Применение генетически модифицированного патогенного вируса кори с улучшенными проапоптотическими свойствами (вирус mv-deltac) в терапии рака - RU2700083C2
Код документа: RU2700083C2
Чертежи

















Описание
ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к генетически модифицированному патогенному вирусу кори,
полученному из штамма живого ослабленного вируса кори, у которого нокаутирован ген,
кодирующий вирусный акцессорный белок С (MV-deltaC). В частности, изобретение
относится к применению указанного генетически модифицированного патогенного MV-deltaC
для лечения злокачественной опухоли или раковых заболеваний, и для приготовления
агентов или композиций для такого лечения.
УРОВЕНЬ ТЕХНИКИ
Злокачественная мезотелиома является редким и очень агрессивным видом рака,
устойчивым к обычным лечебным процедурам. Развитие злокачественной мезотелиомы
плевры в основном связано с длительным воздействием асбестовых волокон и пыли (Kazan-
Allen et al., Lung cancer, 2005, 49SI:S3-S8; Robinson et al., Lancet, 2005, 366:397-408).
Меланома представляет собой злокачественную опухоль, которая развивается из
меланоцитов, и в отсутствии лечения может распространяться по всему телу. Несмотря на то,
что меланома представляет собой одну из наименее часто встречающихся типов рака кожи,
она ответственна за большинство смертей, связанных с раком кожи (Chin, L. et al., Genes Dev.
2006 20: 2149-2182). Аденокарцинома легких является одной из самых распространенных
форм рака легких, как у курящих, так и у некурящих людей, и одной из наиболее
распространенных причин смерти от рака (Travis, W. D. et al., J Thorac Oncol., 2011, 6(2), 244-
285).
В настоящее время ни один способ лечения не предлагает существенных путей лечения
для некоторых агрессивных видов рака, таких как злокачественная мезотелиома, меланома и
аденокарцинома легких.
Данные виды рака имеют плохие прогнозы, и они являются относительно
невосприимчивыми ко всем традиционным методикам лечения, таким как химиотерапия,
лучевая терапия и/или хирургическое вмешательство. Таким образом, существует острая
необходимость в разработке новых клинических подходов.
Виротерапия рака широко известна как новая альтернатива для лечения видов рака,
устойчивых к обычным противораковым терапиям (Boisgerault N et al., Immunotherapy, 2010,
march 2(2), 185-199). Онколитическая виротерапия продемонстрировала многомодальные
противоопухолевые механизмы как в доклинических, так и в нескольких случаях I фазы
клинических противораковых испытаний, (Lech, PJ and Russell, SJ; Expert Review of Vaccines,
2010, 9(11):1275-1302; Galanis et al., Cancer Research, 2010, 70(3):875-882), а также в
исследованиях in vitro (Gauvrit, A et al., Cancer Research, 2008, 68(12), 4882-4892). Вирусные
вакцины имеют преимущество по сравнению с клеточной иммунотерапией, так как
обеспечивают персональным противораковым иммунитетом одновременно с циторедукцией
без необходимости индивидуального изготовления. Кроме того, вирусные вакцины могут
быть сконструированы таким образом, чтобы они удаляли иммуносупрессивные вирусные
компоненты и вставляли трансгены, которые повышают противоопухолевую
цитотоксичность и иммунитет (A. Gauvrit et al., Cancer Research, 2008, 68 (12), 4882-4892).
Вирус кори (MV) представляет собой оболочечный вирус, содержащий
несегментированную одноцепочечную РНК отрицательной полярности, из рода Morbilivirus
семейства Paramyxoviridae. Несегментированный геном MV имеет противоположную
полярность, что проявляется в виде геномной РНК, которая не транслируется ни in vivo, ни in
vitro, и будучи очищенной не является патогенной. Данный вирус был выделен в 1954 году
(Enders, J. F. and Peebles, T.C., 1954, Proc Soc Exp Biol Med, 86(2): 277-286), и с тех пор из
этого вируса были получены живые ослабленные вакцины для создания вакцинных штаммов,
и в частности из штамма Шварца/Моратена.
Транскрипция и репликация вирусов, содержащих несегментированную нить (-) РНК, и
их сборка как вирусных частиц была изучена и описана главным образом в книге
«Fields Virology» (3nd edition, vol 1, 1996, Lippincott-Raven publishers-Fields BN et al.).
Транскрипция и репликация MV не включает ядро инфицированных клеток, а скорее
происходит в цитоплазме указанных инфицированных клеток. Геном MV содержит гены,
кодирующие шесть основных структурных белков из шести генов (обозначенных N, P, M, F,
H и L) и два дополнительных неструктурных белка С и V из гена P. Гены расположены в
следующем порядке: 3', N, P (включая белки С и V), M, F, H и L - большой белок полимеразы
на 5'-конце (Фигура 1А). Геном дополнительно содержит некодирующие фрагменты в
межгенной области M/F; этот некодирующий участок содержит около 1000 нуклеотидов
нетранслируемой РНК. Приведенные гены кодируют соответственно сигнальный пептид (ген
I), белки нуклеокапсида вируса, т.е. нуклеопротеин (N), фосфопротеин (P), и большой белок
(L), который собирается вокруг генома РНК для образования нуклеокапсида. Другие гены
кодируют белки оболочки вируса, включающие гемагглютинин (Н), белок слияния (F) и
матричный белок (M). Белок С вируса кори, кодируемый полицистронным геном P,
представляет собой небольшой (186 аминокислот) и основной белок, локализованный в
цитоплазме и в ядре (Bellini, W.J. et al., J. Virol., 1985, 53:908-919). Роль этого вирусного
белка, описанного как фактор вирулентности MV, еще не очень хорошо ясна. Для
определения роли белка С вируса кори использовали дикий тип рекомбинантного штамма
MV, не экспрессирующий белок С, основанный на высокопатогенном штамме IC-B и
полученный с помощью системы обратной генетики. Было выдвинуто предположение, что
белок С вируса кори может участвовать в сборке вирусных частиц, в экспрессии вирусного
белка и в задержке апоптоза инфицированных клеток для создания стабильных инфекций MV
(Takeuchi, K. et al., J. Virol, June 2005, 7838-7844). И хотя сообщалось, что белок С вируса
кори ингибирует интерферонный противовирусный ответ (Shaffer, J.A. et al., Virology, 2003,
315:389-397), в другом исследовании пришли к противоположному заключению (Takeuchi, K.
et al., J. Virol, June 2005, 7838-7844), тем самым подтверждая, что функция белка С вируса
кори еще точно не установлена.
Среди вирусов человека, подходящих для испытаний в качестве онколитических
агентов, живая ослабленная вакцина MV имеет ряд преимуществ. Введенная сотням
миллионов детей в течение 30 лет, она является самой безопасной и наиболее широко
используемой человеческой вакциной для детей. Ослабленный штамм MV инфицирует много
различных типов клеток и предпочтительно трансформированные раковые клетки. Это
достигается за счет использования вирусом кори CD46, зачастую как рецептора сверх
экспрессии в раковых клетках, для того чтобы противостоять комплемент-зависимой гибели
от естественных клеток-киллеров (Naniche, D. et al., J Virol, 1993, 67(10):6025-6032; Dhiman,
N. et al., Rev Med Virol, 2004, 14(4): 217-229), в то время как дикий тип MV использует
преимущественно SLAM (CD150) (Tatsuo, H. et al., Nature, 2000, 406(6798):893-897; Anderson,
B. D. et al., Cancer Res., 2004, 64: 4919-4926; Schneider, U. et al., J Virol., 2002, 76: 7460-7467).
Интересно то, что MV проявляет природные противоопухолевые свойства посредством
специфического нацеливания на раковые клетки, не заражая здоровые клетки. Таким
образом, MV демонстрирует несомненный профиль безопасности для применения в будущих
терапевтических протоколах.
Онколитические свойства дикого типа MV хорошо известны специалистам в данной
области техники (Mayo Foundation for Medical Education and Research, US07854928). Недавно
были начаты клинические испытания для изучения возможности штамма MV Эдмонстона
лечить рак яичников, глиобластому, немелкоклеточный рак легкого и множественную
миелому (см. http://clinicaltrials.gov, ключевые слова - «measles» и «cancer»). Также было
описано применение как рекомбинантных, так и химерных вакцин MV, в качестве векторов
для вакцинации (WO2004/000876, WO2004/076619, WO2006/136697 и WO2008/078198).
Данная технология также была предложена для иммуно-онколитического лечения
мезотелиомы (Gauvrit, A. et al., Cancer Research, 2008, 68 (12), 4882-4892). Соответственно,
международная заявка на патент WO2009/047331 описывает и онколитические и иммуно-
адъювантные свойства живого ослабленного штамма Шварца вакцинного MV на панели
опухолевых клеток эпителиоидной мезотелиомы. С помощью восстановленного штамма
Шварца вакцинного MV, полученного из патогенного клона кДНК было показано, что клетки
мезотелиомы, инфицированные MV, вызывают спонтанное созревание дендритных клеток из
моноцитов (MoДК) и опухолевый антиген-специфический ответ.
Потенциальные преимущества онковиротерапии, по сравнению с обычными методами
лечения, включают в себя свойство индуцировать иммунный ответ, включающий не только
более точную специфичность против раковых антигенов (опухолеспецифический антиген), а
значит и больший запас безопасности, но также длительный эффект за счет иммунной
памяти, и таким образом, предотвращение рецидивов и возникновения метастазов.
Действительно, было показано, что специфический иммунный ответ и память развиваются
после введения MV в участок с раковыми клетками в присутствии антиген-презентирующих
клеток (Masse, D. et al., Int. J. Cancer, 2004, 111(4), 575-580); Liu et al., Molecular therapy: the
journal of the American Society of Gene Therapy, 2010, 18(6): 1155-1162). Было показано, что
противоопухолевая активность штамма MV Шварца действует посредством нескольких
механизмов, включающих онколизис, индукцию иммуногенного апоптоза опухоли
(экспрессия сигнала опасности связанного с гибелью клеток) и формирование синцития,
опосредованное вирусом (Gauvrit, A. et al., Cancer Res, 2008, 68(12), 4882-4892). Кроме того,
было высказано предположение, что секретируемые опухолеспецифические антигены и
воспаление в результате вирусной репликации позволяют преодолеть иммунологическую
толерантность к опухолям, а также индуцировать противоопухолевый иммунитет.
Несмотря на эффективное заражение, некоторые злокачественные опухоли или раковые
клетки, инфицированные вирусом кори, оказались устойчивы к индукции гибели клеток.
Следовательно, существует необходимость в разработке вирусов, которые помогли бы
преодолеть этот тип устойчивости, и таким образом улучшить и расширить индукцию
специфической гибели клеток злокачественных опухолей или раковых клеток.
Предшественники дендритных клеток (ДК) подразделяются на моноцитарные
дендритные клетки (MoДК) и плазмацитоидные дендритные клетки (пДК), которые
проявляют разные функциональные свойства. пДК представляют собой подгруппу
дендритных клеток, участвующих в противовирусном иммунном ответе в виду экспрессии
ими Толл-подобных рецепторов (TLR), специализирующихся на распознавании вирусных
нуклеиновых кислот (TLR7, TLR9) (Gilliet, M. et al., Nat Rev Immunol., 2008, 8:594-606). Они
реагируют на широкий спектр вирусов (в частности вирус гриппа А, вирус простого герпеса,
ВИЧ) в условиях активации и созревания путем выделения большого количества
интерферона I типа (IFN-α, -β, -ω). Они также могут презентировать вирусные антигены
CD8+ и CD4+ Т-клеткам при инфицировании вирусом (Fonteneau, J. F. et al., Blood, 2003,
101:3520-3526) и кросс-презентировать вирусные антигены из клеток, инфицированных
вирусом, CD8+ Т-лимфоцитам (Di Pucchio, T. et al., Nat Immunol., 2008, 9:551-557; Lui, G. et
al., PLoS One, 2009, 4:e7111). Также было показано, что пДК могут играть положительную
роль в иммунном ответе против опухоли (Drobits, B. et al., J Clin Invest., 2012, I22:575-585;
Liu, C. et al., J Clin Invest., 2008, 118:1165-1175). В качестве примера, наблюдали активацию
пДК и противоопухолевый иммунный ответ на модели меланомы мыши внутри опухолей,
после местного лечения лигандом TLR7, имиквимодом (Drobits, B. et al., J Clin Invest.,2012,
122:575-585). Так как MV представляет собой одноцепочечную РНК (ssRNA), авторы
предположили, что пДК возможно способны обнаружить инфекцию MV опухолевых клеток
из-за их экспрессии TLR7 внутри вакуоли, который распознает одноцепочечную РНК.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы изобретениянеожиданно обнаружили, что генетически модифицированный
патогенный вирус кори, полученный из живого ослабленного штамма MV, у которого
нокаутирован ген, кодирующий вирусный акцессорный белок С (MV-deltaC), вызывает
повышенный ответ против злокачественной опухоли или раковых клеток и, в частности,
имеет улучшенные проапоптотические свойства по сравнению с немодифицированным MV.
Таким образом, настоящее изобретение относится к MV-deltaC и показывает, что данный
вирус может эффективно заражать и убивать злокачественные опухоли или раковые клетки,
такие как злокачественная мезотелиома, меланома и клетки аденокарциномы легких. Авторы
изобретения также показали, что плазмацитоидные дендритные клетки, взаимодействующие
с лизатом злокачественной мезотелиомы, меланомы и клеток аденокарциномы легких,
инфицированных MV, могут активировать CD8 T-клетки против мезотелиомы, меланомы и
против клеток аденокарциномы легких. Таким образом, авторы изобретения предположили,
что отмеченные свойства MV-deltaC могут представлять интерес для применения в качестве
активного соединения против злокачественных опухолей или раковых клеток, когда MVdeltaC
активирует пДК.
Настоящее изобретение относится к патогенному вирусу кори, полученному из штамма
живого ослабленного вируса кори, для применения в лечении агрессивной злокачественной
опухоли или агрессивного рака путем активации плазмацитоидных дендритных клеток
(пДК), при введении индивидууму, у которого дигностирована такая опухоль или раковое
заболевание.
Настоящее изобретение также относится к генетически модифицированному
патогенному вирусу кори, полученному из штамма живого ослабленного вируса кори, у
которого нокаутировали ген, кодирующий вирусный акцессорный белок С (MV-deltaC), для
применения в лечении агрессивной злокачественной опухоли или агрессивного рака путем
активации плазмацитоидных дендритных клеток (пДК), при введении индивидууму, у
которого дигностирована такая опухоль или раковое заболевание.
Для термина «вирус кори» используется сокращение MV, а для выражения «вирус
кори,полученный из штамма живого ослабленного вируса кори, у которого нокаутирован
ген, кодирующий вирусный акцессорный белок С» используется сокращение MV-deltaC.
В настоящем изобретении термин «кодирующий» относится к способности молекул
нуклеиновых кислот к транскрипции и, в случае необходимости, к трансляции для
экспрессии продукта в выбранных клетках или клеточных линиях.
В настоящем изобретении, индивидуумом является предпочтительно млекопитающее,
более предпочтительно человек.
В настоящем изобретении выражение «генетически модифицированный» относится к
тому факту, что синтеза белка С вируса кори был отменен, в частности выключен, при
получении генетически модифицированного патогенного MV-deltaC.
В настоящем изобретении выражение «нокаутирован (нокаутировали) ген, кодирующий
вирусный акцессорный белок C» означает выключение экспрессии гена, кодирующего белок
C.
Ингибирование синтеза белка С, в частности, достигается подавлением экспрессии
открытой рамки считывания белка С (ORF), содержащейся в гене P и кодирующей белок С.
ORF белка С перекрывает часть ORF гена P в рамке +1. Сайленсинг ORF белка C
достигается, в частности, с помощью подходящей мутации последовательности указанной
ORF, в результате мутации второго кодона инициации «ATG» в N-концевом участке гена P. В
дополнение к этой первой мутации, вторая мутации может быть получена путем добавления
стоп-кодона в результате замещения одного нуклеотида ниже по направлению в открытой
рамке считывания белка С (Фигура 1В) (Patterson, J. B. et al., Virology, 2000, 267(1): 80-89).
Согласно конвенции, используемой для Paramyxoviridae, термин «ген» можно
использовать для обозначения геномной нуклеиновой кислоты РНК, кодирующей мРНК. В
контексте настоящего изобретения, данный термин может также относиться к ORF,
содержащейся в указанном мРНК.
Указанная мутация для подавления экспрессии белка C должна сохранять экспрессию
гена P для его способности экспрессировать белок P.
В предпочтительном варианте реализации, мутантная нуклеиновая кислота РНК вируса
кори дополнительно следует так называемому «правилу шести». «Правило шести»
выражается в том, что общее количество нуклеотидов, присутствующих в нуклеиновой
кислоте, кодирующей полноразмерную (+) нить РНК генома MV, является кратным шести. В
конкретном варианте реализации, конструкция нуклеиновой кислоты, содержащая тот же или
мутантный геном MV-deltaC или состоящая из указанного мутантного генома MV-deltaC, и
возможно дополнительных, в частности кодирующих, последовательностей, является
кратной шести. «Правило шести» было признано существующим уровнем техники как
требование в отношении общего количества нуклеотидов в геноме MV, которое обеспечивает
эффективную или оптимизированную репликацию геномной РНК MV в результате
взаимодействия с каждой субъединицей белка MV, содержащей в белковой оболочке 6
рибонуклеотидов в геноме, для формирования нуклеокапсида.
Выключение процессов, отвечающих за экспрессию белка C, в различных штаммах MV,
таких как высокопатогенный штамм IC-B (Takeuchi, K. et al., J. Virol., June 2005, 7838-7844)
или вакцинный штамм Эдмонстона (Radecke, F. et al., Virology, 1996, 217:418-421; Patterson,
J. B. et al., Virology, 2000, 267(1):80-89), было предложено согласно известному уровню
техники с помощью системы обратной генетики, и может быть применено в контексте
настоящего изобретения.
В настоящем изобретении выражение «патогенный вирус кори, полученный из штамма
живого ослабленного вируса кори» относится к вирусу кори, происходящему из
авирулентного штамма или штамма, менее вирулентного чем определенный родительский
штамм в одном и том же хозяине, в частности в организме человека, в то время как он
сохраняет патогенные свойства и иммуногенность и, возможно, адъювантность при введении
в организм хозяина, в частности в организм человека, т.е. сохранение иммунодоминантного T
и B-клеточного ответа к MV и ,возможно, адъювантности, например, индукции ко-
стимуляторных белков Т-клеток или цитокина IL-12. Первичные патогенные штаммы сильно
нарушают гемопоэз (Arneborn, P. et al., Clin Exp Immunol, 1983, 51:165-172; Kim, E.A. et al.,
Radiographics, 2002, 22 Spec No:S137-149; Okada, H. et al., Arch Virol, 2000, 145:905-920) , что
в результате приводит к временной иммуносупрессии, ответственной за большинство
смертей из-за инфекции кори в развивающихся странах. В отличие от первичных штаммов,
живые ослабленные штаммы не вызывают иммуносупрессию (Okada, H. et al., Arch Virol,
2001, 146:859-874).
Живой ослабленный штамм MV соответственно относится к штамму, который был
последовательно пересеян на отдельных клетках и, предпочтительно, адаптирован к другим
клеткам, таким как первичные клетки с IFN α/β ответом, то есть клеткам CEF, для получения
посевных штаммов, пригодных для получения вакцинных штаммов, обладающих
стабильным геномом, который не допускает возврата к патогенности или интеграции в
хромосомы хозяина, в частности в хромосомы хозяина-человека. В конкретном варианте
реализации изобретения, живой ослабленный MV является вирусом, который был выбран из
первичных клеток, таких как клетки CEF.
В частности, «живой ослабленный штамм», утвержденный штамм для вакцины,
используемый для человека, является живым ослабленным штаммом, подходящим для
использования в настоящем изобретении, если он отвечает критериям, определенным FDA
(Управление США по санитарному надзору за качеством пищевых продуктов и
медикаментов), т.е., после тщательных обзоров лабораторных и клинических данных
установлено, что он отвечает критериям безопасности, эффективности, качеству и
воспроизводи мости, (www.fda.gov/cber/vaccine/vacappr.htm).
В настоящем изобретении, конкретный живой ослабленный штамм MV,
представляющий вирус MV, является штаммом вируса кори Шварца или штаммом Моратена,
в частности из вакцины Rouvax® (Аventis). Было показано, что штамм Шварца имеет
абсолютную идентичность последовательности со штаммом Моратен (Parks, C. L. et al., 2001,
J Virol, 75(2):910-920; Schwarz, A. J., 1962, Am J Dis Child, 103, 386-389). В настоящее время
широко применяют штаммы Шварца/Моратена, так как они индуцируют долгосрочный
клеточный и гуморальный иммунный ответы, и обладают необходимой генетической
стабильностью, поскольку никогда не наблюдалось возврата к патогенной форме (Hilleman,
M., 2002, Vaccine, 20:651-665).
Более предпочтительно, генетически модифицированный патогенный MV, описанный
в настоящем изобретении, получают с использованием кДНК штамма MV Шварца,
клонированного в плазмиду pTM-MVSchw, который депонирован в Институте Пастера в
CNCM (Париж, Франция) под номером I- 2889 12 июня 2002 года, последовательность
которого описал Combredet (Combredet, C. et al., 2003, J Virol, 77(21): 11546-11554), а также
описанный в WO2004/000876 и в Примере 1 настоящего изобретения. Плазмида pTM –
MVSchw, полученная из плазмиды Bluescript, содержит полинуклеотид, кодирующий
полноразмерную (+) нить РНК MV штамма Шварца под контролем промотора РНК
полимеразы Т7, и включает 18967 нуклеотидов. кДНКдругих штаммов MV может быть
получена аналогичным образом, на основе нуклеиновой кислоты, очищенной от вирусных
частиц живого ослабленного MV. Для того, чтобы подготовить подходящий кДНК,
кодирующий геном MV-deltaC, плазмиду pTM-MVSchw модифицируют путем замещения
второго стартового кодона "ATG" в гене P, с получением плазмиды pTM-MVSchw-deltaC-
АTU1 (eGFP) (SEQ ID NO: 1). В частности, кодон "ATG" был заменен на кодон "ACG"
посредством замены нуклеотида T на нуклеотид C.
