМР (400 МГц, CD3OD) δ м.д. смесь стереоизомеров.
LC-MS (время удерживания: 2.907 мин), MS m/z 655 (МН+).
Стадия 3: (Схема 2, Стадия 1)
К раствору интермедиата 4 (50 мг, 0.061 ммоля) в СН2Cl2 (2.5 мл) прибавляют 1,2,3,4-тетрагидроизохинолин (0.011 мл, 0.0915 ммоля) и Et3N (0.021 мл, 0.153 ммоля). Реакционную смесь перемешивают при rt в течение ночи и при 40°С в течение 1 дня. Растворитель отгоняют и остаток очищают препаративной ВЭЖХ, получают бесцветное масло. Его растворяют в 4N HCl в диоксане (1 мл) и перемешивают в течение ночи. Упариванием растворителя получают бесцветное масло - солянокислая соль (гидрохлорид) (20 мг, выход 52%).
LC-MS (время удерживания: 1.160 мин), MS m/z 553 (MH+).
Стадия 4: (Схема 2, Стадия 2)
К раствору гидрохлорида (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-амида 4-[2-(3, 4-дигидро-1Н-изохинолин-2-ил)-пиримидин-4-илокси]-пирролидин-2-карбоновой кислоты (20 мг, 0.032 ммоля) в СН3CN (5 мл) прибавляют 2-метоксикарбониламино-3,3-диметилмасляную кислоту (9.1 мг, 0.048 ммоля), DIEA (0.028 мл, 0.16 ммоля) и конденсирующий агент HOBt (7.3 мг, 0.048 ммоля) и HBTU (18.2 мг, 0.048 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Получают желтоватое масло. Его очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль (Соединение 341) в виде бесцветного масла (16 мг, выход 60%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 0.98-1.06 (м, 13Н), 1.13 (м, 1Н), 1.22-1.32 (м, 1Н), 1.35-1.44 (м, 1Н), 1.82 (дд, J=8.24, 5.19 Гц, 0.5Н), 1.90 (дд, J=8.24, 5.49 Гц, 0.5Н), 2.26 (м, 1Н), 2.32-2.43 (м. 1Н), 2.56 (м, 1Н), 2.96 (м, 1Н), 3.11 (м, уш, 2Н), 3.56 (с, 3H), 4.14 (м, 1Н), 4.21 (м, 1Н), 4.38 (м, 1Н), 4.47 (м, 1Н), 5.15 (м, 1Н), 5.31 (м, 1Н), 5.75 (м, 1Н), 5.94 (с, 1Н), 6.47 (д, J=7.02 Гц, 1Н), 7.29 (с, 4Н), 7.49 (м, 1Н), 7.56 (м, 1Н), 7.74 (д, J=8.24 Гц, 1Н), 7.88 (д, J=8.24 Гц, 1Н). 8.11 (д, J=7.02 Гц, 1Н).
LC-MS (время удерживания: 1.517 мин), MS m/z 724 (MH+).
Пример 342: Получение Соединения 342

Соединение 342 получают по Схеме 2 из Примера 341, за исключением того, что вместо 1,2,3,4-тетрагидроизохинолина на Стадии 1 Схемы 2 используют изоиндолин.
Стадия 1:

К раствору интермедиата 4 из Примера 341 (50 мг, 0.061 ммоля) в СН2Cl2 (2.5 мл) прибавляют изоиндолин (0.013 мл, 0.115 ммоля) и Et3N (0.026 мл, 0.19 ммоля). Реакционную смесь перемешивают при rt в течение 2 дней. Растворитель отгоняют в вакууме, а остаток очищают препаративной ВЭЖХ, получают бесцветное масло. Его растворяют в 4N HCl в диоксане (1 мл) и перемешивают в течение ночи. Упариванием растворителя получают сырой продукт, который снова очищают препаративной ВЭЖХ, получают ТФК соль - твердое вещество желтоватого цвета (8.5 мг, выход 14%).
LC-MS (время удерживания: 1.860 мин), MS m/z 539 (MH+).
Стадия 2:

К раствору гидрохлорида (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-амида 4-[2-(1, 3-дигидроизондол-2-ил)-пиримидин-4-илокси]-пирролидин-2-карбоновой кислоты (8.5 мг, 0.0104 ммоля) в CH3CN (5 мл) прибавляют 2-метоксикарбониламино-3,3-диметилмасляную кислоту (3.0 мг, 0.0156 ммоля), DIEA (0.009 мл, 0.052 ммоля) и конденсирующий агент HOBt (2.4 мг, 0.0156 ммоля) и HBTU (5.9 мг, 0.0156 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Получают желтоватое масло. Его очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль (Соединение 342) в виде бесцветного масла (3 мг, выход 35%).
1H ЯМР (300 МГц, CD3OD) δ м.д. смесь диастереомеров.
LC-MS (время удерживания: 2.547 мин), MS m/z 710 (МН+).
Пример 343: Получение Соединения 343
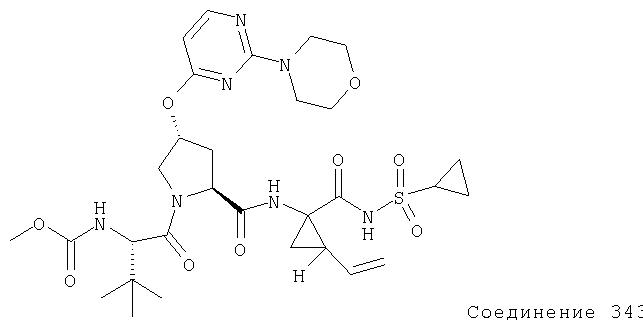
Соединение 343 получают по Схеме 2 из Примера 341, за исключением того, что вместо 1,2,3,4-тетрагидроизохинолина на Стадии 1 Схемы 2 используют морфолин.
Стадия 1:

К раствору интермедиата 4 из Примера 341 (50 мг, 0.061 ммоля) в CH2Cl2 (2.5 мл) прибавляют морфолин (0.008 мл, 0.0915 ммоля) и Et3N (0.021 мл, 0.153 ммоля). Реакционную смесь перемешивают при rt в течение ночи и при 40°С в течение 1 часа. Растворитель отгоняют в вакууме, а остаток очищают препаративной ВЭЖХ, получают бесцветное масло. Его растворяют в 4N HCl в диоксане (1 мл) и перемешивают в течение ночи. Упариванием растворителя получают соль - гидрохлорид в виде бесцветного масла (12.6 мг, выход 36%).
LC-MS (время удерживания: 0.810 мин), MS m/z 507 (МН+).
Стадия 2:

К раствору гидрохлорида (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-амида 4-(2-морфолин-4-ил-пиримидин-4-илокси)-пирролидин-2-карбоновой кислоты (12.6 мг, 0.0217 ммоля) в CH3CN (5 мл) прибавляют 2-метоксикарбониламино-3,3-диметилмасляную кислоту (6.2 мг, 0.0326 ммоля), DIEA (0.019 мл, 0.1085 ммоля) и конденсирующий агент HOBt (5.0 мг, 0.0326 ммоля) и HBTU (12.4 мг, 0.0326 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Получают желтоватое масло. Его очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль (Соединение 343) в виде бесцветного масла (7 мг, выход 41%).
1H ЯМР (300 МГц, CD3OD) δ м.д. смесь диастереомеров.
LC-MS (время удерживания: 1.280 мин), MS m/z 678 (МН+).
Пример 344: Получение Соединения 344
Схема 1:

Схема 2:
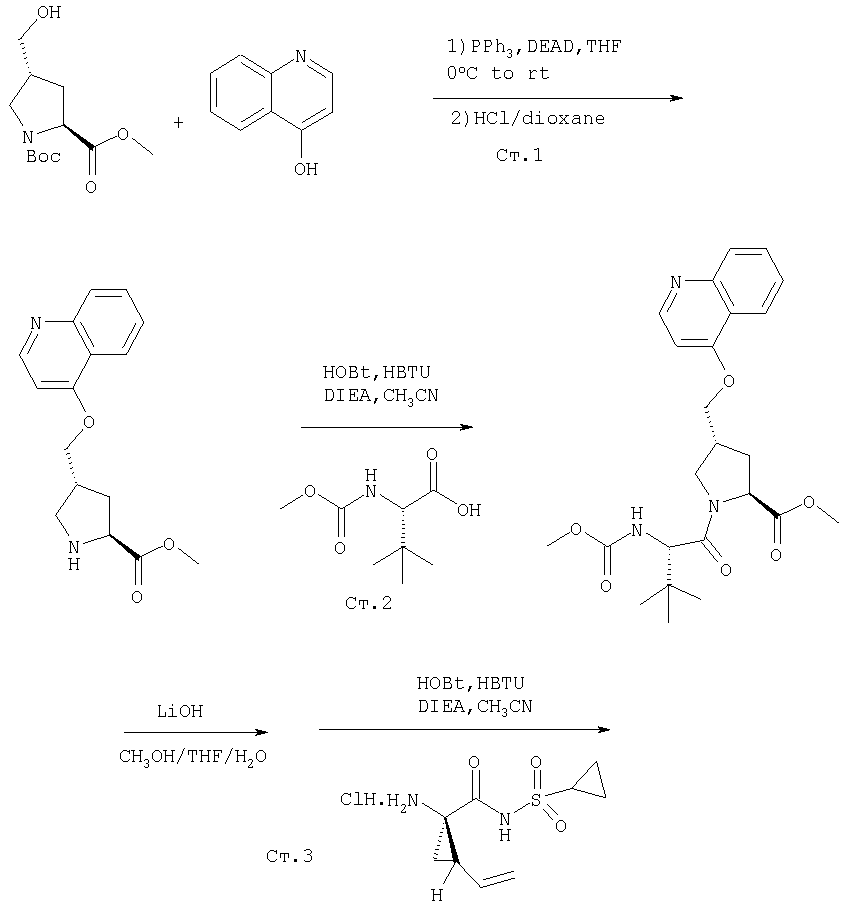

Стадия 1: (Схема 1)
1.43 (м, 9Н), 2.00-2.13 (м, 2Н), 2.46 (м, 1Н), 3.19 (м, 1Н), 3.47-3.53 (м, 2Н), 3.61 (м, 1Н), 3.73 (м, 3H), 4.31 (м, 1Н).
LC-MS (время удерживания: 1.240 мин), MS m/z 282 (M+Na+).
Стадия 2: (Схема 2, Стадия 1)
К раствору интермедиата 5 (80 мг, 0.309 ммоля) в ТГФ (10 мл) при 0°С прибавляют трифенилфосфин (121.4 мг, 0.463 ммоля) и 4-гидроксихинолин (67.2 мг, 0.463 ммоля). Затем прибавляют DEAD (80.6 мг, 0.463 ммоля). Температуру реакционной смеси доводят до комнатной и перемешивают 2 дня. Упаривают растворитель и остаток очищают препаративной ВЭЖХ, получая бесцветное масло. Его растворяют в 4N HCl в диоксане (3 мл) и перемешивают 2 часа. Упариванием растворителя получают бис HCl соль в виде вязкого бесцветного масла (110 мг, выход 99%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 2.52 (м, 1Н), 2.60 (м, 1Н), 3.19 (м, 1Н), 3.45 (м, 1Н), 3.66 (с, 3H), 3.86 (м, 1Н), 4.61-4.75 (м, 3H), 7.56 (д, J=6.7 Гц, 1Н), 7.94 (т, J=7.3 Гц, 1Н), 8.10-8.20 (м, 2Н), 8.55 (д, J=8.2 Гц, 1Н), 9.07 (д, J=6.7 Гц, 1Н).
LC-MS (время удерживания: 0.570 мин), MS m/z 287 (MH+).
Стадия 3: (Схема 2, Стадия 2)
К раствору бис-гидрохлорида метилового эфира 4-(4-хинолин-4-илоксиметил)-пирролидин-2-карбоновой кислоты (110 мг, 0.306 ммоля) в CH3CN (10 мл) прибавляют 2-метоксикарбониламино-3,3-диметилмасляную кислоту (87 мг, 0.46 ммоля), DIEA (0.27 мл, 1.53 ммоля) и конденсирующий агент HOBt (70 мг, 0.46 ммоля) и HBTU (174 мг, 0.46 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Получают желтоватое масло. Его очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль в виде бесцветного масла (105 мг, выход 60%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 1.07 (с, 9Н), 2.34 (м, 1Н), 2.45 (м, 1Н), 3.14 (м, 1Н), 3.27 (с, 3H), 3.75 (с, 3H), 4.05 (м, 1Н), 4.20 (м, 1Н), 4.31 (с, 1Н), 4.57-4.63 (м, 2Н), 4.73 (м,1Н), 7.53 (д, J=6.7 Гц, 1Н), 7.91 (т, J=7.6 Гц, 1Н), 8.06-8.16 (м, 2Н), 8.43 (д, J=8.6 Гц, 1Н), 9.02 (д, J=6.4 Гц, 1Н).
LC-MS (время удерживания: 0.250 мин), MS m/z 458 (МН+).
Стадия 4: (Схема 2, Стадия 3)
К раствору метилового эфира 1-(2-метоксикарбониламино-3,3-диметилбутирил)-4-(хинолин-4-илоксиметил)-пирролидин-2-карбоновой кислоты (100 мг, 0.175 ммоля) в смеси ТГФ (6 мл), метанола (3.25 мл) и воды (1.0 мл) прибавляют моногидрат гидроксида лития (110 мг, 2.62 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем ее подкисляют 1N раствором HCl до рН 3-5 и упаривают. Экстрагируют этилацетатом (3× 40 мл) и объединенные органические вытяжки сушат (MgSO4). Упариванием растворителя получают вязкое бесцветное масло (25 мг, выход 32%).
К раствору вышеуказанного соединения (25 мг, 0.056 ммоля) в CH3CN (5 мл) прибавляют гидрохлорид (1R,2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (22.5 мг, 0.085 ммоля), DIEA (0.05 мл, 0.28 ммоля) и конденсирующий агент HOBt (30 мг, 0.198 ммоля) и HBTU (75 мг, 0.198 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают, получая вязкое бесцветное масло. Его очищают на колонке для препаративной ВЭЖХ и получают в качестве конечного продукта желтую пленку (Соединение 344) (20 мг, выход 46%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 1.02-1.10 (м, 11Н), 1.24 (м, 2Н), 1.40 (дд, J=9.2, 5.5 Гц, 1Н), 1.90 (дд, J=7.9, 5.5 Гц, 1Н), 2.19-2.38 (м, 3H), 2.95 (м, 1Н), 3.19 (м, 1Н), 3.28 (с, 3H), 4.10 (м, 1Н), 4.15 (м, 1Н), 4.34 (с, 1Н), 4.55 (м, 1Н), 4.62 (д, J=4,6 Гц, 2Н), 5.15 (д, J=10.7 Гц, 1Н), 5.30 (д, J=17.1 Гц, 1Н), 5.72 (м, 1Н), 7.54 (д, J=6.7 Гц, 1Н), 7.93 (м, 1Н), 8.07-8.18 (м, 2Н), 8.41 (д, J=8.6 Гц, 1Н), 9.03 (д, J=6.7 Гц. 1Н), 9.09 (с, 1Н).
LC-MS (время удерживания: 1.617 мин), MS m/z 656 (МН+).
Пример 345: Получение Соединения 345

Соединение 345 получают по Схеме 2 из Примера 344 за исключением того, что вместо 4-гидроксихинолина на Стадии 1 Схемы 2 используют 3-бромфенол.
Стадия 1:

К раствору интермедиата 5 из Примера 344 (150 мг, 0.579 ммоля) в ТГФ (15 мл) при 0°С прибавляют трифенилфосфин (228 мг, 0.868 ммоля) и 3-бромфенол (150 мг, 0.868 ммоля). Затем прибавляют DEAD (0.14 мл, 0.868 ммоля). Температуру реакционной смеси доводят до комнатной и перемешивают 2 дня. Упаривают растворитель и остаток очищают препаративной ВЭЖХ, получая бесцветное масло. Получают продукт в виде бесцветного масла (105 мг, выход 44%).
LC-MS (время удерживания: 2.023 мин), MS m/z 434 (M+Na+).
Стадия 2:
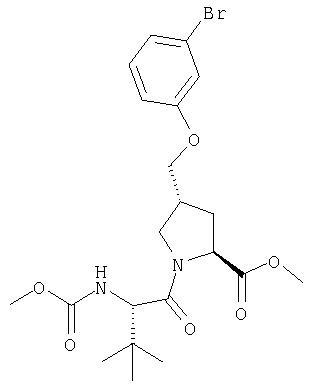
1-трет-Бутиловый эфир 2-метиловый эфир 4-(3-бромфеноксиметил)-пирролидин-1,2-дикарбоновой кислоты (35 мг, 0.085 ммоля) растворяют в 4N HCl в диоксане (1.5 мл) и перемешивают в течение двух часов. Растворитель упаривают, получают вязкое бесцветное масло. К раствору этого масла в CH3CN (10 мл) прибавляют 2-метоксикарбониламино-3, 3-диметилмасляную кислоту (21.9 мг, 0.1155 ммоля), DIEA (0.067 мл, 0.385 ммоля) и конденсирующий агент HOBt (17.7 мг, 0.1155 ммоля) и HBTU (43.8 мг, 0.1155 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают бесцветное масло (20 мг, выход 49%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.03 (с, 9Н), 2.15 (м, 1Н). 2.24 (м, 1Н), 2.83 (м, 1Н), 3.54 (с, 3H), 3.70 (с, 3H), 3.87 (м, 1Н), 3.91-3.98 (м, 3H), 4.31 (с, 1Н), 4.59 (дд, J=8.80, 5.38 Гц, 1Н), 6.89 (д, J=8.32 Гц, 1Н), 7.03-7.10 (м, 2Н), 7.15 (т, J=8.07 Гц, 1Н).
LC-MS (время удерживания: 1.943 мин), MS m/z 485 (МН+).
Стадия 3:
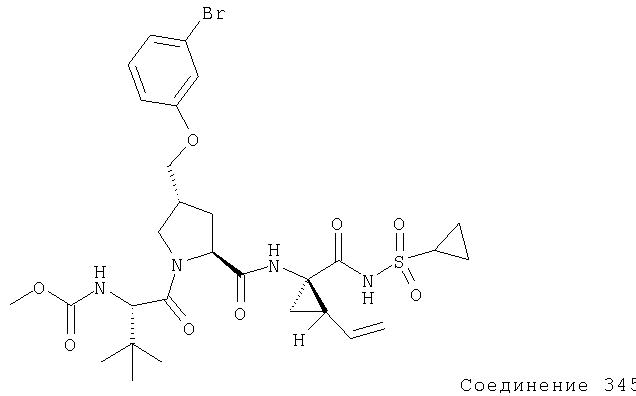
К раствору метилового эфира 4-(бромфеноксиметил)-1-(2-метоксикарбониламино-3,3-диметилбутирил)-пирролидин-2-карбоновой кислоты (17 мг, 0.035 ммоля) в смеси ТГФ (1.5 мл), метанола (0.8 мл) и воды (0.25 мл) прибавляют моногидрат гидроксида лития (22 мг, 0.525 ммоля). Реакционную смесь перемешивают при rt в течение 3 дней. Затем ее подкисляют 1N раствором HCl до рН 3-5 и упаривают. Экстрагируют этилацетатом (2×20 мл) и объединенные органические вытяжки сушат (MgSO4). Упариванием растворителя получают вязкое бесцветное масло (15 мг, выход 91%).
К раствору полученной выше кислоты (15 мг, 0.0318 ммоля) в СН3CN (5 мл) прибавляют гидрохлорид (1R,1S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (12.7 мг, 0.0477 ммоля), DIEA (0.028 мл, 0.159 ммоля) и конденсирующий агент HOBt (7.3 мг, 0.0477 ммоля) и HBTU (18.1 мг, 0.0477 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ и получают в качестве конечного продукта вязкое масло (Соединение 345) (14 мг, выход 64%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00-1.06 (м, 11Н), 1.21 (м, 2Н), 1.37 (дд, J=9.53 Гц, 5.62 Гц, 1Н), 1.86 (дд, J=8.07 Гц, 5.62 Гц, 1Н), 2.06 (м, 1Н), 2.14-2.24 (м, 2Н), 2.81-2.94 (м, 2Н), 3.53 (с, 3H), 3.91-3.97 (м, 4Н), 4.33 (с, 1Н), 4.38 (м, 1Н), 5.11 (дд, J=10.27, 1.47 Гц, 1Н), 5.28 (дд, J=17.12 1.22 Гц, 1Н), 5.70 (м, 1Н), 6.89 (м, 1Н), 7.05-7.11 (м, 2Н), 7.16 (т, J=8.07 Гц, 1Н).
LC-MS (время удерживания: 3.500 мин), MS m/z 683 (МН+).
Пример 346: Получение Соединения 346
Схема 1:

Схема 2:

Стадия 1: (Схема 1, Стадия 1)
К раствору 1-трет-бутилового эфира 2-метилового эфира 4-гидроксиметилпирролидин-1,2-дикарбоновой кислоты (300 мг, 1.157 ммоля) в ТГФ (15 мл) при 0°С прибавляют трифенилфосфин (455 мг, 1.735 ммоля) и 5-бромпиридин-3-ол (полученный по методике: F.E.Ziegler et al., J.Am.Chem.Soc., (1973), 95 7458) (302 мг, 1.735 ммоля). Затем прибавляют DEAD (0.273 мл, 1.735 ммоля). Температуру реакционной смеси доводят до комнатной и перемешивают 2 дня. Упаривают растворитель и остаток очищают препаративной ВЭЖХ, получая бесцветное масло. Его растворяют в 4N HCl в диоксане (3 мл) и перемешивают 4 часа. Упариванием растворителя получают сырой продукт, который далее очищают препаративной ВЭЖХ, получают ТФК соль - масло желтоватого цвета (70 мг, выход 11%).
LC-MS (время удерживания: 0.890 мин), MS m/z 315 (МН+).
Стадия 2: (Схема 1, Стадия 2)
К раствору метилового эфира 4-(5-бромпиридин-3-илоксиметил)-пирролидин-2-карбоновой кислоты (70 мг, 0.129 ммоля) в CH3CN (10 мл) прибавляют 2-метоксикарбониламино-3,3-диметилмасляную кислоту (36.5 мг, 0.193 ммоля), DIEA (0.135 мл, 0.744 ммоля) и конденсирующий агент HOBt (30 мг, 0.193 ммоля) и HBTU (73 мг, 0.193 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ и продукт получают в виде бесцветного масла (80 мг, выход 100%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 1.04 (с, 9Н), 2.17 (м, 1Н), 2.26 (м, 1Н), 2.87 (м, 1Н), 3.50 (с, 3H), 3.70 (с, 3H), 3.88-3.98 (м, 2Н), 4.04-4.12 (м, 2Н), 4.28 (с, 1Н), 4.60 (дд, J=9.05, 5.87 Гц, 1Н), 7.86 (м, 1Н), 8.31-8.35 (м, 2Н).
LC-MS (время удерживания: 1.697 мин), MS m/z 486 (МН+).
Стадия 3: (Схема 1, Стадия 3)
К раствору метилового эфира 4-(5-бромпиридин-3-илоксиметил)-1-(2-метоксикарбониламино-3,3-диметилбутирил)-пирролидин-2-карбоновой кислоты (80 мг, 0.133 ммоля) в смеси ТГФ (5.6 мл), метанола (3 мл) и воды (1 мл) прибавляют моногидрат гидроксида лития (84 мг, 2.0 ммоля). Реакционную смесь перемешивают при rt в течение 3 дней. Затем ее подкисляют 1N раствором HCl до рН 3-5. Экстрагируют этилацетатом (2× 20 мл) и объединенные органические вытяжки сушат (MgSO4). Упариванием растворителя получают продукт в виде вязкого бесцветного масла (интермедиат 6) (50 мг, выход 80%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1 04 (с, 9Н), 2.16-2.30 (м, 2Н). 2 88 (м, 1Н), 3.51 (с, 3H), 3.92 (м, 2Н), 4.07 (м, 2Н), 4.29 (с, 1Н), 4.57 (дд, J=8.56, 5.87 Гц, 1Н), 7.79 (м, 1Н), 8.29 (м, 2Н).
LC-MS (время удерживания: 1.590 мин), MS m/z 472 (МН+).
Стадия 4: (Схема 2)
К раствору 4-(5-бромпиридин-3-илоксиметил)-1-(2-метоксикарбониламино-3,3-диметилбутирил)-пирролидин-2-карбоновой кислоты (5 мг, 0.0106 ммоля) в СН3CN (5 мл) прибавляют гидрохлорид (1R,2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (4.2 мг, 0.0159 ммоля), DIEA (0.009 мл, 0.053 ммоля) и конденсирующий агент HOBt (2.4 мг, 0.0159 ммоля) и HBTU (6.0 мг, 0.0159 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ и получают в качестве конечного продукта вязкое бесцветное масло (Соединение 346) (2 мг, выход 24%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00-1.07 (м, 11Н), 1.20 (м, 2Н). 1.37 (дд, J=9.29 Гц, 5.14 Гц, 1Н), 1.86 (дд, J=8.07 Гц, 5.38 Гц, 1Н), 2.08 (м, 1Н), 2.15-2.25 (м, 2Н), 2.87-2.94 (м, 2Н), 3.51 (с, 3H), 3.92-3.97 (м, 2Н), 4.02-4.07 (м, 2Н), 4.31 (с, 1Н), 4.39 (м, 1Н), 5.10 (дд, J=10.27, 1.47 Гц, 1Н), 5.28 (дд, J=17.12, 1.46 Гц, 1Н), 5.70 (м, 1Н), 7.68 (м, 1Н), 8.24 (м, 2Н).
LC-MS (время удерживания: 1.727 мин), MS m/z 684 (МН+).
Пример 347: Получение Соединения 347

Стадия 1:
К раствору интермедиата 6 из Примера 346 (16 мг, 0.0339 ммоля) в ДМФА (1 мл) прибавляют 3-тиофенбороновую кислоту (5.6 мг, 0.044 ммоля), тетракис(трифенилфосфин)палладий (2.0 мг, 0.017 ммоля) и 2М раствор Na2CO3 (0.051 мл, 0.1017 ммоля). Реакционную смесь нагревают при 110°С в течение 4 час. Затем ее отфильтровывают и промывают метанолом. Фильтрат упаривают и очищают препаративной ВЭЖХ, получая продукт в виде коричневатого масла (6 мг, выход 37%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.05 (с, 9Н), 2.21-2.30 (м, 2Н), 2.95 (м, 1Н), 3.42 (с, 3H), 3.93 (м, 1Н), 4.01 (м, 1Н), 4.20-4.30 (м, 3H), 4.60 (дд, J=8.56, 5.87 Гц, 1Н), 7.64 (м, 2Н), 8.12 (м, 1Н), 8.37 (м, 1Н), 8.45 (м, 1Н), 8.75 (с, 1Н).
LC-MS (время удерживания: 1.353 мин), MS m/z 476 (МН+).
Стадия 2:
К раствору 1-(2-метоксикарбониламино-3,3-диметилбутирил)-4-(5-тиофен-3-илпиридин-3-илоксиметил)-пирролидин-2-карбоновой кислоты (6 мг, 0.0126 ммоля) в CH3CN (5 мл) прибавляют гидрохлорид (1R, 2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (5.0 мг, 0.0189 ммоля), DIEA (0.011 мл, 0.063 ммоля) и конденсирующий агент HOBt (2.9 мг, 0.0189 ммоля) и HBTU (7.2 мг, 0.0189 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ и получают ТФК соль в виде желтоватой пленки (Соединение 347) (2.2 мг, выход 22%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.01-1.07 (м, 11Н), 1.19 (м, 2Н), 1.36 (м, 1Н), 1.88 (дд, J=8.07 Гц, 5.62 Гц, 1Н), 2.09-2.24 (м, 3H), 2.91 (м, 1Н), 3.00 (м, 1Н), 3.45 (с, 3H), 3.98 (д, J=5.86 ГЦ, 2H), 4.20-4.31 (м, 3H), 4.43 (м, 1Н), 5.12 (дд, J=10.27, 1.71 Гц, 1Н), 5.29 (дд, J=17.12, 1.22 Гц, 1Н), 5.69 (м, 1Н), 7.64 (м, 2Н), 8.11 (м, 1Н), 8.31 (м, 1Н), 8.43 (м, 1Н), 8.75 (с, 1Н).
LC-MS (время удерживания: 1.540 мин), MS m/z 688 (МН+).
Пример 348: Получение Соединения 348
Схема 1:

Схема 2:

Схема 3:
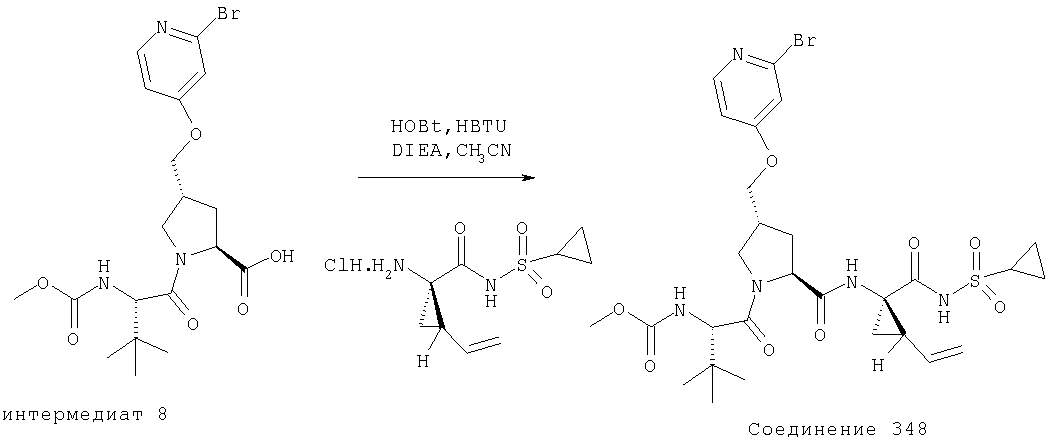
Стадия 1: (Схема 1)
К раствору интермедиата 5 из Примера 344 (700 мг, 2.7 ммоля) в смеси ТГФ (90 мл), метанола (50 мл) и воды (12 мл) прибавляют моногидрат гидроксида лития (1700 мг, 2.0 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем ее подкисляют 1N раствором HCl до рН 3-5. Экстрагируют этилацетатом (2×20 мл) и объединенные органические вытяжки сушат (MgSO4). Упариванием растворителя получают вязкое бесцветное масло (интермедиат 7) (0.58 г, выход 88%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.42 (м, 9Н), 2.00-2.09 (м, 2Н), 2.45 (м, 1Н), 3.17 (м, 1Н), 3.49 (м, 2Н), 3.59 (м, 1Н), 4.24 (м, 1Н).
LC-MS (время удерживания: 1.08 мин), MS m/z 268 (МН+).
Стадия 2: (Схема 2, Стадия 1)
К раствору интермедиата 7 (270 мг, 1.1 ммоля) в ДМСО (10 мл) прибавляют трет-бутоксид калия (309 мг, 2.75 ммоля). Реакционную смесь перемешивают при rt в течение 1 час. Затем прибавляют 2-бром-4-хлорпиридин (254 мг, 1.32 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем прибавляют воду и промывают этилацетатом. Водный слой отделяют и подкисляют 1N раствором HCl до рН 3. Дважды экстрагируют этилацетатом и органические вытяжки объединяют и сушат (MgSO4). Упариванием растворителя получают оранжевое масло. Затем его растворяют в метаноле и в течение 2 мин при -78°С, продувают HCl (газ). Затем температуру реакционной смеси доводят до комнатной и перемешивают в течение ночи. Упариванием растворителя получают сырой продукт в виде оранжевого масла.
LC-MS (время удерживания: 0.65 мин), MS m/z 315 (МН+).
Стадия 3: (Схема 2, Стадия 2)
К раствору сырого метилового эфира 4-(2-бромпиридин-4-илоксиметил)-пирролидин-2-карбоновой кислоты в CH3CN (20 мл) прибавляют 2-метоксикарбониламино-3,3-диметилмасляную кислоту (312 мг, 1.65 ммоля), DIEA (1.15 мл, 6.6 ммоля) и конденсирующий агент HOBt (252 мг, 1.65 ммоля) и HBTU (626 мг, 1.65 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ и конечный продукт получают в виде бесцветного масла (270 мг, выход на две стадии 41%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.03 (с, 9Н), 2.13-2.19 (м, 2Н), 2.87 (м, 1Н), 3.51 (с, 3H), 3.70 (с, 3H), 3.93 (д, J=6.36 Гц, 2Н), 4.11 (м, 2Н), 4.27 (с, 1Н), 4.60 (дд, J=8.80, 5.87 Гц, 1Н), 7.06 (д, J=5.87, 2.20 Гц, 1Н), 7.32 (д, J=2.20 Гц, 1Н), 8.18 (д, J=6.11 Гц, 1Н).
LC-MS (время удерживания: 1.657 мин), MS m/z 486 (МН+).
Стадия 4: (Схема 2, Стадия 3)
К раствору метилового эфира 4-(2-бромпиридин-4-илоксиметил)-1-(2-метоксикарбониламино-3, 3-диметилбутирил)-пирролидин-2-карбоновой кислоты (270 мг, 0.45 ммоля) в смеси ТГФ (18 мл), метанола (10 мл) и воды (3.3 мл) прибавляют моногидрат гидроксида лития (283 мг, 6.75 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем ее упаривают и подкисляют 1N раствором HCl до рН 3-5. В качестве продукта получают твердое вещество не чисто белого цвета (интермедиат 8) (180 мг, выход 85%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 1.06 (с, 9Н), 2 20-2.29 (м, 2Н), 2.89 (м, 1Н), 3.54 (с, 3H), 3.92 (д. J=6.4 Гц, 2Н), 4.06-4.13 (м, 2Н), 4.31 (д, J=8.85 Гц, 1Н), 4.59 (дд, J=8.85, 5.50 Гц, 1Н), 7.00 (дд, J=6.10, 2.24 Гц, 1Н), 7.22 (д, J=1.83 Гц, 1Н), 8.12 (д, J=5.80 Гц, 1Н).
LC-MS (время удерживания: 2.113 мин), MS m/z 472 (МН+).
Стадия 5: (Схема 3)
К раствору интермедиата 8 (10 мг, 0.0212 ммоля) в CH3CN (5 мл) прибавляют гидрохлорид (1R,2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (8.5 мг, 0.0318 ммоля), DIEA (0.018 мл, 0.106 ммоля) и конденсирующий агент HOBt (4.9 мг, 0.0318 ммоля) и HBTU (12.1 мг, 0.0318 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ и получают конечный продукт в виде вязкого масла (Соединение 348) (9 мг, выход 53%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00-1.06 (м, 11Н), 1.20 (м, 2Н), 1.36 (дд, J=9.54 Гц, 5.38 Гц, 1Н), 1.87 (дд, J=8.07 Гц, 5.38 Гц, 1H), 2.04-2.24 (м, 3H), 2.88-2.94 (м, 2Н), 3.52 (с, 3H), 3.93 (д, J=5.87 Гц, 2Н), 4.09 (м, 2Н), 4.30 (с, 1Н), 4.38 (т, J=7.58 Гц, 1Н), 5.11 (дд, J=10.27, 1.47 Гц, 1Н), 5.28 (дд, J=17.12, 1.47 Гц, 1Н), 5.70 (м, 1Н), 7.00 (дд, J=5.87, 2.20 Гц, 1Н), 7.24 (д, J=2.20 Гц, 1Н), 8.14 (д, J=5.87 Гц, 1Н).
LC-MS (время удерживания: 1.670 мин), MS m/z 684 (МН+).
Пример 349: Получение Соединения 349
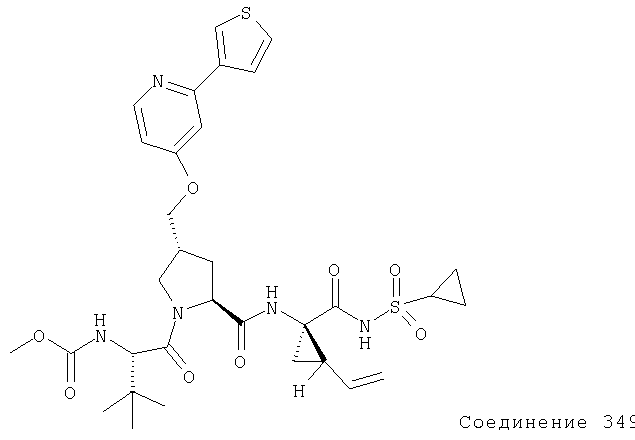
Соединение 349 получают по Схеме из Примера 347 за исключением того, что вместо интермедиата 6 Стадии 1 из Примера 346 используют интермедиат 8 из Примера 348.
Стадия 1:

К раствору интермедиата 8 (20 мг, 0.0423 ммоля) в ДМФА (1 мл) прибавляют 3-тиофенбороновую кислоту (7.0 мг, 0.055 ммоля), тетракис(трифенилфосфин)палладий (2.4 мг, 0.00212 ммоля) и 2М раствор Na2CO3 (0.063 мл, 0.127 ммоля). Реакционную смесь нагревают при 110°С в течение 30 час. Затем ее отфильтровывают и промывают метанолом. Фильтрат упаривают и очищают препаративной ВЭЖХ, получая продукт в виде коричневатого масла (10.5 мг, выход 42%) (50177-165).
LC-MS (время удерживания: 1.690 мин), MS m/z 476 (MH+).
Стадия 2:
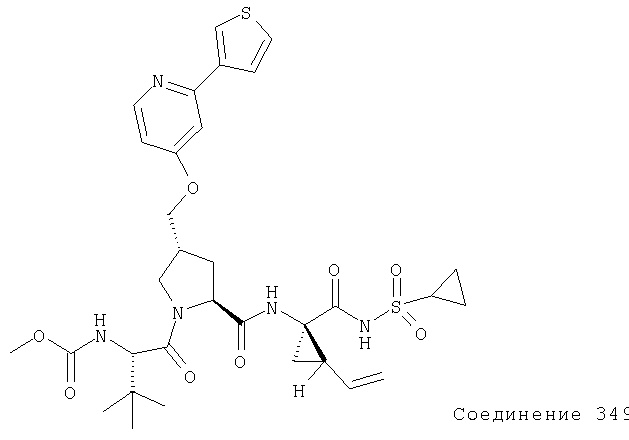
К раствору 1-(2-метоксикарбониламино-3, 3-диметилбутирил)-4-(2-тиофен-3-илпиридин-4-илоксиметил)-пирролидин-2-карбоновой кислоты (10 мг, 0.017 ммоля) в СН3CN (5 мл) прибавляют гидрохлорид (1R,1S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (6.8 мг, 0.0254 ммоля), DIEA (0.015 мл, 0.085 ммоля) и конденсирующий агент HOBt (3.9 мг, 0.0254 ммоля) и HBTU (9.6 мг, 0.0254 ммол). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль (Соединение 349) в виде коричневатой пленки (2.2 мг, выход 16%) (50177-172).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1 00-1.07 (м, 11Н), 1.20 (м, 2Н), 1.37 (дд, J=9.04, 5.38 Гц, 1Н), 1.88 (дд, J=8.07 Гц, 5.38 Гц, 1Н), 2.10-2.25 (м, 3H), 2.90 (м, 1Н), 3.04 (м, 1Н), 3.47 (с, 3H), 3.93-4.02 (м, 2Н), 4.29 (с, 1Н), 4.35-4.45 (м, 3H), 5.12 (д, J=10.51 Гц, 1Н), 5.28 (д, J=17.61 Гц, 1Н), 5.69 (м, 1Н), 7.40 (дд, J=6 84, 2.44 Гц, 1Н), 7.71-7.80 (м, 3H), 8.38 (м, 1Н), 8.51 (д, J=7.09 Гц, 1Н).
LC-MS (время удерживания: 1.443 мин), MS m/z 688 (MH+).
Пример 350: Получение Соединения 350
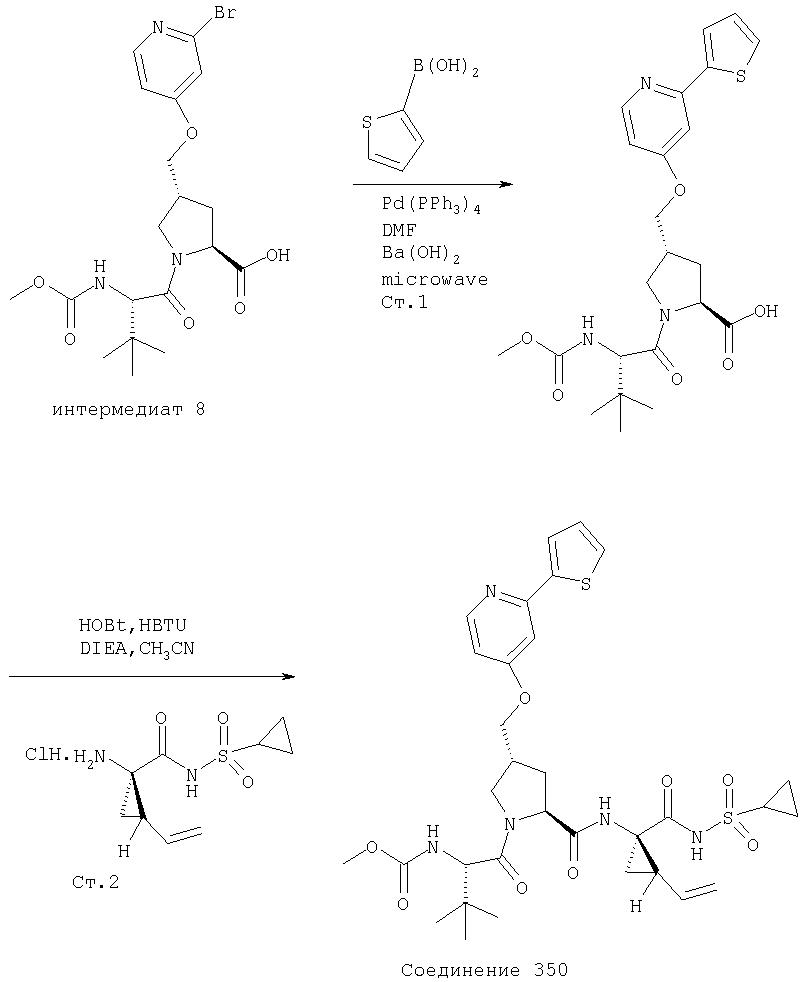
Стадия 1:
К раствору интермедиата 8 из Примера 348 (20 мг, 0.0423 ммоля) в ДМФА (2 мл) прибавляют 2-тиофенбороновую кислоту (7.0 мг, 0.055 ммоля), тетракис(трифенилфосфин)палладий (2.4 мг, 0.00212 ммоля) и гидроксид бария (40 мг, 0.127 ммоля). Реакционную смесь нагревают при 150°С в микроволновом реакторе Смита в течение 110 мин. Затем ее отфильтровывают и промывают метанолом. Фильтрат упаривают и очищают препаративной ВЭЖХ, получая продукт в виде желтоватого масла (5.0 мг, выход 20%).
LC-MS (время удерживания: 2.137 мин), MS m/z 476 (МН+).
Стадия 2:
К раствору полученной выше карбоновой кислоты (5.0 мг, 0.0085 ммоля) в CH3CN (5 мл) прибавляют гидрохлорид (1R,1S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (3.4 мг, 0.0127 ммоля), DDEA (0.007 мл, 0.0424 ммоля) и конденсирующий агент HOBt (1.9 мг, 0.0127 ммоля) и HBTU (4.8 мг, 0.0127 ммол). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль (Соединение 349) в виде желтоватого масла (Соединение 350) (2.6 мг, выход 38%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99-1.07 (м, 11Н), 1.19 (м, 2Н), 1.37 (дд, J=9.54, 5.63 Гц, 1Н), 1.87 (дд, J=8.07 Гц, 5.38 Гц, 1Н), 2.10-2.25 (м, 3H), 2.91 (м, 1Н), 3.03 (м, 1Н), 3.48 (с, 3H), 3.92-4.02 (м, 2Н), 4.30 (с, 1Н), 4.32-4.45 (м, 3H), 5.11 (дд, J=10.27, 1.22 Гц, 1Н), 5.28 (д, J=17.11 Гц, 1Н), 5.69 (м, 1Н), 7.30-7 38 (м, 2Н), 7.66 (д, J=2.45 Гц, 1Н), 7.92 (м, 1Н), 7.95 (м, 1Н), 8.48 (д, J=6.85 Гц, 1Н).
LC-MS (время удерживания: 2.067 мин), MS m/z 688 (MH+).
Пример 351: Получение Соединения 351

Соединение 351 получают по Схеме из Примера 350 за исключением того, что на Стадии 1 вместо 2-тиофенбороновой кислоты используют 3-фуранбороновую кислоту.
Стадия 1:

К раствору интермедиата 8 из Примера 348 (20 мг, 0.0423 ммоля) в ДМФА (2 мл) прибавляют 3-фуранбороновую кислоту (6.2 мг, 0.055 ммоля), тетракис(трифенилфосфин)палладий (2.4 мг, 0.00212 ммоля) и гидроксид бария (40 мг, 0.127 ммоля). Реакционную смесь нагревают при 150°С в микроволновом реакторе Смита в течение 30 мин. Затем ее отфильтровывают и промывают метанолом. Фильтрат упаривают и очищают препаративной ВЭЖХ, получая продукт в виде желтоватого масла (12 мг, выход 49%).
LC-MS (время удерживания: 1.937 мин), MS m/z 460 (MH+).
Стадия 2:

К раствору полученной выше карбоновой кислоты (5.0 мг, 0.0209 ммоля) в CH3CN (5 мл) прибавляют гидрохлорид (1R,1S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (8.4 мг, 0.0314 ммоля), DIEA (0.018 мл, 0.1046 ммоля) и конденсирующий агент HOBt (4.8 мг, 0.0314 ммоля) и HBTU (11.9 мг, 0.0314 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль в виде желтоватого масла (Соединение 351) (4.0 мг, выход 24%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00-1.08 (м, 11H), 1.21 (м, 2Н), 1.37 (дд, J=8.80, 5.62 Гц, 1Н), 1.87 (дд, J=8.32 Гц, 5.38 Гц, 1Н), 2.11-2.24 (м, 3H), 2.91 (м, 1Н), 3.03 (м, 1Н), 3.49 (с, 3H), 3.91-4.03 (м, 2Н), 4.29 (с, 1Н), 4.35-4.46 (м, 3H), 5.12 (дд, J=10.27, 1.47 Гц, 1Н), 5.28 (д, J=17.12 Гц, 1Н), 5.69 (м, 1Н), 7.11 (м, 1Н), 7.38 (дд, J=7.10, 2.69 Гц, 1Н), 7.71 (д, J=2.69 Гц, 1Н), 7.81 (м, 1Н), 8.48 (с, 1Н), 8.50 (д, J=7.09 Гц, 1Н).
LC-MS (время удерживания: 1.410 мин), MS m/z 672 (MH+).
Пример 352: Получение Соединения 352
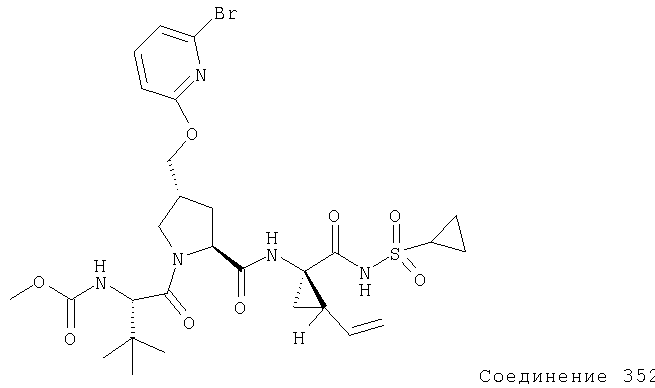
Соединение 352 получают по Схеме 2 и Схеме 3 из Примера 348 за исключением того, что на Стадии 1 Схемы 2 вместо 2-бром-4-хлорпиридина используют 2,6-дибромпиридин.
Стадия 1: (Схема 2, Стадия 1)

К раствору интермедиата 7 из Примера 348 (270 мг, 1.1 ммоля) в ДМСО (10 мл) прибавляют трет-бутоксид калия (309 мг, 2.75 ммоля). Реакционную смесь перемешивают при rt в течение 1 час. Затем прибавляют 2,6-дибромпиридин (313 мг, 1.32 ммоля). Реакционную смесь перемешивают при rt в течение ночи. Затем прибавляют воду и промывают этилацетатом. Водный слой отделяют и подкисляют 1N раствором HCl до рН 3. Дважды экстрагируют этилацетатом и органические вытяжки объединяют и сушат (MgSO4). Упариванием растворителя получают оранжевое масло. Затем его растворяют в метаноле и в течение 2 мин при -78°С продувают HCl (газ). Температуру реакционной смеси доводят до комнатной и перемешивают в течение ночи. Упариванием растворителя получают сырой продукт в виде оранжевого масла.
LC-MS (время удерживания: 1.480 мин), MS m/z 315 (МН+).
Стадия 2: (Схема 2, Стадия 2)

К раствору сырого метилового эфира 4-(6-бромпиридин-2-илоксиметил)-пирролидин-2-карбоновой кислоты в CH3CN (20 мл) прибавляют 2-метоксикарбониламино-3,3-диметилмасляную кислоту (312 мг, 1.65 ммоля), DIEA (1.15 мл, 6.6 ммоля) и конденсирующий агент HOBt (252 мг, 1.65 ммоля) и HBTU (626 мг, 1.65 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для флеш-хроматографии (силикагель, 1:1 смесь гексанов: этилацетат), продукт получают в виде бесцветного масла (340 мг, выход на две стадии 63%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.03 (с, 9Н), 2.10-2.24 (м, 2Н), 2.84 (м, 1Н), 3.55 (с, 3H), 3.70 (с, 3H), 3.83 (м, 1Н), 3.94 (м, 1Н), 4.20-4.29 (м, 2Н), 4.31 (с, 1Н), 4.59 (дд, J=8.80, 5.13 Гц, 1Н), 6.76 (д, J=8.07 Гц, 1Н), 7.11 (д, J=7.58 Гц, 1Н), 7.53 (т, J=7.83 Гц, 1Н).
LC-MS (время удерживания: 1.820 мин), MS m/z 486 (MH+).
Стадия 3: (Схема 2, Стадия 3)
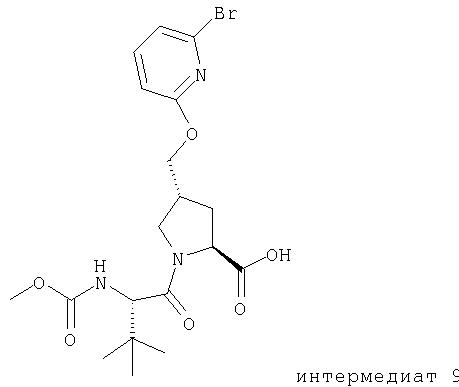
К раствору метилового эфира 4-(6-бромпиридин-2-илоксиметил)-1-(2-метоксикарбониламино-3,3-диметилбутирил)-пирролидин-2-карбоновой кислоты (330 мг, 0.679 ммоля) в смеси ТГФ (28 мл), метанола (15 мл) и воды (5 мл) прибавляют моногидрат гидроксида лития (427 мг, 10.18 ммоля). Реакционную смесь перемешивают при rt в течение двух дней. Затем ее упаривают и подкисляют 1N раствором HCl до рН 3-5. В качестве продукта получают твердое вещество белого цвета (интермедиат 9) (310 мг, выход 97%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 1.06 (с, 9Н), 2.18-2.25 (м, 2Н), 2.88 (м, 1Н), 3.57 (с, 3H), 3.84 (м, 1Н), 3.96 (м, 1Н), 4.25 (м, 1Н), 4.28-4.35 (м, 2Н), 4.58 (м, 1Н), 6.79 (д, J=7.94 Гц, 1Н), 7.13 (д, J=7.32 Гц, 1Н), 7.55 (м, 1Н).
LC-MS (время удерживания: 3.030 мин), MS m/z 472 (MH+).
Стадия 4: (Схема 3)

К раствору интермедиата 9 (10.0 мг, 0.0212 ммоля) в СН3CN (5 мл) прибавляют гидрохлорид (1R,2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (8.5 мг, 0.0318 ммоля), DIEA (0.018 мл, 0.106 ммоля) и конденсирующий агент HOBt (4.9 мг, 0.0318 ммоля) и HBTU (12.1 мг, 0.0318 ммол). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль (Соединение 349) в виде желтоватой пленки (Соединение 352) (10.2 мг, выход 60%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 1.00-1.06 (м, 11Н), 1.20 (м, 2Н), 1.37 (дд, J=9.54 Гц, 5.63 Гц, 1Н), 1.86 (дд, J=8.07 Гц, 5.38 Гц, 1Н), 2.05 (м, 1Н), 2.12-2.25 (м, 2Н), 2.86-2.94 (м, 2Н), 3.54 (с, 3H), 3.87 (м, 1Н), 3.94 (м, 1Н), 4.18-4.27 (м, 2Н), 4.33 (с, 1Н), 4.37 (м, 1Н), 5.11 (дд, J=10.27, 1.72 Гц, 1Н), 5.28 (дд, J=17.12, 1.47 Гц, 1Н), 5.70 (м, 1Н), 6.76 (д, J=8.32 Гц, 1Н), 7.11 (д, J=7.33 Гц, 1Н), 7.53 (т, J=7.82 Гц, 1Н).
LC-MS (время удерживания: 1.837 мин), MS m/z 684 (MH+).
Пример 353: Получение Соединения 353
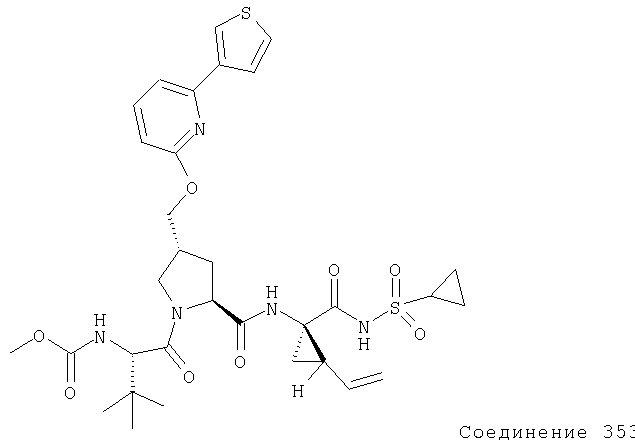
Соединение 353 получают по Схеме из Примера 347 за исключением того, что вместо интермедиата 6 Стадии 1 из Примера 346 используют интермедиат 9 из Примера 352.
Стадия 1:

К раствору интермедиата 9 (25 мг, 0.053 ммоля) в ДМФА (1 мл) прибавляют 3-тиофенбороновую кислоту (8.8 мг, 0.0688 ммоля), тетракис(трифенилфосфин)палладий (3.1 мг, 0.00265 ммоля) и 2М раствор Na2СО3 (0.080 мл, 0.159 ммоля). Реакционную смесь нагревают при 110°С в течение ночи. Затем ее отфильтровывают и промывают метанолом. Фильтрат упаривают и очищают препаративной ВЭЖХ, получая продукт в виде коричневатого масла (15 мг, выход 48%).
1H ЯМР (500 МГц, CD3OD) δ м.д. 1.06 (с, 9Н), 2.20-2.31 (м, 2Н), 2.94 (м, 1Н), 3.55 (с, 3H), 3.91 (м, 1Н), 3.98 (м, 1Н), 4.34 (с, 1Н), 4.37-4.46 (м, 2Н), 4.61 (дд, J=8.85, 5.19 Гц, 1H), 6.77 (д, J=8.24 Гц, 1H), 7.39 (д, J=7.32 Гц, 1Н), 7.48 (дд, J=5.19, 3.05 Гц, 1Н), 7.68 (дд, J=4.88, 1.22 Гц, 1H), 7.77 (т, J=7.93 Гц, 1H), 8.04 (м, 1Н).
LC-MS (время удерживания: 1.857 мин), MS m/z 476 (MH+).
Стадия 2:

К раствору 1-(2-метоксикарбониламино-3,3-диметилбутирил)-4-(6-тиофен-3-илпиридин-2-илоксиметил)-пирролидин-2-карбоновой кислоты (15 мг, 0.0254 ммоля) в CH3CN (5 мл) прибавляют гидрохлорид (1R,2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил))-карбаминовой кислоты (10.2 мг, 0.0382 ммоля), DIEA (0.022 мл, 0.127 ммоля) и конденсирующий агент HOBt (5.8 мг, 0.0382 ммоля) и HBTU (14.5 мг, 0.0382 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают ТФК соль (Соединение 353) в виде желтоватой пленки (6 мг, выход 29%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00-1.06 (м, 11Н), 1.20 (м, 2Н), 1.36 (дд, J=9.29, 5.38 Гц, 1H), 1.86 (дд, J=8.07 Гц, 5.38 Гц, 1H), 2.07 (м, 1H), 2.16-2.25 (м, 2Н), 2.87-2.99 (м, 2Н), 3.54 (с, 3H), 3.87-3.99 (м, 2Н), 4.31-4.44 (м, 4Н), 5.11 (дд, J=10.27, 1.46 Гц, 1H), 5.28 (д, J=17.12 Гц, 1H), 5.70 (м, 1H), 6.67 (д, J=8.31 Гц, 1H), 7.33 (д, J=7.34 Гц, 1H), 7.44 (дд, J=4.89, 2.93 Гц, 1H), 7.63-7.70 (м, 2Н), 7.99 (м, 1Н).
LC-MS (время удерживания: 2.770 мин), MS m/z 688 (МН+).
Пример 354: Получение Соединения 354
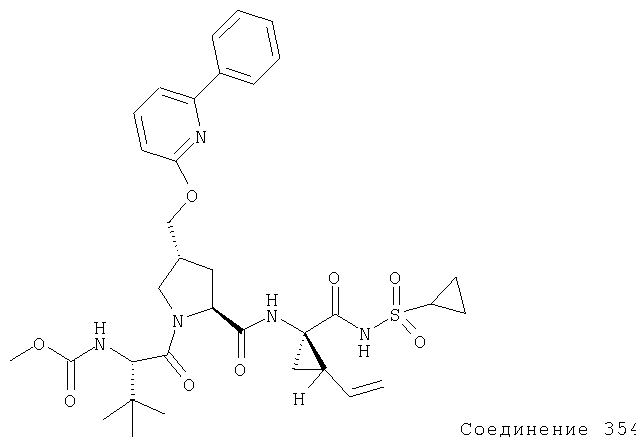
Соединение 354 получают по Схеме из Примера 347 за исключением того, что на Стадии 1 вместо интермедиата 6 из Примера 346 используют интермедиат 9 из Примера 352 и вместо 3-тиофенбороновой кислоты используют фенилбороновую кислоту.
Стадия 1:

К раствору интермедиата 9 (20 мг, 0.0423 ммоля) в ДМФА (1 мл) прибавляют фенилбороновую кислоту (6.7 мг, 0.0688 ммоля), тетракис(трифенилфосфин)палладий (2.4 мг, 0.00212 ммоля) и Cs2СО3 (41 мг, 0.127 ммоля). Реакционную смесь нагревают при 110°С в течение ночи. Затем ее отфильтровывают и промывают метанолом. Фильтрат упаривают и очищают препаративной ВЭЖХ, получая продукт в виде желтоватого масла (12 мг, выход 49%).
LC-MS (время удерживания: 2.733 мин), MS m/z 470 (MH+).
Стадия 2:
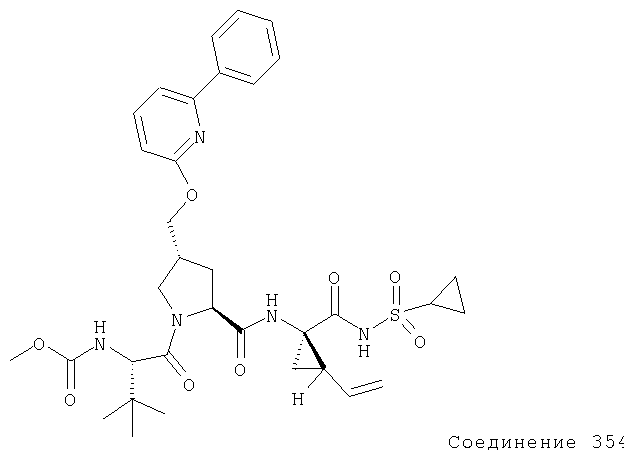
К раствору 1-(2-метоксикарбониламино-3,3-диметилбутирил)-4-(6-фенилпиридин-2-илоксиметил)-пирролидин-2-карбоновой кислоты (12 мг, 0.0206 ммоля) в СН3CN (5 мл) прибавляют гидрохлорид (1R,1S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил))-карбаминовой кислоты (8.2 мг, 0.0308 ммоля), DIEA (0.018 мл, 0.1028 ммоля) и конденсирующий агент HOBt (4.7 мг, 0.0308 ммоля) и HBTU (11.7 мг, 0.0308 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают ТФК соль (Соединение 354) в виде твердого вещества белого цвета (1.5 мг, выход 9%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.00-1.07 (м, 11Н), 1.20 (м, 2Н), 1.36 (м, 1Н), 1.85 (м, 1Н), 2.09 (м, 1Н), 2.17-2.25 (м, 2Н), 2.87-3.00 (м, 2Н), 3.52 (с, 3H), 3.84-4.00 (м, 2Н), 4.33-4.44 (м, 4Н), 5.11 (дд, J=10.27, 1.71 Гц, 1Н), 5.28 (д, J=17.12, 1.22 Гц, 1Н), 5.70 (м, 1Н), 6.71 (д, J=8.31 Гц, 1Н), 6.78 (м, 1Н), 7.34-7.44 (м, 3H), 7.74 (м, 1Н), 7.95 (м, 1Н), 8.01 (д, J=8.31 Гц, 1Н).
LC-MS (время удерживания: 3.553 мин), MS м/z 682 (МН+).
Пример 355: Получение Соединения 355
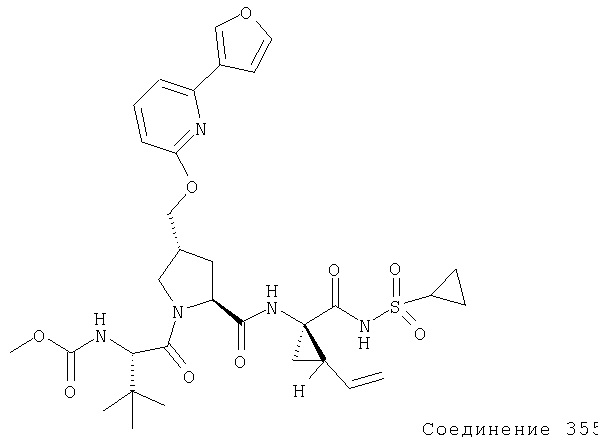
Соединение 355 получают по Схеме из Примера 347 за исключением того, что на Стадии 1 вместо интермедиата 6 из Примера 346 используют интермедиат 9 из Примера 352 и вместо 3-тиофенбороновой кислоты используют 3-фуранбороновую кислоту.
Стадия 1:
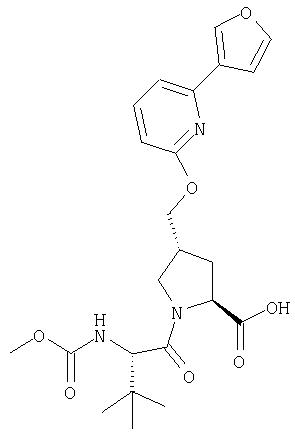
К раствору интермедиата 9 (20 мг, 0.0423 ммоля) в ДМФА (1 мл) прибавляют 3-фуранбороновую кислоту (6.2 мг, 0.055 ммоля), тетракис(трифенилфосфин)палладий (2.4 мг, 0.002115 ммоля) и 2М раствор Na2СО3 (0.064 мл, 0.127 ммоля). Реакционную смесь нагревают при 110°С в течение двух дней. Затем ее отфильтровывают и промывают метанолом. Фильтрат упаривают и очищают препаративной ВЭЖХ, получая продукт в виде желтоватого масла (7.0 мг, выход 29%).
Стадия 2:

К раствору полученной выше карбоновой кислоты (6.0 мг, 0.0109 ммоля) в CH3CN (5 мл) прибавляют гидрохлорид (1R,2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (4.4 мг, 0.0163 ммоля), DIEA (0.0095 мл, 0.0544 ммоля) и конденсирующий агент HOBt (2.5 мг, 0.0163 ммоля) и HBTU (6.2 мг, 0.0163 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают продукт - ТФК соль (Соединение 355) в виде желтоватой пленки (1.5 мг, выход 18%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.98-1.07 (м, 11Н), 1.20 (м, 2Н), 1.35 (дд, J=9.54, 5.87 Гц, 1Н), 1.86 (дд, J=8.07 Гц, 5.62 Гц, 1Н), 2.06 (м, 1Н), 2.15-2.25 (м, 2Н), 2.85-2.98 (м, 2Н), 3.55 (с, 3H), 3.89 (м, 1Н), 3.95 (м, 1Н), 4.28-4.42 (м, 4Н), 5.11 (дд, J=10.27, 1.71 Гц, 1Н), 5.28 (дд, J=17.12, 1.22 Гц, 1Н), 5.69 (м, 1Н), 6.63 (д, J=8.07 Гц, 1Н), 6.90 (м, 1Н), 7.16 (д, J=7.33 Гц, 1Н), 7.53 (м, 1Н), 7.63 (м, 1Н), 8.08 (с, 1Н).
LC-MS (время удерживания: 3.340 мин), MS м/z 672 (МН+).
Пример 356: Получение Соединения 356
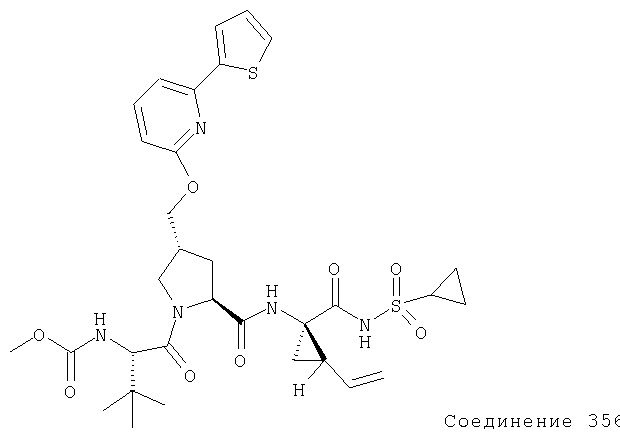
Соединение 356 получают по Схеме из Примера 350 за исключением того, что на Стадии 1 вместо интермедиата 8 из Примера 348 используют интермедиат 9 из Примера 352.
Стадия 1:
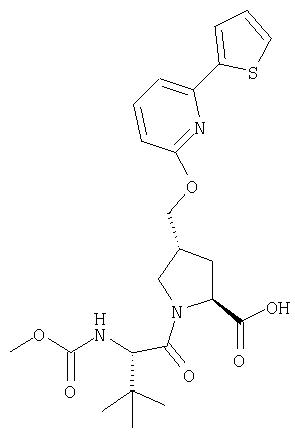
К раствору интермедиата 9 (20 мг, 0.0423 ммоля) в ДМФА (1 мл) прибавляют 2-тиофенбороновую кислоту (7.0 мг, 0.055 ммоля), тетракис(трифенилфосфин)палладий (2.4 мг, 0.00212 ммоля) и гидроксид бария (40 мг, 0.127 ммоля). Реакционную смесь нагревают при 150°С в микроволновом реакторе Смита в течение 30 мин. Затем ее отфильтровывают и промывают метанолом. Фильтрат упаривают и очищают препаративной ВЭЖХ, получая продукт в виде коричневатого масла (13.0 мг, выход 52%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.03 (с, 9Н), 2.18-2.25 (м, 2Н), 2.93 (м, 1Н), 3.55 (с, 3H), 3.83 (м, 1Н), 3.98 (м, 1Н), 4.34 (с, 1Н), 4.38 (м, 2Н), 4.58 (дд, J=8.05, 5.14 Гц, 1Н), 6.63 (д, J=8.07 Гц, 1Н), 7.07 (дд, J=4.89, 3.67 Гц, 1Н), 7.33 (д, J=7.34 Гц, 1Н), 7.42 (д, J=5.14 Гц, 1Н), 7.60-7.66 (м, 2Н).
LC-MS (время удерживания: 3.393 мин), MS m/z 476 (MH+).
Стадия 2:

К раствору 1-(2-метоксикарбониламино-3, 3-диметилбутирил)-4-(6-тиофен-2-илпиридин-2-илоксиметил)-пирролидин-2-карбоновой кислоты (11.5 мг, 0.0195 ммоля) в CH3CN (5 мл) прибавляют гидрохлорид (1R,2S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил))-карбаминовой кислоты (7.8 мг, 0.0293 ммоля), DIEA (0.017 мл, 0.0975 ммоля) и конденсирующий агент HOBt (4.5 мг, 0.0293 ммоля) и HBTU (11.1 мг, 0.0293 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают ТФК соль (Соединение 356) в виде твердого вещества не чисто белого цвета (8.5 мг, выход 54%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 0.99-1.06 (м, 11H), 1.21 (м, 2Н), 1.36 (дд, J=9.54, 5.38 Гц, 1Н), 1.86 (дд, J=8.07 Гц, 5.62 Гц, 1Н), 2.06 (м, 1Н), 2.15-2.25 (м, 2Н), 2.87-2.99 (м, 2Н), 3.54 (с, 3H), 3.89 (м, 1Н), 3.96 (м, 1Н), 4.30-4.44 (м, 4Н), 5.11 (дд, J=10.51, 1.71 Гц, 1Н), 5.28 (дд, J=17.11, 1.22 Гц, 1Н), 5.70 (м, 1Н), 6.62 (д, J=8.07 Гц, 1Н), 7.07 (дд, J=4.89, 3.66 Гц, 1Н), 7.33 (д, J=7.59 Гц, 1Н), 7.42 (д, J=4.89 Гц, 1Н), 7.60-7.66 (м, 2Н).
LC-MS (время удерживания: 1.967 мин), MS m/z 688 (MH+).
Пример 357: Получение Соединения 357

Стадия 1:
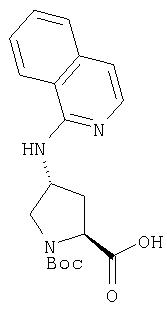
К раствору (2S,4R) Fmoc-4-амино-1-boc-пирролидин-2-карбоновой кислоты (400 мг, 0.884 ммоля) в ацетонитриле (15 мл) прибавляют пять капель пирролидина. Реакционную смесь перемешивают при rt в течение 3 час. Затем ее упаривают и выдерживают в высоком вакууме, получают сырую 4-амино-1-boc-пирролидин-2-карбоновую кислоту. В другой круглодонной колбе раствор Pd2dba3 (40 мг, 5 мольн.%) и рацемического BINAP (56 мг, 10 мольн.%) перемешивают под азотом в дегазированном толуоле (8 мл ) при rt в течение 1 часа. Затем прибавляют 1-хлоризохинолин (216 мг, 1.326 ммоля) трет-бутоксид натрия (340 мг, 3.536 ммоля) и реакционную смесь перемешивают 30 мин. Прибавляют 4-амино-1-boc-пирролидин-2-карбоновую кислоту и реакционную смесь кипятят 1 час. Для прекращения реакции прибавляют воду и водный слой отделяют и фильтруют через бумажный фильтр, упаривают и очищают препаративной ВЭЖХ, получают продукт сочетания в виде ТФК соли (165 мг, выход 40%).
1H ЯМР (400 МГц, CD3OD) δ м.д. 1.44 (м, 9Н), 2.51-2.74 (м, 2Н), 3.64 (м, 1Н), 4.01 (м, 1Н), 4.49 (м, 1Н), 4.64 (м, 1Н), 7.30 (д, J=6.S5 Гц, 1Н), 7.58 (д, J=6.85 Гц, 1Н), 7.79(м, 1Н), 7.91-7.99 (м, 2Н), 8.56 (д, J=8.56 Гц, 1Н).
LC-MS (время удерживания: 1.707 мин), MS m/г 358 (МН+).
Стадия 2:
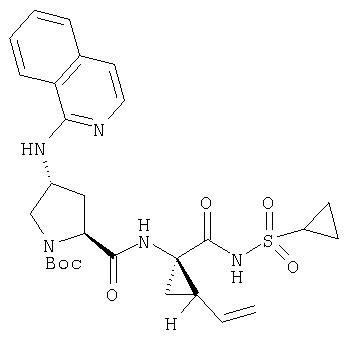
К раствору 1-трет-бутилового эфира 4-(изохинолин-1-иламино)-пирролидин-1,2-дикарбоновой кислоты (115 мг, 0.244 ммоля) в СН3СN (10 мл) прибавляют гидрохлорид (1R,1S) (1-циклопропансульфониламинокарбонил-2-винилциклопропил)-карбаминовой кислоты (97 мг, 0.366 ммоля), DIEA (0.255 мл, 1.464 ммоля) и конденсирующий агент HOBt (56 мг, 0.366 ммоля) и HBTU (139 мг, 0.366 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают, получая желтое масло. Его очищают на колонке для препаративной ВЭЖХ, получают твердое вещество желтоватого цвета - ТФК соль (112 мг, выход 67%).
1H ЯМР (400 МГЦ, CD3OD) δ м.д. 1.05 (м, 2Н), 1.20 (м, 2Н), 1.40-1.48 (м, 10H), 1.87 (дд, J=8.19, 5.50 Гц, 1Н), 2.23 (м, 1Н), 2.39 (м, 1Н), 2.50 (м, 1Н), 2.93 (м, 1Н), 3.65 (м, 1Н), 4.08 (м, 1Н), 4.33 (т, J=7.09 Гц, 1Н), 4.69 (м, 1Н), 5.12 (д, J=10.27 Гц, 1Н), 5.29 (д, J=17.12 Гц, 1Н), 5.74 (м, 1Н), 7.31 (д, J=6.85 Гц, 1Н), 7.60 (д, J=7.09 Гц, 1Н), 7.80 (м, 1Н), 7.93-8.00 (м, 2Н), 8.56 (д, J=8.19 Гц, 1Н).
LC-MS (время удерживания: 2.023 мин), MS m/z 570 (МН+).
Стадия 3:
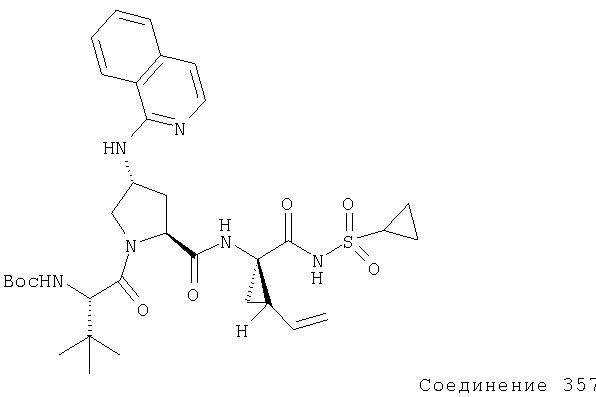
трет-Бутиловый эфир 2-(1-циклопропансульфониламинокарбонил-2-винилциклопропил)-4-(изохинолин-1-иламино)-пирролидин-1-карбоновой кислоты (31 мг, 0.0453 ммоля) растворяют в 4N HCl в диоксане (1.5 мл) и перемешивают при rt в течение двух часов. Растворитель упаривают, получают соль - бисгидрохлорид - в виде желтоватого масла. К раствору бисгидрохлорида в СН3CN (5 мл) прибавляют N-boc-L-лейцин (11.5 мг, 0.0498 ммоля), DIEA (0.047 мл. 0.272 ммоля) и конденсирующий агент HOBt (10.4 мг, 0.068 ммоля) и HBTU (25.8 мг, 0.068 ммоля). Раствор перемешивают при rt в течение ночи. Затем его упаривают, промывают водой и дважды экстрагируют этилацетатом. Объединенные органические вытяжки промывают рассолом, сушат MgSO4 и упаривают. Очищают на колонке для препаративной ВЭЖХ, получают в качестве конечного продукта не чисто белое твердое вещество (Соединение 357) (9 мг, выход 29%).
1 H ЯМР (400 МГц, CD3OD) δ м.д. 0.98 (м, 2Н), 1.05 (с, 9Н), 1.20 (м, 2Н), 1.36-1.43 (м, 10H), 1.84 (м, 1Н), 2.10-2.30 (м, 2Н), 2.52 (м, 1Н), 2.90 (м, 1Н), 4.07 (м, 1Н), 4.17-4.27 (м, 2Н), 4.47 (м, 1Н), 4.79 (м, 1Н), 5.07 (д, J=9.29 Гц, 1Н), 5.24 (д, J=16.87 Гц, 1Н), 5.72 (м, 1Н), 6.62 (м, 1Н), 6.98 (д, J=6.11 Гц, 1Н), 7.47 (м, 1Н), 7.62 (м, 1Н), 7.69 (д, J=8.07 Гц, 1Н), 7.84 (д, J=5.87 Гц, 1Н), 8.20 (д, J=8.56 Гц, 1Н).
LC-MS (время удерживания: 2.043 мин), MS m/z 683 (MH+).
Подраздел Н:
Условия LC-MS в Секции Н
Колонки:
(Метод A) - YMC ODS S7 С18 3.0×50 мм
(Метод В) - YMC ODS-A S7 С18 3.0×50 мм
(Метод С) - YMC S7 С18 3.0×50 мм
(Метод D) - YMC Xterra ODS S7 3.0×50 мм
(Метод Е) - YMC Xterra ODS S7 3.0×50 мм
(Метод F) - YMC ODS-A S7 С18 3.0×50 мм
(Метод Н) - Xterra S7 3.0×50 мм
(Метод I) - Xterra S7 С18 3.0×50 мм
(Метод G) - YMC С18 S5 4.6×50 мм
(Метод J) - Xterra ODS S7 3.0×50 мм
(Метод К) - YMC ODS-A S7 С18 3.0×50 мм
Градиент: от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В
Время градиента: 2 мин (А, В, D, F, G, Н, I); 8 мин (С, Е); 4 мин (J); 3 мин (К)
Время выдерживания: 1 мин (А, В, D, F, G, Н, I, J, К); 2 мин (С, Е)
Скорость потока: 5 мл/мин (А, В, С, D, Е, F, G)
Скорость потока: 4 мл/мин (J, К)
Длина волны детектора: 220 нм
Растворитель А: 10% МеОН /90% Н2O/0.1% ТФК
Растворитель В: 10% H2O /90% МеОН/0.1% ТФК
Пример 370: Получение Соединения 370
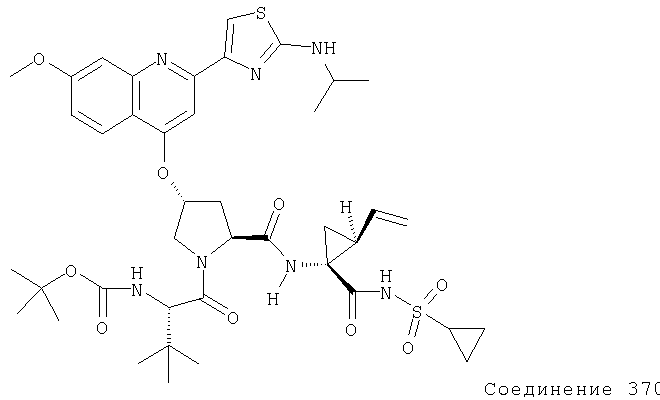
Схема 1

К раствору гидрохлорида этилового эфира (1R,2S)/(1S,2R)-1-амино-2-винилциклопропанкарбоновой кислоты (2.54 г, 12 ммолей) в CH3CN (70 мл) прибавляют раствор диизопропилэтиламина (9.5 мл, 67 ммолей), [(4R)-(2-метоксикарбонил-7-метоксихинолин-4-оксо)-S-пролин]а (5.9 г, 13.2 ммоля) и TBTU (3.89 г, 12.21 ммоля) в CH3CN (50 мл). Реакционную смесь перемешивают 14 час и упаривают. Остаток, растворенный в EtOAc, многократно промывают NaHCO3 (водн.), рассолом, сушат (MgSO4) и упаривают. Остаток очищают на колонке Biotage 65M (EtOAc/гексан: 45-100%), получая стереоизомер с высоким Rf (этиловый эфир Вос-P2[(4R)-(2-метоксикарбонил-7-метоксихинолин-4-оксо)-S-пролин]-P1((1R,2S Vinyl Асса) кислоты 2.0 г (52%) в виде твердого вещества белого цвета:1H ЯМР (CD3OD) δ м.д. 1.24 (т, J=7.02 Гц, 3H), 1.38 (м, 11H), 1.76 (м, 1Н), 2.21 (м, 1Н), 2.45 (м, 1Н), 2.71 (м, 1Н), 3.92 (м, 2Н), 3.96 (с, 3H), 4.03 (с, 3H), 4.16 (кв, J=7.22 Гц, 2Н), 4.42 (м, 1Н), 5.10 (м, 1Н), 5.30 (м, 1Н), 5.44 (с, 1Н), 5.77 (м, 1Н), 7.27 (д, J=9.16 Гц, 1Н), 7.48 (с, 1Н), 7.52 (с, 1Н). 8.05 (с, 1Н).
Схема 2
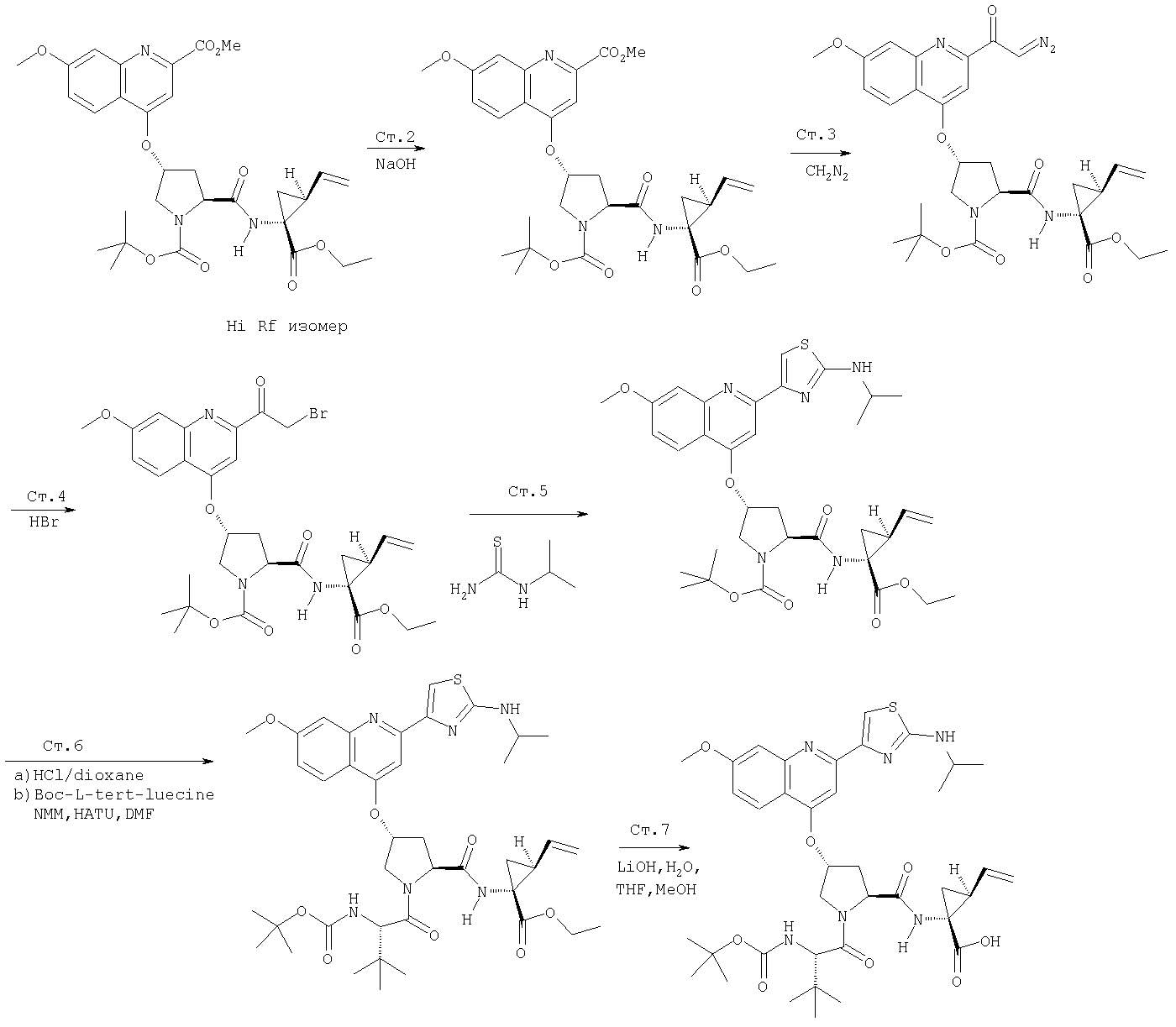
Раствор продукта с высоким Rf (3.16 г, 5.40 ммоля) Стадии 1 из Примера 370 {Boc-Р2[(4R)-(2-метоксикарбонил-7-метоксихинолин-4-оксо)-S-пролин]-Р1((1R,2S Vinyl Acca) COOEt} в МеОН/ТГФ (1/1, 13.2 мл) при 0°С обрабатывают водньм 1.0N раствором NaOH (5.5 мл, 5.5 ммоля), перемешивают 1 час, нейтрализуют, добавляя АсОН. Растворитель отгоняют в вакууме. Остаток снова растворяют в ТГФ/СН2Cl2(1/1, 150 мл), сушат (MgSO4) и упаривают в вакууме, получают продукт, который непосредственно используют на следующей стадии: LC-MS (время удерживания: 1.53 Метод D), MS m/z 570 (M++1).
Стадия 3:
К раствору продукта (предположительно, 5.4 ммоля) Стадии 2 Примера 370 в ТГФ (35 мл) при 0°С прибавляют раствор свежеприготовленного CH2N2 (30 ммолей) в Et2O (80 мл). Реакционную смесь перемешивают при этой температуре в течение 0.5 час и при rt в течение 18.5 час. Продувают азот через реакционную смесь в течение 1 часа и растворитель отгоняют в вакууме. Остаток снова растворяют в EtOAc (1 л), промывают насыщенным водным раствором NaHCO3 (2×200 мл), рассолом (100 мл) и сушат (MgSO4). Растворитель отгоняют в вакууме, получают продукт 3.10 г (97% в расчете на две стадии): LC-MS (время удерживания: 3.06, Метод J), MS m/z 594 (M++1).
Стадия 4:
К раствору продукта (3.03 г, 5.10 ммоля) Стадии 3 из Примера 370 {Вос-P2[(4R)-(2-диазоацетил-7-метоксихинолин-4-оксо)-S-пролин]-Р1((1R,2S Vinyl Асса) COOEt}, при 0°С растворенного в ТГФ (110 мл), прибавляют 2 мл 48% HBr. Смесь перемешивают 1 час, распределяют между EtOAc (500 мл) и насыщенным NaHCO3 (водн.) (100 мл). Слой EtOAc отделяют, сушат (MgSO4). Растворитель отгоняют, получают продукт (3.12g, 95%): LC-MS (время удерживания: 1.56, Метод D). MS m/z 648 (M++1), MS m/z 646 (M--1).
Стадия 5:
Продукт (1.0 г, 1.54 ммоля) Стадии 4 из Примера 370 {Boc-P2[(4R)-(2-бромацетил-7-метоксихинолин-4-оксо)-S-пролин]-Р1((1R,2S Vinyl Асса) COOEt} в течение 2 час обрабатывают изопропилтиомочевиной (0.365 г, 3.09 ммоля) в изопропиловом спирте (57 мл), затем растворитель отгоняют. Остаток растворяют в 1.0N HCl (30 мл) и EtOAc (200 мл), доводят рН до 7, добавляя 1.0N NaOH (водн.). Водный слой экстрагируют EtOAc (2×100 мл) и объединенные вытяжки сушат (MgSO4) и упаривают. Остаток очищают на колонке Biotage 40+M (EtOAc-смесь гексанов: 30-100%), получают 870 мг продукта (84%), пригодного для введения в следующую стадию.
Стадия 6:
Продукт (0.250 г, 0.375 ммоля) стадии 5 из Примера 370 {Boc-P2{(4R)-[2-(2-изопропиламинотиазол-4-ил)-7-метоксихинолин-4-оксо)-S-пролин]-Р1((1R,2S Vinyl Асса) COOEt} обрабатывают 4N раствором HCl в диоксане (2.5 мл, 10 ммолей) в течение 2 5 час и упаривают в вакууме. К остатку прибавляют N-метилморфолин (0.206 мл, 1.875 ммоля) в ДМФА (3 мл), N-Boc-L-трет-лейцин (0.117 г, 0.506 ммоля) и HATU (0.192 г, 0.506 ммоля). Смесь перемешивают в течение ночи и распределяют между EtOAc и буферным раствором с рН 4.0. Слой EtOAc промывают водой, NaHCO3 (водн.), сушат (MgSO4), упаривают. Остаток очищают на колонке Biotage 40M (МеОН-CH2Cl2: 0-8%), получают 0.289 г продукта (99%): LC-MS (время удерживания: 2.53, Метод К). MS m/z 779 (M++1).
Стадия 7:
К суспензии продукта Стадии 6 (274 мг, 0.352 ммоля) из Примера 370 {BOCNH-P3(L-трет-BuGly)-{[2-(2-изопропиламинотиазол-4-ил)-7-метоксихинолин-4-оксо)-S-пролин}-Р1((1R,2S Vinyl Acca) COOEt} в смеси ТГФ (10.6 мл), СН3ОН (2.6 мл) и H2O (5.3 мл) прибавляют LiOH (0.068 г, 2.86 ммоля). Реакционную смесь перемешивают в течение 24 час, доводят рН до 6, отгоняют органические растворители в вакууме. Водный остаток подкисляют до рН 4 и многократно экстрагируют СН2Cl2. Объединенные органические растворы сушат (MgSO4) и упаривают в вакууме, получают 255 мг (95%) нужного продукта: LC-MS (время удерживания: 2.58, Метод К). MS m/z 751 (М++1).
Схема 3
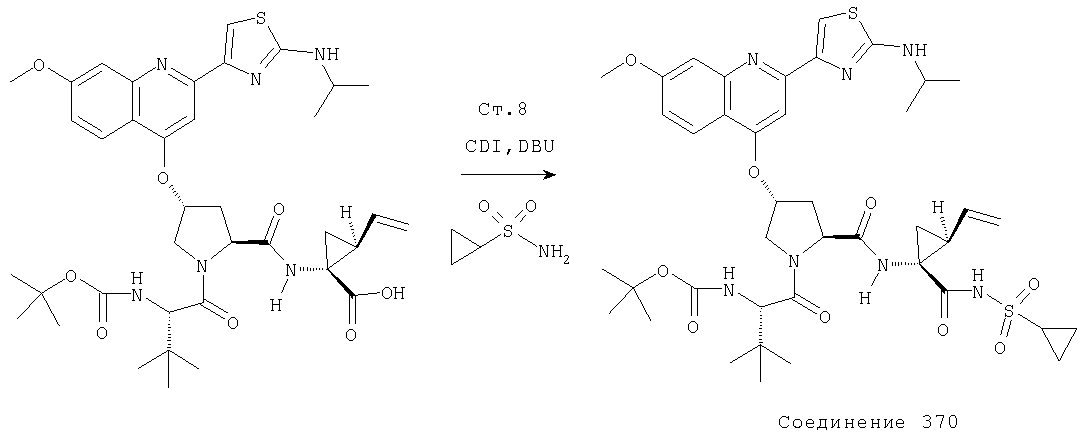
Стадия 8:
Раствор CDI (0.024 г, 0.15 ммоля) и продукта Стадии 7 из Примера 370 (0.0683 г, 0.09 ммоля) {BOCNH-P3(L-трет-BuGly)-{[2-(2-изопропиламинотиазол-4-ил)-7-метоксихинолин-4-оксо)-S-пролин}-Р1((1R,2S Vinyl Acca) COOH} в ТГФ (2 мл) кипятят 60 мин и охлаждают до rt. Прибавляют циклопропансульфонамид (0.022 г, 0.18 ммоля), а затем чистый DBU (0.027 мл, 0.18 ммоля). Реакционную смесь перемешивают в течение ночи, обрабатывают, добавляя EtOAc, и промывают буфером рН 4, сушат (MgSO4) и упаривают. Остаток очищают препаративной ВЭЖХ (0-100% растворителя В) и на пластине 1000 мкМ для препаративной ТСХ Analtech (20×40 см), получают 0.0032 г (4%) нужного продукта (Соединение 370) в виде бледно-желтой лены: LC-MS (время удерживания: 1.71, Метод Г). MS m/z 854 (M++1).
Пример 371: Получение Соединения 371

Схема 1
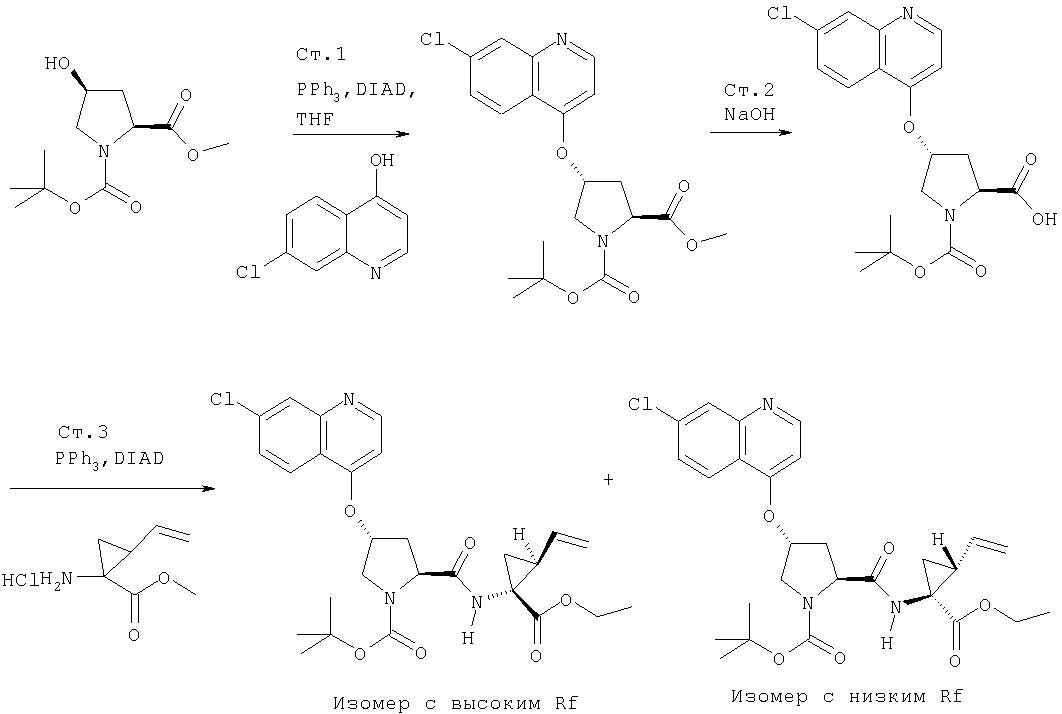
Стадия 1:
К суспензии метилового эфира N-Boc-cis-L-4-гидроксипролина (10 г, 40.7 ммоля) и 7-хлорхинолин-4-ола (8.73 г, 49.0 ммоля) в ТГФ (200 мл ), охлажденной до 0°С, прибавляют PPh3 (12.82 г, 48.9 ммоля) и DIAD (8.80 г, 42.13 ммоля). Смесь медленно доводят до rt в течение ночи, перемешивают всего 30 час. Смесь снова растворяют в EtOAc (800 мл), промывают 1N водной HCl, 5% водным К2СО3 (3×100 мл), рассолом (2×100 мл ), сушат (MgSO4) и упаривают. Остаток несколько раз очищают на колонке Biotage 65M (МеОН-СН2Cl2: 0-10%), всего получают 10.57 г (68%) нужного продукта в виде стекла:1H ЯМР (CDCl3) δ м.д. 1.40 (с, 9Н), 2.33-2.42 (м, 1Н), 2.61-2.72 (м, 1Н), 3.75 (с, 3H), 3.91 (м, 2Н), 4.45-4.59 (м, 1Н), 5.13 (м, 1Н), 6.61-6.64 (м, 1Н), 7.41 (дд, J=9.2 Гц, 1Н), 7.98 (д, J=2 Гц, 1Н), 8.03 (д, J=9 Гц, 1Н), 8.67-8.71 (м, 1Н). LC-MS (время удерживания: 1.39, Метод D). MS m/z 407 (M++1).
Стадия 2:
К раствору продукта (10.57 г, 26.0 ммоля) Стадии 1 из Примера 371 {метилового эфира ВОС-N-P2[(4R)-(7-хлорхинолин-4-оксо) пролина} в МеОН (800 мл), охлажденному до 0°С, прибавляют 1N водный раствор NaOH (44.5 мл, 44.5 ммоля). Через 6 час смесь доводят до rt, перемешивают в течение ночи и, прибавляя 1.0 N водный раствор HCl, доводят значение рН до 7. Раствор упаривают до тех пор, пока не останется только водный слой, доводят рН до 4 с помощью 6N водного раствора HCl и смесь несколько раз экстрагируют (распределяют) EtOAc (3×500 мл). Объединенные органические вытяжки сушат (MgSO4) и упаривают, получают 10.16 г (100%) продукта в виде твердого вещества белого цвета.1H ЯМР (ДМСО-d6) δ м.д. 1.32, 1.34 (два с (ротамеры), 9Н), 2.31-2.40 (м, 1Н), 2.58-2.69 (м, 1Н), 3.65-3.81 (м, 2Н), 4.33-4.40 (м, 1Н), 5.34 (м, 1Н), 7.10-7.11 (м, 1Н), 7.57 (д, J=9 Гц, 1Н), 7.98 (с, 1Н), 8.09-8.14 (м, 1Н), 8.75 (д, J=5 Гц, 1Н), 12.88 (уш с, 1Н).13С ЯМР (ДМСО-d6) δ м.д. 27.82, 35.84, 51.52, 57.75, 76.03, 79.33, 102.95, 119.54, 123.86, 126.34, 127.24, 134.49, 149.32, 152.88, 153.25, 159.08, 173.74. LC-MS (время удерживания: 1.48, Метод D). MS m/z 393 (M++1).
Стадия 3:
К раствору продукта (5.11 г, 13 ммоля) Стадии 2 из Примера 371, {Boc-4(R)-(7-хлорхинолин-4-оксо) пролина}, HCl соли (3.48 г, 18.2 ммоля) vinyl Acca (в виде смеси 1:1 диастереоизомеров (1R,2S/1S,2R, где циклопропилкарбоксиэтильная группа находится в син положении относительно винильного фрагмента) и NMM (7.1 мл, 65 ммоля) в ДМФА (30 мл) прибавляют HATU (6.92 г, 18.2 ммоля). Смесь перемешивают в течение 3 дней. Прибавляют EtOAc (180 мл) и распределяют между ним и буфером с рН 4.0 (3×100 мл). Органический слой промывают насыщенным водным раствором NaHCO3 (2×50 мл), водой (2×50 мл) и рассолом (2×50 мл). Органический раствор сушат (MgSO4) и упаривают. Остаток очищают на колонке Biotage 40M (EtOAc-смесь гексанов: 50%-100%), получают 2.88 г продукта в виде смеси диастереомеров. Эту смесь частично разделяют на колонке Biotage 65M (МеОН-EtOAc: 0%-9%), получают ВОС-NH-Р2[(4R)-(7-хлорхинолин-4-оксо)-S-пролин] Р1(1R,2S vinyl асса P1 фрагмент)-COOEt в качестве элюируемого сначала изомера с высоким Rf (1.20 г, 17.4%).1H ЯМР (CDCl3 /Метанол-d4) δ м.д. 1.16 (т, J=7 Гц, 3H), 1.35 (с, 9Н), 1.37-1.43 (м, 1Н), 1.76-1.84 (м, 1Н), 2.06-2.11 (м, 1Н), 2.35-2.45 (м, 1Н), 2.63 (м, 1Н), 3.72-3.93 (м, 2Н), 4.02-4.15 (м, 1Н), 4.33-4.40 (м, 1Н), 5.06 (д, J=9 Гц, 1Н), 5.16 (м, 1Н), 5.24 (д, J=17 Гц, 1Н), 5.63-5.70 (м, 1Н), 6.74 (м, 1Н), 7.39 (дд, J=9.2 Гц, 1Н), 7.74-7.78 (м, 1Н), 7.89 (д, J=2 Гц, 1Н), 7.97 (д, J=9 Гц, 1Н), 8.60 (д, J=5 Гц, 1Н).1H ЯМР (Метанол-d4, ротамеры 60/40) δ м.д. 1.24 (т, J=7 Гц, 3H), 1.39, 1.43 (2с, 9Н, соотношение 4:6), 1.71-1.74 (м, 0.4Н), 1.78-1.81 (м, 0.6Н), 2.18-2.23 (м, 1Н), 2.65-2.69 (м, 0.4Н), 2.71-2.76 (м, 0.6Н), 3.88-3.96 (м, 2Н), 4.11-4.18 (м, 2H), 4.39-4.45 (м, 1Н), 5.09-5.13 (м, 1Н), 5.28-5.33 (м, 1Н), 5.37 (м, 1Н), 5.73-5.81 (м, 1Н), 7.05 (д, J=5 Гц, 1Н), 7.53 (д, J=8.9 Гц, 1Н), 7.92 (с, 1Н), 8.12 (д, J=8.9 Гц, 1Н), 8.70 (д, J=5 Гц, 1Н). LC-MS (время удерживания: 1.54 мин, Метод A) MS m/z 530 (M++1). Остальное (˜1.66 г, 24%) представляет собой смешанные фракции, в значительной степени обогащенные изомером с низким Rf.
Схема 2:

Стадия 4:
Продукт (0.65 г, 1.22 ммоля) Стадии 3 из Примера 371 {ВОС-P2[(4R)-(7-хлорхинолин-4-оксо)-S-пролин]-Р1(1R,2S Vinyl Acca-COOEt} растворяют в 4N HCl/диоксан (4.5 мл, 18 ммолей) и перемешивают 1 час при rt. Реакционную смесь упаривают и сырой продукт непосредственно используют в следующей стадии. LC-MS (время удерживания: 0.94, Метод A) LC-MS m/z 430 (M++1).
Стадия 5:
К суспензии продукта 1 Стадии 4 (22 ммоля) из Примера 371 {Бис HCl соль NH2 -P2-[(4R)-(7-хлорхинолин-4-оксо)-S-пролин}-Р1((1R,2S Vinyl Acca) COOEt}, N-ВОС-L-трет-лейцин (ВОС L-tBuGly) (0.34 г, 1.47 ммоля), DIPEA (1.0 мл, 5.74 ммоля), HOBt·H2O (0.22 г, 1.47 ммоля) в CH2Cl2 (15 мл) прибавляют HBTU (0.56 г, 1.47 ммоля). Раствор перемешивают при rt в течение ночи, прибавляют СН2Cl2 (50 мл), промывают буфером с рН 4.0 (2×20 мл), насыщенным водным раствором NaHCO3 (50 мл), рассолом (50 мл), сушат (MgSO4) и упаривают. Остаток очищают на колонке Biotage 40M (EtOAc-смесь гексанов: 15%-60%), получают 607 мг (77%) продукта в виде пены.1H ЯМР (CDCl3/Метанол-d4) δ м.д. 1.00 (с, 9Н), 1.19 (т, J=7 Гц, 1Н), 1.30 (с, 9Н), 1.38 (м, 1Н), 1.78-1.83 (м, 1Н), 2.01-2.46 (м, 2Н), 2.73-2.82 (м, 1Н), 3.96-4.03 (м, 1Н), 4.04 (д, J=10 Гц, 1Н), 4.11 (кв, J=7 Гц, 2Н), 4.42 (д, J=12 Гц, 1Н), 4.68-4.73 (м, 1Н), 5.09-5.13 (м, 1Н), 5.23-5.31 (м, 2Н), 5.67-5.79 (м, 1Н), 6.78 (д, J=9 Гц, 1Н), 7.38 (д, J=9 Гц, 1Н), 7.70 (с, 1Н), 7.96 (с, 1Н), 8.08 (д, J=9 Гц, 1Н), 8.68 (д, J=5 Гц, 1Н). LC-MS (время удерживания: 1.64, Метод A). MS m/z 643 (M++1).
Стадия 6:
К суспензии продукта (207 мг, 0.32 ммоля) Стадии 5 из Примера 371 {BOCNH-P3(7-tBuGly)-P2[(4R)-(7-хлорхинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Acca-COOEt} в смеси ТГФ (14 мл), МеОН (2 мл) и Н2О (8 мл) прибавляют LiOH (62 мг, 2.60 ммоля). Реакционную смесь перемешивают один день, доводят значение рН до 7 (нейтрального), упаривают в вакууме до тех пор, пока не останется только водный слой. Доводят рН остатка до 4, добавляя 1.0N водный раствор HCl, а затем насыщают NaCl. Эту водную смесь несколько раз экстрагируют EtOAc (3× 60 мл). Объединенные органические вытяжки сушат (MgSO4) и упаривают, получают 107 мг (54%) продукта, {BOCNH-P3(L-tBuGly)-P2[(4R)-(7-хлорхинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Acca-CO2H} в виде твердого вещества белого цвета.1H ЯМР (CDCl3) δ м.д. 1.06 (с, 9Н), 1.23 (2с, 9Н), 1.31-1.43 (м, 1Н), 1.63-1.70 (м, 1Н), 1.85-1.89 (м, 1Н), 2.19 (м, 1Н), 2.65-2.78 (м, 1Н), 4.03-4.10 (м, 1Н), 4.18-4.21 (м, 1Н), 4.55-4.62 (м, 1Н), 5.03-5.12 (м, 1Н), 5.23-5.31 (м, 1Н), 5.51 (м, 1Н), 5.88-5.95 (м, 1Н), 7.12 (м, 1Н), 7.47-7.50 (m, 1Н), 7.96 (м, 1Н), 8.26 (д, J=9 Гц, 1Н), 8.75 (д, J=5 Гц, 1Н). LC-MS (время удерживания: 1.46, Метод A). MS m/z 615 (M++1).
Стадия 7:
К раствору трипептидной кислоты - продукта Стадии 6 из Примера 371 (0.0453 г, 0.074 ммоля) {BOCNH-P3(L-трет-BuGly)-Р2[(4R)-(7-хлорхинолин-4-оксо)-S-пролин}-Р1((1R,2S Vinyl Acca) COOH} в ТГФ (4 мл) прибавляют CDI (17 мг, 0.10 ммоля), кипятят 45 мин и охлаждают до rt. Прибавляют сразу одной порцией циклопропансульфонамид (0.013 г. 0.10 ммоля), а затем чистый DBU (0.015 мл, 0.10 ммоля). Реакционную смесь перемешивают в течение 18 час, прибавляют EtOAc (200 мл) и промывают буфером рН4 (3×30 мл), водой (2×30 мл), рассолом (30 мл), сушат (MgSO4) и очищают на пластине 20×40 см 1000 мкМ для препаративной ТСХ Analtech (МеОН-СН2Cl2: от 0 до 2%), получают нужный продукт (Соединение 371) в виде пены (0.040 г, 76%):1H ЯМР δ м.д. 0.95-1.23 (м, 4Н), 1.03 (с, 9Н), 1.19 (с, 9Н), 1.40-1.43 (м, 1H), 1.85 (дд, J=8.5 Гц, 1Н), 2.12-2.20 (м, 1Н), 2.43 (м, 1Н), 2.82 (м, 1Н), 4.07-4.19 (м, 2Н), 4.51-4.57 (м, 2Н), 5.07 (д, J=10 Гц, 1Н), 5.25 (д, J=17 Гц, 1Н), 5.85 (м, 1Н), 5.48 (с, 1Н), 7.09 (д, J=5 Гц, 1Н), 7.45 (д, J=9 Гц, 1Н), 7.92 (м, 1Н), 8.20 (д, J=9 Гц, 1Н), 8.72 (д, J=5 Гц, 1Н); LC-MS (время удерживания: 1.52, Метод В), MS m/z 718 (M++1). HRMS m/z (M+H)+ вычислено для C41H51N5SO9. 718.2677, найдено 718.2674.
Пример 372: Получение Соединения 372
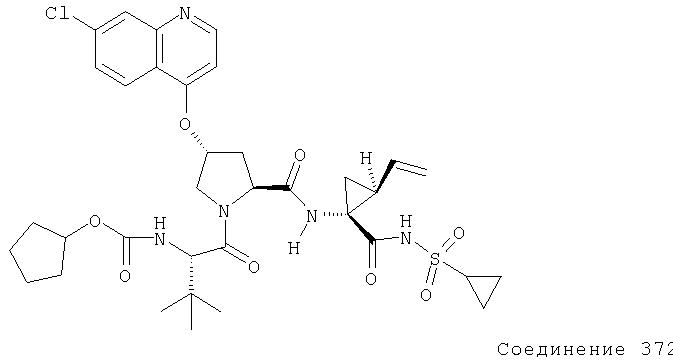
Схема 1
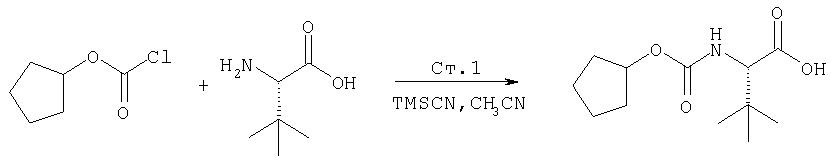
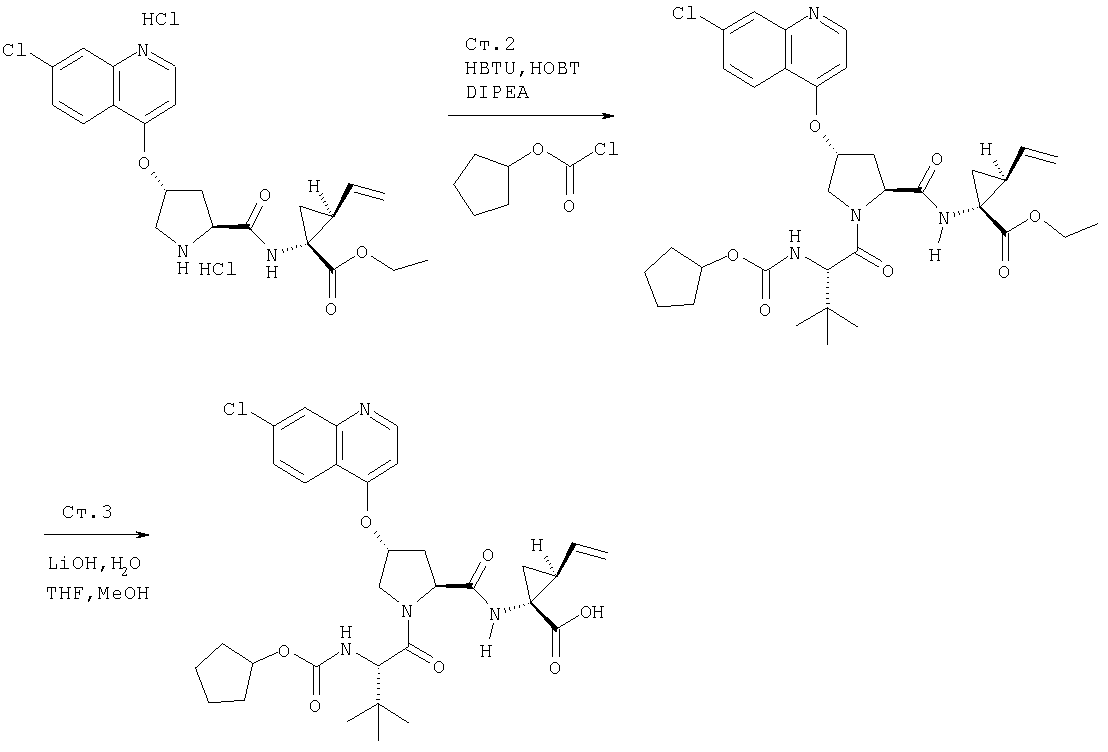
Стадия 1:
К раствору L-трет-лейцина (2 г, 15.25 ммоля) в CH3CN (50 мл) прибавляют TMSCN (7.06 мл, 56.41 ммоля) и перемешивают 15 мин. Реакционную смесь греют при 75°С в течение 30 мин. Прибавляют циклопентилхлорформиат (2.83 г, 19.06 ммоля) и греют при 80°С в течение ночи, упаривают в вакууме. К остатку прибавляют МеОН (40 мл), перемешивают 10 мин и упаривают в вакууме. рН остатка доводят до 8.5 и экстрагируют Et2O (2×200 мл). Водный слой подкисляют до рН 3 и экстрагируют CH2Cl2 (2×200 мл). Объединенные вытяжки сушат (MgSO4) и упаривают в вакууме. Остаток перекристаллизовывают из минимального количества Et2О/смесь гексанов, получают 3.48 г (94%):1H ЯМР (500 МГц, метанол-d4) δ м.д. 1.00 (с, 9Н), 1.59 (с, 2Н), 1.73 (м, 4Н), 1.84 (дд, J=5.95, 3.20 Гц, 2Н), 3.98 (с, 1Н), 5.02 (м, 1Н).
Стадия 2:
К раствору продукта Стадии 4 (530.1 мг, 1.04 ммоля) из Примера 371 {HCl соль Р2[(4R)-(7-хлорхинолин-4-оксо)-S-пролин}-Р1((1R,2S Vinyl Acca) COOEt, продукта (328 мг, 1.35 ммоля) Стадии 1 из Примера 372 {(L)-2-циклопентилоксикарбониламино-3, 3-диметилмасляной кислоты}, НОВТ (146 мг, 1.08 ммоля) и диизопропилэтиламина (0.755 мл, 4.32 ммоля) в CH2Cl2 (7 мл), прибавляют HBTU (512 мг, 1.35 ммоля). Раствор перемешивают при rt в течение ночи, распределяют между CH2Cl2 и буфером с рН 4.0. СН2Cl2 соль промывают водой, насыщенным водным раствором NaHCO3, сушат (MgSO4) и упаривают. Остаток очищают на колонке Biotage 40M (EtOAc-смесь гексанов: 35%-100%), получают 640 мг (92%) продукта в виде пены.1H ЯМР (Метанол-d4) δ м.д. 1.02 (с, 9Н), 1.26 (м, 4Н), 1.56 (м, 10H), 2.19 (кв, J=8.75 Гц, 1Н), 2.41 (м, 1Н), 2.70 (дд, J=14.19, 8.09 Гц, 1Н), 4.01 (дд, J=11.90, 3.05 Гц, 1Н), 4.13 (м, 2Н), 4.20 (с, 1Н), 4.53 (м, 1Н), 4.62 (м, 1Н), 5.09 (д, J=10.38 Гц, 1Н), 5.26 (д, J=17.09 Гц, 1Н), 5.47 (м, 1Н), 5.77 (м, 1Н), 7.07 (д, J=5.49 Гц, 1Н), 7.47 (м, 1Н), 7.94 (м, 1Н), 8.20 (д, J=8.85 Гц, 1Н), 8.72 (д, J=5.49 Гц. 1Н). LC-MS (время удерживания: 1.71, Метод В), MS m/z 655 (M++1).
Стадия 3:
Трипептидную кислоту получают как описано в Стадии 7 Схемы 2 из Примера 370, за исключением того, что вместо продукта Стадии 6 из Примера 370 используют циклопентоксикарбонил-NH-P3(L-трет-BuGly)-Р2[(4R)-7-хлорхинолин-4-оксо)-S-пролин}-Р1((1R,2S Vinyl Acca) COOEt.
Модификация: берут 0.636 г (0.97 ммоля) продукта Стадии 2 из Примера 372, получают 0.424 г (выход 69%).
Продукт:
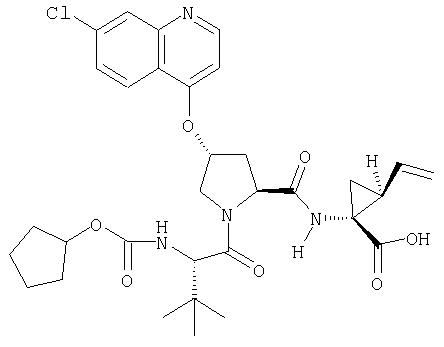
Данные:1H ЯМР (Метанол-d4) δ м.д. 1.02 (с, 9Н), 1.57 (м, 11H), 2.14 (кв, J=9.03 Гц, 1Н), 2.46 (м, 1Н), 2.68 (м, 1Н), 4.02 (дд, J=11.89, 3.11 Гц, 1Н), 4.19 (м, 1Н), 4.50 (д, J=26.35 Гц, 1Н), 4.64 (т, J=8.42 Гц, 1Н), 5.04 (м, 1Н), 5.24 (д, J=17.20 Гц, 1Н), 5.44 (с, 1Н), 5.87 (м, 1Н), 7.05 (д, J=5.12 Гц, 1Н), 7.48 (м, 1Н), 7.92 (м, 1Н), 8.18 (д, J=8.78 Гц, 1Н), 8.71 (д, J=5.49 Гц, 1Н). LC-MS (время удерживания: 2.32, Метод А), MS m/z 627 (M++1).
Схема 2
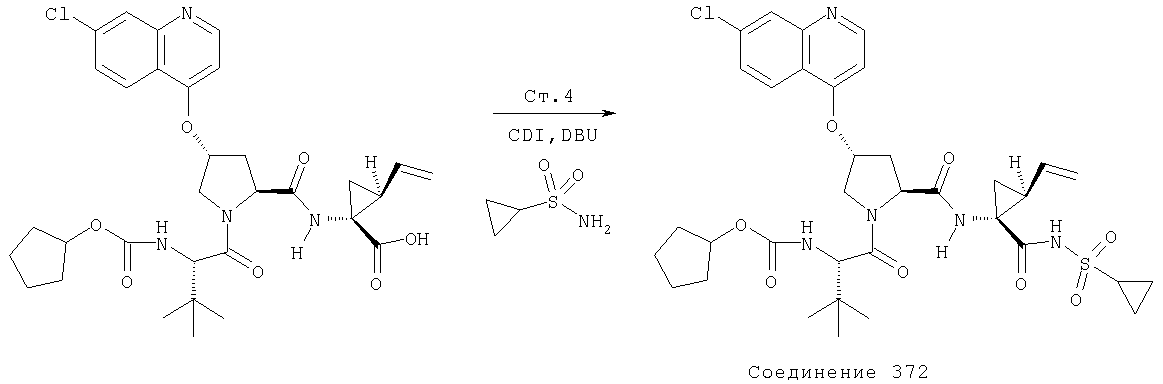
Стадия 4:
Раствор CDI (0.021 г, 0.13 ммоля) и продукта Стадии 3 из Примера 372 (0.058 г, 0.09 ммоля) {BOCNH-P3(L-трет-BuGly)-P2[(4R)-(7-хлорхинолин-4-оксо)-S-пролин}-Р1((1R,2S Vinyl Асса) СООН} в ТГФ (2 мл) кипятят 40 мин и охлаждают до rt. Прибавляют циклопропансульфонамид (всего 0.016 г, 0.13 ммоля), а затем чистый DBU (0.027 мл, 0.18 ммоля). Реакционную смесь перемешивают в течение ночи, прибавляют EtOAc (100 мл) и промывают буфером рН4 (2х), сушат (MgSO4), упаривают и очищают на пластине 1000 мкМ для препаративной ТСХ Analtech (20×40 см, элюент, последовательно 50% до 0% до 2% МеОН в СН2Cl2), получают 27.3 мг (40%) нужного продукта (Соединение 372).1H ЯМР (Метанол-d4) δ м.д. 0.94 (м, 2Н), 1.02 (с, 9Н), 1.14 (м, 1Н), 1.49 (м, 11Н), 1.86 (м, 1Н), 2.14 (м, 1Н), 2.49 (м, 1Н), 2.68 (дд, J=13.89, 7.48 Гц, 1Н), 2.78 (м, 1Н), 4.08 (м, 1Н), 4.22 (с, 1Н), 4.55 (м, 2Н), 5.05 (д, J=10.07 Гц, 1Н), 5.22 (д, J=17.09 Гц, 1Н), 5.46 (м, 1Н), 5.86 (м, 1Н), 7.07 (д, J=5.19 Гц, 1Н), 7.46 (д, J=8.55 Гц, 1Н), 7.91 (с, 1Н), 8.18 (д, J=8.85 Гц, 1Н), 8.72 (д, J=5.19 Гц, 1Н). LC-MS (время удерживания: 1.52, Метод I), MS m/z 730 (M++1).
Пример 373: Получение Соединения 373

Схема 1

Стадия 1:
К раствору 2-амино-4-метоксиацетофенона (4.45 г, 26.94 ммоля) в CH2Cl2 (100 мл) при 0°C прибавляют циклопропанкарбонилхлорид (3.1 мл, 33.68 ммоля), диизопропилэтиламин (19 мл, 107.8 ммоля) и DMAP (0.780 г, 6.4 ммоля). Реакционную смесь перемешивают при rt в течение ночи и упаривают в вакууме. Остаток растворяют в CH2Cl2 (500 мл), промывают 1N водной HCl, водой, NaHCO3(водн.) и сушат (MgSO4). Растворитель отгоняют в вакууме, а твердый остаток обрабатывают EtOAc/смесь гексанов (1/1), получают продукт (5.35 г, 85%):1H ЯМР (Метанол-d4) δ м.д. 0.94 (м, 4Н), 1.69 (м, J=3.97 Гц, 1Н), 2.60 (с, 3H), 3.84 (с, 3H), 6.69 (д, J=7.93 Гц, 1Н), 7.98 (д, J=8.85 Гц, 1Н), 8.23 (с, 1Н).
Стадия 2:
Раствор продукта (5.35 г, 22.72 ммоля) Стадии 1 из Примера 373 {(2-ацетил-5-метоксифенил)-амида циклопропанкарбоновой кислоты} и трет-BuOK (5.45 г, 48.6 ммоля) в трет-бутаноле (130 г) кипятят 6 час. Реакционную смесь охлаждают, выливают в охлажденный льдом холодный буфер и доводят рН до 7, фильтруют. Осадок перекристаллизовывают из MeOH/Et2O, получают продукт (1 г, 20%):1H ЯМР (Метанол-d4) δ м.д. 0.96 (м, 2Н), 1.15 (м, 2Н), 1.94 (м, 1Н), 3.87 (с, 3H), 5.86 (м, 1Н), 6.93 (м, 2Н), 8.04 (д, J=8.85 Гц, 1Н).
Стадия 3:
К раствору N-Boc-L-3-гидроксипролина (1.06 г, 4.32 ммоля) и трифенилфосфина (2.27 г, 8.64 ммоля) в ТГФ (25 мл) при 0°С в течение 30 мин прибавляют раствор продукта (0.93 г, 4.32 ммоля) Стадии 2, Пример 373) {2-циклопропил-7-метоксихинолин-4-ол} и DEAD (1.50 г, 8.64 ммоля) в ТГФ. Реакционную смесь перемешивают в течение ночи и упаривают растворитель. Остаток дважды очищают на колонке Biotage 40+M (EtOAc/смесь гексанов: 20-65%), получают продукт (1.74 г, 90%): LC-MS (время удерживания: 2.56, Метод J), MS m/z 443 (M++1).
Стадия 4:
К суспензии продукта Стадии 3 (1.70 мг, 3.86 ммоля) из Примера 373 {метилового эфира Boc-(4R)-(2-циклопропил-7-метоксихинолин-4-оксо)-S-пролина} в смеси ТГФ (91 мл), СН3ОН (18.2 мл) и Н2О (27 мл) прибавляют LiOH (0.73 г, 30 ммолей). Реакционную смесь перемешивают в течение 16 час, доводят рН до 6, отгоняют органические растворители в вакууме. Водный остаток подкисляют до рН 4 и экстрагируют CH2Cl2 (4×100 мл). Объединенные органические растворы сушат (MgSO4) и упаривают в вакууме, получают 1.64 г (100%) нужного продукта:1H ЯМР (Метанол-d4) δ м.д. 1.32 (м, 13Н), 2.37 (м, 2Н), 2.71 (м, 1Н), 3.86 (м, 1Н), 3.95 (с, 3H), 4.14 (м, 1Н), 4.43 (м, 1Н), 5.41 (с, 1Н), 6.65 (с, 1Н), 7.19 (м, 1Н), 7.30 (м, 1Н), 8.02 (дд, J=12.63, 9.33 Гц, 1Н).
Стадия 5:
Продукт (1.61 г, 2.79 ммоля) Стадии 4 из Примера 373 (Boc-Р2{(4R)-[2-циклопропил-7-метоксихинолин-4-оксо]-S-пролин]-P1(1R,2S Vinyl Acca-COOEt} растворяют в HCl/диоксан (15 мл, 60 ммолей) и перемешивают 3 час при rt. Реакционную смесь упаривают и отгоняют азеотроп с сухим ТГФ, получают продукт (1.58 г, 100%): LC-MS (время удерживания: 2.12, Метод К), MS m/z 566 (M++1).
Схема 2

Стадия 6:
К суспензии продукта Стадии 5 (1.58 г, 2.79 ммоля) из Примера 373 {БисHCl соль Р2{(4R)-[2-циклопропил-7-метоксихинолин-4-оксо)-S-пролин}-Р1((1R,2S Vinyl Асса) COOEt}, диизопропилэтиламина (1.65 мл, 9.25 ммоля), N-Boc-L-трет-лейцина (0.775 г, 3.35 ммоля), НОВТ·Н2О (0.515 г, 3.36 ммоля) в СН2Cl2 (13 мл), прибавляют HBTU (1.28 г, 3.36 ммоля). Смесь перемешивают при rt в течение 14 час, распределяют между EtOAc и буфером с рН 4.0. Слой EtOAc сушат (MgSO4) и упаривают. Остаток очищают на колонке Biotage 40+M (EtOAc-смесь гексанов: 20%-100%, затем МеОН) и дополнительно очищают на пластине 1000 мкМ для препаративной ТСХ Analtech 20×40 см (МеОН-СН2Cl2 2%), получают 1.4 г (63%).1H ЯМР (Метанол-d4) δ м.д. 1.04 (с, 9Н), 1.20 (м, 5Н), 1.28 (с, 9Н), 1.39 (м, 2Н), 1.69 (м, 1Н), 2.19 (м, 2Н), 2.36 (м, 1Н), 2.63 (дд, J=13.54, 7.68 Гц, 1Н), 3.90 (с, 3H), 4.08 (м, 4Н), 4.19 (д, J=11.34 Гц, 1Н), 4.47 (д, J=11.71 Гц, 1Н), 4.56 (т, J=8.60 Гц, 1Н), 5.08 (м, 1Н), 5.24 (м, 1Н), 5.39 (с, 1Н), 5.78 (м, 1Н), 6.56 (с, 1Н), 6.96 (дд, J=9.15, 2.20 Гц, 1Н), 7.21 (д, J=2.56 Гц, 1Н), 7.97 (д, J=9.15 Гц, 1Н). LC-MS (время удерживания: 2.34, Метод К), MS m/z 679 (M++1).
Стадия 7:
К суспензии продукта Стадии 6 (1.28 г, 1.89 ммоля) из Примера 373, Boc-NH-P3(L-трет-BuGly)-P2[(4R)-(2-циклопропил-7-метоксихинолин-4-оксо)-S-пролин]-P1((1R,2S Vinyl Асса)-COOEt в смеси ТГФ (93 мл), СН3ОН (23 мл) и H2O (45 мл) прибавляют LiOH (0.491 г, 20.4 ммоля). Реакционную смесь перемешивают в течение 18.5 час, доводят рН до 4, отгоняют органические растворители в вакууме. Остаток экстрагируют EtOAc (5×100 мл). Объединенные органические растворы сушат (MgSO4) и упаривают в вакууме, получают 1.17 г (97%) нужного продукта:1H ЯМР (Метанол-d4) δ м.д. 1.04 (с, 9Н), 1.24 (с, 9Н), 1.27 (м, 3H), 1.42 (м, 2Н), 1.68 (дд, J=8.05, 5.12 Гц, 1Н), 2.17 (м, 1Н), 2.33 (м, 1Н), 2.47 (м, 1Н), 2.66 (м, 1Н), 3.95 (с, 3H), 4.09 (м, 2Н), 4.51 (д, J=11.71 Гц, 1Н), 4.59 (т, J=8.60 Гц, 1Н), 5.07 (м, 1Н), 5.26 (м, 1Н), 5.52 (с, 1Н), 5.85 (м, 1Н), 6.69 (с, 1Н), 7.10 (дд, J=9.15, 2.20 Гц, 1Н), 7.27 (д, J=2.20 Гц, 1Н), 8.10 (д, J=9.15 Гц, 1Н). LC-MS (время удерживания: 2.21, Метод К), MS m/z 651 (M++1).
Схема 3

Стадия 8:
Раствор CDI (0.058 г, 0.344 ммоля) и продукта Стадии 7 из Примера 373 (0.160 г, 0.246 ммоля) {Boc-NH-P3(L-трет-BuGly)-P2[(4R)-(2-циклопропил-7-метоксихинолин-4-оксо)-S-пролин}-Р1((1R,2S Vinyl Acca) COOH} в ТГФ (2 мл) кипятят 60 мин и охлаждают до rt. Прибавляют циклопропансульфонамид (0.041 г, 0.344 ммоля), а затем чистый DBU (0.051 мл, 0.344 ммоля). Реакционную смесь перемешивают 24 час, обрабатывают, распределяя реакционную смесь между буфером рН 4 и EtOAc. Органический слой сушат (MgSO4), упаривают и очищают препаративной ВЭЖХ (0-100% растворителя В), получают 0.086 г (46%) нужного продукта (Соединение 373).1H ЯМР (CF3COOD) δ м.д. 1.04 (с, 9Н), 1.21 (м, 16Н), 1.41 (м, 1Н), 1.87 (дд, J=8.05, 5.49 Гц, 1Н), 2.26 (м, 3H), 2.61 (дд, J=12.99, 6.77 Гц, 1Н), 2.93 (м, 1Н), 3.92 (с, 3H), 4.09 (м, 1Н), 4.21 (м, 1Н), 4.49 (м, 2Н), 5.11 (д, J=11.71 Гц, 1Н), 5.27 (д, J=17.20 Гц, 1Н), 5.46 (с, 1Н), 5.76 (м, 1Н), 6.62 (м, 2Н), 7.01 (дд, J=8.97. 2.01 Гц, 1Н), 7.23 (д, J=2.56 Гц, 1Н), 8.00 (д, J=8.78 Гц, 1Н).
Пример 374: Получение Соединения 374

Схема 1

Стадия 1:
К раствору м-анизидина (58 г, 471 ммоля) в 800 мл CH3CN прибавляют кислоту Мелдрума (75 г, 518 ммоля) и триметилформиат (60 г, 565 ммоля). Гетерогенную смесь кипятят 2 час. Растворитель отгоняют в вакууме, прибавляют МеОН (30 мл), полученный осадок отфильтровывают и промывают 10-15 мл МеОН. Процедуру прибавления МеОН/фильтрования повторяют с концентрированной (упаренной) маточной жидкостью. Полученный объединенный осадок сушат (˜20 торр (2.7 КПа), 45°С в течение ночи), получают 117.6 г (90%) интермедиата 5-[(3-метоксифениламино)метилен]-2,2-диметил-[1, 3]диоксан-4,6-диона.
Стадия 2:
К раствору Ph2O (500 г), нагретому до 250°С, порциями в течение 30 мин прибавляют 108.7 г (392 ммоля) 5-[(3-метоксифениламино)метилен]-2,2-диметил-[1,3]диоксан-4,6-дион. Смесь нагревают еще в течение 15 мин, охлаждают до rt, прибавляют смесь гексанов (800 мл) и полученную суспензию перемешивают в течение ночи. Смесь гексанов декантируют, твердый остаток при кипячении растворяют в 600 мл МеОН, охлаждают до rt и полученный осадок отфильтровывают и промывают минимальным количеством CH2 Cl2. Всего после аналогичной перекристаллизации получают 20.73 г (30%) 7-метоксихинолин-4-ола в виде светло-коричневого твердого вещества.1Н ЯМР (Метанол-d4) δ м.д. 3.87 (с, 3H), 6.23 (д, J=7.3 Гц, 1Н), 6.68 (д, J=2.4 Гц, 1Н), 6.96 (дд, J=9.0, 2.4 Гц, 1Н), 8.11 (д, J=9 Гц, 1Н); LC-MS (время удерживания: 0.77, Метод D), MS m/z 176 (M++1).
Стадия 3:
К раствору метилового эфира N-Boc-cis-L-4-гидроксипролина (12.24 г, 49.8 ммоля) и PPh3 (26.14 г, 99.7 ммоля) в ТГФ (200 мл), охлажденному до 0°С, в течение 45 мин прибавляют раствор DEAD (17.36 г, 99.7 ммоля) и 7-метоксихинолин-4-ола (8.73 г, 49.8 ммоля) в ТГФ (700 мл). Температуру смеси медленно, в течение ночи, доводят до комнатной, упаривают в вакууме. Остаток очищают на колонке Biotage 65M (МеОН-EtOAc: 0-10%), получают 12.78 г (64%) продукта в виде бесцветного стекла:1H ЯМР (CDCl3) δ м.д. 1.36 (с, 9Н), 2.26-2.35 (м, 1Н), 2.57-2.68 (м, 1Н), 3.71 (с, 3H), 3.75-3.92 (м, 2Н), 3.86, 3.87 (два с (ротамеры), 3H), 4.41-4.53 (м, 1Н), 5.09 (м, 1Н), 6.52 (д, J=5.5 Гц, 1Н), 7.06-7.09 (м, 1Н), 7.24-7.26 (м, 1Н), 7.94 (д, J=9.1 Гц, 1Н), 8.50-8.56 (м, 1Н); LC-MS (время удерживания: 1.34, Метод D), MS m/z 403 (M++1).
Стадия 4:
К раствору продукта (8.54 г, 21.2 ммоля) Стадии 3 из Примера 374 {метилового эфира ВОС-N-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролина} в 600 мл смеси 5:1 ТГФ/МеОН прибавляют раствор LiOH (4.0 г, 167 ммоля) в 150 мл воды. Смесь перемешивают в течение ночи, рН доводят до 7 с помощью 6N водн. HCl и раствор упаривают до тех пор, пока не останется только водный слой. рН остатка доводят до 4, добавляя 1N водн. HCl, насыщают NaCl и смесь несколько раз распределяют между раствором и сначала EtOAc, а затем ТГФ, так как продукт растворим в воде. Объединенные органические вытяжки сушат (MgSO4) и упаривают, получают продукт (8.18 г, 99%) в виде твердого вещества белого цвета.1H ЯМР (CDCl3-Метанол-d4) δ м.д. 1.42 (с, 9Н), 2.40-2.49 (м, 1Н), 2.68-2.77 (м, 1Н), 3.88 (м, 2Н), 3.94 (с, 3H), 4.41-4.53 (м, 1Н), 5.32 (м, 1Н), 6.86-6.92 (м, 1Н), 7.21 (дд, J=9, 2 Гц, 1Н), 7.30 (д, J=2 Hz, 1Н), 8.05-8.10 (м, 1Н), 8.62 (д, J=6 Гц, 1Н); LC-MS (время удерживания: 1.20, Метод A), MS m/z 389 (M++1).
Стадия 5:
К раствору продукта (5.11 г, 13 ммоля) Стадии 2 из Примера 371, {Boc-4(R)-(7-метоксихинолин-4-оксо) пролина}, 2.66 г (13.9 ммоля) HCl соли vinyl Acca (в виде смеси 1:1 диастереоизомеров (1R,2S/1S,2R, где циклопропилкарбоксиэтильная группа находится в син положении относительно винильного фрагмента), 10 мл (57.4 ммоля) DIPEA и 2.13 г (13.9 ммоля) HOBT·H2О в СН2Cl2 (150 мл), прибавляют HBTU (5.27 г, 13.9 ммоля) и смесь перемешивают в течение ночи. Прибавляют CH2Cl2 (200 мл) и распределяют между ним и буфером с рН 4.0 (2×50 мл). Органический слой промывают насыщенным водным раствором NaHCO3 (2×50 мл), водой (2×50 мл) и рассолом (2×50 мл). Органический раствор сушат (MgSO4), упаривают и очищают на колонке Biotage 65M (MeOH/EtOAc: 0%-9%), получают элюируемый сначала изомер Boc-NH-Р2[(4R)-(7-метоксихинолин -4-оксо)-S-пролин]-Р1((1R,2S Vinyl Acca P1 фрагмент)-CO2Et (2.21 г, общий выход 36%), а затем 1.13 г (19%) чистого изомера с низким Rf Boc-NH-P2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-Р1((1R,2S Vinyl Acca PI фрагмент)-CO2Et. Также получают смешанные фракции. Данные для Вос-N(?)-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-Р1((1R,2S Vinyl Acca P1 фрагмент)-COOEt1H ЯМР (CDCl3) δ м.д. 1.16 (т, J=7 Гц, 3H), 1.35 (с, 9Н), 1.37-1.47 (м, 1Н), 1.74-1.88 (м, 1Н), 2.04-2.13 (м, 1Н), 2.32-2.46 (м, 1Н), 2.58-2.69 (м, 1Н), 3.76 (м, 1Н), 3.87 (с, 3H), 4.02-4.13 (м, 2Н), 4.30-4.44 (м, 1Н), 5.05-5.19 (м, 2Н), 5.24 (д, J=17 Гц, 1Н), 5.63-5.71 (м, 1Н), 6.61 (м, 1Н), 7.07 (дд, J=9.2 Гц, 1Н), 7.22 (д, J=2 Гц, 1Н), 7.76-7.83 (м. 1Н), 7.92 (д, J=9 Гц, 1Н), 8.50 (д, J=5 Гц, 1Н). LC-MS (время удерживания: 1.38, Метод A), MS m/z 526 (M+ +1).
Схема 2
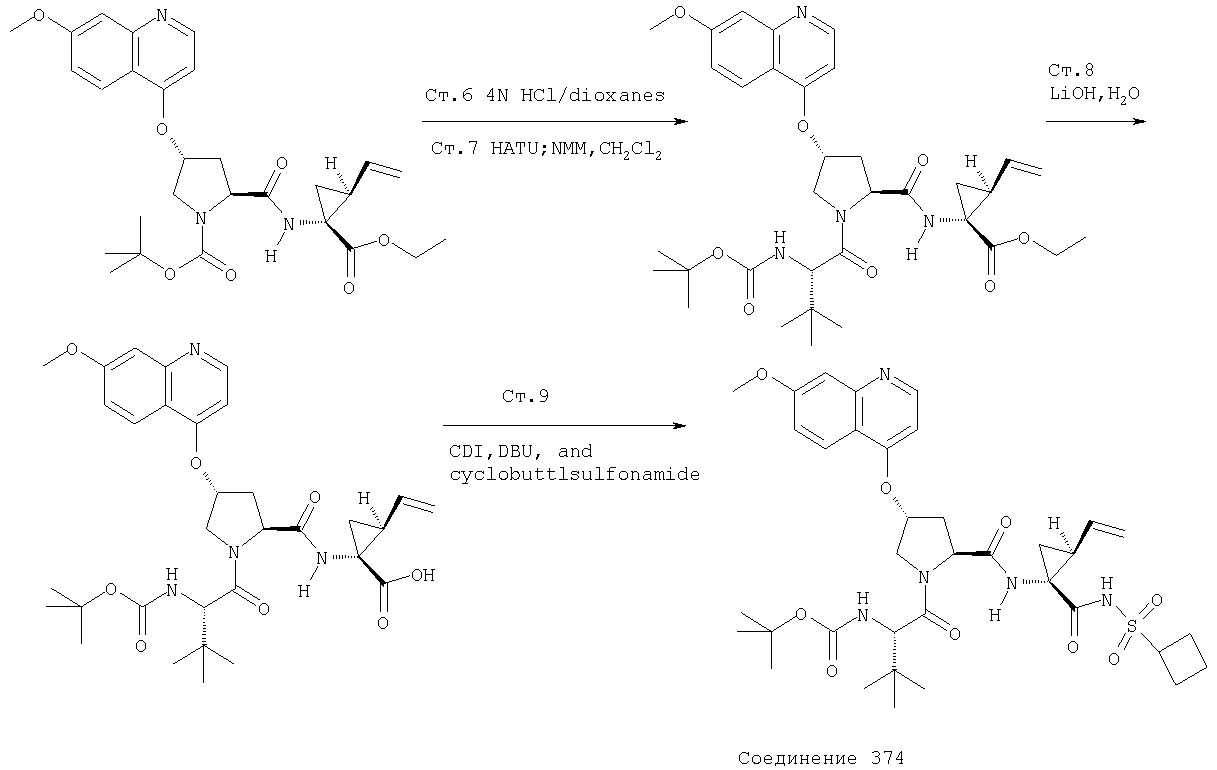
Стадия 6:
Весь продукт (1.35 г, 2.90 ммоля) Стадии 5 из Примера 374 [ВОС-Р2 [(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Acca)-COOEt) растворяют в 4N HCl/диоксан (15 мл, 60 ммоля) и перемешивают 2.5 час при rt. Реакционную смесь упаривают в вакууме, получают 1.3 г (100%) продукта в виде твердого вещества желтовато-коричневого цвета, которое непосредственно используется на следующей стадии.1H ЯМР (Метанол-d4) δ м.д. 1.25 (т, J=7 Гц, 1Н), 1.47-1.52 (м, 1Н), 1.78 (дд, J=8, 5 Гц, 1Н), 2.21-2.32 (м, 1Н), 2.55-2.64 (м, 1Н), 2.99 (дд, J=15.7 Гц, 1Н), 3.96 (с, 2Н), 4.06 (с, 3H), 4.14 (кв, J=7 Гц, 2Н), 4.69-4.75 (м, 1Н), 5.13 (д, J=10 Гц, 1Н), 5.33 (д, J=17 Гц, 1Н), 5.71-5.83 (м, 1Н), 5.89 (м, 1Н), 7.44 (м, 1Н), 7.49-7.52 (м, 1Н), 8.51-8.55 (м, 1Н), 8.94-8.96 (м, 1Н);13С ЯМР (Метанол-d4) δ м.д. 14.62, 23.08, 30.89, 34.73, 36.97, 41.03, 52.42, 57.11, 60.17, 62.70, 81.13, 100.06, 103.07, 117.02, 118.53. 122.70, 126.86, 134.74, 143.15, 146.75, 166.62, 167.71, 169.37, 171.18. LC-MS (время удерживания: 0.94, Метод D), MS m/z 426 (M++1).
Стадия 7:
К суспензии продукта (1.3 г, 2.61 ммоля) Стадии 6 из Примера 374 {БисHCl NH2-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1(1R,2S - Vinyl Acca)-COOEt}, N-ВОС-L-трет-лейцин (ВОС L-tBuGly) (0.94 г, 4.25 ммоля), NMM (1.7 мл, 15.5 ммоля) в ДМФА (20 мл) прибавляют HATU (1.55 г, 3.40 ммоля) при rt. Реакционную смесь перемешивают в течение ночи, прибавляют 75% EtOAc-ТГФ (300 мл), промывают буфером с рН 4.0 (2×50 мл), насыщенным водным раствором NaHCO3 (50 мл), рассолом (50 мл), сушат (MgSO4), очищают на колонке Biotage 40 М (элюент 15% - 100% EtOAc в смеси гексанов), получают продукт 0.702 г (42%) {BOCNH-P3(L-tBuGly)-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1-CO2Et} в виде пены.1H ЯМР (Метанол-d4) δ м.д. 1.06 (с, 9Н), 1.22-1.32 (м, 3H), 1.28 (с, 9Н), 1.42-1.46 (м, 1Н), 1.73 (дд, J=8.5 Гц, 1Н), 2.19-2.25 (м, 1Н), 2.67-2.72 (м, 1Н), 3.95 (с, 3H), 4.03-4.07 (м, 1Н), 4.10-4.18 (м, 2Н), 4.20-4.24 (м, 1Н), 4.54 (д, J=12 Гц, 1Н), 4.60-4.63 (м, 1Н), 5.11 (дд, J=10.2 Гц, 1Н), 5.28-5.30 (м, 1Н), 5.43 (м, 1Н), 5.76-5.83 (м, 1Н), 6.50 (д, J=9 Гц, NH), 6.93 (д, J=5 Гц, 1Н), 7.10 (дд, J=9.2 Гц, 1Н), 7.28 (м, 1Н), 7.99 (м, 1Н), 8.11 (д, J=9 Гц, 1Н), 8.62 (д, J=5H); LC-MS (время удерживания: 0.94, Метод D), MS m/z 639.
Стадия 8:
К суспензии продукта (702 мг, 1.1 ммоля) Стадии 7 из Примера 374 {BOCNH-P3(L-t-BuGly)-P2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1{1R,2S Vinyl Acca-COOEt} в смеси ТГФ (50 мл), МеОН (7 мл) и Н2O (22 мл) прибавляют LiOH (211 мг, 8.80 ммоля). Реакционную смесь перемешивают один день, подкисляют до рН 7 (нейтрального) и упаривают в вакууме до тех пор, пока не останется только водный слой. Доводят рН водного остатка до 4, добавляя 1.0 N водный раствор HCl, а затем насыщают NaCl. Эту водную смесь несколько раз экстрагируют EtOAc и ТГФ. Объединенные органические вытяжки промывают рассолом (50 мл), сушат (MgSO4), фильтруют и упаривают в вакууме, получают 631 мг (92%) продукта, {BOCNH-P3(L-tBuGly)-P2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Acca-CO2H} в виде твердого вещества.1H ЯМР (Метанол-d4) δ м.д. 1.04 (с, 9Н), 1.22 (с, 9Н), 1.34-1.39 (м, 1Н), 1.67 (дд, J=8.5 Гц, 1Н), 2.03-2.13 (м, 1Н), 2.43-2.49 (м, 1Н), 2.67-2.73 (м, 1Н), 3.96 (с, 3H), 4.00-4.05 (м, 1Н), 4.15-4.21 (м, 1Н), 4.56-4.62 (м, 2Н), 5.02 (д, J=10 Гц, 1Н), 5.20 (д, J=17 Гц, 1Н), 5.52 (м, 1Н), 5.87-5.99 (м, 1Н), 6.47 (д, J=8 Гц, 1Н), 6.91 (с, 1Н), 7.12 (д, J=5 Гц, 1Н), 7.19 (дд, J=9.2 Гц, 1Н), 7.31 (д, J=2 Гц, 1Н), 8.22 (д, J=9 Гц, 1Н), 8.72 (д, J=5 Гц, 1Н). LC-MS (время удерживания: 1.44, Метод D), MS m/z 611 (M++1).
Стадия 9:
К раствору трипептидной кислоты - продукта Стадии 8 из Примера 374 (0.120 г, 0.195 ммоля) в ТГФ (2 мл) прибавляют CDI (44.3 мг, 0.27 ммоля) и полученный раствор кипятят 60 мин и охлаждают до rt. Прибавляют сразу одной порцией циклопропансульфонамид (0.037 г. 0.273 ммоля), а затем чистый DBU (0.041 мл, 0.273 ммоля). Реакционную смесь перемешивают в течение 24 час, прибавляют еще один эквивалент CDI и циклопропансульфонамида и смесь перемешивают еще в течение 48 час. Прибавляют 50% ТГФ/EtOAc (200 мл) и промывают буфером рН 4 (30 мл), сушат (MgSO4) и упаривают в вакууме. Остаток растворяют в 2 мл 50% ТГФ-CH2Cl2, прибавляют 75 мг (0.39 ммоля) EDAC, 48 мг (0.39 ммоля) 4-DMAP, 58 мкл (0.39 ммоля DBU и 53 мг (0.39 ммоля) циклобутилсульфонамида и смесь перемешивают 4 дня. Смесь очищают на пластине 20×40 см 1000 мкМ для препаративной ТСХ Analtech (элюция 2% МеОН-CH2Cl2), получают нужный продукт, Соединение 374, BOCNH-P3(L-t-BuGly)-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Acca-CONHSO2-циклобутан} в виде пены (2 мг, 2%):1H ЯМР (Метанол-d4) δ м.д. 1.07, 1.08 (два с (ротамеры) 9Н), 1.20, 1.21 (два с (ротамеры) 9Н), 1.41-1.48 (м, 1Н), 1.64-1.70 (м, 1Н), 1.72-1.91 (м, 2Н), 1.95-2.11 (м, 2Н), 2.23-2.37 (м, 2Н), 2.40-2.58 (т, 2Н), 2.72-2.75 (м, 1Н), 4.06 (с, 3H), 4.12-4.17 (м, 2Н), 4.35-4.38 (м, 1Н), 4.58-4.62 (м, 1Н), 4.65-4.70 (м, 1Н), 5.16-5.18 (м, 1Н), 5.24-5.37 (м, 1Н), 5.69-5.76 (м, 2Н), 7.40-7.46 (м, 3H), 8.35-8.40 (м, 1Н), 8.92 (д, J=7 Гц, 1Н). LC-MS (время удерживания: 1.58, Метод В), MS m/z 728 (M++1).
Пример 375: Получение Соединения 375

Схема 1
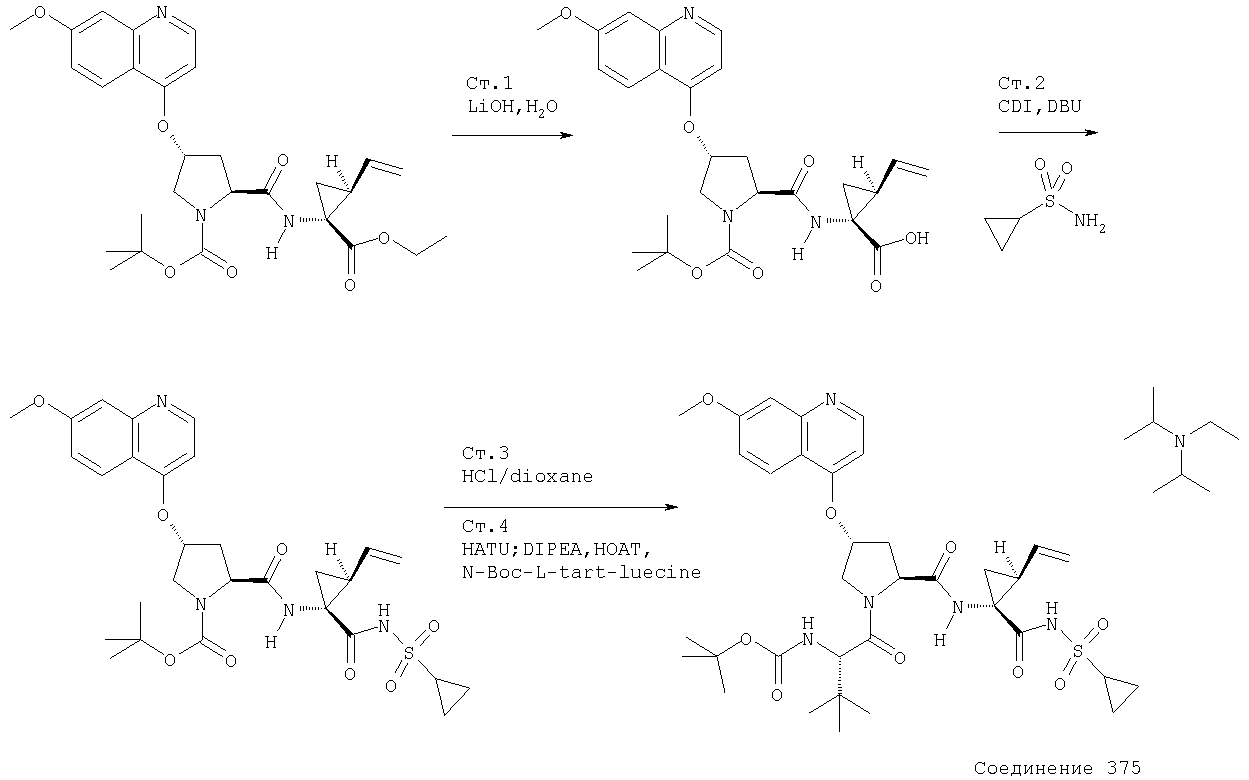
Стадия 1:
К раствору продукта (794 мг, 1.51 ммоля) Стадии 5 из Примера 374 {N-ВОС-P2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин)-P1(1R,2S Vinyl Acca)-CO2Et} в 68 мл of 12% MeOH/THF прибавляют раствор 218 мг (9.08 ммоля) гидроксида лития в 30 мл воды и смесь перемешивают 16 час. Доводят рН до нейтрального, добавляя 6N водн. HCl, упаривают так, чтобы осталась только вода, доводят значение рН до 4, прибавляя 1N HCl, и экстрагируют 50% THF-EtOAc (5×200-мл порции). Объединенные органические вытяжки сушат (MgSO4) и упаривают, получая продукт (752 мг,100%) {N-ВОС-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин)-P1(1R,2S Vinyl Acca)-СО2H):1H ЯМР (Метанол-d4) δ м.д. 1.37-1.43 (м, 1Н), 1.39 (с, 9Н), 1.69-1.78 (м, 1Н), 2.16-2.24 (м, 1Н), 2.44-2.54 (м, 1Н), 2.64-2.74 (м, 1Н), 3.89-3.94 (м, 2Н), 3.96 (с, 3H), 4.40-4.43 (м, 1Н), 5.11 (д, J=10 Гц, 1Н), 5.31 (д, J=17 Гц, 1Н), 5.40 (м, 1Н), 5.79-5.87 (м, 1Н), 6.91 (с, 1Н), 7.04 (д, J=6 Гц, 1Н), 7.25 (дд, J=9.1, 2 Гц, 1Н), 7.29 (м, 1Н), 8.09 (д, J=9.1 Гц, 1Н), 8.66 (д, J=6 Гц, 1Н). LC-MS (время удерживания: 1.05, Метод Н), MS m/z 498 (M++1).
Стадия 2:
К раствору продукта (399.5 мг, 0.668 ммоля) Стадии 1 из Примера 375 {N-ВОС-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин)-P1(1R,2S Vmyl Acca)-CO2H} в ТГФ (4 мл) прибавляют CDI (434 мг, 2.68 ммоля), кипятят 60 мин и охлаждают до rt. Прибавляют сразу одной порцией циклопропансульфонамид (406 мг, 3.35 ммоля), а затем чистый DBU (0.50 мл, 3.35 ммоля). Реакционную смесь перемешивают в течение 16 час, прибавляют 50% ТГФ-EtOAc (200 мл) и промывают рассолом, насыщенным буфером рН 4 (2×40 мл). Органические вытяжки сушат (MgSO4), упаривают, очищают на колонке Biotage 25M (элюция 0-15% МеОН в CH2Cl2), получают 217 мг (54%) нужного продукта, BOCNH-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Асса-CONHSO2-циклопропана}:1H ЯМР (Метанол-d4) δ м.д. 1.01-1.10 (м, 2Н), 1.11-1.18 (м, 1Н), 1.20-1.27 (м, 1Н), 1.39-1.48 (м, 1Н), 1.44 (с, 9Н), 1.87 (дд, J=8,5 Гц, 1Н), 2.01-2.38 (м, 2Н), 2.57 (дд, J=14.7 Гц, 1Н), 2.91-2.96 (м, 1Н), 3.83-3.92 (м, 2Н), 3.94 (с, 3H), 4.36-4.39 (м, 1Н), 5.11 (д, J=10 Гц, 1Н), 5.29 (д, J=17 Гц, 1Н), 5.38 (м, 1Н), 5.74-5.81 (м, 1Н), 6.91 (д, J=5.5 Гц, 1Н), 7.20 (дд, J=9.2, 2.4 Гц, 1Н), 7.29 (м, 1Н). 8.07 (д, J=9.2 Гц, 1Н), 8.60 (д, J=5.5 Гц, 1Н). LC-MS (время удерживания: 1.28, Метод I), MS m/z 601 (M++1).
Стадия 3:
Весь продукт (198 мг, 0.33 ммоля) Стадии 2 из Примера 375 [ВОС-Р2 [(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Асса)-CONHSO2-циклопропан} растворяют в 4N HCl/диоксан (4 мл, 16) и перемешивают 2 час при rt. Реакционную смесь упаривают в вакууме, получают сырой продукт в виде твердого вещества желтовато-коричневого цвета, которое непосредственно используется на следующей стадии.
Стадия 4:
Сырой продукт Стадии 3 из Примера 375 {Бис HCl соль NH-P2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Асса-CONHSO2-циклопропана} суспендируют в 10 мл хлористого метилена. К этой смеси прибавляют N-ВОС-L-трет-лейцин (ВОС L-tBuGly) (120 мг, 0.52 ммоля), НОАТ (30 мг, 0.20 ммоля), DIPEA (0.29 мл, 1.65 ммоля) и HATU (0.56 г, 1.47 ммоля). Реакционную смесь перемешивают в течение 16 час, прибавляют 50% ТГФ-EtOAc (300 мл), промывают рассолом, насыщенным буфером рН4 (3×50 мл). Органические вытяжки сушат (MgSO4), упаривают. Остаток очищают на колонке Isco 35g (элюция 0-15% МеОН в СН2Cl2), получают 130.1 мг (47%) в виде соли основания Хюннига (Hunnig) (Соединение 375):1H ЯМР (Метанол-d4) δ м.д. 1.00-1.48 (м, 29Н), 1.47 (с, 9Н), 1.89 (м, 1Н), 2.26 (м, 1Н), 2.36 (м, 1Н), 2.69 (м, 1Н), 2.97 (м, 1Н), 3.25 (кв, J=7.43 Гц, 2Н), 3.74 (м, 2Н), 3.97 (с, 3H), 4.10 (м, 1Н), 4.23 (дд, J=19.68, 9.92 Гц, 1Н), 4.57 (м, 2Н), 5.15 (м, 1Н), 5.31 (м, 1Н), 5.50 (с, 1Н), 5.77 (м, 1Н), 7.01 (т, J=5.34 Гц, 1Н), 7.16 (д, J=9.16 Гц, 1Н), 7.31 (д, J=1.83 Гц, 1Н), 8.14 (м, 1Н), 8.67 (д, J=5.49 Гц, 1Н). LC-MS (время удерживания: 1.49, Метод D), MS m/z 714 (M++1).
Пример 376: Получение Соединения 376
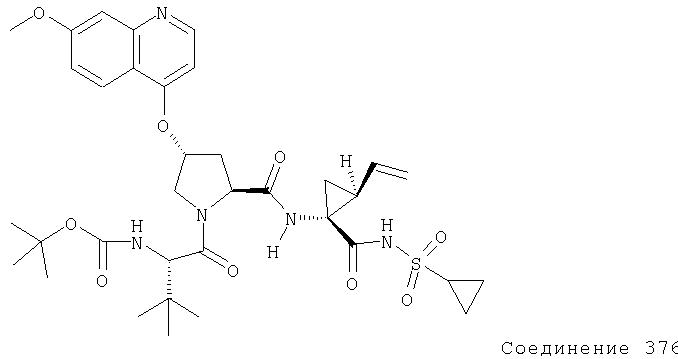
Схема 1
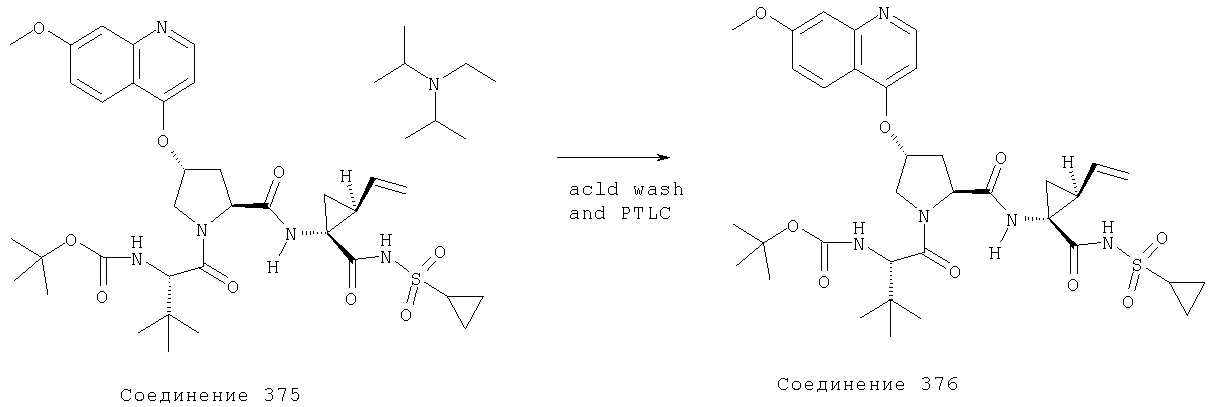
Стадия 1:
Соединение 375 (130 мг) растворяют в EtOAc, еще один раз промывают буфером с рН 4, рассолом, а затем сушат (MgSO4). Сырую смесь очищают на двух пластинах 1000 мкМ для препаративной ТСХ Analtech (20×40 см, элюция 3% МеОН в CH2Cl2), получают 54 мг (выход 23% в расчете на трипептидную кислоту) (Соединение 376).1H ЯМР (Метанол-d4) δ м.д. 0.88-1.00 (м, 2Н), 1.01-1.14 (м, 2Н), 1.03 (с, 9Н), 1.25 (с, 9Н), 1.34 (дд, J=9.5 Гц, 1Н), 1.81-1.89 (м, 1Н), 2.06-2.13 (м, 1Н), 2.45-2.50 (м, 1Н), 2.65-2.75 (м, 1Н), 3.91 (с, 3H), 3.98-4.11 (м, 1Н), 4.21-4.22 (м, 1Н), 4.46-4.50 (м, 1Н), 4.54-4.57 (м, 1Н), 4.97-5.02 (м, 1Н), 5.14-5.22 (м, 1Н), 5.33-5.41 (м, 1Н), 5.81-5.99 (м, 1Н). 6.87-6.95 (м, 1Н), 7.06-7.09 (м, 1Н), 7.25 (м, 1Н), 8.07-8.10 (м, 1Н), 8.59 (д, J=5.2 Гц, 1Н). HRMS m/z (M-H)- вычислено для С35 Н46N5O9S: 712.3016, найдено: 712.3024; LC-MS m/z 714 (время удерживания: 1.42, Метод I).
Пример 377: Получение Соединения 377
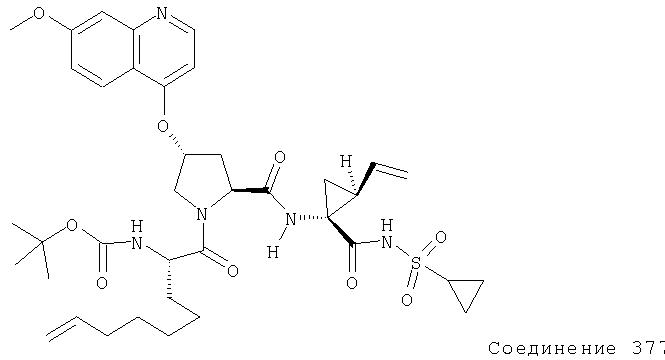
Схема 1

