Клеточный продукт инсулин-продуцирующих клеток млекопитающих и его использование для терапии сахарного диабета - RU2663118C1
Код документа: RU2663118C1
Чертежи
Описание
Область изобретения
[001] Группа изобретений относится к области регенеративной медицины и клеточных технологий.
Уровень техники
[002] Сахарный диабет - заболевание эндокринной системы, характеризующееся нарушениями усвоения глюкозы и возникающее из-за недостатка гормона инсулина. Вследствие этого развивается гипергликемия - устойчивое увеличение уровня глюкозы в крови, что приводит к нарушению всех видов обмена веществ (углеводного, жирового, белкового, минерального, водно-солевого). В организме инсулин продуцируют бета-клетки поджелудочной железы, собранные в анатомические структуры, называемые островками Лангерганса и характеризующееся способностью к глюкозозависимой секреции инсулина. Основные виды сахарного диабета - это диабет 1 и 2 типов. Сахарный диабет 1 типа (или инсулинозависимый диабет) - это аутоиммунное заболевание эндокринной системы, вызванное недостаточностью инсулина, возникающей из-за деструкции бета-клеток иммунными клетками организма вследствие выработки аутоантител против белков бета-клеток. Наиболее часто данным типом диабета заболевают люди молодого возраста: дети, подростки, взрослые люди моложе 30 лет. Сахарный диабет 2 типа (инсулиннезависимый диабет) - метаболическое заболевание, характеризующееся хронической гипергликемией, обусловленной снижением чувствительности тканей к инсулину (инсулинорезистентностью). На начальных этапах данного заболевания инсулин секретируется в повышенных количествах. Со временем избыточная секреция инсулина истощает бета-клетки. Диабет 2 типа составляет 85-90% всех случаев сахарного диабета, наиболее часто развивается у людей после 40 лет [Gavin J.R., Davidson М.В., DeFronzo R.A., Drash A, Gabbe S.G., Genuth S., Harris M.I., Kahn R., Keen H., Knowler W.C., Lebovitz H., Maclaren N.K., Palmer J.P., Raskin P., Rizza R.A., Stern M.P. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 2003. 26(1). s5-s20].
[003] При разработке способов терапии диабета и его последствий обычно используют модели экспериментального диабета у лабораторных животных. Некоторые модели основаны на генетических особенностях линий лабораторных мышей, у которых генетически обусловлено возникновение диабета. [Antoniou A.N., Elliott J., Rosmarakis E., Dyson P.J. MHC class II Ab diabetogenic residue 57 Asp/non-Asp dimorphism influences T-cell recognition and selection. Immunogenetics. 1998. 47(3). 218-225, Driver J.P., Serreze D.V., Chen Y.G. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin. Immunopathol. 2011. 33. 67-87].
[004] Другой группой экспериментальных моделей сахарного диабета являются модели химически индуцированного диабета, при котором воздействием химических веществ селективно разрушают бета-клетки. В частности, может применяться антибиотик стрептозотоцин, вызывающий специфический некроз бета-клеток. [Lenzen S. The mechanisms of alloxan- and streptozotocininduced diabetes. Diabetologia. 2008. 51(2). 216-226].
[005] Один из возможных путей преодоления проблем сахарного диабета - заместительная клеточная терапия, осуществляемая через восполнение функции утраченных бета-клеток по глюкозозависимой секреции инсулина путем введения в организм инсулин-продуцирующих клеток.
[006] Описаны способы трансплантации больным сахарным диабетом донорских клеток человека или животных (ксенотрансплантация). В частности, известна работа по трансплантации донорских островков Лангерганса пациентам с сахарным диабетом [Shapiro AM, et al., N. Engl. J. Med. 2006. 355(13). 1318-1330].
[007] Однако подобные методы имеют серьезные ограничения, связанные с малой доступностью донорских клеток человека из поджелудочной железы и с не до конца решенным вопросом биологической безопасности материала, полученного из животных, в том числе с опасностью заражения человека инфекциями и/или прионами, которые могут содержаться в клетках свиньи. Кроме того, аллогенная и ксеногенная трансплантации требуют использования иммуносупрессии для подавления иммунного ответа на введенный чужеродный биологический материал. В качестве альтернативы иммуносупрессии предложен ряд способов, подразумевающих помещение чужеродных клеток в специальные контейнеры из биосовместимых материалов, которые пропускают питательные вещества и инсулин, но не пропускают внутрь клетки иммунной системы, например, описанные в [Skinner SJM, et al., InTech. 2011. V. 11. 391-408, US 20040197374 A1 от 07.10.2004]. Однако такие контейнеры постепенно зарастают соединительной тканью с образованием фиброзной капсулы, что со временем снижает эффективность снабжения организма инсулином, а клеток питательными веществами и в конечном результате приводит к их гибели.
[008] Предложены методы получения инсулин-продуцирующих клеток из эмбриональных стволовых клеток или из индуцированных плюрипотентных клеток путем эндокринной панкреатической дифференцировки in vitro. В работе [WO 2002092756 А2, 21.11.2002] эмбриональные стволовые клетки изначально культивируют на фидерном слое в безсывороточной среде, содержащей заменитель сыворотки, незаменимые аминокислоты, меркаптоэтанол, основной фактор роста фибробластов (bFGF, basic fibroblast growth factor). Затем клетки подвергают суспензионному культивированию на культуральных чашках с неадгезивной поверхностью в вышеуказанной среде без bFGF. Образовавшиеся эмбриональные тельца дезагрегируют и помещают в покрытые фибронектином культуральные флаконы в безсывороточной среде, к которой добавляют следующие факторы: добавку «инсулин-трансферрин-селенит», добавку В27; добавку N2, bFGF, ламинин и никотинамид. Через 20-30 дней из получившейся культуры выделяют клетки, обладающие способностью секретировать инсулин в глюкозозависимой манере и получают стабильные клеточные линии.
[009] В другом способе [Jiang W, et al., Cell Res. 2007. 17(4). 333-344] эмбриональные стволовые клетки культивируют на безсывороточной среде, затем пересаживают их в культуральные флаконы, покрытые 1% матригелем (B&D Biosciences, США), заменяют среду культивирования на 50% среду Дульбекко, модифицированную по способу Исков (IMDM - Iscove modified Dulbecco's medium) и 50% смеси F12 (F12 Nutrient Mixture), с добавлением добавки «инсулин-трансферрин-селенит», монотиоглицерина, альбумина. Через два дня к клеткам добавляют активин А, через 4 дня - ретиноевую кислоту. Через 4 дня среду культивирования меняют на DMEM/F12 1:1, с добавлением добавки «инсулин-трансферрин-селенит», альбумина, основного фактора роста фибробластов. Через 3 дня к среде добавляют никотинамид. Через 5 дней клетки подвергают сфероидному культивированию для созревания в течение следующих 5 дней. В результате получают клетки, экспрессирующие С-пептид, инсулин, глюкагон, глюкозный транспортер тип 2 (GLUT2). С помощью описанного способа эффективной панкреатической дифференцировке подвергается только около 15% клеток.
[010] Известен способ дифференцировки стволовых клеток в инсулин-продуцирующие клетки [US 20050054102 А1], основанный на применении ряда дифференцировочных сред с целью активации одного или нескольких генов, необходимых для дифференцировки бета-клеток, выбранных из группы: PDX1, РАХ4, РАХ6, NGN3, NKX6.1, NKX6.2, NKX2.2, НВ9, ВЕТА2, NEUROD, ISI1, HNF1-alpha, HNF1-beta, HNF3 или комбинации этих генов. Стволовые клетки сначала культивируют в посуде с неадгезивной поверхностью для образования эмбриоидных телец, затем дифференцируют в среде IMDM с добавлением эмбриональной телячьей сыворотки, L-глутамина, незаменимых аминокислот, эпидермального фактора роста, основного фактора роста фибробластов, прогестерона, фоллистатина и/или активина. В результате получают не менее 20% инсулин-продуцирующих клеток в течение 15 дней, однако полученная культура неоднородна и содержит разные типы клеток.
[011] Описан протокол панкреатической дифференцировки эмбриональных стволовых клеток и плюрипотентных стволовых клеток, позволяющий получить функциональные инсулин-продуцирующие клетки, способные к глюкозозависимой секреции инсулина [Pagliuca FW, et al., Cell. 2014. 159(2). 428-439]. Дифференцировку человеческих плюрипотентных клеток осуществляют течение 28-33 дней по многостадийному протоколу. Полученные в результате проведенной дифференцировки клетки способны к глюкозозависимой секреции инсулина на высоком уровне, сопоставимом со зрелыми бета-клетками. После трансплантации в организм иммунодефицитных мышей с экспериментальным диабетом (SCID/AKITA), эти клетки нормализуют уровень глюкозы в крови.
[012] Группа методов, основанных на панкреатической дифференцировке плюрипотентных клеток (эмбриональных стволовых и индуцированных плюрипотентных клеток), имеет существенные недостатки, ограничивающие их использование: во-первых, для большинства методов это низкая эффективность дифференцировки - способность синтезировать инсулин приобретают около 15-40% клеток. Во-вторых, при использовании эмбриональных стволовых клеток их источником служит материал из человеческих эмбрионов, а его получение сопряжено с этическими проблемами. В-третьих, получаемые в результате клеточные культуры могут быть использованы только для аллогенной трансплантации, то есть время жизни таких клеток ограниченно, и требуются специальные усилия для сохранения введенных инсулин-продуцирующих клеток в организме. Клетки с индуцированной плюрипотентностью можно использовать в аутологичном варианте, однако методы их получения длительны и дорогостоящи. Наконец, эмбриональные стволовые и индуцированные плюрипотентные клетки проявляют туморогенные свойства и склонны к образованию тератом при введении в организм.
[013] Известен способ регулируемой индукции продукции панкреатических гормонов в непанкреатических тканях [US 6774120 В1, 10.08.2004]. Способ основан на получении эктопической экспрессии гена PDX1 (pancreatic and duodenal homeobox). В данном способе введение полипептида или мРНК PDX1 оказывало временный эффект и не приводило к стабильному синтезу инсулина. В то же время введение чужеродной ДНК имеет определенные ограничения для медицинского применения, так как несет за собой риск возникновения мутаций, нарушения жизнедеятельности клеток реципиента и возможность онкотрансформации. Дополнительными проблемами с использованием векторных конструкций, вводимых в организм реципиента, являются: риск их элиминации со временем и метилирование введенной ДНК. Все это приводит к тому, что экспрессия целевого гена со временем снижается или прекращается.
[014] Для преодоления вышеуказанных проблем, предложены подходы, в которых инсулин-продуцирующие клетки получают из легкодоступных типов клеток человека, таких как мезенхимные или эпителиальные клетки.
[015] Известна работа по дифференцировке мезенхимных стволовых клеток пуповинной крови в инсулин-продуцирующие клетки [Prabakar KR, et al., Cell Transplant. 2012. 21(6). 1321-1339]. Авторы использовали протокол дифференцировки, включающий 5 стадий общей продолжительностью более 3-х недель. На первой стадии клетки инкубировали в течение 3-х дней в среде RPMI, добавляли активин А и Wnt3a (wingless-type MMTV integration site family, member 3А). На второй стадии к среде RPMI с 2% эмбриональной телячьей сывороткой добавляли фактор роста фибробластов 10 (FGF-10, fibroblast growth factor 10) и CYC (3-Keto-N-aminoethyl-amino-caproyl-dihydrocinnamoyl Cyclopamine) и инкубировали клетки в течение 4-х дней. На третьей стадии среда была заменена на DMEM с добавлением ретиноевой кислоты, CYC, FGF10 и добавки В27, клетки были инкубированы 4 дня. На четвертой стадии к среде DMEM с добавкой В27 добавляли DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester) и экспендин 4 (Ех4, exendin-4), клетки инкубировали 3 дня. На пятой стадии клетки инкубировали 4 дня в среде CMRL с добавкой В27, с добавлением Ех4, фактора роста гепатоцитов (HGF, hepatocyte growth factor) и инсулиноподобного фактора роста 1 (IGF1, insulin-like growth factor 1). Полученные в результате клетки были способны к глюкозозависимой секреции С-пептида in vitro. После трансплантации иммунодефицитным мышам NOD/SCID, в сыворотке животных также обнаруживали С-пептид человека. Однако данный протокол довольно трудоемок и длителен, так как используемые мезенхимные клетки не принадлежат к железистому эпителию, к которому принадлежат бета-клетки поджелудочной железы.
[016] Известна работа, в которой для получения инсулин-продуцирующих клеток авторы использовали клетки поднижнечелюстной слюнной железы, гистогенетически близкие к бета-клеткам [Sato A, et al., Cloning Stem Cells. 2007. 9(2). 191-205]. Панкреатическую дифференцировку клеток индуцировали методом сфероидного культивирования в среде William's Е с добавлением 10% эмбриональной телячьей сыворотки, 10 мМ никотинамида и 20 нг/мл эпидермального фактора роста. В результате на 6-й день культивирования клетки приобретали способность к экспрессии PDX1, инсулина и С-пептида. Клетки также приобретали способность к глюкозозависимой секреции С-пептида. Однако, используемый протокол дифференцировки эпителиальных клеток, в данном случае, не оптимален и позволяет получить относительно низкий уровень секреции С-пептида: около 0,5 нг/мг белка, сравнимый с его фоновой экспрессией у недифференцированных клеток слюнной железы человека.
[017] Наиболее близким аналогом патентуемого является способ получения инсулин-позитивных клеток из CD49f-позитивных клеток слюнной железы человека, культивируемых in vitro [US 7659121 В2, BIOS RES INST INC. 09.02.2010]. Используемые клетки представляют собой клетки железистого эпителия слюнных желез человека и могут быть дифференцированы в инсулин-позитивные клетки в культуре in vitro методом сфероидного культивирования в присутствии эпидермального фактора роста (EGF, epidermal growth factor), основного фактора роста фибробластов (bFGF, basic fibroblast growth factor) и ингибирующего фактора лейкемии (LIF, leukemia inhibitory factor). Для получения инсулин-позитивных клеток, клетки слюнной железы культивируют в неадгезивных условиях для формирования сфероидов в среде Williams'E с добавлением эмбриональной телячьей сыворотки и глюкагоноподобного пептида-1 (GLP-1, Glucagon-like peptide-1). Через 7 суток в сфероидах детектируют экспрессию инсулина. Данные клетки могут быть использованы для терапии патологий поджелудочной железы. Однако способу-прототипу присущи серьезные недостатки: данный подход не позволяет полностью дифференцировать клетки в нужном направлении, в результате чего получается смешанная культура из глюкагон- и инсулин-позитивных клеток, что говорит о начальных стадиях дифференцировки. В данной работе не продемонстрировано получение инсулин-продуцирующих клеток, способных к глюкозозависимой секреции инсулина, нет количественных данных, демонстрирующих эффективность применяемого способа.
[018] Таким образом, в настоящее время не решена проблема получения инсулин-продуцирующих клеток для заместительной клеточной терапии сахарного диабета с использованием клеток из легкодоступного источника и с применением простых и не длительных дифференцировочных протоколов. Настоящее изобретение направлено на решение этой проблемы.
Сущность изобретения
[019] Настоящее изобретение направлено на способ получения клеточного продукта инсулин-продуцирующих клеток млекопитающего, включающий получение эпителиальных прогениторных клеток и их последующую панкреатическую дифференцировку в клетки, способные к глюкозозависимой секреции инсулина, в котором панкреатическую дифференцировку проводят в две стадии:
(а) на первой стадии клетки дифференцируют в течение 4-15 суток в культуральной среде, содержащей, по меньшей мере, сыворотку крови млекопитающего, глутамин, эпидермальный фактор роста, трансферрин, селенит натрия, ретиноевую кислоту, изопротеренол;
(б) на второй стадии клетки дифференцируют в течение 4-15 суток в культуральной среде, содержащей, по меньшей мере, сыворотку крови млекопитающего, глутамин, эпидермальный фактор роста, ретиноевую кислоту, никотинамид, фактор роста гепатоцитов, дексаметазон.
В преимущественных воплощениях культуральная среда первой стадии панкреатической дифференцировки содержит сыворотку крови - 2-20 объемных %, глутамин - 1-4 мМ, эпидермальный фактор роста - 1-300 нг/мл, трансферрин - 0,1-20 мкг/мл, селенит натрия - 0,1-20 нг/мл, ретиноевую кислоту - 0,1-20 мкМ, изопротеренол - 0,1-10 мкМ.
[020] В преимущественных воплощениях культуральная среда второй стадии содержит сыворотку крови - 2-20 объемных %, глутамин - не менее 1 мМ, эпидермальный фактор роста - 1-300 нг/мл, трансферрин - не менее 0,1 мкг/мл, селенит натрия - 0,1-20 нг/мл, ретиноевую кислоту - 10 нМ - 20 мкМ, никотинамид - 1-100 мМ, фактор роста гепатоцитов - 1-300 нг/мл, дексаметазон - 0,01-5 мкМ.
[021] В некоторых воплощениях культуральная среда первой стадии содержит дополнительно, по крайней мере, инсулиноподобный фактор роста 1, фактор роста фибробластов 10, фактор рост фибробластов 4 и/или фактор роста кератиноцитов.
[022] В некоторых воплощениях культуральная среда на второй стадии содержит дополнительно инсулиноподобный фактор роста 1 и/или бетацеллюлин.
[023] Эпителиальные прогениторные клетки выделяют из биоптата слюнной железы, или тонкой кишки, или желудка, или печени, или поджелудочной железы.
[024] Культивирование на обеих стадиях проводят при температуре 37°С в СО2-инкубаторе в присутствии 5% СО2. В некоторых воплощениях культивирование проводят в присутствии 5% СО2 и 5% О2.
[025] В некоторых воплощениях прогениторные эпителиальные клетки перед панкреатической дифференцировкой дополнительно культивируют для увеличения их биомассы.
[026] В отличие от известных методов, предлагаемый способ дифференцировки позволяет производить инсулин-продуцирующие клетки из эпителиальных прогениторных клеток, которые можно легко получить у взрослого человека. Данные клетки образуют хорошо пролиферирующую культуру in vitro, их легко наращивать с целью получения большой клеточной массы. Сама процедура дифференцировки отличается простотой и не требует больших временных затрат.
[027] Технический результат изобретения состоит в упрощении технологии получения инсулин-продуцирующих клеток, получении не менее 70% функционально активных инсулин-продуцирующих клеток в клеточной культуре, прошедшей дифференцировку, и достигается за счет подбора оптимальных условий дифференцировки. Получаемые клетки продуцируют инсулин в глюкозозависимой манере.
[028] Также обеспечивается клеточный продукт инсулин-продуцирующих клеток млекопитающего, включая человека, полученный с помощью способа настоящего изобретения.
[029] Клеточный продукт настоящего изобретения можно использовать для научных исследований и для заместительной терапии сахарного диабета у млекопитающих, включая человека.
[030] Также обеспечивается способ заместительной терапии сахарного диабета, включающий: трансплантацию клеточного продукта, содержащего 50-200 млн клеток, в организм реципиента, страдающего сахарным диабетом. В преимущественных воплощениях клеточный продукт для заместительной терапии содержит не менее 1 млн клеток на 1 мл изотонического раствора либо не менее 10 тысяч сфероидов на 1 мл изотонического раствора. В качестве изотонического раствора используют стерильные растворы: физиологический раствор для инъекций, фосфатно-солевой буфер, раствор Хенкса, раствор Версена и т.д. В некоторых воплощениях настоящего изобретения клеточный продукт вводят 2-5 раз с интервалом 1-6 месяцев. Клеточный продукт может быть использован как в аутологичном, так и в аллогенном вариантах трансплантации. В некоторых случаях клеточный продукт дополнительно культивируют в трехмерных условиях и вводят в виде сфероидов.
[031] Введение клеточного продукта в организм млекопитающего, включая человека, болеющего сахарным диабетом, приводит к снижению у него уровня глюкозы в крови, уменьшению скачков концентрации глюкозы в крови, а также к регенерации островков Лангерганса поджелудочной железы.