В конкретном варианте реализации, плазмиду pТМ-MVSchw-deltaC-АTU1 (eGFP),
имеющую нуклеотидную последовательность согласно SEQ ID NO: 1, дополнительно
видоизменяют путем замены нуклеотида G в положении 2803 на нуклеотид A, чтобы
образовать стоп-кодон. Данный вариант pTM-MVSchw-deltaC-АTU1 (eGFP) имеет
нуклеотидную последовательность согласно SEQ ID NO: 2.
В предпочтительном варианте реализации настоящего изобретения, генетически
модифицированный патогенный MV-deltaC получают с помощью восстановления.
Выделение не модифицированного MV штамма MV Шварца, было подробно описано в
WO2004/000876, и аналогичный метод может быть применен для получения генетически
модифицированного патогенного MV-deltaC.
В конкретном варианте реализации изобретения, генетически модифицированный
патогенный MV-deltaC экспрессирует GM-CSF (гранулоцитарно-
макрофагальный колониестимулирующий фактор) (Guse, K. et al., 2011, Oncolytic vaccinia
virus for the treatment of cancer, Vol. 11, No. 5, Pages 595-608), и соответственно представляет
собой MV-deltaC-GM-CSF.В настоящем изобретении выражение «для применения в лечении»
относится к генетически модифицированному патогенному MV-deltaC, который может быть
примененв способе введения индивидууму, для того чтобы полностью уничтожить
злокачественную опухоль или раковое заболевание, диагностируемое у индивидуума, в
частности у человека, или, чтобы уменьшить размер опухоли, или для облегчения симптомов
такой злокачественной опухоли или ракового заболевания. В конкретном варианте
реализации, виротерапия, включающая MV-deltaC, описанный в настоящем изобретении,
может быть дополнена другими видами терапии, в частности химиотерапией. Таким образом,
виротерапия, включающая MV-deltaC, может быть использована в комбинации или в
добавлении к терапевтическому режиму.
В настоящем изобретении выражение «агрессивная злокачественная опухоль или
агрессивный рак» относится к злокачественной опухоли или раку, которые не поддаются
известным в настоящее время традиционным методам лечения, таким как химиотерапия,
лучевая терапия и/или хирургическое вмешательство, и, как следствие развиваются, несмотря
на данное традиционное лечение.
В настоящем изобретении выражение «путем активации плазмацитоидных
дендритных клеток (пДК)» относится к опухолевым клеткам, инфицированным MV или MV11
deltaC, которые рекрутируют пДК для противоопухолевого иммунного ответа путем
активации их способности продуцировать большое количество IFN-α и/или кросс-
презентировать ТАА из зараженных опухолевых клеток к опухоль-специфичным CD8+ Т-
лимфоцитам.
В настоящем изобретении термин «плазмацитоидные дендритные клетки (пДК)»
относится к антиген-презентирующим клеткам, которые синтезируют большое количество
альфа/бета интерферонов (IFN-α/β) в ответ на вирусные и бактериальные стимулы, и в
настоящем изобретении признаны способными фагоцитировать инфицированные опухолевые
клетки.
В конкретном варианте реализации указанный MV-deltaC может быть использован для
лечения злокачественной мезотелиомы, в частности злокачественной плевральной
мезотелиомы.
В другом конкретном варианте реализации настоящее изобретение относится к
генетически модифицированному патогенному вирусу кори, полученному из штамма живого
ослабленного вируса кори, в котором нокаутировали ген, кодирующий вирусный
акцессорный белок C (MV-deltaC) для применения в лечении меланомы или аденокарциномы
легкого, при введении индивидууму, у которого дигностировано такое состояние .
В конкретном варианте реализации настоящего изобретения предложен генетически
модифицированный патогенный MV-deltaC, который может быть применен для лечения
злокачественной опухоли или раковых клеток, устойчивых к немодифицированному MV, при
введении индивидууму у которого дигностирована такая опухоль или раковое заболевание.
Выражение "немодифицированный MV" относится к патогенному живому ослабленному
штамму MV, который не был генетически модифицирован, например, вирус штамма Шварца
или Моратена.
В настоящем изобретении выражение «злокачественная опухоль или раковые клетки,
устойчивые к немодифицированному MV» относится к злокачественной опухоли или
раковым клеткам, которые, как известно, устойчивы к индукции гибели клеток, несмотря на
эффективность инфекции с немодифицированным MV, или которые, как известно,
имеютослабленную реакцию на индукцию гибели клеток с немодифицированным MV, по
сравнению с MV-deltaC.
Авторы изобретения показали, что введение MV-deltaC в различные злокачественные
опухоли или раковые клетки обеспечиваетили вызывает ответ, улучшенный по отношению к
ответу, полученному при введении немодифицированного MV, и в частности проявляет
улучшенные апоптотические свойства при низких концентрациях, с более коротким
временем ответа и улучшенными иммуногенными свойствами (Kroemer, G. et al., Annu. Rev.
Immunol., 2012, 31:51-72).
Согласно конкретному варианту реализации изобретения, указанный генетически
модифицированный патогенный MV-deltaC проявляет апоптотические свойства в
злокачественных опухолях или раковых клетках, инфицированных MV-deltaC.
В настоящем изобретении выражение «апоптотические свойства» относится к
способности индуцировать или вызывать апоптоз в клетках, которая может быть
продемонстрирована с помощью in vitro апоптоза в злокачественных опухолях или раковых
клетках, в частности в злокачественной мезотелиоме, меланоме и клетках аденокарциномы
легких, описанных в примерах.
Активность, проявленная вирусом MV-deltaC по настоящему изобретению, может быть
охарактеризована синтезом молекул иммунного ответа или в клеточном стрессе, или в
клеточной гибели. Данные молекулы включают HMGB-1 (группа белков с высокой
подвижностью 1), калретикулин и белок теплового шока (Hsp70), описанные как сигналы
опасности, участвующие в активации иммунного ответа (Zitvogel,L. et al. Cell, 2012, 140:798-
804).
Каспазы-3, как известно, участвуют в заключительной стадии процесса апоптоза
(Duprez, L. et al., Microbes Infect, 2009, 11(13): 1050-1062).
Согласно конкретному варианту реализации изобретения, указанный генетически
модифицированный патогенныйMV-deltaC индуцирует активацию каспазы-3 в
злокачественных опухолях или раковых клетках, инфицированных MV-deltaC.
Белок Hsp70 на наружном слое плазматической мембраны инфицированных клеток
участвует в иммунном ответе, в частности, в распознавании антиген-презентирующими
клетками и эффекторами врожденного клеточного иммунитета (Oglesbee et al.,Viral Immunol,
2002, 15(3): 399-416).
Согласно конкретному варианту реализации изобретения, указанный генетически
модифицированный патогенный MV-deltaC индуцирует экспозицию белка Hsp70 на
наружном слое плазматической мембраны в злокачественных опухолях или раковых клетках,
инфицированных MV-deltaC.
Калретикулин является главным белком эндоплазматического ретикулума, который
может перемещаться в наружный слой плазматической мембраны во время клеточного
стресса (Heal et al., Biochem J, 1998, 329(2), 389-394). В частности, такая экспозиция на
поверхности клетки делает возможным фагоцитоз апоптотических клеток
антигенпрезентирующими клетками (Ogden et al., J Exp Med, 2001, 194(6):781-795). Недавно,
в одном исследовании было выдвинуто предположение, что экспозиция калретикулина на
поверхности клетки обуславливает иммуногенность их гибели (Obeid et al., Nat Med, 2007,
13(1): 54-61).
Согласно конкретному варианту реализации изобретения, указанный генетически
модифицированный патогенный MV-deltaC индуцирует транслокацию калретикулина к
злокачественным опухолям или раковым клеткам, инфицированным MV-deltaC.
Белки HMGB-l (группа белков с высокой подвижностью 1), выделяемые в окружающую
среду во время иммуногенной клеточной смерти действуют на созревание дендритных клеток
путем связывания с различными рецепторами, такими как TLR4 (Apetoh et al., Nat Med, 2007,
13(9):1050-1059) и TLR9 (Tian et al., Nat Immunol, 2007, 8(5): 487-496).
Согласно конкретному варианту реализации изобретения, указанный генетически
модифицированный патогенный MV-deltaC индуцирует выделение HMGB-l во
внеклеточную среду злокачественных опухолей или раковых клеток, инфицированных MVdeltaC.
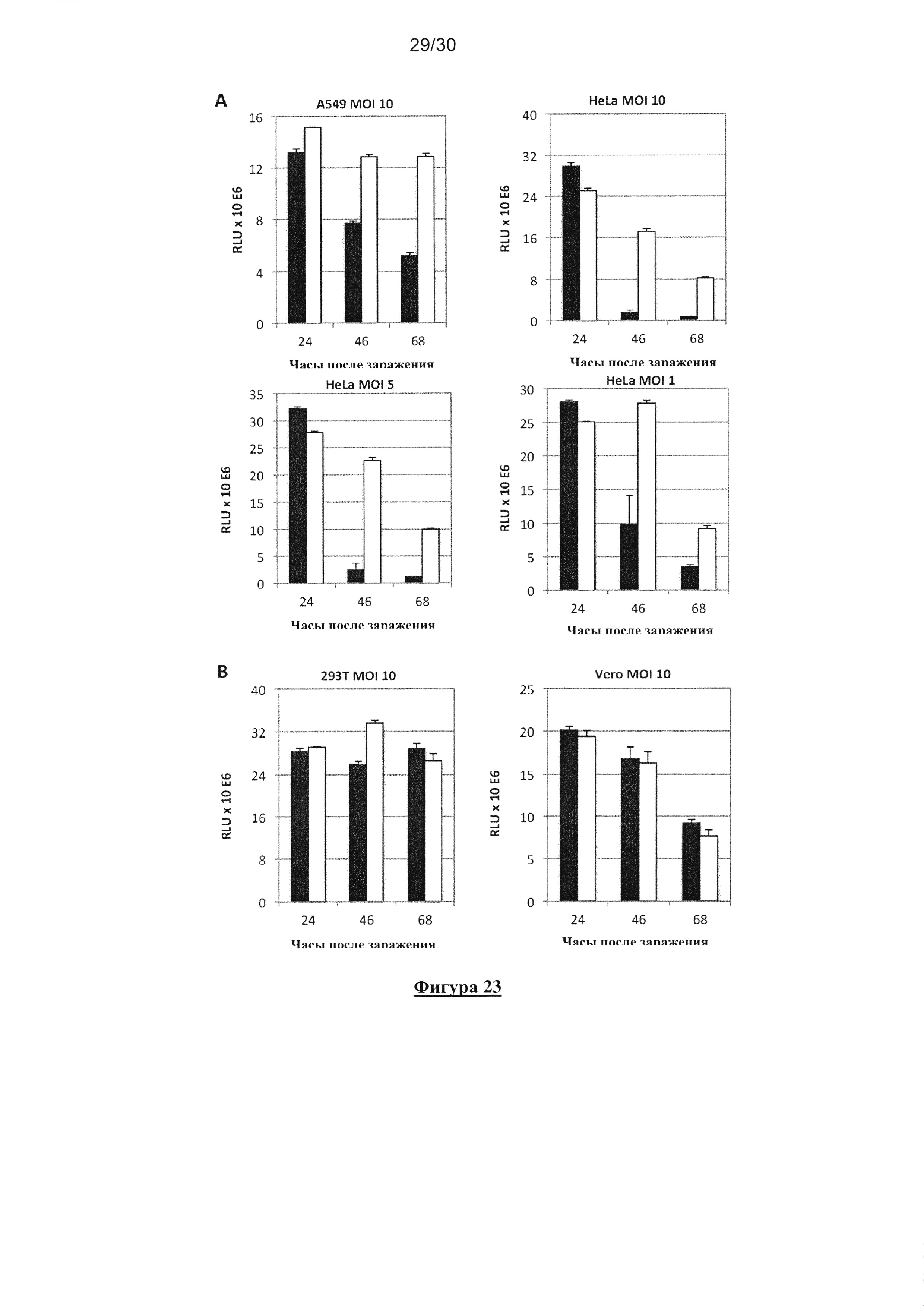
Авторы сравнили индукцию гибели клеток в линиях раковых клеток человека (клетки
аденокарциномы легких человека A549 и Hela-клетки рака шейки матки), и в нераковых
клетках ( эмбриональные клетки почки человека НЕК 293 и Vero-клетки почки
африканской зеленой мартышки). Они показали, что MV-deltaC индуцировал гораздо более
интенсивную и раннюю гибель клеток, чем немодифицированный MV как в А549, так и в
Hela-клетках рака человека, даже при низком MOI (Фигура 23A). Авторы также отметили,
что индукция гибели клеток на Vero-клетках была аналогичной как для
немодифицированного MV, так и для MV-deltaC, в то время как гибель клеток не
наблюдалось на клетках НЕК 293 после 68 часов заражения (Фигура 23B). Таким образом
авторы доказали, что MV-deltaC специфичен к раковым клеткам человека: действительно,
MV-deltaC показал более высокую апоптическую активность, чем немодифицированный MV,
при контакте с раковыми клетками человека, но показал аналогичную апоптотическую
активность по сравнению с немодифицированным MV при контакте с лабораторными
клеточными линиями, т.е. Vero-клетками.
Настоящее изобретение также относится к способу получения вакцинных
плазмацитоидных дендритных клеток (пДК), предназначенных для лечения злокачественной
опухоли или рака у индивидуума, у которого диагностирована такая злокачественная опухоль
или раковое заболевание, включающему следующие стадии:
- in vitro заражение злокачественной опухоли или раковых клеток, предварительно
отобранных у указанного индивидуума, патогенным вирусом кори, полученным из штамма
живого ослабленного вируса кори для получения клеточного лизата;
- приведение пДК с клеточным лизатом для получения вакцинных пДК;
- выделение загруженных пДК.
Настоящее изобретение также относится к способу получения вакцинных
плазмацитоидных дендритных клеток (пДК), предназначенных для лечения злокачественной
опухоли или рака у индивидуума, у которого диагностирована такая злокачественная
опухоль или раковое заболевание, включающему следующие стадии:
- in vitro заражение злокачественной опухоли или раковых клеток, предварительно
отобранных у указанного индивидуума, генетически модифицированным патогенным
вирусом кори, полученным из штамма живого ослабленного вируса кори, у которого
нокаутирован ген, кодирующий вирусный акцессорный белок С (MV-deltaC), для получения
клеточного лизата;
- приведение пДК в контакт с клеточным лизатом для получения вакцинных пДК;
- выделение загруженных пДК.
В настоящем изобретении указанная злокачественная опухоль или рак в определенном
выше способе представляют собой агрессивную злокачественную опухоль или агрессивный
рак, в частности, злокачественную мезотелиому, меланому или аденокарциному легких.
В настоящем изобретении, указанный штамм живого ослабленного вируса кори в
определенном выше способе представляет собой штамм Шварца или штамм Моратена.
Введение MV-deltaC индивидууму возможно через внутриплевральную полость или
интраназально, внутримышечно, внутривенно или подкожно. Если MV-deltaC вводят через
внутриплевральную полость, предпочтительно вводить в непосредственной близости или
непосредственно в злокачественную опухоль или раковые клетки, подлежащие лечению.
Терапевтически эффективное количество MV-deltaC для введения предпочтительно
лежит в диапазоне от 103 до 109 50% инфицирующей дозы для клеточной культуры (TCID50).
Определение TCID50 хорошо известно специалисту в данной области техники и в подробно
описано у Karber (Karber, Arch. Exp. Path. Pharmak, 1931, 162:840-483).
Стадия забора злокачественной опухоли или раковых клеток у индивидуума для
возможности приготовления вакцинных пДК ex vivo предпочтительно не включена в способ
приготовления вакцинных пДК. Данная стадия может быть выполнена согласно любому
способу известному специалисту в данной области техники для забора или взятия образцов
клеток, такие как биопсия или выпот (т.е. плевральный выпот). После забора,
злокачественная опухоль или раковые клетки могут поддерживатьсяв культуре согласно
классическим методам или могут быть заморожены (т.е. при -80°C) для сохранения,
например. Если злокачественная опухоль или раковые клетки не происходят от индивидуума,
подлежащего лечению вакцинными пДК, то они могут быть получены из аллогенных линий
клеток злокачественной мезотелиомы, меланомы или аденокарциномы легких.
В определенном выше способе приготовления in vitro, заражение злокачественной
опухоли или раковых клеток MV-deltaC может быть выполнено непосредственным
контактом клеток и вируса, например, при множественности заражения 1 (MOI), с
инкубацией 2 часа при 37°C. После заражения, гибель инфицированных клеток происходит
спонтанно в результате вирусного воздействия. Сначала как правило формируется синцитий,
затем происходит лизис клеток, таким образом предоставляя клеточный лизат, подходящий
для приготовления вакцины. Данный феномен может быть доказан c помощью прямого
микроскопического наблюдения инфицированных клеток.
В настоящем изобретении термин «клеточный лизат» относится ко всему (или целому)
клеточному лизату, полученному как описано выше, или к фракциям клеточного лизата, в
частности к мембранным фракциям (т.е. цитоплазматические вирусные включения или
апотела).
пДК могут быть получены многочисленными способами хорошо известными
специалисту в данной области техники. В конкретном варианте реализации изобретения, пДК
предпочтительно получают из индивидуума, подлежащего лечению. Весьма предпочтительно
чтобы пДК получали посредством лейкофереза. Получение пДК очень хорошо известно
специалисту в данной области техники. Предпочтительно, пДК могут быть получены
согласно общей методологии, описанной Coulais (Coulais, D et al., Cytotherapy, 2012, 14(7):
887-896). Если пДК происходит из организма индивидуума, подлежащего лечению, пДК
может быть получен посредством лейкофереза от указанного индивидуума.
Как будет понятно специалисту в данной области техники, контакт пДК и клеточного
лизата должен поддерживаться в течение периода времени достаточного для возможности
эффективной загрузки пДК антигенами, находящимися в клеточном лизате. Согласно
настоящему изобретению после загрузки получаются вакцинные пДК. Загрузка может
происходить согласно общей методологии, описанной Gauvrit (Gauvrit, A et al., Cancer Res,
2008, 68(12), 4882-4892). Примерный период контакта между пДК и клеточным лизатом
достаточный для возможности эффективной загрузки пДК составляет около 24 часов.
Активированное состояние пДК как правило достигается после того как пДК были
загружены. Активированное состояние (или зрелое состояние) пДК может быть
подтвержденомногочисленными маркерами хорошо известными специалисту в данной
области техники, такими как мембранные или цитокинные маркеры. Такие маркеры
активированных дендритных клеток в особенности описаны Barchet (Barchet, W et al.,
Seminars in Immunology, 2005, 17(4):253-261) и Marafioti (Marafioti, T et al., Blood, 2008,
111(7):3778-3792).
Таким образом, вакцинные пДК, которые могут быть получены согласно способам
приготовления по изобретению чрезвычайно полезны, так как они являются
сильнодействующими стимуляторами противоопухолевых CD8 T-клеток. В равной степени
полезно, что способ приготовления согласно изобретению, позволяет получить вакцинные
пДК в активированном состоянии.
Настоящее изобретение относится в частности к вакцинным дендритным клеткам,
которые могут быть получены способом приготовления, указанным выше. Согласно
конкретному варианту реализации изобретения, указанные вакцинные пДК могут быть
использованы для лечения агрессивной злокачественной опухоли или агрессивного рака при
введении индивидууму, у которого дигностирована такая опухоль или раковое заболевание.
В частности, вакцинные пДК могут быть использованы для лечения злокачественной
мезотелиомы, меланомы или аденокарциномы легких при введении индивидууму, у которого
диагностировано такое состояние. Описанный вариант реализации, относящийся к
применению MV-deltaC, аналогично релевантный для применения пДК, как описано в
данном описании.
Авторы рассмотрели, in vitro, влияние заражения опухолевой клетки штаммом MV
Шварца на состояние активации человеческих пДК и их способность кросс-презентации
опухолевого антигена специфическому CD8+ T-клеточному клону. Авторы показали, что
несмотря на экспрессию CD46, пДК не были чувствительны к заражению MV. Однако, пДК
были способны отвечать на MV in vitro посредством продуцирования IFN-α, c большей
чувствительностью, при добавлении в культуру IL-3. Авторы также показали, что
опухолевые клетки, зараженные MV, запускали активацию пДК, в частности продуцирование
IFN-α, в то время как опухолевые клетки, облученные УФ, нет. Активация пДК была
вероятно вызвана одноцепочечной РНК MV, которая активировала TLR7 в эндоцитозном
компартменте пДК вслед за фагоцитозом опухолевых клеток, зараженных MV. Интересно
отметить, что авторы проиллюстрировали, впервые, что человеческие пДК,
культивировавшиеся совместно с опухолевыми клетками, зараженными MV, были способны
кросс-презентировать опухолевый антиген NYESO-1 специфическому CD8+ T-клеточному
клону. Данные результатысвидетельствуют о том, что в дополнение к прямому воздействию
на лизис опухоли, противоопухолевая виротерапия, основанная на MV, может запускать
противоопухолевый иммунный ответ посредством активации пДК. Предполагается, что
аналогичные результаты могут быть получены при использовании MV-deltaC, поскольку
заражение опухолевых клеток с помощью мутантного вируса бывает аналогичным.