Стадия 1:
Всего к 1.0 ммоля продукта Стадии 2 из Примера 375 {Бис HCl соль NH-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Acca-CONHSO2-циклопропана}, суспендированного в 20 мл хлористого метилена, при rt прибавляют 351 мг (1.30 ммоля) 2-(S)-трет-бутоксикарбониламино-8-ноненовой кислоты от RSP Aminoacids, НОАТ (82 мг, 0.60 ммоля), DIPEA (0.74 мл, 5.0 ммоля) и HATU (494 мг, 1.30 ммоля). Реакционную смесь перемешивают в течение 16 час, отгоняют основную часть CH2Cl2 в вакууме. К смеси прибавляют насыщенный буфер рН 4 (150 мл) и экстрагируют EtOAc (4×200 мл). Объединенные органические вытяжки сушат (MgSO4), упаривают. Остаток очищают на колонке Biotage 40M (элюция 0-15% МеОН в СН2Cl2, получают 574 мг (76%) продукта (Соединение 377): LC-MS (время удерживания: 1.64, Метод I), MS m/z 754.
Пример 378: Получение Соединения 378.
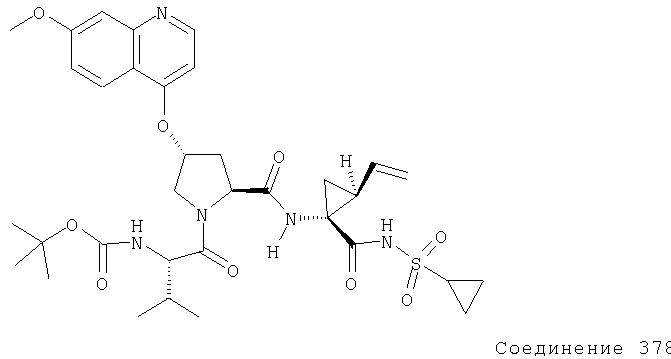
Схема 1
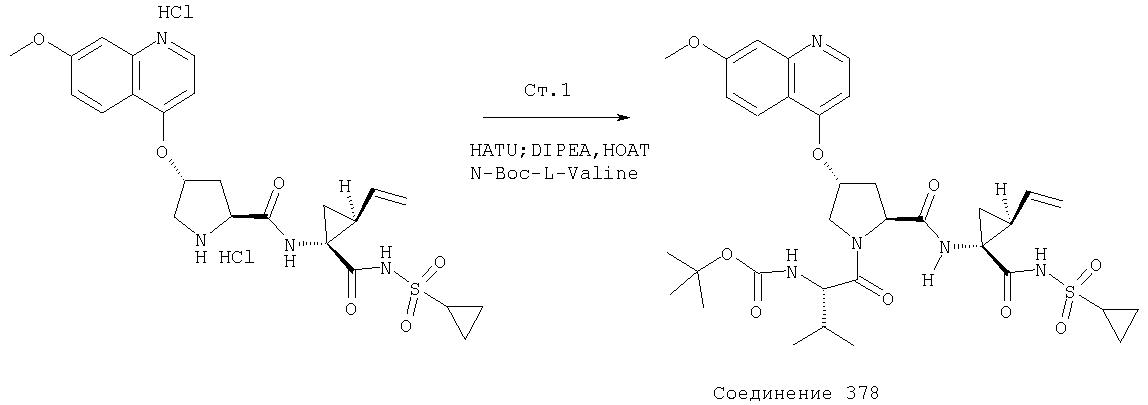
Стадия 1:
Всего 0.34 ммоля продукта Стадии 2 из Примера 375 {Бис HCl соль NH-Р2[(4R)-(7-метоксихинолин-4-оксо)-S-пролин]-Р1(1R,2S Vinyl Acca-CONHSO2-циклопропана} суспендируют в 3 мл хлористого метилена. К нему при rt прибавляют N-ВОС-L-валин (L-Val) (120 мг, 0.55 ммоля), НОАТ (30 мг, 0.20 ммоля), DIPEA (0.29 мл, 1.65 ммоля) и HATU (160 мг, 0.43 ммоля). Реакционную смесь перемешивают в течение 16 час, прибавляют насыщенный буфер рН 4 (150 мл) и экстрагируют EtOAc (3×200 мл). Объединенные органические вытяжки промывают рассолом, сушат (MgSO4) и упаривают. Остаток очищают на колонке Isco 35 g (элюция 0-15% МеОН в СН2Cl2). Затем этот продукт очищают на два δ пластине 1000 мкМ для препаративной ТСХ Analtech (20×40 см, элюция 3% МеОН в СН2Cl2), получают 104.1 мг (44%) продукта (Соединение 378): HRMS m/z (M-H)-, вычислено для С34Н44N5O9S: 698.2860, найдено: 698.2865; LC-MS m/z 700 (время удерживания: 1.60, Метод D).
Пример 379: Получение Соединения 379
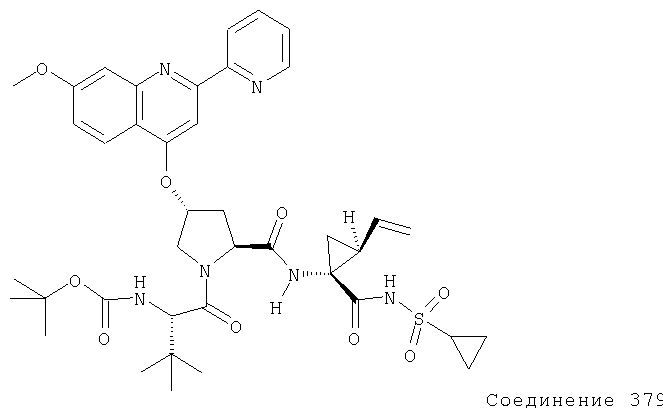
Схема 1:
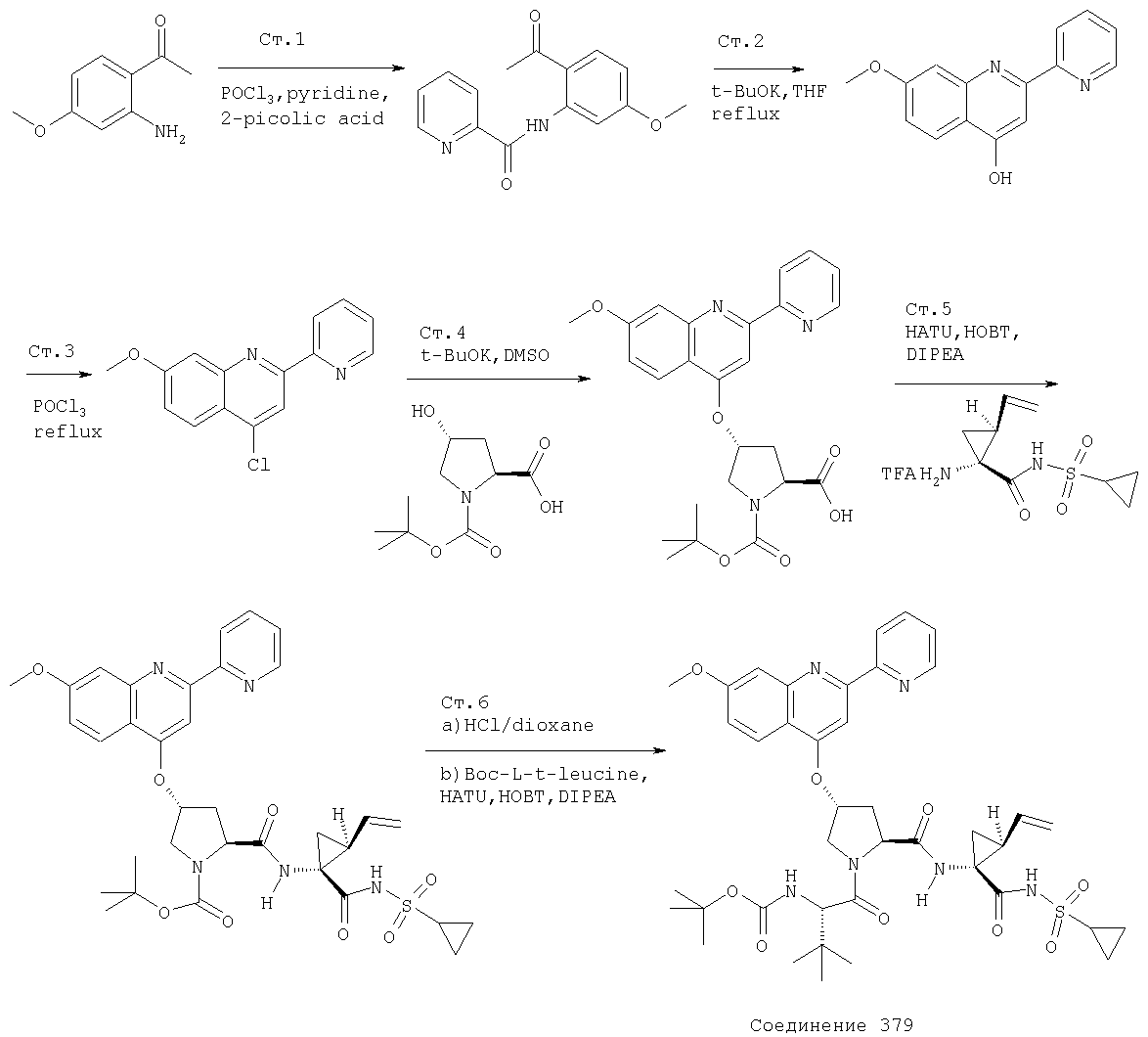
Стадия 1:
К суспензии 2-пиколиновой кислоты (3.73 г, 30.3 ммоля) и 2-амино-4-метоксибензофенона (5.0 г, 30.3 ммоля) в пиридине (150 мл) при -30°С за 5 мин прибавляют POCl3 (3.7 мл, 45.4 ммоля) и реакционную смесь перемешивают при этой температуре в течение 3 час и при rt в течение ночи. Выливают в холодную воду и экстрагируют EtOAc (3х). Объединенные вытяжки сушат, получают продукт (7.67 г, 93%);1H ЯМР (Метанол-d4) δ м.д. 2.65 (с, 3H), 3.92 (с, 3H), 6.78 (м, 1Н), 7.60 (м, 1Н), 8.00 (м, 1Н), 8.06 (м, 1Н), 8.21 (д, J=7.63 Гц, 1Н), 8.59 (т, J=2.29 Гц, 1Н), 8.76 (д, J=3.97 Гц, 1Н). LC-MS (время удерживания: 1.56, Метод D), MS m/z 271 (M++1).
Стадия 2:
К суспензии (2-ацетил-5-метоксифенил)-амида пиридин-2-карбоновой кислоты (2.90 г, 10.7 ммоля) в ТГФ (50 мл) прибавляют трет(t)-BuOK/ТГФ (1М, 24 мл, 24 ммоля). Реакционную смесь нагревают при 70°С в течение 3 час и перемешивают в течение ночи. Растворитель отгоняют в вакууме. К остатку прибавляют холодную воду и доводят рН до 4.6, добавляя 1.0N водн. HCl, отфильтровывают. Осадок очищают на колонке Biotage 65M (элюция 0-15% МеОН в CH2Cl2), получают 2.26 г (84%) продукта: LC-MS (время удерживания: 1.19, Метод D), MS m/z 253 (M++1).
Стадия 3:
Смесь 7-метокси-2-пиридин-2-илхинолин-4-ол (2.2 г, 8.71 ммоля) в POCl3 (92 мл) кипятят 3 час, а затем отгоняют растворитель в вакууме. К остатку прибавляют ледяную воду, прибавляют 1.0N NaOH, получая значение рН>10, и экстрагируют EtOAc (2x). Объединенные органические вытяжки промывают водой, рассолом, сушат (MgSO4), отгоняют растворитель и получают продукт в виде твердого вещества желтого цвета (89%, 2.1 г):1H ЯМР (ДМСО-d6) δ м.д. 3.97 (с, 3H), 7.40 (дд, J=9.16, 2.44 Гц, 1Н), 7.53 (м, 1Н), 8.01 (м, 1Н), 8.09 (д, J=9.16 Гц, 1H), 8.46 (с, 1Н), 8.56 (д, J=7.93 Гц, 1Н), 8.74 (д, J=3.97 Гц, 1Н). LC-MS (время удерживания: 1.50, Метод D), MS m/z 271 (М++1).
Стадия 4:
К раствору N-Boc-4-гидроксипролина (1.6 г, 6.7 ммоля) в ДМСО (20 мл) прибавляют трет-BuOK (1.9 г, 16.8 ммоля). Полученную смесь перемешивают 1.5 час и прибавляют 4-хлор-7-метокси-2-пиридин-2-илхинолин (2.0 г, 7.4 ммоля) и ДМСО (10 мл). Реакционную смесь перемешивают 38 час, прибавляют холодную воду и экстрагируют смесью EtOAc/эфир (1/4, 2x). Водный слой подкисляют до рН 4 и экстрагируют смесью EtOAc/ТГФ (5х). Объединенные органические вытяжки сушат (Na2SO4/MgSO4), отгоняют растворитель в вакууме, а остаток очищают препаративной ВЭЖХ (0-80% растворителя В), получают продукт (1.6 г, 50%): LC-MS (время удерживания: 1.23, Метод I), MS m/z 466 (M++1).
Стадия 5:
Раствор продукта (0.21 г, 0.65 ммоля) Стадии 4 из Примера 379 {этилового эфира N-boc-(1R,2S)-1-амино-2-винилциклопропанкарбоновой кислоты} в растворе HCl/диоксан (4М, 5 мл, 20 ммоля) перемешивают 3 час и растворитель отгоняют в вакууме. К остатку прибавляют СН2Cl2 (10 мл), диизопропилэтиламин (0.4 мл, 3.23 ммоля), НОВТ (0.20 г, 1.35 ммоля), Boc-(4R)-(2-циклопропил-7-метоксихинолин-4-оксо)-S-пролин (0.20 г, 0.5 ммоля) и HATU (0.415 г, 1.07 ммоля). Реакционную смесь перемешивают в течение ночи и прибавляют буфер с рН 4.0, экстрагируют EtOAc. Вытяжки сушат (MgSO4) и очищают на колонке Biotage 40 М, используя в качестве элюента MeOH/CH2Cl2 (0-15%), получают продукт (204.7 мг, 70%):1H ЯМР (Метанол-d4) δ м.д. 0.64 (м, 1Н), 0.96 (м, 2Н), 1.33 (м, 8Н), 1.39 (м, 9Н), 1.90 (м, 2Н), 2.18 (м, 1Н), 2.54 (м, 1Н), 2.81 (м, 1Н), 4.01 (м, 5Н), 4.44 (д, J=28.99 Гц, 1Н), 5.08 (м, 1H), 5.31 (м, 1Н), 5.57 (с, 1Н), 6.03 (м, 1Н), 6.94 (с, 1Н), 7.27 (д, J=8.24 Гц, 1Н), 7.64 (м, 1Н), 7.92 (м, 1Н), 8.14 (м, 2Н), 8.66 (с, 1Н), 8.74 (с, 1Н).
Стадия 6:
Суспензию Р2 Boc-(4R)-(7-метокси-2-пиридин-2-илхинолин-4-оксо)-S-пролин]-P1(1R,2S Vinyl Асса)-CONHSO2(1-циклопропилметилциклопропан-1-ил)а (Стадия 5, Пример 379) (203 мг, 0.3 ммоля) в 4М HCl/диоксан (3,5 мл, 14 ммоля) перемешивают 2 час, растворитель отгоняют в вакууме. К остатку прибавляют CH2Cl2 (2 мл), диизопропилэтиламин (0.63 мл, 3.6 ммоля), Boc-L-лейцин (83 мг, 0.36 ммоля), HOAt (41 мг, 0.3 ммоля) и HATU (148 мг, 0.39 ммоля). Реакционную смесь перемешивают при rt в течение 7 час и растворитель отгоняют в вакууме. Остаток очищают препаративной ВЭЖХ (35-85% растворителя В), получают 25,1 мг (11%) нужного продукта (Соединения 379):1H ЯМР (Метанол-d4) δ м.д. -0.05 (м, 1Н), 0.30 (м, 1Н), 0.66 (м, 1Н), 0.91 (м, 2Н), 1.05 (с, 9Н), 1.28 (с, 9Н), 1.67 (м, 8Н), 2.15 (м, 1Н), 2.58 (м, 1Н), 2.77 (м, 1Н), 3.96 (с, 3H), 4.19 (д, J=40.25 Гц, 2Н), 4.51 (д, J=16.47 Гц, 2Н), 4.95 (м, 1Н), 5.15 (м, 1Н), 5.53 (с, 1Н), 5.89 (дд, J=16,65, 9.33 Гц, 1Н), 7.09 (д, J=8.42 Гц, 1Н), 7.43 (д, J=1.83 Гц, 1Н), 7.50 (м, 1Н), 7.82 (с, 1Н), 7.99 (м, 1Н), 8.10 (д, J=9.15 Гц, 1Н), 8.48 (д, J=7.68 Гц, 1Н), 8.72 (с, 1Н). LC-MS (время удерживания: 1.59, Метод I), MS m/z 791 (M++1).
Пример 380: Получение Соединения 380.

Схема 1

Стадия 1:
Исходное на Схеме 1 данного Примера получают сочетанием продукта Стадии 3 в Примере 11 Секции В с аминоконцом P1(1R,2S Vinyl Acca)-COOEt. Суспензию указанного продукта сочетания, Р2 Boc-(4R)-(6-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S Vinyl Acca)-COOEt (7.88 г, 14.99 ммоля), в 4М HCl/диоксан (120 мл, 480 ммоля) перемешивают в вакууме и сушат азеотропной отгонкой с сухим диоксаном. К остатку прибавляют ДМФА (75 мл), N-метилморфолин (6.27 мл, 57.07 ммоля), Boc-L-трет-лейцин (5.20 г, 22.49 ммоля) и HATU (8.53 г, 22.49 ммоля). Реакционную смесь перемешивают при rt в течение ночи и обрабатывают, выливая реакционную смесь в ледяную воду, доводят до рН 5, добавляя 1.0 N водн. HCl, и экстрагируют EtOAc. Вытяжки промывают NaHCO3 (водн.), рассолом, сушат (MgSO4) и упаривают. Остаток очищают на колонке Biotage 65M (EtOAc-смесь гексанов: 5-100%), получают продукт (8.07 г, 84%): LC-MS (время удерживания: 1.88, Метод I), MS m/z 639 (M++1).
Стадия 2:
К суспензии продукта (4.0 г, 6.26 ммоля) Стадии 1 из Примера 384 {Boc-NH-P3(L-трет-BuGly)-Р2[(4R)-(6-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R, 2S Vinyl Acca)-COOEt} в смеси ТГФ (250 мл), СН3ОН (31 мл) и Н2O (125 мл) прибавляют LiOH (2.4 г, 100.2 ммоля). Реакционную смесь перемешивают в течение ночи, а затем доводят до рН 7, добавляя 1.0 N водя. HCl. Органические растворители отгоняют в вакууме. Водный остаток подкисляют до рН 4 и экстрагируют EtOAc (2х). Объединенные органические вытяжки сушат (Na2SO4/MgSO4) и упаривают в вакууме, получая продукт (3.79 г, 99%):1H ЯМР (Метанол-d4) δ м.д. 1.05 (с, 9Н), 1.25 (м, 1Н), 1.29 (с, 9Н), 1.46 (м, 1Н), 1.72 (дд, J=8.24, 5.19 Гц, 1Н), 2.23 (кв, J=8.55 Гц, 1Н), 2.68 (дд, J=13.89, 7.78 Гц, 1Н), 3.94 (с, 3H), 4.05 (дд, J=11.60, 3.05 Гц, 1Н), 4.23 (д, J=8.85 Гц, 1Н), 4.46 (д, J=11.60 Гц, 1Н), 4.63 (т, J=8.39 Гц, 1Н), 5.10 (д, J=10.38 Гц, 1Н), 5.29 (д, J=17.40 Гц, 1Н), 5.85 (м, 2Н), 7.10 (д, J=9.16 Гц, 1Н), 7.19 (с, 1Н), 7.26 (д, J=5.49 Гц, 1Н), 7.91 (д, J=5.80 Гц, 1Н), 8.12 (д, J=9.16 Гц, 1Н). LC-MS (время удерживания: 1.81, Метод I), MS m/z 611 (M++1).
Стадия 3:
Раствор CDI (0.052 г, 0.32 ммоля) и продукта (0.130 г, 0.21 ммоля) Стадии 2 из Примера 384 {BOCNH-P3(L-трет-BuGly)-Р2[(4R)-6-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S Vinyl Acca)-СО2H) в ТГФ (2 мл) кипятят 60 мин и охлаждают до rt. Прибавляют циклобутансульфонамид (0.043 г, 0.32 ммоля), а затем чистый DBU (0.048 мл, 0.32 ммоля). Реакционную смесь перемешивают в течение ночи, затем фильтруют через шприцевой фильтр и очищают препаративной ВЭЖХ (30%-100% растворителя В), получают 0.1422 г (92%) нужного продукта:1H ЯМР (Метанол-d4) δ м.д. 1.04 (с, 9Н), 1.26 (д, J=13.43 Гц, 9Н), 1.39 (м, 1Н), 1.85 (дд, J=7.63, 5.19 Гц, 1Н), 1.98 (м, 2Н), 2.26 (м, 4 Н), 2.50 (м, 2Н), 2.61 (м, 1Н), 3.92 (с, 3H), 4.05 (м, 1Н), 4.24 (м, 1Н), 4.33 (м, 1Н), 4.43 (д, J=11.60 Гц, 1Н), 4.52 (м, 1Н), 5.13 (м, 1Н), 5.30 (м, 1Н), 5.71 (м, 1Н), 5.82 (с, 1Н), 7.08 (д, J=8.85 Гц, 1Н), 7.18 (с, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.88 (м, 1Н), 8.08 (д, J=9.16 Гц, 1Н). LC-MS (время удерживания: 1.89, Метод I), MS m/z 728 (M++1).
Схема 2
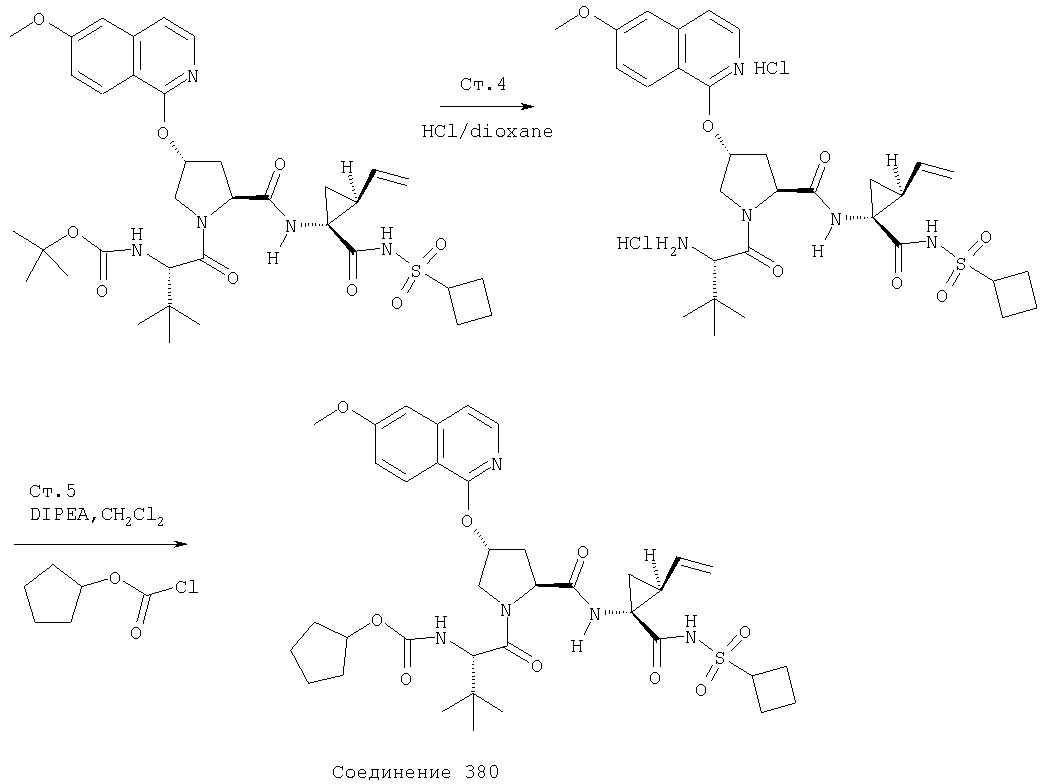
Стадия 4:
Продукт Стадии 3, Пример 380 (0.196 мг, 0.27 ммоля) {(BOCNH-P3(L-трет-BuGly)-P2-(4R)-(6-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S Vinyl Acca)-CONHSO2 циклобутил} растворяют в HCl/диоксане (5 мл; 20 ммолей) и перемешивают в течение 2 час при rt. Отгоняют растворитель в вакууме и получают титульный продукт (0.1887 г, 100%), готовый для применения на следующей стадии.
Стадия 5:
К смеси продукта (0.037 г, 0.053 ммоля) Стадии 4 из Примера 380 {HCl соли NH2-Р3(L-трет-BuGly)-P2-(4R)-(6-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S Vinyl Acca)-CONHSO2 циклобутил} и диизопропилэтиламина (0.046 мл, 0.26 ммоля) в СН2Cl2 (2 мл) прибавляют циклопентилхлорформиат (0.7 М, 0.151 мл, 0.069 ммоля). Реакционную смесь перемешивают в течение ночи и очищают препаративной ВЭЖХ (30% - 100% растворителя В), получают ожидаемый продукт (Соединение 380) (0.0303 г, 77%):1H ЯМР (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.48 (м, 9Н), 1.86 (дд, J=8.24, 5.49 Гц, 1Н), 1.99 (м, 2Н), 2.27 (м, 4Н), 2.51 (м, 2Н), 2.60 (дд, J=13.89, 6.87 Гц, 1Н), 3.92 (с, 3H), 4.05 (дд, J=12.21, 3.97 Гц, 1Н), 4.32 (м, 2Н), 4.41 (д, J=11.90 Гц, 1Н), 4.53 (м, 1Н), 4.69 (м, 1Н), 5.12 (д, J=10.38 Гц, 1Н), 5.29 (д, J=17.09 Гц, 1Н), 5.71 (м, 1Н), 5.83 (с, 1Н), 7.11 (д, J=9.46 Гц, 1Н), 7.19 (с, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.08 (д, J=9.16 Гц, 1Н). LC-MS (время удерживания: 1.85, Метод Н), MS m/z 740 (M++1).
Пример 381: Получение Соединения 381.
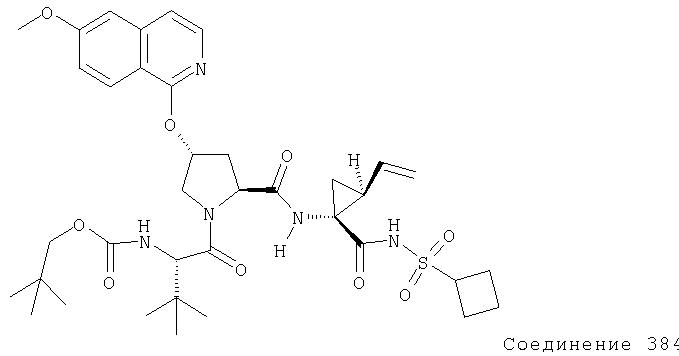
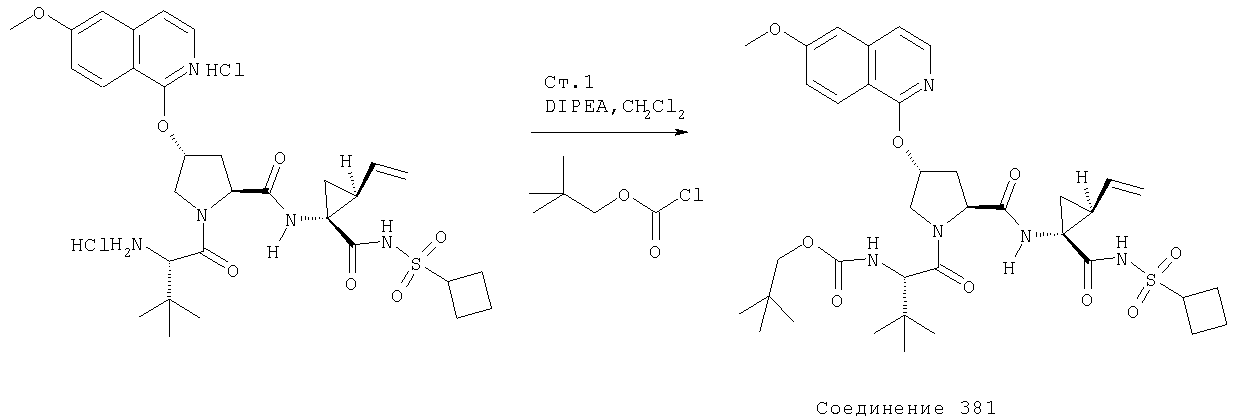
Стадия 1:
К смеси продукта (0.037 г, 0.053 ммоля) Стадии 4 из Примера 380 {HCl соли NH2-P3(L-трет-BuGly)-Р2-(4R)-(6-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S Vinyl Acca)-CONHSO2 циклобутил} и диизопропилэтиламина (0.046 мл, 0.26 ммоля) в CH2Cl2 (2 мл) прибавляют неопентилхлорформиат (0.012 мл, 0.069 ммоля). Реакционную смесь перемешивают в течение ночи и сразу очищают препаративной ВЭЖХ (30%-100% растворителя В), получают ожидаемый продукт (Соединение 381) (0.0252 г, 64%):1H ЯМР (Метанол-d4) δ м.д. 0.84 (с, 9Н), 1.05 (с, 9Н), 1.40 (м, 1Н), 1.86 (м, 1Н), 2.00 (м, 2Н), 2.28 (м, 4Н), 2.51 (м, 2Н), 2.57 (м, 1Н), 3.39 (д, J=10.07 Гц, 1Н), 3.55 (д, J=10.38 Гц, 1Н), 3.92 (с, 3H), 4.05 (м, 1Н), 4,33 (м, 2Н), 4.41 (д, J=11.29 Гц, 1Н), 4.53 (м, 1Н), 5.12 (д, J=10.07 Гц, 1Н), 5.29 (д, J=17.09 Гц, 1Н), 5.71 (м, 1Н), 5.82 (с, 1Н), 7.10 (д, J=9.16 Гц, 1Н), 7.19 (с, 1Н), 7.25 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 7.97 (с, 1Н), 8.07 (д, J=8.85 Гц, 1Н). LC-MS (время удерживания: 1.89, Метод H), MS m/z 742 (M++1).
Пример 382: Получение Соединения 382.
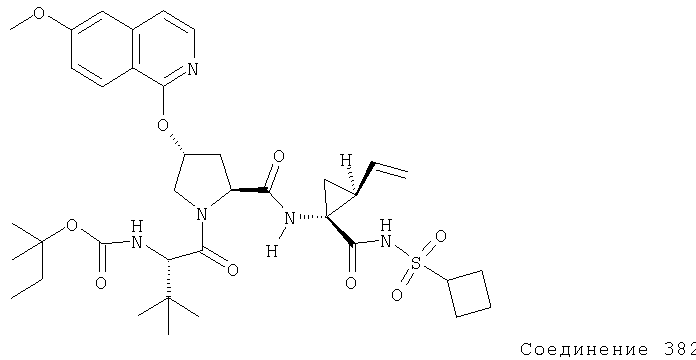
Схема 1

Стадия 1:
К смеси продукта (0.037 г, 0.053 ммоля) Стадии 4 из Примера 380 {HCl соли NH2 -Р3(L-трет-BuGly)-Р2-(4R)-(6-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S Vinyl Acca)-CONHSO2 циклобутил} и диизопропилэтиламина (0.046 мл, 0.26 ммоля) в CH2Cl2 (2 мл) прибавляют ди-трет-амилдикарбонат (0.0169 г, 0.069 ммоля). Реакционную смесь перемешивают в течение ночи и сразу очищают препаративной ВЭЖХ (30%-100% растворителя В), получают ожидаемый продукт (Соединение 382) (0.0175 г, 44%):1H ЯМР (Метанол-d4) δ м.д. 0.79 (т, J=6.87 Гц, 3H), 1.04 (с, 8Н), 1.21 (с, 3H), 1.23 (с, 3H), 1.41 (м, 2Н), 1.64 (м, 2Н), 1.83 (м, 1Н), 2.00 (м, 2Н), 2.26 (м, 4Н), 2.51 (м, 2Н), 2.60 (м, 1Н), 3.92 (с, 3H), 4.07 (м, 1Н), 4.24 (м, 1Н), 4.33 (м, 1Н), 4.43 (д, J=11.60 Гц, 1Н), 4.52 (м, 1Н), 5.13 (м, 1Н), 5.29 (м, 1Н), 5.71 (м, 1Н), 5.82 (с, 1Н), 7.09 (д, J=8.85 Гц, 1Н), 7.18 (с, 1Н), 7.25 (д, J=5.49 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.08 (д, J=8.85 Гц, 1Н). LC-MS (время удерживания: 1.90, Метод H), MS m/z 742 (M++1).
Пример 383: Получение Соединения 383.
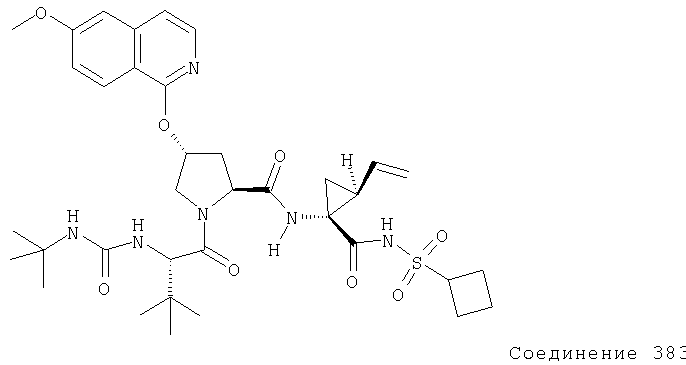
Схема 1

Стадия 1:
К смеси продукта (0.037 г, 0.053 ммоля) Стадии 4 из Примера 380 {HCl соли NH2-P3(L-трет-BuGly)-Р2-(4R)-(6-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S Vinyl Acca)-CONHSO2 циклобутил} и диизопропилэтиламина (0.046 мл, 0.26 ммоля) в СН2Cl2 (2 мл) прибавляют трет-бутилизоцианат (0.008 мл, 0.069 ммоля). Реакционную смесь перемешивают в течение ночи и сразу очищают препаративной ВЭЖХ (30%-100% растворителя В), получают ожидаемый продукт (Соединение 383) (0.024 г, 62%):1H ЯМР (Метанол-d4) δ м.д. 1.05 (с, 9Н), 1.19 (с, 9Н), 1.37 (м, 1Н), 1.85 (дд, J=8.09, 5.34 Гц, 1Н), 2.00 (м, 2Н), 2.26 (м, 4Н), 2.50 (м, 2Н), 2.58 (м, 1Н), 3.92 (с, 3H), 4.06 (м, 1Н), 4.32 (м, 2Н), 4.49 (м, 2Н), 5.11 (д, J=10.38 Гц, 1Н), 5.27 (д, J=17.40 Гц, 1Н), 5.69 (м, 1Н), 5.83 (с, 1Н), 7.08 (дд, J=9.16, 2.44 Гц, 1Н), 7.17 (д, J=2.44 Гц, 1Н), 7.24 (д, J=5.80 Гц, 1Н), 7.87 (д, J=6.10 Гц, 1Н), 8.12 (д, J=8.85 Гц, 1Н). LC-MS (время удерживания: 1.77, Метод Н), MS m/z 727 (M++1).
Пример 384: Получение Соединения 384.

Схема 1

Стадия 1:
Суспензию диизопропилэтиламина (0.031 мл, 0.018 ммоля), N, N'-дисукцинимидилкарбоната (0.0274 г, 0.107 ммоля) и продукта (0.050 г, 0.0714 ммоля) Стадии 4 из Примера 380 {HCl соли NH2 - Р3(L-трет-BuGly)-Р2-(4R)-(6-метоксиизохинолин-1-оксо)-S-пролин]-P1(1R,2S Vinyl Acca)-CONHSO2 циклобутил} в ТГФ (2 мл) подвергают действию ультразвука при 80°С в течение 15 мин. Прибавляют КН (0.046 г, 1.14 ммоля) и 1-метилциклопентанол (0.079 мл, 0.714 ммоля). Реакционную смесь перемешивают в течение 20 мин и обрабатывают, добавляя холодную воду, доводят рН до 4, экстрагируют EtOAc. Органические вытяжки сушат (MgSO4) и остаток очищают препаративной ВЭЖХ (30%-100% растворителя В), получают нужный продукт (Соединение 384) (0.018 г, 33%):1H ЯМР (Метанол-d4) δ м.д. 1.04 (с, 9Н), 1.29-1.79 (м, 10H), 1.84 (м, 2Н), 1.99 (м, 3H), 2.26 (м, 4Н), 2.49 (м, 2Н), 2.60 (дд, J=13.73, 7.02 Гц, 1Н), 3.92 (с, 3H), 4.05 (дд, J=11.29, 2.44 Гц, 1Н), 4.26 (с, 1Н), 4.32 (м, 1Н), 4.44 (д, J=11.90 Гц, 1Н), 4.52 (м, 1Н), 5.12 (д, J=10.07 Гц, 1Н), 5.28 (д, J=16.79 Гц, 1Н), 5.71 (м, 1Н), 5.82 (с, 1Н), 7.10 (д, J=8.85 Гц, 1Н), 7.19 (с, 1Н), 7.26 (д, J=5.80 Гц, 1Н), 7.88 (д, J=5.80 Гц, 1Н), 8.08 (д, J=9.16 Гц, 1Н). LC-MS (время удерживания: 1.91, Метод Н), MS m/z 754 (M++1).
Пример 385: Получение Соединения 385.
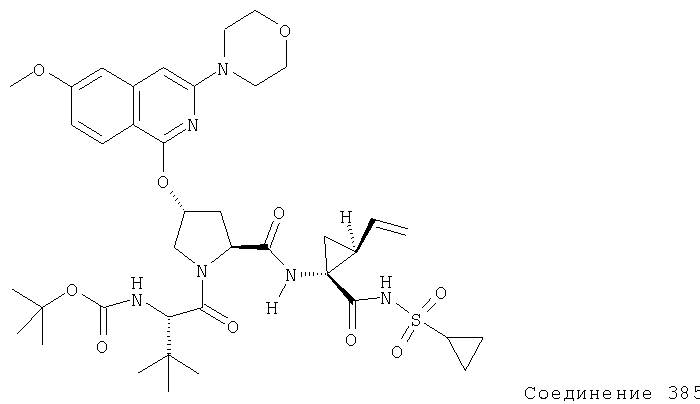
Схема 1

Стадия 1:
Суспензию метилового эфира 2-цианометил-4-метоксибензойной кислоты (1.9 г и TsOH·H2O ( 0.15 г, ммоль) в морфолине (5 мл) кипятят 4 час и отгоняют растворитель в вакууме. Остаток перекристаллизовывают из EtOAc/смесь гексанов с несколькими каплями МеОН, получают продукт (0.43 г, 17%); LC-MS (время удерживания: 1.07, Метод Н), MS m/z 266 (M++1).
Стадия 2:
Смесь 6-метокси-3-морфолин-4-илизохинолин-1-ола (0.298 г, 1.15 ммоля) в POCl3 (20 мл) кипятят в течение 2 час, отгоняют растворитель в вакууме и прибавляют холодную воду. Доводят значение рН до >11, прибавляя 1.0 N NaOH. Водный слой экстрагируют EtOAc. Органические вытяжки сушат (MgSO4), растворитель отгоняют в вакууме, получают продукт (0.299 г, 94%): LC-MS (время удерживания: 1.68, Метод Н), MS m/z 279 (M++1).
Стадия 3:
Смесь 1-хлор-6-метокси-3-морфолин-4-илизохинолина (0.050 г, 0.18 ммоля) и тетрабутилфосфония гидродифторида (0.8 г, 2.8 ммоля) [Synlett 1992, (4), 345-6] нагревают при 140°С в микроволновом реакторе в течение 10 мин, к реакционной смеси прибавляют EtOAc и фильтруют через предварительную колонку ISCO 25 g, в верхнем слое которой находится силикагель, отгоняют растворитель, получают продукт (0.037 г, 77%):1H ЯМР (CDCl3) δ м.д. 3.48 (м, 4Н), 3.84 (м, 4Н), 3.89 (с, 3H), 6.46 (д, J=1.22 Гц, 1Н), 6.85 (с, 1Н), 6.90 (дд, J=9 16, 2.44 Гц, 1Н), 7.82 (д, J=8.85 Гц, 1H). LC-MS (время удерживания: 1.56, Метод Н). MS m/z 263 (M++1).
Стадия 4:
Смесь 1-фтор-6-метокси-3-морфолин-4-илизохинолина (0.037 г, 0.14 ммоля), LaCl3 (0.020 г, 0.8 ммоля), t-BuOK (1М/ТГФ, 0.32 мл, 0.32 ммоля) и Boc-NH-P3(L-трет-BuGly)-Р2 [(4R)-4-гидрокси-S-пролин]-Р1(1R,2S Vinyl Acca)-CONHSO2 циклопропан (0.045 г, 0.08 ммоля) в ТГФ (3 мл) перемешивают 3 дня. К реакционной смеси прибавляют метанол и ее фильтруют через шприцевой фильтр и очищают препаративной ВЭЖХ, получают продукт в виде бледно-желтой пены (0.0158 г, 24%):1H ЯМР (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.24 (м, 4Н), 1.31 (с, 9Н), 1.43 (м, 2Н), 1.88 (м, 1Н), 2.24 (м, 2Н), 2.59 (дд, J=13.43, 6.71 Гц, 1Н), 2.94 (м, 1Н), 3.47 (м, 4Н), 3.83 (м, 4Н), 3.86 (с, 3H), 4.08 (м, 1Н), 4.28 (с, 1Н), 4.48 (м, 1Н), 5.12 (д, J=10.38 Гц, 1Н), 5.29 (д, J=16.48 Гц, 1Н), 5.76 (м, 2Н), 6.74 (д, J=9.16 Гц, 1Н), 6.94 (с, 1Н), 7.85 (д, J=8.85 Гц, 1Н), 9.19 (с, 1Н): LC-MS (время удерживания: 1.86, Метод Н), MS m/z 799 (M++1).
Пример 386: Получение Соединения 386.

Соединение 386 получают методами по данному описанию.
Подраздел I:
Все соединения в Секции I анализируют методом LC/MS в следующих условиях:
Метод A: Xterra C18 S7 3.0×50 мм
Градиент: от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В
Время градиента: 3 мин.
Время выдерживания: 1 мин.
Скорость потока: 4 мл/мин
Длина волны детектора: 220 нм
Растворитель А: 10% МеОН /90% H2O/0.1% ТФК
Растворитель В: 10% H2O /90% МеОН/0.1% ТФК
Пример 410: Получение Соединения 410.