Краткое описание фигур
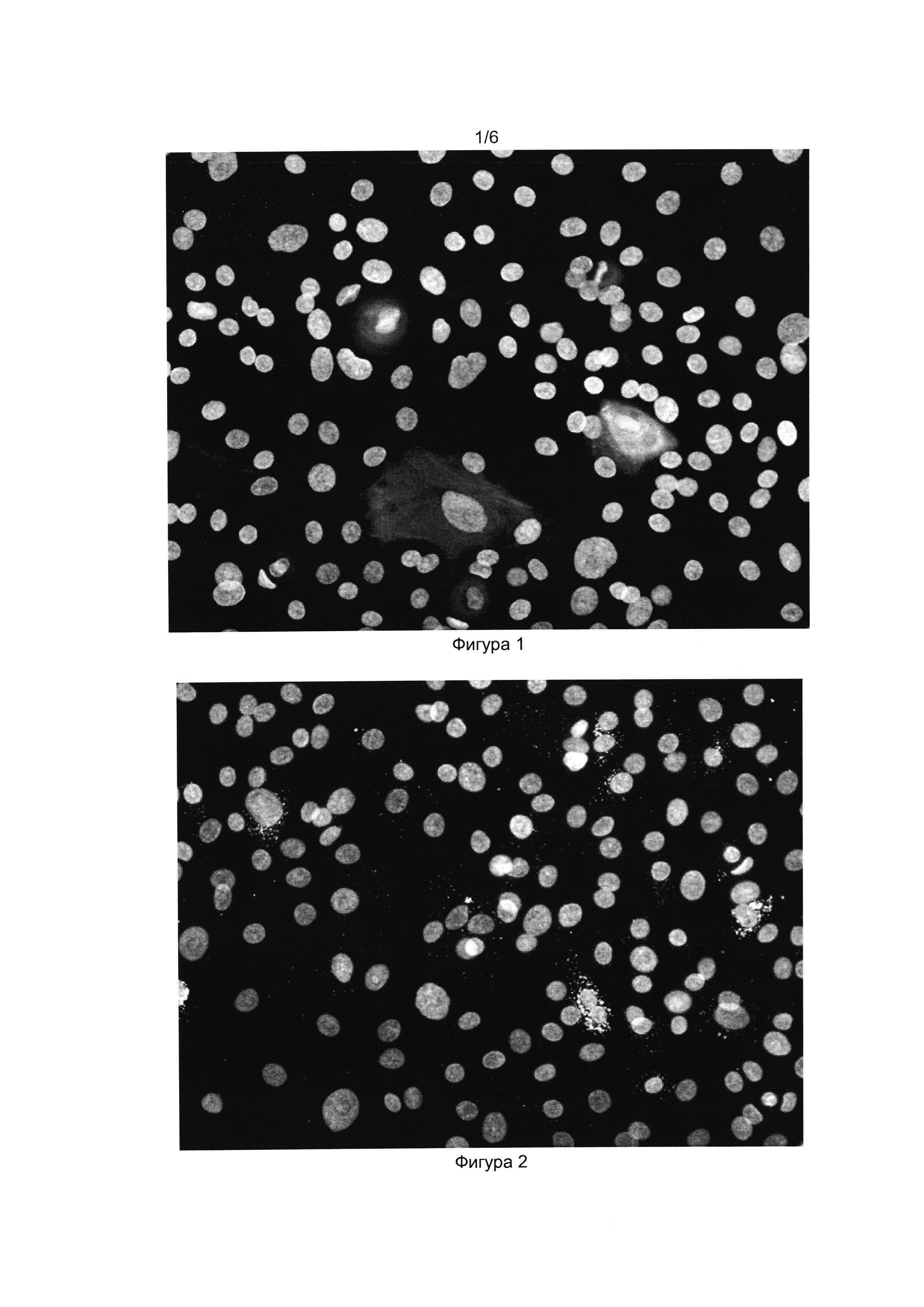
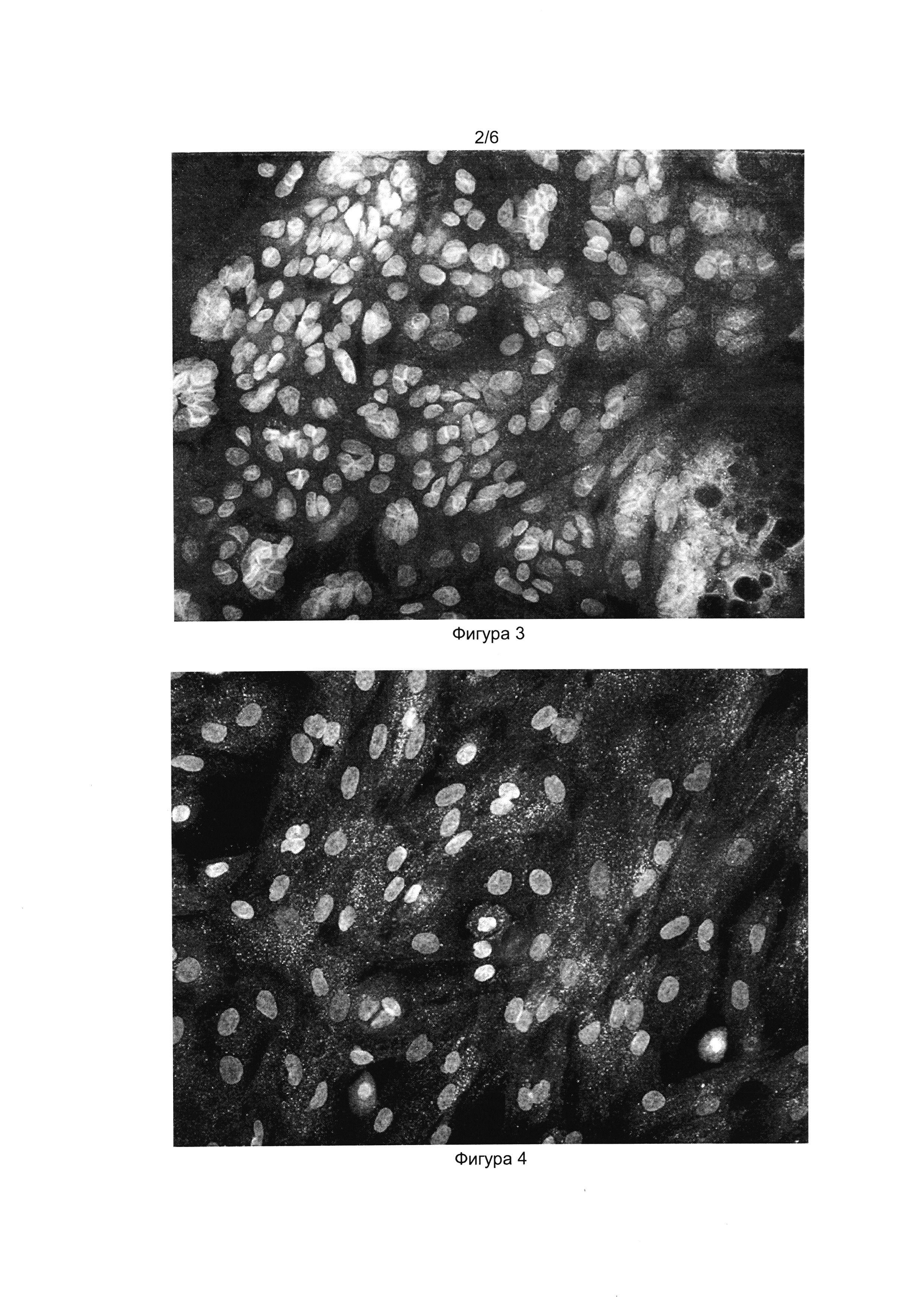
[032] Фигуры 1 и 2 показывают результат иммуноцитохимического окрашивания прогениторных эпителиальных клеток из слюнной железы человека антителами к проинсулину (Фиг. 1) и инсулину (Фиг. 2). Окрашены ядра клеток (краситель DAPI) и проинсулин в цитоплазме (Alexa Fluor 488).
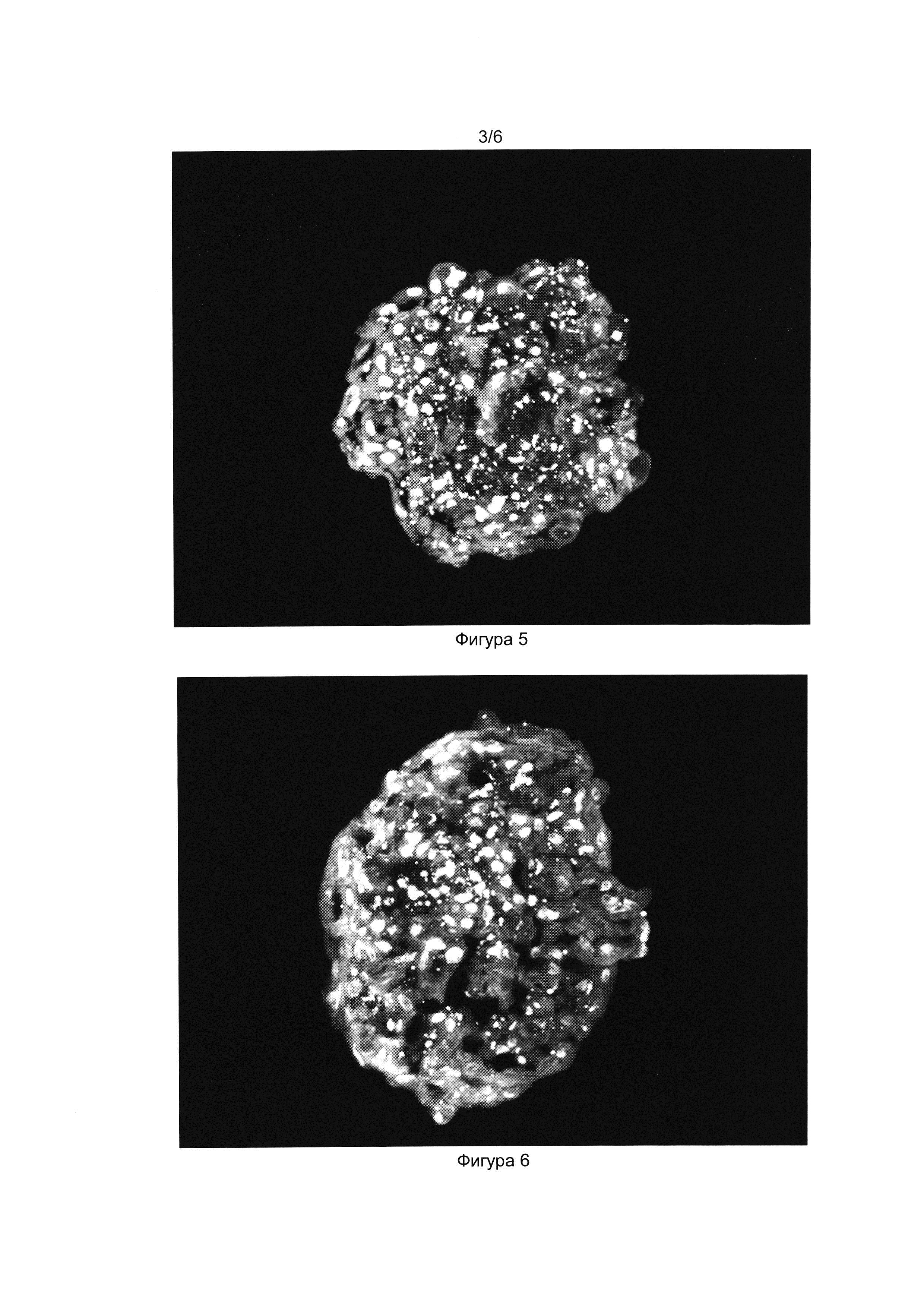
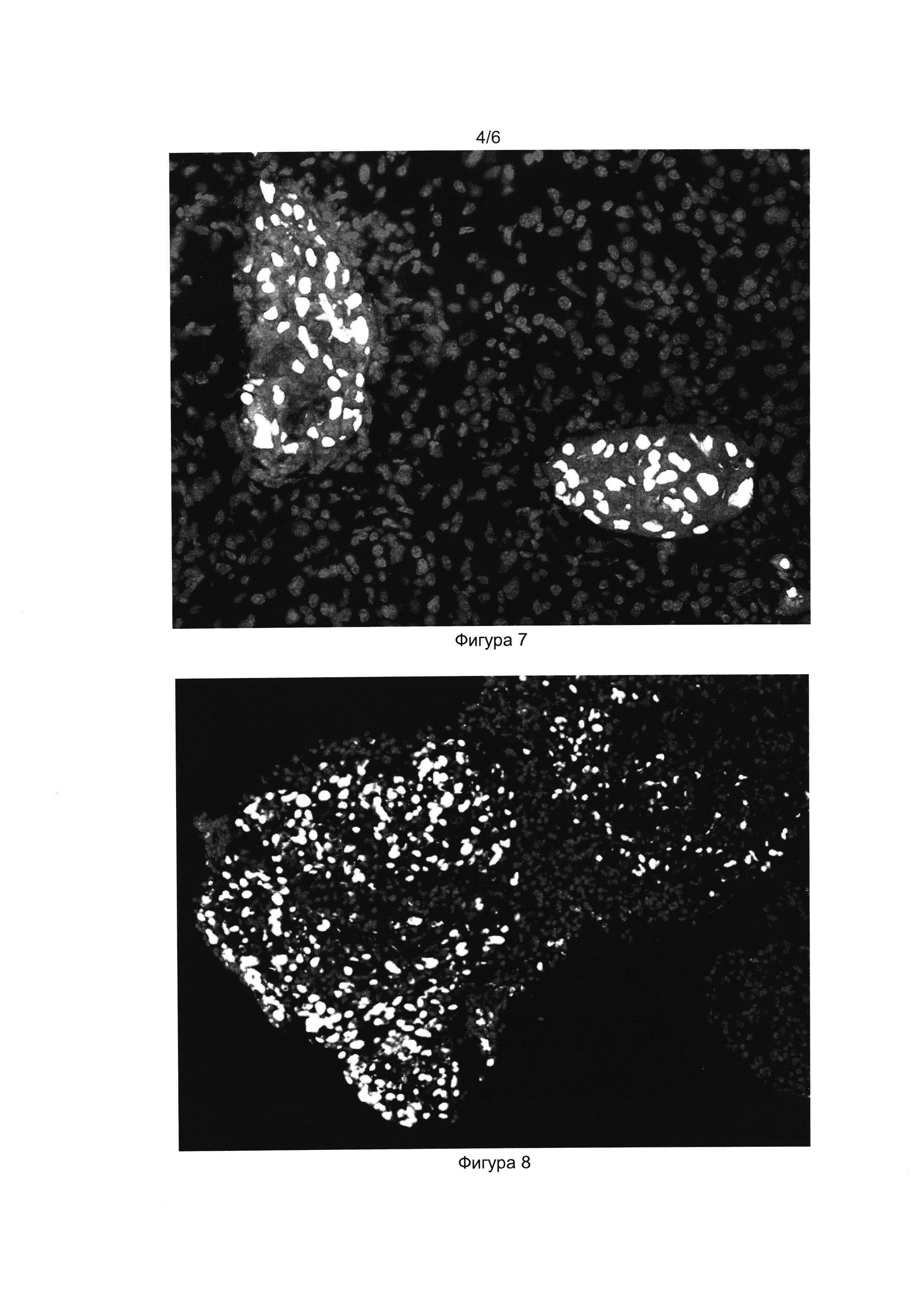
[033] Фигуры 3 и 4 показывают результат иммуноцитохимического окрашивания клеточного продукта инсулин-продуцирующих клеток антителами к проинсулину (Фиг. 3) и инсулину (Фиг. 4). Окрашены ядра клеток (краситель DAPI) и проинсулин в цитоплазме (Alexa Fluor 488).
[034] Фигуры 5 и 6 показывают результаты иммуноцитохимического окрашивания криосрезов сфероида антителами к проинсулину (Фиг. 5) и инсулину (Фиг. 6). Окрашены ядра клеток (краситель DAPI) и проинсулин в цитоплазме (Alexa Fluor 488).
[035] Фигура 7 показывает результат иммуногистохимического окрашивания криосреза поджелудочной железы экспериментальной мыши Nude на третий день после внутрибрюшинной трансплантации. Окрашены ядра клеток (краситель DAPI, серые ядра), ядра клеток человека окрашены антителами к Human nuclei (Alexa Fluor 488, белые ядра), инсулин окрашен в цитоплазме (Alexa Fluor 546).
[036] Фигура 8 показывает результат иммуногистохимического окрашивания криосреза поджелудочной железы мыши Nude на третий день после трансплантации клеточного продукта. Окрашены ядра клеток (краситель DAPI), ядра клеток человека окрашены антителами к Human nuclei (Alexa Fluor 488), маркер макрофагов CD68 окрашен в цитоплазме (Alexa Fluor 546). Толщина криосреза 10 мкм.