Ожидается получение более сильного иммунного ответа, поскольку MV-deltaC приводит к
более интенсивной экспрессии сигналов опасности (калретикулин и Hsp70) после заражения
опухолевых клеток и зараженные клетки также экспрессировали одноцепочечную РНК MVdeltaC.
Настоящее изобретение также относится к фармацевтической композиции, содержащей
генетически модифицированный патогенный MV-deltaC или вакцинные пДК, которые могут
быть получены указанным выше способом, в качестве активного ингредиента, в сочетании с
фармацевтически приемлемым носителем, для применения в лечении агрессивной
злокачественной опухоли или агрессивного рака посредством активации пДК при введении
индивидууму, у которого диагностирована такая злокачественная опухоль или раковое
заболевание.
В данном описании, фармацевтически приемлемый носитель включает в себя любое
вещество, благодаря которому MV-deltaC может входит в состав композиции. Носитель
представляет собой любое физиологически приемлемое вещество или комбинацию веществ,
т.е. подходящее для его применения в композиции по отношению к хозяину, в частности к
человеку, и, следовательно, нетоксичное. Примерами таких носителей являются фосфатно-
буферные солевые растворы, дистиллированная вода, эмульсии, такие как масляные/водные
эмульсии, стерильные растворы различных видов смачивающих веществ, и тому подобное.
Настоящее изобретение также относится к фармацевтической композиции или
комбинации активных ингредиентов, содержащих генетически модифицированный
патогенный MV-deltaC или вакцинные пДК, которые могут быть получены указанным выше
способом, и дополнительно содержащие химиотерапевтический агент и фармацевтически
приемлемый носитель для применения в лечении агрессивной злокачественной опухоли или
агрессивного рака при введении индивидууму, у которого диагностирована такая
злокачественная опухоль или раковое заболевание.
В данном описании, химиотерапевтический агент представляет собой вещество,
которое может быть применено в лечении злокачественной опухоли или рака. Природа
химиотерапевтического агента будет зависеть от типа злокачественной опухоли или рака.
Примеры химиотерапевтических агентов хорошо известны специалисту в данной области
техники.
Настоящее изобретение также относится к комбинации активных ингредиентов,
содержащей (i) живой ослабленный MV или генетически модифицированный патогенный
MV-deltaC и (ii) вакцинные пДК, которые могут быть получены указанным выше способом,
для применения для одновременного или раздельного введения в лечении агрессивной
злокачественной опухоли или агрессивного рака при введении индивидууму, у которого
диагностирована такая злокачественная опухоль или раковое заболевание.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
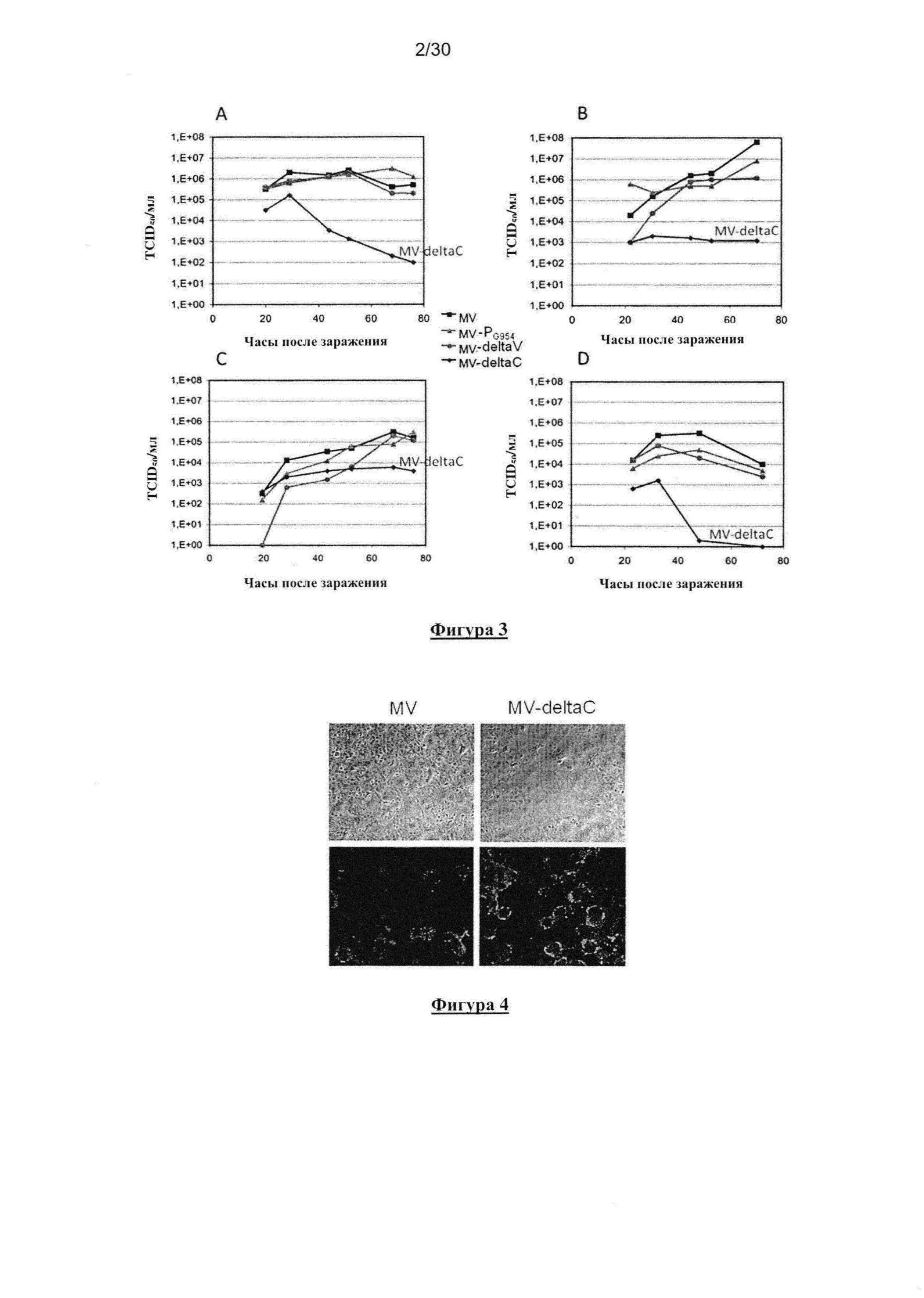
Фигура 1. MV и MV-deltaC. (A) Схематичная диаграмма генома MV и порядок генов,
показывающий, что ген P кодирует белки P, V и С (показаны P, V и С ORFs). (B) Участок
нуклеотидной последовательности плазмид pTM-MVSchw (SEQ ID NO:3) и pTM-MVSchwdeltaC-
ATU1 (eGEP), где нуклеотид T нативной кДНК генома MVSchw в позиции 2788
заменен нуклеотидом С (SEQ ID NO:4); в дополнение к данной первой мутации, вторая
мутация может быть выполнена путем добавления стоп кодона на пути транскрипции в белке
С открытой рамки считывания, т.е. стоп кодона «TAG», который получен замещением
нуклеотида G нативной кДНК генома штамма MVSchw в позиции 2803 на нуклеотид А (SEQ
ID NO:5) (представленные нуклеотидные мутации подчеркнуты).
Фигура 2. Экспрессия белков P, V и С MV, MV-deltaV, MV-deltaC и MV-PG954. Лизаты
Vero-клеток, зараженные различными вирусами, разделяли на фракции в геле SDS-PAGE и
белки P, V и С определяли с помощью Вестерн-блоттинга, используя специфические
моноклональные антитела.
Фигура 3. Кинетика репликации MV, MV-PG954, MV-deltaV и MV-deltaC. Клетки Vero (A),
HeLa (B), Jurkat (C) и U937 (D) заражали различными вирусами при значении
множественности заражения равном 1. Клеточно-ассоциированные титры вируса определяли
с помощью TCID50 анализа.
Фигура 4. Цитопатическое действие MV и MV-deltaC на клетки Vero и HeLa. Верхняя
панель: цитопатическоедействие, индуцированное в Vero-клетках через 24 часа после
заражения MV или MV-deltaC (MOI 0,1). Нижняя панель: иммунофлюоресценция HeLa-
клеток через 24 часа после заражения MV или MV-deltaC. Клетки фиксировали и окрашивали
с моноклональным анти-геммаглютинином (H) MV.
Фигура 5. Кинетика экспрессии вирусного белка в клетках Vero, зараженных MV или
MV-deltaC. Клетки были заражены при MOI равной 1 и затем лизированы в разные моменты
времени. Клеточные лизаты анализировали с помощью Вестерн-блоттинга, и вирусные белки
N и V определяли, используя специфические моноклональные антитела (клон анти-N 120,
Naniche, D. et al., J Gen Virol., 1992, 73(10):2617-2624; анти-V, Takeuchi, K. et al., FEBS Letters,
2003, 545(2), 177-182).
Фигура 6. Анти-MV гуморальный ответ, индуцированный у мышей СD46/IFNAR,
иммунизированных MV-PG954, MV-deltaV или MV-deltaC. Титры антител определяли с
помощью ELISA в сыворотке, собранной через 2 месяца после одной прививки. (А)
Предельное разведение проб сыворотки, объединенной из разных групп мышей. (В)
Отдельные титры каждой мыши. (С) Средние титры каждый группы. Титры антител
определяли, как предельное разведение тестируемой сыворотки, используя дважды значение
абсорбции, рассчитанное против значения абсорбции сыворотки неиммунизированных
мышей.
Фигура 7. Заражение и гибель клеток, индуцированные MV-deltaC. (А) Опухолевые
клетки заразили MV или MV-deltaC (MOI=1,2 часа) и анализировали с помощью проточной
цитометрии после двойного окрашивания FITC-Annexin-V и пропидия иодидом через 72 часа
после заражения. Данные представляют собой проценты Annexin-V клеток.
(В) Опухолевые клетки, зараженные вирусом (MV или MV-deltaC) и незараженные
опухолевые клетки анализировали с помощью проточной цитометрии после окрашивания
анти-активным каспаза-3 антителом (BD Biosciences) через 24 часа после заражения.
Проценты означают количественное соотношение «активированных каспаза-3-
положительных» клеток после заражения MV-deltaC.
Фигура 8. Экспозиция белка Hsp70 на поверхность клетки. Экспрессию мембранного
белка Hsp70 определяли через 24 после заражения для эпителиоидных клеток мезотелиомы
Meso13 и Meso56, или через 72 часа после заражения для клеток аденокарценомы легких
А549 и клеток меланомы M17, c помощью внеклеточного окрашивания и проточной
цитометрии. Данные представляют собой проценты содержания Hsp70 в клетках.
Фигура 9. Мембранная транслокация калретикулина после заражения MV-deltaC.
Незараженные и зараженные (MV или MV-deltaC, MOI=1) опухолевые клетки окрасили анти-
калретикулиновым антителом и конъюгированным анти-мышиным вторичным антителом
Сy5 через 24 часа после заражения для эпителиоидных клеток мезотелиомы Meso13 и
Meso56, или через 72 часа после заражения для клеток аденокарценомы легких А549 и клеток
меланомы M17. Затем клетки анализировали с помощью проточной цитометрии. Данные
представляют собой проценты содержания калретикулина в клетках.
Фигура 10. Высвобождение HMGB-1 во внеклеточную среду. Супернатанты
незараженных и зараженных (MV или MV-deltaC, MOI=1) опухолевых клеток отобрали через
24, 48 или 72 часа после заражения и хранили при -20°C. Затем определяли количество
HMGB-1 в супернатантах с помощью ELISA.
Фигура 11. Экспрессия рецептора MV, чувствительность заражения MV и
выживаемость опухолевых клеток и пДК. (A) Экспрессия СD46 и CD150/SLAM на
поверхности линий опухолевых клеток (M18, Meso13 и A549) и пДК (окрашивание mAb:
серая гистограмма; изотипический контроль: белая гистограмма; значения на гистограммах
представляют собой R-MFI, относительную среднюю интенсивность флюоресценции,
определеннyю с помощью деления MFI окрашивания mAb на MFI изотипического контроля).
(В) Заражение линий опухолевых клеток (М18, Meso13 и А549) и пДК вирусом MV-eGFP
(MOI=1). (C) Заражение пДК вирусом MV-eGFP (MOI=1), в присутствии или отсутствии IL-3.
(D) Заражение пДК вирусом MV-eGFP с увеличением MOI, в присутствии или отсутствии IL-
3. (Е) Выживаемость линий опухолевых клеток после заражения MV или УФ-облучения.
Через три дня после заражения или УФ-облучения, клетки инкубировали с TO-PRO®3,
который окрашивает мертвые клетки. Флюоресценцию анализировали с помощью проточной
цитометрии. Результаты, показанные наФигурах 1А, 1С и 1Е представляют собой результаты
трех независимых экспериментов. Результаты, показанные на Фигурах 1В и 1Е отражают
среднее значение трех независимых экспериментов. Планки погрешностей представляют
собой стандартные отклонения.
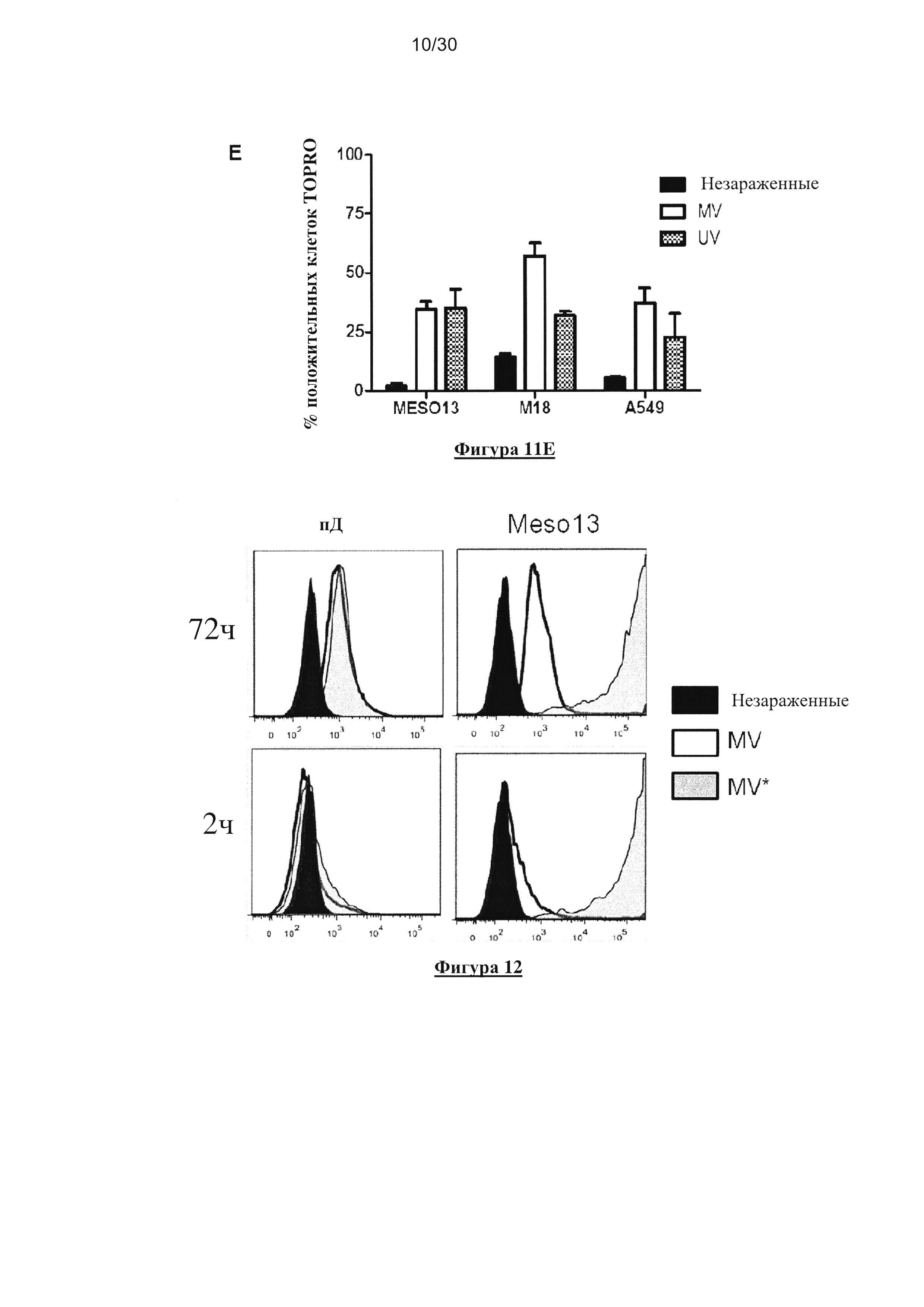
Фигура 12. Заражение пДК вирусом MV-eGFP или облученным УФ MV-eGFP. пДК, в
присутствии IL-3 или клеток Meso13, культивировали без (NI) или совместно с MV-eGFP
(MV), или с облученным УФ (312нм - 100 кДж/м2) MV-eGFP (MV*), при MOI=50 в течение
72 часов (верхняя панель) или в течение 2 часов, и затем культивировали в течение 70 часов
(нижняя панель). Флюоресценцию анализировали с помощью проточной цитометрии.
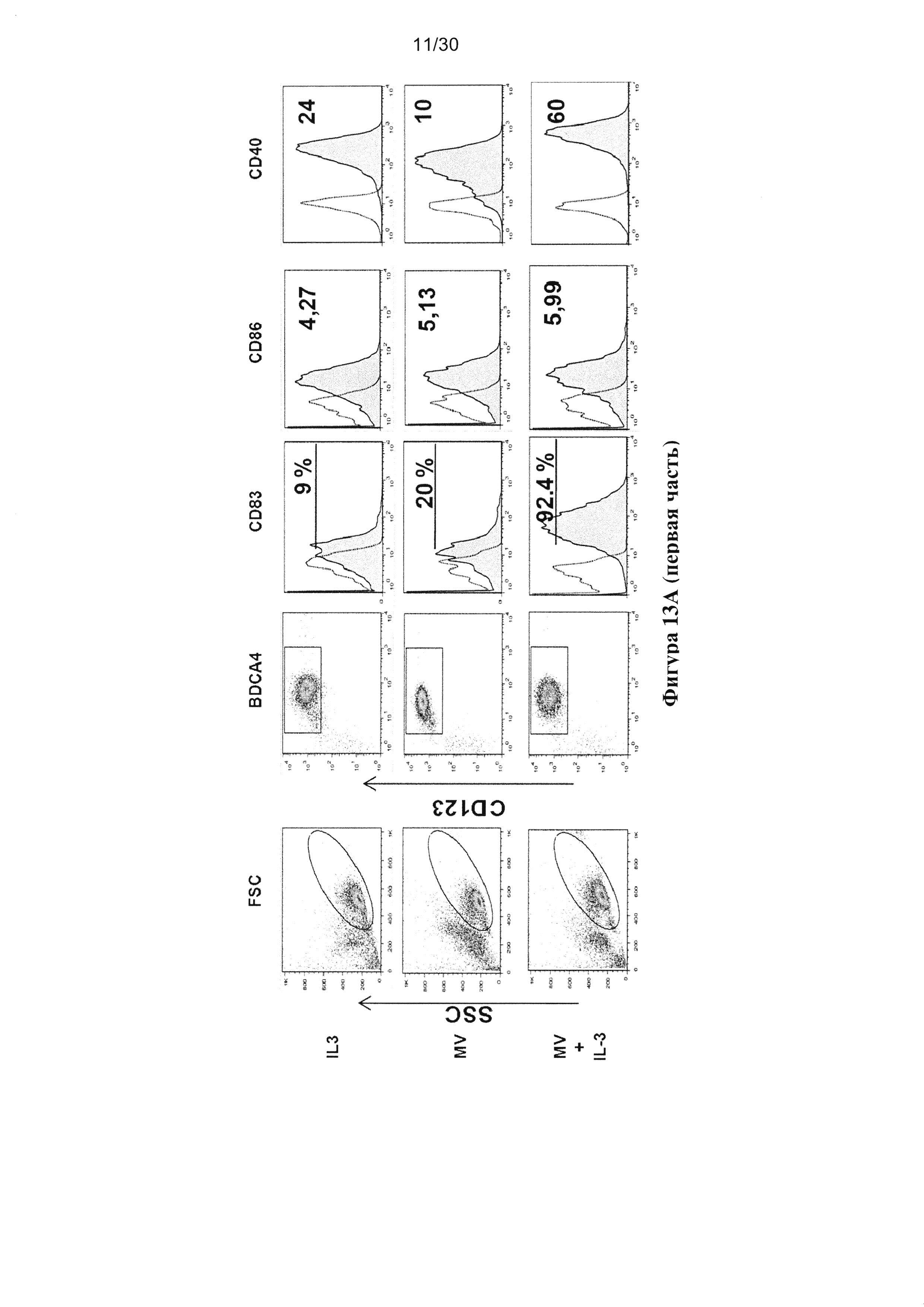
Фигура 13. Опухолевые клетки, зараженные MV, индуцируют созревание пДК. пДК
культивировали в течение 18 часов совместно с IL-3, MV (MOI=1), MV и IL-3, R848,
опухолевыми клетками, облученными УФ или зараженными MV. (A) Экспрессия CD83,
CD86 и СD40 клетками пДК измеряли с помощью проточной цитометрии c гейтом клеток
CD123+/BDCA-4+. (В) Гистограммы получены из трех независимых экспериментов.