Схема 1
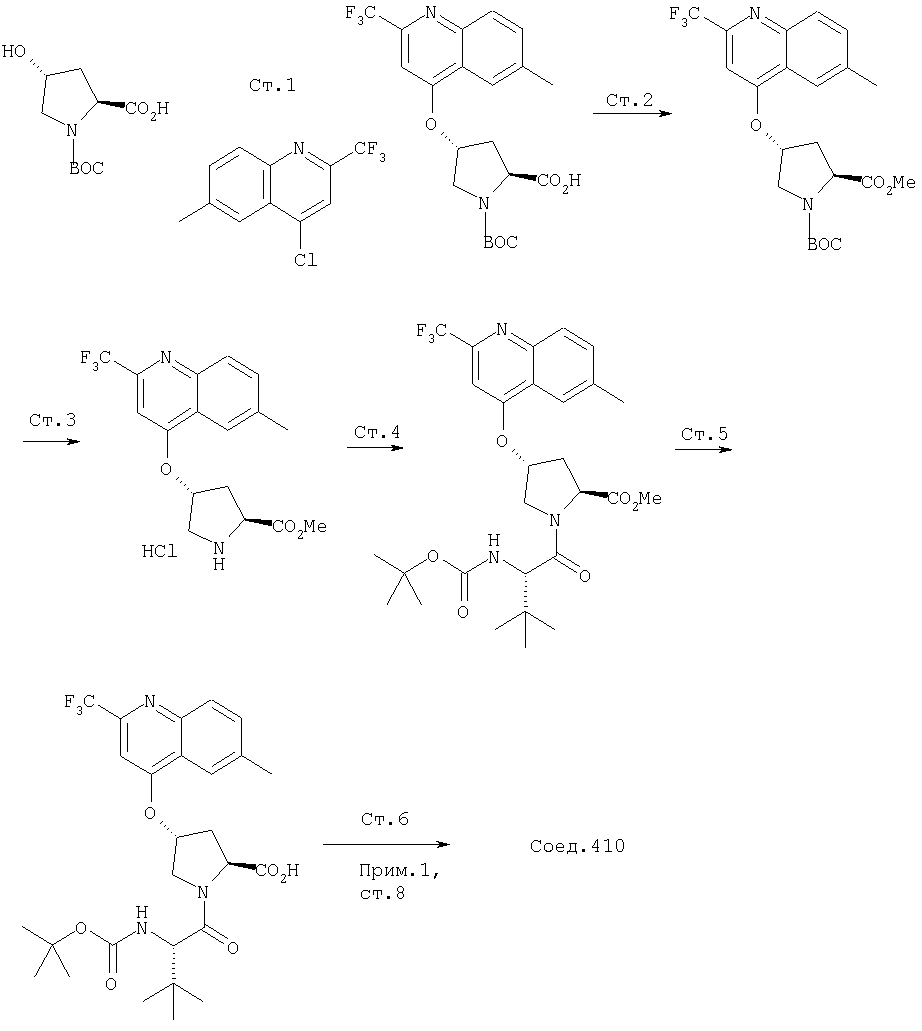
Стадия 1:
К раствору Boc-L-гидроксипролина (0.723 г, 3.13 ммоля) в ДМСО в атмосфере азота прибавляют KOtBu (0.808 г, 7.2 ммоля). Суспензию перемешивают при комнатной температуре в течение 1.5 час и двумя порциями прибавляют 4-хлор-6-метил-2-(трифторметил)хинолин (0.916 г, 3.75 ммоля). Смесь перемешивают при комнатной температуре три часа, для нейтрализации реакционной смеси прибавляют 1.3 эквивалента HCl (1N). Прибавляют буферный раствор с рН 4.0 и доводят рН до 4-5. Водный слой экстрагируют этилацетатом (3×25 мл) и объединенные органические вытяжки промывают рассолом (20 мл) и сушат MgSO4, получают титульное соединение в виде твердого вещества белого цвета (выход сырого продукта не рассчитывают). Сырой продукт используют на следующей стадии.
LC/MS (время удерживания rt. мин.(МН+): 2.48 (441.5) (Метод А).
Стадия 2:
Раствор неочищенного продукта Стадии 1,1-трет-бутилового эфира 4-(6-метил-2-трифторметилхинолин-4-илокси)-пирролидин-1,2-дикарбоновой кислоты (предположительно, 3.13 ммоля) в ТГФ (10 мл) и метаноле (10 мл) охлаждают до 0°С. К раствору при перемешивании под азотом медленно прибавляют TMSCN 2M в смеси гексанов (˜1.3 экв.) до тех пор, пока не прекратится выделение газа. По завершении реакции раствор упаривают в вакууме, очисткой на колонке Biotage 40M (элюпия 10%-30% этилацетатом в смеси гексанов) получают чистое титульное соединение в виде белой пены (976 мг, выход 69% в расчете на Стадии 1 и 2).
LC/MS (время удерживания rt. мин. (Н+): 2.60 (477) (Метод А).
Стадия 3:
Раствор продукта Стадии 2 (0.976 г, 2.15 ммоля) в DCM (7 мл) и ТФК (6.62 мл) перемешивают при комнатной температуре один час. Растворитель отгоняют в вакууме и остаток суспендируют в 1N HCl в диэтиловом эфире (8 мл), осторожно перемешивают и упаривают в вакууме. Эту процедуру повторяют и полученный продукт выдерживают в вакууме масляного насоса в течение ночи, получают твердое вещество белого цвета с количественным выходом.1H ЯМР (ДМСО-d6) δ м.д. 2.50 (с, 3H), 2.57-2.6 (м, 1Н), 2.66-2.71 (м, 1Н), 3.62-3.65 (уш д, J=15 Гц, 1Н), 3.80-3.81 (м, 4Н), 4.8 (уш с, 1Н), 5.7 (с, 1Н), 7.46 (с, 1Н), 7.72-7.75 (д, J=7.5 Гц, 1Н), 7.98-7.8 (д, J=8.5 Гц, 1Н), 8.24 (с, 1Н), 9.54 (уш с, 1Н); LC/MS (время удерживания rt. мин.(MH+): 1.61 (355) (Метод А).
Стадия 4:
В атмосфере азота продукт Стадии 3 (предполагается количественный выход, 2.746 ммоля) прибавляют к раствору ВОС-t-бутил-L-глицин (0.635 г, 2.746 ммоля) в DCM (20 мл). Затем прибавляют HOBt (0.408 г, 3.02 ммоля), DIPEA (3.35 мл, 19.2 ммоля) и HBTU (1.56 г, 4.12 ммоля). Сразу же образуется раствор персикового цвета и реакционную смесь оставляют перемешиваться при комнатной температуре на ночь. С целью увеличения объема по завершении реакции к реакционной смеси прибавляют 10 мл DCM, а затем буферный раствор с рН 4.00 (25 мл). Смесь подкисляют до рН 4.5, добавляя 1N раствор HCl, и водную фазу экстрагируют DCM (3×20 мл). Органические вытяжки дважды промывают буферным раствором рН 4.00 (20 мл), насыщенным раствором NaOH (25 мл) и рассолом (20 мл), а затем сушат MgSO4. Полученный раствор упаривают в вакууме и очищают на колонке Biotage 40M (элюпия 10%-40% этилацетатом в смеси гексанов). Такая очистка дает чистое заглавное соединение в виде твердого вещества белого цвета (1.11 г, 89%).
1H ЯМР (ДМСО-d6) δ м.д. 0.96-1.02 (ротамеры, 3:2, с, 18Н), 2.27-2.33 (м, 1Н), 2.50 (с, 3H), 2.68-2.72 (м, 1Н), 3.67 (с, 3H), 4.02-4.04 (м, 1Н), 4.43-4.45 (уш д, J=15 Гц, 2Н), 4.58-4.61 (т, 1Н), 5.60 (уш с, 1Н), 6.72-6.74 (уш д, J=15 Гц, 1Н), 7.38 (с, 1Н), 7.68-7.73 (м, 1Н), 7.95-7.97 (м, 2Н);
LC/MS rt - мин (MH+): 2.61 (590) (Метод А).
Стадия 5:
LiOH (0.138 г, 5.78 ммоля) растворяют в воде (10 мл) при нагревании и под действием ультразвука. К раствору чистого продукта Стадии 4 (1.09 г, 1.93 ммоля) в ТГФ (10 мл) прибавляют раствор LiOH и МеОН (10 мл). Смесь сразу же приобретает ярко-синюю окраску. Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 3 часов, а затем подкисляют 1N HCl (5.78 мл, 5.78 ммоля). Прибавляют буферный раствор с рН 4.00 и, прибавляя 1N води. раствор NaOH, доводят рН до 4.5. Водный слой экстрагируют EtOAc (3×25 мл), промывают рассолом (20 мл) и сушат MgSO4. Отфильтрованный раствор упаривают в вакууме и оставляют подсоединенным к вакуумной линии на ночь. Сырой продукт (957 мг, выход 90%) используют на следующей стадии.
LC/MS rt-мин (MH+): 2.51 (577) (Метод А).
Стадия 6:
Сырой продукт Стадии 5 (60 мг, 0.11 ммоля) растворяют в DCM (5 мл) и прибавляют гидрохлорид (1(R)-амино-2(S)-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (Пример 1, Стадия 8) (0.029 г, 0.11 ммоля). В атмосфере азота прибавляют DIPEA (0.094 мл мг, 0.541 ммоля), а затем HATU (0.0575 мг, 0.151 ммоля). Реакционную смесь оставляют перемешиваться при комнатной температуре, примерно, на 8 час. С целью увеличения объема к реакционной смеси прибавляют 10 мл DCM, а затем буферный раствор с рН 4.00 (10 мл). Смесь подкисляют до рН 4-5, добавляя 1N раствор HCl, и водную фазу экстрагируют DCM (3×20 мл). Органические вытяжки дважды промывают буферным раствором рН 4.00 (10 мл) и рассолом (10 мл), а затем сушат MgSO4. Полученный раствор упаривают в вакууме и очищают на колонке Biotage 12M (элюция 10%-40% ацетоном в смеси гексанов). Такая очистка дает чистое заглавное соединение 410 в виде белого порошка (29 мг, 35%).
1H ЯМР (ДМСО-d6) δ м.д. 0.976-1.12 (м,24Н), 1.36-1.39 (м, 1Н), 1.70-1.72 (м, 1Н), 2.15-2.25 (м, 2Н), 2.50-2.52 (м, 4Н), 2.91-2.96 (м, 1Н), 3.97-4.01 (м, 2Н), 4.40-4.47 (м, 2Н), 5.09-5.11 (д, J=10 Гц, 1Н), 5.21-5.24 (д, J=15 Гц, 1Н), 5.59-5.66 (м, 2Н), 6.65-6.67 (д, NH), 7.43 (с, 1Н), 7.72-7.74 (д, J=10 Гц, 1Н), 7.90 (с, 1Н), 7.98-8.0 (д, J=10 Гц, 1Н), 8.87 (с, NH), 10.35 (с, NH);
LC/MS rt - мин (MH+): 2.65 (789.61) (Метод А).
Пример 411: Получение Соединения 411.
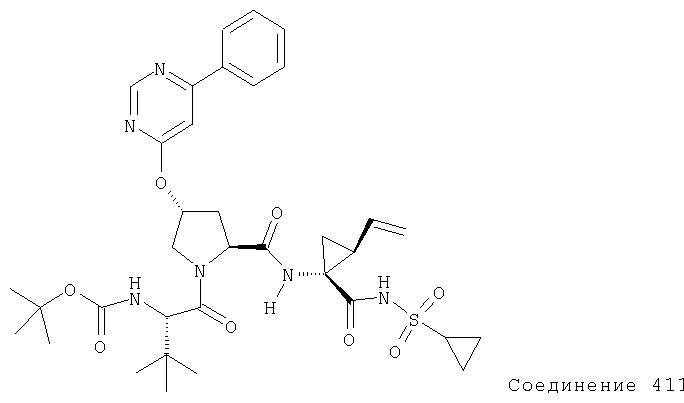
Стадия 1:
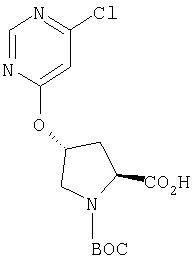
К раствору Boc-L-гидроксипролина (2.00 г, 8.65 ммоля) в ТГФ (25 мл) в атмосфере азота прибавляют NaH (0.795 г. 19.87 ммоля). Суспензию перемешивают при комнатной температуре в течение 15 минут и двумя порциями прибавляют 4,6-дихлорпиримидин (2.58 г, 17.30 ммоля). Смесь перемешивают при комнатной температуре в течение одного часа и для ее нейтрализации прибавляют 1.3 эквивалента HCl (1N). Прибавляют буферный раствор с рН 4.0 и доводят 3Hачение рН до 5. Водную фазу экстрагируют этилацетатом (3×25 мл) и органические вытяжки промывают рассолом (20 мл) и сушат MgSO4, получают титульное соединение в виде твердого вещества белого цвета (выход сырого продукта не рассчитывался). Сырой продукт используют на следующей стадии. LC/MS rt - мин (МН+): 1.91 (366.2) (Метод А).
Стадия 2:

Раствор сырого продукта Стадии 1 (предположительно, 8.65 ммоля) в ТГФ (40 мл) и метаноле (40 мл) охлаждают до 0°С. К раствору при перемешивании в атмосфере азота медленно прибавляют 2М раствор TMSCN в смеси гексанов (˜1.3 экв.) до прекращения выделения газа. Полностью прореагировавший раствор упаривают в вакууме и очищают на колонке Biotage 40M (элюпия 20%-40% этилапетатом в смеси гексанов), получают чистое титульное соединение в виде белой пены (497 мг, выход 16% на Стадии 2а-2b).
1H ЯМР (ДМСО-d6) δ м.д. 1.34-1.38 (ротамеры, 2:1, 9Н), 2.25-2.29 (м, 1Н), 2.53-2.56 (м, 1Н), 3.58-3.75 (м, 2Н), 3.69 (с, 3H), 4.28-4.33 (м, 1Н), 5.59 (с, 1Н), 7.24 (с, 1Н), 8.69 (с, 1Н);
LC/MS rt - мин. (МН+): 2.08 (380.14) (Метод А).
Стадия 3:
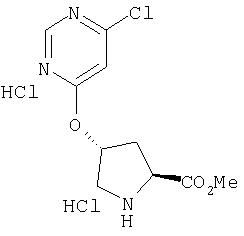
Раствор чистого продукта Стадии 2 (472 мг, 1.83 ммоля) в DCM (3 мл) и ТФК (5.65 мл) перемешивают при комнатной температуре один час. Растворитель отгоняют в вакууме и остаток суспендируют в 1N HCl в диэтиловом эфире (7.33 мл), осторожно перемешивают и упаривают в вакууме. Эту процедуру повторяют и полученный продукт выдерживают в вакууме масляного насоса в течение ночи, получают твердое вещество белого цвета с количественньм выходом.
LC/MS rt. - мин.(MH+): 0.65 (258.35) (Метод А).
Стадия 4:

В атмосфере азота продукт Стадии 3 (предполагается количественный выход, 1.83 ммоля) прибавляют к раствору ВОС-t-бутил-L-глицина (0.424 г, 1.83 ммоля) в DCM (11 мл). Затем прибавляют HOBt (0.272 г, 2.02 ммоля), DIPEA (2.23 мл, 12.82 ммоля) и HBTU (1.04 г, 2.75 ммоля). Реакционную смесь оставляют перемешиваться при комнатной температуре на 15 час. С целью увеличения объема по завершении реакции к реакционной смеси прибавляют 10 мл DCM, затем буферный раствор с рН 4.00 (15 мл). Смесь подкисляют до рН 4.5, добавляя 1N раствор HCl, и водную фазу экстрагируют DCM (3×20 мл). Органические вытяжки дважды промывают буферным раствором рН 4.00 (20 мл), насыщенным раствором NaOH (25 мл), рассолом (20 мл), а затем сушат MgSO4. Полученный раствор упаривают в вакууме и очищают на колонке Biotage 40S (элюция 20%-50% этилацетатом в смеси гексанов). Такая очистка дает чистое заглавное соединение в виде твердого вещества белого цвета (454 мг, 53%).
1H ЯМР (ДМСО-d6) δ м.д. 0.94 (с, 9Н), 1.25 (с, 9Н), 2.21-2.27 (м, 1Н), 2.48-2.55 (м, 1Н), 3.64 (с, 3H), 3.86-4.02 (м, 2Н), 4.29-4.31 (д, J=10 Гц, 1Н), 4.46-4.49 (т, 1Н), 5.75 (уш с, 1Н), 6.72-6.74 (д, NH), 7.12 (с, 1Н), 8.71 (с, 1Н);
LC/MS rt. - мин. (MH+): 2.27 (493.5) (Метод А).
Стадия 5:

LiOH (0.0141 г, 0.589 ммоля) растворяют в воде (7.5 мл) при нагревании и под действием ультразвука (сонификация). К раствору чистого продукта Стадии 4 (252 г, 0.535 ммоля) в ТГФ (7.5 мл) прибавляют раствор LiOH и оставляют перемешиваться при комнатной температуре. Реакция завершается через 3 часа. Прибавляют буфер с рН 4.0 и подкисляют 1N HCl, примерно, до рН 4.5. Водный слой экстрагируют EtOAc (3×25 мл), органические вытяжки промывают рассолом (20 мл) и сушат MgSO4. Отфильтрованный раствор упаривают в вакууме и оставляют подсоединенным к вакуумной линии на ночь. Сырой продукт (231 мг, выход 95%) используют на следующей стадии.
1H ЯМР (ДМСО-d6) δ м.д. 0.94 (с, 9Н), 1.25 (с, 9Н), 2.14-2.22 (м, 1Н), 2.50-2.54 (м, 1Н), 3.84-3.876 (д, J=150 Гц, 1Н), 3.97-3.99 (д, J=10 Гц, 1Н), 4.27-4.30 (д, J=15 Гц, 1Н), 4.37-4.40 (т, 1Н), 5.63 (уш с, 1Н), 6.69-6.71 (д, NH), 7.12 (с, 1Н), 8.71 (с, 1Н), 12.56 (уш с, ОН);
LC/MS rt - мин (МН+): 2.24 (479.5) (Метод А).
Стадия 6:

Чистый продукт Стадии 5 (80 мг, 0.146 ммоля) и фенилбороновую кислоту (0.0178 г, 0.146 ммоля) сольватируют в ДМФА (2 мл). Раствор помещают в атмосферу азота и прибавляют 2М водный раствор Na2СО3 (0.146 мл, 0.292 ммоля). Затем прибавляют пять мольных процентов тетракис(трифенилфосфин)палладия (0) и реакционную смесь нагревают в микроволновом нагревателе Personal Chemistry Emrys Optimizer при 140°С в течение 50 мин. По завершении реакции палладиевая чернь осаждается. Смесь подкисляют одним эквивалентом 1N HCl, фильтруют через шприцевой фильтр, используя МеОН для извлечения продукта. Продукт очищают препаративной ВЭЖХ (колонка 4 Xterra S5 30×75 мм, растворитель - 70% А/30% В-30% А/70% В (где растворитель А представляет собой 10% МеОН, 90% Н2О, 0.1% ТФК, а растворитель В представляет собой 90% МеОН, 10% Н2О, 0.1% ТФК), время градиента-15 мин, время выдерживания - 1 мин, скорость потока - 40 мл/мин, время удерживания чистого продукта - 10.45-11.37). Фракции, содержащие нужный продукт, нейтрализуют 1N NaOH и подсоединяют к роторному испарителю (speed vacuum), примерно, на 4 часа. Фракции объединяют и прибавляют буфер с рН 4.0 (15 мл). рН доводят до 4-5, прибавляя 1N HCl, и водный слой экстрагируют этилацетатом (3×20 мл). Органические вытяжки промывают рассолом (15 мл), сушат MgSO4 и упаривают в вакууме. Продукт сушат в течение ночи в вакууме масляного насоса и получают вязкое масло (37 мг, выход 50%).
LC/MS rt - мин (MH+): 2.37 (499.3) (Метод А).
Стадия 7:
Продукт Стадии 6, Пример 411 (36.7 мг, 0.061 ммоля) растворяют в DCM (4 мл) и прибавляют гидрохлорид (1(R)-амино-2(S)-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (Пример 1, Стадия 8) (0.0164 г, 0.161 ммоля). В атмосфере азота прибавляют DIPEA (0.0534 мл, 0.307 ммоля), а затем HATU (0.0326 г, 0.0858 ммоля). Реакционную смесь оставляют перемешиваться при комнатной температуре на 3 час. С целью увеличения объема к реакционной смеси прибавляют 10 мл DCM, а затем буферный раствор с рН 4.00 (10 мл). Смесь подкисляют до рН 4-5, добавляя 1N раствор HCl, и водную фазу экстрагируют DCM (3×15 мл). Органические вытяжки дважды промывают буферным раствором рН 4.00 (10 мл) и рассолом (10 мл), а затем сушат MgSO4. Полученный раствор упаривают в вакууме и очищают на колонке Biotage 12M (элюция 20%-40% ацетоном в смеси гексанов). Такая очистка дает чистое заглавное соединение 411 в виде белого порошка (7 мг, 15%).
1H ЯМР: δ м.д. 1.07-1.46 (м, 24Н), 1.87-1.90 (м, 1Н), 2.22-2.32 (м, 2Н), 2.51-2.55 (м, 1Н), 2.92-2.97 (м, 1Н), 4.04-4.07 (д, J=15 Гц, 1Н), 4.20-4.22 (д, J=10 Гц, 1Н), 4.36-4.38 (д, J=10 Гц, 1Н), 4.47-4.50 (т, 1Н), 5.12-5.14 (д, J=10 Гц, 1Н), 5.29-5.33 (д, J=20 Гц, 1Н), 5.73-5.82 (м, 2Н), 6.59-6.60 (д, NH), 7.29 (с, 1Н), 7.50-7.51 (м, 3H), 8.03-8.05 (м, 2Н), 8.81 (с, 1Н);
LC/MS rt - мин (MH+): 2.50 (711.4) (Метод А).
Пример 412: Получение Соединения 412.

Стадия 1:
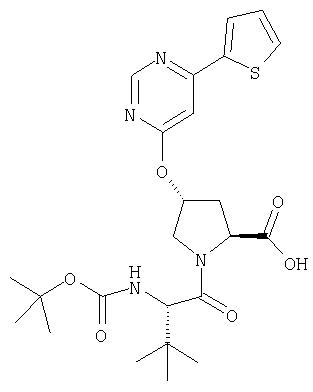
Продукт Стадии 5, Пример 411 (80 мг, 0.146 ммоля) сольватируют в ДМФА (2 мл) и к раствору прибавляют 2-тиофенбороновую кислоту (0.028 г, 0.219 ммоля). Реакционную смесь помещают в атмосферу азота и прибавляют 2М водный раствор Na2СО3 (0.146 мл, 0.292 ммоля) и пять мольных процентов тетракис(трифенилфосфин)палладия (0) (8.44 мг, 0.0073 ммоля). Реакционную смесь нагревают в микроволновом нагревателе Personal Chemistry Emrys Optimizer при 150°С в течение 30 мин. По завершении реакции палладиевая чернь осаждается. Смесь подкисляют одним эквивалентом 1N HCl, фильтруют через шприцевой фильтр, используя МеОН для извлечения продукта. Продукт очищают препаративной ВЭЖХ (колонка 4 Xterra S5 30×75 мм, растворитель - 70% А/30% В-30% А/70% В (где растворитель А представляет собой 10% МеОН, 90% Н2О, 0.1% ТФК, а растворитель В представляет собой 90% МеОН, 10% Н2О, 0.1% ТФК), время градиента - 15 мин, время выдерживания - 1 мин, скорость потока - 40 мл/мин, время удерживания чистого продукта - 10.45-11.37). Фракции, содержащие нужный продукт, нейтрализуют 1N NaOH и подсоединяют к роторному испарителю (speed vacuum), примерно, на 4 часа. Фракции объединяют и прибавляют буфер с рН 4.0 (15 мл). рН доводят до 4-5, прибавляя 1N HCl, и водный слой экстрагируют этилацетатом (3×20 мл). Органические вытяжки промывают рассолом (15 мл), сушат MgSO4 и упаривают в вакууме. Продукт сушат в течение ночи в вакууме масляного насоса и получают маслообразную жидкость (37 мг, выход 50%).
LC/MS rt - мин (MH+): 2.37 (499.3) (Метод А).
Стадия 2:
Продукт Стадии 1, Пример 412 (39 мг, 0.0773 ммоля) растворяют в DCM (4 мл) и прибавляют гидрохлорид (1(R)-амино-2(S)-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (Пример 1, Стадия 8) (0.0206 г, 0.0773 ммоля). В атмосфере азота прибавляют DIPEA (0.015 мл, 0.387 ммоля), а затем HATU (0.0411 г, 0.108 ммоля). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 15 час. С целью увеличения объема к реакционной смеси прибавляют 10 мл DCM, а затем буферный раствор с рН 4.00 (10 мл). Смесь подкисляют до рН 4-5, добавляя 1N раствор HCl, и водную фазу экстрагируют DCM (3× 15 мл). Органические вытяжки дважды промывают буферным раствором рН 4.00 (10 мл) и рассолом (10 мл), а затем сушат MgSO4. Полученный раствор упаривают в вакууме и очищают на колонке Biotage 12M (элюция 20%-50% ацетоном в смеси гексанов). Такая очистка дает чистое заглавное соединение 412 в виде белого порошка (4 мг, 7%).
1H ЯМР: δ м.д. 1.04-1.29 (м, 24Н), 1.45-1.47 (м, 1Н), 1.89-1.91 (м, 1Н), 2.26-2.31 (м, 2Н), 2.51-2.53 (м, 1Н), 2.92-2.95 (м, 1Н), 4.05-4.07 (д, J=10 Гц, 1Н), 4.22-4.24 (д, J=10 Гц, 1Н), 4.38-4.40 (д, J=10 Гц, 1Н), 4.48-4.52 (м, 1Н), 5.15-5.17 (д, J=10 Гц, 1Н), 5.32-5.36 (д, J=20 Гц, 1Н), 5.76-5.83 (м, 2Н), 6.65-6.67 (д, NH), 7.19-7.21 (м, 2Н), 7.66-7.67 (д, J=5 Гц, 1Н), 7.86-7.87 (д, J=5 Гц, 1Н), 8.69 (с, 1Н);
LC/MS rt - мин (MH+): 2.45 (739.4) (Метод А).
Пример 413: Получение Соединения 413.

Стадия 1:
2-Бром-6-хлорпиридин (3.0 г, 15.55 ммоля) и фенилбороновую кислоту (1.896 г, 15.55 ммоля) сольватируют в смеси EtOH, толуола и воды (2:1:1, 120 мл). В атмосфере азота прибавляют 1М водный раствор Na2СО3 (15.55 мл, 31.10 ммоля) и 5 мольных процентов тетракис(трифенилфосфин)палладия (0) (0.896 г, 0.775 ммоля). Реакционную смесь кипятят при 90°С в течение 1 час. Прибавляют воду (20 мл) для прекращения реакции и водный слой экстрагируют диэтиловым эфиром (4×25 мл). Органические вытяжки промывают рассолом, сушат MgSO4 и упаривают в вакууме. Сырую смесь очищают на колонке Biotage 40S (элюент 2%-10% этилацетат в смеси гексанов, получают чистое титульное соединение (1.45 г, выход 74%).
1H ЯМР: δ м.д. 7.24-7.26 (м, 1Н), 7.42-7.48 (м. 3H), 7.63-7.70 (м, 2Н), 7.98-8.00 (м, 2Н);
LC/MS rt - мин (MH+): 2.04 (190.18) (Метод А).
Стадия 2:

К чистому твердому соединению, полученному на Стадии 1 (3.27 г, 17.24 ммоля), прибавляют ТФК (20 мл). К раствору в атмосфере азота при перемешивании медленно, по каплям прибавляют 30% раствор Н2О2 (5.55 мл, 48.9 ммоля). Реакционную смесь кипятят при 100°С в течение 3 часов и прибавляют еще 0.5 эквивалента Н2О2 (2.27 мл, 25 ммолей). Реакционную смесь продолжают перемешивать при 100°С еще в течение 2 часов. Охлаждают до комнатной температуры и раствор упаривают в вакууме, примерно, до половины первоначального объема. Добавляют воду (40 мл) и водный слой экстрагируют этилацетатом (5×30 мл). Органические вытяжки промывают один раз рассолом (20 мл), сушат MgSO4 и упаривают в вакууме. Сырой продукт очищают на колонке Biotage 40M (элюент 10%-75% этилацетат в смеси гексанов), получают бледно-желтую жидкость (0.81 г, 23%). Чистое исходное регенерируют для использования в дальнейшем (1.895 г, 58%).
1H ЯМР: δ м.д. 7.40-7.42 (т, 1Н), 7.49-7.50 (м, 3H), 7.59-7.61 (д, J=10 Гц, 1Н), 7.77-7.82 (м, 3H);
LC/MS rt - мин (МН+): 1.12 (206.37) (Метод А).
Стадия 3:
Очищенный продукт Стадии 2 (0.81 г, 3.94 ммоля) прибавляют к (раствору) SOCl2 (25 мл) и перемешивают при 60°С в течение 2 час. Затем повышают температуру до 80°С, чтобы довести реакцию до конца, и греют еще в течение 30 мин. Раствор упаривают в вакууме и осторожно прибавляют лед. Подставляют баню со льдом и доводят рН до 4-5, прибавляя 10N NaOH. Водный слой экстрагируют эфиром (4×25 мл) и органические вытяжки промывают рассолом (20 мл), сушат MgSO4 и упаривают в вакууме. Сырой продукт, желтую жидкость, очищают на колонке Biotage 40S (элюент 2%-10% этилацетат в смеси гексанов), получают светло-желтую вязкую жидкость (538 мг, 61%).
1H ЯМР: δ м.д. 7.52-7.55 (м, 3H), 7.73 (с, 1Н), 8.10-8.11 (м, 2Н), 8.17 (с, 1Н);
LC/MS rt - мин (МН+): 2.62 (225.33) (Метод А).
Стадия 4:

К раствору Boc-L-гидроксипролина (555 мг, 2.40 ммоля) в ДМСО (4 мл) в атмосфере азота прибавляют KOtBu (0.619 г, 5.52 ммоля). Суспензию перемешивают при комнатной температуре в течение 45 мин и двумя порциями прибавляют 2,4-дихлор-6-фенилпиридин (очищенный на стадии 3) (538 мг, 3.75 ммоля). Смесь перемешивают при комнатной температуре два часа, для нейтрализации реакционной смеси прибавляют 1.3 эквивалента HCl (1N). Прибавляют буферный раствор с рН 4.0 и доводят рН до 4-5. Водный слой экстрагируют этилацетатом (3×25 мл) и объединенные органические вытяжки промывают рассолом (20 мл) и сушат MgSO4, получают титульное соединение в виде твердого вещества белого цвета (выход сырого продукта 962 мг). Сырой продукт используют на следующей стадии.
LC/MS (время удерживания rt. мин. (MH+): 2.55 (419.27) (Метод А).
Стадия 5:

Раствор неочищенного продукта Стадии 4 (предположительно, 2.4 ммоля) в ТГФ (10 мл) и метаноле (10 мл) охлаждают до 0°С. К раствору при перемешивании под азотом медленно прибавляют TMSCN 2M в смеси гексанов (˜1.3 экв.) до тех пор, пока не прекратится выделение газа. По завершении реакции раствор упаривают в вакууме, очисткой на колонке Biotage 25S (элюция 10%-50% этилацетатом в смеси гексанов) получают чистое титульное соединение в виде белой пены (503 мг, выход 69% в расчете на Стадии 4d-4e).
1H ЯМР (ДМСО-d6): δ м.д. 1.35-1.38 (ротамеры 3:2, 9Н), 2.21-2.26 (м, 1Н), 2.50-2.58 (м, 1Н), 3.67-3.70 (м, 5Н), 4.26-4.34 (м, 1Н), 5.35 (уш с, 1Н), 7.14-7.15 (м, 1Н), 7.47-7.55 (м, 4Н), 8.05-8.09 (м, 2Н);
LC/MS (rt. мин. (МН+): 2.69 (455.52) (Метод А).
Стадия 6:
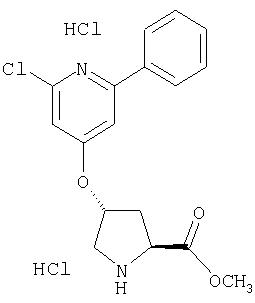
Раствор чистого продукта Стадии 5 (503 мг, 1.16 ммоля) в DCM (2.5 мл) и ТФК (3.58 мл) перемешивают при комнатной температуре один час. Растворитель отгоняют в вакууме и остаток суспендируют в 1N HCl в диэтиловом эфире (6 мл), осторожно перемешивают и упаривают в вакууме. Эту процедуру повторяют и полученный продукт выдерживают в вакууме масляного насоса, получают твердое вещество белого цвета с количественным выходом. Сырой продукт используют на следующей стадии.
Стадия 7:

В атмосфере азота продукт Стадии 6 (предполагается количественный выход, 1.83 ммоля) прибавляют к раствору ВОС-t-бутил-L-глицина (0.424 г, 1.83 ммоля) в DCM (11 мл). Затем прибавляют HOBt (0.272 г, 2.02 ммоля), DIPEA (2.23 мл, 12.82 ммоля) и HBTU (1.04 г, 2.75 ммоля). Реакционную смесь оставляют перемешиваться при комнатной температуре на 15 час. С целью увеличения объема по завершении реакции к реакционной смеси прибавляют 15 мл DCM, затем буферный раствор с рН 4.00 (15 мл). Смесь подкисляют до рН 4.5, добавляя 1N раствор HCl, и водную фазу экстрагируют DCM (3×20 мл). Органические вытяжки дважды промывают буферным раствором рН 4.00 (20 мл), насыщенным раствором NaOH (25 мл), рассолом (20 мл) и сушат MgSO4. Полученный раствор упаривают в вакууме и очищают на колонке Biotage 40S (элюция 20%-50% этилацетатом в смеси гексанов). Такая очистка дает чистое заглавное соединение в виде твердого вещества белого цвета (454 мг, 53%).
1H ЯМР (ДМСО-d6) δ м.д. 0.96 (с, 9Н), 1.19 (с, 9Н), 2.17-2.29 (м, 1Н), 2.48-2.58 (м, 1Н), 3.65 (с, 3H), 3.81-3.89 (м, 1Н), 4.05-4.08 (м, 1Н), 4.21-4.26 (м, 1Н), 4.44-4.50 (т, 1Н), 5.45 (уш с, 1Н), 6.72-6.75 (д, J=15 Гц, 1Н), 7.09 (с, 1Н), 7.46-7.51 (м, 4Н), 8.02-8.06 (м, 2Н);
LC/MS (rt. мин. (MH+): 2.27 (493.5) (Метод А).
Стадия 8:
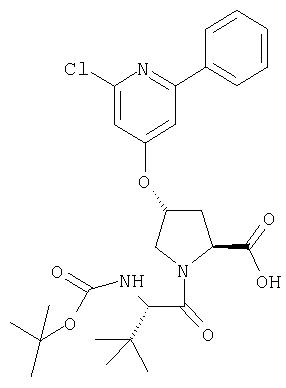
LiOH (0.0233 г, 0.973 ммоля) растворяют в воде (10 мл) при нагревании и под действием ультразвука (сонификация). Раствор LiOH прибавляют к раствору чистого продукта Стадии 7 (483 мг, 0.885 ммоля) в ТГФ (10 мл). Смесь сразу же приобретает бледно-розовую окраску. Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 31 час и подкисляют 1N HCl (0.973 мл, 0.973 ммоля). Прибавляют буферный раствор с рН 4.00 и, прибавляя 1N водн. раствор NaOH, доводят рН до 4-5. Водный слой экстрагируют EtOAc (3×20 мл), промывают рассолом (15 мл) и сушат MgSO4. Отфильтрованный раствор упаривают в вакууме и оставляют подсоединенным к вакуумной линии на ночь. Сырой продукт (480 мг, выход >95%) используют на следующей стадии.
1H ЯМР (ДМСО-d6) δ м.д. 0.94 (с, 9Н), 1.20 (с, 9Н), 1.32-1.34 (м, 1Н), 2.12-2.4 (м, 1Н), 2.51-2.55 (м, 1Н), 3.81-3.83 (д, J=10 Гц, 1Н), 4.21-4.23 (д, J=10 Гц, 1Н), 4.32-4.35 (т, 1Н), 5.4 (уш с, 1Н), 6.63-6.65 (д, NH), 7.09 (с, 1Н), 7.47-7.49 (м, 4Н), 8.03-8.06 (м, 2Н), 12.56 (с, 1Н).
Стадия 9

Дипептид из Стадии 8 (100 мг, 0.188 ммоля) и 2-тиофенбороновую кислоту (0.0481 г, 0.376 ммоля) сольватируют в ДМФА (2 мл). Раствор помещают в атмосферу азота и прибавляют 2М водный раствор KF (0.282 мл, 0.376 ммоля). Затем прибавляют пять мольных процентов тетракис(трифенилфосфин)палладия (0) и реакционную смесь нагревают в микроволновом нагревателе Personal Chemistry Emrys Optimizer при 150°С в течение 30 мин. По завершении реакции палладиевая чернь осаждается. Смесь подкисляют одним эквивалентом 1N HCl, фильтруют через шприцевой фильтр, используя МеОН для извлечения продукта. Продукт очищают препаративной ВЭЖХ (колонка 4 Xterra S5 30×75 мм, растворитель - 85% A/15% В-10% А/90% В (где растворитель А представляет собой 10% МеОН, 90% H2O, 0.1% ТФК, а растворитель В представляет собой 90% МеОН, 10% Н2О, 0.1% ТФК), время градиента - 15 мин, время выдерживания - 1 мин, скорость потока - 40 мл/мин, время удерживания чистого продукта - 16.23). Фракции, содержащие нужный продукт, нейтрализуют 1N NaOH и подсоединяют к роторному испарителю (speed vacuum), примерно, на 2 часа. Фракции объединяют и прибавляют буфер с рН 4.0 (15 мл). рН доводят до 4-5, прибавляя 1N HCl, и водный слой экстрагируют этилацетатом (3×20 мл). Органические вытяжки промывают рассолом (15 мл), сушат MgSO4 и упаривают в вакууме. Продукт сушат в течение ночи в вакууме масляного насоса и получают бледно-желтое масло (44 мг, выход 40%).
LC/MS rt - мин (МН+): 2.7 (580.54) (Метод А).
Стадия 10:
Продукт Стадии 9, Пример 413 (43 мг, 0.0742 ммоля) растворяют в DCM (2 мл) и прибавляют гидрохлорид (1(R)-амино-2(S)-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (Пример 1, Стадия 8) (0.0198 г, 0.0742 ммоля). В атмосфере азота прибавляют DIPEA (0.0646 мл, 0.371 ммоля), а затем HATU (0.039 г, 0.104 ммоля). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 4.5 час. С целью увеличения объема к реакционной смеси прибавляют 10 мл DCM, а затем буферный раствор с рН 4.00 (10 мл). Смесь подкисляют до рН 4-5, добавляя 1N раствор HCl, и водную фазу экстрагируют DCM (3×15 мл). Органические вытяжки дважды промывают буферным раствором рН 4.00 (10 мл) и рассолом (10 мл), а затем сушат MgSO4. Полученный раствор упаривают в вакууме и очищают на колонке Biotage 12S (элюция 10%-50% ацетоном в смеси гексанов). Соединение выделяют и снова очищают препаративной ВЭЖХ (колонка YMC ODS-А 20×50 мм s5, растворитель - 60% А/40% В-10% А/90% В (где растворитель А представляет собой 10% МеОН, 90% Н2О, 0.1% ТФК, а растворитель В представляет собой 90% МеОН, 10% Н2О, 0.1% ТФК), время градиента - 8 мин, время выдерживания - 2 мин, скорость потока - 25 мл/мин, время удерживания чистого продукта - 8.702). Такая очистка дает чистое Соединение 413 в виде масла светло-оранжевого цвета (22.3 мг, 38%).
1H ЯМР: δ м.д. 1.02-1.29 (м, 24Н), 1.42-1.45 (м, 1Н), 1.87-1.89 (м, 1Н), 2.24-2.30 (м, 2Н), 2.53-2.57 (м, 1Н), 2.92-2.97 (м, 1Н), 4.06-4.08 (д, J=10 Гц, 1Н), 4.25 (с, 1Н), 4.31-4.33 (д, J=10 Гц, 1Н), 4.45-4.49 (т, 1Н), 5.12-5.14 (д, J=10 Гц, 1Н), 5.29-5.32 (д, J=15 Гц, 1Н), 5.46 (уш с, 1Н), 5.73-5.80 (м, 1Н), 7.13-7.15 (м, 1Н), 7.29-7.30 (д, J=5 Гц, 2Н), 7.42-7.53 (м, 4Н), 7.75-7.56 (д, J=5 Гц, 1Н), 8.07-8.09 (д, J=10 Гц, 2Н).
LC/MS rt - мин (MH+): 2.79 (792.72) (Метод А).
Пример 414: Получение Соединения 414.
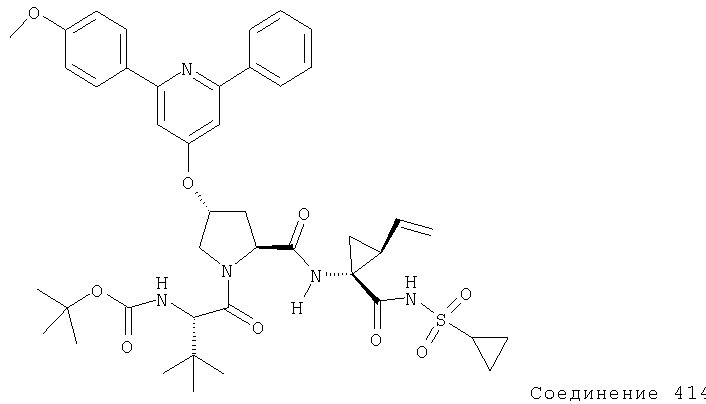
Стадия 1:

Чистый продукт Стадии 8, Пример 413 (100 мг, 0.188 ммоля) и 4-метоксифенилбороновую кислоту (0.0429 г, 0.282 ммоля) сольватируют в ДМФА (2.5 мл). Раствор помещают в атмосферу азота и прибавляют 2М водный раствор Na2СО3 (0.188 мл, 0.376 ммоля). Затем прибавляют пять мольных процентов тетракис(трифенилфосфин)палладия (0) (0.011 мг, 0.094 ммоля) и реакционную смесь нагревают в микроволновом нагревателе Personal Chemistry Emrys Optimizer при 150°С в течение 30 мин. По завершении реакции палладиевая чернь осаждается. Смесь подкисляют одним эквивалентом 1N HCl, фильтруют через шприцевой фильтр, используя МеОН для извлечения продукта. Продукт очищают препаративной ВЭЖХ (колонка 4 Xterra MS С 18 30× 50 мм, растворитель - 90% А/10% В-10% А/90% В (где растворитель А содержит 10% МеОН, 90% Н2О, 0.1% ТФК, а растворитель В содержит 90% МеОН, 10% Н2О, 0.1% ТФК), время градиента - 15 мин, время выдерживания - 1 мин, скорость потока - 45 мл/мин, время удерживания чистого продукта - 13.72). Фракции, содержащие нужный продукт, нейтрализуют 1N NaOH и подсоединяют к роторному испарителю (speed vacuum), примерно, на 2 часа. Фракции объединяют и прибавляют буфер с рН 4.0 (15 мл). рН доводят до 4-5, прибавляя 1N HCl, и водный слой экстрагируют этилацетатом (3×20 мл). Органические вытяжки промывают рассолом (15 мл), сушат MgSO4 и упаривают в вакууме. Продукт сушат в течение ночи в вакууме масляного насоса и получают вязкое масло (44 мг, выход 50%).
LC/MS rt - мин (МН+): 2.23 (604.61) (Метод А).
Стадия 2:
Продукт Стадии 1, Пример 414 (43 мг, 0.0712 ммоля) растворяют в DCM (2 мл) и прибавляют гидрохлорид (1(R)-амино-2(S)-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (Пример 1, Стадия 8) (0.01899 г, 0.0712 ммоля). В атмосфере азота прибавляют DIPEA (0.062 мл, 0.356 ммоля), а затем HATU (0.038 г, 0.0997 ммоля). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 4.5 час. С целью увеличения объема к реакционной смеси прибавляют 10 мл DCM, а затем буферный раствор с рН 4.00 (10 мл). Смесь подкисляют до рН 4-5, добавляя 1N раствор HCl, и водную фазу экстрагируют DCM (3×15 мл). Органические вытяжки дважды промывают буферным раствором рН 4.00 (10 мл) и рассолом (10 мл), а затем сушат MgSO4. Полученный раствор упаривают в вакууме и очищают на колонке Biotage 12S (элюция 20%-50% ацетоном в смеси гексанов). Соединение выделяют и снова очищают препаративной ВЭЖХ (колонка YMC ODS-А 20×50 мм s5, растворитель - 60% А/40% В-10% А/90% В (где растворитель А содержит 10% МеОН, 90% Н2О, 0.1% ТФК, а растворитель В содержит 90% МеОН, 10% H2O, 0.1% ТФК), время градиента - 10 мин, время выдерживания - 2 мин, скорость потока - 25 мл/мин, время удерживания чистого продукта - 7.43-8.24). Такая очистка дает чистое Соединение 414 в виде масла бледно-оранжевого цвета (19.4 мг, 34%).
1H ЯМР: δ м.д. 1.02-1.23 (м, 24Н), 1.29-1.44 (м, 1Н), 1.89-1.95 (м, 1Н), 2.24-2.30 (кв, 1Н), 2.32-2.40 (м, 1Н), 2.59-2.62 (м, 1Н), 3.90 (с, 3H), 4.10-4.12 (д, J=10 Гц, 1Н), 4.21 (с, 1Н), 4.42-4.56 (м, 2Н), 5.12-5.14 (д, J=10 Гц, 1Н), 5.28-5.31 (д, J=15 Гц, 1Н), 5.61 (уш с, 1Н), 5.72-5.80 (м, 1Н), 7.14-7.16 (д, J=10 Гц, 2Н), 7.52-7.54 (д, J=10 Гц, 2Н), 7.61-7.62 (м, 2Н), 7.95-7.98 (м, 4Н);
LC/MS rt - мин (МН+): 2.35 (816.76) (Метод А).
Пример 415: Получение Соединения 415.

Стадия 1:

Дипептид из Стадии 8, Пример 413 (173 мг, 0.327 ммоля) и фенилбороновую кислоту (0.06 г, 0.491 ммоля) сольватируют в ДМФА (4 мл). Раствор помещают в атмосферу азота и прибавляют 2М водный раствор Na2СО3 (0.33 мл, 0.654 ммоля). Затем прибавляют пять мольных процентов тетракис(трифенилфосфин)палладия (0) (0.019 мг, 0.0164 ммоля) и реакционную смесь нагревают в микроволновом нагревателе Personal Chemistry Emrys Optimizer при 150°С в течение 30 мин. По завершении реакции палладиевая чернь осаждается. Смесь подкисляют одним эквивалентом 1N HCl, фильтруют через шприцевой фильтр, используя МеОН для извлечения продукта. Продукт очищают препаративной ВЭЖХ (колонка 5 Xterra С18 5 um 30×100 мм, растворитель - 80% А/20% В-0% А/100% В (где растворитель А содержит 10% МеОН, 90% H2O, 0.1% ТФК, а растворитель В содержит 90% МеОН, 10% Н2O, 0.1% ТФК), время градиента - 20 мин, время выдерживания - 1 мин, скорость потока - 40 мл/мин, время удерживания чистого продукта - 11.28-11.72). Фракции, содержащие нужный продукт, нейтрализуют 1N NaOH и подсоединяют к speed vac, примерно, на 2 часа. Фракции объединяют и прибавляют буфер с рН 4.0 (15 мл). рН доводят до 4-5, прибавляя 1N HCl, и водный слой экстрагируют этилацетатом (4×15 мл). Органические вытяжки промывают рассолом (15 мл), сушат MgSO4 и упаривают в вакууме. Продукт сушат в течение ночи в вакууме масляного насоса и получают вязкое масло (31.5 мг, выход 17%).
LC/MS rt - мин (МН+): 2.54 (574.37) (Метод А).
Стадия 2:
Продукт Стадии 1, Пример 415 (31.5 мг, 0.055 ммоля) растворяют в DCM (3 мл) и прибавляют гидрохлорид (1(R)-амино-2(S)-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты (Пример 1, Стадия 8) (0.0147 г, 0.055 ммоля). В атмосфере азота прибавляют DIPEA (0.048 мл, 0.275 ммоля), а затем HATU (0.029 г, 0.077 ммоля). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 3.5 час и прибавляют еще 0.3 эквивалента гидрохлорида (1(R)-амино-2(S)-винилциклопропанкарбонил)-амида циклопропансульфоновой кислоты. Реакционную смесь перемешивают еще в течение 8 час. С целью увеличения объема к реакционной смеси прибавляют 10 мл DCM, а затем буферный раствор с рН 4.00 (10 мл). Смесь подкисляют до рН 4-5, добавляя 1N раствор HCl, и водную фазу экстрагируют DCM (4×150 мл). Органические вытяжки дважды промывают буферным раствором рН 4.00 (10 мл) и рассолом (10 мл), а затем сушат MgSO4. Полученный раствор упаривают в вакууме и очищают препаративной ВЭЖХ (колонка YMC ODS - А 30×50 мм, растворитель - 80% А/20% В-10% А/90% В (где растворитель А содержит 10% МеОН, 90% H2O, 0.1% ТФК, а растворитель В содержит 90% МеОН, 10% H2O, 0.1% ТФК), время градиента - 20 мин, время выдерживания - 3 мин, скорость потока - 30 мл/мин, время удерживания чистого продукта - 19.6). Такая очистка не дает чистого продукта, поэтому его снова очищают на колонке Biotage 12S (элюция 10% - 50% ацетоном в смеси гексанов). Эта очистка дает чистое титульное соединение в виде масла бледно-оранжевого цвета (22.3 мг, 38%).
1H ЯМР: δ м.д. 1.02-1.45 (м, 24Н), 1.85-1.86 (м, 1Н), 2.03-2.11 (м, 2Н), 2.42-2.46 (м, 1Н), 2.77-2.85 (м, 1Н), 4.10-4.12 (м, 1Н), 4.25 (с, 1Н), 4.31-4.33 (д, J=10 Гц, 1Н), 4.49-4.54 (м, 1Н), 5.03-5.05 (д, J=10 Гц, 1Н), 5.20-5.24 (д, J=20 Гц, 1Н), 5.44 (уш с, 1Н), 5.82-5.94 (м, 1Н), 7.33 (с, 2Н), 7.43-7.50 (м, 6Н), 8.09-8.10 (д, J=5 Гц, 4Н);
LC/MS rt - мин (МН+): 2.37 (786.37) (Метод А).
Подраздел J.
В Секции J применяются следующие условия LC-MS:
Колонки:
Метод А: YMC ODS-А С18 S7 (4.0×33 мм)
Метод В: YMC Xterra ODS S7 (3.0×50 мм)
Метод С: Xterra ms C18 (4.6×33 мм)
Метод D: YMC ODS - A С18 S3 (4.6×33 мм)
Градиент: от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В
Время градиента: 3 мин.
Время выдерживания: 1 мин.
Скорость потока: 5 мл/мин.
Длина волны детектора: 220 нм
Растворитель А: 10% МеОН /90% H2О/0.1% ТФК
Растворитель В: 90% МеОН/10% Н2O/0.1% ТФК
Пример 420: Получение Соединения 420

Схема 1

Стадия 1:
К раствору PPh3 (16.8 мг, 63.9 ммоля) в ТГФ (150 мл) по каплям прибавляют DEAD (10.1 мл, 63.9 ммоля) и раствор перемешивают при комнатной температуре в течение 30 мин. Медленно прибавляют раствор Вос-цис-(L)-Hyp-Оме (10.5 г, 42.6 ммоля) в ТГФ (50 мл), а затем порциями прибавляют 5-бромпиридин-3-ол (полученный по методике: F.E.Ziegler et al., J.Am.Chem.Soc., (1973), 95 7458) (8.90 г, 51,1 ммоля). Полученный раствор перемешивают при 45°С в течение 18 час. Упаривают растворитель в вакууме и остаток распределяют между диэтиловым эфиром (200 мл) и 1N NaOH (50 мл). Органическую фазу сушат (MgSO4), эфирный раствор фильтруют и упаривают до половины объема. Выпавший осадок отфильтровывают. Фильтрат очищают на колонке Biotage 65M (элюент: смесь гексанов: EtOAc: 2:1, 3:2, 1:1), получают титульное соединение (11.9 г, 69%) в виде красного масла.
1H ЯМР: (CDCl3) δ м.д. 1.42, 1.45 (с, 9Н (ротамеры)), 2.25-2.30 (м, 1Н), 2.49-2.58 (м, 1Н), 3.75, 3.81 (с, 3H (ротамеры)), 3.66-3.81 (м, 2Н (наложен.)), 4.42, 4.50 (т, J=8 Гц, 1Н (ротамеры)), 4.92 (м, 1Н), 7.36 (с, 1Н), 8.20 (с, 1Н), 8.32 (с, 1Н).
LC/MS rt - мин (MH+): 2.26 (401, 403) (Метод В).
Стадия 2:
Продукт Стадии 1 (2.34 г, 5.83 ммоля) растворяют в DCM (20 мл) и ТФК (18 мл, 0.23 моля) и перемешивают при комнатной температуре 2 час. Отгоняют летучие в вакууме и остаток распределяют между EtOAc и водой. Органическую фазу экстрагируют 1N HCl (25 мл) и объединенные водные вытяжки нейтрализуют нас. NaHCO3 (40 мл). Водную фазу экстрагируют EtOAc (дважды) и объединенные органические вытяжки промывают рассолом и сушат (MgSO4), получают титульное соединение (1.02 г, 58%) в виде бесцветного масла.
1H ЯМР: (ДМСО-d6) δ м.д. 2.12-2.20 (м. 2Н), 2.94 (д, J=12 Гц, 1Н), 3.19 (дд, J=4.5, 12 Гц, 1Н), 3.64 (с, 3H), 3.90 (т, J=8 Гц, 1Н), 5.07 (м, 1Н), 7.69 (с, 1Н), 8.26 (с, 1Н), 8.29 (с, 1Н).
LC/MS rt - мин (МН+): 0.78 (301, 302) (Метод В).
Стадия 3:
К суспензии продукта Стадии 2 (1.02 г, 3.39 ммоля), N-ВОС-L-t-бутил-лейцина (862 мг, 3.73 ммоля) и HOBt (458 мг, 3.39 ммоля), в DCM (15 мл) прибавляют DIPEA (2.36 мл, 13.6 ммоля), а затем HBTU (1.61 г, 4.24 ммоля). Полученный раствор оставляют перемешиваться при комнатной температуре на 15 час и прибавляют 15 мл DCM, буферный раствор с рН 4 и немного 1N раствора HCl до рН 4-5. Органические вытяжки промывают буферным раствором рН 4, насыщенным раствором NaHCO3 (дважды), рассолом и сушат MgSO4. Очистка на колонке Biotage 40M (элюент: гексан: EtOAc: 3:2, 1:1), получают титульное соединение (1.48 г, 85%) в виде твердого вещества белого цвета.
1H ЯМР (ДМСО-d6) δ м.д. 0.94 (с, 9Н), 1.17 (с, 9Н), 2.16-2.21 (м, 1Н), 2.50 (м, (наложен.), 1Н), 3.64 (с, 3H), 3.82 (д, J=12 Гц, 1Н), 4.06 (д, J=9.5 Гц, 1Н), 4.16 (д, J=12 Гц, 1Н), 4.45 (дд, J=8, 9.5 Гц, 1Н), 5.26 (м, 1Н), 6.71 (д, J=9 Гц, NH), 7.75 (с, 1Н), 8.28 (с, 1Н), 8.32 (с, 1Н).
LC/MS rt - мин (MH+): 2.35 (514, 516) (Метод В).
Стадия 4:
К смеси продукта Стадии 3 (98 мг, 0.19 ммоля), Pd(PPh3)4 (6.6 мг, 0.00573 ммоля), 2М водн. Na2CO3 (0.191 мл, 0.381 ммоля) в толуоле (2 мл) прибавляют раствор фенилбороновой кислоты (29 мг, 0.24 ммоля) в метаноле (0.1 мл). Раствор нагревают при 85°С в течение 6 час в атмосфере азота. По охлаждении до комнатной температуры к смеси прибавляют буфер с рН 4 и экстрагируют EtOAc (2×10 мл), сушат (Na2SO4). Очисткой на колонке Biotage 12М (элюент: гексан - EtOAc 2:3) получают титульное соединение (72 мг, 74%) в виде бесцветного масла.
1H ЯМР: (Метанол-d4) δ м.д. 1.05 (с, 9Н), 1.31 (с, 9Н), 2.29-2.35 (м, 1Н), 2.68-2.72 (м, 1Н), 3.77 (с, 3H), 4.00 (д, J=12 Гц, 1Н), 4.24 (д, J=9.5 Гц, 1Н), 4.41 (д, J=12 Гц, 1Н), 4.67 (дд, J=7.5,10 Гц, 1Н), 5.36 (м, 1Н), 6.51 (д, J=9.5 Гц, NH), 7.79-7.85 (м, 3H), 8.39 (с, 1Н), 8.59 (с, 1Н), 8.67-8.68 (м, 2Н).
LC/MS rt - мин (MH+): 2.16 (512) (Метод В).
Стадия 5:
К раствору продукта Стадии 4 (150 мг, 0.293 ммоля) в ТГФ (2 мл) и метанола (2 мл) прибавляют раствор LiOH (14 мг, 0.59 ммоля) в воде (2 мл). Смесь перемешивают в течение 2 час при комнатной температуре и прибавляют 1N HCl до нейтрального рН. Летучие органические соединения отгоняют в вакууме и к остатку прибавляют буферный раствор с рН 4. Продукт экстрагируют EtOAc (3×10 мл), промывают рассолом/буфером с рН 4 и сушат (MgSO4), после растирания с пентаном получают заглавное соединение в виде твердого вещества белого цвета.
1H ЯМР: (Метанол-d4) δ м.д. 1.05 (с, 9Н), 1.32 (с, 9Н), 2.32-2.38 (м, 1Н), 2.69-2.74 (м, 1Н), 3.99 (дд, J=3,12 Гц, 1Н), 4.25 (с, 1Н), 4.40 (д, J=12 Гц, 1Н), 4.64 (дд, J=7.5, 9.5 Гц, 1Н), 5.36 (м, 1Н), 7.85-7.87 (м, 3H), 8.40 (с, 1Н), 8.60 (с, 1Н), 8.69-8.70 (м, 2Н).
LC/MS rt - мин (MH+): 2.03 (499) (Метод В).
Стадия 6:
К суспензии продукта Стадии 5 (93 мг, 0.19 ммоля) и продукта из Примера 1, Стадия 8 (50 мг, 0.19 ммоля) в DCM (2 мл) прибавляют DIPEA (0.163 мл, 0.935 ммоля), а затем HATU (92 мг, 0.243 ммоля). Полученный раствор оставляют перемешиваться при комнатной температуре на 18 час и прибавляют DCM, буферный раствор с рН 4 и немного 1N раствора HCl до рН 4-5. Слои разделяют и водную фазу экстрагируют DCM (10 мл) и сушат (Na2SO4). Очистка на колонке Biotage 12M (градиент элюента: гексан: ацетон 20-60%), получают титульное соединение (65 мг, 49%) в виде белого порошка.
1H ЯМР (ДМСО-d6) δ м.д. 0.95 (с, 9Н), 1.03-1.05 (м, 2Н), 1.08-1.10 (м, 2Н), 1.26 (с, 9Н), 1.36-1.39 (м, 1Н), 1.69-1.72 (м, 1Н), 2.09-2.21 (м, 2Н), 2.41-2.45 (м, 1Н), 2.92-2.94 (м, 1Н), 3.91 (д, J=11.5 Гц, 1Н), 4.08 (д, J=7.5 Гц, 1Н), 4.16 (д, J=11.5 Гц, 1Н), 4.36 (т, J=8.5 Гц, 1Н), 5.10 (д,J=10 Гц, 1Н), 5.23 (д, J=17.5 Гц, 1Н), 5.38 (м, 1Н), 5.59-5.67 (м, 1Н), 6.54 (с, NH), 7.41-7.46 (м, 1Н). 7.50-7.53 (м, 2Н), 7.65 (с, 1Н), 7.74-7.75 (м, 2Н), 8.28 (с, 1Н), 8.52 (с, 1Н), 8.92 (c, NH).
LC/MS rt - мин (МН+): 2.08 (710) (Метод В).
Пример 421: Получение Соединения 421.