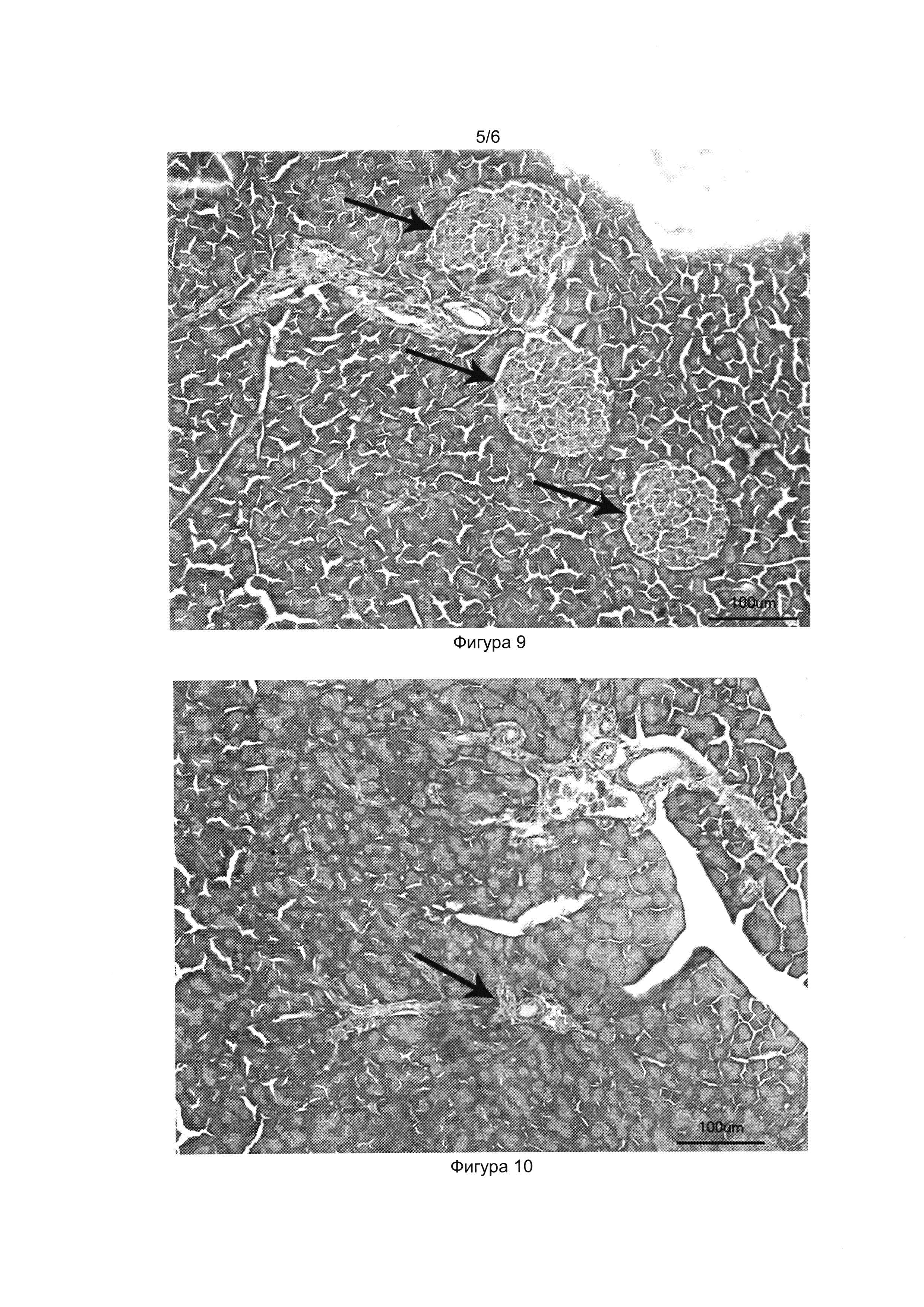
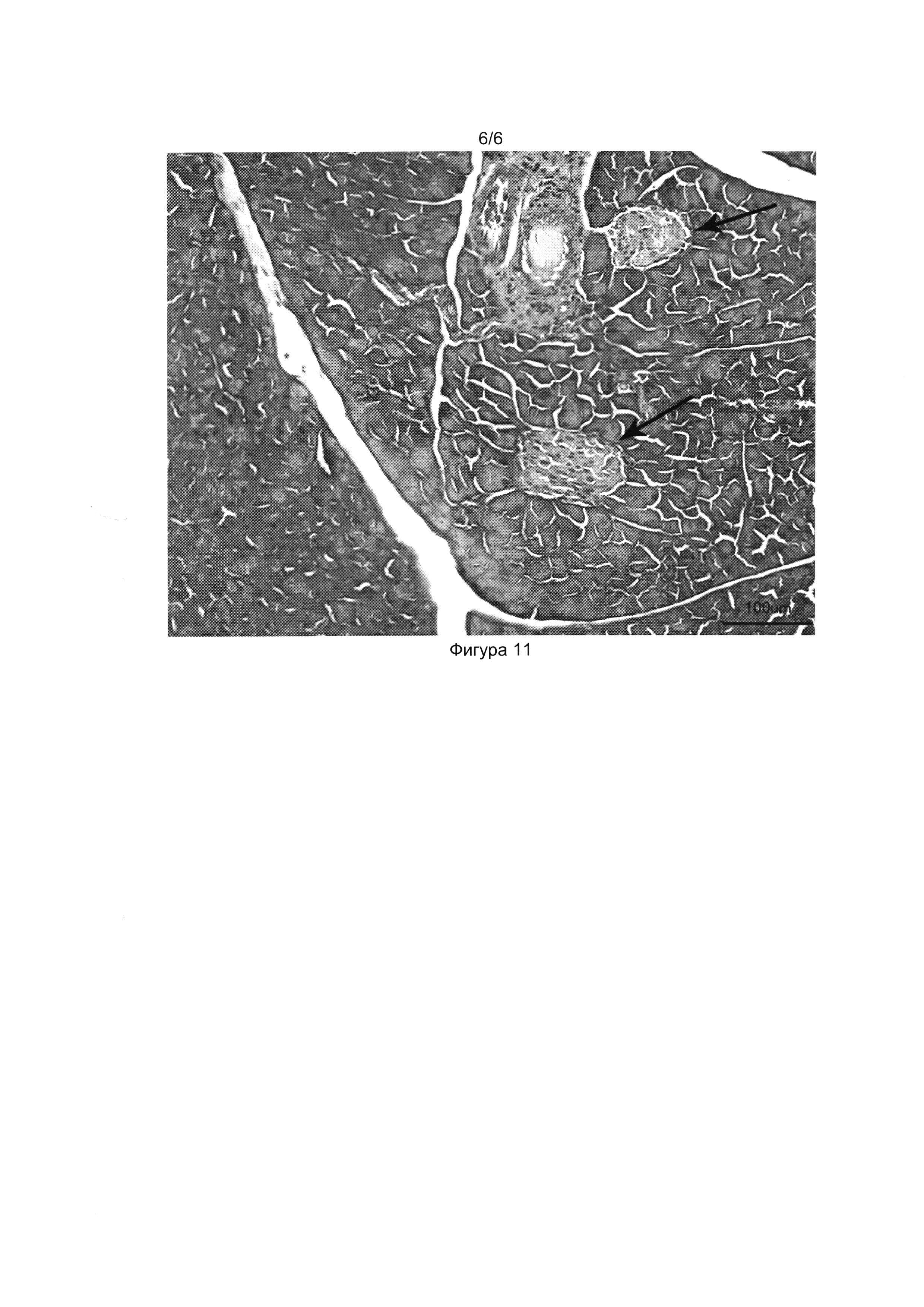
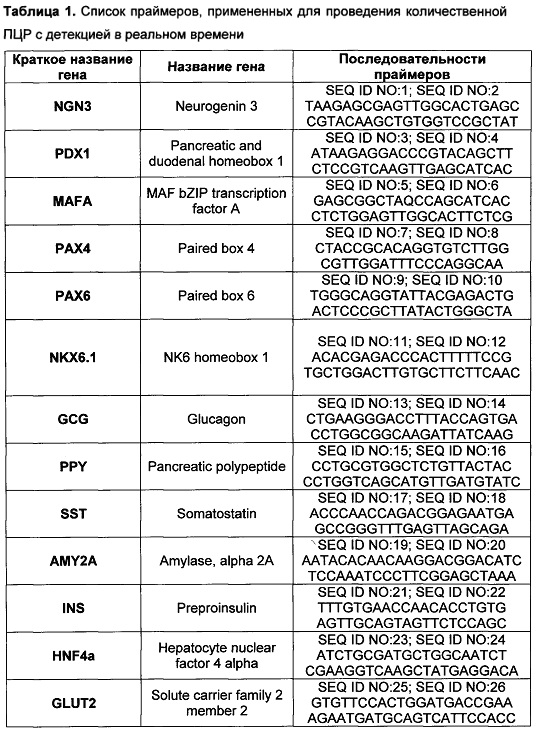
[037] Фигуры 9, 10 и 11 показывают фотографии гистологических срезов поджелудочной железы здоровой мыши (Фиг. 9), мыши со стрептозотоциновым диабетом (Фиг. 10) и мыши со стрептозотоциновым диабетом, получившей трансплантацию клеточного продукта (Фиг.11) на 40-й день после начала эксперимента. Окраска гематоксилин-эозином, световая микроскопия, толщина среза 5 мкм.
Подробное описание изобретения
[038] Как указано выше, настоящее изобретение направлено на способ панкреатической дифференцировки прогениторных эпителиальных клеток млекопитающих, включая человека, получение клеточного продукта, содержащего инсулин-продуцирующие клетки, для заместительной терапии сахарного диабета и способа заместительной терапии сахарного диабета путем коррекции уровня глюкозы в крови при введении в организм клеточного продукта настоящего изобретения.
[039] Способ дифференцировки эпителиальных прогениторных клеток млекопитающих, включая человека, в клетки, способные к глюкозозависимой секреции инсулина, представляет собой новый способ панкреатической дифференцировки in vitro и включает две стадии:
[040] (а) на первой стадии клетки культивируют в течение 4-15 суток в культуральной среде с добавлением, по крайней мере, эмбриональной телячьей сыворотки и глутамина в присутствии, по крайней мере, следующих добавок: эпидермального фактора роста, трансферрина, селенита натрия, ретиноевой кислоты, изопротеренола;
[041] (б) на второй стадии клетки культивируют в течение 4-15 суток в культуральной среде с добавлением, по крайней мере, эмбриональной телячьей сыворотки и глутамина в присутствии, по крайней мере, следующих добавок: эпидермального фактора роста, ретиноевой кислоты, никотинамида, фактора роста гепатоцитов, дексаметазона.
[042] Клеточный продукт представляет собой инсулин-продуцирующие клетки, полученные путем панкреатической дифференцировки, описанной выше, из эпителиальных прогениторных клеток человека, экспрессирующих один или несколько маркеров из списка: c-Kit, Sca-1, ЕрСАМ, LGR-5. После дифференцировки клетки приобретают способность к глюкозозависимой секреции инсулина и меняют свой фенотип, в результате чего экспрессируют один или несколько маркеров из списка: PDX1, NGN3, MAFA, NKX6.1, РАХ4, РАХ6 и инсулин. Клеточный продукт настоящего изобретения содержит не менее 70% инсулин-продуцирующих клеток.
[043] Способ заместительной терапии сахарного диабета включает введение клеточного продукта настоящего изобретения пациентам с сахарным диабетом. Введение клеточного продукта приводит к снижению уровня глюкозы в крови при сахарном диабете, к уменьшению скачков концентрации глюкозы, а также к регенерации островков Лангерганса поджелудочной железы.
Определения
[044] Термин «маркер» или «биомаркер» относится к белку или мРНК, который присутствует в определенном типе клеток, и отличает его от другого типа клеток. Так для клеток, относящихся к настоящему изобретению, характерен определенный набор маркеров.
[045] Термины «прогениторный» или «недифференцированный» или подобный по отношению к клеткам обозначают стволовые клетки, детерминированные на дифференцировку в определенные типы клеток (однако не терминально дифференцированные). Прогениторные клетки обладают высоким пролиферативным потенциалом при культивировании in vitro и имеют биомаркеры, которые позволяют отличить их от клеток других типов.
[046] Термин «эпителиальный» по отношению к клеткам обозначает клетки, полученные из эпителиальных тканей (эпителия) - совокупности дифферонов полярно дифференцированных клеток, тесно расположенных в виде пласта на базальной мембране, на границе с внешней или внутренней средой, а также образующих большинство желез организма. Различают две группы эпителиальных тканей: поверхностные эпителии (покровные и выстилающие) и железистый эпителий, составляющий основную ткань большинства желез.
[047] Термин «дифферон» означает совокупность клеточных форм, составляющих ту или иную линию дифференцировки, включающую несколько различных типов популяций клеток, например, (стволовые клетки, делящиеся клетки, простые транзитные клетки), т.е. гистогенетический ряд.
[048] Термин «мезенхимный» обозначает тип клеток мезодермального происхождения, экспрессирующих по крайней мере следующие маркеры: CD29, CD44, CD73, CD90, CD105, а также способных при определенных условиях культивирования к адипогенной, хондрогенной и остеогенной дифференцировкам in vitro.
[049] Термин «пассаж» или «пассирование» обозначает процедуру снятия адгезивных клеточных культур с культуральной посуды (как правило, с применением протеолитических ферментов), перевод клеток в суспензионное состояние для переноса их в новую культуральную посуду с последующим культивированием до образования адгезивной культуры. В терминах настоящего изобретения «нулевой («0») пассаж» в отношении клеточной культуры означает период инкубации до первого пассажа, «1 пассаж» - период инкубации после 1-го пассажа и до второго пассажа и т.д.
[050] Термин «адгезивная культура» относится к клеткам, которые находятся в прикрепленном к поверхности состоянии.
[051] Термин «биоптат» обозначает биологический материал, полученный путем биопсии от организма донора.
[052] Термин «культивирование» обозначает совокупность методов и протоколов, посредством которых поддерживают жизнеспособность и пролиферативные свойства клеток in vitro.
[053] Культивирование клеток осуществляется в культуральной среде. Культуральная среда - это питательная среда, обычно содержащая композицию незаменимых аминокислот, солей, витаминов, минералов, микроэлементов, сахаров, липидов и нуклеотидов. Среда культивирования обеспечивает клетки компонентами, необходимыми для удовлетворения потребностей в питании и росте клеток. Для различных типов клеток, клеток и клеточных культур различной плотности используют среды, отличающиеся составом питательных веществ, pH и осмолярностью. В литературе описаны многочисленные культуральные среды. Многие среды коммерчески доступны, их идентификация проводится по названию и в ряде случаев каталожному номеру среды. Среды культивирования могут быть дополнены любыми компонентами, необходимыми, чтобы поддержать нужную клетку или культуру клеток. Например, в среду могут быть добавлены стимуляторы роста или ингибиторы роста клеток, гормоны, сыворотка крови млекопитающих, содержащая факторы роста, альбумин, глобулины и другие компоненты.
[054] Термин «культивирование прогениторных эпителиальных клеток» означает процесс культивирования с целью увеличения биомассы указанных клеток, в ходе которого не происходит изменения фенотипа указанных клеток.
[055] Термин «панкреатическая дифференцировка» используется для обозначения процесса культивирования клеток в определенных условиях, в результате которого клетки приобретают сходство с бета-клетками поджелудочной железы. В частности, в них происходит экспрессия характерных генов-маркеров, таких как PDX1, NGN3, MAFA, NKX6.1, РАХ4, РАХ6 и др., а также клетки приобретают способность к синтезу инсулина. Одним из принятых тестов, демонстрирующих продукцию клетками инсулина, является детекция С-пептида, представляющего собой продукт, образующийся в ходе созревания инсулина из молекулы-предшественника. Наличие С-пептида в клетках говорит о том, что они продуцируют инсулин, и что в них происходит его созревание.
[056] Термин «плюрипотентность» обозначает способность клеток дифференцироваться в производные всех трех зародышевых листков (энтодермы, мезодермы, эктодермы). Термин «эмбриональные стволовые клетки» обозначает клетки, полученные из внутренней клеточной массы бластоцисты, образующие культуру клеток, сохраняющих плюрипотентные свойства при длительном культивировании in vitro. Термин «клетки с индуцированной плюрипотентностью» означает клетки, обладающие плюрипотентными свойствами, полученные из соматических клеток путем эпигенетического перепрограммирования.
[057] Термин «сфероидное культивирование» означает культивирование клеток in vitro в неадгезивных условиях, обеспечивающих слипание клеток между собой в трехмерные глобулы, содержащие 500-10000 клеток.
[058] Термин "выделенный" означает молекулу или клетку, которые находятся в среде, отличной от среды, в которой молекула или клетка находятся в естественных условиях.
[059] Термин «изотонические растворы» означает растворы, обеспечивающие следующие свойства: pH: от 7,3 до 7,7; осмолярность: 280+/-20 мосмоль/кг; буферная емкость: не менее 1,4 мл.
[060] Термин «конфлюэнтный монослой» означает монослой, при котором клетки покрывают более 97% поверхности культурального флакона.
[061] Термин «клеточный продукт» означает клеточную культуру, получаемую путем двухстадийной панкреатической дифференцировки in vitro, описанной выше, из эпителиальных прогениторных клеток млекопитающих, включая человека.
Получение прогениторных эпителиальных клеток для панкреатической дифференцировки
[062] Для реализации панкреатической дифференцировки настоящего изобретения используют прогениторные эпителиальные клетки млекопитающих, включая мышь, крысу, свинью, кролика, человека и т.д. Прогениторные эпителиальные клетки могут быть получены от донора клеток из различных органов: слюнных желез, кишечника, желудка, печени, поджелудочной железы.
[063] Биоптат, содержащий эпителиальные прогениторные клетки, получают с помощью биопсии или хирургической операции способами хорошо известными из уровня техники. В преимущественных воплощениях забор клеток и/или тканей при биопсии осуществляется прижизненно. Если дальнейшее использование клеток предполагает медицинские цели, то донор тканевого материала не должен нести инфекционных заболеваний (ВИЧ, гепатиты В и С, сифилис), а также не должен иметь онкологических заболеваний.
[064] Биоптат сразу после забора переносят в стерильных условиях в чашку Петри, содержащую культуральную среду или изотонический раствор и антибиотик. Например, для нужд настоящего изобретения могут быть использованы культуральные среды DMEM/F12 1:1, 199, DMEM, ИГЛА, Alpha-MEM, Ham, F12, IMDM, RPMI-1640 и др., или изотонические растворы: фосфатно-солевой буфер, раствор Хенкса, физиологический раствор, раствор Версена и т.д. Составы изотонических растворов хорошо известны исследователям в данной области. В качестве антибиотика используют гентамицин в конечной концентрации 1-100 мкг/мл (например, в конечной концентрации 40 мкг/мл), или другие антибиотики: пенициллин в конечной концентрации 5-200 ед/мл (например, в конечной концентрации 50 ед/мл), стрептомицин в конечной концентрации 5-200 мкг/мл (например, в конечной концентрации 50 мкг/мл) или иной антибиотик, известный из уровня техники.
[065] Из уровня техники известно, что различия в условиях выделения клеток не оказывают существенного воздействия на качество дальнейшего культивирования и дифференцировки клеток.
[066] Все дальнейшие манипуляции проводят в стерильных условиях, отвечающих требованиям GMP (Good Manufacturing Practice). Эпителиальную ткань механически измельчают стерильными инструментами (скальпелем, пинцетами и др.) до небольших кусочков, например, размером около от 0,5 до 10 мм3. Затем кусочки ткани промывают изотоническим раствором один-три раза, осаждают центрифугированием, инкубируют с 0,1-10 мг/мл коллагеназы IV типа (оптимально 2 мг/мл коллагеназы) в среде DMEM/F12 1:1 с 1-4 мМ глутамина 20-60 минут при 37°С. После этого суспензию клеток пропускают через нейлоновый фильтр с диаметром пор 40-100 мкм и осаждают клетки центрифугированием.
[067] Способы центрифугирования клеток хорошо известны из уровня техники, например, используют центрифугирование в течение 5-15 минут при 100-400 g.
[068] Далее проводят отбор популяции клеток, обогащенных эпителиальными прогениторными клетками. Для этого могут быть использованы различные методики, известные из уровня техники. Например, клетки могут быть отсортированы по маркеру, выбранному из группы: ЕрСАМ, c-Kit, CD49f, LGR5 с помощью магнитной селекции или флуоресцентной селекции. Например, клетки могут быть получены с помощью магнитной сепарации. Для этого суспензию клеток инкубируют с антителами к выбранному маркеру, коньюгированными с магнитными частицами. Как правило, используют антитела к маркерному белку того вида животного, к которому относится донор клеток.
[069] При селекции клеток, полученных из слюнной железы, печени и поджелудочной железы, преимущественно используют маркер ЕрСАМ, для клеток из кишечника - маркер LGR5, из желудка - маркер c-Kit.
[070] Манипуляции проводят по инструкциям производителя антител. Например, инкубацию проводят при температуре около 4°С (на льду) в течение 10-60 минут, как правило, 15-40 минут, используя антитела из расчета 0,1-10 мкг антитела на 106 клеток. Проводят магнитную сепарацию на колонках по инструкциям производителя. Затем отсортированные клетки осаждают центрифугированием, промывают фосфатно-солевым буфером и ресуспендируют в культуральной ростовой среде.
[071] Полученные клетки могут быть использованы для панкреатической дифференцировки или могут пройти через процедуру культивирования для увеличения клеточной массы прогениторных эпителиальных клеток.
[072] Для культивирования клеток может быть использован любой способ, позволяющий увеличить клеточную массу прогениторных эпителиальных клеток, который сохраняет фенотип и характерный набор маркеров для этого типа клеток, обеспечивает однородность клеточной культуры и ее пролиферативный потенциал.
[073] Например, для культивирования клеток из слюнной железы могут быть использованы способы, описанные для клеток крысы [Okumura K. et al., Hepatology. 2003. 38. 104-113], мыши [Hisatomi Y. et al., Hepatology. 2004. 39(3). 667-675], свиньи [Matsumoto S., et al., Cloning and Stem Cells. 2007. 9. 176-190], человека [WO 2014092575 от 19.06.2014 «Means and methods for obtaining salivary gland stem cells and use thereof», Jang S.I. et al., J. Dent. Res. 2015. 94(2). 304-311, WO 2004074465 от 12.03.2009 Human salivary gland-origin stem cell]. Например, может быть использован способ, описанный в заявке [«Способ культивирования клеток слюнной железы человека», регистрационный номер 2016139283, дата поступления 06.10.2016]. В указанном способе клетки культивируют в среде РСТ Epidermal Keratinocyte Medium в культуральных флаконах, обеспечивающих адгезию клеток, при 37°С в присутствии 5% СО2 с заменой среды каждые 2-4 суток до достижения клетками монослоя. После этого, осуществляют пассаж клеток с разведением 1:3-1:5, включающий снятие клеток с поверхности культурального флакона раствором трипсина в ЭДТА и перенос в новые культуральные флаконы и продолжают культивирование с заменой среды каждые 2-4 сутки в ходе культивирования и пассажами по достижении клетками монослоя с разведением не более 1:2-1:3. В преимущественных воплощениях способа инкубацию клеток осуществляют также в присутствии 5% О2. В некоторых воплощениях этого способа клетки сразу после получения инкубируют в течение 6-48 ч в среде DMEM/F12 1:1, содержащей глутамин в конечной концентрации 1-4 мМ и эмбриональную телячью сыворотку в конечной концентрации 5-20%, при 37°С в присутствии 5% СО2. После чего среду меняют на РСТ Epidermal Keratinocyte Medium. Дополнительно в среду культивирования могут быть добавлены инсулин, трансферрин, селенит натрия, эпидермальный фактор роста (EGF).