Непараметрический критерий Манна-Уитни использовали для определения значения белка P,
полученный путем сравнения результата образца с результатом IL-3 пДК (* p<0,05, **p<0,01,
***p<0,0001).
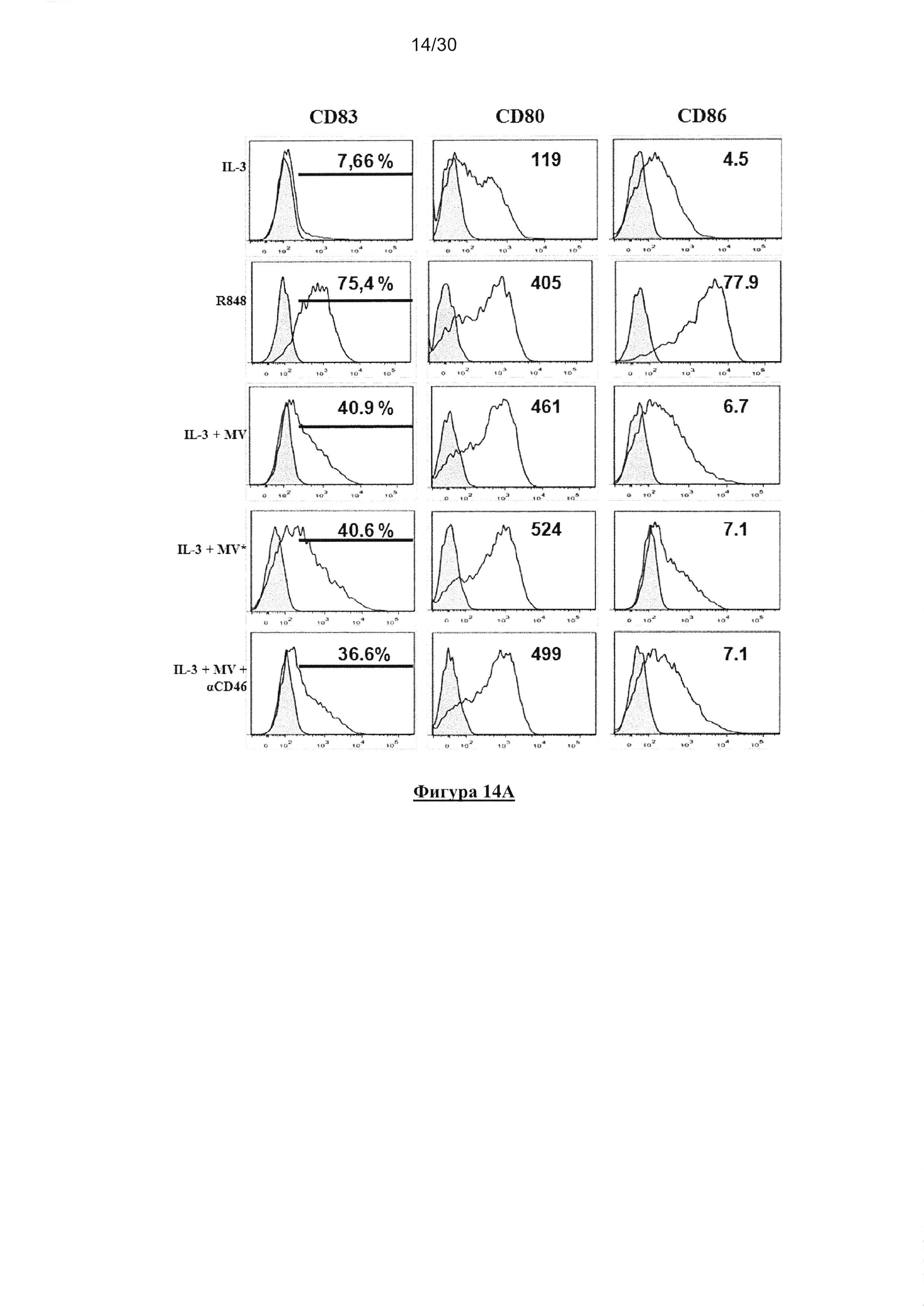
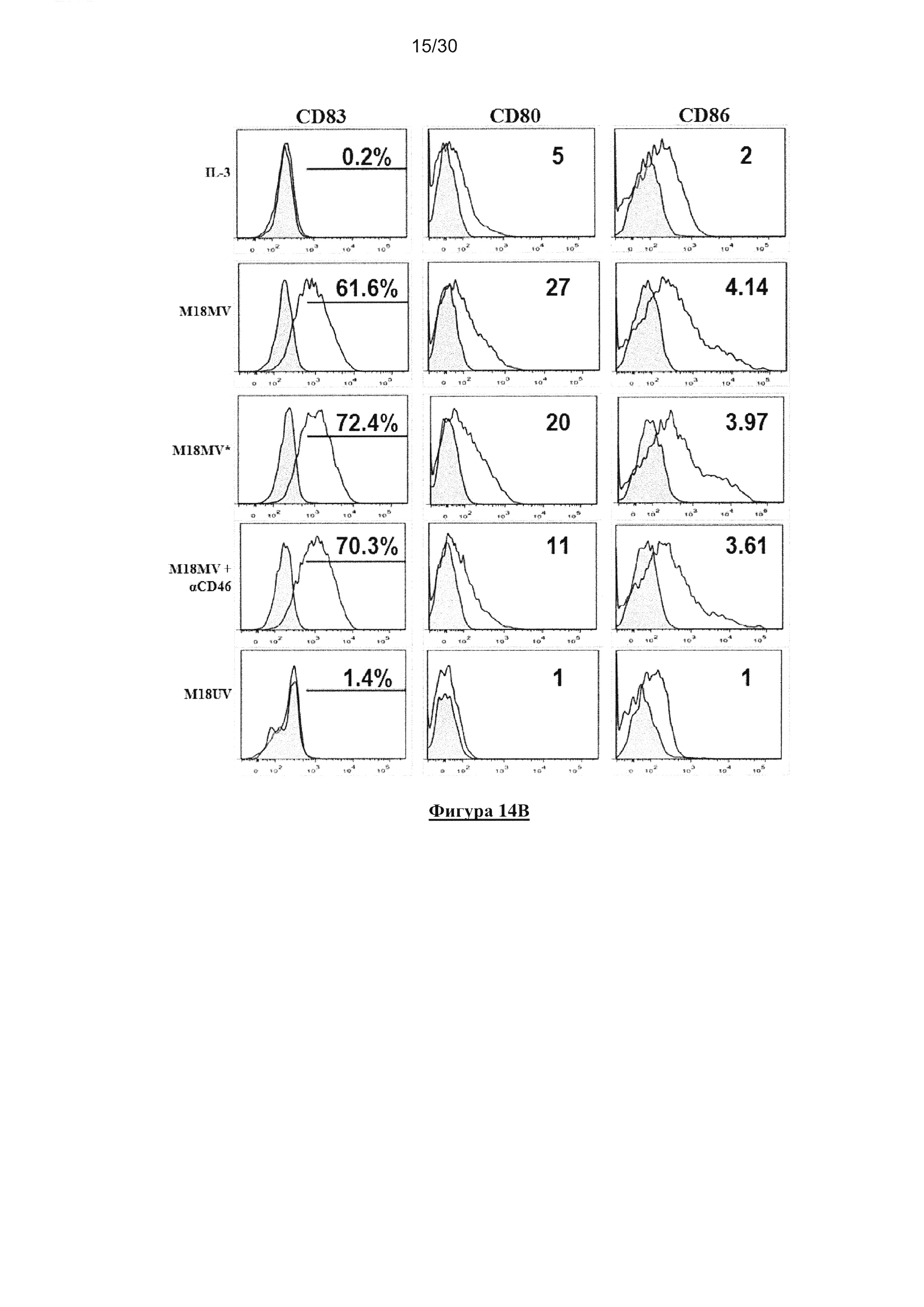
Фигура 14. Активация пДК в ответ на опухолевые клетки, инфицированные MV,
является независимой от репликации MV в пДК и заражения CD46. (A) пДК
культивировали совместно с IL-3, R848, IL-3/MV-eGFP (MV), IL-3/MV-eGFP совместно
c10мг/мл анти-CD46 или IL-3/облученной УФ MV-eGFP (MV*) при MOI=1 в течение 18
часов. Экспрессию CD83, CD80 и СD86 клетками пДК определяли с помощью проточной
цитометрии. (В) пДК культивировали совместно с IL-3, облученными УФ M18, M18,
зараженными MV (M18MV), в присутствии или отсутствии10мг/мл анти-CD46 (Hycult
biotech), или M18, зараженные MV, облученные УФ перед представлением клеткам пДК
(M18MV*). Экспрессию CD83, CD80 и СD86 клетками пДК (гейт BDCA-4+/HLA-DR+
клеток) определяли с помощью проточной цитометрии. (С) Продуцирование IFN-α клетками
пДК измеряли с помощью ELISA. (D) Ингибирование MV-eGFP заражения М18 посредством
10мг/мл анти-CD46 моноклонального антитела через 72 часа культивирования.
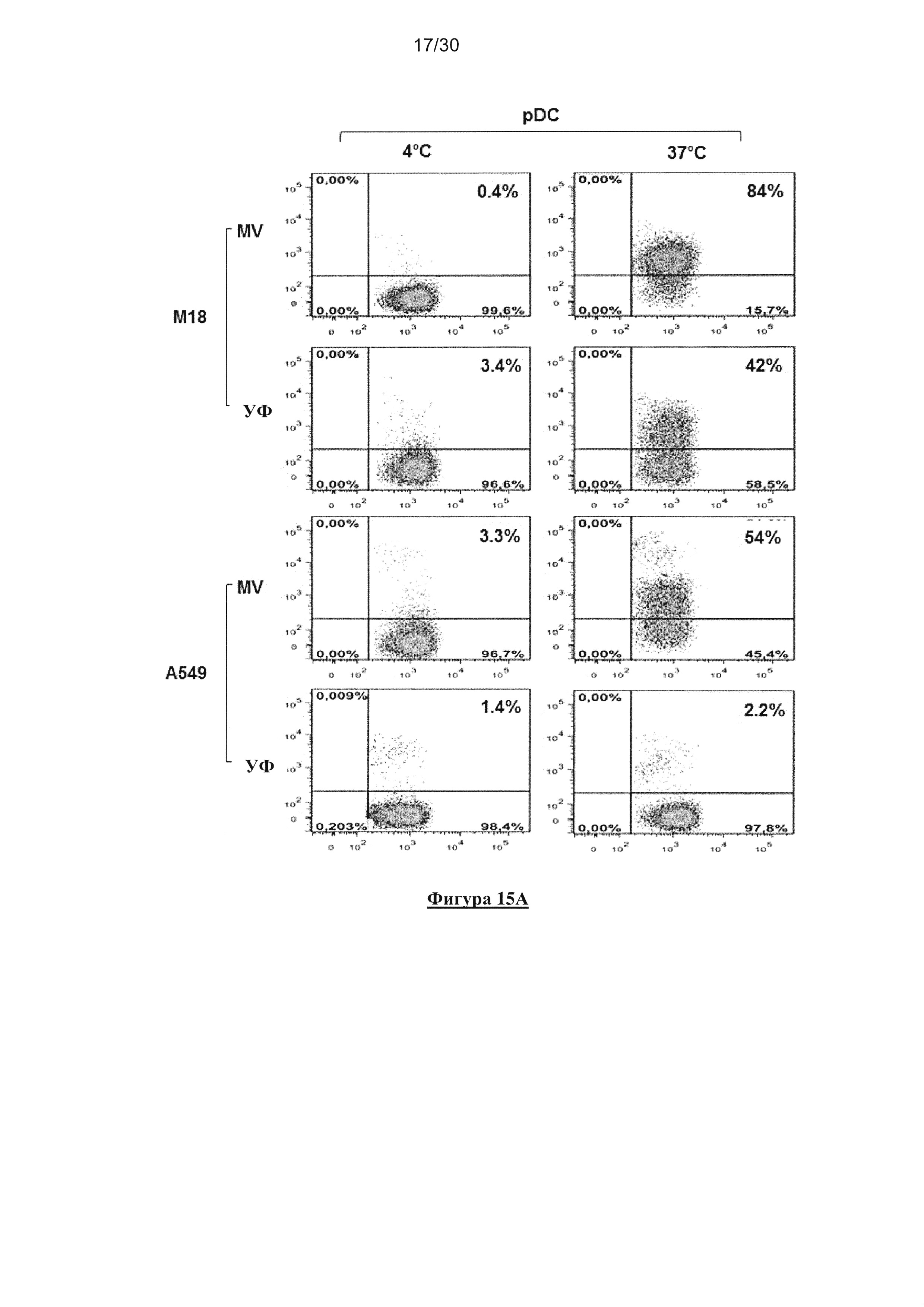
Фигура 15. Фагоцитоз опухолевых клеток, зараженных MV или облученных УФ,
клетками пДК. (А) Опухолевые клетки, зараженные MV и облученные УФ, окрашивали
PKH-67 и культивировали совместно с пДК в течение 18 часов при 4°C или 37°C (1 ДК:1
опухолевая клетка). Клетки окрашивали HLA-DR-специфичным mAb. Флюоресценцию
анализировали с помощью проточной цитометрии. Данный эксперимент проводили в
четырех повторах. (В) Представлен график рассеяния для четырех исследований фагоцитоза.
Планки погрешностей представляют собой стандартные отклонения. (C) Опухолевые клетки,
зараженные MV, окрашивали PKH-67 (зеленый) и культивировали совместно с пДК в
течение 18 часов. Клетки окрашивали HLA-DR-специфичным mAb (красный).
Флюоресценцию анализировали с помощью конфокального микроскопа.
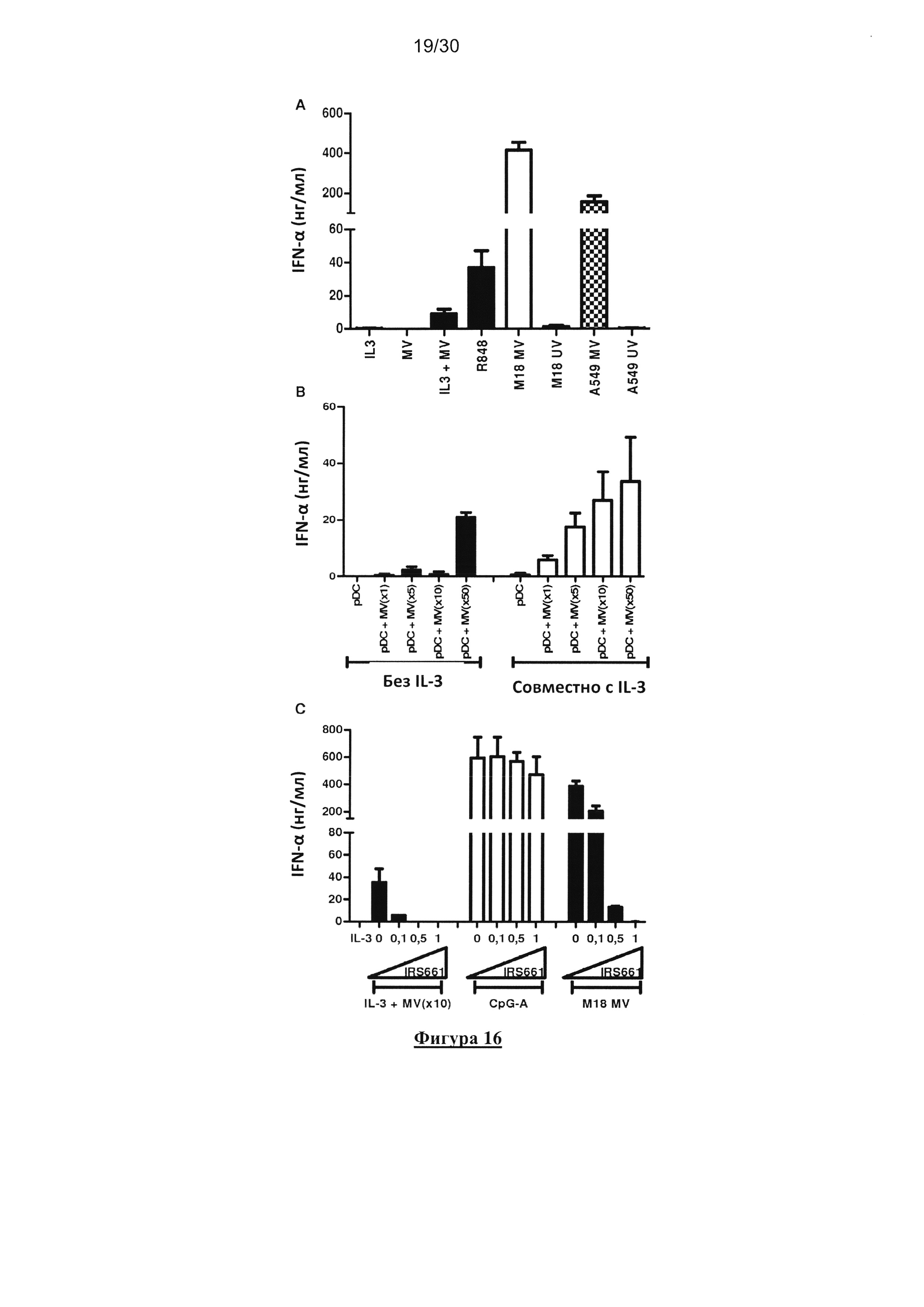
Фигура 16. Продуцирование IFN-α клетками пДК в ответ на MV является TLR7-
зависимым. (А) пДК культивировали в течение 18 часов совместно с IL-3, MV(MOI=1), MV
и IL-3, R848, опухолевыми клетками М18 или А549, облученными УФ или зараженными MV.
Продуцирование IFN-α измеряли с помощью ELISA в культуральных супернатантов. (В) пДК
культивировали в течение 18 часов совместно или без IL-3 и при возрастающих количествах
MV. Продуцирование IFN-α измеряли с помощью ELISA в культуральных супернатантах. (C)
пДК культивировали в течение 18 часов совместно c IL-3 и MV (MOI=10), CpG-A или M18,
зараженная MV, в отсутствии или присутствии разных концентраций IRS661 (TLR7
ингибитор). Продуцирование IFN-α измеряли с помощью ELISA в культуральных
супернатантах. Результаты получены из трех независимых экспериментов.