Схема 1
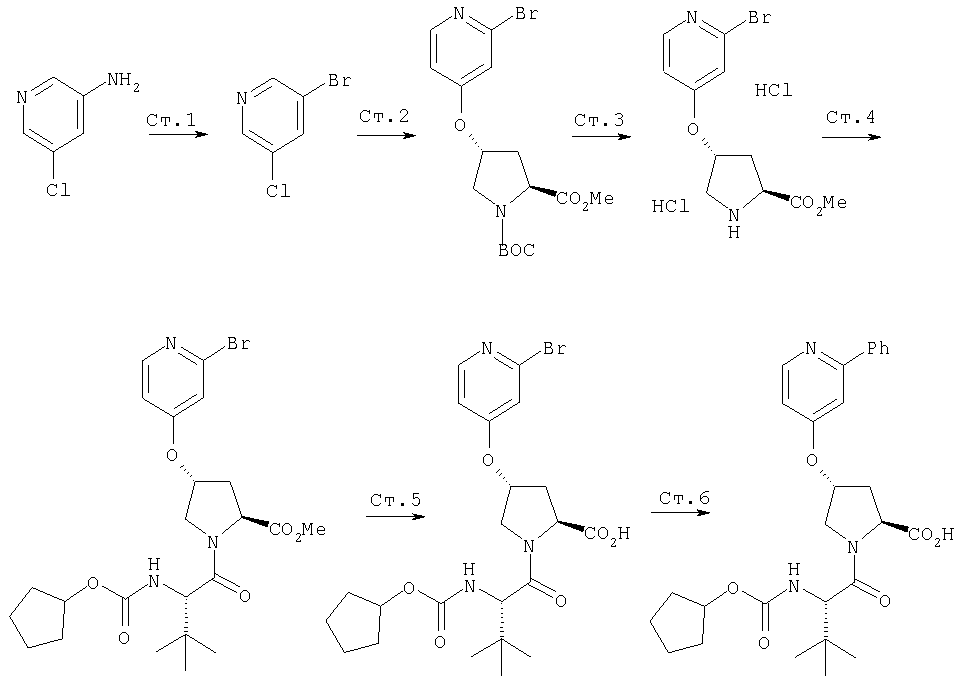
Стадия 7

К холодному (-5°С) 48% водному раствору HBr (35 мл) прибавляют 4-хлорпиридин-2-иламин (4.18 г, 32.5 ммоля), полученный по методике K.S. Gudmundsson et al; Synth. Commun. (1997), 27, 861), а затем медленно прибавляют бром (6.7 мл, 0.13 моля). Через 20 мин при той же температуре прибавляют нитрит натрия (8.63 г, 0.125 моля) в воде (40 мл) и продолжают перемешивать еще в течение 30 мин. При охлаждении льдом прибавляют 10 N NaOH (около 40 мл) до щелочного рН. Продукт экстрагируют EtOAc (2×100 мл) и сушат (Na2SO4). Сырой продукт очищают на колонке Biotage 40 М (элюируют 10% EtOAc в смеси гексанов), получают титульное соединение (2.50 г, 40%) в виде бесцветного масла, затвердевающего при стоянии.
1H ЯМР (ДМСО-d6) δ м.д. 7.64 (дд, J=1.5, 5.5 Гц, 1Н), 7.94 (д, J=1.5 Гц, 1Н), 8.40 (д, J=5.5 Гц, 1Н);
LC/MS rt - мин (MH+): 1.65 (192, 194, 196) (Метод В).
Стадия 2:
К раствору N-ВОС-транс-L-Hyp-ОН (3.22 г, 13.9 ммоля) в ДМСО (30 мл) в атмосфере азота при комнатной температуре порциями прибавляют трет-бутоксид калия (3.90 г, 34.8 ммоля). Через 1.5 час прибавляют продукт Стадии 1 в ДМСО (5 мл). Смесь перемешивают в течение ночи и прибавляют воду (150 мл). Раствор промывают EtOAc (100 мл). Водную фазу подкисляют до рН 4, добавляя 1N HCl. Сырую карбоновую кислоту экстрагируют EtOAc (трижды) и сушат (MgSCO4). Остаток, в виде твердого вещества коричневого цвета, суспендируют в ТГФ (20 мл) и МеОН (20 мл) и охлаждают до 0°С. По каплям прибавляют раствор триметилсилилдиазометана (2 М раствор в гексане, 12 мл) и через 15 мин раствор упаривают в вакууме. Очисткой на колонке Biotage 40 М (элюент: смесь гексанов - EtOAc 2:1, 1:1) получают титульное соединение (3.47 г, 62%) в виде не чисто белого порошка.
1H ЯМР (ДМСО-d6) δ м.д. 1.34, 1.38 (с, 9Н (ротамеры)), 2.24-2.29 (м, 1Н), 2.43-2.50 (м, 1Н), 3.57 (д, J=12.5 Гц, 1Н), 3.64-3.68 (м, 1Н), 3.66, 3.69 (с, 3H (ротамеры)), 4.27-4.34 (м, 1Н), 5.21 (м, 1Н), 7.06 (д, J=6 Гц, 1Н), 7.28 (s, 1H), 8.20 (д, J=6 Гц, 1Н);
LC/MS rt - мин (MH+): 2.59 (401, 403) (Метод В).
Стадия 3:
Продукт Стадии 2 (2.00 г, 4.98 ммоля) суспендируют в смеси: 4N HCl в диоксане (10 мл) и 1N HCl в диэтиловом эфире (40 мл), и перемешивают в течение ночи при комнатной температуре. Полученную суспензию упаривают в вакууме и остаток растирают с пентаном, получают титульное соединение с количественным выходом в виде белого порошка.
LC/MS rt - мин (МН+): 0.24 (301, 303) (Метод В).
Стадия 4:
К раствору продукта Стадии 3 (предположительно, 4.98 ммоля), N-циклопентилоксикарбонил-L-трет-лейцина (1.24 г, 5.10 ммоля) и НОВТ (673 мг, 4.98 ммоля) в DCM (25 мл) прибавляют DIPEA (4.34 мл, 24.9 ммоля), а затем HBTU (2.36 г, 6.23 ммоля). Полученный раствор перемешивают при rt в течение 15 час, прибавляют DCM и буфер с рН 4 и 1N HCl, чтобы довести рН до 4. Водный слой экстрагируют DCM (дважды). Объединенные органические вытяжки промывают насыщенным водным раствором NaHCO3 (дважды), рассолом и сушат (MgSO4). Очищают на колонке Biotage 40M (элюент: смесь гексанов - EtOAc 3:2, 1:1) получают титульное соединение (2.09 г, 80%) в виде белой пены.
1H ЯМР (ДМСО-d6) δ м.д. 0.94 (с, 9Н), 1.43-1.74 (м, 8Н), 2.18-2.23 (м, 1Н), 2.46-2.49 (м, 1Н), 3.64 (с, 3H), 3.87 (д, J=12 Гц, 1Н), 4.08-4.13 (м, 1Н), 4.42 (дд, J=7.5, 9.5 Гц, 1Н), 4.78 (м, 1Н), 5.30 (м, 1Н), 7.03-7.05 (м, 1Н), 7.24 (с, 1Н), 8.120 (д, J=6 Гц, 1Н).
LC/MS rt - мин (MH+): 2.34 (526, 528) (Метод С).
Стадия 5:
К раствору продукта Стадии 4 (206 мг, 0.391 ммоля) в ТГФ (2 мл) и метаноле (2 мл) прибавляют раствор LiOH (28 мг, 1.2 ммоля) в воде (2 мл). Смесь перемешивают в течение 2 час при комнатной температуре и прибавляют 1N HCl до нейтрального рН. Летучие органические соединения отгоняют в вакууме и к остатку прибавляют буферный раствор с рН 4. Продукт экстрагируют EtOAc (2×20 мл) и сушат (MgSO4), получают заглавное соединение (196 мг, 100%) в виде твердого вещества белого цвета.
1H ЯМР: (Метанол-d4) δ м.д. 1.05 (с, 9Н), 1.60-1.85 (м, 8Н), 2.30-2.36 (м, 1Н), 2.62-2.67 (м, 1Н), 3.97 (д, J=12 Гц, 1Н), 4.26 (с, 1Н), 4.32 (д, J=12 Гц, 1Н), 4.58 (дд, J=7.5, 9.5 Гц, 1Н), 4.89 (м (наложен.), 1Н), 5.29 (м, 1Н), 7.03-7.05 (м, 1Н), 7.26 (с, 1Н), 8.19 (д, J=6 Гц, 1Н);
LC/MS rt - мин (MH+): 2.17 (512, 514) (Метод В).
Стадия 6:
Этот продукт получают по методике, описанной в Примере 420, Стадия 4 (время реакции 20 час), с выходом 20% из продукта Стадии 5, Пример 421.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.45-1.74 (м, 8Н), 2.31-2.37 (м, 1Н), 2.65-2.69 (м, 1Н), 3.99 (дд, J=12, 3.0 Гц, 1Н), 4.26 (д, J=9.5 Гц, 1Н), 4.33 (д, J=11.5 Гц, 1Н), 4.60 (дд, J=8.0, 9.5 Гц, 1Н), 4.80 (м, 1Н), 5.35 (м, 1Н), 6.72 (д, J=9.0 Гц, NH), 7.02 (дд, J=2.5, 6.0 Гц, 1Н), 7.39 (с, 1Н), 7.50 (м, 3H (наложен.)), 7.91 (д, J=6.5 Гц, 1Н), 8.46 (д, J=6.0 Гц, 1Н).
LC/MS rt - мин (MH+): 2.03 (510) (Метод А).
Стадия 7:
Соединение 421 получают по методике, описанной в Примере 420, Стадия 6, с выходом 68%. Исходным является продукт Стадии 6, Пример 421.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.06-1.09 (м, 2Н), 1.23-1.26 (м, 2Н), 1.43 (дд, J=5.5, 9.5 Гц, 1Н), 1.47-1.77 (м, 8Н), 1.88 (дд, J=5.5, 8.5 Гц, 1Н), 2.21-2.29 (м, 2Н), 2.50-2.54 (м, 1Н), 2.91-2.96 (м, 1Н), 4.06 (дд, J=3.0, 11.5 Гц, 1Н), 4.28-4.30 (м, 2Н), 4.44 (дд, J=7.0, 10.5 Гц, 1Н). 4.82 (м, 1Н (наложен.)), 5.12 (д, J=10 Гц, 1Н), 5.30 (д, J=17 Гц, 1Н), 5.37 (м, 1Н), 5.73-5.80 (м, 1Н), 6.92 (д, J=9.5 Гц, NH), 6.98 (дд, J=5.5, 2.0 Гц, 1Н), 7.37 (с, 1Н), 7.43-7.50 (м, 3H), 7.90 (д, J=7.0 Гц, 1Н), 8.45 (д, J=5.5 Гц, 1Н).
LC/MS rt - мин (MH+): 2.37 (723) (Метод А).
Пример 422: Получение Соединения 422.
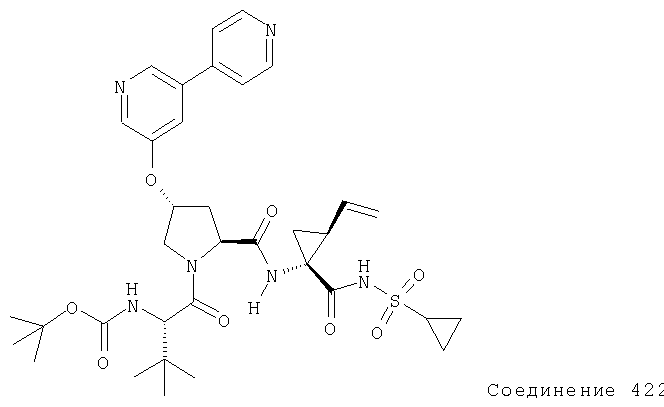
Стадия 1:

К смеси продукта из Примера 420, Стадия 3 (102 мг, 0.198 ммоля) и Pd(PPh3)4 (23 мг, 0.0198 ммоля) в толуоле (2 мл) прибавляют 4-трибутилстаннилпиридин (87 мг, 0.24 ммоля). Раствор нагревают при 105°С в течение 20 час в атмосфере азота. По охлаждении до комнатной температуры к смеси прибавляют насыщенный водный раствор NaHCO3, экстрагируют EtOAc (2×10 мл) и сушат (Na2SO4). Очищают на колонке Biotage 12 М (элюируют в градиенте гексан-ацетон 20-60%), получают титульное соединение (49 мг, 49%) в виде твердого вещества белого цвета.
1H ЯМР: (Метанол-d4) δ м.д. 1.05 (с, 9Н), 1.31 (с, 9Н), 2.29-2.35 (м, 1Н), 2.68-2.72 (м, 1Н), 3.77 (с, 3H), 4.00 (д, J=12 Гц, 1Н), 4.24 (д, J=9.5 Гц, 1Н), 4.41 (д, J=12 Гц, 1Н), 4.67 (дд, J=7.5, 10 Гц, 1Н), 5.36 (м, 1Н), 6.51 (д, J=9.5 Гц, NH), 7.79-7.85 (м, 3H), 8.39 (с, 1Н), 8.59 (с, 1Н), 8.67-8.68 (м, 2Н).
LC/MS rt - мин (MH+): 1.77 (514) (Метод С).
Стадия 2:

Этот продукт получают по методике, описанной в Примере 420, Стадия 5, с количественным выходом, за исключением того, что исходным является продукт Стадии 1, Пример 422.
1H ЯМР: (Метанол-d4) δ м.д. 1.05 (с, 9Н), 1.32 (с, 9Н), 2.32-2.38 (м, 1Н), 2.69-2.74 (м, 1Н), 3.99 (дд, J=3, 12 Гц, 1Н), 4.25 (с, 1Н), 4.40 (д, J=12 Гц, 1Н), 4.64 (дд, J=7.5, 9.5 Гц, 1Н), 5.36 (м, 1Н), 7.85-7.87 (м, 3H), 8.40 (с, 1Н), 8.60 (с, 1Н), 8.69-8.70 (м, 2Н).
LC/MS rt - мин (МН+): 1.57 (500) (Метод В).
Стадия 3:
Соединение 422 получают по методике, описанной в Примере 420, Стадия 6, с выходом 51% в виде твердого вещества белого цвета, но исходным является продукт Стадии 2, Пример 422.
1H ЯМР: (ДМСО-d6) δ м.д. 0.95 (с, 9Н), 1.03 (м, 1Н), 1.08 (м, 1Н), 1.23 (с, 9Н), 1.34-1.37 (м, 1Н), 1.68-1.70 (м, 1Н), 2.12-2.18 (м, 2Н), 2.41-2.45 (м, 1Н), 2.93 (м, 1Н), 3.90 (д, J=11 Гц, 1Н), 4.06 (д, J=9 Гц, 1Н), 4.17 (д, J=11 Гц, 1Н), 4.36 (дд, J=7, 10 Гц, 1Н), 5.10 (д, J=12 Гц, 1Н), 5.23 (д, J=16.5 Гц, 1Н), 5.40 (м, 1Н), 5.59-5.66 (м, 1Н), 6.60 (д, J=9 Гц, NH), 7.81-7.82 (м, 3H), 8.38 (с, 1Н), 8.65 (с, 1Н), 8.68-8.70 (м, 2Н), 8.93 (с, NH), 10.4 (с, NH).
LC/MS rt - мин (MH+): 2.14 (711) (Метод D).
Пример 423: Получение Соединения 423.

Стадия 1:

К раствору продукта Стадии 3, Пример 420 (1.00 г, 1.94 ммоля) в ТГФ (5 мл) и метаноле (5 мл) прибавляют раствор LiOH (140 мг, 5.83 ммоля) в воде (5 мл). Смесь перемешивают в течение 2 час при комнатной температуре и прибавляют 1N HCl до нейтрального рН. Летучие органические соединения отгоняют в вакууме и к остатку прибавляют буферный раствор с рН 4. Продукт экстрагируют EtOAc (3×25 мл) и сушат (MgSO4), получают заглавное соединение (1.0 г, 100%) в виде твердого вещества белого цвета.
1H ЯМР: (ДМСО-d6) δ м.д. 0.95 (с, 9Н), 1.27 (с, 9Н), 2.14-2.19 (м, 1Н), 2.47-2.50 (м, 1Н), 3.80 (д, J=11.5 Гц, 1H), 3.99-4.07 (м, 2Н), 4.15 (д, J=11.5 Гц, 1Н), 4.36 (дд, J=8, 10 Гц, 1Н), 5.25 (м, 1Н), 6.66 (д, J=9 Гц, NH), 7.75 (с, 1Н), 8.28 (с, 1Н), 8.31 (с, 1Н), 12.5 (с, 1Н).
LC/MS rt - мин (MH+): 2.47 (500, 502) (Метод А).
Стадия 2:
Соединение 423 получают по методике, описанной в Примере 420, Стадия 6, с выходом 52%, исходными являются продукты Стадии 1, Пример 423 и Стадии 3, Пример 8 (рацемический P1, (1R,2S) и (1S,2R)).
1H ЯМР: (ДМСО-d6) δ м.д. 0.93, 0.95 (с, 9Н), 1.00-1.09 (м, 4Н), 1.28, 1.29 (с. 9Н), 1.37-1.39 (м. 1Н), 1.69-1.72 (м, 1H), 2.09-2.22 (м, 2Н), 2.36-2.46 (м, 1Н), 2.88-2.94 (м, 1Н), 3.82-3.87 (м, 1Н), 4.02-4.04 (м, 1Н), 4.10-4.15 (м, 1Н), 4.31-4.34 (м, 1Н), 5.10 (д, J=10.5 Гц, 1Н), 5.22-5.28 (м, 1Н), 5.54-5.66 (м, 1Н). 6.56, 6.60 (д. J=9.5 Гц, NH), 7.74, 7.76 (с, 1Н), 8.28-8.29 (м, 1Н), 8.32 (с, 1Н), 8.79, 8.89 (с, 1Н).
LC/MS rt - мин (МН+): 2.42 (712, 714) (Метод В).
Пример 424: Получение Соединения 424.
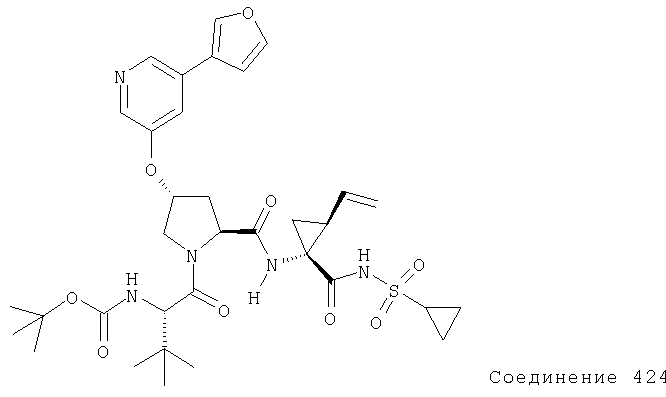
Стадия 1:
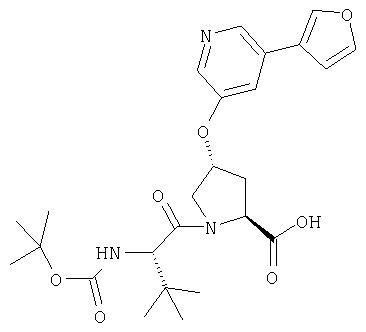
К раствору продукта Стадии 1, Пример 423 (91 мг, 0.18 ммоля), Pd(PPh3)4 (10.5 мг, 0.0091 ммоля) и 3-фурилбороновой кислоты (25.4 мг, 0.227 ммоля) в ДМФА (2 мл), прибавляют 2М водн. раствор Na2СО3 (0.273 мл, 0.546 ммоля). Смесь нагревают при 110°С в течение 2.5 час в атмосфере азота. По охлаждении до комнатной температуры осадок отфильтровывают, фильтрат упаривают в вакууме. Остаток очищают препаративной ВЭЖХ (градиент: 30-80% В), получают титульное соединение (64 мг, 72%) в виде твердого вещества белого цвета.
1H ЯМР: (ДМСО-d6) δ м.д. 0.95 (с, 9Н), 1.25 (с, 9Н), 2.17-2.21 (м, 1Н), 2.50 (м (наложен.), 1Н), 3.84-3.86 (м, 1Н), 4.09-4.15 (м, 2Н), 4.37 (т, J=9 Гц, 1Н), 5.28 (м, 1Н), 6.67 (д, J=9 Гц, NH), 7.08 (с, 1Н), 7.62 (с, 1Н), 7.79 (с, 1Н), 8.17 (с, 1Н), 8.33 (с, 1Н), 8.51 (с, 1Н), 12.6 (с, 1Н).
LC/MS rt - мин (МН+): 1.60 (488) (Метод В).
Стадия 2:
Соединение 424 получают по методике, описанной в Примере 420, Стадия 6, с выходом 55%, исходным является продукт Стадии 1, Пример 424.
1H ЯМР: (Метанол-d4) δ м.д. 1.02 (с, 9Н), 1.05-1.09 (м, 2Н), 1.22-1.25 (м, 2Н), 1.34 (с, 9Н), 1.42-1.45 (м, 1Н), 1.86-1.89 (м, 1Н), 2.21-2.26 (м, 1Н), 2.48-2.52 (м, 1Н), 2.91-2.96 (м, 1Н), 4.02 (д, J=12 Гц, 1Н), 4.24-4.29 (м, 2Н), 4.43-4.47 (дд, J=7.5, 10.5 Гц, 1Н), 5.12 (д, J=10.5 Гц, 1Н), 5.28-5.32 (м, 1Н), 5.73-5.81 (м, 1Н), 6.63 (д, J=9 Гц, NH), 6.90 (с, 1Н), 7.60-7.62 (м, 2Н), 8.07 (с, 1Н), 8.12 (с, 1Н), 8.39 (c, NH).
LC/MS rt - мин (MH+): 2.30 (700) (Метод Е).
Пример 425: Получение Соединения 425.
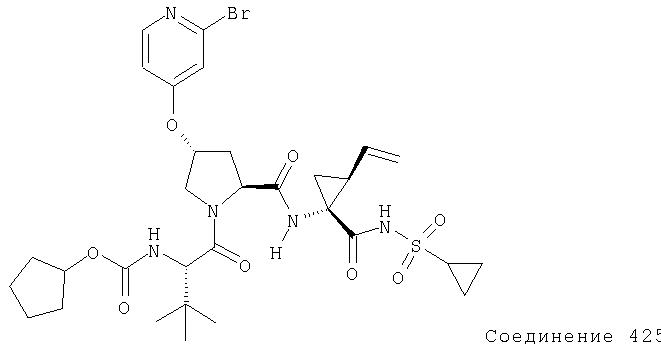
Соединение 425 получают по методике, описанной в Примере 421, Стадия 7, с выходом 45%, исходным является продукт Стадии 5, Пример 421.
1H ЯМР: (ДМСО-d6) δ м.д. 0.95 (с, 9Н), 1.03-1.04 (м, 1Н), 1.08-1.09 (м, 1Н), 1.33-1.36 (м, 1Н), 1.46-1.75 (м, 9Н), 2.08-2.12 (м, 1Н), 2.17 (кв, J=9 Гц, 1Н), 2.35-2.38 (м, 1Н), 2.88-2.93 (м, 1Н), 3.91 (д, J=9.5 Гц, 1Н), 4.05-4.10 (м, 2Н), 4.27 (дд, J=10, 7 Гц, 1Н), 4.80 (м, 1Н), 5.10 (д, J=12 Гц, 1Н), 5.23 (д, J=16.5 Гц, 1Н), 5.32 (м, 1Н), 5.58-5.66 (м, 1Н), 6.94 (д, J=9 Гц, NH), 7.03-7.05 (м, 1Н), 7.26 (с, 1Н), 8.21 (д, J=6 Гц, 1Н), 8.86 (с, NH), 10.4 (с, NH).
LC/MS rt - мин (MH+): 2.56 (724, 726) (Метод D).
Пример 426: Получение Соединения 426.

Стадия 1:

Этот продукт получают по методике, описанной в Примере 421, Стадия 6, с выходом 65%, за исключением того, что используют 3-фуранбороновую кислоту.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.49-1.76 (м, 8Н), 2.30-2 35 (м, 1Н), 2.63-2.67 (м, 1Н), 3.98 (д, J=12 Гц, 1Н), 4.26 (т, J=9 Гц, 1Н), 4.31 (д, J=12 Гц, 1Н), 4.58 (т, J=8.0 Гц, 1Н), 5.32 (м, 1Н), 6.71 (д, J=9.5 Гц, NH), 6.92 (дд, J=2.5, 6.0 Гц, 1Н), 6.98 (с, 1Н), 7.25 (д, J=2.5 Гц, 1Н), 7.60 (с, 1Н), 8.15 (с, 1Н), 8.35 (д, J=5.5 Гц, 1Н).
LC/MS rt - мин (MH+): 1.94 (500) (Метод D).
Стадия 2:
Соединение 426 получают по методике, описанной в Примере 421, Стадия 7, с выходом 57%, исходным является продукт Стадии 1, Пример 426.
1H ЯМР: (ДМСО-d6) δ м.д. 0.95 (с, 9Н), 1.03-1.04 (м, 2Н), 1.08-1.09 (м, 2Н), 1.34-1.71 (м, 8Н), 2.09-2.20 (м, 2Н), 2.37-2.41 (м, 1Н), 2.93 (м, 1Н), 3.96 (д, J=9.5 Гц, 1H), 4.07-4.11 (м, 2Н), 4.29 (м, 1Н), 4.78 (м, 1Н), 5.10 (д, J=10.5 Гц, 1Н), 5.23 (д, J=17 Гц, 1Н), 5.34 (м, 1Н), 5.59-5.66 (м, 1Н), 6.87-6.91 (м, 2Н), 7.08 (с, 1Н), 7.27 (с, 1Н), 7.75 (с, 1Н), 8.33 (с, 1Н), 8.39 (д, J=6 Гц, 1Н), 8.90 (с, NH), 10.4 (с, 1Н).
LC/MS rt - мин (MH+): 2.38 (712) (Метод D).
Пример 427: Получение Соединения 427.
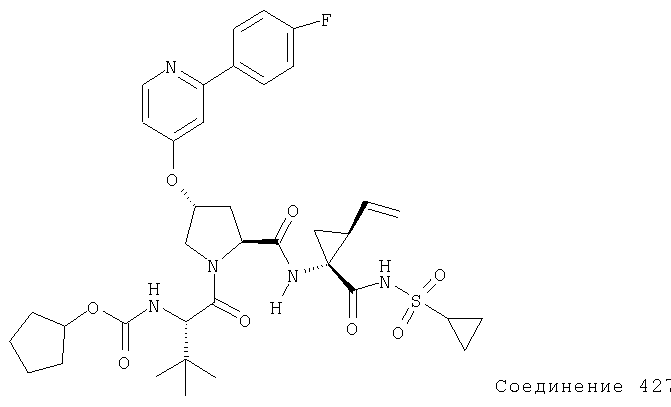
Стадия 1:

Этот продукт получают по методике, описанной в Примере 421, Стадия 6, с выходом 86%, за исключением того, что используют 4-фторфенилбороновую кислоту.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.44-1.75 (м, 8Н), 2.30-2.36 (м, 1Н), 2.64-2.69 (м, 1Н), 3.99 (дд, J=12, 3.5 Гц, 1Н), 4.26 (д, J=9.5 Гц, 1Н), 4.32 (д, J=12 Гц, 1Н), 4.59 (дд, J=8.0, 9.5 Гц, 1Н), 4.81 (м, 1Н), 5.34 (м, 1Н), 6.73 (д, J=9.5 Гц, NH), 6.99 (дд, J=2.5, 6.0 Гц, 1Н), 7.20 (т, 2Н (наложен.)), 7.36 (с, 1Н (наложен.)), 7.94-7.97 (м, 2Н), 8.44 (д, J=6.0 Гц, 1Н).
LC/MS rt - мин (MH+): 2.22 (528) (Метод А).
Стадия 2:
Соединение 427 получают по методике, описанной в Примере 421, Стадия 7, с выходом 53%, исходным является продукт Стадии 1, Пример 427.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.06-1.09 (м, 2Н), 1.23-1.26 (м, 2Н), 1.43 (дд, J=5.0, 9.5 Гц, 1Н), 1.47-1.78 (м, 8Н), 1.88 (дд, J=5.5, 8.0 Гц. 1Н), 2.21-2.29 (м, 2Н), 2.49-2.53 (м, 1Н), 2.91-2.96 (м, 1H), 4.05 (дд, J=3.0, 11.5 Гц, 1Н), 4.27-4.29 (м, 2Н), 4.44 (дд, J=7.0, 10.5 Гц, 1Н), 4.83 (м, 1Н (наложен.)), 5.12 (д, J=10 Гц, 1Н), 5.29 (д, J=17 Гц, 1Н), 5.37 (м, 1Н), 5.73-5.80 (м, 1Н), 6.93 (д, J=9.5 Гц, NH), 6.98 (м, 1Н), 7.21 (т, J=9.0 Гц, 2Н), 7.36 (с, 1Н), 7.94-7.97 (м, 2Н), 8.44 (д, J=6.0 Гц, 1Н).
LC/MS rt - мин (МН+): 2.42 (741) (Метод А).
Пример 428: Получение Соединения 428.
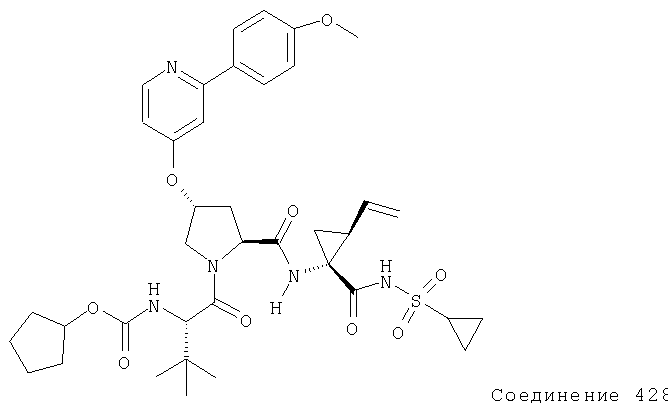
Стадия 1:

Этот продукт получают по методике, описанной в Примере 421, Стадия 6, с выходом 37%, за исключением того, что используют 4-метоксифенилбороновую кислоту.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.45-1.74 (м, 8Н), 2.31-2.36 (м, 1Н), 2.64-2.68 (м, 1Н), 3.85 (с, 3H), 3.98 (дд, J=12, 3.5 Гц, 1Н), 4.25 (д, J=9.0 Гц, 1Н), 4.33 (д, J=11.5 Гц, 1Н), 4.59 (дд, J=8.0, 10 Гц, 1Н), 4.77 (м, 1Н), 5.36 (м, 1Н), 6.72 (д, J=9.0 Гц, NH), 7.01 (м, 1Н), 7.04 (д, J=9.0 Гц, 2Н), 7.37 (д, J=2.0 Гц), 7.86 (д, J=9.0 Гц, 2Н), 8.42 (д, J=6.0 Гц, 1Н).
LC/MS rt - мин (MH+): 2.09 (540) (Метод А).
Стадия 2:
Соединение 428 получают по методике, описанной в Примере 420, Стадия 6, с выходом 72%, исходным является продукт Стадии 1, Пример 428.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.06-1.09 (м, 2Н), 1.23-1.25 (м, 2Н), 1.43 (дд, J=5.5, 9.5 Гц, 1Н), 1.48-1.77 (м, 8Н), 1.88 (дд, J=5.0, 8.0 Гц, 1Н), 2.21-2.28 (м, 2Н), 2.50-2.54 (м, 1Н), 2.91-2.96 (м, 1Н), 3.85 (с, 3H), 4.05 (д, J=12 Гц, 1Н), 4.27-4.29 (м, 2Н), 4.44 (дд, J=7.0, 10.5 Гц, 1Н), 4.84 (м, 1Н (наложен.)), 5.12 (д, J=10 Гц, 1Н), 5.30 (д, J=17 Гц, 1Н), 5.37 (м, 1Н), 5.72-5.80 (м, 1Н), 6.89 (д, J=9.5 Гц, NH), 6.95 (дд, J=2.5, 6.0 Гц, 1Н), 7.03 (д, J=9.0 Гц, 1Н), 7.33 (д, J=2.5 Гц, 1Н), 7.86 (д, J=9.0 Гц, 2Н), 8.41 (д, J=6.0 Гц, 1Н).
LC/MS rt - мин (МН+): 2.40 (753) (Метод А).
Пример 429: Получение Соединения 429.

Стадия 1:

Этот продукт получают по методике, описанной в Примере 421, Стадия 6, с выходом 22%, за исключением того, что используют 2-тиофенбороновую кислоту.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.46-1.74 (м, 8Н), 2.29-2.35 (м, 1Н), 2.63-2.67 (м, 1Н), 3.97 (д, J=12 Гц, 1Н), 4.26 (д, J=8.5 Гц, 1Н), 4.31 (д, J=12 Гц, 1Н), 4.60 (т, J=8.5 Гц, 1Н), 4.81 (м, 1Н (наложен.)), 5.31 (м, 1Н), 6.87-6.89 (м, 2Н), 7.13 (д, J=5.0 Гц, 1Н), 7.52 (с, 1Н (наложен.)), 7.70 (д, J=2.5 Гц, 1Н), 8.32 (д, J=6.0 Гц, 1Н).
LC/MS rt - мин (МН+): 1.96 (516) (Метод А).
Стадия 2:
Соединение 429 получают по методике, описанной в Примере 420, Стадия 6, с выходом 57%, исходным является продукт Стадии 1, Пример 429.
1H ЯМР: (Метанол-d4) δ м.д. 1.02 (с, 9Н), 1.07 (м, 2Н), 1.24 (м, 2Н), 1.43 (дд, J=5.5, 9.5 Гц, 1Н), 1.53-1.78 (м, 8Н), 1.88 (дд, J=5.5, 8.0 Гц, 1Н), 2.21-2.28 (м, 2Н), 2.49-2.53 (м, 1Н), 2.92-2.96 (м, 1Н), 4.04 (д, J=10 Гц, 1Н), 4.26-4.29 (м, 2Н), 4.43 (т, J=9.0 Гц, 1Н), 4.82 (м, 1Н (наложен.)), 5.12 (д, J=10.5 Гц, 1Н), 5.29 (д, J=17.5 Гц, 1Н), 5.35 (м, 1Н), 5.73-5.80 (м, 1Н), 6.89 (д, J=4.5 Гц, 1Н), 6.92 (д, J=4.5 Гц, NH), 7.13 (с, 1Н), 7.35 (с, 1Н), 7.51 (д, J=4.5 Гц, 1Н), 7.70 (с, 1Н), 8.32 (д, J=5.5 Гц, 1Н).
LC/MS rt - мин (MH+): 2.37 (728) (Метод А).
Пример 430: Получение Соединения 430.

Стадия 1:

Этот продукт получают по методике, описанной в Примере 421, Стадия 6, с выходом 45%, за исключением того, что используют 3-тиофенбороновую кислоту.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.43-1.74 (м, 8Н), 2.30-2.36 (м, 1Н), 2.64-2.68 (м, 1Н), 3.98 (дд, J=11.5, 3.0 Гц, 1Н), 4.25 (д, J=9.0 Гц, 1Н), 4.32 (д, J=11.5 Гц, 1Н), 4.60 (дд, J=8.0, 9.5 Гц, 1Н), 4.80 (м, 1Н (наложен.)), 5.33 (м, 1Н), 6.96 (дд, J=2.5, 6.0 Гц, 1Н), 7.36 (с, 1Н (наложен.)), 7.51 (с, 1Н (наложен.)), 7.67 (д, J=5.0 Гц, 1Н), 8.03 (с, 1Н), 8.39 (д, J=6.0 Гц, 1Н)
LC/MS rt - мин (МН+): 1.94 (516) (Метод А).
Стадия 2:
Соединение 430 получают по методике, описанной в Примере 420, Стадия 6, с выходом 45%, исходным является продукт Стадии 1, Пример 430.
1H ЯМР: (Метанол-d4) δ м.д. 1.02 (с, 9Н), 1.06 (м, 2Н), 1.29 (м, 2Н), 1.43 (дд, J=5.5, 9.5 Гц, 1Н), 1.43-1.75 (м, 8Н), 1.87 (дд, J=6.0, 7.5 Гц, 1Н), 2.20-2.27 (м, 2Н), 2.49-2.53 (м, 1Н), 2.93-2.95 (м. 1Н), 4.05 (д, J=9.0 Гц, 1Н), 4.27-4.29 (м, 2Н), 4.42-4.45 (м, 1Н), 4.85 (м, 1Н (наложен.)), 5.12 (д, J=10.0 Гц, 1Н), 5.29 (д, J=17.5 Гц, 1Н), 5.36 (м, 1Н), 5.73-5.80 (м, 1Н), 6.92-6.94 (м, 2Н), 7.35 (с, 1Н), 7.51 (с, 1Н), 7.66 (д, J=4.5 Гц, 1Н), 8.00 (с, 1Н), 8.38 (д, J=5.5 Гц, 1Н).
LC/MS rt - мин (МН+): 2.36 (728) (Метод А).
Пример 431: Получение Соединения 431.
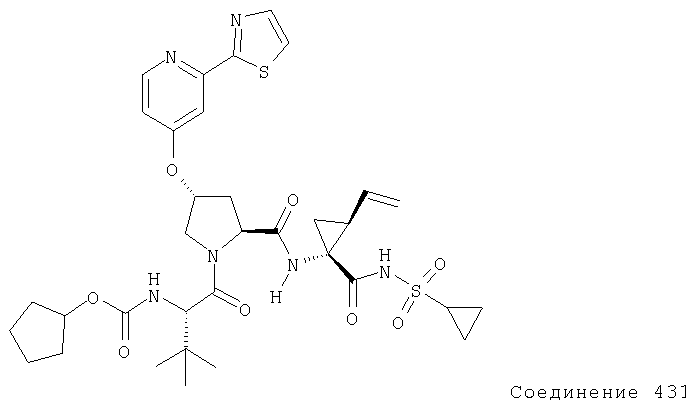
Стадия 1:
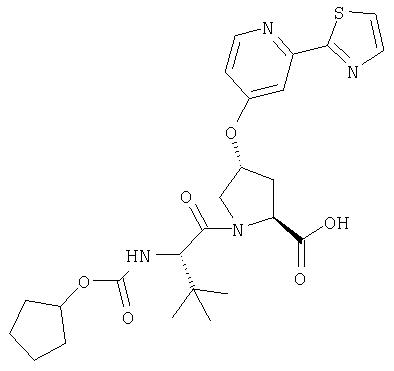
К смеси продукта из Примера 421, Стадия 5 (100 мг, 0.195 ммоля) и Pd(PPh3)4 (23 мг, 0.0195 ммоля) в диоксане (3 мл) прибавляют 2-трибутилстаннилтиазол (95 мг, 0.254 ммоля). Раствор нагревают при 95°С в течение 5 час в атмосфере азота, затем при 105°С в течение 15 час. По охлаждении до комнатной температуры смесь фильтруют, фильтрат упаривают в вакууме. Остаток очищают препаративной ВЭЖХ (градиент: 30-80% В). Объединенные фракции распределяют между буфером с рН 4 и хлористым метиленом. Водную фазу экстрагируют хлористым метиленом и объединенные органические вытяжки промывают рассолом и сушат (MgSO4). Титульное соединение (31 мг) получают в виде бесцветного масла, в значительной степени загрязненного трибутилстаннильным остатком.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.4 (м, 8Н (наложен.)), 2.32-2.36 (м, 1Н), 2.64-2.68 (м, 1Н), 4.00 (д, J=11.5 Гц, 1Н), 4.25 (с, 1Н), 4.33 (д, J=11.5 Гц, 1Н), 4.60 (т, J=8.5 Гц, 1Н), 4.80 (м, 1Н (наложен.)), 5.33 (м, 1Н), 7.03 (уш с, 1Н), 7.70 (с, 1Н), 7.73 (с, 1Н), 7.93 (с, 1Н). 8.41 (д, J=5.5 Гц, 1Н).
LC/MS rt - мин (MH+): 2.25 (517) (Метод А).
Стадия 2:
Соединение 431 получают по методике, описанной в Примере 420, Стадия 6, с выходом 41%, исходным является продукт Стадии 1, Пример 431.
1H ЯМР: (Метанол-d4) δ м.д. 1.02 (с, 9Н), 1.06-1.70 (м, 13Н), 1.86-1.89 (м, 1Н), 2.22-2.31 (м, 2H), 2.51-2.55 (м, 1H), 2.92-2.94 (м, 1Н), 4.06 (д, J=11.5 Гц, 1Н), 4.23-4.32 (м, 2H), 4.44-4.47 (м, 1Н), 4.82 (м, 1Н (наложен.)), 5.12 (д, J=11 Гц, 1Н), 5.30 (д, J=17 Гц, 1Н), 5.36 (м, 1Н), 5.73-5.81 (м, 1Н), 6.90 (д, J=9.0 Гц, NH), 7.04 (м, 1Н), 7.71 (м, 2Н), 7.93 (с, 1Н), 8.42 (д, J=5.5 Гц, 1Н).
LC/MS rt - мин (MH+): 2.46 (729) (Метод А).
Пример 432: Получение Соединения 432.

Стадия 1:
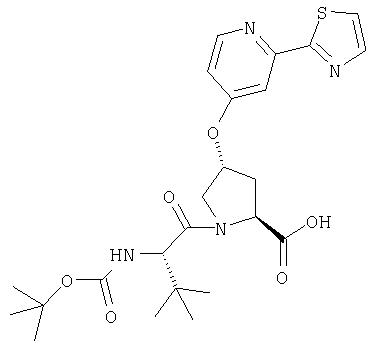
К смеси продукта из Примера 423, Стадия 1 (102 мг, 0.204 ммоля) и Pd(PPh3)4 (24 мг, 0.0204 ммоля) в диоксане (3 мл) прибавляют 2-трибутилстаннилтиазол (99 мг, 0.265 ммоля) и триэтиламин (85 мкл, 0.612 ммоля). Раствор нагревают при 95°С в течение 5 час в атмосфере азота, затем при 105°С в течение 15 час. По охлаждении до комнатной температуры смесь фильтруют, фильтрат упаривают. Остаток очищают препаративной ВЭЖХ (градиент: 30-80% В). Объединенные фракции нейтрализуют концентрированным раствором аммиака и упаривают. Остаток распределяют между буфером с рН 4 и хлористым метиленом. Водную фазу экстрагируют хлористым метиленом и объединенные органические вытяжки промывают рассолом и сушат (MgSCO4). Титульное соединение (33 мг) получают в виде бесцветного масла, в значительной степени загрязненного трибутилстаннилсодержащим остатком.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.30 (с, 9Н), 2.28-2.32 (м, 1Н), 2.62-2.67 (м, 1Н), 3.98 (д,J=11.5 Гц, ГН), 4.21 (с, 1Н), 4.34 (д, J=11.5 Гц, 1Н), 4.58 (т, J=9.0 Гц, 1Н), 5.31 (м, 1Н), 7.71 (д, J=2.5 Гц, 1Н), 7.94-7.96 (м, 2Н), 8.33 (с, 1Н), 8.73 (с, 1Н).
LC/MS rt - мин (MH+): 2.21 (405) (Метод А).
Стадия 2:
Соединение 432 получают по методике, описанной в Примере 420, Стадия 6, с выходом 42%, исходным является продукт Стадии 1, Пример 432.
1H ЯМР: (Метанол-d4) δ м.д. 1.02 (с, 9Н), 1.07-1.08 (м, 2Н), 1.24 (м, 2Н), 1.32 (с, 9Н), 1.44 (м, 1Н (наложен.)), 1.86-1.89 (м, 1Н), 2.21-2.29 (м, 2Н), 2.51-2.55 (м, 1Н), 2.93-2.95 (м, 1Н), 4.04 (д, J=12 Гц, 1Н), 4.23 (д, J=9.5 Гц, 1Н), 4.33 (д, J=12 Гц, 1Н), 4.47 (т, J=9.5 Гц, 1Н), 5.12 (д, J=10.0 Гц, 1Н), 5.30 (д, J=18 Гц, 1Н), 5.36 (м, 1Н), 5.72-5.81 (м, 1Н), 6.62 (д, J=8.5 Гц, NH), 7.73 (с, 1Н), 7.96 (м, 1Н), 8.34 (с, 1Н), 8.74 (с, 1Н).
LC/MS rt - мин (MH+): 2.42 (717) (Метод А).
Пример 433: Получение Соединения 433.
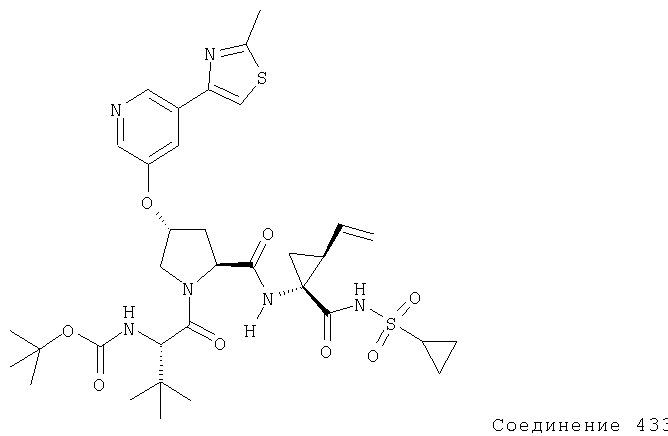
Стадия 1:
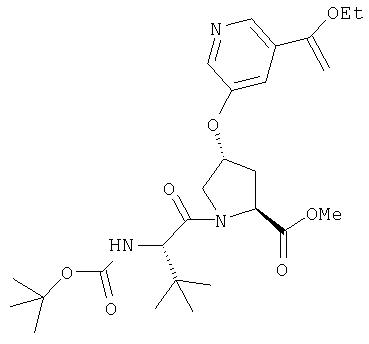
К смеси продукта из Примера 420, Стадия 3 (1.00 г, 1.94 ммоля) и Pd(PPh3)4 (112 мг, 0.097 ммоля) в диоксане (15 мл) прибавляют трибутил(этоксивинил)олово (876 мг, 2.43 ммоля). Раствор нагревают при 105°С в течение 6 час в атмосфере азота. По охлаждении до комнатной температуры смесь фильтруют, фильтрат упаривают. Остаток распределяют между насыщ. раствором NaHCO3 и этилацетатом. Водную фазу экстрагируют этилацетатом и объединенные органические вытяжки промывают 5% водн. KF и рассолом и сушат (MgSO4). Очисткой на колонке Biotage 40 М (элюируют в градиенте гексан - EtOAc 40-70%) получают титульное соединение (624 мг, 64%) в виде желтого масла.
1H ЯМР: (Метанол-d4) δ м.д. 0.95 (с, 9Н), 1.27 (с, 9Н), 1.35 (т, J=7.0 Гц, 3H), 2.16-2.21 (м, 1Н), 2.50 (м, 1Н (наложен.)), 3.64 (с, 3H), 3.85 (д, J=11 Гц, 1Н), 3.90 (кв, J=7.0 Гц, 2Н), 4.09 (д, J=9.0 Гц, 1Н), 4.13 (д, J=11 Гц, 1Н), 4.40 (д, J=2.5 Гц, 1Н), 4.45 (т, J=8.0 Гц, 1Н), 4.91 (д, J=2.5 Гц, 1Н), 5.27 (м, 1Н), 6.69 (д, J=9.0 Гц, NH), 7.48 (с, 1Н), 8.25 (с, 1Н), 8.46 (с, 1Н).
LC/MS rt - мин (MH+): 2.14 (507) (Метод В).
Стадия 2:

К раствору продукта Стадии 1, Пример 433 (125 мг, 0.247 ммоля) в смеси ТГФ (3 мл) и воды (111 мкл, 6.18 ммоля) прибавляют NBS (44 мг, 0.247 ммоля). Перемешивают при комнатной температуре 20 мин и смесь упаривают и распределяют между этилацетатом и рассолом. Органическую фазу сушат (MgSO4), получают промежуточный бромметилкетон. Этот интермедиат растворяют в ДМФА и прибавляют тиоацетамид (24 мг, 0.321 ммоля) и NaHCO3 (31 мг, 0.371 ммоля). Смесь перемешивают 2 часа при комнатной температуре и суспендируют в насыщ. растворе NaHCO3. Продукт экстрагируют этилацетатом (2х), промывают рассолом и сушат (MgSO4). Очищают на колонке Biotage 12 М (элюируют в градиенте hexane-EtOAc 50-70%), получают титульное соединение (50 мг, 38%) в виде бледно-окрашенного масла.
1H ЯМР: (Метанол-d4) δ м.д. 1.02 (с, 9Н), 1.31 (с, 9Н), 2.27-2.31 (м, 1Н), 2.63-2.67 (м, 1Н), 2.77 (с, 3H), 3.74 (с, 3H), 3.98 (д, J=10.0 Гц, 1Н), 4.22 (д, J=9.5 Гц, 1Н), 4.34 (д, J=10.5 Гц, 1Н), 4.64 (т, J=9.0 Гц, 1Н), 5.29 (м, 1Н), 6.38 (с, NH), 7.87 (с, 1Н), 7.91 (с, 1Н), 8.20 (с, 1Н), 8.71 (с, 1Н).
LC/MS rt - мин (МН+): 1.96 (533) (Метод В).
Стадия 3:
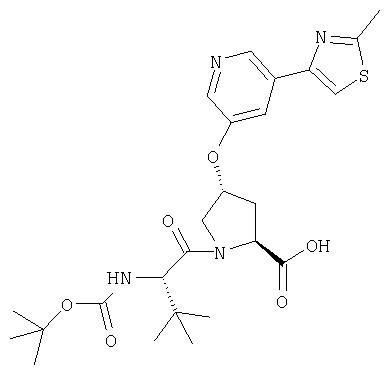
Этот продукт получают по методике, описанной в Примере 420, Стадия 5, за исключением того, что используют продукт Стадии 2, Пример 433.
1H ЯМР: (ДМСО-d6) δ м.д. 0.96 (с, 9Н), 1.25 (с, 9Н), 2.18-2.23 (м, 1Н). 2.50 (м, 1Н (наложен.)), 2.73 (с, 3H), 3.86 (д, J=11.5 Гц, 1Н), 4.10 (д, J=9.0 Гц, 1Н), 4.14 (д, J=11.5 Гц, 1Н), 4.39 (т, J=8.5 Гц, 1Н), 5.30 (м, 1Н), 6.60 (уш с, NH), 7.84 (с, 1Н), 8.14 (с, 1Н), 8.24 (с, 1Н), 8.78 (с, 1Н).
LC/MS rt - мин (МН+): 1.84 (519) (Метод В).
Стадия 4:
Соединение 433 получают по методике, описанной в Примере 420, Стадия 6, с выходом 46%, исходным является продукт Стадии 1, Пример 429.
1H ЯМР: (Метанол-d4) δ м.д. 1.05 (с, 9Н), 1.09-1.11 (м, 2Н), 1.26-1.29 (м, 2Н), 1.36 (с, 9Н), 1.45-1.48 (м, 1Н), 1.89-1.92 (м, 1Н), 2.25-2.30 (м, 2Н), 2.53-2.57 (м, 1Н). 2.80 (с, 3H), 2.94-3.00 (м, 1Н), 4.06 (д, J=10.5 Гц, 1Н), 4.27 (с, 1Н), 4.32 (д, J=12 Гц. 1Н), 4.48 (дд, J=10.5, 7.0 Гц, 1Н), 5.15 (д, J=10.5 Гц, 1Н), 5.33 (д, J=17 Гц, 1Н), 5.35 (м, 1Н), 5.76-5.83 (м, 1Н), 7.93 (с, 1Н), 7.95 (м, 1Н), 8.22 (с, 1Н), 8.74 (с, 1Н).
LC/MS rt - мин (МН+): 2.21 (732) (Метод В).
Пример 434: Получение Соединения 434.
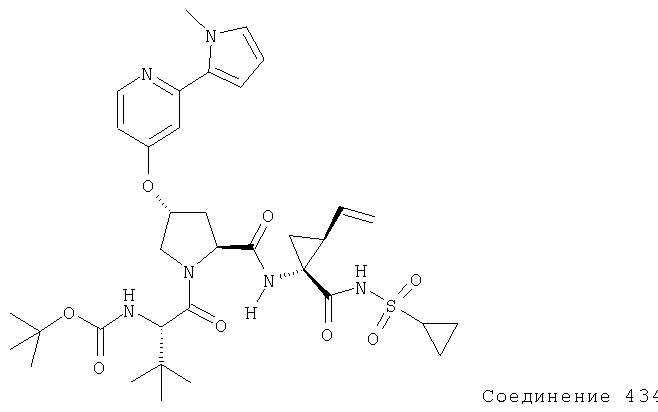
Стадия 1:
К раствору продукта из Примера 421, Стадия 1 (300 мг, 1.56 ммоля) и Pd(PPh3)4 (90 мг, 0.078 ммоля) в ДМФА (6 мл) прибавляют 1-метил-2-(трибутилстаннил)-1Н-пиррол (750 мг, 2.03 ммоля) и триэтиламин (0.435 мл, 3.12 ммоля). Раствор нагревают при 150°С в течение 30 мин в атмосфере азота в микроволновой печи (Emrys, Personal Chemistry). По охлаждении до комнатной температуры к смеси прибавляют диэтиловый эфир и 5% водн. KF и фильтруют. Водную фазу экстрагируют диэтиловым эфиром (2х). Объединенные органические вытяжки промывают 5% водн. KF, водой и рассолом и сушат (MgSO4). Очисткой на колонке Biotage 25 S (элюируют в градиенте гексан-диэтиловый эфир 0-5%) получают титульное соединение (169 мг, 56%) в виде бесцветного масла.
1H ЯМР: (ДМСО-d6) δ м.д. 3.94 (с, 3H), 6.10 (с, 1Н), 6.77 (д, J=2.0 Гц, 1Н), 6.93 (с, 1Н), 7.27 (д, J=4.0 Гц, 1Н), 7.77 (с, 1Н), 8.49 (д, J=5.0 Гц, 1Н),
LC/MS rt - мин (МН+): 1.15 (193,195) (Метод В).
Стадия 2:
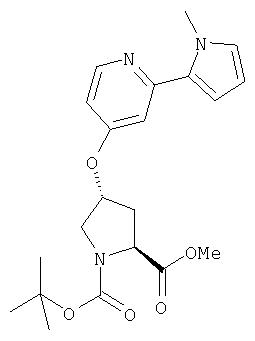
Этот продукт получают по методике, описанной в Примере 421, Стадия 2, с выходом 39%, в качестве исходного используют продукт Стадии 1, Пример 434.
1H ЯМР: (Метанол-d4) δ м.д. 1.42, 1.43 (с, 9Н (ротамеры)), 2.27-2.34 (м, 1Н), 2.55-2.62 (м, 1Н), 3.75 (м, 2Н), 3.76, 3.74 (с, 3H (ротамеры)), 3.84 (с, 3H), 4.39-4.45 (м, 1Н), 5.19 (м, 1Н), 6.10 (м, 1Н), 6.49 (м, 1Н), 6.78 (с, 1Н), 6.80-6.82 (м, 1Н), 7.07 (с, 1Н), 8.35 (д, J=6.0 Гц, 1Н).
LC/MS rt - мин (МН+, карбоновая кислота): 1.49 (389) (Метод В).
Стадия 3:

Продукт Стадии 2, Пример 434 (90 мг, 0.22 ммоля) растворяют в хлористом метилене (1.5 мл) и ТФК (1.0 мл, 9.0 ммоля). Раствор перемешивают при комнатной температуре в течение 45 мин и упаривают. К остатку прибавляют 1N HCl в диэтиловом эфире (5 мл) и упаривают. Титульное соединение получают с количественным выходом в виде бледно окрашенного масла.
LC/MS rt - мин (МН+ ): 0.32 (302) (Метод В).
Стадия 4:

Этот продукт получают по методике, описанной в Примере 420, Стадия 3, с выходом 80%, в качестве исходного используют продукт Стадии 3, Пример 434.
1H ЯМР: (Метанол-d4) δ м.д. 1.02 (s, 9Н), 1.31 (с, 9Н), 2.25-2.31 (м, 1Н), 2.61-2.64 (м, 1Н), 3.73 (с, 3H), 3.84 (с, 3H), 3.97-3.99 (м, 1Н), 4.22 (д, J=9.5 Гц, 1Н), 4.32 (д, J=11.5 Гц, 1Н), 4.61 (т, J=7.5 Гц, 1Н), 5.27 (м, 1Н), 6.10 (м, 1Н), 6.42 (уш д, J=9.0 Гц, NH), 6.49 (м, 1Н), 6.78 (м, 1Н), 6.81 (м, 1Н), 7.07 (с, 1Н), 8.34 (д, J=5.5 Гц, 1Н).
LC/MS rt - мин (МН+): 1.81 (516) (Метод В).
Стадия 5:

К раствору продукта Стадии 4, Пример 434 (92 мг, 0.179 ммоля) в смеси ТГФ (1 мл) и метанола (1 мл) прибавляют раствор LiOH (13 мг, 0.536 ммоля) в воде (1 мл). Смесь перемешивают в течение 1.5 час при комнатной температуре и прибавляют 1N HCl до нейтрального рН. Летучие органические соединения отгоняют в вакууме и остаток очищают препаративной ВЭЖХ (градиент 10-80% В). Объединенные фракции нейтрализуют конц. раствором аммиака и упаривают. Остаток распределяют между буферным раствором с рН 4 и этилацетатом. Водную фазу экстрагируют EtOAc и объединенные органические вытяжки промывают рассолом и сушат (MgSO4). Титульное соединение (56 мг, 62%) получают в виде твердого вещества белого цвета.
1H ЯМР: (Метанол-d4) δ м.д. 1.03 (с, 9Н), 1.32 (с, 9Н), 2.31-2.35 (м, 1Н), 2.62-2.67 (м, 1Н), 3.85 (с, 3H), 3.98 (д, J=11.5 Гц, 1Н), 4.21-4.23 (м, 1Н), 4.33 (д, J=11.5 Гц, 1Н), 4.58 (т, J=8.0 Гц, 1Н), 5.31 (м, 1Н), 6.12 (м, 1Н), 6.40 (уш д, J=8.0 Гц, NH), 6.52 (м, 1Н), 6.82 (м, 1Н), 6.87 (м, 1Н), 7.12 (с, 1Н), 8.36 (д, J=5.5 Гц, 1Н).
LC/MS rt - мин (МН+): 1.73 (502) (Метод В).
Стадия 6:
Соединение 434 получают по методике, описанной в Примере 420, Стадия 6, с выходом 68%, в качестве исходного используют продукт Стадии 5, Пример 434.
1H ЯМР: (Метанол-d4) δ м.д. 1.02 (с, 9Н), 1.06-1.09 (м, 2Н), 1.23-1.26 (м, 2Н), 1.33 (с, 9Н), 1.42-1.45 (м, 1Н), 1.86-1.89 (м, 1Н), 2.21-2.27 (м, 2Н), 2.47-2.51 (м, 1Н), 2.91-2.96 (м, 1Н), 3.85 (с, 3H), 4.04 (д, J=12 Гц, 1Н), 4.24 (д, J=10.0 Гц, 1Н), 4.28 (д, J=12 Гц, 1Н), 4.43 (дд, J=10.0, 7.0 Гц, 1Н), 5.12 (д, J=10.0 Гц, 1Н), 5.30 (д, J=17 Гц, 1Н), 5.32 (м, 1Н), 5.73-5.80 (м, 1Н), 6.10 (м, 1Н), 6.49 (м, 1Н), 6.64 (уш д, J=9.0 Гц, NH), 6.79 (м, 1Н), 6.82 (м, 1Н), 7.08 (с, 1Н), 8.35 (д, J=5.5 Гц, 1Н).
LC/MS rt - мин (MH+): 2.08 (714) (Метод В).
Пример 435: Получение Соединения 435.