[074] Для культивирования прогениторных эпителиальных клеток, полученных из печени, поджелудочной железы, кишечника, желудка может быть использован следующий метод: клетки промывают фосфатно-солевым буфером и ресуспендируют в культуральной среде, например, в DMEM/F12 1:1, содержащей 5-20% эмбриональной телячьей сыворотки, 1% добавку «инсулин-трансферрин-селенит», 1-4 мМ глутамина и 1-300 нг/мл эпидермального фактора роста (EGF). Клетки помещают в покрытые коллагеном I типа культуральные флаконы в количестве 5×103 клеток на 1 см2и инкубируют при 37°С и 5% СО2. Среду культивирования меняют каждые 3 дня. В некоторых воплощениях по достижении клетками конфлюэнтного монослоя (обычно на 10-15 день после выделения), клетки пассируют для дальнейшего культивирования и наращивания клеточной массы. Для этого удаляют среду культивирования, клетки дважды промывают раствором Версена, затем инкубируют 5 минут с 0,05%-0,25% раствором трипсина в ЭДТА в количестве 1 мл на 25 см2 площади культурального флакона. Затем клетки отмывают от трипсина фосфатно-солевым буфером, осаждают центрифугированием 5-10 минут при 200 g, разводят в ростовой среде в соотношении 1:3 и помещают в новые культуральные флаконы, покрытые коллагеном I типа.
[075] Полученная клеточная культура должна содержать эпителиальные прогениторные клетки для дальнейшей панкреатической дифференцировки. Для нужд настоящего изобретения прогениторные эпителиальные клетки могут быть выявлены в культуре с помощью методов иммуноокрашивания или ПЦР на один или несколько маркеров, выбранных из группы: ЕрСАМ, AFP, CD49f, CK18, CK19, LGR5, c-Met. Предпочтительной для медицинского применения является однородная культура, содержащая эпителиальные прогениторные клетки, то есть культура, которая содержит не менее 70% прогениторных эпителиальных клеток, чаще не менее 75% прогениторных эпителиальных клеток, как правило, 80% или более прогениторных эпителиальных клеток, например, 85%, 90%, 95%, 96%, 97%, 98, 99% или более прогениторных эпителиальных клеток.
[076] Прогениторные эпителиальные клетки должны быть способны к пролиферации. Уровень пролиферации клеток можно, например, оценить визуально с помощью световой микроскопии по количеству метафаз в поле зрения микроскопа и по фенотипу клеток. В частности, в активно пролиферирующей культуре количество клеток в состоянии митоза составляет не менее 3-5%. В случае если произошла спонтанная дифференцировка культуры, количество клеток в состоянии митоза падает ниже 3-5%. Кроме того, недифференцированные активно пролиферирующие эпителиальные клетки имеют небольшие размеры (10-30 мкм), высокое ядерно-цитоплазматическое соотношение, полигональную форму, мало отростков, формируют эпителиальный пласт по типу булыжной мостовой. При спонтанной дифференцировке клетки приобретают большие размеры (более 50 мкм), зачастую теряют контакты между собой, приобретают множество отростков, имеют низкое ядерно-цитоплазматическое соотношение и гранулярную цитоплазму. Часто такие клетки имеют несколько ядер.
[077] Скорость пролиферации можно также оценить по скорости достижения клетками конфлюэнтного монослоя. Скорость достижения клетками конфлюэнтного монослоя зависит от исходного разведения клеток, но у активно пролиферирующих клеток она выше, чем у дифференцированных. Например, при разведении клеток 1:3 скорость достижения эпителиальными клетками конфлюэнтного монослоя составляет не более 15 суток, как правило 7-10 суток. У дифференцированной культуры клетки достигают конфлюэнтного монослоя очень медленно (более 15 суток) или не достигают совсем.
Панкреатическая дифференцировка эпителиальных прогениторных клеток
[078] Для осуществления панкреатической дифференцировки по способу настоящего изобретения используют выделенные эпителиальные прогениторные клетки, полученные как описано выше в разделе «Получение прогениторных эпителиальных клеток для панкреатической дифференцировки». Используют клетки, выделенные из организма млекопитающего, включая человека. В некоторых воплощениях эти клетки предварительно проходят процедуру культивирования.
[079] Для осуществления панкреатической дифференцировки клетки помещают в культуральную среду с добавлением сыворотки крови млекопитающих и глутамина. В качестве культуральной среды может быть использована любая жидкая культуральная среда для эукариотических клеток, обеспечивающая концентрацию ионов кальция в пределах от 0,5 мМ до 2,5 мМ, как правило, в пределах 1-2 мМ, например, 1,81 мМ. К пригодным средам в частности относятся DMEM, IMDM, William's Е medium, RPMI, Alpha-MEM, 199, MEM, ВМЕ. Например, может быть использована среда DMEM/F12 1:1.
[080] Для культивирования не подходят низкокальциевые среды, так как они способствуют сохранению недифференцированного состояния эпителиальных прогениторных клеток.
[081] Сыворотку крови млекопитающих, например, эмбриональную телячью сыворотку, или заменитель сыворотки (KSR, KnockOut Serum Replacement), или аутологичную сыворотку, добавляют к среде в конечной концентрации 2-20 объемных %, как правило, 5-15%, например, 10%. Как здесь используется, в понятие «сыворотка крови млекопитающих» включены и ее синтетические заменители.
[082] Глутамин добавляют в среду в конечной концентрации не менее 1 мМ, как правило, 1,5-4 мМ, например, 2 мМ. При этом увеличение концентрации глутамина выше 4 мМ не приводит к заметным изменениям в состоянии клеток и эффективности панкреатической дифференцировки.
[083] Клетки культивируют во флаконах, обеспечивающих адгезию эпителиальных клеток. Например, культуральные флаконы предварительно покрывают коллагеном I типа.
[084] На первой стадии дифференцировки клетки культивируют в присутствии эпидермального фактора роста, трансферрина, селенита натрия, ретиноевой кислоты и изопротеренола. Эпидермальный фактор роста используют в конечной концентрации 1-300 нг/мл, чаще 5-100 нг/мл, как правило, 8-15 нг/мл, например, 10 нг/мл. Трансферрин используют в конечной концентрации не менее 0,1 мкг/мл, как правило, 1-20 мкг/мл, например, 5 мкг/мл. Селенит натрия используют в конечной концентрации 0,1-20 нг/мл, как правило, 1-10 нг/мл, например, 5 нг/мл. Ретиноевую кислоту используют в конечной концентрации 0,1-20 мкМ, как правило, 0,5-5 мкМ, например, 2 мкМ. Изопротеренол используют в конечной концентрации 0,1-10 мкМ, как правило, 0,2-7 мкМ, чаще 0,5-3 мкМ, например, 1 мкМ.
[085] В некоторых воплощениях вместо трансферрина и селенита натрия используют коммерчески доступную добавку «инсулин-трансферрин-селенит», в концентрации, обеспечивающей необходимые концентрации трансферрина и селенита натрия (например, 1 объемный %). При этом культуральная среда содержит также инсулин.
[086] В некоторых воплощениях к среде также добавляют инсулиноподобный фактор роста 1 (IGF-1). Конечная концентрация инсулиноподобного фактора роста 1 не превышает 300 нг/мл, и, как правило, составляет 5-20 нг/мл, например, 10 нг/мл.
[087] В некоторых воплощениях к среде также добавляют один из следующих факторов: фактор роста фибробластов 10, фактор роста фибробластов 4, фактор роста кератиноцитов. Конечная концентрация выбранного фактора роста не превышает 300 нг/мл, и, как правило, составляет 5-20 нг/мл, например, 10 нг/мл.
[088] Клетки культивируют в течение 4-15 дней, как правило 5-10 дней, например, 6-8 дней. Среду культивирования меняют на свежую каждые 1-3 дня.
[089] На второй стадии дифференцировки клетки культивируют в присутствии эпидермального фактора роста, ретиноевой кислоты, никотинамида, фактора роста гепатоцитов и дексаметазона. Эпидермальный фактор роста используют в конечной концентрации 1-300 нг/мл, чаще 5-100 нг/мл, как правило, 8-15 нг/мл, например, 10 нг/мл. Ретиноевую кислоту используют в конечной концентрации 10 нМ - 20 мкМ, как правило, 20 нМ - 1 мкМ, например, 100 нМ. Никотинамид используют в конечной концентрации 1-100 мМ, как правило, 5-50 мМ, например, 10 мМ. Фактор роста гепатоцитов используют в конечной концентрации 1-300 нг/мл, чаще 5-100 нг/мл, как правило 10-50 нг/мл, например, 20 нг/мл. Дексаметазон используют в конечной концентрации 0,01-5 мкМ, как правило, 0,05-1 мкМ, например, 0,1 мкМ.
[090] В некоторых воплощениях к среде также добавляют инсулиноподобный фактор роста 1. Конечная концентрация инсулиноподобного фактора роста 1 не превышает 300 нг/мл, и, как правило, составляет 5-20 нг/мл, например, 10 нг/мл.
[091] В некоторых воплощениях к среде также добавляют бетацеллюлин. Конечная концентрация бетацеллюлина не превышает 300 нг/мл, и, как правило, составляет 5-50 нг/мл, например, 20 нг/мл.
[092] Клетки культивируют в течение 4-15 дней, как правило 5-10 дней, например, 6-8 дней. Среду культивирования меняют на свежую каждые 1-3 дня.
[093] В преимущественных воплощениях способа обе стадии дифференцировки клеток осуществляют при 37°С в присутствии 5% CO2. В некоторых воплощениях обе стадии дифференцировки происходят в присутствии 5% CO2 и 5% О2.
Клеточный продукт для заместительной терапии сахарного диабета
[094] До начала дифференцировки эпителиальные прогениторные клетки экспрессируют маркеры CD49f и KRT18 и один или несколько маркеров прогениторных клеток из списка: c-Kit, Sca-1, ЕрСАМ, LGR-5. После панкреатической дифференцировки по способу настоящего изобретения не менее 99% клеток клеточного продукта продолжают экспрессировать маркеры CD49f и KRT18.
[095] Также клеточный продукт настоящего изобретения после панкреатической дифференцировки содержит изолированные клетки, продуцирующие инсулин и экспрессирующие маркеры, характерные для бета-клеток поджелудочной железы. В частности, в этих клетках происходит экспрессия характерных генов-маркеров, таких как PDX1, NGN3, MAFA, NKX6.1, РАХ4, РАХ6 и др. Одним из принятых тестов, демонстрирующих продукцию клетками инсулина, является детекция инсулина или С-пептида, представляющего собой продукт, образующийся в ходе созревания инсулина из молекулы-предшественника. Наличие С-пептида в клетках говорит о том, что они продуцируют инсулин, и что в них происходит его созревание.
[096] Клеточный продукт настоящего изобретения содержит не менее 70% таких инсулин-продуцирующих клеток, чаще - не менее 75% инсулин-продуцирующих клеток, как правило, 80% или более инсулин-продуцирующих клеток, например, 85%, 88%, 90%, 95%, 96%, 97%, 98%, 99% или более таких клеток.
[097] Инсулин и/или С-пептид секретируются клеточным продуктом в глюкозозависимой манере, то есть, уровень секреции инсулина и/или С-пептида клеточным продуктом возрастает при увеличении концентрации глюкозы в среде культивирования (в буфере, либо во внутренней среде организма после трансплантации клеточного продукта).
[098] Для детекции секретируемого инсулина и/или С-пептида используют, как правило, иммуноферментный анализ (ИФА). Для этого клетки инкубируют в буфере без глюкозы в течение 1-2 часов при 37°С и 5% СО2. В качестве буфера может быть использован изотонический раствор, например, раствор Версена, фосфатно-солевой буфер, физиологический раствор, буфер Кребса без глюкозы (содержащий 59 мМ NaCl, 2,35 мМ KCl, 0,625 мМ CaCl2, 0,6 мМ KH2PO4, 0,6 мМ MgSO4⋅7H2O, 12,5 мМ NaHCO3, pH=7,4). После инкубации изотонический раствор меняют на теплый (37°С) буфер Кребса без глюкозы с добавлением разных концентраций глюкозы (0 мМ, 2 мМ, 5 мМ, 15 мМ, 20 мМ глюкозы). Инкубируют пробы 1 час при 37°С и 5% CO2. Затем отбирают супернатант и хранят при -70°С до проведения ИФА. ИФА проводят по инструкциям производителя, например, с наборами Mercodia Ultrasensitive Insulin ELISA, # 10-1132-01 и/или Mercodia Ultrasensitive C-peptide ELISA, # 10-1141-01.
[099] Для выявления экспрессии генов-маркеров, характерных для того или иного типа клеток, может быть использована иммунодетекция с помощью антител к выбранным белкам, кодируемым указанными генами-маркерами или детекция с помощью полимеразной цепной реакции (ПЦР). Различные варианты детекции с помощью ПЦР хорошо известны из уровня техники.
Способы применения клеточного продукта инсулин-продуцирующих клеток млекопитающих
[0100] Клетки (клеточный продукт), полученные в ходе панкреатической дифференцировки способами настоящего изобретения, могут быть использованы для исследований биохимических и молекулярных механизмов пролиферации и дифференцировки клеток, межклеточных взаимодействий, онкотрансформации, экспрессии цитокинов, анализа цитотоксичности различных веществ и др. Поскольку полученные в изобретении клетки являются не модифицированными клетками организма, с их помощью можно изучать генетическую экспрессию, сигнальные пути, осуществляющиеся при естественных биологических процессах. Таким образом, эти клетки могут быть моделью для изучения механизмов дифференцировки эпителия, исследований межклеточных контактов.
[0101] Поскольку данные клетки можно получить в больших количествах, на них можно тестировать различные вещества, например, для исследования их эффектов на биологические процессы, а также для оценки их влияния на жизнеспособность клеток и для анализа безопасности фармакологических препаратов.
[0102] Клетки, полученные методами настоящего изобретения, могут быть использованы для аутологичной или аллогенной пересадки в целях заместительной клеточной терапии сахарного диабета. В преимущественных воплощениях способ заместительной клеточной терапии сахарного диабета включает забор биоптата эпителиальных прогениторных клеток у донора клеток, культивирование клеток для увеличения биомассы прогениторных эпителиальных клеток как описано в разделе «Получение прогениторных эпителиальных клеток для панкреатической дифференцировки» выше, двухстадийную панкреатическую дифференцировку клеток, как описано в разделе «Панкреатическая дифференцировка эпителиальных прогениторных клеток» выше, и трансплантацию клеток в организм реципиента, страдающего сахарным диабетом.
[0103] Трансплантацию клеток осуществляют способами известными из уровня техники, например, клетки вводят в виде суспензии. Для этого клетки после наращивания необходимой клеточной массы и проведения панкреатической дифференцировки снимают с поверхности культурального флакона: удаляют среду культивирования, клетки дважды промывают раствором Версена, затем инкубируют 5 минут с 0,05%-0,25% раствором трипсина в ЭДТА в количестве 1 мл на 25 см2 площади культурального флакона. Затем клетки трижды промывают фосфатно-солевым буфером, осаждают центрифугированием 5-10 минут при 200 g, разводят в стерильном изотоническом растворе, например, в физиологическом растворе для инъекций или в фосфатно-солевом буфере в количестве 0,5-10 млн кл/мл.
[0104] В преимущественных воплощениях клеточный продукт разводят так, что он содержит не менее 1 млн клеток на 1 мл изотонического раствора либо не менее 10 тысяч сфероидов на 1 мл изотонического раствора. В качестве изотонического раствора используют стерильные растворы: физиологический раствор для инъекций, фосфатно-солевой буфер, раствор Хенкса, раствор Версена и т.д.
[0105] Проверяют жизнеспособность клеток: должно быть не менее 70% живых клеток. Клетки транспортируют и хранят до применения при +4°С не более 24 часов. Вводят клетки в селезенку, либо в воротную вену, либо в большой сальник, либо инъецируют шприцем внутрибрюшинно в проекции поджелудочной железы в количестве 50-200 млн на пациента. Клетки также можно вводить дробно (2-5 раз) в количестве 50-200 млн за одно введение с интервалом 1-6 месяцев. Помимо этого, клетки можно вводить в виде сфероидов. Для этого на 10-15 день дифференцировки клетки снимают с поверхности культуральных флаконов и помещают в неадгезивные условия в среде второго этапа панкреатической дифференцировки в количестве 4-6 тысяч клеток на 20 мкл среды. Клетки инкубируют в данных условиях 3-7 дней до образования сфероидов - конгломератов слипшихся клеток, содержащих 3-6 тысяч клеток. Сфероиды трижды промывают фосфатно-солевым буфером, осаждают центрифугированием 5-10 минут при 100-200 g, разводят в стерильном изотоническом растворе, например, в физиологическом растворе для инъекций или в фосфатно-солевом буфере в количестве 0,5-10 млн кл/мл (около 0,1-5 тыс. сфероидов на 1 мл раствора). Сфероиды транспортируют и хранят до применения при +4°С не более 24 часов. Вводят сфероиды через катетер в воротную вену, либо в селезенку, либо в большой сальник в количестве 50-200 млн клеток на пациента (около 10-50 тыс. сфероидов на пациента). Вводить сфероиды также можно дробно (2-5 раз) в количестве 50-200 млн клеток (около 10-50 тыс. сфероидов) с интервалом 1-6 месяцев.
[0106] В некоторых воплощениях способ заместительной терапии настоящего изобретения включает проверку количества и жизнеспособности клеток в клеточном продукте после панкреатической дифференцировки. В некоторых воплощениях способ заместительной терапии настоящего изобретения включает выявление маркеров и С-пептида в клеточном продукте.
[0107] Подсчет клеток осуществляют с помощью методов, известных специалистам в данной области. Например, количество клеток можно оценить с помощью проточной цитометрии с использованием стандартных антител к специфическим маркерам на поверхности клеток, на клеточном счетчике, в камере Горяева или клеточном сортере.
[0108] Проверку жизнеспособности клеток осуществляют способами, известными из уровня техники, например, окрашиванием трипановым синим. Для этого клетки снимают с поверхности культуральных флаконов трипсином, промывают фосфатно-солевым буфером и разводят в фосфатно-солевом буфере в количестве 0,1-5 млн кл/мл. Отбирают аликвоту суспензии клеток 50-200 мкл и разводят с 4% раствором трипанового синего (BioRad, США) в соотношении 1:1. Через 5 минут проводят подсчет доли живых клеток в пробе на автоматическом счетчике клеток (BioRad, США) либо в камере Горяева (живые клетки не окрашиваются трипановым синим). Либо окраску можно проводить красителем, не проникающим через интактную мембрану клеток, например, DAPI или этидиумом бромидом. Подсчет доли окрашенных (то есть в данном случае, не жизнеспособных) клеток осуществляют визуально под флуоресцентным микроскопом либо на проточном цитофлуориметре.
Экспериментальная часть
[0109] Пример 1. Панкреатическая дифференцировка прогениторных эпителиальных клеток из слюнной железы человека
[0110] Биоптат поднижнечелюстной слюнной железы человека получили от донора мужского пола 37 лет в ходе плановой операции по удалению части слюнной железы вследствие слюннокаменной болезни. Объем железистой ткани биоптата составил около 2 см3. Биоптат перенесли в стерильных условиях в чашку Петри со средой DMEM/F12 1:1 (Gibco, США) и гентамицином 40 мкг/мл (ПанЭко, Россия). Все дальнейшие манипуляции проводили в стерильных условиях, отвечающих требованиям GMP.