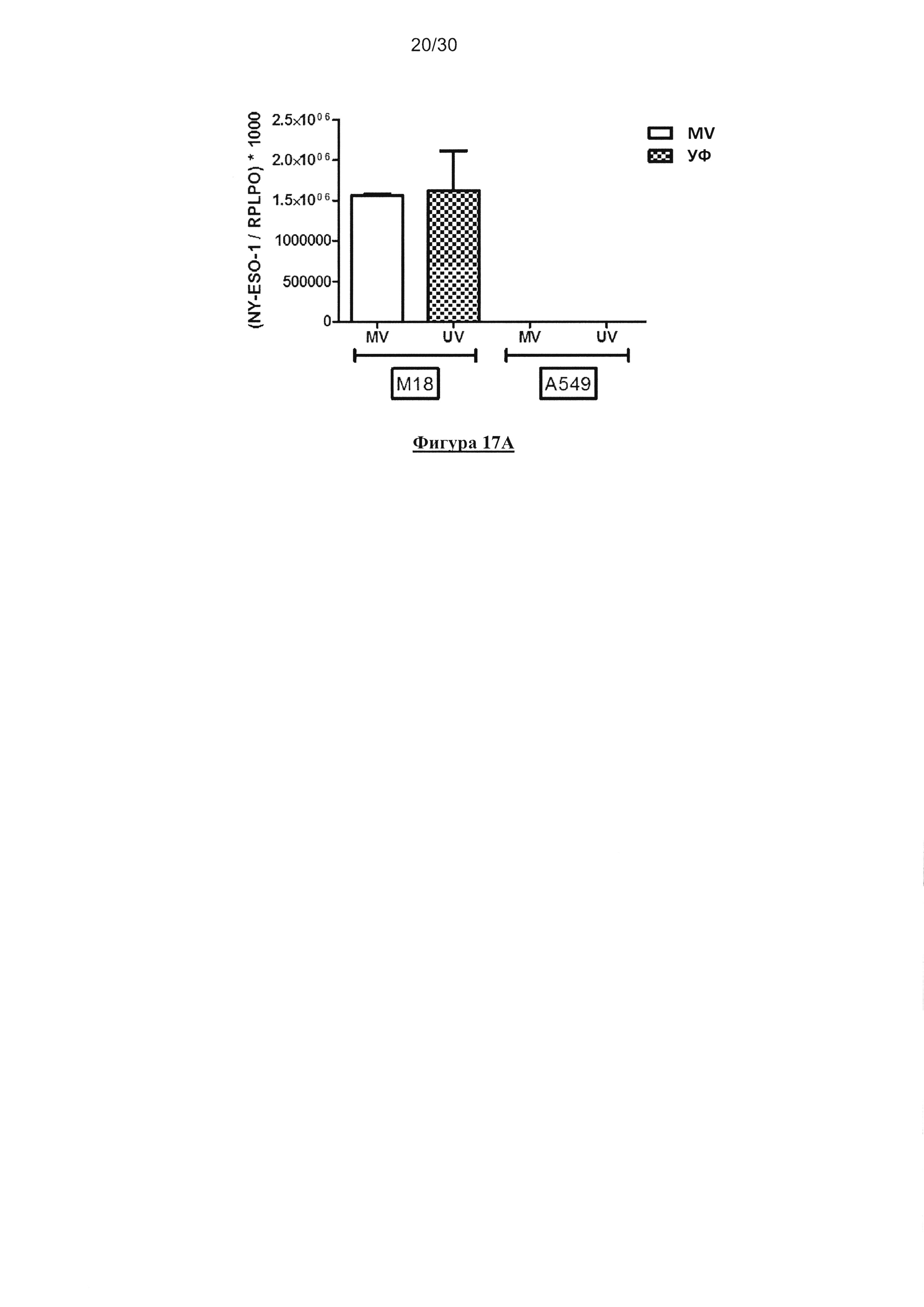
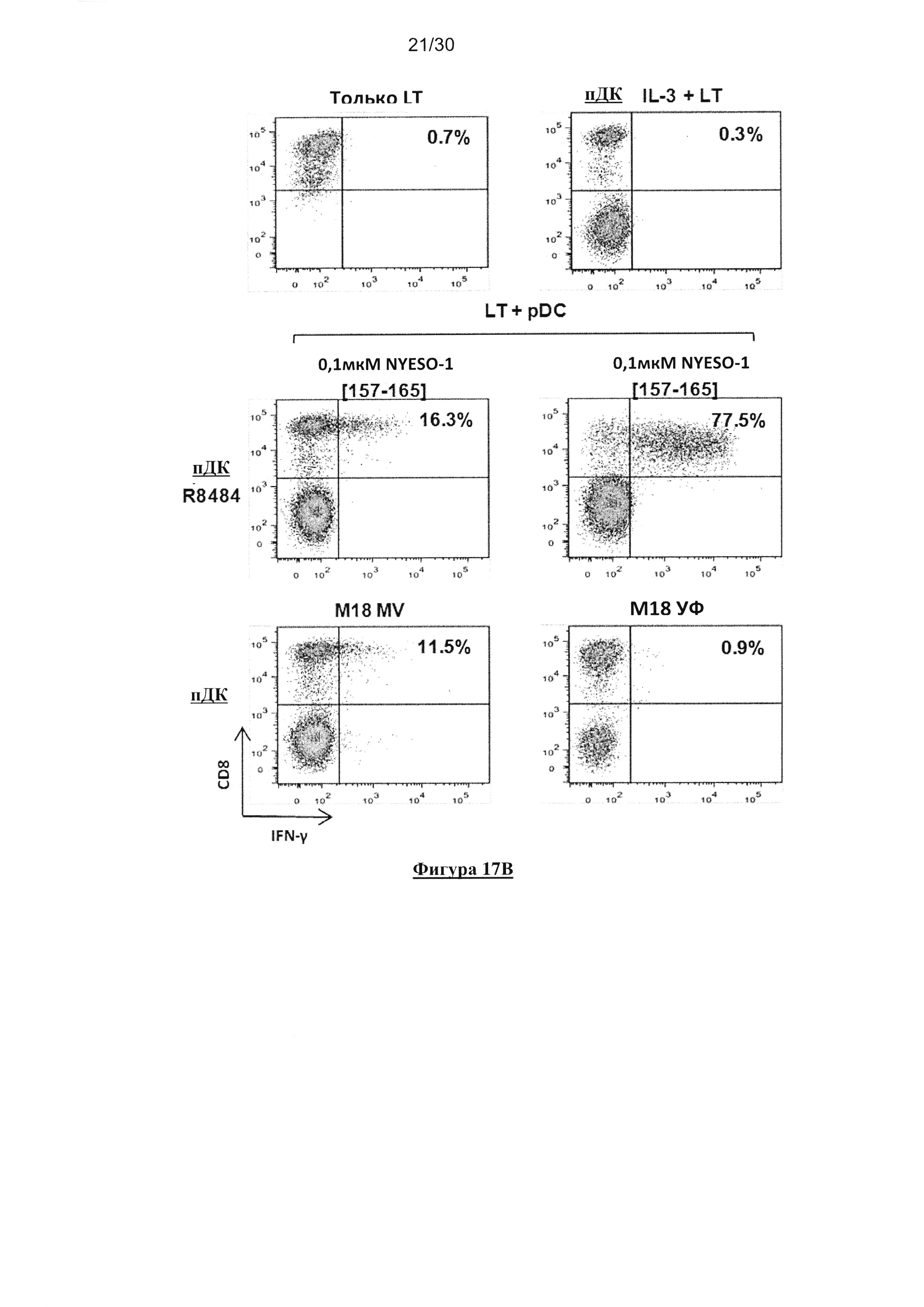
Фигура 17. Кросс-представление NYESO-1 посредством HLA-A*0201+ пДК после
совместного культивирования с NYESO-1/ HLA-A*0201- M18 опухолевыми клетками,
зараженными MV. (А) Экспрессия NYESO-1 линиями опухолевых клеток М18 и А549
определяли с помощью ПЦР в реальном времени (n=3). (В) пДК культивировали в течение 18
часов совместно c IL-3, R848 и с опухолевыми клетками, облученными УФ или зараженными
MV. Некоторые пДК, культивированные совместно с R848, активировали посредством
пептида NYESO-1(157-165) в течение 1 часа и затем промывали. Затем пДК культивировали в
течение 6 часов совместно с M117.167 СD8+ T-клеточным клоном специфичным к HLAA*
0201/ NYESO-1(157-156) (указанный как LT) в присутствии брефелдина А.
Продуцирование IFN-γ M117.167 T-клеточным клоном анализировали с помощью проточной
цитометрии после окрашивания с СD8 и IFN-γ-специфическим mAb. (C) пДК культивировали
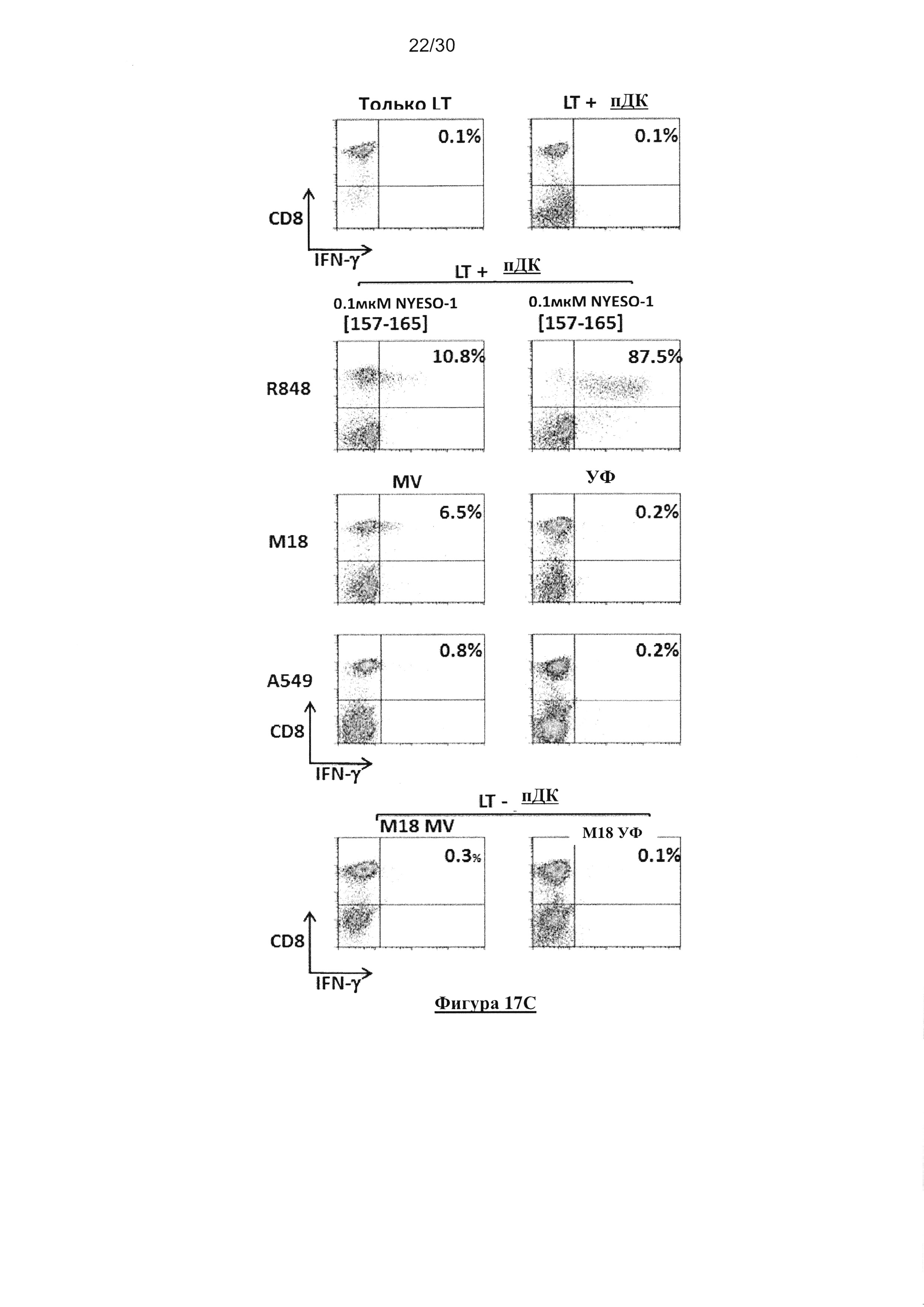
в течение 18 часов совместно c R848, или УФ-облученными или MV-зараженными
опухолевыми клетками M18 (NYESO-1+/ HLA-A*0201-) или А549 (NYESO-1-/ HLA-A*0201-).
Некоторые пДК, культивированные совместно с R848, активировали посредством пептида
NYESO-1(157-165) в течение 1 часа и затем промывали. Затем пДК культивировали в течение
6 часов совместно с M117.167 СD8+ T-клеточным клоном специфичным к HLA-A*0201/
NYESO-1(157-156) в присутствии брефелдина А. Продуцирование IFN-γ M117.167 T-
клеточным клоном анализировали с помощью проточной цитометрии после окрашивания с
СD8- и IFN-γ-специфическим mAb. (D) Представлен график рассеяния для исследования
кросс-представления. «n» представляет собой число проведенных экспериментов. Число «n»
является различным от одного условия к другому, поскольку авторы не смогли провести все
контроли в каждом эксперименте из-за ограниченного количества имеющихся пДК.
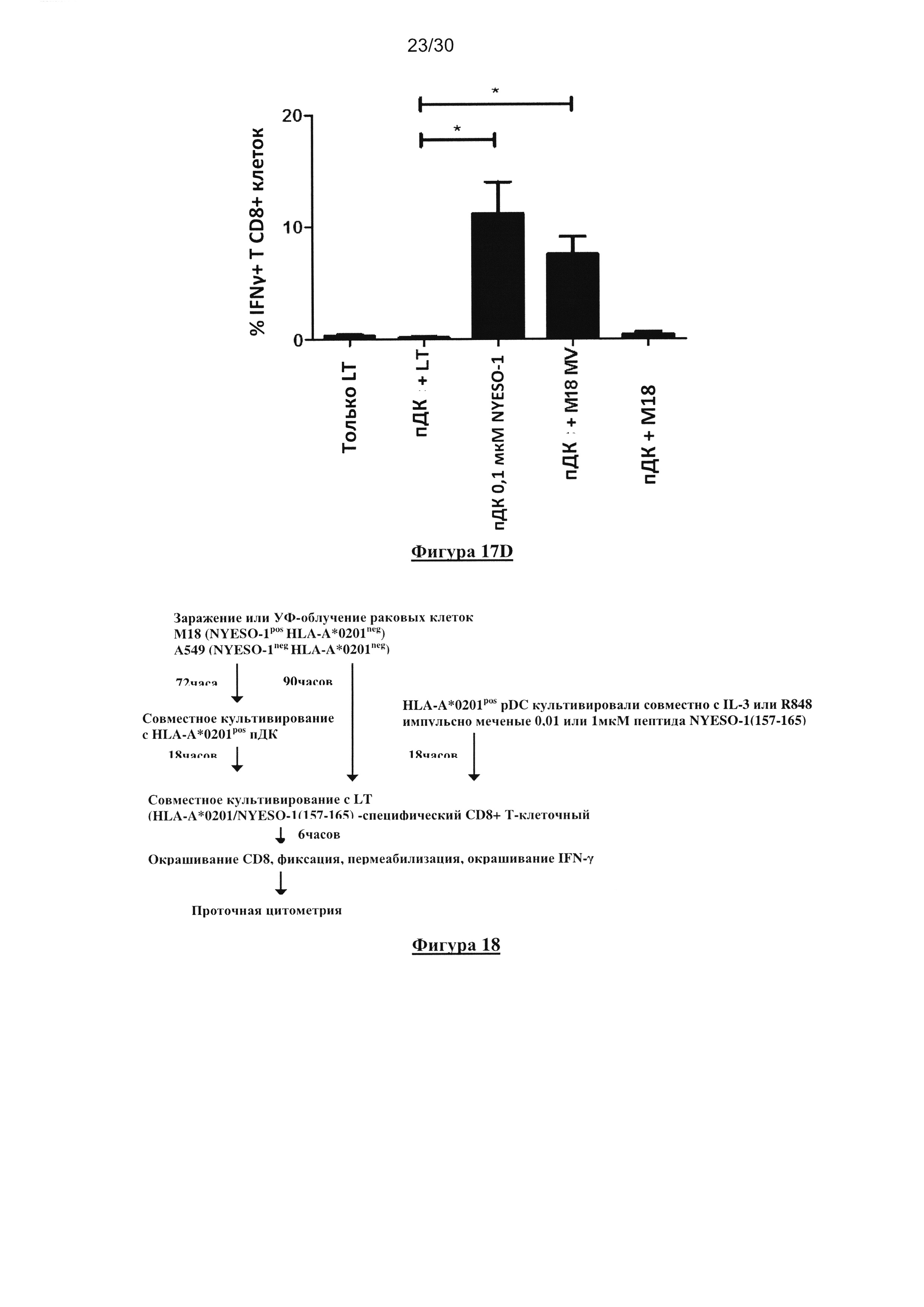
Фигура 18. Диаграмма условий культивирования, использованных в исследованиях
кросс-представления.
Фигура 19. (А) и (В) Заражение и клеточная гибель клеток меланомы посредством
немодифицированного MV или MV-deltaC. Образец анализируют с помощью проточной
цитометрии на уровень заражения и гибель клеток, индуцированную через 24, 48 и 72 часа
после заражения немодифицированным MV или MV-deltaC при MOI равной 1. Вакцинный
штамм MV-deltaC эффективно инфицировал опухолевые клетки, устойчивые к заражению
вакцинным штаммом немодифицированного MV. Опухолевые клетки меланомы были
заражены немодифицированным MV-eGFP или MV-deltaC eGFP при разных MOI в течение 2
часов.
Фигура 20. Клеточная гибель клеток меланомы меланомы посредством
немодифицированного MV или MV-deltaC. Опухолевые клетки меланомы инфицировали
немодифицированным MV- eGFP или MV-deltaC eGFP при разных MOI в течение 2 часов.
Скорость индуцированной гибели клеток (% Topro+ клеток в незараженных клетках - %
Topro+ клеток в зараженных клетках) каждой клеточной линии определяли с помощью
проточной цитометрии через 24, 48 и 72 часа после заражения.
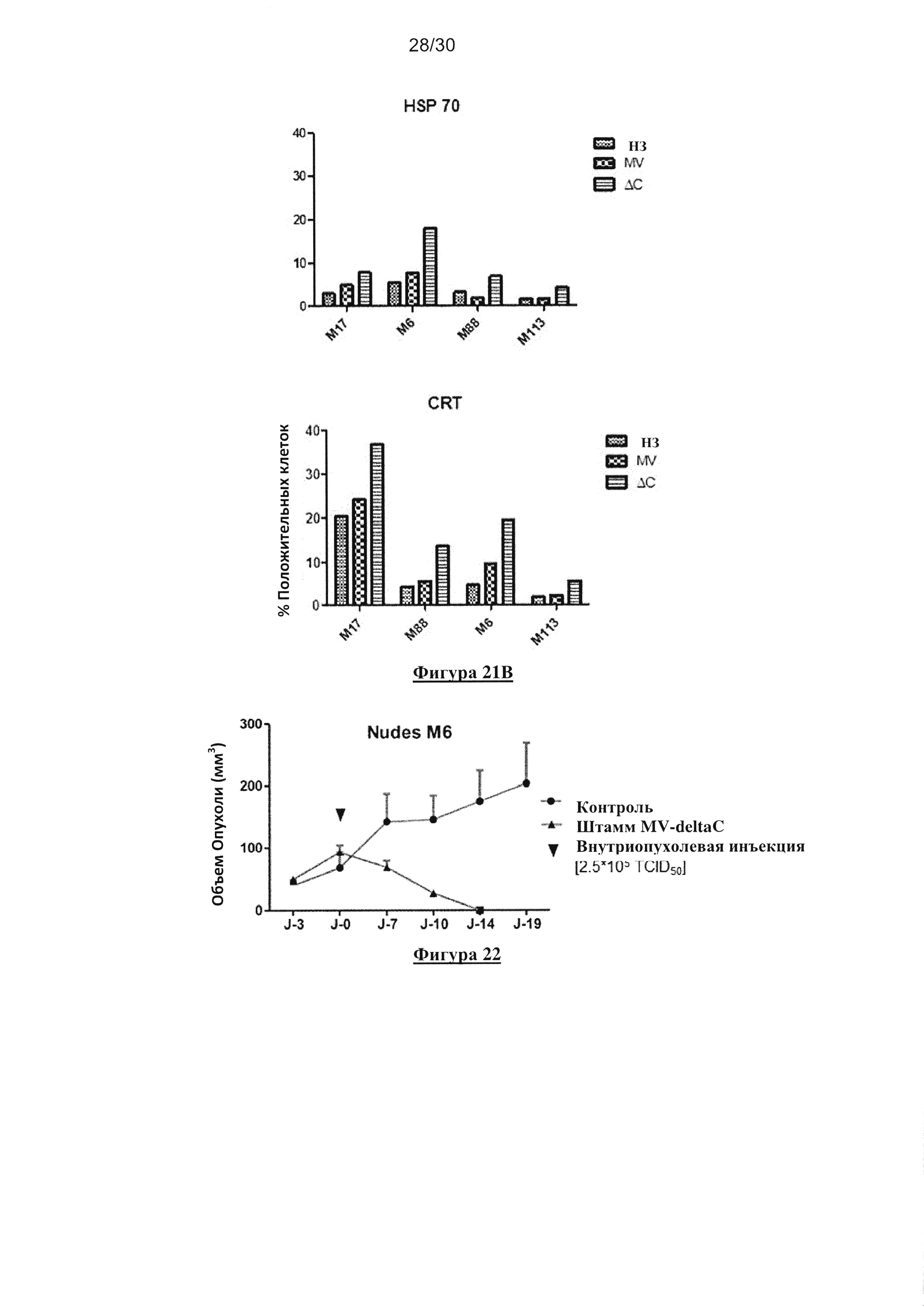
Фигура 21. Экспрессия «сигналов опасности» после заражения клеток меланомы
немодифицированным MV или MV-deltaC. (А) Пример проточного цитометрического
анализа экспрессии HSP70 и калретикулина (CRT) через 24, 48 и 72 часа после заражения
немодифицированным MV или MV-deltaC. (В) Мембранную экспрессию белка HSP70 и CRT
в незараженных опухолевых клетках и в клетках, зараженных немодифицированным MV или
штаммом MV-deltaC (MOI=0,5), определяли через 72 часа после заражения с помощью
внеклеточного маркера и проточной цитометрии.
Фигура 22. Рост опухоли меланомы после внутриопухолевой инъекции MV-deltaC in
vivo. В имплантированную опухоль ввели PBS для того, чтобы визуализировать нормальный
рост опухоли в качестве контроля. В данных, полученных на мышах контрольной группы,
объем опухоли увеличился от 45мм3 до 150мм3 в течение 13 дней. В группе, которую лечили
с помощью MV-deltaC, наблюдали значительное уменьшение опухоли через 10 дней после
заражения, которое достигло 25мм3, и опухоли были элиминированы через 14 дней после
заражения.
Фигура 23. Выживаемость раковых и нераковых клеток после заражения MV-deltaC
или немодифицированным MV. Человеческая линия А549 и раковые клетки Hela (А) и
HEK 293 и нераковые клетки Vero (В) инфицировали MV-deltaC (черные полосы в графике)
или немодифицированным MV (белые полосы в графике) при разных MOI (в трех повторах).
После 24, 46 и 68 часов культивирования, количество живых клеток определяли, используя
реагент CellTiter-GLO, анализ на основе люциферазы, который оценивает посредством
количественного определения АТФ количество метаболически активных клеток в
культуральных ячейках.
Фигура 24. Кинетика репликации немодифицированного MV и MV-deltaC на раковых и
нераковых клетках. Человеческая линия А549 и раковые клетки Hela и HEK 293 и
нераковые клетки Vero инфицировали MV-deltaC или немодифицированным MV при MOI=1
(в трех повторах). Вирусные титры определяли с помощью TCID50.
ПРИМЕРЫ
Пример 1
Сравнительные исследования между немодифицированным MVи MV-deltaC
Заражение немодифицированным MV in vitro. Живой ослабленный MV штамм
Шварца получили от F. Tangy (Институт Пастера, Франция). Штамм MV Шварца был
восстановлен из кДНК плазмиды pTM-MVSchw (сданной на хранение в Институт Пастера в
CNCM (Париж, Франция) под номером I- 2889 12 июня 2002 года) с помощью системы
восстановления на основе клеток-хелперов, описанной Radecke (Radecke et al., EMBO J.,
1995, 14:5773-5784) и измененной Parks (Parks et al., J. Virol., 1999, 73^3560-3566). Вкратце,
клетки-хелперы 293-3-46 трансфицировали 5мкг pTM-MVSchw и 0,02 мкг pEMC-Lschw,
экспрессирующие ген L штамма MV Шварца (Combredet et al., J. Virol., 2003, 77:11546-
11554). После инкубации в течение ночи при 37°C, клетки подвергли тепловому шоку в
течение 2 часов при 43°C, и трансфицированные клетки перенесли на монослой клеток Vero.