Стадия 1:
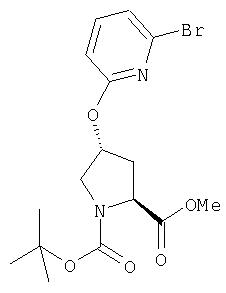
Этот продукт получают по методике, описанной в Примере 421, Стадия 2, с выходом 74%, в качестве исходного используют 2, 6-дибромпиридин.
1H ЯМР: (ДМСО-d6) δ м.д. 1.34, 1.38 (с, 9Н (ротамеры)), 2.23-2.31 (м, 1Н), 2.43-2.47 (м, 1Н (наложен.)), 3.53 (д, J=12 Гц, 1Н), 3.66, 3.69 (с, 3H (ротамеры)), 3.72-3.75 (м, 1Н), 4.29-4.34 (м, 1Н), 5.42 (м, 1Н), 6.89 (д, J=7.5 Гц, 1Н), 7.26 (д, J=7.5 Гц, 1Н), 7.68 (т, J=7.5 Гц, 2Н).
LC/MS rt - мин (MH+): 2.62 (423, 425) (Метод А).
Стадия 2:

Этот продукт получают по методике, описанной в Примере 420, Стадия 2, с количественным выходом из продукта Стадии 1, Пример 435.
LC/MS rt - мин (МН+): 1.42 (301, 303) (Метод А).
Стадия 3:

Этот продукт получают по методике, описанной в Примере 420, Стадия 3, с выходом 96%, в качестве исходного используют продукт из Примера 435, Стадия 2.
1H ЯМР: (ДМСО-d6) δ м.д. 0.95 (с, 9Н), 1.28 (с. 9Н), 2.20-2.26 (м, 1Н), 2.45-2.48 (м, 1Н), 3.64 (с, 3H), 3.94 (д, J=9.5 Гц, 1Н), 4.01-4.08 (м, 2Н), 4.45 (т, J=8.5 Гц, 1Н), 5.53 (м, 1Н), 6.66 (д, J=7.0 Гц, 1Н), 6.82 (д, J=7.0 Гц, 1Н), 7.25 (д, J=7.0 Гц, 1Н), 7.64-7.69 (м, 1Н).
LC/MS rt - мин (МН+): 2.73 (514, 516) (Метод А).
Стадия 3:

Этот продукт получают по методике, описанной в Примере 420, Стадия 5, с количественным выходом, в качестве исходного используют продукт Стадии 2, Пример 435.
1H ЯМР: (ДМСО-d6) δ м.д. 0.95 (с, 9Н), 1.28 (с, 9Н), 2.19-2.25 (м, 1Н), 2.43-2.47 (м, 1Н), 3.94 (м, 1Н), 4.01-4.08 (м, 2Н), 4.36 (т, J=8.5 Гц, 1Н), 5.52 (м, 1Н), 6.65 (д, J=8.0 Гц, 1Н), 6.82 (д, J=7.5 Гц, 1Н), 7.25 (д, J=7.5 Гц. 1Н), 7.67 (т, J=7.5 Гц, 1Н), 12.6 (с, 1Н).
LC/MS rt - мин (MNa+): 2.51 (522, 524) (Метод В).
Стадия 4:

К раствору продукта Стадии 3, Пример 435 (125 мг, 0.250 ммодя), Pd(PPh3)4 (14.4 мг, 0.0125 ммоля) и 3-фурилбороновой кислоты (35 мг, 0.313 ммоля) в смеси ДМФА (2 мл) и воды (0.025 мл) прибавляют Cs2CO3 (244 мг, 0.750 ммоля). Смесь нагревают при 105°С в течение 3 час в атмосфере азота. По охлаждении до комнатной температуры твердое вещество отфильтровывают, фильтрат упаривают в вакууме. Остаток очищают препаративной ВЭЖХ (градиент 30-100% В). Объединенные фракции нейтрализуют конц. раствором аммиака и упаривают. Остаток распределяют между буфером с рН 4 и хлористым метиленом. Водную фазу экстрагируют хлористым метиленом и сушат (MgSO4). Титульное соединение (90 мг, 76%) получают в виде белой пены.
1H ЯМР: (Метанол-d4) δ м.д. 1.06 (с, 9Н), 1.39 (с, 9Н), 2.36-2.42 (м, 1Н), 2.61-2.65 (м, 1Н), 4.12 (дд, J=4.0, 11 Гц, 1Н), 4.19 (д, J=11 Гц, 1Н), 4.28 (с, 1Н), 4.62 (т, J=8.5 Гц, 1Н), 5.79 (м, 1Н), 6.64 (д, J=8.0 Гц, 1Н), 6.96 (с, 1Н), 7.22 (д, J=8.0 Гц, 1Н), 7.58 (м, 1Н), 7.66 (т, J=8.0 Гц, 1Н), 8.13 (с, 1Н).
LC/MS rt - мин (MNa+): 2.53 (511) (Метод В).
Стадия 5:
Соединение 435 получают по методике, описанной в Примере 420, Стадия 6, с выходом 57%, в качестве исходного берут продукт Стадии 4, Пример 435.
1H ЯМР: (ДМСО-d6) δ м.д. 0.96 (с, 9Н), 1.02-1.05 (м, 2Н), 1.09-1.11 (м, 2Н), 1.19 (с, 9Н), 1.35-1.38 (м, 1Н), 1.71 (дд, J=5.5, 8.0 Гц, 1Н), 2.15-2.22 (м, 2Н), 2.37-2.41 (м, 1Н), 2.93 (уш м, 1Н), 4.03-4.08. (м, 3H), 4.35 (уш т, 1Н), 5.10 (д, J=10.5 Гц, 1Н), 5.24 (д, J=17 Гц, 1Н), 5.60-5.67 (м, 1Н), 5.73 (м, 1Н), 6.47 (уш с, 1Н), 6.64 (д, J=7.5 Гц, 1Н), 7.04 (с, 1Н), 7.31 (д, J=7.5 Гц, 1Н), 7.72 (т, J=7.5 Гц, 1Н), 7.77 (с, 1Н), 8.32 (с, 1Н), 8.92 (c, NH), 10.4 (c, NH).
LC/MS rt - мин (MNa+): 2.64 (723) (Метод В).
Пример 436: Получение соединения 436.
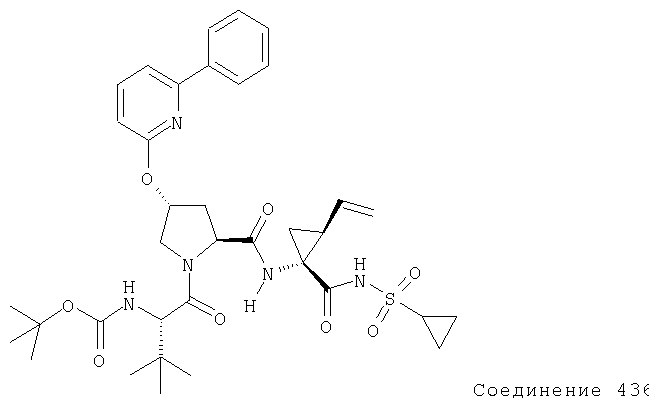
Стадия 1:

Этот продукт получают по методике, описанной в Примере 435, Стадия 4, с выходом 73%, за исключением того, что используют фенилбороновую кислоту.
1H ЯМР: (Метанол-d4) δ м.д. 1.06 (с, 9Н), 1.39 (с, 9Н), 2.39-2.45 (м, 1Н), 2.65-2.77 (м, 1Н), 4.18-4.30 (м, 3H), 4.64 (т, J=8.0 Гц, 1Н), 5.72 (м, 1Н), 6.74 (д, J=8.0 Гц, 1Н), 7.44-7.55 (м, 4Н), 7.75 (т, J=8.0 Гц. 1Н), 8.07 (д, J=7.5 Гц, 2Н).
LC/MS rt - мин (MNa+): 2.72 (521) (Метод В).
Стадия 2:
Соединение 436 получают по методике, описанной в Примере 420, Стадия 6, с выходом 66%, в качестве исходного берут продукт Стадии 1, Пример 436.
1 H ЯМР: (ДМСО-d6) δ м.д. 0.96 (с, 9Н), 1.04-1.05 (м, 2Н), 1.09 (м, 2Н), 1.30 (с, 9Н), 1.36-1.39 (м. 1Н), 1.71 (т, J=7.5 Гц, 1Н), 2.16-2,24 (м, 2Н), 2.41-2.45 (м, 1Н), 2.93 (м, 1Н), 4.06-4.09 (м, 3H), 4.38 (уш т, 1Н), 5.10 (д, J=10.5 Гц, 1Н), 5.30 (д, J=1.7 Гц, 1Н), 5.60-5.67 (м, 1Н), 5.80 (м, 1Н), 6.49 (уш с, 1Н), 6.75 (д, J=7.5 Гц, 1Н), 7.43-7.51 (м, 3H), 7.59 (д, J=7.5 Гц, 1Н), 7.81 (т, J=7.5 Гц, 1Н), 8.09 (д, J=7.0 Гц, 2Н), 8.91 (c, NH), 10.4 (c, NH).
LC/MS rt - мин (MNa+): 2.72 (521) (Метод В).
Пример 437: Получение Соединения 437.
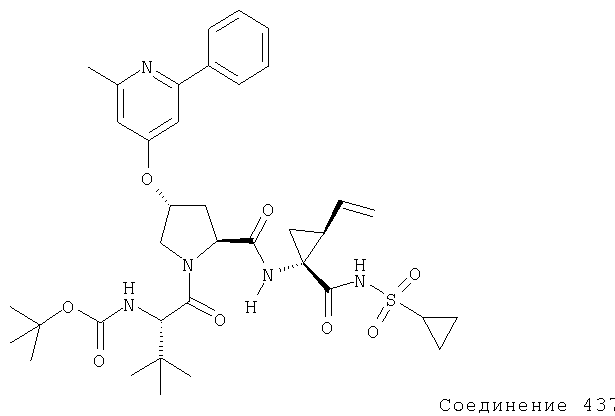
Стадия 1:
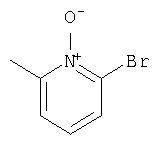
К раствору 2-бром-6-метилпиридина (7.65 г, 44.4 ммоля) в хлористом метилене (50 мл) прибавляют раствор мСРВА (77%, 12.9 г, 57.7 ммоля) в хлористом метилене (100 мл). Раствор перемешивают 18 час при комнатной температуре. Смесь нейтрализуют сухим Na2CO3 и прибавляют воду. Водную фазу экстрагируют хлористым метиленом (2х). Объединенные органические вытяжки промывают 5% Na2S2О3, 5% Na2CO3, рассолом и сушат (MgSO4). Очисткой на колонке Biotage 40 М (элюируют в градиенте гексан-этилацетат 40-70%) получают титульное соединение (5.0 г, 60%) в виде бесцветного масла, затвердевающего при стоянии.
1H ЯМР: (ДМСО-d6) δ м.д. 2.43 (с, 3H), 7.15 (т, J=8 0 Гц, 1Н), 7.50 (с, 1Н), 7.78 (с, 1Н).
LC/MS rt - мин (MH+): 0.32 (188, 190) (Метод В).
Стадия 2:

К раствору продукта Стадии 1, Пример 437 (4.5 г, 24 ммоля) в ДМФА (20 мл) порциями прибавляют POBr3 (8.2 г, 29 ммоля). Наблюдается энергичная экзотермическая реакция и образование осадка. Смесь оставляют на 2 час при комнатной температуре. Прибавляют воду и насыщенный раствор NaHCO3 до нейтрального рН. Водную фазу экстрагируют диэтиловым эфиром (2х). Объединенные органические вытяжки промывают рассолом и сушат (MgSCO4). Очисткой на колонке Biotage 40 М (элюция в градиенте гексан-диэтиловый эфир 0-10%) получают титульное соединение (1.78 г, 30%) в виде бесцветного масла.
1H ЯМР: (ДМСО-d6) δ м.д. 2.45 (с, 3H), 7.64 (с, 1Н), 7.80 (с, 1Н).
LC/MS rt - мин (МН+): 1.85 (250, 252, 254) (Метод В).
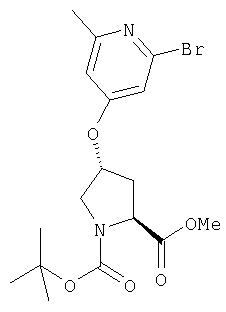
Этот продукт получают по методике, описанной в Примере 421, Стадия 2, с выходом 46%, в качестве исходного используют продукт Стадии 1, Пример 437.
1H ЯМР: (ДМСО-d6) δ м.д. 1.34, 1.38 (с, 9Н (ротамеры)), 2.20-2.27 (м, 1Н), 2.38 (с, 3H), 2.43-2.50 (м, 1Н), 3.55 (д, J=12.5 Гц, 1Н), 3.64-3.67 (м, 1Н), 3.66, 3.69 (с, 3H (ротамеры)), 4.26-4.32 (м, 1Н), 5.17 (м, 1Н), 6.93 (с, 1Н), 7.08 (с, 1Н).
LC/MS rt - мин (МН+): 2.17 (415, 417) (Метод В).
Стадия 4:
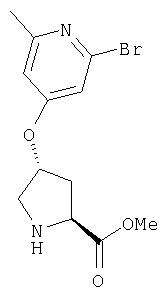
Этот продукт получают по методике, описанной в Примере 420, Стадия 3, с количественным выходом, в качестве исходного используют продукт Стадии 3, Пример 437.
Стадия 4:
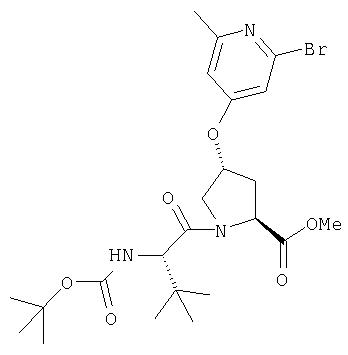
Этот продукт получают по методике, описанной в Примере 420, Стадия 3, с выходом 75%, в качестве исходного используют продукт Стадии 3, Пример 437.
1H ЯМР: (ДМСО-d6) δ м.д. 0.94 (с, 9Н), 1.27 (с, 9Н), 2.16-2.21 (м, 1Н), 2.36 (с, 3H), 2.48 (м, 1Н (наложен.)), 3.64 (с, 3H), 3.83 (д, J=10.5 Гц, 1Н), 4.06 (д, J=8.5 Гц, 1Н), 4.14 (д, J=10.5 Гц, 1Н), 4.42 (т, J=9.0 Гц, 1Н), 5.27 (м, 1Н), 6.68 (уш с, NH), 6.88 (с, 1Н), 7.03 (с, 1Н).
LC/MS rt - мин (MH+): 2.19 (528, 530) (Метод В).
Стадия 5:

Этот продукт получают по методике, описанной в Примере 420, Стадия 5, с выходом 75%, в качестве исходного используют продукт Стадии 4, Пример 437.
1H ЯМР: (ДМСО-d6) δ м.д. 0.95 (с, 9Н), 1.27 (с, 9Н), 2.15-2.19 (м, 1Н), 2.36 (с, 3H), 2.44-2.48 (м, 1Н), 3.81 (д, J=10.5 Гц, 1Н), 4.04-4.06 (м, 1Н), 4.13 (д, J=10.5 Гц, 1Н), 4.33 (т, J=8.5 Гц, 1Н), 5.26 (м, 1Н), 6.66 (уш д, J=9.0 Гц, NH), 6.88 (с, 1Н), 7.03 (с, 1Н).
LC/MS rt - мин (МН+): 2.14 (536, 538) (Метод В).
Стадия 6:
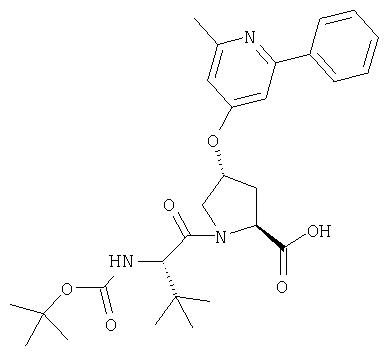
К раствору продукта Стадии 5, Пример 437 (101 мг, 0.196 ммоля), Pd(PPh3)4 (11.3 мг, 0.0098 ммоля) и фенилбороновой кислоты (34 мг, 0.275 ммоля) в ДМФА (2 мл), прибавляют 2М водн. раствор Na2CO3 (0.294 мл, 0.588 ммоля). Ампулу запаивают и нагревают при 150°С в течение 15 мин в атмосфере азота в микроволновой печи (Emrys, Personal Chemistry). По охлаждении до комнатной температуры смесь подкисляют, добавляя 1N HCl (0.5 мл). Осадок отфильтровывают, фильтрат упаривают в вакууме. Остаток очищают препаративной ВЭЖХ (градиент: 20-80% В). Объединенные фракции нейтрализуют конц. раствором аммиака и упаривают. Остаток распределяют между буфером рН 4 и этилацетатом. Водную фазу экстрагируют этилацетатом (2х) и объединенные органические вытяжки промывают рассолом и сушат (MgSO4). Получают титульное соединение (115 мг, >100%) в виде твердого вещества белого цвета.
1H ЯМР: (Метанол-d4) δ м.д. 1.06 (с, 9Н), 1.30 (с, 9Н), 2.36-2.41 (м, 1Н), 2.69 (с, 3H), 2.65-2.76 (м, 1Н), 4.01 (дд, J=3.0, 12 Гц, 1Н), 4.21 (с, 1Н), 4.44 (д, J=12 Гц, 1Н), 4.64 (дд, J=8.O, 10 Гц, 1Н), 5.46 (м, 1Н), 6.91 (с, 1Н), 7.29 (с, 1Н), 7.39 (м, 3H), 7.57 (д, J=7.5 Гц, 2Н).
LC/MS rt - мин (MH+): 1.84 (513) (Метод В).
Стадия 7:
Соединение 437 получают по методике, описанной в Примере 420, Стадия 6, с выходом 31%, в качестве исходного берут продукт Стадии 6, Пример 437.
1H ЯМР: (ДМСО-d6) δ м.д. 0.93 (с, 9Н), 0.99-1.04 (м, 4Н), 1.23 (с, 9Н), 1.34-1.3(5?)7 (м, 1Н), 1.69-1.71 (м, 1Н), 2.11-2.20 (м, 2Н), 2.39-2.43 (м, 1Н), 2.48 (с, 3H), 2.93 (м, 1Н), 3.92 (д, J=8.5 Гц, 1Н), 4.07 (д, J=9.0 Гц, 1Н), 4.11 (д, J=12 Гц, 1Н), 4.32 (т, J=7.0 Гц, 1Н), 5.10 (д, J=10.5 Гц, 1Н), 5.23 (д, J=17.5 Гц, 1Н), 5.39 (м, 1Н), 5.60-5.67 (м, 1Н), 6.58 (д, J=8.5 Гц, 1Н), 6.83 (с, 1Н), 7.27 (с, 1Н), 7.40-7.48 (м, 3H), 8.04-8.06 (м, 2Н), 8.92 (с, NH), 10.4 (с, NH).
LC/MS rt - мин (MH+): 2.10 (725) (Метод В).
Пример 438: Получение Соединения 438.
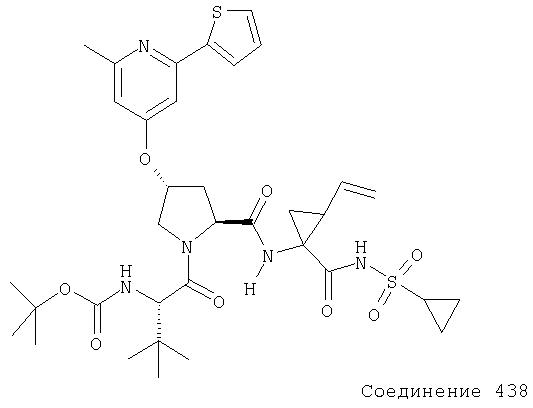
Стадия 1:

Этот продукт получают по методике, описанной в Примере 437, Стадия 6, с количественным выходом, в качестве исходного используют 2-тиофенбороновую кислоту.
1H ЯМР: (Метанол-d4) δ м.д. 1.05 (с, 9Н), 1.32 (с, 9Н), 2.32-2.38 (м, 1Н), 2.56 (с, 3H), 2.66-2.70 (м, 1Н), 3.99 (дд, J=3.0, 12 Гц, 1Н), 4.23 (с, 1Н), 4.36 (д, J=12 Гц, 1Н), 4.61 (т, J=8.5 Гц, 1Н), 5.35 (м, 1Н), 5.86 (с, 1Н), 7.16-7.18 (м, 1Н), 7.21 (с, 1Н), 7.56 (м, 1Н), 7.72 (д, J=3.0 Гц, 1Н).
LC/MS rt - мин (MH+): 1.80 (519) (Метод В).
Стадия 2:
Соединение 438 получают по методике, описанной в Примере 420, Стадия 6, с выходом 41%, в качестве исходного берут продукт Стадии 1, Пример 438.
1H ЯМР: (ДМСО-d6) δ м.д. 0.91 (с, 9Н), 0.99-1.04 (м, 4Н), 1.20 (с, 9Н), 1.35-1.37 (м, 1Н), 1.69-1.71 (м, 1Н), 2.09-2.20 (м, 2Н), 2.33 (м, 1Н), 2.42 (с, 3H), 2.93 (м, 1H), 3.92 (д, J=8.5 Гц, 1Н), 4.07-4.11 (м, 2Н), 4.29-4.32 (м, 1Н), 5.10 (д, J=10.5 Гц, 1Н), 5.23 (д, J=17.0 Гц, 1Н), 5.36 (м, 1Н), 5.60-5.67 (м, 1Н), 6.58 (д, J=8.5 Гц, 1Н), 6.75 (с, 1Н), 7.14 (с, J=4.5 Гц, 1Н), 7.28 (с, 1Н), 7.59 (д, J=5.5 Гц, 1Н), 7.80 (д, J=3.5 Гц, 1Н), 8.92 (с, NH), 10.4 (с, NH).
LC/MS rt - мин (МН+): 2.06 (731) (Метод В).
Подраздел К:
Пример 450: Получение Соединения 450.

Соединение 450 получают по методике, описанной в Примере 8, Стадия 5, за исключением того, что используют 4-хлор-6-фтор-2-трифторметилхинолин.
1H ЯМР: (CD3OD) δ м.д. 0.97-1,04 (м, 12Н), 1.17-1.24 (м, 10Н), 1.39-1.46 (м, 1H), 1.82-1.87 (м, 1Н), 2.20-2.23 (м, 1Н), 2.35-2.39 (м, 1Н), 2.55-2.65 (м, 1Н), 2.91-2.96 (м, 1Н), 4.09-4.11 (м, 1Н), 4.18-4.21 (м. 1Н), 4.56 (уш, 2Н), 5.10-5.14 (м, 1Н), 5.28-5.31 (м, 1Н), 5.60 (уш, 1Н), 5.70-5.80 (м, 1Н), 7.41 (с, 1Н), 7.65-7.68 (м, 1Н), 7.86 (с, 1Н), 8.13-8.15 (м, 1Н).
Пример 451: Получение Соединения 451.

Схема 1
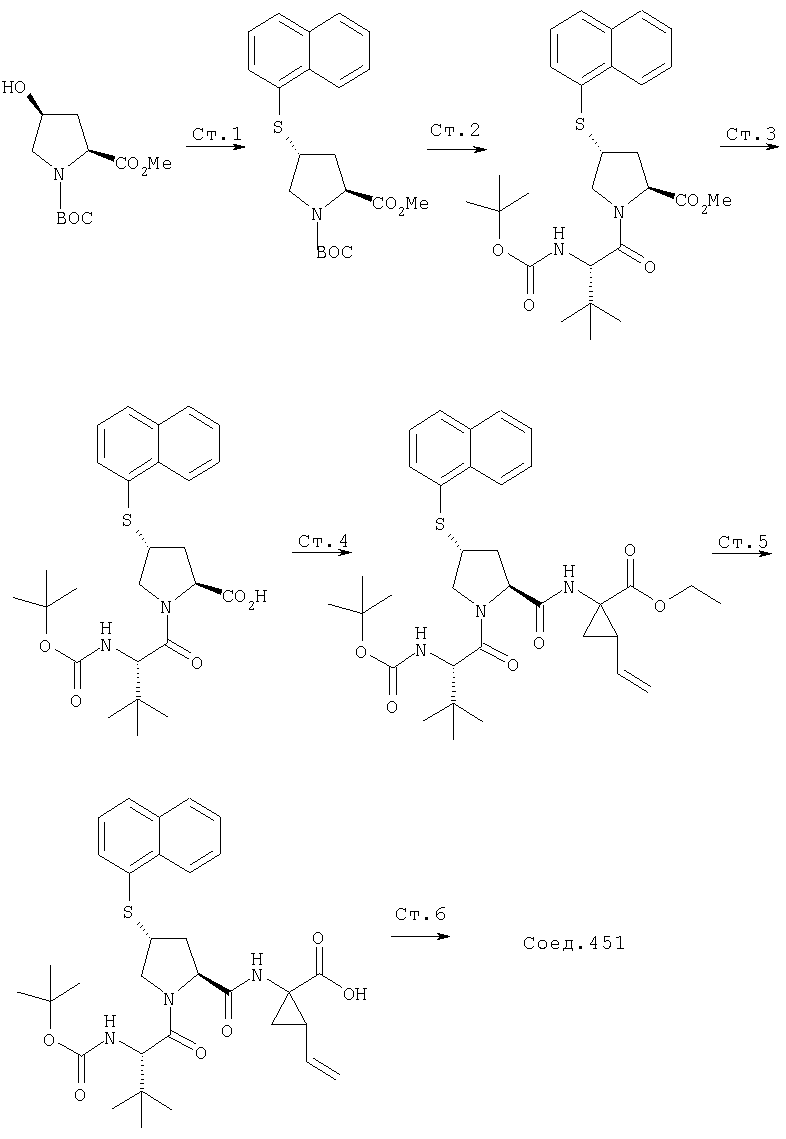
Стадия 1:
Тозилат получают, как описано в литературе (Patchett, A.A.; Witkof, В.J. Am. Chem. Soc. 1957, 185-192) и используют без дополнительной очистки.
К суспензии NaH (76 мг, 1.90 ммоля) в ДМФА (20 мл) прибавляют 1-тионафтол (0.29 мг, 1.80 ммоля) и смесь перемешивают 30 минут. Прибавляют раствор тозилата (0.61 г, 1.80 ммоля) и смесь перемешивают 12 час при 23°С. Смесь упаривают и остаток распределяют между EtOAc/Н2О. Органические вытяжки сушат (MgSO4) и упаривают. Остаток очищают хроматографией на колонке (элюпия от 5% EtOAc/смесь гексанов до 30% EtOAc/смесь гексанов), получают 261 мг (38%) продукта в виде желтого масла.
1H ЯМР: (CDCl3) δ м.д. 1.41 (с, 9Н), 1.44 (с, 9Н), 2.25-2.29 (м, 2Н), 3.69 (с, 3H), 3.35-3.42 (м, 1Н), 3.51-3.53 (м, 1Н), 3.80-3.86 (м, 2Н), 4.38-4.39 (м, 1Н), 4.46-4.48 (м, 1Н), 7.41-7.46 (м, 1Н), 7.42-7.54 (м, 1Н), 7.57-7.59 (м, 1Н), 7.58 (д, J=4 Гц, 1Н), 7.82-7.88 (м, 2Н), 8.46 (д, J=5 Гц, 1Н);
LC-MS (время удерживания: 1.93), MS m/z 388 (M++1).
Стадия 2:
Смесь 1-трет-бутилового эфира 2-метилового эфира 4-(нафталин-1-илсульфанил)-пирролидин-1,2-дикарбоновой кислоты (0.38 г, 0.98 ммоля) и 4N HCl (1.0 мл) перемешивают при 23°С в течение 2 час. Растворитель отгоняют, а остаток растворяют в CH3CN (20 мл) и прибавляют кислоту (0.37 г, 2.16 ммоля), TBTU (0.23 г, 0.98 ммоля) и DIPEA (0.37 г, 2.16 ммоля) и перемешивают 12 час. Смесь упаривают и остаток растворяют в EtOAc и промывают 1 N HCl, насыщенным раствором NaHCO3, затем сушат над MgSO4 и упаривают. Остаток используют без дополнительной очистки.
1H ЯМР: (CDCl3, 1:1 смесь ротамеров) δ м.д. 0.99 (с, 9Н), 1.02 (с, 9Н), 1.44 (с, 9Н), 1.46 (с, 9Н), 2.2-2.25 (м, 2Н), 3.70 (с, 3H), 3.82-3.86 (м, 1Н), 3.89-3.92 (м, 2Н) 4.26 (с, 1Н), 4.28 (с, 1Н), 4.70-4.75 (м, 1Н), 7.40-7.48 (м, 1Н), 7.54-7.55 (м, 1Н), 7.59-7.62 (м, 1Н), 7.72-7.74 (м, 1Н), 7.86-7.89 (м, 2Н), 8.48-8.50 (м, 1Н);
LC-MS (время удерживания: 1.59), MS m/z 523 (М+Na).
Стадия 3:
К смеси метилового эфира 1-(2-трет-бутоксикарбониламино-3,3-диметилбутирил)-4-(нафталин-1-илсульфанил)-пирролидин-2-карбоновой кислоты (Пример 451, Стадия 2) (0.49 г, 0.98 ммоля) в смеси ТГФ/Н2O (2:1) прибавляют LiOH гидрат (0.20 г, 4.9 ммоля) и смесь перемешивают 12 час. Раствор упаривают и промывают EtOAc. Водный слой подкисляют 1N HCl и экстрагируют EtOAc. Продукт обнаруживается в первой EtOAc вытяжке. Первую органическую вытяжку сушат MgSO4 и упаривают, получают 328 мг (71%) твердого вещества желто-коричневого цвета.
1H ЯМР: (ДМСО-d6, 2:1 смесь ротамеров) δ м.д. 0.88 (с, 9Н), 0.92 (с, 9Н), 1.34 (с, 9Н), 1.38 (с. 9Н), 2.18-2.25 (т, 2Н), 3.66-3.75 (м, 1Н), 3.89-4.00 (м, 2Н), 4.10-4.13 (м, 1Н), 4.25-4.32 (м, 1Н), 7.45-7.51 (м, 1Н), 7.56-7.61 (м, 2Н), 7.65-7.69 (м, 1Н), 7.88-7.91 (м, 1Н), 7.97 (д, J=4.8 Гц, 1Н), 8.27-8.35 (м, 1Н);
LC-MS (время удерживания: 1.52), MS m/z 486 (М+1).
Стадия 4:
К раствору кислоты (Пример 451, Стадия 3) (0.32 г, 0.87 ммоля) в смеси CH3CN (10 мл) и ДМФА (2 мл) прибавляют смесь диастереомеров гидрохлорида этилового эфира 1(R)-2(S) и 1(S)-2(R) 1-амино-2-винилциклопропанкарбоновой кислоты (240 мг, 0.87 ммоля), TBTU (201 мг, 0.87 ммоля) и DIPEA (0.32 мл, 0.742 ммоля) и смесь перемешивают при 23°С в течение 12 час. Смесь упаривают и остаток распределяют между EtOAc и водой. Органический слой отделяют, сушат MgSO4 и упаривают. Остаток хроматографируют с 30% EtOAc/смесь гексанов в качестве элюента, получают 240 мг (38%) твердого вещества светло-желтого цвета.
1H ЯМР: (ДМСО-d6, смесь ротамеров и диастереомеров) δ м.д. 0.87 (с, 9Н), 0.88 (с, 9Н), 0.97-1.04 (м, 3H), 1.33 (с, 9Н), 1.38 (с, 9Н), 2.11-2.20 (м, 2Н), 3.74-3.85 (м, 1Н), 3.89-3.96 (м, 2Н), 3.98-4.03 (м, 4Н), 4.00-4.09 (м, 1Н), 4.40-4.42 (м, 1Н), 5.04-5.09 (м, 1Н), 5.17-5.29 (м, 2Н), 5.50-5.70 (м, 1Н), 6.60-6.62 (м, 1Н), 6.72-6.75 (м, 1Н), 7.50-7.56 (м, 1Н), 7.56-7.72 (м, 2Н), 7.70-7.80 (м, 1Н), 7.92-8.00 (м, 1Н), 8.00-8.06 (м, 1Н), 8.29-8.40 (м, 1Н), 8.65 (с, 1Н), 8.79 (с, 1Н);
LC-MS (время удерживания: 1.59), MS m/z 623 (М+1).
Стадия 5:
Кислоту получают как описано ранее, в Примере 451, Стадия 3, с применением LiOH в смеси ТГФ/МеОН/H2O (4/2/1), за исключением того, что используют продукт Стадии 4, Пример 451.
1H ЯМР: (ДМСО-d6, смесь ротамеров и диастереомеров) δ м.д. 0.90 (с, 9Н), 1.17-1.23 (м, 2Н), 1.32-1.37 (м, 9Н), 2.10-2.12 (м, 1Н), 2.20-2.31 (м, 2Н), 3.97-4.05 (м, 2Н), 4.10-4.12 (м, 1Н), 4.32-4.40 (м, 1Н), 4.55-4.61 (м, 1Н), 4.80-4.98 (м, 2Н). 5.03-5.08 (м, 1Н), 5.10-5.20 (м, 1Н), 5.75-5.90 (м. 1Н), 6.55-6.70 (м, 1Н), 7.42-7.57 (м, 1Н), 7.60-7.64 (м, 2Н), 7.70-7.72 (м, 1Н), 7.80-7.97 (м, 1Н), 7.96-7.99 (м, 1Н), 8.20-8.50 (м, 2Н);
LC-MS (время удерживания: 1.52), MS m/z 595 (М+1).
Стадия 6:
Смесь кислоты (Пример 451, Стадия 5) (172 мг, 0.29 ммоля), метансульфонамида (110 мг, 1.16 ммоля), EDAC (110 мг, 0.58 ммоля) и DMAP (71 мг, 0.58 ммоля) растворяют в ТГФ (10 мл) и перемешивают 12 час. Прибавляют DBU (0.087 мл, 0.58 ммоля) и смесь перемешивают 48 час. Растворитель отгоняют и остаток растворяют в EtOAc и промывают водой и 1N HCl, сушат MgSO4 и упаривают. Остаток очищают препаративной тонкослойной хроматографией, получают 15 мг (8%) Соединения 451 в виде твердого вещества желто-коричневого цвета.
1H ЯМР: (ДМСО-d6, смесь ротамеров и диастереомеров) δ м.д. 0.98 (с, 9Н), 1.27-1.42 (м, 2Н), 1.46 (с, 9Н), 1.76-1.79 (м, 1Н), 1.83-1.86 (м, 1Н), 1.92-2.10 (м, 1Н), 2.16-2.25 (м, 2Н), 3.01-3.10 (м, 1Н), 3.80-3,83 (м, 1Н), 3.86-3.89 (м, 1Н), 3.98-3.99 (м, 1Н), 3.99-4.05 (м, 1Н), 4.24-4.29 (м, 1Н), 4.44-4.53 (м, 1Н), 4.86 (с, 3H), 5.08-5.15 (м, 1Н), 5.25-5.29 (м, 1Н), 5.65-5.85 (м, 1Н), 6.5-6.8 (м, 1Н), 7.48 (т, J=7.7 Гц, 1Н), 7.54 (т, J=7.1 Гц, 1Н), 7.59 (т, J=7.1 Гц, 1Н), 7.75-7.77 (м, 1Н), 7.88-7.91 (м, 2Н), 8.46 (м, J=8.25 Гц, 1Н);
LC-MS (время удерживания: 1.52), MS m/z 672 (М+1 минорный), m/z 693 (М+Na основной).
Пример 452: Получение Соединения 452.
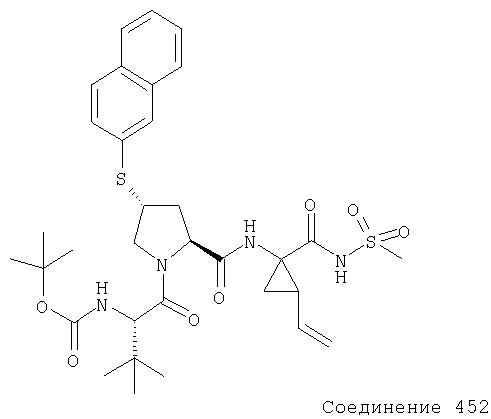
Стадия 1:

К суспензии NaH (76 мг, 1.90 ммоля) в ДМФА (20 мл) прибавляют 2-тионафтол (0.29 мг, 1.80 ммоля) и смесь перемешивают 30 минут. Прибавляют раствор тозилата (Пример 451, Стадия 1) (0.61 г, 1.79 ммоля) в ДМФА (2 мл) и смесь перемешивают 12 час при 23°С. Смесь упаривают, затем (остаток) распределяют между EtOAc/H2O. Органический слой промывают насыщенным раствором NaHCO3, сушат (MgSO4) и упаривают. Остаток хроматографируют, элюируя 30% EtOAc/смесь гексанов, получают 261 мг (38%) продукта в виде прозрачного масла.
1H ЯМР: (ДМСО-d6) δ м.д. 1.32 (с, 9Н), 2.29-2.35 (м, 2Н), 3.33-3.47 (м, 2Н), 3.66 (с, 3H), 3.71-3.81 (м, 1Н), 4.29-4.32 (с, 1Н), 7.49-7.55 (м, 3H), 7.70-7.80 (м, 1Н), 7.81-7.97 (м, 3H);
LC-MS (время удерживания: 1.54), MS m/z 387 (М+1),
Стадия 2:
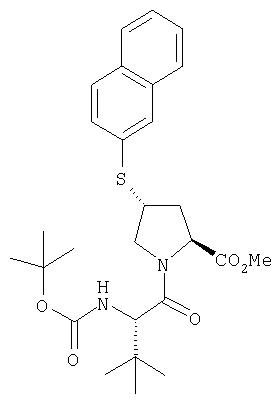
Смесь 1-трет-бутилового эфира 2-метилового эфира 4-(нафталин-2-илсульфанил)-пирролидин-1,2-дикарбоновой кислоты (310 мг, 0.80 ммоля) и 4N HCl в диоксане (1.49 мл, 2.69 ммоля) перемешивают при 23°С в течение 2 час и упаривают. Остаток растворяют в CH3CN (10 мл) и прибавляют N-Boc-трет-бутилглицин (196 мг, 0.85 ммоля), TBTU (0.27 г, 0.85 ммоля) и DIPEA (0.32 мл, 1.85 ммоля) и перемешивают в течение ночи. Смесь упаривают и остаток растворяют в EtOAc, промывают 1N HCl, насыщенным раствором NaHCO3, затем сушат и упаривают. Получают 300 мг (90%) продукта в виде желтого масла.
1H ЯМР: (Метанол-d4) δ м.д. 0.99 (с, 9Н), 1.44 (с, 9Н), 2.20-2.35 (м, 2Н), 3.75 (с, 3H), 3.92-4.08 (м, 2Н), 4.26 (д, J=9.4 Гц, 1Н), 4.57 (т, J=9.5 Гц, 1Н), 6.46 (д, J=9.5 Гц, 1Н), 7.48-7.60 (м, 3H), 7.83-7.90 (м, 3H), 8.02 (с, 1Н)
LC-MS (время удерживания: 1.98), MS m/z 523 (M+Na).
Стадия 3:
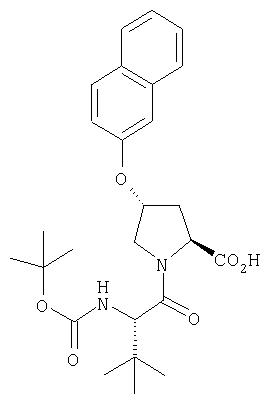
Раствор 1-трет-бутилового эфира 2-метилового эфира 4-(нафталин-2-илсульфанил)-пирролидин-1,2-дикарбоновой кислоты (0.48 г, 0.96 ммоля) растворяют в МеОН (20 мл) и перемешивают с LiOH (0.2 г, 4.8 ммоля) в течение 12 час, раствор упаривают и подкисляют и экстрагируют EtOAc. Органические вытяжки сушат MgSO4 и упаривают, получают 418 мг (91%) твердого вещества желтого цвета.
1H ЯМР: (ДМСО-d6, 1:2 смесь ротамеров) δ м.д. 0.86, 0.93 (с, 9Н) (1:2 смесь ротамеров), 1.35, 1.38 (с, 9Н) (1:2 смесь ротамеров), 2.01-2.18, 2.25-2.35 (м, 2Н), 3.25-3.40 (м, 2Н), 3.70-3.80 (м, 1Н), 4.00-4.20 (м, J=9.4 Гц, 2Н), 4.30-4.40 (с, 1Н), 5.61-5.70, 6.42-6.50 (м, 1Н) (1:2 смесь ротамеров), 7.50-7.54 (м, 3H), 7.87-7.89 (м, 3H), 7.98 (с, 1Н);
LC-MS (время удерживания: 1.93), MS m/z 487 (M+1).
Стадия 4:
Схема 1
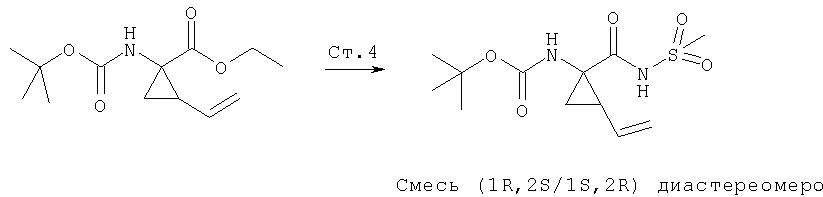
К раствору этилового эфира 1-трет-бутоксикарбониламино-2-винилциклопропанкарбоновой кислоты (4.3 г, 17.8 ммоля) в МеОН (50 мл) прибавляют LiOH (0.84 г, 20.0 ммоля) и воду (5 мл) и смесь перемешивают 12 час. Растворитель отгоняют, а остаток подкисляют и экстрагируют EtOAc. Органические вытяжки сушат MgSCO4, фильтруют и упаривают, получают 2.1 г кислоты (52%) в виде желтого масла. Кислоту (2.1 г, 9.25 ммоля) растворяют в ТГФ и прибавляют CDI (7.25 г, 13.8 ммоля) и кипятят 3 час, затем охлаждают до 23°С. Прибавляют метансульфонамид (1.76 г, 18.5 ммоля), а затем DBU (2.77 мл, 18.5 ммоля) и перемешивают 72 час при 23°С. Смесь упаривают и остаток подкисляют до рН 4 (1N HCl) и экстрагируют EtOAc. Органические вытяжки сушат MgSO4 и упаривают, получают 1.99 г (71%) желтого масла, затвердевающего при стоянии.
1H ЯМР: (ДМСО-d6) δ м.д. 1.16-1.23 (м, 1Н), 1.43 (с, 9Н), 1.65-1.75 (м, 1Н), 2.15-2.25 (м, 2Н), 3.16 (с, 3H), 5.08 (д, J=9.9 Гц, 1Н), 5.22 (д, J=17.1 Гц, 1Н), 5.40-5.52 (м, 1Н);
LC-MS (время удерживания: 1.14), MS m/z 304 (M+1).
Стадия 5:
Раствор продукта Стадии 4, Пример 452 в диоксан/4N HCl (2 мл) перемешивают 2 л, затем упаривают. Остаток растворяют в СН3CN (5 мл) и прибавляют к смеси кислоты (Пример 452, Стадия 3) (120 мг, 0.25 ммоля), TBTU (58 мг, 0.25 ммоля) и DIPEA (0.06 мл, 0.35 ммоля), смесь перемешивают 12 час. Растворитель отгоняют и остаток растворяют в EtOAc и промывают 1N HCl, насыщенным раствором NaHCO3, сушат MgSO4 и упаривают. Остаток очищают препаративной ТСХ (Analtech 20×40 см, 1000 мкМ SiO2), получают 128 мг (70%) Соединения 452 в виде твердого вещества желто-коричневого цвета.
1H ЯМР: (ДМСО-d6, смесь диастереомеров) δ м.д. 0.97, 0.99 (с, 9Н), 1.23-1.43 (м, 2Н), 1.45 (с, 9Н), 1.82-1.83 (м, 1Н), 2.00-2.51 (м, 2Н), 2.90-2.99 (м, 1Н), 3.33 (с, 3H), 3.90-3.99 (м, 1Н), 4.01-4.20 (м, 2Н), 4.25-4.30 (м, 1Н), 4.45-4.55 (м, 1Н), 4.95-5.10 (м, 1Н), 5.12-5.25 (м, 1Н), 5.71-5.85 (м, 1Н), 6.4-6.8 (уш м, 1Н), 7.46-7.53 (м, 3H), 7.80-7.86 (м, 3H), 7.98-7.99 (м, 1Н);
LC-MS (время удерживания: 1.95), MS m/z 672 (M+1).
Пример 453: Получение Соединения 453.

Стадия 1:

Смесь продукта Стадии 3, Пример 452 (110 г, 0.23 ммоля) в DCM (20 мл), 3-хлорбензойной кислоты (121.6 мг, 0.57 ммоля, 85% перкислота (надкислота)), КНРО4 (0.13 г, 0.94 ммоля) и К2НРО4 (0.18 г, 1.05 ммоля) перемешивают при 23°С в течение 12 час. К раствору прибавляют DCM, промывают водой, насыщенным раствором NaHCO3, сушат MgSO4 и упаривают, получают продукт (110 мг, 92%) в виде прозрачного масла.
1H ЯМР: (ДМСО-d6) δ м.д. 0.91 (с, 9Н), 1.48 (с, 9Н), 2.23-2.28 (м, 1H), 2.65-2.80 (м, 1Н), 3.88-3.90 (м, 1Н), 4.12 (т, J=8.0 Гц, 1Н), 4.20 (д, J=9.5 Гц, 1Н), 4.27 (д, J=9.9 Гц, 1Н), 6.76 (д, J=9.3 Гц, 1Н), 7.70-7.80 (м, 2Н), 7.88-7.95 (м, 1Н), 8.11 (д, J=8.1 Гц, 1Н), 8.17 (д, J=8.6 Гц, 1Н), 8.26 (д, J=8.1 Гц, 1Н), 8.71 (с, 1Н);
LC-MS (время удерживания: 1.72), MS m/z 519 (M+1).
Стадия 2:
К смеси продукта Стадии 1, Пример 453 (110 мг, 0.212 ммоля), амина (Пример 452, Стадия 5а) (0.65 мг, 0.212 ммоля) прибавляют TBTU (48.5 мг, 0.21 ммоля) и DIPEA (60.8 мл, 0.35 ммоля) и перемешивают 12 час при 23°С. Растворитель отгоняют, остаток растворяют в EtOAc и промывают 1N HCl, насыщенным раствором NaHCO3, сушат MgSO4 и упаривают. Остаток очищают препаративной ТСХ (элюируют 10% MeOH/CH2Cl2, получают 25 мг (17%) Соединения 453 в виде твердого вещества белого цвета.
1H ЯМР: (ДМСО-d6, смесь диастереомеров) δ м.д. 0.93, 0.96 (с, 9Н), 1.38-1.45 (м, 2Н), 1.53, 1.55 (м, 9Н), 1.76-1.85 (м, 1Н), 2.21-2.40 (м, 2Н), 3.13-3.15 (м, 2Н), 3.34 (с, 3H), 3.91-3.99 (м, 1Н), 4.15 (м, 1Н), 4.25 (м, 1Н), 4.30 (м, 1Н), 5.09-5.12 (м, 1Н), 5.26-5.31 (м, 1Н), 5.72-5.76 (м, 1Н), 6.65-6.68 (м, 1Н), 6.71-6.76 (м, 1Н), 7.67-7.76 (м, 2Н), 7.91-7.95 (м, 1Н), 8.03 (д, J=7.8 Гц, 1Н), 8.13 (д, J=8.5 Гц, 1Н), 8.20 (д, J=7.6 Гц, 1Н), 8.7 (с, 1Н);
LC-MS (время удерживания: 1.76), MS m/z 705 (M+1).
Пример 454: Получение Соединения 454.