[0111] Эпителиальную ткань слюнной железы механически отделяли от жировой и мезенхимной тканей стерильными инструментами под бинокуляром и измельчали скальпелем до небольших кусочков (размером примерно 1-5 мм3). Кусочки ткани дважды промывали фосфатно-солевым буфером, осаждали центрифугированием в течение 5-10 минут при 0,8-1,5 тыс. б/мин, инкубировали 30-60 минут при 37°С в присутствии 2-4 мг/мл раствора коллагеназы IV типа (Gibco, США) в среде DMEM/F12 1:1 (Gibco, США) с 2 мМ глутамином (Invitrogen, США). Каждые 10-15 минут пробирки с кусочками слюнной железы активно встряхивали. После инкубации к клеткам добавляли 10 мл среды DMEM/F12 1:1 (Gibco, США), активно пипетировали в течение 3-5 минут, затем пропускали через нейлоновый фильтр с диаметром пор 40-100 мкм и осаждали клетки центрифугированием в течение 5-10 минут при 1-1,5 тыс. об/мин. Затем проводили магнитную сепарацию клеток по маркеру ЕрСАМ, для чего суспензию клеток промывали 10 мл фосфатно-солевого буфера, ресуспендировали в 0,5 мл фосфатно-солевого буфера и подсчитывали число клеток на приборе для автоматизированного подсчета клеток (Bio-Rad, США). Далее клетки слюнной железы инкубировали с антителами anti-human ЕрСАМ, конъюгированными с магнитными частицами (Miltenyi Biotec GmbH, Германия), в течение 15-40 минут при +4°С. Антитела добавляли из расчета 0,1-5 мкг на 106 клеток. После инкубации клетки промывали 10 мл фосфатно-солевого буфера и проводили магнитную сепарацию на колонках MiniMACS™ Separator (Miltenyi Biotec GmbH, Германия) по инструкциям производителя. Отсортированные клетки осаждали центрифугированием в течение 5-10 минут при 200 g, промывали 10 мл фосфатно-солевого буфера и ресуспендировали в ростовой среде РСТ Epidermal Keratinocyte Medium (1Х, liquid), (CELLnTEC, Швейцария), # CnT-07, содержащей 1× добавку инсулин-трансферрин-селенит (Invitrogen, США) и 10 нг/мл эпидермальный фактор роста (Sigma, США). Клетки помещали в покрытые коллагеном I типа культуральные флаконы в количестве 5×103клеток на 1 см2 и инкубировали при 37°С и 5% CO2. Первые 5 дней среду культивирования меняли ежедневно, затем - каждые 3 дня.
[0112] По достижении клетками конфлюэнтного монослоя (на 10 день после выделения), среду культивирования удаляли, клетки дважды промывали раствором Версена (ПанЭко, Россия), затем инкубировали 5 минут с 0,05% раствором трипсина в ЭДТА (Gibco, США) в количестве 1 мл на 25 см2площади культурального флакона. Затем клетки отмывали от трипсина фосфатно-солевым буфером, осаждали центрифугированием 5-10 минут при 200 g.
[0113] Для проведения панкреатической дифференцировки клетки ресуспендировали в соотношении 1:3 в среде DMEM/F12 1:1 (Gibco, США), содержащей 10% эмбриональной телячьей сыворотки (HyClone, США), 1× добавку инсулин-трансферрин-селенит (Invitrogen, США), 2 мМ глутамина (Invitrogen, США), 10 нг/мл эпидермального фактора роста (Sigma, США), с добавлением 2 мкМ ретиноевой кислоты (Sigma, США), 1 мкМ изопротеринола (Sigma, США), 10 нг/мл фактора роста фибробластов 10 (FGF-10) (Life Technologies, США) и 10 нг/мл инсулиноподобного фактора роста 1 (IGF-1) (R&D, США), помещали в новые культуральные флаконы, покрытые коллагеном I типа и культивировали 7 дней. С 7 по 14 день клетки инкубировали в среде DMEM/F12 1:1 (Gibco, США) с 10% эмбриональной телячьей сыворотки (HyClone, США), 2 мМ глутамина (Invitrogen, США), 10 нг/мл эпидермального фактора роста (Sigma, США), с добавлением 100 нМ ретиноевой кислоты (Sigma, США), 10 мМ никотинамида (Sigma, США), 10 нг/мл инсулиноподобного фактора роста 1 (IGF-1) (R&D, США), 20 нг/мл фактора роста гепатоцитов (HGF) (Gibco, США) и 0,1 мкМ дексаметазона (Sigma, США).
[0114] По окончании инкубирования из клеток выделяли тотальную РНК, которую использовали для синтеза кДНК и количественной ПЦР с детекцией в реальном времени. Тотальную РНК выделяли с помощью набора RNeasy Mini Kit (250) (Qiagen, Германия) по инструкциям производителя. Синтез кДНК проводили с помощью набора для обратной транскриптазы MMLV RT kit (Евроген, Россия) со случайными праймерами по инструкциям производителя. После этого проводили количественную ПЦР с детекцией в реальном времени с применением смеси для ПЦР qPCRmix-HS SYBR (Евроген, Россия) по инструкциям производителя на приборе CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, США) с ген-специфическими праймерами, показанными в Таблице 1. Данные количественной ПЦР были нормализованы по GAPDH.
[0115] Проведенный ПЦР анализ показал, что после панкреатической дифференцировки клеток слюнной железы человека, в них повышается экспрессия ключевых транскрипционных факторов, необходимых для дифференцировки бета-клеток: NGN3, PDX1, MAFA, РАХ4, РАХ6, NKX6.1 (таблица 2). Помимо этого, после панкреатической дифференцировки повышается экспрессия РНК препроинсулина (таблица 2), тогда как экспрессия амилазы уменьшается. Таким образом, был сделан вывод, что предложенный протокол обеспечивает специфично и эффективно эндокринную дифференцировку эпителиальных клеток.
[0116] Для оценки эффективности панкреатической дифференцировки исследовали эпителиальные прогениторные клетки слюнной железы после 1-го пассажа и клетки после панкреатической дифференцировки. Клетки окрашивали антителами к инсулину и проинсулину (список использованных в работе антител представлен в таблице 3). За 48 часов до окраски клетки рассаживали на покрытые коллагеном I типа лунки и инкубировали в среде культивирования (РСТ Epidermal Keratinocyte Medium (1Х, liquid), (CELLnTEC, Швейцария), # CnT-07, содержащей 1× добавку инсулин-трансферрин-селенит (Invitrogen, США) и 10 нг/мл эпидермального фактора роста (Sigma, США) для не дифференцированных клеток и DMEM/F12 1:1 (Gibco, США) с 10% эмбриональной телячьей сыворотки (HyClone, США), 2 мМ глутамина (Invitrogen, США), 10 нг/мл эпидермального фактора роста (Sigma, США), с добавлением 100 нМ ретиноевой кислоты (Sigma, США), 10 мМ никотинамида (Sigma, США), 10 нг/мл инсулиноподобного фактора роста 1 (R&D, США), 20 нг/мл фактора роста гепатоцитов (Gibco, США) и 0,1 мкМ дексаметазона (Sigma, США) для дифференцированных. Затем среду удаляли, клетки промывали фосфатно-солевым буфером, фиксировали 10 минут 4% параформальдегидом (Sigma, США) при комнатной температуре. Клетки трижды промывали фосфатно-солевым буфером и проводили блокировку неспецифического связывания антител в растворе 1% бычьего сывороточного альбумина (Sigma, США) и 0,1% тритона (Sigma, США) в фосфатно-солевом буфере при комнатной температуре в течение 30 минут. С первичными антителами клетки инкубировали в фосфатно-солевом буфере 60 минут при 37°С (или при +4°С в течение ночи) в разведении, рекомендованном производителем (обычно - 1:200-1:500). Клетки отмывали три раза по 10 минут фосфатно-солевым буфером при 37°С, после чего инкубировали с вторичными антителами в фосфатно-солевом буфере (разведение 1:1000) в течение 40-60 минут при 37°С. Снова отмывали три раза по 10 минут фосфатно-солевым буфером при 37°С, добавляя во время последней отмывки 1 мкг/мл DAPI (Sigma, США). Анализировали под флуоресцентным микроскопом Olympus IX51 (Olympus, Япония).
[0117] Было показано возрастание продукции белков проинсулина и инсулина в клеточном продукте после дифференцировки по сравнению с исходной культурой прогениторных эпителиальных клеток (Фиг 1-4).
[0118] Количественную оценку изменения уровня экспрессии инсулина и проинсулина проводили методом проточной цитометрии. Для этого удаляли среду культивирования, клетки дважды промывали раствором Версена (ПанЭко, Россия), затем инкубировали 5 минут с 0,05% раствором трипсина в ЭДТА (Gibco, США). Раствор трипсина добавляли в количестве 1 мл на 25 см2площади культурального флакона. Затем клетки отмывали от трипсина фосфатно-солевым буфером, центрифугировали 5-10 минут при 200 g и тщательно ресуспендировали в фосфатно-солевом буфере с 2% эмбриональной телячьей сыворотки (HyClone, США), добиваясь моноклеточной суспензии. Далее клеточную суспензию разделяли на аликвоты из расчета по 1×106 клеток на одно антитело, кроме того делали аликвоты по 1×106 клеток для каждого изотип-контроля. Инкубировали клетки с первичными антителами в разведении, рекомендованном производителем (1:500-1:1000) в течение 60 минут при комнатной температуре в темноте. Далее клетки трижды промывали фосфатно-солевым буфером (по 10 минут) и в случае, если антитела были конъюгированы с флуорохромом, фиксировали 1% параформальдегидом (Sigma, США) 5 минут в темноте. Затем клетки трижды промывали фосфатно-солевым буфером (по 10 минут), ресуспендировали в 1 мл фосфатно-солевого буфера и анализировали с помощью проточного цитофлуориметра Cell Lab Quanta™ SC MPL (Beckman Coulter, США). В случае если первичные антитела не были конъюгированы с флуорохромом, клетки после отмывки от первичных антител инкубировали с вторичными антителами в течение 40 минут при комнатной температуре в темноте. Затем производили фиксацию клеток 1% параформальдегидом (Sigma, США) и анализ как описано выше. В качестве контроля использовали соответствующий изотип-контроль, анализировали не менее 10000 клеток.
[0119] Методом проточной цитометрии показано, что после дифференцировки клеток слюнной железы человека, доля клеток, экспрессирующих белок проинсулина увеличивается с 32% до 77%, а белок инсулина с 5% до 88%. Полученные изменения в экспрессии генов, происходящие от применения разработанного протокола панкреатической дифференцировки уникальны, и не описаны для других протоколов и других типов клеток.
[0120] Было исследовано влияние различных факторов на эффективность панкреатической дифференцировки. В экспериментах использовали клетки слюнной железы человека, полученные как описано выше. При проведении панкреатической дифференцировки варьировали концентрации отдельных компонентов культуральной среды. По окончании дифференцировки проводили выделение тотальной РНК из клеток, синтез кДНК и анализ эффективности дифференцировки клеток методом количественной ПЦР с детекцией в реальном времени. В качестве контролей использовали клетки слюнной железы человека, не прошедшие дифференцировку и дифференцированные по вышеописанному протоколу.
[0121] Была проверена возможность использования иных жидких культуральных сред для панкреатической дифференцировки. Использовали среды: DMEM (Gibco, США), IMDM (Gibco, США), MEM (Gibco, США), William's E medium (Gibco, США), RPMI 1640 (Gibco, США), Alpha-MEM (Gibco, США), PCT Epidermal Keratinocyte Medium # CnT-07 (1X, liquid), (CELLnTEC, Швейцария). В результате проведенного ПЦР анализа не было выявлено существенной разницы между экспрессией генов-маркеров эндокринной панкреатической дифференцировки при проведении дифференцировки в использованных культуральных средах, за исключением среды Epidermal Keratinocyte Medium # CnT-07, в которой наблюдалось возрастание уровня экспрессии экзокринного маркера амилазы. Был сделан вывод, что среды DMEM, IMDM, MEM, William's E medium, RPMI 1640, Alpha-MEM могут быть использованы для проведения панкреатической дифференцировки.
[0122] Было показано, что при понижении концентрации эмбриональной телячьей сыворотки ниже 2 объемных % в дифференцировочной среде у клеток не наблюдается митотической активности, эффективность дифференцировки падает, что отражается в снижении уровня экспрессии маркеров бета-клеток (PDX1, NGN3, MAFA, NKX6.1, РАХ4, РАХ6, INS). При повышении концентрации эмбриональной телячьей сыворотки выше 20 объемных % в дифференцировочной среде наблюдалась клеточная гибель.
[0123] При снижении концентрации глутамина в среде ниже 1 мМ у клеток не наблюдалось митотической активности и появляются признаки клеточного голодания (крупное светлое ядро, снижение синтеза белков). Эффективность клеточной дифференцировки также снижается. При повышении концентрации глутамина в среде выше 4 мМ значительных изменений в поведении клеток и эффективности клеточной дифференцировки не происходило, поэтому повышать рекомендуемую концентрацию нецелесообразно.
[0124] При снижении концентрации эпидермального фактора роста ниже 1 нг/мл снижалась митотическая активность клеток и эффективность панкреатической дифференцировки, что выражалось в снижении уровня экспрессии маркеров бета-клеток (PDX1, NGN3, MAFA, NKX6.1, РАХ4, РАХ6, INS). При повышении концентрации эпидермального фактора роста выше 300 нг/мл также снижалась митотическая активность клеток и эффективность дифференцировки.
[0125] При снижении концентрации трансферрина ниже 0,1 мкг/мл уменьшалась жизнеспособность клеток, тогда как повышение его концентрации выше 20 мкг/мл не оказывало детектируемого влияния на клетки.
[0126] Снижение концентрации селенита натрия ниже 0,1 нг/мл приводило к понижению пролиферативной активности клеток, а также к увеличению спонтанной дифференцировки, что выражалось в увеличении размеров клеток, появлении гранулярности цитоплазмы. Отмечались признаки деградации клеточной культуры, гибель клеток и снижение эффективности панкреатической дифференцировки. Превышение концентрации селенита натрия выше 20 нг/мл приводило к снижению эффективности панкреатической дифференцировки.
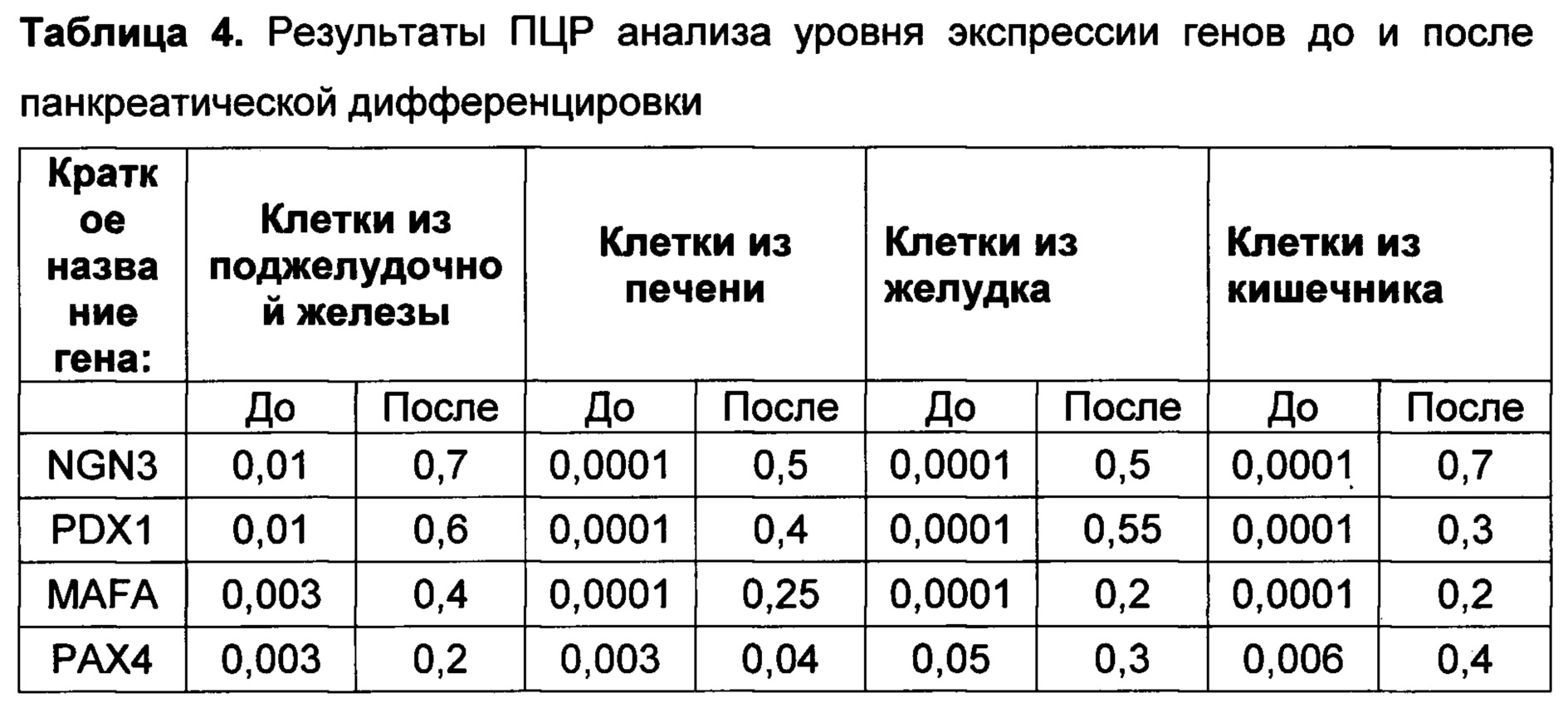
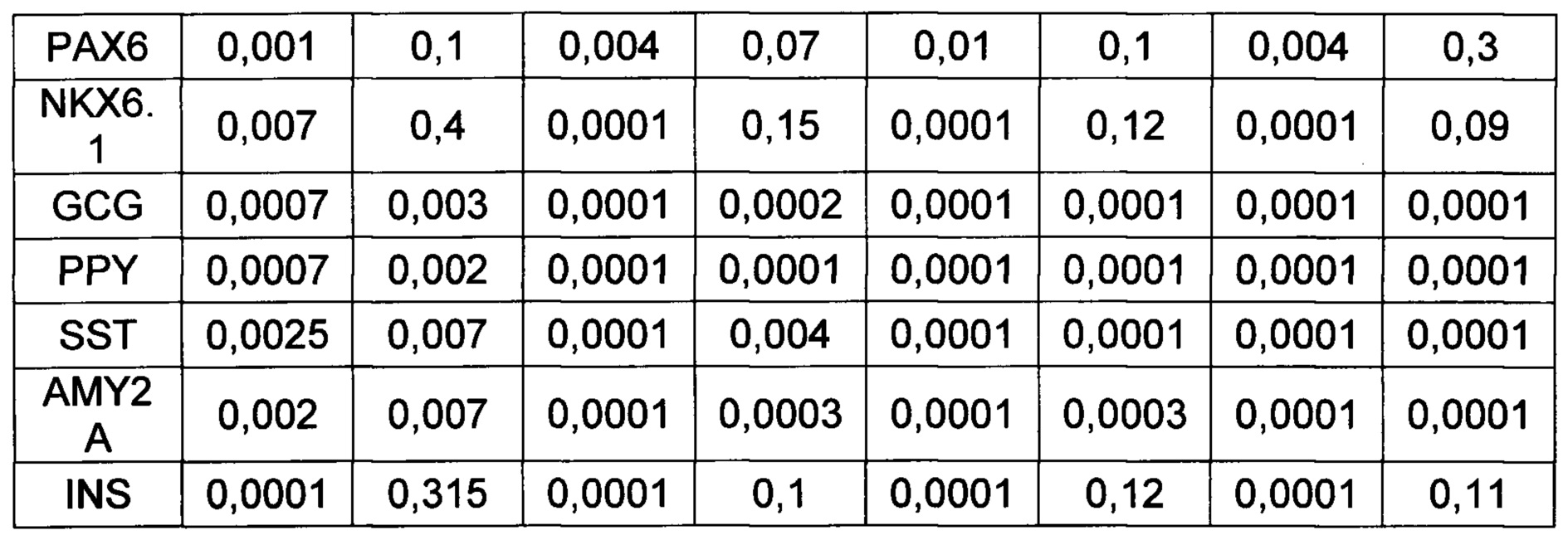
[0127] Как понижение концентрации ретиноевой кислоты ниже 0,1 мкМ, так и повышение ее концентрации выше 20 мкМ на первом этапе дифференцировки приводило к значительному снижению эффективности панкреатической дифференцировки эпителиальных клеток, о чем свидетельствовало отсутствие повышения уровня экспрессии панкреатических маркеров (PDX1, NGN3, MAFA, NKX6.1, РАХ4, РАХ6, INS). На второй стадии панкреатической дифференцировки снижение концентрации ретиноевой кислоты ниже 10 нМ и повышение выше 20 мкМ также значительно снижало эффективность панкреатической дифференцировки.