Синцитий, который появился через 15 дней совместного культивирования перенесли на 35-
мм лунки, и затем использовали в 72-см2 и 150-см2 колбах культуры клеток Vero в 5% FCS
DMEM. Когда синцитий достиг степень смыкания монослоя 80-90%, клетки соскабливали в
небольшой объем OptiMEM и подвергали замораживанию-оттаиванию. После
центрифугирования при низкой скорости, для осаждения клеточных фрагментов,
супернатант, содержащий вирус, хранили при -80°C. Титр исходного раствора
рекомбинантного MV на клетках Vero методом предельных разведений. TCID50 рассчитали с
помощью метода Карбера (Kärber, Arch. Exp. Path. Pharmak., 1931, 162:480-483).
Заражение MV-deltaC in vitro. MV-deltaC был также восстановлен c помощью обратной
генетики на клетках-хелперах HEK293-T7-MVи амплифицирован на клетках Vero
аналогичным способом, описанным в WO2004/000876 для немодифицированных MV.
Подходящий клон кДНК, кодирующий геном MV-deltaC, был соответственно приготовлен из
очищенных вирусных частиц MV, как описано в указанной заявке, или из плазмиды pTMMVSchw,
модифицированной посредством мутации второго стартового кодона «ATG»,
присутствующего в (+1) ORF на N-терминальном участке гена Р, чтобы получить плазмиду
pTM-MVSchw- deltaC-ATU1 (eGFP). В частности, кодон «ATG» заменили на кодон «ACG»
посредством замены T на С (SEQ ID NO: 1) (Фигура 1В). Аналогично вариант MV-deltaC
был также восстановлен с помощью обратной генетики, используя вариант плазмиды pTMMVSchw-
deltaC-ATU1 (eGFP), имеющий нуклеотидную последовательность согласно SEQ
ID NO: 2, где в позиции 2803 провели дополнительное замещение, чтобы заменить нуклеотид
G на нуклеотид A.
Характеристика MV-deltaC. Для подтверждения того, что ген, кодирующий белок С,
был выключен и что белки P и V при этом все еще экспрессировались, был проведен
Вестерн-блот на лизатах зараженных клеток Vero, с использованием специфических
моноклональных антител (анти-Р; анти-V и анти-С, Takeuchi, K. et al., FEBS Letters, 2003,
545(2), 177-182) (Фигура 2).
Таким образом, экспрессию белков P, V и C вирусом MV-deltaC сравнили с
экспрессией, полученной вирусом MV, MV-deltaV (MV в котором выключили белок V) и
MV-PG954 (MV в котором ген Р заменили на ген Р штамма дикого типа (т.е. G954)). Фигура 2
показывает, что MV-deltaC больше не экспрессировал белок С, в то время как белки P и V
экспрессировались корректно. Стабильность мутации, присутствующей в MV-deltaC,
контролировали секвенированием генома после 10 пассажей вируса на клетках Vero:
реверсная мутация не наблюдалась.
Кинетика роста MV-deltaC. Кинетику роста MV-deltaC анализировали на различных
линиях клеток как компетентных, так и не компетентных для ответа IFN типа I (Фигура 3).
Клетки Vero (эпителиальные клетки африканской зеленой мартышки) имеют делецию в гене
IFN-β (Mosca, J.D., Pitha, P.M. Mol Cell Biol., 1986, 6(6), 2279-2283), следовательно, ответ IFN
типа I не может быть инициирован в данных клетках после вирусного заражения. Напротив,
Hela (эпителиальные клетки карциномы человека), Jurkat (T-лимфоциты человека) и U937
(моноциты человека) являются компетентными для инициации ответа IFN типа I. По
сравнению с другими вирусами MV, протестированными на клетках Vero, вирус MV-deltaC
рос быстро в течение первых 24 часов, а затем его рост внезапно снизился. Прекращение
роста подтвердили на других испытанных типах клеток, которые компетентны для ответа IFN
типа I (HeLa, Jurkat и U-937). Таким образом, в отличие от других исследований (Takeuchi, K.
et al., J. Virol., June 2005, 7838-7844; Patterson, J.B. et al., Virology, 2000, 267(1):80-89),
дефицит роста MV-deltaC кажется не связан c присутствием или отсутствием IFN.
Цитопатическое действие MV-deltaC. Можно насчитать несколько объяснений
внезапного прекращения роста штамма MV-deltaC. Фактически, клетки Vero, зараженные
MV-deltaC, характеризуются образованием гигантского синцития (многоядерных клеток) в
результате слияния зараженных клеток, экспрессирующих гликопротеины MV, с соседними
незараженными клетками, экспрессирующими рецептор CD46. Авторы отметили, что MVdeltaC
индуцировал образование синцития гораздо быстрее, чем немодифицированный MV, в
клетках Vero (Фигура 4) и во всех других испытуемых типах клеток. В результате 24 часов
заражения при MOI равной 1, практически все клетки Vero объединились в гигантский
синцитий, который прорвался спустя несколько часов. Это объясняет снижение роста,
отмеченное через 24 часа после заражения: в культуре не осталось ни одной первичной
живой клетки, чтобы поддерживать продуцирование заражения. Преждевременный апоптоз
зараженных клеток вероятно ответственен за отмеченное прекращение вирусного роста.
Недавно было показано, что MV-deltaC, полученный из патогенного штамма MV (Ichinose),
оказывает более сильное цитопатическое действие, чем родительский вирус (Takeuchi, K. et
al., J. Virol., June 2005, 7838-7844).
Усиленное слияние клеток, индуцированное MV-deltaC, может происходить из-за более
высокого или более раннего продуцирования вирусных белков, в частности гликопротеинов
H и F на поверхности зараженных клеток, что может способствовать быстрому и массивному
слиянию клеток. Кинетика экспрессии вирусного геммаглютинина (Н) в клетках HeLa,
инфицированных MV-deltaC или немодифицированным MV (MOI=1) анализировали с
помощью иммунофлюоресценции. Клетки окрашивали моноклональным анти-MV-H
антителом в сочетании с FITC (Фигура 4). Результаты показывают, что через 24 часа после
заражения, MV-deltaC вызывает более массивное заражение и индуцирует гораздо большую
экспрессию, чем немодифицированный MV.
Кинетика экспрессии вирусного белка в клетках Vero, зараженных MV и MVdeltaC.
Для подтверждения увеличенного продуцирования вирусных белков в отсутствии
экспрессии белка С, содержание вирусных белков N и V в лизатах клеток Vero, зараженных
MV-deltaC или немодифицированным MV при MOI равной 1, анализировали в течение
долгого времени (Фигура 5). В результате 6 часов заражения, нуклеопротеин N был
обнаружен в клетках, зараженных MV-deltaC, в то время как он был обнаружен только спустя
21 час после заражения немодифицированным MV. Аналогичное наблюдение было сделано
для белка V. Данные результаты показывают, что вирусные белки экспрессируются гораздо
раньше и в больших количествах посредством MV-deltaC, чем немодифицированным MV.
Иммуногенность _______MV-deltaC. Для оценки влияния подавления экспрессии белка С на
иммуногенность вакцинного вектора MV, иммунизировали CD46+/- IFNAR-/- мышей,
чувствительных к заражению MV (Combredet, C. et al., 2003, J Virol, 77(21): 11546-11554;
Mrkic B. et al., J Virol., 2000, 74(3):1364-1372). При помощи генной инженерии данные мыши
экспрессируют человеческий рецептор CD46 вакцинного штамма MV, а экспрессия
рецептора IFN типа I выключена (IFNAR). Они широко используются для оценки
иммуногенности векторов MV. (Brandler, S. et al., PLoS Neglected Tropical Diseases, 2007,
1(3):e96; Comberet, C. et al., 2003, J Virol, 77(21): 11546-11554). Хотя IFN α/β неэффективны в
этих мышах, данную модель использовали для первоначальной оценки влияния подавления
экспрессии белка С in vivo. MV-deltaC сравнивали с немодифицированным MV, MV-deltaV и
MV-PG954. Однократную дозу 105 TCID50 каждого вируса инокулировали внутрибрюшинно
во всех четырех группах по шесть мышей. Забор сыворотки сделали через 2 месяца после
инокуляции, и анти-MV антитела количественно определяли с помощью ELISA (Trinity
Biotech) (Фигура 6).
В данных мышах, некомпететентных к IFN типа I, уровни антител, индуцированные
модифицированными векторами кори, были сопоставимы с уровнями антител,
индуцированными немодифицированными векторами. Данный результат не удивителен для
векторов MV-deltaV и MV-PG954, которые имеют кинетику роста in vitro аналогичную
немодифицированному MV. Удивительно, что вектор MV-deltaC, имеющий сниженный рост
in vitro, индуцировал титры антител лишь едва ниже, чем те, которые индуцированы
немодифицированным MV. Данный результат показывает, что, либо инокулированная доза
была слишком высокой для того чтобы обнаружить разницу, либо минимальная вирусная
репликация была достаточной, чтобы индуцировать насыщение гуморального ответа.
Недавно было показано на обезьянах, что распространение MV-deltaC, полученное из
патогенного штамма MV (Ichinose), было значительно снижено по сравнению с диким типом
(Takeuchi, K. et al., J. Virol., June 2005, 7838-7844). Эти предварительные данные показывают,
что в отсутствии ответа IFN типа I, подавление белка С не влияет на установление
антивирусного гуморального ответа.
Пример 2
Индуцирование гибели клеток посредством MV или MV-deltaC.
Клеточная культура. Линии клеток эпителиоидной мезотелиомы (Meso11, Meso13 и
Meso56) и линии клеток аденокарциномы легких (А549 и ADK117) были получены и
охарактеризованы (Gueugnon F et al., Am J Pathol, 2011, 178: 1033-1042) из плеврального
выпота посредством торакоцентеза у больных раком, давших информированное согласие.
Линии клеток меланомы (М17 и М18) были синтезированы Б. Дрено и Н. Лабарриер
(Онкологический научный центр, Нантес, Франция). Линия клеток аденокарциномы легких
(А549) были приобретены у ATCC. Линии клеток эпителиоидной мезотелиомы (Meso11,
Meso13 и Meso56) и линия клеток аденокарциномы легких (ADK117) были выделены и
охарактеризованы Ф.Танги (Институт Пастера, Франция). Все линии клеток поддерживали в
среде RPMI-1640 (Gibco-Invitrogen, Cergy-Pontoise, Франция), дополненной 10% (v/v)
инактивированной нагреванием эмбриональной телячьей сывороткой (PAA Laboratories, Ле-
Мюро, Франция), 2 мМ Л-глютамина, 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина
(всё приобретено у Gibco). Клетки культивировали при 37°C во влажной 5% CO2 атмосфере,
и ежедневно проверяли на контаминацию Mycoplasma с помощью ПЦР.
Анализ гибели клеток. Гибель клеток обнаружили через 3 дня после заражения с
помощью набора для обнаружения апоптоза (BD Biosciences). В общем виде, клетки
подвергли двойному окрашиванию FITC-AnnexinV и йодидом пропидия в течение 15 минут и
анализировали с помощью проточной цитометрии в течении 1 часа. Затем определяли MV- и
MV-deltaC-специфическую гибель клеток. Для внеклеточного окрашивания, клетки
инкубировали со специфическим антителом, описанным в следующих примерах. Затем
клетки промывали 3 раза PBS перед анализом с помощью проточной цитометрии
(FACSCalibur, BD Biosciences).
Для сравнения способности к заражению и индукции гибели клеток у MV MV-deltaC,
большую панель трех линий клеток эпителиоидной мезотелиомы (Meso11, Meso13 и Meso56),
две линии клеток меланомы (М17 и М18) и две линии клеток аденокарциномы легких (А549
и ADK117) инфицировали немодифицированным MV или MV-deltaC, при множественности
заражения (MOI) равной 1 в течение 2 часов, инкубация при 37°C. Контрольные линии
клеток не инфицировали MV или MV-deltaC (Фигура 7). Опухолевые клетки анализировали
с помощью проточной цитометрии после двойного окрашивания FITC-AnnexinV и йодидом
пропидия через три дня после заражения.
Клетки эпителиоидной мезотелиомы Meso11 и Meso13, клетки меланомы M18 или
клетки аденокарциномы легких А549 были эффективно заражены немодифицированным MV,
в то время как клетки эпителиоидной мезотелиомы Meso56 и клетки аденокарциномы легких
ADK117 были еженедельно инфицированы немодифицированным MV, а клетки меланомы
М17 были неинфицированы. Даже если наблюдался умеренный или значительный процент
клеток AnnexinV среди клеток, эффективно зараженных немодифицированным MV (клетки
эпителиоидной мезотелиомы Meso11 и Meso13, клетки меланомы M18 и клетки
аденокарциномы легких А549), было обнаружено, что MV-deltaC способен индуцировать
гибель данных опухолевых клеток даже более эффективно. В то время как еженедельно
инфицированные немодифицированным MV (клетки эпителиоидной мезотелиомы Meso56 и
клетки аденокарциномы легких ADK117) или даже неинфицированные (клетки меланомы
М17) опухолевые клетки показали очень низкий процент AnnexinV- клеток, авторы
неожиданно обнаружили, что заражение MV-deltaC вызвало гораздо более интенсивную
индукцию клеточной гибели для двух данных линий клеток (Фигура 7А).
Таким образом, согласно данным in vitro результатам, MV-deltaC индуцировал более
интенсивный апоптоз в зараженных опухолевых клетках, чем немодифицированный MV.
Пример 3
Активация каспазы-3 после заражения MV или MV-deltaC.
Активацию каспазы-3 анализировали и в опухолевых клетках, инфицированных
немодифицированным MV и MV-deltaC (Фигура 7В). Панель двух линий клеток
эпителиоидной мезотелиомы (Meso13 и Meso56), одной линии клеток меланомы (М17) и
одной линии клеток аденокарциномы легких (А459) была заражена немодифицированным
MV или MV-deltaC, при MOI равной 1 в течение 2 часов, инкубация при 37°C. Контрольные
линии клеток не инфицировали MV или MV-deltaC. Зараженные (MV или MV-deltaC) и
незараженные опухолевые клетки анализировали с помощью проточной цитометрии после
окрашивания анти-каспаза-3 антителом (BD Biosciences) через три дня после заражения.
Заражение посредством MV-deltaC индуцировало активацию каспазы-3 в двух разных
испытуемых линиях клеток: 38,2% в клетках эпителиоидной мезотелиомы Meso13, 30,2% в
клетках меланомы М17, 21,8% в клетках аденокарциномы легких А459, и 8,4% в клетках
эпителиоидной мезотелиомы Meso56. С другой стороны, данная активация каспазы-3 не была
или была частично замечена после заражения немодифицированным MV. Данные результаты
предположили, что вирусы: немодифицированный MV и MV-deltaC, могут индуцировать
гибель опухолевых клеток согласно двум различным путям.
Пример 4
Экспозиция белка Hsp70 на поверхность клетки после заражения MV или MV-deltaC.
Заражение опухолевых клеток онколитическими вирусами может вызывать клеточный
стресс (Fabian et al., J Virol, 2007, 81(6): 2817-2830). Заражение, а также гибель клеток,
индуцированная данными вирусами, приводит также к продуцированию и высвобождению в
окружающую среду веществ с иммуногенными свойствами (Wang et al., Viral Immunol, 2006,
19(1): 3-9). Эти эндогенные сигналы опасности, выделенные из инфицированных клеток, в
действительности могут быть распознаны защитными клетками и могут запускать
адаптивный ответ. Моноцитарные и плазмацитоидные дендритные клетки распознают
данные сигналы опасности благодаря экспрессии различных рецепторов. Таким образом,
иммунная система способна работать во взаимодействии с прямой онколитической
активностью вирусов путем активации специфического лимфатического ответа опухолевых
антигенов.
Заражение опухолевых клеток вакцинным штаммом онколитического вируса MV
обеспечивает созревание ДК и активацию аутологичных Т-лимфоцитов (WO2009/047331).
Для того, чтобы охарактеризовать механизм, с помощью которого инфицированные клетки
индуцировали активацию иммунной системы, авторы изучили эксперссию экспрессия,
модификация и/или высвобождение различных клеточных факторов, известных своей
причастностью к иммуногенности гибели клеток. Например, белки семейства HSP70 или
калретикулин могут принимать участие в активации противоопухолевого иммунного ответа.
Провели анализ экспрессии белка Hsp70 на поверхность опухолевых клеток,
зараженных немодифицированным MV и MV-deltaC (Фигура 8). Панель двух линий клеток
эпителиоидной мезотелиомы (Meso13 и Meso56), одной линии клеток меланомы (М17) и
одной линии клеток аденокарциномы легких (А459) была заражена немодифицированным
MV или MV-deltaC, при MOI равной 1,0 в течение 2 часов, инкубация при 37°C. Контрольные
линии клеток не инфицировали MV или MV-deltaC. Экспрессию мембранного белка Hsp70
определяли через 2 дня после заражения для линий клеток эпителиоидной мезотелиомы
Meso13 и Meso56 или через 3 дня после заражения клеток аденокарциномы легких А459 и
клеток меланомы М17, с помощью внеклеточного окрашивания и проточной цитометрии.
Совсем небольшое перемещение белка Hsp70 на поверхность клетки было обнаружено
как для клеток, эффективно зараженных немодифицированным MV (клетки эпителиоидной
мезотелиомы Meso13 и клетки аденокарциномы легких А459), так и для клеток, устойчивых к
MV (клетки эпителиоидной мезотелиомы Meso56 и клетки меланомы М17). C другой
стороны, MV-deltaC неожиданно индуцировал интенсивную экспозицию белка Hsp70 на
наружный слой плазматической мембраны во всех линиях клеток. Данные результаты
предполагают, что гибель клеток, индуцированная MV-deltaC, является признаком его
иммуногенных характеристик.
Пример 5
Мембранная транслокация калретикулина после заражения MV или MV-deltaC
Изучили влияние заражения опухолевых клеток немодифицированным MV и MV-deltaC
на транслокацию калретикулина на поверхность клетки. Для этого анализировали наличие
калретикулина на поверхности зараженных опухолевых клеток с помощью внеклеточного
окрашивания (Фигура 9). Панель двух линий клеток эпителиоидной мезотелиомы (Meso13 и
Meso56), одной линии клеток меланомы (М17) и одной линии клеток аденокарциномы легких
(А459) заразили немодифицированным MV или MV-deltaC, при MOI равной 1,0 в течение 2
часов, инкубация при 37°C. Контрольные линии клеток не инфицировали MV или MV-deltaC.
Незараженные и зараженные (MV или MV-deltaC) опухолевые клетки окрашивали анти-
калретикулиновым антителом и конъюгированным анти-мышиным вторичным антителом
Сy5 через два дня после заражения для клеток эпителиоидной мезотелиомы Meso13 и Meso56
или через 3 дня после заражения для клеток аденокарциномы легких А459 и клеток
меланомы М17. Затем клетки анализировали с помощью проточной цитометрии.
Как и с белком Hsp70, заражение MV-deltaC неожиданно привело к интенсивной
транслокации калретикулина на поверхность клетки по сравнению с заражением
немодифицированным MV, и для клеток, эффективно зараженных немодифицированным MV
(клетки эпителиоидной мезотелиомы Meso13 и клетки аденокарциномы легких А459), так и
для клеток, устойчивых к MV (клетки эпителиоидной мезотелиомы Meso56 и клетки
меланомы М17). Данные результаты также предполагают, что гибель клеток,
индуцированная MV-deltaC, является признаком его иммуногенных характеристик.
Пример 6
Высвобождение HMGB-1 во внеклеточную среду после заражения MV или MV-deltaC
Изучили высвобождение белка HMGB-1 во внеклеточную среду после заражения
опухолевых клеток немодифицированным MV или MV-deltaC (Фигура 10). Две линий клеток
эпителиоидной мезотелиомы (Meso13 и Meso56) инфицировали немодифицированным MV
или MV-deltaC, при MOI равной 1,0 в течение 2 часов, инкубация при 37°C. Контрольные
линии клеток не инфицировали MV или MV-deltaC. Супернатанты незараженных и
зараженных (MV или MV-deltaC) опухолевых клеток отобрали через один, два или три дня
после заражения и хранили при -20°C. Затем количество HMGB-1 в данных супернатантах
определяли с помощью ELISA.
MV-deltaC индуцировал эффективное высвобождение HMGB-1 зараженными опухолевыми
клетками. Заражение вирусом MV-deltaC индуцировало более быстрый апоптоз опухолевых
клеток в культуре; следовательно, MV-deltaC индуцировал более раннее высвобождение
HMGB-1 по сравнению с немодифицированным MV, как показывают результаты клеток
эпителиоидной мезотелиомы Meso13.
Пример 7
Плазмацитоидные дендритные клетки
Культура клеток
Линии клеток культивировали как описано ранее в Примере 2.
Заражение MV и облучение УФ
Живой ослабленный штамм Шварца MV и улучшенный зеленый флуоресцентный белок
рекомбинантного MV были получены способом, описанным ранее (Gauvrit, A. et al., Cancer
Res., 2008, 68: 4882-4892). Заражение опухолевых клеток вирусом кори проводили в течение
2 часов при 37°C при множественности заражения (MOI) равной 1 если не указано иное.
Затем вирусный инокулят заменили на свежую клеточную среду на 72 часа. Для заражения
пДК и достижения экспериментов, MV не промывали и оставляли в среде в течение
культивирования. Измерение скорости заражения выполняли с помощью проточной
цитометрии, используя MV-eGFP, через 24, 48 и 72 часа после заражения. Все остальные
эксперименты проводили, используя MV. Опухолевые клетки облучили УФ-Б (312нм - 100
кДж/м2, УФ-облучатель фирмы Stratagene). Среду обновляли каждые 72 часа.
Выделение и культивирование ДК
пДК были получены из МКПКs здорового донора (Etablissement Français du Sang,
Нантес, Франция) как описано ранее (Coulais, D. et al., Cytotherapy, 2012, 14:887-896).