Стадия 1:
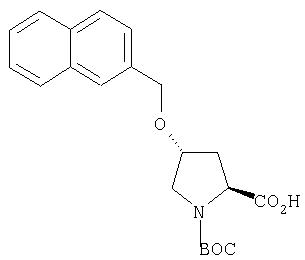
К суспензии гидрида натрия (0.91 г, 22.7 ммоля) в ТГФ (50 мл) прибавляют N-ВОС-транс-4(R)-гидрокси-L-пролин (2.5 г, 10.8 ммоля) и смесь перемешивают при 23°С в течение 1 час. Прибавляют 2-хлорметилнафталин (1.9 г, 10.8 ммоля) и смесь перемешивают 12 час при комнатной температуре. Растворитель отгоняют и остаток выливают в воду и промывают смесью гексанов. Водный слой подкисляют (1N HCl) и экстрагируют EtOAc. EtOAc слой отделяют, сушат (MgSO4) и упаривают, получают светло-желтый остаток. Масло очищают флеш-хроматографией, элюент 1:1 смесь EtOAc/гексаны с добавкой 1% уксусной кислоты. Получают 1.56 г (39%) нужного продукта в виде вязкого масла.
1H ЯМР: (ДМСО-d6, 3:1 смесь ротамеров) δ м.д. 1.35, 1.37 (с, 9Н, основной и минорный, соответственно), 1.92-2.02, 2.15-2.20 (м, 2Н, основной и минорный, соответственно), 2.35-2.50 (м, 2Н), 3.41-3.49 (м, 2Н), 4.12-4.16, 4.20-4.21 (м, 2Н), 4.65-4.68 (м, 2Н), 7.46-7.52 (м, 3H), 7.74-7.91 (м, 4Н), (кислый ОН не виден);
LC-MS (время удерживания: 1.44, колонка YMC ODS-A С 18 S7 3.0×50 мм, градиент 10%/MeOH/H2O 0.1% ТФК-90% МеОН/H2O 0.1% ТФК), MS m/z 394 (M++1+Na).
Стадия 2:

К раствору HCl соли 1:1 смеси диастереомеров (1R,2S/1S,2R, где карбоксигруппа находится син относительно винильного фрагмента) трет-бутилового эфира 2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-4-(нафталин-2-илметокси)-пирролидин-карбоновой кислоты (0.54 г, 1.3 ммоля) [полученного при перемешивании N-Boc амина с HCl (4N) в диоксане в течение 1 час с последующей отгонкой растворителя в вакууме] в СН3CN (50 мл ) прибавляют Boc-4(R)-(2-метилнафтил)пролин (0.5 г, 1.3 ммоля), TBTU (0.45 г, 1.4 ммоля), а затем DIPEA (0.78 мл, 4.5 ммоля). Смесь перемешивают 12 час и упаривают. Остаток растворяют в EtOAc/H2O и промывают насыщенным раствором NaHCO3, насыщенным раствором NaCl, сушат (MgSO4) и упаривают, получают вязкое желтое масло (0.6 г, 91%) - продукт в виде смеси диастереомеров.
1H ЯМР: (ДМСО-d6) δ м.д. 1.08-1.22 (м, 7Н), 1.23-1.39 (м, 9Н), 2.02-2.18 (м, 1Н), 2.25-2.35 (м, 1Н), 3.33-3.53 (м, 2Н), 3.90-4.14 (м, 4Н), 4.45-4.70 (м, 2Н), 5.07-5.11 (м, 1Н), 5.24-5.30 (м. 1Н), 5.58-5.63 (м, 1Н), 7.43-7.51 (м, 4Н), 7.84-7.96 (м, 3H);
MS m/z 531 (M++1+Na).
Стадия 3:

Раствор продукта Стадии 2, Пример 454 (600 мг, 1.18 ммоля) перемешивают с HCl (4N, 3 мл, 11.8 ммоля) в диоксане 1 час, затем растворитель отгоняют в вакууме. Остаток растворяют в CH3CN (10 мл), прибавляют Boc-L-трет-Bu-Gly (0.42 г, 1.38 ммоля), TBTU (0.27 г, 1.18 ммоля), а затем DIPEA (0.71 мл, 4.1 ммоля). Смесь перемешивают 12 час и упаривают. Остаток растворяют в EtOAc/Н2О и промывают 1N HCl, насыщенным раствором NaHCO3, насыщенным раствором NaCl, сушат (MgSO4) и упаривают, получают вязкое желтое масло Продукт очищают флеш-хроматографией, используя градиентную элюцию 5% EtOAc/Гексаны, 10% EtOAc/Гексаны, 30% EtOAc/Гексаны как конечный элюент, получают продукт в виде вязкого масла (0.243 г, 33%), представляющий собой смесь диастереомеров и ротамеров.
1H ЯМР: (ДМСО-d6) δ м.д. 0.83-1.00 (м, 10H), 1.34 (с, 9Н), 1.58-1.59, 1.65-1.67 (м, 2Н), 1,95-1.99, 2.04-2.06, 2.10-2.19, 2.24-2.56 (м, 2Н), 3.97-4.04 (м, 3H), 4.08-4.17 (м, 3H), 4.29-4.31 (м, 2Н), 4.59-4.72 (м, 3H), 5.06-5.10 (м, 1Н), 5.18-5.30 (м, 1Н), 5.60-5.63 (м, 1Н), 6.59-6.65, 6.70-6.74 (м, 1Н), 7.43-7.51 (м, 4Н), 7.84-7.96 (м,3H), 8.66, 8.76 (с, 1Н);
MS m/z 531 (М++1+Na).
Стадия 4:
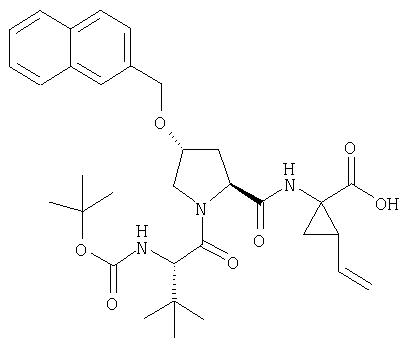
К суспензии продукта Стадии 3, Пример 454 (240 мг, 0.39 ммоля) в смеси ТГФ (15 мл) и H2O (2 мл) прибавляют LiOH (82 мг, 1.95 ммоля). Реакционную смесь перемешивают 12 час, затем упаривают в вакууме до тех пор, пока не останется только водный слой. Полученный водный остаток подкисляют до рН 3.0, добавляя 1.0 N водный раствор HCl, и экстрагируют EtOAc (2×80 мл). Объединенные органические вытяжки сушат (MgSO4), фильтруют и упаривают в вакууме, получают продукт в виде твердого вещества желто-коричневого цвета (200 мг, 0.33 ммоля, 85%):
1H ЯМР: (ДМСО-d6) δ м.д. 0.86. 0.94 (с, 9Н минорный и основной, соответственно), 1.23-1.42 (м, 2Н), 1.34 (с, 9Н), 1.8-2.1 (м, 2Н), 2.18-2.30 (м, 1Н), 3.59-3.73 (м, 3H), 4.0-4.09 (м, 1Н), 4.18-4.34 (м, 3H), 4.56-4.62 (м, 1Н), 4.66-4.67 (м, 1Н), 4.82-4.92 (м, 1Н), 5.0-5.20 (м, 1Н), 5.91-6.08 (м, 1Н), 6.5-6.7 (м, 1Н), 7.45-7.59 (м, 3H), 7.82-7.97 (м, 4Н), 8.2-8.3, 8.3-8.4 (с, 1Н);
LC-MS (время удерживания: 1.50), MS m/z 593 (M++1).
Стадия 5:
К раствору продукта Стадии 4, Пример 454 (190 мг, 0.32 ммоля) и EDAC (122 мг, 0.64 ммоля) и 4-DMAP (78 мг, 0.64 ммоля) в ТГФ (20 мл) прибавляют продажный метансульфонамид (122 мг, 1.28 ммоля). Полученный раствор перемешивают в течение 2 дней, затем прибавляют DBU (95 мкл, 0.64 ммоля). Реакционную смесь перемешивают в течение 24 час, затем упаривают. Остаток распределяют между EtOAc (80 мл) и водой и промывают 1 N HCl, водн. NHCO3 (2×30 мл), сушат (MgSO4) и очищают препаративной ВЭЖХ (65-90% МеОН/Вода/0.1% ТФК), получают 56 мг смеси продукта, в котором отсутствует ВОС группа. Продукт дополнительно очищают препаративной ВЭЖХ (элюция 10% MeOH/CH2Cl2 на пластинах 20×40 см от Analtech), получают Соединение 454 в виде желто-коричневого твердого вещества (12 мг, 6%).
1H ЯМР: (MeOD-d4, 50:50 смесь P1 диастереомеров) δ м.д. 0.88-0.99 (м, 2Н), 1.01, 1.02 (с, 9Н минорный и основной диастереомеры, соответственно), 1.23-1.42 (м, 2Н), 1.38 (с, 9Н), 1.72-1.79 (м, 1Н), 1.86-1.88 (м, 1Н), 2.00-2.10 (м, 2Н), 2.10-2.23 (м, 1Н), 2.3-2.5 (м, 1Н), 3.12, 3.17 (с, 3H), 3.72-3.79 (м, 1Н), 4.26-4.41 (м, 3H), 4.72 (д, J=8.2 Гц, 1Н), 4.76 (д, J=8.2 Гц, 1Н), 5.09-5.12 (т, J=9.3 Гц, 1Н), 5.28 (дд, J=3.5, 17.6 Гц, 1Н), 5.7-5.8 (м, 1Н), 6.55-6.80 (м, 1Н), 7.45-7.47 (м, 3H), 7.79-7.83 (м, 4Н);
LC-MS (время удерживания: 1.48), MS m/z 670 (M++1).
Пример 470: Получение Соединения 470.

Схема 1
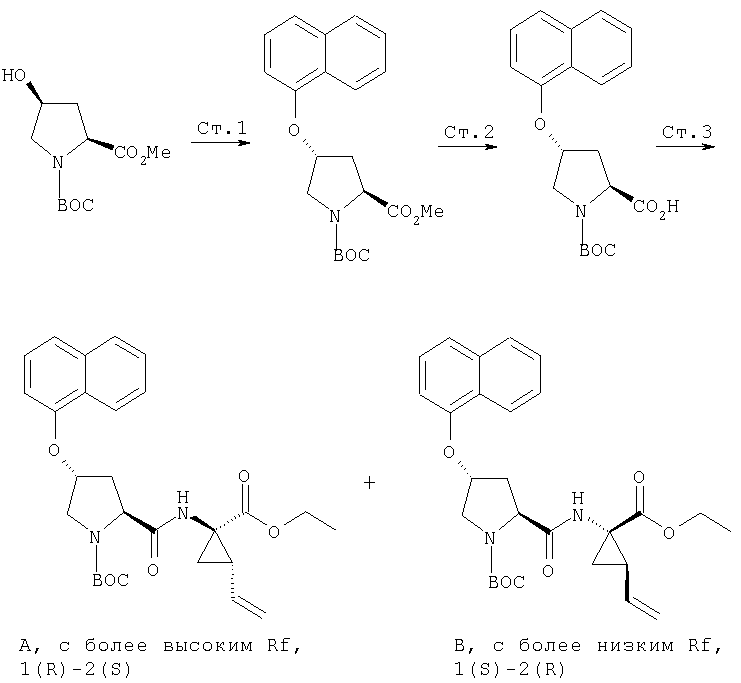
К раствору N-Boc-(4S)-(цис)-гидроксипролин-ОМе (200 мг, 0.82 ммоля), трифенилфосфина (320 мг, 1.22 ммоля) и 1-нафтола (176 мг, 1.22 ммоля) в 2.5 мл в течение 10 минут по каплям прибавляют раствор диэтилдиазодикарбоксилата (190 мкл, 1.22 ммоля) в 1.0 мл ТГФ. Перемешивают в течение 5.5 дней, реакционную смесь упаривают в вакууме. Сырой продукт - желтое масло - хроматографируют на пластине 20×40 см для препаративной тонкослойной хроматографии (Analtech SiO2), элюируя 6-1 смесь гексанов-этилацетат, получают нужный продукт в виде бледно-желтого масла (150 мг, 33%).
1H ЯМР (CDCl3, 500 МГц) δ м.д. 1.44 (с, 9Н), 2.33 (м, 1Н), 2.72 (м, 1Н), 3.77 и 3.38 (2с, 3H, ротамеры), 3.88 (дд, J=4.3, 12.4 Гц, 1Н,), 3.97 (уш д, 1Н), 4.53 и 4.62 (2т, J=7.8 Гц, ротамеры, 1Н,), 5.10 (уш д, 1Н), 6.76 (т, J=9.5 Гц, 1Н), 7.37 (м, 1Н), 7.46 (м, 3H), 7.80 (д, 1Н, J=7.7 Гц), 8.18 (м, 1Н);
LC-MS (время удерживания: 1.86; MS m/z 394 (М+Na)+
Стадия 2:
К раствору Boc-(4R)-нафталин-1-оксо)-Pro-OEt (150 мг, 0.40 ммоля) в 1.5 мл ТГФ и 0.5 мл воды при перемешивании прибавляют гидроксид лития (10 мг). Раствор перемешивают 21 часа при комнатной температуре, а затем прибавляют 0.5N NaHCO3. Основной (щелочной) раствор экстрагируют этилацетатом, а затем водный слой подкисляют до рН 2, прибавляя по каплям конц. HCl. Этот подкисленный раствор снова экстрагируют этилацетатом. Эти вторые EtOAc вытяжки сушат сульфатом магния, фильтруют и упаривают в вакууме, получают Boc-(4R)-нафталин-1-оксо)-Pro-ОН в виде бледно-розового кристаллического вещества (147 мг, 100%).
1H ЯМР (CDCl3, 500 МГц) δ м.д. 1.47 и 1.48 (2с, 9Н, ротамеры), 2.40 и 2.52 (2м, 1Н), 2.68 и 2.78 (2м, 1Н), 3.78-4.07 (м, 2Н), 4.57 и 4.69 (2т, J=7.6 и 8.0 Гц, ротамеры, 1Н), 5.12 (уш д, 1Н), 6.77 (дд, J=7.6. 21.2 Гц, 1Н), 7.37 (м, 1Н), 7.46 (м, 3H), 7.81 (т, J=5.8 Гц, 1Н), 8.19 (м, 1Н);
LC-MS (время удерживания: 1.79; MS m/z 358 (M+H)+
Стадия 3:
К раствору Boc-((4R)-нафталин-1-оксо)-Pro-ОН (147 мг, 0.41 ммоля) и гидрохлорида рацемического этилового эфира (1R/2S)/(1S/2R)-1-амино-2-винилциклопропанкарбоновой кислоты (79 мг, 0.41 ммоля) в 2.8 мл хлористого метилена прибавляют DIPEA (250 мкл, 1.44 ммоля) и TBTU (158 мг, 0.49 ммоля). Полученный раствор перемешивают под азотом в течение 20 часов, а затем прибавляют 40 мл хлористого метилена. Органический слой промывают водой, 1N NaHCO3, 1N HCl, водой и рассолом. Затем раствор сушат сульфатом натрия и упаривают в вакууме. Очистка препаративной ТСХ дает два отдельных диастереомера, диастереомер А с более высоким Rf (P2[Boc(4R)-(нафталин-1-оксо)пролин]-P1(1R,2S Vinyl Acca)-OEt, 78 мг, 38%) и диастереомер В с более низким Rf (P2[Boc(4R)-(нафталин-1-оксо)пролин]-P1(1S,2R Vinyl Acca)-OEt, 91 мг, 45%) в виде твердых веществ белого цвета:
Диастереомер A: P2[Boc(4R)-(нафталин-1-оксо)пролин]-P1(1R,2S Vinyl Acca)-OEt:1H ЯМР (CDCl3, 500 МГц) δ м.д. 1.24 (т, 3H), 1.43 (с, 9Н), 1.52 (м, 1Н), 1.84 (м, 1Н), 2.02 (м, 1Н), 2.14 (м, 1Н), 2.81 (м, 1Н), 3.88 (м, 2Н), 4.11 (кв, J=7.15, 1Н), 4.19 (м, 1Н), 4.54 (м, 1Н), 5.15 (м, 1Н), 5.31 (дд, J=17, 0.8 Гц, 1Н), 5.77 (м, 1Н), 6.83 (м, 1Н), 7.36 (т, J=7.8 Гц, 1Н), 7.46 (м, 3H), 7.78 (д, J=7.6 Гц, 1Н), 8.14 (д, J=8.15 Гц, 1Н);
LC-MS В (время удерживания: 1.85; MS m/z 495 (M+H)+
Диастереомер В: Р2[Вос(4R)-(нафталин-1-оксо)пролин]-P1(1S,2R Vinyl Асса)-OEt:1H ЯМР (CDCl3, 500 МГц) δ м.д. 1.24 (т, 3H), 1.42 (с, 9Н), 1.85 (м, 1Н), 2.15 (кв, J=8.9 Гц, 1Н), 2.40 (м, 1Н), 2.78 (м, 1Н), 3.78 (м, 1Н), 4.12 (м, 2Н), 4.52 (м, 1Н), 5.15 (м, 1Н), 5.31 (м, 1Н), 5.79 (м, 1Н), 6.80 (м, 1Н), 7.35 (т, J=7.6 Гц, 1Н), 7.46 (м, 3H), 7.78 (д, J=7.6 Гц, 1Н), 8.14 (д, J=8.10 Гц, 1Н).
LC-MS В (время удерживания: 1.85; MS m/z 495 (M+H)+

К P2[Boc(4R)-(нафталин-1-оксо)пролин]-P1(1R,2S Vinyl Acca)-OEt (А, с более высоким Rf) (78 мг, 0.16 ммоля) прибавляют 4N HCl в диоксане (2.0 мл) и раствор оставляют перемешиваться в течение 30 минут. Упариванием в вакууме получают HCl соль of P2[(4R)-(нафталин-1-оксо)пролин]-P1(1R,2S Vinyl Acca)-OEt в виде желтого масла, которое используют сразу же без дополнительной очистки.
К раствору ВОС L-tBuGly (73 мг, 0.32 ммоля) и HCl соли P2[Boc(4R)-(нафталин-1-оксо)пролин]-P1(1R,2S Vinyl Acca)-OEt (0,16 ммоля) в 11 мл ацетонитрила прибавляют DIPEA (140 мкл, 0.79 ммоля) и HATU (132 мг, 0.35 ммоля). Полученный раствор перемешивают под азотом в течение 17 час, а затем прибавляют 100 мл этилацетата. Органический слой промывают водой, 1N NaHCO3, 1N HCl, водой и рассолом. Затем раствор сушат сульфатом натрия и упаривают в вакууме, получают титульное соединение в виде бледно-желтой маслянистой пленки (92 мг, 96%).
1H ЯМР (CDCl3, 500 МГц) δ м.д. 1.06 (с, 9Н), 1.22 (т, J=7.1, 3H), 1.38 (с, 9Н), 1.41 (м, 1Н), 1.82 (м, 1Н), 2.13 (м, 1Н), 2.42 (м, 1Н), 2.79 (м, 1Н), 3.92-4.2 (м, 1Н), 4.12 (кв, J=6.6 Гц, 2Н), 4.38 (уш т, 1Н), 5.12 (д, 1Н, J=10.3 Гц), 5.2-5.39 (м, 3H), 5.75 (м, 1Н), 6.82 (д, J=7.5 Гц, 1Н), 7.34-7.46 (м, 4Н), 7.59 (уш, 1Н, NH), 7.76 (д, J=7.9 Гц, 1Н), 8.13 (д, 1Н, J=8.3 Гц);
LC-MS С (время удерживания: 2.82; MS m/z 608 (M+H)+
Стадия 5:
К раствору продукта Стадии 4, Пример 470 (92 мг, 0.15 ммоля) в 750 мл тетрагидрофурана и 250 мл воды прибавляют гидроксид лития (4 мг). Полученный раствор перемешивают 28.5 часов, обрабатывают как обычно, а затем снова повторяют ту же операцию, за исключением того, что прибавляют двойное количество гидроксида лития (8 мг). Через 24 часа к реакционной смеси прибавляют этилацетат и промывают водой. Органический слой сушат сульфатом натрия и упаривают в вакууме. Полученное полужидкое вещество очищают флеш-хроматографией, элюируют 3:1 смесь гексанов: этилацетат, получают BocNH-P3(t-BuGly)-P2[(Boc(4R)-(нафталин-1-оксо)пролин)]-P1(1R,2S Vinyl Acca)-ОН в виде светлого полужидкого вещества (30 мг, 34%).
l H ЯМР(CD3OD, 500 МГц) δ 1.04 (с, 9Н), 1.24 (т, 1Н, J=3.9 Гц), 1.32 (с, 9Н), 1.66 (м, 1Н), 2.07 (м, 1Н), 2.40 (м, 1Н), 2.71 (м, 1Н), 4.04-4.07 (м, 1Н), 4.28 (м, 1Н), 4.42 (м, 1Н), 4.55 (м, 1Н), 5.02 (м, 1Н), 5.18-5.29 (м, 2Н), 5.90 (м, 1Н), 6.54 (м, 1Н), 6.92 (м, 1Н), 4.26 (м, 4Н), 7.77 (м, 1Н), 8.15 (м, 1Н);
LC-MS С (время удерживания: 2.65; MS m/z 580 (M+H)+
Стадия 6:
К раствору BocNH-Р3((трет-BuGly)-P2[(Boc(4R)-(нафталин-1-оксо)пролин)]-P1(1R,2S Vinyl Acca)-ОН (Пример 470, Стадия 5) (65 мг, 0.11 ммоля) в 3.7 мл тетрагидрофурана прибавляют 1,1'-карбонилдиимидазол (22 мг, 0.135 ммоля). Полученную смесь кипятят 30 минут, а затем охлаждают до комнатной температуры. В этот момент прибавляют метансульфонамид (27 мг, 0.28 ммоля) и DBU (34 мкл, 0.224 ммоля). Реакционную смесь перемешивают в течение 2 дней, а затем снова прибавляют DBU (10 мкл) и метансульфонамид (9 мг). Через 24 час к реакционной смеси прибавляют 50 мл этилацетата и промывают 50 мл 0.25N HCl и 50 мл рассола. Раствор сушат сульфатом натрия и упаривают в вакууме. Сырой продукт очищают препаративной ТСХ (3:2 этилацетат: смесь гексанов), получают Соединение 470 (21 мг, 28%) в виде твердой пленки.
1H ЯМР (CD3OD, 500 МГц) δ 1.04 (с, 9Н), 1.36 (с, 9Н), 1.88 (т, 1Н), 2.18 (м, 1Н), 2.31 (м, 1Н), 2.63 (м, 1Н), 3.11 (уш с, 3H), 4.076 (м, 1Н), 4.30 (уш д, 1Н), 4.41 (уш д, 1Н), 4.52 (кажущийся т, 1Н), 5.07 (м, 1Н), 5.24-5.30 (м, 2Н), 5.80 (м, 1Н), 6.92 (д, J=7.45 Гц, 1Н), 7.35-7.46 (м, 4Н), 7.76 (д, J=8.1 Гц, 1Н), 8.13 (д, J=8.3 Гц, 1Н);
LC-MS С (время удерживания: 2.57; MS m/z 657 (M+H)+
Пример 471: Получение Соединения 471.

Схема 2
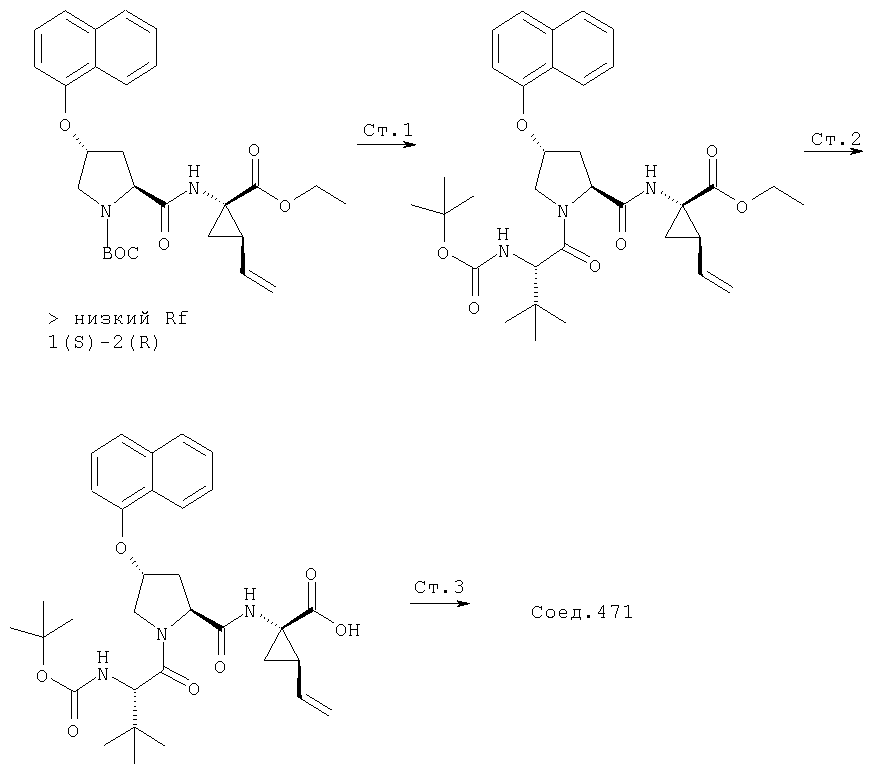
Стадия 1:
К P2[Boc(4R)-(нафталин-1-оксо)пролин]-P1(1S,2R Vinyl Acca)-OEt (Пример 470, Стадия 3, диастереомер с более низким Rf) (91 мг, 0.18 ммоля) прибавляют 4N HCl в диоксане (2.0 мл) и раствор оставляют перемешиваться в течение 30 минут. Упариванием в вакууме получают HCl соль of P2[(4R)-(нафталин-1-оксо)пролин]-P1(1S,2R Vinyl Acca)-OEt в виде желтого масла, которое используют сразу же без дополнительной очистки.
К раствору N-ВОС L-трет-лейцин-ОН (85 мг, 0.32 ммоля) и HCl соли P2[Boc(4R)-(нафталин-1-оксо)пролин]-P1(1S,2R Vinyl Acca)-OEt (продукта, полученного в вышеуказанной реакции) (0.18 ммоля) в 13 мл ацетонитрила прибавляют DIPEA (160 мкл, 0.79 ммоля) и HATU (154 мг, 0.41 ммоля). Полученный раствор перемешивают под азотом в течение 17 час, а затем прибавляют 100 мл этилацетата. Органический слой промывают водой, 1N NaHCO3, 1N HCl, водой и рассолом. Затем раствор сушат сульфатом натрия и упаривают в вакууме, получают титульное соединение в виде светлой пленки (53 мг, 47%).
1H ЯМР (CDCl3, 500 МГц) δ м.д. 1.02 (с, 9Н), 1.22 (т, J=7.0 Гц, 3H), 1.39 (с, 9Н), 1.47 (м, 1H), 1.88 (дд, J=8.0, 5.5 Гц, 1Н), 2.07 (м, 1Н), 2.42 (м, 1Н). 2.80 (дт, J=13.8, 6.0 Гц, 1Н), 3.96 (м, 1Н), 4.14 (м, 2Н), 4.34 (м, 2Н), 4.77 (т, J=7.2 Гц, 1Н). 5.09-5.33 (м, 3H), 5.72 (м, 1H), 6.82 (д, J=7.6 Гц, 1H), 7.34-7.50 (м, 4Н), 7.77 (д, J=8.0 Гц, 1H), 8.15 (д, J=8.25 Гц, 1Н);
LC-MS С (время удерживания: 2.81; MS m/z 608 (М+Н)+
Стадия 2:
Этот продукт получают по методике, описанной в Примере 470, Стадия 5 (5 мг, 10%), за исключением того, что используют продукт Стадии 1, Пример 471.
1H ЯМР (CD3OD, 500 МГц) δ 0.99 (с, 9Н), 1.28 (м, 1H), 1.37 (с, 9Н), 1.60 (м, 1H), 2.06 (м, 1H), 2.28 (м, 1H), 2.66 (м, 1H), 3.91 (м, 1H), 4.33 (м, 2Н), 4.61 (уш т, 1H), 4.97 (д, J=11.0 Гц, 1H), 5.19 (м, 2Н), 6.09 (м, 1H), 6.88 (д, J=7.1 Гц, 1H), 7.35-7.46 (м, 4Н), 7.78 (д, J=8.2 Гц, 1H), 8.12 (д,J=8.3 Гц, 1H);
LC-MS С (время удерживания: 2.60; MS m/z 580 (M+H)+
Стадия 3:
К раствору BocNH-Р3((трет-BuGly)-P2[(Boc(4R)-(нафталин-1-оксо)пролин)]-P1(1S,2R Vinyl Асса)-СООН (38 мг, 0.066 ммоля) (Пример 471, Стадия 2) в 2.2 мл тетрагидрофурана прибавляют 1,1'-карбонилдиимидазол (13 мг, 0.079 ммоля). Полученную смесь кипятят 30 минут, а затем охлаждают до комнатной температуры. В этот момент прибавляют метансульфонамид (16 мг, 0.16 ммоля) и DBU (20 мкл, 0.13 ммоля). Реакционную смесь перемешивают в течение 2 дней, а затем снова прибавляют DBU (10 мкл) и метансульфонамид (9 мг). Через 24 час к реакционной смеси прибавляют 50 мл этилацетата и промывают 50 мл 0.25N HCl и 50 мл рассола. Раствор сушат сульфатом натрия и упаривают в вакууме. Сырой продукт очищают препаративной ТСХ на пластине 20×40 см Analtech (элюент 3:2 этилацетат: смесь гексанов), получают Соединение 471 (25 мг, 58%) в виде пленкообразного твердого соединения.
1H ЯМР (CD3OD, 500 МГц) δ 1.03 (с. 9Н), 1.34 (с, 9Н), 1.80 (м, 1H), 2.18 (м, 1H), 2,31 (м, 1H), 2.68 (м, 1H), 3.09 (уш с, 3H), 4.04 (м, 1H), 4.20-4.44 (м, 2Н), 4,51 (кажущийся т, 1H), 5.08 (м, 1H), 5,25-5.31 (м, 2Н), 5.77 (м, 1H), 6.93 (д, J=7.6 Гц, 1H), 7.36-7.45 (м, 4Н), 7.77 (д, J=8.0 Гц, 1H), 8.15 (m, 1H);
LC-MS С (время удерживания: 2.57; MS m/z 657 (M+H)+
Подраздел L:
Пример 472: Биологические исследования
FRET пептидный анализ комплексов рекомбинантной HCV NS/4A протеазы
Целью данного in vitro анализа является измерение ингибирования комплексов HCV NS3 протеазы, полученных при использовании штаммов BMS, H77C или J416S, описанных ниже, соединениями по данному изобретению. Этот анализ показывает, насколько эффективны соединения по данному изобретению в отношении ингибирования HCV протеолитической активности.
Сыворотку HCV-инфицированного пациента получают от Д-ра Т. Wright, San Francisco Hospital. Генно-инженерную матрицу полноразмерной кДНК (компле(?)ментарной дезоксирибонуклеиновой кислоты) HCV генома (штамм BMS) конструируют при использовании фрагментов ДНК, полученных с помощью обратной транскрипции-ПЦР (PCR) (RT-PCR) сывороточной РНК (рибонуклеиновой кислоты) и при использовании праймеров, выбранных на основе гомологии других штаммов генотипа 1а. На основе определения полной геномной последовательности генотип 1а был отнесен к HCV изоляту по классификации Simmonds et al. (См. Р Simmonds, KA Rose, S Graham, SW Chan, F McOmish, ВС Dow, EA Follett, PL Yap and H Marsden, J. Clin. Microbiol., 31(6), 1493-1503 (1993)). Было показано, что аминокислотная последовательность неструктурной области, NS2-5В, >97% идентична HCV генотипу 1а (H77C) и на 87% идентична генотипу 1b (J4L6S). Инфекционные клоны, H77C (1а генотип) и J4L6S (1b генотип) получены от R. Purcell (NIH), а последовательности опубликованы в Genbank (AAB67036, см. Yanagi, M., Purcell, R.H., Emerson, S.U. and Bukh, J., Proc. Natl. Acad. Sci. U.S.A. 94(16), 8738-8743 (1997); AF054247, см. Yanagi, M., St Claire, M., Shapiro, M., Emerson, S.U., Purcell, R.H. and Bukh, J., Virology 244 (1), 161-172 (1998)).
Штаммы BMS, H77C и J4L6S используют для получения комплексов рекомбинантной NS3/4A протеазы. Манипуляции с ДНК, кодирующими комплекс рекомбинантной HCV NS3/4A протеазы (аминокислоты 1027-1711) для этих штаммов, осуществляют как описано Р. Gallinari et al. (см. Gallinari Р, Paolini С, Brennan D, Nardi С, Steinkuhler С, De Francesco R. Biochemistry. 38(17):5620-32, (1999)). Коротко говоря, на 3'-конце NS4A кодирующей области добавляют солюбилизирующий конец ("хвост") из трех остатков лизина. Цистеин в положении P1 NS4A-NS4B сайта расщепления (аминокислота 1711) заменяют на глицин, чтобы избежать протеолитического расщепления лизинового "хвоста". Далее, для предотвращения автолитического расщепления в домене NS3 геликазы методом ПЦР в положение аминокислоты 1454 вводится мутация цистеина на серин. Фрагмент варианта ДНК клонируют в рЕТ21b бактериальный вектор экспрессии (Novagen) и комплекс NS3/4A экспрессируют в штамме Escherichia coli B1 21 (DE3) (Invitrogen) в соответствии с протоколом, описанным Р.Gallinari et al. (см. Gallinari P, Brennan D, Nardi С, Brunetti M, Tomei L, Steinkuhler C, De Francesco R., J. Virol. 72(8): 6758-69 (1998)), с модификациями. Короче говоря, экспрессию NS3/4A индуцируют с помощью 0.5 мМ изопропил β-D-1-тиогалактопиранозида (IPTG) в течение 22 час при 20°С. Типичная ферментация (10 л) дает около 80 г сырой клеточной массы. Клетки снова суспендируют в буфере для лизиса (10 мл/г), состоящем из 25 мМ N-(2-гидроксиэтил) пиперазин-N'-(2-этансульфоновой кислоты) (HEPES), рН 7.5, 20% глицерина, 500 мМ хлорида натрия (NaCl), 0.5% Triton-Х100, 1 ug/мг лизоцима, 5 мМ хлорида магния (MgCl2), 1 ug/мл Dnasel, 5 mM β-меркаптоэтанола (βМЕ), не содержащем ингибитора протеаз-этилендиаминтетрауксусной кислоты (EDTA) (Roche), гомогенизируют и инкубируют в течение 20 минут при 4°С. Гомогенат подвергают сонификации (ультразвук) и осветляют ультрацентрифугированием при 235000 g в течение 1 час при 4°С. К супернатанту прибавляют имидазол до конечной концентрации 15 мМ и рН доводят до 8.0. Сырой белковый экстракт наносят на колонку с никель-нитрилотриуксусной кислотой (Ni-NTA), предварительно уравновешенную буфером В (25 мМ HEPES, рН 8.0, 20% глицерина, 500 мМ NaCl, 0.5% Triton-Х100, 15 мМ имидазола, 5 мМ βМЕ). Образец наносят при скорости потока 1 мл/мин. Колонку промывают буфером С (таким же, как буфер В, за исключением того, что содержит 0.2% Triton-Х100) в количестве 15 объемов колонки. Белок элюируют буфером D (таким же, как буфер С, но содержащим 200 мМ имидазола) в количестве 5 объемов колонки.
Фракции, содержащие комплекс NS3/4A протеазы, объединяют и загружают в колонку для обессоливания, заполненную Superdex-S200, предварительно уравновешенную буфером D (25 мМ HEPES, рН 7.5, 20% глицерина, 300 мМ NaCl, 0.2% Triton-Х100, 10 мМ βМЕ). Образец загружают при скорости потока 1 мл/мин. Фракции, содержащие NS3/4A протеазный комплекс, объединяют и упаривают, примерно, до 0.5 мг/мл. О том, что чистота NS3/4A протеазных комплексов, полученных при использовании штаммов BMS, H77C и J4L6S, составляет более 90%, судят по анализам SDS-PAGE (электрофорез на полиакриламидном геле) и масс-спектрометрии.
Фермент хранят при -80°С, размораживают на льду и перед употреблением разводят буфером для анализа. Субстрат, используемый для анализа NS3/4A протеазы, представляет собой RET S1 (Депепсипептидный субстрат в методе резонансного переноса энергии; AnaSpec, Inc. cat. # 22991)(FRET пептид), описанный Taliani et al. в Anal. Biochem. 240(2):60-67 (1996). Последовательность этого пептида свободно (нежестко) "размещается" на природном сайте расщепления NS4A/NS4B, за исключением того, что в сайте расщепления находится скорее сложноэфирная, нежели амидная связь. Пептидный субстрат инкубируют с одним из трех рекомбинантных NS3/4A комплексов в отсутствие или в присутствии соединения по данному изобретению и за образованием флуоресцентного продукта реакции следят в реальном времени, используя Cytofluor Series 4000.
Применяются следующие реагенты: HEPES и глицерин (Ultrapure, высокой степени чистоты) получают от GIBCO-BRL. Диметилсульфоксид (ДМСО) получают от Sigma. β-Меркаптоэтанол получают от Bio Rad.
Буфер для анализа: 50 мМ HEPES, рН 7.5; 0.15 М NaCl; 0.1% Triton; 15% глицерина; 10 мМ βМЕ. Субстрат: конечная концентрация 2 мкМ (из 2 мМ исходного раствора в ДМСО, хранящегося при -20°С). HCV NS3/4A типа 1a (1b), конечная концентрация 2-3 нМ (из 5 мкМ исходного раствора в 25 мМ HEPES, рН 7.5, 20% глицерина, 300 мМ NaCl, 0.2% Triton-Х100, 10 мМ βМЕ).
Анализ проводят в 96-луночном черном планшете из полистирола от Falcon. В каждой лунке содержится 25 мкл NS3/4A протеазного комплекса в буфере для анализа, 50 мкл соединения по данному изобретению в 10% ДМСО/буфер для анализа и 25 мкл субстрата в буфере для анализа. В том же планшете для анализа также готовят контрольную пробу (в отсутствие соединения). Комплекс фермента смешивают с раствором соединения или с контрольным раствором за 1 мин до инициирования ферментативной реакции, осуществляемого добавлением субстрата. Планшет для анализа считывают сразу же с помощью Cytofluor Series 4000 (Perspective Biosystems). Прибор настраивают так, чтобы считывать эмиссию при 340 Нм и возбуждение при 490 нм при 25°С. Как правило, реакции следуют, примерно, через 15 минут.
Ингибирование в процентах рассчитывают по следующему уравнению:
100-[(δFinh/δFcon)×100],
где δF обозначает изменение флуоресценции на линейном участке кривой. Аппроксимация на нелинейной кривой применяется к данным ингибирования-концентрации, а концентрацию с эффективностью 50% (IC50) рассчитывают, используя программное обеспечение, соответствующее Excel-X1, по уравнению
y=A+((B-A)/(1+((C/x)^D))).
Найдено, что все испытуемые соединения имеют значение IC50s 10 мкМ или менее. Кроме того, найдено, что соединения по данному изобретению, которые испытывали против более чем одного комплекса NS3/4A, имеют аналогичную ингибиторную активность, хотя соединения одинаково демонстрируют более высокую активность против штаммов 1b по сравнению со штаммами 1а.
Анализы специфичности
Анализы специфичности проводят для того, чтобы продемонстрировать селективность соединений по данному изобретению в отношении ингибирования HCV NS3/4A протеиназы по сравнению с сериновыми или цистеиновыми протеазами.
Определяют специфичность соединений по данному изобретению против различных сериновых протеаз: лейкоцитарной эластазы человека (HLE), свиной панкреатической эластазы (РРЕ) и человеческого панкреатического химотрипсина, и одной цистеиновой протеазы: катепсина В печени человека. Во всех случаях используют протокол в формате 96-луночного планшета с применением колориметрического п-нитроанилинового (pNA) субстрата, специфического в отношении каждого фермента, описанный ранее (Международная заявка WO 00/09543), с некоторыми модификациями при анализе сериновых протеаз. Все ферменты получают от Sigma, тогда как субстраты получают от Bachem.
Каждый анализ включает предварительную инкубацию фермента-ингибитора в течение 2 час при RT с последующим добавлением субстрата и гидролизом с примерной конверсией до 30%, определяемой на ридере для микропланшет Spectramax Pro. Концентрация соединений варьируется от 100 до 0.4 мкМ в зависимости от их активности.
Далее представлены конечные условия каждого анализа:
50 мМ Трис(гидроксиметил)аминометан гидрохлорид (Tris-HCl) рН 8, 0.5М сульфат натрия (Na2SO4), 50 мМ NaCl, 0.1 мМ М EDTA, 3% ДМСО, 0.01% Tween-20 с:
133 мкМ succ-AAA-pNA и 20 нМ HNE или 8 нМ РРЕ; 133 мкМ succ-AAV-pNA и 15 нМ HLE; 100 нМ succ-AAPF-pNA и 250 пМ химотрипсина.
100 мМ NaHPO4 (?) ((Ди?)гидроортофосфат натрия) рН 6, 0.1 мМ EDTA, 3% ДМСО, 1 мМ ТСЕР (Трис(2-карбоксиэтил)фосфин гидрохлорид), 0.01% Tween-20, 30 мкМ Z-FR-pNA и 5 нМ катепсина В (исходный раствор фермента, активированный перед употреблением в буфере, содержащем 20 мМ ТСЕР).
Ингибирование в процентах рассчитывают по формуле:
[1-((UVinh-UVblank )/(UVctl-UVblank))]×100
Аппроксимацию нелинейной кривой применяют для данных ингибирования-концентрации, и концентрацию с 50% эффективностью (IC50) рассчитывают с применением Excel X1 - соответствующего программного обеспечения.
Клеточный анализ HCV репликона
Полную клеточную систему HCV репликона устанавливают, как описано в статье Lohrnann V, Komer F, Koch J, Herian U, Theilmann L, Bartenschlager R., Science 285(5424):110-3 (1999). Эта система позволяет оценить влияние наших HCV протеаз на репликацию РНК HCV.
Коротко говоря, при использовании последовательности HCV штамма 1В, описанной в статье Lohmann (Номер:AJ238799), получены кДНК HCV, кодирующая 5' сайт внутреннего входа рибосом (IRES), ген устойчивости к неомицину, EMCV (вирус энцефаломиокардита)-IRES и HCV неструктурные белки, NS3-NS5B и 3' нетранслируемая область (NTR). In vitro транскрипты кДНК трансфецированы в клеточную линию человеческой гепатомы, Huh7. Селекцию клеток, конститутивно экспрессирующих HCV репликон, осуществляют в присутствии селективного маркера, неомицина (G418). Полученные клеточные линии характеризуют на продуцирование позитивной и негативной нити РНК и продуцирование белка во времени.
Huh7 клетки, конститутивно экспрессирующие HCV репликон, выращивают в модифицированной по способу Дульбекко среде Игла (DMEM), содержащей 10% фетальной телячьей сыворотки (FCS) и 1 мг/мл G418 (Gibco-BRL). Клетки засевают накануне вечером (1.5×104 клеток/лунка) в 96-луночные стерильные планшеты для тканевых культур. Соединение и не содержащие соединения контрольные образцы готовят в DMEM, содержащей 4% FCS, 1:100 пенициллина/стрептомизина, 1:100 L-глутамина и 5% ДМСО, в планшете для разведения (конечная концентрация при анализе составляет 0.5% ДМСО). Смеси соединение/ДМСО прибавляют к клеткам и инкубируют 4 дня при 37°С. Через 4 дня планшеты тщательно отмывают фосфатно-солевым буферным раствором (PBS) (3 раза 150 мкл). Клетки лизируют, используя 25 мкл реагента для тест-лизиса, содержащего FRET пептид (RET S1, описанный для in vitro ферментного анализа). Реагент для тест-лизиса готовят из реагента для лизиса клеточных культур с люциферазой 5Х (Promega #Е153А), который разводят дистиллированной водой до 1X, NaCl, добавляют до конечной концентрации 150 мМ, FRET пептид готовят разведением исходного 2 мМ раствора в 100% ДМСО до конечной концентрации 10 мкМ. Затем планшет помещают в прибор Cytofluor 4000 (флуоресцентный сканер для многолуночных планшетов), который настраивают на 340 нм возбуждение/490 нм эмиссия, автоматический режим 21 циклов и планшет считывают в кинетическом режиме. Определение ЕС50 осуществляют, как описано при определении IC50.
Данные ЕС50, полученные FRET анализом репликона, дополнительно подтверждают количественным анализом РНК. Клетки лизируют, используя набор Rneasy (Qiagen). Очищенную тотальную РНК нормализуют, используя RiboGreen (Jones LJ, Yue ST, Cheung CY, Singer VL, Anal. Chem., 265(2): 368-74 (1998)) и сравнительное количественное определение экспрессии РНК HCV, оцениваемое по методике TaqMan® (Kolykhalov AA, Mihalik К, Feinstone SM, Rice CM, Journal of Virology 74, 2046-2051 (2000)), и набор Platinum Quantitative RT-PCR Thermoscript One-Step (для одностадийного тестирования) (Invitrogen cat # 11731-015). Коротко говоря, РНК, приготовленную в объеме 5 мкл (≤1 нг), прибавляют к 20 мкл Ready-Mix (готовой смеси), содержащей нижеследующее: 1.25Х реакционной смеси Thermoscript (содержащей сульфат магния и 2-дезоксинуклеозид 5'-трифосфаты (dNTPs)), 3 мМ dNTPs, 200 нМ прямого праймера (последовательность: 5'-gggagagccatagtggtctgc-3'), 600 нМ обратного праймера (5'-cccaaatctccaggcattga-3'), 100 нМ зонда (5'-6-FAM-cggaattgccaggacgaccgg-BHQ-1-3')(FAM: Флуоресцеин-аминогексиламидит; BHQ: Black Hole Quencher), 1 мкМ стандартного красителя Rox (1Nvitrogen cat # 12223-012) и смесь Taq-полимеразы от Thermoscript-Plus Platinum. Все праймеры созданы с применением программного обеспечения ABI Prism 7700 software и получены от Biosearch Technologies, Novato, CA. Образцы, содержащие известные концентрации транскрипта РНК НС, используют в качестве стандарта. При использовании в циклическом процессе следующего протокола (50°С, 30 мин; 95°С, 5 мин; 40 циклов 95°С, 15 сек, 60°С, 1 мин), экспрессию РНК HCV рассчитывают, как описано в учебнике Perkin Elmer, используя ABI Prism 7700 Sequence Detector.
Биологические примеры
Представителей соединений по изобретению оценивают с помощью HCV репликон клеточного анализа и/или с помощью нескольких коротко описанных анализов специфичности. Найдено, например, в ферментном анализе, что величина IC50 Соединения 34 составляет 23 наномоля (нМ) против NS3/4A штамма BMS. Аналогичные величины активности получены в отношении штаммов Н77С (IC50 3 нМ) и J4L6S (IC50 2.9 нМ). Величина EC50 в анализе репликона составляет 166 нМ.
Найдено, что в анализах специфичности то же самое соединение проявляет следующую активность: HLE>100 мкМ; РРЕ>200 мкМ; Химотрипсин >200 мкМ; Катепсин В>200 мкМ. Эти результаты показывают, что данный ряд (семейство) соединений является в высшей степени специфическим в отношении NS3 протеазы и многие представители этого ряда ингибируют репликацию HCV репликона.
Проведены испытания соединений по данному изобретению и найдено, что их активность наблюдается в следующих интервалах:
Интервалы IC50 активности (NS3/4A штамм BMS): А обозначает 10-100 микромолей (мкМ); В обозначает 1-10 мкМ; С обозначает 0.1-1 мкМ; D обозначает <0.1 мкМ.
Интервалы EC50 активности (для испытуемых соединений): А обозначает 10-100 мкМ; В обозначает 1-10 мкМ; С обозначает 0.1-1 мкМ; D обозначает <0.1 мкМ.
Замечание: используя номер Примера в Патенте и номер Соединения в Патенте, можно найти структуры приведенных в Таблице Соединений.
В соответствии с настоящим изобретением, предпочтительно, чтобы соединения имели биологическую активность (ЕС50) 10 мкМ или менее, более предпочтительно, 1 мкМ или менее, и, наиболее предпочтительно, 100 нМ или менее.
Подраздел М:
Таблица 2
Ниже приведены соединения, которые можно получать методами по данному описанию, и конкретно, описанию в подразделах А-К раздела Примеров, и более конкретно, в подразделах В, Е, F и G. Кроме того, следует понимать, что каждую из групп В, R3, R2 и R1, изображенных ниже, можно заменить на любые другие группы, приведенные в Примерах Подразделов А-К и в других местах данного описания или определенные в Формуле I. Например, группа R3 в Таблице 2 показана как трет-бутильная группа, но специалист в данной области техники понимает, что в каждом приведенном ниже случае (в каждой позиции в Таблице) эта группа может быть заменена на изопропильную группу, или C1-6 группа может быть заменена на алкоксигруппу. Или изображенная ниже группа В может быть заменена на трет-бутилкарбамидный фрагмент в каждой из приведенных в Таблице позиций.



Реферат
Изобретение относится к ингибиторам вируса гепатита С, имеющим общую формулу
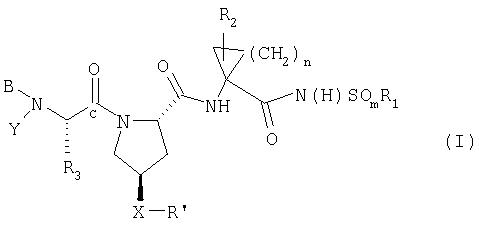
где R1 обозначает C1-8алкил, С3-7циклоалкил; m обозначает 2; n обозначает 1; R2 обозначает Н, C1-6алкил, С2-6алкенил, каждый из которых, необязательно, замещен галогеном; R3 обозначает C1-8алкил, необязательно имеющий следующий заместитель: С6-10арил, C7-14алкиларил, C1-6 алкокси, карбокси, гидрокси, C7-14алкиларилокси, С2-6алкиловый сложный эфир, C8-15алкилариловый сложный эфир; С3-7алкенил, С3-7циклоалкил или C4-10алкилциклоалкил, где циклоалкил или алкилциклоалкил, необязательно, имеют заместитель: C1-6алкил; Y обозначает Н или C1-6алкил, при условии, что если R4 или R5 обозначает Н, тогда Y обозначает Н; В обозначает Н, R4-(C=O)-, R4O(C=O)-, R4-N(R5)-C(=O)-; R4 обозначает (i) C1-10алкил, необязательно имеющий следующий заместитель: фенил, 1-3 атома галогена, гидрокси, -OC(O)C1-6алкил, C1-6алкокси; (ii) C3-7циклоалкил, С3-7циклоалкокси или C4-10алкилциклоалкил; (iii) С6-10арил или C7-16арилалкил, каждый из которых, необязательно, замещенный галогеном; (iv) Het; (v) бицикло(1.1.1)пентан; или (vi) -C(O)OC1-6алкил; R5 обозначает Н; C1-6алкил, необязательно замещенный C1-6алкокси, при условии, что R4 обозначает C1-10алкил; Х обозначает О, S, SO2, СН2О или NH; R' обозначает Het; или С6-10арил, необязательно замещенный Ra; и Ra обозначает C1-6алкил, C1-6алкокси, C3-7цилоалкил, галоген, C1-6алкил, CF3, ди-галоген-C1-6алкокси, циано, галоген, тиоалкил, гидрокси, ди(С1-6)алкил(алкокси)амин, С6-10арил, С7-14алкиларил или 5-7-членный моноциклический гетероцикл; при условии, что X-R' не обозначает

способ лечения HCV инфекции и ингибирования HCV NS3 протеазы, а также композиции, содержащие эти соединения, и применение этих композиций. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 7 н. и 41 з.п. ф-лы, 2 табл.
Формула
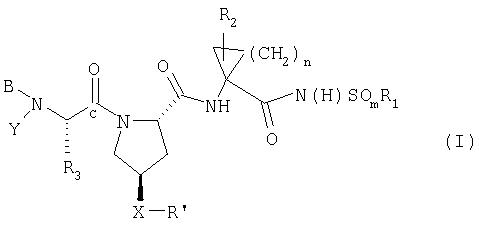
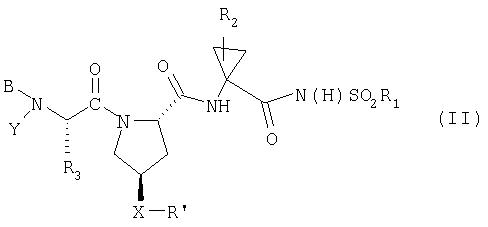
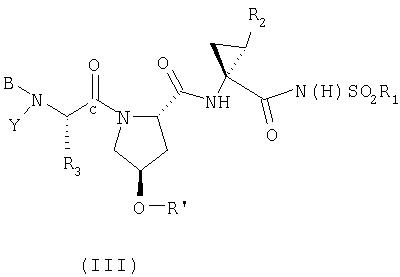

Комментарии