[0128] Снижение концентрации изопротеренола ниже 0,1 мкМ на первой стадии дифференцировки значительно снижало уровень экспрессии РАХ4, необходимого для развития бета-клеток. Повышение концентрации изопротеренола выше 10 мкМ на первой стадии дифференцировки оказывало тормозящий эффект на дифференцировку, что отражалось в снижении уровня экспрессии панкреатических маркеров.
[0129] На второй стадии дифференцировки понижение концентрации никотинамида ниже 1 мМ приводило к снижению уровня экспрессии панкреатических маркеров (PDX1, NGN3, MAFA, NKX6.1, РАХ4, РАХ6, INS). Повышение концентрации никотинамида выше 100 мМ снижало жизнеспособность клеток.
[0130] Понижение концентрации фактора роста гепатоцитов на второй стадии дифференцировки ниже 1 нг/мл, как и понижение концентрации дексаметазона ниже 0,01 мкМ приводило к снижению уровня экспрессии гена препроинсулина (INS). Повышение концентрации фактора роста гепатоцитов выше 300 нг/мл или дексаметазона выше 5 мкМ угнетало клеточную пролиферацию и снижало жизнеспособность клеток.
[0131] Также было показано, что применение добавки инсулин-трансферрин-селенит на первой стадии дифференцировки практически не влияет на экспрессию панкреатических маркеров эпителиальными клетками по сравнению с применением трансферрина и селенита натрия.
[0132] Добавление инсулиноподобного фактора роста 1 в культуральную среду на первой и второй стадиях дифференцировки повышало жизнеспособность эпителиальных клеток. При добавлении фактора роста фибробластов 10, фактора рост фибробластов 4 или фактора роста кератиноцитов; на первой стадии дифференцировки наблюдалось повышение жизнеспособности и митотической активности клеток. Применение бетацеллюлина на второй стадии дифференцировки приводило к небольшому повышению уровня экспрессии препроинсулина. Был сделан вывод о возможности добавления этих компонентов в культуральную среду при проведении панкреатической дифференцировки.
[0133] Было исследовано влияние газовой смеси на дифференцировку эпителиальных клеток: оценивали эффективность дифференцировки в присутствии 5% СО2, а также в присутствии 5% СО2 и 5% О2. Было показано, что панкреатическая дифференцировка осуществляется в обеих исследованных газовых смесях, однако, в присутствии 5% СО2 и 5% О2 повышается жизнеспособность клеток: уменьшается спонтанная клеточная гибель. По этой причине в некоторых воплощениях возможно применение газовой смеси 5% СО2 и 5% О2.
[0134] Было исследовано также влияние длительности стадий дифференцировки. Показано, что при уменьшении длительности первой и второй стадий панкреатической дифференцировки менее 4-х дней, клетки не успевают дифференцироваться, что выражается в низком уровне экспрессии панкреатических маркеров: PDX1, NGN3, MAFA, NKX6.1, РАХ6, INS. При увеличении длительности первой и второй стадий панкреатической дифференцировки больше 15-ти дней, снижается жизнеспособность клеток, уменьшается пролиферативная активность клеток и увеличивается их спонтанная гибель.
[0135] Пример 2. Панкреатическая дифференцировка эпителиальных прогениторных клеток печени, поджелудочной железы, тонкого кишечника и желудка человека
[0136] Биоптаты печени, поджелудочной железы, тонкого кишечника и желудка человека получали в ходе плановых операций по удалению частей органов. Биоптаты органов размером 0,5-5 см3 переносили в стерильных условиях в чашку Петри со средой DMEM/F12 1:1 (Gibco, США) и гентамицином 40 мкг/мл (ПанЭко, Россия). Все дальнейшие манипуляции проводили в стерильных условиях, отвечающих требованиям GMP (Good Manufacturing Practice).
[0137] Биоптаты органов трижды промывали фосфатно-солевым буфером, механически измельчали до кусочков размером около 1-5 мм3 стерильными инструментами (скальпелем и пинцетами). Затем кусочки тканей осаждали центрифугированием в течение 5-10 минут при 0,8-1,5 тыс. об/мин и инкубировали в присутствии 2-4 мг/мл раствора коллагеназы IV типа (Gibco, США) в среде DMEM/F12 1:1 (Gibco, США) с 2 мМ глутамином (Invitrogen, США) (всего - 1 мл раствора на 0,5 см3 ткани). Каждые 10-15 минут пробирки с кусочками тканей активно встряхивали.
[0138] После этого к клеткам добавляли по 10 мл среды DMEM/F12 1:1 (Gibco, США), активно пипетировали в течение 3-5 минут, затем пропускали через нейлоновый фильтр с диаметром пор 40-100 мкм и осаждали клетки центрифугированием в течение 5-10 минут при 0,8-1,5 тыс. об/мин.
[0139] Для получения прогениторных клеток печени и поджелудочной железы проводили магнитную сепарацию клеток по маркеру ЕрСАМ. Для этого суспензию клеток промывали 10 мл фосфатно-солевого буфера, ресуспендировали в 0,5 мл фосфатно-солевого буфера, подсчитывали количество клеток на приборе для автоматизированного подсчета клеток (BioRad, США). Инкубировали клетки с антителами anti-human ЕрСАМ, конъюгированными с магнитными частицами (Miltenyi Biotec GmbH, Германия), в течение 15-40 минут при +4°С, концентрация антител - 0,1-5 мкг на 1 млн клеток. Промывали клетки 10 мл фосфатно-солевого буфера и проводили магнитную сепарацию клеток на колонках MiniMACS™ Separator (Miltenyi Biotec GmbH, Германия) по инструкциям производителя. Отсортированные клетки осаждали центрифугированием в течение 5-10 минут при 200 g.
[0140] Для получения прогениторных клеток кишечника проводили флуоресцентный сортинг клеток по маркеру LGR-5, а для получения прогениторных клеток желудка - по маркеру c-Kit. Суспензию клеток промывали 10 мл фосфатно-солевого буфера, ресуспендировали в 0,5 мл фосфатно-солевого буфера, подсчитывали количество клеток на приборе для автоматизированного подсчета клеток (Bio-Rad, США). Инкубировали клетки с антителами anti-LGR-5 (или anti-c-Kit соответственно), конъюгированными с флуоресцентной меткой Alexa Fluor 488, в течение 15-40 минут при +4°С, концентрация антител - 0,1-5 мкг на 1 млн клеток. Промывали 10 мл фосфатно-солевого буфера и проводили флуоресцентный сортинг на приборе S3e™ Cell Sorter (Bio-Rad, США).
[0141] После сортировки, клетки печени, поджелудочной железы, кишечника и желудка промывали 10 мл фосфатно-солевого буфера и ресуспендировали в ростовой среде DMEM/F12 1:1 (Gibco, США), содержащей 10% эмбриональной телячьей сыворотки (HyClone, США), 1× добавку инсулин-трансферрин-селенит (Invitrogen, США), 2 мМ глутамина (Invitrogen, США) и 10 нг/мл эпидермального фактора роста (Sigma, США). Клетки помещали в покрытые коллагеном I типа культуральные флаконы в количестве 5×103 клеток на 1 см2и инкубировали при 37°С и 5% СО2. Среду культивирования меняли каждые 3 дня.
[0142] После достижения эпителиальными клетками печени, поджелудочной железы, тонкого кишечника и желудка человека монослойной культуры на первом пассаже их подвергали панкреатической дифференцировке по протоколу, описанному в Примере 1.
[0143] На 14-й день дифференцировки проводили выделение тотальной РНК из клеток, синтез кДНК и анализ генов-маркеров панкреатической дифференцировки клеток методом количественной ПЦР с детекцией в реальном времени как описано в Примере 1. В качестве контролей использовали соответствующие эпителиальные клетки человека на первом пассаже, не прошедшие дифференцировку. Данные количественной ПЦР нормализовали по GAPDH.
[0144] Результаты ПЦР анализа панкреатической дифференцировки эпителиальных клеток человека говорят о том, что используемый протокол эффективен для всех изученных типов клеток (таблица 4). После эндокринной панкреатической дифференцировки возрастает экспрессия генов транскрипционных факторов, необходимых для формирования инсулин-продуцирующих клеток (NGN3, PDX1, MAFA, РАХ4, NKX6.1), а также препроинсулина. При этом не происходит значимого увеличения экспрессии амилазы, соматостатина, глюкагона, панкреатического полипептида, что говорит о специфичности применяемого протокола для всех изученных культур.
[0145] Пример 3. Культивирование клеточного продукта в условиях трехмерного культивирования
[0146] Клеточный продукт инсулин-продуцирующих клеток, полученный как описано в Примере 1, после панкреатической дифференцировки дважды промывали раствором Версена (ПанЭко, Россия) и инкубировали 5 минут с 0,05% раствором трипсина в ЭДТА (Gibco, США). Раствор трипсина добавляли в количестве 1 мл на 25 см2 площади культурального флакона. Затем клетки отмывали от трипсина фосфатно-солевым буфером, осаждали центрифугированием 5-10 минут при 200 g и ресуспендировали в той же среде, которую использовали для второй стадии панкреатической дифференцировки. Среду добавляли из расчета 20 мкл среды на каждые 5 тысяч клеток. Пипеткой раскапывали по 20 мкл суспензии клеток на крышку чашки Петри, закрывали чашку и инкубировали клетки в виде висячих капель в стерильных условиях при 37°С и 5% CO2 последующие 5 дней. В результате клетки внутри капель образовывали агрегаты в виде сфероидов размером 250-500 мкм.
[0147] Анализ экспрессии инсулина и проинсулина в сфероидах проводили методом иммуноцитохимии. Сфероиды собирали пипеткой в пробирку, осаждали центрифугированием при 200 g в течение 5 минут, удаляли супернатант, осадок замораживали в среде для приготовления криосрезов OCT (CrioMount Histolab, Швеция). Криосрезы готовили на криостате Leica СМ1900 (Leica, Швейцария) на предметных стеклах Super Frost Plus (Menzel, Германия), толщина срезов 10 мкм. Сразу после приготовления криосрезы фиксировали 4% параформальдегидом (Sigma, США) 10 минут при комнатной температуре. Затем промывали фосфатно-солевым буфером и инкубировали с первичными антителами к инсулину и проинсулину в разведении 1:500 в растворе 0,1% тритона (Sigma, США) и 1% бычьего сывороточного альбумина (Sigma, США) в фосфатно-солевом буфере в течение ночи при +4°С. Отмывали три раза по 10 минут фосфатно-солевым буфером при 37°С и инкубировали с вторичными антителами в разведении 1:1000 в течение 60 минут при 37°С. Криосрезы отмывали три раза по 10 минут фосфатно-солевым буфером и окрашивали ядра клеток 1 мкг/мл DAPI (Sigma, США) 5 минут. Затем криосрезы промывали фосфатно-солевым буфером и заключали под покровное стекло в среде 25% глицерин (Sigma, США) и 25% полиэтиленгликоль (Sigma, США) в фосфатно-солевом буфере. Анализировали срезы под флуоресцентным микроскопом Keyence BZ-9000 (Keyence, Япония). Иммуноцитохимическое окрашивание криосрезов сфероидов клеток слюнной железы показало, что в трехмерных условиях культивирования дифференцированные эпителиальные клетки активно экспрессируют белки проинсулина (Фиг. 5) и инсулина (Фиг. 6).
[0148] Анализ экспрессии генов, необходимых для глюкозозависимой секреции инсулина, методом количественной ПЦР с детекцией в реальном времени был проведен как описано в Примере 1. В качестве контроля использовали недифференцированные эпителиальные прогениторные клетки и клетки после панкреатической дифференцировки, культивированные в двухмерных условиях. Было показано, что прогениторные эпителиальные клетки человека, не прошедшие дифференцировку, практически не экспрессируют мРНК генов, необходимых для осуществления глюкозозависимой секреции инсулина. После дифференцировки происходит увеличение уровня экспрессии этих генов, причем в клетках, которые культивировали в трехмерных условиях, уровень экспрессии этих генов выше (таблица 5). Данные количественной ПЦР нормализовали по GAPDH.

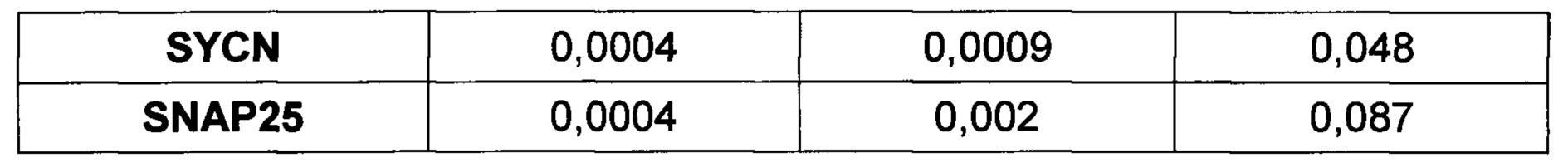
[0149] Исследование секреции инсулина и С-пептида в ответ на разные концентрации глюкозы проводили методом иммуноферментного анализа (ИФА) для клеток слюнной железы, прошедших сфероидное культивирование. Для этого готовили сфероиды по способу, описанному выше. Сфероиды собирали пипеткой с крышек чашек Петри, осаждали центрифугированием 5 минут при 200 g и трижды промывали от остатков среды фосфатно-солевым буфером. После этого инкубировали в Krebs-Ringer буфере (107 мМ NaCl (Sigma, США), 5 мМ KCl (Sigma, США), 3 мМ CaCl2 (Sigma, США), 1 мМ MgSO4*6H2O (Sigma, США), 7 мМ NaHCO3 (Sigma, США), 0,1% BSA (Sigma, США) и 20 мМ HEPES (Sigma, США), pH=7,6) в течение 2 часов при 37°С. Буфер удаляли, а сфероиды делили на 4 аликвоты и инкубировали 1 час при 37°С при разных концентрациях глюкозы (Sigma, США) в буфере Krebs-Ringer: с 0 мМ, 2 мМ, 5 мМ и 15 мМ глюкозы. Супернатант собирали в пробирки для дальнейшего ИФА анализа содержания инсулина и С-пептида с помощью наборов Ultrasensitive Insulin ELISA (Mercodia, Швеция) и Ultrasensitive С-Peptide ELISA (Mercodia, Швеция) по инструкциям производителя. Анализировали результат на планшетном ридере Start Fax-2100 (Awareness Technology, США). Сфероиды анализировали по содержанию белка методом Bradford Protein Assays (Thermo Fisher Scientific, Германия) по инструкциям производителя. Количество белка определяли на планшетном ридере Start Fax-2100 (Awareness Technology, США) при длине волны 595 нм в кювете с толщиной слоя 10 мм. После этого нормировали количество секретированного инсулина и С-пептида по общему количеству белка. Было показано, что после панкреатической дифференцировки в трехмерных условиях культивирования клетки слюнной железы человека приобретают способность к глюкозозависимой секреции инсулина. Секреция инсулина возрастает не менее чем в два раза (таблица 6) при увеличении концентрации глюкозы в среде с 5 мМ до 15 мМ и достигает 14,5 нг/мг белка. Одновременная секреция С-пептида клетками в эквимолярном с инсулином количестве подтверждает эндогенное происхождение детектируемого инсулина и его правильное созревания в клетках слюнной железы после панкреатической дифференцировки (таблица 6). Таким образом, клетки слюнной железы человека после панкреатической дифференцировки обладают способностью к глюкозозависимой секреции in vitro в трехмерных условиях культивирования.
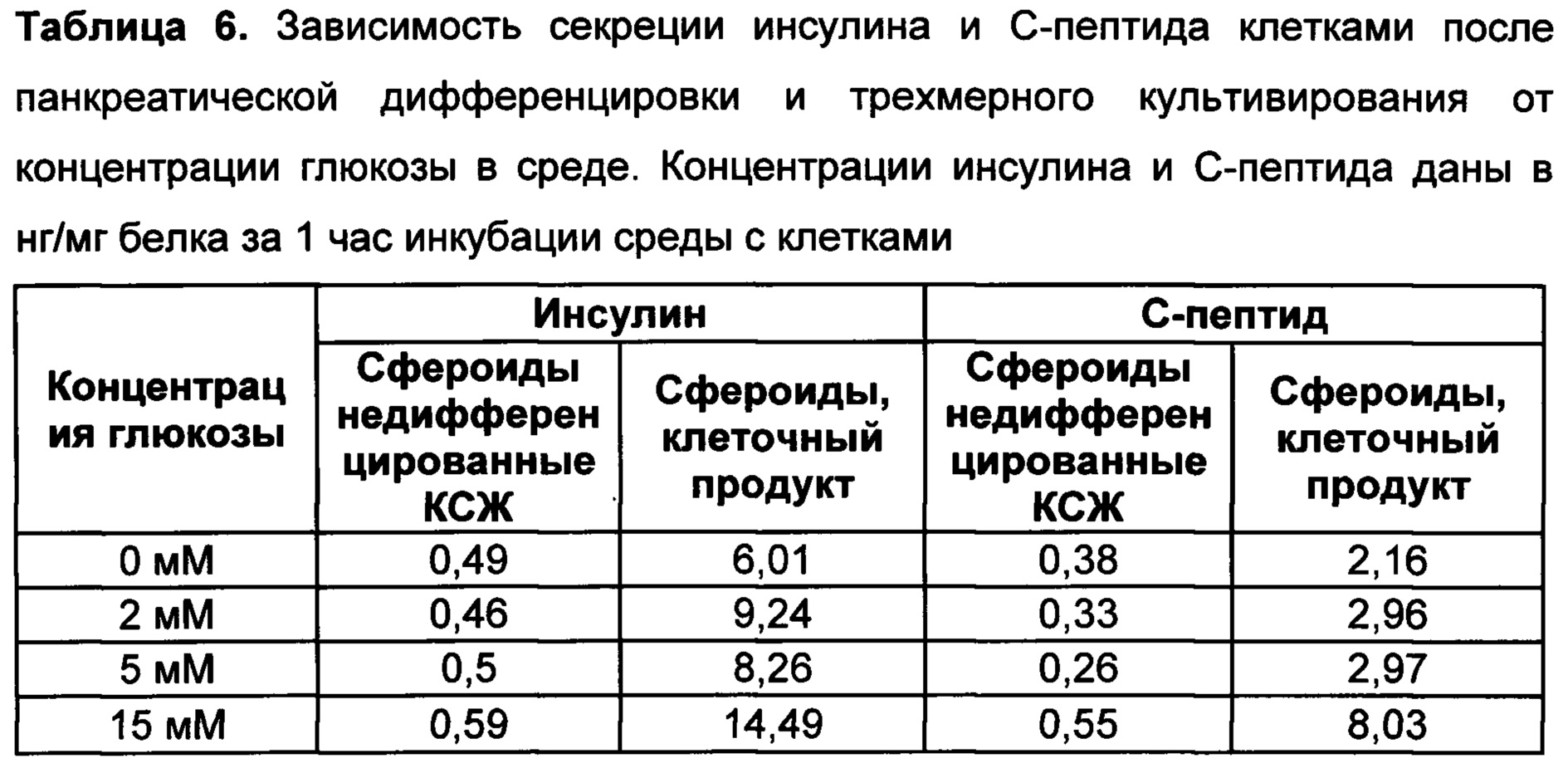
[0150] Пример 4. Анализ функциональной активности клеточного продукта
[0151] Клетки из слюнной железы человека на первом пассаже дифференцировали, как описано в Примере 1, затем дважды промывали раствором Версена, снимали с поверхности культуральных флаконов трипсином как описано в Примере 1, осаждали центрифугированием в течение 5 минут при 200 g, промывали три раза фосфатно-солевым буфером и подсчитывали количество клеток на автоматической счетчике (Bio-Rad, США). Далее к клеткам добавляли фосфатно-солевой буфер из расчета 500 мкл буфера на 5 млн клеток. Аликвоты полученной суспензии, содержащие 5 млн клеток клеточного продукта, инъецировали внутрибрюшинно с помощью стерильных шприцов иммунодефицитным мышам Nude. В качестве контроля использовали мышей Nude, которым проводили инъекцию 500 мкл фосфатно-солевого буфера без клеток. Через 3 дня после инъекции исследовали поджелудочную железу у части мышей: животных наркотизировали изофлураном (Sigma-Aldrich, США), затем смачивали живот спиртом и делали надрез кожи и брюшины. Поджелудочную железу извлекали пинцетами и подвергали криозаморозке в среде для приготовления криосрезов ОСТ (CrioMount Histolab, Швеция). Криосрезы поджелудочной железы готовили на криостате Leica СМ1900 (Leica, Швейцария) на предметных стеклах Super Frost Plus (Menzel, Германия), толщина срезов 10 мкм.