Вкратце, сначала пДК обогащали противоточным центрифугированием, а затем очищали с
помощью негативного отбора магнитными частицами, как рекомендовано в протоколе
производителя (Stemcell Technologies, Гренобль, Франция). Чистота нетронутых пДК была
всегда больше чем 96%. пДК (3х105 на мл) поддерживали в культуре с 20 нг/мл rhIL-3 (Sigma,
Saint Quentin Fallavier, France) или активировали агонистом рецептора TLR-7 in vitro, R848
(InvivoGen, San Diego, USA) (5 мкг/мл). Также пДК культивировали совместно с MV, MV и
IL-3 (MOI=1), или опухолевыми клетками, зараженными MV или облученными УФ
(пДК/опухолевая клетка: 1/1), без rhIL-3 или агента созревания. После 18 часов,
супернатанты культур и пДК отобрали для использования. Для анализа ингибирования TLR-7
использовали иммунорегуляторную последовательность ДНК, которая ингибирует
исключительно передачу сигналов посредством TLR-7 [IRS 661], при концентрациях в
диапазоне от 0,1мкм до 1мкм (Eurofins, Munich, Germany). В качестве контроля использовали
5мкг/мл CpG-A для индуцирования выделения TLR-9 –зависимого IFN-α клетками
пДК(InvivoGen, San Diego, USA).
Иммунофлюоресценция и проточная цитометрия
Фенотипы пДК опеределяли с помощью иммунофлюоресценции c последующей
проточной цитометрией. пДК окрашивали моноклональными антителами, специфичными к
CD40, CD86, HLA-DR (BD Biosciences, San Jose, CA, USA), CD83 (BioLegend, San Diego, CAUSA)
и BDCA-4 (Miltenyi Biotec). пДК селектировали как BDCA-4+/HLA-DR+ клетки, чтобы
дифференцировать их от опухолевых клеток. Гибель опухолевых клеток измеряли с
помощью окрашивания TO-PRO®3 (Invitrogen, Saint Aubin, France) как рекомендовано
производителем. TO-PRO®3 представляет собой карбоцианиновый краситель мономера
нуклеиновой кислоты с крайне красной флюоресценцией, который проникает только в
мертвые клетки и окрашивает ДНК. Флюоресценцию анализировали с помощью FACSantoll
(Becton Dickinson, New Jersey, USA)
Анализ фагоцитоза
Опухолевые клетки, зараженные MV или облученные УФ, окрашивали PKH-67
согласно протоколу производителя (Sigma, Saint Quentin Fallavier, France) и культивировали
совместно с пДК, в течение 18 часов при 4°C или 37°C (1 ДК:1 опухолевая клетка). Со-
культуры промывали PBS-EDTA для того, чтобы отделить клетку-конъюгат. пДК
окрашивали конъюгированными с HorizonV450-, анти-HLA-DR антителами (BD Biosciences,
San Jose, CA, USA) и анализировали с помощью проточной цитометрии (FACSantoll, BD).
Фагоцитоз пДК наблюдали с помощью конфокального микроскопа (Nikon). Опухолевые
клетки, зараженные MV или облученные УФ, окрашивали PKH-67 и затем культивировали
совместно с пДК в 24-луночных планшетах, содержащих предметные стекла с поли-лизином,
в течение 18 часов (пДК:опухолевая клетка в отношении 1:1). пДК окрашивали несвязанным
анти-HLA-DR (BD Biosciences). Окрашивание HLA-DR было выявлено с помощью
вторичного анти-мышиного IgG антитела в сочетании с AlexaFluor 568.
Определение цитокинов
Продуцирование IFN-α (MabTech, Cincinnati, OH-USA) измеряли с помощью ELISA в
супернатантах культуры пДК согласно инструкции производителя.
Анализ кросс-представления
Линии клеток меланомы NYESO-1pos/HLA-A*0201neg (М18) и аденокарциномы легких
NYESO-1 neg/HLA-A*0201neg (A549) инфицировали MV или облучали УФ-Б, культивировали
в течение 72 часов и затем культивировали совместно с HLA-A*0201 pos пДК
(пДК:опухолевая клетка в отношении 1:1). Спустя 18 часов, пДК культивировали совместно с
HLA-A*0201/ NYESO-1 (156-165) -специфическим CD8+ Т-клеточным клоном, М117.167, в
течение 6 часов в присутствии Брефелдина-А (Sigma, Saint Quentin Fallavier, France). Клон
М117.167 был получен путем клонирования методом предельных разведений
инфильтрирующих опухоль лимфоцитов больного меланомой. Клон культивировали как
описано ранее (Fonteneau, J. F. et al., J Immunol Methods, 2001, 258: 111-126). В качестве
контроля авторы использовали пДК, которые активировали в течение 1 часа с 0,1 или 1мкМ
пептида NYESO-1 (156-165) и промывали. Затем клетки фиксировали PBS, содержащим 4%
параформальдегида, в течение 10 мин при комнатной температуре, и пермеабилизировали и
окрашивали IFN-γ и CD8-специфическими антителами (BD Biosciences, SanJose, CA, USA),
как описано ранее (Schlender, J. et al., J Virol., 2005, 79: 5507-5515). Продуцирование IFN-γ
анализировали с помощью проточной цитометрии с использованием гейта CD8+ T-клеток.
ПЦР в реальном времени
Один микрограмм общей РНК подвергали обратной транскрипции c использованием
обратной транскриптазы вируса лейкемии мышей Молони (InVitroGen, Saint Aubin, France).
Реакции ПЦР выполняли с использованием праймеров QuantiTect (Qiagen, Foster City-USA) и
общей реакционной смеси (master mix) RT2 Real-Time SYBR-Green/ROX PCR (Tebu-bio, Le
Perray-en-Yvelines, France), согласно инструкциям производителя.
Статистика
Применяли программное обеспечение GraphPad Prism (Inc., San Diego, CA-USA) с
использованием теста непараметрического сравнения Манна-Уитни. Значения P <0,05
рассматривали как статистически значимые.
Пример 8
Чувствительность опухолевых клеток и пДК к заражению MV
В процессе заражения, MV проникает в клетки в основном посредством CD46 и, в
меньшей степени, CD150/SLAM (Anderson, B. D. et al., Cancer Res., 2004, 64: 4919-4926;
Schneider, U. et al., J Virol, 2002, 76: 7460-7467). В первом эксперименте авторы изучили
экспрессию этих двух главных рецепторов MV, CD46 и CD150/SLAM, на пДК, линиях клеток
меланомы (М18), мезотелиомы (Meso13) и аденокарциномы легких (А549) (Фигура 11А).
Экспрессию CD46 наблюдали во всех типах клеток, с более интенсивной экспрессией на
линиях клеток Meso13 и А549. Касательно экспрессии CD150/SLAM, позитивная экспрессия
была обнаружена на линии клеток меланомы М18. Данные результаты предполагают, что все
данные типы клеток могут быть чувствительными к заражению MV, поскольку все они
экспрессируют CD46.
Затем авторы изучили чувствительность данных четырех типов клеток к заражению
MV, с использованием рекомбинантного MV, кодирующего зеленый флуоресцентный белок
(MV-GFP). Через 72 часа после воздействия MV при MOI=1, три линии опухолевых клеток
были продуктивно заражены вирусом кори, в пределах от 50% клеток А549 положительных
на GFP до 90% клеток Meso13 (Фигура 11В). Более того, образование синцития наблюдали у
трех линий опухолевых клеток, а пДК были непермессивными при MOI=1. Без сигнала
выживания, такого как IL-3, пДК погибли в течение 72 часов культивирования. Таким
образом, эксперименты выполняли где IL-3 был добавлен к пДК, подвергнутым воздействию
MV. (Фигура 11C). В присутствии IL-3, пДК выжили в течение 72 часов, но при этом не
были продуктивно заражены вирусом кори. Для подтверждения данных результатов, MOI
увеличили до 50 в присутствии IL-3, но авторам все же не удалось обнаружить зараженные
пДК (Фигура 11D). Однако, небольшой сдвиг флюоресценции был отмечен при MOI=50,
который произошел вероятно из-за растворимого GFP в процессе 72 часового
культивирования, который занесли при приготовлении MV-GFP. Аналогично, когда авторы
использовали MV-GFP, облученный УФ, неспособный к репликации, эта незначительный
сдвиг флюоресценции все еще наблюдали (Фигура 12). В заключение, когда MV-GFP
проинкубировали в течение 2 часов при MOI=50 совместно с пДК и промыли, авторам не
удалось обнаружить небольшой сдвиг флюоресценции спустя 70 часов (Фигура 12).
Затем авторы измерили гибель опухолевых клеток через 72 часа после заражения и
обнаружили, что около половины опухолевых клеток, зараженные MV, были TO-PRO+ после
72 часов (Фигура 11Е). Аналогичный уровень клеточной гибели был отмечен посредством
облучения опухолевых клеток УФ-Б. Следовательно, заражение MV индуцирует гибель
приблизительно половины опухолевых клеток через 72 часа после инфицирования.
Пример 9
Опухолевые клетки, зараженные MV, индуцируют созревание пДК
Далее авторы исследовали влияние MV и клеток, зараженных MV, на созревание пДК
(Фигура 13). В данных экспериментах авторы оценивали, как заражение MV опухолевых
клеток, по сравнению с облучением УФ, другим индуктором гибели опухолевых клеток,
влияет на созревание пДК. В качестве контроля созревания, пДК подвергали воздействию
агониста TLR7/8 – R848 (Фигура 13A и 13В).
Ранее было показано, что линия опухолевых клеток злокачественной плевральной
мезотелиомы (MPM), зараженная MV, Meso13, индуцировала созревание моноцитарных ДК,
без дополнительных адъювантов, тогда как только вирус или Meso13, облученные УФ, не
индуцировали созревание (Gauvrit, A. et al., Cancer Res., 2008, 68:4882-4892). Авторы провели
ряд экспериментов на пДК, чтобы определить влияние только MV, опухолевых клеток,
зараженных MV или облученных УФ, на статус созревания пДК. Влияние опухолевых
клеток, зараженных MV или облученных УФ, сравнивали на статусе созревания пДК
(Фигура 13). Было обнаружено созревание пДК, культивированных совместно с
опухолевыми клетками, зараженными MV, тогда как опухолевым клеткам, облученным УФ,
не удалось активировать пДК. Действительно, экспрессия маркера созревания CD83 была
индуцирована клетками, зараженными MV, на том же уровне, что и экспрессия,
наблюдаемая, когда пДК подвергались воздействию R848. Авторы отметили, что индукция
экспрессии костимуляторных молекул СD40 и CD86 на пДК, воздействовала на опухолевые
клетки, зараженные MV, хотя данная индукция была низкой по сравнению с уровнями,
запущенными посредством только R848.
Опубликованы два исследования, описывающие противоречивые результаты о
способности MV запускать созревание пДК (Duhen, T. et al., Virus Res., 2010,152: 115-125;
Schlender, J. et al., J. et al., J Virol., 2005, 79: 5507-5515). Однако исследование Duhen et al.,
описывающее, что MV активирует пДК, проводили в присутствии IL-3, фактора
выживаемости пДК, тогда как другое исследование Schlender et al., который отметил что
пДК, культивированный с MV, не индуцирует созревание пДК, было проведено в отсутствии
IL-3. Таким образом, авторы выполнили и сравнили два условия и обнаружили аналогичные
результаты, описанные данными двумя авторами. Действительно, MV при MOI=1
индуцировал созревание IL-3 пДК только в присутствии IL-3 (Фигура 13). Что касается
R848, MV в присутствии IL-3 индуцировал созревание пДК, и в основном отличающийся
значительным увеличением экспрессии CD83 и, в меньшей степени, экспрессии CD40 и
CD86. Авторы также отметили выживаемость и созревание пДК в отсутствии IL-3, когда они
подвергались воздействию высокого количества MV (MOI=50). При более низкой
концентрации вируса в отсутствии IL-3, клетки пДК погибли.
В последней серии исследований, авторы проверяли, действительно ли заражение MV и
репликация в пДК были необходимы для индукции их активации. пДК подвергли
воздействию MV, облученному УФ (MV*), который не способен к репликации, и
обнаружили аналогичный уровень созревания (экспрессии CD83, CD80 и CD86) и
продуцирования IFN-α, как и при необлученных MV (Фигуры 14А и 14С). Присутствие
блокирующего анти-CD46-специфического антитела в культуре пДК, воздействующего на
IL-3 и MV, не повлияло на созревание пДК (Фигура 14А). Аналогичный эксперимент
проводили с пДК, воздействующих на MV-инфицированные опухолевые клетки. Созревание
и продуцирование IFN-α также наблюдалось, когда MV-инфицированные опухолевые клетки
были облучены УФ перед воздействием пДК (Фигуры 14В и 14С). В заключение, авторы
проверили действительно ли CD46-специфическое моноклональное антитело способно
ингибировать созревание пДК в ответ на MV-инфицированные опухолевые клетки (Фигуры
14В и 14С). Ингибирование не обнаружили, тогда как анти-CD46 антитело полностью
ингибировало заражение Meso13 в качестве контроля (Фигура 14D). В целом, данные
результаты предполагают, что заражение MV и репликация в пДК не являются
необходимыми для активации пДК в ответ на MV.
Пример 10
Захват клетками пДК клеточных компонентов из опухолевых клеток, зараженных MV
Благодаря эндо/лизосомальной экспрессии TLR-7 и TLR-9, пДК специализируются на
обнаружении вирусной нуклеотиновой кислоты (Gilliet, M. et al., Nat Rev Immunol., 2008, 8:
594-606). Эти два рецептора являются основными рецепторами врожденного иммунитета,
активирующие пДК (Reizis, B. et al., Nat Rev Immunol., 2011, 11: 558-565). Поскольку MV в
присутствии IL-3 или опухолевые клетки, зараженные MV, способны индуцировать
созревание пДК, вполне вероятно, что стимулятором созревания является одноцепочечная
РНК вируса кори, которая активирует TLR7 в эндо/лизосомальном канале. Данная гипотеза
подтверждается фактом, что MV не индуцирует созревание ДК, так как эти клетки не
экспрессируют TLR7 в человеческих организмах. Это подразумевает, что некоторые частицы
MV подвергаются эндоцитозу дендритными клетками, когда их культивируют c MV и IL-3
или раковыми клетками, зараженными MV. Затем авторы исследовали действительно ли пДК
эффективно захватывали клеточный материал из опухолевых клеток, зараженных MV и
облученных УФ (Фигура 15). Линия клеток М18, зараженная MV и облученная УФ, и
опухолевые клетки А549 пометили PKH67 и культивировали совместно с пДК. Авторы
наблюдали, что пДК эффективно захватывали опухолевые клетки, зараженные MV, при 37°C,
тогда как опухолевые клетки, облученные УФ, были менее эффективно захвачены (Фигура
15А и Фигура 15В). В двух дополнительных экспериментах, авторы наблюдали, что
присутствие в культуре CD46 моноклонального антитела не ингибировало фагоцитоз
опухолевых клеток, зараженных MV.
Данные результаты были подтверждены с помощью конфокального микроскопа
(Фигура 15С). пДК культивировали в течение 18 часов с MV-зараженными опухолевыми
клетками, мечеными PKH-67. Оптические участки показали флуоресцентные фрагменты
опухолевые клеток, зараженных MV, внутри пДК, подтверждающие интернализацию пДК в
MV-инфицированные частицы опухолевых клеток. Интересно, что авторы никогда не
наблюдали образование синцития между пДК и опухолевыми клетками. В целом, данные
результаты предполагают, что некоторые частицы MV, содержащиеся в зараженных
опухолевых клетках, могли достигать каналов, где находятся TLR7.
Пример 11
Опухолевые клетки, зараженные MV, индуцируют сильно выраженную секрецию IFN
типа I посредством запуска TLR7
Известно, что пДК самые сильные продуценты IFN I типа, особенно против вируса, во
время активации TLR-7 или TLR-9 (Gillet, M. et al., Nat Rev Immunol. 2008, 8:594-606). Таким
образом, с помощью ELISA авторы измерили продуцирование IFN-α клетками пДК в
результате контакта с MV, опухолевыми клетками, зараженными MV или облученными УФ.
(Фигура 16A). Непосредственный контакт с MV индуцировал секрецию IFN-α клетками пДК
только в присутствии IL-3, совпадая с созреванием клеток, отмеченным ранее в Фигуре 13.
Количество продуцированного IFN-α в ответ на MV в присутствии IL-3 сравнили с
количеством, индуцированным только R848, сильным агонистом TLR7/8. Неожиданно,
авторы обнаружили большое количество IFN-α в супернатантах после совместного
культивирования в результате воздействия пДК на опухолевые клетки, зараженные MV (в 20-
40 раз больше, чем то, что наблюдали в ответ на MV в присутствии IL-3 или R848). Эти
высокие количества IFN-α вырабатывали пДК, поскольку опухолевые клетки не
продуцировали IFN-α или продуцировали в очень маленьком количестве (в количествах,
измерявшихся в пг/мл) после заражения MV. Опухолевые клетки А549 или М18, облученные
УФ, не индуцировали синтез IFN-α клетками пДК. Данные результаты показывают, что
опухолевые клетки, зараженные MV, способны запускать продуцирование IFN-α в больших
количествах клетками пДК, что значительно больше, чем количество, синтезированное пДК в
результате воздействия MV в присутствии IL-3 или R848.
Авторы ранее показали, что через 3 дня после заражения линии опухолевых клеток
Meso13, было продуцировано большое количество вируса, достигая значения в 1х108
TCID50/мл соответствующее значению MOI более чем 100 от начальной дозы вируса в 1х106
TCID50/мл, соответствующее MOI=1 (Gauvrit, A. et al., Cancer Res., 2008, 68: 4882-4892).
Таким образом, является вероятным, что огромное количество IFN-α, продуцированное
клетками пДК в ответ на опухолевые клетки, зараженные MV, явилось результатом
интенсивной репликации MV в данных опухолевых клетках. Для проверки данной гипотезы,
пДК культивировали при увеличении MOI в диапазоне от 1 до 50, в присутствии или
отсутствии IL-3 (Фигура 16В). В присутствии IL-3, авторы наблюдали, что выработка IFN-α
клетками пДК увеличивалась вместе с MOI. И наоборот, пДК не продуцировали IFN-α в
отсутствии IL-3, за исключением самой высокой MOI (MOI=50). Данные результаты
предполагают, что уровень выработки IFN-α клетками пДК зависит от количества MV и
присутствия либо IL-3 или других сигналов выживаемости, объясняя огромное количество
синтезированного IFN-α в ответ на высокий титр вируса после заражения опухолевых клеток.
Поскольку MV и опухолевые клетки, зараженные MV, содержат вирусную
одноцепочечную РНК, вероятно, что синтез IFN-α клетками пДК происходит в основном за
счет запуска TLR-7. Следовательно, проводили ингибирование TLR7. Применяли
специфическую иммунорегуляторную последовательность ДНК (IRS), ингибирующую
экспрессию IFN-α опосредованную TLR-7 (IRS661) (Barrat, F. J. et al., J Exp Med., 2005, 202:
1131-1139). Авторы показали, что добавление IRS661 ингибирует синтез IFN-α клетками
пДК, культивированными в присутствии MV и IL-3 (Фигура 16С). Аналогичное
ингибирование IFN-α также наблюдали при добавлении IRS661 к пДК, воздействующих на
опухолевые клетки, зараженные MV. В качестве контроля было показано, что IRS661 не
ингибировал СpG-A-индуцированный синтез IFN-α клетками пДК, который является TLR9-
зависимым. В целом, данные результаты демонстрируют, что синтез IFN-α, индуцированный
MV или клетками, зараженными MV, является TLR7-зависимым.
Пример 12
пДК способны к кросс-представлению опухоль-специфического антигена из опухолевых
клеток, зараженных MV.
Сообщалось о способности человеческих пДК к кросс-представлению вирусных
антигенов (Di Pucchio, T. et al., Nat Immunol., 2008, 9: 551-557; Hoeffel, G. et al. Immunity,
2007, 27:481-492; Lui, G. et al., PLos One, 2009, 4: e7111), но кросс-представление опухоль-
специфического антигена (TAAs) еще не было описано. Авторы задались вопросом,
действительно ли человеческие пДК, воздействующие на опухолевые клетки, зараженные
MV, способны к кросс-представлению человеческого TAA, спонтанно экспрессированного
опухолевыми клетками. Авторы с помощью ПЦР в реальном времени показали, что линия
клеток меланомы M18 HLA-A*0201neg экспрессировала антиген рака яичка, NYESO-1, тогда
как линия клеток аденокарциномы легких А549 нет (Фигура 17А).
Для определения действительно ли пДК HLA-A*0201neg способны к кросс-
представлению данного TAA после воздействия на линию опухолевых клеток М18 HLAA*
0201neg/NYESO-1pos, зараженных MV или облученных УФ, использовали CD8+ T-
клеточный клон, М117.167, который является специфичным для комплексов HLAA*
0201/NYESO-1(157-165) (Фигура 17В-D). Диаграмма данного эксперимента показана в
Фигуре 18. T-клеточный клон М117.167 не продуцировал IFN-γ, в присутствии или без IL-3
пДК, но был активирован в присутствии пДК, импульсно меченых пептидами NYESO-1 [157-
161] (Фигура 17В). Клон был активирован, как только 0,1 мкМ пептида внесли в пДК (16,3%
IFN-γ+клетки) и более интенсивно был активирован клетками пДК, импульсно мечеными 1
мкМ пептида (77,5%). В присутствии пДК, культивированных вместе с опухолевыми
клетками М18, зараженными MV, было активировано 11,5% популяции клона, тогда как клон
не продуцировал IFN-γ в ответ на пДК, культивированные вместе с опухолевыми клетками
М18, облученными УФ (Фигура 17В). В ответ на пДК, культивированные совместно с MV-
инфицированной М18, клон продуцировал IFN-γ в количестве сравнимом с количеством,
отмеченным в ответ на пДК, импульсно меченые 0,1мкМ пептида NYESO-1(157-165).