[0152] Криосрезы сразу после приготовления фиксировали 4% параформальдегидом (Sigma, США) 10 минут при комнатной температуре. Затем промывали фосфатно-солевым буфером и инкубировали с первичными антителами anti-Human nuclei, конъюгированными с флуорохромом Alexa Fluor 488 и anti-insulin в разведении 1:500 в растворе 0,3% тритона (Sigma, США) и 2% бычьего сывороточного альбумина (Sigma, США) в фосфатно-солевом буфере в течение ночи при +4°С. Затем отмывали три раза по 10 минут фосфатно-солевым буфером при 37°С и инкубировали с вторичными антителами Alexa Fluor® 546 goat anti-rabbit IgG (H+L) (Invitrogen, США) в разведении 1:1000 в фосфатно-солевом буфере в течение 60 минут при 37°С. Криосрезы отмывали три раза по 10 минут фосфатно-солевым буфером и окрашивали ядра клеток 1 мкг/мл DAPI (Sigma, США) 5 минут. Промывали криосрезы фосфатно-солевым буфером и заключали под покровное стекло в среде 25% глицерин (Sigma, США) и 25% полиэтиленгликоль (Sigma, США) в фосфатно-солевом буфере. Анализировали срезы под флуоресцентным микроскопом Keyence BZ-9000 (Keyence, Япония). Всего было проанализировано 5 экспериментальных мышей в каждой группе животных. Трансплантированные клетки у экспериментальных мышей были обнаружены в поджелудочной железе, где они сформировали агрегаты (Фиг. 7). Окраска антителами к инсулину подтвердила, что эти клетки продуцируют белок инсулин in vivo.
[0153] На 7-й день после трансплантации клеток анализировали содержание человеческого инсулина в сыворотке крови оставшихся мышей. Для этого контрольные мыши, получившие 500 мкл фосфатно-солевого буфера и опытные мыши, получившие трансплантацию 5 млн дифференцированных клеток слюнной железы человека, были лишены корма на 6 часов. После этого у 5 контрольных и у 5 опытных мышей взяли кровь: после наркотизации изофлураном, мышам вскрыли грудную клетку и шприцом, смоченным гепарином, взяли кровь из сердца (0,5-1 мл). Кровь поместили в пробирки с гепарином и осадили центрифугированием в течение 20 минут при 10000 об/мин. Сыворотки крови были отобраны в отдельные пробирки и заморожены при -80°С для дальнейшего анализа. Кроме этого, после 6-часового голодания 5 контрольных и 5 опытных мышей были инъецированы внутрибрюшинно раствором глюкозы количестве 2 мг на кг веса. Через 30 минут после инъекции глюкозы у мышей также была взята кровь и приготовлена сыворотка, как описано выше.
[0154] Был проведен иммуноферментный анализ содержания человеческого инсулина в сыворотках крови мышей с помощью набора Ultrasensitive Insulin ELISA (Mercodia, Швеция) по инструкциям производителя. Для проведения анализа использовали планшетный ридер Start Fax-2100 (Awareness Technology, США). В сыворотке крови всех экспериментальных мышей из опытной группы был выявлен человеческий инсулин. При этом количество инсулина у мышей, получивших инъекцию глюкозы перед взятием крови для анализа, было выше примерно в 2 раза (таблица 7).

[0155] Полученный результат подтверждает, что эпителиальные прогениторные клетки человека после панкреатической дифференцировки способны к глюкозозависимой секреции инсулина in vivo после введения в организм.
[0156] Известно, что прогениторные протоковые клетки слюнных желез обладают потенциалом к дифференцировке в экзокринные клетки, секретирующие амилазу. Поэтому была исследована способность клеток после панкреатической дифференцировки к экзокринной дифференцировке. Клетки слюнной железы человека на первом пассаже были подвергнуты эндокринной панкреатической дифференцировке, как описано в Примере 1. Затем клетки были окрашены иммуноцитохимически антителами к слюнной и панкреатической амилазам методом, описанным выше в Примере 1. Кроме того, были приготовлены криосрезы сфероидов и окрашены антителами к амилазе методами, описанными выше в Примере 3. Антителами к амилазе и к Human nuclei были окрашены криосрезы поджелудочной железы иммунодефицитных мышей nude на третий день после трансплантации 5 млн клеток слюнной железы человека (метод описан выше в Примере 3).
[0157] В результате проведенного иммунофенотипического окрашивания было показано, что в клетках слюнной железы человека амилаза не детектируется ни при культивировании на пластике, ни в трехмерных условиях культивирования. Кроме того, клетки слюнной железы человека не приобретают способность к продукции амилазы in vivo - после трансплантации иммунодефицитным мышам Nude. Эти результаты говорят о специфичности проведенной эндокринной дифференцировки эпителиальных клеток.
[0158] Пример 5. Анализ биологической безопасности клеток слюнной железы человека
[0159] Повышение концентрации амилазы в организме представляет опасность для здоровья. Была исследована способность клеток человека, подвергнутых панкреатической дифференцировке, к ее секреции. Для этого были получены культуры клеток слюнной железы от четырех доноров мужского пола в возрасте от 37 до 55 лет. Культуры названы КСЖ-37М, КСЖ-50М, КСЖ-51М и КСЖ-55М. Клетки слюнной железы человека всех четырех культур на первом пассаже были посажены в 12-луночные культуральные плато в количестве 1 млн клеток на лунку. Через 48 часов среда культивирования была удалена, лунки были промыты три раза фосфатно-солевым буфером, затем к клеткам было добавлено по 1 мл фосфатно-солевого буфера, в котором клетки инкубировали при 37°С в течение 2 часов. Пробы буфера были отобраны и заморожены при -80°С для последующего ИФА. Помимо этого, клетки всех культур были подвергнуты эндокринной панкреатической дифференцировке, как описано выше в Примере 1, а также были приготовлены сфероиды методом, описанным в Примере 3. Аналогичным способом были собраны пробы буфера от дифференцированных клеток слюнной железы и от сфероидов.
[0160] Все пробы были проанализированы методом ИФА на содержание амилазы слюнной железы с помощью набора Salivary Amylase Alpha 1 Human ELISA Kit (MyBioSource, США) и на содержание панкреатической амилазы с помощью набора Pancreatic Amylase Human ELISA Kit (AbCam, Великобритания) по инструкциям производителей. Результат анализировали на планшетном ридере Start Fax-2100 (Awareness Technology, США). В качестве положительного контроля реакции использовали сыворотку крови человека (1 - первый донор, 2 - второй донор).
[0161] Проведенный ИФА по амилазе не выявил секреции панкреатической амилазы и амилазы слюнной железы in vitro клетками слюнной железы всех четырех культур. Секреции амилазы не детектировалось ни после панкреатической дифференцировки клеток, ни в трехмерных условиях культивирования (таблица 8). В лизате КСЖ амилаза также не была обнаружена.
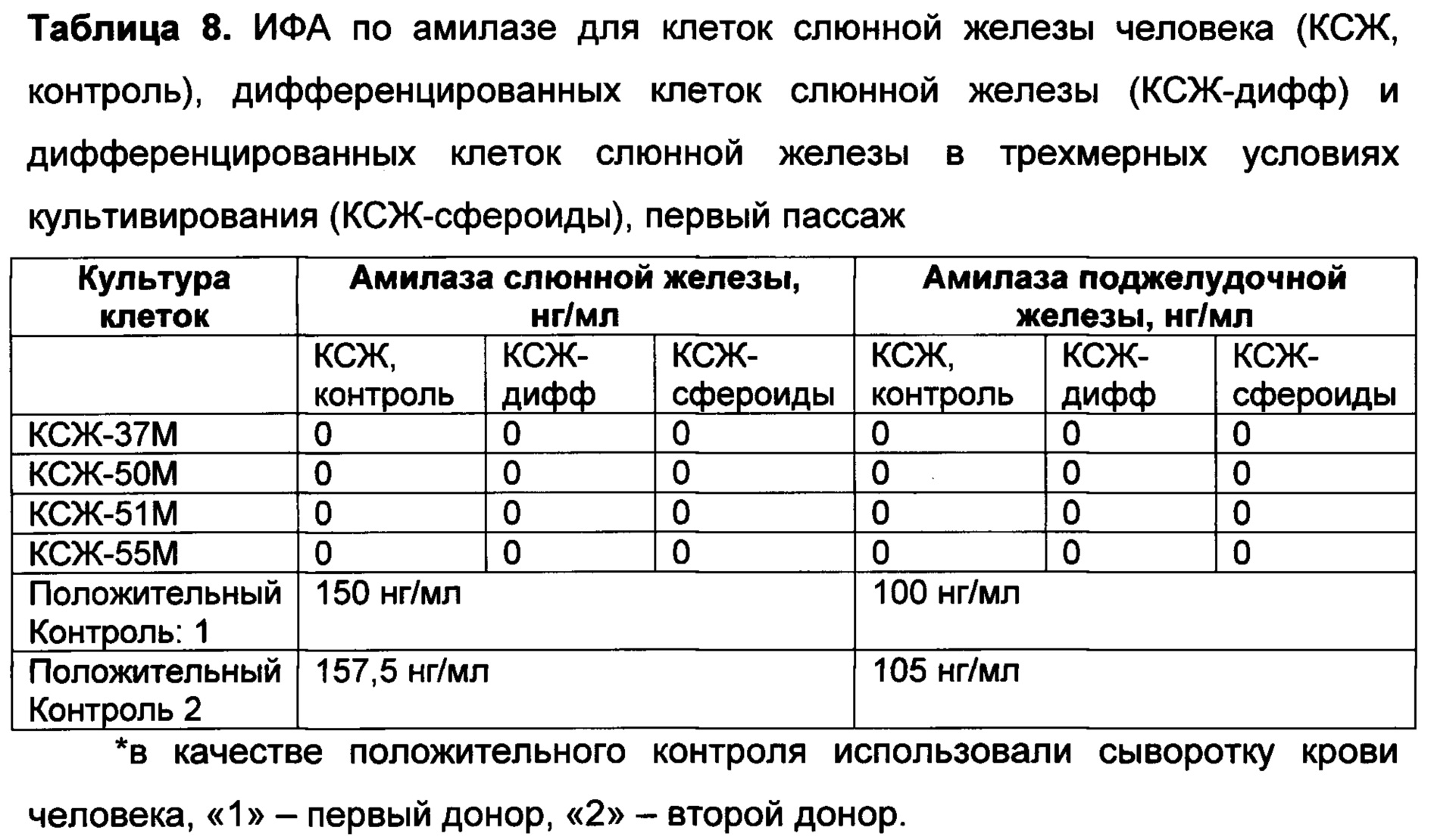
[0162] Для in vivo анализа биологической безопасности эпителиальных клеток человека, 5 млн дифференцированных клеток слюнной железы в виде суспензии в 500 мкл фосфатно-солевого буфера были инъецированы внутрибрюшинно иммунодефицитным мышам Nude. В качестве контроля использовали мышей, которым провели инъекцию 500 мкл фосфатно-солевого буфера. Через 7 дней после трансплантации у мышей взяли кровь для анализа содержания человеческой амилазы в сыворотке крови. Методы получения клеток, их трансплантации и получения сывороток крови мышей описаны выше в Примерах 1 и 4. Для проведения ИФА использовали наборы Salivary Amylase Alpha 1 Human ELISA Kit (MyBioSource, США) и Pancreatic Amylase Human ELISA Kit (AbCam, Великобритания), анализ проводили по инструкциям производителей. Результат анализировали на планшетном ридере Start Fax-2100 (Awareness Technology, США). В качестве положительного контроля реакции использовали сыворотку крови человека (1 - первый донор, 2 - второй донор).
[0163] Проведенный ИФА сывороток крови мышей Nude показал, что через 7 дней после трансплантации дифференцированных клеток слюнной железы человека, в сыворотке крови мышей не наблюдается наличия человеческой амилазы, тогда как в положительном контроле амилаза детектируется в количестве 100-150 нг/мл. Это говорит о том, что клетки слюнной железы человека in vivo не дифференцируются в экзокринном направлении и не продуцируют амилазу.
[0164] На 3-й и 7-й дни после трансплантации клеток слюнной железы человека иммунодефицитным мышам Nude, у них взяли поджелудочную железу на гистологический анализ. Для этого мышей наркотизировали изофлураном (Sigma-Aldrich, США), смачивали живот спиртом и делали надрез кожи и брюшины. Поджелудочную железу извлекали пинцетами и фиксировали 60 минут 4% параформальдегидом (Sigma, США) при +4°С и использовали для приготовления гистологических препаратов: ткань обезвоживали, проводя по спиртам (70% этанол (Sigma, США) в течение 1 часа, 96% этанол в течение 1 часа, 100% этанол в течение 1 часа, 100% этанол и хлороформ (Sigma, США) 1:1 10-15 минут, хлороформ 10-15 минут). Затем ткань помещали в парафин histomix (Биовитрум, Россия), пропитывали в течение 1-2 часов и готовили гистологические срезы парафиновых блоков готовили на микротоме MICROM (Carl Zeiss, Германия), толщина среза 5 мкм. Затем препараты депарафинировали: ксилол (Sigma, США) 2 минуты, 100% этанол (Sigma, США) 2 минуты, 96% этанол 2 минуты, 70% этанол 2 минуты, препараты споласкивали дистиллированной водой. Окрашивали срезы гематоксилином (Sigma, США) 5 минут, после чего споласкивали водой и окрашивали эозином (Sigma, США) в течение 40 секунд. Трижды споласкивали препараты водой и заключали под покровное стекло в среде 25% глицерин (Sigma, США) и 25% полиэтиленгликоль (Sigma, США) в фосфатно-солевом буфере. Анализировали срезы в проходящем свете на микроскопе Keyence BZ-9000 (Keyence, Япония). Гистологический анализ поджелудочных желез мышей Nude, получивших трансплантацию клеток человека, не выявил каких-либо патологических изменений (фиброзных изменений или воспалительных реакций) в поджелудочных железах исследуемых мышей на 3-й и 7-й дни после трансплантации по сравнению с контрольными мышами, получившими инъекцию фосфатно-солевого буфера.
[0165] Поджелудочная железа мышей Nude (10 опытных и 10 контрольных) была исследована на третий день после трансплантации клеток человека на наличие маркера макрофагов CD68 для выявления воспалительной реакции. Криосрезы были приготовлены и окрашены антителами к Human nuclei и CD68 как описано в Примере 4. Проведенное иммуногистохимическое окрашивание не выявило значительной инфильтрации макрофагов вследствие трансплантации дифференцированных клеток слюнной железы человека (Фиг. 8).
[0166] Для исследования способности клеток слюнной железы человека после панкреатической дифференцировки формировать опухоли при трансплантации в организм, 10 млн дифференцированных клеток слюнной железы человека, как описано в Примере 4, на первом пассаже в виде суспензии в 500 мкл фосфатно-солевого буфера были трансплантированы подкожно иммунодефицитным десяти мышам Nude. В качестве контроля использовали десять мышей Nude, которым проводили подкожную инъекцию 500 мкл фосфатно-солевого буфера без клеток. В течение последующих трех месяцев наблюдали выживаемость мышей и обследовали их на наличие опухолей. Показано, что после подкожной трансплантации 10 млн дифференцированных клеток слюнной железы человека иммунодефицитным мышам Nude, у всех мышей не наблюдалось гибели и признаков формирования опухолей в течение всех трех месяцев наблюдения.
[0167] Таким образом, можно заключить, что тестируемые клетки слюнной железы человека отвечают требованиям биологической безопасности: не формируют опухолей, не вызывают воспалительных реакций и патологических изменений в тканях и не продуцируют потенциально опасные для здоровья биологически активные вещества.
[0168] Пример 6. Коррекция экспериментального диабета у мышей
[0169] Экспериментальный диабет вызывали у самцов мышей линии C57Black в возрасте 6-8 недель, весом 20-22 г. Животным после 8 часов голодания вводили ежедневно (в течение 5 дней подряд) внутрибрюшинно стрептозотоцин (Sigma, США) в количестве 40 мкг на кг веса тела, разведенный в 200 мкл цитратного буфера (Sigma, США) ex tempore. Контрольной группе животных вводили цитратный буфер (Sigma, США) в том же объеме (200 мкл). Кормили животных стандартной пищей, поили 10% раствором глюкозы (Sigma, США) в воде на протяжении 5-ти дней. В течение следующей недели животным ежедневно измеряли уровень глюкозы в крови после 6 часов голодания глюкометром OneTouch Select (Johnson&Johnson, США) с помощью тест-полосок OneTouch Ultra (Johnson&Johnson, США). Для измерения глюкозы в крови мышей им делали маленький надрез на конце хвоста, выступающую каплю крови помещали на тест-полоску, измерение глюкометром проводили по инструкциям производителя. Через 1 неделю после последнего введения стрептозотоцина, у животных устанавливается стабильная гипергликемия с концентрацией глюкозы в крови более 25 мМ. После установления стабильной гипергликемии (через 7 дней после последней инъекции стрептозотоцина), мышей разделили на опытную и контрольную группы. Этот день считали нулевым днем эксперимента. В нулевой день эксперимента животным опытной группы внутрибрюшинно трансплантировали 5 млн клеток слюнной железы мыши, дифференцированных по протоколу, описанному в Примере 1, в 500 мкл фосфатно-солевого буфера, как описано в Примере 4. В качестве контроля использовали животных с диабетом, которым инъецировали 500 мкл фосфатно-солевого буфера без клеток, а также здоровых животных без диабета, которым также инъецировали 500 мкл фосфатно-солевого буфера без клеток.
[0170] В последующие 4 недели эксперимента исследовали динамику глюкозы в крови мышей: животным всех групп проводили измерение глюкозы в крови (в одно и тоже время после 4 часов голодания, методом описанным выше). Первое измерение проводили в нулевой день эксперимента, далее - каждые 3 дня. Кроме того, в течение двух месяцев каждый день оценивали выживаемость мышей во всех трех экспериментальных группах.
[0171] Анализ выживаемости мышей показал, что в течение двух месяцев наблюдения здоровые мыши (без диабета) не гибнут, тогда как в группе животных с диабетом наблюдается гибель. В результате, через два месяца наблюдений выжило около 14% диабетных мышей. В то же время в группе диабетных мышей, получивших трансплантацию клеток слюнной железы, выжило около 69% животных (таблица 9). Таким образом, трансплантация дифференцированных клеток слюнной железы значительно повышает выживаемость мышей с экспериментальным диабетом.
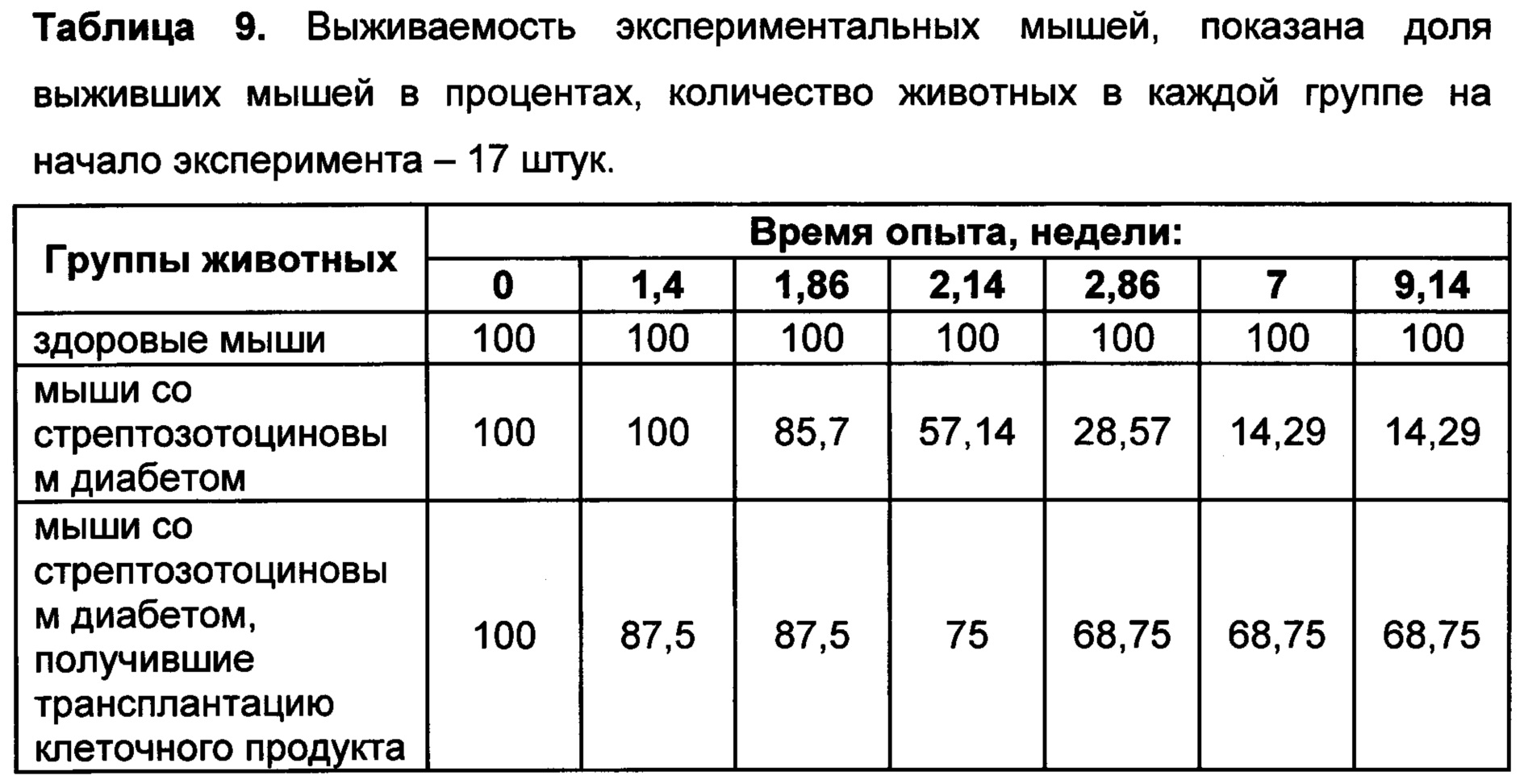
[0172] Концентрацию глюкозы в крови экспериментальных животных оценивали в течение 30 суток, так как стрептозотоциновый диабет стабилен в течение этого времени. Было показано, что на начало эксперимента (0 день) в группе контрольных мышей с диабетом, которым инъецировали 500 мкл фосфатно-солевого буфера без клеток и в опытной группе мышей, получивших трансплантацию клеток, концентрация глюкозы составляет около 27 мМ, тогда как в группе нормальных животных без диабета - около 6 мМ. Трансплантация клеток оказала двоякое действие на течение экспериментального диабета у животных: во-первых, после трансплантации дифференцированных клеток слюнной железы у мышей не наблюдается резких скачков глюкозы, которые есть в группе диабетических животных без трансплантации клеток (таблица 10). Во-вторых, у животных после трансплантации клеток наблюдается постепенное снижение концентрации глюкозы в крови. К концу эксперимента (30 день) у мышей, получивших трансплантацию клеток, статистически достоверно снижается концентрация глюкозы в крови до 11 мМ, тогда как у диабетических мышей без трансплантации клеток концентрация глюкозы остается высокой - около 22 мМ.
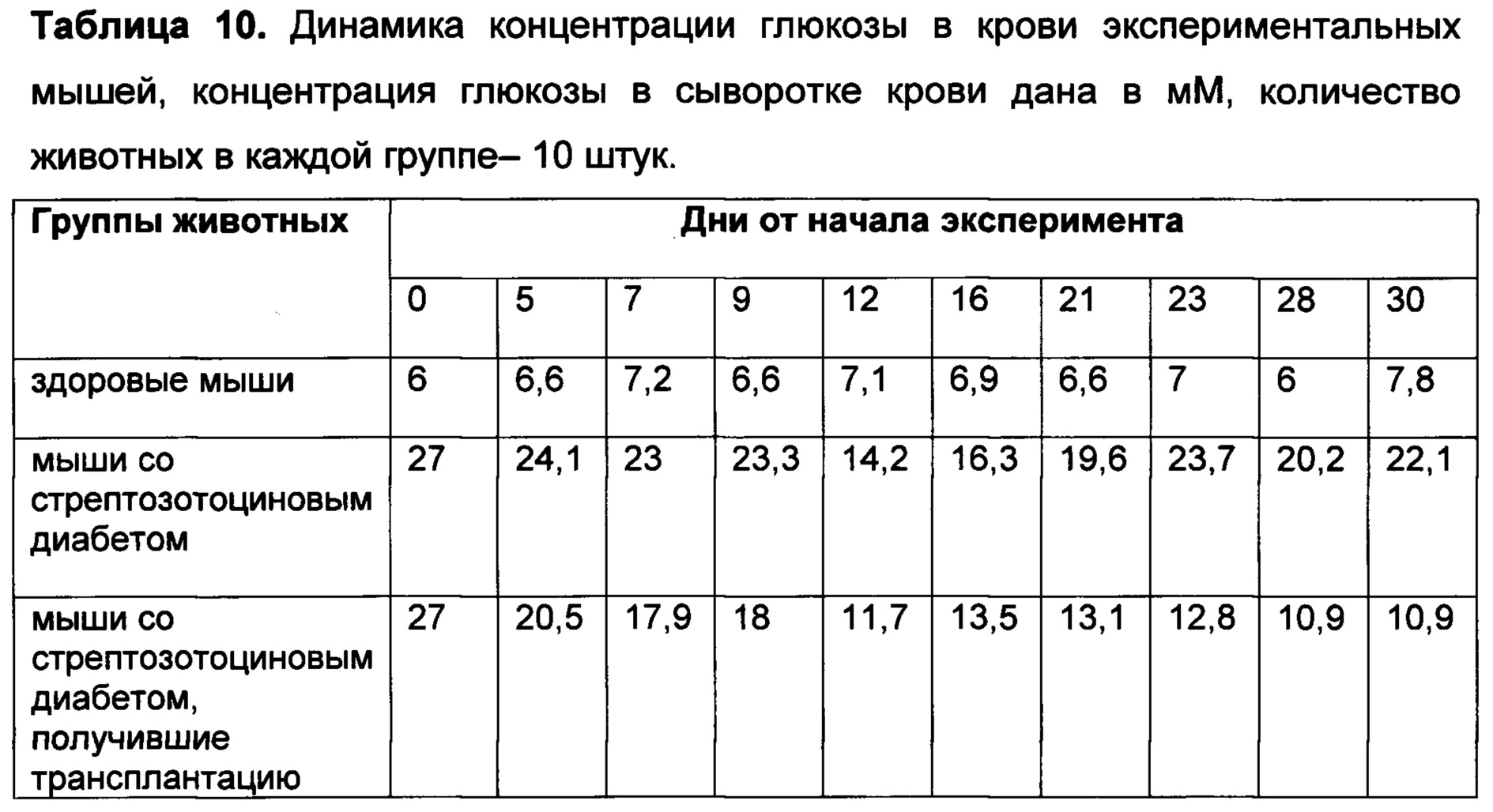
[0173] На 40-й день эксперимента у мышей всех трех групп взяли поджелудочную железу на гистологическое исследование. Были приготовлены гистологические препараты поджелудочной железы как описано в Примере 5. В результате анализа серийных срезов поджелудочных желез мышей было показано, что у мышей с диабетом, не получивших трансплантацию клеток, островки деградируют (Фиг. 10, показано стрелками). Кроме того, у этих животных увеличивается диаметр сосудов. У диабетных мышей, получивших трансплантацию 5 млн дифференцированных клеток слюнной железы, наблюдаются регенерирующие островки на 40-й день после трансплантации (Фиг. 11, показано стрелками). В тоже время у нормальных животных без диабета наблюдаются нормальные островки Лангерганса (Фиг. 9, показано стрелками). Таким образом, трансплантация клеток слюнной железы увеличивает выживаемость мышей с экспериментальным диабетом, снижает гипергликемию и уменьшает скачки концентрации глюкозы в крови диабетных мышей, а также способствует регенерации островков Лангерганса поджелудочной железы. В целом, можно заключить, что дифференцированные клетки слюнной железы в значительной степени способствуют коррекции экспериментального диабета, что показано на примере стрептозотоциновой модели диабета у мышей.
[0174] Пример 7. Культивирование эпителиальных прогениторных клеток и получение клеточного продукта инсулин-продуцирующих клеток
[0175] Биоптат поднижнечелюстной слюнной железы человека объемом 2 см3 был получен в ходе плановой операции по удалению части слюнной железы вследствие слюннокаменной болезни. Донор материала (мужчина, 37 лет) был свободен от инфекций: вирусы гепатита В и С, ВИЧ, сифилис.
[0176] Биоптат поместили в стерильную среду DMEM/F12 1:1 (Gibco, США) с добавлением 40 мкг/мл гентамицина (Sigma, США) и транспортировали в лабораторию в герметичном контейнере. Дальнейшие манипуляции проводили в стерильных условиях по правилам GMP (Good Manufacturing Practice). Биоптат поместили в чашку Петри с фосфатно-солевым буфером, ткань механически измельчали стерильными скальпелем и пинцетом до кусочков размером 5 мм3, при этом механически отделяли и удаляли жировую ткань. Затем кусочки эпителиальной ткани дважды промывали фосфатно-солевым буфером, осаждали центрифугированием в течение 5 минут при 200 g и инкубировали с 2 мг/мл коллагеназы IV типа (Gibco, США) в среде DMEM/F12 1:1 (Gibco, США) с 2 мМ глутамина (Invitrogen, США) 40 минут при 37°С. После этого суспензию клеток пипетировали 5 минут, пропускали через нейлоновый фильтр с диаметром пор 100 мкм и осаждали клетки центрифугированием в течение 10 минут при 200 g. Клетки дважды промывали фосфатно-солевым буфером, осаждали центрифугированием в течение 5 минут при 200 g. Затем проводили магнитную сепарацию клеток по маркеру ЕрСАМ (Miltenyi Biotec GmbH, Германия) по инструкциям производителя. Для этого клетки подсчитывали на приборе для автоматизированного подсчета клеток (Bio-Rad, США) и ресуспендировали в фосфатно-солевом буфере в концентрации 5×106клеток на 1 мл буфера. Инкубировали клетки при температуре 4°С в течение 20 минут с антителами из расчета 5 мкг антител на 106 клеток. Затем проводили магнитную сепарацию клеток на колонках по инструкциям производителя. Отсортированные клетки осаждали центрифугированием в течение 5 минут при 200 g, дважды промывали фосфатно-солевым буфером и ресуспендировали в культуральной ростовой среде РСТ Epidermal Keratinocyte Medium (1Х, liquid), (CELLnTEC, Швейцария), # CnT-07, содержащей 1× добавку инсулин-трансферрин-селенит (Invitrogen, США) и 10 нг/мл эпидермального фактора роста (Sigma, США). Клетки помещали в покрытые коллагеном I типа культуральные флаконы в количестве 5×103 клеток на 1 см2 и инкубировали при 37°С и 5% СО2. Первые 5 дней среду меняли каждый день, при дальнейшем культивировании - раз в 3 дня. Данные клетки являются нулевым пассажем. В результате из 2 см3 биоптата было получено 10 млн эпителиальных прогениторных клеток, экспрессирующих ЕрСАМ.
[0177] Данные клетки также экспрессировали маркеры эпителиальных клеток: KRT18 и CD49f, что было подтверждено методом проточной цитометрии. Для этого, 2 млн клеток были зафиксированы 1% параформальдегидом (Sigma, США) в течение 5 минут при комнатной температуре, клетки дважды промывали фосфатно-солевым буфером и разделяли на 4 аликвоты по 0,5 млн в 200 мкл фосфатно-солевого буфера, содержащего 1% бычьего сывороточного альбумина (Sigma, США) и 0,1% тритона (Sigma, США). Затем к клеткам первой аликвоты добавляли первичные антитела Anti-KRT18 (AbCam, Великобритания) в разведении 1:500. К клеткам второй аликвоты добавляли первичные антитела Anti-CD49f (Millipore, США) в разведении 1:10. К клеткам третьей и четвертой аликвот добавляли соответствующие изотип-контроли. Инкубировали клетки с антителами в течение 60 минут при 37°С. Затем клетки трижды промывали фосфатно-солевым буфером (по 10 минут, при 37°С), ресуспендировали в 200 мкл фосфатно-солевого буфера и инкубировали с вторичными антителами в течение 40 минут при 37°С в темноте. Клетки трижды промывали фосфатно-солевым буфером (по 10 минут, при 37°С), ресуспендировали в 1 мл фосфатно-солевого буфера и анализировали с помощью проточного цитофлуориметра Cell Lab Quanta™ SC MPL (Beckman Coulter, США). В качестве контроля использовали соответствующий изотип-контроль, анализировали не менее 10000 клеток. В результате было показано, что более 99% полученных эпителиальных прогениторных клеток слюнной железы человека экспрессируют маркеры KRT18 и CD49f.
[0178] Через 10 дней культивирования клетки нулевого пассажа достигли состояния монослойной культуры и количества 30 млн. Клетки были сняты с поверхности культуральных флаконов трипсином. Для этого среду культивирования удаляли, клетки дважды промывали раствором Версена, затем инкубировали 5 минут при 37°C с 0,05% раствором трипсина в ЭДТА в количестве 1 мл на 25 см2 площади культурального флакона. Затем клетки отмывали от трипсина фосфатно-солевым буфером, осаждали центрифугированием 5-10 минут при 200 g. 15 млн клеток были подвергнуты криозаморозке для длительного хранения, а другие 15 млн подвергнуты дальнейшему культивированию для наращивания клеточной массы. Для этого делали первый пассаж: клетки разводили в ростовой среде в соотношении 1:5 и помещали в новые культуральные флаконы, покрытые коллагеном I типа. Среду культивирования меняли каждые 3 дня. Через 7 дней клетки первого пассажа достигли состояния монослоя и количества 75 млн. Клетки сняли с поверхности культуральных флаконов трипсином как описано выше, развели в ростовой среде в соотношении 1:3 и поместили в новые культуральные флаконы, покрытые коллагеном I типа. Среду культивирования меняли каждые 3 дня. Через 7 дней клетки второго пассажа достигли состояния монослоя и количества 225 млн. Таким образом, наращивание необходимого количества клеточной массы завершили и приступили к панкреатической дифференцировке клеток.
[0179] Панкреатическую дифференцировку эпителиальных клеток слюнной железы проводили по разработанному протоколу: с 0 по 7 день клетки инкубировали в среде DMEM/F12 1:1 (Gibco, США), содержащей 10% эмбриональной телячьей сыворотки (HyClone, США), 1× добавку инсулин-трансферрин-селенит (Invitrogen, США), 2 мМ глутамина (Invitrogen, США), 10 нг/мл эпидермального фактора роста (Sigma, США), с добавлением 2 мкМ ретиноевой кислоты (Sigma, США), 1 мкМ изопротеринола (Sigma, США), 10 нг/мл фактора роста фибробластов 10 (Life Technologies, США) и 10 нг/мл инсулиноподобного фактора роста 1 (R&D, США). С 7 по 14 день клетки инкубировали в среде DMEM/F12 1:1 (Gibco, США) с 10% эмбриональной телячьей сыворотки (HyClone, США), 2 мМ глутамина (Invitrogen, США), 10 нг/мл эпидермального фактора роста (Sigma, США), с добавлением 100 нМ ретиноевой кислоты (Sigma, США), 10 мМ никотинамида (Sigma, США), 10 нг/мл инсулиноподобного фактора роста 1 (R&D, США), 20 нг/мл фактора роста гепатоцитов (Gibco, США) и 0,1 мкМ дексаметазона (Sigma, США).
[0180] На 14-й день дифференцировки из 2 млн дифференцированных клеток проводили выделение тотальной РНК, синтез кДНК и анализ генов-маркеров панкреатической дифференцировки клеток методом количественной ПЦР с детекцией в реальном времени как описано в Примере 1. В результате было показано, что полученные дифференцированные клетки слюнной железы человека экспрессируют маркеры бета-клеток: NGN3, PDX1, MAFA, РАХ4, NKX6.1, а также препроинсулин.
[0181] Методом проточной цитометрии (описанным выше) было показано, что 99% данных дифференцированных клеток слюнной железы человека экспрессируют маркеры эпителиальных клеток KRT18 и CD49f и 89% экспрессируют инсулин.
[0182] Для анализа глюкозозависимой секреции инсулина, были приготовлены сфероиды дифференцированных клеток слюнной железы человека, как описано в Примере 3 и отобраны пробы буфера Krebs-Ringer: с 0 мМ, 2 мМ, 5 мМ и 15 мМ глюкозы, инкубированные 1 час со сфероидами, как описано в Примере 3. Затем был проведен ИФА содержания инсулина и С-пептида с помощью наборов Ultrasensitive Insulin ELISA (Mercodia, Швеция) и Ultrasensitive C-Peptide ELISA (Mercodia, Швеция) по инструкциям производителя. Было показано, что при увеличении концентрации глюкозы в буфере с 5 мМ до 15 мМ, секреция инсулина и С-пептида увеличивается не менее чем в два раза.
[0183] Таким образом, было получено 225 млн клеток клеточного продукта, содержащего клетки, способные к глюкозозависимой секреции инсулина и экспрессирующие маркеры NGN3, PDX1, MAFA, РАХ4, NKX6.1 и препроинсулин. Продукт содержит 89% клеток, экспрессирующих инсулин, KRT18 и CD49f и 11% клеток, экспрессирующих KRT18 и CD49f.
Реферат
Изобретение относится к биотехнологии. Изобретение представляет собой способ получения клеточного продукта инсулин-продуцирующих клеток человека, включающий получение эпителиальных прогениторных клеток из биоптата эпителиальной ткани и их последующую панкреатическую дифференцировку в клетки, способные к глюкозозависимой секреции инсулина, в котором панкреатическую дифференцировку клеток проводят в две стадии в культуральной среде для эукариотических клеток, обеспечивающей концентрацию ионов кальция в пределах от 0,5 мМ до 2,5 мМ: на первой стадии клетки культивируют в течение 4-15 суток в культуральной среде, содержащей, по крайней мере, сыворотку крови - 2-20 объемных %, глутамин - не менее 1 мМ, эпидермальный фактор роста - 1-300 нг/мл, трансферрин - не менее 0,1 мкг/мл, селенит натрия - 0,1-20 нг/мл, ретиноевую кислоту - 0,1-20 мкМ, изопротеренол - 0,1-10 мкМ; на второй стадии клетки культивируют в течение 4-15 суток в культуральной среде, содержащей, по крайней мере, сыворотку крови - 2-20 объемных %, глутамин - не менее 1 мМ, эпидермальный фактор роста - 1-300 нг/мл, трансферрин - не менее 0,1 мкг/мл, селенит натрия - 0,1-20 нг/мл, ретиноевую кислоту - 10 нМ - 20 мкМ, никотинамид - 1-100 мМ, фактор роста гепатоцитов - 1-300 нг/мл, дексаметазон - 0,01-5 мкМ, где культивирование на обеих стадиях проводят при 37°С в присутствии 5% CO. Группа изобретений включает клеточный продукт инсулин-продуцирующих клеток млекопитающего, а также способ заместительной терапии сахарного диабета с использованием клеточного продукта. Изобретение позволяет упростить технологии получения инсулин-продуцирующих клеток, получить не менее 70% функционально активных инсулин-продуцирующих клеток в клеточной культуре, прошедшей дифференцировку. 3 н. и 11 з.п. ф-лы, 10 табл., 7 пр., 11 ил.
Комментарии