В качестве контроля, авторам не удалось обнаружить активацию T-клеточный клона
М117.167 в ответ на линию опухолевых клеток М18, зараженных MV или облученных УФ
(Фигура 17С). Данный результат был ожидаемым, так как линия опухолевых клеток М18
являлась HLA-A*0201neg, следовательно, не может непосредственно представлять клону
пептид NYESO-1(157-165). Это показывает, что синтез IFN-γ клоном в ответ на HLAA*
0201pos пДК, культивированные совместно с опухолевыми клетками М18, зараженными
MV, происходит благодаря кросс-представлению. Авторы также не наблюдали синтез IFN-γ в
ответ на пДК, культивированные совместно с опухолевыми клетками NYESO-1neg А549,
зараженными MV. В данном представленном эксперименте, клон, продуцировал синтез IFN-γ
в ответ на пДК, культивированные совместно с М18, зараженными MV (6,5% IFN-γ+ клетки),
скорость продуцирования близка к скорости, отмеченной в ответ на пДК, импульсно меченые
0,1мкМ пептида NYESO-1(157-165) (10,8% IFN-γ+ клетки).
В целом, данные результаты показывают, что пДК способны к кросс-представлению
опухолевого антигена, в частности NYESO-1, из опухолевых клеток, зараженных MV, но не
способен к кросс-представлению из клеток, облученных УФ. Таким образом, антиопухолевая
виротерапия, основанная на MV, должна быть в состоянии «нанимать» пДК в
антиопухолевом иммунном ответе посредством активации их способности продуцировать
большие количества IFN-α и кросс-представлять TAA из опухолевых клеток, зараженных
MV, опухоль-специфическим CD8+ T лимфоцитам.
Авторы охарактеризовали, in vitro, последовательность антиопухолевой терапии,
основанной на MV, на функциях человеческих пДК. В первую очередь, они показали, что
пДК не были чувствительны к заражению MV, несмотря на экспрессию CD46. Однако, пДК
смогли обнаружить вирус посредством синтеза IFN-α в ответ на большое количество вируса в
отсутствии сигналов выживаемости, и в ответ на небольшое количество вируса при
добавлении в культуру сигналов выживаемости, в частности IL-3. Кроме того, когда пДК
культивировали совместно с опухолевыми клетками, зараженными MV, пДК претерпели
созревание, отличающееся индукцией экспрессии CD83 и интенсивным синтезом IFN-α, с
едва увеличенной экспрессией костимуляторных молекул. И наоборот, пДК,
культивированные совместно с опухолевыми клетками, облученными УФ, сохранили
незрелый фенотип аналогичный отмеченному при совместном культивировании только с IL-
3. Затем авторы обнаружили TLR7 как рецептор пДК, ответственный за их активацию,
вероятно из-за присутствия одноцепочечной вирусной РНК в эндоцитозном канале пДК
после интернализации фрагментов опухолевых клеток, зараженных MV. В заключение,
используя HLA-A*0201/NYESO-1(157-165) -специфический CD8+ T-клеточный клон, авторы
показали, что HLA-A*0201+ пДК способны к кросс-представлению данного опухоль-
специфического антигена (ТАА) из NYESO-1+ HLA-A*0201neg опухолевых клеток,
зараженных MV, но не способны из клеток, облученных УФ. В первый раз авторы показали
способность человеческих пДК к кросс-представлению ТАА из мертвых опухолевых клеток
CD8+ Т клеткам. В целом, данные результаты предполагают, что антиопухолевая
виротерапия, основанная на MV, в дополнение к непосредственному лизису зараженных
опухолевых клеток, смогла рекрутировать пДК в антиопухолевом иммунном ответе,
активировать их способность продуцировать большое количество IFN типа I и кросс-
представлять ТАА.
Во-первых, авторы показали, что человеческие пДК, воздействующие на MV при
MOI=1 in vitro, не претерпевают созревание без IL-3. При таком условии, без сигналов
выживаемости, пДК подвергались апоптозу и не могли достигнуть MV в эндосомальном
канале, чтобы вступить в процесс созревания посредством лигирования вирусной РНК к
TLR7. Когда пДК подвергали воздействию MV в присутствии IL-3, они выжили и наблюдали
созревание (низкое продуцирование IFN-α и индукция экспреесии СD83). Авторы наблюдали
активацию пДК вирусом кори в отсутствии IL-3, только когда они использовали большое
количество MV (MOI=50). При таких высоких концентрациях MV, авторы предположили,
что достаточно MV достигло эндосомального канала пДК, чтобы обеспечить сигнал
выживаемости/созревания, перед тем как был запущен их апоптоз. Следовательно, когда пДК
подвергли воздействию MV в присутствии IL-3, пДК выжили, интернализовали MV и
позволили запустить TLR7 посредством вирусной одноцепочечной РНК. Когда пДК
подвергли воздействию MV в отсутствии IL-3, они претерпевали апоптоз, за исключением
случаев, когда достаточно MV достигало эндосомального канала для активации и созревания
пДК. Данные результаты объясняют противоречивые публикации в литературе, из-за
различий в экспериментальных параметрах. Действительно, авторы получили аналогичные
результаты с Schlender et al., который сообщил, что небольшое количество MV Шварца не
смогло индуцировать IFN-α клетками пДК, культивированными в отсутствии IL-3 (Schlender,
J. et al., J Virol., 2005, 79:5507-5515), и Duhen et al., который заявил, что MV Шварца
индуцировал большое количество IFN-α клетками пДК в присутствии IL-3 (Duhen et al., Virus
Res., 2010, 152:115-125). Однако, авторы не установили, что MV Шварца ингибировал синтез
IFN-α клетками пДК (31), поскольку пДК продуцировал IFN-α в присутствии IL-3. В
заключение, обе группы описали окрашивание пДК моноклональным антителом к
гемагглютинину (Н) MV, но интерпретировали результат по-разному. Одна группа заявила,
что пДК были заражены и усилены вирусом (Schlender, J. et al., J Virol., 2005, 79:5507-5515),
в то время как другая группа пришла к заключению, что несмотря на окрашивание белка Н в
пДК, репликация MV была низкой (Duhen et al., Virus Res., 2010, 152:115-125). Результаты
настоящего изобретения подтверждают этот последний вывод, поскольку авторы не
наблюдали эффективного заражения, используя MV-eGFP, даже при высокой MOI, ни в
отсутствии ни присутствии IL-3.
Авторы также показали, что в присутствии MV или опухолевых клеток, зараженных
MV, пДК претерпевают созревание, характеризующееся позитивной регуляцией экспрессии
молекулы CD83 на поверхности клетки. В присутствии MV или опухолевых клеток,
зараженных MV, пДК продуцировали большее количество IFN-α в ответ на высокую
вирусную нагрузку, чем пДК, стимулированные только R848. Однако, эти клетки не
экспрессировали в таком же количестве костимуляторные молекулы CD40 и CD86.
Следовательно, данное формирование фенотипа напомнило формирование фенотипа,
индуцированного ВИЧ-инфекцией (Fonteneau, J.F. et al., J Virol., 2004, 78:5223-5232; O’Brien,
M. et al., J Clin Invest., 2011, 121:1088-1101), которая активировала пДК посредством TLR7,
аналогично с MV (Beignon, A.S. et al., J Clin Invest., 2005, 115:3265-3275). Действительно,
стало ясно, что в зависимости от природы использованного агониста TLR7, два основных
пути активации могли быть запущены в человеческих пДК. Данная дихотомия впервые была
сообщена Kerkmann et al., который показал, что два агониста TLR9, CpG-A и CpG-B,
активировали созревание пДК, используя два разных пути (Kerkmann, M. et al., J Immunol.
2203, 170:4465-4474). Совсем недавно, аналогичную дихотомию наблюдали для агониста
TLR7 (O'Brien, M. et al., J Clin Invest., 2011, 121:1088-1101). Действительно, ВИЧ вел себя
аналогично CpG-A, запуская TLR7 и сигнальный путь IRF7 в ранней эндосоме пДК, и
индуцируя интенсивный синтеза IFN-α. Авторы показали, что созревание, индуцированное
посредством MV+IL-3 или клеток, зараженных MV, аналогично активации, индуцированной
ВИЧ, что подтверждает запуск TLR7 в ранней эндосоме посредством одноцепочечной РНК
вируса кори. Был показан данный путь активации в ранней эндосоме, сходный с кросс-
представлением антигена, выраженный клетками, зараженными вирусом, в виде кросс-
представления вирусных антигенов из инфицированных клеток (Hoeffel, G. et al., Immunity,
2007, 27:481-492) и кросс-представление ТАА из клеток, зараженных MV, как описано ранее.
И наоборот, Schnurr et al. cообщил, что in vitro пДК, противоположно миелоидным ДК, не
были способны к кросс-представлению ТАА из одного полноразмерного белка или в качестве
формы иммунного комплекса (Schnurr, M. et al., Blood, 2005, 105:2465-2472). Однако, данные
авторы применяли растворимый белок и не использовали NYESO-1-экспрессирующие
опухолевые клетки как источник антигена. Кросс-представление антигена клетками пДК in
vivo было также спорным. Salio et al. сообщили, что мышиные пДК, стимулированные
посредством CpG, были не способны к кросс-представлению антигенов, тогда как они могли
устанавливать T-клеточный ответ против эндогенных антигенов (Salio, M. et al., J Exp Med.,
2001, 199:567-579). Mouries et al. показали, in vivo и in vitro, также на мышиной модели, что
растворимый белок OVA и агонист TLR (CpG или R848) активировали пДК кросс-
представлять белок OVA специфическим СD8+ Т-клеткам (Mouries, J. et al., Blood, 2008,
112:3713-3722). Аналогично, представление и кросс-представление растворимого пептида
OVA или целого белка, в результате стимуляции TLR9 посредством CpG или инфицирования
вирусом гриппа, содержащего эпитопы OVA, была недавно подтверждена, in vitro, Kool et al.
(Kool et al., J Leukos Biol., 2011, 90:1177-1190). В заключение, Lui et al. сообщили, что
внутриопухолевая инъекция CpG-A-стимулированных пДК мышам, несущим В16 меланому,
индуцировала кросс-представлять опухолевого антигена, но данное кросс-примирование
было выполнено CD11c+ ДК, а не клетками пДК (Lui et al., J Clin Invest., 2008, 118:1165-
1175). Авторы показали, что, in vitro, человеческие пДК, воздействующие на опухолевые
клетки, зараженные MV, были способны кросс-представлять NYESO-1 CD8+ T-клеточному
клону, специфичного к данному ТАА. Авторы продемонстрировали, что опухолевые клетки,
зараженные MV, погибли и затем были фагоцитированы клетками пДК. Данные клетки,
зараженные MV, были способны активировать пДК без добавления адъювантов или агониста
TLR. Эффективность противоопухолевой виротерапии, основанной на MV,
продемонстрировали in vivo в различных моделях ксенотрансплантата опухоли человека в
иммунодефицитных мышах (Peng, K. W. et al., Cancer Res., 2002. 62:4656-4662; McDonald,
C.J. et al., Breast Cancer Res Treat., 2006, 99:177-184; Blechacz, B. et al., Hepatology, 2006,
44:1465-1477). Первые клинические испытания виротерапии, основанной на MV, показали
обнадеживающие результаты (Heinzerling, L. et. al., Blood. 2005. 106:2287-2294; Galanis, E. et
al., Cancer Res., 2010, 70:875-882). Эффективность виротерапии, основанной на MV, скорее
всего связана с лизисом вирусом опухолевых клеток. Однако, также эффективность
виротерапии зависит от способности опухолевых клеток, зараженных MV, активировать
клетки иммунной системы, в особенности пДК. Действительно, активация пДК агонистом
TLR7 в опухоль-несущих мышах показала индукцию противоопухолевого иммунного ответа
и регрессию опухоли (Drobits, B; et al., J Clin Invest., 2012, 122:575-585; Liu. C. et al., J Clin
Invest., 2008, 118:1165-1175; Palamara, F. et al., J Immunol., 2004, 173:3051-3061). Liu et al.
показали, что мышиные пДК, стимулированные агонистом TLR9, индуцировали активацию
NK-клеток и рекрутинг опухолью, запуская кросс-представление опухолевого антигена
посредством CD11c+ДК (Liu. C. et al., J Clin Invest., 2008, 118:1165-1175). Drobits et al.
показали, что актуальное лечение имиквимодом опухоли меланомы у мышей с агонистом
TLR7, индуцировало активацию и рекрутинг пДК в опухоль и вызвало регрессию опухоли
(Drobits, B; et al., J Clin Invest., 2012, 122:575-585). Drobits et al. продемонстрировали, что
пДК приобрели цитотоксическую активность против опухолевых клеток посредством
секреции TRAIL и гранзима В, в IFNAR1-зависимом механизме. Секреция IFN-α клетками
пДК не только индуцировала противоопухолевую цитотоксическую активность в пДК
посредством аутокринного цикла, но и смогла также воздействовать непосредственно на
опухолевые клетки, чтобы индуцировать апоптоз (Thyrell, L. et al., Oncogene, 2002, 21:1251-
1262). IFN типа I также сыграл роль в активации NK и был указан в мышиной модели NK-
зависимого отторжении опухоли (Swann, J.B. et al., J Immunol., 2007, 178:7540-7549). В
заключение необходимо отметить, что данные NK-клетки вероятно тоже участвуют в
ингибировании противоопухолевого ответа путем стимуляции миелоидныхДК, поскольку в
IFNAR1- и STAT1-зависимых мышах не смог развиться противоопухолевый Т-клеточный
ответ (Diamond, M.S. et al., J Exp Med., 2011, 208:1989-2003; Fuertes, M.B. et al., J Exp Med.,
2011, 208:2005-2016). Таким образом, авторы показали, что опухолевые клетки, зараженные
MV, индуцировали большое количество IFN-α посредством пДК, что может быть
благоприятным для развития многоклеточных подмножеств, участвующих в
противоопухолевом иммунном ответе. Более того, другие известные онколитические вирусы
для активации пДК применяют в клинических испытаниях противоопухолевой виротерапии,
в частности коровью оспу (Kim, J.H. et al., Mol Ther., 2006, 14:361-370), вирус простого
герпеса (Kaufman, H.L. et al., Future Oncol., 2010, 6:941-949) и аденовирус (Ramesh, N. et al.,
Clin Cancer Res., 2006, 12:305-313). Опухолевые клетки, зараженные данными вирусами,
также могут индуцировать выработку IFN-α и кросс-представление опухолевого антигена
посредством пДК.
Противоопухолевая виротерапия, основанная на MV, является перспективным
подходом для лечения рака благодаря онколитической активности вируса. Более того, авторы
показали, что опухолевые клетки, зараженные MV, активировали созревание и способность к
кросс-представлению опухолевого антигена человеческих пДК. Следовательно,
противоопухолевая виротерапия, основанная на MV, может представлять собой интересный
подход к рекрутированию пДК в противоопухолевом иммунном ответе.
Пример 13
Сравнительные исследования между немодифицированным MV и MV-deltaC, с
использованием различных линий клеток меланомы
Материалы и Методы
Культура клеток. Испытанные линии опухолевых клеток представляли собой линии
клеток, полученные из меланомы: М6, М17, М117, М88 и М113. Клетки культивировали в
среде RPMI 1640, с добавлением 10% эмбриональной телячьей сыворотки (FCS), 1% L-
глютамина и 1% пенициллина G/стрептомицина. Клетки культивировали при исходной
концентрации 3.106 клеток в колбах 75см2 и поддерживали при 37°C в 5% CO2. Клетки
ежедневно проверяли и получали отрицательное результаты на заражение Mycoplasma.
Заражение опухолевых клеток. Перед инфицированием, клетки засевали на 24 часа в
12-луночные планшеты, в количестве 200.103 клеток на лунку в 1 мл среды с 10% FCS RPMI,
для того, чтобы позволить клеткам прикрепиться. Для заражения опухолевых клеток
применяли два вируса: (I) MV-eGFP: рекомбинантный живой ослабленный вирус кори (MV
Шварца), содержащий ген, кодирующий флюоресцентный белок GFP для исследования
заражения опухолевых клеток; (II) MV-deltaC-eGFP: рекомбинантный модифицированный
вакцинный штамм MV, содержащий ген, кодирующий GFP.
Эффективность заражения была разной для двух вирусов: немодифицированного MV и
MV-deltaC, а также между разными линиями клеток меланомы. Авторы наблюдали, что MVdeltaC
способен инфицировать опухолевые клетки более эффективно, чем
немодифицированный MV через 24 часа после заражения (Фигура 19 и 20).
В дополнение, было показано, что опухолевые клетки экспрессировали больше
«сигналов опасности» на мембране, таких как HSP 70 и CRT, в результате заражения MVdeltaC
по сравнению с немодифицированным MV (Фигура 21). Данный факт предполагает
более эффективную индукцию иммунного ответа, через созревание и активацию дендритных
клеток, после того как они фагоцитируют апоптотические клетки.
In vivo введение вакцины MV-deltaC внутрь опухоли меланомы подтвердило усиленный
эффект MV-deltaC индуцировать быстрый ответ по сравнению с контролем (Фигура 22).
В заключение, вакцинный штамм MV-deltaC показал интересные и более хорошие
проапоптотические свойства по сравнению с обычным вакцинным штаммом
немодифицированного MV.
Пример 14
Сравнение индукции гибели клеток в раковых и нераковых клетках посредством
немодифицированного MV и MV-deltaC
Для того, чтобы оценить действительно ли более сильная индукция гибели клеток,
посредством MV-deltaC по сравнению с немодифицированным MV, была специфична к
раковым клеткам, авторы сравнили их активность на линиях клеток рака человека
(аденокарциномы легких человека А549 и рака шейки матки Hela), и на мертвых нераковых
клетках (человеческие эмбриональные почечные клетки HEK293 и клетки почки
африканской зеленой мартышки Vero). Линии А549 и Hela обычно используют как
прототипы раковых клеток человека. Напротив, в качестве экспериментально
трансформированных с Ad5, клетки HEK 293 не являются раковыми. Родословная линии
клеток Vero является непрерывной и анеуплоидной, т.е. может быть реплицирована в течение
многих циклов деления и при этом не становится стареющей.
Клетки (40 000 на лунку в 96-луночных планшетах) культивировали совместно с
вирусами MV-deltaC и немодифицированным MV при разной MOI (0,1, 1, 5, 10). Заражение
выполняли на неадгезивных клетках в DMEM (0,2 мл). После 0, 24, 46 и 68 часов
культивирования, количество живых клеток определяли с помощью реагента CellTiter-GLO
(Promega). Данный анализ на основе люциферазы, оценивает посредством количественного
опеределения АТФ количество метаболически активных клеток в культуральных ячейках.
Данный анализ подтвердил, что MV-deltaC индуцировал более интенсивную и раннюю
гибель клеток, чем немодифицированный MV, на обоих человеческих раковых клетках А549
и Hela, даже при низкой MOI (Фигура 23А). Следовательно,показана лучшая онколитическая
активность MV-deltaC на всех раковых клетках: мезотелиомы, меланомы, легких и шейки
матки. Напротив, никакую разницу не заметили между двумя вирусамив отношении
индукции гибели клеток на клетках Vero, и не обнаружили гибель клеток на клетках HEK 293
через 68 часов после заражения (Фигура 23В). Данный факт предполагает, что механизм с
помощью которого MV-deltaC ускорил гибель клеток по сравнению с немодифицированным
MV, или возобновил процессы старения, был специфичным к человеческим опухолевым
клеткам. Наблюдение, что клетки Vero не были чувствительны к MV-deltaC, по сравнению с
немодифицированным MV, было ключевым, поскольку MV обычно производят на данной
линии клеток.
Кинетику вирусного роста обоих вирусов одновременно оценивали на одинаковых
клеточных линиях (Фигура 24). Клетки Vero, HEK293, Hela и A549 инфицировали при MOI
равной 1 немодифицированным MV и MV-deltaC в 35 мм культуральных лунках. Титры
вируса определяли как TCID50 в разные моменты времени после заражения. Высокую
скорость репликации наблюдали у обоих вирусов в нераковых клетках Vero и HEK293.
Напротив, титры репликации были ниже в раковых клетках Hela и А549, а репликация MV-deltaC была очень низкой в клетках А549. Данный факт показал, что индукция гибели клеток
в раковых клетках привела к более низкому продуцированию вирусного потомства, что было
преимуществом для безопасности онколитического вируса.
Реферат
Изобретение относится к генной инженерии. Описано применение патогенного вируса кори (MV) из живого ослабленного штамма вируса кори (MV), в котором нокаутирован ген, кодирующий вирусный акцессорный белок С (MV-deltaC), в лечении агрессивной злокачественной опухоли или агрессивного рака при введении индивидууму, у которого диагностирована такая злокачественная опухоль или раковое заболевание. Описан способ получения вакцинных плазмацитоидных дендритных клеток (пДК) для лечения агрессивной злокачественной опухоли или агрессивного рака у индивидуума, у которого диагностирована такая злокачественная опухоль или раковое заболевание, включающий следующие стадии: in vitro заражение злокачественной опухоли или раковых клеток, предварительно отобранных у указанного индивидуума, патогенным вирусом кори (MV), в котором нокаутирован ген, кодирующий вирусный акцессорный белок С (MV-deltaC), для получения клеточного лизата; приведение пДК в контакт с клеточным лизатом для получения вакцинных пДК; выделение загруженных пДК. Изобретение расширяет арсенал средств лечения кори. 5 н. и 10 з.п. ф-лы, 24 ил., 14 пр.
Комментарии